User login
Mitchel is a reporter for MDedge based in the Philadelphia area. He started with the company in 1992, when it was International Medical News Group (IMNG), and has since covered a range of medical specialties. Mitchel trained as a virologist at Roswell Park Memorial Institute in Buffalo, and then worked briefly as a researcher at Boston Children's Hospital before pivoting to journalism as a AAAS Mass Media Fellow in 1980. His first reporting job was with Science Digest magazine, and from the mid-1980s to early-1990s he was a reporter with Medical World News. @mitchelzoler
Ineffective renal denervation blamed for trial’s failure
WASHINGTON – Renal denervation’s not so simple after all.
Highly anticipated data from the large, sham-controlled renal-artery catheter denervation trial, SYMPLICITY HTN-3, gave its investigators reason to believe that inconsistent and inadequate delivery of nerve-ablating radiofrequency energy by inexperienced operators left too many patients undertreated, producing a neutral outcome for the primary endpoint, the study’s top-line result first reported in a company press release in January.
In the SYMPLICITY HTN-3 (Renal Denervation in Patients With Uncontrolled Hypertension) trial, the average systolic blood pressure reduction among the 364 patients randomized to renal denervation was 14 mm Hg, compared with an average 12 mm Hg reduction in 174 control patients who underwent a renal-artery angiogram masked to resemble a denervation procedure, a difference that was not statistically significant, falling substantially short of the prespecified 5 mm Hg between group difference necessary to fulfill superiority, Dr. Deepak L. Bhatt said at the annual meeting of the American College of Cardiology.

Although several factors likely led to the neutral outcome, including an unexpectedly robust blood pressure drop in the control arm, another major factor, many suspect, was inadequate delivery of the denervation treatment.
"It might be that the way we used the catheter wasn’t ideal," said Dr. Bhatt, coprincipal investigator of the study, professor of medicine at Harvard Medical School and executive director of interventional cardiology at Brigham and Women’s Hospital, both in Boston.
Concurrent with Dr. Bhatt’s report at the meeting a published version appeared online (N. Engl. J. Med. 2014 March 29 [doi:10.1056/NEJMoa1402670]).
"There is the concept that renal denervation is easy. As a catheter procedure it is easy, but to achieve denervation is far from easy," said Dr. Murray D. Esler, a professor of medicine at Monash University and senior director of the Baker IDI Heart and Diabetes Institute, both in Melbourne, and a pioneer of renal denervation who was involved in prior SYMPLICITY studies of the same renal-denervation device but was not a coinvestigator for the new trial.
"The SYMPLICITY 3 study made a mistake thinking it could get away with not giving the proceduralists any hands-on experience" before the trial. "There was none. The operators hadn’t done it before, and the trial’s designers would not have done that unless they thought that denervation was easy. Well, it’s not easy. There is a learning curve, and I think that’s the main explanation of what went wrong. "

Exacerbating the operators’ inexperience with denervation (they were all experienced interventional cardiologists) was a second flaw in the trial’s design: No method was used to monitor the efficacy of each denervation procedure, an easy decision because there is still no proven way to measure denervation efficacy during the procedure. Dr. Esler said he believes that he has a solution, one that involves testing for interrupted activity of the parallel, afferent nerves of the renal arteries using adenosine as a surrogate measure for denervation of the target efferent nerves. But while he is optimistic the adenosine approach will work, it is still at least several months from use in a trial.
In the first clinical study of renal denervation, a series of 50 patients treated at five centers in Australia and Europe reported just 5 years ago (Lancet 2009;373:1275-81), Dr. Esler, a lead investigator for that study, and his associates carefully tested the efficacy of denervation in 10 patients by measuring the direct effect of successful denervation, reduction of renal norepinephrine spillover. They reported that, in those 10 patients, renal denervation cut norepinephrine spillover by an average 47%, which correlated with an average reduction in systolic blood pressure of 22 mm Hg after 6 months.
But this effect from denervation was "far from uniform and ranged from zero to 85%," depending on who was doing the denervation. "It’s obviously operator dependent." Dr. Esler said in an interview. This observation of enormous variability in the physiologic impact of denervation "took on a new context with the failed trial. The denervation done by novices explains to some extent the spread [in results] and makes me think the same thing happened in SYMPLICITY 3."

"If you don’t do enough high-frequency burns or if the catheter doesn’t reach enough nerves" because of misplacement within the renal artery, the denervation won’t be very successful, noted Dr. George Bakris, professor of medicine and director of the comprehensive hypertension center at the University of Chicago and coprincipal investigator of SYMPLICITY HTN-3 along with Dr. Bhatt. "It certainly calls into question whether they denervated effectively. One could argue that if denervation were done appropriately" it would add to drug treatment, Dr. Bakris said in an interview. "This issue has come up about the quality of the denervation; the good news is that it can be done correctly," but that will require "a good understanding of what you are doing to the renal nerves and how much energy you need." SYMPLICITY HTN-3 "was a good but expensive learning experience," Dr. Bakris said.
The investigators "were performing a procedure, but they didn’t have the evidence that the procedure was accomplishing what it meant to accomplish," commented Dr. Anthony DeMaria, professor of medicine, the Judith and Jack White chair of cardiology, and director of the cardiovascular center at the University of California, San Diego, who was not involved in the trial. "Most of us feel there are tantalizing data to suggest that denervation can have a benefit, but they need to go back to the physiology to be sure they are denervating effectively."

Though Dr. Bakris and Dr. Esler cautioned that the study’s primary outcome was neutral, they see reason to hope that renal denervation will still prove to have an important and useful clinical effect.
"This report is the tip of the iceberg," Dr. Bakris noted. "When you look at the effect of denervation on blood pressure variability, heart rate variability, and total control over 24 hours using ambulatory blood pressure monitoring, you see a different picture," results that will be reported later this year, he said.
And Dr. Esler said that his experience using renal denervation on about 300 patients with treatment-resistant hypertension has convinced him that denervation can produce real benefits when done properly.
"I’m not changing my practice" using renal denervation on refractory patients, he said. "I accumulated a lot of patients with severe, resistant hypertension, patients whom I couldn’t control with seven or eight different drug classes. But when renal denervation came along, it was like chalk and cheese. Suddenly, I had a therapy that worked. My clinical experience in patients who didn’t budge with medications totally convinces me" that renal denervation works.
SYMPLICITY HTN-3 was sponsored by Medtronic, the company that markets the device tested in the study. Dr. Bhatt said that he has received research grants from Medtronic and five other companies. Dr. Esler said that he has received consulting fees, honoraria, and research grants from Medtronic. Dr. Bakris said that he has received personal fees from Medtronic and five other companies. Dr. DeMaria said that he has received research grants from Angioblast Systems, Cardiovascular Biotherapeutics, General Electric Medical, Gilead, and Lantheus.
On Twitter @mitchelzoler
WASHINGTON – Renal denervation’s not so simple after all.
Highly anticipated data from the large, sham-controlled renal-artery catheter denervation trial, SYMPLICITY HTN-3, gave its investigators reason to believe that inconsistent and inadequate delivery of nerve-ablating radiofrequency energy by inexperienced operators left too many patients undertreated, producing a neutral outcome for the primary endpoint, the study’s top-line result first reported in a company press release in January.
In the SYMPLICITY HTN-3 (Renal Denervation in Patients With Uncontrolled Hypertension) trial, the average systolic blood pressure reduction among the 364 patients randomized to renal denervation was 14 mm Hg, compared with an average 12 mm Hg reduction in 174 control patients who underwent a renal-artery angiogram masked to resemble a denervation procedure, a difference that was not statistically significant, falling substantially short of the prespecified 5 mm Hg between group difference necessary to fulfill superiority, Dr. Deepak L. Bhatt said at the annual meeting of the American College of Cardiology.
Although several factors likely led to the neutral outcome, including an unexpectedly robust blood pressure drop in the control arm, another major factor, many suspect, was inadequate delivery of the denervation treatment.
"It might be that the way we used the catheter wasn’t ideal," said Dr. Bhatt, coprincipal investigator of the study, professor of medicine at Harvard Medical School and executive director of interventional cardiology at Brigham and Women’s Hospital, both in Boston.
Concurrent with Dr. Bhatt’s report at the meeting a published version appeared online (N. Engl. J. Med. 2014 March 29 [doi:10.1056/NEJMoa1402670]).
"There is the concept that renal denervation is easy. As a catheter procedure it is easy, but to achieve denervation is far from easy," said Dr. Murray D. Esler, a professor of medicine at Monash University and senior director of the Baker IDI Heart and Diabetes Institute, both in Melbourne, and a pioneer of renal denervation who was involved in prior SYMPLICITY studies of the same renal-denervation device but was not a coinvestigator for the new trial.
"The SYMPLICITY 3 study made a mistake thinking it could get away with not giving the proceduralists any hands-on experience" before the trial. "There was none. The operators hadn’t done it before, and the trial’s designers would not have done that unless they thought that denervation was easy. Well, it’s not easy. There is a learning curve, and I think that’s the main explanation of what went wrong. "
Exacerbating the operators’ inexperience with denervation (they were all experienced interventional cardiologists) was a second flaw in the trial’s design: No method was used to monitor the efficacy of each denervation procedure, an easy decision because there is still no proven way to measure denervation efficacy during the procedure. Dr. Esler said he believes that he has a solution, one that involves testing for interrupted activity of the parallel, afferent nerves of the renal arteries using adenosine as a surrogate measure for denervation of the target efferent nerves. But while he is optimistic the adenosine approach will work, it is still at least several months from use in a trial.
In the first clinical study of renal denervation, a series of 50 patients treated at five centers in Australia and Europe reported just 5 years ago (Lancet 2009;373:1275-81), Dr. Esler, a lead investigator for that study, and his associates carefully tested the efficacy of denervation in 10 patients by measuring the direct effect of successful denervation, reduction of renal norepinephrine spillover. They reported that, in those 10 patients, renal denervation cut norepinephrine spillover by an average 47%, which correlated with an average reduction in systolic blood pressure of 22 mm Hg after 6 months.
But this effect from denervation was "far from uniform and ranged from zero to 85%," depending on who was doing the denervation. "It’s obviously operator dependent." Dr. Esler said in an interview. This observation of enormous variability in the physiologic impact of denervation "took on a new context with the failed trial. The denervation done by novices explains to some extent the spread [in results] and makes me think the same thing happened in SYMPLICITY 3."
"If you don’t do enough high-frequency burns or if the catheter doesn’t reach enough nerves" because of misplacement within the renal artery, the denervation won’t be very successful, noted Dr. George Bakris, professor of medicine and director of the comprehensive hypertension center at the University of Chicago and coprincipal investigator of SYMPLICITY HTN-3 along with Dr. Bhatt. "It certainly calls into question whether they denervated effectively. One could argue that if denervation were done appropriately" it would add to drug treatment, Dr. Bakris said in an interview. "This issue has come up about the quality of the denervation; the good news is that it can be done correctly," but that will require "a good understanding of what you are doing to the renal nerves and how much energy you need." SYMPLICITY HTN-3 "was a good but expensive learning experience," Dr. Bakris said.
The investigators "were performing a procedure, but they didn’t have the evidence that the procedure was accomplishing what it meant to accomplish," commented Dr. Anthony DeMaria, professor of medicine, the Judith and Jack White chair of cardiology, and director of the cardiovascular center at the University of California, San Diego, who was not involved in the trial. "Most of us feel there are tantalizing data to suggest that denervation can have a benefit, but they need to go back to the physiology to be sure they are denervating effectively."
Though Dr. Bakris and Dr. Esler cautioned that the study’s primary outcome was neutral, they see reason to hope that renal denervation will still prove to have an important and useful clinical effect.
"This report is the tip of the iceberg," Dr. Bakris noted. "When you look at the effect of denervation on blood pressure variability, heart rate variability, and total control over 24 hours using ambulatory blood pressure monitoring, you see a different picture," results that will be reported later this year, he said.
And Dr. Esler said that his experience using renal denervation on about 300 patients with treatment-resistant hypertension has convinced him that denervation can produce real benefits when done properly.
"I’m not changing my practice" using renal denervation on refractory patients, he said. "I accumulated a lot of patients with severe, resistant hypertension, patients whom I couldn’t control with seven or eight different drug classes. But when renal denervation came along, it was like chalk and cheese. Suddenly, I had a therapy that worked. My clinical experience in patients who didn’t budge with medications totally convinces me" that renal denervation works.
SYMPLICITY HTN-3 was sponsored by Medtronic, the company that markets the device tested in the study. Dr. Bhatt said that he has received research grants from Medtronic and five other companies. Dr. Esler said that he has received consulting fees, honoraria, and research grants from Medtronic. Dr. Bakris said that he has received personal fees from Medtronic and five other companies. Dr. DeMaria said that he has received research grants from Angioblast Systems, Cardiovascular Biotherapeutics, General Electric Medical, Gilead, and Lantheus.
On Twitter @mitchelzoler
WASHINGTON – Renal denervation’s not so simple after all.
Highly anticipated data from the large, sham-controlled renal-artery catheter denervation trial, SYMPLICITY HTN-3, gave its investigators reason to believe that inconsistent and inadequate delivery of nerve-ablating radiofrequency energy by inexperienced operators left too many patients undertreated, producing a neutral outcome for the primary endpoint, the study’s top-line result first reported in a company press release in January.
In the SYMPLICITY HTN-3 (Renal Denervation in Patients With Uncontrolled Hypertension) trial, the average systolic blood pressure reduction among the 364 patients randomized to renal denervation was 14 mm Hg, compared with an average 12 mm Hg reduction in 174 control patients who underwent a renal-artery angiogram masked to resemble a denervation procedure, a difference that was not statistically significant, falling substantially short of the prespecified 5 mm Hg between group difference necessary to fulfill superiority, Dr. Deepak L. Bhatt said at the annual meeting of the American College of Cardiology.
Although several factors likely led to the neutral outcome, including an unexpectedly robust blood pressure drop in the control arm, another major factor, many suspect, was inadequate delivery of the denervation treatment.
"It might be that the way we used the catheter wasn’t ideal," said Dr. Bhatt, coprincipal investigator of the study, professor of medicine at Harvard Medical School and executive director of interventional cardiology at Brigham and Women’s Hospital, both in Boston.
Concurrent with Dr. Bhatt’s report at the meeting a published version appeared online (N. Engl. J. Med. 2014 March 29 [doi:10.1056/NEJMoa1402670]).
"There is the concept that renal denervation is easy. As a catheter procedure it is easy, but to achieve denervation is far from easy," said Dr. Murray D. Esler, a professor of medicine at Monash University and senior director of the Baker IDI Heart and Diabetes Institute, both in Melbourne, and a pioneer of renal denervation who was involved in prior SYMPLICITY studies of the same renal-denervation device but was not a coinvestigator for the new trial.
"The SYMPLICITY 3 study made a mistake thinking it could get away with not giving the proceduralists any hands-on experience" before the trial. "There was none. The operators hadn’t done it before, and the trial’s designers would not have done that unless they thought that denervation was easy. Well, it’s not easy. There is a learning curve, and I think that’s the main explanation of what went wrong. "
Exacerbating the operators’ inexperience with denervation (they were all experienced interventional cardiologists) was a second flaw in the trial’s design: No method was used to monitor the efficacy of each denervation procedure, an easy decision because there is still no proven way to measure denervation efficacy during the procedure. Dr. Esler said he believes that he has a solution, one that involves testing for interrupted activity of the parallel, afferent nerves of the renal arteries using adenosine as a surrogate measure for denervation of the target efferent nerves. But while he is optimistic the adenosine approach will work, it is still at least several months from use in a trial.
In the first clinical study of renal denervation, a series of 50 patients treated at five centers in Australia and Europe reported just 5 years ago (Lancet 2009;373:1275-81), Dr. Esler, a lead investigator for that study, and his associates carefully tested the efficacy of denervation in 10 patients by measuring the direct effect of successful denervation, reduction of renal norepinephrine spillover. They reported that, in those 10 patients, renal denervation cut norepinephrine spillover by an average 47%, which correlated with an average reduction in systolic blood pressure of 22 mm Hg after 6 months.
But this effect from denervation was "far from uniform and ranged from zero to 85%," depending on who was doing the denervation. "It’s obviously operator dependent." Dr. Esler said in an interview. This observation of enormous variability in the physiologic impact of denervation "took on a new context with the failed trial. The denervation done by novices explains to some extent the spread [in results] and makes me think the same thing happened in SYMPLICITY 3."
"If you don’t do enough high-frequency burns or if the catheter doesn’t reach enough nerves" because of misplacement within the renal artery, the denervation won’t be very successful, noted Dr. George Bakris, professor of medicine and director of the comprehensive hypertension center at the University of Chicago and coprincipal investigator of SYMPLICITY HTN-3 along with Dr. Bhatt. "It certainly calls into question whether they denervated effectively. One could argue that if denervation were done appropriately" it would add to drug treatment, Dr. Bakris said in an interview. "This issue has come up about the quality of the denervation; the good news is that it can be done correctly," but that will require "a good understanding of what you are doing to the renal nerves and how much energy you need." SYMPLICITY HTN-3 "was a good but expensive learning experience," Dr. Bakris said.
The investigators "were performing a procedure, but they didn’t have the evidence that the procedure was accomplishing what it meant to accomplish," commented Dr. Anthony DeMaria, professor of medicine, the Judith and Jack White chair of cardiology, and director of the cardiovascular center at the University of California, San Diego, who was not involved in the trial. "Most of us feel there are tantalizing data to suggest that denervation can have a benefit, but they need to go back to the physiology to be sure they are denervating effectively."
Though Dr. Bakris and Dr. Esler cautioned that the study’s primary outcome was neutral, they see reason to hope that renal denervation will still prove to have an important and useful clinical effect.
"This report is the tip of the iceberg," Dr. Bakris noted. "When you look at the effect of denervation on blood pressure variability, heart rate variability, and total control over 24 hours using ambulatory blood pressure monitoring, you see a different picture," results that will be reported later this year, he said.
And Dr. Esler said that his experience using renal denervation on about 300 patients with treatment-resistant hypertension has convinced him that denervation can produce real benefits when done properly.
"I’m not changing my practice" using renal denervation on refractory patients, he said. "I accumulated a lot of patients with severe, resistant hypertension, patients whom I couldn’t control with seven or eight different drug classes. But when renal denervation came along, it was like chalk and cheese. Suddenly, I had a therapy that worked. My clinical experience in patients who didn’t budge with medications totally convinces me" that renal denervation works.
SYMPLICITY HTN-3 was sponsored by Medtronic, the company that markets the device tested in the study. Dr. Bhatt said that he has received research grants from Medtronic and five other companies. Dr. Esler said that he has received consulting fees, honoraria, and research grants from Medtronic. Dr. Bakris said that he has received personal fees from Medtronic and five other companies. Dr. DeMaria said that he has received research grants from Angioblast Systems, Cardiovascular Biotherapeutics, General Electric Medical, Gilead, and Lantheus.
On Twitter @mitchelzoler
AT ACC 14
Major finding: Renal denervation produced an average 14 mm Hg drop in systolic blood pressure, compared with an average 12 mm Hg decline in controls.
Data source: SYMPLICITY HTN-3, a randomized trial with 535 patients with uncontrolled hypertension who were treated at 88 U.S. sites.
Disclosures: SYMPLICITY HTN-3 was sponsored by Medtronic, the company that markets the device tested in the study. Dr. Bhatt said that he has received research grants from Medtronic and five other companies. Dr. Esler said that he has received consulting fees, honoraria, and research grants from Medtronic. Dr. Bakris said that he has received personal fees from Medtronic and five other companies. Dr. DeMaria said that he has received research grants from Angioblast Systems, Cardiovascular Biotherapeutics, General Electric Medical, Gilead, and Lantheus.

