User login
Mitchel is a reporter for MDedge based in the Philadelphia area. He started with the company in 1992, when it was International Medical News Group (IMNG), and has since covered a range of medical specialties. Mitchel trained as a virologist at Roswell Park Memorial Institute in Buffalo, and then worked briefly as a researcher at Boston Children's Hospital before pivoting to journalism as a AAAS Mass Media Fellow in 1980. His first reporting job was with Science Digest magazine, and from the mid-1980s to early-1990s he was a reporter with Medical World News. @mitchelzoler
Delayed consent in HEAT-PPCI draws criticism
WASHINGTON – The delayed consent approach that investigators used to run the HEAT-PPCI trial generated almost as much controversy as the surprising finding that unfractionated heparin was safer than bivalirudin for anticoagulating ST-segment elevation myocardial infarction patients undergoing primary coronary stenting.
"I’m extremely bothered by the fact that you entered patients into the study without getting their permission," said Dr. William W. O’Neill, who spoke from the stage following the HEAT-PPCI (How Effective are Antithrombotic Therapies in Primary Percutaneous Coronary Intervention) report as a member of the discussion panel for the session at the annual meeting of the American College of Cardiology.
"There have been people who say that no one [experiencing an acute myocardial infarction] remembers their informed consent, but most of us have a very strong ethical concern. There is a social contract we make with our patients to make sure they are not subject to research without their permission. That’s in the Declaration of Helsinki," said Dr. O’Neill, medical director of the Center for Structural Heart Disease at Henry Ford Hospital in Detroit.
But Dr. Adeel Shahzad, who presented the study’s findings, and Dr. Rod Stables, the study’s principal investigator, unequivocally defended the ethics of their approach, and stressed that the design had been approved by three separate national bodies charged with ethical oversight of U.K. medical studies.
"We’re not advocating the use of delayed consent for all trials. Every trial has different circumstances. We compared two drugs that were in routine use throughout the world and that were used for licensed indications," Dr. Shahzad, an interventional cardiologist at Liverpool (England) Heart and Chest Hospital, said in reply to Dr. O’Neill’s criticism. "In the primary PCI [percutaneous coronary intervention] setting the patient is in pain and possibly under the influence of drugs. In this setting and with an average door-to-balloon time of 29 minutes we don’t believe that informed consent is possible."
"In HEAT-PPCI, 75% of patients were randomized within 9 minutes or arrival. It’s a debatable point whether it is possible to obtain any meaningful consent on that time schedule," said Dr. Stables, director of the cardiac catheterization laboratory at Liverpool Heart and Chest Hospital.
The study’s design called for the informed consent process to occur following intervention, once patients had stabilized. Of the 1,829 patients treated in Liverpool who met the study’s entry criteria, 1,812 (99%) subsequently gave their consent to be included in the trial. Dr. Stables also noted that in addition to the formal approval obtained from three U.K. medical ethics groups, the study’s design also won approval from a panel composed of patients, and the trial also employed a full-time patient advocate.
The ethics of the approach used in HEAT-PPCI were endorsed by both Dr. Spencer B. King III, an interventional cardiologist at St. Joseph’s Health System in Atlanta, and by Dr. David R. Holmes Jr., an interventional cardiologist and professor of medicine at the Mayo Clinic in Rochester, Minn.
Dr. Holmes recalled his own experience when hospitalized on an emergency basis for "massive" gastrointestinal bleeding. A few weeks later he was contacted about a study that he had agreed to participate in while in a very debilitated state before he received treatment; he had absolutely no recollection of ever agreeing to be in the study.
Dr. O’Neill, Dr. Shahzad, Dr. Stables, Dr. King, and Dr. Holmes had no relevant disclosures.
On Twitter @mitchelzoler
WASHINGTON – The delayed consent approach that investigators used to run the HEAT-PPCI trial generated almost as much controversy as the surprising finding that unfractionated heparin was safer than bivalirudin for anticoagulating ST-segment elevation myocardial infarction patients undergoing primary coronary stenting.
"I’m extremely bothered by the fact that you entered patients into the study without getting their permission," said Dr. William W. O’Neill, who spoke from the stage following the HEAT-PPCI (How Effective are Antithrombotic Therapies in Primary Percutaneous Coronary Intervention) report as a member of the discussion panel for the session at the annual meeting of the American College of Cardiology.
"There have been people who say that no one [experiencing an acute myocardial infarction] remembers their informed consent, but most of us have a very strong ethical concern. There is a social contract we make with our patients to make sure they are not subject to research without their permission. That’s in the Declaration of Helsinki," said Dr. O’Neill, medical director of the Center for Structural Heart Disease at Henry Ford Hospital in Detroit.
But Dr. Adeel Shahzad, who presented the study’s findings, and Dr. Rod Stables, the study’s principal investigator, unequivocally defended the ethics of their approach, and stressed that the design had been approved by three separate national bodies charged with ethical oversight of U.K. medical studies.
"We’re not advocating the use of delayed consent for all trials. Every trial has different circumstances. We compared two drugs that were in routine use throughout the world and that were used for licensed indications," Dr. Shahzad, an interventional cardiologist at Liverpool (England) Heart and Chest Hospital, said in reply to Dr. O’Neill’s criticism. "In the primary PCI [percutaneous coronary intervention] setting the patient is in pain and possibly under the influence of drugs. In this setting and with an average door-to-balloon time of 29 minutes we don’t believe that informed consent is possible."
"In HEAT-PPCI, 75% of patients were randomized within 9 minutes or arrival. It’s a debatable point whether it is possible to obtain any meaningful consent on that time schedule," said Dr. Stables, director of the cardiac catheterization laboratory at Liverpool Heart and Chest Hospital.
The study’s design called for the informed consent process to occur following intervention, once patients had stabilized. Of the 1,829 patients treated in Liverpool who met the study’s entry criteria, 1,812 (99%) subsequently gave their consent to be included in the trial. Dr. Stables also noted that in addition to the formal approval obtained from three U.K. medical ethics groups, the study’s design also won approval from a panel composed of patients, and the trial also employed a full-time patient advocate.
The ethics of the approach used in HEAT-PPCI were endorsed by both Dr. Spencer B. King III, an interventional cardiologist at St. Joseph’s Health System in Atlanta, and by Dr. David R. Holmes Jr., an interventional cardiologist and professor of medicine at the Mayo Clinic in Rochester, Minn.
Dr. Holmes recalled his own experience when hospitalized on an emergency basis for "massive" gastrointestinal bleeding. A few weeks later he was contacted about a study that he had agreed to participate in while in a very debilitated state before he received treatment; he had absolutely no recollection of ever agreeing to be in the study.
Dr. O’Neill, Dr. Shahzad, Dr. Stables, Dr. King, and Dr. Holmes had no relevant disclosures.
On Twitter @mitchelzoler
WASHINGTON – The delayed consent approach that investigators used to run the HEAT-PPCI trial generated almost as much controversy as the surprising finding that unfractionated heparin was safer than bivalirudin for anticoagulating ST-segment elevation myocardial infarction patients undergoing primary coronary stenting.
"I’m extremely bothered by the fact that you entered patients into the study without getting their permission," said Dr. William W. O’Neill, who spoke from the stage following the HEAT-PPCI (How Effective are Antithrombotic Therapies in Primary Percutaneous Coronary Intervention) report as a member of the discussion panel for the session at the annual meeting of the American College of Cardiology.
"There have been people who say that no one [experiencing an acute myocardial infarction] remembers their informed consent, but most of us have a very strong ethical concern. There is a social contract we make with our patients to make sure they are not subject to research without their permission. That’s in the Declaration of Helsinki," said Dr. O’Neill, medical director of the Center for Structural Heart Disease at Henry Ford Hospital in Detroit.
But Dr. Adeel Shahzad, who presented the study’s findings, and Dr. Rod Stables, the study’s principal investigator, unequivocally defended the ethics of their approach, and stressed that the design had been approved by three separate national bodies charged with ethical oversight of U.K. medical studies.
"We’re not advocating the use of delayed consent for all trials. Every trial has different circumstances. We compared two drugs that were in routine use throughout the world and that were used for licensed indications," Dr. Shahzad, an interventional cardiologist at Liverpool (England) Heart and Chest Hospital, said in reply to Dr. O’Neill’s criticism. "In the primary PCI [percutaneous coronary intervention] setting the patient is in pain and possibly under the influence of drugs. In this setting and with an average door-to-balloon time of 29 minutes we don’t believe that informed consent is possible."
"In HEAT-PPCI, 75% of patients were randomized within 9 minutes or arrival. It’s a debatable point whether it is possible to obtain any meaningful consent on that time schedule," said Dr. Stables, director of the cardiac catheterization laboratory at Liverpool Heart and Chest Hospital.
The study’s design called for the informed consent process to occur following intervention, once patients had stabilized. Of the 1,829 patients treated in Liverpool who met the study’s entry criteria, 1,812 (99%) subsequently gave their consent to be included in the trial. Dr. Stables also noted that in addition to the formal approval obtained from three U.K. medical ethics groups, the study’s design also won approval from a panel composed of patients, and the trial also employed a full-time patient advocate.
The ethics of the approach used in HEAT-PPCI were endorsed by both Dr. Spencer B. King III, an interventional cardiologist at St. Joseph’s Health System in Atlanta, and by Dr. David R. Holmes Jr., an interventional cardiologist and professor of medicine at the Mayo Clinic in Rochester, Minn.
Dr. Holmes recalled his own experience when hospitalized on an emergency basis for "massive" gastrointestinal bleeding. A few weeks later he was contacted about a study that he had agreed to participate in while in a very debilitated state before he received treatment; he had absolutely no recollection of ever agreeing to be in the study.
Dr. O’Neill, Dr. Shahzad, Dr. Stables, Dr. King, and Dr. Holmes had no relevant disclosures.
On Twitter @mitchelzoler
EXPERT OPINION AT ACC 2014
Heparin surpasses bivalirudin in STEMI primary PCI
WASHINGTON – Unfractionated heparin outperformed bivalirudin for 28-day outcomes in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention in a single-center randomized trial of more than 1,800 patients when use of a glycoprotein IIb/IIIa inhibitor was kept to a minimum.
Use of unfractionated heparin (UFH) compared with bivalirudin (Angiomax) led to a significant reduction in the rate of combined major adverse coronary events after 28 days compared with bivalirudin driven primarily by a significant cut in the rate of acute stent thrombosis and repeat myocardial infarctions with no increased risk for major bleeding events, consistent results across all subgroups examined, and the potential for substantial savings in drug costs, Dr. Adeel Shahzad said at the annual meeting of the American College of Cardiology.

The combined rate of major adverse coronary events was 6% in patients treated with UFH and 9% in those treated with bivalirudin, a statistically significant difference for the study’s primary efficacy endpoint. The rate of major bleeding events, the study’s primary safety endpoint, occurred in 3.5% of patients treated with bivalirudin and in 3.1% of those who received UFH, a difference that was not statistically significant.
The superior outcome with UFH in this study, which contrasted sharply with the results of several prior head-to-head comparisons of the two drugs in patients with a ST-segment elevation myocardial infarction (STEMI) or other acute coronary syndrome events undergoing primary percutaneous coronary intervention (PCI) seemed closely tied to the study’s protocol that restricted coadministration of a glycoprotein IIb/IIIa inhibitor (such as abciximab [ReoPro]) to only those patients experiencing thrombotic complications during PCI ("bailout" use).
"For the first time, unfractionated heparin was used like bivalirudin," with low use of a glycoprotein IIb/IIIa inhibitor, noted the study’s principal investigator, Dr. Rod Stables, director of the cardiac catheterization laboratory at Liverpool (England) Heart and Chest Hospital.
Current STEMI management guidelines from the American College of Cardiology and American Heart Association (J. Am. Coll. Cardiol. 2013;61:e78-140) and from the European Society of Cardiology (Eur. Heart J. 2012;33:2569-619) endorse use of either bivalirudin or UFH as an anticoagulant when patients undergo primary PCI. The European guidelines say that bivalirudin is preferred over UFH when a glycoprotein IIb/IIIa antagonist is administered along with UFH.
"It’s perfectly legitimate with the guidelines to use unfractionated heparin, but most people [in the United States] use bivalirudin or a glycoprotein IIb/IIIa inhibitor with heparin," said Dr. Spencer B. King III, an interventional cardiologist at St. Joseph’s Health System in Atlanta.

Based on the new results, "people will say it’s really interesting, and they may at least think about not using bivalirudin all the time," said Dr. David R. Holmes Jr., an interventional cardiologist and professor of medicine at the Mayo Clinic in Rochester, Minn. "Those who are believers in bivalirudin will continue to use it, but others will say that heparin did just fine [in this trial] and is less expensive.
Dr. Stables predicted that based on these results, interventionalists at his institution who can choose between bivalirudin and UFH will increasingly use heparin, a change that he estimated could save his hospital as much as 500,000 pounds per year (about $800,000).
The HEAT-PPCI (How Effective are Antithrombotic Therapies in Primary PCI) trial randomized 1,829 STEMI patients at Liverpool Heart and Chest Hospital during February 2012–November 2013. The data analysis included the 1,812 patients from the series who agreed to participate in the study, which had a delayed-consent design (see sidebar). A total of 1,917 patients with STEMI presented at Liverpool during this 22-month period, with about 5% of patients excluded because of prespecified exclusion criteria. This produced a highly representative, "real-world" population. "It’s the closest you can get to an all-comers trial," Dr. Stables said.
About 62% of the patients received ticagrelor (Brilinta) as an antiplatelet drug, about 27% received prasugrel (Effient), and the remaining 11% received clopidogrel. About 14% of the bivalirudin-treated patients and about 16% of those randomized to receive UFH were treated with a glycoprotein IIb/IIIa inhibitor. The door-to-balloon time for patients in the study averaged 29 minutes, and the average age of the patients was 63 years. About 81% of patients underwent coronary catheterization by radial-artery access, with the remainder undergoing femoral-artery access; about 82% of all patients had a PCI procedure actually performed. And of those who underwent primary PCI, about 92% of all patients received at least one coronary stent. The 907 consenting patients randomized to UFH received a dose of 70 units/kg prior to catheterization. The 905 consenting patients randomized to bivalirudin received a 0.75-mg/kg bolus and a 1.75-mg/kg/hour infusion during their procedure.
The difference in rates of major adverse coronary events during the 28-day follow-up seemed driven primarily by a difference in the rate of myocardial infarctions, which occurred in 2.3% of the bivalirudin patients and in 0.8% of the UFH patients, Dr. Shahzad said. The rate of acute stent thrombosis was 2.9% in the bivalirudin arm and 0.9% in patients on UFH.
Several prior trials that compared UFH and bivalirudin in STEMI or acute coronary syndrome patients undergoing PCI or other invasive procedures used either mandatory coadministration of a glycoprotein IIb/IIIa inhibitor along with UHF, or a much higher rate of discretionary use, such as in the ACUITY (N. Engl. J. Med. 2006;355:2203-16), HORIZONS (N. Engl. J. Med. 2008;358:2218-30), and EUROMAX (N. Engl. J. Med. 2013;369:2207-17) studies. In all these trials, the rates of major bleeding events were significantly higher in the UFH arm, and this appeared to lead to an increased number of adverse coronary events during the first month following intervention.
"What we’ve seen in every trial before was a reduction in bleeding complications of about 40% to 50%. The National Cardiovascular Data Registry shows a tremendous decrease [in bleeding] when you change from heparin to bivalirudin," said Dr. Roxana Mehran, an interventional cardiologist and professor of medicine at Mount Sinai Hospital in New York.
"All the trials had differential use of glycoprotein IIb/IIIa inhibitors" in the UFH and bivalirudin arms, noted Dr. Shahzad, an interventional cardiologist at Liverpool Heart and Chest Hospital.
Dr. Stables said that the low bleeding rates with UFH in the new study seemed largely dependent on the relatively low rate at which patients received a glycoprotein IIb/IIIa inhibitor and did not correlate with radial-artery access. "We did a prespecified subgroup analysis of patients who underwent radial or femoral access and the outcomes were the same in both groups," he said.
Dr. Gregg W. Stone, who served as lead investigator for ACUITY and HORIZONS, contended that the trialists in HEAT-PPCI underdosed bivalirudin based on the average activated clotting time (ACT) of 270 seconds in the bivalirudin-treated patients in the study. Most patients properly treated with bivalirudin achieve ACTs of 350-450 seconds, said Dr. Stone, professor of medicine and director of cardiovascular research and education at Columbia University in New York. Dr. Stone also noted that the "results of this single-center study were markedly different from three randomized, controlled trials done at 250 centers and involving more than 8,000 patients."
Dr. Shahzad countered that the ACT-measuring device used at his institution was consistently accurate but measures ACT levels that are typically 50 seconds below values obtained with most other devices.
HEAT-PPCI did not receive any commercial funding. Dr. Shahzad, Dr. Stables, Dr. King, and Dr. Holmes had no disclosures. Dr. Mehran has received consultant fees, honoraria, and/or research grants from 10 drug and device companies, but not from The Medicines Company, which markets Angiomax. Dr. Stone has grants or fees from, or has an ownership position in, several drug or device companies, but not The Medicines Company.
On Twitter @mitchelzoler

|
Mitchel L. Zoler/Frontline Medical News
|
This was an extremely well-conducted trial with a very large patient population and meticulous oversight and exceptionally complete follow-up.
There has been a signal of increased stent thrombosis early on with bivalirudin, as was seen in the EUROMAX trial (N. Engl. J. Med. 2013;369:2207-17), but over the longer term these differences in event rates have been mitigated. A surprise in the current study was the absence of a difference in major bleeding complications between the two drug arms. The trial used radial access in 80% of patients, and radial access has been associated with significant decreases in access-site bleeding compared with transfemoral access. Future studies should examine the role of transradial access in leading to reduced bleeding in this setting.
