User login
Helping patients with cystic fibrosis live longer
› Prescribe inhaled dornase alpha and inhaled tobramycin for maintenance pulmonary treatment of moderate to severe cystic fibrosis (CF). A
› Give aggressive nutritional supplementation to maintain a patient’s body mass index and blood sugar control and to attain maximal forced expiratory volume in one second (FEV1). B
› Consider prescribing cystic fibrosis transmembrane conductance regulator modulators, which have demonstrated a 5% to 10% improvement in FEV1 for CF patients. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The focus of treatment. CF is not limited to the classic picture of lung and pancreas destruction with subsequent loss of function. The underlying pathology can occur in body epithelial tissues from the intestinal lining to sweat glands. These tissues contain cystic fibrosis transmembrane conductance regulator (CFTR), a protein that allows for the transport of chloride across epithelial cell membranes.2 In individuals homozygous for mutated CFTR genes, chloride transport can be impaired. In addition to regulating chloride transport, CFTR is part of a larger, complex interaction of ion transport proteins such as the epithelial sodium channel (ENaC) and others that regulate bicarbonate secretion.2 Decreased chloride ion transport in mutant CFTR negatively affects the ion transport complex; the result is a higher-than-normal viscosity of secreted body fluids.
Reason for hope. It is this impairment of chloride ion transport that leads to the classic phenotypic features of CF (eg, pulmonary function decline, pancreatic insufficiency, malnutrition, chronic respiratory infection), and is the target of both established and emerging therapies3—both of which I will review here.
When to consider a CF diagnosis
Cystic fibrosis remains a clinical diagnosis when evidence of at least one phenotypic feature of the disease (TABLE 1) exists in the presence of laboratory evidence of a CFTR abnormality.4 Confirmation of CFTR dysfunction is demonstrated by an abnormality on sweat testing or identification of a CF-causing mutation in each copy of CFTR (ie, one on each chromosome).5 All 50 states now have neonatal laboratory screening programs;4 despite this, 30% of cases in 2012 were still diagnosed in those older than 1 year of age, with 3% to 5% diagnosed after age 18.1
A sweat chloride reading in the abnormal range (>60 mmol/L) is present in 90% of patients diagnosed with CF in adulthood; this test remains the gold standard in the diagnosis of CF and the initial test of choice in suspected cases.4 Newborn screening programs identify those at risk by detecting persistent hypertrypsinogenemia and referring those with positive results for definitive testing with sweat chloride evaluation. Keep CF in mind when evaluating adolescents and adults who have chronic sinusitis, chronic/recurrent pulmonary infections, chronic/recurrent pancreatitis, or infertility from absence of the vas deferens.4 When features of the CF phenotype are present, especially if there is a known positive family history of CF or CF carrier status, order sweat chloride testing.
Traditional therapies
Both maintenance and acute therapies are directed throughout the body at decreasing fluid viscosity, clearing fluid with a high viscosity, or treating the tissue destruction that results from highly-viscous fluid.3 The traditional classic picture of CF is one of lung and pancreas destruction with subsequent loss of function. However, CF is, in reality, a full-body disease.
Respiratory system: Lungs
CFTR dysfunction in the lungs results in thick pulmonary secretions as the aqueous surface layer (ASL) lining the alveolar epithelium becomes dehydrated and creates a prime environment for the development of chronic infection. What ensues is a recurrent cycle of chronic infection, inflammation, and tissue destruction with loss of lung volume and function. Current therapies interrupt this cycle at multiple points.6
Airway clearance is one of the hallmarks of CF therapy, using both chemical and mechanical treatments. Daily, most patients will use either a therapy vest that administers sheering forces to the chest cavity or an airflow device that creates positive expiratory pressure and laminar flow to aid in expectorating pulmonary secretions.7 Because exercise has yielded comparable results to mechanical or airflow clearance devices, it is recommended that all CF patients who are not otherwise prohibited engage in regular, vigorous exercise in accordance with standard recommendations for the general public.7
Mechanical therapies are often preceded by airway dilation with short- and long-acting bronchodilators and inhaled steroids that open airways for optimal airway clearance.4 Thick secretions can be treated directly and enzymatically with nebulized dornase alpha,4,8 which is also best administered before mechanical clearance therapy. Finally, viscosity of airway secretions can be decreased by improving the hydration of the ASL with nebulized 7% hypertonic saline.4,8
Infection suppression. Thickened pulmonary secretions create a fertile environment for the development of chronic infection. By the time most CF patients reach adulthood, many are colonized with mucoid producing strains of Pseudomonas aeruginosa.4,8-10 Many may also have chronic infection with Staphylococcus aureus, some strains of which may be methicillin-resistant. Quarterly culture and sensitivity results can be essential in directing acute antibiotic therapy, both in the hospital and ambulatory settings. In addition, in the case of Pseudomonas, inhaled antibiotics suppress chronic infection, improve lung function, decrease pulmonary secretions, and reduce inflammation.
Formulations are available for tobramycin and aztreonam, both of which are administered every other month to reduce toxicities and to deter antibiotic resistance. Some patients may use a single agent or may alternate agents every month. When acute antibiotic therapy is necessary for a pulmonary exacerbation, the inhaled agent is generally withheld. If outpatient treatment is warranted, the only available oral antibiotics with anti-pseudomonal activity are ciprofloxacin and levofloxacin.4,8-10S aureus can be treated with trimethoprim/sulfamethoxazole or doxycycline.4,8-10
Inflammation reduction is addressed with high-dose ibuprofen twice daily, azithromycin daily or 3 times weekly, or both. Children up to age 18 benefit from ibuprofen, which also improves forced expiratory volume in one second (FEV1) to a greater extent than azithromycin.8 Adults, however, face the risk of gastrointestinal bleeding and renal dysfunction with ibuprofen, which must be weighed against its potential anti-inflammatory benefit. Both populations, however, benefit from chronic azithromycin, whose mechanism of action in this setting is believed to be more anti-inflammatory than bacterial suppression, since it has no direct bactericidal effect on the primary colonizing microbe, P aeruginosa.4,11
Gastrointestinal system: Pancreas
Cystic fibrosis was first comprehensively described in 1938 and was named for the diseased appearance of the pancreas.12 As happens in the lungs, thick pancreatic duct secretions create a cycle of tissue destruction, inflammation, and dysfunction.2 CF patients lack adequate secretion of pancreatic enzymes and bicarbonate into the small bowel, which progressively leads to pancreatic dysfunction in most patients.
As malabsorption of nutrients advances, patients suffer varying degrees of malnutrition and vitamin deficiency, especially of the fat-soluble vitamins A, D, E, and K. Over 85% of CF patients have deficient pancreatic function, requiring pancreatic enzyme supplementation with all food intake and daily vitamin supplementation.2
Ensuring adequate nutrition. Most CF patients experience a chronic mismatch of dietary intake against caloric expenditure and benefit from aggressive nutritional management featuring a high-calorie diet with supplementation in the form of nutrition shakes or bars.2 There is a well-documented linear relationship between BMI and FEV1. Lung function declines in CF when a man’s body mass index (BMI) falls below 23 kg/m2 and a woman’s BMI drops below 22 kg/m2.2 For this reason, the goal for caloric intake can be as high as 200% of the customary recommended daily allowance.2
Watch for CF-related diabetes. Since the pancreas is also the major source of endogenous insulin, nearly half of adults with CF will develop cystic fibrosis-related diabetes (CFRD) as pancreatic deficiency progresses.13 Similar to the relationship between BMI and FEV1, there is a relationship between glucose intolerance and FEV1. For this reason, annual diabetes screening is recommended for all CF patients ages 10 years and older.13 Because glycated hemoglobin (HbA1c) may not accurately reflect low levels of glucose intolerance, screen for CFRD with a 2-hour 75-g oral glucose tolerance test.13 Early insulin therapy can help maintain BMI and lower average blood sugar in support of FEV1. Once CFRD is diagnosed, the goals and recommendations for control are largely the same as those recommended by the American Diabetes Association for other forms of diabetes.13
Cystic Fibrosis Resources
Cystic Fibrosis Foundation
www.cff.org
Consensus report on cystic fibrosis management
Yankaskas JR, Marchall BC, Sufian B, et al. Cystic fibrosis adult care. Chest. 2004;125:1S-39S.
Consensus report on cystic fibrosis diagnostic guidelines
Farrell PM, Rosenstein BJ, White RB. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation Consensus Report. J Pediatr. 2008;153:S4-S14.
Gastrointestinal system: Alimentary canal
CF is often mistakenly believed to be primarily a pulmonary disease since 85% of the mortality is due to lung dysfunction,7 but intermittent abdominal pain is a common experience for most patients, and disorders can range from gastroesophageal reflux disease (GERD) to small bowel bacterial overgrowth (SBBO) to constipation. Up to 85% of adult patients experience symptoms of reflux, with as many as 40% of cases occurring silently.2 Proton pump inhibitors are a first-line treatment, but they can also contribute to intestinal bacterial overgrowth and pulmonary infections.
In SBBO, gram-negative colonic bacteria colonize the small bowel and can contribute to abdominal pain and malabsorption, weight loss, and malnutrition. Treatment requires antibiotics with activity against gram-negative organisms, or non-absorbable agents such as rifamyxin, sometimes on a chronic, recurrent, or rotating basis.2
Chronic constipation is also quite common among CF patients and many require daily administration of poly-ethylene-glycol. Before newborn screening programs were introduced, infants would on occasion present with complete distal intestinal obstruction. Adults are not immune to obstructive complications and may require hospitalization for bowel cleansing.
Gastrointestinal system: Liver
Liver disease is relatively common in CF, with up to 24% of adults experiencing hepatomegaly or persistently elevated liver function tests (LFT).4 Progressive biliary fibrosis and cirrhosis are encountered more often as the median survival age has increased. There is evidence that ursodeoxycholic acid (UDCA) can be a useful adjunct in the treatment of cholestasis, but it is not clear if it alters mortality or progression to cirrhosis. Only CF patients with elevated LFTs should be started on UDCA.4
Other areas of concern: Sinuses, serum sodium levels
Chronic, symptomatic sinus disease in CF patients—chiefly polyposis—is common and may require repeat surgery, although most patients with extensive nasal polyps find symptom relief with daily sinus rinses. Intranasal steroids and intranasal antibiotics are also often employed, and many CF patients need to be in regular contact with an otolaryngologist.14 For symptoms of allergic rhinitis, recommend OTC antihistamines in standard dosages.
Exercise is recommended for all CF patients, as noted earlier, and as life expectancy increases, many are engaging in more strenuous and longer duration activities.15 Due to high sweat sodium loss, CF patients are at risk for hyponatremia, especially when exercising on days with high temperatures and humidity. CF patients need to replace sodium losses in these conditions and when exercising for extended periods.
There are no evidence-based guidelines for sodium replacement. The Cystic Fibrosis Foundation (CFF) recommends that patients increase salt in the diet when under conditions likely to result in increased sodium loss, such as exercise. It has been thought that CF patients can easily dehydrate due to an impaired thirst mechanism and, when exercising, should consume fluids beyond the need to quench thirst.16,17 More recent evidence suggests, however, that the thirst mechanism in those with CF remains normally intact and that overconsumption of fluids beyond the level of thirst may predispose the individual to exercise-associated hyponatremia as serum sodium is diluted.15
New therapies
Small-molecule CFTR-modulating compounds are a promising development in the treatment of CF. The first such available medication was ivacaftor in 2012. Because these molecules are mutation specific, ivacaftor was available at first only for patients with at least one copy of the G551D mutation,18 which means about 5% of patients with CF.3
Ivacaftor increases the likelihood that the CFTR chloride channel will open and patients will exhibit a reduction in sweat chloride levels. In the first reported clinical trial of ivacaftor involving patients with the G551D mutation, FEV1 improvements of 10% occurred by the second week of therapy and persisted for 48 weeks.18 The drug has now been approved by the US Food and Drug Administration (FDA) for patients 12 years of age and older with at least one of the following mutations: R117H, G551D, G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, or G1349D.19
A medication combining ivacaftor with lumacaftor is also now available for patients with a copy of F508del on both chromosomes. F508del is the most common CFTR mutation, with one copy present in almost 87% of people with CF in the United States.1 Since 47% of CF patients have 2 copies of F508del,1 about half of those with CF in the United States are now eligible for small-molecule therapy. Lumacaftor acts by facilitating transport of a misfolded CFTR to the cell membrane where ivacaftor then increases the probability of an open chloride channel. This combination medication has improved lung function by about 5%.
The ivacaftor/lumacaftor combination was approved by the FDA in July 2015. Both ivacaftor and the ivacaftor/lumacaftor combination were deemed by the FDA to demonstrate statistically significant and sustained FEV1 improvements over placebo.
The CFF was instrumental in providing financial support for the development of both ivacaftor and the ivacaftor/lumacaftor combination and continues to provide significant research advancement. According to the CFF (www.cff.org), medications currently in the development pipeline include compounds that provide CFTR modulation, surface airway liquid restoration, anti-inflammation, inhaled anti-infection, and pancreatic enzyme function. For more on CFF, see "The traditional CF care model.”4,20
The traditional CF care model
The Cystic Fibrosis Foundation (CFF) has been a driving force behind the increased life expectancy CF patients have seen over the last 3 decades. Its contributions include the development of medication through the CFF Therapeutics Development Network (TDN) and disease management through a network of CF Care Centers throughout the United States. The CFF recommends a minimum of quarterly visits to a CF Care Center, and the primary care physician can play a critical role alongside the multidisciplinary CF team.20
At every CF Care Center encounter, the entire team (nurse, physician, dietician, social worker, psychologist) interacts with each patient and their families to maximize overall medical care. Respiratory cultures are generally obtained at each visit. Dual-energy x-ray absorptiometry is performed biannually. Lab work (complete blood count, comprehensive metabolic panel, glycated hemoglobin, vitamins A, D, E, and K, 2-hour glucose tolerance test), and chest x-ray are obtained at least annually (TABLE 2).4
Since CF generally involves both restrictive and obstructive lung components, complete spirometry evaluation is performed annually in the pulmonary function lab, with static lung volumes in addition to airflow measurement. Office spirometry to measure airflow alone is performed at each visit. FEV1 is tracked both as an indicator of disease progression and as a measure of current pulmonary status.
The CFF recommends that each patient receive full genetic testing and encourages patient participation in the CFF Registry, where mutation data are documented among other disease parameters to ensure that patients receive mutation specific therapies as they become available.4 The vaccine schedule recommended for CF patients is the same as for the general population.
CORRESPONDENCE
Douglas Lewis, MD, 1121 S. Clifton, Wichita, KS 67218; douglas.lewis@viachristi.org.
1. Cystic Fibrosis Foundation. Patient registry 2012 annual data report. Cystic Fibrosis Foundation Web site. Available at: http://www.cff.org/UploadedFiles/research/ClinicalResearch/PatientRegistryReport/2012-CFF-Patient-Registry.pdf. Accessed August 14, 2014.
2. Haller W, Ledder O, Lewindon PJ, et al. Cystic fibrosis: An update for clinicians. Part 1: Nutrition and gastrointestinal complications. J Gastroenterol Hepatol. 2014;29:1344-1355.
3. Hoffman LR, Ramsey BW. Cystic fibrosis therapeutics: the road ahead. Chest. 2013;143:207-213.
4. Yankaskas JR, Marshall BC, Sufian B, et al. Cystic fibrosis adult care: consensus conference report. Chest. 2004;125:1S-39S.
5. Farrell PM, Rosenstein BJ, White TB, et al; Cystic Fibrosis Foundation. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr. 2008;153:S4-S14.
6. Donaldson SH, Boucher RC. Sodium channels and cystic fibrosis. Chest. 2007;132:1631-1636.
7. Flume PA, Robinson KA, O’Sullivan BP, et al; Clinical Practice Guidelines for Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: airway clearance therapies. Respir Care. 2009;54:522-537.
8. Flume PA, O’Sullivan BP, Robinson KA, et al; Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176:957-969.
9. Döring G, Flume P, Heijerman H, et al; Consensus Study Group. Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J Cyst Fibros. 2012;11:461-479.
10. Flume PA, Mogayzel PJ Jr, Robinson KA, et al; Clinical Practice Guidelines for Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med. 2009;180:802-808.
11. Southern KW, Barker PM. Azithromycin for cystic fibrosis. Eur Respir J. 2004;24:834-838.
12. Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Am J Dis Child. 1938;56:344-399.
13. Moran A, Brunzell C, Cohen RC, et al; CFRD Guidelines Committee. Clinical care guidelines for cystic fibrosis-related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care. 2010;33:2697-2708.
14. Kerem E, Conway S, Elborn S, et al; Consensus Committee. Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros. 2005;4:7-26.
15. Hew-Butler T, Rosner MH, Fowkes-Godek S, et al. Statement of the third international exercise-associated hyponatremia consensus development conference, Carlsbad, California, 2015. Clin J Sport Med. 2015;25:303-320.
16. Brown MB, McCarty NA, Millard-Stafford M. High-sweat Na+ in cystic fibrosis and healthy individuals does not diminish thirst during exercise in the heat. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1177-R1185.
17. Wheatley CM, Wilkins BW, Snyder EM. Exercise is medicine in cystic fibrosis. Exerc Sport Sci Rev. 2011;39:155-160.
18. Ramsey BW, Davies J, McElvaney NG, et al; VX08-770-102 Study Group. ACFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663-1672.
19. Pettit RS, Fellner C. CFTR Modulators for the Treatment of Cystic Fibrosis. P T. 2014;39:500-511.
20. Lewis D. Role of the family physician in the management of cystic fibrosis. Am Fam Physician. 2015;91:822-824.
› Prescribe inhaled dornase alpha and inhaled tobramycin for maintenance pulmonary treatment of moderate to severe cystic fibrosis (CF). A
› Give aggressive nutritional supplementation to maintain a patient’s body mass index and blood sugar control and to attain maximal forced expiratory volume in one second (FEV1). B
› Consider prescribing cystic fibrosis transmembrane conductance regulator modulators, which have demonstrated a 5% to 10% improvement in FEV1 for CF patients. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The focus of treatment. CF is not limited to the classic picture of lung and pancreas destruction with subsequent loss of function. The underlying pathology can occur in body epithelial tissues from the intestinal lining to sweat glands. These tissues contain cystic fibrosis transmembrane conductance regulator (CFTR), a protein that allows for the transport of chloride across epithelial cell membranes.2 In individuals homozygous for mutated CFTR genes, chloride transport can be impaired. In addition to regulating chloride transport, CFTR is part of a larger, complex interaction of ion transport proteins such as the epithelial sodium channel (ENaC) and others that regulate bicarbonate secretion.2 Decreased chloride ion transport in mutant CFTR negatively affects the ion transport complex; the result is a higher-than-normal viscosity of secreted body fluids.
Reason for hope. It is this impairment of chloride ion transport that leads to the classic phenotypic features of CF (eg, pulmonary function decline, pancreatic insufficiency, malnutrition, chronic respiratory infection), and is the target of both established and emerging therapies3—both of which I will review here.
When to consider a CF diagnosis
Cystic fibrosis remains a clinical diagnosis when evidence of at least one phenotypic feature of the disease (TABLE 1) exists in the presence of laboratory evidence of a CFTR abnormality.4 Confirmation of CFTR dysfunction is demonstrated by an abnormality on sweat testing or identification of a CF-causing mutation in each copy of CFTR (ie, one on each chromosome).5 All 50 states now have neonatal laboratory screening programs;4 despite this, 30% of cases in 2012 were still diagnosed in those older than 1 year of age, with 3% to 5% diagnosed after age 18.1
A sweat chloride reading in the abnormal range (>60 mmol/L) is present in 90% of patients diagnosed with CF in adulthood; this test remains the gold standard in the diagnosis of CF and the initial test of choice in suspected cases.4 Newborn screening programs identify those at risk by detecting persistent hypertrypsinogenemia and referring those with positive results for definitive testing with sweat chloride evaluation. Keep CF in mind when evaluating adolescents and adults who have chronic sinusitis, chronic/recurrent pulmonary infections, chronic/recurrent pancreatitis, or infertility from absence of the vas deferens.4 When features of the CF phenotype are present, especially if there is a known positive family history of CF or CF carrier status, order sweat chloride testing.
Traditional therapies
Both maintenance and acute therapies are directed throughout the body at decreasing fluid viscosity, clearing fluid with a high viscosity, or treating the tissue destruction that results from highly-viscous fluid.3 The traditional classic picture of CF is one of lung and pancreas destruction with subsequent loss of function. However, CF is, in reality, a full-body disease.
Respiratory system: Lungs
CFTR dysfunction in the lungs results in thick pulmonary secretions as the aqueous surface layer (ASL) lining the alveolar epithelium becomes dehydrated and creates a prime environment for the development of chronic infection. What ensues is a recurrent cycle of chronic infection, inflammation, and tissue destruction with loss of lung volume and function. Current therapies interrupt this cycle at multiple points.6
Airway clearance is one of the hallmarks of CF therapy, using both chemical and mechanical treatments. Daily, most patients will use either a therapy vest that administers sheering forces to the chest cavity or an airflow device that creates positive expiratory pressure and laminar flow to aid in expectorating pulmonary secretions.7 Because exercise has yielded comparable results to mechanical or airflow clearance devices, it is recommended that all CF patients who are not otherwise prohibited engage in regular, vigorous exercise in accordance with standard recommendations for the general public.7
Mechanical therapies are often preceded by airway dilation with short- and long-acting bronchodilators and inhaled steroids that open airways for optimal airway clearance.4 Thick secretions can be treated directly and enzymatically with nebulized dornase alpha,4,8 which is also best administered before mechanical clearance therapy. Finally, viscosity of airway secretions can be decreased by improving the hydration of the ASL with nebulized 7% hypertonic saline.4,8
Infection suppression. Thickened pulmonary secretions create a fertile environment for the development of chronic infection. By the time most CF patients reach adulthood, many are colonized with mucoid producing strains of Pseudomonas aeruginosa.4,8-10 Many may also have chronic infection with Staphylococcus aureus, some strains of which may be methicillin-resistant. Quarterly culture and sensitivity results can be essential in directing acute antibiotic therapy, both in the hospital and ambulatory settings. In addition, in the case of Pseudomonas, inhaled antibiotics suppress chronic infection, improve lung function, decrease pulmonary secretions, and reduce inflammation.
Formulations are available for tobramycin and aztreonam, both of which are administered every other month to reduce toxicities and to deter antibiotic resistance. Some patients may use a single agent or may alternate agents every month. When acute antibiotic therapy is necessary for a pulmonary exacerbation, the inhaled agent is generally withheld. If outpatient treatment is warranted, the only available oral antibiotics with anti-pseudomonal activity are ciprofloxacin and levofloxacin.4,8-10S aureus can be treated with trimethoprim/sulfamethoxazole or doxycycline.4,8-10
Inflammation reduction is addressed with high-dose ibuprofen twice daily, azithromycin daily or 3 times weekly, or both. Children up to age 18 benefit from ibuprofen, which also improves forced expiratory volume in one second (FEV1) to a greater extent than azithromycin.8 Adults, however, face the risk of gastrointestinal bleeding and renal dysfunction with ibuprofen, which must be weighed against its potential anti-inflammatory benefit. Both populations, however, benefit from chronic azithromycin, whose mechanism of action in this setting is believed to be more anti-inflammatory than bacterial suppression, since it has no direct bactericidal effect on the primary colonizing microbe, P aeruginosa.4,11
Gastrointestinal system: Pancreas
Cystic fibrosis was first comprehensively described in 1938 and was named for the diseased appearance of the pancreas.12 As happens in the lungs, thick pancreatic duct secretions create a cycle of tissue destruction, inflammation, and dysfunction.2 CF patients lack adequate secretion of pancreatic enzymes and bicarbonate into the small bowel, which progressively leads to pancreatic dysfunction in most patients.
As malabsorption of nutrients advances, patients suffer varying degrees of malnutrition and vitamin deficiency, especially of the fat-soluble vitamins A, D, E, and K. Over 85% of CF patients have deficient pancreatic function, requiring pancreatic enzyme supplementation with all food intake and daily vitamin supplementation.2
Ensuring adequate nutrition. Most CF patients experience a chronic mismatch of dietary intake against caloric expenditure and benefit from aggressive nutritional management featuring a high-calorie diet with supplementation in the form of nutrition shakes or bars.2 There is a well-documented linear relationship between BMI and FEV1. Lung function declines in CF when a man’s body mass index (BMI) falls below 23 kg/m2 and a woman’s BMI drops below 22 kg/m2.2 For this reason, the goal for caloric intake can be as high as 200% of the customary recommended daily allowance.2
Watch for CF-related diabetes. Since the pancreas is also the major source of endogenous insulin, nearly half of adults with CF will develop cystic fibrosis-related diabetes (CFRD) as pancreatic deficiency progresses.13 Similar to the relationship between BMI and FEV1, there is a relationship between glucose intolerance and FEV1. For this reason, annual diabetes screening is recommended for all CF patients ages 10 years and older.13 Because glycated hemoglobin (HbA1c) may not accurately reflect low levels of glucose intolerance, screen for CFRD with a 2-hour 75-g oral glucose tolerance test.13 Early insulin therapy can help maintain BMI and lower average blood sugar in support of FEV1. Once CFRD is diagnosed, the goals and recommendations for control are largely the same as those recommended by the American Diabetes Association for other forms of diabetes.13
Cystic Fibrosis Resources
Cystic Fibrosis Foundation
www.cff.org
Consensus report on cystic fibrosis management
Yankaskas JR, Marchall BC, Sufian B, et al. Cystic fibrosis adult care. Chest. 2004;125:1S-39S.
Consensus report on cystic fibrosis diagnostic guidelines
Farrell PM, Rosenstein BJ, White RB. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation Consensus Report. J Pediatr. 2008;153:S4-S14.
Gastrointestinal system: Alimentary canal
CF is often mistakenly believed to be primarily a pulmonary disease since 85% of the mortality is due to lung dysfunction,7 but intermittent abdominal pain is a common experience for most patients, and disorders can range from gastroesophageal reflux disease (GERD) to small bowel bacterial overgrowth (SBBO) to constipation. Up to 85% of adult patients experience symptoms of reflux, with as many as 40% of cases occurring silently.2 Proton pump inhibitors are a first-line treatment, but they can also contribute to intestinal bacterial overgrowth and pulmonary infections.
In SBBO, gram-negative colonic bacteria colonize the small bowel and can contribute to abdominal pain and malabsorption, weight loss, and malnutrition. Treatment requires antibiotics with activity against gram-negative organisms, or non-absorbable agents such as rifamyxin, sometimes on a chronic, recurrent, or rotating basis.2
Chronic constipation is also quite common among CF patients and many require daily administration of poly-ethylene-glycol. Before newborn screening programs were introduced, infants would on occasion present with complete distal intestinal obstruction. Adults are not immune to obstructive complications and may require hospitalization for bowel cleansing.
Gastrointestinal system: Liver
Liver disease is relatively common in CF, with up to 24% of adults experiencing hepatomegaly or persistently elevated liver function tests (LFT).4 Progressive biliary fibrosis and cirrhosis are encountered more often as the median survival age has increased. There is evidence that ursodeoxycholic acid (UDCA) can be a useful adjunct in the treatment of cholestasis, but it is not clear if it alters mortality or progression to cirrhosis. Only CF patients with elevated LFTs should be started on UDCA.4
Other areas of concern: Sinuses, serum sodium levels
Chronic, symptomatic sinus disease in CF patients—chiefly polyposis—is common and may require repeat surgery, although most patients with extensive nasal polyps find symptom relief with daily sinus rinses. Intranasal steroids and intranasal antibiotics are also often employed, and many CF patients need to be in regular contact with an otolaryngologist.14 For symptoms of allergic rhinitis, recommend OTC antihistamines in standard dosages.
Exercise is recommended for all CF patients, as noted earlier, and as life expectancy increases, many are engaging in more strenuous and longer duration activities.15 Due to high sweat sodium loss, CF patients are at risk for hyponatremia, especially when exercising on days with high temperatures and humidity. CF patients need to replace sodium losses in these conditions and when exercising for extended periods.
There are no evidence-based guidelines for sodium replacement. The Cystic Fibrosis Foundation (CFF) recommends that patients increase salt in the diet when under conditions likely to result in increased sodium loss, such as exercise. It has been thought that CF patients can easily dehydrate due to an impaired thirst mechanism and, when exercising, should consume fluids beyond the need to quench thirst.16,17 More recent evidence suggests, however, that the thirst mechanism in those with CF remains normally intact and that overconsumption of fluids beyond the level of thirst may predispose the individual to exercise-associated hyponatremia as serum sodium is diluted.15
New therapies
Small-molecule CFTR-modulating compounds are a promising development in the treatment of CF. The first such available medication was ivacaftor in 2012. Because these molecules are mutation specific, ivacaftor was available at first only for patients with at least one copy of the G551D mutation,18 which means about 5% of patients with CF.3
Ivacaftor increases the likelihood that the CFTR chloride channel will open and patients will exhibit a reduction in sweat chloride levels. In the first reported clinical trial of ivacaftor involving patients with the G551D mutation, FEV1 improvements of 10% occurred by the second week of therapy and persisted for 48 weeks.18 The drug has now been approved by the US Food and Drug Administration (FDA) for patients 12 years of age and older with at least one of the following mutations: R117H, G551D, G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, or G1349D.19
A medication combining ivacaftor with lumacaftor is also now available for patients with a copy of F508del on both chromosomes. F508del is the most common CFTR mutation, with one copy present in almost 87% of people with CF in the United States.1 Since 47% of CF patients have 2 copies of F508del,1 about half of those with CF in the United States are now eligible for small-molecule therapy. Lumacaftor acts by facilitating transport of a misfolded CFTR to the cell membrane where ivacaftor then increases the probability of an open chloride channel. This combination medication has improved lung function by about 5%.
The ivacaftor/lumacaftor combination was approved by the FDA in July 2015. Both ivacaftor and the ivacaftor/lumacaftor combination were deemed by the FDA to demonstrate statistically significant and sustained FEV1 improvements over placebo.
The CFF was instrumental in providing financial support for the development of both ivacaftor and the ivacaftor/lumacaftor combination and continues to provide significant research advancement. According to the CFF (www.cff.org), medications currently in the development pipeline include compounds that provide CFTR modulation, surface airway liquid restoration, anti-inflammation, inhaled anti-infection, and pancreatic enzyme function. For more on CFF, see "The traditional CF care model.”4,20
The traditional CF care model
The Cystic Fibrosis Foundation (CFF) has been a driving force behind the increased life expectancy CF patients have seen over the last 3 decades. Its contributions include the development of medication through the CFF Therapeutics Development Network (TDN) and disease management through a network of CF Care Centers throughout the United States. The CFF recommends a minimum of quarterly visits to a CF Care Center, and the primary care physician can play a critical role alongside the multidisciplinary CF team.20
At every CF Care Center encounter, the entire team (nurse, physician, dietician, social worker, psychologist) interacts with each patient and their families to maximize overall medical care. Respiratory cultures are generally obtained at each visit. Dual-energy x-ray absorptiometry is performed biannually. Lab work (complete blood count, comprehensive metabolic panel, glycated hemoglobin, vitamins A, D, E, and K, 2-hour glucose tolerance test), and chest x-ray are obtained at least annually (TABLE 2).4
Since CF generally involves both restrictive and obstructive lung components, complete spirometry evaluation is performed annually in the pulmonary function lab, with static lung volumes in addition to airflow measurement. Office spirometry to measure airflow alone is performed at each visit. FEV1 is tracked both as an indicator of disease progression and as a measure of current pulmonary status.
The CFF recommends that each patient receive full genetic testing and encourages patient participation in the CFF Registry, where mutation data are documented among other disease parameters to ensure that patients receive mutation specific therapies as they become available.4 The vaccine schedule recommended for CF patients is the same as for the general population.
CORRESPONDENCE
Douglas Lewis, MD, 1121 S. Clifton, Wichita, KS 67218; douglas.lewis@viachristi.org.
› Prescribe inhaled dornase alpha and inhaled tobramycin for maintenance pulmonary treatment of moderate to severe cystic fibrosis (CF). A
› Give aggressive nutritional supplementation to maintain a patient’s body mass index and blood sugar control and to attain maximal forced expiratory volume in one second (FEV1). B
› Consider prescribing cystic fibrosis transmembrane conductance regulator modulators, which have demonstrated a 5% to 10% improvement in FEV1 for CF patients. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The focus of treatment. CF is not limited to the classic picture of lung and pancreas destruction with subsequent loss of function. The underlying pathology can occur in body epithelial tissues from the intestinal lining to sweat glands. These tissues contain cystic fibrosis transmembrane conductance regulator (CFTR), a protein that allows for the transport of chloride across epithelial cell membranes.2 In individuals homozygous for mutated CFTR genes, chloride transport can be impaired. In addition to regulating chloride transport, CFTR is part of a larger, complex interaction of ion transport proteins such as the epithelial sodium channel (ENaC) and others that regulate bicarbonate secretion.2 Decreased chloride ion transport in mutant CFTR negatively affects the ion transport complex; the result is a higher-than-normal viscosity of secreted body fluids.
Reason for hope. It is this impairment of chloride ion transport that leads to the classic phenotypic features of CF (eg, pulmonary function decline, pancreatic insufficiency, malnutrition, chronic respiratory infection), and is the target of both established and emerging therapies3—both of which I will review here.
When to consider a CF diagnosis
Cystic fibrosis remains a clinical diagnosis when evidence of at least one phenotypic feature of the disease (TABLE 1) exists in the presence of laboratory evidence of a CFTR abnormality.4 Confirmation of CFTR dysfunction is demonstrated by an abnormality on sweat testing or identification of a CF-causing mutation in each copy of CFTR (ie, one on each chromosome).5 All 50 states now have neonatal laboratory screening programs;4 despite this, 30% of cases in 2012 were still diagnosed in those older than 1 year of age, with 3% to 5% diagnosed after age 18.1
A sweat chloride reading in the abnormal range (>60 mmol/L) is present in 90% of patients diagnosed with CF in adulthood; this test remains the gold standard in the diagnosis of CF and the initial test of choice in suspected cases.4 Newborn screening programs identify those at risk by detecting persistent hypertrypsinogenemia and referring those with positive results for definitive testing with sweat chloride evaluation. Keep CF in mind when evaluating adolescents and adults who have chronic sinusitis, chronic/recurrent pulmonary infections, chronic/recurrent pancreatitis, or infertility from absence of the vas deferens.4 When features of the CF phenotype are present, especially if there is a known positive family history of CF or CF carrier status, order sweat chloride testing.
Traditional therapies
Both maintenance and acute therapies are directed throughout the body at decreasing fluid viscosity, clearing fluid with a high viscosity, or treating the tissue destruction that results from highly-viscous fluid.3 The traditional classic picture of CF is one of lung and pancreas destruction with subsequent loss of function. However, CF is, in reality, a full-body disease.
Respiratory system: Lungs
CFTR dysfunction in the lungs results in thick pulmonary secretions as the aqueous surface layer (ASL) lining the alveolar epithelium becomes dehydrated and creates a prime environment for the development of chronic infection. What ensues is a recurrent cycle of chronic infection, inflammation, and tissue destruction with loss of lung volume and function. Current therapies interrupt this cycle at multiple points.6
Airway clearance is one of the hallmarks of CF therapy, using both chemical and mechanical treatments. Daily, most patients will use either a therapy vest that administers sheering forces to the chest cavity or an airflow device that creates positive expiratory pressure and laminar flow to aid in expectorating pulmonary secretions.7 Because exercise has yielded comparable results to mechanical or airflow clearance devices, it is recommended that all CF patients who are not otherwise prohibited engage in regular, vigorous exercise in accordance with standard recommendations for the general public.7
Mechanical therapies are often preceded by airway dilation with short- and long-acting bronchodilators and inhaled steroids that open airways for optimal airway clearance.4 Thick secretions can be treated directly and enzymatically with nebulized dornase alpha,4,8 which is also best administered before mechanical clearance therapy. Finally, viscosity of airway secretions can be decreased by improving the hydration of the ASL with nebulized 7% hypertonic saline.4,8
Infection suppression. Thickened pulmonary secretions create a fertile environment for the development of chronic infection. By the time most CF patients reach adulthood, many are colonized with mucoid producing strains of Pseudomonas aeruginosa.4,8-10 Many may also have chronic infection with Staphylococcus aureus, some strains of which may be methicillin-resistant. Quarterly culture and sensitivity results can be essential in directing acute antibiotic therapy, both in the hospital and ambulatory settings. In addition, in the case of Pseudomonas, inhaled antibiotics suppress chronic infection, improve lung function, decrease pulmonary secretions, and reduce inflammation.
Formulations are available for tobramycin and aztreonam, both of which are administered every other month to reduce toxicities and to deter antibiotic resistance. Some patients may use a single agent or may alternate agents every month. When acute antibiotic therapy is necessary for a pulmonary exacerbation, the inhaled agent is generally withheld. If outpatient treatment is warranted, the only available oral antibiotics with anti-pseudomonal activity are ciprofloxacin and levofloxacin.4,8-10S aureus can be treated with trimethoprim/sulfamethoxazole or doxycycline.4,8-10
Inflammation reduction is addressed with high-dose ibuprofen twice daily, azithromycin daily or 3 times weekly, or both. Children up to age 18 benefit from ibuprofen, which also improves forced expiratory volume in one second (FEV1) to a greater extent than azithromycin.8 Adults, however, face the risk of gastrointestinal bleeding and renal dysfunction with ibuprofen, which must be weighed against its potential anti-inflammatory benefit. Both populations, however, benefit from chronic azithromycin, whose mechanism of action in this setting is believed to be more anti-inflammatory than bacterial suppression, since it has no direct bactericidal effect on the primary colonizing microbe, P aeruginosa.4,11
Gastrointestinal system: Pancreas
Cystic fibrosis was first comprehensively described in 1938 and was named for the diseased appearance of the pancreas.12 As happens in the lungs, thick pancreatic duct secretions create a cycle of tissue destruction, inflammation, and dysfunction.2 CF patients lack adequate secretion of pancreatic enzymes and bicarbonate into the small bowel, which progressively leads to pancreatic dysfunction in most patients.
As malabsorption of nutrients advances, patients suffer varying degrees of malnutrition and vitamin deficiency, especially of the fat-soluble vitamins A, D, E, and K. Over 85% of CF patients have deficient pancreatic function, requiring pancreatic enzyme supplementation with all food intake and daily vitamin supplementation.2
Ensuring adequate nutrition. Most CF patients experience a chronic mismatch of dietary intake against caloric expenditure and benefit from aggressive nutritional management featuring a high-calorie diet with supplementation in the form of nutrition shakes or bars.2 There is a well-documented linear relationship between BMI and FEV1. Lung function declines in CF when a man’s body mass index (BMI) falls below 23 kg/m2 and a woman’s BMI drops below 22 kg/m2.2 For this reason, the goal for caloric intake can be as high as 200% of the customary recommended daily allowance.2
Watch for CF-related diabetes. Since the pancreas is also the major source of endogenous insulin, nearly half of adults with CF will develop cystic fibrosis-related diabetes (CFRD) as pancreatic deficiency progresses.13 Similar to the relationship between BMI and FEV1, there is a relationship between glucose intolerance and FEV1. For this reason, annual diabetes screening is recommended for all CF patients ages 10 years and older.13 Because glycated hemoglobin (HbA1c) may not accurately reflect low levels of glucose intolerance, screen for CFRD with a 2-hour 75-g oral glucose tolerance test.13 Early insulin therapy can help maintain BMI and lower average blood sugar in support of FEV1. Once CFRD is diagnosed, the goals and recommendations for control are largely the same as those recommended by the American Diabetes Association for other forms of diabetes.13
Cystic Fibrosis Resources
Cystic Fibrosis Foundation
www.cff.org
Consensus report on cystic fibrosis management
Yankaskas JR, Marchall BC, Sufian B, et al. Cystic fibrosis adult care. Chest. 2004;125:1S-39S.
Consensus report on cystic fibrosis diagnostic guidelines
Farrell PM, Rosenstein BJ, White RB. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation Consensus Report. J Pediatr. 2008;153:S4-S14.
Gastrointestinal system: Alimentary canal
CF is often mistakenly believed to be primarily a pulmonary disease since 85% of the mortality is due to lung dysfunction,7 but intermittent abdominal pain is a common experience for most patients, and disorders can range from gastroesophageal reflux disease (GERD) to small bowel bacterial overgrowth (SBBO) to constipation. Up to 85% of adult patients experience symptoms of reflux, with as many as 40% of cases occurring silently.2 Proton pump inhibitors are a first-line treatment, but they can also contribute to intestinal bacterial overgrowth and pulmonary infections.
In SBBO, gram-negative colonic bacteria colonize the small bowel and can contribute to abdominal pain and malabsorption, weight loss, and malnutrition. Treatment requires antibiotics with activity against gram-negative organisms, or non-absorbable agents such as rifamyxin, sometimes on a chronic, recurrent, or rotating basis.2
Chronic constipation is also quite common among CF patients and many require daily administration of poly-ethylene-glycol. Before newborn screening programs were introduced, infants would on occasion present with complete distal intestinal obstruction. Adults are not immune to obstructive complications and may require hospitalization for bowel cleansing.
Gastrointestinal system: Liver
Liver disease is relatively common in CF, with up to 24% of adults experiencing hepatomegaly or persistently elevated liver function tests (LFT).4 Progressive biliary fibrosis and cirrhosis are encountered more often as the median survival age has increased. There is evidence that ursodeoxycholic acid (UDCA) can be a useful adjunct in the treatment of cholestasis, but it is not clear if it alters mortality or progression to cirrhosis. Only CF patients with elevated LFTs should be started on UDCA.4
Other areas of concern: Sinuses, serum sodium levels
Chronic, symptomatic sinus disease in CF patients—chiefly polyposis—is common and may require repeat surgery, although most patients with extensive nasal polyps find symptom relief with daily sinus rinses. Intranasal steroids and intranasal antibiotics are also often employed, and many CF patients need to be in regular contact with an otolaryngologist.14 For symptoms of allergic rhinitis, recommend OTC antihistamines in standard dosages.
Exercise is recommended for all CF patients, as noted earlier, and as life expectancy increases, many are engaging in more strenuous and longer duration activities.15 Due to high sweat sodium loss, CF patients are at risk for hyponatremia, especially when exercising on days with high temperatures and humidity. CF patients need to replace sodium losses in these conditions and when exercising for extended periods.
There are no evidence-based guidelines for sodium replacement. The Cystic Fibrosis Foundation (CFF) recommends that patients increase salt in the diet when under conditions likely to result in increased sodium loss, such as exercise. It has been thought that CF patients can easily dehydrate due to an impaired thirst mechanism and, when exercising, should consume fluids beyond the need to quench thirst.16,17 More recent evidence suggests, however, that the thirst mechanism in those with CF remains normally intact and that overconsumption of fluids beyond the level of thirst may predispose the individual to exercise-associated hyponatremia as serum sodium is diluted.15
New therapies
Small-molecule CFTR-modulating compounds are a promising development in the treatment of CF. The first such available medication was ivacaftor in 2012. Because these molecules are mutation specific, ivacaftor was available at first only for patients with at least one copy of the G551D mutation,18 which means about 5% of patients with CF.3
Ivacaftor increases the likelihood that the CFTR chloride channel will open and patients will exhibit a reduction in sweat chloride levels. In the first reported clinical trial of ivacaftor involving patients with the G551D mutation, FEV1 improvements of 10% occurred by the second week of therapy and persisted for 48 weeks.18 The drug has now been approved by the US Food and Drug Administration (FDA) for patients 12 years of age and older with at least one of the following mutations: R117H, G551D, G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, or G1349D.19
A medication combining ivacaftor with lumacaftor is also now available for patients with a copy of F508del on both chromosomes. F508del is the most common CFTR mutation, with one copy present in almost 87% of people with CF in the United States.1 Since 47% of CF patients have 2 copies of F508del,1 about half of those with CF in the United States are now eligible for small-molecule therapy. Lumacaftor acts by facilitating transport of a misfolded CFTR to the cell membrane where ivacaftor then increases the probability of an open chloride channel. This combination medication has improved lung function by about 5%.
The ivacaftor/lumacaftor combination was approved by the FDA in July 2015. Both ivacaftor and the ivacaftor/lumacaftor combination were deemed by the FDA to demonstrate statistically significant and sustained FEV1 improvements over placebo.
The CFF was instrumental in providing financial support for the development of both ivacaftor and the ivacaftor/lumacaftor combination and continues to provide significant research advancement. According to the CFF (www.cff.org), medications currently in the development pipeline include compounds that provide CFTR modulation, surface airway liquid restoration, anti-inflammation, inhaled anti-infection, and pancreatic enzyme function. For more on CFF, see "The traditional CF care model.”4,20
The traditional CF care model
The Cystic Fibrosis Foundation (CFF) has been a driving force behind the increased life expectancy CF patients have seen over the last 3 decades. Its contributions include the development of medication through the CFF Therapeutics Development Network (TDN) and disease management through a network of CF Care Centers throughout the United States. The CFF recommends a minimum of quarterly visits to a CF Care Center, and the primary care physician can play a critical role alongside the multidisciplinary CF team.20
At every CF Care Center encounter, the entire team (nurse, physician, dietician, social worker, psychologist) interacts with each patient and their families to maximize overall medical care. Respiratory cultures are generally obtained at each visit. Dual-energy x-ray absorptiometry is performed biannually. Lab work (complete blood count, comprehensive metabolic panel, glycated hemoglobin, vitamins A, D, E, and K, 2-hour glucose tolerance test), and chest x-ray are obtained at least annually (TABLE 2).4
Since CF generally involves both restrictive and obstructive lung components, complete spirometry evaluation is performed annually in the pulmonary function lab, with static lung volumes in addition to airflow measurement. Office spirometry to measure airflow alone is performed at each visit. FEV1 is tracked both as an indicator of disease progression and as a measure of current pulmonary status.
The CFF recommends that each patient receive full genetic testing and encourages patient participation in the CFF Registry, where mutation data are documented among other disease parameters to ensure that patients receive mutation specific therapies as they become available.4 The vaccine schedule recommended for CF patients is the same as for the general population.
CORRESPONDENCE
Douglas Lewis, MD, 1121 S. Clifton, Wichita, KS 67218; douglas.lewis@viachristi.org.
1. Cystic Fibrosis Foundation. Patient registry 2012 annual data report. Cystic Fibrosis Foundation Web site. Available at: http://www.cff.org/UploadedFiles/research/ClinicalResearch/PatientRegistryReport/2012-CFF-Patient-Registry.pdf. Accessed August 14, 2014.
2. Haller W, Ledder O, Lewindon PJ, et al. Cystic fibrosis: An update for clinicians. Part 1: Nutrition and gastrointestinal complications. J Gastroenterol Hepatol. 2014;29:1344-1355.
3. Hoffman LR, Ramsey BW. Cystic fibrosis therapeutics: the road ahead. Chest. 2013;143:207-213.
4. Yankaskas JR, Marshall BC, Sufian B, et al. Cystic fibrosis adult care: consensus conference report. Chest. 2004;125:1S-39S.
5. Farrell PM, Rosenstein BJ, White TB, et al; Cystic Fibrosis Foundation. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr. 2008;153:S4-S14.
6. Donaldson SH, Boucher RC. Sodium channels and cystic fibrosis. Chest. 2007;132:1631-1636.
7. Flume PA, Robinson KA, O’Sullivan BP, et al; Clinical Practice Guidelines for Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: airway clearance therapies. Respir Care. 2009;54:522-537.
8. Flume PA, O’Sullivan BP, Robinson KA, et al; Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176:957-969.
9. Döring G, Flume P, Heijerman H, et al; Consensus Study Group. Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J Cyst Fibros. 2012;11:461-479.
10. Flume PA, Mogayzel PJ Jr, Robinson KA, et al; Clinical Practice Guidelines for Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med. 2009;180:802-808.
11. Southern KW, Barker PM. Azithromycin for cystic fibrosis. Eur Respir J. 2004;24:834-838.
12. Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Am J Dis Child. 1938;56:344-399.
13. Moran A, Brunzell C, Cohen RC, et al; CFRD Guidelines Committee. Clinical care guidelines for cystic fibrosis-related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care. 2010;33:2697-2708.
14. Kerem E, Conway S, Elborn S, et al; Consensus Committee. Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros. 2005;4:7-26.
15. Hew-Butler T, Rosner MH, Fowkes-Godek S, et al. Statement of the third international exercise-associated hyponatremia consensus development conference, Carlsbad, California, 2015. Clin J Sport Med. 2015;25:303-320.
16. Brown MB, McCarty NA, Millard-Stafford M. High-sweat Na+ in cystic fibrosis and healthy individuals does not diminish thirst during exercise in the heat. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1177-R1185.
17. Wheatley CM, Wilkins BW, Snyder EM. Exercise is medicine in cystic fibrosis. Exerc Sport Sci Rev. 2011;39:155-160.
18. Ramsey BW, Davies J, McElvaney NG, et al; VX08-770-102 Study Group. ACFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663-1672.
19. Pettit RS, Fellner C. CFTR Modulators for the Treatment of Cystic Fibrosis. P T. 2014;39:500-511.
20. Lewis D. Role of the family physician in the management of cystic fibrosis. Am Fam Physician. 2015;91:822-824.
1. Cystic Fibrosis Foundation. Patient registry 2012 annual data report. Cystic Fibrosis Foundation Web site. Available at: http://www.cff.org/UploadedFiles/research/ClinicalResearch/PatientRegistryReport/2012-CFF-Patient-Registry.pdf. Accessed August 14, 2014.
2. Haller W, Ledder O, Lewindon PJ, et al. Cystic fibrosis: An update for clinicians. Part 1: Nutrition and gastrointestinal complications. J Gastroenterol Hepatol. 2014;29:1344-1355.
3. Hoffman LR, Ramsey BW. Cystic fibrosis therapeutics: the road ahead. Chest. 2013;143:207-213.
4. Yankaskas JR, Marshall BC, Sufian B, et al. Cystic fibrosis adult care: consensus conference report. Chest. 2004;125:1S-39S.
5. Farrell PM, Rosenstein BJ, White TB, et al; Cystic Fibrosis Foundation. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr. 2008;153:S4-S14.
6. Donaldson SH, Boucher RC. Sodium channels and cystic fibrosis. Chest. 2007;132:1631-1636.
7. Flume PA, Robinson KA, O’Sullivan BP, et al; Clinical Practice Guidelines for Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: airway clearance therapies. Respir Care. 2009;54:522-537.
8. Flume PA, O’Sullivan BP, Robinson KA, et al; Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176:957-969.
9. Döring G, Flume P, Heijerman H, et al; Consensus Study Group. Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J Cyst Fibros. 2012;11:461-479.
10. Flume PA, Mogayzel PJ Jr, Robinson KA, et al; Clinical Practice Guidelines for Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med. 2009;180:802-808.
11. Southern KW, Barker PM. Azithromycin for cystic fibrosis. Eur Respir J. 2004;24:834-838.
12. Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Am J Dis Child. 1938;56:344-399.
13. Moran A, Brunzell C, Cohen RC, et al; CFRD Guidelines Committee. Clinical care guidelines for cystic fibrosis-related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care. 2010;33:2697-2708.
14. Kerem E, Conway S, Elborn S, et al; Consensus Committee. Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros. 2005;4:7-26.
15. Hew-Butler T, Rosner MH, Fowkes-Godek S, et al. Statement of the third international exercise-associated hyponatremia consensus development conference, Carlsbad, California, 2015. Clin J Sport Med. 2015;25:303-320.
16. Brown MB, McCarty NA, Millard-Stafford M. High-sweat Na+ in cystic fibrosis and healthy individuals does not diminish thirst during exercise in the heat. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1177-R1185.
17. Wheatley CM, Wilkins BW, Snyder EM. Exercise is medicine in cystic fibrosis. Exerc Sport Sci Rev. 2011;39:155-160.
18. Ramsey BW, Davies J, McElvaney NG, et al; VX08-770-102 Study Group. ACFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663-1672.
19. Pettit RS, Fellner C. CFTR Modulators for the Treatment of Cystic Fibrosis. P T. 2014;39:500-511.
20. Lewis D. Role of the family physician in the management of cystic fibrosis. Am Fam Physician. 2015;91:822-824.
The PARADIGM-HF trial
To the Editor: Two considerations concerning the interpretation of the Prospective Comparison of ARNI with ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial are not addressed in the article by Sabe et al regarding a new class of drugs for systolic heart failure.1 First of all, the PARADIGM-HF trial compared the maximal dose of sacubitril with a less-than-maximal dose of enalapril. Secondly, sacubitril lowered blood pressure more than enalapril.
The angiotensin receptor blocker dose in sacubitril 200 mg is equivalent to valsartan 160 mg.2 Accordingly, the angiotensin receptor blocker in sacubitril 200 mg twice daily is equivalent to the maximal dosage of valsartan approved by the US Food and Drug Administration. The dosage of enalapril in the PARADIGM-HF trial was 10 mg twice daily. While the target enalapril dosage for heart failure is 10 to 20 mg twice daily,3 the dosage of enalapril in PARADIGM-HF was half the maximal approved dosage.
In the PARADIGM-HF trial, sacubitril 200 mg twice daily reduced the incidence of cardiovascular death by 19% compared with enalapril 10 mg twice daily (the rates were 16.5% vs 13.3%, respectively).2 That sacubitril lowered mean systolic blood pressure 3.2 ± 0.4 mm Hg more than enalapril2,4 may account for much of this benefit.
A 2002 study by Lewington et al5 found that a 2-mm Hg decrease in systolic blood pressure reduces the risk of cardiovascular death by 7% in middle-aged adults. Granted, this study did not involve heart failure patients, but if its results are remotely applicable, a 3.2-mm Hg reduction in systolic blood pressure might be expected to reduce the rate of cardiovascular deaths by 10% to 11%.
Would sacubitril be superior to enalapril if the maximal dose of enalapril were compared to the maximal dose of sacubitril? Would sacubitril be superior to enalapril if blood pressure were lowered comparably between the two groups? These are relevant questions that the PARADIGM-HF trial fails to answer.
- Sabe MA, Jacob MS, Taylor DO. A new class of drugs for systolic heart failure: the PARADIGM-HF study. Cleve Clin J Med 2015; 82:693–701.
- McMurray JJV, Packer M, Desai AS, et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371:993–1004.
- Hunt SA; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure). ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure). J Am Coll Cardiol 2005; 46:e1–e82.
- Jessup J. Neprilysin inhibition—a novel therapy for heart failure. N Engl J Med 2014; 371:1062–1064.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
To the Editor: Two considerations concerning the interpretation of the Prospective Comparison of ARNI with ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial are not addressed in the article by Sabe et al regarding a new class of drugs for systolic heart failure.1 First of all, the PARADIGM-HF trial compared the maximal dose of sacubitril with a less-than-maximal dose of enalapril. Secondly, sacubitril lowered blood pressure more than enalapril.
The angiotensin receptor blocker dose in sacubitril 200 mg is equivalent to valsartan 160 mg.2 Accordingly, the angiotensin receptor blocker in sacubitril 200 mg twice daily is equivalent to the maximal dosage of valsartan approved by the US Food and Drug Administration. The dosage of enalapril in the PARADIGM-HF trial was 10 mg twice daily. While the target enalapril dosage for heart failure is 10 to 20 mg twice daily,3 the dosage of enalapril in PARADIGM-HF was half the maximal approved dosage.
In the PARADIGM-HF trial, sacubitril 200 mg twice daily reduced the incidence of cardiovascular death by 19% compared with enalapril 10 mg twice daily (the rates were 16.5% vs 13.3%, respectively).2 That sacubitril lowered mean systolic blood pressure 3.2 ± 0.4 mm Hg more than enalapril2,4 may account for much of this benefit.
A 2002 study by Lewington et al5 found that a 2-mm Hg decrease in systolic blood pressure reduces the risk of cardiovascular death by 7% in middle-aged adults. Granted, this study did not involve heart failure patients, but if its results are remotely applicable, a 3.2-mm Hg reduction in systolic blood pressure might be expected to reduce the rate of cardiovascular deaths by 10% to 11%.
Would sacubitril be superior to enalapril if the maximal dose of enalapril were compared to the maximal dose of sacubitril? Would sacubitril be superior to enalapril if blood pressure were lowered comparably between the two groups? These are relevant questions that the PARADIGM-HF trial fails to answer.
To the Editor: Two considerations concerning the interpretation of the Prospective Comparison of ARNI with ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial are not addressed in the article by Sabe et al regarding a new class of drugs for systolic heart failure.1 First of all, the PARADIGM-HF trial compared the maximal dose of sacubitril with a less-than-maximal dose of enalapril. Secondly, sacubitril lowered blood pressure more than enalapril.
The angiotensin receptor blocker dose in sacubitril 200 mg is equivalent to valsartan 160 mg.2 Accordingly, the angiotensin receptor blocker in sacubitril 200 mg twice daily is equivalent to the maximal dosage of valsartan approved by the US Food and Drug Administration. The dosage of enalapril in the PARADIGM-HF trial was 10 mg twice daily. While the target enalapril dosage for heart failure is 10 to 20 mg twice daily,3 the dosage of enalapril in PARADIGM-HF was half the maximal approved dosage.
In the PARADIGM-HF trial, sacubitril 200 mg twice daily reduced the incidence of cardiovascular death by 19% compared with enalapril 10 mg twice daily (the rates were 16.5% vs 13.3%, respectively).2 That sacubitril lowered mean systolic blood pressure 3.2 ± 0.4 mm Hg more than enalapril2,4 may account for much of this benefit.
A 2002 study by Lewington et al5 found that a 2-mm Hg decrease in systolic blood pressure reduces the risk of cardiovascular death by 7% in middle-aged adults. Granted, this study did not involve heart failure patients, but if its results are remotely applicable, a 3.2-mm Hg reduction in systolic blood pressure might be expected to reduce the rate of cardiovascular deaths by 10% to 11%.
Would sacubitril be superior to enalapril if the maximal dose of enalapril were compared to the maximal dose of sacubitril? Would sacubitril be superior to enalapril if blood pressure were lowered comparably between the two groups? These are relevant questions that the PARADIGM-HF trial fails to answer.
- Sabe MA, Jacob MS, Taylor DO. A new class of drugs for systolic heart failure: the PARADIGM-HF study. Cleve Clin J Med 2015; 82:693–701.
- McMurray JJV, Packer M, Desai AS, et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371:993–1004.
- Hunt SA; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure). ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure). J Am Coll Cardiol 2005; 46:e1–e82.
- Jessup J. Neprilysin inhibition—a novel therapy for heart failure. N Engl J Med 2014; 371:1062–1064.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- Sabe MA, Jacob MS, Taylor DO. A new class of drugs for systolic heart failure: the PARADIGM-HF study. Cleve Clin J Med 2015; 82:693–701.
- McMurray JJV, Packer M, Desai AS, et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371:993–1004.
- Hunt SA; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure). ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure). J Am Coll Cardiol 2005; 46:e1–e82.
- Jessup J. Neprilysin inhibition—a novel therapy for heart failure. N Engl J Med 2014; 371:1062–1064.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
In reply: The PARADIGM-HF trial
In Reply: We thank Dr. Blankfield for raising these two important points. Although the findings of the PARADIGM-HF study are compelling, the design and results of this trial have incited many questions.
To address his first point, about the differential dosages of the two drugs, we agree, and we did mention in our review that one concern about the results of PARADIGM-HF is the unequal dosages of valsartan and enalapril in the two different arms. We mentioned that this dosage of enalapril was chosen based on its survival benefit in previous trials. However, this still raises the question of whether the benefit seen in the sacubitril-valsartan group was due to greater inhibition of the renin-angiotensin-aldosterone system rather than to the new drug.
To address his second point, the decrease in blood pressure in the sacubitril-valsartan arm was significant, and the patients taking this drug were more likely to have symptomatic hypotension, which may contribute to patient intolerance and difficulty initiating treatment with this drug. Dr. Blankfield brings up an interesting point regarding reduction of blood pressure driving the decrease of events in the sacubitril-valsartan group. In the original trial results section, the authors mentioned that when the difference in blood pressure between the two groups was examined as a time-dependent covariate, it was not a significant predictor of the benefit of sacubitril-valsartan.1
Furthermore, although higher blood pressure is associated with worse cardiovascular outcomes in the general population, higher blood pressure has been shown to be protective in heart failure patients.2 Several studies have shown that the relationship between blood pressure and the mortality rate in patients with heart failure is paradoxical and complex.2–4 Lee et al3 found that this relationship was U-shaped, with increased mortality risk in those with high and low blood pressures (< 120 mm Hg). Ather et al4 also showed that the relationship was U-shaped in patients with a mild to moderate reduction in left ventricular ejection fraction, but linear in those with severely reduced ejection fraction. This study also found that a decrease in systolic blood pressure below 110 mm Hg was associated with increased mortality risk.
The findings of PARADIGM-HF have sparked much conversation and implementation of practice change in the treatment of heart failure patients, and we await additional data on the use and limitations of sacubitril-valsartan in this group of patients.
- McMurray JJV, Packer M, Desai AS, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371:993–1004.
- Raphael CE, Whinnett ZI, Davies JE, et al. Quantifying the paradoxical effect of higher systolic blood pressure on mortality in chronic heart failure. Heart 2009; 95:56–62.
