User login
FDA approves subcutaneous tocilizumab for polyarticular JIA
in patients aged 2 years and older, according to a statement released May 14 by the drug’s manufacturer, Genentech.
While a intravenous formulation of the treatment was approved in 2013, this new delivery method may help make this treatment more accessible to the approximately 30 in every 100,000 children affected by PJIA, according to the release.
Doses were determined based on weight. Patients under 30 kg received 162 mg of tocilizumab every 3 weeks, while those 30 kg and over received 162 mg tocilizumab every 2 weeks.
Overall, safety of the subcutaneous delivery method was consistent with the IV study, as was the efficacy of the drug, the company said. A total of 28.8% of patients reported injection-site reactions – all moderate – and 15.4% reported neutrophil counts below 1 x 109 per liter.Tocilizumab can be taken either by itself or with methotrexate.
in patients aged 2 years and older, according to a statement released May 14 by the drug’s manufacturer, Genentech.
While a intravenous formulation of the treatment was approved in 2013, this new delivery method may help make this treatment more accessible to the approximately 30 in every 100,000 children affected by PJIA, according to the release.
Doses were determined based on weight. Patients under 30 kg received 162 mg of tocilizumab every 3 weeks, while those 30 kg and over received 162 mg tocilizumab every 2 weeks.
Overall, safety of the subcutaneous delivery method was consistent with the IV study, as was the efficacy of the drug, the company said. A total of 28.8% of patients reported injection-site reactions – all moderate – and 15.4% reported neutrophil counts below 1 x 109 per liter.Tocilizumab can be taken either by itself or with methotrexate.
in patients aged 2 years and older, according to a statement released May 14 by the drug’s manufacturer, Genentech.
While a intravenous formulation of the treatment was approved in 2013, this new delivery method may help make this treatment more accessible to the approximately 30 in every 100,000 children affected by PJIA, according to the release.
Doses were determined based on weight. Patients under 30 kg received 162 mg of tocilizumab every 3 weeks, while those 30 kg and over received 162 mg tocilizumab every 2 weeks.
Overall, safety of the subcutaneous delivery method was consistent with the IV study, as was the efficacy of the drug, the company said. A total of 28.8% of patients reported injection-site reactions – all moderate – and 15.4% reported neutrophil counts below 1 x 109 per liter.Tocilizumab can be taken either by itself or with methotrexate.
VIDEO: The effect of removing pregnancy drug category labeling
AUSTIN, TEX. – In a randomized survey, the Food and Drug Administration’s previous letter-category labeling of drugs for pregnant women made prescribers more likely to prescribe appropriate medication than the new labeling standards without the letters.
The FDA removed the letter categories A, B, C, D, and X in 2014 in the belief “that a narrative structure for pregnancy labeling is best able to capture and convey the potential risks of drug exposure based on animal or human data, or both,” as stated in the FDA ruling.
“The [old] FDA categories are actually based upon the evidence related to clinical trials or animal trials and known risks to the fetus or the mother. The categorizations really reflect the evidence that we have or the absence of evidence as to whether medications can be safely used during pregnancy,” Dr. Robinson explained in a video interview. But letters can be perceived as overall “grades,” even though they aren’t.
The researchers sought to evaluate the effect of the letters’ removal by surveying doctors at two centers in New York City and two annual specialty meetings from October 2015 to May 2016.
The survey “included demographic information, followed by four clinical vignettes. Each vignette described a pregnant woman and presented an indication for prescribing a particular drug which was FDA approved. Each vignette was followed by detailed drug information as found in the FDA-approved package insert with the new [Pregnancy and Lactation Labeling Rule] content and formatting,” Dr. Robinson said at the meeting.
The 162 survey respondents estimated their likelihood of prescribing given the information in the vignette. The respondents were randomized to see the letter category or not. Category X (positive evidence of risk that clearly outweighs potential benefits) was not included. For all four remaining categories, the respondents who were shown the letter category were more likely to prescribe. Category B was significantly affected in a mixed linear model, and both categories B and C were significantly affected in a multivariate model.
“The new system of revised labeling may be more ambiguous and present new challenges for clinical decision making that could result in decreased access to medications for pregnant women,” Dr. Robinson said.
Dr. Robinson and her coauthors reported no relevant disclosures.
SOURCE: Robinson A et al. ACOG 2018. Abstract OP5.
AUSTIN, TEX. – In a randomized survey, the Food and Drug Administration’s previous letter-category labeling of drugs for pregnant women made prescribers more likely to prescribe appropriate medication than the new labeling standards without the letters.
The FDA removed the letter categories A, B, C, D, and X in 2014 in the belief “that a narrative structure for pregnancy labeling is best able to capture and convey the potential risks of drug exposure based on animal or human data, or both,” as stated in the FDA ruling.
“The [old] FDA categories are actually based upon the evidence related to clinical trials or animal trials and known risks to the fetus or the mother. The categorizations really reflect the evidence that we have or the absence of evidence as to whether medications can be safely used during pregnancy,” Dr. Robinson explained in a video interview. But letters can be perceived as overall “grades,” even though they aren’t.
The researchers sought to evaluate the effect of the letters’ removal by surveying doctors at two centers in New York City and two annual specialty meetings from October 2015 to May 2016.
The survey “included demographic information, followed by four clinical vignettes. Each vignette described a pregnant woman and presented an indication for prescribing a particular drug which was FDA approved. Each vignette was followed by detailed drug information as found in the FDA-approved package insert with the new [Pregnancy and Lactation Labeling Rule] content and formatting,” Dr. Robinson said at the meeting.
The 162 survey respondents estimated their likelihood of prescribing given the information in the vignette. The respondents were randomized to see the letter category or not. Category X (positive evidence of risk that clearly outweighs potential benefits) was not included. For all four remaining categories, the respondents who were shown the letter category were more likely to prescribe. Category B was significantly affected in a mixed linear model, and both categories B and C were significantly affected in a multivariate model.
“The new system of revised labeling may be more ambiguous and present new challenges for clinical decision making that could result in decreased access to medications for pregnant women,” Dr. Robinson said.
Dr. Robinson and her coauthors reported no relevant disclosures.
SOURCE: Robinson A et al. ACOG 2018. Abstract OP5.
AUSTIN, TEX. – In a randomized survey, the Food and Drug Administration’s previous letter-category labeling of drugs for pregnant women made prescribers more likely to prescribe appropriate medication than the new labeling standards without the letters.
The FDA removed the letter categories A, B, C, D, and X in 2014 in the belief “that a narrative structure for pregnancy labeling is best able to capture and convey the potential risks of drug exposure based on animal or human data, or both,” as stated in the FDA ruling.
“The [old] FDA categories are actually based upon the evidence related to clinical trials or animal trials and known risks to the fetus or the mother. The categorizations really reflect the evidence that we have or the absence of evidence as to whether medications can be safely used during pregnancy,” Dr. Robinson explained in a video interview. But letters can be perceived as overall “grades,” even though they aren’t.
The researchers sought to evaluate the effect of the letters’ removal by surveying doctors at two centers in New York City and two annual specialty meetings from October 2015 to May 2016.
The survey “included demographic information, followed by four clinical vignettes. Each vignette described a pregnant woman and presented an indication for prescribing a particular drug which was FDA approved. Each vignette was followed by detailed drug information as found in the FDA-approved package insert with the new [Pregnancy and Lactation Labeling Rule] content and formatting,” Dr. Robinson said at the meeting.
The 162 survey respondents estimated their likelihood of prescribing given the information in the vignette. The respondents were randomized to see the letter category or not. Category X (positive evidence of risk that clearly outweighs potential benefits) was not included. For all four remaining categories, the respondents who were shown the letter category were more likely to prescribe. Category B was significantly affected in a mixed linear model, and both categories B and C were significantly affected in a multivariate model.
“The new system of revised labeling may be more ambiguous and present new challenges for clinical decision making that could result in decreased access to medications for pregnant women,” Dr. Robinson said.
Dr. Robinson and her coauthors reported no relevant disclosures.
SOURCE: Robinson A et al. ACOG 2018. Abstract OP5.
REPORTING FROM ACOG 2018

FDA expands fingolimod's indications to treat relapsing MS in pediatric patients
Pediatric patients with
The Food and Drug Administration approved on May 11 an expanded indication of the drug to allow it to be used to treat relapsing MS in children and adolescents age 10 and older. Gilenya was first approved by FDA to treat adults with relapsing MS in 2010.
“For the first time, we have an FDA-approved treatment specifically for children and adolescents with multiple sclerosis,” Billy Dunn, MD, director of the Division of Neurology Products in the FDA Center for Drug Evaluation and Research, said in a statement. “This represents an important and needed advance in the care of pediatric patients with multiple sclerosis.”
Side effects for fingolimod in pediatric trial participants were similar to those experienced in adults, the most common of which include headache, liver enzyme elevation, diarrhea, cough, flu, sinusitis, back pain, abdominal pain, and pain in extremities. The drug must be dispensed with a medication guide that describes the product’s more serious risks.
The FDA, which granted the product a priority review and breakthrough designation for this indication, noted that 2%-5% of people with MS have symptoms onset before age 18 and suggested that 8,000-10,000 children and adolescents in the United States suffer from the disease.
The clinical trial evaluating the drug included 214 patients aged 10-17 years and compared fingolimod with interferon beta-1a. In that study, the FDA stated that 86% of patients receiving fingolimod remained relapse-free after 24 months of treatment, compared with 46% of those treated with interferon beta-1a.
