User login
Daily Double! Assessing the Effectiveness of Game-Based Learning on the Pharmacy Knowledge of US Coast Guard Health Services Technicians
Daily Double! Assessing the Effectiveness of Game-Based Learning on the Pharmacy Knowledge of US Coast Guard Health Services Technicians
The US Coast Guard (USCG) operates within the US Department of Homeland Security and represents a force of > 50,000 servicemembers.1 The missions of the service include maritime law enforcement (drug interdiction), search and rescue, and defense readiness.2
The USCG operates 42 clinics and numerous smaller sick bays of varying sizes and medical capabilities throughout the country to provide acute and routine medical services. Health services technicians (HSs) are the most common staffing component and provide much of the support services in each USCG health care setting. The HS rating, colloquially referred to as corpsmen, is achieved through a 22-week course known as “A” school that trains servicemembers in outpatient and acute care, including emergency medical technician training.3 There are about 750 USCG HSs.
Within USCG clinics, HSs conduct ambulatory intakes for outpatient appointments, administer immunizations and blood draws, requisition medical equipment and supplies, serve as a pharmacy technician, complete physical examinations, and manage referrals, among other duties. Their familiarity with different aspects of clinic operations and medical practice must be broad. To that end, corpsmen develop and reinforce their medical knowledge through various trainings, including additional courses to specialize in certain medical skills, such as pharmacy technician “C” school or dental assistant “C” school.
The USCG employs < 15 field pharmacists, most of whom serve in an ambulatory care environment.4 Responsibilities of USCG pharmacists include the routine reinforcement of pharmacy knowledge with HSs. For the corpsmen who are not pharmacy technicians or who have not attended pharmacy technician “C” school, the extent of their pharmacy instruction primarily came from the “A” school curriculum, of which only 1 class is specific to pharmacy. Providing routine pharmacy-related training to the HSs further cultivates their pharmacy knowledge and confidence so that they can practice more holistically. These trainings do not need to follow any specific format.
In this study, 3 pharmacists at 3 separate USCG clinics conducted a training inspired by the Jeopardy! game show with the corpsmen at their respective clinics. This study examined the effectiveness of game-based learning on the pharmacy knowledge retention of HSs at 3 USCG clinics. A secondary objective was to evaluate the baseline pharmacy knowledge of corpsmen based on specific corpsmen demographics.
Methods
As part of a USCG quality improvement study in 2024, 28 HSs at the 3 USCG clinics were provided a preintervention assessment, completed game-based educational program (intervention), and then were assessed again following the intervention.
The HSs were presented with a 25-question assessment that included 10 knowledge questions (3 on over-the-counter medications, 2 on use of medications in pregnancy, 2 on precautions and contraindications, 2 on indications, and 1 on immunizations) and 15 brand-generic matching questions. These questions were developed and reviewed by the 3 participating pharmacists to ensure that their scope was commensurate with the overall pharmacy knowledge that could be reasonably expected of corpsmen spanning various points of their HS career.
One to 7 days after the preintervention assessment, the pharmacists hosted the game-based learning modeled after Jeopardy!. The Jeopardy! categories mirrored the assessment knowledge question categories, and brand-generic nomenclature was freely discussed throughout. About 2 weeks later, the same HSs who completed the preintervention assessment and participated in the game were presented with the same assessment.
In addition to capturing the difference in scores between the 2 assessments, additional demographic data were gathered, including service time as an HS and whether they received formalized pharmacy technician training and if so, how long they have served in that capacity. Demographic data were collected to identify potential correlations between demographic characteristics and results.
Results
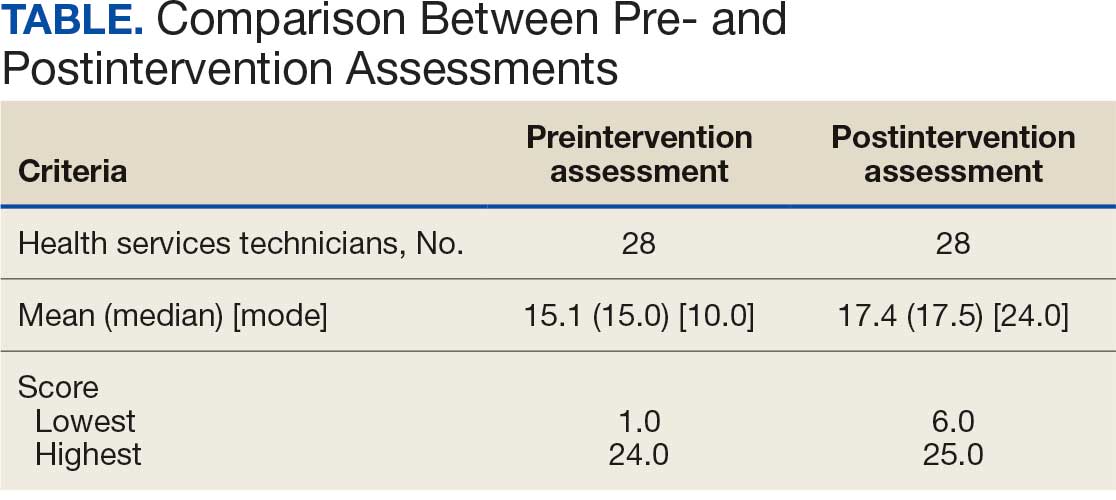
Twenty-eight HSs at the 3 clinics completed the game-based training and both assessments. The mean score increased from 15.1 preintervention to 17.4 postintervention (Table). Preintervention scores ranged from 1 to 24 and postintervention scores ranged from 6 to 25.
There were 19 HSs (68%) whose score increased from preintervention to postintervention and 5 (18%) had decreased scores. The largest score decrease was 4 (from 18 to 14), and the largest score increase was 11 (from 13 to 24). The mean improvement was 3.9 among the 19 HSs with increased scores
Twenty-one HSs reported no formal pharmacy technician training, 3 completed pharmacy technician “C” school, and 4 received informal on-the-job training. The mean score for the “C” school trained HSs was 23.0 preintervention and 23.7 postintervention. The mean score for HSs trained on the job was 16.0 preintervention and 18.5 postintervention. The mean score for HSs with no training was 13.9 preintervention and 16.3 postintervention.
As HSs advance in their careers, they typically assume roles with increasing technical knowledge, responsibility, and oversight, thus aligning with advancement from E-4 (third class petty officer) to E-6 (first class petty officer) and beyond. In this study, there was 1 E-3, 12 E-4s (mean time as an HS, 1.3 years), 8 E-5s (mean time as an HS, 4.8 years), and 7 E-6s (mean time as an HS, 8.6 years). The E-3 had a preintervention score of 1.0 and a postintervention score of 6.0. The E-4s had a mean change in score from pre- to postintervention of 2.4. The E-5s had a mean change in score from pre- to postintervention of 1.6. The E-6s had a mean change in score from pre- to postintervention of 2.3.
Discussion
This study is novel in its examination of the impact of game-based learning on the retention of the pharmacy knowledge of USCG corpsmen. A PubMed literature search of the phrase “((Corpsman) OR (Corpsmen)) AND (Coast Guard)” yields 135 results, though none were relevant to the USCG population described in this study. A PubMed literature search of the phrase “(Jeopardy!) AND (pharmacy)” yields 28 results, only 1 of which discusses using the game-based approach as an instructional tool.5 A PubMed literature search of the phrase “(game) AND (Coast Guard)” yields 55 results, none of which were specifically relevant to game-based learning in the USCG. This study appears to be among the first to discuss results and trends in game-based learning with USCG corpsmen.
The preponderance of literature for game-based learning strategies exists in children; more research in adults is needed.6,7 With studies showing that game-based learning may impact motivation to learn and learning gains, it is unsurprising that there is some research in professional health care education. Games modeled after everything from simulated clinical scenarios to Family Feud and Chutes and Ladders-style games have been compared with traditional learning strategies. However, the results of whether game-based learning strategies improve knowledge, clinical decision-making, and motivation to learn vary, suggesting the need for more research in this field.8
The results of this study suggest that Jeopardy! is likely an effective instructional method for USCG corpsmen on pharmacy topics. While there were some HSs whose postintervention scores decreased, 19 (68%) had increased scores. Because the second assessment was administered about 2 weeks after the game-based learning, the results suggest some level of knowledge retention. Between these results and the informally perceived level of engagement, game-based learning could be a more stimulating alternative training method to a standard slide-based presentation.
Stratifying the data by demographics revealed additional trends, although they should be interpreted with caution due to the small sample size. The baseline results strongly illustrate the value of formalized training. It is generally expected that HSs who have completed the “C” school pharmacy technician training program should have more pharmacy knowledge than those with on-the-job or less training. The results indicate that “C” school trained and on-the-job trained HSs scored higher on the preintervention assessment (mean, 23.0 and 16.0, respectively), than those with no such experiences (mean, 13.9). Such results underscore the value of formalized training—whether as a pharmacy technician or in any other “C” school—in enhancing the medical knowledge of HSs that may allow them to hold roles of increased responsibility and medical scope.
In addition to stratification by pharmacy technician training, stratification by years of HS experience (roughly correlated to rank) yields a similar result. It would be expected that as HSs advance in their careers, they gain more exposure to various medical topics, including pharmacy. That is not always the case, however, as it is possible an HS never rotated through a pharmacy technician position or has not been recently exposed to pharmacy knowledge. Nevertheless, the results suggest that increased HS experience was likely associated with an increased baseline pharmacy knowledge, with mean preintervention scores increasing from 11.9 to 18.1 to 19.3 for E-4, E-5, and E-6, respectively.
While there are many explanations for these results, the authors hypothesize that when HSs are E-4s, they might not yet have exposure to all aspects of the clinic and are perhaps not as well-versed in pharmacy practice. An E-5—now a few years into their career—would have completed pharmacy technician “C” school or on-the-job training (if applicable), which could account for the significant jump in pharmacy knowledge scores. An E-6 can still engage in direct patient care activities but take on leadership and supervisory roles within the clinic, perhaps explaining the smaller increase in score.
In terms of increasing responsibility, many USCG corpsmen complete another schooling opportunity—Independent Duty Health Services Technician (IDHS)—so they can serve in independent duty roles, many of which are on USCG cutters. While cutters are deployed, that IDHS could be the sole medical personnel on the cutter and function in a midlevel practitioner extender role. Formalized training in pharmacy—the benefits of which are suggested through these results—or another field of medical practice would strengthen the skillset and confidence of IDHSs.
Though not formally assessed, the 3 pharmacists noted that the game-based learning was met with overwhelmingly positive feedback in terms of excitement, energy, and overall engagement.
Limitations
This cohort of individuals represents a small proportion of the total number of USCG corpsmen, and it is not fully representative of all practice settings. HSs can be assigned to USCG cutters as IDHSs, which would not be captured in this cohort. Even within a single clinic, the knowledge of HSs varies, as not all HS duties consist solely of clinical skills. Additionally, while the overall game framework was consistent among the 3 sites, there may have been unquantifiable differences in overall teaching style by the 3 pharmacists that may have resulted in different levels of content retention. Given the lack of similar studies in this population, this study can best be described as a quantitative descriptor of results rather than a statistical comparison of what instructional method works best.
Conclusions
The USCG greatly benefits from having trained and experienced HSs fulfilling mission support roles in the organization. In addition to traditional slide-based trainings, game-based learning can be considered to create engaging learning environments to support the knowledge retention of pharmacy and other medical topics for USCG corpsmen.
- US Coast Guard. Organizational overview. About the US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About
- US Coast Guard. Missions. About US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About/Missions/
- US Coast Guard. Health services technician. Accessed October 14, 2025. https://www.gocoastguard.com/careers/enlisted/hs
- Zhou F, Woodward Z. Impact of pharmacist interventions at an outpatient US Coast Guard clinic. Fed Pract. 2023;40(6):174-177. doi:10.12788/fp.0383
- Cusick J. A Jeopardy-style review game using team clickers. MedEdPORTAL. 2016;12:10485. doi:10.15766/mep_2374-8265.10485
- Dahalan F, Alias N, Shaharom MSN. Gamification and game based learning for vocational education and training: a systematic literature review. Educ Inf Technol (Dordr). 2023:1-39. doi:10.1007/s10639-022-11548-w
- Wesselink LA. Testing the Effectiveness of Game-Based Learning for Adults by Designing an Educational Game: A Design and Research Study to Investigate the Effectiveness of Educational Games for Adults to Learn Basic Skills of Microsoft Excel. Master’s thesis. University of Twente; 2020. Accessed October 22, 2025. http://essay.utwentw.nl/88229
- Del Cura-González I, Ariza-Cardiel G, Polentinos-Castro E, et al. Effectiveness of a game-based educational strategy e-EDUCAGUIA for implementing antimicrobial clinical practice guidelines in family medicine residents in Spain: a randomized clinical trial by cluster. BMC Med Educ. 2022;22:893. doi:10.1186/s12909-022-03843-4
The US Coast Guard (USCG) operates within the US Department of Homeland Security and represents a force of > 50,000 servicemembers.1 The missions of the service include maritime law enforcement (drug interdiction), search and rescue, and defense readiness.2
The USCG operates 42 clinics and numerous smaller sick bays of varying sizes and medical capabilities throughout the country to provide acute and routine medical services. Health services technicians (HSs) are the most common staffing component and provide much of the support services in each USCG health care setting. The HS rating, colloquially referred to as corpsmen, is achieved through a 22-week course known as “A” school that trains servicemembers in outpatient and acute care, including emergency medical technician training.3 There are about 750 USCG HSs.
Within USCG clinics, HSs conduct ambulatory intakes for outpatient appointments, administer immunizations and blood draws, requisition medical equipment and supplies, serve as a pharmacy technician, complete physical examinations, and manage referrals, among other duties. Their familiarity with different aspects of clinic operations and medical practice must be broad. To that end, corpsmen develop and reinforce their medical knowledge through various trainings, including additional courses to specialize in certain medical skills, such as pharmacy technician “C” school or dental assistant “C” school.
The USCG employs < 15 field pharmacists, most of whom serve in an ambulatory care environment.4 Responsibilities of USCG pharmacists include the routine reinforcement of pharmacy knowledge with HSs. For the corpsmen who are not pharmacy technicians or who have not attended pharmacy technician “C” school, the extent of their pharmacy instruction primarily came from the “A” school curriculum, of which only 1 class is specific to pharmacy. Providing routine pharmacy-related training to the HSs further cultivates their pharmacy knowledge and confidence so that they can practice more holistically. These trainings do not need to follow any specific format.
In this study, 3 pharmacists at 3 separate USCG clinics conducted a training inspired by the Jeopardy! game show with the corpsmen at their respective clinics. This study examined the effectiveness of game-based learning on the pharmacy knowledge retention of HSs at 3 USCG clinics. A secondary objective was to evaluate the baseline pharmacy knowledge of corpsmen based on specific corpsmen demographics.
Methods
As part of a USCG quality improvement study in 2024, 28 HSs at the 3 USCG clinics were provided a preintervention assessment, completed game-based educational program (intervention), and then were assessed again following the intervention.
The HSs were presented with a 25-question assessment that included 10 knowledge questions (3 on over-the-counter medications, 2 on use of medications in pregnancy, 2 on precautions and contraindications, 2 on indications, and 1 on immunizations) and 15 brand-generic matching questions. These questions were developed and reviewed by the 3 participating pharmacists to ensure that their scope was commensurate with the overall pharmacy knowledge that could be reasonably expected of corpsmen spanning various points of their HS career.
One to 7 days after the preintervention assessment, the pharmacists hosted the game-based learning modeled after Jeopardy!. The Jeopardy! categories mirrored the assessment knowledge question categories, and brand-generic nomenclature was freely discussed throughout. About 2 weeks later, the same HSs who completed the preintervention assessment and participated in the game were presented with the same assessment.
In addition to capturing the difference in scores between the 2 assessments, additional demographic data were gathered, including service time as an HS and whether they received formalized pharmacy technician training and if so, how long they have served in that capacity. Demographic data were collected to identify potential correlations between demographic characteristics and results.
Results
Twenty-eight HSs at the 3 clinics completed the game-based training and both assessments. The mean score increased from 15.1 preintervention to 17.4 postintervention (Table). Preintervention scores ranged from 1 to 24 and postintervention scores ranged from 6 to 25.
There were 19 HSs (68%) whose score increased from preintervention to postintervention and 5 (18%) had decreased scores. The largest score decrease was 4 (from 18 to 14), and the largest score increase was 11 (from 13 to 24). The mean improvement was 3.9 among the 19 HSs with increased scores
Twenty-one HSs reported no formal pharmacy technician training, 3 completed pharmacy technician “C” school, and 4 received informal on-the-job training. The mean score for the “C” school trained HSs was 23.0 preintervention and 23.7 postintervention. The mean score for HSs trained on the job was 16.0 preintervention and 18.5 postintervention. The mean score for HSs with no training was 13.9 preintervention and 16.3 postintervention.
As HSs advance in their careers, they typically assume roles with increasing technical knowledge, responsibility, and oversight, thus aligning with advancement from E-4 (third class petty officer) to E-6 (first class petty officer) and beyond. In this study, there was 1 E-3, 12 E-4s (mean time as an HS, 1.3 years), 8 E-5s (mean time as an HS, 4.8 years), and 7 E-6s (mean time as an HS, 8.6 years). The E-3 had a preintervention score of 1.0 and a postintervention score of 6.0. The E-4s had a mean change in score from pre- to postintervention of 2.4. The E-5s had a mean change in score from pre- to postintervention of 1.6. The E-6s had a mean change in score from pre- to postintervention of 2.3.
Discussion
This study is novel in its examination of the impact of game-based learning on the retention of the pharmacy knowledge of USCG corpsmen. A PubMed literature search of the phrase “((Corpsman) OR (Corpsmen)) AND (Coast Guard)” yields 135 results, though none were relevant to the USCG population described in this study. A PubMed literature search of the phrase “(Jeopardy!) AND (pharmacy)” yields 28 results, only 1 of which discusses using the game-based approach as an instructional tool.5 A PubMed literature search of the phrase “(game) AND (Coast Guard)” yields 55 results, none of which were specifically relevant to game-based learning in the USCG. This study appears to be among the first to discuss results and trends in game-based learning with USCG corpsmen.
The preponderance of literature for game-based learning strategies exists in children; more research in adults is needed.6,7 With studies showing that game-based learning may impact motivation to learn and learning gains, it is unsurprising that there is some research in professional health care education. Games modeled after everything from simulated clinical scenarios to Family Feud and Chutes and Ladders-style games have been compared with traditional learning strategies. However, the results of whether game-based learning strategies improve knowledge, clinical decision-making, and motivation to learn vary, suggesting the need for more research in this field.8
The results of this study suggest that Jeopardy! is likely an effective instructional method for USCG corpsmen on pharmacy topics. While there were some HSs whose postintervention scores decreased, 19 (68%) had increased scores. Because the second assessment was administered about 2 weeks after the game-based learning, the results suggest some level of knowledge retention. Between these results and the informally perceived level of engagement, game-based learning could be a more stimulating alternative training method to a standard slide-based presentation.
Stratifying the data by demographics revealed additional trends, although they should be interpreted with caution due to the small sample size. The baseline results strongly illustrate the value of formalized training. It is generally expected that HSs who have completed the “C” school pharmacy technician training program should have more pharmacy knowledge than those with on-the-job or less training. The results indicate that “C” school trained and on-the-job trained HSs scored higher on the preintervention assessment (mean, 23.0 and 16.0, respectively), than those with no such experiences (mean, 13.9). Such results underscore the value of formalized training—whether as a pharmacy technician or in any other “C” school—in enhancing the medical knowledge of HSs that may allow them to hold roles of increased responsibility and medical scope.
In addition to stratification by pharmacy technician training, stratification by years of HS experience (roughly correlated to rank) yields a similar result. It would be expected that as HSs advance in their careers, they gain more exposure to various medical topics, including pharmacy. That is not always the case, however, as it is possible an HS never rotated through a pharmacy technician position or has not been recently exposed to pharmacy knowledge. Nevertheless, the results suggest that increased HS experience was likely associated with an increased baseline pharmacy knowledge, with mean preintervention scores increasing from 11.9 to 18.1 to 19.3 for E-4, E-5, and E-6, respectively.
While there are many explanations for these results, the authors hypothesize that when HSs are E-4s, they might not yet have exposure to all aspects of the clinic and are perhaps not as well-versed in pharmacy practice. An E-5—now a few years into their career—would have completed pharmacy technician “C” school or on-the-job training (if applicable), which could account for the significant jump in pharmacy knowledge scores. An E-6 can still engage in direct patient care activities but take on leadership and supervisory roles within the clinic, perhaps explaining the smaller increase in score.
In terms of increasing responsibility, many USCG corpsmen complete another schooling opportunity—Independent Duty Health Services Technician (IDHS)—so they can serve in independent duty roles, many of which are on USCG cutters. While cutters are deployed, that IDHS could be the sole medical personnel on the cutter and function in a midlevel practitioner extender role. Formalized training in pharmacy—the benefits of which are suggested through these results—or another field of medical practice would strengthen the skillset and confidence of IDHSs.
Though not formally assessed, the 3 pharmacists noted that the game-based learning was met with overwhelmingly positive feedback in terms of excitement, energy, and overall engagement.
Limitations
This cohort of individuals represents a small proportion of the total number of USCG corpsmen, and it is not fully representative of all practice settings. HSs can be assigned to USCG cutters as IDHSs, which would not be captured in this cohort. Even within a single clinic, the knowledge of HSs varies, as not all HS duties consist solely of clinical skills. Additionally, while the overall game framework was consistent among the 3 sites, there may have been unquantifiable differences in overall teaching style by the 3 pharmacists that may have resulted in different levels of content retention. Given the lack of similar studies in this population, this study can best be described as a quantitative descriptor of results rather than a statistical comparison of what instructional method works best.
Conclusions
The USCG greatly benefits from having trained and experienced HSs fulfilling mission support roles in the organization. In addition to traditional slide-based trainings, game-based learning can be considered to create engaging learning environments to support the knowledge retention of pharmacy and other medical topics for USCG corpsmen.
The US Coast Guard (USCG) operates within the US Department of Homeland Security and represents a force of > 50,000 servicemembers.1 The missions of the service include maritime law enforcement (drug interdiction), search and rescue, and defense readiness.2
The USCG operates 42 clinics and numerous smaller sick bays of varying sizes and medical capabilities throughout the country to provide acute and routine medical services. Health services technicians (HSs) are the most common staffing component and provide much of the support services in each USCG health care setting. The HS rating, colloquially referred to as corpsmen, is achieved through a 22-week course known as “A” school that trains servicemembers in outpatient and acute care, including emergency medical technician training.3 There are about 750 USCG HSs.
Within USCG clinics, HSs conduct ambulatory intakes for outpatient appointments, administer immunizations and blood draws, requisition medical equipment and supplies, serve as a pharmacy technician, complete physical examinations, and manage referrals, among other duties. Their familiarity with different aspects of clinic operations and medical practice must be broad. To that end, corpsmen develop and reinforce their medical knowledge through various trainings, including additional courses to specialize in certain medical skills, such as pharmacy technician “C” school or dental assistant “C” school.
The USCG employs < 15 field pharmacists, most of whom serve in an ambulatory care environment.4 Responsibilities of USCG pharmacists include the routine reinforcement of pharmacy knowledge with HSs. For the corpsmen who are not pharmacy technicians or who have not attended pharmacy technician “C” school, the extent of their pharmacy instruction primarily came from the “A” school curriculum, of which only 1 class is specific to pharmacy. Providing routine pharmacy-related training to the HSs further cultivates their pharmacy knowledge and confidence so that they can practice more holistically. These trainings do not need to follow any specific format.
In this study, 3 pharmacists at 3 separate USCG clinics conducted a training inspired by the Jeopardy! game show with the corpsmen at their respective clinics. This study examined the effectiveness of game-based learning on the pharmacy knowledge retention of HSs at 3 USCG clinics. A secondary objective was to evaluate the baseline pharmacy knowledge of corpsmen based on specific corpsmen demographics.
Methods
As part of a USCG quality improvement study in 2024, 28 HSs at the 3 USCG clinics were provided a preintervention assessment, completed game-based educational program (intervention), and then were assessed again following the intervention.
The HSs were presented with a 25-question assessment that included 10 knowledge questions (3 on over-the-counter medications, 2 on use of medications in pregnancy, 2 on precautions and contraindications, 2 on indications, and 1 on immunizations) and 15 brand-generic matching questions. These questions were developed and reviewed by the 3 participating pharmacists to ensure that their scope was commensurate with the overall pharmacy knowledge that could be reasonably expected of corpsmen spanning various points of their HS career.
One to 7 days after the preintervention assessment, the pharmacists hosted the game-based learning modeled after Jeopardy!. The Jeopardy! categories mirrored the assessment knowledge question categories, and brand-generic nomenclature was freely discussed throughout. About 2 weeks later, the same HSs who completed the preintervention assessment and participated in the game were presented with the same assessment.
In addition to capturing the difference in scores between the 2 assessments, additional demographic data were gathered, including service time as an HS and whether they received formalized pharmacy technician training and if so, how long they have served in that capacity. Demographic data were collected to identify potential correlations between demographic characteristics and results.
Results
Twenty-eight HSs at the 3 clinics completed the game-based training and both assessments. The mean score increased from 15.1 preintervention to 17.4 postintervention (Table). Preintervention scores ranged from 1 to 24 and postintervention scores ranged from 6 to 25.
There were 19 HSs (68%) whose score increased from preintervention to postintervention and 5 (18%) had decreased scores. The largest score decrease was 4 (from 18 to 14), and the largest score increase was 11 (from 13 to 24). The mean improvement was 3.9 among the 19 HSs with increased scores
Twenty-one HSs reported no formal pharmacy technician training, 3 completed pharmacy technician “C” school, and 4 received informal on-the-job training. The mean score for the “C” school trained HSs was 23.0 preintervention and 23.7 postintervention. The mean score for HSs trained on the job was 16.0 preintervention and 18.5 postintervention. The mean score for HSs with no training was 13.9 preintervention and 16.3 postintervention.
As HSs advance in their careers, they typically assume roles with increasing technical knowledge, responsibility, and oversight, thus aligning with advancement from E-4 (third class petty officer) to E-6 (first class petty officer) and beyond. In this study, there was 1 E-3, 12 E-4s (mean time as an HS, 1.3 years), 8 E-5s (mean time as an HS, 4.8 years), and 7 E-6s (mean time as an HS, 8.6 years). The E-3 had a preintervention score of 1.0 and a postintervention score of 6.0. The E-4s had a mean change in score from pre- to postintervention of 2.4. The E-5s had a mean change in score from pre- to postintervention of 1.6. The E-6s had a mean change in score from pre- to postintervention of 2.3.
