User login
Diet Tips for Diabetes Management & Metabolic Health

Can boxing training improve Parkinson reaction times?
NEW YORK – A small pilot study has shown that patients with Parkinson’s disease who participated in the Rock Steady Boxing non-contact training program may have faster reaction times than PD patients who did not participate in the program, according to a poster presented at the International Conference on Parkinson’s Disease and Movement Disorders.

“The novelty of this is that it shows how Rock Steady Boxing and exercise programs that use sequences and the learning of sequences could possibly help slow the decline, or maintain a level of functioning longer, in Parkinson’s disease,” said Christopher McLeod, a second-year medical student at New York Institute of Technology (NYIT) College of Osteopathic Medicine, Old Westbury, N.Y.
Rock Steady Boxing is a non-contact program tailored to Parkinson’s patients founded in 2006 by Scott Newman, an Indiana lawyer who was diagnosed with early onset Parkinson’s at age 40. The regimen involves intense one-on-one training centered around boxing. Rock Steady Boxing offers classes from coast to coast in the United States and in 13 other countries. Mr. McLeod is a volunteer at the NYIT chapter of Rock Steady Boxing in Old Westbury, N.Y.
Mr. McLeod studied 28 PD patients – 14 who had been taking Rock Steady Boxing classes at NYIT for at least 6 months and 14 controls. The goal of the study was to evaluate if the Rock Steady Boxing participants showed any improvement in procedural motor learning. His coauthor was Adena Leder, DO, a faculty neurologist and movement disorder specialist at NYIT,
“What’s new about this research is the procedural memory component and the Rock Steady Boxing program is just more of the vessel, so to speak,” Mr. McLeod said. “This is a pilot study. We wanted to see if Rock Steady Boxing would show benefits in these patients. There are some trends in my research that [indicate] it would; it did not have statistical significance, but we did see trend lines.”
The researchers used a modified Serial Reaction Time Test (SRTT) composed of seven blocks of 10 stimuli each with 30-second breaks between blocks. Blocks consisted of a random familiarization block, four learning blocks repeating the same sequence of stimuli, a transfer block of random stimuli, and a posttransfer block presenting the same sequence of stimuli from the four learning blocks.
They assessed procedural learning by comparing the reduction in response time over the four identical learning blocks as well as by comparing changes in response time when the subjects were subsequently exposed to the random transfer block.
Experienced boxers demonstrated faster reaction time over the four learning blocks, ranging from 795.32 vs. 906.89 ms in the first learning block to 674.79 vs. 787.32 ms in the fourth learning block (P = .19). In the random sequence transfer block, controls showed a 93.5-ms decrease in median reaction time vs. a 27.3-ms increase in reaction time of experienced boxers. One possible explanation the investigators noted is that the controls simply got better at reading the stimuli over time without actually learning the repeated sequence.
Mr. McLeod noted that a typical Rock Steady Boxing session starts with a warmup and stretch, then learning the boxing stance with the nondominant foot back, shoulders over the body and the head over the feet. The boxing moves involve sequences of different punching combinations — jab, jab, cross; left, left, right; jab, cross, hook. Then the class divides into separate circuits for boxing and exercise. The boxing circuit involves punching the speed bag – the small, air-filled, pear-shaped bag attached to a hook at eye level – as well as heavy bag and partner-held focus mitts, all with the aim of reinforcing the learned sequences. The exercise circuit focuses on muscle training and exercise with the goal of improving balance and gait.
“The boxing sequences help not only with cognitive ability but motor control,” Mr. McLeod said. “The program also helps with some of the nonmotor aspects of Parkinson’s disease. Depression is almost synonymous with Parkinson’s disease; this brings people together and builds camaraderie.”
Mr. McLeod said he hopes the research continues. “I’m hoping that this can be a jumping-off point for research going forward with procedural memory, Parkinson’s, and Rock Steady Boxing or programs like it,” he said. Future research should involve more subjects, measure improvement within same subjects who participate in the program, and account for variables such as age and gender.
Mr. McLeod and Dr. Leder reported having no relevant financial disclosures.
NEW YORK – A small pilot study has shown that patients with Parkinson’s disease who participated in the Rock Steady Boxing non-contact training program may have faster reaction times than PD patients who did not participate in the program, according to a poster presented at the International Conference on Parkinson’s Disease and Movement Disorders.
“The novelty of this is that it shows how Rock Steady Boxing and exercise programs that use sequences and the learning of sequences could possibly help slow the decline, or maintain a level of functioning longer, in Parkinson’s disease,” said Christopher McLeod, a second-year medical student at New York Institute of Technology (NYIT) College of Osteopathic Medicine, Old Westbury, N.Y.
Rock Steady Boxing is a non-contact program tailored to Parkinson’s patients founded in 2006 by Scott Newman, an Indiana lawyer who was diagnosed with early onset Parkinson’s at age 40. The regimen involves intense one-on-one training centered around boxing. Rock Steady Boxing offers classes from coast to coast in the United States and in 13 other countries. Mr. McLeod is a volunteer at the NYIT chapter of Rock Steady Boxing in Old Westbury, N.Y.
Mr. McLeod studied 28 PD patients – 14 who had been taking Rock Steady Boxing classes at NYIT for at least 6 months and 14 controls. The goal of the study was to evaluate if the Rock Steady Boxing participants showed any improvement in procedural motor learning. His coauthor was Adena Leder, DO, a faculty neurologist and movement disorder specialist at NYIT,
“What’s new about this research is the procedural memory component and the Rock Steady Boxing program is just more of the vessel, so to speak,” Mr. McLeod said. “This is a pilot study. We wanted to see if Rock Steady Boxing would show benefits in these patients. There are some trends in my research that [indicate] it would; it did not have statistical significance, but we did see trend lines.”
The researchers used a modified Serial Reaction Time Test (SRTT) composed of seven blocks of 10 stimuli each with 30-second breaks between blocks. Blocks consisted of a random familiarization block, four learning blocks repeating the same sequence of stimuli, a transfer block of random stimuli, and a posttransfer block presenting the same sequence of stimuli from the four learning blocks.
They assessed procedural learning by comparing the reduction in response time over the four identical learning blocks as well as by comparing changes in response time when the subjects were subsequently exposed to the random transfer block.
Experienced boxers demonstrated faster reaction time over the four learning blocks, ranging from 795.32 vs. 906.89 ms in the first learning block to 674.79 vs. 787.32 ms in the fourth learning block (P = .19). In the random sequence transfer block, controls showed a 93.5-ms decrease in median reaction time vs. a 27.3-ms increase in reaction time of experienced boxers. One possible explanation the investigators noted is that the controls simply got better at reading the stimuli over time without actually learning the repeated sequence.
Mr. McLeod noted that a typical Rock Steady Boxing session starts with a warmup and stretch, then learning the boxing stance with the nondominant foot back, shoulders over the body and the head over the feet. The boxing moves involve sequences of different punching combinations — jab, jab, cross; left, left, right; jab, cross, hook. Then the class divides into separate circuits for boxing and exercise. The boxing circuit involves punching the speed bag – the small, air-filled, pear-shaped bag attached to a hook at eye level – as well as heavy bag and partner-held focus mitts, all with the aim of reinforcing the learned sequences. The exercise circuit focuses on muscle training and exercise with the goal of improving balance and gait.
“The boxing sequences help not only with cognitive ability but motor control,” Mr. McLeod said. “The program also helps with some of the nonmotor aspects of Parkinson’s disease. Depression is almost synonymous with Parkinson’s disease; this brings people together and builds camaraderie.”
Mr. McLeod said he hopes the research continues. “I’m hoping that this can be a jumping-off point for research going forward with procedural memory, Parkinson’s, and Rock Steady Boxing or programs like it,” he said. Future research should involve more subjects, measure improvement within same subjects who participate in the program, and account for variables such as age and gender.
Mr. McLeod and Dr. Leder reported having no relevant financial disclosures.
NEW YORK – A small pilot study has shown that patients with Parkinson’s disease who participated in the Rock Steady Boxing non-contact training program may have faster reaction times than PD patients who did not participate in the program, according to a poster presented at the International Conference on Parkinson’s Disease and Movement Disorders.
“The novelty of this is that it shows how Rock Steady Boxing and exercise programs that use sequences and the learning of sequences could possibly help slow the decline, or maintain a level of functioning longer, in Parkinson’s disease,” said Christopher McLeod, a second-year medical student at New York Institute of Technology (NYIT) College of Osteopathic Medicine, Old Westbury, N.Y.
Rock Steady Boxing is a non-contact program tailored to Parkinson’s patients founded in 2006 by Scott Newman, an Indiana lawyer who was diagnosed with early onset Parkinson’s at age 40. The regimen involves intense one-on-one training centered around boxing. Rock Steady Boxing offers classes from coast to coast in the United States and in 13 other countries. Mr. McLeod is a volunteer at the NYIT chapter of Rock Steady Boxing in Old Westbury, N.Y.
Mr. McLeod studied 28 PD patients – 14 who had been taking Rock Steady Boxing classes at NYIT for at least 6 months and 14 controls. The goal of the study was to evaluate if the Rock Steady Boxing participants showed any improvement in procedural motor learning. His coauthor was Adena Leder, DO, a faculty neurologist and movement disorder specialist at NYIT,
“What’s new about this research is the procedural memory component and the Rock Steady Boxing program is just more of the vessel, so to speak,” Mr. McLeod said. “This is a pilot study. We wanted to see if Rock Steady Boxing would show benefits in these patients. There are some trends in my research that [indicate] it would; it did not have statistical significance, but we did see trend lines.”
The researchers used a modified Serial Reaction Time Test (SRTT) composed of seven blocks of 10 stimuli each with 30-second breaks between blocks. Blocks consisted of a random familiarization block, four learning blocks repeating the same sequence of stimuli, a transfer block of random stimuli, and a posttransfer block presenting the same sequence of stimuli from the four learning blocks.
They assessed procedural learning by comparing the reduction in response time over the four identical learning blocks as well as by comparing changes in response time when the subjects were subsequently exposed to the random transfer block.
Experienced boxers demonstrated faster reaction time over the four learning blocks, ranging from 795.32 vs. 906.89 ms in the first learning block to 674.79 vs. 787.32 ms in the fourth learning block (P = .19). In the random sequence transfer block, controls showed a 93.5-ms decrease in median reaction time vs. a 27.3-ms increase in reaction time of experienced boxers. One possible explanation the investigators noted is that the controls simply got better at reading the stimuli over time without actually learning the repeated sequence.
Mr. McLeod noted that a typical Rock Steady Boxing session starts with a warmup and stretch, then learning the boxing stance with the nondominant foot back, shoulders over the body and the head over the feet. The boxing moves involve sequences of different punching combinations — jab, jab, cross; left, left, right; jab, cross, hook. Then the class divides into separate circuits for boxing and exercise. The boxing circuit involves punching the speed bag – the small, air-filled, pear-shaped bag attached to a hook at eye level – as well as heavy bag and partner-held focus mitts, all with the aim of reinforcing the learned sequences. The exercise circuit focuses on muscle training and exercise with the goal of improving balance and gait.
“The boxing sequences help not only with cognitive ability but motor control,” Mr. McLeod said. “The program also helps with some of the nonmotor aspects of Parkinson’s disease. Depression is almost synonymous with Parkinson’s disease; this brings people together and builds camaraderie.”
Mr. McLeod said he hopes the research continues. “I’m hoping that this can be a jumping-off point for research going forward with procedural memory, Parkinson’s, and Rock Steady Boxing or programs like it,” he said. Future research should involve more subjects, measure improvement within same subjects who participate in the program, and account for variables such as age and gender.
Mr. McLeod and Dr. Leder reported having no relevant financial disclosures.
REPORTING FROM ICPDMD 2018
Key clinical point: Exercise programs may help improve procedural learning in individuals with Parkinson’s disease.
Major finding: Rock Steady Boxing experienced boxers demonstrated reaction times ranging from 795.32 vs. 906.89 ms to 674.79 vs. 787.32 ms across four test blocks.
Study details: Pilot study of 14 Parkinson’s patients who participated in Rock Steady Boxing vs. 14 controls.
Disclosures: Mr. McLeod reported no relevant financial disclosures.
Changing Public Perception of Vitiligo


Bleeding score could help identify hemophilia
Bleeding scores may be helpful in identifying hemophilia patients, regardless of whether or not clotting factor levels are known, results of a recent investigation suggest.

Both hemophilia A and B patients had significantly higher bleeding scores as assessed by the ISTH-BAT (International Society on Thrombosis and Hemostasis–Bleeding Assessment Tool), compared with control subjects, according to results of the study.
Moreover, hemophilia patients classified as severe had significantly higher ISTH-BAT scores compared with those classified as mild, reported by Munira Borhany, MD, of the National Institute of Blood Disease and Bone Marrow Transplantation, Karachi, Pakistan, and her colleagues.
“The ISTH-BAT can be easily used in the clinics by physicians and can help to identify those patients who should be further investigated,” Dr. Borhany and her coauthors reported in the journal Transfusion and Apheresis Science.
The ISTH-BAT, established to standardize the reporting of bleeding symptoms, scores symptoms from 0, which indicates absent or trivial, to 4, meaning a symptom that requires medical intervention. Total scores considered abnormal are 4 or greater in men, 6 and greater in women, and 3 and greater in children, according to previously published reports.
In the present cross-sectional study, Dr. Borhany and her colleagues evaluated bleeding scores for 115 adult and pediatric patients – 78 with hemophilia A and 37 with hemophilia B – who were treated in Pakistan between 2014 and 2016.
Bleeding scores were a mean of 13.5 and 13.2 for hemophilia A and B patients, respectively, and 0.8 for 100 healthy male controls also included in the study. Scores were significantly higher in hemophilia patients versus controls (P less than .001), but not different between hemophilia A and B patients, the investigators reported.
Further results suggested a correlation between factor levels and clinical presentation of bleeding symptoms, according to the investigators. Statistically significant differences in bleeding scores also were seen between patients with severe and mild disease, and between severe and moderate disease, but not between the mild and moderate groups, they added.
Most studies of bleeding questionnaires to date have been in patients with von Willebrand disease or platelet disorders, with very little data on hemophilia.
“Apart from one recent study using ISTH-BAT in hemophilia carriers as part of assessing quality of life, we are unaware of other studies examining this assessment tool in hemophilia,” the researchers wrote.
This study cohort was unique, according to the investigators, because it included a substantial number of adults who were new patients with bleeding symptoms who had no previous diagnosis of hemophilia. “This allowed assessing whether investigators may tend to apply a higher score when knowing very low factor levels in hemophilia patients,” they said.
In fact, there was no major difference in bleeding scores for those newly diagnosed patients versus already diagnosed patients.
Results of an ongoing study will determine whether the ISTH BAT bleeding score can predict risk of bleeding in hemophilia patients, according to Dr. Borhany and her coauthors.