VIDEO: What explains SYMPLICITY HTN-3’s failure?
WASHINGTON – What can explain the failure of SYMPLICITY HTN-3? After clinical success in reducing blood pressure by 20-30 mm HG in patients with resistant hypertension, the renal denervation treatment showed no significant reduction in the first randomized trial in which control patients underwent sham operations.
According to two top hypertension experts, trial investigator Dr. George Bakris and Dr. Prakash Deedwania, physiology, behavior, and technology come to mind for starters, as explored in Part 1 of our interview.
WASHINGTON – What can explain the failure of SYMPLICITY HTN-3? After clinical success in reducing blood pressure by 20-30 mm HG in patients with resistant hypertension, the renal denervation treatment showed no significant reduction in the first randomized trial in which control patients underwent sham operations.
According to two top hypertension experts, trial investigator Dr. George Bakris and Dr. Prakash Deedwania, physiology, behavior, and technology come to mind for starters, as explored in Part 1 of our interview.
WASHINGTON – What can explain the failure of SYMPLICITY HTN-3? After clinical success in reducing blood pressure by 20-30 mm HG in patients with resistant hypertension, the renal denervation treatment showed no significant reduction in the first randomized trial in which control patients underwent sham operations.
According to two top hypertension experts, trial investigator Dr. George Bakris and Dr. Prakash Deedwania, physiology, behavior, and technology come to mind for starters, as explored in Part 1 of our interview.
At ACC 14

Diet drinks linked to CVD in women
Drinking two or more diet drinks a day was associated with a 29% increased risk of an incident cardiovascular event and a 26% increased risk of all-cause death compared with less diet drink consumption in an observational study of nearly 60,000 postmenopausal American women.
Although the pathogenic mechanisms behind this "hypothesis-generating" finding remain unclear, a link between high use of diet drinks and increased cardiovascular events and death is consistent with prior reports that linked diet drink intake with metabolic syndrome and cardiovascular disease events, said Dr. Ankur Vyas, who summarized the results of the study during a webcast held prior to the annual meeting of the American College of Cardiology, where he is presenting the data.

"This study is very interesting and clinically relevant given the vast number of people who drink diet drinks daily," commented Dr. Jeffrey T. Kuvin of the division of cardiology at Tufts Medical Center in Boston. "We know that drinking sweetened beverages is associated with weight gain, diabetes, and coronary heart disease, and diet drinks have been linked with metabolic syndrome. This report is very provocative and may shed some light on a subject we need to know more about."
Dr. Kuvin, who admitted to often drinking two or more diet drinks a day himself, added, "I’m not ready just yet to give it up, but the data are compelling to take a closer look at why this could be."
The analysis by Dr. Vyas and his associates focused on 59,614 postmenopausal women aged 50-79 from the observational arm of the Women\'s Health Initiative who enrolled during 1993-1998This subgroup excluded women with preexisting cardiovascular disease, no data available on diet drink consumption, or other reasons. The study examined deaths and cardiovascular disease events during an average 9 years of follow-up among the 5% of women who drank an average of two or more diet drinks daily compared with women who had fewer of these beverages. Women in the subgroup with the highest diet drink intake were significantly younger and had a significantly higher rate of obesity than women with lower intake levels.
In an analysis that adjusted for several demographic and clinical factors, women who drank two or more drinks had, during follow-up, 29% more cardiovascular events compared with the other women, a statistically significant difference for the study’s primary endpoint that combined myocardial infarctions, ischemic strokes, coronary artery revascularization, peripheral artery disease, heart failure, and cardiovascular death, reported Dr. Vyas, a fellow in cardiology at the University of Iowa, Iowa City. The highest level of diet drink use also linked with a statistically significant increase of 52% for the single endpoint of cardiovascular death and a 26% higher rate of all-cause death.
Dr. Vyas suggested that higher diet drink consumption may disrupt normal feedback mechanisms that control intake of food and other beverages, or it may link with various elements of an unhealthy lifestyle.
Dr. Vyas also said that the finding could be the result of inadequate adjustment for confounding factors. "In any observational study it’s close to impossible to control for everything and rule out every possible confounder. Our study faces the same limitations as any retrospective observational study," he said during a press briefing.
Dr. Vyas and Dr. Kuvin said that they had no relevant financial disclosures.
On Twitter @mitchelzoler
Drinking two or more diet drinks a day was associated with a 29% increased risk of an incident cardiovascular event and a 26% increased risk of all-cause death compared with less diet drink consumption in an observational study of nearly 60,000 postmenopausal American women.
Although the pathogenic mechanisms behind this "hypothesis-generating" finding remain unclear, a link between high use of diet drinks and increased cardiovascular events and death is consistent with prior reports that linked diet drink intake with metabolic syndrome and cardiovascular disease events, said Dr. Ankur Vyas, who summarized the results of the study during a webcast held prior to the annual meeting of the American College of Cardiology, where he is presenting the data.
"This study is very interesting and clinically relevant given the vast number of people who drink diet drinks daily," commented Dr. Jeffrey T. Kuvin of the division of cardiology at Tufts Medical Center in Boston. "We know that drinking sweetened beverages is associated with weight gain, diabetes, and coronary heart disease, and diet drinks have been linked with metabolic syndrome. This report is very provocative and may shed some light on a subject we need to know more about."
Dr. Kuvin, who admitted to often drinking two or more diet drinks a day himself, added, "I’m not ready just yet to give it up, but the data are compelling to take a closer look at why this could be."
The analysis by Dr. Vyas and his associates focused on 59,614 postmenopausal women aged 50-79 from the observational arm of the Women\'s Health Initiative who enrolled during 1993-1998This subgroup excluded women with preexisting cardiovascular disease, no data available on diet drink consumption, or other reasons. The study examined deaths and cardiovascular disease events during an average 9 years of follow-up among the 5% of women who drank an average of two or more diet drinks daily compared with women who had fewer of these beverages. Women in the subgroup with the highest diet drink intake were significantly younger and had a significantly higher rate of obesity than women with lower intake levels.
In an analysis that adjusted for several demographic and clinical factors, women who drank two or more drinks had, during follow-up, 29% more cardiovascular events compared with the other women, a statistically significant difference for the study’s primary endpoint that combined myocardial infarctions, ischemic strokes, coronary artery revascularization, peripheral artery disease, heart failure, and cardiovascular death, reported Dr. Vyas, a fellow in cardiology at the University of Iowa, Iowa City. The highest level of diet drink use also linked with a statistically significant increase of 52% for the single endpoint of cardiovascular death and a 26% higher rate of all-cause death.
Dr. Vyas suggested that higher diet drink consumption may disrupt normal feedback mechanisms that control intake of food and other beverages, or it may link with various elements of an unhealthy lifestyle.
Dr. Vyas also said that the finding could be the result of inadequate adjustment for confounding factors. "In any observational study it’s close to impossible to control for everything and rule out every possible confounder. Our study faces the same limitations as any retrospective observational study," he said during a press briefing.
Dr. Vyas and Dr. Kuvin said that they had no relevant financial disclosures.
On Twitter @mitchelzoler
Drinking two or more diet drinks a day was associated with a 29% increased risk of an incident cardiovascular event and a 26% increased risk of all-cause death compared with less diet drink consumption in an observational study of nearly 60,000 postmenopausal American women.
Although the pathogenic mechanisms behind this "hypothesis-generating" finding remain unclear, a link between high use of diet drinks and increased cardiovascular events and death is consistent with prior reports that linked diet drink intake with metabolic syndrome and cardiovascular disease events, said Dr. Ankur Vyas, who summarized the results of the study during a webcast held prior to the annual meeting of the American College of Cardiology, where he is presenting the data.
"This study is very interesting and clinically relevant given the vast number of people who drink diet drinks daily," commented Dr. Jeffrey T. Kuvin of the division of cardiology at Tufts Medical Center in Boston. "We know that drinking sweetened beverages is associated with weight gain, diabetes, and coronary heart disease, and diet drinks have been linked with metabolic syndrome. This report is very provocative and may shed some light on a subject we need to know more about."
Dr. Kuvin, who admitted to often drinking two or more diet drinks a day himself, added, "I’m not ready just yet to give it up, but the data are compelling to take a closer look at why this could be."
The analysis by Dr. Vyas and his associates focused on 59,614 postmenopausal women aged 50-79 from the observational arm of the Women\'s Health Initiative who enrolled during 1993-1998This subgroup excluded women with preexisting cardiovascular disease, no data available on diet drink consumption, or other reasons. The study examined deaths and cardiovascular disease events during an average 9 years of follow-up among the 5% of women who drank an average of two or more diet drinks daily compared with women who had fewer of these beverages. Women in the subgroup with the highest diet drink intake were significantly younger and had a significantly higher rate of obesity than women with lower intake levels.
In an analysis that adjusted for several demographic and clinical factors, women who drank two or more drinks had, during follow-up, 29% more cardiovascular events compared with the other women, a statistically significant difference for the study’s primary endpoint that combined myocardial infarctions, ischemic strokes, coronary artery revascularization, peripheral artery disease, heart failure, and cardiovascular death, reported Dr. Vyas, a fellow in cardiology at the University of Iowa, Iowa City. The highest level of diet drink use also linked with a statistically significant increase of 52% for the single endpoint of cardiovascular death and a 26% higher rate of all-cause death.
Dr. Vyas suggested that higher diet drink consumption may disrupt normal feedback mechanisms that control intake of food and other beverages, or it may link with various elements of an unhealthy lifestyle.
Dr. Vyas also said that the finding could be the result of inadequate adjustment for confounding factors. "In any observational study it’s close to impossible to control for everything and rule out every possible confounder. Our study faces the same limitations as any retrospective observational study," he said during a press briefing.
Dr. Vyas and Dr. Kuvin said that they had no relevant financial disclosures.
On Twitter @mitchelzoler
FROM ACC 14
Major finding: .Women who drank at least two diet drinks daily had 29% more cardiovascular events than women having fewer diet drinks.
Data source: A retrospective analysis of data collected from 59,614 postmenopausal American women enrolled in the Women’s Health Initiative.
Disclosures: Dr. Vyas and Dr. Kuvin said that they had no relevant financial disclosures.

Meta-analysis shows statins help erectile function
Men with erectile dysfunction had a better than threefold improvement in erectile function after receiving statin treatment compared with men randomized to placebo in a meta-analysis of 11 controlled studies with a total of more than 600 patients.
Although the result suggests that statin treatment helps men with erectile dysfunction (ED), the evidence is not strong enough to warrant starting statin treatment when the patient has no established indication for the drug, said Dr. John B. Kostis, who summarized the results of the study during a webcast held prior to the annual meeting of the American College of Cardiology, where he is presenting the data.
He called for a multifactorial trial that could assess the roles for statin, treatment, a phosphodiesterase type 5 inhibitor,and testosterone treatment both individually and in combinations.

"Erectile dysfunction is the canary in the coal mine. It can be the first sign of cardiovascular disease," said Dr. Kostis, John G. Detwiler professor of cardiology, professor of medicine and pharmacology, and director of the Cardiovascular Institute at the Rutgers Robert Wood Johnson Medical School in New Brunswick, N.J. "When a man has erectile dysfunction and no explanation like prostatectomy, he should be evaluated for his cardiovascular risk and treated with a statin if that’s justified," he said during the webcast.
"Over the years it’s become apparent that erectile dysfunction is an indication of reduced vascular health in men, and it is considered by many to be a significant cardiovascular disease risk factor," commented Dr. Jeffrey T. Kuvin of the division of cardiology at Tufts Medical Center in Boston. "The results of this meta-analysis strongly show that statin treatment improves erectile dysfunction after only a short duration of treatment. Whether erectile dysfunction improves because of reduced levels of low-density lipoprotein cholesterol or other pleiotropic effects of statins remains unclear," said Dr. Kuvin.
Dr. Kostis and his associate reviewed 11 randomized controlled trials that compared the effect of statin treatment on erectile function with placebo as a primary or secondary endpoint. The 11 studies involved a total of 647 men with ED and an average age of 58 years. The average duration of treatment was 4 months. On statin treatment the average level of LDL cholesterol fell from 138 mg/dL at baseline to 91 mg/dL, while men on placebo had no meaningful change in their LDL cholesterol level.
The meta-analysis showed that after treatment with a statin for an average of 4 months, the level of erectile function increased 3.4-fold compared with patients in the placebo group. "This benefit is clinically relevant. It’s about a third of what’s achieved with a phosphodiesterase type 5 inhibitor, and perhaps slightly more than what is achieved with nondrug treatments," Dr. Kostis said. The analyses he and his associate ran did not examine the impact of individual statin types or dosages, and he said that even though the study combined results from 11 studies, the overall number of patients remained relatively small.
Dr. Kostis noted that three actions of statins may contribute to the effect seen. Improved erectile function could result from the cholesterol-lowering effect and from their pleiotropic effects, such as boosting blood levels of nitric oxide, acting as antioxidants, and in general improving vascular function. "Erectile dysfunction is also endothelial dysfunction," Dr. Kostis said. Statin treatment may also worsen erectile function by reducing testosterone production as a consequence of reduced cholesterol. The net effect of statins likely results from all three of these pathways.
Dr. Kostis said that he had no relevant financial disclosures. Dr. Kuvin had no financial disclosures.
On Twitter @mitchelzoler
Men with erectile dysfunction had a better than threefold improvement in erectile function after receiving statin treatment compared with men randomized to placebo in a meta-analysis of 11 controlled studies with a total of more than 600 patients.
Although the result suggests that statin treatment helps men with erectile dysfunction (ED), the evidence is not strong enough to warrant starting statin treatment when the patient has no established indication for the drug, said Dr. John B. Kostis, who summarized the results of the study during a webcast held prior to the annual meeting of the American College of Cardiology, where he is presenting the data.
He called for a multifactorial trial that could assess the roles for statin, treatment, a phosphodiesterase type 5 inhibitor,and testosterone treatment both individually and in combinations.
"Erectile dysfunction is the canary in the coal mine. It can be the first sign of cardiovascular disease," said Dr. Kostis, John G. Detwiler professor of cardiology, professor of medicine and pharmacology, and director of the Cardiovascular Institute at the Rutgers Robert Wood Johnson Medical School in New Brunswick, N.J. "When a man has erectile dysfunction and no explanation like prostatectomy, he should be evaluated for his cardiovascular risk and treated with a statin if that’s justified," he said during the webcast.
"Over the years it’s become apparent that erectile dysfunction is an indication of reduced vascular health in men, and it is considered by many to be a significant cardiovascular disease risk factor," commented Dr. Jeffrey T. Kuvin of the division of cardiology at Tufts Medical Center in Boston. "The results of this meta-analysis strongly show that statin treatment improves erectile dysfunction after only a short duration of treatment. Whether erectile dysfunction improves because of reduced levels of low-density lipoprotein cholesterol or other pleiotropic effects of statins remains unclear," said Dr. Kuvin.
Dr. Kostis and his associate reviewed 11 randomized controlled trials that compared the effect of statin treatment on erectile function with placebo as a primary or secondary endpoint. The 11 studies involved a total of 647 men with ED and an average age of 58 years. The average duration of treatment was 4 months. On statin treatment the average level of LDL cholesterol fell from 138 mg/dL at baseline to 91 mg/dL, while men on placebo had no meaningful change in their LDL cholesterol level.
The meta-analysis showed that after treatment with a statin for an average of 4 months, the level of erectile function increased 3.4-fold compared with patients in the placebo group. "This benefit is clinically relevant. It’s about a third of what’s achieved with a phosphodiesterase type 5 inhibitor, and perhaps slightly more than what is achieved with nondrug treatments," Dr. Kostis said. The analyses he and his associate ran did not examine the impact of individual statin types or dosages, and he said that even though the study combined results from 11 studies, the overall number of patients remained relatively small.
Dr. Kostis noted that three actions of statins may contribute to the effect seen. Improved erectile function could result from the cholesterol-lowering effect and from their pleiotropic effects, such as boosting blood levels of nitric oxide, acting as antioxidants, and in general improving vascular function. "Erectile dysfunction is also endothelial dysfunction," Dr. Kostis said. Statin treatment may also worsen erectile function by reducing testosterone production as a consequence of reduced cholesterol. The net effect of statins likely results from all three of these pathways.
Dr. Kostis said that he had no relevant financial disclosures. Dr. Kuvin had no financial disclosures.
On Twitter @mitchelzoler
Men with erectile dysfunction had a better than threefold improvement in erectile function after receiving statin treatment compared with men randomized to placebo in a meta-analysis of 11 controlled studies with a total of more than 600 patients.
Although the result suggests that statin treatment helps men with erectile dysfunction (ED), the evidence is not strong enough to warrant starting statin treatment when the patient has no established indication for the drug, said Dr. John B. Kostis, who summarized the results of the study during a webcast held prior to the annual meeting of the American College of Cardiology, where he is presenting the data.
He called for a multifactorial trial that could assess the roles for statin, treatment, a phosphodiesterase type 5 inhibitor,and testosterone treatment both individually and in combinations.
"Erectile dysfunction is the canary in the coal mine. It can be the first sign of cardiovascular disease," said Dr. Kostis, John G. Detwiler professor of cardiology, professor of medicine and pharmacology, and director of the Cardiovascular Institute at the Rutgers Robert Wood Johnson Medical School in New Brunswick, N.J. "When a man has erectile dysfunction and no explanation like prostatectomy, he should be evaluated for his cardiovascular risk and treated with a statin if that’s justified," he said during the webcast.
"Over the years it’s become apparent that erectile dysfunction is an indication of reduced vascular health in men, and it is considered by many to be a significant cardiovascular disease risk factor," commented Dr. Jeffrey T. Kuvin of the division of cardiology at Tufts Medical Center in Boston. "The results of this meta-analysis strongly show that statin treatment improves erectile dysfunction after only a short duration of treatment. Whether erectile dysfunction improves because of reduced levels of low-density lipoprotein cholesterol or other pleiotropic effects of statins remains unclear," said Dr. Kuvin.
Dr. Kostis and his associate reviewed 11 randomized controlled trials that compared the effect of statin treatment on erectile function with placebo as a primary or secondary endpoint. The 11 studies involved a total of 647 men with ED and an average age of 58 years. The average duration of treatment was 4 months. On statin treatment the average level of LDL cholesterol fell from 138 mg/dL at baseline to 91 mg/dL, while men on placebo had no meaningful change in their LDL cholesterol level.
The meta-analysis showed that after treatment with a statin for an average of 4 months, the level of erectile function increased 3.4-fold compared with patients in the placebo group. "This benefit is clinically relevant. It’s about a third of what’s achieved with a phosphodiesterase type 5 inhibitor, and perhaps slightly more than what is achieved with nondrug treatments," Dr. Kostis said. The analyses he and his associate ran did not examine the impact of individual statin types or dosages, and he said that even though the study combined results from 11 studies, the overall number of patients remained relatively small.
Dr. Kostis noted that three actions of statins may contribute to the effect seen. Improved erectile function could result from the cholesterol-lowering effect and from their pleiotropic effects, such as boosting blood levels of nitric oxide, acting as antioxidants, and in general improving vascular function. "Erectile dysfunction is also endothelial dysfunction," Dr. Kostis said. Statin treatment may also worsen erectile function by reducing testosterone production as a consequence of reduced cholesterol. The net effect of statins likely results from all three of these pathways.
Dr. Kostis said that he had no relevant financial disclosures. Dr. Kuvin had no financial disclosures.
On Twitter @mitchelzoler
AT ACC 14
Major finding: Statin-treated patients had a statistically-significant 3.4-fold increased rate of improved erectile function compared with men on placebo.
Data source: A meta-analysis of 11 randomized controlled studies with 647 patients.
Disclosures: Dr. Kostis said he had no relevant financial disclosures. Dr. Kuvin had no financial disclosures.

