User login
Genetic markers may predict response to biologics in rheumatoid arthritis
WASHINGTON – Three gene expression markers appear to predict response to some of the most common and powerful rheumatoid arthritis drugs employed in clinical practice.
In the largest gene-expression study of these drugs to date, researchers from Glasgow found 23 genes that predicted response to TNF inhibitors, and 23 more that predicted response to rituximab. They also found eight genes that predicted positive response to both types of drugs.
Just as important, the team found that 10% of patients in the study had none of the markers, Duncan Porter, MD, said during a press briefing at the annual meeting of the American College of Rheumatology.
If confirmed in independent validation cohorts, the findings could revolutionize medical therapy for rheumatoid arthritis, Dr. Porter said in an interview.
“Right now, the choice of treatment is mostly a flip of the coin,” said Dr. Porter, a rheumatologist at Queen Elizabeth University Hospital in Glasgow. “We don’t have a lot of comparative data to support one drug over the other, and most head-to-head studies show noninferiority.
“So, it would be ideal to be able to identify patients who will respond to one class or the other, and give them the right medicine the first time,” he noted. “If we can do that, we have the beginnings of really meaningful personalized therapy in RA.”
Dr. Porter reported a transcriptome-wide association study of 250 patients with RA who were enrolled in the ORBIT trial (Lancet. 2016 Jul 16;388[10041]:239-47). Dr. Porter was the study’s primary investigator.
ORBIT comprised 295 patients with active, seropositive rheumatoid arthritis who had failed disease-modifying antirheumatic therapy. They were randomized to B-cell depletion with rituximab or to the TNF inhibitors adalimumab or etanercept. ORBIT concluded that clinical response was similar with all three drugs. However, while most patients did well on their assigned drug, no matter which class, 20% failed their initial assignment.
The genetic study examined RNA transcripts in whole blood samples among a subset of the ORBIT patients. Most of the samples (70%) were used to identify markers and created a prediction model; that was then verified in the remaining 30% of samples. Markers were correlated to clinical response associated with rituximab and TNF inhibition. Clinical response was defined as a 1.2-point decline in the Disease Activity Score from baseline to 3 months of therapy.
At least 1 of the 54 genetic response markers was present in 90% of the population; 50% would have responded well to either drug, or 40% to just one of them.
But 10% of the population lacked either of the response markers – a very important finding, Dr. Porter noted, because if identified early, these patients could potentially avoid ineffective medication trials.
The markers’ predictive values were uniformly high, with a sensitivity of 93% and specificity of 91%. The positive predictive value was 96%, and the negative predictive value was 86%. Patients predicted to respond at 3 months were also more likely to have a good response (43% vs. 23%) or remission (23% vs. 10%), as measured by the Disease Activity Score 28 at 12 months.
While Dr. Porter was excited about the findings, he eyed them cautiously.
“The landscape is littered with biomarkers that had great early promise but failed in later validation studies,” he said. “We must be very careful in interpreting this. However, the other response studies have been small, and have not compared two drugs. This study was large with robust results, so we are encouraged.”
Dr Porter is now designing the protocol for a large external validation cohort study, which he hopes to launch in 2017.
Roche funded aspects of the ORBIT trial and the genetic substudy. Dr. Porter has been a consultant for Roche, as well as for AbbVie and Pfizer.
msullivan@frontlinemedcom.comOn Twitter @alz_gal
WASHINGTON – Three gene expression markers appear to predict response to some of the most common and powerful rheumatoid arthritis drugs employed in clinical practice.
In the largest gene-expression study of these drugs to date, researchers from Glasgow found 23 genes that predicted response to TNF inhibitors, and 23 more that predicted response to rituximab. They also found eight genes that predicted positive response to both types of drugs.
Just as important, the team found that 10% of patients in the study had none of the markers, Duncan Porter, MD, said during a press briefing at the annual meeting of the American College of Rheumatology.
If confirmed in independent validation cohorts, the findings could revolutionize medical therapy for rheumatoid arthritis, Dr. Porter said in an interview.
“Right now, the choice of treatment is mostly a flip of the coin,” said Dr. Porter, a rheumatologist at Queen Elizabeth University Hospital in Glasgow. “We don’t have a lot of comparative data to support one drug over the other, and most head-to-head studies show noninferiority.
“So, it would be ideal to be able to identify patients who will respond to one class or the other, and give them the right medicine the first time,” he noted. “If we can do that, we have the beginnings of really meaningful personalized therapy in RA.”
Dr. Porter reported a transcriptome-wide association study of 250 patients with RA who were enrolled in the ORBIT trial (Lancet. 2016 Jul 16;388[10041]:239-47). Dr. Porter was the study’s primary investigator.
ORBIT comprised 295 patients with active, seropositive rheumatoid arthritis who had failed disease-modifying antirheumatic therapy. They were randomized to B-cell depletion with rituximab or to the TNF inhibitors adalimumab or etanercept. ORBIT concluded that clinical response was similar with all three drugs. However, while most patients did well on their assigned drug, no matter which class, 20% failed their initial assignment.
The genetic study examined RNA transcripts in whole blood samples among a subset of the ORBIT patients. Most of the samples (70%) were used to identify markers and created a prediction model; that was then verified in the remaining 30% of samples. Markers were correlated to clinical response associated with rituximab and TNF inhibition. Clinical response was defined as a 1.2-point decline in the Disease Activity Score from baseline to 3 months of therapy.
At least 1 of the 54 genetic response markers was present in 90% of the population; 50% would have responded well to either drug, or 40% to just one of them.
But 10% of the population lacked either of the response markers – a very important finding, Dr. Porter noted, because if identified early, these patients could potentially avoid ineffective medication trials.
The markers’ predictive values were uniformly high, with a sensitivity of 93% and specificity of 91%. The positive predictive value was 96%, and the negative predictive value was 86%. Patients predicted to respond at 3 months were also more likely to have a good response (43% vs. 23%) or remission (23% vs. 10%), as measured by the Disease Activity Score 28 at 12 months.
While Dr. Porter was excited about the findings, he eyed them cautiously.
“The landscape is littered with biomarkers that had great early promise but failed in later validation studies,” he said. “We must be very careful in interpreting this. However, the other response studies have been small, and have not compared two drugs. This study was large with robust results, so we are encouraged.”
Dr Porter is now designing the protocol for a large external validation cohort study, which he hopes to launch in 2017.
Roche funded aspects of the ORBIT trial and the genetic substudy. Dr. Porter has been a consultant for Roche, as well as for AbbVie and Pfizer.
msullivan@frontlinemedcom.comOn Twitter @alz_gal
WASHINGTON – Three gene expression markers appear to predict response to some of the most common and powerful rheumatoid arthritis drugs employed in clinical practice.
In the largest gene-expression study of these drugs to date, researchers from Glasgow found 23 genes that predicted response to TNF inhibitors, and 23 more that predicted response to rituximab. They also found eight genes that predicted positive response to both types of drugs.
Just as important, the team found that 10% of patients in the study had none of the markers, Duncan Porter, MD, said during a press briefing at the annual meeting of the American College of Rheumatology.
If confirmed in independent validation cohorts, the findings could revolutionize medical therapy for rheumatoid arthritis, Dr. Porter said in an interview.
“Right now, the choice of treatment is mostly a flip of the coin,” said Dr. Porter, a rheumatologist at Queen Elizabeth University Hospital in Glasgow. “We don’t have a lot of comparative data to support one drug over the other, and most head-to-head studies show noninferiority.
“So, it would be ideal to be able to identify patients who will respond to one class or the other, and give them the right medicine the first time,” he noted. “If we can do that, we have the beginnings of really meaningful personalized therapy in RA.”
Dr. Porter reported a transcriptome-wide association study of 250 patients with RA who were enrolled in the ORBIT trial (Lancet. 2016 Jul 16;388[10041]:239-47). Dr. Porter was the study’s primary investigator.
ORBIT comprised 295 patients with active, seropositive rheumatoid arthritis who had failed disease-modifying antirheumatic therapy. They were randomized to B-cell depletion with rituximab or to the TNF inhibitors adalimumab or etanercept. ORBIT concluded that clinical response was similar with all three drugs. However, while most patients did well on their assigned drug, no matter which class, 20% failed their initial assignment.
The genetic study examined RNA transcripts in whole blood samples among a subset of the ORBIT patients. Most of the samples (70%) were used to identify markers and created a prediction model; that was then verified in the remaining 30% of samples. Markers were correlated to clinical response associated with rituximab and TNF inhibition. Clinical response was defined as a 1.2-point decline in the Disease Activity Score from baseline to 3 months of therapy.
At least 1 of the 54 genetic response markers was present in 90% of the population; 50% would have responded well to either drug, or 40% to just one of them.
But 10% of the population lacked either of the response markers – a very important finding, Dr. Porter noted, because if identified early, these patients could potentially avoid ineffective medication trials.
The markers’ predictive values were uniformly high, with a sensitivity of 93% and specificity of 91%. The positive predictive value was 96%, and the negative predictive value was 86%. Patients predicted to respond at 3 months were also more likely to have a good response (43% vs. 23%) or remission (23% vs. 10%), as measured by the Disease Activity Score 28 at 12 months.
While Dr. Porter was excited about the findings, he eyed them cautiously.
“The landscape is littered with biomarkers that had great early promise but failed in later validation studies,” he said. “We must be very careful in interpreting this. However, the other response studies have been small, and have not compared two drugs. This study was large with robust results, so we are encouraged.”
Dr Porter is now designing the protocol for a large external validation cohort study, which he hopes to launch in 2017.
Roche funded aspects of the ORBIT trial and the genetic substudy. Dr. Porter has been a consultant for Roche, as well as for AbbVie and Pfizer.
msullivan@frontlinemedcom.comOn Twitter @alz_gal
Key clinical point:
Major finding: The markers’ sensitivity and specificity for clinical response exceeded 90%.
Data source: The genetic-association study comprised 250 patients.
Disclosures: Roche funded aspects of the ORBIT trial and the genetic substudy. Dr. Porter has been a consultant for Roche, as well as for AbbVie and Pfizer.
Study offers reassuring data on certolizumab use in pregnancy
VIENNA – Data from a large prospective registry of pregnancy outcomes in women on certolizumab are reassuring to date, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“This unique dataset of pregnancies exposed to a single agent suggests that exposure is really not a problem, although we’ll continue to collect prospective data and anticipate doing so in women with psoriasis going forward,” said Dr. Kimball, professor of dermatology at Harvard Medical School, Boston.

Certolizumab is a tumor necrosis factor–alpha inhibitor currently approved in the United States for the treatment of Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It is now in clinical trials for psoriasis, and Dr. Kimball said she expects that it will eventually receive an indication for that disease as well. In the interim, she switches her psoriasis patients who are pregnant or plan to become so to either off-label certolizumab or etanercept (Enbrel). She does the same for her psoriatic arthritis patients, although in that situation certolizumab is on-label therapy.
The reason she turns to etanercept or certolizumab in pregnancy or anticipated pregnancy is that these two biologics, unlike others, don’t cross the placenta in the third trimester.
“I’m concerned about the selective uptake of other monoclonal antibodies in the third trimester. We do see in babies born to moms exposed to these other drugs – like ustekinumab, infliximab, and adalimumab – that the baby’s drug blood levels at birth are higher than the mom’s, so you have potentially put them at some risk for infections unnecessarily and maybe have affected how their immune system develops. So if a woman comes to me who is pregnant and on a biologic agent I would either stop treatment in the second trimester or switch to etanercept or certolizumab,” the dermatologist said.
Of the 256 pregnancies prospectively followed in the UCB certolizumab registry, 80.9% resulted in live births, and 10.2% ended in spontaneous abortion or miscarriage. In addition, the induced abortion rate was 8.6%, and there was a single stillbirth.
Of note, the mean age at pregnancy was 31 years, and 29% of the women became pregnant at age 35 or older. In contrast, the mean age at first pregnancy in the general population is 26, and only about 10% are 35 or older. These data are consistent with Dr. Kimball’s own clinical experience, which is that women with moderate to severe psoriasis or psoriatic arthritis often have trouble conceiving, and if they eventually succeed it’s often at a more advanced age.
The rate of maternal complications in this series was unremarkable: preeclampsia in 3.5%, infection in 3.9%, disease flare in 5.1%, and gestational diabetes in 3.1%. The median gestational age at birth was 39 weeks. The rate of early preterm birth before 32 weeks was 3.5%, with 12.6% of babies arriving at 32-36 weeks. Considering these are older moms being treated for serious underlying systemic inflammatory diseases, these numbers look good, according to Dr. Kimball.
Most women were exposed to certolizumab during the first trimester at least, and many were on the drug throughout pregnancy.
She said about one-third of psoriasis patients experience improvement in their skin diseases during pregnancy.
“If they’re doing really well, I see no reason to keep them on systemic therapy during pregnancy. But I will say that I see a lot of very bad postpartum flares, and I’m quite cautious about that. For the women with psoriatic arthritis there may not be a choice; you may need to continue to treat them all the way through their pregnancy to keep from getting permanent joint destruction,” the dermatologist said.
Dr. Kimball reported receiving research grants from and serving as a consultant to UCB and numerous other pharmaceutical companies.
