User login
FDA orders companies to cease all sales of transvaginal mesh for POP repair

The mandate came after Boston Scientific and Coloplast failed to provide adequate safety and efficacy information to the federal regulatory body in the wake of a 2016 reclassification to Class III (high-risk) devices, according to an FDA press statement. Both companies were required to submit a premarket approval application to continue marketing the mesh in the United States. Boston Scientific did file two PMAs, one for each of its transvaginal mesh products, but the FDA said the applications did not contain the required efficacy and safety data.
Both companies will have 10 days to submit their plan to withdraw these products from the market.
“In order for these mesh devices to stay on the market, we determined that we needed evidence that they worked better than surgery without the use of mesh to repair POP. That evidence was lacking in these premarket applications, and we couldn’t assure women that these devices were safe and effective long term,” said Jeffrey Shuren, MD, director of FDA’s Center for Devices and Radiological Health. “Patient safety is our highest priority, and women must have access to safe medical devices that provide relief from symptoms and better management of their medical conditions. The FDA has committed to taking forceful new actions to enhance device safety and encourage innovations that lead to safer medical devices, so that patients have access to safe and effective medical devices and the information they need to make informed decisions about their care.”
The deadline for submitting premarket approval applications for POP repair with transvaginal mesh was July 5, 2018. Manufacturers that did not file PMAs were required to pull their devices from the market. Those that did could keep selling the mesh while FDA reviewed their PMAs.
Boston Scientific submitted PMAs for its two devices, the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair System. Coloplast filed a PMA for its device, Restorelle DirectFix Anterior. But in February, the FDA convened an advisory panel to discuss just how to evaluate the safety and efficacy of the products.
To prove efficacy, the panel concluded, transvaginal POP repair with mesh should be better than repair with native tissue at 36 months, and the safety should be superior to repair with native tissue repair. The FDA agreed. However, the submitted premarket approval application did not include these kinds of data. Therefore, the agency declined to approve the devices.
In addition to stopping U.S. sales, FDA has required Boston Scientific and Coloplast to continue safety and efficacy follow-up of all women included in their 522 studies.
Coloplast did not have a press or public statement on its website as of April 16. Boston Scientific did have one.
“Up to 50% of women in the U.S. will suffer from POP during their lives, and we believe these women should have access to safe and effective treatment options,” according to the statement. “As a global leader in the pelvic floor space, we remain steadfast in our commitment to helping women live better and healthier lives. We also remain confident in the benefits and safety of our treatments for POP, and we look forward to continuing to work with the FDA on our PMAs for the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair Matrix, which are currently under review.”
The FDA statement also included advice to women who have had the mesh procedure for POP, and for their physicians
“Women who have had transvaginal mesh placed for the surgical repair of POP should continue with their annual and other routine check-ups and follow-up care. There is no need to take additional action if they are satisfied with their surgery and are not having complications or symptoms. Patients should notify their health care professionals if they have complications or symptoms, including persistent vaginal bleeding or discharge, pelvic or groin pain, or pain with sex. They should also let their health care professional know if they have surgical mesh, especially if they plan to have another surgery or other medical procedures. Women who were planning to have mesh placed transvaginally for the repair of POP should discuss other treatment options with their doctors.”
The Food and Drug Administration’s decision ordering manufacturers to remove mesh for transvaginal repair of prolapse from the market was based on the products’ effectiveness and safety profile, compared with vaginal native tissue repairs. Previous studies have shown that polypropylene mesh for anterior repair had similar or slightly higher success, compared with native tissue repairs. This was not a sufficient benefit considering the potential adverse events that include mesh exposure, and the pelvic pain and dyspareunia associated with using these products. There is no additional benefit of using polypropylene mesh in the posterior compartment.
It would be interesting to review the information provided by manufacturers as part of the premarket approval. What were the primary endpoints for efficacy that were used? What were the rates of complications for mesh exposure, pelvic pain, and dyspareunia? How did the rates of pelvic pain and dyspareunia compare with native tissue repair.
Gynecologic surgeons still have a number of options for treating vaginal prolapse, which include vaginal native tissue repairs, and laparoscopic and abdominal surgeries that involve native tissue or polypropylene mesh. It will be interesting to see how the FDA’s Medical Device Safety action plan will affect future innovations for treating vaginal prolapse, while at the same time providing women and their physicians with products that are safe and effective.
Jose S. Maceda, MD, is a urogynecologist at Axia Women’s Health in King of Prussia, Penn. Dr. Maceda, who was asked to comment on the FDA decision, has no relevant financial disclosures.
The Food and Drug Administration’s decision ordering manufacturers to remove mesh for transvaginal repair of prolapse from the market was based on the products’ effectiveness and safety profile, compared with vaginal native tissue repairs. Previous studies have shown that polypropylene mesh for anterior repair had similar or slightly higher success, compared with native tissue repairs. This was not a sufficient benefit considering the potential adverse events that include mesh exposure, and the pelvic pain and dyspareunia associated with using these products. There is no additional benefit of using polypropylene mesh in the posterior compartment.
It would be interesting to review the information provided by manufacturers as part of the premarket approval. What were the primary endpoints for efficacy that were used? What were the rates of complications for mesh exposure, pelvic pain, and dyspareunia? How did the rates of pelvic pain and dyspareunia compare with native tissue repair.
Gynecologic surgeons still have a number of options for treating vaginal prolapse, which include vaginal native tissue repairs, and laparoscopic and abdominal surgeries that involve native tissue or polypropylene mesh. It will be interesting to see how the FDA’s Medical Device Safety action plan will affect future innovations for treating vaginal prolapse, while at the same time providing women and their physicians with products that are safe and effective.
Jose S. Maceda, MD, is a urogynecologist at Axia Women’s Health in King of Prussia, Penn. Dr. Maceda, who was asked to comment on the FDA decision, has no relevant financial disclosures.
The Food and Drug Administration’s decision ordering manufacturers to remove mesh for transvaginal repair of prolapse from the market was based on the products’ effectiveness and safety profile, compared with vaginal native tissue repairs. Previous studies have shown that polypropylene mesh for anterior repair had similar or slightly higher success, compared with native tissue repairs. This was not a sufficient benefit considering the potential adverse events that include mesh exposure, and the pelvic pain and dyspareunia associated with using these products. There is no additional benefit of using polypropylene mesh in the posterior compartment.
It would be interesting to review the information provided by manufacturers as part of the premarket approval. What were the primary endpoints for efficacy that were used? What were the rates of complications for mesh exposure, pelvic pain, and dyspareunia? How did the rates of pelvic pain and dyspareunia compare with native tissue repair.
Gynecologic surgeons still have a number of options for treating vaginal prolapse, which include vaginal native tissue repairs, and laparoscopic and abdominal surgeries that involve native tissue or polypropylene mesh. It will be interesting to see how the FDA’s Medical Device Safety action plan will affect future innovations for treating vaginal prolapse, while at the same time providing women and their physicians with products that are safe and effective.
Jose S. Maceda, MD, is a urogynecologist at Axia Women’s Health in King of Prussia, Penn. Dr. Maceda, who was asked to comment on the FDA decision, has no relevant financial disclosures.
The mandate came after Boston Scientific and Coloplast failed to provide adequate safety and efficacy information to the federal regulatory body in the wake of a 2016 reclassification to Class III (high-risk) devices, according to an FDA press statement. Both companies were required to submit a premarket approval application to continue marketing the mesh in the United States. Boston Scientific did file two PMAs, one for each of its transvaginal mesh products, but the FDA said the applications did not contain the required efficacy and safety data.
Both companies will have 10 days to submit their plan to withdraw these products from the market.