AAN calls oral cannabinoids effective for MS pain, spasticity
An expert panel organized by the American Academy of Neurology called oral cannabis extract the only complementary and alternative medicine unequivocally effective for helping patients with multiple sclerosis, specifically easing their pain and symptoms of spasticity, possibly for as long as 1 year of treatment.
The academy’s Guideline Development Subcommittee also found existing evidence "insufficient to support or refute the effectiveness" of 25 other complementary and alternative medicine (CAM) treatments, including acupuncture, chelation therapy, mindfulness training, and muscle-relaxation therapy. The panel noted that two of these inadequately assessed treatments – dental amalgam removal and transdermal histamine – have received substantial media attention despite having "little or no evidence to support recommendations."

Aside from various forms and delivery methods for cannabinoids, the nine-member panel found six other treatments with adequate evidence to develop practice recommendations that either endorsed their efficacy or lack of effect. Ginkgo biloba, reflexology, and magnetic therapy all had some proven level of efficacy, while bee venom, low-fat diet with omega-3 supplementation, and lofepramine plus L-phenylalanine with B12 were all found ineffective, the subcommittee said in guidelines released on March 24 (Neurology 2014;82:1083-92).
The efficacy of CAM therapies in patients with multiple sclerosis (MS) is an important clinical issue. Ten reports cited by the subcommittee and published during 1999-2009 documented that anywhere from a third to 80% of MS patients – particularly women, patients with higher education levels, and patients who report poorer health – used one or more CAM therapies, according to the panel, which was led by Dr. Vijayshree Yadav of the department of neurology at Oregon Health and Science University, Portland, and the Portland VA Medical Center.
The group also determined that oral cannabis extract and another orally delivered cannabinoid, synthetic tetrahydrocannabinol (THC), were possibly effective for reducing symptoms and objective measures of spasticity during treatment beyond 1 year, and that THC is probably effective for reducing symptoms of spasticity and pain during the first year of treatment. The panel decided that, based on existing evidence, both of these oral agents are "probably ineffective" for reducing both objective spasticity measures and MS-related tremor symptoms.
The subcommittee reviewed two other delivery forms of cannabinoids. The members concluded that Sativex oromucosal cannabinoid spray is probably effective for improving subjective spasticity symptoms for periods of 5-10 weeks and possibly ineffective when used for longer periods or for reducing MS-related tremor. When it came to smoked cannabis, the panel decided that the data were inadequate to draw any conclusions on safety or efficacy.
It also deemed the evidence inadequate to draw conclusions about oral cannabis extract or THC for bladder-urge incontinence or for treating overall symptoms; synthetic THC for central neuropathic pain; and Sativex spray for overall bladder symptoms, anxiety, sleep problems, cognitive symptoms, quality of life, or fatigue.
In addition, cannabinoid studies have been of short duration (6-15 weeks), and central side effects may have caused unblinding in studies. The panel cautioned clinicians to counsel patients about potential psychopathologic effects, cognitive effects, or both with cannabinoid use, and cautioned against extrapolating from findings with standardized oral cannabis extract to other, nonstandardized cannabis extracts.
For other treatments with an adequate evidence base, the panel concluded that magnetic therapy is probably effective for reducing fatigue and probably ineffective for reducing depression, with inadequate data to support or refute other effects in MS patients.
The subcommittee said that study findings established Ginkgo biloba as ineffective for improving cognitive function in patients with MS but possibly effective during 4 weeks of treatment to reduce fatigue. The members also warned that Ginkgo biloba and other supplements not regulated by the Food and Drug Administration may vary considerably in efficacy and adverse effects and may interact with other medications, especially disease-modifying therapies for MS.
The panel called low-fat diet with omega-3 fatty acid supplementation probably ineffective for reducing MS relapses, disability, or MRI lesions, or for improving fatigue or quality of life. It found lofepramine plus L-phenylalanine and vitamin B12 possibly ineffective for treating disability, symptoms, depression, or fatigue, and bee-sting therapy possibly ineffective for reducing relapses, disability, fatigue, total MRI-lesion burden, and gadolinium-enhancing lesion volume, or for improving health-related quality of life.
The subcommittee said that reflexology is possibly effective for reducing MS-associated paresthesia during 11 weeks of treatment, but that data were inadequate to support or refute its use for pain, spasticity, fatigue, anxiety, or several other MS manifestations.
The guidelines were funded by the American Academy of Neurology. Most of the panel members reported some potential conflicts of interest in relationships with pharmaceutical companies that market drugs for MS as well as ties to MS medical societies.
On Twitter @mitchelzoler
An expert panel organized by the American Academy of Neurology called oral cannabis extract the only complementary and alternative medicine unequivocally effective for helping patients with multiple sclerosis, specifically easing their pain and symptoms of spasticity, possibly for as long as 1 year of treatment.
The academy’s Guideline Development Subcommittee also found existing evidence "insufficient to support or refute the effectiveness" of 25 other complementary and alternative medicine (CAM) treatments, including acupuncture, chelation therapy, mindfulness training, and muscle-relaxation therapy. The panel noted that two of these inadequately assessed treatments – dental amalgam removal and transdermal histamine – have received substantial media attention despite having "little or no evidence to support recommendations."
Aside from various forms and delivery methods for cannabinoids, the nine-member panel found six other treatments with adequate evidence to develop practice recommendations that either endorsed their efficacy or lack of effect. Ginkgo biloba, reflexology, and magnetic therapy all had some proven level of efficacy, while bee venom, low-fat diet with omega-3 supplementation, and lofepramine plus L-phenylalanine with B12 were all found ineffective, the subcommittee said in guidelines released on March 24 (Neurology 2014;82:1083-92).
The efficacy of CAM therapies in patients with multiple sclerosis (MS) is an important clinical issue. Ten reports cited by the subcommittee and published during 1999-2009 documented that anywhere from a third to 80% of MS patients – particularly women, patients with higher education levels, and patients who report poorer health – used one or more CAM therapies, according to the panel, which was led by Dr. Vijayshree Yadav of the department of neurology at Oregon Health and Science University, Portland, and the Portland VA Medical Center.
The group also determined that oral cannabis extract and another orally delivered cannabinoid, synthetic tetrahydrocannabinol (THC), were possibly effective for reducing symptoms and objective measures of spasticity during treatment beyond 1 year, and that THC is probably effective for reducing symptoms of spasticity and pain during the first year of treatment. The panel decided that, based on existing evidence, both of these oral agents are "probably ineffective" for reducing both objective spasticity measures and MS-related tremor symptoms.
The subcommittee reviewed two other delivery forms of cannabinoids. The members concluded that Sativex oromucosal cannabinoid spray is probably effective for improving subjective spasticity symptoms for periods of 5-10 weeks and possibly ineffective when used for longer periods or for reducing MS-related tremor. When it came to smoked cannabis, the panel decided that the data were inadequate to draw any conclusions on safety or efficacy.
It also deemed the evidence inadequate to draw conclusions about oral cannabis extract or THC for bladder-urge incontinence or for treating overall symptoms; synthetic THC for central neuropathic pain; and Sativex spray for overall bladder symptoms, anxiety, sleep problems, cognitive symptoms, quality of life, or fatigue.
In addition, cannabinoid studies have been of short duration (6-15 weeks), and central side effects may have caused unblinding in studies. The panel cautioned clinicians to counsel patients about potential psychopathologic effects, cognitive effects, or both with cannabinoid use, and cautioned against extrapolating from findings with standardized oral cannabis extract to other, nonstandardized cannabis extracts.
For other treatments with an adequate evidence base, the panel concluded that magnetic therapy is probably effective for reducing fatigue and probably ineffective for reducing depression, with inadequate data to support or refute other effects in MS patients.
The subcommittee said that study findings established Ginkgo biloba as ineffective for improving cognitive function in patients with MS but possibly effective during 4 weeks of treatment to reduce fatigue. The members also warned that Ginkgo biloba and other supplements not regulated by the Food and Drug Administration may vary considerably in efficacy and adverse effects and may interact with other medications, especially disease-modifying therapies for MS.
The panel called low-fat diet with omega-3 fatty acid supplementation probably ineffective for reducing MS relapses, disability, or MRI lesions, or for improving fatigue or quality of life. It found lofepramine plus L-phenylalanine and vitamin B12 possibly ineffective for treating disability, symptoms, depression, or fatigue, and bee-sting therapy possibly ineffective for reducing relapses, disability, fatigue, total MRI-lesion burden, and gadolinium-enhancing lesion volume, or for improving health-related quality of life.
The subcommittee said that reflexology is possibly effective for reducing MS-associated paresthesia during 11 weeks of treatment, but that data were inadequate to support or refute its use for pain, spasticity, fatigue, anxiety, or several other MS manifestations.
The guidelines were funded by the American Academy of Neurology. Most of the panel members reported some potential conflicts of interest in relationships with pharmaceutical companies that market drugs for MS as well as ties to MS medical societies.
On Twitter @mitchelzoler
An expert panel organized by the American Academy of Neurology called oral cannabis extract the only complementary and alternative medicine unequivocally effective for helping patients with multiple sclerosis, specifically easing their pain and symptoms of spasticity, possibly for as long as 1 year of treatment.
The academy’s Guideline Development Subcommittee also found existing evidence "insufficient to support or refute the effectiveness" of 25 other complementary and alternative medicine (CAM) treatments, including acupuncture, chelation therapy, mindfulness training, and muscle-relaxation therapy. The panel noted that two of these inadequately assessed treatments – dental amalgam removal and transdermal histamine – have received substantial media attention despite having "little or no evidence to support recommendations."
Aside from various forms and delivery methods for cannabinoids, the nine-member panel found six other treatments with adequate evidence to develop practice recommendations that either endorsed their efficacy or lack of effect. Ginkgo biloba, reflexology, and magnetic therapy all had some proven level of efficacy, while bee venom, low-fat diet with omega-3 supplementation, and lofepramine plus L-phenylalanine with B12 were all found ineffective, the subcommittee said in guidelines released on March 24 (Neurology 2014;82:1083-92).
The efficacy of CAM therapies in patients with multiple sclerosis (MS) is an important clinical issue. Ten reports cited by the subcommittee and published during 1999-2009 documented that anywhere from a third to 80% of MS patients – particularly women, patients with higher education levels, and patients who report poorer health – used one or more CAM therapies, according to the panel, which was led by Dr. Vijayshree Yadav of the department of neurology at Oregon Health and Science University, Portland, and the Portland VA Medical Center.
The group also determined that oral cannabis extract and another orally delivered cannabinoid, synthetic tetrahydrocannabinol (THC), were possibly effective for reducing symptoms and objective measures of spasticity during treatment beyond 1 year, and that THC is probably effective for reducing symptoms of spasticity and pain during the first year of treatment. The panel decided that, based on existing evidence, both of these oral agents are "probably ineffective" for reducing both objective spasticity measures and MS-related tremor symptoms.
The subcommittee reviewed two other delivery forms of cannabinoids. The members concluded that Sativex oromucosal cannabinoid spray is probably effective for improving subjective spasticity symptoms for periods of 5-10 weeks and possibly ineffective when used for longer periods or for reducing MS-related tremor. When it came to smoked cannabis, the panel decided that the data were inadequate to draw any conclusions on safety or efficacy.
It also deemed the evidence inadequate to draw conclusions about oral cannabis extract or THC for bladder-urge incontinence or for treating overall symptoms; synthetic THC for central neuropathic pain; and Sativex spray for overall bladder symptoms, anxiety, sleep problems, cognitive symptoms, quality of life, or fatigue.
In addition, cannabinoid studies have been of short duration (6-15 weeks), and central side effects may have caused unblinding in studies. The panel cautioned clinicians to counsel patients about potential psychopathologic effects, cognitive effects, or both with cannabinoid use, and cautioned against extrapolating from findings with standardized oral cannabis extract to other, nonstandardized cannabis extracts.
For other treatments with an adequate evidence base, the panel concluded that magnetic therapy is probably effective for reducing fatigue and probably ineffective for reducing depression, with inadequate data to support or refute other effects in MS patients.
The subcommittee said that study findings established Ginkgo biloba as ineffective for improving cognitive function in patients with MS but possibly effective during 4 weeks of treatment to reduce fatigue. The members also warned that Ginkgo biloba and other supplements not regulated by the Food and Drug Administration may vary considerably in efficacy and adverse effects and may interact with other medications, especially disease-modifying therapies for MS.
The panel called low-fat diet with omega-3 fatty acid supplementation probably ineffective for reducing MS relapses, disability, or MRI lesions, or for improving fatigue or quality of life. It found lofepramine plus L-phenylalanine and vitamin B12 possibly ineffective for treating disability, symptoms, depression, or fatigue, and bee-sting therapy possibly ineffective for reducing relapses, disability, fatigue, total MRI-lesion burden, and gadolinium-enhancing lesion volume, or for improving health-related quality of life.
The subcommittee said that reflexology is possibly effective for reducing MS-associated paresthesia during 11 weeks of treatment, but that data were inadequate to support or refute its use for pain, spasticity, fatigue, anxiety, or several other MS manifestations.
The guidelines were funded by the American Academy of Neurology. Most of the panel members reported some potential conflicts of interest in relationships with pharmaceutical companies that market drugs for MS as well as ties to MS medical societies.
On Twitter @mitchelzoler
FROM NEUROLOGY

Pazopanib helps significant minority of sarcoma patients
MILAN – Pazopanib, which received U.S. approval in 2012 for treating advanced soft-tissue sarcomas, can be very effective for durably halting tumor progression in a significant minority of sarcoma patients but requires close monitoring for adverse effects.
"Although the overall response rate is low, some patients experience important palliation of symptoms and prolonged disease control" from treatment with pazotinib (Votrient), Dr. Ian R. Judson said at Sarcoma and GIST 2014, hosted by the European Society for Medical Oncology.
"We see a number of adverse effects [from pazopanib treatment] that need careful monitoring, particularly fatigue, diarrhea, nausea, and weight loss," adverse effects that had previously been seen in patients with other tumor types treated with the drug, said Dr. Judson, professor of cancer pharmacology and head of the sarcoma unit at the Royal Marsden Hospital, London. Results from the phase III trial of pazopanib in patients with advanced soft-tissue sarcoma with a history of chemotherapy, also showed that the drug can cause other, previously unreported adverse effects: myocardial dysfunction, an increased risk for venous thromboembolism, and the possibility for some patients to develop pneumothorax (Lancet 2012;379:1879-86).
The upside of pazopanib treatment is that it can produce "clear and dramatic" increases in progression-free survival and "durable, stable disease" in certain patients, said Dr. Judson.
Pazopanib became the first tyrosine kinase inhibitor to receive approval from the Food and Drug Administration and other regulatory agencies for treating soft tissue sarcomas (STS), although it has not been proven effective for treating adipocyte STS and is also not indicated for gastrointestinal stromal tumors. But it remains unclear which patients with other types of STS will respond to pazopanib and which won’t. "I wish we knew what the molecular target for this drug really is," Dr. Judson said.

