User login
Sorrows hidden in plain sight
“The greatest hazard of all, losing one’s self, can occur very quietly in the world, as if it were nothing at all. No other loss can occur so quietly”
—Søren Kierkegaard
In our interactions with patients we often focus on the medical issue at hand, forgetting the other dimensions of our patients’ lives. Yet these invisible dimensions can have a huge impact on their humanity. I humbly submit that it can be profoundly meaningful, for them and for us, if we paid close attention.

1. JV has rheumatoid arthritis. He is not the most compliant, and I had not seen him in over a year. He was well controlled on Enbrel.
He came to the office one day last week to make an appointment. We gave him one for the next day, but he missed the appointment. When he finally came a week later, I got the real story. He had gotten arrested.
The day he came into my office to ask for an appointment after a prolonged absence, it was because his insurance was denying coverage for his Enbrel. His next stop, after making his appointment, was to the Social Security office to try to sort out his coverage. When asked about whether or not he needed special accommodations, he made a point to mention his hearing loss. However, the person ultimately assigned to help him was not a native English speaker. This made for a very confusing and loud exchange that led to a heated argument with security. It ended with him being led out of the office in handcuffs, and spending the night in jail.
2. Tyler has inflammatory arthritis. He received his diagnosis in Arizona 5 years ago. He was stable on Plaquenil when I met him a year ago, and I only see him every 6 months.
On our third visit, I noticed he had gained weight. He attributed this to the discontinuation of his Dexedrine, in the interest of adjusting his medications for bipolar disorder. I couldn’t remember our second visit very well, but he apologized profusely for having been “lit.” He apparently was so manic that it drove his family crazy. In an effort to get him to take medications for the problem, his sister said: “It’s like living with Vincent van Gogh, except now we have meds for it.”
Now he takes his medications. As a result, his body feels foreign. In the summer, he ran around the backyard all day and night constructing an obstacle course for his dog that he knew he would never get right. Now he is lucky if he gets a few days a week of productive work on his art. He feels dulled; he is not as quick-witted. It is sad, but, he says, this is what society expects of him.
3. Judy is in her mid-70s. That’s not old these days, but she is somewhat crippled by her rheumatoid arthritis. She lived in Manhattan with her husband and enjoyed the city immensely. After her husband passed away, she managed to live in her Manhattan apartment independently for a little bit, but after suffering a fall, she let her children move her to Rhode Island to be closer to them. She has struggled to find her own apartment with amenities that will allow her to remain independent. For the past 3 years she has been in an assisted living facility.
She is so unhappy there. She doesn’t like forced interactions, doesn’t like to gossip, doesn’t like when her neighbors behave like they’re in middle school. It has taken 3 years for the people around her to finally respect her desire to be left alone, to spend her time reading and listening to the opera rather than engaging in idle chitchat that does not interest her at all.
Dr. Chan practices rheumatology in Pawtucket, R.I.
“The greatest hazard of all, losing one’s self, can occur very quietly in the world, as if it were nothing at all. No other loss can occur so quietly”
—Søren Kierkegaard
In our interactions with patients we often focus on the medical issue at hand, forgetting the other dimensions of our patients’ lives. Yet these invisible dimensions can have a huge impact on their humanity. I humbly submit that it can be profoundly meaningful, for them and for us, if we paid close attention.
1. JV has rheumatoid arthritis. He is not the most compliant, and I had not seen him in over a year. He was well controlled on Enbrel.
He came to the office one day last week to make an appointment. We gave him one for the next day, but he missed the appointment. When he finally came a week later, I got the real story. He had gotten arrested.
The day he came into my office to ask for an appointment after a prolonged absence, it was because his insurance was denying coverage for his Enbrel. His next stop, after making his appointment, was to the Social Security office to try to sort out his coverage. When asked about whether or not he needed special accommodations, he made a point to mention his hearing loss. However, the person ultimately assigned to help him was not a native English speaker. This made for a very confusing and loud exchange that led to a heated argument with security. It ended with him being led out of the office in handcuffs, and spending the night in jail.
2. Tyler has inflammatory arthritis. He received his diagnosis in Arizona 5 years ago. He was stable on Plaquenil when I met him a year ago, and I only see him every 6 months.
On our third visit, I noticed he had gained weight. He attributed this to the discontinuation of his Dexedrine, in the interest of adjusting his medications for bipolar disorder. I couldn’t remember our second visit very well, but he apologized profusely for having been “lit.” He apparently was so manic that it drove his family crazy. In an effort to get him to take medications for the problem, his sister said: “It’s like living with Vincent van Gogh, except now we have meds for it.”
Now he takes his medications. As a result, his body feels foreign. In the summer, he ran around the backyard all day and night constructing an obstacle course for his dog that he knew he would never get right. Now he is lucky if he gets a few days a week of productive work on his art. He feels dulled; he is not as quick-witted. It is sad, but, he says, this is what society expects of him.
3. Judy is in her mid-70s. That’s not old these days, but she is somewhat crippled by her rheumatoid arthritis. She lived in Manhattan with her husband and enjoyed the city immensely. After her husband passed away, she managed to live in her Manhattan apartment independently for a little bit, but after suffering a fall, she let her children move her to Rhode Island to be closer to them. She has struggled to find her own apartment with amenities that will allow her to remain independent. For the past 3 years she has been in an assisted living facility.
She is so unhappy there. She doesn’t like forced interactions, doesn’t like to gossip, doesn’t like when her neighbors behave like they’re in middle school. It has taken 3 years for the people around her to finally respect her desire to be left alone, to spend her time reading and listening to the opera rather than engaging in idle chitchat that does not interest her at all.
Dr. Chan practices rheumatology in Pawtucket, R.I.
“The greatest hazard of all, losing one’s self, can occur very quietly in the world, as if it were nothing at all. No other loss can occur so quietly”
—Søren Kierkegaard
In our interactions with patients we often focus on the medical issue at hand, forgetting the other dimensions of our patients’ lives. Yet these invisible dimensions can have a huge impact on their humanity. I humbly submit that it can be profoundly meaningful, for them and for us, if we paid close attention.
1. JV has rheumatoid arthritis. He is not the most compliant, and I had not seen him in over a year. He was well controlled on Enbrel.
He came to the office one day last week to make an appointment. We gave him one for the next day, but he missed the appointment. When he finally came a week later, I got the real story. He had gotten arrested.
The day he came into my office to ask for an appointment after a prolonged absence, it was because his insurance was denying coverage for his Enbrel. His next stop, after making his appointment, was to the Social Security office to try to sort out his coverage. When asked about whether or not he needed special accommodations, he made a point to mention his hearing loss. However, the person ultimately assigned to help him was not a native English speaker. This made for a very confusing and loud exchange that led to a heated argument with security. It ended with him being led out of the office in handcuffs, and spending the night in jail.
2. Tyler has inflammatory arthritis. He received his diagnosis in Arizona 5 years ago. He was stable on Plaquenil when I met him a year ago, and I only see him every 6 months.
On our third visit, I noticed he had gained weight. He attributed this to the discontinuation of his Dexedrine, in the interest of adjusting his medications for bipolar disorder. I couldn’t remember our second visit very well, but he apologized profusely for having been “lit.” He apparently was so manic that it drove his family crazy. In an effort to get him to take medications for the problem, his sister said: “It’s like living with Vincent van Gogh, except now we have meds for it.”
Now he takes his medications. As a result, his body feels foreign. In the summer, he ran around the backyard all day and night constructing an obstacle course for his dog that he knew he would never get right. Now he is lucky if he gets a few days a week of productive work on his art. He feels dulled; he is not as quick-witted. It is sad, but, he says, this is what society expects of him.
3. Judy is in her mid-70s. That’s not old these days, but she is somewhat crippled by her rheumatoid arthritis. She lived in Manhattan with her husband and enjoyed the city immensely. After her husband passed away, she managed to live in her Manhattan apartment independently for a little bit, but after suffering a fall, she let her children move her to Rhode Island to be closer to them. She has struggled to find her own apartment with amenities that will allow her to remain independent. For the past 3 years she has been in an assisted living facility.
She is so unhappy there. She doesn’t like forced interactions, doesn’t like to gossip, doesn’t like when her neighbors behave like they’re in middle school. It has taken 3 years for the people around her to finally respect her desire to be left alone, to spend her time reading and listening to the opera rather than engaging in idle chitchat that does not interest her at all.
Dr. Chan practices rheumatology in Pawtucket, R.I.

2015 ACR RA guidelines criticized on steroid, HBV screening, cancer history advice
Rheumatologists have criticized what they described as omissions and problems with the 2015 American College of Rheumatology guidelines for treating rheumatoid arthritis.
Although the authors (Arthritis Care Res. 2016;68[1]:1-25) evaluated evidence with GRADE (Grading of Recommendations Assessment, Development, and Evaluation) methods and solicited public feedback on core questions, the final document minimized the use of early low-dose glucocorticoids, failed to address screening for hepatitis B virus (HBV) infection before starting biologics, and underestimated their risks in patients with a history of cancer or lymphoproliferative disease, according to separate editorial letters published in Arthritis Care & Research.
Although the guidelines recommend initial methotrexate monotherapy regardless of disease activity level, “experienced clinicians know that before any effect of the methotrexate is realized, the patient will experience joint pain, swelling, functional impairment, and morning stiffness, making the patient quite miserable,” wrote Dr. Doyt L. Conn of Emory University, Atlanta. Patients will then self-treat with NSAIDs, he added (Arthritis Care Res. 2016 Feb 11. doi: 10.1002/acr.22864). “Do the authors of the 2015 ACR guidelines think that NSAIDs are as effective as and safer than low dose prednisone?” Furthermore, the guidelines seem to rank tumor necrosis factor (TNF) inhibitors and non-TNF biologics above short-term, low-dose prednisone, he wrote. “Do the authors really mean that?’’

In a reply letter (Arthritis Care Res. Feb 11. doi: 10.1002/acr.22862), Dr. Jasvinder A. Singh of the University of Alabama at Birmingham and guideline coauthors denied recommending a “rigid” treatment sequence. They recommend short-term, low-dose steroids any time after initial disease-modifying antirheumatic drug (DMARD) therapy if disease activity remains moderate or high, or during flares, they wrote. “The guideline does not mandate that patients must fail any particular therapy before glucocorticoid can be given. It merely suggests that a DMARD should be tried first.”
Regarding HBV screening, Dr. Leonard H. Calabrese was “disappointed” to see no changes since the 2008 ACR RA guidance (Arthritis Care Res. Feb 11. doi: 10.1002/acr.22865). One biologic, rituximab, now has a black box warning for potentially fatal HBV reactivation, mandating HBV screening before its use, said Dr. Calabrese, of the Cleveland Clinic. “We suggest that the standards of screening for and prevention of HBV reactivation have changed dramatically, and assert that the ACR guidelines are accordingly overdue for changes,” he wrote.
Dr. Singh and colleagues replied that viral hepatitis screening was “vital,” but that “it was not our charge – nor was it feasible – to cover every topic relevant to the care of RA patients in this guideline.” The field lacks data on the cost-effectiveness of universally screening RA patients for HBV before starting biologics, they added. They recommended working with hepatology/gastroenterology providers to manage RA patients with chronic viral hepatitis who need potentially immunosuppressive therapies.

Finally, the guidelines cite relatively small observational studies when addressing the use of biologics in patients with a history of cancer or lymphoproliferative disorders, wrote Dr. Hasan Yazici of Academic Hospital, Istanbul, Turkey, and his associates. Interpreting these studies as negating risk is especially concerning because of “depletion of the susceptibles bias,” when one takes into consideration that one-sixth of patients with cancer have a second or higher order primary cancer; that most national registries do not differentiate between a first primary cancer and second or higher order primary cancer in their cancer incidence rates in the general population; and that RA patients on biologics probably have no cancer history. This leads to skewed results when comparing standardized incidence ratios for cancer in TNF inhibitor users versus the general population, they added (Arthritis Care Res. 2016 Feb 11. doi: 10.1002/acr.22863).
Dr. Singh and colleagues responded that “the 2015 recommendations for RA treatment in persons with past lymphoproliferative disorders or solid cancers, are conditional, not strong.” Because of the lack of evidence establishing risk in these patients, the voting panel recommended treating them the same way as other patients, they added. “The panel recognized, and the readership of these guidelines should also understand, that lack of evidence of difference does not imply an evidence of lack of difference.” They called for better studies of DMARDs and biologics in RA patients with current or historical hepatitis, cancer, heart failure, and serious infections.
Going forward, the ACR is considering how to better publicize its 30-day public comment period when developing future guidelines to better capture perspectives at the beginning, instead of at the end when GRADE protocols preclude further adjustments, Dr. Singh and his associates wrote. In addition, the ACR will broaden its RA guidelines, post drafts on its website for comment, and use a “dynamic” process “to ensure timeliness, more frequent updating, online updates, and approaches to harmonize with recommendations of other professional organizations, as needed.”
None of these authors reported funding sources or conflicts of interest.
Rheumatologists have criticized what they described as omissions and problems with the 2015 American College of Rheumatology guidelines for treating rheumatoid arthritis.
Although the authors (Arthritis Care Res. 2016;68[1]:1-25) evaluated evidence with GRADE (Grading of Recommendations Assessment, Development, and Evaluation) methods and solicited public feedback on core questions, the final document minimized the use of early low-dose glucocorticoids, failed to address screening for hepatitis B virus (HBV) infection before starting biologics, and underestimated their risks in patients with a history of cancer or lymphoproliferative disease, according to separate editorial letters published in Arthritis Care & Research.