They reported having no conflicts of interest.
SOURCE: Borhany M et al. Transfus Apher Sci. 2018 Aug;57(4):556-60.
Bleeding scores may be helpful in identifying hemophilia patients, regardless of whether or not clotting factor levels are known, results of a recent investigation suggest.
Both hemophilia A and B patients had significantly higher bleeding scores as assessed by the ISTH-BAT (International Society on Thrombosis and Hemostasis–Bleeding Assessment Tool), compared with control subjects, according to results of the study.
Moreover, hemophilia patients classified as severe had significantly higher ISTH-BAT scores compared with those classified as mild, reported by Munira Borhany, MD, of the National Institute of Blood Disease and Bone Marrow Transplantation, Karachi, Pakistan, and her colleagues.
“The ISTH-BAT can be easily used in the clinics by physicians and can help to identify those patients who should be further investigated,” Dr. Borhany and her coauthors reported in the journal Transfusion and Apheresis Science.
The ISTH-BAT, established to standardize the reporting of bleeding symptoms, scores symptoms from 0, which indicates absent or trivial, to 4, meaning a symptom that requires medical intervention. Total scores considered abnormal are 4 or greater in men, 6 and greater in women, and 3 and greater in children, according to previously published reports.
In the present cross-sectional study, Dr. Borhany and her colleagues evaluated bleeding scores for 115 adult and pediatric patients – 78 with hemophilia A and 37 with hemophilia B – who were treated in Pakistan between 2014 and 2016.
Bleeding scores were a mean of 13.5 and 13.2 for hemophilia A and B patients, respectively, and 0.8 for 100 healthy male controls also included in the study. Scores were significantly higher in hemophilia patients versus controls (P less than .001), but not different between hemophilia A and B patients, the investigators reported.
Further results suggested a correlation between factor levels and clinical presentation of bleeding symptoms, according to the investigators. Statistically significant differences in bleeding scores also were seen between patients with severe and mild disease, and between severe and moderate disease, but not between the mild and moderate groups, they added.
Most studies of bleeding questionnaires to date have been in patients with von Willebrand disease or platelet disorders, with very little data on hemophilia.
“Apart from one recent study using ISTH-BAT in hemophilia carriers as part of assessing quality of life, we are unaware of other studies examining this assessment tool in hemophilia,” the researchers wrote.
This study cohort was unique, according to the investigators, because it included a substantial number of adults who were new patients with bleeding symptoms who had no previous diagnosis of hemophilia. “This allowed assessing whether investigators may tend to apply a higher score when knowing very low factor levels in hemophilia patients,” they said.
In fact, there was no major difference in bleeding scores for those newly diagnosed patients versus already diagnosed patients.
Results of an ongoing study will determine whether the ISTH BAT bleeding score can predict risk of bleeding in hemophilia patients, according to Dr. Borhany and her coauthors.
They reported having no conflicts of interest.
SOURCE: Borhany M et al. Transfus Apher Sci. 2018 Aug;57(4):556-60.
Bleeding scores may be helpful in identifying hemophilia patients, regardless of whether or not clotting factor levels are known, results of a recent investigation suggest.
Both hemophilia A and B patients had significantly higher bleeding scores as assessed by the ISTH-BAT (International Society on Thrombosis and Hemostasis–Bleeding Assessment Tool), compared with control subjects, according to results of the study.
Moreover, hemophilia patients classified as severe had significantly higher ISTH-BAT scores compared with those classified as mild, reported by Munira Borhany, MD, of the National Institute of Blood Disease and Bone Marrow Transplantation, Karachi, Pakistan, and her colleagues.
“The ISTH-BAT can be easily used in the clinics by physicians and can help to identify those patients who should be further investigated,” Dr. Borhany and her coauthors reported in the journal Transfusion and Apheresis Science.
The ISTH-BAT, established to standardize the reporting of bleeding symptoms, scores symptoms from 0, which indicates absent or trivial, to 4, meaning a symptom that requires medical intervention. Total scores considered abnormal are 4 or greater in men, 6 and greater in women, and 3 and greater in children, according to previously published reports.
In the present cross-sectional study, Dr. Borhany and her colleagues evaluated bleeding scores for 115 adult and pediatric patients – 78 with hemophilia A and 37 with hemophilia B – who were treated in Pakistan between 2014 and 2016.
Bleeding scores were a mean of 13.5 and 13.2 for hemophilia A and B patients, respectively, and 0.8 for 100 healthy male controls also included in the study. Scores were significantly higher in hemophilia patients versus controls (P less than .001), but not different between hemophilia A and B patients, the investigators reported.
Further results suggested a correlation between factor levels and clinical presentation of bleeding symptoms, according to the investigators. Statistically significant differences in bleeding scores also were seen between patients with severe and mild disease, and between severe and moderate disease, but not between the mild and moderate groups, they added.
Most studies of bleeding questionnaires to date have been in patients with von Willebrand disease or platelet disorders, with very little data on hemophilia.
“Apart from one recent study using ISTH-BAT in hemophilia carriers as part of assessing quality of life, we are unaware of other studies examining this assessment tool in hemophilia,” the researchers wrote.
This study cohort was unique, according to the investigators, because it included a substantial number of adults who were new patients with bleeding symptoms who had no previous diagnosis of hemophilia. “This allowed assessing whether investigators may tend to apply a higher score when knowing very low factor levels in hemophilia patients,” they said.
In fact, there was no major difference in bleeding scores for those newly diagnosed patients versus already diagnosed patients.
Results of an ongoing study will determine whether the ISTH BAT bleeding score can predict risk of bleeding in hemophilia patients, according to Dr. Borhany and her coauthors.
They reported having no conflicts of interest.
SOURCE: Borhany M et al. Transfus Apher Sci. 2018 Aug;57(4):556-60.
FROM TRANSFUSION AND APHERESIS SCIENCE
Key clinical point:
Major finding: Bleeding scores were a mean of 13.5 and 13.2 for hemophilia A and B patients, respectively, and 0.8 for healthy male controls (P less than .001).
Study details: A cross-sectional study included 115 adult and pediatric patients with hemophilia A or B treated in Pakistan between 2014 and 2016.
Disclosures: The authors reported having no conflicts of interest.
Source: Borhany M et al. Transfus Apher Sci. 2018 Aug;57(4):556-60.
Psoriasis adds to increased risk of cardiovascular procedures, surgery in patients with hypertension
compared with patients with hypertension alone.
“The results suggested that hypertensive patients with concurrent psoriasis experienced an earlier and more aggressive disease progression of hypertension, compared with general hypertensive patients,” Hsien-Yi Chiu, MD, PhD, from the department of dermatology at the National Taiwan University Hospital in Hsinchu, Taiwan, and his colleagues wrote in the Journal of Dermatology. “Thus, patients with hypertension and psoriasis should be considered for more aggressive strategies for prevention of primary [cardiovascular disease] and more intense assessments for cardiovascular interventions needed to improve [cardiovascular disease] outcome in these patients.”
They performed a nationwide cohort study of patients in the Taiwan National Health Insurance Research Database with new onset hypertension from 2005 to 2006. Those with psoriasis (4,039 patients) were matched by age and sex to patients in the database who were diagnosed with hypertension but not psoriasis; the mean follow-up was 5.62 years. Their mean age was 58 years and about 31% of the psoriasis cohort were female. They were divided into groups based on psoriasis severity (mild and severe psoriasis) and type (psoriasis with and without arthritis). Researchers noted patients with both psoriasis and hypertension also had higher rates of cerebrovascular disease, coronary heart disease, hyperlipidemia, and diabetes mellitus during the year prior to the study.
The outcome measured was having a cardiovascular procedure (percutaneous coronary intervention with/without stenting or percutaneous transluminal coronary angioplasty and transcatheter radiofrequency ablation for arrhythmia) and cardiovascular surgery (coronary artery bypass grafting and other surgery for heart valves, arrhythmia, cerebrovascular disease, peripheral vessels, and the aorta).
Patients with both psoriasis and hypertension were at an increased risk for having a cardiovascular procedure and surgery (adjusted hazard ratio, 1.28; 95% confidence interval, 1.07-1.53), compared with patients with only hypertension. The risk of this outcome was also increased among patients with severe psoriasis or psoriatic arthritis, compared with patients who had mild psoriasis (aHR, 1.22; 95% CI, 0.98-1.51) and with patients with psoriasis but not arthritis (aHR, 1.15; 95% CI, 0.84-1.58); however, the results did not reach statistical significance after adjustment, which the researchers attributed to the small subgroup size.
“Another possible explanation was that the observed increased requirement for cardiovascular procedure and surgery in patients with severe psoriasis was mediated by a complex interplay among inflammation, traditional risk factors for [cardiovascular disease], and antirheumatic drugs, which probably attenuate the effects conferred by psoriasis,” the authors wrote.
Limitations in the study included reliance on administrative claims data for psoriasis diagnosis, unavailability of some details of the cardiovascular procedures and surgery, lack of blood pressure data to identify hypertension severity, as well as unmeasured factors and confounders. Further, “comparative occurrence of a requirement for cardiovascular procedure and surgery in the two groups may be influenced by a competing risk for death,” the researchers noted.
This study was supported in part through grants by the National Taiwan University Hospital, Asia-Pacific La Roche–Posay Foundation 2014, and the Ministry of Science and Technology. Dr. Chiu is on the speaker’s bureau for AbbVie, Janssen Pharmaceuticals, Novartis, Eli Lilly and Pfizer. Another author has conducted clinical trials for or received fees for being a consultant or speaker for companies that include Abbvie, Boehringer Ingelheim, and Celgene. The remaining authors reported no relevant conflicts of interest.
SOURCE: Chiu H-Y et al. J Dermatol. 2018 Oct 16. doi: 10.1111/1346-8138.14654.
compared with patients with hypertension alone.
“The results suggested that hypertensive patients with concurrent psoriasis experienced an earlier and more aggressive disease progression of hypertension, compared with general hypertensive patients,” Hsien-Yi Chiu, MD, PhD, from the department of dermatology at the National Taiwan University Hospital in Hsinchu, Taiwan, and his colleagues wrote in the Journal of Dermatology. “Thus, patients with hypertension and psoriasis should be considered for more aggressive strategies for prevention of primary [cardiovascular disease] and more intense assessments for cardiovascular interventions needed to improve [cardiovascular disease] outcome in these patients.”
They performed a nationwide cohort study of patients in the Taiwan National Health Insurance Research Database with new onset hypertension from 2005 to 2006. Those with psoriasis (4,039 patients) were matched by age and sex to patients in the database who were diagnosed with hypertension but not psoriasis; the mean follow-up was 5.62 years. Their mean age was 58 years and about 31% of the psoriasis cohort were female. They were divided into groups based on psoriasis severity (mild and severe psoriasis) and type (psoriasis with and without arthritis). Researchers noted patients with both psoriasis and hypertension also had higher rates of cerebrovascular disease, coronary heart disease, hyperlipidemia, and diabetes mellitus during the year prior to the study.
The outcome measured was having a cardiovascular procedure (percutaneous coronary intervention with/without stenting or percutaneous transluminal coronary angioplasty and transcatheter radiofrequency ablation for arrhythmia) and cardiovascular surgery (coronary artery bypass grafting and other surgery for heart valves, arrhythmia, cerebrovascular disease, peripheral vessels, and the aorta).
Patients with both psoriasis and hypertension were at an increased risk for having a cardiovascular procedure and surgery (adjusted hazard ratio, 1.28; 95% confidence interval, 1.07-1.53), compared with patients with only hypertension. The risk of this outcome was also increased among patients with severe psoriasis or psoriatic arthritis, compared with patients who had mild psoriasis (aHR, 1.22; 95% CI, 0.98-1.51) and with patients with psoriasis but not arthritis (aHR, 1.15; 95% CI, 0.84-1.58); however, the results did not reach statistical significance after adjustment, which the researchers attributed to the small subgroup size.
“Another possible explanation was that the observed increased requirement for cardiovascular procedure and surgery in patients with severe psoriasis was mediated by a complex interplay among inflammation, traditional risk factors for [cardiovascular disease], and antirheumatic drugs, which probably attenuate the effects conferred by psoriasis,” the authors wrote.
Limitations in the study included reliance on administrative claims data for psoriasis diagnosis, unavailability of some details of the cardiovascular procedures and surgery, lack of blood pressure data to identify hypertension severity, as well as unmeasured factors and confounders. Further, “comparative occurrence of a requirement for cardiovascular procedure and surgery in the two groups may be influenced by a competing risk for death,” the researchers noted.
This study was supported in part through grants by the National Taiwan University Hospital, Asia-Pacific La Roche–Posay Foundation 2014, and the Ministry of Science and Technology. Dr. Chiu is on the speaker’s bureau for AbbVie, Janssen Pharmaceuticals, Novartis, Eli Lilly and Pfizer. Another author has conducted clinical trials for or received fees for being a consultant or speaker for companies that include Abbvie, Boehringer Ingelheim, and Celgene. The remaining authors reported no relevant conflicts of interest.
SOURCE: Chiu H-Y et al. J Dermatol. 2018 Oct 16. doi: 10.1111/1346-8138.14654.
compared with patients with hypertension alone.
“The results suggested that hypertensive patients with concurrent psoriasis experienced an earlier and more aggressive disease progression of hypertension, compared with general hypertensive patients,” Hsien-Yi Chiu, MD, PhD, from the department of dermatology at the National Taiwan University Hospital in Hsinchu, Taiwan, and his colleagues wrote in the Journal of Dermatology. “Thus, patients with hypertension and psoriasis should be considered for more aggressive strategies for prevention of primary [cardiovascular disease] and more intense assessments for cardiovascular interventions needed to improve [cardiovascular disease] outcome in these patients.”
They performed a nationwide cohort study of patients in the Taiwan National Health Insurance Research Database with new onset hypertension from 2005 to 2006. Those with psoriasis (4,039 patients) were matched by age and sex to patients in the database who were diagnosed with hypertension but not psoriasis; the mean follow-up was 5.62 years. Their mean age was 58 years and about 31% of the psoriasis cohort were female. They were divided into groups based on psoriasis severity (mild and severe psoriasis) and type (psoriasis with and without arthritis). Researchers noted patients with both psoriasis and hypertension also had higher rates of cerebrovascular disease, coronary heart disease, hyperlipidemia, and diabetes mellitus during the year prior to the study.
The outcome measured was having a cardiovascular procedure (percutaneous coronary intervention with/without stenting or percutaneous transluminal coronary angioplasty and transcatheter radiofrequency ablation for arrhythmia) and cardiovascular surgery (coronary artery bypass grafting and other surgery for heart valves, arrhythmia, cerebrovascular disease, peripheral vessels, and the aorta).