Discussion
This study is novel in its examination of the impact of game-based learning on the retention of the pharmacy knowledge of USCG corpsmen. A PubMed literature search of the phrase “((Corpsman) OR (Corpsmen)) AND (Coast Guard)” yields 135 results, though none were relevant to the USCG population described in this study. A PubMed literature search of the phrase “(Jeopardy!) AND (pharmacy)” yields 28 results, only 1 of which discusses using the game-based approach as an instructional tool.5 A PubMed literature search of the phrase “(game) AND (Coast Guard)” yields 55 results, none of which were specifically relevant to game-based learning in the USCG. This study appears to be among the first to discuss results and trends in game-based learning with USCG corpsmen.
The preponderance of literature for game-based learning strategies exists in children; more research in adults is needed.6,7 With studies showing that game-based learning may impact motivation to learn and learning gains, it is unsurprising that there is some research in professional health care education. Games modeled after everything from simulated clinical scenarios to Family Feud and Chutes and Ladders-style games have been compared with traditional learning strategies. However, the results of whether game-based learning strategies improve knowledge, clinical decision-making, and motivation to learn vary, suggesting the need for more research in this field.8
The results of this study suggest that Jeopardy! is likely an effective instructional method for USCG corpsmen on pharmacy topics. While there were some HSs whose postintervention scores decreased, 19 (68%) had increased scores. Because the second assessment was administered about 2 weeks after the game-based learning, the results suggest some level of knowledge retention. Between these results and the informally perceived level of engagement, game-based learning could be a more stimulating alternative training method to a standard slide-based presentation.
Stratifying the data by demographics revealed additional trends, although they should be interpreted with caution due to the small sample size. The baseline results strongly illustrate the value of formalized training. It is generally expected that HSs who have completed the “C” school pharmacy technician training program should have more pharmacy knowledge than those with on-the-job or less training. The results indicate that “C” school trained and on-the-job trained HSs scored higher on the preintervention assessment (mean, 23.0 and 16.0, respectively), than those with no such experiences (mean, 13.9). Such results underscore the value of formalized training—whether as a pharmacy technician or in any other “C” school—in enhancing the medical knowledge of HSs that may allow them to hold roles of increased responsibility and medical scope.
In addition to stratification by pharmacy technician training, stratification by years of HS experience (roughly correlated to rank) yields a similar result. It would be expected that as HSs advance in their careers, they gain more exposure to various medical topics, including pharmacy. That is not always the case, however, as it is possible an HS never rotated through a pharmacy technician position or has not been recently exposed to pharmacy knowledge. Nevertheless, the results suggest that increased HS experience was likely associated with an increased baseline pharmacy knowledge, with mean preintervention scores increasing from 11.9 to 18.1 to 19.3 for E-4, E-5, and E-6, respectively.
While there are many explanations for these results, the authors hypothesize that when HSs are E-4s, they might not yet have exposure to all aspects of the clinic and are perhaps not as well-versed in pharmacy practice. An E-5—now a few years into their career—would have completed pharmacy technician “C” school or on-the-job training (if applicable), which could account for the significant jump in pharmacy knowledge scores. An E-6 can still engage in direct patient care activities but take on leadership and supervisory roles within the clinic, perhaps explaining the smaller increase in score.
In terms of increasing responsibility, many USCG corpsmen complete another schooling opportunity—Independent Duty Health Services Technician (IDHS)—so they can serve in independent duty roles, many of which are on USCG cutters. While cutters are deployed, that IDHS could be the sole medical personnel on the cutter and function in a midlevel practitioner extender role. Formalized training in pharmacy—the benefits of which are suggested through these results—or another field of medical practice would strengthen the skillset and confidence of IDHSs.
Though not formally assessed, the 3 pharmacists noted that the game-based learning was met with overwhelmingly positive feedback in terms of excitement, energy, and overall engagement.
Limitations
This cohort of individuals represents a small proportion of the total number of USCG corpsmen, and it is not fully representative of all practice settings. HSs can be assigned to USCG cutters as IDHSs, which would not be captured in this cohort. Even within a single clinic, the knowledge of HSs varies, as not all HS duties consist solely of clinical skills. Additionally, while the overall game framework was consistent among the 3 sites, there may have been unquantifiable differences in overall teaching style by the 3 pharmacists that may have resulted in different levels of content retention. Given the lack of similar studies in this population, this study can best be described as a quantitative descriptor of results rather than a statistical comparison of what instructional method works best.
Conclusions
The USCG greatly benefits from having trained and experienced HSs fulfilling mission support roles in the organization. In addition to traditional slide-based trainings, game-based learning can be considered to create engaging learning environments to support the knowledge retention of pharmacy and other medical topics for USCG corpsmen.
- US Coast Guard. Organizational overview. About the US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About
- US Coast Guard. Missions. About US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About/Missions/
- US Coast Guard. Health services technician. Accessed October 14, 2025. https://www.gocoastguard.com/careers/enlisted/hs
- Zhou F, Woodward Z. Impact of pharmacist interventions at an outpatient US Coast Guard clinic. Fed Pract. 2023;40(6):174-177. doi:10.12788/fp.0383
- Cusick J. A Jeopardy-style review game using team clickers. MedEdPORTAL. 2016;12:10485. doi:10.15766/mep_2374-8265.10485
- Dahalan F, Alias N, Shaharom MSN. Gamification and game based learning for vocational education and training: a systematic literature review. Educ Inf Technol (Dordr). 2023:1-39. doi:10.1007/s10639-022-11548-w
- Wesselink LA. Testing the Effectiveness of Game-Based Learning for Adults by Designing an Educational Game: A Design and Research Study to Investigate the Effectiveness of Educational Games for Adults to Learn Basic Skills of Microsoft Excel. Master’s thesis. University of Twente; 2020. Accessed October 22, 2025. http://essay.utwentw.nl/88229
- Del Cura-González I, Ariza-Cardiel G, Polentinos-Castro E, et al. Effectiveness of a game-based educational strategy e-EDUCAGUIA for implementing antimicrobial clinical practice guidelines in family medicine residents in Spain: a randomized clinical trial by cluster. BMC Med Educ. 2022;22:893. doi:10.1186/s12909-022-03843-4
- US Coast Guard. Organizational overview. About the US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About
- US Coast Guard. Missions. About US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About/Missions/
- US Coast Guard. Health services technician. Accessed October 14, 2025. https://www.gocoastguard.com/careers/enlisted/hs
- Zhou F, Woodward Z. Impact of pharmacist interventions at an outpatient US Coast Guard clinic. Fed Pract. 2023;40(6):174-177. doi:10.12788/fp.0383
- Cusick J. A Jeopardy-style review game using team clickers. MedEdPORTAL. 2016;12:10485. doi:10.15766/mep_2374-8265.10485
- Dahalan F, Alias N, Shaharom MSN. Gamification and game based learning for vocational education and training: a systematic literature review. Educ Inf Technol (Dordr). 2023:1-39. doi:10.1007/s10639-022-11548-w
- Wesselink LA. Testing the Effectiveness of Game-Based Learning for Adults by Designing an Educational Game: A Design and Research Study to Investigate the Effectiveness of Educational Games for Adults to Learn Basic Skills of Microsoft Excel. Master’s thesis. University of Twente; 2020. Accessed October 22, 2025. http://essay.utwentw.nl/88229
- Del Cura-González I, Ariza-Cardiel G, Polentinos-Castro E, et al. Effectiveness of a game-based educational strategy e-EDUCAGUIA for implementing antimicrobial clinical practice guidelines in family medicine residents in Spain: a randomized clinical trial by cluster. BMC Med Educ. 2022;22:893. doi:10.1186/s12909-022-03843-4
Daily Double! Assessing the Effectiveness of Game-Based Learning on the Pharmacy Knowledge of US Coast Guard Health Services Technicians
Daily Double! Assessing the Effectiveness of Game-Based Learning on the Pharmacy Knowledge of US Coast Guard Health Services Technicians
Evaluation of Pharmacist-Driven Inhaled Corticosteroid De-escalation in Veterans
Evaluation of Pharmacist-Driven Inhaled Corticosteroid De-escalation in Veterans
Systemic glucocorticoids play an important role in the treatment of chronic obstructive pulmonary disease (COPD) exacerbations. They are recommended to shorten recovery time and increase forced expiratory volume in 1 second (FEV1) during exacerbations.1 However, the role of the chronic use of inhaled corticosteroids (ICSs) in the treatment of COPD is less clear.
When added to inhaled β-2 agonists and muscarinic antagonists, ICSs can decrease the risk of exacerbations.1 However, not all patients with COPD benefit from ICS therapy. The degree of benefit an ICS can provide has been shown to correlate with eosinophil count—a marker of inflammation. The expected benefit of using an ICS increases as the eosinophil count increases.1 Maximum benefit can be observed with eosinophil counts ≥ 300 cells/µL, and minimal benefit is observed with eosinophil counts < 100 cells/µL. Adverse effects (AEs) of ICSs include a hoarse voice, oral candidiasis, and an increased risk of pneumonia.1 Given the risk of AEs, it is important to limit ICS use in patients who are unlikely to reap any benefits.
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines suggest the use of ICSs in patients who experience exacerbations while using long-acting β agonist (LABA) plus long-acting muscarinic antagonist (LAMA) therapy and have an eosinophil count ≥ 100 cells/µL. Switching from LABA or LAMA monotherapy to triple therapy with LAMA/LABA/ICS may be considered if patients have continued exacerbations and an eosinophil count ≥ 300 cells/µL. De-escalation of ICS therapy should be considered if patients do not meet these criteria or if patients experience ICS AEs, such as pneumonia. The patients most likely to have increased exacerbations or decreased FEV1 with ICS withdrawal are those with eosinophil counts ≥ 300 cells/µL.1,2
Several studies have explored the effects of ICS de-escalation in real-world clinical settings. A systematic review of 11 studies indicated that de-escalation of ICS in COPD does not result in increased exacerbations.3 A prospective study by Rossi et al found that in a 6-month period, 141 of 482 patients on ICS therapy (29%) had an exacerbation. In the opposing arm of the study, 88 of 334 patients (26%) with deprescribed ICS experienced an exacerbation. The difference between these 2 groups was not statistically significant.4 The researchers concluded that in real-world practice, ICS withdrawal can be safe in patients at low risk of exacerbation.
About 25% of veterans (1.25 million) have been diagnosed with COPD.5 To address this, the US Department of Veterans Affairs (VA) and US Department of Defense published updated COPD guidelines in 2021 that specify criteria for de-escalation of ICS.6 Guidelines, however, may not be reflected in common clinical practice for several years following publication. The VA Academic Detailing Service (ADS) provides tools to help clinicians identify patients who may benefit from changes in treatment plans. A recent ADS focus was the implementation of a COPD dashboard, which identifies patients with COPD who are candidates for ICS de-escalation based on comorbid diagnoses, exacerbation history, and eosinophil count. VA pharmacists have an expanded role in the management of primary care disease states and are therefore well-positioned to increase adherence to guideline-directed therapy. The objective of this quality improvement project was to determine the impact of pharmacist-driven de-escalation on ICS usage in veterans with COPD.
Methods
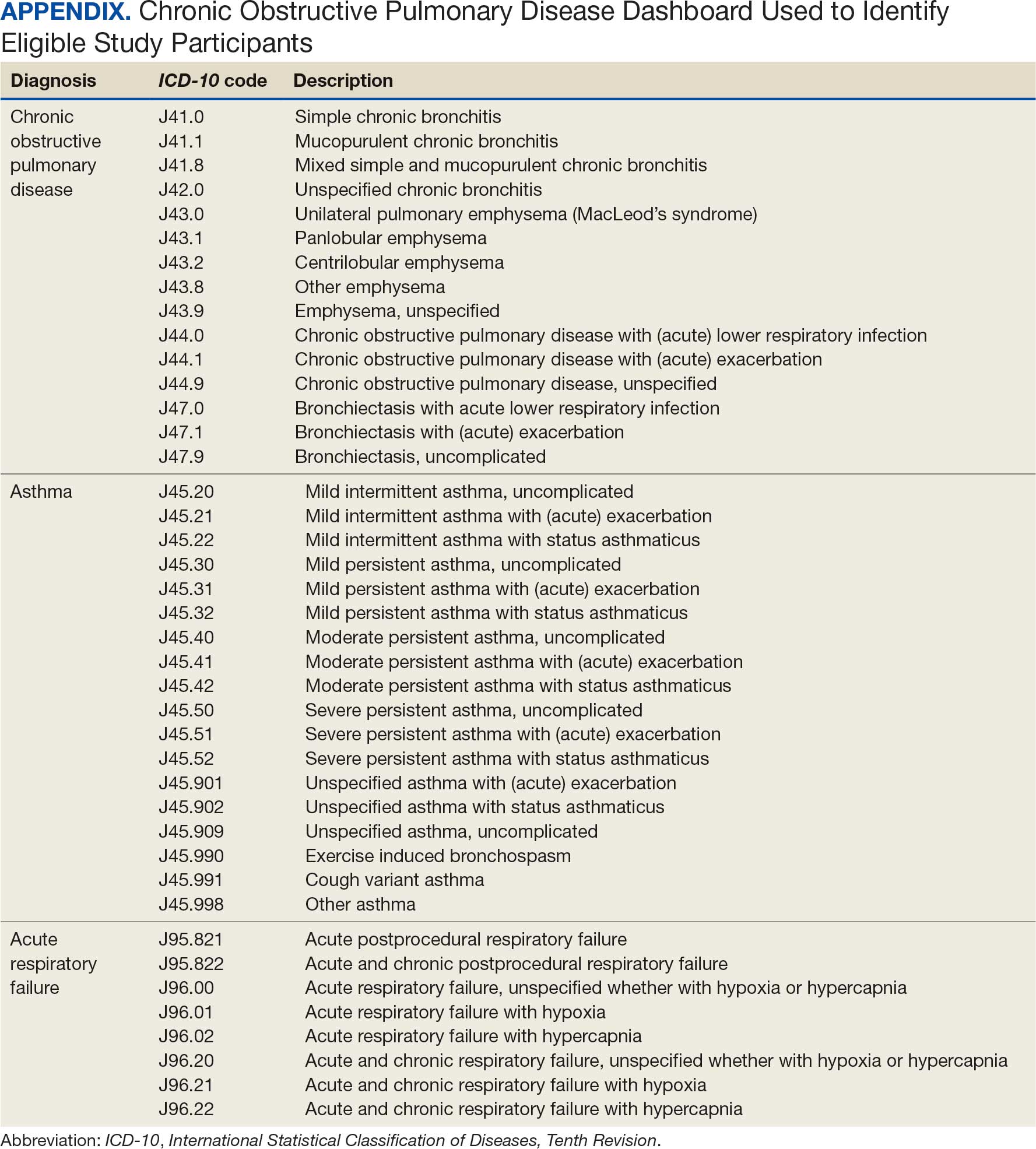
This project was conducted in an outpatient clinic at the Robley Rex VA Medical Center beginning September 21, 2023, with a progress note in the Computerized Patient Record System (CPRS). Eligible patients were selected using the COPD Dashboard provided by ADS. The COPD Dashboard defined patients with COPD as those with ≥ 2 outpatient COPD diagnoses in the past 2 years, 1 inpatient discharge COPD diagnosis in the past year, or COPD listed as an active problem. COPD diagnoses were identified using International Statistical Classification of Disease, Tenth Revision (ICD-10) codes
Candidates identified for ICS de-escalation by the dashboard were excluded if they had a history of COPD exacerbation in the previous 2 years. The dashboard identified COPD exacerbations via ICD-10 codes for COPD or acute respiratory failure for inpatient discharges, emergency department (ED) visits, urgent care visits, and community care consults with 1 of the following terms: emergency, inpatient, hospital, urgent, ED (self). The COPD dashboard excluded patients with a diagnosis of asthma.
After patients were selected, they were screened for additional exclusion criteria. Patients were excluded if a pulmonary care practitioner managed their COPD; if identified via an active pulmonary consult in CPRS; if a non-VA clinician prescribed their ICS; or if they were being treated with roflumilast, theophylline, or chronic azithromycin. Individuals taking these 3 drugs were excluded due to potential severe and/or refractory COPD. Patients also were excluded if they: (1) had prior ICS de-escalation failure (defined as a COPD exacerbation following ICS de-escalation that resulted in ICS resumption); (2) had a COPD exacerbation requiring systemic corticosteroids or antibiotics in the previous year; (3) had active lung cancer; (4) did not have any eosinophil levels in CPRS within the previous 2 years; or (5) had any eosinophil levels ≥ 300 cells/µL in the previous year.
Each patient who met the inclusion criteria and was not excluded received a focused medication review by a pharmacist who created a templated progress note, with patient-specific recommendations, that was entered in the CPRS (eAppendix). The recommendations were also attached as an addendum to the patient’s last primary care visit note, and the primary care practitioner (PCP) was alerted via CPRS to consider ICS de-escalation and non-ICS alternatives. Tapering of ICS therapy was offered as an option to de-escalate if abrupt discontinuation was deemed inappropriate. PCPs were also prompted to consider referral to a primary care clinical pharmacy specialist for management and follow-up of ICS de-escalation.
The primary outcome was the number of patients with de-escalated ICS at 3 and 6 months following the recommendation. Secondary outcomes included the number of: patients who were no longer prescribed an ICS or who had a non-ICS alternative initiated at a pharmacist’s recommendation; patients who were referred to a primary care clinical pharmacy specialist for ICS de-escalation; COPD exacerbations requiring systemic steroids or antibiotics, or requiring an ED visit, inpatient admission, or urgent-care clinic visit; and cases of pneumonia or oral candidiasis. Primary and secondary outcomes were evaluated via chart review in CPRS. For secondary outcomes of pneumonia and COPD exacerbation, identification was made by documented diagnosis in CPRS. For continuous data such as age, the mean was calculated.
Results
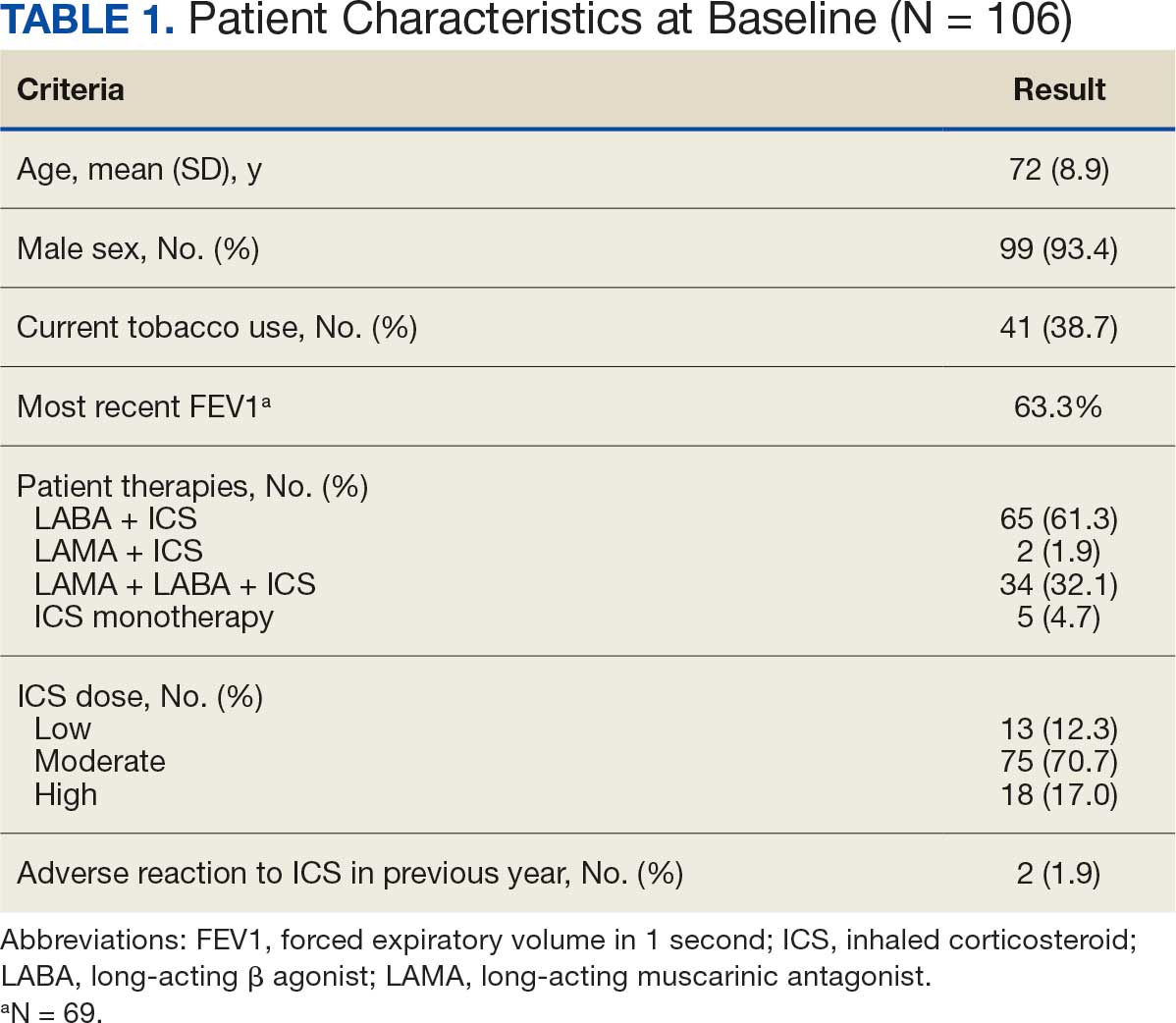
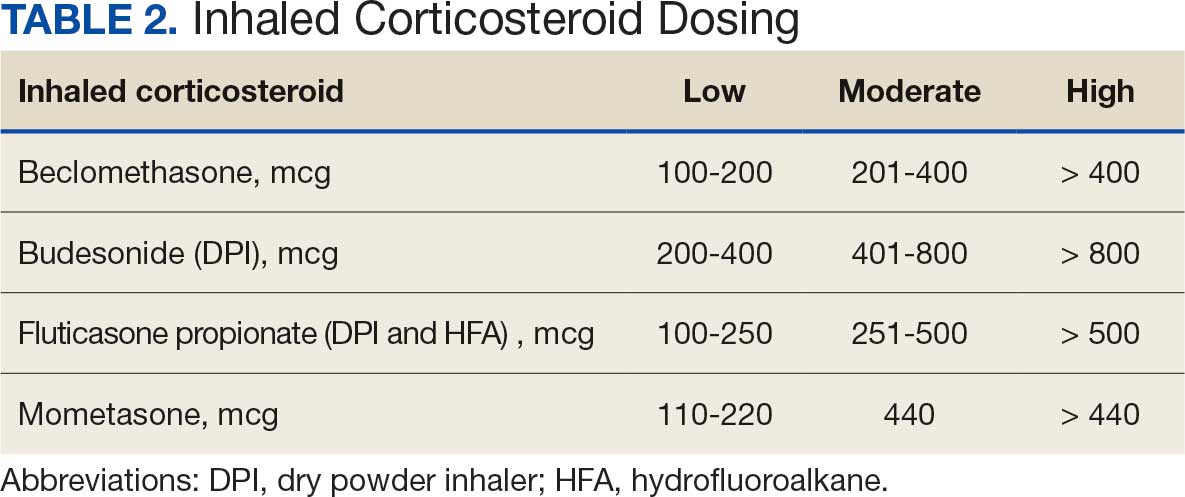
Pharmacist ICS de-escalation recommendations were made between September 21, 2023, and November 19, 2023, for 106 patients. The mean age was 72 years and 99 (93%) patients were male (Table 1). Forty-one (39%) of the patients used tobacco at the time of the study. FEV1 was available for 69 patients with a mean of 63% (GOLD grade 2).1 Based on FEV1 values, 16 patients had mild COPD (GOLD grade 1), 37 patients had moderate COPD (GOLD grade 2), 14 patients had severe COPD (GOLD grade 3), and 2 patients had very severe COPD (GOLD grade 4).1 Thirty-four patients received LABA + LAMA + ICS, 65 received LABA + ICS, 2 received LAMA + ICS, and 5 received ICS monotherapy. The most common dose of ICS was a moderate dose (Table 2). Only 2 patients had an ICS AE in the previous year.
ICS de-escalation recommendations resulted in ICS de-escalation in 50 (47.2%) and 62 (58.5%) patients at 3 and 6 months, respectively. The 6-month ICS de-escalation rate by ICS dose at baseline was 72.2% (high dose), 60.0% (moderate), and 30.8% (low). De-escalation at 6 months by GOLD grade at baseline was 56.3% (9 of 16 patients, GOLD 1), 64.9% (24 of 37 patients, GOLD 2), 50% (7 of 14 patients, GOLD 3), and 50% (1 of 2 patients, GOLD 4). Six months after the ICS de-escalation recommendation appeared in the CPRS, the percentage of patients on LABA + ICS therapy dropped from 65 patients (61.3%) at baseline to 25 patients (23.6%).
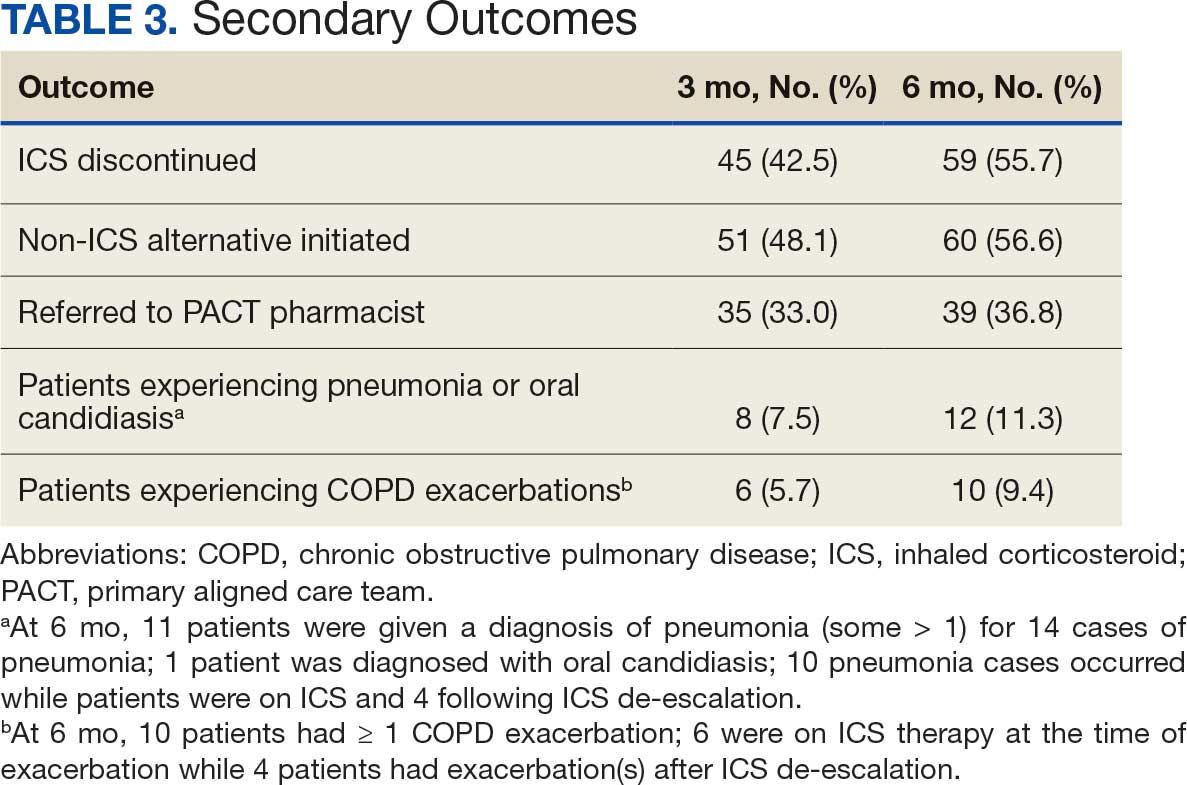
Secondary outcomes were assessed at 3 and 6 months following the recommendation. Most patients with de-escalated ICS had their ICS discontinued and a non-ICS alternative initiated per pharmacist recommendations. At 6 months, 39 patients (36.8%) patients were referred to a patient aligned care team (PACT) pharmacist for de-escalation. Of the 39 patients referred to pharmacists, 69.2% (27 patients) were de-escalated; this compared to 52.2% (35 patients) who were not referred to pharmacists (Table 3).
