User login
FDA approves first treatment for advanced epithelioid sarcoma
The Food and Drug Administration has granted accelerated approval to tazemetostat (Tazverik) for the treatment of adults and pediatric patients aged 16 years and older with metastatic or locally advanced epithelioid sarcoma not eligible for complete resection.

Approval was based on overall response rate in a trial enrolling 62 patients with metastatic or locally advanced epithelioid sarcoma. The overall response rate was 15%, with 1.6% of patients having a complete response and 13% having a partial response. Of the nine patients that had a response, six (67%) had a response lasting 6 months or longer, the FDA said in a press statement.
The most common side effects for patients taking tazemetostat were pain, fatigue, nausea, decreased appetite, vomiting, and constipation. Patients treated with tazemetostat are at increased risk of developing secondary malignancies, including T-cell lymphoblastic lymphoma, myelodysplastic syndrome, and acute myeloid leukemia.
“Epithelioid sarcoma accounts for less than 1% of all soft-tissue sarcomas,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the Center for Drug Evaluation and Research. “Until today, there were no treatment options specifically for patients with epithelioid sarcoma. The approval of Tazverik provides a treatment option that specifically targets this disease.”
Tazemetostat must be dispensed with a patient medication guide that describes important information about the drug’s uses and risks, the FDA said.
The Food and Drug Administration has granted accelerated approval to tazemetostat (Tazverik) for the treatment of adults and pediatric patients aged 16 years and older with metastatic or locally advanced epithelioid sarcoma not eligible for complete resection.
Approval was based on overall response rate in a trial enrolling 62 patients with metastatic or locally advanced epithelioid sarcoma. The overall response rate was 15%, with 1.6% of patients having a complete response and 13% having a partial response. Of the nine patients that had a response, six (67%) had a response lasting 6 months or longer, the FDA said in a press statement.
The most common side effects for patients taking tazemetostat were pain, fatigue, nausea, decreased appetite, vomiting, and constipation. Patients treated with tazemetostat are at increased risk of developing secondary malignancies, including T-cell lymphoblastic lymphoma, myelodysplastic syndrome, and acute myeloid leukemia.
“Epithelioid sarcoma accounts for less than 1% of all soft-tissue sarcomas,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the Center for Drug Evaluation and Research. “Until today, there were no treatment options specifically for patients with epithelioid sarcoma. The approval of Tazverik provides a treatment option that specifically targets this disease.”
Tazemetostat must be dispensed with a patient medication guide that describes important information about the drug’s uses and risks, the FDA said.
The Food and Drug Administration has granted accelerated approval to tazemetostat (Tazverik) for the treatment of adults and pediatric patients aged 16 years and older with metastatic or locally advanced epithelioid sarcoma not eligible for complete resection.
Approval was based on overall response rate in a trial enrolling 62 patients with metastatic or locally advanced epithelioid sarcoma. The overall response rate was 15%, with 1.6% of patients having a complete response and 13% having a partial response. Of the nine patients that had a response, six (67%) had a response lasting 6 months or longer, the FDA said in a press statement.
The most common side effects for patients taking tazemetostat were pain, fatigue, nausea, decreased appetite, vomiting, and constipation. Patients treated with tazemetostat are at increased risk of developing secondary malignancies, including T-cell lymphoblastic lymphoma, myelodysplastic syndrome, and acute myeloid leukemia.
“Epithelioid sarcoma accounts for less than 1% of all soft-tissue sarcomas,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the Center for Drug Evaluation and Research. “Until today, there were no treatment options specifically for patients with epithelioid sarcoma. The approval of Tazverik provides a treatment option that specifically targets this disease.”
Tazemetostat must be dispensed with a patient medication guide that describes important information about the drug’s uses and risks, the FDA said.
T-VEC plus pembrolizumab yields promising response rate in phase 2 sarcoma study
The combination of oncolytic immunotherapy and an immune checkpoint inhibitor had promising activity in a phase 2 study including patients with previously treated advanced sarcoma, investigators report.
Responses were seen in about one-third of the patients (30%) at 24 weeks after starting treatment with talimogene laherparepvec (T-VEC) and pembrolizumab, according to the investigators, led by Ciara M. Kelly, MBBChBAO, MD, of the department of sarcoma oncology at Memorial Sloan Kettering Cancer Center, New York.
“To our knowledge, this is one of the highest ORRs [objective response rates] reported in an unselected sarcoma-specific study population evaluating the efficacy of combination immunotherapy,” Dr. Kelly and coauthors wrote in JAMA Oncology.
Two of the patients who responded to the combination had previously failed regimens that included an immune checkpoint inhibitor, suggesting T-VEC might be augmenting the efficacy of pembrolizumab, according to the authors.
Among responders, the mean number of prior treatments was one, while by contrast, most of the study cohort (60%) had three or more prior treatments. “This finding supports the rationale to enroll patients with sarcoma in immunotherapy trials earlier in their treatment course,” Dr. Kelly and colleagues wrote.
In studies of immunotherapy in Merkel cell carcinoma and other cancer types, higher response rates have been seen in patients who were naive to chemotherapy, as opposed to those who were refractory to it.
The present single-institution, phase 2 study by Dr. Kelly and colleagues included 20 adult patients with histologically confirmed locally advanced or metastatic sarcoma. The patients received a fixed dose of pembrolizumab administered intravenously, and had T-VEC injected into palpable lesions, on day 1 of 21-day cycles.
While the ORR was 30% at 24 weeks (7 of 35 patients), an additional patient had a response at 32 weeks, bringing the ORR to 35%, according to the report.
“Maximal response to therapy may take a prolonged period to achieve,” the investigators wrote, noting that the median time to response was 14.4 weeks.
Median duration of response was 56.1 weeks, suggesting the combination provided durable disease control, they added.
Safety was consistent with what was seen in a previous study of T-VEC and pembrolizumab in melanoma. In this sarcoma study, the incidence of grade 3 treatment-related adverse events was 20%, while in studies of conventional chemotherapy in this setting, the rate of those events has been “comparable but generally higher,” the investigators wrote.
Dr. Kelly and colleagues wrote that a randomized clinical trial is the “ideal next step” in the development of the regimen, adding that an expansion of the present phase 2 study is in progress.
The study was funded by Amgen and Merck, with additional grant support from Cycle for Survival and the National Institutes of Health/National Cancer Institute. Dr. Kelly reported disclosures related to Merck, Amgen, Agios Pharmaceuticals, and Exicure. Her coauthors reported disclosures with Regeneron, Novartis, Takeda, and Nektar, among others.
SOURCE: Kelly CM et al. JAMA Oncol. 2020 Jan 23. doi: 10.1001/jamaoncol.2019.6152.
The combination of oncolytic immunotherapy and an immune checkpoint inhibitor had promising activity in a phase 2 study including patients with previously treated advanced sarcoma, investigators report.
Responses were seen in about one-third of the patients (30%) at 24 weeks after starting treatment with talimogene laherparepvec (T-VEC) and pembrolizumab, according to the investigators, led by Ciara M. Kelly, MBBChBAO, MD, of the department of sarcoma oncology at Memorial Sloan Kettering Cancer Center, New York.
“To our knowledge, this is one of the highest ORRs [objective response rates] reported in an unselected sarcoma-specific study population evaluating the efficacy of combination immunotherapy,” Dr. Kelly and coauthors wrote in JAMA Oncology.
Two of the patients who responded to the combination had previously failed regimens that included an immune checkpoint inhibitor, suggesting T-VEC might be augmenting the efficacy of pembrolizumab, according to the authors.
Among responders, the mean number of prior treatments was one, while by contrast, most of the study cohort (60%) had three or more prior treatments. “This finding supports the rationale to enroll patients with sarcoma in immunotherapy trials earlier in their treatment course,” Dr. Kelly and colleagues wrote.
In studies of immunotherapy in Merkel cell carcinoma and other cancer types, higher response rates have been seen in patients who were naive to chemotherapy, as opposed to those who were refractory to it.
The present single-institution, phase 2 study by Dr. Kelly and colleagues included 20 adult patients with histologically confirmed locally advanced or metastatic sarcoma. The patients received a fixed dose of pembrolizumab administered intravenously, and had T-VEC injected into palpable lesions, on day 1 of 21-day cycles.
While the ORR was 30% at 24 weeks (7 of 35 patients), an additional patient had a response at 32 weeks, bringing the ORR to 35%, according to the report.
“Maximal response to therapy may take a prolonged period to achieve,” the investigators wrote, noting that the median time to response was 14.4 weeks.
Median duration of response was 56.1 weeks, suggesting the combination provided durable disease control, they added.
Safety was consistent with what was seen in a previous study of T-VEC and pembrolizumab in melanoma. In this sarcoma study, the incidence of grade 3 treatment-related adverse events was 20%, while in studies of conventional chemotherapy in this setting, the rate of those events has been “comparable but generally higher,” the investigators wrote.
Dr. Kelly and colleagues wrote that a randomized clinical trial is the “ideal next step” in the development of the regimen, adding that an expansion of the present phase 2 study is in progress.
The study was funded by Amgen and Merck, with additional grant support from Cycle for Survival and the National Institutes of Health/National Cancer Institute. Dr. Kelly reported disclosures related to Merck, Amgen, Agios Pharmaceuticals, and Exicure. Her coauthors reported disclosures with Regeneron, Novartis, Takeda, and Nektar, among others.
SOURCE: Kelly CM et al. JAMA Oncol. 2020 Jan 23. doi: 10.1001/jamaoncol.2019.6152.
The combination of oncolytic immunotherapy and an immune checkpoint inhibitor had promising activity in a phase 2 study including patients with previously treated advanced sarcoma, investigators report.
Responses were seen in about one-third of the patients (30%) at 24 weeks after starting treatment with talimogene laherparepvec (T-VEC) and pembrolizumab, according to the investigators, led by Ciara M. Kelly, MBBChBAO, MD, of the department of sarcoma oncology at Memorial Sloan Kettering Cancer Center, New York.
“To our knowledge, this is one of the highest ORRs [objective response rates] reported in an unselected sarcoma-specific study population evaluating the efficacy of combination immunotherapy,” Dr. Kelly and coauthors wrote in JAMA Oncology.
Two of the patients who responded to the combination had previously failed regimens that included an immune checkpoint inhibitor, suggesting T-VEC might be augmenting the efficacy of pembrolizumab, according to the authors.
Among responders, the mean number of prior treatments was one, while by contrast, most of the study cohort (60%) had three or more prior treatments. “This finding supports the rationale to enroll patients with sarcoma in immunotherapy trials earlier in their treatment course,” Dr. Kelly and colleagues wrote.
In studies of immunotherapy in Merkel cell carcinoma and other cancer types, higher response rates have been seen in patients who were naive to chemotherapy, as opposed to those who were refractory to it.
The present single-institution, phase 2 study by Dr. Kelly and colleagues included 20 adult patients with histologically confirmed locally advanced or metastatic sarcoma. The patients received a fixed dose of pembrolizumab administered intravenously, and had T-VEC injected into palpable lesions, on day 1 of 21-day cycles.
While the ORR was 30% at 24 weeks (7 of 35 patients), an additional patient had a response at 32 weeks, bringing the ORR to 35%, according to the report.
“Maximal response to therapy may take a prolonged period to achieve,” the investigators wrote, noting that the median time to response was 14.4 weeks.
Median duration of response was 56.1 weeks, suggesting the combination provided durable disease control, they added.
Safety was consistent with what was seen in a previous study of T-VEC and pembrolizumab in melanoma. In this sarcoma study, the incidence of grade 3 treatment-related adverse events was 20%, while in studies of conventional chemotherapy in this setting, the rate of those events has been “comparable but generally higher,” the investigators wrote.
Dr. Kelly and colleagues wrote that a randomized clinical trial is the “ideal next step” in the development of the regimen, adding that an expansion of the present phase 2 study is in progress.
The study was funded by Amgen and Merck, with additional grant support from Cycle for Survival and the National Institutes of Health/National Cancer Institute. Dr. Kelly reported disclosures related to Merck, Amgen, Agios Pharmaceuticals, and Exicure. Her coauthors reported disclosures with Regeneron, Novartis, Takeda, and Nektar, among others.
SOURCE: Kelly CM et al. JAMA Oncol. 2020 Jan 23. doi: 10.1001/jamaoncol.2019.6152.
FROM JAMA ONCOLOGY
FDA approves avapritinib for adults with GIST with PDGFRA mutation
The Food and Drug Administration has approved avapritinib (Ayvakit) for the treatment of adults with unresectable or metastatic gastrointestinal stromal tumors (GIST) with a platelet-derived growth factor receptor–alpha (PDGFRA) exon 18 mutation.
Approval was based on results from a clinical trial of 43 patients with PDGFRA exon 18 mutations, including 38 patients with a PDGFRA D842V mutation, who received 300 mg avapritinib once daily, the FDA said in a statement.
The overall response rate was 84% (7% with complete response, 77% with partial response); the response rate in patients with a D842V mutation was 89% (8% with complete response, 82% with partial response). Median response duration was not reached, but 61% of patients had a response lasting longer than 6 months.
The most common adverse events associated with avapritinib include edema, nausea, fatigue/asthenia, cognitive impairment, vomiting, decreased appetite, diarrhea, hair color changes, increased lacrimation, abdominal pain, constipation, rash, and dizziness. The drug also can cause intracranial hemorrhage and have effects on the central nervous system.
“GIST harboring a PDGFRA exon 18 mutation do not respond to standard therapies for GIST. However, today’s approval provides patients with the first drug specifically approved for GIST harboring this mutation,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the Center for Drug Evaluation and Research, said in the statement.
The Food and Drug Administration has approved avapritinib (Ayvakit) for the treatment of adults with unresectable or metastatic gastrointestinal stromal tumors (GIST) with a platelet-derived growth factor receptor–alpha (PDGFRA) exon 18 mutation.
Approval was based on results from a clinical trial of 43 patients with PDGFRA exon 18 mutations, including 38 patients with a PDGFRA D842V mutation, who received 300 mg avapritinib once daily, the FDA said in a statement.
The overall response rate was 84% (7% with complete response, 77% with partial response); the response rate in patients with a D842V mutation was 89% (8% with complete response, 82% with partial response). Median response duration was not reached, but 61% of patients had a response lasting longer than 6 months.
The most common adverse events associated with avapritinib include edema, nausea, fatigue/asthenia, cognitive impairment, vomiting, decreased appetite, diarrhea, hair color changes, increased lacrimation, abdominal pain, constipation, rash, and dizziness. The drug also can cause intracranial hemorrhage and have effects on the central nervous system.
“GIST harboring a PDGFRA exon 18 mutation do not respond to standard therapies for GIST. However, today’s approval provides patients with the first drug specifically approved for GIST harboring this mutation,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the Center for Drug Evaluation and Research, said in the statement.
The Food and Drug Administration has approved avapritinib (Ayvakit) for the treatment of adults with unresectable or metastatic gastrointestinal stromal tumors (GIST) with a platelet-derived growth factor receptor–alpha (PDGFRA) exon 18 mutation.
Approval was based on results from a clinical trial of 43 patients with PDGFRA exon 18 mutations, including 38 patients with a PDGFRA D842V mutation, who received 300 mg avapritinib once daily, the FDA said in a statement.
The overall response rate was 84% (7% with complete response, 77% with partial response); the response rate in patients with a D842V mutation was 89% (8% with complete response, 82% with partial response). Median response duration was not reached, but 61% of patients had a response lasting longer than 6 months.
The most common adverse events associated with avapritinib include edema, nausea, fatigue/asthenia, cognitive impairment, vomiting, decreased appetite, diarrhea, hair color changes, increased lacrimation, abdominal pain, constipation, rash, and dizziness. The drug also can cause intracranial hemorrhage and have effects on the central nervous system.
“GIST harboring a PDGFRA exon 18 mutation do not respond to standard therapies for GIST. However, today’s approval provides patients with the first drug specifically approved for GIST harboring this mutation,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the Center for Drug Evaluation and Research, said in the statement.
FDA Awards Grant To Study Temozolomide in Gist
A phase 2 study of temozolomide in gastrointestinal stromal tumors (GIST) received one of the 12 grants awarded in October by the US Food and Drug Administration (FDA) to enhance the development of medical products for patients with rare diseases. Jason Sicklick, MD, and the University of California San Diego in La Jolla will receive $1.5 million over 3 years to conduct the phase 2 study (NCT03556384). The objective of the study is to determine the efficacy at 6 months of temozolomide therapy in patients with the SDH-mutant/deficient subtype. Temozolomide is approved by the FDA to treat newly diagnosed glioblastoma multiforme and refractory anaplastic astrocytomas. It is not approved for the treatment of SDH-mutant/deficient GIST.
The FDA awarded the grants through the Orphan Products Clinical Trials Grants Program. Other orphan diseases receiving grants included glomerulopathy, gliomas, Fanconi anemia, sickle cell respiratory complications, Duchenne muscular dystrophy, HPV associated respiratory papillomatosis, refractory viral infections and T-cell immunodeficiency, oral cancer, retinoblastoma, cerebellar brain tumors, and acute myeloid leukemia.
A phase 2 study of temozolomide in gastrointestinal stromal tumors (GIST) received one of the 12 grants awarded in October by the US Food and Drug Administration (FDA) to enhance the development of medical products for patients with rare diseases. Jason Sicklick, MD, and the University of California San Diego in La Jolla will receive $1.5 million over 3 years to conduct the phase 2 study (NCT03556384). The objective of the study is to determine the efficacy at 6 months of temozolomide therapy in patients with the SDH-mutant/deficient subtype. Temozolomide is approved by the FDA to treat newly diagnosed glioblastoma multiforme and refractory anaplastic astrocytomas. It is not approved for the treatment of SDH-mutant/deficient GIST.
The FDA awarded the grants through the Orphan Products Clinical Trials Grants Program. Other orphan diseases receiving grants included glomerulopathy, gliomas, Fanconi anemia, sickle cell respiratory complications, Duchenne muscular dystrophy, HPV associated respiratory papillomatosis, refractory viral infections and T-cell immunodeficiency, oral cancer, retinoblastoma, cerebellar brain tumors, and acute myeloid leukemia.
A phase 2 study of temozolomide in gastrointestinal stromal tumors (GIST) received one of the 12 grants awarded in October by the US Food and Drug Administration (FDA) to enhance the development of medical products for patients with rare diseases. Jason Sicklick, MD, and the University of California San Diego in La Jolla will receive $1.5 million over 3 years to conduct the phase 2 study (NCT03556384). The objective of the study is to determine the efficacy at 6 months of temozolomide therapy in patients with the SDH-mutant/deficient subtype. Temozolomide is approved by the FDA to treat newly diagnosed glioblastoma multiforme and refractory anaplastic astrocytomas. It is not approved for the treatment of SDH-mutant/deficient GIST.
The FDA awarded the grants through the Orphan Products Clinical Trials Grants Program. Other orphan diseases receiving grants included glomerulopathy, gliomas, Fanconi anemia, sickle cell respiratory complications, Duchenne muscular dystrophy, HPV associated respiratory papillomatosis, refractory viral infections and T-cell immunodeficiency, oral cancer, retinoblastoma, cerebellar brain tumors, and acute myeloid leukemia.
Scientific Roundtable Focuses Efforts on Leiomyosarcoma Research
Research clinicians from the US and abroad participated in a scientific research roundtable this past September to establish the most important issues facing leiomyosarcoma (LMS) research and clinical trials. The workshop, expected to be an annual event, is a joint effort of the National Leiomyosarcoma Foundation (NLMSF) and Sarcoma Patients EuroNet (SPAEN).
The roundtable’s mission is to bring together sarcoma experts for a meeting dedicated to LMS, where participants discuss the present state of LMS and the continued challenges of diagnosis and treatment. Its goal is to develop working plans to close the gaps in LMS patient care and improve LMS patient-care protocols. They also advise the NLMSF on worthy and important research projects that deserve the foundation’s future funding efforts.
