User login
Surprising new findings on hypertension in elderly blacks
CHICAGO– Older black Americans are more likely to have hypertension and less likely to have it under control than are their white counterparts, yet paradoxically they are also on more antihypertensive medications, according to a new analysis from the landmark ARIC study.
This finding is at odds with the conventional wisdom, which holds that the higher rate of poorly controlled hypertension in black patients is due to racial disparities in treatment, with blacks receiving less adequate treatment.

“In our study it appeared they were very well treated, yet they still didn’t achieve the same blood pressure as the older white patients,” Dr. Michael D. Miedema said in presenting the ARIC (Atherosclerosis Risk in Communities) findings at the American Heart Association Scientific Sessions.
Indeed, 88% of elderly black hypertensive patients were on at least one antihypertensive medication, compared with 71% of white hypertensives patients. Black hypertensive patients were also more likely to be on three or more antihypertensive drugs, by a margin of 27% to 16%.
“So it’s not for lack of antihypertensive medication use,” according to Dr. Miedema, a cardiologist at the Minneapolis Heart Institute.
The ARIC study is an ongoing longitudinal study of cardiovascular disease in older black and white men and women. The National Heart, Lung, and Blood Institute–funded study began in the late 1980s. Dr. Miedema’s analysis included 6,088 participants in the fifth clinical visit, which took place in 2011-2013. The subjects’ mean age was 75.6 years, and 23% were black. A total of 82% of subjects had hypertension as defined by blood pressures greater than 140/90 mm Hg; 81% of those with hypertension were aware of that fact. Put another way, nearly 20% of elderly hypertensive subjects were unaware they had hypertension, he noted.
One-third of ARIC participants had diabetes, 30% had cardiovascular disease, and 37% had chronic kidney disease. The prevalence of hypertension in subjects with diabetes or chronic kidney disease was 92%; in those without either comorbidity, it was 69%.
Of the total study population, 63% were at the blood pressure goals defined by JNC-7. This figure shot up to 79% using the less aggressive blood pressure goals recommended in the 2014 expert panel report (JAMA 2014;311:502-20), namely, targets of less than 150/90 mm Hg in individuals aged 60 years or older, and 140/90 in those with diabetes or chronic kidney disease. Thus, one in six elderly subjects in ARIC were reclassified from having high blood pressure to normal blood pressure through the use of the 2014 guidelines.
“Despite the more lenient 2014 blood pressure goals and a high rate of antihypertensive medication use, almost 20% of our total sample were not at goal blood pressure as defined by the 2014 expert committee. This high rate of uncontrolled blood pressure may be caused by a lack of awareness, treatment inertia, or medication nonadherence; it’s hard to say. But further efforts aimed at improving detection and control of hypertension in older individuals remain warranted,” Dr. Miedema commented.
Regardless of whether the 2003 JNC-7 guidelines or the 2014 expert panel recommendations were applied as the yardstick, elderly black patients with hypertension were an absolute 10%-15% less likely to be at target blood pressure, compared with their white counterparts.
It’s possible that the worse blood pressure control in black patients was brought about in part to less-appropriate drug prescribing. The most commonly used class of antihypertensive medications in both black and white hypertensives was ACE inhibitors/angiotensin receptor blockers (ARBs), even though both JNC-7 and the 2014 expert panel guidelines recommend diuretics or calcium channel blockers as first-line antihypertensive therapy in black patients. That’s because black patients with hypertension are often in a low renin state, in which ACE inhibitors and ARBs are less effective. Confusing the picture, however, is the fact that diabetes and chronic kidney disease were particularly prevalent among older black hypertensive patients, and ACE inhibitors and ARBs are preferentially guideline-recommended in patients with those diseases.
Dr. Miedema reported having no financial conflicts of interest.
CHICAGO– Older black Americans are more likely to have hypertension and less likely to have it under control than are their white counterparts, yet paradoxically they are also on more antihypertensive medications, according to a new analysis from the landmark ARIC study.
This finding is at odds with the conventional wisdom, which holds that the higher rate of poorly controlled hypertension in black patients is due to racial disparities in treatment, with blacks receiving less adequate treatment.
“In our study it appeared they were very well treated, yet they still didn’t achieve the same blood pressure as the older white patients,” Dr. Michael D. Miedema said in presenting the ARIC (Atherosclerosis Risk in Communities) findings at the American Heart Association Scientific Sessions.
Indeed, 88% of elderly black hypertensive patients were on at least one antihypertensive medication, compared with 71% of white hypertensives patients. Black hypertensive patients were also more likely to be on three or more antihypertensive drugs, by a margin of 27% to 16%.
“So it’s not for lack of antihypertensive medication use,” according to Dr. Miedema, a cardiologist at the Minneapolis Heart Institute.
The ARIC study is an ongoing longitudinal study of cardiovascular disease in older black and white men and women. The National Heart, Lung, and Blood Institute–funded study began in the late 1980s. Dr. Miedema’s analysis included 6,088 participants in the fifth clinical visit, which took place in 2011-2013. The subjects’ mean age was 75.6 years, and 23% were black. A total of 82% of subjects had hypertension as defined by blood pressures greater than 140/90 mm Hg; 81% of those with hypertension were aware of that fact. Put another way, nearly 20% of elderly hypertensive subjects were unaware they had hypertension, he noted.
One-third of ARIC participants had diabetes, 30% had cardiovascular disease, and 37% had chronic kidney disease. The prevalence of hypertension in subjects with diabetes or chronic kidney disease was 92%; in those without either comorbidity, it was 69%.
Of the total study population, 63% were at the blood pressure goals defined by JNC-7. This figure shot up to 79% using the less aggressive blood pressure goals recommended in the 2014 expert panel report (JAMA 2014;311:502-20), namely, targets of less than 150/90 mm Hg in individuals aged 60 years or older, and 140/90 in those with diabetes or chronic kidney disease. Thus, one in six elderly subjects in ARIC were reclassified from having high blood pressure to normal blood pressure through the use of the 2014 guidelines.
“Despite the more lenient 2014 blood pressure goals and a high rate of antihypertensive medication use, almost 20% of our total sample were not at goal blood pressure as defined by the 2014 expert committee. This high rate of uncontrolled blood pressure may be caused by a lack of awareness, treatment inertia, or medication nonadherence; it’s hard to say. But further efforts aimed at improving detection and control of hypertension in older individuals remain warranted,” Dr. Miedema commented.
Regardless of whether the 2003 JNC-7 guidelines or the 2014 expert panel recommendations were applied as the yardstick, elderly black patients with hypertension were an absolute 10%-15% less likely to be at target blood pressure, compared with their white counterparts.
It’s possible that the worse blood pressure control in black patients was brought about in part to less-appropriate drug prescribing. The most commonly used class of antihypertensive medications in both black and white hypertensives was ACE inhibitors/angiotensin receptor blockers (ARBs), even though both JNC-7 and the 2014 expert panel guidelines recommend diuretics or calcium channel blockers as first-line antihypertensive therapy in black patients. That’s because black patients with hypertension are often in a low renin state, in which ACE inhibitors and ARBs are less effective. Confusing the picture, however, is the fact that diabetes and chronic kidney disease were particularly prevalent among older black hypertensive patients, and ACE inhibitors and ARBs are preferentially guideline-recommended in patients with those diseases.
Dr. Miedema reported having no financial conflicts of interest.
CHICAGO– Older black Americans are more likely to have hypertension and less likely to have it under control than are their white counterparts, yet paradoxically they are also on more antihypertensive medications, according to a new analysis from the landmark ARIC study.
This finding is at odds with the conventional wisdom, which holds that the higher rate of poorly controlled hypertension in black patients is due to racial disparities in treatment, with blacks receiving less adequate treatment.
“In our study it appeared they were very well treated, yet they still didn’t achieve the same blood pressure as the older white patients,” Dr. Michael D. Miedema said in presenting the ARIC (Atherosclerosis Risk in Communities) findings at the American Heart Association Scientific Sessions.
Indeed, 88% of elderly black hypertensive patients were on at least one antihypertensive medication, compared with 71% of white hypertensives patients. Black hypertensive patients were also more likely to be on three or more antihypertensive drugs, by a margin of 27% to 16%.
“So it’s not for lack of antihypertensive medication use,” according to Dr. Miedema, a cardiologist at the Minneapolis Heart Institute.
The ARIC study is an ongoing longitudinal study of cardiovascular disease in older black and white men and women. The National Heart, Lung, and Blood Institute–funded study began in the late 1980s. Dr. Miedema’s analysis included 6,088 participants in the fifth clinical visit, which took place in 2011-2013. The subjects’ mean age was 75.6 years, and 23% were black. A total of 82% of subjects had hypertension as defined by blood pressures greater than 140/90 mm Hg; 81% of those with hypertension were aware of that fact. Put another way, nearly 20% of elderly hypertensive subjects were unaware they had hypertension, he noted.
One-third of ARIC participants had diabetes, 30% had cardiovascular disease, and 37% had chronic kidney disease. The prevalence of hypertension in subjects with diabetes or chronic kidney disease was 92%; in those without either comorbidity, it was 69%.
Of the total study population, 63% were at the blood pressure goals defined by JNC-7. This figure shot up to 79% using the less aggressive blood pressure goals recommended in the 2014 expert panel report (JAMA 2014;311:502-20), namely, targets of less than 150/90 mm Hg in individuals aged 60 years or older, and 140/90 in those with diabetes or chronic kidney disease. Thus, one in six elderly subjects in ARIC were reclassified from having high blood pressure to normal blood pressure through the use of the 2014 guidelines.
“Despite the more lenient 2014 blood pressure goals and a high rate of antihypertensive medication use, almost 20% of our total sample were not at goal blood pressure as defined by the 2014 expert committee. This high rate of uncontrolled blood pressure may be caused by a lack of awareness, treatment inertia, or medication nonadherence; it’s hard to say. But further efforts aimed at improving detection and control of hypertension in older individuals remain warranted,” Dr. Miedema commented.
Regardless of whether the 2003 JNC-7 guidelines or the 2014 expert panel recommendations were applied as the yardstick, elderly black patients with hypertension were an absolute 10%-15% less likely to be at target blood pressure, compared with their white counterparts.
It’s possible that the worse blood pressure control in black patients was brought about in part to less-appropriate drug prescribing. The most commonly used class of antihypertensive medications in both black and white hypertensives was ACE inhibitors/angiotensin receptor blockers (ARBs), even though both JNC-7 and the 2014 expert panel guidelines recommend diuretics or calcium channel blockers as first-line antihypertensive therapy in black patients. That’s because black patients with hypertension are often in a low renin state, in which ACE inhibitors and ARBs are less effective. Confusing the picture, however, is the fact that diabetes and chronic kidney disease were particularly prevalent among older black hypertensive patients, and ACE inhibitors and ARBs are preferentially guideline-recommended in patients with those diseases.
Dr. Miedema reported having no financial conflicts of interest.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: Older black patients with hypertension are less likely, by an absolute 10%-15%, to be at goal blood pressure than are their white hypertensive counterparts, despite taking more antihypertensive drugs.
Major finding: 88% of elderly black individuals with hypertension were on at least one antihypertensive agent and 27% were on three or more, compared with rates of 71% and 16%, respectively, in older white hypertensive subjects.
Data source: The Atherosclerosis Risk In Communities (ARIC) study is an ongoing longitudinal observational study.
Disclosures: The ARIC study is funded by the National Heart, Lung, and Blood Institute. The presenter reported having no financial conflicts.

TOPCAT reconsidered: Say ‘nyet’ to Russian data
CHICAGO – Something smells fishy about the results from Russia and Georgia in the TOPCAT trial, according to a study reappraisal conducted by the trial’s leaders.
TOPCAT (Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist) was a randomized, double-blind, placebo-controlled trial of spironolactone for the treatment of heart failure with preserved ejection fraction (HFpEF) in 3,445 patients in six countries. The primary outcome was negative, as presented at last year’s AHA scientific sessions and later published (N. Engl. J. Med. 2014;370:1383-92). However, a new post hoc analysis casts doubt on the validity of the results reported from Russia and Georgia, countries that contributed 49% of TOPCAT participants, Dr. Marc A. Pfeffer reported at the American Heart Association scientific sessions.

