User login
Gene variants linked to drug intolerance

Photo courtesy of the CDC
New research has revealed inherited genetic variations that may predispose patients to severe toxicity from thiopurines, a class of medications used as anticancer and immunosuppressive drugs.
Investigators identified 4 variations in the NUDT15 gene that alter thiopurine metabolism, leaving patients particularly sensitive to the drugs and at risk for toxicity.
One in 3 Japanese patients in this study carried the variations.
And evidence suggests the variations are common in other populations across Asia and in individuals of Hispanic ethnicity.
Jun J. Yang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues reported these findings in Nature Genetics.
In 2015, Dr Yang and his colleagues published evidence linking a NUDT15 variant to reduced tolerance of mercaptopurine and reported the variant was more common in patients of East Asian ancestry.
With the current study, the investigators identified 3 additional NUDT15 variants and found that all 4 variants—p.Arg139Cys, p.Arg139His, p.Val18Ile, and p.Val18_Val19insGlyVal—were associated with lower levels of enzymatic activity and imbalance of thiopurine metabolism.
In a group of 270 children with acute lymphoblastic leukemia (ALL), the variants caused a 74.4% to 100% loss of NUDT15 function. They also predicted enzyme activity and mercaptopurine tolerance. In Singapore and Japan, for example, patients with the 2 highest risk variants had the lowest level of enzyme activity.
“These patients had excessive levels of the active drug metabolites per mercaptopurine dose, which suggests we may reduce the drug dose to achieve the level necessary to kill leukemia cells without causing toxicity,” Dr Yang said, adding that the NUDT15 variants have no other known health consequences.
The investigators also checked leukemic cells from 285 children newly diagnosed with ALL and found that patients with NUDT15 variants were more sensitive to thiopurines.
“That suggests we can screen for NUDT15 variants and potentially plan mercaptopurine doses according to each patient’s genotype before the therapy starts,” Dr Yang said. “This way, we hope to avoid toxicity without compromising treatment effectiveness.”
The investigators noted that future studies are needed to determine optimal thiopurine doses for patients with different NUDT15 variants.
Meanwhile, the search continues for variants in NUDT15 or other genes that influence chemotherapy effectiveness and safety. The NUDT15 variants and previously identified TPMT variants could not fully explain why Guatemalan patients in this study tolerated the lowest doses of mercaptopurine. ![]()
Photo courtesy of the CDC
New research has revealed inherited genetic variations that may predispose patients to severe toxicity from thiopurines, a class of medications used as anticancer and immunosuppressive drugs.
Investigators identified 4 variations in the NUDT15 gene that alter thiopurine metabolism, leaving patients particularly sensitive to the drugs and at risk for toxicity.
One in 3 Japanese patients in this study carried the variations.
And evidence suggests the variations are common in other populations across Asia and in individuals of Hispanic ethnicity.
Jun J. Yang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues reported these findings in Nature Genetics.
In 2015, Dr Yang and his colleagues published evidence linking a NUDT15 variant to reduced tolerance of mercaptopurine and reported the variant was more common in patients of East Asian ancestry.
With the current study, the investigators identified 3 additional NUDT15 variants and found that all 4 variants—p.Arg139Cys, p.Arg139His, p.Val18Ile, and p.Val18_Val19insGlyVal—were associated with lower levels of enzymatic activity and imbalance of thiopurine metabolism.
In a group of 270 children with acute lymphoblastic leukemia (ALL), the variants caused a 74.4% to 100% loss of NUDT15 function. They also predicted enzyme activity and mercaptopurine tolerance. In Singapore and Japan, for example, patients with the 2 highest risk variants had the lowest level of enzyme activity.
“These patients had excessive levels of the active drug metabolites per mercaptopurine dose, which suggests we may reduce the drug dose to achieve the level necessary to kill leukemia cells without causing toxicity,” Dr Yang said, adding that the NUDT15 variants have no other known health consequences.
The investigators also checked leukemic cells from 285 children newly diagnosed with ALL and found that patients with NUDT15 variants were more sensitive to thiopurines.
“That suggests we can screen for NUDT15 variants and potentially plan mercaptopurine doses according to each patient’s genotype before the therapy starts,” Dr Yang said. “This way, we hope to avoid toxicity without compromising treatment effectiveness.”
The investigators noted that future studies are needed to determine optimal thiopurine doses for patients with different NUDT15 variants.
Meanwhile, the search continues for variants in NUDT15 or other genes that influence chemotherapy effectiveness and safety. The NUDT15 variants and previously identified TPMT variants could not fully explain why Guatemalan patients in this study tolerated the lowest doses of mercaptopurine. ![]()
Photo courtesy of the CDC
New research has revealed inherited genetic variations that may predispose patients to severe toxicity from thiopurines, a class of medications used as anticancer and immunosuppressive drugs.
Investigators identified 4 variations in the NUDT15 gene that alter thiopurine metabolism, leaving patients particularly sensitive to the drugs and at risk for toxicity.
One in 3 Japanese patients in this study carried the variations.
And evidence suggests the variations are common in other populations across Asia and in individuals of Hispanic ethnicity.
Jun J. Yang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues reported these findings in Nature Genetics.
In 2015, Dr Yang and his colleagues published evidence linking a NUDT15 variant to reduced tolerance of mercaptopurine and reported the variant was more common in patients of East Asian ancestry.
With the current study, the investigators identified 3 additional NUDT15 variants and found that all 4 variants—p.Arg139Cys, p.Arg139His, p.Val18Ile, and p.Val18_Val19insGlyVal—were associated with lower levels of enzymatic activity and imbalance of thiopurine metabolism.
In a group of 270 children with acute lymphoblastic leukemia (ALL), the variants caused a 74.4% to 100% loss of NUDT15 function. They also predicted enzyme activity and mercaptopurine tolerance. In Singapore and Japan, for example, patients with the 2 highest risk variants had the lowest level of enzyme activity.
“These patients had excessive levels of the active drug metabolites per mercaptopurine dose, which suggests we may reduce the drug dose to achieve the level necessary to kill leukemia cells without causing toxicity,” Dr Yang said, adding that the NUDT15 variants have no other known health consequences.
The investigators also checked leukemic cells from 285 children newly diagnosed with ALL and found that patients with NUDT15 variants were more sensitive to thiopurines.
“That suggests we can screen for NUDT15 variants and potentially plan mercaptopurine doses according to each patient’s genotype before the therapy starts,” Dr Yang said. “This way, we hope to avoid toxicity without compromising treatment effectiveness.”
The investigators noted that future studies are needed to determine optimal thiopurine doses for patients with different NUDT15 variants.
Meanwhile, the search continues for variants in NUDT15 or other genes that influence chemotherapy effectiveness and safety. The NUDT15 variants and previously identified TPMT variants could not fully explain why Guatemalan patients in this study tolerated the lowest doses of mercaptopurine. ![]()
The Clinical Learning Environment Review as a Model for Impactful Self-directed Quality Control Initiatives in Clinical Practice
As part of its Next Accreditation System, the Accreditation Council for Graduate Medical Education (ACGME) has introduced the Clinical Learning Environment Review (CLER) program, designed to assess the learning environment of institutions that have ACGME residency and fellowship programs.1 The CLER program emphasizes the responsibility of these hospitals, multispecialty groups, and other organizations to focus on quality and safety in the health care environment of resident learning and patient care. The expectation is that emphasis on quality of care in a residency training program will influence these physicians’ approach to quality of care after graduation.2,3 The Department of Dermatology at the University of Mississippi Medical Center (UMMC)(Jackson, Mississippi) saw CLER as an opportunity to demonstrate leadership in the patient safety movement.
CLER Program at UMMC
As a model CLER program at our institution, our project at the outset concentrated resident efforts on the focus areas specified by the ACGME (Table 1). We also were aware that our ACGME committee would need to answer questions during CLER site visits (Table 2). Because the data generated would not be used for accreditation decisions, there was no concern that exposing errors would jeopardize our postgraduate training certification.

The first 15 minutes of monthly faculty meetings were devoted to the presentation of a resident project, called a QA/QI (quality assurance/quality improvement) moment, that addressed ACGME focus areas 1, 2, 3, or 6 (Table 1). (Transitions in care [focus area 4] and work hours and fatigue [focus area 5] generally are less important issues in a predominantly outpatient specialty such as dermatology.) The residents were encouraged to identify areas where patient harm could occur due to poorly designed systems and to report situations in which patients actually were harmed.

Each project had to be approved by the department chairperson based on the following 4 requirements: First, the initiative must have the potential to notably impact patient safety and reduce harm. Second, residents with faculty support had to design methods to assess the identified problem. Third, participants had to design (to the best of their abilities) cost-effective and achievable interventions in a manner that would not produce unintended consequences. Fourth, residents were asked to devise a system to close the loop, ensuring that the effort put into the process was not wasted.
Findings From the CLER Program
The CLER program generates data on program and institutional attributes that have a salutatory effect on quality and safety, specifically involving 6 focus areas highlighted in Table 1. Putting residents at the center of efforts to improve the quality of care in our department proved critical to improving patient safety.
Involving residents in a series of QA/QI initiatives was logical because they rotate with faculty members. They also are in a position to view inconsistencies and to work to establish consistent patterns of patient care. In addition, our busy faculty members are charged with a variety of other clinical, educational, and administrative duties complicated by requirements in the design of a new residency training program. Faculty and residents working together were able to find problem areas in our department and devise solutions to improve those problems.
The CLER program involved a series of steps. Residents were charged with identifying errors (QA) and then devising a system to prevent similar errors from being repeated (QI)(Table 3). Efforts focused on preventing needless harm in our department. Initiatives developed by residents, who are closest to patients, have advantages over safety programs developed by the hospital’s administration. Residents became passionate about error prevention when they determined that their efforts could make a difference to patients.
Forward Thinking for Dermatology Practices
Perhaps there are lessons here that could apply to safety promotion in the practicing dermatologist’s office. The American Board of Dermatology, within the framework established by the American Board of Medical Specialties, requires physicians seeking recertification to participate in preapproved practice assessment QI exercises twice every 10 years.17 Six programs sponsored by the American Academy of Dermatology have now been approved in the areas of melanoma, biopsy follow-up measure, psoriasis, chronic urticaria, venous insufficiency, and laser- and light-based therapy for rejuvenation.18 An additional program has been approved for dermatopathologists through the American Society of Dermatopathology.19 None of these programs match the topics chosen by our residents in consultation with faculty to meet safety gaps identified in clinics at UMMC. Perhaps the next generation of performance improvement continuing medical education programs could include a pilot program for part 4 of Maintenance of Certification credit that is nonpunitive, patient focused, and allows dermatologists to design specific error-prevention solutions tailored to their individual practice in the same way residency programs are taking up this task.
- Nasca TJ, Philibert I, Brigham T, et al. The Next GME accreditation system—rationale and benefits. N Engl J Med. 2012;366:1051-1056.
- Philibert I, Gonzalez del Rey JA, Lannon C, et al. Quality improvement skills for pediatric residents: from lecture to implementation and sustainability. Acad Pediatr. 2014;14:40-46.
- Vidyarthi AR, Green AL, Rosenbluth G, et al. Engaging residents and fellows to improve institution-wide quality: the first six years of a novel financial incentive program. Acad Med. 2014;89:460-468.
- Brodell RT, Elewski B. Antifungal drug interactions. avoidance requires more than memorization. Postgrad Med. 2000;107:41-43.
- Kerr IG, Jolivet J, Collin JM, et al. Test dose for predicting high-dose methotrexate infusions. Clin Pharmacol Ther. 1983;33:44-51.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451-485.
- Saporito FC, Menter MA. Methotrexate and psoriasis in the era of new biologic agents. J Am Acad Dermatol. 2004;50:301-309.
- Van Der Sijs H, Aarts J, Vulto A, et al. Overriding of drug safety alerts in computerized physician order entry. J Am Med Inform Assoc. 2006;13:138-147.
- Hunter KM. Implementation of an electronic medication administration record and bedside verification system. Online J Nurs Inform (OJNI). 2011;15:672.
- Nanji KC, Slight SP, Seger DL, et al. Overrides of medication-related clinical decision support alerts in outpatients. J Am Med Inform Assoc. 2014;21:487-491.
- Schedlbauer A, Prasad V, Mulvaney C, et al. What evidence supports the use of computerized alerts and prompts to improve clinicians’ prescribing behavior? J Am Med Inform Assoc. 2009;16:531-538.
- Lee EK, Mejia AF, Senior T, et al. Improving patient safety through medical alert management: an automated decision tool to reduce alert fatigue. AMIA Annu Symp Proc. 2010;2010:417-421.
- Brenner AB. Physician and nurse relationships, a key to patient safety. J Ky Med Assoc. 2007;105:165-169.
- Rush JL, Flowers RH, Casamiquela KM, et al. Research letter: the knock: an adjunct to education opening the door to improved outpatient hand hygiene. J Am Acad Dermatol. In press.
- Lee SL. The extended surgical time-out: does it improve quality and prevent wrong-site surgery? Perm J. 2010;14:19-23.
- Altpeter T, Luckhardt K, Lewis JN, et al. Expanded surgical time out: a key to real-time data collection and quality improvement. J Am Coll Surg. 2007;204:527-532.
- MOC requirements. American Board of Dermatology Web site. https://www.abderm.org/diplomates/fulfilling-moc-requirements/moc-requirements.aspx#PI. Accessed January 18, 2016.
- How AAD develops measures. American Academy of Dermatology Web site. https://www.aad.org/practice-tools/quality-care/quality-measures. Accessed January 20, 2016.
- Quality assurance programs. The American Society of Dermatopathology Web site. http://www.asdp.org/education/quality-assurance-programs. Accessed January 20, 2016.
As part of its Next Accreditation System, the Accreditation Council for Graduate Medical Education (ACGME) has introduced the Clinical Learning Environment Review (CLER) program, designed to assess the learning environment of institutions that have ACGME residency and fellowship programs.1 The CLER program emphasizes the responsibility of these hospitals, multispecialty groups, and other organizations to focus on quality and safety in the health care environment of resident learning and patient care. The expectation is that emphasis on quality of care in a residency training program will influence these physicians’ approach to quality of care after graduation.2,3 The Department of Dermatology at the University of Mississippi Medical Center (UMMC)(Jackson, Mississippi) saw CLER as an opportunity to demonstrate leadership in the patient safety movement.
CLER Program at UMMC
As a model CLER program at our institution, our project at the outset concentrated resident efforts on the focus areas specified by the ACGME (Table 1). We also were aware that our ACGME committee would need to answer questions during CLER site visits (Table 2). Because the data generated would not be used for accreditation decisions, there was no concern that exposing errors would jeopardize our postgraduate training certification.
The first 15 minutes of monthly faculty meetings were devoted to the presentation of a resident project, called a QA/QI (quality assurance/quality improvement) moment, that addressed ACGME focus areas 1, 2, 3, or 6 (Table 1). (Transitions in care [focus area 4] and work hours and fatigue [focus area 5] generally are less important issues in a predominantly outpatient specialty such as dermatology.) The residents were encouraged to identify areas where patient harm could occur due to poorly designed systems and to report situations in which patients actually were harmed.
Each project had to be approved by the department chairperson based on the following 4 requirements: First, the initiative must have the potential to notably impact patient safety and reduce harm. Second, residents with faculty support had to design methods to assess the identified problem. Third, participants had to design (to the best of their abilities) cost-effective and achievable interventions in a manner that would not produce unintended consequences. Fourth, residents were asked to devise a system to close the loop, ensuring that the effort put into the process was not wasted.