VIENNA – Data from a large prospective registry of pregnancy outcomes in women on certolizumab are reassuring to date, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“This unique dataset of pregnancies exposed to a single agent suggests that exposure is really not a problem, although we’ll continue to collect prospective data and anticipate doing so in women with psoriasis going forward,” said Dr. Kimball, professor of dermatology at Harvard Medical School, Boston.
Certolizumab is a tumor necrosis factor–alpha inhibitor currently approved in the United States for the treatment of Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It is now in clinical trials for psoriasis, and Dr. Kimball said she expects that it will eventually receive an indication for that disease as well. In the interim, she switches her psoriasis patients who are pregnant or plan to become so to either off-label certolizumab or etanercept (Enbrel). She does the same for her psoriatic arthritis patients, although in that situation certolizumab is on-label therapy.
The reason she turns to etanercept or certolizumab in pregnancy or anticipated pregnancy is that these two biologics, unlike others, don’t cross the placenta in the third trimester.
“I’m concerned about the selective uptake of other monoclonal antibodies in the third trimester. We do see in babies born to moms exposed to these other drugs – like ustekinumab, infliximab, and adalimumab – that the baby’s drug blood levels at birth are higher than the mom’s, so you have potentially put them at some risk for infections unnecessarily and maybe have affected how their immune system develops. So if a woman comes to me who is pregnant and on a biologic agent I would either stop treatment in the second trimester or switch to etanercept or certolizumab,” the dermatologist said.
Of the 256 pregnancies prospectively followed in the UCB certolizumab registry, 80.9% resulted in live births, and 10.2% ended in spontaneous abortion or miscarriage. In addition, the induced abortion rate was 8.6%, and there was a single stillbirth.
Of note, the mean age at pregnancy was 31 years, and 29% of the women became pregnant at age 35 or older. In contrast, the mean age at first pregnancy in the general population is 26, and only about 10% are 35 or older. These data are consistent with Dr. Kimball’s own clinical experience, which is that women with moderate to severe psoriasis or psoriatic arthritis often have trouble conceiving, and if they eventually succeed it’s often at a more advanced age.
The rate of maternal complications in this series was unremarkable: preeclampsia in 3.5%, infection in 3.9%, disease flare in 5.1%, and gestational diabetes in 3.1%. The median gestational age at birth was 39 weeks. The rate of early preterm birth before 32 weeks was 3.5%, with 12.6% of babies arriving at 32-36 weeks. Considering these are older moms being treated for serious underlying systemic inflammatory diseases, these numbers look good, according to Dr. Kimball.
Most women were exposed to certolizumab during the first trimester at least, and many were on the drug throughout pregnancy.
She said about one-third of psoriasis patients experience improvement in their skin diseases during pregnancy.
“If they’re doing really well, I see no reason to keep them on systemic therapy during pregnancy. But I will say that I see a lot of very bad postpartum flares, and I’m quite cautious about that. For the women with psoriatic arthritis there may not be a choice; you may need to continue to treat them all the way through their pregnancy to keep from getting permanent joint destruction,” the dermatologist said.
Dr. Kimball reported receiving research grants from and serving as a consultant to UCB and numerous other pharmaceutical companies.
VIENNA – Data from a large prospective registry of pregnancy outcomes in women on certolizumab are reassuring to date, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“This unique dataset of pregnancies exposed to a single agent suggests that exposure is really not a problem, although we’ll continue to collect prospective data and anticipate doing so in women with psoriasis going forward,” said Dr. Kimball, professor of dermatology at Harvard Medical School, Boston.
Certolizumab is a tumor necrosis factor–alpha inhibitor currently approved in the United States for the treatment of Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It is now in clinical trials for psoriasis, and Dr. Kimball said she expects that it will eventually receive an indication for that disease as well. In the interim, she switches her psoriasis patients who are pregnant or plan to become so to either off-label certolizumab or etanercept (Enbrel). She does the same for her psoriatic arthritis patients, although in that situation certolizumab is on-label therapy.
The reason she turns to etanercept or certolizumab in pregnancy or anticipated pregnancy is that these two biologics, unlike others, don’t cross the placenta in the third trimester.
“I’m concerned about the selective uptake of other monoclonal antibodies in the third trimester. We do see in babies born to moms exposed to these other drugs – like ustekinumab, infliximab, and adalimumab – that the baby’s drug blood levels at birth are higher than the mom’s, so you have potentially put them at some risk for infections unnecessarily and maybe have affected how their immune system develops. So if a woman comes to me who is pregnant and on a biologic agent I would either stop treatment in the second trimester or switch to etanercept or certolizumab,” the dermatologist said.
Of the 256 pregnancies prospectively followed in the UCB certolizumab registry, 80.9% resulted in live births, and 10.2% ended in spontaneous abortion or miscarriage. In addition, the induced abortion rate was 8.6%, and there was a single stillbirth.
Of note, the mean age at pregnancy was 31 years, and 29% of the women became pregnant at age 35 or older. In contrast, the mean age at first pregnancy in the general population is 26, and only about 10% are 35 or older. These data are consistent with Dr. Kimball’s own clinical experience, which is that women with moderate to severe psoriasis or psoriatic arthritis often have trouble conceiving, and if they eventually succeed it’s often at a more advanced age.
The rate of maternal complications in this series was unremarkable: preeclampsia in 3.5%, infection in 3.9%, disease flare in 5.1%, and gestational diabetes in 3.1%. The median gestational age at birth was 39 weeks. The rate of early preterm birth before 32 weeks was 3.5%, with 12.6% of babies arriving at 32-36 weeks. Considering these are older moms being treated for serious underlying systemic inflammatory diseases, these numbers look good, according to Dr. Kimball.
Most women were exposed to certolizumab during the first trimester at least, and many were on the drug throughout pregnancy.
She said about one-third of psoriasis patients experience improvement in their skin diseases during pregnancy.
“If they’re doing really well, I see no reason to keep them on systemic therapy during pregnancy. But I will say that I see a lot of very bad postpartum flares, and I’m quite cautious about that. For the women with psoriatic arthritis there may not be a choice; you may need to continue to treat them all the way through their pregnancy to keep from getting permanent joint destruction,” the dermatologist said.
Dr. Kimball reported receiving research grants from and serving as a consultant to UCB and numerous other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point:
Major finding: The rate of major congenital malformations in a large prospective series of pregnancies in women on certolizumab was reassuringly low at 4.2%, with no pattern of malformations being seen.
Data source: This was a report on maternal and fetal outcomes of 256 prospectively followed pregnancies in women on certolizumab.
Disclosures: The presenter reported receiving research funds from and serving as a consultant to UCB, which markets certolizumab and maintains the pregnancy registry.
Abatacept may benefit ACPA-negative undifferentiated arthritis
Abatacept may benefit ACPA-negative undifferentiated arthritis, which generally has a poor prognosis and for which there is no proven therapy currently available, according to a report published in Rheumatology.
A 1-year course of abatacept (Orencia) reduced disease activity and an ultrasound measure of synovial inflammation in a manufacturer-funded, open-label, proof-of-concept study. However, it did not achieve the primary composite endpoint of remission on the DAS44 (44-joint Disease Activity Score), a maximum of one swollen joint for at least 3 consecutive months, and no radiographic progression after 6 months of treatment.
They assessed 20 adults with undifferentiated arthritis that had lasted 12 weeks to 18 months who showed definite, active synovitis in at least 1 of 20 scanned joints and had no previous disease-modifying antirheumatic drug (DMARD) therapy; these study subjects were considered likely to progress to rheumatoid arthritis. They received 14 doses of IV abatacept for 1 year.
Two patients withdrew from the study at 6 months and 12 months because of adverse events, and another three were lost to follow-up. Only 2 of the 20 patients (10%) achieved the composite primary endpoint. The majority of patients met two of the individual components – 15 patients showed no radiographic progression and 12 had a maximum of one swollen joint – but only 6 achieved DAS44 remission, the investigators said (Rheumatology [Oxford]. 2016 Oct 22. doi: 10.1093/rheumatology/kew357).
The treatment appeared to suppress C-reactive protein levels immediately, from a median of 9 mg/L at baseline to 0 mg/L at 3 and 6 months. This implies a clear biologic effect. The median number of swollen joints also decreased from a median of 2 at baseline to 0 at 6 and 12 months. There were small reductions in median scores on a disability index and a small decrease in patient-reported visual analog scale disease activity from a median of 52 at baseline to 28 at 6 months and 24 at 12 months. Median ultrasound scores of synovitis decreased from 10 at baseline to 3 at both 6 and 12 months.
Most of these benefits persisted for 1 year after abatacept therapy was stopped, but approximately half of patients required a synthetic DMARD to maintain the benefits. “Overall, these data suggest that in the vast majority of patients (18 of 20), abatacept therapy prevented further progression of disease, but on cessation, additional therapy was indicated to maintain this,” Dr. Buch and her associates said.
There were no serious adverse events during treatment, including no infections and no abnormal liver function tests. A total of 131 nonserious adverse events were reported.
This study was supported by a research grant from Bristol-Myers Squibb and by institution-level grants from Arthritis UK and the U.K. National Institute for Health Research. Dr. Buch reported ties to Bristol-Myers Squibb, AbbVie, AstraZeneca, Pfizer, and Roche-Chugai; her associates reported ties to numerous industry sources.
Abatacept may benefit ACPA-negative undifferentiated arthritis, which generally has a poor prognosis and for which there is no proven therapy currently available, according to a report published in Rheumatology.
A 1-year course of abatacept (Orencia) reduced disease activity and an ultrasound measure of synovial inflammation in a manufacturer-funded, open-label, proof-of-concept study. However, it did not achieve the primary composite endpoint of remission on the DAS44 (44-joint Disease Activity Score), a maximum of one swollen joint for at least 3 consecutive months, and no radiographic progression after 6 months of treatment.
They assessed 20 adults with undifferentiated arthritis that had lasted 12 weeks to 18 months who showed definite, active synovitis in at least 1 of 20 scanned joints and had no previous disease-modifying antirheumatic drug (DMARD) therapy; these study subjects were considered likely to progress to rheumatoid arthritis. They received 14 doses of IV abatacept for 1 year.
Two patients withdrew from the study at 6 months and 12 months because of adverse events, and another three were lost to follow-up. Only 2 of the 20 patients (10%) achieved the composite primary endpoint. The majority of patients met two of the individual components – 15 patients showed no radiographic progression and 12 had a maximum of one swollen joint – but only 6 achieved DAS44 remission, the investigators said (Rheumatology [Oxford]. 2016 Oct 22. doi: 10.1093/rheumatology/kew357).
The treatment appeared to suppress C-reactive protein levels immediately, from a median of 9 mg/L at baseline to 0 mg/L at 3 and 6 months. This implies a clear biologic effect. The median number of swollen joints also decreased from a median of 2 at baseline to 0 at 6 and 12 months. There were small reductions in median scores on a disability index and a small decrease in patient-reported visual analog scale disease activity from a median of 52 at baseline to 28 at 6 months and 24 at 12 months. Median ultrasound scores of synovitis decreased from 10 at baseline to 3 at both 6 and 12 months.
Most of these benefits persisted for 1 year after abatacept therapy was stopped, but approximately half of patients required a synthetic DMARD to maintain the benefits. “Overall, these data suggest that in the vast majority of patients (18 of 20), abatacept therapy prevented further progression of disease, but on cessation, additional therapy was indicated to maintain this,” Dr. Buch and her associates said.
There were no serious adverse events during treatment, including no infections and no abnormal liver function tests. A total of 131 nonserious adverse events were reported.
This study was supported by a research grant from Bristol-Myers Squibb and by institution-level grants from Arthritis UK and the U.K. National Institute for Health Research. Dr. Buch reported ties to Bristol-Myers Squibb, AbbVie, AstraZeneca, Pfizer, and Roche-Chugai; her associates reported ties to numerous industry sources.
Abatacept may benefit ACPA-negative undifferentiated arthritis, which generally has a poor prognosis and for which there is no proven therapy currently available, according to a report published in Rheumatology.
A 1-year course of abatacept (Orencia) reduced disease activity and an ultrasound measure of synovial inflammation in a manufacturer-funded, open-label, proof-of-concept study. However, it did not achieve the primary composite endpoint of remission on the DAS44 (44-joint Disease Activity Score), a maximum of one swollen joint for at least 3 consecutive months, and no radiographic progression after 6 months of treatment.
They assessed 20 adults with undifferentiated arthritis that had lasted 12 weeks to 18 months who showed definite, active synovitis in at least 1 of 20 scanned joints and had no previous disease-modifying antirheumatic drug (DMARD) therapy; these study subjects were considered likely to progress to rheumatoid arthritis. They received 14 doses of IV abatacept for 1 year.
Two patients withdrew from the study at 6 months and 12 months because of adverse events, and another three were lost to follow-up. Only 2 of the 20 patients (10%) achieved the composite primary endpoint. The majority of patients met two of the individual components – 15 patients showed no radiographic progression and 12 had a maximum of one swollen joint – but only 6 achieved DAS44 remission, the investigators said (Rheumatology [Oxford]. 2016 Oct 22. doi: 10.1093/rheumatology/kew357).
The treatment appeared to suppress C-reactive protein levels immediately, from a median of 9 mg/L at baseline to 0 mg/L at 3 and 6 months. This implies a clear biologic effect. The median number of swollen joints also decreased from a median of 2 at baseline to 0 at 6 and 12 months. There were small reductions in median scores on a disability index and a small decrease in patient-reported visual analog scale disease activity from a median of 52 at baseline to 28 at 6 months and 24 at 12 months. Median ultrasound scores of synovitis decreased from 10 at baseline to 3 at both 6 and 12 months.