“In order for these mesh devices to stay on the market, we determined that we needed evidence that they worked better than surgery without the use of mesh to repair POP. That evidence was lacking in these premarket applications, and we couldn’t assure women that these devices were safe and effective long term,” said Jeffrey Shuren, MD, director of FDA’s Center for Devices and Radiological Health. “Patient safety is our highest priority, and women must have access to safe medical devices that provide relief from symptoms and better management of their medical conditions. The FDA has committed to taking forceful new actions to enhance device safety and encourage innovations that lead to safer medical devices, so that patients have access to safe and effective medical devices and the information they need to make informed decisions about their care.”
The deadline for submitting premarket approval applications for POP repair with transvaginal mesh was July 5, 2018. Manufacturers that did not file PMAs were required to pull their devices from the market. Those that did could keep selling the mesh while FDA reviewed their PMAs.
Boston Scientific submitted PMAs for its two devices, the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair System. Coloplast filed a PMA for its device, Restorelle DirectFix Anterior. But in February, the FDA convened an advisory panel to discuss just how to evaluate the safety and efficacy of the products.
To prove efficacy, the panel concluded, transvaginal POP repair with mesh should be better than repair with native tissue at 36 months, and the safety should be superior to repair with native tissue repair. The FDA agreed. However, the submitted premarket approval application did not include these kinds of data. Therefore, the agency declined to approve the devices.
In addition to stopping U.S. sales, FDA has required Boston Scientific and Coloplast to continue safety and efficacy follow-up of all women included in their 522 studies.
Coloplast did not have a press or public statement on its website as of April 16. Boston Scientific did have one.
“Up to 50% of women in the U.S. will suffer from POP during their lives, and we believe these women should have access to safe and effective treatment options,” according to the statement. “As a global leader in the pelvic floor space, we remain steadfast in our commitment to helping women live better and healthier lives. We also remain confident in the benefits and safety of our treatments for POP, and we look forward to continuing to work with the FDA on our PMAs for the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair Matrix, which are currently under review.”
The FDA statement also included advice to women who have had the mesh procedure for POP, and for their physicians
“Women who have had transvaginal mesh placed for the surgical repair of POP should continue with their annual and other routine check-ups and follow-up care. There is no need to take additional action if they are satisfied with their surgery and are not having complications or symptoms. Patients should notify their health care professionals if they have complications or symptoms, including persistent vaginal bleeding or discharge, pelvic or groin pain, or pain with sex. They should also let their health care professional know if they have surgical mesh, especially if they plan to have another surgery or other medical procedures. Women who were planning to have mesh placed transvaginally for the repair of POP should discuss other treatment options with their doctors.”
The mandate came after Boston Scientific and Coloplast failed to provide adequate safety and efficacy information to the federal regulatory body in the wake of a 2016 reclassification to Class III (high-risk) devices, according to an FDA press statement. Both companies were required to submit a premarket approval application to continue marketing the mesh in the United States. Boston Scientific did file two PMAs, one for each of its transvaginal mesh products, but the FDA said the applications did not contain the required efficacy and safety data.
Both companies will have 10 days to submit their plan to withdraw these products from the market.
“In order for these mesh devices to stay on the market, we determined that we needed evidence that they worked better than surgery without the use of mesh to repair POP. That evidence was lacking in these premarket applications, and we couldn’t assure women that these devices were safe and effective long term,” said Jeffrey Shuren, MD, director of FDA’s Center for Devices and Radiological Health. “Patient safety is our highest priority, and women must have access to safe medical devices that provide relief from symptoms and better management of their medical conditions. The FDA has committed to taking forceful new actions to enhance device safety and encourage innovations that lead to safer medical devices, so that patients have access to safe and effective medical devices and the information they need to make informed decisions about their care.”
The deadline for submitting premarket approval applications for POP repair with transvaginal mesh was July 5, 2018. Manufacturers that did not file PMAs were required to pull their devices from the market. Those that did could keep selling the mesh while FDA reviewed their PMAs.
Boston Scientific submitted PMAs for its two devices, the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair System. Coloplast filed a PMA for its device, Restorelle DirectFix Anterior. But in February, the FDA convened an advisory panel to discuss just how to evaluate the safety and efficacy of the products.
To prove efficacy, the panel concluded, transvaginal POP repair with mesh should be better than repair with native tissue at 36 months, and the safety should be superior to repair with native tissue repair. The FDA agreed. However, the submitted premarket approval application did not include these kinds of data. Therefore, the agency declined to approve the devices.
In addition to stopping U.S. sales, FDA has required Boston Scientific and Coloplast to continue safety and efficacy follow-up of all women included in their 522 studies.
Coloplast did not have a press or public statement on its website as of April 16. Boston Scientific did have one.
“Up to 50% of women in the U.S. will suffer from POP during their lives, and we believe these women should have access to safe and effective treatment options,” according to the statement. “As a global leader in the pelvic floor space, we remain steadfast in our commitment to helping women live better and healthier lives. We also remain confident in the benefits and safety of our treatments for POP, and we look forward to continuing to work with the FDA on our PMAs for the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair Matrix, which are currently under review.”
The FDA statement also included advice to women who have had the mesh procedure for POP, and for their physicians
“Women who have had transvaginal mesh placed for the surgical repair of POP should continue with their annual and other routine check-ups and follow-up care. There is no need to take additional action if they are satisfied with their surgery and are not having complications or symptoms. Patients should notify their health care professionals if they have complications or symptoms, including persistent vaginal bleeding or discharge, pelvic or groin pain, or pain with sex. They should also let their health care professional know if they have surgical mesh, especially if they plan to have another surgery or other medical procedures. Women who were planning to have mesh placed transvaginally for the repair of POP should discuss other treatment options with their doctors.”
Direct-to-consumer genetic testing fraught with validity concerns
PHILADELPHIA – There are real concerns about whether today’s direct-to-consumer (DTC) genetic tests actually test the mutations they claim to test and of how well a tested DNA sequence tracks actual disease, Andrew D. Coyle, MD, said during a podium presentation at the annual meeting of the American College of Physicians.
said Dr. Coyle, assistant professor of medicine and medical education in the general internal medicine division at the Icahn School of Medicine at Mount Sinai, New York.
“I wouldn’t recommend to patients that they do [DTC testing], if they asked,” he said. “I think we probably do need to be better about asking our patients whether they’re doing this on their own, to make sure that we help them interpret it correctly, that it’s a change in risk – not a diagnosis, and not an assurance they won’t get that disease process.”
High false-positive rates have been seen in recent studies that sought to confirm genotyping data from DTC genetic test results, according to Dr. Coyle. In one 2018 study in Genetics and Medicine (2018;20:1515-21) of 49 patient samples tested for previously identified genetic variants found in raw DTC data, investigators found a 40% false positive rate, which they said underscored the importance of clinical confirmation testing to assure proper patient care, he noted.
To support his claim that many patients aren’t even telling their doctors about the DTC testing they are having done in the first place, Dr. Coyle mentioned results of a survey in Annals of Internal Medicine (2016;164[8]:513-22), which showed that only 19% of patients shared their DTC results with their primary care physicians.
He also pointed out a potential problem regarding the discussions between patients and their physicians about these test results, based on another finding reported in the paper. Of those who did tell their physicians they had DTC testing, 35% said they were “very satisfied” with how that discussion went, Dr. Coyle said.
“So they don’t tell us, and they aren’t very happy when they do tell us,” he told his audience.
The 23andMe test and the FDA
The 23andMe test, which was first directly marketed to consumers in 2006, was one of the DTC genetic tests discussed by Dr. Coyle. The Food and Drug Administration later halted the company’s Personal Genome Service in 2013 because of a lack of demonstrated clinical validity, Dr. Coyle noted.
Subsequently, the FDA classified carrier-screening tests as medical devices, allowing such services to come back, leading to what Dr. Coyle described as an explosion over the past few years of DTC evaluation of a variety of conditions, including celiac disease, Alzheimer’s disease, hemochromatosis, and then more recently, screening for cancer risk factors.
In 2018, 23andMe began offering DTC BRCA testing, but for 3 BRCA1/2 mutations seen in individuals of Ashkenazi Jewish descent, Dr. Coyle said.