The patient outcomes reported from Russia and Georgia were spectacularly at odds with those reported from the United States, Canada, Argentina, and Brazil. Upon careful scrutiny, it appears likely that many Russian and Georgian patients either did not actually have HFpEF or were not taking their spironolactone. And if the results from the two Eastern European countries are put aside, then spironolactone markedly reduced the rate of the primary composite outcome – cardiovascular death or hospitalization for management of heart failure – in the 1,767 study participants in the Americas.
Indeed, the primary composite outcome occurred in 27.3% of spironolactone-treated subjects in the Western Hemisphere during a mean 3.3 years of follow-up, for an event rate of 10.4 per 100 patient-years, compared with rates of 31.8% and 12.6 per 100 patient-years in placebo-treated controls. That translates to a highly significant, 18% relative risk reduction (P = .026) in the primary outcome in spironolactone-treated HFpEF patients in the Americas. Cardiovascular mortality was reduced by 26% and heart failure hospitalization was reduced by 18%, according to Dr. Pfeffer, professor of medicine at Harvard University, Boston.
A post hoc analysis such as this would ordinarily be viewed as hypothesis-generating and nondefinitive. But this is a special situation, according to the cardiologist, who noted that guidelines offer no recommendations for the treatment of HFpEF, which now accounts for roughly 50% of all cases of heart failure.
“HFpEF is a growing part of the heart failure syndrome; it’s a frustrating part of the heart failure syndrome. And if we can improve the prognosis of the 40%-50% of people with symptomatic HFpEF by stating that our observation in the Americas was that spironolactone was associated with reduced cardiovascular deaths as well as hospitalizations for heart failure, then this should be taken into account. Since we don’t have other things we can do for these patients, I bring this to your attention,” Dr. Pfeffer said.
Among the major tip-offs that the Russian/Georgian TOPCAT data were dodgy was the post hoc finding that all-cause mortality, irrespective of treatment, was 21.8% in the Americas but a mere 8.4% in Eastern Europe.
“When I look at anyone’s clinical trial, if I want to ask about the severity of illness, I go to all-cause mortality. And here it was markedly different,” he observed.
Indeed, life-table analyses showed that the Russian/Georgian HFpEF subjects had a life expectancy typical of the region’s general population, whereas death rates for HFpEF enrollees from the Americas were several-fold higher than expected for age- and gender-matched controls.
Discussant Dr. Judith S. Hochman issued the usual caveats about the hazards of drawing conclusions from post hoc analyses of overall negative clinical trials, but then went on to agree with Dr. Pfeffer’s conclusions.
“I conclude, as does Dr. Pfeffer, that it’s reasonable to try mineralocorticoid receptor antagonists for symptomatic HFpEF patients with anticipated risks similar to those enrolled in the Americas. ... This is a growing condition and we have no other treatments,” said Dr. Hochman, professor and associate director of cardiology at New York University.
A key factor in her thinking was the mechanistic plausibility of the differential subgroup treatment effects, she added. For example, hyperkalemia – a well-known side effect of spironolactone – was 3.5-fold more common in patients randomized to spironolactone than in placebo patients in the Americas, as would be expected; but hyperkalemia was equally infrequent in both treatment arms in Russia and Georgia. Conversely, hypokalemia was 49% less likely with spironolactone than placebo in the Americas, yet there was no difference in the incidence of this side effect according to treatment status in Eastern Europe.
Moreover, systolic blood pressure fell by a placebo-subtracted 4.2 mm Hg at 1 year in spironolactone-treated patients in the Americas but was unchanged in Russian and Georgian patients. And while doubling of creatinine levels to above normal range was 60% more common with spironolactone than placebo in the Americas, rates were identical in the two treatment arms in Russia and Georgia.
“There was a physiologic response to spironolactone that paralleled an apparent outcome response,” Dr. Hochman concluded.
As for the discordant Eastern European data, she observed that confirming the diagnosis of HFpEF may be difficult: “Dyspnea, orthopnea, fatigue, lower extremity edema – all of these may be caused by other conditions. So it’s very complicated.”
In contrast to heart failure with reduced ejection fraction, which has seen enormous treatment advances in recent years, not much progress has been made in HFpEF. For that to occur, Dr. Hochman said, it will be essential to come up with refined, objective diagnostic criteria for use in clinical trials. Perhaps echocardiographic findings or elevated natriuretic peptide levels will fill that role, she added.
However, Dr. Pfeffer said that, much to his disappointment, he and other investigators have not seen a consistent correlation between higher baseline brain natriuretic peptide levels and greater clinical response to mineralocorticoid receptor antagonist therapy.
Asked what sort of oversight he and the other TOPCAT leaders had over the Russian and Georgian study sites, Dr. Pfeffer replied that the National Institutes of Health–sponsored study was underfunded for such monitoring.
“As one of the leaders of the trial, there are a lot of things that I would have wished to have done differently,” he said. “I have to stand here and say the amount that you get is inadequate to do what happens in industry-sponsored trials if there’s a perceived problem.”
Both Dr. Pfeffer and Dr. Hochman emphasized the critical importance of careful monitoring of serum potassium and creatinine when prescribing spironolactone in patients with HFpEF. But with regular monitoring, Dr. Pfeffer observed, the risk of major elevations is reassuringly low. For example, with monitoring of serum creatinine at every clinic visit and dose change as per TOPCAT protocol, the incidence of a level of 3.0 mg/dL or more was 10% with spironolactone and not significantly different at 9% with placebo in the Americas.
Dr. Pfeffer reported having received consultant fees from 20 pharmaceutical or medical device companies.
CHICAGO – Something smells fishy about the results from Russia and Georgia in the TOPCAT trial, according to a study reappraisal conducted by the trial’s leaders.
TOPCAT (Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist) was a randomized, double-blind, placebo-controlled trial of spironolactone for the treatment of heart failure with preserved ejection fraction (HFpEF) in 3,445 patients in six countries. The primary outcome was negative, as presented at last year’s AHA scientific sessions and later published (N. Engl. J. Med. 2014;370:1383-92). However, a new post hoc analysis casts doubt on the validity of the results reported from Russia and Georgia, countries that contributed 49% of TOPCAT participants, Dr. Marc A. Pfeffer reported at the American Heart Association scientific sessions.
The patient outcomes reported from Russia and Georgia were spectacularly at odds with those reported from the United States, Canada, Argentina, and Brazil. Upon careful scrutiny, it appears likely that many Russian and Georgian patients either did not actually have HFpEF or were not taking their spironolactone. And if the results from the two Eastern European countries are put aside, then spironolactone markedly reduced the rate of the primary composite outcome – cardiovascular death or hospitalization for management of heart failure – in the 1,767 study participants in the Americas.
Indeed, the primary composite outcome occurred in 27.3% of spironolactone-treated subjects in the Western Hemisphere during a mean 3.3 years of follow-up, for an event rate of 10.4 per 100 patient-years, compared with rates of 31.8% and 12.6 per 100 patient-years in placebo-treated controls. That translates to a highly significant, 18% relative risk reduction (P = .026) in the primary outcome in spironolactone-treated HFpEF patients in the Americas. Cardiovascular mortality was reduced by 26% and heart failure hospitalization was reduced by 18%, according to Dr. Pfeffer, professor of medicine at Harvard University, Boston.
A post hoc analysis such as this would ordinarily be viewed as hypothesis-generating and nondefinitive. But this is a special situation, according to the cardiologist, who noted that guidelines offer no recommendations for the treatment of HFpEF, which now accounts for roughly 50% of all cases of heart failure.
“HFpEF is a growing part of the heart failure syndrome; it’s a frustrating part of the heart failure syndrome. And if we can improve the prognosis of the 40%-50% of people with symptomatic HFpEF by stating that our observation in the Americas was that spironolactone was associated with reduced cardiovascular deaths as well as hospitalizations for heart failure, then this should be taken into account. Since we don’t have other things we can do for these patients, I bring this to your attention,” Dr. Pfeffer said.
Among the major tip-offs that the Russian/Georgian TOPCAT data were dodgy was the post hoc finding that all-cause mortality, irrespective of treatment, was 21.8% in the Americas but a mere 8.4% in Eastern Europe.
“When I look at anyone’s clinical trial, if I want to ask about the severity of illness, I go to all-cause mortality. And here it was markedly different,” he observed.
Indeed, life-table analyses showed that the Russian/Georgian HFpEF subjects had a life expectancy typical of the region’s general population, whereas death rates for HFpEF enrollees from the Americas were several-fold higher than expected for age- and gender-matched controls.
Discussant Dr. Judith S. Hochman issued the usual caveats about the hazards of drawing conclusions from post hoc analyses of overall negative clinical trials, but then went on to agree with Dr. Pfeffer’s conclusions.
“I conclude, as does Dr. Pfeffer, that it’s reasonable to try mineralocorticoid receptor antagonists for symptomatic HFpEF patients with anticipated risks similar to those enrolled in the Americas. ... This is a growing condition and we have no other treatments,” said Dr. Hochman, professor and associate director of cardiology at New York University.
A key factor in her thinking was the mechanistic plausibility of the differential subgroup treatment effects, she added. For example, hyperkalemia – a well-known side effect of spironolactone – was 3.5-fold more common in patients randomized to spironolactone than in placebo patients in the Americas, as would be expected; but hyperkalemia was equally infrequent in both treatment arms in Russia and Georgia. Conversely, hypokalemia was 49% less likely with spironolactone than placebo in the Americas, yet there was no difference in the incidence of this side effect according to treatment status in Eastern Europe.
Moreover, systolic blood pressure fell by a placebo-subtracted 4.2 mm Hg at 1 year in spironolactone-treated patients in the Americas but was unchanged in Russian and Georgian patients. And while doubling of creatinine levels to above normal range was 60% more common with spironolactone than placebo in the Americas, rates were identical in the two treatment arms in Russia and Georgia.
“There was a physiologic response to spironolactone that paralleled an apparent outcome response,” Dr. Hochman concluded.
As for the discordant Eastern European data, she observed that confirming the diagnosis of HFpEF may be difficult: “Dyspnea, orthopnea, fatigue, lower extremity edema – all of these may be caused by other conditions. So it’s very complicated.”
In contrast to heart failure with reduced ejection fraction, which has seen enormous treatment advances in recent years, not much progress has been made in HFpEF. For that to occur, Dr. Hochman said, it will be essential to come up with refined, objective diagnostic criteria for use in clinical trials. Perhaps echocardiographic findings or elevated natriuretic peptide levels will fill that role, she added.
However, Dr. Pfeffer said that, much to his disappointment, he and other investigators have not seen a consistent correlation between higher baseline brain natriuretic peptide levels and greater clinical response to mineralocorticoid receptor antagonist therapy.
Asked what sort of oversight he and the other TOPCAT leaders had over the Russian and Georgian study sites, Dr. Pfeffer replied that the National Institutes of Health–sponsored study was underfunded for such monitoring.
“As one of the leaders of the trial, there are a lot of things that I would have wished to have done differently,” he said. “I have to stand here and say the amount that you get is inadequate to do what happens in industry-sponsored trials if there’s a perceived problem.”
Both Dr. Pfeffer and Dr. Hochman emphasized the critical importance of careful monitoring of serum potassium and creatinine when prescribing spironolactone in patients with HFpEF. But with regular monitoring, Dr. Pfeffer observed, the risk of major elevations is reassuringly low. For example, with monitoring of serum creatinine at every clinic visit and dose change as per TOPCAT protocol, the incidence of a level of 3.0 mg/dL or more was 10% with spironolactone and not significantly different at 9% with placebo in the Americas.
Dr. Pfeffer reported having received consultant fees from 20 pharmaceutical or medical device companies.
CHICAGO – Something smells fishy about the results from Russia and Georgia in the TOPCAT trial, according to a study reappraisal conducted by the trial’s leaders.
TOPCAT (Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist) was a randomized, double-blind, placebo-controlled trial of spironolactone for the treatment of heart failure with preserved ejection fraction (HFpEF) in 3,445 patients in six countries. The primary outcome was negative, as presented at last year’s AHA scientific sessions and later published (N. Engl. J. Med. 2014;370:1383-92). However, a new post hoc analysis casts doubt on the validity of the results reported from Russia and Georgia, countries that contributed 49% of TOPCAT participants, Dr. Marc A. Pfeffer reported at the American Heart Association scientific sessions.
The patient outcomes reported from Russia and Georgia were spectacularly at odds with those reported from the United States, Canada, Argentina, and Brazil. Upon careful scrutiny, it appears likely that many Russian and Georgian patients either did not actually have HFpEF or were not taking their spironolactone. And if the results from the two Eastern European countries are put aside, then spironolactone markedly reduced the rate of the primary composite outcome – cardiovascular death or hospitalization for management of heart failure – in the 1,767 study participants in the Americas.
Indeed, the primary composite outcome occurred in 27.3% of spironolactone-treated subjects in the Western Hemisphere during a mean 3.3 years of follow-up, for an event rate of 10.4 per 100 patient-years, compared with rates of 31.8% and 12.6 per 100 patient-years in placebo-treated controls. That translates to a highly significant, 18% relative risk reduction (P = .026) in the primary outcome in spironolactone-treated HFpEF patients in the Americas. Cardiovascular mortality was reduced by 26% and heart failure hospitalization was reduced by 18%, according to Dr. Pfeffer, professor of medicine at Harvard University, Boston.
A post hoc analysis such as this would ordinarily be viewed as hypothesis-generating and nondefinitive. But this is a special situation, according to the cardiologist, who noted that guidelines offer no recommendations for the treatment of HFpEF, which now accounts for roughly 50% of all cases of heart failure.
“HFpEF is a growing part of the heart failure syndrome; it’s a frustrating part of the heart failure syndrome. And if we can improve the prognosis of the 40%-50% of people with symptomatic HFpEF by stating that our observation in the Americas was that spironolactone was associated with reduced cardiovascular deaths as well as hospitalizations for heart failure, then this should be taken into account. Since we don’t have other things we can do for these patients, I bring this to your attention,” Dr. Pfeffer said.
Among the major tip-offs that the Russian/Georgian TOPCAT data were dodgy was the post hoc finding that all-cause mortality, irrespective of treatment, was 21.8% in the Americas but a mere 8.4% in Eastern Europe.
“When I look at anyone’s clinical trial, if I want to ask about the severity of illness, I go to all-cause mortality. And here it was markedly different,” he observed.
Indeed, life-table analyses showed that the Russian/Georgian HFpEF subjects had a life expectancy typical of the region’s general population, whereas death rates for HFpEF enrollees from the Americas were several-fold higher than expected for age- and gender-matched controls.
Discussant Dr. Judith S. Hochman issued the usual caveats about the hazards of drawing conclusions from post hoc analyses of overall negative clinical trials, but then went on to agree with Dr. Pfeffer’s conclusions.
“I conclude, as does Dr. Pfeffer, that it’s reasonable to try mineralocorticoid receptor antagonists for symptomatic HFpEF patients with anticipated risks similar to those enrolled in the Americas. ... This is a growing condition and we have no other treatments,” said Dr. Hochman, professor and associate director of cardiology at New York University.
A key factor in her thinking was the mechanistic plausibility of the differential subgroup treatment effects, she added. For example, hyperkalemia – a well-known side effect of spironolactone – was 3.5-fold more common in patients randomized to spironolactone than in placebo patients in the Americas, as would be expected; but hyperkalemia was equally infrequent in both treatment arms in Russia and Georgia. Conversely, hypokalemia was 49% less likely with spironolactone than placebo in the Americas, yet there was no difference in the incidence of this side effect according to treatment status in Eastern Europe.
Moreover, systolic blood pressure fell by a placebo-subtracted 4.2 mm Hg at 1 year in spironolactone-treated patients in the Americas but was unchanged in Russian and Georgian patients. And while doubling of creatinine levels to above normal range was 60% more common with spironolactone than placebo in the Americas, rates were identical in the two treatment arms in Russia and Georgia.
“There was a physiologic response to spironolactone that paralleled an apparent outcome response,” Dr. Hochman concluded.
As for the discordant Eastern European data, she observed that confirming the diagnosis of HFpEF may be difficult: “Dyspnea, orthopnea, fatigue, lower extremity edema – all of these may be caused by other conditions. So it’s very complicated.”
In contrast to heart failure with reduced ejection fraction, which has seen enormous treatment advances in recent years, not much progress has been made in HFpEF. For that to occur, Dr. Hochman said, it will be essential to come up with refined, objective diagnostic criteria for use in clinical trials. Perhaps echocardiographic findings or elevated natriuretic peptide levels will fill that role, she added.
However, Dr. Pfeffer said that, much to his disappointment, he and other investigators have not seen a consistent correlation between higher baseline brain natriuretic peptide levels and greater clinical response to mineralocorticoid receptor antagonist therapy.
Asked what sort of oversight he and the other TOPCAT leaders had over the Russian and Georgian study sites, Dr. Pfeffer replied that the National Institutes of Health–sponsored study was underfunded for such monitoring.
“As one of the leaders of the trial, there are a lot of things that I would have wished to have done differently,” he said. “I have to stand here and say the amount that you get is inadequate to do what happens in industry-sponsored trials if there’s a perceived problem.”
Both Dr. Pfeffer and Dr. Hochman emphasized the critical importance of careful monitoring of serum potassium and creatinine when prescribing spironolactone in patients with HFpEF. But with regular monitoring, Dr. Pfeffer observed, the risk of major elevations is reassuringly low. For example, with monitoring of serum creatinine at every clinic visit and dose change as per TOPCAT protocol, the incidence of a level of 3.0 mg/dL or more was 10% with spironolactone and not significantly different at 9% with placebo in the Americas.
Dr. Pfeffer reported having received consultant fees from 20 pharmaceutical or medical device companies.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: Prescribing spironolactone for patients with symptomatic heart failure with preserved ejection fraction may reduce their risks of cardiovascular death and hospitalizations for heart failure.
Major finding: After exclusion of highly suspect Russian and Georgian data on 1,678 patients, the primary composite outcome of cardiovascular death or heart failure hospitalization in the remaining 1,767 patients with heart failure with preserved ejection fraction in four Western Hemisphere countries occurred in 27.3% of patients on spironolactone, for a significant 18% relative risk reduction compared with the 31.8% rate in placebo-treated controls.
Data source: TOPCAT, a randomized, double-blind, placebo-controlled, six-nation study involving 3,445 patients with heart failure with preserved ejection fraction treated for a mean of 3.3 years.
Disclosures: TOPCAT was sponsored by the National Heart, Lung, and Blood Institute. The presenter has received consulting fees from 20 pharmaceutical or medical device companies.

Heart failure device shown safe in PARACHUTE III
LAS VEGAS – Percutaneous ventricular restoration using the Parachute device in patients with ischemic dilated cardiomyopathy had a promisingly low 26% combined rate of all-cause mortality and heart failure hospitalization at 1 year in the PARACHUTE III trial, according to Dr. William T. Abraham
PARACHUTE III was a European postmarketing study. And while the composite endpoint of all-cause mortality and heart failure hospitalization was a prespecified secondary outcome in that study, it’s the primary endpoint in the ongoing pivotal U.S. phase III PARACHUTE IV trial headed by Dr. Abraham.

What’s encouraging about the PARACHUTE III result is that the 26% rate of the combined endpoint is in line with the PARACHUTE IV investigators’ assumption for outcomes in the ongoing U.S. trial, which will randomize roughly 500 patients to device therapy plus optimal medical therapy or optimal medical therapy alone, he reported at the annual meeting of the Heart Failure Society of America.
“The PARACHUTE III results are reassuring. In historical controls, the expected rate [of all-cause mortality and heart failure hospitalization] in this kind of population at 1 year is about 35%,” according to Dr. Abraham, professor of internal medicine, physiology, and cell biology and director of the division of cardiovascular medicine at Ohio State University, Columbus.
Plus, technical improvements in device design and percutaneous implantation technique have been made since PARACHUTE III and are incorporated in PARACHUTE IV. These improvements could drive the composite endpoint rate in the ongoing U.S. trial even lower, he continued. For example, while the 1-year major complication rate in the European postmarketing study was 11%, thus far it’s less than 8% in the U.S. experience.
The proprietary Parachute device is so named because, when deployed, it looks like an upside-down parachute. It is intended for patients with ischemic dilated cardiomyopathy secondary to anteroapical MI. The goal of this device therapy is to attenuate or reverse progression of heart failure in this high-risk population.
The Parachute device has two proposed mechanisms of action, the cardiologist explained. One involves ventricular restoration via ventricular partitioning to reduce wall stress in the upper chamber as well as fostering a return to a more normal, elliptical shape to the heart. Dr. Abraham termed the other mechanism a trampoline effect, in which the compliant device encourages replacement of eccentric apical wall motion with more synchronized wall motion throughout the cardiac cycle.
The device, comprised mainly of a fluoropolymer membrane and a nitinol frame, is implanted percutaneously retrograde across the aortic valve. It is placed in the apex of the left ventricle.

