User login
Perspective from the heartland: Cancer care and research during a public health crisis
I have no knowledge of, or experience with, managing a cancer patient during a pandemic. However, from the published and otherwise shared experience of others, we should not allow ourselves to underestimate the voracity of the coronavirus pandemic on our patients, communities, and health care systems.

Data from China suggest cancer patients infected with SARS-CoV-2 face a 3.5 times higher risk of mechanical ventilation, intensive care unit admission, or death, compared with infected patients without cancer (Lancet Oncol 2020;21:335-7).
Health care workers in Seattle have also shared their experiences battling coronavirus infections in cancer patients (J Natl Compr Canc Netw. 2020 Mar 20. doi: 10.6004/jnccn.2020.7560). Masumi Ueda, MD, of Seattle Cancer Care Alliance, and colleagues reviewed their decisions in multiple domains over a 7-week period, during which the state of Washington went from a single case of SARS-CoV-2 infection to nearly 650 cases and 40 deaths.
Making tough treatment decisions
Dr. Ueda and colleagues contrasted their customary resource-rich, innovation-oriented, cancer-combatting environment with their current circumstance, in which they must prioritize treatment for patients for whom the risk-reward balance has tilted substantially toward “risk.”
The authors noted that their most difficult decisions were those regarding delay of cancer treatment. They suggested that plans for potentially curative adjuvant therapy should likely proceed, but, for patients with metastatic disease, the equation is more nuanced.
In some cases, treatment should be delayed or interrupted with recognition of how that could result in worsening performance status and admission for symptom palliation, further stressing inpatient resources.
The authors suggested scenarios for prioritizing cancer surgery. For example, several months of systemic therapy (ideally, low-risk systemic therapy such as hormone therapy for breast or prostate cancer) and surgical delay may be worthwhile, without compromising patient care.
Patients with aggressive hematologic malignancy requiring urgent systemic treatment (potentially stem cell transplantation and cellular immunotherapies) should be treated promptly. However, even in those cases, opportunities should be sought to lessen immunosuppression and transition care as quickly as possible to the outpatient clinic, according to guidelines from the American Society of Transplantation and Cellular Therapy.
See one, do one, teach one
Rendering patient care during a pandemic would be unique for me. However, I, like all physicians, am familiar with feelings of inadequacy at times of professional challenge. On countless occasions, I have started my day or walked into a patient’s room wondering whether I will have the fortitude, knowledge, creativity, or help I need to get through that day or make that patient “better” by any definition of that word.
We all know the formula: “Work hard. Make evidence-based, personalized decisions for those who have entrusted their care to us. Learn from those encounters. Teach from our knowledge and experience – that is, ‘See one, do one, teach one.’ ”
The Seattle oncologists are living the lives of first responders and deserve our admiration for putting pen to paper so we can learn from their considerable, relevant experience.
Similar admiration is due to Giuseppe Curigliano, MD, of the European Institute of Oncology in Milan. In the ASCO Daily News, Dr. Curigliano described an epidemic that, within 3 weeks, overloaded the health care system across northern Italy.
Hospitalization was needed for over 60% of infected patients, and nearly 15% of those patients needed intensive care unit services for respiratory distress. The Italians centralized oncology care in specialized hubs, with spokes of institutions working in parallel to provide cancer-specific care in a COVID-free environment.
To build upon cancer-specific information from Italy and other areas hard-hit by COVID-19, more than 30 cancer centers have joined together to form the COVID-19 and Cancer Consortium. The consortium’s website hosts a survey designed to “capture details related to cancer patients presumed to have COVID-19.”
Calculating deaths and long-term consequences for cancer care delivery
It is proper that the authors from China, Italy, and Seattle did not focus attention on the case fatality rate from the COVID-19 pandemic among cancer patients. To say the least, it would be complicated to tally the direct mortality – either overall or in clinically important subsets of patients, including country-specific cohorts.
What we know from published reports is that, in Italy, cancer patients account for about 20% of deaths from coronavirus. In China, the case-fatality rate for patients with cancer was 5.6% (JAMA. 2020 Feb 24. doi: 10.1001/jama.2020.2648).
However, we know nothing about the indirect death toll from malignancy (without coronavirus infection) that was untreated or managed less than optimally because of personnel and physical resources that were diverted to COVID-19–associated cases.
Similarly, we cannot begin to estimate indirect consequences of the pandemic to oncology practices, such as accelerated burnout and posttraumatic stress disorder, as well as the long-range effects of economic turmoil on patients, health care workers, and provider organizations.
What happens to cancer trials?
From China, Italy, and Seattle, thus far, there is little information about how the pandemic will affect the vital clinical research endeavor. The Seattle physicians did say they plan to enroll patients on clinical trials only when the trial offers a high chance of benefiting the patient over standard therapy alone.
Fortunately, the National Institutes of Health and Food and Drug Administration have released guidance documents related to clinical trials.
The National Cancer Institute (NCI) has also released guidance documents (March 13 guidance; March 23 guidance) for patients on clinical trials supported by the NCI Cancer Therapy Evaluation Program (CTEP) and the NCI Community Oncology Research Program (NCORP).
CTEP and NCORP are making reasonable accommodations to suspend monitoring visits and audits, allow tele–follow-up visits for patients, and permit local physicians to provide care for patients on study. In addition, with appropriate procedural adherence and documentation, CTEP and NCORP will allow oral investigational medicines to be mailed directly to patients’ homes.
Planned NCI National Clinical Trials Network meetings will be conducted via remote access webinars, conference calls, and similar technology. These adjustments – and probably many more to come – are geared toward facilitating ongoing care to proceed safely and with minimal risk for patients currently receiving investigational therapies and for the sites and investigators engaged in those studies.
Each of us has probably faced a personal “defining professional moment,” when we had to utilize every skill in our arsenal and examine the motivations that led us to a career in oncology. However, it is clear from the forgoing clinical and research processes and guidelines that the COVID-19 pandemic is such a defining professional moment for each of us, in every community we serve.
Critical junctures like this cause more rapid behavior change and innovation than the slow-moving pace that characterizes our idealized preferences. As oncologists who embrace new data and behavioral change, we stand to learn processes that will facilitate more perfected systems of care than the one that preceded this unprecedented crisis, promote more efficient sharing of high-quality information, and improve the outcome for our future patients.
Dr. Lyss was an oncologist and researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
I have no knowledge of, or experience with, managing a cancer patient during a pandemic. However, from the published and otherwise shared experience of others, we should not allow ourselves to underestimate the voracity of the coronavirus pandemic on our patients, communities, and health care systems.
Data from China suggest cancer patients infected with SARS-CoV-2 face a 3.5 times higher risk of mechanical ventilation, intensive care unit admission, or death, compared with infected patients without cancer (Lancet Oncol 2020;21:335-7).
Health care workers in Seattle have also shared their experiences battling coronavirus infections in cancer patients (J Natl Compr Canc Netw. 2020 Mar 20. doi: 10.6004/jnccn.2020.7560). Masumi Ueda, MD, of Seattle Cancer Care Alliance, and colleagues reviewed their decisions in multiple domains over a 7-week period, during which the state of Washington went from a single case of SARS-CoV-2 infection to nearly 650 cases and 40 deaths.
Making tough treatment decisions
Dr. Ueda and colleagues contrasted their customary resource-rich, innovation-oriented, cancer-combatting environment with their current circumstance, in which they must prioritize treatment for patients for whom the risk-reward balance has tilted substantially toward “risk.”
The authors noted that their most difficult decisions were those regarding delay of cancer treatment. They suggested that plans for potentially curative adjuvant therapy should likely proceed, but, for patients with metastatic disease, the equation is more nuanced.
In some cases, treatment should be delayed or interrupted with recognition of how that could result in worsening performance status and admission for symptom palliation, further stressing inpatient resources.
The authors suggested scenarios for prioritizing cancer surgery. For example, several months of systemic therapy (ideally, low-risk systemic therapy such as hormone therapy for breast or prostate cancer) and surgical delay may be worthwhile, without compromising patient care.
Patients with aggressive hematologic malignancy requiring urgent systemic treatment (potentially stem cell transplantation and cellular immunotherapies) should be treated promptly. However, even in those cases, opportunities should be sought to lessen immunosuppression and transition care as quickly as possible to the outpatient clinic, according to guidelines from the American Society of Transplantation and Cellular Therapy.
See one, do one, teach one
Rendering patient care during a pandemic would be unique for me. However, I, like all physicians, am familiar with feelings of inadequacy at times of professional challenge. On countless occasions, I have started my day or walked into a patient’s room wondering whether I will have the fortitude, knowledge, creativity, or help I need to get through that day or make that patient “better” by any definition of that word.
We all know the formula: “Work hard. Make evidence-based, personalized decisions for those who have entrusted their care to us. Learn from those encounters. Teach from our knowledge and experience – that is, ‘See one, do one, teach one.’ ”
The Seattle oncologists are living the lives of first responders and deserve our admiration for putting pen to paper so we can learn from their considerable, relevant experience.
Similar admiration is due to Giuseppe Curigliano, MD, of the European Institute of Oncology in Milan. In the ASCO Daily News, Dr. Curigliano described an epidemic that, within 3 weeks, overloaded the health care system across northern Italy.
Hospitalization was needed for over 60% of infected patients, and nearly 15% of those patients needed intensive care unit services for respiratory distress. The Italians centralized oncology care in specialized hubs, with spokes of institutions working in parallel to provide cancer-specific care in a COVID-free environment.
To build upon cancer-specific information from Italy and other areas hard-hit by COVID-19, more than 30 cancer centers have joined together to form the COVID-19 and Cancer Consortium. The consortium’s website hosts a survey designed to “capture details related to cancer patients presumed to have COVID-19.”
Calculating deaths and long-term consequences for cancer care delivery
It is proper that the authors from China, Italy, and Seattle did not focus attention on the case fatality rate from the COVID-19 pandemic among cancer patients. To say the least, it would be complicated to tally the direct mortality – either overall or in clinically important subsets of patients, including country-specific cohorts.
What we know from published reports is that, in Italy, cancer patients account for about 20% of deaths from coronavirus. In China, the case-fatality rate for patients with cancer was 5.6% (JAMA. 2020 Feb 24. doi: 10.1001/jama.2020.2648).
However, we know nothing about the indirect death toll from malignancy (without coronavirus infection) that was untreated or managed less than optimally because of personnel and physical resources that were diverted to COVID-19–associated cases.
Similarly, we cannot begin to estimate indirect consequences of the pandemic to oncology practices, such as accelerated burnout and posttraumatic stress disorder, as well as the long-range effects of economic turmoil on patients, health care workers, and provider organizations.
What happens to cancer trials?
From China, Italy, and Seattle, thus far, there is little information about how the pandemic will affect the vital clinical research endeavor. The Seattle physicians did say they plan to enroll patients on clinical trials only when the trial offers a high chance of benefiting the patient over standard therapy alone.
Fortunately, the National Institutes of Health and Food and Drug Administration have released guidance documents related to clinical trials.
The National Cancer Institute (NCI) has also released guidance documents (March 13 guidance; March 23 guidance) for patients on clinical trials supported by the NCI Cancer Therapy Evaluation Program (CTEP) and the NCI Community Oncology Research Program (NCORP).
CTEP and NCORP are making reasonable accommodations to suspend monitoring visits and audits, allow tele–follow-up visits for patients, and permit local physicians to provide care for patients on study. In addition, with appropriate procedural adherence and documentation, CTEP and NCORP will allow oral investigational medicines to be mailed directly to patients’ homes.
Planned NCI National Clinical Trials Network meetings will be conducted via remote access webinars, conference calls, and similar technology. These adjustments – and probably many more to come – are geared toward facilitating ongoing care to proceed safely and with minimal risk for patients currently receiving investigational therapies and for the sites and investigators engaged in those studies.
Each of us has probably faced a personal “defining professional moment,” when we had to utilize every skill in our arsenal and examine the motivations that led us to a career in oncology. However, it is clear from the forgoing clinical and research processes and guidelines that the COVID-19 pandemic is such a defining professional moment for each of us, in every community we serve.
Critical junctures like this cause more rapid behavior change and innovation than the slow-moving pace that characterizes our idealized preferences. As oncologists who embrace new data and behavioral change, we stand to learn processes that will facilitate more perfected systems of care than the one that preceded this unprecedented crisis, promote more efficient sharing of high-quality information, and improve the outcome for our future patients.
Dr. Lyss was an oncologist and researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
I have no knowledge of, or experience with, managing a cancer patient during a pandemic. However, from the published and otherwise shared experience of others, we should not allow ourselves to underestimate the voracity of the coronavirus pandemic on our patients, communities, and health care systems.
Data from China suggest cancer patients infected with SARS-CoV-2 face a 3.5 times higher risk of mechanical ventilation, intensive care unit admission, or death, compared with infected patients without cancer (Lancet Oncol 2020;21:335-7).
Health care workers in Seattle have also shared their experiences battling coronavirus infections in cancer patients (J Natl Compr Canc Netw. 2020 Mar 20. doi: 10.6004/jnccn.2020.7560). Masumi Ueda, MD, of Seattle Cancer Care Alliance, and colleagues reviewed their decisions in multiple domains over a 7-week period, during which the state of Washington went from a single case of SARS-CoV-2 infection to nearly 650 cases and 40 deaths.
Making tough treatment decisions
Dr. Ueda and colleagues contrasted their customary resource-rich, innovation-oriented, cancer-combatting environment with their current circumstance, in which they must prioritize treatment for patients for whom the risk-reward balance has tilted substantially toward “risk.”
The authors noted that their most difficult decisions were those regarding delay of cancer treatment. They suggested that plans for potentially curative adjuvant therapy should likely proceed, but, for patients with metastatic disease, the equation is more nuanced.
In some cases, treatment should be delayed or interrupted with recognition of how that could result in worsening performance status and admission for symptom palliation, further stressing inpatient resources.
The authors suggested scenarios for prioritizing cancer surgery. For example, several months of systemic therapy (ideally, low-risk systemic therapy such as hormone therapy for breast or prostate cancer) and surgical delay may be worthwhile, without compromising patient care.
Patients with aggressive hematologic malignancy requiring urgent systemic treatment (potentially stem cell transplantation and cellular immunotherapies) should be treated promptly. However, even in those cases, opportunities should be sought to lessen immunosuppression and transition care as quickly as possible to the outpatient clinic, according to guidelines from the American Society of Transplantation and Cellular Therapy.
See one, do one, teach one
Rendering patient care during a pandemic would be unique for me. However, I, like all physicians, am familiar with feelings of inadequacy at times of professional challenge. On countless occasions, I have started my day or walked into a patient’s room wondering whether I will have the fortitude, knowledge, creativity, or help I need to get through that day or make that patient “better” by any definition of that word.
We all know the formula: “Work hard. Make evidence-based, personalized decisions for those who have entrusted their care to us. Learn from those encounters. Teach from our knowledge and experience – that is, ‘See one, do one, teach one.’ ”
The Seattle oncologists are living the lives of first responders and deserve our admiration for putting pen to paper so we can learn from their considerable, relevant experience.
Similar admiration is due to Giuseppe Curigliano, MD, of the European Institute of Oncology in Milan. In the ASCO Daily News, Dr. Curigliano described an epidemic that, within 3 weeks, overloaded the health care system across northern Italy.
Hospitalization was needed for over 60% of infected patients, and nearly 15% of those patients needed intensive care unit services for respiratory distress. The Italians centralized oncology care in specialized hubs, with spokes of institutions working in parallel to provide cancer-specific care in a COVID-free environment.
To build upon cancer-specific information from Italy and other areas hard-hit by COVID-19, more than 30 cancer centers have joined together to form the COVID-19 and Cancer Consortium. The consortium’s website hosts a survey designed to “capture details related to cancer patients presumed to have COVID-19.”
Calculating deaths and long-term consequences for cancer care delivery
It is proper that the authors from China, Italy, and Seattle did not focus attention on the case fatality rate from the COVID-19 pandemic among cancer patients. To say the least, it would be complicated to tally the direct mortality – either overall or in clinically important subsets of patients, including country-specific cohorts.
What we know from published reports is that, in Italy, cancer patients account for about 20% of deaths from coronavirus. In China, the case-fatality rate for patients with cancer was 5.6% (JAMA. 2020 Feb 24. doi: 10.1001/jama.2020.2648).
However, we know nothing about the indirect death toll from malignancy (without coronavirus infection) that was untreated or managed less than optimally because of personnel and physical resources that were diverted to COVID-19–associated cases.
Similarly, we cannot begin to estimate indirect consequences of the pandemic to oncology practices, such as accelerated burnout and posttraumatic stress disorder, as well as the long-range effects of economic turmoil on patients, health care workers, and provider organizations.
What happens to cancer trials?
From China, Italy, and Seattle, thus far, there is little information about how the pandemic will affect the vital clinical research endeavor. The Seattle physicians did say they plan to enroll patients on clinical trials only when the trial offers a high chance of benefiting the patient over standard therapy alone.
Fortunately, the National Institutes of Health and Food and Drug Administration have released guidance documents related to clinical trials.
The National Cancer Institute (NCI) has also released guidance documents (March 13 guidance; March 23 guidance) for patients on clinical trials supported by the NCI Cancer Therapy Evaluation Program (CTEP) and the NCI Community Oncology Research Program (NCORP).
CTEP and NCORP are making reasonable accommodations to suspend monitoring visits and audits, allow tele–follow-up visits for patients, and permit local physicians to provide care for patients on study. In addition, with appropriate procedural adherence and documentation, CTEP and NCORP will allow oral investigational medicines to be mailed directly to patients’ homes.
Planned NCI National Clinical Trials Network meetings will be conducted via remote access webinars, conference calls, and similar technology. These adjustments – and probably many more to come – are geared toward facilitating ongoing care to proceed safely and with minimal risk for patients currently receiving investigational therapies and for the sites and investigators engaged in those studies.
Each of us has probably faced a personal “defining professional moment,” when we had to utilize every skill in our arsenal and examine the motivations that led us to a career in oncology. However, it is clear from the forgoing clinical and research processes and guidelines that the COVID-19 pandemic is such a defining professional moment for each of us, in every community we serve.
Critical junctures like this cause more rapid behavior change and innovation than the slow-moving pace that characterizes our idealized preferences. As oncologists who embrace new data and behavioral change, we stand to learn processes that will facilitate more perfected systems of care than the one that preceded this unprecedented crisis, promote more efficient sharing of high-quality information, and improve the outcome for our future patients.
Dr. Lyss was an oncologist and researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
Cancer care and COVID-19 in Seattle, the first U.S. epicenter
Two months after the first patient with COVID-19 was identified in China, the first case was reported in the United States in the Seattle, Washington, metropolitan area.
