User login
What is the best approach to treat an upper-extremity DVT?
Case
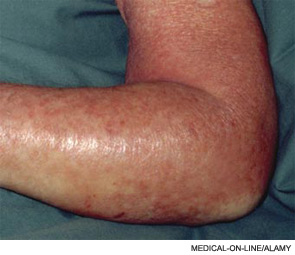
A 45-year-old female with a history of cellulitis requiring peripheral inserted central catheter (PICC) line placement for intravenous antibiotics presents two weeks after line removal with persistent, dull, aching pain in her right shoulder and difficulty removing the rings on her right hand. The pain worsens with exercise and is relieved with rest. The physical exam reveals nonpitting edema of her hand. The ultrasound shows subclavian vein thrombosis. What is the best approach to treating her upper extremity deep venous thrombosis (UEDVT)?
Background
DVT and pulmonary embolism (PE) have been subject to increased publicity recently, and both conditions are recognized as serious entities with life-threatening consequences. In fact, more people die annually from blood clots than breast cancer and AIDS combined.1,2 Still, the increased DVT and PE awareness is primarily focused on lower extremity DVT (LEDVT), while UEDVT is thought of as a more benign entity. However, current data suggest that UEDVT is associated with equally significant morbidity and mortality.
UEDVT prevalence has increased in step with the increased use of central venous catheters (CVCs) and pacemakers. Although most patients present with pain, swelling, parathesias, and prominent veins throughout the arm or shoulder, many patients will not display any local DVT symptoms. For example, Kabani et al recently presented data for 1,275 patients admitted to the surgical ICU over a 12-month period. They found the incidence of UEDVT was higher than that of LEDVT (17% vs. 11%; P=0.11). They also determined that scanning all four extremities diagnosed more DVT than two-extremity scans (33% vs. 7%; P<0.001).3
While current medical literature has pushed for increased UEDVT attention, there is no consensus on its treatment. Recent American College of Chest Physicians (ACCP) guidelines addressed UEDVT treatment specifically and recommended analogous treatment to LEDVT with heparin and warfarin.4 This follows prospective studies that have shown patients with UEDVT and LEDVT have similar three-month clinical outcomes. The ACCP guidelines do not specifically recommend different treatment courses based on whether the UEDVT is catheter-related or not. Furthermore, while one might assume that removal of an associated catheter might reduce the treatment duration, there is limited data to support shorter courses in this scenario.
Review of the Data
Incidence: UEDVT is becoming more common secondary to increased interventions in the upper extremity (CVC, pacemaker), and is more easily recognized due to improvement in noninvasive ultrasound technology. UEDVT accounts for up to 10% of all DVT, with an incidence of approximately three per 100,000 persons in the general population.5-8 Because UEDVT can also be asymptomatic, it is believed that the incidence likely is higher than previously reported, but prospective data are lacking.
Risk factors: UEDVT is further categorized as either primary or secondary, depending upon the cause. First described in the late 1800s, spontaneous primary thrombosis of the upper extremity, or Paget-Schroetter syndrome, accounts for approximately 20% of UEDVT.9 Primary UEDVT includes both idiopathic and “effort-related” thrombosis. Effort-related thrombosis usually develops among young people after strenuous or repetitive exercise, such as pitching a baseball. Some hypothesize that effort-related thrombosis is related to a hypercoaguable state or anatomic abnormalities, although a specific cause, such as thoracic outlet syndrome, is found in only 5% of these cases.10,11
Secondary UEDVT characterizes thrombosis in which an endogenous or exogenous risk factor is present. Endogenous risk factors include coagulation abnormalities, such as antithrombin, protein C and protein S deficiencies; factor V Leiden gene mutation; hyperhomocysteinemia; and antiphospholipid antibody syndrome. Exogenous risk factors include CVC pacemakers, intracardiac defibrillators, malignancy, previous or concurrent LEDVT, oral contraceptives, some artificial reproductive technologies (women can develop ovarian hyperstimulation syndrome, which is associated with increased hypercoaguability), trauma, and IV drug use (especially cocaine).5,12-14
Clinical presentation and diagnosis: Swelling (80% of patients) and pain (40% of patients) are the most common UEDVT symptoms at presentation.2 Other clinical features include new, prominent veins throughout the shoulder girdle, erythema, increased warmth, functional impairment, parathesias, and non-specific feelings of arm heaviness or discomfort. Symptoms typically worsen with arm use and improve with rest and elevation.15 Patients with UEDVT related to CVC are more likely to be asymptomatic and may present only with PE.16 The differential diagnosis includes superficial phlebitis, lymphatic edema, hematoma, contusions, venous compression, and muscle tears.17
Contrast venography is the gold standard for the UEDVT diagnosis. However, it is more expensive and invasive than ultrasound, and thus serial compression ultrasound is now the standard test in UEDVT evaluation. Then again, contrast venography remains the test of choice in patients with high pre-test probability and negative ultrasound results.18,19
Prevention: Nearly 70% of secondary UEDVT is associated with a CVC.5 Further, CVC use is the most powerful predictor of UEDVT (adjusted odds ratio (OR), 9.7; 95% CI, 7.8 to 12.2).2 Despite the association between CVCs and UEDVT, anticoagulant prophylaxis is not recommended. Studies evaluating the results of 1-mg warfarin conflict and include small populations. Warfarin’s potential interaction with antibiotics and dosing variance based on nutritional intake logically prompted studies on the potential benefit of low-weight molecular heparain (LWMH); however, these studies have failed to show benefit.20,21
Treatment: ACCP guidelines recommend treating UEDVT patients with unfractionated heparin (UFH) or LMWH and warfarin, with an INR goal of 2 to 3 for at least three months depending upon the overall clinical scenario. Two small studies evaluating catheter-related thrombosis (15 patients in each trial) reported no subsequent embolic phenomenon.22,23 Some authors interpreted this data to mean UEDVT was not as morbid as LEDVT and, subsequently, that catheter-related UEDVTs require only one month of therapy. Since the small studies were published, the increasing incidence and relevance of UEDVT have become more widely recognized, and most authors are recommending three months of treatment.

Still, it’s important to note that there aren’t any published data directly comparing the one-month and the three-month anticoagulation therapies. The RIETE registry, which is the largest ongoing published registry of patients with confirmed DVT or PE, reports similar three-month clinical outcomes between those with UEDVT and LEDVT.
Small, single-center trials have shown that such active interventions as thrombolysis, surgery, or multi-staged approaches are associated with increased vein patency and decreased rates of post-thrombotic syndrome.24,25 However, ACCP has withheld general recommendations for these interventions based on a lack of sufficient data to comment on their overall safety and efficacy, as well as comparable rates of post-thrombotic syndrome (15% to 50%) in studies that directly compared surgical and medical intervention. In fact, the ACCP recommends against interventional treatments unless the patient has failed anticoagulation therapy, has severe symptoms, and expertise is available.4
Superior vena cava filters are available at some centers for patients in whom anticoagulation is contraindicated, but efficacy data is limited. While the data for filter use in UEDVT is limited, its use should be considered in patients who have a contraindication to anticoagulation and remain high risk for UEDVT (e.g., prolonged central line placement).
Complications: Post-thrombotic syndrome (PTS) is the most significant local complication of UEDVT. PTS characteristics are edema, pain, venous ulcers, and skin pigmentation changes, and it is the result of chronic venous insufficiency due to the clot. A meta-analysis of clinical studies on UEDVT noted that PTS occurs in 7% to 46% (mean 15%) of patients.26 One hypothesis for the wide range in frequency is the lack of clear diagnostic criteria for PTS.27 No clear beneficial treatment or prevention for PTS exists, but many recommend graduated compression stockings for the arm.
Residual and recurrent thrombosis are associated with increased PTS risk, which emphasizes the need for further study of interventional treatment because preliminary work has shown increased rates of vein patency in comparison to anticoagulants alone. Recurrent venous thromboembolism (VTE), another local complication, appears to occur less often than it does in patients with LEDVTs, but reaches 8% after five years of followup.28
PE is less common on presentation among patients with UEDVT when compared to patients with LEDVT, but when PE occurs, the three-month outcome is similar.3 PE appears to be more frequent in patients who have a CVC, with an incidence as high as 36% of DVT patients.4,13,21,29
Increased mortality: The mortality among UEDVT patients has been described as 10% to 50% in the 12 months after diagnosis, which is much higher than the ratio in LEDVT patients.21,30 This in part is due to sicker cohorts getting UEDVT. For example, patients with distant metastasis are more likely to develop UEDVT than those with confined malignancy (adjusted OR 11.5; 95% CI, 1.6 to 80.2).31
Occult malignancy, most commonly lung cancer or lymphoma, has been found in as many as 24% of UEDVT patients.32 The high rate of mortality associated with UEDVT appears to be related more with the patient's overall poor clinical condition rather than directly related to complications from the DVT.
However, its presence should alert hospitalists to the patient's potentially poorer prognosis and prompt evaluation for occult malignancy if no risk factor is present.
Back to the Case
This patient should be started on either UFH or LMWH while simultaneously beginning warfarin. She should continue warfarin treatment for at least three months, with a goal INR of 2.0 to 3.0, similar to treatment for LEDVT. The ultimate treatment duration with warfarin follows the same guidelines as treatment with a LEDVT. Although prophylaxis is not routinely recommended, dosing 1 mg of warfarin beginning three days before subsequent CVC placement should be considered if this patient requires a future CVC.
Additionally, an evaluation for occult malignancy should be considered in this patient.
Bottom Line
Upper extremity DVT is not a benign condition, and is associated with a general increase in mortality. It should be treated similarly to LEDVT in order to decrease PTS, recurrent DVT, and pumonary embolism.
Dr. Hollberg is an assistant professor of medicine, Emory University School of Medicine, Atlanta, and medical director for information services, Emory Healthcare.
References
- Hirsh J, Hoak J. Management of deep vein thrombosis and pulmonary embolism. A statement for healthcare professionals. Council on Thrombosis, American Heart Association. Circulation. 1996;93(12):2212-2245.
- Gerotziafas GT, Samama MM. Prophylaxis of venous thromboembolism in medical patients. Curr Opin Pulm Med. 2004;10(5):356-365.
- Kabani L, et al. Upper extremity DVT as prevalent as lower extremity DVT in ICU patients. Society of Critical Care Medicine (SCCM) 38th annual Critical Care Congress: Abstract 305. Presented Feb. 2, 2009.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Therapy for venous thromboembolic disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6Suppl):454S-545S.
- Joffe HV, Kucher N, Tapson VF, Goldhaber SZ. Upper extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110:1605.
- Munoz FJ, Mismetti P, Poggio R, et al. Clinical outcome of patients with an upper-extremity deep vein thrombosis: results from the RIETE registry. Chest. 2008,133:143-148.
- Coon WW, Willis PW. Thrombosis of axillary and subclavian veins. Arch Surg. 1967;94(5):657-663.
- Horattas MC, Wright DJ, Fenton AH, et al. Changing concepts of deep venous thrombosis of the upper extremity—a report of a series and review of the literature. Surgery. 1988;104(3):561-567.
- Bernardi E, Piccioli A, Marchiori A, Girolami B, Prandoni P. Upper extremity deep vein thrombosis: risk factors, diagnosis, and management. Semin Vasc Med. 2001;1(1):105;110.
- Heron E, Lozinguez O, Alhenc-Gelas M, Emmerich J, Flessinger JN. Hypercoagulable states in primary upper-extremity deep vein thrombosis. Arch Intern Med. 2000;160:382-386.
- Ninet J, Demolombe-Rague S, Bureau Du Colombier P, Coppere B. Les thromboses veineuses profondes des members superieurs. Sang Thromb Vaisseaux. 1994;6:103-114.
- Painter TD, Kerpf M. Deep venous thrombosis of the upper extremity five years experience at a university hospital. Angiology. 1984;35(35):743-749.
- Chan WS, Ginsberg JS. A review of upper extremity deep vein thrombosis in pregnancy: unmasking the “ART” behind the clot. J Thromb Haemost. 2006; 4(8):1673-1677.
- Hughes MJ, D’Agostino JC. Upper extremity deep vein thrombosis: a case report and review of current diagnostic/therapeutic modalities. Am J Emerg Med. 1994;12(6):631-635.
- Prandoni P, Polistena P, Bernardi E, et al. Upper extremity deep vein thrombosis. Risk factors, diagnosis, and complications. Arch Intern Med. 1997;157:57-62.
- Van Rooden CJ, Tesslar ME, Osanto S, Rosendal FR, Huisman MV. Deep vein thrombosis associated with central venous catheters—a review. J Thromb Haemost. 2005;3:2049-2419.
- Horattas MC, Wright DJ, Fenton AH, et al. Changing concepts of deep venous thrombosis of the upper extremity—report of a series and review of the literature. Surgery. 1988;104(3):561-567.
- Bernardi E, Pesavento R, Prandoni P. Upper extremity deep venous thrombosis. Semin Thromb Hemost. 2006;32(7):729-736.
- Baxter GM, McKechnie S, Duffy P. Colour Doppler ultrasound in deep venous thrombosis: a comparison with venography. Clin Radiol. 1990;42(1):32-36.
- Bern MM, Lokich JJ, Wallach SR, et al. Very low doses of warfarin can prevent thrombosis in central venous catheters. A randomized prospective trial. Ann Intern Med. 1990;112(6):423-428.
- Couban S, Goodyear M, Burnell M, et al. Randomized placebo-controlled study of low-dose warfarin for the prevention of central venous catheter-associated thrombosis in patients with cancer. J Clin Oncol. 2005;23(18):4063-4069.
- Lokich JJ, Both A, Benotti P. Complications and management of implanted central venous catheters. J Clin Oncol. 1985;3:710-717.
- Moss JF, Wagman LD, Rijhmaki DU, Terz JJ. Central venous thrombosis related to the silastic Hickman-Broviac catheter in an oncologic population. J Parenter Enteral Nutr. 1989;13:397.
- Machleder HI. Evaluation of a new treatment strategy for Paget-Schroetter syndrome: spontaneous thrombosis of the axillary-subclavian vein. J Vasc Surg. 1993;17:305-315.
- Malcynski J, O’Donnell TF, Mackey WC. Long-term results of treatment for axillary subclavian vein thrombosis. Can J Surg. 1993;36:365-371.
- Elman EE, Kahn SR. The post-thrombotic syndrome after upper extremity deep vein thrombosis in adults: a systematic review. Thromb Res. 2006;117(6):609-614.
- Baarslag HJ, Koopman MM, Hutten BA, et al. Long-term follow up of patients with suspected deep vein thrombosis of the upper extremity: survival, risk factors and post-thrombotic syndrome. Eur J Intern Med. 2004;15:503-507.
- Prandoni P, Bernardi E, Marchiori A, et al. The long term clinical consequence of acute deep venous thrombosis of the arm: prospective cohort study. BMJ. 2004;329:484-485.
- Monreal M, Raventos A, Lerma R, et al. Pulmonary embolism in patients with upper extremity DVT associated to venous central lines—a prospective study. Thromb Haemost. 1994;72(4):548-550.
- Hingorani A, Ascher E, Lorenson E, et al. Upper extremity deep venous thrombosis and its impact on morbidity and mortality rates in a hospital-based population. J Vasc Surg. 1997;26:853-860.
- Blom JW, Doggen CM, Osanto S, Rosendaal FR. Old and new risk factors for upper extremity deep vein thrombosis. J Thromb Haemost. 2005;3:2471-2478.
- Girolami A, Prandoni P, Zanon E, Bagatella P, Girolami B. Venous thromboses of upper limbs are more frequently associated with occult cancer as compared with those of lower limbs. Blood Coagul Fibrinolysis. 1999;10(8):455-457.
Case
A 45-year-old female with a history of cellulitis requiring peripheral inserted central catheter (PICC) line placement for intravenous antibiotics presents two weeks after line removal with persistent, dull, aching pain in her right shoulder and difficulty removing the rings on her right hand. The pain worsens with exercise and is relieved with rest. The physical exam reveals nonpitting edema of her hand. The ultrasound shows subclavian vein thrombosis. What is the best approach to treating her upper extremity deep venous thrombosis (UEDVT)?
Background
DVT and pulmonary embolism (PE) have been subject to increased publicity recently, and both conditions are recognized as serious entities with life-threatening consequences. In fact, more people die annually from blood clots than breast cancer and AIDS combined.1,2 Still, the increased DVT and PE awareness is primarily focused on lower extremity DVT (LEDVT), while UEDVT is thought of as a more benign entity. However, current data suggest that UEDVT is associated with equally significant morbidity and mortality.
UEDVT prevalence has increased in step with the increased use of central venous catheters (CVCs) and pacemakers. Although most patients present with pain, swelling, parathesias, and prominent veins throughout the arm or shoulder, many patients will not display any local DVT symptoms. For example, Kabani et al recently presented data for 1,275 patients admitted to the surgical ICU over a 12-month period. They found the incidence of UEDVT was higher than that of LEDVT (17% vs. 11%; P=0.11). They also determined that scanning all four extremities diagnosed more DVT than two-extremity scans (33% vs. 7%; P<0.001).3
While current medical literature has pushed for increased UEDVT attention, there is no consensus on its treatment. Recent American College of Chest Physicians (ACCP) guidelines addressed UEDVT treatment specifically and recommended analogous treatment to LEDVT with heparin and warfarin.4 This follows prospective studies that have shown patients with UEDVT and LEDVT have similar three-month clinical outcomes. The ACCP guidelines do not specifically recommend different treatment courses based on whether the UEDVT is catheter-related or not. Furthermore, while one might assume that removal of an associated catheter might reduce the treatment duration, there is limited data to support shorter courses in this scenario.
Review of the Data
Incidence: UEDVT is becoming more common secondary to increased interventions in the upper extremity (CVC, pacemaker), and is more easily recognized due to improvement in noninvasive ultrasound technology. UEDVT accounts for up to 10% of all DVT, with an incidence of approximately three per 100,000 persons in the general population.5-8 Because UEDVT can also be asymptomatic, it is believed that the incidence likely is higher than previously reported, but prospective data are lacking.
Risk factors: UEDVT is further categorized as either primary or secondary, depending upon the cause. First described in the late 1800s, spontaneous primary thrombosis of the upper extremity, or Paget-Schroetter syndrome, accounts for approximately 20% of UEDVT.9 Primary UEDVT includes both idiopathic and “effort-related” thrombosis. Effort-related thrombosis usually develops among young people after strenuous or repetitive exercise, such as pitching a baseball. Some hypothesize that effort-related thrombosis is related to a hypercoaguable state or anatomic abnormalities, although a specific cause, such as thoracic outlet syndrome, is found in only 5% of these cases.10,11
Secondary UEDVT characterizes thrombosis in which an endogenous or exogenous risk factor is present. Endogenous risk factors include coagulation abnormalities, such as antithrombin, protein C and protein S deficiencies; factor V Leiden gene mutation; hyperhomocysteinemia; and antiphospholipid antibody syndrome. Exogenous risk factors include CVC pacemakers, intracardiac defibrillators, malignancy, previous or concurrent LEDVT, oral contraceptives, some artificial reproductive technologies (women can develop ovarian hyperstimulation syndrome, which is associated with increased hypercoaguability), trauma, and IV drug use (especially cocaine).5,12-14
Clinical presentation and diagnosis: Swelling (80% of patients) and pain (40% of patients) are the most common UEDVT symptoms at presentation.2 Other clinical features include new, prominent veins throughout the shoulder girdle, erythema, increased warmth, functional impairment, parathesias, and non-specific feelings of arm heaviness or discomfort. Symptoms typically worsen with arm use and improve with rest and elevation.15 Patients with UEDVT related to CVC are more likely to be asymptomatic and may present only with PE.16 The differential diagnosis includes superficial phlebitis, lymphatic edema, hematoma, contusions, venous compression, and muscle tears.17
Contrast venography is the gold standard for the UEDVT diagnosis. However, it is more expensive and invasive than ultrasound, and thus serial compression ultrasound is now the standard test in UEDVT evaluation. Then again, contrast venography remains the test of choice in patients with high pre-test probability and negative ultrasound results.18,19
Prevention: Nearly 70% of secondary UEDVT is associated with a CVC.5 Further, CVC use is the most powerful predictor of UEDVT (adjusted odds ratio (OR), 9.7; 95% CI, 7.8 to 12.2).2 Despite the association between CVCs and UEDVT, anticoagulant prophylaxis is not recommended. Studies evaluating the results of 1-mg warfarin conflict and include small populations. Warfarin’s potential interaction with antibiotics and dosing variance based on nutritional intake logically prompted studies on the potential benefit of low-weight molecular heparain (LWMH); however, these studies have failed to show benefit.20,21
Treatment: ACCP guidelines recommend treating UEDVT patients with unfractionated heparin (UFH) or LMWH and warfarin, with an INR goal of 2 to 3 for at least three months depending upon the overall clinical scenario. Two small studies evaluating catheter-related thrombosis (15 patients in each trial) reported no subsequent embolic phenomenon.22,23 Some authors interpreted this data to mean UEDVT was not as morbid as LEDVT and, subsequently, that catheter-related UEDVTs require only one month of therapy. Since the small studies were published, the increasing incidence and relevance of UEDVT have become more widely recognized, and most authors are recommending three months of treatment.
Still, it’s important to note that there aren’t any published data directly comparing the one-month and the three-month anticoagulation therapies. The RIETE registry, which is the largest ongoing published registry of patients with confirmed DVT or PE, reports similar three-month clinical outcomes between those with UEDVT and LEDVT.
Small, single-center trials have shown that such active interventions as thrombolysis, surgery, or multi-staged approaches are associated with increased vein patency and decreased rates of post-thrombotic syndrome.24,25 However, ACCP has withheld general recommendations for these interventions based on a lack of sufficient data to comment on their overall safety and efficacy, as well as comparable rates of post-thrombotic syndrome (15% to 50%) in studies that directly compared surgical and medical intervention. In fact, the ACCP recommends against interventional treatments unless the patient has failed anticoagulation therapy, has severe symptoms, and expertise is available.4
Superior vena cava filters are available at some centers for patients in whom anticoagulation is contraindicated, but efficacy data is limited. While the data for filter use in UEDVT is limited, its use should be considered in patients who have a contraindication to anticoagulation and remain high risk for UEDVT (e.g., prolonged central line placement).
Complications: Post-thrombotic syndrome (PTS) is the most significant local complication of UEDVT. PTS characteristics are edema, pain, venous ulcers, and skin pigmentation changes, and it is the result of chronic venous insufficiency due to the clot. A meta-analysis of clinical studies on UEDVT noted that PTS occurs in 7% to 46% (mean 15%) of patients.26 One hypothesis for the wide range in frequency is the lack of clear diagnostic criteria for PTS.27 No clear beneficial treatment or prevention for PTS exists, but many recommend graduated compression stockings for the arm.
Residual and recurrent thrombosis are associated with increased PTS risk, which emphasizes the need for further study of interventional treatment because preliminary work has shown increased rates of vein patency in comparison to anticoagulants alone. Recurrent venous thromboembolism (VTE), another local complication, appears to occur less often than it does in patients with LEDVTs, but reaches 8% after five years of followup.28
PE is less common on presentation among patients with UEDVT when compared to patients with LEDVT, but when PE occurs, the three-month outcome is similar.3 PE appears to be more frequent in patients who have a CVC, with an incidence as high as 36% of DVT patients.4,13,21,29
Increased mortality: The mortality among UEDVT patients has been described as 10% to 50% in the 12 months after diagnosis, which is much higher than the ratio in LEDVT patients.21,30 This in part is due to sicker cohorts getting UEDVT. For example, patients with distant metastasis are more likely to develop UEDVT than those with confined malignancy (adjusted OR 11.5; 95% CI, 1.6 to 80.2).31
Occult malignancy, most commonly lung cancer or lymphoma, has been found in as many as 24% of UEDVT patients.32 The high rate of mortality associated with UEDVT appears to be related more with the patient's overall poor clinical condition rather than directly related to complications from the DVT.
However, its presence should alert hospitalists to the patient's potentially poorer prognosis and prompt evaluation for occult malignancy if no risk factor is present.
Back to the Case
This patient should be started on either UFH or LMWH while simultaneously beginning warfarin. She should continue warfarin treatment for at least three months, with a goal INR of 2.0 to 3.0, similar to treatment for LEDVT. The ultimate treatment duration with warfarin follows the same guidelines as treatment with a LEDVT. Although prophylaxis is not routinely recommended, dosing 1 mg of warfarin beginning three days before subsequent CVC placement should be considered if this patient requires a future CVC.
Additionally, an evaluation for occult malignancy should be considered in this patient.
Bottom Line
Upper extremity DVT is not a benign condition, and is associated with a general increase in mortality. It should be treated similarly to LEDVT in order to decrease PTS, recurrent DVT, and pumonary embolism.
Dr. Hollberg is an assistant professor of medicine, Emory University School of Medicine, Atlanta, and medical director for information services, Emory Healthcare.
References
- Hirsh J, Hoak J. Management of deep vein thrombosis and pulmonary embolism. A statement for healthcare professionals. Council on Thrombosis, American Heart Association. Circulation. 1996;93(12):2212-2245.
- Gerotziafas GT, Samama MM. Prophylaxis of venous thromboembolism in medical patients. Curr Opin Pulm Med. 2004;10(5):356-365.
- Kabani L, et al. Upper extremity DVT as prevalent as lower extremity DVT in ICU patients. Society of Critical Care Medicine (SCCM) 38th annual Critical Care Congress: Abstract 305. Presented Feb. 2, 2009.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Therapy for venous thromboembolic disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6Suppl):454S-545S.
- Joffe HV, Kucher N, Tapson VF, Goldhaber SZ. Upper extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110:1605.
- Munoz FJ, Mismetti P, Poggio R, et al. Clinical outcome of patients with an upper-extremity deep vein thrombosis: results from the RIETE registry. Chest. 2008,133:143-148.
- Coon WW, Willis PW. Thrombosis of axillary and subclavian veins. Arch Surg. 1967;94(5):657-663.
- Horattas MC, Wright DJ, Fenton AH, et al. Changing concepts of deep venous thrombosis of the upper extremity—a report of a series and review of the literature. Surgery. 1988;104(3):561-567.
- Bernardi E, Piccioli A, Marchiori A, Girolami B, Prandoni P. Upper extremity deep vein thrombosis: risk factors, diagnosis, and management. Semin Vasc Med. 2001;1(1):105;110.
- Heron E, Lozinguez O, Alhenc-Gelas M, Emmerich J, Flessinger JN. Hypercoagulable states in primary upper-extremity deep vein thrombosis. Arch Intern Med. 2000;160:382-386.
- Ninet J, Demolombe-Rague S, Bureau Du Colombier P, Coppere B. Les thromboses veineuses profondes des members superieurs. Sang Thromb Vaisseaux. 1994;6:103-114.
- Painter TD, Kerpf M. Deep venous thrombosis of the upper extremity five years experience at a university hospital. Angiology. 1984;35(35):743-749.
- Chan WS, Ginsberg JS. A review of upper extremity deep vein thrombosis in pregnancy: unmasking the “ART” behind the clot. J Thromb Haemost. 2006; 4(8):1673-1677.
- Hughes MJ, D’Agostino JC. Upper extremity deep vein thrombosis: a case report and review of current diagnostic/therapeutic modalities. Am J Emerg Med. 1994;12(6):631-635.
- Prandoni P, Polistena P, Bernardi E, et al. Upper extremity deep vein thrombosis. Risk factors, diagnosis, and complications. Arch Intern Med. 1997;157:57-62.
- Van Rooden CJ, Tesslar ME, Osanto S, Rosendal FR, Huisman MV. Deep vein thrombosis associated with central venous catheters—a review. J Thromb Haemost. 2005;3:2049-2419.
- Horattas MC, Wright DJ, Fenton AH, et al. Changing concepts of deep venous thrombosis of the upper extremity—report of a series and review of the literature. Surgery. 1988;104(3):561-567.
- Bernardi E, Pesavento R, Prandoni P. Upper extremity deep venous thrombosis. Semin Thromb Hemost. 2006;32(7):729-736.
- Baxter GM, McKechnie S, Duffy P. Colour Doppler ultrasound in deep venous thrombosis: a comparison with venography. Clin Radiol. 1990;42(1):32-36.
- Bern MM, Lokich JJ, Wallach SR, et al. Very low doses of warfarin can prevent thrombosis in central venous catheters. A randomized prospective trial. Ann Intern Med. 1990;112(6):423-428.
- Couban S, Goodyear M, Burnell M, et al. Randomized placebo-controlled study of low-dose warfarin for the prevention of central venous catheter-associated thrombosis in patients with cancer. J Clin Oncol. 2005;23(18):4063-4069.
- Lokich JJ, Both A, Benotti P. Complications and management of implanted central venous catheters. J Clin Oncol. 1985;3:710-717.
- Moss JF, Wagman LD, Rijhmaki DU, Terz JJ. Central venous thrombosis related to the silastic Hickman-Broviac catheter in an oncologic population. J Parenter Enteral Nutr. 1989;13:397.
- Machleder HI. Evaluation of a new treatment strategy for Paget-Schroetter syndrome: spontaneous thrombosis of the axillary-subclavian vein. J Vasc Surg. 1993;17:305-315.
- Malcynski J, O’Donnell TF, Mackey WC. Long-term results of treatment for axillary subclavian vein thrombosis. Can J Surg. 1993;36:365-371.