Findings From the CLER Program
The CLER program generates data on program and institutional attributes that have a salutatory effect on quality and safety, specifically involving 6 focus areas highlighted in Table 1. Putting residents at the center of efforts to improve the quality of care in our department proved critical to improving patient safety.
Involving residents in a series of QA/QI initiatives was logical because they rotate with faculty members. They also are in a position to view inconsistencies and to work to establish consistent patterns of patient care. In addition, our busy faculty members are charged with a variety of other clinical, educational, and administrative duties complicated by requirements in the design of a new residency training program. Faculty and residents working together were able to find problem areas in our department and devise solutions to improve those problems.
The CLER program involved a series of steps. Residents were charged with identifying errors (QA) and then devising a system to prevent similar errors from being repeated (QI)(Table 3). Efforts focused on preventing needless harm in our department. Initiatives developed by residents, who are closest to patients, have advantages over safety programs developed by the hospital’s administration. Residents became passionate about error prevention when they determined that their efforts could make a difference to patients.
Forward Thinking for Dermatology Practices
Perhaps there are lessons here that could apply to safety promotion in the practicing dermatologist’s office. The American Board of Dermatology, within the framework established by the American Board of Medical Specialties, requires physicians seeking recertification to participate in preapproved practice assessment QI exercises twice every 10 years.17 Six programs sponsored by the American Academy of Dermatology have now been approved in the areas of melanoma, biopsy follow-up measure, psoriasis, chronic urticaria, venous insufficiency, and laser- and light-based therapy for rejuvenation.18 An additional program has been approved for dermatopathologists through the American Society of Dermatopathology.19 None of these programs match the topics chosen by our residents in consultation with faculty to meet safety gaps identified in clinics at UMMC. Perhaps the next generation of performance improvement continuing medical education programs could include a pilot program for part 4 of Maintenance of Certification credit that is nonpunitive, patient focused, and allows dermatologists to design specific error-prevention solutions tailored to their individual practice in the same way residency programs are taking up this task.
As part of its Next Accreditation System, the Accreditation Council for Graduate Medical Education (ACGME) has introduced the Clinical Learning Environment Review (CLER) program, designed to assess the learning environment of institutions that have ACGME residency and fellowship programs.1 The CLER program emphasizes the responsibility of these hospitals, multispecialty groups, and other organizations to focus on quality and safety in the health care environment of resident learning and patient care. The expectation is that emphasis on quality of care in a residency training program will influence these physicians’ approach to quality of care after graduation.2,3 The Department of Dermatology at the University of Mississippi Medical Center (UMMC)(Jackson, Mississippi) saw CLER as an opportunity to demonstrate leadership in the patient safety movement.
CLER Program at UMMC
As a model CLER program at our institution, our project at the outset concentrated resident efforts on the focus areas specified by the ACGME (Table 1). We also were aware that our ACGME committee would need to answer questions during CLER site visits (Table 2). Because the data generated would not be used for accreditation decisions, there was no concern that exposing errors would jeopardize our postgraduate training certification.
The first 15 minutes of monthly faculty meetings were devoted to the presentation of a resident project, called a QA/QI (quality assurance/quality improvement) moment, that addressed ACGME focus areas 1, 2, 3, or 6 (Table 1). (Transitions in care [focus area 4] and work hours and fatigue [focus area 5] generally are less important issues in a predominantly outpatient specialty such as dermatology.) The residents were encouraged to identify areas where patient harm could occur due to poorly designed systems and to report situations in which patients actually were harmed.
Each project had to be approved by the department chairperson based on the following 4 requirements: First, the initiative must have the potential to notably impact patient safety and reduce harm. Second, residents with faculty support had to design methods to assess the identified problem. Third, participants had to design (to the best of their abilities) cost-effective and achievable interventions in a manner that would not produce unintended consequences. Fourth, residents were asked to devise a system to close the loop, ensuring that the effort put into the process was not wasted.
Findings From the CLER Program
The CLER program generates data on program and institutional attributes that have a salutatory effect on quality and safety, specifically involving 6 focus areas highlighted in Table 1. Putting residents at the center of efforts to improve the quality of care in our department proved critical to improving patient safety.
Involving residents in a series of QA/QI initiatives was logical because they rotate with faculty members. They also are in a position to view inconsistencies and to work to establish consistent patterns of patient care. In addition, our busy faculty members are charged with a variety of other clinical, educational, and administrative duties complicated by requirements in the design of a new residency training program. Faculty and residents working together were able to find problem areas in our department and devise solutions to improve those problems.
The CLER program involved a series of steps. Residents were charged with identifying errors (QA) and then devising a system to prevent similar errors from being repeated (QI)(Table 3). Efforts focused on preventing needless harm in our department. Initiatives developed by residents, who are closest to patients, have advantages over safety programs developed by the hospital’s administration. Residents became passionate about error prevention when they determined that their efforts could make a difference to patients.
Forward Thinking for Dermatology Practices
Perhaps there are lessons here that could apply to safety promotion in the practicing dermatologist’s office. The American Board of Dermatology, within the framework established by the American Board of Medical Specialties, requires physicians seeking recertification to participate in preapproved practice assessment QI exercises twice every 10 years.17 Six programs sponsored by the American Academy of Dermatology have now been approved in the areas of melanoma, biopsy follow-up measure, psoriasis, chronic urticaria, venous insufficiency, and laser- and light-based therapy for rejuvenation.18 An additional program has been approved for dermatopathologists through the American Society of Dermatopathology.19 None of these programs match the topics chosen by our residents in consultation with faculty to meet safety gaps identified in clinics at UMMC. Perhaps the next generation of performance improvement continuing medical education programs could include a pilot program for part 4 of Maintenance of Certification credit that is nonpunitive, patient focused, and allows dermatologists to design specific error-prevention solutions tailored to their individual practice in the same way residency programs are taking up this task.
- Nasca TJ, Philibert I, Brigham T, et al. The Next GME accreditation system—rationale and benefits. N Engl J Med. 2012;366:1051-1056.
- Philibert I, Gonzalez del Rey JA, Lannon C, et al. Quality improvement skills for pediatric residents: from lecture to implementation and sustainability. Acad Pediatr. 2014;14:40-46.
- Vidyarthi AR, Green AL, Rosenbluth G, et al. Engaging residents and fellows to improve institution-wide quality: the first six years of a novel financial incentive program. Acad Med. 2014;89:460-468.
- Brodell RT, Elewski B. Antifungal drug interactions. avoidance requires more than memorization. Postgrad Med. 2000;107:41-43.
- Kerr IG, Jolivet J, Collin JM, et al. Test dose for predicting high-dose methotrexate infusions. Clin Pharmacol Ther. 1983;33:44-51.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451-485.
- Saporito FC, Menter MA. Methotrexate and psoriasis in the era of new biologic agents. J Am Acad Dermatol. 2004;50:301-309.
- Van Der Sijs H, Aarts J, Vulto A, et al. Overriding of drug safety alerts in computerized physician order entry. J Am Med Inform Assoc. 2006;13:138-147.
- Hunter KM. Implementation of an electronic medication administration record and bedside verification system. Online J Nurs Inform (OJNI). 2011;15:672.
- Nanji KC, Slight SP, Seger DL, et al. Overrides of medication-related clinical decision support alerts in outpatients. J Am Med Inform Assoc. 2014;21:487-491.
- Schedlbauer A, Prasad V, Mulvaney C, et al. What evidence supports the use of computerized alerts and prompts to improve clinicians’ prescribing behavior? J Am Med Inform Assoc. 2009;16:531-538.
- Lee EK, Mejia AF, Senior T, et al. Improving patient safety through medical alert management: an automated decision tool to reduce alert fatigue. AMIA Annu Symp Proc. 2010;2010:417-421.
- Brenner AB. Physician and nurse relationships, a key to patient safety. J Ky Med Assoc. 2007;105:165-169.
- Rush JL, Flowers RH, Casamiquela KM, et al. Research letter: the knock: an adjunct to education opening the door to improved outpatient hand hygiene. J Am Acad Dermatol. In press.
- Lee SL. The extended surgical time-out: does it improve quality and prevent wrong-site surgery? Perm J. 2010;14:19-23.
- Altpeter T, Luckhardt K, Lewis JN, et al. Expanded surgical time out: a key to real-time data collection and quality improvement. J Am Coll Surg. 2007;204:527-532.
- MOC requirements. American Board of Dermatology Web site. https://www.abderm.org/diplomates/fulfilling-moc-requirements/moc-requirements.aspx#PI. Accessed January 18, 2016.
- How AAD develops measures. American Academy of Dermatology Web site. https://www.aad.org/practice-tools/quality-care/quality-measures. Accessed January 20, 2016.
- Quality assurance programs. The American Society of Dermatopathology Web site. http://www.asdp.org/education/quality-assurance-programs. Accessed January 20, 2016.
- Nasca TJ, Philibert I, Brigham T, et al. The Next GME accreditation system—rationale and benefits. N Engl J Med. 2012;366:1051-1056.
- Philibert I, Gonzalez del Rey JA, Lannon C, et al. Quality improvement skills for pediatric residents: from lecture to implementation and sustainability. Acad Pediatr. 2014;14:40-46.
- Vidyarthi AR, Green AL, Rosenbluth G, et al. Engaging residents and fellows to improve institution-wide quality: the first six years of a novel financial incentive program. Acad Med. 2014;89:460-468.
- Brodell RT, Elewski B. Antifungal drug interactions. avoidance requires more than memorization. Postgrad Med. 2000;107:41-43.
- Kerr IG, Jolivet J, Collin JM, et al. Test dose for predicting high-dose methotrexate infusions. Clin Pharmacol Ther. 1983;33:44-51.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451-485.
- Saporito FC, Menter MA. Methotrexate and psoriasis in the era of new biologic agents. J Am Acad Dermatol. 2004;50:301-309.
- Van Der Sijs H, Aarts J, Vulto A, et al. Overriding of drug safety alerts in computerized physician order entry. J Am Med Inform Assoc. 2006;13:138-147.
- Hunter KM. Implementation of an electronic medication administration record and bedside verification system. Online J Nurs Inform (OJNI). 2011;15:672.
- Nanji KC, Slight SP, Seger DL, et al. Overrides of medication-related clinical decision support alerts in outpatients. J Am Med Inform Assoc. 2014;21:487-491.
- Schedlbauer A, Prasad V, Mulvaney C, et al. What evidence supports the use of computerized alerts and prompts to improve clinicians’ prescribing behavior? J Am Med Inform Assoc. 2009;16:531-538.
- Lee EK, Mejia AF, Senior T, et al. Improving patient safety through medical alert management: an automated decision tool to reduce alert fatigue. AMIA Annu Symp Proc. 2010;2010:417-421.
- Brenner AB. Physician and nurse relationships, a key to patient safety. J Ky Med Assoc. 2007;105:165-169.
- Rush JL, Flowers RH, Casamiquela KM, et al. Research letter: the knock: an adjunct to education opening the door to improved outpatient hand hygiene. J Am Acad Dermatol. In press.
- Lee SL. The extended surgical time-out: does it improve quality and prevent wrong-site surgery? Perm J. 2010;14:19-23.
- Altpeter T, Luckhardt K, Lewis JN, et al. Expanded surgical time out: a key to real-time data collection and quality improvement. J Am Coll Surg. 2007;204:527-532.
- MOC requirements. American Board of Dermatology Web site. https://www.abderm.org/diplomates/fulfilling-moc-requirements/moc-requirements.aspx#PI. Accessed January 18, 2016.
- How AAD develops measures. American Academy of Dermatology Web site. https://www.aad.org/practice-tools/quality-care/quality-measures. Accessed January 20, 2016.
- Quality assurance programs. The American Society of Dermatopathology Web site. http://www.asdp.org/education/quality-assurance-programs. Accessed January 20, 2016.
Practice Points
- The Clinical Learning Environment Review mobilizes residency and fellowship training programs in the movement to improve the quality of patient care.
- Quality assessment/quality improvement (QA/QI) projects enhance communication between residents and faculty and promote systems that improve patient safety.
- Emphasis on resident-initiated QA/QI impacts quality of care in clinical practice long after graduation.

Electronic Assessment of Mental Status
Altered mental status (AMS) is a complex spectrum of cognitive deficits that includes orientation, memory, language, visuospatial ability, and perception.[1] The clinical definitions of both delirium and dementia include AMS as a hallmark clinical prerequisite. Regardless of etiology, this broader AMS definition is particularly salient in the hospital setting, where AMS is present in up to 60% of inpatients and is associated with longer hospital stay as well as increased morbidity and mortality.[2, 3] Not surprisingly, due to the complexity of identifying and assessing changes in mental status, clinically relevant AMS is often undetected among inpatients.[2] However, when detected, the most common causes of AMS (infection, polypharmacy, and pain) are treatable, suggesting that early AMS identification could alert clinicians to early signs of clinical decompensation, potentially improving clinical outcomes.[4]
Because rapid and systemic clinical detection of AMS is limited by the complexity of mental status, a number of assessments have been created, each with their own advantages, limitations, and target populations. These assessments are often limited by time‐intensive administration, subjectivity of mental status assessment, and lack of sensitivity in general medicine patients. Time‐intensive measures, such as the Short Portable Mental Status Questionnaire (SPMSQ) have utility in the research setting, whereas current common clinical risk stratification tools (eg, National Early Warning Score) utilize simpler measures such as the Alert, Voice, Pain, Unresponsive (AVPU) and Glasgow Coma Scale (GCS) as measures of mental status.[2, 5, 6, 7, 8, 9]
To address the need for a brief, clinically feasible, accurate tool in clinical detection of AMS, our group developed a mobile application for working memory testing, the Functional Assessment of Mentation (FAMTM). In this study, we aimed to identify baseline scoring distributions of the FAMTM in a nonhospitalized subgroup, as well as assess the correlation of the FAMTM to discharge disposition and compare it to the SPMSQ in inpatients.
METHODS
Study Design
We conducted a prospective observational study. Data were collected from both hospitalized and nonhospitalized adult participants as 2 distinct subgroups. Nonhospitalized adult subjects were recruited from a university medical campus (June 2013July 2013; IRB‐12‐0175). Hospitalized participants were recruited from the general medicine service as part of an ongoing study measuring quality of care and resource allocation at the same academic medical center (June 2014August 2014; IRB‐9967).[10]
FAMTM Application
The FAMTM application is a bedside tool for working memory assessment developed for the iPhone mobile operating system (Apple Inc., Cupertino, CA) and presented on an iPad mini (Apple). The application interface displays 4 colored rectangles individually labeled with a number (see Supporting Figure 1 in the online version of this article). The testing portion of the application presents a sequence of numbered rectangles, illuminated 1 at a time in random order. Subjects are prompted first to watch and remember the sequence and then repeat the sequence by touching the screen within each numbered rectangle. Successful reproduction of the sequence is followed by a distinct and longer sequence, whereas unsuccessful attempts are followed by a shorter sequence. The final FAMTM score corresponds to the longest sequence of rectangles successfully repeated by the subject.
Data Collection
In the nonhospitalized subject population, research assistants collected demographic data immediately prior to FAMTM administration. Among hospitalized subjects, GCS information was collected by nursing staff as part of standard clinical care. One research assistant administered the SPMSQ while a second assistant, blinded to the SPMSQ and GCS scores, administered the FAMTM. Clinical data were obtained from medical records (EPIC Systems Corp., Verona, WI). Discharge disposition was dichotomized as discharged home or not.