Patients with both psoriasis and hypertension were at an increased risk for having a cardiovascular procedure and surgery (adjusted hazard ratio, 1.28; 95% confidence interval, 1.07-1.53), compared with patients with only hypertension. The risk of this outcome was also increased among patients with severe psoriasis or psoriatic arthritis, compared with patients who had mild psoriasis (aHR, 1.22; 95% CI, 0.98-1.51) and with patients with psoriasis but not arthritis (aHR, 1.15; 95% CI, 0.84-1.58); however, the results did not reach statistical significance after adjustment, which the researchers attributed to the small subgroup size.
“Another possible explanation was that the observed increased requirement for cardiovascular procedure and surgery in patients with severe psoriasis was mediated by a complex interplay among inflammation, traditional risk factors for [cardiovascular disease], and antirheumatic drugs, which probably attenuate the effects conferred by psoriasis,” the authors wrote.
Limitations in the study included reliance on administrative claims data for psoriasis diagnosis, unavailability of some details of the cardiovascular procedures and surgery, lack of blood pressure data to identify hypertension severity, as well as unmeasured factors and confounders. Further, “comparative occurrence of a requirement for cardiovascular procedure and surgery in the two groups may be influenced by a competing risk for death,” the researchers noted.
This study was supported in part through grants by the National Taiwan University Hospital, Asia-Pacific La Roche–Posay Foundation 2014, and the Ministry of Science and Technology. Dr. Chiu is on the speaker’s bureau for AbbVie, Janssen Pharmaceuticals, Novartis, Eli Lilly and Pfizer. Another author has conducted clinical trials for or received fees for being a consultant or speaker for companies that include Abbvie, Boehringer Ingelheim, and Celgene. The remaining authors reported no relevant conflicts of interest.
SOURCE: Chiu H-Y et al. J Dermatol. 2018 Oct 16. doi: 10.1111/1346-8138.14654.
FROM THE JOURNAL OF DERMATOLOGY
Key clinical point: More aggressive cardiovascular disease preventive strategies should be considered in patients with hypertension who also have psoriasis.
Major finding: Patients with both psoriasis and hypertension were at an increased risk for requiring a cardiovascular procedure and surgery (adjusted hazard ratio, 1.28), compared with patients with hypertension alone.
Study details: A retrospective cohort study evaluated risk of this outcome in 4,039 patients with psoriasis and hypertension, compared with patients who had hypertension, matched for age and sex.
Disclosures: This study was supported in part through grants by the National Taiwan University Hospital Hsin-Chu Branch, Asia-Pacific La Roche–Posay Foundation 2014, and the Ministry of Science and Technology. Dr. Chiu is on the speaker’s bureau for companies including AbbVie, Novartis, and Eli Lilly. Another author has conducted clinical trials for or received fees for being a consultant or speaker for Abbvie, Boehringer Ingelheim, Celgene, Janssen Pharmaceuticals, Eli Lilly, Galderma, Novartis, and Pfizer. The other authors reported no relevant conflicts of interest.
Source: Chiu H-Y et al. J Dermatol. 2018 Oct 16. doi:10.1111/1346-8138.14654.
Pathologic superstition
When you believe in things that you don’t understand
Then you suffer
Superstition ain’t the way
– Stevie Wonder

I have always found it odd that airplanes don’t have a 13th row and hotels don’t have a 13th floor. Well, of course they do, but they are not labeled that way. Many people would hesitate to sit in the 13th row of an airplane since 13 is such an unlucky number. At least many people in the United States think the number 13 is unlucky. Thirteen is just a number in much of Asia. There, the number 4 is just as threatening as 13 is to us.
Superstitions like these are familiar to all of us.
One of my favorites is the belief that vacuum cups attached to the skin will somehow draw out toxins and generally improve health. “Cupping,” as the practice is known, is endorsed by several celebrities and famous athletes. After the treatment, a cupped patient exhibits circles of hyperemia, and no other apparent harm. I suspect that about a third of cupped patients truly think they have benefited from a good cupping, about the same number that would benefit from an orally administered placebo.
Superstitions are everywhere. Whether it is a black cat in the United States, infinite reflecting mirrors in Mexico, going back to your house after a wake in the Philippines, or whistling indoors in Lithuania, superstitions are pervasive, deeply held, and generally harmless. They are good for a good laugh as we recognize how ludicrous these unfounded fears are.
Some superstitions, though, are no laughing matter. They can be quite harmful. They are pathologic superstitions.
For example, some people believe vaccines cause autism in children. That pathologic superstition has consequences. A recent CDC report revealed that the population of unvaccinated children in the United States has quadrupled since 2001. This comes as no surprise as we hear about more measles outbreaks – and the deaths associated with them – in populations of unvaccinated children every year. A similar and pervasive pathologic superstition is the fear that an influenza vaccine will cause the flu. I wonder how many people die from this misconception.
Other people believe that their cancer can be treated, if not cured, with unproven, unconventional treatments. I cannot understand how this pathologic superstition developed. The purveyors of unconventional treatment hold much of the blame, but gullibility and ignorance may play a larger role. The consequences are tragic. A recent report demonstrated an approximately twofold increased risk of death in patients who used complementary therapies, compared with those who did not (JAMA Oncol. 2018 Oct 1;4[10]:1375-81).
These are sobering data for those of us who have in the past relented when our patients asked if they could take this or that supplement because we did not think they would cause significant harm.
Superstitions apparently are part of the human condition, evolved to attribute causation and provide order. They are a learned phenomenon. They are learned by reasonable people with normal intelligence and rational thinking. A superstition is born when someone is exposed to a false statement by someone or something they trust – a trusted other.
Trusted others exude certainty. Once established, superstitions are regrettably difficult to remove by those who are less certain, like physicians. How willing are we to say that the flu vaccine is 100% safe? Without certainty, how can a physician debunk a superstition? The techniques that we have been taught usually work, but not when faced with a pathologic superstition.
Science and experience teach us that firmly held superstitions cannot be broken with logical, stepwise reasoning. Jonathan Haidt provides a useful metaphor for this problem in his book “The Happiness Hypothesis” (Basic Books, 2006). He describes a rider on an elephant. The rider represents our rational thought and the elephant represents our emotional foundation. The rider thinks he controls the elephant, but the opposite is more likely true. In order to move the elephant in a certain direction, the rider needs to make the elephant want to turn in that direction. Otherwise, all the cajoling and arguing in the world won’t make the elephant turn. A rational argument made to someone emotionally invested in the counter argument will fail. That is why we cannot convince antivaccine parents to vaccinate their children by trying to persuade them with facts. Neither can we convince global warming skeptics to stop burning coal, gun advocates to vote for restrictions on gun ownership, or cancer patients to accept curative treatment if their values and morals are being challenged.
In a later book, “The Righteous Mind: Why Good People Are Divided by Politics and Religion” (Vintage Books, 2012), Mr. Haidt expands his hypothesis to declare that to change minds, we must appeal to underlying moral values. The challenge is to identify those moral underpinnings in our patients in order to develop an appeal likely to resonate with their emotions and values.
Superstition derives from something people learn either from trusted others or from personal experience. It does no good for physicians to deride patient beliefs and denigrate their agency in an attempt to persuade them to abandon what we consider irrational beliefs. For physicians to penetrate pathologic superstitions, they will have to become the trusted other, to understand moral foundations, to emotionally connect. That does not usually happen the first day we meet a new patient, especially a skeptical one. It takes time, and effort, to reach out and bond with the patient and their family. Only then can pathologic superstitions dissolve and a better patient-doctor relationship evolve.
During this season rife with superstition, remember that your patient’s own superstitions are part of their belief system, and your belief system may be threatening to them. Make your beliefs less threatening, become a trusted other, and appeal to their foundational values, and you can successfully break a pathologic superstition.
Dr. Kalaycio is editor in chief of Hematology News. He chairs the department of hematologic oncology and blood disorders at Cleveland Clinic Taussig Cancer Institute. Contact him at kalaycm@ccf.org.
When you believe in things that you don’t understand
Then you suffer
Superstition ain’t the way
– Stevie Wonder
I have always found it odd that airplanes don’t have a 13th row and hotels don’t have a 13th floor. Well, of course they do, but they are not labeled that way. Many people would hesitate to sit in the 13th row of an airplane since 13 is such an unlucky number. At least many people in the United States think the number 13 is unlucky. Thirteen is just a number in much of Asia. There, the number 4 is just as threatening as 13 is to us.
Superstitions like these are familiar to all of us.
One of my favorites is the belief that vacuum cups attached to the skin will somehow draw out toxins and generally improve health. “Cupping,” as the practice is known, is endorsed by several celebrities and famous athletes. After the treatment, a cupped patient exhibits circles of hyperemia, and no other apparent harm. I suspect that about a third of cupped patients truly think they have benefited from a good cupping, about the same number that would benefit from an orally administered placebo.
Superstitions are everywhere. Whether it is a black cat in the United States, infinite reflecting mirrors in Mexico, going back to your house after a wake in the Philippines, or whistling indoors in Lithuania, superstitions are pervasive, deeply held, and generally harmless. They are good for a good laugh as we recognize how ludicrous these unfounded fears are.
Some superstitions, though, are no laughing matter. They can be quite harmful. They are pathologic superstitions.
For example, some people believe vaccines cause autism in children. That pathologic superstition has consequences. A recent CDC report revealed that the population of unvaccinated children in the United States has quadrupled since 2001. This comes as no surprise as we hear about more measles outbreaks – and the deaths associated with them – in populations of unvaccinated children every year. A similar and pervasive pathologic superstition is the fear that an influenza vaccine will cause the flu. I wonder how many people die from this misconception.
Other people believe that their cancer can be treated, if not cured, with unproven, unconventional treatments. I cannot understand how this pathologic superstition developed. The purveyors of unconventional treatment hold much of the blame, but gullibility and ignorance may play a larger role. The consequences are tragic. A recent report demonstrated an approximately twofold increased risk of death in patients who used complementary therapies, compared with those who did not (JAMA Oncol. 2018 Oct 1;4[10]:1375-81).
These are sobering data for those of us who have in the past relented when our patients asked if they could take this or that supplement because we did not think they would cause significant harm.
Superstitions apparently are part of the human condition, evolved to attribute causation and provide order. They are a learned phenomenon. They are learned by reasonable people with normal intelligence and rational thinking. A superstition is born when someone is exposed to a false statement by someone or something they trust – a trusted other.
Trusted others exude certainty. Once established, superstitions are regrettably difficult to remove by those who are less certain, like physicians. How willing are we to say that the flu vaccine is 100% safe? Without certainty, how can a physician debunk a superstition? The techniques that we have been taught usually work, but not when faced with a pathologic superstition.
Science and experience teach us that firmly held superstitions cannot be broken with logical, stepwise reasoning. Jonathan Haidt provides a useful metaphor for this problem in his book “The Happiness Hypothesis” (Basic Books, 2006). He describes a rider on an elephant. The rider represents our rational thought and the elephant represents our emotional foundation. The rider thinks he controls the elephant, but the opposite is more likely true. In order to move the elephant in a certain direction, the rider needs to make the elephant want to turn in that direction. Otherwise, all the cajoling and arguing in the world won’t make the elephant turn. A rational argument made to someone emotionally invested in the counter argument will fail. That is why we cannot convince antivaccine parents to vaccinate their children by trying to persuade them with facts. Neither can we convince global warming skeptics to stop burning coal, gun advocates to vote for restrictions on gun ownership, or cancer patients to accept curative treatment if their values and morals are being challenged.
In a later book, “The Righteous Mind: Why Good People Are Divided by Politics and Religion” (Vintage Books, 2012), Mr. Haidt expands his hypothesis to declare that to change minds, we must appeal to underlying moral values. The challenge is to identify those moral underpinnings in our patients in order to develop an appeal likely to resonate with their emotions and values.
Superstition derives from something people learn either from trusted others or from personal experience. It does no good for physicians to deride patient beliefs and denigrate their agency in an attempt to persuade them to abandon what we consider irrational beliefs. For physicians to penetrate pathologic superstitions, they will have to become the trusted other, to understand moral foundations, to emotionally connect. That does not usually happen the first day we meet a new patient, especially a skeptical one. It takes time, and effort, to reach out and bond with the patient and their family. Only then can pathologic superstitions dissolve and a better patient-doctor relationship evolve.
During this season rife with superstition, remember that your patient’s own superstitions are part of their belief system, and your belief system may be threatening to them. Make your beliefs less threatening, become a trusted other, and appeal to their foundational values, and you can successfully break a pathologic superstition.
Dr. Kalaycio is editor in chief of Hematology News. He chairs the department of hematologic oncology and blood disorders at Cleveland Clinic Taussig Cancer Institute. Contact him at kalaycm@ccf.org.
When you believe in things that you don’t understand
Then you suffer
Superstition ain’t the way
– Stevie Wonder
I have always found it odd that airplanes don’t have a 13th row and hotels don’t have a 13th floor. Well, of course they do, but they are not labeled that way. Many people would hesitate to sit in the 13th row of an airplane since 13 is such an unlucky number. At least many people in the United States think the number 13 is unlucky. Thirteen is just a number in much of Asia. There, the number 4 is just as threatening as 13 is to us.
Superstitions like these are familiar to all of us.
One of my favorites is the belief that vacuum cups attached to the skin will somehow draw out toxins and generally improve health. “Cupping,” as the practice is known, is endorsed by several celebrities and famous athletes. After the treatment, a cupped patient exhibits circles of hyperemia, and no other apparent harm. I suspect that about a third of cupped patients truly think they have benefited from a good cupping, about the same number that would benefit from an orally administered placebo.
Superstitions are everywhere. Whether it is a black cat in the United States, infinite reflecting mirrors in Mexico, going back to your house after a wake in the Philippines, or whistling indoors in Lithuania, superstitions are pervasive, deeply held, and generally harmless. They are good for a good laugh as we recognize how ludicrous these unfounded fears are.
Some superstitions, though, are no laughing matter. They can be quite harmful. They are pathologic superstitions.
For example, some people believe vaccines cause autism in children. That pathologic superstition has consequences. A recent CDC report revealed that the population of unvaccinated children in the United States has quadrupled since 2001. This comes as no surprise as we hear about more measles outbreaks – and the deaths associated with them – in populations of unvaccinated children every year. A similar and pervasive pathologic superstition is the fear that an influenza vaccine will cause the flu. I wonder how many people die from this misconception.
Other people believe that their cancer can be treated, if not cured, with unproven, unconventional treatments. I cannot understand how this pathologic superstition developed. The purveyors of unconventional treatment hold much of the blame, but gullibility and ignorance may play a larger role. The consequences are tragic. A recent report demonstrated an approximately twofold increased risk of death in patients who used complementary therapies, compared with those who did not (JAMA Oncol. 2018 Oct 1;4[10]:1375-81).
These are sobering data for those of us who have in the past relented when our patients asked if they could take this or that supplement because we did not think they would cause significant harm.
