User login
Dupilumab advances for severe atopic dermatitis
AMSTERDAM – The novel biologic agent dupilumab showed strong dose-dependent efficacy in adults with moderate to severe atopic dermatitis not adequately controlled with topical medications in a 380-patient phase IIb dose-ranging study.
“Based on the results of this study, we’ll take the top two doses further to our phase III program. We’re also planning to conduct a maintenance study. At the end of 16 weeks of treatment, we’ll investigate lower-dose regimens that may be capable of maintaining clinical response,” Dr. Marius Ardeleanu said at the annual congress of the European Academy of Dermatology and Venereology.

Dupilumab is an investigational fully human monoclonal antibody that addresses a novel target: It is directed against the interleukin-4 receptor alpha subunit (IL-4Ra). Through this effect it blocks IL-4 and IL-13, the drivers of the type 2 helper T-cell–mediated inflammation responsible for the hallmark symptoms of atopic dermatitis (AD), explained Dr. Ardeleanu of Regeneron Pharmaceuticals in Tarrytown, N.Y.
Participants in this 16-week, double-blind, international phase IIb study were randomized to placebo or one of five dupilumab dosing regimens ranging from a low of 100 mg given subcutaneously every 4 weeks to a maximum of 300 mg once weekly or every 2 weeks.
These patients had a significant disease burden. They were typically in their mid- to late 30s and had a 27-year disease history, a mean baseline SCORAD of 67 on a 0-100 scale, a baseline Eczema Area and Severity Index (EASI) score of 32, an Investigator’s Global Assessment of disease severity score of 3.5 on a 0-4 scale, and 50% body surface area involvement. Their mean average weekly self-rated itching score was 6.8 on a 0-10 scale.
The primary study endpoint was change in the EASI score from baseline to 16 weeks. The score dropped by 20% in placebo-treated controls and by significantly greater margins in all five dupilumab arms. The largest reduction in EASI score – nearly 80% – occurred in the group on 300 mg/wk, with the 300 mg every 2 weeks group showing about a 70% reduction.
Roughly 80% of patients on 300 mg/wk or every 2 weeks showed an EASI 50 response at week 16, meaning a 50% reduction from baseline in their score, which is considered clinically meaningful improvement. One-third of patients on either of these top two–performing regimens achieved an Investigator’s Global Assessment score of 0 or 1, which is virtual remission; none of the controls did. Overall weekly average pruritus scores dropped by more than 60% with weekly treatment at 300 mg and by slightly less with biweekly therapy at 300 mg.
Safety data were similar to those from a recently published earlier phase IIa study (N. Engl. J. Med. 2014; 371:130-9). There were no dose-limiting toxicities. Headache and injection site reactions were the only adverse events more common with dupilumab than with placebo in the phase IIb trial, with the incidence of injection site reactions showing a possible dose-response relationship.
In addition to the large phase III studies now being planned, which will also evaluate step-down maintenance therapy, another study has been scheduled to investigate the use of dupilumab in combination with topical corticosteroid therapy. The earlier phase IIa study provided evidence to suggest this combination has even greater efficacy than dupilumab alone, and with modest use of the topical agent, according to Dr. Ardeleanu.
Dupilumab is also being developed as a treatment for tough-to-control moderate to severe asthma. It showed positive results in a phase II study, with reduced asthma exacerbations and improved lung function, compared with placebo (N. Engl. J. Med. 2013; 368:2455-66).
AMSTERDAM – The novel biologic agent dupilumab showed strong dose-dependent efficacy in adults with moderate to severe atopic dermatitis not adequately controlled with topical medications in a 380-patient phase IIb dose-ranging study.
“Based on the results of this study, we’ll take the top two doses further to our phase III program. We’re also planning to conduct a maintenance study. At the end of 16 weeks of treatment, we’ll investigate lower-dose regimens that may be capable of maintaining clinical response,” Dr. Marius Ardeleanu said at the annual congress of the European Academy of Dermatology and Venereology.
Dupilumab is an investigational fully human monoclonal antibody that addresses a novel target: It is directed against the interleukin-4 receptor alpha subunit (IL-4Ra). Through this effect it blocks IL-4 and IL-13, the drivers of the type 2 helper T-cell–mediated inflammation responsible for the hallmark symptoms of atopic dermatitis (AD), explained Dr. Ardeleanu of Regeneron Pharmaceuticals in Tarrytown, N.Y.
Participants in this 16-week, double-blind, international phase IIb study were randomized to placebo or one of five dupilumab dosing regimens ranging from a low of 100 mg given subcutaneously every 4 weeks to a maximum of 300 mg once weekly or every 2 weeks.
These patients had a significant disease burden. They were typically in their mid- to late 30s and had a 27-year disease history, a mean baseline SCORAD of 67 on a 0-100 scale, a baseline Eczema Area and Severity Index (EASI) score of 32, an Investigator’s Global Assessment of disease severity score of 3.5 on a 0-4 scale, and 50% body surface area involvement. Their mean average weekly self-rated itching score was 6.8 on a 0-10 scale.
The primary study endpoint was change in the EASI score from baseline to 16 weeks. The score dropped by 20% in placebo-treated controls and by significantly greater margins in all five dupilumab arms. The largest reduction in EASI score – nearly 80% – occurred in the group on 300 mg/wk, with the 300 mg every 2 weeks group showing about a 70% reduction.
Roughly 80% of patients on 300 mg/wk or every 2 weeks showed an EASI 50 response at week 16, meaning a 50% reduction from baseline in their score, which is considered clinically meaningful improvement. One-third of patients on either of these top two–performing regimens achieved an Investigator’s Global Assessment score of 0 or 1, which is virtual remission; none of the controls did. Overall weekly average pruritus scores dropped by more than 60% with weekly treatment at 300 mg and by slightly less with biweekly therapy at 300 mg.
Safety data were similar to those from a recently published earlier phase IIa study (N. Engl. J. Med. 2014; 371:130-9). There were no dose-limiting toxicities. Headache and injection site reactions were the only adverse events more common with dupilumab than with placebo in the phase IIb trial, with the incidence of injection site reactions showing a possible dose-response relationship.
In addition to the large phase III studies now being planned, which will also evaluate step-down maintenance therapy, another study has been scheduled to investigate the use of dupilumab in combination with topical corticosteroid therapy. The earlier phase IIa study provided evidence to suggest this combination has even greater efficacy than dupilumab alone, and with modest use of the topical agent, according to Dr. Ardeleanu.
Dupilumab is also being developed as a treatment for tough-to-control moderate to severe asthma. It showed positive results in a phase II study, with reduced asthma exacerbations and improved lung function, compared with placebo (N. Engl. J. Med. 2013; 368:2455-66).
AMSTERDAM – The novel biologic agent dupilumab showed strong dose-dependent efficacy in adults with moderate to severe atopic dermatitis not adequately controlled with topical medications in a 380-patient phase IIb dose-ranging study.
“Based on the results of this study, we’ll take the top two doses further to our phase III program. We’re also planning to conduct a maintenance study. At the end of 16 weeks of treatment, we’ll investigate lower-dose regimens that may be capable of maintaining clinical response,” Dr. Marius Ardeleanu said at the annual congress of the European Academy of Dermatology and Venereology.
Dupilumab is an investigational fully human monoclonal antibody that addresses a novel target: It is directed against the interleukin-4 receptor alpha subunit (IL-4Ra). Through this effect it blocks IL-4 and IL-13, the drivers of the type 2 helper T-cell–mediated inflammation responsible for the hallmark symptoms of atopic dermatitis (AD), explained Dr. Ardeleanu of Regeneron Pharmaceuticals in Tarrytown, N.Y.
Participants in this 16-week, double-blind, international phase IIb study were randomized to placebo or one of five dupilumab dosing regimens ranging from a low of 100 mg given subcutaneously every 4 weeks to a maximum of 300 mg once weekly or every 2 weeks.
These patients had a significant disease burden. They were typically in their mid- to late 30s and had a 27-year disease history, a mean baseline SCORAD of 67 on a 0-100 scale, a baseline Eczema Area and Severity Index (EASI) score of 32, an Investigator’s Global Assessment of disease severity score of 3.5 on a 0-4 scale, and 50% body surface area involvement. Their mean average weekly self-rated itching score was 6.8 on a 0-10 scale.
The primary study endpoint was change in the EASI score from baseline to 16 weeks. The score dropped by 20% in placebo-treated controls and by significantly greater margins in all five dupilumab arms. The largest reduction in EASI score – nearly 80% – occurred in the group on 300 mg/wk, with the 300 mg every 2 weeks group showing about a 70% reduction.
Roughly 80% of patients on 300 mg/wk or every 2 weeks showed an EASI 50 response at week 16, meaning a 50% reduction from baseline in their score, which is considered clinically meaningful improvement. One-third of patients on either of these top two–performing regimens achieved an Investigator’s Global Assessment score of 0 or 1, which is virtual remission; none of the controls did. Overall weekly average pruritus scores dropped by more than 60% with weekly treatment at 300 mg and by slightly less with biweekly therapy at 300 mg.
Safety data were similar to those from a recently published earlier phase IIa study (N. Engl. J. Med. 2014; 371:130-9). There were no dose-limiting toxicities. Headache and injection site reactions were the only adverse events more common with dupilumab than with placebo in the phase IIb trial, with the incidence of injection site reactions showing a possible dose-response relationship.
In addition to the large phase III studies now being planned, which will also evaluate step-down maintenance therapy, another study has been scheduled to investigate the use of dupilumab in combination with topical corticosteroid therapy. The earlier phase IIa study provided evidence to suggest this combination has even greater efficacy than dupilumab alone, and with modest use of the topical agent, according to Dr. Ardeleanu.
Dupilumab is also being developed as a treatment for tough-to-control moderate to severe asthma. It showed positive results in a phase II study, with reduced asthma exacerbations and improved lung function, compared with placebo (N. Engl. J. Med. 2013; 368:2455-66).
AT THE EADV CONGRESS
Key clinical point: A promising new therapy with a novel mechanism of action is progressing through the pipeline for adults with refractory atopic dermatitis.
Major finding: Patients on subcutaneous dupilumab at 300 mg once weekly showed nearly an 80% reduction from baseline in Eczema Area and Severity Index scores after 16 weeks of treatment.
Data source: A randomized, double-blind, placebo-controlled, international, 16-week phase IIb study of 380 adults with moderate to severe atopic dermatitis inadequately controlled by topical therapies.
Disclosures: The study was sponsored by Regeneron Pharmaceuticals and presented by the company’s senior director of clinical sciences.

IMPROVE-IT: Ezetimibe/simvastatin further reduces cardiovascular events
CHICAGO – Adding ezetimibe to a midpotency statin in high-risk cardiovascular patients to drive their LDL cholesterol into the low 50s resulted in a further significant reduction in major cardiovascular events, compared with statin alone, in the 18,144-patient IMPROVE-IT study.
In IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial), 7 years of double-blind therapy with ezetimibe 10 mg/simvastatin 40 mg (Vytorin) achieved a 6.4% lower relative risk of the primary composite endpoint, compared with patients randomized to simvastatin alone (P = 0.016).

The 7-year rate of this composite outcome – comprised of cardiovascular death, nonfatal MI, rehospitalization for unstable angina, stroke, or coronary revascularization – was 32.7% with ezetimibe/simvastatin and 34.7% with simvastatin alone, for an absolute difference of 2%, Dr. Christopher P. Cannon reported at the American Heart Association scientific sessions.
Patients on dual therapy showed relative risk reductions of 13% for MI and 21% for ischemic stroke. The number needed to treat was 50, meaning that treating 100 patients with ezetimibe/simvastatin for 7 years should prevent 2 patients from having an ischemic stroke or MI who would have had such an event on simvastatin alone. That NNT is “well within the range of our standard therapies,” noted Dr. Cannon, professor of medicine at Harvard Medical School, Boston.
Patients in IMPROVE-IT began double-blind therapy within 10 days of hospitalization for an ST-elevation MI or non-STEMI/unstable angina. Participants also had to have at least one additional high-risk feature, such as a previous MI, diabetes, or coronary artery bypass surgery.
Observers hailed IMPROVE-IT as a major study for several reasons. It’s the first-ever study to demonstrate added clinical benefit when a nonstatin having a different mechanism of action is added to a statin. Thus, it provides physicians with a new, solidly evidence-based treatment option for high-risk acute coronary syndrome patients. Also, the trial emphatically affirms the LDL hypothesis that lowering LDL prevents cardiovascular events: The mean LDL at 1 year was 53.7 mg/dL in the dual-treatment group and 69.9 mg/dL in the simvastatin arm. And, as Dr. Karol Watson noted, IMPROVE-IT, with 7-plus years of careful follow-up, confirms the safety of ezetimibe, which had been questioned earlier on the basis of smaller studies that were far smaller.
“That’s one of the most important things to me to come from this study,” said Dr. Watson, professor of medicine and codirector of the program in preventive cardiology at the University of California, Los Angeles.
Once IMPROVE-IT has been published in a peer-reviewed journal it will be reviewed by the expert panel responsible for the AHA/American College of Cardiology cholesterol management practice guidelines, which in 2013 famously abolished specific LDL target goals.
Just what impact IMPROVE-IT ought to have on the guidelines was a point of considerable contention at the Chicago AHA meeting. Late-breaking clinical trials session cochair Dr. Sidney C. Smith noted that the current guidelines (J. Am. Coll. Cardiol. 2014;63:2889-934), on the basis of very solid evidence, call for high-intensity statin therapy, such as atorvastatin at 80 mg/day, as the first-line therapy for high-risk patients such as those with acute coronary syndrome.