A recently published analysis retrospectively pooled data from 118 STS patients enrolled in a phase II study of pazopanib and 226 patients from the phase III study PALETTE (Pazopanib for Metastatic Soft Tissue Sarcoma). The analysis showed that 36% of the entire group of patients on pazopanib were long-term responders to the drug, defined as having progression-free survival for at least 6 months following the start of pazopanib treatment, and 34% of patients on the drug were long-term survivors on the drug, defined as living for at least 18 months on treatment, noted Dr. Shreyaskumar R. Patel in a talk at the conference (Ann. Oncol. 2014;25:719-24).
During an overall median follow-up of 2.3 years in the two studies, 76 patients (22%) were both long-term responders and long-term survivors. Twelve patients remained on pazopanib treatment for more than 2 years, with a median time on treatment of 2.4 years, and 1 patient from the combined groups stayed on pazopanib for as long as 3.7 years, said Dr. Patel, professor and deputy chair of the department of sarcoma medical oncology at M.D. Anderson Cancer Center, Houston.
"Pazopanib is probably my second-line choice" for treating advanced STS, "particularly synovial sarcomas" after treatment with doxorubicin (Adriamycin) and ifosfamide (Ifex) fails, said Dr. Robert S. Benjamin, professor and chair of sarcoma medical oncology at M.D. Anderson.
The pazopanib trials were sponsored by GlaxoSmithKline, which markets pazopanib. Dr. Judson said that he has received honoraria from GlaxoSmithKline and Novartis, and research support from GlaxoSmithKline, AstraZeneca, and other companies. Dr. Patel said that he has received honoraria or consulting fees from GlaxoSmithKline, Novartis, and Johnson & Johnson, and research support from Johnson & Johnson, PharmaMar, and other companies. Dr. Benjamin said that he has received research support from Johnson & Johnson, Merck, and Pfizer.
mzoler@frontlinemedcom.com Twitter: @mitchelzoler
MILAN – Pazopanib, which received U.S. approval in 2012 for treating advanced soft-tissue sarcomas, can be very effective for durably halting tumor progression in a significant minority of sarcoma patients but requires close monitoring for adverse effects.
"Although the overall response rate is low, some patients experience important palliation of symptoms and prolonged disease control" from treatment with pazotinib (Votrient), Dr. Ian R. Judson said at Sarcoma and GIST 2014, hosted by the European Society for Medical Oncology.
"We see a number of adverse effects [from pazopanib treatment] that need careful monitoring, particularly fatigue, diarrhea, nausea, and weight loss," adverse effects that had previously been seen in patients with other tumor types treated with the drug, said Dr. Judson, professor of cancer pharmacology and head of the sarcoma unit at the Royal Marsden Hospital, London. Results from the phase III trial of pazopanib in patients with advanced soft-tissue sarcoma with a history of chemotherapy, also showed that the drug can cause other, previously unreported adverse effects: myocardial dysfunction, an increased risk for venous thromboembolism, and the possibility for some patients to develop pneumothorax (Lancet 2012;379:1879-86).
The upside of pazopanib treatment is that it can produce "clear and dramatic" increases in progression-free survival and "durable, stable disease" in certain patients, said Dr. Judson.
Pazopanib became the first tyrosine kinase inhibitor to receive approval from the Food and Drug Administration and other regulatory agencies for treating soft tissue sarcomas (STS), although it has not been proven effective for treating adipocyte STS and is also not indicated for gastrointestinal stromal tumors. But it remains unclear which patients with other types of STS will respond to pazopanib and which won’t. "I wish we knew what the molecular target for this drug really is," Dr. Judson said.
A recently published analysis retrospectively pooled data from 118 STS patients enrolled in a phase II study of pazopanib and 226 patients from the phase III study PALETTE (Pazopanib for Metastatic Soft Tissue Sarcoma). The analysis showed that 36% of the entire group of patients on pazopanib were long-term responders to the drug, defined as having progression-free survival for at least 6 months following the start of pazopanib treatment, and 34% of patients on the drug were long-term survivors on the drug, defined as living for at least 18 months on treatment, noted Dr. Shreyaskumar R. Patel in a talk at the conference (Ann. Oncol. 2014;25:719-24).
During an overall median follow-up of 2.3 years in the two studies, 76 patients (22%) were both long-term responders and long-term survivors. Twelve patients remained on pazopanib treatment for more than 2 years, with a median time on treatment of 2.4 years, and 1 patient from the combined groups stayed on pazopanib for as long as 3.7 years, said Dr. Patel, professor and deputy chair of the department of sarcoma medical oncology at M.D. Anderson Cancer Center, Houston.
"Pazopanib is probably my second-line choice" for treating advanced STS, "particularly synovial sarcomas" after treatment with doxorubicin (Adriamycin) and ifosfamide (Ifex) fails, said Dr. Robert S. Benjamin, professor and chair of sarcoma medical oncology at M.D. Anderson.
The pazopanib trials were sponsored by GlaxoSmithKline, which markets pazopanib. Dr. Judson said that he has received honoraria from GlaxoSmithKline and Novartis, and research support from GlaxoSmithKline, AstraZeneca, and other companies. Dr. Patel said that he has received honoraria or consulting fees from GlaxoSmithKline, Novartis, and Johnson & Johnson, and research support from Johnson & Johnson, PharmaMar, and other companies. Dr. Benjamin said that he has received research support from Johnson & Johnson, Merck, and Pfizer.
mzoler@frontlinemedcom.com Twitter: @mitchelzoler
MILAN – Pazopanib, which received U.S. approval in 2012 for treating advanced soft-tissue sarcomas, can be very effective for durably halting tumor progression in a significant minority of sarcoma patients but requires close monitoring for adverse effects.
"Although the overall response rate is low, some patients experience important palliation of symptoms and prolonged disease control" from treatment with pazotinib (Votrient), Dr. Ian R. Judson said at Sarcoma and GIST 2014, hosted by the European Society for Medical Oncology.
"We see a number of adverse effects [from pazopanib treatment] that need careful monitoring, particularly fatigue, diarrhea, nausea, and weight loss," adverse effects that had previously been seen in patients with other tumor types treated with the drug, said Dr. Judson, professor of cancer pharmacology and head of the sarcoma unit at the Royal Marsden Hospital, London. Results from the phase III trial of pazopanib in patients with advanced soft-tissue sarcoma with a history of chemotherapy, also showed that the drug can cause other, previously unreported adverse effects: myocardial dysfunction, an increased risk for venous thromboembolism, and the possibility for some patients to develop pneumothorax (Lancet 2012;379:1879-86).
The upside of pazopanib treatment is that it can produce "clear and dramatic" increases in progression-free survival and "durable, stable disease" in certain patients, said Dr. Judson.
Pazopanib became the first tyrosine kinase inhibitor to receive approval from the Food and Drug Administration and other regulatory agencies for treating soft tissue sarcomas (STS), although it has not been proven effective for treating adipocyte STS and is also not indicated for gastrointestinal stromal tumors. But it remains unclear which patients with other types of STS will respond to pazopanib and which won’t. "I wish we knew what the molecular target for this drug really is," Dr. Judson said.
A recently published analysis retrospectively pooled data from 118 STS patients enrolled in a phase II study of pazopanib and 226 patients from the phase III study PALETTE (Pazopanib for Metastatic Soft Tissue Sarcoma). The analysis showed that 36% of the entire group of patients on pazopanib were long-term responders to the drug, defined as having progression-free survival for at least 6 months following the start of pazopanib treatment, and 34% of patients on the drug were long-term survivors on the drug, defined as living for at least 18 months on treatment, noted Dr. Shreyaskumar R. Patel in a talk at the conference (Ann. Oncol. 2014;25:719-24).
During an overall median follow-up of 2.3 years in the two studies, 76 patients (22%) were both long-term responders and long-term survivors. Twelve patients remained on pazopanib treatment for more than 2 years, with a median time on treatment of 2.4 years, and 1 patient from the combined groups stayed on pazopanib for as long as 3.7 years, said Dr. Patel, professor and deputy chair of the department of sarcoma medical oncology at M.D. Anderson Cancer Center, Houston.
"Pazopanib is probably my second-line choice" for treating advanced STS, "particularly synovial sarcomas" after treatment with doxorubicin (Adriamycin) and ifosfamide (Ifex) fails, said Dr. Robert S. Benjamin, professor and chair of sarcoma medical oncology at M.D. Anderson.
The pazopanib trials were sponsored by GlaxoSmithKline, which markets pazopanib. Dr. Judson said that he has received honoraria from GlaxoSmithKline and Novartis, and research support from GlaxoSmithKline, AstraZeneca, and other companies. Dr. Patel said that he has received honoraria or consulting fees from GlaxoSmithKline, Novartis, and Johnson & Johnson, and research support from Johnson & Johnson, PharmaMar, and other companies. Dr. Benjamin said that he has received research support from Johnson & Johnson, Merck, and Pfizer.
mzoler@frontlinemedcom.com Twitter: @mitchelzoler
EXPERT ANALYSIS FROM SARCOMA AND GIST 2014

CoreValve holds size advantage for U.S. TAVR
When the Food and Drug Administration in January granted marketing approval to a second transcatheter aortic valve replacement system for inoperable patients with aortic stenosis, the CoreValve marketed by Medtronic, the new valve conceded a greater than 2-year head start to the first system on the U.S. market, Sapien marketed by Edwards.
But cardiologists see that 2-year edge in familiarity eclipsed for at least some patients by two major advantages that CoreValve currently holds over Sapien: delivery via a significantly thinner sheath, and the option of larger-diameter valves that allow replacement in patients with a wider aortic annulus.
The CoreValve delivery sheath is 18 French, compared with a 22F or 24F size for the Sapien transcatheter aortic valve replacement (TAVR) with U.S. approval, and the Sapien valves come in diameters of 23 and 26 mm, compared with options of 23, 26, 29, and 31 mm for the CoreValve.
"CoreValve is the device of choice for patients with smaller vessel sizes. Sapien has been a wonderful device to use, and we have so much experience with it, but the smaller CoreValve size will allow many more patients to be done with a transfemoral approach," said Dr. Peter C. Block, a professor of medicine at Emory University in Atlanta and an interventional cardiologist who performs TAVR.
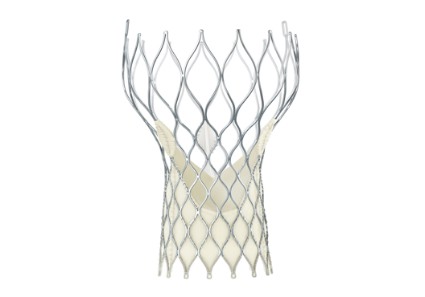
"More patients will qualify for TAVR and will be treated transfemorally with the larger valve diameters and smaller sheath size," agreed Dr. Mauricio G. Cohen, director of cardiac catheterization at the University of Miami and TAVR interventionalist. Another potential positive of having CoreValve on the U.S. market is that competition between the two options will likely drive down the cost of a TAVR system, which until now has run more than $30,000, Dr. Cohen said in an interview.
CoreValve received FDA approval less than 3 months after researchers first reported data from the company’s U.S. Pivotal Trial Extreme Risk Iliofemoral Study last October at the TCT (Transcatheter Cardiovascular Therapeutics) annual meeting. In that study, 471 inoperable aortic stenosis patients had a 26% 1-year rate of death or major stroke, substantially surpassing the 43% rate that the study set up as the target for superiority, reported Dr. Jeffrey J. Popma, lead investigator on the study.
Dr. Popma warned against comparing CoreValve’s efficacy and safety performance in the trial and the Sapien system’s performance in its pivotal trial in inoperable patients, the PARTNER cohort B trial (N. Engl. J. Med. 2010;363:1597-607). "It’s very difficult to make cross-trial comparison," he said in an interview, a limitation also noted by Dr. Cohen and Dr. Block. But Dr. Popma highlighted the 2.4% 30-day stroke rate in the pivotal trial, and a 1.8% 30-day stroke rate seen with CoreValve in inoperable patients in a continued access program at the trial’s study sites. He also highlighted the 11% rate of moderate paravalvular aortic regurgitation after 30 days that dropped to a 4% rate after 1 year.
Perhaps the biggest downside of CoreValve’s performance in the pivotal trial was that 22% of patients required a permanent pacemaker implant within the first 30 days, increasing to 27% of patients with 1-year follow-up. Increased risk for a pacemaker is an inherent downside of CoreValve because of its longer size compared with the Sapien valve and how the CoreValve sits in the aortic annulus. The CoreValve is designed for supravalvular placement and anchoring in the left ventricular outflow tract near the left bundle branch that can result in mechanical irritation and arrhythmia with the need for pacing, explained Dr. Popma, professor of medicine at Harvard University and an interventional cardiologist at Beth Israel Deaconess Medical Center, Boston.
"I think our pacemaker rate was very acceptable. I don’t think it will ever be as low as with Sapien, but it’s a worthwhile trade-off because the CoreValve functions well and results in a low rate of paravalvular regurgitation," he said.
Dr. Popma also stressed that 1-year mortality was no greater among the patients who required a pacemaker implant in the pivotal trial. A subgroup analysis of results from the trial to try to identify which patients had the greatest risk for needing a pacemaker after a CoreValve implant has not yet finished, he said. It’s possible that certain patients with preexisting conduction abnormalities, such as a right bundle branch block coupled with a left anterior fascicular block, have the greatest vulnerability.
Patients for whom the Sapien system remains ideal are those with a narrow sinus of Valsalva, because the longer CoreValve frame crosses the sinus and may compromise coronary blood flow in patients with a narrow sinus, Dr. Popma said.
The choice between CoreValve and Sapien systems will grow even more complicated for U.S. cardiologists and surgeons when the Sapien XT valve system receives FDA marketing approval, likely later this year. The Sapien XT delivery sheath matches the 18F size of CoreValve and will also come in a 29-mm size, blunting two of CoreValves main advantages.
Medtronic also faces charges of patent infringement by its CoreValve in a court case initiated by Edwards. In mid-January, a jury in a U.S. District Court assessed a penalty of $394 million against Medtronic. Edwards is also seeking a court-ordered halt to U.S. marketing of CoreValve. But Medtronic is appealing the jury verdict and continues to fight the injunction, and a company spokesperson said in an interview that the legal maneuverings will likely take at least another year to fully resolve. In the meantime, Medtronic began U.S. distribution of the CoreValve on Jan. 17.
Dr. Block said that his institution received a research grant to participate in Sapien trials. Dr. Cohen said that he has been a consultant to Medtronic and Edwards. Dr. Popma said that his institution received research support from Medtronic and that he has been a consultant to and received research support from Boston Scientific.
When the Food and Drug Administration in January granted marketing approval to a second transcatheter aortic valve replacement system for inoperable patients with aortic stenosis, the CoreValve marketed by Medtronic, the new valve conceded a greater than 2-year head start to the first system on the U.S. market, Sapien marketed by Edwards.
But cardiologists see that 2-year edge in familiarity eclipsed for at least some patients by two major advantages that CoreValve currently holds over Sapien: delivery via a significantly thinner sheath, and the option of larger-diameter valves that allow replacement in patients with a wider aortic annulus.
The CoreValve delivery sheath is 18 French, compared with a 22F or 24F size for the Sapien transcatheter aortic valve replacement (TAVR) with U.S. approval, and the Sapien valves come in diameters of 23 and 26 mm, compared with options of 23, 26, 29, and 31 mm for the CoreValve.
"CoreValve is the device of choice for patients with smaller vessel sizes. Sapien has been a wonderful device to use, and we have so much experience with it, but the smaller CoreValve size will allow many more patients to be done with a transfemoral approach," said Dr. Peter C. Block, a professor of medicine at Emory University in Atlanta and an interventional cardiologist who performs TAVR.
"More patients will qualify for TAVR and will be treated transfemorally with the larger valve diameters and smaller sheath size," agreed Dr. Mauricio G. Cohen, director of cardiac catheterization at the University of Miami and TAVR interventionalist. Another potential positive of having CoreValve on the U.S. market is that competition between the two options will likely drive down the cost of a TAVR system, which until now has run more than $30,000, Dr. Cohen said in an interview.
CoreValve received FDA approval less than 3 months after researchers first reported data from the company’s U.S. Pivotal Trial Extreme Risk Iliofemoral Study last October at the TCT (Transcatheter Cardiovascular Therapeutics) annual meeting. In that study, 471 inoperable aortic stenosis patients had a 26% 1-year rate of death or major stroke, substantially surpassing the 43% rate that the study set up as the target for superiority, reported Dr. Jeffrey J. Popma, lead investigator on the study.
Dr. Popma warned against comparing CoreValve’s efficacy and safety performance in the trial and the Sapien system’s performance in its pivotal trial in inoperable patients, the PARTNER cohort B trial (N. Engl. J. Med. 2010;363:1597-607). "It’s very difficult to make cross-trial comparison," he said in an interview, a limitation also noted by Dr. Cohen and Dr. Block. But Dr. Popma highlighted the 2.4% 30-day stroke rate in the pivotal trial, and a 1.8% 30-day stroke rate seen with CoreValve in inoperable patients in a continued access program at the trial’s study sites. He also highlighted the 11% rate of moderate paravalvular aortic regurgitation after 30 days that dropped to a 4% rate after 1 year.
Perhaps the biggest downside of CoreValve’s performance in the pivotal trial was that 22% of patients required a permanent pacemaker implant within the first 30 days, increasing to 27% of patients with 1-year follow-up. Increased risk for a pacemaker is an inherent downside of CoreValve because of its longer size compared with the Sapien valve and how the CoreValve sits in the aortic annulus. The CoreValve is designed for supravalvular placement and anchoring in the left ventricular outflow tract near the left bundle branch that can result in mechanical irritation and arrhythmia with the need for pacing, explained Dr. Popma, professor of medicine at Harvard University and an interventional cardiologist at Beth Israel Deaconess Medical Center, Boston.
"I think our pacemaker rate was very acceptable. I don’t think it will ever be as low as with Sapien, but it’s a worthwhile trade-off because the CoreValve functions well and results in a low rate of paravalvular regurgitation," he said.
Dr. Popma also stressed that 1-year mortality was no greater among the patients who required a pacemaker implant in the pivotal trial. A subgroup analysis of results from the trial to try to identify which patients had the greatest risk for needing a pacemaker after a CoreValve implant has not yet finished, he said. It’s possible that certain patients with preexisting conduction abnormalities, such as a right bundle branch block coupled with a left anterior fascicular block, have the greatest vulnerability.
Patients for whom the Sapien system remains ideal are those with a narrow sinus of Valsalva, because the longer CoreValve frame crosses the sinus and may compromise coronary blood flow in patients with a narrow sinus, Dr. Popma said.
The choice between CoreValve and Sapien systems will grow even more complicated for U.S. cardiologists and surgeons when the Sapien XT valve system receives FDA marketing approval, likely later this year. The Sapien XT delivery sheath matches the 18F size of CoreValve and will also come in a 29-mm size, blunting two of CoreValves main advantages.
Medtronic also faces charges of patent infringement by its CoreValve in a court case initiated by Edwards. In mid-January, a jury in a U.S. District Court assessed a penalty of $394 million against Medtronic. Edwards is also seeking a court-ordered halt to U.S. marketing of CoreValve. But Medtronic is appealing the jury verdict and continues to fight the injunction, and a company spokesperson said in an interview that the legal maneuverings will likely take at least another year to fully resolve. In the meantime, Medtronic began U.S. distribution of the CoreValve on Jan. 17.
Dr. Block said that his institution received a research grant to participate in Sapien trials. Dr. Cohen said that he has been a consultant to Medtronic and Edwards. Dr. Popma said that his institution received research support from Medtronic and that he has been a consultant to and received research support from Boston Scientific.
When the Food and Drug Administration in January granted marketing approval to a second transcatheter aortic valve replacement system for inoperable patients with aortic stenosis, the CoreValve marketed by Medtronic, the new valve conceded a greater than 2-year head start to the first system on the U.S. market, Sapien marketed by Edwards.
But cardiologists see that 2-year edge in familiarity eclipsed for at least some patients by two major advantages that CoreValve currently holds over Sapien: delivery via a significantly thinner sheath, and the option of larger-diameter valves that allow replacement in patients with a wider aortic annulus.
The CoreValve delivery sheath is 18 French, compared with a 22F or 24F size for the Sapien transcatheter aortic valve replacement (TAVR) with U.S. approval, and the Sapien valves come in diameters of 23 and 26 mm, compared with options of 23, 26, 29, and 31 mm for the CoreValve.
"CoreValve is the device of choice for patients with smaller vessel sizes. Sapien has been a wonderful device to use, and we have so much experience with it, but the smaller CoreValve size will allow many more patients to be done with a transfemoral approach," said Dr. Peter C. Block, a professor of medicine at Emory University in Atlanta and an interventional cardiologist who performs TAVR.
"More patients will qualify for TAVR and will be treated transfemorally with the larger valve diameters and smaller sheath size," agreed Dr. Mauricio G. Cohen, director of cardiac catheterization at the University of Miami and TAVR interventionalist. Another potential positive of having CoreValve on the U.S. market is that competition between the two options will likely drive down the cost of a TAVR system, which until now has run more than $30,000, Dr. Cohen said in an interview.
CoreValve received FDA approval less than 3 months after researchers first reported data from the company’s U.S. Pivotal Trial Extreme Risk Iliofemoral Study last October at the TCT (Transcatheter Cardiovascular Therapeutics) annual meeting. In that study, 471 inoperable aortic stenosis patients had a 26% 1-year rate of death or major stroke, substantially surpassing the 43% rate that the study set up as the target for superiority, reported Dr. Jeffrey J. Popma, lead investigator on the study.
Dr. Popma warned against comparing CoreValve’s efficacy and safety performance in the trial and the Sapien system’s performance in its pivotal trial in inoperable patients, the PARTNER cohort B trial (N. Engl. J. Med. 2010;363:1597-607). "It’s very difficult to make cross-trial comparison," he said in an interview, a limitation also noted by Dr. Cohen and Dr. Block. But Dr. Popma highlighted the 2.4% 30-day stroke rate in the pivotal trial, and a 1.8% 30-day stroke rate seen with CoreValve in inoperable patients in a continued access program at the trial’s study sites. He also highlighted the 11% rate of moderate paravalvular aortic regurgitation after 30 days that dropped to a 4% rate after 1 year.
Perhaps the biggest downside of CoreValve’s performance in the pivotal trial was that 22% of patients required a permanent pacemaker implant within the first 30 days, increasing to 27% of patients with 1-year follow-up. Increased risk for a pacemaker is an inherent downside of CoreValve because of its longer size compared with the Sapien valve and how the CoreValve sits in the aortic annulus. The CoreValve is designed for supravalvular placement and anchoring in the left ventricular outflow tract near the left bundle branch that can result in mechanical irritation and arrhythmia with the need for pacing, explained Dr. Popma, professor of medicine at Harvard University and an interventional cardiologist at Beth Israel Deaconess Medical Center, Boston.
"I think our pacemaker rate was very acceptable. I don’t think it will ever be as low as with Sapien, but it’s a worthwhile trade-off because the CoreValve functions well and results in a low rate of paravalvular regurgitation," he said.
Dr. Popma also stressed that 1-year mortality was no greater among the patients who required a pacemaker implant in the pivotal trial. A subgroup analysis of results from the trial to try to identify which patients had the greatest risk for needing a pacemaker after a CoreValve implant has not yet finished, he said. It’s possible that certain patients with preexisting conduction abnormalities, such as a right bundle branch block coupled with a left anterior fascicular block, have the greatest vulnerability.
Patients for whom the Sapien system remains ideal are those with a narrow sinus of Valsalva, because the longer CoreValve frame crosses the sinus and may compromise coronary blood flow in patients with a narrow sinus, Dr. Popma said.
The choice between CoreValve and Sapien systems will grow even more complicated for U.S. cardiologists and surgeons when the Sapien XT valve system receives FDA marketing approval, likely later this year. The Sapien XT delivery sheath matches the 18F size of CoreValve and will also come in a 29-mm size, blunting two of CoreValves main advantages.
Medtronic also faces charges of patent infringement by its CoreValve in a court case initiated by Edwards. In mid-January, a jury in a U.S. District Court assessed a penalty of $394 million against Medtronic. Edwards is also seeking a court-ordered halt to U.S. marketing of CoreValve. But Medtronic is appealing the jury verdict and continues to fight the injunction, and a company spokesperson said in an interview that the legal maneuverings will likely take at least another year to fully resolve. In the meantime, Medtronic began U.S. distribution of the CoreValve on Jan. 17.
Dr. Block said that his institution received a research grant to participate in Sapien trials. Dr. Cohen said that he has been a consultant to Medtronic and Edwards. Dr. Popma said that his institution received research support from Medtronic and that he has been a consultant to and received research support from Boston Scientific.
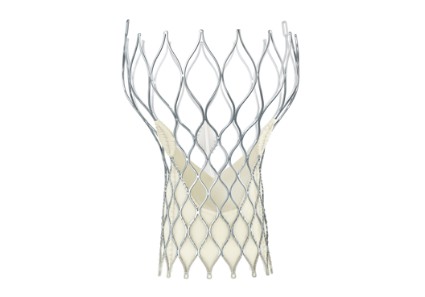
Doxorubicin, ifosfamide combination touted for advanced sarcomas
MILAN – The combination of doxorubicin and ifosfamide is the best treatment for most patients with a sarcoma that does not have a proven targeted therapy, although a recent report from a large randomized study failed to prove that the combination surpassed doxorubicin alone for improving overall survival, said Dr. Robert S. Benjamin.
"From my point of view, this was clearly a positive study, but it was interpreted as a negative study" by the investigators who ran it, Dr. Benjamin said at Sarcoma and GIST 2014, hosted by the European Society for Medical Oncology.
Among patients who received doxorubicin (Adriamycin) plus ifosfamide (Ifex), compared with those randomized to doxorubicin only, "the incidence of progressive disease was cut by more than half, the objective response rate was twice as high, and the time to progression was increased by more than 50%. Overall survival was also better" but did not reach statistical significance. "It was supposed to be 10% better for overall survival [based on the study’s design], but it was only 9% better and they considered that the same as zero," noted Dr. Benjamin, professor and chairman of sarcoma medical oncology at the University of Texas M.D. Anderson Cancer Center, Houston.