- Elman EE, Kahn SR. The post-thrombotic syndrome after upper extremity deep vein thrombosis in adults: a systematic review. Thromb Res. 2006;117(6):609-614.
- Baarslag HJ, Koopman MM, Hutten BA, et al. Long-term follow up of patients with suspected deep vein thrombosis of the upper extremity: survival, risk factors and post-thrombotic syndrome. Eur J Intern Med. 2004;15:503-507.
- Prandoni P, Bernardi E, Marchiori A, et al. The long term clinical consequence of acute deep venous thrombosis of the arm: prospective cohort study. BMJ. 2004;329:484-485.
- Monreal M, Raventos A, Lerma R, et al. Pulmonary embolism in patients with upper extremity DVT associated to venous central lines—a prospective study. Thromb Haemost. 1994;72(4):548-550.
- Hingorani A, Ascher E, Lorenson E, et al. Upper extremity deep venous thrombosis and its impact on morbidity and mortality rates in a hospital-based population. J Vasc Surg. 1997;26:853-860.
- Blom JW, Doggen CM, Osanto S, Rosendaal FR. Old and new risk factors for upper extremity deep vein thrombosis. J Thromb Haemost. 2005;3:2471-2478.
- Girolami A, Prandoni P, Zanon E, Bagatella P, Girolami B. Venous thromboses of upper limbs are more frequently associated with occult cancer as compared with those of lower limbs. Blood Coagul Fibrinolysis. 1999;10(8):455-457.
Case
A 45-year-old female with a history of cellulitis requiring peripheral inserted central catheter (PICC) line placement for intravenous antibiotics presents two weeks after line removal with persistent, dull, aching pain in her right shoulder and difficulty removing the rings on her right hand. The pain worsens with exercise and is relieved with rest. The physical exam reveals nonpitting edema of her hand. The ultrasound shows subclavian vein thrombosis. What is the best approach to treating her upper extremity deep venous thrombosis (UEDVT)?
Background
DVT and pulmonary embolism (PE) have been subject to increased publicity recently, and both conditions are recognized as serious entities with life-threatening consequences. In fact, more people die annually from blood clots than breast cancer and AIDS combined.1,2 Still, the increased DVT and PE awareness is primarily focused on lower extremity DVT (LEDVT), while UEDVT is thought of as a more benign entity. However, current data suggest that UEDVT is associated with equally significant morbidity and mortality.
UEDVT prevalence has increased in step with the increased use of central venous catheters (CVCs) and pacemakers. Although most patients present with pain, swelling, parathesias, and prominent veins throughout the arm or shoulder, many patients will not display any local DVT symptoms. For example, Kabani et al recently presented data for 1,275 patients admitted to the surgical ICU over a 12-month period. They found the incidence of UEDVT was higher than that of LEDVT (17% vs. 11%; P=0.11). They also determined that scanning all four extremities diagnosed more DVT than two-extremity scans (33% vs. 7%; P<0.001).3
While current medical literature has pushed for increased UEDVT attention, there is no consensus on its treatment. Recent American College of Chest Physicians (ACCP) guidelines addressed UEDVT treatment specifically and recommended analogous treatment to LEDVT with heparin and warfarin.4 This follows prospective studies that have shown patients with UEDVT and LEDVT have similar three-month clinical outcomes. The ACCP guidelines do not specifically recommend different treatment courses based on whether the UEDVT is catheter-related or not. Furthermore, while one might assume that removal of an associated catheter might reduce the treatment duration, there is limited data to support shorter courses in this scenario.
Review of the Data
Incidence: UEDVT is becoming more common secondary to increased interventions in the upper extremity (CVC, pacemaker), and is more easily recognized due to improvement in noninvasive ultrasound technology. UEDVT accounts for up to 10% of all DVT, with an incidence of approximately three per 100,000 persons in the general population.5-8 Because UEDVT can also be asymptomatic, it is believed that the incidence likely is higher than previously reported, but prospective data are lacking.
Risk factors: UEDVT is further categorized as either primary or secondary, depending upon the cause. First described in the late 1800s, spontaneous primary thrombosis of the upper extremity, or Paget-Schroetter syndrome, accounts for approximately 20% of UEDVT.9 Primary UEDVT includes both idiopathic and “effort-related” thrombosis. Effort-related thrombosis usually develops among young people after strenuous or repetitive exercise, such as pitching a baseball. Some hypothesize that effort-related thrombosis is related to a hypercoaguable state or anatomic abnormalities, although a specific cause, such as thoracic outlet syndrome, is found in only 5% of these cases.10,11
Secondary UEDVT characterizes thrombosis in which an endogenous or exogenous risk factor is present. Endogenous risk factors include coagulation abnormalities, such as antithrombin, protein C and protein S deficiencies; factor V Leiden gene mutation; hyperhomocysteinemia; and antiphospholipid antibody syndrome. Exogenous risk factors include CVC pacemakers, intracardiac defibrillators, malignancy, previous or concurrent LEDVT, oral contraceptives, some artificial reproductive technologies (women can develop ovarian hyperstimulation syndrome, which is associated with increased hypercoaguability), trauma, and IV drug use (especially cocaine).5,12-14
Clinical presentation and diagnosis: Swelling (80% of patients) and pain (40% of patients) are the most common UEDVT symptoms at presentation.2 Other clinical features include new, prominent veins throughout the shoulder girdle, erythema, increased warmth, functional impairment, parathesias, and non-specific feelings of arm heaviness or discomfort. Symptoms typically worsen with arm use and improve with rest and elevation.15 Patients with UEDVT related to CVC are more likely to be asymptomatic and may present only with PE.16 The differential diagnosis includes superficial phlebitis, lymphatic edema, hematoma, contusions, venous compression, and muscle tears.17
Contrast venography is the gold standard for the UEDVT diagnosis. However, it is more expensive and invasive than ultrasound, and thus serial compression ultrasound is now the standard test in UEDVT evaluation. Then again, contrast venography remains the test of choice in patients with high pre-test probability and negative ultrasound results.18,19
Prevention: Nearly 70% of secondary UEDVT is associated with a CVC.5 Further, CVC use is the most powerful predictor of UEDVT (adjusted odds ratio (OR), 9.7; 95% CI, 7.8 to 12.2).2 Despite the association between CVCs and UEDVT, anticoagulant prophylaxis is not recommended. Studies evaluating the results of 1-mg warfarin conflict and include small populations. Warfarin’s potential interaction with antibiotics and dosing variance based on nutritional intake logically prompted studies on the potential benefit of low-weight molecular heparain (LWMH); however, these studies have failed to show benefit.20,21
Treatment: ACCP guidelines recommend treating UEDVT patients with unfractionated heparin (UFH) or LMWH and warfarin, with an INR goal of 2 to 3 for at least three months depending upon the overall clinical scenario. Two small studies evaluating catheter-related thrombosis (15 patients in each trial) reported no subsequent embolic phenomenon.22,23 Some authors interpreted this data to mean UEDVT was not as morbid as LEDVT and, subsequently, that catheter-related UEDVTs require only one month of therapy. Since the small studies were published, the increasing incidence and relevance of UEDVT have become more widely recognized, and most authors are recommending three months of treatment.
Still, it’s important to note that there aren’t any published data directly comparing the one-month and the three-month anticoagulation therapies. The RIETE registry, which is the largest ongoing published registry of patients with confirmed DVT or PE, reports similar three-month clinical outcomes between those with UEDVT and LEDVT.
Small, single-center trials have shown that such active interventions as thrombolysis, surgery, or multi-staged approaches are associated with increased vein patency and decreased rates of post-thrombotic syndrome.24,25 However, ACCP has withheld general recommendations for these interventions based on a lack of sufficient data to comment on their overall safety and efficacy, as well as comparable rates of post-thrombotic syndrome (15% to 50%) in studies that directly compared surgical and medical intervention. In fact, the ACCP recommends against interventional treatments unless the patient has failed anticoagulation therapy, has severe symptoms, and expertise is available.4
Superior vena cava filters are available at some centers for patients in whom anticoagulation is contraindicated, but efficacy data is limited. While the data for filter use in UEDVT is limited, its use should be considered in patients who have a contraindication to anticoagulation and remain high risk for UEDVT (e.g., prolonged central line placement).
Complications: Post-thrombotic syndrome (PTS) is the most significant local complication of UEDVT. PTS characteristics are edema, pain, venous ulcers, and skin pigmentation changes, and it is the result of chronic venous insufficiency due to the clot. A meta-analysis of clinical studies on UEDVT noted that PTS occurs in 7% to 46% (mean 15%) of patients.26 One hypothesis for the wide range in frequency is the lack of clear diagnostic criteria for PTS.27 No clear beneficial treatment or prevention for PTS exists, but many recommend graduated compression stockings for the arm.
Residual and recurrent thrombosis are associated with increased PTS risk, which emphasizes the need for further study of interventional treatment because preliminary work has shown increased rates of vein patency in comparison to anticoagulants alone. Recurrent venous thromboembolism (VTE), another local complication, appears to occur less often than it does in patients with LEDVTs, but reaches 8% after five years of followup.28
PE is less common on presentation among patients with UEDVT when compared to patients with LEDVT, but when PE occurs, the three-month outcome is similar.3 PE appears to be more frequent in patients who have a CVC, with an incidence as high as 36% of DVT patients.4,13,21,29
Increased mortality: The mortality among UEDVT patients has been described as 10% to 50% in the 12 months after diagnosis, which is much higher than the ratio in LEDVT patients.21,30 This in part is due to sicker cohorts getting UEDVT. For example, patients with distant metastasis are more likely to develop UEDVT than those with confined malignancy (adjusted OR 11.5; 95% CI, 1.6 to 80.2).31
Occult malignancy, most commonly lung cancer or lymphoma, has been found in as many as 24% of UEDVT patients.32 The high rate of mortality associated with UEDVT appears to be related more with the patient's overall poor clinical condition rather than directly related to complications from the DVT.
However, its presence should alert hospitalists to the patient's potentially poorer prognosis and prompt evaluation for occult malignancy if no risk factor is present.
Back to the Case
This patient should be started on either UFH or LMWH while simultaneously beginning warfarin. She should continue warfarin treatment for at least three months, with a goal INR of 2.0 to 3.0, similar to treatment for LEDVT. The ultimate treatment duration with warfarin follows the same guidelines as treatment with a LEDVT. Although prophylaxis is not routinely recommended, dosing 1 mg of warfarin beginning three days before subsequent CVC placement should be considered if this patient requires a future CVC.
Additionally, an evaluation for occult malignancy should be considered in this patient.
Bottom Line
Upper extremity DVT is not a benign condition, and is associated with a general increase in mortality. It should be treated similarly to LEDVT in order to decrease PTS, recurrent DVT, and pumonary embolism.
Dr. Hollberg is an assistant professor of medicine, Emory University School of Medicine, Atlanta, and medical director for information services, Emory Healthcare.
References
- Hirsh J, Hoak J. Management of deep vein thrombosis and pulmonary embolism. A statement for healthcare professionals. Council on Thrombosis, American Heart Association. Circulation. 1996;93(12):2212-2245.
- Gerotziafas GT, Samama MM. Prophylaxis of venous thromboembolism in medical patients. Curr Opin Pulm Med. 2004;10(5):356-365.
- Kabani L, et al. Upper extremity DVT as prevalent as lower extremity DVT in ICU patients. Society of Critical Care Medicine (SCCM) 38th annual Critical Care Congress: Abstract 305. Presented Feb. 2, 2009.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Therapy for venous thromboembolic disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6Suppl):454S-545S.
- Joffe HV, Kucher N, Tapson VF, Goldhaber SZ. Upper extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110:1605.
- Munoz FJ, Mismetti P, Poggio R, et al. Clinical outcome of patients with an upper-extremity deep vein thrombosis: results from the RIETE registry. Chest. 2008,133:143-148.
- Coon WW, Willis PW. Thrombosis of axillary and subclavian veins. Arch Surg. 1967;94(5):657-663.
- Horattas MC, Wright DJ, Fenton AH, et al. Changing concepts of deep venous thrombosis of the upper extremity—a report of a series and review of the literature. Surgery. 1988;104(3):561-567.
- Bernardi E, Piccioli A, Marchiori A, Girolami B, Prandoni P. Upper extremity deep vein thrombosis: risk factors, diagnosis, and management. Semin Vasc Med. 2001;1(1):105;110.
- Heron E, Lozinguez O, Alhenc-Gelas M, Emmerich J, Flessinger JN. Hypercoagulable states in primary upper-extremity deep vein thrombosis. Arch Intern Med. 2000;160:382-386.
- Ninet J, Demolombe-Rague S, Bureau Du Colombier P, Coppere B. Les thromboses veineuses profondes des members superieurs. Sang Thromb Vaisseaux. 1994;6:103-114.
- Painter TD, Kerpf M. Deep venous thrombosis of the upper extremity five years experience at a university hospital. Angiology. 1984;35(35):743-749.
- Chan WS, Ginsberg JS. A review of upper extremity deep vein thrombosis in pregnancy: unmasking the “ART” behind the clot. J Thromb Haemost. 2006; 4(8):1673-1677.
- Hughes MJ, D’Agostino JC. Upper extremity deep vein thrombosis: a case report and review of current diagnostic/therapeutic modalities. Am J Emerg Med. 1994;12(6):631-635.
- Prandoni P, Polistena P, Bernardi E, et al. Upper extremity deep vein thrombosis. Risk factors, diagnosis, and complications. Arch Intern Med. 1997;157:57-62.
- Van Rooden CJ, Tesslar ME, Osanto S, Rosendal FR, Huisman MV. Deep vein thrombosis associated with central venous catheters—a review. J Thromb Haemost. 2005;3:2049-2419.
- Horattas MC, Wright DJ, Fenton AH, et al. Changing concepts of deep venous thrombosis of the upper extremity—report of a series and review of the literature. Surgery. 1988;104(3):561-567.
- Bernardi E, Pesavento R, Prandoni P. Upper extremity deep venous thrombosis. Semin Thromb Hemost. 2006;32(7):729-736.
- Baxter GM, McKechnie S, Duffy P. Colour Doppler ultrasound in deep venous thrombosis: a comparison with venography. Clin Radiol. 1990;42(1):32-36.
- Bern MM, Lokich JJ, Wallach SR, et al. Very low doses of warfarin can prevent thrombosis in central venous catheters. A randomized prospective trial. Ann Intern Med. 1990;112(6):423-428.
- Couban S, Goodyear M, Burnell M, et al. Randomized placebo-controlled study of low-dose warfarin for the prevention of central venous catheter-associated thrombosis in patients with cancer. J Clin Oncol. 2005;23(18):4063-4069.
- Lokich JJ, Both A, Benotti P. Complications and management of implanted central venous catheters. J Clin Oncol. 1985;3:710-717.
- Moss JF, Wagman LD, Rijhmaki DU, Terz JJ. Central venous thrombosis related to the silastic Hickman-Broviac catheter in an oncologic population. J Parenter Enteral Nutr. 1989;13:397.
- Machleder HI. Evaluation of a new treatment strategy for Paget-Schroetter syndrome: spontaneous thrombosis of the axillary-subclavian vein. J Vasc Surg. 1993;17:305-315.
- Malcynski J, O’Donnell TF, Mackey WC. Long-term results of treatment for axillary subclavian vein thrombosis. Can J Surg. 1993;36:365-371.
- Elman EE, Kahn SR. The post-thrombotic syndrome after upper extremity deep vein thrombosis in adults: a systematic review. Thromb Res. 2006;117(6):609-614.
- Baarslag HJ, Koopman MM, Hutten BA, et al. Long-term follow up of patients with suspected deep vein thrombosis of the upper extremity: survival, risk factors and post-thrombotic syndrome. Eur J Intern Med. 2004;15:503-507.
- Prandoni P, Bernardi E, Marchiori A, et al. The long term clinical consequence of acute deep venous thrombosis of the arm: prospective cohort study. BMJ. 2004;329:484-485.
- Monreal M, Raventos A, Lerma R, et al. Pulmonary embolism in patients with upper extremity DVT associated to venous central lines—a prospective study. Thromb Haemost. 1994;72(4):548-550.
- Hingorani A, Ascher E, Lorenson E, et al. Upper extremity deep venous thrombosis and its impact on morbidity and mortality rates in a hospital-based population. J Vasc Surg. 1997;26:853-860.
- Blom JW, Doggen CM, Osanto S, Rosendaal FR. Old and new risk factors for upper extremity deep vein thrombosis. J Thromb Haemost. 2005;3:2471-2478.
- Girolami A, Prandoni P, Zanon E, Bagatella P, Girolami B. Venous thromboses of upper limbs are more frequently associated with occult cancer as compared with those of lower limbs. Blood Coagul Fibrinolysis. 1999;10(8):455-457.
Small-Town Tonic
In mid-August, the White House released its “Jobs and Economic Security for Rural America” report (www.whitehouse.gov), which underlines what most hospitalists already know: Rural healthcare is ailing. As the report points out, rural residents are more likely to be uninsured or be covered through public sources, while mortality rates have dropped more slowly in rural areas than in urban ones.
One troubling statistic in particular highlights the disparity in access: In 2008, the report notes, rural counties had 62 primary-care physicians (PCPs) per 100,000 residents, while urban areas counted an average of 79.5 PCPs (28% more). Although a number of initiatives have specifically sought to narrow that gap, a lesser-known dynamic between primary care and HM might be exacerbating the shortage.
Over the past few years, several reports and media accounts have suggested that medical students increasingly want practices that are either hospital-based or office-based, but not both. The presence of hospitalists, then, helps rural facilities create an attractive office-hospital divide and place PCPs in practices frequently owned by the hospital. Hospitalists, in other words, might be necessary prerequisites to help lure and retain PCPs.

—Louis J. O’Boyle, DO, FACP, FHM, medical director, Advanced Inpatient Medicine, P.C., Honesdale, Pa.
Meanwhile, many physicians already in private rural practices are burning out. According to the 2009 Rural Hospitalist Study by the Illinois Critical Access Health Network, “primary-care physicians in rural areas are throwing in the towel of managing their hospitalized patients. More and more, these PCPs unilaterally are announcing to their patients and to the local hospitals they will neither continue to take responsibility for hospitalized patients nor continue to ‘take call.’ ”
Ome Nwanze, MD, one of two hospitalists at the 42-bed Greenville Regional Hospital in Greenville, Ill., says the biggest benefit to being a rural hospitalist is the ability to make a difference in the lives of everyone in the community. Along with patients, Dr. Nwanze includes other doctors as beneficiaries: “The primary-care physicians and specialists are very happy with the program and the difference it makes in their lives.”
Competitive Business
If hospitalists are a natural solution, though, there’s a key problem: Rural communities are struggling to attract them as well. One sign of the difficulty is median salary. Similar to what surveys consistently show for other specialties, rural hospitalists outpace their urban counterparts in median annual salary, at roughly $206,000 versus $187,000, according to Becker’s Hospital Review (overall, hospitalists rank behind most other specialties in salary). The rural-urban divide can be attributed to that old real estate adage: location, location, location. Competition for hospitalist jobs in large cities is generally fierce, while rural communities often have to offer more incentives to attract and retain the doctors they need.
“The two biggest issues that I can see are recruitment and night coverage,” says Louis J. O’Boyle, DO, FACP, FHM, medical director of Advanced Inpatient Medicine (AIM), P.C., in Honesdale, Pa. He and AIM’s four other hospitalists work exclusively with the town’s 98-bed Wayne Memorial Hospital. “It is easier to recruit to a larger city, closer to more activities and residency programs,” Dr. O’Boyle says. “To get someone to come to our area almost always requires some form of local connection. That makes retention paramount.”
Night call can be a particular sticking point: Most rural hospitals aren’t busy enough to justify an FTE nocturnist, he says, putting the onus of night call on full-time hospitalists. Wayne Memorial Hospital is fortunate in that regard, as it averages only one or two admissions a day after 10 p.m., leaving the hospitalists “fresh enough to round the next day,” Dr. O’Boyle says. “However, this still makes rural programs less attractive compared to places that can boast a nocturnist team that eliminates night call.”
Government Assistance
So what has the government done to help address the growing need for more rural hospitalists and other healthcare providers? If the Affordable Care Act’s (ACA) measures proceed as expected, most experts predict a significant drop in the number of uninsured individuals—meaning a surge in both rural and urban demand for care.
According to the White House report, the Department of Health and Human Services has funded 444 rural community health centers since 2009. The ACA has expanded and extended the Medicare Rural Community Hospital Demonstration, providing “an estimated $52 million in enhanced reimbursement for inpatient services at 25 rural hospitals.” And the administration has expanded funding for the National Health Service Corps, which offers doctors scholarships and loan repayment in exchange for a commitment to practice medicine at underserved communities. The corps website boasts that more than 8,000 clinicians are in place, but it also notes that there are “more than 9,000 job vacancies for NHSC primary care medical, dental, and mental health clinicians.” (View the full report at http://nhsc.hrsa.gov/about.) Clearly, loan forgiveness isn’t enough.
Furthermore, the government might be facing a perception problem. Dr. Nwanze describes government support to rural programs as “poor,” while Dr. O’Boyle says he’s not aware of any specific efforts to support rural hospitalists. “There may be some areas, such as giving grants for telemedicine and other tertiary support, but I don’t think those of us in rural programs can sense any impact,” Dr. O’Boyle says. Wayne Memorial Hospital is in an underserved area, he says, and PCPs there do receive loan forgiveness. “However, I was disappointed to learn that those programs are not open to hospitalists.”
Meanwhile, many rural hospitalists face daunting responsibilities. Dr. Nwanze cites “the need to be a jack-of-all-trades and master of all,” and notes the pressure of providing a wide range of services and handling almost all situations with little or no specialist support.
But Dr. O’Boyle also sees opportunity in the autonomy, such as the ability to play a larger role in hospital management and more independence. “We don’t have a plethora of subspecialists looking for business,” he says. “That means much greater responsibility for our hospitalists, who will take care of much sicker patients without specialist backup being readily available.” As a result, advanced duties like ventilator management and the care of complex patients with such diagnoses as acute renal failure or new malignancies are all within the realm of the hospitalist.
“This is an attractive prospect for certain hospitalists who like the idea of taking care of patients without feeling like a captain who merely delegates to multiple specialists,” Dr. O’Boyle says. “Also, the group integrates into hospital committees at every level, and has an overall much larger say in the day-to-day operations, something largely out of the control of a hospitalist group at a large tertiary facility.”
Tech Solutions
Despite the challenges, many rural hospitals are gaining new tools to help them survive, and tech-savvy hospitalists might be big assets. Smaller facilities are increasingly gaining access to electronic health records, while many also are using video links to allow specialists hundreds of miles away to help with diagnoses without having to transfer the patients.
Recent research also suggests that hospital discharges could be better in rural communities.
Bryn Nelson is a freelance medical writer based in Seattle.
In mid-August, the White House released its “Jobs and Economic Security for Rural America” report (www.whitehouse.gov), which underlines what most hospitalists already know: Rural healthcare is ailing. As the report points out, rural residents are more likely to be uninsured or be covered through public sources, while mortality rates have dropped more slowly in rural areas than in urban ones.
One troubling statistic in particular highlights the disparity in access: In 2008, the report notes, rural counties had 62 primary-care physicians (PCPs) per 100,000 residents, while urban areas counted an average of 79.5 PCPs (28% more). Although a number of initiatives have specifically sought to narrow that gap, a lesser-known dynamic between primary care and HM might be exacerbating the shortage.
Over the past few years, several reports and media accounts have suggested that medical students increasingly want practices that are either hospital-based or office-based, but not both. The presence of hospitalists, then, helps rural facilities create an attractive office-hospital divide and place PCPs in practices frequently owned by the hospital. Hospitalists, in other words, might be necessary prerequisites to help lure and retain PCPs.
—Louis J. O’Boyle, DO, FACP, FHM, medical director, Advanced Inpatient Medicine, P.C., Honesdale, Pa.
Meanwhile, many physicians already in private rural practices are burning out. According to the 2009 Rural Hospitalist Study by the Illinois Critical Access Health Network, “primary-care physicians in rural areas are throwing in the towel of managing their hospitalized patients. More and more, these PCPs unilaterally are announcing to their patients and to the local hospitals they will neither continue to take responsibility for hospitalized patients nor continue to ‘take call.’ ”
Ome Nwanze, MD, one of two hospitalists at the 42-bed Greenville Regional Hospital in Greenville, Ill., says the biggest benefit to being a rural hospitalist is the ability to make a difference in the lives of everyone in the community. Along with patients, Dr. Nwanze includes other doctors as beneficiaries: “The primary-care physicians and specialists are very happy with the program and the difference it makes in their lives.”
Competitive Business
If hospitalists are a natural solution, though, there’s a key problem: Rural communities are struggling to attract them as well. One sign of the difficulty is median salary. Similar to what surveys consistently show for other specialties, rural hospitalists outpace their urban counterparts in median annual salary, at roughly $206,000 versus $187,000, according to Becker’s Hospital Review (overall, hospitalists rank behind most other specialties in salary). The rural-urban divide can be attributed to that old real estate adage: location, location, location. Competition for hospitalist jobs in large cities is generally fierce, while rural communities often have to offer more incentives to attract and retain the doctors they need.
“The two biggest issues that I can see are recruitment and night coverage,” says Louis J. O’Boyle, DO, FACP, FHM, medical director of Advanced Inpatient Medicine (AIM), P.C., in Honesdale, Pa. He and AIM’s four other hospitalists work exclusively with the town’s 98-bed Wayne Memorial Hospital. “It is easier to recruit to a larger city, closer to more activities and residency programs,” Dr. O’Boyle says. “To get someone to come to our area almost always requires some form of local connection. That makes retention paramount.”
Night call can be a particular sticking point: Most rural hospitals aren’t busy enough to justify an FTE nocturnist, he says, putting the onus of night call on full-time hospitalists. Wayne Memorial Hospital is fortunate in that regard, as it averages only one or two admissions a day after 10 p.m., leaving the hospitalists “fresh enough to round the next day,” Dr. O’Boyle says. “However, this still makes rural programs less attractive compared to places that can boast a nocturnist team that eliminates night call.”
Government Assistance
So what has the government done to help address the growing need for more rural hospitalists and other healthcare providers? If the Affordable Care Act’s (ACA) measures proceed as expected, most experts predict a significant drop in the number of uninsured individuals—meaning a surge in both rural and urban demand for care.
According to the White House report, the Department of Health and Human Services has funded 444 rural community health centers since 2009. The ACA has expanded and extended the Medicare Rural Community Hospital Demonstration, providing “an estimated $52 million in enhanced reimbursement for inpatient services at 25 rural hospitals.” And the administration has expanded funding for the National Health Service Corps, which offers doctors scholarships and loan repayment in exchange for a commitment to practice medicine at underserved communities. The corps website boasts that more than 8,000 clinicians are in place, but it also notes that there are “more than 9,000 job vacancies for NHSC primary care medical, dental, and mental health clinicians.” (View the full report at http://nhsc.hrsa.gov/about.) Clearly, loan forgiveness isn’t enough.
Furthermore, the government might be facing a perception problem. Dr. Nwanze describes government support to rural programs as “poor,” while Dr. O’Boyle says he’s not aware of any specific efforts to support rural hospitalists. “There may be some areas, such as giving grants for telemedicine and other tertiary support, but I don’t think those of us in rural programs can sense any impact,” Dr. O’Boyle says. Wayne Memorial Hospital is in an underserved area, he says, and PCPs there do receive loan forgiveness. “However, I was disappointed to learn that those programs are not open to hospitalists.”
Meanwhile, many rural hospitalists face daunting responsibilities. Dr. Nwanze cites “the need to be a jack-of-all-trades and master of all,” and notes the pressure of providing a wide range of services and handling almost all situations with little or no specialist support.
But Dr. O’Boyle also sees opportunity in the autonomy, such as the ability to play a larger role in hospital management and more independence. “We don’t have a plethora of subspecialists looking for business,” he says. “That means much greater responsibility for our hospitalists, who will take care of much sicker patients without specialist backup being readily available.” As a result, advanced duties like ventilator management and the care of complex patients with such diagnoses as acute renal failure or new malignancies are all within the realm of the hospitalist.
“This is an attractive prospect for certain hospitalists who like the idea of taking care of patients without feeling like a captain who merely delegates to multiple specialists,” Dr. O’Boyle says. “Also, the group integrates into hospital committees at every level, and has an overall much larger say in the day-to-day operations, something largely out of the control of a hospitalist group at a large tertiary facility.”
Tech Solutions
Despite the challenges, many rural hospitals are gaining new tools to help them survive, and tech-savvy hospitalists might be big assets. Smaller facilities are increasingly gaining access to electronic health records, while many also are using video links to allow specialists hundreds of miles away to help with diagnoses without having to transfer the patients.
Recent research also suggests that hospital discharges could be better in rural communities.
Bryn Nelson is a freelance medical writer based in Seattle.
In mid-August, the White House released its “Jobs and Economic Security for Rural America” report (www.whitehouse.gov), which underlines what most hospitalists already know: Rural healthcare is ailing. As the report points out, rural residents are more likely to be uninsured or be covered through public sources, while mortality rates have dropped more slowly in rural areas than in urban ones.