- Lee DS, Ghosh N, Floras JS, et al. Association of blood pressure at hospital discharge with mortality in patients diagnosed with heart failure. Circ Heart Fail 2009; 2:616-623.
- Ather S, Chan W, Chillar A, et al. Association of systolic blood pressure with mortality in patients with heart failure with reduced ejection fraction: a complex relationship. Am Heart J 2011; 161:567–573.
In Reply: We thank Dr. Blankfield for raising these two important points. Although the findings of the PARADIGM-HF study are compelling, the design and results of this trial have incited many questions.
To address his first point, about the differential dosages of the two drugs, we agree, and we did mention in our review that one concern about the results of PARADIGM-HF is the unequal dosages of valsartan and enalapril in the two different arms. We mentioned that this dosage of enalapril was chosen based on its survival benefit in previous trials. However, this still raises the question of whether the benefit seen in the sacubitril-valsartan group was due to greater inhibition of the renin-angiotensin-aldosterone system rather than to the new drug.
To address his second point, the decrease in blood pressure in the sacubitril-valsartan arm was significant, and the patients taking this drug were more likely to have symptomatic hypotension, which may contribute to patient intolerance and difficulty initiating treatment with this drug. Dr. Blankfield brings up an interesting point regarding reduction of blood pressure driving the decrease of events in the sacubitril-valsartan group. In the original trial results section, the authors mentioned that when the difference in blood pressure between the two groups was examined as a time-dependent covariate, it was not a significant predictor of the benefit of sacubitril-valsartan.1
Furthermore, although higher blood pressure is associated with worse cardiovascular outcomes in the general population, higher blood pressure has been shown to be protective in heart failure patients.2 Several studies have shown that the relationship between blood pressure and the mortality rate in patients with heart failure is paradoxical and complex.2–4 Lee et al3 found that this relationship was U-shaped, with increased mortality risk in those with high and low blood pressures (< 120 mm Hg). Ather et al4 also showed that the relationship was U-shaped in patients with a mild to moderate reduction in left ventricular ejection fraction, but linear in those with severely reduced ejection fraction. This study also found that a decrease in systolic blood pressure below 110 mm Hg was associated with increased mortality risk.
The findings of PARADIGM-HF have sparked much conversation and implementation of practice change in the treatment of heart failure patients, and we await additional data on the use and limitations of sacubitril-valsartan in this group of patients.
In Reply: We thank Dr. Blankfield for raising these two important points. Although the findings of the PARADIGM-HF study are compelling, the design and results of this trial have incited many questions.
To address his first point, about the differential dosages of the two drugs, we agree, and we did mention in our review that one concern about the results of PARADIGM-HF is the unequal dosages of valsartan and enalapril in the two different arms. We mentioned that this dosage of enalapril was chosen based on its survival benefit in previous trials. However, this still raises the question of whether the benefit seen in the sacubitril-valsartan group was due to greater inhibition of the renin-angiotensin-aldosterone system rather than to the new drug.
To address his second point, the decrease in blood pressure in the sacubitril-valsartan arm was significant, and the patients taking this drug were more likely to have symptomatic hypotension, which may contribute to patient intolerance and difficulty initiating treatment with this drug. Dr. Blankfield brings up an interesting point regarding reduction of blood pressure driving the decrease of events in the sacubitril-valsartan group. In the original trial results section, the authors mentioned that when the difference in blood pressure between the two groups was examined as a time-dependent covariate, it was not a significant predictor of the benefit of sacubitril-valsartan.1
Furthermore, although higher blood pressure is associated with worse cardiovascular outcomes in the general population, higher blood pressure has been shown to be protective in heart failure patients.2 Several studies have shown that the relationship between blood pressure and the mortality rate in patients with heart failure is paradoxical and complex.2–4 Lee et al3 found that this relationship was U-shaped, with increased mortality risk in those with high and low blood pressures (< 120 mm Hg). Ather et al4 also showed that the relationship was U-shaped in patients with a mild to moderate reduction in left ventricular ejection fraction, but linear in those with severely reduced ejection fraction. This study also found that a decrease in systolic blood pressure below 110 mm Hg was associated with increased mortality risk.
The findings of PARADIGM-HF have sparked much conversation and implementation of practice change in the treatment of heart failure patients, and we await additional data on the use and limitations of sacubitril-valsartan in this group of patients.
- McMurray JJV, Packer M, Desai AS, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371:993–1004.
- Raphael CE, Whinnett ZI, Davies JE, et al. Quantifying the paradoxical effect of higher systolic blood pressure on mortality in chronic heart failure. Heart 2009; 95:56–62.
- Lee DS, Ghosh N, Floras JS, et al. Association of blood pressure at hospital discharge with mortality in patients diagnosed with heart failure. Circ Heart Fail 2009; 2:616-623.
- Ather S, Chan W, Chillar A, et al. Association of systolic blood pressure with mortality in patients with heart failure with reduced ejection fraction: a complex relationship. Am Heart J 2011; 161:567–573.
- McMurray JJV, Packer M, Desai AS, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371:993–1004.
- Raphael CE, Whinnett ZI, Davies JE, et al. Quantifying the paradoxical effect of higher systolic blood pressure on mortality in chronic heart failure. Heart 2009; 95:56–62.
- Lee DS, Ghosh N, Floras JS, et al. Association of blood pressure at hospital discharge with mortality in patients diagnosed with heart failure. Circ Heart Fail 2009; 2:616-623.
- Ather S, Chan W, Chillar A, et al. Association of systolic blood pressure with mortality in patients with heart failure with reduced ejection fraction: a complex relationship. Am Heart J 2011; 161:567–573.
The microbiome in celiac disease: Beyond diet-genetic interactions
Inheriting the wrong genes and eating the wrong food (ie, gluten) are necessary for celiac disease to develop, but are not enough by themselves. Something else must be contributing, and evidence is pointing to the mix of bacteria that make our guts their home, collectively called the microbiome.
Celiac disease is a highly prevalent, chronic, immune-mediated form of enteropathy.1 It affects 0.5% to 1% of the population, and although it is mostly seen in people of northern European descent, those in other populations can develop the disease as well. Historically, celiac disease was classified as an infant condition. However, it now commonly presents later in life (between ages 10 and 40) and often with extraintestinal manifestations.2
In this issue of Cleveland Clinic Journal of Medicine, Kochhar et al provide a comprehensive updated review of celiac disease.3
GENES AND GLUTEN ARE NECESSARY BUT NOT SUFFICIENT
Although genetic factors and exposure to gluten in the diet are proven to be necessary for celiac disease to develop, they are not sufficient. Evidence of this is in the numbers; although one-third of the general population carries the HLA susceptibility genes (specifically HLA-DQ2 and DQ8),4 only 2% to 5% of people with these genes develop clinically evident celiac disease.
Additional environmental factors must be contributing to disease development, but these other factors are poorly understood. Some of the possible culprits that might influence the risk of disease occurrence and the timing of its onset include5:
- The amount and quality of gluten ingested—the higher the concentration of gluten, the higher the risk, and different grains have gluten varieties with more or less immunogenic capabilities, ie, T-cell activation properties
- The pattern of infant feeding—the risk may be lower with breastfeeding than with formula
- The age at which gluten is introduced into the diet—the risk may be higher if gluten is introduced earlier.6
More recently, studies of the pathogenesis of celiac disease and gene-environmental interactions have expanded beyond host predisposition and dietary factors.
OUR BODIES, OUR MICROBIOMES: A SYMBIOTIC RELATIONSHIP
The role of the human microbiome in autoimmune disease is now being elucidated.7 Remarkably, the microorganisms living in our bodies outnumber our body cells by a factor of 10, and their genomes vastly exceed our own protein-coding genome capabilities by a factor of 100.
The gut microbiome is now considered a true bioreactor with enzymatic and immunologic capabilities beyond (and complementary to) those of its host. The commensal microbiome of the host intestine provides benefits that can be broken down into three broad categories:
- Nutritional—producing essential amino acids and vitamins
- Metabolic—degrading complex polysaccharides from dietary fibers
- Immunologic—shaping the host immune system while cooperating with it against pathogenic microorganisms.
The immunologic function is highly relevant. We have coevolved with our bacteria in a mutually beneficial, symbiotic relationship in which we maintain an active state of low inflammation so that a constant bacterial and dietary antigenic load can be tolerated.
Is there a core human microbiome shared by all individuals? And what is the impact of altering the relative microbial composition (dysbiosis) in physiologic and disease states? To find out, the National Institutes of Health launched the Human Microbiome Project8 in 2008. Important tools in this work include novel culture-independent approaches (high-throughput DNA sequencing and whole-microbiome “shotgun” sequencing with metagenomic analysis) and computational analytical tools.9
An accumulating body of evidence is now available from animal models and human studies correlating states of intestinal dysbiosis (disruption in homeostatic community composition) with various disease processes. These have ranged from inflammatory bowel disease to systemic autoimmune disorders such as psoriasis, inflammatory arthropathies, and demyelinating central nervous system diseases.10–14
RESEARCH INTO THE MICROBIOME IN CELIAC DISEASE
Celiac disease has also served as a unique model for studying this biologic relationship, and the microbiome has been postulated to have a role in its pathogenesis.15 Multiple clinical studies demonstrate that a state of intestinal dysbiosis is indeed associated with celiac disease.
Specifically, decreases in the abundance of Firmicutes spp and increases in Proteobacteria spp have been detected in both children and adults with active celiac disease.16,17 Intriguingly, overrepresentation of Proteobacteria was also correlated with disease activity. Other studies have reported decreases in the proportion of reportedly protective, anti-inflammatory bacteria such as Bifidobacterium and increases in the proportion of Bacteroides and Escherichia coli in patients with active disease.18,19 Altered diversity and altered metabolic function, ie, decreased concentration of protective short-chain fatty acids of the microbiota, have also been reported in patients with celiac disease.19,20
To move beyond correlative studies and mechanistically address the possibility of causation, multiple groups have used a gnotobiotic approach, ie, maintaining animals under germ-free conditions and incorporating microbes of interest. This approach is highly relevant in studying whether the bacterial community composition is capable of modulating loss of tolerance to gluten in genetically susceptible hosts. A few notable examples have been published.
In germ-free rats, long-term feeding of gliadin, but not albumin, from birth until 2 months of age induced moderate small-intestinal damage.21 Similarly, germ-free nonobese diabetic-DQ8 mice developed more severe gluten-induced disease than mice with normal intestinal bacteria.22
These findings suggest that the normal gut microbiome may have intrinsic beneficial properties capable of reducing the inflammatory effects associated with gluten ingestion. Notably, the specific composition of the intestinal microbiome can define the fate of gluten-induced pathology. Mice colonized with commensal microbiota are indeed protected from gluten-induced pathology, while mice colonized with Proteobacteria spp develop a moderate degree of gluten-induced disease. When Escherichia coli derived from patients with celiac disease is added to commensal colonization, the celiac disease-like phenotype develops.23
Taken together, these studies support the hypothesis that the intestinal microbiome may be another environmental factor involved in the development of celiac disease.
QUESTIONS AND CHALLENGES REMAIN
The results of clinical studies are not necessarily consistent at the taxonomy level. The fields of metagenomics, which investigates all genes and their enzymatic function in a given community, and metabolomics, which identifies bacterial end-products, characterizing their functional capabilities, are still in their infancy and will be required to further investigate functionality of the altered microbiome in celiac disease.
Second, the directionality—the causality or consequences of this dysbiosis—and timing—the moment at which changes occur, ie, after introducing gluten or at the time when symptoms appear—remain elusive, and prospective studies in humans will be essential.
Finally, more mechanistic studies in animal models are needed to dissect the host immune response to dietary gluten and perturbation of intestinal community composition. This may lead to the possibility of future interventions in the form of prebiotics, probiotics, or specific metabolites, complementary to gluten avoidance.
In the meantime, increasing disease awareness and rapid diagnosis and treatment continue to be of utmost importance to address the clinical consequences of celiac disease in both children and adults.
- Guandalini S, Assiri A. Celiac disease: a review. JAMA Pediatr 2014; 168:272–278.
- Green PH, Cellier C. Celiac disease. N Engl J Med 2007; 357:1731–1743.
- Kochhar GS, Singh T, Gill A, Kirby DF. Celiac disease: an internist’s perspective. Cleve Clin J Med 2016; 83:217–227.
- Gutierrez-Achury J, Zhernakova A, Pulit SL, et al. Fine mapping in the MHC region accounts for 18% additional genetic risk for celiac disease. Nat Genet 2015; 47:577–578.
- Catassi C, Kryszak D, Bhatti B, et al. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann Med 2010; 42:530–538.
- Norris JM, Barriga K, Hoffenberg EJ, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA 2005; 293:2343–2351.
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature 2007; 449:804–810.
- NIH HMP Working Group; Peterson J, Garges S, Giovanni M, et al. The NIH Human Microbiome Project. Genome Res 2009; 19:2317–2323.
- Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59–65.
- Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013; 2:e01202.
- Scher JU, Ubeda C, Artacho A, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol 2015; 67:128–139.
- Gao Z, Tseng CH, Strober BE, Pei Z, Blaser MJ. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS One 2008; 3:e2719.
- Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013; 155:1451–1463.
- Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn‘s disease. Cell Host Microbe 2014; 15:382–392.
- Verdu EF, Galipeau HJ, Jabri B. Novel players in coeliac disease pathogenesis: role of the gut microbiota. Nat Rev Gastroenterol Hepatol 2015; 12:497–506.
- Sanchez E, Donat E, Ribes-Koninckx C, Fernandez-Murga ML, Sanz Y. Duodenal-mucosal bacteria associated with celiac disease in children. Appl Environ Microbiol 2013; 79:5472–5479.
- Wacklin P, Kaukinen K, Tuovinen E, et al. The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm Bowel Dis 2013; 19:934–941.
- Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J Clin Pathol 2009; 62:264–269.
- Di Cagno R, De Angelis M, De Pasquale I, et al. Duodenal and faecal microbiota of celiac children: molecular, phenotype and metabolome characterization. BMC Microbiol 2011; 11:219.
- Schippa S, Iebba V, Barbato M, et al. A distinctive ‘microbial signature’ in celiac pediatric patients. BMC Microbiol 2010; 10:175.
- Stepankova R, Tlaskalova-Hogenova H, Sinkora J, Jodl J, Fric P. Changes in jejunal mucosa after long-term feeding of germfree rats with gluten. Scand J Gastroenterol 1996; 31:551–557.
- Galipeau HJ, Rulli NE, Jury J, et al. Sensitization to gliadin induces moderate enteropathy and insulitis in nonobese diabetic-DQ8 mice. J Immunol 2011; 187:4338–4346.
- Galipeau HJ, Verdu EF. Gut microbes and adverse food reactions: focus on gluten related disorders. Gut Microbes 2014; 5:594–605.
Inheriting the wrong genes and eating the wrong food (ie, gluten) are necessary for celiac disease to develop, but are not enough by themselves. Something else must be contributing, and evidence is pointing to the mix of bacteria that make our guts their home, collectively called the microbiome.
Celiac disease is a highly prevalent, chronic, immune-mediated form of enteropathy.1 It affects 0.5% to 1% of the population, and although it is mostly seen in people of northern European descent, those in other populations can develop the disease as well. Historically, celiac disease was classified as an infant condition. However, it now commonly presents later in life (between ages 10 and 40) and often with extraintestinal manifestations.2
In this issue of Cleveland Clinic Journal of Medicine, Kochhar et al provide a comprehensive updated review of celiac disease.3
GENES AND GLUTEN ARE NECESSARY BUT NOT SUFFICIENT
Although genetic factors and exposure to gluten in the diet are proven to be necessary for celiac disease to develop, they are not sufficient. Evidence of this is in the numbers; although one-third of the general population carries the HLA susceptibility genes (specifically HLA-DQ2 and DQ8),4 only 2% to 5% of people with these genes develop clinically evident celiac disease.
Additional environmental factors must be contributing to disease development, but these other factors are poorly understood. Some of the possible culprits that might influence the risk of disease occurrence and the timing of its onset include5:
- The amount and quality of gluten ingested—the higher the concentration of gluten, the higher the risk, and different grains have gluten varieties with more or less immunogenic capabilities, ie, T-cell activation properties
- The pattern of infant feeding—the risk may be lower with breastfeeding than with formula
- The age at which gluten is introduced into the diet—the risk may be higher if gluten is introduced earlier.6
More recently, studies of the pathogenesis of celiac disease and gene-environmental interactions have expanded beyond host predisposition and dietary factors.
OUR BODIES, OUR MICROBIOMES: A SYMBIOTIC RELATIONSHIP
The role of the human microbiome in autoimmune disease is now being elucidated.7 Remarkably, the microorganisms living in our bodies outnumber our body cells by a factor of 10, and their genomes vastly exceed our own protein-coding genome capabilities by a factor of 100.
The gut microbiome is now considered a true bioreactor with enzymatic and immunologic capabilities beyond (and complementary to) those of its host. The commensal microbiome of the host intestine provides benefits that can be broken down into three broad categories:
- Nutritional—producing essential amino acids and vitamins
- Metabolic—degrading complex polysaccharides from dietary fibers
- Immunologic—shaping the host immune system while cooperating with it against pathogenic microorganisms.
The immunologic function is highly relevant. We have coevolved with our bacteria in a mutually beneficial, symbiotic relationship in which we maintain an active state of low inflammation so that a constant bacterial and dietary antigenic load can be tolerated.
Is there a core human microbiome shared by all individuals? And what is the impact of altering the relative microbial composition (dysbiosis) in physiologic and disease states? To find out, the National Institutes of Health launched the Human Microbiome Project8 in 2008. Important tools in this work include novel culture-independent approaches (high-throughput DNA sequencing and whole-microbiome “shotgun” sequencing with metagenomic analysis) and computational analytical tools.9
An accumulating body of evidence is now available from animal models and human studies correlating states of intestinal dysbiosis (disruption in homeostatic community composition) with various disease processes. These have ranged from inflammatory bowel disease to systemic autoimmune disorders such as psoriasis, inflammatory arthropathies, and demyelinating central nervous system diseases.10–14
RESEARCH INTO THE MICROBIOME IN CELIAC DISEASE
Celiac disease has also served as a unique model for studying this biologic relationship, and the microbiome has been postulated to have a role in its pathogenesis.15 Multiple clinical studies demonstrate that a state of intestinal dysbiosis is indeed associated with celiac disease.
Specifically, decreases in the abundance of Firmicutes spp and increases in Proteobacteria spp have been detected in both children and adults with active celiac disease.16,17 Intriguingly, overrepresentation of Proteobacteria was also correlated with disease activity. Other studies have reported decreases in the proportion of reportedly protective, anti-inflammatory bacteria such as Bifidobacterium and increases in the proportion of Bacteroides and Escherichia coli in patients with active disease.18,19 Altered diversity and altered metabolic function, ie, decreased concentration of protective short-chain fatty acids of the microbiota, have also been reported in patients with celiac disease.19,20
To move beyond correlative studies and mechanistically address the possibility of causation, multiple groups have used a gnotobiotic approach, ie, maintaining animals under germ-free conditions and incorporating microbes of interest. This approach is highly relevant in studying whether the bacterial community composition is capable of modulating loss of tolerance to gluten in genetically susceptible hosts. A few notable examples have been published.
In germ-free rats, long-term feeding of gliadin, but not albumin, from birth until 2 months of age induced moderate small-intestinal damage.21 Similarly, germ-free nonobese diabetic-DQ8 mice developed more severe gluten-induced disease than mice with normal intestinal bacteria.22
These findings suggest that the normal gut microbiome may have intrinsic beneficial properties capable of reducing the inflammatory effects associated with gluten ingestion. Notably, the specific composition of the intestinal microbiome can define the fate of gluten-induced pathology. Mice colonized with commensal microbiota are indeed protected from gluten-induced pathology, while mice colonized with Proteobacteria spp develop a moderate degree of gluten-induced disease. When Escherichia coli derived from patients with celiac disease is added to commensal colonization, the celiac disease-like phenotype develops.23
Taken together, these studies support the hypothesis that the intestinal microbiome may be another environmental factor involved in the development of celiac disease.
QUESTIONS AND CHALLENGES REMAIN
The results of clinical studies are not necessarily consistent at the taxonomy level. The fields of metagenomics, which investigates all genes and their enzymatic function in a given community, and metabolomics, which identifies bacterial end-products, characterizing their functional capabilities, are still in their infancy and will be required to further investigate functionality of the altered microbiome in celiac disease.
Second, the directionality—the causality or consequences of this dysbiosis—and timing—the moment at which changes occur, ie, after introducing gluten or at the time when symptoms appear—remain elusive, and prospective studies in humans will be essential.
Finally, more mechanistic studies in animal models are needed to dissect the host immune response to dietary gluten and perturbation of intestinal community composition. This may lead to the possibility of future interventions in the form of prebiotics, probiotics, or specific metabolites, complementary to gluten avoidance.
In the meantime, increasing disease awareness and rapid diagnosis and treatment continue to be of utmost importance to address the clinical consequences of celiac disease in both children and adults.
Inheriting the wrong genes and eating the wrong food (ie, gluten) are necessary for celiac disease to develop, but are not enough by themselves. Something else must be contributing, and evidence is pointing to the mix of bacteria that make our guts their home, collectively called the microbiome.
Celiac disease is a highly prevalent, chronic, immune-mediated form of enteropathy.1 It affects 0.5% to 1% of the population, and although it is mostly seen in people of northern European descent, those in other populations can develop the disease as well. Historically, celiac disease was classified as an infant condition. However, it now commonly presents later in life (between ages 10 and 40) and often with extraintestinal manifestations.2
In this issue of Cleveland Clinic Journal of Medicine, Kochhar et al provide a comprehensive updated review of celiac disease.3
GENES AND GLUTEN ARE NECESSARY BUT NOT SUFFICIENT
Although genetic factors and exposure to gluten in the diet are proven to be necessary for celiac disease to develop, they are not sufficient. Evidence of this is in the numbers; although one-third of the general population carries the HLA susceptibility genes (specifically HLA-DQ2 and DQ8),4 only 2% to 5% of people with these genes develop clinically evident celiac disease.
Additional environmental factors must be contributing to disease development, but these other factors are poorly understood. Some of the possible culprits that might influence the risk of disease occurrence and the timing of its onset include5:
- The amount and quality of gluten ingested—the higher the concentration of gluten, the higher the risk, and different grains have gluten varieties with more or less immunogenic capabilities, ie, T-cell activation properties
- The pattern of infant feeding—the risk may be lower with breastfeeding than with formula
- The age at which gluten is introduced into the diet—the risk may be higher if gluten is introduced earlier.6
More recently, studies of the pathogenesis of celiac disease and gene-environmental interactions have expanded beyond host predisposition and dietary factors.
OUR BODIES, OUR MICROBIOMES: A SYMBIOTIC RELATIONSHIP
The role of the human microbiome in autoimmune disease is now being elucidated.7 Remarkably, the microorganisms living in our bodies outnumber our body cells by a factor of 10, and their genomes vastly exceed our own protein-coding genome capabilities by a factor of 100.
The gut microbiome is now considered a true bioreactor with enzymatic and immunologic capabilities beyond (and complementary to) those of its host. The commensal microbiome of the host intestine provides benefits that can be broken down into three broad categories:
- Nutritional—producing essential amino acids and vitamins
- Metabolic—degrading complex polysaccharides from dietary fibers
- Immunologic—shaping the host immune system while cooperating with it against pathogenic microorganisms.
The immunologic function is highly relevant. We have coevolved with our bacteria in a mutually beneficial, symbiotic relationship in which we maintain an active state of low inflammation so that a constant bacterial and dietary antigenic load can be tolerated.
Is there a core human microbiome shared by all individuals? And what is the impact of altering the relative microbial composition (dysbiosis) in physiologic and disease states? To find out, the National Institutes of Health launched the Human Microbiome Project8 in 2008. Important tools in this work include novel culture-independent approaches (high-throughput DNA sequencing and whole-microbiome “shotgun” sequencing with metagenomic analysis) and computational analytical tools.9
An accumulating body of evidence is now available from animal models and human studies correlating states of intestinal dysbiosis (disruption in homeostatic community composition) with various disease processes. These have ranged from inflammatory bowel disease to systemic autoimmune disorders such as psoriasis, inflammatory arthropathies, and demyelinating central nervous system diseases.10–14
RESEARCH INTO THE MICROBIOME IN CELIAC DISEASE
Celiac disease has also served as a unique model for studying this biologic relationship, and the microbiome has been postulated to have a role in its pathogenesis.15 Multiple clinical studies demonstrate that a state of intestinal dysbiosis is indeed associated with celiac disease.
Specifically, decreases in the abundance of Firmicutes spp and increases in Proteobacteria spp have been detected in both children and adults with active celiac disease.16,17 Intriguingly, overrepresentation of Proteobacteria was also correlated with disease activity. Other studies have reported decreases in the proportion of reportedly protective, anti-inflammatory bacteria such as Bifidobacterium and increases in the proportion of Bacteroides and Escherichia coli in patients with active disease.18,19 Altered diversity and altered metabolic function, ie, decreased concentration of protective short-chain fatty acids of the microbiota, have also been reported in patients with celiac disease.19,20
To move beyond correlative studies and mechanistically address the possibility of causation, multiple groups have used a gnotobiotic approach, ie, maintaining animals under germ-free conditions and incorporating microbes of interest. This approach is highly relevant in studying whether the bacterial community composition is capable of modulating loss of tolerance to gluten in genetically susceptible hosts. A few notable examples have been published.
In germ-free rats, long-term feeding of gliadin, but not albumin, from birth until 2 months of age induced moderate small-intestinal damage.21 Similarly, germ-free nonobese diabetic-DQ8 mice developed more severe gluten-induced disease than mice with normal intestinal bacteria.22
These findings suggest that the normal gut microbiome may have intrinsic beneficial properties capable of reducing the inflammatory effects associated with gluten ingestion. Notably, the specific composition of the intestinal microbiome can define the fate of gluten-induced pathology. Mice colonized with commensal microbiota are indeed protected from gluten-induced pathology, while mice colonized with Proteobacteria spp develop a moderate degree of gluten-induced disease. When Escherichia coli derived from patients with celiac disease is added to commensal colonization, the celiac disease-like phenotype develops.23
Taken together, these studies support the hypothesis that the intestinal microbiome may be another environmental factor involved in the development of celiac disease.
QUESTIONS AND CHALLENGES REMAIN
The results of clinical studies are not necessarily consistent at the taxonomy level. The fields of metagenomics, which investigates all genes and their enzymatic function in a given community, and metabolomics, which identifies bacterial end-products, characterizing their functional capabilities, are still in their infancy and will be required to further investigate functionality of the altered microbiome in celiac disease.
Second, the directionality—the causality or consequences of this dysbiosis—and timing—the moment at which changes occur, ie, after introducing gluten or at the time when symptoms appear—remain elusive, and prospective studies in humans will be essential.
Finally, more mechanistic studies in animal models are needed to dissect the host immune response to dietary gluten and perturbation of intestinal community composition. This may lead to the possibility of future interventions in the form of prebiotics, probiotics, or specific metabolites, complementary to gluten avoidance.
In the meantime, increasing disease awareness and rapid diagnosis and treatment continue to be of utmost importance to address the clinical consequences of celiac disease in both children and adults.
- Guandalini S, Assiri A. Celiac disease: a review. JAMA Pediatr 2014; 168:272–278.
- Green PH, Cellier C. Celiac disease. N Engl J Med 2007; 357:1731–1743.
- Kochhar GS, Singh T, Gill A, Kirby DF. Celiac disease: an internist’s perspective. Cleve Clin J Med 2016; 83:217–227.
- Gutierrez-Achury J, Zhernakova A, Pulit SL, et al. Fine mapping in the MHC region accounts for 18% additional genetic risk for celiac disease. Nat Genet 2015; 47:577–578.
- Catassi C, Kryszak D, Bhatti B, et al. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann Med 2010; 42:530–538.
- Norris JM, Barriga K, Hoffenberg EJ, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA 2005; 293:2343–2351.
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature 2007; 449:804–810.
- NIH HMP Working Group; Peterson J, Garges S, Giovanni M, et al. The NIH Human Microbiome Project. Genome Res 2009; 19:2317–2323.
- Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59–65.
- Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013; 2:e01202.
- Scher JU, Ubeda C, Artacho A, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol 2015; 67:128–139.
- Gao Z, Tseng CH, Strober BE, Pei Z, Blaser MJ. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS One 2008; 3:e2719.
- Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013; 155:1451–1463.
- Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn‘s disease. Cell Host Microbe 2014; 15:382–392.
- Verdu EF, Galipeau HJ, Jabri B. Novel players in coeliac disease pathogenesis: role of the gut microbiota. Nat Rev Gastroenterol Hepatol 2015; 12:497–506.
- Sanchez E, Donat E, Ribes-Koninckx C, Fernandez-Murga ML, Sanz Y. Duodenal-mucosal bacteria associated with celiac disease in children. Appl Environ Microbiol 2013; 79:5472–5479.
- Wacklin P, Kaukinen K, Tuovinen E, et al. The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm Bowel Dis 2013; 19:934–941.
- Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J Clin Pathol 2009; 62:264–269.
- Di Cagno R, De Angelis M, De Pasquale I, et al. Duodenal and faecal microbiota of celiac children: molecular, phenotype and metabolome characterization. BMC Microbiol 2011; 11:219.
- Schippa S, Iebba V, Barbato M, et al. A distinctive ‘microbial signature’ in celiac pediatric patients. BMC Microbiol 2010; 10:175.
- Stepankova R, Tlaskalova-Hogenova H, Sinkora J, Jodl J, Fric P. Changes in jejunal mucosa after long-term feeding of germfree rats with gluten. Scand J Gastroenterol 1996; 31:551–557.
- Galipeau HJ, Rulli NE, Jury J, et al. Sensitization to gliadin induces moderate enteropathy and insulitis in nonobese diabetic-DQ8 mice. J Immunol 2011; 187:4338–4346.
- Galipeau HJ, Verdu EF. Gut microbes and adverse food reactions: focus on gluten related disorders. Gut Microbes 2014; 5:594–605.
- Guandalini S, Assiri A. Celiac disease: a review. JAMA Pediatr 2014; 168:272–278.
- Green PH, Cellier C. Celiac disease. N Engl J Med 2007; 357:1731–1743.
- Kochhar GS, Singh T, Gill A, Kirby DF. Celiac disease: an internist’s perspective. Cleve Clin J Med 2016; 83:217–227.
- Gutierrez-Achury J, Zhernakova A, Pulit SL, et al. Fine mapping in the MHC region accounts for 18% additional genetic risk for celiac disease. Nat Genet 2015; 47:577–578.
- Catassi C, Kryszak D, Bhatti B, et al. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann Med 2010; 42:530–538.
- Norris JM, Barriga K, Hoffenberg EJ, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA 2005; 293:2343–2351.
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature 2007; 449:804–810.
- NIH HMP Working Group; Peterson J, Garges S, Giovanni M, et al. The NIH Human Microbiome Project. Genome Res 2009; 19:2317–2323.
- Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59–65.
- Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013; 2:e01202.
- Scher JU, Ubeda C, Artacho A, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol 2015; 67:128–139.
- Gao Z, Tseng CH, Strober BE, Pei Z, Blaser MJ. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS One 2008; 3:e2719.
- Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013; 155:1451–1463.
- Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn‘s disease. Cell Host Microbe 2014; 15:382–392.
- Verdu EF, Galipeau HJ, Jabri B. Novel players in coeliac disease pathogenesis: role of the gut microbiota. Nat Rev Gastroenterol Hepatol 2015; 12:497–506.
- Sanchez E, Donat E, Ribes-Koninckx C, Fernandez-Murga ML, Sanz Y. Duodenal-mucosal bacteria associated with celiac disease in children. Appl Environ Microbiol 2013; 79:5472–5479.
- Wacklin P, Kaukinen K, Tuovinen E, et al. The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm Bowel Dis 2013; 19:934–941.
- Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J Clin Pathol 2009; 62:264–269.
- Di Cagno R, De Angelis M, De Pasquale I, et al. Duodenal and faecal microbiota of celiac children: molecular, phenotype and metabolome characterization. BMC Microbiol 2011; 11:219.
- Schippa S, Iebba V, Barbato M, et al. A distinctive ‘microbial signature’ in celiac pediatric patients. BMC Microbiol 2010; 10:175.
- Stepankova R, Tlaskalova-Hogenova H, Sinkora J, Jodl J, Fric P. Changes in jejunal mucosa after long-term feeding of germfree rats with gluten. Scand J Gastroenterol 1996; 31:551–557.
- Galipeau HJ, Rulli NE, Jury J, et al. Sensitization to gliadin induces moderate enteropathy and insulitis in nonobese diabetic-DQ8 mice. J Immunol 2011; 187:4338–4346.
- Galipeau HJ, Verdu EF. Gut microbes and adverse food reactions: focus on gluten related disorders. Gut Microbes 2014; 5:594–605.
Celiac disease: Managing a multisystem disorder
Celiac disease is an autoimmune disorder that occurs in genetically predisposed individuals in response to ingestion of gluten. Its prevalence is about 0.7% of the US population.1
The gold standard for diagnosis is duodenal biopsy, in which the histologic features may include varying gradations of flattening of intestinal villi, crypt hyperplasia, and infiltration of the lamina propria by lymphocytes. Many patients have no symptoms at the time of diagnosis, but presenting symptoms can include diarrhea along with features of malabsorption,2 and, in about 25% of patients (mainly adults), a bullous cutaneous disorder called dermatitis herpetiformis.3,4 The pathogenesis of celiac disease and that of dermatitis herpetiformis are similar in that in both, ingestion of gluten induces an inflammatory reaction leading to the clinical manifestations.
The mainstay of treatment of celiac disease remains avoidance of gluten in the diet.
GENETIC PREDISPOSITION AND DIETARY TRIGGER
The pathogenesis of celiac disease has been well studied in both humans and animals. The disease is thought to develop by an interplay of genetic and autoimmune factors and the ingestion of gluten (ie, an environmental factor).
Celiac disease occurs in genetically predisposed individuals, ie, those who carry the HLA alleles DQ2 (DQA1*05, DQB1*02), DQ8 (DQA1*03, DQB1*0302), or both.5
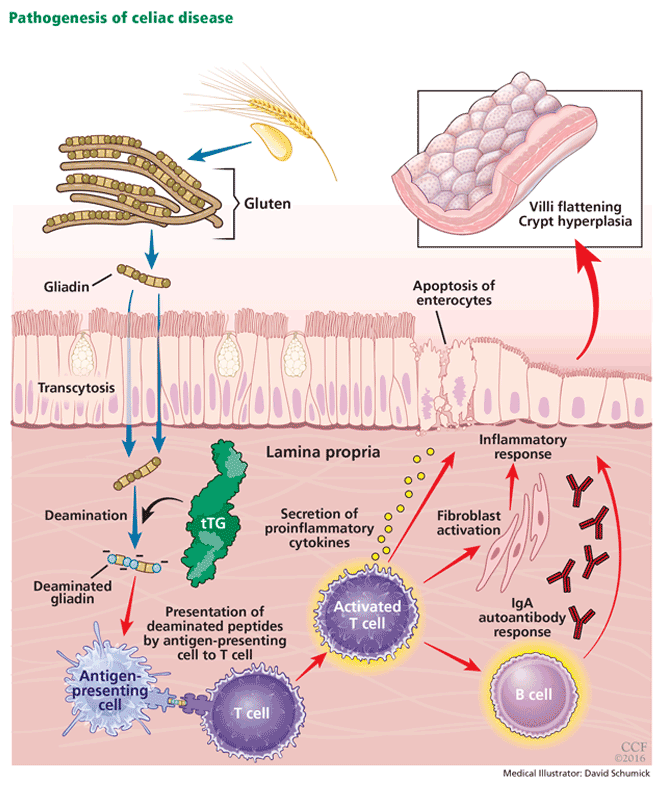
Ingestion of gluten is necessary for the disease to develop. Gluten, the protein component of wheat, barley, and rye, contains proteins called prolamins, which vary among the different types of grain. In wheat, the prolamin is gliadin, which is alcohol-soluble. In barley the prolamin is hordein, and in rye it is secalin.4 The prolamin content in gluten makes it resistant to degradation by gastric, pancreatic, and intestinal brush border proteases.6 Gluten crosses the epithelial barrier and promotes an inflammatory reaction by both the innate and adaptive immune systems that can ultimately result in flattening of villi and crypt hyperplasia (Figure 1).7
Tissue transglutaminase also plays a central role in the pathogenesis, as it further deaminates gliadin and increases its immunogenicity by causing it to bind to receptors on antigen-presenting cells with stronger affinity. Furthermore, gliadin-tissue transglutaminase complexes formed by protein cross-linkages generate an autoantibody response (predominantly immunoglobulin A [IgA] type) that can exacerbate the inflammatory process.8,9
Certain viral infections during childhood, such as rotavirus and adenovirus infection, can increase the risk of celiac disease.10–13 Although earlier studies reported that breast-feeding seemed to have a protective effect,14 as did introducing grains in the diet in the 4th to 6th months of life as opposed to earlier or later,15 more recent studies have not confirmed these benefits.16,17
CLINICAL FEATURES
Most adults diagnosed with celiac disease are in their 30s, 40s, or 50s, and most are women.
Diarrhea remains a common presenting symptom, although the percentage of patients with celiac disease who present with diarrhea has decreased over time.18,19
Abdominal pain and weight loss are also common.20
Pallor or decreased exercise tolerance can develop due to anemia from iron malabsorption, and some patients have easy bruising due to vitamin K malabsorption.
Gynecologic and obstetric complications associated with celiac disease include delayed menarche, amenorrhea, spontaneous abortion, intrauterine growth retardation, preterm delivery, and low-birth-weight babies.21,22 Patients who follow a gluten-free diet tend to have a lower incidence of intrauterine growth retardation, preterm delivery, and low-birth-weight babies compared with untreated patients.21,22
Osteoporosis and osteopenia due to malabsorption of vitamin D are common and are seen in two-thirds of patients presenting with celiac disease.23 A meta-analysis and position statement from Canada concluded that dual-energy x-ray absorptiometry should be done at the time of diagnosis of celiac disease if the patient is at risk of osteoporosis.24 If the scan is abnormal, it should be repeated 1 to 2 years after initiation of a gluten-free diet and vitamin D supplementation to ensure that the osteopenia has improved.24
OTHER DISEASE ASSOCIATIONS

Celiac disease is associated with various other autoimmune diseases (Table 1), including Hashimoto thyroiditis,25 type 1 diabetes mellitus,26 primary biliary cirrhosis,27 primary sclerosing cholangitis,28 and Addison disease.29
Dermatitis herpetiformis
Dermatitis herpetiformis is one of the most common cutaneous manifestations of celiac disease. It presents between ages 10 and 50, and unlike celiac disease, it is more common in males.30
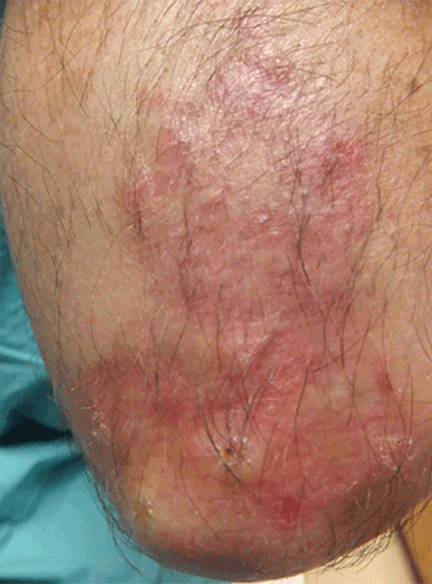
The characteristic lesions are pruritic, grouped erythematous papules surmounted by vesicles distributed symmetrically over the extensor surfaces of the upper and lower extremities, elbows, knees, scalp, nuchal area, and buttocks31 (Figures 2 and 3). In addition, some patients also present with vesicles, erythematous macules, and erosions in the oral mucosa32 or purpura on the palms and soles.33–35
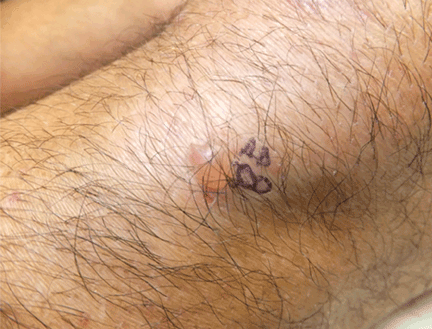
The pathogenesis of dermatitis herpetiformis in the skin is related to the pathogenesis of celiac disease in the gut. Like celiac disease, dermatitis herpetiformis is more common in genetically predisposed individuals carrying either the HLA-DQ2 or the HLA-DQ8 haplotype. In the skin, there is an analogue of tissue transglutaminase called epidermal transglutaminase, which helps in maintaining the integrity of cornified epithelium.36 In patients with celiac disease, along with formation of IgA antibodies to tissue transglutaminase, there is also formation of IgA antibodies to epidermal transglutaminase. IgA antibodies are deposit- ed in the tips of dermal papillae and along the basement membrane.37–39 These deposits then initiate an inflammatory response that is predominantly neutrophilic and results in formation of vesicles and bullae in the skin.40 Also supporting the linkage between celiac disease and dermatitis herpetiformis, if patients adhere to a gluten-free diet, the deposits of immune complexes in the skin disappear.41
CELIAC DISEASE-ASSOCIATED MALIGNANCY
Patients with celiac disease have a higher risk of developing enteric malignancies, particularly intestinal T-cell lymphoma, and they have smaller increased risk of colon, oropharyngeal, esophageal, pancreatic, and hepatobiliary cancer.42–45 For all of these cancers, the risk is higher than in the general public in the first year after celiac disease is diagnosed, but after the first year, the risk is increased only for small-bowel and hepatobiliary malignancies.46
T-cell lymphoma
T-cell lymphoma is a rare but serious complication that has a poor prognosis.47 Its prevalence has been increasing with time and is currently estimated to be around 0.01 to 0.02 per 100,000 people in the population as a whole.48,49 The risk of developing lymphoma is 2.5 times higher in people with celiac disease than in the general population.50 T-cell lymphoma is seen more commonly in patients with refractory celiac disease and DQ2 homozygosity.51
This disease is difficult to detect clinically, but sometimes it presents as an acute exacerbation of celiac disease symptoms despite strict adherence to a gluten-free diet. Associated alarm symptoms include fever, night sweats, and laboratory abnormalities such as low albumin and high lactate dehydrogenase levels.
Strict adherence to a gluten-free diet remains the only way to prevent intestinal T-cell lymphoma.52
Other malignancies
Some earlier studies reported an increased risk of thyroid cancer and malignant melanoma, but two newer studies have refuted this finding.53,54 Conversely, celiac disease appears to have a protective effect against breast, ovarian, and endometrial cancers.55
DIAGNOSIS: SEROLOGY, BIOPSY, GENETIC TESTING
Serologic tests
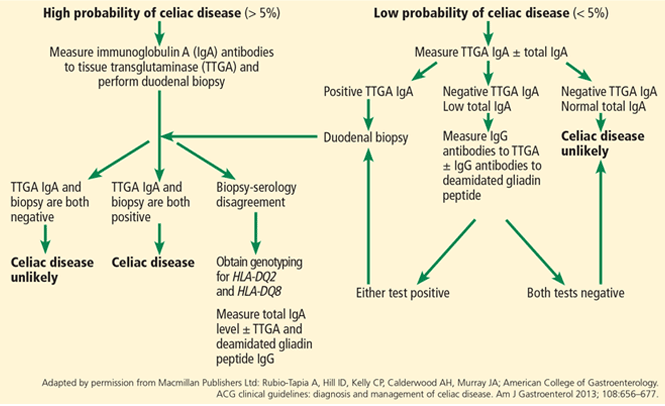
Patients strongly suspected of having celiac disease should be screened for IgA antibodies to tissue transglutaminase while on a gluten-containing diet, according to recommendations of the American College of Gastroenterology (Figure 4).56 The sensitivity and specificity of this test are around 95%. If the patient has an IgA deficiency, screening should be done by checking the level of IgG antibodies to tissue transglutaminase.
Biopsy for confirmation
If testing for IgA to tissue transglutaminase is positive, upper endoscopy with biopsy is needed. Ideally, one to two samples should be taken from the duodenal bulb and at least four samples from the rest of the duodenum, preferably from two different locations.56
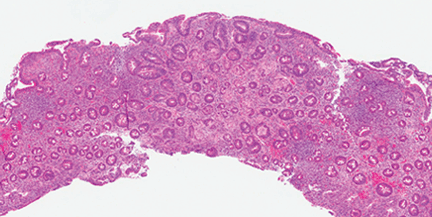
Celiac disease has a broad spectrum of pathologic expressions, from mild distortion of crypt architecture to total villous atrophy and infiltration of lamina propria by lymphocytes57 (Figures 5 and 6). Because these changes can be seen in a variety of diarrheal diseases, their reversal after adherence to a gluten-free diet is part of the current diagnostic criteria for the diagnosis of celiac disease.56
Genetic testing
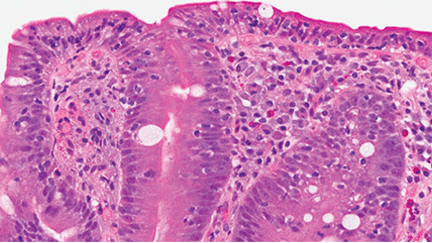
Although the combination of positive serologic tests and pathologic changes confirms the diagnosis of celiac disease, in some cases one type of test is positive and the other is negative. In this situation, genetic testing for HLA-DQ2 and HLA-DQ8 can help rule out the diagnosis, as a negative genetic test rules out celiac disease in more than 99% of cases.58
Genetic testing is also useful in patients who are already adhering to a gluten-free diet at the time of presentation to the clinic and who have had no testing done for celiac disease in the past. Here again, a negative test for both HLA-DQ2 and HLA-DQ8 makes a diagnosis of celiac disease highly unlikely.
If the test is positive, further testing needs to be done, as a positive genetic test cannot differentiate celiac disease from nonceliac gluten sensitivity. In this case, a gluten challenge needs to be done, ideally for 8 weeks, but for at least 2 weeks if the patient cannot tolerate gluten-containing food for a longer period of time. The gluten challenge is to be followed by testing for antibodies to tissue transglutaminase or obtaining duodenal biopsies to confirm the presence or absence of celiac disease.
Standard laboratory tests
Standard laboratory tests do not help much in diagnosing celiac disease, but they should include a complete blood chemistry along with a complete metabolic panel. Usually, serum albumin levels are normal.
Due to malabsorption of iron, patients may have iron deficiency anemia,59 but anemia can also be due to a deficiency of folate or vitamin B12. In patients undergoing endoscopic evaluation of iron deficiency anemia of unknown cause, celiac disease was discovered in approximately 15%.60 Therefore, some experts believe that any patient presenting with unexplained iron deficiency anemia should be screened for celiac disease.
Because of malabsorption of vitamin D, levels of vitamin D can be low.
Elevations in levels of aminotransferases are also fairly common and usually resolve after the start of a gluten-free diet. If they persist despite adherence to a gluten-free diet, then an alternate cause of liver disease should be sought.61
Diagnosis of dermatitis herpetiformis
When trying to diagnose dermatitis herpetiformis, antibodies against epidermal transglutaminase can also be checked if testing for antibody against tissue transglutaminase is negative. A significant number of patients with biopsy-confirmed dermatitis herpetiformis are positive for epidermal transglutaminase antibodies but not for tissue transglutaminase antibodies.62
The confirmatory test for dermatitis herpetiformis remains skin biopsy. Ideally, the sample should be taken while the patient is on a gluten-containing diet and from an area of normal-appearing skin around the lesions.63 On histopathologic study, neutrophilic infiltrates are seen in dermal papillae and a perivascular lymphocytic infiltrate can also be seen in the superficial zones.64 This presentation can also be seen in other bullous disorders, however. To differentiate dermatitis herpetiformis from other disorders, direct immunofluorescence is needed, which will detect granular IgA deposits in the dermal papillae or along the basement membrane, a finding pathognomic of dermatitis herpetiformis.63
A GLUTEN-FREE DIET IS THE MAINSTAY OF TREATMENT
The mainstay of treatment is lifelong adherence to a gluten-free diet. Most patients report improvement in abdominal pain within days of starting this diet and improvement of diarrhea within 4 weeks.65
The maximum amount of gluten that can be tolerated is debatable. A study established that intake of less than 10 mg a day is associated with fewer histologic abnormalities,66 and an earlier study noted that intake of less than 50 mg a day was clinically well tolerated.67 But patients differ in their tolerance for gluten, and it is hard to predict what the threshold of tolerance for gluten will be for a particular individual. Thus, it is better to avoid gluten completely.
Gluten-free if it is inherently gluten-free. If the food has a gluten-containing grain, then it should be processed to remove the gluten, and the resultant food product should not contain more than 20 parts per million of gluten. Gluten-free products that have gluten-containing grain that has been processed usually have a label indicating the gluten content in the food in parts per million.
Patients who understand the need to adhere to a gluten-free diet and the implications of not adhering to it are generally more compliant. Thus, patients need to be strongly educated that they need to adhere to a gluten-free diet and that nonadherence can cause further damage to the gut and can pose a higher risk of malignancy. Even though patients are usually concerned about the cost of gluten-free food and worry about adherence to the diet, these factors do not generally limit diet adherence.68 All patients diagnosed with celiac disease should meet with a registered dietitian to discuss diet options based on their food preferences and to better address all their concerns.
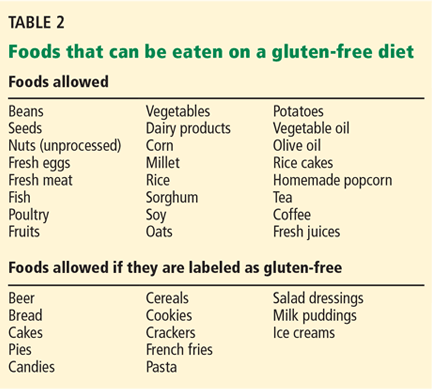
With increasing awareness of celiac disease and with increasing numbers of patients being diagnosed with it, the food industry has recognized the need to produce gluten-free items. There are now plenty of food products available for these patients, who no longer have to forgo cakes, cookies, and other such items. Table 2 lists some common foods that patients with celiac disease can consume.
Nutritional supplements for some
If anemia is due purely to iron deficiency, it may resolve after starting a gluten-free diet, and no additional supplementation may be needed. However, if it is due to a combination of iron plus folate or vitamin B12 deficiency, then folate, vitamin B12, or both should be given.
In addition, if the patient is found to have a deficiency of vitamin D, then a vitamin D supplement should be given.69 At the time of diagnosis, all patients with celiac disease should be screened for deficiencies of vitamins A, B12, D, E, and K, as well as copper, zinc, folic acid, and iron.
Follow-up at 3 to 6 months
A follow-up visit should be scheduled for 3 to 6 months after the diagnosis and after that on an annual basis, and many of the abnormal laboratory tests will need to be repeated.
If intestinal or extraintestinal symptoms or nutrient deficiencies persist, then the patient’s adherence to the gluten-free diet needs to be checked. Adherence to a gluten-free diet can be assessed by checking for serologic markers of celiac disease. A decrease in baseline values can be seen within a few months of starting the diet.70 Failure of serologic markers to decrease by the end of 1 year of a gluten-free diet usually indicates gluten contamination.71 If adherence is confirmed (ie, if baseline values fall) but symptoms persist, then further workup needs to be done to find the cause of refractory disease.
Skin lesions should also respond to a gluten-free diet
The first and foremost therapy for the skin lesions in dermatitis herpetiformis is the same as that for the intestinal manifestations in celiac disease, ie, adherence to a gluten-free diet. Soon after patients begin a gluten-free diet, the itching around the skin lesions goes away, and over time, most patients have complete resolution of the skin manifestations.
Dapsone is also frequently used to treat dermatitis herpetiformis if there is an incomplete response to a gluten-free diet or as an adjunct to diet to treat the pruritus. Patients often have a good response to dapsone.72
The recommended starting dosage is 100 to 200 mg a day, and a response is usually seen within a few days. If the symptoms do not improve, the dose can be increased. Once the lesions resolve, the dose can be tapered and patients may not require any further medication. In some cases, patients may need to be chronically maintained on the lowest dose possible, due to the side effects of the drug.3
Dapsone is associated with significant adverse effects. Methemoglobinemia is the most common and is seen particularly in dosages exceeding 200 mg a day. Hemolytic anemia, another common adverse effect, is seen with dosages of more than 100 mg a day. Patients with a deficiency of glucose-6-phosphate dehydrogenase (G6PD) are at increased risk of hemolysis, and screening for G6PD deficiency is usually done before starting dapsone. Other rare adverse effects of dapsone include agranulocytosis, peripheral neuropathy, psychosis,73 pancreatitis, cholestatic jaundice, bullous and exfoliative dermatitis, Stevens-Johnson syndrome, toxic epidermal necrolysis, nephrotic syndrome, and renal papillary necrosis.
Besides testing for G6PD deficiency, a complete blood cell count, a reticulocyte count, a hepatic function panel, renal function tests, and urinalysis should be done before starting dapsone therapy and repeated while on therapy. The complete blood cell count and reticulocyte count should be checked weekly for the first month, twice a month for the next 2 months, and then once every 3 months. Liver and renal function tests are to be done once every 3 months.74
NOVEL THERAPIES BEING TESTED
Research is under way for other treatments for celiac disease besides a gluten-free diet.
Larazotide (Alba Therapeutics, Baltimore, MD) is being tested in a randomized, placebo-controlled trial. Early results indicate that it is effective in controlling both gastrointestinal and nongastrointestinal symptoms of celiac disease, but it still has to undergo phase 3 clinical trials.
Sorghum is a grain commonly used in Asia and Africa. The gluten in sorghum is different from that in wheat and is not immunogenic. In a small case series in patients with known celiac disease, sorghum did not induce diarrhea or change in levels of antibodies to tissue transglutaminase.75
Nonimmunogenic wheat that does not contain the immunogenic gluten is being developed.
Oral enzyme supplements called glutenases are being developed. Glutenases can cleave gluten, particularly the proline and glutamine residues that make gluten resistant to degradation by gastric, pancreatic, and intestinal brush border proteases. A phase 2 trial of one of these oral enzyme supplements showed that it appeared to attenuate mucosal injury in patients with biopsy-proven celiac disease.76
These novel therapies look promising, but for now the best treatment is lifelong adherence to the gluten-free diet.
NONRESPONSIVE AND REFRACTORY CELIAC DISEASE
Celiac disease is considered nonresponsive if its symptoms or laboratory abnormalities persist after the patient is on a gluten-free diet for 6 to 12 months. It is considered refractory if symptoms persist or recur along with villous atrophy despite adherence to the diet for more than 12 months in the absence of other causes of the symptoms. Refractory celiac disease can be further classified either as type 1 if there are typical intraepithelial lymphocytes, or as type 2 if there are atypical intraepithelial lymphocytes.
Celiac disease is nonresponsive in about 10% to 19% of cases,76 and it is refractory in 1% to 2%.77
Managing nonresponsive celiac disease
The first step in managing a patient with nonresponsive celiac disease is to confirm the diagnosis by reviewing the serologic tests and the biopsy samples from the time of diagnosis. If celiac disease is confirmed, then one should re-evaluate for gluten ingestion, the most common cause of nonresponsiveness.78 If strict adherence is confirmed, then check for other causes of symptoms such as lactose or fructose intolerance. If no other cause is found, then repeat the duodenal biopsies with flow cytometry to look for CD3 and CD8 expression in T cells in the small-bowel mucosa.79 Presence or absence of villous atrophy can point to possible other causes of malabsorption including pancreatic insufficiency, small intestinal bowel overgrowth, and microscopic colitis.
Managing refractory celiac disease
Traditionally, corticosteroids have been shown to be beneficial in alleviating symptoms in patients with refractory celiac disease but do not improve the histologic findings.80 Because of the adverse effects associated with long-term corticosteroid use, azathioprine has been successfully used to maintain remission of the disease after induction with corticosteroids in patients with type 1 refractory celiac disease.81
Cladribine, a chemotherapeutic agent used to treat hairy cell leukemia, has shown some benefit in treating type 2 refractory celiac disease.82
In type 2 refractory celiac disease, use of an immunomodulator agent carries an increased risk of transformation to lymphoma.
Because of the lack of a satisfactory response to the agents available so far to treat refractory celiac disease, more treatment options acting at the molecular level are being explored.
NONCELIAC GLUTEN SENSITIVITY DISORDER
Nonceliac gluten sensitivity disorder is an evolving concept. The clinical presentation of this disorder is similar to celiac disease in that patients may have diarrhea or other extraintestinal symptoms when on a regular diet and have resolution of symptoms on a gluten-free diet. But unlike celiac disease, there is no serologic or histologic evidence of celiac disease even when patients are on a regular diet.
One of every 17 patients who presents with clinical features suggestive of celiac disease is found to have nonceliac gluten sensitivity disorder, not celiac disease.83 In contrast to celiac disease, in which the adaptive immune system is thought to contribute to the disease process, in nonceliac gluten sensitivity disorder the innate immune system is believed to play the dominant role,84 but the exact pathogenesis of the disease is still unclear.
The diagnosis of nonceliac gluten sensitivity disorder is one of exclusion. Celiac disease needs to be ruled out by serologic testing and by duodenal biopsy while the patient is on a regular diet, and then a trial of a gluten-free diet needs to be done to confirm resolution of symptoms before the diagnosis of nonceliac gluten sensitivity disorder can be established.
As with celiac disease, the treatment involves adhering to a gluten-free diet, but it is still not known if patients need to stay on it for the rest of their life, or if they will be able to tolerate gluten-containing products after a few years.
- Rubio-Tapia A, Ludvigsson JF, Bratner TL, Murray JA, Everhart JE. The prevalence of celiac disease in the United States. Am J Gastroenterol 2012; 107:1538–1544.
- Dewar DH, Ciclitira PJ. Clinical features and diagnosis of celiac disease. Gastroenterology 2005; 128(suppl 1):S19–S24.
- Mendes FB, Hissa-Elian A, Abreu MA, Goncalves VS. Review: dermatitis herpetiformis. An Bras Dermatol 2013; 88:594–599.
- Lauret E, Rodrigo L. Celiac disease and autoimmune-associated conditions. Biomed Res Int 2013; 2013:127589.
- Sollid LM, Lie BA. Celiac disease genetics: current concepts and practical applications. Clin Gastroenterol Hepatol 2005; 3:843–851.
- Hausch F, Shan L, Santiago NA, Gray GM, Khosla C. Intestinal digestive resistance of immunodominant gliadin peptides. Am J Physiol Gastrointest Liver Physiol 2002; 283:G996–G1003.
- Green PH, Cellier C. Celiac disease. N Engl J Med 2007; 357:1731–1743.
- Caputo I, Barone MV, Martucciello S, Lepretti M, Esposito C. Tissue transglutaminase in celiac disease: role of autoantibodies. Amino Acids 2009; 36:693–699.
- Schuppan D, Dieterich W, Riecken EO. Exposing gliadin as a tasty food for lymphocytes. Nat Med 1998; 4:666–667.
- Stene LC, Honeyman MC, Hoffenberg EJ, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol 2006; 101:2333–2340.
- Kagnoff MF, Austin RK, Hubert JJ, Bernardin JE, Kasarda DD. Possible role for a human adenovirus in the pathogenesis of celiac disease. J Exp Med 1984; 160:1544–1557.
- Ruggeri C, LaMasa AT, Rudi S, et al. Celiac disease and non-organ-specific autoantibodies in patients with chronic hepatitis C virus infection. Dig Dis Sci 2008; 53:2151–2155.
- Sjoberg K, Lindgren S, Eriksson S. Frequent occurrence of non-specific gliadin antibodies in chronic liver disease. Endomysial but not gliadin antibodies predict coelic disease in patients with chronic liver disease. Scand J Gastroenterol 1997; 32:1162–1167.
- Persson LA, Ivarsson A, Hernell O. Breast-feeding protects against celiac disease in childhood—epidemiological evidence. Adv Exp Med Biol 2002; 503:115–123.
- Norris JM, Barriga K, Hoffenberg EJ, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA 2005; 293:2343–2351.
- Vriezinga SL, Auricchio R, Bravi E, et al. Randomized feeding intervention in infants at high risk for celiac disease. N Engl J Med 2014; 371:1304–1315.
- Lionetti E, Castelaneta S, Francavilla R, et al. Introduction of gluten, HLA status, and the risk of celiac disease in children. N Engl J Med 2014; 371:1295–1303
- Green PH. The many faces of celiac disease: clinical presentation of celiac disease in the adult population. Gastroenterology 2005; 128:S74–S78.
- Rampertab SD, Pooran N, Brar P, Singh P, Green PH. Trends in the presentation of celiac disease. Am J Med 2006; 119:355 e9–e14.
- Rashid M, Cranney A, Zarkadas M, et al. Celiac disease: evaluation of the diagnosis and dietary compliance in Canadian children. Pediatrics 2005; 116:e754–e759.
- Molteni N, Bardella MT, Bianchi PA. Obstetric and gynecological problems in women with untreated celiac sprue. J Clin Gastroenterol 1990; 12:37–39.
- Tersigni C, Castellani R, de Waure C, et al. Celiac disease and reproductive disorders: meta-analysis of epidemiologic associations and potential pathogenic mechanisms. Hum Reprod Update 2014; 20:582–593.