David Kandzari, M.D., is an interventional cardiologist with Piedmont Healthcare in Atlanta. He has been a consultant to, and has received honoraria from, Micell Technologies, Biotronik, Boston Scientific, Thoratec, and Medtronic. He made these comments as a discussant for the trial.
|
|
Mitchel L. Zoler/Frontline Medical News
|
This was an extremely well-conducted trial with a very large patient population and meticulous oversight and exceptionally complete follow-up.
There has been a signal of increased stent thrombosis early on with bivalirudin, as was seen in the EUROMAX trial (N. Engl. J. Med. 2013;369:2207-17), but over the longer term these differences in event rates have been mitigated. A surprise in the current study was the absence of a difference in major bleeding complications between the two drug arms. The trial used radial access in 80% of patients, and radial access has been associated with significant decreases in access-site bleeding compared with transfemoral access. Future studies should examine the role of transradial access in leading to reduced bleeding in this setting.
David Kandzari, M.D., is an interventional cardiologist with Piedmont Healthcare in Atlanta. He has been a consultant to, and has received honoraria from, Micell Technologies, Biotronik, Boston Scientific, Thoratec, and Medtronic. He made these comments as a discussant for the trial.
|
|
Mitchel L. Zoler/Frontline Medical News
|
This was an extremely well-conducted trial with a very large patient population and meticulous oversight and exceptionally complete follow-up.
There has been a signal of increased stent thrombosis early on with bivalirudin, as was seen in the EUROMAX trial (N. Engl. J. Med. 2013;369:2207-17), but over the longer term these differences in event rates have been mitigated. A surprise in the current study was the absence of a difference in major bleeding complications between the two drug arms. The trial used radial access in 80% of patients, and radial access has been associated with significant decreases in access-site bleeding compared with transfemoral access. Future studies should examine the role of transradial access in leading to reduced bleeding in this setting.
David Kandzari, M.D., is an interventional cardiologist with Piedmont Healthcare in Atlanta. He has been a consultant to, and has received honoraria from, Micell Technologies, Biotronik, Boston Scientific, Thoratec, and Medtronic. He made these comments as a discussant for the trial.
WASHINGTON – Unfractionated heparin outperformed bivalirudin for 28-day outcomes in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention in a single-center randomized trial of more than 1,800 patients when use of a glycoprotein IIb/IIIa inhibitor was kept to a minimum.
Use of unfractionated heparin (UFH) compared with bivalirudin (Angiomax) led to a significant reduction in the rate of combined major adverse coronary events after 28 days compared with bivalirudin driven primarily by a significant cut in the rate of acute stent thrombosis and repeat myocardial infarctions with no increased risk for major bleeding events, consistent results across all subgroups examined, and the potential for substantial savings in drug costs, Dr. Adeel Shahzad said at the annual meeting of the American College of Cardiology.
The combined rate of major adverse coronary events was 6% in patients treated with UFH and 9% in those treated with bivalirudin, a statistically significant difference for the study’s primary efficacy endpoint. The rate of major bleeding events, the study’s primary safety endpoint, occurred in 3.5% of patients treated with bivalirudin and in 3.1% of those who received UFH, a difference that was not statistically significant.
The superior outcome with UFH in this study, which contrasted sharply with the results of several prior head-to-head comparisons of the two drugs in patients with a ST-segment elevation myocardial infarction (STEMI) or other acute coronary syndrome events undergoing primary percutaneous coronary intervention (PCI) seemed closely tied to the study’s protocol that restricted coadministration of a glycoprotein IIb/IIIa inhibitor (such as abciximab [ReoPro]) to only those patients experiencing thrombotic complications during PCI ("bailout" use).
"For the first time, unfractionated heparin was used like bivalirudin," with low use of a glycoprotein IIb/IIIa inhibitor, noted the study’s principal investigator, Dr. Rod Stables, director of the cardiac catheterization laboratory at Liverpool (England) Heart and Chest Hospital.
Current STEMI management guidelines from the American College of Cardiology and American Heart Association (J. Am. Coll. Cardiol. 2013;61:e78-140) and from the European Society of Cardiology (Eur. Heart J. 2012;33:2569-619) endorse use of either bivalirudin or UFH as an anticoagulant when patients undergo primary PCI. The European guidelines say that bivalirudin is preferred over UFH when a glycoprotein IIb/IIIa antagonist is administered along with UFH.
"It’s perfectly legitimate with the guidelines to use unfractionated heparin, but most people [in the United States] use bivalirudin or a glycoprotein IIb/IIIa inhibitor with heparin," said Dr. Spencer B. King III, an interventional cardiologist at St. Joseph’s Health System in Atlanta.
Based on the new results, "people will say it’s really interesting, and they may at least think about not using bivalirudin all the time," said Dr. David R. Holmes Jr., an interventional cardiologist and professor of medicine at the Mayo Clinic in Rochester, Minn. "Those who are believers in bivalirudin will continue to use it, but others will say that heparin did just fine [in this trial] and is less expensive.
Dr. Stables predicted that based on these results, interventionalists at his institution who can choose between bivalirudin and UFH will increasingly use heparin, a change that he estimated could save his hospital as much as 500,000 pounds per year (about $800,000).
The HEAT-PPCI (How Effective are Antithrombotic Therapies in Primary PCI) trial randomized 1,829 STEMI patients at Liverpool Heart and Chest Hospital during February 2012–November 2013. The data analysis included the 1,812 patients from the series who agreed to participate in the study, which had a delayed-consent design (see sidebar). A total of 1,917 patients with STEMI presented at Liverpool during this 22-month period, with about 5% of patients excluded because of prespecified exclusion criteria. This produced a highly representative, "real-world" population. "It’s the closest you can get to an all-comers trial," Dr. Stables said.
About 62% of the patients received ticagrelor (Brilinta) as an antiplatelet drug, about 27% received prasugrel (Effient), and the remaining 11% received clopidogrel. About 14% of the bivalirudin-treated patients and about 16% of those randomized to receive UFH were treated with a glycoprotein IIb/IIIa inhibitor. The door-to-balloon time for patients in the study averaged 29 minutes, and the average age of the patients was 63 years. About 81% of patients underwent coronary catheterization by radial-artery access, with the remainder undergoing femoral-artery access; about 82% of all patients had a PCI procedure actually performed. And of those who underwent primary PCI, about 92% of all patients received at least one coronary stent. The 907 consenting patients randomized to UFH received a dose of 70 units/kg prior to catheterization. The 905 consenting patients randomized to bivalirudin received a 0.75-mg/kg bolus and a 1.75-mg/kg/hour infusion during their procedure.
The difference in rates of major adverse coronary events during the 28-day follow-up seemed driven primarily by a difference in the rate of myocardial infarctions, which occurred in 2.3% of the bivalirudin patients and in 0.8% of the UFH patients, Dr. Shahzad said. The rate of acute stent thrombosis was 2.9% in the bivalirudin arm and 0.9% in patients on UFH.
Several prior trials that compared UFH and bivalirudin in STEMI or acute coronary syndrome patients undergoing PCI or other invasive procedures used either mandatory coadministration of a glycoprotein IIb/IIIa inhibitor along with UHF, or a much higher rate of discretionary use, such as in the ACUITY (N. Engl. J. Med. 2006;355:2203-16), HORIZONS (N. Engl. J. Med. 2008;358:2218-30), and EUROMAX (N. Engl. J. Med. 2013;369:2207-17) studies. In all these trials, the rates of major bleeding events were significantly higher in the UFH arm, and this appeared to lead to an increased number of adverse coronary events during the first month following intervention.
"What we’ve seen in every trial before was a reduction in bleeding complications of about 40% to 50%. The National Cardiovascular Data Registry shows a tremendous decrease [in bleeding] when you change from heparin to bivalirudin," said Dr. Roxana Mehran, an interventional cardiologist and professor of medicine at Mount Sinai Hospital in New York.
"All the trials had differential use of glycoprotein IIb/IIIa inhibitors" in the UFH and bivalirudin arms, noted Dr. Shahzad, an interventional cardiologist at Liverpool Heart and Chest Hospital.
Dr. Stables said that the low bleeding rates with UFH in the new study seemed largely dependent on the relatively low rate at which patients received a glycoprotein IIb/IIIa inhibitor and did not correlate with radial-artery access. "We did a prespecified subgroup analysis of patients who underwent radial or femoral access and the outcomes were the same in both groups," he said.
Dr. Gregg W. Stone, who served as lead investigator for ACUITY and HORIZONS, contended that the trialists in HEAT-PPCI underdosed bivalirudin based on the average activated clotting time (ACT) of 270 seconds in the bivalirudin-treated patients in the study. Most patients properly treated with bivalirudin achieve ACTs of 350-450 seconds, said Dr. Stone, professor of medicine and director of cardiovascular research and education at Columbia University in New York. Dr. Stone also noted that the "results of this single-center study were markedly different from three randomized, controlled trials done at 250 centers and involving more than 8,000 patients."
Dr. Shahzad countered that the ACT-measuring device used at his institution was consistently accurate but measures ACT levels that are typically 50 seconds below values obtained with most other devices.
HEAT-PPCI did not receive any commercial funding. Dr. Shahzad, Dr. Stables, Dr. King, and Dr. Holmes had no disclosures. Dr. Mehran has received consultant fees, honoraria, and/or research grants from 10 drug and device companies, but not from The Medicines Company, which markets Angiomax. Dr. Stone has grants or fees from, or has an ownership position in, several drug or device companies, but not The Medicines Company.
On Twitter @mitchelzoler
WASHINGTON – Unfractionated heparin outperformed bivalirudin for 28-day outcomes in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention in a single-center randomized trial of more than 1,800 patients when use of a glycoprotein IIb/IIIa inhibitor was kept to a minimum.
Use of unfractionated heparin (UFH) compared with bivalirudin (Angiomax) led to a significant reduction in the rate of combined major adverse coronary events after 28 days compared with bivalirudin driven primarily by a significant cut in the rate of acute stent thrombosis and repeat myocardial infarctions with no increased risk for major bleeding events, consistent results across all subgroups examined, and the potential for substantial savings in drug costs, Dr. Adeel Shahzad said at the annual meeting of the American College of Cardiology.
The combined rate of major adverse coronary events was 6% in patients treated with UFH and 9% in those treated with bivalirudin, a statistically significant difference for the study’s primary efficacy endpoint. The rate of major bleeding events, the study’s primary safety endpoint, occurred in 3.5% of patients treated with bivalirudin and in 3.1% of those who received UFH, a difference that was not statistically significant.
The superior outcome with UFH in this study, which contrasted sharply with the results of several prior head-to-head comparisons of the two drugs in patients with a ST-segment elevation myocardial infarction (STEMI) or other acute coronary syndrome events undergoing primary percutaneous coronary intervention (PCI) seemed closely tied to the study’s protocol that restricted coadministration of a glycoprotein IIb/IIIa inhibitor (such as abciximab [ReoPro]) to only those patients experiencing thrombotic complications during PCI ("bailout" use).
"For the first time, unfractionated heparin was used like bivalirudin," with low use of a glycoprotein IIb/IIIa inhibitor, noted the study’s principal investigator, Dr. Rod Stables, director of the cardiac catheterization laboratory at Liverpool (England) Heart and Chest Hospital.
Current STEMI management guidelines from the American College of Cardiology and American Heart Association (J. Am. Coll. Cardiol. 2013;61:e78-140) and from the European Society of Cardiology (Eur. Heart J. 2012;33:2569-619) endorse use of either bivalirudin or UFH as an anticoagulant when patients undergo primary PCI. The European guidelines say that bivalirudin is preferred over UFH when a glycoprotein IIb/IIIa antagonist is administered along with UFH.
"It’s perfectly legitimate with the guidelines to use unfractionated heparin, but most people [in the United States] use bivalirudin or a glycoprotein IIb/IIIa inhibitor with heparin," said Dr. Spencer B. King III, an interventional cardiologist at St. Joseph’s Health System in Atlanta.
Based on the new results, "people will say it’s really interesting, and they may at least think about not using bivalirudin all the time," said Dr. David R. Holmes Jr., an interventional cardiologist and professor of medicine at the Mayo Clinic in Rochester, Minn. "Those who are believers in bivalirudin will continue to use it, but others will say that heparin did just fine [in this trial] and is less expensive.
Dr. Stables predicted that based on these results, interventionalists at his institution who can choose between bivalirudin and UFH will increasingly use heparin, a change that he estimated could save his hospital as much as 500,000 pounds per year (about $800,000).
The HEAT-PPCI (How Effective are Antithrombotic Therapies in Primary PCI) trial randomized 1,829 STEMI patients at Liverpool Heart and Chest Hospital during February 2012–November 2013. The data analysis included the 1,812 patients from the series who agreed to participate in the study, which had a delayed-consent design (see sidebar). A total of 1,917 patients with STEMI presented at Liverpool during this 22-month period, with about 5% of patients excluded because of prespecified exclusion criteria. This produced a highly representative, "real-world" population. "It’s the closest you can get to an all-comers trial," Dr. Stables said.
About 62% of the patients received ticagrelor (Brilinta) as an antiplatelet drug, about 27% received prasugrel (Effient), and the remaining 11% received clopidogrel. About 14% of the bivalirudin-treated patients and about 16% of those randomized to receive UFH were treated with a glycoprotein IIb/IIIa inhibitor. The door-to-balloon time for patients in the study averaged 29 minutes, and the average age of the patients was 63 years. About 81% of patients underwent coronary catheterization by radial-artery access, with the remainder undergoing femoral-artery access; about 82% of all patients had a PCI procedure actually performed. And of those who underwent primary PCI, about 92% of all patients received at least one coronary stent. The 907 consenting patients randomized to UFH received a dose of 70 units/kg prior to catheterization. The 905 consenting patients randomized to bivalirudin received a 0.75-mg/kg bolus and a 1.75-mg/kg/hour infusion during their procedure.
The difference in rates of major adverse coronary events during the 28-day follow-up seemed driven primarily by a difference in the rate of myocardial infarctions, which occurred in 2.3% of the bivalirudin patients and in 0.8% of the UFH patients, Dr. Shahzad said. The rate of acute stent thrombosis was 2.9% in the bivalirudin arm and 0.9% in patients on UFH.
Several prior trials that compared UFH and bivalirudin in STEMI or acute coronary syndrome patients undergoing PCI or other invasive procedures used either mandatory coadministration of a glycoprotein IIb/IIIa inhibitor along with UHF, or a much higher rate of discretionary use, such as in the ACUITY (N. Engl. J. Med. 2006;355:2203-16), HORIZONS (N. Engl. J. Med. 2008;358:2218-30), and EUROMAX (N. Engl. J. Med. 2013;369:2207-17) studies. In all these trials, the rates of major bleeding events were significantly higher in the UFH arm, and this appeared to lead to an increased number of adverse coronary events during the first month following intervention.
"What we’ve seen in every trial before was a reduction in bleeding complications of about 40% to 50%. The National Cardiovascular Data Registry shows a tremendous decrease [in bleeding] when you change from heparin to bivalirudin," said Dr. Roxana Mehran, an interventional cardiologist and professor of medicine at Mount Sinai Hospital in New York.
"All the trials had differential use of glycoprotein IIb/IIIa inhibitors" in the UFH and bivalirudin arms, noted Dr. Shahzad, an interventional cardiologist at Liverpool Heart and Chest Hospital.
Dr. Stables said that the low bleeding rates with UFH in the new study seemed largely dependent on the relatively low rate at which patients received a glycoprotein IIb/IIIa inhibitor and did not correlate with radial-artery access. "We did a prespecified subgroup analysis of patients who underwent radial or femoral access and the outcomes were the same in both groups," he said.
Dr. Gregg W. Stone, who served as lead investigator for ACUITY and HORIZONS, contended that the trialists in HEAT-PPCI underdosed bivalirudin based on the average activated clotting time (ACT) of 270 seconds in the bivalirudin-treated patients in the study. Most patients properly treated with bivalirudin achieve ACTs of 350-450 seconds, said Dr. Stone, professor of medicine and director of cardiovascular research and education at Columbia University in New York. Dr. Stone also noted that the "results of this single-center study were markedly different from three randomized, controlled trials done at 250 centers and involving more than 8,000 patients."
Dr. Shahzad countered that the ACT-measuring device used at his institution was consistently accurate but measures ACT levels that are typically 50 seconds below values obtained with most other devices.
HEAT-PPCI did not receive any commercial funding. Dr. Shahzad, Dr. Stables, Dr. King, and Dr. Holmes had no disclosures. Dr. Mehran has received consultant fees, honoraria, and/or research grants from 10 drug and device companies, but not from The Medicines Company, which markets Angiomax. Dr. Stone has grants or fees from, or has an ownership position in, several drug or device companies, but not The Medicines Company.
On Twitter @mitchelzoler
AT ACC 14

A fib–associated stroke jumped 42% in 2000s
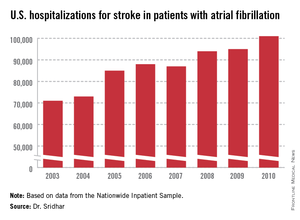
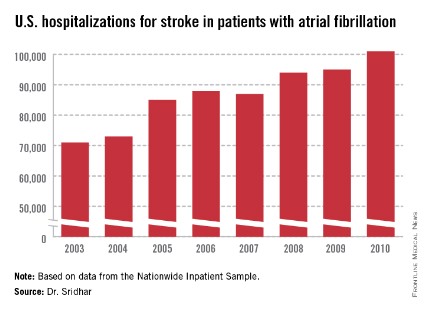
WASHINGTON – A third more Americans were hospitalized for ischemic stroke in 2010 than 7 years before, and the incidence of stroke associated with atrial fibrillation grew even more sharply, up by 42% in 2010 relative to the 2003 rate, according to nationwide data collected in the Nationwide Inpatient Sample.