Pediatric patients with
The Food and Drug Administration approved on May 11 an expanded indication of the drug to allow it to be used to treat relapsing MS in children and adolescents age 10 and older. Gilenya was first approved by FDA to treat adults with relapsing MS in 2010.
“For the first time, we have an FDA-approved treatment specifically for children and adolescents with multiple sclerosis,” Billy Dunn, MD, director of the Division of Neurology Products in the FDA Center for Drug Evaluation and Research, said in a statement. “This represents an important and needed advance in the care of pediatric patients with multiple sclerosis.”
Side effects for fingolimod in pediatric trial participants were similar to those experienced in adults, the most common of which include headache, liver enzyme elevation, diarrhea, cough, flu, sinusitis, back pain, abdominal pain, and pain in extremities. The drug must be dispensed with a medication guide that describes the product’s more serious risks.
The FDA, which granted the product a priority review and breakthrough designation for this indication, noted that 2%-5% of people with MS have symptoms onset before age 18 and suggested that 8,000-10,000 children and adolescents in the United States suffer from the disease.
The clinical trial evaluating the drug included 214 patients aged 10-17 years and compared fingolimod with interferon beta-1a. In that study, the FDA stated that 86% of patients receiving fingolimod remained relapse-free after 24 months of treatment, compared with 46% of those treated with interferon beta-1a.
Pediatric patients with
The Food and Drug Administration approved on May 11 an expanded indication of the drug to allow it to be used to treat relapsing MS in children and adolescents age 10 and older. Gilenya was first approved by FDA to treat adults with relapsing MS in 2010.
“For the first time, we have an FDA-approved treatment specifically for children and adolescents with multiple sclerosis,” Billy Dunn, MD, director of the Division of Neurology Products in the FDA Center for Drug Evaluation and Research, said in a statement. “This represents an important and needed advance in the care of pediatric patients with multiple sclerosis.”
Side effects for fingolimod in pediatric trial participants were similar to those experienced in adults, the most common of which include headache, liver enzyme elevation, diarrhea, cough, flu, sinusitis, back pain, abdominal pain, and pain in extremities. The drug must be dispensed with a medication guide that describes the product’s more serious risks.
The FDA, which granted the product a priority review and breakthrough designation for this indication, noted that 2%-5% of people with MS have symptoms onset before age 18 and suggested that 8,000-10,000 children and adolescents in the United States suffer from the disease.
The clinical trial evaluating the drug included 214 patients aged 10-17 years and compared fingolimod with interferon beta-1a. In that study, the FDA stated that 86% of patients receiving fingolimod remained relapse-free after 24 months of treatment, compared with 46% of those treated with interferon beta-1a.
FDA advisory committee recommends volanesorsen for rare triglyceride disorder
SILVER SPRING, Md. – A novel drug to treat a rare metabolic disorder was recommended for approval by the Endocrinologic and Metabolic Drugs Advisory Committee in a May 10 meeting.
Advisors voted 12 to 8 to recommend volanesorsen, a novel antisense drug, as an adjunct to diet for the treatment of familial chylomicronemia syndrome (FCS).
“The sponsor provided compelling evidence that the drug lowers triglyceride levels, substantially,” said panelist Jean-Pierre Raufman, MD, of the University of Maryland. I think that there is clearly a group of patients with this disease that can benefit from this agent.”

FCS is an extremely rare genetic disorder characterized by severe hypertriglyceridemia and recurrent pancreatitis caused by a deficiency in lipoprotein lipase. Pancreatitis in the setting of hypertriglyceridemia is particularly severe, often leading to multi-organ failure, pancreatic necrosis, and death.
Volanesorsen targets apolipoprotein C-III (apoC-III), a key regulator of lipoprotein lipase, the essential enzyme involved in chylomicron and triglyceride clearance.
Because of the small number of patients affected, estimated at just 3,000-5,000 patients worldwide, the FDA considers FCS to be an orphan disease.
The application, submitted by Akcea Pharmaceuticals, was based on the results of several phase 3 clinical trials conducted both in FCS patients and, because of the rarity of the disorder, patients with high triglycerides. The APPROACH study looked at patients with FCS. The COMPASS study examined patients with hypertriglyceridemia. Additionally, an ongoing, open-label extension of the APPROACH study examined patients with FCS who had completed APPROACH, COMPASS, and FCS patients who had not participated in a previous volanesorsen trial.
APPROACH APPROACH was a phase 3, double-blind clinical trial to assess the safety and efficacy of volanesorsen in patients with FCS. Because of the extremely small number of patients affected by FCS, non-FCS patients with hypertriglyceridemia were included in the trial.
All patients who had completed a 6 week diet, lifestyle, and medication stabilization period were enrolled in the study. In total, 67 patients were randomized to receive a weekly, subcutaneous 300 mg dose of volanesorsen or a placebo for 52 weeks. Due to the risk of platelet reduction, the study allowed for dosing schedule interruptions or pauses, if needed. To assess efficacy, the study looked at the percent change in fasting triglycerides at month 3 of the study in all randomized patients who received at least one dose of the study drug and who had baseline fasting plasma trigylcerides recorded.
Volanesorsen proved quite effective in reducing fasting triglyceride levels, with a 94% (P < 0.0001) reduction in plasma concentrations at 3 months, compared with placebo.
The effectiveness of volanesorsen extended beyond month 3, with statistically significant reductions in triglyceride concentrations at both month 6 and month 12 compared with placebo. Patients taking volanesorsen had 53% and 40% reductions at month 6 and 12 compared to baseline. Conversely, patients taking placebo experienced increases of 25% and 9%, respectively. The differences between volanesorsen and placebo patients at month 6 (–78%) and month 12 (–49%) were both statistically significant.
Another important finding from this study was that 77% of patients responded to treatment, evidenced by a reduction of triglyceride levels greater than 750 mg/dL, a much better response than the 10% of placebo patients (P =.0001).
Nine patients (27%) discontinued the study due to adverse events (AE). Five of the discontinuations were related to platelet reductions with the other four related to nonplatelet-related adverse events.
COMPASS
The COMPASS study primarily looked at patients without FCS, but severe hypertriglyceridemia, to evaluate the safety and efficacy of volanesorsen in a similarly ill, but less severe, population over a 6 month study period. Patients with FCS had baseline fasting plasma triglyceride levels of 2,644 mg/dL and 2,134 mg/dL in the placebo and volanesorsen groups, respectively. This is nearly double what was seen in non-FCS patients in the study, and is 14-17 higher than the upper limit of normal (ULN).
Researchers enrolled 114 patients and randomized them 2:1 to receive volanesorsen or placebo. This led to 75 patients taking volanesorsen and 34 placebo.
The efficacy results were similar to those in the FCS-focused APPROACH trial, with volanesorsen treatment leading to reductions in triglyceride levels after 3 months, with further reductions over the 6-month study treatment.
Weekly treatment with volanesorsen reduced triglycerides by nearly 71% in the total patient population, compared to placebo, which only achieved a 0.9% decrease in triglycerides. These reductions were conserved at the end of week 26 in patients who had to reduce their dosing to biweekly (62%) and patients who maintained weekly dosing treatments (78%). Overall, a greater than 40% reduction of fasting triglyceride levels after 3 months of treatment was achieved in 87% of volanesorsen-treated patients, compared to 13% of placebo-treated patients (P < 0.0001).
Serious adverse events (SAEs) occurred in nine patients (8%). In the placebo group, two patients (5%) reported acute pancreatitis. Apart from the bouts of pancreatitis, some of the most common AEs were decreased platelet count, thrombocytopenia, and nasopharyngitis.
APPROACH OPEN LABEL
The open label portion of the APPROACH study is ongoing and was designed to assess the safety and efficacy of extended treatment with volanesorsen in patients with FCS who had previously completed APPROACH or COMPASS studies or had never received volanesorsen treatment. Patients received 300 mg of volanesorsen once weekly for 52 weeks. Patients who completed the trial were eligible to continue treatment for an additional 52 weeks or until a product is available. As of August 31, 2017, after the data cutoff, 60 patients were enrolled.
The results of this open label portion of the APPROACH trial were similar to that of the APPROACH trial itself. Patients who transitioned from APPROACH or COMPASS showed decreases in their open label extension (OLE) baseline triglyceride measurements to OLE month 3 of 48.1% and 52.1%, respectively. Treatment-naive patients displayed a nearly 60% decrease in their triglyceride levels 3 months.
Of the 60 patients who started the study, 12 have discontinued the study prematurely. The majority of patients, eight, who discontinued did so because of adverse events and four withdrew voluntarily.
Adverse events were not uncommon, with 56 patients (93%) experiencing an AE during the course of the study. Some of the most common AEs were decreased platelet count, thrombocytopenia, and nasopharyngitis.
Akcea has developed a Risk Evaluation and Mitigation program to mitigate the risk of serious bleeding related to severe thrombocytopenia with volanesorsen use in patients with FCS.
Panelist Susan Z. Yanovksi, MD, of the National Institutes of Health, said she voted no over safety concerns, but felt conflicted.
“There’s no question that volanesorsen is effective in dramatically reducing triglycerides, but I had a lot of concerns whether the data presented by the sponsor actually established favorable risk-benefit ratio.”
The FDA is expected to decide on the application by August 30. The FDA usually follows the recommendations of its advisory panels, which are not binding.
ilacy@mdedge.com
SILVER SPRING, Md. – A novel drug to treat a rare metabolic disorder was recommended for approval by the Endocrinologic and Metabolic Drugs Advisory Committee in a May 10 meeting.