The European Union PARACHUTE III postmarketing study was a single-arm study including 100 consecutive symptomatic patients in 10 countries. Their baseline left ventricular ejection fraction was 28%. Device implantation was accomplished in 97 of the 100 patients, with a mean 94-minute procedure duration and 23 minutes of fluoroscopy time.
The primary study endpoint was a safety outcome: the 1-year rate of procedure- and/or device-related adverse events. The 7% rate included several cases of left ventricular perforation as well as aortic or mitral valve damage.
“The rates and distribution of these events are comparable to other structural heart disease interventions, such as transcatheter aortic valve replacement, and might be considered an acceptable postmarketing result,” Dr. Abraham commented.
There was a 3.2% stroke rate through 1 year and a 9.5% mortality rate.
Hemodynamic improvements were seen in both systolic and diastolic function. The left atrial volume index improved from 42.5 to 38.3 mL/m2, indicative of favorable remodeling. The improvement in systolic function was expressed in an increased contractility index.
Moreover, left ventricular end-systolic volume index improved over the course of the year from 84 to 70.5 mL/m2, while left ventricular end-diastolic volume index also improved, from 117.3 to 99.1 mL/m2.
Eighty percent of patients maintained or improved their 6-minute walk distance. On average, patients improved their walk distance by 25 m from a mean baseline of 372 m. Similarly, 80% of patients maintained or improved their baseline New York Heart Association functional class.
Discussant Dr. James C. Fang called the Parachute device “an important investigational therapeutic option. ” He urged cardiologists to enroll patients in PARACHUTE IV in order to learn whether the percutaneous device therapy will indeed enable patients to live longer and/or avoid hospitalizations for heart failure.
The improvements in myocardial contractility and left atrial volume documented in PARACHUTE III are particularly encouraging, added Dr. Fang, professor of internal medicine and chief of the division of cardiovascular medicine at the University of Utah, Salt Lake City.
Dr. Abraham reported serving as a consultant to CardioKinetix, which sponsors the PARACHUTE IV trial.
LAS VEGAS – Percutaneous ventricular restoration using the Parachute device in patients with ischemic dilated cardiomyopathy had a promisingly low 26% combined rate of all-cause mortality and heart failure hospitalization at 1 year in the PARACHUTE III trial, according to Dr. William T. Abraham
PARACHUTE III was a European postmarketing study. And while the composite endpoint of all-cause mortality and heart failure hospitalization was a prespecified secondary outcome in that study, it’s the primary endpoint in the ongoing pivotal U.S. phase III PARACHUTE IV trial headed by Dr. Abraham.
What’s encouraging about the PARACHUTE III result is that the 26% rate of the combined endpoint is in line with the PARACHUTE IV investigators’ assumption for outcomes in the ongoing U.S. trial, which will randomize roughly 500 patients to device therapy plus optimal medical therapy or optimal medical therapy alone, he reported at the annual meeting of the Heart Failure Society of America.
“The PARACHUTE III results are reassuring. In historical controls, the expected rate [of all-cause mortality and heart failure hospitalization] in this kind of population at 1 year is about 35%,” according to Dr. Abraham, professor of internal medicine, physiology, and cell biology and director of the division of cardiovascular medicine at Ohio State University, Columbus.
Plus, technical improvements in device design and percutaneous implantation technique have been made since PARACHUTE III and are incorporated in PARACHUTE IV. These improvements could drive the composite endpoint rate in the ongoing U.S. trial even lower, he continued. For example, while the 1-year major complication rate in the European postmarketing study was 11%, thus far it’s less than 8% in the U.S. experience.
The proprietary Parachute device is so named because, when deployed, it looks like an upside-down parachute. It is intended for patients with ischemic dilated cardiomyopathy secondary to anteroapical MI. The goal of this device therapy is to attenuate or reverse progression of heart failure in this high-risk population.
The Parachute device has two proposed mechanisms of action, the cardiologist explained. One involves ventricular restoration via ventricular partitioning to reduce wall stress in the upper chamber as well as fostering a return to a more normal, elliptical shape to the heart. Dr. Abraham termed the other mechanism a trampoline effect, in which the compliant device encourages replacement of eccentric apical wall motion with more synchronized wall motion throughout the cardiac cycle.
The device, comprised mainly of a fluoropolymer membrane and a nitinol frame, is implanted percutaneously retrograde across the aortic valve. It is placed in the apex of the left ventricle.
The European Union PARACHUTE III postmarketing study was a single-arm study including 100 consecutive symptomatic patients in 10 countries. Their baseline left ventricular ejection fraction was 28%. Device implantation was accomplished in 97 of the 100 patients, with a mean 94-minute procedure duration and 23 minutes of fluoroscopy time.
The primary study endpoint was a safety outcome: the 1-year rate of procedure- and/or device-related adverse events. The 7% rate included several cases of left ventricular perforation as well as aortic or mitral valve damage.
“The rates and distribution of these events are comparable to other structural heart disease interventions, such as transcatheter aortic valve replacement, and might be considered an acceptable postmarketing result,” Dr. Abraham commented.
There was a 3.2% stroke rate through 1 year and a 9.5% mortality rate.
Hemodynamic improvements were seen in both systolic and diastolic function. The left atrial volume index improved from 42.5 to 38.3 mL/m2, indicative of favorable remodeling. The improvement in systolic function was expressed in an increased contractility index.
Moreover, left ventricular end-systolic volume index improved over the course of the year from 84 to 70.5 mL/m2, while left ventricular end-diastolic volume index also improved, from 117.3 to 99.1 mL/m2.
Eighty percent of patients maintained or improved their 6-minute walk distance. On average, patients improved their walk distance by 25 m from a mean baseline of 372 m. Similarly, 80% of patients maintained or improved their baseline New York Heart Association functional class.
Discussant Dr. James C. Fang called the Parachute device “an important investigational therapeutic option. ” He urged cardiologists to enroll patients in PARACHUTE IV in order to learn whether the percutaneous device therapy will indeed enable patients to live longer and/or avoid hospitalizations for heart failure.
The improvements in myocardial contractility and left atrial volume documented in PARACHUTE III are particularly encouraging, added Dr. Fang, professor of internal medicine and chief of the division of cardiovascular medicine at the University of Utah, Salt Lake City.
Dr. Abraham reported serving as a consultant to CardioKinetix, which sponsors the PARACHUTE IV trial.
LAS VEGAS – Percutaneous ventricular restoration using the Parachute device in patients with ischemic dilated cardiomyopathy had a promisingly low 26% combined rate of all-cause mortality and heart failure hospitalization at 1 year in the PARACHUTE III trial, according to Dr. William T. Abraham
PARACHUTE III was a European postmarketing study. And while the composite endpoint of all-cause mortality and heart failure hospitalization was a prespecified secondary outcome in that study, it’s the primary endpoint in the ongoing pivotal U.S. phase III PARACHUTE IV trial headed by Dr. Abraham.
What’s encouraging about the PARACHUTE III result is that the 26% rate of the combined endpoint is in line with the PARACHUTE IV investigators’ assumption for outcomes in the ongoing U.S. trial, which will randomize roughly 500 patients to device therapy plus optimal medical therapy or optimal medical therapy alone, he reported at the annual meeting of the Heart Failure Society of America.
“The PARACHUTE III results are reassuring. In historical controls, the expected rate [of all-cause mortality and heart failure hospitalization] in this kind of population at 1 year is about 35%,” according to Dr. Abraham, professor of internal medicine, physiology, and cell biology and director of the division of cardiovascular medicine at Ohio State University, Columbus.
Plus, technical improvements in device design and percutaneous implantation technique have been made since PARACHUTE III and are incorporated in PARACHUTE IV. These improvements could drive the composite endpoint rate in the ongoing U.S. trial even lower, he continued. For example, while the 1-year major complication rate in the European postmarketing study was 11%, thus far it’s less than 8% in the U.S. experience.
The proprietary Parachute device is so named because, when deployed, it looks like an upside-down parachute. It is intended for patients with ischemic dilated cardiomyopathy secondary to anteroapical MI. The goal of this device therapy is to attenuate or reverse progression of heart failure in this high-risk population.
The Parachute device has two proposed mechanisms of action, the cardiologist explained. One involves ventricular restoration via ventricular partitioning to reduce wall stress in the upper chamber as well as fostering a return to a more normal, elliptical shape to the heart. Dr. Abraham termed the other mechanism a trampoline effect, in which the compliant device encourages replacement of eccentric apical wall motion with more synchronized wall motion throughout the cardiac cycle.
The device, comprised mainly of a fluoropolymer membrane and a nitinol frame, is implanted percutaneously retrograde across the aortic valve. It is placed in the apex of the left ventricle.
The European Union PARACHUTE III postmarketing study was a single-arm study including 100 consecutive symptomatic patients in 10 countries. Their baseline left ventricular ejection fraction was 28%. Device implantation was accomplished in 97 of the 100 patients, with a mean 94-minute procedure duration and 23 minutes of fluoroscopy time.
The primary study endpoint was a safety outcome: the 1-year rate of procedure- and/or device-related adverse events. The 7% rate included several cases of left ventricular perforation as well as aortic or mitral valve damage.
“The rates and distribution of these events are comparable to other structural heart disease interventions, such as transcatheter aortic valve replacement, and might be considered an acceptable postmarketing result,” Dr. Abraham commented.
There was a 3.2% stroke rate through 1 year and a 9.5% mortality rate.
Hemodynamic improvements were seen in both systolic and diastolic function. The left atrial volume index improved from 42.5 to 38.3 mL/m2, indicative of favorable remodeling. The improvement in systolic function was expressed in an increased contractility index.
Moreover, left ventricular end-systolic volume index improved over the course of the year from 84 to 70.5 mL/m2, while left ventricular end-diastolic volume index also improved, from 117.3 to 99.1 mL/m2.
Eighty percent of patients maintained or improved their 6-minute walk distance. On average, patients improved their walk distance by 25 m from a mean baseline of 372 m. Similarly, 80% of patients maintained or improved their baseline New York Heart Association functional class.
Discussant Dr. James C. Fang called the Parachute device “an important investigational therapeutic option. ” He urged cardiologists to enroll patients in PARACHUTE IV in order to learn whether the percutaneous device therapy will indeed enable patients to live longer and/or avoid hospitalizations for heart failure.
The improvements in myocardial contractility and left atrial volume documented in PARACHUTE III are particularly encouraging, added Dr. Fang, professor of internal medicine and chief of the division of cardiovascular medicine at the University of Utah, Salt Lake City.
Dr. Abraham reported serving as a consultant to CardioKinetix, which sponsors the PARACHUTE IV trial.
AT THE HFSA ANNUAL SCIENTIFIC MEETING
Key clinical point: A percutaneous device known as the Parachute, when placed in the apex of the left ventricle, may slow or reverse progression of heart failure in patients with ischemic dilated cardiomyopathy.
Major finding: The 1-year rate of the composite endpoint of all-cause mortality and heart failure hospitalization following Parachute placement was 26%.
Data source: The PARACHUTE III study was a single-arm European postmarketing study involving 100 consecutive symptomatic patients.
Disclosures: PARACHUTE III as well as the ongoing pivotal U.S. PARACHUTE IV study were sponsored by CardioKinetix. The presenter serves as a consultant to the company and principal investigator of PARACHUTE IV.

Redefined schizoaffective disorder in DSM-5 seen as problematic
BERLIN – The redefinition of schizoaffective disorder unveiled in the DSM-5 leaves much to be desired, experts opined at the annual congress of the European College of Neuropsychopharmacology
“We all thought we had the solution for making new criteria for schizoaffective disorder,” recalled Dr. Jim van Os, who served on the DSM-5 committee charged with reexamining the disorder. “But in the end, nothing much changed.”
Indeed, despite the lofty ambitions he and many of his fellow DSM-5 committee members held for thoroughly overhauling the criteria for what he called “this enigmatic combination of affective and psychotic dysregulation,” the difference between the DSM-IV and DSM-5 boiled down to small potatoes: Whereas the DSM-IV stated that the diagnosis of schizoaffective disorder required that the mood episode must be present for “a substantial duration of the illness,” the DSM-5 requires that the mood episode be present for “the majority” of the illness.

“The change was made to improve the reliability and stability of the diagnosis,” explained Dr. van Os, professor of psychiatric epidemiology at Maastricht (the Netherlands) University.
He was an advocate on the committee for introducing a dimensional approach to the diagnosis of schizoaffective disorder, one that would incorporate consideration of clustering of symptoms across a continuum.
“This would have been the best solution to the conundrum of the ‘fuzzy set’ problem of mental disorders. It would have been a great way of recognizing the continuity of all mental phenomena seen in mental disorders. However, it did not happen, unfortunately.”
The psychiatrist noted that committee members debated “for many hours” how best to overhaul the criteria for schizoaffective disorder in the DSM-5 before deciding to scale back their scope.
“We realized that any measure of precision in the criteria we tried to introduce for the admixture of affective dysregulation and psychosis is spurious. We really haven’t got the tools yet to make valid distinctions. That’s why we said in DSM-5 that the clinician can make the diagnosis using clinical intuition that the majority of time in the illness there is affective dysregulation. So, not spurious precision, but clinical intuition. I don’t know if that’s a good decision, but that’s what we did,” Dr. van Os said.
At the same symposium on schizoaffective disorder held during the ECNP congress, Dr. Heinz Grunze declared: “I would have welcomed as a development in DSM-5, as we’ve seen with some other disorders, basically abolishing schizoaffective disorder on its own and assigning it either to bipolar disorder with psychotic features specified or to schizophrenia with a strong affective specifier. I think that would be better suited to characterizing these patients.”

“Clearly, schizoaffective disorder has quite a polymorphic course. It’s definitely not one illness; it’s several illnesses which we put together in this random category,” added Dr. Grunze, professor of clinical psychiatry at University of Newcastle (England).
His own view is that the disorder belongs under the umbrella of bipolar disorder. A recent Spanish functional magnetic resonance brain imaging study demonstrated that schizoaffective disorder exhibits features that more closely resemble those of bipolar disorder than schizophrenia, including the finding that patients in clinical remission show failure of deactivation in the medial frontal gyrus, compared with healthy controls, Dr. Grunze noted.
“I personally can’t see a difference between a schizoaffective acute episode, manic type, and a psychotic manic episode. So my personal opinion is that schizoaffective disorder belongs under the heading of bipolar disorder. That’s obviously different from the opinion of those conducting treatment trials and the pharmaceutical industry, where schizoaffective disorder is never a subgroup in bipolar disorder trials, but a subgroup in schizophrenia trials,” Dr. Grunze said.
One audience member rose to state that while all this wrangling over how best to subclassify these patients is interesting, why not take a pragmatic approach and simply treat them symptomatically?
“Well, I think that’s what everyone in this room who’s a clinician is doing,” Dr. Grunze replied. “Because even if we assume that the diagnosis itself might be unstable, I think the greatest variance in patients switching around between bipolar, schizophrenia, and schizoaffective comes from the different doctors seeing them, each having a different understanding of schizoaffective disorder. I see a lot of patients who are classified as schizoaffective disorder because they have what I would have considered just as mood-congruent psychotic symptoms while manic. So I absolutely agree with you that this is more of an academic discussion, really, because at the moment we don’t have any consequences for treatment. We don’t have good evidence about how to treat these patients; we just extrapolate from whatever we think their symptoms come closest to.”
He reported receiving research grants from the National Institute for Health Research and the U.K. Medical Research Council, as well as more than a half-dozen pharmaceutical companies.
Dr. van Os reported having no financial conflicts.
BERLIN – The redefinition of schizoaffective disorder unveiled in the DSM-5 leaves much to be desired, experts opined at the annual congress of the European College of Neuropsychopharmacology
“We all thought we had the solution for making new criteria for schizoaffective disorder,” recalled Dr. Jim van Os, who served on the DSM-5 committee charged with reexamining the disorder. “But in the end, nothing much changed.”
Indeed, despite the lofty ambitions he and many of his fellow DSM-5 committee members held for thoroughly overhauling the criteria for what he called “this enigmatic combination of affective and psychotic dysregulation,” the difference between the DSM-IV and DSM-5 boiled down to small potatoes: Whereas the DSM-IV stated that the diagnosis of schizoaffective disorder required that the mood episode must be present for “a substantial duration of the illness,” the DSM-5 requires that the mood episode be present for “the majority” of the illness.
“The change was made to improve the reliability and stability of the diagnosis,” explained Dr. van Os, professor of psychiatric epidemiology at Maastricht (the Netherlands) University.
He was an advocate on the committee for introducing a dimensional approach to the diagnosis of schizoaffective disorder, one that would incorporate consideration of clustering of symptoms across a continuum.
“This would have been the best solution to the conundrum of the ‘fuzzy set’ problem of mental disorders. It would have been a great way of recognizing the continuity of all mental phenomena seen in mental disorders. However, it did not happen, unfortunately.”
The psychiatrist noted that committee members debated “for many hours” how best to overhaul the criteria for schizoaffective disorder in the DSM-5 before deciding to scale back their scope.
“We realized that any measure of precision in the criteria we tried to introduce for the admixture of affective dysregulation and psychosis is spurious. We really haven’t got the tools yet to make valid distinctions. That’s why we said in DSM-5 that the clinician can make the diagnosis using clinical intuition that the majority of time in the illness there is affective dysregulation. So, not spurious precision, but clinical intuition. I don’t know if that’s a good decision, but that’s what we did,” Dr. van Os said.
At the same symposium on schizoaffective disorder held during the ECNP congress, Dr. Heinz Grunze declared: “I would have welcomed as a development in DSM-5, as we’ve seen with some other disorders, basically abolishing schizoaffective disorder on its own and assigning it either to bipolar disorder with psychotic features specified or to schizophrenia with a strong affective specifier. I think that would be better suited to characterizing these patients.”
“Clearly, schizoaffective disorder has quite a polymorphic course. It’s definitely not one illness; it’s several illnesses which we put together in this random category,” added Dr. Grunze, professor of clinical psychiatry at University of Newcastle (England).
His own view is that the disorder belongs under the umbrella of bipolar disorder. A recent Spanish functional magnetic resonance brain imaging study demonstrated that schizoaffective disorder exhibits features that more closely resemble those of bipolar disorder than schizophrenia, including the finding that patients in clinical remission show failure of deactivation in the medial frontal gyrus, compared with healthy controls, Dr. Grunze noted.
“I personally can’t see a difference between a schizoaffective acute episode, manic type, and a psychotic manic episode. So my personal opinion is that schizoaffective disorder belongs under the heading of bipolar disorder. That’s obviously different from the opinion of those conducting treatment trials and the pharmaceutical industry, where schizoaffective disorder is never a subgroup in bipolar disorder trials, but a subgroup in schizophrenia trials,” Dr. Grunze said.
One audience member rose to state that while all this wrangling over how best to subclassify these patients is interesting, why not take a pragmatic approach and simply treat them symptomatically?
“Well, I think that’s what everyone in this room who’s a clinician is doing,” Dr. Grunze replied. “Because even if we assume that the diagnosis itself might be unstable, I think the greatest variance in patients switching around between bipolar, schizophrenia, and schizoaffective comes from the different doctors seeing them, each having a different understanding of schizoaffective disorder. I see a lot of patients who are classified as schizoaffective disorder because they have what I would have considered just as mood-congruent psychotic symptoms while manic. So I absolutely agree with you that this is more of an academic discussion, really, because at the moment we don’t have any consequences for treatment. We don’t have good evidence about how to treat these patients; we just extrapolate from whatever we think their symptoms come closest to.”
He reported receiving research grants from the National Institute for Health Research and the U.K. Medical Research Council, as well as more than a half-dozen pharmaceutical companies.
Dr. van Os reported having no financial conflicts.
BERLIN – The redefinition of schizoaffective disorder unveiled in the DSM-5 leaves much to be desired, experts opined at the annual congress of the European College of Neuropsychopharmacology
“We all thought we had the solution for making new criteria for schizoaffective disorder,” recalled Dr. Jim van Os, who served on the DSM-5 committee charged with reexamining the disorder. “But in the end, nothing much changed.”
Indeed, despite the lofty ambitions he and many of his fellow DSM-5 committee members held for thoroughly overhauling the criteria for what he called “this enigmatic combination of affective and psychotic dysregulation,” the difference between the DSM-IV and DSM-5 boiled down to small potatoes: Whereas the DSM-IV stated that the diagnosis of schizoaffective disorder required that the mood episode must be present for “a substantial duration of the illness,” the DSM-5 requires that the mood episode be present for “the majority” of the illness.
“The change was made to improve the reliability and stability of the diagnosis,” explained Dr. van Os, professor of psychiatric epidemiology at Maastricht (the Netherlands) University.
He was an advocate on the committee for introducing a dimensional approach to the diagnosis of schizoaffective disorder, one that would incorporate consideration of clustering of symptoms across a continuum.
“This would have been the best solution to the conundrum of the ‘fuzzy set’ problem of mental disorders. It would have been a great way of recognizing the continuity of all mental phenomena seen in mental disorders. However, it did not happen, unfortunately.”
The psychiatrist noted that committee members debated “for many hours” how best to overhaul the criteria for schizoaffective disorder in the DSM-5 before deciding to scale back their scope.
“We realized that any measure of precision in the criteria we tried to introduce for the admixture of affective dysregulation and psychosis is spurious. We really haven’t got the tools yet to make valid distinctions. That’s why we said in DSM-5 that the clinician can make the diagnosis using clinical intuition that the majority of time in the illness there is affective dysregulation. So, not spurious precision, but clinical intuition. I don’t know if that’s a good decision, but that’s what we did,” Dr. van Os said.
At the same symposium on schizoaffective disorder held during the ECNP congress, Dr. Heinz Grunze declared: “I would have welcomed as a development in DSM-5, as we’ve seen with some other disorders, basically abolishing schizoaffective disorder on its own and assigning it either to bipolar disorder with psychotic features specified or to schizophrenia with a strong affective specifier. I think that would be better suited to characterizing these patients.”
“Clearly, schizoaffective disorder has quite a polymorphic course. It’s definitely not one illness; it’s several illnesses which we put together in this random category,” added Dr. Grunze, professor of clinical psychiatry at University of Newcastle (England).
His own view is that the disorder belongs under the umbrella of bipolar disorder. A recent Spanish functional magnetic resonance brain imaging study demonstrated that schizoaffective disorder exhibits features that more closely resemble those of bipolar disorder than schizophrenia, including the finding that patients in clinical remission show failure of deactivation in the medial frontal gyrus, compared with healthy controls, Dr. Grunze noted.
“I personally can’t see a difference between a schizoaffective acute episode, manic type, and a psychotic manic episode. So my personal opinion is that schizoaffective disorder belongs under the heading of bipolar disorder. That’s obviously different from the opinion of those conducting treatment trials and the pharmaceutical industry, where schizoaffective disorder is never a subgroup in bipolar disorder trials, but a subgroup in schizophrenia trials,” Dr. Grunze said.
One audience member rose to state that while all this wrangling over how best to subclassify these patients is interesting, why not take a pragmatic approach and simply treat them symptomatically?
“Well, I think that’s what everyone in this room who’s a clinician is doing,” Dr. Grunze replied. “Because even if we assume that the diagnosis itself might be unstable, I think the greatest variance in patients switching around between bipolar, schizophrenia, and schizoaffective comes from the different doctors seeing them, each having a different understanding of schizoaffective disorder. I see a lot of patients who are classified as schizoaffective disorder because they have what I would have considered just as mood-congruent psychotic symptoms while manic. So I absolutely agree with you that this is more of an academic discussion, really, because at the moment we don’t have any consequences for treatment. We don’t have good evidence about how to treat these patients; we just extrapolate from whatever we think their symptoms come closest to.”
He reported receiving research grants from the National Institute for Health Research and the U.K. Medical Research Council, as well as more than a half-dozen pharmaceutical companies.
Dr. van Os reported having no financial conflicts.
EXPERT ANALYSIS FROM THE ECNP CONGRESS