Although the guidelines recommend initial methotrexate monotherapy regardless of disease activity level, “experienced clinicians know that before any effect of the methotrexate is realized, the patient will experience joint pain, swelling, functional impairment, and morning stiffness, making the patient quite miserable,” wrote Dr. Doyt L. Conn of Emory University, Atlanta. Patients will then self-treat with NSAIDs, he added (Arthritis Care Res. 2016 Feb 11. doi: 10.1002/acr.22864). “Do the authors of the 2015 ACR guidelines think that NSAIDs are as effective as and safer than low dose prednisone?” Furthermore, the guidelines seem to rank tumor necrosis factor (TNF) inhibitors and non-TNF biologics above short-term, low-dose prednisone, he wrote. “Do the authors really mean that?’’
In a reply letter (Arthritis Care Res. Feb 11. doi: 10.1002/acr.22862), Dr. Jasvinder A. Singh of the University of Alabama at Birmingham and guideline coauthors denied recommending a “rigid” treatment sequence. They recommend short-term, low-dose steroids any time after initial disease-modifying antirheumatic drug (DMARD) therapy if disease activity remains moderate or high, or during flares, they wrote. “The guideline does not mandate that patients must fail any particular therapy before glucocorticoid can be given. It merely suggests that a DMARD should be tried first.”
Regarding HBV screening, Dr. Leonard H. Calabrese was “disappointed” to see no changes since the 2008 ACR RA guidance (Arthritis Care Res. Feb 11. doi: 10.1002/acr.22865). One biologic, rituximab, now has a black box warning for potentially fatal HBV reactivation, mandating HBV screening before its use, said Dr. Calabrese, of the Cleveland Clinic. “We suggest that the standards of screening for and prevention of HBV reactivation have changed dramatically, and assert that the ACR guidelines are accordingly overdue for changes,” he wrote.
Dr. Singh and colleagues replied that viral hepatitis screening was “vital,” but that “it was not our charge – nor was it feasible – to cover every topic relevant to the care of RA patients in this guideline.” The field lacks data on the cost-effectiveness of universally screening RA patients for HBV before starting biologics, they added. They recommended working with hepatology/gastroenterology providers to manage RA patients with chronic viral hepatitis who need potentially immunosuppressive therapies.
Finally, the guidelines cite relatively small observational studies when addressing the use of biologics in patients with a history of cancer or lymphoproliferative disorders, wrote Dr. Hasan Yazici of Academic Hospital, Istanbul, Turkey, and his associates. Interpreting these studies as negating risk is especially concerning because of “depletion of the susceptibles bias,” when one takes into consideration that one-sixth of patients with cancer have a second or higher order primary cancer; that most national registries do not differentiate between a first primary cancer and second or higher order primary cancer in their cancer incidence rates in the general population; and that RA patients on biologics probably have no cancer history. This leads to skewed results when comparing standardized incidence ratios for cancer in TNF inhibitor users versus the general population, they added (Arthritis Care Res. 2016 Feb 11. doi: 10.1002/acr.22863).
Dr. Singh and colleagues responded that “the 2015 recommendations for RA treatment in persons with past lymphoproliferative disorders or solid cancers, are conditional, not strong.” Because of the lack of evidence establishing risk in these patients, the voting panel recommended treating them the same way as other patients, they added. “The panel recognized, and the readership of these guidelines should also understand, that lack of evidence of difference does not imply an evidence of lack of difference.” They called for better studies of DMARDs and biologics in RA patients with current or historical hepatitis, cancer, heart failure, and serious infections.
Going forward, the ACR is considering how to better publicize its 30-day public comment period when developing future guidelines to better capture perspectives at the beginning, instead of at the end when GRADE protocols preclude further adjustments, Dr. Singh and his associates wrote. In addition, the ACR will broaden its RA guidelines, post drafts on its website for comment, and use a “dynamic” process “to ensure timeliness, more frequent updating, online updates, and approaches to harmonize with recommendations of other professional organizations, as needed.”
None of these authors reported funding sources or conflicts of interest.
Rheumatologists have criticized what they described as omissions and problems with the 2015 American College of Rheumatology guidelines for treating rheumatoid arthritis.
Although the authors (Arthritis Care Res. 2016;68[1]:1-25) evaluated evidence with GRADE (Grading of Recommendations Assessment, Development, and Evaluation) methods and solicited public feedback on core questions, the final document minimized the use of early low-dose glucocorticoids, failed to address screening for hepatitis B virus (HBV) infection before starting biologics, and underestimated their risks in patients with a history of cancer or lymphoproliferative disease, according to separate editorial letters published in Arthritis Care & Research.
Although the guidelines recommend initial methotrexate monotherapy regardless of disease activity level, “experienced clinicians know that before any effect of the methotrexate is realized, the patient will experience joint pain, swelling, functional impairment, and morning stiffness, making the patient quite miserable,” wrote Dr. Doyt L. Conn of Emory University, Atlanta. Patients will then self-treat with NSAIDs, he added (Arthritis Care Res. 2016 Feb 11. doi: 10.1002/acr.22864). “Do the authors of the 2015 ACR guidelines think that NSAIDs are as effective as and safer than low dose prednisone?” Furthermore, the guidelines seem to rank tumor necrosis factor (TNF) inhibitors and non-TNF biologics above short-term, low-dose prednisone, he wrote. “Do the authors really mean that?’’
In a reply letter (Arthritis Care Res. Feb 11. doi: 10.1002/acr.22862), Dr. Jasvinder A. Singh of the University of Alabama at Birmingham and guideline coauthors denied recommending a “rigid” treatment sequence. They recommend short-term, low-dose steroids any time after initial disease-modifying antirheumatic drug (DMARD) therapy if disease activity remains moderate or high, or during flares, they wrote. “The guideline does not mandate that patients must fail any particular therapy before glucocorticoid can be given. It merely suggests that a DMARD should be tried first.”
Regarding HBV screening, Dr. Leonard H. Calabrese was “disappointed” to see no changes since the 2008 ACR RA guidance (Arthritis Care Res. Feb 11. doi: 10.1002/acr.22865). One biologic, rituximab, now has a black box warning for potentially fatal HBV reactivation, mandating HBV screening before its use, said Dr. Calabrese, of the Cleveland Clinic. “We suggest that the standards of screening for and prevention of HBV reactivation have changed dramatically, and assert that the ACR guidelines are accordingly overdue for changes,” he wrote.
Dr. Singh and colleagues replied that viral hepatitis screening was “vital,” but that “it was not our charge – nor was it feasible – to cover every topic relevant to the care of RA patients in this guideline.” The field lacks data on the cost-effectiveness of universally screening RA patients for HBV before starting biologics, they added. They recommended working with hepatology/gastroenterology providers to manage RA patients with chronic viral hepatitis who need potentially immunosuppressive therapies.
Finally, the guidelines cite relatively small observational studies when addressing the use of biologics in patients with a history of cancer or lymphoproliferative disorders, wrote Dr. Hasan Yazici of Academic Hospital, Istanbul, Turkey, and his associates. Interpreting these studies as negating risk is especially concerning because of “depletion of the susceptibles bias,” when one takes into consideration that one-sixth of patients with cancer have a second or higher order primary cancer; that most national registries do not differentiate between a first primary cancer and second or higher order primary cancer in their cancer incidence rates in the general population; and that RA patients on biologics probably have no cancer history. This leads to skewed results when comparing standardized incidence ratios for cancer in TNF inhibitor users versus the general population, they added (Arthritis Care Res. 2016 Feb 11. doi: 10.1002/acr.22863).
Dr. Singh and colleagues responded that “the 2015 recommendations for RA treatment in persons with past lymphoproliferative disorders or solid cancers, are conditional, not strong.” Because of the lack of evidence establishing risk in these patients, the voting panel recommended treating them the same way as other patients, they added. “The panel recognized, and the readership of these guidelines should also understand, that lack of evidence of difference does not imply an evidence of lack of difference.” They called for better studies of DMARDs and biologics in RA patients with current or historical hepatitis, cancer, heart failure, and serious infections.
Going forward, the ACR is considering how to better publicize its 30-day public comment period when developing future guidelines to better capture perspectives at the beginning, instead of at the end when GRADE protocols preclude further adjustments, Dr. Singh and his associates wrote. In addition, the ACR will broaden its RA guidelines, post drafts on its website for comment, and use a “dynamic” process “to ensure timeliness, more frequent updating, online updates, and approaches to harmonize with recommendations of other professional organizations, as needed.”
None of these authors reported funding sources or conflicts of interest.

Once-daily tofacitinib formulation gets FDA clearance for RA
The Food and Drug Administration has approved a once-daily, extended-release tablet formulation of tofacitinib 11 mg, the drug’s manufacturer, Pfizer, announced.
The Janus kinase inhibitor, to be marketed as Xeljanz XR, is indicated for the treatment of moderate to severe rheumatoid arthritis (RA) in patients who have had an inadequate response or intolerance to methotrexate, but it can be used with methotrexate.

“The availability of Xeljanz XR provides physicians with a new treatment option for people with RA who may prefer an oral once-daily treatment,” Dr. Roy Fleischmann, clinical professor in the department of internal medicine at the University of Texas Southwestern Medical Center, Dallas, said in Pfizer’s announcement.
The original 5-mg tofacitinib tablet, approved in 2012, is taken twice daily.
According to Pfizer, the global clinical development program for tofacitinib has evaluated its safety and efficacy in approximately 6,200 patients with moderate to severe RA, amounting to more than 19,400 patient-years of drug exposure.
The new labeling can be found here.
The Food and Drug Administration has approved a once-daily, extended-release tablet formulation of tofacitinib 11 mg, the drug’s manufacturer, Pfizer, announced.
The Janus kinase inhibitor, to be marketed as Xeljanz XR, is indicated for the treatment of moderate to severe rheumatoid arthritis (RA) in patients who have had an inadequate response or intolerance to methotrexate, but it can be used with methotrexate.
“The availability of Xeljanz XR provides physicians with a new treatment option for people with RA who may prefer an oral once-daily treatment,” Dr. Roy Fleischmann, clinical professor in the department of internal medicine at the University of Texas Southwestern Medical Center, Dallas, said in Pfizer’s announcement.
The original 5-mg tofacitinib tablet, approved in 2012, is taken twice daily.
According to Pfizer, the global clinical development program for tofacitinib has evaluated its safety and efficacy in approximately 6,200 patients with moderate to severe RA, amounting to more than 19,400 patient-years of drug exposure.
The new labeling can be found here.
The Food and Drug Administration has approved a once-daily, extended-release tablet formulation of tofacitinib 11 mg, the drug’s manufacturer, Pfizer, announced.
The Janus kinase inhibitor, to be marketed as Xeljanz XR, is indicated for the treatment of moderate to severe rheumatoid arthritis (RA) in patients who have had an inadequate response or intolerance to methotrexate, but it can be used with methotrexate.
“The availability of Xeljanz XR provides physicians with a new treatment option for people with RA who may prefer an oral once-daily treatment,” Dr. Roy Fleischmann, clinical professor in the department of internal medicine at the University of Texas Southwestern Medical Center, Dallas, said in Pfizer’s announcement.
The original 5-mg tofacitinib tablet, approved in 2012, is taken twice daily.
According to Pfizer, the global clinical development program for tofacitinib has evaluated its safety and efficacy in approximately 6,200 patients with moderate to severe RA, amounting to more than 19,400 patient-years of drug exposure.
The new labeling can be found here.

Ultrasound could help identify patients suitable for biologic tapering
Ultrasound evaluation at the time of clinical remission could be a useful tool to select the most appropriate rheumatoid arthritis (RA) patients to undergo biologic therapy tapering and discontinuation, Italian researchers say.
In a study involving 42 RA patients in clinical remission who tapered their anti–tumor necrosis factor–alpha (anti-TNF-alpha) therapy according to ultrasound criteria, 69.1 % maintained remission at 12 weeks.
Furthermore, 26 of the patients (89.7 %) maintained disease remission after 6 months of follow-up, reported the research team led by Dr. Gianfranco Ferraccioli and Dr. Stefano Alivernini of the Institute of Rheumatology, Catholic University of the Sacred Heart, Rome (Arthritis Res Ther. 2016. doi: 10.1186/s13075-016-0927-z).
The 30% of patients who relapsed (n = 13) were retreated and reached a good European League Against Rheumatism (EULAR) response within 3 months, results from the observational study showed.
According to the researchers, the daily management of patients receiving long-term biologic treatment remains a matter of debate, and it is currently unclear how to select the most appropriate patients for discontinuing biologic treatment.
People with RA, even when in remission, tend to have residual synovitis. Previous research had shown that patients with negative signaling detected on power Doppler (PD) ultrasound were less likely to have disease flares.
To determine if the detection of residual synovitis with PD signaling could help in selecting patients suitable for anti-TNF discontinuation, the researchers selected 42 RA patients with disease duration of more than 12 months who were in sustained remission (Disease Activity Score less than 1.6 at three visits 3 months apart) who were receiving anti-TNF-alpha treatment plus methotrexate.
Patients were first tapered on anti-TNF-alpha therapy (adalimumab 40 mg/4 weeks or etanercept 50 mg/2 weeks).
Each patient underwent ultrasound evaluation of synovial hypertrophy (SH) and PD signal presence in the second and third metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints, the wrist (radiocarpal-intercarpal), bilateral knee, and second to fifth metatarsophalangeal (MTP) joints.
After 3 months, patients with no power Doppler signaling on ultrasound discontinued anti-TNF-alpha therapy and were followed every 3 months while maintaining stable doses of methotrexate.
Disease flares after anti-TNF-alpha discontinuation occurred in the joints with higher SH scores that were clinically involved at disease onset, despite the fact that no SH cut-off discriminated patients who relapsed from those who did not.
In particular, the fifth MTP joint was informative (in both the tapering and discontinuation groups) and the second MCP joint was informative for the tapering group only.
“This finding suggests the possible utility of following US [ultrasound] with indices of joints initially involved at disease onset with higher likelihood of relapse,” the researchers said.
Results from subgroup of five patients who also underwent ultrasound-guided knee synovial tissue biopsy to assess histologic features of residual synovitis revealed that the absence of ultrasound activity was associated with almost normal findings at the synovial level, they reported.