Plans for roundtable meetings in 2020 and 2021 are already underway. Between the annual meetings, workgroups continue to take steps toward addressing the issues identified by the roundtable. For more information on the roundtable and NLMSF, visit: https://nlmsf.org.
Research clinicians from the US and abroad participated in a scientific research roundtable this past September to establish the most important issues facing leiomyosarcoma (LMS) research and clinical trials. The workshop, expected to be an annual event, is a joint effort of the National Leiomyosarcoma Foundation (NLMSF) and Sarcoma Patients EuroNet (SPAEN).
The roundtable’s mission is to bring together sarcoma experts for a meeting dedicated to LMS, where participants discuss the present state of LMS and the continued challenges of diagnosis and treatment. Its goal is to develop working plans to close the gaps in LMS patient care and improve LMS patient-care protocols. They also advise the NLMSF on worthy and important research projects that deserve the foundation’s future funding efforts.
Plans for roundtable meetings in 2020 and 2021 are already underway. Between the annual meetings, workgroups continue to take steps toward addressing the issues identified by the roundtable. For more information on the roundtable and NLMSF, visit: https://nlmsf.org.
Research clinicians from the US and abroad participated in a scientific research roundtable this past September to establish the most important issues facing leiomyosarcoma (LMS) research and clinical trials. The workshop, expected to be an annual event, is a joint effort of the National Leiomyosarcoma Foundation (NLMSF) and Sarcoma Patients EuroNet (SPAEN).
The roundtable’s mission is to bring together sarcoma experts for a meeting dedicated to LMS, where participants discuss the present state of LMS and the continued challenges of diagnosis and treatment. Its goal is to develop working plans to close the gaps in LMS patient care and improve LMS patient-care protocols. They also advise the NLMSF on worthy and important research projects that deserve the foundation’s future funding efforts.
Plans for roundtable meetings in 2020 and 2021 are already underway. Between the annual meetings, workgroups continue to take steps toward addressing the issues identified by the roundtable. For more information on the roundtable and NLMSF, visit: https://nlmsf.org.
FB Support Groups Enable Rapid Access to Large Numbers of Patients With Rare Disease
Investigators conducted a survey study of 214 patients with dermatofibrosarcoma protuberans (DFSP) or their family members using in part existing Facebook patient support groups (FBSG) to recruit respondents. They found the approach provides a “powerful” tool to collect relevant disease information from large numbers of patients with rare diseases.
A team of medical practitioners and patients developed the multiple-choice survey, and after testing the survey twice, posted a survey announcement on FBSGs for DFSP. The survey was live for 3 weeks in 2015. The investigators rapidly collected disease statistics, including information on recurrence, metastasis, surgical outcomes, diagnostic delay, and more, suggesting that FBSGs are useful medical research tools.
One hundred ninety-nine respondents were patients and 15 were family members. The respondents reported a median of 4 years to receive a correct diagnosis after noticing a lesion, ranging from less than 1 year to 42 years. About half the patients (52.3%) believed they received a misdiagnosis at some point, either from a dermatologist, primary care clinician, or another type of physician. Patients first noticed DFSP at a median age of 29.6 years. Many of their lesions appeared initially as flat plaques that eventually became protuberant. Because of this disconnect between the disease name and its clinical presentation, the investigators proposed the alternative term, dermatofibrosarcoma, often protuberant, be adopted. The investigators concluded that “FBSGs appear to be powerful tools to synergize effective and rapid research collaborations with large numbers of international patients with rare disease.” TSJ
Investigators conducted a survey study of 214 patients with dermatofibrosarcoma protuberans (DFSP) or their family members using in part existing Facebook patient support groups (FBSG) to recruit respondents. They found the approach provides a “powerful” tool to collect relevant disease information from large numbers of patients with rare diseases.
A team of medical practitioners and patients developed the multiple-choice survey, and after testing the survey twice, posted a survey announcement on FBSGs for DFSP. The survey was live for 3 weeks in 2015. The investigators rapidly collected disease statistics, including information on recurrence, metastasis, surgical outcomes, diagnostic delay, and more, suggesting that FBSGs are useful medical research tools.
One hundred ninety-nine respondents were patients and 15 were family members. The respondents reported a median of 4 years to receive a correct diagnosis after noticing a lesion, ranging from less than 1 year to 42 years. About half the patients (52.3%) believed they received a misdiagnosis at some point, either from a dermatologist, primary care clinician, or another type of physician. Patients first noticed DFSP at a median age of 29.6 years. Many of their lesions appeared initially as flat plaques that eventually became protuberant. Because of this disconnect between the disease name and its clinical presentation, the investigators proposed the alternative term, dermatofibrosarcoma, often protuberant, be adopted. The investigators concluded that “FBSGs appear to be powerful tools to synergize effective and rapid research collaborations with large numbers of international patients with rare disease.” TSJ
Investigators conducted a survey study of 214 patients with dermatofibrosarcoma protuberans (DFSP) or their family members using in part existing Facebook patient support groups (FBSG) to recruit respondents. They found the approach provides a “powerful” tool to collect relevant disease information from large numbers of patients with rare diseases.
A team of medical practitioners and patients developed the multiple-choice survey, and after testing the survey twice, posted a survey announcement on FBSGs for DFSP. The survey was live for 3 weeks in 2015. The investigators rapidly collected disease statistics, including information on recurrence, metastasis, surgical outcomes, diagnostic delay, and more, suggesting that FBSGs are useful medical research tools.
One hundred ninety-nine respondents were patients and 15 were family members. The respondents reported a median of 4 years to receive a correct diagnosis after noticing a lesion, ranging from less than 1 year to 42 years. About half the patients (52.3%) believed they received a misdiagnosis at some point, either from a dermatologist, primary care clinician, or another type of physician. Patients first noticed DFSP at a median age of 29.6 years. Many of their lesions appeared initially as flat plaques that eventually became protuberant. Because of this disconnect between the disease name and its clinical presentation, the investigators proposed the alternative term, dermatofibrosarcoma, often protuberant, be adopted. The investigators concluded that “FBSGs appear to be powerful tools to synergize effective and rapid research collaborations with large numbers of international patients with rare disease.” TSJ
Pexidartinib Receives Category 1 Recommendation from NCCN
Pexidartinib, the newly approved agent to treat patients with tenosynovial giant cell tumor (TGCT), received a category 1 recommendation from the National Comprehensive Cancer Network (NCCN) in the recent update of its Clinical Practice Guidelines in Oncology, Soft Tissue Sarcoma (Version 4.2019). A category 1 recommendation is based on a high level of evidence with uniform consensus that the intervention is appropriate.
The NCCN based its recommendation on the randomized, placebo-controlled phase 3 ENLIVEN study (NCT02371369) published in The Lancet (Tap WD, Gelderblom H, Palmerini E, et al. Lancet. 2019;394:478-487). The placebo-controlled portion of the study showed that patients treated with pexidartinib achieved a significantly higher overall response than patients in the placebo arm, 39% compared to none, respectively. The investigators identified mixed or cholestatic hepatotoxicity to be a risk of systemic therapy with the agent. Nevertheless, the “robust tumour response,” they wrote, “with improved patient symptoms and functional outcomes” establish pexidartinib as a potential treatment for TGCT in cases not amenable to improvement with surgery.
Pexidartinib, the newly approved agent to treat patients with tenosynovial giant cell tumor (TGCT), received a category 1 recommendation from the National Comprehensive Cancer Network (NCCN) in the recent update of its Clinical Practice Guidelines in Oncology, Soft Tissue Sarcoma (Version 4.2019). A category 1 recommendation is based on a high level of evidence with uniform consensus that the intervention is appropriate.
The NCCN based its recommendation on the randomized, placebo-controlled phase 3 ENLIVEN study (NCT02371369) published in The Lancet (Tap WD, Gelderblom H, Palmerini E, et al. Lancet. 2019;394:478-487). The placebo-controlled portion of the study showed that patients treated with pexidartinib achieved a significantly higher overall response than patients in the placebo arm, 39% compared to none, respectively. The investigators identified mixed or cholestatic hepatotoxicity to be a risk of systemic therapy with the agent. Nevertheless, the “robust tumour response,” they wrote, “with improved patient symptoms and functional outcomes” establish pexidartinib as a potential treatment for TGCT in cases not amenable to improvement with surgery.
Pexidartinib, the newly approved agent to treat patients with tenosynovial giant cell tumor (TGCT), received a category 1 recommendation from the National Comprehensive Cancer Network (NCCN) in the recent update of its Clinical Practice Guidelines in Oncology, Soft Tissue Sarcoma (Version 4.2019). A category 1 recommendation is based on a high level of evidence with uniform consensus that the intervention is appropriate.
The NCCN based its recommendation on the randomized, placebo-controlled phase 3 ENLIVEN study (NCT02371369) published in The Lancet (Tap WD, Gelderblom H, Palmerini E, et al. Lancet. 2019;394:478-487). The placebo-controlled portion of the study showed that patients treated with pexidartinib achieved a significantly higher overall response than patients in the placebo arm, 39% compared to none, respectively. The investigators identified mixed or cholestatic hepatotoxicity to be a risk of systemic therapy with the agent. Nevertheless, the “robust tumour response,” they wrote, “with improved patient symptoms and functional outcomes” establish pexidartinib as a potential treatment for TGCT in cases not amenable to improvement with surgery.
Metastatic angiosarcoma arising in a patient with long-standing treatment-refractory hemangioma
Angiosarcomas are malignant tumors of the vascular endothelium and are typically idiopathic. These tumors comprise 2% of all soft tissue sarcomas and have an estimated incidence of 2 per million.1,2 Known causes of angiosarcoma include genetic syndromes—such as von Hippel- Lindau, Chuvash polycythemia, Bannayan- Riley-Ruvalcaba, Cowden, and hamartomatous polyposis syndromes— chronic lymphedema, and exposure to radiation.3 Vinyl chloride, arsenicals, and thorotrast are known to increase the incidence of liver angiosarcoma.4
Malignant transformation of hemangioma is rare. We describe metastatic angiosarcoma in a patient with a large, longterm treatment-resistant subcutaneous hemangioma, illustrating such a possibility. We review similar cases and discuss the value of determining pathogenesis in such patients.
Case Presentation and Summary
A 55-year-old female with a long-standing childhood hemangioma of the left lower extremity was referred to Ochsner Medical Center for tissue diagnosis of new pulmonary nodules. Her medical history included a 7 pack-year smoking history; she had quit 3 years prior. Her family history included a sister who died from breast cancer. The patient initially had a progressive, intermittently bleeding tumor in the left foot at age 7. She was diagnosed with hemangioma in her twenties. At that point, her tumor began to involve the posterior calf and femur, causing deformity. She had multiple surgical resections but reportedly all pathology demonstrated benign hemangioma. She received radiation for pain, a routine treatment at the time, but developed a focus of progression in the heel. Above-knee amputation was considered but could not be performed when hemangioma was discovered in the hip area. She was lost to follow-up between 2001 and 2015. Lower extremity magnetic resonance imaging in 2015 was stable with imaging prior to 2001. A repeat biopsy in 2016 demonstrated hemangioma. The patient then received radiation to a wider field, including the femur, with minimal response. She completed a course of steroids as well. Bevacizumab was started in 2017 and improved foot deformity. She also briefly trialed pazopanib for 4 weeks in 2018 in an attempt to switch to oral medications. Despite partial response, she discontinued both agents in July 2018 because of toxicity and the burden of recurrent infusions.
Four months later, she presented with 2 months of intermittent hemoptysis and 18 months of metallic odors. Additionally, she lost 25 pounds in 3 months, which she attributed to a diet plan. At this visit, her left lower extremity exhibited multiple subcutaneous tumors and nodules.
Computed tomography (CT) with contrast demonstrated innumerable pulmonary nodules, the largest measuring 2.2 cm in the right lower lobe superior segment. Positron emission tomography (PET)/CT revealed 2 nodules with mild hypermetabolic activity; the largest nodule had a maximum standardized uptake value of 2.7. Bronchoalveolar lavage studies showed intra-alveolar hemorrhage with hemosiderin-laden macrophages. No malignancy, granuloma, or dysplasia was found in transbronchial needle aspirate of the largest nodule. The patient had no lymphadenopathy.
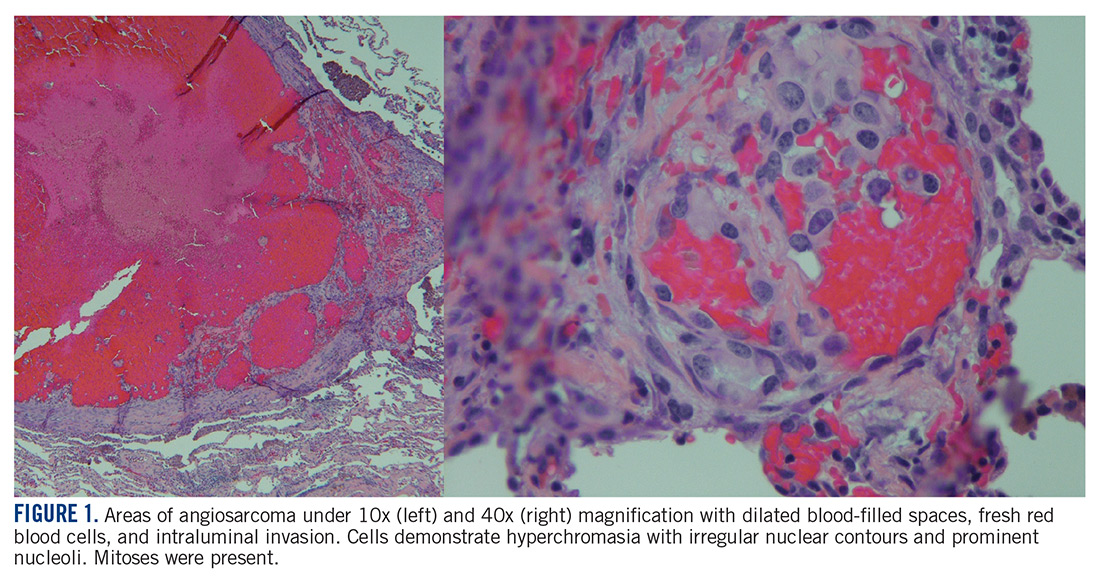
At this hospital, surgical resection by video-assisted thoracoscopic surgery confirmed multifocal malignant epithelioid neoplasm suspicious for angiosarcoma. Multiple areas showed proliferation of atypical epithelioid-to-spindle cells. There were prominent associated hemosiderin-laden macrophages, fresh red blood cells, and dilated blood-filled spaces. Cells demonstrated hyperchromasia with irregular nuclear contours, prominent nucleoli, and mitoses (FIGURE 1). Additionally, there were areas of focal organizing pneumonia. For atypical cells, staining was CD31-positive and CD34-negative. Staining was strongly positive for ERG. There was increased Ki-67 with retained INI expression and patchy weak reactivity for Fli-1.
Next-generation sequencing was performed. Specimen tumor content was 15%. Genomic findings included IDH1 p.R132C mutation, with variant allele frequency <10%. Testing was inconclusive for MSI and TMB mutations. PD-L1 assessment could not be performed. Unfortunately, the patient did not qualify for any clinical trials, as there were no matching alterations. This patient was lost to follow-up.
Discussion
Angiosarcoma accounts for 2% of soft tissue sarcomas.1 Cutaneous angiosarcomas most commonly occur in the face and scalp of the elderly, or in sites of chronic lymphedema. Angiosarcoma also develops following radiation therapy.5 For breast cancers and tumors of the head and neck, irradiation has <1% risk of inducing secondary malignancy, including angiosarcoma.6
This patient had a new diagnosis of angiosarcoma in the setting of long-standing benign hemangioma with history of radiation treatment. Thus, it is unclear whether this angiosarcoma was primary, radiation-induced, or secondary to transformation from the preexisting vascular tumor. Post-irradiation sarcoma carries a less favorable prognosis compared to de novo sarcoma; however, reports conflict on whether this holds for angiosarcoma subtypes.6 Determining etiology may benefit patients for prognostication and possibly inform future selection of treatment modalities.
The mutational signature in radiation- associated sarcomas differs from that of sporadic sarcomas. First, radiation- associated sarcomas demonstrate more frequent small deletions and balanced translocations. TP53 mutations are found in up to 1/3 of radiation-associated sarcomas and are more often due to small deletions than in sporadic sarcomas.7 High-level MYC amplification occurs in 54%-100% of secondary angiosarcomas, compared to 0-7% in sporadic angiosarcomas. Co-amplification of FLT4 occurs in 11%-25% of secondary angiosarcomas.8 Additionally, transcriptome analysis revealed differential expression of a 135-gene signature compared to non-radiation- induced sarcomas.7 Although this patient was not specifically analyzed for such alterations, such tests may differentiate post-irradiation angiosarcoma from sporadic etiologies.
In this patient, the R132C IDH1 mutation was identified and may be the first reported case in angiosarcoma. Typically, this mutation occurs in chondrosarcoma, myeloid neoplasms, gliomas, and cholangiocarcinomas. It is also found in spindle cell hemangiomas but not in other vascular tumors.9 The clinical significance of this mutation is uncertain at this time.