Most of these benefits persisted for 1 year after abatacept therapy was stopped, but approximately half of patients required a synthetic DMARD to maintain the benefits. “Overall, these data suggest that in the vast majority of patients (18 of 20), abatacept therapy prevented further progression of disease, but on cessation, additional therapy was indicated to maintain this,” Dr. Buch and her associates said.
There were no serious adverse events during treatment, including no infections and no abnormal liver function tests. A total of 131 nonserious adverse events were reported.
This study was supported by a research grant from Bristol-Myers Squibb and by institution-level grants from Arthritis UK and the U.K. National Institute for Health Research. Dr. Buch reported ties to Bristol-Myers Squibb, AbbVie, AstraZeneca, Pfizer, and Roche-Chugai; her associates reported ties to numerous industry sources.
Key clinical point:
Major finding: Only 2 of the 20 patients (10%) achieved the composite primary endpoint of DAS44 remission, a maximum of 1 swollen joint for at least 3 consecutive months, and no radiographic progression at 6-month follow-up.
Data source: A manufacturer-supported, open-label, proof-of-concept study involving 20 adults treated for 1 year and followed for 1 further year.
Disclosures: This study was supported by a research grant from Bristol-Myers Squibb and by institution-level grants from Arthritis UK and the U.K. National Institute for Health Research. Dr. Buch reported ties to Bristol-Myers Squibb, AbbVie, AstraZeneca, Pfizer, and Roche-Chugai; her associates reported ties to numerous industry sources.
What good are biosimilars if patients won’t use them?
BOSTON – Biosimilar versions of disease-modifying antirheumatic drugs have arrived in the United States, but even the best, most efficacious drugs are worthless if patients don’t want to take them.
“The science is important, the medicine is important, but at the end of the day, acceptance and use is what’s going to measure success,” said Seth D. Ginsberg, at a biosimilars symposium sponsored by Corrona, a business that provides registry data and consulting services to biopharmaceutical companies.
He illustrated the value of biologic agents with this anecdote: “When we got started long, long ago, we used to hold patient events,” he said “and we usually set up for 100. The instructions to meeting planners were right before the event that it was protocol to pull the front 25, the front-right quarter of chairs. Why? To make room for those who can’t walk, to make room for the wheelchairs,” he said.
“Today, if we have one wheelchair at an event, it’s an outlier, and I can’t think of a better way to summarize the impact that biologics have had on our lives,” he said.
Biosimilar confidence
His group has launched “Operation: Biosimilar Confidence” which is designed to educate patients and physicians about the clinical value and scientific underpinnings of biosimilars, as well as the thorough development, review, and regulatory processes involved.
The goal of the project is to instill confidence in patients by helping them to understand the manufacturer’s safety track record, reliability of the biosimilar supply chain, and the availability to them of support services, if they make the switch to a biosimilar.
“Generics don’t have equivalent patient-support programs, and the projection is theoretically that [biosimilar] manufacturers won’t either. We will not accept that. We are going to do everything we can for those patients, to advocate for the continuation of the support programs that we rely on as patients,” he said.
Patient concerns
Surveys of patient concerns about biosimilars have highlighted four key areas:
- What is the manufacturer’s overall safety record in both biologic agents and small-molecule therapies?
- Supply-chain logistic – Will the manufacturer commit to consistent production and supply?
- Will biosimilar manufacturers provide patient support at levels equal to those offered by innovator biologic makers, and what kind of support will be available – phone, websites, social media, copays, etc.?
- Payer ethics – Will payers offer lower copays, deductibles, or premiums, and are payers as concerned as patients about product safety, supply chain, and support?
The implementation strategy for the campaign will focus on speaking directly to patients through CreakyJoints.org, partner Global Healthy Living Foundation, patient and physician organizations, social and conventional media, advertising, and one-on-one encounters.
“We have to talk directly and indirectly to employers and employee-advocacy groups. We have to let these big self-insured employers understand what the perspective of the patient is and what life is like thanks to these medicine, and why biosimilars are a critical component to the success of living with these conditions,” he said.
Advocates also have to work with the media to create “a surround-sound message that reaches all audiences with additional frequency.”
“We cannot allow Wall Street Journal business analysts to dictate the conversations about biosimilars. Why? They’re looking at one thing, and only one thing, and they’re ignoring the patient perspective,” Ginsberg said.
Lastly, patient groups need to work closely with payers, physician groups, and manufacturers to ensure that biosimilars can be smoothly integrated into the healthcare system, he emphasized.
“I want to be crystal clear here: We can’t wait for biosimilars. Bring it on! We want them,” he said.
BOSTON – Biosimilar versions of disease-modifying antirheumatic drugs have arrived in the United States, but even the best, most efficacious drugs are worthless if patients don’t want to take them.
“The science is important, the medicine is important, but at the end of the day, acceptance and use is what’s going to measure success,” said Seth D. Ginsberg, at a biosimilars symposium sponsored by Corrona, a business that provides registry data and consulting services to biopharmaceutical companies.
He illustrated the value of biologic agents with this anecdote: “When we got started long, long ago, we used to hold patient events,” he said “and we usually set up for 100. The instructions to meeting planners were right before the event that it was protocol to pull the front 25, the front-right quarter of chairs. Why? To make room for those who can’t walk, to make room for the wheelchairs,” he said.
“Today, if we have one wheelchair at an event, it’s an outlier, and I can’t think of a better way to summarize the impact that biologics have had on our lives,” he said.
Biosimilar confidence
His group has launched “Operation: Biosimilar Confidence” which is designed to educate patients and physicians about the clinical value and scientific underpinnings of biosimilars, as well as the thorough development, review, and regulatory processes involved.
The goal of the project is to instill confidence in patients by helping them to understand the manufacturer’s safety track record, reliability of the biosimilar supply chain, and the availability to them of support services, if they make the switch to a biosimilar.
“Generics don’t have equivalent patient-support programs, and the projection is theoretically that [biosimilar] manufacturers won’t either. We will not accept that. We are going to do everything we can for those patients, to advocate for the continuation of the support programs that we rely on as patients,” he said.
Patient concerns
Surveys of patient concerns about biosimilars have highlighted four key areas:
- What is the manufacturer’s overall safety record in both biologic agents and small-molecule therapies?
- Supply-chain logistic – Will the manufacturer commit to consistent production and supply?
- Will biosimilar manufacturers provide patient support at levels equal to those offered by innovator biologic makers, and what kind of support will be available – phone, websites, social media, copays, etc.?
- Payer ethics – Will payers offer lower copays, deductibles, or premiums, and are payers as concerned as patients about product safety, supply chain, and support?
The implementation strategy for the campaign will focus on speaking directly to patients through CreakyJoints.org, partner Global Healthy Living Foundation, patient and physician organizations, social and conventional media, advertising, and one-on-one encounters.
“We have to talk directly and indirectly to employers and employee-advocacy groups. We have to let these big self-insured employers understand what the perspective of the patient is and what life is like thanks to these medicine, and why biosimilars are a critical component to the success of living with these conditions,” he said.
Advocates also have to work with the media to create “a surround-sound message that reaches all audiences with additional frequency.”
“We cannot allow Wall Street Journal business analysts to dictate the conversations about biosimilars. Why? They’re looking at one thing, and only one thing, and they’re ignoring the patient perspective,” Ginsberg said.
Lastly, patient groups need to work closely with payers, physician groups, and manufacturers to ensure that biosimilars can be smoothly integrated into the healthcare system, he emphasized.
“I want to be crystal clear here: We can’t wait for biosimilars. Bring it on! We want them,” he said.
BOSTON – Biosimilar versions of disease-modifying antirheumatic drugs have arrived in the United States, but even the best, most efficacious drugs are worthless if patients don’t want to take them.
“The science is important, the medicine is important, but at the end of the day, acceptance and use is what’s going to measure success,” said Seth D. Ginsberg, at a biosimilars symposium sponsored by Corrona, a business that provides registry data and consulting services to biopharmaceutical companies.
He illustrated the value of biologic agents with this anecdote: “When we got started long, long ago, we used to hold patient events,” he said “and we usually set up for 100. The instructions to meeting planners were right before the event that it was protocol to pull the front 25, the front-right quarter of chairs. Why? To make room for those who can’t walk, to make room for the wheelchairs,” he said.
“Today, if we have one wheelchair at an event, it’s an outlier, and I can’t think of a better way to summarize the impact that biologics have had on our lives,” he said.
Biosimilar confidence
His group has launched “Operation: Biosimilar Confidence” which is designed to educate patients and physicians about the clinical value and scientific underpinnings of biosimilars, as well as the thorough development, review, and regulatory processes involved.
The goal of the project is to instill confidence in patients by helping them to understand the manufacturer’s safety track record, reliability of the biosimilar supply chain, and the availability to them of support services, if they make the switch to a biosimilar.
“Generics don’t have equivalent patient-support programs, and the projection is theoretically that [biosimilar] manufacturers won’t either. We will not accept that. We are going to do everything we can for those patients, to advocate for the continuation of the support programs that we rely on as patients,” he said.
Patient concerns
Surveys of patient concerns about biosimilars have highlighted four key areas:
- What is the manufacturer’s overall safety record in both biologic agents and small-molecule therapies?
- Supply-chain logistic – Will the manufacturer commit to consistent production and supply?
- Will biosimilar manufacturers provide patient support at levels equal to those offered by innovator biologic makers, and what kind of support will be available – phone, websites, social media, copays, etc.?
- Payer ethics – Will payers offer lower copays, deductibles, or premiums, and are payers as concerned as patients about product safety, supply chain, and support?
The implementation strategy for the campaign will focus on speaking directly to patients through CreakyJoints.org, partner Global Healthy Living Foundation, patient and physician organizations, social and conventional media, advertising, and one-on-one encounters.
“We have to talk directly and indirectly to employers and employee-advocacy groups. We have to let these big self-insured employers understand what the perspective of the patient is and what life is like thanks to these medicine, and why biosimilars are a critical component to the success of living with these conditions,” he said.
Advocates also have to work with the media to create “a surround-sound message that reaches all audiences with additional frequency.”
“We cannot allow Wall Street Journal business analysts to dictate the conversations about biosimilars. Why? They’re looking at one thing, and only one thing, and they’re ignoring the patient perspective,” Ginsberg said.
Lastly, patient groups need to work closely with payers, physician groups, and manufacturers to ensure that biosimilars can be smoothly integrated into the healthcare system, he emphasized.
“I want to be crystal clear here: We can’t wait for biosimilars. Bring it on! We want them,” he said.
EXPERT ANALYSIS FROM A BIOSIMILARS IN RHEUMATOLOGY SYMPOSIUM

FDA’s Woodcock: Give biosimilars a chance
BOSTON – Biosimilar drugs are not identical twins of original biologic agents, but there are very strong family ties, and the newcomer is expected to look and behave very much like its older relative, said Janet Woodcock, MD, director of the Center for Drug Evaluation and Research at the Food and Drug Administration.
Although biosimilars differ from generic, small-molecule drugs, concerns about the development of biosimilars mirrors the kerfuffle over generic drugs surrounding the passage of the Hatch-Waxman (Drug Price Competition and Patent Term Restoration) Act in 1984.

She noted that clinicians today are asking the same questions about biosimilars that were asked about generics three decades ago:
- Are biosimilars as effective and as safe as the originally licensed biopharmaceuticals?
- If pharmacists substitute biosimilars for prescribed biologics, will patients be adversely affected?
- Can biosimilars reduce the high cost of biologic therapy?
“In certain specialties, this skepticism has persisted to this very day,” she said.
ACA mandate
Biosimilars owe their existence in large measure to the Biologics Price Competition and Innovation Act of 2009 (BPCI), passed as a part of the Affordable Care Act and signed into law by President Obama in 2010.
The act created an abbreviated licensure pathway for biologic products that can be shown to be either biosimilar to or interchangeable with an FDA-licensed reference drug.
A biosimilar is defined as a biological product that is highly similar to the reference product “notwithstanding minor differences in clinically inactive components,” and with no clinically meaningful differences between it and the reference product in terms of purity, safety, and potency.
To be interchangeable, a biosimilar must be expected to produce the same clinical results as the reference drug in any given patient, and “for a product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the product and its reference product is not greater than the risk of using the reference product without such alternation or switch.”
The definition of interchangeability includes the understanding that the prescriber’s approval is not necessary for substitution of a biosimilar for its reference product.
“If we’re going to have that kind of switching, if they are going to be interchangeable, then we have to have a very high bar,” Dr. Woodcock said.
She added that “any pressure people are feeling to push their patients to biosimilars is from the reimbursement system. There is no non-prescriber switching allowed currently; however, that doesn’t say there isn’t pressure on prescribers to write a different prescription.”
Faster track and bridge to approval
The biosimilar development and approval requires only convincing demonstration of biosimilarity to an existing agent, rather than an independent finding of safety or effectiveness, and the purpose of clinical studies in this case is to address “residual uncertainties,” Dr. Woodcock said.
Drug developers and regulatory authorities alike “are having trouble getting their mind around this concept,” she said.
The FDA requires manufacturers to provide data in their biosimilar drug license applications demonstrating biosimilarity based on analytical studies, animal studies that include toxicity assessments, and one or more clinical studies that include information on immunogenicity and pharmacokinetics (PK) or pharmacodynamics (PD) that is sufficient to demonstrate the safety, purity, and potency of the candidate biosimilar.