Overall, the findings suggested there was a “meaningful, large patient population with established RA in remission for whom the anti-TNF-alpha dose can be decreased without clinical and functional worsening,” the researchers wrote.
They suggested the combination of PD-US evaluation and American College of Rheumatology/EULAR remission criteria could help identify patients on biologics who are likely to achieve drug-free remission.
Use of three sequential ultrasound evaluations might identify an even higher proportion of patients likely to reach persistent drug-free remission, compared with current clinical methods of disease activity assessment, they added.
Ultrasound evaluation at the time of clinical remission could be a useful tool to select the most appropriate rheumatoid arthritis (RA) patients to undergo biologic therapy tapering and discontinuation, Italian researchers say.
In a study involving 42 RA patients in clinical remission who tapered their anti–tumor necrosis factor–alpha (anti-TNF-alpha) therapy according to ultrasound criteria, 69.1 % maintained remission at 12 weeks.
Furthermore, 26 of the patients (89.7 %) maintained disease remission after 6 months of follow-up, reported the research team led by Dr. Gianfranco Ferraccioli and Dr. Stefano Alivernini of the Institute of Rheumatology, Catholic University of the Sacred Heart, Rome (Arthritis Res Ther. 2016. doi: 10.1186/s13075-016-0927-z).
The 30% of patients who relapsed (n = 13) were retreated and reached a good European League Against Rheumatism (EULAR) response within 3 months, results from the observational study showed.
According to the researchers, the daily management of patients receiving long-term biologic treatment remains a matter of debate, and it is currently unclear how to select the most appropriate patients for discontinuing biologic treatment.
People with RA, even when in remission, tend to have residual synovitis. Previous research had shown that patients with negative signaling detected on power Doppler (PD) ultrasound were less likely to have disease flares.
To determine if the detection of residual synovitis with PD signaling could help in selecting patients suitable for anti-TNF discontinuation, the researchers selected 42 RA patients with disease duration of more than 12 months who were in sustained remission (Disease Activity Score less than 1.6 at three visits 3 months apart) who were receiving anti-TNF-alpha treatment plus methotrexate.
Patients were first tapered on anti-TNF-alpha therapy (adalimumab 40 mg/4 weeks or etanercept 50 mg/2 weeks).
Each patient underwent ultrasound evaluation of synovial hypertrophy (SH) and PD signal presence in the second and third metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints, the wrist (radiocarpal-intercarpal), bilateral knee, and second to fifth metatarsophalangeal (MTP) joints.
After 3 months, patients with no power Doppler signaling on ultrasound discontinued anti-TNF-alpha therapy and were followed every 3 months while maintaining stable doses of methotrexate.
Disease flares after anti-TNF-alpha discontinuation occurred in the joints with higher SH scores that were clinically involved at disease onset, despite the fact that no SH cut-off discriminated patients who relapsed from those who did not.
In particular, the fifth MTP joint was informative (in both the tapering and discontinuation groups) and the second MCP joint was informative for the tapering group only.
“This finding suggests the possible utility of following US [ultrasound] with indices of joints initially involved at disease onset with higher likelihood of relapse,” the researchers said.
Results from subgroup of five patients who also underwent ultrasound-guided knee synovial tissue biopsy to assess histologic features of residual synovitis revealed that the absence of ultrasound activity was associated with almost normal findings at the synovial level, they reported.
Overall, the findings suggested there was a “meaningful, large patient population with established RA in remission for whom the anti-TNF-alpha dose can be decreased without clinical and functional worsening,” the researchers wrote.
They suggested the combination of PD-US evaluation and American College of Rheumatology/EULAR remission criteria could help identify patients on biologics who are likely to achieve drug-free remission.
Use of three sequential ultrasound evaluations might identify an even higher proportion of patients likely to reach persistent drug-free remission, compared with current clinical methods of disease activity assessment, they added.
Ultrasound evaluation at the time of clinical remission could be a useful tool to select the most appropriate rheumatoid arthritis (RA) patients to undergo biologic therapy tapering and discontinuation, Italian researchers say.
In a study involving 42 RA patients in clinical remission who tapered their anti–tumor necrosis factor–alpha (anti-TNF-alpha) therapy according to ultrasound criteria, 69.1 % maintained remission at 12 weeks.
Furthermore, 26 of the patients (89.7 %) maintained disease remission after 6 months of follow-up, reported the research team led by Dr. Gianfranco Ferraccioli and Dr. Stefano Alivernini of the Institute of Rheumatology, Catholic University of the Sacred Heart, Rome (Arthritis Res Ther. 2016. doi: 10.1186/s13075-016-0927-z).
The 30% of patients who relapsed (n = 13) were retreated and reached a good European League Against Rheumatism (EULAR) response within 3 months, results from the observational study showed.
According to the researchers, the daily management of patients receiving long-term biologic treatment remains a matter of debate, and it is currently unclear how to select the most appropriate patients for discontinuing biologic treatment.
People with RA, even when in remission, tend to have residual synovitis. Previous research had shown that patients with negative signaling detected on power Doppler (PD) ultrasound were less likely to have disease flares.
To determine if the detection of residual synovitis with PD signaling could help in selecting patients suitable for anti-TNF discontinuation, the researchers selected 42 RA patients with disease duration of more than 12 months who were in sustained remission (Disease Activity Score less than 1.6 at three visits 3 months apart) who were receiving anti-TNF-alpha treatment plus methotrexate.
Patients were first tapered on anti-TNF-alpha therapy (adalimumab 40 mg/4 weeks or etanercept 50 mg/2 weeks).
Each patient underwent ultrasound evaluation of synovial hypertrophy (SH) and PD signal presence in the second and third metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints, the wrist (radiocarpal-intercarpal), bilateral knee, and second to fifth metatarsophalangeal (MTP) joints.
After 3 months, patients with no power Doppler signaling on ultrasound discontinued anti-TNF-alpha therapy and were followed every 3 months while maintaining stable doses of methotrexate.
Disease flares after anti-TNF-alpha discontinuation occurred in the joints with higher SH scores that were clinically involved at disease onset, despite the fact that no SH cut-off discriminated patients who relapsed from those who did not.
In particular, the fifth MTP joint was informative (in both the tapering and discontinuation groups) and the second MCP joint was informative for the tapering group only.
“This finding suggests the possible utility of following US [ultrasound] with indices of joints initially involved at disease onset with higher likelihood of relapse,” the researchers said.
Results from subgroup of five patients who also underwent ultrasound-guided knee synovial tissue biopsy to assess histologic features of residual synovitis revealed that the absence of ultrasound activity was associated with almost normal findings at the synovial level, they reported.
Overall, the findings suggested there was a “meaningful, large patient population with established RA in remission for whom the anti-TNF-alpha dose can be decreased without clinical and functional worsening,” the researchers wrote.
They suggested the combination of PD-US evaluation and American College of Rheumatology/EULAR remission criteria could help identify patients on biologics who are likely to achieve drug-free remission.
Use of three sequential ultrasound evaluations might identify an even higher proportion of patients likely to reach persistent drug-free remission, compared with current clinical methods of disease activity assessment, they added.
FROM ARTHRITIS RESEARCH & THERAPY
Key clinical point: Ultrasound evaluation of joints, particularly those involved at disease onset, could be a useful tool to select the most appropriate RA patients in remission to undergo biologic therapy tapering.
Major finding: Of the 42 patients who tapered anti-TNF-alpha therapy according to ultrasound selection criteria, 69% maintained remission at 12 weeks; almost 90% of these patients retained remission at 6 months.
Data source: An observational, prospective study cohort of 42 consecutive patients with RA with disease duration of more than 12 months who were in clinical remission.
Disclosures: Gianfranco Ferraccioli declared receiving consulting fees and speaking fees from Wyeth, Roche, AbbVie, and Bristol-Myers Squibb. The other authors said that they have no competing interests.
Biosimilar program reshapes FDA’s objectivity
The U.S. program to develop biosimilar agents – somewhat akin to generic drugs for complex, biologic molecules that have come off patent protection – is gathering momentum, with the first U.S. biosimilar, Zarxio, approved by the Food and Drug Administration in March 2015 and with the second, a biosimilar to infliximab, recommended by an FDA advisory committee on Feb. 9 of this year.
What’s striking about the burgeoning biosimilar development process, created by the Affordable Care Act, is how it has morphed the FDA from its traditional role as an objective arbiter of a drug’s safety and efficacy into an active partner in shepherding biosimilars onto the market.

As explained on Feb. 4 in testimony before a Congressional committee by Dr. Janet Woodcock, director of the FDA Center for Drug Evaluation and Research, the Biologic Price Competition and Innovation Act that was part of the Affordable Care Act launched a new U.S. drug-development pathway expressly for biosimilars. To implement that law, the FDA created an entirely new infrastructure within the agency – the Biosimilar Product Development Program – to help guide prospective manufacturers (called sponsors) of biosimilars through the regulatory and research hurdles to get a new biosimilar approved and into the hands of U.S. patients.
According to Dr. Woodcock, this program involves many steps where FDA staffers provide “review” and “advice” to sponsors on the studies they need to conduct and the analysis they need to perform to get their new products to market. The sponsor joins this program by paying an upfront fee that the FDA uses to keep the program running. Once a sponsor of a prospective biosimilar is in the program, the FDA’s staff helps guide the biosimilar development to a smooth conclusion.
To some extent, the FDA staff fills a similar role for conventional drug-development enterprises, conferring with manufacturers from the outset on matters such as the types and design of studies needed to insure success. What’s different about the biosimilar program is that conventional-drug development went on well before the FDA (or its predecessor) entered the scene, and the U.S. government created the FDA to police and regulate the drug production industry and protect the public against unscrupulous manufacturers of ineffective or dangerous drugs.
In contrast, the FDA itself created this new biosimilar development structure, and Dr. Woodcock noted that the in-depth review and advice meetings that the FDA offers to prospective biosimilar sponsors “has no counterpart in the Prescription Drug User Fee Act program and is unique” to the biosimilar program.
The consequence of having the FDA create the biosimilar development program from the ground up and structure it to provide such intimate input from the agency to sponsors at every step of the way seems to give the agency a notable and somewhat unnerving investment in the program’s success.
Dr. Woodcock called the approval of Zarxio an “exciting accomplishment,” and in her testimony before Congress she trumpeted the fact that as of January 2016 the biosimilar program was working on 59 proposed products that would mimic 18 different reference-product biologics. She also said that the FDA is “excited about the growing demand” for biosimilar-oriented meetings and marketing applications.
Don’t get me wrong: I think that the biosimilar concept is great, and has the potential to make what have become life-changing treatments more affordable and more available. And making the FDA such an active participant in getting biosimilar drugs created and approved is undoubtedly the most efficient way to accomplish this.
But in the process, the biosimilar program has changed the FDA from its more disengaged role as objective pharmaceutical judge into an active and seemingly not completely neutral codeveloper, risking at least the appearance of lost impartiality. Given that the FDA now wears two very different hats, we need to trust that the integrity and dedication of its staff will keep them from confusing their roles as proponent and gatekeeper.
On Twitter @mitchelzoler
The U.S. program to develop biosimilar agents – somewhat akin to generic drugs for complex, biologic molecules that have come off patent protection – is gathering momentum, with the first U.S. biosimilar, Zarxio, approved by the Food and Drug Administration in March 2015 and with the second, a biosimilar to infliximab, recommended by an FDA advisory committee on Feb. 9 of this year.
What’s striking about the burgeoning biosimilar development process, created by the Affordable Care Act, is how it has morphed the FDA from its traditional role as an objective arbiter of a drug’s safety and efficacy into an active partner in shepherding biosimilars onto the market.
As explained on Feb. 4 in testimony before a Congressional committee by Dr. Janet Woodcock, director of the FDA Center for Drug Evaluation and Research, the Biologic Price Competition and Innovation Act that was part of the Affordable Care Act launched a new U.S. drug-development pathway expressly for biosimilars. To implement that law, the FDA created an entirely new infrastructure within the agency – the Biosimilar Product Development Program – to help guide prospective manufacturers (called sponsors) of biosimilars through the regulatory and research hurdles to get a new biosimilar approved and into the hands of U.S. patients.
According to Dr. Woodcock, this program involves many steps where FDA staffers provide “review” and “advice” to sponsors on the studies they need to conduct and the analysis they need to perform to get their new products to market. The sponsor joins this program by paying an upfront fee that the FDA uses to keep the program running. Once a sponsor of a prospective biosimilar is in the program, the FDA’s staff helps guide the biosimilar development to a smooth conclusion.
To some extent, the FDA staff fills a similar role for conventional drug-development enterprises, conferring with manufacturers from the outset on matters such as the types and design of studies needed to insure success. What’s different about the biosimilar program is that conventional-drug development went on well before the FDA (or its predecessor) entered the scene, and the U.S. government created the FDA to police and regulate the drug production industry and protect the public against unscrupulous manufacturers of ineffective or dangerous drugs.
In contrast, the FDA itself created this new biosimilar development structure, and Dr. Woodcock noted that the in-depth review and advice meetings that the FDA offers to prospective biosimilar sponsors “has no counterpart in the Prescription Drug User Fee Act program and is unique” to the biosimilar program.
The consequence of having the FDA create the biosimilar development program from the ground up and structure it to provide such intimate input from the agency to sponsors at every step of the way seems to give the agency a notable and somewhat unnerving investment in the program’s success.
Dr. Woodcock called the approval of Zarxio an “exciting accomplishment,” and in her testimony before Congress she trumpeted the fact that as of January 2016 the biosimilar program was working on 59 proposed products that would mimic 18 different reference-product biologics. She also said that the FDA is “excited about the growing demand” for biosimilar-oriented meetings and marketing applications.