ICS use increases the risk of pneumonia.1 At 6 months, 11 patients were diagnosed with pneumonia; 3 patients were diagnosed with pneumonia twice, resulting in a total of 14 cases. Ten cases occurred while patients were on ICS and 4 cases occurred following ICS de-escalation. One patient had a documented case of oral candidiasis that occurred while on ICS therapy; no patients with discontinued ICS were diagnosed with oral candidiasis. In addition, 10 patients had COPD exacerbations; however no patients had exacerbations both before and after de-escalation. Six patients were on ICS therapy when they experienced an exacerbation, and 4 patients had an exacerbation after ICS de-escalation.
Discussion
More than half of patients receiving the pharmacist intervention achieved the primary outcome of ICS de-escalation at 6 months. Furthermore, a larger percentage of patients referred to pharmacists for the management of ICS de-escalation successfully achieved de-escalation compared to those who were not referred. These outcomes reflect the important role pharmacists can play in identifying appropriate candidates for ICS de-escalation and assisting in the management of ICS de-escalation. Patients referred to pharmacists also received other services such as smoking cessation pharmacotherapy and counseling on inhaler technique and adherence. These interventions can support improved COPD clinical outcomes.
The purpose of de-escalating ICS therapy is to reduce the risk of AEs such as pneumonia and oral candidiasis.1 The secondary outcomes of this study support previous evidence that patients who have de-escalated ICS therapy may have reduced risk of AEs compared to those who remain on ICS therapy.3 Specifically, of the 14 cases of pneumonia that occurred during the study, 10 cases occurred while patients were on ICS and 4 cases occurred following ICS de-escalation.
ICS de-escalation may increase risk of increased COPD exacerbations.1 However, the secondary outcomes of this study do not indicate that those with de-escalated ICS had more COPD exacerbations compared to those who continued on ICS. Pharmacists’ recommendations were more effective for patients with less severe COPD based on baseline FEV1.
The previous GOLD Guidelines for COPD suggested LABA + ICS therapy as an option for patients with a high symptom and exacerbation burden (previously known as GOLD Group D). Guidelines no longer recommend LABA + ICS therapy due to the superiority of triple inhaled therapy for exacerbations and the superiority of LAMA + LABA therapy for dyspnea.7 A majority of identified patients in this project were on LABA + ICS therapy alone at baseline. The ICS de-escalation recommendation resulted in a 61.5% reduction in patients on LABA + ICS therapy at 6 months. By decreasing the number of patients on LABA + ICS without LAMA, recommendations increased the number of patients on guideline-directed therapy.
Limitations
This study lacked a control group, and the rate of ICS de-escalation in patients who did not receive a pharmacist recommendation was not assessed. Therefore, it could not be determined whether the pharmacist recommendation is more effective than no recommendation. Another limitation was our inability to access records from non-VA health care facilities. This may have resulted in missed COPD exacerbations, pneumonia, and oral candidiasis prior to or following the pharmacist recommendation.
In addition, the method used to notify PCPs of the pharmacist recommendation was a CPRS alert. Clinicians often receive multiple daily alerts and may not always pay close attention to them due to alert fatigue. Early in the study, some PCPs were unknowingly omitted from the alert of the pharmacist recommendation for 10 patients due to human error. For 8 of these 10 patients, the PCP was notified of the recommendations during the 3-month follow-up period. However, 2 patients had COPD exacerbations during the 3-month follow-up period. In these cases, the PCP was not alerted to de-escalate ICS. The data for these patients were collected at 3 and 6 months in the same manner as all other patients. Also, 7 of 35 patients who were referred to a pharmacist for ICS de-escalation did not have a scheduled appointment. These patients were considered to be lost to follow-up and this may have resulted in an underestimation of the ability of pharmacists to successfully de-escalate ICS in patients with COPD.
Other studies have evaluated the efficacy of a pharmacy-driven ICS de-escalation.8,9 Hegland et al reported ICS de-escalation for 22% of 141 eligible ambulatory patients with COPD on triple inhaled therapy following pharmacist appointments.8 A study by Hahn et al resulted in 63.8% of 58 patients with COPD being maintained off ICS following a pharmacist de-escalation initiative.9 However, these studies relied upon more time-consuming de-escalation interventions, including at least 1 phone, video, or in-person patient visit.8,9
This project used a single chart review and templated progress note to recommend ICS de-escalation and achieved similar or improved de-escalation rates compared to previous studies.8,9 Previous studies were conducted prior to the updated 2023 GOLD guidelines for COPD which no longer recommend LABA + ICS therapy. This project addressed ICS de-escalation in patients on LABA + ICS therapy in addition to those on triple inhaled therapy. Additionally, previous studies did not address rates of moderate to severe COPD exacerbation and adverse events to ICS following the pharmacist intervention.8,9
This study included COPD exacerbations and cases of pneumonia or oral candidiasis as secondary outcomes to assess the safety and efficacy of the ICS de-escalation. It appeared there were similar or lower rates of COPD exacerbations, pneumonia, and oral candidiasis in those with de-escalated ICS therapy in this study. However, these secondary outcomes are exploratory and would need to be confirmed by larger studies powered to address these outcomes.
CONCLUSIONS
Pharmacist-driven ICS de-escalation may be an effective method for reducing ICS usage in veterans as seen in this study. Additional controlled studies are required to evaluate the efficacy and safety of pharmacist-driven ICS de-escalation.
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2024 Report). Accessed October 14, 2025. https://goldcopd.org/2024-gold-report/
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2025 Report). Accessed November 14, 2025. https://goldcopd.org/2025-gold-report/
- Rogliani P, Ritondo BL, Gabriele M, et al. Optimizing de-escalation of inhaled corticosteroids in COPD: a systematic review of real-world findings. Expert Rev Clin Pharmacol. 2020;13(9):977-990. doi:10.1080/17512433.2020.1817739
- Rossi A, Guerriero M, Corrado A; OPTIMO/AIPO Study Group. Withdrawal of inhaled corticosteroids can be safe in COPD patients at low risk of exacerbation: a real-life study on the appropriateness of treatment in moderate COPD patients (OPTIMO). Respir Res. 2014;15(1):77. doi:10.1186/1465-9921-15-77
- Anderson E, Wiener RS, Resnick K, et al. Care coordination for veterans with COPD: a positive deviance study. Am J Manag Care. 2020;26(2):63-68. doi:10.37765/ajmc.2020.42394
- US Department of Veterans Affairs, US Department of Defense. VA/DoD Clinical Practice Guideline for the Management of Chronic Obstructive Pulmonary Disease. 2021. Accessed October 14, 2025. https://www.healthquality.va.gov/guidelines/CD/copd/
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2023 Report). Accessed October 14, 2025. https://goldcopd.org/wp-content/uploads/2023/03/GOLD-2023-ver-1.3-17Feb2023_WMV.pdf
- Hegland AJ, Bolduc J, Jones L, Kunisaki KM, Melzer AC. Pharmacist-driven deprescribing of inhaled corticosteroids in patients with stable chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2021;18(4):730-733. doi:10.1513/AnnalsATS.202007-871RL
- Hahn NM, Nagy MW. Implementation of a targeted inhaled corticosteroid de-escalation process in patients with chronic obstructive pulmonary disease in the primary care setting. Innov Pharm. 2022;13(1):10.24926/iip.v13i1.4349. doi:10.24926/iip.v13i1.4349
Systemic glucocorticoids play an important role in the treatment of chronic obstructive pulmonary disease (COPD) exacerbations. They are recommended to shorten recovery time and increase forced expiratory volume in 1 second (FEV1) during exacerbations.1 However, the role of the chronic use of inhaled corticosteroids (ICSs) in the treatment of COPD is less clear.
When added to inhaled β-2 agonists and muscarinic antagonists, ICSs can decrease the risk of exacerbations.1 However, not all patients with COPD benefit from ICS therapy. The degree of benefit an ICS can provide has been shown to correlate with eosinophil count—a marker of inflammation. The expected benefit of using an ICS increases as the eosinophil count increases.1 Maximum benefit can be observed with eosinophil counts ≥ 300 cells/µL, and minimal benefit is observed with eosinophil counts < 100 cells/µL. Adverse effects (AEs) of ICSs include a hoarse voice, oral candidiasis, and an increased risk of pneumonia.1 Given the risk of AEs, it is important to limit ICS use in patients who are unlikely to reap any benefits.
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines suggest the use of ICSs in patients who experience exacerbations while using long-acting β agonist (LABA) plus long-acting muscarinic antagonist (LAMA) therapy and have an eosinophil count ≥ 100 cells/µL. Switching from LABA or LAMA monotherapy to triple therapy with LAMA/LABA/ICS may be considered if patients have continued exacerbations and an eosinophil count ≥ 300 cells/µL. De-escalation of ICS therapy should be considered if patients do not meet these criteria or if patients experience ICS AEs, such as pneumonia. The patients most likely to have increased exacerbations or decreased FEV1 with ICS withdrawal are those with eosinophil counts ≥ 300 cells/µL.1,2
Several studies have explored the effects of ICS de-escalation in real-world clinical settings. A systematic review of 11 studies indicated that de-escalation of ICS in COPD does not result in increased exacerbations.3 A prospective study by Rossi et al found that in a 6-month period, 141 of 482 patients on ICS therapy (29%) had an exacerbation. In the opposing arm of the study, 88 of 334 patients (26%) with deprescribed ICS experienced an exacerbation. The difference between these 2 groups was not statistically significant.4 The researchers concluded that in real-world practice, ICS withdrawal can be safe in patients at low risk of exacerbation.
About 25% of veterans (1.25 million) have been diagnosed with COPD.5 To address this, the US Department of Veterans Affairs (VA) and US Department of Defense published updated COPD guidelines in 2021 that specify criteria for de-escalation of ICS.6 Guidelines, however, may not be reflected in common clinical practice for several years following publication. The VA Academic Detailing Service (ADS) provides tools to help clinicians identify patients who may benefit from changes in treatment plans. A recent ADS focus was the implementation of a COPD dashboard, which identifies patients with COPD who are candidates for ICS de-escalation based on comorbid diagnoses, exacerbation history, and eosinophil count. VA pharmacists have an expanded role in the management of primary care disease states and are therefore well-positioned to increase adherence to guideline-directed therapy. The objective of this quality improvement project was to determine the impact of pharmacist-driven de-escalation on ICS usage in veterans with COPD.
Methods
This project was conducted in an outpatient clinic at the Robley Rex VA Medical Center beginning September 21, 2023, with a progress note in the Computerized Patient Record System (CPRS). Eligible patients were selected using the COPD Dashboard provided by ADS. The COPD Dashboard defined patients with COPD as those with ≥ 2 outpatient COPD diagnoses in the past 2 years, 1 inpatient discharge COPD diagnosis in the past year, or COPD listed as an active problem. COPD diagnoses were identified using International Statistical Classification of Disease, Tenth Revision (ICD-10) codes
Candidates identified for ICS de-escalation by the dashboard were excluded if they had a history of COPD exacerbation in the previous 2 years. The dashboard identified COPD exacerbations via ICD-10 codes for COPD or acute respiratory failure for inpatient discharges, emergency department (ED) visits, urgent care visits, and community care consults with 1 of the following terms: emergency, inpatient, hospital, urgent, ED (self). The COPD dashboard excluded patients with a diagnosis of asthma.
After patients were selected, they were screened for additional exclusion criteria. Patients were excluded if a pulmonary care practitioner managed their COPD; if identified via an active pulmonary consult in CPRS; if a non-VA clinician prescribed their ICS; or if they were being treated with roflumilast, theophylline, or chronic azithromycin. Individuals taking these 3 drugs were excluded due to potential severe and/or refractory COPD. Patients also were excluded if they: (1) had prior ICS de-escalation failure (defined as a COPD exacerbation following ICS de-escalation that resulted in ICS resumption); (2) had a COPD exacerbation requiring systemic corticosteroids or antibiotics in the previous year; (3) had active lung cancer; (4) did not have any eosinophil levels in CPRS within the previous 2 years; or (5) had any eosinophil levels ≥ 300 cells/µL in the previous year.
Each patient who met the inclusion criteria and was not excluded received a focused medication review by a pharmacist who created a templated progress note, with patient-specific recommendations, that was entered in the CPRS (eAppendix). The recommendations were also attached as an addendum to the patient’s last primary care visit note, and the primary care practitioner (PCP) was alerted via CPRS to consider ICS de-escalation and non-ICS alternatives. Tapering of ICS therapy was offered as an option to de-escalate if abrupt discontinuation was deemed inappropriate. PCPs were also prompted to consider referral to a primary care clinical pharmacy specialist for management and follow-up of ICS de-escalation.
The primary outcome was the number of patients with de-escalated ICS at 3 and 6 months following the recommendation. Secondary outcomes included the number of: patients who were no longer prescribed an ICS or who had a non-ICS alternative initiated at a pharmacist’s recommendation; patients who were referred to a primary care clinical pharmacy specialist for ICS de-escalation; COPD exacerbations requiring systemic steroids or antibiotics, or requiring an ED visit, inpatient admission, or urgent-care clinic visit; and cases of pneumonia or oral candidiasis. Primary and secondary outcomes were evaluated via chart review in CPRS. For secondary outcomes of pneumonia and COPD exacerbation, identification was made by documented diagnosis in CPRS. For continuous data such as age, the mean was calculated.
Results
Pharmacist ICS de-escalation recommendations were made between September 21, 2023, and November 19, 2023, for 106 patients. The mean age was 72 years and 99 (93%) patients were male (Table 1). Forty-one (39%) of the patients used tobacco at the time of the study. FEV1 was available for 69 patients with a mean of 63% (GOLD grade 2).1 Based on FEV1 values, 16 patients had mild COPD (GOLD grade 1), 37 patients had moderate COPD (GOLD grade 2), 14 patients had severe COPD (GOLD grade 3), and 2 patients had very severe COPD (GOLD grade 4).1 Thirty-four patients received LABA + LAMA + ICS, 65 received LABA + ICS, 2 received LAMA + ICS, and 5 received ICS monotherapy. The most common dose of ICS was a moderate dose (Table 2). Only 2 patients had an ICS AE in the previous year.
ICS de-escalation recommendations resulted in ICS de-escalation in 50 (47.2%) and 62 (58.5%) patients at 3 and 6 months, respectively. The 6-month ICS de-escalation rate by ICS dose at baseline was 72.2% (high dose), 60.0% (moderate), and 30.8% (low). De-escalation at 6 months by GOLD grade at baseline was 56.3% (9 of 16 patients, GOLD 1), 64.9% (24 of 37 patients, GOLD 2), 50% (7 of 14 patients, GOLD 3), and 50% (1 of 2 patients, GOLD 4). Six months after the ICS de-escalation recommendation appeared in the CPRS, the percentage of patients on LABA + ICS therapy dropped from 65 patients (61.3%) at baseline to 25 patients (23.6%).
Secondary outcomes were assessed at 3 and 6 months following the recommendation. Most patients with de-escalated ICS had their ICS discontinued and a non-ICS alternative initiated per pharmacist recommendations. At 6 months, 39 patients (36.8%) patients were referred to a patient aligned care team (PACT) pharmacist for de-escalation. Of the 39 patients referred to pharmacists, 69.2% (27 patients) were de-escalated; this compared to 52.2% (35 patients) who were not referred to pharmacists (Table 3).
ICS use increases the risk of pneumonia.1 At 6 months, 11 patients were diagnosed with pneumonia; 3 patients were diagnosed with pneumonia twice, resulting in a total of 14 cases. Ten cases occurred while patients were on ICS and 4 cases occurred following ICS de-escalation. One patient had a documented case of oral candidiasis that occurred while on ICS therapy; no patients with discontinued ICS were diagnosed with oral candidiasis. In addition, 10 patients had COPD exacerbations; however no patients had exacerbations both before and after de-escalation. Six patients were on ICS therapy when they experienced an exacerbation, and 4 patients had an exacerbation after ICS de-escalation.
Discussion
More than half of patients receiving the pharmacist intervention achieved the primary outcome of ICS de-escalation at 6 months. Furthermore, a larger percentage of patients referred to pharmacists for the management of ICS de-escalation successfully achieved de-escalation compared to those who were not referred. These outcomes reflect the important role pharmacists can play in identifying appropriate candidates for ICS de-escalation and assisting in the management of ICS de-escalation. Patients referred to pharmacists also received other services such as smoking cessation pharmacotherapy and counseling on inhaler technique and adherence. These interventions can support improved COPD clinical outcomes.
The purpose of de-escalating ICS therapy is to reduce the risk of AEs such as pneumonia and oral candidiasis.1 The secondary outcomes of this study support previous evidence that patients who have de-escalated ICS therapy may have reduced risk of AEs compared to those who remain on ICS therapy.3 Specifically, of the 14 cases of pneumonia that occurred during the study, 10 cases occurred while patients were on ICS and 4 cases occurred following ICS de-escalation.
ICS de-escalation may increase risk of increased COPD exacerbations.1 However, the secondary outcomes of this study do not indicate that those with de-escalated ICS had more COPD exacerbations compared to those who continued on ICS. Pharmacists’ recommendations were more effective for patients with less severe COPD based on baseline FEV1.
The previous GOLD Guidelines for COPD suggested LABA + ICS therapy as an option for patients with a high symptom and exacerbation burden (previously known as GOLD Group D). Guidelines no longer recommend LABA + ICS therapy due to the superiority of triple inhaled therapy for exacerbations and the superiority of LAMA + LABA therapy for dyspnea.7 A majority of identified patients in this project were on LABA + ICS therapy alone at baseline. The ICS de-escalation recommendation resulted in a 61.5% reduction in patients on LABA + ICS therapy at 6 months. By decreasing the number of patients on LABA + ICS without LAMA, recommendations increased the number of patients on guideline-directed therapy.
Limitations
This study lacked a control group, and the rate of ICS de-escalation in patients who did not receive a pharmacist recommendation was not assessed. Therefore, it could not be determined whether the pharmacist recommendation is more effective than no recommendation. Another limitation was our inability to access records from non-VA health care facilities. This may have resulted in missed COPD exacerbations, pneumonia, and oral candidiasis prior to or following the pharmacist recommendation.
In addition, the method used to notify PCPs of the pharmacist recommendation was a CPRS alert. Clinicians often receive multiple daily alerts and may not always pay close attention to them due to alert fatigue. Early in the study, some PCPs were unknowingly omitted from the alert of the pharmacist recommendation for 10 patients due to human error. For 8 of these 10 patients, the PCP was notified of the recommendations during the 3-month follow-up period. However, 2 patients had COPD exacerbations during the 3-month follow-up period. In these cases, the PCP was not alerted to de-escalate ICS. The data for these patients were collected at 3 and 6 months in the same manner as all other patients. Also, 7 of 35 patients who were referred to a pharmacist for ICS de-escalation did not have a scheduled appointment. These patients were considered to be lost to follow-up and this may have resulted in an underestimation of the ability of pharmacists to successfully de-escalate ICS in patients with COPD.
Other studies have evaluated the efficacy of a pharmacy-driven ICS de-escalation.8,9 Hegland et al reported ICS de-escalation for 22% of 141 eligible ambulatory patients with COPD on triple inhaled therapy following pharmacist appointments.8 A study by Hahn et al resulted in 63.8% of 58 patients with COPD being maintained off ICS following a pharmacist de-escalation initiative.9 However, these studies relied upon more time-consuming de-escalation interventions, including at least 1 phone, video, or in-person patient visit.8,9
This project used a single chart review and templated progress note to recommend ICS de-escalation and achieved similar or improved de-escalation rates compared to previous studies.8,9 Previous studies were conducted prior to the updated 2023 GOLD guidelines for COPD which no longer recommend LABA + ICS therapy. This project addressed ICS de-escalation in patients on LABA + ICS therapy in addition to those on triple inhaled therapy. Additionally, previous studies did not address rates of moderate to severe COPD exacerbation and adverse events to ICS following the pharmacist intervention.8,9
This study included COPD exacerbations and cases of pneumonia or oral candidiasis as secondary outcomes to assess the safety and efficacy of the ICS de-escalation. It appeared there were similar or lower rates of COPD exacerbations, pneumonia, and oral candidiasis in those with de-escalated ICS therapy in this study. However, these secondary outcomes are exploratory and would need to be confirmed by larger studies powered to address these outcomes.
CONCLUSIONS
Pharmacist-driven ICS de-escalation may be an effective method for reducing ICS usage in veterans as seen in this study. Additional controlled studies are required to evaluate the efficacy and safety of pharmacist-driven ICS de-escalation.
Systemic glucocorticoids play an important role in the treatment of chronic obstructive pulmonary disease (COPD) exacerbations. They are recommended to shorten recovery time and increase forced expiratory volume in 1 second (FEV1) during exacerbations.1 However, the role of the chronic use of inhaled corticosteroids (ICSs) in the treatment of COPD is less clear.
When added to inhaled β-2 agonists and muscarinic antagonists, ICSs can decrease the risk of exacerbations.1 However, not all patients with COPD benefit from ICS therapy. The degree of benefit an ICS can provide has been shown to correlate with eosinophil count—a marker of inflammation. The expected benefit of using an ICS increases as the eosinophil count increases.1 Maximum benefit can be observed with eosinophil counts ≥ 300 cells/µL, and minimal benefit is observed with eosinophil counts < 100 cells/µL. Adverse effects (AEs) of ICSs include a hoarse voice, oral candidiasis, and an increased risk of pneumonia.1 Given the risk of AEs, it is important to limit ICS use in patients who are unlikely to reap any benefits.
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines suggest the use of ICSs in patients who experience exacerbations while using long-acting β agonist (LABA) plus long-acting muscarinic antagonist (LAMA) therapy and have an eosinophil count ≥ 100 cells/µL. Switching from LABA or LAMA monotherapy to triple therapy with LAMA/LABA/ICS may be considered if patients have continued exacerbations and an eosinophil count ≥ 300 cells/µL. De-escalation of ICS therapy should be considered if patients do not meet these criteria or if patients experience ICS AEs, such as pneumonia. The patients most likely to have increased exacerbations or decreased FEV1 with ICS withdrawal are those with eosinophil counts ≥ 300 cells/µL.1,2
Several studies have explored the effects of ICS de-escalation in real-world clinical settings. A systematic review of 11 studies indicated that de-escalation of ICS in COPD does not result in increased exacerbations.3 A prospective study by Rossi et al found that in a 6-month period, 141 of 482 patients on ICS therapy (29%) had an exacerbation. In the opposing arm of the study, 88 of 334 patients (26%) with deprescribed ICS experienced an exacerbation. The difference between these 2 groups was not statistically significant.4 The researchers concluded that in real-world practice, ICS withdrawal can be safe in patients at low risk of exacerbation.
About 25% of veterans (1.25 million) have been diagnosed with COPD.5 To address this, the US Department of Veterans Affairs (VA) and US Department of Defense published updated COPD guidelines in 2021 that specify criteria for de-escalation of ICS.6 Guidelines, however, may not be reflected in common clinical practice for several years following publication. The VA Academic Detailing Service (ADS) provides tools to help clinicians identify patients who may benefit from changes in treatment plans. A recent ADS focus was the implementation of a COPD dashboard, which identifies patients with COPD who are candidates for ICS de-escalation based on comorbid diagnoses, exacerbation history, and eosinophil count. VA pharmacists have an expanded role in the management of primary care disease states and are therefore well-positioned to increase adherence to guideline-directed therapy. The objective of this quality improvement project was to determine the impact of pharmacist-driven de-escalation on ICS usage in veterans with COPD.
Methods
This project was conducted in an outpatient clinic at the Robley Rex VA Medical Center beginning September 21, 2023, with a progress note in the Computerized Patient Record System (CPRS). Eligible patients were selected using the COPD Dashboard provided by ADS. The COPD Dashboard defined patients with COPD as those with ≥ 2 outpatient COPD diagnoses in the past 2 years, 1 inpatient discharge COPD diagnosis in the past year, or COPD listed as an active problem. COPD diagnoses were identified using International Statistical Classification of Disease, Tenth Revision (ICD-10) codes
Candidates identified for ICS de-escalation by the dashboard were excluded if they had a history of COPD exacerbation in the previous 2 years. The dashboard identified COPD exacerbations via ICD-10 codes for COPD or acute respiratory failure for inpatient discharges, emergency department (ED) visits, urgent care visits, and community care consults with 1 of the following terms: emergency, inpatient, hospital, urgent, ED (self). The COPD dashboard excluded patients with a diagnosis of asthma.
After patients were selected, they were screened for additional exclusion criteria. Patients were excluded if a pulmonary care practitioner managed their COPD; if identified via an active pulmonary consult in CPRS; if a non-VA clinician prescribed their ICS; or if they were being treated with roflumilast, theophylline, or chronic azithromycin. Individuals taking these 3 drugs were excluded due to potential severe and/or refractory COPD. Patients also were excluded if they: (1) had prior ICS de-escalation failure (defined as a COPD exacerbation following ICS de-escalation that resulted in ICS resumption); (2) had a COPD exacerbation requiring systemic corticosteroids or antibiotics in the previous year; (3) had active lung cancer; (4) did not have any eosinophil levels in CPRS within the previous 2 years; or (5) had any eosinophil levels ≥ 300 cells/µL in the previous year.