One troubling statistic in particular highlights the disparity in access: In 2008, the report notes, rural counties had 62 primary-care physicians (PCPs) per 100,000 residents, while urban areas counted an average of 79.5 PCPs (28% more). Although a number of initiatives have specifically sought to narrow that gap, a lesser-known dynamic between primary care and HM might be exacerbating the shortage.
Over the past few years, several reports and media accounts have suggested that medical students increasingly want practices that are either hospital-based or office-based, but not both. The presence of hospitalists, then, helps rural facilities create an attractive office-hospital divide and place PCPs in practices frequently owned by the hospital. Hospitalists, in other words, might be necessary prerequisites to help lure and retain PCPs.
—Louis J. O’Boyle, DO, FACP, FHM, medical director, Advanced Inpatient Medicine, P.C., Honesdale, Pa.
Meanwhile, many physicians already in private rural practices are burning out. According to the 2009 Rural Hospitalist Study by the Illinois Critical Access Health Network, “primary-care physicians in rural areas are throwing in the towel of managing their hospitalized patients. More and more, these PCPs unilaterally are announcing to their patients and to the local hospitals they will neither continue to take responsibility for hospitalized patients nor continue to ‘take call.’ ”
Ome Nwanze, MD, one of two hospitalists at the 42-bed Greenville Regional Hospital in Greenville, Ill., says the biggest benefit to being a rural hospitalist is the ability to make a difference in the lives of everyone in the community. Along with patients, Dr. Nwanze includes other doctors as beneficiaries: “The primary-care physicians and specialists are very happy with the program and the difference it makes in their lives.”
Competitive Business
If hospitalists are a natural solution, though, there’s a key problem: Rural communities are struggling to attract them as well. One sign of the difficulty is median salary. Similar to what surveys consistently show for other specialties, rural hospitalists outpace their urban counterparts in median annual salary, at roughly $206,000 versus $187,000, according to Becker’s Hospital Review (overall, hospitalists rank behind most other specialties in salary). The rural-urban divide can be attributed to that old real estate adage: location, location, location. Competition for hospitalist jobs in large cities is generally fierce, while rural communities often have to offer more incentives to attract and retain the doctors they need.
“The two biggest issues that I can see are recruitment and night coverage,” says Louis J. O’Boyle, DO, FACP, FHM, medical director of Advanced Inpatient Medicine (AIM), P.C., in Honesdale, Pa. He and AIM’s four other hospitalists work exclusively with the town’s 98-bed Wayne Memorial Hospital. “It is easier to recruit to a larger city, closer to more activities and residency programs,” Dr. O’Boyle says. “To get someone to come to our area almost always requires some form of local connection. That makes retention paramount.”
Night call can be a particular sticking point: Most rural hospitals aren’t busy enough to justify an FTE nocturnist, he says, putting the onus of night call on full-time hospitalists. Wayne Memorial Hospital is fortunate in that regard, as it averages only one or two admissions a day after 10 p.m., leaving the hospitalists “fresh enough to round the next day,” Dr. O’Boyle says. “However, this still makes rural programs less attractive compared to places that can boast a nocturnist team that eliminates night call.”
Government Assistance
So what has the government done to help address the growing need for more rural hospitalists and other healthcare providers? If the Affordable Care Act’s (ACA) measures proceed as expected, most experts predict a significant drop in the number of uninsured individuals—meaning a surge in both rural and urban demand for care.
According to the White House report, the Department of Health and Human Services has funded 444 rural community health centers since 2009. The ACA has expanded and extended the Medicare Rural Community Hospital Demonstration, providing “an estimated $52 million in enhanced reimbursement for inpatient services at 25 rural hospitals.” And the administration has expanded funding for the National Health Service Corps, which offers doctors scholarships and loan repayment in exchange for a commitment to practice medicine at underserved communities. The corps website boasts that more than 8,000 clinicians are in place, but it also notes that there are “more than 9,000 job vacancies for NHSC primary care medical, dental, and mental health clinicians.” (View the full report at http://nhsc.hrsa.gov/about.) Clearly, loan forgiveness isn’t enough.
Furthermore, the government might be facing a perception problem. Dr. Nwanze describes government support to rural programs as “poor,” while Dr. O’Boyle says he’s not aware of any specific efforts to support rural hospitalists. “There may be some areas, such as giving grants for telemedicine and other tertiary support, but I don’t think those of us in rural programs can sense any impact,” Dr. O’Boyle says. Wayne Memorial Hospital is in an underserved area, he says, and PCPs there do receive loan forgiveness. “However, I was disappointed to learn that those programs are not open to hospitalists.”
Meanwhile, many rural hospitalists face daunting responsibilities. Dr. Nwanze cites “the need to be a jack-of-all-trades and master of all,” and notes the pressure of providing a wide range of services and handling almost all situations with little or no specialist support.
But Dr. O’Boyle also sees opportunity in the autonomy, such as the ability to play a larger role in hospital management and more independence. “We don’t have a plethora of subspecialists looking for business,” he says. “That means much greater responsibility for our hospitalists, who will take care of much sicker patients without specialist backup being readily available.” As a result, advanced duties like ventilator management and the care of complex patients with such diagnoses as acute renal failure or new malignancies are all within the realm of the hospitalist.
“This is an attractive prospect for certain hospitalists who like the idea of taking care of patients without feeling like a captain who merely delegates to multiple specialists,” Dr. O’Boyle says. “Also, the group integrates into hospital committees at every level, and has an overall much larger say in the day-to-day operations, something largely out of the control of a hospitalist group at a large tertiary facility.”
Tech Solutions
Despite the challenges, many rural hospitals are gaining new tools to help them survive, and tech-savvy hospitalists might be big assets. Smaller facilities are increasingly gaining access to electronic health records, while many also are using video links to allow specialists hundreds of miles away to help with diagnoses without having to transfer the patients.
Recent research also suggests that hospital discharges could be better in rural communities.
Bryn Nelson is a freelance medical writer based in Seattle.
A Brief History
Each visit category and level of service has corresponding documentation requirements.1 Selecting an evaluation and management (E/M) level is based upon 1) the content of the three “key” components: history, exam, and decision-making, or 2) time, but only when counseling or coordination of care dominates more than 50% of the physician’s total visit time. Failure to document any essential element in a given visit level (e.g. family history required but missing for 99222 and 99223) could result in downcoding or service denial. Be aware of what an auditor expects when reviewing the key component of “history.”
Documentation Options
Auditors recognize two sets of documentation guidelines: “1995” and “1997” guidelines.2,3,4 Each set of guidelines has received valid criticism. The 1995 guidelines undoubtedly are vague and subjective in some areas, whereas the 1997 guidelines are known for arduous specificity.
However, to benefit all physicians and specialties, both sets of guidelines apply to visit-level selection. In other words, physicians can utilize either set when documenting their services, and auditors must review provider records against both styles. The final audited outcome reflects the highest visit level supported upon comparison.
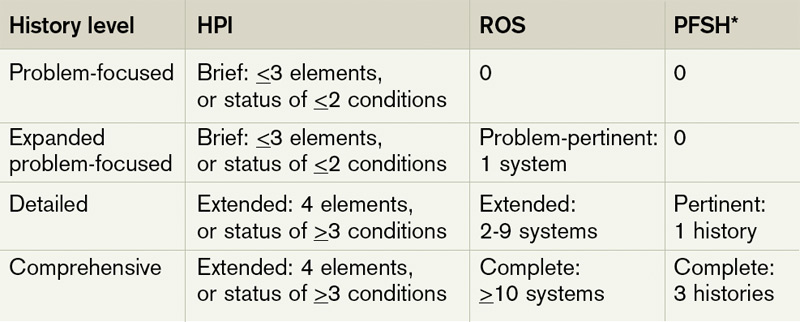
Elements of History2,3,4
Chief complaint. The chief complaint (CC) is the reason for the visit, as stated in the patient’s own words. Every encounter, regardless of visit type, must include a CC. The physician must personally document and/or validate the CC with reference to a specific condition or symptom (e.g. patient complains of abdominal pain).
History of present illness (HPI). The HPI is a description of the patient’s present illness as it developed. It characteristically is referenced as location, quality, severity, timing, context, modifying factors, and associated signs/symptoms, as related to the chief complaint. The 1997 guidelines allow physicians to receive HPI credit for providing the status of the patient’s chronic or inactive conditions, such as “extrinsic asthma without acute exacerbation in past six months.” An auditor will not assign HPI credit to a chronic or inactive condition that does not have a corresponding status (e.g. “asthma”). This will be considered “past medical history.”
The HPI is classified as brief (a comment on <3 HPI elements, or the status of <2 conditions) or extended (a comment on >4 HPI elements, or the status of >3 conditions). Consider these examples of an extended HPI:
- “The patient has intermittent (duration), sharp (quality) pain in the right upper quadrant (location) without associated nausea, vomiting, or diarrhea (associated signs/symptoms).”
- “Diabetes controlled by oral medication; hyperlipidemia stable on simvastatin with increased dietary efforts; hypertension stable with pressures ranging from 130-140/80-90.” (Status of three chronic conditions.)
Physicians receive credit for confirming and personally documenting the HPI, or linking to documentation recorded by residents (residents, fellows, interns) or nonphysician providers (NPPs) when performing services according to the Teaching Physician Rules or Split-Shared Billing Rules, respectively. An auditor will not assign physician credit for HPI elements documented by ancillary staff (registered nurses, medical assistants) or students.
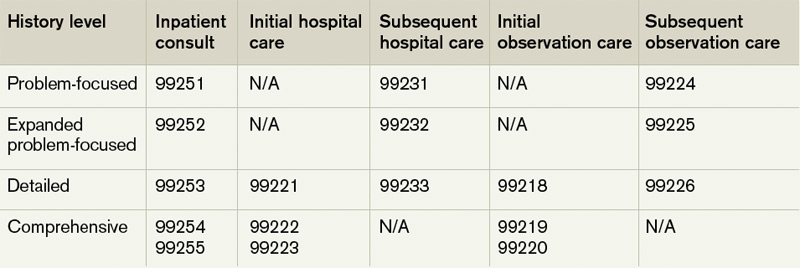
Review of systems (ROS). The ROS is a series of questions used to elicit information about additional signs, symptoms, or problems currently or previously experienced by the patient: constitutional; eyes, ears, nose, mouth, throat; cardiovascular; respiratory; gastrointestinal; genitourinary; musculoskeletal; integumentary (including skin and/or breast); neurological; psychiatric; endocrine; hematologic/ lymphatic; and allergic/immunologic. Auditors classify the ROS as brief (a comment on one system), extended (a comment on two to nine systems), or complete (a comment on >10 systems). Physicians can document a complete ROS by noting individual systems: “no fever/chills (constitutional) or blurred vision (eyes); no chest pain (cardiovascular) or shortness of breath (respiratory); intermittent nausea (gastrointestinal); and occasional runny nose (ears, nose, mouth, throat),” or by eliciting a complete system review but documenting only the positive and pertinent negative findings related to the chief complaint, along with an additional comment that “all other systems are negative.”
Although the latter method is formally included in Medicare’s documentation guidelines and accepted by some Medicare contractors (e.g. Highmark, WPS), be aware that it is not universally accepted.5,6
Documentation involving the ROS can be provided by anyone, including the patient. The physician should reference ROS information that is completed by individuals other than residents or NPPs during services provided under the Teaching Physician Rules or Split-Shared Billing Rules. Physician duplication of ROS information is unnecessary unless an update or revision is required.
Past, family, and social history (PFSH). The PFSH involves data obtained about the patient’s previous illness or medical conditions/therapies, family occurrences with illness, and relevant patient activities. The PFSH could be classified as pertinent (a comment on one history) or complete (a comment in each of the three histories). The physician merely needs a single comment associated with each history for the PFSH to be regarded as complete. Refrain from using “noncontributory” to describe any of the histories, as previous misuse of this term has resulted in its prohibition. An example of a complete PFSH documentation includes: “Patient currently on Prilosec 20 mg daily; family history of Barrett’s esophagus; no tobacco or alcohol use.”
Similar to the ROS, PFSH documentation can be provided by anyone, including the patient, and the physician should reference the documented PFSH in his own progress note. Redocumentation of the PFSH is not necessary unless a revision is required.
PFSH documentation is only required for initial care services (i.e. initial hospital care, initial observation care, consultations). It is not warranted in subsequent care services unless additional, pertinent information is obtained during the hospital stay that impacts care.
Considerations
When a physician cannot elicit historical information from the patient directly, and no other source is available, they should document “unable to obtain” the history. A comment regarding the circumstances surrounding this problem (e.g. patient confused, no caregiver present) should be provided, along with the available information from the limited resources (e.g. emergency medical technicians, previous hospitalizations at the same facility). Some contractors will not penalize the physician for the inability to ascertain complete historical information, as long as a proven attempt to obtain the information is evident.
Never document any item for the purpose of “getting paid.” Only document information that is clinically relevant, lends to the quality of care provided, or demonstrates the delivery of healthcare services. This prevents accusations of fraud and abuse, promotes billing compliance, and supports medical necessity for the services provided.
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center in Philadelphia. She is faculty for SHM’s inpatient coding course.
References
- Pohlig, C. Documentation and Coding Evaluation and Management Services. In: Coding for Chest Medicine 2010. Northbrook, IL: American College of Chest Physicians, 2009; 87-118.
- Centers for Medicare & Medicaid Services. 1995 Documentation Guidelines for Evaluation & Management Services. CMS website. Available at: www.cms.hhs.gov/MLNProducts/Downloads/1995dg.pdf. Accessed July 7, 2011.
- Centers for Medicare & Medicaid Services. 1997 Documentation Guidelines for Evaluation & Management Services. CMS website. Available at: http://www.cms.hhs.gov/MLNProducts/Downloads/MASTER1.pdf. Accessed July 7, 2011.
- Abraham M, Ahlman J, Boudreau A, Connelly J, Evans D. Current Procedural Terminology Professional Edition. Chicago: American Medical Association Press; 2011.
- History of E/M (Q&As). WPS Health Insurance website. Available at: http://www.wpsmedicare.com/j5macpartb/resources/provider_types/2009_0526_emqahistory.shtml. Accessed July 11, 2011.
- Frequently Asked Questions: Evaluation and Management Services (Part B). Highmark Medicare Services website. Available at: www.highmarkmedicareservices.com/faq/partb/pet/lpet-evaluation_management_services.html. Accessed on July 11, 2011.
Each visit category and level of service has corresponding documentation requirements.1 Selecting an evaluation and management (E/M) level is based upon 1) the content of the three “key” components: history, exam, and decision-making, or 2) time, but only when counseling or coordination of care dominates more than 50% of the physician’s total visit time. Failure to document any essential element in a given visit level (e.g. family history required but missing for 99222 and 99223) could result in downcoding or service denial. Be aware of what an auditor expects when reviewing the key component of “history.”
Documentation Options
Auditors recognize two sets of documentation guidelines: “1995” and “1997” guidelines.2,3,4 Each set of guidelines has received valid criticism. The 1995 guidelines undoubtedly are vague and subjective in some areas, whereas the 1997 guidelines are known for arduous specificity.
However, to benefit all physicians and specialties, both sets of guidelines apply to visit-level selection. In other words, physicians can utilize either set when documenting their services, and auditors must review provider records against both styles. The final audited outcome reflects the highest visit level supported upon comparison.
Elements of History2,3,4
Chief complaint. The chief complaint (CC) is the reason for the visit, as stated in the patient’s own words. Every encounter, regardless of visit type, must include a CC. The physician must personally document and/or validate the CC with reference to a specific condition or symptom (e.g. patient complains of abdominal pain).
History of present illness (HPI). The HPI is a description of the patient’s present illness as it developed. It characteristically is referenced as location, quality, severity, timing, context, modifying factors, and associated signs/symptoms, as related to the chief complaint. The 1997 guidelines allow physicians to receive HPI credit for providing the status of the patient’s chronic or inactive conditions, such as “extrinsic asthma without acute exacerbation in past six months.” An auditor will not assign HPI credit to a chronic or inactive condition that does not have a corresponding status (e.g. “asthma”). This will be considered “past medical history.”
The HPI is classified as brief (a comment on <3 HPI elements, or the status of <2 conditions) or extended (a comment on >4 HPI elements, or the status of >3 conditions). Consider these examples of an extended HPI:
- “The patient has intermittent (duration), sharp (quality) pain in the right upper quadrant (location) without associated nausea, vomiting, or diarrhea (associated signs/symptoms).”
- “Diabetes controlled by oral medication; hyperlipidemia stable on simvastatin with increased dietary efforts; hypertension stable with pressures ranging from 130-140/80-90.” (Status of three chronic conditions.)
Physicians receive credit for confirming and personally documenting the HPI, or linking to documentation recorded by residents (residents, fellows, interns) or nonphysician providers (NPPs) when performing services according to the Teaching Physician Rules or Split-Shared Billing Rules, respectively. An auditor will not assign physician credit for HPI elements documented by ancillary staff (registered nurses, medical assistants) or students.
Review of systems (ROS). The ROS is a series of questions used to elicit information about additional signs, symptoms, or problems currently or previously experienced by the patient: constitutional; eyes, ears, nose, mouth, throat; cardiovascular; respiratory; gastrointestinal; genitourinary; musculoskeletal; integumentary (including skin and/or breast); neurological; psychiatric; endocrine; hematologic/ lymphatic; and allergic/immunologic. Auditors classify the ROS as brief (a comment on one system), extended (a comment on two to nine systems), or complete (a comment on >10 systems). Physicians can document a complete ROS by noting individual systems: “no fever/chills (constitutional) or blurred vision (eyes); no chest pain (cardiovascular) or shortness of breath (respiratory); intermittent nausea (gastrointestinal); and occasional runny nose (ears, nose, mouth, throat),” or by eliciting a complete system review but documenting only the positive and pertinent negative findings related to the chief complaint, along with an additional comment that “all other systems are negative.”
Although the latter method is formally included in Medicare’s documentation guidelines and accepted by some Medicare contractors (e.g. Highmark, WPS), be aware that it is not universally accepted.5,6
Documentation involving the ROS can be provided by anyone, including the patient. The physician should reference ROS information that is completed by individuals other than residents or NPPs during services provided under the Teaching Physician Rules or Split-Shared Billing Rules. Physician duplication of ROS information is unnecessary unless an update or revision is required.
Past, family, and social history (PFSH). The PFSH involves data obtained about the patient’s previous illness or medical conditions/therapies, family occurrences with illness, and relevant patient activities. The PFSH could be classified as pertinent (a comment on one history) or complete (a comment in each of the three histories). The physician merely needs a single comment associated with each history for the PFSH to be regarded as complete. Refrain from using “noncontributory” to describe any of the histories, as previous misuse of this term has resulted in its prohibition. An example of a complete PFSH documentation includes: “Patient currently on Prilosec 20 mg daily; family history of Barrett’s esophagus; no tobacco or alcohol use.”
Similar to the ROS, PFSH documentation can be provided by anyone, including the patient, and the physician should reference the documented PFSH in his own progress note. Redocumentation of the PFSH is not necessary unless a revision is required.
PFSH documentation is only required for initial care services (i.e. initial hospital care, initial observation care, consultations). It is not warranted in subsequent care services unless additional, pertinent information is obtained during the hospital stay that impacts care.
Considerations
When a physician cannot elicit historical information from the patient directly, and no other source is available, they should document “unable to obtain” the history. A comment regarding the circumstances surrounding this problem (e.g. patient confused, no caregiver present) should be provided, along with the available information from the limited resources (e.g. emergency medical technicians, previous hospitalizations at the same facility). Some contractors will not penalize the physician for the inability to ascertain complete historical information, as long as a proven attempt to obtain the information is evident.
Never document any item for the purpose of “getting paid.” Only document information that is clinically relevant, lends to the quality of care provided, or demonstrates the delivery of healthcare services. This prevents accusations of fraud and abuse, promotes billing compliance, and supports medical necessity for the services provided.
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center in Philadelphia. She is faculty for SHM’s inpatient coding course.
References
- Pohlig, C. Documentation and Coding Evaluation and Management Services. In: Coding for Chest Medicine 2010. Northbrook, IL: American College of Chest Physicians, 2009; 87-118.
- Centers for Medicare & Medicaid Services. 1995 Documentation Guidelines for Evaluation & Management Services. CMS website. Available at: www.cms.hhs.gov/MLNProducts/Downloads/1995dg.pdf. Accessed July 7, 2011.
- Centers for Medicare & Medicaid Services. 1997 Documentation Guidelines for Evaluation & Management Services. CMS website. Available at: http://www.cms.hhs.gov/MLNProducts/Downloads/MASTER1.pdf. Accessed July 7, 2011.
- Abraham M, Ahlman J, Boudreau A, Connelly J, Evans D. Current Procedural Terminology Professional Edition. Chicago: American Medical Association Press; 2011.
- History of E/M (Q&As). WPS Health Insurance website. Available at: http://www.wpsmedicare.com/j5macpartb/resources/provider_types/2009_0526_emqahistory.shtml. Accessed July 11, 2011.
- Frequently Asked Questions: Evaluation and Management Services (Part B). Highmark Medicare Services website. Available at: www.highmarkmedicareservices.com/faq/partb/pet/lpet-evaluation_management_services.html. Accessed on July 11, 2011.
Each visit category and level of service has corresponding documentation requirements.1 Selecting an evaluation and management (E/M) level is based upon 1) the content of the three “key” components: history, exam, and decision-making, or 2) time, but only when counseling or coordination of care dominates more than 50% of the physician’s total visit time. Failure to document any essential element in a given visit level (e.g. family history required but missing for 99222 and 99223) could result in downcoding or service denial. Be aware of what an auditor expects when reviewing the key component of “history.”
Documentation Options
Auditors recognize two sets of documentation guidelines: “1995” and “1997” guidelines.2,3,4 Each set of guidelines has received valid criticism. The 1995 guidelines undoubtedly are vague and subjective in some areas, whereas the 1997 guidelines are known for arduous specificity.
However, to benefit all physicians and specialties, both sets of guidelines apply to visit-level selection. In other words, physicians can utilize either set when documenting their services, and auditors must review provider records against both styles. The final audited outcome reflects the highest visit level supported upon comparison.
Elements of History2,3,4
Chief complaint. The chief complaint (CC) is the reason for the visit, as stated in the patient’s own words. Every encounter, regardless of visit type, must include a CC. The physician must personally document and/or validate the CC with reference to a specific condition or symptom (e.g. patient complains of abdominal pain).
History of present illness (HPI). The HPI is a description of the patient’s present illness as it developed. It characteristically is referenced as location, quality, severity, timing, context, modifying factors, and associated signs/symptoms, as related to the chief complaint. The 1997 guidelines allow physicians to receive HPI credit for providing the status of the patient’s chronic or inactive conditions, such as “extrinsic asthma without acute exacerbation in past six months.” An auditor will not assign HPI credit to a chronic or inactive condition that does not have a corresponding status (e.g. “asthma”). This will be considered “past medical history.”
The HPI is classified as brief (a comment on <3 HPI elements, or the status of <2 conditions) or extended (a comment on >4 HPI elements, or the status of >3 conditions). Consider these examples of an extended HPI:
- “The patient has intermittent (duration), sharp (quality) pain in the right upper quadrant (location) without associated nausea, vomiting, or diarrhea (associated signs/symptoms).”
- “Diabetes controlled by oral medication; hyperlipidemia stable on simvastatin with increased dietary efforts; hypertension stable with pressures ranging from 130-140/80-90.” (Status of three chronic conditions.)
Physicians receive credit for confirming and personally documenting the HPI, or linking to documentation recorded by residents (residents, fellows, interns) or nonphysician providers (NPPs) when performing services according to the Teaching Physician Rules or Split-Shared Billing Rules, respectively. An auditor will not assign physician credit for HPI elements documented by ancillary staff (registered nurses, medical assistants) or students.
Review of systems (ROS). The ROS is a series of questions used to elicit information about additional signs, symptoms, or problems currently or previously experienced by the patient: constitutional; eyes, ears, nose, mouth, throat; cardiovascular; respiratory; gastrointestinal; genitourinary; musculoskeletal; integumentary (including skin and/or breast); neurological; psychiatric; endocrine; hematologic/ lymphatic; and allergic/immunologic. Auditors classify the ROS as brief (a comment on one system), extended (a comment on two to nine systems), or complete (a comment on >10 systems). Physicians can document a complete ROS by noting individual systems: “no fever/chills (constitutional) or blurred vision (eyes); no chest pain (cardiovascular) or shortness of breath (respiratory); intermittent nausea (gastrointestinal); and occasional runny nose (ears, nose, mouth, throat),” or by eliciting a complete system review but documenting only the positive and pertinent negative findings related to the chief complaint, along with an additional comment that “all other systems are negative.”
Although the latter method is formally included in Medicare’s documentation guidelines and accepted by some Medicare contractors (e.g. Highmark, WPS), be aware that it is not universally accepted.5,6
Documentation involving the ROS can be provided by anyone, including the patient. The physician should reference ROS information that is completed by individuals other than residents or NPPs during services provided under the Teaching Physician Rules or Split-Shared Billing Rules. Physician duplication of ROS information is unnecessary unless an update or revision is required.
Past, family, and social history (PFSH). The PFSH involves data obtained about the patient’s previous illness or medical conditions/therapies, family occurrences with illness, and relevant patient activities. The PFSH could be classified as pertinent (a comment on one history) or complete (a comment in each of the three histories). The physician merely needs a single comment associated with each history for the PFSH to be regarded as complete. Refrain from using “noncontributory” to describe any of the histories, as previous misuse of this term has resulted in its prohibition. An example of a complete PFSH documentation includes: “Patient currently on Prilosec 20 mg daily; family history of Barrett’s esophagus; no tobacco or alcohol use.”
Similar to the ROS, PFSH documentation can be provided by anyone, including the patient, and the physician should reference the documented PFSH in his own progress note. Redocumentation of the PFSH is not necessary unless a revision is required.
PFSH documentation is only required for initial care services (i.e. initial hospital care, initial observation care, consultations). It is not warranted in subsequent care services unless additional, pertinent information is obtained during the hospital stay that impacts care.
Considerations
When a physician cannot elicit historical information from the patient directly, and no other source is available, they should document “unable to obtain” the history. A comment regarding the circumstances surrounding this problem (e.g. patient confused, no caregiver present) should be provided, along with the available information from the limited resources (e.g. emergency medical technicians, previous hospitalizations at the same facility). Some contractors will not penalize the physician for the inability to ascertain complete historical information, as long as a proven attempt to obtain the information is evident.
Never document any item for the purpose of “getting paid.” Only document information that is clinically relevant, lends to the quality of care provided, or demonstrates the delivery of healthcare services. This prevents accusations of fraud and abuse, promotes billing compliance, and supports medical necessity for the services provided.
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center in Philadelphia. She is faculty for SHM’s inpatient coding course.
References
- Pohlig, C. Documentation and Coding Evaluation and Management Services. In: Coding for Chest Medicine 2010. Northbrook, IL: American College of Chest Physicians, 2009; 87-118.
- Centers for Medicare & Medicaid Services. 1995 Documentation Guidelines for Evaluation & Management Services. CMS website. Available at: www.cms.hhs.gov/MLNProducts/Downloads/1995dg.pdf. Accessed July 7, 2011.
- Centers for Medicare & Medicaid Services. 1997 Documentation Guidelines for Evaluation & Management Services. CMS website. Available at: http://www.cms.hhs.gov/MLNProducts/Downloads/MASTER1.pdf. Accessed July 7, 2011.
- Abraham M, Ahlman J, Boudreau A, Connelly J, Evans D. Current Procedural Terminology Professional Edition. Chicago: American Medical Association Press; 2011.
- History of E/M (Q&As). WPS Health Insurance website. Available at: http://www.wpsmedicare.com/j5macpartb/resources/provider_types/2009_0526_emqahistory.shtml. Accessed July 11, 2011.
- Frequently Asked Questions: Evaluation and Management Services (Part B). Highmark Medicare Services website. Available at: www.highmarkmedicareservices.com/faq/partb/pet/lpet-evaluation_management_services.html. Accessed on July 11, 2011.
21st-Century Trainer
When Joshua Lenchus, DO, RPh, FACP, FHM, discussed his love for chemistry with his high school guidance counselors, they told him it could take him in one of two directions: teaching or a career in pharmacy.
Intrigued by the latter option, he decided to become a volunteer in the pharmacy department at a local hospital. Soon after, he became a full-time pharmacy technician and eventually enrolled in pharmacy school at the University of Florida.
“I always knew I wanted to be a physician, and everybody needs a bachelor’s degree in something,” Dr. Lenchus says. “I thought, ‘What better way to do it than to get a bachelor’s degree in pharmacy and then move into medicine?’ ”
After college, he worked as a retail pharmacist, then moved to the institutional setting, creating the position of clinical pharmacist at Wellington (Fla.) Regional Medical Center. Three years later, he entered medical school and ultimately pursued a career as an academic hospitalist.
Dr. Lenchus now serves as associate professor of clinical medicine at the University of Miami’s (UM) Miller School of Medicine, associate program director of Jackson Memorial Hospital’s (JMH) internal medicine residency training program, and associate director of the UM-JMH Center for Patient Safety, which trains about 1,000 medical students, residents, and interns each year.