The FDA is also allowing manufacturers to submit data from animal studies and specified clinical studies comparing a proposed biosimilar product with a product not licensed in the United States, “as long as we are convinced that the reference product is equivalent to the U.S. product,” Dr. Woodcock said.
This “analytical bridge” process was requested by manufacturers who began development of biosimilars in Europe. The European Medicines Agency approved a biosimilar process in 2005, and gave the nod to the first biosimilar agents to existing erythropoietin products in 2007.
Current and pending
As of early October 2016, 66 programs were enrolled in FDA’s Biosimilar Product Development Program, and CDER has received requests for meetings with manufacturers to discuss what tests and documents are required for the development of biosimilars to 20 different reference products.
The FDA is prohibited from publicly discussing the existence of a pending application unless it has been previously disclosed or acknowledged publicly with the manufacturer’s permission, Dr. Woodcock noted, but as of Oct. 10, 2016, seven companies have announced a total of 10 biologic license applications for biosimilars to etanercept (Enbrel), adalimumab (Humira), pegfilgrastim (Neulasta), epoetin alfa (Epogen/Procrit), filgrastim (Neupogen), and infliximab (Remicade).
The FDA has granted licenses to four biosimilars to date (the four-letter suffix is intended to differentiate biosimilars agents from other biosimilars to the same reference product):
- Zarxio (filgrastim-sndz).
- Inflectra (infliximab-dyyb).
- Erelzi (etanercept-szzs).
- Amjevita (adalimumab-atto).
Physician perspective
“Everything I have heard suggests that biosimilars will be useful, but the scientist in me is a skeptic,” commented Donald Massenburg, MD, PhD, a rheumatologist at Wheaton Franciscan Healthcare in Franklin, Wisc., in an interview.
“I feel better now after learning that current biosimilars are not considered interchangeable, meaning that there can’t be substitutions made without the treating physician’s consent,” he said.
He acknowledged that “I wouldn’t be that excited” if a specific biosimilar was approved for interchangeability and was given to his patients without his knowledge.
Dr. Massenburg pointed to data from the NOR-SWITCH trial comparing the biosimilar Remsima to the reference product Remicade for treatment of rheumatic diseases, psoriasis, and inflammatory bowel disease. The trial showed that Remsima was noninferior to the reference product.

“I would like to be able to say whether a patient should be switched to a biosimilar or not just because of that potential risk,” Dr. Massenburg said.
A rheumatologist in private practice in New England said that what’s really needed in rheumatology is not the availability of more drugs that act like other drugs, but innovative research into therapies with better targeted mechanism of action.
“We’ve been through the ‘me-too’ hype; we did that with nonsteroidal anti-inflammatory drugs,” said J. Scott Toder, MD, director of the Toder Rheumatology and Osteoporosis Center, Providence, R.I.
“I think we need to concentrate on innovative therapies, and we may be able to do something about the escalating price of the biologics on the market by creating drugs with new mechanism of action to actually increase competition and hopefully control prices. I don’t think that having multiple drugs with the same mechanism of action is in the best interest of our patients,” he said in an interview.
Dr. Woodcock, Dr. Massenburg, and Dr. Toder reported having no relevant disclosures.
BOSTON – Biosimilar drugs are not identical twins of original biologic agents, but there are very strong family ties, and the newcomer is expected to look and behave very much like its older relative, said Janet Woodcock, MD, director of the Center for Drug Evaluation and Research at the Food and Drug Administration.
Although biosimilars differ from generic, small-molecule drugs, concerns about the development of biosimilars mirrors the kerfuffle over generic drugs surrounding the passage of the Hatch-Waxman (Drug Price Competition and Patent Term Restoration) Act in 1984.
She noted that clinicians today are asking the same questions about biosimilars that were asked about generics three decades ago:
- Are biosimilars as effective and as safe as the originally licensed biopharmaceuticals?
- If pharmacists substitute biosimilars for prescribed biologics, will patients be adversely affected?
- Can biosimilars reduce the high cost of biologic therapy?
“In certain specialties, this skepticism has persisted to this very day,” she said.
ACA mandate
Biosimilars owe their existence in large measure to the Biologics Price Competition and Innovation Act of 2009 (BPCI), passed as a part of the Affordable Care Act and signed into law by President Obama in 2010.
The act created an abbreviated licensure pathway for biologic products that can be shown to be either biosimilar to or interchangeable with an FDA-licensed reference drug.
A biosimilar is defined as a biological product that is highly similar to the reference product “notwithstanding minor differences in clinically inactive components,” and with no clinically meaningful differences between it and the reference product in terms of purity, safety, and potency.
To be interchangeable, a biosimilar must be expected to produce the same clinical results as the reference drug in any given patient, and “for a product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the product and its reference product is not greater than the risk of using the reference product without such alternation or switch.”
The definition of interchangeability includes the understanding that the prescriber’s approval is not necessary for substitution of a biosimilar for its reference product.
“If we’re going to have that kind of switching, if they are going to be interchangeable, then we have to have a very high bar,” Dr. Woodcock said.
She added that “any pressure people are feeling to push their patients to biosimilars is from the reimbursement system. There is no non-prescriber switching allowed currently; however, that doesn’t say there isn’t pressure on prescribers to write a different prescription.”
Faster track and bridge to approval
The biosimilar development and approval requires only convincing demonstration of biosimilarity to an existing agent, rather than an independent finding of safety or effectiveness, and the purpose of clinical studies in this case is to address “residual uncertainties,” Dr. Woodcock said.
Drug developers and regulatory authorities alike “are having trouble getting their mind around this concept,” she said.
The FDA requires manufacturers to provide data in their biosimilar drug license applications demonstrating biosimilarity based on analytical studies, animal studies that include toxicity assessments, and one or more clinical studies that include information on immunogenicity and pharmacokinetics (PK) or pharmacodynamics (PD) that is sufficient to demonstrate the safety, purity, and potency of the candidate biosimilar.
The FDA is also allowing manufacturers to submit data from animal studies and specified clinical studies comparing a proposed biosimilar product with a product not licensed in the United States, “as long as we are convinced that the reference product is equivalent to the U.S. product,” Dr. Woodcock said.
This “analytical bridge” process was requested by manufacturers who began development of biosimilars in Europe. The European Medicines Agency approved a biosimilar process in 2005, and gave the nod to the first biosimilar agents to existing erythropoietin products in 2007.
Current and pending
As of early October 2016, 66 programs were enrolled in FDA’s Biosimilar Product Development Program, and CDER has received requests for meetings with manufacturers to discuss what tests and documents are required for the development of biosimilars to 20 different reference products.
The FDA is prohibited from publicly discussing the existence of a pending application unless it has been previously disclosed or acknowledged publicly with the manufacturer’s permission, Dr. Woodcock noted, but as of Oct. 10, 2016, seven companies have announced a total of 10 biologic license applications for biosimilars to etanercept (Enbrel), adalimumab (Humira), pegfilgrastim (Neulasta), epoetin alfa (Epogen/Procrit), filgrastim (Neupogen), and infliximab (Remicade).
The FDA has granted licenses to four biosimilars to date (the four-letter suffix is intended to differentiate biosimilars agents from other biosimilars to the same reference product):
- Zarxio (filgrastim-sndz).
- Inflectra (infliximab-dyyb).
- Erelzi (etanercept-szzs).
- Amjevita (adalimumab-atto).
Physician perspective
“Everything I have heard suggests that biosimilars will be useful, but the scientist in me is a skeptic,” commented Donald Massenburg, MD, PhD, a rheumatologist at Wheaton Franciscan Healthcare in Franklin, Wisc., in an interview.
“I feel better now after learning that current biosimilars are not considered interchangeable, meaning that there can’t be substitutions made without the treating physician’s consent,” he said.
He acknowledged that “I wouldn’t be that excited” if a specific biosimilar was approved for interchangeability and was given to his patients without his knowledge.
Dr. Massenburg pointed to data from the NOR-SWITCH trial comparing the biosimilar Remsima to the reference product Remicade for treatment of rheumatic diseases, psoriasis, and inflammatory bowel disease. The trial showed that Remsima was noninferior to the reference product.
“I would like to be able to say whether a patient should be switched to a biosimilar or not just because of that potential risk,” Dr. Massenburg said.
A rheumatologist in private practice in New England said that what’s really needed in rheumatology is not the availability of more drugs that act like other drugs, but innovative research into therapies with better targeted mechanism of action.
“We’ve been through the ‘me-too’ hype; we did that with nonsteroidal anti-inflammatory drugs,” said J. Scott Toder, MD, director of the Toder Rheumatology and Osteoporosis Center, Providence, R.I.
“I think we need to concentrate on innovative therapies, and we may be able to do something about the escalating price of the biologics on the market by creating drugs with new mechanism of action to actually increase competition and hopefully control prices. I don’t think that having multiple drugs with the same mechanism of action is in the best interest of our patients,” he said in an interview.
Dr. Woodcock, Dr. Massenburg, and Dr. Toder reported having no relevant disclosures.
BOSTON – Biosimilar drugs are not identical twins of original biologic agents, but there are very strong family ties, and the newcomer is expected to look and behave very much like its older relative, said Janet Woodcock, MD, director of the Center for Drug Evaluation and Research at the Food and Drug Administration.
Although biosimilars differ from generic, small-molecule drugs, concerns about the development of biosimilars mirrors the kerfuffle over generic drugs surrounding the passage of the Hatch-Waxman (Drug Price Competition and Patent Term Restoration) Act in 1984.
She noted that clinicians today are asking the same questions about biosimilars that were asked about generics three decades ago:
- Are biosimilars as effective and as safe as the originally licensed biopharmaceuticals?
- If pharmacists substitute biosimilars for prescribed biologics, will patients be adversely affected?
- Can biosimilars reduce the high cost of biologic therapy?
“In certain specialties, this skepticism has persisted to this very day,” she said.
ACA mandate
Biosimilars owe their existence in large measure to the Biologics Price Competition and Innovation Act of 2009 (BPCI), passed as a part of the Affordable Care Act and signed into law by President Obama in 2010.
The act created an abbreviated licensure pathway for biologic products that can be shown to be either biosimilar to or interchangeable with an FDA-licensed reference drug.
A biosimilar is defined as a biological product that is highly similar to the reference product “notwithstanding minor differences in clinically inactive components,” and with no clinically meaningful differences between it and the reference product in terms of purity, safety, and potency.
To be interchangeable, a biosimilar must be expected to produce the same clinical results as the reference drug in any given patient, and “for a product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the product and its reference product is not greater than the risk of using the reference product without such alternation or switch.”
The definition of interchangeability includes the understanding that the prescriber’s approval is not necessary for substitution of a biosimilar for its reference product.
“If we’re going to have that kind of switching, if they are going to be interchangeable, then we have to have a very high bar,” Dr. Woodcock said.
She added that “any pressure people are feeling to push their patients to biosimilars is from the reimbursement system. There is no non-prescriber switching allowed currently; however, that doesn’t say there isn’t pressure on prescribers to write a different prescription.”
Faster track and bridge to approval
The biosimilar development and approval requires only convincing demonstration of biosimilarity to an existing agent, rather than an independent finding of safety or effectiveness, and the purpose of clinical studies in this case is to address “residual uncertainties,” Dr. Woodcock said.
Drug developers and regulatory authorities alike “are having trouble getting their mind around this concept,” she said.
The FDA requires manufacturers to provide data in their biosimilar drug license applications demonstrating biosimilarity based on analytical studies, animal studies that include toxicity assessments, and one or more clinical studies that include information on immunogenicity and pharmacokinetics (PK) or pharmacodynamics (PD) that is sufficient to demonstrate the safety, purity, and potency of the candidate biosimilar.
The FDA is also allowing manufacturers to submit data from animal studies and specified clinical studies comparing a proposed biosimilar product with a product not licensed in the United States, “as long as we are convinced that the reference product is equivalent to the U.S. product,” Dr. Woodcock said.
This “analytical bridge” process was requested by manufacturers who began development of biosimilars in Europe. The European Medicines Agency approved a biosimilar process in 2005, and gave the nod to the first biosimilar agents to existing erythropoietin products in 2007.
Current and pending
As of early October 2016, 66 programs were enrolled in FDA’s Biosimilar Product Development Program, and CDER has received requests for meetings with manufacturers to discuss what tests and documents are required for the development of biosimilars to 20 different reference products.
The FDA is prohibited from publicly discussing the existence of a pending application unless it has been previously disclosed or acknowledged publicly with the manufacturer’s permission, Dr. Woodcock noted, but as of Oct. 10, 2016, seven companies have announced a total of 10 biologic license applications for biosimilars to etanercept (Enbrel), adalimumab (Humira), pegfilgrastim (Neulasta), epoetin alfa (Epogen/Procrit), filgrastim (Neupogen), and infliximab (Remicade).
The FDA has granted licenses to four biosimilars to date (the four-letter suffix is intended to differentiate biosimilars agents from other biosimilars to the same reference product):
- Zarxio (filgrastim-sndz).
- Inflectra (infliximab-dyyb).
- Erelzi (etanercept-szzs).
- Amjevita (adalimumab-atto).