Each patient who met the inclusion criteria and was not excluded received a focused medication review by a pharmacist who created a templated progress note, with patient-specific recommendations, that was entered in the CPRS (eAppendix). The recommendations were also attached as an addendum to the patient’s last primary care visit note, and the primary care practitioner (PCP) was alerted via CPRS to consider ICS de-escalation and non-ICS alternatives. Tapering of ICS therapy was offered as an option to de-escalate if abrupt discontinuation was deemed inappropriate. PCPs were also prompted to consider referral to a primary care clinical pharmacy specialist for management and follow-up of ICS de-escalation.
The primary outcome was the number of patients with de-escalated ICS at 3 and 6 months following the recommendation. Secondary outcomes included the number of: patients who were no longer prescribed an ICS or who had a non-ICS alternative initiated at a pharmacist’s recommendation; patients who were referred to a primary care clinical pharmacy specialist for ICS de-escalation; COPD exacerbations requiring systemic steroids or antibiotics, or requiring an ED visit, inpatient admission, or urgent-care clinic visit; and cases of pneumonia or oral candidiasis. Primary and secondary outcomes were evaluated via chart review in CPRS. For secondary outcomes of pneumonia and COPD exacerbation, identification was made by documented diagnosis in CPRS. For continuous data such as age, the mean was calculated.
Results
Pharmacist ICS de-escalation recommendations were made between September 21, 2023, and November 19, 2023, for 106 patients. The mean age was 72 years and 99 (93%) patients were male (Table 1). Forty-one (39%) of the patients used tobacco at the time of the study. FEV1 was available for 69 patients with a mean of 63% (GOLD grade 2).1 Based on FEV1 values, 16 patients had mild COPD (GOLD grade 1), 37 patients had moderate COPD (GOLD grade 2), 14 patients had severe COPD (GOLD grade 3), and 2 patients had very severe COPD (GOLD grade 4).1 Thirty-four patients received LABA + LAMA + ICS, 65 received LABA + ICS, 2 received LAMA + ICS, and 5 received ICS monotherapy. The most common dose of ICS was a moderate dose (Table 2). Only 2 patients had an ICS AE in the previous year.
ICS de-escalation recommendations resulted in ICS de-escalation in 50 (47.2%) and 62 (58.5%) patients at 3 and 6 months, respectively. The 6-month ICS de-escalation rate by ICS dose at baseline was 72.2% (high dose), 60.0% (moderate), and 30.8% (low). De-escalation at 6 months by GOLD grade at baseline was 56.3% (9 of 16 patients, GOLD 1), 64.9% (24 of 37 patients, GOLD 2), 50% (7 of 14 patients, GOLD 3), and 50% (1 of 2 patients, GOLD 4). Six months after the ICS de-escalation recommendation appeared in the CPRS, the percentage of patients on LABA + ICS therapy dropped from 65 patients (61.3%) at baseline to 25 patients (23.6%).
Secondary outcomes were assessed at 3 and 6 months following the recommendation. Most patients with de-escalated ICS had their ICS discontinued and a non-ICS alternative initiated per pharmacist recommendations. At 6 months, 39 patients (36.8%) patients were referred to a patient aligned care team (PACT) pharmacist for de-escalation. Of the 39 patients referred to pharmacists, 69.2% (27 patients) were de-escalated; this compared to 52.2% (35 patients) who were not referred to pharmacists (Table 3).
ICS use increases the risk of pneumonia.1 At 6 months, 11 patients were diagnosed with pneumonia; 3 patients were diagnosed with pneumonia twice, resulting in a total of 14 cases. Ten cases occurred while patients were on ICS and 4 cases occurred following ICS de-escalation. One patient had a documented case of oral candidiasis that occurred while on ICS therapy; no patients with discontinued ICS were diagnosed with oral candidiasis. In addition, 10 patients had COPD exacerbations; however no patients had exacerbations both before and after de-escalation. Six patients were on ICS therapy when they experienced an exacerbation, and 4 patients had an exacerbation after ICS de-escalation.
Discussion
More than half of patients receiving the pharmacist intervention achieved the primary outcome of ICS de-escalation at 6 months. Furthermore, a larger percentage of patients referred to pharmacists for the management of ICS de-escalation successfully achieved de-escalation compared to those who were not referred. These outcomes reflect the important role pharmacists can play in identifying appropriate candidates for ICS de-escalation and assisting in the management of ICS de-escalation. Patients referred to pharmacists also received other services such as smoking cessation pharmacotherapy and counseling on inhaler technique and adherence. These interventions can support improved COPD clinical outcomes.
The purpose of de-escalating ICS therapy is to reduce the risk of AEs such as pneumonia and oral candidiasis.1 The secondary outcomes of this study support previous evidence that patients who have de-escalated ICS therapy may have reduced risk of AEs compared to those who remain on ICS therapy.3 Specifically, of the 14 cases of pneumonia that occurred during the study, 10 cases occurred while patients were on ICS and 4 cases occurred following ICS de-escalation.
ICS de-escalation may increase risk of increased COPD exacerbations.1 However, the secondary outcomes of this study do not indicate that those with de-escalated ICS had more COPD exacerbations compared to those who continued on ICS. Pharmacists’ recommendations were more effective for patients with less severe COPD based on baseline FEV1.
The previous GOLD Guidelines for COPD suggested LABA + ICS therapy as an option for patients with a high symptom and exacerbation burden (previously known as GOLD Group D). Guidelines no longer recommend LABA + ICS therapy due to the superiority of triple inhaled therapy for exacerbations and the superiority of LAMA + LABA therapy for dyspnea.7 A majority of identified patients in this project were on LABA + ICS therapy alone at baseline. The ICS de-escalation recommendation resulted in a 61.5% reduction in patients on LABA + ICS therapy at 6 months. By decreasing the number of patients on LABA + ICS without LAMA, recommendations increased the number of patients on guideline-directed therapy.
Limitations
This study lacked a control group, and the rate of ICS de-escalation in patients who did not receive a pharmacist recommendation was not assessed. Therefore, it could not be determined whether the pharmacist recommendation is more effective than no recommendation. Another limitation was our inability to access records from non-VA health care facilities. This may have resulted in missed COPD exacerbations, pneumonia, and oral candidiasis prior to or following the pharmacist recommendation.
In addition, the method used to notify PCPs of the pharmacist recommendation was a CPRS alert. Clinicians often receive multiple daily alerts and may not always pay close attention to them due to alert fatigue. Early in the study, some PCPs were unknowingly omitted from the alert of the pharmacist recommendation for 10 patients due to human error. For 8 of these 10 patients, the PCP was notified of the recommendations during the 3-month follow-up period. However, 2 patients had COPD exacerbations during the 3-month follow-up period. In these cases, the PCP was not alerted to de-escalate ICS. The data for these patients were collected at 3 and 6 months in the same manner as all other patients. Also, 7 of 35 patients who were referred to a pharmacist for ICS de-escalation did not have a scheduled appointment. These patients were considered to be lost to follow-up and this may have resulted in an underestimation of the ability of pharmacists to successfully de-escalate ICS in patients with COPD.
Other studies have evaluated the efficacy of a pharmacy-driven ICS de-escalation.8,9 Hegland et al reported ICS de-escalation for 22% of 141 eligible ambulatory patients with COPD on triple inhaled therapy following pharmacist appointments.8 A study by Hahn et al resulted in 63.8% of 58 patients with COPD being maintained off ICS following a pharmacist de-escalation initiative.9 However, these studies relied upon more time-consuming de-escalation interventions, including at least 1 phone, video, or in-person patient visit.8,9
This project used a single chart review and templated progress note to recommend ICS de-escalation and achieved similar or improved de-escalation rates compared to previous studies.8,9 Previous studies were conducted prior to the updated 2023 GOLD guidelines for COPD which no longer recommend LABA + ICS therapy. This project addressed ICS de-escalation in patients on LABA + ICS therapy in addition to those on triple inhaled therapy. Additionally, previous studies did not address rates of moderate to severe COPD exacerbation and adverse events to ICS following the pharmacist intervention.8,9
This study included COPD exacerbations and cases of pneumonia or oral candidiasis as secondary outcomes to assess the safety and efficacy of the ICS de-escalation. It appeared there were similar or lower rates of COPD exacerbations, pneumonia, and oral candidiasis in those with de-escalated ICS therapy in this study. However, these secondary outcomes are exploratory and would need to be confirmed by larger studies powered to address these outcomes.
CONCLUSIONS
Pharmacist-driven ICS de-escalation may be an effective method for reducing ICS usage in veterans as seen in this study. Additional controlled studies are required to evaluate the efficacy and safety of pharmacist-driven ICS de-escalation.
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2024 Report). Accessed October 14, 2025. https://goldcopd.org/2024-gold-report/
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2025 Report). Accessed November 14, 2025. https://goldcopd.org/2025-gold-report/
- Rogliani P, Ritondo BL, Gabriele M, et al. Optimizing de-escalation of inhaled corticosteroids in COPD: a systematic review of real-world findings. Expert Rev Clin Pharmacol. 2020;13(9):977-990. doi:10.1080/17512433.2020.1817739
- Rossi A, Guerriero M, Corrado A; OPTIMO/AIPO Study Group. Withdrawal of inhaled corticosteroids can be safe in COPD patients at low risk of exacerbation: a real-life study on the appropriateness of treatment in moderate COPD patients (OPTIMO). Respir Res. 2014;15(1):77. doi:10.1186/1465-9921-15-77
- Anderson E, Wiener RS, Resnick K, et al. Care coordination for veterans with COPD: a positive deviance study. Am J Manag Care. 2020;26(2):63-68. doi:10.37765/ajmc.2020.42394
- US Department of Veterans Affairs, US Department of Defense. VA/DoD Clinical Practice Guideline for the Management of Chronic Obstructive Pulmonary Disease. 2021. Accessed October 14, 2025. https://www.healthquality.va.gov/guidelines/CD/copd/
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2023 Report). Accessed October 14, 2025. https://goldcopd.org/wp-content/uploads/2023/03/GOLD-2023-ver-1.3-17Feb2023_WMV.pdf
- Hegland AJ, Bolduc J, Jones L, Kunisaki KM, Melzer AC. Pharmacist-driven deprescribing of inhaled corticosteroids in patients with stable chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2021;18(4):730-733. doi:10.1513/AnnalsATS.202007-871RL
- Hahn NM, Nagy MW. Implementation of a targeted inhaled corticosteroid de-escalation process in patients with chronic obstructive pulmonary disease in the primary care setting. Innov Pharm. 2022;13(1):10.24926/iip.v13i1.4349. doi:10.24926/iip.v13i1.4349
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2024 Report). Accessed October 14, 2025. https://goldcopd.org/2024-gold-report/
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2025 Report). Accessed November 14, 2025. https://goldcopd.org/2025-gold-report/
- Rogliani P, Ritondo BL, Gabriele M, et al. Optimizing de-escalation of inhaled corticosteroids in COPD: a systematic review of real-world findings. Expert Rev Clin Pharmacol. 2020;13(9):977-990. doi:10.1080/17512433.2020.1817739
- Rossi A, Guerriero M, Corrado A; OPTIMO/AIPO Study Group. Withdrawal of inhaled corticosteroids can be safe in COPD patients at low risk of exacerbation: a real-life study on the appropriateness of treatment in moderate COPD patients (OPTIMO). Respir Res. 2014;15(1):77. doi:10.1186/1465-9921-15-77
- Anderson E, Wiener RS, Resnick K, et al. Care coordination for veterans with COPD: a positive deviance study. Am J Manag Care. 2020;26(2):63-68. doi:10.37765/ajmc.2020.42394
- US Department of Veterans Affairs, US Department of Defense. VA/DoD Clinical Practice Guideline for the Management of Chronic Obstructive Pulmonary Disease. 2021. Accessed October 14, 2025. https://www.healthquality.va.gov/guidelines/CD/copd/
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2023 Report). Accessed October 14, 2025. https://goldcopd.org/wp-content/uploads/2023/03/GOLD-2023-ver-1.3-17Feb2023_WMV.pdf
- Hegland AJ, Bolduc J, Jones L, Kunisaki KM, Melzer AC. Pharmacist-driven deprescribing of inhaled corticosteroids in patients with stable chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2021;18(4):730-733. doi:10.1513/AnnalsATS.202007-871RL
- Hahn NM, Nagy MW. Implementation of a targeted inhaled corticosteroid de-escalation process in patients with chronic obstructive pulmonary disease in the primary care setting. Innov Pharm. 2022;13(1):10.24926/iip.v13i1.4349. doi:10.24926/iip.v13i1.4349
Evaluation of Pharmacist-Driven Inhaled Corticosteroid De-escalation in Veterans
Evaluation of Pharmacist-Driven Inhaled Corticosteroid De-escalation in Veterans
New Drug Eases Side Effects of Weight-Loss Meds
A new drug currently known as NG101 reduced nausea and vomiting in patients with obesity using GLP-1s by 40% and 67%, respectively, based on data from a phase 2 trial presented at the Obesity Society’s Obesity Week 2025 in Atlanta.
Previous research published in JAMA Network Open showed a nearly 65% discontinuation rate for three GLP-1s (liraglutide, semaglutide, or tirzepatide) among adults with overweight or obesity and without type 2 diabetes. Gastrointestinal (GI) side effects topped the list of reasons for dropping the medications.
Given the impact of nausea and vomiting on discontinuation, there is an unmet need for therapies to manage GI symptoms, said Kimberley Cummings, PhD, of Neurogastrx, Inc., in her presentation.
In the new study, Cummings and colleagues randomly assigned 90 adults aged 18-55 years with overweight or obesity (defined as a BMI ranging from 22.0 to 35.0) to receive a single subcutaneous dose of semaglutide (0.5 mg) plus 5 days of NG101 at 20 mg twice daily, or a placebo.
NG101 is a peripherally acting D2 antagonist designed to reduce nausea and vomiting associated with GLP-1 use, Cummings said. NG101 targets the nausea center of the brain but is peripherally restricted to prevent central nervous system side effects, she explained.
Compared with placebo, NG101 significantly reduced the incidence of nausea and vomiting by 40% and 67%, respectively. Use of NG101 also was associated with a significant reduction in the duration of nausea and vomiting; GI events lasting longer than 1 day were reported in 22% and 51% of the NG101 patients and placebo patients, respectively.
In addition, participants who received NG101 reported a 70% decrease in nausea severity from baseline.
Overall, patients in the NG101 group also reported significantly fewer adverse events than those in the placebo group (74 vs 135), suggesting an improved safety profile when semaglutide is administered in conjunction with NG101, the researchers noted. No serious adverse events related to the study drug were reported in either group.
The findings were limited by several factors including the relatively small sample size. Additional research is needed with other GLP-1 agonists in larger populations with longer follow-up periods, Cummings said. However, the results suggest that NG101 was safe and effectively improved side effects associated with GLP-1 agonists.
“We know there are receptors for GLP-1 in the area postrema (nausea center of the brain), and that NG101 works on this area to reduce nausea and vomiting, so the study findings were not unexpected,” said Jim O’Mara, president and CEO of Neurogastrx, in an interview.
The study was a single-dose study designed to show proof of concept, and future studies would involve treating patients going through the recommended titration schedule for their GLP-1s, O’Mara said. However, NG101 offers an opportunity to keep more patients on GLP-1 therapy and help them reach their long-term therapeutic goals, he said.
Decrease Side Effects for Weight-Loss Success
“GI side effects are often the rate-limiting step in implementing an effective medication that patients want to take but may not be able to tolerate,” said Sean Wharton, MD, PharmD, medical director of the Wharton Medical Clinic for Weight and Diabetes Management, Burlington, Ontario, Canada, in an interview. “If we can decrease side effects, these medications could improve patients’ lives,” said Wharton, who was not involved in the study.
The improvement after a single dose of NG101 in patients receiving a single dose of semaglutide was impressive and in keeping with the mechanism of the drug action, said Wharton. “I was not surprised by the result but pleased that this single dose was shown to reduce the overall incidence of nausea and vomiting, the duration of nausea, the severity of nausea as rated by the study participants compared to placebo,” he said.
Ultimately, the clinical implications for NG101 are improved patient tolerance for GLP-1s and the ability to titrate and stay on them long term, incurring greater cardiometabolic benefit, Wharton told this news organization.
The current trial was limited to GLP1-1s on the market; newer medications may have fewer side effects, Wharton noted. “In clinical practice, patients often decrease the medication or titrate slower, and this could be the comparator,” he added.
The study was funded by Neurogastrx.
Wharton disclosed serving as a consultant for Neurogastrx but not as an investigator on the current study. He also reported having disclosed research on various GLP-1 medications.
A version of this article first appeared on Medscape.com.
A new drug currently known as NG101 reduced nausea and vomiting in patients with obesity using GLP-1s by 40% and 67%, respectively, based on data from a phase 2 trial presented at the Obesity Society’s Obesity Week 2025 in Atlanta.
Previous research published in JAMA Network Open showed a nearly 65% discontinuation rate for three GLP-1s (liraglutide, semaglutide, or tirzepatide) among adults with overweight or obesity and without type 2 diabetes. Gastrointestinal (GI) side effects topped the list of reasons for dropping the medications.
Given the impact of nausea and vomiting on discontinuation, there is an unmet need for therapies to manage GI symptoms, said Kimberley Cummings, PhD, of Neurogastrx, Inc., in her presentation.
In the new study, Cummings and colleagues randomly assigned 90 adults aged 18-55 years with overweight or obesity (defined as a BMI ranging from 22.0 to 35.0) to receive a single subcutaneous dose of semaglutide (0.5 mg) plus 5 days of NG101 at 20 mg twice daily, or a placebo.
NG101 is a peripherally acting D2 antagonist designed to reduce nausea and vomiting associated with GLP-1 use, Cummings said. NG101 targets the nausea center of the brain but is peripherally restricted to prevent central nervous system side effects, she explained.
Compared with placebo, NG101 significantly reduced the incidence of nausea and vomiting by 40% and 67%, respectively. Use of NG101 also was associated with a significant reduction in the duration of nausea and vomiting; GI events lasting longer than 1 day were reported in 22% and 51% of the NG101 patients and placebo patients, respectively.
In addition, participants who received NG101 reported a 70% decrease in nausea severity from baseline.
Overall, patients in the NG101 group also reported significantly fewer adverse events than those in the placebo group (74 vs 135), suggesting an improved safety profile when semaglutide is administered in conjunction with NG101, the researchers noted. No serious adverse events related to the study drug were reported in either group.
The findings were limited by several factors including the relatively small sample size. Additional research is needed with other GLP-1 agonists in larger populations with longer follow-up periods, Cummings said. However, the results suggest that NG101 was safe and effectively improved side effects associated with GLP-1 agonists.
“We know there are receptors for GLP-1 in the area postrema (nausea center of the brain), and that NG101 works on this area to reduce nausea and vomiting, so the study findings were not unexpected,” said Jim O’Mara, president and CEO of Neurogastrx, in an interview.
The study was a single-dose study designed to show proof of concept, and future studies would involve treating patients going through the recommended titration schedule for their GLP-1s, O’Mara said. However, NG101 offers an opportunity to keep more patients on GLP-1 therapy and help them reach their long-term therapeutic goals, he said.
Decrease Side Effects for Weight-Loss Success
“GI side effects are often the rate-limiting step in implementing an effective medication that patients want to take but may not be able to tolerate,” said Sean Wharton, MD, PharmD, medical director of the Wharton Medical Clinic for Weight and Diabetes Management, Burlington, Ontario, Canada, in an interview. “If we can decrease side effects, these medications could improve patients’ lives,” said Wharton, who was not involved in the study.
The improvement after a single dose of NG101 in patients receiving a single dose of semaglutide was impressive and in keeping with the mechanism of the drug action, said Wharton. “I was not surprised by the result but pleased that this single dose was shown to reduce the overall incidence of nausea and vomiting, the duration of nausea, the severity of nausea as rated by the study participants compared to placebo,” he said.
Ultimately, the clinical implications for NG101 are improved patient tolerance for GLP-1s and the ability to titrate and stay on them long term, incurring greater cardiometabolic benefit, Wharton told this news organization.
The current trial was limited to GLP1-1s on the market; newer medications may have fewer side effects, Wharton noted. “In clinical practice, patients often decrease the medication or titrate slower, and this could be the comparator,” he added.
The study was funded by Neurogastrx.
Wharton disclosed serving as a consultant for Neurogastrx but not as an investigator on the current study. He also reported having disclosed research on various GLP-1 medications.
A version of this article first appeared on Medscape.com.
A new drug currently known as NG101 reduced nausea and vomiting in patients with obesity using GLP-1s by 40% and 67%, respectively, based on data from a phase 2 trial presented at the Obesity Society’s Obesity Week 2025 in Atlanta.
Previous research published in JAMA Network Open showed a nearly 65% discontinuation rate for three GLP-1s (liraglutide, semaglutide, or tirzepatide) among adults with overweight or obesity and without type 2 diabetes. Gastrointestinal (GI) side effects topped the list of reasons for dropping the medications.
Given the impact of nausea and vomiting on discontinuation, there is an unmet need for therapies to manage GI symptoms, said Kimberley Cummings, PhD, of Neurogastrx, Inc., in her presentation.
In the new study, Cummings and colleagues randomly assigned 90 adults aged 18-55 years with overweight or obesity (defined as a BMI ranging from 22.0 to 35.0) to receive a single subcutaneous dose of semaglutide (0.5 mg) plus 5 days of NG101 at 20 mg twice daily, or a placebo.
NG101 is a peripherally acting D2 antagonist designed to reduce nausea and vomiting associated with GLP-1 use, Cummings said. NG101 targets the nausea center of the brain but is peripherally restricted to prevent central nervous system side effects, she explained.
Compared with placebo, NG101 significantly reduced the incidence of nausea and vomiting by 40% and 67%, respectively. Use of NG101 also was associated with a significant reduction in the duration of nausea and vomiting; GI events lasting longer than 1 day were reported in 22% and 51% of the NG101 patients and placebo patients, respectively.
In addition, participants who received NG101 reported a 70% decrease in nausea severity from baseline.
Overall, patients in the NG101 group also reported significantly fewer adverse events than those in the placebo group (74 vs 135), suggesting an improved safety profile when semaglutide is administered in conjunction with NG101, the researchers noted. No serious adverse events related to the study drug were reported in either group.
The findings were limited by several factors including the relatively small sample size. Additional research is needed with other GLP-1 agonists in larger populations with longer follow-up periods, Cummings said. However, the results suggest that NG101 was safe and effectively improved side effects associated with GLP-1 agonists.
“We know there are receptors for GLP-1 in the area postrema (nausea center of the brain), and that NG101 works on this area to reduce nausea and vomiting, so the study findings were not unexpected,” said Jim O’Mara, president and CEO of Neurogastrx, in an interview.
The study was a single-dose study designed to show proof of concept, and future studies would involve treating patients going through the recommended titration schedule for their GLP-1s, O’Mara said. However, NG101 offers an opportunity to keep more patients on GLP-1 therapy and help them reach their long-term therapeutic goals, he said.
Decrease Side Effects for Weight-Loss Success
“GI side effects are often the rate-limiting step in implementing an effective medication that patients want to take but may not be able to tolerate,” said Sean Wharton, MD, PharmD, medical director of the Wharton Medical Clinic for Weight and Diabetes Management, Burlington, Ontario, Canada, in an interview. “If we can decrease side effects, these medications could improve patients’ lives,” said Wharton, who was not involved in the study.
The improvement after a single dose of NG101 in patients receiving a single dose of semaglutide was impressive and in keeping with the mechanism of the drug action, said Wharton. “I was not surprised by the result but pleased that this single dose was shown to reduce the overall incidence of nausea and vomiting, the duration of nausea, the severity of nausea as rated by the study participants compared to placebo,” he said.
Ultimately, the clinical implications for NG101 are improved patient tolerance for GLP-1s and the ability to titrate and stay on them long term, incurring greater cardiometabolic benefit, Wharton told this news organization.
The current trial was limited to GLP1-1s on the market; newer medications may have fewer side effects, Wharton noted. “In clinical practice, patients often decrease the medication or titrate slower, and this could be the comparator,” he added.
The study was funded by Neurogastrx.
Wharton disclosed serving as a consultant for Neurogastrx but not as an investigator on the current study. He also reported having disclosed research on various GLP-1 medications.
A version of this article first appeared on Medscape.com.
FROM OBESITY WEEK 2025

The Litter Olympics: Addressing Individual Critical Tasks Lists Requirements in a Forward-Deployed Setting
The Litter Olympics: Addressing Individual Critical Tasks Lists Requirements in a Forward-Deployed Setting
Military medical personnel rely on individual critical tasks lists (ICTLs) to maintain proficiency in essential medical skills during deployments. However, sustaining these competencies in a low-casualty operational setting presents unique challenges. Traditional training methods, such as lectures or simulations outside operational contexts, may lack engagement and fail to replicate the stressors of real-world scenarios. Previous research has emphasized the importance of continuous medical readiness training in austere environments, highlighting the need for innovative approaches.1,2
The Litter Olympics was developed as an in-theater training exercise designed to enhance medical readiness, foster interdisciplinary teamwork, and incorporate physical exertion into skill maintenance. By requiring teams to carry a patient litter through multiple “events,” the exercise reinforced teamwork within a medical readiness-focused series inspired by an Olympic decathlon. This article discusses the feasibility, effectiveness, and potential impact of the Litter Olympics as a training tool for maintaining ICTLs in a deployed environment.
Program
The Litter Olympics were implemented at a Role 3 medical facility in Baghdad, Iraq, where teams composed of individuals from military occupational specialties (MOSs) and areas of concentration (AOCs) participated. Role 3 facilities provide specialty surgical and critical care capabilities, enabling a robust medical training environment.3 The event was designed to reflect the interdisciplinary nature of deployed medical teams and incorporated hands-on training stations covering critical medical skills such as traction splinting, spinal precautions, patient movement, hemorrhage control, airway management, and tactical evacuation procedures.
Tasks were selected based on their relevance to deployed medical care and their inclusion in ICTLs, ensuring alignment with mission-essential skills. Participants were evaluated on task completion, efficiency, and teamwork by experienced medical personnel. Postexercise surveys assessed skill improvement, confidence levels, and areas for refinement. Future studies should incorporate structured performance metrics, such as pre- and postevent evaluations, to quantify proficiency gains (Table 1).
Five mixed MOS/AOC teams participated in the event, completing the exercise in an average time of 50 minutes (Table 2). Participants reported increased confidence in performing ICTs, particularly in patient movement, hemorrhage control, and airway management. The interdisciplinary nature of the teams facilitated peer teaching and cross-training, allowing individuals to better understand each other’s roles and responsibilities. This mirrors findings in previous studies on predeployment training that emphasize the importance of collaborative, hands-on learning.4 The physical aspect of the exercise was well received, as it simulated operational conditions and reinforced endurance in high-stress environments. Some tasks, such as cricothyroidotomy and satellite radio setup, required additional instruction, highlighting areas for improvement in future iterations.