“There are people who can’t take atorvastatin at 80 mg or atorvastatin at all. It seems to me that these IMPROVE-IT data fit very nicely with the guidelines: In such patients, you would start with 40 mg of simvastatin along with ezetimibe because of the proven outcome you’ve demonstrated,” suggested Dr. Smith, professor of medicine at the University of North Carolina, Chapel Hill.
Dr. Cannon indicated that he sees a broader role than that for statin/ezetimibe combination therapy. Since IMPROVE-IT has now shown that the lower a patient’s LDL the better the clinical outcomes, it makes sense to consider starting high-risk patients on maximal statin therapy – atorvastatin 80 mg/day or its equivalent – along with ezetimibe, he said.
“If you’re starting out at a high baseline risk, you might go straight to both. That’s what I hope the revised guidelines will say. Whereas, if someone was at a more moderate baseline LDL level they might get a very good response with just the high-intensity statin,” the cardiologist asserted.
Dr. Neil J. Stone, chair of the cholesterol guidelines panel, wasn’t buying that argument.
“That strategy simply wasn’t tested in terms of safety and efficacy in this trial. I’m not willing to make a leap of faith. I’d like to stay as close to the evidence as we can,” said Dr. Stone, professor of medicine at Northwestern University in Chicago. He cited several fairly recent examples in which lower proved not to be better in terms of LDL reduction: Niacin, fenofibrate, estrogen/progestin for women, and torcetrapib all went down in flames in randomized trials.

“If we had made a leap of faith that lower is better, we wouldn’t have done those trials,” he observed.
Also, it’s important to recognize that the IMPROVE-IT data don’t speak to the use of ezetimibe for primary prevention in lower-risk patients. “This study is not a signal that everyone should be on ezetimibe,” Dr. Stone emphasized.
It’s estimated that, in clinical practice, up to 25% of patients are statin intolerant to varying degrees. Dr. Watson said that, after seeing the IMPROVE-IT results, “I would absolutely try to persist with statin therapy, but there are some patients who are truly statin intolerant, and for them I might add ezetimibe to a low-dose statin.”
Indeed, like many other physicians, she has been doing that all along in selected cases on the basis of clinical judgment and biologic plausibility while anxiously awaiting the IMPROVE-IT findings, which come as a great relief, she added.
IMPROVE-IT was initially planned to include 10,000 randomized patients and a minimum of 2.5 years of on-treatment follow-up. Over the years, however, it underwent four protocol amendments. As IMPROVE-IT kept growing larger and longer, some observers quipped that it appeared the organizers would just keep the trial going until it produced a positive result, although, in fact, the study leaders remained blinded to the interim safety and efficacy data.
Merck, which sponsored the study, announced that it will use the data in petitioning the Food and Drug Administration next year for a new indication for reduction of major cardiovascular events for Vytorin and ezetimibe (Zetia).
Dr. Cannon reported receiving research grants from Merck and half a dozen other pharmaceutical companies. Dr. Watson reported participating in the clinical trials adjudication committee for Merck. Dr. Smith and Dr. Stone had no disclosures.
*Correction, 11/18/14: An earlier version of this article carried an incorrect photo credit.
CHICAGO – Adding ezetimibe to a midpotency statin in high-risk cardiovascular patients to drive their LDL cholesterol into the low 50s resulted in a further significant reduction in major cardiovascular events, compared with statin alone, in the 18,144-patient IMPROVE-IT study.
In IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial), 7 years of double-blind therapy with ezetimibe 10 mg/simvastatin 40 mg (Vytorin) achieved a 6.4% lower relative risk of the primary composite endpoint, compared with patients randomized to simvastatin alone (P = 0.016).
The 7-year rate of this composite outcome – comprised of cardiovascular death, nonfatal MI, rehospitalization for unstable angina, stroke, or coronary revascularization – was 32.7% with ezetimibe/simvastatin and 34.7% with simvastatin alone, for an absolute difference of 2%, Dr. Christopher P. Cannon reported at the American Heart Association scientific sessions.
Patients on dual therapy showed relative risk reductions of 13% for MI and 21% for ischemic stroke. The number needed to treat was 50, meaning that treating 100 patients with ezetimibe/simvastatin for 7 years should prevent 2 patients from having an ischemic stroke or MI who would have had such an event on simvastatin alone. That NNT is “well within the range of our standard therapies,” noted Dr. Cannon, professor of medicine at Harvard Medical School, Boston.
Patients in IMPROVE-IT began double-blind therapy within 10 days of hospitalization for an ST-elevation MI or non-STEMI/unstable angina. Participants also had to have at least one additional high-risk feature, such as a previous MI, diabetes, or coronary artery bypass surgery.
Observers hailed IMPROVE-IT as a major study for several reasons. It’s the first-ever study to demonstrate added clinical benefit when a nonstatin having a different mechanism of action is added to a statin. Thus, it provides physicians with a new, solidly evidence-based treatment option for high-risk acute coronary syndrome patients. Also, the trial emphatically affirms the LDL hypothesis that lowering LDL prevents cardiovascular events: The mean LDL at 1 year was 53.7 mg/dL in the dual-treatment group and 69.9 mg/dL in the simvastatin arm. And, as Dr. Karol Watson noted, IMPROVE-IT, with 7-plus years of careful follow-up, confirms the safety of ezetimibe, which had been questioned earlier on the basis of smaller studies that were far smaller.
“That’s one of the most important things to me to come from this study,” said Dr. Watson, professor of medicine and codirector of the program in preventive cardiology at the University of California, Los Angeles.
Once IMPROVE-IT has been published in a peer-reviewed journal it will be reviewed by the expert panel responsible for the AHA/American College of Cardiology cholesterol management practice guidelines, which in 2013 famously abolished specific LDL target goals.
Just what impact IMPROVE-IT ought to have on the guidelines was a point of considerable contention at the Chicago AHA meeting. Late-breaking clinical trials session cochair Dr. Sidney C. Smith noted that the current guidelines (J. Am. Coll. Cardiol. 2014;63:2889-934), on the basis of very solid evidence, call for high-intensity statin therapy, such as atorvastatin at 80 mg/day, as the first-line therapy for high-risk patients such as those with acute coronary syndrome.
“There are people who can’t take atorvastatin at 80 mg or atorvastatin at all. It seems to me that these IMPROVE-IT data fit very nicely with the guidelines: In such patients, you would start with 40 mg of simvastatin along with ezetimibe because of the proven outcome you’ve demonstrated,” suggested Dr. Smith, professor of medicine at the University of North Carolina, Chapel Hill.
Dr. Cannon indicated that he sees a broader role than that for statin/ezetimibe combination therapy. Since IMPROVE-IT has now shown that the lower a patient’s LDL the better the clinical outcomes, it makes sense to consider starting high-risk patients on maximal statin therapy – atorvastatin 80 mg/day or its equivalent – along with ezetimibe, he said.
“If you’re starting out at a high baseline risk, you might go straight to both. That’s what I hope the revised guidelines will say. Whereas, if someone was at a more moderate baseline LDL level they might get a very good response with just the high-intensity statin,” the cardiologist asserted.
Dr. Neil J. Stone, chair of the cholesterol guidelines panel, wasn’t buying that argument.
“That strategy simply wasn’t tested in terms of safety and efficacy in this trial. I’m not willing to make a leap of faith. I’d like to stay as close to the evidence as we can,” said Dr. Stone, professor of medicine at Northwestern University in Chicago. He cited several fairly recent examples in which lower proved not to be better in terms of LDL reduction: Niacin, fenofibrate, estrogen/progestin for women, and torcetrapib all went down in flames in randomized trials.
“If we had made a leap of faith that lower is better, we wouldn’t have done those trials,” he observed.
Also, it’s important to recognize that the IMPROVE-IT data don’t speak to the use of ezetimibe for primary prevention in lower-risk patients. “This study is not a signal that everyone should be on ezetimibe,” Dr. Stone emphasized.
It’s estimated that, in clinical practice, up to 25% of patients are statin intolerant to varying degrees. Dr. Watson said that, after seeing the IMPROVE-IT results, “I would absolutely try to persist with statin therapy, but there are some patients who are truly statin intolerant, and for them I might add ezetimibe to a low-dose statin.”
Indeed, like many other physicians, she has been doing that all along in selected cases on the basis of clinical judgment and biologic plausibility while anxiously awaiting the IMPROVE-IT findings, which come as a great relief, she added.
IMPROVE-IT was initially planned to include 10,000 randomized patients and a minimum of 2.5 years of on-treatment follow-up. Over the years, however, it underwent four protocol amendments. As IMPROVE-IT kept growing larger and longer, some observers quipped that it appeared the organizers would just keep the trial going until it produced a positive result, although, in fact, the study leaders remained blinded to the interim safety and efficacy data.
Merck, which sponsored the study, announced that it will use the data in petitioning the Food and Drug Administration next year for a new indication for reduction of major cardiovascular events for Vytorin and ezetimibe (Zetia).
Dr. Cannon reported receiving research grants from Merck and half a dozen other pharmaceutical companies. Dr. Watson reported participating in the clinical trials adjudication committee for Merck. Dr. Smith and Dr. Stone had no disclosures.
*Correction, 11/18/14: An earlier version of this article carried an incorrect photo credit.
CHICAGO – Adding ezetimibe to a midpotency statin in high-risk cardiovascular patients to drive their LDL cholesterol into the low 50s resulted in a further significant reduction in major cardiovascular events, compared with statin alone, in the 18,144-patient IMPROVE-IT study.
In IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial), 7 years of double-blind therapy with ezetimibe 10 mg/simvastatin 40 mg (Vytorin) achieved a 6.4% lower relative risk of the primary composite endpoint, compared with patients randomized to simvastatin alone (P = 0.016).
The 7-year rate of this composite outcome – comprised of cardiovascular death, nonfatal MI, rehospitalization for unstable angina, stroke, or coronary revascularization – was 32.7% with ezetimibe/simvastatin and 34.7% with simvastatin alone, for an absolute difference of 2%, Dr. Christopher P. Cannon reported at the American Heart Association scientific sessions.
Patients on dual therapy showed relative risk reductions of 13% for MI and 21% for ischemic stroke. The number needed to treat was 50, meaning that treating 100 patients with ezetimibe/simvastatin for 7 years should prevent 2 patients from having an ischemic stroke or MI who would have had such an event on simvastatin alone. That NNT is “well within the range of our standard therapies,” noted Dr. Cannon, professor of medicine at Harvard Medical School, Boston.
Patients in IMPROVE-IT began double-blind therapy within 10 days of hospitalization for an ST-elevation MI or non-STEMI/unstable angina. Participants also had to have at least one additional high-risk feature, such as a previous MI, diabetes, or coronary artery bypass surgery.
Observers hailed IMPROVE-IT as a major study for several reasons. It’s the first-ever study to demonstrate added clinical benefit when a nonstatin having a different mechanism of action is added to a statin. Thus, it provides physicians with a new, solidly evidence-based treatment option for high-risk acute coronary syndrome patients. Also, the trial emphatically affirms the LDL hypothesis that lowering LDL prevents cardiovascular events: The mean LDL at 1 year was 53.7 mg/dL in the dual-treatment group and 69.9 mg/dL in the simvastatin arm. And, as Dr. Karol Watson noted, IMPROVE-IT, with 7-plus years of careful follow-up, confirms the safety of ezetimibe, which had been questioned earlier on the basis of smaller studies that were far smaller.
“That’s one of the most important things to me to come from this study,” said Dr. Watson, professor of medicine and codirector of the program in preventive cardiology at the University of California, Los Angeles.
Once IMPROVE-IT has been published in a peer-reviewed journal it will be reviewed by the expert panel responsible for the AHA/American College of Cardiology cholesterol management practice guidelines, which in 2013 famously abolished specific LDL target goals.
Just what impact IMPROVE-IT ought to have on the guidelines was a point of considerable contention at the Chicago AHA meeting. Late-breaking clinical trials session cochair Dr. Sidney C. Smith noted that the current guidelines (J. Am. Coll. Cardiol. 2014;63:2889-934), on the basis of very solid evidence, call for high-intensity statin therapy, such as atorvastatin at 80 mg/day, as the first-line therapy for high-risk patients such as those with acute coronary syndrome.
“There are people who can’t take atorvastatin at 80 mg or atorvastatin at all. It seems to me that these IMPROVE-IT data fit very nicely with the guidelines: In such patients, you would start with 40 mg of simvastatin along with ezetimibe because of the proven outcome you’ve demonstrated,” suggested Dr. Smith, professor of medicine at the University of North Carolina, Chapel Hill.
Dr. Cannon indicated that he sees a broader role than that for statin/ezetimibe combination therapy. Since IMPROVE-IT has now shown that the lower a patient’s LDL the better the clinical outcomes, it makes sense to consider starting high-risk patients on maximal statin therapy – atorvastatin 80 mg/day or its equivalent – along with ezetimibe, he said.
“If you’re starting out at a high baseline risk, you might go straight to both. That’s what I hope the revised guidelines will say. Whereas, if someone was at a more moderate baseline LDL level they might get a very good response with just the high-intensity statin,” the cardiologist asserted.
Dr. Neil J. Stone, chair of the cholesterol guidelines panel, wasn’t buying that argument.
“That strategy simply wasn’t tested in terms of safety and efficacy in this trial. I’m not willing to make a leap of faith. I’d like to stay as close to the evidence as we can,” said Dr. Stone, professor of medicine at Northwestern University in Chicago. He cited several fairly recent examples in which lower proved not to be better in terms of LDL reduction: Niacin, fenofibrate, estrogen/progestin for women, and torcetrapib all went down in flames in randomized trials.