Seattle rapidly became the first US epicenter for COVID-19, and local experts are now offering their expertise and advice on how to provide optimal cancer care during the pandemic in a special feature published online March 20 in the Journal of the National Comprehensive Cancer Network.
“We began implementing measures in early March, including infection control and screening of visitors, staff, and patients at the door,” said lead author Masumi Ueda, MD, who holds positions at the Seattle Cancer Care Alliance, the University of Washington, and the Fred Hutchinson Research Center.
“A lot of changes have been implemented, and it changes on a daily basis. We are responding to the growing rate of COVID-19 infection in the community,” she told Medscape Medical News.
Ueda notes that as a result of the quick implementation of new procedures, so far, very few cancer patients at their facilities have been infected by the virus. “It has not hit our cancer population hard, which is a good thing,” she said.
Create “Incident Command Structure”
In sharing their experience, the authors emphasize the importance of keeping channels of communication open between all stakeholders ― administrators and staff, patients, caregivers, and the general public. They also recommend that each facility create an “incident command structure” that can provide early coordination of institution-wide efforts and that can rapidly respond to changing information.
Ueda noted that their command structure was set up very early on, “so we could get communication set up and start building an infrastructure for response.”
Several areas of care that required new strategies were addressed, both to protect patients and to work around staff shortages caused by possible exposure and/or school closings, as well as projected shortages of supplies and hospital resources.
First and foremost was to identify patients and visitors who had respiratory symptoms and to provide them with masks. Although this is always routine practice during the respiratory virus season, screening has now been initiated at entry points throughout the system.
“We were lucky in Seattle and Washington state in that the University of Washington virology lab developed PCR [polymerase chain reaction] testing early on for COVID-19, which subsequently got FDA approval,” said Ueda. “So we were able to have local testing and didn’t have to rely on the state lab. Testing has also been rapidly scaled up.”
Initiating a comprehensive policy for testing staff, tracking results and exposures for persons under investigation, and defining when it is possible to return to work are essential elements for maintaining a stable workforce. In addition, reinforcing a strict “stay at home when ill” policy and providing access to testing for symptomatic staff have been key to limiting exposures.
“What is unique to our region is that we had testing early on, and we are turning it around in 24 hours,” she pointed out. “This is important for staff to be able to return to work.” Currently, staff, patients, and visitors are being tested only if they show the cardinal symptoms associated with COVID-19: fever, shortness of breath, and cough, although muscle aches have recently been added to their testing protocol.
“I think if we had unlimited capacity, we might consider testing people who are asymptomatic,” Ueda noted, “although if you don’t have symptoms, you may not have the viral load needed for an accurate test.”
Educational materials explaining infection control were also needed for patients and families, along with signs and a website to provide COVID-19 education. These were quickly developed.
In addition, a telephone triage line was established for patients with mild symptoms in order to minimize exposures in clinics and to lessen the number of patients presenting at emergency departments.
Outpatient Care
Because theirs is a referral center, many cancer patients come from out of town, and so there is concern about exposing nonlocal patients to COVID-19 as the virus spreads in the Seattle area. In addition, staffing shortages due to factors such as illness, exposure, and school closures are anticipated.
To address these problems, an initial priority was to establish a “multilayer” coverage system for the clinics in the event that practitioners had to be quarantined on short notice, the authors explain.
One decision was to reschedule all wellness visits for current patients or to use telemedicine. Capacity for that option expanded quickly, which was greatly helped by the recent decision by the Centers for Medicare & Medicaid Services to lift Medicare restrictions on the use of certain telemedicine services.
Another approach is to defer all consultations for second opinions for patients who were already undergoing treatment and to increase clinic hours of operations and capabilities for acute evaluations. This helps reserve emergency departments and hospital resources for patients who require higher-level care, the authors comment.
Treatment Decisions
Treatment decisions were more challenging to make, the authors note. One decision was that, despite the risk for COVID-19 for patients with solid tumors, adjuvant therapy with curative intent should proceed, they note. Similarly, patients with metastatic disease might lose the window of opportunity for treatment if it is delayed.
Treatment for aggressive hematologic malignancies is usually urgent, and stem cell transplant and cellular immunotherapies that provide curative treatments cannot be delayed in many cases.
Enrollment in clinical trials will most likely be limited to those trials that are most likely to benefit the patient.
Ueda noted that, because their patients come from all over the country, they are now conducting consultations for stem cell transplant by telephone so that nonlocal patients do not have to travel to Seattle. “If there is some way we can delay the treatment, we have taken that approach,” Ueda told Medscape Medical News. “If we can divert a patient to an area that is not as heavily affected, that’s another option we are taking.”
Although cancer surgery is not considered elective, surgical intervention needs to be prioritized, the authors comment. In the Seattle system, there is currently a 2-week ban on elective surgery in the healthcare system, owing to limited availability of personal protective equipment (PPE), staffing, and beds.
The oncology teams are currently reviewing treatment regimens to determine which treatments might lessen immunosuppression and which treatment options can be moved from the inpatient to the outpatient setting or can be delayed.
Inpatient Care
For hospitalized patients, several issues are being addressed. The priority is to prepare for an upcoming shortage of beds and resources because of the surge of patients with COVID-19 that is predicted. For both clinic and hospitalized patients, shortages of blood products have necessitated stricter adherence to thresholds for transfusion, and consideration is being given to lowering those thresholds.
Another important problem is the need to conserve PPE, which includes masks, gowns, gloves, and other products. The Seattle teams have implemented solutions such as favoring handwashing with soap and water over the use of hand gel for standard-precaution rooms, limiting the number of personnel entering patient rooms (so as to use less PPE), and reducing nursing procedures that require PPE, such as measuring urine output, unless they are necessary.
In addition, a no-visitor policy has been adopted in inpatient units to conserve PPE, with the exception of end-of-life situations.
The Future
The future trajectory of the COVID-19 pandemic is uncertain, Ueda commented. She emphasized that “we must continue to prepare for its widespread impact. The unknown is what we are looking at. We are expecting it to evolve, and the number of infections cannot go down.”
Ueda and coauthors end their article on a positive note. “To many of us, this has become the health care challenge of our generation, one that modern cancer therapy has never had to face. We will prevail, and when the pandemic ends, we will all be proud of what we did for our patients and each other in this critical moment for humanity.”
Two months after the first patient with COVID-19 was identified in China, the first case was reported in the United States in the Seattle, Washington, metropolitan area.
Seattle rapidly became the first US epicenter for COVID-19, and local experts are now offering their expertise and advice on how to provide optimal cancer care during the pandemic in a special feature published online March 20 in the Journal of the National Comprehensive Cancer Network.
“We began implementing measures in early March, including infection control and screening of visitors, staff, and patients at the door,” said lead author Masumi Ueda, MD, who holds positions at the Seattle Cancer Care Alliance, the University of Washington, and the Fred Hutchinson Research Center.
“A lot of changes have been implemented, and it changes on a daily basis. We are responding to the growing rate of COVID-19 infection in the community,” she told Medscape Medical News.
Ueda notes that as a result of the quick implementation of new procedures, so far, very few cancer patients at their facilities have been infected by the virus. “It has not hit our cancer population hard, which is a good thing,” she said.
Create “Incident Command Structure”
In sharing their experience, the authors emphasize the importance of keeping channels of communication open between all stakeholders ― administrators and staff, patients, caregivers, and the general public. They also recommend that each facility create an “incident command structure” that can provide early coordination of institution-wide efforts and that can rapidly respond to changing information.
Ueda noted that their command structure was set up very early on, “so we could get communication set up and start building an infrastructure for response.”
Several areas of care that required new strategies were addressed, both to protect patients and to work around staff shortages caused by possible exposure and/or school closings, as well as projected shortages of supplies and hospital resources.
First and foremost was to identify patients and visitors who had respiratory symptoms and to provide them with masks. Although this is always routine practice during the respiratory virus season, screening has now been initiated at entry points throughout the system.
“We were lucky in Seattle and Washington state in that the University of Washington virology lab developed PCR [polymerase chain reaction] testing early on for COVID-19, which subsequently got FDA approval,” said Ueda. “So we were able to have local testing and didn’t have to rely on the state lab. Testing has also been rapidly scaled up.”
Initiating a comprehensive policy for testing staff, tracking results and exposures for persons under investigation, and defining when it is possible to return to work are essential elements for maintaining a stable workforce. In addition, reinforcing a strict “stay at home when ill” policy and providing access to testing for symptomatic staff have been key to limiting exposures.
“What is unique to our region is that we had testing early on, and we are turning it around in 24 hours,” she pointed out. “This is important for staff to be able to return to work.” Currently, staff, patients, and visitors are being tested only if they show the cardinal symptoms associated with COVID-19: fever, shortness of breath, and cough, although muscle aches have recently been added to their testing protocol.
“I think if we had unlimited capacity, we might consider testing people who are asymptomatic,” Ueda noted, “although if you don’t have symptoms, you may not have the viral load needed for an accurate test.”
Educational materials explaining infection control were also needed for patients and families, along with signs and a website to provide COVID-19 education. These were quickly developed.
In addition, a telephone triage line was established for patients with mild symptoms in order to minimize exposures in clinics and to lessen the number of patients presenting at emergency departments.
Outpatient Care
Because theirs is a referral center, many cancer patients come from out of town, and so there is concern about exposing nonlocal patients to COVID-19 as the virus spreads in the Seattle area. In addition, staffing shortages due to factors such as illness, exposure, and school closures are anticipated.
To address these problems, an initial priority was to establish a “multilayer” coverage system for the clinics in the event that practitioners had to be quarantined on short notice, the authors explain.
One decision was to reschedule all wellness visits for current patients or to use telemedicine. Capacity for that option expanded quickly, which was greatly helped by the recent decision by the Centers for Medicare & Medicaid Services to lift Medicare restrictions on the use of certain telemedicine services.
Another approach is to defer all consultations for second opinions for patients who were already undergoing treatment and to increase clinic hours of operations and capabilities for acute evaluations. This helps reserve emergency departments and hospital resources for patients who require higher-level care, the authors comment.
Treatment Decisions
Treatment decisions were more challenging to make, the authors note. One decision was that, despite the risk for COVID-19 for patients with solid tumors, adjuvant therapy with curative intent should proceed, they note. Similarly, patients with metastatic disease might lose the window of opportunity for treatment if it is delayed.
Treatment for aggressive hematologic malignancies is usually urgent, and stem cell transplant and cellular immunotherapies that provide curative treatments cannot be delayed in many cases.
Enrollment in clinical trials will most likely be limited to those trials that are most likely to benefit the patient.
Ueda noted that, because their patients come from all over the country, they are now conducting consultations for stem cell transplant by telephone so that nonlocal patients do not have to travel to Seattle. “If there is some way we can delay the treatment, we have taken that approach,” Ueda told Medscape Medical News. “If we can divert a patient to an area that is not as heavily affected, that’s another option we are taking.”
Although cancer surgery is not considered elective, surgical intervention needs to be prioritized, the authors comment. In the Seattle system, there is currently a 2-week ban on elective surgery in the healthcare system, owing to limited availability of personal protective equipment (PPE), staffing, and beds.
The oncology teams are currently reviewing treatment regimens to determine which treatments might lessen immunosuppression and which treatment options can be moved from the inpatient to the outpatient setting or can be delayed.
Inpatient Care
For hospitalized patients, several issues are being addressed. The priority is to prepare for an upcoming shortage of beds and resources because of the surge of patients with COVID-19 that is predicted. For both clinic and hospitalized patients, shortages of blood products have necessitated stricter adherence to thresholds for transfusion, and consideration is being given to lowering those thresholds.
Another important problem is the need to conserve PPE, which includes masks, gowns, gloves, and other products. The Seattle teams have implemented solutions such as favoring handwashing with soap and water over the use of hand gel for standard-precaution rooms, limiting the number of personnel entering patient rooms (so as to use less PPE), and reducing nursing procedures that require PPE, such as measuring urine output, unless they are necessary.
In addition, a no-visitor policy has been adopted in inpatient units to conserve PPE, with the exception of end-of-life situations.
The Future
The future trajectory of the COVID-19 pandemic is uncertain, Ueda commented. She emphasized that “we must continue to prepare for its widespread impact. The unknown is what we are looking at. We are expecting it to evolve, and the number of infections cannot go down.”
Ueda and coauthors end their article on a positive note. “To many of us, this has become the health care challenge of our generation, one that modern cancer therapy has never had to face. We will prevail, and when the pandemic ends, we will all be proud of what we did for our patients and each other in this critical moment for humanity.”
Two months after the first patient with COVID-19 was identified in China, the first case was reported in the United States in the Seattle, Washington, metropolitan area.
Seattle rapidly became the first US epicenter for COVID-19, and local experts are now offering their expertise and advice on how to provide optimal cancer care during the pandemic in a special feature published online March 20 in the Journal of the National Comprehensive Cancer Network.
“We began implementing measures in early March, including infection control and screening of visitors, staff, and patients at the door,” said lead author Masumi Ueda, MD, who holds positions at the Seattle Cancer Care Alliance, the University of Washington, and the Fred Hutchinson Research Center.
“A lot of changes have been implemented, and it changes on a daily basis. We are responding to the growing rate of COVID-19 infection in the community,” she told Medscape Medical News.
Ueda notes that as a result of the quick implementation of new procedures, so far, very few cancer patients at their facilities have been infected by the virus. “It has not hit our cancer population hard, which is a good thing,” she said.
Create “Incident Command Structure”
In sharing their experience, the authors emphasize the importance of keeping channels of communication open between all stakeholders ― administrators and staff, patients, caregivers, and the general public. They also recommend that each facility create an “incident command structure” that can provide early coordination of institution-wide efforts and that can rapidly respond to changing information.
Ueda noted that their command structure was set up very early on, “so we could get communication set up and start building an infrastructure for response.”
Several areas of care that required new strategies were addressed, both to protect patients and to work around staff shortages caused by possible exposure and/or school closings, as well as projected shortages of supplies and hospital resources.
First and foremost was to identify patients and visitors who had respiratory symptoms and to provide them with masks. Although this is always routine practice during the respiratory virus season, screening has now been initiated at entry points throughout the system.
“We were lucky in Seattle and Washington state in that the University of Washington virology lab developed PCR [polymerase chain reaction] testing early on for COVID-19, which subsequently got FDA approval,” said Ueda. “So we were able to have local testing and didn’t have to rely on the state lab. Testing has also been rapidly scaled up.”
Initiating a comprehensive policy for testing staff, tracking results and exposures for persons under investigation, and defining when it is possible to return to work are essential elements for maintaining a stable workforce. In addition, reinforcing a strict “stay at home when ill” policy and providing access to testing for symptomatic staff have been key to limiting exposures.
“What is unique to our region is that we had testing early on, and we are turning it around in 24 hours,” she pointed out. “This is important for staff to be able to return to work.” Currently, staff, patients, and visitors are being tested only if they show the cardinal symptoms associated with COVID-19: fever, shortness of breath, and cough, although muscle aches have recently been added to their testing protocol.
“I think if we had unlimited capacity, we might consider testing people who are asymptomatic,” Ueda noted, “although if you don’t have symptoms, you may not have the viral load needed for an accurate test.”
Educational materials explaining infection control were also needed for patients and families, along with signs and a website to provide COVID-19 education. These were quickly developed.
In addition, a telephone triage line was established for patients with mild symptoms in order to minimize exposures in clinics and to lessen the number of patients presenting at emergency departments.
Outpatient Care
Because theirs is a referral center, many cancer patients come from out of town, and so there is concern about exposing nonlocal patients to COVID-19 as the virus spreads in the Seattle area. In addition, staffing shortages due to factors such as illness, exposure, and school closures are anticipated.
To address these problems, an initial priority was to establish a “multilayer” coverage system for the clinics in the event that practitioners had to be quarantined on short notice, the authors explain.
One decision was to reschedule all wellness visits for current patients or to use telemedicine. Capacity for that option expanded quickly, which was greatly helped by the recent decision by the Centers for Medicare & Medicaid Services to lift Medicare restrictions on the use of certain telemedicine services.
Another approach is to defer all consultations for second opinions for patients who were already undergoing treatment and to increase clinic hours of operations and capabilities for acute evaluations. This helps reserve emergency departments and hospital resources for patients who require higher-level care, the authors comment.
Treatment Decisions
Treatment decisions were more challenging to make, the authors note. One decision was that, despite the risk for COVID-19 for patients with solid tumors, adjuvant therapy with curative intent should proceed, they note. Similarly, patients with metastatic disease might lose the window of opportunity for treatment if it is delayed.
Treatment for aggressive hematologic malignancies is usually urgent, and stem cell transplant and cellular immunotherapies that provide curative treatments cannot be delayed in many cases.
Enrollment in clinical trials will most likely be limited to those trials that are most likely to benefit the patient.
Ueda noted that, because their patients come from all over the country, they are now conducting consultations for stem cell transplant by telephone so that nonlocal patients do not have to travel to Seattle. “If there is some way we can delay the treatment, we have taken that approach,” Ueda told Medscape Medical News. “If we can divert a patient to an area that is not as heavily affected, that’s another option we are taking.”
Although cancer surgery is not considered elective, surgical intervention needs to be prioritized, the authors comment. In the Seattle system, there is currently a 2-week ban on elective surgery in the healthcare system, owing to limited availability of personal protective equipment (PPE), staffing, and beds.
The oncology teams are currently reviewing treatment regimens to determine which treatments might lessen immunosuppression and which treatment options can be moved from the inpatient to the outpatient setting or can be delayed.
Inpatient Care
For hospitalized patients, several issues are being addressed. The priority is to prepare for an upcoming shortage of beds and resources because of the surge of patients with COVID-19 that is predicted. For both clinic and hospitalized patients, shortages of blood products have necessitated stricter adherence to thresholds for transfusion, and consideration is being given to lowering those thresholds.
Another important problem is the need to conserve PPE, which includes masks, gowns, gloves, and other products. The Seattle teams have implemented solutions such as favoring handwashing with soap and water over the use of hand gel for standard-precaution rooms, limiting the number of personnel entering patient rooms (so as to use less PPE), and reducing nursing procedures that require PPE, such as measuring urine output, unless they are necessary.
In addition, a no-visitor policy has been adopted in inpatient units to conserve PPE, with the exception of end-of-life situations.
The Future
The future trajectory of the COVID-19 pandemic is uncertain, Ueda commented. She emphasized that “we must continue to prepare for its widespread impact. The unknown is what we are looking at. We are expecting it to evolve, and the number of infections cannot go down.”
Ueda and coauthors end their article on a positive note. “To many of us, this has become the health care challenge of our generation, one that modern cancer therapy has never had to face. We will prevail, and when the pandemic ends, we will all be proud of what we did for our patients and each other in this critical moment for humanity.”
How is oncology adapting to COVID-19?