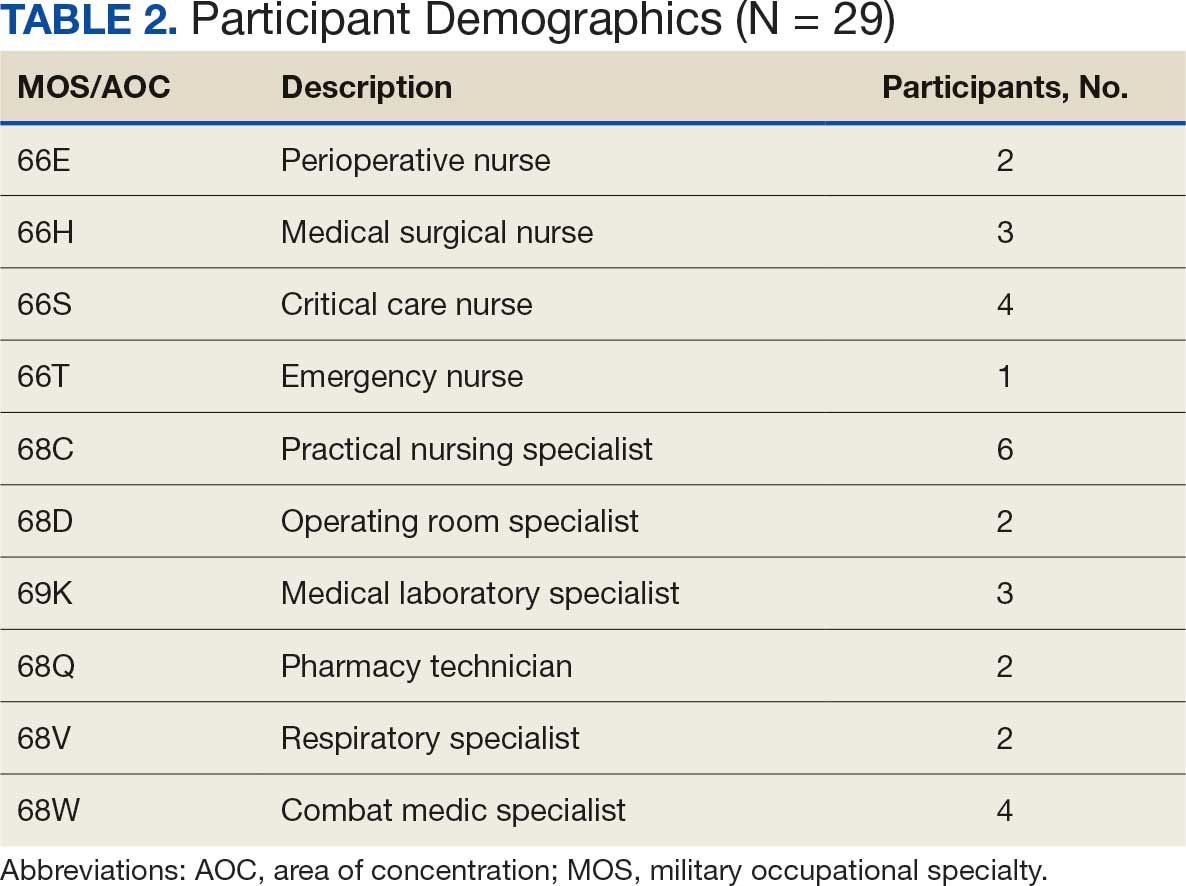
Discussion
The Litter Olympics provide a dynamic alternative to traditional classroom instruction by integrating realistic, scenario-based training. However, several limitations were identified. The most significant was the lack of formalized outcome metrics. While qualitative feedback was overwhelmingly positive, no structured performance assessment tool, such as pre- and postevent skill evaluations, was used. Future studies should incorporate objective measures of competency to strengthen the evidence base for this training model. Additionally, participant feedback suggested that more structured debriefing sessions postexercise would enhance learning retention and provide actionable insights for future program modifications.
Another consideration is the scalability and adaptability of the exercise. While effective in a Role 3 setting, modifications may be required for smaller units or lower levels of care. Future iterations could adapt the format for Role 1 or 2 environments by reducing the number of stations while preserving the core training elements. Furthermore, the event relied on access to specialized personnel and equipment, which may not always be feasible in austere settings. Developing a streamlined version focusing on essential tasks could improve accessibility and sustainability across different operational environments.
Participants expressed a preference for this hands-on, competitive training model over traditional didactic instruction. However, further research should compare skill retention rates between the Litter Olympics and other training modalities to validate effectiveness. While peer teaching was a notable strength of the event, structured mentorship from senior medical personnel could further enhance skill acquisition and reinforce best practices.
Conclusions
The Litter Olympics present a reproducible, engaging, and effective method for sustaining medical readiness in a deployed Role 3 setting. By fostering interdisciplinary collaboration and incorporating physical and cognitive stressors, it enhances both individual and team preparedness. Future research should develop standardized, measurable outcome assessments, explore application in diverse deployment settings, and optimize scalability for broader military medical training programs. Standardized evaluation tools should be developed to quantify performance improvements, and the training model should be expanded to include lower levels of care and nonmedical personnel. Structured debriefing sessions would also provide valuable insight into lessons learned and potential refinements. By integrating these enhancements, the Litter Olympics can serve as a cornerstone for maintaining operational medical readiness in deployed environments.
- Suresh MR, Valdez-Delgado KK, Staudt AM, et al. An assessment of pre-deployment training for army nurses and medics. Mil Med. 2021;186:203-211. doi:10.1093/milmed/usaa291
- Mead KC, Tennent DJ, Stinner DJ. The importance of medical readiness training exercises: maintaining medical readiness in a low-volume combat casualty flow era. Mil Med. 2017;182:e1734-e1737. doi:10.7205/milmed-d-16-00335
- Brisebois R, Hennecke P, Kao R, et al. The Role 3 multinational medical nit at Kandahar airfield 2005–2010. Can J Surg. 2011;54:S124-S129. doi:10.1503/cjs.024811
- Huh J, Brockmeyer JR, Bertsch SR, et al. Conducting pre-deployment training in Honduras: the 240th forward resuscitative surgical team experience. Mil Med. 2021;187:e690-e695. doi:10.1093/milmed/usaa545
Military medical personnel rely on individual critical tasks lists (ICTLs) to maintain proficiency in essential medical skills during deployments. However, sustaining these competencies in a low-casualty operational setting presents unique challenges. Traditional training methods, such as lectures or simulations outside operational contexts, may lack engagement and fail to replicate the stressors of real-world scenarios. Previous research has emphasized the importance of continuous medical readiness training in austere environments, highlighting the need for innovative approaches.1,2
The Litter Olympics was developed as an in-theater training exercise designed to enhance medical readiness, foster interdisciplinary teamwork, and incorporate physical exertion into skill maintenance. By requiring teams to carry a patient litter through multiple “events,” the exercise reinforced teamwork within a medical readiness-focused series inspired by an Olympic decathlon. This article discusses the feasibility, effectiveness, and potential impact of the Litter Olympics as a training tool for maintaining ICTLs in a deployed environment.
Program
The Litter Olympics were implemented at a Role 3 medical facility in Baghdad, Iraq, where teams composed of individuals from military occupational specialties (MOSs) and areas of concentration (AOCs) participated. Role 3 facilities provide specialty surgical and critical care capabilities, enabling a robust medical training environment.3 The event was designed to reflect the interdisciplinary nature of deployed medical teams and incorporated hands-on training stations covering critical medical skills such as traction splinting, spinal precautions, patient movement, hemorrhage control, airway management, and tactical evacuation procedures.
Tasks were selected based on their relevance to deployed medical care and their inclusion in ICTLs, ensuring alignment with mission-essential skills. Participants were evaluated on task completion, efficiency, and teamwork by experienced medical personnel. Postexercise surveys assessed skill improvement, confidence levels, and areas for refinement. Future studies should incorporate structured performance metrics, such as pre- and postevent evaluations, to quantify proficiency gains (Table 1).
Five mixed MOS/AOC teams participated in the event, completing the exercise in an average time of 50 minutes (Table 2). Participants reported increased confidence in performing ICTs, particularly in patient movement, hemorrhage control, and airway management. The interdisciplinary nature of the teams facilitated peer teaching and cross-training, allowing individuals to better understand each other’s roles and responsibilities. This mirrors findings in previous studies on predeployment training that emphasize the importance of collaborative, hands-on learning.4 The physical aspect of the exercise was well received, as it simulated operational conditions and reinforced endurance in high-stress environments. Some tasks, such as cricothyroidotomy and satellite radio setup, required additional instruction, highlighting areas for improvement in future iterations.
Discussion
The Litter Olympics provide a dynamic alternative to traditional classroom instruction by integrating realistic, scenario-based training. However, several limitations were identified. The most significant was the lack of formalized outcome metrics. While qualitative feedback was overwhelmingly positive, no structured performance assessment tool, such as pre- and postevent skill evaluations, was used. Future studies should incorporate objective measures of competency to strengthen the evidence base for this training model. Additionally, participant feedback suggested that more structured debriefing sessions postexercise would enhance learning retention and provide actionable insights for future program modifications.
Another consideration is the scalability and adaptability of the exercise. While effective in a Role 3 setting, modifications may be required for smaller units or lower levels of care. Future iterations could adapt the format for Role 1 or 2 environments by reducing the number of stations while preserving the core training elements. Furthermore, the event relied on access to specialized personnel and equipment, which may not always be feasible in austere settings. Developing a streamlined version focusing on essential tasks could improve accessibility and sustainability across different operational environments.
Participants expressed a preference for this hands-on, competitive training model over traditional didactic instruction. However, further research should compare skill retention rates between the Litter Olympics and other training modalities to validate effectiveness. While peer teaching was a notable strength of the event, structured mentorship from senior medical personnel could further enhance skill acquisition and reinforce best practices.
Conclusions
The Litter Olympics present a reproducible, engaging, and effective method for sustaining medical readiness in a deployed Role 3 setting. By fostering interdisciplinary collaboration and incorporating physical and cognitive stressors, it enhances both individual and team preparedness. Future research should develop standardized, measurable outcome assessments, explore application in diverse deployment settings, and optimize scalability for broader military medical training programs. Standardized evaluation tools should be developed to quantify performance improvements, and the training model should be expanded to include lower levels of care and nonmedical personnel. Structured debriefing sessions would also provide valuable insight into lessons learned and potential refinements. By integrating these enhancements, the Litter Olympics can serve as a cornerstone for maintaining operational medical readiness in deployed environments.
Military medical personnel rely on individual critical tasks lists (ICTLs) to maintain proficiency in essential medical skills during deployments. However, sustaining these competencies in a low-casualty operational setting presents unique challenges. Traditional training methods, such as lectures or simulations outside operational contexts, may lack engagement and fail to replicate the stressors of real-world scenarios. Previous research has emphasized the importance of continuous medical readiness training in austere environments, highlighting the need for innovative approaches.1,2
The Litter Olympics was developed as an in-theater training exercise designed to enhance medical readiness, foster interdisciplinary teamwork, and incorporate physical exertion into skill maintenance. By requiring teams to carry a patient litter through multiple “events,” the exercise reinforced teamwork within a medical readiness-focused series inspired by an Olympic decathlon. This article discusses the feasibility, effectiveness, and potential impact of the Litter Olympics as a training tool for maintaining ICTLs in a deployed environment.
Program
The Litter Olympics were implemented at a Role 3 medical facility in Baghdad, Iraq, where teams composed of individuals from military occupational specialties (MOSs) and areas of concentration (AOCs) participated. Role 3 facilities provide specialty surgical and critical care capabilities, enabling a robust medical training environment.3 The event was designed to reflect the interdisciplinary nature of deployed medical teams and incorporated hands-on training stations covering critical medical skills such as traction splinting, spinal precautions, patient movement, hemorrhage control, airway management, and tactical evacuation procedures.
Tasks were selected based on their relevance to deployed medical care and their inclusion in ICTLs, ensuring alignment with mission-essential skills. Participants were evaluated on task completion, efficiency, and teamwork by experienced medical personnel. Postexercise surveys assessed skill improvement, confidence levels, and areas for refinement. Future studies should incorporate structured performance metrics, such as pre- and postevent evaluations, to quantify proficiency gains (Table 1).
Five mixed MOS/AOC teams participated in the event, completing the exercise in an average time of 50 minutes (Table 2). Participants reported increased confidence in performing ICTs, particularly in patient movement, hemorrhage control, and airway management. The interdisciplinary nature of the teams facilitated peer teaching and cross-training, allowing individuals to better understand each other’s roles and responsibilities. This mirrors findings in previous studies on predeployment training that emphasize the importance of collaborative, hands-on learning.4 The physical aspect of the exercise was well received, as it simulated operational conditions and reinforced endurance in high-stress environments. Some tasks, such as cricothyroidotomy and satellite radio setup, required additional instruction, highlighting areas for improvement in future iterations.
Discussion
The Litter Olympics provide a dynamic alternative to traditional classroom instruction by integrating realistic, scenario-based training. However, several limitations were identified. The most significant was the lack of formalized outcome metrics. While qualitative feedback was overwhelmingly positive, no structured performance assessment tool, such as pre- and postevent skill evaluations, was used. Future studies should incorporate objective measures of competency to strengthen the evidence base for this training model. Additionally, participant feedback suggested that more structured debriefing sessions postexercise would enhance learning retention and provide actionable insights for future program modifications.
Another consideration is the scalability and adaptability of the exercise. While effective in a Role 3 setting, modifications may be required for smaller units or lower levels of care. Future iterations could adapt the format for Role 1 or 2 environments by reducing the number of stations while preserving the core training elements. Furthermore, the event relied on access to specialized personnel and equipment, which may not always be feasible in austere settings. Developing a streamlined version focusing on essential tasks could improve accessibility and sustainability across different operational environments.
Participants expressed a preference for this hands-on, competitive training model over traditional didactic instruction. However, further research should compare skill retention rates between the Litter Olympics and other training modalities to validate effectiveness. While peer teaching was a notable strength of the event, structured mentorship from senior medical personnel could further enhance skill acquisition and reinforce best practices.
Conclusions
The Litter Olympics present a reproducible, engaging, and effective method for sustaining medical readiness in a deployed Role 3 setting. By fostering interdisciplinary collaboration and incorporating physical and cognitive stressors, it enhances both individual and team preparedness. Future research should develop standardized, measurable outcome assessments, explore application in diverse deployment settings, and optimize scalability for broader military medical training programs. Standardized evaluation tools should be developed to quantify performance improvements, and the training model should be expanded to include lower levels of care and nonmedical personnel. Structured debriefing sessions would also provide valuable insight into lessons learned and potential refinements. By integrating these enhancements, the Litter Olympics can serve as a cornerstone for maintaining operational medical readiness in deployed environments.
- Suresh MR, Valdez-Delgado KK, Staudt AM, et al. An assessment of pre-deployment training for army nurses and medics. Mil Med. 2021;186:203-211. doi:10.1093/milmed/usaa291
- Mead KC, Tennent DJ, Stinner DJ. The importance of medical readiness training exercises: maintaining medical readiness in a low-volume combat casualty flow era. Mil Med. 2017;182:e1734-e1737. doi:10.7205/milmed-d-16-00335
- Brisebois R, Hennecke P, Kao R, et al. The Role 3 multinational medical nit at Kandahar airfield 2005–2010. Can J Surg. 2011;54:S124-S129. doi:10.1503/cjs.024811
- Huh J, Brockmeyer JR, Bertsch SR, et al. Conducting pre-deployment training in Honduras: the 240th forward resuscitative surgical team experience. Mil Med. 2021;187:e690-e695. doi:10.1093/milmed/usaa545
- Suresh MR, Valdez-Delgado KK, Staudt AM, et al. An assessment of pre-deployment training for army nurses and medics. Mil Med. 2021;186:203-211. doi:10.1093/milmed/usaa291
- Mead KC, Tennent DJ, Stinner DJ. The importance of medical readiness training exercises: maintaining medical readiness in a low-volume combat casualty flow era. Mil Med. 2017;182:e1734-e1737. doi:10.7205/milmed-d-16-00335
- Brisebois R, Hennecke P, Kao R, et al. The Role 3 multinational medical nit at Kandahar airfield 2005–2010. Can J Surg. 2011;54:S124-S129. doi:10.1503/cjs.024811
- Huh J, Brockmeyer JR, Bertsch SR, et al. Conducting pre-deployment training in Honduras: the 240th forward resuscitative surgical team experience. Mil Med. 2021;187:e690-e695. doi:10.1093/milmed/usaa545
The Litter Olympics: Addressing Individual Critical Tasks Lists Requirements in a Forward-Deployed Setting
The Litter Olympics: Addressing Individual Critical Tasks Lists Requirements in a Forward-Deployed Setting
Preoperative Diabetes Management for Patients Undergoing Elective Surgeries at a Veterans Affairs Medical Center
Preoperative Diabetes Management for Patients Undergoing Elective Surgeries at a Veterans Affairs Medical Center
More than 38 million people in the United States (12%) have diabetes mellitus (DM), though 1 in 5 are unaware they have DM.1 The prevalence among veterans is even more substantial, impacting nearly 25% of those who received care from the US Department of Veterans Affairs (VA).2 DM can lead to increased health care costs in addition to various complications (eg, cardiovascular, renal), especially if left uncontrolled.1,3 similar impact is found in the perioperative period (defined as at or around the time of an operation), as multiple studies have found that uncontrolled preoperative DM can result in worsened surgical outcomes, including longer hospital stays, more infectious complications, and higher perioperative mortality.4-6
In contrast, adequate glycemic control assessed with blood glucose levels has been shown to decrease the incidence of postoperative infections.7 Optimizing glycemic control during hospital stays, especially postsurgery, has become the standard of care, with most health systems establishing specific protocols. In current literature, most studies examining DM management in the perioperative period are focused on postoperative care, with little attention to the preoperative period.4,6,7
One study found that patients with poor presurgery glycemic control assessed by hemoglobin A1c (HbA1c) levels were more likely to remain hyperglycemic during and after surgery. 8 Blood glucose levels < 200 mg/dL can lead to an increased risk of infection and impaired wound healing, meaning a well-controlled HbA1c before a procedure serves as a potential factor for success.9 The 2025 American Diabetes Association (ADA) Standards of Care (SOC) recommendation is to target HbA1c < 8% whenever possible, and some health systems require lower levels (eg, < 7% or 7.5%).10 With that goal in mind and knowing that preoperative hyperglycemia has been shown to be a contributing factor in the delay or cancellation of surgical cases, an argument can be made that attention to preoperative DM management also should be a focus for health care systems performing surgeries.8,9,11
Attention to glucose control during preoperative care offers an opportunity to screen for DM in patients who may not have been screened otherwise and to standardize perioperative DM management. Since DM disproportionately impacts veterans, this is a pertinent issue to the VA. Veterans can be more susceptible to complications if DM is left uncontrolled prior to surgery. To determine readiness for surgery and control of comorbid conditions such as DM before a planned surgery, facilities often perform a preoperative clinic assessment, often in a multidisciplinary clinic.
At Veteran Health Indiana (VHI), a presurgery clinic visit involving the primary surgery service (physician, nurse practitioner, and/or a physician assistant) is conducted 1 to 2 months prior to the planned procedure to determine whether a patient is ready for surgery. During this visit, patients receive a packet with instructions for various tasks and medications, such as applying topical antibiotic prophylaxis on the anticipated surgical site. This is documented in the form of a note in the VHI Computerized Patient Record System (CPRS). The medication instructions are provided according to the preferences of the surgical team. These may be templated notes that contain general directions on the timing and dosing of specific medications, in addition to instructions for holding or reducing doses when appropriate. The instructions can be tailored by the team conducting the preoperative visit (eg, “Take 20 units of insulin glargine the day before surgery” vs “Take half of your long-acting insulin the night before surgery”). Specific to DM, VHI has a nurse-driven day of surgery glucose assessment where point-of-care blood glucose is collected during preoperative holding for most patients.
There is limited research assessing the level of preoperative glycemic control and the incidence of complications in a veteran population. The objective of this study was to gain a baseline understanding of what, if any, standardization exists for preoperative instructions for DM medications and to assess the level of preoperative glycemic control and postoperative complications in patients with DM undergoing major elective surgical procedures.
Methods
This retrospective, single-center chart review was conducted at VHI. The Indiana University and VHI institutional review boards determined that this quality improvement project was exempt from review.
The primary outcome was the number of patients with surgical procedures delayed or canceled due to hyperglycemia or hypoglycemia. Hyperglycemia was defined as blood glucose > 180 mg/dL and hypoglycemia was defined as < 70 mg/dL, slight variations from the current ADA SOC preoperative specific recommendation of a blood glucose reading of 100 to 180 mg/dL within 4 hours of surgery.10 The standard outpatient hypoglycemia definition of blood glucose < 70 mg/dL was chosen because the current goal (< 100 mg/dL) was not the standard in previous ADA SOCs that were in place during the study period. Specifically, the 2018 ADA SOC did not provide preoperative recommendations and the 2019-2021 ADA SOC recommended 80 to 180 mg/dL.10,12-18 For patients who had multiple preoperative blood glucose measurements, the first recorded glucose on the day of the procedure was used.
The secondary outcomes of this study were focused on the preoperative process/care at VHI and postoperative glycemic control. The preoperative process included examining whether medication instructions were given and their quality. Additionally, the number of interventions for hyperglycemia and hypoglycemia were required immediately prior to surgery and the average preoperative HbA1c (measured within 3 months prior to surgery) were collected and analyzed. For postoperative glycemic control, average blood glucose measurements and number of hypoglycemic (< 70 mg/dL) and hyperglycemic (> 180 mg/dL) events were measured in addition to the frequency of changes made at discharge to patients’ DM medication regimens.
The safety outcome of this study assessed commonly observed postoperative complications and was examined up to 30 days postsurgery. These included acute kidney injury (defined using Kidney Disease: Improving Global Outcomes 2012, the standard during the study period), nonfatal myocardial infarction, nonfatal stroke, and surgical site infections, which were identified from the discharge summary written by the primary surgery service.19 All-cause mortality also was collected.
Patients were included if they were admitted for major elective surgeries and had a diagnosis of either type 1 or type 2 DM on their problem list, determined by International Classification of Diseases, Tenth Revision codes. Major elective surgery was defined as a procedure that would likely result in a hospital admission of > 24 hours. Of note, patients may have been included in this study more than once if they had > 1 procedure at least 30 days apart and met inclusion criteria within the time frame. Patients were excluded if they were taking no DM medications or chronic steroids (at any dose), residing in a long-term care facility, being managed by a non-VA clinician prior to surgery, or missing a preoperative blood glucose measurement.
All data were collected from the CPRS. A list of surgical cases involving patients with DM who were scheduled to undergo major elective surgeries from January 1, 2018, to December 31, 2021, at VHI was generated. The list was randomized to a smaller number (N = 394) for data collection due to the time and resource constraints for a pharmacy residency project. All data were deidentified and stored in a secured VA server to protect patient confidentiality. Descriptive statistics were used for all results.
Results
Initially, 2362 surgeries were identified. A randomized sample of 394 charts were reviewed and 131 cases met inclusion criteria. Each case involved a unique patient (Figure). The most common reasons for exclusion were 143 patients with diet-controlled DM and 78 nonelective surgeries. The mean (SD) age of patients was 68 (8) years, and the most were male (98.5%) and White (76.3%) (Table 1).
At baseline, 45 of 131 patients (34.4%) had coronary artery disease and 29 (22.1%) each had autonomic neuropathy and chronic kidney disease. Most surgeries were conducted by orthopedic (32.1%) and peripheral vascular (21.4%) specialties. The mean (SD) length of surgery was 4.6 (2.6) hours and of hospital length of stay was 4 (4) days. No patients stayed longer than the 30-day safety outcome follow-up period. All patients had type 2 DM and took a mean 2 DM medications. The 63 patients taking insulin had a mean (SD) total daily dose of 99 (77) U (Table 2). A preoperative HbA1c was collected in 116 patients within 3 months of surgery, with a mean HbA1c of 7.0% (range, 5.3-10.7).
No patients had surgeries delayed or canceled because of uncontrolled DM on the day of surgery. The mean preoperative blood glucose level was 146 mg/dL (range, 73-365) (Table 3). No patients had a preoperative blood glucose level of < 70 mg/dL and 19 (14.5%) had a blood glucose level > 180 mg/dL. Among patients with hyperglycemia immediately prior to surgery, 6 (31.6%) had documentation of insulin being provided.
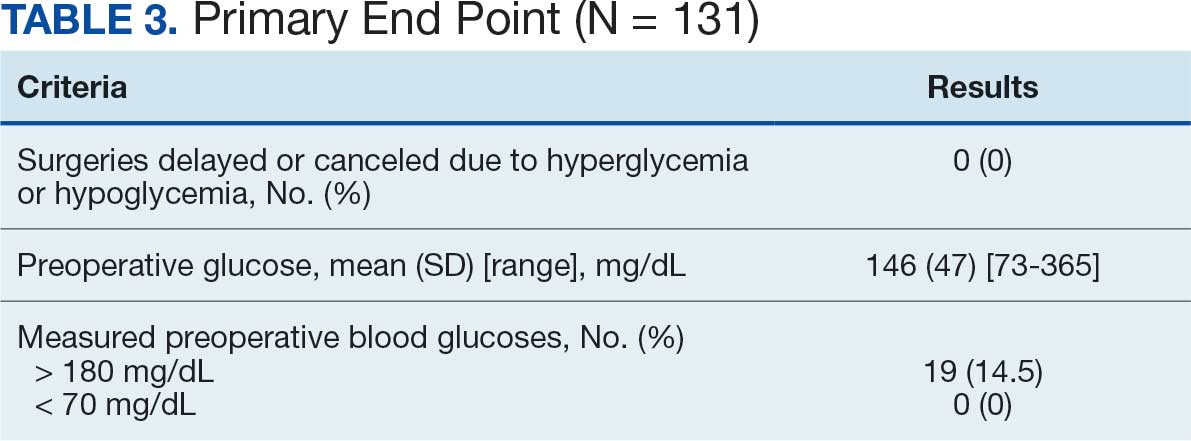
For this sample of patients, the preoperative clinic visit was conducted a mean 22 days prior to the planned surgery date. Among the 131 included patients, 122 (93.1%) had documentation of receiving instructions for DM medications. Among patients who had documented receipt of instructions, only 30 (24.6%) had instructions specifically tailored to their regimen rather than a generic templated form. The mean (SD) preoperative blood glucose was similar for those who received specific perioperative DM instructions at 146 (50) mg/dL when compared with those who did not at 147 (45) mg/dL. The mean (SD) preoperative blood glucose reading for those who had no documentation of receipt of perioperative instructions was 126 (54) mg/dL compared with 147 (46) mg/dL for those who did.