Superstitions apparently are part of the human condition, evolved to attribute causation and provide order. They are a learned phenomenon. They are learned by reasonable people with normal intelligence and rational thinking. A superstition is born when someone is exposed to a false statement by someone or something they trust – a trusted other.
Trusted others exude certainty. Once established, superstitions are regrettably difficult to remove by those who are less certain, like physicians. How willing are we to say that the flu vaccine is 100% safe? Without certainty, how can a physician debunk a superstition? The techniques that we have been taught usually work, but not when faced with a pathologic superstition.
Science and experience teach us that firmly held superstitions cannot be broken with logical, stepwise reasoning. Jonathan Haidt provides a useful metaphor for this problem in his book “The Happiness Hypothesis” (Basic Books, 2006). He describes a rider on an elephant. The rider represents our rational thought and the elephant represents our emotional foundation. The rider thinks he controls the elephant, but the opposite is more likely true. In order to move the elephant in a certain direction, the rider needs to make the elephant want to turn in that direction. Otherwise, all the cajoling and arguing in the world won’t make the elephant turn. A rational argument made to someone emotionally invested in the counter argument will fail. That is why we cannot convince antivaccine parents to vaccinate their children by trying to persuade them with facts. Neither can we convince global warming skeptics to stop burning coal, gun advocates to vote for restrictions on gun ownership, or cancer patients to accept curative treatment if their values and morals are being challenged.
In a later book, “The Righteous Mind: Why Good People Are Divided by Politics and Religion” (Vintage Books, 2012), Mr. Haidt expands his hypothesis to declare that to change minds, we must appeal to underlying moral values. The challenge is to identify those moral underpinnings in our patients in order to develop an appeal likely to resonate with their emotions and values.
Superstition derives from something people learn either from trusted others or from personal experience. It does no good for physicians to deride patient beliefs and denigrate their agency in an attempt to persuade them to abandon what we consider irrational beliefs. For physicians to penetrate pathologic superstitions, they will have to become the trusted other, to understand moral foundations, to emotionally connect. That does not usually happen the first day we meet a new patient, especially a skeptical one. It takes time, and effort, to reach out and bond with the patient and their family. Only then can pathologic superstitions dissolve and a better patient-doctor relationship evolve.
During this season rife with superstition, remember that your patient’s own superstitions are part of their belief system, and your belief system may be threatening to them. Make your beliefs less threatening, become a trusted other, and appeal to their foundational values, and you can successfully break a pathologic superstition.
Dr. Kalaycio is editor in chief of Hematology News. He chairs the department of hematologic oncology and blood disorders at Cleveland Clinic Taussig Cancer Institute. Contact him at kalaycm@ccf.org.
Palliative care update highlights role of nonspecialists
The new edition of providing care for critically ill patients, not just those clinicians actively specialized in palliative care.
The Clinical Practice Guidelines for Quality Palliative Care, 4th Edition, emphasizes the importance of palliative care provided by “clinicians in primary care and specialty care practices, such as oncologists,” the guideline authors stated.
The latest revision of the guideline aims to establish a foundation for “gold-standard” palliative care for people living with serious illness, regardless of diagnosis, prognosis, setting, or age, according to the National Coalition for Hospice and Palliative Care, which published the clinical practice guidelines.
The update was developed by the National Consensus Project for Quality Palliative Care (NCP), which includes 16 national organizations with palliative care and hospice expertise, and is endorsed by more than 80 national organizations, including the American Society of Hematology and the Oncology Nurses Society.
One key reason for the update, according to the NCP, was to acknowledge that today’s health care system may not be meeting patients’ palliative care needs.
Specifically, the guidelines call on all clinicians who are not palliative specialists to integrate palliative care principles into their routine assessment of seriously ill patients with conditions such as heart failure, lung disease, and cancer.
This approach differs from the way palliative care is traditionally practiced, often by fellowship-trained physicians, trained nurses, and other specialists who provide that support.
The guidelines are organized into sections covering palliative care structure and processes, care for the patient nearing the end of life, and specific aspects of palliative care, including physical, psychological, and psychiatric; social; cultural, ethical, and legal; and spiritual, religious, and existential aspects.
“The expectation is that all clinicians caring for seriously ill patients will integrate palliative care competencies, such as safe and effective pain and symptom management and expert communication skills in their practice, and palliative care specialists will provide expertise for those with the most complex needs,” the guideline authors wrote.
Implications for treatment of oncology patients
These new guidelines represent a “blueprint for what it looks like to provide high-quality, comprehensive palliative care to people with serious illness,” said Thomas W. LeBlanc, MD, who is a medical oncologist, palliative care physician, and patient experience researcher at Duke University, Durham, N.C.
“Part of this report to is about trying to raise the game of everybody in medicine and provide a higher basic level of primary palliative care to all people with serious illness, but then also to figure out who has higher levels of needs where the specialists should be applied, since they are a scarce resource,” said Dr. LeBlanc.
An issue with that traditional model is a shortage of specialized clinicians to meet palliative care needs, said Dr. LeBlanc, whose clinical practice and research focuses on palliative care needs of patients with hematologic malignancies.
“Palliative care has matured as a field such that we are now actually facing workforce shortage issues and really fundamental questions about who needs us the most, and how we increase our reach to improve the lives of more patients and families facing serious illness,” he said in an interview.
That’s a major driver behind the emphasis in these latest guidelines on providing palliative care in the community, coordinating care, and dealing with care transitions, he added.
“I hope that this document will help to demonstrate the value and the need for palliative care specialists, and for improvements in primary care in the care of patients with hematologic diseases in general,” he said. “To me, this adds increasing legitimacy to this whole field.”
Palliative care in surgical care
These guidelines are particularly useful to surgeons in part because of their focus on what’s known as primary palliative care, said to Geoffrey P. Dunn, MD, former chair of the American College of Surgeons Committee on Surgical Palliative Care. Palliative care, the new guidelines suggest, can be implemented by nonspecialists.
Primary palliative care includes diverse skills such as breaking adverse news to patients, managing uncomplicated pain, and being able to recognize signs and symptoms of imminent demise. “These are the minimum deliverables for all people dealing with seriously ill patients,” Dr. Dunn said in an interview. “It’s palliative care that any practicing physician should be able to handle.”
Dr. Dunn concurred with Dr. LaBlanc about the workforce shortage in the palliative field. The traditional model has created a shortage of specialized clinicians to meet palliative care needs. Across the board, “staffing for palliative teams is very inconsistent,” said Dr. Dunn. “It’s a classic unfunded mandate.”
While these guidelines are a step forward in recognizing the importance of palliative care outside of the palliative care specialty, there is no reference to surgery anywhere in the text of the 141-page prepublication draft provided by the NCP, Dr. Dunn noted in the interview.
“There’s still a danger of parallel universes, where surgery is developing its own understanding of this in parallel with the more general national palliative care movement,” he said. Despite that, there is a growing connection between surgery and the broader palliative care community. That linkage is especially important given the number of seriously ill patients with high symptom burden that are seen in surgery.
“I think where surgeons are beginning to find [palliative principles] very helpful is dealing with these protracted serial discussions with families in difficult circumstances, such as how long is the life support going to be prolonged in someone with a devastating head injury, or multiple system organ failure in the elderly,” Dr. Dunn added.
The new edition of providing care for critically ill patients, not just those clinicians actively specialized in palliative care.
The Clinical Practice Guidelines for Quality Palliative Care, 4th Edition, emphasizes the importance of palliative care provided by “clinicians in primary care and specialty care practices, such as oncologists,” the guideline authors stated.
The latest revision of the guideline aims to establish a foundation for “gold-standard” palliative care for people living with serious illness, regardless of diagnosis, prognosis, setting, or age, according to the National Coalition for Hospice and Palliative Care, which published the clinical practice guidelines.
The update was developed by the National Consensus Project for Quality Palliative Care (NCP), which includes 16 national organizations with palliative care and hospice expertise, and is endorsed by more than 80 national organizations, including the American Society of Hematology and the Oncology Nurses Society.
One key reason for the update, according to the NCP, was to acknowledge that today’s health care system may not be meeting patients’ palliative care needs.
Specifically, the guidelines call on all clinicians who are not palliative specialists to integrate palliative care principles into their routine assessment of seriously ill patients with conditions such as heart failure, lung disease, and cancer.
This approach differs from the way palliative care is traditionally practiced, often by fellowship-trained physicians, trained nurses, and other specialists who provide that support.
The guidelines are organized into sections covering palliative care structure and processes, care for the patient nearing the end of life, and specific aspects of palliative care, including physical, psychological, and psychiatric; social; cultural, ethical, and legal; and spiritual, religious, and existential aspects.
“The expectation is that all clinicians caring for seriously ill patients will integrate palliative care competencies, such as safe and effective pain and symptom management and expert communication skills in their practice, and palliative care specialists will provide expertise for those with the most complex needs,” the guideline authors wrote.
Implications for treatment of oncology patients
These new guidelines represent a “blueprint for what it looks like to provide high-quality, comprehensive palliative care to people with serious illness,” said Thomas W. LeBlanc, MD, who is a medical oncologist, palliative care physician, and patient experience researcher at Duke University, Durham, N.C.
“Part of this report to is about trying to raise the game of everybody in medicine and provide a higher basic level of primary palliative care to all people with serious illness, but then also to figure out who has higher levels of needs where the specialists should be applied, since they are a scarce resource,” said Dr. LeBlanc.
An issue with that traditional model is a shortage of specialized clinicians to meet palliative care needs, said Dr. LeBlanc, whose clinical practice and research focuses on palliative care needs of patients with hematologic malignancies.
“Palliative care has matured as a field such that we are now actually facing workforce shortage issues and really fundamental questions about who needs us the most, and how we increase our reach to improve the lives of more patients and families facing serious illness,” he said in an interview.
That’s a major driver behind the emphasis in these latest guidelines on providing palliative care in the community, coordinating care, and dealing with care transitions, he added.
“I hope that this document will help to demonstrate the value and the need for palliative care specialists, and for improvements in primary care in the care of patients with hematologic diseases in general,” he said. “To me, this adds increasing legitimacy to this whole field.”
Palliative care in surgical care
These guidelines are particularly useful to surgeons in part because of their focus on what’s known as primary palliative care, said to Geoffrey P. Dunn, MD, former chair of the American College of Surgeons Committee on Surgical Palliative Care. Palliative care, the new guidelines suggest, can be implemented by nonspecialists.
Primary palliative care includes diverse skills such as breaking adverse news to patients, managing uncomplicated pain, and being able to recognize signs and symptoms of imminent demise. “These are the minimum deliverables for all people dealing with seriously ill patients,” Dr. Dunn said in an interview. “It’s palliative care that any practicing physician should be able to handle.”
Dr. Dunn concurred with Dr. LaBlanc about the workforce shortage in the palliative field. The traditional model has created a shortage of specialized clinicians to meet palliative care needs. Across the board, “staffing for palliative teams is very inconsistent,” said Dr. Dunn. “It’s a classic unfunded mandate.”
While these guidelines are a step forward in recognizing the importance of palliative care outside of the palliative care specialty, there is no reference to surgery anywhere in the text of the 141-page prepublication draft provided by the NCP, Dr. Dunn noted in the interview.
“There’s still a danger of parallel universes, where surgery is developing its own understanding of this in parallel with the more general national palliative care movement,” he said. Despite that, there is a growing connection between surgery and the broader palliative care community. That linkage is especially important given the number of seriously ill patients with high symptom burden that are seen in surgery.
“I think where surgeons are beginning to find [palliative principles] very helpful is dealing with these protracted serial discussions with families in difficult circumstances, such as how long is the life support going to be prolonged in someone with a devastating head injury, or multiple system organ failure in the elderly,” Dr. Dunn added.
The new edition of providing care for critically ill patients, not just those clinicians actively specialized in palliative care.
The Clinical Practice Guidelines for Quality Palliative Care, 4th Edition, emphasizes the importance of palliative care provided by “clinicians in primary care and specialty care practices, such as oncologists,” the guideline authors stated.
The latest revision of the guideline aims to establish a foundation for “gold-standard” palliative care for people living with serious illness, regardless of diagnosis, prognosis, setting, or age, according to the National Coalition for Hospice and Palliative Care, which published the clinical practice guidelines.
The update was developed by the National Consensus Project for Quality Palliative Care (NCP), which includes 16 national organizations with palliative care and hospice expertise, and is endorsed by more than 80 national organizations, including the American Society of Hematology and the Oncology Nurses Society.
One key reason for the update, according to the NCP, was to acknowledge that today’s health care system may not be meeting patients’ palliative care needs.
Specifically, the guidelines call on all clinicians who are not palliative specialists to integrate palliative care principles into their routine assessment of seriously ill patients with conditions such as heart failure, lung disease, and cancer.
This approach differs from the way palliative care is traditionally practiced, often by fellowship-trained physicians, trained nurses, and other specialists who provide that support.
The guidelines are organized into sections covering palliative care structure and processes, care for the patient nearing the end of life, and specific aspects of palliative care, including physical, psychological, and psychiatric; social; cultural, ethical, and legal; and spiritual, religious, and existential aspects.
“The expectation is that all clinicians caring for seriously ill patients will integrate palliative care competencies, such as safe and effective pain and symptom management and expert communication skills in their practice, and palliative care specialists will provide expertise for those with the most complex needs,” the guideline authors wrote.
Implications for treatment of oncology patients
These new guidelines represent a “blueprint for what it looks like to provide high-quality, comprehensive palliative care to people with serious illness,” said Thomas W. LeBlanc, MD, who is a medical oncologist, palliative care physician, and patient experience researcher at Duke University, Durham, N.C.
“Part of this report to is about trying to raise the game of everybody in medicine and provide a higher basic level of primary palliative care to all people with serious illness, but then also to figure out who has higher levels of needs where the specialists should be applied, since they are a scarce resource,” said Dr. LeBlanc.
An issue with that traditional model is a shortage of specialized clinicians to meet palliative care needs, said Dr. LeBlanc, whose clinical practice and research focuses on palliative care needs of patients with hematologic malignancies.
“Palliative care has matured as a field such that we are now actually facing workforce shortage issues and really fundamental questions about who needs us the most, and how we increase our reach to improve the lives of more patients and families facing serious illness,” he said in an interview.
That’s a major driver behind the emphasis in these latest guidelines on providing palliative care in the community, coordinating care, and dealing with care transitions, he added.
“I hope that this document will help to demonstrate the value and the need for palliative care specialists, and for improvements in primary care in the care of patients with hematologic diseases in general,” he said. “To me, this adds increasing legitimacy to this whole field.”
Palliative care in surgical care
These guidelines are particularly useful to surgeons in part because of their focus on what’s known as primary palliative care, said to Geoffrey P. Dunn, MD, former chair of the American College of Surgeons Committee on Surgical Palliative Care. Palliative care, the new guidelines suggest, can be implemented by nonspecialists.