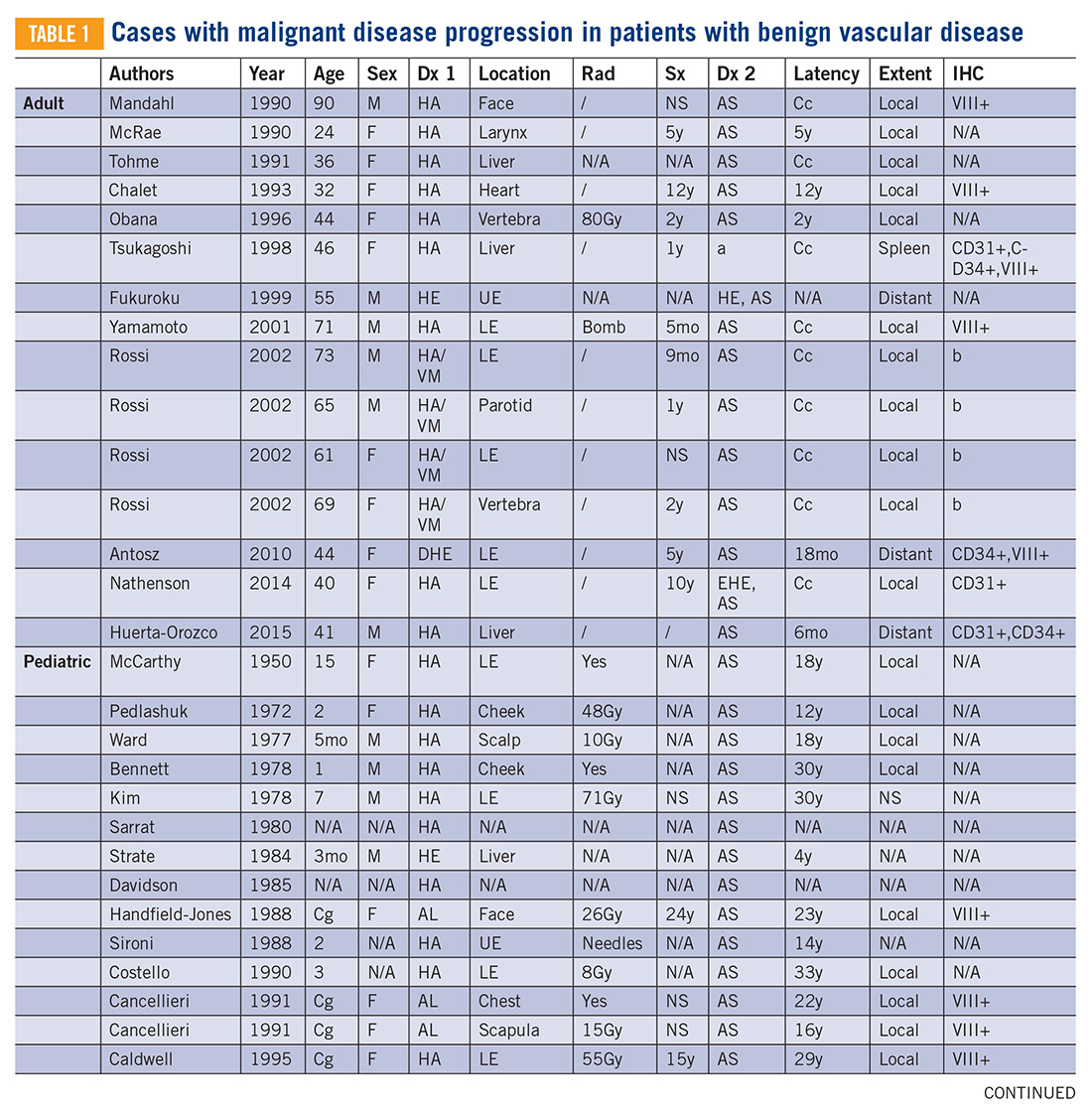
There are approximately 36 reported cases of malignant disease arising in patients with less aggressive vascular tumors (TABLE 1). Of these, 25 of 36 involve angiosarcoma arising in patients with hemangioma. Four cases of angiosarcoma were reported in patients with hemangioendothelioma, 1 case of hemangioendothelioma in a patient with hemangioma, 1 case of Dabska tumor in a patient with hemangioma, and 1 case of angiosarcoma in a patient with Dabska tumor. Fifteen cases involved initial disease with adult onset and 21 involved initial disease with pediatric onset, suggesting even distribution. Malignant disease mostly occurred in adulthood, in 26 out of 33 cases. Latency to malignancy ranged from concurrent discovery to 54 years. Mean latency, excluding cases with concurrent discovery, was shorter with adult-onset initial disease, at 4.2 years, compared to 16 years among patients with onset of initial disease in childhood. Longer latency in the pediatric-onset population correlated with longer latent periods for radiation-induced angiosarcoma following benign disease, which is reported to average 23 years.10 Thirteen of 19 cases with pediatric onset disease had a history of radiotherapy, while 2 of 13 cases with adult onset disease did. Sixteen cases involved tumor in the bone and soft tissue, as in this patient. Notably, 4 of these cases involved long-standing hemangioma for 10 years or more, as in this patient, suggesting a possible correlation between long-standing vascular tumors and malignant transformation. Angiosarcoma arising in non-irradiated patients suggests that malignant transformation and de novo transformation may compete with radiation-induced mutation in tumorigenesis. Further, 8 cases involved angiosarcoma growing within another vascular tumor, demonstrating the possibility of malignant transformation. Dehner and Ishak described a histological model for quantifying such a risk; a validated model may be particularly useful in patients with long-standing hemangioma.11
Etiology of tumorigenesis in cases of angiosarcoma arising in patients with a history of benign hemangioma may benefit prognostication and inform treatment selection in the future. Owing to long latent periods, radiation-associated angiosarcoma incidence may rise, as radiation therapy for benign hemangioma was recently routine. Future research may provide insight into disease progression and possibly predict the risk of angiosarcoma in patients with long-standing benign disease. TSJ
1. Tambe SA, Nayak CS. Metastatic angiosarcoma of lower extremity. Indian Dermatol Online J. 2018;9(3)177-181.
2. Cioffi A, Reichert S, Antonescu CR, Maki RG. Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am.2013;27(5):975-988.
3. Cohen SM, Storer RD, Criswell KA, et al. Hemangiosarcoma in rodents: mode-of-action evaluation and human relevance. Toxicol Sci. 2009;111(1):4-18.
4. Popper H, Thomas LB, Telles NC, Falk H, Selikoff IJ. Development of hepatic angiosarcoma in man induced by vinyl chloride, thorotrast, and arsenic. Comparison with cases of unknown etiology. Am J Pathol. 1978;92(2):349- 376.
5. Mark RJ, Bailet JW, Poen J, et al. Postirradiation sarcoma of the head and neck. Cancer. 1993;72(3):887-893.
6. Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20(4):1267-1274.
7. Mito JK, Mitra D, Doyle LA. Radiation-associated sarcomas: an update on clinical, histologic, and molecular features. Surg Pathol Clin. 2019;12(1):139-148.
8. Weidema ME, Versleijen-Jonkers YMH, Flucke UE, Desar IME, van der Graaf WTA. Targeting angiosarcomas of the soft tissues: A challenging effort in a heterogeneous and rare disease. Crit Rev Oncol Hematol. 2019;138:120-131.
9. Kurek KC, Pansuriya TC, van Ruler MAJH, et al. R132C IDH1 mutations are found in spindle cell hemangiomas and not in other vascular tumors or malformations. Am J Pathol. 2013;182(5):1494-1500.
10. Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
11. Dehner LP, Ishak KG. Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol. 1971;92(2):101-111.
Angiosarcomas are malignant tumors of the vascular endothelium and are typically idiopathic. These tumors comprise 2% of all soft tissue sarcomas and have an estimated incidence of 2 per million.1,2 Known causes of angiosarcoma include genetic syndromes—such as von Hippel- Lindau, Chuvash polycythemia, Bannayan- Riley-Ruvalcaba, Cowden, and hamartomatous polyposis syndromes— chronic lymphedema, and exposure to radiation.3 Vinyl chloride, arsenicals, and thorotrast are known to increase the incidence of liver angiosarcoma.4
Malignant transformation of hemangioma is rare. We describe metastatic angiosarcoma in a patient with a large, longterm treatment-resistant subcutaneous hemangioma, illustrating such a possibility. We review similar cases and discuss the value of determining pathogenesis in such patients.
Case Presentation and Summary
A 55-year-old female with a long-standing childhood hemangioma of the left lower extremity was referred to Ochsner Medical Center for tissue diagnosis of new pulmonary nodules. Her medical history included a 7 pack-year smoking history; she had quit 3 years prior. Her family history included a sister who died from breast cancer. The patient initially had a progressive, intermittently bleeding tumor in the left foot at age 7. She was diagnosed with hemangioma in her twenties. At that point, her tumor began to involve the posterior calf and femur, causing deformity. She had multiple surgical resections but reportedly all pathology demonstrated benign hemangioma. She received radiation for pain, a routine treatment at the time, but developed a focus of progression in the heel. Above-knee amputation was considered but could not be performed when hemangioma was discovered in the hip area. She was lost to follow-up between 2001 and 2015. Lower extremity magnetic resonance imaging in 2015 was stable with imaging prior to 2001. A repeat biopsy in 2016 demonstrated hemangioma. The patient then received radiation to a wider field, including the femur, with minimal response. She completed a course of steroids as well. Bevacizumab was started in 2017 and improved foot deformity. She also briefly trialed pazopanib for 4 weeks in 2018 in an attempt to switch to oral medications. Despite partial response, she discontinued both agents in July 2018 because of toxicity and the burden of recurrent infusions.
Four months later, she presented with 2 months of intermittent hemoptysis and 18 months of metallic odors. Additionally, she lost 25 pounds in 3 months, which she attributed to a diet plan. At this visit, her left lower extremity exhibited multiple subcutaneous tumors and nodules.
Computed tomography (CT) with contrast demonstrated innumerable pulmonary nodules, the largest measuring 2.2 cm in the right lower lobe superior segment. Positron emission tomography (PET)/CT revealed 2 nodules with mild hypermetabolic activity; the largest nodule had a maximum standardized uptake value of 2.7. Bronchoalveolar lavage studies showed intra-alveolar hemorrhage with hemosiderin-laden macrophages. No malignancy, granuloma, or dysplasia was found in transbronchial needle aspirate of the largest nodule. The patient had no lymphadenopathy.
At this hospital, surgical resection by video-assisted thoracoscopic surgery confirmed multifocal malignant epithelioid neoplasm suspicious for angiosarcoma. Multiple areas showed proliferation of atypical epithelioid-to-spindle cells. There were prominent associated hemosiderin-laden macrophages, fresh red blood cells, and dilated blood-filled spaces. Cells demonstrated hyperchromasia with irregular nuclear contours, prominent nucleoli, and mitoses (FIGURE 1). Additionally, there were areas of focal organizing pneumonia. For atypical cells, staining was CD31-positive and CD34-negative. Staining was strongly positive for ERG. There was increased Ki-67 with retained INI expression and patchy weak reactivity for Fli-1.
Next-generation sequencing was performed. Specimen tumor content was 15%. Genomic findings included IDH1 p.R132C mutation, with variant allele frequency <10%. Testing was inconclusive for MSI and TMB mutations. PD-L1 assessment could not be performed. Unfortunately, the patient did not qualify for any clinical trials, as there were no matching alterations. This patient was lost to follow-up.
Discussion
Angiosarcoma accounts for 2% of soft tissue sarcomas.1 Cutaneous angiosarcomas most commonly occur in the face and scalp of the elderly, or in sites of chronic lymphedema. Angiosarcoma also develops following radiation therapy.5 For breast cancers and tumors of the head and neck, irradiation has <1% risk of inducing secondary malignancy, including angiosarcoma.6
This patient had a new diagnosis of angiosarcoma in the setting of long-standing benign hemangioma with history of radiation treatment. Thus, it is unclear whether this angiosarcoma was primary, radiation-induced, or secondary to transformation from the preexisting vascular tumor. Post-irradiation sarcoma carries a less favorable prognosis compared to de novo sarcoma; however, reports conflict on whether this holds for angiosarcoma subtypes.6 Determining etiology may benefit patients for prognostication and possibly inform future selection of treatment modalities.
The mutational signature in radiation- associated sarcomas differs from that of sporadic sarcomas. First, radiation- associated sarcomas demonstrate more frequent small deletions and balanced translocations. TP53 mutations are found in up to 1/3 of radiation-associated sarcomas and are more often due to small deletions than in sporadic sarcomas.7 High-level MYC amplification occurs in 54%-100% of secondary angiosarcomas, compared to 0-7% in sporadic angiosarcomas. Co-amplification of FLT4 occurs in 11%-25% of secondary angiosarcomas.8 Additionally, transcriptome analysis revealed differential expression of a 135-gene signature compared to non-radiation- induced sarcomas.7 Although this patient was not specifically analyzed for such alterations, such tests may differentiate post-irradiation angiosarcoma from sporadic etiologies.
In this patient, the R132C IDH1 mutation was identified and may be the first reported case in angiosarcoma. Typically, this mutation occurs in chondrosarcoma, myeloid neoplasms, gliomas, and cholangiocarcinomas. It is also found in spindle cell hemangiomas but not in other vascular tumors.9 The clinical significance of this mutation is uncertain at this time.
There are approximately 36 reported cases of malignant disease arising in patients with less aggressive vascular tumors (TABLE 1). Of these, 25 of 36 involve angiosarcoma arising in patients with hemangioma. Four cases of angiosarcoma were reported in patients with hemangioendothelioma, 1 case of hemangioendothelioma in a patient with hemangioma, 1 case of Dabska tumor in a patient with hemangioma, and 1 case of angiosarcoma in a patient with Dabska tumor. Fifteen cases involved initial disease with adult onset and 21 involved initial disease with pediatric onset, suggesting even distribution. Malignant disease mostly occurred in adulthood, in 26 out of 33 cases. Latency to malignancy ranged from concurrent discovery to 54 years. Mean latency, excluding cases with concurrent discovery, was shorter with adult-onset initial disease, at 4.2 years, compared to 16 years among patients with onset of initial disease in childhood. Longer latency in the pediatric-onset population correlated with longer latent periods for radiation-induced angiosarcoma following benign disease, which is reported to average 23 years.10 Thirteen of 19 cases with pediatric onset disease had a history of radiotherapy, while 2 of 13 cases with adult onset disease did. Sixteen cases involved tumor in the bone and soft tissue, as in this patient. Notably, 4 of these cases involved long-standing hemangioma for 10 years or more, as in this patient, suggesting a possible correlation between long-standing vascular tumors and malignant transformation. Angiosarcoma arising in non-irradiated patients suggests that malignant transformation and de novo transformation may compete with radiation-induced mutation in tumorigenesis. Further, 8 cases involved angiosarcoma growing within another vascular tumor, demonstrating the possibility of malignant transformation. Dehner and Ishak described a histological model for quantifying such a risk; a validated model may be particularly useful in patients with long-standing hemangioma.11
Etiology of tumorigenesis in cases of angiosarcoma arising in patients with a history of benign hemangioma may benefit prognostication and inform treatment selection in the future. Owing to long latent periods, radiation-associated angiosarcoma incidence may rise, as radiation therapy for benign hemangioma was recently routine. Future research may provide insight into disease progression and possibly predict the risk of angiosarcoma in patients with long-standing benign disease. TSJ
Angiosarcomas are malignant tumors of the vascular endothelium and are typically idiopathic. These tumors comprise 2% of all soft tissue sarcomas and have an estimated incidence of 2 per million.1,2 Known causes of angiosarcoma include genetic syndromes—such as von Hippel- Lindau, Chuvash polycythemia, Bannayan- Riley-Ruvalcaba, Cowden, and hamartomatous polyposis syndromes— chronic lymphedema, and exposure to radiation.3 Vinyl chloride, arsenicals, and thorotrast are known to increase the incidence of liver angiosarcoma.4
Malignant transformation of hemangioma is rare. We describe metastatic angiosarcoma in a patient with a large, longterm treatment-resistant subcutaneous hemangioma, illustrating such a possibility. We review similar cases and discuss the value of determining pathogenesis in such patients.
Case Presentation and Summary
A 55-year-old female with a long-standing childhood hemangioma of the left lower extremity was referred to Ochsner Medical Center for tissue diagnosis of new pulmonary nodules. Her medical history included a 7 pack-year smoking history; she had quit 3 years prior. Her family history included a sister who died from breast cancer. The patient initially had a progressive, intermittently bleeding tumor in the left foot at age 7. She was diagnosed with hemangioma in her twenties. At that point, her tumor began to involve the posterior calf and femur, causing deformity. She had multiple surgical resections but reportedly all pathology demonstrated benign hemangioma. She received radiation for pain, a routine treatment at the time, but developed a focus of progression in the heel. Above-knee amputation was considered but could not be performed when hemangioma was discovered in the hip area. She was lost to follow-up between 2001 and 2015. Lower extremity magnetic resonance imaging in 2015 was stable with imaging prior to 2001. A repeat biopsy in 2016 demonstrated hemangioma. The patient then received radiation to a wider field, including the femur, with minimal response. She completed a course of steroids as well. Bevacizumab was started in 2017 and improved foot deformity. She also briefly trialed pazopanib for 4 weeks in 2018 in an attempt to switch to oral medications. Despite partial response, she discontinued both agents in July 2018 because of toxicity and the burden of recurrent infusions.
Four months later, she presented with 2 months of intermittent hemoptysis and 18 months of metallic odors. Additionally, she lost 25 pounds in 3 months, which she attributed to a diet plan. At this visit, her left lower extremity exhibited multiple subcutaneous tumors and nodules.
Computed tomography (CT) with contrast demonstrated innumerable pulmonary nodules, the largest measuring 2.2 cm in the right lower lobe superior segment. Positron emission tomography (PET)/CT revealed 2 nodules with mild hypermetabolic activity; the largest nodule had a maximum standardized uptake value of 2.7. Bronchoalveolar lavage studies showed intra-alveolar hemorrhage with hemosiderin-laden macrophages. No malignancy, granuloma, or dysplasia was found in transbronchial needle aspirate of the largest nodule. The patient had no lymphadenopathy.
At this hospital, surgical resection by video-assisted thoracoscopic surgery confirmed multifocal malignant epithelioid neoplasm suspicious for angiosarcoma. Multiple areas showed proliferation of atypical epithelioid-to-spindle cells. There were prominent associated hemosiderin-laden macrophages, fresh red blood cells, and dilated blood-filled spaces. Cells demonstrated hyperchromasia with irregular nuclear contours, prominent nucleoli, and mitoses (FIGURE 1). Additionally, there were areas of focal organizing pneumonia. For atypical cells, staining was CD31-positive and CD34-negative. Staining was strongly positive for ERG. There was increased Ki-67 with retained INI expression and patchy weak reactivity for Fli-1.
Next-generation sequencing was performed. Specimen tumor content was 15%. Genomic findings included IDH1 p.R132C mutation, with variant allele frequency <10%. Testing was inconclusive for MSI and TMB mutations. PD-L1 assessment could not be performed. Unfortunately, the patient did not qualify for any clinical trials, as there were no matching alterations. This patient was lost to follow-up.
Discussion
Angiosarcoma accounts for 2% of soft tissue sarcomas.1 Cutaneous angiosarcomas most commonly occur in the face and scalp of the elderly, or in sites of chronic lymphedema. Angiosarcoma also develops following radiation therapy.5 For breast cancers and tumors of the head and neck, irradiation has <1% risk of inducing secondary malignancy, including angiosarcoma.6
This patient had a new diagnosis of angiosarcoma in the setting of long-standing benign hemangioma with history of radiation treatment. Thus, it is unclear whether this angiosarcoma was primary, radiation-induced, or secondary to transformation from the preexisting vascular tumor. Post-irradiation sarcoma carries a less favorable prognosis compared to de novo sarcoma; however, reports conflict on whether this holds for angiosarcoma subtypes.6 Determining etiology may benefit patients for prognostication and possibly inform future selection of treatment modalities.
The mutational signature in radiation- associated sarcomas differs from that of sporadic sarcomas. First, radiation- associated sarcomas demonstrate more frequent small deletions and balanced translocations. TP53 mutations are found in up to 1/3 of radiation-associated sarcomas and are more often due to small deletions than in sporadic sarcomas.7 High-level MYC amplification occurs in 54%-100% of secondary angiosarcomas, compared to 0-7% in sporadic angiosarcomas. Co-amplification of FLT4 occurs in 11%-25% of secondary angiosarcomas.8 Additionally, transcriptome analysis revealed differential expression of a 135-gene signature compared to non-radiation- induced sarcomas.7 Although this patient was not specifically analyzed for such alterations, such tests may differentiate post-irradiation angiosarcoma from sporadic etiologies.
In this patient, the R132C IDH1 mutation was identified and may be the first reported case in angiosarcoma. Typically, this mutation occurs in chondrosarcoma, myeloid neoplasms, gliomas, and cholangiocarcinomas. It is also found in spindle cell hemangiomas but not in other vascular tumors.9 The clinical significance of this mutation is uncertain at this time.
There are approximately 36 reported cases of malignant disease arising in patients with less aggressive vascular tumors (TABLE 1). Of these, 25 of 36 involve angiosarcoma arising in patients with hemangioma. Four cases of angiosarcoma were reported in patients with hemangioendothelioma, 1 case of hemangioendothelioma in a patient with hemangioma, 1 case of Dabska tumor in a patient with hemangioma, and 1 case of angiosarcoma in a patient with Dabska tumor. Fifteen cases involved initial disease with adult onset and 21 involved initial disease with pediatric onset, suggesting even distribution. Malignant disease mostly occurred in adulthood, in 26 out of 33 cases. Latency to malignancy ranged from concurrent discovery to 54 years. Mean latency, excluding cases with concurrent discovery, was shorter with adult-onset initial disease, at 4.2 years, compared to 16 years among patients with onset of initial disease in childhood. Longer latency in the pediatric-onset population correlated with longer latent periods for radiation-induced angiosarcoma following benign disease, which is reported to average 23 years.10 Thirteen of 19 cases with pediatric onset disease had a history of radiotherapy, while 2 of 13 cases with adult onset disease did. Sixteen cases involved tumor in the bone and soft tissue, as in this patient. Notably, 4 of these cases involved long-standing hemangioma for 10 years or more, as in this patient, suggesting a possible correlation between long-standing vascular tumors and malignant transformation. Angiosarcoma arising in non-irradiated patients suggests that malignant transformation and de novo transformation may compete with radiation-induced mutation in tumorigenesis. Further, 8 cases involved angiosarcoma growing within another vascular tumor, demonstrating the possibility of malignant transformation. Dehner and Ishak described a histological model for quantifying such a risk; a validated model may be particularly useful in patients with long-standing hemangioma.11
Etiology of tumorigenesis in cases of angiosarcoma arising in patients with a history of benign hemangioma may benefit prognostication and inform treatment selection in the future. Owing to long latent periods, radiation-associated angiosarcoma incidence may rise, as radiation therapy for benign hemangioma was recently routine. Future research may provide insight into disease progression and possibly predict the risk of angiosarcoma in patients with long-standing benign disease. TSJ
1. Tambe SA, Nayak CS. Metastatic angiosarcoma of lower extremity. Indian Dermatol Online J. 2018;9(3)177-181.
2. Cioffi A, Reichert S, Antonescu CR, Maki RG. Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am.2013;27(5):975-988.