“If we had made a leap of faith that lower is better, we wouldn’t have done those trials,” he observed.
Also, it’s important to recognize that the IMPROVE-IT data don’t speak to the use of ezetimibe for primary prevention in lower-risk patients. “This study is not a signal that everyone should be on ezetimibe,” Dr. Stone emphasized.
It’s estimated that, in clinical practice, up to 25% of patients are statin intolerant to varying degrees. Dr. Watson said that, after seeing the IMPROVE-IT results, “I would absolutely try to persist with statin therapy, but there are some patients who are truly statin intolerant, and for them I might add ezetimibe to a low-dose statin.”
Indeed, like many other physicians, she has been doing that all along in selected cases on the basis of clinical judgment and biologic plausibility while anxiously awaiting the IMPROVE-IT findings, which come as a great relief, she added.
IMPROVE-IT was initially planned to include 10,000 randomized patients and a minimum of 2.5 years of on-treatment follow-up. Over the years, however, it underwent four protocol amendments. As IMPROVE-IT kept growing larger and longer, some observers quipped that it appeared the organizers would just keep the trial going until it produced a positive result, although, in fact, the study leaders remained blinded to the interim safety and efficacy data.
Merck, which sponsored the study, announced that it will use the data in petitioning the Food and Drug Administration next year for a new indication for reduction of major cardiovascular events for Vytorin and ezetimibe (Zetia).
Dr. Cannon reported receiving research grants from Merck and half a dozen other pharmaceutical companies. Dr. Watson reported participating in the clinical trials adjudication committee for Merck. Dr. Smith and Dr. Stone had no disclosures.
*Correction, 11/18/14: An earlier version of this article carried an incorrect photo credit.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: Combined therapy with ezetimibe and simvastatin reduces cardiovascular events more than simvastatin alone in high-risk patients.
Major finding: The number needed to treat with the combination therapy to prevent one MI or ischemic stroke was 50 patients for 7 years.
Data source: IMPROVE-IT was a randomized, double-blind study involving 18,144 patients with acute coronary syndrome at 1,158 centers in 39 countries.
Disclosures: The study was sponsored by Merck. The presenter reported having received research grants from Merck and several other pharmaceutical companies.

VIDEO: IMPROVE-IT exonerates ezetimibe, proves LDL hypothesis
CHICAGO – Results of the IMPROVE-IT megatrial not only show that adding ezetimibe to statin therapy lowered the risk of all cardiovascular events in high-risk patients, they also prove the LDL hypothesis, two experts commented at the American Heart Association scientific sessions.
IMPROVE-IT compared simvastatin plus placebo with simvastatin plus ezetimibe in more than 18,000 patients within 10 days of an acute coronary syndrome event. The dual-therapy patients’ LDL cholesterol went down to an average of 54 mg/dL, while the statin/placebo patients’ LDL reached 69 mg/dL.
The patients taking ezetimibe in addition to simvastatin had a 6.4% lower risk of all cardiovascular events, a 14% reduced risk of all heart attacks, and a 21% lower risk of ischemic stroke. There was no difference in mortality between groups.
Dr. Lori Mosca, professor of medicine at Columbia University, New York, and director of preventive cardiology at New York-Presbyterian Hospital, and Dr. Prakash Deedwania, professor of medicine at the University of California, San Francisco, Fresno, told us how these “exciting” results will change practice for treating very high-risk patients.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
CHICAGO – Results of the IMPROVE-IT megatrial not only show that adding ezetimibe to statin therapy lowered the risk of all cardiovascular events in high-risk patients, they also prove the LDL hypothesis, two experts commented at the American Heart Association scientific sessions.
IMPROVE-IT compared simvastatin plus placebo with simvastatin plus ezetimibe in more than 18,000 patients within 10 days of an acute coronary syndrome event. The dual-therapy patients’ LDL cholesterol went down to an average of 54 mg/dL, while the statin/placebo patients’ LDL reached 69 mg/dL.
The patients taking ezetimibe in addition to simvastatin had a 6.4% lower risk of all cardiovascular events, a 14% reduced risk of all heart attacks, and a 21% lower risk of ischemic stroke. There was no difference in mortality between groups.
Dr. Lori Mosca, professor of medicine at Columbia University, New York, and director of preventive cardiology at New York-Presbyterian Hospital, and Dr. Prakash Deedwania, professor of medicine at the University of California, San Francisco, Fresno, told us how these “exciting” results will change practice for treating very high-risk patients.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
CHICAGO – Results of the IMPROVE-IT megatrial not only show that adding ezetimibe to statin therapy lowered the risk of all cardiovascular events in high-risk patients, they also prove the LDL hypothesis, two experts commented at the American Heart Association scientific sessions.
IMPROVE-IT compared simvastatin plus placebo with simvastatin plus ezetimibe in more than 18,000 patients within 10 days of an acute coronary syndrome event. The dual-therapy patients’ LDL cholesterol went down to an average of 54 mg/dL, while the statin/placebo patients’ LDL reached 69 mg/dL.
The patients taking ezetimibe in addition to simvastatin had a 6.4% lower risk of all cardiovascular events, a 14% reduced risk of all heart attacks, and a 21% lower risk of ischemic stroke. There was no difference in mortality between groups.
Dr. Lori Mosca, professor of medicine at Columbia University, New York, and director of preventive cardiology at New York-Presbyterian Hospital, and Dr. Prakash Deedwania, professor of medicine at the University of California, San Francisco, Fresno, told us how these “exciting” results will change practice for treating very high-risk patients.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Increased Alzheimer’s risk with long-term benzodiazepine use
BERLIN – Chronic use of benzodiazepines by elderly patients is associated with a 43%-51% increased risk of being diagnosed with Alzheimer’s disease 5-10 years later, according to a large case-control study. “Considering the extent to which benzodiazepines are prescribed in the elderly population and the growing incidence of dementia, unwarranted chronic use of benzodiazepines in the elderly should be viewed as a public health issue,” Sophie Billioti de Gage said at the annual congress of the European College of Neuropsychopharmacology.
Her case-control study used the Quebec health insurance database. The subjects were 1,796 elderly individuals diagnosed with Alzheimer’s disease, each matched with 4 controls based upon age, gender, and duration of follow-up. The Quebec database permitted identification of all subjects with prescriptions for benzodiazepines during 2000-2009, a period 5-10 years prior to diagnosis of Alzheimer’s disease. This substantial time lag was chosen because previous studies reporting a link between benzodiazepines and dementia have often been criticized for possible confounding due to reverse causality. That is, because many earlier studies featured a shorter interval between medication use and dementia diagnosis, skeptics argued that benzodiazepines might not have caused the dementia but rather were prescribed to treat early manifestations of the disease, such as anxiety, depressive symptoms, and insomnia, explained Ms. Billioti de Gage, a PhD student at the University of Bordeaux (France).

The database enabled her to determine how many cumulative days worth of benzodiazepine prescriptions participants filled during the study years, as well as whether the medications had a short or long half-life.
In a multivariate analysis adjusted for stroke, MI, hypertension, diabetes, and use of antiplatelet agents or anticoagulants during the period 5-10 years prior to diagnosis of Alzheimer’s disease, the use of benzodiazepines was independently associated with a 51% increased risk of subsequent Alzheimer’s compared with nonusers.
A dose-response relationship was apparent. Individuals with prescriptions for up to 90 days worth of benzodiazepines were not at significantly greater risk for future Alzheimer’s disease than nonusers. Those with a cumulative 91- to 180-day exposure had a 32% increased risk, compared with nonusers, however, and patients with more than 180 days of benzodiazepine use had an 84% increased risk.
The association with later Alzheimer’s disease was stronger in patients who used benzodiazepines with a half-life of 20 hours or longer. In a multivariate analysis they had a 70% greater risk of Alzheimer’s, compared with nonusers. Patients who used a benzodiazepine having a half-life of less than 20 hours had a 43% increase in risk.
When the multivariate analyses were further adjusted for anxiety, depressive symptoms, and insomnia – all of which can be prodromes of dementia – the results were not meaningfully altered, she added.
This case-control study confirms the results of an earlier prospective population-based study by Ms. Billioti de Gage and coworkers. That French study also found a roughly 50% increased risk of dementia in elderly patients who initiated chronic benzodiazepine therapy (BMJ 2012;345:e6231 [doi: 10.1136/bmj.e6231]). However, with only 1,063 participants, 253 of whom were diagnosed with dementia during 15 years of follow-up, the sample size was too small to draw firm conclusions. The current case-control study, with 8,980 subjects representative of the Quebec community-dwelling elderly population, is more persuasive, the investigator said.
Guidelines recommend preferential use of short half-life benzodiazepines and short durations of use in the elderly; however, in clinical practice the medications are often used long-term.
The biologic mechanism by which chronic use of benzodiazepines by elderly individuals might predispose to Alzheimer’s disease hasn’t been worked out, but the medications’ short-term adverse impact on memory and cognition are well recognized.
The case-control study was funded by the French Ministry of Health and the Funding Agency for Health Research of Quebec as well as by university grants.
BERLIN – Chronic use of benzodiazepines by elderly patients is associated with a 43%-51% increased risk of being diagnosed with Alzheimer’s disease 5-10 years later, according to a large case-control study. “Considering the extent to which benzodiazepines are prescribed in the elderly population and the growing incidence of dementia, unwarranted chronic use of benzodiazepines in the elderly should be viewed as a public health issue,” Sophie Billioti de Gage said at the annual congress of the European College of Neuropsychopharmacology.
Her case-control study used the Quebec health insurance database. The subjects were 1,796 elderly individuals diagnosed with Alzheimer’s disease, each matched with 4 controls based upon age, gender, and duration of follow-up. The Quebec database permitted identification of all subjects with prescriptions for benzodiazepines during 2000-2009, a period 5-10 years prior to diagnosis of Alzheimer’s disease. This substantial time lag was chosen because previous studies reporting a link between benzodiazepines and dementia have often been criticized for possible confounding due to reverse causality. That is, because many earlier studies featured a shorter interval between medication use and dementia diagnosis, skeptics argued that benzodiazepines might not have caused the dementia but rather were prescribed to treat early manifestations of the disease, such as anxiety, depressive symptoms, and insomnia, explained Ms. Billioti de Gage, a PhD student at the University of Bordeaux (France).
The database enabled her to determine how many cumulative days worth of benzodiazepine prescriptions participants filled during the study years, as well as whether the medications had a short or long half-life.
In a multivariate analysis adjusted for stroke, MI, hypertension, diabetes, and use of antiplatelet agents or anticoagulants during the period 5-10 years prior to diagnosis of Alzheimer’s disease, the use of benzodiazepines was independently associated with a 51% increased risk of subsequent Alzheimer’s compared with nonusers.
A dose-response relationship was apparent. Individuals with prescriptions for up to 90 days worth of benzodiazepines were not at significantly greater risk for future Alzheimer’s disease than nonusers. Those with a cumulative 91- to 180-day exposure had a 32% increased risk, compared with nonusers, however, and patients with more than 180 days of benzodiazepine use had an 84% increased risk.
The association with later Alzheimer’s disease was stronger in patients who used benzodiazepines with a half-life of 20 hours or longer. In a multivariate analysis they had a 70% greater risk of Alzheimer’s, compared with nonusers. Patients who used a benzodiazepine having a half-life of less than 20 hours had a 43% increase in risk.
When the multivariate analyses were further adjusted for anxiety, depressive symptoms, and insomnia – all of which can be prodromes of dementia – the results were not meaningfully altered, she added.
This case-control study confirms the results of an earlier prospective population-based study by Ms. Billioti de Gage and coworkers. That French study also found a roughly 50% increased risk of dementia in elderly patients who initiated chronic benzodiazepine therapy (BMJ 2012;345:e6231 [doi: 10.1136/bmj.e6231]). However, with only 1,063 participants, 253 of whom were diagnosed with dementia during 15 years of follow-up, the sample size was too small to draw firm conclusions. The current case-control study, with 8,980 subjects representative of the Quebec community-dwelling elderly population, is more persuasive, the investigator said.
Guidelines recommend preferential use of short half-life benzodiazepines and short durations of use in the elderly; however, in clinical practice the medications are often used long-term.
The biologic mechanism by which chronic use of benzodiazepines by elderly individuals might predispose to Alzheimer’s disease hasn’t been worked out, but the medications’ short-term adverse impact on memory and cognition are well recognized.
The case-control study was funded by the French Ministry of Health and the Funding Agency for Health Research of Quebec as well as by university grants.
BERLIN – Chronic use of benzodiazepines by elderly patients is associated with a 43%-51% increased risk of being diagnosed with Alzheimer’s disease 5-10 years later, according to a large case-control study. “Considering the extent to which benzodiazepines are prescribed in the elderly population and the growing incidence of dementia, unwarranted chronic use of benzodiazepines in the elderly should be viewed as a public health issue,” Sophie Billioti de Gage said at the annual congress of the European College of Neuropsychopharmacology.
Her case-control study used the Quebec health insurance database. The subjects were 1,796 elderly individuals diagnosed with Alzheimer’s disease, each matched with 4 controls based upon age, gender, and duration of follow-up. The Quebec database permitted identification of all subjects with prescriptions for benzodiazepines during 2000-2009, a period 5-10 years prior to diagnosis of Alzheimer’s disease. This substantial time lag was chosen because previous studies reporting a link between benzodiazepines and dementia have often been criticized for possible confounding due to reverse causality. That is, because many earlier studies featured a shorter interval between medication use and dementia diagnosis, skeptics argued that benzodiazepines might not have caused the dementia but rather were prescribed to treat early manifestations of the disease, such as anxiety, depressive symptoms, and insomnia, explained Ms. Billioti de Gage, a PhD student at the University of Bordeaux (France).