Don’t get me wrong: I think that the biosimilar concept is great, and has the potential to make what have become life-changing treatments more affordable and more available. And making the FDA such an active participant in getting biosimilar drugs created and approved is undoubtedly the most efficient way to accomplish this.
But in the process, the biosimilar program has changed the FDA from its more disengaged role as objective pharmaceutical judge into an active and seemingly not completely neutral codeveloper, risking at least the appearance of lost impartiality. Given that the FDA now wears two very different hats, we need to trust that the integrity and dedication of its staff will keep them from confusing their roles as proponent and gatekeeper.
On Twitter @mitchelzoler
The U.S. program to develop biosimilar agents – somewhat akin to generic drugs for complex, biologic molecules that have come off patent protection – is gathering momentum, with the first U.S. biosimilar, Zarxio, approved by the Food and Drug Administration in March 2015 and with the second, a biosimilar to infliximab, recommended by an FDA advisory committee on Feb. 9 of this year.
What’s striking about the burgeoning biosimilar development process, created by the Affordable Care Act, is how it has morphed the FDA from its traditional role as an objective arbiter of a drug’s safety and efficacy into an active partner in shepherding biosimilars onto the market.
As explained on Feb. 4 in testimony before a Congressional committee by Dr. Janet Woodcock, director of the FDA Center for Drug Evaluation and Research, the Biologic Price Competition and Innovation Act that was part of the Affordable Care Act launched a new U.S. drug-development pathway expressly for biosimilars. To implement that law, the FDA created an entirely new infrastructure within the agency – the Biosimilar Product Development Program – to help guide prospective manufacturers (called sponsors) of biosimilars through the regulatory and research hurdles to get a new biosimilar approved and into the hands of U.S. patients.
According to Dr. Woodcock, this program involves many steps where FDA staffers provide “review” and “advice” to sponsors on the studies they need to conduct and the analysis they need to perform to get their new products to market. The sponsor joins this program by paying an upfront fee that the FDA uses to keep the program running. Once a sponsor of a prospective biosimilar is in the program, the FDA’s staff helps guide the biosimilar development to a smooth conclusion.
To some extent, the FDA staff fills a similar role for conventional drug-development enterprises, conferring with manufacturers from the outset on matters such as the types and design of studies needed to insure success. What’s different about the biosimilar program is that conventional-drug development went on well before the FDA (or its predecessor) entered the scene, and the U.S. government created the FDA to police and regulate the drug production industry and protect the public against unscrupulous manufacturers of ineffective or dangerous drugs.
In contrast, the FDA itself created this new biosimilar development structure, and Dr. Woodcock noted that the in-depth review and advice meetings that the FDA offers to prospective biosimilar sponsors “has no counterpart in the Prescription Drug User Fee Act program and is unique” to the biosimilar program.
The consequence of having the FDA create the biosimilar development program from the ground up and structure it to provide such intimate input from the agency to sponsors at every step of the way seems to give the agency a notable and somewhat unnerving investment in the program’s success.
Dr. Woodcock called the approval of Zarxio an “exciting accomplishment,” and in her testimony before Congress she trumpeted the fact that as of January 2016 the biosimilar program was working on 59 proposed products that would mimic 18 different reference-product biologics. She also said that the FDA is “excited about the growing demand” for biosimilar-oriented meetings and marketing applications.
Don’t get me wrong: I think that the biosimilar concept is great, and has the potential to make what have become life-changing treatments more affordable and more available. And making the FDA such an active participant in getting biosimilar drugs created and approved is undoubtedly the most efficient way to accomplish this.
But in the process, the biosimilar program has changed the FDA from its more disengaged role as objective pharmaceutical judge into an active and seemingly not completely neutral codeveloper, risking at least the appearance of lost impartiality. Given that the FDA now wears two very different hats, we need to trust that the integrity and dedication of its staff will keep them from confusing their roles as proponent and gatekeeper.
On Twitter @mitchelzoler

Greater myocardial inflammation found in RA patients after heart attack
The presence of rheumatoid arthritis in individuals who died from a myocardial infarction was associated with greater inflammatory burden at different levels of myocardial tissue than in those without the disease in a small, retrospective, case-control autopsy study.
“These findings support the hypothesis that RA [rheumatoid arthritis] patients are prone to develop more vulnerable plaques due to more inflammation in the coronary arteries, but also develop increased intramyocardial inflammation indicative of a higher risk of myocardial tissue damage post MI. This might not only explain the higher incidence of cardiovascular events in this population, but also the higher fatality rate after myocardial infarction,” wrote Dr. Inge A.M. van den Oever of the Amsterdam Rheumatology and Immunology Center and colleagues (Int J Cardiol. 2016 Feb 3. doi: 10.1016/j.ijcard.2016.02.065).
The investigators took tissue samples from the infarcted left ventricle and microscopically determined infarct border area of five patients with RA and MI and five controls with MI but without RA. They found that RA patients had a greater burden of inflammatory cells in the adventitia of infarct-related epicardial coronary arteries, the intramyocardial vasculature, and the border area of the infarcted heart, compared with controls matched for age, sex, year of death, grade of stenosis, and infarct phase.
Specifically, the adventitial layer of the infarct-related epicardial coronary artery of RA patients had significantly more lymphocytes and mast cells. Intramyocardial arteries in infarct and infarct border areas of RA patients, compared with controls, showed significantly greater staining for advanced glycation end product N-epsilon-(carboxymethyl) lysine (CML), which is thought to reflect oxidative damage to cells, as well as noninfarcted tissues taken from the right ventricle.
The researchers included cases and controls from a postmortem tissue database of subjects who underwent autopsy within 24 hours after death at the VU University Medical Centre, Amsterdam, between January 1990 and December 2010.
The research was partly funded by a grant from the Dutch Society for Rheumatology. The authors had no conflicts of interest to declare.
The presence of rheumatoid arthritis in individuals who died from a myocardial infarction was associated with greater inflammatory burden at different levels of myocardial tissue than in those without the disease in a small, retrospective, case-control autopsy study.
“These findings support the hypothesis that RA [rheumatoid arthritis] patients are prone to develop more vulnerable plaques due to more inflammation in the coronary arteries, but also develop increased intramyocardial inflammation indicative of a higher risk of myocardial tissue damage post MI. This might not only explain the higher incidence of cardiovascular events in this population, but also the higher fatality rate after myocardial infarction,” wrote Dr. Inge A.M. van den Oever of the Amsterdam Rheumatology and Immunology Center and colleagues (Int J Cardiol. 2016 Feb 3. doi: 10.1016/j.ijcard.2016.02.065).
The investigators took tissue samples from the infarcted left ventricle and microscopically determined infarct border area of five patients with RA and MI and five controls with MI but without RA. They found that RA patients had a greater burden of inflammatory cells in the adventitia of infarct-related epicardial coronary arteries, the intramyocardial vasculature, and the border area of the infarcted heart, compared with controls matched for age, sex, year of death, grade of stenosis, and infarct phase.
Specifically, the adventitial layer of the infarct-related epicardial coronary artery of RA patients had significantly more lymphocytes and mast cells. Intramyocardial arteries in infarct and infarct border areas of RA patients, compared with controls, showed significantly greater staining for advanced glycation end product N-epsilon-(carboxymethyl) lysine (CML), which is thought to reflect oxidative damage to cells, as well as noninfarcted tissues taken from the right ventricle.
The researchers included cases and controls from a postmortem tissue database of subjects who underwent autopsy within 24 hours after death at the VU University Medical Centre, Amsterdam, between January 1990 and December 2010.
The research was partly funded by a grant from the Dutch Society for Rheumatology. The authors had no conflicts of interest to declare.
The presence of rheumatoid arthritis in individuals who died from a myocardial infarction was associated with greater inflammatory burden at different levels of myocardial tissue than in those without the disease in a small, retrospective, case-control autopsy study.
“These findings support the hypothesis that RA [rheumatoid arthritis] patients are prone to develop more vulnerable plaques due to more inflammation in the coronary arteries, but also develop increased intramyocardial inflammation indicative of a higher risk of myocardial tissue damage post MI. This might not only explain the higher incidence of cardiovascular events in this population, but also the higher fatality rate after myocardial infarction,” wrote Dr. Inge A.M. van den Oever of the Amsterdam Rheumatology and Immunology Center and colleagues (Int J Cardiol. 2016 Feb 3. doi: 10.1016/j.ijcard.2016.02.065).
The investigators took tissue samples from the infarcted left ventricle and microscopically determined infarct border area of five patients with RA and MI and five controls with MI but without RA. They found that RA patients had a greater burden of inflammatory cells in the adventitia of infarct-related epicardial coronary arteries, the intramyocardial vasculature, and the border area of the infarcted heart, compared with controls matched for age, sex, year of death, grade of stenosis, and infarct phase.
Specifically, the adventitial layer of the infarct-related epicardial coronary artery of RA patients had significantly more lymphocytes and mast cells. Intramyocardial arteries in infarct and infarct border areas of RA patients, compared with controls, showed significantly greater staining for advanced glycation end product N-epsilon-(carboxymethyl) lysine (CML), which is thought to reflect oxidative damage to cells, as well as noninfarcted tissues taken from the right ventricle.
The researchers included cases and controls from a postmortem tissue database of subjects who underwent autopsy within 24 hours after death at the VU University Medical Centre, Amsterdam, between January 1990 and December 2010.
The research was partly funded by a grant from the Dutch Society for Rheumatology. The authors had no conflicts of interest to declare.
FROM THE INTERNATIONAL JOURNAL OF CARDIOLOGY
Key clinical point:Post-MI autopsy data support the view that RA patients have greater intramyocardial inflammatory burden than do patients without the disease.
Major finding: The adventitial layer of the infarct-related epicardial coronary artery of RA patients had significantly more lymphocytes and mast cells than did controls.
Data source: A retrospective, case-control, autopsy study of five patients with RA and MI and five controls with MI but without RA.
Disclosures: The research was partly funded by a grant from the Dutch Society for Rheumatology. The authors had no conflicts of interest to declare.
Biosimilar infliximab gains FDA Advisory Committee endorsement
A biosimilar agent to Remicade, the brand-name and reference form of infliximab, stayed on track to become the second biosimilar drug to enter the U.S. market when the Arthritis Advisory Committee of the Food and Drug Administration voted overwhelmingly in favor of licensure of the biosimilar at a meeting on Feb. 9.
The vote was 21 in favor and 3 against, with no abstentions.
Because of the way the FDA staff worded the question that the Advisory Committee voted on, the panel not only was in favor of approving biosimilar licensure but also recommended that license for six of the seven diverse indications that Remicade currently has: treatment of rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, plaque psoriasis, adult and pediatric Crohn’s disease, and adult ulcerative colitis. The panel did not vote on licensing the biosimilar for treatment of pediatric ulcerative colitis because that specific indication for Remicade remains on patent for a few more years.
The broad range of indications for which the Committee recommended approval was notable because the formulation of biosimilar infliximab under review, manufactured by Celltrion and known in the United States as CT-P13, had been clinically studied only in patients with rheumatoid arthritis or ankylosing spondylitis. The other four recommended indications represented extrapolations, based on the totality of biosimilar evidence presented at the meeting by both Celltrion staffers and consultants as well as analyses presented by FDA staff members.
The overall thrust of the extrapolation issue was that if biosimilarity to Remicade was proven by a range of preclinical and clinical testing, and if safety and efficacy similar to Remicade was shown in trials that enrolled only patients with rheumatoid arthritis or ankylosing spondylitis, then the safety and efficacy previously proven for Remicade for the other indications could be reasonably extrapolated to apply to CT-P13 also, even though CT-P13 was never tested on patients with those conditions. This turned out to often be the key issue that panel members grappled with as they decided whether to vote in favor of the question the FDA asked them to address.
“Many of us are uncomfortable with this new pathway” of extrapolation, said panel member Dr. Beth L. Jonas, a rheumatologist at the University of North Carolina at Chapel Hill.
“I feel we’re taking a risk” with the extrapolations, said Dr. Mary E. Maloney, professor of medicine and chief of dermatology at the University of Massachusetts in Worcester. “We have a responsibility to take a risk to provide biosimilars to patients and to reduce their cost” for needed treatments, she said during the Committee’s discussion of their votes.
“Biosimilar is a new concept, but it’s the future of how we will look at drugs,” explained panel member Dr. Wilma Bergfeld, professor of dermatology at the Cleveland Clinic.
CT-P13 is currently marketed in many other countries worldwide under the brand names Remsima or Inflectra.
The FDA’s staff was clearly behind this application. After summarizing the agency’s internal analysis of the data submitted by Celltrion, Dr. Nikolay Nikolov, clinical team leader for the FDA’s Division of Pulmonary, Allergy and Rheumatology Products, concluded that “the totality of evidence provided by the applicant supports a conclusion that CT-P13 is biosimilar to U.S.-licensed Remicade,” and that “scientific justification for extrapolating the clinical data supports a finding of biosimilarity for all indications for which U.S.-licensed Remicade is licensed.” The FDA’s position makes it seem very likely that the agency will accept the Advisory Committee’s vote and grant CT-P13 license for U.S. marketing in the near future.
CT-P13 also received support during the public comment period of the Committee’s deliberations. At that time, Dr. Gideon P. Smith, a dermatologist at Massachusetts General Hospital in Boston spoke on behalf of the American Academy of Dermatology Association. “Biologics are some of the most important recent developments in treating plaque psoriasis, but cost is an important issue. We hope that biosimilars will decrease the cost of this treatment,” Dr. Smith said. “Infliximab is a complex molecule with a complex production process. We are concerned about the safety and efficacy of treatment. The AADA supports approval based on reducing cost and improving patient access. However, we strongly recommend caution through long-term postmarketing surveillance and using registry data to identify issues of immunogenicity, efficacy, and safety that were not seen in the clinical trials.”