3. Cohen SM, Storer RD, Criswell KA, et al. Hemangiosarcoma in rodents: mode-of-action evaluation and human relevance. Toxicol Sci. 2009;111(1):4-18.
4. Popper H, Thomas LB, Telles NC, Falk H, Selikoff IJ. Development of hepatic angiosarcoma in man induced by vinyl chloride, thorotrast, and arsenic. Comparison with cases of unknown etiology. Am J Pathol. 1978;92(2):349- 376.
5. Mark RJ, Bailet JW, Poen J, et al. Postirradiation sarcoma of the head and neck. Cancer. 1993;72(3):887-893.
6. Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20(4):1267-1274.
7. Mito JK, Mitra D, Doyle LA. Radiation-associated sarcomas: an update on clinical, histologic, and molecular features. Surg Pathol Clin. 2019;12(1):139-148.
8. Weidema ME, Versleijen-Jonkers YMH, Flucke UE, Desar IME, van der Graaf WTA. Targeting angiosarcomas of the soft tissues: A challenging effort in a heterogeneous and rare disease. Crit Rev Oncol Hematol. 2019;138:120-131.
9. Kurek KC, Pansuriya TC, van Ruler MAJH, et al. R132C IDH1 mutations are found in spindle cell hemangiomas and not in other vascular tumors or malformations. Am J Pathol. 2013;182(5):1494-1500.
10. Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
11. Dehner LP, Ishak KG. Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol. 1971;92(2):101-111.
1. Tambe SA, Nayak CS. Metastatic angiosarcoma of lower extremity. Indian Dermatol Online J. 2018;9(3)177-181.
2. Cioffi A, Reichert S, Antonescu CR, Maki RG. Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am.2013;27(5):975-988.
3. Cohen SM, Storer RD, Criswell KA, et al. Hemangiosarcoma in rodents: mode-of-action evaluation and human relevance. Toxicol Sci. 2009;111(1):4-18.
4. Popper H, Thomas LB, Telles NC, Falk H, Selikoff IJ. Development of hepatic angiosarcoma in man induced by vinyl chloride, thorotrast, and arsenic. Comparison with cases of unknown etiology. Am J Pathol. 1978;92(2):349- 376.
5. Mark RJ, Bailet JW, Poen J, et al. Postirradiation sarcoma of the head and neck. Cancer. 1993;72(3):887-893.
6. Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20(4):1267-1274.
7. Mito JK, Mitra D, Doyle LA. Radiation-associated sarcomas: an update on clinical, histologic, and molecular features. Surg Pathol Clin. 2019;12(1):139-148.
8. Weidema ME, Versleijen-Jonkers YMH, Flucke UE, Desar IME, van der Graaf WTA. Targeting angiosarcomas of the soft tissues: A challenging effort in a heterogeneous and rare disease. Crit Rev Oncol Hematol. 2019;138:120-131.
9. Kurek KC, Pansuriya TC, van Ruler MAJH, et al. R132C IDH1 mutations are found in spindle cell hemangiomas and not in other vascular tumors or malformations. Am J Pathol. 2013;182(5):1494-1500.
10. Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
11. Dehner LP, Ishak KG. Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol. 1971;92(2):101-111.
Becoming the paradigm for clinical trial enrollment
The previous issue of The Sarcoma Journal focused on findings from numerous clinical trials in sarcomas of various histologies presented at ASCO’s annual meeting. This issue features a study on enrollment issues that surround clinical trials in sarcoma and sheds light on patient perceptions on clinical trial enrollment.
Clinical trials and their investigators are frequently impacted by enrollment issues, such as the limited number of eligible patients and the wide variations in time it can take to reach complete enrollment. For example, the phase 3 ANNOUNCE trial of olaratumab in soft tissue sarcoma completed its accrual of 509 patients in a record 10 months, while the trial of temozolomide by the European Pediatric Soft Tissue Sarcoma Study Group took 6 years to enroll 120 patients. Recruitment difficulties may even hamper the investigators’ and sponsors’ ability to bring a trial to a meaningful conclusion.
An interesting finding from the study published in this issue is the correlation between knowledge about trials and the positive attitude towards participating in them. People who had participated in clinical trials had higher levels of knowledge and developed more favorable attitudes towards clinical trials. One of the goals of the Sarcoma Foundation of America (curesarcoma.org) is to increase awareness of the numbers and types of ongoing clinical trials in sarcoma, benefitting patients and investigators alike. The SFA operates the Clinical Trial Navigating Service, which offers patients, caregivers, and health care professionals up-to-date information about sarcoma clinical trials throughout the United States and Canada. The service, provided in collaboration with EmergingMed, helps patients search for clinical trial options that match their specific diagnosis and treatment history.
The paper published in this issue suggests that, through patient education and careful trial design, sarcoma could become a paradigm for trial enrollment in other therapeutic areas. Together—as physicians, investigators, patients, trial sponsors, and anyone interested in curing sarcoma—we may be able to accomplish this. It’s certainly worth a try.
William D. Tap, MD
Editor-in-Chief
The previous issue of The Sarcoma Journal focused on findings from numerous clinical trials in sarcomas of various histologies presented at ASCO’s annual meeting. This issue features a study on enrollment issues that surround clinical trials in sarcoma and sheds light on patient perceptions on clinical trial enrollment.
Clinical trials and their investigators are frequently impacted by enrollment issues, such as the limited number of eligible patients and the wide variations in time it can take to reach complete enrollment. For example, the phase 3 ANNOUNCE trial of olaratumab in soft tissue sarcoma completed its accrual of 509 patients in a record 10 months, while the trial of temozolomide by the European Pediatric Soft Tissue Sarcoma Study Group took 6 years to enroll 120 patients. Recruitment difficulties may even hamper the investigators’ and sponsors’ ability to bring a trial to a meaningful conclusion.
An interesting finding from the study published in this issue is the correlation between knowledge about trials and the positive attitude towards participating in them. People who had participated in clinical trials had higher levels of knowledge and developed more favorable attitudes towards clinical trials. One of the goals of the Sarcoma Foundation of America (curesarcoma.org) is to increase awareness of the numbers and types of ongoing clinical trials in sarcoma, benefitting patients and investigators alike. The SFA operates the Clinical Trial Navigating Service, which offers patients, caregivers, and health care professionals up-to-date information about sarcoma clinical trials throughout the United States and Canada. The service, provided in collaboration with EmergingMed, helps patients search for clinical trial options that match their specific diagnosis and treatment history.
The paper published in this issue suggests that, through patient education and careful trial design, sarcoma could become a paradigm for trial enrollment in other therapeutic areas. Together—as physicians, investigators, patients, trial sponsors, and anyone interested in curing sarcoma—we may be able to accomplish this. It’s certainly worth a try.
William D. Tap, MD
Editor-in-Chief
The previous issue of The Sarcoma Journal focused on findings from numerous clinical trials in sarcomas of various histologies presented at ASCO’s annual meeting. This issue features a study on enrollment issues that surround clinical trials in sarcoma and sheds light on patient perceptions on clinical trial enrollment.
Clinical trials and their investigators are frequently impacted by enrollment issues, such as the limited number of eligible patients and the wide variations in time it can take to reach complete enrollment. For example, the phase 3 ANNOUNCE trial of olaratumab in soft tissue sarcoma completed its accrual of 509 patients in a record 10 months, while the trial of temozolomide by the European Pediatric Soft Tissue Sarcoma Study Group took 6 years to enroll 120 patients. Recruitment difficulties may even hamper the investigators’ and sponsors’ ability to bring a trial to a meaningful conclusion.
An interesting finding from the study published in this issue is the correlation between knowledge about trials and the positive attitude towards participating in them. People who had participated in clinical trials had higher levels of knowledge and developed more favorable attitudes towards clinical trials. One of the goals of the Sarcoma Foundation of America (curesarcoma.org) is to increase awareness of the numbers and types of ongoing clinical trials in sarcoma, benefitting patients and investigators alike. The SFA operates the Clinical Trial Navigating Service, which offers patients, caregivers, and health care professionals up-to-date information about sarcoma clinical trials throughout the United States and Canada. The service, provided in collaboration with EmergingMed, helps patients search for clinical trial options that match their specific diagnosis and treatment history.
The paper published in this issue suggests that, through patient education and careful trial design, sarcoma could become a paradigm for trial enrollment in other therapeutic areas. Together—as physicians, investigators, patients, trial sponsors, and anyone interested in curing sarcoma—we may be able to accomplish this. It’s certainly worth a try.
William D. Tap, MD
Editor-in-Chief
Overcoming barriers to clinical trial enrollment in patients with bone and soft tissue sarcoma: a paradigm for an increasingly heterogeneous cancer population
Introduction
The development of new cancer therapies relies on the successful development and completion of clinical trials. While clinical trials have led to significant improvements in cancer treatment, the success is dependent upon patient enrollment and participation. Unfortunately, fewer than 5% of adult patients enroll in trials.1-3 This represents a significant barrier to the development and approval of new cancer treatments. Reasons for low accrual into trials are multifactorial, but include structural barriers (eg, clinic access), clinical barriers (eg, eligibility criteria), and physician and patient attitudes towards trial enrollment.4,5 One study at the University of California Davis Cancer Center reported 49% of patients declined participation despite meeting eligibility criteria,3,6 suggesting that psychosocial barriers such as knowledge of trials and attitudes towards clinical research are a major impediment to accrual.7-9
Bone and soft tissue sarcoma represent a heterogeneous group of tumors of mesenchymal origin that are an important cause of morbidity and mortality. Local disease is often treated with a multidisciplinary approach including surgery, radiation, and systemic therapy. Metastatic disease is predominantly treated palliatively with systemic therapy.10 Given its rarity and heterogeneity, trial accrual is of particular importance in sarcoma and often requires multiple sites to enroll adequate numbers of patients. While sarcoma represents <1% of adult malignancies overall, it constitutes ~15% of malignancies in the adolescent and young adult (AYA) population (15- 39 years old).11,12 Sarcoma represents a patient population in which low trial accrual has been correlated with lack of progress in cancer-related outcomes in both the adult and AYA populations.13 The reasons for low accrual rates among patients with sarcoma are poorly understood.
Sarcomas represent a molecularly and biologically heterogeneous group of malignancies with over 100 different subtypes.12 As a result, there has been significant interest in performing molecular profiling, or genetic sequencing, to identify “targetable” mutations. Targetable mutations refer to a specific genetic change identified within the tumor molecular profile for which there is a specific drug that may demonstrate activity against a particular tumor. Given the widespread utilization of this technology in sarcoma, identifying and understanding patient perceptions with regard to molecular profiling is critically important in this disease.14
In this study, we use a cross-sectional design to describe patient perceptions of trial enrollment among patients with bone and soft tissue sarcoma through validated measures, including attitudes towards clinical trials, knowledge of clinical trials, and perceived ability (ie, self-efficacy) to carry out actions involved in making an informed decision about clinical trial participation, receptivity to learning more about clinical trials, and willingness to participate in clinical trials.6 In addition, we describe this patient cohort’s perceptions of molecular profiling, as current and future trials are increasingly driven by molecular or other biomarkers.
Methods
This was a cross-sectional electronic survey study of patients with bone and soft tissue sarcoma treated at Northwestern Medicine (NM) over a 5-year period. NM Enterprise Data Warehouse (NMEDW) is a single, comprehensive, and integrated repository of all clinical and research data sources within NM. The study was approved by the Northwestern University Institutional Review Board.
Survey
The investigators designed a self-administered, online survey, which was built using Research Electronic Data Capture (REDCap). The survey consisted of three sections that were answered using skip logic—a custom path through the survey that varied based on patients’ answers: (1) Patient demographic information and trial perceptions (answered by all patients); (2) Thoughts about molecular profiling (answered by patients who answered “yes” to the question, “Have you heard about molecular profiling of tumors?”); and (3) Considerations to undergo molecular profiling (answered by patients who answered “yes” to the question, “Have you undergone profiling of your cancer?”).
Clinical trial perceptions included questions assessing (1) patient knowledge about trials; (2) patient attitudes toward trials; (3) perceived ability (ie, self-efficacy) to carry out actions involved in making an informed decision about trial participation; (4) receptivity to learning more about trials; and (5) willingness to participate in trials. These outcome measures had been previously developed and pilot tested for reliability and validity (TABLE 1).6
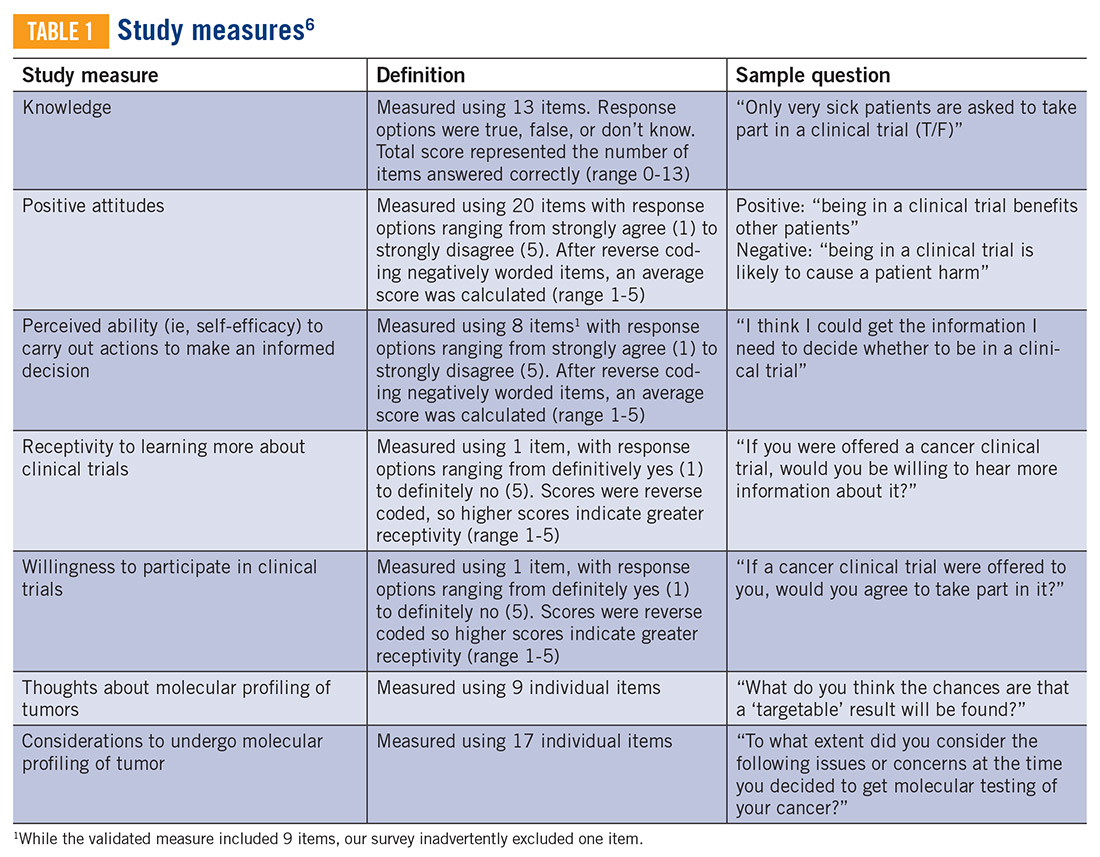
Thoughts about molecular profiling of tumors were assessed using nine items (TABLE 1). Of these, items assessing potential benefit or harm of molecular profiling were assessed using a 7-step Likert scale. Items assessing maximal benefit or harm of therapy, importance of quality vs length of life, and concern about the cost of molecular testing were assessed using a 5-step Likert scale. The study team developed and piloted these questions because there is no validated survey assessing these domains.
Considerations to undergo molecular profiling were assessed using 17 items. Items were in response to the question, “To what extent did you consider the following issues or concerns at the time you decided to get molecular testing of your cancer?” Responses were assessed using a 5-point Likert scale.
Data Collection
Patients 18 years and older evaluated at NM between November 20, 2012, and November 20, 2017, with a diagnosis of sarcoma were identified by query of the NMEDW by ICD-10 codes (C40, C41.9, C44.99, C45-49, C55, C71.9, D48, D49.9, and M12.20) or equivalent ICD-9 codes. Patients were subsequently excluded if they did not have a diagnosis of bone or soft tissue sarcoma, no e-mail address listed, had died, or had not been evaluated at an NM clinic in the previous 5 years. Patients with a diagnosis of gastrointestinal stromal tumor and Kaposi’s sarcoma were also excluded.
A personalized contact e-mail was sent to patients containing an explanation of the survey and an internet link to the electronic survey through REDCap from January 2018 to March 2018. If patients did not respond to the survey, two follow-up reminder e-mails were sent 2 and 4 days following the initial survey. The link was protected so that each patient could complete the survey only once. Responses were collected through the REDCap platform. Patients read and signed an electronic consent form prior to completing the survey.
Upon completion of the survey, patients were offered a $50 VISA gift card as compensation, with an option to donate their compensation to the Robert H. Lurie Comprehensive Cancer Center Sarcoma Research Fund.
Over the described survey period, open clinical trials for patients with bone and soft tissue sarcoma available at NM were evaluated. The number of patients screened and accrued to each trial were recorded.
Statistical analysis
Responses were separated from the personal data for complete anonymization. Descriptive statistical analysis was performed for demographics and disease variables and were summarized using frequencies and percentages. Median and range were used for age. Correlations between continuous variables were analyzed using Spearman correlations. Scores were compared between subgroups using the Mann-Whitney test. Descriptive statistics for knowledge, attitude, and ability scores include means and 95% confidence intervals. Correlations were interpreted as small (r=0.10), medium (r=0.30), or large (r=0.50).15 Statistical significance was indicated when P<0.05.
Results
Patients
Seven hundred fifty patients were eligible to participate in the survey and received the initial and two follow-up e-mails. Twenty e-mailed surveys bounced back. Three hundred nine patients opened the initial e-mail and 283 patients (37.7% of total and 91.6% of opened) completed at least a portion of the survey, with 182 patients completing the entire survey (FIGURE 1). Data for analysis were used from patients who completed at least a portion of the survey.