“Pharmacy has provided an invaluable background for becoming a physician,” says Dr. Lenchus, who was appointed a member of Team Hospitalist in May. “Many physicians order a medication and have no idea what the other half of the equation entails. My experience gave me a solid footing from which I could springboard.”
Q: You spend considerable time mentoring the next generation of physicians. What’s the best advice you can give them?
A: Physicians have these altruistic notions about wanting to help people, but you really have to do what you love. There’s another hospital a mile and a half away from my house, whereas Jackson is 35 miles away and it takes me an hour in transit time each way. But I couldn’t do what I’m doing now at any other facility. I stay because I love what I’m doing.
Q: Why is the UM-JMH Center for Patient Safety so beneficial?
A: The greatest benefit is the ability to be exposed to and tackle real-life scenarios in a risk-free environment. We use life-size mannequins to re-create scenarios that medical personnel will see during their training. We try to re-create the chaos that will ensue.
Q: So it’s similar to a pilot using a flight simulator.
A: Exactly. When a plane crashes and the NTSB goes to see what happened, they perform what we in medicine call a root-cause analysis. They’re not blaming an individual; they want to see what they can change on a system level to prevent an error like that from happening again. We culminate our training with a debriefing that we approach the same way, so nobody walks away thinking they failed.
Q: How effective can simulation-based education be?
A: There will be some limitations because the technology simply cannot account for every aspect of a human. But there’s a wealth of data that supports this as a pretty good surrogate. The technology provides for an incredible amount of experience and exposure without any potential harm to a patient, and it provides [trainees] an opportunity to do things they otherwise would have to wait to do until a clinical scenario demanded it.
Q: Do you think this is the wave of the future?
A: Absolutely. And as the Accreditation Council for Graduate Medical Education promulgates new rules that limit the hours trainees can work, it’s going to be incumbent on training programs to be creative in providing equal or near-equal experience in a much shorter time. Simulation can help fill that bill.
Q: You created a crisis-management simulation course for IM residents. How did that come about?
A: When we have a crisis like a code blue, I witnessed the chaos that ensued. I thought some of the paltry resuscitation rate could be due to the fact there was no meaningful communication in that scenario.
Using full-scale mannequins, I put nurses and residents into those types of situations and videotaped what ensued. Frequently we saw the same chaos we see in reality, and many rather basic, commonsensical concepts went out the window.
Q: Can you offer an example?
A: A big one is situational awareness. If the head of the gurney is in a seated position, that’s not a conducive way to do chest compressions. If the side rails of the bed are up, you can lower them so you aren’t reaching over them. Was a team leader assigned or were roles delegated? These aren’t novel concepts, but when faced with a crisis, everybody tends to focus on their own thing. In a crisis, you need to break those silos down and operate as a team.
Q: How effective is the training?
A: After the first scenario, we show the video and debrief them for 10 or 15 minutes, keying in on some behaviors that can be employed in a crisis. Then we expose them to a different crisis scenario immediately thereafter. Often we see an immediate change in their behavior.
Q: You developed a curriculum through which residents are taught in a standardized manner how to perform invasive bedside procedures. How does it work?
A: They have 12 hours of hands-on instruction using fluid-filled, ultrasound-capable mannequins. A faculty attending teaches these procedures. We took it a step further and made a four-week rotation as a mandatory component of the residency program. They carry a beeper, and any service within the hospital can call the procedure team to do one of the procedures on which they were already trained.
Q: How successful is the effort?
A: This is the beginning of our fifth year, and we’ve been called more than 4,000 times to do procedures on hospitalized patients. We’ve published our curriculum. We’ve shown a significant improvement in knowledge, technical skills, and confidence level, and we have data we’re going to publish later this year that shows our complication rates are better than complication rates that are published elsewhere.
Q: What is your biggest professional reward?
A: The ability to impact the next generation. With the procedural training alone, we have just trained our 1,000th person. Each one of them is going to take care of thousands of patients in their professional careers. That’s an expansive influence.
Q: What is your biggest professional challenge?
A: The culture of medicine. It is infused with hundreds of years of tradition and, at times, it feels like trying to move a mountain. It may take a generation to do it, but there will come a time—at least within the field of patient safety—when more people are attuned to it and understand the concepts really are lifesaving. That doesn’t happen as fast as I would like it to, but if we keep plugging away one year at a time, we will be able to make an impact.
Mark Leiser is a freelance writer based in New Jersey.
When Joshua Lenchus, DO, RPh, FACP, FHM, discussed his love for chemistry with his high school guidance counselors, they told him it could take him in one of two directions: teaching or a career in pharmacy.
Intrigued by the latter option, he decided to become a volunteer in the pharmacy department at a local hospital. Soon after, he became a full-time pharmacy technician and eventually enrolled in pharmacy school at the University of Florida.
“I always knew I wanted to be a physician, and everybody needs a bachelor’s degree in something,” Dr. Lenchus says. “I thought, ‘What better way to do it than to get a bachelor’s degree in pharmacy and then move into medicine?’ ”
After college, he worked as a retail pharmacist, then moved to the institutional setting, creating the position of clinical pharmacist at Wellington (Fla.) Regional Medical Center. Three years later, he entered medical school and ultimately pursued a career as an academic hospitalist.
Dr. Lenchus now serves as associate professor of clinical medicine at the University of Miami’s (UM) Miller School of Medicine, associate program director of Jackson Memorial Hospital’s (JMH) internal medicine residency training program, and associate director of the UM-JMH Center for Patient Safety, which trains about 1,000 medical students, residents, and interns each year.
“Pharmacy has provided an invaluable background for becoming a physician,” says Dr. Lenchus, who was appointed a member of Team Hospitalist in May. “Many physicians order a medication and have no idea what the other half of the equation entails. My experience gave me a solid footing from which I could springboard.”
Q: You spend considerable time mentoring the next generation of physicians. What’s the best advice you can give them?
A: Physicians have these altruistic notions about wanting to help people, but you really have to do what you love. There’s another hospital a mile and a half away from my house, whereas Jackson is 35 miles away and it takes me an hour in transit time each way. But I couldn’t do what I’m doing now at any other facility. I stay because I love what I’m doing.
Q: Why is the UM-JMH Center for Patient Safety so beneficial?
A: The greatest benefit is the ability to be exposed to and tackle real-life scenarios in a risk-free environment. We use life-size mannequins to re-create scenarios that medical personnel will see during their training. We try to re-create the chaos that will ensue.
Q: So it’s similar to a pilot using a flight simulator.
A: Exactly. When a plane crashes and the NTSB goes to see what happened, they perform what we in medicine call a root-cause analysis. They’re not blaming an individual; they want to see what they can change on a system level to prevent an error like that from happening again. We culminate our training with a debriefing that we approach the same way, so nobody walks away thinking they failed.
Q: How effective can simulation-based education be?
A: There will be some limitations because the technology simply cannot account for every aspect of a human. But there’s a wealth of data that supports this as a pretty good surrogate. The technology provides for an incredible amount of experience and exposure without any potential harm to a patient, and it provides [trainees] an opportunity to do things they otherwise would have to wait to do until a clinical scenario demanded it.
Q: Do you think this is the wave of the future?
A: Absolutely. And as the Accreditation Council for Graduate Medical Education promulgates new rules that limit the hours trainees can work, it’s going to be incumbent on training programs to be creative in providing equal or near-equal experience in a much shorter time. Simulation can help fill that bill.
Q: You created a crisis-management simulation course for IM residents. How did that come about?
A: When we have a crisis like a code blue, I witnessed the chaos that ensued. I thought some of the paltry resuscitation rate could be due to the fact there was no meaningful communication in that scenario.
Using full-scale mannequins, I put nurses and residents into those types of situations and videotaped what ensued. Frequently we saw the same chaos we see in reality, and many rather basic, commonsensical concepts went out the window.
Q: Can you offer an example?
A: A big one is situational awareness. If the head of the gurney is in a seated position, that’s not a conducive way to do chest compressions. If the side rails of the bed are up, you can lower them so you aren’t reaching over them. Was a team leader assigned or were roles delegated? These aren’t novel concepts, but when faced with a crisis, everybody tends to focus on their own thing. In a crisis, you need to break those silos down and operate as a team.
Q: How effective is the training?
A: After the first scenario, we show the video and debrief them for 10 or 15 minutes, keying in on some behaviors that can be employed in a crisis. Then we expose them to a different crisis scenario immediately thereafter. Often we see an immediate change in their behavior.
Q: You developed a curriculum through which residents are taught in a standardized manner how to perform invasive bedside procedures. How does it work?
A: They have 12 hours of hands-on instruction using fluid-filled, ultrasound-capable mannequins. A faculty attending teaches these procedures. We took it a step further and made a four-week rotation as a mandatory component of the residency program. They carry a beeper, and any service within the hospital can call the procedure team to do one of the procedures on which they were already trained.
Q: How successful is the effort?
A: This is the beginning of our fifth year, and we’ve been called more than 4,000 times to do procedures on hospitalized patients. We’ve published our curriculum. We’ve shown a significant improvement in knowledge, technical skills, and confidence level, and we have data we’re going to publish later this year that shows our complication rates are better than complication rates that are published elsewhere.
Q: What is your biggest professional reward?
A: The ability to impact the next generation. With the procedural training alone, we have just trained our 1,000th person. Each one of them is going to take care of thousands of patients in their professional careers. That’s an expansive influence.
Q: What is your biggest professional challenge?
A: The culture of medicine. It is infused with hundreds of years of tradition and, at times, it feels like trying to move a mountain. It may take a generation to do it, but there will come a time—at least within the field of patient safety—when more people are attuned to it and understand the concepts really are lifesaving. That doesn’t happen as fast as I would like it to, but if we keep plugging away one year at a time, we will be able to make an impact.
Mark Leiser is a freelance writer based in New Jersey.
When Joshua Lenchus, DO, RPh, FACP, FHM, discussed his love for chemistry with his high school guidance counselors, they told him it could take him in one of two directions: teaching or a career in pharmacy.
Intrigued by the latter option, he decided to become a volunteer in the pharmacy department at a local hospital. Soon after, he became a full-time pharmacy technician and eventually enrolled in pharmacy school at the University of Florida.
“I always knew I wanted to be a physician, and everybody needs a bachelor’s degree in something,” Dr. Lenchus says. “I thought, ‘What better way to do it than to get a bachelor’s degree in pharmacy and then move into medicine?’ ”
After college, he worked as a retail pharmacist, then moved to the institutional setting, creating the position of clinical pharmacist at Wellington (Fla.) Regional Medical Center. Three years later, he entered medical school and ultimately pursued a career as an academic hospitalist.
Dr. Lenchus now serves as associate professor of clinical medicine at the University of Miami’s (UM) Miller School of Medicine, associate program director of Jackson Memorial Hospital’s (JMH) internal medicine residency training program, and associate director of the UM-JMH Center for Patient Safety, which trains about 1,000 medical students, residents, and interns each year.
“Pharmacy has provided an invaluable background for becoming a physician,” says Dr. Lenchus, who was appointed a member of Team Hospitalist in May. “Many physicians order a medication and have no idea what the other half of the equation entails. My experience gave me a solid footing from which I could springboard.”
Q: You spend considerable time mentoring the next generation of physicians. What’s the best advice you can give them?
A: Physicians have these altruistic notions about wanting to help people, but you really have to do what you love. There’s another hospital a mile and a half away from my house, whereas Jackson is 35 miles away and it takes me an hour in transit time each way. But I couldn’t do what I’m doing now at any other facility. I stay because I love what I’m doing.
Q: Why is the UM-JMH Center for Patient Safety so beneficial?
A: The greatest benefit is the ability to be exposed to and tackle real-life scenarios in a risk-free environment. We use life-size mannequins to re-create scenarios that medical personnel will see during their training. We try to re-create the chaos that will ensue.
Q: So it’s similar to a pilot using a flight simulator.
A: Exactly. When a plane crashes and the NTSB goes to see what happened, they perform what we in medicine call a root-cause analysis. They’re not blaming an individual; they want to see what they can change on a system level to prevent an error like that from happening again. We culminate our training with a debriefing that we approach the same way, so nobody walks away thinking they failed.
Q: How effective can simulation-based education be?
A: There will be some limitations because the technology simply cannot account for every aspect of a human. But there’s a wealth of data that supports this as a pretty good surrogate. The technology provides for an incredible amount of experience and exposure without any potential harm to a patient, and it provides [trainees] an opportunity to do things they otherwise would have to wait to do until a clinical scenario demanded it.
Q: Do you think this is the wave of the future?
A: Absolutely. And as the Accreditation Council for Graduate Medical Education promulgates new rules that limit the hours trainees can work, it’s going to be incumbent on training programs to be creative in providing equal or near-equal experience in a much shorter time. Simulation can help fill that bill.
Q: You created a crisis-management simulation course for IM residents. How did that come about?
A: When we have a crisis like a code blue, I witnessed the chaos that ensued. I thought some of the paltry resuscitation rate could be due to the fact there was no meaningful communication in that scenario.
Using full-scale mannequins, I put nurses and residents into those types of situations and videotaped what ensued. Frequently we saw the same chaos we see in reality, and many rather basic, commonsensical concepts went out the window.
Q: Can you offer an example?
A: A big one is situational awareness. If the head of the gurney is in a seated position, that’s not a conducive way to do chest compressions. If the side rails of the bed are up, you can lower them so you aren’t reaching over them. Was a team leader assigned or were roles delegated? These aren’t novel concepts, but when faced with a crisis, everybody tends to focus on their own thing. In a crisis, you need to break those silos down and operate as a team.
Q: How effective is the training?
A: After the first scenario, we show the video and debrief them for 10 or 15 minutes, keying in on some behaviors that can be employed in a crisis. Then we expose them to a different crisis scenario immediately thereafter. Often we see an immediate change in their behavior.
Q: You developed a curriculum through which residents are taught in a standardized manner how to perform invasive bedside procedures. How does it work?
A: They have 12 hours of hands-on instruction using fluid-filled, ultrasound-capable mannequins. A faculty attending teaches these procedures. We took it a step further and made a four-week rotation as a mandatory component of the residency program. They carry a beeper, and any service within the hospital can call the procedure team to do one of the procedures on which they were already trained.
Q: How successful is the effort?
A: This is the beginning of our fifth year, and we’ve been called more than 4,000 times to do procedures on hospitalized patients. We’ve published our curriculum. We’ve shown a significant improvement in knowledge, technical skills, and confidence level, and we have data we’re going to publish later this year that shows our complication rates are better than complication rates that are published elsewhere.
Q: What is your biggest professional reward?
A: The ability to impact the next generation. With the procedural training alone, we have just trained our 1,000th person. Each one of them is going to take care of thousands of patients in their professional careers. That’s an expansive influence.
Q: What is your biggest professional challenge?
A: The culture of medicine. It is infused with hundreds of years of tradition and, at times, it feels like trying to move a mountain. It may take a generation to do it, but there will come a time—at least within the field of patient safety—when more people are attuned to it and understand the concepts really are lifesaving. That doesn’t happen as fast as I would like it to, but if we keep plugging away one year at a time, we will be able to make an impact.
Mark Leiser is a freelance writer based in New Jersey.
Study: Rural Hospitals Behind IT Curve
Only a sliver of rural hospitals would meet the Center for Medicare & Medicaid Services’ (CMS) criteria to qualify for “meaningful use” of health information technology (HIT), according to a new study, but that could be a window for HM group leaders to take the reins of technology projects.
“[Hospitalists] could be very useful as a champion,” says Brock Slabach, MPH, FACHE, senior vice president for member services at the National Rural Health Association.
The new report showed that 5% of rural hospitals could demonstrate meaningful use of an electronic health record (EHR) system, as opposed to 9% of urban hospitals (J Rural Health. 2011;27(3):329-337). The number dips to 3% for critical-access hospitals. EHR usage often is used as a benchmark for HIT implementation.
CMS has allotted $20 billion for physicians and hospitals to adopt new technologies, but entities must prove they have met “meaningful use” requirements.
Slabach, who spent 20 years as an administrator at Field Memorial Community Hospital in Centreville, Miss., says the major hurdle for HIT implementation at rural hospitals is a lack of knowledge. But if hospitalists can show other physicians the value of HIT, others will follow, he adds.
“Somebody who may not have any informatics background, but is willing to grab a hold of the system, learn its applications, develop methods to spread the knowledge to the rest of the medical staff, is critical,” Slabach says. “It just takes that one or two [people] to get the momentum starting, in terms of a transition to what for a lot of middle-aged and older physicians is a completely new world.”
Only a sliver of rural hospitals would meet the Center for Medicare & Medicaid Services’ (CMS) criteria to qualify for “meaningful use” of health information technology (HIT), according to a new study, but that could be a window for HM group leaders to take the reins of technology projects.
“[Hospitalists] could be very useful as a champion,” says Brock Slabach, MPH, FACHE, senior vice president for member services at the National Rural Health Association.
The new report showed that 5% of rural hospitals could demonstrate meaningful use of an electronic health record (EHR) system, as opposed to 9% of urban hospitals (J Rural Health. 2011;27(3):329-337). The number dips to 3% for critical-access hospitals. EHR usage often is used as a benchmark for HIT implementation.
CMS has allotted $20 billion for physicians and hospitals to adopt new technologies, but entities must prove they have met “meaningful use” requirements.
Slabach, who spent 20 years as an administrator at Field Memorial Community Hospital in Centreville, Miss., says the major hurdle for HIT implementation at rural hospitals is a lack of knowledge. But if hospitalists can show other physicians the value of HIT, others will follow, he adds.
“Somebody who may not have any informatics background, but is willing to grab a hold of the system, learn its applications, develop methods to spread the knowledge to the rest of the medical staff, is critical,” Slabach says. “It just takes that one or two [people] to get the momentum starting, in terms of a transition to what for a lot of middle-aged and older physicians is a completely new world.”
Only a sliver of rural hospitals would meet the Center for Medicare & Medicaid Services’ (CMS) criteria to qualify for “meaningful use” of health information technology (HIT), according to a new study, but that could be a window for HM group leaders to take the reins of technology projects.
“[Hospitalists] could be very useful as a champion,” says Brock Slabach, MPH, FACHE, senior vice president for member services at the National Rural Health Association.
The new report showed that 5% of rural hospitals could demonstrate meaningful use of an electronic health record (EHR) system, as opposed to 9% of urban hospitals (J Rural Health. 2011;27(3):329-337). The number dips to 3% for critical-access hospitals. EHR usage often is used as a benchmark for HIT implementation.
CMS has allotted $20 billion for physicians and hospitals to adopt new technologies, but entities must prove they have met “meaningful use” requirements.
Slabach, who spent 20 years as an administrator at Field Memorial Community Hospital in Centreville, Miss., says the major hurdle for HIT implementation at rural hospitals is a lack of knowledge. But if hospitalists can show other physicians the value of HIT, others will follow, he adds.
“Somebody who may not have any informatics background, but is willing to grab a hold of the system, learn its applications, develop methods to spread the knowledge to the rest of the medical staff, is critical,” Slabach says. “It just takes that one or two [people] to get the momentum starting, in terms of a transition to what for a lot of middle-aged and older physicians is a completely new world.”
HM@15 - Myriad Points of View
HM’s evolution the past 15 years has helped to reshape patient care in the hospital. Hospitalists near and far, young and old, are most proud of their work.
But how do others view hospitalists? What do nurses, pharmacists, and surgical specialists—professionals who work with hospitalists on a daily basis—say about hospitalists and their daily contributions to medicine and the U.S. healthcare system?
The Hospitalist talked with an array of medical professionals to develop a 360-degree sense of how HM is regarded in the medical community, speaking with sources affiliated with organizations as those sources are inclined to have a more panoramic understanding of how their field views hospitalists. The views presented are those of the individuals and do not necessarily represent the stances of their organizations.
Pharmacy
Stan Kent, president of the American Society of Health-System Pharmacists, says he always thought that the idea of having doctors who worked exclusively in the hospital would be good idea—even before there was such a thing as a hospitalist.
“I witnessed the movement of internists and surgeons transformed from being hospital-based to more office-based,” says Kent, who also is an assistant vice president at Northshore University Health System in Evanston, Ill., where he oversees pharmacy services. “I always wished that there could be more consistency on the part of those physicians in taking care of the patients in the hospital.”
Once hospitalists became a fixture in hospitals, their familiarity with the hospital and knowledge helped pharmacists do their jobs better, according to Kent. With hospitals becoming more and more complex, with electronic medical records and the handling of cases that are more and more difficult, doctors generally are less efficient if they’re not intimately involved in the system.
Kristi Killelea, an inpatient pharmacist at Northshore, says that it’s easier to develop working relationships with hospitalists whom you frequently see in the hospital.
“From the inpatient perspective, I think the nice part about hospitalists is they are more familiar with inpatient medicine, which typically involves more intravenous-type medications,” she says. “It just makes it easier to deal with them because they see that a little bit more frequently.”
There are times when the gap between inpatient care and outpatient care shows, she notes, but that is uncommon. “Sometimes, if you’re looking for historical knowledge about the patient, about why they are doing what they’re doing with the medication, [hospitalists] can’t always contribute that because they’re not following the patient in their office,” she says. “But I think that’s more rare than the norm.”
Even as medication reconciliation continues to be an issue throughout the healthcare landscape, Kent and Killelea agree it’s not due to hospitalists. “Sometimes patients tell their PCP that they’re taking Lipitor, for example, but they don’t give them the strength and they don’t tell them how many times they’re taking it. Those instances become more cumbersome from a medication reconciliation standpoint,” Kent says. “Whereas if this information is gathered by the hospitalist, they are more accurate and complete, I think, in getting that history, and then doing the reconciliation.”
Quality Control
To date, there is no definitive data to show what effect hospitalists have on the quality of care at hospitals, says Robert Wise, MD, medical advisor to the Joint Commission’s Division of Healthcare Quality Evaluation in Washington, D.C.
He says a hospitalist can’t be judged on his or her own but has to be seen in the context of the system in which he or she is working. Hospitalists have in-depth knowledge of the complex processes and technology special to hospital care, but their work is only part of the entire “episode of care” for a patient.
“While the physician in the hospital is highly trained to deal with the unique clinical needs of that patient, it is also important that the team treating the patient has all relevant information from all clinicians who may have treated the patient prior to the acute episode,” he says.
“It is also critical that when the patient is discharged that there is as seamless transition back to the system that will continue to care for that patient. Those handoffs may or may not be working well.”
The handoff, to and from the hospital, is one of the most risk-fraught areas for patients. So what is gained from the specialized skills of hospitalists might be lost if transitions from the hospital are not done well, Dr. Wise explains. “The hospitalist concept, while adding a new level of expertise, also increases the fragmentation of care and, therefore, can lead to some increased risk,” he says. “That risk is mitigated by well-functioning systems that can both initiate and accept the transfers.”
The use and mastery of the electronic medical record is crucial to the successful handoff, he adds.
“Another issue that is often discussed is whether, as the number of [hospital]-employed physicians increase, that will impact the medical staff’s freedom to constructively challenge hospital administration or the board concerning issues of quality and safety,” Dr. Wise says. “While this remains a theoretical issue, as the number of medical staff members employed by the hospital increase, [it is important] that their voices on the issues of quality and safety of medical care remain unimpeded.”
He also says that the speed of the growth of the hospitalist field comes with a certain amount of risk.
“The current hospitalist system attempts to assure that seriously ill patients are being treated by physicians who are current and competent in the complicated, high-tech environment of the 21st-century hospital,” he explains. “It will take time to develop a number of the supporting systems. If the speed of growth is very rapid, it is possible that the supporting systems, both inside and outside of the hospital, will not be able to keep up. None of these possible problems are insurmountable, but all will take a significant amount of attention and resources to support this method to deliver care.”
—Robert Wise, MD, medical advisor, Division of Healthcare Quality Evaluation, The Joint Commission, Washington, D.C.
Orthopedic Surgery
Older orthopedic patients are at serious risk after surgery, but their chances are improved by the work of hospitalists, says Alexandra Page, MD, a member of the American Academy of Orthopaedic Surgeons’ National Health Care Systems Committee and a surgeon with Kaiser Permanente in La Jolla, Calif., who works with geriatric patients.
A major role of hospitalists in support of orthopedic surgeons is to help patients be “as tuned up as they can be prior to surgery,” she says.
For octogenarians, there is a 25% mortality rate in the year after a hip fracture. For a nonagenarian, the one-year mortality rate is 50%.
“That’s a real high risk, and we don’t even in orthopedics have a good sense of what those factors are that make them so high-risk,” says Dr. Page, adding that it is known that optimal levels of glycemic control can minimize perioperative complications like infection.
That makes it all the more important for hospitalists to get patients into the best shape possible. After the operation, hospitalists help control blood pressure and blood sugar, and take steps to minimize post-operative delirium.
“It doesn’t affect our ability to perform the surgery at a technical level, but ultimately it gives our patients better outcomes,” Dr. Page says. “That’s really what it’s all about.”
Dr. Page’s role as an examiner for the orthopedic boards gives her insight into how different hospital systems work. She says she hopes there can be more consistency in the role that hospitalists have in helping with orthopedic surgery patients, with patients being routinely admitted through the hospitalist service. “I think there’s still a lot of variability, in terms of who’s managing these patients,” she says.
Continued below...

HM@15 - Patient-Care Partners
Relationships with other medical professionals are evolving, longtime hospitalist says
Twenty-five years ago, healthcare experts were forecasting something that Janet Nagamine, MD, RN, SFHM, thought was highly unlikely. “When I was an ICU nurse back in the 1980s, people projected that the hospital would become one big ICU,” recalls Dr. Nagamine, who has worked as a hospitalist since 1999 and worked in hospitals since 1986. “And at that time, I thought that was a crazy notion. How could the entire hospital be an intensive care [unit]?”
When she looks around now, she sees much more complex care being provided at hospitals—patients who would have died are in the ICU, those who would have been in the ICU are now on stepdown and telemetry units, and patients who would have been on the floors are being cared for at home.
“It really does look like an ICU,” says Dr. Nagamine, an SHM board member who works at Kaiser Permanente in Santa Clara, Calif.
That shift in acuity has helped carve a niche for hospitalist physicians—a role that has become more and more embraced by the array of medical professionals working in hospitals. With patients as sick as they are in hospitals, it’s much harder to manage their care from an office-based practice.
Dr. Nagamine says that at first there was some tension between PCPs and hospitalists, with PCPs wanting to continue seeing their hospitalized patients.
“Initially, that was a difficult challenge,” she says. Now, though, she says most of the tension has evaporated. “It’s really interesting how people respond to change,” she says. “In a relatively short time, it’s like that battle never happened.”
Hospitalists’ relationships with nurses, she says, were smooth from the beginning.
“It was almost an immediate partnership because, as a nurse who’s been at the bedside at 2 a.m. without an attending physician in-house, it was scary,” she says. “You have a partner in-house for the first time.”
Hospitalist comanaging of complex cases with specialists has evolved, too, but Dr. Nagamine says it remains an area in need of improvement, particularly on weekends and other off hours when a hospitalist might get “sideswiped” with patients.
“Just because we happen to be in the hospital does not mean that we should be the attending on certain types of patients,” she says. “We want to be nice. We want to help everybody. But sometimes we end up with patients that really aren’t appropriate for us to manage.”
Family Medicine
When one of his patients is admitted to the hospital and comes under the care of a hospitalist, his involvement doesn’t end, says Glen Stream, MD, president-elect of the American Academy of Family Physicians, who works with Rockwood Clinic in Spokane, Wash.
Dr. Stream continues to keep in touch with patients, and that has made for a good working relationship with hospitalists. It helps put patients at ease and helps with handoffs to and from the hospital, he says. “I don’t think you can overcommunicate in either direction,” he says. “The most complete medical information enables the best-informed decision-making for treatment decisions.” Such levels of involvement usually are welcomed by hospitalists, he says, adding “I’ve been able to be the hospital physician’s advocate.”
Meanwhile, HM has made his office-based practice more flexible and more accessible. “In my medical group, a number of my partners actually start seeing patients [in the office] as early as seven in the morning,” Dr. Stream says. “They can commit to being there for patients at that early hour.”
He points out that handoffs to and from primary-care doctors and hospitalists has improved, but it’s still a work in progress. “I think it’s gotten better over time,” he says. “I think there’s recognition—on both sides of those handoffs—that things could be improved. I think the commitment is there both for the ambulatory physicians, the primary-care doctor, the family doctor, and the hospitalist taking care of them.”
Although hospitalists generally are better compensation than family doctors, Dr. Stream says he isn’t aware of “any friction” from family physicians. “Our academy, our members, family physicians, believe that the work that [we] do is undervalued in our current healthcare system. But that doesn’t mean that we have to compare ourselves to hospitalists,” he says.
Nursing
Even as fragmentation of medical care has increased, the emergence of the hospitalist has helped to streamline care, says Joanne Disch, PhD, RN, president-elect of the American Academy of Nursing and clinical professor at the University of Minnesota School of Nursing in Minneapolis.
“There has become such increasing fragmentation of who is the team around the patient,” she says. But, she notes, “the hospitalist really provided a mechanism to promote continuity of care.”
Nurses, she says, have found hospitalists to be “somebody who can cover your back.” “When the system works right, the nurses do not have to seek out a physician and hope that they can either grab somebody or somebody makes rounds,” Disch says, noting a general frustration amongst her peers as to a lack of clarity in regard to who’s in charge. “What hospitalists inherently do, structurally, is provide a main physician who will be the accountable one in the hospital setting. You have a named person that the nurse knows, ‘Ah, this is who I need to go to.’ ”
Although most nurses welcomed hospitalists from the very beginning, she continues, the addition of MDs into the hospital setting did cause confusion, most notably over the roles of PCPs, referring physicians, and hospitalists.