Primary palliative care includes diverse skills such as breaking adverse news to patients, managing uncomplicated pain, and being able to recognize signs and symptoms of imminent demise. “These are the minimum deliverables for all people dealing with seriously ill patients,” Dr. Dunn said in an interview. “It’s palliative care that any practicing physician should be able to handle.”
Dr. Dunn concurred with Dr. LaBlanc about the workforce shortage in the palliative field. The traditional model has created a shortage of specialized clinicians to meet palliative care needs. Across the board, “staffing for palliative teams is very inconsistent,” said Dr. Dunn. “It’s a classic unfunded mandate.”
While these guidelines are a step forward in recognizing the importance of palliative care outside of the palliative care specialty, there is no reference to surgery anywhere in the text of the 141-page prepublication draft provided by the NCP, Dr. Dunn noted in the interview.
“There’s still a danger of parallel universes, where surgery is developing its own understanding of this in parallel with the more general national palliative care movement,” he said. Despite that, there is a growing connection between surgery and the broader palliative care community. That linkage is especially important given the number of seriously ill patients with high symptom burden that are seen in surgery.
“I think where surgeons are beginning to find [palliative principles] very helpful is dealing with these protracted serial discussions with families in difficult circumstances, such as how long is the life support going to be prolonged in someone with a devastating head injury, or multiple system organ failure in the elderly,” Dr. Dunn added.
Jack Rozel: Gun violence
Dr. Rozel makes his home and practices in Pittsburgh, the site of the tragic mass shooting at the Tree of Life temple.
Dr. Rozel makes his home and practices in Pittsburgh, the site of the tragic mass shooting at the Tree of Life temple.
Dr. Rozel makes his home and practices in Pittsburgh, the site of the tragic mass shooting at the Tree of Life temple.

NSAID risk score validated for CV events
Also today, the ACIP resuscitates pertussis working group, it’s important to talk to adolescents about sexual assault, and costs increase for gun injuries in children.
Amazon Alexa
Apple Podcasts
Spotify
Also today, the ACIP resuscitates pertussis working group, it’s important to talk to adolescents about sexual assault, and costs increase for gun injuries in children.
Amazon Alexa
Apple Podcasts
Spotify
Also today, the ACIP resuscitates pertussis working group, it’s important to talk to adolescents about sexual assault, and costs increase for gun injuries in children.
Amazon Alexa
Apple Podcasts
Spotify

Game changers in pediatric cancer
Although there have been significant improvements in patient outcomes for some forms of pediatric cancer, progress has been painfully slow for others. An increasing understanding of pediatric cancers is highlighting the unique molecular drivers and challenging the assumption that drugs developed in adults can be applied to children and young adults. Here, we discuss game-changing therapeutic advances and a shifting view of childhood cancers.
Unique genomic background
Although pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, they are the second leading cause of death in children aged 1 to 14 years. There are many different histological tumor types under the umbrella of childhood cancers, of which the most common are leukemias, central nervous system tumors, and lymphomas (Figure 1).1,2
Significant progress has been made in the treatment of certain pediatric cancers in recent decades, exemplified by pediatric acute lymphoblastic leukemia (ALL), which has been transformed from a virtually incurable cancer to one in which 5-year survival rates now reach up to 90%. In other forms of pediatric cancer, however, survival rates have stagnated and little progress has been made in the development of effective new therapies.3
Because of their rarity, pediatric cancers are difficult to study and adequate enrollment of children in clinical trials can be challenging. Pharmaceutical companies are often hesitant to test drugs in the pediatric population in patients who often cannot advocate for themselves. As a result, the activity of drugs developed in adult patients has often been inferred in pediatric patients with the same tumor type or molecular aberrations. However, as researchers have gathered more information about pediatric cancers, there has been increasing recognition of their unique attributes and the need for dedicated clinical trials in this patient population.
Pediatric cancers tend to be found in the developing mesodermic tissue, whereas adult cancers are more prevalent in the epithelial tissues. Genome sequencing studies have revealed a much lower mutational burden in pediatric cancers and the mechanisms of oncogenesis are also quite different; adult tumors can develop from a series of acquired gene mutations, but pediatric tumors tend to develop from a single catastrophic event.4,5
Even the same type of cancer in a pediatric and adult patient can be quite different, with very different underlying molecular mechanisms. In a recent genomic analysis of different types of pediatric cancer by researchers at St Jude’s Children’s Research Hospital, less than half of the identified mutated genes were found to be similar to those found in adult patients.6
A ‘magic bullet’?
Chromosomal rearrangements are common in pediatric cancers. This type of molecular abnormality can result in a fusion of 2 different genes when the chromosome breaks apart and the pieces join back together in a muddled order. If the genetic code fuses in a manner that is “readable” by the cell, then it can drive aberrant activation of one or both genes.7 Gene fusions often involve kinase enzymes that are essential players in cell signaling pathways regulating hallmark cancer processes, such as unchecked cell proliferation. The fusion drives the constitutive activation of the kinase and, thus, these downstream signaling pathways.
One of the first chromosomal rearrangements linked to cancer, BCR-ABL1 – more commonly known as the Philadelphia chromosome – results in aberrant activation of the ABL1 kinase. It is present in nearly all cases of chronic myeloid leukemia (CML) and 3% to 5% of patients with ALL, and thus became the central focus of targeted drug development. Imatinib was initially approved by the US Food and Drug Administration (FDA) in 2001 for the treatment of adult patients with CML and had such a significant impact on the treatment landscape that it made the cover of Time magazine as a “magic bullet” in the war on cancer.8
Approval was expanded into pediatric patients in 2006 and for pediatric patients with ALL in 2013. However, as with the use of most kinase inhibitors, tumors can evolve under the selective pressure of treatment, developing additional molecular abnormalities that drive resistance.9
Next-generation multikinase inhibitors that more potently inhibit the BCR-ABL1 fusion protein have been developed to provide additional treatment options for patients who become resistant to imatinib. Dasatinib and nilotinib are among several drugs that have recently been approved for pediatric cancer therapy (Table 1). Both therapies were approved to treat children with Philadelphia chromosome-positive CML in the chronic phase in either the front- or second-line setting after failure of imatinib.
The approval of dasatinib was based on data from 97 patients across 2 trials, 51 of whom were newly diagnosed and 46 previously treated with imatinib. Most of the patients were treated with dasatinib 60 mg/m2 once daily. After 2 years of follow-up, more than 95% of newly diagnosed patients and 82.6% of relapsed/refractory patients had complete cytogenetic response.10
Nilotinib was approved on the basis of findings from 2 clinical trials including 69 patients – 1 trial involving patients who were refractory to or relapsed after dasatinib and imatinib treatment, and 1 that included both relapsed/refractory and newly diagnosed patients. Patients received nilotinib 230 mg/m2 twice daily, rounded to the nearest 50-mg dose, in 28-day cycles. By cycle 12, the cumulative major molecular response rate (MMR) was 47.7% in patients with relapsed/refractory disease, and 64% in newly diagnosed patients.11 Clinical trials of both drugs in the pediatric setting are ongoing.
Other prominent gene fusions
Gene fusions involving the anaplastic lymphoma kinase (ALK) occur in patients with non–small-cell lung cancer and ALK inhibitors have provided an effective new treatment option for patients whose tumors display this abnormality.
ALK fusions are also a prominent feature of several kinds of pediatric cancers and ALK inhibitors offer promise in this setting.7,12 An NPM-ALK fusion is found in 90% of pediatric anaplastic large cell lymphoma (ALCL) cases,13 whereas a variety of ALK fusions are found in up to half of patients with inflammatory myofibroblastic tumor (IMT), a rare form of soft tissue sarcoma.14 ALK inhibitors are being tested in a variety of clinical trials in pediatric patients (Table 2).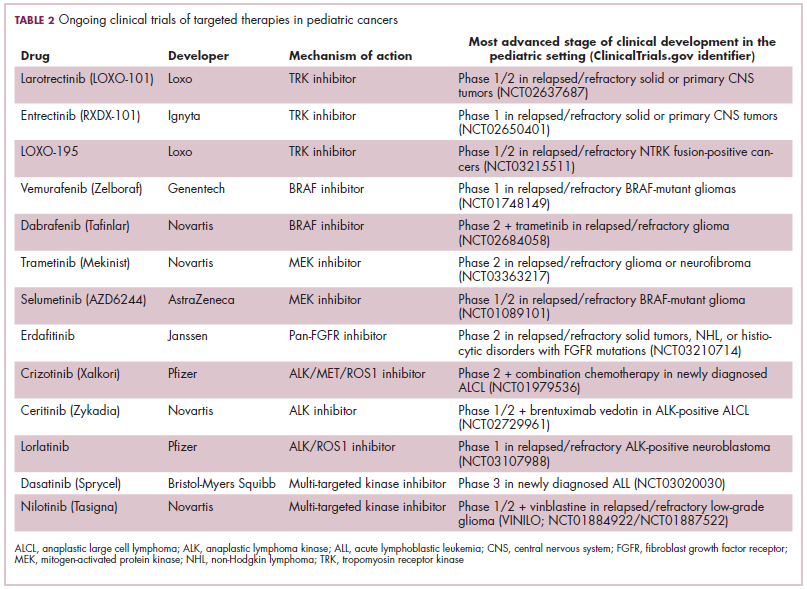
The results of a small phase 1 study of crizotinib in pediatric patients with ALK-positive ALCL (n = 26) or IMT (n = 14) were recently published. ALCL patients received crizotinib at a dose of 165 mg/m2, while IMT patients were given 100, 165, or 280 mg/m2. For the latter, the results were presented as a pooled cohort since safety and efficacy data were similar across dose levels. The overall response rate (ORR) was 83% for patients with ALCL and 86% for those with IMT. Grade 3/4 adverse events occurred in 83% and 71% of patients, respectively, and most commonly involved reduced neutrophil count.15
Most recently and perhaps most promisingly, fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) gene have generated significant buzz. There are 3 NTRK genes, NTRK1, 2, and 3, which encode the TRKA, TRKB, and TRKC proteins, respectively.
To date, 22 different partner genes have been identified that can fuse with the NTRK genes and, as with other kinase fusions, drive constitutive activation of the receptor proteins and downstream oncogenic signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway (Figure 2).
NTRK fusions are being identified in an ever-growing number of cancer types, but are typically found in a small percentage of patients. However, in certain rare pediatric tumors, including congenital infantile fibrosarcoma and papillary thyroid cancer, they are found at much higher frequencies.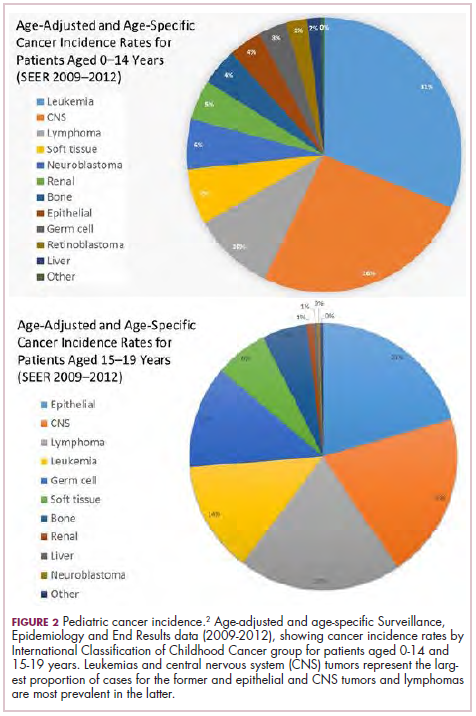
TRK inhibitors have been developed to target the fusion proteins and, given the spread of NTRK fusions across different types of cancers, they offer the most substantial promise as the next tumor agnostic cancer therapy – to treat patients based on the shared presence of a molecular aberration, irrespective of the type of cancer.16
The ongoing SCOUT trial is evaluating larotrectinib (LOXO-101) in pediatric patients. Among 24 patients (17 with NTRK fusions and 7 without) with infantile fibrosarcoma (47%), soft tissue sarcoma (41%) or papillary thyroid cancer (12%), the ORR was 93%, including complete response (CR) in 13% of patients.17
Preliminary results from an ongoing phase 1/2 study of entrectinib in pediatric patients with extracranial solid tumors were also recently presented at the annual meeting of the American Society for Clinical Oncology (ASCO). Among 15 evaluable patients enrolled to date, 3 have NTRK fusions and all experienced an objective response, with 1 (a patient with IMT) ongoing at 10 months.18
CAR T cells transformative in ALL
A variety of different types of immunotherapy have been tested in patients with pediatric cancers. In general, immunotherapy has proved less effective than in adult cancers, possibly because of the lower tumor mutation burden in pediatric cancers, which means there are likely fewer cancer antigens to provoke an anti-tumor immune response.
There are notable exceptions among the disappointments, however, and most exciting is the development of chimeric antigen receptor (CAR) T cells. CAR T cells fall into a category of immunotherapy known as adoptive cell therapy (ACT), in which immune cells are harvested from a patient and grown outside the body to increase their numbers before being reinfused into the patient.