New evidence suggests 2014 hypertension guidelines could backfire
CHICAGO – Nearly one in seven patients in U.S. ambulatory cardiology practices who would have been recommended for initiation or intensification of antihypertensive drug therapy under the 2003 Seventh Joint National Committee guidelines are no longer treatment candidates under the 2014 expert panel recommendations.
These patients who no longer qualify for antihypertensive therapy under the 2014 guidelines turn out to have a disturbingly high average estimated 10-year risk of cardiovascular events. As a result, widespread adoption of the 2014 expert panel recommendations could have major adverse consequences for cardiovascular health, Dr. William B. Borden cautioned at the American Heart Association scientific sessions.
“Given the size and underlying cardiovascular risk of the population affected by the changes in the 2014 panel recommendations, close monitoring will be required to assess changes in practice patterns, blood pressure control, and – importantly – any changes in cardiovascular morbidity and mortality,” said Dr. Borden, a cardiologist at George Washington University in Washington.
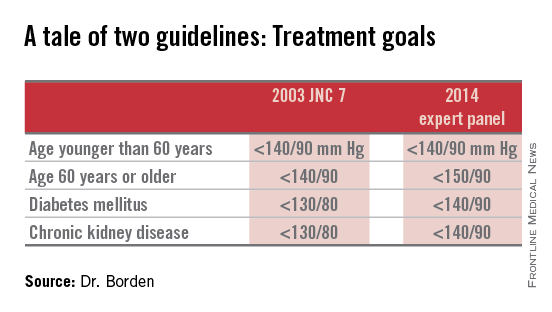
Because the 2014 expert panel guidelines represent a major shift in hypertension management, Dr. Borden and coinvestigators sought to quantify the potential cardiovascular health impact of this more lenient treatment approach. For this purpose they turned to the National Cardiovascular Data Registry Practice Innovation and Clinical Excellence (NCDR PINNACLE) Registry, a voluntary quality improvement project involving outpatient cardiology practices.
Of 1,185,253 patients with hypertension as identified in their chart by a recorded diagnosis or notation of blood pressure greater than 140/90 mm Hg, 60% met the 2003 JNC 7 goals (JAMA 2003;289:2560-72), meaning the other 40% were candidates for initiation or intensification of antihypertensive therapy in order to achieve those goals (see chart). In contrast, 74% of hypertensive patients in U.S. cardiology practices met the less aggressive targets recommended in the 2014 expert panel report (JAMA 2014;311:502-20).
Thus, fewer than two-thirds of hypertensive patients in outpatient cardiology practices met the 2003 JNC 7 blood pressure targets, while three-quarters met the liberalized 2014 targets.
Dr. Borden and coworkers zeroed in on the 15% of hypertensive patients – that’s fully 173,519 individuals in cardiology practices participating in the PINNACLE Registry – who would have been eligible for treatment under the JNC 7 recommendations but not the 2014 expert panel guidelines. Interestingly, that 15% figure was closely similar to the 17% rate reported by Dr. Michael D. Miedema of the Minneapolis Heart Institute in an analysis of a more primary care population of older patients in the Atherosclerosis Risk in Communities (ARIC) study he presented in the same session.