The period 2003-2010 also witnessed other notable changes in hospitalized U.S. patients with stroke or atrial fibrillation (AF) or both. The in-hospital mortality rate of hospitalized stroke patients with AF dropped from 12% in 2003 to 9% in 2010. During the same period the prevalence of hospitalized AF patients with a high stroke risk, identified by a CHADS (congestive heart failure, hypertension, age > 75 years, diabetes, and prior stroke or transient ischemic attack) score greater than two rose from 20% of hospitalized AF patients in 2003 to 29% in 2010, reflecting an increased prevalence of the comorbidities that drive the CHADS score. The prevalence of a prior stroke or transient ischemic attack (TIA) among hospitalized American AF patients jumped from 5% in 2003 to 12% in 2010, Dr. Arun R.M. Sridhar said at the annual meeting of the American College of Cardiology.
In a multivariate analysis, prior stroke or TIA was the strongest risk predictor for a subsequent stroke among the hospitalized AF cases in the data base, said Dr. Sridhar, a physician at the University of Kansas Medical Center in Kansas City.
But increases in the prevalence of prior stroke and TIA as well as in other measured comorbidities and the growing elderly population with AF and stroke were together unable to fully explain the rising rate of AF-associated strokes. "The rise in strokes persists despite accounting for the changes in demographics. The reasons are not very clear," Dr. Sridhar said.
The Nationwide Inpatient Sample data, collected annually by the Agency for Healthcare Research and Quality, showed that total U.S. hospitalizations for stroke rose from about 335,000 in 2003 to 448,000 in 2010, a 34% jump over 8 years. During the same time stroke hospitalizations for patients with AF increased from about 71,000 in 2003 to 101,000 in 2010, a 42% relative spike. This means the percent of hospitalized stroke patients who also had AF inched up from 21.2% in 2003 to 22.5% in 2010.
One of the clear drivers of the increase in AF-associated strokes was an aging population. In 2003, 37% of AF associated, hospitalized strokes were in patients aged 85 or older. By 2010, the 85 and older segment had swelled to 42%. The statistics also showed discernable but slower growth in patients aged 65-84 years, as well as in women and whites.
Mortality among hospitalized stroke patients fell across the board, not just among those with coexistent AF. For hospitalized stroke patients without AF in-hospital mortality fell from 5.1% in 2003 to 4.1% in 2010. During the 8 years, the average length of hospitalization dropped by about a day for all stroke patients, but despite that inflation-adjusted hospitalization charges jumped by 27% for patients with stroke and AF and by 39% for those with stroke and no AF.
Dr. Sridhar and his associates also analyzed data from more than 27 million Americans hospitalized with AF during the 8-year period. During this time the prevalence of several comorbidities in AF patients all rose and drove up their CHADS scores, starting with the increased prevalence of a history of stroke or TIA. The prevalence of hypertension in hospitalized AF patients rose from 45% to 48%, the prevalence of concomitant heart failure rose from 31% to 37%, and the prevalence of diabetes rose from 24% to 29%. But all these increases, as well as older patient ages, could not fully account for the 42% jump in the total of hospitalized stroke patients with AF from 2003 to 2010, Dr. Sridhar said.
Dr. Sridhar and his associates said that they had no disclosures.
On Twitter @mitchelzoler
WASHINGTON – A third more Americans were hospitalized for ischemic stroke in 2010 than 7 years before, and the incidence of stroke associated with atrial fibrillation grew even more sharply, up by 42% in 2010 relative to the 2003 rate, according to nationwide data collected in the Nationwide Inpatient Sample.
The period 2003-2010 also witnessed other notable changes in hospitalized U.S. patients with stroke or atrial fibrillation (AF) or both. The in-hospital mortality rate of hospitalized stroke patients with AF dropped from 12% in 2003 to 9% in 2010. During the same period the prevalence of hospitalized AF patients with a high stroke risk, identified by a CHADS (congestive heart failure, hypertension, age > 75 years, diabetes, and prior stroke or transient ischemic attack) score greater than two rose from 20% of hospitalized AF patients in 2003 to 29% in 2010, reflecting an increased prevalence of the comorbidities that drive the CHADS score. The prevalence of a prior stroke or transient ischemic attack (TIA) among hospitalized American AF patients jumped from 5% in 2003 to 12% in 2010, Dr. Arun R.M. Sridhar said at the annual meeting of the American College of Cardiology.
In a multivariate analysis, prior stroke or TIA was the strongest risk predictor for a subsequent stroke among the hospitalized AF cases in the data base, said Dr. Sridhar, a physician at the University of Kansas Medical Center in Kansas City.
But increases in the prevalence of prior stroke and TIA as well as in other measured comorbidities and the growing elderly population with AF and stroke were together unable to fully explain the rising rate of AF-associated strokes. "The rise in strokes persists despite accounting for the changes in demographics. The reasons are not very clear," Dr. Sridhar said.
The Nationwide Inpatient Sample data, collected annually by the Agency for Healthcare Research and Quality, showed that total U.S. hospitalizations for stroke rose from about 335,000 in 2003 to 448,000 in 2010, a 34% jump over 8 years. During the same time stroke hospitalizations for patients with AF increased from about 71,000 in 2003 to 101,000 in 2010, a 42% relative spike. This means the percent of hospitalized stroke patients who also had AF inched up from 21.2% in 2003 to 22.5% in 2010.
One of the clear drivers of the increase in AF-associated strokes was an aging population. In 2003, 37% of AF associated, hospitalized strokes were in patients aged 85 or older. By 2010, the 85 and older segment had swelled to 42%. The statistics also showed discernable but slower growth in patients aged 65-84 years, as well as in women and whites.
Mortality among hospitalized stroke patients fell across the board, not just among those with coexistent AF. For hospitalized stroke patients without AF in-hospital mortality fell from 5.1% in 2003 to 4.1% in 2010. During the 8 years, the average length of hospitalization dropped by about a day for all stroke patients, but despite that inflation-adjusted hospitalization charges jumped by 27% for patients with stroke and AF and by 39% for those with stroke and no AF.
Dr. Sridhar and his associates also analyzed data from more than 27 million Americans hospitalized with AF during the 8-year period. During this time the prevalence of several comorbidities in AF patients all rose and drove up their CHADS scores, starting with the increased prevalence of a history of stroke or TIA. The prevalence of hypertension in hospitalized AF patients rose from 45% to 48%, the prevalence of concomitant heart failure rose from 31% to 37%, and the prevalence of diabetes rose from 24% to 29%. But all these increases, as well as older patient ages, could not fully account for the 42% jump in the total of hospitalized stroke patients with AF from 2003 to 2010, Dr. Sridhar said.
Dr. Sridhar and his associates said that they had no disclosures.
On Twitter @mitchelzoler
WASHINGTON – A third more Americans were hospitalized for ischemic stroke in 2010 than 7 years before, and the incidence of stroke associated with atrial fibrillation grew even more sharply, up by 42% in 2010 relative to the 2003 rate, according to nationwide data collected in the Nationwide Inpatient Sample.
The period 2003-2010 also witnessed other notable changes in hospitalized U.S. patients with stroke or atrial fibrillation (AF) or both. The in-hospital mortality rate of hospitalized stroke patients with AF dropped from 12% in 2003 to 9% in 2010. During the same period the prevalence of hospitalized AF patients with a high stroke risk, identified by a CHADS (congestive heart failure, hypertension, age > 75 years, diabetes, and prior stroke or transient ischemic attack) score greater than two rose from 20% of hospitalized AF patients in 2003 to 29% in 2010, reflecting an increased prevalence of the comorbidities that drive the CHADS score. The prevalence of a prior stroke or transient ischemic attack (TIA) among hospitalized American AF patients jumped from 5% in 2003 to 12% in 2010, Dr. Arun R.M. Sridhar said at the annual meeting of the American College of Cardiology.
In a multivariate analysis, prior stroke or TIA was the strongest risk predictor for a subsequent stroke among the hospitalized AF cases in the data base, said Dr. Sridhar, a physician at the University of Kansas Medical Center in Kansas City.
But increases in the prevalence of prior stroke and TIA as well as in other measured comorbidities and the growing elderly population with AF and stroke were together unable to fully explain the rising rate of AF-associated strokes. "The rise in strokes persists despite accounting for the changes in demographics. The reasons are not very clear," Dr. Sridhar said.
The Nationwide Inpatient Sample data, collected annually by the Agency for Healthcare Research and Quality, showed that total U.S. hospitalizations for stroke rose from about 335,000 in 2003 to 448,000 in 2010, a 34% jump over 8 years. During the same time stroke hospitalizations for patients with AF increased from about 71,000 in 2003 to 101,000 in 2010, a 42% relative spike. This means the percent of hospitalized stroke patients who also had AF inched up from 21.2% in 2003 to 22.5% in 2010.
One of the clear drivers of the increase in AF-associated strokes was an aging population. In 2003, 37% of AF associated, hospitalized strokes were in patients aged 85 or older. By 2010, the 85 and older segment had swelled to 42%. The statistics also showed discernable but slower growth in patients aged 65-84 years, as well as in women and whites.
Mortality among hospitalized stroke patients fell across the board, not just among those with coexistent AF. For hospitalized stroke patients without AF in-hospital mortality fell from 5.1% in 2003 to 4.1% in 2010. During the 8 years, the average length of hospitalization dropped by about a day for all stroke patients, but despite that inflation-adjusted hospitalization charges jumped by 27% for patients with stroke and AF and by 39% for those with stroke and no AF.
Dr. Sridhar and his associates also analyzed data from more than 27 million Americans hospitalized with AF during the 8-year period. During this time the prevalence of several comorbidities in AF patients all rose and drove up their CHADS scores, starting with the increased prevalence of a history of stroke or TIA. The prevalence of hypertension in hospitalized AF patients rose from 45% to 48%, the prevalence of concomitant heart failure rose from 31% to 37%, and the prevalence of diabetes rose from 24% to 29%. But all these increases, as well as older patient ages, could not fully account for the 42% jump in the total of hospitalized stroke patients with AF from 2003 to 2010, Dr. Sridhar said.
Dr. Sridhar and his associates said that they had no disclosures.
On Twitter @mitchelzoler
AT ACC 2014
Major finding: During 2003-2010, annual stroke hospitalizations in U.S. atrial fibrillation patients rose from 71,000 to 101,000, a 42% increase.
Data source: Review of U.S. stroke hospitalizations during 2003-2010 recorded by the Nationwide Inpatient Sample.
Disclosures: Dr. Sridhar and his associates said that they had no disclosures.
Stopping biologics in RA remission remains uncertain
Once a rheumatoid arthritis patient maintains relatively long-term remission on a regimen that combines a synthetic and a biologic disease-modifying drug, temptation mounts to try withdrawing one of the two drugs. Cost considerations alone can make it irresistible to patients to taper down or stop an expensive biologic drug to see whether the patient’s remission can sustain dose reduction or elimination.
But several American experts cautioned that despite growing evidence that at least some rheumatoid arthritis (RA) patients will stay in remission after their biologic disease-modifying antirheumatic drug (DMARD) is gone, the overall efficacy and safety of this approach remain uncertain and it’s not fully clear which patients are the best candidates. Biologic-drug withdrawal has not repeatedly been shown to be fully safe and worth attempting with results from rigorously controlled and adequately powered studies, an evidence base that may still be several years off.

"We can try withdrawing a drug," and many American RA patients do just that, a step more often than not initiated by the patient. "But we don’t know if it’s benign for radiographic progression or for cardiovascular disease events," said Dr. Arthur Weinstein, director of rheumatology at Washington (D.C.) Hospital Center. Even though RA patients in remission who taper down or fully stop their biologic DMARD are usually closely monitored for flare, "there is some evidence for a disconnect between radiographic progression and clinical status," raising the danger that a patient may appear to remain in remission when off the biologic drug while joint deterioration occurs. That, coupled with concern that an RA patient’s cardiovascular risk may worsen off their biologic, can make patients and physicians hesitate when faced with the prospect of messing with a stable remission on a combined biologic and synthetic DMARD regimen, Dr. Weinstein said in an interview.
Biologic withdrawal can succeed
Despite that, it’s undeniable that a substantial minority or even a majority in some series of RA patients in remission or low disease activity on both a biologic and synthetic DMARD can taper down or stop the biologic without a flare occurring. The flip side of that is, of course, that in many series a good number of patients who stop their biologic have a flare and need to have the drug restarted. The good news for these scenarios is that prompt restart of the biologic once a flare occurs generally seems capable of restoring remission with no long-term deleterious effects.
In its 2013 RA treatment guidelines, the European League Against Rheumatism (EULAR) said that "if a patient is in persistent remission after having tapered glucocorticoids, one can consider tapering biologic DMARDs, especially if this treatment is combined with a synthetic DMARD" (Ann. Rheum. Dis. 2014;73:492-509). The most recent RA management guidelines from the American College of Rheumatology, published in 2012, don’t address tapering down or withdrawing biologics. Dr. Weinstein noted that the EULAR recommendation is largely based on consensus expert opinion without a definitive evidence base to justify this approach.
Recent evidence documenting the success of biologic withdrawal includes a report earlier this year from a prespecified analysis in the OPTIMA (Optimal Protocol for Treatment Initiation with Methotrexate and Adalimumab) trial that detailed the outcomes of 207 patients who had stable, low disease activity on combined treatment with adalimumab (Humira) and methotrexate and were randomized to either adalimumab withdrawal or continuation. A year later, 70% of patients who maintained the two-drug regimen and 54% of those randomized to stop adalimumab remained at low disease activity and had no sign of radiographic progression, a between-group difference that was not statistically significant (Lancet 2014;383:321-32).
But in other studies, patients withdrawn from a biologic did not fare nearly as well. The PRESERVE (Maintenance, induction, or withdrawal of etanercept after treatment with etanercept and methotrexate in patients with moderate rheumatoid arthritis) trial randomized 604 RA patients with low disease activity on a combination of etanercept (Enbrel) and methotrexate to continue to receive 50 mg of etanercept plus methotrexate weekly, step down to 25 mg of etanercept plus methotrexate weekly, or completely stop etanercept and continue on methotrexate alone. A year later, the percentage of patients who remained in low disease activity tallied 83% in those who received at least one dose of 50 mg etanercept, 79% of patients in the 25-mg etanercept group, and 43% in the group maintained on methotrexate monotherapy, statistically significant differences between each of the two etanercept groups and the monotherapy arm (Lancet 2013;381:918-29).

A more real-world assessment included 717 RA patients at low disease activity who discontinued their first biologic drug while enrolled in the CORRONA (Consortium of Rheumatology Researchers of North America) registry. The results showed that after 1 year on monotherapy without a biologic, 73% of patients remained free from a flare, and that state persisted in 56% to 18 months, 42% after 2 years, and 28% after 3 years on monotherapy, Dr. Arthur F. Kavanaugh and his associates reported last year at the annual meeting of the American College of Rheumatology (Arthritis Rheum. 2013;65:S603).
Finding the right patients
Despite evidence like this, the right time and the right patient for withdrawing a biologic remain murky. "At this point it is investigational," said Dr. Kavanaugh in an interview. "In the United States, it is mostly initiated by patients. There are many factors to consider, including the duration of RA, how well the patients are doing, is the patient on another drug that should be stopped first such as prednisone, and can the synthetic DMARD like methotrexate sustain the response," said Dr. Kavanaugh, a rheumatologist who is professor of clinical medicine and director of the Center for Innovative Therapy at the University of California, San Diego.
Routinely attempting biologic taper-down or withdrawal from RA patients in remission or at low disease activity "is not yet standard practice, but many patients do it," said Dr. Joel M. Kremer, a rheumatologist and professor of medicine at Albany (N.Y.) Medical College. When he attempts withdrawing a biologic, he prefers to use a gradual taper-down of the dosage, and he said he is more reluctant to attempt this for patients who began treatment with severe RA, and that he is more likely to try it for patients who began treatment with early RA, although he admitted that this remains uncertain. Like all experts interviewed, he said the type of biologic a patient is on doesn’t seem to matter.
"I think this entire area needs more study," Dr. Kremer said in an interview. For example, "are there biomarkers associated with the ability to withdraw? I don’t believe we know much about who can or should withdraw." Once withdrawal occurs, he recommended careful follow-up to detect a flare quickly, and restarting the biologic if even a mild flare occurs.

Biologic withdrawal "is really more investigational" right now, said Dr. Eric L. Matteson, professor and chairman of rheumatology at the Mayo Clinic in Rochester, Minn. "No guideline establishes it as standard of practice. I like to see patients in full remission for a year before considering such a move," he said in an interview. Withdrawal depends on whether the RA is well controlled and not on whether treatment started on early or established RA, he added; nor did he think that a flare after withdrawal precludes future withdrawal attempts as long as the patient returns to a durable, full remission. But Dr. Matteson did caution about also considering extra-articular disease like vasculitis or iritis that may be active even when a patient’s joints are not. "Any evidence of recurrent disease should cause the patient and physician to consider ramping the therapy back up."
Like others, Dr. Matteson highlighted the lingering unknowns about biologic withdrawal that keep it from being standard practice. "We don’t know whether the biologic or synthetic DMARD should get withdrawn first. We don’t have a great handle on what biomarkers to use to help decide if there is subclinical disease activity that necessitates reimplementation of the biologic. We don’t know which patients may violently flare following withdrawal and which might not. We also don’t know if retreatment of the disease is as good when a biologic is withdrawn and then restarted. Finally, I think we overestimate the percent of our patients who are actually in remission or a low-disease-activity state and underestimate who really should continue their medications."
The STARA trial
Dr. Weinstein and his associates recently planned a U.S.-based trial that has come close to launching to address several of these issues, the STARA (Stopping TNF-Alpha Inhibitors in Rheumatoid Arthritis) trial.