The mean number of postoperative blood glucose events per day was negligible for hypoglycemia and more frequent for hyperglycemia with a mean of 2 events per day. The mean postoperative blood glucose range was 121 to 247 mg/dL with most readings < 180 mg/dL. Upon discharge, most patients continued their home DM regimen with 5 patients (3.8%) having changes made to their regimen upon discharge.
Very few postoperative complications were identified from chart review. The most frequently observed postoperative complications were acute kidney injury, surgical site infections, and nonfatal stroke. There were no documented nonfatal myocardial infarctions. Two patients (1.5%) died within 30 days of the surgery; neither death was deemed to have been related to poor perioperative glycemic control.
Discussion
To our knowledge, this retrospective chart review was the first study to assess preoperative DM management and postoperative complications in a veteran population. VHI is a large, tertiary, level 1a, academic medical center that serves approximately 62,000 veterans annually and performs about 5000 to 6000 surgeries annually, a total that is increasing following the COVID-19 pandemic.20 This study found that the current process of a presurgery clinic visit and day of surgery glucose assessment has prevented surgical delays or cancellations.
Most patients included in this study were well controlled at baseline in accordance with the 2025 ADA SOC HbA1c recommendation of a preoperative HbA1c of < 8%, which may have contributed to no surgical delays or cancellations.10 However, not all patients had HbA1c collected within 3 months of surgery or even had one collected at all. Despite the ADA SOC providing no explicit recommendation for universal HbA1c screening prior to elective procedures, its importance cannot be understated given the body of evidence demonstrating poor outcomes with uncontrolled preoperative DM.8,10 The glycemic control at baseline may have contributed to the very few postsurgical complications observed in this study.
Although the current process at VHI prevented surgical delays and cancellations in this sample, there are still identified areas for improvement. One area is the instructions the patients received. Patients with DM are often prescribed ≥ 1 medication or a combination of insulins, noninsulin injectables, and oral DM medications, and this study population was no different. Because these medications may influence the anesthesia and perioperative periods, the ADA has specific guidance for altering administration schedules in the days leading up to surgery.10
Inappropriate administration of DM medications could lead to perioperative hypoglycemia or hyperglycemia, possibly causing surgical delays, case cancellations, and/or postoperative complications.21 Although these data reveal the specificity and documented receipt that the preoperative DM instructions did not impact the first recorded preoperative blood glucose, future studies should examine patient confidence in how to properly administer their DM medications prior to surgery. It is vital that patients receive clear instructions in accordance with the ADA SOC on whether to continue, hold, or adjust the dose of their medications to prevent fluctuations in blood glucose levels in the perioperative period, ensure safety with anesthesia, and prevent postoperative complications such as acute kidney injury. Of note, compliance with guideline recommendations for medication instructions was not examined because the data collection time frame expanded over multiple years and the recommendations have evolved each year as new data emerge.
Preoperative DM Management
The first key takeaway from this study is to ensure patients are ready for surgery with a formal assessment (typically in the form of a clinic visit) prior to the surgery. One private sector health system published their approach to this by administering an automatic preoperative HbA1c screening for those with a DM diagnosis and all patients with a random plasma glucose ≥ 200 mg/dL.22 Additionally, if the patient's HbA1c level was not at goal prior to surgery (≥ 8% for those with known DM and ≥ 6.5% with no known DM), patients were referred to endocrinology for further management. Increasing attention to the preoperative visit and extending HbA1c testing to all patients regardless of DM status also provides an opportunity to identify individuals living with undiagnosed DM.1
Even though there was no difference in the mean preoperative blood glucose level based on receipt or specificity of preoperative DM instructions, a second takeaway from this study is the importance of ensuring patients receive clear instructions on their DM medication schedule in the perioperative period. A practical first step may be updating the templates used by the primary surgery teams and providing education to the clinicians in the clinic on how to personalize the visits. Because the current preoperative DM process at VHI is managed by the primary surgical team in a clinic visit, there is an opportunity to shift this responsibility to other health care professionals, such as pharmacists—a change shown to reduce unintended omission of home medications following surgery during hospitalization and reduce costs.23,24
Limitations
This study relied on data included in the patient chart. These data include medication interventions made immediately prior to surgery, which can sometimes be inaccurately charted or difficult to find as they are not documented in the typical medication administration record. Also, the safety outcomes were collected from a discharge summary written by different clinicians, which may lead to information bias. Special attention was taken to ensure these data points were collected as accurately as possible, but it is possible some data may be inaccurate from unintentional human error. Additionally, the safety outcome was limited to a 30-day follow-up, but encompassed the entire length of postoperative stay for all included patients. Finally, given this study was retrospective with no comparison group and the intent was to improve processes at VHI, only hypotheses and potential interventions can be generated from this study. Future prospective studies with larger sample sizes and comparator groups are needed to draw further conclusions.
Conclusions
This study found that the current presurgery process at VHI appears to be successful in preventing surgical delays or cancellations due to hyperglycemia or hypoglycemia. Optimizing DM management can improve surgical outcomes by decreasing rates of postoperative complications, and this study added additional evidence in support of that in a unique population: veterans. Insight on the awareness of preoperative blood glucose management should be gleaned from this study, and based on this sample and site, the preadmission screening process and instructions provided to patients can serve as 2 starting points for optimizing elective surgery.
- Centers for Disease Control and Prevention. Diabetes basics. May 15, 2024. Accessed September 24, 2025. https://www.cdc.gov/diabetes/about/index.html
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- Farmaki P, Damaskos C, Garmpis N, et al . Complications of the Type 2 Diabetes Mellitus. Curr Cardiol Rev. 2020;16(4):249-251. doi:10.2174/1573403X1604201229115531
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care. 2010;33:1783-1788. doi:10.2337/dc10-0304
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol. 2007;156:137 -142. doi:10.1530/eje.1.02321
- Pomposelli JJ, Baxter JK 3rd, Babineau TJ, et al. Early postoperative glucose control predicts nosocomial infection rate in diabetic patients. JPEN J Parenter Enteral Nutr. 1998;22:77-81. doi:10.1177/01486071980220027
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care. 2011;34:256-261. doi:10.2337/dc10-1407
- Pasquel FJ, Gomez-Huelgas R, Anzola I, et al. Predictive value of admission hemoglobin A1c on inpatient glycemic control and response to insulin therapy in medicine and surgery patients with type 2 diabetes. Diabetes Care. 2015;38:e202-e203. doi:10.2337/dc15-1835
- Alexiewicz JM, Kumar D, Smogorzewski M, et al. Polymorphonuclear leukocytes in non-insulin-dependent diabetes mellitus: abnormalities in metabolism and function. Ann Intern Med. 1995;123:919-924. doi:10.7326/0003-4819-123-12-199512150-00004
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2025. Diabetes Care. 2025;48(1 suppl 1):S321-S334. doi:10.2337/dc25-S016
- Kumar R, Gandhi R. Reasons for cancellation of operation on the day of intended surgery in a multidisciplinary 500 bedded hospital. J Anaesthesiol Clin Pharmacol. 2012;28:66-69. doi:10.4103/0970-9185.92442
- American Diabetes Association. 14. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2018. Diabetes Care. 2018;41(1 suppl 1):S144- S151. doi:10.2337/dc18-S014
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2019. Diabetes Care. 2019;42(suppl 1):S173- S181. doi:10.2337/dc19-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2020. Diabetes Care. 2020;43(suppl 1):S193- S202. doi:10.2337/dc20-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2021. Diabetes Care. 2021;44(suppl 1):S211- S220. doi:10.2337/dc21-S015
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2022. Diabetes Care. 2022;45(suppl 1):S244-S253. doi:10.2337/dc22-S016
- ElSayed NA, Aleppo G, Aroda VR, et al. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2023. Diabetes Care. 2023;46(suppl 1):S267-S278. doi:10.2337/dc23-S016
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2024. Diabetes Care. 2024;47(suppl 1):S295-S306. doi:10.2337/dc24-S016
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1-138. Accessed September 24, 2025. https:// www.kisupplements.org/issue/S2157-1716(12)X7200-9
- US Department of Veterans Affairs. VA Indiana Healthcare: about us. Accessed September 24, 2025. https:// www.va.gov/indiana-health-care/about-us/
- Koh WX, Phelan R, Hopman WM, et al. Cancellation of elective surgery: rates, reasons and effect on patient satisfaction. Can J Surg. 2021;64:E155-E161. doi:10.1503/cjs.008119
- Pai S-L, Haehn DA, Pitruzzello NE, et al. Reducing infection rates with enhanced preoperative diabetes mellitus diagnosis and optimization processes. South Med J. 2023;116:215-219. doi:10.14423/SMJ.0000000000001507
- Forrester TG, Sullivan S, Snoswell CL, et al. Integrating a pharmacist into the perioperative setting. Aust Health Rev. 2020;44:563-568. doi:10.1071/AH19126
- Hale AR, Coombes ID, Stokes J, et al. Perioperative medication management: expanding the role of the preadmission clinic pharmacist in a single centre, randomised controlled trial of collaborative prescribing. BMJ Open. 2013;3:e003027. doi:10.1136/bmjopen-2013-003027
More than 38 million people in the United States (12%) have diabetes mellitus (DM), though 1 in 5 are unaware they have DM.1 The prevalence among veterans is even more substantial, impacting nearly 25% of those who received care from the US Department of Veterans Affairs (VA).2 DM can lead to increased health care costs in addition to various complications (eg, cardiovascular, renal), especially if left uncontrolled.1,3 similar impact is found in the perioperative period (defined as at or around the time of an operation), as multiple studies have found that uncontrolled preoperative DM can result in worsened surgical outcomes, including longer hospital stays, more infectious complications, and higher perioperative mortality.4-6
In contrast, adequate glycemic control assessed with blood glucose levels has been shown to decrease the incidence of postoperative infections.7 Optimizing glycemic control during hospital stays, especially postsurgery, has become the standard of care, with most health systems establishing specific protocols. In current literature, most studies examining DM management in the perioperative period are focused on postoperative care, with little attention to the preoperative period.4,6,7
One study found that patients with poor presurgery glycemic control assessed by hemoglobin A1c (HbA1c) levels were more likely to remain hyperglycemic during and after surgery. 8 Blood glucose levels < 200 mg/dL can lead to an increased risk of infection and impaired wound healing, meaning a well-controlled HbA1c before a procedure serves as a potential factor for success.9 The 2025 American Diabetes Association (ADA) Standards of Care (SOC) recommendation is to target HbA1c < 8% whenever possible, and some health systems require lower levels (eg, < 7% or 7.5%).10 With that goal in mind and knowing that preoperative hyperglycemia has been shown to be a contributing factor in the delay or cancellation of surgical cases, an argument can be made that attention to preoperative DM management also should be a focus for health care systems performing surgeries.8,9,11
Attention to glucose control during preoperative care offers an opportunity to screen for DM in patients who may not have been screened otherwise and to standardize perioperative DM management. Since DM disproportionately impacts veterans, this is a pertinent issue to the VA. Veterans can be more susceptible to complications if DM is left uncontrolled prior to surgery. To determine readiness for surgery and control of comorbid conditions such as DM before a planned surgery, facilities often perform a preoperative clinic assessment, often in a multidisciplinary clinic.
At Veteran Health Indiana (VHI), a presurgery clinic visit involving the primary surgery service (physician, nurse practitioner, and/or a physician assistant) is conducted 1 to 2 months prior to the planned procedure to determine whether a patient is ready for surgery. During this visit, patients receive a packet with instructions for various tasks and medications, such as applying topical antibiotic prophylaxis on the anticipated surgical site. This is documented in the form of a note in the VHI Computerized Patient Record System (CPRS). The medication instructions are provided according to the preferences of the surgical team. These may be templated notes that contain general directions on the timing and dosing of specific medications, in addition to instructions for holding or reducing doses when appropriate. The instructions can be tailored by the team conducting the preoperative visit (eg, “Take 20 units of insulin glargine the day before surgery” vs “Take half of your long-acting insulin the night before surgery”). Specific to DM, VHI has a nurse-driven day of surgery glucose assessment where point-of-care blood glucose is collected during preoperative holding for most patients.
There is limited research assessing the level of preoperative glycemic control and the incidence of complications in a veteran population. The objective of this study was to gain a baseline understanding of what, if any, standardization exists for preoperative instructions for DM medications and to assess the level of preoperative glycemic control and postoperative complications in patients with DM undergoing major elective surgical procedures.
Methods
This retrospective, single-center chart review was conducted at VHI. The Indiana University and VHI institutional review boards determined that this quality improvement project was exempt from review.
The primary outcome was the number of patients with surgical procedures delayed or canceled due to hyperglycemia or hypoglycemia. Hyperglycemia was defined as blood glucose > 180 mg/dL and hypoglycemia was defined as < 70 mg/dL, slight variations from the current ADA SOC preoperative specific recommendation of a blood glucose reading of 100 to 180 mg/dL within 4 hours of surgery.10 The standard outpatient hypoglycemia definition of blood glucose < 70 mg/dL was chosen because the current goal (< 100 mg/dL) was not the standard in previous ADA SOCs that were in place during the study period. Specifically, the 2018 ADA SOC did not provide preoperative recommendations and the 2019-2021 ADA SOC recommended 80 to 180 mg/dL.10,12-18 For patients who had multiple preoperative blood glucose measurements, the first recorded glucose on the day of the procedure was used.
The secondary outcomes of this study were focused on the preoperative process/care at VHI and postoperative glycemic control. The preoperative process included examining whether medication instructions were given and their quality. Additionally, the number of interventions for hyperglycemia and hypoglycemia were required immediately prior to surgery and the average preoperative HbA1c (measured within 3 months prior to surgery) were collected and analyzed. For postoperative glycemic control, average blood glucose measurements and number of hypoglycemic (< 70 mg/dL) and hyperglycemic (> 180 mg/dL) events were measured in addition to the frequency of changes made at discharge to patients’ DM medication regimens.
The safety outcome of this study assessed commonly observed postoperative complications and was examined up to 30 days postsurgery. These included acute kidney injury (defined using Kidney Disease: Improving Global Outcomes 2012, the standard during the study period), nonfatal myocardial infarction, nonfatal stroke, and surgical site infections, which were identified from the discharge summary written by the primary surgery service.19 All-cause mortality also was collected.
Patients were included if they were admitted for major elective surgeries and had a diagnosis of either type 1 or type 2 DM on their problem list, determined by International Classification of Diseases, Tenth Revision codes. Major elective surgery was defined as a procedure that would likely result in a hospital admission of > 24 hours. Of note, patients may have been included in this study more than once if they had > 1 procedure at least 30 days apart and met inclusion criteria within the time frame. Patients were excluded if they were taking no DM medications or chronic steroids (at any dose), residing in a long-term care facility, being managed by a non-VA clinician prior to surgery, or missing a preoperative blood glucose measurement.
All data were collected from the CPRS. A list of surgical cases involving patients with DM who were scheduled to undergo major elective surgeries from January 1, 2018, to December 31, 2021, at VHI was generated. The list was randomized to a smaller number (N = 394) for data collection due to the time and resource constraints for a pharmacy residency project. All data were deidentified and stored in a secured VA server to protect patient confidentiality. Descriptive statistics were used for all results.
Results
Initially, 2362 surgeries were identified. A randomized sample of 394 charts were reviewed and 131 cases met inclusion criteria. Each case involved a unique patient (Figure). The most common reasons for exclusion were 143 patients with diet-controlled DM and 78 nonelective surgeries. The mean (SD) age of patients was 68 (8) years, and the most were male (98.5%) and White (76.3%) (Table 1).
At baseline, 45 of 131 patients (34.4%) had coronary artery disease and 29 (22.1%) each had autonomic neuropathy and chronic kidney disease. Most surgeries were conducted by orthopedic (32.1%) and peripheral vascular (21.4%) specialties. The mean (SD) length of surgery was 4.6 (2.6) hours and of hospital length of stay was 4 (4) days. No patients stayed longer than the 30-day safety outcome follow-up period. All patients had type 2 DM and took a mean 2 DM medications. The 63 patients taking insulin had a mean (SD) total daily dose of 99 (77) U (Table 2). A preoperative HbA1c was collected in 116 patients within 3 months of surgery, with a mean HbA1c of 7.0% (range, 5.3-10.7).
No patients had surgeries delayed or canceled because of uncontrolled DM on the day of surgery. The mean preoperative blood glucose level was 146 mg/dL (range, 73-365) (Table 3). No patients had a preoperative blood glucose level of < 70 mg/dL and 19 (14.5%) had a blood glucose level > 180 mg/dL. Among patients with hyperglycemia immediately prior to surgery, 6 (31.6%) had documentation of insulin being provided.
For this sample of patients, the preoperative clinic visit was conducted a mean 22 days prior to the planned surgery date. Among the 131 included patients, 122 (93.1%) had documentation of receiving instructions for DM medications. Among patients who had documented receipt of instructions, only 30 (24.6%) had instructions specifically tailored to their regimen rather than a generic templated form. The mean (SD) preoperative blood glucose was similar for those who received specific perioperative DM instructions at 146 (50) mg/dL when compared with those who did not at 147 (45) mg/dL. The mean (SD) preoperative blood glucose reading for those who had no documentation of receipt of perioperative instructions was 126 (54) mg/dL compared with 147 (46) mg/dL for those who did.
The mean number of postoperative blood glucose events per day was negligible for hypoglycemia and more frequent for hyperglycemia with a mean of 2 events per day. The mean postoperative blood glucose range was 121 to 247 mg/dL with most readings < 180 mg/dL. Upon discharge, most patients continued their home DM regimen with 5 patients (3.8%) having changes made to their regimen upon discharge.
Very few postoperative complications were identified from chart review. The most frequently observed postoperative complications were acute kidney injury, surgical site infections, and nonfatal stroke. There were no documented nonfatal myocardial infarctions. Two patients (1.5%) died within 30 days of the surgery; neither death was deemed to have been related to poor perioperative glycemic control.
Discussion
To our knowledge, this retrospective chart review was the first study to assess preoperative DM management and postoperative complications in a veteran population. VHI is a large, tertiary, level 1a, academic medical center that serves approximately 62,000 veterans annually and performs about 5000 to 6000 surgeries annually, a total that is increasing following the COVID-19 pandemic.20 This study found that the current process of a presurgery clinic visit and day of surgery glucose assessment has prevented surgical delays or cancellations.
Most patients included in this study were well controlled at baseline in accordance with the 2025 ADA SOC HbA1c recommendation of a preoperative HbA1c of < 8%, which may have contributed to no surgical delays or cancellations.10 However, not all patients had HbA1c collected within 3 months of surgery or even had one collected at all. Despite the ADA SOC providing no explicit recommendation for universal HbA1c screening prior to elective procedures, its importance cannot be understated given the body of evidence demonstrating poor outcomes with uncontrolled preoperative DM.8,10 The glycemic control at baseline may have contributed to the very few postsurgical complications observed in this study.
Although the current process at VHI prevented surgical delays and cancellations in this sample, there are still identified areas for improvement. One area is the instructions the patients received. Patients with DM are often prescribed ≥ 1 medication or a combination of insulins, noninsulin injectables, and oral DM medications, and this study population was no different. Because these medications may influence the anesthesia and perioperative periods, the ADA has specific guidance for altering administration schedules in the days leading up to surgery.10
Inappropriate administration of DM medications could lead to perioperative hypoglycemia or hyperglycemia, possibly causing surgical delays, case cancellations, and/or postoperative complications.21 Although these data reveal the specificity and documented receipt that the preoperative DM instructions did not impact the first recorded preoperative blood glucose, future studies should examine patient confidence in how to properly administer their DM medications prior to surgery. It is vital that patients receive clear instructions in accordance with the ADA SOC on whether to continue, hold, or adjust the dose of their medications to prevent fluctuations in blood glucose levels in the perioperative period, ensure safety with anesthesia, and prevent postoperative complications such as acute kidney injury. Of note, compliance with guideline recommendations for medication instructions was not examined because the data collection time frame expanded over multiple years and the recommendations have evolved each year as new data emerge.
Preoperative DM Management
The first key takeaway from this study is to ensure patients are ready for surgery with a formal assessment (typically in the form of a clinic visit) prior to the surgery. One private sector health system published their approach to this by administering an automatic preoperative HbA1c screening for those with a DM diagnosis and all patients with a random plasma glucose ≥ 200 mg/dL.22 Additionally, if the patient's HbA1c level was not at goal prior to surgery (≥ 8% for those with known DM and ≥ 6.5% with no known DM), patients were referred to endocrinology for further management. Increasing attention to the preoperative visit and extending HbA1c testing to all patients regardless of DM status also provides an opportunity to identify individuals living with undiagnosed DM.1
Even though there was no difference in the mean preoperative blood glucose level based on receipt or specificity of preoperative DM instructions, a second takeaway from this study is the importance of ensuring patients receive clear instructions on their DM medication schedule in the perioperative period. A practical first step may be updating the templates used by the primary surgery teams and providing education to the clinicians in the clinic on how to personalize the visits. Because the current preoperative DM process at VHI is managed by the primary surgical team in a clinic visit, there is an opportunity to shift this responsibility to other health care professionals, such as pharmacists—a change shown to reduce unintended omission of home medications following surgery during hospitalization and reduce costs.23,24
Limitations
This study relied on data included in the patient chart. These data include medication interventions made immediately prior to surgery, which can sometimes be inaccurately charted or difficult to find as they are not documented in the typical medication administration record. Also, the safety outcomes were collected from a discharge summary written by different clinicians, which may lead to information bias. Special attention was taken to ensure these data points were collected as accurately as possible, but it is possible some data may be inaccurate from unintentional human error. Additionally, the safety outcome was limited to a 30-day follow-up, but encompassed the entire length of postoperative stay for all included patients. Finally, given this study was retrospective with no comparison group and the intent was to improve processes at VHI, only hypotheses and potential interventions can be generated from this study. Future prospective studies with larger sample sizes and comparator groups are needed to draw further conclusions.
Conclusions
This study found that the current presurgery process at VHI appears to be successful in preventing surgical delays or cancellations due to hyperglycemia or hypoglycemia. Optimizing DM management can improve surgical outcomes by decreasing rates of postoperative complications, and this study added additional evidence in support of that in a unique population: veterans. Insight on the awareness of preoperative blood glucose management should be gleaned from this study, and based on this sample and site, the preadmission screening process and instructions provided to patients can serve as 2 starting points for optimizing elective surgery.
More than 38 million people in the United States (12%) have diabetes mellitus (DM), though 1 in 5 are unaware they have DM.1 The prevalence among veterans is even more substantial, impacting nearly 25% of those who received care from the US Department of Veterans Affairs (VA).2 DM can lead to increased health care costs in addition to various complications (eg, cardiovascular, renal), especially if left uncontrolled.1,3 similar impact is found in the perioperative period (defined as at or around the time of an operation), as multiple studies have found that uncontrolled preoperative DM can result in worsened surgical outcomes, including longer hospital stays, more infectious complications, and higher perioperative mortality.4-6
In contrast, adequate glycemic control assessed with blood glucose levels has been shown to decrease the incidence of postoperative infections.7 Optimizing glycemic control during hospital stays, especially postsurgery, has become the standard of care, with most health systems establishing specific protocols. In current literature, most studies examining DM management in the perioperative period are focused on postoperative care, with little attention to the preoperative period.4,6,7
One study found that patients with poor presurgery glycemic control assessed by hemoglobin A1c (HbA1c) levels were more likely to remain hyperglycemic during and after surgery. 8 Blood glucose levels < 200 mg/dL can lead to an increased risk of infection and impaired wound healing, meaning a well-controlled HbA1c before a procedure serves as a potential factor for success.9 The 2025 American Diabetes Association (ADA) Standards of Care (SOC) recommendation is to target HbA1c < 8% whenever possible, and some health systems require lower levels (eg, < 7% or 7.5%).10 With that goal in mind and knowing that preoperative hyperglycemia has been shown to be a contributing factor in the delay or cancellation of surgical cases, an argument can be made that attention to preoperative DM management also should be a focus for health care systems performing surgeries.8,9,11
Attention to glucose control during preoperative care offers an opportunity to screen for DM in patients who may not have been screened otherwise and to standardize perioperative DM management. Since DM disproportionately impacts veterans, this is a pertinent issue to the VA. Veterans can be more susceptible to complications if DM is left uncontrolled prior to surgery. To determine readiness for surgery and control of comorbid conditions such as DM before a planned surgery, facilities often perform a preoperative clinic assessment, often in a multidisciplinary clinic.
At Veteran Health Indiana (VHI), a presurgery clinic visit involving the primary surgery service (physician, nurse practitioner, and/or a physician assistant) is conducted 1 to 2 months prior to the planned procedure to determine whether a patient is ready for surgery. During this visit, patients receive a packet with instructions for various tasks and medications, such as applying topical antibiotic prophylaxis on the anticipated surgical site. This is documented in the form of a note in the VHI Computerized Patient Record System (CPRS). The medication instructions are provided according to the preferences of the surgical team. These may be templated notes that contain general directions on the timing and dosing of specific medications, in addition to instructions for holding or reducing doses when appropriate. The instructions can be tailored by the team conducting the preoperative visit (eg, “Take 20 units of insulin glargine the day before surgery” vs “Take half of your long-acting insulin the night before surgery”). Specific to DM, VHI has a nurse-driven day of surgery glucose assessment where point-of-care blood glucose is collected during preoperative holding for most patients.
There is limited research assessing the level of preoperative glycemic control and the incidence of complications in a veteran population. The objective of this study was to gain a baseline understanding of what, if any, standardization exists for preoperative instructions for DM medications and to assess the level of preoperative glycemic control and postoperative complications in patients with DM undergoing major elective surgical procedures.
Methods
This retrospective, single-center chart review was conducted at VHI. The Indiana University and VHI institutional review boards determined that this quality improvement project was exempt from review.
The primary outcome was the number of patients with surgical procedures delayed or canceled due to hyperglycemia or hypoglycemia. Hyperglycemia was defined as blood glucose > 180 mg/dL and hypoglycemia was defined as < 70 mg/dL, slight variations from the current ADA SOC preoperative specific recommendation of a blood glucose reading of 100 to 180 mg/dL within 4 hours of surgery.10 The standard outpatient hypoglycemia definition of blood glucose < 70 mg/dL was chosen because the current goal (< 100 mg/dL) was not the standard in previous ADA SOCs that were in place during the study period. Specifically, the 2018 ADA SOC did not provide preoperative recommendations and the 2019-2021 ADA SOC recommended 80 to 180 mg/dL.10,12-18 For patients who had multiple preoperative blood glucose measurements, the first recorded glucose on the day of the procedure was used.