As the coronavirus pandemic escalates in the United States, Medscape Oncology reached out to a group of our contributors and asked them to provide their perspective on how their oncology departments and centers are preparing. Here are their responses to a number of issues facing oncologists in the US and around the world.
Have you shifted nonurgent follow-up visits to telemedicine, either via video or phone?
Kathy Miller, MD, Associate Director of Indiana University Simon Cancer Center: We are reviewing our clinic schedules and identifying “routine” follow-up patients who can be rescheduled. When patients are contacted to reschedule, they are asked if they have any urgent, immediate concerns that need to be addressed before the new appointment. If yes, they are offered a virtual visit.
Don Dizon, MD, Director of Women’s Cancers, Lifespan Cancer Institute; Director of Medical Oncology, Rhode Island Hospital: We have started to do this in preparation for a surge of people with COVID-19. Patients who are in long-term follow-up (no evidence of disease at 3 years or longer, being seen annually) or those in routine surveillance after curative treatment (that is, seen every 3 months) as well as those being seen for supportive care–type visits, like sexual health or survivorship, are all being contacted and visits are being moved to telehealth.
Jeffrey S. Weber, MD, PhD, Deputy Director of the Laura and Isaac Perlmutter Cancer Center at NYU Langone Medical Center: Yes. Any follow-up, nontreatment visits are done by phone or video if the patient agrees. (They all have).
Have you delayed or canceled cancer surgeries?
Ravi B. Parikh, MD, MPP, Medical oncologist at the University of Pennsylvania and the Philadelphia VA Medical Center: The University of Pennsylvania has taken this seriously. We’ve canceled all elective surgeries, have ramped up our telemedicine (video and phone) capabilities significantly, are limiting our appointments mostly to on-treatment visits, and have been asked to reconsider regular scans and reviews.
Dizon: We have not done this. There are apparently differences in interpretation in what institutions might mean as “elective surgeries.” At our institution, surgery for invasive malignancies is not elective. However, this may (or will) change if resources become an issue.
Lidia Schapira, MD, Associate Professor of Medicine and Director of Cancer Survivorship at the Stanford Comprehensive Cancer Institute: Delaying elective surgery is something that hospitals here have already implemented, and I imagine that this trend will spread. But it may be difficult to decide in situations that are not exactly “life-saving” but where an earlier intervention could preserve function or improve quality of life.
Mark A. Lewis, MD, Director of Gastrointestinal Oncology at Intermountain Healthcare in Utah: Cancer surgeries have not been deemed elective or delayed.
Have you delayed or altered the delivery of potentially immune-comprising treatments?
David Kerr, MD, Professor of Cancer Medicine at the University of Oxford in England: We are considering delaying initiation of our adjuvant colorectal cancer treatments, as we have data from our own QUASAR trials suggesting that patients who commence chemotherapy between 2 and 6 weeks do equally as well as those who begin 6-12 weeks after surgery.
Parikh: I personally haven’t delayed giving chemotherapy to avoid immune compromise, but I believe some others may have. It’s a delicate balance between wanting to ensure cancer control and making sure we are flattening the curve. As an example, though, I delayed three on-treatment visits for my clinic last Monday, and I converted 70% of my visits to telemedicine. However, I’m a genitourinary cancer specialist and the treatments I give are very different from others.
Lewis: The most difficult calculus is around adjuvant therapy. For metastatic patients, I am trying to use the least immunosuppressive regimen possible that will still control their disease. As you can imagine, it’s an assessment of competing risks.
Schapira: Patients who need essential anticancer therapy should still get it, but attempts to deintensify therapy should continue—for example, holding or postponing treatment without harm (based on evidence, not opinion). This may be possible for patients considering hormonal therapies for breast or prostate cancer.
Patients who need radiation should discuss the timing with their radiation oncologist. In some cases, it may be possible to delay treatment without affecting outcomes, but these decisions should be made carefully. Alternatively, shorter courses of radiation may be appropriate.
Have you advised your own patients differently given the high risk to cancer patients?
Kerr: We have factored potential infection with the virus into discussions where the benefits of chemotherapy are very marginal. This could tip the balance toward the patient deciding not to pursue chemotherapy.
Dizon: The data from China are not entirely crystal-clear. While they noted that people with active cancer and those who had a history of cancer are at increased risk for more severe infections and worse outcomes, the Chinese cohort was small, and compared with people without cancer, it tended to be much older and to be smokers (former or current). Having said this, we are counseling everyone about the importance of social distancing, washing hands, and not touching your face.
Lewis: If I have a complete blood count with a differential that includes lymphocytes, I can advise my lymphopenic patients (who are particularly vulnerable to viral infection) to take special precautions regarding social distancing in their own families.
Have any of your hospitalized patients been affected by policy changes to prepare beds/departments for the expected increase in COVID-19–positive patients?
Weber: Not yet.
Dizon: No, not at the moment.
Have you been asked to assist with other services or COVID-19 task forces?
Dizon: I am keenly involved in the preparations and modifications to procedures, including staffing decisions in outpatient, movement to telehealth, and work-from-home policies.
Lewis: I am engaged in system-wide COVID-19 efforts around oncology.
Kerr: Perhaps oddest of all, I am learning with some of our junior doctors to care for ventilated patients. I still consider myself enough of a general physician that I would hope to be able to contribute to the truly sick, but I accept that I do need an appropriate refresher course.
Bishal Gyawali, MD, PhD, medical oncologist at Queen’s University Cancer Research Institute: Queen’s Hospital medical students are now volunteering to help with daycare, groceries, and other tasks for staff who are working in the hospital.
Are you experiencing any shortages in personal protective equipment (PPE) at your center?
Miller: Some supplies are running short, though none are frankly out at this point. However, rationing and controls are in place to stretch the supplies as far as possible, including reusing some PPE.
Dizon: We are rationing face masks and N95 respirators, eye shields, and even surgical scrubs. We are talking about postponing elective surgery to save PPE but are not yet to that point. We’re asking that face masks be reused for at least 2 days, maybe longer. PPEs are one per day. Scrubs are kept secure.
Lewis: We are being very careful not to overuse PPE but currently have an adequate inventory. We have had to move gloves and masks to areas where they are not accessible to the general public, as otherwise they were being stolen (this started weeks ago).
Kerr: Our National Health System has an adequate supply of PPE equipment centrally, but there seems to be a problem with distribution, as some hospitals are reporting shortages.
Weber: Masks are in short supply, so they are being used for several days if not wet. We are short of plastic gowns and are using paper chemo gowns. Similar story at many places.
This article first appeared on Medscape.com.
As the coronavirus pandemic escalates in the United States, Medscape Oncology reached out to a group of our contributors and asked them to provide their perspective on how their oncology departments and centers are preparing. Here are their responses to a number of issues facing oncologists in the US and around the world.
Have you shifted nonurgent follow-up visits to telemedicine, either via video or phone?
Kathy Miller, MD, Associate Director of Indiana University Simon Cancer Center: We are reviewing our clinic schedules and identifying “routine” follow-up patients who can be rescheduled. When patients are contacted to reschedule, they are asked if they have any urgent, immediate concerns that need to be addressed before the new appointment. If yes, they are offered a virtual visit.
Don Dizon, MD, Director of Women’s Cancers, Lifespan Cancer Institute; Director of Medical Oncology, Rhode Island Hospital: We have started to do this in preparation for a surge of people with COVID-19. Patients who are in long-term follow-up (no evidence of disease at 3 years or longer, being seen annually) or those in routine surveillance after curative treatment (that is, seen every 3 months) as well as those being seen for supportive care–type visits, like sexual health or survivorship, are all being contacted and visits are being moved to telehealth.
Jeffrey S. Weber, MD, PhD, Deputy Director of the Laura and Isaac Perlmutter Cancer Center at NYU Langone Medical Center: Yes. Any follow-up, nontreatment visits are done by phone or video if the patient agrees. (They all have).
Have you delayed or canceled cancer surgeries?
Ravi B. Parikh, MD, MPP, Medical oncologist at the University of Pennsylvania and the Philadelphia VA Medical Center: The University of Pennsylvania has taken this seriously. We’ve canceled all elective surgeries, have ramped up our telemedicine (video and phone) capabilities significantly, are limiting our appointments mostly to on-treatment visits, and have been asked to reconsider regular scans and reviews.
Dizon: We have not done this. There are apparently differences in interpretation in what institutions might mean as “elective surgeries.” At our institution, surgery for invasive malignancies is not elective. However, this may (or will) change if resources become an issue.
Lidia Schapira, MD, Associate Professor of Medicine and Director of Cancer Survivorship at the Stanford Comprehensive Cancer Institute: Delaying elective surgery is something that hospitals here have already implemented, and I imagine that this trend will spread. But it may be difficult to decide in situations that are not exactly “life-saving” but where an earlier intervention could preserve function or improve quality of life.
Mark A. Lewis, MD, Director of Gastrointestinal Oncology at Intermountain Healthcare in Utah: Cancer surgeries have not been deemed elective or delayed.
Have you delayed or altered the delivery of potentially immune-comprising treatments?
David Kerr, MD, Professor of Cancer Medicine at the University of Oxford in England: We are considering delaying initiation of our adjuvant colorectal cancer treatments, as we have data from our own QUASAR trials suggesting that patients who commence chemotherapy between 2 and 6 weeks do equally as well as those who begin 6-12 weeks after surgery.
Parikh: I personally haven’t delayed giving chemotherapy to avoid immune compromise, but I believe some others may have. It’s a delicate balance between wanting to ensure cancer control and making sure we are flattening the curve. As an example, though, I delayed three on-treatment visits for my clinic last Monday, and I converted 70% of my visits to telemedicine. However, I’m a genitourinary cancer specialist and the treatments I give are very different from others.
Lewis: The most difficult calculus is around adjuvant therapy. For metastatic patients, I am trying to use the least immunosuppressive regimen possible that will still control their disease. As you can imagine, it’s an assessment of competing risks.
Schapira: Patients who need essential anticancer therapy should still get it, but attempts to deintensify therapy should continue—for example, holding or postponing treatment without harm (based on evidence, not opinion). This may be possible for patients considering hormonal therapies for breast or prostate cancer.
Patients who need radiation should discuss the timing with their radiation oncologist. In some cases, it may be possible to delay treatment without affecting outcomes, but these decisions should be made carefully. Alternatively, shorter courses of radiation may be appropriate.
Have you advised your own patients differently given the high risk to cancer patients?
Kerr: We have factored potential infection with the virus into discussions where the benefits of chemotherapy are very marginal. This could tip the balance toward the patient deciding not to pursue chemotherapy.
Dizon: The data from China are not entirely crystal-clear. While they noted that people with active cancer and those who had a history of cancer are at increased risk for more severe infections and worse outcomes, the Chinese cohort was small, and compared with people without cancer, it tended to be much older and to be smokers (former or current). Having said this, we are counseling everyone about the importance of social distancing, washing hands, and not touching your face.
Lewis: If I have a complete blood count with a differential that includes lymphocytes, I can advise my lymphopenic patients (who are particularly vulnerable to viral infection) to take special precautions regarding social distancing in their own families.
Have any of your hospitalized patients been affected by policy changes to prepare beds/departments for the expected increase in COVID-19–positive patients?
Weber: Not yet.
Dizon: No, not at the moment.
Have you been asked to assist with other services or COVID-19 task forces?
Dizon: I am keenly involved in the preparations and modifications to procedures, including staffing decisions in outpatient, movement to telehealth, and work-from-home policies.
Lewis: I am engaged in system-wide COVID-19 efforts around oncology.
Kerr: Perhaps oddest of all, I am learning with some of our junior doctors to care for ventilated patients. I still consider myself enough of a general physician that I would hope to be able to contribute to the truly sick, but I accept that I do need an appropriate refresher course.
Bishal Gyawali, MD, PhD, medical oncologist at Queen’s University Cancer Research Institute: Queen’s Hospital medical students are now volunteering to help with daycare, groceries, and other tasks for staff who are working in the hospital.
Are you experiencing any shortages in personal protective equipment (PPE) at your center?
Miller: Some supplies are running short, though none are frankly out at this point. However, rationing and controls are in place to stretch the supplies as far as possible, including reusing some PPE.
Dizon: We are rationing face masks and N95 respirators, eye shields, and even surgical scrubs. We are talking about postponing elective surgery to save PPE but are not yet to that point. We’re asking that face masks be reused for at least 2 days, maybe longer. PPEs are one per day. Scrubs are kept secure.
Lewis: We are being very careful not to overuse PPE but currently have an adequate inventory. We have had to move gloves and masks to areas where they are not accessible to the general public, as otherwise they were being stolen (this started weeks ago).
Kerr: Our National Health System has an adequate supply of PPE equipment centrally, but there seems to be a problem with distribution, as some hospitals are reporting shortages.
Weber: Masks are in short supply, so they are being used for several days if not wet. We are short of plastic gowns and are using paper chemo gowns. Similar story at many places.
This article first appeared on Medscape.com.
As the coronavirus pandemic escalates in the United States, Medscape Oncology reached out to a group of our contributors and asked them to provide their perspective on how their oncology departments and centers are preparing. Here are their responses to a number of issues facing oncologists in the US and around the world.
Have you shifted nonurgent follow-up visits to telemedicine, either via video or phone?
Kathy Miller, MD, Associate Director of Indiana University Simon Cancer Center: We are reviewing our clinic schedules and identifying “routine” follow-up patients who can be rescheduled. When patients are contacted to reschedule, they are asked if they have any urgent, immediate concerns that need to be addressed before the new appointment. If yes, they are offered a virtual visit.
Don Dizon, MD, Director of Women’s Cancers, Lifespan Cancer Institute; Director of Medical Oncology, Rhode Island Hospital: We have started to do this in preparation for a surge of people with COVID-19. Patients who are in long-term follow-up (no evidence of disease at 3 years or longer, being seen annually) or those in routine surveillance after curative treatment (that is, seen every 3 months) as well as those being seen for supportive care–type visits, like sexual health or survivorship, are all being contacted and visits are being moved to telehealth.
Jeffrey S. Weber, MD, PhD, Deputy Director of the Laura and Isaac Perlmutter Cancer Center at NYU Langone Medical Center: Yes. Any follow-up, nontreatment visits are done by phone or video if the patient agrees. (They all have).
Have you delayed or canceled cancer surgeries?
Ravi B. Parikh, MD, MPP, Medical oncologist at the University of Pennsylvania and the Philadelphia VA Medical Center: The University of Pennsylvania has taken this seriously. We’ve canceled all elective surgeries, have ramped up our telemedicine (video and phone) capabilities significantly, are limiting our appointments mostly to on-treatment visits, and have been asked to reconsider regular scans and reviews.
Dizon: We have not done this. There are apparently differences in interpretation in what institutions might mean as “elective surgeries.” At our institution, surgery for invasive malignancies is not elective. However, this may (or will) change if resources become an issue.
Lidia Schapira, MD, Associate Professor of Medicine and Director of Cancer Survivorship at the Stanford Comprehensive Cancer Institute: Delaying elective surgery is something that hospitals here have already implemented, and I imagine that this trend will spread. But it may be difficult to decide in situations that are not exactly “life-saving” but where an earlier intervention could preserve function or improve quality of life.
Mark A. Lewis, MD, Director of Gastrointestinal Oncology at Intermountain Healthcare in Utah: Cancer surgeries have not been deemed elective or delayed.
Have you delayed or altered the delivery of potentially immune-comprising treatments?
David Kerr, MD, Professor of Cancer Medicine at the University of Oxford in England: We are considering delaying initiation of our adjuvant colorectal cancer treatments, as we have data from our own QUASAR trials suggesting that patients who commence chemotherapy between 2 and 6 weeks do equally as well as those who begin 6-12 weeks after surgery.
Parikh: I personally haven’t delayed giving chemotherapy to avoid immune compromise, but I believe some others may have. It’s a delicate balance between wanting to ensure cancer control and making sure we are flattening the curve. As an example, though, I delayed three on-treatment visits for my clinic last Monday, and I converted 70% of my visits to telemedicine. However, I’m a genitourinary cancer specialist and the treatments I give are very different from others.
Lewis: The most difficult calculus is around adjuvant therapy. For metastatic patients, I am trying to use the least immunosuppressive regimen possible that will still control their disease. As you can imagine, it’s an assessment of competing risks.
Schapira: Patients who need essential anticancer therapy should still get it, but attempts to deintensify therapy should continue—for example, holding or postponing treatment without harm (based on evidence, not opinion). This may be possible for patients considering hormonal therapies for breast or prostate cancer.
Patients who need radiation should discuss the timing with their radiation oncologist. In some cases, it may be possible to delay treatment without affecting outcomes, but these decisions should be made carefully. Alternatively, shorter courses of radiation may be appropriate.
Have you advised your own patients differently given the high risk to cancer patients?
Kerr: We have factored potential infection with the virus into discussions where the benefits of chemotherapy are very marginal. This could tip the balance toward the patient deciding not to pursue chemotherapy.
Dizon: The data from China are not entirely crystal-clear. While they noted that people with active cancer and those who had a history of cancer are at increased risk for more severe infections and worse outcomes, the Chinese cohort was small, and compared with people without cancer, it tended to be much older and to be smokers (former or current). Having said this, we are counseling everyone about the importance of social distancing, washing hands, and not touching your face.
Lewis: If I have a complete blood count with a differential that includes lymphocytes, I can advise my lymphopenic patients (who are particularly vulnerable to viral infection) to take special precautions regarding social distancing in their own families.
Have any of your hospitalized patients been affected by policy changes to prepare beds/departments for the expected increase in COVID-19–positive patients?
Weber: Not yet.
Dizon: No, not at the moment.
Have you been asked to assist with other services or COVID-19 task forces?
Dizon: I am keenly involved in the preparations and modifications to procedures, including staffing decisions in outpatient, movement to telehealth, and work-from-home policies.
Lewis: I am engaged in system-wide COVID-19 efforts around oncology.
Kerr: Perhaps oddest of all, I am learning with some of our junior doctors to care for ventilated patients. I still consider myself enough of a general physician that I would hope to be able to contribute to the truly sick, but I accept that I do need an appropriate refresher course.
Bishal Gyawali, MD, PhD, medical oncologist at Queen’s University Cancer Research Institute: Queen’s Hospital medical students are now volunteering to help with daycare, groceries, and other tasks for staff who are working in the hospital.
Are you experiencing any shortages in personal protective equipment (PPE) at your center?
Miller: Some supplies are running short, though none are frankly out at this point. However, rationing and controls are in place to stretch the supplies as far as possible, including reusing some PPE.
Dizon: We are rationing face masks and N95 respirators, eye shields, and even surgical scrubs. We are talking about postponing elective surgery to save PPE but are not yet to that point. We’re asking that face masks be reused for at least 2 days, maybe longer. PPEs are one per day. Scrubs are kept secure.