Physician perspective
“Everything I have heard suggests that biosimilars will be useful, but the scientist in me is a skeptic,” commented Donald Massenburg, MD, PhD, a rheumatologist at Wheaton Franciscan Healthcare in Franklin, Wisc., in an interview.
“I feel better now after learning that current biosimilars are not considered interchangeable, meaning that there can’t be substitutions made without the treating physician’s consent,” he said.
He acknowledged that “I wouldn’t be that excited” if a specific biosimilar was approved for interchangeability and was given to his patients without his knowledge.
Dr. Massenburg pointed to data from the NOR-SWITCH trial comparing the biosimilar Remsima to the reference product Remicade for treatment of rheumatic diseases, psoriasis, and inflammatory bowel disease. The trial showed that Remsima was noninferior to the reference product.
“I would like to be able to say whether a patient should be switched to a biosimilar or not just because of that potential risk,” Dr. Massenburg said.
A rheumatologist in private practice in New England said that what’s really needed in rheumatology is not the availability of more drugs that act like other drugs, but innovative research into therapies with better targeted mechanism of action.
“We’ve been through the ‘me-too’ hype; we did that with nonsteroidal anti-inflammatory drugs,” said J. Scott Toder, MD, director of the Toder Rheumatology and Osteoporosis Center, Providence, R.I.
“I think we need to concentrate on innovative therapies, and we may be able to do something about the escalating price of the biologics on the market by creating drugs with new mechanism of action to actually increase competition and hopefully control prices. I don’t think that having multiple drugs with the same mechanism of action is in the best interest of our patients,” he said in an interview.
Dr. Woodcock, Dr. Massenburg, and Dr. Toder reported having no relevant disclosures.
EXPERT ANALYSIS FROM A BIOSIMILARS IN RHEUMATOLOGY SYMPOSIUM
ACR 2016 continues big buffet of basic and clinical science sessions
This year’s annual meeting of the American College of Rheumatology will feature cutting-edge research and results of studies that directly affect how attendees will manage patients once they are back in the clinical setting, according to both Richard Loeser, MD, program chair of the Annual Meeting Planning Committee (AMPC), and Gregory Gardner, MD, clinical subchair of the AMPC, who suggested special sessions of interest culled from the more than 450 sessions to be presented.
“It is an exciting time in rheumatology. Basic research is being translated into new therapies before our very eyes. Areas on the program this year that have translational potential include immunometabolism, blocking interleukin-1 (IL-1), T-cell receptor signaling, and meta-analysis of gene expression data. The meeting will also feature trials that refine and advance the management of rheumatologic diseases, including results on studies of new biologics,” Dr. Loeser said.

Hot sessions
Luke O’Neill, MD, will talk about immunometabolism Monday at 7:30 a.m. This session will explore a newly described connection between energy metabolism and the immune system and the link with inflammation.
Charles Dinarello, MD, will give the Philip Hensch Memorial Lecture Sunday at 8:30 a.m. on blocking IL-1 in inflammatory diseases. He will cover a host of diseases from gout to cancer, Dr. Loeser noted.
Another hot topic, T-cell receptor signaling in autoimmune diseases and the development of new therapies, will be discussed by Arthur Weiss, MD, Tuesday morning at 7:30 a.m.
Tuesday at 11:00 a.m., Peter Lipsky, MD, will tackle big data mining, presenting a meta-analysis of gene expression datasets to identify novel pathways and targets in systemic lupus erythematosus (SLE).
“SLE lags behind rheumatoid arthritis in therapeutic advances. A number of trials of biologics have failed in SLE, whereas they have been found effective in rheumatoid arthritis,” Dr. Loeser explained.
Clinical slant
Sunday’s Plenary session at 11:00 a.m. will feature several top-rated abstracts, among them results of a phase III study on tocilizumab in giant cell arteritis to be presented by John Stone, MD. “Tocilizumab is a major breakthrough as a steroid-sparing treatment for the most common form of vasculitis that affects older adults,” Dr. Loeser said.
At 2:30 p.m. on Sunday at The Great Debate, Paul Emery, MD, and Arthur Kavanaugh, MD, will tackle the very important clinical topic of “To Taper or Not to Taper? – Biologic DMARDs in Low Rheumatoid Arthritis Disease Activity.”
“People aren’t sure what to do. The fear with tapering is rebound, with the disease coming back even more forcefully. There is new evidence to suggest that tapering may be safe under certain circumstances. This session should inform attendees on how to make the decision to taper and on the best way to do it,” Dr. Loeser commented.
The Late-Breaking Abstract session on Tuesday at 4:30 p.m. will feature six clinical trials. Dr. Loeser singled out a study to be presented by Elaine Husni, MD, on “Vascular Safety of Celecoxib versus Ibuprofen or Naproxen” in more than 20,000 patients with osteoarthritis or rheumatoid arthritis.
“The fear is that COX-2 inhibitors have increased cardiovascular risk. The data from this study that will be presented at the meeting should answer the question of whether or not this is true in patients with arthritis,” Dr. Loeser explained.
Wednesday at 7:30 a.m. Candida Fratazzi, MD, will talk about “Emerging Biosimilars in Therapeutic Management,” a subject of great interest since they have the potential to be equally effective and less expensive than current biologics.
Two “bookends” of the meeting will frame the opening and closing. Sunday at 7:30 a.m., the “Year in Review” session will feature the best published studies on rheumatologic diseases from the past year, based on the judgment of two experts. Ingrid Lundberg, MD, will present the best clinical studies and Bruce Cronstein, MD, will present the best basic science studies. Wednesday at 7:30 a.m., John Cush, MD, and Dr. Kavanaugh will present the “Rheumatology Roundup” of the best abstracts and put them into context. “This session is usually quite entertaining,” Dr. Loeser said.

More sessions of clinical import
“In keeping with our meeting theme of fine-tuning our care of patients with rheumatic disease, I want to point out several sessions,” Dr. Gardner said.
Attendees interested in sessions on clinical applicability will have to choose between two different sessions Monday at 4:30 p.m.: one on dermatomyositis, a relatively rare but difficult-to-treat entity, and the other about treatment of the patient with rheumatoid arthritis when the patient is not well and suffering from comorbidities.
Monday at 8:30 a.m., an “Osteoporosis Update” will give listeners perspective on current and future therapies.
Sunday at 2:30 p.m., new guidelines for steroid-induced osteoporosis will be presented.
“Four or five sessions on the Tech Track will show rheumatologists how they can improve their practice by using technology,” Dr. Gardner said. “Several high-quality sessions are important to educators, including ‘Flipped Classroom, Technology, and Reflection’ [Monday at 12:30 p.m.] and ‘Year in Review’ [Sunday at 1:00 p.m.].”
Monday at 11:00 a.m., the Plenary session will feature Workforce Study results on how many rheumatologists will be needed in the year 2030, and in which geographic locations. This session will also include a discussion of the impact of part-time rheumatologists.
“Two sessions I am excited about are ‘Treat to Target in 2016,’ Tuesday at 4:30 p.m., and ‘Rheumatic Diseases in Native Americans,’ Sunday at 11:00 a.m.,” Dr. Gardner noted. “Concurrent abstract sessions throughout the meeting will feature discussions on new biologics, small molecules, and gene therapy.”
This year’s annual meeting of the American College of Rheumatology will feature cutting-edge research and results of studies that directly affect how attendees will manage patients once they are back in the clinical setting, according to both Richard Loeser, MD, program chair of the Annual Meeting Planning Committee (AMPC), and Gregory Gardner, MD, clinical subchair of the AMPC, who suggested special sessions of interest culled from the more than 450 sessions to be presented.
“It is an exciting time in rheumatology. Basic research is being translated into new therapies before our very eyes. Areas on the program this year that have translational potential include immunometabolism, blocking interleukin-1 (IL-1), T-cell receptor signaling, and meta-analysis of gene expression data. The meeting will also feature trials that refine and advance the management of rheumatologic diseases, including results on studies of new biologics,” Dr. Loeser said.
Hot sessions
Luke O’Neill, MD, will talk about immunometabolism Monday at 7:30 a.m. This session will explore a newly described connection between energy metabolism and the immune system and the link with inflammation.
Charles Dinarello, MD, will give the Philip Hensch Memorial Lecture Sunday at 8:30 a.m. on blocking IL-1 in inflammatory diseases. He will cover a host of diseases from gout to cancer, Dr. Loeser noted.
Another hot topic, T-cell receptor signaling in autoimmune diseases and the development of new therapies, will be discussed by Arthur Weiss, MD, Tuesday morning at 7:30 a.m.
Tuesday at 11:00 a.m., Peter Lipsky, MD, will tackle big data mining, presenting a meta-analysis of gene expression datasets to identify novel pathways and targets in systemic lupus erythematosus (SLE).
“SLE lags behind rheumatoid arthritis in therapeutic advances. A number of trials of biologics have failed in SLE, whereas they have been found effective in rheumatoid arthritis,” Dr. Loeser explained.
Clinical slant
Sunday’s Plenary session at 11:00 a.m. will feature several top-rated abstracts, among them results of a phase III study on tocilizumab in giant cell arteritis to be presented by John Stone, MD. “Tocilizumab is a major breakthrough as a steroid-sparing treatment for the most common form of vasculitis that affects older adults,” Dr. Loeser said.
At 2:30 p.m. on Sunday at The Great Debate, Paul Emery, MD, and Arthur Kavanaugh, MD, will tackle the very important clinical topic of “To Taper or Not to Taper? – Biologic DMARDs in Low Rheumatoid Arthritis Disease Activity.”
“People aren’t sure what to do. The fear with tapering is rebound, with the disease coming back even more forcefully. There is new evidence to suggest that tapering may be safe under certain circumstances. This session should inform attendees on how to make the decision to taper and on the best way to do it,” Dr. Loeser commented.
The Late-Breaking Abstract session on Tuesday at 4:30 p.m. will feature six clinical trials. Dr. Loeser singled out a study to be presented by Elaine Husni, MD, on “Vascular Safety of Celecoxib versus Ibuprofen or Naproxen” in more than 20,000 patients with osteoarthritis or rheumatoid arthritis.
“The fear is that COX-2 inhibitors have increased cardiovascular risk. The data from this study that will be presented at the meeting should answer the question of whether or not this is true in patients with arthritis,” Dr. Loeser explained.
Wednesday at 7:30 a.m. Candida Fratazzi, MD, will talk about “Emerging Biosimilars in Therapeutic Management,” a subject of great interest since they have the potential to be equally effective and less expensive than current biologics.
Two “bookends” of the meeting will frame the opening and closing. Sunday at 7:30 a.m., the “Year in Review” session will feature the best published studies on rheumatologic diseases from the past year, based on the judgment of two experts. Ingrid Lundberg, MD, will present the best clinical studies and Bruce Cronstein, MD, will present the best basic science studies. Wednesday at 7:30 a.m., John Cush, MD, and Dr. Kavanaugh will present the “Rheumatology Roundup” of the best abstracts and put them into context. “This session is usually quite entertaining,” Dr. Loeser said.
More sessions of clinical import
“In keeping with our meeting theme of fine-tuning our care of patients with rheumatic disease, I want to point out several sessions,” Dr. Gardner said.
Attendees interested in sessions on clinical applicability will have to choose between two different sessions Monday at 4:30 p.m.: one on dermatomyositis, a relatively rare but difficult-to-treat entity, and the other about treatment of the patient with rheumatoid arthritis when the patient is not well and suffering from comorbidities.
Monday at 8:30 a.m., an “Osteoporosis Update” will give listeners perspective on current and future therapies.
Sunday at 2:30 p.m., new guidelines for steroid-induced osteoporosis will be presented.
“Four or five sessions on the Tech Track will show rheumatologists how they can improve their practice by using technology,” Dr. Gardner said. “Several high-quality sessions are important to educators, including ‘Flipped Classroom, Technology, and Reflection’ [Monday at 12:30 p.m.] and ‘Year in Review’ [Sunday at 1:00 p.m.].”
Monday at 11:00 a.m., the Plenary session will feature Workforce Study results on how many rheumatologists will be needed in the year 2030, and in which geographic locations. This session will also include a discussion of the impact of part-time rheumatologists.
“Two sessions I am excited about are ‘Treat to Target in 2016,’ Tuesday at 4:30 p.m., and ‘Rheumatic Diseases in Native Americans,’ Sunday at 11:00 a.m.,” Dr. Gardner noted. “Concurrent abstract sessions throughout the meeting will feature discussions on new biologics, small molecules, and gene therapy.”
This year’s annual meeting of the American College of Rheumatology will feature cutting-edge research and results of studies that directly affect how attendees will manage patients once they are back in the clinical setting, according to both Richard Loeser, MD, program chair of the Annual Meeting Planning Committee (AMPC), and Gregory Gardner, MD, clinical subchair of the AMPC, who suggested special sessions of interest culled from the more than 450 sessions to be presented.
“It is an exciting time in rheumatology. Basic research is being translated into new therapies before our very eyes. Areas on the program this year that have translational potential include immunometabolism, blocking interleukin-1 (IL-1), T-cell receptor signaling, and meta-analysis of gene expression data. The meeting will also feature trials that refine and advance the management of rheumatologic diseases, including results on studies of new biologics,” Dr. Loeser said.
Hot sessions
Luke O’Neill, MD, will talk about immunometabolism Monday at 7:30 a.m. This session will explore a newly described connection between energy metabolism and the immune system and the link with inflammation.