"One aspect of STARA is to determine whether there is a profile of clinical, imaging, and laboratory factors that will reliably distinguish patients who will flare from those who will remain in remission" following withdrawal of a biologic DMARD, said Dr. Weinstein, STARA’s lead investigator and also a professor of medicine at Georgetown University in Washington. "Another aim is to find predictors of good candidates for withdrawal. The advantage of STARA is patients are drawn from real-world practice and not from a clinical trial." Dr. Weinstein faulted some of the published studies of biologic withdrawal, such as OPTIMA and PRESERVE, because they included trial patients exclusively and not patients managed in routine practice.
STARA’s design uses a sudden, full withdrawal of the biologic to allow a standardized approach to patients who could enter the study on etanercept, adalimumab, or infliximab (Remicade). For routine practice, gradual withdrawal is more common, though there is no evidence this produces better long-term outcomes. An important withdrawal question that STARA won’t address is: Which is the better drug to remove, the biologic or the synthetic DMARD? In most cases, the more expensive biologic is the patient’s choice, but ideally this issue should be addressed in a randomized trial, Dr. Weinstein said.
But as of late March, STARA’s status was in doubt, as anticipated funding was withdrawn. If STARA isn’t run, Dr. Weinstein predicts that attempts to cut biologics from dual regimens will continue and likely expand, but with unknown implications for long-term safety that he believes can only be addressed in a prospective, randomized trial.
Dr. Weinstein said that he had no relevant disclosures. Dr. Matteson had no disclosures. Dr. Kremer has received research support and been a consultant to more than 10 drug companies. Dr. Kavanaugh has received research support from more than 10 drug companies.
On Twitter @mitchelzoler
Once a rheumatoid arthritis patient maintains relatively long-term remission on a regimen that combines a synthetic and a biologic disease-modifying drug, temptation mounts to try withdrawing one of the two drugs. Cost considerations alone can make it irresistible to patients to taper down or stop an expensive biologic drug to see whether the patient’s remission can sustain dose reduction or elimination.
But several American experts cautioned that despite growing evidence that at least some rheumatoid arthritis (RA) patients will stay in remission after their biologic disease-modifying antirheumatic drug (DMARD) is gone, the overall efficacy and safety of this approach remain uncertain and it’s not fully clear which patients are the best candidates. Biologic-drug withdrawal has not repeatedly been shown to be fully safe and worth attempting with results from rigorously controlled and adequately powered studies, an evidence base that may still be several years off.
"We can try withdrawing a drug," and many American RA patients do just that, a step more often than not initiated by the patient. "But we don’t know if it’s benign for radiographic progression or for cardiovascular disease events," said Dr. Arthur Weinstein, director of rheumatology at Washington (D.C.) Hospital Center. Even though RA patients in remission who taper down or fully stop their biologic DMARD are usually closely monitored for flare, "there is some evidence for a disconnect between radiographic progression and clinical status," raising the danger that a patient may appear to remain in remission when off the biologic drug while joint deterioration occurs. That, coupled with concern that an RA patient’s cardiovascular risk may worsen off their biologic, can make patients and physicians hesitate when faced with the prospect of messing with a stable remission on a combined biologic and synthetic DMARD regimen, Dr. Weinstein said in an interview.
Biologic withdrawal can succeed
Despite that, it’s undeniable that a substantial minority or even a majority in some series of RA patients in remission or low disease activity on both a biologic and synthetic DMARD can taper down or stop the biologic without a flare occurring. The flip side of that is, of course, that in many series a good number of patients who stop their biologic have a flare and need to have the drug restarted. The good news for these scenarios is that prompt restart of the biologic once a flare occurs generally seems capable of restoring remission with no long-term deleterious effects.
In its 2013 RA treatment guidelines, the European League Against Rheumatism (EULAR) said that "if a patient is in persistent remission after having tapered glucocorticoids, one can consider tapering biologic DMARDs, especially if this treatment is combined with a synthetic DMARD" (Ann. Rheum. Dis. 2014;73:492-509). The most recent RA management guidelines from the American College of Rheumatology, published in 2012, don’t address tapering down or withdrawing biologics. Dr. Weinstein noted that the EULAR recommendation is largely based on consensus expert opinion without a definitive evidence base to justify this approach.
Recent evidence documenting the success of biologic withdrawal includes a report earlier this year from a prespecified analysis in the OPTIMA (Optimal Protocol for Treatment Initiation with Methotrexate and Adalimumab) trial that detailed the outcomes of 207 patients who had stable, low disease activity on combined treatment with adalimumab (Humira) and methotrexate and were randomized to either adalimumab withdrawal or continuation. A year later, 70% of patients who maintained the two-drug regimen and 54% of those randomized to stop adalimumab remained at low disease activity and had no sign of radiographic progression, a between-group difference that was not statistically significant (Lancet 2014;383:321-32).
But in other studies, patients withdrawn from a biologic did not fare nearly as well. The PRESERVE (Maintenance, induction, or withdrawal of etanercept after treatment with etanercept and methotrexate in patients with moderate rheumatoid arthritis) trial randomized 604 RA patients with low disease activity on a combination of etanercept (Enbrel) and methotrexate to continue to receive 50 mg of etanercept plus methotrexate weekly, step down to 25 mg of etanercept plus methotrexate weekly, or completely stop etanercept and continue on methotrexate alone. A year later, the percentage of patients who remained in low disease activity tallied 83% in those who received at least one dose of 50 mg etanercept, 79% of patients in the 25-mg etanercept group, and 43% in the group maintained on methotrexate monotherapy, statistically significant differences between each of the two etanercept groups and the monotherapy arm (Lancet 2013;381:918-29).
A more real-world assessment included 717 RA patients at low disease activity who discontinued their first biologic drug while enrolled in the CORRONA (Consortium of Rheumatology Researchers of North America) registry. The results showed that after 1 year on monotherapy without a biologic, 73% of patients remained free from a flare, and that state persisted in 56% to 18 months, 42% after 2 years, and 28% after 3 years on monotherapy, Dr. Arthur F. Kavanaugh and his associates reported last year at the annual meeting of the American College of Rheumatology (Arthritis Rheum. 2013;65:S603).
Finding the right patients
Despite evidence like this, the right time and the right patient for withdrawing a biologic remain murky. "At this point it is investigational," said Dr. Kavanaugh in an interview. "In the United States, it is mostly initiated by patients. There are many factors to consider, including the duration of RA, how well the patients are doing, is the patient on another drug that should be stopped first such as prednisone, and can the synthetic DMARD like methotrexate sustain the response," said Dr. Kavanaugh, a rheumatologist who is professor of clinical medicine and director of the Center for Innovative Therapy at the University of California, San Diego.
Routinely attempting biologic taper-down or withdrawal from RA patients in remission or at low disease activity "is not yet standard practice, but many patients do it," said Dr. Joel M. Kremer, a rheumatologist and professor of medicine at Albany (N.Y.) Medical College. When he attempts withdrawing a biologic, he prefers to use a gradual taper-down of the dosage, and he said he is more reluctant to attempt this for patients who began treatment with severe RA, and that he is more likely to try it for patients who began treatment with early RA, although he admitted that this remains uncertain. Like all experts interviewed, he said the type of biologic a patient is on doesn’t seem to matter.
"I think this entire area needs more study," Dr. Kremer said in an interview. For example, "are there biomarkers associated with the ability to withdraw? I don’t believe we know much about who can or should withdraw." Once withdrawal occurs, he recommended careful follow-up to detect a flare quickly, and restarting the biologic if even a mild flare occurs.
Biologic withdrawal "is really more investigational" right now, said Dr. Eric L. Matteson, professor and chairman of rheumatology at the Mayo Clinic in Rochester, Minn. "No guideline establishes it as standard of practice. I like to see patients in full remission for a year before considering such a move," he said in an interview. Withdrawal depends on whether the RA is well controlled and not on whether treatment started on early or established RA, he added; nor did he think that a flare after withdrawal precludes future withdrawal attempts as long as the patient returns to a durable, full remission. But Dr. Matteson did caution about also considering extra-articular disease like vasculitis or iritis that may be active even when a patient’s joints are not. "Any evidence of recurrent disease should cause the patient and physician to consider ramping the therapy back up."
Like others, Dr. Matteson highlighted the lingering unknowns about biologic withdrawal that keep it from being standard practice. "We don’t know whether the biologic or synthetic DMARD should get withdrawn first. We don’t have a great handle on what biomarkers to use to help decide if there is subclinical disease activity that necessitates reimplementation of the biologic. We don’t know which patients may violently flare following withdrawal and which might not. We also don’t know if retreatment of the disease is as good when a biologic is withdrawn and then restarted. Finally, I think we overestimate the percent of our patients who are actually in remission or a low-disease-activity state and underestimate who really should continue their medications."
The STARA trial
Dr. Weinstein and his associates recently planned a U.S.-based trial that has come close to launching to address several of these issues, the STARA (Stopping TNF-Alpha Inhibitors in Rheumatoid Arthritis) trial.
"One aspect of STARA is to determine whether there is a profile of clinical, imaging, and laboratory factors that will reliably distinguish patients who will flare from those who will remain in remission" following withdrawal of a biologic DMARD, said Dr. Weinstein, STARA’s lead investigator and also a professor of medicine at Georgetown University in Washington. "Another aim is to find predictors of good candidates for withdrawal. The advantage of STARA is patients are drawn from real-world practice and not from a clinical trial." Dr. Weinstein faulted some of the published studies of biologic withdrawal, such as OPTIMA and PRESERVE, because they included trial patients exclusively and not patients managed in routine practice.
STARA’s design uses a sudden, full withdrawal of the biologic to allow a standardized approach to patients who could enter the study on etanercept, adalimumab, or infliximab (Remicade). For routine practice, gradual withdrawal is more common, though there is no evidence this produces better long-term outcomes. An important withdrawal question that STARA won’t address is: Which is the better drug to remove, the biologic or the synthetic DMARD? In most cases, the more expensive biologic is the patient’s choice, but ideally this issue should be addressed in a randomized trial, Dr. Weinstein said.
But as of late March, STARA’s status was in doubt, as anticipated funding was withdrawn. If STARA isn’t run, Dr. Weinstein predicts that attempts to cut biologics from dual regimens will continue and likely expand, but with unknown implications for long-term safety that he believes can only be addressed in a prospective, randomized trial.
Dr. Weinstein said that he had no relevant disclosures. Dr. Matteson had no disclosures. Dr. Kremer has received research support and been a consultant to more than 10 drug companies. Dr. Kavanaugh has received research support from more than 10 drug companies.
On Twitter @mitchelzoler
Once a rheumatoid arthritis patient maintains relatively long-term remission on a regimen that combines a synthetic and a biologic disease-modifying drug, temptation mounts to try withdrawing one of the two drugs. Cost considerations alone can make it irresistible to patients to taper down or stop an expensive biologic drug to see whether the patient’s remission can sustain dose reduction or elimination.
But several American experts cautioned that despite growing evidence that at least some rheumatoid arthritis (RA) patients will stay in remission after their biologic disease-modifying antirheumatic drug (DMARD) is gone, the overall efficacy and safety of this approach remain uncertain and it’s not fully clear which patients are the best candidates. Biologic-drug withdrawal has not repeatedly been shown to be fully safe and worth attempting with results from rigorously controlled and adequately powered studies, an evidence base that may still be several years off.
"We can try withdrawing a drug," and many American RA patients do just that, a step more often than not initiated by the patient. "But we don’t know if it’s benign for radiographic progression or for cardiovascular disease events," said Dr. Arthur Weinstein, director of rheumatology at Washington (D.C.) Hospital Center. Even though RA patients in remission who taper down or fully stop their biologic DMARD are usually closely monitored for flare, "there is some evidence for a disconnect between radiographic progression and clinical status," raising the danger that a patient may appear to remain in remission when off the biologic drug while joint deterioration occurs. That, coupled with concern that an RA patient’s cardiovascular risk may worsen off their biologic, can make patients and physicians hesitate when faced with the prospect of messing with a stable remission on a combined biologic and synthetic DMARD regimen, Dr. Weinstein said in an interview.
Biologic withdrawal can succeed
Despite that, it’s undeniable that a substantial minority or even a majority in some series of RA patients in remission or low disease activity on both a biologic and synthetic DMARD can taper down or stop the biologic without a flare occurring. The flip side of that is, of course, that in many series a good number of patients who stop their biologic have a flare and need to have the drug restarted. The good news for these scenarios is that prompt restart of the biologic once a flare occurs generally seems capable of restoring remission with no long-term deleterious effects.
In its 2013 RA treatment guidelines, the European League Against Rheumatism (EULAR) said that "if a patient is in persistent remission after having tapered glucocorticoids, one can consider tapering biologic DMARDs, especially if this treatment is combined with a synthetic DMARD" (Ann. Rheum. Dis. 2014;73:492-509). The most recent RA management guidelines from the American College of Rheumatology, published in 2012, don’t address tapering down or withdrawing biologics. Dr. Weinstein noted that the EULAR recommendation is largely based on consensus expert opinion without a definitive evidence base to justify this approach.
Recent evidence documenting the success of biologic withdrawal includes a report earlier this year from a prespecified analysis in the OPTIMA (Optimal Protocol for Treatment Initiation with Methotrexate and Adalimumab) trial that detailed the outcomes of 207 patients who had stable, low disease activity on combined treatment with adalimumab (Humira) and methotrexate and were randomized to either adalimumab withdrawal or continuation. A year later, 70% of patients who maintained the two-drug regimen and 54% of those randomized to stop adalimumab remained at low disease activity and had no sign of radiographic progression, a between-group difference that was not statistically significant (Lancet 2014;383:321-32).
But in other studies, patients withdrawn from a biologic did not fare nearly as well. The PRESERVE (Maintenance, induction, or withdrawal of etanercept after treatment with etanercept and methotrexate in patients with moderate rheumatoid arthritis) trial randomized 604 RA patients with low disease activity on a combination of etanercept (Enbrel) and methotrexate to continue to receive 50 mg of etanercept plus methotrexate weekly, step down to 25 mg of etanercept plus methotrexate weekly, or completely stop etanercept and continue on methotrexate alone. A year later, the percentage of patients who remained in low disease activity tallied 83% in those who received at least one dose of 50 mg etanercept, 79% of patients in the 25-mg etanercept group, and 43% in the group maintained on methotrexate monotherapy, statistically significant differences between each of the two etanercept groups and the monotherapy arm (Lancet 2013;381:918-29).
A more real-world assessment included 717 RA patients at low disease activity who discontinued their first biologic drug while enrolled in the CORRONA (Consortium of Rheumatology Researchers of North America) registry. The results showed that after 1 year on monotherapy without a biologic, 73% of patients remained free from a flare, and that state persisted in 56% to 18 months, 42% after 2 years, and 28% after 3 years on monotherapy, Dr. Arthur F. Kavanaugh and his associates reported last year at the annual meeting of the American College of Rheumatology (Arthritis Rheum. 2013;65:S603).
Finding the right patients
Despite evidence like this, the right time and the right patient for withdrawing a biologic remain murky. "At this point it is investigational," said Dr. Kavanaugh in an interview. "In the United States, it is mostly initiated by patients. There are many factors to consider, including the duration of RA, how well the patients are doing, is the patient on another drug that should be stopped first such as prednisone, and can the synthetic DMARD like methotrexate sustain the response," said Dr. Kavanaugh, a rheumatologist who is professor of clinical medicine and director of the Center for Innovative Therapy at the University of California, San Diego.
Routinely attempting biologic taper-down or withdrawal from RA patients in remission or at low disease activity "is not yet standard practice, but many patients do it," said Dr. Joel M. Kremer, a rheumatologist and professor of medicine at Albany (N.Y.) Medical College. When he attempts withdrawing a biologic, he prefers to use a gradual taper-down of the dosage, and he said he is more reluctant to attempt this for patients who began treatment with severe RA, and that he is more likely to try it for patients who began treatment with early RA, although he admitted that this remains uncertain. Like all experts interviewed, he said the type of biologic a patient is on doesn’t seem to matter.
"I think this entire area needs more study," Dr. Kremer said in an interview. For example, "are there biomarkers associated with the ability to withdraw? I don’t believe we know much about who can or should withdraw." Once withdrawal occurs, he recommended careful follow-up to detect a flare quickly, and restarting the biologic if even a mild flare occurs.
Biologic withdrawal "is really more investigational" right now, said Dr. Eric L. Matteson, professor and chairman of rheumatology at the Mayo Clinic in Rochester, Minn. "No guideline establishes it as standard of practice. I like to see patients in full remission for a year before considering such a move," he said in an interview. Withdrawal depends on whether the RA is well controlled and not on whether treatment started on early or established RA, he added; nor did he think that a flare after withdrawal precludes future withdrawal attempts as long as the patient returns to a durable, full remission. But Dr. Matteson did caution about also considering extra-articular disease like vasculitis or iritis that may be active even when a patient’s joints are not. "Any evidence of recurrent disease should cause the patient and physician to consider ramping the therapy back up."
Like others, Dr. Matteson highlighted the lingering unknowns about biologic withdrawal that keep it from being standard practice. "We don’t know whether the biologic or synthetic DMARD should get withdrawn first. We don’t have a great handle on what biomarkers to use to help decide if there is subclinical disease activity that necessitates reimplementation of the biologic. We don’t know which patients may violently flare following withdrawal and which might not. We also don’t know if retreatment of the disease is as good when a biologic is withdrawn and then restarted. Finally, I think we overestimate the percent of our patients who are actually in remission or a low-disease-activity state and underestimate who really should continue their medications."
The STARA trial
Dr. Weinstein and his associates recently planned a U.S.-based trial that has come close to launching to address several of these issues, the STARA (Stopping TNF-Alpha Inhibitors in Rheumatoid Arthritis) trial.
"One aspect of STARA is to determine whether there is a profile of clinical, imaging, and laboratory factors that will reliably distinguish patients who will flare from those who will remain in remission" following withdrawal of a biologic DMARD, said Dr. Weinstein, STARA’s lead investigator and also a professor of medicine at Georgetown University in Washington. "Another aim is to find predictors of good candidates for withdrawal. The advantage of STARA is patients are drawn from real-world practice and not from a clinical trial." Dr. Weinstein faulted some of the published studies of biologic withdrawal, such as OPTIMA and PRESERVE, because they included trial patients exclusively and not patients managed in routine practice.