Lewis: We are being very careful not to overuse PPE but currently have an adequate inventory. We have had to move gloves and masks to areas where they are not accessible to the general public, as otherwise they were being stolen (this started weeks ago).
Kerr: Our National Health System has an adequate supply of PPE equipment centrally, but there seems to be a problem with distribution, as some hospitals are reporting shortages.
Weber: Masks are in short supply, so they are being used for several days if not wet. We are short of plastic gowns and are using paper chemo gowns. Similar story at many places.
This article first appeared on Medscape.com.
Disruptions in cancer care in the era of COVID-19
Editor’s note: Find the latest COVID-19 news and guidance in Medscape’s Coronavirus Resource Center.
Even in the midst of the COVID-19 pandemic, cancer care must go on, but changes may need to be made in the way some care is delivered.

“We’re headed for a time when there will be significant disruptions in the care of patients with cancer,” said Len Lichtenfeld, MD, deputy chief medical officer of the American Cancer Society (ACS), in a statement. “For some it may be as straightforward as a delay in having elective surgery. For others it may be delaying preventive care or adjuvant chemotherapy that’s meant to keep cancer from returning or rescheduling appointments.”
Lichtenfeld emphasized that cancer care teams are going to do the best they can to deliver care to those most in need. However, even in those circumstances, it won’t be life as usual. “It will require patience on everyone’s part as we go through this pandemic,” he said.
“The way we treat cancer over the next few months will change enormously,” writes a British oncologist in an article published in the Guardian.
“As oncologists, we will have to find a tenuous balance between undertreating people with cancer, resulting in more deaths from the disease in the medium to long term, and increasing deaths from COVID-19 in a vulnerable patient population. Alongside our patients we will have to make difficult decisions regarding treatments, with only low-quality evidence to guide us,” writes Lucy Gossage, MD, consultant oncologist at Nottingham University Hospital, UK.
The evidence to date (from reports from China in Lancet Oncology) suggests that people with cancer have a significantly higher risk of severe illness resulting in intensive care admissions or death when infected with COVID-19, particularly if they recently had chemotherapy or surgery.
“Many of the oncology treatments we currently use, especially those given after surgery to reduce risk of cancer recurrence, have relatively small benefits,” she writes.
“In the current climate, the balance of offering these treatments may shift; a small reduction in risk of cancer recurrence over the next 5 years may be outweighed by the potential for a short-term increase in risk of death from COVID-19. In the long term, more people’s cancer will return if we aren’t able to offer these treatments,” she adds.
Postpone Routine Screening
One thing that can go on the back burner for now is routine cancer screening, which can be postponed for now in order to conserve health system resources and reduce contact with healthcare facilities, says the ACS.
“Patients seeking routine cancer screenings should delay those until further notice,” said Lichtenfeld. “While timely screening is important, the need to prevent the spread of coronavirus and to reduce the strain on the medical system is more important right now.”
But as soon as restrictions to slow the spread of COVID-19 are lifted and routine visits to health facilities are safe, regular screening tests should be rescheduled.
Guidance From ASCO
The American Society of Clinical Oncology (ASCO) has issued new guidance on caring for patients with cancer during the COVID-19 outbreak.
First and foremost, ASCO encourages providers, facilities, and anyone caring for patients with cancer to follow the existing guidelines from the Center for Disease Control and Prevention when possible.
ASCO highlights the CDC’s general recommendation for healthcare facilities that suggests “elective surgeries” at inpatient facilities be rescheduled if possible, which has also been recommended by the American College of Surgeons.
However, in many cases, cancer surgery is not elective but essential, it points out. So this is largely an individual determination that clinicians and patients will need to make, taking into account the potential harms of delaying needed cancer-related surgery.
Systemic treatments, including chemotherapy and immunotherapy, leave cancer patients vulnerable to infection, but ASCO says there is no direct evidence to support changes in regimens during the pandemic. Therefore, routinely stopping anticancer or immunosuppressive therapy is not recommended, as the balance of potential harms that may result from delaying or interrupting treatment versus the potential benefits of possibly preventing or delaying COVID-19 infection remains very unclear.
Clinical decisions must be individualized, ASCO emphasized, and suggested the following practice points be considered:
- For patients already in deep remission who are receiving maintenance therapy, stopping treatment may be an option.
- Some patients may be able to switch from IV to oral therapies, which would decrease the frequency of clinic visits.
- Decisions on modifying or withholding chemotherapy need to consider both the indication and goals of care, as well as where the patient is in the treatment regimen and tolerance to the therapy. As an example, the risk–benefit assessment for proceeding with chemotherapy in patients with untreated extensive small-cell lung cancer is quite different than proceeding with maintenance pemetrexed for metastatic non–small cell lung cancer.
- If local coronavirus transmission is an issue at a particular cancer center, reasonable options may include taking a 2-week treatment break or arranging treatment at a different facility.
- Evaluate if home infusion is medically and logistically feasible.
- In some settings, delaying or modifying adjuvant treatment presents a higher risk of compromised disease control and long-term survival than in others, but in cases where the absolute benefit of adjuvant chemotherapy may be quite small and other options are available, the risk of COVID-19 may be considered an additional factor when evaluating care.
Delay Stem Cell Transplants
For patients who are candidates for allogeneic stem cell transplantation, a delay may be reasonable if the patient is currently well controlled with conventional treatment, ASCO comments. It also directs clinicians to follow the recommendations provided by the American Society of Transplantation and Cellular Therapy and from the European Society for Blood and Marrow Transplantation regarding this issue.
Finally, there is also the question of prophylactic antiviral therapy: Should it be considered for cancer patients undergoing active therapy?
The answer to that question is currently unknown, says ASCO, but “this is an active area of research and evidence may be available at any time.”
This article first appeared on Medscape.com.
Editor’s note: Find the latest COVID-19 news and guidance in Medscape’s Coronavirus Resource Center.
Even in the midst of the COVID-19 pandemic, cancer care must go on, but changes may need to be made in the way some care is delivered.
“We’re headed for a time when there will be significant disruptions in the care of patients with cancer,” said Len Lichtenfeld, MD, deputy chief medical officer of the American Cancer Society (ACS), in a statement. “For some it may be as straightforward as a delay in having elective surgery. For others it may be delaying preventive care or adjuvant chemotherapy that’s meant to keep cancer from returning or rescheduling appointments.”
Lichtenfeld emphasized that cancer care teams are going to do the best they can to deliver care to those most in need. However, even in those circumstances, it won’t be life as usual. “It will require patience on everyone’s part as we go through this pandemic,” he said.
“The way we treat cancer over the next few months will change enormously,” writes a British oncologist in an article published in the Guardian.
“As oncologists, we will have to find a tenuous balance between undertreating people with cancer, resulting in more deaths from the disease in the medium to long term, and increasing deaths from COVID-19 in a vulnerable patient population. Alongside our patients we will have to make difficult decisions regarding treatments, with only low-quality evidence to guide us,” writes Lucy Gossage, MD, consultant oncologist at Nottingham University Hospital, UK.
The evidence to date (from reports from China in Lancet Oncology) suggests that people with cancer have a significantly higher risk of severe illness resulting in intensive care admissions or death when infected with COVID-19, particularly if they recently had chemotherapy or surgery.
“Many of the oncology treatments we currently use, especially those given after surgery to reduce risk of cancer recurrence, have relatively small benefits,” she writes.
“In the current climate, the balance of offering these treatments may shift; a small reduction in risk of cancer recurrence over the next 5 years may be outweighed by the potential for a short-term increase in risk of death from COVID-19. In the long term, more people’s cancer will return if we aren’t able to offer these treatments,” she adds.
Postpone Routine Screening
One thing that can go on the back burner for now is routine cancer screening, which can be postponed for now in order to conserve health system resources and reduce contact with healthcare facilities, says the ACS.
“Patients seeking routine cancer screenings should delay those until further notice,” said Lichtenfeld. “While timely screening is important, the need to prevent the spread of coronavirus and to reduce the strain on the medical system is more important right now.”
But as soon as restrictions to slow the spread of COVID-19 are lifted and routine visits to health facilities are safe, regular screening tests should be rescheduled.
Guidance From ASCO
The American Society of Clinical Oncology (ASCO) has issued new guidance on caring for patients with cancer during the COVID-19 outbreak.
First and foremost, ASCO encourages providers, facilities, and anyone caring for patients with cancer to follow the existing guidelines from the Center for Disease Control and Prevention when possible.
ASCO highlights the CDC’s general recommendation for healthcare facilities that suggests “elective surgeries” at inpatient facilities be rescheduled if possible, which has also been recommended by the American College of Surgeons.
However, in many cases, cancer surgery is not elective but essential, it points out. So this is largely an individual determination that clinicians and patients will need to make, taking into account the potential harms of delaying needed cancer-related surgery.
Systemic treatments, including chemotherapy and immunotherapy, leave cancer patients vulnerable to infection, but ASCO says there is no direct evidence to support changes in regimens during the pandemic. Therefore, routinely stopping anticancer or immunosuppressive therapy is not recommended, as the balance of potential harms that may result from delaying or interrupting treatment versus the potential benefits of possibly preventing or delaying COVID-19 infection remains very unclear.
Clinical decisions must be individualized, ASCO emphasized, and suggested the following practice points be considered:
- For patients already in deep remission who are receiving maintenance therapy, stopping treatment may be an option.
- Some patients may be able to switch from IV to oral therapies, which would decrease the frequency of clinic visits.
- Decisions on modifying or withholding chemotherapy need to consider both the indication and goals of care, as well as where the patient is in the treatment regimen and tolerance to the therapy. As an example, the risk–benefit assessment for proceeding with chemotherapy in patients with untreated extensive small-cell lung cancer is quite different than proceeding with maintenance pemetrexed for metastatic non–small cell lung cancer.
- If local coronavirus transmission is an issue at a particular cancer center, reasonable options may include taking a 2-week treatment break or arranging treatment at a different facility.
- Evaluate if home infusion is medically and logistically feasible.
- In some settings, delaying or modifying adjuvant treatment presents a higher risk of compromised disease control and long-term survival than in others, but in cases where the absolute benefit of adjuvant chemotherapy may be quite small and other options are available, the risk of COVID-19 may be considered an additional factor when evaluating care.
Delay Stem Cell Transplants
For patients who are candidates for allogeneic stem cell transplantation, a delay may be reasonable if the patient is currently well controlled with conventional treatment, ASCO comments. It also directs clinicians to follow the recommendations provided by the American Society of Transplantation and Cellular Therapy and from the European Society for Blood and Marrow Transplantation regarding this issue.
Finally, there is also the question of prophylactic antiviral therapy: Should it be considered for cancer patients undergoing active therapy?
The answer to that question is currently unknown, says ASCO, but “this is an active area of research and evidence may be available at any time.”
This article first appeared on Medscape.com.
Editor’s note: Find the latest COVID-19 news and guidance in Medscape’s Coronavirus Resource Center.
Even in the midst of the COVID-19 pandemic, cancer care must go on, but changes may need to be made in the way some care is delivered.
“We’re headed for a time when there will be significant disruptions in the care of patients with cancer,” said Len Lichtenfeld, MD, deputy chief medical officer of the American Cancer Society (ACS), in a statement. “For some it may be as straightforward as a delay in having elective surgery. For others it may be delaying preventive care or adjuvant chemotherapy that’s meant to keep cancer from returning or rescheduling appointments.”
Lichtenfeld emphasized that cancer care teams are going to do the best they can to deliver care to those most in need. However, even in those circumstances, it won’t be life as usual. “It will require patience on everyone’s part as we go through this pandemic,” he said.
“The way we treat cancer over the next few months will change enormously,” writes a British oncologist in an article published in the Guardian.
“As oncologists, we will have to find a tenuous balance between undertreating people with cancer, resulting in more deaths from the disease in the medium to long term, and increasing deaths from COVID-19 in a vulnerable patient population. Alongside our patients we will have to make difficult decisions regarding treatments, with only low-quality evidence to guide us,” writes Lucy Gossage, MD, consultant oncologist at Nottingham University Hospital, UK.
The evidence to date (from reports from China in Lancet Oncology) suggests that people with cancer have a significantly higher risk of severe illness resulting in intensive care admissions or death when infected with COVID-19, particularly if they recently had chemotherapy or surgery.
“Many of the oncology treatments we currently use, especially those given after surgery to reduce risk of cancer recurrence, have relatively small benefits,” she writes.
“In the current climate, the balance of offering these treatments may shift; a small reduction in risk of cancer recurrence over the next 5 years may be outweighed by the potential for a short-term increase in risk of death from COVID-19. In the long term, more people’s cancer will return if we aren’t able to offer these treatments,” she adds.
Postpone Routine Screening
One thing that can go on the back burner for now is routine cancer screening, which can be postponed for now in order to conserve health system resources and reduce contact with healthcare facilities, says the ACS.
“Patients seeking routine cancer screenings should delay those until further notice,” said Lichtenfeld. “While timely screening is important, the need to prevent the spread of coronavirus and to reduce the strain on the medical system is more important right now.”
But as soon as restrictions to slow the spread of COVID-19 are lifted and routine visits to health facilities are safe, regular screening tests should be rescheduled.
Guidance From ASCO
The American Society of Clinical Oncology (ASCO) has issued new guidance on caring for patients with cancer during the COVID-19 outbreak.
First and foremost, ASCO encourages providers, facilities, and anyone caring for patients with cancer to follow the existing guidelines from the Center for Disease Control and Prevention when possible.
ASCO highlights the CDC’s general recommendation for healthcare facilities that suggests “elective surgeries” at inpatient facilities be rescheduled if possible, which has also been recommended by the American College of Surgeons.
However, in many cases, cancer surgery is not elective but essential, it points out. So this is largely an individual determination that clinicians and patients will need to make, taking into account the potential harms of delaying needed cancer-related surgery.
Systemic treatments, including chemotherapy and immunotherapy, leave cancer patients vulnerable to infection, but ASCO says there is no direct evidence to support changes in regimens during the pandemic. Therefore, routinely stopping anticancer or immunosuppressive therapy is not recommended, as the balance of potential harms that may result from delaying or interrupting treatment versus the potential benefits of possibly preventing or delaying COVID-19 infection remains very unclear.
Clinical decisions must be individualized, ASCO emphasized, and suggested the following practice points be considered:
- For patients already in deep remission who are receiving maintenance therapy, stopping treatment may be an option.
- Some patients may be able to switch from IV to oral therapies, which would decrease the frequency of clinic visits.
- Decisions on modifying or withholding chemotherapy need to consider both the indication and goals of care, as well as where the patient is in the treatment regimen and tolerance to the therapy. As an example, the risk–benefit assessment for proceeding with chemotherapy in patients with untreated extensive small-cell lung cancer is quite different than proceeding with maintenance pemetrexed for metastatic non–small cell lung cancer.
- If local coronavirus transmission is an issue at a particular cancer center, reasonable options may include taking a 2-week treatment break or arranging treatment at a different facility.
- Evaluate if home infusion is medically and logistically feasible.
- In some settings, delaying or modifying adjuvant treatment presents a higher risk of compromised disease control and long-term survival than in others, but in cases where the absolute benefit of adjuvant chemotherapy may be quite small and other options are available, the risk of COVID-19 may be considered an additional factor when evaluating care.
Delay Stem Cell Transplants
For patients who are candidates for allogeneic stem cell transplantation, a delay may be reasonable if the patient is currently well controlled with conventional treatment, ASCO comments. It also directs clinicians to follow the recommendations provided by the American Society of Transplantation and Cellular Therapy and from the European Society for Blood and Marrow Transplantation regarding this issue.
Finally, there is also the question of prophylactic antiviral therapy: Should it be considered for cancer patients undergoing active therapy?
The answer to that question is currently unknown, says ASCO, but “this is an active area of research and evidence may be available at any time.”
This article first appeared on Medscape.com.
Largest meeting on cancer research canceled: AACR
The biggest cancer research meeting of the year has been canceled as a reaction to the novel coronavirus (COVID-19) outbreak, which has also led to many other medical conferences being canceled or postponed.
The annual meeting of the American Association for Cancer Research (AACR) was due to take place April 24-29 in San Diego, California. More than 24,000 delegates from 80 countries and more than 500 exhibitors were expected to attend.
There are plans to reschedule it for later this year.
This has been a “difficult decision,” said the AACR board of directors, but “we believe that the decision to postpone the meeting is absolutely the correct one to safeguard our meeting participants from further potential exposure to the coronavirus.”
The board goes on to explain that “this evidence-based decision was made after a thorough review and discussion of all factors impacting the annual meeting, including the US government’s enforcement of restrictions on international travelers to enter the US; the imposition of travel restrictions issued by US government agencies, cancer centers, academic institutions, and pharmaceutical and biotech companies; and the counsel of infectious disease experts. It is clear that all of these elements significantly affect the ability of delegates, speakers, presenters of proffered papers, and exhibitors to participate fully in the annual meeting.”
Other cancer conferences that were planned for March and that have been canceled include the following:
- European Breast Cancer Conference (EBCC), Barcelona, Spain, which was to have taken place March 18-20. This conference has been postponed and will now take place September 30 to October 2 at the same venue. Abstracts that have been accepted for the initial conference will remain in the program, and organizers will reopen abstract submissions in May.
- National Comprehensive Cancer Network (NCCN), Orlando, Florida, was scheduled for March 19-22. This conference has been postponed. No new dates have been provided, but the society notes that “NCCN staff is working as quickly as possible to notify all conference registrants about the postponement and further information regarding the refund process.”
- European Association of Urology (EAU), Amsterdam, the Netherlands, at which there is always new research presented on prostate, kidney, and bladder cancer, was due to take place March 20-24. This conference has been postponed to July 2020.
- Society of Gynecologic Oncology (SGO), in Toronto, Canada, which was scheduled for March 28-31. SGO is “exploring alternatives for delivering the science and education.”
Overall, the move to cancel medical conferences over the next few months is a good idea, commented F. Perry Wilson, MD, MSCE, associate professor of medicine and director of Yale’s Program of Applied Translational Research, in a Medscape Medical News commentary.
“There’s a pretty straightforward case here,” he argued. “Medical professionals are at higher risk for exposure to coronavirus because we come into contact with lots and lots of patients. Gathering a large group of medical professionals in a single place increases the risk for exposure further. Factor in airplane flights to and from the conferences, and the chance that infection is spread is significant.”
This article first appeared on Medscape.com.
The biggest cancer research meeting of the year has been canceled as a reaction to the novel coronavirus (COVID-19) outbreak, which has also led to many other medical conferences being canceled or postponed.
The annual meeting of the American Association for Cancer Research (AACR) was due to take place April 24-29 in San Diego, California. More than 24,000 delegates from 80 countries and more than 500 exhibitors were expected to attend.
There are plans to reschedule it for later this year.