“If someone did a BRCA test which is only testing three specific mutations seen mostly in Ashkenazi Jewish populations, they may be falsely reassured that the risk of breast cancer is low, when in fact they may have other BRCA mutations,” he said.
In real life, however, many people who order genetic tests online may not even act on the results. In a 2017 study in the Journal of Clinical Oncology, customers whose DTC genetic testing results showed elevated cancer risk were no more likely than were customers without elevated risk to change diet or exercise, engage in advanced planning behaviors, or get screened.
Dr. Coyle had no relevant disclosures to report.
PHILADELPHIA – There are real concerns about whether today’s direct-to-consumer (DTC) genetic tests actually test the mutations they claim to test and of how well a tested DNA sequence tracks actual disease, Andrew D. Coyle, MD, said during a podium presentation at the annual meeting of the American College of Physicians.
said Dr. Coyle, assistant professor of medicine and medical education in the general internal medicine division at the Icahn School of Medicine at Mount Sinai, New York.
“I wouldn’t recommend to patients that they do [DTC testing], if they asked,” he said. “I think we probably do need to be better about asking our patients whether they’re doing this on their own, to make sure that we help them interpret it correctly, that it’s a change in risk – not a diagnosis, and not an assurance they won’t get that disease process.”
High false-positive rates have been seen in recent studies that sought to confirm genotyping data from DTC genetic test results, according to Dr. Coyle. In one 2018 study in Genetics and Medicine (2018;20:1515-21) of 49 patient samples tested for previously identified genetic variants found in raw DTC data, investigators found a 40% false positive rate, which they said underscored the importance of clinical confirmation testing to assure proper patient care, he noted.
To support his claim that many patients aren’t even telling their doctors about the DTC testing they are having done in the first place, Dr. Coyle mentioned results of a survey in Annals of Internal Medicine (2016;164[8]:513-22), which showed that only 19% of patients shared their DTC results with their primary care physicians.
He also pointed out a potential problem regarding the discussions between patients and their physicians about these test results, based on another finding reported in the paper. Of those who did tell their physicians they had DTC testing, 35% said they were “very satisfied” with how that discussion went, Dr. Coyle said.
“So they don’t tell us, and they aren’t very happy when they do tell us,” he told his audience.
The 23andMe test and the FDA
The 23andMe test, which was first directly marketed to consumers in 2006, was one of the DTC genetic tests discussed by Dr. Coyle. The Food and Drug Administration later halted the company’s Personal Genome Service in 2013 because of a lack of demonstrated clinical validity, Dr. Coyle noted.
Subsequently, the FDA classified carrier-screening tests as medical devices, allowing such services to come back, leading to what Dr. Coyle described as an explosion over the past few years of DTC evaluation of a variety of conditions, including celiac disease, Alzheimer’s disease, hemochromatosis, and then more recently, screening for cancer risk factors.
In 2018, 23andMe began offering DTC BRCA testing, but for 3 BRCA1/2 mutations seen in individuals of Ashkenazi Jewish descent, Dr. Coyle said.
“If someone did a BRCA test which is only testing three specific mutations seen mostly in Ashkenazi Jewish populations, they may be falsely reassured that the risk of breast cancer is low, when in fact they may have other BRCA mutations,” he said.
In real life, however, many people who order genetic tests online may not even act on the results. In a 2017 study in the Journal of Clinical Oncology, customers whose DTC genetic testing results showed elevated cancer risk were no more likely than were customers without elevated risk to change diet or exercise, engage in advanced planning behaviors, or get screened.
Dr. Coyle had no relevant disclosures to report.
PHILADELPHIA – There are real concerns about whether today’s direct-to-consumer (DTC) genetic tests actually test the mutations they claim to test and of how well a tested DNA sequence tracks actual disease, Andrew D. Coyle, MD, said during a podium presentation at the annual meeting of the American College of Physicians.
said Dr. Coyle, assistant professor of medicine and medical education in the general internal medicine division at the Icahn School of Medicine at Mount Sinai, New York.
“I wouldn’t recommend to patients that they do [DTC testing], if they asked,” he said. “I think we probably do need to be better about asking our patients whether they’re doing this on their own, to make sure that we help them interpret it correctly, that it’s a change in risk – not a diagnosis, and not an assurance they won’t get that disease process.”
High false-positive rates have been seen in recent studies that sought to confirm genotyping data from DTC genetic test results, according to Dr. Coyle. In one 2018 study in Genetics and Medicine (2018;20:1515-21) of 49 patient samples tested for previously identified genetic variants found in raw DTC data, investigators found a 40% false positive rate, which they said underscored the importance of clinical confirmation testing to assure proper patient care, he noted.
To support his claim that many patients aren’t even telling their doctors about the DTC testing they are having done in the first place, Dr. Coyle mentioned results of a survey in Annals of Internal Medicine (2016;164[8]:513-22), which showed that only 19% of patients shared their DTC results with their primary care physicians.
He also pointed out a potential problem regarding the discussions between patients and their physicians about these test results, based on another finding reported in the paper. Of those who did tell their physicians they had DTC testing, 35% said they were “very satisfied” with how that discussion went, Dr. Coyle said.
“So they don’t tell us, and they aren’t very happy when they do tell us,” he told his audience.
The 23andMe test and the FDA
The 23andMe test, which was first directly marketed to consumers in 2006, was one of the DTC genetic tests discussed by Dr. Coyle. The Food and Drug Administration later halted the company’s Personal Genome Service in 2013 because of a lack of demonstrated clinical validity, Dr. Coyle noted.
Subsequently, the FDA classified carrier-screening tests as medical devices, allowing such services to come back, leading to what Dr. Coyle described as an explosion over the past few years of DTC evaluation of a variety of conditions, including celiac disease, Alzheimer’s disease, hemochromatosis, and then more recently, screening for cancer risk factors.
In 2018, 23andMe began offering DTC BRCA testing, but for 3 BRCA1/2 mutations seen in individuals of Ashkenazi Jewish descent, Dr. Coyle said.
“If someone did a BRCA test which is only testing three specific mutations seen mostly in Ashkenazi Jewish populations, they may be falsely reassured that the risk of breast cancer is low, when in fact they may have other BRCA mutations,” he said.
In real life, however, many people who order genetic tests online may not even act on the results. In a 2017 study in the Journal of Clinical Oncology, customers whose DTC genetic testing results showed elevated cancer risk were no more likely than were customers without elevated risk to change diet or exercise, engage in advanced planning behaviors, or get screened.
Dr. Coyle had no relevant disclosures to report.
EXPERT ANALYSIS FROM INTERNAL MEDICINE 2019
CDC clarifies opioid prescribing guidelines in cancer, sickle cell disease
Officials at the Centers for Disease Control and Prevention have clarified the agency’s guidelines on opioid prescribing after a trio of organizations raised concerns that insurers were inappropriately applying the recommendations to active cancer patients when making coverage determinations.

The CDC guidelines, released in March 2016, address when to initiate or continue opioids for chronic pain, opioid selection, dosage, duration, follow-up, and discontinuation, and assess risk and harms of opioid use. Although the guidelines clearly state they are intended for clinicians prescribing opioids outside of active cancer treatment, insurance companies are still applying the guidelines to opioid coverage decisions for patients with active cancer, according to a Feb. 13, 2019, letter sent to the CDC from leaders at the American Society of Clinical Oncology, the National Comprehensive Cancer Network, and the American Society of Hematology.
Additionally, the associations wrote that the CDC’s recommendations pose coverage problems for sickle cell patients and select groups of cancer survivors who may benefit from opioids for pain management. The groups asked the CDC to issue a clarification to ensure appropriate implementation of the opioid recommendations.
In a Feb. 28, 2019, letter to ASCO, NCCN, and ASH, Deborah Dowell, MD, chief medical officer for the CDC’s National Center for Injury Prevention and Control took note of the concerns, clarifying that the recommendations are not intended to deny clinically appropriate opioid therapy to any patients who suffer chronic pain, but rather to ensure that physicians and patients consider all safe and effective treatment options.