STARA’s design uses a sudden, full withdrawal of the biologic to allow a standardized approach to patients who could enter the study on etanercept, adalimumab, or infliximab (Remicade). For routine practice, gradual withdrawal is more common, though there is no evidence this produces better long-term outcomes. An important withdrawal question that STARA won’t address is: Which is the better drug to remove, the biologic or the synthetic DMARD? In most cases, the more expensive biologic is the patient’s choice, but ideally this issue should be addressed in a randomized trial, Dr. Weinstein said.
But as of late March, STARA’s status was in doubt, as anticipated funding was withdrawn. If STARA isn’t run, Dr. Weinstein predicts that attempts to cut biologics from dual regimens will continue and likely expand, but with unknown implications for long-term safety that he believes can only be addressed in a prospective, randomized trial.
Dr. Weinstein said that he had no relevant disclosures. Dr. Matteson had no disclosures. Dr. Kremer has received research support and been a consultant to more than 10 drug companies. Dr. Kavanaugh has received research support from more than 10 drug companies.
On Twitter @mitchelzoler

New data question oseltamivir’s influenza efficacy, safety
A new systematic review of clinical studies of the oral anti-influenza drug oseltamivir that included a substantial amount of clinical data that were not part of previously published assessments showed that significant questions exist about oseltamivir’s role in treating and preventing influenza and in pandemic preparedness.
"Our results show that oseltamivir reduces the proportion of participants with symptomatic influenza when used for prophylaxis and has modest symptomatic effects when used for treatment, but it also causes nausea and vomiting and increases the risk of headache and renal and psychiatric syndromes," Tom Jefferson, Ph.D., wrote in an article published online on April 9 (BMJ 2014;348:g2545 doi:10.1136/bmj.g2545).
"We believe these findings provide reason to question the stockpiling of oseltamivir, its inclusion on the [World Health Organization] list of essential drugs, and its use in clinical practice as an anti-influenza drug," concluded Dr. Jefferson, an epidemiologist with the Cochrane Library and head of the Italian Health Technology Assessment program in Rome, and his coauthors. "The trade-off between benefits and harms should be borne in mind when making decisions to use oseltamivir for treatment, prophylaxis, or stockpiling," they wrote.
A second, simultaneously-published study by the same Cochrane-based group using previously unavailable clinical-study data for zanamivir, an inhaled anti-influenza drug, found that, while zanamivir treatment safely reduced by half a day the time to improvement in adults with symptomatic influenza, it had no effect on reducing the risk of complications of influenza, particularly pneumonia, or the risk for hospitalization or death, nor did it show any efficacy for reducing asymptomatic influenza and the subsequent risk for transmission (BMJ 2014;348:g2547 doi:10.1136/bmj.g2547).
The newly available data used by the Cochrane researchers in both reviews came from the manufacturers of the two drugs, Roche in the case of oseltamivir (Tamiflu) and GlaxoSmithKline in the case of zanamivir (Relenza). "Owing to the risk of reporting bias there are legitimate reasons to doubt the stated benefits of oseltamivir and the results of previous Cochrane reviews of neuraminidase inhibitors in adults and children," wrote Dr. Jefferson and his associates in the oseltamivir report.
"To tackle these problems, we have conducted a 4-year campaign to obtain full study reports of the oseltamivir trial program" as well as a parallel campaign that yielded full study reports for zanamivir. The 4-year campaign noted in the review involved an unusual collaboration between Dr. Jefferson and his Cochrane associates and editors at BMJ to put ongoing pressure on Roche and GlaxoSmithKline to promise and then follow through on delivering the full clinical reports from the drugs’ studies, data that finally reached the Cochrane researchers in late 2013.
"For the first time, a Cochrane review [on neuraminidase inhibitors] is based on all relevant full clinical study reports of a family of drugs, integrated by regulatory comments," the authors wrote. "We are pretty confident that this is the closest you’ll get to an unbiased data set," Dr. Jefferson added in an interview.
Despite the unprecedented completeness of the data behind the two reports and the questions they raise about the two drugs, Dr. Jefferson expressed skepticism that public health policy in the United States or United Kingdom will change as a result of the publications.
"I don’t think the people who made the decision to stockpile these drugs for influenza can make an objective decision with any new evidence. [Oseltamivir] was stockpiled but the evidence of its toxicity wasn’t ‘discovered’ by us. It’s been known since the 1990s. I’m skeptical [there will be change now] because the whole system needs to be overhauled. Individual physicians are very loath to prescribe these drugs. But for public health decisions I’m not holding my breath," Dr. Jefferson said.
In reply to a request for comment on the new reports, Gregory Härtl, a spokesperson for the WHO in Geneva, made this statement: "WHO has not seen the paper so cannot comment on specifics. We welcome a new and rigorous analysis of available data and look forward to consideration of its findings after it appears. WHO is committed to an evidence-based approach to determining the best advice for patient care."
Jason McDonald, a spokesperson for the Centers for Disease Control and Prevention in Atlanta made this statement: "CDC supports efforts to make public previously unpublished data from clinical trials. CDC and its advisory committee on immunization practices review all relevant scientific literature when making policy decisions about public health. Clinical trials are just one component of the overall body of scientific evidence that CDC and ACIP [Advisory Committee on Immunization Practices] review when making clinical recommendations on antiviral drugs. Observational trials are another such component, and CDC and ACIP have reviewed a considerable and ever growing number of such studies that show the benefits of antiviral medications in not only reducing and shortening symptoms of disease but also preventing hospitalizations and deaths, including in high risk children and adults. While there are limitations and possible biases that must be considered and controlled in such studies, these studies are easier to conduct due to ethical considerations and more of them are available.
"CDC welcomes the public release of all such data for academic review and discussion. These data have not changed CDC and ACIP’s recommendations for the clinical use of antivirals in the United States. Decisions regarding the purchase of antivirals for use in the U.S. Strategic National Stockpile in the event of an emergency are not made by CDC.
"It’s important to acknowledge that the neuraminidase inhibitor class of antiviral drugs, which includes Tamiflu and zanamivir, are a second line of defense against the flu. These drugs represent an important tool in the event of a flu outbreak, when a vaccine isn’t yet available. Such drugs are often the only treatment option available for flu, and while we all support the development of new and better drugs in the future, we must make the best use of the tools we have at our disposal to promote public health."
In June 2013, a multidisciplinary and international group of researchers developed an independent statistical plan for analyzing the full oseltamivir dataset with an eye toward determining the efficacy and safety of oseltamivir for treating and preventing influenza in both individual patients and for public health. The Multidisciplinary Group for Advice on Science (MUGAS) plans to present the results of its analysis in September at a meeting of the European Scientific Working Group on Influenza, according to Dr. Richard J. Whitely, professor and director of pediatric infectious diseases at the University of Alabama in Birmingham, and one of the organizers of the MUGAS project on oseltamivir.
Dr. Jefferson receives royalties from Blackwells and Il Pensiero Scientifico Editore, Rome, and gives interviews anonymously to market research companies about phase I or II pharmaceutical products. In 2011, he was an expert witness in a litigation case related to oseltamivir phosphate and in a labor case on influenza vaccines in health care workers in Canada. He has been a consultant for Roche, GlaxoSmithKline, and Sanofi-Synthelabo, and is currently a consultant for IMS Health.
BMJ support key
Dr. Jefferson called his 4-year effort to get full clinical-testing data on oseltamivir and zanamivir from the manufacturers a "campaign," and cited key support from the staff of the U.K.-based BMJ medical journal.
"If BMJ had not become involved neither Roche nor GlaxoSmithKline would have cooperated. BMJ pledged their support when we asked for the full clinical reports" in 2009, recalled Dr. Jefferson, who first raised the issue of getting wider data access and also led the Cochrane analysis on the new data.
The story began in 2009, during the H1N1 pandemic, which led to the Cochrane group’s receiving funding to perform an update to a neuraminidase-inhibitor review they had done in 2006. In the intervening years, information emerged suggesting a large cache of unpublished data had never been seen during the 2006 review, which focused on published materials.
"In 2011, we decided not to accept anything that had been published because it was biased," Dr. Jefferson said in an interview.
BMJ editors compiled details and links on this web page to the thrusts and parries between themselves and Cochrane staffers and the two companies involved, Roche and GlaxoSmithKline, during the several years it took to receive and evaluate the full data: www.bmj.com/tamiflu.
In an editorial accompanying the two reports from Cochrane on oseltamivir and zanamivir, Dr. Fiona Godlee, editor-in-chief at BMJ, Dr. Elizabeth Loder, another BMJ editor, and Dr. David Tovey, editor-in-chief of the Cochrane Library, called the Cochrane reviewers and their associates who authored the two papers "pioneers" for "navigating the uncharted territory" of the data analyses they performed using huge numbers of clinical study reports (BMJ 201;348:g2630 [doi: 10.1136/bmj.g2630]).
Dr. Loder, Dr. Tovey, and Dr. Godlee also said in their editorial that they saw the effort to get more data on the neuraminidase inhibitors as part of a larger effort to remake the way that drugs are assessed and reviewed.
The process required to obtain and analyze the neuraminidase inhibitor data showed "with greater clarity than ever that the current system is broken," they wrote. "The review’s conclusions should lead to serious soul-searching among policy makers. So, too, should the story behind the review, illustrating as it does the entrenched flaws in the current system. Why did no one else demand this level of scrutiny before spending such huge sums of money on one drug?" they asked.
Dr. Godlee said that she and BMJ have been closely involved from the outset in the campaign for access to clinical trial data on oseltamivir. She coedited a book on peer review with one of the Cochrane authors, Dr. Tom Jefferson. She and BMJ work closely with another of the Cochrane authors, Carl Heneghan, on jointly hosted conferences. She recently recruited a third Cochrane author, Peter Doshi, to the staff of BMJ as an associate editor. She was not directly involved in the decisions to accept the neuraminidase inhibitor papers for publication in BMJ but will undoubtedly have communicated to her editorial colleagues her keenness that BMJ should publish them if at all possible.
*This article was updated April 10, 2014.
On Twitter @mitchelzoler
This publication of the most recent iteration of Cochrane reviews on the benefits and harms of oseltamivir and zanamivir marks the first time that reviews of these products included information from all the pertinent clinical trials conducted by the manufacturers. With these comprehensive new reviews based on all the data, the perspective on these drugs has changed substantially. With all the evidence available from treatment and prophylaxis studies it has become clear that convincing trial evidence of a reduction in the risk of hospitalization or complications is lacking.
In addition, the reviews found that oseltamivir causes nausea and vomiting, and probably causes heachaches, renal problems, and psychiatric syndromes. Zanamivir had fewer adverse effects but also had no demonstrated effect on complications or hospitalizations. Zanamivir reduced the duration of symptoms by about half a day, but the reviewers found that it may be no better than other symptom-relief medications. For prophylaxis, zanamivir significantly reduced the risk of symptomatic influenza, but there was heterogeneity among the studies and sample sizes were small. In addition there was no difference in asymptomatic influenza.
Based on these findings it is difficult to conceive that many patients would actively seek these treatments. The benefits involve a shortening of symptoms that few patients would find worth the risk of incurring the harms of treatment. From a health-system perspective, enormous expenditures on these drugs do not appear to have commensurate benefit.
Despite questions raised several years ago about full transparency on the risks and benefits of these two drugs, the medical establishment embraced them. Governments stockpiled the drugs, presuming that their potential public-health benefit merited a substantial financial investment. During this period, when not all of the trial results were fully and widely available, the Centers for Disease Control and Prevention, the U.K. National Institute for Health Care Excellence, the American Academy of Pediatrics, and the World Health Organization all suggested that oseltamivir and zanamivir provided benefits with minimal risks.
Of primary importance for decision makers is that now, with all of the data available for others to evaluate, the knowns and unknowns can be laid out clearly and reveal the weakness of the evidentiary support for these products and what we need to learn about them in the future.
Dr. Harlan M. Krumholz is a cardiologist and professor of medicine and director of the Center for Outcomes Research and Evaluation at Yale University, New Haven, Conn. He said that he has received research grants from Medtronic and Johnson & Johnson and that he chairs a scientific advisory board for UnitedHealthcare. He made these comments in an editorial (BMJ 2014;348:g2548 doi:10.1136/bmj.g2548).
This publication of the most recent iteration of Cochrane reviews on the benefits and harms of oseltamivir and zanamivir marks the first time that reviews of these products included information from all the pertinent clinical trials conducted by the manufacturers. With these comprehensive new reviews based on all the data, the perspective on these drugs has changed substantially. With all the evidence available from treatment and prophylaxis studies it has become clear that convincing trial evidence of a reduction in the risk of hospitalization or complications is lacking.
In addition, the reviews found that oseltamivir causes nausea and vomiting, and probably causes heachaches, renal problems, and psychiatric syndromes. Zanamivir had fewer adverse effects but also had no demonstrated effect on complications or hospitalizations. Zanamivir reduced the duration of symptoms by about half a day, but the reviewers found that it may be no better than other symptom-relief medications. For prophylaxis, zanamivir significantly reduced the risk of symptomatic influenza, but there was heterogeneity among the studies and sample sizes were small. In addition there was no difference in asymptomatic influenza.
Based on these findings it is difficult to conceive that many patients would actively seek these treatments. The benefits involve a shortening of symptoms that few patients would find worth the risk of incurring the harms of treatment. From a health-system perspective, enormous expenditures on these drugs do not appear to have commensurate benefit.
Despite questions raised several years ago about full transparency on the risks and benefits of these two drugs, the medical establishment embraced them. Governments stockpiled the drugs, presuming that their potential public-health benefit merited a substantial financial investment. During this period, when not all of the trial results were fully and widely available, the Centers for Disease Control and Prevention, the U.K. National Institute for Health Care Excellence, the American Academy of Pediatrics, and the World Health Organization all suggested that oseltamivir and zanamivir provided benefits with minimal risks.
Of primary importance for decision makers is that now, with all of the data available for others to evaluate, the knowns and unknowns can be laid out clearly and reveal the weakness of the evidentiary support for these products and what we need to learn about them in the future.
Dr. Harlan M. Krumholz is a cardiologist and professor of medicine and director of the Center for Outcomes Research and Evaluation at Yale University, New Haven, Conn. He said that he has received research grants from Medtronic and Johnson & Johnson and that he chairs a scientific advisory board for UnitedHealthcare. He made these comments in an editorial (BMJ 2014;348:g2548 doi:10.1136/bmj.g2548).
This publication of the most recent iteration of Cochrane reviews on the benefits and harms of oseltamivir and zanamivir marks the first time that reviews of these products included information from all the pertinent clinical trials conducted by the manufacturers. With these comprehensive new reviews based on all the data, the perspective on these drugs has changed substantially. With all the evidence available from treatment and prophylaxis studies it has become clear that convincing trial evidence of a reduction in the risk of hospitalization or complications is lacking.
In addition, the reviews found that oseltamivir causes nausea and vomiting, and probably causes heachaches, renal problems, and psychiatric syndromes. Zanamivir had fewer adverse effects but also had no demonstrated effect on complications or hospitalizations. Zanamivir reduced the duration of symptoms by about half a day, but the reviewers found that it may be no better than other symptom-relief medications. For prophylaxis, zanamivir significantly reduced the risk of symptomatic influenza, but there was heterogeneity among the studies and sample sizes were small. In addition there was no difference in asymptomatic influenza.
Based on these findings it is difficult to conceive that many patients would actively seek these treatments. The benefits involve a shortening of symptoms that few patients would find worth the risk of incurring the harms of treatment. From a health-system perspective, enormous expenditures on these drugs do not appear to have commensurate benefit.
Despite questions raised several years ago about full transparency on the risks and benefits of these two drugs, the medical establishment embraced them. Governments stockpiled the drugs, presuming that their potential public-health benefit merited a substantial financial investment. During this period, when not all of the trial results were fully and widely available, the Centers for Disease Control and Prevention, the U.K. National Institute for Health Care Excellence, the American Academy of Pediatrics, and the World Health Organization all suggested that oseltamivir and zanamivir provided benefits with minimal risks.
Of primary importance for decision makers is that now, with all of the data available for others to evaluate, the knowns and unknowns can be laid out clearly and reveal the weakness of the evidentiary support for these products and what we need to learn about them in the future.
Dr. Harlan M. Krumholz is a cardiologist and professor of medicine and director of the Center for Outcomes Research and Evaluation at Yale University, New Haven, Conn. He said that he has received research grants from Medtronic and Johnson & Johnson and that he chairs a scientific advisory board for UnitedHealthcare. He made these comments in an editorial (BMJ 2014;348:g2548 doi:10.1136/bmj.g2548).
A new systematic review of clinical studies of the oral anti-influenza drug oseltamivir that included a substantial amount of clinical data that were not part of previously published assessments showed that significant questions exist about oseltamivir’s role in treating and preventing influenza and in pandemic preparedness.
"Our results show that oseltamivir reduces the proportion of participants with symptomatic influenza when used for prophylaxis and has modest symptomatic effects when used for treatment, but it also causes nausea and vomiting and increases the risk of headache and renal and psychiatric syndromes," Tom Jefferson, Ph.D., wrote in an article published online on April 9 (BMJ 2014;348:g2545 doi:10.1136/bmj.g2545).
"We believe these findings provide reason to question the stockpiling of oseltamivir, its inclusion on the [World Health Organization] list of essential drugs, and its use in clinical practice as an anti-influenza drug," concluded Dr. Jefferson, an epidemiologist with the Cochrane Library and head of the Italian Health Technology Assessment program in Rome, and his coauthors. "The trade-off between benefits and harms should be borne in mind when making decisions to use oseltamivir for treatment, prophylaxis, or stockpiling," they wrote.