This has been a “difficult decision,” said the AACR board of directors, but “we believe that the decision to postpone the meeting is absolutely the correct one to safeguard our meeting participants from further potential exposure to the coronavirus.”
The board goes on to explain that “this evidence-based decision was made after a thorough review and discussion of all factors impacting the annual meeting, including the US government’s enforcement of restrictions on international travelers to enter the US; the imposition of travel restrictions issued by US government agencies, cancer centers, academic institutions, and pharmaceutical and biotech companies; and the counsel of infectious disease experts. It is clear that all of these elements significantly affect the ability of delegates, speakers, presenters of proffered papers, and exhibitors to participate fully in the annual meeting.”
Other cancer conferences that were planned for March and that have been canceled include the following:
- European Breast Cancer Conference (EBCC), Barcelona, Spain, which was to have taken place March 18-20. This conference has been postponed and will now take place September 30 to October 2 at the same venue. Abstracts that have been accepted for the initial conference will remain in the program, and organizers will reopen abstract submissions in May.
- National Comprehensive Cancer Network (NCCN), Orlando, Florida, was scheduled for March 19-22. This conference has been postponed. No new dates have been provided, but the society notes that “NCCN staff is working as quickly as possible to notify all conference registrants about the postponement and further information regarding the refund process.”
- European Association of Urology (EAU), Amsterdam, the Netherlands, at which there is always new research presented on prostate, kidney, and bladder cancer, was due to take place March 20-24. This conference has been postponed to July 2020.
- Society of Gynecologic Oncology (SGO), in Toronto, Canada, which was scheduled for March 28-31. SGO is “exploring alternatives for delivering the science and education.”
Overall, the move to cancel medical conferences over the next few months is a good idea, commented F. Perry Wilson, MD, MSCE, associate professor of medicine and director of Yale’s Program of Applied Translational Research, in a Medscape Medical News commentary.
“There’s a pretty straightforward case here,” he argued. “Medical professionals are at higher risk for exposure to coronavirus because we come into contact with lots and lots of patients. Gathering a large group of medical professionals in a single place increases the risk for exposure further. Factor in airplane flights to and from the conferences, and the chance that infection is spread is significant.”
This article first appeared on Medscape.com.
The biggest cancer research meeting of the year has been canceled as a reaction to the novel coronavirus (COVID-19) outbreak, which has also led to many other medical conferences being canceled or postponed.
The annual meeting of the American Association for Cancer Research (AACR) was due to take place April 24-29 in San Diego, California. More than 24,000 delegates from 80 countries and more than 500 exhibitors were expected to attend.
There are plans to reschedule it for later this year.
This has been a “difficult decision,” said the AACR board of directors, but “we believe that the decision to postpone the meeting is absolutely the correct one to safeguard our meeting participants from further potential exposure to the coronavirus.”
The board goes on to explain that “this evidence-based decision was made after a thorough review and discussion of all factors impacting the annual meeting, including the US government’s enforcement of restrictions on international travelers to enter the US; the imposition of travel restrictions issued by US government agencies, cancer centers, academic institutions, and pharmaceutical and biotech companies; and the counsel of infectious disease experts. It is clear that all of these elements significantly affect the ability of delegates, speakers, presenters of proffered papers, and exhibitors to participate fully in the annual meeting.”
Other cancer conferences that were planned for March and that have been canceled include the following:
- European Breast Cancer Conference (EBCC), Barcelona, Spain, which was to have taken place March 18-20. This conference has been postponed and will now take place September 30 to October 2 at the same venue. Abstracts that have been accepted for the initial conference will remain in the program, and organizers will reopen abstract submissions in May.
- National Comprehensive Cancer Network (NCCN), Orlando, Florida, was scheduled for March 19-22. This conference has been postponed. No new dates have been provided, but the society notes that “NCCN staff is working as quickly as possible to notify all conference registrants about the postponement and further information regarding the refund process.”
- European Association of Urology (EAU), Amsterdam, the Netherlands, at which there is always new research presented on prostate, kidney, and bladder cancer, was due to take place March 20-24. This conference has been postponed to July 2020.
- Society of Gynecologic Oncology (SGO), in Toronto, Canada, which was scheduled for March 28-31. SGO is “exploring alternatives for delivering the science and education.”
Overall, the move to cancel medical conferences over the next few months is a good idea, commented F. Perry Wilson, MD, MSCE, associate professor of medicine and director of Yale’s Program of Applied Translational Research, in a Medscape Medical News commentary.
“There’s a pretty straightforward case here,” he argued. “Medical professionals are at higher risk for exposure to coronavirus because we come into contact with lots and lots of patients. Gathering a large group of medical professionals in a single place increases the risk for exposure further. Factor in airplane flights to and from the conferences, and the chance that infection is spread is significant.”
This article first appeared on Medscape.com.
Late effects in young cancer survivors underscore importance of high-risk screening
according to data from the Childhood Cancer Survivor Study.
At a median follow-up of 21 years, the SMR for all-cause mortality was 5.9 among survivors aged 15-20 years and 6.2 among diagnosis-matched children under 15 years, compared with expected rates at the same ages in the general population. For health-related causes – excluding primary cancer recurrence or progression but including late effects of cancer therapy – the SMRs were 4.8 in the older group and 6.8 in the younger group.
Eugene Suh, MD, of Loyola University Chicago Medical Center, Maywood, Ill., and colleagues reported these results in Lancet Oncology.
The difference between the older and younger survivors (n = 5,804 in each group) was most evident at least 20 years after cancer diagnosis, the authors noted.
For both groups, but more so for childhood cancer survivors, the risk of developing any chronic health condition and any grade 3-5 health condition was greater than for siblings of the same age who did not have cancer (hazard ratios, 4.2 for adolescents/young adults and 5.6 for childhood survivors). The same was true for grade 3-5 cardiac conditions (HRs, 4.3 and 5.6, respectively), endocrine conditions (HRs, 3.9 and 6.4, respectively), and musculoskeletal conditions (HRs, 6.5 and 8.0, respectively).
These findings, which confirm those of previous studies suggesting that younger children might be more vulnerable to the adverse effects of cancer treatment, “underscore that focused efforts are needed to ensure early-adolescent and young adult cancer survivors are receiving recommended risk-based care, with a focus on high-risk cancer screening, to reduce morbidity and premature mortality,” the researchers concluded, noting that “studies to date indicate that adherence to such high-risk screening is poor.”
In a related editorial, Päivi Lähteenmäki, MD, PhD, of University of Turku (Finland) and Turku University Hospital, wrote that these findings warrant long-term follow-up of adolescent and young adult cancer survivors. She also argued that the results “might not be fully generalizable to patients treated today who might be on different treatment regimens to those treated in previous decades” and that “[m]ore prospectively collected objective data focusing on survivors ... are needed.”
Accurate characterization of patients at high risk who would benefit from a tailored screening program is most important, and identifying underlying genetic or molecular factors that confer higher risk for late sequelae would be useful for “planning approaches to survivorship,” Dr. Lähteenmäki added.
This study was funded by the National Cancer Institute and American Lebanese-Syrian Associated Charities. Dr. Suh and Dr. Lähteenmäki reported having no competing interests.
SOURCES: Suh E et al. Lancet Oncology. 2020 Feb 14. doi: 10.1016/S1470-2045(19)30800-9;Lähteenmäki P. Lancet Oncol. 2020 Feb 14. doi: 10.106/S1470-2045(19)30858-7.
according to data from the Childhood Cancer Survivor Study.
At a median follow-up of 21 years, the SMR for all-cause mortality was 5.9 among survivors aged 15-20 years and 6.2 among diagnosis-matched children under 15 years, compared with expected rates at the same ages in the general population. For health-related causes – excluding primary cancer recurrence or progression but including late effects of cancer therapy – the SMRs were 4.8 in the older group and 6.8 in the younger group.
Eugene Suh, MD, of Loyola University Chicago Medical Center, Maywood, Ill., and colleagues reported these results in Lancet Oncology.
The difference between the older and younger survivors (n = 5,804 in each group) was most evident at least 20 years after cancer diagnosis, the authors noted.
For both groups, but more so for childhood cancer survivors, the risk of developing any chronic health condition and any grade 3-5 health condition was greater than for siblings of the same age who did not have cancer (hazard ratios, 4.2 for adolescents/young adults and 5.6 for childhood survivors). The same was true for grade 3-5 cardiac conditions (HRs, 4.3 and 5.6, respectively), endocrine conditions (HRs, 3.9 and 6.4, respectively), and musculoskeletal conditions (HRs, 6.5 and 8.0, respectively).
These findings, which confirm those of previous studies suggesting that younger children might be more vulnerable to the adverse effects of cancer treatment, “underscore that focused efforts are needed to ensure early-adolescent and young adult cancer survivors are receiving recommended risk-based care, with a focus on high-risk cancer screening, to reduce morbidity and premature mortality,” the researchers concluded, noting that “studies to date indicate that adherence to such high-risk screening is poor.”
In a related editorial, Päivi Lähteenmäki, MD, PhD, of University of Turku (Finland) and Turku University Hospital, wrote that these findings warrant long-term follow-up of adolescent and young adult cancer survivors. She also argued that the results “might not be fully generalizable to patients treated today who might be on different treatment regimens to those treated in previous decades” and that “[m]ore prospectively collected objective data focusing on survivors ... are needed.”
Accurate characterization of patients at high risk who would benefit from a tailored screening program is most important, and identifying underlying genetic or molecular factors that confer higher risk for late sequelae would be useful for “planning approaches to survivorship,” Dr. Lähteenmäki added.
This study was funded by the National Cancer Institute and American Lebanese-Syrian Associated Charities. Dr. Suh and Dr. Lähteenmäki reported having no competing interests.
SOURCES: Suh E et al. Lancet Oncology. 2020 Feb 14. doi: 10.1016/S1470-2045(19)30800-9;Lähteenmäki P. Lancet Oncol. 2020 Feb 14. doi: 10.106/S1470-2045(19)30858-7.
according to data from the Childhood Cancer Survivor Study.
At a median follow-up of 21 years, the SMR for all-cause mortality was 5.9 among survivors aged 15-20 years and 6.2 among diagnosis-matched children under 15 years, compared with expected rates at the same ages in the general population. For health-related causes – excluding primary cancer recurrence or progression but including late effects of cancer therapy – the SMRs were 4.8 in the older group and 6.8 in the younger group.
Eugene Suh, MD, of Loyola University Chicago Medical Center, Maywood, Ill., and colleagues reported these results in Lancet Oncology.
The difference between the older and younger survivors (n = 5,804 in each group) was most evident at least 20 years after cancer diagnosis, the authors noted.
For both groups, but more so for childhood cancer survivors, the risk of developing any chronic health condition and any grade 3-5 health condition was greater than for siblings of the same age who did not have cancer (hazard ratios, 4.2 for adolescents/young adults and 5.6 for childhood survivors). The same was true for grade 3-5 cardiac conditions (HRs, 4.3 and 5.6, respectively), endocrine conditions (HRs, 3.9 and 6.4, respectively), and musculoskeletal conditions (HRs, 6.5 and 8.0, respectively).
These findings, which confirm those of previous studies suggesting that younger children might be more vulnerable to the adverse effects of cancer treatment, “underscore that focused efforts are needed to ensure early-adolescent and young adult cancer survivors are receiving recommended risk-based care, with a focus on high-risk cancer screening, to reduce morbidity and premature mortality,” the researchers concluded, noting that “studies to date indicate that adherence to such high-risk screening is poor.”
In a related editorial, Päivi Lähteenmäki, MD, PhD, of University of Turku (Finland) and Turku University Hospital, wrote that these findings warrant long-term follow-up of adolescent and young adult cancer survivors. She also argued that the results “might not be fully generalizable to patients treated today who might be on different treatment regimens to those treated in previous decades” and that “[m]ore prospectively collected objective data focusing on survivors ... are needed.”
Accurate characterization of patients at high risk who would benefit from a tailored screening program is most important, and identifying underlying genetic or molecular factors that confer higher risk for late sequelae would be useful for “planning approaches to survivorship,” Dr. Lähteenmäki added.
This study was funded by the National Cancer Institute and American Lebanese-Syrian Associated Charities. Dr. Suh and Dr. Lähteenmäki reported having no competing interests.
SOURCES: Suh E et al. Lancet Oncology. 2020 Feb 14. doi: 10.1016/S1470-2045(19)30800-9;Lähteenmäki P. Lancet Oncol. 2020 Feb 14. doi: 10.106/S1470-2045(19)30858-7.
FROM LANCET ONCOLOGY
FDA: Cell phones still look safe
according to a review by the Food and Drug Administration.

The FDA reviewed the published literature from 2008 to 2018 and concluded that the data don’t support any quantifiable adverse health risks from RFR. However, the evidence is not without limitations.
The FDA’s evaluation included evidence from in vivo animal studies from Jan. 1, 2008, to Aug. 1, 2018, and epidemiologic studies in humans from Jan. 1, 2008, to May 8, 2018. Both kinds of evidence had limitations, but neither produced strong indications of any causal risks from cell phone use.
The FDA noted that in vivo animal studies are limited by variability of methods and RFR exposure, which make comparisons of results difficult. These studies are also impacted by the indirect effects of temperature increases (the only currently established biological effect of RFR) and stress experienced by the animals, which make teasing out the direct effects of RFR difficult.
The FDA noted that strong epidemiologic studies can provide more relevant and accurate information than in vivo studies, but epidemiologic studies are not without limitations. For example, most have participants track and self-report their cell phone use. There’s also no way to directly track certain factors of RFR exposure, such as frequency, duration, or intensity.
Even with those caveats in mind, the FDA wrote that, “based on the studies that are described in detail in this report, there is insufficient evidence to support a causal association between RFR exposure and tumorigenesis. There is a lack of clear dose-response relationship, a lack of consistent findings or specificity, and a lack of biological mechanistic plausibility.”
The full review is available on the FDA website.
according to a review by the Food and Drug Administration.
The FDA reviewed the published literature from 2008 to 2018 and concluded that the data don’t support any quantifiable adverse health risks from RFR. However, the evidence is not without limitations.
The FDA’s evaluation included evidence from in vivo animal studies from Jan. 1, 2008, to Aug. 1, 2018, and epidemiologic studies in humans from Jan. 1, 2008, to May 8, 2018. Both kinds of evidence had limitations, but neither produced strong indications of any causal risks from cell phone use.
The FDA noted that in vivo animal studies are limited by variability of methods and RFR exposure, which make comparisons of results difficult. These studies are also impacted by the indirect effects of temperature increases (the only currently established biological effect of RFR) and stress experienced by the animals, which make teasing out the direct effects of RFR difficult.
The FDA noted that strong epidemiologic studies can provide more relevant and accurate information than in vivo studies, but epidemiologic studies are not without limitations. For example, most have participants track and self-report their cell phone use. There’s also no way to directly track certain factors of RFR exposure, such as frequency, duration, or intensity.
Even with those caveats in mind, the FDA wrote that, “based on the studies that are described in detail in this report, there is insufficient evidence to support a causal association between RFR exposure and tumorigenesis. There is a lack of clear dose-response relationship, a lack of consistent findings or specificity, and a lack of biological mechanistic plausibility.”
The full review is available on the FDA website.
according to a review by the Food and Drug Administration.
The FDA reviewed the published literature from 2008 to 2018 and concluded that the data don’t support any quantifiable adverse health risks from RFR. However, the evidence is not without limitations.
The FDA’s evaluation included evidence from in vivo animal studies from Jan. 1, 2008, to Aug. 1, 2018, and epidemiologic studies in humans from Jan. 1, 2008, to May 8, 2018. Both kinds of evidence had limitations, but neither produced strong indications of any causal risks from cell phone use.
The FDA noted that in vivo animal studies are limited by variability of methods and RFR exposure, which make comparisons of results difficult. These studies are also impacted by the indirect effects of temperature increases (the only currently established biological effect of RFR) and stress experienced by the animals, which make teasing out the direct effects of RFR difficult.
The FDA noted that strong epidemiologic studies can provide more relevant and accurate information than in vivo studies, but epidemiologic studies are not without limitations. For example, most have participants track and self-report their cell phone use. There’s also no way to directly track certain factors of RFR exposure, such as frequency, duration, or intensity.
Even with those caveats in mind, the FDA wrote that, “based on the studies that are described in detail in this report, there is insufficient evidence to support a causal association between RFR exposure and tumorigenesis. There is a lack of clear dose-response relationship, a lack of consistent findings or specificity, and a lack of biological mechanistic plausibility.”
The full review is available on the FDA website.
CRISPR-engineered T cells may be safe for cancer, but do they work?
according to a report in Science.
The results of no harm support this “promising” area of cancer immunotherapy, according to study investigator Edward A. Stadtmauer, MD, of the University of Pennsylvania in Philadelphia and colleagues.
However, there was no evidence of benefit in this trial. One patient transfused with CRISPR-engineered T cells has since died, and the other two have moved on to other treatments.
“The big question that remains unanswered by this study is whether gene-edited, engineered T cells are effective against advanced cancer,” Jennifer Hamilton, PhD, and Jennifer Doudna, PhD, both of the University of California, Berkeley, wrote in an accompanying editorial.
The study enrolled six patients with refractory cancer, and three of them received CRISPR-engineered T cells. Two patients had multiple myeloma, and one had metastatic sarcoma.
Dr. Stadtmauer and colleagues drew blood from the patients, isolated the T cells, and used CRISPR-Cas9 to modify the cells. The T cells were transfected with Cas9 protein complexed with single guide RNAs against TRAC and TRBC (genes encoding the T-cell receptor chains TCR-alpha and TCR-beta) as well as PDCD1 (a gene encoding programmed cell death protein 1). The T cells were then transduced with a lentiviral vector to express a transgenic NY-ESO-1 cancer-specific T-cell receptor.
The investigators expanded the cell lines and infused them back into the patients after administering lymphodepleting chemotherapy. The sarcoma patient initially had a 50% decrease in a large abdominal mass, but all three patients ultimately progressed.
The editorialists noted that gene disruption efficiencies in this study were “modest,” ranging from 15% to 45%, but the investigators used a protocol from 2016, when the study was given the go-ahead by the National Institutes of Health and the Food and Drug Administration. With current protocols, gene disruption efficiencies can exceed 90%, which means patients might do better in subsequent trials.
There was no more than mild toxicity in this trial, and most adverse events were attributed to the lymphodepleting chemotherapy.
There was concern about potential rejection of infused cells because of preexisting immune responses to Cas9, but it doesn’t seem “to be a barrier to the application of this promising technology,” the investigators said.
They noted that “the stable engraftment of our engineered T cells is remarkably different from previously reported trials ... where the half-life of the cells in blood was [about] 1 week. Biopsy specimens of bone marrow in the myeloma patients and tumor in the sarcoma patient demonstrated trafficking of the engineered T cells to the tumor in all three patients” beyond that point. The decay half-life of the transduced cells was 20.3 days, 121.8 days, and 293.5 days in these patients.