The CDC guidance may apply to cancer survivors in certain conditions, Dr. Dowell wrote, namely when survivors experience chronic pain after cancer treatment completion, are in clinical remission, and are under cancer surveillance only. However, she agreed that, for select groups of cancer survivors with persistent pain caused by past cancer, the ratio of opioid benefits to risks for chronic pain is unique. She referred health providers to guidelines by ASCO on chronic pain management for adult cancer survivors and NCCN guidance on managing adult cancer pain when considering opioids for pain control in such populations.
Special considerations in sickle cell disease may also change the balance of opioid risks to benefits for pain management, Dr. Dowell wrote, referring providers and insurers to additional guidance on sickle cell disease from the National Institute of Health when making treatment and reimbursement decisions.
“Clinical decision making should be based on the relationship between the clinician and patient, with an understanding of the patient’s clinical situation, functioning, and life context, as well as careful consideration of the benefits and risk of all treatment options, including opioid therapy,” Dr. Dowell wrote. “CDC encourages physicians to continue using their clinical judgment and base treatment on what they know about their patients, including the use of opioids if determined to be the best course of treatment.”
Clifford A. Hudis, MD, CEO of ASCO, praised the clarification, calling the letter necessary to clear up confusion and prevent inappropriate coverage decisions.
“This clarification from CDC is critically important because, while the agency’s guideline clearly states that it is not intended to apply to patients during active cancer and sickle cell disease treatment, many payers have been inappropriately using it to make opioid coverage determinations for those exact populations,” Dr. Hudis said in a statement.
Sickle cell patients suffer from severe, chronic pain, which is debilitating on its own without the added burden of having to constantly appeal coverage denials, added ASH President Roy Silverstein, MD.
“We appreciate CDC’s acknowledgment that the challenges of managing severe and chronic pain in conditions, such as sickle cell disease, require special consideration, and we hope payers will take the CDC’s clarification into account to ensure that patients’ pain management needs are covered,” he said in the same statement.
Officials at the Centers for Disease Control and Prevention have clarified the agency’s guidelines on opioid prescribing after a trio of organizations raised concerns that insurers were inappropriately applying the recommendations to active cancer patients when making coverage determinations.
The CDC guidelines, released in March 2016, address when to initiate or continue opioids for chronic pain, opioid selection, dosage, duration, follow-up, and discontinuation, and assess risk and harms of opioid use. Although the guidelines clearly state they are intended for clinicians prescribing opioids outside of active cancer treatment, insurance companies are still applying the guidelines to opioid coverage decisions for patients with active cancer, according to a Feb. 13, 2019, letter sent to the CDC from leaders at the American Society of Clinical Oncology, the National Comprehensive Cancer Network, and the American Society of Hematology.
Additionally, the associations wrote that the CDC’s recommendations pose coverage problems for sickle cell patients and select groups of cancer survivors who may benefit from opioids for pain management. The groups asked the CDC to issue a clarification to ensure appropriate implementation of the opioid recommendations.
In a Feb. 28, 2019, letter to ASCO, NCCN, and ASH, Deborah Dowell, MD, chief medical officer for the CDC’s National Center for Injury Prevention and Control took note of the concerns, clarifying that the recommendations are not intended to deny clinically appropriate opioid therapy to any patients who suffer chronic pain, but rather to ensure that physicians and patients consider all safe and effective treatment options.
The CDC guidance may apply to cancer survivors in certain conditions, Dr. Dowell wrote, namely when survivors experience chronic pain after cancer treatment completion, are in clinical remission, and are under cancer surveillance only. However, she agreed that, for select groups of cancer survivors with persistent pain caused by past cancer, the ratio of opioid benefits to risks for chronic pain is unique. She referred health providers to guidelines by ASCO on chronic pain management for adult cancer survivors and NCCN guidance on managing adult cancer pain when considering opioids for pain control in such populations.
Special considerations in sickle cell disease may also change the balance of opioid risks to benefits for pain management, Dr. Dowell wrote, referring providers and insurers to additional guidance on sickle cell disease from the National Institute of Health when making treatment and reimbursement decisions.
“Clinical decision making should be based on the relationship between the clinician and patient, with an understanding of the patient’s clinical situation, functioning, and life context, as well as careful consideration of the benefits and risk of all treatment options, including opioid therapy,” Dr. Dowell wrote. “CDC encourages physicians to continue using their clinical judgment and base treatment on what they know about their patients, including the use of opioids if determined to be the best course of treatment.”
Clifford A. Hudis, MD, CEO of ASCO, praised the clarification, calling the letter necessary to clear up confusion and prevent inappropriate coverage decisions.
“This clarification from CDC is critically important because, while the agency’s guideline clearly states that it is not intended to apply to patients during active cancer and sickle cell disease treatment, many payers have been inappropriately using it to make opioid coverage determinations for those exact populations,” Dr. Hudis said in a statement.
Sickle cell patients suffer from severe, chronic pain, which is debilitating on its own without the added burden of having to constantly appeal coverage denials, added ASH President Roy Silverstein, MD.
“We appreciate CDC’s acknowledgment that the challenges of managing severe and chronic pain in conditions, such as sickle cell disease, require special consideration, and we hope payers will take the CDC’s clarification into account to ensure that patients’ pain management needs are covered,” he said in the same statement.
Officials at the Centers for Disease Control and Prevention have clarified the agency’s guidelines on opioid prescribing after a trio of organizations raised concerns that insurers were inappropriately applying the recommendations to active cancer patients when making coverage determinations.
The CDC guidelines, released in March 2016, address when to initiate or continue opioids for chronic pain, opioid selection, dosage, duration, follow-up, and discontinuation, and assess risk and harms of opioid use. Although the guidelines clearly state they are intended for clinicians prescribing opioids outside of active cancer treatment, insurance companies are still applying the guidelines to opioid coverage decisions for patients with active cancer, according to a Feb. 13, 2019, letter sent to the CDC from leaders at the American Society of Clinical Oncology, the National Comprehensive Cancer Network, and the American Society of Hematology.
Additionally, the associations wrote that the CDC’s recommendations pose coverage problems for sickle cell patients and select groups of cancer survivors who may benefit from opioids for pain management. The groups asked the CDC to issue a clarification to ensure appropriate implementation of the opioid recommendations.
In a Feb. 28, 2019, letter to ASCO, NCCN, and ASH, Deborah Dowell, MD, chief medical officer for the CDC’s National Center for Injury Prevention and Control took note of the concerns, clarifying that the recommendations are not intended to deny clinically appropriate opioid therapy to any patients who suffer chronic pain, but rather to ensure that physicians and patients consider all safe and effective treatment options.
The CDC guidance may apply to cancer survivors in certain conditions, Dr. Dowell wrote, namely when survivors experience chronic pain after cancer treatment completion, are in clinical remission, and are under cancer surveillance only. However, she agreed that, for select groups of cancer survivors with persistent pain caused by past cancer, the ratio of opioid benefits to risks for chronic pain is unique. She referred health providers to guidelines by ASCO on chronic pain management for adult cancer survivors and NCCN guidance on managing adult cancer pain when considering opioids for pain control in such populations.
Special considerations in sickle cell disease may also change the balance of opioid risks to benefits for pain management, Dr. Dowell wrote, referring providers and insurers to additional guidance on sickle cell disease from the National Institute of Health when making treatment and reimbursement decisions.
“Clinical decision making should be based on the relationship between the clinician and patient, with an understanding of the patient’s clinical situation, functioning, and life context, as well as careful consideration of the benefits and risk of all treatment options, including opioid therapy,” Dr. Dowell wrote. “CDC encourages physicians to continue using their clinical judgment and base treatment on what they know about their patients, including the use of opioids if determined to be the best course of treatment.”
Clifford A. Hudis, MD, CEO of ASCO, praised the clarification, calling the letter necessary to clear up confusion and prevent inappropriate coverage decisions.