Statistical Analyses
Demographic characteristics of the 2 subject populations were compared using Student t tests (continuous variables) and 2 tests (categorical variables). Score distribution and discharge disposition comparison was conducted with the Mann‐Whitney U test and area under receiver operating characteristic curve (AUC) analysis, using the trapezoidal rule.[11] Multivariable linear regression was used to investigate the impact of age, race, education, discharge disposition, and hospitalization status on patient scores and times. Correlations between the FAMTM and SPMSQ scores and between the GCS and SPMSQ scores were calculated using the Spearman rank test. Significance was set at a 2‐sided P value of 0.05. Analyses were conducted using Stata version 13.1 (StataCorp, College Station, TX).
RESULTS
A total of 931 subjects were enrolled in the study. In the nonhospitalized subgroup, 651 consented to study participation and 612 were included in final analysis. Subjects were excluded if they started but did not complete the application (n = 36) or were under the age of 18 years (n = 3). Of the 363 hospitalized subjects approached for enrollment, 319 were included in the final analysis. Subjects were excluded if they refused to participate (n = 23), were under the age of 18 (n = 2), had technical failures (n = 5), or had physical or visual limitations that precluded them from participation (n = 14). Within the hospitalized subgroup, 268 subjects were discharged home (85%). The table displays demographics and score distributions by subgroup.1
| Nonhospitalized Subjects, n = 612 | Hospitalized Subjects Discharged Home, n = 268 | Hospitalized Subjects Discharged Elsewhere, n = 48 | P Value | |
|---|---|---|---|---|
| ||||
| Age, y | 52 18 | 52 19 | 62 17 | 0.001 |
| Female sex | 343 (56%) | 158 (59%) | 26 (54%) | 0.63 |
| Education | 0.001 | |||
| Less than high school graduate | 31 (5%) | 32 (12%) | 7 (15%) | |
| High school graduate | 312 (51%) | 153 (57%) | 26 (54%) | |
| College graduate | 263 (43%) | 43 (16%) | 8 (17%) | |
| Missing | 6 (1%) | 40 (15%) | 7 (15%) | |
| Race | 0.001 | |||
| Black | 196 (32%) | 185 (69%) | 34 (71%) | |
| White | 324 (53%) | 75 (28%) | 13 (27%) | |
| Other | 86 (14%) | 4 (1%) | 4 (1%) | |
| Missing | 6 (1%) | 4 (1%) | 0 (0%) | |
| FAMTM score, median (IQR) | 5 (47) | 5 (36) | 3 (15) | 0.001 |
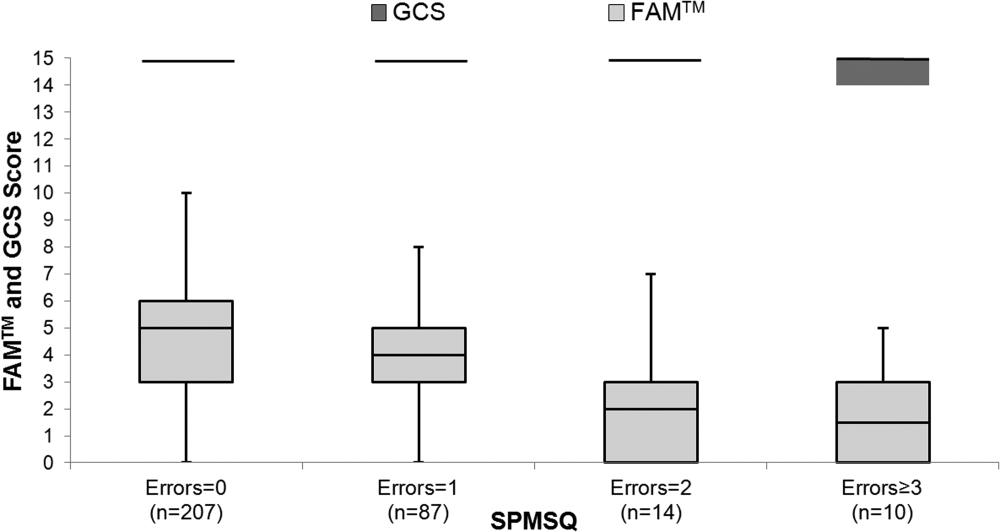
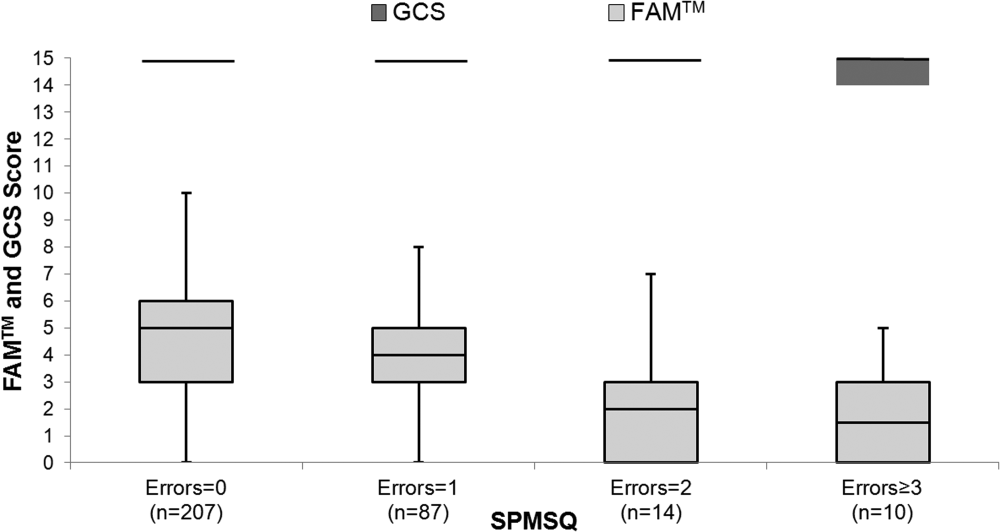
The median FAMTM score for the combined study population was 5 (interquartile range [IQR] 36), and median time to completion was 55 seconds (IQR 4567 seconds). A graded reduction was found in the FAMTM score for all stepwise comparisons between nonhospitalized subjects, hospitalized subjects discharged home, and hospitalized subjects not discharged home (median 5 [IQR 47] vs 5 [IQR 36] vs 3 [IQR 15]; P 0.001 for all pairwise comparisons). The AUC for the FAMTM predicting discharge disposition (home vs not) was 0.66 (95% confidence interval [CI]: 0.58‐0.74]. After adjusting for confounders, higher FAMTM scores were independently associated with not being hospitalized, being discharged home, higher levels of education, younger age, and white race (see Supporting Table 1 in the online version of this article). Additionally, in the hospitalized subgroup, decreasing FAMTM score was significantly correlated with increasing errors on the SPMSQ (Spearman = 0.27, P 0.001), whereas the GCS score was not correlated with the SPMSQ (Spearman = 0.05, P = 0.40) (Figure 1).
DISCUSSION
We demonstrated the utility of a rapid and accurate mobile application for assessment of mental status. The FAMTM was able to be quickly administered with a median time to completion of approximately 1 minute. The ability to detect mild alterations in mental status was shown through concurrent validity by FAMTM correlation with the SPMSQ and predictive validity with the association between the FAMTM and discharge disposition. Our study highlights the potential for the FAMTM to be used as a sensitive marker of AMS.
The novel design of the FAMTM presents unique advantages compared to current mental status testing. First, the FAMTM could allow patients with hearing impairment or language barriers to complete a mental status assessment. Additionally, the approximately 1‐minute median time to completion is much faster than other established mental status assessments including the SPMSQ (510 minutes). Compared to the SPMSQ taking 5 minutes, in a 400‐bed hospital, taken once per nursing shift, the FAMTM would save approximately 20,000 hours and 10 nursing full‐time equivalents per year.[5] Finally, many current mental status tests such as the Confusion Assessment Model utilize subjective mental status assessments.[2] However, the FAMTM is designed to be conducted through self‐assessment and, thus, could theoretically be free of observer bias. This potential for self‐administration expands beyond other proposed alternative testing mechanisms of the AMS such as ultrabrief assessments that include items such as asking subjects the months of the year backwards, and what is the day of the week?, and assessing arousal.[12, 13, 14]
In research settings and commonly in hospitals, the GCS and AVPU are used clinically for mental status assessment of hospitalized patients.[6, 15] However, similar to previous literature, our study found that the vast majority of hospitalized patients were defined as neurologically intact by the GCS, which is the more accurate predictor of the 2.[7] One major strength of the FAMTM was that it identified an extensive gradation of scores for patients previously labeled as merely alert, providing greater resolution than the GCS in quantifying mental status.
One of the key benefits of the FAMTM is that it can be measured longitudinally over the course of a patient's hospital stay. Therefore, once a baseline FAMTM score is established, variation from the patient's personal baseline could indicate mental status deterioration, which would not be affected by the patient's demographics, health status, or underlying neurocognitive deficits.
There were important limitations to this study. First, limited generalizability of these data may exist due to the single‐center setting and patient population. However, this initial study provides pilot data for further expansion into the potential broad applicability of the FAMTM to other patient populations and settings. Additionally, the cost of large‐scale implementation of the FAMTM is unknown and was beyond the scope of this pilot study. However, to reduce costs, the FAMTM technology could be integrated into existing hospital technology infrastructure. Finally, the scope of this study prevented a complete assessment of all validity measures or comparison to other mental status assessments such as the digit span or serial sevens tests. However, predictive and concurrent validity were assessed with comparison by discharge disposition, SPMSQ, and GCS scores.
In conclusion, this pilot study identifies the FAMTM application as a potentially clinically useful, novel, rapid, and feasible assessment tool of mental status in a general medicine inpatient setting.
Acknowledgements
The authors thank Frank Zadravecz, MPH, for his support with this project.
Disclosures: This research was supported in part by a grant from the National Institutes of Health (NIA 2T35AG029795‐07) and in part by career development awards granted to Dr. Churpek, Dr. Edelson, and Dr. Press by the National Heart, Lung, and Blood Institute (K08 HL121080, K23 HL097157, and K23 HL118151, respectively). Dr. Churpek has received honoraria from Chest for invited speaking engagements. Drs. Churpek and Edelson have a patent pending (ARCD. P0535US.P2) for risk stratification algorithms for hospitalized patients. In addition, Dr. Edelson has received research support from Philips Healthcare (Andover, MA), the American Heart Association (Dallas, TX), and Laerdal Medical (Stavanger, Norway). She has ownership interest in Quant HC (Chicago, IL), which is developing products for risk stratification of hospitalized patients. All other authors report no potential conflicts of interest.
- , . Altered mental status in older patients in the emergency department. Clin Geriatr Med. 2013;29(1):101–136.
- , , , , , . Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113(12):941–948.
- , , , , . Association between clinically abnormal observations and subsequent in‐hospital mortality: a prospective study. Resuscitation. 2004;62(2):137–141.
- , , . Early recognition of delirium: review of the literature. J Clin Nurs. 2001;10(6):721–729.
- . A short portable mental status questionnaire for the assessment of organic brain deficit in elderly patients. J Am Geriatr Soc. 1975;23(10):433–441.
- , , , , . The ability of the National Early Warning Score (NEWS) to discriminate patients at risk of early cardiac arrest, unanticipated intensive care unit admission, and death. Resuscitation. 2013;84(4):465–470.
- , , , et al. Comparison of mental‐status scales for predicting mortality on the general wards. J Hosp Med. 2015;10(10):658–663.
- , . Assessment of coma and impaired consciousness: a practical scale. Lancet. 1974;304(7872):81–84.
- , , , . Short Portable Mental Status Questionnaire as a Screening Test for Dementia and Delirium Among the Elderly. J Am Geriatr Soc. 1987;35(5):412–416.
- , , , et al. Effects of physician experience on costs and outcomes on an academic general medicine service: results of a trial of hospitalists. Ann Intern Med. 2002;137(11):866–874.
- , , . Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44(3):837–845.
- , , , et al. Preliminary development of an ultrabrief two‐item bedside test for delirium. J Hosp Med. 2015;10(10):645–650.
- , , , , , . The association between an ultrabrief cognitive screening in older adults and hospital outcomes. J Hosp Med. 2015;10(10):651–657.
- , , , et al. Selecting optimal screening items for delirium: an application of item response theory. BMC Med Res Methodol. 2013;13:8.
- , , . Variability in agreement between physicians and nurses when measuring the Glasgow Coma Scale in the emergency department limits its clinical usefulness. Emerg Med Australas. 2006;18(4):379–384.
Altered mental status (AMS) is a complex spectrum of cognitive deficits that includes orientation, memory, language, visuospatial ability, and perception.[1] The clinical definitions of both delirium and dementia include AMS as a hallmark clinical prerequisite. Regardless of etiology, this broader AMS definition is particularly salient in the hospital setting, where AMS is present in up to 60% of inpatients and is associated with longer hospital stay as well as increased morbidity and mortality.[2, 3] Not surprisingly, due to the complexity of identifying and assessing changes in mental status, clinically relevant AMS is often undetected among inpatients.[2] However, when detected, the most common causes of AMS (infection, polypharmacy, and pain) are treatable, suggesting that early AMS identification could alert clinicians to early signs of clinical decompensation, potentially improving clinical outcomes.[4]
Because rapid and systemic clinical detection of AMS is limited by the complexity of mental status, a number of assessments have been created, each with their own advantages, limitations, and target populations. These assessments are often limited by time‐intensive administration, subjectivity of mental status assessment, and lack of sensitivity in general medicine patients. Time‐intensive measures, such as the Short Portable Mental Status Questionnaire (SPMSQ) have utility in the research setting, whereas current common clinical risk stratification tools (eg, National Early Warning Score) utilize simpler measures such as the Alert, Voice, Pain, Unresponsive (AVPU) and Glasgow Coma Scale (GCS) as measures of mental status.[2, 5, 6, 7, 8, 9]
To address the need for a brief, clinically feasible, accurate tool in clinical detection of AMS, our group developed a mobile application for working memory testing, the Functional Assessment of Mentation (FAMTM). In this study, we aimed to identify baseline scoring distributions of the FAMTM in a nonhospitalized subgroup, as well as assess the correlation of the FAMTM to discharge disposition and compare it to the SPMSQ in inpatients.
METHODS
Study Design
We conducted a prospective observational study. Data were collected from both hospitalized and nonhospitalized adult participants as 2 distinct subgroups. Nonhospitalized adult subjects were recruited from a university medical campus (June 2013July 2013; IRB‐12‐0175). Hospitalized participants were recruited from the general medicine service as part of an ongoing study measuring quality of care and resource allocation at the same academic medical center (June 2014August 2014; IRB‐9967).[10]
FAMTM Application
The FAMTM application is a bedside tool for working memory assessment developed for the iPhone mobile operating system (Apple Inc., Cupertino, CA) and presented on an iPad mini (Apple). The application interface displays 4 colored rectangles individually labeled with a number (see Supporting Figure 1 in the online version of this article). The testing portion of the application presents a sequence of numbered rectangles, illuminated 1 at a time in random order. Subjects are prompted first to watch and remember the sequence and then repeat the sequence by touching the screen within each numbered rectangle. Successful reproduction of the sequence is followed by a distinct and longer sequence, whereas unsuccessful attempts are followed by a shorter sequence. The final FAMTM score corresponds to the longest sequence of rectangles successfully repeated by the subject.
Data Collection
In the nonhospitalized subject population, research assistants collected demographic data immediately prior to FAMTM administration. Among hospitalized subjects, GCS information was collected by nursing staff as part of standard clinical care. One research assistant administered the SPMSQ while a second assistant, blinded to the SPMSQ and GCS scores, administered the FAMTM. Clinical data were obtained from medical records (EPIC Systems Corp., Verona, WI). Discharge disposition was dichotomized as discharged home or not.