"Overall survival is a lousy endpoint," he said. "It measures everything that happens when you treat, but also everything that happens after" treatment is finished. Dr. Benjamin said his center was invited by the organizers of this phase III trial to participate, but he declined because the trial used overall survival as its primary endpoint.
The trial enrolled 455 patients with a locally-advanced, unresectable, or metastatic high-grade soft-tissue sarcoma, aged 18-60 years at 38 centers in nine European countries and Canada. It included patients with more than 10 different sarcoma types including liposarcomas, synovial sarcomas, leiomyosarcomas, undifferentiated pleomorphic sarcomas, and unclassified high-grade sarcomas. The study randomized patients to either doxorubicin alone or intensified doxorubicin plus ifosfamide as first-line treatment. Patients were treated every 3 weeks for up to six cycles and were followed for a median of 56-59 months (Lancet Oncol. 2014 [doi:10.1016/S1470-2045(14)70063-4]).
The study’s primary endpoint, overall survival, averaged 12.8 months in the monotherapy arm and 14.3 months in the dual-agent arm, a hazard ratio of 0.83 that failed to meet statistical significance (P =.076), reported Dr. Ian Judson, professor and head of the sarcoma unit at the Royal Marsden Hospital in London, and his associates. However, median progression-free survival was 4.6 months in the doxorubicin-only arm and 7.4 months for the combined-treatment arm, a statistically significant difference (P = .003). Progressive disease occurred in 32% of the patients on monotherapy and in 13% of those on combined treatment.
The study’s authors concluded that if chemotherapy is used palliatively to relieve acute symptoms, then sequential treatment with single drugs would probably be less toxic without significantly impairing survival. For patients in whom symptoms could be relieved by tumor shrinkage, or if debulking of the tumor before surgery or radiotherapy is the goal, or if delaying disease progression as long as possible is the goal, then combination treatment is justified. They also cautioned against extrapolating the findings to patients older than 60 years, a limitation seconded by Dr. Benjamin. He cautioned against administering the combination to any patient older than 65 years or to patients with substantially impaired renal function.
But he disagreed with using doxorubicin as monotherapy unless tolerance was an issue. "Doxorubicin is a very good drug; it just shouldn’t be used by itself," he said.
Dr. Benjamin offered a few tips for using the doxorubicin and ifosfamide combination in practice: It’s important to use doxorubicin at the recommended dosage of 75 mg/m2. If a patient cannot tolerate that dosage in combination with ifosfamide then the best option is to change to doxorubicin monotherapy to be sure the patient receives 75 mg/m2. He recommended starting patients first on the doxorubicin and ifosfamide combination because it is the most toxic, with a less toxic pairing like gemcitabine (Gemzar) and docetaxel (Dosefirez) as second-line therapy because it’s harder for patients to switch to a more toxic regimen, he said. Dacarbazine also can pair effectively with doxorubicin. This combination is "almost as good as doxorubicin plus ifosfamide and is less toxic." He also noted the efficacy of trabectedin (Yondelis), which is available in European countries but not in the United States.
"These drugs all work, and the patient will need everything you have. You need to put it all together," Dr. Benjamin advised. "All the chemotherapy we currently have is grossly inadequate, but given what we have, use doxorubicin and ifosfamide for most patients with soft tissue sarcomas."
On Twitter @mitchelzoler
MILAN – The combination of doxorubicin and ifosfamide is the best treatment for most patients with a sarcoma that does not have a proven targeted therapy, although a recent report from a large randomized study failed to prove that the combination surpassed doxorubicin alone for improving overall survival, said Dr. Robert S. Benjamin.
"From my point of view, this was clearly a positive study, but it was interpreted as a negative study" by the investigators who ran it, Dr. Benjamin said at Sarcoma and GIST 2014, hosted by the European Society for Medical Oncology.
Among patients who received doxorubicin (Adriamycin) plus ifosfamide (Ifex), compared with those randomized to doxorubicin only, "the incidence of progressive disease was cut by more than half, the objective response rate was twice as high, and the time to progression was increased by more than 50%. Overall survival was also better" but did not reach statistical significance. "It was supposed to be 10% better for overall survival [based on the study’s design], but it was only 9% better and they considered that the same as zero," noted Dr. Benjamin, professor and chairman of sarcoma medical oncology at the University of Texas M.D. Anderson Cancer Center, Houston.
"Overall survival is a lousy endpoint," he said. "It measures everything that happens when you treat, but also everything that happens after" treatment is finished. Dr. Benjamin said his center was invited by the organizers of this phase III trial to participate, but he declined because the trial used overall survival as its primary endpoint.
The trial enrolled 455 patients with a locally-advanced, unresectable, or metastatic high-grade soft-tissue sarcoma, aged 18-60 years at 38 centers in nine European countries and Canada. It included patients with more than 10 different sarcoma types including liposarcomas, synovial sarcomas, leiomyosarcomas, undifferentiated pleomorphic sarcomas, and unclassified high-grade sarcomas. The study randomized patients to either doxorubicin alone or intensified doxorubicin plus ifosfamide as first-line treatment. Patients were treated every 3 weeks for up to six cycles and were followed for a median of 56-59 months (Lancet Oncol. 2014 [doi:10.1016/S1470-2045(14)70063-4]).
The study’s primary endpoint, overall survival, averaged 12.8 months in the monotherapy arm and 14.3 months in the dual-agent arm, a hazard ratio of 0.83 that failed to meet statistical significance (P =.076), reported Dr. Ian Judson, professor and head of the sarcoma unit at the Royal Marsden Hospital in London, and his associates. However, median progression-free survival was 4.6 months in the doxorubicin-only arm and 7.4 months for the combined-treatment arm, a statistically significant difference (P = .003). Progressive disease occurred in 32% of the patients on monotherapy and in 13% of those on combined treatment.
The study’s authors concluded that if chemotherapy is used palliatively to relieve acute symptoms, then sequential treatment with single drugs would probably be less toxic without significantly impairing survival. For patients in whom symptoms could be relieved by tumor shrinkage, or if debulking of the tumor before surgery or radiotherapy is the goal, or if delaying disease progression as long as possible is the goal, then combination treatment is justified. They also cautioned against extrapolating the findings to patients older than 60 years, a limitation seconded by Dr. Benjamin. He cautioned against administering the combination to any patient older than 65 years or to patients with substantially impaired renal function.
But he disagreed with using doxorubicin as monotherapy unless tolerance was an issue. "Doxorubicin is a very good drug; it just shouldn’t be used by itself," he said.
Dr. Benjamin offered a few tips for using the doxorubicin and ifosfamide combination in practice: It’s important to use doxorubicin at the recommended dosage of 75 mg/m2. If a patient cannot tolerate that dosage in combination with ifosfamide then the best option is to change to doxorubicin monotherapy to be sure the patient receives 75 mg/m2. He recommended starting patients first on the doxorubicin and ifosfamide combination because it is the most toxic, with a less toxic pairing like gemcitabine (Gemzar) and docetaxel (Dosefirez) as second-line therapy because it’s harder for patients to switch to a more toxic regimen, he said. Dacarbazine also can pair effectively with doxorubicin. This combination is "almost as good as doxorubicin plus ifosfamide and is less toxic." He also noted the efficacy of trabectedin (Yondelis), which is available in European countries but not in the United States.
"These drugs all work, and the patient will need everything you have. You need to put it all together," Dr. Benjamin advised. "All the chemotherapy we currently have is grossly inadequate, but given what we have, use doxorubicin and ifosfamide for most patients with soft tissue sarcomas."
On Twitter @mitchelzoler
MILAN – The combination of doxorubicin and ifosfamide is the best treatment for most patients with a sarcoma that does not have a proven targeted therapy, although a recent report from a large randomized study failed to prove that the combination surpassed doxorubicin alone for improving overall survival, said Dr. Robert S. Benjamin.
"From my point of view, this was clearly a positive study, but it was interpreted as a negative study" by the investigators who ran it, Dr. Benjamin said at Sarcoma and GIST 2014, hosted by the European Society for Medical Oncology.
Among patients who received doxorubicin (Adriamycin) plus ifosfamide (Ifex), compared with those randomized to doxorubicin only, "the incidence of progressive disease was cut by more than half, the objective response rate was twice as high, and the time to progression was increased by more than 50%. Overall survival was also better" but did not reach statistical significance. "It was supposed to be 10% better for overall survival [based on the study’s design], but it was only 9% better and they considered that the same as zero," noted Dr. Benjamin, professor and chairman of sarcoma medical oncology at the University of Texas M.D. Anderson Cancer Center, Houston.
"Overall survival is a lousy endpoint," he said. "It measures everything that happens when you treat, but also everything that happens after" treatment is finished. Dr. Benjamin said his center was invited by the organizers of this phase III trial to participate, but he declined because the trial used overall survival as its primary endpoint.
The trial enrolled 455 patients with a locally-advanced, unresectable, or metastatic high-grade soft-tissue sarcoma, aged 18-60 years at 38 centers in nine European countries and Canada. It included patients with more than 10 different sarcoma types including liposarcomas, synovial sarcomas, leiomyosarcomas, undifferentiated pleomorphic sarcomas, and unclassified high-grade sarcomas. The study randomized patients to either doxorubicin alone or intensified doxorubicin plus ifosfamide as first-line treatment. Patients were treated every 3 weeks for up to six cycles and were followed for a median of 56-59 months (Lancet Oncol. 2014 [doi:10.1016/S1470-2045(14)70063-4]).
The study’s primary endpoint, overall survival, averaged 12.8 months in the monotherapy arm and 14.3 months in the dual-agent arm, a hazard ratio of 0.83 that failed to meet statistical significance (P =.076), reported Dr. Ian Judson, professor and head of the sarcoma unit at the Royal Marsden Hospital in London, and his associates. However, median progression-free survival was 4.6 months in the doxorubicin-only arm and 7.4 months for the combined-treatment arm, a statistically significant difference (P = .003). Progressive disease occurred in 32% of the patients on monotherapy and in 13% of those on combined treatment.
The study’s authors concluded that if chemotherapy is used palliatively to relieve acute symptoms, then sequential treatment with single drugs would probably be less toxic without significantly impairing survival. For patients in whom symptoms could be relieved by tumor shrinkage, or if debulking of the tumor before surgery or radiotherapy is the goal, or if delaying disease progression as long as possible is the goal, then combination treatment is justified. They also cautioned against extrapolating the findings to patients older than 60 years, a limitation seconded by Dr. Benjamin. He cautioned against administering the combination to any patient older than 65 years or to patients with substantially impaired renal function.
But he disagreed with using doxorubicin as monotherapy unless tolerance was an issue. "Doxorubicin is a very good drug; it just shouldn’t be used by itself," he said.
Dr. Benjamin offered a few tips for using the doxorubicin and ifosfamide combination in practice: It’s important to use doxorubicin at the recommended dosage of 75 mg/m2. If a patient cannot tolerate that dosage in combination with ifosfamide then the best option is to change to doxorubicin monotherapy to be sure the patient receives 75 mg/m2. He recommended starting patients first on the doxorubicin and ifosfamide combination because it is the most toxic, with a less toxic pairing like gemcitabine (Gemzar) and docetaxel (Dosefirez) as second-line therapy because it’s harder for patients to switch to a more toxic regimen, he said. Dacarbazine also can pair effectively with doxorubicin. This combination is "almost as good as doxorubicin plus ifosfamide and is less toxic." He also noted the efficacy of trabectedin (Yondelis), which is available in European countries but not in the United States.
"These drugs all work, and the patient will need everything you have. You need to put it all together," Dr. Benjamin advised. "All the chemotherapy we currently have is grossly inadequate, but given what we have, use doxorubicin and ifosfamide for most patients with soft tissue sarcomas."
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM SARCOMA AND GIST 2014

Some consider denosumab standard treatment for unresectable giant cell tumor of bone
MILAN – Denosumab, a drug originally developed to treat patients with osteoporosis, can halt progression of giant cell tumor of the bone and is now considered by some experts to be standard treatment for patients with tumors that cannot be surgically removed without causing significant morbidity.
The Food and Drug Administration added this indication to the labeling for denosumab (Xgeva) in June 2013, and current management recommendations from the National Comprehensive Cancer Network list denosumab treatment as an option for patients with unresectable giant cell tumor of the bone (GCTB).