- Meyer D, Stravropolous S, Diamond B, Shane E, Green PH. Osteoporosis in a North American adult population with celiac disease. Am J Gastroenterol 2001; 96:112–119.
- Fouda MA, Khan AA, Sultan MS, Rios LP, McAssey K, Armstrong D. Evaluation and management of skeletal health in celiac disease: position statement. Can J Gastroenterol 2012; 26:819–829.
- van der Pals M, Ivarsson A, Norström F, Högberg L, Svensson J, Carlsson A. Prevalence of thyroid autoimmunity in children with celiac disease compared to healthy 12-year olds. Autoimmune Dis 2014; 2014:417356.
- Mahmud FH, Murray JA, Kudva YC, et al. Celiac disease in type 1 diabetes mellitus in a North American community: prevalence, serologic screening, and clinical features. Mayo Clin Proc 2005; 80:1429–1434.
- Sorensen HT, Thulstrup AM, Blomqvist P, Nørgaard B, Fonager K, Ekbom A. Risk of primary biliary liver cirrhosis in patients with coeliac disease: Danish and Swedish cohort data. Gut 1999; 44:736–738.
- Volta U, Rodrigo L, Granito A, et al. Celiac disease in autoimmune cholestatic liver disorders. Am J Gastroenterol 2002; 97:2609–2613.
- Elfstrom P, Montgomery SM, Kämpe O, Ekbom A, Ludvigsson JF. Risk of primary adrenal insufficiency in patients with celiac disease. J Clin Endocrinol Metab 2007; 92:3595–3598.
- Younus J, Ahmed AR. Clinical features of dermatitis herpetiformis. Clin Dermatol 1991; 9:279–281.
- Bolotin D, Petronic-Rosic V. Dermatitis herpetiformis. Part I. Epidemiology, pathogenesis, and clinical presentation. J Am Acad Dermatol 2011; 64:1017–1026.
- Lahteenoja H, Irjala K, Viander M, Vainio E, Toivanen A, Syrjänen S. Oral mucosa is frequently affected in patients with dermatitis herpetiformis. Arch Dermatol 1998; 134:756–758.
- Marks R, Jones EW. Purpura in dermatitis herpetiformis. Br J Dermatol 1971; 84:386–388.
- McGovern TW, Bennion SD. Palmar purpura: an atypical presentation of childhood dermatitis herpetiformis. Pediatr Dermatol 1994; 11:319–322.
- Pierce DK, Purcell SM, Spielvogel RL. Purpuric papules and vesicles of the palms in dermatitis herpetiformis. J Am Acad Dermatol 1987; 16:1274–1276.
- Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 2003; 4:140–156.
- Hull CM, Liddle M, Hansen N, et al. Elevation of IgA anti-epidermal transglutaminase antibodies in dermatitis herpetiformis. Br J Dermatol 2008; 159:120–124.
- Kawana S, Segawa A. Confocal laser scanning microscopic and immunoelectron microscopic studies of the anatomical distribution of fibrillar IgA deposits in dermatitis herpetiformis. Arch Dermatol 1993; 129:456–459.
- Sárdy M, Kárpáti S, Merkl B, Paulsson M, Smyth N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J Exp Med 2002; 195:747–757.
- Nicolas ME, Krause PK, Gibson LE, Murray JA. Dermatitis herpetiformis. Int J Dermatol 2003; 42:588–600.
- Leonard J, Haffenden G, Tucker W, et al. Gluten challenge in dermatitis herpetiformis. N Engl J Med 1983; 308:816–819.
- Summaries for patients. Risk for lymphoma and the results of follow-up gut biopsies in patients with celiac disease. Ann Intern Med 2013; 159:I–20.
- Lebwohl B, Granath F, Ekbom A, et al. Mucosal healing and risk for lymphoproliferative malignancy in celiac disease: a population-based cohort study. Ann Intern Med 2013; 159:169–175.
- Volta U, Vincentini O, Quintarelli F, Felli C, Silano M; Collaborating Centres of the Italian Registry of the Complications of Celiac Disease. Low risk of colon cancer in patients with celiac disease. Scand J Gastroenterol 2014; 49:564–568.
- Askling J, Linet M, Gridley G, Halstensen TS, Ekström K, Ekbom A. Cancer incidence in a population-based cohort of individuals hospitalized with celiac disease or dermatitis herpetiformis. Gastroenterology 2002; 123:1428–1435.
- Elfström P, Granath F, Ye W, Ludvigsson JF. Low risk of gastrointestinal cancer among patients with celiac disease, inflammation, or latent celiac disease. Clin Gastroenterol Hepatol 2012; 10:30–36.
- Al-Toma A, Verbeek WH, Hadithi M, von Blomberg BM, Mulder CJ. Survival in refractory coeliac disease and enteropathy-associated T-cell lymphoma: retrospective evaluation of single-centre experience. Gut 2007; 56:1373–1378.
- Verbeek WH, Van De Water JM, Al-Toma A, Oudejans JJ, Mulder CJ, Coupé VM. Incidence of enteropathy—associated T-cell lymphoma: a nation-wide study of a population-based registry in The Netherlands. Scand J Gastroenterol 2008; 43:1322–1328.
- Sharaiha RZ, Lebwohl B, Reimers L, Bhagat G, Green PH, Neugut AI. Increasing incidence of enteropathy-associated T-cell lymphoma in the United States, 1973-2008. Cancer 2012; 118:3786–3792.
- Mearin ML, Catassi C, Brousse N, et al; Biomed Study Group on Coeliac Disease and Non-Hodgkin Lymphoma. European multi-centre study on coeliac disease and non-Hodgkin lymphoma. Eur J Gastroenterol Hepatol 2006; 18:187–194.
- Al-Toma A, Goerres MS, Meijer JW, Pena AS, Crusius JB, Mulder CJ. Human leukocyte antigen-DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clin Gastroenterol Hepatol 2006; 4:315–319.
- Sieniawski MK, Lennard AL. Enteropathy-associated T-cell lymphoma: epidemiology, clinical features, and current treatment strategies. Curr Hematol Malig Rep 2011; 6:231–240.
- Lebwohl B, Eriksson H, Hansson J, Green PH, Ludvigsson JF. Risk of cutaneous malignant melanoma in patients with celiac disease: a population-based study. J Am Acad Dermatol 2014; 71:245–248.
- Ludvigsson JF, Lebwohl B, Kämpe O, Murray JA, Green PH, Ekbom A. Risk of thyroid cancer in a nationwide cohort of patients with biopsy-verified celiac disease. Thyroid 2013; 23:971–976.
- Ludvigsson JF, West J, Ekbom A, Stephansson O. Reduced risk of breast, endometrial and ovarian cancer in women with celiac disease. Int J Cancer 2012; 13:E244–E250.
- Rubio-Tapia A, Hill ID, Kelly CP, Calderwood AH, Murray JA; American College of Gastroenterology. ACG clinical guidelines: diagnosis and management of celiac disease. Am J Gastroenterol 2013; 108:656–677.
- Marsh MN. Gluten, major histocompatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity (‘celiac sprue’). Gastroenterology 1992; 102:330–354.
- Hadithi M, von Blomberg BM, Crusius JB, et al. Accuracy of serologic tests and HLA-DQ typing for diagnosing celiac disease. Ann Intern Med 2007; 147:294–302.
- Lo W, Sano K, Lebwohl B, Diamond B, Green PH. Changing presentation of adult celiac disease. Dig Dis Sci 2003; 48:395–398.
- Oxentenko AS, Grisolano SW, Murray JA, Burgart LJ, Dierkhising RA, Alexander JA. The insensitivity of endoscopic markers in celiac disease. Am J Gastroenterol 2002; 97:933–938.
- Casella G, Antonelli E, Di Bella C, et al. Prevalence and causes of abnormal liver function in patients with coeliac disease. Liver Int 2013; 33:1128–1131.
- Jaskowski TD, Hamblin T, Wilson AR, et al. IgA anti-epidermal transglutaminase antibodies in dermatitis herpetiformis and pediatric celiac disease. J Invest Dermatol 2009; 129:2728–2730.
- Zone JJ, Meyer LJ, Petersen MJ. Deposition of granular IgA relative to clinical lesions in dermatitis herpetiformis. Arch Dermatol 1996; 132:912–918.
- Plotnikova N, Miller JL. Dermatitis herpetiformis. Skin Ther Lett 2013; 18:1–3.
- Murray JA, Watson T, Clearman B, Mitros F. Effect of a gluten-free diet on gastrointestinal symptoms in celiac disease. Am J Clin Nutr 2004; 79:669–673.
- Akobeng AK, Thomas AG. Systematic review: tolerable amount of gluten for people with coeliac disease. Aliment Pharmacol Ther 2008; 27:1044–1052.
- Catassi C, Fabiani E, Iacono G, et al. A prospective, double-blind, placebo-controlled trial to establish a safe gluten threshold for patients with celiac disease. Am J Clin Nutr 2007; 85:160–166.
- Leffler DA, Edwards-George J, Dennis M, et al. Factors that influence adherence to a gluten-free diet in adults with celiac disease. Dig Dis Sci 2008; 53:1573–1581.
- Caruso R, Pallone F, Stasi E, Romeo S, Monteleone G. Appropriate nutrient supplementation in celiac disease. Ann Med 2013; 45:522–531.
- Nachman F, Sugai E, Vazquez H, et al. Serological tests for celiac disease as indicators of long-term compliance with the gluten-free diet. Eur J Gastroenterol Hepatol 2011; 23:473–480.
- Abdulkarim AS, Burgart LJ, See J, Murray JA. Etiology of nonresponsive celiac disease: results of a systemic approach. Am J Gastroenterol 2002; 97:2016–2021.
- Fry L, Seah PP, Hoffbrand AV. Dermatitis herpetiformis. Clin Gastroenterol 1974; 3:145–157.
- Zhu YI, Stiller MJ. Dapsone and sulfones in dermatology: overview and update. J Am Acad Dermatol 2001; 45:420-434.
- Wolf R, Matz H, Orion E, Tuzun B, Tuzun Y. Dapsone. Dermatol Online J 2002; 8:2.
- Ciacci C, Maiuri L, Caporaso N, et al. Celiac disease: in vitro and in vivo safety and palatability of wheat-free sorghum food products. Clin Nutr 2007; 26:799–805.
- Lähdeaho ML, Kaukinen K, Laurila K, et al. Glutenase ALV003 attenuates gluten-induced mucosal injury in patients with celiac disease. Gastroenterology 2014; 146:1649–1658.
- Roshan B, Leffler DA, Jamma S, et al. The incidence and clinical spectrum of refractory celiac disease in a North American referral center. Am J Gastroenterol 2011; 106:923–928.
- Leffler DA, Dennis M, Hyett B, Kelly E, Schuppan D, Kelly CP. Etiologies and predictors of diagnosis in nonresponsive celiac disease. Clin Gastroenterol Hepatol 2007; 5:445–450.
- Cellier C, Delabesse E, Helmer C, et al. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. French Coeliac Disease Study Group. Lancet 2000; 356:203–208.
- Malamut G, Afchain P, Verkarre V, et al. Presentation and long-term follow-up of refractory celiac disease: comparison of type I with type II. Gastroenterology 2009; 136:81–90.
- Goerres MS, Meijer JW, Wahab PJ, et al. Azathioprine and prednisone combination therapy in refractory celiac disease. Aliment Pharmacol Ther 2003; 18:487–494.
- Tack GJ, Verbeek WH, Al-Toma A, et al. Evaluation of cladribine treatment in refractory celiac disease type II. World J Gastroenterol 2011; 17:506–513.
- Sapone A, Bai JC, Dolinsek J, et al. Spectrum of gluten-related disorders: consensus on new nomenclature and classification. BMC Med 2012; 7:10–13.
- Sapone A, Lammers KM, Casolaro V, et al. Divergence of gut permeability and mucosal immune gene expression in two gluten-associated conditions: celiac disease and gluten sensitivity. BMC Med 2011; 9:9–23.
Celiac disease is an autoimmune disorder that occurs in genetically predisposed individuals in response to ingestion of gluten. Its prevalence is about 0.7% of the US population.1
The gold standard for diagnosis is duodenal biopsy, in which the histologic features may include varying gradations of flattening of intestinal villi, crypt hyperplasia, and infiltration of the lamina propria by lymphocytes. Many patients have no symptoms at the time of diagnosis, but presenting symptoms can include diarrhea along with features of malabsorption,2 and, in about 25% of patients (mainly adults), a bullous cutaneous disorder called dermatitis herpetiformis.3,4 The pathogenesis of celiac disease and that of dermatitis herpetiformis are similar in that in both, ingestion of gluten induces an inflammatory reaction leading to the clinical manifestations.
The mainstay of treatment of celiac disease remains avoidance of gluten in the diet.
GENETIC PREDISPOSITION AND DIETARY TRIGGER
The pathogenesis of celiac disease has been well studied in both humans and animals. The disease is thought to develop by an interplay of genetic and autoimmune factors and the ingestion of gluten (ie, an environmental factor).
Celiac disease occurs in genetically predisposed individuals, ie, those who carry the HLA alleles DQ2 (DQA1*05, DQB1*02), DQ8 (DQA1*03, DQB1*0302), or both.5
Ingestion of gluten is necessary for the disease to develop. Gluten, the protein component of wheat, barley, and rye, contains proteins called prolamins, which vary among the different types of grain. In wheat, the prolamin is gliadin, which is alcohol-soluble. In barley the prolamin is hordein, and in rye it is secalin.4 The prolamin content in gluten makes it resistant to degradation by gastric, pancreatic, and intestinal brush border proteases.6 Gluten crosses the epithelial barrier and promotes an inflammatory reaction by both the innate and adaptive immune systems that can ultimately result in flattening of villi and crypt hyperplasia (Figure 1).7
Tissue transglutaminase also plays a central role in the pathogenesis, as it further deaminates gliadin and increases its immunogenicity by causing it to bind to receptors on antigen-presenting cells with stronger affinity. Furthermore, gliadin-tissue transglutaminase complexes formed by protein cross-linkages generate an autoantibody response (predominantly immunoglobulin A [IgA] type) that can exacerbate the inflammatory process.8,9
Certain viral infections during childhood, such as rotavirus and adenovirus infection, can increase the risk of celiac disease.10–13 Although earlier studies reported that breast-feeding seemed to have a protective effect,14 as did introducing grains in the diet in the 4th to 6th months of life as opposed to earlier or later,15 more recent studies have not confirmed these benefits.16,17
CLINICAL FEATURES
Most adults diagnosed with celiac disease are in their 30s, 40s, or 50s, and most are women.
Diarrhea remains a common presenting symptom, although the percentage of patients with celiac disease who present with diarrhea has decreased over time.18,19
Abdominal pain and weight loss are also common.20
Pallor or decreased exercise tolerance can develop due to anemia from iron malabsorption, and some patients have easy bruising due to vitamin K malabsorption.
Gynecologic and obstetric complications associated with celiac disease include delayed menarche, amenorrhea, spontaneous abortion, intrauterine growth retardation, preterm delivery, and low-birth-weight babies.21,22 Patients who follow a gluten-free diet tend to have a lower incidence of intrauterine growth retardation, preterm delivery, and low-birth-weight babies compared with untreated patients.21,22
Osteoporosis and osteopenia due to malabsorption of vitamin D are common and are seen in two-thirds of patients presenting with celiac disease.23 A meta-analysis and position statement from Canada concluded that dual-energy x-ray absorptiometry should be done at the time of diagnosis of celiac disease if the patient is at risk of osteoporosis.24 If the scan is abnormal, it should be repeated 1 to 2 years after initiation of a gluten-free diet and vitamin D supplementation to ensure that the osteopenia has improved.24
OTHER DISEASE ASSOCIATIONS
Celiac disease is associated with various other autoimmune diseases (Table 1), including Hashimoto thyroiditis,25 type 1 diabetes mellitus,26 primary biliary cirrhosis,27 primary sclerosing cholangitis,28 and Addison disease.29
Dermatitis herpetiformis
Dermatitis herpetiformis is one of the most common cutaneous manifestations of celiac disease. It presents between ages 10 and 50, and unlike celiac disease, it is more common in males.30
The characteristic lesions are pruritic, grouped erythematous papules surmounted by vesicles distributed symmetrically over the extensor surfaces of the upper and lower extremities, elbows, knees, scalp, nuchal area, and buttocks31 (Figures 2 and 3). In addition, some patients also present with vesicles, erythematous macules, and erosions in the oral mucosa32 or purpura on the palms and soles.33–35
The pathogenesis of dermatitis herpetiformis in the skin is related to the pathogenesis of celiac disease in the gut. Like celiac disease, dermatitis herpetiformis is more common in genetically predisposed individuals carrying either the HLA-DQ2 or the HLA-DQ8 haplotype. In the skin, there is an analogue of tissue transglutaminase called epidermal transglutaminase, which helps in maintaining the integrity of cornified epithelium.36 In patients with celiac disease, along with formation of IgA antibodies to tissue transglutaminase, there is also formation of IgA antibodies to epidermal transglutaminase. IgA antibodies are deposit- ed in the tips of dermal papillae and along the basement membrane.37–39 These deposits then initiate an inflammatory response that is predominantly neutrophilic and results in formation of vesicles and bullae in the skin.40 Also supporting the linkage between celiac disease and dermatitis herpetiformis, if patients adhere to a gluten-free diet, the deposits of immune complexes in the skin disappear.41
CELIAC DISEASE-ASSOCIATED MALIGNANCY
Patients with celiac disease have a higher risk of developing enteric malignancies, particularly intestinal T-cell lymphoma, and they have smaller increased risk of colon, oropharyngeal, esophageal, pancreatic, and hepatobiliary cancer.42–45 For all of these cancers, the risk is higher than in the general public in the first year after celiac disease is diagnosed, but after the first year, the risk is increased only for small-bowel and hepatobiliary malignancies.46
T-cell lymphoma
T-cell lymphoma is a rare but serious complication that has a poor prognosis.47 Its prevalence has been increasing with time and is currently estimated to be around 0.01 to 0.02 per 100,000 people in the population as a whole.48,49 The risk of developing lymphoma is 2.5 times higher in people with celiac disease than in the general population.50 T-cell lymphoma is seen more commonly in patients with refractory celiac disease and DQ2 homozygosity.51
This disease is difficult to detect clinically, but sometimes it presents as an acute exacerbation of celiac disease symptoms despite strict adherence to a gluten-free diet. Associated alarm symptoms include fever, night sweats, and laboratory abnormalities such as low albumin and high lactate dehydrogenase levels.
Strict adherence to a gluten-free diet remains the only way to prevent intestinal T-cell lymphoma.52
Other malignancies
Some earlier studies reported an increased risk of thyroid cancer and malignant melanoma, but two newer studies have refuted this finding.53,54 Conversely, celiac disease appears to have a protective effect against breast, ovarian, and endometrial cancers.55
DIAGNOSIS: SEROLOGY, BIOPSY, GENETIC TESTING
Serologic tests
Patients strongly suspected of having celiac disease should be screened for IgA antibodies to tissue transglutaminase while on a gluten-containing diet, according to recommendations of the American College of Gastroenterology (Figure 4).56 The sensitivity and specificity of this test are around 95%. If the patient has an IgA deficiency, screening should be done by checking the level of IgG antibodies to tissue transglutaminase.
Biopsy for confirmation
If testing for IgA to tissue transglutaminase is positive, upper endoscopy with biopsy is needed. Ideally, one to two samples should be taken from the duodenal bulb and at least four samples from the rest of the duodenum, preferably from two different locations.56
Celiac disease has a broad spectrum of pathologic expressions, from mild distortion of crypt architecture to total villous atrophy and infiltration of lamina propria by lymphocytes57 (Figures 5 and 6). Because these changes can be seen in a variety of diarrheal diseases, their reversal after adherence to a gluten-free diet is part of the current diagnostic criteria for the diagnosis of celiac disease.56
Genetic testing
Although the combination of positive serologic tests and pathologic changes confirms the diagnosis of celiac disease, in some cases one type of test is positive and the other is negative. In this situation, genetic testing for HLA-DQ2 and HLA-DQ8 can help rule out the diagnosis, as a negative genetic test rules out celiac disease in more than 99% of cases.58
Genetic testing is also useful in patients who are already adhering to a gluten-free diet at the time of presentation to the clinic and who have had no testing done for celiac disease in the past. Here again, a negative test for both HLA-DQ2 and HLA-DQ8 makes a diagnosis of celiac disease highly unlikely.
If the test is positive, further testing needs to be done, as a positive genetic test cannot differentiate celiac disease from nonceliac gluten sensitivity. In this case, a gluten challenge needs to be done, ideally for 8 weeks, but for at least 2 weeks if the patient cannot tolerate gluten-containing food for a longer period of time. The gluten challenge is to be followed by testing for antibodies to tissue transglutaminase or obtaining duodenal biopsies to confirm the presence or absence of celiac disease.
Standard laboratory tests
Standard laboratory tests do not help much in diagnosing celiac disease, but they should include a complete blood chemistry along with a complete metabolic panel. Usually, serum albumin levels are normal.
Due to malabsorption of iron, patients may have iron deficiency anemia,59 but anemia can also be due to a deficiency of folate or vitamin B12. In patients undergoing endoscopic evaluation of iron deficiency anemia of unknown cause, celiac disease was discovered in approximately 15%.60 Therefore, some experts believe that any patient presenting with unexplained iron deficiency anemia should be screened for celiac disease.
Because of malabsorption of vitamin D, levels of vitamin D can be low.
Elevations in levels of aminotransferases are also fairly common and usually resolve after the start of a gluten-free diet. If they persist despite adherence to a gluten-free diet, then an alternate cause of liver disease should be sought.61
Diagnosis of dermatitis herpetiformis
When trying to diagnose dermatitis herpetiformis, antibodies against epidermal transglutaminase can also be checked if testing for antibody against tissue transglutaminase is negative. A significant number of patients with biopsy-confirmed dermatitis herpetiformis are positive for epidermal transglutaminase antibodies but not for tissue transglutaminase antibodies.62
The confirmatory test for dermatitis herpetiformis remains skin biopsy. Ideally, the sample should be taken while the patient is on a gluten-containing diet and from an area of normal-appearing skin around the lesions.63 On histopathologic study, neutrophilic infiltrates are seen in dermal papillae and a perivascular lymphocytic infiltrate can also be seen in the superficial zones.64 This presentation can also be seen in other bullous disorders, however. To differentiate dermatitis herpetiformis from other disorders, direct immunofluorescence is needed, which will detect granular IgA deposits in the dermal papillae or along the basement membrane, a finding pathognomic of dermatitis herpetiformis.63
A GLUTEN-FREE DIET IS THE MAINSTAY OF TREATMENT
The mainstay of treatment is lifelong adherence to a gluten-free diet. Most patients report improvement in abdominal pain within days of starting this diet and improvement of diarrhea within 4 weeks.65
The maximum amount of gluten that can be tolerated is debatable. A study established that intake of less than 10 mg a day is associated with fewer histologic abnormalities,66 and an earlier study noted that intake of less than 50 mg a day was clinically well tolerated.67 But patients differ in their tolerance for gluten, and it is hard to predict what the threshold of tolerance for gluten will be for a particular individual. Thus, it is better to avoid gluten completely.
Gluten-free if it is inherently gluten-free. If the food has a gluten-containing grain, then it should be processed to remove the gluten, and the resultant food product should not contain more than 20 parts per million of gluten. Gluten-free products that have gluten-containing grain that has been processed usually have a label indicating the gluten content in the food in parts per million.
Patients who understand the need to adhere to a gluten-free diet and the implications of not adhering to it are generally more compliant. Thus, patients need to be strongly educated that they need to adhere to a gluten-free diet and that nonadherence can cause further damage to the gut and can pose a higher risk of malignancy. Even though patients are usually concerned about the cost of gluten-free food and worry about adherence to the diet, these factors do not generally limit diet adherence.68 All patients diagnosed with celiac disease should meet with a registered dietitian to discuss diet options based on their food preferences and to better address all their concerns.
With increasing awareness of celiac disease and with increasing numbers of patients being diagnosed with it, the food industry has recognized the need to produce gluten-free items. There are now plenty of food products available for these patients, who no longer have to forgo cakes, cookies, and other such items. Table 2 lists some common foods that patients with celiac disease can consume.
Nutritional supplements for some
If anemia is due purely to iron deficiency, it may resolve after starting a gluten-free diet, and no additional supplementation may be needed. However, if it is due to a combination of iron plus folate or vitamin B12 deficiency, then folate, vitamin B12, or both should be given.
In addition, if the patient is found to have a deficiency of vitamin D, then a vitamin D supplement should be given.69 At the time of diagnosis, all patients with celiac disease should be screened for deficiencies of vitamins A, B12, D, E, and K, as well as copper, zinc, folic acid, and iron.
Follow-up at 3 to 6 months
A follow-up visit should be scheduled for 3 to 6 months after the diagnosis and after that on an annual basis, and many of the abnormal laboratory tests will need to be repeated.
If intestinal or extraintestinal symptoms or nutrient deficiencies persist, then the patient’s adherence to the gluten-free diet needs to be checked. Adherence to a gluten-free diet can be assessed by checking for serologic markers of celiac disease. A decrease in baseline values can be seen within a few months of starting the diet.70 Failure of serologic markers to decrease by the end of 1 year of a gluten-free diet usually indicates gluten contamination.71 If adherence is confirmed (ie, if baseline values fall) but symptoms persist, then further workup needs to be done to find the cause of refractory disease.
Skin lesions should also respond to a gluten-free diet
The first and foremost therapy for the skin lesions in dermatitis herpetiformis is the same as that for the intestinal manifestations in celiac disease, ie, adherence to a gluten-free diet. Soon after patients begin a gluten-free diet, the itching around the skin lesions goes away, and over time, most patients have complete resolution of the skin manifestations.
Dapsone is also frequently used to treat dermatitis herpetiformis if there is an incomplete response to a gluten-free diet or as an adjunct to diet to treat the pruritus. Patients often have a good response to dapsone.72
The recommended starting dosage is 100 to 200 mg a day, and a response is usually seen within a few days. If the symptoms do not improve, the dose can be increased. Once the lesions resolve, the dose can be tapered and patients may not require any further medication. In some cases, patients may need to be chronically maintained on the lowest dose possible, due to the side effects of the drug.3
Dapsone is associated with significant adverse effects. Methemoglobinemia is the most common and is seen particularly in dosages exceeding 200 mg a day. Hemolytic anemia, another common adverse effect, is seen with dosages of more than 100 mg a day. Patients with a deficiency of glucose-6-phosphate dehydrogenase (G6PD) are at increased risk of hemolysis, and screening for G6PD deficiency is usually done before starting dapsone. Other rare adverse effects of dapsone include agranulocytosis, peripheral neuropathy, psychosis,73 pancreatitis, cholestatic jaundice, bullous and exfoliative dermatitis, Stevens-Johnson syndrome, toxic epidermal necrolysis, nephrotic syndrome, and renal papillary necrosis.
Besides testing for G6PD deficiency, a complete blood cell count, a reticulocyte count, a hepatic function panel, renal function tests, and urinalysis should be done before starting dapsone therapy and repeated while on therapy. The complete blood cell count and reticulocyte count should be checked weekly for the first month, twice a month for the next 2 months, and then once every 3 months. Liver and renal function tests are to be done once every 3 months.74
NOVEL THERAPIES BEING TESTED
Research is under way for other treatments for celiac disease besides a gluten-free diet.
Larazotide (Alba Therapeutics, Baltimore, MD) is being tested in a randomized, placebo-controlled trial. Early results indicate that it is effective in controlling both gastrointestinal and nongastrointestinal symptoms of celiac disease, but it still has to undergo phase 3 clinical trials.
Sorghum is a grain commonly used in Asia and Africa. The gluten in sorghum is different from that in wheat and is not immunogenic. In a small case series in patients with known celiac disease, sorghum did not induce diarrhea or change in levels of antibodies to tissue transglutaminase.75
Nonimmunogenic wheat that does not contain the immunogenic gluten is being developed.
Oral enzyme supplements called glutenases are being developed. Glutenases can cleave gluten, particularly the proline and glutamine residues that make gluten resistant to degradation by gastric, pancreatic, and intestinal brush border proteases. A phase 2 trial of one of these oral enzyme supplements showed that it appeared to attenuate mucosal injury in patients with biopsy-proven celiac disease.76
These novel therapies look promising, but for now the best treatment is lifelong adherence to the gluten-free diet.
NONRESPONSIVE AND REFRACTORY CELIAC DISEASE
Celiac disease is considered nonresponsive if its symptoms or laboratory abnormalities persist after the patient is on a gluten-free diet for 6 to 12 months. It is considered refractory if symptoms persist or recur along with villous atrophy despite adherence to the diet for more than 12 months in the absence of other causes of the symptoms. Refractory celiac disease can be further classified either as type 1 if there are typical intraepithelial lymphocytes, or as type 2 if there are atypical intraepithelial lymphocytes.
Celiac disease is nonresponsive in about 10% to 19% of cases,76 and it is refractory in 1% to 2%.77
Managing nonresponsive celiac disease
The first step in managing a patient with nonresponsive celiac disease is to confirm the diagnosis by reviewing the serologic tests and the biopsy samples from the time of diagnosis. If celiac disease is confirmed, then one should re-evaluate for gluten ingestion, the most common cause of nonresponsiveness.78 If strict adherence is confirmed, then check for other causes of symptoms such as lactose or fructose intolerance. If no other cause is found, then repeat the duodenal biopsies with flow cytometry to look for CD3 and CD8 expression in T cells in the small-bowel mucosa.79 Presence or absence of villous atrophy can point to possible other causes of malabsorption including pancreatic insufficiency, small intestinal bowel overgrowth, and microscopic colitis.
Managing refractory celiac disease
Traditionally, corticosteroids have been shown to be beneficial in alleviating symptoms in patients with refractory celiac disease but do not improve the histologic findings.80 Because of the adverse effects associated with long-term corticosteroid use, azathioprine has been successfully used to maintain remission of the disease after induction with corticosteroids in patients with type 1 refractory celiac disease.81
Cladribine, a chemotherapeutic agent used to treat hairy cell leukemia, has shown some benefit in treating type 2 refractory celiac disease.82
In type 2 refractory celiac disease, use of an immunomodulator agent carries an increased risk of transformation to lymphoma.
Because of the lack of a satisfactory response to the agents available so far to treat refractory celiac disease, more treatment options acting at the molecular level are being explored.
NONCELIAC GLUTEN SENSITIVITY DISORDER
Nonceliac gluten sensitivity disorder is an evolving concept. The clinical presentation of this disorder is similar to celiac disease in that patients may have diarrhea or other extraintestinal symptoms when on a regular diet and have resolution of symptoms on a gluten-free diet. But unlike celiac disease, there is no serologic or histologic evidence of celiac disease even when patients are on a regular diet.
One of every 17 patients who presents with clinical features suggestive of celiac disease is found to have nonceliac gluten sensitivity disorder, not celiac disease.83 In contrast to celiac disease, in which the adaptive immune system is thought to contribute to the disease process, in nonceliac gluten sensitivity disorder the innate immune system is believed to play the dominant role,84 but the exact pathogenesis of the disease is still unclear.
The diagnosis of nonceliac gluten sensitivity disorder is one of exclusion. Celiac disease needs to be ruled out by serologic testing and by duodenal biopsy while the patient is on a regular diet, and then a trial of a gluten-free diet needs to be done to confirm resolution of symptoms before the diagnosis of nonceliac gluten sensitivity disorder can be established.
As with celiac disease, the treatment involves adhering to a gluten-free diet, but it is still not known if patients need to stay on it for the rest of their life, or if they will be able to tolerate gluten-containing products after a few years.
Celiac disease is an autoimmune disorder that occurs in genetically predisposed individuals in response to ingestion of gluten. Its prevalence is about 0.7% of the US population.1
The gold standard for diagnosis is duodenal biopsy, in which the histologic features may include varying gradations of flattening of intestinal villi, crypt hyperplasia, and infiltration of the lamina propria by lymphocytes. Many patients have no symptoms at the time of diagnosis, but presenting symptoms can include diarrhea along with features of malabsorption,2 and, in about 25% of patients (mainly adults), a bullous cutaneous disorder called dermatitis herpetiformis.3,4 The pathogenesis of celiac disease and that of dermatitis herpetiformis are similar in that in both, ingestion of gluten induces an inflammatory reaction leading to the clinical manifestations.
The mainstay of treatment of celiac disease remains avoidance of gluten in the diet.
GENETIC PREDISPOSITION AND DIETARY TRIGGER
The pathogenesis of celiac disease has been well studied in both humans and animals. The disease is thought to develop by an interplay of genetic and autoimmune factors and the ingestion of gluten (ie, an environmental factor).
Celiac disease occurs in genetically predisposed individuals, ie, those who carry the HLA alleles DQ2 (DQA1*05, DQB1*02), DQ8 (DQA1*03, DQB1*0302), or both.5
Ingestion of gluten is necessary for the disease to develop. Gluten, the protein component of wheat, barley, and rye, contains proteins called prolamins, which vary among the different types of grain. In wheat, the prolamin is gliadin, which is alcohol-soluble. In barley the prolamin is hordein, and in rye it is secalin.4 The prolamin content in gluten makes it resistant to degradation by gastric, pancreatic, and intestinal brush border proteases.6 Gluten crosses the epithelial barrier and promotes an inflammatory reaction by both the innate and adaptive immune systems that can ultimately result in flattening of villi and crypt hyperplasia (Figure 1).7
Tissue transglutaminase also plays a central role in the pathogenesis, as it further deaminates gliadin and increases its immunogenicity by causing it to bind to receptors on antigen-presenting cells with stronger affinity. Furthermore, gliadin-tissue transglutaminase complexes formed by protein cross-linkages generate an autoantibody response (predominantly immunoglobulin A [IgA] type) that can exacerbate the inflammatory process.8,9
Certain viral infections during childhood, such as rotavirus and adenovirus infection, can increase the risk of celiac disease.10–13 Although earlier studies reported that breast-feeding seemed to have a protective effect,14 as did introducing grains in the diet in the 4th to 6th months of life as opposed to earlier or later,15 more recent studies have not confirmed these benefits.16,17
CLINICAL FEATURES
Most adults diagnosed with celiac disease are in their 30s, 40s, or 50s, and most are women.
Diarrhea remains a common presenting symptom, although the percentage of patients with celiac disease who present with diarrhea has decreased over time.18,19
Abdominal pain and weight loss are also common.20
Pallor or decreased exercise tolerance can develop due to anemia from iron malabsorption, and some patients have easy bruising due to vitamin K malabsorption.
Gynecologic and obstetric complications associated with celiac disease include delayed menarche, amenorrhea, spontaneous abortion, intrauterine growth retardation, preterm delivery, and low-birth-weight babies.21,22 Patients who follow a gluten-free diet tend to have a lower incidence of intrauterine growth retardation, preterm delivery, and low-birth-weight babies compared with untreated patients.21,22
Osteoporosis and osteopenia due to malabsorption of vitamin D are common and are seen in two-thirds of patients presenting with celiac disease.23 A meta-analysis and position statement from Canada concluded that dual-energy x-ray absorptiometry should be done at the time of diagnosis of celiac disease if the patient is at risk of osteoporosis.24 If the scan is abnormal, it should be repeated 1 to 2 years after initiation of a gluten-free diet and vitamin D supplementation to ensure that the osteopenia has improved.24
OTHER DISEASE ASSOCIATIONS
Celiac disease is associated with various other autoimmune diseases (Table 1), including Hashimoto thyroiditis,25 type 1 diabetes mellitus,26 primary biliary cirrhosis,27 primary sclerosing cholangitis,28 and Addison disease.29
Dermatitis herpetiformis
Dermatitis herpetiformis is one of the most common cutaneous manifestations of celiac disease. It presents between ages 10 and 50, and unlike celiac disease, it is more common in males.30
The characteristic lesions are pruritic, grouped erythematous papules surmounted by vesicles distributed symmetrically over the extensor surfaces of the upper and lower extremities, elbows, knees, scalp, nuchal area, and buttocks31 (Figures 2 and 3). In addition, some patients also present with vesicles, erythematous macules, and erosions in the oral mucosa32 or purpura on the palms and soles.33–35
The pathogenesis of dermatitis herpetiformis in the skin is related to the pathogenesis of celiac disease in the gut. Like celiac disease, dermatitis herpetiformis is more common in genetically predisposed individuals carrying either the HLA-DQ2 or the HLA-DQ8 haplotype. In the skin, there is an analogue of tissue transglutaminase called epidermal transglutaminase, which helps in maintaining the integrity of cornified epithelium.36 In patients with celiac disease, along with formation of IgA antibodies to tissue transglutaminase, there is also formation of IgA antibodies to epidermal transglutaminase. IgA antibodies are deposit- ed in the tips of dermal papillae and along the basement membrane.37–39 These deposits then initiate an inflammatory response that is predominantly neutrophilic and results in formation of vesicles and bullae in the skin.40 Also supporting the linkage between celiac disease and dermatitis herpetiformis, if patients adhere to a gluten-free diet, the deposits of immune complexes in the skin disappear.41
CELIAC DISEASE-ASSOCIATED MALIGNANCY
Patients with celiac disease have a higher risk of developing enteric malignancies, particularly intestinal T-cell lymphoma, and they have smaller increased risk of colon, oropharyngeal, esophageal, pancreatic, and hepatobiliary cancer.42–45 For all of these cancers, the risk is higher than in the general public in the first year after celiac disease is diagnosed, but after the first year, the risk is increased only for small-bowel and hepatobiliary malignancies.46
T-cell lymphoma
T-cell lymphoma is a rare but serious complication that has a poor prognosis.47 Its prevalence has been increasing with time and is currently estimated to be around 0.01 to 0.02 per 100,000 people in the population as a whole.48,49 The risk of developing lymphoma is 2.5 times higher in people with celiac disease than in the general population.50 T-cell lymphoma is seen more commonly in patients with refractory celiac disease and DQ2 homozygosity.51
This disease is difficult to detect clinically, but sometimes it presents as an acute exacerbation of celiac disease symptoms despite strict adherence to a gluten-free diet. Associated alarm symptoms include fever, night sweats, and laboratory abnormalities such as low albumin and high lactate dehydrogenase levels.
Strict adherence to a gluten-free diet remains the only way to prevent intestinal T-cell lymphoma.52
Other malignancies
Some earlier studies reported an increased risk of thyroid cancer and malignant melanoma, but two newer studies have refuted this finding.53,54 Conversely, celiac disease appears to have a protective effect against breast, ovarian, and endometrial cancers.55
DIAGNOSIS: SEROLOGY, BIOPSY, GENETIC TESTING
Serologic tests
Patients strongly suspected of having celiac disease should be screened for IgA antibodies to tissue transglutaminase while on a gluten-containing diet, according to recommendations of the American College of Gastroenterology (Figure 4).56 The sensitivity and specificity of this test are around 95%. If the patient has an IgA deficiency, screening should be done by checking the level of IgG antibodies to tissue transglutaminase.
Biopsy for confirmation
If testing for IgA to tissue transglutaminase is positive, upper endoscopy with biopsy is needed. Ideally, one to two samples should be taken from the duodenal bulb and at least four samples from the rest of the duodenum, preferably from two different locations.56
Celiac disease has a broad spectrum of pathologic expressions, from mild distortion of crypt architecture to total villous atrophy and infiltration of lamina propria by lymphocytes57 (Figures 5 and 6). Because these changes can be seen in a variety of diarrheal diseases, their reversal after adherence to a gluten-free diet is part of the current diagnostic criteria for the diagnosis of celiac disease.56
Genetic testing
Although the combination of positive serologic tests and pathologic changes confirms the diagnosis of celiac disease, in some cases one type of test is positive and the other is negative. In this situation, genetic testing for HLA-DQ2 and HLA-DQ8 can help rule out the diagnosis, as a negative genetic test rules out celiac disease in more than 99% of cases.58
Genetic testing is also useful in patients who are already adhering to a gluten-free diet at the time of presentation to the clinic and who have had no testing done for celiac disease in the past. Here again, a negative test for both HLA-DQ2 and HLA-DQ8 makes a diagnosis of celiac disease highly unlikely.
If the test is positive, further testing needs to be done, as a positive genetic test cannot differentiate celiac disease from nonceliac gluten sensitivity. In this case, a gluten challenge needs to be done, ideally for 8 weeks, but for at least 2 weeks if the patient cannot tolerate gluten-containing food for a longer period of time. The gluten challenge is to be followed by testing for antibodies to tissue transglutaminase or obtaining duodenal biopsies to confirm the presence or absence of celiac disease.
Standard laboratory tests
Standard laboratory tests do not help much in diagnosing celiac disease, but they should include a complete blood chemistry along with a complete metabolic panel. Usually, serum albumin levels are normal.
Due to malabsorption of iron, patients may have iron deficiency anemia,59 but anemia can also be due to a deficiency of folate or vitamin B12. In patients undergoing endoscopic evaluation of iron deficiency anemia of unknown cause, celiac disease was discovered in approximately 15%.60 Therefore, some experts believe that any patient presenting with unexplained iron deficiency anemia should be screened for celiac disease.
Because of malabsorption of vitamin D, levels of vitamin D can be low.
Elevations in levels of aminotransferases are also fairly common and usually resolve after the start of a gluten-free diet. If they persist despite adherence to a gluten-free diet, then an alternate cause of liver disease should be sought.61
Diagnosis of dermatitis herpetiformis
When trying to diagnose dermatitis herpetiformis, antibodies against epidermal transglutaminase can also be checked if testing for antibody against tissue transglutaminase is negative. A significant number of patients with biopsy-confirmed dermatitis herpetiformis are positive for epidermal transglutaminase antibodies but not for tissue transglutaminase antibodies.62
The confirmatory test for dermatitis herpetiformis remains skin biopsy. Ideally, the sample should be taken while the patient is on a gluten-containing diet and from an area of normal-appearing skin around the lesions.63 On histopathologic study, neutrophilic infiltrates are seen in dermal papillae and a perivascular lymphocytic infiltrate can also be seen in the superficial zones.64 This presentation can also be seen in other bullous disorders, however. To differentiate dermatitis herpetiformis from other disorders, direct immunofluorescence is needed, which will detect granular IgA deposits in the dermal papillae or along the basement membrane, a finding pathognomic of dermatitis herpetiformis.63
A GLUTEN-FREE DIET IS THE MAINSTAY OF TREATMENT
The mainstay of treatment is lifelong adherence to a gluten-free diet. Most patients report improvement in abdominal pain within days of starting this diet and improvement of diarrhea within 4 weeks.65
The maximum amount of gluten that can be tolerated is debatable. A study established that intake of less than 10 mg a day is associated with fewer histologic abnormalities,66 and an earlier study noted that intake of less than 50 mg a day was clinically well tolerated.67 But patients differ in their tolerance for gluten, and it is hard to predict what the threshold of tolerance for gluten will be for a particular individual. Thus, it is better to avoid gluten completely.
Gluten-free if it is inherently gluten-free. If the food has a gluten-containing grain, then it should be processed to remove the gluten, and the resultant food product should not contain more than 20 parts per million of gluten. Gluten-free products that have gluten-containing grain that has been processed usually have a label indicating the gluten content in the food in parts per million.
Patients who understand the need to adhere to a gluten-free diet and the implications of not adhering to it are generally more compliant. Thus, patients need to be strongly educated that they need to adhere to a gluten-free diet and that nonadherence can cause further damage to the gut and can pose a higher risk of malignancy. Even though patients are usually concerned about the cost of gluten-free food and worry about adherence to the diet, these factors do not generally limit diet adherence.68 All patients diagnosed with celiac disease should meet with a registered dietitian to discuss diet options based on their food preferences and to better address all their concerns.
With increasing awareness of celiac disease and with increasing numbers of patients being diagnosed with it, the food industry has recognized the need to produce gluten-free items. There are now plenty of food products available for these patients, who no longer have to forgo cakes, cookies, and other such items. Table 2 lists some common foods that patients with celiac disease can consume.
Nutritional supplements for some
If anemia is due purely to iron deficiency, it may resolve after starting a gluten-free diet, and no additional supplementation may be needed. However, if it is due to a combination of iron plus folate or vitamin B12 deficiency, then folate, vitamin B12, or both should be given.
In addition, if the patient is found to have a deficiency of vitamin D, then a vitamin D supplement should be given.69 At the time of diagnosis, all patients with celiac disease should be screened for deficiencies of vitamins A, B12, D, E, and K, as well as copper, zinc, folic acid, and iron.
Follow-up at 3 to 6 months
A follow-up visit should be scheduled for 3 to 6 months after the diagnosis and after that on an annual basis, and many of the abnormal laboratory tests will need to be repeated.
If intestinal or extraintestinal symptoms or nutrient deficiencies persist, then the patient’s adherence to the gluten-free diet needs to be checked. Adherence to a gluten-free diet can be assessed by checking for serologic markers of celiac disease. A decrease in baseline values can be seen within a few months of starting the diet.70 Failure of serologic markers to decrease by the end of 1 year of a gluten-free diet usually indicates gluten contamination.71 If adherence is confirmed (ie, if baseline values fall) but symptoms persist, then further workup needs to be done to find the cause of refractory disease.
Skin lesions should also respond to a gluten-free diet
The first and foremost therapy for the skin lesions in dermatitis herpetiformis is the same as that for the intestinal manifestations in celiac disease, ie, adherence to a gluten-free diet. Soon after patients begin a gluten-free diet, the itching around the skin lesions goes away, and over time, most patients have complete resolution of the skin manifestations.
Dapsone is also frequently used to treat dermatitis herpetiformis if there is an incomplete response to a gluten-free diet or as an adjunct to diet to treat the pruritus. Patients often have a good response to dapsone.72
The recommended starting dosage is 100 to 200 mg a day, and a response is usually seen within a few days. If the symptoms do not improve, the dose can be increased. Once the lesions resolve, the dose can be tapered and patients may not require any further medication. In some cases, patients may need to be chronically maintained on the lowest dose possible, due to the side effects of the drug.3
Dapsone is associated with significant adverse effects. Methemoglobinemia is the most common and is seen particularly in dosages exceeding 200 mg a day. Hemolytic anemia, another common adverse effect, is seen with dosages of more than 100 mg a day. Patients with a deficiency of glucose-6-phosphate dehydrogenase (G6PD) are at increased risk of hemolysis, and screening for G6PD deficiency is usually done before starting dapsone. Other rare adverse effects of dapsone include agranulocytosis, peripheral neuropathy, psychosis,73 pancreatitis, cholestatic jaundice, bullous and exfoliative dermatitis, Stevens-Johnson syndrome, toxic epidermal necrolysis, nephrotic syndrome, and renal papillary necrosis.
Besides testing for G6PD deficiency, a complete blood cell count, a reticulocyte count, a hepatic function panel, renal function tests, and urinalysis should be done before starting dapsone therapy and repeated while on therapy. The complete blood cell count and reticulocyte count should be checked weekly for the first month, twice a month for the next 2 months, and then once every 3 months. Liver and renal function tests are to be done once every 3 months.74
NOVEL THERAPIES BEING TESTED
Research is under way for other treatments for celiac disease besides a gluten-free diet.
Larazotide (Alba Therapeutics, Baltimore, MD) is being tested in a randomized, placebo-controlled trial. Early results indicate that it is effective in controlling both gastrointestinal and nongastrointestinal symptoms of celiac disease, but it still has to undergo phase 3 clinical trials.
Sorghum is a grain commonly used in Asia and Africa. The gluten in sorghum is different from that in wheat and is not immunogenic. In a small case series in patients with known celiac disease, sorghum did not induce diarrhea or change in levels of antibodies to tissue transglutaminase.75
Nonimmunogenic wheat that does not contain the immunogenic gluten is being developed.
Oral enzyme supplements called glutenases are being developed. Glutenases can cleave gluten, particularly the proline and glutamine residues that make gluten resistant to degradation by gastric, pancreatic, and intestinal brush border proteases. A phase 2 trial of one of these oral enzyme supplements showed that it appeared to attenuate mucosal injury in patients with biopsy-proven celiac disease.76
These novel therapies look promising, but for now the best treatment is lifelong adherence to the gluten-free diet.
NONRESPONSIVE AND REFRACTORY CELIAC DISEASE
Celiac disease is considered nonresponsive if its symptoms or laboratory abnormalities persist after the patient is on a gluten-free diet for 6 to 12 months. It is considered refractory if symptoms persist or recur along with villous atrophy despite adherence to the diet for more than 12 months in the absence of other causes of the symptoms. Refractory celiac disease can be further classified either as type 1 if there are typical intraepithelial lymphocytes, or as type 2 if there are atypical intraepithelial lymphocytes.
Celiac disease is nonresponsive in about 10% to 19% of cases,76 and it is refractory in 1% to 2%.77
Managing nonresponsive celiac disease
The first step in managing a patient with nonresponsive celiac disease is to confirm the diagnosis by reviewing the serologic tests and the biopsy samples from the time of diagnosis. If celiac disease is confirmed, then one should re-evaluate for gluten ingestion, the most common cause of nonresponsiveness.78 If strict adherence is confirmed, then check for other causes of symptoms such as lactose or fructose intolerance. If no other cause is found, then repeat the duodenal biopsies with flow cytometry to look for CD3 and CD8 expression in T cells in the small-bowel mucosa.79 Presence or absence of villous atrophy can point to possible other causes of malabsorption including pancreatic insufficiency, small intestinal bowel overgrowth, and microscopic colitis.
Managing refractory celiac disease
Traditionally, corticosteroids have been shown to be beneficial in alleviating symptoms in patients with refractory celiac disease but do not improve the histologic findings.80 Because of the adverse effects associated with long-term corticosteroid use, azathioprine has been successfully used to maintain remission of the disease after induction with corticosteroids in patients with type 1 refractory celiac disease.81
Cladribine, a chemotherapeutic agent used to treat hairy cell leukemia, has shown some benefit in treating type 2 refractory celiac disease.82
In type 2 refractory celiac disease, use of an immunomodulator agent carries an increased risk of transformation to lymphoma.
Because of the lack of a satisfactory response to the agents available so far to treat refractory celiac disease, more treatment options acting at the molecular level are being explored.
NONCELIAC GLUTEN SENSITIVITY DISORDER
Nonceliac gluten sensitivity disorder is an evolving concept. The clinical presentation of this disorder is similar to celiac disease in that patients may have diarrhea or other extraintestinal symptoms when on a regular diet and have resolution of symptoms on a gluten-free diet. But unlike celiac disease, there is no serologic or histologic evidence of celiac disease even when patients are on a regular diet.
One of every 17 patients who presents with clinical features suggestive of celiac disease is found to have nonceliac gluten sensitivity disorder, not celiac disease.83 In contrast to celiac disease, in which the adaptive immune system is thought to contribute to the disease process, in nonceliac gluten sensitivity disorder the innate immune system is believed to play the dominant role,84 but the exact pathogenesis of the disease is still unclear.
The diagnosis of nonceliac gluten sensitivity disorder is one of exclusion. Celiac disease needs to be ruled out by serologic testing and by duodenal biopsy while the patient is on a regular diet, and then a trial of a gluten-free diet needs to be done to confirm resolution of symptoms before the diagnosis of nonceliac gluten sensitivity disorder can be established.
As with celiac disease, the treatment involves adhering to a gluten-free diet, but it is still not known if patients need to stay on it for the rest of their life, or if they will be able to tolerate gluten-containing products after a few years.
- Rubio-Tapia A, Ludvigsson JF, Bratner TL, Murray JA, Everhart JE. The prevalence of celiac disease in the United States. Am J Gastroenterol 2012; 107:1538–1544.
- Dewar DH, Ciclitira PJ. Clinical features and diagnosis of celiac disease. Gastroenterology 2005; 128(suppl 1):S19–S24.
- Mendes FB, Hissa-Elian A, Abreu MA, Goncalves VS. Review: dermatitis herpetiformis. An Bras Dermatol 2013; 88:594–599.
- Lauret E, Rodrigo L. Celiac disease and autoimmune-associated conditions. Biomed Res Int 2013; 2013:127589.
- Sollid LM, Lie BA. Celiac disease genetics: current concepts and practical applications. Clin Gastroenterol Hepatol 2005; 3:843–851.
- Hausch F, Shan L, Santiago NA, Gray GM, Khosla C. Intestinal digestive resistance of immunodominant gliadin peptides. Am J Physiol Gastrointest Liver Physiol 2002; 283:G996–G1003.
- Green PH, Cellier C. Celiac disease. N Engl J Med 2007; 357:1731–1743.
- Caputo I, Barone MV, Martucciello S, Lepretti M, Esposito C. Tissue transglutaminase in celiac disease: role of autoantibodies. Amino Acids 2009; 36:693–699.
- Schuppan D, Dieterich W, Riecken EO. Exposing gliadin as a tasty food for lymphocytes. Nat Med 1998; 4:666–667.
- Stene LC, Honeyman MC, Hoffenberg EJ, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol 2006; 101:2333–2340.
- Kagnoff MF, Austin RK, Hubert JJ, Bernardin JE, Kasarda DD. Possible role for a human adenovirus in the pathogenesis of celiac disease. J Exp Med 1984; 160:1544–1557.
- Ruggeri C, LaMasa AT, Rudi S, et al. Celiac disease and non-organ-specific autoantibodies in patients with chronic hepatitis C virus infection. Dig Dis Sci 2008; 53:2151–2155.
- Sjoberg K, Lindgren S, Eriksson S. Frequent occurrence of non-specific gliadin antibodies in chronic liver disease. Endomysial but not gliadin antibodies predict coelic disease in patients with chronic liver disease. Scand J Gastroenterol 1997; 32:1162–1167.
- Persson LA, Ivarsson A, Hernell O. Breast-feeding protects against celiac disease in childhood—epidemiological evidence. Adv Exp Med Biol 2002; 503:115–123.
- Norris JM, Barriga K, Hoffenberg EJ, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA 2005; 293:2343–2351.
- Vriezinga SL, Auricchio R, Bravi E, et al. Randomized feeding intervention in infants at high risk for celiac disease. N Engl J Med 2014; 371:1304–1315.
- Lionetti E, Castelaneta S, Francavilla R, et al. Introduction of gluten, HLA status, and the risk of celiac disease in children. N Engl J Med 2014; 371:1295–1303
- Green PH. The many faces of celiac disease: clinical presentation of celiac disease in the adult population. Gastroenterology 2005; 128:S74–S78.
- Rampertab SD, Pooran N, Brar P, Singh P, Green PH. Trends in the presentation of celiac disease. Am J Med 2006; 119:355 e9–e14.
- Rashid M, Cranney A, Zarkadas M, et al. Celiac disease: evaluation of the diagnosis and dietary compliance in Canadian children. Pediatrics 2005; 116:e754–e759.
- Molteni N, Bardella MT, Bianchi PA. Obstetric and gynecological problems in women with untreated celiac sprue. J Clin Gastroenterol 1990; 12:37–39.
- Tersigni C, Castellani R, de Waure C, et al. Celiac disease and reproductive disorders: meta-analysis of epidemiologic associations and potential pathogenic mechanisms. Hum Reprod Update 2014; 20:582–593.
- Meyer D, Stravropolous S, Diamond B, Shane E, Green PH. Osteoporosis in a North American adult population with celiac disease. Am J Gastroenterol 2001; 96:112–119.
- Fouda MA, Khan AA, Sultan MS, Rios LP, McAssey K, Armstrong D. Evaluation and management of skeletal health in celiac disease: position statement. Can J Gastroenterol 2012; 26:819–829.
- van der Pals M, Ivarsson A, Norström F, Högberg L, Svensson J, Carlsson A. Prevalence of thyroid autoimmunity in children with celiac disease compared to healthy 12-year olds. Autoimmune Dis 2014; 2014:417356.
- Mahmud FH, Murray JA, Kudva YC, et al. Celiac disease in type 1 diabetes mellitus in a North American community: prevalence, serologic screening, and clinical features. Mayo Clin Proc 2005; 80:1429–1434.
- Sorensen HT, Thulstrup AM, Blomqvist P, Nørgaard B, Fonager K, Ekbom A. Risk of primary biliary liver cirrhosis in patients with coeliac disease: Danish and Swedish cohort data. Gut 1999; 44:736–738.
- Volta U, Rodrigo L, Granito A, et al. Celiac disease in autoimmune cholestatic liver disorders. Am J Gastroenterol 2002; 97:2609–2613.
- Elfstrom P, Montgomery SM, Kämpe O, Ekbom A, Ludvigsson JF. Risk of primary adrenal insufficiency in patients with celiac disease. J Clin Endocrinol Metab 2007; 92:3595–3598.
- Younus J, Ahmed AR. Clinical features of dermatitis herpetiformis. Clin Dermatol 1991; 9:279–281.
- Bolotin D, Petronic-Rosic V. Dermatitis herpetiformis. Part I. Epidemiology, pathogenesis, and clinical presentation. J Am Acad Dermatol 2011; 64:1017–1026.
- Lahteenoja H, Irjala K, Viander M, Vainio E, Toivanen A, Syrjänen S. Oral mucosa is frequently affected in patients with dermatitis herpetiformis. Arch Dermatol 1998; 134:756–758.
- Marks R, Jones EW. Purpura in dermatitis herpetiformis. Br J Dermatol 1971; 84:386–388.
- McGovern TW, Bennion SD. Palmar purpura: an atypical presentation of childhood dermatitis herpetiformis. Pediatr Dermatol 1994; 11:319–322.
- Pierce DK, Purcell SM, Spielvogel RL. Purpuric papules and vesicles of the palms in dermatitis herpetiformis. J Am Acad Dermatol 1987; 16:1274–1276.
- Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 2003; 4:140–156.
- Hull CM, Liddle M, Hansen N, et al. Elevation of IgA anti-epidermal transglutaminase antibodies in dermatitis herpetiformis. Br J Dermatol 2008; 159:120–124.
- Kawana S, Segawa A. Confocal laser scanning microscopic and immunoelectron microscopic studies of the anatomical distribution of fibrillar IgA deposits in dermatitis herpetiformis. Arch Dermatol 1993; 129:456–459.
- Sárdy M, Kárpáti S, Merkl B, Paulsson M, Smyth N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J Exp Med 2002; 195:747–757.
- Nicolas ME, Krause PK, Gibson LE, Murray JA. Dermatitis herpetiformis. Int J Dermatol 2003; 42:588–600.
- Leonard J, Haffenden G, Tucker W, et al. Gluten challenge in dermatitis herpetiformis. N Engl J Med 1983; 308:816–819.
- Summaries for patients. Risk for lymphoma and the results of follow-up gut biopsies in patients with celiac disease. Ann Intern Med 2013; 159:I–20.
- Lebwohl B, Granath F, Ekbom A, et al. Mucosal healing and risk for lymphoproliferative malignancy in celiac disease: a population-based cohort study. Ann Intern Med 2013; 159:169–175.
- Volta U, Vincentini O, Quintarelli F, Felli C, Silano M; Collaborating Centres of the Italian Registry of the Complications of Celiac Disease. Low risk of colon cancer in patients with celiac disease. Scand J Gastroenterol 2014; 49:564–568.
- Askling J, Linet M, Gridley G, Halstensen TS, Ekström K, Ekbom A. Cancer incidence in a population-based cohort of individuals hospitalized with celiac disease or dermatitis herpetiformis. Gastroenterology 2002; 123:1428–1435.
- Elfström P, Granath F, Ye W, Ludvigsson JF. Low risk of gastrointestinal cancer among patients with celiac disease, inflammation, or latent celiac disease. Clin Gastroenterol Hepatol 2012; 10:30–36.
- Al-Toma A, Verbeek WH, Hadithi M, von Blomberg BM, Mulder CJ. Survival in refractory coeliac disease and enteropathy-associated T-cell lymphoma: retrospective evaluation of single-centre experience. Gut 2007; 56:1373–1378.
- Verbeek WH, Van De Water JM, Al-Toma A, Oudejans JJ, Mulder CJ, Coupé VM. Incidence of enteropathy—associated T-cell lymphoma: a nation-wide study of a population-based registry in The Netherlands. Scand J Gastroenterol 2008; 43:1322–1328.
- Sharaiha RZ, Lebwohl B, Reimers L, Bhagat G, Green PH, Neugut AI. Increasing incidence of enteropathy-associated T-cell lymphoma in the United States, 1973-2008. Cancer 2012; 118:3786–3792.
- Mearin ML, Catassi C, Brousse N, et al; Biomed Study Group on Coeliac Disease and Non-Hodgkin Lymphoma. European multi-centre study on coeliac disease and non-Hodgkin lymphoma. Eur J Gastroenterol Hepatol 2006; 18:187–194.
- Al-Toma A, Goerres MS, Meijer JW, Pena AS, Crusius JB, Mulder CJ. Human leukocyte antigen-DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clin Gastroenterol Hepatol 2006; 4:315–319.
- Sieniawski MK, Lennard AL. Enteropathy-associated T-cell lymphoma: epidemiology, clinical features, and current treatment strategies. Curr Hematol Malig Rep 2011; 6:231–240.
- Lebwohl B, Eriksson H, Hansson J, Green PH, Ludvigsson JF. Risk of cutaneous malignant melanoma in patients with celiac disease: a population-based study. J Am Acad Dermatol 2014; 71:245–248.