Dr. Borden and coinvestigators determined from medical records that the PINNACLE Registry group whose antihypertensive therapy treatment status changed between the two guidelines was at substantial baseline cardiovascular risk: Nearly two-thirds had been diagnosed with CAD, 54% had diabetes, 27% had a history of heart failure, 25% had a prior MI, and 23% had a prior transient ischemic attack or stroke.
This large group of patients who fell through the cracks between two conflicting sets of guidelines turned out to have a mean 10-year Framingham Risk Score of 8.5%. Upon incorporating the patients’ stroke risk using the atherosclerotic cardiovascular disease (ASCVD) risk score embedded in the 2013 ACC/AHA cholesterol management guidelines, their 10-year risk shot up to 28%.
The investigators then conducted a modeling exercise aimed at estimating the clinical impact of lowering systolic blood pressure in the elderly from about 150 mm Hg, as recommended in the 2014 expert panel guidelines, to about 140 mm Hg, as was the goal in JNC 7. To do so they extrapolated from the results of two randomized controlled clinical trials: the Systolic Hypertension in the Elderly Program (SHEP) and the Hypertension in the Very Elderly Trial (HYVET).
The result? Extrapolating from SHEP data, the 10-year ASCVD risk in these real-world elderly hypertensive patients caught between two conflicting sets of guidelines would drop from 28% to 19%. Using HYVET data, the average 10-year ASCVD risk would fall to 18.4%.
“This is equivalent to a number-needed-to-treat of 10-11 patients for 10 years in order to prevent one cardiovascular event,” according to Dr. Borden.
For the more than 80,000 patients over age 60 in the study population, that works out to roughly 8,000 cardiovascular events averted over the course of 10 years, he added.
The 2014 expert panel recommendations were based on a strict evidence-based review of published randomized controlled trials. The guidelines are new enough that it remains unclear if they will be embraced by clinicians or incorporated into performance measures and value-based health care purchasing programs.
The 2014 guidelines are considered highly controversial. The guideline committee comprising some of the nation’s top hypertension researchers was initially convened to come up with what was intended to be the long-awaited JNC 8 report; however, in the midst of the process the sponsoring National Heart, Lung, and Blood Institute declared it was getting out of the guideline-writing business altogether. As a result, the guidelines ultimately published carried the imprimatur of “the 2014 expert panel,” rather than the more prestigious official stamp of JNC 8.
Indeed, five members of the guideline panel felt strongly enough to break away and issued a minority report (Ann. Intern. Med. 2014;160:499-503) in which they argued there is insufficient evidence of harm stemming from the JNC 7 goal of 140/90 mm Hg in patients over age 60 to justify revising the target to 150/90. They warned that this step could reverse the impressive reductions in cardiovascular and cerebrovascular morbidity and mortality realized in recent decades. And they concluded that the burden of proof should be on those who advocate raising the treatment threshold to 150/90 mm Hg to demonstrate that it has benefit in patients over age 60, which they haven’t done.
“I’m very concerned about the [2014 expert panel] guidelines. Older individuals have the highest prevalence of hypertension, they’re the least adequately controlled, and based on the available data I’m concerned that if people follow the new guidelines there’s going to be an increase in cardiovascular events,” said Dr. Wilbert F. Aronow of New York Medical College, Valhalla, who chaired the writing committee for the first-ever ACC/AHA clinical guidelines for controlling high blood pressure in the elderly (J. Am. Coll. Cardiol. 2011;57:2037-114).
The NCDR PINNACLE Registry and this study were supported by the American College of Cardiology Foundation. Dr. Borden and Dr. Aronow reported having no financial conflicts.
CHICAGO – Nearly one in seven patients in U.S. ambulatory cardiology practices who would have been recommended for initiation or intensification of antihypertensive drug therapy under the 2003 Seventh Joint National Committee guidelines are no longer treatment candidates under the 2014 expert panel recommendations.
These patients who no longer qualify for antihypertensive therapy under the 2014 guidelines turn out to have a disturbingly high average estimated 10-year risk of cardiovascular events. As a result, widespread adoption of the 2014 expert panel recommendations could have major adverse consequences for cardiovascular health, Dr. William B. Borden cautioned at the American Heart Association scientific sessions.
“Given the size and underlying cardiovascular risk of the population affected by the changes in the 2014 panel recommendations, close monitoring will be required to assess changes in practice patterns, blood pressure control, and – importantly – any changes in cardiovascular morbidity and mortality,” said Dr. Borden, a cardiologist at George Washington University in Washington.
Because the 2014 expert panel guidelines represent a major shift in hypertension management, Dr. Borden and coinvestigators sought to quantify the potential cardiovascular health impact of this more lenient treatment approach. For this purpose they turned to the National Cardiovascular Data Registry Practice Innovation and Clinical Excellence (NCDR PINNACLE) Registry, a voluntary quality improvement project involving outpatient cardiology practices.
Of 1,185,253 patients with hypertension as identified in their chart by a recorded diagnosis or notation of blood pressure greater than 140/90 mm Hg, 60% met the 2003 JNC 7 goals (JAMA 2003;289:2560-72), meaning the other 40% were candidates for initiation or intensification of antihypertensive therapy in order to achieve those goals (see chart). In contrast, 74% of hypertensive patients in U.S. cardiology practices met the less aggressive targets recommended in the 2014 expert panel report (JAMA 2014;311:502-20).
Thus, fewer than two-thirds of hypertensive patients in outpatient cardiology practices met the 2003 JNC 7 blood pressure targets, while three-quarters met the liberalized 2014 targets.
Dr. Borden and coworkers zeroed in on the 15% of hypertensive patients – that’s fully 173,519 individuals in cardiology practices participating in the PINNACLE Registry – who would have been eligible for treatment under the JNC 7 recommendations but not the 2014 expert panel guidelines. Interestingly, that 15% figure was closely similar to the 17% rate reported by Dr. Michael D. Miedema of the Minneapolis Heart Institute in an analysis of a more primary care population of older patients in the Atherosclerosis Risk in Communities (ARIC) study he presented in the same session.
Dr. Borden and coinvestigators determined from medical records that the PINNACLE Registry group whose antihypertensive therapy treatment status changed between the two guidelines was at substantial baseline cardiovascular risk: Nearly two-thirds had been diagnosed with CAD, 54% had diabetes, 27% had a history of heart failure, 25% had a prior MI, and 23% had a prior transient ischemic attack or stroke.
This large group of patients who fell through the cracks between two conflicting sets of guidelines turned out to have a mean 10-year Framingham Risk Score of 8.5%. Upon incorporating the patients’ stroke risk using the atherosclerotic cardiovascular disease (ASCVD) risk score embedded in the 2013 ACC/AHA cholesterol management guidelines, their 10-year risk shot up to 28%.
The investigators then conducted a modeling exercise aimed at estimating the clinical impact of lowering systolic blood pressure in the elderly from about 150 mm Hg, as recommended in the 2014 expert panel guidelines, to about 140 mm Hg, as was the goal in JNC 7. To do so they extrapolated from the results of two randomized controlled clinical trials: the Systolic Hypertension in the Elderly Program (SHEP) and the Hypertension in the Very Elderly Trial (HYVET).
The result? Extrapolating from SHEP data, the 10-year ASCVD risk in these real-world elderly hypertensive patients caught between two conflicting sets of guidelines would drop from 28% to 19%. Using HYVET data, the average 10-year ASCVD risk would fall to 18.4%.
“This is equivalent to a number-needed-to-treat of 10-11 patients for 10 years in order to prevent one cardiovascular event,” according to Dr. Borden.
For the more than 80,000 patients over age 60 in the study population, that works out to roughly 8,000 cardiovascular events averted over the course of 10 years, he added.
The 2014 expert panel recommendations were based on a strict evidence-based review of published randomized controlled trials. The guidelines are new enough that it remains unclear if they will be embraced by clinicians or incorporated into performance measures and value-based health care purchasing programs.
The 2014 guidelines are considered highly controversial. The guideline committee comprising some of the nation’s top hypertension researchers was initially convened to come up with what was intended to be the long-awaited JNC 8 report; however, in the midst of the process the sponsoring National Heart, Lung, and Blood Institute declared it was getting out of the guideline-writing business altogether. As a result, the guidelines ultimately published carried the imprimatur of “the 2014 expert panel,” rather than the more prestigious official stamp of JNC 8.
Indeed, five members of the guideline panel felt strongly enough to break away and issued a minority report (Ann. Intern. Med. 2014;160:499-503) in which they argued there is insufficient evidence of harm stemming from the JNC 7 goal of 140/90 mm Hg in patients over age 60 to justify revising the target to 150/90. They warned that this step could reverse the impressive reductions in cardiovascular and cerebrovascular morbidity and mortality realized in recent decades. And they concluded that the burden of proof should be on those who advocate raising the treatment threshold to 150/90 mm Hg to demonstrate that it has benefit in patients over age 60, which they haven’t done.
“I’m very concerned about the [2014 expert panel] guidelines. Older individuals have the highest prevalence of hypertension, they’re the least adequately controlled, and based on the available data I’m concerned that if people follow the new guidelines there’s going to be an increase in cardiovascular events,” said Dr. Wilbert F. Aronow of New York Medical College, Valhalla, who chaired the writing committee for the first-ever ACC/AHA clinical guidelines for controlling high blood pressure in the elderly (J. Am. Coll. Cardiol. 2011;57:2037-114).
The NCDR PINNACLE Registry and this study were supported by the American College of Cardiology Foundation. Dr. Borden and Dr. Aronow reported having no financial conflicts.
CHICAGO – Nearly one in seven patients in U.S. ambulatory cardiology practices who would have been recommended for initiation or intensification of antihypertensive drug therapy under the 2003 Seventh Joint National Committee guidelines are no longer treatment candidates under the 2014 expert panel recommendations.
These patients who no longer qualify for antihypertensive therapy under the 2014 guidelines turn out to have a disturbingly high average estimated 10-year risk of cardiovascular events. As a result, widespread adoption of the 2014 expert panel recommendations could have major adverse consequences for cardiovascular health, Dr. William B. Borden cautioned at the American Heart Association scientific sessions.
“Given the size and underlying cardiovascular risk of the population affected by the changes in the 2014 panel recommendations, close monitoring will be required to assess changes in practice patterns, blood pressure control, and – importantly – any changes in cardiovascular morbidity and mortality,” said Dr. Borden, a cardiologist at George Washington University in Washington.
Because the 2014 expert panel guidelines represent a major shift in hypertension management, Dr. Borden and coinvestigators sought to quantify the potential cardiovascular health impact of this more lenient treatment approach. For this purpose they turned to the National Cardiovascular Data Registry Practice Innovation and Clinical Excellence (NCDR PINNACLE) Registry, a voluntary quality improvement project involving outpatient cardiology practices.
Of 1,185,253 patients with hypertension as identified in their chart by a recorded diagnosis or notation of blood pressure greater than 140/90 mm Hg, 60% met the 2003 JNC 7 goals (JAMA 2003;289:2560-72), meaning the other 40% were candidates for initiation or intensification of antihypertensive therapy in order to achieve those goals (see chart). In contrast, 74% of hypertensive patients in U.S. cardiology practices met the less aggressive targets recommended in the 2014 expert panel report (JAMA 2014;311:502-20).
Thus, fewer than two-thirds of hypertensive patients in outpatient cardiology practices met the 2003 JNC 7 blood pressure targets, while three-quarters met the liberalized 2014 targets.
Dr. Borden and coworkers zeroed in on the 15% of hypertensive patients – that’s fully 173,519 individuals in cardiology practices participating in the PINNACLE Registry – who would have been eligible for treatment under the JNC 7 recommendations but not the 2014 expert panel guidelines. Interestingly, that 15% figure was closely similar to the 17% rate reported by Dr. Michael D. Miedema of the Minneapolis Heart Institute in an analysis of a more primary care population of older patients in the Atherosclerosis Risk in Communities (ARIC) study he presented in the same session.
Dr. Borden and coinvestigators determined from medical records that the PINNACLE Registry group whose antihypertensive therapy treatment status changed between the two guidelines was at substantial baseline cardiovascular risk: Nearly two-thirds had been diagnosed with CAD, 54% had diabetes, 27% had a history of heart failure, 25% had a prior MI, and 23% had a prior transient ischemic attack or stroke.
This large group of patients who fell through the cracks between two conflicting sets of guidelines turned out to have a mean 10-year Framingham Risk Score of 8.5%. Upon incorporating the patients’ stroke risk using the atherosclerotic cardiovascular disease (ASCVD) risk score embedded in the 2013 ACC/AHA cholesterol management guidelines, their 10-year risk shot up to 28%.
The investigators then conducted a modeling exercise aimed at estimating the clinical impact of lowering systolic blood pressure in the elderly from about 150 mm Hg, as recommended in the 2014 expert panel guidelines, to about 140 mm Hg, as was the goal in JNC 7. To do so they extrapolated from the results of two randomized controlled clinical trials: the Systolic Hypertension in the Elderly Program (SHEP) and the Hypertension in the Very Elderly Trial (HYVET).
The result? Extrapolating from SHEP data, the 10-year ASCVD risk in these real-world elderly hypertensive patients caught between two conflicting sets of guidelines would drop from 28% to 19%. Using HYVET data, the average 10-year ASCVD risk would fall to 18.4%.
“This is equivalent to a number-needed-to-treat of 10-11 patients for 10 years in order to prevent one cardiovascular event,” according to Dr. Borden.
For the more than 80,000 patients over age 60 in the study population, that works out to roughly 8,000 cardiovascular events averted over the course of 10 years, he added.
The 2014 expert panel recommendations were based on a strict evidence-based review of published randomized controlled trials. The guidelines are new enough that it remains unclear if they will be embraced by clinicians or incorporated into performance measures and value-based health care purchasing programs.
The 2014 guidelines are considered highly controversial. The guideline committee comprising some of the nation’s top hypertension researchers was initially convened to come up with what was intended to be the long-awaited JNC 8 report; however, in the midst of the process the sponsoring National Heart, Lung, and Blood Institute declared it was getting out of the guideline-writing business altogether. As a result, the guidelines ultimately published carried the imprimatur of “the 2014 expert panel,” rather than the more prestigious official stamp of JNC 8.
Indeed, five members of the guideline panel felt strongly enough to break away and issued a minority report (Ann. Intern. Med. 2014;160:499-503) in which they argued there is insufficient evidence of harm stemming from the JNC 7 goal of 140/90 mm Hg in patients over age 60 to justify revising the target to 150/90. They warned that this step could reverse the impressive reductions in cardiovascular and cerebrovascular morbidity and mortality realized in recent decades. And they concluded that the burden of proof should be on those who advocate raising the treatment threshold to 150/90 mm Hg to demonstrate that it has benefit in patients over age 60, which they haven’t done.
“I’m very concerned about the [2014 expert panel] guidelines. Older individuals have the highest prevalence of hypertension, they’re the least adequately controlled, and based on the available data I’m concerned that if people follow the new guidelines there’s going to be an increase in cardiovascular events,” said Dr. Wilbert F. Aronow of New York Medical College, Valhalla, who chaired the writing committee for the first-ever ACC/AHA clinical guidelines for controlling high blood pressure in the elderly (J. Am. Coll. Cardiol. 2011;57:2037-114).
The NCDR PINNACLE Registry and this study were supported by the American College of Cardiology Foundation. Dr. Borden and Dr. Aronow reported having no financial conflicts.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: Adoption of the less aggressive blood pressure goal of 150/90 mm Hg for patients age 60 and older as recommended in the 2014 expert panel guidelines could result in significantly more harm than good.
Major finding: Patients in cardiology practices who qualified for antihypertensive therapy under the 2003 JNC 7 guidelines but not under the 2014 expert panel guidelines have a baseline 10-year 28% risk of cardiovascular events or stroke using the risk calculator included in the 2013 ACC/AHA cholesterol management guidelines.
Data source: An analysis of 1,185,253 patients in U.S. ambulatory cardiology practices.
Disclosures: The NCDR PINNACLE Registry is supported by the American College of Cardiology Foundation. The presenter reported having no financial conflicts of interest.
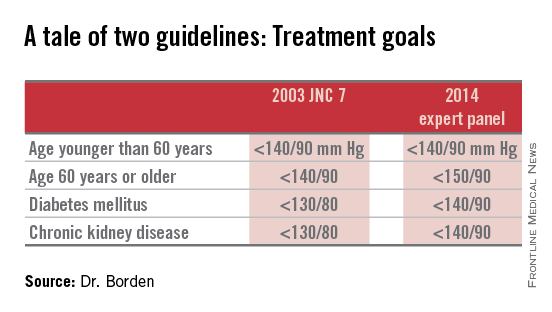
Pushing LDL below 25 mg/dL with alirocumab safe ‘so far’
CHICAGO – Driving LDL cholesterol below 25 mg/dL for a year or more in high-cardiovascular-risk patients using the investigational human monoclonal antibody alirocumab proved well tolerated and displayed a treatment-emergent adverse event profile reassuringly comparable to that of placebo in an interim analysis of the largest double-blind phase III clinical trial to date involving any PCSK9 inhibitor.
While this is encouraging preliminary evidence that it may prove safe to pharmacologically maintain unnaturally low LDL concentrations – levels below those seen in newborns – in high-risk patients, Dr. Jennifer G. Robinson nonetheless urged her cardiology colleagues to curb their enthusiasm as she presented an interim analysis of the 2,341-patient ODYSSEY Long Term trial at the American Heart Association scientific sessions.
“We’re all concerned about the long-term safety of very low LDL levels. The fact that there are no signals so far is good, but it’s only been 52 weeks. In my own mind, we’re treating people with very-high-risk cardiovascular disease who are older and have comorbidities, and I think we just have to establish that there really aren’t any adverse effects of prolonged low levels of LDL,” said Dr. Robinson, professor of medicine at the University of Iowa in Iowa City.
Session cochair Dr. David C. Goff applauded her cautionary stance.
“The 1-year data on safety are promising, although these are still relatively small numbers of patients who have been exposed to the medication for at least a year. So rare adverse events will still be difficult to detect in just a couple thousand people treated for that length of time,” observed Dr. Goff, a cardiovascular epidemiologist and dean of the University of Colorado School of Public Health, Denver.
He was particularly intrigued by a different part of her presentation, a new post hoc pooled analysis of hard cardiovascular outcomes in ODYSSEY Long Term and two other ongoing, phase III, placebo-controlled alirocumab trials totaling 3,459 high-risk patients with follow-up of 52 weeks or more. In this Cox regression analysis, the relative risk of the composite endpoint of adjudicated death from coronary heart disease, nonfatal myocardial infarction, fatal or nonfatal ischemic stroke, or unstable angina requiring hospitalization was 35% lower in alirocumab- than placebo-treated patients. While this hefty-looking difference in risk didn’t achieve statistical significance a mere year into follow-up, the fact that the two event curves diverged from the very beginning of follow-up and the separation continues to expand bodes well indeed.
“Dr. Robinson has shown us a first glimpse of a promise of cardiovascular disease prevention from the pooled data. I think we’re all now looking forward to the results of the ongoing ODYSSEY Outcomes trial, with 18,000 randomized ACS [acute coronary syndrome] patients being followed for 5-6 years, which will provide us with much better evidence with regard to safety and cardiovascular event prevention. But so far so good, from what I’ve seen here today,” Dr. Goff commented.
ODYSSEY Long Term included 2,341 patients with high cardiovascular risk, either because of known coronary heart disease or, in the case of 415 subjects, heterozygous familial hypercholesterolemia (FH). At entry, participants were on maximum tolerated statin therapy, often accompanied by additional lipid-lowering agents, yet their mean baseline LDL was 122 mg/dL. While remaining on their maximum tolerated statin and other LDL-lowering drugs throughout the trial, they were randomized 2:1 to 150 mg of alirocumab by self-administered subcutaneous injection every 2 weeks or placebo. Alirocumab is a human monoclonal antibody designed to inhibit PCSK9 (proprotein convertase subtilisin/kexin type 9), an enzyme that helps govern cholesterol homeostasis.
Seventy-nine percent of alirocumab-treated patients achieved an LDL below 70 mg/dL at week 24. At week 52, mean LDL in the alirocumab group was 53 mg/dL, compared with 123 mg/dL in the control group.