Charles Dinarello, MD, will give the Philip Hensch Memorial Lecture Sunday at 8:30 a.m. on blocking IL-1 in inflammatory diseases. He will cover a host of diseases from gout to cancer, Dr. Loeser noted.
Another hot topic, T-cell receptor signaling in autoimmune diseases and the development of new therapies, will be discussed by Arthur Weiss, MD, Tuesday morning at 7:30 a.m.
Tuesday at 11:00 a.m., Peter Lipsky, MD, will tackle big data mining, presenting a meta-analysis of gene expression datasets to identify novel pathways and targets in systemic lupus erythematosus (SLE).
“SLE lags behind rheumatoid arthritis in therapeutic advances. A number of trials of biologics have failed in SLE, whereas they have been found effective in rheumatoid arthritis,” Dr. Loeser explained.
Clinical slant
Sunday’s Plenary session at 11:00 a.m. will feature several top-rated abstracts, among them results of a phase III study on tocilizumab in giant cell arteritis to be presented by John Stone, MD. “Tocilizumab is a major breakthrough as a steroid-sparing treatment for the most common form of vasculitis that affects older adults,” Dr. Loeser said.
At 2:30 p.m. on Sunday at The Great Debate, Paul Emery, MD, and Arthur Kavanaugh, MD, will tackle the very important clinical topic of “To Taper or Not to Taper? – Biologic DMARDs in Low Rheumatoid Arthritis Disease Activity.”
“People aren’t sure what to do. The fear with tapering is rebound, with the disease coming back even more forcefully. There is new evidence to suggest that tapering may be safe under certain circumstances. This session should inform attendees on how to make the decision to taper and on the best way to do it,” Dr. Loeser commented.
The Late-Breaking Abstract session on Tuesday at 4:30 p.m. will feature six clinical trials. Dr. Loeser singled out a study to be presented by Elaine Husni, MD, on “Vascular Safety of Celecoxib versus Ibuprofen or Naproxen” in more than 20,000 patients with osteoarthritis or rheumatoid arthritis.
“The fear is that COX-2 inhibitors have increased cardiovascular risk. The data from this study that will be presented at the meeting should answer the question of whether or not this is true in patients with arthritis,” Dr. Loeser explained.
Wednesday at 7:30 a.m. Candida Fratazzi, MD, will talk about “Emerging Biosimilars in Therapeutic Management,” a subject of great interest since they have the potential to be equally effective and less expensive than current biologics.
Two “bookends” of the meeting will frame the opening and closing. Sunday at 7:30 a.m., the “Year in Review” session will feature the best published studies on rheumatologic diseases from the past year, based on the judgment of two experts. Ingrid Lundberg, MD, will present the best clinical studies and Bruce Cronstein, MD, will present the best basic science studies. Wednesday at 7:30 a.m., John Cush, MD, and Dr. Kavanaugh will present the “Rheumatology Roundup” of the best abstracts and put them into context. “This session is usually quite entertaining,” Dr. Loeser said.
More sessions of clinical import
“In keeping with our meeting theme of fine-tuning our care of patients with rheumatic disease, I want to point out several sessions,” Dr. Gardner said.
Attendees interested in sessions on clinical applicability will have to choose between two different sessions Monday at 4:30 p.m.: one on dermatomyositis, a relatively rare but difficult-to-treat entity, and the other about treatment of the patient with rheumatoid arthritis when the patient is not well and suffering from comorbidities.
Monday at 8:30 a.m., an “Osteoporosis Update” will give listeners perspective on current and future therapies.
Sunday at 2:30 p.m., new guidelines for steroid-induced osteoporosis will be presented.
“Four or five sessions on the Tech Track will show rheumatologists how they can improve their practice by using technology,” Dr. Gardner said. “Several high-quality sessions are important to educators, including ‘Flipped Classroom, Technology, and Reflection’ [Monday at 12:30 p.m.] and ‘Year in Review’ [Sunday at 1:00 p.m.].”
Monday at 11:00 a.m., the Plenary session will feature Workforce Study results on how many rheumatologists will be needed in the year 2030, and in which geographic locations. This session will also include a discussion of the impact of part-time rheumatologists.
“Two sessions I am excited about are ‘Treat to Target in 2016,’ Tuesday at 4:30 p.m., and ‘Rheumatic Diseases in Native Americans,’ Sunday at 11:00 a.m.,” Dr. Gardner noted. “Concurrent abstract sessions throughout the meeting will feature discussions on new biologics, small molecules, and gene therapy.”
FROM THE ACR ANNUAL MEETING
Biosimilars face barriers to broader acceptance
BOSTON – For rheumatologists, the primary selling points of biosimilar agents are their presumed efficacy and safety, and their promised cost savings, compared with the reference product, an original biologic agent.
But market forces, patient worries, and provider concerns threaten to file off at least some of biosimilars’ presumed competitive edge, says a rheumatologist involved in the development of biosimilars to treat rheumatic diseases.
“The availability of lower-priced biosimilars, if they’re truly lower priced, should decrease the cost of treating patients; if they’re not lower cost, then this advantage is not holding true,” said Jonathan Kay, MD, professor of medicine at UMass Memorial Medical Center in Worcester.

But the world is far from perfect, and there are a host of educational, financial, and societal barriers to more widespread acceptance of these agents, he said.
Misperceptions
A common concern among patients and uninformed providers is that biosimilars are the same as biomimics – cheap, usually foreign-made knockoffs of existing drugs that have not undergone regulatory scrutiny and may be neither effective nor safe.
“Biomimics scare patients and scare physicians,” he said, “and misunderstanding of the regulatory approval process, or lack of understanding of the regulatory review or approval process, also intimidates individuals about biosimilars.”
Acceptance of biosimilars is also hampered by a lack of long-term, real-world efficacy and safety data. This is due, in part, to the “inverted pyramid” nature of the biosimilar development and approval process, which starts with myriad analytical, chemical, and functional characterization studies; nonclinical studies; and pharmacokinetic and pharmacodynamic analyses, but only one or two clinical trials. In contrast, the development process for innovator biologics starts with molecular characterization, then moves through the traditional drug development stages of laboratory and preclinical studies, phase I and II trials, and finally phase III registration trials and post-marketing studies.
Because most of the drug development for biosimilars happens behind the scenes, patients and their physicians are often leery about the finished product, no matter how rigorously it was developed. Similarly, there is a paucity of data on long-term immunogenicity of biosimilars, Dr. Kay noted.
Extrapolating about extrapolation
The idea of extrapolation of indications for biosimilars is also an alien concept to many. Simply put, if a reference product is approved for rheumatoid arthritis and has additional indications for treatment of psoriatic arthritis, the approved biosimilar to the original can be extrapolated to be effective against the same indications, even if it has only been tested against one of the indications.
“We have a tremendous amount of clinical experience not only in randomized, controlled clinical trials but also years of marketing experience showing that the reference product is effective and safe for each of the indications for which it’s been approved, and knowing that the biosimilar is essentially like another [manufacturing] run of the reference product allows us to extrapolate indications. But until you become comfortable with accepting that concept, it is certainly a barrier to biosimilars,” he said.
Patients are also scared by the issue of interchangeability, which would allow pharmacists to substitute one product for another without approval of either the patient or the prescribing physician. However, there are currently no biosimilar agents approved as interchangeable by the Food and Drug Administration.
What cost savings?
In the United States, the price of marketed biosimilars has been only 15% lower than that of the reference product, Dr. Kay noted, and multiple discount programs and rebates offered by marketers of the reference product make it difficult to figure out the actual costs of the drugs to payers and whether biosimilars are actually any cheaper.
Additionally, clinicians may be reluctant to adopt biosimilars because of a Centers for Medicare & Medicaid Services billing quirk that currently offers only a single “J” code with a blended reimbursement for all biosimilar agents of a reference product, regardless of cost differences.
Nordic competition model
Dr. Kay pointed to the Norwegian Tender System as a model for using biosimilars to drive down drug costs.
The Norwegian Drug Procurement Cooperation (LIS) annually procures drugs for all publicly funded hospitals, which ensures that prices offered to hospitals for patent-protected medicines are lower than those offered for distribution to wholesalers or pharmacies. Under this system, cost is the driving factor, assuming that drugs in a similar class have similar efficacy and safety, he said.
In 2015, when Norway was purchasing infliximab, it chose Remsima, the biosimilar, over the reference product Remicade, resulting in a 69% cost saving.
The potential interchangeability of Remsima and Remicade in patients with rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, ulcerative colitis, Crohn’s disease, and chronic plaque psoriasis is currently being explored in the NOR-SWITCH trial.
Dr. Kay disclosed receiving institutional grants from AbbVie, Genentech, Roche Laboratories, and UCB, and serving as a consultant for those companies.
BOSTON – For rheumatologists, the primary selling points of biosimilar agents are their presumed efficacy and safety, and their promised cost savings, compared with the reference product, an original biologic agent.
But market forces, patient worries, and provider concerns threaten to file off at least some of biosimilars’ presumed competitive edge, says a rheumatologist involved in the development of biosimilars to treat rheumatic diseases.
“The availability of lower-priced biosimilars, if they’re truly lower priced, should decrease the cost of treating patients; if they’re not lower cost, then this advantage is not holding true,” said Jonathan Kay, MD, professor of medicine at UMass Memorial Medical Center in Worcester.
But the world is far from perfect, and there are a host of educational, financial, and societal barriers to more widespread acceptance of these agents, he said.
Misperceptions
A common concern among patients and uninformed providers is that biosimilars are the same as biomimics – cheap, usually foreign-made knockoffs of existing drugs that have not undergone regulatory scrutiny and may be neither effective nor safe.
“Biomimics scare patients and scare physicians,” he said, “and misunderstanding of the regulatory approval process, or lack of understanding of the regulatory review or approval process, also intimidates individuals about biosimilars.”
Acceptance of biosimilars is also hampered by a lack of long-term, real-world efficacy and safety data. This is due, in part, to the “inverted pyramid” nature of the biosimilar development and approval process, which starts with myriad analytical, chemical, and functional characterization studies; nonclinical studies; and pharmacokinetic and pharmacodynamic analyses, but only one or two clinical trials. In contrast, the development process for innovator biologics starts with molecular characterization, then moves through the traditional drug development stages of laboratory and preclinical studies, phase I and II trials, and finally phase III registration trials and post-marketing studies.
Because most of the drug development for biosimilars happens behind the scenes, patients and their physicians are often leery about the finished product, no matter how rigorously it was developed. Similarly, there is a paucity of data on long-term immunogenicity of biosimilars, Dr. Kay noted.
Extrapolating about extrapolation
The idea of extrapolation of indications for biosimilars is also an alien concept to many. Simply put, if a reference product is approved for rheumatoid arthritis and has additional indications for treatment of psoriatic arthritis, the approved biosimilar to the original can be extrapolated to be effective against the same indications, even if it has only been tested against one of the indications.
“We have a tremendous amount of clinical experience not only in randomized, controlled clinical trials but also years of marketing experience showing that the reference product is effective and safe for each of the indications for which it’s been approved, and knowing that the biosimilar is essentially like another [manufacturing] run of the reference product allows us to extrapolate indications. But until you become comfortable with accepting that concept, it is certainly a barrier to biosimilars,” he said.
Patients are also scared by the issue of interchangeability, which would allow pharmacists to substitute one product for another without approval of either the patient or the prescribing physician. However, there are currently no biosimilar agents approved as interchangeable by the Food and Drug Administration.
What cost savings?
In the United States, the price of marketed biosimilars has been only 15% lower than that of the reference product, Dr. Kay noted, and multiple discount programs and rebates offered by marketers of the reference product make it difficult to figure out the actual costs of the drugs to payers and whether biosimilars are actually any cheaper.
Additionally, clinicians may be reluctant to adopt biosimilars because of a Centers for Medicare & Medicaid Services billing quirk that currently offers only a single “J” code with a blended reimbursement for all biosimilar agents of a reference product, regardless of cost differences.
Nordic competition model
Dr. Kay pointed to the Norwegian Tender System as a model for using biosimilars to drive down drug costs.
The Norwegian Drug Procurement Cooperation (LIS) annually procures drugs for all publicly funded hospitals, which ensures that prices offered to hospitals for patent-protected medicines are lower than those offered for distribution to wholesalers or pharmacies. Under this system, cost is the driving factor, assuming that drugs in a similar class have similar efficacy and safety, he said.
In 2015, when Norway was purchasing infliximab, it chose Remsima, the biosimilar, over the reference product Remicade, resulting in a 69% cost saving.
The potential interchangeability of Remsima and Remicade in patients with rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, ulcerative colitis, Crohn’s disease, and chronic plaque psoriasis is currently being explored in the NOR-SWITCH trial.
Dr. Kay disclosed receiving institutional grants from AbbVie, Genentech, Roche Laboratories, and UCB, and serving as a consultant for those companies.
BOSTON – For rheumatologists, the primary selling points of biosimilar agents are their presumed efficacy and safety, and their promised cost savings, compared with the reference product, an original biologic agent.
But market forces, patient worries, and provider concerns threaten to file off at least some of biosimilars’ presumed competitive edge, says a rheumatologist involved in the development of biosimilars to treat rheumatic diseases.
“The availability of lower-priced biosimilars, if they’re truly lower priced, should decrease the cost of treating patients; if they’re not lower cost, then this advantage is not holding true,” said Jonathan Kay, MD, professor of medicine at UMass Memorial Medical Center in Worcester.