The editorialists said the details in the report are a model for other researchers to follow, but “as more gene-based therapies are demonstrated to be safe and effective, the barrier to clinical translation will become cell manufacturing and administration.”
This work was funded by the National Institutes of Health and others. Dr. Stadtmauer didn’t report any disclosures, but other investigators disclosed patent applications and commercialization efforts. Dr. Doudna disclosed that she is a cofounder or adviser for several companies developing gene-editing therapeutics.
SOURCE: Stadtmauer EA et al. Science. 2020 Feb 6. doi: 10.1126/science.aba7365.
according to a report in Science.
The results of no harm support this “promising” area of cancer immunotherapy, according to study investigator Edward A. Stadtmauer, MD, of the University of Pennsylvania in Philadelphia and colleagues.
However, there was no evidence of benefit in this trial. One patient transfused with CRISPR-engineered T cells has since died, and the other two have moved on to other treatments.
“The big question that remains unanswered by this study is whether gene-edited, engineered T cells are effective against advanced cancer,” Jennifer Hamilton, PhD, and Jennifer Doudna, PhD, both of the University of California, Berkeley, wrote in an accompanying editorial.
The study enrolled six patients with refractory cancer, and three of them received CRISPR-engineered T cells. Two patients had multiple myeloma, and one had metastatic sarcoma.
Dr. Stadtmauer and colleagues drew blood from the patients, isolated the T cells, and used CRISPR-Cas9 to modify the cells. The T cells were transfected with Cas9 protein complexed with single guide RNAs against TRAC and TRBC (genes encoding the T-cell receptor chains TCR-alpha and TCR-beta) as well as PDCD1 (a gene encoding programmed cell death protein 1). The T cells were then transduced with a lentiviral vector to express a transgenic NY-ESO-1 cancer-specific T-cell receptor.
The investigators expanded the cell lines and infused them back into the patients after administering lymphodepleting chemotherapy. The sarcoma patient initially had a 50% decrease in a large abdominal mass, but all three patients ultimately progressed.
The editorialists noted that gene disruption efficiencies in this study were “modest,” ranging from 15% to 45%, but the investigators used a protocol from 2016, when the study was given the go-ahead by the National Institutes of Health and the Food and Drug Administration. With current protocols, gene disruption efficiencies can exceed 90%, which means patients might do better in subsequent trials.
There was no more than mild toxicity in this trial, and most adverse events were attributed to the lymphodepleting chemotherapy.
There was concern about potential rejection of infused cells because of preexisting immune responses to Cas9, but it doesn’t seem “to be a barrier to the application of this promising technology,” the investigators said.
They noted that “the stable engraftment of our engineered T cells is remarkably different from previously reported trials ... where the half-life of the cells in blood was [about] 1 week. Biopsy specimens of bone marrow in the myeloma patients and tumor in the sarcoma patient demonstrated trafficking of the engineered T cells to the tumor in all three patients” beyond that point. The decay half-life of the transduced cells was 20.3 days, 121.8 days, and 293.5 days in these patients.
The editorialists said the details in the report are a model for other researchers to follow, but “as more gene-based therapies are demonstrated to be safe and effective, the barrier to clinical translation will become cell manufacturing and administration.”
This work was funded by the National Institutes of Health and others. Dr. Stadtmauer didn’t report any disclosures, but other investigators disclosed patent applications and commercialization efforts. Dr. Doudna disclosed that she is a cofounder or adviser for several companies developing gene-editing therapeutics.
SOURCE: Stadtmauer EA et al. Science. 2020 Feb 6. doi: 10.1126/science.aba7365.
according to a report in Science.
The results of no harm support this “promising” area of cancer immunotherapy, according to study investigator Edward A. Stadtmauer, MD, of the University of Pennsylvania in Philadelphia and colleagues.
However, there was no evidence of benefit in this trial. One patient transfused with CRISPR-engineered T cells has since died, and the other two have moved on to other treatments.
“The big question that remains unanswered by this study is whether gene-edited, engineered T cells are effective against advanced cancer,” Jennifer Hamilton, PhD, and Jennifer Doudna, PhD, both of the University of California, Berkeley, wrote in an accompanying editorial.
The study enrolled six patients with refractory cancer, and three of them received CRISPR-engineered T cells. Two patients had multiple myeloma, and one had metastatic sarcoma.
Dr. Stadtmauer and colleagues drew blood from the patients, isolated the T cells, and used CRISPR-Cas9 to modify the cells. The T cells were transfected with Cas9 protein complexed with single guide RNAs against TRAC and TRBC (genes encoding the T-cell receptor chains TCR-alpha and TCR-beta) as well as PDCD1 (a gene encoding programmed cell death protein 1). The T cells were then transduced with a lentiviral vector to express a transgenic NY-ESO-1 cancer-specific T-cell receptor.
The investigators expanded the cell lines and infused them back into the patients after administering lymphodepleting chemotherapy. The sarcoma patient initially had a 50% decrease in a large abdominal mass, but all three patients ultimately progressed.
The editorialists noted that gene disruption efficiencies in this study were “modest,” ranging from 15% to 45%, but the investigators used a protocol from 2016, when the study was given the go-ahead by the National Institutes of Health and the Food and Drug Administration. With current protocols, gene disruption efficiencies can exceed 90%, which means patients might do better in subsequent trials.
There was no more than mild toxicity in this trial, and most adverse events were attributed to the lymphodepleting chemotherapy.
There was concern about potential rejection of infused cells because of preexisting immune responses to Cas9, but it doesn’t seem “to be a barrier to the application of this promising technology,” the investigators said.
They noted that “the stable engraftment of our engineered T cells is remarkably different from previously reported trials ... where the half-life of the cells in blood was [about] 1 week. Biopsy specimens of bone marrow in the myeloma patients and tumor in the sarcoma patient demonstrated trafficking of the engineered T cells to the tumor in all three patients” beyond that point. The decay half-life of the transduced cells was 20.3 days, 121.8 days, and 293.5 days in these patients.
The editorialists said the details in the report are a model for other researchers to follow, but “as more gene-based therapies are demonstrated to be safe and effective, the barrier to clinical translation will become cell manufacturing and administration.”
This work was funded by the National Institutes of Health and others. Dr. Stadtmauer didn’t report any disclosures, but other investigators disclosed patent applications and commercialization efforts. Dr. Doudna disclosed that she is a cofounder or adviser for several companies developing gene-editing therapeutics.
SOURCE: Stadtmauer EA et al. Science. 2020 Feb 6. doi: 10.1126/science.aba7365.
FROM SCIENCE
Global project reveals cancer’s genomic playbook
A massive collaborative project spanning four continents and 744 research centers has revealed driver mutations in both protein-coding and noncoding regions of 38 cancer types.

The Pan-Cancer Analysis of Whole Genomes (PCAWG) is an integrative analysis of the whole-genome sequences from 2,658 donors across 38 common tumor types. The findings are expected to add exponentially to what’s currently known about the complex genetics of cancer, and they point to possible strategies for improving cancer prevention, diagnosis, and care.
Six articles summarizing the findings are presented in a series of papers in Nature, and 16 more appear in affiliated publications.
“It’s humbling that it was only 14 years ago that the genomics community sequenced its very first cancer exome, and it was able to identify mutations within the roughly 20,000 protein-coding genes in the human cell,” investigator Lincoln Stein, MD, PhD, of the Ontario Institute for Cancer Research in Toronto, said in a telephone briefing.
Exome sequencing, however, covers only protein-coding genomic regions, which constitute only about 1% of the entire genome, “so assembling an accurate portrait of the cancer genome using just the exome data is like trying to put together a 100,000-piece jigsaw puzzle when you’re missing 99% of the pieces and there’s no puzzle box with a completed picture to guide you,” Dr. Stein said.
Members of the PCAWG from centers in North America, Europe, Asia, and Australia screened 2,658 whole-cancer genomes and matched samples of noncancerous tissues from the same individuals, along with 1,188 transcriptomes cataloging the sequences and expression of RNA transcripts in a given tumor. The 6-year project netted more than 800 terabytes of genomic data, roughly equivalent to the digital holdings of the U.S. Library of Congress multiplied by 11.
The findings are summarized in papers focusing on cancer drivers, noncoding changes, mutational signatures, structural variants, cancer evolution over time, and RNA alterations.
Driver mutations
Investigators found that the average cancer genome contains four or five driver mutations located in both coding and noncoding regions. They also found, however, that in approximately 5% of cases no driver mutations could be identified.
A substantial proportion of tumors displayed “hallmarks of genomic catastrophes.” About 22% of tumors exhibited chromothripsis, a mutational process marked by hundreds or even thousands of clustered chromosomal rearrangements. About 18% showed chromoplexy, which is characterized by scattering and rearrangement of multiple strands of DNA from one or more chromosomes.
Analyzing driver point mutations and structural variants in noncoding regions, the investigators found the usual suspects – previously reported culprits – as well as novel candidates.
For example, they identified point mutations in the five prime region of the tumor suppressor gene TP53 and the three prime untranslated regions of NFKBIZ (a nuclear factor kappa B inhibitor) and TOB1 (an antiproliferative protein), focal deletion in BRD4 (a transcriptional and epigenetic regulator), and rearrangements in chromosomal loci in members of the AKR1C family of enzymes thought to play a role in disease progression.
In addition, investigators identified mutations in noncoding regions of TERT, a telomerase gene. These mutations result in ramped-up expression of telomerase, which in turn promotes uncontrollable division of tumor cells.
Mutational signatures
In a related line of research, PCAWG investigators identified new DNA mutational signatures ranging from single nucleotide polymorphisms to insertions and deletions, as well as to structural variants – rearrangements of large sections of the genome.
“The substantial size of our dataset, compared with previous analyses, enabled the discovery of new signatures, the separation of overlapping signatures, and the decomposition of signatures into components that may represent associated – but distinct – DNA damage, repair, and/or replication mechanisms. By estimating the contribution of each signature to the mutational catalogs of individual cancer genomes, we revealed associations of signatures to exogenous or endogenous exposures, as well as to defective DNA maintenance processes,” the investigators wrote.
They also acknowledged, however, that “many signatures are of unknown cause.”
Cancer evolution
One of the six main studies focused on the evolution of cancer over time. Instead of providing a “snapshot” of the genome as captured by sequencing tissue from a single biopsy, consortium investigators created full-length features of the “life history and evolution of mutational processes and driver mutation sequences.”
They found that early cancer development was marked by relatively few mutations in driver genes and by identifiable copy-number gains, including trisomy 7 in glioblastoma, and an abnormal mirroring of the arms (isochromosome) of chromosome 17 in medulloblastoma.
In 40% of the samples, however, there were significant changes in the mutational spectrum as the cancers grew, leading to a near quadrupling of driver genes and increased genomic instability in later-stage tumors.
“Copy-number alterations often occur in mitotic crises and lead to simultaneous gains of chromosomal segments,” the investigators wrote. “Timing analyses suggest that driver mutations often precede diagnosis by many years, if not decades. Together, these results determine the evolutionary trajectories of cancer and highlight opportunities for early cancer detection.”
Implications for cancer care
“When I used to treat patients with cancer, I was always completely amazed and puzzled by how two patients could have what looked like the same tumor. It would look the same under the microscope, have the same size, and the two patients would receive exactly the same treatment, but the two patients would have completely opposite outcomes; one would survive, and one would die. What this analysis … has done is really laid bare the reasons for that unpredictability in clinical outcomes,” Peter Campbell, MD, PhD, of the Wellcome Sanger Institute in Hinxton, England, said during the telebriefing.
“The most striking finding out of all of the suite of papers is just how different one person’s cancer genome is from another person’s. We see thousands of different combinations of mutations that can cause the cancer, and more than 80 different underlying processes generating the mutations in a cancer, and that leads to very different shapes and patterns in the genome that result,” he added.
On a positive note, the research shows that one or more driver mutations can be identified in about 95% of all cancer patients, and it elucidates the sequence of events leading to oncogenesis and tumor evolution, providing opportunities for earlier identification and potential interventions to prevent cancer, Dr. Campbell said.
The PCAWG was a collaborative multinational effort with multiple funding sources and many investigators.
SOURCE: Nature. 2020 Feb 5. https://www.nature.com/collections/pcawg/
A massive collaborative project spanning four continents and 744 research centers has revealed driver mutations in both protein-coding and noncoding regions of 38 cancer types.
The Pan-Cancer Analysis of Whole Genomes (PCAWG) is an integrative analysis of the whole-genome sequences from 2,658 donors across 38 common tumor types. The findings are expected to add exponentially to what’s currently known about the complex genetics of cancer, and they point to possible strategies for improving cancer prevention, diagnosis, and care.
Six articles summarizing the findings are presented in a series of papers in Nature, and 16 more appear in affiliated publications.
“It’s humbling that it was only 14 years ago that the genomics community sequenced its very first cancer exome, and it was able to identify mutations within the roughly 20,000 protein-coding genes in the human cell,” investigator Lincoln Stein, MD, PhD, of the Ontario Institute for Cancer Research in Toronto, said in a telephone briefing.
Exome sequencing, however, covers only protein-coding genomic regions, which constitute only about 1% of the entire genome, “so assembling an accurate portrait of the cancer genome using just the exome data is like trying to put together a 100,000-piece jigsaw puzzle when you’re missing 99% of the pieces and there’s no puzzle box with a completed picture to guide you,” Dr. Stein said.
Members of the PCAWG from centers in North America, Europe, Asia, and Australia screened 2,658 whole-cancer genomes and matched samples of noncancerous tissues from the same individuals, along with 1,188 transcriptomes cataloging the sequences and expression of RNA transcripts in a given tumor. The 6-year project netted more than 800 terabytes of genomic data, roughly equivalent to the digital holdings of the U.S. Library of Congress multiplied by 11.
The findings are summarized in papers focusing on cancer drivers, noncoding changes, mutational signatures, structural variants, cancer evolution over time, and RNA alterations.
Driver mutations
Investigators found that the average cancer genome contains four or five driver mutations located in both coding and noncoding regions. They also found, however, that in approximately 5% of cases no driver mutations could be identified.
A substantial proportion of tumors displayed “hallmarks of genomic catastrophes.” About 22% of tumors exhibited chromothripsis, a mutational process marked by hundreds or even thousands of clustered chromosomal rearrangements. About 18% showed chromoplexy, which is characterized by scattering and rearrangement of multiple strands of DNA from one or more chromosomes.
Analyzing driver point mutations and structural variants in noncoding regions, the investigators found the usual suspects – previously reported culprits – as well as novel candidates.
For example, they identified point mutations in the five prime region of the tumor suppressor gene TP53 and the three prime untranslated regions of NFKBIZ (a nuclear factor kappa B inhibitor) and TOB1 (an antiproliferative protein), focal deletion in BRD4 (a transcriptional and epigenetic regulator), and rearrangements in chromosomal loci in members of the AKR1C family of enzymes thought to play a role in disease progression.
In addition, investigators identified mutations in noncoding regions of TERT, a telomerase gene. These mutations result in ramped-up expression of telomerase, which in turn promotes uncontrollable division of tumor cells.
Mutational signatures
In a related line of research, PCAWG investigators identified new DNA mutational signatures ranging from single nucleotide polymorphisms to insertions and deletions, as well as to structural variants – rearrangements of large sections of the genome.
“The substantial size of our dataset, compared with previous analyses, enabled the discovery of new signatures, the separation of overlapping signatures, and the decomposition of signatures into components that may represent associated – but distinct – DNA damage, repair, and/or replication mechanisms. By estimating the contribution of each signature to the mutational catalogs of individual cancer genomes, we revealed associations of signatures to exogenous or endogenous exposures, as well as to defective DNA maintenance processes,” the investigators wrote.
They also acknowledged, however, that “many signatures are of unknown cause.”
Cancer evolution
One of the six main studies focused on the evolution of cancer over time. Instead of providing a “snapshot” of the genome as captured by sequencing tissue from a single biopsy, consortium investigators created full-length features of the “life history and evolution of mutational processes and driver mutation sequences.”
They found that early cancer development was marked by relatively few mutations in driver genes and by identifiable copy-number gains, including trisomy 7 in glioblastoma, and an abnormal mirroring of the arms (isochromosome) of chromosome 17 in medulloblastoma.
In 40% of the samples, however, there were significant changes in the mutational spectrum as the cancers grew, leading to a near quadrupling of driver genes and increased genomic instability in later-stage tumors.
“Copy-number alterations often occur in mitotic crises and lead to simultaneous gains of chromosomal segments,” the investigators wrote. “Timing analyses suggest that driver mutations often precede diagnosis by many years, if not decades. Together, these results determine the evolutionary trajectories of cancer and highlight opportunities for early cancer detection.”
Implications for cancer care
“When I used to treat patients with cancer, I was always completely amazed and puzzled by how two patients could have what looked like the same tumor. It would look the same under the microscope, have the same size, and the two patients would receive exactly the same treatment, but the two patients would have completely opposite outcomes; one would survive, and one would die. What this analysis … has done is really laid bare the reasons for that unpredictability in clinical outcomes,” Peter Campbell, MD, PhD, of the Wellcome Sanger Institute in Hinxton, England, said during the telebriefing.
“The most striking finding out of all of the suite of papers is just how different one person’s cancer genome is from another person’s. We see thousands of different combinations of mutations that can cause the cancer, and more than 80 different underlying processes generating the mutations in a cancer, and that leads to very different shapes and patterns in the genome that result,” he added.
On a positive note, the research shows that one or more driver mutations can be identified in about 95% of all cancer patients, and it elucidates the sequence of events leading to oncogenesis and tumor evolution, providing opportunities for earlier identification and potential interventions to prevent cancer, Dr. Campbell said.
The PCAWG was a collaborative multinational effort with multiple funding sources and many investigators.
SOURCE: Nature. 2020 Feb 5. https://www.nature.com/collections/pcawg/
A massive collaborative project spanning four continents and 744 research centers has revealed driver mutations in both protein-coding and noncoding regions of 38 cancer types.
The Pan-Cancer Analysis of Whole Genomes (PCAWG) is an integrative analysis of the whole-genome sequences from 2,658 donors across 38 common tumor types. The findings are expected to add exponentially to what’s currently known about the complex genetics of cancer, and they point to possible strategies for improving cancer prevention, diagnosis, and care.
Six articles summarizing the findings are presented in a series of papers in Nature, and 16 more appear in affiliated publications.
“It’s humbling that it was only 14 years ago that the genomics community sequenced its very first cancer exome, and it was able to identify mutations within the roughly 20,000 protein-coding genes in the human cell,” investigator Lincoln Stein, MD, PhD, of the Ontario Institute for Cancer Research in Toronto, said in a telephone briefing.