The drug also received support from Dr. Angus B. Worthing, who represented the American College of Rheumatology. “Biosimilars may be the only tool to keep prices of biologics within reason,” said Dr. Worthing, a rheumatologist in Washington. But he also stressed that “extrapolation should be done with caution and not routinely granted.”
CT-P13 has the potential to make a fairly widely used biologic significantly more affordable. In countries where it has come onto the market, it’s been priced at roughly 30% below the prevailing cost of Remicade prior to this competition.
“Infliximab is an extremely important tool in our armamentarium for treatment of both ulcerative colitis and Crohn’s disease,” commented Dr. Stephen B. Hanauer, professor of gastroenterology and hepatology at Northwestern University in Chicago. “Biologic therapies account for an increasing proportion of health care costs for chronic diseases such as inflammatory bowel disease and reducing these costs will be important as increasing numbers of patients are benefiting from long-term biologic therapies. Having reviewed the extensive preclinical and clinical data with CT-P13, I am comfortable with potential substitution or switching as long as physicians are aware of the change and can track any potential reactions to the administered product,” he said in an interview.
“Infliximab is currently used by U.S. rheumatologists to treat certain patients with rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It is not the most-widely used tumor necrosis factor inhibitor, which is adalimumab, but it is often used. After FDA approval, biosimilar infliximab is anticipated to be priced lower than Remicade and that would likely increase use of infliximab for rheumatologic conditions,” said Dr. Jonathan Kay, a rheumatologist and professor of medicine at the University of Massachusetts in Worcester. “The clinical experience with CT-P13 in trials and in routine use in other countries show no significant loss of efficacy or any other major problem when changing patients from Remicade to CT-P13. All the data suggest that CT-P13 is highly similar to the reference product. It’s almost akin to comparing one lot of Remicade to another lot of Remicade. I personally would not have a problem initiating a patient on CT-P13 if infliximab was the appropriate drug to use,” Dr. Kay said in an interview.
Dr. Hanauer has been a consultant to Celltrion. Dr. Kay has been a consultant to several drug companies.
On Twitter @mitchelzoler
A biosimilar agent to Remicade, the brand-name and reference form of infliximab, stayed on track to become the second biosimilar drug to enter the U.S. market when the Arthritis Advisory Committee of the Food and Drug Administration voted overwhelmingly in favor of licensure of the biosimilar at a meeting on Feb. 9.
The vote was 21 in favor and 3 against, with no abstentions.
Because of the way the FDA staff worded the question that the Advisory Committee voted on, the panel not only was in favor of approving biosimilar licensure but also recommended that license for six of the seven diverse indications that Remicade currently has: treatment of rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, plaque psoriasis, adult and pediatric Crohn’s disease, and adult ulcerative colitis. The panel did not vote on licensing the biosimilar for treatment of pediatric ulcerative colitis because that specific indication for Remicade remains on patent for a few more years.
The broad range of indications for which the Committee recommended approval was notable because the formulation of biosimilar infliximab under review, manufactured by Celltrion and known in the United States as CT-P13, had been clinically studied only in patients with rheumatoid arthritis or ankylosing spondylitis. The other four recommended indications represented extrapolations, based on the totality of biosimilar evidence presented at the meeting by both Celltrion staffers and consultants as well as analyses presented by FDA staff members.
The overall thrust of the extrapolation issue was that if biosimilarity to Remicade was proven by a range of preclinical and clinical testing, and if safety and efficacy similar to Remicade was shown in trials that enrolled only patients with rheumatoid arthritis or ankylosing spondylitis, then the safety and efficacy previously proven for Remicade for the other indications could be reasonably extrapolated to apply to CT-P13 also, even though CT-P13 was never tested on patients with those conditions. This turned out to often be the key issue that panel members grappled with as they decided whether to vote in favor of the question the FDA asked them to address.
“Many of us are uncomfortable with this new pathway” of extrapolation, said panel member Dr. Beth L. Jonas, a rheumatologist at the University of North Carolina at Chapel Hill.
“I feel we’re taking a risk” with the extrapolations, said Dr. Mary E. Maloney, professor of medicine and chief of dermatology at the University of Massachusetts in Worcester. “We have a responsibility to take a risk to provide biosimilars to patients and to reduce their cost” for needed treatments, she said during the Committee’s discussion of their votes.
“Biosimilar is a new concept, but it’s the future of how we will look at drugs,” explained panel member Dr. Wilma Bergfeld, professor of dermatology at the Cleveland Clinic.
CT-P13 is currently marketed in many other countries worldwide under the brand names Remsima or Inflectra.
The FDA’s staff was clearly behind this application. After summarizing the agency’s internal analysis of the data submitted by Celltrion, Dr. Nikolay Nikolov, clinical team leader for the FDA’s Division of Pulmonary, Allergy and Rheumatology Products, concluded that “the totality of evidence provided by the applicant supports a conclusion that CT-P13 is biosimilar to U.S.-licensed Remicade,” and that “scientific justification for extrapolating the clinical data supports a finding of biosimilarity for all indications for which U.S.-licensed Remicade is licensed.” The FDA’s position makes it seem very likely that the agency will accept the Advisory Committee’s vote and grant CT-P13 license for U.S. marketing in the near future.
CT-P13 also received support during the public comment period of the Committee’s deliberations. At that time, Dr. Gideon P. Smith, a dermatologist at Massachusetts General Hospital in Boston spoke on behalf of the American Academy of Dermatology Association. “Biologics are some of the most important recent developments in treating plaque psoriasis, but cost is an important issue. We hope that biosimilars will decrease the cost of this treatment,” Dr. Smith said. “Infliximab is a complex molecule with a complex production process. We are concerned about the safety and efficacy of treatment. The AADA supports approval based on reducing cost and improving patient access. However, we strongly recommend caution through long-term postmarketing surveillance and using registry data to identify issues of immunogenicity, efficacy, and safety that were not seen in the clinical trials.”
The drug also received support from Dr. Angus B. Worthing, who represented the American College of Rheumatology. “Biosimilars may be the only tool to keep prices of biologics within reason,” said Dr. Worthing, a rheumatologist in Washington. But he also stressed that “extrapolation should be done with caution and not routinely granted.”
CT-P13 has the potential to make a fairly widely used biologic significantly more affordable. In countries where it has come onto the market, it’s been priced at roughly 30% below the prevailing cost of Remicade prior to this competition.
“Infliximab is an extremely important tool in our armamentarium for treatment of both ulcerative colitis and Crohn’s disease,” commented Dr. Stephen B. Hanauer, professor of gastroenterology and hepatology at Northwestern University in Chicago. “Biologic therapies account for an increasing proportion of health care costs for chronic diseases such as inflammatory bowel disease and reducing these costs will be important as increasing numbers of patients are benefiting from long-term biologic therapies. Having reviewed the extensive preclinical and clinical data with CT-P13, I am comfortable with potential substitution or switching as long as physicians are aware of the change and can track any potential reactions to the administered product,” he said in an interview.
“Infliximab is currently used by U.S. rheumatologists to treat certain patients with rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It is not the most-widely used tumor necrosis factor inhibitor, which is adalimumab, but it is often used. After FDA approval, biosimilar infliximab is anticipated to be priced lower than Remicade and that would likely increase use of infliximab for rheumatologic conditions,” said Dr. Jonathan Kay, a rheumatologist and professor of medicine at the University of Massachusetts in Worcester. “The clinical experience with CT-P13 in trials and in routine use in other countries show no significant loss of efficacy or any other major problem when changing patients from Remicade to CT-P13. All the data suggest that CT-P13 is highly similar to the reference product. It’s almost akin to comparing one lot of Remicade to another lot of Remicade. I personally would not have a problem initiating a patient on CT-P13 if infliximab was the appropriate drug to use,” Dr. Kay said in an interview.
Dr. Hanauer has been a consultant to Celltrion. Dr. Kay has been a consultant to several drug companies.
On Twitter @mitchelzoler
A biosimilar agent to Remicade, the brand-name and reference form of infliximab, stayed on track to become the second biosimilar drug to enter the U.S. market when the Arthritis Advisory Committee of the Food and Drug Administration voted overwhelmingly in favor of licensure of the biosimilar at a meeting on Feb. 9.
The vote was 21 in favor and 3 against, with no abstentions.
Because of the way the FDA staff worded the question that the Advisory Committee voted on, the panel not only was in favor of approving biosimilar licensure but also recommended that license for six of the seven diverse indications that Remicade currently has: treatment of rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, plaque psoriasis, adult and pediatric Crohn’s disease, and adult ulcerative colitis. The panel did not vote on licensing the biosimilar for treatment of pediatric ulcerative colitis because that specific indication for Remicade remains on patent for a few more years.
The broad range of indications for which the Committee recommended approval was notable because the formulation of biosimilar infliximab under review, manufactured by Celltrion and known in the United States as CT-P13, had been clinically studied only in patients with rheumatoid arthritis or ankylosing spondylitis. The other four recommended indications represented extrapolations, based on the totality of biosimilar evidence presented at the meeting by both Celltrion staffers and consultants as well as analyses presented by FDA staff members.
The overall thrust of the extrapolation issue was that if biosimilarity to Remicade was proven by a range of preclinical and clinical testing, and if safety and efficacy similar to Remicade was shown in trials that enrolled only patients with rheumatoid arthritis or ankylosing spondylitis, then the safety and efficacy previously proven for Remicade for the other indications could be reasonably extrapolated to apply to CT-P13 also, even though CT-P13 was never tested on patients with those conditions. This turned out to often be the key issue that panel members grappled with as they decided whether to vote in favor of the question the FDA asked them to address.
“Many of us are uncomfortable with this new pathway” of extrapolation, said panel member Dr. Beth L. Jonas, a rheumatologist at the University of North Carolina at Chapel Hill.
“I feel we’re taking a risk” with the extrapolations, said Dr. Mary E. Maloney, professor of medicine and chief of dermatology at the University of Massachusetts in Worcester. “We have a responsibility to take a risk to provide biosimilars to patients and to reduce their cost” for needed treatments, she said during the Committee’s discussion of their votes.
“Biosimilar is a new concept, but it’s the future of how we will look at drugs,” explained panel member Dr. Wilma Bergfeld, professor of dermatology at the Cleveland Clinic.
CT-P13 is currently marketed in many other countries worldwide under the brand names Remsima or Inflectra.
The FDA’s staff was clearly behind this application. After summarizing the agency’s internal analysis of the data submitted by Celltrion, Dr. Nikolay Nikolov, clinical team leader for the FDA’s Division of Pulmonary, Allergy and Rheumatology Products, concluded that “the totality of evidence provided by the applicant supports a conclusion that CT-P13 is biosimilar to U.S.-licensed Remicade,” and that “scientific justification for extrapolating the clinical data supports a finding of biosimilarity for all indications for which U.S.-licensed Remicade is licensed.” The FDA’s position makes it seem very likely that the agency will accept the Advisory Committee’s vote and grant CT-P13 license for U.S. marketing in the near future.
CT-P13 also received support during the public comment period of the Committee’s deliberations. At that time, Dr. Gideon P. Smith, a dermatologist at Massachusetts General Hospital in Boston spoke on behalf of the American Academy of Dermatology Association. “Biologics are some of the most important recent developments in treating plaque psoriasis, but cost is an important issue. We hope that biosimilars will decrease the cost of this treatment,” Dr. Smith said. “Infliximab is a complex molecule with a complex production process. We are concerned about the safety and efficacy of treatment. The AADA supports approval based on reducing cost and improving patient access. However, we strongly recommend caution through long-term postmarketing surveillance and using registry data to identify issues of immunogenicity, efficacy, and safety that were not seen in the clinical trials.”
The drug also received support from Dr. Angus B. Worthing, who represented the American College of Rheumatology. “Biosimilars may be the only tool to keep prices of biologics within reason,” said Dr. Worthing, a rheumatologist in Washington. But he also stressed that “extrapolation should be done with caution and not routinely granted.”
CT-P13 has the potential to make a fairly widely used biologic significantly more affordable. In countries where it has come onto the market, it’s been priced at roughly 30% below the prevailing cost of Remicade prior to this competition.
“Infliximab is an extremely important tool in our armamentarium for treatment of both ulcerative colitis and Crohn’s disease,” commented Dr. Stephen B. Hanauer, professor of gastroenterology and hepatology at Northwestern University in Chicago. “Biologic therapies account for an increasing proportion of health care costs for chronic diseases such as inflammatory bowel disease and reducing these costs will be important as increasing numbers of patients are benefiting from long-term biologic therapies. Having reviewed the extensive preclinical and clinical data with CT-P13, I am comfortable with potential substitution or switching as long as physicians are aware of the change and can track any potential reactions to the administered product,” he said in an interview.
“Infliximab is currently used by U.S. rheumatologists to treat certain patients with rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It is not the most-widely used tumor necrosis factor inhibitor, which is adalimumab, but it is often used. After FDA approval, biosimilar infliximab is anticipated to be priced lower than Remicade and that would likely increase use of infliximab for rheumatologic conditions,” said Dr. Jonathan Kay, a rheumatologist and professor of medicine at the University of Massachusetts in Worcester. “The clinical experience with CT-P13 in trials and in routine use in other countries show no significant loss of efficacy or any other major problem when changing patients from Remicade to CT-P13. All the data suggest that CT-P13 is highly similar to the reference product. It’s almost akin to comparing one lot of Remicade to another lot of Remicade. I personally would not have a problem initiating a patient on CT-P13 if infliximab was the appropriate drug to use,” Dr. Kay said in an interview.
Dr. Hanauer has been a consultant to Celltrion. Dr. Kay has been a consultant to several drug companies.
On Twitter @mitchelzoler
Potential etanercept response biomarker identified
Researchers have identified DNA methylation as a potential biomarker of response to etanercept in an epigenome-wide association study of a longitudinal cohort of patients with rheumatoid arthritis .