- Ludvigsson JF, Lebwohl B, Kämpe O, Murray JA, Green PH, Ekbom A. Risk of thyroid cancer in a nationwide cohort of patients with biopsy-verified celiac disease. Thyroid 2013; 23:971–976.
- Ludvigsson JF, West J, Ekbom A, Stephansson O. Reduced risk of breast, endometrial and ovarian cancer in women with celiac disease. Int J Cancer 2012; 13:E244–E250.
- Rubio-Tapia A, Hill ID, Kelly CP, Calderwood AH, Murray JA; American College of Gastroenterology. ACG clinical guidelines: diagnosis and management of celiac disease. Am J Gastroenterol 2013; 108:656–677.
- Marsh MN. Gluten, major histocompatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity (‘celiac sprue’). Gastroenterology 1992; 102:330–354.
- Hadithi M, von Blomberg BM, Crusius JB, et al. Accuracy of serologic tests and HLA-DQ typing for diagnosing celiac disease. Ann Intern Med 2007; 147:294–302.
- Lo W, Sano K, Lebwohl B, Diamond B, Green PH. Changing presentation of adult celiac disease. Dig Dis Sci 2003; 48:395–398.
- Oxentenko AS, Grisolano SW, Murray JA, Burgart LJ, Dierkhising RA, Alexander JA. The insensitivity of endoscopic markers in celiac disease. Am J Gastroenterol 2002; 97:933–938.
- Casella G, Antonelli E, Di Bella C, et al. Prevalence and causes of abnormal liver function in patients with coeliac disease. Liver Int 2013; 33:1128–1131.
- Jaskowski TD, Hamblin T, Wilson AR, et al. IgA anti-epidermal transglutaminase antibodies in dermatitis herpetiformis and pediatric celiac disease. J Invest Dermatol 2009; 129:2728–2730.
- Zone JJ, Meyer LJ, Petersen MJ. Deposition of granular IgA relative to clinical lesions in dermatitis herpetiformis. Arch Dermatol 1996; 132:912–918.
- Plotnikova N, Miller JL. Dermatitis herpetiformis. Skin Ther Lett 2013; 18:1–3.
- Murray JA, Watson T, Clearman B, Mitros F. Effect of a gluten-free diet on gastrointestinal symptoms in celiac disease. Am J Clin Nutr 2004; 79:669–673.
- Akobeng AK, Thomas AG. Systematic review: tolerable amount of gluten for people with coeliac disease. Aliment Pharmacol Ther 2008; 27:1044–1052.
- Catassi C, Fabiani E, Iacono G, et al. A prospective, double-blind, placebo-controlled trial to establish a safe gluten threshold for patients with celiac disease. Am J Clin Nutr 2007; 85:160–166.
- Leffler DA, Edwards-George J, Dennis M, et al. Factors that influence adherence to a gluten-free diet in adults with celiac disease. Dig Dis Sci 2008; 53:1573–1581.
- Caruso R, Pallone F, Stasi E, Romeo S, Monteleone G. Appropriate nutrient supplementation in celiac disease. Ann Med 2013; 45:522–531.
- Nachman F, Sugai E, Vazquez H, et al. Serological tests for celiac disease as indicators of long-term compliance with the gluten-free diet. Eur J Gastroenterol Hepatol 2011; 23:473–480.
- Abdulkarim AS, Burgart LJ, See J, Murray JA. Etiology of nonresponsive celiac disease: results of a systemic approach. Am J Gastroenterol 2002; 97:2016–2021.
- Fry L, Seah PP, Hoffbrand AV. Dermatitis herpetiformis. Clin Gastroenterol 1974; 3:145–157.
- Zhu YI, Stiller MJ. Dapsone and sulfones in dermatology: overview and update. J Am Acad Dermatol 2001; 45:420-434.
- Wolf R, Matz H, Orion E, Tuzun B, Tuzun Y. Dapsone. Dermatol Online J 2002; 8:2.
- Ciacci C, Maiuri L, Caporaso N, et al. Celiac disease: in vitro and in vivo safety and palatability of wheat-free sorghum food products. Clin Nutr 2007; 26:799–805.
- Lähdeaho ML, Kaukinen K, Laurila K, et al. Glutenase ALV003 attenuates gluten-induced mucosal injury in patients with celiac disease. Gastroenterology 2014; 146:1649–1658.
- Roshan B, Leffler DA, Jamma S, et al. The incidence and clinical spectrum of refractory celiac disease in a North American referral center. Am J Gastroenterol 2011; 106:923–928.
- Leffler DA, Dennis M, Hyett B, Kelly E, Schuppan D, Kelly CP. Etiologies and predictors of diagnosis in nonresponsive celiac disease. Clin Gastroenterol Hepatol 2007; 5:445–450.
- Cellier C, Delabesse E, Helmer C, et al. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. French Coeliac Disease Study Group. Lancet 2000; 356:203–208.
- Malamut G, Afchain P, Verkarre V, et al. Presentation and long-term follow-up of refractory celiac disease: comparison of type I with type II. Gastroenterology 2009; 136:81–90.
- Goerres MS, Meijer JW, Wahab PJ, et al. Azathioprine and prednisone combination therapy in refractory celiac disease. Aliment Pharmacol Ther 2003; 18:487–494.
- Tack GJ, Verbeek WH, Al-Toma A, et al. Evaluation of cladribine treatment in refractory celiac disease type II. World J Gastroenterol 2011; 17:506–513.
- Sapone A, Bai JC, Dolinsek J, et al. Spectrum of gluten-related disorders: consensus on new nomenclature and classification. BMC Med 2012; 7:10–13.
- Sapone A, Lammers KM, Casolaro V, et al. Divergence of gut permeability and mucosal immune gene expression in two gluten-associated conditions: celiac disease and gluten sensitivity. BMC Med 2011; 9:9–23.
- Rubio-Tapia A, Ludvigsson JF, Bratner TL, Murray JA, Everhart JE. The prevalence of celiac disease in the United States. Am J Gastroenterol 2012; 107:1538–1544.
- Dewar DH, Ciclitira PJ. Clinical features and diagnosis of celiac disease. Gastroenterology 2005; 128(suppl 1):S19–S24.
- Mendes FB, Hissa-Elian A, Abreu MA, Goncalves VS. Review: dermatitis herpetiformis. An Bras Dermatol 2013; 88:594–599.
- Lauret E, Rodrigo L. Celiac disease and autoimmune-associated conditions. Biomed Res Int 2013; 2013:127589.
- Sollid LM, Lie BA. Celiac disease genetics: current concepts and practical applications. Clin Gastroenterol Hepatol 2005; 3:843–851.
- Hausch F, Shan L, Santiago NA, Gray GM, Khosla C. Intestinal digestive resistance of immunodominant gliadin peptides. Am J Physiol Gastrointest Liver Physiol 2002; 283:G996–G1003.
- Green PH, Cellier C. Celiac disease. N Engl J Med 2007; 357:1731–1743.
- Caputo I, Barone MV, Martucciello S, Lepretti M, Esposito C. Tissue transglutaminase in celiac disease: role of autoantibodies. Amino Acids 2009; 36:693–699.
- Schuppan D, Dieterich W, Riecken EO. Exposing gliadin as a tasty food for lymphocytes. Nat Med 1998; 4:666–667.
- Stene LC, Honeyman MC, Hoffenberg EJ, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol 2006; 101:2333–2340.
- Kagnoff MF, Austin RK, Hubert JJ, Bernardin JE, Kasarda DD. Possible role for a human adenovirus in the pathogenesis of celiac disease. J Exp Med 1984; 160:1544–1557.
- Ruggeri C, LaMasa AT, Rudi S, et al. Celiac disease and non-organ-specific autoantibodies in patients with chronic hepatitis C virus infection. Dig Dis Sci 2008; 53:2151–2155.
- Sjoberg K, Lindgren S, Eriksson S. Frequent occurrence of non-specific gliadin antibodies in chronic liver disease. Endomysial but not gliadin antibodies predict coelic disease in patients with chronic liver disease. Scand J Gastroenterol 1997; 32:1162–1167.
- Persson LA, Ivarsson A, Hernell O. Breast-feeding protects against celiac disease in childhood—epidemiological evidence. Adv Exp Med Biol 2002; 503:115–123.
- Norris JM, Barriga K, Hoffenberg EJ, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA 2005; 293:2343–2351.
- Vriezinga SL, Auricchio R, Bravi E, et al. Randomized feeding intervention in infants at high risk for celiac disease. N Engl J Med 2014; 371:1304–1315.
- Lionetti E, Castelaneta S, Francavilla R, et al. Introduction of gluten, HLA status, and the risk of celiac disease in children. N Engl J Med 2014; 371:1295–1303
- Green PH. The many faces of celiac disease: clinical presentation of celiac disease in the adult population. Gastroenterology 2005; 128:S74–S78.
- Rampertab SD, Pooran N, Brar P, Singh P, Green PH. Trends in the presentation of celiac disease. Am J Med 2006; 119:355 e9–e14.
- Rashid M, Cranney A, Zarkadas M, et al. Celiac disease: evaluation of the diagnosis and dietary compliance in Canadian children. Pediatrics 2005; 116:e754–e759.
- Molteni N, Bardella MT, Bianchi PA. Obstetric and gynecological problems in women with untreated celiac sprue. J Clin Gastroenterol 1990; 12:37–39.
- Tersigni C, Castellani R, de Waure C, et al. Celiac disease and reproductive disorders: meta-analysis of epidemiologic associations and potential pathogenic mechanisms. Hum Reprod Update 2014; 20:582–593.
- Meyer D, Stravropolous S, Diamond B, Shane E, Green PH. Osteoporosis in a North American adult population with celiac disease. Am J Gastroenterol 2001; 96:112–119.
- Fouda MA, Khan AA, Sultan MS, Rios LP, McAssey K, Armstrong D. Evaluation and management of skeletal health in celiac disease: position statement. Can J Gastroenterol 2012; 26:819–829.
- van der Pals M, Ivarsson A, Norström F, Högberg L, Svensson J, Carlsson A. Prevalence of thyroid autoimmunity in children with celiac disease compared to healthy 12-year olds. Autoimmune Dis 2014; 2014:417356.
- Mahmud FH, Murray JA, Kudva YC, et al. Celiac disease in type 1 diabetes mellitus in a North American community: prevalence, serologic screening, and clinical features. Mayo Clin Proc 2005; 80:1429–1434.
- Sorensen HT, Thulstrup AM, Blomqvist P, Nørgaard B, Fonager K, Ekbom A. Risk of primary biliary liver cirrhosis in patients with coeliac disease: Danish and Swedish cohort data. Gut 1999; 44:736–738.
- Volta U, Rodrigo L, Granito A, et al. Celiac disease in autoimmune cholestatic liver disorders. Am J Gastroenterol 2002; 97:2609–2613.
- Elfstrom P, Montgomery SM, Kämpe O, Ekbom A, Ludvigsson JF. Risk of primary adrenal insufficiency in patients with celiac disease. J Clin Endocrinol Metab 2007; 92:3595–3598.
- Younus J, Ahmed AR. Clinical features of dermatitis herpetiformis. Clin Dermatol 1991; 9:279–281.
- Bolotin D, Petronic-Rosic V. Dermatitis herpetiformis. Part I. Epidemiology, pathogenesis, and clinical presentation. J Am Acad Dermatol 2011; 64:1017–1026.
- Lahteenoja H, Irjala K, Viander M, Vainio E, Toivanen A, Syrjänen S. Oral mucosa is frequently affected in patients with dermatitis herpetiformis. Arch Dermatol 1998; 134:756–758.
- Marks R, Jones EW. Purpura in dermatitis herpetiformis. Br J Dermatol 1971; 84:386–388.
- McGovern TW, Bennion SD. Palmar purpura: an atypical presentation of childhood dermatitis herpetiformis. Pediatr Dermatol 1994; 11:319–322.
- Pierce DK, Purcell SM, Spielvogel RL. Purpuric papules and vesicles of the palms in dermatitis herpetiformis. J Am Acad Dermatol 1987; 16:1274–1276.
- Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 2003; 4:140–156.
- Hull CM, Liddle M, Hansen N, et al. Elevation of IgA anti-epidermal transglutaminase antibodies in dermatitis herpetiformis. Br J Dermatol 2008; 159:120–124.
- Kawana S, Segawa A. Confocal laser scanning microscopic and immunoelectron microscopic studies of the anatomical distribution of fibrillar IgA deposits in dermatitis herpetiformis. Arch Dermatol 1993; 129:456–459.
- Sárdy M, Kárpáti S, Merkl B, Paulsson M, Smyth N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J Exp Med 2002; 195:747–757.
- Nicolas ME, Krause PK, Gibson LE, Murray JA. Dermatitis herpetiformis. Int J Dermatol 2003; 42:588–600.
- Leonard J, Haffenden G, Tucker W, et al. Gluten challenge in dermatitis herpetiformis. N Engl J Med 1983; 308:816–819.
- Summaries for patients. Risk for lymphoma and the results of follow-up gut biopsies in patients with celiac disease. Ann Intern Med 2013; 159:I–20.
- Lebwohl B, Granath F, Ekbom A, et al. Mucosal healing and risk for lymphoproliferative malignancy in celiac disease: a population-based cohort study. Ann Intern Med 2013; 159:169–175.
- Volta U, Vincentini O, Quintarelli F, Felli C, Silano M; Collaborating Centres of the Italian Registry of the Complications of Celiac Disease. Low risk of colon cancer in patients with celiac disease. Scand J Gastroenterol 2014; 49:564–568.
- Askling J, Linet M, Gridley G, Halstensen TS, Ekström K, Ekbom A. Cancer incidence in a population-based cohort of individuals hospitalized with celiac disease or dermatitis herpetiformis. Gastroenterology 2002; 123:1428–1435.
- Elfström P, Granath F, Ye W, Ludvigsson JF. Low risk of gastrointestinal cancer among patients with celiac disease, inflammation, or latent celiac disease. Clin Gastroenterol Hepatol 2012; 10:30–36.
- Al-Toma A, Verbeek WH, Hadithi M, von Blomberg BM, Mulder CJ. Survival in refractory coeliac disease and enteropathy-associated T-cell lymphoma: retrospective evaluation of single-centre experience. Gut 2007; 56:1373–1378.
- Verbeek WH, Van De Water JM, Al-Toma A, Oudejans JJ, Mulder CJ, Coupé VM. Incidence of enteropathy—associated T-cell lymphoma: a nation-wide study of a population-based registry in The Netherlands. Scand J Gastroenterol 2008; 43:1322–1328.
- Sharaiha RZ, Lebwohl B, Reimers L, Bhagat G, Green PH, Neugut AI. Increasing incidence of enteropathy-associated T-cell lymphoma in the United States, 1973-2008. Cancer 2012; 118:3786–3792.
- Mearin ML, Catassi C, Brousse N, et al; Biomed Study Group on Coeliac Disease and Non-Hodgkin Lymphoma. European multi-centre study on coeliac disease and non-Hodgkin lymphoma. Eur J Gastroenterol Hepatol 2006; 18:187–194.
- Al-Toma A, Goerres MS, Meijer JW, Pena AS, Crusius JB, Mulder CJ. Human leukocyte antigen-DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clin Gastroenterol Hepatol 2006; 4:315–319.
- Sieniawski MK, Lennard AL. Enteropathy-associated T-cell lymphoma: epidemiology, clinical features, and current treatment strategies. Curr Hematol Malig Rep 2011; 6:231–240.
- Lebwohl B, Eriksson H, Hansson J, Green PH, Ludvigsson JF. Risk of cutaneous malignant melanoma in patients with celiac disease: a population-based study. J Am Acad Dermatol 2014; 71:245–248.
- Ludvigsson JF, Lebwohl B, Kämpe O, Murray JA, Green PH, Ekbom A. Risk of thyroid cancer in a nationwide cohort of patients with biopsy-verified celiac disease. Thyroid 2013; 23:971–976.
- Ludvigsson JF, West J, Ekbom A, Stephansson O. Reduced risk of breast, endometrial and ovarian cancer in women with celiac disease. Int J Cancer 2012; 13:E244–E250.
- Rubio-Tapia A, Hill ID, Kelly CP, Calderwood AH, Murray JA; American College of Gastroenterology. ACG clinical guidelines: diagnosis and management of celiac disease. Am J Gastroenterol 2013; 108:656–677.
- Marsh MN. Gluten, major histocompatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity (‘celiac sprue’). Gastroenterology 1992; 102:330–354.
- Hadithi M, von Blomberg BM, Crusius JB, et al. Accuracy of serologic tests and HLA-DQ typing for diagnosing celiac disease. Ann Intern Med 2007; 147:294–302.
- Lo W, Sano K, Lebwohl B, Diamond B, Green PH. Changing presentation of adult celiac disease. Dig Dis Sci 2003; 48:395–398.
- Oxentenko AS, Grisolano SW, Murray JA, Burgart LJ, Dierkhising RA, Alexander JA. The insensitivity of endoscopic markers in celiac disease. Am J Gastroenterol 2002; 97:933–938.
- Casella G, Antonelli E, Di Bella C, et al. Prevalence and causes of abnormal liver function in patients with coeliac disease. Liver Int 2013; 33:1128–1131.
- Jaskowski TD, Hamblin T, Wilson AR, et al. IgA anti-epidermal transglutaminase antibodies in dermatitis herpetiformis and pediatric celiac disease. J Invest Dermatol 2009; 129:2728–2730.
- Zone JJ, Meyer LJ, Petersen MJ. Deposition of granular IgA relative to clinical lesions in dermatitis herpetiformis. Arch Dermatol 1996; 132:912–918.
- Plotnikova N, Miller JL. Dermatitis herpetiformis. Skin Ther Lett 2013; 18:1–3.
- Murray JA, Watson T, Clearman B, Mitros F. Effect of a gluten-free diet on gastrointestinal symptoms in celiac disease. Am J Clin Nutr 2004; 79:669–673.
- Akobeng AK, Thomas AG. Systematic review: tolerable amount of gluten for people with coeliac disease. Aliment Pharmacol Ther 2008; 27:1044–1052.
- Catassi C, Fabiani E, Iacono G, et al. A prospective, double-blind, placebo-controlled trial to establish a safe gluten threshold for patients with celiac disease. Am J Clin Nutr 2007; 85:160–166.
- Leffler DA, Edwards-George J, Dennis M, et al. Factors that influence adherence to a gluten-free diet in adults with celiac disease. Dig Dis Sci 2008; 53:1573–1581.
- Caruso R, Pallone F, Stasi E, Romeo S, Monteleone G. Appropriate nutrient supplementation in celiac disease. Ann Med 2013; 45:522–531.
- Nachman F, Sugai E, Vazquez H, et al. Serological tests for celiac disease as indicators of long-term compliance with the gluten-free diet. Eur J Gastroenterol Hepatol 2011; 23:473–480.
- Abdulkarim AS, Burgart LJ, See J, Murray JA. Etiology of nonresponsive celiac disease: results of a systemic approach. Am J Gastroenterol 2002; 97:2016–2021.
- Fry L, Seah PP, Hoffbrand AV. Dermatitis herpetiformis. Clin Gastroenterol 1974; 3:145–157.
- Zhu YI, Stiller MJ. Dapsone and sulfones in dermatology: overview and update. J Am Acad Dermatol 2001; 45:420-434.
- Wolf R, Matz H, Orion E, Tuzun B, Tuzun Y. Dapsone. Dermatol Online J 2002; 8:2.
- Ciacci C, Maiuri L, Caporaso N, et al. Celiac disease: in vitro and in vivo safety and palatability of wheat-free sorghum food products. Clin Nutr 2007; 26:799–805.
- Lähdeaho ML, Kaukinen K, Laurila K, et al. Glutenase ALV003 attenuates gluten-induced mucosal injury in patients with celiac disease. Gastroenterology 2014; 146:1649–1658.
- Roshan B, Leffler DA, Jamma S, et al. The incidence and clinical spectrum of refractory celiac disease in a North American referral center. Am J Gastroenterol 2011; 106:923–928.
- Leffler DA, Dennis M, Hyett B, Kelly E, Schuppan D, Kelly CP. Etiologies and predictors of diagnosis in nonresponsive celiac disease. Clin Gastroenterol Hepatol 2007; 5:445–450.
- Cellier C, Delabesse E, Helmer C, et al. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. French Coeliac Disease Study Group. Lancet 2000; 356:203–208.
- Malamut G, Afchain P, Verkarre V, et al. Presentation and long-term follow-up of refractory celiac disease: comparison of type I with type II. Gastroenterology 2009; 136:81–90.
- Goerres MS, Meijer JW, Wahab PJ, et al. Azathioprine and prednisone combination therapy in refractory celiac disease. Aliment Pharmacol Ther 2003; 18:487–494.
- Tack GJ, Verbeek WH, Al-Toma A, et al. Evaluation of cladribine treatment in refractory celiac disease type II. World J Gastroenterol 2011; 17:506–513.
- Sapone A, Bai JC, Dolinsek J, et al. Spectrum of gluten-related disorders: consensus on new nomenclature and classification. BMC Med 2012; 7:10–13.
- Sapone A, Lammers KM, Casolaro V, et al. Divergence of gut permeability and mucosal immune gene expression in two gluten-associated conditions: celiac disease and gluten sensitivity. BMC Med 2011; 9:9–23.
KEY POINTS
- Besides gastrointestinal symptoms, celiac disease is associated with a variety of diseases, including dermatitis herpetiformis, malabsorption of several nutrients (potentially leading to osteoporosis, iron deficiency anemia, and other disorders), and intestinal malignancies.
- While serologic testing for immunoglobulin A antibodies to tissue transglutaminase can be used as an initial screening test for this condition, the confirmatory tests are invasive, involving upper endoscopy for duodenal biopsy in celiac disease and skin biopsy in dermatitis herpetiformis.
- The only effective treatment is lifelong adherence to a gluten-free diet, and nonadherence is a common cause of refractory disease.
- Concomitant conditions such as anemia and vitamin deficiency often require nutritional supplements. In addition, patients with dermatitis herpetiformis often require treatment with dapsone.
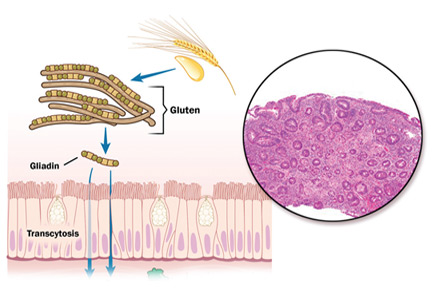
Prescribing opioids in primary care: Safely starting, monitoring, and stopping
Chronic pain affects an estimated 100 million Americans, at a cost of $635 billion each year in medical expenses, lost wages, and reduced productivity.1 It is often managed in primary care settings with opioids by clinicians who have little or no formal training in pain management.2,3 Some primary care providers may seek assistance from board-certified pain specialists, but with only four such experts for every 100,000 patients with chronic pain, primary care providers are typically on their own.4
Although opioids may help in some chronic pain syndromes, they also carry the risk of serious harm, including unintentional overdose and death. In 2009, unintentional drug overdoses, most commonly with opioids, surpassed motor vehicle accidents as the leading cause of accidental death in the United States.5 Additionally, nonmedical use of prescription drugs is the third most common category of drug abuse, after marijuana and alcohol.6
Unfortunately, clinicians cannot accurately predict future medication misuse.7 And while the potential harms of opioids are many, the long-term benefits are questionable.8,9
For these reasons, providers need to understand the indications for and potential benefits of opioids, as well as the potential harms and how to monitor their safe use. Also important to know is how and when to discontinue opioids while preserving the therapeutic relationship.
This paper offers practical strategies to primary care providers and their care teams on how to safely initiate, monitor, and discontinue chronic opioid therapy.
STARTING OPIOID THERAPY FOR CHRONIC PAIN
Guidelines recommend considering starting patients on opioid therapy when the benefits are likely to outweigh the risks, when pain is moderate to severe, and when other multimodal treatment strategies have not achieved functional goals.10 Unfortunately, few studies have examined or demonstrated long-term benefit, and those that did examine this outcome reported reduction of pain severity but did not assess functional improvement.9 Meanwhile, data are increasingly clear that long-term use increases the risk of harm, both acute (eg, overdose) and chronic (eg, osteoporosis), especially with high doses.
Tools have been developed to predict the risk of misuse,11–13 but few have been validated in primary care, where most opioids are prescribed. This limitation aside, consensus guidelines state that untreated substance use disorders, poorly controlled psychiatric disease, and erratic treatment adherence are contraindications to opioid therapy, at least until these other issues are treated.10
Faced with the benefit-harm conundrum, we recommend a generally conservative approach to opioid initiation. With long-term functional benefit questionable and toxicity relatively common, we are increasingly avoiding chronic opioid therapy in younger patients with chronic pain.
Empathize and partner with your patient
Chronic pain care can be fraught with frustration and mutual distrust between patient and provider.14 Empathy and a collaborative stance help signal to the patient that the provider has the patient’s best interest in mind,15 whether initiating or deciding not to initiate opioids.
Optimize nonopioid therapy
In light of the risks associated with chronic opioid therapy, the clinician is urged to review and optimize nonopioid therapy before starting a patient on opioid treatment, and to maintain this approach if opioid therapy is started. Whenever possible, nonopioid treatment should include disease-modifying therapy and nondrug modalities such as physical therapy.
Judicious use of adequately dosed analgesics such as acetaminophen and nonsteroidal anti-inflammatory drugs may be sufficient to achieve analgesic goals if not contraindicated, and in some patients the addition of a topical analgesic (eg, diclofenac gel, lidocaine patches), a tricyclic or serotonin-norepinephrine reuptake inhibitor antidepressant, an anticonvulsant (eg, gabapentin), or a combination of the above can effectively address underlying pain-generating mechanisms.16 As with opioids, the risks and benefits of nonopioid pharmacotherapy should be reviewed both at initiation and periodically thereafter.
Frame the opioid treatment plan as a ‘therapeutic trial’
Starting an opioid should be framed as a “therapeutic trial.” These drugs should be continued only if safe and effective, at the lowest effective dose, and as one component of a multimodal pain treatment plan. Concurrent use of nonpharmacologic therapies (eg, physical therapy, structured exercise, yoga, relaxation training, biofeedback, cognitive behavioral therapy) and rational pharmacotherapy while promoting patient self-care is the standard of pain management called for by the Institute of Medicine.1
Set functional goals
We recommend clearly defining functional goals with each patient before starting therapy. These goals should be written into the treatment plan as a way for patient and provider to evaluate the effectiveness of chronic opioid therapy. A useful mnemonic to help identify such goals is SMART, an acronym for specific, measurable, action-oriented, realistic, and time-bound. Specific goals will depend on pain severity, but examples could include being able to do grocery shopping without assistance, to play on the floor with grandchildren, or to engage in healthy exercise habits such as 20 minutes of moderately brisk walking 3 days per week.
Obtain informed consent, and document it thoroughly
Providers must communicate risks, potential benefits, and safe medication-taking practices, including how to safely store and dispose of unused opioids, and document this conversation clearly in the medical record. From a medicolegal perspective, if it wasn’t documented, it did not happen.17
Informed consent can be further advanced with the use of a controlled substance agreement that outlines the treatment plan as well as potential risks, benefits, and practice policies in a structured way. Most states now either recommend or mandate the use of such agreements.18
Controlled substance agreements give providers a greater sense of mastery and comfort when prescribing opioids,19 but they have important limitations. In particular, there is a lack of consensus on what the agreement should say and relatively weak evidence that these agreements are efficacious. Additionally, a poorly written agreement can be stigmatizing and can erode trust.20 However, we believe that when the agreement is written in an appropriate framework of safety at an appropriate level of health literacy and with a focus on shared decision-making, it can be very helpful and should be used.
Employ safe, rational pharmacotherapy
Considerations when choosing an opioid include its potency, onset of action, and half-life. Comorbid conditions (eg, advanced age,21 sleep-disordered breathing22) and concurrent medications (eg, benzodiazepines, anticonvulsants, muscle relaxants) also affect decisions about the formulation, starting dose, rapidity of titration, and ceiling dose. Risk of harm increases in patients with such comorbid factors, and it is prudent to start with lower doses of shorter-acting medications until patients can demonstrate safe use. Risk of unintentional overdose is higher with higher prescribed doses.23 Pharmacologically there is no analgesic dose ceiling, but we urge caution, particularly in opioid-naive patients.
A patient’s response to any particular opioid is idiosyncratic and variable. There are more than 100 known polymorphisms in the human opioid mu-receptor gene, and thus differences in receptor affinity and activation as well as in metabolism make it difficult to predict which opioid will work best for a particular patient.24 However, a less potent opioid receptor agonist with less addictive potential, such as tramadol or codeine, should generally be tried first before escalating to a riskier, more potent opioid such as hydrocodone, oxycodone, or morphine. This “analgesic ladder,” a concept introduced by the World Health Organization in 1986 to provide a framework for managing cancer pain, has been adapted to a variety of chronic pain syndromes.25
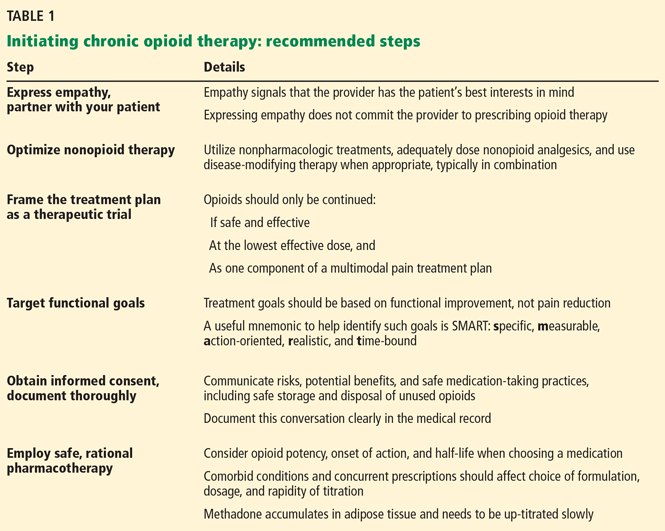
Methadone deserves special mention. A strongly lipophilic molecule with a long and variable half-life, it accumulates in fat,26 and long after the analgesic effect has worn off, methadone will still be present. Repeated dosing or rapid dose escalation in an attempt to achieve adequate analgesia may result in inadvertent overdose. Additionally, methadone can prolong the QT interval, and periodic electrocardiographic monitoring is recommended.27 For these reasons, we recommend avoiding the use of methadone in most cases unless the provider has significant experience, expertise, or support in the safe use of this medication.
Table 1 summarizes these recommendations.
MONITORING AND SAFETY
Providers must periodically reassess the safety and efficacy of chronic opioid therapy to be sure that it is still indicated.10 Since we cannot accurately predict which patients will suffer adverse reactions or demonstrate aberrant behaviors,7 it is important to be transparent and consistent with monitoring practices for all patients on chronic opioid therapy.17 By framing monitoring in terms of safety and employing it universally, providers can minimize miscommunication and accidental stigmatization.
Prescription monitoring programs
In 2002, Congress appropriated funding to the US Department of Justice to support prescription monitoring programs nationally.28 At the time of this writing, Missouri is the only state without an approved monitoring program.29
Although the design and function of the programs vary from state to state, they require pharmacies to collect and report data on controlled substances for individual patients and prescribers. These data are sometimes shared across state lines, and the programs enhance the capacity of regulatory and law enforcement agencies to analyze controlled substance use.
Prescribers can (and are sometimes required to) register for access in their state and use this resource to assess the opioid refill history of their patients. This powerful tool improves detection of “doctor-shopping” and other common scams.30
Additionally, recognizing that the risk of death from overdose increases as the total daily dose of opioids increases,23 some states provide data on their composite report expressing the morphine equivalent daily dose or daily morphine milligram equivalents of the opioids prescribed. This information is valuable to the busy clinician; at a glance the prescriber can quickly discern the total daily opioid dose and use that information to assess risk and manage change. Furthermore, some states restrict further dose escalation when the morphine equivalent daily dose exceeds a predetermined amount (typically 100 to 120 morphine milligram equivalents).
Tamper-resistant prescribing
To minimize the risk of prescription tampering, simple techniques such as writing out the number of tablets dispensed can help, and use of tamper-resistant prescription paper has been required for Medicaid recipients since 2008.31
When possible, we recommend products with abuse-deterrent properties. Although the science of abuse deterrence is relatively new and few products are labeled as such, a number of opioids are formulated to resist deformation, vaporization, dissolving, or other physical tampering. Additionally, some abuse-deterrent opioid formulations contain naloxone, which is released only when the drug is deformed in some way, thereby decreasing the user’s response to an abused substance or resulting in opioid withdrawal.32
Urine drug testing
Although complex and nuanced, guidelines recommend urine drug testing to confirm the presence or absence of prescribed and illicit substances in the body.10 There is no consensus on when or how often to test, but it should be done randomly and without forewarning to foil efforts to defeat testing such as provision of synthetic, adulterated, or substituted urine.
Providers underuse urine drug testing.33 We recommend that it be done at the start of opioid therapy, sporadically thereafter, when therapy is changed, and whenever the provider is concerned about possible aberrant drug use.
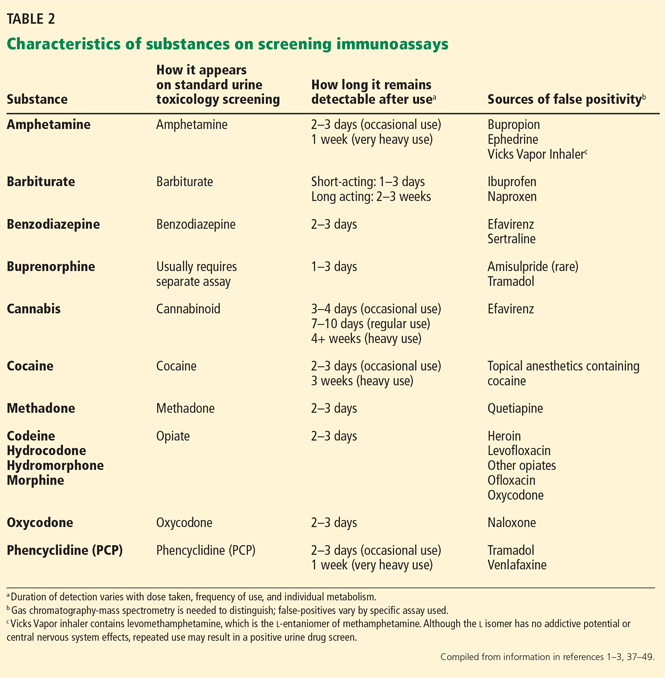
Understanding opioid metabolism, cross-reactivity, and the types of tests available will help avoid misinterpretation of results.34 For example, a positive “opiate” result in most screening immunoassay tests does not reflect oxycodone use, since tests for synthetic opioids often need to be ordered separately; the commonly used Cedia opiate assay cross-reacts with oxycodone at a concentration of 10,000 ng/mL only 3.1% of the time.35 Immunoassay screening tests are widely available, sensitive, inexpensive, and fast, but they are qualitative, have limited specificity, and are subject to false-positive and false-negative results.36 Table 2 outlines some common characteristics of substances on screening immunoassays, including reported causes of false-positive results.37–39
Confirmatory testing using gas chromatography or mass spectroscopy is more expensive and slower to process, but is highly sensitive and specific, quantitative, and useful when screening results are difficult to interpret.
Knowing how and when to order the right urine drug test and knowing how to interpret the results are skills prescribers should master.
DISCONTINUING OPIOIDS
When opioids are no longer safe or effective, they should be stopped. The decision can be difficult for both the patient and provider, and a certain degree of equanimity is needed to approach it rationally.
Strong indications for discontinuation
Respiratory depression, cognitive impairment, falls, and motor vehicle accidents mean harm is already apparent. At a minimum, dose reduction is warranted and discontinuation should be strongly considered. Similarly, overdose (intentional or accidental) and active suicidal ideation contraindicate ongoing opioid prescribing unless the ongoing risk can be decisively mitigated.
Certain aberrant behaviors such as prescription forgery or theft, threats of violence to obtain analgesics, and diversion (transfer of the drug to another person for nonmedical use) also warrant immediate discontinuation. Continuing to prescribe an opioid while knowing diversion is taking place may be a violation of federal or state law or both.40
Another reason to stop is failure to achieve the expected benefit from chronic opioid therapy (ie, agreed-upon functional goals) despite appropriate dose adjustment. In these cases, ongoing risk by definition outweighs observed benefit.
Relative indications for discontinuation
Opioid therapy has many potential adverse effects. Depending on the severity and duration of the symptom and its response to either dose reduction or adjunctive management, opioids may need to be discontinued.
For example, pruritus, constipation, urinary retention, nausea, sedation, and sexual dysfunction may all be reasons to stop chronic opioid therapy. Similarly, chronic opioid therapy may paradoxically worsen pain in some susceptible patients, a complication known as opioid-induced hyperalgesia; in these cases, tapering off opioids should be considered as well.41 Aberrant behaviors should prompt reconsideration of chronic opioid therapy; these include hazardous alcohol consumption, use of illicit drugs, pill hoarding, and use of opioids in a manner different than intended by the prescriber.
Another relative indication for discontinuation is receipt of controlled substances from other providers. A well-written controlled substance agreement and adequate counseling may help mitigate this risk; poor communication between providers, lack of integration of electronic medical record systems, urgent or emergency room care, and poor health literacy may all lead to redundant prescribing in some circumstances. While unintentional use of controlled substances from different providers is no less dangerous than intentional misuse, the specifics of each case need to be considered before opioids are reflexively discontinued.
How to discontinue opioids
In most cases, opioids should be tapered to reduce the risk and severity of withdrawal symptoms. Decreasing the dose by 10% of the original dose per week is usually well tolerated with minimal adverse effects.42 Tapering can be done much faster, and numerous rapid detoxification protocols are available. In general, a patient needs 20% of the previous day’s dose to prevent withdrawal symptoms.43
Withdrawal symptoms are rarely life-threatening but can be very uncomfortable. Some providers add clonidine to attenuate associated autonomic symptoms such as hypertension, nausea, cramps, diaphoresis, and tachycardia if they occur. Other adjunctive medications include nonsteroidal anti-inflammatory drugs for body aches, antiemetics for nausea and vomiting, bismuth subsalicylate for diarrhea, and trazodone for insomnia.
It is unlawful for primary care physicians to use another opioid to treat symptoms of withdrawal in the outpatient setting unless it is issued through a federally certified narcotic treatment program or prescribed by a qualified clinician registered with the US Drug Enforcement Administration to prescribe buprenorphine-naloxone.44
In some circumstances, it may be appropriate to abruptly discontinue opioids without a taper, such as when diversion is evident. However, a decision to discontinue opioids due to misuse should not equate to an automatic decision to terminate a patient from the practice. Instead, providers should use this opportunity to offer empathy and referral to drug treatment counseling and rehabilitation. A decision to discontinue opioids because they are no longer safe or effective does not mean that the patient’s pain is not real—it is “real” for them, even if caused by the pain of addiction—or that shared decision-making is no longer possible or appropriate.
Handling difficult conversations when discontinuing opioids
The conversation between patient and provider when discontinuing opioids can be difficult. Misaligned expectations of both parties, patient fear of uncontrolled pain, and provider concern about causing suffering are frequent contributing factors. Patients abusing prescription drugs may also have a stronger relationship with their medication than with their provider and may use manipulative strategies including overt hostility and threats to obtain a prescription. Providers need to maintain their composure to de-escalate these potentially upsetting confrontations.
Table 3 outlines some specific suggestions that may be helpful, including the following:
- Frame the discussion in terms of safety—opioids are being discontinued because the benefit no longer outweighs the risk

- Don’t debate your decision with the patient, but present your reasoning in a considered manner
- Focus on the appropriateness of the treatment and not on the patient’s character
- Avoid the use of labels (eg, “drug addict”)
- Emphasize your commitment to the patient’s well-being and an alternative treatment plan (ie, nonabandonment)
- Respond to emotional distress with empathy, but do not let that change your decision to discontinue opioids.
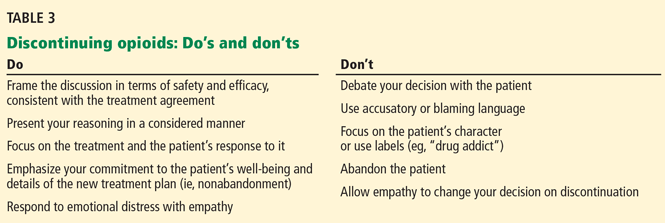
Finally, we strongly encourage providers to insist on being treated respectfully. When safety cannot be ensured, providers should remove themselves from the room until the patient can calm down or the provider can ask for assistance from colleagues.
Maintaining empathy by understanding grief
Discontinuing opioids may trigger in a patient an emotional response similar to grief. When considered in this framework, it may empower an otherwise frustrated provider to remain empathetic even in the midst of a difficult confrontation. Paralleling Kübler-Ross’s five stages of grief,45 we propose a similar model we call the “five stages of opioid loss”; this model has been successfully used in the residency continuity clinic at the University of Connecticut as a training aid.
Hopelessness and helplessness. During the first stage of the discussion the patient struggles with how to move forward. This conversation is frequently characterized by tearfulness and explanations to account for aberrant behavior or willingness to continue to suffer side effects. Active listening, empathy, and a focus on the factors that led to discontinuation of opioids while still validating pain are important.
Demanding and indignant. During the second stage, patients frequently push the limits of “no.” Accusations of abandonment and lack of empathy may accompany this stage and can be quite upsetting for the unprepared provider. A novice clinician can use role-play as a tool to better prepare for this type of encounter. Patients should be allowed to express their frustration but ultimatums and threats of violence should not be tolerated. Reassuring patients that their pain will be addressed using nonopioid therapy can be helpful, and a simple offer of continued care can help to preserve the therapeutic relationship.
Bargaining, the third stage of this model, is characterized by attempts to negotiate continued prescribing. While it can be frustrating, this push and pull is the beginning of real conversation and identification of a treatment plan for the future.
Resignation. The fourth stage begins when the patient has resigned himself or herself to your decision, but may not have accepted the available treatment options. At this point the patient may return for care or seek out a new provider. Empathy is again the element most crucial to success; this stage carries an opportunity to develop mutual respect.
Acceptance. The patients who choose to continue care with you have progressed to the final phase. They begin to look toward the future, having chosen the better of the two paths: partnering with a caring provider to develop a shared therapeutic plan.
A CONSISTENT AND TRANSPARENT APPROACH
Opioids can be useful for selected patients when they are carefully prescribed, but the prescriber must fully consider the risks and benefits specific to each patient and mitigate risk whenever possible.
Collaborating with patients to use opioids rationally is easier when it is part of a multimodal pain management plan and is initiated with clear functional goals and parameters for discontinuation. Presenting risks and benefits in a framework of safety and educating patients will help to reduce the stigma that may otherwise accompany safety monitoring using tools such as controlled substance agreements and urine toxicology testing.
Despite these efforts, patients may become psychologically dependent on opioids and discontinuation may prove difficult. However, a consistent and transparent approach to prescribing with special efforts to empathize with suffering patients may empower providers to navigate this process effectively.
- Institute of Medicine of the National Academies. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. http://iom.nationalacademies.org/reports/2011/relieving-pain-in-america-a-blueprint-for-transforming-prevention-care-education-research.aspx. Accessed February 8, 2016.
- McCarberg BH, Nicholson BD, Todd KH, Palmer T, Penles L. The impact of pain on quality of life and the unmet needs of pain management: results from pain sufferers and physicians participating in an Internet survey. Am J Ther 2008; 15:312–320.
- Roehr B. US needs new strategy to help 116 million patients in chronic pain. BMJ 2011; 343:d4206.
- Breuer B, Pappagallo M, Tai JY, Portenoy RK. US board-certified pain physician practices: uniformity and census data of their locations. J Pain 2007; 8:244–250.
- Paulozzi L, Dellinger A, Degutis L. Lessons from the past. Inj Prev 2012; 18:70.
- US Department of Health and Human Services; Substance Abuse and Mental Health Services Administration. Results from the 2013 national survey on drug use and health: Summary of national findings, NSDUH series H-48, HHS publication no. (SMA) 14-4863. www.samhsa.gov/data/sites/default/files/NSDUHresultsPDFWHTML2013/Web/NSDUHresults2013.htm. Accessed February 8, 2016.
- Bronstein K, Passik S, Munitz L, Leider H. Can clinicians accurately predict which patients are misusing their medications? J Pain 2011; 12(suppl):P3.
- Von Korff M, Kolodny A, Deyo RA, Chou R. Long-term opioid therapy reconsidered. Ann Intern Med 2011; 155:325–328.
- Chou R, Turner JA, Devine EB, et al. The effectiveness and risks of long-term opioid therapy for chronic pain: a systematic review for a National Institutes of Health Pathways to Prevention Workshop. Ann Intern Med 2015; 162:276–286.
- Chou R, Fanciullo GJ, Fine PG, et al; American Pain Society-American Academy of Pain Medicine Opioids Guidelines Panel. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain 2009; 10:113–130.
- Butler SF, Budman SH, Fernandez K, Jamison RN. Validation of a screener and opioid assessment measure for patients with chronic pain. Pain 2004; 112:65–75.
- Compton PA, Wu SM, Schieffer B, Pham Q, Naliboff BD. Introduction of a self-report version of the prescription drug use questionnaire and relationship to medication agreement noncompliance. J Pain Symptom Manage 2008; 36:383–395.
- Webster LR, Webster RM. Predicting aberrant behaviors in opioid-treated patients: preliminary validation of the opioid risk tool. Pain Med 2005; 6:432–442.
- Chen JT, Fagan MJ, Diaz JA, Reinert SE. Is treating chronic pain torture? Internal medicine residents’ experience with patients with chronic nonmalignant pain. Teach Learn Med 2007; 19:101–105.
- Gallagher RM. Empathy: a timeless skill for the pain medicine toolbox. Pain Med 2006; 7:213–214.
- Woolf CJ; American College of Physicians; American Physiological Society. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med 2004; 140:441–451.
- Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med 2005; 6:107–112.
- Medscape. A guide to state opioid prescribing policies resource center news. www.medscape.com/index/list_5657_1. Accessed February 8, 2016.
- Penko J, Mattson J, Miaskowski C, Kushel M. Do patients know they are on pain medication agreements? Results from a sample of high-risk patients on chronic opioid therapy. Pain Med 2012; 13:1174–1180.
- McGee S, Silverman RD. Treatment agreements, informed consent, and the role of state medical boards in opioid prescribing. Pain Med 2015; 16:25–29.
- Solomon DH, Rassen JA, Glynn RJ, Lee J, Levin R, Schneeweiss S. The comparative safety of analgesics in older adults with arthritis. Arch Intern Med 2010; 170:1968–1976.
- Wang D, Teichtahl H. Opioids, sleep architecture and sleep-disordered breathing. Sleep Med Rev 2007; 11:35–46.
- Bohnert AS, Valenstein M, Bair MJ, et al. Association between opioid prescribing patterns and opioid overdose-related deaths. JAMA 2011; 305:1315–1321.
- Smith HS. Variations in opioid responsiveness. Pain Physician 2008; 11:237–248.
- Vargas-Schaffer G. Is the WHO analgesic ladder still valid? Twenty-four years of experience. Can Fam Physician 2010; 56:514-517.
- Inturrisi CE. Clinical pharmacology of opioids for pain. Clin J Pain 2002; 18(suppl 4):S3–S13.
- Krantz MJ, Martin J, Stimmel B, Mehta D, Haigney MC. QTc interval screening in methadone treatment. Ann Intern Med 2009; 150:387–395.
- 107th Congress Public Law 77. US Government Printing Office. Departments of Commerce, Justice, and State, the Judiciary, and Related Agencies Appropriations Act, 2002. https://www.gpo.gov/fdsys/pkg/PLAW-107publ77/html/PLAW-107publ77.htm. Accessed February 8, 2016.
- Missouri Prescription Drug Monitoring Program NOW Coalition. http://mopdmpnow.org/. Accessed February 8, 2016.
- Prescription Drug Monitoring Program Center of Excellence at Brandeis. www.pdmpexcellence.org/sites/all/pdfs/Briefing%20on%20PDMP%20Effectiveness%203rd%20revision.pdf. Accessed February 8, 2016.
- Centers for Medicare and Medicaid Services. Tamper Resistant Prescriptions. www.cms.gov/Medicare-Medicaid-Coordination/Fraud-Prevention/FraudAbuseforProfs/TRP.html. Accessed February 8, 2016.
- Moorman-Li R, Motycka CA, Inge LD, Congdon JM, Hobson S, Pokropski B. A review of abuse-deterrent opioids for chronic nonmalignant pain. P T 2012; 37:412–418.
- Starrels JL, Becker WC, Weiner MG, Li X, Heo M, Turner BJ. Low use of opioid risk reduction strategies in primary care even for high risk patients with chronic pain. J Gen Intern Med 2011; 26:958–964.
- Herring C, Muzyk AJ, Johnston C. Interferences with urine drug screens. J Pharm Pract 2011; 24:102–108.
- Thermo Fisher Scientific. Cedia opiate 2K drugs of abuse assays. http://www.thermoscientific.com/en/product/cedia-opiate-2k-drugs-abuse-assays.html. Accessed February 8, 2016.
- Markway EC, Baker SN. A review of the methods, interpretation, and limitations of the urine drug screen. Orthopedics 2011; 34:877–881.
- Saitman A, Park HD, Fitzgerald RL. False-positive interferences of common urine drug screen immunoassays: a review. J Anal Toxicol 2014; 38:387–396.
- Standridge JB, Adams SM, Zotos AP. Urine drug screening: a valuable office procedure. Am Fam Physician 2010; 81:635–640.
- National Highway Traffic Safety Administration. Drugs and human performance fact sheet. www.nhtsa.gov/staticfiles/nti/pdf/809725-DrugsHumanPerformFS.pdf. Accessed February 8, 2016.
- US Department of Health and Human Services; Centers for Medicare and Medicaid Services. Partners in Integrity: What is the Prescriber's Role in Preventing the Diversion of Prescription Drugs. www.cms.gov/Medicare-Medicaid-Coordination/Fraud-Prevention/Medicaid-Integrity-Education/Provider-Education-Toolkits/Downloads/prescriber-role-drugdiversion.pdf. Accessed February 8, 2016.
- Silverman SM. Opioid induced hyperalgesia: clinical implications for the pain practitioner. Pain Physician 2009; 12:679–684.
- US Department of Health and Human Services; Agency for Healthcare Research and Quality (AHRQ); National Guideline Clearinghouse. Interagency guideline on opioid dosing for chronic non-cancer pain: an educational aid to improve care and safety with opioid therapy. www.guideline.gov/content.aspx?id=23792. Accessed February 8, 2016.
- Department of Veterans Affairs/Department of Defense. Tapering and discontinuing opioids factsheet. www.healthquality.va.gov/guidelines/Pain/cot/OpioidTaperingFactSheet23May2013v1.pdf. Accessed February 8, 2016.
- US Department of Justice Drug Enforcement Administration: Office of Diversion Control. Title 21 Code of Federal Regulations, Part 1306, Section 1306.04. Purpose of issue of prescription. www.deadiversion.usdoj.gov/21cfr/cfr/1306/1306_04.htm. Accessed February 8, 2016.
- Kübler-Ross E, Wessler S, Avioli LV. On death and dying. JAMA 1972; 221:174–179.
Chronic pain affects an estimated 100 million Americans, at a cost of $635 billion each year in medical expenses, lost wages, and reduced productivity.1 It is often managed in primary care settings with opioids by clinicians who have little or no formal training in pain management.2,3 Some primary care providers may seek assistance from board-certified pain specialists, but with only four such experts for every 100,000 patients with chronic pain, primary care providers are typically on their own.4
Although opioids may help in some chronic pain syndromes, they also carry the risk of serious harm, including unintentional overdose and death. In 2009, unintentional drug overdoses, most commonly with opioids, surpassed motor vehicle accidents as the leading cause of accidental death in the United States.5 Additionally, nonmedical use of prescription drugs is the third most common category of drug abuse, after marijuana and alcohol.6
Unfortunately, clinicians cannot accurately predict future medication misuse.7 And while the potential harms of opioids are many, the long-term benefits are questionable.8,9
For these reasons, providers need to understand the indications for and potential benefits of opioids, as well as the potential harms and how to monitor their safe use. Also important to know is how and when to discontinue opioids while preserving the therapeutic relationship.
This paper offers practical strategies to primary care providers and their care teams on how to safely initiate, monitor, and discontinue chronic opioid therapy.
STARTING OPIOID THERAPY FOR CHRONIC PAIN
Guidelines recommend considering starting patients on opioid therapy when the benefits are likely to outweigh the risks, when pain is moderate to severe, and when other multimodal treatment strategies have not achieved functional goals.10 Unfortunately, few studies have examined or demonstrated long-term benefit, and those that did examine this outcome reported reduction of pain severity but did not assess functional improvement.9 Meanwhile, data are increasingly clear that long-term use increases the risk of harm, both acute (eg, overdose) and chronic (eg, osteoporosis), especially with high doses.
Tools have been developed to predict the risk of misuse,11–13 but few have been validated in primary care, where most opioids are prescribed. This limitation aside, consensus guidelines state that untreated substance use disorders, poorly controlled psychiatric disease, and erratic treatment adherence are contraindications to opioid therapy, at least until these other issues are treated.10
Faced with the benefit-harm conundrum, we recommend a generally conservative approach to opioid initiation. With long-term functional benefit questionable and toxicity relatively common, we are increasingly avoiding chronic opioid therapy in younger patients with chronic pain.
Empathize and partner with your patient
Chronic pain care can be fraught with frustration and mutual distrust between patient and provider.14 Empathy and a collaborative stance help signal to the patient that the provider has the patient’s best interest in mind,15 whether initiating or deciding not to initiate opioids.
Optimize nonopioid therapy
In light of the risks associated with chronic opioid therapy, the clinician is urged to review and optimize nonopioid therapy before starting a patient on opioid treatment, and to maintain this approach if opioid therapy is started. Whenever possible, nonopioid treatment should include disease-modifying therapy and nondrug modalities such as physical therapy.
Judicious use of adequately dosed analgesics such as acetaminophen and nonsteroidal anti-inflammatory drugs may be sufficient to achieve analgesic goals if not contraindicated, and in some patients the addition of a topical analgesic (eg, diclofenac gel, lidocaine patches), a tricyclic or serotonin-norepinephrine reuptake inhibitor antidepressant, an anticonvulsant (eg, gabapentin), or a combination of the above can effectively address underlying pain-generating mechanisms.16 As with opioids, the risks and benefits of nonopioid pharmacotherapy should be reviewed both at initiation and periodically thereafter.
Frame the opioid treatment plan as a ‘therapeutic trial’
Starting an opioid should be framed as a “therapeutic trial.” These drugs should be continued only if safe and effective, at the lowest effective dose, and as one component of a multimodal pain treatment plan. Concurrent use of nonpharmacologic therapies (eg, physical therapy, structured exercise, yoga, relaxation training, biofeedback, cognitive behavioral therapy) and rational pharmacotherapy while promoting patient self-care is the standard of pain management called for by the Institute of Medicine.1
Set functional goals
We recommend clearly defining functional goals with each patient before starting therapy. These goals should be written into the treatment plan as a way for patient and provider to evaluate the effectiveness of chronic opioid therapy. A useful mnemonic to help identify such goals is SMART, an acronym for specific, measurable, action-oriented, realistic, and time-bound. Specific goals will depend on pain severity, but examples could include being able to do grocery shopping without assistance, to play on the floor with grandchildren, or to engage in healthy exercise habits such as 20 minutes of moderately brisk walking 3 days per week.
Obtain informed consent, and document it thoroughly
Providers must communicate risks, potential benefits, and safe medication-taking practices, including how to safely store and dispose of unused opioids, and document this conversation clearly in the medical record. From a medicolegal perspective, if it wasn’t documented, it did not happen.17
Informed consent can be further advanced with the use of a controlled substance agreement that outlines the treatment plan as well as potential risks, benefits, and practice policies in a structured way. Most states now either recommend or mandate the use of such agreements.18
Controlled substance agreements give providers a greater sense of mastery and comfort when prescribing opioids,19 but they have important limitations. In particular, there is a lack of consensus on what the agreement should say and relatively weak evidence that these agreements are efficacious. Additionally, a poorly written agreement can be stigmatizing and can erode trust.20 However, we believe that when the agreement is written in an appropriate framework of safety at an appropriate level of health literacy and with a focus on shared decision-making, it can be very helpful and should be used.
Employ safe, rational pharmacotherapy
Considerations when choosing an opioid include its potency, onset of action, and half-life. Comorbid conditions (eg, advanced age,21 sleep-disordered breathing22) and concurrent medications (eg, benzodiazepines, anticonvulsants, muscle relaxants) also affect decisions about the formulation, starting dose, rapidity of titration, and ceiling dose. Risk of harm increases in patients with such comorbid factors, and it is prudent to start with lower doses of shorter-acting medications until patients can demonstrate safe use. Risk of unintentional overdose is higher with higher prescribed doses.23 Pharmacologically there is no analgesic dose ceiling, but we urge caution, particularly in opioid-naive patients.
A patient’s response to any particular opioid is idiosyncratic and variable. There are more than 100 known polymorphisms in the human opioid mu-receptor gene, and thus differences in receptor affinity and activation as well as in metabolism make it difficult to predict which opioid will work best for a particular patient.24 However, a less potent opioid receptor agonist with less addictive potential, such as tramadol or codeine, should generally be tried first before escalating to a riskier, more potent opioid such as hydrocodone, oxycodone, or morphine. This “analgesic ladder,” a concept introduced by the World Health Organization in 1986 to provide a framework for managing cancer pain, has been adapted to a variety of chronic pain syndromes.25
Methadone deserves special mention. A strongly lipophilic molecule with a long and variable half-life, it accumulates in fat,26 and long after the analgesic effect has worn off, methadone will still be present. Repeated dosing or rapid dose escalation in an attempt to achieve adequate analgesia may result in inadvertent overdose. Additionally, methadone can prolong the QT interval, and periodic electrocardiographic monitoring is recommended.27 For these reasons, we recommend avoiding the use of methadone in most cases unless the provider has significant experience, expertise, or support in the safe use of this medication.
Table 1 summarizes these recommendations.
MONITORING AND SAFETY
Providers must periodically reassess the safety and efficacy of chronic opioid therapy to be sure that it is still indicated.10 Since we cannot accurately predict which patients will suffer adverse reactions or demonstrate aberrant behaviors,7 it is important to be transparent and consistent with monitoring practices for all patients on chronic opioid therapy.17 By framing monitoring in terms of safety and employing it universally, providers can minimize miscommunication and accidental stigmatization.
Prescription monitoring programs
In 2002, Congress appropriated funding to the US Department of Justice to support prescription monitoring programs nationally.28 At the time of this writing, Missouri is the only state without an approved monitoring program.29
Although the design and function of the programs vary from state to state, they require pharmacies to collect and report data on controlled substances for individual patients and prescribers. These data are sometimes shared across state lines, and the programs enhance the capacity of regulatory and law enforcement agencies to analyze controlled substance use.
Prescribers can (and are sometimes required to) register for access in their state and use this resource to assess the opioid refill history of their patients. This powerful tool improves detection of “doctor-shopping” and other common scams.30
Additionally, recognizing that the risk of death from overdose increases as the total daily dose of opioids increases,23 some states provide data on their composite report expressing the morphine equivalent daily dose or daily morphine milligram equivalents of the opioids prescribed. This information is valuable to the busy clinician; at a glance the prescriber can quickly discern the total daily opioid dose and use that information to assess risk and manage change. Furthermore, some states restrict further dose escalation when the morphine equivalent daily dose exceeds a predetermined amount (typically 100 to 120 morphine milligram equivalents).
Tamper-resistant prescribing
To minimize the risk of prescription tampering, simple techniques such as writing out the number of tablets dispensed can help, and use of tamper-resistant prescription paper has been required for Medicaid recipients since 2008.31
When possible, we recommend products with abuse-deterrent properties. Although the science of abuse deterrence is relatively new and few products are labeled as such, a number of opioids are formulated to resist deformation, vaporization, dissolving, or other physical tampering. Additionally, some abuse-deterrent opioid formulations contain naloxone, which is released only when the drug is deformed in some way, thereby decreasing the user’s response to an abused substance or resulting in opioid withdrawal.32
Urine drug testing
Although complex and nuanced, guidelines recommend urine drug testing to confirm the presence or absence of prescribed and illicit substances in the body.10 There is no consensus on when or how often to test, but it should be done randomly and without forewarning to foil efforts to defeat testing such as provision of synthetic, adulterated, or substituted urine.
Providers underuse urine drug testing.33 We recommend that it be done at the start of opioid therapy, sporadically thereafter, when therapy is changed, and whenever the provider is concerned about possible aberrant drug use.
Understanding opioid metabolism, cross-reactivity, and the types of tests available will help avoid misinterpretation of results.34 For example, a positive “opiate” result in most screening immunoassay tests does not reflect oxycodone use, since tests for synthetic opioids often need to be ordered separately; the commonly used Cedia opiate assay cross-reacts with oxycodone at a concentration of 10,000 ng/mL only 3.1% of the time.35 Immunoassay screening tests are widely available, sensitive, inexpensive, and fast, but they are qualitative, have limited specificity, and are subject to false-positive and false-negative results.36 Table 2 outlines some common characteristics of substances on screening immunoassays, including reported causes of false-positive results.37–39
Confirmatory testing using gas chromatography or mass spectroscopy is more expensive and slower to process, but is highly sensitive and specific, quantitative, and useful when screening results are difficult to interpret.