But experience with denosumab for this indication is limited and of relatively short duration, and questions remain about its optimal use.
"Does treatment need to be lifelong, if so what is the optimal schedule, and what are the long-term sequelae?" Dr. David Thomas said at Sarcoma and GIST 2014, hosted by the European Society for Medical Oncology. "If surgical resection is an option that is absolutely the first option. This is not an antitumor drug. It contains the tumor’s progression, and it does that very well," said Dr. Thomas, head of the sarcoma genomics and genetics laboratory at Peter MacCallum Cancer Centre in Melbourne.
"It’s clearly a good idea for patients with GCTB, where you would have to do a mutilating operation" to remove the tumor. But you’d be crazy to use it for a GCTB of the proximal tibia that is curable by resection and curettage and packing with cement," said Dr. Robert S. Benjamin, professor and chairman of sarcoma medical oncology at University of Texas M.D. Anderson Cancer Center, Houston. About 90% of GCTB are resectable in a way that is acceptable to patients, he estimated during an interview.
"For the 10% for whom surgery is not a good option, denosumab is fantastic, and it’s being used, but most medical oncologists never see these patients" because GCTB is relatively rare, Dr. Benjamin said.

The largest reported experience with denosumab in patients with GCTB was an international, multicenter, open-label study with 282 patients and a 13-month median follow-up reported last year by Dr. Thomas and his associates (Lancet Onc. 2013;14:901-8). Although this experience provided evidence for the drug’s efficacy and reasonable safety for many patients, the documented experience remains relatively short for a drug that may need to be taken lifelong to keep the tumor in check. And many patients would start treatment relatively early in life because when GCTB occurs, it is often in adolescents or young adults.
"We really don’t know what will happen long term," Dr. Thomas cautioned in an interview.
The published experience also highlighted some potential adverse effects from denosumab that warrant close monitoring of patients. Three of the 281 patients (1%) available for the safety analysis developed osteonecrosis of the jaw; 15 patients (5%) developed hypocalcemia, although none were considered serious; 5 patients (2%) had a serious infection; and 3 patients (1%) had a new primary malignancy. In addition, women should not become pregnant while on denosumab. So far, follow-up produced no suggestion of an increased rate of malignant transformation of the GCTB, he said.
"Because you may need to treat for a lifetime, it’s a big decision to start treatment. But before we had this, we couldn’t have a discussion," of a treatment option when surgery wasn’t possible, he said. "I had a patient in 2004, before denosumab was available, with multiple, recurrent giant cell tumors that spread up his spine" and hence were inoperable and the patient died from the "enormous morbidity" the tumor caused. "Denosumab would have likely saved his life," Dr. Thomas said.
The denosumab study was sponsored by Amgen, maker of the drug. Dr. Thomas said that he has received consulting fees and research support from Amgen. Dr. Benjamin said that he has been a consultant to Johnson & Johnson, Merck, and Pfizer.
On Twitter @mitchelzoler
MILAN – Denosumab, a drug originally developed to treat patients with osteoporosis, can halt progression of giant cell tumor of the bone and is now considered by some experts to be standard treatment for patients with tumors that cannot be surgically removed without causing significant morbidity.
The Food and Drug Administration added this indication to the labeling for denosumab (Xgeva) in June 2013, and current management recommendations from the National Comprehensive Cancer Network list denosumab treatment as an option for patients with unresectable giant cell tumor of the bone (GCTB).
But experience with denosumab for this indication is limited and of relatively short duration, and questions remain about its optimal use.
"Does treatment need to be lifelong, if so what is the optimal schedule, and what are the long-term sequelae?" Dr. David Thomas said at Sarcoma and GIST 2014, hosted by the European Society for Medical Oncology. "If surgical resection is an option that is absolutely the first option. This is not an antitumor drug. It contains the tumor’s progression, and it does that very well," said Dr. Thomas, head of the sarcoma genomics and genetics laboratory at Peter MacCallum Cancer Centre in Melbourne.
"It’s clearly a good idea for patients with GCTB, where you would have to do a mutilating operation" to remove the tumor. But you’d be crazy to use it for a GCTB of the proximal tibia that is curable by resection and curettage and packing with cement," said Dr. Robert S. Benjamin, professor and chairman of sarcoma medical oncology at University of Texas M.D. Anderson Cancer Center, Houston. About 90% of GCTB are resectable in a way that is acceptable to patients, he estimated during an interview.
"For the 10% for whom surgery is not a good option, denosumab is fantastic, and it’s being used, but most medical oncologists never see these patients" because GCTB is relatively rare, Dr. Benjamin said.
The largest reported experience with denosumab in patients with GCTB was an international, multicenter, open-label study with 282 patients and a 13-month median follow-up reported last year by Dr. Thomas and his associates (Lancet Onc. 2013;14:901-8). Although this experience provided evidence for the drug’s efficacy and reasonable safety for many patients, the documented experience remains relatively short for a drug that may need to be taken lifelong to keep the tumor in check. And many patients would start treatment relatively early in life because when GCTB occurs, it is often in adolescents or young adults.
"We really don’t know what will happen long term," Dr. Thomas cautioned in an interview.
The published experience also highlighted some potential adverse effects from denosumab that warrant close monitoring of patients. Three of the 281 patients (1%) available for the safety analysis developed osteonecrosis of the jaw; 15 patients (5%) developed hypocalcemia, although none were considered serious; 5 patients (2%) had a serious infection; and 3 patients (1%) had a new primary malignancy. In addition, women should not become pregnant while on denosumab. So far, follow-up produced no suggestion of an increased rate of malignant transformation of the GCTB, he said.
"Because you may need to treat for a lifetime, it’s a big decision to start treatment. But before we had this, we couldn’t have a discussion," of a treatment option when surgery wasn’t possible, he said. "I had a patient in 2004, before denosumab was available, with multiple, recurrent giant cell tumors that spread up his spine" and hence were inoperable and the patient died from the "enormous morbidity" the tumor caused. "Denosumab would have likely saved his life," Dr. Thomas said.
The denosumab study was sponsored by Amgen, maker of the drug. Dr. Thomas said that he has received consulting fees and research support from Amgen. Dr. Benjamin said that he has been a consultant to Johnson & Johnson, Merck, and Pfizer.
On Twitter @mitchelzoler
MILAN – Denosumab, a drug originally developed to treat patients with osteoporosis, can halt progression of giant cell tumor of the bone and is now considered by some experts to be standard treatment for patients with tumors that cannot be surgically removed without causing significant morbidity.
The Food and Drug Administration added this indication to the labeling for denosumab (Xgeva) in June 2013, and current management recommendations from the National Comprehensive Cancer Network list denosumab treatment as an option for patients with unresectable giant cell tumor of the bone (GCTB).
But experience with denosumab for this indication is limited and of relatively short duration, and questions remain about its optimal use.
"Does treatment need to be lifelong, if so what is the optimal schedule, and what are the long-term sequelae?" Dr. David Thomas said at Sarcoma and GIST 2014, hosted by the European Society for Medical Oncology. "If surgical resection is an option that is absolutely the first option. This is not an antitumor drug. It contains the tumor’s progression, and it does that very well," said Dr. Thomas, head of the sarcoma genomics and genetics laboratory at Peter MacCallum Cancer Centre in Melbourne.
"It’s clearly a good idea for patients with GCTB, where you would have to do a mutilating operation" to remove the tumor. But you’d be crazy to use it for a GCTB of the proximal tibia that is curable by resection and curettage and packing with cement," said Dr. Robert S. Benjamin, professor and chairman of sarcoma medical oncology at University of Texas M.D. Anderson Cancer Center, Houston. About 90% of GCTB are resectable in a way that is acceptable to patients, he estimated during an interview.
"For the 10% for whom surgery is not a good option, denosumab is fantastic, and it’s being used, but most medical oncologists never see these patients" because GCTB is relatively rare, Dr. Benjamin said.
The largest reported experience with denosumab in patients with GCTB was an international, multicenter, open-label study with 282 patients and a 13-month median follow-up reported last year by Dr. Thomas and his associates (Lancet Onc. 2013;14:901-8). Although this experience provided evidence for the drug’s efficacy and reasonable safety for many patients, the documented experience remains relatively short for a drug that may need to be taken lifelong to keep the tumor in check. And many patients would start treatment relatively early in life because when GCTB occurs, it is often in adolescents or young adults.
"We really don’t know what will happen long term," Dr. Thomas cautioned in an interview.
The published experience also highlighted some potential adverse effects from denosumab that warrant close monitoring of patients. Three of the 281 patients (1%) available for the safety analysis developed osteonecrosis of the jaw; 15 patients (5%) developed hypocalcemia, although none were considered serious; 5 patients (2%) had a serious infection; and 3 patients (1%) had a new primary malignancy. In addition, women should not become pregnant while on denosumab. So far, follow-up produced no suggestion of an increased rate of malignant transformation of the GCTB, he said.
"Because you may need to treat for a lifetime, it’s a big decision to start treatment. But before we had this, we couldn’t have a discussion," of a treatment option when surgery wasn’t possible, he said. "I had a patient in 2004, before denosumab was available, with multiple, recurrent giant cell tumors that spread up his spine" and hence were inoperable and the patient died from the "enormous morbidity" the tumor caused. "Denosumab would have likely saved his life," Dr. Thomas said.
The denosumab study was sponsored by Amgen, maker of the drug. Dr. Thomas said that he has received consulting fees and research support from Amgen. Dr. Benjamin said that he has been a consultant to Johnson & Johnson, Merck, and Pfizer.
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM SARCOMA AND GIST 2014

HFPEF’s diverse features pose management and trial challenges
When the highly-anticipated results from the TOPCAT trial reported last November failed to show a statistically significant efficacy benefit from spironolactone treatment in patients with heart failure and preserved left ventricular ejection fraction for the study’s primary endpoint of death or heart failure hospitalization, the neutral result represented more than another failed treatment trial in this rapidly expanding patient population that remains bereft of treatments with proven efficacy.
TOPCAT’s results also underscored the heterogeneity of patients diagnosed with heart failure with preserved ejection fraction (HFPEF), a signature feature of the syndrome. Heterogeneity is believed to have played a large role in the lack of success in TOPCAT as well as in several other major HFPEF trials.

Although HFPEF is defined as patients who exhibit clear clinical symptoms of heart failure but with a LVEF of at least 45%, and more recently at least 50%, what’s become increasingly clear as neutral trial results accumulate is that HFPEF is a syndrome. Far from a single pathologic entity, it can have different etiologies and present as different subtypes in a spectrum of abnormalities and severities.
HFPEF "is not just a simple disease of diastolic function and a stiff ventricle. It’s a very complicated, highly integrated, multisystem failure of cardiovascular and peripheral reserve," said Dr. Barry A. Borlaug, a cardiologist at the Mayo Clinic in Rochester, Minn. "A lot of us are coming to the conclusion that there are different subphenotypes of HFPEF, each with unique mechanisms and clinical trajectories that probably need unique treatment," he said in an interview.
"This is a very heterogeneous syndrome with no one etiology and a wide range of patients, some with marked ventricular hypertrophy, some with diastolic dysfunction, some who are fairly asymptomatic but become symptomatic with exercise, some who are compensated and stable, and others who are uncompensated. The spectrum of disease is very broad, and it may be many diseases," said Dr. Scott D. Solomon, professor of medicine at Harvard Medical School and director of noninvasive cardiology at Brigham and Women’s Hospital, Boston.
Last year, the heart failure literature featured two editorials spelling out proposed visions of the HFPEF spectrum. One of these proposed three characteristic types of HFPEF patients (J. Am. Coll. Cardiol. 2013;62:1339-42): those with exercise-induced diastolic dysfunction with no symptoms at rest, minimal fluid retention, and never hospitalized for heart failure but with long-standing hypertension and exercise intolerance; patients with volume overload and edema, recently hospitalized heart failure with dyspnea on exertion, and moderately severe heart failure symptoms; and the worst form, patients who have developed pulmonary hypertension and right ventricular failure as a consequence of their HFPEF and now have frequent heart failure hospitalizations. But patients do not necessarily progress from one severity stage to the next, noted Dr. Sanjiv J. Shah, a cardiologist at Northwestern University, Chicago, who wrote the editorial.

"Patients can progress back and forth across the spectrum" or remain in one stage, he said in an interview. "The syndrome is quite heterogeneous, not only in clinical presentation, but in etiology, and pathophysiology. There is still much to learn about the natural history of HFPEF , how it develops, and the trajectory of patients."
The second editorial described several relatively common HFPEF phenotypes that can appear individually or in combination, including filling limitation, ejection limitation, cardioacceleration, and vasoregulation (Eur. Heart J. 2013;34:1393-5). "Patients with heart failure caused by severe mitral insufficiency or aortic stenosis will clearly behave differently and respond to treatments differently from patients with hypertropic cardiomyopathy, constrictive pericarditis, or high-output heart failure. However, despite this heterogeneity these entities continue currently to be lumped together into the category of HFPEF," Dr. Borlaug wrote in this editorial.
The perils of heterogeneity
In addition to providing a better framework for understanding the causes and consequences of HFPEF, the paradigm of HFPEF as a heterogeneous syndrome also helps explain why the major intervention trials that focused on HFPEF patients, starting a decade ago with studies such as CHARM Preserved (Lancet 2003;362:777-81) and I-PRESERVE (N. Engl. J. Med. 2008;359:2456-67), and continuing through to the recent TOPCAT, have all failed to produce a statistically significant benefit for their primary endpoints.
"Heterogeneity confounds our ability to identify effective treatments. It’s the most likely single explanation" for the neutral HFPEF trials, Dr. Borlaug said. "I don’t think it’s as simple as one thing, but my speculation is that it’s the dominant reason."

"Many trials such as CHARM Preserved and TOPCAT seemed to enroll some patients who in retrospect did not have HFPEF," noted Dr. Gregg C. Fonarow, professor of medicine and associate chief of cardiology at UCLA in Los Angeles. "The key to future clinical trials will be better classification of HFPEF" and better matching of HFPEF patients to their treatment, said Dr. Shah.
"In the neutral megatrials some of the patients did not have HFPEF but had other disorders such as deconditioning and obesity – things that cannot be helped by treatments directed at the heart. We need to identify the patients who will respond to the treatment," said Dr. Burkert M. Pieske, professor and director of cardiology at the Medical University of Graz, Austria.
The TOPCAT (Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist) results reported last November exemplified the problem of patient heterogeneity. The study enrolled more than 3,400 patients in six countries: Argentina, Brazil, Canada, Russia, the Republic of Georgia, and the United States. Retrospective analysis showed that among patients in the placebo group, the primary endpoint of death or heart failure hospitalization during follow-up occurred at a rate of 12.6 events/100 patient-years among the 881 patients treated in the four Western Hemisphere countries, and at a rate of 2.3 events/100 patient-years among the 842 patients treated in Russia or Georgia, a greater than fivefold difference between the two subgroups.
"As a whole, the patients in Russia and Georgia had a lower event rate and were certainly less severely ill than the other patients, but they supposedly still had heart failure. Defining this disorder is difficult, and when a patient has signs and symptoms of heart failure and preserved ejection fraction you may not be certain that heart failure is causing the symptoms. That’s why many people think that we should use another criterion" to define HFPEF in trials, such as elevated serum level of some form of natriuretic peptide, said Dr. Solomon, a TOPCAT coinvestigator. By design, patients could enter TOPCAT either because of a recent heart failure hospitalization, which is how 72% of patients got in, or by having a threshold level of natriuretic peptide, the way the remaining 28% entered the study. Within the subgroup enrolled by natriuretic peptide level, spironolactone treatment had a statistically significant effect in reducing the primary endpoint, while in the other 72% the drug produced no discernable benefit over placebo.

"TOPCAT had funding problems, recruitment problems, and monitoring problems. There was not enough money for good monitoring," said Dr. Pieske, who did not participate in TOPCAT. The patients enrolled in Russia and Georgia were "unbelievably stable," with their 2.3/100 patient-years event rate. "Even if the drug works, it can’t exert an effect if there are no events."
Dr. Pieske ran a much smaller study of spironolactone involving 422 patients with HFPEF, the Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF) trial, which showed efficacy for the endpoint of improved diastolic function but not for the co-endpoint of improved exercise capacity (JAMA 2013;309:781-91).
"If you look at TOPCAT and Aldo-DHF together and ask, does spironolactone work, the answer is yes if you select the right patients, those with clear evidence of cardiac functional abnormalities and increased natriuretic peptide levels or documented hospitalization for heart failure. The drug clearly works in these patients, but there were enough inconsistencies in these trials so that this will not get into the treatment guidelines," Dr. Pieske said in an interview.
"Spironolactone provides most benefit in patients who are volume overloaded, have right-heart failure, or both. These are also the patients most likely to have elevated natriuretic peptide. Spironolactone does not seem to benefit as much early-stage HFPEF," noted Dr. Shah.

Several experts cautioned that because the TOPCAT results failed to produce a statistically significant benefit for the study’s primary endpoint, any other conclusions from the results must be made very cautiously, and several also said that they wanted to see the full, published results which had not appeared as of late February. They also noted that many patients with HFPEF, most likely a majority, do not have elevated blood levels of natriuretic peptide.
Where HFPEF management stands now
Evidence-based guidelines for the specific treatment of HFPEF are simple. There aren’t any.
The American Heart Association/American College of Cardiology heart failure management guidelines released last October made no disease-specific treatment recommendations (J. Amer. Coll. Cardiol. 2013;62:e147-e239). They call for controlling hypertension in patients who need intervention by prevailing standards, treatment with a diuretic in patients with volume overload, coronary revascularization if coronary disease is present, and management of atrial fibrillation if that’s present. The European Society of Cardiology’s guidelines issued in 2012 say pretty much the same (Eur. J. Cardiol. 2012;33:1787-1847), except they add this statement: "No treatment has yet been shown, convincingly, to reduce morbidity and mortality in patients with HFPEF."
If nothing else, TOPCAT’s results further solidified spironolactone’s role as a reasonably safe drug for blood pressure lowering in HFPEF patients who are also hypertensive. Hypertension control is a must for patients with HFPEF as it’s believed to significantly contribute to HFPEF in most patients. "Hypertension is the most common underlying cause of HFPEF," said Dr. John R. Teerlink, professor of medicine and director of the heart failure program at the San Francisco Veterans Affairs Medical Center.