Advisors voted 12 to 8 to recommend volanesorsen, a novel antisense drug, as an adjunct to diet for the treatment of familial chylomicronemia syndrome (FCS).
“The sponsor provided compelling evidence that the drug lowers triglyceride levels, substantially,” said panelist Jean-Pierre Raufman, MD, of the University of Maryland. I think that there is clearly a group of patients with this disease that can benefit from this agent.”
FCS is an extremely rare genetic disorder characterized by severe hypertriglyceridemia and recurrent pancreatitis caused by a deficiency in lipoprotein lipase. Pancreatitis in the setting of hypertriglyceridemia is particularly severe, often leading to multi-organ failure, pancreatic necrosis, and death.
Volanesorsen targets apolipoprotein C-III (apoC-III), a key regulator of lipoprotein lipase, the essential enzyme involved in chylomicron and triglyceride clearance.
Because of the small number of patients affected, estimated at just 3,000-5,000 patients worldwide, the FDA considers FCS to be an orphan disease.
The application, submitted by Akcea Pharmaceuticals, was based on the results of several phase 3 clinical trials conducted both in FCS patients and, because of the rarity of the disorder, patients with high triglycerides. The APPROACH study looked at patients with FCS. The COMPASS study examined patients with hypertriglyceridemia. Additionally, an ongoing, open-label extension of the APPROACH study examined patients with FCS who had completed APPROACH, COMPASS, and FCS patients who had not participated in a previous volanesorsen trial.
APPROACH APPROACH was a phase 3, double-blind clinical trial to assess the safety and efficacy of volanesorsen in patients with FCS. Because of the extremely small number of patients affected by FCS, non-FCS patients with hypertriglyceridemia were included in the trial.
All patients who had completed a 6 week diet, lifestyle, and medication stabilization period were enrolled in the study. In total, 67 patients were randomized to receive a weekly, subcutaneous 300 mg dose of volanesorsen or a placebo for 52 weeks. Due to the risk of platelet reduction, the study allowed for dosing schedule interruptions or pauses, if needed. To assess efficacy, the study looked at the percent change in fasting triglycerides at month 3 of the study in all randomized patients who received at least one dose of the study drug and who had baseline fasting plasma trigylcerides recorded.
Volanesorsen proved quite effective in reducing fasting triglyceride levels, with a 94% (P < 0.0001) reduction in plasma concentrations at 3 months, compared with placebo.
The effectiveness of volanesorsen extended beyond month 3, with statistically significant reductions in triglyceride concentrations at both month 6 and month 12 compared with placebo. Patients taking volanesorsen had 53% and 40% reductions at month 6 and 12 compared to baseline. Conversely, patients taking placebo experienced increases of 25% and 9%, respectively. The differences between volanesorsen and placebo patients at month 6 (–78%) and month 12 (–49%) were both statistically significant.
Another important finding from this study was that 77% of patients responded to treatment, evidenced by a reduction of triglyceride levels greater than 750 mg/dL, a much better response than the 10% of placebo patients (P =.0001).
Nine patients (27%) discontinued the study due to adverse events (AE). Five of the discontinuations were related to platelet reductions with the other four related to nonplatelet-related adverse events.
COMPASS
The COMPASS study primarily looked at patients without FCS, but severe hypertriglyceridemia, to evaluate the safety and efficacy of volanesorsen in a similarly ill, but less severe, population over a 6 month study period. Patients with FCS had baseline fasting plasma triglyceride levels of 2,644 mg/dL and 2,134 mg/dL in the placebo and volanesorsen groups, respectively. This is nearly double what was seen in non-FCS patients in the study, and is 14-17 higher than the upper limit of normal (ULN).
Researchers enrolled 114 patients and randomized them 2:1 to receive volanesorsen or placebo. This led to 75 patients taking volanesorsen and 34 placebo.
The efficacy results were similar to those in the FCS-focused APPROACH trial, with volanesorsen treatment leading to reductions in triglyceride levels after 3 months, with further reductions over the 6-month study treatment.
Weekly treatment with volanesorsen reduced triglycerides by nearly 71% in the total patient population, compared to placebo, which only achieved a 0.9% decrease in triglycerides. These reductions were conserved at the end of week 26 in patients who had to reduce their dosing to biweekly (62%) and patients who maintained weekly dosing treatments (78%). Overall, a greater than 40% reduction of fasting triglyceride levels after 3 months of treatment was achieved in 87% of volanesorsen-treated patients, compared to 13% of placebo-treated patients (P < 0.0001).
Serious adverse events (SAEs) occurred in nine patients (8%). In the placebo group, two patients (5%) reported acute pancreatitis. Apart from the bouts of pancreatitis, some of the most common AEs were decreased platelet count, thrombocytopenia, and nasopharyngitis.
APPROACH OPEN LABEL
The open label portion of the APPROACH study is ongoing and was designed to assess the safety and efficacy of extended treatment with volanesorsen in patients with FCS who had previously completed APPROACH or COMPASS studies or had never received volanesorsen treatment. Patients received 300 mg of volanesorsen once weekly for 52 weeks. Patients who completed the trial were eligible to continue treatment for an additional 52 weeks or until a product is available. As of August 31, 2017, after the data cutoff, 60 patients were enrolled.
The results of this open label portion of the APPROACH trial were similar to that of the APPROACH trial itself. Patients who transitioned from APPROACH or COMPASS showed decreases in their open label extension (OLE) baseline triglyceride measurements to OLE month 3 of 48.1% and 52.1%, respectively. Treatment-naive patients displayed a nearly 60% decrease in their triglyceride levels 3 months.
Of the 60 patients who started the study, 12 have discontinued the study prematurely. The majority of patients, eight, who discontinued did so because of adverse events and four withdrew voluntarily.
Adverse events were not uncommon, with 56 patients (93%) experiencing an AE during the course of the study. Some of the most common AEs were decreased platelet count, thrombocytopenia, and nasopharyngitis.
Akcea has developed a Risk Evaluation and Mitigation program to mitigate the risk of serious bleeding related to severe thrombocytopenia with volanesorsen use in patients with FCS.
Panelist Susan Z. Yanovksi, MD, of the National Institutes of Health, said she voted no over safety concerns, but felt conflicted.
“There’s no question that volanesorsen is effective in dramatically reducing triglycerides, but I had a lot of concerns whether the data presented by the sponsor actually established favorable risk-benefit ratio.”
The FDA is expected to decide on the application by August 30. The FDA usually follows the recommendations of its advisory panels, which are not binding.
ilacy@mdedge.com
SILVER SPRING, Md. – A novel drug to treat a rare metabolic disorder was recommended for approval by the Endocrinologic and Metabolic Drugs Advisory Committee in a May 10 meeting.
Advisors voted 12 to 8 to recommend volanesorsen, a novel antisense drug, as an adjunct to diet for the treatment of familial chylomicronemia syndrome (FCS).
“The sponsor provided compelling evidence that the drug lowers triglyceride levels, substantially,” said panelist Jean-Pierre Raufman, MD, of the University of Maryland. I think that there is clearly a group of patients with this disease that can benefit from this agent.”
FCS is an extremely rare genetic disorder characterized by severe hypertriglyceridemia and recurrent pancreatitis caused by a deficiency in lipoprotein lipase. Pancreatitis in the setting of hypertriglyceridemia is particularly severe, often leading to multi-organ failure, pancreatic necrosis, and death.
Volanesorsen targets apolipoprotein C-III (apoC-III), a key regulator of lipoprotein lipase, the essential enzyme involved in chylomicron and triglyceride clearance.
Because of the small number of patients affected, estimated at just 3,000-5,000 patients worldwide, the FDA considers FCS to be an orphan disease.
The application, submitted by Akcea Pharmaceuticals, was based on the results of several phase 3 clinical trials conducted both in FCS patients and, because of the rarity of the disorder, patients with high triglycerides. The APPROACH study looked at patients with FCS. The COMPASS study examined patients with hypertriglyceridemia. Additionally, an ongoing, open-label extension of the APPROACH study examined patients with FCS who had completed APPROACH, COMPASS, and FCS patients who had not participated in a previous volanesorsen trial.
APPROACH APPROACH was a phase 3, double-blind clinical trial to assess the safety and efficacy of volanesorsen in patients with FCS. Because of the extremely small number of patients affected by FCS, non-FCS patients with hypertriglyceridemia were included in the trial.
All patients who had completed a 6 week diet, lifestyle, and medication stabilization period were enrolled in the study. In total, 67 patients were randomized to receive a weekly, subcutaneous 300 mg dose of volanesorsen or a placebo for 52 weeks. Due to the risk of platelet reduction, the study allowed for dosing schedule interruptions or pauses, if needed. To assess efficacy, the study looked at the percent change in fasting triglycerides at month 3 of the study in all randomized patients who received at least one dose of the study drug and who had baseline fasting plasma trigylcerides recorded.
Volanesorsen proved quite effective in reducing fasting triglyceride levels, with a 94% (P < 0.0001) reduction in plasma concentrations at 3 months, compared with placebo.
The effectiveness of volanesorsen extended beyond month 3, with statistically significant reductions in triglyceride concentrations at both month 6 and month 12 compared with placebo. Patients taking volanesorsen had 53% and 40% reductions at month 6 and 12 compared to baseline. Conversely, patients taking placebo experienced increases of 25% and 9%, respectively. The differences between volanesorsen and placebo patients at month 6 (–78%) and month 12 (–49%) were both statistically significant.