Knowing how and when to order the right urine drug test and knowing how to interpret the results are skills prescribers should master.
DISCONTINUING OPIOIDS
When opioids are no longer safe or effective, they should be stopped. The decision can be difficult for both the patient and provider, and a certain degree of equanimity is needed to approach it rationally.
Strong indications for discontinuation
Respiratory depression, cognitive impairment, falls, and motor vehicle accidents mean harm is already apparent. At a minimum, dose reduction is warranted and discontinuation should be strongly considered. Similarly, overdose (intentional or accidental) and active suicidal ideation contraindicate ongoing opioid prescribing unless the ongoing risk can be decisively mitigated.
Certain aberrant behaviors such as prescription forgery or theft, threats of violence to obtain analgesics, and diversion (transfer of the drug to another person for nonmedical use) also warrant immediate discontinuation. Continuing to prescribe an opioid while knowing diversion is taking place may be a violation of federal or state law or both.40
Another reason to stop is failure to achieve the expected benefit from chronic opioid therapy (ie, agreed-upon functional goals) despite appropriate dose adjustment. In these cases, ongoing risk by definition outweighs observed benefit.
Relative indications for discontinuation
Opioid therapy has many potential adverse effects. Depending on the severity and duration of the symptom and its response to either dose reduction or adjunctive management, opioids may need to be discontinued.
For example, pruritus, constipation, urinary retention, nausea, sedation, and sexual dysfunction may all be reasons to stop chronic opioid therapy. Similarly, chronic opioid therapy may paradoxically worsen pain in some susceptible patients, a complication known as opioid-induced hyperalgesia; in these cases, tapering off opioids should be considered as well.41 Aberrant behaviors should prompt reconsideration of chronic opioid therapy; these include hazardous alcohol consumption, use of illicit drugs, pill hoarding, and use of opioids in a manner different than intended by the prescriber.
Another relative indication for discontinuation is receipt of controlled substances from other providers. A well-written controlled substance agreement and adequate counseling may help mitigate this risk; poor communication between providers, lack of integration of electronic medical record systems, urgent or emergency room care, and poor health literacy may all lead to redundant prescribing in some circumstances. While unintentional use of controlled substances from different providers is no less dangerous than intentional misuse, the specifics of each case need to be considered before opioids are reflexively discontinued.
How to discontinue opioids
In most cases, opioids should be tapered to reduce the risk and severity of withdrawal symptoms. Decreasing the dose by 10% of the original dose per week is usually well tolerated with minimal adverse effects.42 Tapering can be done much faster, and numerous rapid detoxification protocols are available. In general, a patient needs 20% of the previous day’s dose to prevent withdrawal symptoms.43
Withdrawal symptoms are rarely life-threatening but can be very uncomfortable. Some providers add clonidine to attenuate associated autonomic symptoms such as hypertension, nausea, cramps, diaphoresis, and tachycardia if they occur. Other adjunctive medications include nonsteroidal anti-inflammatory drugs for body aches, antiemetics for nausea and vomiting, bismuth subsalicylate for diarrhea, and trazodone for insomnia.
It is unlawful for primary care physicians to use another opioid to treat symptoms of withdrawal in the outpatient setting unless it is issued through a federally certified narcotic treatment program or prescribed by a qualified clinician registered with the US Drug Enforcement Administration to prescribe buprenorphine-naloxone.44
In some circumstances, it may be appropriate to abruptly discontinue opioids without a taper, such as when diversion is evident. However, a decision to discontinue opioids due to misuse should not equate to an automatic decision to terminate a patient from the practice. Instead, providers should use this opportunity to offer empathy and referral to drug treatment counseling and rehabilitation. A decision to discontinue opioids because they are no longer safe or effective does not mean that the patient’s pain is not real—it is “real” for them, even if caused by the pain of addiction—or that shared decision-making is no longer possible or appropriate.
Handling difficult conversations when discontinuing opioids
The conversation between patient and provider when discontinuing opioids can be difficult. Misaligned expectations of both parties, patient fear of uncontrolled pain, and provider concern about causing suffering are frequent contributing factors. Patients abusing prescription drugs may also have a stronger relationship with their medication than with their provider and may use manipulative strategies including overt hostility and threats to obtain a prescription. Providers need to maintain their composure to de-escalate these potentially upsetting confrontations.
Table 3 outlines some specific suggestions that may be helpful, including the following:
- Frame the discussion in terms of safety—opioids are being discontinued because the benefit no longer outweighs the risk
- Don’t debate your decision with the patient, but present your reasoning in a considered manner
- Focus on the appropriateness of the treatment and not on the patient’s character
- Avoid the use of labels (eg, “drug addict”)
- Emphasize your commitment to the patient’s well-being and an alternative treatment plan (ie, nonabandonment)
- Respond to emotional distress with empathy, but do not let that change your decision to discontinue opioids.
Finally, we strongly encourage providers to insist on being treated respectfully. When safety cannot be ensured, providers should remove themselves from the room until the patient can calm down or the provider can ask for assistance from colleagues.
Maintaining empathy by understanding grief
Discontinuing opioids may trigger in a patient an emotional response similar to grief. When considered in this framework, it may empower an otherwise frustrated provider to remain empathetic even in the midst of a difficult confrontation. Paralleling Kübler-Ross’s five stages of grief,45 we propose a similar model we call the “five stages of opioid loss”; this model has been successfully used in the residency continuity clinic at the University of Connecticut as a training aid.
Hopelessness and helplessness. During the first stage of the discussion the patient struggles with how to move forward. This conversation is frequently characterized by tearfulness and explanations to account for aberrant behavior or willingness to continue to suffer side effects. Active listening, empathy, and a focus on the factors that led to discontinuation of opioids while still validating pain are important.
Demanding and indignant. During the second stage, patients frequently push the limits of “no.” Accusations of abandonment and lack of empathy may accompany this stage and can be quite upsetting for the unprepared provider. A novice clinician can use role-play as a tool to better prepare for this type of encounter. Patients should be allowed to express their frustration but ultimatums and threats of violence should not be tolerated. Reassuring patients that their pain will be addressed using nonopioid therapy can be helpful, and a simple offer of continued care can help to preserve the therapeutic relationship.
Bargaining, the third stage of this model, is characterized by attempts to negotiate continued prescribing. While it can be frustrating, this push and pull is the beginning of real conversation and identification of a treatment plan for the future.
Resignation. The fourth stage begins when the patient has resigned himself or herself to your decision, but may not have accepted the available treatment options. At this point the patient may return for care or seek out a new provider. Empathy is again the element most crucial to success; this stage carries an opportunity to develop mutual respect.
Acceptance. The patients who choose to continue care with you have progressed to the final phase. They begin to look toward the future, having chosen the better of the two paths: partnering with a caring provider to develop a shared therapeutic plan.
A CONSISTENT AND TRANSPARENT APPROACH
Opioids can be useful for selected patients when they are carefully prescribed, but the prescriber must fully consider the risks and benefits specific to each patient and mitigate risk whenever possible.
Collaborating with patients to use opioids rationally is easier when it is part of a multimodal pain management plan and is initiated with clear functional goals and parameters for discontinuation. Presenting risks and benefits in a framework of safety and educating patients will help to reduce the stigma that may otherwise accompany safety monitoring using tools such as controlled substance agreements and urine toxicology testing.
Despite these efforts, patients may become psychologically dependent on opioids and discontinuation may prove difficult. However, a consistent and transparent approach to prescribing with special efforts to empathize with suffering patients may empower providers to navigate this process effectively.
Chronic pain affects an estimated 100 million Americans, at a cost of $635 billion each year in medical expenses, lost wages, and reduced productivity.1 It is often managed in primary care settings with opioids by clinicians who have little or no formal training in pain management.2,3 Some primary care providers may seek assistance from board-certified pain specialists, but with only four such experts for every 100,000 patients with chronic pain, primary care providers are typically on their own.4
Although opioids may help in some chronic pain syndromes, they also carry the risk of serious harm, including unintentional overdose and death. In 2009, unintentional drug overdoses, most commonly with opioids, surpassed motor vehicle accidents as the leading cause of accidental death in the United States.5 Additionally, nonmedical use of prescription drugs is the third most common category of drug abuse, after marijuana and alcohol.6
Unfortunately, clinicians cannot accurately predict future medication misuse.7 And while the potential harms of opioids are many, the long-term benefits are questionable.8,9
For these reasons, providers need to understand the indications for and potential benefits of opioids, as well as the potential harms and how to monitor their safe use. Also important to know is how and when to discontinue opioids while preserving the therapeutic relationship.
This paper offers practical strategies to primary care providers and their care teams on how to safely initiate, monitor, and discontinue chronic opioid therapy.
STARTING OPIOID THERAPY FOR CHRONIC PAIN
Guidelines recommend considering starting patients on opioid therapy when the benefits are likely to outweigh the risks, when pain is moderate to severe, and when other multimodal treatment strategies have not achieved functional goals.10 Unfortunately, few studies have examined or demonstrated long-term benefit, and those that did examine this outcome reported reduction of pain severity but did not assess functional improvement.9 Meanwhile, data are increasingly clear that long-term use increases the risk of harm, both acute (eg, overdose) and chronic (eg, osteoporosis), especially with high doses.
Tools have been developed to predict the risk of misuse,11–13 but few have been validated in primary care, where most opioids are prescribed. This limitation aside, consensus guidelines state that untreated substance use disorders, poorly controlled psychiatric disease, and erratic treatment adherence are contraindications to opioid therapy, at least until these other issues are treated.10
Faced with the benefit-harm conundrum, we recommend a generally conservative approach to opioid initiation. With long-term functional benefit questionable and toxicity relatively common, we are increasingly avoiding chronic opioid therapy in younger patients with chronic pain.
Empathize and partner with your patient
Chronic pain care can be fraught with frustration and mutual distrust between patient and provider.14 Empathy and a collaborative stance help signal to the patient that the provider has the patient’s best interest in mind,15 whether initiating or deciding not to initiate opioids.
Optimize nonopioid therapy
In light of the risks associated with chronic opioid therapy, the clinician is urged to review and optimize nonopioid therapy before starting a patient on opioid treatment, and to maintain this approach if opioid therapy is started. Whenever possible, nonopioid treatment should include disease-modifying therapy and nondrug modalities such as physical therapy.
Judicious use of adequately dosed analgesics such as acetaminophen and nonsteroidal anti-inflammatory drugs may be sufficient to achieve analgesic goals if not contraindicated, and in some patients the addition of a topical analgesic (eg, diclofenac gel, lidocaine patches), a tricyclic or serotonin-norepinephrine reuptake inhibitor antidepressant, an anticonvulsant (eg, gabapentin), or a combination of the above can effectively address underlying pain-generating mechanisms.16 As with opioids, the risks and benefits of nonopioid pharmacotherapy should be reviewed both at initiation and periodically thereafter.
Frame the opioid treatment plan as a ‘therapeutic trial’
Starting an opioid should be framed as a “therapeutic trial.” These drugs should be continued only if safe and effective, at the lowest effective dose, and as one component of a multimodal pain treatment plan. Concurrent use of nonpharmacologic therapies (eg, physical therapy, structured exercise, yoga, relaxation training, biofeedback, cognitive behavioral therapy) and rational pharmacotherapy while promoting patient self-care is the standard of pain management called for by the Institute of Medicine.1
Set functional goals
We recommend clearly defining functional goals with each patient before starting therapy. These goals should be written into the treatment plan as a way for patient and provider to evaluate the effectiveness of chronic opioid therapy. A useful mnemonic to help identify such goals is SMART, an acronym for specific, measurable, action-oriented, realistic, and time-bound. Specific goals will depend on pain severity, but examples could include being able to do grocery shopping without assistance, to play on the floor with grandchildren, or to engage in healthy exercise habits such as 20 minutes of moderately brisk walking 3 days per week.
Obtain informed consent, and document it thoroughly
Providers must communicate risks, potential benefits, and safe medication-taking practices, including how to safely store and dispose of unused opioids, and document this conversation clearly in the medical record. From a medicolegal perspective, if it wasn’t documented, it did not happen.17
Informed consent can be further advanced with the use of a controlled substance agreement that outlines the treatment plan as well as potential risks, benefits, and practice policies in a structured way. Most states now either recommend or mandate the use of such agreements.18
Controlled substance agreements give providers a greater sense of mastery and comfort when prescribing opioids,19 but they have important limitations. In particular, there is a lack of consensus on what the agreement should say and relatively weak evidence that these agreements are efficacious. Additionally, a poorly written agreement can be stigmatizing and can erode trust.20 However, we believe that when the agreement is written in an appropriate framework of safety at an appropriate level of health literacy and with a focus on shared decision-making, it can be very helpful and should be used.
Employ safe, rational pharmacotherapy
Considerations when choosing an opioid include its potency, onset of action, and half-life. Comorbid conditions (eg, advanced age,21 sleep-disordered breathing22) and concurrent medications (eg, benzodiazepines, anticonvulsants, muscle relaxants) also affect decisions about the formulation, starting dose, rapidity of titration, and ceiling dose. Risk of harm increases in patients with such comorbid factors, and it is prudent to start with lower doses of shorter-acting medications until patients can demonstrate safe use. Risk of unintentional overdose is higher with higher prescribed doses.23 Pharmacologically there is no analgesic dose ceiling, but we urge caution, particularly in opioid-naive patients.
A patient’s response to any particular opioid is idiosyncratic and variable. There are more than 100 known polymorphisms in the human opioid mu-receptor gene, and thus differences in receptor affinity and activation as well as in metabolism make it difficult to predict which opioid will work best for a particular patient.24 However, a less potent opioid receptor agonist with less addictive potential, such as tramadol or codeine, should generally be tried first before escalating to a riskier, more potent opioid such as hydrocodone, oxycodone, or morphine. This “analgesic ladder,” a concept introduced by the World Health Organization in 1986 to provide a framework for managing cancer pain, has been adapted to a variety of chronic pain syndromes.25
Methadone deserves special mention. A strongly lipophilic molecule with a long and variable half-life, it accumulates in fat,26 and long after the analgesic effect has worn off, methadone will still be present. Repeated dosing or rapid dose escalation in an attempt to achieve adequate analgesia may result in inadvertent overdose. Additionally, methadone can prolong the QT interval, and periodic electrocardiographic monitoring is recommended.27 For these reasons, we recommend avoiding the use of methadone in most cases unless the provider has significant experience, expertise, or support in the safe use of this medication.
Table 1 summarizes these recommendations.
MONITORING AND SAFETY
Providers must periodically reassess the safety and efficacy of chronic opioid therapy to be sure that it is still indicated.10 Since we cannot accurately predict which patients will suffer adverse reactions or demonstrate aberrant behaviors,7 it is important to be transparent and consistent with monitoring practices for all patients on chronic opioid therapy.17 By framing monitoring in terms of safety and employing it universally, providers can minimize miscommunication and accidental stigmatization.
Prescription monitoring programs
In 2002, Congress appropriated funding to the US Department of Justice to support prescription monitoring programs nationally.28 At the time of this writing, Missouri is the only state without an approved monitoring program.29
Although the design and function of the programs vary from state to state, they require pharmacies to collect and report data on controlled substances for individual patients and prescribers. These data are sometimes shared across state lines, and the programs enhance the capacity of regulatory and law enforcement agencies to analyze controlled substance use.
Prescribers can (and are sometimes required to) register for access in their state and use this resource to assess the opioid refill history of their patients. This powerful tool improves detection of “doctor-shopping” and other common scams.30
Additionally, recognizing that the risk of death from overdose increases as the total daily dose of opioids increases,23 some states provide data on their composite report expressing the morphine equivalent daily dose or daily morphine milligram equivalents of the opioids prescribed. This information is valuable to the busy clinician; at a glance the prescriber can quickly discern the total daily opioid dose and use that information to assess risk and manage change. Furthermore, some states restrict further dose escalation when the morphine equivalent daily dose exceeds a predetermined amount (typically 100 to 120 morphine milligram equivalents).
Tamper-resistant prescribing
To minimize the risk of prescription tampering, simple techniques such as writing out the number of tablets dispensed can help, and use of tamper-resistant prescription paper has been required for Medicaid recipients since 2008.31
When possible, we recommend products with abuse-deterrent properties. Although the science of abuse deterrence is relatively new and few products are labeled as such, a number of opioids are formulated to resist deformation, vaporization, dissolving, or other physical tampering. Additionally, some abuse-deterrent opioid formulations contain naloxone, which is released only when the drug is deformed in some way, thereby decreasing the user’s response to an abused substance or resulting in opioid withdrawal.32
Urine drug testing
Although complex and nuanced, guidelines recommend urine drug testing to confirm the presence or absence of prescribed and illicit substances in the body.10 There is no consensus on when or how often to test, but it should be done randomly and without forewarning to foil efforts to defeat testing such as provision of synthetic, adulterated, or substituted urine.
Providers underuse urine drug testing.33 We recommend that it be done at the start of opioid therapy, sporadically thereafter, when therapy is changed, and whenever the provider is concerned about possible aberrant drug use.
Understanding opioid metabolism, cross-reactivity, and the types of tests available will help avoid misinterpretation of results.34 For example, a positive “opiate” result in most screening immunoassay tests does not reflect oxycodone use, since tests for synthetic opioids often need to be ordered separately; the commonly used Cedia opiate assay cross-reacts with oxycodone at a concentration of 10,000 ng/mL only 3.1% of the time.35 Immunoassay screening tests are widely available, sensitive, inexpensive, and fast, but they are qualitative, have limited specificity, and are subject to false-positive and false-negative results.36 Table 2 outlines some common characteristics of substances on screening immunoassays, including reported causes of false-positive results.37–39
Confirmatory testing using gas chromatography or mass spectroscopy is more expensive and slower to process, but is highly sensitive and specific, quantitative, and useful when screening results are difficult to interpret.
Knowing how and when to order the right urine drug test and knowing how to interpret the results are skills prescribers should master.
DISCONTINUING OPIOIDS
When opioids are no longer safe or effective, they should be stopped. The decision can be difficult for both the patient and provider, and a certain degree of equanimity is needed to approach it rationally.
Strong indications for discontinuation
Respiratory depression, cognitive impairment, falls, and motor vehicle accidents mean harm is already apparent. At a minimum, dose reduction is warranted and discontinuation should be strongly considered. Similarly, overdose (intentional or accidental) and active suicidal ideation contraindicate ongoing opioid prescribing unless the ongoing risk can be decisively mitigated.
Certain aberrant behaviors such as prescription forgery or theft, threats of violence to obtain analgesics, and diversion (transfer of the drug to another person for nonmedical use) also warrant immediate discontinuation. Continuing to prescribe an opioid while knowing diversion is taking place may be a violation of federal or state law or both.40
Another reason to stop is failure to achieve the expected benefit from chronic opioid therapy (ie, agreed-upon functional goals) despite appropriate dose adjustment. In these cases, ongoing risk by definition outweighs observed benefit.
Relative indications for discontinuation
Opioid therapy has many potential adverse effects. Depending on the severity and duration of the symptom and its response to either dose reduction or adjunctive management, opioids may need to be discontinued.
For example, pruritus, constipation, urinary retention, nausea, sedation, and sexual dysfunction may all be reasons to stop chronic opioid therapy. Similarly, chronic opioid therapy may paradoxically worsen pain in some susceptible patients, a complication known as opioid-induced hyperalgesia; in these cases, tapering off opioids should be considered as well.41 Aberrant behaviors should prompt reconsideration of chronic opioid therapy; these include hazardous alcohol consumption, use of illicit drugs, pill hoarding, and use of opioids in a manner different than intended by the prescriber.
Another relative indication for discontinuation is receipt of controlled substances from other providers. A well-written controlled substance agreement and adequate counseling may help mitigate this risk; poor communication between providers, lack of integration of electronic medical record systems, urgent or emergency room care, and poor health literacy may all lead to redundant prescribing in some circumstances. While unintentional use of controlled substances from different providers is no less dangerous than intentional misuse, the specifics of each case need to be considered before opioids are reflexively discontinued.
How to discontinue opioids
In most cases, opioids should be tapered to reduce the risk and severity of withdrawal symptoms. Decreasing the dose by 10% of the original dose per week is usually well tolerated with minimal adverse effects.42 Tapering can be done much faster, and numerous rapid detoxification protocols are available. In general, a patient needs 20% of the previous day’s dose to prevent withdrawal symptoms.43
Withdrawal symptoms are rarely life-threatening but can be very uncomfortable. Some providers add clonidine to attenuate associated autonomic symptoms such as hypertension, nausea, cramps, diaphoresis, and tachycardia if they occur. Other adjunctive medications include nonsteroidal anti-inflammatory drugs for body aches, antiemetics for nausea and vomiting, bismuth subsalicylate for diarrhea, and trazodone for insomnia.
It is unlawful for primary care physicians to use another opioid to treat symptoms of withdrawal in the outpatient setting unless it is issued through a federally certified narcotic treatment program or prescribed by a qualified clinician registered with the US Drug Enforcement Administration to prescribe buprenorphine-naloxone.44
In some circumstances, it may be appropriate to abruptly discontinue opioids without a taper, such as when diversion is evident. However, a decision to discontinue opioids due to misuse should not equate to an automatic decision to terminate a patient from the practice. Instead, providers should use this opportunity to offer empathy and referral to drug treatment counseling and rehabilitation. A decision to discontinue opioids because they are no longer safe or effective does not mean that the patient’s pain is not real—it is “real” for them, even if caused by the pain of addiction—or that shared decision-making is no longer possible or appropriate.
Handling difficult conversations when discontinuing opioids
The conversation between patient and provider when discontinuing opioids can be difficult. Misaligned expectations of both parties, patient fear of uncontrolled pain, and provider concern about causing suffering are frequent contributing factors. Patients abusing prescription drugs may also have a stronger relationship with their medication than with their provider and may use manipulative strategies including overt hostility and threats to obtain a prescription. Providers need to maintain their composure to de-escalate these potentially upsetting confrontations.
Table 3 outlines some specific suggestions that may be helpful, including the following:
- Frame the discussion in terms of safety—opioids are being discontinued because the benefit no longer outweighs the risk
- Don’t debate your decision with the patient, but present your reasoning in a considered manner
- Focus on the appropriateness of the treatment and not on the patient’s character
- Avoid the use of labels (eg, “drug addict”)
- Emphasize your commitment to the patient’s well-being and an alternative treatment plan (ie, nonabandonment)
- Respond to emotional distress with empathy, but do not let that change your decision to discontinue opioids.
Finally, we strongly encourage providers to insist on being treated respectfully. When safety cannot be ensured, providers should remove themselves from the room until the patient can calm down or the provider can ask for assistance from colleagues.
Maintaining empathy by understanding grief
Discontinuing opioids may trigger in a patient an emotional response similar to grief. When considered in this framework, it may empower an otherwise frustrated provider to remain empathetic even in the midst of a difficult confrontation. Paralleling Kübler-Ross’s five stages of grief,45 we propose a similar model we call the “five stages of opioid loss”; this model has been successfully used in the residency continuity clinic at the University of Connecticut as a training aid.
Hopelessness and helplessness. During the first stage of the discussion the patient struggles with how to move forward. This conversation is frequently characterized by tearfulness and explanations to account for aberrant behavior or willingness to continue to suffer side effects. Active listening, empathy, and a focus on the factors that led to discontinuation of opioids while still validating pain are important.
Demanding and indignant. During the second stage, patients frequently push the limits of “no.” Accusations of abandonment and lack of empathy may accompany this stage and can be quite upsetting for the unprepared provider. A novice clinician can use role-play as a tool to better prepare for this type of encounter. Patients should be allowed to express their frustration but ultimatums and threats of violence should not be tolerated. Reassuring patients that their pain will be addressed using nonopioid therapy can be helpful, and a simple offer of continued care can help to preserve the therapeutic relationship.
Bargaining, the third stage of this model, is characterized by attempts to negotiate continued prescribing. While it can be frustrating, this push and pull is the beginning of real conversation and identification of a treatment plan for the future.
Resignation. The fourth stage begins when the patient has resigned himself or herself to your decision, but may not have accepted the available treatment options. At this point the patient may return for care or seek out a new provider. Empathy is again the element most crucial to success; this stage carries an opportunity to develop mutual respect.
Acceptance. The patients who choose to continue care with you have progressed to the final phase. They begin to look toward the future, having chosen the better of the two paths: partnering with a caring provider to develop a shared therapeutic plan.
A CONSISTENT AND TRANSPARENT APPROACH
Opioids can be useful for selected patients when they are carefully prescribed, but the prescriber must fully consider the risks and benefits specific to each patient and mitigate risk whenever possible.
Collaborating with patients to use opioids rationally is easier when it is part of a multimodal pain management plan and is initiated with clear functional goals and parameters for discontinuation. Presenting risks and benefits in a framework of safety and educating patients will help to reduce the stigma that may otherwise accompany safety monitoring using tools such as controlled substance agreements and urine toxicology testing.
Despite these efforts, patients may become psychologically dependent on opioids and discontinuation may prove difficult. However, a consistent and transparent approach to prescribing with special efforts to empathize with suffering patients may empower providers to navigate this process effectively.
- Institute of Medicine of the National Academies. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. http://iom.nationalacademies.org/reports/2011/relieving-pain-in-america-a-blueprint-for-transforming-prevention-care-education-research.aspx. Accessed February 8, 2016.
- McCarberg BH, Nicholson BD, Todd KH, Palmer T, Penles L. The impact of pain on quality of life and the unmet needs of pain management: results from pain sufferers and physicians participating in an Internet survey. Am J Ther 2008; 15:312–320.
- Roehr B. US needs new strategy to help 116 million patients in chronic pain. BMJ 2011; 343:d4206.
- Breuer B, Pappagallo M, Tai JY, Portenoy RK. US board-certified pain physician practices: uniformity and census data of their locations. J Pain 2007; 8:244–250.
- Paulozzi L, Dellinger A, Degutis L. Lessons from the past. Inj Prev 2012; 18:70.
- US Department of Health and Human Services; Substance Abuse and Mental Health Services Administration. Results from the 2013 national survey on drug use and health: Summary of national findings, NSDUH series H-48, HHS publication no. (SMA) 14-4863. www.samhsa.gov/data/sites/default/files/NSDUHresultsPDFWHTML2013/Web/NSDUHresults2013.htm. Accessed February 8, 2016.
- Bronstein K, Passik S, Munitz L, Leider H. Can clinicians accurately predict which patients are misusing their medications? J Pain 2011; 12(suppl):P3.
- Von Korff M, Kolodny A, Deyo RA, Chou R. Long-term opioid therapy reconsidered. Ann Intern Med 2011; 155:325–328.
- Chou R, Turner JA, Devine EB, et al. The effectiveness and risks of long-term opioid therapy for chronic pain: a systematic review for a National Institutes of Health Pathways to Prevention Workshop. Ann Intern Med 2015; 162:276–286.
- Chou R, Fanciullo GJ, Fine PG, et al; American Pain Society-American Academy of Pain Medicine Opioids Guidelines Panel. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain 2009; 10:113–130.
- Butler SF, Budman SH, Fernandez K, Jamison RN. Validation of a screener and opioid assessment measure for patients with chronic pain. Pain 2004; 112:65–75.
- Compton PA, Wu SM, Schieffer B, Pham Q, Naliboff BD. Introduction of a self-report version of the prescription drug use questionnaire and relationship to medication agreement noncompliance. J Pain Symptom Manage 2008; 36:383–395.
- Webster LR, Webster RM. Predicting aberrant behaviors in opioid-treated patients: preliminary validation of the opioid risk tool. Pain Med 2005; 6:432–442.
- Chen JT, Fagan MJ, Diaz JA, Reinert SE. Is treating chronic pain torture? Internal medicine residents’ experience with patients with chronic nonmalignant pain. Teach Learn Med 2007; 19:101–105.
- Gallagher RM. Empathy: a timeless skill for the pain medicine toolbox. Pain Med 2006; 7:213–214.
- Woolf CJ; American College of Physicians; American Physiological Society. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med 2004; 140:441–451.
- Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med 2005; 6:107–112.
- Medscape. A guide to state opioid prescribing policies resource center news. www.medscape.com/index/list_5657_1. Accessed February 8, 2016.
- Penko J, Mattson J, Miaskowski C, Kushel M. Do patients know they are on pain medication agreements? Results from a sample of high-risk patients on chronic opioid therapy. Pain Med 2012; 13:1174–1180.
- McGee S, Silverman RD. Treatment agreements, informed consent, and the role of state medical boards in opioid prescribing. Pain Med 2015; 16:25–29.
- Solomon DH, Rassen JA, Glynn RJ, Lee J, Levin R, Schneeweiss S. The comparative safety of analgesics in older adults with arthritis. Arch Intern Med 2010; 170:1968–1976.
- Wang D, Teichtahl H. Opioids, sleep architecture and sleep-disordered breathing. Sleep Med Rev 2007; 11:35–46.
- Bohnert AS, Valenstein M, Bair MJ, et al. Association between opioid prescribing patterns and opioid overdose-related deaths. JAMA 2011; 305:1315–1321.
- Smith HS. Variations in opioid responsiveness. Pain Physician 2008; 11:237–248.
- Vargas-Schaffer G. Is the WHO analgesic ladder still valid? Twenty-four years of experience. Can Fam Physician 2010; 56:514-517.
- Inturrisi CE. Clinical pharmacology of opioids for pain. Clin J Pain 2002; 18(suppl 4):S3–S13.
- Krantz MJ, Martin J, Stimmel B, Mehta D, Haigney MC. QTc interval screening in methadone treatment. Ann Intern Med 2009; 150:387–395.
- 107th Congress Public Law 77. US Government Printing Office. Departments of Commerce, Justice, and State, the Judiciary, and Related Agencies Appropriations Act, 2002. https://www.gpo.gov/fdsys/pkg/PLAW-107publ77/html/PLAW-107publ77.htm. Accessed February 8, 2016.
- Missouri Prescription Drug Monitoring Program NOW Coalition. http://mopdmpnow.org/. Accessed February 8, 2016.
- Prescription Drug Monitoring Program Center of Excellence at Brandeis. www.pdmpexcellence.org/sites/all/pdfs/Briefing%20on%20PDMP%20Effectiveness%203rd%20revision.pdf. Accessed February 8, 2016.
- Centers for Medicare and Medicaid Services. Tamper Resistant Prescriptions. www.cms.gov/Medicare-Medicaid-Coordination/Fraud-Prevention/FraudAbuseforProfs/TRP.html. Accessed February 8, 2016.
- Moorman-Li R, Motycka CA, Inge LD, Congdon JM, Hobson S, Pokropski B. A review of abuse-deterrent opioids for chronic nonmalignant pain. P T 2012; 37:412–418.
- Starrels JL, Becker WC, Weiner MG, Li X, Heo M, Turner BJ. Low use of opioid risk reduction strategies in primary care even for high risk patients with chronic pain. J Gen Intern Med 2011; 26:958–964.
- Herring C, Muzyk AJ, Johnston C. Interferences with urine drug screens. J Pharm Pract 2011; 24:102–108.
- Thermo Fisher Scientific. Cedia opiate 2K drugs of abuse assays. http://www.thermoscientific.com/en/product/cedia-opiate-2k-drugs-abuse-assays.html. Accessed February 8, 2016.
- Markway EC, Baker SN. A review of the methods, interpretation, and limitations of the urine drug screen. Orthopedics 2011; 34:877–881.
- Saitman A, Park HD, Fitzgerald RL. False-positive interferences of common urine drug screen immunoassays: a review. J Anal Toxicol 2014; 38:387–396.
- Standridge JB, Adams SM, Zotos AP. Urine drug screening: a valuable office procedure. Am Fam Physician 2010; 81:635–640.
- National Highway Traffic Safety Administration. Drugs and human performance fact sheet. www.nhtsa.gov/staticfiles/nti/pdf/809725-DrugsHumanPerformFS.pdf. Accessed February 8, 2016.
- US Department of Health and Human Services; Centers for Medicare and Medicaid Services. Partners in Integrity: What is the Prescriber's Role in Preventing the Diversion of Prescription Drugs. www.cms.gov/Medicare-Medicaid-Coordination/Fraud-Prevention/Medicaid-Integrity-Education/Provider-Education-Toolkits/Downloads/prescriber-role-drugdiversion.pdf. Accessed February 8, 2016.
- Silverman SM. Opioid induced hyperalgesia: clinical implications for the pain practitioner. Pain Physician 2009; 12:679–684.
- US Department of Health and Human Services; Agency for Healthcare Research and Quality (AHRQ); National Guideline Clearinghouse. Interagency guideline on opioid dosing for chronic non-cancer pain: an educational aid to improve care and safety with opioid therapy. www.guideline.gov/content.aspx?id=23792. Accessed February 8, 2016.
- Department of Veterans Affairs/Department of Defense. Tapering and discontinuing opioids factsheet. www.healthquality.va.gov/guidelines/Pain/cot/OpioidTaperingFactSheet23May2013v1.pdf. Accessed February 8, 2016.
- US Department of Justice Drug Enforcement Administration: Office of Diversion Control. Title 21 Code of Federal Regulations, Part 1306, Section 1306.04. Purpose of issue of prescription. www.deadiversion.usdoj.gov/21cfr/cfr/1306/1306_04.htm. Accessed February 8, 2016.
- Kübler-Ross E, Wessler S, Avioli LV. On death and dying. JAMA 1972; 221:174–179.
- Institute of Medicine of the National Academies. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. http://iom.nationalacademies.org/reports/2011/relieving-pain-in-america-a-blueprint-for-transforming-prevention-care-education-research.aspx. Accessed February 8, 2016.
- McCarberg BH, Nicholson BD, Todd KH, Palmer T, Penles L. The impact of pain on quality of life and the unmet needs of pain management: results from pain sufferers and physicians participating in an Internet survey. Am J Ther 2008; 15:312–320.
- Roehr B. US needs new strategy to help 116 million patients in chronic pain. BMJ 2011; 343:d4206.
- Breuer B, Pappagallo M, Tai JY, Portenoy RK. US board-certified pain physician practices: uniformity and census data of their locations. J Pain 2007; 8:244–250.
- Paulozzi L, Dellinger A, Degutis L. Lessons from the past. Inj Prev 2012; 18:70.
- US Department of Health and Human Services; Substance Abuse and Mental Health Services Administration. Results from the 2013 national survey on drug use and health: Summary of national findings, NSDUH series H-48, HHS publication no. (SMA) 14-4863. www.samhsa.gov/data/sites/default/files/NSDUHresultsPDFWHTML2013/Web/NSDUHresults2013.htm. Accessed February 8, 2016.
- Bronstein K, Passik S, Munitz L, Leider H. Can clinicians accurately predict which patients are misusing their medications? J Pain 2011; 12(suppl):P3.
- Von Korff M, Kolodny A, Deyo RA, Chou R. Long-term opioid therapy reconsidered. Ann Intern Med 2011; 155:325–328.
- Chou R, Turner JA, Devine EB, et al. The effectiveness and risks of long-term opioid therapy for chronic pain: a systematic review for a National Institutes of Health Pathways to Prevention Workshop. Ann Intern Med 2015; 162:276–286.
- Chou R, Fanciullo GJ, Fine PG, et al; American Pain Society-American Academy of Pain Medicine Opioids Guidelines Panel. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain 2009; 10:113–130.
- Butler SF, Budman SH, Fernandez K, Jamison RN. Validation of a screener and opioid assessment measure for patients with chronic pain. Pain 2004; 112:65–75.
- Compton PA, Wu SM, Schieffer B, Pham Q, Naliboff BD. Introduction of a self-report version of the prescription drug use questionnaire and relationship to medication agreement noncompliance. J Pain Symptom Manage 2008; 36:383–395.
- Webster LR, Webster RM. Predicting aberrant behaviors in opioid-treated patients: preliminary validation of the opioid risk tool. Pain Med 2005; 6:432–442.
- Chen JT, Fagan MJ, Diaz JA, Reinert SE. Is treating chronic pain torture? Internal medicine residents’ experience with patients with chronic nonmalignant pain. Teach Learn Med 2007; 19:101–105.
- Gallagher RM. Empathy: a timeless skill for the pain medicine toolbox. Pain Med 2006; 7:213–214.
- Woolf CJ; American College of Physicians; American Physiological Society. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med 2004; 140:441–451.
- Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med 2005; 6:107–112.
- Medscape. A guide to state opioid prescribing policies resource center news. www.medscape.com/index/list_5657_1. Accessed February 8, 2016.
- Penko J, Mattson J, Miaskowski C, Kushel M. Do patients know they are on pain medication agreements? Results from a sample of high-risk patients on chronic opioid therapy. Pain Med 2012; 13:1174–1180.
- McGee S, Silverman RD. Treatment agreements, informed consent, and the role of state medical boards in opioid prescribing. Pain Med 2015; 16:25–29.
- Solomon DH, Rassen JA, Glynn RJ, Lee J, Levin R, Schneeweiss S. The comparative safety of analgesics in older adults with arthritis. Arch Intern Med 2010; 170:1968–1976.
- Wang D, Teichtahl H. Opioids, sleep architecture and sleep-disordered breathing. Sleep Med Rev 2007; 11:35–46.
- Bohnert AS, Valenstein M, Bair MJ, et al. Association between opioid prescribing patterns and opioid overdose-related deaths. JAMA 2011; 305:1315–1321.
- Smith HS. Variations in opioid responsiveness. Pain Physician 2008; 11:237–248.
- Vargas-Schaffer G. Is the WHO analgesic ladder still valid? Twenty-four years of experience. Can Fam Physician 2010; 56:514-517.
- Inturrisi CE. Clinical pharmacology of opioids for pain. Clin J Pain 2002; 18(suppl 4):S3–S13.
- Krantz MJ, Martin J, Stimmel B, Mehta D, Haigney MC. QTc interval screening in methadone treatment. Ann Intern Med 2009; 150:387–395.
- 107th Congress Public Law 77. US Government Printing Office. Departments of Commerce, Justice, and State, the Judiciary, and Related Agencies Appropriations Act, 2002. https://www.gpo.gov/fdsys/pkg/PLAW-107publ77/html/PLAW-107publ77.htm. Accessed February 8, 2016.
- Missouri Prescription Drug Monitoring Program NOW Coalition. http://mopdmpnow.org/. Accessed February 8, 2016.
- Prescription Drug Monitoring Program Center of Excellence at Brandeis. www.pdmpexcellence.org/sites/all/pdfs/Briefing%20on%20PDMP%20Effectiveness%203rd%20revision.pdf. Accessed February 8, 2016.
- Centers for Medicare and Medicaid Services. Tamper Resistant Prescriptions. www.cms.gov/Medicare-Medicaid-Coordination/Fraud-Prevention/FraudAbuseforProfs/TRP.html. Accessed February 8, 2016.
- Moorman-Li R, Motycka CA, Inge LD, Congdon JM, Hobson S, Pokropski B. A review of abuse-deterrent opioids for chronic nonmalignant pain. P T 2012; 37:412–418.
- Starrels JL, Becker WC, Weiner MG, Li X, Heo M, Turner BJ. Low use of opioid risk reduction strategies in primary care even for high risk patients with chronic pain. J Gen Intern Med 2011; 26:958–964.
- Herring C, Muzyk AJ, Johnston C. Interferences with urine drug screens. J Pharm Pract 2011; 24:102–108.
- Thermo Fisher Scientific. Cedia opiate 2K drugs of abuse assays. http://www.thermoscientific.com/en/product/cedia-opiate-2k-drugs-abuse-assays.html. Accessed February 8, 2016.
- Markway EC, Baker SN. A review of the methods, interpretation, and limitations of the urine drug screen. Orthopedics 2011; 34:877–881.
- Saitman A, Park HD, Fitzgerald RL. False-positive interferences of common urine drug screen immunoassays: a review. J Anal Toxicol 2014; 38:387–396.
- Standridge JB, Adams SM, Zotos AP. Urine drug screening: a valuable office procedure. Am Fam Physician 2010; 81:635–640.
- National Highway Traffic Safety Administration. Drugs and human performance fact sheet. www.nhtsa.gov/staticfiles/nti/pdf/809725-DrugsHumanPerformFS.pdf. Accessed February 8, 2016.
- US Department of Health and Human Services; Centers for Medicare and Medicaid Services. Partners in Integrity: What is the Prescriber's Role in Preventing the Diversion of Prescription Drugs. www.cms.gov/Medicare-Medicaid-Coordination/Fraud-Prevention/Medicaid-Integrity-Education/Provider-Education-Toolkits/Downloads/prescriber-role-drugdiversion.pdf. Accessed February 8, 2016.
- Silverman SM. Opioid induced hyperalgesia: clinical implications for the pain practitioner. Pain Physician 2009; 12:679–684.
- US Department of Health and Human Services; Agency for Healthcare Research and Quality (AHRQ); National Guideline Clearinghouse. Interagency guideline on opioid dosing for chronic non-cancer pain: an educational aid to improve care and safety with opioid therapy. www.guideline.gov/content.aspx?id=23792. Accessed February 8, 2016.
- Department of Veterans Affairs/Department of Defense. Tapering and discontinuing opioids factsheet. www.healthquality.va.gov/guidelines/Pain/cot/OpioidTaperingFactSheet23May2013v1.pdf. Accessed February 8, 2016.
- US Department of Justice Drug Enforcement Administration: Office of Diversion Control. Title 21 Code of Federal Regulations, Part 1306, Section 1306.04. Purpose of issue of prescription. www.deadiversion.usdoj.gov/21cfr/cfr/1306/1306_04.htm. Accessed February 8, 2016.
- Kübler-Ross E, Wessler S, Avioli LV. On death and dying. JAMA 1972; 221:174–179.
KEY POINTS
- Predicting which patients will benefit and which ones will be harmed is difficult. We generally recommend a conservative approach to starting opioid treatment.
- Providers must periodically reassess the safety and efficacy of opioid therapy to be sure it is still indicated.
- Monitoring should be transparent and consistent. By framing monitoring in terms of safety and employing it universally, providers can minimize miscommunication and accidental stigmatization.
- When opioids are no longer safe or effective, they should be stopped. The decision can be difficult for the patient and the provider.

Managing patients at genetic risk of breast cancer
While most cases of breast cancer are sporadic (ie, not inherited), up to 10% are attributable to single-gene hereditary cancer syndromes.1–4 People with these syndromes have a lifetime risk of breast cancer much higher than in the general population, and the cancers often occur at a much earlier age.
With genetic testing becoming more common, primary care physicians need to be familiar with the known syndromes, associated risks, and evidence-based recommendations for management. Here, we review the management of cancer risk in the most common hereditary breast cancer syndromes, ie:
- Hereditary breast and ovarian cancer syndrome5
- Hereditary diffuse gastric cancer
- Cowden syndrome (PTEN hamartoma tumor syndrome)
- Peutz-Jeghers syndrome
- Li-Fraumeni syndrome.
IT TAKES A TEAM, BUT PRIMARY CARE PHYSICIANS ARE CENTRAL
Women who have a hereditary predisposition to breast cancer face complex and emotional decisions about the best ways to manage and reduce their risks. Their management includes close clinical surveillance, chemoprevention, and surgical risk reduction.1,4
Referral to multiple subspecialists is an important component of these patients’ preventive care. They may need referrals to a cancer genetic counselor, a high-risk breast clinic, a gynecologic oncologist, and counseling services. They may also require referrals to gastroenterologists, colorectal surgeons, endocrinologists, and endocrine surgeons, depending on the syndrome identified.
Consultation with a certified genetic counselor is critical for patients harboring mutations associated with cancer risk. The National Society of Genetic Counselors maintains a directory of genetic counselors by location and practice specialty at www.nsgc.org. The counselor’s evaluation will provide patients with a detailed explanation of the cancer risks and management guidelines for their particular condition, along with offering diagnostic genetic testing if appropriate. Women with germline mutations who plan to have children should be informed about preimplantation genetic diagnosis and about fertility specialists who can perform this service if they are interested in pursuing it.6
Screening and management guidelines for hereditary breast cancer syndromes are evolving. While subspecialists may be involved in enhanced surveillance and preventive care, the primary care physician is the central player, with both a broader perspective and knowledge of the patient’s competing medical issues, risks, and preferences.
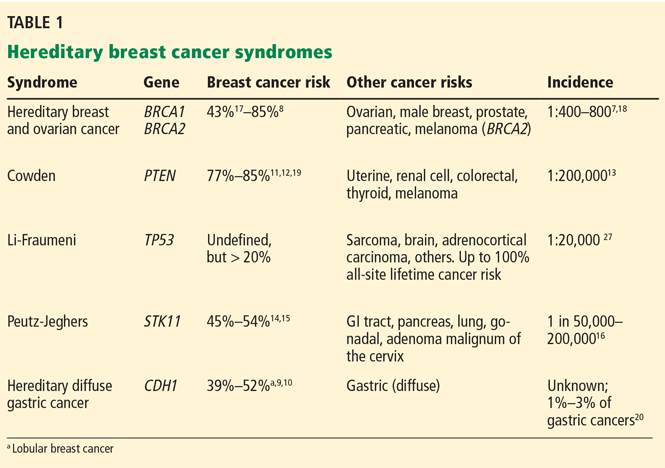
In addition to breast cancer, the risk of other malignancies is also higher, with the pattern varying by syndrome (Table 1).7–20 The management of these additional risks is beyond the scope of this review; however, primary care physicians need to be familiar with these risks to provide adequate referrals.
WHO IS AT INCREASED RISK OF BREAST CANCER?
In considering recommendations to reduce the risk of breast cancer, it is useful to think of a patient as being at either high risk or average risk.
The risk of breast cancer in women in the general population is about 12%, and most cases of breast cancer occur in patients who have no known risk factors for it. “High risk” of breast cancer generally means having more than a 20% lifetime risk (ie, before age 70) of developing the condition.
Even without a hereditary cancer syndrome, a combination of reproductive, environmental, personal, and family history factors can confer a 20% lifetime risk. But for women with hereditary syndromes, the risk far exceeds 20% regardless of such risk factors. It is likely that interactions with reproductive, environmental, and personal risk factors likely affect the individual risk of a woman with a known genetic mutation, and evidence is emerging with regard to further risk stratification.
In an earlier article in this journal, Smith and colleagues21 reviewed how to recognize heritable breast cancer syndromes. In general, referral for genetic counseling should be considered for patients and their families who have:
- Early-onset breast cancers (before age 50)
- Bilateral breast cancers at any age
- Ovarian cancers at any age
- “Triple-negative” breast cancers (ie, estrogen receptor-negative, progesterone receptor-negative, and human epidermal growth factor receptor 2-nonamplified (HER2-negative)
- Male breast cancer at any age
- Cancers affecting multiple individuals and in multiple generations.
- Breast, ovarian, pancreatic or prostate cancer in families with Ashkenazi Jewish ancestry
HEREDITARY BREAST CANCER SYNDROMES
Hereditary breast and ovarian cancer syndrome
The most common of these syndromes is hereditary breast and ovarian cancer syndrome, caused by germline mutations in the tumor-suppressor genes BRCA1 or BRCA2.7 The estimated prevalence of BRCA1 mutations is 1 in 250 to 300, and the prevalence of BRCA2 mutations is 1 in 800.1,4 However, in families of Ashkenazi Jewish ancestry, the population frequency of either a BRCA1 or BRCA2 mutation is approximately 1 in 40.1,4,6
Women with BRCA1 or BRCA2 mutations have a lifetime risk of breast cancer of up to 87%, or 5 to 7 times higher than in the general population, with the risk rising steeply beginning at age 30.1,5,8 In addition, the lifetime risk of ovarian cancer is nearly 59% in BRCA1 mutation carriers and 17% in BRCA2 mutation carriers.22
A meta-analysis found that BRCA1 mutation carriers diagnosed with cancer in one breast have a 5-year risk of developing cancer in the other breast of 15%, and BRCA2 mutation carriers have a risk of 9%.23 Overall, the risk of contralateral breast cancer is about 3% per year.3,4,24
BRCA1 mutations are strongly associated with triple-negative breast cancers.1,3,4
Hereditary diffuse gastric cancer
Hereditary diffuse gastric cancer is an autosomal-dominant syndrome associated with mutations in the CDH1 gene, although up to 75% of patients with this syndrome do not have an identifiable CDH1 mutation.9,25,26 In cases in which there is no identifiable CDH1 mutation, the diagnosis is made on the basis of the patient’s medical and family history.
Hereditary diffuse gastric cancer is associated with an increased risk of the lobular subtype of breast cancer as well as diffuse gastric cancer. The cumulative lifetime risk of breast cancer in women with CDH1 mutations is 39% to 52%,6,9–11,25 and their lifetime risk of diffuse gastric cancer is 83%.9 The combined risk of breast cancer and gastric cancer in women with this syndrome is 90% by age 80.9
Cowden syndrome (PTEN hamartoma tumor syndrome)
Cowden syndrome (PTEN hamartoma tumor syndrome) is caused by mutations in PTEN, another tumor-suppressor gene.11 The primary clinical concerns are melanoma and breast, endometrial, thyroid (follicular or papillary), colon, and renal cell cancers. Women with a PTEN mutation have a twofold greater risk of developing any type of cancer than men with a PTEN mutation.12 The cumulative lifetime risk of invasive breast cancer in women with this syndrome is 70% to 85%.11–13
Peutz-Jeghers syndrome
Peutz-Jeghers syndrome is an autosomal dominant polyposis disorder caused, in most patients, by a mutation in the serine/threonine kinase tumor-suppressor gene STK11.14
Patients with Peutz-Jeghers syndrome have higher risks of gastrointestinal, breast, gynecologic (uterine, ovarian, and cervical), pancreatic, and lung cancers. In women, the lifetime risk of breast cancer is 44% to 50% by age 70, regardless of the type of mutation.6,14,15 Breast cancers associated with Peutz-Jeghers syndrome are usually ductal, and the mean age at diagnosis is 37 years.16
Li-Fraumeni syndrome
Li-Fraumeni syndrome is an autosomal-dominant disorder caused by germline mutations in the TP53 gene, which codes for a transcription factor associated with cell proliferation and apoptosis.27
These mutations confer a lifetime cancer risk of 93% in women (mainly breast cancer) and 68% in men.1,27 Other cancers associated with TP53 mutations include sarcomas, brain cancer, leukemia, and adrenocortical tumors. Germline TP53 mutations are responsible for approximately 1% of all breast cancers.1,4
Breast cancers can occur at a young age in patients with a TP53 mutation. Women with TP53 mutations are 18 times more likely to develop breast cancer before age 45 compared with the general population.4
It is important to consider a TP53 mutation in premenopausal women or women less than 30 years of age with breast cancer who have no mutations in BRCA1 and BRCA2.1
MANAGING PATIENTS WITH GENETIC PREDISPOSITION TO BREAST CANCER
Management for patients at high risk fall into three broad categories: clinical surveillance, chemoprevention, and surgical risk reduction. The utility and benefit of each depend to a large degree on the patient’s specific mutation, family history, and comorbidities. Decisions must be shared with the patient.
CLOSE CLINICAL SURVEILLANCE
Consensus guidelines for cancer screening in the syndromes described here are available from the National Comprehensive Cancer Network at www.nccn.org and are summarized in Table 2.26,28 While the guidelines are broadly applicable to all women with these conditions, some individualization is required based on personal and family medical history.
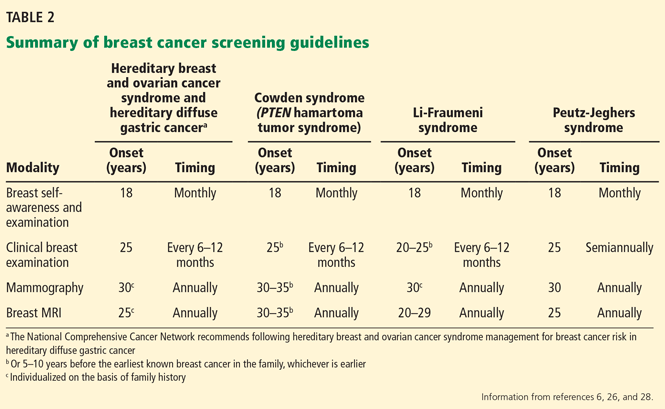
In general, screening begins at the ages listed in Table 2 or 10 years earlier than the age at which cancer developed in the first affected relative, whichever is earlier. However, screening decisions are shared with the patient and are sometimes affected by significant out-of-pocket costs for the patient and anxiety resulting from the test or subsequent test findings, which must all be considered.
Breast self-awareness and clinical breast examination
Although controversial in the general population, breast self-examination is recommended for patients carrying mutations that increase risk.6
A discussion about breast self-awareness is recommended for all women at the age of 18. It should include the signs and symptoms of breast cancer, what feels “normal” to the patient, and what is known about modifiable risk factors for breast cancer. The patient should also be told to report any changes in her personal or family history.
Clinical breast examinations should be done every 6 months, as some cancers are found clinically, particularly in young women with dense tissue, and confirmed by diagnostic imaging and targeted ultrasonography.
Radiographic surveillance
Mammography and magnetic resonance imaging (MRI) are also important components of a breast cancer surveillance regimen in women at high risk. Adherence to a well-formulated plan of clinical and radiographic examinations increases early detection in patients who have a hereditary predisposition to breast cancer.
MRI is more sensitive than mammography and reduces the likelihood of finding advanced cancers by up to 70% compared with mammography in women at high risk of breast cancer.29–31 The sensitivity of breast MRI alone ranges from 71% to 100%, and the sensitivity increases to 89% to 100% when combined with mammography. In contrast, the sensitivity of mammography alone is 25% to 59%.29 MRI has also been shown to be cost-effective when added to mammography and physical examination in women at high risk.5,32
Adding MRI to the breast cancer screening regimen has been under discussion and has been endorsed by the American Cancer Society in formal recommendations set forth in 2007 for patients with known hereditary cancer syndromes, in untested first-degree relatives of identified genetic mutation carriers, or in women who have an estimated lifetime risk of breast cancer of 20% or more, as determined by models largely dependent on family history.33
But MRI has a downside—it is less specific than mammography.29,33 Its lower specificity (77% to 90% vs 95% with mammography alone) leads to additional radiographic studies and tissue samplings for the “suspicious” lesions discovered. From 3% to 15% of screening breast MRIs result in a biopsy, and the proportion of biopsies that reveal cancer is 13% to 40%.33 Furthermore, by itself, MRI has not been shown to reduce mortality in any high-risk group.
Mammography remains useful in conjunction with MRI due to its ability to detect breast calcifications, which may be the earliest sign of breast cancer, and ability to detect changes in breast architecture. A typical screening program (Table 2) should incorporate both modalities, commonly offset by 6 months (eg, mammography at baseline, then MRI 6 months later, then mammography again 6 months after that, and so on) to increase the detection of interval cancer development.
Chemoprevention
Chemoprevention means taking medications to reduce the risk. Certain selective estrogen receptor modulators and aromatase inhibitors decrease the risk of invasive breast cancer in healthy women at high risk. These drugs include tamoxifen, which can be used before menopause, and raloxifene, anastrozole, and exemestane, which must be used only after menopause.
Because data are limited, we cannot make any generalized recommendations about chemoprevention in patients with hereditary breast cancer syndromes. Decisions about chemoprevention should take into account the patient’s personal and family histories. Often, a medical oncologist or medical breast specialist can help by discussing the risks and benefits for the individual patient.
Tamoxifen has been the most studied, mainly in BRCA mutation carriers.6,34–37 As in the general population, tamoxifen reduces the incidence of estrogen receptor-positive breast cancers by 50%.36–38 It has not been shown to significantly reduce breast cancer risk in premenopausal women with BRCA1 mutations,37 most likely because most cancers that occur in this group are estrogen receptor-negative. In patients with a history of breast cancer, however, tamoxifen has been shown to reduce the risk of developing contralateral breast cancer by 45% to 60% in both BRCA1 and BRCA2 mutation carriers.6,35
There is also little evidence that giving a chemopreventive agent after bilateral salpingo-oophorectomy reduces the risk further in premenopausal BRCA mutation carriers.35 These patients often receive hormonal therapy with estrogen, which currently would preclude the use of tamoxifen. Tamoxifen in postmenopausal women is associated with a small increased risk of venous thromboembolic disease and endometrial cancer.38
Oral contraceptives reduce the risk of ovarian cancer by up to 50% in BRCA1 mutation carriers and up to 60% in BRCA2 mutation carriers.6 However, data conflict on their effect on the risk of breast cancer in BRCA1 and BRCA2 mutation carriers.39
Decisions about chemoprevention with agents other than tamoxifen and in syndromes other than hereditary breast and ovarian cancer syndrome must take into consideration the existing lack of data in this area.
SURGICAL PROPHYLAXIS
Surgical prophylactic options for patients at genetic risk of breast cancer are bilateral mastectomy and bilateral salpingo-oophorectomy.
Prophylactic mastectomy
Bilateral risk-reducing mastectomy reduces the risk of breast cancer by at least 90%24,39,40 and greatly reduces the need for complex surveillance. Patients are often followed annually clinically, with single-view mammography if they have tissue flap reconstruction.
Nipple-sparing and skin-sparing mastectomies, which facilitate reconstruction and cosmetic outcomes, are an option in the risk-reduction setting and have been shown thus far to be safe.41–43 In patients with breast cancer, the overall breast cancer recurrence rates with nipple-sparing mastectomy are similar to those of traditional mastectomy and breast conservation treatment.41
In patients at very high risk of breast cancer, risk-reducing operations also reduce the risk of ultimately needing chemotherapy and radiation to treat breast cancer, as the risk of developing breast cancer is significantly lowered.
The timing of risk-reducing mastectomy depends largely on personal and family medical history and personal choice. Bilateral mastectomy at age 25 results in the greatest survival gain for patients with hereditary breast and ovarian cancer syndrome.5 Such precise data are not available for other hereditary cancer syndromes, but it is reasonable to consider bilateral mastectomy as an option for any woman with a highly penetrant genetic mutation that predisposes her to breast cancer. Special consideration in the timing of risk-reducing mastectomy must be given to women with Li-Fraumeni syndrome, as this condition is often associated with an earlier age at breast cancer diagnosis (before age 30).1
Family planning, sexuality, self-image, and the anxiety associated with both cancer risk and surveillance are all factors women consider when deciding whether and when to undergo mastectomy. A survey of 12 high-risk women who elected prophylactic mastectomy elicited feelings of some regret in 3 of them, while all expressed a sense of relief and reduced anxiety related to both cancer risk and screenings.24 Another group of 14 women surveyed after the surgery reported initial distress related to physical appearance, self-image, and intimacy but also reported a significant decrease in anxiety related to breast cancer risk and were largely satisfied with their decision.44
Prophylactic salpingo-oophorectomy
In patients who have pathogenic mutations in BRCA1 or 2, prophylactic salpingo-oophorectomy before age 40 decreases the risk of ovarian cancer by up to 96% and breast cancer by 50%.1,37,45 This operation, in fact, is the only intervention that has been shown to reduce the mortality rate in patients with a hereditary predisposition to cancer.46
We recommend that women with hereditary breast and ovarian cancer syndrome strongly consider prophylactic salpingo-oophorectomy by age 40 or when childbearing is complete for the greatest reduction in risk.1,5 In 2006, Domchek et al46 reported an overall decrease in the mortality rate in BRCA1/2-positive patients who underwent this surgery, but not in breast cancer-specific or ovarian cancer-specific mortality.
On the other hand, removing the ovaries before menopause places women at risk of serious complications associated with premature loss of gonadal hormones, including cardiovascular disease, decreased bone density, reduced sexual satisfaction, dyspareunia, hot flashes, and night sweats.47 Therefore, it is generally reserved for women who are also at risk of ovarian cancer.
Hormonal therapy, ie, estrogen therapy for patients who choose complete hysterectomy, and estrogen-progesterone therapy for patients who choose to keep their uterus, reduces menopausal symptoms and symptoms of sexual dissatisfaction and has not thus far been shown to increase breast cancer risk.1,34 However, this information is from nonrandomized studies, which are inherently limited.
It is important to address and modify risk factors for heart disease and osteoporosis in women with premature surgical menopause, as they may be particularly vulnerable to these conditions.
HEREDITARY BREAST CANCER IN MEN
Fewer than 1% of cases of breast cancer arise in men, and fewer than 1% of cases of cancer in men are breast cancer.
Male breast cancer is more likely than female breast cancer to be estrogen receptor- and progesterone receptor-positive. In an analysis of the Surveillance, Epidemiology, and End Results registry between 1973 and 2005, triple-negative breast cancer was found in 23% of female patients but only 7.6% of male patients.2
Male breast cancer is most common in families with BRCA2, and to a lesser degree, BRCA1 mutations. Other genetic disorders including Li-Fraumeni syndrome, hereditary nonpolyposis colorectal cancer, and Klinefelter syndrome also increase the risk of male breast cancer. A genetic predisposition for breast cancer is present in approximately 10% of male breast cancer patients.2 Any man with breast cancer, therefore, should be referred for genetic counseling.