“This clarification from CDC is critically important because, while the agency’s guideline clearly states that it is not intended to apply to patients during active cancer and sickle cell disease treatment, many payers have been inappropriately using it to make opioid coverage determinations for those exact populations,” Dr. Hudis said in a statement.
Sickle cell patients suffer from severe, chronic pain, which is debilitating on its own without the added burden of having to constantly appeal coverage denials, added ASH President Roy Silverstein, MD.
“We appreciate CDC’s acknowledgment that the challenges of managing severe and chronic pain in conditions, such as sickle cell disease, require special consideration, and we hope payers will take the CDC’s clarification into account to ensure that patients’ pain management needs are covered,” he said in the same statement.
FDA approves pembrolizumab for first-line stage III NSCLC
The who are not candidates for surgical resection or definitive chemoradiation, and for stage IV NSCLC.

Patients’ tumors must express programmed death-ligand 1 (PD-L1) as determined by an FDA-approved test (tumor proportion score ≥1%) and have no epidermal growth factor receptor or anaplastic lymphoma kinase mutations.
The checkpoint inhibitor was previously approved as a single agent for the first-line treatment of patients with metastatic disease with PD-L1 expression at a higher level (TPS ≥50%), the FDA said in a press statement.
Approval was based on statistically significant overall survival improvement with pembrolizumab, compared with investigator’s choice of a carboplatin-containing regimen with either pemetrexed or paclitaxel in KEYNOTE‑042. The trial enrolled 1,274 patients with stage III or IV NSCLC who had not received prior systemic treatment for metastatic NSCLC and whose tumors expressed PD-L1 (TPS ≥1%).
Overall survival was improved in all three subgroups for pembrolizumab, compared with chemotherapy: in the TPS ≥50% subgroup, the TPS ≥20% subgroup, and the overall population (TPS ≥1%). The median overall survival in the TPS ≥1% population was 16.7 for pembrolizumab and 12.1 months for the chemotherapy arms (hazard ratio, 0.81; 95% confidence interval, 0.71-0.93; P = .0036). For the TPS ≥50% subgroup, the estimated median overall survival was 20 months for pembrolizumab and 12.2 months for the chemotherapy arm (HR, 0.69; 95% CI, 0.56-0.85; P = .0006).
The most common adverse reactions reported for patients who received pembrolizumab included fatigue, decreased appetite, dyspnea, cough, rash, constipation, diarrhea, nausea, hypothyroidism, pneumonia, pyrexia, and weight loss, the FDA said.
The recommended dose for NSCLC is 200 mg as an IV infusion over 30 minutes every 3 weeks.
The who are not candidates for surgical resection or definitive chemoradiation, and for stage IV NSCLC.
Patients’ tumors must express programmed death-ligand 1 (PD-L1) as determined by an FDA-approved test (tumor proportion score ≥1%) and have no epidermal growth factor receptor or anaplastic lymphoma kinase mutations.
The checkpoint inhibitor was previously approved as a single agent for the first-line treatment of patients with metastatic disease with PD-L1 expression at a higher level (TPS ≥50%), the FDA said in a press statement.
Approval was based on statistically significant overall survival improvement with pembrolizumab, compared with investigator’s choice of a carboplatin-containing regimen with either pemetrexed or paclitaxel in KEYNOTE‑042. The trial enrolled 1,274 patients with stage III or IV NSCLC who had not received prior systemic treatment for metastatic NSCLC and whose tumors expressed PD-L1 (TPS ≥1%).
Overall survival was improved in all three subgroups for pembrolizumab, compared with chemotherapy: in the TPS ≥50% subgroup, the TPS ≥20% subgroup, and the overall population (TPS ≥1%). The median overall survival in the TPS ≥1% population was 16.7 for pembrolizumab and 12.1 months for the chemotherapy arms (hazard ratio, 0.81; 95% confidence interval, 0.71-0.93; P = .0036). For the TPS ≥50% subgroup, the estimated median overall survival was 20 months for pembrolizumab and 12.2 months for the chemotherapy arm (HR, 0.69; 95% CI, 0.56-0.85; P = .0006).
The most common adverse reactions reported for patients who received pembrolizumab included fatigue, decreased appetite, dyspnea, cough, rash, constipation, diarrhea, nausea, hypothyroidism, pneumonia, pyrexia, and weight loss, the FDA said.
The recommended dose for NSCLC is 200 mg as an IV infusion over 30 minutes every 3 weeks.
The who are not candidates for surgical resection or definitive chemoradiation, and for stage IV NSCLC.
Patients’ tumors must express programmed death-ligand 1 (PD-L1) as determined by an FDA-approved test (tumor proportion score ≥1%) and have no epidermal growth factor receptor or anaplastic lymphoma kinase mutations.
The checkpoint inhibitor was previously approved as a single agent for the first-line treatment of patients with metastatic disease with PD-L1 expression at a higher level (TPS ≥50%), the FDA said in a press statement.
Approval was based on statistically significant overall survival improvement with pembrolizumab, compared with investigator’s choice of a carboplatin-containing regimen with either pemetrexed or paclitaxel in KEYNOTE‑042. The trial enrolled 1,274 patients with stage III or IV NSCLC who had not received prior systemic treatment for metastatic NSCLC and whose tumors expressed PD-L1 (TPS ≥1%).
Overall survival was improved in all three subgroups for pembrolizumab, compared with chemotherapy: in the TPS ≥50% subgroup, the TPS ≥20% subgroup, and the overall population (TPS ≥1%). The median overall survival in the TPS ≥1% population was 16.7 for pembrolizumab and 12.1 months for the chemotherapy arms (hazard ratio, 0.81; 95% confidence interval, 0.71-0.93; P = .0036). For the TPS ≥50% subgroup, the estimated median overall survival was 20 months for pembrolizumab and 12.2 months for the chemotherapy arm (HR, 0.69; 95% CI, 0.56-0.85; P = .0006).
The most common adverse reactions reported for patients who received pembrolizumab included fatigue, decreased appetite, dyspnea, cough, rash, constipation, diarrhea, nausea, hypothyroidism, pneumonia, pyrexia, and weight loss, the FDA said.
The recommended dose for NSCLC is 200 mg as an IV infusion over 30 minutes every 3 weeks.
FDA modifies safety label for Addyi
The Food and Drug Administration has issued a safety labeling change for flibanserin (Addyi), a treatment for premenopausal women with acquired, generalized hypoactive sexual desire disorder, according to a press release issued April 11 by the agency. Previously, the warning said women should abstain from alcohol entirely.
According to the release, the manufacturer, Sprout, had hoped the FDA would remove the boxed warning and contraindication entirely. However, based on a review of two postmarket research studies, the agency chose to order these modifications to the warnings instead.
The first postmarket study was missing information related to participants’ blood pressure, which FDA officials thought was critical in determining risk; it appeared that this resulted from safety precautions built into the trial. The concern was that not only did this absent information provide further evidence of an interaction but that women at home would not have the benefit of these safety precautions and could suffer serious outcomes, including falls, accidents, and bodily harm. The other postmarketing trial showed that delaying administration of flibanserin until at least 2 hours after consuming alcohol reduced the risk of serious hypotension and syncope.
It is recommended that flibanserin be taken at bedtime because of risks associated with hypotension and syncope, as well as risks associated with central nervous system depression (such as sleepiness). Furthermore, patients are encouraged to discontinue treatment with flibanserin if their hypoactive sexual desire disorder does not improve after 8 weeks. The most common adverse reactions include dizziness, sleepiness, nausea, fatigue, insomnia, and dry mouth.
Full prescribing information is available on the FDA website, as is the full release regarding these safety label modifications.
The Food and Drug Administration has issued a safety labeling change for flibanserin (Addyi), a treatment for premenopausal women with acquired, generalized hypoactive sexual desire disorder, according to a press release issued April 11 by the agency. Previously, the warning said women should abstain from alcohol entirely.