The database enabled her to determine how many cumulative days worth of benzodiazepine prescriptions participants filled during the study years, as well as whether the medications had a short or long half-life.
In a multivariate analysis adjusted for stroke, MI, hypertension, diabetes, and use of antiplatelet agents or anticoagulants during the period 5-10 years prior to diagnosis of Alzheimer’s disease, the use of benzodiazepines was independently associated with a 51% increased risk of subsequent Alzheimer’s compared with nonusers.
A dose-response relationship was apparent. Individuals with prescriptions for up to 90 days worth of benzodiazepines were not at significantly greater risk for future Alzheimer’s disease than nonusers. Those with a cumulative 91- to 180-day exposure had a 32% increased risk, compared with nonusers, however, and patients with more than 180 days of benzodiazepine use had an 84% increased risk.
The association with later Alzheimer’s disease was stronger in patients who used benzodiazepines with a half-life of 20 hours or longer. In a multivariate analysis they had a 70% greater risk of Alzheimer’s, compared with nonusers. Patients who used a benzodiazepine having a half-life of less than 20 hours had a 43% increase in risk.
When the multivariate analyses were further adjusted for anxiety, depressive symptoms, and insomnia – all of which can be prodromes of dementia – the results were not meaningfully altered, she added.
This case-control study confirms the results of an earlier prospective population-based study by Ms. Billioti de Gage and coworkers. That French study also found a roughly 50% increased risk of dementia in elderly patients who initiated chronic benzodiazepine therapy (BMJ 2012;345:e6231 [doi: 10.1136/bmj.e6231]). However, with only 1,063 participants, 253 of whom were diagnosed with dementia during 15 years of follow-up, the sample size was too small to draw firm conclusions. The current case-control study, with 8,980 subjects representative of the Quebec community-dwelling elderly population, is more persuasive, the investigator said.
Guidelines recommend preferential use of short half-life benzodiazepines and short durations of use in the elderly; however, in clinical practice the medications are often used long-term.
The biologic mechanism by which chronic use of benzodiazepines by elderly individuals might predispose to Alzheimer’s disease hasn’t been worked out, but the medications’ short-term adverse impact on memory and cognition are well recognized.
The case-control study was funded by the French Ministry of Health and the Funding Agency for Health Research of Quebec as well as by university grants.
AT THE ECNP CONGRESS
Key clinical point: Chronic use of benzodiazepines by the elderly may boost their risk of Alzheimer’s disease in a dose-dependent fashion.
Major finding: Individuals who used benzodiazepines for more than 180 days during a 6-year period had an 84% greater risk of being diagnosed with Alzheimer’s disease 5-10 years later than nonusers.
Data source: This was a case-control study involving 1,796 elderly Quebec residents diagnosed with Alzheimer’s disease and 7,184 matched controls.
Disclosures:This study was funded primarily by the French Ministry of Health and the Funding Agency for Health Research of Quebec.

Less daytime sleepiness with lurasidone for schizophrenia
BERLIN – Daytime sleepiness was reduced when patients with schizophrenia were placed on lurasidone but increased when assigned to quetiapine extended-release, in a randomized trial.
This improvement in daytime sleepiness in patients treated with lurasidone had important clinical consequences: namely, significantly better scores on measures of cognition and functional capacity, Dr. Antony Loebel reported at the annual congress of the European College of Neuropsychopharmacology.
He presented a post hoc analysis of a 6-week, double-blind, placebo-controlled randomized trial. The study population comprised 486 patients with an acute exacerbation of schizophrenia who were randomized to fixed-dose therapy with lurasidone (Latuda) 80 mg, lurasidone 160 mg, quetiapine extended release (Seroquel XR) 600 mg, or placebo, all dosed once daily with food in the evening.
Daytime sleepiness as assessed by the Epworth Sleepiness Scale (ESS) improved significantly from baseline in the two lurasidone groups and in placebo-treated controls while worsening significantly in the quetiapine group. The mean total ESS score decreased by 0.7 points in the lower-dose lurasidone group, 1.1 points in patients on lurasidone at 160 mg/day, and 0.9 points in controls. In contrast, the mean score rose by 0.6 points in the quetiapine group.
Daytime sleepiness was associated with reduced agitation as assessed by the PANSS (Positive and Negative Syndrome Scale) excitement subscale. However, this came at the cost of reduced functional capacity.
Specifically, the quetiapine group showed significantly increased sleepiness in five of the eight daytime scenarios assessed in the ESS: dozing when talking, sitting and reading, watching television, sitting quietly after lunch without alcohol, and during afternoon resting.
Moreover, as daytime sleepiness increased, functional capacity evaluated via the UCSD Performance-Based Skills Assessment, Brief, worsened, as did cognitive performance as assessed by the CogState Computerized Schizophrenia Battery, according to Dr. Loebel, executive vice president and chief medical officer at Sunovion Pharmaceuticals in Fort Lee, N.J.
Lurasidone at 160 mg/day not only yielded the greatest improvement from baseline in daytime sleepiness, it also demonstrated a significant improvement in overall cognitive performance when compared to quetiapine and placebo.
The psychiatrist concluded that the impact of daytime sleepiness on key treatment outcomes is underappreciated. Daytime sleepiness is generally not assessed with a validated scale. The ESS is a quick, practical, well-validated tool for this purpose that’s well suited for use in clinical practice, he added.
BERLIN – Daytime sleepiness was reduced when patients with schizophrenia were placed on lurasidone but increased when assigned to quetiapine extended-release, in a randomized trial.
This improvement in daytime sleepiness in patients treated with lurasidone had important clinical consequences: namely, significantly better scores on measures of cognition and functional capacity, Dr. Antony Loebel reported at the annual congress of the European College of Neuropsychopharmacology.
He presented a post hoc analysis of a 6-week, double-blind, placebo-controlled randomized trial. The study population comprised 486 patients with an acute exacerbation of schizophrenia who were randomized to fixed-dose therapy with lurasidone (Latuda) 80 mg, lurasidone 160 mg, quetiapine extended release (Seroquel XR) 600 mg, or placebo, all dosed once daily with food in the evening.
Daytime sleepiness as assessed by the Epworth Sleepiness Scale (ESS) improved significantly from baseline in the two lurasidone groups and in placebo-treated controls while worsening significantly in the quetiapine group. The mean total ESS score decreased by 0.7 points in the lower-dose lurasidone group, 1.1 points in patients on lurasidone at 160 mg/day, and 0.9 points in controls. In contrast, the mean score rose by 0.6 points in the quetiapine group.
Daytime sleepiness was associated with reduced agitation as assessed by the PANSS (Positive and Negative Syndrome Scale) excitement subscale. However, this came at the cost of reduced functional capacity.
Specifically, the quetiapine group showed significantly increased sleepiness in five of the eight daytime scenarios assessed in the ESS: dozing when talking, sitting and reading, watching television, sitting quietly after lunch without alcohol, and during afternoon resting.
Moreover, as daytime sleepiness increased, functional capacity evaluated via the UCSD Performance-Based Skills Assessment, Brief, worsened, as did cognitive performance as assessed by the CogState Computerized Schizophrenia Battery, according to Dr. Loebel, executive vice president and chief medical officer at Sunovion Pharmaceuticals in Fort Lee, N.J.
Lurasidone at 160 mg/day not only yielded the greatest improvement from baseline in daytime sleepiness, it also demonstrated a significant improvement in overall cognitive performance when compared to quetiapine and placebo.
The psychiatrist concluded that the impact of daytime sleepiness on key treatment outcomes is underappreciated. Daytime sleepiness is generally not assessed with a validated scale. The ESS is a quick, practical, well-validated tool for this purpose that’s well suited for use in clinical practice, he added.
BERLIN – Daytime sleepiness was reduced when patients with schizophrenia were placed on lurasidone but increased when assigned to quetiapine extended-release, in a randomized trial.
This improvement in daytime sleepiness in patients treated with lurasidone had important clinical consequences: namely, significantly better scores on measures of cognition and functional capacity, Dr. Antony Loebel reported at the annual congress of the European College of Neuropsychopharmacology.
He presented a post hoc analysis of a 6-week, double-blind, placebo-controlled randomized trial. The study population comprised 486 patients with an acute exacerbation of schizophrenia who were randomized to fixed-dose therapy with lurasidone (Latuda) 80 mg, lurasidone 160 mg, quetiapine extended release (Seroquel XR) 600 mg, or placebo, all dosed once daily with food in the evening.
Daytime sleepiness as assessed by the Epworth Sleepiness Scale (ESS) improved significantly from baseline in the two lurasidone groups and in placebo-treated controls while worsening significantly in the quetiapine group. The mean total ESS score decreased by 0.7 points in the lower-dose lurasidone group, 1.1 points in patients on lurasidone at 160 mg/day, and 0.9 points in controls. In contrast, the mean score rose by 0.6 points in the quetiapine group.
Daytime sleepiness was associated with reduced agitation as assessed by the PANSS (Positive and Negative Syndrome Scale) excitement subscale. However, this came at the cost of reduced functional capacity.
Specifically, the quetiapine group showed significantly increased sleepiness in five of the eight daytime scenarios assessed in the ESS: dozing when talking, sitting and reading, watching television, sitting quietly after lunch without alcohol, and during afternoon resting.
Moreover, as daytime sleepiness increased, functional capacity evaluated via the UCSD Performance-Based Skills Assessment, Brief, worsened, as did cognitive performance as assessed by the CogState Computerized Schizophrenia Battery, according to Dr. Loebel, executive vice president and chief medical officer at Sunovion Pharmaceuticals in Fort Lee, N.J.
Lurasidone at 160 mg/day not only yielded the greatest improvement from baseline in daytime sleepiness, it also demonstrated a significant improvement in overall cognitive performance when compared to quetiapine and placebo.
The psychiatrist concluded that the impact of daytime sleepiness on key treatment outcomes is underappreciated. Daytime sleepiness is generally not assessed with a validated scale. The ESS is a quick, practical, well-validated tool for this purpose that’s well suited for use in clinical practice, he added.
AT THE ECNP CONGRESS
Key clinical point: Patients with an acute exacerbation of schizophrenia treated with lurasidone experience less sleepiness during waking hours, with associated significant improvements in functional capacity and cognition, compared with patients on quetiapine extended release.
Major finding: Patients on lurasidone at 160 mg/day showed a mean 1.1-point improvement on the Epworth Sleepiness Scale total score, compared with a 0.6-point worsening on quetiapine extended release at 600 mg/day.
Data source: A post hoc analysis of a 486-patient, 6-week, randomized, double-blind, placebo-controlled clinical trial.
Disclosures: The presenter is chief medical officer of Sunovion Pharmaceuticals, which sponsored the study.
Certolizumab achieves sustained skin improvement in psoriatic arthritis
AMSTERDAM – Certolizumab pegol maintained significant improvement in dermatologic outcomes in psoriatic arthritis patients through 96 weeks of treatment in the phase III RAPID-PsA trial.
Moreover, the safety profile of this tumor necrosis factor (TNF) inhibitor was in line with findings from shorter-term studies, including the week 24 report from RAPID-PsA. Treatment-emergent adverse events were similar in type and frequency to those in placebo-treated controls, with the exception of an increased rate of minor upper respiratory tract infections. No cases of tuberculosis occurred.
“There were no new safety issues despite the increased exposure time out to 96 weeks,” Dr. Owen Davies reported at the annual congress of the European Academy of Dermatology and Venereology.
RAPID-PsA is an ongoing 216-week phase III study. It was double-blind and placebo-controlled through the first 24 weeks. The study started out with 409 psoriatic arthritis patients, half of whom had previously failed to response to one nonbiologic disease-modifying antirheumatic drug (DMARD), while the other half had been nonresponders to two or more. Of the 273 patients placed on certolizumab, 80% completed both 48 and 96 weeks of the study, explained Dr. Davies of UCB Pharma in Slough, England.
He focused on the dermatologic outcomes because the arthritis outcomes have previously been reported and served to support certolizumab’s regulatory approval for the treatment of psoriatic arthritis. The biologic is also approved for treatment of rheumatoid arthritis, ankylosing spondylitis, and Crohn’s disease. However, certolizumab’s durability of effect on the psoriatic skin manifestations of psoriatic arthritis hasn’t previously been addressed.
Briefly, at week 12 – the primary endpoint for the joint-related outcomes – 55% of patients achieved an ACR 20 response, compared with 24% on placebo. Moreover, 35% of certolizumab-treated patients had an ACR 50 response at that point, and 20% had an ACR 70 response. The ACR response rate in certolizumab-treated patients was similar regardless of whether or not they had previously been on another anti-TNF biologic.
Dr. Davies addressed in detail the dermatologic outcomes in the 166 psoriatic arthritis patients with at least 3% psoriasis body surface area involvement at baseline. They had an average 10-year disease duration, 24% body surface area involvement, and a baseline Psoriasis Area Severity Index (PASI) score of 12.0.
Dermatologic responses to certolizumab were comparable regardless of whether patients had been randomized to the biologic at 200 mg subcutaneously every 2 weeks or 400 mg once every 4 weeks. As was true for the joint-related responses to certolizumab, the skin responses were similar both in anti-TNF–naive and anti-TNF–experienced patients, he noted.
The PASI 75 response rate in this group of psoriatic arthritis patients with significant skin involvement was 61% at week 24, 65% at week 48, and 53% at week 96. The improvement was even greater in the 71 patients with more severe skin involvement as defined by a baseline PASI score of 10 or more. Certolizumab-treated patients also showed important improvements on the Physician Global Assessment and Dermatology Life Quality Index.