Statistical Analyses
Demographic characteristics of the 2 subject populations were compared using Student t tests (continuous variables) and 2 tests (categorical variables). Score distribution and discharge disposition comparison was conducted with the Mann‐Whitney U test and area under receiver operating characteristic curve (AUC) analysis, using the trapezoidal rule.[11] Multivariable linear regression was used to investigate the impact of age, race, education, discharge disposition, and hospitalization status on patient scores and times. Correlations between the FAMTM and SPMSQ scores and between the GCS and SPMSQ scores were calculated using the Spearman rank test. Significance was set at a 2‐sided P value of 0.05. Analyses were conducted using Stata version 13.1 (StataCorp, College Station, TX).
RESULTS
A total of 931 subjects were enrolled in the study. In the nonhospitalized subgroup, 651 consented to study participation and 612 were included in final analysis. Subjects were excluded if they started but did not complete the application (n = 36) or were under the age of 18 years (n = 3). Of the 363 hospitalized subjects approached for enrollment, 319 were included in the final analysis. Subjects were excluded if they refused to participate (n = 23), were under the age of 18 (n = 2), had technical failures (n = 5), or had physical or visual limitations that precluded them from participation (n = 14). Within the hospitalized subgroup, 268 subjects were discharged home (85%). The table displays demographics and score distributions by subgroup.1
| Nonhospitalized Subjects, n = 612 | Hospitalized Subjects Discharged Home, n = 268 | Hospitalized Subjects Discharged Elsewhere, n = 48 | P Value | |
|---|---|---|---|---|
| ||||
| Age, y | 52 18 | 52 19 | 62 17 | 0.001 |
| Female sex | 343 (56%) | 158 (59%) | 26 (54%) | 0.63 |
| Education | 0.001 | |||
| Less than high school graduate | 31 (5%) | 32 (12%) | 7 (15%) | |
| High school graduate | 312 (51%) | 153 (57%) | 26 (54%) | |
| College graduate | 263 (43%) | 43 (16%) | 8 (17%) | |
| Missing | 6 (1%) | 40 (15%) | 7 (15%) | |
| Race | 0.001 | |||
| Black | 196 (32%) | 185 (69%) | 34 (71%) | |
| White | 324 (53%) | 75 (28%) | 13 (27%) | |
| Other | 86 (14%) | 4 (1%) | 4 (1%) | |
| Missing | 6 (1%) | 4 (1%) | 0 (0%) | |
| FAMTM score, median (IQR) | 5 (47) | 5 (36) | 3 (15) | 0.001 |
The median FAMTM score for the combined study population was 5 (interquartile range [IQR] 36), and median time to completion was 55 seconds (IQR 4567 seconds). A graded reduction was found in the FAMTM score for all stepwise comparisons between nonhospitalized subjects, hospitalized subjects discharged home, and hospitalized subjects not discharged home (median 5 [IQR 47] vs 5 [IQR 36] vs 3 [IQR 15]; P 0.001 for all pairwise comparisons). The AUC for the FAMTM predicting discharge disposition (home vs not) was 0.66 (95% confidence interval [CI]: 0.58‐0.74]. After adjusting for confounders, higher FAMTM scores were independently associated with not being hospitalized, being discharged home, higher levels of education, younger age, and white race (see Supporting Table 1 in the online version of this article). Additionally, in the hospitalized subgroup, decreasing FAMTM score was significantly correlated with increasing errors on the SPMSQ (Spearman = 0.27, P 0.001), whereas the GCS score was not correlated with the SPMSQ (Spearman = 0.05, P = 0.40) (Figure 1).
DISCUSSION
We demonstrated the utility of a rapid and accurate mobile application for assessment of mental status. The FAMTM was able to be quickly administered with a median time to completion of approximately 1 minute. The ability to detect mild alterations in mental status was shown through concurrent validity by FAMTM correlation with the SPMSQ and predictive validity with the association between the FAMTM and discharge disposition. Our study highlights the potential for the FAMTM to be used as a sensitive marker of AMS.
The novel design of the FAMTM presents unique advantages compared to current mental status testing. First, the FAMTM could allow patients with hearing impairment or language barriers to complete a mental status assessment. Additionally, the approximately 1‐minute median time to completion is much faster than other established mental status assessments including the SPMSQ (510 minutes). Compared to the SPMSQ taking 5 minutes, in a 400‐bed hospital, taken once per nursing shift, the FAMTM would save approximately 20,000 hours and 10 nursing full‐time equivalents per year.[5] Finally, many current mental status tests such as the Confusion Assessment Model utilize subjective mental status assessments.[2] However, the FAMTM is designed to be conducted through self‐assessment and, thus, could theoretically be free of observer bias. This potential for self‐administration expands beyond other proposed alternative testing mechanisms of the AMS such as ultrabrief assessments that include items such as asking subjects the months of the year backwards, and what is the day of the week?, and assessing arousal.[12, 13, 14]
In research settings and commonly in hospitals, the GCS and AVPU are used clinically for mental status assessment of hospitalized patients.[6, 15] However, similar to previous literature, our study found that the vast majority of hospitalized patients were defined as neurologically intact by the GCS, which is the more accurate predictor of the 2.[7] One major strength of the FAMTM was that it identified an extensive gradation of scores for patients previously labeled as merely alert, providing greater resolution than the GCS in quantifying mental status.
One of the key benefits of the FAMTM is that it can be measured longitudinally over the course of a patient's hospital stay. Therefore, once a baseline FAMTM score is established, variation from the patient's personal baseline could indicate mental status deterioration, which would not be affected by the patient's demographics, health status, or underlying neurocognitive deficits.
There were important limitations to this study. First, limited generalizability of these data may exist due to the single‐center setting and patient population. However, this initial study provides pilot data for further expansion into the potential broad applicability of the FAMTM to other patient populations and settings. Additionally, the cost of large‐scale implementation of the FAMTM is unknown and was beyond the scope of this pilot study. However, to reduce costs, the FAMTM technology could be integrated into existing hospital technology infrastructure. Finally, the scope of this study prevented a complete assessment of all validity measures or comparison to other mental status assessments such as the digit span or serial sevens tests. However, predictive and concurrent validity were assessed with comparison by discharge disposition, SPMSQ, and GCS scores.
In conclusion, this pilot study identifies the FAMTM application as a potentially clinically useful, novel, rapid, and feasible assessment tool of mental status in a general medicine inpatient setting.
Acknowledgements
The authors thank Frank Zadravecz, MPH, for his support with this project.
Disclosures: This research was supported in part by a grant from the National Institutes of Health (NIA 2T35AG029795‐07) and in part by career development awards granted to Dr. Churpek, Dr. Edelson, and Dr. Press by the National Heart, Lung, and Blood Institute (K08 HL121080, K23 HL097157, and K23 HL118151, respectively). Dr. Churpek has received honoraria from Chest for invited speaking engagements. Drs. Churpek and Edelson have a patent pending (ARCD. P0535US.P2) for risk stratification algorithms for hospitalized patients. In addition, Dr. Edelson has received research support from Philips Healthcare (Andover, MA), the American Heart Association (Dallas, TX), and Laerdal Medical (Stavanger, Norway). She has ownership interest in Quant HC (Chicago, IL), which is developing products for risk stratification of hospitalized patients. All other authors report no potential conflicts of interest.
Altered mental status (AMS) is a complex spectrum of cognitive deficits that includes orientation, memory, language, visuospatial ability, and perception.[1] The clinical definitions of both delirium and dementia include AMS as a hallmark clinical prerequisite. Regardless of etiology, this broader AMS definition is particularly salient in the hospital setting, where AMS is present in up to 60% of inpatients and is associated with longer hospital stay as well as increased morbidity and mortality.[2, 3] Not surprisingly, due to the complexity of identifying and assessing changes in mental status, clinically relevant AMS is often undetected among inpatients.[2] However, when detected, the most common causes of AMS (infection, polypharmacy, and pain) are treatable, suggesting that early AMS identification could alert clinicians to early signs of clinical decompensation, potentially improving clinical outcomes.[4]
Because rapid and systemic clinical detection of AMS is limited by the complexity of mental status, a number of assessments have been created, each with their own advantages, limitations, and target populations. These assessments are often limited by time‐intensive administration, subjectivity of mental status assessment, and lack of sensitivity in general medicine patients. Time‐intensive measures, such as the Short Portable Mental Status Questionnaire (SPMSQ) have utility in the research setting, whereas current common clinical risk stratification tools (eg, National Early Warning Score) utilize simpler measures such as the Alert, Voice, Pain, Unresponsive (AVPU) and Glasgow Coma Scale (GCS) as measures of mental status.[2, 5, 6, 7, 8, 9]
To address the need for a brief, clinically feasible, accurate tool in clinical detection of AMS, our group developed a mobile application for working memory testing, the Functional Assessment of Mentation (FAMTM). In this study, we aimed to identify baseline scoring distributions of the FAMTM in a nonhospitalized subgroup, as well as assess the correlation of the FAMTM to discharge disposition and compare it to the SPMSQ in inpatients.
METHODS
Study Design
We conducted a prospective observational study. Data were collected from both hospitalized and nonhospitalized adult participants as 2 distinct subgroups. Nonhospitalized adult subjects were recruited from a university medical campus (June 2013July 2013; IRB‐12‐0175). Hospitalized participants were recruited from the general medicine service as part of an ongoing study measuring quality of care and resource allocation at the same academic medical center (June 2014August 2014; IRB‐9967).[10]
FAMTM Application
The FAMTM application is a bedside tool for working memory assessment developed for the iPhone mobile operating system (Apple Inc., Cupertino, CA) and presented on an iPad mini (Apple). The application interface displays 4 colored rectangles individually labeled with a number (see Supporting Figure 1 in the online version of this article). The testing portion of the application presents a sequence of numbered rectangles, illuminated 1 at a time in random order. Subjects are prompted first to watch and remember the sequence and then repeat the sequence by touching the screen within each numbered rectangle. Successful reproduction of the sequence is followed by a distinct and longer sequence, whereas unsuccessful attempts are followed by a shorter sequence. The final FAMTM score corresponds to the longest sequence of rectangles successfully repeated by the subject.
Data Collection
In the nonhospitalized subject population, research assistants collected demographic data immediately prior to FAMTM administration. Among hospitalized subjects, GCS information was collected by nursing staff as part of standard clinical care. One research assistant administered the SPMSQ while a second assistant, blinded to the SPMSQ and GCS scores, administered the FAMTM. Clinical data were obtained from medical records (EPIC Systems Corp., Verona, WI). Discharge disposition was dichotomized as discharged home or not.
Statistical Analyses
Demographic characteristics of the 2 subject populations were compared using Student t tests (continuous variables) and 2 tests (categorical variables). Score distribution and discharge disposition comparison was conducted with the Mann‐Whitney U test and area under receiver operating characteristic curve (AUC) analysis, using the trapezoidal rule.[11] Multivariable linear regression was used to investigate the impact of age, race, education, discharge disposition, and hospitalization status on patient scores and times. Correlations between the FAMTM and SPMSQ scores and between the GCS and SPMSQ scores were calculated using the Spearman rank test. Significance was set at a 2‐sided P value of 0.05. Analyses were conducted using Stata version 13.1 (StataCorp, College Station, TX).
RESULTS
A total of 931 subjects were enrolled in the study. In the nonhospitalized subgroup, 651 consented to study participation and 612 were included in final analysis. Subjects were excluded if they started but did not complete the application (n = 36) or were under the age of 18 years (n = 3). Of the 363 hospitalized subjects approached for enrollment, 319 were included in the final analysis. Subjects were excluded if they refused to participate (n = 23), were under the age of 18 (n = 2), had technical failures (n = 5), or had physical or visual limitations that precluded them from participation (n = 14). Within the hospitalized subgroup, 268 subjects were discharged home (85%). The table displays demographics and score distributions by subgroup.1
| Nonhospitalized Subjects, n = 612 | Hospitalized Subjects Discharged Home, n = 268 | Hospitalized Subjects Discharged Elsewhere, n = 48 | P Value | |
|---|---|---|---|---|
| ||||
| Age, y | 52 18 | 52 19 | 62 17 | 0.001 |
| Female sex | 343 (56%) | 158 (59%) | 26 (54%) | 0.63 |
| Education | 0.001 | |||
| Less than high school graduate | 31 (5%) | 32 (12%) | 7 (15%) | |
| High school graduate | 312 (51%) | 153 (57%) | 26 (54%) | |
| College graduate | 263 (43%) | 43 (16%) | 8 (17%) | |
| Missing | 6 (1%) | 40 (15%) | 7 (15%) | |
| Race | 0.001 | |||
| Black | 196 (32%) | 185 (69%) | 34 (71%) | |
| White | 324 (53%) | 75 (28%) | 13 (27%) | |
| Other | 86 (14%) | 4 (1%) | 4 (1%) | |
| Missing | 6 (1%) | 4 (1%) | 0 (0%) | |
| FAMTM score, median (IQR) | 5 (47) | 5 (36) | 3 (15) | 0.001 |
The median FAMTM score for the combined study population was 5 (interquartile range [IQR] 36), and median time to completion was 55 seconds (IQR 4567 seconds). A graded reduction was found in the FAMTM score for all stepwise comparisons between nonhospitalized subjects, hospitalized subjects discharged home, and hospitalized subjects not discharged home (median 5 [IQR 47] vs 5 [IQR 36] vs 3 [IQR 15]; P 0.001 for all pairwise comparisons). The AUC for the FAMTM predicting discharge disposition (home vs not) was 0.66 (95% confidence interval [CI]: 0.58‐0.74]. After adjusting for confounders, higher FAMTM scores were independently associated with not being hospitalized, being discharged home, higher levels of education, younger age, and white race (see Supporting Table 1 in the online version of this article). Additionally, in the hospitalized subgroup, decreasing FAMTM score was significantly correlated with increasing errors on the SPMSQ (Spearman = 0.27, P 0.001), whereas the GCS score was not correlated with the SPMSQ (Spearman = 0.05, P = 0.40) (Figure 1).
DISCUSSION
We demonstrated the utility of a rapid and accurate mobile application for assessment of mental status. The FAMTM was able to be quickly administered with a median time to completion of approximately 1 minute. The ability to detect mild alterations in mental status was shown through concurrent validity by FAMTM correlation with the SPMSQ and predictive validity with the association between the FAMTM and discharge disposition. Our study highlights the potential for the FAMTM to be used as a sensitive marker of AMS.
The novel design of the FAMTM presents unique advantages compared to current mental status testing. First, the FAMTM could allow patients with hearing impairment or language barriers to complete a mental status assessment. Additionally, the approximately 1‐minute median time to completion is much faster than other established mental status assessments including the SPMSQ (510 minutes). Compared to the SPMSQ taking 5 minutes, in a 400‐bed hospital, taken once per nursing shift, the FAMTM would save approximately 20,000 hours and 10 nursing full‐time equivalents per year.[5] Finally, many current mental status tests such as the Confusion Assessment Model utilize subjective mental status assessments.[2] However, the FAMTM is designed to be conducted through self‐assessment and, thus, could theoretically be free of observer bias. This potential for self‐administration expands beyond other proposed alternative testing mechanisms of the AMS such as ultrabrief assessments that include items such as asking subjects the months of the year backwards, and what is the day of the week?, and assessing arousal.[12, 13, 14]
In research settings and commonly in hospitals, the GCS and AVPU are used clinically for mental status assessment of hospitalized patients.[6, 15] However, similar to previous literature, our study found that the vast majority of hospitalized patients were defined as neurologically intact by the GCS, which is the more accurate predictor of the 2.[7] One major strength of the FAMTM was that it identified an extensive gradation of scores for patients previously labeled as merely alert, providing greater resolution than the GCS in quantifying mental status.