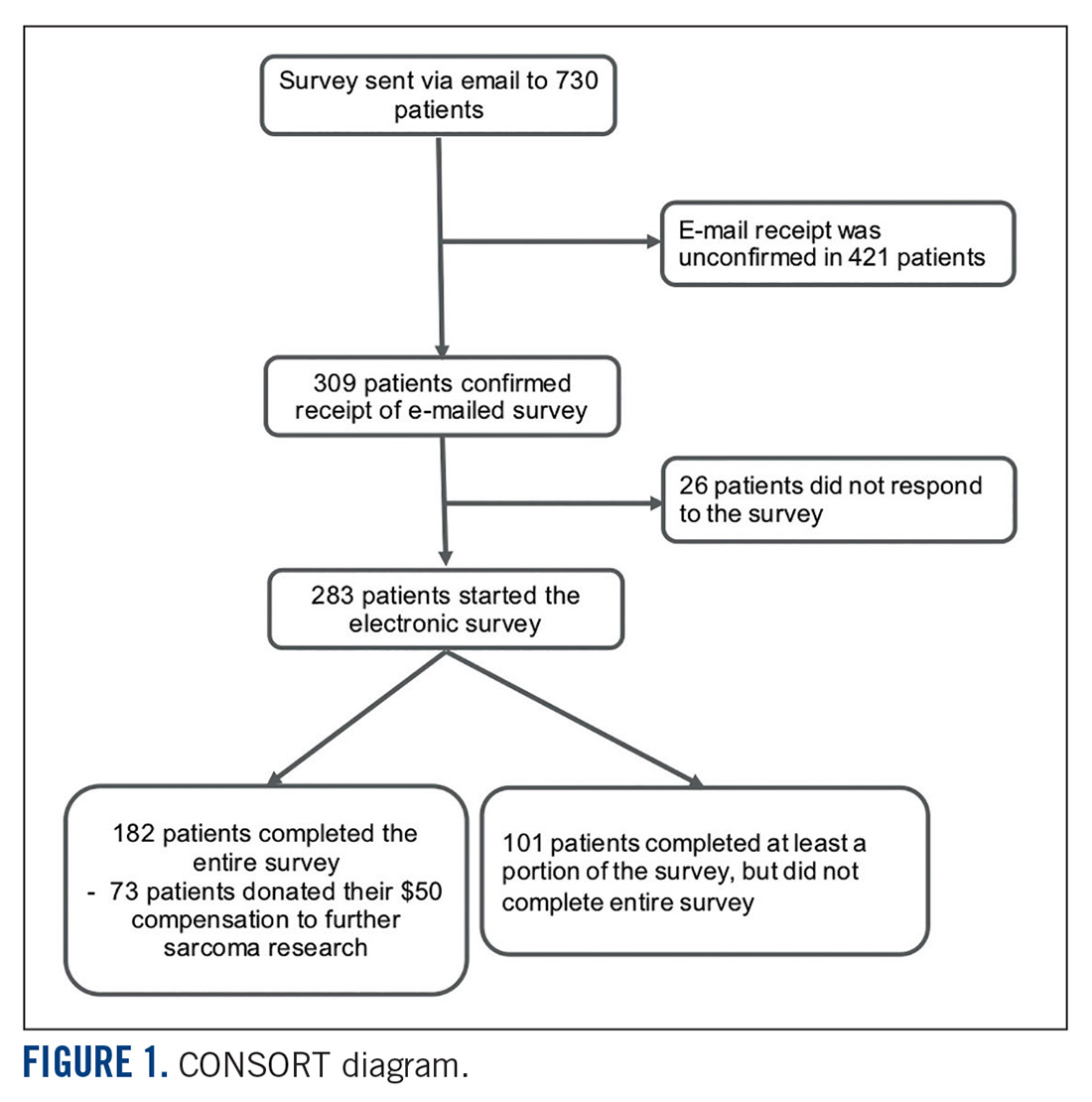
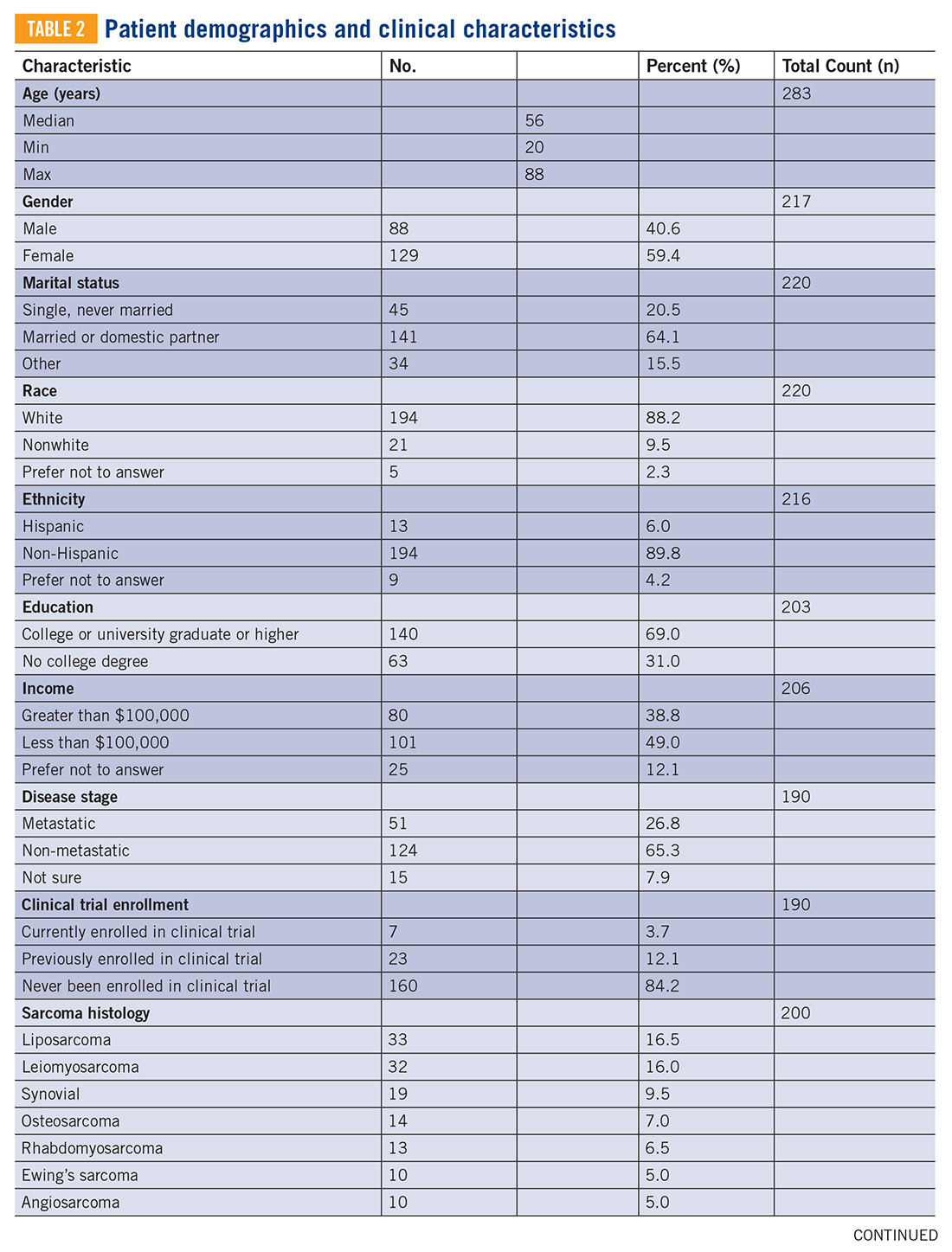
Baseline characteristics of patients who responded can be seen in TABLE 2. Patients had a median age of 56, the majority were female (59.4%), white (88.2%), and most had college or university graduate degrees or higher educational level (69.0%). Patients had various different histological subtypes, with the most common being liposarcoma (16.5%) and leiomyosarcoma (16.0%). Slightly more than a quarter (26.8%) of patients had metastatic disease, and 84.2% had never been enrolled in a clinical trial. Previous treatments included surgery (91.1%), radiation (53.2%), and chemotherapy (51.6%). Prior to completing the survey, 85.4% reported being receptive to a cancer clinical trial, while 60.7% of patients reported willingness to participate in a clinical trial.
Knowledge, attitudes, and perceived ability
A statistically significant correlation was observed between greater knowledge of trials and more positive attitudes towards trials (P<0.001; r=0.5, FIGURE 2A). In relating patient attitudes with perceived ability, again a significant correlation was seen (P<0.001; r=0.4, FIGURE 2B). In contrast, knowledge had a weak correlation with perceived ability (P=0.024; r=0.2, FIGURE 2C). There was no difference regarding patient knowledge, attitudes, or perceived ability by age, gender, race, or income.
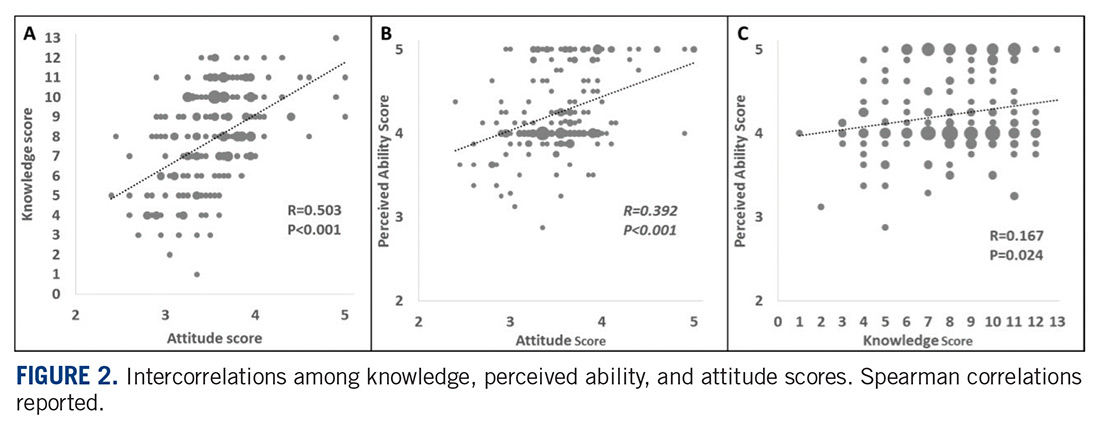
Knowledge, attitudes, perceived ability, and clinical trial enrollment
Thirty patients reported clinical trial experience (either previously or currently enrolled in trials) and 160 patients were never enrolled. Of the 30 patients with trial experience, 7 reported being currently enrolled, while 23 reported previous enrollment. Of these patients, 16 had metastatic disease, while 12 had non-metastatic disease, and 2 were unsure whether or not they had metastatic disease.
Patients with previous clinical trial exposure (currently or previously enrolled in clinical trials) demonstrated significantly greater trial knowledge, with a mean knowledge score of 9.3 (CI 8.5-10.0) compared with 7.7 (CI 7.3-8.1) among patients without trial exposure (P=0.002; FIGURE 3A). Similarly, patients with trial experience also had statistically significant more positive attitudes towards trials as compared with patients with no trial experience, with a mean attitude score of 3.8 (CI 3.6-4.0) and 3.5 (CI 3.4-3.6), respectively (P=0.001; FIGURE 3B). While numerically patients with trial experience have greater perceived ability compared with patients with no trial experience, with a mean score of 4.4 (CI 4.2-4.6) and 4.2 (CI 4.1-4.3), respectively, this difference did not reach statistical significance (FIGURE 3C).
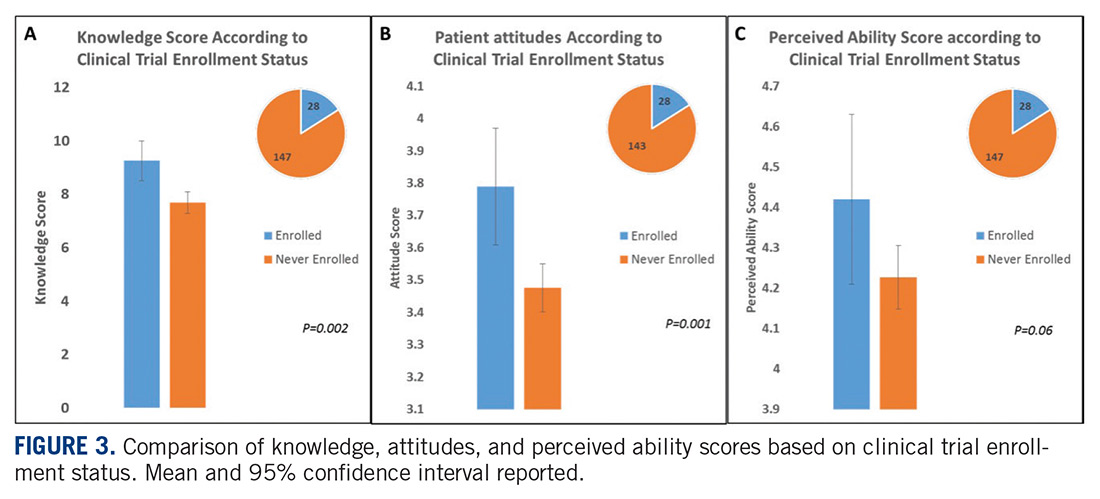
Knowledge, attitudes, perceived ability, and disease stage
An analysis was performed comparing patients with metastatic vs non-metastatic disease. It was observed that patients with metastatic disease had similar knowledge of trials compared with non-metastatic patients, with a mean knowledge score of 8.4 (CI 7.7- 9.1) and 7.9 (CI 7.5-8.4), respectively, (P=0.3; FIGURE 4A). In contrast, patients with metastatic disease had more positive attitudes compared with non-metastatic patients, with a mean score of 3.7 (CI 3.5-3.8) and 3.5 (CI 3.4-3.6), respectively, which was statistically significant (P=0.03; FIGURE 4B). There was no difference in perceived ability in metastatic vs non-metastatic patients (FIGURE 4C).
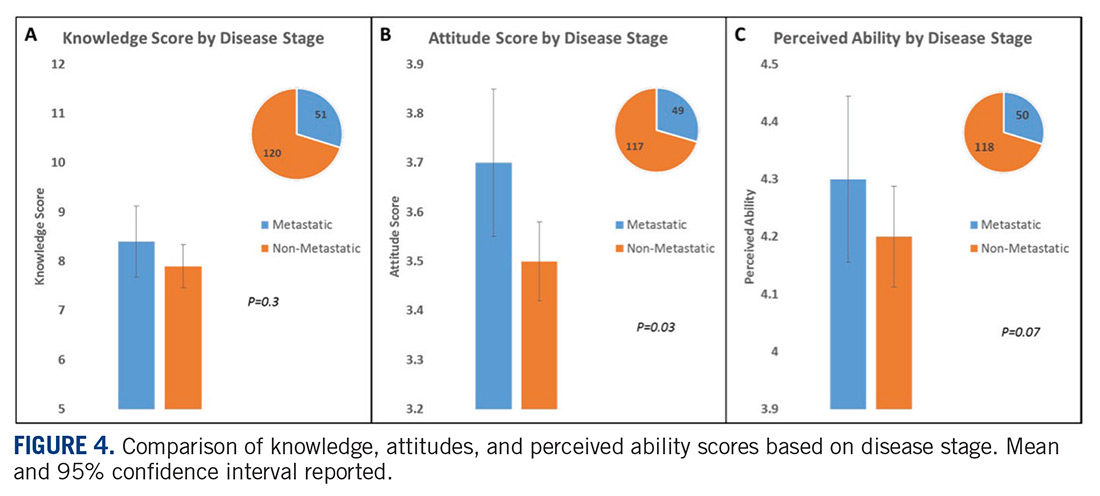
Thoughts about molecular profiling
Of the total number of patients, 46 patients had heard of molecular profiling and were presented with questions regarding their thoughts (TABLE 3). Approximately two-thirds (65.2%) thought there would be a 50% or greater likelihood of finding a targetable result in their tumor molecular profile. A majority (71.7%) of patients thought that a new experimental therapy chosen based on a patient’s tumor molecular profile would have at least a 50% chance of controlling the cancer. Somewhat less than a third (30.4%) of patients thought that total cure is the maximal benefit a patient could experience as a result of a treatment on a clinical trial using a drug chosen based on molecular tests. About half (52.2%) of patients agreed with the statement, “I am concerned about the cost of the test to molecularly profile my cancer.”
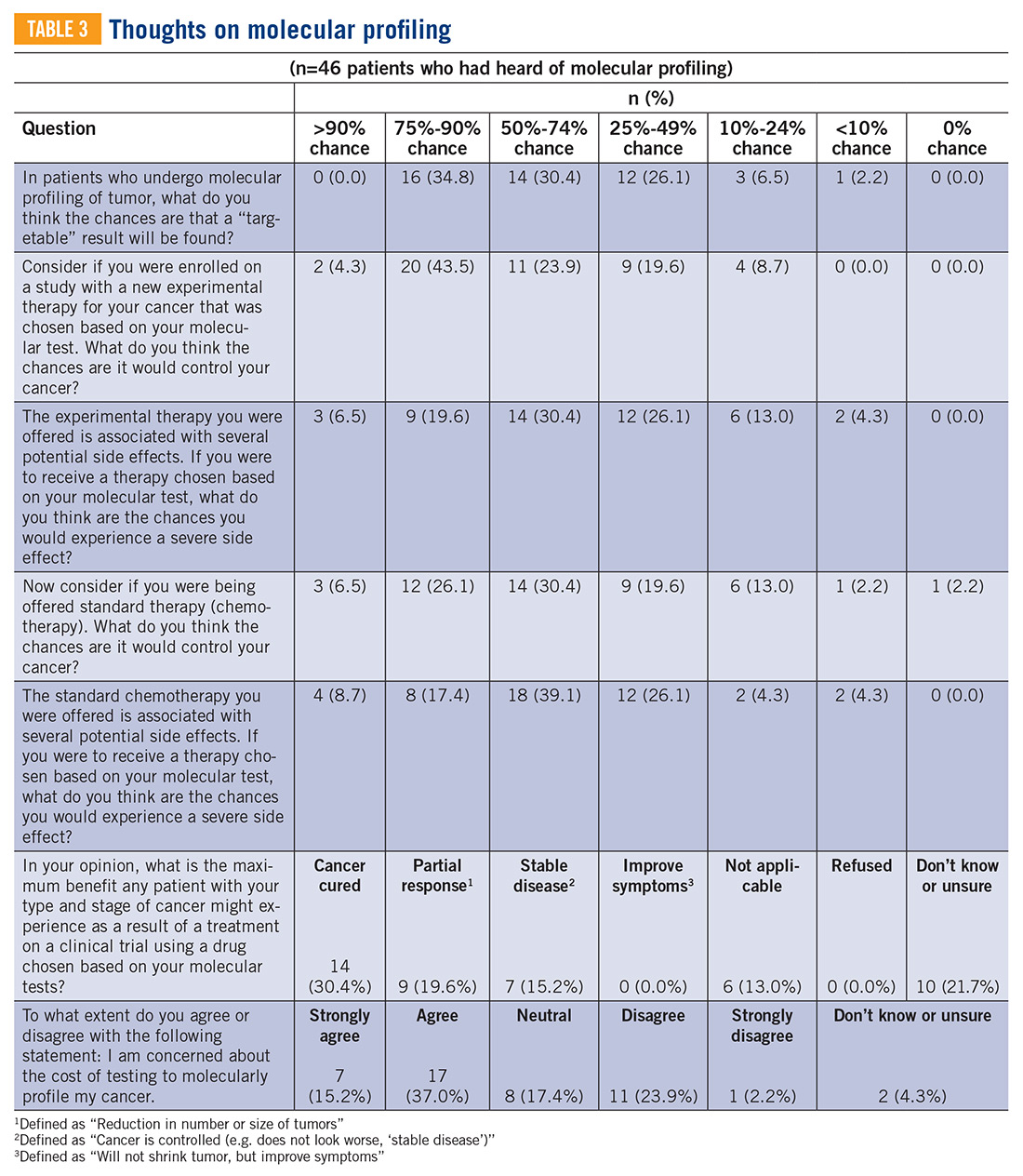
Considerations to undergo molecular profiling
Eighteen patients had undergone molecular profiling of their tumor. These patients were posed the question, “To what extent did you consider the following issues or concerns at the time you decided to get molecular testing of your cancer?” (TABLE 4). A majority (83.3%) of patients stated that wanting to live as long as possible was important, and 72.2% of patients stated that quality of life was important. A majority (83.3%) of patients stated that hope for a cure was an extremely or quite a bit important consideration. Helping future cancer patients was extremely or quite a bit important for 77.8% of patients, while wanting to be a part of research was not at all or of little importance in 50.0% of patients.