Certolizumab is a pegylated Fab’ fragment of a humanized TNF inhibitor monoclonal antibody.
“Certolizumab is structurally different from other currently available anti-TNF agents, which are either IgG1 monoclonal antibodies or, in the case of etanercept, a receptor fusion protein. Whether or not these structural differences will translate into clinical differences is a question being addressed in ongoing clinical trials,” Dr. Davies said.
The RAPID-PsA study is sponsored by UCB Pharma, where Dr. Davies is employed.
AMSTERDAM – Certolizumab pegol maintained significant improvement in dermatologic outcomes in psoriatic arthritis patients through 96 weeks of treatment in the phase III RAPID-PsA trial.
Moreover, the safety profile of this tumor necrosis factor (TNF) inhibitor was in line with findings from shorter-term studies, including the week 24 report from RAPID-PsA. Treatment-emergent adverse events were similar in type and frequency to those in placebo-treated controls, with the exception of an increased rate of minor upper respiratory tract infections. No cases of tuberculosis occurred.
“There were no new safety issues despite the increased exposure time out to 96 weeks,” Dr. Owen Davies reported at the annual congress of the European Academy of Dermatology and Venereology.
RAPID-PsA is an ongoing 216-week phase III study. It was double-blind and placebo-controlled through the first 24 weeks. The study started out with 409 psoriatic arthritis patients, half of whom had previously failed to response to one nonbiologic disease-modifying antirheumatic drug (DMARD), while the other half had been nonresponders to two or more. Of the 273 patients placed on certolizumab, 80% completed both 48 and 96 weeks of the study, explained Dr. Davies of UCB Pharma in Slough, England.
He focused on the dermatologic outcomes because the arthritis outcomes have previously been reported and served to support certolizumab’s regulatory approval for the treatment of psoriatic arthritis. The biologic is also approved for treatment of rheumatoid arthritis, ankylosing spondylitis, and Crohn’s disease. However, certolizumab’s durability of effect on the psoriatic skin manifestations of psoriatic arthritis hasn’t previously been addressed.
Briefly, at week 12 – the primary endpoint for the joint-related outcomes – 55% of patients achieved an ACR 20 response, compared with 24% on placebo. Moreover, 35% of certolizumab-treated patients had an ACR 50 response at that point, and 20% had an ACR 70 response. The ACR response rate in certolizumab-treated patients was similar regardless of whether or not they had previously been on another anti-TNF biologic.
Dr. Davies addressed in detail the dermatologic outcomes in the 166 psoriatic arthritis patients with at least 3% psoriasis body surface area involvement at baseline. They had an average 10-year disease duration, 24% body surface area involvement, and a baseline Psoriasis Area Severity Index (PASI) score of 12.0.
Dermatologic responses to certolizumab were comparable regardless of whether patients had been randomized to the biologic at 200 mg subcutaneously every 2 weeks or 400 mg once every 4 weeks. As was true for the joint-related responses to certolizumab, the skin responses were similar both in anti-TNF–naive and anti-TNF–experienced patients, he noted.
The PASI 75 response rate in this group of psoriatic arthritis patients with significant skin involvement was 61% at week 24, 65% at week 48, and 53% at week 96. The improvement was even greater in the 71 patients with more severe skin involvement as defined by a baseline PASI score of 10 or more. Certolizumab-treated patients also showed important improvements on the Physician Global Assessment and Dermatology Life Quality Index.
Certolizumab is a pegylated Fab’ fragment of a humanized TNF inhibitor monoclonal antibody.
“Certolizumab is structurally different from other currently available anti-TNF agents, which are either IgG1 monoclonal antibodies or, in the case of etanercept, a receptor fusion protein. Whether or not these structural differences will translate into clinical differences is a question being addressed in ongoing clinical trials,” Dr. Davies said.
The RAPID-PsA study is sponsored by UCB Pharma, where Dr. Davies is employed.
AMSTERDAM – Certolizumab pegol maintained significant improvement in dermatologic outcomes in psoriatic arthritis patients through 96 weeks of treatment in the phase III RAPID-PsA trial.
Moreover, the safety profile of this tumor necrosis factor (TNF) inhibitor was in line with findings from shorter-term studies, including the week 24 report from RAPID-PsA. Treatment-emergent adverse events were similar in type and frequency to those in placebo-treated controls, with the exception of an increased rate of minor upper respiratory tract infections. No cases of tuberculosis occurred.
“There were no new safety issues despite the increased exposure time out to 96 weeks,” Dr. Owen Davies reported at the annual congress of the European Academy of Dermatology and Venereology.
RAPID-PsA is an ongoing 216-week phase III study. It was double-blind and placebo-controlled through the first 24 weeks. The study started out with 409 psoriatic arthritis patients, half of whom had previously failed to response to one nonbiologic disease-modifying antirheumatic drug (DMARD), while the other half had been nonresponders to two or more. Of the 273 patients placed on certolizumab, 80% completed both 48 and 96 weeks of the study, explained Dr. Davies of UCB Pharma in Slough, England.
He focused on the dermatologic outcomes because the arthritis outcomes have previously been reported and served to support certolizumab’s regulatory approval for the treatment of psoriatic arthritis. The biologic is also approved for treatment of rheumatoid arthritis, ankylosing spondylitis, and Crohn’s disease. However, certolizumab’s durability of effect on the psoriatic skin manifestations of psoriatic arthritis hasn’t previously been addressed.
Briefly, at week 12 – the primary endpoint for the joint-related outcomes – 55% of patients achieved an ACR 20 response, compared with 24% on placebo. Moreover, 35% of certolizumab-treated patients had an ACR 50 response at that point, and 20% had an ACR 70 response. The ACR response rate in certolizumab-treated patients was similar regardless of whether or not they had previously been on another anti-TNF biologic.
Dr. Davies addressed in detail the dermatologic outcomes in the 166 psoriatic arthritis patients with at least 3% psoriasis body surface area involvement at baseline. They had an average 10-year disease duration, 24% body surface area involvement, and a baseline Psoriasis Area Severity Index (PASI) score of 12.0.
Dermatologic responses to certolizumab were comparable regardless of whether patients had been randomized to the biologic at 200 mg subcutaneously every 2 weeks or 400 mg once every 4 weeks. As was true for the joint-related responses to certolizumab, the skin responses were similar both in anti-TNF–naive and anti-TNF–experienced patients, he noted.
The PASI 75 response rate in this group of psoriatic arthritis patients with significant skin involvement was 61% at week 24, 65% at week 48, and 53% at week 96. The improvement was even greater in the 71 patients with more severe skin involvement as defined by a baseline PASI score of 10 or more. Certolizumab-treated patients also showed important improvements on the Physician Global Assessment and Dermatology Life Quality Index.
Certolizumab is a pegylated Fab’ fragment of a humanized TNF inhibitor monoclonal antibody.
“Certolizumab is structurally different from other currently available anti-TNF agents, which are either IgG1 monoclonal antibodies or, in the case of etanercept, a receptor fusion protein. Whether or not these structural differences will translate into clinical differences is a question being addressed in ongoing clinical trials,” Dr. Davies said.
The RAPID-PsA study is sponsored by UCB Pharma, where Dr. Davies is employed.
AT THE EADV CONGRESS
Key clinical point: Certolizumab pegol maintains sustained improvement in the dermatologic manifestations of psoriatic arthritis through 96 weeks.
Major finding: Sixty-two percent of psoriatic arthritis patients with at least 3% body surface area involvement at a baseline PASI score of 10 or more still had a PASI 75 dermatologic response after 96 weeks on certolizumab.
Data source: The RAPID-PsA study is an ongoing 216-week, prospective, randomized, multicenter trial involving 409 psoriatic arthritis patients, including 166 with significant skin involvement at baseline.
Disclosures: The study is sponsored by UCB Pharma. The presenter is a full-time company employee.
Telephone CPR training boosts cardiac arrest survival
CHICAGO – Systematic implementation of a comprehensive telephone CPR bundle of care targeting EMS dispatch services resulted in substantial improvements in rates of survival to hospital discharge with good neurologic outcomes in patients with out-of-hospital cardiac arrest in a major Arizona statewide public health initiative.
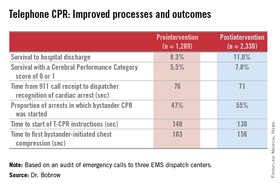
How big was the intervention’s impact? The rate of survival to hospital discharge showed a 33% relative increase compared to preintervention, and survival with a favorable Cerebral Performance Category score of 0 or 1 increased by 42%, Dr. Bentley J. Bobrow reported at the American Heart Association Scientific Sessions.
“These results suggest that when deliberately implemented and measured, telephone CPR is a targeted, effective method to increase bystander CPR and survival on a vast scale with minimal capital expense. This is why we believe telephone CPR along with public training may be the most efficient way to move the needle on cardiac arrest survival,” declared Dr. Bobrow, professor of emergency medicine at the University of Arizona College of Medicine-Phoenix Campus and chair of the AHA Basic Life Support Subcommittee.
Telephone CPR (T-CPR) entails the provision of CPR instruction to bystanders who have called 911 regarding an out of hospital cardiac arrest (OHCA). It’s well established that bystander CPR commenced before EMS personnel arrive on the scene doubles or even triples OHCA survival, but it is provided in only about one-third of OHCA events. And while T-CPR is independently associated with increased rates of bystander CPR as well as patient survival, its utilization varies widely throughout the country and few EMS services measure performance.
Dr. Bobrow reported on an ambitious undertaking that involved systematic training in T-CPR for dispatchers, 911 managers, and medical directors at all nine of the regional emergency dispatch centers in Arizona, which together with 190 EMS agencies and 40 cardiac care hospitals participate in a statewide resuscitation program.
The training was designed to implement the latest AHA guidelines on T-CPR (Circulation 2012;125:648-55). The program entailed a half-day in-person training session plus completion of a 1-hour web-based interactive video. The protocol emphasizes asking two key questions of the 911 caller: “Is the patient conscious?” and “Is the patient breathing normally?” If the response is no to both, the dispatcher is to start issuing bystander CPR instructions without delay – no further questions – and continue the coaching until EMS personnel arrive on the scene to take over.
A core aspect of the T-CPR bundle is performance measurement for quality improvement, with auditing of 911 calls to learn the time from the start of the call to the bystander’s first chest compression and five other key performance metrics. Feedback is provided to the 911 call center regarding system- and case-level performance reports in a continuing education, quality improvement process. Individual dispatchers are singled out for exemplary performance, Dr. Bobrow explained.
He presented a prospective before-and-after study conducted at the three EMS dispatch centers serving Arizona’s Maricopa County, home to two-thirds of the state’s population. The study entailed auditing nearly 6,000 911 calls, each averaging 6.5 minutes in length. After excluding calls where CPR wasn’t indicated or the OHCA involved a patient less than 8 years old, investigators were left with two groups for comparison comprised of 1,289 pre- and 2,330 post-intervention events.

The improvements in process and clinical outcomes were dramatic. In 2012, after introduction of the T-CPR training program, the bystander CPR rate crossed the 50% threshold for the first time ever in Maricopa County. The rate of survival of OHCA to hospital discharge improved from 8.3% to 11%, a highly statistically significant 33% relative increase. Survival with a Cerebral Performance Category score of 0 or 1 climbed from 5.5% to 7.8%, a 42% relative increase. In a multivariate analysis adjusted for potential confounders, the adjusted odds ratio for survival of OHCA was 2.25-fold greater for all cases after implementation of the T-CPR program and similarly increased for arrests of cardiac origin.
Dr. Bobrow observed that this was not a randomized trial, which he considered would be both unethical and impractical.
“We controlled for known risk factors and confounders, and while we cannot prove that better outcomes resulted directly from the process improvements, the two are independently associated in this controlled study,” said the emergency physician, who is medical director of the Bureau of Emergency Medical Services and Trauma Systems at the Arizona Dept. of Health Services.
Audience members rose to praise the “fantastic” achievements in Arizona and ask why they’re not having similar success rates in their own districts, given that the AHA guidelines are readily available.
“A lot of places say they’re doing this,” Dr. Bobrow replied, “but when they realize what ‘this’ is, they understand that they really weren’t doing it in this type of depth. When we showed them the data on how marginal their performance was, I think that really made the difference.”
“I think that once most dispatch centers really understand the issues and their importance and the power that they have, and you can engage them, I’m confident that people would see the same changes,” he added.
His study was honored as the best oral abstract presentation at the AHA resuscitation science symposium.
Dr. Bobrow reported serving as co-principal investigator of the HeartRescue Project, funded by Medtronic Philanthropy.
CHICAGO – Systematic implementation of a comprehensive telephone CPR bundle of care targeting EMS dispatch services resulted in substantial improvements in rates of survival to hospital discharge with good neurologic outcomes in patients with out-of-hospital cardiac arrest in a major Arizona statewide public health initiative.
How big was the intervention’s impact? The rate of survival to hospital discharge showed a 33% relative increase compared to preintervention, and survival with a favorable Cerebral Performance Category score of 0 or 1 increased by 42%, Dr. Bentley J. Bobrow reported at the American Heart Association Scientific Sessions.
“These results suggest that when deliberately implemented and measured, telephone CPR is a targeted, effective method to increase bystander CPR and survival on a vast scale with minimal capital expense. This is why we believe telephone CPR along with public training may be the most efficient way to move the needle on cardiac arrest survival,” declared Dr. Bobrow, professor of emergency medicine at the University of Arizona College of Medicine-Phoenix Campus and chair of the AHA Basic Life Support Subcommittee.