One of the key benefits of the FAMTM is that it can be measured longitudinally over the course of a patient's hospital stay. Therefore, once a baseline FAMTM score is established, variation from the patient's personal baseline could indicate mental status deterioration, which would not be affected by the patient's demographics, health status, or underlying neurocognitive deficits.
There were important limitations to this study. First, limited generalizability of these data may exist due to the single‐center setting and patient population. However, this initial study provides pilot data for further expansion into the potential broad applicability of the FAMTM to other patient populations and settings. Additionally, the cost of large‐scale implementation of the FAMTM is unknown and was beyond the scope of this pilot study. However, to reduce costs, the FAMTM technology could be integrated into existing hospital technology infrastructure. Finally, the scope of this study prevented a complete assessment of all validity measures or comparison to other mental status assessments such as the digit span or serial sevens tests. However, predictive and concurrent validity were assessed with comparison by discharge disposition, SPMSQ, and GCS scores.
In conclusion, this pilot study identifies the FAMTM application as a potentially clinically useful, novel, rapid, and feasible assessment tool of mental status in a general medicine inpatient setting.
Acknowledgements
The authors thank Frank Zadravecz, MPH, for his support with this project.
Disclosures: This research was supported in part by a grant from the National Institutes of Health (NIA 2T35AG029795‐07) and in part by career development awards granted to Dr. Churpek, Dr. Edelson, and Dr. Press by the National Heart, Lung, and Blood Institute (K08 HL121080, K23 HL097157, and K23 HL118151, respectively). Dr. Churpek has received honoraria from Chest for invited speaking engagements. Drs. Churpek and Edelson have a patent pending (ARCD. P0535US.P2) for risk stratification algorithms for hospitalized patients. In addition, Dr. Edelson has received research support from Philips Healthcare (Andover, MA), the American Heart Association (Dallas, TX), and Laerdal Medical (Stavanger, Norway). She has ownership interest in Quant HC (Chicago, IL), which is developing products for risk stratification of hospitalized patients. All other authors report no potential conflicts of interest.
- , . Altered mental status in older patients in the emergency department. Clin Geriatr Med. 2013;29(1):101–136.
- , , , , , . Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113(12):941–948.
- , , , , . Association between clinically abnormal observations and subsequent in‐hospital mortality: a prospective study. Resuscitation. 2004;62(2):137–141.
- , , . Early recognition of delirium: review of the literature. J Clin Nurs. 2001;10(6):721–729.
- . A short portable mental status questionnaire for the assessment of organic brain deficit in elderly patients. J Am Geriatr Soc. 1975;23(10):433–441.
- , , , , . The ability of the National Early Warning Score (NEWS) to discriminate patients at risk of early cardiac arrest, unanticipated intensive care unit admission, and death. Resuscitation. 2013;84(4):465–470.
- , , , et al. Comparison of mental‐status scales for predicting mortality on the general wards. J Hosp Med. 2015;10(10):658–663.
- , . Assessment of coma and impaired consciousness: a practical scale. Lancet. 1974;304(7872):81–84.
- , , , . Short Portable Mental Status Questionnaire as a Screening Test for Dementia and Delirium Among the Elderly. J Am Geriatr Soc. 1987;35(5):412–416.
- , , , et al. Effects of physician experience on costs and outcomes on an academic general medicine service: results of a trial of hospitalists. Ann Intern Med. 2002;137(11):866–874.
- , , . Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44(3):837–845.
- , , , et al. Preliminary development of an ultrabrief two‐item bedside test for delirium. J Hosp Med. 2015;10(10):645–650.
- , , , , , . The association between an ultrabrief cognitive screening in older adults and hospital outcomes. J Hosp Med. 2015;10(10):651–657.
- , , , et al. Selecting optimal screening items for delirium: an application of item response theory. BMC Med Res Methodol. 2013;13:8.
- , , . Variability in agreement between physicians and nurses when measuring the Glasgow Coma Scale in the emergency department limits its clinical usefulness. Emerg Med Australas. 2006;18(4):379–384.
- , . Altered mental status in older patients in the emergency department. Clin Geriatr Med. 2013;29(1):101–136.
- , , , , , . Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113(12):941–948.
- , , , , . Association between clinically abnormal observations and subsequent in‐hospital mortality: a prospective study. Resuscitation. 2004;62(2):137–141.
- , , . Early recognition of delirium: review of the literature. J Clin Nurs. 2001;10(6):721–729.
- . A short portable mental status questionnaire for the assessment of organic brain deficit in elderly patients. J Am Geriatr Soc. 1975;23(10):433–441.
- , , , , . The ability of the National Early Warning Score (NEWS) to discriminate patients at risk of early cardiac arrest, unanticipated intensive care unit admission, and death. Resuscitation. 2013;84(4):465–470.
- , , , et al. Comparison of mental‐status scales for predicting mortality on the general wards. J Hosp Med. 2015;10(10):658–663.
- , . Assessment of coma and impaired consciousness: a practical scale. Lancet. 1974;304(7872):81–84.
- , , , . Short Portable Mental Status Questionnaire as a Screening Test for Dementia and Delirium Among the Elderly. J Am Geriatr Soc. 1987;35(5):412–416.
- , , , et al. Effects of physician experience on costs and outcomes on an academic general medicine service: results of a trial of hospitalists. Ann Intern Med. 2002;137(11):866–874.
- , , . Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44(3):837–845.
- , , , et al. Preliminary development of an ultrabrief two‐item bedside test for delirium. J Hosp Med. 2015;10(10):645–650.
- , , , , , . The association between an ultrabrief cognitive screening in older adults and hospital outcomes. J Hosp Med. 2015;10(10):651–657.
- , , , et al. Selecting optimal screening items for delirium: an application of item response theory. BMC Med Res Methodol. 2013;13:8.
- , , . Variability in agreement between physicians and nurses when measuring the Glasgow Coma Scale in the emergency department limits its clinical usefulness. Emerg Med Australas. 2006;18(4):379–384.
SDEF: Severe acne responds to fixed-combo gel
A convenient, once-daily fixed combination of 0.3% adapalene plus 2.5% benzoyl peroxide gel significantly improved lesion counts over the course of 12 weeks in patients aged 12 years and older with moderate or severe acne.
Investigators enrolled just over 500 patients from 31 sites in the United States and Canada. About half of patients were rated as having severe acne and half as having moderate acne on the investigator’s global assessment (IGA) scale, Dr. Linda F. Stein Gold said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.

Patients were randomized to three treatment groups: adapalene 0.3%/benzoyl peroxide 2.5% gel (A-BPO-0.3%), adapalene 0.1%/benzoyl peroxide 2.5% (A-BPO-0.1%), or vehicle. Patients in each group had approximately the same total lesion count, and about half in each group had truncal acne lesions, said Dr. Stein Gold, director of clinical research in the department of dermatology at Henry Ford Hospital, Detroit.
Patients were instructed to use their study medications once daily at night after washing with a provided cleanser. They were provided with a standardized moisturizer and cleaners.
Treatment with A-BPO-0.3% was judged as successful (IGA of 1 or almost clear) at 12 weeks in 31% of patients with severe acne. By contrast, 13.3% of patients with severe acne were judged as almost clear. In patients with severe acne, A-BPO-1% was not statistically superior to vehicle (J Drugs Dermatol. 2015 Dec 1;14[12]:1427-35).
“Topical treatment is still the cornerstone of acne therapy, and it is great to have additional options, especially for our more severe acne patients,” Dr. Stein Gold said.
Patients noted dryness, scaling, erythema, and stinging/burning with A-BPO-0.3%, especially between weeks 1 and 2.
Dr. Stein Gold disclosed that she serves as a consultant and scientific advisory board member to Galderma, which markets A-BPO-0.3% as Epiduo Forte.
SDEF and this news organization are owned by the same parent company.
On Twitter @denisefulton
A convenient, once-daily fixed combination of 0.3% adapalene plus 2.5% benzoyl peroxide gel significantly improved lesion counts over the course of 12 weeks in patients aged 12 years and older with moderate or severe acne.
Investigators enrolled just over 500 patients from 31 sites in the United States and Canada. About half of patients were rated as having severe acne and half as having moderate acne on the investigator’s global assessment (IGA) scale, Dr. Linda F. Stein Gold said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
Patients were randomized to three treatment groups: adapalene 0.3%/benzoyl peroxide 2.5% gel (A-BPO-0.3%), adapalene 0.1%/benzoyl peroxide 2.5% (A-BPO-0.1%), or vehicle. Patients in each group had approximately the same total lesion count, and about half in each group had truncal acne lesions, said Dr. Stein Gold, director of clinical research in the department of dermatology at Henry Ford Hospital, Detroit.
Patients were instructed to use their study medications once daily at night after washing with a provided cleanser. They were provided with a standardized moisturizer and cleaners.
Treatment with A-BPO-0.3% was judged as successful (IGA of 1 or almost clear) at 12 weeks in 31% of patients with severe acne. By contrast, 13.3% of patients with severe acne were judged as almost clear. In patients with severe acne, A-BPO-1% was not statistically superior to vehicle (J Drugs Dermatol. 2015 Dec 1;14[12]:1427-35).
“Topical treatment is still the cornerstone of acne therapy, and it is great to have additional options, especially for our more severe acne patients,” Dr. Stein Gold said.
Patients noted dryness, scaling, erythema, and stinging/burning with A-BPO-0.3%, especially between weeks 1 and 2.
Dr. Stein Gold disclosed that she serves as a consultant and scientific advisory board member to Galderma, which markets A-BPO-0.3% as Epiduo Forte.
SDEF and this news organization are owned by the same parent company.
On Twitter @denisefulton
A convenient, once-daily fixed combination of 0.3% adapalene plus 2.5% benzoyl peroxide gel significantly improved lesion counts over the course of 12 weeks in patients aged 12 years and older with moderate or severe acne.
Investigators enrolled just over 500 patients from 31 sites in the United States and Canada. About half of patients were rated as having severe acne and half as having moderate acne on the investigator’s global assessment (IGA) scale, Dr. Linda F. Stein Gold said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
Patients were randomized to three treatment groups: adapalene 0.3%/benzoyl peroxide 2.5% gel (A-BPO-0.3%), adapalene 0.1%/benzoyl peroxide 2.5% (A-BPO-0.1%), or vehicle. Patients in each group had approximately the same total lesion count, and about half in each group had truncal acne lesions, said Dr. Stein Gold, director of clinical research in the department of dermatology at Henry Ford Hospital, Detroit.
Patients were instructed to use their study medications once daily at night after washing with a provided cleanser. They were provided with a standardized moisturizer and cleaners.
Treatment with A-BPO-0.3% was judged as successful (IGA of 1 or almost clear) at 12 weeks in 31% of patients with severe acne. By contrast, 13.3% of patients with severe acne were judged as almost clear. In patients with severe acne, A-BPO-1% was not statistically superior to vehicle (J Drugs Dermatol. 2015 Dec 1;14[12]:1427-35).
“Topical treatment is still the cornerstone of acne therapy, and it is great to have additional options, especially for our more severe acne patients,” Dr. Stein Gold said.
Patients noted dryness, scaling, erythema, and stinging/burning with A-BPO-0.3%, especially between weeks 1 and 2.
Dr. Stein Gold disclosed that she serves as a consultant and scientific advisory board member to Galderma, which markets A-BPO-0.3% as Epiduo Forte.
SDEF and this news organization are owned by the same parent company.
On Twitter @denisefulton
EXPERT ANALYSIS FROM SDEF HAWAII DERMATOLOGY SEMINAR

Study Shows Best Performing Hospitals Manage Pain Best
NEW YORK (Reuters Health) - Hospital differences in pain management are tied to different patient-reported pain scores after colorectal resection, according to a study from the Michigan Surgical Quality Collaborative (MSQC).
"The best-performing hospitals achieved superior pain control through the use of analgesia regimens that more often used local anesthesia blocks in the operating room,non-steroidal anti-inflammatory drugs, and patient-controlled analgesia rather than intermittent narcotics," said Dr. Scott E. Regenbogen from the University of Michigan, Ann Arbor.
"This suggests that efforts to implement multimodal analgesia regimens may improve pain control in the immediate postoperative period," he told Reuters Health by email.
Despite increasing attention to postoperative pain management, most patients continue to experience severe pain after major surgery, Dr. Regenbogen and colleagues note in Annals of Surgery, online January 7.
The researchers used MSQC data from 52 hospitals to evaluate the extent to which multimodal pain management practices are used after major surgery and how hospitals' perioperative practices might affect patient-reported pain levels in real-world surgical practice. Their study included 7,221 patients who underwent colorectal resection between 2012 and 2014.
Nine hospitals had average adjusted pain scores significantly worse and eight hospitals had average adjusted pain scores significantly better than the overall average. The "best" hospitals were somewhat larger and had higher annual volumes of colorectal resection than the "worst" hospitals.
Patients in hospitals with best pain scores were significantly more likely to receive local anesthesia and epidural anesthesia during the operation and to have patient-controlled analgesia (PCA), nonsteroidal anti-inflammatory drugs (NSAIDs), or a combination of PCA and NSAIDs and significantly less likely to receive intermittent postoperative narcotics, compared with patients in hospitals with worst pain scores.
Hospitals with the lowest pain scores had a significantly higher proportion of operations with a minimally invasive approach than did hospitals with the highest pain scores.
Patients whose operations took place in hospitals in the lowest quartile of pain scores had significantly shorter mean postoperative length of stay and were significantly less likely to have a postoperative complication, emergency department visit, or readmission.
Patient factors contributing to worse pain scores included being younger than 50 (versus age over 75), a woman, black (versus white), a smoker, and uninsured or insured by Medicaid (versus Medicare or privately insured).
"Most likely," the researchers note, "both pain scores and clinical outcomes reflect more global features of the quality of care in hospitals' surgical performance. Thus, hospitals with the most streamlined, high-quality perioperative care pathways experience the best pain scores, as well as improved clinical outcomes."
"Early postoperative analgesia regimens are an essential component of efforts to improve the efficiency and quality of postoperative recovery," Dr. Regenbogen said."Effective pain control, even in the first postoperative day, is associated with reduced postoperative length of stay and fewer major complications and readmissions. Thus, effective multimodal analgesia is an essential component of high-value perioperative care around colorectal surgery."
"This study is hopefully just one example of a growing emphasis on patient-reported outcomes in surgery," Dr.Regenbogen said. "Within a statewide quality collaborative, we have begun to prioritize engagement of patients, in addition to our partnering surgeons, hospitals, and mayors, in efforts to improve surgical care in Michigan. In this study, we used patient-reported pain measures to evaluate quality of care. In the near future, we will elicit patient-reported measures of functional recovery, psychosocial support, and other outcomes to validate the perioperative outcomes we have relied on traditionally. We hope this study will serve as a model for those novel areas of investigation."
Dr. Antoni Sabate from Hospital Universitari de Bellvitge in Barcelona, Spain, who has researched postoperative analgesia told Reuters Health by email, "Pain is largely influenced by magnitude of the surgical procedure, surgical technique (minimally invasive), analgesia protocol (the use of local anesthetic infiltration in open and laparoscopic, the use of epidural in open procedures, implementation of PCA and NSAID in both laparoscopic and open procedures."