In men, a BRCA2 mutation confers a lifetime risk of breast cancer of 5% to 10%.2 This is similar to the lifetime risk of breast cancer for the average woman but it is still significant, as the lifetime risk of breast cancer for the average man is 0.1%.1,2
Five-year survival rates in male breast cancer range from only 36% to 66%, most likely because it is usually diagnosed in later stages, as men are not routinely screened for breast cancer. In men with known hereditary susceptibility, National Comprehensive Cancer Network guidelines recommend that they be educated about and begin breast self-examination at the age of 35 and be clinically examined every 12 months starting at age 35.48 There are limited data to support breast imaging in men. High-risk surveillance with MRI screening in this group is not recommended. Prostate cancer screening is recommended for men with BRCA2 mutations starting at age 40, and should be considered for men with BRCA1 mutations starting at age 40.
No specific guidelines exist for pancreatic cancer and melanoma, but screening may be individualized based on cancers observed in the family.
- Daly MB, Axilbund JE, Buys S, et al; National Comprehensive Cancer Network. Genetic/familial high-risk assessment: breast and ovarian. J Natl Compr Canc Netw 2010; 8:562–594.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med 2008; 359:2143–2153.
- Schwartz GF, Hughes KS, Lynch HT, et al. Proceedings of the international consensus conference on breast cancer risk, genetics, and risk management, April 2007. Breast J 2009; 15:4–16.
- Kurian AW, Sigal BM, Plevritis SK. Survival analysis of cancer risk reduction strategies for BRCA1/2 mutation carriers. J Clin Oncol 2010; 28:222–231.
- National Comprehensive Cancer Network Guidelines Version 2.2014. Genetic/familial high risk assessment: breast and ovarian. www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed January 22, 2016.
- Ford D, Easton DF, Peto J. Estimates of the gene frequency of BRCA1 and its contribution to breast and ovarian cancer incidence. Am J Hum Genet 1995; 57:1457–1462.
- Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62:676–689.
- Pharoah PD, Guilford P, Caldas C; International Gastric Cancer Linkage Consortium. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001; 121:1348–1353.
- Kaurah P, MacMillan A, Boyd N, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 2007; 297:2360–2372.
- Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012; 18:400–407.
- Bubien V, Bonnet F, Brouste V, et al; French Cowden Disease Network. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet 2013; 50:255–263.
- Nelen MR, Kremer H, Konings IB, et al. Novel PTEN mutations in patients with Cowden disease: absence of clear genotype-phenotype correlations. Eur J Hum Genet 1999; 7:267–273.
- Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 2006; 12:3209–3215.
- Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000; 119:1447–1453.
- Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut 2010; 59:975–986.
- Chen S, Iversen ES, Friebel T, et al. Characterization of BRCA1 and BRCA2 mutations in a large United States sample. J Clin Oncol 2006; 24:863–871.
- Claus EB, Schildkraut JM, Thompson WD, Risch NJ. The genetic attributable risk of breast and ovarian cancer. Cancer 1996; 77:2318–2324.
- Riegert-Johnson DL, Gleeson FC, Roberts M, et al. Cancer and Lhermitte-Duclos disease are common in Cowden syndrome patients. Hered Cancer Clin Pract 2010; 8:6.
- Stone J, Bevan S, Cunningham D, et al. Low frequency of germline E-cadherin mutations in familial and nonfamilial gastric cancer. Br J Cancer 1999; 79:1935–1937.
- Smith M, Mester J, Eng C. How to spot heritable breast cancer: a primary care physician’s guide. Cleve Clin J Med 2014; 81:31–40.
- Mavaddat N, Peock S, Frost D, et al; EMBRACE. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst 2013; 105:812–822.
- Molina-Montes E, Pérez-Nevot B, Pollán M, Sánchez-Cantalejo E, Espín J, Sánchez MJ. Cumulative risk of second primary contralateral breast cancer in BRCA1/BRCA2 mutation carriers with a first breast cancer: a systematic review and meta-analysis. Breast 2014; 23:721–742.
- Kwong A, Chu AT. What made her give up her breasts: a qualitative study on decisional considerations for contralateral prophylactic mastectomy among breast cancer survivors undergoing BRCA1/2 genetic testing. Asian Pac J Cancer Prev 2012; 13:2241–2247.
- Dixon M, Seevaratnam R, Wirtzfeld D, et al. A RAND/UCLA appropriateness study of the management of familial gastric cancer. Ann Surg Oncol 2013; 20:533–541.
- Fitzgerald RC, Hardwick R, Huntsman D, et al; International Gastric Cancer Linkage Consortium. Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet 2010; 47:436–444.
- Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009; 27:1250–1256.
- National Comprehensive Cancer Network Guidelines Version 1. 2015. Gastric Cancer. www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed January 22, 2016.
- Warner, E. Impact of MRI surveillance and breast cancer detection in young women with BRCA mutations. Ann Oncol 2011; 22(suppl 1):i44–i49.
- Kriege M, Brekelmans CT, Boetes C, et al; Magnetic Resonance Imaging Screening Study Group. Efficacy of MRI and mammography for breast-cancer screening in women with a familial or genetic predisposition. N Engl J Med 2004; 351:427–437.
- Pederson HJ, O’Rourke C, Lyons J, Patrick RJ, Crowe JP Jr, Grobmyer SR. Time-related changes in yield and harms of screening breast magnetic resonance imaging. Clin Breast Cancer 2015 Jan 21: S1526-8209(15)00024–00025. Epub ahead of print.
- Grann VR, Patel PR, Jacobson JS, et al. Comparative effectiveness of screening and prevention strategies among BRCA1/2-affected mutation carriers. Breast Cancer Res Treat 2011; 125:837–847.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Rebbeck TR, Friebel T, Wagner T, et al; PROSE Study Group. Effect of short term hormone replacement therapy on breast cancer risk reduction after bilateral prophylactic oophorectomy in BRCA1 and BRCA2 mutation carriers: the PROSE study group. J Clin Oncol 2005; 23:7804–7610.
- Narod SA, Brunet JS, Ghadirian P, et al; Hereditary Breast Cancer Clinical Study Group. Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: a case control study. Lancet 2000; 356:1876–1881.
- Njiaju UO, Olopade OI. Genetic determinants of breast cancer risk: a review of the current literature and issues pertaining to clinical application. Breast J 2012; 18:436–442.
- King MC, Wieand S, Hale K, et al; National Surgical Adjuvant Breast and Bowel Project. Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: National Surgical Adjuvant Breast and Bowel Project (NSABP-P1) Breast Cancer Prevention trial. JAMA 2001; 286:2251–2256.
- Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998; 90:1371–1388.
- Rebbeck TR, Friebel T, Lynch HT, et al. Bilateral prophylactic mastectomy reduces breast cancer risk in BRCA1 and BRCA2 mutation carriers: the PROSE Study Group. J Clin Oncol 2004; 22:1055–1062.
- Hartmann LC, Schaid DJ, Woods JE, et al. Efficacy of bilateral prophylactic mastectomy in women with a family history of breast cancer. N Engl J Med 1999; 340:77–84.
- Mallon P, Feron JG, Couturaud B, et al. The role of nipple-sparing mastectomy in breast cancer: a comprehensive review of the literature. Plast Reconstr Surg 2013; 131:969–984.
- Stanec Z, Žic R, Budi S, et al. Skin and nipple-areola complex sparing mastectomy in breast cancer patients: 15-year experience. Ann Plast Surg 2014; 73:485–491.
- Eisenberg RE, Chan JS, Swistel AJ, Hoda SA. Pathological evaluation of nipple-sparing mastectomies with emphasis on occult nipple involvement: the Weill-Cornell experience with 325 cases. Breast J 2014; 20:15–21.
- Lodder LN, Frets PG, Trijsburg RW, et al. One year follow-up of women opting for presymptomatic testing for BRCA1 and BRCA2: emotional impact of the test outcome and decisions on risk management (surveillance or prophylactic surgery). Breast Cancer Res Treat 2002; 73:97–112.
- Rebbeck TR, Lynch HT, Neuhausen SL, et al; Prevention and Observation of Surgical End Points Study Group. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 2002; 346:1616–1622.
- Domchek SM, Friebel TM, Neuhausen SL, et al. Mortality after bilateral salpingo-oophorectomy in BRCA1 and BRCA2 mutation carriers: a prospective cohort study. Lancet Oncol 2006; 7:223–229.
- Finch A, Evans G, Narod SA. BRCA carriers, prophylactic salpingo-oophorectomy and menopause: clinical management considerations and recommendations. Womens Health (Lond Engl) 2012; 8:543–555.
- National Comprehensive Cancer Network Guidelines. Version 2.2015. Genetic/Familial High-Risk Assessment Breast and Ovarian. www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed February 8, 2016.
While most cases of breast cancer are sporadic (ie, not inherited), up to 10% are attributable to single-gene hereditary cancer syndromes.1–4 People with these syndromes have a lifetime risk of breast cancer much higher than in the general population, and the cancers often occur at a much earlier age.
With genetic testing becoming more common, primary care physicians need to be familiar with the known syndromes, associated risks, and evidence-based recommendations for management. Here, we review the management of cancer risk in the most common hereditary breast cancer syndromes, ie:
- Hereditary breast and ovarian cancer syndrome5
- Hereditary diffuse gastric cancer
- Cowden syndrome (PTEN hamartoma tumor syndrome)
- Peutz-Jeghers syndrome
- Li-Fraumeni syndrome.
IT TAKES A TEAM, BUT PRIMARY CARE PHYSICIANS ARE CENTRAL
Women who have a hereditary predisposition to breast cancer face complex and emotional decisions about the best ways to manage and reduce their risks. Their management includes close clinical surveillance, chemoprevention, and surgical risk reduction.1,4
Referral to multiple subspecialists is an important component of these patients’ preventive care. They may need referrals to a cancer genetic counselor, a high-risk breast clinic, a gynecologic oncologist, and counseling services. They may also require referrals to gastroenterologists, colorectal surgeons, endocrinologists, and endocrine surgeons, depending on the syndrome identified.
Consultation with a certified genetic counselor is critical for patients harboring mutations associated with cancer risk. The National Society of Genetic Counselors maintains a directory of genetic counselors by location and practice specialty at www.nsgc.org. The counselor’s evaluation will provide patients with a detailed explanation of the cancer risks and management guidelines for their particular condition, along with offering diagnostic genetic testing if appropriate. Women with germline mutations who plan to have children should be informed about preimplantation genetic diagnosis and about fertility specialists who can perform this service if they are interested in pursuing it.6
Screening and management guidelines for hereditary breast cancer syndromes are evolving. While subspecialists may be involved in enhanced surveillance and preventive care, the primary care physician is the central player, with both a broader perspective and knowledge of the patient’s competing medical issues, risks, and preferences.
In addition to breast cancer, the risk of other malignancies is also higher, with the pattern varying by syndrome (Table 1).7–20 The management of these additional risks is beyond the scope of this review; however, primary care physicians need to be familiar with these risks to provide adequate referrals.
WHO IS AT INCREASED RISK OF BREAST CANCER?
In considering recommendations to reduce the risk of breast cancer, it is useful to think of a patient as being at either high risk or average risk.
The risk of breast cancer in women in the general population is about 12%, and most cases of breast cancer occur in patients who have no known risk factors for it. “High risk” of breast cancer generally means having more than a 20% lifetime risk (ie, before age 70) of developing the condition.
Even without a hereditary cancer syndrome, a combination of reproductive, environmental, personal, and family history factors can confer a 20% lifetime risk. But for women with hereditary syndromes, the risk far exceeds 20% regardless of such risk factors. It is likely that interactions with reproductive, environmental, and personal risk factors likely affect the individual risk of a woman with a known genetic mutation, and evidence is emerging with regard to further risk stratification.
In an earlier article in this journal, Smith and colleagues21 reviewed how to recognize heritable breast cancer syndromes. In general, referral for genetic counseling should be considered for patients and their families who have:
- Early-onset breast cancers (before age 50)
- Bilateral breast cancers at any age
- Ovarian cancers at any age
- “Triple-negative” breast cancers (ie, estrogen receptor-negative, progesterone receptor-negative, and human epidermal growth factor receptor 2-nonamplified (HER2-negative)
- Male breast cancer at any age
- Cancers affecting multiple individuals and in multiple generations.
- Breast, ovarian, pancreatic or prostate cancer in families with Ashkenazi Jewish ancestry
HEREDITARY BREAST CANCER SYNDROMES
Hereditary breast and ovarian cancer syndrome
The most common of these syndromes is hereditary breast and ovarian cancer syndrome, caused by germline mutations in the tumor-suppressor genes BRCA1 or BRCA2.7 The estimated prevalence of BRCA1 mutations is 1 in 250 to 300, and the prevalence of BRCA2 mutations is 1 in 800.1,4 However, in families of Ashkenazi Jewish ancestry, the population frequency of either a BRCA1 or BRCA2 mutation is approximately 1 in 40.1,4,6
Women with BRCA1 or BRCA2 mutations have a lifetime risk of breast cancer of up to 87%, or 5 to 7 times higher than in the general population, with the risk rising steeply beginning at age 30.1,5,8 In addition, the lifetime risk of ovarian cancer is nearly 59% in BRCA1 mutation carriers and 17% in BRCA2 mutation carriers.22
A meta-analysis found that BRCA1 mutation carriers diagnosed with cancer in one breast have a 5-year risk of developing cancer in the other breast of 15%, and BRCA2 mutation carriers have a risk of 9%.23 Overall, the risk of contralateral breast cancer is about 3% per year.3,4,24
BRCA1 mutations are strongly associated with triple-negative breast cancers.1,3,4
Hereditary diffuse gastric cancer
Hereditary diffuse gastric cancer is an autosomal-dominant syndrome associated with mutations in the CDH1 gene, although up to 75% of patients with this syndrome do not have an identifiable CDH1 mutation.9,25,26 In cases in which there is no identifiable CDH1 mutation, the diagnosis is made on the basis of the patient’s medical and family history.
Hereditary diffuse gastric cancer is associated with an increased risk of the lobular subtype of breast cancer as well as diffuse gastric cancer. The cumulative lifetime risk of breast cancer in women with CDH1 mutations is 39% to 52%,6,9–11,25 and their lifetime risk of diffuse gastric cancer is 83%.9 The combined risk of breast cancer and gastric cancer in women with this syndrome is 90% by age 80.9
Cowden syndrome (PTEN hamartoma tumor syndrome)
Cowden syndrome (PTEN hamartoma tumor syndrome) is caused by mutations in PTEN, another tumor-suppressor gene.11 The primary clinical concerns are melanoma and breast, endometrial, thyroid (follicular or papillary), colon, and renal cell cancers. Women with a PTEN mutation have a twofold greater risk of developing any type of cancer than men with a PTEN mutation.12 The cumulative lifetime risk of invasive breast cancer in women with this syndrome is 70% to 85%.11–13
Peutz-Jeghers syndrome
Peutz-Jeghers syndrome is an autosomal dominant polyposis disorder caused, in most patients, by a mutation in the serine/threonine kinase tumor-suppressor gene STK11.14
Patients with Peutz-Jeghers syndrome have higher risks of gastrointestinal, breast, gynecologic (uterine, ovarian, and cervical), pancreatic, and lung cancers. In women, the lifetime risk of breast cancer is 44% to 50% by age 70, regardless of the type of mutation.6,14,15 Breast cancers associated with Peutz-Jeghers syndrome are usually ductal, and the mean age at diagnosis is 37 years.16
Li-Fraumeni syndrome
Li-Fraumeni syndrome is an autosomal-dominant disorder caused by germline mutations in the TP53 gene, which codes for a transcription factor associated with cell proliferation and apoptosis.27
These mutations confer a lifetime cancer risk of 93% in women (mainly breast cancer) and 68% in men.1,27 Other cancers associated with TP53 mutations include sarcomas, brain cancer, leukemia, and adrenocortical tumors. Germline TP53 mutations are responsible for approximately 1% of all breast cancers.1,4
Breast cancers can occur at a young age in patients with a TP53 mutation. Women with TP53 mutations are 18 times more likely to develop breast cancer before age 45 compared with the general population.4
It is important to consider a TP53 mutation in premenopausal women or women less than 30 years of age with breast cancer who have no mutations in BRCA1 and BRCA2.1
MANAGING PATIENTS WITH GENETIC PREDISPOSITION TO BREAST CANCER
Management for patients at high risk fall into three broad categories: clinical surveillance, chemoprevention, and surgical risk reduction. The utility and benefit of each depend to a large degree on the patient’s specific mutation, family history, and comorbidities. Decisions must be shared with the patient.
CLOSE CLINICAL SURVEILLANCE
Consensus guidelines for cancer screening in the syndromes described here are available from the National Comprehensive Cancer Network at www.nccn.org and are summarized in Table 2.26,28 While the guidelines are broadly applicable to all women with these conditions, some individualization is required based on personal and family medical history.
In general, screening begins at the ages listed in Table 2 or 10 years earlier than the age at which cancer developed in the first affected relative, whichever is earlier. However, screening decisions are shared with the patient and are sometimes affected by significant out-of-pocket costs for the patient and anxiety resulting from the test or subsequent test findings, which must all be considered.
Breast self-awareness and clinical breast examination
Although controversial in the general population, breast self-examination is recommended for patients carrying mutations that increase risk.6
A discussion about breast self-awareness is recommended for all women at the age of 18. It should include the signs and symptoms of breast cancer, what feels “normal” to the patient, and what is known about modifiable risk factors for breast cancer. The patient should also be told to report any changes in her personal or family history.
Clinical breast examinations should be done every 6 months, as some cancers are found clinically, particularly in young women with dense tissue, and confirmed by diagnostic imaging and targeted ultrasonography.
Radiographic surveillance
Mammography and magnetic resonance imaging (MRI) are also important components of a breast cancer surveillance regimen in women at high risk. Adherence to a well-formulated plan of clinical and radiographic examinations increases early detection in patients who have a hereditary predisposition to breast cancer.
MRI is more sensitive than mammography and reduces the likelihood of finding advanced cancers by up to 70% compared with mammography in women at high risk of breast cancer.29–31 The sensitivity of breast MRI alone ranges from 71% to 100%, and the sensitivity increases to 89% to 100% when combined with mammography. In contrast, the sensitivity of mammography alone is 25% to 59%.29 MRI has also been shown to be cost-effective when added to mammography and physical examination in women at high risk.5,32
Adding MRI to the breast cancer screening regimen has been under discussion and has been endorsed by the American Cancer Society in formal recommendations set forth in 2007 for patients with known hereditary cancer syndromes, in untested first-degree relatives of identified genetic mutation carriers, or in women who have an estimated lifetime risk of breast cancer of 20% or more, as determined by models largely dependent on family history.33
But MRI has a downside—it is less specific than mammography.29,33 Its lower specificity (77% to 90% vs 95% with mammography alone) leads to additional radiographic studies and tissue samplings for the “suspicious” lesions discovered. From 3% to 15% of screening breast MRIs result in a biopsy, and the proportion of biopsies that reveal cancer is 13% to 40%.33 Furthermore, by itself, MRI has not been shown to reduce mortality in any high-risk group.
Mammography remains useful in conjunction with MRI due to its ability to detect breast calcifications, which may be the earliest sign of breast cancer, and ability to detect changes in breast architecture. A typical screening program (Table 2) should incorporate both modalities, commonly offset by 6 months (eg, mammography at baseline, then MRI 6 months later, then mammography again 6 months after that, and so on) to increase the detection of interval cancer development.
Chemoprevention
Chemoprevention means taking medications to reduce the risk. Certain selective estrogen receptor modulators and aromatase inhibitors decrease the risk of invasive breast cancer in healthy women at high risk. These drugs include tamoxifen, which can be used before menopause, and raloxifene, anastrozole, and exemestane, which must be used only after menopause.
Because data are limited, we cannot make any generalized recommendations about chemoprevention in patients with hereditary breast cancer syndromes. Decisions about chemoprevention should take into account the patient’s personal and family histories. Often, a medical oncologist or medical breast specialist can help by discussing the risks and benefits for the individual patient.
Tamoxifen has been the most studied, mainly in BRCA mutation carriers.6,34–37 As in the general population, tamoxifen reduces the incidence of estrogen receptor-positive breast cancers by 50%.36–38 It has not been shown to significantly reduce breast cancer risk in premenopausal women with BRCA1 mutations,37 most likely because most cancers that occur in this group are estrogen receptor-negative. In patients with a history of breast cancer, however, tamoxifen has been shown to reduce the risk of developing contralateral breast cancer by 45% to 60% in both BRCA1 and BRCA2 mutation carriers.6,35
There is also little evidence that giving a chemopreventive agent after bilateral salpingo-oophorectomy reduces the risk further in premenopausal BRCA mutation carriers.35 These patients often receive hormonal therapy with estrogen, which currently would preclude the use of tamoxifen. Tamoxifen in postmenopausal women is associated with a small increased risk of venous thromboembolic disease and endometrial cancer.38
Oral contraceptives reduce the risk of ovarian cancer by up to 50% in BRCA1 mutation carriers and up to 60% in BRCA2 mutation carriers.6 However, data conflict on their effect on the risk of breast cancer in BRCA1 and BRCA2 mutation carriers.39
Decisions about chemoprevention with agents other than tamoxifen and in syndromes other than hereditary breast and ovarian cancer syndrome must take into consideration the existing lack of data in this area.
SURGICAL PROPHYLAXIS
Surgical prophylactic options for patients at genetic risk of breast cancer are bilateral mastectomy and bilateral salpingo-oophorectomy.
Prophylactic mastectomy
Bilateral risk-reducing mastectomy reduces the risk of breast cancer by at least 90%24,39,40 and greatly reduces the need for complex surveillance. Patients are often followed annually clinically, with single-view mammography if they have tissue flap reconstruction.
Nipple-sparing and skin-sparing mastectomies, which facilitate reconstruction and cosmetic outcomes, are an option in the risk-reduction setting and have been shown thus far to be safe.41–43 In patients with breast cancer, the overall breast cancer recurrence rates with nipple-sparing mastectomy are similar to those of traditional mastectomy and breast conservation treatment.41
In patients at very high risk of breast cancer, risk-reducing operations also reduce the risk of ultimately needing chemotherapy and radiation to treat breast cancer, as the risk of developing breast cancer is significantly lowered.
The timing of risk-reducing mastectomy depends largely on personal and family medical history and personal choice. Bilateral mastectomy at age 25 results in the greatest survival gain for patients with hereditary breast and ovarian cancer syndrome.5 Such precise data are not available for other hereditary cancer syndromes, but it is reasonable to consider bilateral mastectomy as an option for any woman with a highly penetrant genetic mutation that predisposes her to breast cancer. Special consideration in the timing of risk-reducing mastectomy must be given to women with Li-Fraumeni syndrome, as this condition is often associated with an earlier age at breast cancer diagnosis (before age 30).1
Family planning, sexuality, self-image, and the anxiety associated with both cancer risk and surveillance are all factors women consider when deciding whether and when to undergo mastectomy. A survey of 12 high-risk women who elected prophylactic mastectomy elicited feelings of some regret in 3 of them, while all expressed a sense of relief and reduced anxiety related to both cancer risk and screenings.24 Another group of 14 women surveyed after the surgery reported initial distress related to physical appearance, self-image, and intimacy but also reported a significant decrease in anxiety related to breast cancer risk and were largely satisfied with their decision.44
Prophylactic salpingo-oophorectomy
In patients who have pathogenic mutations in BRCA1 or 2, prophylactic salpingo-oophorectomy before age 40 decreases the risk of ovarian cancer by up to 96% and breast cancer by 50%.1,37,45 This operation, in fact, is the only intervention that has been shown to reduce the mortality rate in patients with a hereditary predisposition to cancer.46
We recommend that women with hereditary breast and ovarian cancer syndrome strongly consider prophylactic salpingo-oophorectomy by age 40 or when childbearing is complete for the greatest reduction in risk.1,5 In 2006, Domchek et al46 reported an overall decrease in the mortality rate in BRCA1/2-positive patients who underwent this surgery, but not in breast cancer-specific or ovarian cancer-specific mortality.
On the other hand, removing the ovaries before menopause places women at risk of serious complications associated with premature loss of gonadal hormones, including cardiovascular disease, decreased bone density, reduced sexual satisfaction, dyspareunia, hot flashes, and night sweats.47 Therefore, it is generally reserved for women who are also at risk of ovarian cancer.
Hormonal therapy, ie, estrogen therapy for patients who choose complete hysterectomy, and estrogen-progesterone therapy for patients who choose to keep their uterus, reduces menopausal symptoms and symptoms of sexual dissatisfaction and has not thus far been shown to increase breast cancer risk.1,34 However, this information is from nonrandomized studies, which are inherently limited.
It is important to address and modify risk factors for heart disease and osteoporosis in women with premature surgical menopause, as they may be particularly vulnerable to these conditions.
HEREDITARY BREAST CANCER IN MEN
Fewer than 1% of cases of breast cancer arise in men, and fewer than 1% of cases of cancer in men are breast cancer.
Male breast cancer is more likely than female breast cancer to be estrogen receptor- and progesterone receptor-positive. In an analysis of the Surveillance, Epidemiology, and End Results registry between 1973 and 2005, triple-negative breast cancer was found in 23% of female patients but only 7.6% of male patients.2
Male breast cancer is most common in families with BRCA2, and to a lesser degree, BRCA1 mutations. Other genetic disorders including Li-Fraumeni syndrome, hereditary nonpolyposis colorectal cancer, and Klinefelter syndrome also increase the risk of male breast cancer. A genetic predisposition for breast cancer is present in approximately 10% of male breast cancer patients.2 Any man with breast cancer, therefore, should be referred for genetic counseling.
In men, a BRCA2 mutation confers a lifetime risk of breast cancer of 5% to 10%.2 This is similar to the lifetime risk of breast cancer for the average woman but it is still significant, as the lifetime risk of breast cancer for the average man is 0.1%.1,2
Five-year survival rates in male breast cancer range from only 36% to 66%, most likely because it is usually diagnosed in later stages, as men are not routinely screened for breast cancer. In men with known hereditary susceptibility, National Comprehensive Cancer Network guidelines recommend that they be educated about and begin breast self-examination at the age of 35 and be clinically examined every 12 months starting at age 35.48 There are limited data to support breast imaging in men. High-risk surveillance with MRI screening in this group is not recommended. Prostate cancer screening is recommended for men with BRCA2 mutations starting at age 40, and should be considered for men with BRCA1 mutations starting at age 40.
No specific guidelines exist for pancreatic cancer and melanoma, but screening may be individualized based on cancers observed in the family.
While most cases of breast cancer are sporadic (ie, not inherited), up to 10% are attributable to single-gene hereditary cancer syndromes.1–4 People with these syndromes have a lifetime risk of breast cancer much higher than in the general population, and the cancers often occur at a much earlier age.
With genetic testing becoming more common, primary care physicians need to be familiar with the known syndromes, associated risks, and evidence-based recommendations for management. Here, we review the management of cancer risk in the most common hereditary breast cancer syndromes, ie:
- Hereditary breast and ovarian cancer syndrome5
- Hereditary diffuse gastric cancer
- Cowden syndrome (PTEN hamartoma tumor syndrome)
- Peutz-Jeghers syndrome
- Li-Fraumeni syndrome.
IT TAKES A TEAM, BUT PRIMARY CARE PHYSICIANS ARE CENTRAL
Women who have a hereditary predisposition to breast cancer face complex and emotional decisions about the best ways to manage and reduce their risks. Their management includes close clinical surveillance, chemoprevention, and surgical risk reduction.1,4
Referral to multiple subspecialists is an important component of these patients’ preventive care. They may need referrals to a cancer genetic counselor, a high-risk breast clinic, a gynecologic oncologist, and counseling services. They may also require referrals to gastroenterologists, colorectal surgeons, endocrinologists, and endocrine surgeons, depending on the syndrome identified.
Consultation with a certified genetic counselor is critical for patients harboring mutations associated with cancer risk. The National Society of Genetic Counselors maintains a directory of genetic counselors by location and practice specialty at www.nsgc.org. The counselor’s evaluation will provide patients with a detailed explanation of the cancer risks and management guidelines for their particular condition, along with offering diagnostic genetic testing if appropriate. Women with germline mutations who plan to have children should be informed about preimplantation genetic diagnosis and about fertility specialists who can perform this service if they are interested in pursuing it.6
Screening and management guidelines for hereditary breast cancer syndromes are evolving. While subspecialists may be involved in enhanced surveillance and preventive care, the primary care physician is the central player, with both a broader perspective and knowledge of the patient’s competing medical issues, risks, and preferences.
In addition to breast cancer, the risk of other malignancies is also higher, with the pattern varying by syndrome (Table 1).7–20 The management of these additional risks is beyond the scope of this review; however, primary care physicians need to be familiar with these risks to provide adequate referrals.
WHO IS AT INCREASED RISK OF BREAST CANCER?
In considering recommendations to reduce the risk of breast cancer, it is useful to think of a patient as being at either high risk or average risk.
The risk of breast cancer in women in the general population is about 12%, and most cases of breast cancer occur in patients who have no known risk factors for it. “High risk” of breast cancer generally means having more than a 20% lifetime risk (ie, before age 70) of developing the condition.
Even without a hereditary cancer syndrome, a combination of reproductive, environmental, personal, and family history factors can confer a 20% lifetime risk. But for women with hereditary syndromes, the risk far exceeds 20% regardless of such risk factors. It is likely that interactions with reproductive, environmental, and personal risk factors likely affect the individual risk of a woman with a known genetic mutation, and evidence is emerging with regard to further risk stratification.
In an earlier article in this journal, Smith and colleagues21 reviewed how to recognize heritable breast cancer syndromes. In general, referral for genetic counseling should be considered for patients and their families who have:
- Early-onset breast cancers (before age 50)
- Bilateral breast cancers at any age
- Ovarian cancers at any age
- “Triple-negative” breast cancers (ie, estrogen receptor-negative, progesterone receptor-negative, and human epidermal growth factor receptor 2-nonamplified (HER2-negative)
- Male breast cancer at any age
- Cancers affecting multiple individuals and in multiple generations.
- Breast, ovarian, pancreatic or prostate cancer in families with Ashkenazi Jewish ancestry
HEREDITARY BREAST CANCER SYNDROMES
Hereditary breast and ovarian cancer syndrome
The most common of these syndromes is hereditary breast and ovarian cancer syndrome, caused by germline mutations in the tumor-suppressor genes BRCA1 or BRCA2.7 The estimated prevalence of BRCA1 mutations is 1 in 250 to 300, and the prevalence of BRCA2 mutations is 1 in 800.1,4 However, in families of Ashkenazi Jewish ancestry, the population frequency of either a BRCA1 or BRCA2 mutation is approximately 1 in 40.1,4,6
Women with BRCA1 or BRCA2 mutations have a lifetime risk of breast cancer of up to 87%, or 5 to 7 times higher than in the general population, with the risk rising steeply beginning at age 30.1,5,8 In addition, the lifetime risk of ovarian cancer is nearly 59% in BRCA1 mutation carriers and 17% in BRCA2 mutation carriers.22
A meta-analysis found that BRCA1 mutation carriers diagnosed with cancer in one breast have a 5-year risk of developing cancer in the other breast of 15%, and BRCA2 mutation carriers have a risk of 9%.23 Overall, the risk of contralateral breast cancer is about 3% per year.3,4,24
BRCA1 mutations are strongly associated with triple-negative breast cancers.1,3,4
Hereditary diffuse gastric cancer
Hereditary diffuse gastric cancer is an autosomal-dominant syndrome associated with mutations in the CDH1 gene, although up to 75% of patients with this syndrome do not have an identifiable CDH1 mutation.9,25,26 In cases in which there is no identifiable CDH1 mutation, the diagnosis is made on the basis of the patient’s medical and family history.
Hereditary diffuse gastric cancer is associated with an increased risk of the lobular subtype of breast cancer as well as diffuse gastric cancer. The cumulative lifetime risk of breast cancer in women with CDH1 mutations is 39% to 52%,6,9–11,25 and their lifetime risk of diffuse gastric cancer is 83%.9 The combined risk of breast cancer and gastric cancer in women with this syndrome is 90% by age 80.9
Cowden syndrome (PTEN hamartoma tumor syndrome)
Cowden syndrome (PTEN hamartoma tumor syndrome) is caused by mutations in PTEN, another tumor-suppressor gene.11 The primary clinical concerns are melanoma and breast, endometrial, thyroid (follicular or papillary), colon, and renal cell cancers. Women with a PTEN mutation have a twofold greater risk of developing any type of cancer than men with a PTEN mutation.12 The cumulative lifetime risk of invasive breast cancer in women with this syndrome is 70% to 85%.11–13
Peutz-Jeghers syndrome
Peutz-Jeghers syndrome is an autosomal dominant polyposis disorder caused, in most patients, by a mutation in the serine/threonine kinase tumor-suppressor gene STK11.14
Patients with Peutz-Jeghers syndrome have higher risks of gastrointestinal, breast, gynecologic (uterine, ovarian, and cervical), pancreatic, and lung cancers. In women, the lifetime risk of breast cancer is 44% to 50% by age 70, regardless of the type of mutation.6,14,15 Breast cancers associated with Peutz-Jeghers syndrome are usually ductal, and the mean age at diagnosis is 37 years.16
Li-Fraumeni syndrome
Li-Fraumeni syndrome is an autosomal-dominant disorder caused by germline mutations in the TP53 gene, which codes for a transcription factor associated with cell proliferation and apoptosis.27
These mutations confer a lifetime cancer risk of 93% in women (mainly breast cancer) and 68% in men.1,27 Other cancers associated with TP53 mutations include sarcomas, brain cancer, leukemia, and adrenocortical tumors. Germline TP53 mutations are responsible for approximately 1% of all breast cancers.1,4
Breast cancers can occur at a young age in patients with a TP53 mutation. Women with TP53 mutations are 18 times more likely to develop breast cancer before age 45 compared with the general population.4
It is important to consider a TP53 mutation in premenopausal women or women less than 30 years of age with breast cancer who have no mutations in BRCA1 and BRCA2.1
MANAGING PATIENTS WITH GENETIC PREDISPOSITION TO BREAST CANCER
Management for patients at high risk fall into three broad categories: clinical surveillance, chemoprevention, and surgical risk reduction. The utility and benefit of each depend to a large degree on the patient’s specific mutation, family history, and comorbidities. Decisions must be shared with the patient.
CLOSE CLINICAL SURVEILLANCE
Consensus guidelines for cancer screening in the syndromes described here are available from the National Comprehensive Cancer Network at www.nccn.org and are summarized in Table 2.26,28 While the guidelines are broadly applicable to all women with these conditions, some individualization is required based on personal and family medical history.
In general, screening begins at the ages listed in Table 2 or 10 years earlier than the age at which cancer developed in the first affected relative, whichever is earlier. However, screening decisions are shared with the patient and are sometimes affected by significant out-of-pocket costs for the patient and anxiety resulting from the test or subsequent test findings, which must all be considered.
Breast self-awareness and clinical breast examination
Although controversial in the general population, breast self-examination is recommended for patients carrying mutations that increase risk.6
A discussion about breast self-awareness is recommended for all women at the age of 18. It should include the signs and symptoms of breast cancer, what feels “normal” to the patient, and what is known about modifiable risk factors for breast cancer. The patient should also be told to report any changes in her personal or family history.
Clinical breast examinations should be done every 6 months, as some cancers are found clinically, particularly in young women with dense tissue, and confirmed by diagnostic imaging and targeted ultrasonography.
Radiographic surveillance
Mammography and magnetic resonance imaging (MRI) are also important components of a breast cancer surveillance regimen in women at high risk. Adherence to a well-formulated plan of clinical and radiographic examinations increases early detection in patients who have a hereditary predisposition to breast cancer.
MRI is more sensitive than mammography and reduces the likelihood of finding advanced cancers by up to 70% compared with mammography in women at high risk of breast cancer.29–31 The sensitivity of breast MRI alone ranges from 71% to 100%, and the sensitivity increases to 89% to 100% when combined with mammography. In contrast, the sensitivity of mammography alone is 25% to 59%.29 MRI has also been shown to be cost-effective when added to mammography and physical examination in women at high risk.5,32
Adding MRI to the breast cancer screening regimen has been under discussion and has been endorsed by the American Cancer Society in formal recommendations set forth in 2007 for patients with known hereditary cancer syndromes, in untested first-degree relatives of identified genetic mutation carriers, or in women who have an estimated lifetime risk of breast cancer of 20% or more, as determined by models largely dependent on family history.33
But MRI has a downside—it is less specific than mammography.29,33 Its lower specificity (77% to 90% vs 95% with mammography alone) leads to additional radiographic studies and tissue samplings for the “suspicious” lesions discovered. From 3% to 15% of screening breast MRIs result in a biopsy, and the proportion of biopsies that reveal cancer is 13% to 40%.33 Furthermore, by itself, MRI has not been shown to reduce mortality in any high-risk group.
Mammography remains useful in conjunction with MRI due to its ability to detect breast calcifications, which may be the earliest sign of breast cancer, and ability to detect changes in breast architecture. A typical screening program (Table 2) should incorporate both modalities, commonly offset by 6 months (eg, mammography at baseline, then MRI 6 months later, then mammography again 6 months after that, and so on) to increase the detection of interval cancer development.
Chemoprevention
Chemoprevention means taking medications to reduce the risk. Certain selective estrogen receptor modulators and aromatase inhibitors decrease the risk of invasive breast cancer in healthy women at high risk. These drugs include tamoxifen, which can be used before menopause, and raloxifene, anastrozole, and exemestane, which must be used only after menopause.
Because data are limited, we cannot make any generalized recommendations about chemoprevention in patients with hereditary breast cancer syndromes. Decisions about chemoprevention should take into account the patient’s personal and family histories. Often, a medical oncologist or medical breast specialist can help by discussing the risks and benefits for the individual patient.
Tamoxifen has been the most studied, mainly in BRCA mutation carriers.6,34–37 As in the general population, tamoxifen reduces the incidence of estrogen receptor-positive breast cancers by 50%.36–38 It has not been shown to significantly reduce breast cancer risk in premenopausal women with BRCA1 mutations,37 most likely because most cancers that occur in this group are estrogen receptor-negative. In patients with a history of breast cancer, however, tamoxifen has been shown to reduce the risk of developing contralateral breast cancer by 45% to 60% in both BRCA1 and BRCA2 mutation carriers.6,35
There is also little evidence that giving a chemopreventive agent after bilateral salpingo-oophorectomy reduces the risk further in premenopausal BRCA mutation carriers.35 These patients often receive hormonal therapy with estrogen, which currently would preclude the use of tamoxifen. Tamoxifen in postmenopausal women is associated with a small increased risk of venous thromboembolic disease and endometrial cancer.38
Oral contraceptives reduce the risk of ovarian cancer by up to 50% in BRCA1 mutation carriers and up to 60% in BRCA2 mutation carriers.6 However, data conflict on their effect on the risk of breast cancer in BRCA1 and BRCA2 mutation carriers.39
Decisions about chemoprevention with agents other than tamoxifen and in syndromes other than hereditary breast and ovarian cancer syndrome must take into consideration the existing lack of data in this area.
SURGICAL PROPHYLAXIS
Surgical prophylactic options for patients at genetic risk of breast cancer are bilateral mastectomy and bilateral salpingo-oophorectomy.
Prophylactic mastectomy
Bilateral risk-reducing mastectomy reduces the risk of breast cancer by at least 90%24,39,40 and greatly reduces the need for complex surveillance. Patients are often followed annually clinically, with single-view mammography if they have tissue flap reconstruction.
Nipple-sparing and skin-sparing mastectomies, which facilitate reconstruction and cosmetic outcomes, are an option in the risk-reduction setting and have been shown thus far to be safe.41–43 In patients with breast cancer, the overall breast cancer recurrence rates with nipple-sparing mastectomy are similar to those of traditional mastectomy and breast conservation treatment.41
In patients at very high risk of breast cancer, risk-reducing operations also reduce the risk of ultimately needing chemotherapy and radiation to treat breast cancer, as the risk of developing breast cancer is significantly lowered.
The timing of risk-reducing mastectomy depends largely on personal and family medical history and personal choice. Bilateral mastectomy at age 25 results in the greatest survival gain for patients with hereditary breast and ovarian cancer syndrome.5 Such precise data are not available for other hereditary cancer syndromes, but it is reasonable to consider bilateral mastectomy as an option for any woman with a highly penetrant genetic mutation that predisposes her to breast cancer. Special consideration in the timing of risk-reducing mastectomy must be given to women with Li-Fraumeni syndrome, as this condition is often associated with an earlier age at breast cancer diagnosis (before age 30).1
Family planning, sexuality, self-image, and the anxiety associated with both cancer risk and surveillance are all factors women consider when deciding whether and when to undergo mastectomy. A survey of 12 high-risk women who elected prophylactic mastectomy elicited feelings of some regret in 3 of them, while all expressed a sense of relief and reduced anxiety related to both cancer risk and screenings.24 Another group of 14 women surveyed after the surgery reported initial distress related to physical appearance, self-image, and intimacy but also reported a significant decrease in anxiety related to breast cancer risk and were largely satisfied with their decision.44
Prophylactic salpingo-oophorectomy
In patients who have pathogenic mutations in BRCA1 or 2, prophylactic salpingo-oophorectomy before age 40 decreases the risk of ovarian cancer by up to 96% and breast cancer by 50%.1,37,45 This operation, in fact, is the only intervention that has been shown to reduce the mortality rate in patients with a hereditary predisposition to cancer.46
We recommend that women with hereditary breast and ovarian cancer syndrome strongly consider prophylactic salpingo-oophorectomy by age 40 or when childbearing is complete for the greatest reduction in risk.1,5 In 2006, Domchek et al46 reported an overall decrease in the mortality rate in BRCA1/2-positive patients who underwent this surgery, but not in breast cancer-specific or ovarian cancer-specific mortality.
On the other hand, removing the ovaries before menopause places women at risk of serious complications associated with premature loss of gonadal hormones, including cardiovascular disease, decreased bone density, reduced sexual satisfaction, dyspareunia, hot flashes, and night sweats.47 Therefore, it is generally reserved for women who are also at risk of ovarian cancer.
Hormonal therapy, ie, estrogen therapy for patients who choose complete hysterectomy, and estrogen-progesterone therapy for patients who choose to keep their uterus, reduces menopausal symptoms and symptoms of sexual dissatisfaction and has not thus far been shown to increase breast cancer risk.1,34 However, this information is from nonrandomized studies, which are inherently limited.
It is important to address and modify risk factors for heart disease and osteoporosis in women with premature surgical menopause, as they may be particularly vulnerable to these conditions.
HEREDITARY BREAST CANCER IN MEN
Fewer than 1% of cases of breast cancer arise in men, and fewer than 1% of cases of cancer in men are breast cancer.
Male breast cancer is more likely than female breast cancer to be estrogen receptor- and progesterone receptor-positive. In an analysis of the Surveillance, Epidemiology, and End Results registry between 1973 and 2005, triple-negative breast cancer was found in 23% of female patients but only 7.6% of male patients.2
Male breast cancer is most common in families with BRCA2, and to a lesser degree, BRCA1 mutations. Other genetic disorders including Li-Fraumeni syndrome, hereditary nonpolyposis colorectal cancer, and Klinefelter syndrome also increase the risk of male breast cancer. A genetic predisposition for breast cancer is present in approximately 10% of male breast cancer patients.2 Any man with breast cancer, therefore, should be referred for genetic counseling.
In men, a BRCA2 mutation confers a lifetime risk of breast cancer of 5% to 10%.2 This is similar to the lifetime risk of breast cancer for the average woman but it is still significant, as the lifetime risk of breast cancer for the average man is 0.1%.1,2
Five-year survival rates in male breast cancer range from only 36% to 66%, most likely because it is usually diagnosed in later stages, as men are not routinely screened for breast cancer. In men with known hereditary susceptibility, National Comprehensive Cancer Network guidelines recommend that they be educated about and begin breast self-examination at the age of 35 and be clinically examined every 12 months starting at age 35.48 There are limited data to support breast imaging in men. High-risk surveillance with MRI screening in this group is not recommended. Prostate cancer screening is recommended for men with BRCA2 mutations starting at age 40, and should be considered for men with BRCA1 mutations starting at age 40.
No specific guidelines exist for pancreatic cancer and melanoma, but screening may be individualized based on cancers observed in the family.
- Daly MB, Axilbund JE, Buys S, et al; National Comprehensive Cancer Network. Genetic/familial high-risk assessment: breast and ovarian. J Natl Compr Canc Netw 2010; 8:562–594.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med 2008; 359:2143–2153.
- Schwartz GF, Hughes KS, Lynch HT, et al. Proceedings of the international consensus conference on breast cancer risk, genetics, and risk management, April 2007. Breast J 2009; 15:4–16.
- Kurian AW, Sigal BM, Plevritis SK. Survival analysis of cancer risk reduction strategies for BRCA1/2 mutation carriers. J Clin Oncol 2010; 28:222–231.
- National Comprehensive Cancer Network Guidelines Version 2.2014. Genetic/familial high risk assessment: breast and ovarian. www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed January 22, 2016.
- Ford D, Easton DF, Peto J. Estimates of the gene frequency of BRCA1 and its contribution to breast and ovarian cancer incidence. Am J Hum Genet 1995; 57:1457–1462.
- Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62:676–689.
- Pharoah PD, Guilford P, Caldas C; International Gastric Cancer Linkage Consortium. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001; 121:1348–1353.
- Kaurah P, MacMillan A, Boyd N, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 2007; 297:2360–2372.
- Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012; 18:400–407.
- Bubien V, Bonnet F, Brouste V, et al; French Cowden Disease Network. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet 2013; 50:255–263.
- Nelen MR, Kremer H, Konings IB, et al. Novel PTEN mutations in patients with Cowden disease: absence of clear genotype-phenotype correlations. Eur J Hum Genet 1999; 7:267–273.
- Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 2006; 12:3209–3215.
- Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000; 119:1447–1453.
- Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut 2010; 59:975–986.
- Chen S, Iversen ES, Friebel T, et al. Characterization of BRCA1 and BRCA2 mutations in a large United States sample. J Clin Oncol 2006; 24:863–871.
- Claus EB, Schildkraut JM, Thompson WD, Risch NJ. The genetic attributable risk of breast and ovarian cancer. Cancer 1996; 77:2318–2324.
- Riegert-Johnson DL, Gleeson FC, Roberts M, et al. Cancer and Lhermitte-Duclos disease are common in Cowden syndrome patients. Hered Cancer Clin Pract 2010; 8:6.
- Stone J, Bevan S, Cunningham D, et al. Low frequency of germline E-cadherin mutations in familial and nonfamilial gastric cancer. Br J Cancer 1999; 79:1935–1937.
- Smith M, Mester J, Eng C. How to spot heritable breast cancer: a primary care physician’s guide. Cleve Clin J Med 2014; 81:31–40.
- Mavaddat N, Peock S, Frost D, et al; EMBRACE. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst 2013; 105:812–822.
- Molina-Montes E, Pérez-Nevot B, Pollán M, Sánchez-Cantalejo E, Espín J, Sánchez MJ. Cumulative risk of second primary contralateral breast cancer in BRCA1/BRCA2 mutation carriers with a first breast cancer: a systematic review and meta-analysis. Breast 2014; 23:721–742.
- Kwong A, Chu AT. What made her give up her breasts: a qualitative study on decisional considerations for contralateral prophylactic mastectomy among breast cancer survivors undergoing BRCA1/2 genetic testing. Asian Pac J Cancer Prev 2012; 13:2241–2247.
- Dixon M, Seevaratnam R, Wirtzfeld D, et al. A RAND/UCLA appropriateness study of the management of familial gastric cancer. Ann Surg Oncol 2013; 20:533–541.
- Fitzgerald RC, Hardwick R, Huntsman D, et al; International Gastric Cancer Linkage Consortium. Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet 2010; 47:436–444.
- Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009; 27:1250–1256.
- National Comprehensive Cancer Network Guidelines Version 1. 2015. Gastric Cancer. www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed January 22, 2016.
- Warner, E. Impact of MRI surveillance and breast cancer detection in young women with BRCA mutations. Ann Oncol 2011; 22(suppl 1):i44–i49.
- Kriege M, Brekelmans CT, Boetes C, et al; Magnetic Resonance Imaging Screening Study Group. Efficacy of MRI and mammography for breast-cancer screening in women with a familial or genetic predisposition. N Engl J Med 2004; 351:427–437.
- Pederson HJ, O’Rourke C, Lyons J, Patrick RJ, Crowe JP Jr, Grobmyer SR. Time-related changes in yield and harms of screening breast magnetic resonance imaging. Clin Breast Cancer 2015 Jan 21: S1526-8209(15)00024–00025. Epub ahead of print.
- Grann VR, Patel PR, Jacobson JS, et al. Comparative effectiveness of screening and prevention strategies among BRCA1/2-affected mutation carriers. Breast Cancer Res Treat 2011; 125:837–847.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Rebbeck TR, Friebel T, Wagner T, et al; PROSE Study Group. Effect of short term hormone replacement therapy on breast cancer risk reduction after bilateral prophylactic oophorectomy in BRCA1 and BRCA2 mutation carriers: the PROSE study group. J Clin Oncol 2005; 23:7804–7610.
- Narod SA, Brunet JS, Ghadirian P, et al; Hereditary Breast Cancer Clinical Study Group. Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: a case control study. Lancet 2000; 356:1876–1881.
- Njiaju UO, Olopade OI. Genetic determinants of breast cancer risk: a review of the current literature and issues pertaining to clinical application. Breast J 2012; 18:436–442.
- King MC, Wieand S, Hale K, et al; National Surgical Adjuvant Breast and Bowel Project. Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: National Surgical Adjuvant Breast and Bowel Project (NSABP-P1) Breast Cancer Prevention trial. JAMA 2001; 286:2251–2256.
- Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998; 90:1371–1388.
- Rebbeck TR, Friebel T, Lynch HT, et al. Bilateral prophylactic mastectomy reduces breast cancer risk in BRCA1 and BRCA2 mutation carriers: the PROSE Study Group. J Clin Oncol 2004; 22:1055–1062.
- Hartmann LC, Schaid DJ, Woods JE, et al. Efficacy of bilateral prophylactic mastectomy in women with a family history of breast cancer. N Engl J Med 1999; 340:77–84.
- Mallon P, Feron JG, Couturaud B, et al. The role of nipple-sparing mastectomy in breast cancer: a comprehensive review of the literature. Plast Reconstr Surg 2013; 131:969–984.
- Stanec Z, Žic R, Budi S, et al. Skin and nipple-areola complex sparing mastectomy in breast cancer patients: 15-year experience. Ann Plast Surg 2014; 73:485–491.
- Eisenberg RE, Chan JS, Swistel AJ, Hoda SA. Pathological evaluation of nipple-sparing mastectomies with emphasis on occult nipple involvement: the Weill-Cornell experience with 325 cases. Breast J 2014; 20:15–21.
- Lodder LN, Frets PG, Trijsburg RW, et al. One year follow-up of women opting for presymptomatic testing for BRCA1 and BRCA2: emotional impact of the test outcome and decisions on risk management (surveillance or prophylactic surgery). Breast Cancer Res Treat 2002; 73:97–112.
- Rebbeck TR, Lynch HT, Neuhausen SL, et al; Prevention and Observation of Surgical End Points Study Group. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 2002; 346:1616–1622.
- Domchek SM, Friebel TM, Neuhausen SL, et al. Mortality after bilateral salpingo-oophorectomy in BRCA1 and BRCA2 mutation carriers: a prospective cohort study. Lancet Oncol 2006; 7:223–229.
- Finch A, Evans G, Narod SA. BRCA carriers, prophylactic salpingo-oophorectomy and menopause: clinical management considerations and recommendations. Womens Health (Lond Engl) 2012; 8:543–555.
- National Comprehensive Cancer Network Guidelines. Version 2.2015. Genetic/Familial High-Risk Assessment Breast and Ovarian. www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed February 8, 2016.
- Daly MB, Axilbund JE, Buys S, et al; National Comprehensive Cancer Network. Genetic/familial high-risk assessment: breast and ovarian. J Natl Compr Canc Netw 2010; 8:562–594.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med 2008; 359:2143–2153.
- Schwartz GF, Hughes KS, Lynch HT, et al. Proceedings of the international consensus conference on breast cancer risk, genetics, and risk management, April 2007. Breast J 2009; 15:4–16.
- Kurian AW, Sigal BM, Plevritis SK. Survival analysis of cancer risk reduction strategies for BRCA1/2 mutation carriers. J Clin Oncol 2010; 28:222–231.
- National Comprehensive Cancer Network Guidelines Version 2.2014. Genetic/familial high risk assessment: breast and ovarian. www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed January 22, 2016.
- Ford D, Easton DF, Peto J. Estimates of the gene frequency of BRCA1 and its contribution to breast and ovarian cancer incidence. Am J Hum Genet 1995; 57:1457–1462.
- Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62:676–689.
- Pharoah PD, Guilford P, Caldas C; International Gastric Cancer Linkage Consortium. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001; 121:1348–1353.
- Kaurah P, MacMillan A, Boyd N, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 2007; 297:2360–2372.
- Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012; 18:400–407.
- Bubien V, Bonnet F, Brouste V, et al; French Cowden Disease Network. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet 2013; 50:255–263.
- Nelen MR, Kremer H, Konings IB, et al. Novel PTEN mutations in patients with Cowden disease: absence of clear genotype-phenotype correlations. Eur J Hum Genet 1999; 7:267–273.
- Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 2006; 12:3209–3215.
- Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000; 119:1447–1453.
- Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut 2010; 59:975–986.
- Chen S, Iversen ES, Friebel T, et al. Characterization of BRCA1 and BRCA2 mutations in a large United States sample. J Clin Oncol 2006; 24:863–871.
- Claus EB, Schildkraut JM, Thompson WD, Risch NJ. The genetic attributable risk of breast and ovarian cancer. Cancer 1996; 77:2318–2324.
- Riegert-Johnson DL, Gleeson FC, Roberts M, et al. Cancer and Lhermitte-Duclos disease are common in Cowden syndrome patients. Hered Cancer Clin Pract 2010; 8:6.
- Stone J, Bevan S, Cunningham D, et al. Low frequency of germline E-cadherin mutations in familial and nonfamilial gastric cancer. Br J Cancer 1999; 79:1935–1937.
- Smith M, Mester J, Eng C. How to spot heritable breast cancer: a primary care physician’s guide. Cleve Clin J Med 2014; 81:31–40.
- Mavaddat N, Peock S, Frost D, et al; EMBRACE. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst 2013; 105:812–822.
- Molina-Montes E, Pérez-Nevot B, Pollán M, Sánchez-Cantalejo E, Espín J, Sánchez MJ. Cumulative risk of second primary contralateral breast cancer in BRCA1/BRCA2 mutation carriers with a first breast cancer: a systematic review and meta-analysis. Breast 2014; 23:721–742.
- Kwong A, Chu AT. What made her give up her breasts: a qualitative study on decisional considerations for contralateral prophylactic mastectomy among breast cancer survivors undergoing BRCA1/2 genetic testing. Asian Pac J Cancer Prev 2012; 13:2241–2247.
- Dixon M, Seevaratnam R, Wirtzfeld D, et al. A RAND/UCLA appropriateness study of the management of familial gastric cancer. Ann Surg Oncol 2013; 20:533–541.
- Fitzgerald RC, Hardwick R, Huntsman D, et al; International Gastric Cancer Linkage Consortium. Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet 2010; 47:436–444.
- Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009; 27:1250–1256.
- National Comprehensive Cancer Network Guidelines Version 1. 2015. Gastric Cancer. www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed January 22, 2016.
- Warner, E. Impact of MRI surveillance and breast cancer detection in young women with BRCA mutations. Ann Oncol 2011; 22(suppl 1):i44–i49.
- Kriege M, Brekelmans CT, Boetes C, et al; Magnetic Resonance Imaging Screening Study Group. Efficacy of MRI and mammography for breast-cancer screening in women with a familial or genetic predisposition. N Engl J Med 2004; 351:427–437.
- Pederson HJ, O’Rourke C, Lyons J, Patrick RJ, Crowe JP Jr, Grobmyer SR. Time-related changes in yield and harms of screening breast magnetic resonance imaging. Clin Breast Cancer 2015 Jan 21: S1526-8209(15)00024–00025. Epub ahead of print.
- Grann VR, Patel PR, Jacobson JS, et al. Comparative effectiveness of screening and prevention strategies among BRCA1/2-affected mutation carriers. Breast Cancer Res Treat 2011; 125:837–847.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Rebbeck TR, Friebel T, Wagner T, et al; PROSE Study Group. Effect of short term hormone replacement therapy on breast cancer risk reduction after bilateral prophylactic oophorectomy in BRCA1 and BRCA2 mutation carriers: the PROSE study group. J Clin Oncol 2005; 23:7804–7610.
- Narod SA, Brunet JS, Ghadirian P, et al; Hereditary Breast Cancer Clinical Study Group. Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: a case control study. Lancet 2000; 356:1876–1881.
- Njiaju UO, Olopade OI. Genetic determinants of breast cancer risk: a review of the current literature and issues pertaining to clinical application. Breast J 2012; 18:436–442.
- King MC, Wieand S, Hale K, et al; National Surgical Adjuvant Breast and Bowel Project. Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: National Surgical Adjuvant Breast and Bowel Project (NSABP-P1) Breast Cancer Prevention trial. JAMA 2001; 286:2251–2256.
- Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998; 90:1371–1388.
- Rebbeck TR, Friebel T, Lynch HT, et al. Bilateral prophylactic mastectomy reduces breast cancer risk in BRCA1 and BRCA2 mutation carriers: the PROSE Study Group. J Clin Oncol 2004; 22:1055–1062.
- Hartmann LC, Schaid DJ, Woods JE, et al. Efficacy of bilateral prophylactic mastectomy in women with a family history of breast cancer. N Engl J Med 1999; 340:77–84.
- Mallon P, Feron JG, Couturaud B, et al. The role of nipple-sparing mastectomy in breast cancer: a comprehensive review of the literature. Plast Reconstr Surg 2013; 131:969–984.
- Stanec Z, Žic R, Budi S, et al. Skin and nipple-areola complex sparing mastectomy in breast cancer patients: 15-year experience. Ann Plast Surg 2014; 73:485–491.
- Eisenberg RE, Chan JS, Swistel AJ, Hoda SA. Pathological evaluation of nipple-sparing mastectomies with emphasis on occult nipple involvement: the Weill-Cornell experience with 325 cases. Breast J 2014; 20:15–21.
- Lodder LN, Frets PG, Trijsburg RW, et al. One year follow-up of women opting for presymptomatic testing for BRCA1 and BRCA2: emotional impact of the test outcome and decisions on risk management (surveillance or prophylactic surgery). Breast Cancer Res Treat 2002; 73:97–112.
- Rebbeck TR, Lynch HT, Neuhausen SL, et al; Prevention and Observation of Surgical End Points Study Group. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 2002; 346:1616–1622.
- Domchek SM, Friebel TM, Neuhausen SL, et al. Mortality after bilateral salpingo-oophorectomy in BRCA1 and BRCA2 mutation carriers: a prospective cohort study. Lancet Oncol 2006; 7:223–229.
- Finch A, Evans G, Narod SA. BRCA carriers, prophylactic salpingo-oophorectomy and menopause: clinical management considerations and recommendations. Womens Health (Lond Engl) 2012; 8:543–555.
- National Comprehensive Cancer Network Guidelines. Version 2.2015. Genetic/Familial High-Risk Assessment Breast and Ovarian. www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed February 8, 2016.
KEY POINTS
- In addition to breast cancer, hereditary cancer syndromes increase the risk of other malignancies, with the patterns of malignancy varying by causative genetic mutation.
- Genetic counselors, medical breast specialists, surgical breast specialists, gynecologic oncologists, and others can help, but the primary care provider is the nucleus of the multidisciplinary team.
- Management of these patients often includes surveillance, chemoprevention, and prophylactic surgery.
- All decisions about surveillance, chemoprevention, and surgical risk reduction should be shared with the patient.

Blood pressure management in the wake of SPRINT
High blood pressure is a major cause of morbidity and death worldwide.1 Observational data from the general population show a log-linear relationship between both systolic and diastolic blood pressure and the rate of cardiovascular death.2 Placebo-controlled trials have shown a clear-cut benefit in treating moderate to severe hypertension based on diastolic pressure in initial trials, and systolic pressure subsequently.3 What remains uncertain is the optimal target for a particular patient, and whether other factors such as number of medications, starting blood pressure, and other comorbidities should influence this target.
Publication of the Systolic Blood Pressure Intervention Trial (SPRINT) furthered the debate regarding the optimal blood pressure target in hypertension treatment.4 SPRINT randomized 9,361 nondiabetic persons with systolic pressure higher than 130 mm Hg and increased cardiovascular risk but without prior stroke to intensive therapy (goal systolic pressure < 120 mm Hg) or standard therapy as control (goal systolic pressure < 140 mm Hg) and showed a significant reduction in the composite end point and all-cause mortality—at the expense of an increase in serious adverse events.
EARLIER TRIALS WERE GENERALLY NEGATIVE
Before SPRINT, approximately 20 randomized controlled trials attempted to define whether a more intensive target was better than standard control. These included the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial restricted to patients with diabetes5 and the Secondary Prevention of Small Subcortical Strokes (SPS3) trial restricted to patients with lacunar infarcts.6 These two groups of patients were specifically excluded from SPRINT.6 Many of the other trials had primary renal end points, although several had primary cardiovascular end points.