“It wasn’t clear the extent of this individual’s responsibility and how to use them effectively, but over time my sense is that people … really find this helpful,” she says.
An area that might have room for improvement is hospitalist-nurse communication, with more “huddling” and discussions at shift change. Better communication with patients’ families also could be improved, she says. “[It] gets a little confusing sometimes,” she says. “Either everybody, or nobody, is talking with the patient and the family.”
Hospital Administration
The reaction of Craig Becker, a member of the American Hospital Association board and president of the Tennessee Hospital Association, was, at first, fairly dismissive. An idea being discussed in the industry—inpatient physicians working full-time in hospitals—would not be worth it, he thought. He couldn’t get past the notion that such an arrangement would be “a waste of money,” and that if someone tried it, it would just be in the clinical-care units.
Once a couple of hospitals started hospitalist services, he was more inclined to listen. “I was getting feedback from them, and they were saying: ‘Boy, this has made a big difference, both in patient care and financially,’ ” Becker explains. Once he noticed HM programs popping up in small, rural hospitals, Becker knew “this was a movement whose time had come.”
In Tennessee, where hospitalists were almost unheard of a decade ago, hospitalists now work in every shape and size of hospital, some with fewer than 100 beds. At one hospital that employs its own hospitalist, there are just 58 beds and an attached nursing home, Becker says.
Showing that hospitalists have been worth the cost is really as simple as looking at the length of stay, he says. “If you can knock six-tenths of a day off a stay, that’s pretty significant savings,” Becker says.
Becker notes other positives the HM model has brought to Tennessee hospitals: They make the jobs of hospital administrators easier because specialists and referring physicians are happier.
“They can spend more time doing whatever they want to do on a personal basis or in their offices,” he says. “So I think just in terms of improving relationships with the medical staffs, hospitalists have been a real plus.”
Tom Collins is a freelance writer based in Florida.

—Janet Nagamine, MD, RN, SFHM, Kaiser Permanente Medical Center, Santa Clara, Calif., SHM board member
HM@15 - Patients Benefit from Honed Relations Between Hospitalists, Staff
As the working relationships between hospitalists and other medical professionals have been refined through the years, so has the experience of the patients under their care, those working in the hospital say.
Glen Stream, MD, president-elect of the American Academy of Family Physicians who works with Rockwood Clinic in Spokane, Wash., says he likes to help bridge the gap between the patient and a hospitalist whom the patient probably never met before.
For example, he tells patients, “ ‘Oh, Dr. Jones is the hospitalist looking after you. He’s a really excellent physician and I agree with the things that they’re doing. I look forward to seeing you when you come home,’ ” Dr. Stream explains. “And, because I stop by, I’m going to be familiar with what was going on and what the issues were and what the follow-up should be. I think that that helps me, as their doctor, but also think that it’s a positive thing for the patient.”
Alexandra Page, MD, a member of the American Academy of Orthopaedic Surgeons’ National Health Care Systems Committee and a surgeon with Kaiser Permanente in La Jolla, Calif., says that while no hard data is available, she thinks hospitalist involvement in orthopedic procedures improves patient care.
She says her “gut feeling” is that the mortality rate would tend to fall where hospitalists are more involved. But she also says that there might be room for hospitalists to become more involved in those procedures, to become familiar with the patient at an earlier stage.
“Would it make sense for a hospitalist … since a hospitalist team would be managing them post-operatively, to consider seeing them pre-operatively? That would be the other area where I think there may be potential growth,” she says.
Dr. Nagamine says more effort is being put into familiarizing patients with hospitalists.
“I think that patients more and more understand our role,” she says. “Part of it is the communication. When the primary-care physician or whoever refers them to the hospital, it’s nice that they explain that someone else will be managing their care. When they arrive at the hospital, we explain our role. In order to gain a patient’s trust, you have to show that you know something about them, you’ve read the chart, you’ve talked to Dr. Smith.”
That transition is something that has received more attention over time, she says, with doctors increasingly providing patients business cards with photos so that they can keep track of who’s who.
“It’s something we could still work on,” Dr. Nagamine says. “But we’re very focused on patient satisfaction and communication. There’s a lot of work going on in that regard.”
HM’s evolution the past 15 years has helped to reshape patient care in the hospital. Hospitalists near and far, young and old, are most proud of their work.
But how do others view hospitalists? What do nurses, pharmacists, and surgical specialists—professionals who work with hospitalists on a daily basis—say about hospitalists and their daily contributions to medicine and the U.S. healthcare system?
The Hospitalist talked with an array of medical professionals to develop a 360-degree sense of how HM is regarded in the medical community, speaking with sources affiliated with organizations as those sources are inclined to have a more panoramic understanding of how their field views hospitalists. The views presented are those of the individuals and do not necessarily represent the stances of their organizations.
Pharmacy
Stan Kent, president of the American Society of Health-System Pharmacists, says he always thought that the idea of having doctors who worked exclusively in the hospital would be good idea—even before there was such a thing as a hospitalist.
“I witnessed the movement of internists and surgeons transformed from being hospital-based to more office-based,” says Kent, who also is an assistant vice president at Northshore University Health System in Evanston, Ill., where he oversees pharmacy services. “I always wished that there could be more consistency on the part of those physicians in taking care of the patients in the hospital.”
Once hospitalists became a fixture in hospitals, their familiarity with the hospital and knowledge helped pharmacists do their jobs better, according to Kent. With hospitals becoming more and more complex, with electronic medical records and the handling of cases that are more and more difficult, doctors generally are less efficient if they’re not intimately involved in the system.
Kristi Killelea, an inpatient pharmacist at Northshore, says that it’s easier to develop working relationships with hospitalists whom you frequently see in the hospital.
“From the inpatient perspective, I think the nice part about hospitalists is they are more familiar with inpatient medicine, which typically involves more intravenous-type medications,” she says. “It just makes it easier to deal with them because they see that a little bit more frequently.”
There are times when the gap between inpatient care and outpatient care shows, she notes, but that is uncommon. “Sometimes, if you’re looking for historical knowledge about the patient, about why they are doing what they’re doing with the medication, [hospitalists] can’t always contribute that because they’re not following the patient in their office,” she says. “But I think that’s more rare than the norm.”
Even as medication reconciliation continues to be an issue throughout the healthcare landscape, Kent and Killelea agree it’s not due to hospitalists. “Sometimes patients tell their PCP that they’re taking Lipitor, for example, but they don’t give them the strength and they don’t tell them how many times they’re taking it. Those instances become more cumbersome from a medication reconciliation standpoint,” Kent says. “Whereas if this information is gathered by the hospitalist, they are more accurate and complete, I think, in getting that history, and then doing the reconciliation.”
Quality Control
To date, there is no definitive data to show what effect hospitalists have on the quality of care at hospitals, says Robert Wise, MD, medical advisor to the Joint Commission’s Division of Healthcare Quality Evaluation in Washington, D.C.
He says a hospitalist can’t be judged on his or her own but has to be seen in the context of the system in which he or she is working. Hospitalists have in-depth knowledge of the complex processes and technology special to hospital care, but their work is only part of the entire “episode of care” for a patient.
“While the physician in the hospital is highly trained to deal with the unique clinical needs of that patient, it is also important that the team treating the patient has all relevant information from all clinicians who may have treated the patient prior to the acute episode,” he says.
“It is also critical that when the patient is discharged that there is as seamless transition back to the system that will continue to care for that patient. Those handoffs may or may not be working well.”
The handoff, to and from the hospital, is one of the most risk-fraught areas for patients. So what is gained from the specialized skills of hospitalists might be lost if transitions from the hospital are not done well, Dr. Wise explains. “The hospitalist concept, while adding a new level of expertise, also increases the fragmentation of care and, therefore, can lead to some increased risk,” he says. “That risk is mitigated by well-functioning systems that can both initiate and accept the transfers.”
The use and mastery of the electronic medical record is crucial to the successful handoff, he adds.
“Another issue that is often discussed is whether, as the number of [hospital]-employed physicians increase, that will impact the medical staff’s freedom to constructively challenge hospital administration or the board concerning issues of quality and safety,” Dr. Wise says. “While this remains a theoretical issue, as the number of medical staff members employed by the hospital increase, [it is important] that their voices on the issues of quality and safety of medical care remain unimpeded.”
He also says that the speed of the growth of the hospitalist field comes with a certain amount of risk.
“The current hospitalist system attempts to assure that seriously ill patients are being treated by physicians who are current and competent in the complicated, high-tech environment of the 21st-century hospital,” he explains. “It will take time to develop a number of the supporting systems. If the speed of growth is very rapid, it is possible that the supporting systems, both inside and outside of the hospital, will not be able to keep up. None of these possible problems are insurmountable, but all will take a significant amount of attention and resources to support this method to deliver care.”
—Robert Wise, MD, medical advisor, Division of Healthcare Quality Evaluation, The Joint Commission, Washington, D.C.
Orthopedic Surgery
Older orthopedic patients are at serious risk after surgery, but their chances are improved by the work of hospitalists, says Alexandra Page, MD, a member of the American Academy of Orthopaedic Surgeons’ National Health Care Systems Committee and a surgeon with Kaiser Permanente in La Jolla, Calif., who works with geriatric patients.
A major role of hospitalists in support of orthopedic surgeons is to help patients be “as tuned up as they can be prior to surgery,” she says.
For octogenarians, there is a 25% mortality rate in the year after a hip fracture. For a nonagenarian, the one-year mortality rate is 50%.
“That’s a real high risk, and we don’t even in orthopedics have a good sense of what those factors are that make them so high-risk,” says Dr. Page, adding that it is known that optimal levels of glycemic control can minimize perioperative complications like infection.
That makes it all the more important for hospitalists to get patients into the best shape possible. After the operation, hospitalists help control blood pressure and blood sugar, and take steps to minimize post-operative delirium.
“It doesn’t affect our ability to perform the surgery at a technical level, but ultimately it gives our patients better outcomes,” Dr. Page says. “That’s really what it’s all about.”
Dr. Page’s role as an examiner for the orthopedic boards gives her insight into how different hospital systems work. She says she hopes there can be more consistency in the role that hospitalists have in helping with orthopedic surgery patients, with patients being routinely admitted through the hospitalist service. “I think there’s still a lot of variability, in terms of who’s managing these patients,” she says.
Continued below...
HM@15 - Patient-Care Partners
Relationships with other medical professionals are evolving, longtime hospitalist says
Twenty-five years ago, healthcare experts were forecasting something that Janet Nagamine, MD, RN, SFHM, thought was highly unlikely. “When I was an ICU nurse back in the 1980s, people projected that the hospital would become one big ICU,” recalls Dr. Nagamine, who has worked as a hospitalist since 1999 and worked in hospitals since 1986. “And at that time, I thought that was a crazy notion. How could the entire hospital be an intensive care [unit]?”
When she looks around now, she sees much more complex care being provided at hospitals—patients who would have died are in the ICU, those who would have been in the ICU are now on stepdown and telemetry units, and patients who would have been on the floors are being cared for at home.
“It really does look like an ICU,” says Dr. Nagamine, an SHM board member who works at Kaiser Permanente in Santa Clara, Calif.
That shift in acuity has helped carve a niche for hospitalist physicians—a role that has become more and more embraced by the array of medical professionals working in hospitals. With patients as sick as they are in hospitals, it’s much harder to manage their care from an office-based practice.
Dr. Nagamine says that at first there was some tension between PCPs and hospitalists, with PCPs wanting to continue seeing their hospitalized patients.
“Initially, that was a difficult challenge,” she says. Now, though, she says most of the tension has evaporated. “It’s really interesting how people respond to change,” she says. “In a relatively short time, it’s like that battle never happened.”
Hospitalists’ relationships with nurses, she says, were smooth from the beginning.
“It was almost an immediate partnership because, as a nurse who’s been at the bedside at 2 a.m. without an attending physician in-house, it was scary,” she says. “You have a partner in-house for the first time.”
Hospitalist comanaging of complex cases with specialists has evolved, too, but Dr. Nagamine says it remains an area in need of improvement, particularly on weekends and other off hours when a hospitalist might get “sideswiped” with patients.
“Just because we happen to be in the hospital does not mean that we should be the attending on certain types of patients,” she says. “We want to be nice. We want to help everybody. But sometimes we end up with patients that really aren’t appropriate for us to manage.”
Family Medicine
When one of his patients is admitted to the hospital and comes under the care of a hospitalist, his involvement doesn’t end, says Glen Stream, MD, president-elect of the American Academy of Family Physicians, who works with Rockwood Clinic in Spokane, Wash.
Dr. Stream continues to keep in touch with patients, and that has made for a good working relationship with hospitalists. It helps put patients at ease and helps with handoffs to and from the hospital, he says. “I don’t think you can overcommunicate in either direction,” he says. “The most complete medical information enables the best-informed decision-making for treatment decisions.” Such levels of involvement usually are welcomed by hospitalists, he says, adding “I’ve been able to be the hospital physician’s advocate.”
Meanwhile, HM has made his office-based practice more flexible and more accessible. “In my medical group, a number of my partners actually start seeing patients [in the office] as early as seven in the morning,” Dr. Stream says. “They can commit to being there for patients at that early hour.”
He points out that handoffs to and from primary-care doctors and hospitalists has improved, but it’s still a work in progress. “I think it’s gotten better over time,” he says. “I think there’s recognition—on both sides of those handoffs—that things could be improved. I think the commitment is there both for the ambulatory physicians, the primary-care doctor, the family doctor, and the hospitalist taking care of them.”
Although hospitalists generally are better compensation than family doctors, Dr. Stream says he isn’t aware of “any friction” from family physicians. “Our academy, our members, family physicians, believe that the work that [we] do is undervalued in our current healthcare system. But that doesn’t mean that we have to compare ourselves to hospitalists,” he says.
Nursing
Even as fragmentation of medical care has increased, the emergence of the hospitalist has helped to streamline care, says Joanne Disch, PhD, RN, president-elect of the American Academy of Nursing and clinical professor at the University of Minnesota School of Nursing in Minneapolis.
“There has become such increasing fragmentation of who is the team around the patient,” she says. But, she notes, “the hospitalist really provided a mechanism to promote continuity of care.”
Nurses, she says, have found hospitalists to be “somebody who can cover your back.” “When the system works right, the nurses do not have to seek out a physician and hope that they can either grab somebody or somebody makes rounds,” Disch says, noting a general frustration amongst her peers as to a lack of clarity in regard to who’s in charge. “What hospitalists inherently do, structurally, is provide a main physician who will be the accountable one in the hospital setting. You have a named person that the nurse knows, ‘Ah, this is who I need to go to.’ ”
Although most nurses welcomed hospitalists from the very beginning, she continues, the addition of MDs into the hospital setting did cause confusion, most notably over the roles of PCPs, referring physicians, and hospitalists.
“It wasn’t clear the extent of this individual’s responsibility and how to use them effectively, but over time my sense is that people … really find this helpful,” she says.
An area that might have room for improvement is hospitalist-nurse communication, with more “huddling” and discussions at shift change. Better communication with patients’ families also could be improved, she says. “[It] gets a little confusing sometimes,” she says. “Either everybody, or nobody, is talking with the patient and the family.”
Hospital Administration
The reaction of Craig Becker, a member of the American Hospital Association board and president of the Tennessee Hospital Association, was, at first, fairly dismissive. An idea being discussed in the industry—inpatient physicians working full-time in hospitals—would not be worth it, he thought. He couldn’t get past the notion that such an arrangement would be “a waste of money,” and that if someone tried it, it would just be in the clinical-care units.
Once a couple of hospitals started hospitalist services, he was more inclined to listen. “I was getting feedback from them, and they were saying: ‘Boy, this has made a big difference, both in patient care and financially,’ ” Becker explains. Once he noticed HM programs popping up in small, rural hospitals, Becker knew “this was a movement whose time had come.”
In Tennessee, where hospitalists were almost unheard of a decade ago, hospitalists now work in every shape and size of hospital, some with fewer than 100 beds. At one hospital that employs its own hospitalist, there are just 58 beds and an attached nursing home, Becker says.
Showing that hospitalists have been worth the cost is really as simple as looking at the length of stay, he says. “If you can knock six-tenths of a day off a stay, that’s pretty significant savings,” Becker says.
Becker notes other positives the HM model has brought to Tennessee hospitals: They make the jobs of hospital administrators easier because specialists and referring physicians are happier.
“They can spend more time doing whatever they want to do on a personal basis or in their offices,” he says. “So I think just in terms of improving relationships with the medical staffs, hospitalists have been a real plus.”
Tom Collins is a freelance writer based in Florida.
—Janet Nagamine, MD, RN, SFHM, Kaiser Permanente Medical Center, Santa Clara, Calif., SHM board member
HM@15 - Patients Benefit from Honed Relations Between Hospitalists, Staff
As the working relationships between hospitalists and other medical professionals have been refined through the years, so has the experience of the patients under their care, those working in the hospital say.
Glen Stream, MD, president-elect of the American Academy of Family Physicians who works with Rockwood Clinic in Spokane, Wash., says he likes to help bridge the gap between the patient and a hospitalist whom the patient probably never met before.
For example, he tells patients, “ ‘Oh, Dr. Jones is the hospitalist looking after you. He’s a really excellent physician and I agree with the things that they’re doing. I look forward to seeing you when you come home,’ ” Dr. Stream explains. “And, because I stop by, I’m going to be familiar with what was going on and what the issues were and what the follow-up should be. I think that that helps me, as their doctor, but also think that it’s a positive thing for the patient.”
Alexandra Page, MD, a member of the American Academy of Orthopaedic Surgeons’ National Health Care Systems Committee and a surgeon with Kaiser Permanente in La Jolla, Calif., says that while no hard data is available, she thinks hospitalist involvement in orthopedic procedures improves patient care.
She says her “gut feeling” is that the mortality rate would tend to fall where hospitalists are more involved. But she also says that there might be room for hospitalists to become more involved in those procedures, to become familiar with the patient at an earlier stage.
“Would it make sense for a hospitalist … since a hospitalist team would be managing them post-operatively, to consider seeing them pre-operatively? That would be the other area where I think there may be potential growth,” she says.
Dr. Nagamine says more effort is being put into familiarizing patients with hospitalists.
“I think that patients more and more understand our role,” she says. “Part of it is the communication. When the primary-care physician or whoever refers them to the hospital, it’s nice that they explain that someone else will be managing their care. When they arrive at the hospital, we explain our role. In order to gain a patient’s trust, you have to show that you know something about them, you’ve read the chart, you’ve talked to Dr. Smith.”
That transition is something that has received more attention over time, she says, with doctors increasingly providing patients business cards with photos so that they can keep track of who’s who.
“It’s something we could still work on,” Dr. Nagamine says. “But we’re very focused on patient satisfaction and communication. There’s a lot of work going on in that regard.”
HM’s evolution the past 15 years has helped to reshape patient care in the hospital. Hospitalists near and far, young and old, are most proud of their work.
But how do others view hospitalists? What do nurses, pharmacists, and surgical specialists—professionals who work with hospitalists on a daily basis—say about hospitalists and their daily contributions to medicine and the U.S. healthcare system?
The Hospitalist talked with an array of medical professionals to develop a 360-degree sense of how HM is regarded in the medical community, speaking with sources affiliated with organizations as those sources are inclined to have a more panoramic understanding of how their field views hospitalists. The views presented are those of the individuals and do not necessarily represent the stances of their organizations.
Pharmacy
Stan Kent, president of the American Society of Health-System Pharmacists, says he always thought that the idea of having doctors who worked exclusively in the hospital would be good idea—even before there was such a thing as a hospitalist.
“I witnessed the movement of internists and surgeons transformed from being hospital-based to more office-based,” says Kent, who also is an assistant vice president at Northshore University Health System in Evanston, Ill., where he oversees pharmacy services. “I always wished that there could be more consistency on the part of those physicians in taking care of the patients in the hospital.”
Once hospitalists became a fixture in hospitals, their familiarity with the hospital and knowledge helped pharmacists do their jobs better, according to Kent. With hospitals becoming more and more complex, with electronic medical records and the handling of cases that are more and more difficult, doctors generally are less efficient if they’re not intimately involved in the system.
Kristi Killelea, an inpatient pharmacist at Northshore, says that it’s easier to develop working relationships with hospitalists whom you frequently see in the hospital.
“From the inpatient perspective, I think the nice part about hospitalists is they are more familiar with inpatient medicine, which typically involves more intravenous-type medications,” she says. “It just makes it easier to deal with them because they see that a little bit more frequently.”
There are times when the gap between inpatient care and outpatient care shows, she notes, but that is uncommon. “Sometimes, if you’re looking for historical knowledge about the patient, about why they are doing what they’re doing with the medication, [hospitalists] can’t always contribute that because they’re not following the patient in their office,” she says. “But I think that’s more rare than the norm.”
Even as medication reconciliation continues to be an issue throughout the healthcare landscape, Kent and Killelea agree it’s not due to hospitalists. “Sometimes patients tell their PCP that they’re taking Lipitor, for example, but they don’t give them the strength and they don’t tell them how many times they’re taking it. Those instances become more cumbersome from a medication reconciliation standpoint,” Kent says. “Whereas if this information is gathered by the hospitalist, they are more accurate and complete, I think, in getting that history, and then doing the reconciliation.”
Quality Control
To date, there is no definitive data to show what effect hospitalists have on the quality of care at hospitals, says Robert Wise, MD, medical advisor to the Joint Commission’s Division of Healthcare Quality Evaluation in Washington, D.C.
He says a hospitalist can’t be judged on his or her own but has to be seen in the context of the system in which he or she is working. Hospitalists have in-depth knowledge of the complex processes and technology special to hospital care, but their work is only part of the entire “episode of care” for a patient.
“While the physician in the hospital is highly trained to deal with the unique clinical needs of that patient, it is also important that the team treating the patient has all relevant information from all clinicians who may have treated the patient prior to the acute episode,” he says.
“It is also critical that when the patient is discharged that there is as seamless transition back to the system that will continue to care for that patient. Those handoffs may or may not be working well.”
The handoff, to and from the hospital, is one of the most risk-fraught areas for patients. So what is gained from the specialized skills of hospitalists might be lost if transitions from the hospital are not done well, Dr. Wise explains. “The hospitalist concept, while adding a new level of expertise, also increases the fragmentation of care and, therefore, can lead to some increased risk,” he says. “That risk is mitigated by well-functioning systems that can both initiate and accept the transfers.”
The use and mastery of the electronic medical record is crucial to the successful handoff, he adds.
“Another issue that is often discussed is whether, as the number of [hospital]-employed physicians increase, that will impact the medical staff’s freedom to constructively challenge hospital administration or the board concerning issues of quality and safety,” Dr. Wise says. “While this remains a theoretical issue, as the number of medical staff members employed by the hospital increase, [it is important] that their voices on the issues of quality and safety of medical care remain unimpeded.”
He also says that the speed of the growth of the hospitalist field comes with a certain amount of risk.
“The current hospitalist system attempts to assure that seriously ill patients are being treated by physicians who are current and competent in the complicated, high-tech environment of the 21st-century hospital,” he explains. “It will take time to develop a number of the supporting systems. If the speed of growth is very rapid, it is possible that the supporting systems, both inside and outside of the hospital, will not be able to keep up. None of these possible problems are insurmountable, but all will take a significant amount of attention and resources to support this method to deliver care.”
—Robert Wise, MD, medical advisor, Division of Healthcare Quality Evaluation, The Joint Commission, Washington, D.C.
Orthopedic Surgery
Older orthopedic patients are at serious risk after surgery, but their chances are improved by the work of hospitalists, says Alexandra Page, MD, a member of the American Academy of Orthopaedic Surgeons’ National Health Care Systems Committee and a surgeon with Kaiser Permanente in La Jolla, Calif., who works with geriatric patients.
A major role of hospitalists in support of orthopedic surgeons is to help patients be “as tuned up as they can be prior to surgery,” she says.
For octogenarians, there is a 25% mortality rate in the year after a hip fracture. For a nonagenarian, the one-year mortality rate is 50%.
“That’s a real high risk, and we don’t even in orthopedics have a good sense of what those factors are that make them so high-risk,” says Dr. Page, adding that it is known that optimal levels of glycemic control can minimize perioperative complications like infection.
That makes it all the more important for hospitalists to get patients into the best shape possible. After the operation, hospitalists help control blood pressure and blood sugar, and take steps to minimize post-operative delirium.
“It doesn’t affect our ability to perform the surgery at a technical level, but ultimately it gives our patients better outcomes,” Dr. Page says. “That’s really what it’s all about.”
Dr. Page’s role as an examiner for the orthopedic boards gives her insight into how different hospital systems work. She says she hopes there can be more consistency in the role that hospitalists have in helping with orthopedic surgery patients, with patients being routinely admitted through the hospitalist service. “I think there’s still a lot of variability, in terms of who’s managing these patients,” she says.
Continued below...
HM@15 - Patient-Care Partners
Relationships with other medical professionals are evolving, longtime hospitalist says
Twenty-five years ago, healthcare experts were forecasting something that Janet Nagamine, MD, RN, SFHM, thought was highly unlikely. “When I was an ICU nurse back in the 1980s, people projected that the hospital would become one big ICU,” recalls Dr. Nagamine, who has worked as a hospitalist since 1999 and worked in hospitals since 1986. “And at that time, I thought that was a crazy notion. How could the entire hospital be an intensive care [unit]?”
When she looks around now, she sees much more complex care being provided at hospitals—patients who would have died are in the ICU, those who would have been in the ICU are now on stepdown and telemetry units, and patients who would have been on the floors are being cared for at home.
“It really does look like an ICU,” says Dr. Nagamine, an SHM board member who works at Kaiser Permanente in Santa Clara, Calif.
That shift in acuity has helped carve a niche for hospitalist physicians—a role that has become more and more embraced by the array of medical professionals working in hospitals. With patients as sick as they are in hospitals, it’s much harder to manage their care from an office-based practice.
Dr. Nagamine says that at first there was some tension between PCPs and hospitalists, with PCPs wanting to continue seeing their hospitalized patients.
“Initially, that was a difficult challenge,” she says. Now, though, she says most of the tension has evaporated. “It’s really interesting how people respond to change,” she says. “In a relatively short time, it’s like that battle never happened.”
Hospitalists’ relationships with nurses, she says, were smooth from the beginning.
“It was almost an immediate partnership because, as a nurse who’s been at the bedside at 2 a.m. without an attending physician in-house, it was scary,” she says. “You have a partner in-house for the first time.”
Hospitalist comanaging of complex cases with specialists has evolved, too, but Dr. Nagamine says it remains an area in need of improvement, particularly on weekends and other off hours when a hospitalist might get “sideswiped” with patients.
“Just because we happen to be in the hospital does not mean that we should be the attending on certain types of patients,” she says. “We want to be nice. We want to help everybody. But sometimes we end up with patients that really aren’t appropriate for us to manage.”
Family Medicine
When one of his patients is admitted to the hospital and comes under the care of a hospitalist, his involvement doesn’t end, says Glen Stream, MD, president-elect of the American Academy of Family Physicians, who works with Rockwood Clinic in Spokane, Wash.
Dr. Stream continues to keep in touch with patients, and that has made for a good working relationship with hospitalists. It helps put patients at ease and helps with handoffs to and from the hospital, he says. “I don’t think you can overcommunicate in either direction,” he says. “The most complete medical information enables the best-informed decision-making for treatment decisions.” Such levels of involvement usually are welcomed by hospitalists, he says, adding “I’ve been able to be the hospital physician’s advocate.”
Meanwhile, HM has made his office-based practice more flexible and more accessible. “In my medical group, a number of my partners actually start seeing patients [in the office] as early as seven in the morning,” Dr. Stream says. “They can commit to being there for patients at that early hour.”
He points out that handoffs to and from primary-care doctors and hospitalists has improved, but it’s still a work in progress. “I think it’s gotten better over time,” he says. “I think there’s recognition—on both sides of those handoffs—that things could be improved. I think the commitment is there both for the ambulatory physicians, the primary-care doctor, the family doctor, and the hospitalist taking care of them.”
Although hospitalists generally are better compensation than family doctors, Dr. Stream says he isn’t aware of “any friction” from family physicians. “Our academy, our members, family physicians, believe that the work that [we] do is undervalued in our current healthcare system. But that doesn’t mean that we have to compare ourselves to hospitalists,” he says.
Nursing
Even as fragmentation of medical care has increased, the emergence of the hospitalist has helped to streamline care, says Joanne Disch, PhD, RN, president-elect of the American Academy of Nursing and clinical professor at the University of Minnesota School of Nursing in Minneapolis.
“There has become such increasing fragmentation of who is the team around the patient,” she says. But, she notes, “the hospitalist really provided a mechanism to promote continuity of care.”
Nurses, she says, have found hospitalists to be “somebody who can cover your back.” “When the system works right, the nurses do not have to seek out a physician and hope that they can either grab somebody or somebody makes rounds,” Disch says, noting a general frustration amongst her peers as to a lack of clarity in regard to who’s in charge. “What hospitalists inherently do, structurally, is provide a main physician who will be the accountable one in the hospital setting. You have a named person that the nurse knows, ‘Ah, this is who I need to go to.’ ”
Although most nurses welcomed hospitalists from the very beginning, she continues, the addition of MDs into the hospital setting did cause confusion, most notably over the roles of PCPs, referring physicians, and hospitalists.