Selecting the right therapy the first time for patients with RA is a research priority because very good disease control is only achieved with etanercept (Enbrel) in 30% of patients and other biologic therapies targeting other pathways besides tumor necrosis factor are available, wrote researchers led by Dr. Darren Plant of the National Institute for Health Research Manchester Musculoskeletal Biomedical Research Unit at the Manchester (England) Academy of Health Sciences (Arthritis Rheumatol. 2016 Jan 27. doi: 10.1002/art.39590).
Their study selected 72 patients from the Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate (BRAGGSS) longitudinal cohort based on having an extreme response phenotype after 3 months of treatment with etanercept. DNA from each patient was sampled before therapy was initiated.
A total of 36 patients were very good responders to etanercept and in clinical remission, defined by having a 28-joint Disease Activity Score (DAS28) of less than 2.6.
The 36 patients classified as nonresponders to etanercept had a DAS28 improvement of less than 0.6 or an endpoint DAS28 of greater than 5.1.
Following bisulfite conversion of the DNA, the research team used the HumanMethylation450 BeadChip to determine unmethylated versus methylated cytosines in the DNA and found five differentially methylated sites associated with response to etanercept that passed the 5% false discovery rate threshold.
The most compelling evidence for differential methylation was observed in the LRPAP1 gene located on chromosome 4. This gene encodes a chaperone of low density lipoprotein receptor-related protein 1 (LRP1), which is known to influence TGF-beta activity.
Four CpG sites within exon 7 of the LRPAP1 gene were more methylated in nonresponders, results showed.
“The results indicate that DNA methylation profiling may provide a new biomarker of response to etanercept in patients with RA,” concluded the researchers, who said that the study is the first to investigate the influence of epigenetic variation on response to biologic therapies. Validation in a larger independent cohort is needed before transfer to clinical practice would be possible, they said.
Researchers have identified DNA methylation as a potential biomarker of response to etanercept in an epigenome-wide association study of a longitudinal cohort of patients with rheumatoid arthritis .
Selecting the right therapy the first time for patients with RA is a research priority because very good disease control is only achieved with etanercept (Enbrel) in 30% of patients and other biologic therapies targeting other pathways besides tumor necrosis factor are available, wrote researchers led by Dr. Darren Plant of the National Institute for Health Research Manchester Musculoskeletal Biomedical Research Unit at the Manchester (England) Academy of Health Sciences (Arthritis Rheumatol. 2016 Jan 27. doi: 10.1002/art.39590).
Their study selected 72 patients from the Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate (BRAGGSS) longitudinal cohort based on having an extreme response phenotype after 3 months of treatment with etanercept. DNA from each patient was sampled before therapy was initiated.
A total of 36 patients were very good responders to etanercept and in clinical remission, defined by having a 28-joint Disease Activity Score (DAS28) of less than 2.6.
The 36 patients classified as nonresponders to etanercept had a DAS28 improvement of less than 0.6 or an endpoint DAS28 of greater than 5.1.
Following bisulfite conversion of the DNA, the research team used the HumanMethylation450 BeadChip to determine unmethylated versus methylated cytosines in the DNA and found five differentially methylated sites associated with response to etanercept that passed the 5% false discovery rate threshold.
The most compelling evidence for differential methylation was observed in the LRPAP1 gene located on chromosome 4. This gene encodes a chaperone of low density lipoprotein receptor-related protein 1 (LRP1), which is known to influence TGF-beta activity.
Four CpG sites within exon 7 of the LRPAP1 gene were more methylated in nonresponders, results showed.
“The results indicate that DNA methylation profiling may provide a new biomarker of response to etanercept in patients with RA,” concluded the researchers, who said that the study is the first to investigate the influence of epigenetic variation on response to biologic therapies. Validation in a larger independent cohort is needed before transfer to clinical practice would be possible, they said.
Researchers have identified DNA methylation as a potential biomarker of response to etanercept in an epigenome-wide association study of a longitudinal cohort of patients with rheumatoid arthritis .
Selecting the right therapy the first time for patients with RA is a research priority because very good disease control is only achieved with etanercept (Enbrel) in 30% of patients and other biologic therapies targeting other pathways besides tumor necrosis factor are available, wrote researchers led by Dr. Darren Plant of the National Institute for Health Research Manchester Musculoskeletal Biomedical Research Unit at the Manchester (England) Academy of Health Sciences (Arthritis Rheumatol. 2016 Jan 27. doi: 10.1002/art.39590).
Their study selected 72 patients from the Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate (BRAGGSS) longitudinal cohort based on having an extreme response phenotype after 3 months of treatment with etanercept. DNA from each patient was sampled before therapy was initiated.
A total of 36 patients were very good responders to etanercept and in clinical remission, defined by having a 28-joint Disease Activity Score (DAS28) of less than 2.6.
The 36 patients classified as nonresponders to etanercept had a DAS28 improvement of less than 0.6 or an endpoint DAS28 of greater than 5.1.
Following bisulfite conversion of the DNA, the research team used the HumanMethylation450 BeadChip to determine unmethylated versus methylated cytosines in the DNA and found five differentially methylated sites associated with response to etanercept that passed the 5% false discovery rate threshold.
The most compelling evidence for differential methylation was observed in the LRPAP1 gene located on chromosome 4. This gene encodes a chaperone of low density lipoprotein receptor-related protein 1 (LRP1), which is known to influence TGF-beta activity.
Four CpG sites within exon 7 of the LRPAP1 gene were more methylated in nonresponders, results showed.
“The results indicate that DNA methylation profiling may provide a new biomarker of response to etanercept in patients with RA,” concluded the researchers, who said that the study is the first to investigate the influence of epigenetic variation on response to biologic therapies. Validation in a larger independent cohort is needed before transfer to clinical practice would be possible, they said.
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point: Researchers have identified a potential biomarker of response to etanercept in patients with rheumatoid arthritis.
Major finding: The most compelling evidence for differential methylation was observed in etanercept patients mapped to the LRPAP1 gene that is known to influence TGF-beta activity.
Data source: An epigenome-wide association study involving 72 selected patients from the Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate (BRAGGSS) longitudinal cohort.
Disclosures: The study was supported by the NIHR Manchester Musculoskeletal Biomedical Research Unit and Arthritis Research UK.
Rituxan led Medicare part B drug spending in 2014
In 2014, Medicare part B drug spending was led by the $1.5 billion cost of Rituxan (rituximab), the Centers for Medicare & Medicaid Services reported.
That made Rituxan – approved to treat non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis – one of six drugs to exceed $1 billion in spending by Medicare part B for the year. Second and third on the list were the macular degeneration/macular edema/diabetic retinopathy drugs Lucentis (ranibizumab) at $1.33 billion and Eylea (aflibercept) at $1.3 billion, according to the Medicare drug spending dashboard.
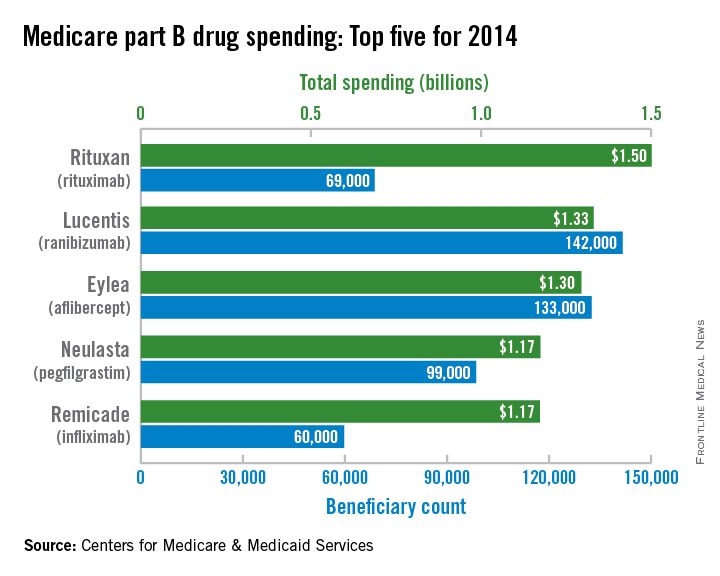
The other three members of the part B $1-billion club were Neulasta (pegfilgrastim), which prevents chemotherapy-induced neutropenia, at $1.174 billion; the rheumatoid and psoriatic arthritis/Crohn’s disease/ulcerative colitis/ankylosing spondylitis/psoriasis drug Remicade (infliximab) at $1.173 billion; and the chemotherapy drug Avastin (bevacizumab) at $1.06 billion, the CMS said.
Average cost per unit varied widely among the six drugs: Neulasta was $3,200 per unit, followed by Eylea ($960), Rituxan ($690), Lucentis ($390), Remicade ($71), and Avastin ($65).
Part B drug costs in 2014 totaled $21.5 billion for 606 different drug products, with 60% of that total cost going to the 21 drugs with spending over $250 million each, the CMS noted.
In 2014, Medicare part B drug spending was led by the $1.5 billion cost of Rituxan (rituximab), the Centers for Medicare & Medicaid Services reported.
That made Rituxan – approved to treat non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis – one of six drugs to exceed $1 billion in spending by Medicare part B for the year. Second and third on the list were the macular degeneration/macular edema/diabetic retinopathy drugs Lucentis (ranibizumab) at $1.33 billion and Eylea (aflibercept) at $1.3 billion, according to the Medicare drug spending dashboard.
The other three members of the part B $1-billion club were Neulasta (pegfilgrastim), which prevents chemotherapy-induced neutropenia, at $1.174 billion; the rheumatoid and psoriatic arthritis/Crohn’s disease/ulcerative colitis/ankylosing spondylitis/psoriasis drug Remicade (infliximab) at $1.173 billion; and the chemotherapy drug Avastin (bevacizumab) at $1.06 billion, the CMS said.
Average cost per unit varied widely among the six drugs: Neulasta was $3,200 per unit, followed by Eylea ($960), Rituxan ($690), Lucentis ($390), Remicade ($71), and Avastin ($65).
Part B drug costs in 2014 totaled $21.5 billion for 606 different drug products, with 60% of that total cost going to the 21 drugs with spending over $250 million each, the CMS noted.
In 2014, Medicare part B drug spending was led by the $1.5 billion cost of Rituxan (rituximab), the Centers for Medicare & Medicaid Services reported.
That made Rituxan – approved to treat non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis – one of six drugs to exceed $1 billion in spending by Medicare part B for the year. Second and third on the list were the macular degeneration/macular edema/diabetic retinopathy drugs Lucentis (ranibizumab) at $1.33 billion and Eylea (aflibercept) at $1.3 billion, according to the Medicare drug spending dashboard.
The other three members of the part B $1-billion club were Neulasta (pegfilgrastim), which prevents chemotherapy-induced neutropenia, at $1.174 billion; the rheumatoid and psoriatic arthritis/Crohn’s disease/ulcerative colitis/ankylosing spondylitis/psoriasis drug Remicade (infliximab) at $1.173 billion; and the chemotherapy drug Avastin (bevacizumab) at $1.06 billion, the CMS said.
Average cost per unit varied widely among the six drugs: Neulasta was $3,200 per unit, followed by Eylea ($960), Rituxan ($690), Lucentis ($390), Remicade ($71), and Avastin ($65).
Part B drug costs in 2014 totaled $21.5 billion for 606 different drug products, with 60% of that total cost going to the 21 drugs with spending over $250 million each, the CMS noted.
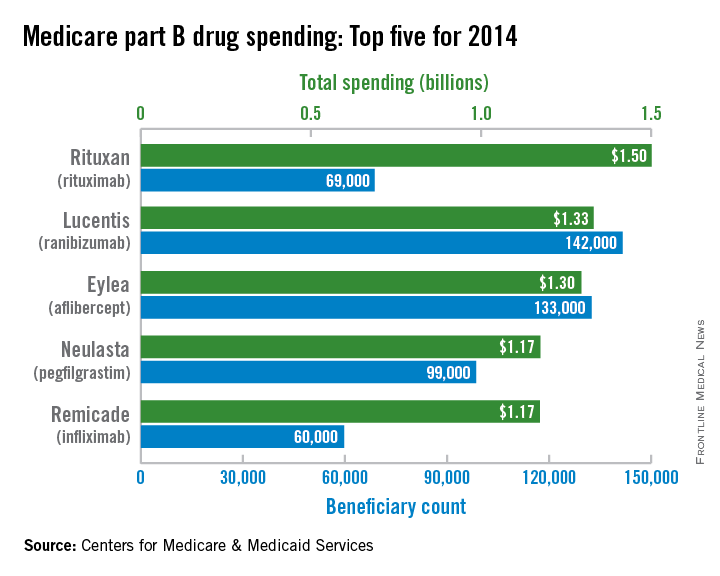
Step therapy and biologics: An easier road ahead?
Laws recently passed or under consideration in state legislatures may offer some relief to physicians and patients dogged by the “step” or “fail-first” therapy protocols mandated by insurers, but until better clinical evidence is available to support treatment decisions and biosimilars reduce costs, clinicians must strategize to get patients through the step pathways as fast as possible.

Rheumatologists, gastroenterologists, and dermatologists all confront fail-first policies in their practices, particularly when prescribing the biologic agents that have been game changers in treating rheumatoid arthritis (RA), inflammatory bowel disease (IBD), and psoriasis, among other diseases.
In RA, for example, a patient might be required to fail a series of disease-modifying antirheumatic drugs (DMARDs), including methotrexate, before starting a biologic. In Crohn’s disease, patients might have to first fail on steroids and immunosuppressants.
Most clinicians consider cost concerns fair as a basis for insurance decisions. But they can also have strong rationales for making exceptions. This may mean starting patients on a biologic early, particularly those they deem unlikely to respond to first- or second-line treatments – which may be cheaper but are not necessarily safer.

In egregious cases, a patient already stable on a biologic who has changed insurance plans may be forced to go backwards in the treatment pathway, and fail first- and second-line therapies all over again before resuming – a process unlikely to be cost-effective in the long term, and also rife with ethical concerns, say clinicians.