A second, simultaneously-published study by the same Cochrane-based group using previously unavailable clinical-study data for zanamivir, an inhaled anti-influenza drug, found that, while zanamivir treatment safely reduced by half a day the time to improvement in adults with symptomatic influenza, it had no effect on reducing the risk of complications of influenza, particularly pneumonia, or the risk for hospitalization or death, nor did it show any efficacy for reducing asymptomatic influenza and the subsequent risk for transmission (BMJ 2014;348:g2547 doi:10.1136/bmj.g2547).
The newly available data used by the Cochrane researchers in both reviews came from the manufacturers of the two drugs, Roche in the case of oseltamivir (Tamiflu) and GlaxoSmithKline in the case of zanamivir (Relenza). "Owing to the risk of reporting bias there are legitimate reasons to doubt the stated benefits of oseltamivir and the results of previous Cochrane reviews of neuraminidase inhibitors in adults and children," wrote Dr. Jefferson and his associates in the oseltamivir report.
"To tackle these problems, we have conducted a 4-year campaign to obtain full study reports of the oseltamivir trial program" as well as a parallel campaign that yielded full study reports for zanamivir. The 4-year campaign noted in the review involved an unusual collaboration between Dr. Jefferson and his Cochrane associates and editors at BMJ to put ongoing pressure on Roche and GlaxoSmithKline to promise and then follow through on delivering the full clinical reports from the drugs’ studies, data that finally reached the Cochrane researchers in late 2013.
"For the first time, a Cochrane review [on neuraminidase inhibitors] is based on all relevant full clinical study reports of a family of drugs, integrated by regulatory comments," the authors wrote. "We are pretty confident that this is the closest you’ll get to an unbiased data set," Dr. Jefferson added in an interview.
Despite the unprecedented completeness of the data behind the two reports and the questions they raise about the two drugs, Dr. Jefferson expressed skepticism that public health policy in the United States or United Kingdom will change as a result of the publications.
"I don’t think the people who made the decision to stockpile these drugs for influenza can make an objective decision with any new evidence. [Oseltamivir] was stockpiled but the evidence of its toxicity wasn’t ‘discovered’ by us. It’s been known since the 1990s. I’m skeptical [there will be change now] because the whole system needs to be overhauled. Individual physicians are very loath to prescribe these drugs. But for public health decisions I’m not holding my breath," Dr. Jefferson said.
In reply to a request for comment on the new reports, Gregory Härtl, a spokesperson for the WHO in Geneva, made this statement: "WHO has not seen the paper so cannot comment on specifics. We welcome a new and rigorous analysis of available data and look forward to consideration of its findings after it appears. WHO is committed to an evidence-based approach to determining the best advice for patient care."
Jason McDonald, a spokesperson for the Centers for Disease Control and Prevention in Atlanta made this statement: "CDC supports efforts to make public previously unpublished data from clinical trials. CDC and its advisory committee on immunization practices review all relevant scientific literature when making policy decisions about public health. Clinical trials are just one component of the overall body of scientific evidence that CDC and ACIP [Advisory Committee on Immunization Practices] review when making clinical recommendations on antiviral drugs. Observational trials are another such component, and CDC and ACIP have reviewed a considerable and ever growing number of such studies that show the benefits of antiviral medications in not only reducing and shortening symptoms of disease but also preventing hospitalizations and deaths, including in high risk children and adults. While there are limitations and possible biases that must be considered and controlled in such studies, these studies are easier to conduct due to ethical considerations and more of them are available.
"CDC welcomes the public release of all such data for academic review and discussion. These data have not changed CDC and ACIP’s recommendations for the clinical use of antivirals in the United States. Decisions regarding the purchase of antivirals for use in the U.S. Strategic National Stockpile in the event of an emergency are not made by CDC.
"It’s important to acknowledge that the neuraminidase inhibitor class of antiviral drugs, which includes Tamiflu and zanamivir, are a second line of defense against the flu. These drugs represent an important tool in the event of a flu outbreak, when a vaccine isn’t yet available. Such drugs are often the only treatment option available for flu, and while we all support the development of new and better drugs in the future, we must make the best use of the tools we have at our disposal to promote public health."
In June 2013, a multidisciplinary and international group of researchers developed an independent statistical plan for analyzing the full oseltamivir dataset with an eye toward determining the efficacy and safety of oseltamivir for treating and preventing influenza in both individual patients and for public health. The Multidisciplinary Group for Advice on Science (MUGAS) plans to present the results of its analysis in September at a meeting of the European Scientific Working Group on Influenza, according to Dr. Richard J. Whitely, professor and director of pediatric infectious diseases at the University of Alabama in Birmingham, and one of the organizers of the MUGAS project on oseltamivir.
Dr. Jefferson receives royalties from Blackwells and Il Pensiero Scientifico Editore, Rome, and gives interviews anonymously to market research companies about phase I or II pharmaceutical products. In 2011, he was an expert witness in a litigation case related to oseltamivir phosphate and in a labor case on influenza vaccines in health care workers in Canada. He has been a consultant for Roche, GlaxoSmithKline, and Sanofi-Synthelabo, and is currently a consultant for IMS Health.
BMJ support key
Dr. Jefferson called his 4-year effort to get full clinical-testing data on oseltamivir and zanamivir from the manufacturers a "campaign," and cited key support from the staff of the U.K.-based BMJ medical journal.
"If BMJ had not become involved neither Roche nor GlaxoSmithKline would have cooperated. BMJ pledged their support when we asked for the full clinical reports" in 2009, recalled Dr. Jefferson, who first raised the issue of getting wider data access and also led the Cochrane analysis on the new data.
The story began in 2009, during the H1N1 pandemic, which led to the Cochrane group’s receiving funding to perform an update to a neuraminidase-inhibitor review they had done in 2006. In the intervening years, information emerged suggesting a large cache of unpublished data had never been seen during the 2006 review, which focused on published materials.
"In 2011, we decided not to accept anything that had been published because it was biased," Dr. Jefferson said in an interview.
BMJ editors compiled details and links on this web page to the thrusts and parries between themselves and Cochrane staffers and the two companies involved, Roche and GlaxoSmithKline, during the several years it took to receive and evaluate the full data: www.bmj.com/tamiflu.
In an editorial accompanying the two reports from Cochrane on oseltamivir and zanamivir, Dr. Fiona Godlee, editor-in-chief at BMJ, Dr. Elizabeth Loder, another BMJ editor, and Dr. David Tovey, editor-in-chief of the Cochrane Library, called the Cochrane reviewers and their associates who authored the two papers "pioneers" for "navigating the uncharted territory" of the data analyses they performed using huge numbers of clinical study reports (BMJ 201;348:g2630 [doi: 10.1136/bmj.g2630]).
Dr. Loder, Dr. Tovey, and Dr. Godlee also said in their editorial that they saw the effort to get more data on the neuraminidase inhibitors as part of a larger effort to remake the way that drugs are assessed and reviewed.
The process required to obtain and analyze the neuraminidase inhibitor data showed "with greater clarity than ever that the current system is broken," they wrote. "The review’s conclusions should lead to serious soul-searching among policy makers. So, too, should the story behind the review, illustrating as it does the entrenched flaws in the current system. Why did no one else demand this level of scrutiny before spending such huge sums of money on one drug?" they asked.
Dr. Godlee said that she and BMJ have been closely involved from the outset in the campaign for access to clinical trial data on oseltamivir. She coedited a book on peer review with one of the Cochrane authors, Dr. Tom Jefferson. She and BMJ work closely with another of the Cochrane authors, Carl Heneghan, on jointly hosted conferences. She recently recruited a third Cochrane author, Peter Doshi, to the staff of BMJ as an associate editor. She was not directly involved in the decisions to accept the neuraminidase inhibitor papers for publication in BMJ but will undoubtedly have communicated to her editorial colleagues her keenness that BMJ should publish them if at all possible.
*This article was updated April 10, 2014.
On Twitter @mitchelzoler
A new systematic review of clinical studies of the oral anti-influenza drug oseltamivir that included a substantial amount of clinical data that were not part of previously published assessments showed that significant questions exist about oseltamivir’s role in treating and preventing influenza and in pandemic preparedness.
"Our results show that oseltamivir reduces the proportion of participants with symptomatic influenza when used for prophylaxis and has modest symptomatic effects when used for treatment, but it also causes nausea and vomiting and increases the risk of headache and renal and psychiatric syndromes," Tom Jefferson, Ph.D., wrote in an article published online on April 9 (BMJ 2014;348:g2545 doi:10.1136/bmj.g2545).
"We believe these findings provide reason to question the stockpiling of oseltamivir, its inclusion on the [World Health Organization] list of essential drugs, and its use in clinical practice as an anti-influenza drug," concluded Dr. Jefferson, an epidemiologist with the Cochrane Library and head of the Italian Health Technology Assessment program in Rome, and his coauthors. "The trade-off between benefits and harms should be borne in mind when making decisions to use oseltamivir for treatment, prophylaxis, or stockpiling," they wrote.
A second, simultaneously-published study by the same Cochrane-based group using previously unavailable clinical-study data for zanamivir, an inhaled anti-influenza drug, found that, while zanamivir treatment safely reduced by half a day the time to improvement in adults with symptomatic influenza, it had no effect on reducing the risk of complications of influenza, particularly pneumonia, or the risk for hospitalization or death, nor did it show any efficacy for reducing asymptomatic influenza and the subsequent risk for transmission (BMJ 2014;348:g2547 doi:10.1136/bmj.g2547).
The newly available data used by the Cochrane researchers in both reviews came from the manufacturers of the two drugs, Roche in the case of oseltamivir (Tamiflu) and GlaxoSmithKline in the case of zanamivir (Relenza). "Owing to the risk of reporting bias there are legitimate reasons to doubt the stated benefits of oseltamivir and the results of previous Cochrane reviews of neuraminidase inhibitors in adults and children," wrote Dr. Jefferson and his associates in the oseltamivir report.
"To tackle these problems, we have conducted a 4-year campaign to obtain full study reports of the oseltamivir trial program" as well as a parallel campaign that yielded full study reports for zanamivir. The 4-year campaign noted in the review involved an unusual collaboration between Dr. Jefferson and his Cochrane associates and editors at BMJ to put ongoing pressure on Roche and GlaxoSmithKline to promise and then follow through on delivering the full clinical reports from the drugs’ studies, data that finally reached the Cochrane researchers in late 2013.
"For the first time, a Cochrane review [on neuraminidase inhibitors] is based on all relevant full clinical study reports of a family of drugs, integrated by regulatory comments," the authors wrote. "We are pretty confident that this is the closest you’ll get to an unbiased data set," Dr. Jefferson added in an interview.
Despite the unprecedented completeness of the data behind the two reports and the questions they raise about the two drugs, Dr. Jefferson expressed skepticism that public health policy in the United States or United Kingdom will change as a result of the publications.
"I don’t think the people who made the decision to stockpile these drugs for influenza can make an objective decision with any new evidence. [Oseltamivir] was stockpiled but the evidence of its toxicity wasn’t ‘discovered’ by us. It’s been known since the 1990s. I’m skeptical [there will be change now] because the whole system needs to be overhauled. Individual physicians are very loath to prescribe these drugs. But for public health decisions I’m not holding my breath," Dr. Jefferson said.
In reply to a request for comment on the new reports, Gregory Härtl, a spokesperson for the WHO in Geneva, made this statement: "WHO has not seen the paper so cannot comment on specifics. We welcome a new and rigorous analysis of available data and look forward to consideration of its findings after it appears. WHO is committed to an evidence-based approach to determining the best advice for patient care."
Jason McDonald, a spokesperson for the Centers for Disease Control and Prevention in Atlanta made this statement: "CDC supports efforts to make public previously unpublished data from clinical trials. CDC and its advisory committee on immunization practices review all relevant scientific literature when making policy decisions about public health. Clinical trials are just one component of the overall body of scientific evidence that CDC and ACIP [Advisory Committee on Immunization Practices] review when making clinical recommendations on antiviral drugs. Observational trials are another such component, and CDC and ACIP have reviewed a considerable and ever growing number of such studies that show the benefits of antiviral medications in not only reducing and shortening symptoms of disease but also preventing hospitalizations and deaths, including in high risk children and adults. While there are limitations and possible biases that must be considered and controlled in such studies, these studies are easier to conduct due to ethical considerations and more of them are available.
"CDC welcomes the public release of all such data for academic review and discussion. These data have not changed CDC and ACIP’s recommendations for the clinical use of antivirals in the United States. Decisions regarding the purchase of antivirals for use in the U.S. Strategic National Stockpile in the event of an emergency are not made by CDC.
"It’s important to acknowledge that the neuraminidase inhibitor class of antiviral drugs, which includes Tamiflu and zanamivir, are a second line of defense against the flu. These drugs represent an important tool in the event of a flu outbreak, when a vaccine isn’t yet available. Such drugs are often the only treatment option available for flu, and while we all support the development of new and better drugs in the future, we must make the best use of the tools we have at our disposal to promote public health."
In June 2013, a multidisciplinary and international group of researchers developed an independent statistical plan for analyzing the full oseltamivir dataset with an eye toward determining the efficacy and safety of oseltamivir for treating and preventing influenza in both individual patients and for public health. The Multidisciplinary Group for Advice on Science (MUGAS) plans to present the results of its analysis in September at a meeting of the European Scientific Working Group on Influenza, according to Dr. Richard J. Whitely, professor and director of pediatric infectious diseases at the University of Alabama in Birmingham, and one of the organizers of the MUGAS project on oseltamivir.
Dr. Jefferson receives royalties from Blackwells and Il Pensiero Scientifico Editore, Rome, and gives interviews anonymously to market research companies about phase I or II pharmaceutical products. In 2011, he was an expert witness in a litigation case related to oseltamivir phosphate and in a labor case on influenza vaccines in health care workers in Canada. He has been a consultant for Roche, GlaxoSmithKline, and Sanofi-Synthelabo, and is currently a consultant for IMS Health.
BMJ support key
Dr. Jefferson called his 4-year effort to get full clinical-testing data on oseltamivir and zanamivir from the manufacturers a "campaign," and cited key support from the staff of the U.K.-based BMJ medical journal.
"If BMJ had not become involved neither Roche nor GlaxoSmithKline would have cooperated. BMJ pledged their support when we asked for the full clinical reports" in 2009, recalled Dr. Jefferson, who first raised the issue of getting wider data access and also led the Cochrane analysis on the new data.
The story began in 2009, during the H1N1 pandemic, which led to the Cochrane group’s receiving funding to perform an update to a neuraminidase-inhibitor review they had done in 2006. In the intervening years, information emerged suggesting a large cache of unpublished data had never been seen during the 2006 review, which focused on published materials.
"In 2011, we decided not to accept anything that had been published because it was biased," Dr. Jefferson said in an interview.
BMJ editors compiled details and links on this web page to the thrusts and parries between themselves and Cochrane staffers and the two companies involved, Roche and GlaxoSmithKline, during the several years it took to receive and evaluate the full data: www.bmj.com/tamiflu.
In an editorial accompanying the two reports from Cochrane on oseltamivir and zanamivir, Dr. Fiona Godlee, editor-in-chief at BMJ, Dr. Elizabeth Loder, another BMJ editor, and Dr. David Tovey, editor-in-chief of the Cochrane Library, called the Cochrane reviewers and their associates who authored the two papers "pioneers" for "navigating the uncharted territory" of the data analyses they performed using huge numbers of clinical study reports (BMJ 201;348:g2630 [doi: 10.1136/bmj.g2630]).
Dr. Loder, Dr. Tovey, and Dr. Godlee also said in their editorial that they saw the effort to get more data on the neuraminidase inhibitors as part of a larger effort to remake the way that drugs are assessed and reviewed.
The process required to obtain and analyze the neuraminidase inhibitor data showed "with greater clarity than ever that the current system is broken," they wrote. "The review’s conclusions should lead to serious soul-searching among policy makers. So, too, should the story behind the review, illustrating as it does the entrenched flaws in the current system. Why did no one else demand this level of scrutiny before spending such huge sums of money on one drug?" they asked.
Dr. Godlee said that she and BMJ have been closely involved from the outset in the campaign for access to clinical trial data on oseltamivir. She coedited a book on peer review with one of the Cochrane authors, Dr. Tom Jefferson. She and BMJ work closely with another of the Cochrane authors, Carl Heneghan, on jointly hosted conferences. She recently recruited a third Cochrane author, Peter Doshi, to the staff of BMJ as an associate editor. She was not directly involved in the decisions to accept the neuraminidase inhibitor papers for publication in BMJ but will undoubtedly have communicated to her editorial colleagues her keenness that BMJ should publish them if at all possible.
*This article was updated April 10, 2014.
On Twitter @mitchelzoler
FROM BMJ
Key clinical point: The benefit of oseltamivir may not outweigh the risks for influenza prophylaxis or for treatment. While zanamivir reduced the length of influenza symptoms, it did not reduce the rate of complications.
Major finding: New clinical data on safety and efficacy of oseltamivir show that in prophylaxis trials, it reduced symptomatic influenza by 55% and zanamivir reduced the time to first alleviation of symptoms of influenza-like illness by an average 14.4 hours, raising questions on risk-to-benefit of both drugs.
Data source: Review of 20 clinical studies of oseltamivir and 26 studies of zanamivir.
Disclosures: Dr. Jefferson receives royalties from Blackwells and Il Pensiero Scientifico Editore, Rome, and gives interviews anonymously to market research companies about phase I or II pharmaceutical products. In 2011, he was an expert witness in a litigation case related to oseltamivir phosphate and in a labor case on influenza vaccines in health care workers in Canada. He has been a consultant for Roche, GlaxoSmithKline, and Sanofi-Synthelabo, and is currently a consultant for IMS Health.
Symple is as simple does
After the surprising, top-line result from the SYMPLICITY HTN-3 trial came out in a press release from Medtronic last January, the question remained of what went wrong: Why did renal denervation fail to outperform a sham procedure in reducing blood pressure?