Telephone CPR (T-CPR) entails the provision of CPR instruction to bystanders who have called 911 regarding an out of hospital cardiac arrest (OHCA). It’s well established that bystander CPR commenced before EMS personnel arrive on the scene doubles or even triples OHCA survival, but it is provided in only about one-third of OHCA events. And while T-CPR is independently associated with increased rates of bystander CPR as well as patient survival, its utilization varies widely throughout the country and few EMS services measure performance.
Dr. Bobrow reported on an ambitious undertaking that involved systematic training in T-CPR for dispatchers, 911 managers, and medical directors at all nine of the regional emergency dispatch centers in Arizona, which together with 190 EMS agencies and 40 cardiac care hospitals participate in a statewide resuscitation program.
The training was designed to implement the latest AHA guidelines on T-CPR (Circulation 2012;125:648-55). The program entailed a half-day in-person training session plus completion of a 1-hour web-based interactive video. The protocol emphasizes asking two key questions of the 911 caller: “Is the patient conscious?” and “Is the patient breathing normally?” If the response is no to both, the dispatcher is to start issuing bystander CPR instructions without delay – no further questions – and continue the coaching until EMS personnel arrive on the scene to take over.
A core aspect of the T-CPR bundle is performance measurement for quality improvement, with auditing of 911 calls to learn the time from the start of the call to the bystander’s first chest compression and five other key performance metrics. Feedback is provided to the 911 call center regarding system- and case-level performance reports in a continuing education, quality improvement process. Individual dispatchers are singled out for exemplary performance, Dr. Bobrow explained.
He presented a prospective before-and-after study conducted at the three EMS dispatch centers serving Arizona’s Maricopa County, home to two-thirds of the state’s population. The study entailed auditing nearly 6,000 911 calls, each averaging 6.5 minutes in length. After excluding calls where CPR wasn’t indicated or the OHCA involved a patient less than 8 years old, investigators were left with two groups for comparison comprised of 1,289 pre- and 2,330 post-intervention events.
The improvements in process and clinical outcomes were dramatic. In 2012, after introduction of the T-CPR training program, the bystander CPR rate crossed the 50% threshold for the first time ever in Maricopa County. The rate of survival of OHCA to hospital discharge improved from 8.3% to 11%, a highly statistically significant 33% relative increase. Survival with a Cerebral Performance Category score of 0 or 1 climbed from 5.5% to 7.8%, a 42% relative increase. In a multivariate analysis adjusted for potential confounders, the adjusted odds ratio for survival of OHCA was 2.25-fold greater for all cases after implementation of the T-CPR program and similarly increased for arrests of cardiac origin.
Dr. Bobrow observed that this was not a randomized trial, which he considered would be both unethical and impractical.
“We controlled for known risk factors and confounders, and while we cannot prove that better outcomes resulted directly from the process improvements, the two are independently associated in this controlled study,” said the emergency physician, who is medical director of the Bureau of Emergency Medical Services and Trauma Systems at the Arizona Dept. of Health Services.
Audience members rose to praise the “fantastic” achievements in Arizona and ask why they’re not having similar success rates in their own districts, given that the AHA guidelines are readily available.
“A lot of places say they’re doing this,” Dr. Bobrow replied, “but when they realize what ‘this’ is, they understand that they really weren’t doing it in this type of depth. When we showed them the data on how marginal their performance was, I think that really made the difference.”
“I think that once most dispatch centers really understand the issues and their importance and the power that they have, and you can engage them, I’m confident that people would see the same changes,” he added.
His study was honored as the best oral abstract presentation at the AHA resuscitation science symposium.
Dr. Bobrow reported serving as co-principal investigator of the HeartRescue Project, funded by Medtronic Philanthropy.
CHICAGO – Systematic implementation of a comprehensive telephone CPR bundle of care targeting EMS dispatch services resulted in substantial improvements in rates of survival to hospital discharge with good neurologic outcomes in patients with out-of-hospital cardiac arrest in a major Arizona statewide public health initiative.
How big was the intervention’s impact? The rate of survival to hospital discharge showed a 33% relative increase compared to preintervention, and survival with a favorable Cerebral Performance Category score of 0 or 1 increased by 42%, Dr. Bentley J. Bobrow reported at the American Heart Association Scientific Sessions.
“These results suggest that when deliberately implemented and measured, telephone CPR is a targeted, effective method to increase bystander CPR and survival on a vast scale with minimal capital expense. This is why we believe telephone CPR along with public training may be the most efficient way to move the needle on cardiac arrest survival,” declared Dr. Bobrow, professor of emergency medicine at the University of Arizona College of Medicine-Phoenix Campus and chair of the AHA Basic Life Support Subcommittee.
Telephone CPR (T-CPR) entails the provision of CPR instruction to bystanders who have called 911 regarding an out of hospital cardiac arrest (OHCA). It’s well established that bystander CPR commenced before EMS personnel arrive on the scene doubles or even triples OHCA survival, but it is provided in only about one-third of OHCA events. And while T-CPR is independently associated with increased rates of bystander CPR as well as patient survival, its utilization varies widely throughout the country and few EMS services measure performance.
Dr. Bobrow reported on an ambitious undertaking that involved systematic training in T-CPR for dispatchers, 911 managers, and medical directors at all nine of the regional emergency dispatch centers in Arizona, which together with 190 EMS agencies and 40 cardiac care hospitals participate in a statewide resuscitation program.
The training was designed to implement the latest AHA guidelines on T-CPR (Circulation 2012;125:648-55). The program entailed a half-day in-person training session plus completion of a 1-hour web-based interactive video. The protocol emphasizes asking two key questions of the 911 caller: “Is the patient conscious?” and “Is the patient breathing normally?” If the response is no to both, the dispatcher is to start issuing bystander CPR instructions without delay – no further questions – and continue the coaching until EMS personnel arrive on the scene to take over.
A core aspect of the T-CPR bundle is performance measurement for quality improvement, with auditing of 911 calls to learn the time from the start of the call to the bystander’s first chest compression and five other key performance metrics. Feedback is provided to the 911 call center regarding system- and case-level performance reports in a continuing education, quality improvement process. Individual dispatchers are singled out for exemplary performance, Dr. Bobrow explained.
He presented a prospective before-and-after study conducted at the three EMS dispatch centers serving Arizona’s Maricopa County, home to two-thirds of the state’s population. The study entailed auditing nearly 6,000 911 calls, each averaging 6.5 minutes in length. After excluding calls where CPR wasn’t indicated or the OHCA involved a patient less than 8 years old, investigators were left with two groups for comparison comprised of 1,289 pre- and 2,330 post-intervention events.
The improvements in process and clinical outcomes were dramatic. In 2012, after introduction of the T-CPR training program, the bystander CPR rate crossed the 50% threshold for the first time ever in Maricopa County. The rate of survival of OHCA to hospital discharge improved from 8.3% to 11%, a highly statistically significant 33% relative increase. Survival with a Cerebral Performance Category score of 0 or 1 climbed from 5.5% to 7.8%, a 42% relative increase. In a multivariate analysis adjusted for potential confounders, the adjusted odds ratio for survival of OHCA was 2.25-fold greater for all cases after implementation of the T-CPR program and similarly increased for arrests of cardiac origin.
Dr. Bobrow observed that this was not a randomized trial, which he considered would be both unethical and impractical.
“We controlled for known risk factors and confounders, and while we cannot prove that better outcomes resulted directly from the process improvements, the two are independently associated in this controlled study,” said the emergency physician, who is medical director of the Bureau of Emergency Medical Services and Trauma Systems at the Arizona Dept. of Health Services.
Audience members rose to praise the “fantastic” achievements in Arizona and ask why they’re not having similar success rates in their own districts, given that the AHA guidelines are readily available.
“A lot of places say they’re doing this,” Dr. Bobrow replied, “but when they realize what ‘this’ is, they understand that they really weren’t doing it in this type of depth. When we showed them the data on how marginal their performance was, I think that really made the difference.”
“I think that once most dispatch centers really understand the issues and their importance and the power that they have, and you can engage them, I’m confident that people would see the same changes,” he added.
His study was honored as the best oral abstract presentation at the AHA resuscitation science symposium.
Dr. Bobrow reported serving as co-principal investigator of the HeartRescue Project, funded by Medtronic Philanthropy.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: Adoption of the most recent AHA guidelines on telephone CPR by EMS dispatchers will lead to vast improvement in survival for patients with out-of-hospital cardiac arrest.
Major finding: Following implementation of an Arizona statewide program to improve telephone CPR by 911 dispatchers to bystanders at the scene of out-of-hospital cardiac arrest, survival to hospital discharge climbed from 8.3% to 11.0%.
Data source: This was a prospective study comparing the outcomes of 1,289 calls to Arizona 911 centers regarding out-of-hospital cardiac arrests before introduction of a comprehensive statewide telephone CPR program to 2,330 calls received post-intervention.
Disclosures: The study was financially supported by Medtronic Philanthropy as part of the HeartRescue Project. The presenter is co-principal investigator of the project.

Switch to long-acting antipsychotics improved treatment attitude
BERLIN – A switch to long-acting second-generation injectable antipsychotics brought improved attitudes and beliefs regarding antipsychotic therapy among patients with schizoaffective disorder who already were stabilized on a single oral antipsychotic agent, Dr. Francesco Pietrini reported at the annual congress of the European College of Neuropsychopharmacology.
The conventional view has been that long-acting injectable antipsychotics are most appropriate as an alternative option for patients with poor adherence to oral antipsychotics. But this small observational study suggests that the switch is beneficial in a broader population, according to Dr. Pietrini of the University of Florence (Italy).
The 18 outpatients who participated in this 12-month, prospective, observational study showed significant improvements in a variety of measures of psychopathology after the switch to long-acting injectable therapy. These benefits were accompanied by a steadily improving attitude toward treatment as reflected in rising scores on the Drug Attitude Inventory-10 (DAI-10).
All patients were stable at baseline on either oral olanzapine or oral paliperidone. They were then switched to the corresponding dose of the long-acting injectable formulation of the same drug and followed prospectively for 12 months.
Mean scores on the 0-10 DAI-10 rose from 3.22 at baseline to 6.56 after 6 months on a long-acting injectable antipsychotic and 8.11 at 12 months. These improvements in scores reflect the patients’ growing endorsement of inventory items such as “taking medication will prevent me from having a breakdown,” “my thoughts are clearer on medication,” and “for me, the good things about medication outweigh the bad.”
Scores on most measures of psychopathology improved significantly from baseline during the first 6 months on long-acting therapy. The improvement was maintained but not expanded upon during the second 6 months. The exception was negative psychotic symptoms as assessed by the negative symptom scale of the Positive and Negative Syndrome Scale (PANSS), which didn’t show significant improvement during the first 6 months but did in the second half of the year-long study (see chart).
Dr. Pietrini reported having no financial conflicts regarding this study, which was conducted free of commercial support.
BERLIN – A switch to long-acting second-generation injectable antipsychotics brought improved attitudes and beliefs regarding antipsychotic therapy among patients with schizoaffective disorder who already were stabilized on a single oral antipsychotic agent, Dr. Francesco Pietrini reported at the annual congress of the European College of Neuropsychopharmacology.
The conventional view has been that long-acting injectable antipsychotics are most appropriate as an alternative option for patients with poor adherence to oral antipsychotics. But this small observational study suggests that the switch is beneficial in a broader population, according to Dr. Pietrini of the University of Florence (Italy).
The 18 outpatients who participated in this 12-month, prospective, observational study showed significant improvements in a variety of measures of psychopathology after the switch to long-acting injectable therapy. These benefits were accompanied by a steadily improving attitude toward treatment as reflected in rising scores on the Drug Attitude Inventory-10 (DAI-10).
All patients were stable at baseline on either oral olanzapine or oral paliperidone. They were then switched to the corresponding dose of the long-acting injectable formulation of the same drug and followed prospectively for 12 months.
Mean scores on the 0-10 DAI-10 rose from 3.22 at baseline to 6.56 after 6 months on a long-acting injectable antipsychotic and 8.11 at 12 months. These improvements in scores reflect the patients’ growing endorsement of inventory items such as “taking medication will prevent me from having a breakdown,” “my thoughts are clearer on medication,” and “for me, the good things about medication outweigh the bad.”
Scores on most measures of psychopathology improved significantly from baseline during the first 6 months on long-acting therapy. The improvement was maintained but not expanded upon during the second 6 months. The exception was negative psychotic symptoms as assessed by the negative symptom scale of the Positive and Negative Syndrome Scale (PANSS), which didn’t show significant improvement during the first 6 months but did in the second half of the year-long study (see chart).
Dr. Pietrini reported having no financial conflicts regarding this study, which was conducted free of commercial support.
BERLIN – A switch to long-acting second-generation injectable antipsychotics brought improved attitudes and beliefs regarding antipsychotic therapy among patients with schizoaffective disorder who already were stabilized on a single oral antipsychotic agent, Dr. Francesco Pietrini reported at the annual congress of the European College of Neuropsychopharmacology.
The conventional view has been that long-acting injectable antipsychotics are most appropriate as an alternative option for patients with poor adherence to oral antipsychotics. But this small observational study suggests that the switch is beneficial in a broader population, according to Dr. Pietrini of the University of Florence (Italy).
The 18 outpatients who participated in this 12-month, prospective, observational study showed significant improvements in a variety of measures of psychopathology after the switch to long-acting injectable therapy. These benefits were accompanied by a steadily improving attitude toward treatment as reflected in rising scores on the Drug Attitude Inventory-10 (DAI-10).
All patients were stable at baseline on either oral olanzapine or oral paliperidone. They were then switched to the corresponding dose of the long-acting injectable formulation of the same drug and followed prospectively for 12 months.