The potential for adverse neurocognitive effects with very low achieved LDL levels has been of particular concern with regard to the PCSK9 inhibitors. The issue is quite relevant because patients on alirocumab commonly develop LDL levels below 25 mg/dL. In ODYSSEY Long Term, for example, 36% of alirocumab-treated patients did so. Lipidologists have been eager to learn more about the impact of maintaining patients in this state of subphysiologic LDL, and Dr. Robinson’s data provided the first meaningful opportunity to do so.
Reassuringly, the rate of treatment-emergent neurocognitive disorders was 1.2% in the total alirocumab-treated population, 0.5% in those with an on-treatment LDL below 25 mg/dL, and 0.5% in placebo-treated controls who, it must be remembered, were on maximum-tolerated therapy with statins and other conventional lipid-lowering agents. Other treatment-emergent adverse events – and more than 30 categories of such events were tracked – followed a similar pattern (see graphic).
There was one event of possible concern that has been reported to the Food and Drug Administration. An alirocumab-treated patient was diagnosed with Miller Fisher syndrome, a rare variant of Guillain-Barre syndrome, at week 27 after a prodromal severe gastroenteritis. At week 24, just 3 weeks earlier, the patient had an LDL of 1.5 mg/dL. The double vision and other symptoms of Miller Fisher syndrome resolved completely after treatment discontinuation.
During a panel discussion of the ODYSSEY clinical trial series, which includes 14 phase III studies, Dr. Henry N. Ginsburg predicted that further experience will eventually show that the dramatic LDL-lowering achieved with alirocumab not only doesn’t promote neurocognitive dysfunction, but actually protects against it.
“I think an important point to make is that, despite our neurology colleagues having tried for about a decade to completely separate the clinical phenotype of Alzheimer’s disease from vascular dementia, we all know that if you have both you’ll do much worse. So if we reduce cerebrovascular atherosclerosis and you have any predisposition to Alzheimer’s disease, you’ll do better. I think there will be studies to show that,” said Dr. Ginsburg, professor of medicine and director of the Irving Institute for Clinical and Translational Research at Columbia University in New York, who earlier in the session presented the findings from the phase III ODYSSEY High FH trial.
Another potential safety concern emerged during Dr. Dean J. Kereiakes’ presentation of the results of the phase III ODYSSEY Combo I study. Thirteen of 199 alirocumab-treated patients (6.6%) developed treatment-emergent, low-titer antialirocumab antibodies. The median time to their detection was 12 weeks. Four of the 13 patients had neutralizing antibodies, and all 4 became antibody negative by 24 weeks.
The presence of these antidrug antibodies had no discernible effect on treatment safety or efficacy. However, Combo I was a 52-week study. Whether the prevalence of these antibodies will increase over time and interfere with what is expected to be lifelong treatment is an unanswered question, noted Dr. Kereiakes, medical director for the Christ Hospital Heart and Vascular Center, Cincinnati.
Discussant Dr. Joshua W. Knowles, medical director of the FH Foundation, commented that “it’s really incredible to see the reductions in LDL that we’re getting with PCSK9 inhibitors in FH patients, the vast majority of whom require more than two standard medications to get to optimal LDL cholesterol levels.”
“The PCSK9 inhibitors are really the poster child for drug discovery in the modern genetic era. It has been a beautiful 10-year journey for them,” declared Dr. Knowles, a cardiologist at Stanford (Calif.) University.
In light of the powerful LDL-lowering effect of these agents, it’s not too soon to start thinking about how to carry out studies of atherosclerotic plaque stabilization and/or regression in treated patients, he added.
Other PCSK9 inhibitors in phase III clinical trials, besides alirocumab, are Amgen’s evolocumab and Pfizer/Renat’s bococizumab. In addition, Eli Lilly’s LY3015014 is in phase II studies, as is Roche/Genentech’s RG7652.
The ODYSSEY studies are funded by Sanofi and Regeneron Pharmaceuticals. Dr. Robinson, Dr. Ginsburg, and Dr. Kereiakes reported serving as consultants to those and other pharmaceutical companies.
CHICAGO – Driving LDL cholesterol below 25 mg/dL for a year or more in high-cardiovascular-risk patients using the investigational human monoclonal antibody alirocumab proved well tolerated and displayed a treatment-emergent adverse event profile reassuringly comparable to that of placebo in an interim analysis of the largest double-blind phase III clinical trial to date involving any PCSK9 inhibitor.
While this is encouraging preliminary evidence that it may prove safe to pharmacologically maintain unnaturally low LDL concentrations – levels below those seen in newborns – in high-risk patients, Dr. Jennifer G. Robinson nonetheless urged her cardiology colleagues to curb their enthusiasm as she presented an interim analysis of the 2,341-patient ODYSSEY Long Term trial at the American Heart Association scientific sessions.
“We’re all concerned about the long-term safety of very low LDL levels. The fact that there are no signals so far is good, but it’s only been 52 weeks. In my own mind, we’re treating people with very-high-risk cardiovascular disease who are older and have comorbidities, and I think we just have to establish that there really aren’t any adverse effects of prolonged low levels of LDL,” said Dr. Robinson, professor of medicine at the University of Iowa in Iowa City.
Session cochair Dr. David C. Goff applauded her cautionary stance.
“The 1-year data on safety are promising, although these are still relatively small numbers of patients who have been exposed to the medication for at least a year. So rare adverse events will still be difficult to detect in just a couple thousand people treated for that length of time,” observed Dr. Goff, a cardiovascular epidemiologist and dean of the University of Colorado School of Public Health, Denver.
He was particularly intrigued by a different part of her presentation, a new post hoc pooled analysis of hard cardiovascular outcomes in ODYSSEY Long Term and two other ongoing, phase III, placebo-controlled alirocumab trials totaling 3,459 high-risk patients with follow-up of 52 weeks or more. In this Cox regression analysis, the relative risk of the composite endpoint of adjudicated death from coronary heart disease, nonfatal myocardial infarction, fatal or nonfatal ischemic stroke, or unstable angina requiring hospitalization was 35% lower in alirocumab- than placebo-treated patients. While this hefty-looking difference in risk didn’t achieve statistical significance a mere year into follow-up, the fact that the two event curves diverged from the very beginning of follow-up and the separation continues to expand bodes well indeed.
“Dr. Robinson has shown us a first glimpse of a promise of cardiovascular disease prevention from the pooled data. I think we’re all now looking forward to the results of the ongoing ODYSSEY Outcomes trial, with 18,000 randomized ACS [acute coronary syndrome] patients being followed for 5-6 years, which will provide us with much better evidence with regard to safety and cardiovascular event prevention. But so far so good, from what I’ve seen here today,” Dr. Goff commented.
ODYSSEY Long Term included 2,341 patients with high cardiovascular risk, either because of known coronary heart disease or, in the case of 415 subjects, heterozygous familial hypercholesterolemia (FH). At entry, participants were on maximum tolerated statin therapy, often accompanied by additional lipid-lowering agents, yet their mean baseline LDL was 122 mg/dL. While remaining on their maximum tolerated statin and other LDL-lowering drugs throughout the trial, they were randomized 2:1 to 150 mg of alirocumab by self-administered subcutaneous injection every 2 weeks or placebo. Alirocumab is a human monoclonal antibody designed to inhibit PCSK9 (proprotein convertase subtilisin/kexin type 9), an enzyme that helps govern cholesterol homeostasis.
Seventy-nine percent of alirocumab-treated patients achieved an LDL below 70 mg/dL at week 24. At week 52, mean LDL in the alirocumab group was 53 mg/dL, compared with 123 mg/dL in the control group.
The potential for adverse neurocognitive effects with very low achieved LDL levels has been of particular concern with regard to the PCSK9 inhibitors. The issue is quite relevant because patients on alirocumab commonly develop LDL levels below 25 mg/dL. In ODYSSEY Long Term, for example, 36% of alirocumab-treated patients did so. Lipidologists have been eager to learn more about the impact of maintaining patients in this state of subphysiologic LDL, and Dr. Robinson’s data provided the first meaningful opportunity to do so.
Reassuringly, the rate of treatment-emergent neurocognitive disorders was 1.2% in the total alirocumab-treated population, 0.5% in those with an on-treatment LDL below 25 mg/dL, and 0.5% in placebo-treated controls who, it must be remembered, were on maximum-tolerated therapy with statins and other conventional lipid-lowering agents. Other treatment-emergent adverse events – and more than 30 categories of such events were tracked – followed a similar pattern (see graphic).
There was one event of possible concern that has been reported to the Food and Drug Administration. An alirocumab-treated patient was diagnosed with Miller Fisher syndrome, a rare variant of Guillain-Barre syndrome, at week 27 after a prodromal severe gastroenteritis. At week 24, just 3 weeks earlier, the patient had an LDL of 1.5 mg/dL. The double vision and other symptoms of Miller Fisher syndrome resolved completely after treatment discontinuation.
During a panel discussion of the ODYSSEY clinical trial series, which includes 14 phase III studies, Dr. Henry N. Ginsburg predicted that further experience will eventually show that the dramatic LDL-lowering achieved with alirocumab not only doesn’t promote neurocognitive dysfunction, but actually protects against it.
“I think an important point to make is that, despite our neurology colleagues having tried for about a decade to completely separate the clinical phenotype of Alzheimer’s disease from vascular dementia, we all know that if you have both you’ll do much worse. So if we reduce cerebrovascular atherosclerosis and you have any predisposition to Alzheimer’s disease, you’ll do better. I think there will be studies to show that,” said Dr. Ginsburg, professor of medicine and director of the Irving Institute for Clinical and Translational Research at Columbia University in New York, who earlier in the session presented the findings from the phase III ODYSSEY High FH trial.
Another potential safety concern emerged during Dr. Dean J. Kereiakes’ presentation of the results of the phase III ODYSSEY Combo I study. Thirteen of 199 alirocumab-treated patients (6.6%) developed treatment-emergent, low-titer antialirocumab antibodies. The median time to their detection was 12 weeks. Four of the 13 patients had neutralizing antibodies, and all 4 became antibody negative by 24 weeks.
The presence of these antidrug antibodies had no discernible effect on treatment safety or efficacy. However, Combo I was a 52-week study. Whether the prevalence of these antibodies will increase over time and interfere with what is expected to be lifelong treatment is an unanswered question, noted Dr. Kereiakes, medical director for the Christ Hospital Heart and Vascular Center, Cincinnati.
Discussant Dr. Joshua W. Knowles, medical director of the FH Foundation, commented that “it’s really incredible to see the reductions in LDL that we’re getting with PCSK9 inhibitors in FH patients, the vast majority of whom require more than two standard medications to get to optimal LDL cholesterol levels.”
“The PCSK9 inhibitors are really the poster child for drug discovery in the modern genetic era. It has been a beautiful 10-year journey for them,” declared Dr. Knowles, a cardiologist at Stanford (Calif.) University.
In light of the powerful LDL-lowering effect of these agents, it’s not too soon to start thinking about how to carry out studies of atherosclerotic plaque stabilization and/or regression in treated patients, he added.
Other PCSK9 inhibitors in phase III clinical trials, besides alirocumab, are Amgen’s evolocumab and Pfizer/Renat’s bococizumab. In addition, Eli Lilly’s LY3015014 is in phase II studies, as is Roche/Genentech’s RG7652.
The ODYSSEY studies are funded by Sanofi and Regeneron Pharmaceuticals. Dr. Robinson, Dr. Ginsburg, and Dr. Kereiakes reported serving as consultants to those and other pharmaceutical companies.
CHICAGO – Driving LDL cholesterol below 25 mg/dL for a year or more in high-cardiovascular-risk patients using the investigational human monoclonal antibody alirocumab proved well tolerated and displayed a treatment-emergent adverse event profile reassuringly comparable to that of placebo in an interim analysis of the largest double-blind phase III clinical trial to date involving any PCSK9 inhibitor.
While this is encouraging preliminary evidence that it may prove safe to pharmacologically maintain unnaturally low LDL concentrations – levels below those seen in newborns – in high-risk patients, Dr. Jennifer G. Robinson nonetheless urged her cardiology colleagues to curb their enthusiasm as she presented an interim analysis of the 2,341-patient ODYSSEY Long Term trial at the American Heart Association scientific sessions.
“We’re all concerned about the long-term safety of very low LDL levels. The fact that there are no signals so far is good, but it’s only been 52 weeks. In my own mind, we’re treating people with very-high-risk cardiovascular disease who are older and have comorbidities, and I think we just have to establish that there really aren’t any adverse effects of prolonged low levels of LDL,” said Dr. Robinson, professor of medicine at the University of Iowa in Iowa City.
Session cochair Dr. David C. Goff applauded her cautionary stance.
“The 1-year data on safety are promising, although these are still relatively small numbers of patients who have been exposed to the medication for at least a year. So rare adverse events will still be difficult to detect in just a couple thousand people treated for that length of time,” observed Dr. Goff, a cardiovascular epidemiologist and dean of the University of Colorado School of Public Health, Denver.
He was particularly intrigued by a different part of her presentation, a new post hoc pooled analysis of hard cardiovascular outcomes in ODYSSEY Long Term and two other ongoing, phase III, placebo-controlled alirocumab trials totaling 3,459 high-risk patients with follow-up of 52 weeks or more. In this Cox regression analysis, the relative risk of the composite endpoint of adjudicated death from coronary heart disease, nonfatal myocardial infarction, fatal or nonfatal ischemic stroke, or unstable angina requiring hospitalization was 35% lower in alirocumab- than placebo-treated patients. While this hefty-looking difference in risk didn’t achieve statistical significance a mere year into follow-up, the fact that the two event curves diverged from the very beginning of follow-up and the separation continues to expand bodes well indeed.
“Dr. Robinson has shown us a first glimpse of a promise of cardiovascular disease prevention from the pooled data. I think we’re all now looking forward to the results of the ongoing ODYSSEY Outcomes trial, with 18,000 randomized ACS [acute coronary syndrome] patients being followed for 5-6 years, which will provide us with much better evidence with regard to safety and cardiovascular event prevention. But so far so good, from what I’ve seen here today,” Dr. Goff commented.
ODYSSEY Long Term included 2,341 patients with high cardiovascular risk, either because of known coronary heart disease or, in the case of 415 subjects, heterozygous familial hypercholesterolemia (FH). At entry, participants were on maximum tolerated statin therapy, often accompanied by additional lipid-lowering agents, yet their mean baseline LDL was 122 mg/dL. While remaining on their maximum tolerated statin and other LDL-lowering drugs throughout the trial, they were randomized 2:1 to 150 mg of alirocumab by self-administered subcutaneous injection every 2 weeks or placebo. Alirocumab is a human monoclonal antibody designed to inhibit PCSK9 (proprotein convertase subtilisin/kexin type 9), an enzyme that helps govern cholesterol homeostasis.
Seventy-nine percent of alirocumab-treated patients achieved an LDL below 70 mg/dL at week 24. At week 52, mean LDL in the alirocumab group was 53 mg/dL, compared with 123 mg/dL in the control group.
The potential for adverse neurocognitive effects with very low achieved LDL levels has been of particular concern with regard to the PCSK9 inhibitors. The issue is quite relevant because patients on alirocumab commonly develop LDL levels below 25 mg/dL. In ODYSSEY Long Term, for example, 36% of alirocumab-treated patients did so. Lipidologists have been eager to learn more about the impact of maintaining patients in this state of subphysiologic LDL, and Dr. Robinson’s data provided the first meaningful opportunity to do so.
Reassuringly, the rate of treatment-emergent neurocognitive disorders was 1.2% in the total alirocumab-treated population, 0.5% in those with an on-treatment LDL below 25 mg/dL, and 0.5% in placebo-treated controls who, it must be remembered, were on maximum-tolerated therapy with statins and other conventional lipid-lowering agents. Other treatment-emergent adverse events – and more than 30 categories of such events were tracked – followed a similar pattern (see graphic).
There was one event of possible concern that has been reported to the Food and Drug Administration. An alirocumab-treated patient was diagnosed with Miller Fisher syndrome, a rare variant of Guillain-Barre syndrome, at week 27 after a prodromal severe gastroenteritis. At week 24, just 3 weeks earlier, the patient had an LDL of 1.5 mg/dL. The double vision and other symptoms of Miller Fisher syndrome resolved completely after treatment discontinuation.
During a panel discussion of the ODYSSEY clinical trial series, which includes 14 phase III studies, Dr. Henry N. Ginsburg predicted that further experience will eventually show that the dramatic LDL-lowering achieved with alirocumab not only doesn’t promote neurocognitive dysfunction, but actually protects against it.
“I think an important point to make is that, despite our neurology colleagues having tried for about a decade to completely separate the clinical phenotype of Alzheimer’s disease from vascular dementia, we all know that if you have both you’ll do much worse. So if we reduce cerebrovascular atherosclerosis and you have any predisposition to Alzheimer’s disease, you’ll do better. I think there will be studies to show that,” said Dr. Ginsburg, professor of medicine and director of the Irving Institute for Clinical and Translational Research at Columbia University in New York, who earlier in the session presented the findings from the phase III ODYSSEY High FH trial.
Another potential safety concern emerged during Dr. Dean J. Kereiakes’ presentation of the results of the phase III ODYSSEY Combo I study. Thirteen of 199 alirocumab-treated patients (6.6%) developed treatment-emergent, low-titer antialirocumab antibodies. The median time to their detection was 12 weeks. Four of the 13 patients had neutralizing antibodies, and all 4 became antibody negative by 24 weeks.
The presence of these antidrug antibodies had no discernible effect on treatment safety or efficacy. However, Combo I was a 52-week study. Whether the prevalence of these antibodies will increase over time and interfere with what is expected to be lifelong treatment is an unanswered question, noted Dr. Kereiakes, medical director for the Christ Hospital Heart and Vascular Center, Cincinnati.
Discussant Dr. Joshua W. Knowles, medical director of the FH Foundation, commented that “it’s really incredible to see the reductions in LDL that we’re getting with PCSK9 inhibitors in FH patients, the vast majority of whom require more than two standard medications to get to optimal LDL cholesterol levels.”
“The PCSK9 inhibitors are really the poster child for drug discovery in the modern genetic era. It has been a beautiful 10-year journey for them,” declared Dr. Knowles, a cardiologist at Stanford (Calif.) University.
In light of the powerful LDL-lowering effect of these agents, it’s not too soon to start thinking about how to carry out studies of atherosclerotic plaque stabilization and/or regression in treated patients, he added.
Other PCSK9 inhibitors in phase III clinical trials, besides alirocumab, are Amgen’s evolocumab and Pfizer/Renat’s bococizumab. In addition, Eli Lilly’s LY3015014 is in phase II studies, as is Roche/Genentech’s RG7652.
The ODYSSEY studies are funded by Sanofi and Regeneron Pharmaceuticals. Dr. Robinson, Dr. Ginsburg, and Dr. Kereiakes reported serving as consultants to those and other pharmaceutical companies.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: LDL cholesterol levels below 25 mg/dL induced by a superpotent investigational lipid-lowering agent appear to be safe through year 1 of follow-up.
Major finding: Thirty-six percent of high-cardiovascular-risk patients with a mean baseline LDL of 122 mg/dL despite maximum tolerated statin therapy responded to alirocumab with an LDL below 25 mg/dL, with treatment-emergent adverse events similar to those of placebo.
Data source: A prespecified interim analysis at week 52 of the ongoing double-blind, phase III ODYSSEY Long Term study, involving 2,341 high-cardiovascular-risk patients randomized 2:1 to self-administered subcutaneous injections of alirocumab at 150 mg or placebo every 2 weeks.
Disclosures: The study is sponsored by Sanofi and Regeneron Pharmaceuticals. Dr. Robinson reported serving as a consultant to those and other pharmaceutical companies.
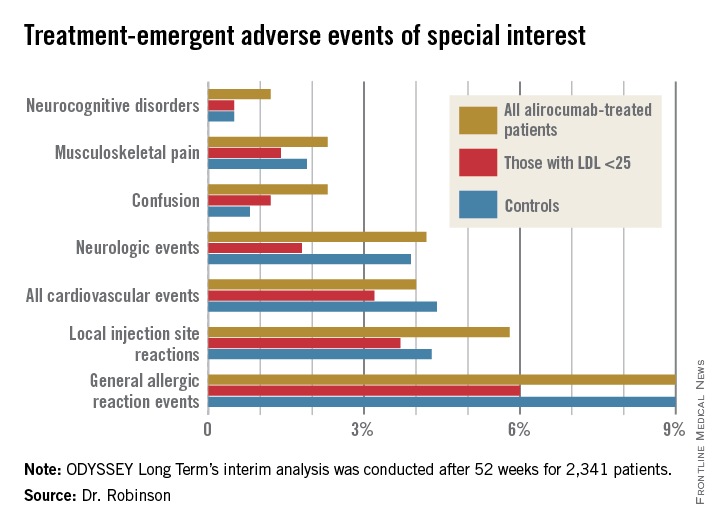
Adapalene/benzoyl peroxide + doxycycline outscores oral isotretinoin
AMSTERDAM – The combination of fixed-dose adapalene/benzoyl peroxide plus oral doxycyline is a reasonable alternative to oral isotretinoin in patients with severe nodular acne, according to the findings of a phase IIIb randomized head-to-head comparative study.
The topical/oral combination is a particularly attractive option for those patients who aren’t candidates for the potent oral retinoid, are unwilling to take it, or can’t tolerate its side effects, Dr. Jerry Tan said at the annual congress of the European Academy of Dermatology and Venereology.