NEW YORK (Reuters Health) - Hospital differences in pain management are tied to different patient-reported pain scores after colorectal resection, according to a study from the Michigan Surgical Quality Collaborative (MSQC).
"The best-performing hospitals achieved superior pain control through the use of analgesia regimens that more often used local anesthesia blocks in the operating room,non-steroidal anti-inflammatory drugs, and patient-controlled analgesia rather than intermittent narcotics," said Dr. Scott E. Regenbogen from the University of Michigan, Ann Arbor.
"This suggests that efforts to implement multimodal analgesia regimens may improve pain control in the immediate postoperative period," he told Reuters Health by email.
Despite increasing attention to postoperative pain management, most patients continue to experience severe pain after major surgery, Dr. Regenbogen and colleagues note in Annals of Surgery, online January 7.
The researchers used MSQC data from 52 hospitals to evaluate the extent to which multimodal pain management practices are used after major surgery and how hospitals' perioperative practices might affect patient-reported pain levels in real-world surgical practice. Their study included 7,221 patients who underwent colorectal resection between 2012 and 2014.
Nine hospitals had average adjusted pain scores significantly worse and eight hospitals had average adjusted pain scores significantly better than the overall average. The "best" hospitals were somewhat larger and had higher annual volumes of colorectal resection than the "worst" hospitals.
Patients in hospitals with best pain scores were significantly more likely to receive local anesthesia and epidural anesthesia during the operation and to have patient-controlled analgesia (PCA), nonsteroidal anti-inflammatory drugs (NSAIDs), or a combination of PCA and NSAIDs and significantly less likely to receive intermittent postoperative narcotics, compared with patients in hospitals with worst pain scores.
Hospitals with the lowest pain scores had a significantly higher proportion of operations with a minimally invasive approach than did hospitals with the highest pain scores.
Patients whose operations took place in hospitals in the lowest quartile of pain scores had significantly shorter mean postoperative length of stay and were significantly less likely to have a postoperative complication, emergency department visit, or readmission.
Patient factors contributing to worse pain scores included being younger than 50 (versus age over 75), a woman, black (versus white), a smoker, and uninsured or insured by Medicaid (versus Medicare or privately insured).
"Most likely," the researchers note, "both pain scores and clinical outcomes reflect more global features of the quality of care in hospitals' surgical performance. Thus, hospitals with the most streamlined, high-quality perioperative care pathways experience the best pain scores, as well as improved clinical outcomes."
"Early postoperative analgesia regimens are an essential component of efforts to improve the efficiency and quality of postoperative recovery," Dr. Regenbogen said."Effective pain control, even in the first postoperative day, is associated with reduced postoperative length of stay and fewer major complications and readmissions. Thus, effective multimodal analgesia is an essential component of high-value perioperative care around colorectal surgery."
"This study is hopefully just one example of a growing emphasis on patient-reported outcomes in surgery," Dr.Regenbogen said. "Within a statewide quality collaborative, we have begun to prioritize engagement of patients, in addition to our partnering surgeons, hospitals, and mayors, in efforts to improve surgical care in Michigan. In this study, we used patient-reported pain measures to evaluate quality of care. In the near future, we will elicit patient-reported measures of functional recovery, psychosocial support, and other outcomes to validate the perioperative outcomes we have relied on traditionally. We hope this study will serve as a model for those novel areas of investigation."
Dr. Antoni Sabate from Hospital Universitari de Bellvitge in Barcelona, Spain, who has researched postoperative analgesia told Reuters Health by email, "Pain is largely influenced by magnitude of the surgical procedure, surgical technique (minimally invasive), analgesia protocol (the use of local anesthetic infiltration in open and laparoscopic, the use of epidural in open procedures, implementation of PCA and NSAID in both laparoscopic and open procedures."
NEW YORK (Reuters Health) - Hospital differences in pain management are tied to different patient-reported pain scores after colorectal resection, according to a study from the Michigan Surgical Quality Collaborative (MSQC).
"The best-performing hospitals achieved superior pain control through the use of analgesia regimens that more often used local anesthesia blocks in the operating room,non-steroidal anti-inflammatory drugs, and patient-controlled analgesia rather than intermittent narcotics," said Dr. Scott E. Regenbogen from the University of Michigan, Ann Arbor.
"This suggests that efforts to implement multimodal analgesia regimens may improve pain control in the immediate postoperative period," he told Reuters Health by email.
Despite increasing attention to postoperative pain management, most patients continue to experience severe pain after major surgery, Dr. Regenbogen and colleagues note in Annals of Surgery, online January 7.
The researchers used MSQC data from 52 hospitals to evaluate the extent to which multimodal pain management practices are used after major surgery and how hospitals' perioperative practices might affect patient-reported pain levels in real-world surgical practice. Their study included 7,221 patients who underwent colorectal resection between 2012 and 2014.
Nine hospitals had average adjusted pain scores significantly worse and eight hospitals had average adjusted pain scores significantly better than the overall average. The "best" hospitals were somewhat larger and had higher annual volumes of colorectal resection than the "worst" hospitals.
Patients in hospitals with best pain scores were significantly more likely to receive local anesthesia and epidural anesthesia during the operation and to have patient-controlled analgesia (PCA), nonsteroidal anti-inflammatory drugs (NSAIDs), or a combination of PCA and NSAIDs and significantly less likely to receive intermittent postoperative narcotics, compared with patients in hospitals with worst pain scores.
Hospitals with the lowest pain scores had a significantly higher proportion of operations with a minimally invasive approach than did hospitals with the highest pain scores.
Patients whose operations took place in hospitals in the lowest quartile of pain scores had significantly shorter mean postoperative length of stay and were significantly less likely to have a postoperative complication, emergency department visit, or readmission.
Patient factors contributing to worse pain scores included being younger than 50 (versus age over 75), a woman, black (versus white), a smoker, and uninsured or insured by Medicaid (versus Medicare or privately insured).
"Most likely," the researchers note, "both pain scores and clinical outcomes reflect more global features of the quality of care in hospitals' surgical performance. Thus, hospitals with the most streamlined, high-quality perioperative care pathways experience the best pain scores, as well as improved clinical outcomes."
"Early postoperative analgesia regimens are an essential component of efforts to improve the efficiency and quality of postoperative recovery," Dr. Regenbogen said."Effective pain control, even in the first postoperative day, is associated with reduced postoperative length of stay and fewer major complications and readmissions. Thus, effective multimodal analgesia is an essential component of high-value perioperative care around colorectal surgery."
"This study is hopefully just one example of a growing emphasis on patient-reported outcomes in surgery," Dr.Regenbogen said. "Within a statewide quality collaborative, we have begun to prioritize engagement of patients, in addition to our partnering surgeons, hospitals, and mayors, in efforts to improve surgical care in Michigan. In this study, we used patient-reported pain measures to evaluate quality of care. In the near future, we will elicit patient-reported measures of functional recovery, psychosocial support, and other outcomes to validate the perioperative outcomes we have relied on traditionally. We hope this study will serve as a model for those novel areas of investigation."
Dr. Antoni Sabate from Hospital Universitari de Bellvitge in Barcelona, Spain, who has researched postoperative analgesia told Reuters Health by email, "Pain is largely influenced by magnitude of the surgical procedure, surgical technique (minimally invasive), analgesia protocol (the use of local anesthetic infiltration in open and laparoscopic, the use of epidural in open procedures, implementation of PCA and NSAID in both laparoscopic and open procedures."
NICE says it can’t recommend sepsis tests

Photo by Juan D. Alfonso
The National Institute for Health and Care Excellence (NICE) has said there is not enough evidence to recommend the routine use of 3 new tests to help identify the cause of sepsis.
The agency issued a final diagnostics guidance recommending further research on the tests—the LightCycler SeptiFast Test MGRADE (Roche Diagnostics), SepsiTest (Molzym Molecular Diagnostics), and IRIDICA BAC BSI assay (Abbott Laboratories).
All 3 tests analyze whole blood samples to identify bacterial and fungal DNA. The tests aim to identify the causes of infection much quicker than traditional microbiology techniques, which require blood samples to be incubated and cultured before pathogens can be identified.
“Rapid molecular tests that can identify the cause of an infection in hours rather than the days typically needed for traditional microbiology tests could ensure the most appropriate antibiotics are used much earlier,” said Carole Longson, NICE Health Technology Evaluation Centre Director.
“This, in turn, could improve outcomes for patients with suspected bloodstream infections, as well as help to reduce the spread of resistant microbes. The independent diagnostics advisory committee [advising NICE on the tests] concluded that, although the tests show promise, there is currently not enough evidence to recommend their routine adoption in the NHS [National Health Service].”
“They felt the tests may offer clinical benefit by providing results more quickly, but there was currently too much uncertainty in their accuracy for clinicians to be able to use them as the basis for clinical decision-making in people with suspected bloodstream infections, who can be acutely unwell.”
“The committee therefore decided that further research should be encouraged to provide robust evidence, particularly around demonstrating the value of using the test results in clinical decision-making.”
The diagnostics guidance on the tests is available on the NICE website. ![]()
Photo by Juan D. Alfonso
The National Institute for Health and Care Excellence (NICE) has said there is not enough evidence to recommend the routine use of 3 new tests to help identify the cause of sepsis.
The agency issued a final diagnostics guidance recommending further research on the tests—the LightCycler SeptiFast Test MGRADE (Roche Diagnostics), SepsiTest (Molzym Molecular Diagnostics), and IRIDICA BAC BSI assay (Abbott Laboratories).
All 3 tests analyze whole blood samples to identify bacterial and fungal DNA. The tests aim to identify the causes of infection much quicker than traditional microbiology techniques, which require blood samples to be incubated and cultured before pathogens can be identified.
“Rapid molecular tests that can identify the cause of an infection in hours rather than the days typically needed for traditional microbiology tests could ensure the most appropriate antibiotics are used much earlier,” said Carole Longson, NICE Health Technology Evaluation Centre Director.
“This, in turn, could improve outcomes for patients with suspected bloodstream infections, as well as help to reduce the spread of resistant microbes. The independent diagnostics advisory committee [advising NICE on the tests] concluded that, although the tests show promise, there is currently not enough evidence to recommend their routine adoption in the NHS [National Health Service].”
“They felt the tests may offer clinical benefit by providing results more quickly, but there was currently too much uncertainty in their accuracy for clinicians to be able to use them as the basis for clinical decision-making in people with suspected bloodstream infections, who can be acutely unwell.”
“The committee therefore decided that further research should be encouraged to provide robust evidence, particularly around demonstrating the value of using the test results in clinical decision-making.”
The diagnostics guidance on the tests is available on the NICE website. ![]()
Photo by Juan D. Alfonso
The National Institute for Health and Care Excellence (NICE) has said there is not enough evidence to recommend the routine use of 3 new tests to help identify the cause of sepsis.
The agency issued a final diagnostics guidance recommending further research on the tests—the LightCycler SeptiFast Test MGRADE (Roche Diagnostics), SepsiTest (Molzym Molecular Diagnostics), and IRIDICA BAC BSI assay (Abbott Laboratories).
All 3 tests analyze whole blood samples to identify bacterial and fungal DNA. The tests aim to identify the causes of infection much quicker than traditional microbiology techniques, which require blood samples to be incubated and cultured before pathogens can be identified.
“Rapid molecular tests that can identify the cause of an infection in hours rather than the days typically needed for traditional microbiology tests could ensure the most appropriate antibiotics are used much earlier,” said Carole Longson, NICE Health Technology Evaluation Centre Director.
“This, in turn, could improve outcomes for patients with suspected bloodstream infections, as well as help to reduce the spread of resistant microbes. The independent diagnostics advisory committee [advising NICE on the tests] concluded that, although the tests show promise, there is currently not enough evidence to recommend their routine adoption in the NHS [National Health Service].”
“They felt the tests may offer clinical benefit by providing results more quickly, but there was currently too much uncertainty in their accuracy for clinicians to be able to use them as the basis for clinical decision-making in people with suspected bloodstream infections, who can be acutely unwell.”
“The committee therefore decided that further research should be encouraged to provide robust evidence, particularly around demonstrating the value of using the test results in clinical decision-making.”
The diagnostics guidance on the tests is available on the NICE website. ![]()
SDEF: New, aggressive strategies show promise in alopecia areata
Alopecia areata’s mysterious appearances, regressions, and recurrences frustrate patients and stymie physicians, but new treatments may be around the corner.
Tofacitinib, along with other medications that target the autoimmune etiology of alopecia areata, have shown complete alopecia reversal in case studies, Dr. Maria Hordinsky said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation. “There’s a lot of excitement bubbling up in hair disease research because of these new potential topical and oral treatments.”

Janus kinase (JAK) inhibitors, including tofacitinib, baricitinib, and ruxolitinib, have also been reported to reverse alopecia areata.
“There’s been a surge of enthusiasm for using more aggressive systemic therapies, including not only tobacitinib and ruxolitinib but also methotrexate and interleukin-2,” Dr. Hordinsky said, noting that these are still investigational uses.
The new treatment targets are welcome for physicians treating patients with alopecia areata, since currently there are no FDA-approved treatments, Dr. Hordinsky said.
A review by Dr. Hordinsky and colleague found a total of 29 trials investigating more than a dozen topical and oral treatments. Most trials were of moderate or lower quality, and most had major limitations. Treatments that were effective included topical and oral corticosteroids, as well as the sensitizing agents diphenylcyclopropenone and dinitrochlorobenzene (Am J Clin Dermatol. 2014;15:231-46).
In the absence of high-quality evidence for effective treatments, patient characteristics and preference, as well as disease activity and location, can guide treatment. In some cases, a scalp biopsy can give more information about follicle differentiation, inflammation, and the stage of the hair cycle at the time of assessment, Dr. Hordinsky said.
It’s important to set expectations for patients, so they know that treatments will take time, she said. Providers should be alert to the possibility that hair loss may also be associated with an underlying medical problem, so a thorough workup is indicated.
Patients should be given the opportunity to enroll in clinical trials, where available, and should also be directed to the National Alopecia Areata Foundation (NAAF). Their website provides information and resources for patients and families, information for local support groups, and information on a national registry.
Dr. Hordinsky reported receiving grant or research support from a number of pharmaceutical and consumer product companies in the dermatology space. She serves on the scientific advisory board of the National Alopecia Areata Foundation.
This news organization and SDEF are owned by the same parent company.
On Twitter @karioakes
Alopecia areata’s mysterious appearances, regressions, and recurrences frustrate patients and stymie physicians, but new treatments may be around the corner.
Tofacitinib, along with other medications that target the autoimmune etiology of alopecia areata, have shown complete alopecia reversal in case studies, Dr. Maria Hordinsky said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation. “There’s a lot of excitement bubbling up in hair disease research because of these new potential topical and oral treatments.”
Janus kinase (JAK) inhibitors, including tofacitinib, baricitinib, and ruxolitinib, have also been reported to reverse alopecia areata.
“There’s been a surge of enthusiasm for using more aggressive systemic therapies, including not only tobacitinib and ruxolitinib but also methotrexate and interleukin-2,” Dr. Hordinsky said, noting that these are still investigational uses.
The new treatment targets are welcome for physicians treating patients with alopecia areata, since currently there are no FDA-approved treatments, Dr. Hordinsky said.
A review by Dr. Hordinsky and colleague found a total of 29 trials investigating more than a dozen topical and oral treatments. Most trials were of moderate or lower quality, and most had major limitations. Treatments that were effective included topical and oral corticosteroids, as well as the sensitizing agents diphenylcyclopropenone and dinitrochlorobenzene (Am J Clin Dermatol. 2014;15:231-46).