The secondary outcomes of this study were focused on the preoperative process/care at VHI and postoperative glycemic control. The preoperative process included examining whether medication instructions were given and their quality. Additionally, the number of interventions for hyperglycemia and hypoglycemia were required immediately prior to surgery and the average preoperative HbA1c (measured within 3 months prior to surgery) were collected and analyzed. For postoperative glycemic control, average blood glucose measurements and number of hypoglycemic (< 70 mg/dL) and hyperglycemic (> 180 mg/dL) events were measured in addition to the frequency of changes made at discharge to patients’ DM medication regimens.
The safety outcome of this study assessed commonly observed postoperative complications and was examined up to 30 days postsurgery. These included acute kidney injury (defined using Kidney Disease: Improving Global Outcomes 2012, the standard during the study period), nonfatal myocardial infarction, nonfatal stroke, and surgical site infections, which were identified from the discharge summary written by the primary surgery service.19 All-cause mortality also was collected.
Patients were included if they were admitted for major elective surgeries and had a diagnosis of either type 1 or type 2 DM on their problem list, determined by International Classification of Diseases, Tenth Revision codes. Major elective surgery was defined as a procedure that would likely result in a hospital admission of > 24 hours. Of note, patients may have been included in this study more than once if they had > 1 procedure at least 30 days apart and met inclusion criteria within the time frame. Patients were excluded if they were taking no DM medications or chronic steroids (at any dose), residing in a long-term care facility, being managed by a non-VA clinician prior to surgery, or missing a preoperative blood glucose measurement.
All data were collected from the CPRS. A list of surgical cases involving patients with DM who were scheduled to undergo major elective surgeries from January 1, 2018, to December 31, 2021, at VHI was generated. The list was randomized to a smaller number (N = 394) for data collection due to the time and resource constraints for a pharmacy residency project. All data were deidentified and stored in a secured VA server to protect patient confidentiality. Descriptive statistics were used for all results.
Results
Initially, 2362 surgeries were identified. A randomized sample of 394 charts were reviewed and 131 cases met inclusion criteria. Each case involved a unique patient (Figure). The most common reasons for exclusion were 143 patients with diet-controlled DM and 78 nonelective surgeries. The mean (SD) age of patients was 68 (8) years, and the most were male (98.5%) and White (76.3%) (Table 1).
At baseline, 45 of 131 patients (34.4%) had coronary artery disease and 29 (22.1%) each had autonomic neuropathy and chronic kidney disease. Most surgeries were conducted by orthopedic (32.1%) and peripheral vascular (21.4%) specialties. The mean (SD) length of surgery was 4.6 (2.6) hours and of hospital length of stay was 4 (4) days. No patients stayed longer than the 30-day safety outcome follow-up period. All patients had type 2 DM and took a mean 2 DM medications. The 63 patients taking insulin had a mean (SD) total daily dose of 99 (77) U (Table 2). A preoperative HbA1c was collected in 116 patients within 3 months of surgery, with a mean HbA1c of 7.0% (range, 5.3-10.7).
No patients had surgeries delayed or canceled because of uncontrolled DM on the day of surgery. The mean preoperative blood glucose level was 146 mg/dL (range, 73-365) (Table 3). No patients had a preoperative blood glucose level of < 70 mg/dL and 19 (14.5%) had a blood glucose level > 180 mg/dL. Among patients with hyperglycemia immediately prior to surgery, 6 (31.6%) had documentation of insulin being provided.
For this sample of patients, the preoperative clinic visit was conducted a mean 22 days prior to the planned surgery date. Among the 131 included patients, 122 (93.1%) had documentation of receiving instructions for DM medications. Among patients who had documented receipt of instructions, only 30 (24.6%) had instructions specifically tailored to their regimen rather than a generic templated form. The mean (SD) preoperative blood glucose was similar for those who received specific perioperative DM instructions at 146 (50) mg/dL when compared with those who did not at 147 (45) mg/dL. The mean (SD) preoperative blood glucose reading for those who had no documentation of receipt of perioperative instructions was 126 (54) mg/dL compared with 147 (46) mg/dL for those who did.
The mean number of postoperative blood glucose events per day was negligible for hypoglycemia and more frequent for hyperglycemia with a mean of 2 events per day. The mean postoperative blood glucose range was 121 to 247 mg/dL with most readings < 180 mg/dL. Upon discharge, most patients continued their home DM regimen with 5 patients (3.8%) having changes made to their regimen upon discharge.
Very few postoperative complications were identified from chart review. The most frequently observed postoperative complications were acute kidney injury, surgical site infections, and nonfatal stroke. There were no documented nonfatal myocardial infarctions. Two patients (1.5%) died within 30 days of the surgery; neither death was deemed to have been related to poor perioperative glycemic control.
Discussion
To our knowledge, this retrospective chart review was the first study to assess preoperative DM management and postoperative complications in a veteran population. VHI is a large, tertiary, level 1a, academic medical center that serves approximately 62,000 veterans annually and performs about 5000 to 6000 surgeries annually, a total that is increasing following the COVID-19 pandemic.20 This study found that the current process of a presurgery clinic visit and day of surgery glucose assessment has prevented surgical delays or cancellations.
Most patients included in this study were well controlled at baseline in accordance with the 2025 ADA SOC HbA1c recommendation of a preoperative HbA1c of < 8%, which may have contributed to no surgical delays or cancellations.10 However, not all patients had HbA1c collected within 3 months of surgery or even had one collected at all. Despite the ADA SOC providing no explicit recommendation for universal HbA1c screening prior to elective procedures, its importance cannot be understated given the body of evidence demonstrating poor outcomes with uncontrolled preoperative DM.8,10 The glycemic control at baseline may have contributed to the very few postsurgical complications observed in this study.
Although the current process at VHI prevented surgical delays and cancellations in this sample, there are still identified areas for improvement. One area is the instructions the patients received. Patients with DM are often prescribed ≥ 1 medication or a combination of insulins, noninsulin injectables, and oral DM medications, and this study population was no different. Because these medications may influence the anesthesia and perioperative periods, the ADA has specific guidance for altering administration schedules in the days leading up to surgery.10
Inappropriate administration of DM medications could lead to perioperative hypoglycemia or hyperglycemia, possibly causing surgical delays, case cancellations, and/or postoperative complications.21 Although these data reveal the specificity and documented receipt that the preoperative DM instructions did not impact the first recorded preoperative blood glucose, future studies should examine patient confidence in how to properly administer their DM medications prior to surgery. It is vital that patients receive clear instructions in accordance with the ADA SOC on whether to continue, hold, or adjust the dose of their medications to prevent fluctuations in blood glucose levels in the perioperative period, ensure safety with anesthesia, and prevent postoperative complications such as acute kidney injury. Of note, compliance with guideline recommendations for medication instructions was not examined because the data collection time frame expanded over multiple years and the recommendations have evolved each year as new data emerge.
Preoperative DM Management
The first key takeaway from this study is to ensure patients are ready for surgery with a formal assessment (typically in the form of a clinic visit) prior to the surgery. One private sector health system published their approach to this by administering an automatic preoperative HbA1c screening for those with a DM diagnosis and all patients with a random plasma glucose ≥ 200 mg/dL.22 Additionally, if the patient's HbA1c level was not at goal prior to surgery (≥ 8% for those with known DM and ≥ 6.5% with no known DM), patients were referred to endocrinology for further management. Increasing attention to the preoperative visit and extending HbA1c testing to all patients regardless of DM status also provides an opportunity to identify individuals living with undiagnosed DM.1
Even though there was no difference in the mean preoperative blood glucose level based on receipt or specificity of preoperative DM instructions, a second takeaway from this study is the importance of ensuring patients receive clear instructions on their DM medication schedule in the perioperative period. A practical first step may be updating the templates used by the primary surgery teams and providing education to the clinicians in the clinic on how to personalize the visits. Because the current preoperative DM process at VHI is managed by the primary surgical team in a clinic visit, there is an opportunity to shift this responsibility to other health care professionals, such as pharmacists—a change shown to reduce unintended omission of home medications following surgery during hospitalization and reduce costs.23,24
Limitations
This study relied on data included in the patient chart. These data include medication interventions made immediately prior to surgery, which can sometimes be inaccurately charted or difficult to find as they are not documented in the typical medication administration record. Also, the safety outcomes were collected from a discharge summary written by different clinicians, which may lead to information bias. Special attention was taken to ensure these data points were collected as accurately as possible, but it is possible some data may be inaccurate from unintentional human error. Additionally, the safety outcome was limited to a 30-day follow-up, but encompassed the entire length of postoperative stay for all included patients. Finally, given this study was retrospective with no comparison group and the intent was to improve processes at VHI, only hypotheses and potential interventions can be generated from this study. Future prospective studies with larger sample sizes and comparator groups are needed to draw further conclusions.
Conclusions
This study found that the current presurgery process at VHI appears to be successful in preventing surgical delays or cancellations due to hyperglycemia or hypoglycemia. Optimizing DM management can improve surgical outcomes by decreasing rates of postoperative complications, and this study added additional evidence in support of that in a unique population: veterans. Insight on the awareness of preoperative blood glucose management should be gleaned from this study, and based on this sample and site, the preadmission screening process and instructions provided to patients can serve as 2 starting points for optimizing elective surgery.
- Centers for Disease Control and Prevention. Diabetes basics. May 15, 2024. Accessed September 24, 2025. https://www.cdc.gov/diabetes/about/index.html
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- Farmaki P, Damaskos C, Garmpis N, et al . Complications of the Type 2 Diabetes Mellitus. Curr Cardiol Rev. 2020;16(4):249-251. doi:10.2174/1573403X1604201229115531
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care. 2010;33:1783-1788. doi:10.2337/dc10-0304
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol. 2007;156:137 -142. doi:10.1530/eje.1.02321
- Pomposelli JJ, Baxter JK 3rd, Babineau TJ, et al. Early postoperative glucose control predicts nosocomial infection rate in diabetic patients. JPEN J Parenter Enteral Nutr. 1998;22:77-81. doi:10.1177/01486071980220027
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care. 2011;34:256-261. doi:10.2337/dc10-1407
- Pasquel FJ, Gomez-Huelgas R, Anzola I, et al. Predictive value of admission hemoglobin A1c on inpatient glycemic control and response to insulin therapy in medicine and surgery patients with type 2 diabetes. Diabetes Care. 2015;38:e202-e203. doi:10.2337/dc15-1835
- Alexiewicz JM, Kumar D, Smogorzewski M, et al. Polymorphonuclear leukocytes in non-insulin-dependent diabetes mellitus: abnormalities in metabolism and function. Ann Intern Med. 1995;123:919-924. doi:10.7326/0003-4819-123-12-199512150-00004
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2025. Diabetes Care. 2025;48(1 suppl 1):S321-S334. doi:10.2337/dc25-S016
- Kumar R, Gandhi R. Reasons for cancellation of operation on the day of intended surgery in a multidisciplinary 500 bedded hospital. J Anaesthesiol Clin Pharmacol. 2012;28:66-69. doi:10.4103/0970-9185.92442
- American Diabetes Association. 14. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2018. Diabetes Care. 2018;41(1 suppl 1):S144- S151. doi:10.2337/dc18-S014
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2019. Diabetes Care. 2019;42(suppl 1):S173- S181. doi:10.2337/dc19-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2020. Diabetes Care. 2020;43(suppl 1):S193- S202. doi:10.2337/dc20-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2021. Diabetes Care. 2021;44(suppl 1):S211- S220. doi:10.2337/dc21-S015
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2022. Diabetes Care. 2022;45(suppl 1):S244-S253. doi:10.2337/dc22-S016
- ElSayed NA, Aleppo G, Aroda VR, et al. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2023. Diabetes Care. 2023;46(suppl 1):S267-S278. doi:10.2337/dc23-S016
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2024. Diabetes Care. 2024;47(suppl 1):S295-S306. doi:10.2337/dc24-S016
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1-138. Accessed September 24, 2025. https:// www.kisupplements.org/issue/S2157-1716(12)X7200-9
- US Department of Veterans Affairs. VA Indiana Healthcare: about us. Accessed September 24, 2025. https:// www.va.gov/indiana-health-care/about-us/
- Koh WX, Phelan R, Hopman WM, et al. Cancellation of elective surgery: rates, reasons and effect on patient satisfaction. Can J Surg. 2021;64:E155-E161. doi:10.1503/cjs.008119
- Pai S-L, Haehn DA, Pitruzzello NE, et al. Reducing infection rates with enhanced preoperative diabetes mellitus diagnosis and optimization processes. South Med J. 2023;116:215-219. doi:10.14423/SMJ.0000000000001507
- Forrester TG, Sullivan S, Snoswell CL, et al. Integrating a pharmacist into the perioperative setting. Aust Health Rev. 2020;44:563-568. doi:10.1071/AH19126
- Hale AR, Coombes ID, Stokes J, et al. Perioperative medication management: expanding the role of the preadmission clinic pharmacist in a single centre, randomised controlled trial of collaborative prescribing. BMJ Open. 2013;3:e003027. doi:10.1136/bmjopen-2013-003027
- Centers for Disease Control and Prevention. Diabetes basics. May 15, 2024. Accessed September 24, 2025. https://www.cdc.gov/diabetes/about/index.html
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- Farmaki P, Damaskos C, Garmpis N, et al . Complications of the Type 2 Diabetes Mellitus. Curr Cardiol Rev. 2020;16(4):249-251. doi:10.2174/1573403X1604201229115531
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care. 2010;33:1783-1788. doi:10.2337/dc10-0304
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol. 2007;156:137 -142. doi:10.1530/eje.1.02321
- Pomposelli JJ, Baxter JK 3rd, Babineau TJ, et al. Early postoperative glucose control predicts nosocomial infection rate in diabetic patients. JPEN J Parenter Enteral Nutr. 1998;22:77-81. doi:10.1177/01486071980220027
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care. 2011;34:256-261. doi:10.2337/dc10-1407
- Pasquel FJ, Gomez-Huelgas R, Anzola I, et al. Predictive value of admission hemoglobin A1c on inpatient glycemic control and response to insulin therapy in medicine and surgery patients with type 2 diabetes. Diabetes Care. 2015;38:e202-e203. doi:10.2337/dc15-1835
- Alexiewicz JM, Kumar D, Smogorzewski M, et al. Polymorphonuclear leukocytes in non-insulin-dependent diabetes mellitus: abnormalities in metabolism and function. Ann Intern Med. 1995;123:919-924. doi:10.7326/0003-4819-123-12-199512150-00004
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2025. Diabetes Care. 2025;48(1 suppl 1):S321-S334. doi:10.2337/dc25-S016
- Kumar R, Gandhi R. Reasons for cancellation of operation on the day of intended surgery in a multidisciplinary 500 bedded hospital. J Anaesthesiol Clin Pharmacol. 2012;28:66-69. doi:10.4103/0970-9185.92442
- American Diabetes Association. 14. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2018. Diabetes Care. 2018;41(1 suppl 1):S144- S151. doi:10.2337/dc18-S014
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2019. Diabetes Care. 2019;42(suppl 1):S173- S181. doi:10.2337/dc19-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2020. Diabetes Care. 2020;43(suppl 1):S193- S202. doi:10.2337/dc20-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2021. Diabetes Care. 2021;44(suppl 1):S211- S220. doi:10.2337/dc21-S015
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2022. Diabetes Care. 2022;45(suppl 1):S244-S253. doi:10.2337/dc22-S016
- ElSayed NA, Aleppo G, Aroda VR, et al. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2023. Diabetes Care. 2023;46(suppl 1):S267-S278. doi:10.2337/dc23-S016
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2024. Diabetes Care. 2024;47(suppl 1):S295-S306. doi:10.2337/dc24-S016
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1-138. Accessed September 24, 2025. https:// www.kisupplements.org/issue/S2157-1716(12)X7200-9
- US Department of Veterans Affairs. VA Indiana Healthcare: about us. Accessed September 24, 2025. https:// www.va.gov/indiana-health-care/about-us/
- Koh WX, Phelan R, Hopman WM, et al. Cancellation of elective surgery: rates, reasons and effect on patient satisfaction. Can J Surg. 2021;64:E155-E161. doi:10.1503/cjs.008119
- Pai S-L, Haehn DA, Pitruzzello NE, et al. Reducing infection rates with enhanced preoperative diabetes mellitus diagnosis and optimization processes. South Med J. 2023;116:215-219. doi:10.14423/SMJ.0000000000001507
- Forrester TG, Sullivan S, Snoswell CL, et al. Integrating a pharmacist into the perioperative setting. Aust Health Rev. 2020;44:563-568. doi:10.1071/AH19126
- Hale AR, Coombes ID, Stokes J, et al. Perioperative medication management: expanding the role of the preadmission clinic pharmacist in a single centre, randomised controlled trial of collaborative prescribing. BMJ Open. 2013;3:e003027. doi:10.1136/bmjopen-2013-003027
Preoperative Diabetes Management for Patients Undergoing Elective Surgeries at a Veterans Affairs Medical Center
Preoperative Diabetes Management for Patients Undergoing Elective Surgeries at a Veterans Affairs Medical Center
Efficacy of Subcutaneous Semaglutide Dose Escalation in Reducing Insulin in Patients With Type 2 Diabetes
Efficacy of Subcutaneous Semaglutide Dose Escalation in Reducing Insulin in Patients With Type 2 Diabetes
Type 2 diabetes mellitus (T2DM) is a chronic disease becoming more prevalent each year and is the seventh-leading cause of death in the United States.1 The most common reason for hospitalization for patients with T2DM is uncontrolled glycemic levels.2 Nearly 25% of the US Department of Veterans Affairs (VA) patient population has T2DM.3 T2DM is the leading cause of blindness, end-stage renal disease, and amputation for VA patients.4
According to the 2023 American Diabetes Association (ADA) guidelines, treatment goals of T2DM include eliminating symptoms, preventing or delaying complications, and attaining glycemic goals. A typical hemoglobin A1c (HbA1c) goal range is < 7%, but individual goals can vary up to < 9% due to a multitude of factors, including patient comorbidities and clinical status.5
Initial treatment recommendations are nonpharmacologic and include comprehensive lifestyle interventions such as optimizing nutrition, physical activity, and behavioral therapy. When pharmacologic therapy is required, metformin is the preferred first-line treatment for the majority of newly diagnosed patients with T2DM and should be added to continued lifestyle management.5 If HbA1c levels remains above goal, the 2023 ADA guidelines recommend adding a second medication, including but not limited to insulin, a glucagonlike peptide-1 receptor agonist (GLP-1RA), or a sodium-glucose cotransporter 2 inhibitor. Medication choice is largely based on the patient’s concomitant conditions (eg, atherosclerotic cardiovascular disease, heart failure, or chronic kidney disease). The 2023 ADA guidelines suggest initiating insulin therapy when a patient's blood glucose ≥ 300 mg/dL, HbA1c > 10%, or if the patient has symptoms of hyperglycemia, even at initial diagnosis. Initiating medications to minimize or avoid hypoglycemia is a priority, especially in high-risk individuals.5
Clinical evidence shows that GLP-1RAs may provide similar glycemic control to insulin with lower risk of hypoglycemia.6 Other reported benefits of GLP-1RAs include weight loss, blood pressure reduction, and improved lipid levels. The most common adverse events (AEs) with GLP-1RAs are gastrointestinal. Including GLP-1RAs in T2DM pharmacotherapy may lower the risk of hypoglycemia, especially in patients at high risk of hypoglycemia.
The 2023 ADA guidelines indicate that it is appropriate to initiate GLP-]1RAs in patients on insulin.5 However, while GLP-1RAs do not increase the risk of hypoglycemia independently, combination treatment with GLP-1RAs and insulin can still result in hypoglycemia.6 Insulin is the key suspect of this hypoglycemic risk.7 Thus, if insulin dosage can be reduced or discontinued, this might reduce the risk of hypoglycemia.
The literature is limited on how the addition of a GLP-1RA to insulin treatment will affect the patient's daily insulin doses, particularly for the veteran population. The goal of this study is to examine this gap in current research by examining semaglutide, which is the current formulary preferred GLP-1RA at the VA.
Semaglutide is subcutaneously initiated at a dose of 0.25 mg once weekly for 4 weeks to reduce gastrointestinal symptoms, then increased to 0.5 mg weekly. Additional increases to a maintenance dose of 1 mg or 2 mg weekly can occur to achieve glycemic goals. The SUSTAIN-FORTE randomized controlled trial sought to determine whether there was a difference in HbA1c level reduction and significant weight loss with the 2-mg vs 1-mg dose.8 Patients in the trial were taking metformin but needed additional medication to control their HbA1c. They were not using insulin and may or may not have been taking sulfonylureas prior to semaglutide initiation. Semaglutide 2 mg was found to significantly improve HbA1c control and promote weight loss compared with semaglutide 1 mg, while maintaining a similar safety profile.
Because this study involved patients who required additional HbA1c control, although semaglutide reduced HbA1c, not all patients were able to reduce their other diabetes medications, which depended on the baseline HbA1c level and the level upon completion of semaglutide titration. Dose reductions for the patients’ other T2DM medications were not reported at trial end. SUSTAIN-FORTE established titration up to semaglutide 2 mg as effective for HbA1c reduction, although it did not study patients also on insulin.8
Insulin is associated with hypoglycemic risk, weight gain, and other AEs.7,8 This study analyzed whether increasing semaglutide could reduce insulin doses and therefore reduce risk of AEs in patients with T2DM.
Methods
A retrospective, single-center, chart review was conducted at VA Sioux Falls Health Care System (VASFHCS). Data were collected through manual review of VASFHCS electronic medical records. Patients aged ≥ 18 years with active prescriptions for at least once-daily insulin who were initiated on 2-mg weekly dose of semaglutide at the VASFHCS clinical pharmacy practitioner medication management clinic between January 1, 2021, and September 1, 2023, were included. VASFHCS clinical pharmacy practitioners have a scope of practice that allows them to initiate, modify, or discontinue medication therapy within medication management clinics.
The most frequently used prandial insulin at VASFHCS is insulin aspart, and the most frequently used basal insulin is insulin glargine. Patients were retrospectively monitored as they progressed from baseline (the point in time where semaglutide 0.5 mg was initiated) to ≥ 3 months on semaglutide 2-mg therapy. Patients were excluded if they previously used a GLP-1RA or if they were on sliding scale insulin without an exact daily dosage.
The primary endpoint was the percent change in total daily insulin dose from baseline to each dose increase after receiving semaglutide 2 mg for ≥ 3 months. Secondary endpoints included changes in daily prandial insulin dose, daily basal insulin dose, HbA1c, and number of hypoglycemic events reported. Data collected included age, race, weight, body mass index, total daily prandial insulin dose, total daily basal insulin dose, HbA1c, and hypoglycemic events reported at the visit when semaglutide was initiated.
Statistical Analysis
The sample size was calculated prior to data collection, and it was determined that for α = .05, 47 patients were needed to achieve 95% power. The primary endpoint was assessed using a paired t test, as were each secondary endpoint. Results with P < .05 were considered statistically significant.
Results
Sixty-two patients were included. The mean HbA1c level at baseline was 7.7%, the baseline mean prandial and insulin daily doses were 41.5 units and 85.1 units, respectively (Table 1) From baseline to initiation of a semaglutide 1-mg dose, the daily insulin dose changed –5.6% (95% CI, 2.2-14.0; P = .008). From baseline to 2-mg dose initiation daily insulin changed -22.2% (95% CI, 22.0-35.1; P < .001) and for patients receiving semaglutide 2 mg for ≥ 3 months it changed -36.9% (95% CI, 37.4-56.5; P < .001) (Figure).

After receiving the 2-mg dose for ≥ 3 months, the mean daily dose of prandial insulin decreased from 41.5 units to 24.6 units (95% CI, 12.6-21.2; P < .001); mean daily dose of basal insulin decreased from 85.1 units to 52.1 units (95% CI, 23.9-42.0; P < .001); and mean HbA1c level decreased from 7.7% to 7.1% (95% CI, 0.3-0.8; P < .001). Mean number of hypoglycemic events reported was not statistically significant, changing from 3.6 to 3.2 (95% CI, –0.6 to 0.1; P = .21) (Table 2).
Discussion
This study investigated the effect of subcutaneous semaglutide dose escalation on total daily insulin dose for patients with T2DM. There was a statistically significant decrease in total daily insulin dose from baseline to 1 mg initiation; this decrease continued with further insulin dose reduction seen at the 2-mg dose initiation and additional insulin dose reduction at ≥ 3 months at this dose. It was hypothesized there would be a significant total daily insulin dose reduction at some point, especially when transitioning from the semaglutide 1-mg to the 2-mg dose, based on previous research. 9,10 The additional reduction in daily insulin dose when continuing on semaglutide 2 mg for ≥ 3 months was an unanticipated but added benefit, showing that if tolerated, maintaining the 2-mg dose will help patients reduce their insulin doses.
In terms of secondary endpoints, there was a statistically significant decrease in mean total daily dose individually for prandial and basal insulin from baseline to ≥ 3 months after semaglutide 2 mg initiation. The change in HbA1c level was also statistically significant and decreased from baseline, even as insulin doses were reduced. This change in HbA1c level was expected; previous literature has shown a significant link between improving HbA1c control when semaglutide doses are increased to 2 mg weekly.10 Due to having been shown in previous trials, it was expected that HbA1c levels would decrease even when the insulin doses were being reduced.10 Insulin dose reduction can potentially be added to the growing evidence of semaglutide benefits. The change in the number of hypoglycemic events was not statistically significant, which was unexpected since previous research show a trend in patients taking GLP-1RAs having fewer hypoglycemic events than those taking insulin.6 Further investigation with a larger sample size and prospective trial could determine whether this result is an outlier. In this study, there was no increase in HbA1c or hypoglycemic events reported with increasing semaglutide doses, which provides further evidence of the safety of semaglutide even at higher doses.
These data suggest that for a patient with T2DM who is already taking insulin, the recommended titration of semaglutide is to start with 0.5 mg and titrate up to a 2-mg subcutaneous weekly dose and to then continue at that dose. As long as the 2-mg dose is tolerated, it will provide patients with the most HbA1c control and lead to a reduction of their total daily insulin doses according to these results.
Strengths and Limitations
This study compared patient data at different points. This method did not require a second distinct control group, which would potentially introduce confounding factors, such as different baseline characteristics. Another strength is that documentation was available for all patients throughout the study so no one was lost to follow-up. This allowed comprehensive data collection and provided a stronger conclusion given the completeness of the data from baseline to follow-up.
Limitations include the retrospective design and small sample size. In addition, the study design did not allow for randomization. There is no documentation of adherence to medication regimen, which was difficult to determine due to the retrospective nature. Other changes to the patients’ medication regimen were not collected in aggregate and thus, it is possible the total daily insulin dose was impacted by other medication changes. There is also potential for inconsistent documentation of the patients’ true total daily insulin dose in the medical record, thus leading to inaccuracy of recorded data.