Dr. Tan presented the results of the phase IIIb POWER trial, a 20-week, multicenter, randomized investigator-blinded study involving 266 patients with an average age of 19 years. Participants were randomized to adapalene 0.1%/benzoyl peroxide 2.5% gel (Epiduo)plus oral doxycyline hyclate at 200 mg/day or to placebo gel plus oral isotretinoin at 0.5 mg/kg per day for the first 4 weeks and 1.0 mg/kg per day for the remaining 16 weeks.
At 2 weeks, patients in the combination treatment group averaged a 40% reduction in facial nodules, compared with baseline, versus a 24% decrease with isotretinoin. The combination treatment group also averaged a 27% reduction in facial papules and pustules, an effect size twice that seen in the isotretinoin group at that point. The combination therapy group had a 13% decrease in comedones, compared with 6% with isotretinoin, reported Dr. Tan, a dermatologist at the University of Western Ontario, London.

By the study’s end at week 20, however, isotretinoin showed superior efficacy. But this came at the cost of a significantly worse safety profile. Indeed, the only truly serious treatment-related adverse event seen in the study – a case of Stevens-Johnson syndrome requiring hospitalization – occurred in a patient on isotretinoin.
In an effort to capture both the strengths and drawbacks of each treatment strategy in a single metric, Dr. Tan and his coinvestigators came up with a prespecified novel endpoint they termed “the composite success rate.” They defined it as at least a 75% reduction in a patient’s nodule count, plus the absence of treatment-related, medically relevant adverse events. Based on this measure, the fixed-dose topical plus oral doxycycline regimen was more effective, with a 74% composite success rate, compared with 58% for isotretinoin.
The trial was funded by Galderma. Dr. Tan disclosed serving as a paid researcher for and consultant to Galderma and other pharmaceutical companies.
AMSTERDAM – The combination of fixed-dose adapalene/benzoyl peroxide plus oral doxycyline is a reasonable alternative to oral isotretinoin in patients with severe nodular acne, according to the findings of a phase IIIb randomized head-to-head comparative study.
The topical/oral combination is a particularly attractive option for those patients who aren’t candidates for the potent oral retinoid, are unwilling to take it, or can’t tolerate its side effects, Dr. Jerry Tan said at the annual congress of the European Academy of Dermatology and Venereology.
Dr. Tan presented the results of the phase IIIb POWER trial, a 20-week, multicenter, randomized investigator-blinded study involving 266 patients with an average age of 19 years. Participants were randomized to adapalene 0.1%/benzoyl peroxide 2.5% gel (Epiduo)plus oral doxycyline hyclate at 200 mg/day or to placebo gel plus oral isotretinoin at 0.5 mg/kg per day for the first 4 weeks and 1.0 mg/kg per day for the remaining 16 weeks.
At 2 weeks, patients in the combination treatment group averaged a 40% reduction in facial nodules, compared with baseline, versus a 24% decrease with isotretinoin. The combination treatment group also averaged a 27% reduction in facial papules and pustules, an effect size twice that seen in the isotretinoin group at that point. The combination therapy group had a 13% decrease in comedones, compared with 6% with isotretinoin, reported Dr. Tan, a dermatologist at the University of Western Ontario, London.
By the study’s end at week 20, however, isotretinoin showed superior efficacy. But this came at the cost of a significantly worse safety profile. Indeed, the only truly serious treatment-related adverse event seen in the study – a case of Stevens-Johnson syndrome requiring hospitalization – occurred in a patient on isotretinoin.
In an effort to capture both the strengths and drawbacks of each treatment strategy in a single metric, Dr. Tan and his coinvestigators came up with a prespecified novel endpoint they termed “the composite success rate.” They defined it as at least a 75% reduction in a patient’s nodule count, plus the absence of treatment-related, medically relevant adverse events. Based on this measure, the fixed-dose topical plus oral doxycycline regimen was more effective, with a 74% composite success rate, compared with 58% for isotretinoin.
The trial was funded by Galderma. Dr. Tan disclosed serving as a paid researcher for and consultant to Galderma and other pharmaceutical companies.
AMSTERDAM – The combination of fixed-dose adapalene/benzoyl peroxide plus oral doxycyline is a reasonable alternative to oral isotretinoin in patients with severe nodular acne, according to the findings of a phase IIIb randomized head-to-head comparative study.
The topical/oral combination is a particularly attractive option for those patients who aren’t candidates for the potent oral retinoid, are unwilling to take it, or can’t tolerate its side effects, Dr. Jerry Tan said at the annual congress of the European Academy of Dermatology and Venereology.
Dr. Tan presented the results of the phase IIIb POWER trial, a 20-week, multicenter, randomized investigator-blinded study involving 266 patients with an average age of 19 years. Participants were randomized to adapalene 0.1%/benzoyl peroxide 2.5% gel (Epiduo)plus oral doxycyline hyclate at 200 mg/day or to placebo gel plus oral isotretinoin at 0.5 mg/kg per day for the first 4 weeks and 1.0 mg/kg per day for the remaining 16 weeks.
At 2 weeks, patients in the combination treatment group averaged a 40% reduction in facial nodules, compared with baseline, versus a 24% decrease with isotretinoin. The combination treatment group also averaged a 27% reduction in facial papules and pustules, an effect size twice that seen in the isotretinoin group at that point. The combination therapy group had a 13% decrease in comedones, compared with 6% with isotretinoin, reported Dr. Tan, a dermatologist at the University of Western Ontario, London.
By the study’s end at week 20, however, isotretinoin showed superior efficacy. But this came at the cost of a significantly worse safety profile. Indeed, the only truly serious treatment-related adverse event seen in the study – a case of Stevens-Johnson syndrome requiring hospitalization – occurred in a patient on isotretinoin.
In an effort to capture both the strengths and drawbacks of each treatment strategy in a single metric, Dr. Tan and his coinvestigators came up with a prespecified novel endpoint they termed “the composite success rate.” They defined it as at least a 75% reduction in a patient’s nodule count, plus the absence of treatment-related, medically relevant adverse events. Based on this measure, the fixed-dose topical plus oral doxycycline regimen was more effective, with a 74% composite success rate, compared with 58% for isotretinoin.
The trial was funded by Galderma. Dr. Tan disclosed serving as a paid researcher for and consultant to Galderma and other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point: The combination of fixed-dose adapalene/benzoyl peroxide plus oral doxycyline is an alternative to oral isotretinoin in patients with severe nodular acne.
Major finding: The composite success rate – a novel metric balancing efficacy and safety – was 74% in acne patients treated for 20 weeks with fixed-dose adapalene/benzoyl peroxide gel plus oral doxycycline, compared with 58% in those treated with oral isotretinoin.
Data source: A 20-week, multicenter, randomized, placebo-controlled, investigator-blinded, phase IIIb clinical trial involving 266 patients with severe nodular acne.
Disclosures: The POWER study was funded by Galderma. The presenter has received research grants from and serves as a consultant to Galderma and other pharmaceutical companies.

Simple risk score predicts dementia risk in type 2 diabetes
BERLIN – The first-ever dementia risk score designed specifically for patients with type 2 diabetes has successfully undergone external validation and is ready for everyday use by clinicians, Rachel A. Whitmer, Ph.D., said at the annual congress of the European College of Neuropsychopharmacology.
Patients with type 2 diabetes are on average twice as likely to develop dementia as are nondiabetics. But within the diabetes population, the magnitude of risk varies enormously. The Diabetes-Specific Dementia Risk Score pins down an individual’s 10-year risk more precisely. The predictive factors are easily obtained from a patient’s medical history, enabling primary care physicians, endocrinologists, psychiatrists, and neurologists to readily calculate an individualized risk score without resorting to cognitive function testing or other labor-intensive measures.

“This is a risk score that can easily be determined in a primary care setting. It can be done by self-report. It can be done using an electronic medical record. This is a way to tell someone what their risk is. Knowing your risk might encourage patients to take steps to reduce that risk. And if their risk is low you can show it to them and say it’s all the more reason to avoid getting diabetes complications – because it’s not just about your diabetes, it’s also for your brain health,” said Dr. Whitmer, a research scientist at the division of research, Kaiser Permanente Northern California, Oakland.
She and her coinvestigators harnessed the Kaiser Permanente Northern California Diabetes Registry to develop the risk score. They evaluated 45 candidate predictors in nearly 30,000 registry participants age 60 or older with type 2 diabetes, 5,173 of whom developed dementia during 10 years of follow-up. Once they’d developed the risk score, which is based upon the eight strongest predictors, they validated it in a cohort of 2,413 type 2 diabetic patients at Group Health of Puget Sound. The validation study was published last year (Lancet Diabetes Endocrinol. 2013;1:183-90).
The predictive variables are age, education, depression, microvascular disease, acute metabolic events, cerebrovascular disease, cardiovascular disease, and having a diabetic foot.
Here’s how the Diabetes-Specific Dementia Risk Score works: A patient gets 0 points for being age 60-64, 3 for being 65-69, and so on up to a maximum of 10 points for being age 85 or more. A history of an acute metabolic event is worth 2 points, as is cerebrovascular disease or depression. Microvascular disease or cardiovascular disease are 1 point each. A college education is worth minus 1 point, a high school education or less gets 0 points. The points awarded for the eight predictors are added up. In the validation study, the 10-year risk of dementia ranged from a low of 5% in patients with a net score of minus 1 to 73% in patients with a score of 12-19.
Dr. Whitmer sees the risk score as being particularly well-suited as a tool for dementia screening of type 2 diabetes patients by primary care physicians. She noted that, today, well-managed diabetic patients have a podiatrist, dietician, and dentist, as well as a physician, all working to prevent the physical complications of diabetes. But there’s not typically anyone looking out for the diabetes patient’s brain health.
“I think it’s time now when we’re taking care of diabetic individuals that we need to think about the brain,” she said.
Numerous earlier studies conducted using the Kaiser Permanente Northern California Diabetes Registry have been instrumental in helping to establish vascular complications, depression, and hypoglycemic episodes as events that markedly elevate the risk of dementia among people with type 2 diabetes.
“The question is, if we can treat depression more effectively and manage glycemic control and prevent vascular complications, can we lower the risk of dementia in these people who are at particularly high risk? That’s where the field is right now. There are trials ongoing in people with diabetes and prediabetes looking at this question,” Dr. Whitmer noted.
The question takes on some urgency, she continued, because the intersection of type 2 diabetes and dementia looms ahead as an enormous public health problem. The International Diabetes Federation estimates that there are now 392 million people worldwide with diabetes, and that by 2035 this figure will climb to 592 million. Meanwhile, other projections are that the worldwide population of individuals with dementia will jump from just under 50 million today to more than 125 million by 2050, with most of the growth coming from low- and middle-income countries where the average lifespan is increasing.
The risk score project was supported by Kaiser Permanente and the National Institutes of Health. Dr. Whitmer reported having no financial conflicts.
BERLIN – The first-ever dementia risk score designed specifically for patients with type 2 diabetes has successfully undergone external validation and is ready for everyday use by clinicians, Rachel A. Whitmer, Ph.D., said at the annual congress of the European College of Neuropsychopharmacology.
Patients with type 2 diabetes are on average twice as likely to develop dementia as are nondiabetics. But within the diabetes population, the magnitude of risk varies enormously. The Diabetes-Specific Dementia Risk Score pins down an individual’s 10-year risk more precisely. The predictive factors are easily obtained from a patient’s medical history, enabling primary care physicians, endocrinologists, psychiatrists, and neurologists to readily calculate an individualized risk score without resorting to cognitive function testing or other labor-intensive measures.
“This is a risk score that can easily be determined in a primary care setting. It can be done by self-report. It can be done using an electronic medical record. This is a way to tell someone what their risk is. Knowing your risk might encourage patients to take steps to reduce that risk. And if their risk is low you can show it to them and say it’s all the more reason to avoid getting diabetes complications – because it’s not just about your diabetes, it’s also for your brain health,” said Dr. Whitmer, a research scientist at the division of research, Kaiser Permanente Northern California, Oakland.
She and her coinvestigators harnessed the Kaiser Permanente Northern California Diabetes Registry to develop the risk score. They evaluated 45 candidate predictors in nearly 30,000 registry participants age 60 or older with type 2 diabetes, 5,173 of whom developed dementia during 10 years of follow-up. Once they’d developed the risk score, which is based upon the eight strongest predictors, they validated it in a cohort of 2,413 type 2 diabetic patients at Group Health of Puget Sound. The validation study was published last year (Lancet Diabetes Endocrinol. 2013;1:183-90).
The predictive variables are age, education, depression, microvascular disease, acute metabolic events, cerebrovascular disease, cardiovascular disease, and having a diabetic foot.
Here’s how the Diabetes-Specific Dementia Risk Score works: A patient gets 0 points for being age 60-64, 3 for being 65-69, and so on up to a maximum of 10 points for being age 85 or more. A history of an acute metabolic event is worth 2 points, as is cerebrovascular disease or depression. Microvascular disease or cardiovascular disease are 1 point each. A college education is worth minus 1 point, a high school education or less gets 0 points. The points awarded for the eight predictors are added up. In the validation study, the 10-year risk of dementia ranged from a low of 5% in patients with a net score of minus 1 to 73% in patients with a score of 12-19.
Dr. Whitmer sees the risk score as being particularly well-suited as a tool for dementia screening of type 2 diabetes patients by primary care physicians. She noted that, today, well-managed diabetic patients have a podiatrist, dietician, and dentist, as well as a physician, all working to prevent the physical complications of diabetes. But there’s not typically anyone looking out for the diabetes patient’s brain health.
“I think it’s time now when we’re taking care of diabetic individuals that we need to think about the brain,” she said.
Numerous earlier studies conducted using the Kaiser Permanente Northern California Diabetes Registry have been instrumental in helping to establish vascular complications, depression, and hypoglycemic episodes as events that markedly elevate the risk of dementia among people with type 2 diabetes.
“The question is, if we can treat depression more effectively and manage glycemic control and prevent vascular complications, can we lower the risk of dementia in these people who are at particularly high risk? That’s where the field is right now. There are trials ongoing in people with diabetes and prediabetes looking at this question,” Dr. Whitmer noted.
The question takes on some urgency, she continued, because the intersection of type 2 diabetes and dementia looms ahead as an enormous public health problem. The International Diabetes Federation estimates that there are now 392 million people worldwide with diabetes, and that by 2035 this figure will climb to 592 million. Meanwhile, other projections are that the worldwide population of individuals with dementia will jump from just under 50 million today to more than 125 million by 2050, with most of the growth coming from low- and middle-income countries where the average lifespan is increasing.
The risk score project was supported by Kaiser Permanente and the National Institutes of Health. Dr. Whitmer reported having no financial conflicts.
BERLIN – The first-ever dementia risk score designed specifically for patients with type 2 diabetes has successfully undergone external validation and is ready for everyday use by clinicians, Rachel A. Whitmer, Ph.D., said at the annual congress of the European College of Neuropsychopharmacology.
Patients with type 2 diabetes are on average twice as likely to develop dementia as are nondiabetics. But within the diabetes population, the magnitude of risk varies enormously. The Diabetes-Specific Dementia Risk Score pins down an individual’s 10-year risk more precisely. The predictive factors are easily obtained from a patient’s medical history, enabling primary care physicians, endocrinologists, psychiatrists, and neurologists to readily calculate an individualized risk score without resorting to cognitive function testing or other labor-intensive measures.
“This is a risk score that can easily be determined in a primary care setting. It can be done by self-report. It can be done using an electronic medical record. This is a way to tell someone what their risk is. Knowing your risk might encourage patients to take steps to reduce that risk. And if their risk is low you can show it to them and say it’s all the more reason to avoid getting diabetes complications – because it’s not just about your diabetes, it’s also for your brain health,” said Dr. Whitmer, a research scientist at the division of research, Kaiser Permanente Northern California, Oakland.
She and her coinvestigators harnessed the Kaiser Permanente Northern California Diabetes Registry to develop the risk score. They evaluated 45 candidate predictors in nearly 30,000 registry participants age 60 or older with type 2 diabetes, 5,173 of whom developed dementia during 10 years of follow-up. Once they’d developed the risk score, which is based upon the eight strongest predictors, they validated it in a cohort of 2,413 type 2 diabetic patients at Group Health of Puget Sound. The validation study was published last year (Lancet Diabetes Endocrinol. 2013;1:183-90).
The predictive variables are age, education, depression, microvascular disease, acute metabolic events, cerebrovascular disease, cardiovascular disease, and having a diabetic foot.
Here’s how the Diabetes-Specific Dementia Risk Score works: A patient gets 0 points for being age 60-64, 3 for being 65-69, and so on up to a maximum of 10 points for being age 85 or more. A history of an acute metabolic event is worth 2 points, as is cerebrovascular disease or depression. Microvascular disease or cardiovascular disease are 1 point each. A college education is worth minus 1 point, a high school education or less gets 0 points. The points awarded for the eight predictors are added up. In the validation study, the 10-year risk of dementia ranged from a low of 5% in patients with a net score of minus 1 to 73% in patients with a score of 12-19.
Dr. Whitmer sees the risk score as being particularly well-suited as a tool for dementia screening of type 2 diabetes patients by primary care physicians. She noted that, today, well-managed diabetic patients have a podiatrist, dietician, and dentist, as well as a physician, all working to prevent the physical complications of diabetes. But there’s not typically anyone looking out for the diabetes patient’s brain health.
“I think it’s time now when we’re taking care of diabetic individuals that we need to think about the brain,” she said.
Numerous earlier studies conducted using the Kaiser Permanente Northern California Diabetes Registry have been instrumental in helping to establish vascular complications, depression, and hypoglycemic episodes as events that markedly elevate the risk of dementia among people with type 2 diabetes.
“The question is, if we can treat depression more effectively and manage glycemic control and prevent vascular complications, can we lower the risk of dementia in these people who are at particularly high risk? That’s where the field is right now. There are trials ongoing in people with diabetes and prediabetes looking at this question,” Dr. Whitmer noted.
The question takes on some urgency, she continued, because the intersection of type 2 diabetes and dementia looms ahead as an enormous public health problem. The International Diabetes Federation estimates that there are now 392 million people worldwide with diabetes, and that by 2035 this figure will climb to 592 million. Meanwhile, other projections are that the worldwide population of individuals with dementia will jump from just under 50 million today to more than 125 million by 2050, with most of the growth coming from low- and middle-income countries where the average lifespan is increasing.
The risk score project was supported by Kaiser Permanente and the National Institutes of Health. Dr. Whitmer reported having no financial conflicts.
AT THE ECNP CONGRESS
Key clinical point: It’s now readily possible to predict the 10-year dementia risk for individuals with type 2 diabetes.
Major finding: The 10-year risk of dementia in patients with type 2 diabetes ranged from 5% to 73% depending upon their Diabetes-Specific Dementia Risk Score, on the basis of eight variables easily obtained from a patient’s medical history.
Data source: The risk score was developed by evaluating 45 candidate predictive variables in a longitudinal cohort of 29,961 type 2 diabetes patients, 5,173 of whom developed dementia during 10 years of follow-up.
Disclosures: The study was funded by Kaiser Permanente and the National Institutes of Health. The presenter reported having no financial conflicts.