But the world is far from perfect, and there are a host of educational, financial, and societal barriers to more widespread acceptance of these agents, he said.
Misperceptions
A common concern among patients and uninformed providers is that biosimilars are the same as biomimics – cheap, usually foreign-made knockoffs of existing drugs that have not undergone regulatory scrutiny and may be neither effective nor safe.
“Biomimics scare patients and scare physicians,” he said, “and misunderstanding of the regulatory approval process, or lack of understanding of the regulatory review or approval process, also intimidates individuals about biosimilars.”
Acceptance of biosimilars is also hampered by a lack of long-term, real-world efficacy and safety data. This is due, in part, to the “inverted pyramid” nature of the biosimilar development and approval process, which starts with myriad analytical, chemical, and functional characterization studies; nonclinical studies; and pharmacokinetic and pharmacodynamic analyses, but only one or two clinical trials. In contrast, the development process for innovator biologics starts with molecular characterization, then moves through the traditional drug development stages of laboratory and preclinical studies, phase I and II trials, and finally phase III registration trials and post-marketing studies.
Because most of the drug development for biosimilars happens behind the scenes, patients and their physicians are often leery about the finished product, no matter how rigorously it was developed. Similarly, there is a paucity of data on long-term immunogenicity of biosimilars, Dr. Kay noted.
Extrapolating about extrapolation
The idea of extrapolation of indications for biosimilars is also an alien concept to many. Simply put, if a reference product is approved for rheumatoid arthritis and has additional indications for treatment of psoriatic arthritis, the approved biosimilar to the original can be extrapolated to be effective against the same indications, even if it has only been tested against one of the indications.
“We have a tremendous amount of clinical experience not only in randomized, controlled clinical trials but also years of marketing experience showing that the reference product is effective and safe for each of the indications for which it’s been approved, and knowing that the biosimilar is essentially like another [manufacturing] run of the reference product allows us to extrapolate indications. But until you become comfortable with accepting that concept, it is certainly a barrier to biosimilars,” he said.
Patients are also scared by the issue of interchangeability, which would allow pharmacists to substitute one product for another without approval of either the patient or the prescribing physician. However, there are currently no biosimilar agents approved as interchangeable by the Food and Drug Administration.
What cost savings?
In the United States, the price of marketed biosimilars has been only 15% lower than that of the reference product, Dr. Kay noted, and multiple discount programs and rebates offered by marketers of the reference product make it difficult to figure out the actual costs of the drugs to payers and whether biosimilars are actually any cheaper.
Additionally, clinicians may be reluctant to adopt biosimilars because of a Centers for Medicare & Medicaid Services billing quirk that currently offers only a single “J” code with a blended reimbursement for all biosimilar agents of a reference product, regardless of cost differences.
Nordic competition model
Dr. Kay pointed to the Norwegian Tender System as a model for using biosimilars to drive down drug costs.
The Norwegian Drug Procurement Cooperation (LIS) annually procures drugs for all publicly funded hospitals, which ensures that prices offered to hospitals for patent-protected medicines are lower than those offered for distribution to wholesalers or pharmacies. Under this system, cost is the driving factor, assuming that drugs in a similar class have similar efficacy and safety, he said.
In 2015, when Norway was purchasing infliximab, it chose Remsima, the biosimilar, over the reference product Remicade, resulting in a 69% cost saving.
The potential interchangeability of Remsima and Remicade in patients with rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, ulcerative colitis, Crohn’s disease, and chronic plaque psoriasis is currently being explored in the NOR-SWITCH trial.
Dr. Kay disclosed receiving institutional grants from AbbVie, Genentech, Roche Laboratories, and UCB, and serving as a consultant for those companies.
EXPERT ANALYSIS FROM A BIOSIMILARS IN RHEUMATOLOGY SYMPOSIUM
Rheumatoid arthritis increases heart failure risk
ROME – Rheumatoid arthritis is associated with a significantly increased risk of hospitalization for heart failure, according to a nationwide Danish study, Usman Khalid, MD, reported at the annual congress of the European Society of Cardiology.
As a chronic systemic inflammatory disease, rheumatoid arthritis (RA) has been associated with increased risk of a variety of comorbid conditions, including cardiovascular disease. But RA’s relationship specifically with heart failure hasn’t previously been looked at in the comprehensive way that’s possible in Denmark, where linked national registries enable researchers to follow health issues in the entire population from birth to death, noted Dr. Khalid of the University of Copenhagen.

In an analysis adjusted for age, sex, and calendar year, individuals with RA were 83% more likely to be hospitalized for heart failure than were the non-RA Danish population. In a fully adjusted analysis that controlled for those potential confounders as well as comorbid conditions, smoking, alcohol intake, socioeconomic status, and prescription medications, patients with RA remained at a statistically significant and clinically meaningful 38% increased risk of heart failure hospitalization.
Further studies are planned to determine the underlying mechanisms of this association, Dr. Khalid added.
He reported having no financial conflicts of interest regarding this study, which was supported by an unrestricted grant from Leo Pharma.
ROME – Rheumatoid arthritis is associated with a significantly increased risk of hospitalization for heart failure, according to a nationwide Danish study, Usman Khalid, MD, reported at the annual congress of the European Society of Cardiology.
As a chronic systemic inflammatory disease, rheumatoid arthritis (RA) has been associated with increased risk of a variety of comorbid conditions, including cardiovascular disease. But RA’s relationship specifically with heart failure hasn’t previously been looked at in the comprehensive way that’s possible in Denmark, where linked national registries enable researchers to follow health issues in the entire population from birth to death, noted Dr. Khalid of the University of Copenhagen.
In an analysis adjusted for age, sex, and calendar year, individuals with RA were 83% more likely to be hospitalized for heart failure than were the non-RA Danish population. In a fully adjusted analysis that controlled for those potential confounders as well as comorbid conditions, smoking, alcohol intake, socioeconomic status, and prescription medications, patients with RA remained at a statistically significant and clinically meaningful 38% increased risk of heart failure hospitalization.
Further studies are planned to determine the underlying mechanisms of this association, Dr. Khalid added.
He reported having no financial conflicts of interest regarding this study, which was supported by an unrestricted grant from Leo Pharma.
ROME – Rheumatoid arthritis is associated with a significantly increased risk of hospitalization for heart failure, according to a nationwide Danish study, Usman Khalid, MD, reported at the annual congress of the European Society of Cardiology.
As a chronic systemic inflammatory disease, rheumatoid arthritis (RA) has been associated with increased risk of a variety of comorbid conditions, including cardiovascular disease. But RA’s relationship specifically with heart failure hasn’t previously been looked at in the comprehensive way that’s possible in Denmark, where linked national registries enable researchers to follow health issues in the entire population from birth to death, noted Dr. Khalid of the University of Copenhagen.
In an analysis adjusted for age, sex, and calendar year, individuals with RA were 83% more likely to be hospitalized for heart failure than were the non-RA Danish population. In a fully adjusted analysis that controlled for those potential confounders as well as comorbid conditions, smoking, alcohol intake, socioeconomic status, and prescription medications, patients with RA remained at a statistically significant and clinically meaningful 38% increased risk of heart failure hospitalization.
Further studies are planned to determine the underlying mechanisms of this association, Dr. Khalid added.
He reported having no financial conflicts of interest regarding this study, which was supported by an unrestricted grant from Leo Pharma.
AT THE ESC CONGRESS 2016
Key clinical point:
Major finding: The incidence rate for heart failure hospitalization was 7.37 per 1,000 person-years in Danish adults with rheumatoid arthritis, compared with 2.45 per 1,000 in the general population.
Data source: This study utilized Danish comprehensive national registries to determine the rate at which 13,800 Danes with rheumatoid arthritis and no baseline history of heart failure were hospitalized for heart failure during 15 years of follow-up.
Disclosures: The study was supported by an unrestricted grant from Leo Pharma. The presenter reported having no financial conflicts of interest.
Unexplained subfertility in RA linked to periconceptional NSAID use
Women with rheumatoid arthritis are more often diagnosed with unexplained subfertility, compared with women in the general population, according to a new study published in Arthritis Care & Research.
This finding may imply that fertility in female RA patients is influenced by disease-related factors, specifically the use of periconceptional NSAIDs, according to the investigators.
In addition, when women with RA try to conceive, their antirheumatic treatment regimens need to be adjusted, which increases risk for permanent joint damage, the investigators wrote, adding that “understanding the underlying mechanisms of subfertility in RA, and treatment of these mechanisms whenever possible, would be an important step forward in the care for these patients.”
To study the outcome of fertility assessments in women with RA and subfertility, Dr. Brouwer and her associates performed a cross-sectional study of 260 female RA patients who were recruited from the Pregnancy-induced Amelioration of RA study, a Dutch nationwide prospective observational trial of women diagnosed with RA who were in their first trimester of pregnancy or who were trying to conceive.
Each eligible participant received a questionnaire that included questions regarding reproductive history, time to pregnancy, mode of conception for prior pregnancies, fertility assessments, and fertility treatments. For the 178 (68%) women who returned completed questionnaires, additional gynecologic histories and diagnoses were collected from medical histories and/or patient files.
Analysis of these data revealed that 82 women (46%; 95% confidence interval, 39%-53%) with RA were considered subfertile. Subfertility was most often unexplained (48% of known diagnoses) or caused by anovulation (28%) or semen abnormalities (16%).
“In comparison to the general population, female RA patients appear to be more often diagnosed with unexplained subfertility, whereas the percentage of subfertile women with anovulation was equal or slightly increased compared to percentages found in the general population,” the investigators wrote.
The majority of subfertile RA patients received fertility treatments, and “a considerable number of all pregnancies were conceived after women had been treated for subfertility,” according to the researchers.
The significant association between periconceptional NSAIDs use and unexplained subfertility “is in concordance with a previous study within the PARA cohort where we have shown that a longer [time to pregnancy] was associated with the periconceptional use of NSAIDs,” Dr. Brouwer and her associates wrote.
“In daily practice, when an RA patient wishes to conceive, NSAIDs should be avoided, and early consultation with an expert rheumatologist and a fertility specialist should be considered to optimize the patient’s chance of a complete family,” the investigators recommended.
This study was funded by the Dutch Arthritis Foundation. One investigator reported received financial compensation from UCB Pharma; the other investigators reported having no relevant disclosures.
jcraig@frontlinemedcom.com
On Twitter @jessnicolecraig
Women with rheumatoid arthritis are more often diagnosed with unexplained subfertility, compared with women in the general population, according to a new study published in Arthritis Care & Research.
This finding may imply that fertility in female RA patients is influenced by disease-related factors, specifically the use of periconceptional NSAIDs, according to the investigators.
In addition, when women with RA try to conceive, their antirheumatic treatment regimens need to be adjusted, which increases risk for permanent joint damage, the investigators wrote, adding that “understanding the underlying mechanisms of subfertility in RA, and treatment of these mechanisms whenever possible, would be an important step forward in the care for these patients.”
To study the outcome of fertility assessments in women with RA and subfertility, Dr. Brouwer and her associates performed a cross-sectional study of 260 female RA patients who were recruited from the Pregnancy-induced Amelioration of RA study, a Dutch nationwide prospective observational trial of women diagnosed with RA who were in their first trimester of pregnancy or who were trying to conceive.
Each eligible participant received a questionnaire that included questions regarding reproductive history, time to pregnancy, mode of conception for prior pregnancies, fertility assessments, and fertility treatments. For the 178 (68%) women who returned completed questionnaires, additional gynecologic histories and diagnoses were collected from medical histories and/or patient files.
Analysis of these data revealed that 82 women (46%; 95% confidence interval, 39%-53%) with RA were considered subfertile. Subfertility was most often unexplained (48% of known diagnoses) or caused by anovulation (28%) or semen abnormalities (16%).
“In comparison to the general population, female RA patients appear to be more often diagnosed with unexplained subfertility, whereas the percentage of subfertile women with anovulation was equal or slightly increased compared to percentages found in the general population,” the investigators wrote.
The majority of subfertile RA patients received fertility treatments, and “a considerable number of all pregnancies were conceived after women had been treated for subfertility,” according to the researchers.
The significant association between periconceptional NSAIDs use and unexplained subfertility “is in concordance with a previous study within the PARA cohort where we have shown that a longer [time to pregnancy] was associated with the periconceptional use of NSAIDs,” Dr. Brouwer and her associates wrote.
“In daily practice, when an RA patient wishes to conceive, NSAIDs should be avoided, and early consultation with an expert rheumatologist and a fertility specialist should be considered to optimize the patient’s chance of a complete family,” the investigators recommended.
This study was funded by the Dutch Arthritis Foundation. One investigator reported received financial compensation from UCB Pharma; the other investigators reported having no relevant disclosures.
jcraig@frontlinemedcom.com
On Twitter @jessnicolecraig
Women with rheumatoid arthritis are more often diagnosed with unexplained subfertility, compared with women in the general population, according to a new study published in Arthritis Care & Research.
This finding may imply that fertility in female RA patients is influenced by disease-related factors, specifically the use of periconceptional NSAIDs, according to the investigators.
In addition, when women with RA try to conceive, their antirheumatic treatment regimens need to be adjusted, which increases risk for permanent joint damage, the investigators wrote, adding that “understanding the underlying mechanisms of subfertility in RA, and treatment of these mechanisms whenever possible, would be an important step forward in the care for these patients.”