As we reviewed previously in this Journal, individually these trials were generally inconclusive.7 When analyzed by meta-analysis, a significant benefit was found for cardiovascular events, stroke, and end-stage renal disease, with a marginal benefit for myocardial infarction.8 The validity of such analysis may be questioned due to heterogeneous populations, lack of individual patient data, different blood pressure targets and medication regimens, and different primary end points.
Together, ACCORD in patients with diabetes, SPS3 in patients with stroke, and SPRINT in patients at increased cardiovascular risk but without diabetes or stroke cover most hypertensive patients with more than low cardiovascular risk. All three trials were government-funded, and ACCORD and SPRINT used the same blood pressure targets and treatment algorithm. It remains speculative why ACCORD was essentially negative and SPRINT was positive.
CAUTION IN GENERALIZING THE RESULTS
In this issue of the Journal, Thomas and colleagues9 review the SPRINT results in detail and attempt to reconcile the disparity with ACCORD.
We agree with their interpretation that risks and benefits of a more intensive blood pressure target (ie, < 120 mm Hg systolic) need to be addressed in the individual patient and do not apply across the board to all hypertensive patients. This more intensive target would be appropriate for patients fulfilling criteria for entry into SPRINT, ie, no diabetes or prior stroke. They must be able to tolerate more intensive therapy and should not be frail or at risk for falls. Furthermore, the increased hypertension medication burden required for stricter control will increase side effects and complexity of overall medication regimens, and will possibly foster noncompliance.
In our opinion, one must be careful in generalizing the results of SPRINT to more than the type of patient enrolled. At best, one can say that a lower target is acceptable in a patient over age 50 at increased cardiovascular risk but without diabetes or stroke.
SPRINT may not even be representative of all such patients, however. Patients requiring more than four medications were excluded from the trial, as were patients with systolic pressure higher than 180 mm Hg, or with pressure higher than 170 mm Hg requiring two medications, or with pressure higher than 160 mm Hg requiring three medications, or with pressure higher than 150 mm Hg requiring four medications. Hence, SPRINT has not determined the appropriate approach to the patient with a systolic pressure between 150 and 180 mm Hg already on multiple medications above these cutoffs. It is not hard to envision the potential for adverse events and drug interactions using four or more antihypertensive medications to achieve a lower target, in addition to other classes of medications that many patients need.
The average systolic pressure on entry into SPRINT was 139 mm Hg, and patients were taking an average of 1.8 medications. In fact, one-third of patients had systolic pressures between 130 and 132 mm Hg, a range where most physicians would probably not want to intensify therapy. By protocol, such patients in the standard treatment group in SPRINT would actually have had their baseline antihypertensive therapy reduced if the systolic pressure fell below 130 mm Hg on one occasion or below 135 mm Hg on two consecutive visits. Reduction of therapy would seem to bias the trial against the standard treatment. An identical algorithm was used in ACCORD.
We are unable to reconcile the differences in outcome between ACCORD and SPRINT, although they were congruent in one important aspect: significantly higher rates of serious adverse events with more intensive therapy. ACCORD had fewer patients, but they were at higher risk since all had diabetes, and more had previous cardiovascular events (34% vs 17% in SPRINT). This is reflected in higher event rates:
- Myocardial infarction occurred in 1.13% per year in the intensive therapy group, and 1.28% per year with standard therapy in ACCORD, compared with 0.65% and 0.78% per year, respectively, in SPRINT.
- Cardiovascular death occurred in 0.52% per year with intensive therapy and 0.49% per year with standard therapy in ACCORD, compared with 0.25% and 0.43% per year, respectively, in SPRINT. Event rates for stroke were similar.
Overall, 445 primary end points occurred in ACCORD compared with 562 with SPRINT. After subtracting heart failure from the SPRINT data (not included in the primary end point of ACCORD), 400 events occurred, actually less than in ACCORD. The early termination of SPRINT may be partly to blame. In our opinion ACCORD and SPRINT were equally powered. While cardiovascular event risk reductions in ACCORD trended in the same direction as those in SPRINT, the total mortality rate trended in the opposite direction. Perhaps the play of chance is the best explanation.
ONE TARGET DOES NOT FIT ALL
SPRINT clearly added much needed data, but results should be interpreted in the context of previous trials as well as of the specific inclusion and exclusion criteria. One target does not fit all, and systolic pressure of less than 120 mm Hg should not automatically be the target for all hypertensive patients.
Should patients with diabetes be targeted to systolic pressure of less than 140 mm Hg based on the ACCORD results, and patients with stroke to systolic pressure of less than 130 mm Hg based on the SPS3 results? We are unsure. More data are clearly required, especially in patients already on multiple antihypertensive medications with unacceptable blood pressure.
As pointed out by Thomas and colleagues, lower systolic pressure may be better in select patients, but only as long as adverse events can be avoided or managed.
- Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012; 380:2224–2260.
- Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- SPS3 Study Group; Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013; 382:507–515.
- Filippone EJ, Foy A, Newman E. Goal-directed antihypertensive therapy: lower may not always be better. Cleve Clin J Med 2011; 78:123–133.
- Lv J, Neal B, Ehteshami P, et al. Effects of intensive blood pressure lowering on cardiovascular and renal outcomes: a systematic review and meta-analysis. PLoS Med 2012; 9:e1001293.
- Thomas G, Nally JV, Pohl MA. Interpreting SPRINT: how low should you go? Cleve Clin J Med 2016; 83:187–195.
High blood pressure is a major cause of morbidity and death worldwide.1 Observational data from the general population show a log-linear relationship between both systolic and diastolic blood pressure and the rate of cardiovascular death.2 Placebo-controlled trials have shown a clear-cut benefit in treating moderate to severe hypertension based on diastolic pressure in initial trials, and systolic pressure subsequently.3 What remains uncertain is the optimal target for a particular patient, and whether other factors such as number of medications, starting blood pressure, and other comorbidities should influence this target.
Publication of the Systolic Blood Pressure Intervention Trial (SPRINT) furthered the debate regarding the optimal blood pressure target in hypertension treatment.4 SPRINT randomized 9,361 nondiabetic persons with systolic pressure higher than 130 mm Hg and increased cardiovascular risk but without prior stroke to intensive therapy (goal systolic pressure < 120 mm Hg) or standard therapy as control (goal systolic pressure < 140 mm Hg) and showed a significant reduction in the composite end point and all-cause mortality—at the expense of an increase in serious adverse events.
EARLIER TRIALS WERE GENERALLY NEGATIVE
Before SPRINT, approximately 20 randomized controlled trials attempted to define whether a more intensive target was better than standard control. These included the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial restricted to patients with diabetes5 and the Secondary Prevention of Small Subcortical Strokes (SPS3) trial restricted to patients with lacunar infarcts.6 These two groups of patients were specifically excluded from SPRINT.6 Many of the other trials had primary renal end points, although several had primary cardiovascular end points.
As we reviewed previously in this Journal, individually these trials were generally inconclusive.7 When analyzed by meta-analysis, a significant benefit was found for cardiovascular events, stroke, and end-stage renal disease, with a marginal benefit for myocardial infarction.8 The validity of such analysis may be questioned due to heterogeneous populations, lack of individual patient data, different blood pressure targets and medication regimens, and different primary end points.
Together, ACCORD in patients with diabetes, SPS3 in patients with stroke, and SPRINT in patients at increased cardiovascular risk but without diabetes or stroke cover most hypertensive patients with more than low cardiovascular risk. All three trials were government-funded, and ACCORD and SPRINT used the same blood pressure targets and treatment algorithm. It remains speculative why ACCORD was essentially negative and SPRINT was positive.
CAUTION IN GENERALIZING THE RESULTS
In this issue of the Journal, Thomas and colleagues9 review the SPRINT results in detail and attempt to reconcile the disparity with ACCORD.
We agree with their interpretation that risks and benefits of a more intensive blood pressure target (ie, < 120 mm Hg systolic) need to be addressed in the individual patient and do not apply across the board to all hypertensive patients. This more intensive target would be appropriate for patients fulfilling criteria for entry into SPRINT, ie, no diabetes or prior stroke. They must be able to tolerate more intensive therapy and should not be frail or at risk for falls. Furthermore, the increased hypertension medication burden required for stricter control will increase side effects and complexity of overall medication regimens, and will possibly foster noncompliance.
In our opinion, one must be careful in generalizing the results of SPRINT to more than the type of patient enrolled. At best, one can say that a lower target is acceptable in a patient over age 50 at increased cardiovascular risk but without diabetes or stroke.
SPRINT may not even be representative of all such patients, however. Patients requiring more than four medications were excluded from the trial, as were patients with systolic pressure higher than 180 mm Hg, or with pressure higher than 170 mm Hg requiring two medications, or with pressure higher than 160 mm Hg requiring three medications, or with pressure higher than 150 mm Hg requiring four medications. Hence, SPRINT has not determined the appropriate approach to the patient with a systolic pressure between 150 and 180 mm Hg already on multiple medications above these cutoffs. It is not hard to envision the potential for adverse events and drug interactions using four or more antihypertensive medications to achieve a lower target, in addition to other classes of medications that many patients need.
The average systolic pressure on entry into SPRINT was 139 mm Hg, and patients were taking an average of 1.8 medications. In fact, one-third of patients had systolic pressures between 130 and 132 mm Hg, a range where most physicians would probably not want to intensify therapy. By protocol, such patients in the standard treatment group in SPRINT would actually have had their baseline antihypertensive therapy reduced if the systolic pressure fell below 130 mm Hg on one occasion or below 135 mm Hg on two consecutive visits. Reduction of therapy would seem to bias the trial against the standard treatment. An identical algorithm was used in ACCORD.
We are unable to reconcile the differences in outcome between ACCORD and SPRINT, although they were congruent in one important aspect: significantly higher rates of serious adverse events with more intensive therapy. ACCORD had fewer patients, but they were at higher risk since all had diabetes, and more had previous cardiovascular events (34% vs 17% in SPRINT). This is reflected in higher event rates:
- Myocardial infarction occurred in 1.13% per year in the intensive therapy group, and 1.28% per year with standard therapy in ACCORD, compared with 0.65% and 0.78% per year, respectively, in SPRINT.
- Cardiovascular death occurred in 0.52% per year with intensive therapy and 0.49% per year with standard therapy in ACCORD, compared with 0.25% and 0.43% per year, respectively, in SPRINT. Event rates for stroke were similar.
Overall, 445 primary end points occurred in ACCORD compared with 562 with SPRINT. After subtracting heart failure from the SPRINT data (not included in the primary end point of ACCORD), 400 events occurred, actually less than in ACCORD. The early termination of SPRINT may be partly to blame. In our opinion ACCORD and SPRINT were equally powered. While cardiovascular event risk reductions in ACCORD trended in the same direction as those in SPRINT, the total mortality rate trended in the opposite direction. Perhaps the play of chance is the best explanation.
ONE TARGET DOES NOT FIT ALL
SPRINT clearly added much needed data, but results should be interpreted in the context of previous trials as well as of the specific inclusion and exclusion criteria. One target does not fit all, and systolic pressure of less than 120 mm Hg should not automatically be the target for all hypertensive patients.
Should patients with diabetes be targeted to systolic pressure of less than 140 mm Hg based on the ACCORD results, and patients with stroke to systolic pressure of less than 130 mm Hg based on the SPS3 results? We are unsure. More data are clearly required, especially in patients already on multiple antihypertensive medications with unacceptable blood pressure.
As pointed out by Thomas and colleagues, lower systolic pressure may be better in select patients, but only as long as adverse events can be avoided or managed.
High blood pressure is a major cause of morbidity and death worldwide.1 Observational data from the general population show a log-linear relationship between both systolic and diastolic blood pressure and the rate of cardiovascular death.2 Placebo-controlled trials have shown a clear-cut benefit in treating moderate to severe hypertension based on diastolic pressure in initial trials, and systolic pressure subsequently.3 What remains uncertain is the optimal target for a particular patient, and whether other factors such as number of medications, starting blood pressure, and other comorbidities should influence this target.
Publication of the Systolic Blood Pressure Intervention Trial (SPRINT) furthered the debate regarding the optimal blood pressure target in hypertension treatment.4 SPRINT randomized 9,361 nondiabetic persons with systolic pressure higher than 130 mm Hg and increased cardiovascular risk but without prior stroke to intensive therapy (goal systolic pressure < 120 mm Hg) or standard therapy as control (goal systolic pressure < 140 mm Hg) and showed a significant reduction in the composite end point and all-cause mortality—at the expense of an increase in serious adverse events.
EARLIER TRIALS WERE GENERALLY NEGATIVE
Before SPRINT, approximately 20 randomized controlled trials attempted to define whether a more intensive target was better than standard control. These included the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial restricted to patients with diabetes5 and the Secondary Prevention of Small Subcortical Strokes (SPS3) trial restricted to patients with lacunar infarcts.6 These two groups of patients were specifically excluded from SPRINT.6 Many of the other trials had primary renal end points, although several had primary cardiovascular end points.
As we reviewed previously in this Journal, individually these trials were generally inconclusive.7 When analyzed by meta-analysis, a significant benefit was found for cardiovascular events, stroke, and end-stage renal disease, with a marginal benefit for myocardial infarction.8 The validity of such analysis may be questioned due to heterogeneous populations, lack of individual patient data, different blood pressure targets and medication regimens, and different primary end points.
Together, ACCORD in patients with diabetes, SPS3 in patients with stroke, and SPRINT in patients at increased cardiovascular risk but without diabetes or stroke cover most hypertensive patients with more than low cardiovascular risk. All three trials were government-funded, and ACCORD and SPRINT used the same blood pressure targets and treatment algorithm. It remains speculative why ACCORD was essentially negative and SPRINT was positive.
CAUTION IN GENERALIZING THE RESULTS
In this issue of the Journal, Thomas and colleagues9 review the SPRINT results in detail and attempt to reconcile the disparity with ACCORD.
We agree with their interpretation that risks and benefits of a more intensive blood pressure target (ie, < 120 mm Hg systolic) need to be addressed in the individual patient and do not apply across the board to all hypertensive patients. This more intensive target would be appropriate for patients fulfilling criteria for entry into SPRINT, ie, no diabetes or prior stroke. They must be able to tolerate more intensive therapy and should not be frail or at risk for falls. Furthermore, the increased hypertension medication burden required for stricter control will increase side effects and complexity of overall medication regimens, and will possibly foster noncompliance.
In our opinion, one must be careful in generalizing the results of SPRINT to more than the type of patient enrolled. At best, one can say that a lower target is acceptable in a patient over age 50 at increased cardiovascular risk but without diabetes or stroke.
SPRINT may not even be representative of all such patients, however. Patients requiring more than four medications were excluded from the trial, as were patients with systolic pressure higher than 180 mm Hg, or with pressure higher than 170 mm Hg requiring two medications, or with pressure higher than 160 mm Hg requiring three medications, or with pressure higher than 150 mm Hg requiring four medications. Hence, SPRINT has not determined the appropriate approach to the patient with a systolic pressure between 150 and 180 mm Hg already on multiple medications above these cutoffs. It is not hard to envision the potential for adverse events and drug interactions using four or more antihypertensive medications to achieve a lower target, in addition to other classes of medications that many patients need.
The average systolic pressure on entry into SPRINT was 139 mm Hg, and patients were taking an average of 1.8 medications. In fact, one-third of patients had systolic pressures between 130 and 132 mm Hg, a range where most physicians would probably not want to intensify therapy. By protocol, such patients in the standard treatment group in SPRINT would actually have had their baseline antihypertensive therapy reduced if the systolic pressure fell below 130 mm Hg on one occasion or below 135 mm Hg on two consecutive visits. Reduction of therapy would seem to bias the trial against the standard treatment. An identical algorithm was used in ACCORD.
We are unable to reconcile the differences in outcome between ACCORD and SPRINT, although they were congruent in one important aspect: significantly higher rates of serious adverse events with more intensive therapy. ACCORD had fewer patients, but they were at higher risk since all had diabetes, and more had previous cardiovascular events (34% vs 17% in SPRINT). This is reflected in higher event rates:
- Myocardial infarction occurred in 1.13% per year in the intensive therapy group, and 1.28% per year with standard therapy in ACCORD, compared with 0.65% and 0.78% per year, respectively, in SPRINT.
- Cardiovascular death occurred in 0.52% per year with intensive therapy and 0.49% per year with standard therapy in ACCORD, compared with 0.25% and 0.43% per year, respectively, in SPRINT. Event rates for stroke were similar.
Overall, 445 primary end points occurred in ACCORD compared with 562 with SPRINT. After subtracting heart failure from the SPRINT data (not included in the primary end point of ACCORD), 400 events occurred, actually less than in ACCORD. The early termination of SPRINT may be partly to blame. In our opinion ACCORD and SPRINT were equally powered. While cardiovascular event risk reductions in ACCORD trended in the same direction as those in SPRINT, the total mortality rate trended in the opposite direction. Perhaps the play of chance is the best explanation.
ONE TARGET DOES NOT FIT ALL
SPRINT clearly added much needed data, but results should be interpreted in the context of previous trials as well as of the specific inclusion and exclusion criteria. One target does not fit all, and systolic pressure of less than 120 mm Hg should not automatically be the target for all hypertensive patients.
Should patients with diabetes be targeted to systolic pressure of less than 140 mm Hg based on the ACCORD results, and patients with stroke to systolic pressure of less than 130 mm Hg based on the SPS3 results? We are unsure. More data are clearly required, especially in patients already on multiple antihypertensive medications with unacceptable blood pressure.
As pointed out by Thomas and colleagues, lower systolic pressure may be better in select patients, but only as long as adverse events can be avoided or managed.
- Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012; 380:2224–2260.
- Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- SPS3 Study Group; Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013; 382:507–515.
- Filippone EJ, Foy A, Newman E. Goal-directed antihypertensive therapy: lower may not always be better. Cleve Clin J Med 2011; 78:123–133.
- Lv J, Neal B, Ehteshami P, et al. Effects of intensive blood pressure lowering on cardiovascular and renal outcomes: a systematic review and meta-analysis. PLoS Med 2012; 9:e1001293.
- Thomas G, Nally JV, Pohl MA. Interpreting SPRINT: how low should you go? Cleve Clin J Med 2016; 83:187–195.
- Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012; 380:2224–2260.
- Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- SPS3 Study Group; Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013; 382:507–515.
- Filippone EJ, Foy A, Newman E. Goal-directed antihypertensive therapy: lower may not always be better. Cleve Clin J Med 2011; 78:123–133.
- Lv J, Neal B, Ehteshami P, et al. Effects of intensive blood pressure lowering on cardiovascular and renal outcomes: a systematic review and meta-analysis. PLoS Med 2012; 9:e1001293.
- Thomas G, Nally JV, Pohl MA. Interpreting SPRINT: how low should you go? Cleve Clin J Med 2016; 83:187–195.
Interpreting SPRINT: How low should you go?
In treating hypertension, lower systolic pressure is better than higher—with caveats. This is the message of the Systolic Blood Pressure Intervention Trial (SPRINT),1 a large, federally funded study that was halted early when patients at high cardiovascular risk who were randomized to a goal systolic pressure of less than 120 mm Hg were found to have better outcomes, including lower rates of heart failure, death from cardiovascular causes, and death from any cause, than patients randomized to a goal of less than 140 mm Hg.
The caveats: the benefit came at a price of more adverse events. Also, the trial excluded patients who had diabetes mellitus or previous strokes, so it is uncertain if these subgroups would also benefit from intensive lowering of systolic pressure—and in earlier trials they did not.
This article reviews the trial design and protocol, summarizes the results, and briefly discusses the implications of these results.
BEFORE SPRINT
Hypertension is very common in adults in the United States, and is a risk factor for heart disease, stroke, heart failure, and kidney disease. The estimated prevalence of hypertension in the 2011–2014 National Health and Nutrition Examination Survey (NHANES) was 29%, and the prevalence increases with age (7.3% in those ages 18 to 39, 32.2% in those ages 40 to 59, and 64.9% in those ages 60 and older).2 Isolated systolic hypertension (ie, systolic blood pressure > 140 mm Hg with diastolic pressure < 90 mm Hg) is the most common form of hypertension after age 50.3
Clinical trials have provided substantial evidence that treating hypertension reduces the incidence of stroke, myocardial infarction, and heart failure.4,5 Although observational studies show a progressive and linear rise in cardiovascular risk as systolic blood pressure rises above 115 mm Hg,6 clinical trials in the general population have not documented benefits of lowering systolic pressure to this level.7–11 However, clinical trials that directly evaluated two different blood pressure goals in the general population showed benefit with achieving systolic blood pressure less than 150 mm Hg,7,9 with limited data on lower blood pressure targets.10–12
No benefit found in intensive systolic lowering in diabetes or after stroke
The Action to Control Cardiovascular Risk in Diabetes-Blood Pressure (ACCORD BP) trial13 in patients with type 2 diabetes found no benefit in lowering systolic pressure to less than 120 mm Hg compared with less than 140 mm Hg in terms of the trial’s primary composite cardiovascular outcome (ie, nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes). However, the intensively treated group in this trial did enjoy a benefit in terms of fewer stroke events.
The Secondary Prevention of Small Subcortical Strokes (SPS3) trial14 in patients with stroke found no significant benefit in lowering systolic pressure to less than 130 mm Hg compared with less than 150 mm Hg for overall risk of another stroke, but a significant benefit was noted in reduced risk of intracerebral hemorrhage.
Current guidelines, based on available evidence, advocate treatment to a systolic goal of less than 140 mm Hg in most patients, and recommend relaxing this goal to less than 150 mm Hg in the elderly.15,16
Given the uncertainty surrounding optimal systolic targets, SPRINT was designed to test the hypothesis that a goal of less than 120 mm Hg would reduce the risk of cardiovascular events more than the generally accepted systolic goal of less than 140 mm Hg.17 Patients with diabetes and stroke were excluded because a similar hypothesis was tested in the ACCORD BP and SPS3 trials, which included patients with these conditions.
SPRINT DESIGN
SPRINT was a randomized, controlled, open-label trial sponsored by the National Institutes of Health and conducted at 102 US sites.
Inclusion criteria. Participants had to be at least 50 years old, with systolic pressure of 130 to 180 mm Hg, and had to have at least one cardiovascular risk factor, eg:
- Clinical or subclinical cardiovascular disease (other than stroke)
- Chronic kidney disease, defined as estimated glomerular filtration rate (eGFR), calculated by the Modification of Diet in Renal Disease (MDRD) study equation, of 20 to less than 60 mL/min/1.73 m2
- Framingham risk score of 15% of more
- Age 75 or older.
Major exclusion criteria included:
- Diabetes
- Stroke
- Polycystic kidney disease
- Chronic kidney disease with an eGFR less than 20 mL/min/1.73 m2
- Proteinuria (excretion > 1 g/day).
Intensive vs standard treatment
Participants were randomized to receive intensive treatment (systolic goal < 120 mm Hg) or standard treatment (systolic goal < 140 mm Hg). Baseline antihypertensive medications were adjusted to achieve blood pressure goals based on randomization assignment.
Doses of medications were adjusted on the basis of an average of three seated office blood pressure measurements after a 5-minute period of rest, taken with an automated monitor (Omron Healthcare Model 907); the same monitor was used and the same protocol was followed at all participating sites. Blood pressure was also measured after standing for 1 minute to assess orthostatic change.
Lifestyle modifications were encouraged in both groups. There was no restriction on using any antihypertensive medication, and this was at the discretion of individual investigators. Thiazide-type diuretics were encouraged as first-line agents (with chlorthalidone encouraged as the primary thiazide-type diuretic).
Outcomes measured
The primary outcome was a composite of myocardial infarction, acute coronary syndrome not resulting in myocardial infarction, stroke, acute decompensated heart failure, and cardiovascular mortality.
Secondary outcomes included individual components of the primary composite outcome, all-cause mortality, and the composite of primary outcome and all-cause mortality.
Renal outcomes were assessed as:
- Incident albuminuria (doubling of the urinary albumin-to-creatinine ratio from less than 10 mg/g to more than 10 mg/g)
- Composite of a 50% decrease in eGFR or development of end-stage renal disease requiring long-term dialysis or kidney transplantation (in those with baseline chronic kidney disease)
- A 30% decrease in eGFR (in those without chronic kidney disease).1,17
SPRINT also recruited participants to two nested substudies: SPRINT MIND and SPRINT MIND MRI, to study differences in cognitive outcomes and small-vessel ischemic disease between intensive treatment and standard treatment.
STUDY RESULTS
Older patients at risk, but without diabetes
Of 14,692 participants screened, 9,361 were enrolled in the study between 2010 and 2013. Baseline characteristics were comparable in both groups.
Demographics. The mean age of the participants was 67.9, and about 28% were 75 or older. About 36% were women, 58% white, 30% black, and 11% Hispanic.
Cardiovascular risk. The mean Framingham risk score was 20% (ie, they had a 20% risk of having a cardiovascular event within 10 years), and 61% of the participants had a risk score of at least 15%. Twenty percent already had cardiovascular disease.
Blood pressure. The average baseline blood pressure was 139.7/78.2 mm Hg. One-third of the participants had baseline systolic pressures of 132 mm Hg or less, another third had pressures in the range of 132 to 145, and the rest had 145 mm Hg or higher.
Renal function. The mean serum creatinine level was about 1.1 mg/dL. The mean eGFR was about 71 mL/min/1.73 m2 as calculated by the MDRD equation, and about 28% had eGFRs less than 60. The mean ratio of urinary albumin to creatinine was 44.1 mg/g in the intensive treatment group and 41.1 in the standard treatment group.
Other. The mean total cholesterol level was 190 mg/dL, fasting plasma glucose 99 mg/dL, and body mass index nearly 30 kg/m2.
Blood pressure during treatment
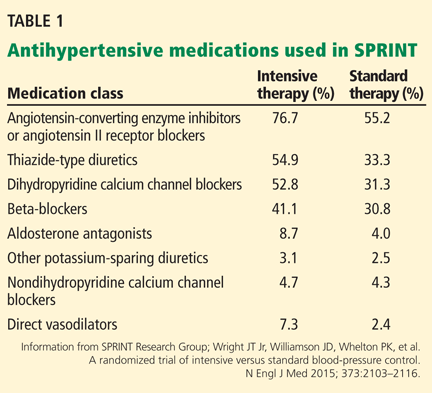
People in the intensive treatment group were taking a mean of 2.8 antihypertensive medications, and those in the standard treatment group were taking 1.8. Patients in the intensive group required greater use of all classes of medications to achieve goal systolic pressure (Table 1).
Study halted early due to efficacy
Throughout the 3.26 years of follow-up, the average difference in systolic pressure between the two groups was 13.1 mm Hg, with a mean systolic pressure of 121.5 mm Hg in the intensive treatment group and 134.6 mm Hg in the standard treatment group. The mean diastolic blood pressure was 68.7 mm Hg in the intensive treatment group and 76.3 mm Hg in the standard treatment group.
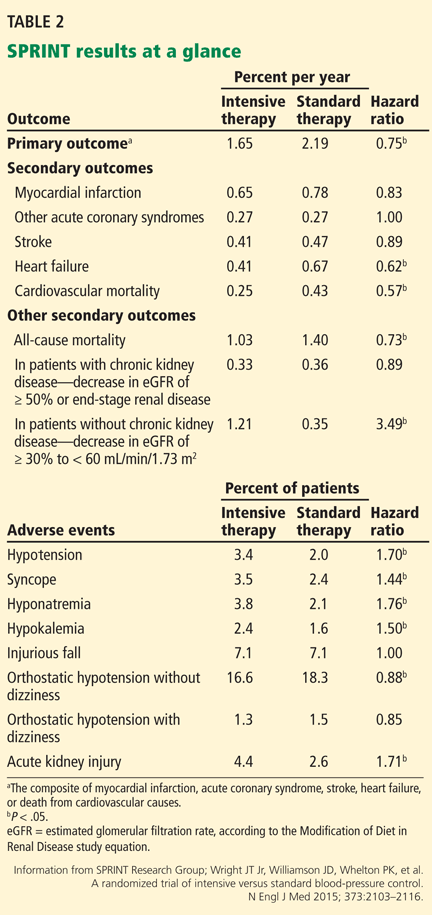
Although the study was planned to run for an average follow-up of 5 years, the National Heart, Lung, and Blood Institute terminated it early at a median of 3.26 years in view of lower rates of the primary outcome and of heart failure and death in the intensive treatment group (Table 2).
The effects on the primary outcome and mortality were consistent across the prespecified subgroups of age (< 75 vs ≥ 75), sex (female vs male), race (black vs nonblack), cardiovascular disease (presence or absence at baseline), prior chronic kidney disease (presence or absence at baseline), and across blood pressure tertiles (≤ 132 mm Hg, > 132 to < 145 mm Hg, ≥ 145 mm Hg).
Follow-up for assessment of cognitive outcomes (SPRINT MIND) and small-vessel ischemic disease (SPRINT MIND MRI) is ongoing.
WHAT DOES THIS MEAN?
SPRINT is the first large, adequately powered, randomized trial to demonstrate cardiovascular and mortality benefit from lowering the systolic blood pressure (goal < 120 mm Hg) in older patients at cardiovascular risk but without a history of diabetes mellitus or stroke.1
Most SPRINT patients had reasonably controlled blood pressure at baseline (the mean systolic pressure was 139.7 mm Hg, and two-thirds of participants had systolic pressure < 145 mm Hg). Of note, however, this trial excluded patients with systolic pressure higher than 180 mm Hg. There was excellent separation of systolic pressure between the two groups beginning at 1 year, which was consistent through the course of the trial.
The cardiovascular benefit in the intensive treatment group was predominantly driven by lower rates of heart failure (a 38% reduction in the intensive treatment group, P = .0002) and cardiovascular mortality (a 43% reduction in the intensive treatment group, P = .005), while there was no significant difference between the two groups in myocardial infarction or stroke. The beneficial effect on heart failure events is consistent with results from other trials including the Systolic Hypertension in the Elderly Program,7 Systolic Hypertension in Europe,8 and Hypertension in the Very Elderly Trial,9 all of which showed greatest risk reduction for heart failure events with systolic pressure-lowering (although to higher systolic levels than SPRINT).7–9 It is unclear why there was no beneficial effect on stroke events. The reduction in all-cause mortality in the intensive treatment group in SPRINT was greater than the reduction in cardiovascular deaths, which is also unexplained.
Although the study was terminated early due to efficacy (which introduces the possible bias that the estimated effect size will be too high), the number of primary end points reached was large (562 in the two groups combined), providing reassurance that the findings are valid. There was no blinding in the study (both participants and study investigators were aware of treatment assignment and study medications), but there was a structured assessment of outcomes and adverse events, with adjudication done by blinded reviewers.
SPRINT used an automated device for blood pressure measurement, which is known to reduce the “white coat” effect and correlates tightly with average daytime blood pressure done by ambulatory blood pressure monitoring.18 However, in clinical practice automated devices may not be available and a strict protocol for correct measurement may not be followed, with the possible result that blood pressure may be overestimated and overtreated.
What about diastolic pressure?
The trial, by design, focused on lowering systolic pressure (given the greater prevalence of isolated systolic hypertension with age), and the implications of lowering diastolic pressure are unclear. The issue of a J-shaped relationship between diastolic pressure and cardiovascular risk is debated in the literature: patients with a diastolic pressure of 60 to 65 mm Hg, especially those with existing coronary artery disease, may not tolerate aggressive blood pressure-lowering.19,20 Further analysis of this association (if any) from SPRINT will be helpful.
What about patients with diabetes?
Patients were excluded from SPRINT if they were under age 50, were at low cardiovascular risk, or had diabetes, raising the question of whether the results apply to these groups as well.

The question is particularly relevant in diabetes, as the ACCORD BP study, which used the same blood pressure targets as SPRINT, did not show a significant difference in the primary cardiovascular outcome between the intensive and standard treatments in patients with diabetes (Table 3).13 In ACCORD BP, the rate of the primary outcome was 12% lower in the intensive treatment group than in the standard treatment group, but the 95% confidence interval was –27% to +6%, so the finding was not statistically significant. However, the wide confidence interval does not exclude the possibility of a benefit that was comparable to that observed in SPRINT.
It has been speculated that ACCORD BP was underpowered to detect significant differences in the primary outcome.21 An analysis combining data from both trials indicated that effects on individual outcomes were generally consistent in both trials (with no significant heterogeneity noted).22 Also, the primary composite outcome in ACCORD did not include heart failure, which is particularly sensitive to blood pressure reduction.
Additionally, ACCORD BP had a 2 × 2 factorial design involving a simultaneous comparison of intensive vs standard glycemic control, which may have influenced the effects due to blood pressure. Indeed, a post hoc analysis showed that there was a significant 26% lower risk of the primary outcome in ACCORD BP patients who received intensive systolic pressure control plus standard glycemic control than in those receiving standard systolic control plus standard glycemic control.23
Are more adverse events an acceptable trade-off?
Adverse events, including acute kidney injury, were more frequent in the intensive therapy group in SPRINT.
Acute kidney injury was coded as an adverse event on the basis of this diagnosis being included in the hospital discharge summary (as a primary or main secondary diagnosis) and if considered by the safety officer to be one of the top three reasons for admission or continued hospitalization. Further analysis of renal events should be forthcoming.
People in the intensive treatment group, on average, needed one more medication than those in the standard treatment group. Some of the adverse events may be related to the antihypertensive medications taken (eg, electrolyte abnormalities such as hyponatremia and hypokalemia due to diuretic use), and others may be related to blood pressure-lowering (eg, acute kidney injury due to renal hypoperfusion).
At this point, the long-term effects of these adverse events, especially on kidney function, are not known. Patients enrolled in clinical trials tend to be healthier than patients seen in clinical practice; thus, the rate of adverse events reported in the trial may be lower than one would see in the real world.
Does lower systolic pressure protect or harm the kidneys?
SPRINT included patients with stage 3 and 4 chronic kidney disease (ie, with eGFR 20–50 mL/min/1.73 m2), but it was designed to assess cardiovascular outcomes, not the progression of chronic kidney disease. The trial excluded patients with diabetic nephropathy or high degrees of proteinuria.
Earlier randomized trials that focused on chronic kidney disease progression, including the MDRD24 and the African American Study of Kidney Disease and Hypertension,25 did not show benefit with more aggressive blood pressure-lowering (except in patients with higher degrees of proteinuria), and these trials were not powered to assess effects on cardiovascular outcomes.24,25
The Irbesartan Diabetic Nephropathy Trial,26,27 which was done in patients with overt diabetic nephropathy, showed that a progressively lower achieved systolic pressure down to 120 mm Hg predicted lower rates of heart failure, cardiovascular mortality, and renal events (although the trial target was ≤ 130/85 mm Hg and few participants achieved systolic pressure lower than 120 mm Hg).
IMPLICATIONS FOR MANAGEMENT
The recent estimates of hypertension prevalence and control from NHANES show that only about 53% of hypertensive adults have their blood pressure under control (defined as systolic pressure < 140 mm Hg and diastolic pressure < 90 mm Hg).2 Analysis of the NHANES 2007–2012 data showed that 16.7% or 8.2 million US adults with treated hypertension meet the eligibility criteria for SPRINT.28
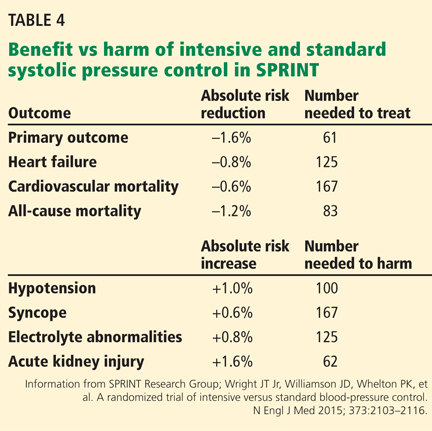
Although the SPRINT results support the notion that “lower is better,” the risks and benefits of intensive control will need to be balanced in individual patients. Table 4 shows the number needed to treat and number needed to harm in the trial.
More aggressive management of hypertension is challenging. The median systolic pressure achieved in the intensive group in SPRINT was just over 120 mm Hg, which implies that at least half of the participants in the intensive group did not achieve the goal of less than 120 mm Hg. While it may be reasonable to aim for systolic pressure of less than 120 or 125 mm Hg in patients who fit the SPRINT criteria and can tolerate intensive blood pressure lowering, it would be prudent to aim for a more conservative goal in elderly patients who are frail and at risk for falls, considering the higher incidence of specified adverse events in the intensive group.
Results of cognitive outcomes, as well as data related to quality of life, are still awaited. Long-term renal outcomes are also unclear.
As noted above, the question of generalizability of SPRINT results to patients with diabetes is open to debate. In our opinion, with currently available evidence, it is difficult to conclusively answer the question of whether a lower systolic target provides cardiovascular benefit in diabetes. It is also unclear whether similar beneficial results would be seen with intensive treatment in a population at low cardiovascular risk. The American Heart Association and the American College of Cardiology are in the process of formulating new hypertension guidelines, and evidence from SPRINT will inform any new recommendations.
As more medications will likely be needed for intensive systolic blood pressure control, side effects and tolerability of medications with polypharmacy and potential nonadherence with increasing complexity of medication regimens should be kept in mind. Lifestyle modifications will need to be emphasized and reinforced, with greater use of combination antihypertensive therapy.
The data from SPRINT indicate that lower systolic pressure is better, as long as untoward clinical events can be monitored and avoided or easily managed. Careful monitoring will likely entail more frequent clinic visits and more frequent assessment of renal function and electrolyte levels (participants in the intensive group in the trial were seen every month until goal was achieved). A team approach that includes pharmacists and nurse practitioners, along with optimal use of best practice algorithms and remote monitoring technology, will need to be implemented for efficient and effective care.
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- Yoon SS, Fryar CD, Carroll MD. Hypertension prevalence and control among adults: United States, 2011–2014. NCHS data brief, no. 220. Hyattsville, MD: National Center for Health Statistics. 2015.
- Franklin SS, Jacobs MJ, Wong ND, L’Italien GJ, Lapuerta P. Predominance of isolated systolic hypertension among middle-aged and elderly US hypertensives: analysis based on National Health and Nutrition Examination Survey (NHANES) III. Hypertension 2001; 37:869–874.
- Neal B, MacMahon S, Chapman N; Blood Pressure Lowering Treatment Trialists’ Collaboration. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials. Blood Pressure Lowering Treatment Trialists’ Collaboration. Lancet 2000; 356:1955–1964.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Staessen JA, Fagard R, Thijs L, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet 1997; 350:757–764.
- Beckett NS, Peters R, Fletcher AE, et al; HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res 2008; 31:2115–2127.
- Ogihara T, Saruta T, Rakugi H, et al; Valsartan in Elderly Isolated Systolic Hypertension Study Group. Target blood pressure for treatment of isolated systolic hypertension in the elderly: Valsartan in Elderly Isolated Systolic Hypertension study. Hypertension 2010; 56:196–202.
- Liu L, Zhang Y, Liu G, Li W, Zhang X, Zanchetti A; FEVER Study Group. The Felodipine Event Reduction (FEVER) Study: a randomized long-term placebo-controlled trial in Chinese hypertensive patients. J Hypertens 2005; 23:2157–2172.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- SPS3 Study Group; Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013; 382:507–515.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520. Erratum in: JAMA. 2014; 311:1809.
- Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens (Greenwich) 2014; 16:14–26.
- Ambrosius WT, Sink KM, Foy CG, et al; SPRINT Study Research Group. The design and rationale of a multicenter clinical trial comparing two strategies for control of systolic blood pressure: the Systolic Blood Pressure Intervention Trial (SPRINT). Clin Trials 2014; 11:532–546.
- Myers MG, Godwin M, Dawes M, Kiss A, Tobe SW, Kaczorowski J. Conventional versus automated measurement of blood pressure in the office (CAMBO) trial. Fam Pract 2012; 29:376–382.
- Messerli FH, Mancia G, Conti CR, et al. Dogma disputed: can aggressively lowering blood pressure in hypertensive patients with coronary artery disease be dangerous? Ann Intern Med 2006; 144:884–893.
- Boutitie F, Gueyffier F, Pocock S, Fagard R, Boissel JP; INDANA Project Steering Committee; INdividual Data ANalysis of Antihypertensive intervention. J-shaped relationship between blood pressure and mortality in hypertensive patients: new insights from a meta-analysis of individual-patient data. Ann Intern Med 2002; 136:438–448.
- Mancia G. Effects of intensive blood pressure control in the management of patients with type 2 diabetes mellitus in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Circulation 2010; 122:847–849.
- Perkovic V, Rodgers A. Redefining blood-pressure targets—SPRINT starts the marathon. N Engl J Med 2015; 373:2175–2178.
- Margolis KL, O’Connor PJ, Morgan TM, et al. Outcomes of combined cardiovascular risk factor management strategies in type 2 diabetes: the ACCORD randomized trial. Diabetes Care 2014; 37:1721–1728.
- Peterson JC, Adler S, Burkart JM, et al. Blood pressure control, proteinuria, and the progression of renal disease. The Modification of Diet in Renal Disease study. Ann Intern Med 1995; 123:754–762.
- Agodoa LY, Appel L, Bakris GL, et al; African American Study of Kidney Disease and Hypertension (AASK) Study Group. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA 2001; 285:2719–2728.
- Berl T, Hunsicker LG, Lewis JB, et al; Collaborative Study Group. Impact of achieved blood pressure on cardiovascular outcomes in the Irbesartan Diabetic Nephropathy Trial. J Am Soc Nephrol 2005; 16:2170–2179.
- Pohl MA, Blumenthal S, Cordonnier DJ, et al. Independent and additive impact of blood pressure control and angiotensin II receptor blockade on renal outcomes in the Irbesartan Diabetic Nephropathy Trial: clinical implications and limitations. J Am Soc Nephrol 2005; 16:3027–3037.
- Bress AP, Tanner RM, Hess R, Colantonio LD, Shimbo D, Muntner P. Generalizability of results from the Systolic Blood Pressure Intervention Trial (SPRINT) to the US adult population. J Am Coll Cardiol 2015 Oct 31. doi: 10.1016/j.jacc.2015.10.037. Epub ahead of print.
In treating hypertension, lower systolic pressure is better than higher—with caveats. This is the message of the Systolic Blood Pressure Intervention Trial (SPRINT),1 a large, federally funded study that was halted early when patients at high cardiovascular risk who were randomized to a goal systolic pressure of less than 120 mm Hg were found to have better outcomes, including lower rates of heart failure, death from cardiovascular causes, and death from any cause, than patients randomized to a goal of less than 140 mm Hg.
The caveats: the benefit came at a price of more adverse events. Also, the trial excluded patients who had diabetes mellitus or previous strokes, so it is uncertain if these subgroups would also benefit from intensive lowering of systolic pressure—and in earlier trials they did not.
This article reviews the trial design and protocol, summarizes the results, and briefly discusses the implications of these results.
BEFORE SPRINT
Hypertension is very common in adults in the United States, and is a risk factor for heart disease, stroke, heart failure, and kidney disease. The estimated prevalence of hypertension in the 2011–2014 National Health and Nutrition Examination Survey (NHANES) was 29%, and the prevalence increases with age (7.3% in those ages 18 to 39, 32.2% in those ages 40 to 59, and 64.9% in those ages 60 and older).2 Isolated systolic hypertension (ie, systolic blood pressure > 140 mm Hg with diastolic pressure < 90 mm Hg) is the most common form of hypertension after age 50.3
Clinical trials have provided substantial evidence that treating hypertension reduces the incidence of stroke, myocardial infarction, and heart failure.4,5 Although observational studies show a progressive and linear rise in cardiovascular risk as systolic blood pressure rises above 115 mm Hg,6 clinical trials in the general population have not documented benefits of lowering systolic pressure to this level.7–11 However, clinical trials that directly evaluated two different blood pressure goals in the general population showed benefit with achieving systolic blood pressure less than 150 mm Hg,7,9 with limited data on lower blood pressure targets.10–12
No benefit found in intensive systolic lowering in diabetes or after stroke
The Action to Control Cardiovascular Risk in Diabetes-Blood Pressure (ACCORD BP) trial13 in patients with type 2 diabetes found no benefit in lowering systolic pressure to less than 120 mm Hg compared with less than 140 mm Hg in terms of the trial’s primary composite cardiovascular outcome (ie, nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes). However, the intensively treated group in this trial did enjoy a benefit in terms of fewer stroke events.
The Secondary Prevention of Small Subcortical Strokes (SPS3) trial14 in patients with stroke found no significant benefit in lowering systolic pressure to less than 130 mm Hg compared with less than 150 mm Hg for overall risk of another stroke, but a significant benefit was noted in reduced risk of intracerebral hemorrhage.
Current guidelines, based on available evidence, advocate treatment to a systolic goal of less than 140 mm Hg in most patients, and recommend relaxing this goal to less than 150 mm Hg in the elderly.15,16
Given the uncertainty surrounding optimal systolic targets, SPRINT was designed to test the hypothesis that a goal of less than 120 mm Hg would reduce the risk of cardiovascular events more than the generally accepted systolic goal of less than 140 mm Hg.17 Patients with diabetes and stroke were excluded because a similar hypothesis was tested in the ACCORD BP and SPS3 trials, which included patients with these conditions.
SPRINT DESIGN
SPRINT was a randomized, controlled, open-label trial sponsored by the National Institutes of Health and conducted at 102 US sites.
Inclusion criteria. Participants had to be at least 50 years old, with systolic pressure of 130 to 180 mm Hg, and had to have at least one cardiovascular risk factor, eg:
- Clinical or subclinical cardiovascular disease (other than stroke)
- Chronic kidney disease, defined as estimated glomerular filtration rate (eGFR), calculated by the Modification of Diet in Renal Disease (MDRD) study equation, of 20 to less than 60 mL/min/1.73 m2
- Framingham risk score of 15% of more
- Age 75 or older.
Major exclusion criteria included:
- Diabetes
- Stroke
- Polycystic kidney disease
- Chronic kidney disease with an eGFR less than 20 mL/min/1.73 m2
- Proteinuria (excretion > 1 g/day).
Intensive vs standard treatment
Participants were randomized to receive intensive treatment (systolic goal < 120 mm Hg) or standard treatment (systolic goal < 140 mm Hg). Baseline antihypertensive medications were adjusted to achieve blood pressure goals based on randomization assignment.
Doses of medications were adjusted on the basis of an average of three seated office blood pressure measurements after a 5-minute period of rest, taken with an automated monitor (Omron Healthcare Model 907); the same monitor was used and the same protocol was followed at all participating sites. Blood pressure was also measured after standing for 1 minute to assess orthostatic change.
Lifestyle modifications were encouraged in both groups. There was no restriction on using any antihypertensive medication, and this was at the discretion of individual investigators. Thiazide-type diuretics were encouraged as first-line agents (with chlorthalidone encouraged as the primary thiazide-type diuretic).
Outcomes measured
The primary outcome was a composite of myocardial infarction, acute coronary syndrome not resulting in myocardial infarction, stroke, acute decompensated heart failure, and cardiovascular mortality.
Secondary outcomes included individual components of the primary composite outcome, all-cause mortality, and the composite of primary outcome and all-cause mortality.
Renal outcomes were assessed as:
- Incident albuminuria (doubling of the urinary albumin-to-creatinine ratio from less than 10 mg/g to more than 10 mg/g)
- Composite of a 50% decrease in eGFR or development of end-stage renal disease requiring long-term dialysis or kidney transplantation (in those with baseline chronic kidney disease)
- A 30% decrease in eGFR (in those without chronic kidney disease).1,17
SPRINT also recruited participants to two nested substudies: SPRINT MIND and SPRINT MIND MRI, to study differences in cognitive outcomes and small-vessel ischemic disease between intensive treatment and standard treatment.
STUDY RESULTS
Older patients at risk, but without diabetes
Of 14,692 participants screened, 9,361 were enrolled in the study between 2010 and 2013. Baseline characteristics were comparable in both groups.
Demographics. The mean age of the participants was 67.9, and about 28% were 75 or older. About 36% were women, 58% white, 30% black, and 11% Hispanic.
Cardiovascular risk. The mean Framingham risk score was 20% (ie, they had a 20% risk of having a cardiovascular event within 10 years), and 61% of the participants had a risk score of at least 15%. Twenty percent already had cardiovascular disease.
Blood pressure. The average baseline blood pressure was 139.7/78.2 mm Hg. One-third of the participants had baseline systolic pressures of 132 mm Hg or less, another third had pressures in the range of 132 to 145, and the rest had 145 mm Hg or higher.
Renal function. The mean serum creatinine level was about 1.1 mg/dL. The mean eGFR was about 71 mL/min/1.73 m2 as calculated by the MDRD equation, and about 28% had eGFRs less than 60. The mean ratio of urinary albumin to creatinine was 44.1 mg/g in the intensive treatment group and 41.1 in the standard treatment group.
Other. The mean total cholesterol level was 190 mg/dL, fasting plasma glucose 99 mg/dL, and body mass index nearly 30 kg/m2.
Blood pressure during treatment
People in the intensive treatment group were taking a mean of 2.8 antihypertensive medications, and those in the standard treatment group were taking 1.8. Patients in the intensive group required greater use of all classes of medications to achieve goal systolic pressure (Table 1).
Study halted early due to efficacy
Throughout the 3.26 years of follow-up, the average difference in systolic pressure between the two groups was 13.1 mm Hg, with a mean systolic pressure of 121.5 mm Hg in the intensive treatment group and 134.6 mm Hg in the standard treatment group. The mean diastolic blood pressure was 68.7 mm Hg in the intensive treatment group and 76.3 mm Hg in the standard treatment group.
Although the study was planned to run for an average follow-up of 5 years, the National Heart, Lung, and Blood Institute terminated it early at a median of 3.26 years in view of lower rates of the primary outcome and of heart failure and death in the intensive treatment group (Table 2).
The effects on the primary outcome and mortality were consistent across the prespecified subgroups of age (< 75 vs ≥ 75), sex (female vs male), race (black vs nonblack), cardiovascular disease (presence or absence at baseline), prior chronic kidney disease (presence or absence at baseline), and across blood pressure tertiles (≤ 132 mm Hg, > 132 to < 145 mm Hg, ≥ 145 mm Hg).
Follow-up for assessment of cognitive outcomes (SPRINT MIND) and small-vessel ischemic disease (SPRINT MIND MRI) is ongoing.
WHAT DOES THIS MEAN?
SPRINT is the first large, adequately powered, randomized trial to demonstrate cardiovascular and mortality benefit from lowering the systolic blood pressure (goal < 120 mm Hg) in older patients at cardiovascular risk but without a history of diabetes mellitus or stroke.1
Most SPRINT patients had reasonably controlled blood pressure at baseline (the mean systolic pressure was 139.7 mm Hg, and two-thirds of participants had systolic pressure < 145 mm Hg). Of note, however, this trial excluded patients with systolic pressure higher than 180 mm Hg. There was excellent separation of systolic pressure between the two groups beginning at 1 year, which was consistent through the course of the trial.
The cardiovascular benefit in the intensive treatment group was predominantly driven by lower rates of heart failure (a 38% reduction in the intensive treatment group, P = .0002) and cardiovascular mortality (a 43% reduction in the intensive treatment group, P = .005), while there was no significant difference between the two groups in myocardial infarction or stroke. The beneficial effect on heart failure events is consistent with results from other trials including the Systolic Hypertension in the Elderly Program,7 Systolic Hypertension in Europe,8 and Hypertension in the Very Elderly Trial,9 all of which showed greatest risk reduction for heart failure events with systolic pressure-lowering (although to higher systolic levels than SPRINT).7–9 It is unclear why there was no beneficial effect on stroke events. The reduction in all-cause mortality in the intensive treatment group in SPRINT was greater than the reduction in cardiovascular deaths, which is also unexplained.
Although the study was terminated early due to efficacy (which introduces the possible bias that the estimated effect size will be too high), the number of primary end points reached was large (562 in the two groups combined), providing reassurance that the findings are valid. There was no blinding in the study (both participants and study investigators were aware of treatment assignment and study medications), but there was a structured assessment of outcomes and adverse events, with adjudication done by blinded reviewers.
SPRINT used an automated device for blood pressure measurement, which is known to reduce the “white coat” effect and correlates tightly with average daytime blood pressure done by ambulatory blood pressure monitoring.18 However, in clinical practice automated devices may not be available and a strict protocol for correct measurement may not be followed, with the possible result that blood pressure may be overestimated and overtreated.
What about diastolic pressure?
The trial, by design, focused on lowering systolic pressure (given the greater prevalence of isolated systolic hypertension with age), and the implications of lowering diastolic pressure are unclear. The issue of a J-shaped relationship between diastolic pressure and cardiovascular risk is debated in the literature: patients with a diastolic pressure of 60 to 65 mm Hg, especially those with existing coronary artery disease, may not tolerate aggressive blood pressure-lowering.19,20 Further analysis of this association (if any) from SPRINT will be helpful.
What about patients with diabetes?
Patients were excluded from SPRINT if they were under age 50, were at low cardiovascular risk, or had diabetes, raising the question of whether the results apply to these groups as well.
The question is particularly relevant in diabetes, as the ACCORD BP study, which used the same blood pressure targets as SPRINT, did not show a significant difference in the primary cardiovascular outcome between the intensive and standard treatments in patients with diabetes (Table 3).13 In ACCORD BP, the rate of the primary outcome was 12% lower in the intensive treatment group than in the standard treatment group, but the 95% confidence interval was –27% to +6%, so the finding was not statistically significant. However, the wide confidence interval does not exclude the possibility of a benefit that was comparable to that observed in SPRINT.
It has been speculated that ACCORD BP was underpowered to detect significant differences in the primary outcome.21 An analysis combining data from both trials indicated that effects on individual outcomes were generally consistent in both trials (with no significant heterogeneity noted).22 Also, the primary composite outcome in ACCORD did not include heart failure, which is particularly sensitive to blood pressure reduction.
Additionally, ACCORD BP had a 2 × 2 factorial design involving a simultaneous comparison of intensive vs standard glycemic control, which may have influenced the effects due to blood pressure. Indeed, a post hoc analysis showed that there was a significant 26% lower risk of the primary outcome in ACCORD BP patients who received intensive systolic pressure control plus standard glycemic control than in those receiving standard systolic control plus standard glycemic control.23
Are more adverse events an acceptable trade-off?
Adverse events, including acute kidney injury, were more frequent in the intensive therapy group in SPRINT.
Acute kidney injury was coded as an adverse event on the basis of this diagnosis being included in the hospital discharge summary (as a primary or main secondary diagnosis) and if considered by the safety officer to be one of the top three reasons for admission or continued hospitalization. Further analysis of renal events should be forthcoming.
People in the intensive treatment group, on average, needed one more medication than those in the standard treatment group. Some of the adverse events may be related to the antihypertensive medications taken (eg, electrolyte abnormalities such as hyponatremia and hypokalemia due to diuretic use), and others may be related to blood pressure-lowering (eg, acute kidney injury due to renal hypoperfusion).
At this point, the long-term effects of these adverse events, especially on kidney function, are not known. Patients enrolled in clinical trials tend to be healthier than patients seen in clinical practice; thus, the rate of adverse events reported in the trial may be lower than one would see in the real world.
Does lower systolic pressure protect or harm the kidneys?
SPRINT included patients with stage 3 and 4 chronic kidney disease (ie, with eGFR 20–50 mL/min/1.73 m2), but it was designed to assess cardiovascular outcomes, not the progression of chronic kidney disease. The trial excluded patients with diabetic nephropathy or high degrees of proteinuria.
Earlier randomized trials that focused on chronic kidney disease progression, including the MDRD24 and the African American Study of Kidney Disease and Hypertension,25 did not show benefit with more aggressive blood pressure-lowering (except in patients with higher degrees of proteinuria), and these trials were not powered to assess effects on cardiovascular outcomes.24,25
The Irbesartan Diabetic Nephropathy Trial,26,27 which was done in patients with overt diabetic nephropathy, showed that a progressively lower achieved systolic pressure down to 120 mm Hg predicted lower rates of heart failure, cardiovascular mortality, and renal events (although the trial target was ≤ 130/85 mm Hg and few participants achieved systolic pressure lower than 120 mm Hg).
IMPLICATIONS FOR MANAGEMENT
The recent estimates of hypertension prevalence and control from NHANES show that only about 53% of hypertensive adults have their blood pressure under control (defined as systolic pressure < 140 mm Hg and diastolic pressure < 90 mm Hg).2 Analysis of the NHANES 2007–2012 data showed that 16.7% or 8.2 million US adults with treated hypertension meet the eligibility criteria for SPRINT.28
Although the SPRINT results support the notion that “lower is better,” the risks and benefits of intensive control will need to be balanced in individual patients. Table 4 shows the number needed to treat and number needed to harm in the trial.
More aggressive management of hypertension is challenging. The median systolic pressure achieved in the intensive group in SPRINT was just over 120 mm Hg, which implies that at least half of the participants in the intensive group did not achieve the goal of less than 120 mm Hg. While it may be reasonable to aim for systolic pressure of less than 120 or 125 mm Hg in patients who fit the SPRINT criteria and can tolerate intensive blood pressure lowering, it would be prudent to aim for a more conservative goal in elderly patients who are frail and at risk for falls, considering the higher incidence of specified adverse events in the intensive group.
Results of cognitive outcomes, as well as data related to quality of life, are still awaited. Long-term renal outcomes are also unclear.
As noted above, the question of generalizability of SPRINT results to patients with diabetes is open to debate. In our opinion, with currently available evidence, it is difficult to conclusively answer the question of whether a lower systolic target provides cardiovascular benefit in diabetes. It is also unclear whether similar beneficial results would be seen with intensive treatment in a population at low cardiovascular risk. The American Heart Association and the American College of Cardiology are in the process of formulating new hypertension guidelines, and evidence from SPRINT will inform any new recommendations.
As more medications will likely be needed for intensive systolic blood pressure control, side effects and tolerability of medications with polypharmacy and potential nonadherence with increasing complexity of medication regimens should be kept in mind. Lifestyle modifications will need to be emphasized and reinforced, with greater use of combination antihypertensive therapy.
The data from SPRINT indicate that lower systolic pressure is better, as long as untoward clinical events can be monitored and avoided or easily managed. Careful monitoring will likely entail more frequent clinic visits and more frequent assessment of renal function and electrolyte levels (participants in the intensive group in the trial were seen every month until goal was achieved). A team approach that includes pharmacists and nurse practitioners, along with optimal use of best practice algorithms and remote monitoring technology, will need to be implemented for efficient and effective care.
In treating hypertension, lower systolic pressure is better than higher—with caveats. This is the message of the Systolic Blood Pressure Intervention Trial (SPRINT),1 a large, federally funded study that was halted early when patients at high cardiovascular risk who were randomized to a goal systolic pressure of less than 120 mm Hg were found to have better outcomes, including lower rates of heart failure, death from cardiovascular causes, and death from any cause, than patients randomized to a goal of less than 140 mm Hg.
The caveats: the benefit came at a price of more adverse events. Also, the trial excluded patients who had diabetes mellitus or previous strokes, so it is uncertain if these subgroups would also benefit from intensive lowering of systolic pressure—and in earlier trials they did not.
This article reviews the trial design and protocol, summarizes the results, and briefly discusses the implications of these results.
BEFORE SPRINT
Hypertension is very common in adults in the United States, and is a risk factor for heart disease, stroke, heart failure, and kidney disease. The estimated prevalence of hypertension in the 2011–2014 National Health and Nutrition Examination Survey (NHANES) was 29%, and the prevalence increases with age (7.3% in those ages 18 to 39, 32.2% in those ages 40 to 59, and 64.9% in those ages 60 and older).2 Isolated systolic hypertension (ie, systolic blood pressure > 140 mm Hg with diastolic pressure < 90 mm Hg) is the most common form of hypertension after age 50.3
Clinical trials have provided substantial evidence that treating hypertension reduces the incidence of stroke, myocardial infarction, and heart failure.4,5 Although observational studies show a progressive and linear rise in cardiovascular risk as systolic blood pressure rises above 115 mm Hg,6 clinical trials in the general population have not documented benefits of lowering systolic pressure to this level.7–11 However, clinical trials that directly evaluated two different blood pressure goals in the general population showed benefit with achieving systolic blood pressure less than 150 mm Hg,7,9 with limited data on lower blood pressure targets.10–12
No benefit found in intensive systolic lowering in diabetes or after stroke
The Action to Control Cardiovascular Risk in Diabetes-Blood Pressure (ACCORD BP) trial13 in patients with type 2 diabetes found no benefit in lowering systolic pressure to less than 120 mm Hg compared with less than 140 mm Hg in terms of the trial’s primary composite cardiovascular outcome (ie, nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes). However, the intensively treated group in this trial did enjoy a benefit in terms of fewer stroke events.
The Secondary Prevention of Small Subcortical Strokes (SPS3) trial14 in patients with stroke found no significant benefit in lowering systolic pressure to less than 130 mm Hg compared with less than 150 mm Hg for overall risk of another stroke, but a significant benefit was noted in reduced risk of intracerebral hemorrhage.
Current guidelines, based on available evidence, advocate treatment to a systolic goal of less than 140 mm Hg in most patients, and recommend relaxing this goal to less than 150 mm Hg in the elderly.15,16
Given the uncertainty surrounding optimal systolic targets, SPRINT was designed to test the hypothesis that a goal of less than 120 mm Hg would reduce the risk of cardiovascular events more than the generally accepted systolic goal of less than 140 mm Hg.17 Patients with diabetes and stroke were excluded because a similar hypothesis was tested in the ACCORD BP and SPS3 trials, which included patients with these conditions.