“It wasn’t clear the extent of this individual’s responsibility and how to use them effectively, but over time my sense is that people … really find this helpful,” she says.
An area that might have room for improvement is hospitalist-nurse communication, with more “huddling” and discussions at shift change. Better communication with patients’ families also could be improved, she says. “[It] gets a little confusing sometimes,” she says. “Either everybody, or nobody, is talking with the patient and the family.”
Hospital Administration
The reaction of Craig Becker, a member of the American Hospital Association board and president of the Tennessee Hospital Association, was, at first, fairly dismissive. An idea being discussed in the industry—inpatient physicians working full-time in hospitals—would not be worth it, he thought. He couldn’t get past the notion that such an arrangement would be “a waste of money,” and that if someone tried it, it would just be in the clinical-care units.
Once a couple of hospitals started hospitalist services, he was more inclined to listen. “I was getting feedback from them, and they were saying: ‘Boy, this has made a big difference, both in patient care and financially,’ ” Becker explains. Once he noticed HM programs popping up in small, rural hospitals, Becker knew “this was a movement whose time had come.”
In Tennessee, where hospitalists were almost unheard of a decade ago, hospitalists now work in every shape and size of hospital, some with fewer than 100 beds. At one hospital that employs its own hospitalist, there are just 58 beds and an attached nursing home, Becker says.
Showing that hospitalists have been worth the cost is really as simple as looking at the length of stay, he says. “If you can knock six-tenths of a day off a stay, that’s pretty significant savings,” Becker says.
Becker notes other positives the HM model has brought to Tennessee hospitals: They make the jobs of hospital administrators easier because specialists and referring physicians are happier.
“They can spend more time doing whatever they want to do on a personal basis or in their offices,” he says. “So I think just in terms of improving relationships with the medical staffs, hospitalists have been a real plus.”
Tom Collins is a freelance writer based in Florida.
—Janet Nagamine, MD, RN, SFHM, Kaiser Permanente Medical Center, Santa Clara, Calif., SHM board member
HM@15 - Patients Benefit from Honed Relations Between Hospitalists, Staff
As the working relationships between hospitalists and other medical professionals have been refined through the years, so has the experience of the patients under their care, those working in the hospital say.
Glen Stream, MD, president-elect of the American Academy of Family Physicians who works with Rockwood Clinic in Spokane, Wash., says he likes to help bridge the gap between the patient and a hospitalist whom the patient probably never met before.
For example, he tells patients, “ ‘Oh, Dr. Jones is the hospitalist looking after you. He’s a really excellent physician and I agree with the things that they’re doing. I look forward to seeing you when you come home,’ ” Dr. Stream explains. “And, because I stop by, I’m going to be familiar with what was going on and what the issues were and what the follow-up should be. I think that that helps me, as their doctor, but also think that it’s a positive thing for the patient.”
Alexandra Page, MD, a member of the American Academy of Orthopaedic Surgeons’ National Health Care Systems Committee and a surgeon with Kaiser Permanente in La Jolla, Calif., says that while no hard data is available, she thinks hospitalist involvement in orthopedic procedures improves patient care.
She says her “gut feeling” is that the mortality rate would tend to fall where hospitalists are more involved. But she also says that there might be room for hospitalists to become more involved in those procedures, to become familiar with the patient at an earlier stage.
“Would it make sense for a hospitalist … since a hospitalist team would be managing them post-operatively, to consider seeing them pre-operatively? That would be the other area where I think there may be potential growth,” she says.
Dr. Nagamine says more effort is being put into familiarizing patients with hospitalists.
“I think that patients more and more understand our role,” she says. “Part of it is the communication. When the primary-care physician or whoever refers them to the hospital, it’s nice that they explain that someone else will be managing their care. When they arrive at the hospital, we explain our role. In order to gain a patient’s trust, you have to show that you know something about them, you’ve read the chart, you’ve talked to Dr. Smith.”
That transition is something that has received more attention over time, she says, with doctors increasingly providing patients business cards with photos so that they can keep track of who’s who.
“It’s something we could still work on,” Dr. Nagamine says. “But we’re very focused on patient satisfaction and communication. There’s a lot of work going on in that regard.”
Super-Commuters
A “long commute” once meant 60 minutes of drive time or a long haul on public transit from the suburbs to city centers. That definition has changed quite a bit as the nation’s workforce becomes more mobile.
Take, for instance, hospitalist Yun Namkung, MD, who lives in Queens, N.Y., but works at Leflore Hospital, a 248-bed regional medical center in Greenwood, Miss., about 130 miles south of Memphis. “I’m something called a ‘firefighter’ within the company,” says Dr. Namkung, who’s been traveling long distances to work for his employer, Brentwood, Tenn.-based Cogent-HMG.
Dr. Namkung’s first long-distance commute was an interim assignment: He was an HMG program director in upstate New York anticipating a move to California. The move didn’t materialize, and now, after two years as a “super-commuter,” he says, “Traveling is actually fulfilling. You get to meet different people and supporting staff. You get exposed to a variety of patients, so clinically, you get better. I think I can continue to do this for a while.”
Super-commuters go by various names and monikers—“firefighters,” “travelers,” “vagabonds”—but they share a common reality: one or two weeks a month, and in some cases every week, they’re traveling long distances from home to work. And while it might not be for every hospitalist, this mega-commute phenomenon has pros and cons, hidden costs, and unexpected perks.

—Charles Barnett, MD, Knoxville, Tenn.
An Upward Trend?
Transportation policy consultant Alan E. Pisarski, author of “Commuting in America (Vols. 1-3),” often testifies before Congress on transportation issues for policy planning and investment requirements. The third volume of his “Commuting in America” series, published in 2006, found that the number of workers with commutes of more than 60 minutes increased almost 50% from 1990 to 2000.1 That duration probably rose even more following the economic downturn that began in 2008, he says, as the notion of an “acceptable” commute changes when the job market is tight.
The long-distance commuting trend is likely to increase, he says, because highly skilled workers (e.g. physicians) are in short supply. In our mobile society, he adds, “professionals are more willing to accept long distance separation from their families, on at least some kind of scheduled basis.”
In addition, as millions of baby boomers retire, replacing their skill sets is proving difficult. Companies are trying to hold boomers in the labor force longer, offering attractive perks so that they will stay.
Many jobs, even in a telecommuting society, still require in-person deliveries. And for some, super-commuting is a better alternative to relocation. For others, it might be the only alternative, given the poor housing market. That’s the way Anthony Venturato sees it.
“In my business, [we] have to be where the project is,” says Venturato, a project manager for passenger rail projects for STV Inc., a leading architectural, engineering, and construction management firm. “We have virtual meeting rooms, but we’ve got a long way to go before working closely together and being physically far away are equivalent—like that great scene in “Star Wars” where holograms of ‘attendees’ were interacting around a conference table. To run a project, at least in the early 21st century, you’ve gotta be there.” (see “Nomadic Lifestyle Works for Some,”)
—Yun Namkung, MD, Queens, N.Y.
Models Differ
Mark Dotson, vice president of recruiting at Cogent-HMG, says his company instituted a “travelers” model in October of 2009 to reduce its locum tenens usage. Travelers, he says, are hospitalists licensed in several states who can be placed in different programs, most within driving distance. Some request a remote location, such as one Cogent-HMG hospitalist who resides in Dallas and has been commuting to Great Falls, Tenn., for more two years.
Dotson explains that the company’s travelers “are not typical locums who may just say, ‘I’ll be here for two months and then I’m out of here.’ They are employed by us, get full benefits [plus a 10% premium over regular employees] and training from our academy,” he says. “They are looked upon as part of the team when we place them in a program, and not an interim solution.”
Travelers contribute to program stability and improved quality and productivity metrics, Dotson adds. In Great Falls, for instance, the hospitalist team, which includes a traveler on every rotation, has regularly met its quality performance measures and RVU requirements since being fully staffed. Dotson estimates that 10% of the hospitalists hired by Cogent-HMG last year were travelers, and he’d like to see that percentage grow to 25% to meet increasing demand.
EmCare Inpatient Services in Dallas takes a different approach. They use super-commuters only for short-term startups, says CEO Mark Hamm, who’s “never been an advocate of flying people in and out. You don’t ever get the continuity that you need within the practice.”
To establish trust with referring primary-care physicians (PCPs), hospitalist programs need to comprise 80% to 90% of residential hospitalists, he says. Otherwise, EmCare becomes “just a staffing company and not a partner” with client hospitals. This is especially essential when it comes to hiring medical directors, he says, who must be present for meetings and administering program operations.

A Good Fit
So who are the super-commuter hospitalists? Dotson, of Cogent-HMG, says that the majority of those willing to travel tend to be single. Hospitalists who are in between residency and starting a fellowship find this type of assignment provides consistent scheduling, income, and benefits to them and their families. Another contingent: mature career hospitalists with grown children.
Eric Kerley, MD, FAAP, FACP lives and works primarily in eastern Tennessee, where he is a full-time medical director. He saw his friend and colleague Charles Barnett, MD, taking assignments in Wyoming, and thought traveling for work “sounded interesting.”
“I’m a Southern boy who has lived my entire life between Orlando [Fla.], Tennessee, and Texas,” he says, “so I picked my locations based on places I would want to go.”
Dr. Kerley’s first yearlong assignment, in 2009-2010, was in central Alaska at a 75-bed facility. He worked as a nocturnist. “To see minus-20-degree Fahrenheit temperatures and frozen rivers, and days that are 22 hours long, that was pretty amazing,” he says. Being away for one week a month is really not much different than a week of day shifts at home, he adds.
Dr. Barnett began super-commuting four years ago from his home in Knoxville, Tenn., to Gillette, Wyo. Traveling to Wyoming is his regular commuter gig—he stays at the hospital—and he enjoys working in another environment.
The away time also works for his marriage, he says. “Just before I leave for an assignment, my wife’s ready to see me go,” he says. “And then, when I come home, she’s anxious for me to be there, so it’s sort of like a honeymoon once a month for both of us.”
Continued below...

Nomadic lifestyle works for some
Anthony Venturato has traveled extensively throughout his career as a project manager for Douglassville, Pa.-based engineering consulting firm STV Inc. “I’m considered the company vagabond,” he says. “I will take assignments wherever I can reasonably fly to.”
He’s currently managing construction of a rail project in Southern California. His longest extended commute was from his home in Tampa, Fla., to London. When it became clear he was needed full-time, he and his wife relocated their family to England.
While his children were growing, Venturato and his wife moved nine times to different states. “To move your household lock, stock, and barrel is really very difficult,” he says. “When the kids lived at home, it was tough on them.”

—Anthony Venturato, project manager, STV Inc.
Finally, in the late 1990s, he and his wife decided to pick a location near a hub airport from which he could readily commute.
Even though his weekends are short—he’s home every other weekend for about a day and a half—it’s a better alternative to constant relocation, he says.
“I would guess that 10% or less of people in my business are willing to super-commute, and people willing to relocate is probably even lower than that,” he explains.—GH
Pros and Cons
Although he misses his family when he’s traveling, Dr. Namkung now spends more quality time with them, “because I realize how precious that time is.” His wife, a pharmacist, makes it a point to take time off when he’s home, and they do more things together as a family.
Another bonus: “I meet different docs, nursing staffs, and administrators,” Dr. Namkung says. “Since I’m here alone, we have the chance to have dinner together and spend time. In that way, I bond with a lot more people than I would normally if I stayed in one place.”
Dr. Kerley racked up the frequent-flier miles during his one-year assignment to Alaska, which was a plus when it came to financing family vacations.
Working in other states entails meeting state-specific licensing requirements. Some companies, such as Cogent-HMG, pay the costs of obtaining those state licenses. Others do not, and the paperwork, says Dr. Barnett, can be “a nightmare.” Locum Leaders CEO Will Drescher, MD, says his company pays for licenses in some states and assists with paperwork in others.

—Eric Kerley, MD, medical director, Morristown, Tenn, nocturnist, PeaceHealth Medical Group, Ketchikan, Ak.
Unless hospitalists are full-time employees of the organization, such as Dr. Namkung with Cogent-HMG, their income likely will be considered independent contracting by the IRS. That means you’ll be filing an extra form (1099) with your return, and you may have to pay quarterly estimated self-employment tax. Hospitalists are encouraged to consult their financial advisors to make sure they are set up properly. Hospitalists who live in one state and work in another also need to beware of state and municipal tax guidelines.
One hidden cost of super-commuting is less time for household upkeep. Tony Venturato does not have the luxury of a week-on/week-off schedule, and with travel, his weekends are cut down to a day or a day and a half twice a month. That doesn’t leave much time for household chores and home improvement projects.
“The same way that you cannot run a project from the road, it’s also pretty hard to run a household from remote, and that puts a burden on your spouse,” he says. “That leaky faucet that might have been a small fix-it project? Now my wife has to find a plumber to come fix it. Do-it-yourself home improvement projects? Fuhgeddaboudit.”
Dr. Kerley nearly missed the birth of his first grandchild the first week he had agreed to work in Alaska. However, his state license to practice was delayed, so he was there for the important event. “After that, I realized that I did need to be more intentional about dates and scheduling,” he says. “Since then, the scheduling has become more rhythmic.”
Good Career Move?
Super-commuting adds to the bank account, widens travel experiences, and sharpens clinical skills. But does it work for career advancement? Dotson believes that working with various types of teams in different settings helps hospitalists mature quickly.
Venturato thinks that accepting long-distance assignments will become even more necessary for career-building. “There’s still the aggravation of flying,” he admits. “But the jobs you get, the opportunities that you have, make it all worthwhile. If you limit yourself to not going to these interesting projects, you’re limiting your career.”
Dotson seconds that notion. “If people are willing to do the traveling, and they are good people, there are lots of opportunities for them,” he says.

The Life of an HM Traveler
From her ranch in North Platte, Neb., Susan Schuckert, MD, is almost equidistant between her two hospitalist jobs. One week a month, she drives 4 1/2 hours to Nebraska Medical Center in Omaha, where she works for IMI Hospitalists. She bunks with friends, whom she’s known since she completed medical training there. Just recently, she began a locum tenens assignment in Scottsbluff, Neb.—a 3 1/2-hour drive to the opposite side of the state.
Dr. Schuckert is building a private practice in North Platte with a partner. She began working as a hospitalist to keep her skills current, and says she is realizing a professional net gain. Being a hospitalist, she says, “is fun. I enjoy getting to know the nurses, seeing different ways of doing things, and the variety.”

—Susan Schuckert, MD, North Platte, Neb.
In addition to the stimulation and camaraderie of a large medical center, Dr. Schuckert has found that her patients back home in North Platte embrace the fact that she moonlights at Nebraska Medical Center. “If I have to send patients to Omaha, I personally know who I’m sending them to,” she says. “It also makes referrals smoother. Other physicians in town have called the medical center when I’m on as a hospitalist, so it makes it possible for me to make sure that their patients get what they need.”
There are times, of course, when she misses being at home. She recently was working in Omaha and missed the birth of a new colt. Not to worry: Her husband, who is “very supportive” of her super-commuting, texted her photos of little Rudy every day. So for the moment, the mixture of a home practice and working as a traveling hospitalist is a good one.
And the long drives? With a hands-free phone and books on CD, “It’s no big deal,” she says.
Gretchen Henkel is a freelance writer in Southern California.
References
- Pisarski, AE. Commuting in America III: The Third National Reporter on Commuting Patterns and Trends. 2006: Transportation Research Board of the National Academies; Washington, D.C.
- Sandow E. Till work do us part: The social fallacy of long-distance commuting [dissertation]. Available at: http://umu.diva-portal.org. Accessed June 22, 2011.
A “long commute” once meant 60 minutes of drive time or a long haul on public transit from the suburbs to city centers. That definition has changed quite a bit as the nation’s workforce becomes more mobile.
Take, for instance, hospitalist Yun Namkung, MD, who lives in Queens, N.Y., but works at Leflore Hospital, a 248-bed regional medical center in Greenwood, Miss., about 130 miles south of Memphis. “I’m something called a ‘firefighter’ within the company,” says Dr. Namkung, who’s been traveling long distances to work for his employer, Brentwood, Tenn.-based Cogent-HMG.
Dr. Namkung’s first long-distance commute was an interim assignment: He was an HMG program director in upstate New York anticipating a move to California. The move didn’t materialize, and now, after two years as a “super-commuter,” he says, “Traveling is actually fulfilling. You get to meet different people and supporting staff. You get exposed to a variety of patients, so clinically, you get better. I think I can continue to do this for a while.”
Super-commuters go by various names and monikers—“firefighters,” “travelers,” “vagabonds”—but they share a common reality: one or two weeks a month, and in some cases every week, they’re traveling long distances from home to work. And while it might not be for every hospitalist, this mega-commute phenomenon has pros and cons, hidden costs, and unexpected perks.
—Charles Barnett, MD, Knoxville, Tenn.
An Upward Trend?
Transportation policy consultant Alan E. Pisarski, author of “Commuting in America (Vols. 1-3),” often testifies before Congress on transportation issues for policy planning and investment requirements. The third volume of his “Commuting in America” series, published in 2006, found that the number of workers with commutes of more than 60 minutes increased almost 50% from 1990 to 2000.1 That duration probably rose even more following the economic downturn that began in 2008, he says, as the notion of an “acceptable” commute changes when the job market is tight.
The long-distance commuting trend is likely to increase, he says, because highly skilled workers (e.g. physicians) are in short supply. In our mobile society, he adds, “professionals are more willing to accept long distance separation from their families, on at least some kind of scheduled basis.”
In addition, as millions of baby boomers retire, replacing their skill sets is proving difficult. Companies are trying to hold boomers in the labor force longer, offering attractive perks so that they will stay.
Many jobs, even in a telecommuting society, still require in-person deliveries. And for some, super-commuting is a better alternative to relocation. For others, it might be the only alternative, given the poor housing market. That’s the way Anthony Venturato sees it.
“In my business, [we] have to be where the project is,” says Venturato, a project manager for passenger rail projects for STV Inc., a leading architectural, engineering, and construction management firm. “We have virtual meeting rooms, but we’ve got a long way to go before working closely together and being physically far away are equivalent—like that great scene in “Star Wars” where holograms of ‘attendees’ were interacting around a conference table. To run a project, at least in the early 21st century, you’ve gotta be there.” (see “Nomadic Lifestyle Works for Some,”)
—Yun Namkung, MD, Queens, N.Y.
Models Differ
Mark Dotson, vice president of recruiting at Cogent-HMG, says his company instituted a “travelers” model in October of 2009 to reduce its locum tenens usage. Travelers, he says, are hospitalists licensed in several states who can be placed in different programs, most within driving distance. Some request a remote location, such as one Cogent-HMG hospitalist who resides in Dallas and has been commuting to Great Falls, Tenn., for more two years.
Dotson explains that the company’s travelers “are not typical locums who may just say, ‘I’ll be here for two months and then I’m out of here.’ They are employed by us, get full benefits [plus a 10% premium over regular employees] and training from our academy,” he says. “They are looked upon as part of the team when we place them in a program, and not an interim solution.”
Travelers contribute to program stability and improved quality and productivity metrics, Dotson adds. In Great Falls, for instance, the hospitalist team, which includes a traveler on every rotation, has regularly met its quality performance measures and RVU requirements since being fully staffed. Dotson estimates that 10% of the hospitalists hired by Cogent-HMG last year were travelers, and he’d like to see that percentage grow to 25% to meet increasing demand.
EmCare Inpatient Services in Dallas takes a different approach. They use super-commuters only for short-term startups, says CEO Mark Hamm, who’s “never been an advocate of flying people in and out. You don’t ever get the continuity that you need within the practice.”
To establish trust with referring primary-care physicians (PCPs), hospitalist programs need to comprise 80% to 90% of residential hospitalists, he says. Otherwise, EmCare becomes “just a staffing company and not a partner” with client hospitals. This is especially essential when it comes to hiring medical directors, he says, who must be present for meetings and administering program operations.
A Good Fit
So who are the super-commuter hospitalists? Dotson, of Cogent-HMG, says that the majority of those willing to travel tend to be single. Hospitalists who are in between residency and starting a fellowship find this type of assignment provides consistent scheduling, income, and benefits to them and their families. Another contingent: mature career hospitalists with grown children.
Eric Kerley, MD, FAAP, FACP lives and works primarily in eastern Tennessee, where he is a full-time medical director. He saw his friend and colleague Charles Barnett, MD, taking assignments in Wyoming, and thought traveling for work “sounded interesting.”
“I’m a Southern boy who has lived my entire life between Orlando [Fla.], Tennessee, and Texas,” he says, “so I picked my locations based on places I would want to go.”
Dr. Kerley’s first yearlong assignment, in 2009-2010, was in central Alaska at a 75-bed facility. He worked as a nocturnist. “To see minus-20-degree Fahrenheit temperatures and frozen rivers, and days that are 22 hours long, that was pretty amazing,” he says. Being away for one week a month is really not much different than a week of day shifts at home, he adds.
Dr. Barnett began super-commuting four years ago from his home in Knoxville, Tenn., to Gillette, Wyo. Traveling to Wyoming is his regular commuter gig—he stays at the hospital—and he enjoys working in another environment.
The away time also works for his marriage, he says. “Just before I leave for an assignment, my wife’s ready to see me go,” he says. “And then, when I come home, she’s anxious for me to be there, so it’s sort of like a honeymoon once a month for both of us.”
Continued below...
Nomadic lifestyle works for some
Anthony Venturato has traveled extensively throughout his career as a project manager for Douglassville, Pa.-based engineering consulting firm STV Inc. “I’m considered the company vagabond,” he says. “I will take assignments wherever I can reasonably fly to.”
He’s currently managing construction of a rail project in Southern California. His longest extended commute was from his home in Tampa, Fla., to London. When it became clear he was needed full-time, he and his wife relocated their family to England.
While his children were growing, Venturato and his wife moved nine times to different states. “To move your household lock, stock, and barrel is really very difficult,” he says. “When the kids lived at home, it was tough on them.”
—Anthony Venturato, project manager, STV Inc.
Finally, in the late 1990s, he and his wife decided to pick a location near a hub airport from which he could readily commute.
Even though his weekends are short—he’s home every other weekend for about a day and a half—it’s a better alternative to constant relocation, he says.
“I would guess that 10% or less of people in my business are willing to super-commute, and people willing to relocate is probably even lower than that,” he explains.—GH
Pros and Cons
Although he misses his family when he’s traveling, Dr. Namkung now spends more quality time with them, “because I realize how precious that time is.” His wife, a pharmacist, makes it a point to take time off when he’s home, and they do more things together as a family.
Another bonus: “I meet different docs, nursing staffs, and administrators,” Dr. Namkung says. “Since I’m here alone, we have the chance to have dinner together and spend time. In that way, I bond with a lot more people than I would normally if I stayed in one place.”
Dr. Kerley racked up the frequent-flier miles during his one-year assignment to Alaska, which was a plus when it came to financing family vacations.
Working in other states entails meeting state-specific licensing requirements. Some companies, such as Cogent-HMG, pay the costs of obtaining those state licenses. Others do not, and the paperwork, says Dr. Barnett, can be “a nightmare.” Locum Leaders CEO Will Drescher, MD, says his company pays for licenses in some states and assists with paperwork in others.
—Eric Kerley, MD, medical director, Morristown, Tenn, nocturnist, PeaceHealth Medical Group, Ketchikan, Ak.
Unless hospitalists are full-time employees of the organization, such as Dr. Namkung with Cogent-HMG, their income likely will be considered independent contracting by the IRS. That means you’ll be filing an extra form (1099) with your return, and you may have to pay quarterly estimated self-employment tax. Hospitalists are encouraged to consult their financial advisors to make sure they are set up properly. Hospitalists who live in one state and work in another also need to beware of state and municipal tax guidelines.
One hidden cost of super-commuting is less time for household upkeep. Tony Venturato does not have the luxury of a week-on/week-off schedule, and with travel, his weekends are cut down to a day or a day and a half twice a month. That doesn’t leave much time for household chores and home improvement projects.
“The same way that you cannot run a project from the road, it’s also pretty hard to run a household from remote, and that puts a burden on your spouse,” he says. “That leaky faucet that might have been a small fix-it project? Now my wife has to find a plumber to come fix it. Do-it-yourself home improvement projects? Fuhgeddaboudit.”
Dr. Kerley nearly missed the birth of his first grandchild the first week he had agreed to work in Alaska. However, his state license to practice was delayed, so he was there for the important event. “After that, I realized that I did need to be more intentional about dates and scheduling,” he says. “Since then, the scheduling has become more rhythmic.”
Good Career Move?
Super-commuting adds to the bank account, widens travel experiences, and sharpens clinical skills. But does it work for career advancement? Dotson believes that working with various types of teams in different settings helps hospitalists mature quickly.
Venturato thinks that accepting long-distance assignments will become even more necessary for career-building. “There’s still the aggravation of flying,” he admits. “But the jobs you get, the opportunities that you have, make it all worthwhile. If you limit yourself to not going to these interesting projects, you’re limiting your career.”
Dotson seconds that notion. “If people are willing to do the traveling, and they are good people, there are lots of opportunities for them,” he says.
The Life of an HM Traveler
From her ranch in North Platte, Neb., Susan Schuckert, MD, is almost equidistant between her two hospitalist jobs. One week a month, she drives 4 1/2 hours to Nebraska Medical Center in Omaha, where she works for IMI Hospitalists. She bunks with friends, whom she’s known since she completed medical training there. Just recently, she began a locum tenens assignment in Scottsbluff, Neb.—a 3 1/2-hour drive to the opposite side of the state.
Dr. Schuckert is building a private practice in North Platte with a partner. She began working as a hospitalist to keep her skills current, and says she is realizing a professional net gain. Being a hospitalist, she says, “is fun. I enjoy getting to know the nurses, seeing different ways of doing things, and the variety.”
—Susan Schuckert, MD, North Platte, Neb.
In addition to the stimulation and camaraderie of a large medical center, Dr. Schuckert has found that her patients back home in North Platte embrace the fact that she moonlights at Nebraska Medical Center. “If I have to send patients to Omaha, I personally know who I’m sending them to,” she says. “It also makes referrals smoother. Other physicians in town have called the medical center when I’m on as a hospitalist, so it makes it possible for me to make sure that their patients get what they need.”
There are times, of course, when she misses being at home. She recently was working in Omaha and missed the birth of a new colt. Not to worry: Her husband, who is “very supportive” of her super-commuting, texted her photos of little Rudy every day. So for the moment, the mixture of a home practice and working as a traveling hospitalist is a good one.
And the long drives? With a hands-free phone and books on CD, “It’s no big deal,” she says.
Gretchen Henkel is a freelance writer in Southern California.
References
- Pisarski, AE. Commuting in America III: The Third National Reporter on Commuting Patterns and Trends. 2006: Transportation Research Board of the National Academies; Washington, D.C.
- Sandow E. Till work do us part: The social fallacy of long-distance commuting [dissertation]. Available at: http://umu.diva-portal.org. Accessed June 22, 2011.
A “long commute” once meant 60 minutes of drive time or a long haul on public transit from the suburbs to city centers. That definition has changed quite a bit as the nation’s workforce becomes more mobile.
Take, for instance, hospitalist Yun Namkung, MD, who lives in Queens, N.Y., but works at Leflore Hospital, a 248-bed regional medical center in Greenwood, Miss., about 130 miles south of Memphis. “I’m something called a ‘firefighter’ within the company,” says Dr. Namkung, who’s been traveling long distances to work for his employer, Brentwood, Tenn.-based Cogent-HMG.
Dr. Namkung’s first long-distance commute was an interim assignment: He was an HMG program director in upstate New York anticipating a move to California. The move didn’t materialize, and now, after two years as a “super-commuter,” he says, “Traveling is actually fulfilling. You get to meet different people and supporting staff. You get exposed to a variety of patients, so clinically, you get better. I think I can continue to do this for a while.”
Super-commuters go by various names and monikers—“firefighters,” “travelers,” “vagabonds”—but they share a common reality: one or two weeks a month, and in some cases every week, they’re traveling long distances from home to work. And while it might not be for every hospitalist, this mega-commute phenomenon has pros and cons, hidden costs, and unexpected perks.
—Charles Barnett, MD, Knoxville, Tenn.
An Upward Trend?
Transportation policy consultant Alan E. Pisarski, author of “Commuting in America (Vols. 1-3),” often testifies before Congress on transportation issues for policy planning and investment requirements. The third volume of his “Commuting in America” series, published in 2006, found that the number of workers with commutes of more than 60 minutes increased almost 50% from 1990 to 2000.1 That duration probably rose even more following the economic downturn that began in 2008, he says, as the notion of an “acceptable” commute changes when the job market is tight.
The long-distance commuting trend is likely to increase, he says, because highly skilled workers (e.g. physicians) are in short supply. In our mobile society, he adds, “professionals are more willing to accept long distance separation from their families, on at least some kind of scheduled basis.”
In addition, as millions of baby boomers retire, replacing their skill sets is proving difficult. Companies are trying to hold boomers in the labor force longer, offering attractive perks so that they will stay.