Exome sequencing, however, covers only protein-coding genomic regions, which constitute only about 1% of the entire genome, “so assembling an accurate portrait of the cancer genome using just the exome data is like trying to put together a 100,000-piece jigsaw puzzle when you’re missing 99% of the pieces and there’s no puzzle box with a completed picture to guide you,” Dr. Stein said.
Members of the PCAWG from centers in North America, Europe, Asia, and Australia screened 2,658 whole-cancer genomes and matched samples of noncancerous tissues from the same individuals, along with 1,188 transcriptomes cataloging the sequences and expression of RNA transcripts in a given tumor. The 6-year project netted more than 800 terabytes of genomic data, roughly equivalent to the digital holdings of the U.S. Library of Congress multiplied by 11.
The findings are summarized in papers focusing on cancer drivers, noncoding changes, mutational signatures, structural variants, cancer evolution over time, and RNA alterations.
Driver mutations
Investigators found that the average cancer genome contains four or five driver mutations located in both coding and noncoding regions. They also found, however, that in approximately 5% of cases no driver mutations could be identified.
A substantial proportion of tumors displayed “hallmarks of genomic catastrophes.” About 22% of tumors exhibited chromothripsis, a mutational process marked by hundreds or even thousands of clustered chromosomal rearrangements. About 18% showed chromoplexy, which is characterized by scattering and rearrangement of multiple strands of DNA from one or more chromosomes.
Analyzing driver point mutations and structural variants in noncoding regions, the investigators found the usual suspects – previously reported culprits – as well as novel candidates.
For example, they identified point mutations in the five prime region of the tumor suppressor gene TP53 and the three prime untranslated regions of NFKBIZ (a nuclear factor kappa B inhibitor) and TOB1 (an antiproliferative protein), focal deletion in BRD4 (a transcriptional and epigenetic regulator), and rearrangements in chromosomal loci in members of the AKR1C family of enzymes thought to play a role in disease progression.
In addition, investigators identified mutations in noncoding regions of TERT, a telomerase gene. These mutations result in ramped-up expression of telomerase, which in turn promotes uncontrollable division of tumor cells.
Mutational signatures
In a related line of research, PCAWG investigators identified new DNA mutational signatures ranging from single nucleotide polymorphisms to insertions and deletions, as well as to structural variants – rearrangements of large sections of the genome.
“The substantial size of our dataset, compared with previous analyses, enabled the discovery of new signatures, the separation of overlapping signatures, and the decomposition of signatures into components that may represent associated – but distinct – DNA damage, repair, and/or replication mechanisms. By estimating the contribution of each signature to the mutational catalogs of individual cancer genomes, we revealed associations of signatures to exogenous or endogenous exposures, as well as to defective DNA maintenance processes,” the investigators wrote.
They also acknowledged, however, that “many signatures are of unknown cause.”
Cancer evolution
One of the six main studies focused on the evolution of cancer over time. Instead of providing a “snapshot” of the genome as captured by sequencing tissue from a single biopsy, consortium investigators created full-length features of the “life history and evolution of mutational processes and driver mutation sequences.”
They found that early cancer development was marked by relatively few mutations in driver genes and by identifiable copy-number gains, including trisomy 7 in glioblastoma, and an abnormal mirroring of the arms (isochromosome) of chromosome 17 in medulloblastoma.
In 40% of the samples, however, there were significant changes in the mutational spectrum as the cancers grew, leading to a near quadrupling of driver genes and increased genomic instability in later-stage tumors.
“Copy-number alterations often occur in mitotic crises and lead to simultaneous gains of chromosomal segments,” the investigators wrote. “Timing analyses suggest that driver mutations often precede diagnosis by many years, if not decades. Together, these results determine the evolutionary trajectories of cancer and highlight opportunities for early cancer detection.”
Implications for cancer care
“When I used to treat patients with cancer, I was always completely amazed and puzzled by how two patients could have what looked like the same tumor. It would look the same under the microscope, have the same size, and the two patients would receive exactly the same treatment, but the two patients would have completely opposite outcomes; one would survive, and one would die. What this analysis … has done is really laid bare the reasons for that unpredictability in clinical outcomes,” Peter Campbell, MD, PhD, of the Wellcome Sanger Institute in Hinxton, England, said during the telebriefing.
“The most striking finding out of all of the suite of papers is just how different one person’s cancer genome is from another person’s. We see thousands of different combinations of mutations that can cause the cancer, and more than 80 different underlying processes generating the mutations in a cancer, and that leads to very different shapes and patterns in the genome that result,” he added.
On a positive note, the research shows that one or more driver mutations can be identified in about 95% of all cancer patients, and it elucidates the sequence of events leading to oncogenesis and tumor evolution, providing opportunities for earlier identification and potential interventions to prevent cancer, Dr. Campbell said.
The PCAWG was a collaborative multinational effort with multiple funding sources and many investigators.
SOURCE: Nature. 2020 Feb 5. https://www.nature.com/collections/pcawg/
FROM NATURE
Presentation of a Rare Malignancy: Leiomyosarcoma of the Prostate (FULL)
Prostatic leiomyosarcoma is an aggressive malignancy with a high risk of metastasis and a poor prognosis that poses unique diagnostic and treatment challenges.
Prostatic leiomyosarcoma is a rare tumor.1 This neoplasm is composed of highly aggressive prostatic smooth muscle cells that present with nonspecific signs and symptoms mimicking other forms of prostatic pathology. Of the primary prostatic sarcomas, leiomyosarcoma represents the most common subtype in adults and is found in 38% to 52% of newly diagnosed prostate sarcoma.1,2 The prognosis is poor, and no clear guidelines exist regarding the optimal treatment approach. We report a case of prostate leiomyosarcoma and describe the disease characteristics, diagnostic modalities, and treatment approach regarding these rare malignancies.
Case Presentation
A 72-year-old male presented with 6 months of progressive severe lower urinary tract symptoms (LUTS) secondary to bladder outlet obstruction. The patient was refractory to medical management with combination α-blocker and 5-α-reductase inhibitor therapy and continued to require multiple emergent bladder catheterizations. Workup with urinalysis, blood biochemistry, and prostate specific antigen (PSA) levels were persistently normal. He reported no hematuria, weight loss, or perineal pain. The patient reported no history of tobacco use, exposure to hazardous chemicals, and had no family history of genitourinary cancers. On rectal exam, the prostate was firm and nodular, with induration noted along the right upper lobe of the prostate.
The patient was referred for a urology consultation and subsequently underwent transurethral resection of the prostate (TURP) for suspected severe benign prostatic hypertrophy (BPH). A histopathologic examination demonstrated atypical cytology consistent with high- grade leiomyosarcoma. Immunohistochemical analysis revealed positive staining for vimentin, smooth muscle actin, desmin (partial), cytokeratin, smooth muscle myosin, muscle specific actin, and Ki-67 (50%-60% expression).
Fluorodeoxyglucose positron emission tomography (FDG-PET) scan revealed a 5.7 x 5.9 cm tumor with a maximum standardized uptake value (SUVmax) of 12.6 in the right posterior prostate, without evidence of metastatic disease (Figures 1A and 1B). 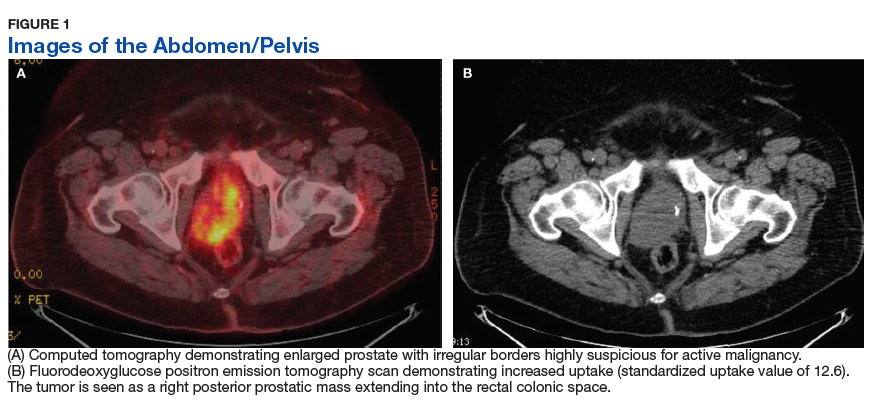
Discussion
Originating from prostatic interstitial cells, prostatic leiomyosarcoma is a rare tumor that accounts for < 0.1% of all primary prostatic malignancies.1 Since its first description in 1950 by Riba and colleagues, < 200 cases have been reported worldwide.2 Among the sarcomas of the prostate, it is the most common tumor, accounting for around 38% to 52% of prostate sarcoma presentations.1,2
Patients typically present between the ages of 41 and 78 years (mean age 61 years).2,3 Signs and symptoms at presentation may vary; however, the most common symptoms are related to lower urinary tract obstruction (89.4% of patients). These symptoms include urinary frequency, urgency, nocturia, and may mimic the presentation of BPH.
Symptoms commonly associated with other malignancies, including constitutional symptoms such as weight loss, tend to occur less frequently or may be absent. Perineal or rectal pain may only be present in 25.6% of patients. Hematuria, burning on ejaculation, and constitutional symptoms are a less common presentation (< 10% of patients).3,4 PSA levels typically do not rise and are found to be within normal limits. The lack of PSA elevation is related to the tumors nonepithelial origin and may contribute to a delay in diagnosis.2,4,5
Diagnosis
Diagnosis may be further eluded as digital rectal exam (DRE) findings tend to reveal nonspecific enlargement of the prostate, resembling that of BPH. DRE may show a hard and firm prostate with nodular induration at the base or over the lobes of the prostate.6 At this stage a urology consultation is useful, as diagnosis is most commonly achieved using transrectal ultrasound (TRUS) with ultrasound-guided needle biopsy or after a TURP procedure.3
Prostate sarcoma is associated with markedly enlarged prostate volume, irregular margins with invasion, or heterogenous hypoechoic lesions on TRUS.7 Transperineal biopsy, computed tomography (CT)-guided biopsy, or suprapubic prostatectomy have been less frequently employed for diagnosis in previously reported cases.8 Specialized imaging modalities, such as CT scan or bone scan, do not show any specific findings with regards to these tumors; their role is limited to evaluation of the local and distant metastasis and for follow-up assessments.9 Transabdominal ultrasound may assess hydronephrosis or enlarged prostate and its relation to nearby structures, although it has not been shown to be helpful in establishing a specific diagnosis.6
Histologically, prostatic leiomyosarcoma is a distinct subtype of prostatic sarcoma. Other subtypes include stromal tumors such as rhabdomyosarcoma, fibrosarcoma, and spindle cell sarcoma.2 The majority of leiomyosarcomas are high-grade lesions demonstrating neoplastic spindle cells with nuclear atypia, multifocal necrosis, and cystic degeneration. Low-grade leiomyosarcomas are very rare.10 Immunohistochemistry is characteristically positive for vimentin, smooth muscle actin, and desmin expression. Cytokeratin may be positive in up to 25% of cases, whereas S-100, CD34, CD117, and PSA are negative.2,3 These histopathological findings help to differentiate leiomyosarcoma from other prostatic tumors.
Tumor size may vary greatly, and measurements have been reported to range from 3 cm to 21 cm, frequently presenting with invasion of local structures.11 Advanced stage disease is commonly found at initial diagnosis and is thought to be due to the lack of early specific symptoms. Metastatic disease at presentation may be found in up to one-third of patients, with the lungs being the most common site of metastasis followed by the liver. Local extent and distant spread of disease may be determined by CT or magnetic resonance imaging (MRI) scans, which provide clear delineation of neoplastic and nonneoplastic tissues. 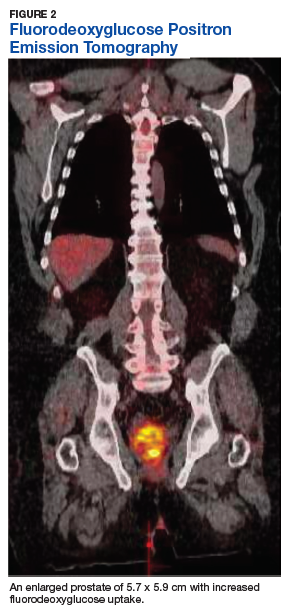
Treatment
Treatment regimens may include a multimodal approach of combination surgery, radiation, and chemotherapy. However, there are currently no standardized guidelines for treatment and the optimal therapy remains unknown.2,3,6 Surgery remains the mainstay of treatment, and patients with surgically resectable tumors are treated with curative intent. Surgeries performed include radical retropubic prostatectomy, radical cystoprostatectomy, suprapubic prostatectomy, and pelvic exenteration.2,5,8,12 These operations may be preceded or followed by radiation therapy and/or chemotherapy depending on extent of disease.
It has been reported that neo-adjuvant chemotherapy and/or radiotherapy can aid in decreasing tumor burden to facilitate a complete resection.2,8,13,14 Patients who are determined to not be candidates for surgery or whom have widespread disease may be offered systemic chemotherapy. Chemotherapy regimens vary, but common regimens include anthracyclines (doxorubicin or epirubicin), alkylating agents (cyclophosphamide, ifosfamide, dacarbazine), and/or vinca alkaloids (vinblastine or vincristine). Patients who do not receive surgical intervention rarely achieve a sustained remission.3,5,8
The long-term prognosis of prostatic leiomyosarcoma is poor due to the aggressive nature of the neoplasm and the high chance of disease recurrence or metastasis. Median survival is estimated at 17 months, and from 50% to 75% of patients die within 2 to 5 years of diagnosis.2,3 Prognosis may be improved in patients with localized disease at diagnosis who are candidates for complete surgical resection with negative margins.13 Adverse prognostic factors include metastatic disease at presentation and the presence of positive surgical margins after surgery.
Overall survival is very poor, and it is estimated that the 1-, 3-, and 5-year survival rates are 68%, 34%, and 26%, respectively.3 However, some studies estimate the 5-year survival to be anywhere from 0 to 60%.8,9 Due to the substantially high risk of death, prostatic leiomyosarcoma may be one of the most aggressive and poorly prognostic malignancies involving the prostate.
Conclusion
Prostatic leiomyosarcoma poses a unique diagnostic challenge, as clinical presentation alone may not always be suggestive of underlying malignancy. This challenge is further exacerbated by its aggressive nature, high risk of metastasis, and difficulties with unclear treatment. Proper history and physical examination, differential diagnosis, and a multidisciplinary approach to patient care are the foundation for early detection and promoting improved survival.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Miedler JD, MacLennan GT. Leiomyosarcoma of the prostate. J Urol. 2007;178(2):668.
2. Zazzara M, Divenuto L, Scarcia M, Cardo G, Maselli FP, Ludovico GM. Leiomyosarcoma of prostate: case report and literature review. Urol Case Rep. 2018;17:4-6.
3. Vandoros GP, Manolidis T, Karamouzis MV, et al. Leiomyosarcoma of the prostate: case report and review of 54 previously published cases. Sarcoma. 2008;2008:458709.
4. Talapatra K, Nemade B, Bhutani R, et al. Recurrent episodes of hematuria: a rare presentation of leiomyosarcoma of prostate. J Cancer Res Ther. 2006;2(4):212-214.
5. Cheville JC, Dundore PA, Nascimento AG, et al. Leiomyosarcoma of the prostate. Report of 23 cases. Cancer. 1995;76(8):1422-1427.
6. Venyo AK. A review of the literature on primary leiomyosarcoma of the prostate gland. Adv Urol. 2015;2015:485786.
7. Stilgenbauer R, Benedict M, Bamshad R, Viduetsky A. Sarcoma of the prostate: sonographic findings and pathologic correlation. J Ultrasound Med. 2007;26(12):1789-1793.
8. Sexton WJ, Lance RE, Reyes AO, Pisters PW, Tu SM, Pisters LL. Adult prostate sarcoma: the M.D. Anderson Cancer Center experience. J Urol. 2001;166(2):521-525.
9. Singh JP, Chakraborty D, Bera MK, Pal D. Leiomyosarcoma of prostate: a rare, aggressive tumor. J Cancer Res Ther. 2013;9(4):743-745.
10. Hansel DE, Herawi M, Montgomery E, Epstein JI. Spindle cell lesions of the adult prostate. Mod Pathol. 2007;20(1):148-158.
11. Punt SE, Eary JF, O'Sullivan J, Conrad EU. Fluorodeoxyglucose positron emission tomography in leiomyosarcoma: imaging characteristics. Nucl Med Commun. 2009;30(7):546-549.
12. Dotan ZA, Tal R, Golijanin D, et al. Adult genitourinary sarcoma: the 25-year Memorial Sloan-Kettering experience. J Urol. 2006;176(5):2033-2038.
13. Musser JE, Assel M, Mashni JW, Sjoberg DD, Russo P. Adult prostate sarcoma: the Memorial Sloan Kettering experience. Urology. 2014;84(3):624-628.
14. Janet NL, May AW, Akins RS. Sarcoma of the prostate: a single institutional review. Am J Clin Oncol. 2009;32:27-29
Prostatic leiomyosarcoma is an aggressive malignancy with a high risk of metastasis and a poor prognosis that poses unique diagnostic and treatment challenges.
Prostatic leiomyosarcoma is an aggressive malignancy with a high risk of metastasis and a poor prognosis that poses unique diagnostic and treatment challenges.
Prostatic leiomyosarcoma is a rare tumor.1 This neoplasm is composed of highly aggressive prostatic smooth muscle cells that present with nonspecific signs and symptoms mimicking other forms of prostatic pathology. Of the primary prostatic sarcomas, leiomyosarcoma represents the most common subtype in adults and is found in 38% to 52% of newly diagnosed prostate sarcoma.1,2 The prognosis is poor, and no clear guidelines exist regarding the optimal treatment approach. We report a case of prostate leiomyosarcoma and describe the disease characteristics, diagnostic modalities, and treatment approach regarding these rare malignancies.
Case Presentation
A 72-year-old male presented with 6 months of progressive severe lower urinary tract symptoms (LUTS) secondary to bladder outlet obstruction. The patient was refractory to medical management with combination α-blocker and 5-α-reductase inhibitor therapy and continued to require multiple emergent bladder catheterizations. Workup with urinalysis, blood biochemistry, and prostate specific antigen (PSA) levels were persistently normal. He reported no hematuria, weight loss, or perineal pain. The patient reported no history of tobacco use, exposure to hazardous chemicals, and had no family history of genitourinary cancers. On rectal exam, the prostate was firm and nodular, with induration noted along the right upper lobe of the prostate.
The patient was referred for a urology consultation and subsequently underwent transurethral resection of the prostate (TURP) for suspected severe benign prostatic hypertrophy (BPH). A histopathologic examination demonstrated atypical cytology consistent with high- grade leiomyosarcoma. Immunohistochemical analysis revealed positive staining for vimentin, smooth muscle actin, desmin (partial), cytokeratin, smooth muscle myosin, muscle specific actin, and Ki-67 (50%-60% expression).