According to the release, the manufacturer, Sprout, had hoped the FDA would remove the boxed warning and contraindication entirely. However, based on a review of two postmarket research studies, the agency chose to order these modifications to the warnings instead.
The first postmarket study was missing information related to participants’ blood pressure, which FDA officials thought was critical in determining risk; it appeared that this resulted from safety precautions built into the trial. The concern was that not only did this absent information provide further evidence of an interaction but that women at home would not have the benefit of these safety precautions and could suffer serious outcomes, including falls, accidents, and bodily harm. The other postmarketing trial showed that delaying administration of flibanserin until at least 2 hours after consuming alcohol reduced the risk of serious hypotension and syncope.
It is recommended that flibanserin be taken at bedtime because of risks associated with hypotension and syncope, as well as risks associated with central nervous system depression (such as sleepiness). Furthermore, patients are encouraged to discontinue treatment with flibanserin if their hypoactive sexual desire disorder does not improve after 8 weeks. The most common adverse reactions include dizziness, sleepiness, nausea, fatigue, insomnia, and dry mouth.
Full prescribing information is available on the FDA website, as is the full release regarding these safety label modifications.
The Food and Drug Administration has issued a safety labeling change for flibanserin (Addyi), a treatment for premenopausal women with acquired, generalized hypoactive sexual desire disorder, according to a press release issued April 11 by the agency. Previously, the warning said women should abstain from alcohol entirely.
According to the release, the manufacturer, Sprout, had hoped the FDA would remove the boxed warning and contraindication entirely. However, based on a review of two postmarket research studies, the agency chose to order these modifications to the warnings instead.
The first postmarket study was missing information related to participants’ blood pressure, which FDA officials thought was critical in determining risk; it appeared that this resulted from safety precautions built into the trial. The concern was that not only did this absent information provide further evidence of an interaction but that women at home would not have the benefit of these safety precautions and could suffer serious outcomes, including falls, accidents, and bodily harm. The other postmarketing trial showed that delaying administration of flibanserin until at least 2 hours after consuming alcohol reduced the risk of serious hypotension and syncope.
It is recommended that flibanserin be taken at bedtime because of risks associated with hypotension and syncope, as well as risks associated with central nervous system depression (such as sleepiness). Furthermore, patients are encouraged to discontinue treatment with flibanserin if their hypoactive sexual desire disorder does not improve after 8 weeks. The most common adverse reactions include dizziness, sleepiness, nausea, fatigue, insomnia, and dry mouth.
Full prescribing information is available on the FDA website, as is the full release regarding these safety label modifications.
FDA to expand opioid labeling with instructions on proper tapering
The Food and Drug Administration is making changes to opioid analgesic labeling to give better information to clinicians on how to properly taper patients dependent on opioid use, according to Douglas Throckmorton, MD, deputy director for regulatory programs in the FDA’s Center for Drug Evaluation and Research.
, such as withdrawal symptoms, uncontrolled pain, and suicide. Both the FDA and the Centers for Disease Control and Prevention offer guidelines on how to properly taper opioids, Dr. Throckmorton said, but more needs to be done to ensure that patients are being provided with the correct advice and care.
The changes to the labels will include expanded information to health care clinicians and are intended to be used when both the clinician and patient have agreed to reduce the opioid dosage. When this is discussed, factors that should be considered include the dose of the drug, the duration of treatment, the type of pain being treated, and the physical and psychological attributes of the patient.
Other actions the FDA is pursuing to combat opioid use disorder include working with the National Academies of Sciences, Engineering, and Medicine on guidelines for the proper opioid analgesic prescribing for acute pain resulting from specific conditions or procedures, and advancing policies that make immediate-release opioid formulations available in fixed-quantity packaging for 1 or 2 days.
“The FDA remains committed to addressing the opioid crisis on all fronts, with a significant focus on decreasing unnecessary exposure to opioids and preventing new addiction; supporting the treatment of those with opioid use disorder; fostering the development of novel pain treatment therapies and opioids more resistant to abuse and misuse; and taking action against those involved in the illegal importation and sale of opioids,” Dr. Throckmorton said.
Find the full statement by Dr. Throckmorton on the FDA website.
The Food and Drug Administration is making changes to opioid analgesic labeling to give better information to clinicians on how to properly taper patients dependent on opioid use, according to Douglas Throckmorton, MD, deputy director for regulatory programs in the FDA’s Center for Drug Evaluation and Research.
, such as withdrawal symptoms, uncontrolled pain, and suicide. Both the FDA and the Centers for Disease Control and Prevention offer guidelines on how to properly taper opioids, Dr. Throckmorton said, but more needs to be done to ensure that patients are being provided with the correct advice and care.
The changes to the labels will include expanded information to health care clinicians and are intended to be used when both the clinician and patient have agreed to reduce the opioid dosage. When this is discussed, factors that should be considered include the dose of the drug, the duration of treatment, the type of pain being treated, and the physical and psychological attributes of the patient.
Other actions the FDA is pursuing to combat opioid use disorder include working with the National Academies of Sciences, Engineering, and Medicine on guidelines for the proper opioid analgesic prescribing for acute pain resulting from specific conditions or procedures, and advancing policies that make immediate-release opioid formulations available in fixed-quantity packaging for 1 or 2 days.
“The FDA remains committed to addressing the opioid crisis on all fronts, with a significant focus on decreasing unnecessary exposure to opioids and preventing new addiction; supporting the treatment of those with opioid use disorder; fostering the development of novel pain treatment therapies and opioids more resistant to abuse and misuse; and taking action against those involved in the illegal importation and sale of opioids,” Dr. Throckmorton said.
Find the full statement by Dr. Throckmorton on the FDA website.
The Food and Drug Administration is making changes to opioid analgesic labeling to give better information to clinicians on how to properly taper patients dependent on opioid use, according to Douglas Throckmorton, MD, deputy director for regulatory programs in the FDA’s Center for Drug Evaluation and Research.
, such as withdrawal symptoms, uncontrolled pain, and suicide. Both the FDA and the Centers for Disease Control and Prevention offer guidelines on how to properly taper opioids, Dr. Throckmorton said, but more needs to be done to ensure that patients are being provided with the correct advice and care.
The changes to the labels will include expanded information to health care clinicians and are intended to be used when both the clinician and patient have agreed to reduce the opioid dosage. When this is discussed, factors that should be considered include the dose of the drug, the duration of treatment, the type of pain being treated, and the physical and psychological attributes of the patient.
Other actions the FDA is pursuing to combat opioid use disorder include working with the National Academies of Sciences, Engineering, and Medicine on guidelines for the proper opioid analgesic prescribing for acute pain resulting from specific conditions or procedures, and advancing policies that make immediate-release opioid formulations available in fixed-quantity packaging for 1 or 2 days.
“The FDA remains committed to addressing the opioid crisis on all fronts, with a significant focus on decreasing unnecessary exposure to opioids and preventing new addiction; supporting the treatment of those with opioid use disorder; fostering the development of novel pain treatment therapies and opioids more resistant to abuse and misuse; and taking action against those involved in the illegal importation and sale of opioids,” Dr. Throckmorton said.
Find the full statement by Dr. Throckmorton on the FDA website.
Romosozumab gets FDA approval for treating osteoporosis
“These are women who have a history of osteoporotic fracture or multiple risk factors or have failed other treatments for osteoporosis,” according to a news release from the agency.
The monthly treatment of two injections (given one after the other at one visit) mainly works by increasing new bone formation, but these effects wane after 12 doses. If patients still need osteoporosis therapy after that maximum of 12 doses, it’s recommended they are put on treatments that reduce bone breakdown. Romosozumab-aqqg is “a monoclonal antibody that blocks the effects of the protein sclerostin,” according to the news release.