Many jobs, even in a telecommuting society, still require in-person deliveries. And for some, super-commuting is a better alternative to relocation. For others, it might be the only alternative, given the poor housing market. That’s the way Anthony Venturato sees it.
“In my business, [we] have to be where the project is,” says Venturato, a project manager for passenger rail projects for STV Inc., a leading architectural, engineering, and construction management firm. “We have virtual meeting rooms, but we’ve got a long way to go before working closely together and being physically far away are equivalent—like that great scene in “Star Wars” where holograms of ‘attendees’ were interacting around a conference table. To run a project, at least in the early 21st century, you’ve gotta be there.” (see “Nomadic Lifestyle Works for Some,”)
—Yun Namkung, MD, Queens, N.Y.
Models Differ
Mark Dotson, vice president of recruiting at Cogent-HMG, says his company instituted a “travelers” model in October of 2009 to reduce its locum tenens usage. Travelers, he says, are hospitalists licensed in several states who can be placed in different programs, most within driving distance. Some request a remote location, such as one Cogent-HMG hospitalist who resides in Dallas and has been commuting to Great Falls, Tenn., for more two years.
Dotson explains that the company’s travelers “are not typical locums who may just say, ‘I’ll be here for two months and then I’m out of here.’ They are employed by us, get full benefits [plus a 10% premium over regular employees] and training from our academy,” he says. “They are looked upon as part of the team when we place them in a program, and not an interim solution.”
Travelers contribute to program stability and improved quality and productivity metrics, Dotson adds. In Great Falls, for instance, the hospitalist team, which includes a traveler on every rotation, has regularly met its quality performance measures and RVU requirements since being fully staffed. Dotson estimates that 10% of the hospitalists hired by Cogent-HMG last year were travelers, and he’d like to see that percentage grow to 25% to meet increasing demand.
EmCare Inpatient Services in Dallas takes a different approach. They use super-commuters only for short-term startups, says CEO Mark Hamm, who’s “never been an advocate of flying people in and out. You don’t ever get the continuity that you need within the practice.”
To establish trust with referring primary-care physicians (PCPs), hospitalist programs need to comprise 80% to 90% of residential hospitalists, he says. Otherwise, EmCare becomes “just a staffing company and not a partner” with client hospitals. This is especially essential when it comes to hiring medical directors, he says, who must be present for meetings and administering program operations.
A Good Fit
So who are the super-commuter hospitalists? Dotson, of Cogent-HMG, says that the majority of those willing to travel tend to be single. Hospitalists who are in between residency and starting a fellowship find this type of assignment provides consistent scheduling, income, and benefits to them and their families. Another contingent: mature career hospitalists with grown children.
Eric Kerley, MD, FAAP, FACP lives and works primarily in eastern Tennessee, where he is a full-time medical director. He saw his friend and colleague Charles Barnett, MD, taking assignments in Wyoming, and thought traveling for work “sounded interesting.”
“I’m a Southern boy who has lived my entire life between Orlando [Fla.], Tennessee, and Texas,” he says, “so I picked my locations based on places I would want to go.”
Dr. Kerley’s first yearlong assignment, in 2009-2010, was in central Alaska at a 75-bed facility. He worked as a nocturnist. “To see minus-20-degree Fahrenheit temperatures and frozen rivers, and days that are 22 hours long, that was pretty amazing,” he says. Being away for one week a month is really not much different than a week of day shifts at home, he adds.
Dr. Barnett began super-commuting four years ago from his home in Knoxville, Tenn., to Gillette, Wyo. Traveling to Wyoming is his regular commuter gig—he stays at the hospital—and he enjoys working in another environment.
The away time also works for his marriage, he says. “Just before I leave for an assignment, my wife’s ready to see me go,” he says. “And then, when I come home, she’s anxious for me to be there, so it’s sort of like a honeymoon once a month for both of us.”
Continued below...
Nomadic lifestyle works for some
Anthony Venturato has traveled extensively throughout his career as a project manager for Douglassville, Pa.-based engineering consulting firm STV Inc. “I’m considered the company vagabond,” he says. “I will take assignments wherever I can reasonably fly to.”
He’s currently managing construction of a rail project in Southern California. His longest extended commute was from his home in Tampa, Fla., to London. When it became clear he was needed full-time, he and his wife relocated their family to England.
While his children were growing, Venturato and his wife moved nine times to different states. “To move your household lock, stock, and barrel is really very difficult,” he says. “When the kids lived at home, it was tough on them.”
—Anthony Venturato, project manager, STV Inc.
Finally, in the late 1990s, he and his wife decided to pick a location near a hub airport from which he could readily commute.
Even though his weekends are short—he’s home every other weekend for about a day and a half—it’s a better alternative to constant relocation, he says.
“I would guess that 10% or less of people in my business are willing to super-commute, and people willing to relocate is probably even lower than that,” he explains.—GH
Pros and Cons
Although he misses his family when he’s traveling, Dr. Namkung now spends more quality time with them, “because I realize how precious that time is.” His wife, a pharmacist, makes it a point to take time off when he’s home, and they do more things together as a family.
Another bonus: “I meet different docs, nursing staffs, and administrators,” Dr. Namkung says. “Since I’m here alone, we have the chance to have dinner together and spend time. In that way, I bond with a lot more people than I would normally if I stayed in one place.”
Dr. Kerley racked up the frequent-flier miles during his one-year assignment to Alaska, which was a plus when it came to financing family vacations.
Working in other states entails meeting state-specific licensing requirements. Some companies, such as Cogent-HMG, pay the costs of obtaining those state licenses. Others do not, and the paperwork, says Dr. Barnett, can be “a nightmare.” Locum Leaders CEO Will Drescher, MD, says his company pays for licenses in some states and assists with paperwork in others.
—Eric Kerley, MD, medical director, Morristown, Tenn, nocturnist, PeaceHealth Medical Group, Ketchikan, Ak.
Unless hospitalists are full-time employees of the organization, such as Dr. Namkung with Cogent-HMG, their income likely will be considered independent contracting by the IRS. That means you’ll be filing an extra form (1099) with your return, and you may have to pay quarterly estimated self-employment tax. Hospitalists are encouraged to consult their financial advisors to make sure they are set up properly. Hospitalists who live in one state and work in another also need to beware of state and municipal tax guidelines.
One hidden cost of super-commuting is less time for household upkeep. Tony Venturato does not have the luxury of a week-on/week-off schedule, and with travel, his weekends are cut down to a day or a day and a half twice a month. That doesn’t leave much time for household chores and home improvement projects.
“The same way that you cannot run a project from the road, it’s also pretty hard to run a household from remote, and that puts a burden on your spouse,” he says. “That leaky faucet that might have been a small fix-it project? Now my wife has to find a plumber to come fix it. Do-it-yourself home improvement projects? Fuhgeddaboudit.”
Dr. Kerley nearly missed the birth of his first grandchild the first week he had agreed to work in Alaska. However, his state license to practice was delayed, so he was there for the important event. “After that, I realized that I did need to be more intentional about dates and scheduling,” he says. “Since then, the scheduling has become more rhythmic.”
Good Career Move?
Super-commuting adds to the bank account, widens travel experiences, and sharpens clinical skills. But does it work for career advancement? Dotson believes that working with various types of teams in different settings helps hospitalists mature quickly.
Venturato thinks that accepting long-distance assignments will become even more necessary for career-building. “There’s still the aggravation of flying,” he admits. “But the jobs you get, the opportunities that you have, make it all worthwhile. If you limit yourself to not going to these interesting projects, you’re limiting your career.”
Dotson seconds that notion. “If people are willing to do the traveling, and they are good people, there are lots of opportunities for them,” he says.
The Life of an HM Traveler
From her ranch in North Platte, Neb., Susan Schuckert, MD, is almost equidistant between her two hospitalist jobs. One week a month, she drives 4 1/2 hours to Nebraska Medical Center in Omaha, where she works for IMI Hospitalists. She bunks with friends, whom she’s known since she completed medical training there. Just recently, she began a locum tenens assignment in Scottsbluff, Neb.—a 3 1/2-hour drive to the opposite side of the state.
Dr. Schuckert is building a private practice in North Platte with a partner. She began working as a hospitalist to keep her skills current, and says she is realizing a professional net gain. Being a hospitalist, she says, “is fun. I enjoy getting to know the nurses, seeing different ways of doing things, and the variety.”
—Susan Schuckert, MD, North Platte, Neb.
In addition to the stimulation and camaraderie of a large medical center, Dr. Schuckert has found that her patients back home in North Platte embrace the fact that she moonlights at Nebraska Medical Center. “If I have to send patients to Omaha, I personally know who I’m sending them to,” she says. “It also makes referrals smoother. Other physicians in town have called the medical center when I’m on as a hospitalist, so it makes it possible for me to make sure that their patients get what they need.”
There are times, of course, when she misses being at home. She recently was working in Omaha and missed the birth of a new colt. Not to worry: Her husband, who is “very supportive” of her super-commuting, texted her photos of little Rudy every day. So for the moment, the mixture of a home practice and working as a traveling hospitalist is a good one.
And the long drives? With a hands-free phone and books on CD, “It’s no big deal,” she says.
Gretchen Henkel is a freelance writer in Southern California.
References
- Pisarski, AE. Commuting in America III: The Third National Reporter on Commuting Patterns and Trends. 2006: Transportation Research Board of the National Academies; Washington, D.C.
- Sandow E. Till work do us part: The social fallacy of long-distance commuting [dissertation]. Available at: http://umu.diva-portal.org. Accessed June 22, 2011.
Our Wake-Up Call

For those who say they would pay $50 more per patient if the quality is better, here’s the problem: Show me the data that say hospitalist care is higher-quality.
I suspect most of you have reviewed the study or at least heard about it. Bob Wachter, MD, MHM, blogged about the study. An article about the study appeared in American Medical Association News. Even National Public Radio ran a piece about the study on their show “Morning Edition.”
I am, of course, referring to the study by Kuo and Goodwin, which was published in the Annals of Internal Medicine in early August.1
In this study, the authors looked at a sample of patients (5%) with primary-care physicians (PCPs) enrolled in Medicare who were cared for by their PCP or a hospitalist during a period from 2001 to 2006. The authors stated their underlying hypotheses as:
- Hospitalist care would be associated with costs shifting from the hospital to the post-hospital setting;
- Hospitalist care would be associated with a decrease in discharges directly to home; and
- Discontinuities of care associated with hospitalist care would lead to a greater rate of visits to the emergency room and readmissions to the hospital, resulting in increased Medicare costs.
Did the authors say hospitalist care cost more? They can’t possibly be correct, can they? Don’t all the hospitalist studies show that hospitalists provide the same quality of care as primary-care doctors, except the costs are lower and the hospital length of stay (LOS) is shorter when hospitalists care for patients?
The point here is that these investigators look at the care not only during a patient’s hospital stay, but also for 30 days after discharge. This is something that had not been done previously—at least not on this scale.
Focus on Facts
And what did the authors find? Patients cared for by hospitalists, as compared to their PCPs, had a shorter LOS and lower in-hospital costs, but these patients also were less likely to be discharged directly to home, less likely to see their PCPs post-discharge, and had more hospital readmissions, ED visits, and nursing home visits after discharge.
Since its release two months ago, I have heard a lot of discussion about the study. Here are a few of the comments I’ve heard:
- “This was an observational study. You can’t possibly remove all confounders in an observational study.”
- “The authors looked at a time period early in the hospitalist movement. If they did the study today, the results would be different.”
- “The additional costs hospitalists incurred were only $50 per patient. Wouldn’t you pay $50 more if the care was better?”
- “This is why hospitals hired hospitalists. They save money for the hospitals. What did they expect to find?”
I agree that observational studies have limitations (even the authors acknowledged this), but this doesn’t mean results from observational studies are invalid. Some of us don’t want to hear this, but this actually was a pretty well-done study with a robust statistical analysis. We should recognize the study has limitations and think about the results.
Kuo and Goodwin looked at data during a period of time early in the hospitalist movement; the results could be different if the study were to be repeated today. But we don’t know what the data would be today. I suppose the data could be better, worse, or about the same. The fact of the matter is that HM leaders—and most of the rest of us—knew that transitions of care, under the hospitalist model, were a potential weakness. How many times have you heard Win Whitcomb, MD, MHM, and John Nelson, MD, MHM, talk about the potential “voltage drop” with handoffs?
The good news is that leaders in our field have done something about this. Project BOOST (Better Outcomes for Older Adults through Safer Transitions) is a program SHM has helped implement at dozens of hospitals across the country to address the issue of unnecessary hospital readmissions (www.hospitalmedicine. org/boost). Improving transitions of care and preventing unnecessary readmissions should be on the minds of all hospitalists. If your program and your hospital have not yet taken steps to address this issue, please let this be your wake-up call.
Show Me the Money
For those who say they would pay $50 more per patient if the quality is better, here’s the problem: Show me the data that say hospitalist care is higher-quality. I agree with you that it is hard to look at costs without looking at quality. Therein lies the basis for our nation’s move toward value-based purchasing of healthcare (see “Value-Based Purchasing Raises the Stakes,” May 2011).
When I hear hospitalists explain why the role of hospitalists was developed, the explanation often involves some discussion of cost and LOS reduction. Don’t get me wrong; it’s not that I believe HM has focused too much attention on cost reduction. I believe we have not focused enough on improving quality. This should not be surprising. Moving the bar on cost reduction is a lot easier than moving the bar on quality and patient safety. The first step toward improvement is an understanding of what you are doing currently. If your hospitalist group has not implemented a program to help its hospitalists measure the quality of care being provided, again, this is your wake-up call.
Last, but not least, for those of you who are not “surprised” by the results because of the belief that hospitalists were created to help the hospital save money and nothing more, I could not disagree with you more. I look at the roles that hospitalists have taken on in our nation’s hospitals, and I am incredibly proud to call myself a hospitalist.
Hospitalists are providing timely care when patients need it. Hospitalists are caring for patients without PCPs. Not only do hospitalists allow PCPs to provide more care in their outpatient clinics, but hospitalists also are caring for patients in ICUs in many places where there are not enough doctors sufficiently trained in critical care.
Rather than acting as an indictment on HM, I believe the Annals article makes a comment on the misalignment of incentives in our healthcare system.
It is 2011, not 1996; HM is here to stay. Most acute-care hospitals in America could not function without hospitalists. I applaud Kuo and Goodwin for doing the research and publishing their results. Let this be an opportunity for hospitalists around the country to think about how to implement systems to improve transitions of care and the quality of care we provide.
Dr. Li is president of SHM.
Reference
For those who say they would pay $50 more per patient if the quality is better, here’s the problem: Show me the data that say hospitalist care is higher-quality.
I suspect most of you have reviewed the study or at least heard about it. Bob Wachter, MD, MHM, blogged about the study. An article about the study appeared in American Medical Association News. Even National Public Radio ran a piece about the study on their show “Morning Edition.”
I am, of course, referring to the study by Kuo and Goodwin, which was published in the Annals of Internal Medicine in early August.1
In this study, the authors looked at a sample of patients (5%) with primary-care physicians (PCPs) enrolled in Medicare who were cared for by their PCP or a hospitalist during a period from 2001 to 2006. The authors stated their underlying hypotheses as:
- Hospitalist care would be associated with costs shifting from the hospital to the post-hospital setting;
- Hospitalist care would be associated with a decrease in discharges directly to home; and
- Discontinuities of care associated with hospitalist care would lead to a greater rate of visits to the emergency room and readmissions to the hospital, resulting in increased Medicare costs.
Did the authors say hospitalist care cost more? They can’t possibly be correct, can they? Don’t all the hospitalist studies show that hospitalists provide the same quality of care as primary-care doctors, except the costs are lower and the hospital length of stay (LOS) is shorter when hospitalists care for patients?
The point here is that these investigators look at the care not only during a patient’s hospital stay, but also for 30 days after discharge. This is something that had not been done previously—at least not on this scale.
Focus on Facts
And what did the authors find? Patients cared for by hospitalists, as compared to their PCPs, had a shorter LOS and lower in-hospital costs, but these patients also were less likely to be discharged directly to home, less likely to see their PCPs post-discharge, and had more hospital readmissions, ED visits, and nursing home visits after discharge.
Since its release two months ago, I have heard a lot of discussion about the study. Here are a few of the comments I’ve heard:
- “This was an observational study. You can’t possibly remove all confounders in an observational study.”
- “The authors looked at a time period early in the hospitalist movement. If they did the study today, the results would be different.”
- “The additional costs hospitalists incurred were only $50 per patient. Wouldn’t you pay $50 more if the care was better?”
- “This is why hospitals hired hospitalists. They save money for the hospitals. What did they expect to find?”
I agree that observational studies have limitations (even the authors acknowledged this), but this doesn’t mean results from observational studies are invalid. Some of us don’t want to hear this, but this actually was a pretty well-done study with a robust statistical analysis. We should recognize the study has limitations and think about the results.
Kuo and Goodwin looked at data during a period of time early in the hospitalist movement; the results could be different if the study were to be repeated today. But we don’t know what the data would be today. I suppose the data could be better, worse, or about the same. The fact of the matter is that HM leaders—and most of the rest of us—knew that transitions of care, under the hospitalist model, were a potential weakness. How many times have you heard Win Whitcomb, MD, MHM, and John Nelson, MD, MHM, talk about the potential “voltage drop” with handoffs?
The good news is that leaders in our field have done something about this. Project BOOST (Better Outcomes for Older Adults through Safer Transitions) is a program SHM has helped implement at dozens of hospitals across the country to address the issue of unnecessary hospital readmissions (www.hospitalmedicine. org/boost). Improving transitions of care and preventing unnecessary readmissions should be on the minds of all hospitalists. If your program and your hospital have not yet taken steps to address this issue, please let this be your wake-up call.
Show Me the Money
For those who say they would pay $50 more per patient if the quality is better, here’s the problem: Show me the data that say hospitalist care is higher-quality. I agree with you that it is hard to look at costs without looking at quality. Therein lies the basis for our nation’s move toward value-based purchasing of healthcare (see “Value-Based Purchasing Raises the Stakes,” May 2011).
When I hear hospitalists explain why the role of hospitalists was developed, the explanation often involves some discussion of cost and LOS reduction. Don’t get me wrong; it’s not that I believe HM has focused too much attention on cost reduction. I believe we have not focused enough on improving quality. This should not be surprising. Moving the bar on cost reduction is a lot easier than moving the bar on quality and patient safety. The first step toward improvement is an understanding of what you are doing currently. If your hospitalist group has not implemented a program to help its hospitalists measure the quality of care being provided, again, this is your wake-up call.
Last, but not least, for those of you who are not “surprised” by the results because of the belief that hospitalists were created to help the hospital save money and nothing more, I could not disagree with you more. I look at the roles that hospitalists have taken on in our nation’s hospitals, and I am incredibly proud to call myself a hospitalist.
Hospitalists are providing timely care when patients need it. Hospitalists are caring for patients without PCPs. Not only do hospitalists allow PCPs to provide more care in their outpatient clinics, but hospitalists also are caring for patients in ICUs in many places where there are not enough doctors sufficiently trained in critical care.
Rather than acting as an indictment on HM, I believe the Annals article makes a comment on the misalignment of incentives in our healthcare system.
It is 2011, not 1996; HM is here to stay. Most acute-care hospitals in America could not function without hospitalists. I applaud Kuo and Goodwin for doing the research and publishing their results. Let this be an opportunity for hospitalists around the country to think about how to implement systems to improve transitions of care and the quality of care we provide.
Dr. Li is president of SHM.
Reference
For those who say they would pay $50 more per patient if the quality is better, here’s the problem: Show me the data that say hospitalist care is higher-quality.
I suspect most of you have reviewed the study or at least heard about it. Bob Wachter, MD, MHM, blogged about the study. An article about the study appeared in American Medical Association News. Even National Public Radio ran a piece about the study on their show “Morning Edition.”
I am, of course, referring to the study by Kuo and Goodwin, which was published in the Annals of Internal Medicine in early August.1
In this study, the authors looked at a sample of patients (5%) with primary-care physicians (PCPs) enrolled in Medicare who were cared for by their PCP or a hospitalist during a period from 2001 to 2006. The authors stated their underlying hypotheses as:
- Hospitalist care would be associated with costs shifting from the hospital to the post-hospital setting;
- Hospitalist care would be associated with a decrease in discharges directly to home; and
- Discontinuities of care associated with hospitalist care would lead to a greater rate of visits to the emergency room and readmissions to the hospital, resulting in increased Medicare costs.
Did the authors say hospitalist care cost more? They can’t possibly be correct, can they? Don’t all the hospitalist studies show that hospitalists provide the same quality of care as primary-care doctors, except the costs are lower and the hospital length of stay (LOS) is shorter when hospitalists care for patients?
The point here is that these investigators look at the care not only during a patient’s hospital stay, but also for 30 days after discharge. This is something that had not been done previously—at least not on this scale.
Focus on Facts
And what did the authors find? Patients cared for by hospitalists, as compared to their PCPs, had a shorter LOS and lower in-hospital costs, but these patients also were less likely to be discharged directly to home, less likely to see their PCPs post-discharge, and had more hospital readmissions, ED visits, and nursing home visits after discharge.
Since its release two months ago, I have heard a lot of discussion about the study. Here are a few of the comments I’ve heard:
- “This was an observational study. You can’t possibly remove all confounders in an observational study.”
- “The authors looked at a time period early in the hospitalist movement. If they did the study today, the results would be different.”
- “The additional costs hospitalists incurred were only $50 per patient. Wouldn’t you pay $50 more if the care was better?”
- “This is why hospitals hired hospitalists. They save money for the hospitals. What did they expect to find?”
I agree that observational studies have limitations (even the authors acknowledged this), but this doesn’t mean results from observational studies are invalid. Some of us don’t want to hear this, but this actually was a pretty well-done study with a robust statistical analysis. We should recognize the study has limitations and think about the results.
Kuo and Goodwin looked at data during a period of time early in the hospitalist movement; the results could be different if the study were to be repeated today. But we don’t know what the data would be today. I suppose the data could be better, worse, or about the same. The fact of the matter is that HM leaders—and most of the rest of us—knew that transitions of care, under the hospitalist model, were a potential weakness. How many times have you heard Win Whitcomb, MD, MHM, and John Nelson, MD, MHM, talk about the potential “voltage drop” with handoffs?
The good news is that leaders in our field have done something about this. Project BOOST (Better Outcomes for Older Adults through Safer Transitions) is a program SHM has helped implement at dozens of hospitals across the country to address the issue of unnecessary hospital readmissions (www.hospitalmedicine. org/boost). Improving transitions of care and preventing unnecessary readmissions should be on the minds of all hospitalists. If your program and your hospital have not yet taken steps to address this issue, please let this be your wake-up call.
Show Me the Money
For those who say they would pay $50 more per patient if the quality is better, here’s the problem: Show me the data that say hospitalist care is higher-quality. I agree with you that it is hard to look at costs without looking at quality. Therein lies the basis for our nation’s move toward value-based purchasing of healthcare (see “Value-Based Purchasing Raises the Stakes,” May 2011).
When I hear hospitalists explain why the role of hospitalists was developed, the explanation often involves some discussion of cost and LOS reduction. Don’t get me wrong; it’s not that I believe HM has focused too much attention on cost reduction. I believe we have not focused enough on improving quality. This should not be surprising. Moving the bar on cost reduction is a lot easier than moving the bar on quality and patient safety. The first step toward improvement is an understanding of what you are doing currently. If your hospitalist group has not implemented a program to help its hospitalists measure the quality of care being provided, again, this is your wake-up call.
Last, but not least, for those of you who are not “surprised” by the results because of the belief that hospitalists were created to help the hospital save money and nothing more, I could not disagree with you more. I look at the roles that hospitalists have taken on in our nation’s hospitals, and I am incredibly proud to call myself a hospitalist.
Hospitalists are providing timely care when patients need it. Hospitalists are caring for patients without PCPs. Not only do hospitalists allow PCPs to provide more care in their outpatient clinics, but hospitalists also are caring for patients in ICUs in many places where there are not enough doctors sufficiently trained in critical care.
Rather than acting as an indictment on HM, I believe the Annals article makes a comment on the misalignment of incentives in our healthcare system.
It is 2011, not 1996; HM is here to stay. Most acute-care hospitals in America could not function without hospitalists. I applaud Kuo and Goodwin for doing the research and publishing their results. Let this be an opportunity for hospitalists around the country to think about how to implement systems to improve transitions of care and the quality of care we provide.
Dr. Li is president of SHM.
Reference
A Run for Safety

It was when my lung fell out that it hit me. No, come to think of it, it was before that, when a scorpion struck my left calf. Or it might have been when my heart exploded. No, it was earlier than that—probably around the time my right lower abdominal quadrant was gutted by that wild boar. No, actually, it was even earlier than that. Somewhere around the time I pulled my 204th muscle. Yeah, that was it. That’s when I first wondered why there is no “fun run” for safety.
Truth be told, there was no de-lunging, scorpions, cardiac explosion, or wild-boar-goring. It just felt like that. The reason: I was running. The occasion: an annual fun (?) run to support Crohn’s disease and ulcerative colitis. Why I, an out-of-shape specimen blessed with a superhero-like affinity for both chips and the couch, should be pounding the pavement early on a Saturday morning is a case study in wifely nagging.
And misplaced healthcare priorities.
You see, I have neither Crohn’s nor ulcerative colitis, have no friends or family members with them, and, frankly, rarely even provide care for patients with these diseases. What I do have is a gastroenterologist for a wife. A gastroenterologist who passionately supports gastroenterological problems; a gastroenterologist who doesn’t herself like to participate in fun runs; a gastroenterologist who relishes, apparently, seeing her husband sweat lactate while testing the anaerobic limits of the human organism. This was the nagging part.
As the gun reported at 7:30 a.m., there I was, fidgeting nervously at the starting line in a moth-eaten cotton tee from the last road race I had run—in 1989—while those around me gave my too-short, reversible, blue-and-white gym shorts the up-and-down. Cotton socks crotched, feet pre-blistered, I departed, feeling good—for the first four meters.
The next 4.99 kilometers proved slightly more daunting—providing an abundance of K’s to ponder the misplaced healthcare priorities part.
Running in The Trees
After I expertly buried the first 100-meter downhill, the race entered a well-worn, tree-lined footpath. I was shocked by both the splendor of the environs as well as the hordes of people passing me. I was comfortable with the concept of the taut young adults leaving me in their dust and, even, sort of, the superiorly fit elders. The pre-teens were more unsettling. As were the walkers—especially the walker using a walker.
It’s interesting, the relationship between road races and medical diseases. It’s not surprising, really, that generally healthy specimens would band together and use exercise as a weapon against disease—it’s actually quite noble. And common. My guess is your hometown counts numerous foot, bike, and foot-and-bike races supporting the eradication of myriad medical maladies.
In the span of just a few months, I’ve noted local races raising awareness of neurologic disorders (multiple sclerosis, Alzheimer’s, stroke, spinal muscular atrophy), cancer (breast, prostate, lung, leukemia, lymphoma, colon, skin, sarcoma, carcinoid), infectious disease (HIV/AIDS), and other medical conditions or causes (cystic fibrosis, cleft palate, pre-eclampsia, transplant, veterans).
Now, don’t get me wrong: I fully support any fund- or awareness-raising events targeting specific diseases or causes. In fact, if I were only slightly less slothlike, I’d participate in more of them. It’s just that in the grand scheme of things, it seems we are missing the forest through the trees. Finding a cure for cancer will matter little if we can’t deliver that cure in a safe, efficient, high-quality manner. Put another way, we can’t cure cancer patients if our health delivery system kills them first.
Seeing the Forest
And kill them we do. Now, you may not like the word “kill,” and certainly it makes me uncomfortable, but what other word better characterizes the situation? Medical errors result in up to 200,000 preventable deaths per year, according to the recent HealthGrades patient safety report.1 This study reviewed Medicare data from all 50 states and found a mortality rate that was nearly double that reported in the seminal 1999 Institute of Medicine report (44,000-98,000; extrapolated from data in three states).2
And these are just deaths in hospitals; no mention is made of community or residential deaths from medical error. These data also don’t account for the pain and suffering left in the wake of the estimated 15 million annual episodes of harm (that’s 40,000 per day!).
In the end, the World Health Organization (WHO) estimates that 10% of hospital stays involve a serious, preventable, adverse event. Which of the 10 patients you’ll see tomorrow will suffer that serious, PREVENTABLE harm?
Using a conservative average of the two reports, roughly 100,000 people die annually from hospital-based medical errors. This slots medical error as the sixth-most-common cause of death in the U.S., trailing only heart disease (616,067), cancer (562,875), stroke (135,952), chronic lower respiratory disease (127,924), and accidents (123,706). If we use the 200,000 estimate, then error trails only the heart and cancer as a cause of death. And, in terms of individual cancers, only the lung (156,940) kills as many Americans as medical errors. Colorectal (49,380), breast (39,970), and prostate (33,660) don’t even come close.
Yet these data appear to be lost on the legions of race organizers. A Web search uncovered not a single organized race event trying to counter the perils of medical error. No Lance Armstrong, no Katie Couric, no Jerry Lewis. Nothing.