Another important finding from this study was that 77% of patients responded to treatment, evidenced by a reduction of triglyceride levels greater than 750 mg/dL, a much better response than the 10% of placebo patients (P =.0001).
Nine patients (27%) discontinued the study due to adverse events (AE). Five of the discontinuations were related to platelet reductions with the other four related to nonplatelet-related adverse events.
COMPASS
The COMPASS study primarily looked at patients without FCS, but severe hypertriglyceridemia, to evaluate the safety and efficacy of volanesorsen in a similarly ill, but less severe, population over a 6 month study period. Patients with FCS had baseline fasting plasma triglyceride levels of 2,644 mg/dL and 2,134 mg/dL in the placebo and volanesorsen groups, respectively. This is nearly double what was seen in non-FCS patients in the study, and is 14-17 higher than the upper limit of normal (ULN).
Researchers enrolled 114 patients and randomized them 2:1 to receive volanesorsen or placebo. This led to 75 patients taking volanesorsen and 34 placebo.
The efficacy results were similar to those in the FCS-focused APPROACH trial, with volanesorsen treatment leading to reductions in triglyceride levels after 3 months, with further reductions over the 6-month study treatment.
Weekly treatment with volanesorsen reduced triglycerides by nearly 71% in the total patient population, compared to placebo, which only achieved a 0.9% decrease in triglycerides. These reductions were conserved at the end of week 26 in patients who had to reduce their dosing to biweekly (62%) and patients who maintained weekly dosing treatments (78%). Overall, a greater than 40% reduction of fasting triglyceride levels after 3 months of treatment was achieved in 87% of volanesorsen-treated patients, compared to 13% of placebo-treated patients (P < 0.0001).
Serious adverse events (SAEs) occurred in nine patients (8%). In the placebo group, two patients (5%) reported acute pancreatitis. Apart from the bouts of pancreatitis, some of the most common AEs were decreased platelet count, thrombocytopenia, and nasopharyngitis.
APPROACH OPEN LABEL
The open label portion of the APPROACH study is ongoing and was designed to assess the safety and efficacy of extended treatment with volanesorsen in patients with FCS who had previously completed APPROACH or COMPASS studies or had never received volanesorsen treatment. Patients received 300 mg of volanesorsen once weekly for 52 weeks. Patients who completed the trial were eligible to continue treatment for an additional 52 weeks or until a product is available. As of August 31, 2017, after the data cutoff, 60 patients were enrolled.
The results of this open label portion of the APPROACH trial were similar to that of the APPROACH trial itself. Patients who transitioned from APPROACH or COMPASS showed decreases in their open label extension (OLE) baseline triglyceride measurements to OLE month 3 of 48.1% and 52.1%, respectively. Treatment-naive patients displayed a nearly 60% decrease in their triglyceride levels 3 months.
Of the 60 patients who started the study, 12 have discontinued the study prematurely. The majority of patients, eight, who discontinued did so because of adverse events and four withdrew voluntarily.
Adverse events were not uncommon, with 56 patients (93%) experiencing an AE during the course of the study. Some of the most common AEs were decreased platelet count, thrombocytopenia, and nasopharyngitis.
Akcea has developed a Risk Evaluation and Mitigation program to mitigate the risk of serious bleeding related to severe thrombocytopenia with volanesorsen use in patients with FCS.
Panelist Susan Z. Yanovksi, MD, of the National Institutes of Health, said she voted no over safety concerns, but felt conflicted.
“There’s no question that volanesorsen is effective in dramatically reducing triglycerides, but I had a lot of concerns whether the data presented by the sponsor actually established favorable risk-benefit ratio.”
The FDA is expected to decide on the application by August 30. The FDA usually follows the recommendations of its advisory panels, which are not binding.
ilacy@mdedge.com
REPORTING FROM an fda advisory committee meeting
Zika topped Lyme in 2016
Ticks are the arthropod ride of choice for vector-borne diseases in the United States, but the Zika virus and its mosquito minions gave the ticks and their bacterial passengers a run for their money in 2016, according to the Centers for Disease Control and Prevention.
There were 41,680 cases of Zika virus that year, more than any other vector-borne disease, including Lyme disease, which had been the most common transmissible pathogen going back to at least 2004, when arthropod-borne viral diseases became nationally notifiable, said Ronald Rosenberg, ScD, and his associates at the CDC’s National Center for Emerging and Zoonotic Infectious Diseases in Fort Collins, Colo.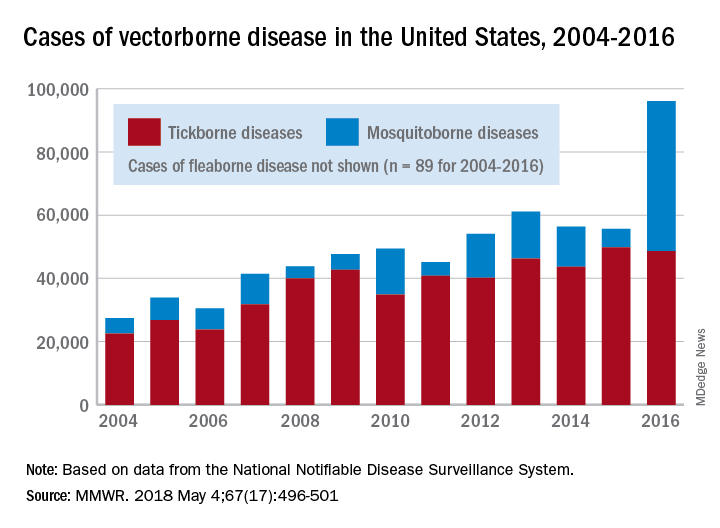
Since 2004, there have been 643,000 reported cases of vector-borne disease in the United States: 492,000 cases of tick-borne disease, of which over 402,000 were Lyme disease; 151,000 cases of mosquito-borne disease; and 89 cases of plague carried by the third type of vector, fleas, Dr. Rosenberg and his associates said based on data from the National Notifiable Disease Surveillance System.
In 2004, there were 22,527 cases of tick-borne disease and 4,858 cases of mosquito-borne disease, and the increases since then reflect the dynamics of the pathogens and vectors involved. Growth of tick-borne disease has been gradual: “Tick-borne pathogens rarely cause sudden epidemics because humans are typically incidental hosts who do not transmit further, and tick mobility is mostly limited to that of its animal hosts,” the researchers explained.
The number of mosquito-borne disease cases, on the other hand, varies considerably from year to year: There were 5,800 cases in 2015, almost 15,000 in 2013, and only 4,400 in 2011. Unlike ticks, which may feed on blood only once in a year, the more mobile mosquitoes feed every 48-72 hours and transmit their pathogens “directly between humans … resulting in explosive epidemics,” the investigators wrote.
SOURCE: Rosenberg R et al. MMWR 2018 May 4;67(17):496-501.
Ticks are the arthropod ride of choice for vector-borne diseases in the United States, but the Zika virus and its mosquito minions gave the ticks and their bacterial passengers a run for their money in 2016, according to the Centers for Disease Control and Prevention.
There were 41,680 cases of Zika virus that year, more than any other vector-borne disease, including Lyme disease, which had been the most common transmissible pathogen going back to at least 2004, when arthropod-borne viral diseases became nationally notifiable, said Ronald Rosenberg, ScD, and his associates at the CDC’s National Center for Emerging and Zoonotic Infectious Diseases in Fort Collins, Colo.
Since 2004, there have been 643,000 reported cases of vector-borne disease in the United States: 492,000 cases of tick-borne disease, of which over 402,000 were Lyme disease; 151,000 cases of mosquito-borne disease; and 89 cases of plague carried by the third type of vector, fleas, Dr. Rosenberg and his associates said based on data from the National Notifiable Disease Surveillance System.
In 2004, there were 22,527 cases of tick-borne disease and 4,858 cases of mosquito-borne disease, and the increases since then reflect the dynamics of the pathogens and vectors involved. Growth of tick-borne disease has been gradual: “Tick-borne pathogens rarely cause sudden epidemics because humans are typically incidental hosts who do not transmit further, and tick mobility is mostly limited to that of its animal hosts,” the researchers explained.
The number of mosquito-borne disease cases, on the other hand, varies considerably from year to year: There were 5,800 cases in 2015, almost 15,000 in 2013, and only 4,400 in 2011. Unlike ticks, which may feed on blood only once in a year, the more mobile mosquitoes feed every 48-72 hours and transmit their pathogens “directly between humans … resulting in explosive epidemics,” the investigators wrote.
SOURCE: Rosenberg R et al. MMWR 2018 May 4;67(17):496-501.
Ticks are the arthropod ride of choice for vector-borne diseases in the United States, but the Zika virus and its mosquito minions gave the ticks and their bacterial passengers a run for their money in 2016, according to the Centers for Disease Control and Prevention.
There were 41,680 cases of Zika virus that year, more than any other vector-borne disease, including Lyme disease, which had been the most common transmissible pathogen going back to at least 2004, when arthropod-borne viral diseases became nationally notifiable, said Ronald Rosenberg, ScD, and his associates at the CDC’s National Center for Emerging and Zoonotic Infectious Diseases in Fort Collins, Colo.
Since 2004, there have been 643,000 reported cases of vector-borne disease in the United States: 492,000 cases of tick-borne disease, of which over 402,000 were Lyme disease; 151,000 cases of mosquito-borne disease; and 89 cases of plague carried by the third type of vector, fleas, Dr. Rosenberg and his associates said based on data from the National Notifiable Disease Surveillance System.