In the case of CAR T-cell therapy, the cells are genetically engineered to express a CAR that endows them with tumor-targeting capabilities. To date, the development of CAR T cells has focused on the use of the CD19 antigen as a target, which is highly expressed on a variety of B-cell malignancies, including several of the most common forms of pediatric cancer. ASCO shined the spotlight on CAR T-cell therapy this year, naming it the Advance of the Year for 2018, saying that the treatment is “poised to transform childhood ALL.”19
Two CD19-targeted CAR T-cell therapies – tisagenlecleucel and axicabtagene ciloleucel – were brought to market in 2017. Only tisagenlecleucel is approved in the pediatric ALL population, however, having been awarded approval for the treatment of patients aged up to 25 years whose disease is refractory to or relapsed after receiving at least 2 prior therapies. In the pivotal trial, complete responses were observed in more than 60% of patients.20 Clinical trials of both CAR T-cell therapies in pediatric ALL and non-Hodgkin lymphoma are ongoing (Table 3).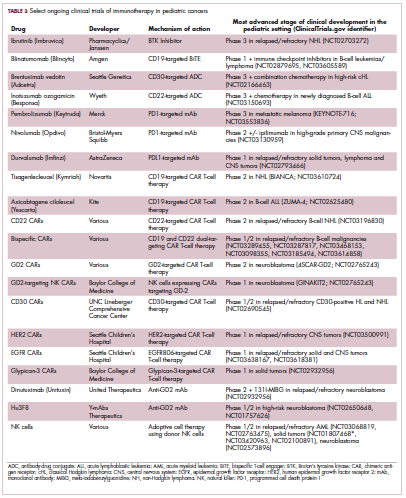
CD19 has also proven to be a promising target for other forms of immunotherapy, including a new type of antibody known as a bispecific T-cell engager (BiTE). In 2014, blinatumomab became the first BiTE to receive regulatory approval, for the treatment of adult patients with relapsed/refractory ALL. Blinatumomab also targets the CD3 protein on T cells and helps to bring cancer cells and cytotoxic immune cells into close enough proximity that an immunological synapse can be formed between the two, facilitating tumor cell killing.21
In 2016, the approved indication was expanded into the pediatric population based on the results of a phase 1/2 study in which the safety and efficacy of blinatumomab were evaluated in 93 pediatric patients with relapsed/refractory ALL. Among the 70 patients who received the recommended dose of 5µg/m2 a day for the first 7 days, followed by 15µg/m2 a day thereafter, 51% achieved complete remission within the first 2 cycles, 52% of whom achieved minimal residual disease (MRD).22 Most recently, the FDA expanded the indication for blinatumomab to include patients (both adults and children) who are in remission, but MRD positive.23Despite the dramatic responses, many patients relapse after treatment with CD19-targeted CAR T cells, and researchers have uncovered numerous mechanisms of resistance. Among them is the loss of the CD19 antigen on the surface of target cells, such that a CD19-positive tumor becomes CD19-negative after treatment, driving relapse.24-26Several strategies for overcoming CD19-negative relapse are already being investigated, including the development of CD22-targeted CAR T cells and bispecific CAR T cells that target both CD19 and CD22. The results of a first-in-human trial of anti-CD22 CAR T-cell therapy were recently published. Among 21 pediatric and adult patients with relapsed/refractory B-cell ALL who were treated with either 3 x 105 cells/kg, 1 x 106 cells/kg, or 3 x 106 cells/kg, complete responses were observed in 57%.27
Results from 15 pediatric patients enrolled in a trial evaluating CD22-targeted CAR T cells as salvage therapy for those who relapse after CD19-targeted CAR T cell therapy were presented at the recent Congress of the European Hematology Association in Stockholm, Sweden. Patients who had undergone a stem cell transplant received the CAR T cells at a dose of 0.9 x 105 cell/kg and those who had not undergone a transplant received a dose of 8.2 x 105 cells/kg. At 30 days after CAR T cell infusion, the CR rate was 80% and the treatment was well tolerated.28
More immunotherapy approvals
The immune checkpoint inhibitors, which work by blocking inhibitory receptors on the surface of T cells, have also had recent approvals in pediatric patient populations. Pembrolizumab and nivolumab, inhibitors of the programmed cell death receptor 1 (PD-1) protein, have both been approved for use in adult and pediatric patients (older than 12 years) with relapsed/refractory metastatic colorectal cancer (and other solid tumors in the case of pembrolizumab) that display defects in the mismatch repair pathway that fixes damaged DNA or in patients that have high levels of microsatellite instability. Both deficient mismatch repair and microsatellite instability–high can indicate a high mutation burden in a tumor, which may predict increased sensitivity to immunotherapy.29
The approval in pediatric patients in both of those instances, however, was not based on data in pediatric patient populations but extrapolated from adult patients. Pembrolizumab is also approved for the treatment of adults and pediatric patients with classical Hodgkin lymphoma (cHL) after 3 or more previous treatments, but once again efficacy in the pediatric population was inferred from clinical trials performed in adults. Most recently, pembrolizumab was approved for the treatment of adult and pediatric patients with relapsed or refractory primary mediastinal large B-cell lymphoma.30Ipilimumab, which targets a different T cell receptor – cytotoxic T lymphocyte antigen-4 (CTLA-4) – has been approved for the treatment of pediatric patients aged 12 years and older with metastatic melanoma. This expanded indication, following on from its approval in adult patients in 2011, was based on data from 2 trials in which objective responses were observed in 2 out of 17 patients, including 1 partial response that lasted 16 months.31Finally, antibody-drug conjugates (ADC), in which tumor antigen-targeting monoclonal antibodies are conjugated to cytotoxic payloads to combine the specificity of an antibody with the cell-killing potency of chemotherapy, have also generated some recent successes in pediatric cancers.
Gemtuzumab ozogamicin is an ADC that targets the CD33 protein, which is highly expressed on 85%-90% of cases of acute myeloid leukemia (AML). In 2000, it was the first ADC to be brought to market in the United States, but it was subsequently voluntarily withdrawn by the manufacturer in 2010 after confirmatory trials failed to show a survival benefit.
Recently, a meta-analysis of gemtuzumab ozogamicin trials suggested that the drug likely does improve long-term overall survival (OS) and reduce the risk of relapse and researchers developed an intermittent dosing schedule to help mitigate toxicity.32 This new dosing regimen received FDA approval in 2017 for the treatment of pediatric patients aged 2 years and older on the basis of 2 clinical trials.
In the MyloFrance-1 trial, 57 patients were administered 3 mg/m2 gemtuzumab ozogamicin on days 1, 4, and 7 followed by cytarabine consolidation therapy and demonstrated a 26% CR rate and median recurrence-free survival of 11.6 months. In the phase 3 AML-19 trial, 237 patients received gemtuzumab ozogamicin at a dose of 6 mg/m2 on day 1 and 3 mg/m2 on day 8 or best supportive care. Gemtuzumab ozogamicin improved OS from 3.6 to 4.9 months.33,34
Inotuzumab ozogamicin is a CD22-targeting ADC that has been FDA approved for the treatment of adult patients with relapsed/refractory B-cell precursor ALL since last year. The therapy has been available to pediatric patients through a compassionate access program, but it has not been extensively evaluated in this population. The results of a retrospective analysis of pediatric patients who received at least 1 dose of inotuzumab ozogamicin were presented at ASCO in 2017. Among 29 patients with heavily pretreated disease the CR rate was 62%, 72% of whom achieved MRD negativity.35
1. American Cancer Society. Key statistics for childhood cancers. https://www.cancer.org/cancer/cancer-in-children/key-statistics.html. Last revised September 10, 2018. Accessed September 16, 2018.
2. NHI/National Cancer Institute website. Unusual cancers of childhood treatment (PDQ) - Health Professional Version. https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Last updated August 28, 2018. Accessed September 8, 2018.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
4. Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277-289.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
6. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371.
7. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad-Massade L. Relevance of fusion genes in pediatric cancers: toward precision medicine. Molec Ther Nucleic Acids. 2017;6:315-326.
8. Lemonick MD, Park A. New hope for cancer. http://content.time.com/time/world/article/0,8599,2047900-2,00.html. Published May 28, 2001. Last accessed September 13, 2018.
9. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. https://www.hindawi.com/journals/cherp/2014/357027/. Published May 19, 2014. Accessed September 16, 2018.
10. Gore L, Kearns PR, Martino MLd, et al. Dasatinib in pediatric patients with chronic myeloid leukemia in chronic phase: results from a phase II trial. J Clin Oncol. 2018;36(13):1330-1338.
11. Novartis press release. Novartis drug Tasigna approved by FDA to treat children with rare form of leukemia. 2018; https://www.novartis.com/news/media-releases/novartis-drug-tasignar-approved-fda-treat-children-rare-form-leukemia. Released March 22, 2018. Accessed September 16, 2018.
12. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913-1920.
13. Turner SD, Lamant L, Kenner L, Brugieres L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol. 2016;173(4):560-572.
14. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am J Surg Pathol. 2015;39(7):957-967.
15. Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a children's oncology group study. J Clin Oncol. 2017;35(28):3215-3221.
16. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070277/. Published online March 18, 2016. Accessed September 16, 2018.
17. [Behind paywall.] Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705-714.
18. Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. J Clin Oncol. 2018;36(suppl;):abstr 10536.
19. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2018;36(10):1020-1044.
20. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. NEJM. 2018;378(5):439-448.
21. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
22. Stackelberg Av, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
23. Gokbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
24. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195.
25. Fousek K, Watanabe J, George A, et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J Clin Oncol. 2018;36(5_suppl):121-121.
26. Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659.
27. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
28. Pan J, Deng B, Liu S, et al. Efficacy and safety of CD22-directed CAR T-cell therapy in 15 pediatric refractory or relapsed b acute lymphoblastic leukemia patients. Paper presented at 23rd Congress of the European Hematology Association 2018; Stockholm, Sweden.
29. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35.
30. Drugs.com. Keytruda approval history. 2018; https://www.drugs.com/history/keytruda.html. Last update information not given. Accessed September 16, 2018.
31. Bristol Myers Squibb press release. US Food and Drug Administration expands approval of Yervoy (ipilimumab) to include pediatric patients 12 years and older with unresectable or metastatic melanoma. https://news.bms.com/press-release/corporatefinancial-news/us-food-and-drug-administration-expands-approval-yervoy-ipilim. Released July 24, 2017. Accessed September 16, 2018.
32. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
33. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972-979.
34. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66-71.
35. Bhojwani D, Sposto R, Shah N, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). J Clin Oncol. 2017;35(15_suppl):10512-10512.
Although there have been significant improvements in patient outcomes for some forms of pediatric cancer, progress has been painfully slow for others. An increasing understanding of pediatric cancers is highlighting the unique molecular drivers and challenging the assumption that drugs developed in adults can be applied to children and young adults. Here, we discuss game-changing therapeutic advances and a shifting view of childhood cancers.
Unique genomic background
Although pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, they are the second leading cause of death in children aged 1 to 14 years. There are many different histological tumor types under the umbrella of childhood cancers, of which the most common are leukemias, central nervous system tumors, and lymphomas (Figure 1).1,2
Significant progress has been made in the treatment of certain pediatric cancers in recent decades, exemplified by pediatric acute lymphoblastic leukemia (ALL), which has been transformed from a virtually incurable cancer to one in which 5-year survival rates now reach up to 90%. In other forms of pediatric cancer, however, survival rates have stagnated and little progress has been made in the development of effective new therapies.3
Because of their rarity, pediatric cancers are difficult to study and adequate enrollment of children in clinical trials can be challenging. Pharmaceutical companies are often hesitant to test drugs in the pediatric population in patients who often cannot advocate for themselves. As a result, the activity of drugs developed in adult patients has often been inferred in pediatric patients with the same tumor type or molecular aberrations. However, as researchers have gathered more information about pediatric cancers, there has been increasing recognition of their unique attributes and the need for dedicated clinical trials in this patient population.
Pediatric cancers tend to be found in the developing mesodermic tissue, whereas adult cancers are more prevalent in the epithelial tissues. Genome sequencing studies have revealed a much lower mutational burden in pediatric cancers and the mechanisms of oncogenesis are also quite different; adult tumors can develop from a series of acquired gene mutations, but pediatric tumors tend to develop from a single catastrophic event.4,5
Even the same type of cancer in a pediatric and adult patient can be quite different, with very different underlying molecular mechanisms. In a recent genomic analysis of different types of pediatric cancer by researchers at St Jude’s Children’s Research Hospital, less than half of the identified mutated genes were found to be similar to those found in adult patients.6
A ‘magic bullet’?
Chromosomal rearrangements are common in pediatric cancers. This type of molecular abnormality can result in a fusion of 2 different genes when the chromosome breaks apart and the pieces join back together in a muddled order. If the genetic code fuses in a manner that is “readable” by the cell, then it can drive aberrant activation of one or both genes.7 Gene fusions often involve kinase enzymes that are essential players in cell signaling pathways regulating hallmark cancer processes, such as unchecked cell proliferation. The fusion drives the constitutive activation of the kinase and, thus, these downstream signaling pathways.
One of the first chromosomal rearrangements linked to cancer, BCR-ABL1 – more commonly known as the Philadelphia chromosome – results in aberrant activation of the ABL1 kinase. It is present in nearly all cases of chronic myeloid leukemia (CML) and 3% to 5% of patients with ALL, and thus became the central focus of targeted drug development. Imatinib was initially approved by the US Food and Drug Administration (FDA) in 2001 for the treatment of adult patients with CML and had such a significant impact on the treatment landscape that it made the cover of Time magazine as a “magic bullet” in the war on cancer.8
Approval was expanded into pediatric patients in 2006 and for pediatric patients with ALL in 2013. However, as with the use of most kinase inhibitors, tumors can evolve under the selective pressure of treatment, developing additional molecular abnormalities that drive resistance.9
Next-generation multikinase inhibitors that more potently inhibit the BCR-ABL1 fusion protein have been developed to provide additional treatment options for patients who become resistant to imatinib. Dasatinib and nilotinib are among several drugs that have recently been approved for pediatric cancer therapy (Table 1). Both therapies were approved to treat children with Philadelphia chromosome-positive CML in the chronic phase in either the front- or second-line setting after failure of imatinib.
The approval of dasatinib was based on data from 97 patients across 2 trials, 51 of whom were newly diagnosed and 46 previously treated with imatinib. Most of the patients were treated with dasatinib 60 mg/m2 once daily. After 2 years of follow-up, more than 95% of newly diagnosed patients and 82.6% of relapsed/refractory patients had complete cytogenetic response.10
Nilotinib was approved on the basis of findings from 2 clinical trials including 69 patients – 1 trial involving patients who were refractory to or relapsed after dasatinib and imatinib treatment, and 1 that included both relapsed/refractory and newly diagnosed patients. Patients received nilotinib 230 mg/m2 twice daily, rounded to the nearest 50-mg dose, in 28-day cycles. By cycle 12, the cumulative major molecular response rate (MMR) was 47.7% in patients with relapsed/refractory disease, and 64% in newly diagnosed patients.11 Clinical trials of both drugs in the pediatric setting are ongoing.
Other prominent gene fusions
Gene fusions involving the anaplastic lymphoma kinase (ALK) occur in patients with non–small-cell lung cancer and ALK inhibitors have provided an effective new treatment option for patients whose tumors display this abnormality.
ALK fusions are also a prominent feature of several kinds of pediatric cancers and ALK inhibitors offer promise in this setting.7,12 An NPM-ALK fusion is found in 90% of pediatric anaplastic large cell lymphoma (ALCL) cases,13 whereas a variety of ALK fusions are found in up to half of patients with inflammatory myofibroblastic tumor (IMT), a rare form of soft tissue sarcoma.14 ALK inhibitors are being tested in a variety of clinical trials in pediatric patients (Table 2).
The results of a small phase 1 study of crizotinib in pediatric patients with ALK-positive ALCL (n = 26) or IMT (n = 14) were recently published. ALCL patients received crizotinib at a dose of 165 mg/m2, while IMT patients were given 100, 165, or 280 mg/m2. For the latter, the results were presented as a pooled cohort since safety and efficacy data were similar across dose levels. The overall response rate (ORR) was 83% for patients with ALCL and 86% for those with IMT. Grade 3/4 adverse events occurred in 83% and 71% of patients, respectively, and most commonly involved reduced neutrophil count.15
Most recently and perhaps most promisingly, fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) gene have generated significant buzz. There are 3 NTRK genes, NTRK1, 2, and 3, which encode the TRKA, TRKB, and TRKC proteins, respectively.