The apparent answer emerged in late March when the full results finally went public in a report at the annual meeting of the American College of Cardiology and in an article in the New England Journal of Medicine. Many of the 364 patients who underwent active denervation – which involves zapping the efferent nerves that run along the outer wall of the renal arteries with a few brief pulses of radiofrequency energy – probably failed to receive adequate treatment, so their renal innervation remained mostly intact. There’s no proof that’s what happened, but it seems plausible given that all the U.S. operators in the trial had no prior experience performing the procedure, as well as the observation several years ago that denervation can produce highly variable results and is very operator dependent.
This variability in success had been documented back in the 2000s by one of the pioneers of renal denervation, Dr. Murray Esler of Melbourne, yet the people who designed SYMPLICITY HTN-3 didn’t pay attention. Their failure to apply what earlier findings had taught about the variability of denervation proved especially egregious, as the interventionalists also couldn’t gauge their procedural success because no easy way exists right now to do this.
But these details didn’t slow the first controlled clinical trial. The concept was so ... simple: Insert catheter into renal artery, throw switch and zap, remove catheter. Easy peasy.
I first heard about renal denervation more than 2 years ago, and marveled at the unmitigated hubris to name the first catheter developed for denervation Symplicity, as well as giving that moniker to a series of uncontrolled and controlled studies that tested the technique. The Symplicity crowd seemed very sure of themselves, of this catheter, and of this procedure.
Fast forward a couple of years and the name morphs into the ironic butt of an expensive, failed trial.
If there is anything I’ve learned during more than 3 decades of covering medicine, it’s that the discipline is hardly ever simple. Think of signaling-pathway diagrams, the ones with all the arrows, boxes, and small fonts. The most reliable reaction when confronted in medicine by something that appears simple is to ask: What am I missing here? Hopefully, when this still-promising technology resurrects, its developers will have learned that lesson.
On Twitter @mitchelzoler
After the surprising, top-line result from the SYMPLICITY HTN-3 trial came out in a press release from Medtronic last January, the question remained of what went wrong: Why did renal denervation fail to outperform a sham procedure in reducing blood pressure?
The apparent answer emerged in late March when the full results finally went public in a report at the annual meeting of the American College of Cardiology and in an article in the New England Journal of Medicine. Many of the 364 patients who underwent active denervation – which involves zapping the efferent nerves that run along the outer wall of the renal arteries with a few brief pulses of radiofrequency energy – probably failed to receive adequate treatment, so their renal innervation remained mostly intact. There’s no proof that’s what happened, but it seems plausible given that all the U.S. operators in the trial had no prior experience performing the procedure, as well as the observation several years ago that denervation can produce highly variable results and is very operator dependent.
This variability in success had been documented back in the 2000s by one of the pioneers of renal denervation, Dr. Murray Esler of Melbourne, yet the people who designed SYMPLICITY HTN-3 didn’t pay attention. Their failure to apply what earlier findings had taught about the variability of denervation proved especially egregious, as the interventionalists also couldn’t gauge their procedural success because no easy way exists right now to do this.
But these details didn’t slow the first controlled clinical trial. The concept was so ... simple: Insert catheter into renal artery, throw switch and zap, remove catheter. Easy peasy.
I first heard about renal denervation more than 2 years ago, and marveled at the unmitigated hubris to name the first catheter developed for denervation Symplicity, as well as giving that moniker to a series of uncontrolled and controlled studies that tested the technique. The Symplicity crowd seemed very sure of themselves, of this catheter, and of this procedure.
Fast forward a couple of years and the name morphs into the ironic butt of an expensive, failed trial.
If there is anything I’ve learned during more than 3 decades of covering medicine, it’s that the discipline is hardly ever simple. Think of signaling-pathway diagrams, the ones with all the arrows, boxes, and small fonts. The most reliable reaction when confronted in medicine by something that appears simple is to ask: What am I missing here? Hopefully, when this still-promising technology resurrects, its developers will have learned that lesson.
On Twitter @mitchelzoler
After the surprising, top-line result from the SYMPLICITY HTN-3 trial came out in a press release from Medtronic last January, the question remained of what went wrong: Why did renal denervation fail to outperform a sham procedure in reducing blood pressure?
The apparent answer emerged in late March when the full results finally went public in a report at the annual meeting of the American College of Cardiology and in an article in the New England Journal of Medicine. Many of the 364 patients who underwent active denervation – which involves zapping the efferent nerves that run along the outer wall of the renal arteries with a few brief pulses of radiofrequency energy – probably failed to receive adequate treatment, so their renal innervation remained mostly intact. There’s no proof that’s what happened, but it seems plausible given that all the U.S. operators in the trial had no prior experience performing the procedure, as well as the observation several years ago that denervation can produce highly variable results and is very operator dependent.
This variability in success had been documented back in the 2000s by one of the pioneers of renal denervation, Dr. Murray Esler of Melbourne, yet the people who designed SYMPLICITY HTN-3 didn’t pay attention. Their failure to apply what earlier findings had taught about the variability of denervation proved especially egregious, as the interventionalists also couldn’t gauge their procedural success because no easy way exists right now to do this.
But these details didn’t slow the first controlled clinical trial. The concept was so ... simple: Insert catheter into renal artery, throw switch and zap, remove catheter. Easy peasy.
I first heard about renal denervation more than 2 years ago, and marveled at the unmitigated hubris to name the first catheter developed for denervation Symplicity, as well as giving that moniker to a series of uncontrolled and controlled studies that tested the technique. The Symplicity crowd seemed very sure of themselves, of this catheter, and of this procedure.
Fast forward a couple of years and the name morphs into the ironic butt of an expensive, failed trial.
If there is anything I’ve learned during more than 3 decades of covering medicine, it’s that the discipline is hardly ever simple. Think of signaling-pathway diagrams, the ones with all the arrows, boxes, and small fonts. The most reliable reaction when confronted in medicine by something that appears simple is to ask: What am I missing here? Hopefully, when this still-promising technology resurrects, its developers will have learned that lesson.
On Twitter @mitchelzoler
Launch of rare-cancer trial spurs many more
MILAN – An ongoing phase III trial launched in 2012 that is testing whether adjuvant therapy aids women following removal of high-risk uterine leiomyosarcomas has also blazed a path for the International Rare Cancer Initiative, which has launched two other trials in rare cancers and is planning to start several more.
The advanced uterine leiomyosarcoma trial, which is testing an adjuvant regimen of up to four courses of gemcitabine (Gemzar) plus docetaxel (Taxotere) followed by doxorubicin (Adriamycin) for four courses should "answer the adjuvant chemotherapy question" for these patients, Dr. Martee L. Hensley said at Sarcoma and GIST 2014, hosted by the European Society for Medical Oncology.
The trial, known as Gynecologic Oncology Group (GOG) 0277, was the first study organized by the International Rare Cancer Initiative (IRCI) to activate. The study involves more than 200 U.S. centers and will open in many more European centers once regulatory approvals occur, said Dr. Hensley, professor of medicine at Weill Cornell Medical College and a gynecologic medical oncologist at Memorial Sloan- Kettering Cancer Center, New York.

Oncologists diagnose about 1,200 uterine sarcomas annually in the United States, most of which are uterine-limited and histologically high grade. "Successful conduct of this study in this rare but high-risk disease will establish the standard of care for managing women who have undergone complete resection," she said in an interview.
"Conducting prospective randomized trials in rare cancers is a significant challenge. International collaboration is considered a key factor in success" by speeding patient accrual, identifying research questions of international importance, and designing a trial that is internationally accepted and generalizable, Dr. Hensley said. In 2011, five cancer organizations formed the IRCI: the U.S. National Cancer Institute, the European Organization for the Research and Treatment of Cancer, Cancer Research UK, the U.K. National Cancer Research Network, and the French Institut National du Cancer.
The IRCI defines rare cancers as generally having an incidence below 2 cases per 100,000 population, and it is charged to develop intervention trials for these cancers, especially randomized trials.
"Creation of the IRCI has provided some needed infrastructure and has been critical to the success of GOG 0277," Dr. Hensley said. "But one could also say that GOG 0277 is also key to the IRCI’s success. The work we have done for GOG 0277 will inform the design and conduct of future international studies" in rare cancers.
The IRCI includes nine committees, each of which develops trials for different rare-cancer types. These include the gynecologic sarcoma committee that Dr. Hensley serves on and which helped organize GOG 0277, and other committees for small bowel adenocarcinoma, salivary gland cancer, thymoma, ocular melanoma, relapsed or metastatic anal cancer, rare brain cancer, desmoplastic small-round-cell tumor, and penile cancer. The committees include representatives appointed by the founding organizations, which also appoint the members of the IRCI board, the body that determines which committees to form, explained Nicola Keat, a staffer at Cancer Research UK in London who serves as the IRCI coordinator.
The gynecologic sarcoma committee decided that the question of whether adjuvant chemotherapy following complete resection helps patients with uterus-limited, high-grade leiomyosarcoma had "primary importance," said Dr. Hensley. "We recognized that the IRCI provided an ideal opportunity."
The gynecologic sarcoma group also plans to open a prospective study of doxorubicin for chemotherapy-naive patients with advanced, high-grade undifferentiated sarcoma of the uterus, a rare and aggressive cancer with no standard treatment. The study would also assess whether cabozantinib (Cometriq) can further prolong progression-free survival, compared with placebo, in patients with stable disease or an objective response to doxorubicin. In addition, the committee would like to launch a trial of aromatase inhibition for patients with low-grade endometrial stromal sarcoma through the IRCI, she said.
Following the launch of GOG 0277, the IRCI opened enrollment of patients into a trial focused on advanced uveal melanoma at U.S. centers, with U.K. recruitment anticipated to start later this year, Ms. Keat said in an interview. A third IRCI-organized trial, for patients with advanced anal cancer, recently opened for enrollment at participating U.K. centers, she added.
GOG 0277 began in June 2012 and aims to enroll 216 patients. As of February 2014, it had accrued seven patients, but Dr. Hensley said she believed the study was on track to its targeted finish date in 2018, as patients will soon start to enroll in Europe. "We expect the study to open in the U.K. in the next couple of months," Ms. Keat said in late March.
Dr. Hensley said that her spouse is a Sanofi employee. Ms. Keat had no disclosures.
On Twitter @mitchelzoler
MILAN – An ongoing phase III trial launched in 2012 that is testing whether adjuvant therapy aids women following removal of high-risk uterine leiomyosarcomas has also blazed a path for the International Rare Cancer Initiative, which has launched two other trials in rare cancers and is planning to start several more.
The advanced uterine leiomyosarcoma trial, which is testing an adjuvant regimen of up to four courses of gemcitabine (Gemzar) plus docetaxel (Taxotere) followed by doxorubicin (Adriamycin) for four courses should "answer the adjuvant chemotherapy question" for these patients, Dr. Martee L. Hensley said at Sarcoma and GIST 2014, hosted by the European Society for Medical Oncology.
The trial, known as Gynecologic Oncology Group (GOG) 0277, was the first study organized by the International Rare Cancer Initiative (IRCI) to activate. The study involves more than 200 U.S. centers and will open in many more European centers once regulatory approvals occur, said Dr. Hensley, professor of medicine at Weill Cornell Medical College and a gynecologic medical oncologist at Memorial Sloan- Kettering Cancer Center, New York.
Oncologists diagnose about 1,200 uterine sarcomas annually in the United States, most of which are uterine-limited and histologically high grade. "Successful conduct of this study in this rare but high-risk disease will establish the standard of care for managing women who have undergone complete resection," she said in an interview.
"Conducting prospective randomized trials in rare cancers is a significant challenge. International collaboration is considered a key factor in success" by speeding patient accrual, identifying research questions of international importance, and designing a trial that is internationally accepted and generalizable, Dr. Hensley said. In 2011, five cancer organizations formed the IRCI: the U.S. National Cancer Institute, the European Organization for the Research and Treatment of Cancer, Cancer Research UK, the U.K. National Cancer Research Network, and the French Institut National du Cancer.
The IRCI defines rare cancers as generally having an incidence below 2 cases per 100,000 population, and it is charged to develop intervention trials for these cancers, especially randomized trials.
"Creation of the IRCI has provided some needed infrastructure and has been critical to the success of GOG 0277," Dr. Hensley said. "But one could also say that GOG 0277 is also key to the IRCI’s success. The work we have done for GOG 0277 will inform the design and conduct of future international studies" in rare cancers.
The IRCI includes nine committees, each of which develops trials for different rare-cancer types. These include the gynecologic sarcoma committee that Dr. Hensley serves on and which helped organize GOG 0277, and other committees for small bowel adenocarcinoma, salivary gland cancer, thymoma, ocular melanoma, relapsed or metastatic anal cancer, rare brain cancer, desmoplastic small-round-cell tumor, and penile cancer. The committees include representatives appointed by the founding organizations, which also appoint the members of the IRCI board, the body that determines which committees to form, explained Nicola Keat, a staffer at Cancer Research UK in London who serves as the IRCI coordinator.
The gynecologic sarcoma committee decided that the question of whether adjuvant chemotherapy following complete resection helps patients with uterus-limited, high-grade leiomyosarcoma had "primary importance," said Dr. Hensley. "We recognized that the IRCI provided an ideal opportunity."
The gynecologic sarcoma group also plans to open a prospective study of doxorubicin for chemotherapy-naive patients with advanced, high-grade undifferentiated sarcoma of the uterus, a rare and aggressive cancer with no standard treatment. The study would also assess whether cabozantinib (Cometriq) can further prolong progression-free survival, compared with placebo, in patients with stable disease or an objective response to doxorubicin. In addition, the committee would like to launch a trial of aromatase inhibition for patients with low-grade endometrial stromal sarcoma through the IRCI, she said.
Following the launch of GOG 0277, the IRCI opened enrollment of patients into a trial focused on advanced uveal melanoma at U.S. centers, with U.K. recruitment anticipated to start later this year, Ms. Keat said in an interview. A third IRCI-organized trial, for patients with advanced anal cancer, recently opened for enrollment at participating U.K. centers, she added.
GOG 0277 began in June 2012 and aims to enroll 216 patients. As of February 2014, it had accrued seven patients, but Dr. Hensley said she believed the study was on track to its targeted finish date in 2018, as patients will soon start to enroll in Europe. "We expect the study to open in the U.K. in the next couple of months," Ms. Keat said in late March.
Dr. Hensley said that her spouse is a Sanofi employee. Ms. Keat had no disclosures.
On Twitter @mitchelzoler
MILAN – An ongoing phase III trial launched in 2012 that is testing whether adjuvant therapy aids women following removal of high-risk uterine leiomyosarcomas has also blazed a path for the International Rare Cancer Initiative, which has launched two other trials in rare cancers and is planning to start several more.
The advanced uterine leiomyosarcoma trial, which is testing an adjuvant regimen of up to four courses of gemcitabine (Gemzar) plus docetaxel (Taxotere) followed by doxorubicin (Adriamycin) for four courses should "answer the adjuvant chemotherapy question" for these patients, Dr. Martee L. Hensley said at Sarcoma and GIST 2014, hosted by the European Society for Medical Oncology.
The trial, known as Gynecologic Oncology Group (GOG) 0277, was the first study organized by the International Rare Cancer Initiative (IRCI) to activate. The study involves more than 200 U.S. centers and will open in many more European centers once regulatory approvals occur, said Dr. Hensley, professor of medicine at Weill Cornell Medical College and a gynecologic medical oncologist at Memorial Sloan- Kettering Cancer Center, New York.
Oncologists diagnose about 1,200 uterine sarcomas annually in the United States, most of which are uterine-limited and histologically high grade. "Successful conduct of this study in this rare but high-risk disease will establish the standard of care for managing women who have undergone complete resection," she said in an interview.
"Conducting prospective randomized trials in rare cancers is a significant challenge. International collaboration is considered a key factor in success" by speeding patient accrual, identifying research questions of international importance, and designing a trial that is internationally accepted and generalizable, Dr. Hensley said. In 2011, five cancer organizations formed the IRCI: the U.S. National Cancer Institute, the European Organization for the Research and Treatment of Cancer, Cancer Research UK, the U.K. National Cancer Research Network, and the French Institut National du Cancer.
The IRCI defines rare cancers as generally having an incidence below 2 cases per 100,000 population, and it is charged to develop intervention trials for these cancers, especially randomized trials.
"Creation of the IRCI has provided some needed infrastructure and has been critical to the success of GOG 0277," Dr. Hensley said. "But one could also say that GOG 0277 is also key to the IRCI’s success. The work we have done for GOG 0277 will inform the design and conduct of future international studies" in rare cancers.
The IRCI includes nine committees, each of which develops trials for different rare-cancer types. These include the gynecologic sarcoma committee that Dr. Hensley serves on and which helped organize GOG 0277, and other committees for small bowel adenocarcinoma, salivary gland cancer, thymoma, ocular melanoma, relapsed or metastatic anal cancer, rare brain cancer, desmoplastic small-round-cell tumor, and penile cancer. The committees include representatives appointed by the founding organizations, which also appoint the members of the IRCI board, the body that determines which committees to form, explained Nicola Keat, a staffer at Cancer Research UK in London who serves as the IRCI coordinator.
The gynecologic sarcoma committee decided that the question of whether adjuvant chemotherapy following complete resection helps patients with uterus-limited, high-grade leiomyosarcoma had "primary importance," said Dr. Hensley. "We recognized that the IRCI provided an ideal opportunity."
The gynecologic sarcoma group also plans to open a prospective study of doxorubicin for chemotherapy-naive patients with advanced, high-grade undifferentiated sarcoma of the uterus, a rare and aggressive cancer with no standard treatment. The study would also assess whether cabozantinib (Cometriq) can further prolong progression-free survival, compared with placebo, in patients with stable disease or an objective response to doxorubicin. In addition, the committee would like to launch a trial of aromatase inhibition for patients with low-grade endometrial stromal sarcoma through the IRCI, she said.