In 2004, there were 22,527 cases of tick-borne disease and 4,858 cases of mosquito-borne disease, and the increases since then reflect the dynamics of the pathogens and vectors involved. Growth of tick-borne disease has been gradual: “Tick-borne pathogens rarely cause sudden epidemics because humans are typically incidental hosts who do not transmit further, and tick mobility is mostly limited to that of its animal hosts,” the researchers explained.
The number of mosquito-borne disease cases, on the other hand, varies considerably from year to year: There were 5,800 cases in 2015, almost 15,000 in 2013, and only 4,400 in 2011. Unlike ticks, which may feed on blood only once in a year, the more mobile mosquitoes feed every 48-72 hours and transmit their pathogens “directly between humans … resulting in explosive epidemics,” the investigators wrote.
SOURCE: Rosenberg R et al. MMWR 2018 May 4;67(17):496-501.
FROM MMWR
FDA approves dabrafenib/trametinib for BRAF-positive anaplastic thyroid cancer
FDA approval was based on results from an open-label clinical trial of patients with various rare, BRAF V600E–positive cancers. In a group of 23 patients, 57% experienced a partial response and 4% experienced a full response. In the response group, nine patients had no significant tumor growth for a period of at least 6 months.
The most common side effects of dabrafenib/trametinib are fever, rash, chills, headache, joint pain, cough, fatigue, nausea, vomiting, diarrhea, myalgia, dry skin, decreased appetite, edema, hemorrhage, high blood pressure, and difficulty breathing. Both drugs can cause damage to developing fetuses, and women should be advised to use proper contraception.
“This approval demonstrates that targeting the same molecular pathway in diverse diseases is an effective way to expedite the development of treatments that may help more patients,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in the press release.
Find the full press release on the FDA website.
FDA approval was based on results from an open-label clinical trial of patients with various rare, BRAF V600E–positive cancers. In a group of 23 patients, 57% experienced a partial response and 4% experienced a full response. In the response group, nine patients had no significant tumor growth for a period of at least 6 months.
The most common side effects of dabrafenib/trametinib are fever, rash, chills, headache, joint pain, cough, fatigue, nausea, vomiting, diarrhea, myalgia, dry skin, decreased appetite, edema, hemorrhage, high blood pressure, and difficulty breathing. Both drugs can cause damage to developing fetuses, and women should be advised to use proper contraception.
“This approval demonstrates that targeting the same molecular pathway in diverse diseases is an effective way to expedite the development of treatments that may help more patients,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in the press release.
Find the full press release on the FDA website.
FDA approval was based on results from an open-label clinical trial of patients with various rare, BRAF V600E–positive cancers. In a group of 23 patients, 57% experienced a partial response and 4% experienced a full response. In the response group, nine patients had no significant tumor growth for a period of at least 6 months.
The most common side effects of dabrafenib/trametinib are fever, rash, chills, headache, joint pain, cough, fatigue, nausea, vomiting, diarrhea, myalgia, dry skin, decreased appetite, edema, hemorrhage, high blood pressure, and difficulty breathing. Both drugs can cause damage to developing fetuses, and women should be advised to use proper contraception.
“This approval demonstrates that targeting the same molecular pathway in diverse diseases is an effective way to expedite the development of treatments that may help more patients,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in the press release.
Find the full press release on the FDA website.
FDA approves anti-CD38 with VMP in myeloma
The who are ineligible for autologous stem cell transplant (ASCT).
The drug is approved in combination with a standard VMP regimen – bortezomib (Velcade), melphalan, and prednisone. The FDA had granted priority review to the drug application in January 2018 based on the results of the phase 3 ALCYONE study (NCT02195479).
Daratumumab, an anti-CD38 monoclonal antibody, reduced the risk of disease progression or death by 50%, compared with VMP alone in the ALCYONE study. The median progression-free survival had not yet been reached in the daratumumab arm; the median progression-free survival was 18.1 months in the VMP-only arm (N Engl J Med. 2018;378:518-28).
Daratumumab is marketed by Janssen Biotech as Darzalex.
The who are ineligible for autologous stem cell transplant (ASCT).
The drug is approved in combination with a standard VMP regimen – bortezomib (Velcade), melphalan, and prednisone. The FDA had granted priority review to the drug application in January 2018 based on the results of the phase 3 ALCYONE study (NCT02195479).
Daratumumab, an anti-CD38 monoclonal antibody, reduced the risk of disease progression or death by 50%, compared with VMP alone in the ALCYONE study. The median progression-free survival had not yet been reached in the daratumumab arm; the median progression-free survival was 18.1 months in the VMP-only arm (N Engl J Med. 2018;378:518-28).
Daratumumab is marketed by Janssen Biotech as Darzalex.
The who are ineligible for autologous stem cell transplant (ASCT).
The drug is approved in combination with a standard VMP regimen – bortezomib (Velcade), melphalan, and prednisone. The FDA had granted priority review to the drug application in January 2018 based on the results of the phase 3 ALCYONE study (NCT02195479).
Daratumumab, an anti-CD38 monoclonal antibody, reduced the risk of disease progression or death by 50%, compared with VMP alone in the ALCYONE study. The median progression-free survival had not yet been reached in the daratumumab arm; the median progression-free survival was 18.1 months in the VMP-only arm (N Engl J Med. 2018;378:518-28).
Daratumumab is marketed by Janssen Biotech as Darzalex.
First reversal agent for apixaban and rivaroxaban gets fast-track approval
, according to a May 3 statement from Portola Pharmaceuticals.
It is approved for use in patients treated with these factor Xa inhibitors when reversal of anticoagulation is needed because of life-threatening or uncontrolled bleeding, according to the company.

Andexanet alfa (Andexxa, Portola) received both U.S. Orphan Drug and FDA Breakthrough Therapy designations and was approved under the FDA’s Accelerated Approval pathway.
“Today’s approval represents a significant step forward in patient care and one that the medical community has been eagerly anticipating,” said Stuart J. Connolly, MD, professor of medicine and an electrophysiologist at McMaster University in Hamilton, Ont., who is chair of the ANNEXA-4 executive committee. “Andexxa’s rapid reversal of the anticoagulating effects of rivaroxaban and apixaban will help clinicians treat life-threatening bleeds, where every minute counts,” he added in the statement.
The approval was supported by two phase 3 trials in the ANNEXA series, which showed acceptable change from baseline in anti-Factor Xa activity in healthy volunteers. But the strongest data came from interim results from ANNEXA-4, a single-arm cohort study with 227 patients who were receiving a factor Xa inhibitor and were experiencing an acute major bleeding event.
Clinicians administered andexanet alfa as a bolus followed by a 2-hour continuous infusion, with hemostatic efficacy assessed 12 hours after the start of treatment. The results showed that factor Xa inhibition fell by a median 90% for rivaroxaban and 93% for apixaban.
Andexanet alfa is a factor Xa “decoy” molecule that acts by latching onto the inhibitor molecules and thereby preventing them from interacting with actual factor Xa, but andexanet also has a short half life and hence the effect quickly reduces once treatment stops, Dr. Connelly reported at the American College of Cardiology annual meeting in March when presenting ANNEXA-4.
He noted at the time the results placed andexanet in the same ballpark for efficacy and safety as idarucizumab (Praxbind) approved in 2015 for reversing the anticoagulant dabigatran (Pradaxa)
“The expansion of available reversal agents for people prescribed newer oral anticoagulant therapies is crucial,” Randy Fenninger, chief executive officer of the National Blood Clot Alliance, said in the Portola statement. “The availability now of a reversal agent specific to rivaroxaban and apixaban expands choice and enables patients and providers to consider these treatment options with greater confidence.”
The prescribing information for andexanet states that treated patients should be monitored for signs and symptoms of arterial and venous thromboembolic events, ischemic events, and cardiac arrest. Further, anticoagulant therapy should be resumed as soon as medically appropriate following andexanet treatment to reduce thromboembolic risk.
The most common adverse reactions, occurring in at least 5% of patients, were urinary tract infections and pneumonia.
Portola intends to bring Andexxa to limited markets in early June; a broader commercial launch is anticipated in early 2019.*
The FDA is requiring a postmarketing clinical trial that randomizes patients to either andexanet or usual care. The study is scheduled to begin in 2019 and report outcomes in 2023.
*This article was updated on May 7, 2018.
, according to a May 3 statement from Portola Pharmaceuticals.
It is approved for use in patients treated with these factor Xa inhibitors when reversal of anticoagulation is needed because of life-threatening or uncontrolled bleeding, according to the company.
Andexanet alfa (Andexxa, Portola) received both U.S. Orphan Drug and FDA Breakthrough Therapy designations and was approved under the FDA’s Accelerated Approval pathway.
“Today’s approval represents a significant step forward in patient care and one that the medical community has been eagerly anticipating,” said Stuart J. Connolly, MD, professor of medicine and an electrophysiologist at McMaster University in Hamilton, Ont., who is chair of the ANNEXA-4 executive committee. “Andexxa’s rapid reversal of the anticoagulating effects of rivaroxaban and apixaban will help clinicians treat life-threatening bleeds, where every minute counts,” he added in the statement.
The approval was supported by two phase 3 trials in the ANNEXA series, which showed acceptable change from baseline in anti-Factor Xa activity in healthy volunteers. But the strongest data came from interim results from ANNEXA-4, a single-arm cohort study with 227 patients who were receiving a factor Xa inhibitor and were experiencing an acute major bleeding event.