Mean scores on the 0-10 DAI-10 rose from 3.22 at baseline to 6.56 after 6 months on a long-acting injectable antipsychotic and 8.11 at 12 months. These improvements in scores reflect the patients’ growing endorsement of inventory items such as “taking medication will prevent me from having a breakdown,” “my thoughts are clearer on medication,” and “for me, the good things about medication outweigh the bad.”
Scores on most measures of psychopathology improved significantly from baseline during the first 6 months on long-acting therapy. The improvement was maintained but not expanded upon during the second 6 months. The exception was negative psychotic symptoms as assessed by the negative symptom scale of the Positive and Negative Syndrome Scale (PANSS), which didn’t show significant improvement during the first 6 months but did in the second half of the year-long study (see chart).
Dr. Pietrini reported having no financial conflicts regarding this study, which was conducted free of commercial support.
AT THE ECNP CONGRESS
Key clinical point: A switch to a long-acting injectable antipsychotic proved beneficial in multiple ways in patients with schizoaffective disorder who were stable on a single oral agent.
Major finding: Mean scores on the Drug Attitude Inventory-10 improved from 3.22 when patients were stabilized on an oral antipsychotic to 6.56 after 6 months on the long-term injectable formulation of the same drug and 8.11 after 12 months.
Data source: This was a 12-month observational study of 18 outpatients with schizoaffective disorder.
Disclosures: Dr. Pietrini reported having no financial conflicts regarding this study, which was conducted free of commercial support.
Injectable paliperidone palmitate reduces incarcerations in schizophrenia
BERLIN – Once-monthly intramuscular paliperidone palmitate significantly delayed the unwelcome real world consequences of schizophrenia – including contact with the criminal justice system as well as psychiatric hospitalization – compared with oral antipsychotics, in the randomized PRIDE study.
PRIDE, or Paliperidone Palmitate Research in Demonstrating Effectiveness, deliberately enrolled the sort of real world patients with schizophrenia who are familiar to every clinician and jail warden but normally excluded from participation in clinical trials, including patients with a history of arrest and incarceration, and comorbid substance abuse.
Indeed, release from incarceration within the past 90 days was a prerequisite for eligibility for the PRIDE study, Dr. H. Lynn Starr explained at the annual congress of the European College of Neuropsychopharmacology.
Meanwhile, Janssen announced Nov. 13 that the Food and Drug Administration has approved the supplemental new drug applications for the long-acting drug for treating schizoaffective disorder.
PRIDE was a prospective, open-label, multicenter single-blind study in which 450 U.S. schizophrenia patients were randomized to treatment with once-monthly injectable paliperidone palmitate (Invega Sustenna) or daily oral antipsychotics for up to 15 months. Willingness to accept the long-acting injectable medication was a precondition for eligibility. Prior to randomization, patients sat down individually with a clinician, reviewed seven widely prescribed oral antipsychotics available in this study, and eliminated up to six of them from further consideration. They were then randomly assigned to flexibly dosed therapy with either an oral antipsychotic agent or injectable paliperidone palmitate.
The primary study endpoint was time to treatment failure, broadly defined in this study as a composite of arrest or incarceration; psychiatric hospitalization; suicide; dropout because of insufficient efficacy; discontinuation because of safety or tolerability issues; and the need for treatment supplementation with an additional antipsychotic because of inadequate efficacy, suicide risk, or increased use of psychiatric services in order to prevent imminent psychiatric hospitalization.
Median time to treatment failure was 226 days in the oral antipsychotic group and nearly twice as long at 416 days in the injectable paliperidone palmitate group. The treatment failure rate was 39.8% with paliperidone palmitate, compared with 53.7% with oral antipsychotics. This translated to a 43% greater risk of treatment failure in the oral therapy group, according to Dr. Starr of Janssen Pharmaceuticals in Titusville, N.J.
A prespecified key secondary endpoint was time to first psychiatric hospitalization or arrest/incarceration, as these outcomes are associated with considerable public health impact. The risk of either event proved to be 43% higher in the oral antipsychotic group. Arrest/incarceration and psychiatric hospitalization turned out to be the two most frequently occurring elements of the primary composite endpoint. Arrest/incarceration occurred in 21.2% of the paliperidone group, compared with 29.4% on oral antipsychotics. Eight percent of patients on long-term injectable therapy underwent psychiatric hospitalization, as did 11.9% on oral antipsychotics.
Patients on oral antipsychotic therapy not only experienced arrest/incarceration or psychiatric hospitalization sooner, they also had a significantly greater cumulative number of such events during follow-up.
The study drug was discontinued because of safety or tolerability issues in 6.6% of the paliperidone palmitate group and 3.7% on an oral antipsychotic. Erectile dysfunction was reported by 8.8% of patients on long-acting therapy but none on oral agents. Among women, amenorrhea occurred in 15.2% on paliperidone palmitate, vs. 3.6% on oral antipsychotics.
Thirty-two percent of patients on paliperidone palmitate had at least a 7% weight gain, as did 14% of the oral antipsychotic group.
In the oral antipsychotic group, 22% of patients were on paliperidone; 17%, on risperidone; 16.5%, on olanzapine; 15.1%, on aripiprazole; 13.3%, on quetiapine; 9.2%, on perphenazine; and 6.9%, on haloperidol.
The PRIDE study was funded by Janssen Scientific Affairs, where Dr. Starr serves as director of clinical development.
BERLIN – Once-monthly intramuscular paliperidone palmitate significantly delayed the unwelcome real world consequences of schizophrenia – including contact with the criminal justice system as well as psychiatric hospitalization – compared with oral antipsychotics, in the randomized PRIDE study.
PRIDE, or Paliperidone Palmitate Research in Demonstrating Effectiveness, deliberately enrolled the sort of real world patients with schizophrenia who are familiar to every clinician and jail warden but normally excluded from participation in clinical trials, including patients with a history of arrest and incarceration, and comorbid substance abuse.
Indeed, release from incarceration within the past 90 days was a prerequisite for eligibility for the PRIDE study, Dr. H. Lynn Starr explained at the annual congress of the European College of Neuropsychopharmacology.
Meanwhile, Janssen announced Nov. 13 that the Food and Drug Administration has approved the supplemental new drug applications for the long-acting drug for treating schizoaffective disorder.
PRIDE was a prospective, open-label, multicenter single-blind study in which 450 U.S. schizophrenia patients were randomized to treatment with once-monthly injectable paliperidone palmitate (Invega Sustenna) or daily oral antipsychotics for up to 15 months. Willingness to accept the long-acting injectable medication was a precondition for eligibility. Prior to randomization, patients sat down individually with a clinician, reviewed seven widely prescribed oral antipsychotics available in this study, and eliminated up to six of them from further consideration. They were then randomly assigned to flexibly dosed therapy with either an oral antipsychotic agent or injectable paliperidone palmitate.
The primary study endpoint was time to treatment failure, broadly defined in this study as a composite of arrest or incarceration; psychiatric hospitalization; suicide; dropout because of insufficient efficacy; discontinuation because of safety or tolerability issues; and the need for treatment supplementation with an additional antipsychotic because of inadequate efficacy, suicide risk, or increased use of psychiatric services in order to prevent imminent psychiatric hospitalization.
Median time to treatment failure was 226 days in the oral antipsychotic group and nearly twice as long at 416 days in the injectable paliperidone palmitate group. The treatment failure rate was 39.8% with paliperidone palmitate, compared with 53.7% with oral antipsychotics. This translated to a 43% greater risk of treatment failure in the oral therapy group, according to Dr. Starr of Janssen Pharmaceuticals in Titusville, N.J.
A prespecified key secondary endpoint was time to first psychiatric hospitalization or arrest/incarceration, as these outcomes are associated with considerable public health impact. The risk of either event proved to be 43% higher in the oral antipsychotic group. Arrest/incarceration and psychiatric hospitalization turned out to be the two most frequently occurring elements of the primary composite endpoint. Arrest/incarceration occurred in 21.2% of the paliperidone group, compared with 29.4% on oral antipsychotics. Eight percent of patients on long-term injectable therapy underwent psychiatric hospitalization, as did 11.9% on oral antipsychotics.
Patients on oral antipsychotic therapy not only experienced arrest/incarceration or psychiatric hospitalization sooner, they also had a significantly greater cumulative number of such events during follow-up.
The study drug was discontinued because of safety or tolerability issues in 6.6% of the paliperidone palmitate group and 3.7% on an oral antipsychotic. Erectile dysfunction was reported by 8.8% of patients on long-acting therapy but none on oral agents. Among women, amenorrhea occurred in 15.2% on paliperidone palmitate, vs. 3.6% on oral antipsychotics.
Thirty-two percent of patients on paliperidone palmitate had at least a 7% weight gain, as did 14% of the oral antipsychotic group.
In the oral antipsychotic group, 22% of patients were on paliperidone; 17%, on risperidone; 16.5%, on olanzapine; 15.1%, on aripiprazole; 13.3%, on quetiapine; 9.2%, on perphenazine; and 6.9%, on haloperidol.
The PRIDE study was funded by Janssen Scientific Affairs, where Dr. Starr serves as director of clinical development.
BERLIN – Once-monthly intramuscular paliperidone palmitate significantly delayed the unwelcome real world consequences of schizophrenia – including contact with the criminal justice system as well as psychiatric hospitalization – compared with oral antipsychotics, in the randomized PRIDE study.
PRIDE, or Paliperidone Palmitate Research in Demonstrating Effectiveness, deliberately enrolled the sort of real world patients with schizophrenia who are familiar to every clinician and jail warden but normally excluded from participation in clinical trials, including patients with a history of arrest and incarceration, and comorbid substance abuse.
Indeed, release from incarceration within the past 90 days was a prerequisite for eligibility for the PRIDE study, Dr. H. Lynn Starr explained at the annual congress of the European College of Neuropsychopharmacology.
Meanwhile, Janssen announced Nov. 13 that the Food and Drug Administration has approved the supplemental new drug applications for the long-acting drug for treating schizoaffective disorder.
PRIDE was a prospective, open-label, multicenter single-blind study in which 450 U.S. schizophrenia patients were randomized to treatment with once-monthly injectable paliperidone palmitate (Invega Sustenna) or daily oral antipsychotics for up to 15 months. Willingness to accept the long-acting injectable medication was a precondition for eligibility. Prior to randomization, patients sat down individually with a clinician, reviewed seven widely prescribed oral antipsychotics available in this study, and eliminated up to six of them from further consideration. They were then randomly assigned to flexibly dosed therapy with either an oral antipsychotic agent or injectable paliperidone palmitate.
The primary study endpoint was time to treatment failure, broadly defined in this study as a composite of arrest or incarceration; psychiatric hospitalization; suicide; dropout because of insufficient efficacy; discontinuation because of safety or tolerability issues; and the need for treatment supplementation with an additional antipsychotic because of inadequate efficacy, suicide risk, or increased use of psychiatric services in order to prevent imminent psychiatric hospitalization.
Median time to treatment failure was 226 days in the oral antipsychotic group and nearly twice as long at 416 days in the injectable paliperidone palmitate group. The treatment failure rate was 39.8% with paliperidone palmitate, compared with 53.7% with oral antipsychotics. This translated to a 43% greater risk of treatment failure in the oral therapy group, according to Dr. Starr of Janssen Pharmaceuticals in Titusville, N.J.
A prespecified key secondary endpoint was time to first psychiatric hospitalization or arrest/incarceration, as these outcomes are associated with considerable public health impact. The risk of either event proved to be 43% higher in the oral antipsychotic group. Arrest/incarceration and psychiatric hospitalization turned out to be the two most frequently occurring elements of the primary composite endpoint. Arrest/incarceration occurred in 21.2% of the paliperidone group, compared with 29.4% on oral antipsychotics. Eight percent of patients on long-term injectable therapy underwent psychiatric hospitalization, as did 11.9% on oral antipsychotics.
Patients on oral antipsychotic therapy not only experienced arrest/incarceration or psychiatric hospitalization sooner, they also had a significantly greater cumulative number of such events during follow-up.
The study drug was discontinued because of safety or tolerability issues in 6.6% of the paliperidone palmitate group and 3.7% on an oral antipsychotic. Erectile dysfunction was reported by 8.8% of patients on long-acting therapy but none on oral agents. Among women, amenorrhea occurred in 15.2% on paliperidone palmitate, vs. 3.6% on oral antipsychotics.
Thirty-two percent of patients on paliperidone palmitate had at least a 7% weight gain, as did 14% of the oral antipsychotic group.
In the oral antipsychotic group, 22% of patients were on paliperidone; 17%, on risperidone; 16.5%, on olanzapine; 15.1%, on aripiprazole; 13.3%, on quetiapine; 9.2%, on perphenazine; and 6.9%, on haloperidol.
The PRIDE study was funded by Janssen Scientific Affairs, where Dr. Starr serves as director of clinical development.
AT THE ECNP CONGRESS
Key clinical point: Real world schizophrenia patients with a history of incarceration are less likely to return to jail if they’re on long-acting intramuscular paliperidone palmitate than an oral antipsychotic.
Major finding: The median time to treatment failure, a composite endpoint including arrest/incarceration or psychiatric hospitalization, was 416 days in patients randomized to paliperidone palmitate and 226 days in those assigned to an oral antipsychotic.
Data source: The PRIDE study was a prospective, multicenter, open-label single-blind study in which 450 schizophrenia patients with contact with the criminal justice system within the last 90 days were randomized to long-acting injectable paliperidone palmitate or daily oral antipsychotic therapy.
Disclosures: The PRIDE study was sponsored by Janssen Pharmaceuticals. The presenter is a company employee.