“Making a patient fail to get a less toxic drug sort of violates our ‘do no harm’ principle,” Dr. Stephen B. Hanauer, medical director of the Digestive Health Center at Northwestern University, Chicago, said in an interview.
“I always say that if biologics cost a dollar, we’d be using them for everybody. If you take away the steroids and the immunosuppressants, these are very safe drugs for IBD, far safer than steroids – but steroids are cheap,” Dr. Hanauer said.
And with some debilitating disease presentations, such as severe Crohn’s, “being told that we have to try conventional therapies and the patient has to fail them can mean putting the patient through progression of their disease, and suffering,” Dr. David T. Rubin, codirector of the Digestive Diseases Center at the University of Chicago, said in an interview. “We really struggle with this.”

Dr. Joseph S. Eastern, a dermatologist practicing in Belleville, N.J., said his specialty faces similar challenges with step therapy. “Dermatologists as a group are pretty risk averse. When given the opportunity, we do an excellent job of prescribing conventional medications, ultraviolet therapy, and biologics in the most cost-efficient possible way,” he said in an email.
Yet “third-party payers tell us, for example, that a patient must fail methotrexate before we can use a biologic, when the whole advantage of biologic therapy for many of these patients is the avoidance of organotoxicity and other serious risks.”
As a result, Dr. Eastern said, “I write a lot more vehement letters to payers about the biologics these days.”
Choice vs. cost
Rheumatologists are among the clinicians most affected by fail-first and step therapy mandates, as the diseases they treat – particularly RA – are the most established indications for biologic therapies, and for which the largest number of these are approved.
Though Medicare and Medicaid allow physicians considerable leeway, private insurers often tightly circumscribe the timing and choice of biologics in RA. As insurers’ first-choice biologic drug changes frequently, and varies from plan to plan, a patient who is stable on one agent might be asked to switch to another, a phenomenon known as nonmedical switching.
Insurers are seldom transparent about their reasons for establishing certain biologic drugs as their go-to agents, clinicians say, while making it difficult for physicians to prescribe others. “There’s a tremendous amount of discounting going on that we are oblivious to as physicians. I’ve seen situations where drug A is the first one you have to use this month and the next month, drug C,” said Dr. Norman Gaylis, a rheumatologist practicing in Aventura, Fla.
Attempting to start a patient on a nonpreferential biologic will generate paperwork and delays, Dr. Gaylis said, which can cost patients valuable time. “There’s a window of opportunity to treat these diseases, and by creating a step therapy pathway we’re closing the window at least partially.”
“As an example, rituximab in most payer plans is not tiered as a first-line biologic treatment option despite the fact that there are frequent scenarios where the clinical and serological presentation of a patient would suggest it to be preferable as a first-line treatment choice over an anti-TNF [tumor necrosis factor],” Dr. Gaylis said.
“There is no room for clinical decision making based upon the unique presentation of each different patient when choosing the best treatment option,” he said.
Rheumatologists often become overwhelmed with authorization paperwork, “and still in many instances end up with a denial of their request.”

Dr. Karen Kolba, a rheumatologist in private practice in Santa Maria, Calif., said that she agreed in principle with the way step therapy protocols have been established, and that some of the frustration with step therapy amounts to a tendency among specialist clinicians to bristle at being told what to do.
“Physicians hate protocol,” Dr. Kolba said. “But comparing one protocol to another is the only way we are going to make advances.” It took the rheumatology community about 30 years to come to terms with the use of methotrexate in RA, she noted, and the stepped approach grew naturally from the treatment of methotrexate failures with biologic agents when these first emerged in the late 1990s.
A majority of RA patients started on a stepped approach using DMARDs will respond, Dr. Kolba said, and for those who must move into the biologic realm, the vast majority will succeed on the first anti-TNF agent prescribed. And there is little science to establish that one TNF inhibitor is superior – only that patients can sometimes succeed with one and fail another.
“As far as which biologic to initiate, my personal opinion is I don’t care, and I tell the patients that I don’t mind if the insurer picks out of this category because I’m flipping a coin as well,” she said.
Step mandates become objectionable, Dr. Kolba said, when they are purportedly based in science that doesn’t exist, or when they seem to exist only to wear down the provider.
“With private insurance not only do they have the drug of the year, they’re going to make me battle for every single prescription. When I say I have tried this patient on maximal tolerable doses of all these DMARDs, they ought to believe me. Yet I get six-page forms back saying, ‘Give me the start and stop dates of all the drugs you’ve used.’”
States constrain fail first
For many specialists treating patients with biologics, some of these hurdles are already getting lowered.
Concerns about physician choice, a lack of transparency in insurer decision making, and the ethics of forcing patients to fail have led advocacy groups to press hard in recent years for legislation limiting step therapy – with successes in a dozen states.
While the state legislation is not disease or drug specific, it has important implications for clinicians treating with biologics. “Step therapy in its genesis was a good idea – it’s OK to try to reduce costs in the health care system,” said Patrick Stone, state government relations manager at the National Psoriasis Foundation in Annapolis, Md., a group that works extensively on step therapy issues. “But when these protocols were first crafted, medications like biologics weren’t in use.”
Jeff Okazaki, associate director of the Coalition of State Rheumatology Organizations, a group based in Schaumberg, Ill., said lawmakers are starting to accept that in terms of cost of care, “somebody not being treated appropriately and down the line has organ damage or comorbidity because of incorrect treatment decisions due to step therapy is a higher burden.”
Moreover, he said, “we’d seen protocols requiring five or more steps, and for each step you have to try it at least 90 days.” For a patient with rheumatic or autoimmune disease, “getting through something like that can just be devastating.”
In 2011, Connecticut, Mississippi, and Arkansas became the first states to pass legislation limiting some aspect of step therapy. Since then, nine additional states have passed legislation varying in focus and scope.
In Kentucky, for example, patients cannot be forced by their insurer to remain on an ineffective therapy for more than 30 days, and insurers must respond to physician requests for an override within 2 days. Mississippi allows physicians to override insurer decisions with proof of clinical evidence. In California, legislation passed last year aims to reduce bureaucracy and speed up response to physician requests for overrides.
Mr. Stone and Mr. Okazaki are working in a coalition with other dermatology, rheumatology, and GI groups to push bills in seven more states, including New York, North Carolina, and Ohio.
While all the bills differ in what they attempt to limit, the model legislation has three basic objectives, Mr. Okazaki said. “We want a clear set of clinical guidelines, a quick review process, and overrides that allow for exceptions in cases where patients shouldn’t have to go through step therapy.”
Clinical strategies and research gaps
New legislation undoubtedly will help providers and patients get access to their choice of treatment agents. But so long as biologics are expensive – and it will be a while before the first biosimilar drugs, which will have efficacy and safety similar to their reference biologics, reduce prices in any meaningful way – step therapy will likely remain the norm.
One of the key difficulties providers face when pushing back on an insurer in favor of a biologic drug is insufficient clinical evidence.
With IBD, Dr. Rubin said, “we need a need more longitudinal understanding” and better prognostic indicators “in order to justify spending the extra money or going to one of these therapies.”
Dr. Hanauer said one of the limitations he faces in practice is insufficient clinical evidence for biologics early in the treatment pathway for IBD.
RA “is much more common than Crohn’s disease is. In trials, it’s much easier to recruit hundreds of patients [for an RA trial], while with Crohn’s it’s very hard to enroll more than a couple a year at most sites,” he said. “And as you move earlier in the treatment pathway that becomes somewhat more difficult as well.”
His solution for now, he said, is to follow established step pathways in an accelerated way, for “a rapid transition toward highly effective therapies” without having to face extensive pushback from insurers.
“The idea is to initiate immunosuppressants for any patients with sufficient disease activity to justify steroids,” Dr. Hanauer said. “Their steroids are then tapered, and while on immunosuppressants, patients are in a perfect setup to get combination therapy with an immunosuppressive and a biologic – and that’s a 2- to 3-month transition, not 2-3 years.”
Dr. Kolba said that despite the wide array of options for treating RA, the specialty suffers from a dearth of understanding as to why some patients fail drugs while others succeed, even within the same drug class.
Rheumatologists’ prescribing choices would be highly influenced by better biomarkers, were they to become available, she said. And they’d have far better arguments when confronted with payer pushback.
“We’re all looking for that magic biologic marker to tell me which drug to use,” Dr. Kolba said, “because God knows if I had a blood test that said ‘this is the drug,’ I would go to the mat with the insurer.”
Laws recently passed or under consideration in state legislatures may offer some relief to physicians and patients dogged by the “step” or “fail-first” therapy protocols mandated by insurers, but until better clinical evidence is available to support treatment decisions and biosimilars reduce costs, clinicians must strategize to get patients through the step pathways as fast as possible.
Rheumatologists, gastroenterologists, and dermatologists all confront fail-first policies in their practices, particularly when prescribing the biologic agents that have been game changers in treating rheumatoid arthritis (RA), inflammatory bowel disease (IBD), and psoriasis, among other diseases.
In RA, for example, a patient might be required to fail a series of disease-modifying antirheumatic drugs (DMARDs), including methotrexate, before starting a biologic. In Crohn’s disease, patients might have to first fail on steroids and immunosuppressants.
Most clinicians consider cost concerns fair as a basis for insurance decisions. But they can also have strong rationales for making exceptions. This may mean starting patients on a biologic early, particularly those they deem unlikely to respond to first- or second-line treatments – which may be cheaper but are not necessarily safer.
In egregious cases, a patient already stable on a biologic who has changed insurance plans may be forced to go backwards in the treatment pathway, and fail first- and second-line therapies all over again before resuming – a process unlikely to be cost-effective in the long term, and also rife with ethical concerns, say clinicians.
“Making a patient fail to get a less toxic drug sort of violates our ‘do no harm’ principle,” Dr. Stephen B. Hanauer, medical director of the Digestive Health Center at Northwestern University, Chicago, said in an interview.
“I always say that if biologics cost a dollar, we’d be using them for everybody. If you take away the steroids and the immunosuppressants, these are very safe drugs for IBD, far safer than steroids – but steroids are cheap,” Dr. Hanauer said.
And with some debilitating disease presentations, such as severe Crohn’s, “being told that we have to try conventional therapies and the patient has to fail them can mean putting the patient through progression of their disease, and suffering,” Dr. David T. Rubin, codirector of the Digestive Diseases Center at the University of Chicago, said in an interview. “We really struggle with this.”
Dr. Joseph S. Eastern, a dermatologist practicing in Belleville, N.J., said his specialty faces similar challenges with step therapy. “Dermatologists as a group are pretty risk averse. When given the opportunity, we do an excellent job of prescribing conventional medications, ultraviolet therapy, and biologics in the most cost-efficient possible way,” he said in an email.
Yet “third-party payers tell us, for example, that a patient must fail methotrexate before we can use a biologic, when the whole advantage of biologic therapy for many of these patients is the avoidance of organotoxicity and other serious risks.”
As a result, Dr. Eastern said, “I write a lot more vehement letters to payers about the biologics these days.”
Choice vs. cost
Rheumatologists are among the clinicians most affected by fail-first and step therapy mandates, as the diseases they treat – particularly RA – are the most established indications for biologic therapies, and for which the largest number of these are approved.
Though Medicare and Medicaid allow physicians considerable leeway, private insurers often tightly circumscribe the timing and choice of biologics in RA. As insurers’ first-choice biologic drug changes frequently, and varies from plan to plan, a patient who is stable on one agent might be asked to switch to another, a phenomenon known as nonmedical switching.
Insurers are seldom transparent about their reasons for establishing certain biologic drugs as their go-to agents, clinicians say, while making it difficult for physicians to prescribe others. “There’s a tremendous amount of discounting going on that we are oblivious to as physicians. I’ve seen situations where drug A is the first one you have to use this month and the next month, drug C,” said Dr. Norman Gaylis, a rheumatologist practicing in Aventura, Fla.
Attempting to start a patient on a nonpreferential biologic will generate paperwork and delays, Dr. Gaylis said, which can cost patients valuable time. “There’s a window of opportunity to treat these diseases, and by creating a step therapy pathway we’re closing the window at least partially.”
“As an example, rituximab in most payer plans is not tiered as a first-line biologic treatment option despite the fact that there are frequent scenarios where the clinical and serological presentation of a patient would suggest it to be preferable as a first-line treatment choice over an anti-TNF [tumor necrosis factor],” Dr. Gaylis said.
“There is no room for clinical decision making based upon the unique presentation of each different patient when choosing the best treatment option,” he said.
Rheumatologists often become overwhelmed with authorization paperwork, “and still in many instances end up with a denial of their request.”
Dr. Karen Kolba, a rheumatologist in private practice in Santa Maria, Calif., said that she agreed in principle with the way step therapy protocols have been established, and that some of the frustration with step therapy amounts to a tendency among specialist clinicians to bristle at being told what to do.
“Physicians hate protocol,” Dr. Kolba said. “But comparing one protocol to another is the only way we are going to make advances.” It took the rheumatology community about 30 years to come to terms with the use of methotrexate in RA, she noted, and the stepped approach grew naturally from the treatment of methotrexate failures with biologic agents when these first emerged in the late 1990s.
A majority of RA patients started on a stepped approach using DMARDs will respond, Dr. Kolba said, and for those who must move into the biologic realm, the vast majority will succeed on the first anti-TNF agent prescribed. And there is little science to establish that one TNF inhibitor is superior – only that patients can sometimes succeed with one and fail another.
“As far as which biologic to initiate, my personal opinion is I don’t care, and I tell the patients that I don’t mind if the insurer picks out of this category because I’m flipping a coin as well,” she said.
Step mandates become objectionable, Dr. Kolba said, when they are purportedly based in science that doesn’t exist, or when they seem to exist only to wear down the provider.