Clinicians administered andexanet alfa as a bolus followed by a 2-hour continuous infusion, with hemostatic efficacy assessed 12 hours after the start of treatment. The results showed that factor Xa inhibition fell by a median 90% for rivaroxaban and 93% for apixaban.
Andexanet alfa is a factor Xa “decoy” molecule that acts by latching onto the inhibitor molecules and thereby preventing them from interacting with actual factor Xa, but andexanet also has a short half life and hence the effect quickly reduces once treatment stops, Dr. Connelly reported at the American College of Cardiology annual meeting in March when presenting ANNEXA-4.
He noted at the time the results placed andexanet in the same ballpark for efficacy and safety as idarucizumab (Praxbind) approved in 2015 for reversing the anticoagulant dabigatran (Pradaxa)
“The expansion of available reversal agents for people prescribed newer oral anticoagulant therapies is crucial,” Randy Fenninger, chief executive officer of the National Blood Clot Alliance, said in the Portola statement. “The availability now of a reversal agent specific to rivaroxaban and apixaban expands choice and enables patients and providers to consider these treatment options with greater confidence.”
The prescribing information for andexanet states that treated patients should be monitored for signs and symptoms of arterial and venous thromboembolic events, ischemic events, and cardiac arrest. Further, anticoagulant therapy should be resumed as soon as medically appropriate following andexanet treatment to reduce thromboembolic risk.
The most common adverse reactions, occurring in at least 5% of patients, were urinary tract infections and pneumonia.
Portola intends to bring Andexxa to limited markets in early June; a broader commercial launch is anticipated in early 2019.*
The FDA is requiring a postmarketing clinical trial that randomizes patients to either andexanet or usual care. The study is scheduled to begin in 2019 and report outcomes in 2023.
*This article was updated on May 7, 2018.
, according to a May 3 statement from Portola Pharmaceuticals.
It is approved for use in patients treated with these factor Xa inhibitors when reversal of anticoagulation is needed because of life-threatening or uncontrolled bleeding, according to the company.
Andexanet alfa (Andexxa, Portola) received both U.S. Orphan Drug and FDA Breakthrough Therapy designations and was approved under the FDA’s Accelerated Approval pathway.
“Today’s approval represents a significant step forward in patient care and one that the medical community has been eagerly anticipating,” said Stuart J. Connolly, MD, professor of medicine and an electrophysiologist at McMaster University in Hamilton, Ont., who is chair of the ANNEXA-4 executive committee. “Andexxa’s rapid reversal of the anticoagulating effects of rivaroxaban and apixaban will help clinicians treat life-threatening bleeds, where every minute counts,” he added in the statement.
The approval was supported by two phase 3 trials in the ANNEXA series, which showed acceptable change from baseline in anti-Factor Xa activity in healthy volunteers. But the strongest data came from interim results from ANNEXA-4, a single-arm cohort study with 227 patients who were receiving a factor Xa inhibitor and were experiencing an acute major bleeding event.
Clinicians administered andexanet alfa as a bolus followed by a 2-hour continuous infusion, with hemostatic efficacy assessed 12 hours after the start of treatment. The results showed that factor Xa inhibition fell by a median 90% for rivaroxaban and 93% for apixaban.
Andexanet alfa is a factor Xa “decoy” molecule that acts by latching onto the inhibitor molecules and thereby preventing them from interacting with actual factor Xa, but andexanet also has a short half life and hence the effect quickly reduces once treatment stops, Dr. Connelly reported at the American College of Cardiology annual meeting in March when presenting ANNEXA-4.
He noted at the time the results placed andexanet in the same ballpark for efficacy and safety as idarucizumab (Praxbind) approved in 2015 for reversing the anticoagulant dabigatran (Pradaxa)
“The expansion of available reversal agents for people prescribed newer oral anticoagulant therapies is crucial,” Randy Fenninger, chief executive officer of the National Blood Clot Alliance, said in the Portola statement. “The availability now of a reversal agent specific to rivaroxaban and apixaban expands choice and enables patients and providers to consider these treatment options with greater confidence.”
The prescribing information for andexanet states that treated patients should be monitored for signs and symptoms of arterial and venous thromboembolic events, ischemic events, and cardiac arrest. Further, anticoagulant therapy should be resumed as soon as medically appropriate following andexanet treatment to reduce thromboembolic risk.
The most common adverse reactions, occurring in at least 5% of patients, were urinary tract infections and pneumonia.
Portola intends to bring Andexxa to limited markets in early June; a broader commercial launch is anticipated in early 2019.*
The FDA is requiring a postmarketing clinical trial that randomizes patients to either andexanet or usual care. The study is scheduled to begin in 2019 and report outcomes in 2023.
*This article was updated on May 7, 2018.
Novartis CAR T-cell therapy adds a lymphoma indication
Novartis’s after failure of two or more lines of systemic therapy.
The Food and Drug Administration approved the expanded indication on May 1. The chimeric antigen receptor (CAR) T-cell therapy was initially approved in Aug. 2017 for refractory or relapsed B-cell precursor acute lymphoblastic leukemia (ALL) in patients up to 25 years old. The new approval brings tisagenlecleucel into direct competition with Gilead Science’s CAR T-cell therapy axicabtagene ciloleucel (Yescarta), which was approved in Oct. 2017 for B-cell lymphoma.
Besides matching the competition, she said the lower price is because tisagenlecleucel takes longer to work for lymphoma, and the response isn’t as potent as for childhood ALL. Novartis is looking into chronic lymphocytic leukemia, multiple myeloma, and solid tumor indications for tisagenlecleucel and other CAR T-cell agents, she added.
The Centers for Medicare & Medicaid Services recently committed to covering outpatient administration of both agents for their initial indications; Novartis is working with CMS for coverage of the new lymphoma indication.
With both products, T cells are collected then shipped off to a company facility where a CAR gene is spliced into their DNA, essentially programming the T cells to attack the targeted cancer. The cells are then infused back into the patient.
In the phase 2 JULIET trial, tisagenlecleucel showed an overall response rate of 50% among 68 B-cell lymphoma patients, with 32% achieving complete response (CR) and 18% achieving partial response (PR). The median duration of response was not reached.
Axicabtagene ciloleucel’s label reports an objective response rate of 72% among 101 patients, with CR in 51% and PR in 21%. Median duration of response was 9.2 months but was also not reached among complete responders.
“Different trials. Different CARTs. Different levels of disease. Our drug is cryopreserved and theirs is not. No way to compare them,” the Novartis spokeswoman said when asked about the response differences.
T-cell reprogramming isn’t clean at this point in medical history; both agents carry black box warnings of potentially fatal cytokine release syndrome and neurologic toxicity, and both are subject to Risk Evaluation and Mitigation Strategy programs.
The B-cell lymphoma indication for both therapies includes diffuse large B-cell lymphoma (DLBCL), high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The Gilead product carries an additional indication for primary mediastinal large B-cell lymphoma. Neither agent is indicated for primary central nervous system lymphoma. Both labels say that patients should not donate blood, organs, or tissues after treatment. Tisagenlecleucel labeling also notes that some commercial HIV nucleic acid tests may yield false positives after treatment.
Novartis said in a press release that T cells are treated at the company’s Morris Plains, N.J., facility with a turnaround time of about 22 days. Cryopreservation of the harvested cells gives providers some flexibility in treatment timing.
Novartis’s after failure of two or more lines of systemic therapy.
The Food and Drug Administration approved the expanded indication on May 1. The chimeric antigen receptor (CAR) T-cell therapy was initially approved in Aug. 2017 for refractory or relapsed B-cell precursor acute lymphoblastic leukemia (ALL) in patients up to 25 years old. The new approval brings tisagenlecleucel into direct competition with Gilead Science’s CAR T-cell therapy axicabtagene ciloleucel (Yescarta), which was approved in Oct. 2017 for B-cell lymphoma.
Besides matching the competition, she said the lower price is because tisagenlecleucel takes longer to work for lymphoma, and the response isn’t as potent as for childhood ALL. Novartis is looking into chronic lymphocytic leukemia, multiple myeloma, and solid tumor indications for tisagenlecleucel and other CAR T-cell agents, she added.
The Centers for Medicare & Medicaid Services recently committed to covering outpatient administration of both agents for their initial indications; Novartis is working with CMS for coverage of the new lymphoma indication.
With both products, T cells are collected then shipped off to a company facility where a CAR gene is spliced into their DNA, essentially programming the T cells to attack the targeted cancer. The cells are then infused back into the patient.
In the phase 2 JULIET trial, tisagenlecleucel showed an overall response rate of 50% among 68 B-cell lymphoma patients, with 32% achieving complete response (CR) and 18% achieving partial response (PR). The median duration of response was not reached.
Axicabtagene ciloleucel’s label reports an objective response rate of 72% among 101 patients, with CR in 51% and PR in 21%. Median duration of response was 9.2 months but was also not reached among complete responders.
“Different trials. Different CARTs. Different levels of disease. Our drug is cryopreserved and theirs is not. No way to compare them,” the Novartis spokeswoman said when asked about the response differences.
T-cell reprogramming isn’t clean at this point in medical history; both agents carry black box warnings of potentially fatal cytokine release syndrome and neurologic toxicity, and both are subject to Risk Evaluation and Mitigation Strategy programs.
The B-cell lymphoma indication for both therapies includes diffuse large B-cell lymphoma (DLBCL), high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The Gilead product carries an additional indication for primary mediastinal large B-cell lymphoma. Neither agent is indicated for primary central nervous system lymphoma. Both labels say that patients should not donate blood, organs, or tissues after treatment. Tisagenlecleucel labeling also notes that some commercial HIV nucleic acid tests may yield false positives after treatment.