To date, 22 different partner genes have been identified that can fuse with the NTRK genes and, as with other kinase fusions, drive constitutive activation of the receptor proteins and downstream oncogenic signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway (Figure 2).
NTRK fusions are being identified in an ever-growing number of cancer types, but are typically found in a small percentage of patients. However, in certain rare pediatric tumors, including congenital infantile fibrosarcoma and papillary thyroid cancer, they are found at much higher frequencies.
TRK inhibitors have been developed to target the fusion proteins and, given the spread of NTRK fusions across different types of cancers, they offer the most substantial promise as the next tumor agnostic cancer therapy – to treat patients based on the shared presence of a molecular aberration, irrespective of the type of cancer.16
The ongoing SCOUT trial is evaluating larotrectinib (LOXO-101) in pediatric patients. Among 24 patients (17 with NTRK fusions and 7 without) with infantile fibrosarcoma (47%), soft tissue sarcoma (41%) or papillary thyroid cancer (12%), the ORR was 93%, including complete response (CR) in 13% of patients.17
Preliminary results from an ongoing phase 1/2 study of entrectinib in pediatric patients with extracranial solid tumors were also recently presented at the annual meeting of the American Society for Clinical Oncology (ASCO). Among 15 evaluable patients enrolled to date, 3 have NTRK fusions and all experienced an objective response, with 1 (a patient with IMT) ongoing at 10 months.18
CAR T cells transformative in ALL
A variety of different types of immunotherapy have been tested in patients with pediatric cancers. In general, immunotherapy has proved less effective than in adult cancers, possibly because of the lower tumor mutation burden in pediatric cancers, which means there are likely fewer cancer antigens to provoke an anti-tumor immune response.
There are notable exceptions among the disappointments, however, and most exciting is the development of chimeric antigen receptor (CAR) T cells. CAR T cells fall into a category of immunotherapy known as adoptive cell therapy (ACT), in which immune cells are harvested from a patient and grown outside the body to increase their numbers before being reinfused into the patient.
In the case of CAR T-cell therapy, the cells are genetically engineered to express a CAR that endows them with tumor-targeting capabilities. To date, the development of CAR T cells has focused on the use of the CD19 antigen as a target, which is highly expressed on a variety of B-cell malignancies, including several of the most common forms of pediatric cancer. ASCO shined the spotlight on CAR T-cell therapy this year, naming it the Advance of the Year for 2018, saying that the treatment is “poised to transform childhood ALL.”19
Two CD19-targeted CAR T-cell therapies – tisagenlecleucel and axicabtagene ciloleucel – were brought to market in 2017. Only tisagenlecleucel is approved in the pediatric ALL population, however, having been awarded approval for the treatment of patients aged up to 25 years whose disease is refractory to or relapsed after receiving at least 2 prior therapies. In the pivotal trial, complete responses were observed in more than 60% of patients.20 Clinical trials of both CAR T-cell therapies in pediatric ALL and non-Hodgkin lymphoma are ongoing (Table 3).
CD19 has also proven to be a promising target for other forms of immunotherapy, including a new type of antibody known as a bispecific T-cell engager (BiTE). In 2014, blinatumomab became the first BiTE to receive regulatory approval, for the treatment of adult patients with relapsed/refractory ALL. Blinatumomab also targets the CD3 protein on T cells and helps to bring cancer cells and cytotoxic immune cells into close enough proximity that an immunological synapse can be formed between the two, facilitating tumor cell killing.21
In 2016, the approved indication was expanded into the pediatric population based on the results of a phase 1/2 study in which the safety and efficacy of blinatumomab were evaluated in 93 pediatric patients with relapsed/refractory ALL. Among the 70 patients who received the recommended dose of 5µg/m2 a day for the first 7 days, followed by 15µg/m2 a day thereafter, 51% achieved complete remission within the first 2 cycles, 52% of whom achieved minimal residual disease (MRD).22 Most recently, the FDA expanded the indication for blinatumomab to include patients (both adults and children) who are in remission, but MRD positive.23Despite the dramatic responses, many patients relapse after treatment with CD19-targeted CAR T cells, and researchers have uncovered numerous mechanisms of resistance. Among them is the loss of the CD19 antigen on the surface of target cells, such that a CD19-positive tumor becomes CD19-negative after treatment, driving relapse.24-26Several strategies for overcoming CD19-negative relapse are already being investigated, including the development of CD22-targeted CAR T cells and bispecific CAR T cells that target both CD19 and CD22. The results of a first-in-human trial of anti-CD22 CAR T-cell therapy were recently published. Among 21 pediatric and adult patients with relapsed/refractory B-cell ALL who were treated with either 3 x 105 cells/kg, 1 x 106 cells/kg, or 3 x 106 cells/kg, complete responses were observed in 57%.27
Results from 15 pediatric patients enrolled in a trial evaluating CD22-targeted CAR T cells as salvage therapy for those who relapse after CD19-targeted CAR T cell therapy were presented at the recent Congress of the European Hematology Association in Stockholm, Sweden. Patients who had undergone a stem cell transplant received the CAR T cells at a dose of 0.9 x 105 cell/kg and those who had not undergone a transplant received a dose of 8.2 x 105 cells/kg. At 30 days after CAR T cell infusion, the CR rate was 80% and the treatment was well tolerated.28
More immunotherapy approvals
The immune checkpoint inhibitors, which work by blocking inhibitory receptors on the surface of T cells, have also had recent approvals in pediatric patient populations. Pembrolizumab and nivolumab, inhibitors of the programmed cell death receptor 1 (PD-1) protein, have both been approved for use in adult and pediatric patients (older than 12 years) with relapsed/refractory metastatic colorectal cancer (and other solid tumors in the case of pembrolizumab) that display defects in the mismatch repair pathway that fixes damaged DNA or in patients that have high levels of microsatellite instability. Both deficient mismatch repair and microsatellite instability–high can indicate a high mutation burden in a tumor, which may predict increased sensitivity to immunotherapy.29
The approval in pediatric patients in both of those instances, however, was not based on data in pediatric patient populations but extrapolated from adult patients. Pembrolizumab is also approved for the treatment of adults and pediatric patients with classical Hodgkin lymphoma (cHL) after 3 or more previous treatments, but once again efficacy in the pediatric population was inferred from clinical trials performed in adults. Most recently, pembrolizumab was approved for the treatment of adult and pediatric patients with relapsed or refractory primary mediastinal large B-cell lymphoma.30Ipilimumab, which targets a different T cell receptor – cytotoxic T lymphocyte antigen-4 (CTLA-4) – has been approved for the treatment of pediatric patients aged 12 years and older with metastatic melanoma. This expanded indication, following on from its approval in adult patients in 2011, was based on data from 2 trials in which objective responses were observed in 2 out of 17 patients, including 1 partial response that lasted 16 months.31Finally, antibody-drug conjugates (ADC), in which tumor antigen-targeting monoclonal antibodies are conjugated to cytotoxic payloads to combine the specificity of an antibody with the cell-killing potency of chemotherapy, have also generated some recent successes in pediatric cancers.
Gemtuzumab ozogamicin is an ADC that targets the CD33 protein, which is highly expressed on 85%-90% of cases of acute myeloid leukemia (AML). In 2000, it was the first ADC to be brought to market in the United States, but it was subsequently voluntarily withdrawn by the manufacturer in 2010 after confirmatory trials failed to show a survival benefit.
Recently, a meta-analysis of gemtuzumab ozogamicin trials suggested that the drug likely does improve long-term overall survival (OS) and reduce the risk of relapse and researchers developed an intermittent dosing schedule to help mitigate toxicity.32 This new dosing regimen received FDA approval in 2017 for the treatment of pediatric patients aged 2 years and older on the basis of 2 clinical trials.
In the MyloFrance-1 trial, 57 patients were administered 3 mg/m2 gemtuzumab ozogamicin on days 1, 4, and 7 followed by cytarabine consolidation therapy and demonstrated a 26% CR rate and median recurrence-free survival of 11.6 months. In the phase 3 AML-19 trial, 237 patients received gemtuzumab ozogamicin at a dose of 6 mg/m2 on day 1 and 3 mg/m2 on day 8 or best supportive care. Gemtuzumab ozogamicin improved OS from 3.6 to 4.9 months.33,34
Inotuzumab ozogamicin is a CD22-targeting ADC that has been FDA approved for the treatment of adult patients with relapsed/refractory B-cell precursor ALL since last year. The therapy has been available to pediatric patients through a compassionate access program, but it has not been extensively evaluated in this population. The results of a retrospective analysis of pediatric patients who received at least 1 dose of inotuzumab ozogamicin were presented at ASCO in 2017. Among 29 patients with heavily pretreated disease the CR rate was 62%, 72% of whom achieved MRD negativity.35
Although there have been significant improvements in patient outcomes for some forms of pediatric cancer, progress has been painfully slow for others. An increasing understanding of pediatric cancers is highlighting the unique molecular drivers and challenging the assumption that drugs developed in adults can be applied to children and young adults. Here, we discuss game-changing therapeutic advances and a shifting view of childhood cancers.
Unique genomic background
Although pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, they are the second leading cause of death in children aged 1 to 14 years. There are many different histological tumor types under the umbrella of childhood cancers, of which the most common are leukemias, central nervous system tumors, and lymphomas (Figure 1).1,2
Significant progress has been made in the treatment of certain pediatric cancers in recent decades, exemplified by pediatric acute lymphoblastic leukemia (ALL), which has been transformed from a virtually incurable cancer to one in which 5-year survival rates now reach up to 90%. In other forms of pediatric cancer, however, survival rates have stagnated and little progress has been made in the development of effective new therapies.3
Because of their rarity, pediatric cancers are difficult to study and adequate enrollment of children in clinical trials can be challenging. Pharmaceutical companies are often hesitant to test drugs in the pediatric population in patients who often cannot advocate for themselves. As a result, the activity of drugs developed in adult patients has often been inferred in pediatric patients with the same tumor type or molecular aberrations. However, as researchers have gathered more information about pediatric cancers, there has been increasing recognition of their unique attributes and the need for dedicated clinical trials in this patient population.
Pediatric cancers tend to be found in the developing mesodermic tissue, whereas adult cancers are more prevalent in the epithelial tissues. Genome sequencing studies have revealed a much lower mutational burden in pediatric cancers and the mechanisms of oncogenesis are also quite different; adult tumors can develop from a series of acquired gene mutations, but pediatric tumors tend to develop from a single catastrophic event.4,5
Even the same type of cancer in a pediatric and adult patient can be quite different, with very different underlying molecular mechanisms. In a recent genomic analysis of different types of pediatric cancer by researchers at St Jude’s Children’s Research Hospital, less than half of the identified mutated genes were found to be similar to those found in adult patients.6
A ‘magic bullet’?
Chromosomal rearrangements are common in pediatric cancers. This type of molecular abnormality can result in a fusion of 2 different genes when the chromosome breaks apart and the pieces join back together in a muddled order. If the genetic code fuses in a manner that is “readable” by the cell, then it can drive aberrant activation of one or both genes.7 Gene fusions often involve kinase enzymes that are essential players in cell signaling pathways regulating hallmark cancer processes, such as unchecked cell proliferation. The fusion drives the constitutive activation of the kinase and, thus, these downstream signaling pathways.
One of the first chromosomal rearrangements linked to cancer, BCR-ABL1 – more commonly known as the Philadelphia chromosome – results in aberrant activation of the ABL1 kinase. It is present in nearly all cases of chronic myeloid leukemia (CML) and 3% to 5% of patients with ALL, and thus became the central focus of targeted drug development. Imatinib was initially approved by the US Food and Drug Administration (FDA) in 2001 for the treatment of adult patients with CML and had such a significant impact on the treatment landscape that it made the cover of Time magazine as a “magic bullet” in the war on cancer.8
Approval was expanded into pediatric patients in 2006 and for pediatric patients with ALL in 2013. However, as with the use of most kinase inhibitors, tumors can evolve under the selective pressure of treatment, developing additional molecular abnormalities that drive resistance.9
Next-generation multikinase inhibitors that more potently inhibit the BCR-ABL1 fusion protein have been developed to provide additional treatment options for patients who become resistant to imatinib. Dasatinib and nilotinib are among several drugs that have recently been approved for pediatric cancer therapy (Table 1). Both therapies were approved to treat children with Philadelphia chromosome-positive CML in the chronic phase in either the front- or second-line setting after failure of imatinib.
The approval of dasatinib was based on data from 97 patients across 2 trials, 51 of whom were newly diagnosed and 46 previously treated with imatinib. Most of the patients were treated with dasatinib 60 mg/m2 once daily. After 2 years of follow-up, more than 95% of newly diagnosed patients and 82.6% of relapsed/refractory patients had complete cytogenetic response.10
Nilotinib was approved on the basis of findings from 2 clinical trials including 69 patients – 1 trial involving patients who were refractory to or relapsed after dasatinib and imatinib treatment, and 1 that included both relapsed/refractory and newly diagnosed patients. Patients received nilotinib 230 mg/m2 twice daily, rounded to the nearest 50-mg dose, in 28-day cycles. By cycle 12, the cumulative major molecular response rate (MMR) was 47.7% in patients with relapsed/refractory disease, and 64% in newly diagnosed patients.11 Clinical trials of both drugs in the pediatric setting are ongoing.
Other prominent gene fusions
Gene fusions involving the anaplastic lymphoma kinase (ALK) occur in patients with non–small-cell lung cancer and ALK inhibitors have provided an effective new treatment option for patients whose tumors display this abnormality.
ALK fusions are also a prominent feature of several kinds of pediatric cancers and ALK inhibitors offer promise in this setting.7,12 An NPM-ALK fusion is found in 90% of pediatric anaplastic large cell lymphoma (ALCL) cases,13 whereas a variety of ALK fusions are found in up to half of patients with inflammatory myofibroblastic tumor (IMT), a rare form of soft tissue sarcoma.14 ALK inhibitors are being tested in a variety of clinical trials in pediatric patients (Table 2).
The results of a small phase 1 study of crizotinib in pediatric patients with ALK-positive ALCL (n = 26) or IMT (n = 14) were recently published. ALCL patients received crizotinib at a dose of 165 mg/m2, while IMT patients were given 100, 165, or 280 mg/m2. For the latter, the results were presented as a pooled cohort since safety and efficacy data were similar across dose levels. The overall response rate (ORR) was 83% for patients with ALCL and 86% for those with IMT. Grade 3/4 adverse events occurred in 83% and 71% of patients, respectively, and most commonly involved reduced neutrophil count.15
Most recently and perhaps most promisingly, fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) gene have generated significant buzz. There are 3 NTRK genes, NTRK1, 2, and 3, which encode the TRKA, TRKB, and TRKC proteins, respectively.
To date, 22 different partner genes have been identified that can fuse with the NTRK genes and, as with other kinase fusions, drive constitutive activation of the receptor proteins and downstream oncogenic signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway (Figure 2).