The treatment’s efficacy and safety was evaluated in two clinical trials of more than 11,000 women with postmenopausal osteoporosis. In one trial, women received 12 months of either romosozumab-aqqg or placebo. The treatment arm had a 73% lower risk of vertebral fracture than did the placebo arm, and this benefit was maintained over a second year when both groups were switched to denosumab, another osteoporosis therapy. In the second trial, one group received romosozumab-aqqg for 1 year and then a year of alendronate, and the other group received 2 years of alendronate, another osteoporosis therapy, according to the news release. In this trial, the romosozumab-aqqg arm had 50% less risk of vertebral fractures than did the alendronate-only arm, as well as reduced risk of nonvertebral fractures.
Romosozumab-aqqg was associated with higher risks of cardiovascular death, heart attack, and stroke in the alendronate trial, so the treatment comes with a boxed warning regarding those risks and recommends that the drug not be used in patients who have had a heart attack or stroke within the previous year, according to the news release. Common side effects include joint pain and headache, as well as injection-site reactions.
“These are women who have a history of osteoporotic fracture or multiple risk factors or have failed other treatments for osteoporosis,” according to a news release from the agency.
The monthly treatment of two injections (given one after the other at one visit) mainly works by increasing new bone formation, but these effects wane after 12 doses. If patients still need osteoporosis therapy after that maximum of 12 doses, it’s recommended they are put on treatments that reduce bone breakdown. Romosozumab-aqqg is “a monoclonal antibody that blocks the effects of the protein sclerostin,” according to the news release.
The treatment’s efficacy and safety was evaluated in two clinical trials of more than 11,000 women with postmenopausal osteoporosis. In one trial, women received 12 months of either romosozumab-aqqg or placebo. The treatment arm had a 73% lower risk of vertebral fracture than did the placebo arm, and this benefit was maintained over a second year when both groups were switched to denosumab, another osteoporosis therapy. In the second trial, one group received romosozumab-aqqg for 1 year and then a year of alendronate, and the other group received 2 years of alendronate, another osteoporosis therapy, according to the news release. In this trial, the romosozumab-aqqg arm had 50% less risk of vertebral fractures than did the alendronate-only arm, as well as reduced risk of nonvertebral fractures.
Romosozumab-aqqg was associated with higher risks of cardiovascular death, heart attack, and stroke in the alendronate trial, so the treatment comes with a boxed warning regarding those risks and recommends that the drug not be used in patients who have had a heart attack or stroke within the previous year, according to the news release. Common side effects include joint pain and headache, as well as injection-site reactions.
“These are women who have a history of osteoporotic fracture or multiple risk factors or have failed other treatments for osteoporosis,” according to a news release from the agency.
The monthly treatment of two injections (given one after the other at one visit) mainly works by increasing new bone formation, but these effects wane after 12 doses. If patients still need osteoporosis therapy after that maximum of 12 doses, it’s recommended they are put on treatments that reduce bone breakdown. Romosozumab-aqqg is “a monoclonal antibody that blocks the effects of the protein sclerostin,” according to the news release.
The treatment’s efficacy and safety was evaluated in two clinical trials of more than 11,000 women with postmenopausal osteoporosis. In one trial, women received 12 months of either romosozumab-aqqg or placebo. The treatment arm had a 73% lower risk of vertebral fracture than did the placebo arm, and this benefit was maintained over a second year when both groups were switched to denosumab, another osteoporosis therapy. In the second trial, one group received romosozumab-aqqg for 1 year and then a year of alendronate, and the other group received 2 years of alendronate, another osteoporosis therapy, according to the news release. In this trial, the romosozumab-aqqg arm had 50% less risk of vertebral fractures than did the alendronate-only arm, as well as reduced risk of nonvertebral fractures.
Romosozumab-aqqg was associated with higher risks of cardiovascular death, heart attack, and stroke in the alendronate trial, so the treatment comes with a boxed warning regarding those risks and recommends that the drug not be used in patients who have had a heart attack or stroke within the previous year, according to the news release. Common side effects include joint pain and headache, as well as injection-site reactions.
FDA approves first two-drug tablet for HIV
The U.S. Food and Drug Administration has approved the first two-drug, fixed-dose, complete regimen for HIV-infected adults, according to an FDA press announcement.
Dovato (dolutegravir and lamivudine), a product of ViiV Healthcare, is intended to serve “as a complete regimen” for the treatment of HIV-1 infection in adults who have had no previous antiretroviral treatment and who have an infection with no known or suspected genetic substitutions associated with resistance to the individual components of Dovato.
“With this approval, patients who have never been treated have the option of taking a two-drug regimen in a single tablet while eliminating additional toxicity and potential drug interactions from a third drug,” said Debra Birnkrant, MD, director of the FDA’s Division of Antiviral Products.
The Dovato labeling includes a Boxed Warning that patients infected with both HIV and hepatitis B should add additional treatment for their HBV or consider a different drug regimen. The most common adverse reactions with Dovato were headache, diarrhea, nausea, insomnia, and fatigue. In addition, the FDA warned that, as there is a known risk for neural tube defects with dolutegravir, patients are advised to avoid use of Dovato at the time of conception through the first trimester of pregnancy.
mlesney@mdedge.com
The U.S. Food and Drug Administration has approved the first two-drug, fixed-dose, complete regimen for HIV-infected adults, according to an FDA press announcement.
Dovato (dolutegravir and lamivudine), a product of ViiV Healthcare, is intended to serve “as a complete regimen” for the treatment of HIV-1 infection in adults who have had no previous antiretroviral treatment and who have an infection with no known or suspected genetic substitutions associated with resistance to the individual components of Dovato.
“With this approval, patients who have never been treated have the option of taking a two-drug regimen in a single tablet while eliminating additional toxicity and potential drug interactions from a third drug,” said Debra Birnkrant, MD, director of the FDA’s Division of Antiviral Products.
The Dovato labeling includes a Boxed Warning that patients infected with both HIV and hepatitis B should add additional treatment for their HBV or consider a different drug regimen. The most common adverse reactions with Dovato were headache, diarrhea, nausea, insomnia, and fatigue. In addition, the FDA warned that, as there is a known risk for neural tube defects with dolutegravir, patients are advised to avoid use of Dovato at the time of conception through the first trimester of pregnancy.
mlesney@mdedge.com
The U.S. Food and Drug Administration has approved the first two-drug, fixed-dose, complete regimen for HIV-infected adults, according to an FDA press announcement.
Dovato (dolutegravir and lamivudine), a product of ViiV Healthcare, is intended to serve “as a complete regimen” for the treatment of HIV-1 infection in adults who have had no previous antiretroviral treatment and who have an infection with no known or suspected genetic substitutions associated with resistance to the individual components of Dovato.
“With this approval, patients who have never been treated have the option of taking a two-drug regimen in a single tablet while eliminating additional toxicity and potential drug interactions from a third drug,” said Debra Birnkrant, MD, director of the FDA’s Division of Antiviral Products.
The Dovato labeling includes a Boxed Warning that patients infected with both HIV and hepatitis B should add additional treatment for their HBV or consider a different drug regimen. The most common adverse reactions with Dovato were headache, diarrhea, nausea, insomnia, and fatigue. In addition, the FDA warned that, as there is a known risk for neural tube defects with dolutegravir, patients are advised to avoid use of Dovato at the time of conception through the first trimester of pregnancy.
mlesney@mdedge.com
Flu activity falling but still elevated
Measures of influenza activity fell again as the flu season continues to make its later-than-usual departure this year, according to the Centers for Disease Control and Prevention.