Psoriasis: Brodalumab maintains efficacy through 144 weeks
AMSTERDAM – The majority of psoriasis patients placed on the investigational biologic agent brodalumab maintained a PASI 100 response throughout 144 weeks in a long-term, open-label extension of a phase II study.
“The benefit/risk ratio of brodalumab remains very favorable, and warrants continued development of this anti–interleukin-17 receptor A human monoclonal antibody as a potential treatment for psoriasis,” Dr. Kim Papp declared in presenting the study results at the annual congress of the European Academy of Dermatology and Venereology.
Large phase III clinical trials of brodalumab for the treatment of moderate to severe plaque psoriasis are ongoing.
The parent phase II study was 16 weeks long, double blinded, and placebo controlled. At the study’s end, 181 participants enrolled in the open-label extension, in which they received subcutaneous brodalumab at 210 mg every 2 weeks. The study protocol was amended after about a year, with the dose reduced to 140 mg every 2 weeks in the 119 patients weighing 100 kg or less. Six of those patients subsequently had an inadequate response to the lower dose, and were returned to the higher-dose regimen, explained Dr. Papp of Probity Medical Research in Waterloo, Ont.
Roughly 95% of patients became PASI 75 responders within the first few weeks. There was very little drop-off over time. The PASI 75 response rate was 95% at week 12, 93% at week 48, and 85% at week 144.
At week 144, roughly two-thirds of subjects were deemed clear or almost clear according to Physician Global Assessment.
No new safety signals emerged during this long-term study. The most common adverse events were minor upper respiratory infections. Two percent of patients developed a grade 2 absolute neutrophil count of less than 1,500 per 109/L; however, these were transitory events that resolved without a change in treatment.
In response to an audience question, Dr. Papp said antidrug antibodies did develop in some patients during the course of 144 weeks of treatment, but he had no information as to the clinical effect, if any.
“This is a fairly small study population. I think it makes sense to wait for the phase III results to see if the antibodies affect safety and/or efficacy,” he added.
Dr. Papp reported receiving research support from Amgen, which is codeveloping brodalumab with AstraZeneca/MedImmune.
AMSTERDAM – The majority of psoriasis patients placed on the investigational biologic agent brodalumab maintained a PASI 100 response throughout 144 weeks in a long-term, open-label extension of a phase II study.
“The benefit/risk ratio of brodalumab remains very favorable, and warrants continued development of this anti–interleukin-17 receptor A human monoclonal antibody as a potential treatment for psoriasis,” Dr. Kim Papp declared in presenting the study results at the annual congress of the European Academy of Dermatology and Venereology.
Large phase III clinical trials of brodalumab for the treatment of moderate to severe plaque psoriasis are ongoing.
The parent phase II study was 16 weeks long, double blinded, and placebo controlled. At the study’s end, 181 participants enrolled in the open-label extension, in which they received subcutaneous brodalumab at 210 mg every 2 weeks. The study protocol was amended after about a year, with the dose reduced to 140 mg every 2 weeks in the 119 patients weighing 100 kg or less. Six of those patients subsequently had an inadequate response to the lower dose, and were returned to the higher-dose regimen, explained Dr. Papp of Probity Medical Research in Waterloo, Ont.
Roughly 95% of patients became PASI 75 responders within the first few weeks. There was very little drop-off over time. The PASI 75 response rate was 95% at week 12, 93% at week 48, and 85% at week 144.
At week 144, roughly two-thirds of subjects were deemed clear or almost clear according to Physician Global Assessment.
No new safety signals emerged during this long-term study. The most common adverse events were minor upper respiratory infections. Two percent of patients developed a grade 2 absolute neutrophil count of less than 1,500 per 109/L; however, these were transitory events that resolved without a change in treatment.
In response to an audience question, Dr. Papp said antidrug antibodies did develop in some patients during the course of 144 weeks of treatment, but he had no information as to the clinical effect, if any.
“This is a fairly small study population. I think it makes sense to wait for the phase III results to see if the antibodies affect safety and/or efficacy,” he added.
Dr. Papp reported receiving research support from Amgen, which is codeveloping brodalumab with AstraZeneca/MedImmune.
AMSTERDAM – The majority of psoriasis patients placed on the investigational biologic agent brodalumab maintained a PASI 100 response throughout 144 weeks in a long-term, open-label extension of a phase II study.
“The benefit/risk ratio of brodalumab remains very favorable, and warrants continued development of this anti–interleukin-17 receptor A human monoclonal antibody as a potential treatment for psoriasis,” Dr. Kim Papp declared in presenting the study results at the annual congress of the European Academy of Dermatology and Venereology.
Large phase III clinical trials of brodalumab for the treatment of moderate to severe plaque psoriasis are ongoing.
The parent phase II study was 16 weeks long, double blinded, and placebo controlled. At the study’s end, 181 participants enrolled in the open-label extension, in which they received subcutaneous brodalumab at 210 mg every 2 weeks. The study protocol was amended after about a year, with the dose reduced to 140 mg every 2 weeks in the 119 patients weighing 100 kg or less. Six of those patients subsequently had an inadequate response to the lower dose, and were returned to the higher-dose regimen, explained Dr. Papp of Probity Medical Research in Waterloo, Ont.
Roughly 95% of patients became PASI 75 responders within the first few weeks. There was very little drop-off over time. The PASI 75 response rate was 95% at week 12, 93% at week 48, and 85% at week 144.
At week 144, roughly two-thirds of subjects were deemed clear or almost clear according to Physician Global Assessment.
No new safety signals emerged during this long-term study. The most common adverse events were minor upper respiratory infections. Two percent of patients developed a grade 2 absolute neutrophil count of less than 1,500 per 109/L; however, these were transitory events that resolved without a change in treatment.
In response to an audience question, Dr. Papp said antidrug antibodies did develop in some patients during the course of 144 weeks of treatment, but he had no information as to the clinical effect, if any.
“This is a fairly small study population. I think it makes sense to wait for the phase III results to see if the antibodies affect safety and/or efficacy,” he added.
Dr. Papp reported receiving research support from Amgen, which is codeveloping brodalumab with AstraZeneca/MedImmune.
AT THE EADV CONGRESS
Key clinical point: The investigational interleukin-17 receptor A inhibitor brodalumab maintained strong clinical efficacy throughout 144 weeks of psoriasis treatment.
Major finding: The PASI 90 response rate to brodalumab was 85% at week 12, 83% at week 48, and 74% at week 144.
Data source: A 181-patient, prospective, open-label extension of a phase II study.
Disclosures: Dr. Papp reported receiving financial support from Amgen, which is codeveloping brodalumab with AstraZeneca/MedImmune.
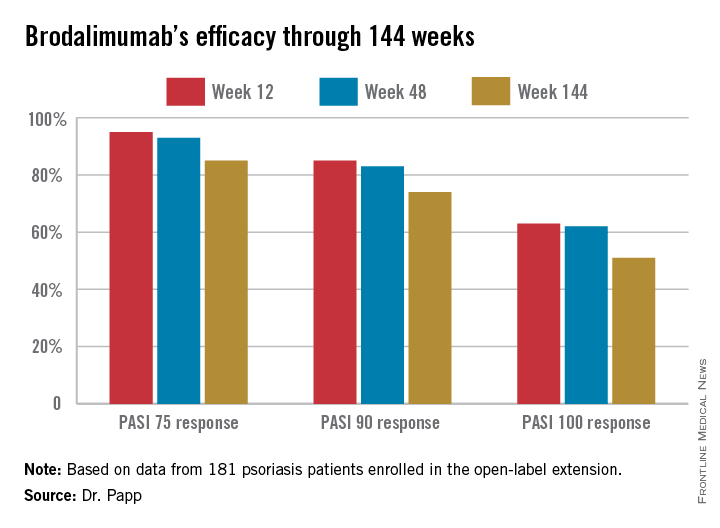
Dupilumab Advances for Severe Atopic Dermatitis
AMSTERDAM – The novel biologic agent dupilumab showed strong dose-dependent efficacy in adults with moderate to severe atopic dermatitis not adequately controlled with topical medications in a 380-patient phase IIb dose-ranging study.
“Based on the results of this study, we’ll take the top two doses further to our phase III program. We’re also planning to conduct a maintenance study. At the end of 16 weeks of treatment, we’ll investigate lower-dose regimens that may be capable of maintaining clinical response,” Dr. Marius Ardeleanu said at the annual congress of the European Academy of Dermatology and Venereology.
Dipilumab is an investigational fully human monoclonal antibody that addresses a novel target: It is directed against the interleukin-4 receptor alpha subunit (IL-4Ra). Through this effect it blocks IL-4 and IL-17, the drivers of the type 2 helper T-cell–mediated inflammation responsible for the hallmark symptoms of atopic dermatitis (AD), explained Dr. Ardeleanu of Regeneron Pharmaceuticals in Tarrytown, N.Y.
Participants in this 16-week, double-blind, international phase IIb study were randomized to placebo or one of five dupilumab dosing regimens ranging from a low of 100 mg given subcutaneously every 4 weeks to a maximum of 300 mg once weekly or every 2 weeks.
These patients had a significant disease burden. They were typically in their mid- to late 30s and had a 27-year disease history, a mean baseline SCORAD of 67 on a 0-100 scale, a baseline Eczema Area and Severity Index (EASI) score of 32, an Investigator’s Global Assessment of disease severity score of 3.5 on a 0-4 scale, and 50% body surface area involvement. Their mean average weekly self-rated itching score was 6.8 on a 0-10 scale.
The primary study endpoint was change in the EASI score from baseline to 16 weeks. The score dropped by 20% in placebo-treated controls and by significantly greater margins in all five dupilumab arms. The largest reduction in EASI score – nearly 80% – occurred in the group on 300 mg/wk, with the 300 mg every 2 weeks group showing about a 70% reduction.
Roughly 80% of patients on 300 mg/wk or every 2 weeks showed an EASI 50 response at week 16, meaning a 50% reduction from baseline in their score, which is considered clinically meaningful improvement. One-third of patients on either of these top two–performing regimens achieved an Investigator’s Global Assessment score of 0 or 1, which is virtual remission; none of the controls did. Overall weekly average pruritus scores dropped by more than 60% with weekly treatment at 300 mg and by slightly less with biweekly therapy at 300 mg.
Safety data were similar to those from a recently published earlier phase IIa study (N. Engl. J. Med. 2014; 371:130-9). There were no dose-limiting toxicities. Headache and injection site reactions were the only adverse events more common with dupilumab than with placebo in the phase IIb trial, with the incidence of injection site reactions showing a possible dose-response relationship.
In addition to the large phase III studies now being planned, which will also evaluate step-down maintenance therapy, another study has been scheduled to investigate the use of dupilumab in combination with topical corticosteroid therapy. The earlier phase IIa study provided evidence to suggest this combination has even greater efficacy than dupilumab alone, and with modest use of the topical agent, according to Dr. Ardeleanu.
Dupilumab is also being developed as a treatment for tough-to-control moderate to severe asthma. It showed positive results in a phase II study, with reduced asthma exacerbations and improved lung function, compared with placebo (N. Engl. J. Med. 2013; 368:2455-66).
AMSTERDAM – The novel biologic agent dupilumab showed strong dose-dependent efficacy in adults with moderate to severe atopic dermatitis not adequately controlled with topical medications in a 380-patient phase IIb dose-ranging study.
“Based on the results of this study, we’ll take the top two doses further to our phase III program. We’re also planning to conduct a maintenance study. At the end of 16 weeks of treatment, we’ll investigate lower-dose regimens that may be capable of maintaining clinical response,” Dr. Marius Ardeleanu said at the annual congress of the European Academy of Dermatology and Venereology.
Dipilumab is an investigational fully human monoclonal antibody that addresses a novel target: It is directed against the interleukin-4 receptor alpha subunit (IL-4Ra). Through this effect it blocks IL-4 and IL-17, the drivers of the type 2 helper T-cell–mediated inflammation responsible for the hallmark symptoms of atopic dermatitis (AD), explained Dr. Ardeleanu of Regeneron Pharmaceuticals in Tarrytown, N.Y.
Participants in this 16-week, double-blind, international phase IIb study were randomized to placebo or one of five dupilumab dosing regimens ranging from a low of 100 mg given subcutaneously every 4 weeks to a maximum of 300 mg once weekly or every 2 weeks.
These patients had a significant disease burden. They were typically in their mid- to late 30s and had a 27-year disease history, a mean baseline SCORAD of 67 on a 0-100 scale, a baseline Eczema Area and Severity Index (EASI) score of 32, an Investigator’s Global Assessment of disease severity score of 3.5 on a 0-4 scale, and 50% body surface area involvement. Their mean average weekly self-rated itching score was 6.8 on a 0-10 scale.
The primary study endpoint was change in the EASI score from baseline to 16 weeks. The score dropped by 20% in placebo-treated controls and by significantly greater margins in all five dupilumab arms. The largest reduction in EASI score – nearly 80% – occurred in the group on 300 mg/wk, with the 300 mg every 2 weeks group showing about a 70% reduction.
Roughly 80% of patients on 300 mg/wk or every 2 weeks showed an EASI 50 response at week 16, meaning a 50% reduction from baseline in their score, which is considered clinically meaningful improvement. One-third of patients on either of these top two–performing regimens achieved an Investigator’s Global Assessment score of 0 or 1, which is virtual remission; none of the controls did. Overall weekly average pruritus scores dropped by more than 60% with weekly treatment at 300 mg and by slightly less with biweekly therapy at 300 mg.
Safety data were similar to those from a recently published earlier phase IIa study (N. Engl. J. Med. 2014; 371:130-9). There were no dose-limiting toxicities. Headache and injection site reactions were the only adverse events more common with dupilumab than with placebo in the phase IIb trial, with the incidence of injection site reactions showing a possible dose-response relationship.
In addition to the large phase III studies now being planned, which will also evaluate step-down maintenance therapy, another study has been scheduled to investigate the use of dupilumab in combination with topical corticosteroid therapy. The earlier phase IIa study provided evidence to suggest this combination has even greater efficacy than dupilumab alone, and with modest use of the topical agent, according to Dr. Ardeleanu.
Dupilumab is also being developed as a treatment for tough-to-control moderate to severe asthma. It showed positive results in a phase II study, with reduced asthma exacerbations and improved lung function, compared with placebo (N. Engl. J. Med. 2013; 368:2455-66).
AMSTERDAM – The novel biologic agent dupilumab showed strong dose-dependent efficacy in adults with moderate to severe atopic dermatitis not adequately controlled with topical medications in a 380-patient phase IIb dose-ranging study.
“Based on the results of this study, we’ll take the top two doses further to our phase III program. We’re also planning to conduct a maintenance study. At the end of 16 weeks of treatment, we’ll investigate lower-dose regimens that may be capable of maintaining clinical response,” Dr. Marius Ardeleanu said at the annual congress of the European Academy of Dermatology and Venereology.
Dipilumab is an investigational fully human monoclonal antibody that addresses a novel target: It is directed against the interleukin-4 receptor alpha subunit (IL-4Ra). Through this effect it blocks IL-4 and IL-17, the drivers of the type 2 helper T-cell–mediated inflammation responsible for the hallmark symptoms of atopic dermatitis (AD), explained Dr. Ardeleanu of Regeneron Pharmaceuticals in Tarrytown, N.Y.
Participants in this 16-week, double-blind, international phase IIb study were randomized to placebo or one of five dupilumab dosing regimens ranging from a low of 100 mg given subcutaneously every 4 weeks to a maximum of 300 mg once weekly or every 2 weeks.
These patients had a significant disease burden. They were typically in their mid- to late 30s and had a 27-year disease history, a mean baseline SCORAD of 67 on a 0-100 scale, a baseline Eczema Area and Severity Index (EASI) score of 32, an Investigator’s Global Assessment of disease severity score of 3.5 on a 0-4 scale, and 50% body surface area involvement. Their mean average weekly self-rated itching score was 6.8 on a 0-10 scale.
The primary study endpoint was change in the EASI score from baseline to 16 weeks. The score dropped by 20% in placebo-treated controls and by significantly greater margins in all five dupilumab arms. The largest reduction in EASI score – nearly 80% – occurred in the group on 300 mg/wk, with the 300 mg every 2 weeks group showing about a 70% reduction.
Roughly 80% of patients on 300 mg/wk or every 2 weeks showed an EASI 50 response at week 16, meaning a 50% reduction from baseline in their score, which is considered clinically meaningful improvement. One-third of patients on either of these top two–performing regimens achieved an Investigator’s Global Assessment score of 0 or 1, which is virtual remission; none of the controls did. Overall weekly average pruritus scores dropped by more than 60% with weekly treatment at 300 mg and by slightly less with biweekly therapy at 300 mg.
Safety data were similar to those from a recently published earlier phase IIa study (N. Engl. J. Med. 2014; 371:130-9). There were no dose-limiting toxicities. Headache and injection site reactions were the only adverse events more common with dupilumab than with placebo in the phase IIb trial, with the incidence of injection site reactions showing a possible dose-response relationship.
In addition to the large phase III studies now being planned, which will also evaluate step-down maintenance therapy, another study has been scheduled to investigate the use of dupilumab in combination with topical corticosteroid therapy. The earlier phase IIa study provided evidence to suggest this combination has even greater efficacy than dupilumab alone, and with modest use of the topical agent, according to Dr. Ardeleanu.
Dupilumab is also being developed as a treatment for tough-to-control moderate to severe asthma. It showed positive results in a phase II study, with reduced asthma exacerbations and improved lung function, compared with placebo (N. Engl. J. Med. 2013; 368:2455-66).
AT THE EADV CONGRESS