NTRK fusions are being identified in an ever-growing number of cancer types, but are typically found in a small percentage of patients. However, in certain rare pediatric tumors, including congenital infantile fibrosarcoma and papillary thyroid cancer, they are found at much higher frequencies.
TRK inhibitors have been developed to target the fusion proteins and, given the spread of NTRK fusions across different types of cancers, they offer the most substantial promise as the next tumor agnostic cancer therapy – to treat patients based on the shared presence of a molecular aberration, irrespective of the type of cancer.16
The ongoing SCOUT trial is evaluating larotrectinib (LOXO-101) in pediatric patients. Among 24 patients (17 with NTRK fusions and 7 without) with infantile fibrosarcoma (47%), soft tissue sarcoma (41%) or papillary thyroid cancer (12%), the ORR was 93%, including complete response (CR) in 13% of patients.17
Preliminary results from an ongoing phase 1/2 study of entrectinib in pediatric patients with extracranial solid tumors were also recently presented at the annual meeting of the American Society for Clinical Oncology (ASCO). Among 15 evaluable patients enrolled to date, 3 have NTRK fusions and all experienced an objective response, with 1 (a patient with IMT) ongoing at 10 months.18
CAR T cells transformative in ALL
A variety of different types of immunotherapy have been tested in patients with pediatric cancers. In general, immunotherapy has proved less effective than in adult cancers, possibly because of the lower tumor mutation burden in pediatric cancers, which means there are likely fewer cancer antigens to provoke an anti-tumor immune response.
There are notable exceptions among the disappointments, however, and most exciting is the development of chimeric antigen receptor (CAR) T cells. CAR T cells fall into a category of immunotherapy known as adoptive cell therapy (ACT), in which immune cells are harvested from a patient and grown outside the body to increase their numbers before being reinfused into the patient.
In the case of CAR T-cell therapy, the cells are genetically engineered to express a CAR that endows them with tumor-targeting capabilities. To date, the development of CAR T cells has focused on the use of the CD19 antigen as a target, which is highly expressed on a variety of B-cell malignancies, including several of the most common forms of pediatric cancer. ASCO shined the spotlight on CAR T-cell therapy this year, naming it the Advance of the Year for 2018, saying that the treatment is “poised to transform childhood ALL.”19
Two CD19-targeted CAR T-cell therapies – tisagenlecleucel and axicabtagene ciloleucel – were brought to market in 2017. Only tisagenlecleucel is approved in the pediatric ALL population, however, having been awarded approval for the treatment of patients aged up to 25 years whose disease is refractory to or relapsed after receiving at least 2 prior therapies. In the pivotal trial, complete responses were observed in more than 60% of patients.20 Clinical trials of both CAR T-cell therapies in pediatric ALL and non-Hodgkin lymphoma are ongoing (Table 3).
CD19 has also proven to be a promising target for other forms of immunotherapy, including a new type of antibody known as a bispecific T-cell engager (BiTE). In 2014, blinatumomab became the first BiTE to receive regulatory approval, for the treatment of adult patients with relapsed/refractory ALL. Blinatumomab also targets the CD3 protein on T cells and helps to bring cancer cells and cytotoxic immune cells into close enough proximity that an immunological synapse can be formed between the two, facilitating tumor cell killing.21
In 2016, the approved indication was expanded into the pediatric population based on the results of a phase 1/2 study in which the safety and efficacy of blinatumomab were evaluated in 93 pediatric patients with relapsed/refractory ALL. Among the 70 patients who received the recommended dose of 5µg/m2 a day for the first 7 days, followed by 15µg/m2 a day thereafter, 51% achieved complete remission within the first 2 cycles, 52% of whom achieved minimal residual disease (MRD).22 Most recently, the FDA expanded the indication for blinatumomab to include patients (both adults and children) who are in remission, but MRD positive.23Despite the dramatic responses, many patients relapse after treatment with CD19-targeted CAR T cells, and researchers have uncovered numerous mechanisms of resistance. Among them is the loss of the CD19 antigen on the surface of target cells, such that a CD19-positive tumor becomes CD19-negative after treatment, driving relapse.24-26Several strategies for overcoming CD19-negative relapse are already being investigated, including the development of CD22-targeted CAR T cells and bispecific CAR T cells that target both CD19 and CD22. The results of a first-in-human trial of anti-CD22 CAR T-cell therapy were recently published. Among 21 pediatric and adult patients with relapsed/refractory B-cell ALL who were treated with either 3 x 105 cells/kg, 1 x 106 cells/kg, or 3 x 106 cells/kg, complete responses were observed in 57%.27
Results from 15 pediatric patients enrolled in a trial evaluating CD22-targeted CAR T cells as salvage therapy for those who relapse after CD19-targeted CAR T cell therapy were presented at the recent Congress of the European Hematology Association in Stockholm, Sweden. Patients who had undergone a stem cell transplant received the CAR T cells at a dose of 0.9 x 105 cell/kg and those who had not undergone a transplant received a dose of 8.2 x 105 cells/kg. At 30 days after CAR T cell infusion, the CR rate was 80% and the treatment was well tolerated.28
More immunotherapy approvals
The immune checkpoint inhibitors, which work by blocking inhibitory receptors on the surface of T cells, have also had recent approvals in pediatric patient populations. Pembrolizumab and nivolumab, inhibitors of the programmed cell death receptor 1 (PD-1) protein, have both been approved for use in adult and pediatric patients (older than 12 years) with relapsed/refractory metastatic colorectal cancer (and other solid tumors in the case of pembrolizumab) that display defects in the mismatch repair pathway that fixes damaged DNA or in patients that have high levels of microsatellite instability. Both deficient mismatch repair and microsatellite instability–high can indicate a high mutation burden in a tumor, which may predict increased sensitivity to immunotherapy.29
The approval in pediatric patients in both of those instances, however, was not based on data in pediatric patient populations but extrapolated from adult patients. Pembrolizumab is also approved for the treatment of adults and pediatric patients with classical Hodgkin lymphoma (cHL) after 3 or more previous treatments, but once again efficacy in the pediatric population was inferred from clinical trials performed in adults. Most recently, pembrolizumab was approved for the treatment of adult and pediatric patients with relapsed or refractory primary mediastinal large B-cell lymphoma.30Ipilimumab, which targets a different T cell receptor – cytotoxic T lymphocyte antigen-4 (CTLA-4) – has been approved for the treatment of pediatric patients aged 12 years and older with metastatic melanoma. This expanded indication, following on from its approval in adult patients in 2011, was based on data from 2 trials in which objective responses were observed in 2 out of 17 patients, including 1 partial response that lasted 16 months.31Finally, antibody-drug conjugates (ADC), in which tumor antigen-targeting monoclonal antibodies are conjugated to cytotoxic payloads to combine the specificity of an antibody with the cell-killing potency of chemotherapy, have also generated some recent successes in pediatric cancers.
Gemtuzumab ozogamicin is an ADC that targets the CD33 protein, which is highly expressed on 85%-90% of cases of acute myeloid leukemia (AML). In 2000, it was the first ADC to be brought to market in the United States, but it was subsequently voluntarily withdrawn by the manufacturer in 2010 after confirmatory trials failed to show a survival benefit.
Recently, a meta-analysis of gemtuzumab ozogamicin trials suggested that the drug likely does improve long-term overall survival (OS) and reduce the risk of relapse and researchers developed an intermittent dosing schedule to help mitigate toxicity.32 This new dosing regimen received FDA approval in 2017 for the treatment of pediatric patients aged 2 years and older on the basis of 2 clinical trials.
In the MyloFrance-1 trial, 57 patients were administered 3 mg/m2 gemtuzumab ozogamicin on days 1, 4, and 7 followed by cytarabine consolidation therapy and demonstrated a 26% CR rate and median recurrence-free survival of 11.6 months. In the phase 3 AML-19 trial, 237 patients received gemtuzumab ozogamicin at a dose of 6 mg/m2 on day 1 and 3 mg/m2 on day 8 or best supportive care. Gemtuzumab ozogamicin improved OS from 3.6 to 4.9 months.33,34
Inotuzumab ozogamicin is a CD22-targeting ADC that has been FDA approved for the treatment of adult patients with relapsed/refractory B-cell precursor ALL since last year. The therapy has been available to pediatric patients through a compassionate access program, but it has not been extensively evaluated in this population. The results of a retrospective analysis of pediatric patients who received at least 1 dose of inotuzumab ozogamicin were presented at ASCO in 2017. Among 29 patients with heavily pretreated disease the CR rate was 62%, 72% of whom achieved MRD negativity.35
1. American Cancer Society. Key statistics for childhood cancers. https://www.cancer.org/cancer/cancer-in-children/key-statistics.html. Last revised September 10, 2018. Accessed September 16, 2018.
2. NHI/National Cancer Institute website. Unusual cancers of childhood treatment (PDQ) - Health Professional Version. https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Last updated August 28, 2018. Accessed September 8, 2018.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
4. Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277-289.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
6. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371.
7. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad-Massade L. Relevance of fusion genes in pediatric cancers: toward precision medicine. Molec Ther Nucleic Acids. 2017;6:315-326.
8. Lemonick MD, Park A. New hope for cancer. http://content.time.com/time/world/article/0,8599,2047900-2,00.html. Published May 28, 2001. Last accessed September 13, 2018.
9. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. https://www.hindawi.com/journals/cherp/2014/357027/. Published May 19, 2014. Accessed September 16, 2018.
10. Gore L, Kearns PR, Martino MLd, et al. Dasatinib in pediatric patients with chronic myeloid leukemia in chronic phase: results from a phase II trial. J Clin Oncol. 2018;36(13):1330-1338.
11. Novartis press release. Novartis drug Tasigna approved by FDA to treat children with rare form of leukemia. 2018; https://www.novartis.com/news/media-releases/novartis-drug-tasignar-approved-fda-treat-children-rare-form-leukemia. Released March 22, 2018. Accessed September 16, 2018.
12. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913-1920.
13. Turner SD, Lamant L, Kenner L, Brugieres L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol. 2016;173(4):560-572.
14. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am J Surg Pathol. 2015;39(7):957-967.
15. Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a children's oncology group study. J Clin Oncol. 2017;35(28):3215-3221.
16. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070277/. Published online March 18, 2016. Accessed September 16, 2018.
17. [Behind paywall.] Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705-714.
18. Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. J Clin Oncol. 2018;36(suppl;):abstr 10536.
19. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2018;36(10):1020-1044.
20. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. NEJM. 2018;378(5):439-448.
21. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
22. Stackelberg Av, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
23. Gokbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
24. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195.
25. Fousek K, Watanabe J, George A, et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J Clin Oncol. 2018;36(5_suppl):121-121.
26. Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659.
27. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
28. Pan J, Deng B, Liu S, et al. Efficacy and safety of CD22-directed CAR T-cell therapy in 15 pediatric refractory or relapsed b acute lymphoblastic leukemia patients. Paper presented at 23rd Congress of the European Hematology Association 2018; Stockholm, Sweden.
29. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35.
30. Drugs.com. Keytruda approval history. 2018; https://www.drugs.com/history/keytruda.html. Last update information not given. Accessed September 16, 2018.
31. Bristol Myers Squibb press release. US Food and Drug Administration expands approval of Yervoy (ipilimumab) to include pediatric patients 12 years and older with unresectable or metastatic melanoma. https://news.bms.com/press-release/corporatefinancial-news/us-food-and-drug-administration-expands-approval-yervoy-ipilim. Released July 24, 2017. Accessed September 16, 2018.
32. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
33. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972-979.
34. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66-71.
35. Bhojwani D, Sposto R, Shah N, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). J Clin Oncol. 2017;35(15_suppl):10512-10512.
1. American Cancer Society. Key statistics for childhood cancers. https://www.cancer.org/cancer/cancer-in-children/key-statistics.html. Last revised September 10, 2018. Accessed September 16, 2018.
2. NHI/National Cancer Institute website. Unusual cancers of childhood treatment (PDQ) - Health Professional Version. https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Last updated August 28, 2018. Accessed September 8, 2018.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
4. Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277-289.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
6. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371.
7. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad-Massade L. Relevance of fusion genes in pediatric cancers: toward precision medicine. Molec Ther Nucleic Acids. 2017;6:315-326.
8. Lemonick MD, Park A. New hope for cancer. http://content.time.com/time/world/article/0,8599,2047900-2,00.html. Published May 28, 2001. Last accessed September 13, 2018.
9. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. https://www.hindawi.com/journals/cherp/2014/357027/. Published May 19, 2014. Accessed September 16, 2018.
10. Gore L, Kearns PR, Martino MLd, et al. Dasatinib in pediatric patients with chronic myeloid leukemia in chronic phase: results from a phase II trial. J Clin Oncol. 2018;36(13):1330-1338.
11. Novartis press release. Novartis drug Tasigna approved by FDA to treat children with rare form of leukemia. 2018; https://www.novartis.com/news/media-releases/novartis-drug-tasignar-approved-fda-treat-children-rare-form-leukemia. Released March 22, 2018. Accessed September 16, 2018.
12. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913-1920.
13. Turner SD, Lamant L, Kenner L, Brugieres L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol. 2016;173(4):560-572.
14. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am J Surg Pathol. 2015;39(7):957-967.
15. Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a children's oncology group study. J Clin Oncol. 2017;35(28):3215-3221.
16. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070277/. Published online March 18, 2016. Accessed September 16, 2018.
17. [Behind paywall.] Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705-714.
18. Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. J Clin Oncol. 2018;36(suppl;):abstr 10536.
19. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2018;36(10):1020-1044.
20. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. NEJM. 2018;378(5):439-448.
21. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
22. Stackelberg Av, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
23. Gokbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
24. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195.
25. Fousek K, Watanabe J, George A, et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J Clin Oncol. 2018;36(5_suppl):121-121.
26. Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659.
27. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
28. Pan J, Deng B, Liu S, et al. Efficacy and safety of CD22-directed CAR T-cell therapy in 15 pediatric refractory or relapsed b acute lymphoblastic leukemia patients. Paper presented at 23rd Congress of the European Hematology Association 2018; Stockholm, Sweden.
29. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35.
30. Drugs.com. Keytruda approval history. 2018; https://www.drugs.com/history/keytruda.html. Last update information not given. Accessed September 16, 2018.
31. Bristol Myers Squibb press release. US Food and Drug Administration expands approval of Yervoy (ipilimumab) to include pediatric patients 12 years and older with unresectable or metastatic melanoma. https://news.bms.com/press-release/corporatefinancial-news/us-food-and-drug-administration-expands-approval-yervoy-ipilim. Released July 24, 2017. Accessed September 16, 2018.
32. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
33. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972-979.
34. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66-71.
35. Bhojwani D, Sposto R, Shah N, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). J Clin Oncol. 2017;35(15_suppl):10512-10512.