Low-risk actinic keratosis? No such thing
AMSTERDAM – The majority of cutaneous infiltrating squamous cell carcinomas arise via direct transformation of grade 1 actinic keratoses – that is, differentiated dysplasia of the basal one-third of the epidermis – via extension of atypical keratinocytes and ultimately of tumor cells along the adnexal epithelium rather than through the classical pathway most dermatologists know, Dr. Maria Teresa Fernandez-Figueras said at the annual congress of the European Academy of Dermatology and Venereology.
This direct transformation by means of what pathologists call the differentiated pathway has important implications for clinical practice, she added.

“For me, this is something that should change our way of seeing these lesions. When you’re facing an actinic keratosis-1 (AK 1), I think you can no longer call it a low-grade lesion, an early lesion, or a low-risk lesion, because you don’t know if it’s going to follow the differentiated pathway. It can infiltrate at any moment. So from this point of view, all AKs carry a risk of immediate transformation – a low risk, it’s true – and thus maybe all of them require treatment,” explained Dr. Fernandez-Figueras, a pathologist at the University of Barcelona.
The classical pathway by which an AK 1 transforms into an infiltrating squamous cell carcinoma (SCC) is as follows: Before an AK 1 can become a malignant lesion, it first must transition to an AK 2, marked by progression of dysplasia to the lower two-thirds of the epidermis, and then to AK 3, with full-thickness epidermal dysplasia. That’s how most dermatologists understand the process. But there are precedents elsewhere in the body for the existence of a separate differentiated pathway.
The most notable example is vulvar carcinoma. Human papillomavirus–related vulvar malignancy is known to arise from two different pathways. In the classical pathway, vulvar intraepithelial neoplasia 1 (VIN 1) must transition to VIN 2 and then VIN 3 before transformation to SCC. But the cancer can also emerge directly from areas of differentiated dysplastic VIN 1 epidermis, bypassing VINs 2 and 3 altogether.
Dr. Fernandez-Figueras sought to study the relative importance of the two pathways in the formation of cutaneous SCC. To do so, she and two other pathologists examined pathologic specimens and reports for 503 consecutive cases of cutaneous infiltrating SCC. The majority were unsuitable for thorough evaluation because they were fragmented or curettaged, leaving a final study sample of 196 cases.
Two-thirds of all the SCCs had only AK 1 overlying the cutaneous malignancy. Another 15% had overlying AK 2, and 18% had overlying AK 3. A total of 79% of the SCCs had only AK 1 in the epidermis adjacent to the SCC, while 7% had AK 2, and 8% had AK 3; the remaining 6% were unevaluable on this score.
Proliferative growth extending along the sweat ducts and hair follicles of the adnexal epithelium over the SCC was identified in 32% of the cancerous lesions with overlying AK 1, compared with 23% of cancers with overlying AK 2 and 14% with overlying AK 3. Thirty-eight percent of SCCs with AK 1 in the adjacent epithelium had differentiated dysplastic growth migrating along the adnexal epithelium, as did 25% of SCCs with adjacent AK 2 and 11% with neighboring AK 2.
These observations suggest that the differentiated pathway is by far the more common of the two mechanisms of malignant transformation, according to Dr. Fernandez-Figueras.
“Another interesting observation was that very often the areas of invasion were coincident with the focus of adnexal extension,” she continued. “We believe that this tumor advance along the adnexal structures plays a pivotal role in transformation and inversion, especially in AK 1.”
One dermatologist in the audience challenged her. “What do you want us to do – aggressively excise all AKs?”
“I’m a pathologist; I don’t have a suggestion regarding treatment,” Dr. Fernandez-Figueras replied. “I’m just saying, don’t think that AK 1 is better than AK 3, and don’t think that making this distinction gives you any security that you can tell the patient, ‘Don’t worry, nothing is going to happen, we’ll wait until it’s AK 3.’ In my own case, I know that if I had an AK 1, I would treat it. I don’t want to wait to see if it’s going to follow the classical or the differentiated pathway. All lesions can be high grade.”
Session cochair Dr. Michael Reusch pronounced her findings and interpretation of them “completely consistent” with his own experience.
“I think from a histologic point of view, I completely share your view. Still, for the dermatologist part of us, it creates a problem as to what to do. It’s a major issue. The number of AKs out there is incredible,” commented Dr. Reusch of the University Clinics of Hamburg-Eppendorf, Germany.
Dr. Fernandez-Figueras reported that her study received financial support from Almirall.
AMSTERDAM – The majority of cutaneous infiltrating squamous cell carcinomas arise via direct transformation of grade 1 actinic keratoses – that is, differentiated dysplasia of the basal one-third of the epidermis – via extension of atypical keratinocytes and ultimately of tumor cells along the adnexal epithelium rather than through the classical pathway most dermatologists know, Dr. Maria Teresa Fernandez-Figueras said at the annual congress of the European Academy of Dermatology and Venereology.
This direct transformation by means of what pathologists call the differentiated pathway has important implications for clinical practice, she added.
“For me, this is something that should change our way of seeing these lesions. When you’re facing an actinic keratosis-1 (AK 1), I think you can no longer call it a low-grade lesion, an early lesion, or a low-risk lesion, because you don’t know if it’s going to follow the differentiated pathway. It can infiltrate at any moment. So from this point of view, all AKs carry a risk of immediate transformation – a low risk, it’s true – and thus maybe all of them require treatment,” explained Dr. Fernandez-Figueras, a pathologist at the University of Barcelona.
The classical pathway by which an AK 1 transforms into an infiltrating squamous cell carcinoma (SCC) is as follows: Before an AK 1 can become a malignant lesion, it first must transition to an AK 2, marked by progression of dysplasia to the lower two-thirds of the epidermis, and then to AK 3, with full-thickness epidermal dysplasia. That’s how most dermatologists understand the process. But there are precedents elsewhere in the body for the existence of a separate differentiated pathway.
The most notable example is vulvar carcinoma. Human papillomavirus–related vulvar malignancy is known to arise from two different pathways. In the classical pathway, vulvar intraepithelial neoplasia 1 (VIN 1) must transition to VIN 2 and then VIN 3 before transformation to SCC. But the cancer can also emerge directly from areas of differentiated dysplastic VIN 1 epidermis, bypassing VINs 2 and 3 altogether.
Dr. Fernandez-Figueras sought to study the relative importance of the two pathways in the formation of cutaneous SCC. To do so, she and two other pathologists examined pathologic specimens and reports for 503 consecutive cases of cutaneous infiltrating SCC. The majority were unsuitable for thorough evaluation because they were fragmented or curettaged, leaving a final study sample of 196 cases.
Two-thirds of all the SCCs had only AK 1 overlying the cutaneous malignancy. Another 15% had overlying AK 2, and 18% had overlying AK 3. A total of 79% of the SCCs had only AK 1 in the epidermis adjacent to the SCC, while 7% had AK 2, and 8% had AK 3; the remaining 6% were unevaluable on this score.
Proliferative growth extending along the sweat ducts and hair follicles of the adnexal epithelium over the SCC was identified in 32% of the cancerous lesions with overlying AK 1, compared with 23% of cancers with overlying AK 2 and 14% with overlying AK 3. Thirty-eight percent of SCCs with AK 1 in the adjacent epithelium had differentiated dysplastic growth migrating along the adnexal epithelium, as did 25% of SCCs with adjacent AK 2 and 11% with neighboring AK 2.
These observations suggest that the differentiated pathway is by far the more common of the two mechanisms of malignant transformation, according to Dr. Fernandez-Figueras.
“Another interesting observation was that very often the areas of invasion were coincident with the focus of adnexal extension,” she continued. “We believe that this tumor advance along the adnexal structures plays a pivotal role in transformation and inversion, especially in AK 1.”
One dermatologist in the audience challenged her. “What do you want us to do – aggressively excise all AKs?”
“I’m a pathologist; I don’t have a suggestion regarding treatment,” Dr. Fernandez-Figueras replied. “I’m just saying, don’t think that AK 1 is better than AK 3, and don’t think that making this distinction gives you any security that you can tell the patient, ‘Don’t worry, nothing is going to happen, we’ll wait until it’s AK 3.’ In my own case, I know that if I had an AK 1, I would treat it. I don’t want to wait to see if it’s going to follow the classical or the differentiated pathway. All lesions can be high grade.”
Session cochair Dr. Michael Reusch pronounced her findings and interpretation of them “completely consistent” with his own experience.
“I think from a histologic point of view, I completely share your view. Still, for the dermatologist part of us, it creates a problem as to what to do. It’s a major issue. The number of AKs out there is incredible,” commented Dr. Reusch of the University Clinics of Hamburg-Eppendorf, Germany.
Dr. Fernandez-Figueras reported that her study received financial support from Almirall.
AMSTERDAM – The majority of cutaneous infiltrating squamous cell carcinomas arise via direct transformation of grade 1 actinic keratoses – that is, differentiated dysplasia of the basal one-third of the epidermis – via extension of atypical keratinocytes and ultimately of tumor cells along the adnexal epithelium rather than through the classical pathway most dermatologists know, Dr. Maria Teresa Fernandez-Figueras said at the annual congress of the European Academy of Dermatology and Venereology.
This direct transformation by means of what pathologists call the differentiated pathway has important implications for clinical practice, she added.
“For me, this is something that should change our way of seeing these lesions. When you’re facing an actinic keratosis-1 (AK 1), I think you can no longer call it a low-grade lesion, an early lesion, or a low-risk lesion, because you don’t know if it’s going to follow the differentiated pathway. It can infiltrate at any moment. So from this point of view, all AKs carry a risk of immediate transformation – a low risk, it’s true – and thus maybe all of them require treatment,” explained Dr. Fernandez-Figueras, a pathologist at the University of Barcelona.
The classical pathway by which an AK 1 transforms into an infiltrating squamous cell carcinoma (SCC) is as follows: Before an AK 1 can become a malignant lesion, it first must transition to an AK 2, marked by progression of dysplasia to the lower two-thirds of the epidermis, and then to AK 3, with full-thickness epidermal dysplasia. That’s how most dermatologists understand the process. But there are precedents elsewhere in the body for the existence of a separate differentiated pathway.
The most notable example is vulvar carcinoma. Human papillomavirus–related vulvar malignancy is known to arise from two different pathways. In the classical pathway, vulvar intraepithelial neoplasia 1 (VIN 1) must transition to VIN 2 and then VIN 3 before transformation to SCC. But the cancer can also emerge directly from areas of differentiated dysplastic VIN 1 epidermis, bypassing VINs 2 and 3 altogether.
Dr. Fernandez-Figueras sought to study the relative importance of the two pathways in the formation of cutaneous SCC. To do so, she and two other pathologists examined pathologic specimens and reports for 503 consecutive cases of cutaneous infiltrating SCC. The majority were unsuitable for thorough evaluation because they were fragmented or curettaged, leaving a final study sample of 196 cases.
Two-thirds of all the SCCs had only AK 1 overlying the cutaneous malignancy. Another 15% had overlying AK 2, and 18% had overlying AK 3. A total of 79% of the SCCs had only AK 1 in the epidermis adjacent to the SCC, while 7% had AK 2, and 8% had AK 3; the remaining 6% were unevaluable on this score.
Proliferative growth extending along the sweat ducts and hair follicles of the adnexal epithelium over the SCC was identified in 32% of the cancerous lesions with overlying AK 1, compared with 23% of cancers with overlying AK 2 and 14% with overlying AK 3. Thirty-eight percent of SCCs with AK 1 in the adjacent epithelium had differentiated dysplastic growth migrating along the adnexal epithelium, as did 25% of SCCs with adjacent AK 2 and 11% with neighboring AK 2.
These observations suggest that the differentiated pathway is by far the more common of the two mechanisms of malignant transformation, according to Dr. Fernandez-Figueras.
“Another interesting observation was that very often the areas of invasion were coincident with the focus of adnexal extension,” she continued. “We believe that this tumor advance along the adnexal structures plays a pivotal role in transformation and inversion, especially in AK 1.”
One dermatologist in the audience challenged her. “What do you want us to do – aggressively excise all AKs?”
“I’m a pathologist; I don’t have a suggestion regarding treatment,” Dr. Fernandez-Figueras replied. “I’m just saying, don’t think that AK 1 is better than AK 3, and don’t think that making this distinction gives you any security that you can tell the patient, ‘Don’t worry, nothing is going to happen, we’ll wait until it’s AK 3.’ In my own case, I know that if I had an AK 1, I would treat it. I don’t want to wait to see if it’s going to follow the classical or the differentiated pathway. All lesions can be high grade.”
Session cochair Dr. Michael Reusch pronounced her findings and interpretation of them “completely consistent” with his own experience.
“I think from a histologic point of view, I completely share your view. Still, for the dermatologist part of us, it creates a problem as to what to do. It’s a major issue. The number of AKs out there is incredible,” commented Dr. Reusch of the University Clinics of Hamburg-Eppendorf, Germany.
Dr. Fernandez-Figueras reported that her study received financial support from Almirall.
AT THE EADV CONGRESS
Key clinical point: Actinic keratoses traditionally considered low-risk AK 1s with dysplasia confined to the basal one-third of the epidermis can undergo immediate transformation to infiltrating squamous cell carcinoma without mandatory sequential passage through AK 2 and 3.
Major finding: Two-thirds of SCCs had only AK 1 overlying the malignancy, 15% had overlying AK 2, and only 8% had AK 3.
Data source: A retrospective study, in which three pathologists examined the pathologic specimens and supporting reports for 196 cutaneous infiltrating SCCs.
Disclosures: The study received funding support from Almirall.