Thankfully, these data are not lost on the ones who bear the brunt of these errors. A Commonwealth survey reported that 22% of respondents were aware of a medical error in care provided to them or their family. Another paper following the release of the IOM report put the number at 42%.3,4
Still, nary a race “K” has been devoted to reducing medical errors.
Harriers Against Harm
As the finish line draws near, I note that the overhead scoreboard has taken on the appearance of the national debt clock in Manhattan—a large number rapidly getting larger. The replenishment table is littered with crumpled Dixie cups, the music has drifted, and the crowd has dwindled to a handful of volunteers, many of whom tap their toes awaiting my finish.
I wonder what it’ll take. If 12,000 people with spinal muscular atrophy is enough to convene a race, what of the millions of people harmed annually by medical errors? How many more have to die before patient safety becomes an issue, becomes it’s own cause, gets it own fun run?
Dr. Glasheen is associate professor of medicine at the University of Colorado at Denver,where he serves as director of the Hospital Medicine Program and the Hospitalist Training Program, and as associate program director of the Internal Medicine Residency Program.
References
- HealthGrades Eighth Annual Report on Patient Safety in American Hospitals Study. Available at: www.healthgrades.com. Published March 2011. Accessed Aug. 31, 2011.
- Kohn LT, Corrigan JM, Donaldson MS, et al. To Err Is Human: Building a Safer Health System. Washington, D.C.: National Academies Press, 2000.
- The Commonwealth Fund 2002 Annual Report. The Commonwealth Fund website. Available at: http://www.commonwealthfund.org/Content/Annual-Reports/2002-Annual-Report.aspx. Accessed Sept. 9, 2011.
- Blendon RJ, DesRoches CM, Brodie M, et al. Views of practicing physicians and the public on medical errors. N Engl J Med. 2002;347:1933-1940.
It was when my lung fell out that it hit me. No, come to think of it, it was before that, when a scorpion struck my left calf. Or it might have been when my heart exploded. No, it was earlier than that—probably around the time my right lower abdominal quadrant was gutted by that wild boar. No, actually, it was even earlier than that. Somewhere around the time I pulled my 204th muscle. Yeah, that was it. That’s when I first wondered why there is no “fun run” for safety.
Truth be told, there was no de-lunging, scorpions, cardiac explosion, or wild-boar-goring. It just felt like that. The reason: I was running. The occasion: an annual fun (?) run to support Crohn’s disease and ulcerative colitis. Why I, an out-of-shape specimen blessed with a superhero-like affinity for both chips and the couch, should be pounding the pavement early on a Saturday morning is a case study in wifely nagging.
And misplaced healthcare priorities.
You see, I have neither Crohn’s nor ulcerative colitis, have no friends or family members with them, and, frankly, rarely even provide care for patients with these diseases. What I do have is a gastroenterologist for a wife. A gastroenterologist who passionately supports gastroenterological problems; a gastroenterologist who doesn’t herself like to participate in fun runs; a gastroenterologist who relishes, apparently, seeing her husband sweat lactate while testing the anaerobic limits of the human organism. This was the nagging part.
As the gun reported at 7:30 a.m., there I was, fidgeting nervously at the starting line in a moth-eaten cotton tee from the last road race I had run—in 1989—while those around me gave my too-short, reversible, blue-and-white gym shorts the up-and-down. Cotton socks crotched, feet pre-blistered, I departed, feeling good—for the first four meters.
The next 4.99 kilometers proved slightly more daunting—providing an abundance of K’s to ponder the misplaced healthcare priorities part.
Running in The Trees
After I expertly buried the first 100-meter downhill, the race entered a well-worn, tree-lined footpath. I was shocked by both the splendor of the environs as well as the hordes of people passing me. I was comfortable with the concept of the taut young adults leaving me in their dust and, even, sort of, the superiorly fit elders. The pre-teens were more unsettling. As were the walkers—especially the walker using a walker.
It’s interesting, the relationship between road races and medical diseases. It’s not surprising, really, that generally healthy specimens would band together and use exercise as a weapon against disease—it’s actually quite noble. And common. My guess is your hometown counts numerous foot, bike, and foot-and-bike races supporting the eradication of myriad medical maladies.
In the span of just a few months, I’ve noted local races raising awareness of neurologic disorders (multiple sclerosis, Alzheimer’s, stroke, spinal muscular atrophy), cancer (breast, prostate, lung, leukemia, lymphoma, colon, skin, sarcoma, carcinoid), infectious disease (HIV/AIDS), and other medical conditions or causes (cystic fibrosis, cleft palate, pre-eclampsia, transplant, veterans).
Now, don’t get me wrong: I fully support any fund- or awareness-raising events targeting specific diseases or causes. In fact, if I were only slightly less slothlike, I’d participate in more of them. It’s just that in the grand scheme of things, it seems we are missing the forest through the trees. Finding a cure for cancer will matter little if we can’t deliver that cure in a safe, efficient, high-quality manner. Put another way, we can’t cure cancer patients if our health delivery system kills them first.
Seeing the Forest
And kill them we do. Now, you may not like the word “kill,” and certainly it makes me uncomfortable, but what other word better characterizes the situation? Medical errors result in up to 200,000 preventable deaths per year, according to the recent HealthGrades patient safety report.1 This study reviewed Medicare data from all 50 states and found a mortality rate that was nearly double that reported in the seminal 1999 Institute of Medicine report (44,000-98,000; extrapolated from data in three states).2
And these are just deaths in hospitals; no mention is made of community or residential deaths from medical error. These data also don’t account for the pain and suffering left in the wake of the estimated 15 million annual episodes of harm (that’s 40,000 per day!).
In the end, the World Health Organization (WHO) estimates that 10% of hospital stays involve a serious, preventable, adverse event. Which of the 10 patients you’ll see tomorrow will suffer that serious, PREVENTABLE harm?
Using a conservative average of the two reports, roughly 100,000 people die annually from hospital-based medical errors. This slots medical error as the sixth-most-common cause of death in the U.S., trailing only heart disease (616,067), cancer (562,875), stroke (135,952), chronic lower respiratory disease (127,924), and accidents (123,706). If we use the 200,000 estimate, then error trails only the heart and cancer as a cause of death. And, in terms of individual cancers, only the lung (156,940) kills as many Americans as medical errors. Colorectal (49,380), breast (39,970), and prostate (33,660) don’t even come close.
Yet these data appear to be lost on the legions of race organizers. A Web search uncovered not a single organized race event trying to counter the perils of medical error. No Lance Armstrong, no Katie Couric, no Jerry Lewis. Nothing.
Thankfully, these data are not lost on the ones who bear the brunt of these errors. A Commonwealth survey reported that 22% of respondents were aware of a medical error in care provided to them or their family. Another paper following the release of the IOM report put the number at 42%.3,4
Still, nary a race “K” has been devoted to reducing medical errors.
Harriers Against Harm
As the finish line draws near, I note that the overhead scoreboard has taken on the appearance of the national debt clock in Manhattan—a large number rapidly getting larger. The replenishment table is littered with crumpled Dixie cups, the music has drifted, and the crowd has dwindled to a handful of volunteers, many of whom tap their toes awaiting my finish.
I wonder what it’ll take. If 12,000 people with spinal muscular atrophy is enough to convene a race, what of the millions of people harmed annually by medical errors? How many more have to die before patient safety becomes an issue, becomes it’s own cause, gets it own fun run?
Dr. Glasheen is associate professor of medicine at the University of Colorado at Denver,where he serves as director of the Hospital Medicine Program and the Hospitalist Training Program, and as associate program director of the Internal Medicine Residency Program.
References
- HealthGrades Eighth Annual Report on Patient Safety in American Hospitals Study. Available at: www.healthgrades.com. Published March 2011. Accessed Aug. 31, 2011.
- Kohn LT, Corrigan JM, Donaldson MS, et al. To Err Is Human: Building a Safer Health System. Washington, D.C.: National Academies Press, 2000.
- The Commonwealth Fund 2002 Annual Report. The Commonwealth Fund website. Available at: http://www.commonwealthfund.org/Content/Annual-Reports/2002-Annual-Report.aspx. Accessed Sept. 9, 2011.
- Blendon RJ, DesRoches CM, Brodie M, et al. Views of practicing physicians and the public on medical errors. N Engl J Med. 2002;347:1933-1940.
It was when my lung fell out that it hit me. No, come to think of it, it was before that, when a scorpion struck my left calf. Or it might have been when my heart exploded. No, it was earlier than that—probably around the time my right lower abdominal quadrant was gutted by that wild boar. No, actually, it was even earlier than that. Somewhere around the time I pulled my 204th muscle. Yeah, that was it. That’s when I first wondered why there is no “fun run” for safety.
Truth be told, there was no de-lunging, scorpions, cardiac explosion, or wild-boar-goring. It just felt like that. The reason: I was running. The occasion: an annual fun (?) run to support Crohn’s disease and ulcerative colitis. Why I, an out-of-shape specimen blessed with a superhero-like affinity for both chips and the couch, should be pounding the pavement early on a Saturday morning is a case study in wifely nagging.
And misplaced healthcare priorities.
You see, I have neither Crohn’s nor ulcerative colitis, have no friends or family members with them, and, frankly, rarely even provide care for patients with these diseases. What I do have is a gastroenterologist for a wife. A gastroenterologist who passionately supports gastroenterological problems; a gastroenterologist who doesn’t herself like to participate in fun runs; a gastroenterologist who relishes, apparently, seeing her husband sweat lactate while testing the anaerobic limits of the human organism. This was the nagging part.
As the gun reported at 7:30 a.m., there I was, fidgeting nervously at the starting line in a moth-eaten cotton tee from the last road race I had run—in 1989—while those around me gave my too-short, reversible, blue-and-white gym shorts the up-and-down. Cotton socks crotched, feet pre-blistered, I departed, feeling good—for the first four meters.
The next 4.99 kilometers proved slightly more daunting—providing an abundance of K’s to ponder the misplaced healthcare priorities part.
Running in The Trees
After I expertly buried the first 100-meter downhill, the race entered a well-worn, tree-lined footpath. I was shocked by both the splendor of the environs as well as the hordes of people passing me. I was comfortable with the concept of the taut young adults leaving me in their dust and, even, sort of, the superiorly fit elders. The pre-teens were more unsettling. As were the walkers—especially the walker using a walker.
It’s interesting, the relationship between road races and medical diseases. It’s not surprising, really, that generally healthy specimens would band together and use exercise as a weapon against disease—it’s actually quite noble. And common. My guess is your hometown counts numerous foot, bike, and foot-and-bike races supporting the eradication of myriad medical maladies.
In the span of just a few months, I’ve noted local races raising awareness of neurologic disorders (multiple sclerosis, Alzheimer’s, stroke, spinal muscular atrophy), cancer (breast, prostate, lung, leukemia, lymphoma, colon, skin, sarcoma, carcinoid), infectious disease (HIV/AIDS), and other medical conditions or causes (cystic fibrosis, cleft palate, pre-eclampsia, transplant, veterans).
Now, don’t get me wrong: I fully support any fund- or awareness-raising events targeting specific diseases or causes. In fact, if I were only slightly less slothlike, I’d participate in more of them. It’s just that in the grand scheme of things, it seems we are missing the forest through the trees. Finding a cure for cancer will matter little if we can’t deliver that cure in a safe, efficient, high-quality manner. Put another way, we can’t cure cancer patients if our health delivery system kills them first.
Seeing the Forest
And kill them we do. Now, you may not like the word “kill,” and certainly it makes me uncomfortable, but what other word better characterizes the situation? Medical errors result in up to 200,000 preventable deaths per year, according to the recent HealthGrades patient safety report.1 This study reviewed Medicare data from all 50 states and found a mortality rate that was nearly double that reported in the seminal 1999 Institute of Medicine report (44,000-98,000; extrapolated from data in three states).2
And these are just deaths in hospitals; no mention is made of community or residential deaths from medical error. These data also don’t account for the pain and suffering left in the wake of the estimated 15 million annual episodes of harm (that’s 40,000 per day!).
In the end, the World Health Organization (WHO) estimates that 10% of hospital stays involve a serious, preventable, adverse event. Which of the 10 patients you’ll see tomorrow will suffer that serious, PREVENTABLE harm?
Using a conservative average of the two reports, roughly 100,000 people die annually from hospital-based medical errors. This slots medical error as the sixth-most-common cause of death in the U.S., trailing only heart disease (616,067), cancer (562,875), stroke (135,952), chronic lower respiratory disease (127,924), and accidents (123,706). If we use the 200,000 estimate, then error trails only the heart and cancer as a cause of death. And, in terms of individual cancers, only the lung (156,940) kills as many Americans as medical errors. Colorectal (49,380), breast (39,970), and prostate (33,660) don’t even come close.
Yet these data appear to be lost on the legions of race organizers. A Web search uncovered not a single organized race event trying to counter the perils of medical error. No Lance Armstrong, no Katie Couric, no Jerry Lewis. Nothing.
Thankfully, these data are not lost on the ones who bear the brunt of these errors. A Commonwealth survey reported that 22% of respondents were aware of a medical error in care provided to them or their family. Another paper following the release of the IOM report put the number at 42%.3,4
Still, nary a race “K” has been devoted to reducing medical errors.
Harriers Against Harm
As the finish line draws near, I note that the overhead scoreboard has taken on the appearance of the national debt clock in Manhattan—a large number rapidly getting larger. The replenishment table is littered with crumpled Dixie cups, the music has drifted, and the crowd has dwindled to a handful of volunteers, many of whom tap their toes awaiting my finish.
I wonder what it’ll take. If 12,000 people with spinal muscular atrophy is enough to convene a race, what of the millions of people harmed annually by medical errors? How many more have to die before patient safety becomes an issue, becomes it’s own cause, gets it own fun run?
Dr. Glasheen is associate professor of medicine at the University of Colorado at Denver,where he serves as director of the Hospital Medicine Program and the Hospitalist Training Program, and as associate program director of the Internal Medicine Residency Program.
References
- HealthGrades Eighth Annual Report on Patient Safety in American Hospitals Study. Available at: www.healthgrades.com. Published March 2011. Accessed Aug. 31, 2011.
- Kohn LT, Corrigan JM, Donaldson MS, et al. To Err Is Human: Building a Safer Health System. Washington, D.C.: National Academies Press, 2000.
- The Commonwealth Fund 2002 Annual Report. The Commonwealth Fund website. Available at: http://www.commonwealthfund.org/Content/Annual-Reports/2002-Annual-Report.aspx. Accessed Sept. 9, 2011.
- Blendon RJ, DesRoches CM, Brodie M, et al. Views of practicing physicians and the public on medical errors. N Engl J Med. 2002;347:1933-1940.
Laborists, Defined

Last month (see “Hospital-Focused Practice,” Septem-ber 2011, p. 61), I discussed the adoption of the hospitalist model of practice by many specialties, some of the common issues they face, and highlighted a national meeting to examine this phenomenon (for more information on the meeting, visit www.hospitalmedicine/hfpm). This month, relying mostly on my own experience with this practice model, I’ll drill deeper into OB hospitalists (also known as laborists). While there are a lot of ways in which hospitalist practice in many specialties are the same, laborists differ from those in other fields in important and interesting ways.
Prevalence
One of the most informative sources about the “laborist movement” is ObGynHospitalist.com, a website started and managed by Dr. Rob Olson, an enterprising laborist in Bellingham, Wash. As of July, the site listed 132 laborist programs nationwide (and that figure likely underestimates the actual number in operation). A survey of registered users of the website in April yielded 106 responses, representing a 24% response rate. Seventy-five of the respondents indicated they were full-time laborists.
Unique Drivers
Because obstetric malpractice costs are so high, and many lawsuits are related to delayed response to obstetric emergencies, there is hope (not much hard proof yet) that outcomes will be better, and lawsuits less common or less costly.1 So the hope of reduced malpractice costs figures more prominently into the cost-benefit analysis of the OB hospitalist model than most other types of HM practice.
Financial Model
It appears that all hospitalist models require financial support over and above professional fee revenue. Hospitals usually are willing (happy?) to provide this money because they can make back even more as a result of increased patient volume/market share or lower costs. And, as is the case for hospitalists in other specialties, laborist presence can be an asset in recruitment and retention of other OBGYNs.
I think the most interesting feature of laborist practice is that in many settings, it has the potential to open new sources of revenue—both hospital “facility fee” and professional fee revenue. A common practice in many hospitals is for obstetricians to send patients, or for them to self-present, to labor and delivery to be checked for a cold, vomiting, or whether labor has started. Many times, a nurse performs these checks, communicates with a doctor, then discharges the patient—and no bill is generated. An on-site laborist can see the same patients (presumably making for a higher-quality visit for the patient) and, assuming the visit is medically necessary, both a facility and professional charge can be submitted. Revenue from such visits can go a long way toward making up the difference between the total cost of the laborist program and fee collections. This adds to patient safety, as each patient is evaluated in person by a physician rather than only a nurse.
In most settings, the laborist submits a charge for delivery only for unassigned patients. For those patients who “belong to” another OB who provided prenatal care, it is often most practical for that doctor to submit the global fee for prenatal care and delivery, and to pay the laborist program an agreed-upon rate for each service provided.
Compensation
Laborists often are paid an hourly rate, and they typically don’t have a salary component tied to work relative-value unit (wRVU) production or other productivity metrics. Total annual compensation is typically lower than private-practice OBGYN physicians. It also varies widely, depending on local market forces, job description, and workload. Most programs are trying to implement meaningful quality bonuses for laborists.
Scope of Practice
Laborists typically provide care to all unassigned patients who present to labor and delivery, and perform deliveries, C-sections, and other services on patients when requested by OBs in traditional practice. Requests arise when an OB simply needs to be relieved of being on call for their private patients, or when an emergency arises. (These “as-needed” referrals are different from the most common arrangement for “medical hospitalist” practices that ask other doctors to refer all or none of their patients, not just when they are otherwise occupied.)
Lastly, the laborist might serve as surgical assistant to other OBGYNs. In nearly all settings, there is no need to require that any physicians refer to the laborist, and the other OBs are free to decide when to refer.
A reasonably common scenario is that, to avoid disruption of scheduled office hours, an OB in traditional practice might ask that the laborist manage a patient who presents in labor. But if still undelivered at the close of office hours, the traditional OB might assume care from that point on or have the laborist remain responsible through delivery. The traditional OB usually will make post-partum “rounding” visits on all of their patients but could rely on the laborist for these visits.
In most cases, the laborist does not have any scheduled gynecologic procedures, though he or she may see GYN consults throughout the hospital as time permits. Laborists typically have no outpatient responsibilities, but some OBGYN hospitalists cover GYN in the ED.
Operational Structure
Although models vary significantly, the single most common arrangement is for laborists to work 24-hour, in-house shifts. Rarely is there a need or justification to have more than one laborist on at a time. For a single physician, seven or eight 24-hour shifts per month is considered full-time. My experience is that most laborists are employed by the hospital in which they work.
As is the case in every specialty, some large OBGYN groups adopt a rotating laborist model, in which one member of their group becomes the laborist for 24 hours at a time, during which they are relieved of all other responsibilities.
Recruitment
ObGynHospitalist.com shows that, as of July, 40 of the 132 laborist programs that had identified themselves on the site were recruiting. My experience is that unlike “medical hospitalist” practices, which tend to successfully recruit those very early in their career, or “surgical hospitalist” programs, which target mid- to late-career general surgeons, laborist candidates come from any point in their careers. Most programs prefer that a laborist has several years of post-residency experience, but they generally have no other preference.
Dr. Nelson has been a practicing hospitalist since 1988 and is cofounder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is course codirector and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
Reference
Last month (see “Hospital-Focused Practice,” Septem-ber 2011, p. 61), I discussed the adoption of the hospitalist model of practice by many specialties, some of the common issues they face, and highlighted a national meeting to examine this phenomenon (for more information on the meeting, visit www.hospitalmedicine/hfpm). This month, relying mostly on my own experience with this practice model, I’ll drill deeper into OB hospitalists (also known as laborists). While there are a lot of ways in which hospitalist practice in many specialties are the same, laborists differ from those in other fields in important and interesting ways.
Prevalence
One of the most informative sources about the “laborist movement” is ObGynHospitalist.com, a website started and managed by Dr. Rob Olson, an enterprising laborist in Bellingham, Wash. As of July, the site listed 132 laborist programs nationwide (and that figure likely underestimates the actual number in operation). A survey of registered users of the website in April yielded 106 responses, representing a 24% response rate. Seventy-five of the respondents indicated they were full-time laborists.
Unique Drivers
Because obstetric malpractice costs are so high, and many lawsuits are related to delayed response to obstetric emergencies, there is hope (not much hard proof yet) that outcomes will be better, and lawsuits less common or less costly.1 So the hope of reduced malpractice costs figures more prominently into the cost-benefit analysis of the OB hospitalist model than most other types of HM practice.
Financial Model
It appears that all hospitalist models require financial support over and above professional fee revenue. Hospitals usually are willing (happy?) to provide this money because they can make back even more as a result of increased patient volume/market share or lower costs. And, as is the case for hospitalists in other specialties, laborist presence can be an asset in recruitment and retention of other OBGYNs.
I think the most interesting feature of laborist practice is that in many settings, it has the potential to open new sources of revenue—both hospital “facility fee” and professional fee revenue. A common practice in many hospitals is for obstetricians to send patients, or for them to self-present, to labor and delivery to be checked for a cold, vomiting, or whether labor has started. Many times, a nurse performs these checks, communicates with a doctor, then discharges the patient—and no bill is generated. An on-site laborist can see the same patients (presumably making for a higher-quality visit for the patient) and, assuming the visit is medically necessary, both a facility and professional charge can be submitted. Revenue from such visits can go a long way toward making up the difference between the total cost of the laborist program and fee collections. This adds to patient safety, as each patient is evaluated in person by a physician rather than only a nurse.
In most settings, the laborist submits a charge for delivery only for unassigned patients. For those patients who “belong to” another OB who provided prenatal care, it is often most practical for that doctor to submit the global fee for prenatal care and delivery, and to pay the laborist program an agreed-upon rate for each service provided.
Compensation
Laborists often are paid an hourly rate, and they typically don’t have a salary component tied to work relative-value unit (wRVU) production or other productivity metrics. Total annual compensation is typically lower than private-practice OBGYN physicians. It also varies widely, depending on local market forces, job description, and workload. Most programs are trying to implement meaningful quality bonuses for laborists.
Scope of Practice
Laborists typically provide care to all unassigned patients who present to labor and delivery, and perform deliveries, C-sections, and other services on patients when requested by OBs in traditional practice. Requests arise when an OB simply needs to be relieved of being on call for their private patients, or when an emergency arises. (These “as-needed” referrals are different from the most common arrangement for “medical hospitalist” practices that ask other doctors to refer all or none of their patients, not just when they are otherwise occupied.)
Lastly, the laborist might serve as surgical assistant to other OBGYNs. In nearly all settings, there is no need to require that any physicians refer to the laborist, and the other OBs are free to decide when to refer.
A reasonably common scenario is that, to avoid disruption of scheduled office hours, an OB in traditional practice might ask that the laborist manage a patient who presents in labor. But if still undelivered at the close of office hours, the traditional OB might assume care from that point on or have the laborist remain responsible through delivery. The traditional OB usually will make post-partum “rounding” visits on all of their patients but could rely on the laborist for these visits.
In most cases, the laborist does not have any scheduled gynecologic procedures, though he or she may see GYN consults throughout the hospital as time permits. Laborists typically have no outpatient responsibilities, but some OBGYN hospitalists cover GYN in the ED.
Operational Structure
Although models vary significantly, the single most common arrangement is for laborists to work 24-hour, in-house shifts. Rarely is there a need or justification to have more than one laborist on at a time. For a single physician, seven or eight 24-hour shifts per month is considered full-time. My experience is that most laborists are employed by the hospital in which they work.
As is the case in every specialty, some large OBGYN groups adopt a rotating laborist model, in which one member of their group becomes the laborist for 24 hours at a time, during which they are relieved of all other responsibilities.
Recruitment
ObGynHospitalist.com shows that, as of July, 40 of the 132 laborist programs that had identified themselves on the site were recruiting. My experience is that unlike “medical hospitalist” practices, which tend to successfully recruit those very early in their career, or “surgical hospitalist” programs, which target mid- to late-career general surgeons, laborist candidates come from any point in their careers. Most programs prefer that a laborist has several years of post-residency experience, but they generally have no other preference.
Dr. Nelson has been a practicing hospitalist since 1988 and is cofounder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is course codirector and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
Reference
Last month (see “Hospital-Focused Practice,” Septem-ber 2011, p. 61), I discussed the adoption of the hospitalist model of practice by many specialties, some of the common issues they face, and highlighted a national meeting to examine this phenomenon (for more information on the meeting, visit www.hospitalmedicine/hfpm). This month, relying mostly on my own experience with this practice model, I’ll drill deeper into OB hospitalists (also known as laborists). While there are a lot of ways in which hospitalist practice in many specialties are the same, laborists differ from those in other fields in important and interesting ways.
Prevalence
One of the most informative sources about the “laborist movement” is ObGynHospitalist.com, a website started and managed by Dr. Rob Olson, an enterprising laborist in Bellingham, Wash. As of July, the site listed 132 laborist programs nationwide (and that figure likely underestimates the actual number in operation). A survey of registered users of the website in April yielded 106 responses, representing a 24% response rate. Seventy-five of the respondents indicated they were full-time laborists.
Unique Drivers
Because obstetric malpractice costs are so high, and many lawsuits are related to delayed response to obstetric emergencies, there is hope (not much hard proof yet) that outcomes will be better, and lawsuits less common or less costly.1 So the hope of reduced malpractice costs figures more prominently into the cost-benefit analysis of the OB hospitalist model than most other types of HM practice.
Financial Model
It appears that all hospitalist models require financial support over and above professional fee revenue. Hospitals usually are willing (happy?) to provide this money because they can make back even more as a result of increased patient volume/market share or lower costs. And, as is the case for hospitalists in other specialties, laborist presence can be an asset in recruitment and retention of other OBGYNs.
I think the most interesting feature of laborist practice is that in many settings, it has the potential to open new sources of revenue—both hospital “facility fee” and professional fee revenue. A common practice in many hospitals is for obstetricians to send patients, or for them to self-present, to labor and delivery to be checked for a cold, vomiting, or whether labor has started. Many times, a nurse performs these checks, communicates with a doctor, then discharges the patient—and no bill is generated. An on-site laborist can see the same patients (presumably making for a higher-quality visit for the patient) and, assuming the visit is medically necessary, both a facility and professional charge can be submitted. Revenue from such visits can go a long way toward making up the difference between the total cost of the laborist program and fee collections. This adds to patient safety, as each patient is evaluated in person by a physician rather than only a nurse.
In most settings, the laborist submits a charge for delivery only for unassigned patients. For those patients who “belong to” another OB who provided prenatal care, it is often most practical for that doctor to submit the global fee for prenatal care and delivery, and to pay the laborist program an agreed-upon rate for each service provided.
Compensation
Laborists often are paid an hourly rate, and they typically don’t have a salary component tied to work relative-value unit (wRVU) production or other productivity metrics. Total annual compensation is typically lower than private-practice OBGYN physicians. It also varies widely, depending on local market forces, job description, and workload. Most programs are trying to implement meaningful quality bonuses for laborists.
Scope of Practice
Laborists typically provide care to all unassigned patients who present to labor and delivery, and perform deliveries, C-sections, and other services on patients when requested by OBs in traditional practice. Requests arise when an OB simply needs to be relieved of being on call for their private patients, or when an emergency arises. (These “as-needed” referrals are different from the most common arrangement for “medical hospitalist” practices that ask other doctors to refer all or none of their patients, not just when they are otherwise occupied.)
Lastly, the laborist might serve as surgical assistant to other OBGYNs. In nearly all settings, there is no need to require that any physicians refer to the laborist, and the other OBs are free to decide when to refer.
A reasonably common scenario is that, to avoid disruption of scheduled office hours, an OB in traditional practice might ask that the laborist manage a patient who presents in labor. But if still undelivered at the close of office hours, the traditional OB might assume care from that point on or have the laborist remain responsible through delivery. The traditional OB usually will make post-partum “rounding” visits on all of their patients but could rely on the laborist for these visits.
In most cases, the laborist does not have any scheduled gynecologic procedures, though he or she may see GYN consults throughout the hospital as time permits. Laborists typically have no outpatient responsibilities, but some OBGYN hospitalists cover GYN in the ED.
Operational Structure
Although models vary significantly, the single most common arrangement is for laborists to work 24-hour, in-house shifts. Rarely is there a need or justification to have more than one laborist on at a time. For a single physician, seven or eight 24-hour shifts per month is considered full-time. My experience is that most laborists are employed by the hospital in which they work.
As is the case in every specialty, some large OBGYN groups adopt a rotating laborist model, in which one member of their group becomes the laborist for 24 hours at a time, during which they are relieved of all other responsibilities.
Recruitment
ObGynHospitalist.com shows that, as of July, 40 of the 132 laborist programs that had identified themselves on the site were recruiting. My experience is that unlike “medical hospitalist” practices, which tend to successfully recruit those very early in their career, or “surgical hospitalist” programs, which target mid- to late-career general surgeons, laborist candidates come from any point in their careers. Most programs prefer that a laborist has several years of post-residency experience, but they generally have no other preference.
Dr. Nelson has been a practicing hospitalist since 1988 and is cofounder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is course codirector and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.