When it comes to treating hypertension in HFPEF patients, "unless there is a contraindication, spironolactone should be one of the first drugs to try, but I’m not sure you can say it’s the first choice" based on current evidence, Dr. Borlaug said.
A diuretic is an obvious antihypertensive for HFPEF patients with fluid overload. And there were suggestions of benefit from the ACE inhibitor perindopril in elderly patients in the PEP-CHF trial (Eur. Heart J. 2006;27:2338-45), another large HFPEF-treatment trial that failed to show significant benefit for the primary endpoint with perindopril treatment but had positive results in some secondary endpoints.
Patients "get these drugs to treat their hypertension, but we believe they may also help their heart failure. It’s a belief system. My colleagues and I already use a lot of spironolactone to treat hypertension, and the TOPCAT results won’t change my practice. I’m not comfortable telling people that you should use an aldosterone antagonist because of TOPCAT," Dr. Teerlink said in an interview.
"An angiotensin-converting enzyme inhibitor will probably work; spironolactone probably works if you get the diagnosis right; we don’t know about beta-blockers; and there is some evidence for using digoxin," said Dr. John G.F. Cleland, professor of cardiology at the University of Hull, Kingston-upon-Hull, England. "There must also be good treatment of hypertension, and judicious use of diuretics."
"I’m careful with beta-blockers; I’ve had some patients who felt miserable on them," said Dr. Borlaug. "Some HFPEF patients don’t have a stroke volume, they don’t have diastole, all they have is heart rate, and if you take that away they are left with no cardiac output."
Aside from controlling blood pressure, experts advise good management of other comorbidities such as coronary disease, atrial fibrillation, diabetes, sleep disordered breathing, and renal disease. "Clinicians should make sure that they are not missing severe coronary artery disease, infiltrative cardiomyopathy, constrictive pericarditis, or other causes of HFPEF that have specific treatments," said Dr. Shah. Some drugs have shown promise in early-phase studies – such as ivabradine and neprilysin – but phase III trials are needed. "My advice on how to manage patients with HFPEF is to make every effort to enroll them in a randomized clinical trial," said Dr. Fonarow.

Another key is making the diagnosis, ideally to prevent development of irreversible cardiovascular damage. "HFPEF is difficult to diagnose with certainty unless you do a cardiac catheterization to measure filling pressures, but in patients with early-stage HFPEF, even their invasive hemodynamics look pretty normal," said Dr. Borlaug. Another approach is an exercise echo, which is noninvasive and can identify stress-induced diastolic dysfunction, but the sensitivity and specificity of this approach remains uncertain, he said. And an elevated natriuretic peptide level can help nail the HFPEF diagnosis in some patients but many other patients have levels within the normal range.
To find HFPEF patients, apply a low index of suspicion and look for breathlessness, loss of functional capacity, signs of congestion, lung crackles, echocardiographic signs, pulmonary artery hypertension, and an enlarged left atrium, he suggested.
"Finding patients is a big challenge," said Dr. Pieske. HFPEF patients tend to be elderly, women, and people who are obese who have hypertension and perhaps diabetes. "They complain of dyspnea and fatigue and many physicians think this is just how it is. They will not consider that there is a true diagnosis behind these symptoms and complaints," especially if the patient has preserved left ventricular ejection fraction and a normal natriuretic peptide level." A diastolic stress test using exercise and an echo exam may identify stable patients with early-stage diastolic dysfunction but this requires confirmation, he said.
HFPEF dominates heart failure, lacks good treatment
Experts say the onus on physicians to diagnose and manage HFPEF will grow substantially, since HFPEF is not only highly prevalent but also increasing faster than heart failure with reduced ejection fraction (HFREF). Results from 13 community-based studies published during 1997-2006 showed that HFPEF represented an average of 55% of all heart failure cases, Dr. Carolyn S.P. Lam said last November during a talk at the American Heart Association’s scientific sessions.