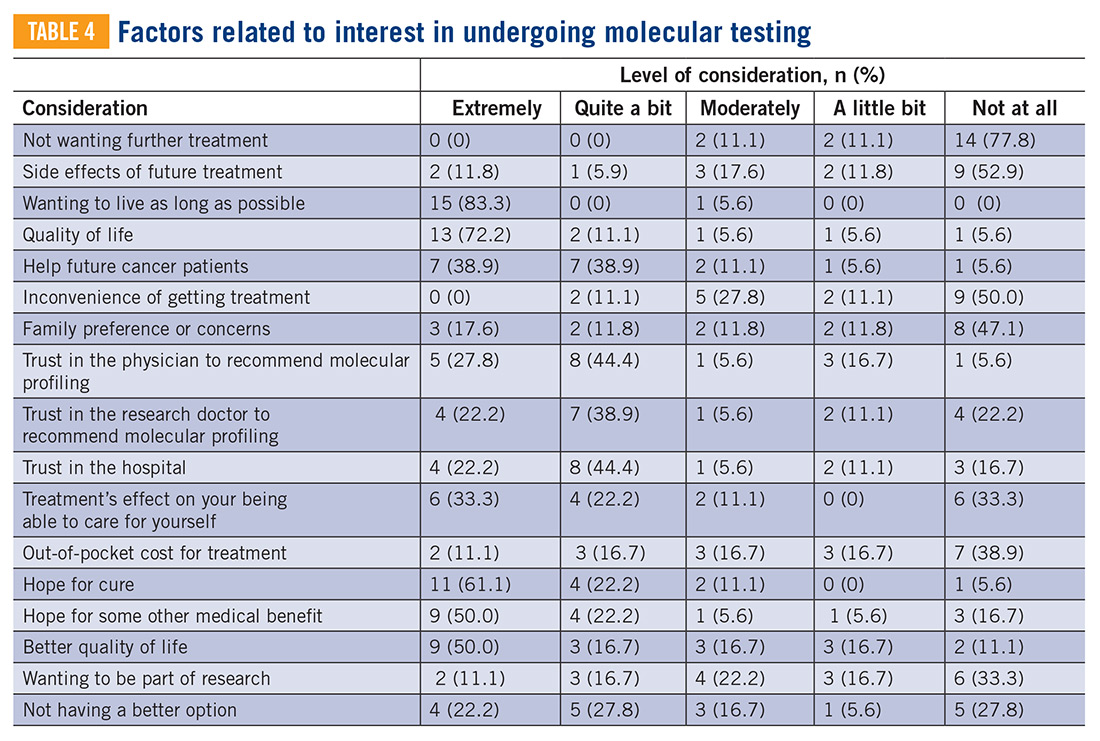
Clinical trials at NM
Twenty-four clinical trials were available at NM between the years 2012 and 2017 for patients with bone and soft tissue sarcoma. Of these trials, 3 of 24 were for non-metastatic patients, while the remaining 21 were open only to metastatic patients. The median number of patients screened per trial was 11 (range 0-66) and the median number of patients accrued per trial was 9 (range 0-58). Of the 24 trials, 17 were not subtype specific (13 included soft tissue sarcoma alone while 4 included both bone and soft tissue sarcoma). The remaining 7 trials were sarcoma subtype specific (eg, angiosarcoma, liposarcoma, etc). Trials available at NM during this period are included in TABLE 5. There were 318 patients screened and 262 patients accrued to sarcoma trials over this time period, with a screen failure rate of 17.6% overall.
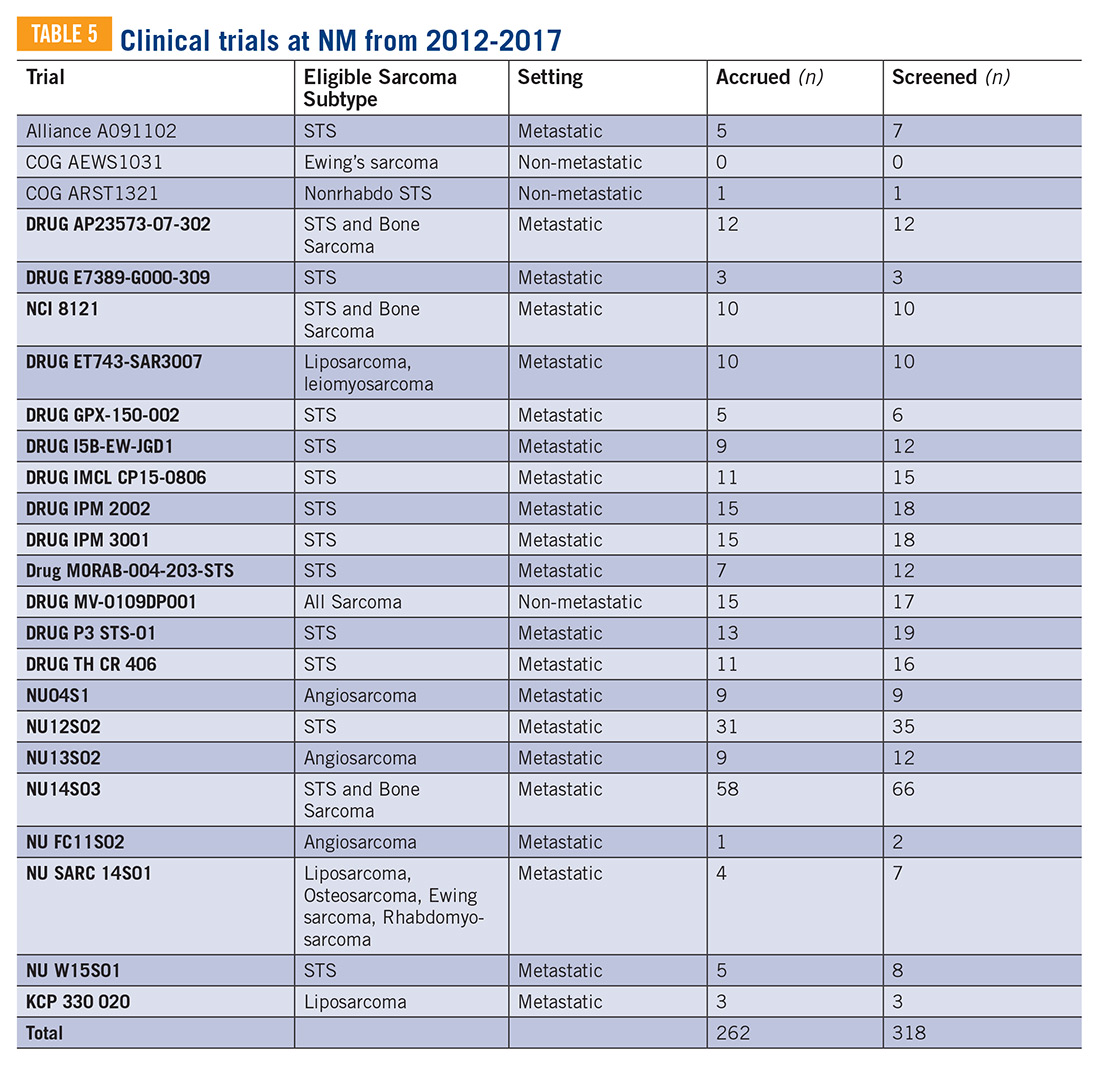
Discussion
Our study sought to describe perceptions of clinical trial enrollment among patients with bone and soft tissue sarcoma in order to elucidate and overcome barriers to enrollment, which to the best of our knowledge had not been previously described. Using previously validated patient- reported outcomes in the literature,6 our data reveal a correlation between knowledge of trials and more positive attitudes towards trials. This underscores the importance of awareness and educational strategies in this cancer population as a whole. Interventions should focus on patient perceptions that contribute to lack of participation, such as fear of side effects, loss of control (eg, idea of placebo or randomization), logistical challenges (eg, additional time or convenient location), and cost.3,5,7,16,17 For example, patients concerned about randomization should be educated on equipoise and other ethical considerations in trial design.5 Previous research has suggested that a multimedia psychoeducational intervention was effective in improving attitudes toward trials.6 Educating patients on the essential role of trials in oncology care, as demonstrated by the vast number of new drug approvals in recent years, is an essential strategy to improve attitudes, and subsequently leads to higher patient accrual rates.
In our study, both knowledge and at titudes were increased in patients with previous trial exposure. This suggests that either patients with greater knowledge and more positive attitudes are more likely to enroll in trials, or that patients with direct trial exposure are more knowledgeable and develop more positive attitudes. Our patient population was overall receptive to learning more about clinical trials (85.4%) and willing to participate (65.8%). At the same time, our study demonstrated low to medium correlations between attitudes and perceived ability to take steps towards making an informed decision to enroll in a trial (r=0.4), which may partially be explained by the absence of a tangible trial opportunity. While we did not assess specifically whether patients in our study were offered a trial, there was a substantial trial menu at NM with limited screen failures and decent trial accrual over the time frame of our study. This underscores the importance not only of patient-focused strategies to increase educational and attitudinal resources, but also a need to focus on research-site optimization that includes opening of multiple trials in various settings, systematic pre-screening of patients, and eligibility criteria that are inclusive and rational.5
The patient population with metastatic disease demonstrated more positive attitudes towards enrolling in clinical trials. This cohort accrued well to clinical trials, with 21 of 24 trials enrolling specifically patients with metastatic disease. Of the patients who responded to our survey, patients with previous trial exposure were enriched for patients with metastatic disease (53.3% metastatic among previous trial exposure versus 26.8% metastatic overall). These observations are likely reflective of the need for novel therapies in this disease setting. At the same time, approximately 25% of patients with localized soft tissue sarcoma will develop distant metastatic disease after successful treatment of their primary tumor, which increases to 40% to 50% in larger and higher-grade tumors.18 Three of 24 trials were open for patients with non-metastatic disease, of which one managed to accrue patients. Patients with non-metastatic disease had more negative attitudes towards trial enrollment. These disproportionate findings suggest a need for interventions to increase patient awareness and attitudes towards trial enrollment among this patient population and the importance of research-site optimization for trial opportunities across disease states.
Molecular profiling of tumors and biomarker identification has become a critical component of further characterizing cancer subtypes. In our study, a majority (65.2%) thought there would be a 50% or greater likelihood of finding a targetable result in their molecular profile. Molecular data on 5,749 bone and soft tissue sarcomas suggested that 9.5% of tumors demonstrate a “targetable result,” defined as a new molecular finding for which there is an FDA-approved drug for malignancies other than sarcoma (eg, BRAF V600E, Her2, etc.),19 suggesting an overestimation in our patient cohort of the likelihood of benefit of molecular profiling. These results highlight that the growing use of molecular profiling has increased the need for educational and supportive resources to help patients understand the utility of molecular profiling and aid in shared decision-making surrounding the results.
At the same time, the importance of identifying a targetable mutation in patients with sarcoma cannot be understated. As a recent example of this paradigm, tumors that harbor fusions with the neurotrophic receptor tyrosine kinase 1, 2, or 3 (NTRK1, 2, and 3) have a high response rate (~75%) to drugs that target these fusions, such as larotrectinib and entrectinib.13 Molecular profiling and identification of predictive biomarkers in small patient subsets has led to great challenges in trial design and research-site optimization. Novel designs that incorporate molecular profiling,20 such as the Lung- MAP trial21 and NCI’s Molecular Analysis for Therapy Choice (MATCH) trial,22 are emerging to identify new therapies for small patient subsets. As a rare and increasingly heterogeneous cancer, sarcoma represents a paradigm to provide insight into optimizing patient perceptions and research enterprises to maximize clinical trial enrollment.
Some limitations of our study include a homogeneous and selected patient population that was predominantly Caucasian and highly educated. Therefore, these findings should not be extrapolated to other populations with barriers to trial accrual, such as lower socioeconomic or minority populations. The low response rate and failure of some to complete the survey may have introduced some bias. Additionally, our data include self-reported outcomes, which could have affected our results. Finally, the limited number of patients who had undergone or heard of molecular profiling limited our ability to draw definitive conclusions, and should be assessed in larger patient cohorts.
While our paper addresses a unique population—the sarcoma patient—similar themes and issues pertain to all oncology patients. A recent review was published in the American Society of Clinical Oncology Educational Book23 looking at methods to overcome barriers to clinical trial enrollment. Their paper clearly illustrates mechanisms to assist with overcoming financial burdens associated with cancer clinical trials, overcoming barriers as they relate to patient and clinician difficulty in coping with the uncertainty inherent in clinical trial participation, and highlight the role of a patient navigator in clinical trial participation.
Conclusions
Interventions aimed at increasing awareness, knowledge, and attitudes towards clinical trials among sarcoma patients may lead to increased trial enrollment and greater progress in cancer treatment in this population. In addition to patient- focused interventions, thoughtful and strategic clinical trial designs that allow for the development of biomarker- driven therapeutics, while at the same time optimizing patient accrual rates, should be developed. Evaluation of barriers to clinical trial enrollment and molecular profiling of tumors among bone and soft tissue sarcoma patients at an academic center can serve as a paradigm to overcome barriers to enrollment in the era of an increasingly heterogeneous cancer population. TSJ
1. Go RS, Frisby KA, Lee JA, et al. Clinical trial accrual among new cancer patients at a community-based cancer center. Cancer. 2006;106(2):426-433.
2. Murthy VH, Krumholz HM, Gross CP. Participation in cancer clinical trials: Race-, sex-, and age-based disparities. JAMA. 2004; 291(22):2720-2726.
3. Lara PN Jr, Higdon R, Lim N, et al. Prospective evaluation of cancer clinical trial accrual patterns: identifying potential barriers to enrollment. J Clin Oncol. 2001;19(6):1728-1733.
4. Unger JM, Cook E, Tai E, Bleyer A. The role of clinical trial participation in cancer research: barriers, evidence, and strategies. Am Soc Clin Oncol Educ Book. 2016;35:185-198.
5. Cancer Action Network American Cancer Society. Barriers to patient enrollment in therapeutic clinical trials for cancer. 2018: https:// www.fightcancer.org/policy-resources/clinical- trial-barriers#figures. Accessed March 14, 2019.
6. Jacobsen PB, Wells KJ, Meade CD, et al. Effects of a brief multimedia psychoeducational intervention on the attitudes and interest of patients with cancer regarding clinical trial participation: a multicenter randomized controlled trial. J Clin Oncol. 2012;30(20):2516-2521.
7. Meropol NJ, Buzaglo JS, Millard J, et al. Barriers to clinical trial participation as perceived by oncologists and patients. J Natl Compr Canc Netw. 2007;5(8):655-664.
8. Cox K, McGarry J. Why patients don’t take part in cancer clinical trials: an overview of the literature. Eur J Cancer Care (Engl). 2003;12(2): 114-122.
9. Mills EJ, Seely D, Rachlis B, et al. Barriers to participation in clinical trials of cancer: a meta- analysis and systematic review of patient-reported factors. Lancet Oncol. 2006;7(2):141-148.
10. National Comprehensive Cancer Network. Soft Tissue Sarcoma (Version 4.2019). https:// www.nccn.org/professionals/physician_gls/ pdf/sarcoma.pdf. Accessed October 30, 2019.
11. Nass SJ, Beaupin LK, Demark-Wahnefried W, et al. Identifying and addressing the needs of adolescents and young adults with cancer: summary of an Institute of Medicine workshop. Oncologist. 2015;20(2):186-195.
12. Wilky BA, Villalobos VM. Emerging role for precision therapy through next-generation sequencing for sarcomas. JCO Precision Oncol. 2018(2):1-4.
13. Bleyer A, Montello M, Budd T, Saxman S. National survival trends of young adults with sarcoma: lack of progress is associated with lack of clinical trial participation. Cancer. 2005;103(9):1891-1897.
14. Gornick MC, Cobain E, Le LQ, et al. Oncologists’ use of genomic sequencing data to inform clinical management. JCO Precision Oncol. 2018(2):1-13.
15. Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2nd ed. Hillsdale, NJ: L. Erlbaum Associates; 1988.
16. Unger JM, Hershman DL, Albain KS, et al. Patient income level and cancer clinical trial participation. J Clin Oncol. 2013;31(5):536- 542.
17. Javid SH, Unger JM, Gralow JR, et al. A prospective analysis of the influence of older age on physician and patient decision-making when considering enrollment in breast cancer clinical trials (SWOG S0316). Oncologist. 2012;17(9):1180-1190.
18. Singhi EK, Moore DC, Muslimani A. Metastatic soft tissue sarcomas: a review of treatment and new pharmacotherapies. P T. 2018;43(7): 410-429.
19. Gounder MM, Ali SM, Robinson V, et al. Impact of next-generation sequencing (NGS) on diagnostic and therapeutic options in soft-tissue and bone sarcoma. J Clin Oncol. 2017;35(15_suppl):abstr 11001.
20. Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med. 2017;377(1):62-70.
21. Steuer CE, Papadimitrakopoulou V, Herbst RS, et al. Innovative clinical trials: the LUNGMAP study. Clin Pharmacol Ther. 2015;97(5): 488-491.
22. McNeil C. NCI-MATCH launch highlights new trial design in precision-medicine era. J Natl Cancer Inst. 2015;107(7).
23. Nipp RD, Hong K, Paskett ED. Overcoming barriers to clinical trial enrollment. Am Soc Clin Oncol Educ Book. 2019;39:105-114.
Introduction
The development of new cancer therapies relies on the successful development and completion of clinical trials. While clinical trials have led to significant improvements in cancer treatment, the success is dependent upon patient enrollment and participation. Unfortunately, fewer than 5% of adult patients enroll in trials.1-3 This represents a significant barrier to the development and approval of new cancer treatments. Reasons for low accrual into trials are multifactorial, but include structural barriers (eg, clinic access), clinical barriers (eg, eligibility criteria), and physician and patient attitudes towards trial enrollment.4,5 One study at the University of California Davis Cancer Center reported 49% of patients declined participation despite meeting eligibility criteria,3,6 suggesting that psychosocial barriers such as knowledge of trials and attitudes towards clinical research are a major impediment to accrual.7-9
Bone and soft tissue sarcoma represent a heterogeneous group of tumors of mesenchymal origin that are an important cause of morbidity and mortality. Local disease is often treated with a multidisciplinary approach including surgery, radiation, and systemic therapy. Metastatic disease is predominantly treated palliatively with systemic therapy.10 Given its rarity and heterogeneity, trial accrual is of particular importance in sarcoma and often requires multiple sites to enroll adequate numbers of patients. While sarcoma represents <1% of adult malignancies overall, it constitutes ~15% of malignancies in the adolescent and young adult (AYA) population (15- 39 years old).11,12 Sarcoma represents a patient population in which low trial accrual has been correlated with lack of progress in cancer-related outcomes in both the adult and AYA populations.13 The reasons for low accrual rates among patients with sarcoma are poorly understood.
Sarcomas represent a molecularly and biologically heterogeneous group of malignancies with over 100 different subtypes.12 As a result, there has been significant interest in performing molecular profiling, or genetic sequencing, to identify “targetable” mutations. Targetable mutations refer to a specific genetic change identified within the tumor molecular profile for which there is a specific drug that may demonstrate activity against a particular tumor. Given the widespread utilization of this technology in sarcoma, identifying and understanding patient perceptions with regard to molecular profiling is critically important in this disease.14
In this study, we use a cross-sectional design to describe patient perceptions of trial enrollment among patients with bone and soft tissue sarcoma through validated measures, including attitudes towards clinical trials, knowledge of clinical trials, and perceived ability (ie, self-efficacy) to carry out actions involved in making an informed decision about clinical trial participation, receptivity to learning more about clinical trials, and willingness to participate in clinical trials.6 In addition, we describe this patient cohort’s perceptions of molecular profiling, as current and future trials are increasingly driven by molecular or other biomarkers.
Methods
This was a cross-sectional electronic survey study of patients with bone and soft tissue sarcoma treated at Northwestern Medicine (NM) over a 5-year period. NM Enterprise Data Warehouse (NMEDW) is a single, comprehensive, and integrated repository of all clinical and research data sources within NM. The study was approved by the Northwestern University Institutional Review Board.
Survey
The investigators designed a self-administered, online survey, which was built using Research Electronic Data Capture (REDCap). The survey consisted of three sections that were answered using skip logic—a custom path through the survey that varied based on patients’ answers: (1) Patient demographic information and trial perceptions (answered by all patients); (2) Thoughts about molecular profiling (answered by patients who answered “yes” to the question, “Have you heard about molecular profiling of tumors?”); and (3) Considerations to undergo molecular profiling (answered by patients who answered “yes” to the question, “Have you undergone profiling of your cancer?”).