“With private insurance not only do they have the drug of the year, they’re going to make me battle for every single prescription. When I say I have tried this patient on maximal tolerable doses of all these DMARDs, they ought to believe me. Yet I get six-page forms back saying, ‘Give me the start and stop dates of all the drugs you’ve used.’”
States constrain fail first
For many specialists treating patients with biologics, some of these hurdles are already getting lowered.
Concerns about physician choice, a lack of transparency in insurer decision making, and the ethics of forcing patients to fail have led advocacy groups to press hard in recent years for legislation limiting step therapy – with successes in a dozen states.
While the state legislation is not disease or drug specific, it has important implications for clinicians treating with biologics. “Step therapy in its genesis was a good idea – it’s OK to try to reduce costs in the health care system,” said Patrick Stone, state government relations manager at the National Psoriasis Foundation in Annapolis, Md., a group that works extensively on step therapy issues. “But when these protocols were first crafted, medications like biologics weren’t in use.”
Jeff Okazaki, associate director of the Coalition of State Rheumatology Organizations, a group based in Schaumberg, Ill., said lawmakers are starting to accept that in terms of cost of care, “somebody not being treated appropriately and down the line has organ damage or comorbidity because of incorrect treatment decisions due to step therapy is a higher burden.”
Moreover, he said, “we’d seen protocols requiring five or more steps, and for each step you have to try it at least 90 days.” For a patient with rheumatic or autoimmune disease, “getting through something like that can just be devastating.”
In 2011, Connecticut, Mississippi, and Arkansas became the first states to pass legislation limiting some aspect of step therapy. Since then, nine additional states have passed legislation varying in focus and scope.
In Kentucky, for example, patients cannot be forced by their insurer to remain on an ineffective therapy for more than 30 days, and insurers must respond to physician requests for an override within 2 days. Mississippi allows physicians to override insurer decisions with proof of clinical evidence. In California, legislation passed last year aims to reduce bureaucracy and speed up response to physician requests for overrides.
Mr. Stone and Mr. Okazaki are working in a coalition with other dermatology, rheumatology, and GI groups to push bills in seven more states, including New York, North Carolina, and Ohio.
While all the bills differ in what they attempt to limit, the model legislation has three basic objectives, Mr. Okazaki said. “We want a clear set of clinical guidelines, a quick review process, and overrides that allow for exceptions in cases where patients shouldn’t have to go through step therapy.”
Clinical strategies and research gaps
New legislation undoubtedly will help providers and patients get access to their choice of treatment agents. But so long as biologics are expensive – and it will be a while before the first biosimilar drugs, which will have efficacy and safety similar to their reference biologics, reduce prices in any meaningful way – step therapy will likely remain the norm.
One of the key difficulties providers face when pushing back on an insurer in favor of a biologic drug is insufficient clinical evidence.
With IBD, Dr. Rubin said, “we need a need more longitudinal understanding” and better prognostic indicators “in order to justify spending the extra money or going to one of these therapies.”
Dr. Hanauer said one of the limitations he faces in practice is insufficient clinical evidence for biologics early in the treatment pathway for IBD.
RA “is much more common than Crohn’s disease is. In trials, it’s much easier to recruit hundreds of patients [for an RA trial], while with Crohn’s it’s very hard to enroll more than a couple a year at most sites,” he said. “And as you move earlier in the treatment pathway that becomes somewhat more difficult as well.”
His solution for now, he said, is to follow established step pathways in an accelerated way, for “a rapid transition toward highly effective therapies” without having to face extensive pushback from insurers.
“The idea is to initiate immunosuppressants for any patients with sufficient disease activity to justify steroids,” Dr. Hanauer said. “Their steroids are then tapered, and while on immunosuppressants, patients are in a perfect setup to get combination therapy with an immunosuppressive and a biologic – and that’s a 2- to 3-month transition, not 2-3 years.”
Dr. Kolba said that despite the wide array of options for treating RA, the specialty suffers from a dearth of understanding as to why some patients fail drugs while others succeed, even within the same drug class.
Rheumatologists’ prescribing choices would be highly influenced by better biomarkers, were they to become available, she said. And they’d have far better arguments when confronted with payer pushback.
“We’re all looking for that magic biologic marker to tell me which drug to use,” Dr. Kolba said, “because God knows if I had a blood test that said ‘this is the drug,’ I would go to the mat with the insurer.”
Laws recently passed or under consideration in state legislatures may offer some relief to physicians and patients dogged by the “step” or “fail-first” therapy protocols mandated by insurers, but until better clinical evidence is available to support treatment decisions and biosimilars reduce costs, clinicians must strategize to get patients through the step pathways as fast as possible.
Rheumatologists, gastroenterologists, and dermatologists all confront fail-first policies in their practices, particularly when prescribing the biologic agents that have been game changers in treating rheumatoid arthritis (RA), inflammatory bowel disease (IBD), and psoriasis, among other diseases.
In RA, for example, a patient might be required to fail a series of disease-modifying antirheumatic drugs (DMARDs), including methotrexate, before starting a biologic. In Crohn’s disease, patients might have to first fail on steroids and immunosuppressants.
Most clinicians consider cost concerns fair as a basis for insurance decisions. But they can also have strong rationales for making exceptions. This may mean starting patients on a biologic early, particularly those they deem unlikely to respond to first- or second-line treatments – which may be cheaper but are not necessarily safer.
In egregious cases, a patient already stable on a biologic who has changed insurance plans may be forced to go backwards in the treatment pathway, and fail first- and second-line therapies all over again before resuming – a process unlikely to be cost-effective in the long term, and also rife with ethical concerns, say clinicians.
“Making a patient fail to get a less toxic drug sort of violates our ‘do no harm’ principle,” Dr. Stephen B. Hanauer, medical director of the Digestive Health Center at Northwestern University, Chicago, said in an interview.
“I always say that if biologics cost a dollar, we’d be using them for everybody. If you take away the steroids and the immunosuppressants, these are very safe drugs for IBD, far safer than steroids – but steroids are cheap,” Dr. Hanauer said.
And with some debilitating disease presentations, such as severe Crohn’s, “being told that we have to try conventional therapies and the patient has to fail them can mean putting the patient through progression of their disease, and suffering,” Dr. David T. Rubin, codirector of the Digestive Diseases Center at the University of Chicago, said in an interview. “We really struggle with this.”
Dr. Joseph S. Eastern, a dermatologist practicing in Belleville, N.J., said his specialty faces similar challenges with step therapy. “Dermatologists as a group are pretty risk averse. When given the opportunity, we do an excellent job of prescribing conventional medications, ultraviolet therapy, and biologics in the most cost-efficient possible way,” he said in an email.
Yet “third-party payers tell us, for example, that a patient must fail methotrexate before we can use a biologic, when the whole advantage of biologic therapy for many of these patients is the avoidance of organotoxicity and other serious risks.”
As a result, Dr. Eastern said, “I write a lot more vehement letters to payers about the biologics these days.”
Choice vs. cost
Rheumatologists are among the clinicians most affected by fail-first and step therapy mandates, as the diseases they treat – particularly RA – are the most established indications for biologic therapies, and for which the largest number of these are approved.
Though Medicare and Medicaid allow physicians considerable leeway, private insurers often tightly circumscribe the timing and choice of biologics in RA. As insurers’ first-choice biologic drug changes frequently, and varies from plan to plan, a patient who is stable on one agent might be asked to switch to another, a phenomenon known as nonmedical switching.
Insurers are seldom transparent about their reasons for establishing certain biologic drugs as their go-to agents, clinicians say, while making it difficult for physicians to prescribe others. “There’s a tremendous amount of discounting going on that we are oblivious to as physicians. I’ve seen situations where drug A is the first one you have to use this month and the next month, drug C,” said Dr. Norman Gaylis, a rheumatologist practicing in Aventura, Fla.
Attempting to start a patient on a nonpreferential biologic will generate paperwork and delays, Dr. Gaylis said, which can cost patients valuable time. “There’s a window of opportunity to treat these diseases, and by creating a step therapy pathway we’re closing the window at least partially.”
“As an example, rituximab in most payer plans is not tiered as a first-line biologic treatment option despite the fact that there are frequent scenarios where the clinical and serological presentation of a patient would suggest it to be preferable as a first-line treatment choice over an anti-TNF [tumor necrosis factor],” Dr. Gaylis said.
“There is no room for clinical decision making based upon the unique presentation of each different patient when choosing the best treatment option,” he said.
Rheumatologists often become overwhelmed with authorization paperwork, “and still in many instances end up with a denial of their request.”
Dr. Karen Kolba, a rheumatologist in private practice in Santa Maria, Calif., said that she agreed in principle with the way step therapy protocols have been established, and that some of the frustration with step therapy amounts to a tendency among specialist clinicians to bristle at being told what to do.
“Physicians hate protocol,” Dr. Kolba said. “But comparing one protocol to another is the only way we are going to make advances.” It took the rheumatology community about 30 years to come to terms with the use of methotrexate in RA, she noted, and the stepped approach grew naturally from the treatment of methotrexate failures with biologic agents when these first emerged in the late 1990s.
A majority of RA patients started on a stepped approach using DMARDs will respond, Dr. Kolba said, and for those who must move into the biologic realm, the vast majority will succeed on the first anti-TNF agent prescribed. And there is little science to establish that one TNF inhibitor is superior – only that patients can sometimes succeed with one and fail another.
“As far as which biologic to initiate, my personal opinion is I don’t care, and I tell the patients that I don’t mind if the insurer picks out of this category because I’m flipping a coin as well,” she said.
Step mandates become objectionable, Dr. Kolba said, when they are purportedly based in science that doesn’t exist, or when they seem to exist only to wear down the provider.
“With private insurance not only do they have the drug of the year, they’re going to make me battle for every single prescription. When I say I have tried this patient on maximal tolerable doses of all these DMARDs, they ought to believe me. Yet I get six-page forms back saying, ‘Give me the start and stop dates of all the drugs you’ve used.’”
States constrain fail first
For many specialists treating patients with biologics, some of these hurdles are already getting lowered.
Concerns about physician choice, a lack of transparency in insurer decision making, and the ethics of forcing patients to fail have led advocacy groups to press hard in recent years for legislation limiting step therapy – with successes in a dozen states.
While the state legislation is not disease or drug specific, it has important implications for clinicians treating with biologics. “Step therapy in its genesis was a good idea – it’s OK to try to reduce costs in the health care system,” said Patrick Stone, state government relations manager at the National Psoriasis Foundation in Annapolis, Md., a group that works extensively on step therapy issues. “But when these protocols were first crafted, medications like biologics weren’t in use.”
Jeff Okazaki, associate director of the Coalition of State Rheumatology Organizations, a group based in Schaumberg, Ill., said lawmakers are starting to accept that in terms of cost of care, “somebody not being treated appropriately and down the line has organ damage or comorbidity because of incorrect treatment decisions due to step therapy is a higher burden.”
Moreover, he said, “we’d seen protocols requiring five or more steps, and for each step you have to try it at least 90 days.” For a patient with rheumatic or autoimmune disease, “getting through something like that can just be devastating.”
In 2011, Connecticut, Mississippi, and Arkansas became the first states to pass legislation limiting some aspect of step therapy. Since then, nine additional states have passed legislation varying in focus and scope.
In Kentucky, for example, patients cannot be forced by their insurer to remain on an ineffective therapy for more than 30 days, and insurers must respond to physician requests for an override within 2 days. Mississippi allows physicians to override insurer decisions with proof of clinical evidence. In California, legislation passed last year aims to reduce bureaucracy and speed up response to physician requests for overrides.
Mr. Stone and Mr. Okazaki are working in a coalition with other dermatology, rheumatology, and GI groups to push bills in seven more states, including New York, North Carolina, and Ohio.
While all the bills differ in what they attempt to limit, the model legislation has three basic objectives, Mr. Okazaki said. “We want a clear set of clinical guidelines, a quick review process, and overrides that allow for exceptions in cases where patients shouldn’t have to go through step therapy.”
Clinical strategies and research gaps
New legislation undoubtedly will help providers and patients get access to their choice of treatment agents. But so long as biologics are expensive – and it will be a while before the first biosimilar drugs, which will have efficacy and safety similar to their reference biologics, reduce prices in any meaningful way – step therapy will likely remain the norm.
One of the key difficulties providers face when pushing back on an insurer in favor of a biologic drug is insufficient clinical evidence.
With IBD, Dr. Rubin said, “we need a need more longitudinal understanding” and better prognostic indicators “in order to justify spending the extra money or going to one of these therapies.”
Dr. Hanauer said one of the limitations he faces in practice is insufficient clinical evidence for biologics early in the treatment pathway for IBD.
RA “is much more common than Crohn’s disease is. In trials, it’s much easier to recruit hundreds of patients [for an RA trial], while with Crohn’s it’s very hard to enroll more than a couple a year at most sites,” he said. “And as you move earlier in the treatment pathway that becomes somewhat more difficult as well.”
His solution for now, he said, is to follow established step pathways in an accelerated way, for “a rapid transition toward highly effective therapies” without having to face extensive pushback from insurers.
“The idea is to initiate immunosuppressants for any patients with sufficient disease activity to justify steroids,” Dr. Hanauer said. “Their steroids are then tapered, and while on immunosuppressants, patients are in a perfect setup to get combination therapy with an immunosuppressive and a biologic – and that’s a 2- to 3-month transition, not 2-3 years.”
Dr. Kolba said that despite the wide array of options for treating RA, the specialty suffers from a dearth of understanding as to why some patients fail drugs while others succeed, even within the same drug class.
Rheumatologists’ prescribing choices would be highly influenced by better biomarkers, were they to become available, she said. And they’d have far better arguments when confronted with payer pushback.
“We’re all looking for that magic biologic marker to tell me which drug to use,” Dr. Kolba said, “because God knows if I had a blood test that said ‘this is the drug,’ I would go to the mat with the insurer.”