In the absence of high-quality evidence for effective treatments, patient characteristics and preference, as well as disease activity and location, can guide treatment. In some cases, a scalp biopsy can give more information about follicle differentiation, inflammation, and the stage of the hair cycle at the time of assessment, Dr. Hordinsky said.
It’s important to set expectations for patients, so they know that treatments will take time, she said. Providers should be alert to the possibility that hair loss may also be associated with an underlying medical problem, so a thorough workup is indicated.
Patients should be given the opportunity to enroll in clinical trials, where available, and should also be directed to the National Alopecia Areata Foundation (NAAF). Their website provides information and resources for patients and families, information for local support groups, and information on a national registry.
Dr. Hordinsky reported receiving grant or research support from a number of pharmaceutical and consumer product companies in the dermatology space. She serves on the scientific advisory board of the National Alopecia Areata Foundation.
This news organization and SDEF are owned by the same parent company.
On Twitter @karioakes
Alopecia areata’s mysterious appearances, regressions, and recurrences frustrate patients and stymie physicians, but new treatments may be around the corner.
Tofacitinib, along with other medications that target the autoimmune etiology of alopecia areata, have shown complete alopecia reversal in case studies, Dr. Maria Hordinsky said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation. “There’s a lot of excitement bubbling up in hair disease research because of these new potential topical and oral treatments.”
Janus kinase (JAK) inhibitors, including tofacitinib, baricitinib, and ruxolitinib, have also been reported to reverse alopecia areata.
“There’s been a surge of enthusiasm for using more aggressive systemic therapies, including not only tobacitinib and ruxolitinib but also methotrexate and interleukin-2,” Dr. Hordinsky said, noting that these are still investigational uses.
The new treatment targets are welcome for physicians treating patients with alopecia areata, since currently there are no FDA-approved treatments, Dr. Hordinsky said.
A review by Dr. Hordinsky and colleague found a total of 29 trials investigating more than a dozen topical and oral treatments. Most trials were of moderate or lower quality, and most had major limitations. Treatments that were effective included topical and oral corticosteroids, as well as the sensitizing agents diphenylcyclopropenone and dinitrochlorobenzene (Am J Clin Dermatol. 2014;15:231-46).
In the absence of high-quality evidence for effective treatments, patient characteristics and preference, as well as disease activity and location, can guide treatment. In some cases, a scalp biopsy can give more information about follicle differentiation, inflammation, and the stage of the hair cycle at the time of assessment, Dr. Hordinsky said.
It’s important to set expectations for patients, so they know that treatments will take time, she said. Providers should be alert to the possibility that hair loss may also be associated with an underlying medical problem, so a thorough workup is indicated.
Patients should be given the opportunity to enroll in clinical trials, where available, and should also be directed to the National Alopecia Areata Foundation (NAAF). Their website provides information and resources for patients and families, information for local support groups, and information on a national registry.
Dr. Hordinsky reported receiving grant or research support from a number of pharmaceutical and consumer product companies in the dermatology space. She serves on the scientific advisory board of the National Alopecia Areata Foundation.
This news organization and SDEF are owned by the same parent company.
On Twitter @karioakes
EXPERT ANALYSIS FROM SDEF HAWAII DERMATOLOGY SEMINAR

SDEF: New clues emerge in scarring alopecias
Some kinds of scarring alopecia may also be related to lipid metabolism, pointing the way to novel therapeutic targets for this class of hair diseases, Dr. Maria Hordinsky said at the Hawaii Dermatology Symposium.
While the scarring alopecias are broadly divided into lymphocytic and neutrophilic alopecias, clinically, they are a heterogeneous group, said Dr. Hordinsky, chair of the department of dermatology at the University of Minnesota, Minneapolis.

Two lymphocytic cicatricial alopecias, lichen planopilaris and frontal fibrosing alopecia, have been associated with peroxisome proliferator-activated receptor gamma (PPARG) deficiency. This finding suggests that they may be a type of lipid metabolism disorder; the theory is bolstered by case reports of improvement with the use of pioglitazone, Dr. Hordinsky said at the meeting provided by Global Academy for Medical Education/Skin Disease Education Foundation.
In both of these cicatricial alopecias, patients may also have significant discomfort including scalp burning, pain, paresthesias, and itching. Patients may even say they feel as though their scalp is on fire.
Dr. Hordinsky and her collaborators are currently conducting a clinical trial of the efficacy of a compounded formulation of 6% topical gabapentin to address the scalp discomfort associated with the cicatricial alopecias. The study is ongoing and recruiting patients. “FFA has been described as an emerging disease, and participation of FFA patients in a national registry focused on improving our understanding of the epidemiology of this disease is highly recommended,” said Dr. Hordinsky.
Currently, stepwise treatment for the lymphocytic cicatricial alopecias can be divided into 3 tiers:
• Tier 1: Topical high potency corticosteroids and/or intralesional steroids, as well as non-steroidal topical anti-inflammatory medications, such as tacrolimus and pemicrolimus.
• Tier 2: Hydroxychloroquine and acetretin, as well as low-dose antibiotics, which are used for anti-inflammatory effect.
• Tier 3: Cyclosporin, mycophenolate mofetil, and prednisone.
Low-level light therapy – approved by the Food and Drug Administration to treat thinning hair in men and women – may also be of benefit for those with inflammatory scalp diseases, Dr. Hordinsky added.
Dr. Hordinsky reported financial relationships with a number of pharmaceutical and consumer product companies in the dermatologic space.
This news organization and SDEF are owned by the same parent company.
On Twitter @karioakes
Some kinds of scarring alopecia may also be related to lipid metabolism, pointing the way to novel therapeutic targets for this class of hair diseases, Dr. Maria Hordinsky said at the Hawaii Dermatology Symposium.
While the scarring alopecias are broadly divided into lymphocytic and neutrophilic alopecias, clinically, they are a heterogeneous group, said Dr. Hordinsky, chair of the department of dermatology at the University of Minnesota, Minneapolis.
Two lymphocytic cicatricial alopecias, lichen planopilaris and frontal fibrosing alopecia, have been associated with peroxisome proliferator-activated receptor gamma (PPARG) deficiency. This finding suggests that they may be a type of lipid metabolism disorder; the theory is bolstered by case reports of improvement with the use of pioglitazone, Dr. Hordinsky said at the meeting provided by Global Academy for Medical Education/Skin Disease Education Foundation.
In both of these cicatricial alopecias, patients may also have significant discomfort including scalp burning, pain, paresthesias, and itching. Patients may even say they feel as though their scalp is on fire.
Dr. Hordinsky and her collaborators are currently conducting a clinical trial of the efficacy of a compounded formulation of 6% topical gabapentin to address the scalp discomfort associated with the cicatricial alopecias. The study is ongoing and recruiting patients. “FFA has been described as an emerging disease, and participation of FFA patients in a national registry focused on improving our understanding of the epidemiology of this disease is highly recommended,” said Dr. Hordinsky.
Currently, stepwise treatment for the lymphocytic cicatricial alopecias can be divided into 3 tiers:
• Tier 1: Topical high potency corticosteroids and/or intralesional steroids, as well as non-steroidal topical anti-inflammatory medications, such as tacrolimus and pemicrolimus.
• Tier 2: Hydroxychloroquine and acetretin, as well as low-dose antibiotics, which are used for anti-inflammatory effect.
• Tier 3: Cyclosporin, mycophenolate mofetil, and prednisone.
Low-level light therapy – approved by the Food and Drug Administration to treat thinning hair in men and women – may also be of benefit for those with inflammatory scalp diseases, Dr. Hordinsky added.
Dr. Hordinsky reported financial relationships with a number of pharmaceutical and consumer product companies in the dermatologic space.
This news organization and SDEF are owned by the same parent company.
On Twitter @karioakes
Some kinds of scarring alopecia may also be related to lipid metabolism, pointing the way to novel therapeutic targets for this class of hair diseases, Dr. Maria Hordinsky said at the Hawaii Dermatology Symposium.
While the scarring alopecias are broadly divided into lymphocytic and neutrophilic alopecias, clinically, they are a heterogeneous group, said Dr. Hordinsky, chair of the department of dermatology at the University of Minnesota, Minneapolis.
Two lymphocytic cicatricial alopecias, lichen planopilaris and frontal fibrosing alopecia, have been associated with peroxisome proliferator-activated receptor gamma (PPARG) deficiency. This finding suggests that they may be a type of lipid metabolism disorder; the theory is bolstered by case reports of improvement with the use of pioglitazone, Dr. Hordinsky said at the meeting provided by Global Academy for Medical Education/Skin Disease Education Foundation.
In both of these cicatricial alopecias, patients may also have significant discomfort including scalp burning, pain, paresthesias, and itching. Patients may even say they feel as though their scalp is on fire.
Dr. Hordinsky and her collaborators are currently conducting a clinical trial of the efficacy of a compounded formulation of 6% topical gabapentin to address the scalp discomfort associated with the cicatricial alopecias. The study is ongoing and recruiting patients. “FFA has been described as an emerging disease, and participation of FFA patients in a national registry focused on improving our understanding of the epidemiology of this disease is highly recommended,” said Dr. Hordinsky.
Currently, stepwise treatment for the lymphocytic cicatricial alopecias can be divided into 3 tiers:
• Tier 1: Topical high potency corticosteroids and/or intralesional steroids, as well as non-steroidal topical anti-inflammatory medications, such as tacrolimus and pemicrolimus.
• Tier 2: Hydroxychloroquine and acetretin, as well as low-dose antibiotics, which are used for anti-inflammatory effect.
• Tier 3: Cyclosporin, mycophenolate mofetil, and prednisone.
Low-level light therapy – approved by the Food and Drug Administration to treat thinning hair in men and women – may also be of benefit for those with inflammatory scalp diseases, Dr. Hordinsky added.
Dr. Hordinsky reported financial relationships with a number of pharmaceutical and consumer product companies in the dermatologic space.
This news organization and SDEF are owned by the same parent company.
On Twitter @karioakes
EXPERT ANALYSIS FROM SDEF HAWAII DERMATOLOGY SYMPOSIUM

Team finds potential marker of long-term HSCs

in the bone marrow
Nearly 30 years after the discovery of the hematopoietic stem cell (HSC), researchers believe they have found a marker specific to long-term HSCs—the gene Hoxb5.
If confirmed, this finding would help settle long-standing debates about the identity of long-term HSCs and their support cells.
It may also pave the way for understanding how long-term HSCs maintain themselves and provide scientists with the necessary information to grow HSCs in the lab.
Irving Weissman, MD, of Stanford University School of Medicine in California, and his colleagues described this work in a letter to Nature.
In 1988, Dr Weissman and his colleagues isolated the HSC. Since that time, researchers have had mixed success in their attempts to get a detailed picture of how HSCs maintain themselves and grow in the body.
Over the years, it became clear why. HSCs come in 2 forms—short-term HSCs that lose their powers of replication over time and long-term HSCs that can replicate indefinitely.
With the new study, Dr Weissman and his colleagues believe they have found a reliable way to tell the difference between long-term and short-term HSCs. Namely, the presence of Hoxb5.
“In this paper, we have found a single marker that, in the entire bone marrow, is only found in these long-term stem cells,” Dr Weissman said.
The researchers hope this finding will enable them to look at how nearby cells create a niche where the long-term HSCs are supported and maintained.
“For nearly 30 years, people have been trying to grow HSCs outside the body and have not been able to do it; it’s arguably the ‘holy grail’ in this field,” said James Y. Chen, an MD/PhD candidate at Stanford University School of Medicine.
“Now that we have an anchor, a way to look at long-term HSCs, we can look at the cells around them to understand and, ideally, recreate the niche.”
An extensive search
Over the years, scientists have proposed various markers that they felt were unique to long-term HSCs, but the reliability of each proposed marker has been heatedly debated by other research groups, said Masanori Miyanishi, MD, PhD, also of Stanford University School of Medicine.
In an attempt to settle the issue, Chen and Dr Miyanishi examined a list of more than 100 genes that are expressed in the bone marrow and seemed like good candidates to be unique markers of long-term HSCs.
The researchers eliminated genes that are turned on in areas of the bone that don’t involve the creation of new blood and immune cells. That narrowed the field to 45 genes.
The team then performed an analysis to determine how much protein these genes were making in various cells. They found that only 3 proteins were produced at a high enough level to mark HSCs.
Finally, the researchers needed to find if 1 of these 3 was turned on in long-term HSCs and turned off in short-term HSCs.
Although they couldn’t yet identify which cells were long-term HSCs, the team knew that any collection of HSCs should have both long-term and short-term HSCs, so they expected to find the candidate gene turned off in some cells and on in others. Only 1 gene fit that bill—Hoxb5.
The researchers pointed out that there may be other unique markers of long-term HSCs, such as genes that weren’t among the initial group of genes screened. But among the screened genes, only Hoxb5 was a unique identifier of the long-term HSC.
Finding the niche
The researchers were also able to solve another mystery by showing where in the bone marrow long-term HSCs reside.
Satoshi Yamazaki, PhD, and Hiromitsu Nakauchi, MD, PhD, both from the University of Tokyo in Japan, used new technology to prepare bone marrow tissue and do computational analyses that validated the location and architecture of the HSC niche.
“More than 90% of these cells reside on a particular type of blood vessel called venous sinusoids,” Dr Nakauchi said.
The researchers believe the ability to identify long-term HSCs will give scientists a powerful tool for further study.
“This opens the way to observe long-term HSCs and other cells in the niche as they exist in the body, without transplanting them,” Dr Weissman said. “This is how science works, by getting down to the purest irreducible element—in this case, blood stem cells—in order to develop new tools and understandings.” ![]()
in the bone marrow
Nearly 30 years after the discovery of the hematopoietic stem cell (HSC), researchers believe they have found a marker specific to long-term HSCs—the gene Hoxb5.
If confirmed, this finding would help settle long-standing debates about the identity of long-term HSCs and their support cells.
It may also pave the way for understanding how long-term HSCs maintain themselves and provide scientists with the necessary information to grow HSCs in the lab.
Irving Weissman, MD, of Stanford University School of Medicine in California, and his colleagues described this work in a letter to Nature.
In 1988, Dr Weissman and his colleagues isolated the HSC. Since that time, researchers have had mixed success in their attempts to get a detailed picture of how HSCs maintain themselves and grow in the body.
Over the years, it became clear why. HSCs come in 2 forms—short-term HSCs that lose their powers of replication over time and long-term HSCs that can replicate indefinitely.
With the new study, Dr Weissman and his colleagues believe they have found a reliable way to tell the difference between long-term and short-term HSCs. Namely, the presence of Hoxb5.
“In this paper, we have found a single marker that, in the entire bone marrow, is only found in these long-term stem cells,” Dr Weissman said.
The researchers hope this finding will enable them to look at how nearby cells create a niche where the long-term HSCs are supported and maintained.
“For nearly 30 years, people have been trying to grow HSCs outside the body and have not been able to do it; it’s arguably the ‘holy grail’ in this field,” said James Y. Chen, an MD/PhD candidate at Stanford University School of Medicine.
“Now that we have an anchor, a way to look at long-term HSCs, we can look at the cells around them to understand and, ideally, recreate the niche.”
An extensive search
Over the years, scientists have proposed various markers that they felt were unique to long-term HSCs, but the reliability of each proposed marker has been heatedly debated by other research groups, said Masanori Miyanishi, MD, PhD, also of Stanford University School of Medicine.