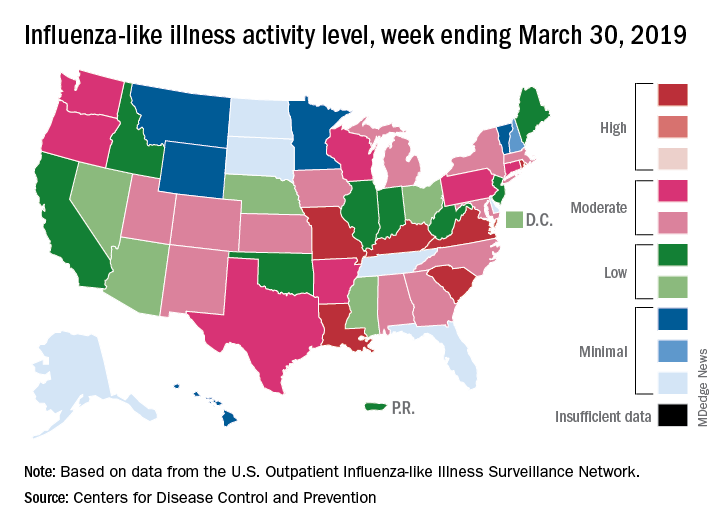
On the geographic front, the map of influenza-like illness (ILI) activity for the week ending March 30 shows that only 6 states are at level 10 on the CDC’s 1-10 scale, compared with 11 for the previous week, and that those same 6 states make up the entire membership of the high range of levels 8-10, which is down from 20 states a week ago, data from the CDC’s Outpatient ILI Surveillance Network show.
The proportion of outpatient visits for ILI, now at 3.2%, dropped for the sixth consecutive week after reaching its season high of 5.1% back in mid-February. The outpatient rate has now been at or above the national baseline of 2.2% for 19 weeks this season, the CDC’s influenza division said April 5, noting that the average for the past five seasons is 16 weeks.
Six flu-related pediatric deaths were reported in the week ending March 30, and the total is now 82 for the 2018-2019 season. Five of the six occurred during previous weeks of this season, and one occurred in the 2017-2018 season, the CDC said.
Measures of influenza activity fell again as the flu season continues to make its later-than-usual departure this year, according to the Centers for Disease Control and Prevention.
On the geographic front, the map of influenza-like illness (ILI) activity for the week ending March 30 shows that only 6 states are at level 10 on the CDC’s 1-10 scale, compared with 11 for the previous week, and that those same 6 states make up the entire membership of the high range of levels 8-10, which is down from 20 states a week ago, data from the CDC’s Outpatient ILI Surveillance Network show.
The proportion of outpatient visits for ILI, now at 3.2%, dropped for the sixth consecutive week after reaching its season high of 5.1% back in mid-February. The outpatient rate has now been at or above the national baseline of 2.2% for 19 weeks this season, the CDC’s influenza division said April 5, noting that the average for the past five seasons is 16 weeks.
Six flu-related pediatric deaths were reported in the week ending March 30, and the total is now 82 for the 2018-2019 season. Five of the six occurred during previous weeks of this season, and one occurred in the 2017-2018 season, the CDC said.
Measures of influenza activity fell again as the flu season continues to make its later-than-usual departure this year, according to the Centers for Disease Control and Prevention.
On the geographic front, the map of influenza-like illness (ILI) activity for the week ending March 30 shows that only 6 states are at level 10 on the CDC’s 1-10 scale, compared with 11 for the previous week, and that those same 6 states make up the entire membership of the high range of levels 8-10, which is down from 20 states a week ago, data from the CDC’s Outpatient ILI Surveillance Network show.
The proportion of outpatient visits for ILI, now at 3.2%, dropped for the sixth consecutive week after reaching its season high of 5.1% back in mid-February. The outpatient rate has now been at or above the national baseline of 2.2% for 19 weeks this season, the CDC’s influenza division said April 5, noting that the average for the past five seasons is 16 weeks.
Six flu-related pediatric deaths were reported in the week ending March 30, and the total is now 82 for the 2018-2019 season. Five of the six occurred during previous weeks of this season, and one occurred in the 2017-2018 season, the CDC said.
FDA names 40 ARBs that are free of nitrosamines
The Food and Drug Administration has identified 40 angiotensin II receptor blockers (ARBs) that do not contain the environmental contaminants, nitrosamines.
since impurities in these antihypertensive drugs were discovered last summer, according to a statement from the regulatory agency.
Among the drugs on this list are Accord Healthcare’s amlodipine and olmesartan medoxomil, Alembic Pharmaceuticals’ valsartan and hydrochlorothiazide, and Hisun Pharmaceuticals USA’s telmisartan.
Despite the FDA’s findings, the agency recommends patients continue taking the ARBs they have been prescribed until their pharmacists or physicians change their prescriptions to a safe replacement or different treatment option.
“We want to reassure patients that we strongly believe the risks, such as stroke, of abruptly discontinuing these important medications far outweighs the low risk associated with continuing the medications with these impurities,” says the statement.
The FDA noted that it is “continuing to work with manufacturers to swiftly remove medications from the market if they contain a nitrosamine impurity at levels higher than the interim acceptable intake limits,” and that this effort has resulted in shortages of valsartan products. In anticipation of more shortages, the FDA “is not objecting to temporary distribution” of specific lots of losartan containing impurities at levels exceeding the regulatory agency’s aforementioned standards.
The FDA’s scientists said that using ARBs with impurity levels above the interim acceptable intake limits over the time it should take to get impurity-free losartan to market will not result in an increased risk for cancer.
More information, including the full statement, is available on the FDA’s website.
cpalmer@mdedge.com
The Food and Drug Administration has identified 40 angiotensin II receptor blockers (ARBs) that do not contain the environmental contaminants, nitrosamines.
since impurities in these antihypertensive drugs were discovered last summer, according to a statement from the regulatory agency.
Among the drugs on this list are Accord Healthcare’s amlodipine and olmesartan medoxomil, Alembic Pharmaceuticals’ valsartan and hydrochlorothiazide, and Hisun Pharmaceuticals USA’s telmisartan.
Despite the FDA’s findings, the agency recommends patients continue taking the ARBs they have been prescribed until their pharmacists or physicians change their prescriptions to a safe replacement or different treatment option.
“We want to reassure patients that we strongly believe the risks, such as stroke, of abruptly discontinuing these important medications far outweighs the low risk associated with continuing the medications with these impurities,” says the statement.
The FDA noted that it is “continuing to work with manufacturers to swiftly remove medications from the market if they contain a nitrosamine impurity at levels higher than the interim acceptable intake limits,” and that this effort has resulted in shortages of valsartan products. In anticipation of more shortages, the FDA “is not objecting to temporary distribution” of specific lots of losartan containing impurities at levels exceeding the regulatory agency’s aforementioned standards.
The FDA’s scientists said that using ARBs with impurity levels above the interim acceptable intake limits over the time it should take to get impurity-free losartan to market will not result in an increased risk for cancer.
More information, including the full statement, is available on the FDA’s website.
cpalmer@mdedge.com
The Food and Drug Administration has identified 40 angiotensin II receptor blockers (ARBs) that do not contain the environmental contaminants, nitrosamines.
since impurities in these antihypertensive drugs were discovered last summer, according to a statement from the regulatory agency.
Among the drugs on this list are Accord Healthcare’s amlodipine and olmesartan medoxomil, Alembic Pharmaceuticals’ valsartan and hydrochlorothiazide, and Hisun Pharmaceuticals USA’s telmisartan.
Despite the FDA’s findings, the agency recommends patients continue taking the ARBs they have been prescribed until their pharmacists or physicians change their prescriptions to a safe replacement or different treatment option.
“We want to reassure patients that we strongly believe the risks, such as stroke, of abruptly discontinuing these important medications far outweighs the low risk associated with continuing the medications with these impurities,” says the statement.
The FDA noted that it is “continuing to work with manufacturers to swiftly remove medications from the market if they contain a nitrosamine impurity at levels higher than the interim acceptable intake limits,” and that this effort has resulted in shortages of valsartan products. In anticipation of more shortages, the FDA “is not objecting to temporary distribution” of specific lots of losartan containing impurities at levels exceeding the regulatory agency’s aforementioned standards.
The FDA’s scientists said that using ARBs with impurity levels above the interim acceptable intake limits over the time it should take to get impurity-free losartan to market will not result in an increased risk for cancer.
More information, including the full statement, is available on the FDA’s website.
cpalmer@mdedge.com