To study the outcome of fertility assessments in women with RA and subfertility, Dr. Brouwer and her associates performed a cross-sectional study of 260 female RA patients who were recruited from the Pregnancy-induced Amelioration of RA study, a Dutch nationwide prospective observational trial of women diagnosed with RA who were in their first trimester of pregnancy or who were trying to conceive.
Each eligible participant received a questionnaire that included questions regarding reproductive history, time to pregnancy, mode of conception for prior pregnancies, fertility assessments, and fertility treatments. For the 178 (68%) women who returned completed questionnaires, additional gynecologic histories and diagnoses were collected from medical histories and/or patient files.
Analysis of these data revealed that 82 women (46%; 95% confidence interval, 39%-53%) with RA were considered subfertile. Subfertility was most often unexplained (48% of known diagnoses) or caused by anovulation (28%) or semen abnormalities (16%).
“In comparison to the general population, female RA patients appear to be more often diagnosed with unexplained subfertility, whereas the percentage of subfertile women with anovulation was equal or slightly increased compared to percentages found in the general population,” the investigators wrote.
The majority of subfertile RA patients received fertility treatments, and “a considerable number of all pregnancies were conceived after women had been treated for subfertility,” according to the researchers.
The significant association between periconceptional NSAIDs use and unexplained subfertility “is in concordance with a previous study within the PARA cohort where we have shown that a longer [time to pregnancy] was associated with the periconceptional use of NSAIDs,” Dr. Brouwer and her associates wrote.
“In daily practice, when an RA patient wishes to conceive, NSAIDs should be avoided, and early consultation with an expert rheumatologist and a fertility specialist should be considered to optimize the patient’s chance of a complete family,” the investigators recommended.
This study was funded by the Dutch Arthritis Foundation. One investigator reported received financial compensation from UCB Pharma; the other investigators reported having no relevant disclosures.
jcraig@frontlinemedcom.com
On Twitter @jessnicolecraig
FROM ARTHRITIS CARE & RESEARCH
Key clinical point:
Major finding: Of 178 women with RA, 46% were considered subfertile. Subfertility was most often unexplained (48% of known diagnoses) or caused by anovulation (28%).
Data source: Cross-sectional study of 260 female RA patients.
Disclosures: This study was funded by the Dutch Arthritis Foundation. One investigator reported received financial compensation from UCB Pharma; the other investigators reported having no relevant disclosures.
Silicone joint arthroplasty for RA shows sustained improvements at 7 years
Rheumatoid arthritis patients who underwent silicone metacarpophalangeal joint replacement maintained significant improvement in ulnar drift and extensor lag after 7 years of follow-up in the prospective, multicenter Silicone Arthroplasty in Rheumatoid Arthritis (SARA) study.
The silicone metacarpophalangeal joint arthroplasty (SMPA) group also showed significantly better metacarpophalangeal (MCP) joint arc of motion, as well as function, aesthetics, and satisfaction scores than did patients in the cohort who chose nonsurgical management.
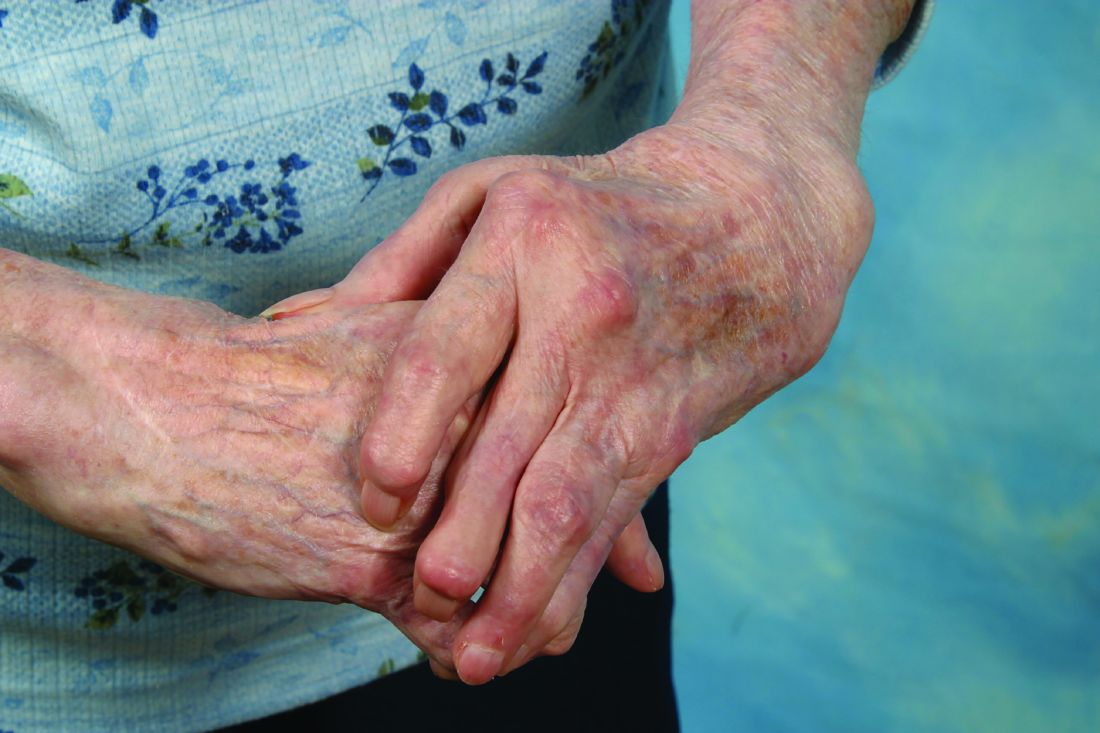
The practice of replacing deformed MCP joints with a hinged silicone implant has been around for almost half a century, the investigators noted, but despite widespread use, there is a lack of high-quality surgical outcomes data for the procedure. The authors suggested that the scarcity of data may create a barrier for surgical referrals from rheumatologists and could account for the declining rate of surgical intervention for RA joint deformities.
The significant improvement in ulnar drift that occurred in patients who underwent SMPA versus no surgery remained even after adjustment for baseline severity, age, sex, and use of biologics. The adjustment was especially important because the nonsurgical group had better function at baseline, with significantly stronger grip and pinch strength, better Michigan Hand Questionnaire (MHQ) scores, and significantly better ulnar drift, extensor lag, and arc of motion than in the surgical group (Arthritis Care Res. 2016 Oct 1. doi: 10.1002/acr.23105).
The nonsurgical group largely maintained its baseline functional state during the 7-year follow-up period except for a decline in pinch strength.
“When the SMPA cohort was compared with the non-SMPA cohort, the covariate-adjusted difference showed significant benefits associated with SMPA hand outcome as measured by the MHQ function, aesthetics, and satisfaction scores, with no measures showing significantly better outcomes for the non-SMPA group,” the researchers wrote. “Although the average treatment effect estimate (ATT estimate) showed a decline over time for the average benefit from SMPA in those treated, the function score benefit as shown by the ATT estimate remained significant at year 5.”
Patients in the nonsurgical group were also given the option to crossover and receive surgery after 1 year, which two did. Eleven patients in the surgical group also decided to undergo surgery on their other hand.
Researchers saw one mild and three moderate implant-related adverse events during the study, including one patient who experienced ulnar deviation and needed a new joint replacement, one who dislocated the implant of the little finger a few weeks after insertion, and one who experienced sepsis in the joints 6 years after insertion and needed replacements for two implants. The fracture incidence of about 10% fits into the mid-range of previously reported fracture rates with this implant.
The study experienced significant losses to follow-up, particularly in the surgical group in which 7-year data were only available for 43% of participants in this group. The authors suggested this could have been because the surgical patients achieved their goals and were less inclined to the follow-up visits.
The study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. No conflicts of interest were declared.
Rheumatoid arthritis patients who underwent silicone metacarpophalangeal joint replacement maintained significant improvement in ulnar drift and extensor lag after 7 years of follow-up in the prospective, multicenter Silicone Arthroplasty in Rheumatoid Arthritis (SARA) study.
The silicone metacarpophalangeal joint arthroplasty (SMPA) group also showed significantly better metacarpophalangeal (MCP) joint arc of motion, as well as function, aesthetics, and satisfaction scores than did patients in the cohort who chose nonsurgical management.
The practice of replacing deformed MCP joints with a hinged silicone implant has been around for almost half a century, the investigators noted, but despite widespread use, there is a lack of high-quality surgical outcomes data for the procedure. The authors suggested that the scarcity of data may create a barrier for surgical referrals from rheumatologists and could account for the declining rate of surgical intervention for RA joint deformities.
The significant improvement in ulnar drift that occurred in patients who underwent SMPA versus no surgery remained even after adjustment for baseline severity, age, sex, and use of biologics. The adjustment was especially important because the nonsurgical group had better function at baseline, with significantly stronger grip and pinch strength, better Michigan Hand Questionnaire (MHQ) scores, and significantly better ulnar drift, extensor lag, and arc of motion than in the surgical group (Arthritis Care Res. 2016 Oct 1. doi: 10.1002/acr.23105).
The nonsurgical group largely maintained its baseline functional state during the 7-year follow-up period except for a decline in pinch strength.
“When the SMPA cohort was compared with the non-SMPA cohort, the covariate-adjusted difference showed significant benefits associated with SMPA hand outcome as measured by the MHQ function, aesthetics, and satisfaction scores, with no measures showing significantly better outcomes for the non-SMPA group,” the researchers wrote. “Although the average treatment effect estimate (ATT estimate) showed a decline over time for the average benefit from SMPA in those treated, the function score benefit as shown by the ATT estimate remained significant at year 5.”
Patients in the nonsurgical group were also given the option to crossover and receive surgery after 1 year, which two did. Eleven patients in the surgical group also decided to undergo surgery on their other hand.
Researchers saw one mild and three moderate implant-related adverse events during the study, including one patient who experienced ulnar deviation and needed a new joint replacement, one who dislocated the implant of the little finger a few weeks after insertion, and one who experienced sepsis in the joints 6 years after insertion and needed replacements for two implants. The fracture incidence of about 10% fits into the mid-range of previously reported fracture rates with this implant.
The study experienced significant losses to follow-up, particularly in the surgical group in which 7-year data were only available for 43% of participants in this group. The authors suggested this could have been because the surgical patients achieved their goals and were less inclined to the follow-up visits.
The study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. No conflicts of interest were declared.
Rheumatoid arthritis patients who underwent silicone metacarpophalangeal joint replacement maintained significant improvement in ulnar drift and extensor lag after 7 years of follow-up in the prospective, multicenter Silicone Arthroplasty in Rheumatoid Arthritis (SARA) study.
The silicone metacarpophalangeal joint arthroplasty (SMPA) group also showed significantly better metacarpophalangeal (MCP) joint arc of motion, as well as function, aesthetics, and satisfaction scores than did patients in the cohort who chose nonsurgical management.
The practice of replacing deformed MCP joints with a hinged silicone implant has been around for almost half a century, the investigators noted, but despite widespread use, there is a lack of high-quality surgical outcomes data for the procedure. The authors suggested that the scarcity of data may create a barrier for surgical referrals from rheumatologists and could account for the declining rate of surgical intervention for RA joint deformities.
The significant improvement in ulnar drift that occurred in patients who underwent SMPA versus no surgery remained even after adjustment for baseline severity, age, sex, and use of biologics. The adjustment was especially important because the nonsurgical group had better function at baseline, with significantly stronger grip and pinch strength, better Michigan Hand Questionnaire (MHQ) scores, and significantly better ulnar drift, extensor lag, and arc of motion than in the surgical group (Arthritis Care Res. 2016 Oct 1. doi: 10.1002/acr.23105).
The nonsurgical group largely maintained its baseline functional state during the 7-year follow-up period except for a decline in pinch strength.
“When the SMPA cohort was compared with the non-SMPA cohort, the covariate-adjusted difference showed significant benefits associated with SMPA hand outcome as measured by the MHQ function, aesthetics, and satisfaction scores, with no measures showing significantly better outcomes for the non-SMPA group,” the researchers wrote. “Although the average treatment effect estimate (ATT estimate) showed a decline over time for the average benefit from SMPA in those treated, the function score benefit as shown by the ATT estimate remained significant at year 5.”
Patients in the nonsurgical group were also given the option to crossover and receive surgery after 1 year, which two did. Eleven patients in the surgical group also decided to undergo surgery on their other hand.
Researchers saw one mild and three moderate implant-related adverse events during the study, including one patient who experienced ulnar deviation and needed a new joint replacement, one who dislocated the implant of the little finger a few weeks after insertion, and one who experienced sepsis in the joints 6 years after insertion and needed replacements for two implants. The fracture incidence of about 10% fits into the mid-range of previously reported fracture rates with this implant.
The study experienced significant losses to follow-up, particularly in the surgical group in which 7-year data were only available for 43% of participants in this group. The authors suggested this could have been because the surgical patients achieved their goals and were less inclined to the follow-up visits.
The study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. No conflicts of interest were declared.
Key clinical point:
Major finding: Patients who elected to undergo silicone MCP joint replacement showed significant improvements in ulnar drift and extensor lag, as well as in function, aesthetics, and satisfaction scores at 7 years after the procedure.
Data source: Cohort study of 170 patients with rheumatoid arthritis–related severe deformity at the metacarpophalangeal joints.
Disclosures: The study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. No conflicts of interest were declared.