Fluorodeoxyglucose positron emission tomography (FDG-PET) scan revealed a 5.7 x 5.9 cm tumor with a maximum standardized uptake value (SUVmax) of 12.6 in the right posterior prostate, without evidence of metastatic disease (Figures 1A and 1B).
Discussion
Originating from prostatic interstitial cells, prostatic leiomyosarcoma is a rare tumor that accounts for < 0.1% of all primary prostatic malignancies.1 Since its first description in 1950 by Riba and colleagues, < 200 cases have been reported worldwide.2 Among the sarcomas of the prostate, it is the most common tumor, accounting for around 38% to 52% of prostate sarcoma presentations.1,2
Patients typically present between the ages of 41 and 78 years (mean age 61 years).2,3 Signs and symptoms at presentation may vary; however, the most common symptoms are related to lower urinary tract obstruction (89.4% of patients). These symptoms include urinary frequency, urgency, nocturia, and may mimic the presentation of BPH.
Symptoms commonly associated with other malignancies, including constitutional symptoms such as weight loss, tend to occur less frequently or may be absent. Perineal or rectal pain may only be present in 25.6% of patients. Hematuria, burning on ejaculation, and constitutional symptoms are a less common presentation (< 10% of patients).3,4 PSA levels typically do not rise and are found to be within normal limits. The lack of PSA elevation is related to the tumors nonepithelial origin and may contribute to a delay in diagnosis.2,4,5
Diagnosis
Diagnosis may be further eluded as digital rectal exam (DRE) findings tend to reveal nonspecific enlargement of the prostate, resembling that of BPH. DRE may show a hard and firm prostate with nodular induration at the base or over the lobes of the prostate.6 At this stage a urology consultation is useful, as diagnosis is most commonly achieved using transrectal ultrasound (TRUS) with ultrasound-guided needle biopsy or after a TURP procedure.3
Prostate sarcoma is associated with markedly enlarged prostate volume, irregular margins with invasion, or heterogenous hypoechoic lesions on TRUS.7 Transperineal biopsy, computed tomography (CT)-guided biopsy, or suprapubic prostatectomy have been less frequently employed for diagnosis in previously reported cases.8 Specialized imaging modalities, such as CT scan or bone scan, do not show any specific findings with regards to these tumors; their role is limited to evaluation of the local and distant metastasis and for follow-up assessments.9 Transabdominal ultrasound may assess hydronephrosis or enlarged prostate and its relation to nearby structures, although it has not been shown to be helpful in establishing a specific diagnosis.6
Histologically, prostatic leiomyosarcoma is a distinct subtype of prostatic sarcoma. Other subtypes include stromal tumors such as rhabdomyosarcoma, fibrosarcoma, and spindle cell sarcoma.2 The majority of leiomyosarcomas are high-grade lesions demonstrating neoplastic spindle cells with nuclear atypia, multifocal necrosis, and cystic degeneration. Low-grade leiomyosarcomas are very rare.10 Immunohistochemistry is characteristically positive for vimentin, smooth muscle actin, and desmin expression. Cytokeratin may be positive in up to 25% of cases, whereas S-100, CD34, CD117, and PSA are negative.2,3 These histopathological findings help to differentiate leiomyosarcoma from other prostatic tumors.
Tumor size may vary greatly, and measurements have been reported to range from 3 cm to 21 cm, frequently presenting with invasion of local structures.11 Advanced stage disease is commonly found at initial diagnosis and is thought to be due to the lack of early specific symptoms. Metastatic disease at presentation may be found in up to one-third of patients, with the lungs being the most common site of metastasis followed by the liver. Local extent and distant spread of disease may be determined by CT or magnetic resonance imaging (MRI) scans, which provide clear delineation of neoplastic and nonneoplastic tissues.
Treatment
Treatment regimens may include a multimodal approach of combination surgery, radiation, and chemotherapy. However, there are currently no standardized guidelines for treatment and the optimal therapy remains unknown.2,3,6 Surgery remains the mainstay of treatment, and patients with surgically resectable tumors are treated with curative intent. Surgeries performed include radical retropubic prostatectomy, radical cystoprostatectomy, suprapubic prostatectomy, and pelvic exenteration.2,5,8,12 These operations may be preceded or followed by radiation therapy and/or chemotherapy depending on extent of disease.
It has been reported that neo-adjuvant chemotherapy and/or radiotherapy can aid in decreasing tumor burden to facilitate a complete resection.2,8,13,14 Patients who are determined to not be candidates for surgery or whom have widespread disease may be offered systemic chemotherapy. Chemotherapy regimens vary, but common regimens include anthracyclines (doxorubicin or epirubicin), alkylating agents (cyclophosphamide, ifosfamide, dacarbazine), and/or vinca alkaloids (vinblastine or vincristine). Patients who do not receive surgical intervention rarely achieve a sustained remission.3,5,8
The long-term prognosis of prostatic leiomyosarcoma is poor due to the aggressive nature of the neoplasm and the high chance of disease recurrence or metastasis. Median survival is estimated at 17 months, and from 50% to 75% of patients die within 2 to 5 years of diagnosis.2,3 Prognosis may be improved in patients with localized disease at diagnosis who are candidates for complete surgical resection with negative margins.13 Adverse prognostic factors include metastatic disease at presentation and the presence of positive surgical margins after surgery.
Overall survival is very poor, and it is estimated that the 1-, 3-, and 5-year survival rates are 68%, 34%, and 26%, respectively.3 However, some studies estimate the 5-year survival to be anywhere from 0 to 60%.8,9 Due to the substantially high risk of death, prostatic leiomyosarcoma may be one of the most aggressive and poorly prognostic malignancies involving the prostate.
Conclusion
Prostatic leiomyosarcoma poses a unique diagnostic challenge, as clinical presentation alone may not always be suggestive of underlying malignancy. This challenge is further exacerbated by its aggressive nature, high risk of metastasis, and difficulties with unclear treatment. Proper history and physical examination, differential diagnosis, and a multidisciplinary approach to patient care are the foundation for early detection and promoting improved survival.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Prostatic leiomyosarcoma is a rare tumor.1 This neoplasm is composed of highly aggressive prostatic smooth muscle cells that present with nonspecific signs and symptoms mimicking other forms of prostatic pathology. Of the primary prostatic sarcomas, leiomyosarcoma represents the most common subtype in adults and is found in 38% to 52% of newly diagnosed prostate sarcoma.1,2 The prognosis is poor, and no clear guidelines exist regarding the optimal treatment approach. We report a case of prostate leiomyosarcoma and describe the disease characteristics, diagnostic modalities, and treatment approach regarding these rare malignancies.
Case Presentation
A 72-year-old male presented with 6 months of progressive severe lower urinary tract symptoms (LUTS) secondary to bladder outlet obstruction. The patient was refractory to medical management with combination α-blocker and 5-α-reductase inhibitor therapy and continued to require multiple emergent bladder catheterizations. Workup with urinalysis, blood biochemistry, and prostate specific antigen (PSA) levels were persistently normal. He reported no hematuria, weight loss, or perineal pain. The patient reported no history of tobacco use, exposure to hazardous chemicals, and had no family history of genitourinary cancers. On rectal exam, the prostate was firm and nodular, with induration noted along the right upper lobe of the prostate.
The patient was referred for a urology consultation and subsequently underwent transurethral resection of the prostate (TURP) for suspected severe benign prostatic hypertrophy (BPH). A histopathologic examination demonstrated atypical cytology consistent with high- grade leiomyosarcoma. Immunohistochemical analysis revealed positive staining for vimentin, smooth muscle actin, desmin (partial), cytokeratin, smooth muscle myosin, muscle specific actin, and Ki-67 (50%-60% expression).
Fluorodeoxyglucose positron emission tomography (FDG-PET) scan revealed a 5.7 x 5.9 cm tumor with a maximum standardized uptake value (SUVmax) of 12.6 in the right posterior prostate, without evidence of metastatic disease (Figures 1A and 1B).
Discussion
Originating from prostatic interstitial cells, prostatic leiomyosarcoma is a rare tumor that accounts for < 0.1% of all primary prostatic malignancies.1 Since its first description in 1950 by Riba and colleagues, < 200 cases have been reported worldwide.2 Among the sarcomas of the prostate, it is the most common tumor, accounting for around 38% to 52% of prostate sarcoma presentations.1,2
Patients typically present between the ages of 41 and 78 years (mean age 61 years).2,3 Signs and symptoms at presentation may vary; however, the most common symptoms are related to lower urinary tract obstruction (89.4% of patients). These symptoms include urinary frequency, urgency, nocturia, and may mimic the presentation of BPH.
Symptoms commonly associated with other malignancies, including constitutional symptoms such as weight loss, tend to occur less frequently or may be absent. Perineal or rectal pain may only be present in 25.6% of patients. Hematuria, burning on ejaculation, and constitutional symptoms are a less common presentation (< 10% of patients).3,4 PSA levels typically do not rise and are found to be within normal limits. The lack of PSA elevation is related to the tumors nonepithelial origin and may contribute to a delay in diagnosis.2,4,5
Diagnosis
Diagnosis may be further eluded as digital rectal exam (DRE) findings tend to reveal nonspecific enlargement of the prostate, resembling that of BPH. DRE may show a hard and firm prostate with nodular induration at the base or over the lobes of the prostate.6 At this stage a urology consultation is useful, as diagnosis is most commonly achieved using transrectal ultrasound (TRUS) with ultrasound-guided needle biopsy or after a TURP procedure.3
Prostate sarcoma is associated with markedly enlarged prostate volume, irregular margins with invasion, or heterogenous hypoechoic lesions on TRUS.7 Transperineal biopsy, computed tomography (CT)-guided biopsy, or suprapubic prostatectomy have been less frequently employed for diagnosis in previously reported cases.8 Specialized imaging modalities, such as CT scan or bone scan, do not show any specific findings with regards to these tumors; their role is limited to evaluation of the local and distant metastasis and for follow-up assessments.9 Transabdominal ultrasound may assess hydronephrosis or enlarged prostate and its relation to nearby structures, although it has not been shown to be helpful in establishing a specific diagnosis.6
Histologically, prostatic leiomyosarcoma is a distinct subtype of prostatic sarcoma. Other subtypes include stromal tumors such as rhabdomyosarcoma, fibrosarcoma, and spindle cell sarcoma.2 The majority of leiomyosarcomas are high-grade lesions demonstrating neoplastic spindle cells with nuclear atypia, multifocal necrosis, and cystic degeneration. Low-grade leiomyosarcomas are very rare.10 Immunohistochemistry is characteristically positive for vimentin, smooth muscle actin, and desmin expression. Cytokeratin may be positive in up to 25% of cases, whereas S-100, CD34, CD117, and PSA are negative.2,3 These histopathological findings help to differentiate leiomyosarcoma from other prostatic tumors.
Tumor size may vary greatly, and measurements have been reported to range from 3 cm to 21 cm, frequently presenting with invasion of local structures.11 Advanced stage disease is commonly found at initial diagnosis and is thought to be due to the lack of early specific symptoms. Metastatic disease at presentation may be found in up to one-third of patients, with the lungs being the most common site of metastasis followed by the liver. Local extent and distant spread of disease may be determined by CT or magnetic resonance imaging (MRI) scans, which provide clear delineation of neoplastic and nonneoplastic tissues.
Treatment
Treatment regimens may include a multimodal approach of combination surgery, radiation, and chemotherapy. However, there are currently no standardized guidelines for treatment and the optimal therapy remains unknown.2,3,6 Surgery remains the mainstay of treatment, and patients with surgically resectable tumors are treated with curative intent. Surgeries performed include radical retropubic prostatectomy, radical cystoprostatectomy, suprapubic prostatectomy, and pelvic exenteration.2,5,8,12 These operations may be preceded or followed by radiation therapy and/or chemotherapy depending on extent of disease.
It has been reported that neo-adjuvant chemotherapy and/or radiotherapy can aid in decreasing tumor burden to facilitate a complete resection.2,8,13,14 Patients who are determined to not be candidates for surgery or whom have widespread disease may be offered systemic chemotherapy. Chemotherapy regimens vary, but common regimens include anthracyclines (doxorubicin or epirubicin), alkylating agents (cyclophosphamide, ifosfamide, dacarbazine), and/or vinca alkaloids (vinblastine or vincristine). Patients who do not receive surgical intervention rarely achieve a sustained remission.3,5,8
The long-term prognosis of prostatic leiomyosarcoma is poor due to the aggressive nature of the neoplasm and the high chance of disease recurrence or metastasis. Median survival is estimated at 17 months, and from 50% to 75% of patients die within 2 to 5 years of diagnosis.2,3 Prognosis may be improved in patients with localized disease at diagnosis who are candidates for complete surgical resection with negative margins.13 Adverse prognostic factors include metastatic disease at presentation and the presence of positive surgical margins after surgery.
Overall survival is very poor, and it is estimated that the 1-, 3-, and 5-year survival rates are 68%, 34%, and 26%, respectively.3 However, some studies estimate the 5-year survival to be anywhere from 0 to 60%.8,9 Due to the substantially high risk of death, prostatic leiomyosarcoma may be one of the most aggressive and poorly prognostic malignancies involving the prostate.
Conclusion
Prostatic leiomyosarcoma poses a unique diagnostic challenge, as clinical presentation alone may not always be suggestive of underlying malignancy. This challenge is further exacerbated by its aggressive nature, high risk of metastasis, and difficulties with unclear treatment. Proper history and physical examination, differential diagnosis, and a multidisciplinary approach to patient care are the foundation for early detection and promoting improved survival.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Miedler JD, MacLennan GT. Leiomyosarcoma of the prostate. J Urol. 2007;178(2):668.
2. Zazzara M, Divenuto L, Scarcia M, Cardo G, Maselli FP, Ludovico GM. Leiomyosarcoma of prostate: case report and literature review. Urol Case Rep. 2018;17:4-6.
3. Vandoros GP, Manolidis T, Karamouzis MV, et al. Leiomyosarcoma of the prostate: case report and review of 54 previously published cases. Sarcoma. 2008;2008:458709.
4. Talapatra K, Nemade B, Bhutani R, et al. Recurrent episodes of hematuria: a rare presentation of leiomyosarcoma of prostate. J Cancer Res Ther. 2006;2(4):212-214.
5. Cheville JC, Dundore PA, Nascimento AG, et al. Leiomyosarcoma of the prostate. Report of 23 cases. Cancer. 1995;76(8):1422-1427.
6. Venyo AK. A review of the literature on primary leiomyosarcoma of the prostate gland. Adv Urol. 2015;2015:485786.
7. Stilgenbauer R, Benedict M, Bamshad R, Viduetsky A. Sarcoma of the prostate: sonographic findings and pathologic correlation. J Ultrasound Med. 2007;26(12):1789-1793.
8. Sexton WJ, Lance RE, Reyes AO, Pisters PW, Tu SM, Pisters LL. Adult prostate sarcoma: the M.D. Anderson Cancer Center experience. J Urol. 2001;166(2):521-525.
9. Singh JP, Chakraborty D, Bera MK, Pal D. Leiomyosarcoma of prostate: a rare, aggressive tumor. J Cancer Res Ther. 2013;9(4):743-745.
10. Hansel DE, Herawi M, Montgomery E, Epstein JI. Spindle cell lesions of the adult prostate. Mod Pathol. 2007;20(1):148-158.
11. Punt SE, Eary JF, O'Sullivan J, Conrad EU. Fluorodeoxyglucose positron emission tomography in leiomyosarcoma: imaging characteristics. Nucl Med Commun. 2009;30(7):546-549.
12. Dotan ZA, Tal R, Golijanin D, et al. Adult genitourinary sarcoma: the 25-year Memorial Sloan-Kettering experience. J Urol. 2006;176(5):2033-2038.
13. Musser JE, Assel M, Mashni JW, Sjoberg DD, Russo P. Adult prostate sarcoma: the Memorial Sloan Kettering experience. Urology. 2014;84(3):624-628.
14. Janet NL, May AW, Akins RS. Sarcoma of the prostate: a single institutional review. Am J Clin Oncol. 2009;32:27-29
1. Miedler JD, MacLennan GT. Leiomyosarcoma of the prostate. J Urol. 2007;178(2):668.
2. Zazzara M, Divenuto L, Scarcia M, Cardo G, Maselli FP, Ludovico GM. Leiomyosarcoma of prostate: case report and literature review. Urol Case Rep. 2018;17:4-6.
3. Vandoros GP, Manolidis T, Karamouzis MV, et al. Leiomyosarcoma of the prostate: case report and review of 54 previously published cases. Sarcoma. 2008;2008:458709.
4. Talapatra K, Nemade B, Bhutani R, et al. Recurrent episodes of hematuria: a rare presentation of leiomyosarcoma of prostate. J Cancer Res Ther. 2006;2(4):212-214.
5. Cheville JC, Dundore PA, Nascimento AG, et al. Leiomyosarcoma of the prostate. Report of 23 cases. Cancer. 1995;76(8):1422-1427.
6. Venyo AK. A review of the literature on primary leiomyosarcoma of the prostate gland. Adv Urol. 2015;2015:485786.
7. Stilgenbauer R, Benedict M, Bamshad R, Viduetsky A. Sarcoma of the prostate: sonographic findings and pathologic correlation. J Ultrasound Med. 2007;26(12):1789-1793.
8. Sexton WJ, Lance RE, Reyes AO, Pisters PW, Tu SM, Pisters LL. Adult prostate sarcoma: the M.D. Anderson Cancer Center experience. J Urol. 2001;166(2):521-525.
9. Singh JP, Chakraborty D, Bera MK, Pal D. Leiomyosarcoma of prostate: a rare, aggressive tumor. J Cancer Res Ther. 2013;9(4):743-745.
10. Hansel DE, Herawi M, Montgomery E, Epstein JI. Spindle cell lesions of the adult prostate. Mod Pathol. 2007;20(1):148-158.
11. Punt SE, Eary JF, O'Sullivan J, Conrad EU. Fluorodeoxyglucose positron emission tomography in leiomyosarcoma: imaging characteristics. Nucl Med Commun. 2009;30(7):546-549.
12. Dotan ZA, Tal R, Golijanin D, et al. Adult genitourinary sarcoma: the 25-year Memorial Sloan-Kettering experience. J Urol. 2006;176(5):2033-2038.
13. Musser JE, Assel M, Mashni JW, Sjoberg DD, Russo P. Adult prostate sarcoma: the Memorial Sloan Kettering experience. Urology. 2014;84(3):624-628.
14. Janet NL, May AW, Akins RS. Sarcoma of the prostate: a single institutional review. Am J Clin Oncol. 2009;32:27-29