Clinical trial perceptions included questions assessing (1) patient knowledge about trials; (2) patient attitudes toward trials; (3) perceived ability (ie, self-efficacy) to carry out actions involved in making an informed decision about trial participation; (4) receptivity to learning more about trials; and (5) willingness to participate in trials. These outcome measures had been previously developed and pilot tested for reliability and validity (TABLE 1).6
Thoughts about molecular profiling of tumors were assessed using nine items (TABLE 1). Of these, items assessing potential benefit or harm of molecular profiling were assessed using a 7-step Likert scale. Items assessing maximal benefit or harm of therapy, importance of quality vs length of life, and concern about the cost of molecular testing were assessed using a 5-step Likert scale. The study team developed and piloted these questions because there is no validated survey assessing these domains.
Considerations to undergo molecular profiling were assessed using 17 items. Items were in response to the question, “To what extent did you consider the following issues or concerns at the time you decided to get molecular testing of your cancer?” Responses were assessed using a 5-point Likert scale.
Data Collection
Patients 18 years and older evaluated at NM between November 20, 2012, and November 20, 2017, with a diagnosis of sarcoma were identified by query of the NMEDW by ICD-10 codes (C40, C41.9, C44.99, C45-49, C55, C71.9, D48, D49.9, and M12.20) or equivalent ICD-9 codes. Patients were subsequently excluded if they did not have a diagnosis of bone or soft tissue sarcoma, no e-mail address listed, had died, or had not been evaluated at an NM clinic in the previous 5 years. Patients with a diagnosis of gastrointestinal stromal tumor and Kaposi’s sarcoma were also excluded.
A personalized contact e-mail was sent to patients containing an explanation of the survey and an internet link to the electronic survey through REDCap from January 2018 to March 2018. If patients did not respond to the survey, two follow-up reminder e-mails were sent 2 and 4 days following the initial survey. The link was protected so that each patient could complete the survey only once. Responses were collected through the REDCap platform. Patients read and signed an electronic consent form prior to completing the survey.
Upon completion of the survey, patients were offered a $50 VISA gift card as compensation, with an option to donate their compensation to the Robert H. Lurie Comprehensive Cancer Center Sarcoma Research Fund.
Over the described survey period, open clinical trials for patients with bone and soft tissue sarcoma available at NM were evaluated. The number of patients screened and accrued to each trial were recorded.
Statistical analysis
Responses were separated from the personal data for complete anonymization. Descriptive statistical analysis was performed for demographics and disease variables and were summarized using frequencies and percentages. Median and range were used for age. Correlations between continuous variables were analyzed using Spearman correlations. Scores were compared between subgroups using the Mann-Whitney test. Descriptive statistics for knowledge, attitude, and ability scores include means and 95% confidence intervals. Correlations were interpreted as small (r=0.10), medium (r=0.30), or large (r=0.50).15 Statistical significance was indicated when P<0.05.
Results
Patients
Seven hundred fifty patients were eligible to participate in the survey and received the initial and two follow-up e-mails. Twenty e-mailed surveys bounced back. Three hundred nine patients opened the initial e-mail and 283 patients (37.7% of total and 91.6% of opened) completed at least a portion of the survey, with 182 patients completing the entire survey (FIGURE 1). Data for analysis were used from patients who completed at least a portion of the survey.
Baseline characteristics of patients who responded can be seen in TABLE 2. Patients had a median age of 56, the majority were female (59.4%), white (88.2%), and most had college or university graduate degrees or higher educational level (69.0%). Patients had various different histological subtypes, with the most common being liposarcoma (16.5%) and leiomyosarcoma (16.0%). Slightly more than a quarter (26.8%) of patients had metastatic disease, and 84.2% had never been enrolled in a clinical trial. Previous treatments included surgery (91.1%), radiation (53.2%), and chemotherapy (51.6%). Prior to completing the survey, 85.4% reported being receptive to a cancer clinical trial, while 60.7% of patients reported willingness to participate in a clinical trial.
Knowledge, attitudes, and perceived ability
A statistically significant correlation was observed between greater knowledge of trials and more positive attitudes towards trials (P<0.001; r=0.5, FIGURE 2A). In relating patient attitudes with perceived ability, again a significant correlation was seen (P<0.001; r=0.4, FIGURE 2B). In contrast, knowledge had a weak correlation with perceived ability (P=0.024; r=0.2, FIGURE 2C). There was no difference regarding patient knowledge, attitudes, or perceived ability by age, gender, race, or income.
Knowledge, attitudes, perceived ability, and clinical trial enrollment
Thirty patients reported clinical trial experience (either previously or currently enrolled in trials) and 160 patients were never enrolled. Of the 30 patients with trial experience, 7 reported being currently enrolled, while 23 reported previous enrollment. Of these patients, 16 had metastatic disease, while 12 had non-metastatic disease, and 2 were unsure whether or not they had metastatic disease.
Patients with previous clinical trial exposure (currently or previously enrolled in clinical trials) demonstrated significantly greater trial knowledge, with a mean knowledge score of 9.3 (CI 8.5-10.0) compared with 7.7 (CI 7.3-8.1) among patients without trial exposure (P=0.002; FIGURE 3A). Similarly, patients with trial experience also had statistically significant more positive attitudes towards trials as compared with patients with no trial experience, with a mean attitude score of 3.8 (CI 3.6-4.0) and 3.5 (CI 3.4-3.6), respectively (P=0.001; FIGURE 3B). While numerically patients with trial experience have greater perceived ability compared with patients with no trial experience, with a mean score of 4.4 (CI 4.2-4.6) and 4.2 (CI 4.1-4.3), respectively, this difference did not reach statistical significance (FIGURE 3C).
Knowledge, attitudes, perceived ability, and disease stage
An analysis was performed comparing patients with metastatic vs non-metastatic disease. It was observed that patients with metastatic disease had similar knowledge of trials compared with non-metastatic patients, with a mean knowledge score of 8.4 (CI 7.7- 9.1) and 7.9 (CI 7.5-8.4), respectively, (P=0.3; FIGURE 4A). In contrast, patients with metastatic disease had more positive attitudes compared with non-metastatic patients, with a mean score of 3.7 (CI 3.5-3.8) and 3.5 (CI 3.4-3.6), respectively, which was statistically significant (P=0.03; FIGURE 4B). There was no difference in perceived ability in metastatic vs non-metastatic patients (FIGURE 4C).
Thoughts about molecular profiling
Of the total number of patients, 46 patients had heard of molecular profiling and were presented with questions regarding their thoughts (TABLE 3). Approximately two-thirds (65.2%) thought there would be a 50% or greater likelihood of finding a targetable result in their tumor molecular profile. A majority (71.7%) of patients thought that a new experimental therapy chosen based on a patient’s tumor molecular profile would have at least a 50% chance of controlling the cancer. Somewhat less than a third (30.4%) of patients thought that total cure is the maximal benefit a patient could experience as a result of a treatment on a clinical trial using a drug chosen based on molecular tests. About half (52.2%) of patients agreed with the statement, “I am concerned about the cost of the test to molecularly profile my cancer.”
Considerations to undergo molecular profiling
Eighteen patients had undergone molecular profiling of their tumor. These patients were posed the question, “To what extent did you consider the following issues or concerns at the time you decided to get molecular testing of your cancer?” (TABLE 4). A majority (83.3%) of patients stated that wanting to live as long as possible was important, and 72.2% of patients stated that quality of life was important. A majority (83.3%) of patients stated that hope for a cure was an extremely or quite a bit important consideration. Helping future cancer patients was extremely or quite a bit important for 77.8% of patients, while wanting to be a part of research was not at all or of little importance in 50.0% of patients.
Clinical trials at NM
Twenty-four clinical trials were available at NM between the years 2012 and 2017 for patients with bone and soft tissue sarcoma. Of these trials, 3 of 24 were for non-metastatic patients, while the remaining 21 were open only to metastatic patients. The median number of patients screened per trial was 11 (range 0-66) and the median number of patients accrued per trial was 9 (range 0-58). Of the 24 trials, 17 were not subtype specific (13 included soft tissue sarcoma alone while 4 included both bone and soft tissue sarcoma). The remaining 7 trials were sarcoma subtype specific (eg, angiosarcoma, liposarcoma, etc). Trials available at NM during this period are included in TABLE 5. There were 318 patients screened and 262 patients accrued to sarcoma trials over this time period, with a screen failure rate of 17.6% overall.
Discussion
Our study sought to describe perceptions of clinical trial enrollment among patients with bone and soft tissue sarcoma in order to elucidate and overcome barriers to enrollment, which to the best of our knowledge had not been previously described. Using previously validated patient- reported outcomes in the literature,6 our data reveal a correlation between knowledge of trials and more positive attitudes towards trials. This underscores the importance of awareness and educational strategies in this cancer population as a whole. Interventions should focus on patient perceptions that contribute to lack of participation, such as fear of side effects, loss of control (eg, idea of placebo or randomization), logistical challenges (eg, additional time or convenient location), and cost.3,5,7,16,17 For example, patients concerned about randomization should be educated on equipoise and other ethical considerations in trial design.5 Previous research has suggested that a multimedia psychoeducational intervention was effective in improving attitudes toward trials.6 Educating patients on the essential role of trials in oncology care, as demonstrated by the vast number of new drug approvals in recent years, is an essential strategy to improve attitudes, and subsequently leads to higher patient accrual rates.
In our study, both knowledge and at titudes were increased in patients with previous trial exposure. This suggests that either patients with greater knowledge and more positive attitudes are more likely to enroll in trials, or that patients with direct trial exposure are more knowledgeable and develop more positive attitudes. Our patient population was overall receptive to learning more about clinical trials (85.4%) and willing to participate (65.8%). At the same time, our study demonstrated low to medium correlations between attitudes and perceived ability to take steps towards making an informed decision to enroll in a trial (r=0.4), which may partially be explained by the absence of a tangible trial opportunity. While we did not assess specifically whether patients in our study were offered a trial, there was a substantial trial menu at NM with limited screen failures and decent trial accrual over the time frame of our study. This underscores the importance not only of patient-focused strategies to increase educational and attitudinal resources, but also a need to focus on research-site optimization that includes opening of multiple trials in various settings, systematic pre-screening of patients, and eligibility criteria that are inclusive and rational.5
The patient population with metastatic disease demonstrated more positive attitudes towards enrolling in clinical trials. This cohort accrued well to clinical trials, with 21 of 24 trials enrolling specifically patients with metastatic disease. Of the patients who responded to our survey, patients with previous trial exposure were enriched for patients with metastatic disease (53.3% metastatic among previous trial exposure versus 26.8% metastatic overall). These observations are likely reflective of the need for novel therapies in this disease setting. At the same time, approximately 25% of patients with localized soft tissue sarcoma will develop distant metastatic disease after successful treatment of their primary tumor, which increases to 40% to 50% in larger and higher-grade tumors.18 Three of 24 trials were open for patients with non-metastatic disease, of which one managed to accrue patients. Patients with non-metastatic disease had more negative attitudes towards trial enrollment. These disproportionate findings suggest a need for interventions to increase patient awareness and attitudes towards trial enrollment among this patient population and the importance of research-site optimization for trial opportunities across disease states.
Molecular profiling of tumors and biomarker identification has become a critical component of further characterizing cancer subtypes. In our study, a majority (65.2%) thought there would be a 50% or greater likelihood of finding a targetable result in their molecular profile. Molecular data on 5,749 bone and soft tissue sarcomas suggested that 9.5% of tumors demonstrate a “targetable result,” defined as a new molecular finding for which there is an FDA-approved drug for malignancies other than sarcoma (eg, BRAF V600E, Her2, etc.),19 suggesting an overestimation in our patient cohort of the likelihood of benefit of molecular profiling. These results highlight that the growing use of molecular profiling has increased the need for educational and supportive resources to help patients understand the utility of molecular profiling and aid in shared decision-making surrounding the results.
At the same time, the importance of identifying a targetable mutation in patients with sarcoma cannot be understated. As a recent example of this paradigm, tumors that harbor fusions with the neurotrophic receptor tyrosine kinase 1, 2, or 3 (NTRK1, 2, and 3) have a high response rate (~75%) to drugs that target these fusions, such as larotrectinib and entrectinib.13 Molecular profiling and identification of predictive biomarkers in small patient subsets has led to great challenges in trial design and research-site optimization. Novel designs that incorporate molecular profiling,20 such as the Lung- MAP trial21 and NCI’s Molecular Analysis for Therapy Choice (MATCH) trial,22 are emerging to identify new therapies for small patient subsets. As a rare and increasingly heterogeneous cancer, sarcoma represents a paradigm to provide insight into optimizing patient perceptions and research enterprises to maximize clinical trial enrollment.
Some limitations of our study include a homogeneous and selected patient population that was predominantly Caucasian and highly educated. Therefore, these findings should not be extrapolated to other populations with barriers to trial accrual, such as lower socioeconomic or minority populations. The low response rate and failure of some to complete the survey may have introduced some bias. Additionally, our data include self-reported outcomes, which could have affected our results. Finally, the limited number of patients who had undergone or heard of molecular profiling limited our ability to draw definitive conclusions, and should be assessed in larger patient cohorts.
While our paper addresses a unique population—the sarcoma patient—similar themes and issues pertain to all oncology patients. A recent review was published in the American Society of Clinical Oncology Educational Book23 looking at methods to overcome barriers to clinical trial enrollment. Their paper clearly illustrates mechanisms to assist with overcoming financial burdens associated with cancer clinical trials, overcoming barriers as they relate to patient and clinician difficulty in coping with the uncertainty inherent in clinical trial participation, and highlight the role of a patient navigator in clinical trial participation.
Conclusions
Interventions aimed at increasing awareness, knowledge, and attitudes towards clinical trials among sarcoma patients may lead to increased trial enrollment and greater progress in cancer treatment in this population. In addition to patient- focused interventions, thoughtful and strategic clinical trial designs that allow for the development of biomarker- driven therapeutics, while at the same time optimizing patient accrual rates, should be developed. Evaluation of barriers to clinical trial enrollment and molecular profiling of tumors among bone and soft tissue sarcoma patients at an academic center can serve as a paradigm to overcome barriers to enrollment in the era of an increasingly heterogeneous cancer population. TSJ
Introduction
The development of new cancer therapies relies on the successful development and completion of clinical trials. While clinical trials have led to significant improvements in cancer treatment, the success is dependent upon patient enrollment and participation. Unfortunately, fewer than 5% of adult patients enroll in trials.1-3 This represents a significant barrier to the development and approval of new cancer treatments. Reasons for low accrual into trials are multifactorial, but include structural barriers (eg, clinic access), clinical barriers (eg, eligibility criteria), and physician and patient attitudes towards trial enrollment.4,5 One study at the University of California Davis Cancer Center reported 49% of patients declined participation despite meeting eligibility criteria,3,6 suggesting that psychosocial barriers such as knowledge of trials and attitudes towards clinical research are a major impediment to accrual.7-9
Bone and soft tissue sarcoma represent a heterogeneous group of tumors of mesenchymal origin that are an important cause of morbidity and mortality. Local disease is often treated with a multidisciplinary approach including surgery, radiation, and systemic therapy. Metastatic disease is predominantly treated palliatively with systemic therapy.10 Given its rarity and heterogeneity, trial accrual is of particular importance in sarcoma and often requires multiple sites to enroll adequate numbers of patients. While sarcoma represents <1% of adult malignancies overall, it constitutes ~15% of malignancies in the adolescent and young adult (AYA) population (15- 39 years old).11,12 Sarcoma represents a patient population in which low trial accrual has been correlated with lack of progress in cancer-related outcomes in both the adult and AYA populations.13 The reasons for low accrual rates among patients with sarcoma are poorly understood.
Sarcomas represent a molecularly and biologically heterogeneous group of malignancies with over 100 different subtypes.12 As a result, there has been significant interest in performing molecular profiling, or genetic sequencing, to identify “targetable” mutations. Targetable mutations refer to a specific genetic change identified within the tumor molecular profile for which there is a specific drug that may demonstrate activity against a particular tumor. Given the widespread utilization of this technology in sarcoma, identifying and understanding patient perceptions with regard to molecular profiling is critically important in this disease.14
In this study, we use a cross-sectional design to describe patient perceptions of trial enrollment among patients with bone and soft tissue sarcoma through validated measures, including attitudes towards clinical trials, knowledge of clinical trials, and perceived ability (ie, self-efficacy) to carry out actions involved in making an informed decision about clinical trial participation, receptivity to learning more about clinical trials, and willingness to participate in clinical trials.6 In addition, we describe this patient cohort’s perceptions of molecular profiling, as current and future trials are increasingly driven by molecular or other biomarkers.
Methods
This was a cross-sectional electronic survey study of patients with bone and soft tissue sarcoma treated at Northwestern Medicine (NM) over a 5-year period. NM Enterprise Data Warehouse (NMEDW) is a single, comprehensive, and integrated repository of all clinical and research data sources within NM. The study was approved by the Northwestern University Institutional Review Board.
Survey
The investigators designed a self-administered, online survey, which was built using Research Electronic Data Capture (REDCap). The survey consisted of three sections that were answered using skip logic—a custom path through the survey that varied based on patients’ answers: (1) Patient demographic information and trial perceptions (answered by all patients); (2) Thoughts about molecular profiling (answered by patients who answered “yes” to the question, “Have you heard about molecular profiling of tumors?”); and (3) Considerations to undergo molecular profiling (answered by patients who answered “yes” to the question, “Have you undergone profiling of your cancer?”).
Clinical trial perceptions included questions assessing (1) patient knowledge about trials; (2) patient attitudes toward trials; (3) perceived ability (ie, self-efficacy) to carry out actions involved in making an informed decision about trial participation; (4) receptivity to learning more about trials; and (5) willingness to participate in trials. These outcome measures had been previously developed and pilot tested for reliability and validity (TABLE 1).6
Thoughts about molecular profiling of tumors were assessed using nine items (TABLE 1). Of these, items assessing potential benefit or harm of molecular profiling were assessed using a 7-step Likert scale. Items assessing maximal benefit or harm of therapy, importance of quality vs length of life, and concern about the cost of molecular testing were assessed using a 5-step Likert scale. The study team developed and piloted these questions because there is no validated survey assessing these domains.
Considerations to undergo molecular profiling were assessed using 17 items. Items were in response to the question, “To what extent did you consider the following issues or concerns at the time you decided to get molecular testing of your cancer?” Responses were assessed using a 5-point Likert scale.
Data Collection
Patients 18 years and older evaluated at NM between November 20, 2012, and November 20, 2017, with a diagnosis of sarcoma were identified by query of the NMEDW by ICD-10 codes (C40, C41.9, C44.99, C45-49, C55, C71.9, D48, D49.9, and M12.20) or equivalent ICD-9 codes. Patients were subsequently excluded if they did not have a diagnosis of bone or soft tissue sarcoma, no e-mail address listed, had died, or had not been evaluated at an NM clinic in the previous 5 years. Patients with a diagnosis of gastrointestinal stromal tumor and Kaposi’s sarcoma were also excluded.
A personalized contact e-mail was sent to patients containing an explanation of the survey and an internet link to the electronic survey through REDCap from January 2018 to March 2018. If patients did not respond to the survey, two follow-up reminder e-mails were sent 2 and 4 days following the initial survey. The link was protected so that each patient could complete the survey only once. Responses were collected through the REDCap platform. Patients read and signed an electronic consent form prior to completing the survey.
Upon completion of the survey, patients were offered a $50 VISA gift card as compensation, with an option to donate their compensation to the Robert H. Lurie Comprehensive Cancer Center Sarcoma Research Fund.
Over the described survey period, open clinical trials for patients with bone and soft tissue sarcoma available at NM were evaluated. The number of patients screened and accrued to each trial were recorded.
Statistical analysis
Responses were separated from the personal data for complete anonymization. Descriptive statistical analysis was performed for demographics and disease variables and were summarized using frequencies and percentages. Median and range were used for age. Correlations between continuous variables were analyzed using Spearman correlations. Scores were compared between subgroups using the Mann-Whitney test. Descriptive statistics for knowledge, attitude, and ability scores include means and 95% confidence intervals. Correlations were interpreted as small (r=0.10), medium (r=0.30), or large (r=0.50).15 Statistical significance was indicated when P<0.05.