Following the launch of GOG 0277, the IRCI opened enrollment of patients into a trial focused on advanced uveal melanoma at U.S. centers, with U.K. recruitment anticipated to start later this year, Ms. Keat said in an interview. A third IRCI-organized trial, for patients with advanced anal cancer, recently opened for enrollment at participating U.K. centers, she added.
GOG 0277 began in June 2012 and aims to enroll 216 patients. As of February 2014, it had accrued seven patients, but Dr. Hensley said she believed the study was on track to its targeted finish date in 2018, as patients will soon start to enroll in Europe. "We expect the study to open in the U.K. in the next couple of months," Ms. Keat said in late March.
Dr. Hensley said that her spouse is a Sanofi employee. Ms. Keat had no disclosures.
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM SARCOMA AND GIST 2014

VIDEO: Exploring SYMPLICITY’s failure, Part 2
WASHINGTON – Far from being doomed after the failure SYMPLICITY HTN-3, renal denervation for refractory hypertension will continue to be explored. But it can’t go very far without finding a way to measure its effectiveness.
In Part 2 of our interview, Dr. Prakash Deedwania and Dr. George Bakris explore the options, and don’t miss the chance to talk about the controversial new hypertension guidelines.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
WASHINGTON – Far from being doomed after the failure SYMPLICITY HTN-3, renal denervation for refractory hypertension will continue to be explored. But it can’t go very far without finding a way to measure its effectiveness.
In Part 2 of our interview, Dr. Prakash Deedwania and Dr. George Bakris explore the options, and don’t miss the chance to talk about the controversial new hypertension guidelines.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
WASHINGTON – Far from being doomed after the failure SYMPLICITY HTN-3, renal denervation for refractory hypertension will continue to be explored. But it can’t go very far without finding a way to measure its effectiveness.
In Part 2 of our interview, Dr. Prakash Deedwania and Dr. George Bakris explore the options, and don’t miss the chance to talk about the controversial new hypertension guidelines.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
AT ACC 14

Colchicine called first-line pericarditis treatment
WASHINGTON – Adding colchicine to standard anti-inflammatory treatment for patients with recurrent pericarditis led to significantly fewer subsequent recurrences than did standard treatment alone in a randomized trial with 240 patients.
The findings cement colchicine as a first-line treatment for pericarditis patients who need drug treatment, Dr. Massimo Imazio said at the annual meeting of the American College of Cardiology. "Taken together with results from other randomized controlled trials, these findings suggest that colchicine should probably be regarded as a first-line treatment for either acute or recurrent pericarditis in the absence of contraindications."
Concurrent with Dr. Imazio’s report at the meeting, the results of the study were published online. (Lancet 2014 [doi:10.1016/S0140-6736(13)62709-9]).

The CORP 2 (Colchicine for Recurrent Pericarditis 2) trial was conducted at four centers in Italy during 2005-2012 and enrolled 240 adults with an episode of pericarditis and a documented prior pericarditis episode that had been followed by a symptom-free interval of at least 6 weeks.
For the study, Dr. Imazio and his associates randomized patients to received colchicine, either at 0.5 mg b.i.d. or once daily in patients weighing 70 kg or less, with no loading dose to avoid potential gastrointestinal adverse effects. Patients also received standard anti-inflammatory treatment with aspirin, ibuprofen, or indomethacin, and select patients could receive corticosteroid treatment. Treatment was continued for 6 months. Patients averaged 49 years of age; more than 80% had idiopathic pericarditis with the rest divided nearly equally between cardiac injury syndrome and connective tissue disease. Enrollment excluded patients with bacterial or neoplastic etiologies for their pericarditis.
Pericarditis occurred in 22% of patients treated with colchicine and 43% of those on placebo during an average follow-up of 20 months, a statistically significant difference for the study’s primary endpoint, reported Dr. Imazio, a cardiologist at Maria Vittoria Hospital in Torino, Italy. This benefit translated into a number needed to treat of five patients to prevent one pericarditis recurrence during 18 months of follow-up, he said. Colchicine was as effective in patients previously treated with the drug as in colchicine-naive patients, and was equally effective in all of the etiologies studied.
The dosage of colchicine used was well tolerated with no excess adverse effects or treatment discontinuations compared with placebo and no serious adverse effects. Overall, colchicine cut recurrence rates by about half based on a meta-analysis of these results along with those from six prior reports from randomized controlled trials of colchicine in a total of 1,275 patients with either a first episode or recurrent pericarditis, Dr. Imazio said. The meta-analysis also showed a consistent safety profile, with no excess adverse events or need for treatment withdrawal. The 6-month duration of treatment was selected arbitrarily; a future study could evaluate the drug for a longer treatment period, he added.
A concern with colchicine is that it morphed a few years ago from a cheap generic drug to a rebranded trade drug, Colcrys, with a large rise in price. Takeda, the company that now solely markets colchicine in the United States, had no role in the CORP 2 study.
Dr. Imazio said that he had no relevant financial disclosures.
On Twitter @mitchelzoler
These results are a major and important advance. Patients with recurrent pericarditis often require treatment with a corticosteroid that is effective, but patients frequently relapse when you try to taper the dose. It can be a very difficult down titration, and persistent corticosteroid treatment can cause salt retention and hypertension.

|
Mitchel L. Zoler/Frontline Medical News
|
In my practice colchicine is the drug of choice for patients with either a first episode or a recurrence of pericarditis. The dosing schedule used in the current study avoided the adverse effects that had been seen in prior studies with colchicine.
Colchicine is better than standard anti-inflammatory drugs, but it is not widely used for pericarditis in the United States, in part because the price has risen dramatically in recent years. But even at its current price, I use colchicine for all pericarditis patients, except those with a mild, brief episode that can be treated with aspirin alone. Patients with persistent and recurrent pericarditis can have persistent pain and can really suffer. Pericarditis is difficult to treat, but colchicine markedly helps. It ought to be used more than it currently is, although it has no labeling for this indication.
Dr. Allan S. Jaffe is a cardiologist and professor of medicine at the Mayo Clinic in Rochester, Minn. He said that he has been a consultant to or received honoraria from 10 drug or device companies but has no relationship with Takeda, the company that markets colchicine. He made these remarks in an interview and as a discussant for the report at the meeting.
These results are a major and important advance. Patients with recurrent pericarditis often require treatment with a corticosteroid that is effective, but patients frequently relapse when you try to taper the dose. It can be a very difficult down titration, and persistent corticosteroid treatment can cause salt retention and hypertension.
|
|
Mitchel L. Zoler/Frontline Medical News
|
In my practice colchicine is the drug of choice for patients with either a first episode or a recurrence of pericarditis. The dosing schedule used in the current study avoided the adverse effects that had been seen in prior studies with colchicine.
Colchicine is better than standard anti-inflammatory drugs, but it is not widely used for pericarditis in the United States, in part because the price has risen dramatically in recent years. But even at its current price, I use colchicine for all pericarditis patients, except those with a mild, brief episode that can be treated with aspirin alone. Patients with persistent and recurrent pericarditis can have persistent pain and can really suffer. Pericarditis is difficult to treat, but colchicine markedly helps. It ought to be used more than it currently is, although it has no labeling for this indication.
Dr. Allan S. Jaffe is a cardiologist and professor of medicine at the Mayo Clinic in Rochester, Minn. He said that he has been a consultant to or received honoraria from 10 drug or device companies but has no relationship with Takeda, the company that markets colchicine. He made these remarks in an interview and as a discussant for the report at the meeting.
These results are a major and important advance. Patients with recurrent pericarditis often require treatment with a corticosteroid that is effective, but patients frequently relapse when you try to taper the dose. It can be a very difficult down titration, and persistent corticosteroid treatment can cause salt retention and hypertension.
|
|
Mitchel L. Zoler/Frontline Medical News
|
In my practice colchicine is the drug of choice for patients with either a first episode or a recurrence of pericarditis. The dosing schedule used in the current study avoided the adverse effects that had been seen in prior studies with colchicine.
Colchicine is better than standard anti-inflammatory drugs, but it is not widely used for pericarditis in the United States, in part because the price has risen dramatically in recent years. But even at its current price, I use colchicine for all pericarditis patients, except those with a mild, brief episode that can be treated with aspirin alone. Patients with persistent and recurrent pericarditis can have persistent pain and can really suffer. Pericarditis is difficult to treat, but colchicine markedly helps. It ought to be used more than it currently is, although it has no labeling for this indication.
Dr. Allan S. Jaffe is a cardiologist and professor of medicine at the Mayo Clinic in Rochester, Minn. He said that he has been a consultant to or received honoraria from 10 drug or device companies but has no relationship with Takeda, the company that markets colchicine. He made these remarks in an interview and as a discussant for the report at the meeting.
WASHINGTON – Adding colchicine to standard anti-inflammatory treatment for patients with recurrent pericarditis led to significantly fewer subsequent recurrences than did standard treatment alone in a randomized trial with 240 patients.
The findings cement colchicine as a first-line treatment for pericarditis patients who need drug treatment, Dr. Massimo Imazio said at the annual meeting of the American College of Cardiology. "Taken together with results from other randomized controlled trials, these findings suggest that colchicine should probably be regarded as a first-line treatment for either acute or recurrent pericarditis in the absence of contraindications."
Concurrent with Dr. Imazio’s report at the meeting, the results of the study were published online. (Lancet 2014 [doi:10.1016/S0140-6736(13)62709-9]).
The CORP 2 (Colchicine for Recurrent Pericarditis 2) trial was conducted at four centers in Italy during 2005-2012 and enrolled 240 adults with an episode of pericarditis and a documented prior pericarditis episode that had been followed by a symptom-free interval of at least 6 weeks.
For the study, Dr. Imazio and his associates randomized patients to received colchicine, either at 0.5 mg b.i.d. or once daily in patients weighing 70 kg or less, with no loading dose to avoid potential gastrointestinal adverse effects. Patients also received standard anti-inflammatory treatment with aspirin, ibuprofen, or indomethacin, and select patients could receive corticosteroid treatment. Treatment was continued for 6 months. Patients averaged 49 years of age; more than 80% had idiopathic pericarditis with the rest divided nearly equally between cardiac injury syndrome and connective tissue disease. Enrollment excluded patients with bacterial or neoplastic etiologies for their pericarditis.
Pericarditis occurred in 22% of patients treated with colchicine and 43% of those on placebo during an average follow-up of 20 months, a statistically significant difference for the study’s primary endpoint, reported Dr. Imazio, a cardiologist at Maria Vittoria Hospital in Torino, Italy. This benefit translated into a number needed to treat of five patients to prevent one pericarditis recurrence during 18 months of follow-up, he said. Colchicine was as effective in patients previously treated with the drug as in colchicine-naive patients, and was equally effective in all of the etiologies studied.
The dosage of colchicine used was well tolerated with no excess adverse effects or treatment discontinuations compared with placebo and no serious adverse effects. Overall, colchicine cut recurrence rates by about half based on a meta-analysis of these results along with those from six prior reports from randomized controlled trials of colchicine in a total of 1,275 patients with either a first episode or recurrent pericarditis, Dr. Imazio said. The meta-analysis also showed a consistent safety profile, with no excess adverse events or need for treatment withdrawal. The 6-month duration of treatment was selected arbitrarily; a future study could evaluate the drug for a longer treatment period, he added.
A concern with colchicine is that it morphed a few years ago from a cheap generic drug to a rebranded trade drug, Colcrys, with a large rise in price. Takeda, the company that now solely markets colchicine in the United States, had no role in the CORP 2 study.
Dr. Imazio said that he had no relevant financial disclosures.
On Twitter @mitchelzoler
WASHINGTON – Adding colchicine to standard anti-inflammatory treatment for patients with recurrent pericarditis led to significantly fewer subsequent recurrences than did standard treatment alone in a randomized trial with 240 patients.
The findings cement colchicine as a first-line treatment for pericarditis patients who need drug treatment, Dr. Massimo Imazio said at the annual meeting of the American College of Cardiology. "Taken together with results from other randomized controlled trials, these findings suggest that colchicine should probably be regarded as a first-line treatment for either acute or recurrent pericarditis in the absence of contraindications."
Concurrent with Dr. Imazio’s report at the meeting, the results of the study were published online. (Lancet 2014 [doi:10.1016/S0140-6736(13)62709-9]).
The CORP 2 (Colchicine for Recurrent Pericarditis 2) trial was conducted at four centers in Italy during 2005-2012 and enrolled 240 adults with an episode of pericarditis and a documented prior pericarditis episode that had been followed by a symptom-free interval of at least 6 weeks.
For the study, Dr. Imazio and his associates randomized patients to received colchicine, either at 0.5 mg b.i.d. or once daily in patients weighing 70 kg or less, with no loading dose to avoid potential gastrointestinal adverse effects. Patients also received standard anti-inflammatory treatment with aspirin, ibuprofen, or indomethacin, and select patients could receive corticosteroid treatment. Treatment was continued for 6 months. Patients averaged 49 years of age; more than 80% had idiopathic pericarditis with the rest divided nearly equally between cardiac injury syndrome and connective tissue disease. Enrollment excluded patients with bacterial or neoplastic etiologies for their pericarditis.
Pericarditis occurred in 22% of patients treated with colchicine and 43% of those on placebo during an average follow-up of 20 months, a statistically significant difference for the study’s primary endpoint, reported Dr. Imazio, a cardiologist at Maria Vittoria Hospital in Torino, Italy. This benefit translated into a number needed to treat of five patients to prevent one pericarditis recurrence during 18 months of follow-up, he said. Colchicine was as effective in patients previously treated with the drug as in colchicine-naive patients, and was equally effective in all of the etiologies studied.
The dosage of colchicine used was well tolerated with no excess adverse effects or treatment discontinuations compared with placebo and no serious adverse effects. Overall, colchicine cut recurrence rates by about half based on a meta-analysis of these results along with those from six prior reports from randomized controlled trials of colchicine in a total of 1,275 patients with either a first episode or recurrent pericarditis, Dr. Imazio said. The meta-analysis also showed a consistent safety profile, with no excess adverse events or need for treatment withdrawal. The 6-month duration of treatment was selected arbitrarily; a future study could evaluate the drug for a longer treatment period, he added.
A concern with colchicine is that it morphed a few years ago from a cheap generic drug to a rebranded trade drug, Colcrys, with a large rise in price. Takeda, the company that now solely markets colchicine in the United States, had no role in the CORP 2 study.
Dr. Imazio said that he had no relevant financial disclosures.
On Twitter @mitchelzoler
AT ACC 14
Major finding: Patients with recurrent pericarditis treated with colchicine had a 22% recurrence rate compared with a 43% rate in control patients.
Data source: A randomized placebo-controlled trial with 240 patients enrolled at four centers in Italy.
Disclosures: Dr. Imazio said he had no relevant financial disclosures. Takeda, the sole company marketing colchicine (Colcrys) in the United States, played no role in the CORP 2 study. Dr. Jaffe said that he has been a consultant to or received honoraria from 10 drug or device companies, but he has no relationship with Takeda.

VIDEO: Study of hs-cTnT in chest pain intriguing, but flawed
WASHINGTON – The lack of detectable high-sensitivity cardiac troponin T in patients presenting with chest pain may be a marker of minimal risk of myocardial infarction within 30 days, according to Dr. Nadia Bandstein of the department of medicine at the Karolinska Institute, Solna, Sweden.
She and her colleagues looked at data from all emergency department visits for chest pain during 2010-2012 in Sweden. They found that the negative predictive value of myocardial infarction at 30 days for patients reporting with an hs-cTnT level of less than 5 ng/L, in addition to an ECG showing no blood flow-related damage, was 99.8%. That negative predictive value for death, meaning the patients are not at risk, was 100%.
Discussant Dr. Allan Jaffe of the Mayo Clinic in Rochester, Minn., said in an interview that although these findings are promising, the documentation in this study is not sufficient to use in clinical practice to let patients with these characteristics leave the emergency department.
*Update (3/31/14): This video has been updated.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
emergency department, Dr. Allan Jaffe, ACC
WASHINGTON – The lack of detectable high-sensitivity cardiac troponin T in patients presenting with chest pain may be a marker of minimal risk of myocardial infarction within 30 days, according to Dr. Nadia Bandstein of the department of medicine at the Karolinska Institute, Solna, Sweden.
She and her colleagues looked at data from all emergency department visits for chest pain during 2010-2012 in Sweden. They found that the negative predictive value of myocardial infarction at 30 days for patients reporting with an hs-cTnT level of less than 5 ng/L, in addition to an ECG showing no blood flow-related damage, was 99.8%. That negative predictive value for death, meaning the patients are not at risk, was 100%.
Discussant Dr. Allan Jaffe of the Mayo Clinic in Rochester, Minn., said in an interview that although these findings are promising, the documentation in this study is not sufficient to use in clinical practice to let patients with these characteristics leave the emergency department.
*Update (3/31/14): This video has been updated.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WASHINGTON – The lack of detectable high-sensitivity cardiac troponin T in patients presenting with chest pain may be a marker of minimal risk of myocardial infarction within 30 days, according to Dr. Nadia Bandstein of the department of medicine at the Karolinska Institute, Solna, Sweden.
She and her colleagues looked at data from all emergency department visits for chest pain during 2010-2012 in Sweden. They found that the negative predictive value of myocardial infarction at 30 days for patients reporting with an hs-cTnT level of less than 5 ng/L, in addition to an ECG showing no blood flow-related damage, was 99.8%. That negative predictive value for death, meaning the patients are not at risk, was 100%.
Discussant Dr. Allan Jaffe of the Mayo Clinic in Rochester, Minn., said in an interview that although these findings are promising, the documentation in this study is not sufficient to use in clinical practice to let patients with these characteristics leave the emergency department.
*Update (3/31/14): This video has been updated.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
emergency department, Dr. Allan Jaffe, ACC
emergency department, Dr. Allan Jaffe, ACC
AT ACC 14