In an attempt to settle the issue, Chen and Dr Miyanishi examined a list of more than 100 genes that are expressed in the bone marrow and seemed like good candidates to be unique markers of long-term HSCs.
The researchers eliminated genes that are turned on in areas of the bone that don’t involve the creation of new blood and immune cells. That narrowed the field to 45 genes.
The team then performed an analysis to determine how much protein these genes were making in various cells. They found that only 3 proteins were produced at a high enough level to mark HSCs.
Finally, the researchers needed to find if 1 of these 3 was turned on in long-term HSCs and turned off in short-term HSCs.
Although they couldn’t yet identify which cells were long-term HSCs, the team knew that any collection of HSCs should have both long-term and short-term HSCs, so they expected to find the candidate gene turned off in some cells and on in others. Only 1 gene fit that bill—Hoxb5.
The researchers pointed out that there may be other unique markers of long-term HSCs, such as genes that weren’t among the initial group of genes screened. But among the screened genes, only Hoxb5 was a unique identifier of the long-term HSC.
Finding the niche
The researchers were also able to solve another mystery by showing where in the bone marrow long-term HSCs reside.
Satoshi Yamazaki, PhD, and Hiromitsu Nakauchi, MD, PhD, both from the University of Tokyo in Japan, used new technology to prepare bone marrow tissue and do computational analyses that validated the location and architecture of the HSC niche.
“More than 90% of these cells reside on a particular type of blood vessel called venous sinusoids,” Dr Nakauchi said.
The researchers believe the ability to identify long-term HSCs will give scientists a powerful tool for further study.
“This opens the way to observe long-term HSCs and other cells in the niche as they exist in the body, without transplanting them,” Dr Weissman said. “This is how science works, by getting down to the purest irreducible element—in this case, blood stem cells—in order to develop new tools and understandings.” ![]()
in the bone marrow
Nearly 30 years after the discovery of the hematopoietic stem cell (HSC), researchers believe they have found a marker specific to long-term HSCs—the gene Hoxb5.
If confirmed, this finding would help settle long-standing debates about the identity of long-term HSCs and their support cells.
It may also pave the way for understanding how long-term HSCs maintain themselves and provide scientists with the necessary information to grow HSCs in the lab.
Irving Weissman, MD, of Stanford University School of Medicine in California, and his colleagues described this work in a letter to Nature.
In 1988, Dr Weissman and his colleagues isolated the HSC. Since that time, researchers have had mixed success in their attempts to get a detailed picture of how HSCs maintain themselves and grow in the body.
Over the years, it became clear why. HSCs come in 2 forms—short-term HSCs that lose their powers of replication over time and long-term HSCs that can replicate indefinitely.
With the new study, Dr Weissman and his colleagues believe they have found a reliable way to tell the difference between long-term and short-term HSCs. Namely, the presence of Hoxb5.
“In this paper, we have found a single marker that, in the entire bone marrow, is only found in these long-term stem cells,” Dr Weissman said.
The researchers hope this finding will enable them to look at how nearby cells create a niche where the long-term HSCs are supported and maintained.
“For nearly 30 years, people have been trying to grow HSCs outside the body and have not been able to do it; it’s arguably the ‘holy grail’ in this field,” said James Y. Chen, an MD/PhD candidate at Stanford University School of Medicine.
“Now that we have an anchor, a way to look at long-term HSCs, we can look at the cells around them to understand and, ideally, recreate the niche.”
An extensive search
Over the years, scientists have proposed various markers that they felt were unique to long-term HSCs, but the reliability of each proposed marker has been heatedly debated by other research groups, said Masanori Miyanishi, MD, PhD, also of Stanford University School of Medicine.
In an attempt to settle the issue, Chen and Dr Miyanishi examined a list of more than 100 genes that are expressed in the bone marrow and seemed like good candidates to be unique markers of long-term HSCs.
The researchers eliminated genes that are turned on in areas of the bone that don’t involve the creation of new blood and immune cells. That narrowed the field to 45 genes.
The team then performed an analysis to determine how much protein these genes were making in various cells. They found that only 3 proteins were produced at a high enough level to mark HSCs.
Finally, the researchers needed to find if 1 of these 3 was turned on in long-term HSCs and turned off in short-term HSCs.
Although they couldn’t yet identify which cells were long-term HSCs, the team knew that any collection of HSCs should have both long-term and short-term HSCs, so they expected to find the candidate gene turned off in some cells and on in others. Only 1 gene fit that bill—Hoxb5.
The researchers pointed out that there may be other unique markers of long-term HSCs, such as genes that weren’t among the initial group of genes screened. But among the screened genes, only Hoxb5 was a unique identifier of the long-term HSC.
Finding the niche
The researchers were also able to solve another mystery by showing where in the bone marrow long-term HSCs reside.
Satoshi Yamazaki, PhD, and Hiromitsu Nakauchi, MD, PhD, both from the University of Tokyo in Japan, used new technology to prepare bone marrow tissue and do computational analyses that validated the location and architecture of the HSC niche.
“More than 90% of these cells reside on a particular type of blood vessel called venous sinusoids,” Dr Nakauchi said.
The researchers believe the ability to identify long-term HSCs will give scientists a powerful tool for further study.
“This opens the way to observe long-term HSCs and other cells in the niche as they exist in the body, without transplanting them,” Dr Weissman said. “This is how science works, by getting down to the purest irreducible element—in this case, blood stem cells—in order to develop new tools and understandings.” ![]()
Advancing the fight against drug-resistant malaria

Image by Ute Frevert
and Margaret Shear
Researchers say they have designed a compound that kills malaria parasites—even those resistant to current antimalarial therapy—but
avoids harming human cells.
The compound exploits tiny structural differences between the parasitic and human versions of the proteasome.
In preclinical experiments, this proteasome inhibitor was able to kill artemisinin-resistant malaria parasites and further sensitize parasites to artemisinin.
Matthew Bogyo, PhD, of Stanford University School of Medicine in California, and his colleagues conducted this research and recounted the results in a letter to Nature.
Previous research has shown that proteasome inhibitors can be toxic to the malaria parasite Plasmodium falciparum. But the drugs have tended to inhibit the human version of the proteasome too, resulting in toxicity that would be unacceptable in a malaria drug.
Dr Bogyo and his colleagues wanted to overcome this problem, so they produced highly purified preparations of both human and P falciparum proteasomes. The team then “fed” those 2 preparations a set of protein fragments containing a variety of amino-acid linkages to see which amino-acid linkages the proteasomes would cleave.
The researchers identified 113 amino-acid linkages that are readily cleaved by P falciparum proteasomes but not so well by human proteasomes, and 153 amino-acid linkages where the reverse is the case.
The team used this information to design tiny protein snippets that failed to interact with human proteasomes but inhibited parts of the P falciparum proteasomes responsible for cleaving certain amino-acid links.
The researchers investigated the basis for this selectivity by using high-resolution electron microscopy to map the detailed structure of the parasite and human proteasomes. This allowed them to optimize the protein snippets they were using as parasite-selective proteasome inhibitors.
The 3-amino-acid snippet they ultimately focused on, called WLL, was able to inhibit 2 different catalytic regions in P falciparum proteasomes without having any effect on those of cultured human cells. There was a 600-fold difference in WLL’s potency at killing the parasitic cells over the human cells.
In experiments with mice, the researchers saw a nearly complete reduction of malaria parasites with both single and multiple doses of WLL.
Other tests, performed on artemisinin-resistant parasites infecting human red blood cells, suggested the WLL compound was equally effective at killing artemisinin-resistant parasites and artemisinin-sensitive parasites.
Dr Bogyo pointed out that the artemisinin family of drugs work by modifying proteins in the parasite. Resistance occurs when the parasites’ proteasomes are able to recycle those modified proteins. But this means that artemisinin-treated parasites are particularly sensitive to disruption of normal protein function.
“The compounds we’ve derived can kill artemisinin-resistant parasites because those parasites have an increased need for highly efficient proteasomes,” he said.
“So combining the proteasome inhibitor with artemisinin should make it possible to block the onset of resistance. That will, in turn, allow the continued use of that front-line malaria treatment, which has been so effective up until now.”
Clinical trials of compounds derived from this research remain several years away, Dr Bogyo added.
Study author Leann Tilley, PhD, of the University of Melbourne in Victoria, Australia, and her team are working with experts from Takeda Pharmaceutical Company Limited and Medicines for Malaria Venture to identify additional classes of parasite-specific proteasome inhibitors that could be advanced to clinical trials.
“The next step is screening the Takeda libraries to find a similar drug that doesn’t affect the human proteasome,” Dr Tilley said. “The current drug is a good start, but it’s not yet suitable for humans. It needs to be able to be administered orally and needs to last a long time in the blood stream.”
Dr Tilley said if they can find an existing drug in Takeda’s libraries that matches the structure of the new malaria drug, they could move it toward human trials very quickly. ![]()
Image by Ute Frevert
and Margaret Shear
Researchers say they have designed a compound that kills malaria parasites—even those resistant to current antimalarial therapy—but
avoids harming human cells.
The compound exploits tiny structural differences between the parasitic and human versions of the proteasome.
In preclinical experiments, this proteasome inhibitor was able to kill artemisinin-resistant malaria parasites and further sensitize parasites to artemisinin.
Matthew Bogyo, PhD, of Stanford University School of Medicine in California, and his colleagues conducted this research and recounted the results in a letter to Nature.
Previous research has shown that proteasome inhibitors can be toxic to the malaria parasite Plasmodium falciparum. But the drugs have tended to inhibit the human version of the proteasome too, resulting in toxicity that would be unacceptable in a malaria drug.
Dr Bogyo and his colleagues wanted to overcome this problem, so they produced highly purified preparations of both human and P falciparum proteasomes. The team then “fed” those 2 preparations a set of protein fragments containing a variety of amino-acid linkages to see which amino-acid linkages the proteasomes would cleave.
The researchers identified 113 amino-acid linkages that are readily cleaved by P falciparum proteasomes but not so well by human proteasomes, and 153 amino-acid linkages where the reverse is the case.
The team used this information to design tiny protein snippets that failed to interact with human proteasomes but inhibited parts of the P falciparum proteasomes responsible for cleaving certain amino-acid links.
The researchers investigated the basis for this selectivity by using high-resolution electron microscopy to map the detailed structure of the parasite and human proteasomes. This allowed them to optimize the protein snippets they were using as parasite-selective proteasome inhibitors.
The 3-amino-acid snippet they ultimately focused on, called WLL, was able to inhibit 2 different catalytic regions in P falciparum proteasomes without having any effect on those of cultured human cells. There was a 600-fold difference in WLL’s potency at killing the parasitic cells over the human cells.
In experiments with mice, the researchers saw a nearly complete reduction of malaria parasites with both single and multiple doses of WLL.
Other tests, performed on artemisinin-resistant parasites infecting human red blood cells, suggested the WLL compound was equally effective at killing artemisinin-resistant parasites and artemisinin-sensitive parasites.
Dr Bogyo pointed out that the artemisinin family of drugs work by modifying proteins in the parasite. Resistance occurs when the parasites’ proteasomes are able to recycle those modified proteins. But this means that artemisinin-treated parasites are particularly sensitive to disruption of normal protein function.
“The compounds we’ve derived can kill artemisinin-resistant parasites because those parasites have an increased need for highly efficient proteasomes,” he said.
“So combining the proteasome inhibitor with artemisinin should make it possible to block the onset of resistance. That will, in turn, allow the continued use of that front-line malaria treatment, which has been so effective up until now.”
Clinical trials of compounds derived from this research remain several years away, Dr Bogyo added.
Study author Leann Tilley, PhD, of the University of Melbourne in Victoria, Australia, and her team are working with experts from Takeda Pharmaceutical Company Limited and Medicines for Malaria Venture to identify additional classes of parasite-specific proteasome inhibitors that could be advanced to clinical trials.
“The next step is screening the Takeda libraries to find a similar drug that doesn’t affect the human proteasome,” Dr Tilley said. “The current drug is a good start, but it’s not yet suitable for humans. It needs to be able to be administered orally and needs to last a long time in the blood stream.”
Dr Tilley said if they can find an existing drug in Takeda’s libraries that matches the structure of the new malaria drug, they could move it toward human trials very quickly. ![]()
Image by Ute Frevert
and Margaret Shear
Researchers say they have designed a compound that kills malaria parasites—even those resistant to current antimalarial therapy—but
avoids harming human cells.
The compound exploits tiny structural differences between the parasitic and human versions of the proteasome.
In preclinical experiments, this proteasome inhibitor was able to kill artemisinin-resistant malaria parasites and further sensitize parasites to artemisinin.
Matthew Bogyo, PhD, of Stanford University School of Medicine in California, and his colleagues conducted this research and recounted the results in a letter to Nature.
Previous research has shown that proteasome inhibitors can be toxic to the malaria parasite Plasmodium falciparum. But the drugs have tended to inhibit the human version of the proteasome too, resulting in toxicity that would be unacceptable in a malaria drug.
Dr Bogyo and his colleagues wanted to overcome this problem, so they produced highly purified preparations of both human and P falciparum proteasomes. The team then “fed” those 2 preparations a set of protein fragments containing a variety of amino-acid linkages to see which amino-acid linkages the proteasomes would cleave.
The researchers identified 113 amino-acid linkages that are readily cleaved by P falciparum proteasomes but not so well by human proteasomes, and 153 amino-acid linkages where the reverse is the case.
The team used this information to design tiny protein snippets that failed to interact with human proteasomes but inhibited parts of the P falciparum proteasomes responsible for cleaving certain amino-acid links.
The researchers investigated the basis for this selectivity by using high-resolution electron microscopy to map the detailed structure of the parasite and human proteasomes. This allowed them to optimize the protein snippets they were using as parasite-selective proteasome inhibitors.
The 3-amino-acid snippet they ultimately focused on, called WLL, was able to inhibit 2 different catalytic regions in P falciparum proteasomes without having any effect on those of cultured human cells. There was a 600-fold difference in WLL’s potency at killing the parasitic cells over the human cells.
In experiments with mice, the researchers saw a nearly complete reduction of malaria parasites with both single and multiple doses of WLL.
Other tests, performed on artemisinin-resistant parasites infecting human red blood cells, suggested the WLL compound was equally effective at killing artemisinin-resistant parasites and artemisinin-sensitive parasites.
Dr Bogyo pointed out that the artemisinin family of drugs work by modifying proteins in the parasite. Resistance occurs when the parasites’ proteasomes are able to recycle those modified proteins. But this means that artemisinin-treated parasites are particularly sensitive to disruption of normal protein function.
“The compounds we’ve derived can kill artemisinin-resistant parasites because those parasites have an increased need for highly efficient proteasomes,” he said.
“So combining the proteasome inhibitor with artemisinin should make it possible to block the onset of resistance. That will, in turn, allow the continued use of that front-line malaria treatment, which has been so effective up until now.”
Clinical trials of compounds derived from this research remain several years away, Dr Bogyo added.
Study author Leann Tilley, PhD, of the University of Melbourne in Victoria, Australia, and her team are working with experts from Takeda Pharmaceutical Company Limited and Medicines for Malaria Venture to identify additional classes of parasite-specific proteasome inhibitors that could be advanced to clinical trials.
“The next step is screening the Takeda libraries to find a similar drug that doesn’t affect the human proteasome,” Dr Tilley said. “The current drug is a good start, but it’s not yet suitable for humans. It needs to be able to be administered orally and needs to last a long time in the blood stream.”
Dr Tilley said if they can find an existing drug in Takeda’s libraries that matches the structure of the new malaria drug, they could move it toward human trials very quickly. ![]()