Results
Patients
Seven hundred fifty patients were eligible to participate in the survey and received the initial and two follow-up e-mails. Twenty e-mailed surveys bounced back. Three hundred nine patients opened the initial e-mail and 283 patients (37.7% of total and 91.6% of opened) completed at least a portion of the survey, with 182 patients completing the entire survey (FIGURE 1). Data for analysis were used from patients who completed at least a portion of the survey.
Baseline characteristics of patients who responded can be seen in TABLE 2. Patients had a median age of 56, the majority were female (59.4%), white (88.2%), and most had college or university graduate degrees or higher educational level (69.0%). Patients had various different histological subtypes, with the most common being liposarcoma (16.5%) and leiomyosarcoma (16.0%). Slightly more than a quarter (26.8%) of patients had metastatic disease, and 84.2% had never been enrolled in a clinical trial. Previous treatments included surgery (91.1%), radiation (53.2%), and chemotherapy (51.6%). Prior to completing the survey, 85.4% reported being receptive to a cancer clinical trial, while 60.7% of patients reported willingness to participate in a clinical trial.
Knowledge, attitudes, and perceived ability
A statistically significant correlation was observed between greater knowledge of trials and more positive attitudes towards trials (P<0.001; r=0.5, FIGURE 2A). In relating patient attitudes with perceived ability, again a significant correlation was seen (P<0.001; r=0.4, FIGURE 2B). In contrast, knowledge had a weak correlation with perceived ability (P=0.024; r=0.2, FIGURE 2C). There was no difference regarding patient knowledge, attitudes, or perceived ability by age, gender, race, or income.
Knowledge, attitudes, perceived ability, and clinical trial enrollment
Thirty patients reported clinical trial experience (either previously or currently enrolled in trials) and 160 patients were never enrolled. Of the 30 patients with trial experience, 7 reported being currently enrolled, while 23 reported previous enrollment. Of these patients, 16 had metastatic disease, while 12 had non-metastatic disease, and 2 were unsure whether or not they had metastatic disease.
Patients with previous clinical trial exposure (currently or previously enrolled in clinical trials) demonstrated significantly greater trial knowledge, with a mean knowledge score of 9.3 (CI 8.5-10.0) compared with 7.7 (CI 7.3-8.1) among patients without trial exposure (P=0.002; FIGURE 3A). Similarly, patients with trial experience also had statistically significant more positive attitudes towards trials as compared with patients with no trial experience, with a mean attitude score of 3.8 (CI 3.6-4.0) and 3.5 (CI 3.4-3.6), respectively (P=0.001; FIGURE 3B). While numerically patients with trial experience have greater perceived ability compared with patients with no trial experience, with a mean score of 4.4 (CI 4.2-4.6) and 4.2 (CI 4.1-4.3), respectively, this difference did not reach statistical significance (FIGURE 3C).
Knowledge, attitudes, perceived ability, and disease stage
An analysis was performed comparing patients with metastatic vs non-metastatic disease. It was observed that patients with metastatic disease had similar knowledge of trials compared with non-metastatic patients, with a mean knowledge score of 8.4 (CI 7.7- 9.1) and 7.9 (CI 7.5-8.4), respectively, (P=0.3; FIGURE 4A). In contrast, patients with metastatic disease had more positive attitudes compared with non-metastatic patients, with a mean score of 3.7 (CI 3.5-3.8) and 3.5 (CI 3.4-3.6), respectively, which was statistically significant (P=0.03; FIGURE 4B). There was no difference in perceived ability in metastatic vs non-metastatic patients (FIGURE 4C).
Thoughts about molecular profiling
Of the total number of patients, 46 patients had heard of molecular profiling and were presented with questions regarding their thoughts (TABLE 3). Approximately two-thirds (65.2%) thought there would be a 50% or greater likelihood of finding a targetable result in their tumor molecular profile. A majority (71.7%) of patients thought that a new experimental therapy chosen based on a patient’s tumor molecular profile would have at least a 50% chance of controlling the cancer. Somewhat less than a third (30.4%) of patients thought that total cure is the maximal benefit a patient could experience as a result of a treatment on a clinical trial using a drug chosen based on molecular tests. About half (52.2%) of patients agreed with the statement, “I am concerned about the cost of the test to molecularly profile my cancer.”
Considerations to undergo molecular profiling
Eighteen patients had undergone molecular profiling of their tumor. These patients were posed the question, “To what extent did you consider the following issues or concerns at the time you decided to get molecular testing of your cancer?” (TABLE 4). A majority (83.3%) of patients stated that wanting to live as long as possible was important, and 72.2% of patients stated that quality of life was important. A majority (83.3%) of patients stated that hope for a cure was an extremely or quite a bit important consideration. Helping future cancer patients was extremely or quite a bit important for 77.8% of patients, while wanting to be a part of research was not at all or of little importance in 50.0% of patients.
Clinical trials at NM
Twenty-four clinical trials were available at NM between the years 2012 and 2017 for patients with bone and soft tissue sarcoma. Of these trials, 3 of 24 were for non-metastatic patients, while the remaining 21 were open only to metastatic patients. The median number of patients screened per trial was 11 (range 0-66) and the median number of patients accrued per trial was 9 (range 0-58). Of the 24 trials, 17 were not subtype specific (13 included soft tissue sarcoma alone while 4 included both bone and soft tissue sarcoma). The remaining 7 trials were sarcoma subtype specific (eg, angiosarcoma, liposarcoma, etc). Trials available at NM during this period are included in TABLE 5. There were 318 patients screened and 262 patients accrued to sarcoma trials over this time period, with a screen failure rate of 17.6% overall.
Discussion
Our study sought to describe perceptions of clinical trial enrollment among patients with bone and soft tissue sarcoma in order to elucidate and overcome barriers to enrollment, which to the best of our knowledge had not been previously described. Using previously validated patient- reported outcomes in the literature,6 our data reveal a correlation between knowledge of trials and more positive attitudes towards trials. This underscores the importance of awareness and educational strategies in this cancer population as a whole. Interventions should focus on patient perceptions that contribute to lack of participation, such as fear of side effects, loss of control (eg, idea of placebo or randomization), logistical challenges (eg, additional time or convenient location), and cost.3,5,7,16,17 For example, patients concerned about randomization should be educated on equipoise and other ethical considerations in trial design.5 Previous research has suggested that a multimedia psychoeducational intervention was effective in improving attitudes toward trials.6 Educating patients on the essential role of trials in oncology care, as demonstrated by the vast number of new drug approvals in recent years, is an essential strategy to improve attitudes, and subsequently leads to higher patient accrual rates.
In our study, both knowledge and at titudes were increased in patients with previous trial exposure. This suggests that either patients with greater knowledge and more positive attitudes are more likely to enroll in trials, or that patients with direct trial exposure are more knowledgeable and develop more positive attitudes. Our patient population was overall receptive to learning more about clinical trials (85.4%) and willing to participate (65.8%). At the same time, our study demonstrated low to medium correlations between attitudes and perceived ability to take steps towards making an informed decision to enroll in a trial (r=0.4), which may partially be explained by the absence of a tangible trial opportunity. While we did not assess specifically whether patients in our study were offered a trial, there was a substantial trial menu at NM with limited screen failures and decent trial accrual over the time frame of our study. This underscores the importance not only of patient-focused strategies to increase educational and attitudinal resources, but also a need to focus on research-site optimization that includes opening of multiple trials in various settings, systematic pre-screening of patients, and eligibility criteria that are inclusive and rational.5
The patient population with metastatic disease demonstrated more positive attitudes towards enrolling in clinical trials. This cohort accrued well to clinical trials, with 21 of 24 trials enrolling specifically patients with metastatic disease. Of the patients who responded to our survey, patients with previous trial exposure were enriched for patients with metastatic disease (53.3% metastatic among previous trial exposure versus 26.8% metastatic overall). These observations are likely reflective of the need for novel therapies in this disease setting. At the same time, approximately 25% of patients with localized soft tissue sarcoma will develop distant metastatic disease after successful treatment of their primary tumor, which increases to 40% to 50% in larger and higher-grade tumors.18 Three of 24 trials were open for patients with non-metastatic disease, of which one managed to accrue patients. Patients with non-metastatic disease had more negative attitudes towards trial enrollment. These disproportionate findings suggest a need for interventions to increase patient awareness and attitudes towards trial enrollment among this patient population and the importance of research-site optimization for trial opportunities across disease states.
Molecular profiling of tumors and biomarker identification has become a critical component of further characterizing cancer subtypes. In our study, a majority (65.2%) thought there would be a 50% or greater likelihood of finding a targetable result in their molecular profile. Molecular data on 5,749 bone and soft tissue sarcomas suggested that 9.5% of tumors demonstrate a “targetable result,” defined as a new molecular finding for which there is an FDA-approved drug for malignancies other than sarcoma (eg, BRAF V600E, Her2, etc.),19 suggesting an overestimation in our patient cohort of the likelihood of benefit of molecular profiling. These results highlight that the growing use of molecular profiling has increased the need for educational and supportive resources to help patients understand the utility of molecular profiling and aid in shared decision-making surrounding the results.
At the same time, the importance of identifying a targetable mutation in patients with sarcoma cannot be understated. As a recent example of this paradigm, tumors that harbor fusions with the neurotrophic receptor tyrosine kinase 1, 2, or 3 (NTRK1, 2, and 3) have a high response rate (~75%) to drugs that target these fusions, such as larotrectinib and entrectinib.13 Molecular profiling and identification of predictive biomarkers in small patient subsets has led to great challenges in trial design and research-site optimization. Novel designs that incorporate molecular profiling,20 such as the Lung- MAP trial21 and NCI’s Molecular Analysis for Therapy Choice (MATCH) trial,22 are emerging to identify new therapies for small patient subsets. As a rare and increasingly heterogeneous cancer, sarcoma represents a paradigm to provide insight into optimizing patient perceptions and research enterprises to maximize clinical trial enrollment.
Some limitations of our study include a homogeneous and selected patient population that was predominantly Caucasian and highly educated. Therefore, these findings should not be extrapolated to other populations with barriers to trial accrual, such as lower socioeconomic or minority populations. The low response rate and failure of some to complete the survey may have introduced some bias. Additionally, our data include self-reported outcomes, which could have affected our results. Finally, the limited number of patients who had undergone or heard of molecular profiling limited our ability to draw definitive conclusions, and should be assessed in larger patient cohorts.
While our paper addresses a unique population—the sarcoma patient—similar themes and issues pertain to all oncology patients. A recent review was published in the American Society of Clinical Oncology Educational Book23 looking at methods to overcome barriers to clinical trial enrollment. Their paper clearly illustrates mechanisms to assist with overcoming financial burdens associated with cancer clinical trials, overcoming barriers as they relate to patient and clinician difficulty in coping with the uncertainty inherent in clinical trial participation, and highlight the role of a patient navigator in clinical trial participation.
Conclusions
Interventions aimed at increasing awareness, knowledge, and attitudes towards clinical trials among sarcoma patients may lead to increased trial enrollment and greater progress in cancer treatment in this population. In addition to patient- focused interventions, thoughtful and strategic clinical trial designs that allow for the development of biomarker- driven therapeutics, while at the same time optimizing patient accrual rates, should be developed. Evaluation of barriers to clinical trial enrollment and molecular profiling of tumors among bone and soft tissue sarcoma patients at an academic center can serve as a paradigm to overcome barriers to enrollment in the era of an increasingly heterogeneous cancer population. TSJ
1. Go RS, Frisby KA, Lee JA, et al. Clinical trial accrual among new cancer patients at a community-based cancer center. Cancer. 2006;106(2):426-433.
2. Murthy VH, Krumholz HM, Gross CP. Participation in cancer clinical trials: Race-, sex-, and age-based disparities. JAMA. 2004; 291(22):2720-2726.
3. Lara PN Jr, Higdon R, Lim N, et al. Prospective evaluation of cancer clinical trial accrual patterns: identifying potential barriers to enrollment. J Clin Oncol. 2001;19(6):1728-1733.
4. Unger JM, Cook E, Tai E, Bleyer A. The role of clinical trial participation in cancer research: barriers, evidence, and strategies. Am Soc Clin Oncol Educ Book. 2016;35:185-198.
5. Cancer Action Network American Cancer Society. Barriers to patient enrollment in therapeutic clinical trials for cancer. 2018: https:// www.fightcancer.org/policy-resources/clinical- trial-barriers#figures. Accessed March 14, 2019.
6. Jacobsen PB, Wells KJ, Meade CD, et al. Effects of a brief multimedia psychoeducational intervention on the attitudes and interest of patients with cancer regarding clinical trial participation: a multicenter randomized controlled trial. J Clin Oncol. 2012;30(20):2516-2521.
7. Meropol NJ, Buzaglo JS, Millard J, et al. Barriers to clinical trial participation as perceived by oncologists and patients. J Natl Compr Canc Netw. 2007;5(8):655-664.
8. Cox K, McGarry J. Why patients don’t take part in cancer clinical trials: an overview of the literature. Eur J Cancer Care (Engl). 2003;12(2): 114-122.
9. Mills EJ, Seely D, Rachlis B, et al. Barriers to participation in clinical trials of cancer: a meta- analysis and systematic review of patient-reported factors. Lancet Oncol. 2006;7(2):141-148.
10. National Comprehensive Cancer Network. Soft Tissue Sarcoma (Version 4.2019). https:// www.nccn.org/professionals/physician_gls/ pdf/sarcoma.pdf. Accessed October 30, 2019.
11. Nass SJ, Beaupin LK, Demark-Wahnefried W, et al. Identifying and addressing the needs of adolescents and young adults with cancer: summary of an Institute of Medicine workshop. Oncologist. 2015;20(2):186-195.
12. Wilky BA, Villalobos VM. Emerging role for precision therapy through next-generation sequencing for sarcomas. JCO Precision Oncol. 2018(2):1-4.
13. Bleyer A, Montello M, Budd T, Saxman S. National survival trends of young adults with sarcoma: lack of progress is associated with lack of clinical trial participation. Cancer. 2005;103(9):1891-1897.
14. Gornick MC, Cobain E, Le LQ, et al. Oncologists’ use of genomic sequencing data to inform clinical management. JCO Precision Oncol. 2018(2):1-13.
15. Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2nd ed. Hillsdale, NJ: L. Erlbaum Associates; 1988.
16. Unger JM, Hershman DL, Albain KS, et al. Patient income level and cancer clinical trial participation. J Clin Oncol. 2013;31(5):536- 542.
17. Javid SH, Unger JM, Gralow JR, et al. A prospective analysis of the influence of older age on physician and patient decision-making when considering enrollment in breast cancer clinical trials (SWOG S0316). Oncologist. 2012;17(9):1180-1190.
18. Singhi EK, Moore DC, Muslimani A. Metastatic soft tissue sarcomas: a review of treatment and new pharmacotherapies. P T. 2018;43(7): 410-429.
19. Gounder MM, Ali SM, Robinson V, et al. Impact of next-generation sequencing (NGS) on diagnostic and therapeutic options in soft-tissue and bone sarcoma. J Clin Oncol. 2017;35(15_suppl):abstr 11001.
20. Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med. 2017;377(1):62-70.
21. Steuer CE, Papadimitrakopoulou V, Herbst RS, et al. Innovative clinical trials: the LUNGMAP study. Clin Pharmacol Ther. 2015;97(5): 488-491.
22. McNeil C. NCI-MATCH launch highlights new trial design in precision-medicine era. J Natl Cancer Inst. 2015;107(7).
23. Nipp RD, Hong K, Paskett ED. Overcoming barriers to clinical trial enrollment. Am Soc Clin Oncol Educ Book. 2019;39:105-114.
1. Go RS, Frisby KA, Lee JA, et al. Clinical trial accrual among new cancer patients at a community-based cancer center. Cancer. 2006;106(2):426-433.
2. Murthy VH, Krumholz HM, Gross CP. Participation in cancer clinical trials: Race-, sex-, and age-based disparities. JAMA. 2004; 291(22):2720-2726.
3. Lara PN Jr, Higdon R, Lim N, et al. Prospective evaluation of cancer clinical trial accrual patterns: identifying potential barriers to enrollment. J Clin Oncol. 2001;19(6):1728-1733.
4. Unger JM, Cook E, Tai E, Bleyer A. The role of clinical trial participation in cancer research: barriers, evidence, and strategies. Am Soc Clin Oncol Educ Book. 2016;35:185-198.
5. Cancer Action Network American Cancer Society. Barriers to patient enrollment in therapeutic clinical trials for cancer. 2018: https:// www.fightcancer.org/policy-resources/clinical- trial-barriers#figures. Accessed March 14, 2019.
6. Jacobsen PB, Wells KJ, Meade CD, et al. Effects of a brief multimedia psychoeducational intervention on the attitudes and interest of patients with cancer regarding clinical trial participation: a multicenter randomized controlled trial. J Clin Oncol. 2012;30(20):2516-2521.
7. Meropol NJ, Buzaglo JS, Millard J, et al. Barriers to clinical trial participation as perceived by oncologists and patients. J Natl Compr Canc Netw. 2007;5(8):655-664.
8. Cox K, McGarry J. Why patients don’t take part in cancer clinical trials: an overview of the literature. Eur J Cancer Care (Engl). 2003;12(2): 114-122.
9. Mills EJ, Seely D, Rachlis B, et al. Barriers to participation in clinical trials of cancer: a meta- analysis and systematic review of patient-reported factors. Lancet Oncol. 2006;7(2):141-148.
10. National Comprehensive Cancer Network. Soft Tissue Sarcoma (Version 4.2019). https:// www.nccn.org/professionals/physician_gls/ pdf/sarcoma.pdf. Accessed October 30, 2019.
11. Nass SJ, Beaupin LK, Demark-Wahnefried W, et al. Identifying and addressing the needs of adolescents and young adults with cancer: summary of an Institute of Medicine workshop. Oncologist. 2015;20(2):186-195.
12. Wilky BA, Villalobos VM. Emerging role for precision therapy through next-generation sequencing for sarcomas. JCO Precision Oncol. 2018(2):1-4.
13. Bleyer A, Montello M, Budd T, Saxman S. National survival trends of young adults with sarcoma: lack of progress is associated with lack of clinical trial participation. Cancer. 2005;103(9):1891-1897.
14. Gornick MC, Cobain E, Le LQ, et al. Oncologists’ use of genomic sequencing data to inform clinical management. JCO Precision Oncol. 2018(2):1-13.
15. Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2nd ed. Hillsdale, NJ: L. Erlbaum Associates; 1988.
16. Unger JM, Hershman DL, Albain KS, et al. Patient income level and cancer clinical trial participation. J Clin Oncol. 2013;31(5):536- 542.
17. Javid SH, Unger JM, Gralow JR, et al. A prospective analysis of the influence of older age on physician and patient decision-making when considering enrollment in breast cancer clinical trials (SWOG S0316). Oncologist. 2012;17(9):1180-1190.
18. Singhi EK, Moore DC, Muslimani A. Metastatic soft tissue sarcomas: a review of treatment and new pharmacotherapies. P T. 2018;43(7): 410-429.
19. Gounder MM, Ali SM, Robinson V, et al. Impact of next-generation sequencing (NGS) on diagnostic and therapeutic options in soft-tissue and bone sarcoma. J Clin Oncol. 2017;35(15_suppl):abstr 11001.
20. Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med. 2017;377(1):62-70.
21. Steuer CE, Papadimitrakopoulou V, Herbst RS, et al. Innovative clinical trials: the LUNGMAP study. Clin Pharmacol Ther. 2015;97(5): 488-491.
22. McNeil C. NCI-MATCH launch highlights new trial design in precision-medicine era. J Natl Cancer Inst. 2015;107(7).
23. Nipp RD, Hong K, Paskett ED. Overcoming barriers to clinical trial enrollment. Am Soc Clin Oncol Educ Book. 2019;39:105-114.