SPRINT DESIGN
SPRINT was a randomized, controlled, open-label trial sponsored by the National Institutes of Health and conducted at 102 US sites.
Inclusion criteria. Participants had to be at least 50 years old, with systolic pressure of 130 to 180 mm Hg, and had to have at least one cardiovascular risk factor, eg:
- Clinical or subclinical cardiovascular disease (other than stroke)
- Chronic kidney disease, defined as estimated glomerular filtration rate (eGFR), calculated by the Modification of Diet in Renal Disease (MDRD) study equation, of 20 to less than 60 mL/min/1.73 m2
- Framingham risk score of 15% of more
- Age 75 or older.
Major exclusion criteria included:
- Diabetes
- Stroke
- Polycystic kidney disease
- Chronic kidney disease with an eGFR less than 20 mL/min/1.73 m2
- Proteinuria (excretion > 1 g/day).
Intensive vs standard treatment
Participants were randomized to receive intensive treatment (systolic goal < 120 mm Hg) or standard treatment (systolic goal < 140 mm Hg). Baseline antihypertensive medications were adjusted to achieve blood pressure goals based on randomization assignment.
Doses of medications were adjusted on the basis of an average of three seated office blood pressure measurements after a 5-minute period of rest, taken with an automated monitor (Omron Healthcare Model 907); the same monitor was used and the same protocol was followed at all participating sites. Blood pressure was also measured after standing for 1 minute to assess orthostatic change.
Lifestyle modifications were encouraged in both groups. There was no restriction on using any antihypertensive medication, and this was at the discretion of individual investigators. Thiazide-type diuretics were encouraged as first-line agents (with chlorthalidone encouraged as the primary thiazide-type diuretic).
Outcomes measured
The primary outcome was a composite of myocardial infarction, acute coronary syndrome not resulting in myocardial infarction, stroke, acute decompensated heart failure, and cardiovascular mortality.
Secondary outcomes included individual components of the primary composite outcome, all-cause mortality, and the composite of primary outcome and all-cause mortality.
Renal outcomes were assessed as:
- Incident albuminuria (doubling of the urinary albumin-to-creatinine ratio from less than 10 mg/g to more than 10 mg/g)
- Composite of a 50% decrease in eGFR or development of end-stage renal disease requiring long-term dialysis or kidney transplantation (in those with baseline chronic kidney disease)
- A 30% decrease in eGFR (in those without chronic kidney disease).1,17
SPRINT also recruited participants to two nested substudies: SPRINT MIND and SPRINT MIND MRI, to study differences in cognitive outcomes and small-vessel ischemic disease between intensive treatment and standard treatment.
STUDY RESULTS
Older patients at risk, but without diabetes
Of 14,692 participants screened, 9,361 were enrolled in the study between 2010 and 2013. Baseline characteristics were comparable in both groups.
Demographics. The mean age of the participants was 67.9, and about 28% were 75 or older. About 36% were women, 58% white, 30% black, and 11% Hispanic.
Cardiovascular risk. The mean Framingham risk score was 20% (ie, they had a 20% risk of having a cardiovascular event within 10 years), and 61% of the participants had a risk score of at least 15%. Twenty percent already had cardiovascular disease.
Blood pressure. The average baseline blood pressure was 139.7/78.2 mm Hg. One-third of the participants had baseline systolic pressures of 132 mm Hg or less, another third had pressures in the range of 132 to 145, and the rest had 145 mm Hg or higher.
Renal function. The mean serum creatinine level was about 1.1 mg/dL. The mean eGFR was about 71 mL/min/1.73 m2 as calculated by the MDRD equation, and about 28% had eGFRs less than 60. The mean ratio of urinary albumin to creatinine was 44.1 mg/g in the intensive treatment group and 41.1 in the standard treatment group.
Other. The mean total cholesterol level was 190 mg/dL, fasting plasma glucose 99 mg/dL, and body mass index nearly 30 kg/m2.
Blood pressure during treatment
People in the intensive treatment group were taking a mean of 2.8 antihypertensive medications, and those in the standard treatment group were taking 1.8. Patients in the intensive group required greater use of all classes of medications to achieve goal systolic pressure (Table 1).
Study halted early due to efficacy
Throughout the 3.26 years of follow-up, the average difference in systolic pressure between the two groups was 13.1 mm Hg, with a mean systolic pressure of 121.5 mm Hg in the intensive treatment group and 134.6 mm Hg in the standard treatment group. The mean diastolic blood pressure was 68.7 mm Hg in the intensive treatment group and 76.3 mm Hg in the standard treatment group.
Although the study was planned to run for an average follow-up of 5 years, the National Heart, Lung, and Blood Institute terminated it early at a median of 3.26 years in view of lower rates of the primary outcome and of heart failure and death in the intensive treatment group (Table 2).
The effects on the primary outcome and mortality were consistent across the prespecified subgroups of age (< 75 vs ≥ 75), sex (female vs male), race (black vs nonblack), cardiovascular disease (presence or absence at baseline), prior chronic kidney disease (presence or absence at baseline), and across blood pressure tertiles (≤ 132 mm Hg, > 132 to < 145 mm Hg, ≥ 145 mm Hg).
Follow-up for assessment of cognitive outcomes (SPRINT MIND) and small-vessel ischemic disease (SPRINT MIND MRI) is ongoing.
WHAT DOES THIS MEAN?
SPRINT is the first large, adequately powered, randomized trial to demonstrate cardiovascular and mortality benefit from lowering the systolic blood pressure (goal < 120 mm Hg) in older patients at cardiovascular risk but without a history of diabetes mellitus or stroke.1
Most SPRINT patients had reasonably controlled blood pressure at baseline (the mean systolic pressure was 139.7 mm Hg, and two-thirds of participants had systolic pressure < 145 mm Hg). Of note, however, this trial excluded patients with systolic pressure higher than 180 mm Hg. There was excellent separation of systolic pressure between the two groups beginning at 1 year, which was consistent through the course of the trial.
The cardiovascular benefit in the intensive treatment group was predominantly driven by lower rates of heart failure (a 38% reduction in the intensive treatment group, P = .0002) and cardiovascular mortality (a 43% reduction in the intensive treatment group, P = .005), while there was no significant difference between the two groups in myocardial infarction or stroke. The beneficial effect on heart failure events is consistent with results from other trials including the Systolic Hypertension in the Elderly Program,7 Systolic Hypertension in Europe,8 and Hypertension in the Very Elderly Trial,9 all of which showed greatest risk reduction for heart failure events with systolic pressure-lowering (although to higher systolic levels than SPRINT).7–9 It is unclear why there was no beneficial effect on stroke events. The reduction in all-cause mortality in the intensive treatment group in SPRINT was greater than the reduction in cardiovascular deaths, which is also unexplained.
Although the study was terminated early due to efficacy (which introduces the possible bias that the estimated effect size will be too high), the number of primary end points reached was large (562 in the two groups combined), providing reassurance that the findings are valid. There was no blinding in the study (both participants and study investigators were aware of treatment assignment and study medications), but there was a structured assessment of outcomes and adverse events, with adjudication done by blinded reviewers.
SPRINT used an automated device for blood pressure measurement, which is known to reduce the “white coat” effect and correlates tightly with average daytime blood pressure done by ambulatory blood pressure monitoring.18 However, in clinical practice automated devices may not be available and a strict protocol for correct measurement may not be followed, with the possible result that blood pressure may be overestimated and overtreated.
What about diastolic pressure?
The trial, by design, focused on lowering systolic pressure (given the greater prevalence of isolated systolic hypertension with age), and the implications of lowering diastolic pressure are unclear. The issue of a J-shaped relationship between diastolic pressure and cardiovascular risk is debated in the literature: patients with a diastolic pressure of 60 to 65 mm Hg, especially those with existing coronary artery disease, may not tolerate aggressive blood pressure-lowering.19,20 Further analysis of this association (if any) from SPRINT will be helpful.
What about patients with diabetes?
Patients were excluded from SPRINT if they were under age 50, were at low cardiovascular risk, or had diabetes, raising the question of whether the results apply to these groups as well.
The question is particularly relevant in diabetes, as the ACCORD BP study, which used the same blood pressure targets as SPRINT, did not show a significant difference in the primary cardiovascular outcome between the intensive and standard treatments in patients with diabetes (Table 3).13 In ACCORD BP, the rate of the primary outcome was 12% lower in the intensive treatment group than in the standard treatment group, but the 95% confidence interval was –27% to +6%, so the finding was not statistically significant. However, the wide confidence interval does not exclude the possibility of a benefit that was comparable to that observed in SPRINT.
It has been speculated that ACCORD BP was underpowered to detect significant differences in the primary outcome.21 An analysis combining data from both trials indicated that effects on individual outcomes were generally consistent in both trials (with no significant heterogeneity noted).22 Also, the primary composite outcome in ACCORD did not include heart failure, which is particularly sensitive to blood pressure reduction.
Additionally, ACCORD BP had a 2 × 2 factorial design involving a simultaneous comparison of intensive vs standard glycemic control, which may have influenced the effects due to blood pressure. Indeed, a post hoc analysis showed that there was a significant 26% lower risk of the primary outcome in ACCORD BP patients who received intensive systolic pressure control plus standard glycemic control than in those receiving standard systolic control plus standard glycemic control.23
Are more adverse events an acceptable trade-off?
Adverse events, including acute kidney injury, were more frequent in the intensive therapy group in SPRINT.
Acute kidney injury was coded as an adverse event on the basis of this diagnosis being included in the hospital discharge summary (as a primary or main secondary diagnosis) and if considered by the safety officer to be one of the top three reasons for admission or continued hospitalization. Further analysis of renal events should be forthcoming.
People in the intensive treatment group, on average, needed one more medication than those in the standard treatment group. Some of the adverse events may be related to the antihypertensive medications taken (eg, electrolyte abnormalities such as hyponatremia and hypokalemia due to diuretic use), and others may be related to blood pressure-lowering (eg, acute kidney injury due to renal hypoperfusion).
At this point, the long-term effects of these adverse events, especially on kidney function, are not known. Patients enrolled in clinical trials tend to be healthier than patients seen in clinical practice; thus, the rate of adverse events reported in the trial may be lower than one would see in the real world.
Does lower systolic pressure protect or harm the kidneys?
SPRINT included patients with stage 3 and 4 chronic kidney disease (ie, with eGFR 20–50 mL/min/1.73 m2), but it was designed to assess cardiovascular outcomes, not the progression of chronic kidney disease. The trial excluded patients with diabetic nephropathy or high degrees of proteinuria.
Earlier randomized trials that focused on chronic kidney disease progression, including the MDRD24 and the African American Study of Kidney Disease and Hypertension,25 did not show benefit with more aggressive blood pressure-lowering (except in patients with higher degrees of proteinuria), and these trials were not powered to assess effects on cardiovascular outcomes.24,25
The Irbesartan Diabetic Nephropathy Trial,26,27 which was done in patients with overt diabetic nephropathy, showed that a progressively lower achieved systolic pressure down to 120 mm Hg predicted lower rates of heart failure, cardiovascular mortality, and renal events (although the trial target was ≤ 130/85 mm Hg and few participants achieved systolic pressure lower than 120 mm Hg).
IMPLICATIONS FOR MANAGEMENT
The recent estimates of hypertension prevalence and control from NHANES show that only about 53% of hypertensive adults have their blood pressure under control (defined as systolic pressure < 140 mm Hg and diastolic pressure < 90 mm Hg).2 Analysis of the NHANES 2007–2012 data showed that 16.7% or 8.2 million US adults with treated hypertension meet the eligibility criteria for SPRINT.28
Although the SPRINT results support the notion that “lower is better,” the risks and benefits of intensive control will need to be balanced in individual patients. Table 4 shows the number needed to treat and number needed to harm in the trial.
More aggressive management of hypertension is challenging. The median systolic pressure achieved in the intensive group in SPRINT was just over 120 mm Hg, which implies that at least half of the participants in the intensive group did not achieve the goal of less than 120 mm Hg. While it may be reasonable to aim for systolic pressure of less than 120 or 125 mm Hg in patients who fit the SPRINT criteria and can tolerate intensive blood pressure lowering, it would be prudent to aim for a more conservative goal in elderly patients who are frail and at risk for falls, considering the higher incidence of specified adverse events in the intensive group.
Results of cognitive outcomes, as well as data related to quality of life, are still awaited. Long-term renal outcomes are also unclear.
As noted above, the question of generalizability of SPRINT results to patients with diabetes is open to debate. In our opinion, with currently available evidence, it is difficult to conclusively answer the question of whether a lower systolic target provides cardiovascular benefit in diabetes. It is also unclear whether similar beneficial results would be seen with intensive treatment in a population at low cardiovascular risk. The American Heart Association and the American College of Cardiology are in the process of formulating new hypertension guidelines, and evidence from SPRINT will inform any new recommendations.
As more medications will likely be needed for intensive systolic blood pressure control, side effects and tolerability of medications with polypharmacy and potential nonadherence with increasing complexity of medication regimens should be kept in mind. Lifestyle modifications will need to be emphasized and reinforced, with greater use of combination antihypertensive therapy.
The data from SPRINT indicate that lower systolic pressure is better, as long as untoward clinical events can be monitored and avoided or easily managed. Careful monitoring will likely entail more frequent clinic visits and more frequent assessment of renal function and electrolyte levels (participants in the intensive group in the trial were seen every month until goal was achieved). A team approach that includes pharmacists and nurse practitioners, along with optimal use of best practice algorithms and remote monitoring technology, will need to be implemented for efficient and effective care.
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- Yoon SS, Fryar CD, Carroll MD. Hypertension prevalence and control among adults: United States, 2011–2014. NCHS data brief, no. 220. Hyattsville, MD: National Center for Health Statistics. 2015.
- Franklin SS, Jacobs MJ, Wong ND, L’Italien GJ, Lapuerta P. Predominance of isolated systolic hypertension among middle-aged and elderly US hypertensives: analysis based on National Health and Nutrition Examination Survey (NHANES) III. Hypertension 2001; 37:869–874.
- Neal B, MacMahon S, Chapman N; Blood Pressure Lowering Treatment Trialists’ Collaboration. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials. Blood Pressure Lowering Treatment Trialists’ Collaboration. Lancet 2000; 356:1955–1964.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Staessen JA, Fagard R, Thijs L, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet 1997; 350:757–764.
- Beckett NS, Peters R, Fletcher AE, et al; HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res 2008; 31:2115–2127.
- Ogihara T, Saruta T, Rakugi H, et al; Valsartan in Elderly Isolated Systolic Hypertension Study Group. Target blood pressure for treatment of isolated systolic hypertension in the elderly: Valsartan in Elderly Isolated Systolic Hypertension study. Hypertension 2010; 56:196–202.
- Liu L, Zhang Y, Liu G, Li W, Zhang X, Zanchetti A; FEVER Study Group. The Felodipine Event Reduction (FEVER) Study: a randomized long-term placebo-controlled trial in Chinese hypertensive patients. J Hypertens 2005; 23:2157–2172.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- SPS3 Study Group; Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013; 382:507–515.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520. Erratum in: JAMA. 2014; 311:1809.
- Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens (Greenwich) 2014; 16:14–26.
- Ambrosius WT, Sink KM, Foy CG, et al; SPRINT Study Research Group. The design and rationale of a multicenter clinical trial comparing two strategies for control of systolic blood pressure: the Systolic Blood Pressure Intervention Trial (SPRINT). Clin Trials 2014; 11:532–546.
- Myers MG, Godwin M, Dawes M, Kiss A, Tobe SW, Kaczorowski J. Conventional versus automated measurement of blood pressure in the office (CAMBO) trial. Fam Pract 2012; 29:376–382.
- Messerli FH, Mancia G, Conti CR, et al. Dogma disputed: can aggressively lowering blood pressure in hypertensive patients with coronary artery disease be dangerous? Ann Intern Med 2006; 144:884–893.
- Boutitie F, Gueyffier F, Pocock S, Fagard R, Boissel JP; INDANA Project Steering Committee; INdividual Data ANalysis of Antihypertensive intervention. J-shaped relationship between blood pressure and mortality in hypertensive patients: new insights from a meta-analysis of individual-patient data. Ann Intern Med 2002; 136:438–448.
- Mancia G. Effects of intensive blood pressure control in the management of patients with type 2 diabetes mellitus in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Circulation 2010; 122:847–849.
- Perkovic V, Rodgers A. Redefining blood-pressure targets—SPRINT starts the marathon. N Engl J Med 2015; 373:2175–2178.
- Margolis KL, O’Connor PJ, Morgan TM, et al. Outcomes of combined cardiovascular risk factor management strategies in type 2 diabetes: the ACCORD randomized trial. Diabetes Care 2014; 37:1721–1728.
- Peterson JC, Adler S, Burkart JM, et al. Blood pressure control, proteinuria, and the progression of renal disease. The Modification of Diet in Renal Disease study. Ann Intern Med 1995; 123:754–762.
- Agodoa LY, Appel L, Bakris GL, et al; African American Study of Kidney Disease and Hypertension (AASK) Study Group. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA 2001; 285:2719–2728.
- Berl T, Hunsicker LG, Lewis JB, et al; Collaborative Study Group. Impact of achieved blood pressure on cardiovascular outcomes in the Irbesartan Diabetic Nephropathy Trial. J Am Soc Nephrol 2005; 16:2170–2179.
- Pohl MA, Blumenthal S, Cordonnier DJ, et al. Independent and additive impact of blood pressure control and angiotensin II receptor blockade on renal outcomes in the Irbesartan Diabetic Nephropathy Trial: clinical implications and limitations. J Am Soc Nephrol 2005; 16:3027–3037.
- Bress AP, Tanner RM, Hess R, Colantonio LD, Shimbo D, Muntner P. Generalizability of results from the Systolic Blood Pressure Intervention Trial (SPRINT) to the US adult population. J Am Coll Cardiol 2015 Oct 31. doi: 10.1016/j.jacc.2015.10.037. Epub ahead of print.
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- Yoon SS, Fryar CD, Carroll MD. Hypertension prevalence and control among adults: United States, 2011–2014. NCHS data brief, no. 220. Hyattsville, MD: National Center for Health Statistics. 2015.
- Franklin SS, Jacobs MJ, Wong ND, L’Italien GJ, Lapuerta P. Predominance of isolated systolic hypertension among middle-aged and elderly US hypertensives: analysis based on National Health and Nutrition Examination Survey (NHANES) III. Hypertension 2001; 37:869–874.
- Neal B, MacMahon S, Chapman N; Blood Pressure Lowering Treatment Trialists’ Collaboration. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials. Blood Pressure Lowering Treatment Trialists’ Collaboration. Lancet 2000; 356:1955–1964.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Staessen JA, Fagard R, Thijs L, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet 1997; 350:757–764.
- Beckett NS, Peters R, Fletcher AE, et al; HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res 2008; 31:2115–2127.
- Ogihara T, Saruta T, Rakugi H, et al; Valsartan in Elderly Isolated Systolic Hypertension Study Group. Target blood pressure for treatment of isolated systolic hypertension in the elderly: Valsartan in Elderly Isolated Systolic Hypertension study. Hypertension 2010; 56:196–202.
- Liu L, Zhang Y, Liu G, Li W, Zhang X, Zanchetti A; FEVER Study Group. The Felodipine Event Reduction (FEVER) Study: a randomized long-term placebo-controlled trial in Chinese hypertensive patients. J Hypertens 2005; 23:2157–2172.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- SPS3 Study Group; Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013; 382:507–515.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520. Erratum in: JAMA. 2014; 311:1809.
- Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens (Greenwich) 2014; 16:14–26.
- Ambrosius WT, Sink KM, Foy CG, et al; SPRINT Study Research Group. The design and rationale of a multicenter clinical trial comparing two strategies for control of systolic blood pressure: the Systolic Blood Pressure Intervention Trial (SPRINT). Clin Trials 2014; 11:532–546.
- Myers MG, Godwin M, Dawes M, Kiss A, Tobe SW, Kaczorowski J. Conventional versus automated measurement of blood pressure in the office (CAMBO) trial. Fam Pract 2012; 29:376–382.
- Messerli FH, Mancia G, Conti CR, et al. Dogma disputed: can aggressively lowering blood pressure in hypertensive patients with coronary artery disease be dangerous? Ann Intern Med 2006; 144:884–893.
- Boutitie F, Gueyffier F, Pocock S, Fagard R, Boissel JP; INDANA Project Steering Committee; INdividual Data ANalysis of Antihypertensive intervention. J-shaped relationship between blood pressure and mortality in hypertensive patients: new insights from a meta-analysis of individual-patient data. Ann Intern Med 2002; 136:438–448.
- Mancia G. Effects of intensive blood pressure control in the management of patients with type 2 diabetes mellitus in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Circulation 2010; 122:847–849.
- Perkovic V, Rodgers A. Redefining blood-pressure targets—SPRINT starts the marathon. N Engl J Med 2015; 373:2175–2178.
- Margolis KL, O’Connor PJ, Morgan TM, et al. Outcomes of combined cardiovascular risk factor management strategies in type 2 diabetes: the ACCORD randomized trial. Diabetes Care 2014; 37:1721–1728.
- Peterson JC, Adler S, Burkart JM, et al. Blood pressure control, proteinuria, and the progression of renal disease. The Modification of Diet in Renal Disease study. Ann Intern Med 1995; 123:754–762.
- Agodoa LY, Appel L, Bakris GL, et al; African American Study of Kidney Disease and Hypertension (AASK) Study Group. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA 2001; 285:2719–2728.
- Berl T, Hunsicker LG, Lewis JB, et al; Collaborative Study Group. Impact of achieved blood pressure on cardiovascular outcomes in the Irbesartan Diabetic Nephropathy Trial. J Am Soc Nephrol 2005; 16:2170–2179.
- Pohl MA, Blumenthal S, Cordonnier DJ, et al. Independent and additive impact of blood pressure control and angiotensin II receptor blockade on renal outcomes in the Irbesartan Diabetic Nephropathy Trial: clinical implications and limitations. J Am Soc Nephrol 2005; 16:3027–3037.
- Bress AP, Tanner RM, Hess R, Colantonio LD, Shimbo D, Muntner P. Generalizability of results from the Systolic Blood Pressure Intervention Trial (SPRINT) to the US adult population. J Am Coll Cardiol 2015 Oct 31. doi: 10.1016/j.jacc.2015.10.037. Epub ahead of print.
KEY POINTS
- SPRINT is the first large prospective randomized trial to show evidence of cardiovascular and mortality benefit for intensive lowering of systolic blood pressure (goal < 120 mm Hg) in older patients at cardiovascular risk, but without a history of diabetes mellitus or stroke.
- A similar trial in patients with type 2 diabetes mellitus did not show significant benefit of intensive treatment.
- Intensive treatment was associated with more adverse events, including hypotension, syncope, electrolyte abnormalities, and acute kidney injury.
- It is unclear if these results can be extrapolated to patients with a history of diabetes or stroke, younger patients, or those with low cardiovascular risk.
- Healthcare providers should engage patients in a shared decision-making process, with discussion of the benefits and risks associated with intensive lowering of blood pressure.
Advances in the treatment of dyslipidemia
The 2013 joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA)1 on the treatment of blood cholesterol to reduce cardiovascular risk recommend high-intensity statin therapy for secondary prevention of cardiovascular events. The question of primary prevention is not so straightforward, and the recommended strategy has come under fire. In addition, the guidelines focus on statins and not on LDL-C levels, and the role of nonstatin lipid-lowering drugs and the value of reducing LDL-C levels to well below levels currently regarded as “normal” remain unclear.
This article comments on the 2013 ACC/AHA guidelines, reviews the data on optimal LDL-C levels, and discusses new nonstatin agents.
ACC/AHA GUIDELINES: A MIXED MESSAGE
The 2013 ACC/AHA cholesterol guidelines1 can be characterized by the title from the famous Western film “The Good, the Bad, and the Ugly.”
The good: A clear message to treat
The guidelines deliver an unambiguous message to treat patients at high risk with high-intensity statin therapy. This mandate is very helpful as it should reduce the undertreatment of patients.
The seemingly bad
Two common misconceptions regarding the guidelines:

They abandon LDL-C targets. Actually, the guidelines do not argue for or against targets; they simply remain silent, citing that randomized trials have not been conducted with LDL-C targets as specific goals. Technically, this statement is true. However, it seems contrived to argue, for example, that the benefit of atorvastatin 80 mg over 10 mg in the Treating to New Targets trial could not be reliably ascribed to the lower LDL-C achieved with the higher dose, but rather to some undefined benefit of high-intensity statin therapy, especially as the guidelines define the intensity of statins by the degree of LDL-C lowering. In fact, by correlating the incidence of coronary heart disease events with the levels of LDL-C achieved in those trials, conclusions can reasonably be drawn from such data (Figure 1).2
The guidelines do not recommend nonstatin drugs. Actually, the guidelines note that clinicians are free to consider other therapies, especially those proven to reduce the risk of cardiovascular events, a central principle of medicine. Since the guidelines were published, data have emerged indicating that the role of nonstatin drugs also needs consideration.
The ugly: Risk calculator untested
The guidelines promote the use of a risk calculator developed by the ACC/AHA to estimate the 10-year risk of an atherosclerotic event for people whose LDL-C levels are between 70 and 189 mg/dL to help decide whether to initiate statin therapy for primary prevention of atherosclerotic cardiovascular disease. Such an approach is reasonable, although the risk score was promulgated without evidence to support its utility.
Media coverage of the risk calculator was fierce. Some physicians found imperfections in the risk score (as is true for all risk scores), resulting in public mistrust of the guidelines and of the medical community as a whole. This needless controversy may have compromised the main message—that LDL-C should be lowered in many people—a message backed by strong evidence.
Alternative strategies proposed
Ridker et al3 have proposed a hybrid strategy to guide statin use for apparently healthy people that combines the ACC/AHA guideline approach with entry criteria for randomized clinical trials that showed statin efficacy for primary prevention.
Genetic analysis may offer another approach. Mega et al4 stratified more than 48,000 people by a genetic risk score based on 27 genetic variants and found a significant association with risk of coronary events. Targeting therapy to people found to be at higher risk on this basis offers greater risk reduction than expected for the general population. Biomarkers and imaging tests are other potentially useful risk determinants.
LDL-C: LOWER IS BETTER
Although no clinical trial has yet targeted specific LDL-C levels, there is plenty of evidence that lower LDL-C levels offer greater benefit (Figure 1).2
In 1994, the Scandinavian Simvastatin Survival Study5 established the benefit of statins in patients with known vascular disease. The mean LDL-C level achieved in the active treatment group was 120 mg/dL. More trials followed supporting the benefits of statins and of reducing LDL-C from average levels in the 120s down to 100 mg/dL.
In 2004, the Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 trial6 observed an even greater risk reduction in patients with known risk by treating with statins; the mean LDL-C level achieved in the group randomized to an intensive regimen of atorvastatin 80 mg per day was 62 mg/dL. The same year, the Adult Treatment Panel III of the National Cholesterol Education Program7 issued updated guidelines including an optional goal of LDL-C less than 70 mg/dL for patients at very high risk.
In 2008, the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER)8 found a significantly lower incidence of major cardiovascular events at 2 years in apparently healthy men and women with baseline LDL-C levels of less than 130 mg/dL after treatment with rosuvastatin 20 mg daily, with an achieved median LDL-C of 55 mg/dL.
How low should LDL-C go?
Evidence from clinical trials indicates a 20% to 25% reduction in the risk of cardiovascular events for every 39-mg/dL decrease in LDL-C. Extrapolating the data, cardiovascular disease risk would be reduced to zero if LDL-C were brought down below 40 mg/dL.
Brown and Goldstein,9 who won the 1985 Nobel Prize in medicine for their work in cholesterol metabolism, estimated that a plasma level of LDL-C of only 25 mg/dL would be sufficient to nourish cells with cholesterol. Cells can synthesize all the cholesterol they need, underscoring that LDL-C is simply the final end-product that the liver removes from circulation.
Other evidence that lower LDL-C does not have adverse effects comes from non-Western populations as well as from other mammals. Total cholesterol levels range in the low 100s mg/dL in Native American and African tribal populations, with LDL-C estimated to be about 50 to 75 mg/dL. Elephants, baboons, and foxes have even lower levels.10
Clinical trial data also support that LDL-C levels below the current “normal” are better. The Cholesterol Treatment Trialists’ Collaboration11 analyzed data from more than 160,000 patients in 26 trials that evaluated either more- vs less-intensive statin regimens or statin treatment vs control. No baseline level below which lowering LDL-C further was not beneficial was found. Patients who started out with an LDL-C level of less than 77 mg/dL had the same risk reduction of major vascular events when the level was dropped to 50 mg/dL as those who started at higher levels and reduced their LDL-C by the same amount. In the JUPITER trial, even those with a baseline LDL-C of less than 60 mg/dL benefited from statin therapy.12
BEYOND STATINS
Ezetimibe further lowers risk
Ezetimibe is a nonstatin drug that reduces LDL-C by about 15% to 20%. The Improved Reduction of Outcomes: Vytorin Efficacy International Trial13 registered more than 18,000 patients with a baseline LDL-C level of less than 125 mg/dL (or 100 mg/dL if already on lipid-lowering therapy) who had been stabilized shortly after an acute cardiovascular event. They were randomized to receive either simvastatin 40 mg or combined simvastatin 40 mg and ezetimibe 10 mg. The study intended to determine two things: whether ezetimibe could further lower LDL-C when combined with a statin, and whether risk could be reduced further by driving the LDL-C below 70 mg/dL and down to the mid-50s.
After 1 year, the average LDL-C level was 70 mg/dL in the simvastatin group and 53 mg/dL in the combined simvastatin and ezetimibe group. At 7 years, for the primary end point (cardiovascular death, myocardial infarction, unstable angina requiring hospitalization, coronary revascularization, or stroke), there was a 6% reduction of events in the combined drug treatment group, with the number of people needed to treat being 50 to prevent one event. For the narrower end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke, there was a 10% risk reduction in the combined drug treatment arm.14
The amount of risk reduction is exactly what was predicted by the Cholesterol Treatment Trialists’ Collaboration’s plot of reduction in events vs reduction in LDL-C based on the analysis of 26 trials, adding further evidence that it is the LDL-C reduction itself, rather than the means by which LDL-C is reduced, that is critical for benefit.
PCSK9 inhibitors: A new approach
Mutations in the gene for proprotein convertase subtilisin kexin type 9 (PCSK9) have become a new focus of interest for reducing LDL-C and cardiovascular risk.15 PCSK9 binds to the LDL-C receptor on the surface of hepatocytes and escorts it to its destruction in the lysosomes, rather than allowing it to return to the cell surface to take more LDL-C out of circulation.
People with a gain-of-function mutation (conferring too much PCSK9, resulting in fewer LDL-C receptors and more LDL-C in circulation) are a more recently recognized subset of those with autosomal-dominant familial hypercholesterolemia. They have total cholesterol levels in the 90th percentile, tendon xanthomas, and a high risk of myocardial infarction and stroke at a young age.
Conversely, those with a nonsense mutation in PCSK9—leading to loss of function—have a 28% reduction in mean LDL-C and 88% reduction in risk of coronary heart disease compared with those without the mutation.16 Two women (ages 32 and 21, fertile) have been found who have inactivating mutations in both PCSK9 alleles, and both are in apparent good health, with LDL-C levels of 14 mg/dL and 15 mg/dL, respectively.17,18
Dramatic reduction in LDL-C
Monoclonal antibodies have been developed that bind PCSK9 and block its action with the goal of developing new LDL-C–lowering treatments. Phase 2 clinical trials of varying doses of evolocumab (Repatha), a drug in this class, combined with standard therapy (a statin with or without ezetimibe), found a 66% reduction of LDL-C at high doses at 12 weeks compared with standard therapy alone, with concomitant reductions in other atherogenic lipoproteins.19 Patients who could not tolerate statins because of myalgia responded well to evolocumab.20
Patients with heterozygous familial hypercholesterolemia also had a substantial reduction in LDL-C (55% at the highest dosage), even though they have fewer LDL-C receptors for the drug to act upon.21 People with homozygous familial hypercholesterolemia and no LDL-C receptors had a lesser relative reduction in LDL-C that depended on the type of mutations they had. Nonetheless, given how high LDL-C levels are in this population, the absolute decreases in LDL-C level were quite impressive.
Cardiovascular risk reduced
Data at nearly 1 year showed continued reduction of LDL-C by about 60% (absolute reduction: 73 mg/dL), as well as a lower incidence of cardiovascular events starting at just 3 months, much sooner than observed in some statin trials.22 Benefits were found regardless of subgroup (sex, age, statin use, baseline LDL-C level, or known vascular disease). No difference was found in the safety profile between the evolocumab and control arms. Only 2.4% of participants discontinued evolocumab because of adverse events, and the incidence of adverse effects did not correlate with LDL-C level achieved.
Neurocognitive effects occurred in 0.9% of the evolocumab arm vs 0.3% in the control arm. This difference has not been explained: although there is cholesterol in the central nervous system, it is generated locally, and lipoproteins—and evolocumab—are not thought to cross the blood-brain barrier.
Long-term trials of evolocumab are currently under way for patients with cardiovascular disease, as are trials of two other PCSK9 inhibitors, alirocumab and bococizumab, in addition to standard statin therapy.
On July 24, 2015, the US Food and Drug Administration (FDA) approved the first PCSK9 inhibitor, alirocumab (Praluent) for patients with heterozygous familial hypercholesterolemia or those with clinical atherosclerotic cardiovascular disease who require additional lowering of LDL-C. The starting dosage is 75 mg subcutaneously every 2 weeks, which can be increased up to 150 mg every 2 weeks.
Evolocumab was approved by the FDA on August 27, 2015, for the same indications. The dosage is 140 mg subcutaneously every 2 weeks or 420 mg every month.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129:S1-S45. Erratum in: Circulation 2014; 129:S46–S48.
- Raymond C, Cho L, Rocco M, Hazen SL. New cholesterol guidelines: worth the wait? Cleve Clin J Med 2014; 81:11–19.
- Ridker PM, Rose L, Cook NR. A proposal to incorporate trial data into a hybrid ACC/AHA algorithm for the allocation of statin therapy in primary prevention. J Am Coll Cardiol 2015; 65:942–948.
- Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 2015; 385:2264–2271.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Grundy SM, Cleeman JI, Merz CN, et al; National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239. Erratum in Circulation 2004; 110:763.
- Ridker PM, Danielson E, Fonseca FAH, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232:34–47.
- Hochholzer W, Giugliano RP. Lipid lowering goals: back to nature? Ther Adv Cardiovasc Dis 2010; 4:185–191.
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- Hsia J, MacFadyen JG, Monyak J, Ridker PM. Cardiovascular event reduction and adverse events among subjects attaining low-density lipoprotein cholesterol <50 mg/dl with rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J Am Coll Cardiol 2011; 57:1666–1675.
- Cannon CP, Giugliano RP, Blazing MA, et al; IMPROVE-IT Investigators. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J 2008; 156:826–832.
- Cannon CP, Blazing MA, Giugliano RP, et al for the IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015; 372:2387–2397.
- Giugliano RP, Sabatine MS. Are PCSK9 Inhibitors the next breakthrough in the cardiovascular field? J Am Coll Cardiol 2015; 65:2638–2651.
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272.
- Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 2006; 79:514-523.
- Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007; 193:445–448.
- Giugliano RP, Desai NR, Kohli P, et al; LAPLACE-TIMI 57 Investigators. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet 2012; 380:2007–2017.
- Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA 2012; 308:2497–2506.
- Raal F, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012; 126:2408–2417.
- Sabatine MS, Giugliano RP, Wiviott SD, et al; Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500–1509.
The 2013 joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA)1 on the treatment of blood cholesterol to reduce cardiovascular risk recommend high-intensity statin therapy for secondary prevention of cardiovascular events. The question of primary prevention is not so straightforward, and the recommended strategy has come under fire. In addition, the guidelines focus on statins and not on LDL-C levels, and the role of nonstatin lipid-lowering drugs and the value of reducing LDL-C levels to well below levels currently regarded as “normal” remain unclear.
This article comments on the 2013 ACC/AHA guidelines, reviews the data on optimal LDL-C levels, and discusses new nonstatin agents.
ACC/AHA GUIDELINES: A MIXED MESSAGE
The 2013 ACC/AHA cholesterol guidelines1 can be characterized by the title from the famous Western film “The Good, the Bad, and the Ugly.”
The good: A clear message to treat
The guidelines deliver an unambiguous message to treat patients at high risk with high-intensity statin therapy. This mandate is very helpful as it should reduce the undertreatment of patients.
The seemingly bad
Two common misconceptions regarding the guidelines:
They abandon LDL-C targets. Actually, the guidelines do not argue for or against targets; they simply remain silent, citing that randomized trials have not been conducted with LDL-C targets as specific goals. Technically, this statement is true. However, it seems contrived to argue, for example, that the benefit of atorvastatin 80 mg over 10 mg in the Treating to New Targets trial could not be reliably ascribed to the lower LDL-C achieved with the higher dose, but rather to some undefined benefit of high-intensity statin therapy, especially as the guidelines define the intensity of statins by the degree of LDL-C lowering. In fact, by correlating the incidence of coronary heart disease events with the levels of LDL-C achieved in those trials, conclusions can reasonably be drawn from such data (Figure 1).2
The guidelines do not recommend nonstatin drugs. Actually, the guidelines note that clinicians are free to consider other therapies, especially those proven to reduce the risk of cardiovascular events, a central principle of medicine. Since the guidelines were published, data have emerged indicating that the role of nonstatin drugs also needs consideration.
The ugly: Risk calculator untested
The guidelines promote the use of a risk calculator developed by the ACC/AHA to estimate the 10-year risk of an atherosclerotic event for people whose LDL-C levels are between 70 and 189 mg/dL to help decide whether to initiate statin therapy for primary prevention of atherosclerotic cardiovascular disease. Such an approach is reasonable, although the risk score was promulgated without evidence to support its utility.
Media coverage of the risk calculator was fierce. Some physicians found imperfections in the risk score (as is true for all risk scores), resulting in public mistrust of the guidelines and of the medical community as a whole. This needless controversy may have compromised the main message—that LDL-C should be lowered in many people—a message backed by strong evidence.
Alternative strategies proposed
Ridker et al3 have proposed a hybrid strategy to guide statin use for apparently healthy people that combines the ACC/AHA guideline approach with entry criteria for randomized clinical trials that showed statin efficacy for primary prevention.
Genetic analysis may offer another approach. Mega et al4 stratified more than 48,000 people by a genetic risk score based on 27 genetic variants and found a significant association with risk of coronary events. Targeting therapy to people found to be at higher risk on this basis offers greater risk reduction than expected for the general population. Biomarkers and imaging tests are other potentially useful risk determinants.
LDL-C: LOWER IS BETTER
Although no clinical trial has yet targeted specific LDL-C levels, there is plenty of evidence that lower LDL-C levels offer greater benefit (Figure 1).2
In 1994, the Scandinavian Simvastatin Survival Study5 established the benefit of statins in patients with known vascular disease. The mean LDL-C level achieved in the active treatment group was 120 mg/dL. More trials followed supporting the benefits of statins and of reducing LDL-C from average levels in the 120s down to 100 mg/dL.
In 2004, the Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 trial6 observed an even greater risk reduction in patients with known risk by treating with statins; the mean LDL-C level achieved in the group randomized to an intensive regimen of atorvastatin 80 mg per day was 62 mg/dL. The same year, the Adult Treatment Panel III of the National Cholesterol Education Program7 issued updated guidelines including an optional goal of LDL-C less than 70 mg/dL for patients at very high risk.
In 2008, the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER)8 found a significantly lower incidence of major cardiovascular events at 2 years in apparently healthy men and women with baseline LDL-C levels of less than 130 mg/dL after treatment with rosuvastatin 20 mg daily, with an achieved median LDL-C of 55 mg/dL.
How low should LDL-C go?
Evidence from clinical trials indicates a 20% to 25% reduction in the risk of cardiovascular events for every 39-mg/dL decrease in LDL-C. Extrapolating the data, cardiovascular disease risk would be reduced to zero if LDL-C were brought down below 40 mg/dL.
Brown and Goldstein,9 who won the 1985 Nobel Prize in medicine for their work in cholesterol metabolism, estimated that a plasma level of LDL-C of only 25 mg/dL would be sufficient to nourish cells with cholesterol. Cells can synthesize all the cholesterol they need, underscoring that LDL-C is simply the final end-product that the liver removes from circulation.
Other evidence that lower LDL-C does not have adverse effects comes from non-Western populations as well as from other mammals. Total cholesterol levels range in the low 100s mg/dL in Native American and African tribal populations, with LDL-C estimated to be about 50 to 75 mg/dL. Elephants, baboons, and foxes have even lower levels.10
Clinical trial data also support that LDL-C levels below the current “normal” are better. The Cholesterol Treatment Trialists’ Collaboration11 analyzed data from more than 160,000 patients in 26 trials that evaluated either more- vs less-intensive statin regimens or statin treatment vs control. No baseline level below which lowering LDL-C further was not beneficial was found. Patients who started out with an LDL-C level of less than 77 mg/dL had the same risk reduction of major vascular events when the level was dropped to 50 mg/dL as those who started at higher levels and reduced their LDL-C by the same amount. In the JUPITER trial, even those with a baseline LDL-C of less than 60 mg/dL benefited from statin therapy.12
BEYOND STATINS
Ezetimibe further lowers risk
Ezetimibe is a nonstatin drug that reduces LDL-C by about 15% to 20%. The Improved Reduction of Outcomes: Vytorin Efficacy International Trial13 registered more than 18,000 patients with a baseline LDL-C level of less than 125 mg/dL (or 100 mg/dL if already on lipid-lowering therapy) who had been stabilized shortly after an acute cardiovascular event. They were randomized to receive either simvastatin 40 mg or combined simvastatin 40 mg and ezetimibe 10 mg. The study intended to determine two things: whether ezetimibe could further lower LDL-C when combined with a statin, and whether risk could be reduced further by driving the LDL-C below 70 mg/dL and down to the mid-50s.
After 1 year, the average LDL-C level was 70 mg/dL in the simvastatin group and 53 mg/dL in the combined simvastatin and ezetimibe group. At 7 years, for the primary end point (cardiovascular death, myocardial infarction, unstable angina requiring hospitalization, coronary revascularization, or stroke), there was a 6% reduction of events in the combined drug treatment group, with the number of people needed to treat being 50 to prevent one event. For the narrower end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke, there was a 10% risk reduction in the combined drug treatment arm.14
The amount of risk reduction is exactly what was predicted by the Cholesterol Treatment Trialists’ Collaboration’s plot of reduction in events vs reduction in LDL-C based on the analysis of 26 trials, adding further evidence that it is the LDL-C reduction itself, rather than the means by which LDL-C is reduced, that is critical for benefit.
PCSK9 inhibitors: A new approach
Mutations in the gene for proprotein convertase subtilisin kexin type 9 (PCSK9) have become a new focus of interest for reducing LDL-C and cardiovascular risk.15 PCSK9 binds to the LDL-C receptor on the surface of hepatocytes and escorts it to its destruction in the lysosomes, rather than allowing it to return to the cell surface to take more LDL-C out of circulation.
People with a gain-of-function mutation (conferring too much PCSK9, resulting in fewer LDL-C receptors and more LDL-C in circulation) are a more recently recognized subset of those with autosomal-dominant familial hypercholesterolemia. They have total cholesterol levels in the 90th percentile, tendon xanthomas, and a high risk of myocardial infarction and stroke at a young age.
Conversely, those with a nonsense mutation in PCSK9—leading to loss of function—have a 28% reduction in mean LDL-C and 88% reduction in risk of coronary heart disease compared with those without the mutation.16 Two women (ages 32 and 21, fertile) have been found who have inactivating mutations in both PCSK9 alleles, and both are in apparent good health, with LDL-C levels of 14 mg/dL and 15 mg/dL, respectively.17,18
Dramatic reduction in LDL-C
Monoclonal antibodies have been developed that bind PCSK9 and block its action with the goal of developing new LDL-C–lowering treatments. Phase 2 clinical trials of varying doses of evolocumab (Repatha), a drug in this class, combined with standard therapy (a statin with or without ezetimibe), found a 66% reduction of LDL-C at high doses at 12 weeks compared with standard therapy alone, with concomitant reductions in other atherogenic lipoproteins.19 Patients who could not tolerate statins because of myalgia responded well to evolocumab.20
Patients with heterozygous familial hypercholesterolemia also had a substantial reduction in LDL-C (55% at the highest dosage), even though they have fewer LDL-C receptors for the drug to act upon.21 People with homozygous familial hypercholesterolemia and no LDL-C receptors had a lesser relative reduction in LDL-C that depended on the type of mutations they had. Nonetheless, given how high LDL-C levels are in this population, the absolute decreases in LDL-C level were quite impressive.
Cardiovascular risk reduced
Data at nearly 1 year showed continued reduction of LDL-C by about 60% (absolute reduction: 73 mg/dL), as well as a lower incidence of cardiovascular events starting at just 3 months, much sooner than observed in some statin trials.22 Benefits were found regardless of subgroup (sex, age, statin use, baseline LDL-C level, or known vascular disease). No difference was found in the safety profile between the evolocumab and control arms. Only 2.4% of participants discontinued evolocumab because of adverse events, and the incidence of adverse effects did not correlate with LDL-C level achieved.
Neurocognitive effects occurred in 0.9% of the evolocumab arm vs 0.3% in the control arm. This difference has not been explained: although there is cholesterol in the central nervous system, it is generated locally, and lipoproteins—and evolocumab—are not thought to cross the blood-brain barrier.
Long-term trials of evolocumab are currently under way for patients with cardiovascular disease, as are trials of two other PCSK9 inhibitors, alirocumab and bococizumab, in addition to standard statin therapy.
On July 24, 2015, the US Food and Drug Administration (FDA) approved the first PCSK9 inhibitor, alirocumab (Praluent) for patients with heterozygous familial hypercholesterolemia or those with clinical atherosclerotic cardiovascular disease who require additional lowering of LDL-C. The starting dosage is 75 mg subcutaneously every 2 weeks, which can be increased up to 150 mg every 2 weeks.
Evolocumab was approved by the FDA on August 27, 2015, for the same indications. The dosage is 140 mg subcutaneously every 2 weeks or 420 mg every month.
The 2013 joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA)1 on the treatment of blood cholesterol to reduce cardiovascular risk recommend high-intensity statin therapy for secondary prevention of cardiovascular events. The question of primary prevention is not so straightforward, and the recommended strategy has come under fire. In addition, the guidelines focus on statins and not on LDL-C levels, and the role of nonstatin lipid-lowering drugs and the value of reducing LDL-C levels to well below levels currently regarded as “normal” remain unclear.
This article comments on the 2013 ACC/AHA guidelines, reviews the data on optimal LDL-C levels, and discusses new nonstatin agents.
ACC/AHA GUIDELINES: A MIXED MESSAGE
The 2013 ACC/AHA cholesterol guidelines1 can be characterized by the title from the famous Western film “The Good, the Bad, and the Ugly.”
The good: A clear message to treat
The guidelines deliver an unambiguous message to treat patients at high risk with high-intensity statin therapy. This mandate is very helpful as it should reduce the undertreatment of patients.
The seemingly bad
Two common misconceptions regarding the guidelines:
They abandon LDL-C targets. Actually, the guidelines do not argue for or against targets; they simply remain silent, citing that randomized trials have not been conducted with LDL-C targets as specific goals. Technically, this statement is true. However, it seems contrived to argue, for example, that the benefit of atorvastatin 80 mg over 10 mg in the Treating to New Targets trial could not be reliably ascribed to the lower LDL-C achieved with the higher dose, but rather to some undefined benefit of high-intensity statin therapy, especially as the guidelines define the intensity of statins by the degree of LDL-C lowering. In fact, by correlating the incidence of coronary heart disease events with the levels of LDL-C achieved in those trials, conclusions can reasonably be drawn from such data (Figure 1).2
The guidelines do not recommend nonstatin drugs. Actually, the guidelines note that clinicians are free to consider other therapies, especially those proven to reduce the risk of cardiovascular events, a central principle of medicine. Since the guidelines were published, data have emerged indicating that the role of nonstatin drugs also needs consideration.
The ugly: Risk calculator untested
The guidelines promote the use of a risk calculator developed by the ACC/AHA to estimate the 10-year risk of an atherosclerotic event for people whose LDL-C levels are between 70 and 189 mg/dL to help decide whether to initiate statin therapy for primary prevention of atherosclerotic cardiovascular disease. Such an approach is reasonable, although the risk score was promulgated without evidence to support its utility.
Media coverage of the risk calculator was fierce. Some physicians found imperfections in the risk score (as is true for all risk scores), resulting in public mistrust of the guidelines and of the medical community as a whole. This needless controversy may have compromised the main message—that LDL-C should be lowered in many people—a message backed by strong evidence.
Alternative strategies proposed
Ridker et al3 have proposed a hybrid strategy to guide statin use for apparently healthy people that combines the ACC/AHA guideline approach with entry criteria for randomized clinical trials that showed statin efficacy for primary prevention.
Genetic analysis may offer another approach. Mega et al4 stratified more than 48,000 people by a genetic risk score based on 27 genetic variants and found a significant association with risk of coronary events. Targeting therapy to people found to be at higher risk on this basis offers greater risk reduction than expected for the general population. Biomarkers and imaging tests are other potentially useful risk determinants.
LDL-C: LOWER IS BETTER
Although no clinical trial has yet targeted specific LDL-C levels, there is plenty of evidence that lower LDL-C levels offer greater benefit (Figure 1).2
In 1994, the Scandinavian Simvastatin Survival Study5 established the benefit of statins in patients with known vascular disease. The mean LDL-C level achieved in the active treatment group was 120 mg/dL. More trials followed supporting the benefits of statins and of reducing LDL-C from average levels in the 120s down to 100 mg/dL.
In 2004, the Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 trial6 observed an even greater risk reduction in patients with known risk by treating with statins; the mean LDL-C level achieved in the group randomized to an intensive regimen of atorvastatin 80 mg per day was 62 mg/dL. The same year, the Adult Treatment Panel III of the National Cholesterol Education Program7 issued updated guidelines including an optional goal of LDL-C less than 70 mg/dL for patients at very high risk.
In 2008, the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER)8 found a significantly lower incidence of major cardiovascular events at 2 years in apparently healthy men and women with baseline LDL-C levels of less than 130 mg/dL after treatment with rosuvastatin 20 mg daily, with an achieved median LDL-C of 55 mg/dL.
How low should LDL-C go?
Evidence from clinical trials indicates a 20% to 25% reduction in the risk of cardiovascular events for every 39-mg/dL decrease in LDL-C. Extrapolating the data, cardiovascular disease risk would be reduced to zero if LDL-C were brought down below 40 mg/dL.
Brown and Goldstein,9 who won the 1985 Nobel Prize in medicine for their work in cholesterol metabolism, estimated that a plasma level of LDL-C of only 25 mg/dL would be sufficient to nourish cells with cholesterol. Cells can synthesize all the cholesterol they need, underscoring that LDL-C is simply the final end-product that the liver removes from circulation.
Other evidence that lower LDL-C does not have adverse effects comes from non-Western populations as well as from other mammals. Total cholesterol levels range in the low 100s mg/dL in Native American and African tribal populations, with LDL-C estimated to be about 50 to 75 mg/dL. Elephants, baboons, and foxes have even lower levels.10
Clinical trial data also support that LDL-C levels below the current “normal” are better. The Cholesterol Treatment Trialists’ Collaboration11 analyzed data from more than 160,000 patients in 26 trials that evaluated either more- vs less-intensive statin regimens or statin treatment vs control. No baseline level below which lowering LDL-C further was not beneficial was found. Patients who started out with an LDL-C level of less than 77 mg/dL had the same risk reduction of major vascular events when the level was dropped to 50 mg/dL as those who started at higher levels and reduced their LDL-C by the same amount. In the JUPITER trial, even those with a baseline LDL-C of less than 60 mg/dL benefited from statin therapy.12
BEYOND STATINS
Ezetimibe further lowers risk
Ezetimibe is a nonstatin drug that reduces LDL-C by about 15% to 20%. The Improved Reduction of Outcomes: Vytorin Efficacy International Trial13 registered more than 18,000 patients with a baseline LDL-C level of less than 125 mg/dL (or 100 mg/dL if already on lipid-lowering therapy) who had been stabilized shortly after an acute cardiovascular event. They were randomized to receive either simvastatin 40 mg or combined simvastatin 40 mg and ezetimibe 10 mg. The study intended to determine two things: whether ezetimibe could further lower LDL-C when combined with a statin, and whether risk could be reduced further by driving the LDL-C below 70 mg/dL and down to the mid-50s.
After 1 year, the average LDL-C level was 70 mg/dL in the simvastatin group and 53 mg/dL in the combined simvastatin and ezetimibe group. At 7 years, for the primary end point (cardiovascular death, myocardial infarction, unstable angina requiring hospitalization, coronary revascularization, or stroke), there was a 6% reduction of events in the combined drug treatment group, with the number of people needed to treat being 50 to prevent one event. For the narrower end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke, there was a 10% risk reduction in the combined drug treatment arm.14
The amount of risk reduction is exactly what was predicted by the Cholesterol Treatment Trialists’ Collaboration’s plot of reduction in events vs reduction in LDL-C based on the analysis of 26 trials, adding further evidence that it is the LDL-C reduction itself, rather than the means by which LDL-C is reduced, that is critical for benefit.
PCSK9 inhibitors: A new approach
Mutations in the gene for proprotein convertase subtilisin kexin type 9 (PCSK9) have become a new focus of interest for reducing LDL-C and cardiovascular risk.15 PCSK9 binds to the LDL-C receptor on the surface of hepatocytes and escorts it to its destruction in the lysosomes, rather than allowing it to return to the cell surface to take more LDL-C out of circulation.
People with a gain-of-function mutation (conferring too much PCSK9, resulting in fewer LDL-C receptors and more LDL-C in circulation) are a more recently recognized subset of those with autosomal-dominant familial hypercholesterolemia. They have total cholesterol levels in the 90th percentile, tendon xanthomas, and a high risk of myocardial infarction and stroke at a young age.
Conversely, those with a nonsense mutation in PCSK9—leading to loss of function—have a 28% reduction in mean LDL-C and 88% reduction in risk of coronary heart disease compared with those without the mutation.16 Two women (ages 32 and 21, fertile) have been found who have inactivating mutations in both PCSK9 alleles, and both are in apparent good health, with LDL-C levels of 14 mg/dL and 15 mg/dL, respectively.17,18
Dramatic reduction in LDL-C
Monoclonal antibodies have been developed that bind PCSK9 and block its action with the goal of developing new LDL-C–lowering treatments. Phase 2 clinical trials of varying doses of evolocumab (Repatha), a drug in this class, combined with standard therapy (a statin with or without ezetimibe), found a 66% reduction of LDL-C at high doses at 12 weeks compared with standard therapy alone, with concomitant reductions in other atherogenic lipoproteins.19 Patients who could not tolerate statins because of myalgia responded well to evolocumab.20
Patients with heterozygous familial hypercholesterolemia also had a substantial reduction in LDL-C (55% at the highest dosage), even though they have fewer LDL-C receptors for the drug to act upon.21 People with homozygous familial hypercholesterolemia and no LDL-C receptors had a lesser relative reduction in LDL-C that depended on the type of mutations they had. Nonetheless, given how high LDL-C levels are in this population, the absolute decreases in LDL-C level were quite impressive.
Cardiovascular risk reduced
Data at nearly 1 year showed continued reduction of LDL-C by about 60% (absolute reduction: 73 mg/dL), as well as a lower incidence of cardiovascular events starting at just 3 months, much sooner than observed in some statin trials.22 Benefits were found regardless of subgroup (sex, age, statin use, baseline LDL-C level, or known vascular disease). No difference was found in the safety profile between the evolocumab and control arms. Only 2.4% of participants discontinued evolocumab because of adverse events, and the incidence of adverse effects did not correlate with LDL-C level achieved.
Neurocognitive effects occurred in 0.9% of the evolocumab arm vs 0.3% in the control arm. This difference has not been explained: although there is cholesterol in the central nervous system, it is generated locally, and lipoproteins—and evolocumab—are not thought to cross the blood-brain barrier.
Long-term trials of evolocumab are currently under way for patients with cardiovascular disease, as are trials of two other PCSK9 inhibitors, alirocumab and bococizumab, in addition to standard statin therapy.
On July 24, 2015, the US Food and Drug Administration (FDA) approved the first PCSK9 inhibitor, alirocumab (Praluent) for patients with heterozygous familial hypercholesterolemia or those with clinical atherosclerotic cardiovascular disease who require additional lowering of LDL-C. The starting dosage is 75 mg subcutaneously every 2 weeks, which can be increased up to 150 mg every 2 weeks.
Evolocumab was approved by the FDA on August 27, 2015, for the same indications. The dosage is 140 mg subcutaneously every 2 weeks or 420 mg every month.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129:S1-S45. Erratum in: Circulation 2014; 129:S46–S48.
- Raymond C, Cho L, Rocco M, Hazen SL. New cholesterol guidelines: worth the wait? Cleve Clin J Med 2014; 81:11–19.
- Ridker PM, Rose L, Cook NR. A proposal to incorporate trial data into a hybrid ACC/AHA algorithm for the allocation of statin therapy in primary prevention. J Am Coll Cardiol 2015; 65:942–948.
- Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 2015; 385:2264–2271.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Grundy SM, Cleeman JI, Merz CN, et al; National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239. Erratum in Circulation 2004; 110:763.
- Ridker PM, Danielson E, Fonseca FAH, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232:34–47.
- Hochholzer W, Giugliano RP. Lipid lowering goals: back to nature? Ther Adv Cardiovasc Dis 2010; 4:185–191.
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- Hsia J, MacFadyen JG, Monyak J, Ridker PM. Cardiovascular event reduction and adverse events among subjects attaining low-density lipoprotein cholesterol <50 mg/dl with rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J Am Coll Cardiol 2011; 57:1666–1675.
- Cannon CP, Giugliano RP, Blazing MA, et al; IMPROVE-IT Investigators. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J 2008; 156:826–832.
- Cannon CP, Blazing MA, Giugliano RP, et al for the IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015; 372:2387–2397.
- Giugliano RP, Sabatine MS. Are PCSK9 Inhibitors the next breakthrough in the cardiovascular field? J Am Coll Cardiol 2015; 65:2638–2651.
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272.
- Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 2006; 79:514-523.
- Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007; 193:445–448.
- Giugliano RP, Desai NR, Kohli P, et al; LAPLACE-TIMI 57 Investigators. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet 2012; 380:2007–2017.
- Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA 2012; 308:2497–2506.
- Raal F, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012; 126:2408–2417.
- Sabatine MS, Giugliano RP, Wiviott SD, et al; Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500–1509.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129:S1-S45. Erratum in: Circulation 2014; 129:S46–S48.
- Raymond C, Cho L, Rocco M, Hazen SL. New cholesterol guidelines: worth the wait? Cleve Clin J Med 2014; 81:11–19.
- Ridker PM, Rose L, Cook NR. A proposal to incorporate trial data into a hybrid ACC/AHA algorithm for the allocation of statin therapy in primary prevention. J Am Coll Cardiol 2015; 65:942–948.
- Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 2015; 385:2264–2271.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Grundy SM, Cleeman JI, Merz CN, et al; National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239. Erratum in Circulation 2004; 110:763.
- Ridker PM, Danielson E, Fonseca FAH, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232:34–47.
- Hochholzer W, Giugliano RP. Lipid lowering goals: back to nature? Ther Adv Cardiovasc Dis 2010; 4:185–191.
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- Hsia J, MacFadyen JG, Monyak J, Ridker PM. Cardiovascular event reduction and adverse events among subjects attaining low-density lipoprotein cholesterol <50 mg/dl with rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J Am Coll Cardiol 2011; 57:1666–1675.
- Cannon CP, Giugliano RP, Blazing MA, et al; IMPROVE-IT Investigators. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J 2008; 156:826–832.
- Cannon CP, Blazing MA, Giugliano RP, et al for the IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015; 372:2387–2397.
- Giugliano RP, Sabatine MS. Are PCSK9 Inhibitors the next breakthrough in the cardiovascular field? J Am Coll Cardiol 2015; 65:2638–2651.
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272.
- Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 2006; 79:514-523.
- Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007; 193:445–448.
- Giugliano RP, Desai NR, Kohli P, et al; LAPLACE-TIMI 57 Investigators. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet 2012; 380:2007–2017.
- Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA 2012; 308:2497–2506.
- Raal F, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012; 126:2408–2417.
- Sabatine MS, Giugliano RP, Wiviott SD, et al; Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500–1509.
KEY POINTS
- Patients at high risk of atherosclerotic cardiovascular disease should be treated with high-intensity statin therapy.
- To date, no baseline level has been identified beneath which lowering LDL-C does not provide clinical benefit.
- The benefits of lower LDL-C are seen with a variety of pharmacologic interventions and in people who have naturally low levels due to genetic variants.
- Clinical trial evidence supports that ezetimibe reduces the risk of cardiovascular events.
- Proprotein convertase subtilisin kexin type 9 (PCSK9) inhibitors reduce LDL-C by approximately 60%, and preliminary data show that they reduce the risk of cardiovascular events.