Two recent reports of U.S. data documented that the increasing prevalence of HFPEF is outpacing that of HFREF by 1% per year, driven by the aging of the American population (older age is a risk factor for HFPEF) and by the increasing prevalence of comorbidities that contribute to HFPEF. One of the reports, based on U.S. national data collected by the Get With the Guidelines Program, extrapolated that by the end of this decade about 65% of all patients hospitalized for heart failure will have a left ventricular ejection fraction of 40% or greater, the vast majority with HFPEF (Curr. Heart Fail. Rep. 2013;10:401-10). What’s coming is a HFPEF "epidemic," said Dr. Lam, a cardiologist at the National University Heart Centre in Singapore.
The fact that HFPEF is now at least as prevalent as HFREF, that it is about as lethal independent of what comorbidities might contribute, and that it is growing more prevalent than HFREF sharply contrasts with the absence of treatments with proven efficacy. How did that happen?
"Research into HFPEF is decades behind research into HFREF," said Dr. Fonarow. "HFPEF was largely ignored because the prevailing wisdom [20 or so years ago] was that it represented only a small proportion of cases and that outcomes were much better compared with HFREF." Only with registry data and community-based studies run more recently did cardiologists realize that outcomes from HFPEF were as bad as from HFREF. Progress was further hampered by enrollment of patients who did not have HFPEF or a broad spectrum of patients unable to respond equally well to the treatment under study.
"There are large gaps in knowledge of the pathophysiology of HFPEF and little investment has been made to identify potential therapeutic targets. This creates cascading levels of challenges for developing effective treatments despite the massive unmet need," said Dr. Fonarow.
"We don’t know what to treat yet," was Dr. Teerlink’s summation of the HFPEF dilemma.
Dr. Borlaug said that he has been a consultant to GlaxoSmithKline, Merck, Amgen, CardioKinetix, and DC Devices and has received research support from Atcor. Dr. Solomon said he has been a consultant to and received research support from Novartis and more than 10 other drug and device companies. Dr. Pieske said that he has received honoraria from Bayer, Boehringer Ingelheim, Servier, Medtronic, Bristol-Myers Squibb, Menarini, and Novartis and received research support from Bayer and Medtronic. Dr. Shah said that he has been a consultant to Novartis, Bayer Schering, and the Pulmonary Hypertension Association. Dr. Fonarow said that he has been a consultant to Medtronic, Novartis, and Pfizer. Dr. Teerlink said that he has been an adviser to and received research support from Novartis. Dr. Cleland said that he has received honoraria from Novartis and research funding from Pfizer. Dr. Lam said that she has been a consultant to Bayer, Novartis, and DC Devices and has received research support from Boston Scientific, Medtronic, and Vifor Pharma.
On Twitter @mitchelzoler
When the highly-anticipated results from the TOPCAT trial reported last November failed to show a statistically significant efficacy benefit from spironolactone treatment in patients with heart failure and preserved left ventricular ejection fraction for the study’s primary endpoint of death or heart failure hospitalization, the neutral result represented more than another failed treatment trial in this rapidly expanding patient population that remains bereft of treatments with proven efficacy.
TOPCAT’s results also underscored the heterogeneity of patients diagnosed with heart failure with preserved ejection fraction (HFPEF), a signature feature of the syndrome. Heterogeneity is believed to have played a large role in the lack of success in TOPCAT as well as in several other major HFPEF trials.
Although HFPEF is defined as patients who exhibit clear clinical symptoms of heart failure but with a LVEF of at least 45%, and more recently at least 50%, what’s become increasingly clear as neutral trial results accumulate is that HFPEF is a syndrome. Far from a single pathologic entity, it can have different etiologies and present as different subtypes in a spectrum of abnormalities and severities.
HFPEF "is not just a simple disease of diastolic function and a stiff ventricle. It’s a very complicated, highly integrated, multisystem failure of cardiovascular and peripheral reserve," said Dr. Barry A. Borlaug, a cardiologist at the Mayo Clinic in Rochester, Minn. "A lot of us are coming to the conclusion that there are different subphenotypes of HFPEF, each with unique mechanisms and clinical trajectories that probably need unique treatment," he said in an interview.
"This is a very heterogeneous syndrome with no one etiology and a wide range of patients, some with marked ventricular hypertrophy, some with diastolic dysfunction, some who are fairly asymptomatic but become symptomatic with exercise, some who are compensated and stable, and others who are uncompensated. The spectrum of disease is very broad, and it may be many diseases," said Dr. Scott D. Solomon, professor of medicine at Harvard Medical School and director of noninvasive cardiology at Brigham and Women’s Hospital, Boston.
Last year, the heart failure literature featured two editorials spelling out proposed visions of the HFPEF spectrum. One of these proposed three characteristic types of HFPEF patients (J. Am. Coll. Cardiol. 2013;62:1339-42): those with exercise-induced diastolic dysfunction with no symptoms at rest, minimal fluid retention, and never hospitalized for heart failure but with long-standing hypertension and exercise intolerance; patients with volume overload and edema, recently hospitalized heart failure with dyspnea on exertion, and moderately severe heart failure symptoms; and the worst form, patients who have developed pulmonary hypertension and right ventricular failure as a consequence of their HFPEF and now have frequent heart failure hospitalizations. But patients do not necessarily progress from one severity stage to the next, noted Dr. Sanjiv J. Shah, a cardiologist at Northwestern University, Chicago, who wrote the editorial.
"Patients can progress back and forth across the spectrum" or remain in one stage, he said in an interview. "The syndrome is quite heterogeneous, not only in clinical presentation, but in etiology, and pathophysiology. There is still much to learn about the natural history of HFPEF , how it develops, and the trajectory of patients."
The second editorial described several relatively common HFPEF phenotypes that can appear individually or in combination, including filling limitation, ejection limitation, cardioacceleration, and vasoregulation (Eur. Heart J. 2013;34:1393-5). "Patients with heart failure caused by severe mitral insufficiency or aortic stenosis will clearly behave differently and respond to treatments differently from patients with hypertropic cardiomyopathy, constrictive pericarditis, or high-output heart failure. However, despite this heterogeneity these entities continue currently to be lumped together into the category of HFPEF," Dr. Borlaug wrote in this editorial.
The perils of heterogeneity
In addition to providing a better framework for understanding the causes and consequences of HFPEF, the paradigm of HFPEF as a heterogeneous syndrome also helps explain why the major intervention trials that focused on HFPEF patients, starting a decade ago with studies such as CHARM Preserved (Lancet 2003;362:777-81) and I-PRESERVE (N. Engl. J. Med. 2008;359:2456-67), and continuing through to the recent TOPCAT, have all failed to produce a statistically significant benefit for their primary endpoints.
"Heterogeneity confounds our ability to identify effective treatments. It’s the most likely single explanation" for the neutral HFPEF trials, Dr. Borlaug said. "I don’t think it’s as simple as one thing, but my speculation is that it’s the dominant reason."
"Many trials such as CHARM Preserved and TOPCAT seemed to enroll some patients who in retrospect did not have HFPEF," noted Dr. Gregg C. Fonarow, professor of medicine and associate chief of cardiology at UCLA in Los Angeles. "The key to future clinical trials will be better classification of HFPEF" and better matching of HFPEF patients to their treatment, said Dr. Shah.
"In the neutral megatrials some of the patients did not have HFPEF but had other disorders such as deconditioning and obesity – things that cannot be helped by treatments directed at the heart. We need to identify the patients who will respond to the treatment," said Dr. Burkert M. Pieske, professor and director of cardiology at the Medical University of Graz, Austria.
The TOPCAT (Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist) results reported last November exemplified the problem of patient heterogeneity. The study enrolled more than 3,400 patients in six countries: Argentina, Brazil, Canada, Russia, the Republic of Georgia, and the United States. Retrospective analysis showed that among patients in the placebo group, the primary endpoint of death or heart failure hospitalization during follow-up occurred at a rate of 12.6 events/100 patient-years among the 881 patients treated in the four Western Hemisphere countries, and at a rate of 2.3 events/100 patient-years among the 842 patients treated in Russia or Georgia, a greater than fivefold difference between the two subgroups.
"As a whole, the patients in Russia and Georgia had a lower event rate and were certainly less severely ill than the other patients, but they supposedly still had heart failure. Defining this disorder is difficult, and when a patient has signs and symptoms of heart failure and preserved ejection fraction you may not be certain that heart failure is causing the symptoms. That’s why many people think that we should use another criterion" to define HFPEF in trials, such as elevated serum level of some form of natriuretic peptide, said Dr. Solomon, a TOPCAT coinvestigator. By design, patients could enter TOPCAT either because of a recent heart failure hospitalization, which is how 72% of patients got in, or by having a threshold level of natriuretic peptide, the way the remaining 28% entered the study. Within the subgroup enrolled by natriuretic peptide level, spironolactone treatment had a statistically significant effect in reducing the primary endpoint, while in the other 72% the drug produced no discernable benefit over placebo.
"TOPCAT had funding problems, recruitment problems, and monitoring problems. There was not enough money for good monitoring," said Dr. Pieske, who did not participate in TOPCAT. The patients enrolled in Russia and Georgia were "unbelievably stable," with their 2.3/100 patient-years event rate. "Even if the drug works, it can’t exert an effect if there are no events."
Dr. Pieske ran a much smaller study of spironolactone involving 422 patients with HFPEF, the Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF) trial, which showed efficacy for the endpoint of improved diastolic function but not for the co-endpoint of improved exercise capacity (JAMA 2013;309:781-91).
"If you look at TOPCAT and Aldo-DHF together and ask, does spironolactone work, the answer is yes if you select the right patients, those with clear evidence of cardiac functional abnormalities and increased natriuretic peptide levels or documented hospitalization for heart failure. The drug clearly works in these patients, but there were enough inconsistencies in these trials so that this will not get into the treatment guidelines," Dr. Pieske said in an interview.
"Spironolactone provides most benefit in patients who are volume overloaded, have right-heart failure, or both. These are also the patients most likely to have elevated natriuretic peptide. Spironolactone does not seem to benefit as much early-stage HFPEF," noted Dr. Shah.
Several experts cautioned that because the TOPCAT results failed to produce a statistically significant benefit for the study’s primary endpoint, any other conclusions from the results must be made very cautiously, and several also said that they wanted to see the full, published results which had not appeared as of late February. They also noted that many patients with HFPEF, most likely a majority, do not have elevated blood levels of natriuretic peptide.
Where HFPEF management stands now
Evidence-based guidelines for the specific treatment of HFPEF are simple. There aren’t any.
The American Heart Association/American College of Cardiology heart failure management guidelines released last October made no disease-specific treatment recommendations (J. Amer. Coll. Cardiol. 2013;62:e147-e239). They call for controlling hypertension in patients who need intervention by prevailing standards, treatment with a diuretic in patients with volume overload, coronary revascularization if coronary disease is present, and management of atrial fibrillation if that’s present. The European Society of Cardiology’s guidelines issued in 2012 say pretty much the same (Eur. J. Cardiol. 2012;33:1787-1847), except they add this statement: "No treatment has yet been shown, convincingly, to reduce morbidity and mortality in patients with HFPEF."
If nothing else, TOPCAT’s results further solidified spironolactone’s role as a reasonably safe drug for blood pressure lowering in HFPEF patients who are also hypertensive. Hypertension control is a must for patients with HFPEF as it’s believed to significantly contribute to HFPEF in most patients. "Hypertension is the most common underlying cause of HFPEF," said Dr. John R. Teerlink, professor of medicine and director of the heart failure program at the San Francisco Veterans Affairs Medical Center.
When it comes to treating hypertension in HFPEF patients, "unless there is a contraindication, spironolactone should be one of the first drugs to try, but I’m not sure you can say it’s the first choice" based on current evidence, Dr. Borlaug said.
A diuretic is an obvious antihypertensive for HFPEF patients with fluid overload. And there were suggestions of benefit from the ACE inhibitor perindopril in elderly patients in the PEP-CHF trial (Eur. Heart J. 2006;27:2338-45), another large HFPEF-treatment trial that failed to show significant benefit for the primary endpoint with perindopril treatment but had positive results in some secondary endpoints.
Patients "get these drugs to treat their hypertension, but we believe they may also help their heart failure. It’s a belief system. My colleagues and I already use a lot of spironolactone to treat hypertension, and the TOPCAT results won’t change my practice. I’m not comfortable telling people that you should use an aldosterone antagonist because of TOPCAT," Dr. Teerlink said in an interview.
"An angiotensin-converting enzyme inhibitor will probably work; spironolactone probably works if you get the diagnosis right; we don’t know about beta-blockers; and there is some evidence for using digoxin," said Dr. John G.F. Cleland, professor of cardiology at the University of Hull, Kingston-upon-Hull, England. "There must also be good treatment of hypertension, and judicious use of diuretics."
"I’m careful with beta-blockers; I’ve had some patients who felt miserable on them," said Dr. Borlaug. "Some HFPEF patients don’t have a stroke volume, they don’t have diastole, all they have is heart rate, and if you take that away they are left with no cardiac output."
Aside from controlling blood pressure, experts advise good management of other comorbidities such as coronary disease, atrial fibrillation, diabetes, sleep disordered breathing, and renal disease. "Clinicians should make sure that they are not missing severe coronary artery disease, infiltrative cardiomyopathy, constrictive pericarditis, or other causes of HFPEF that have specific treatments," said Dr. Shah. Some drugs have shown promise in early-phase studies – such as ivabradine and neprilysin – but phase III trials are needed. "My advice on how to manage patients with HFPEF is to make every effort to enroll them in a randomized clinical trial," said Dr. Fonarow.
Another key is making the diagnosis, ideally to prevent development of irreversible cardiovascular damage. "HFPEF is difficult to diagnose with certainty unless you do a cardiac catheterization to measure filling pressures, but in patients with early-stage HFPEF, even their invasive hemodynamics look pretty normal," said Dr. Borlaug. Another approach is an exercise echo, which is noninvasive and can identify stress-induced diastolic dysfunction, but the sensitivity and specificity of this approach remains uncertain, he said. And an elevated natriuretic peptide level can help nail the HFPEF diagnosis in some patients but many other patients have levels within the normal range.
To find HFPEF patients, apply a low index of suspicion and look for breathlessness, loss of functional capacity, signs of congestion, lung crackles, echocardiographic signs, pulmonary artery hypertension, and an enlarged left atrium, he suggested.
"Finding patients is a big challenge," said Dr. Pieske. HFPEF patients tend to be elderly, women, and people who are obese who have hypertension and perhaps diabetes. "They complain of dyspnea and fatigue and many physicians think this is just how it is. They will not consider that there is a true diagnosis behind these symptoms and complaints," especially if the patient has preserved left ventricular ejection fraction and a normal natriuretic peptide level." A diastolic stress test using exercise and an echo exam may identify stable patients with early-stage diastolic dysfunction but this requires confirmation, he said.
HFPEF dominates heart failure, lacks good treatment
Experts say the onus on physicians to diagnose and manage HFPEF will grow substantially, since HFPEF is not only highly prevalent but also increasing faster than heart failure with reduced ejection fraction (HFREF). Results from 13 community-based studies published during 1997-2006 showed that HFPEF represented an average of 55% of all heart failure cases, Dr. Carolyn S.P. Lam said last November during a talk at the American Heart Association’s scientific sessions.
Two recent reports of U.S. data documented that the increasing prevalence of HFPEF is outpacing that of HFREF by 1% per year, driven by the aging of the American population (older age is a risk factor for HFPEF) and by the increasing prevalence of comorbidities that contribute to HFPEF. One of the reports, based on U.S. national data collected by the Get With the Guidelines Program, extrapolated that by the end of this decade about 65% of all patients hospitalized for heart failure will have a left ventricular ejection fraction of 40% or greater, the vast majority with HFPEF (Curr. Heart Fail. Rep. 2013;10:401-10). What’s coming is a HFPEF "epidemic," said Dr. Lam, a cardiologist at the National University Heart Centre in Singapore.
The fact that HFPEF is now at least as prevalent as HFREF, that it is about as lethal independent of what comorbidities might contribute, and that it is growing more prevalent than HFREF sharply contrasts with the absence of treatments with proven efficacy. How did that happen?
"Research into HFPEF is decades behind research into HFREF," said Dr. Fonarow. "HFPEF was largely ignored because the prevailing wisdom [20 or so years ago] was that it represented only a small proportion of cases and that outcomes were much better compared with HFREF." Only with registry data and community-based studies run more recently did cardiologists realize that outcomes from HFPEF were as bad as from HFREF. Progress was further hampered by enrollment of patients who did not have HFPEF or a broad spectrum of patients unable to respond equally well to the treatment under study.
"There are large gaps in knowledge of the pathophysiology of HFPEF and little investment has been made to identify potential therapeutic targets. This creates cascading levels of challenges for developing effective treatments despite the massive unmet need," said Dr. Fonarow.
"We don’t know what to treat yet," was Dr. Teerlink’s summation of the HFPEF dilemma.
Dr. Borlaug said that he has been a consultant to GlaxoSmithKline, Merck, Amgen, CardioKinetix, and DC Devices and has received research support from Atcor. Dr. Solomon said he has been a consultant to and received research support from Novartis and more than 10 other drug and device companies. Dr. Pieske said that he has received honoraria from Bayer, Boehringer Ingelheim, Servier, Medtronic, Bristol-Myers Squibb, Menarini, and Novartis and received research support from Bayer and Medtronic. Dr. Shah said that he has been a consultant to Novartis, Bayer Schering, and the Pulmonary Hypertension Association. Dr. Fonarow said that he has been a consultant to Medtronic, Novartis, and Pfizer. Dr. Teerlink said that he has been an adviser to and received research support from Novartis. Dr. Cleland said that he has received honoraria from Novartis and research funding from Pfizer. Dr. Lam said that she has been a consultant to Bayer, Novartis, and DC Devices and has received research support from Boston Scientific, Medtronic, and Vifor Pharma.
On Twitter @mitchelzoler
When the highly-anticipated results from the TOPCAT trial reported last November failed to show a statistically significant efficacy benefit from spironolactone treatment in patients with heart failure and preserved left ventricular ejection fraction for the study’s primary endpoint of death or heart failure hospitalization, the neutral result represented more than another failed treatment trial in this rapidly expanding patient population that remains bereft of treatments with proven efficacy.
TOPCAT’s results also underscored the heterogeneity of patients diagnosed with heart failure with preserved ejection fraction (HFPEF), a signature feature of the syndrome. Heterogeneity is believed to have played a large role in the lack of success in TOPCAT as well as in several other major HFPEF trials.
Although HFPEF is defined as patients who exhibit clear clinical symptoms of heart failure but with a LVEF of at least 45%, and more recently at least 50%, what’s become increasingly clear as neutral trial results accumulate is that HFPEF is a syndrome. Far from a single pathologic entity, it can have different etiologies and present as different subtypes in a spectrum of abnormalities and severities.
HFPEF "is not just a simple disease of diastolic function and a stiff ventricle. It’s a very complicated, highly integrated, multisystem failure of cardiovascular and peripheral reserve," said Dr. Barry A. Borlaug, a cardiologist at the Mayo Clinic in Rochester, Minn. "A lot of us are coming to the conclusion that there are different subphenotypes of HFPEF, each with unique mechanisms and clinical trajectories that probably need unique treatment," he said in an interview.
"This is a very heterogeneous syndrome with no one etiology and a wide range of patients, some with marked ventricular hypertrophy, some with diastolic dysfunction, some who are fairly asymptomatic but become symptomatic with exercise, some who are compensated and stable, and others who are uncompensated. The spectrum of disease is very broad, and it may be many diseases," said Dr. Scott D. Solomon, professor of medicine at Harvard Medical School and director of noninvasive cardiology at Brigham and Women’s Hospital, Boston.
Last year, the heart failure literature featured two editorials spelling out proposed visions of the HFPEF spectrum. One of these proposed three characteristic types of HFPEF patients (J. Am. Coll. Cardiol. 2013;62:1339-42): those with exercise-induced diastolic dysfunction with no symptoms at rest, minimal fluid retention, and never hospitalized for heart failure but with long-standing hypertension and exercise intolerance; patients with volume overload and edema, recently hospitalized heart failure with dyspnea on exertion, and moderately severe heart failure symptoms; and the worst form, patients who have developed pulmonary hypertension and right ventricular failure as a consequence of their HFPEF and now have frequent heart failure hospitalizations. But patients do not necessarily progress from one severity stage to the next, noted Dr. Sanjiv J. Shah, a cardiologist at Northwestern University, Chicago, who wrote the editorial.
"Patients can progress back and forth across the spectrum" or remain in one stage, he said in an interview. "The syndrome is quite heterogeneous, not only in clinical presentation, but in etiology, and pathophysiology. There is still much to learn about the natural history of HFPEF , how it develops, and the trajectory of patients."
The second editorial described several relatively common HFPEF phenotypes that can appear individually or in combination, including filling limitation, ejection limitation, cardioacceleration, and vasoregulation (Eur. Heart J. 2013;34:1393-5). "Patients with heart failure caused by severe mitral insufficiency or aortic stenosis will clearly behave differently and respond to treatments differently from patients with hypertropic cardiomyopathy, constrictive pericarditis, or high-output heart failure. However, despite this heterogeneity these entities continue currently to be lumped together into the category of HFPEF," Dr. Borlaug wrote in this editorial.
The perils of heterogeneity
In addition to providing a better framework for understanding the causes and consequences of HFPEF, the paradigm of HFPEF as a heterogeneous syndrome also helps explain why the major intervention trials that focused on HFPEF patients, starting a decade ago with studies such as CHARM Preserved (Lancet 2003;362:777-81) and I-PRESERVE (N. Engl. J. Med. 2008;359:2456-67), and continuing through to the recent TOPCAT, have all failed to produce a statistically significant benefit for their primary endpoints.
"Heterogeneity confounds our ability to identify effective treatments. It’s the most likely single explanation" for the neutral HFPEF trials, Dr. Borlaug said. "I don’t think it’s as simple as one thing, but my speculation is that it’s the dominant reason."
"Many trials such as CHARM Preserved and TOPCAT seemed to enroll some patients who in retrospect did not have HFPEF," noted Dr. Gregg C. Fonarow, professor of medicine and associate chief of cardiology at UCLA in Los Angeles. "The key to future clinical trials will be better classification of HFPEF" and better matching of HFPEF patients to their treatment, said Dr. Shah.
"In the neutral megatrials some of the patients did not have HFPEF but had other disorders such as deconditioning and obesity – things that cannot be helped by treatments directed at the heart. We need to identify the patients who will respond to the treatment," said Dr. Burkert M. Pieske, professor and director of cardiology at the Medical University of Graz, Austria.
The TOPCAT (Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist) results reported last November exemplified the problem of patient heterogeneity. The study enrolled more than 3,400 patients in six countries: Argentina, Brazil, Canada, Russia, the Republic of Georgia, and the United States. Retrospective analysis showed that among patients in the placebo group, the primary endpoint of death or heart failure hospitalization during follow-up occurred at a rate of 12.6 events/100 patient-years among the 881 patients treated in the four Western Hemisphere countries, and at a rate of 2.3 events/100 patient-years among the 842 patients treated in Russia or Georgia, a greater than fivefold difference between the two subgroups.
"As a whole, the patients in Russia and Georgia had a lower event rate and were certainly less severely ill than the other patients, but they supposedly still had heart failure. Defining this disorder is difficult, and when a patient has signs and symptoms of heart failure and preserved ejection fraction you may not be certain that heart failure is causing the symptoms. That’s why many people think that we should use another criterion" to define HFPEF in trials, such as elevated serum level of some form of natriuretic peptide, said Dr. Solomon, a TOPCAT coinvestigator. By design, patients could enter TOPCAT either because of a recent heart failure hospitalization, which is how 72% of patients got in, or by having a threshold level of natriuretic peptide, the way the remaining 28% entered the study. Within the subgroup enrolled by natriuretic peptide level, spironolactone treatment had a statistically significant effect in reducing the primary endpoint, while in the other 72% the drug produced no discernable benefit over placebo.
"TOPCAT had funding problems, recruitment problems, and monitoring problems. There was not enough money for good monitoring," said Dr. Pieske, who did not participate in TOPCAT. The patients enrolled in Russia and Georgia were "unbelievably stable," with their 2.3/100 patient-years event rate. "Even if the drug works, it can’t exert an effect if there are no events."
Dr. Pieske ran a much smaller study of spironolactone involving 422 patients with HFPEF, the Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF) trial, which showed efficacy for the endpoint of improved diastolic function but not for the co-endpoint of improved exercise capacity (JAMA 2013;309:781-91).
"If you look at TOPCAT and Aldo-DHF together and ask, does spironolactone work, the answer is yes if you select the right patients, those with clear evidence of cardiac functional abnormalities and increased natriuretic peptide levels or documented hospitalization for heart failure. The drug clearly works in these patients, but there were enough inconsistencies in these trials so that this will not get into the treatment guidelines," Dr. Pieske said in an interview.
"Spironolactone provides most benefit in patients who are volume overloaded, have right-heart failure, or both. These are also the patients most likely to have elevated natriuretic peptide. Spironolactone does not seem to benefit as much early-stage HFPEF," noted Dr. Shah.
Several experts cautioned that because the TOPCAT results failed to produce a statistically significant benefit for the study’s primary endpoint, any other conclusions from the results must be made very cautiously, and several also said that they wanted to see the full, published results which had not appeared as of late February. They also noted that many patients with HFPEF, most likely a majority, do not have elevated blood levels of natriuretic peptide.
Where HFPEF management stands now
Evidence-based guidelines for the specific treatment of HFPEF are simple. There aren’t any.
The American Heart Association/American College of Cardiology heart failure management guidelines released last October made no disease-specific treatment recommendations (J. Amer. Coll. Cardiol. 2013;62:e147-e239). They call for controlling hypertension in patients who need intervention by prevailing standards, treatment with a diuretic in patients with volume overload, coronary revascularization if coronary disease is present, and management of atrial fibrillation if that’s present. The European Society of Cardiology’s guidelines issued in 2012 say pretty much the same (Eur. J. Cardiol. 2012;33:1787-1847), except they add this statement: "No treatment has yet been shown, convincingly, to reduce morbidity and mortality in patients with HFPEF."
If nothing else, TOPCAT’s results further solidified spironolactone’s role as a reasonably safe drug for blood pressure lowering in HFPEF patients who are also hypertensive. Hypertension control is a must for patients with HFPEF as it’s believed to significantly contribute to HFPEF in most patients. "Hypertension is the most common underlying cause of HFPEF," said Dr. John R. Teerlink, professor of medicine and director of the heart failure program at the San Francisco Veterans Affairs Medical Center.
When it comes to treating hypertension in HFPEF patients, "unless there is a contraindication, spironolactone should be one of the first drugs to try, but I’m not sure you can say it’s the first choice" based on current evidence, Dr. Borlaug said.
A diuretic is an obvious antihypertensive for HFPEF patients with fluid overload. And there were suggestions of benefit from the ACE inhibitor perindopril in elderly patients in the PEP-CHF trial (Eur. Heart J. 2006;27:2338-45), another large HFPEF-treatment trial that failed to show significant benefit for the primary endpoint with perindopril treatment but had positive results in some secondary endpoints.
Patients "get these drugs to treat their hypertension, but we believe they may also help their heart failure. It’s a belief system. My colleagues and I already use a lot of spironolactone to treat hypertension, and the TOPCAT results won’t change my practice. I’m not comfortable telling people that you should use an aldosterone antagonist because of TOPCAT," Dr. Teerlink said in an interview.
"An angiotensin-converting enzyme inhibitor will probably work; spironolactone probably works if you get the diagnosis right; we don’t know about beta-blockers; and there is some evidence for using digoxin," said Dr. John G.F. Cleland, professor of cardiology at the University of Hull, Kingston-upon-Hull, England. "There must also be good treatment of hypertension, and judicious use of diuretics."
"I’m careful with beta-blockers; I’ve had some patients who felt miserable on them," said Dr. Borlaug. "Some HFPEF patients don’t have a stroke volume, they don’t have diastole, all they have is heart rate, and if you take that away they are left with no cardiac output."
Aside from controlling blood pressure, experts advise good management of other comorbidities such as coronary disease, atrial fibrillation, diabetes, sleep disordered breathing, and renal disease. "Clinicians should make sure that they are not missing severe coronary artery disease, infiltrative cardiomyopathy, constrictive pericarditis, or other causes of HFPEF that have specific treatments," said Dr. Shah. Some drugs have shown promise in early-phase studies – such as ivabradine and neprilysin – but phase III trials are needed. "My advice on how to manage patients with HFPEF is to make every effort to enroll them in a randomized clinical trial," said Dr. Fonarow.
Another key is making the diagnosis, ideally to prevent development of irreversible cardiovascular damage. "HFPEF is difficult to diagnose with certainty unless you do a cardiac catheterization to measure filling pressures, but in patients with early-stage HFPEF, even their invasive hemodynamics look pretty normal," said Dr. Borlaug. Another approach is an exercise echo, which is noninvasive and can identify stress-induced diastolic dysfunction, but the sensitivity and specificity of this approach remains uncertain, he said. And an elevated natriuretic peptide level can help nail the HFPEF diagnosis in some patients but many other patients have levels within the normal range.
To find HFPEF patients, apply a low index of suspicion and look for breathlessness, loss of functional capacity, signs of congestion, lung crackles, echocardiographic signs, pulmonary artery hypertension, and an enlarged left atrium, he suggested.
"Finding patients is a big challenge," said Dr. Pieske. HFPEF patients tend to be elderly, women, and people who are obese who have hypertension and perhaps diabetes. "They complain of dyspnea and fatigue and many physicians think this is just how it is. They will not consider that there is a true diagnosis behind these symptoms and complaints," especially if the patient has preserved left ventricular ejection fraction and a normal natriuretic peptide level." A diastolic stress test using exercise and an echo exam may identify stable patients with early-stage diastolic dysfunction but this requires confirmation, he said.
HFPEF dominates heart failure, lacks good treatment
Experts say the onus on physicians to diagnose and manage HFPEF will grow substantially, since HFPEF is not only highly prevalent but also increasing faster than heart failure with reduced ejection fraction (HFREF). Results from 13 community-based studies published during 1997-2006 showed that HFPEF represented an average of 55% of all heart failure cases, Dr. Carolyn S.P. Lam said last November during a talk at the American Heart Association’s scientific sessions.
Two recent reports of U.S. data documented that the increasing prevalence of HFPEF is outpacing that of HFREF by 1% per year, driven by the aging of the American population (older age is a risk factor for HFPEF) and by the increasing prevalence of comorbidities that contribute to HFPEF. One of the reports, based on U.S. national data collected by the Get With the Guidelines Program, extrapolated that by the end of this decade about 65% of all patients hospitalized for heart failure will have a left ventricular ejection fraction of 40% or greater, the vast majority with HFPEF (Curr. Heart Fail. Rep. 2013;10:401-10). What’s coming is a HFPEF "epidemic," said Dr. Lam, a cardiologist at the National University Heart Centre in Singapore.
The fact that HFPEF is now at least as prevalent as HFREF, that it is about as lethal independent of what comorbidities might contribute, and that it is growing more prevalent than HFREF sharply contrasts with the absence of treatments with proven efficacy. How did that happen?
"Research into HFPEF is decades behind research into HFREF," said Dr. Fonarow. "HFPEF was largely ignored because the prevailing wisdom [20 or so years ago] was that it represented only a small proportion of cases and that outcomes were much better compared with HFREF." Only with registry data and community-based studies run more recently did cardiologists realize that outcomes from HFPEF were as bad as from HFREF. Progress was further hampered by enrollment of patients who did not have HFPEF or a broad spectrum of patients unable to respond equally well to the treatment under study.
"There are large gaps in knowledge of the pathophysiology of HFPEF and little investment has been made to identify potential therapeutic targets. This creates cascading levels of challenges for developing effective treatments despite the massive unmet need," said Dr. Fonarow.
"We don’t know what to treat yet," was Dr. Teerlink’s summation of the HFPEF dilemma.
Dr. Borlaug said that he has been a consultant to GlaxoSmithKline, Merck, Amgen, CardioKinetix, and DC Devices and has received research support from Atcor. Dr. Solomon said he has been a consultant to and received research support from Novartis and more than 10 other drug and device companies. Dr. Pieske said that he has received honoraria from Bayer, Boehringer Ingelheim, Servier, Medtronic, Bristol-Myers Squibb, Menarini, and Novartis and received research support from Bayer and Medtronic. Dr. Shah said that he has been a consultant to Novartis, Bayer Schering, and the Pulmonary Hypertension Association. Dr. Fonarow said that he has been a consultant to Medtronic, Novartis, and Pfizer. Dr. Teerlink said that he has been an adviser to and received research support from Novartis. Dr. Cleland said that he has received honoraria from Novartis and research funding from Pfizer. Dr. Lam said that she has been a consultant to Bayer, Novartis, and DC Devices and has received research support from Boston Scientific, Medtronic, and Vifor Pharma.
On Twitter @mitchelzoler