Novartis said in a press release that T cells are treated at the company’s Morris Plains, N.J., facility with a turnaround time of about 22 days. Cryopreservation of the harvested cells gives providers some flexibility in treatment timing.
Novartis’s after failure of two or more lines of systemic therapy.
The Food and Drug Administration approved the expanded indication on May 1. The chimeric antigen receptor (CAR) T-cell therapy was initially approved in Aug. 2017 for refractory or relapsed B-cell precursor acute lymphoblastic leukemia (ALL) in patients up to 25 years old. The new approval brings tisagenlecleucel into direct competition with Gilead Science’s CAR T-cell therapy axicabtagene ciloleucel (Yescarta), which was approved in Oct. 2017 for B-cell lymphoma.
Besides matching the competition, she said the lower price is because tisagenlecleucel takes longer to work for lymphoma, and the response isn’t as potent as for childhood ALL. Novartis is looking into chronic lymphocytic leukemia, multiple myeloma, and solid tumor indications for tisagenlecleucel and other CAR T-cell agents, she added.
The Centers for Medicare & Medicaid Services recently committed to covering outpatient administration of both agents for their initial indications; Novartis is working with CMS for coverage of the new lymphoma indication.
With both products, T cells are collected then shipped off to a company facility where a CAR gene is spliced into their DNA, essentially programming the T cells to attack the targeted cancer. The cells are then infused back into the patient.
In the phase 2 JULIET trial, tisagenlecleucel showed an overall response rate of 50% among 68 B-cell lymphoma patients, with 32% achieving complete response (CR) and 18% achieving partial response (PR). The median duration of response was not reached.
Axicabtagene ciloleucel’s label reports an objective response rate of 72% among 101 patients, with CR in 51% and PR in 21%. Median duration of response was 9.2 months but was also not reached among complete responders.
“Different trials. Different CARTs. Different levels of disease. Our drug is cryopreserved and theirs is not. No way to compare them,” the Novartis spokeswoman said when asked about the response differences.
T-cell reprogramming isn’t clean at this point in medical history; both agents carry black box warnings of potentially fatal cytokine release syndrome and neurologic toxicity, and both are subject to Risk Evaluation and Mitigation Strategy programs.
The B-cell lymphoma indication for both therapies includes diffuse large B-cell lymphoma (DLBCL), high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The Gilead product carries an additional indication for primary mediastinal large B-cell lymphoma. Neither agent is indicated for primary central nervous system lymphoma. Both labels say that patients should not donate blood, organs, or tissues after treatment. Tisagenlecleucel labeling also notes that some commercial HIV nucleic acid tests may yield false positives after treatment.
Novartis said in a press release that T cells are treated at the company’s Morris Plains, N.J., facility with a turnaround time of about 22 days. Cryopreservation of the harvested cells gives providers some flexibility in treatment timing.
FDA advisory committee votes to recommend first once-daily aminoglycoside antibiotic
The Antimicrobial Drugs Advisory Committee of the Food and Drug Administration has voted to recommend plazomicin, a new aminoglycoside, for systemic use in the treatment of complicated urinary tract infections (cUTI) but rejected it for treatment of blood stream infections (BSIs) that are caused by multidrug resistant (MDR) Enterobacteriaceae.
Advisers voted unanimously to recommend plazomicin for cUTI, but rejected the drug for BSIs in an 11-4 no vote, based on the results of two phase 3 clinical trials: EPIC and CARE.
“Today’s meeting brought the committee face to face with the crisis of multidrug resistant bacteria,” he said. “Results of the 009 [EPIC] study, in my mind, clearly showed plazomicin met the noninferiority endpoints.”
EPIC study
EPIC was a phase 3 clinical trial to assess the noninferiority of plazomicin to meropenem in patients with cUTI and/or acute pyelonephritis (AP). Many patients with drug resistant infections have limited treatment options, so plazomicin was reviewed under the Limited Population Antibacterial Drug pathway.
Patients in the study were stratified by geographical region and infection type – cUTI or acute pyelonephritis (AP). In total, 609 patients were randomized in the intent-to-treat (ITT) group with 306 and 303 receiving plazomicin or meropenem, respectively. Using the coprimary efficacy endpoints of microbiological eradication and clinical cure, a measure known as composite cure was developed to assess efficacy at Day 5 and the test of cure (TOC) visit in the microbiological modified intent-to-treat (mMITT) population. The mMITT group consisted of all patients who had received any dose of study drug and had at least one qualified baseline pathogen with 105 or more colony-forming units/mL that was susceptible to both meropenem and plazomicin.Plazomicin was noninferior to meropenem with a margin of 15%. At day 5, the composite cure rate was 88% in the plazomicin group, compared with 91.4% in the meropenem group. Similar results were seen at the test of cure visit, with composite cure rates of 81.7% and 70.1% in the plazomicin and meropenem groups, respectively.
In a secondary analysis of microbiologically evaluable populations, or patients who had an appropriately collected urine specimen yielding interpretable culture results, plazomicin again showed noninferiority. Composite cure rates at Day 5 were 89.4% in the plazomicin group, compared with 94.2% in the meropenem group. TOC composite scores also were similar, at 89.4% and 75.1%, respectively.An analysis of composite cure scores at the end of the intravenous therapy visit revealed that Day 5 scores were comparable between the plazomicin and meropenem groups at 93.7% and 94.9%, respectively, demonstrating that high cure rates were achieved with IV treatment, irrespective of the drug used.
In a pooled safety analysis of a phase 2 trial and the EPIC trial, 22.5% experienced a treatment-emergent adverse event (TEAE); of these, 2.9% had a severe TEAE and 2.7% experienced a TEAE that led to discontinuation of the intravenous study drug. All adverse events were related to renal function, diarrhea, headache, nausea, and vomiting.
CARE study
CARE was an open-label trial to assess the safety and efficacy of plazomicin as compared with colistin in patients with hospital-acquired bacterial pneumonia or ventilator-associated bacterial pneumonia (HABP/VABP) or bloodstream infections caused by CRE. The primary endpoint in the study was all-cause mortality at Day 28 or significant disease-related complications in the mMITT population, which included all patients with a confirmed CRE pathogen who had at least one dose of the study drug.
Overall, CARE enrolled 69 patients and split them into two cohorts. In Cohort 1, there were 39 randomized patients; 30 with blood stream infections and 9 with HABP or VABP. Cohort 2 was uncontrolled and consisted of 30 total patients; 27 patients who were in the mMITT population included 14 patients with BSI, 9 with HABP/VABP, and 4 with cUTI, all of whom received plazomicin.
Overall in Cohort 1, all-cause mortality at Day 28 or significant disease-related complications were lower with plazomicin than with colistin (23.5% vs. 50.0%). Because of the small patient HABP/VABP population in Cohort 1, the trial focused on patients with blood stream infections.
The rates of all-cause mortality and significant disease-related complications at Day 28 were much lower with plazomicin therapy than with colistin for patients with blood infections (14.3% vs. 53.3%, respectively). All-cause mortality alone at Day 28 was 7.1% in the plazomicin patients and 40.0% in colistin patients.
In the BSI subgroup, plazomicin reduced the rate of death by 86% and 63% on days 28 and 60, respectively, compared with colistin.
The uncontrolled data from Cohort 2 were supportive of the all-cause mortality rate in Cohort 1, with a rate of 14.3% at Day 28.
Because this was a severely ill patient population, there was a higher rate of adverse events. In fact, nearly all the patients in this study (16/18) experienced one treatment-emergent adverse event; 61.1% experienced severe TEAEs, with 11.1% severe enough to discontinue the study drug use.
Despite the study results, many of the panel members were not comfortable recommending it for approval.
Dr. Green, who supported plazomicin for use in cUTI but rejected it for use in BSIs, shared some of his concerns: “Because of the clear need, I was really tempted to vote yes and I actually came here today, thinking that I was going to vote yes. But this study clearly had a number of limitations that impacted the interpretation of results to support the approval for bloodstream infection,” he said. “The limitation that I could just not overcome was the small numbers [of participants].”
Plazomicin has a Prescription Drug User Fee Act date of June 25 of this year by which time the FDA will decide on its approval. While the FDA does not always follow the recommendations of these committees, they usually accept them and proceed accordingly.
The Antimicrobial Drugs Advisory Committee of the Food and Drug Administration has voted to recommend plazomicin, a new aminoglycoside, for systemic use in the treatment of complicated urinary tract infections (cUTI) but rejected it for treatment of blood stream infections (BSIs) that are caused by multidrug resistant (MDR) Enterobacteriaceae.
Advisers voted unanimously to recommend plazomicin for cUTI, but rejected the drug for BSIs in an 11-4 no vote, based on the results of two phase 3 clinical trials: EPIC and CARE.
“Today’s meeting brought the committee face to face with the crisis of multidrug resistant bacteria,” he said. “Results of the 009 [EPIC] study, in my mind, clearly showed plazomicin met the noninferiority endpoints.”
EPIC study
EPIC was a phase 3 clinical trial to assess the noninferiority of plazomicin to meropenem in patients with cUTI and/or acute pyelonephritis (AP). Many patients with drug resistant infections have limited treatment options, so plazomicin was reviewed under the Limited Population Antibacterial Drug pathway.
Patients in the study were stratified by geographical region and infection type – cUTI or acute pyelonephritis (AP). In total, 609 patients were randomized in the intent-to-treat (ITT) group with 306 and 303 receiving plazomicin or meropenem, respectively. Using the coprimary efficacy endpoints of microbiological eradication and clinical cure, a measure known as composite cure was developed to assess efficacy at Day 5 and the test of cure (TOC) visit in the microbiological modified intent-to-treat (mMITT) population. The mMITT group consisted of all patients who had received any dose of study drug and had at least one qualified baseline pathogen with 105 or more colony-forming units/mL that was susceptible to both meropenem and plazomicin.Plazomicin was noninferior to meropenem with a margin of 15%. At day 5, the composite cure rate was 88% in the plazomicin group, compared with 91.4% in the meropenem group. Similar results were seen at the test of cure visit, with composite cure rates of 81.7% and 70.1% in the plazomicin and meropenem groups, respectively.
In a secondary analysis of microbiologically evaluable populations, or patients who had an appropriately collected urine specimen yielding interpretable culture results, plazomicin again showed noninferiority. Composite cure rates at Day 5 were 89.4% in the plazomicin group, compared with 94.2% in the meropenem group. TOC composite scores also were similar, at 89.4% and 75.1%, respectively.An analysis of composite cure scores at the end of the intravenous therapy visit revealed that Day 5 scores were comparable between the plazomicin and meropenem groups at 93.7% and 94.9%, respectively, demonstrating that high cure rates were achieved with IV treatment, irrespective of the drug used.
In a pooled safety analysis of a phase 2 trial and the EPIC trial, 22.5% experienced a treatment-emergent adverse event (TEAE); of these, 2.9% had a severe TEAE and 2.7% experienced a TEAE that led to discontinuation of the intravenous study drug. All adverse events were related to renal function, diarrhea, headache, nausea, and vomiting.
CARE study
CARE was an open-label trial to assess the safety and efficacy of plazomicin as compared with colistin in patients with hospital-acquired bacterial pneumonia or ventilator-associated bacterial pneumonia (HABP/VABP) or bloodstream infections caused by CRE. The primary endpoint in the study was all-cause mortality at Day 28 or significant disease-related complications in the mMITT population, which included all patients with a confirmed CRE pathogen who had at least one dose of the study drug.
Overall, CARE enrolled 69 patients and split them into two cohorts. In Cohort 1, there were 39 randomized patients; 30 with blood stream infections and 9 with HABP or VABP. Cohort 2 was uncontrolled and consisted of 30 total patients; 27 patients who were in the mMITT population included 14 patients with BSI, 9 with HABP/VABP, and 4 with cUTI, all of whom received plazomicin.
Overall in Cohort 1, all-cause mortality at Day 28 or significant disease-related complications were lower with plazomicin than with colistin (23.5% vs. 50.0%). Because of the small patient HABP/VABP population in Cohort 1, the trial focused on patients with blood stream infections.
The rates of all-cause mortality and significant disease-related complications at Day 28 were much lower with plazomicin therapy than with colistin for patients with blood infections (14.3% vs. 53.3%, respectively). All-cause mortality alone at Day 28 was 7.1% in the plazomicin patients and 40.0% in colistin patients.
In the BSI subgroup, plazomicin reduced the rate of death by 86% and 63% on days 28 and 60, respectively, compared with colistin.
The uncontrolled data from Cohort 2 were supportive of the all-cause mortality rate in Cohort 1, with a rate of 14.3% at Day 28.
Because this was a severely ill patient population, there was a higher rate of adverse events. In fact, nearly all the patients in this study (16/18) experienced one treatment-emergent adverse event; 61.1% experienced severe TEAEs, with 11.1% severe enough to discontinue the study drug use.
Despite the study results, many of the panel members were not comfortable recommending it for approval.
Dr. Green, who supported plazomicin for use in cUTI but rejected it for use in BSIs, shared some of his concerns: “Because of the clear need, I was really tempted to vote yes and I actually came here today, thinking that I was going to vote yes. But this study clearly had a number of limitations that impacted the interpretation of results to support the approval for bloodstream infection,” he said. “The limitation that I could just not overcome was the small numbers [of participants].”
Plazomicin has a Prescription Drug User Fee Act date of June 25 of this year by which time the FDA will decide on its approval. While the FDA does not always follow the recommendations of these committees, they usually accept them and proceed accordingly.
The Antimicrobial Drugs Advisory Committee of the Food and Drug Administration has voted to recommend plazomicin, a new aminoglycoside, for systemic use in the treatment of complicated urinary tract infections (cUTI) but rejected it for treatment of blood stream infections (BSIs) that are caused by multidrug resistant (MDR) Enterobacteriaceae.
Advisers voted unanimously to recommend plazomicin for cUTI, but rejected the drug for BSIs in an 11-4 no vote, based on the results of two phase 3 clinical trials: EPIC and CARE.
“Today’s meeting brought the committee face to face with the crisis of multidrug resistant bacteria,” he said. “Results of the 009 [EPIC] study, in my mind, clearly showed plazomicin met the noninferiority endpoints.”
EPIC study
EPIC was a phase 3 clinical trial to assess the noninferiority of plazomicin to meropenem in patients with cUTI and/or acute pyelonephritis (AP). Many patients with drug resistant infections have limited treatment options, so plazomicin was reviewed under the Limited Population Antibacterial Drug pathway.
Patients in the study were stratified by geographical region and infection type – cUTI or acute pyelonephritis (AP). In total, 609 patients were randomized in the intent-to-treat (ITT) group with 306 and 303 receiving plazomicin or meropenem, respectively. Using the coprimary efficacy endpoints of microbiological eradication and clinical cure, a measure known as composite cure was developed to assess efficacy at Day 5 and the test of cure (TOC) visit in the microbiological modified intent-to-treat (mMITT) population. The mMITT group consisted of all patients who had received any dose of study drug and had at least one qualified baseline pathogen with 105 or more colony-forming units/mL that was susceptible to both meropenem and plazomicin.Plazomicin was noninferior to meropenem with a margin of 15%. At day 5, the composite cure rate was 88% in the plazomicin group, compared with 91.4% in the meropenem group. Similar results were seen at the test of cure visit, with composite cure rates of 81.7% and 70.1% in the plazomicin and meropenem groups, respectively.
In a secondary analysis of microbiologically evaluable populations, or patients who had an appropriately collected urine specimen yielding interpretable culture results, plazomicin again showed noninferiority. Composite cure rates at Day 5 were 89.4% in the plazomicin group, compared with 94.2% in the meropenem group. TOC composite scores also were similar, at 89.4% and 75.1%, respectively.An analysis of composite cure scores at the end of the intravenous therapy visit revealed that Day 5 scores were comparable between the plazomicin and meropenem groups at 93.7% and 94.9%, respectively, demonstrating that high cure rates were achieved with IV treatment, irrespective of the drug used.
In a pooled safety analysis of a phase 2 trial and the EPIC trial, 22.5% experienced a treatment-emergent adverse event (TEAE); of these, 2.9% had a severe TEAE and 2.7% experienced a TEAE that led to discontinuation of the intravenous study drug. All adverse events were related to renal function, diarrhea, headache, nausea, and vomiting.
CARE study
CARE was an open-label trial to assess the safety and efficacy of plazomicin as compared with colistin in patients with hospital-acquired bacterial pneumonia or ventilator-associated bacterial pneumonia (HABP/VABP) or bloodstream infections caused by CRE. The primary endpoint in the study was all-cause mortality at Day 28 or significant disease-related complications in the mMITT population, which included all patients with a confirmed CRE pathogen who had at least one dose of the study drug.
Overall, CARE enrolled 69 patients and split them into two cohorts. In Cohort 1, there were 39 randomized patients; 30 with blood stream infections and 9 with HABP or VABP. Cohort 2 was uncontrolled and consisted of 30 total patients; 27 patients who were in the mMITT population included 14 patients with BSI, 9 with HABP/VABP, and 4 with cUTI, all of whom received plazomicin.
Overall in Cohort 1, all-cause mortality at Day 28 or significant disease-related complications were lower with plazomicin than with colistin (23.5% vs. 50.0%). Because of the small patient HABP/VABP population in Cohort 1, the trial focused on patients with blood stream infections.
The rates of all-cause mortality and significant disease-related complications at Day 28 were much lower with plazomicin therapy than with colistin for patients with blood infections (14.3% vs. 53.3%, respectively). All-cause mortality alone at Day 28 was 7.1% in the plazomicin patients and 40.0% in colistin patients.
In the BSI subgroup, plazomicin reduced the rate of death by 86% and 63% on days 28 and 60, respectively, compared with colistin.
The uncontrolled data from Cohort 2 were supportive of the all-cause mortality rate in Cohort 1, with a rate of 14.3% at Day 28.
Because this was a severely ill patient population, there was a higher rate of adverse events. In fact, nearly all the patients in this study (16/18) experienced one treatment-emergent adverse event; 61.1% experienced severe TEAEs, with 11.1% severe enough to discontinue the study drug use.
Despite the study results, many of the panel members were not comfortable recommending it for approval.
Dr. Green, who supported plazomicin for use in cUTI but rejected it for use in BSIs, shared some of his concerns: “Because of the clear need, I was really tempted to vote yes and I actually came here today, thinking that I was going to vote yes. But this study clearly had a number of limitations that impacted the interpretation of results to support the approval for bloodstream infection,” he said. “The limitation that I could just not overcome was the small numbers [of participants].”
Plazomicin has a Prescription Drug User Fee Act date of June 25 of this year by which time the FDA will decide on its approval. While the FDA does not always follow the recommendations of these committees, they usually accept them and proceed accordingly.