Conclusions
A small sample of veterans with T2DM had statistically significant reductions in total daily insulin dose when subcutaneous semaglutide was initiated, as well as after each dose increase. There was also a statistically significant reduction in HbA1c levels from baseline even as patient insulin doses were reduced. These results support the current practice of using semaglutide to treat T2DM, suggesting it may be safe and effective at reducing HbA1c levels as the dose is titrated up to 2 mg. There was no statistically significant change in the number of hypoglycemic events reported as semaglutide was titrated up. Thus, when semaglutide is increased to the maximum recommended dose of 2 mg for T2DM, patients may experience a reduction of their total daily dose of insulin and HbA1c levels. These benefits may reduce the risk of insulin-related AEs while maintaining appropriate glycemic control.
- Diabetes mellitus: in federal health care data trends 2017. Fed Pract. 2017:S20. Accessed August 6, 2025. https://www.fedprac-digital.com/federalpractitioner/data_trends_2017
- Centers for Disease Control and Prevention. National diabetes statistics report. May 15, 2024. Accessed September 17, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- US Department of Veterans Affairs. VA research on diabetes. Updated January 15, 2021. Accessed August 6, 2025. https://www.research.va.gov/topics/diabetes.cfm
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- American Diabetes Association. Standards of care in diabetes— 2023 abridged for primary care providers. Clin Diabetes. 2022;41:4-31. doi:10.2337/cd23-as01
- Zhao Z, Tang Y, Hu Y, Zhu H, Chen X, Zhao B. Hypoglycemia following the use of glucagon-like peptide-1 receptor agonists: a real-world analysis of post-marketing surveillance data. Ann Transl Med. 2021;9:1482. doi:10.21037/atm-21-4162
- Workgroup on Hypoglycemia, American Diabetes Association. Defining and reporting hypoglycemia in diabetes: a report from the American Diabetes Association Workgroup on Hypoglycemia. Diabetes Care. 2005;28:1245-1249. doi:10.2337/diacare.28.5.1245
- Frías JP, Auerbach P, Bajaj HS, et al. Efficacy and safety of once-weekly semaglutide 2.0 mg versus 1.0 mg in patients with type 2 diabetes (SUSTAIN FORTE): a double-blind, randomised, phase 3B trial. Lancet Diabetes Endocrinol. 2021;9:563-574. doi:10.1016/S2213-8587(21)00174-1
- Garber AJ, Handelsman Y, Grunberger G, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2020 executive summary. Endocr Pract. 2020;26:107-139. doi:10.4158/CS-2019-0472
- Miles KE, Kerr JL. Semaglutide for the treatment of type 2 diabetes mellitus. J Pharm Technol. 2018;34:281-289. doi:10.1177/8755122518790925
Type 2 diabetes mellitus (T2DM) is a chronic disease becoming more prevalent each year and is the seventh-leading cause of death in the United States.1 The most common reason for hospitalization for patients with T2DM is uncontrolled glycemic levels.2 Nearly 25% of the US Department of Veterans Affairs (VA) patient population has T2DM.3 T2DM is the leading cause of blindness, end-stage renal disease, and amputation for VA patients.4
According to the 2023 American Diabetes Association (ADA) guidelines, treatment goals of T2DM include eliminating symptoms, preventing or delaying complications, and attaining glycemic goals. A typical hemoglobin A1c (HbA1c) goal range is < 7%, but individual goals can vary up to < 9% due to a multitude of factors, including patient comorbidities and clinical status.5
Initial treatment recommendations are nonpharmacologic and include comprehensive lifestyle interventions such as optimizing nutrition, physical activity, and behavioral therapy. When pharmacologic therapy is required, metformin is the preferred first-line treatment for the majority of newly diagnosed patients with T2DM and should be added to continued lifestyle management.5 If HbA1c levels remains above goal, the 2023 ADA guidelines recommend adding a second medication, including but not limited to insulin, a glucagonlike peptide-1 receptor agonist (GLP-1RA), or a sodium-glucose cotransporter 2 inhibitor. Medication choice is largely based on the patient’s concomitant conditions (eg, atherosclerotic cardiovascular disease, heart failure, or chronic kidney disease). The 2023 ADA guidelines suggest initiating insulin therapy when a patient's blood glucose ≥ 300 mg/dL, HbA1c > 10%, or if the patient has symptoms of hyperglycemia, even at initial diagnosis. Initiating medications to minimize or avoid hypoglycemia is a priority, especially in high-risk individuals.5
Clinical evidence shows that GLP-1RAs may provide similar glycemic control to insulin with lower risk of hypoglycemia.6 Other reported benefits of GLP-1RAs include weight loss, blood pressure reduction, and improved lipid levels. The most common adverse events (AEs) with GLP-1RAs are gastrointestinal. Including GLP-1RAs in T2DM pharmacotherapy may lower the risk of hypoglycemia, especially in patients at high risk of hypoglycemia.
The 2023 ADA guidelines indicate that it is appropriate to initiate GLP-]1RAs in patients on insulin.5 However, while GLP-1RAs do not increase the risk of hypoglycemia independently, combination treatment with GLP-1RAs and insulin can still result in hypoglycemia.6 Insulin is the key suspect of this hypoglycemic risk.7 Thus, if insulin dosage can be reduced or discontinued, this might reduce the risk of hypoglycemia.
The literature is limited on how the addition of a GLP-1RA to insulin treatment will affect the patient's daily insulin doses, particularly for the veteran population. The goal of this study is to examine this gap in current research by examining semaglutide, which is the current formulary preferred GLP-1RA at the VA.
Semaglutide is subcutaneously initiated at a dose of 0.25 mg once weekly for 4 weeks to reduce gastrointestinal symptoms, then increased to 0.5 mg weekly. Additional increases to a maintenance dose of 1 mg or 2 mg weekly can occur to achieve glycemic goals. The SUSTAIN-FORTE randomized controlled trial sought to determine whether there was a difference in HbA1c level reduction and significant weight loss with the 2-mg vs 1-mg dose.8 Patients in the trial were taking metformin but needed additional medication to control their HbA1c. They were not using insulin and may or may not have been taking sulfonylureas prior to semaglutide initiation. Semaglutide 2 mg was found to significantly improve HbA1c control and promote weight loss compared with semaglutide 1 mg, while maintaining a similar safety profile.
Because this study involved patients who required additional HbA1c control, although semaglutide reduced HbA1c, not all patients were able to reduce their other diabetes medications, which depended on the baseline HbA1c level and the level upon completion of semaglutide titration. Dose reductions for the patients’ other T2DM medications were not reported at trial end. SUSTAIN-FORTE established titration up to semaglutide 2 mg as effective for HbA1c reduction, although it did not study patients also on insulin.8
Insulin is associated with hypoglycemic risk, weight gain, and other AEs.7,8 This study analyzed whether increasing semaglutide could reduce insulin doses and therefore reduce risk of AEs in patients with T2DM.
Methods
A retrospective, single-center, chart review was conducted at VA Sioux Falls Health Care System (VASFHCS). Data were collected through manual review of VASFHCS electronic medical records. Patients aged ≥ 18 years with active prescriptions for at least once-daily insulin who were initiated on 2-mg weekly dose of semaglutide at the VASFHCS clinical pharmacy practitioner medication management clinic between January 1, 2021, and September 1, 2023, were included. VASFHCS clinical pharmacy practitioners have a scope of practice that allows them to initiate, modify, or discontinue medication therapy within medication management clinics.
The most frequently used prandial insulin at VASFHCS is insulin aspart, and the most frequently used basal insulin is insulin glargine. Patients were retrospectively monitored as they progressed from baseline (the point in time where semaglutide 0.5 mg was initiated) to ≥ 3 months on semaglutide 2-mg therapy. Patients were excluded if they previously used a GLP-1RA or if they were on sliding scale insulin without an exact daily dosage.
The primary endpoint was the percent change in total daily insulin dose from baseline to each dose increase after receiving semaglutide 2 mg for ≥ 3 months. Secondary endpoints included changes in daily prandial insulin dose, daily basal insulin dose, HbA1c, and number of hypoglycemic events reported. Data collected included age, race, weight, body mass index, total daily prandial insulin dose, total daily basal insulin dose, HbA1c, and hypoglycemic events reported at the visit when semaglutide was initiated.
Statistical Analysis
The sample size was calculated prior to data collection, and it was determined that for α = .05, 47 patients were needed to achieve 95% power. The primary endpoint was assessed using a paired t test, as were each secondary endpoint. Results with P < .05 were considered statistically significant.
Results
Sixty-two patients were included. The mean HbA1c level at baseline was 7.7%, the baseline mean prandial and insulin daily doses were 41.5 units and 85.1 units, respectively (Table 1) From baseline to initiation of a semaglutide 1-mg dose, the daily insulin dose changed –5.6% (95% CI, 2.2-14.0; P = .008). From baseline to 2-mg dose initiation daily insulin changed -22.2% (95% CI, 22.0-35.1; P < .001) and for patients receiving semaglutide 2 mg for ≥ 3 months it changed -36.9% (95% CI, 37.4-56.5; P < .001) (Figure).
After receiving the 2-mg dose for ≥ 3 months, the mean daily dose of prandial insulin decreased from 41.5 units to 24.6 units (95% CI, 12.6-21.2; P < .001); mean daily dose of basal insulin decreased from 85.1 units to 52.1 units (95% CI, 23.9-42.0; P < .001); and mean HbA1c level decreased from 7.7% to 7.1% (95% CI, 0.3-0.8; P < .001). Mean number of hypoglycemic events reported was not statistically significant, changing from 3.6 to 3.2 (95% CI, –0.6 to 0.1; P = .21) (Table 2).
Discussion
This study investigated the effect of subcutaneous semaglutide dose escalation on total daily insulin dose for patients with T2DM. There was a statistically significant decrease in total daily insulin dose from baseline to 1 mg initiation; this decrease continued with further insulin dose reduction seen at the 2-mg dose initiation and additional insulin dose reduction at ≥ 3 months at this dose. It was hypothesized there would be a significant total daily insulin dose reduction at some point, especially when transitioning from the semaglutide 1-mg to the 2-mg dose, based on previous research. 9,10 The additional reduction in daily insulin dose when continuing on semaglutide 2 mg for ≥ 3 months was an unanticipated but added benefit, showing that if tolerated, maintaining the 2-mg dose will help patients reduce their insulin doses.
In terms of secondary endpoints, there was a statistically significant decrease in mean total daily dose individually for prandial and basal insulin from baseline to ≥ 3 months after semaglutide 2 mg initiation. The change in HbA1c level was also statistically significant and decreased from baseline, even as insulin doses were reduced. This change in HbA1c level was expected; previous literature has shown a significant link between improving HbA1c control when semaglutide doses are increased to 2 mg weekly.10 Due to having been shown in previous trials, it was expected that HbA1c levels would decrease even when the insulin doses were being reduced.10 Insulin dose reduction can potentially be added to the growing evidence of semaglutide benefits. The change in the number of hypoglycemic events was not statistically significant, which was unexpected since previous research show a trend in patients taking GLP-1RAs having fewer hypoglycemic events than those taking insulin.6 Further investigation with a larger sample size and prospective trial could determine whether this result is an outlier. In this study, there was no increase in HbA1c or hypoglycemic events reported with increasing semaglutide doses, which provides further evidence of the safety of semaglutide even at higher doses.
These data suggest that for a patient with T2DM who is already taking insulin, the recommended titration of semaglutide is to start with 0.5 mg and titrate up to a 2-mg subcutaneous weekly dose and to then continue at that dose. As long as the 2-mg dose is tolerated, it will provide patients with the most HbA1c control and lead to a reduction of their total daily insulin doses according to these results.
Strengths and Limitations
This study compared patient data at different points. This method did not require a second distinct control group, which would potentially introduce confounding factors, such as different baseline characteristics. Another strength is that documentation was available for all patients throughout the study so no one was lost to follow-up. This allowed comprehensive data collection and provided a stronger conclusion given the completeness of the data from baseline to follow-up.
Limitations include the retrospective design and small sample size. In addition, the study design did not allow for randomization. There is no documentation of adherence to medication regimen, which was difficult to determine due to the retrospective nature. Other changes to the patients’ medication regimen were not collected in aggregate and thus, it is possible the total daily insulin dose was impacted by other medication changes. There is also potential for inconsistent documentation of the patients’ true total daily insulin dose in the medical record, thus leading to inaccuracy of recorded data.
Conclusions
A small sample of veterans with T2DM had statistically significant reductions in total daily insulin dose when subcutaneous semaglutide was initiated, as well as after each dose increase. There was also a statistically significant reduction in HbA1c levels from baseline even as patient insulin doses were reduced. These results support the current practice of using semaglutide to treat T2DM, suggesting it may be safe and effective at reducing HbA1c levels as the dose is titrated up to 2 mg. There was no statistically significant change in the number of hypoglycemic events reported as semaglutide was titrated up. Thus, when semaglutide is increased to the maximum recommended dose of 2 mg for T2DM, patients may experience a reduction of their total daily dose of insulin and HbA1c levels. These benefits may reduce the risk of insulin-related AEs while maintaining appropriate glycemic control.
Type 2 diabetes mellitus (T2DM) is a chronic disease becoming more prevalent each year and is the seventh-leading cause of death in the United States.1 The most common reason for hospitalization for patients with T2DM is uncontrolled glycemic levels.2 Nearly 25% of the US Department of Veterans Affairs (VA) patient population has T2DM.3 T2DM is the leading cause of blindness, end-stage renal disease, and amputation for VA patients.4
According to the 2023 American Diabetes Association (ADA) guidelines, treatment goals of T2DM include eliminating symptoms, preventing or delaying complications, and attaining glycemic goals. A typical hemoglobin A1c (HbA1c) goal range is < 7%, but individual goals can vary up to < 9% due to a multitude of factors, including patient comorbidities and clinical status.5
Initial treatment recommendations are nonpharmacologic and include comprehensive lifestyle interventions such as optimizing nutrition, physical activity, and behavioral therapy. When pharmacologic therapy is required, metformin is the preferred first-line treatment for the majority of newly diagnosed patients with T2DM and should be added to continued lifestyle management.5 If HbA1c levels remains above goal, the 2023 ADA guidelines recommend adding a second medication, including but not limited to insulin, a glucagonlike peptide-1 receptor agonist (GLP-1RA), or a sodium-glucose cotransporter 2 inhibitor. Medication choice is largely based on the patient’s concomitant conditions (eg, atherosclerotic cardiovascular disease, heart failure, or chronic kidney disease). The 2023 ADA guidelines suggest initiating insulin therapy when a patient's blood glucose ≥ 300 mg/dL, HbA1c > 10%, or if the patient has symptoms of hyperglycemia, even at initial diagnosis. Initiating medications to minimize or avoid hypoglycemia is a priority, especially in high-risk individuals.5
Clinical evidence shows that GLP-1RAs may provide similar glycemic control to insulin with lower risk of hypoglycemia.6 Other reported benefits of GLP-1RAs include weight loss, blood pressure reduction, and improved lipid levels. The most common adverse events (AEs) with GLP-1RAs are gastrointestinal. Including GLP-1RAs in T2DM pharmacotherapy may lower the risk of hypoglycemia, especially in patients at high risk of hypoglycemia.
The 2023 ADA guidelines indicate that it is appropriate to initiate GLP-]1RAs in patients on insulin.5 However, while GLP-1RAs do not increase the risk of hypoglycemia independently, combination treatment with GLP-1RAs and insulin can still result in hypoglycemia.6 Insulin is the key suspect of this hypoglycemic risk.7 Thus, if insulin dosage can be reduced or discontinued, this might reduce the risk of hypoglycemia.
The literature is limited on how the addition of a GLP-1RA to insulin treatment will affect the patient's daily insulin doses, particularly for the veteran population. The goal of this study is to examine this gap in current research by examining semaglutide, which is the current formulary preferred GLP-1RA at the VA.
Semaglutide is subcutaneously initiated at a dose of 0.25 mg once weekly for 4 weeks to reduce gastrointestinal symptoms, then increased to 0.5 mg weekly. Additional increases to a maintenance dose of 1 mg or 2 mg weekly can occur to achieve glycemic goals. The SUSTAIN-FORTE randomized controlled trial sought to determine whether there was a difference in HbA1c level reduction and significant weight loss with the 2-mg vs 1-mg dose.8 Patients in the trial were taking metformin but needed additional medication to control their HbA1c. They were not using insulin and may or may not have been taking sulfonylureas prior to semaglutide initiation. Semaglutide 2 mg was found to significantly improve HbA1c control and promote weight loss compared with semaglutide 1 mg, while maintaining a similar safety profile.
Because this study involved patients who required additional HbA1c control, although semaglutide reduced HbA1c, not all patients were able to reduce their other diabetes medications, which depended on the baseline HbA1c level and the level upon completion of semaglutide titration. Dose reductions for the patients’ other T2DM medications were not reported at trial end. SUSTAIN-FORTE established titration up to semaglutide 2 mg as effective for HbA1c reduction, although it did not study patients also on insulin.8
Insulin is associated with hypoglycemic risk, weight gain, and other AEs.7,8 This study analyzed whether increasing semaglutide could reduce insulin doses and therefore reduce risk of AEs in patients with T2DM.
Methods
A retrospective, single-center, chart review was conducted at VA Sioux Falls Health Care System (VASFHCS). Data were collected through manual review of VASFHCS electronic medical records. Patients aged ≥ 18 years with active prescriptions for at least once-daily insulin who were initiated on 2-mg weekly dose of semaglutide at the VASFHCS clinical pharmacy practitioner medication management clinic between January 1, 2021, and September 1, 2023, were included. VASFHCS clinical pharmacy practitioners have a scope of practice that allows them to initiate, modify, or discontinue medication therapy within medication management clinics.
The most frequently used prandial insulin at VASFHCS is insulin aspart, and the most frequently used basal insulin is insulin glargine. Patients were retrospectively monitored as they progressed from baseline (the point in time where semaglutide 0.5 mg was initiated) to ≥ 3 months on semaglutide 2-mg therapy. Patients were excluded if they previously used a GLP-1RA or if they were on sliding scale insulin without an exact daily dosage.
The primary endpoint was the percent change in total daily insulin dose from baseline to each dose increase after receiving semaglutide 2 mg for ≥ 3 months. Secondary endpoints included changes in daily prandial insulin dose, daily basal insulin dose, HbA1c, and number of hypoglycemic events reported. Data collected included age, race, weight, body mass index, total daily prandial insulin dose, total daily basal insulin dose, HbA1c, and hypoglycemic events reported at the visit when semaglutide was initiated.
Statistical Analysis
The sample size was calculated prior to data collection, and it was determined that for α = .05, 47 patients were needed to achieve 95% power. The primary endpoint was assessed using a paired t test, as were each secondary endpoint. Results with P < .05 were considered statistically significant.
Results
Sixty-two patients were included. The mean HbA1c level at baseline was 7.7%, the baseline mean prandial and insulin daily doses were 41.5 units and 85.1 units, respectively (Table 1) From baseline to initiation of a semaglutide 1-mg dose, the daily insulin dose changed –5.6% (95% CI, 2.2-14.0; P = .008). From baseline to 2-mg dose initiation daily insulin changed -22.2% (95% CI, 22.0-35.1; P < .001) and for patients receiving semaglutide 2 mg for ≥ 3 months it changed -36.9% (95% CI, 37.4-56.5; P < .001) (Figure).
After receiving the 2-mg dose for ≥ 3 months, the mean daily dose of prandial insulin decreased from 41.5 units to 24.6 units (95% CI, 12.6-21.2; P < .001); mean daily dose of basal insulin decreased from 85.1 units to 52.1 units (95% CI, 23.9-42.0; P < .001); and mean HbA1c level decreased from 7.7% to 7.1% (95% CI, 0.3-0.8; P < .001). Mean number of hypoglycemic events reported was not statistically significant, changing from 3.6 to 3.2 (95% CI, –0.6 to 0.1; P = .21) (Table 2).
Discussion
This study investigated the effect of subcutaneous semaglutide dose escalation on total daily insulin dose for patients with T2DM. There was a statistically significant decrease in total daily insulin dose from baseline to 1 mg initiation; this decrease continued with further insulin dose reduction seen at the 2-mg dose initiation and additional insulin dose reduction at ≥ 3 months at this dose. It was hypothesized there would be a significant total daily insulin dose reduction at some point, especially when transitioning from the semaglutide 1-mg to the 2-mg dose, based on previous research. 9,10 The additional reduction in daily insulin dose when continuing on semaglutide 2 mg for ≥ 3 months was an unanticipated but added benefit, showing that if tolerated, maintaining the 2-mg dose will help patients reduce their insulin doses.
In terms of secondary endpoints, there was a statistically significant decrease in mean total daily dose individually for prandial and basal insulin from baseline to ≥ 3 months after semaglutide 2 mg initiation. The change in HbA1c level was also statistically significant and decreased from baseline, even as insulin doses were reduced. This change in HbA1c level was expected; previous literature has shown a significant link between improving HbA1c control when semaglutide doses are increased to 2 mg weekly.10 Due to having been shown in previous trials, it was expected that HbA1c levels would decrease even when the insulin doses were being reduced.10 Insulin dose reduction can potentially be added to the growing evidence of semaglutide benefits. The change in the number of hypoglycemic events was not statistically significant, which was unexpected since previous research show a trend in patients taking GLP-1RAs having fewer hypoglycemic events than those taking insulin.6 Further investigation with a larger sample size and prospective trial could determine whether this result is an outlier. In this study, there was no increase in HbA1c or hypoglycemic events reported with increasing semaglutide doses, which provides further evidence of the safety of semaglutide even at higher doses.
These data suggest that for a patient with T2DM who is already taking insulin, the recommended titration of semaglutide is to start with 0.5 mg and titrate up to a 2-mg subcutaneous weekly dose and to then continue at that dose. As long as the 2-mg dose is tolerated, it will provide patients with the most HbA1c control and lead to a reduction of their total daily insulin doses according to these results.
Strengths and Limitations
This study compared patient data at different points. This method did not require a second distinct control group, which would potentially introduce confounding factors, such as different baseline characteristics. Another strength is that documentation was available for all patients throughout the study so no one was lost to follow-up. This allowed comprehensive data collection and provided a stronger conclusion given the completeness of the data from baseline to follow-up.
Limitations include the retrospective design and small sample size. In addition, the study design did not allow for randomization. There is no documentation of adherence to medication regimen, which was difficult to determine due to the retrospective nature. Other changes to the patients’ medication regimen were not collected in aggregate and thus, it is possible the total daily insulin dose was impacted by other medication changes. There is also potential for inconsistent documentation of the patients’ true total daily insulin dose in the medical record, thus leading to inaccuracy of recorded data.
Conclusions
A small sample of veterans with T2DM had statistically significant reductions in total daily insulin dose when subcutaneous semaglutide was initiated, as well as after each dose increase. There was also a statistically significant reduction in HbA1c levels from baseline even as patient insulin doses were reduced. These results support the current practice of using semaglutide to treat T2DM, suggesting it may be safe and effective at reducing HbA1c levels as the dose is titrated up to 2 mg. There was no statistically significant change in the number of hypoglycemic events reported as semaglutide was titrated up. Thus, when semaglutide is increased to the maximum recommended dose of 2 mg for T2DM, patients may experience a reduction of their total daily dose of insulin and HbA1c levels. These benefits may reduce the risk of insulin-related AEs while maintaining appropriate glycemic control.
- Diabetes mellitus: in federal health care data trends 2017. Fed Pract. 2017:S20. Accessed August 6, 2025. https://www.fedprac-digital.com/federalpractitioner/data_trends_2017
- Centers for Disease Control and Prevention. National diabetes statistics report. May 15, 2024. Accessed September 17, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- US Department of Veterans Affairs. VA research on diabetes. Updated January 15, 2021. Accessed August 6, 2025. https://www.research.va.gov/topics/diabetes.cfm
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- American Diabetes Association. Standards of care in diabetes— 2023 abridged for primary care providers. Clin Diabetes. 2022;41:4-31. doi:10.2337/cd23-as01
- Zhao Z, Tang Y, Hu Y, Zhu H, Chen X, Zhao B. Hypoglycemia following the use of glucagon-like peptide-1 receptor agonists: a real-world analysis of post-marketing surveillance data. Ann Transl Med. 2021;9:1482. doi:10.21037/atm-21-4162
- Workgroup on Hypoglycemia, American Diabetes Association. Defining and reporting hypoglycemia in diabetes: a report from the American Diabetes Association Workgroup on Hypoglycemia. Diabetes Care. 2005;28:1245-1249. doi:10.2337/diacare.28.5.1245
- Frías JP, Auerbach P, Bajaj HS, et al. Efficacy and safety of once-weekly semaglutide 2.0 mg versus 1.0 mg in patients with type 2 diabetes (SUSTAIN FORTE): a double-blind, randomised, phase 3B trial. Lancet Diabetes Endocrinol. 2021;9:563-574. doi:10.1016/S2213-8587(21)00174-1
- Garber AJ, Handelsman Y, Grunberger G, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2020 executive summary. Endocr Pract. 2020;26:107-139. doi:10.4158/CS-2019-0472
- Miles KE, Kerr JL. Semaglutide for the treatment of type 2 diabetes mellitus. J Pharm Technol. 2018;34:281-289. doi:10.1177/8755122518790925
- Diabetes mellitus: in federal health care data trends 2017. Fed Pract. 2017:S20. Accessed August 6, 2025. https://www.fedprac-digital.com/federalpractitioner/data_trends_2017
- Centers for Disease Control and Prevention. National diabetes statistics report. May 15, 2024. Accessed September 17, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- US Department of Veterans Affairs. VA research on diabetes. Updated January 15, 2021. Accessed August 6, 2025. https://www.research.va.gov/topics/diabetes.cfm
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- American Diabetes Association. Standards of care in diabetes— 2023 abridged for primary care providers. Clin Diabetes. 2022;41:4-31. doi:10.2337/cd23-as01
- Zhao Z, Tang Y, Hu Y, Zhu H, Chen X, Zhao B. Hypoglycemia following the use of glucagon-like peptide-1 receptor agonists: a real-world analysis of post-marketing surveillance data. Ann Transl Med. 2021;9:1482. doi:10.21037/atm-21-4162
- Workgroup on Hypoglycemia, American Diabetes Association. Defining and reporting hypoglycemia in diabetes: a report from the American Diabetes Association Workgroup on Hypoglycemia. Diabetes Care. 2005;28:1245-1249. doi:10.2337/diacare.28.5.1245
- Frías JP, Auerbach P, Bajaj HS, et al. Efficacy and safety of once-weekly semaglutide 2.0 mg versus 1.0 mg in patients with type 2 diabetes (SUSTAIN FORTE): a double-blind, randomised, phase 3B trial. Lancet Diabetes Endocrinol. 2021;9:563-574. doi:10.1016/S2213-8587(21)00174-1
- Garber AJ, Handelsman Y, Grunberger G, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2020 executive summary. Endocr Pract. 2020;26:107-139. doi:10.4158/CS-2019-0472
- Miles KE, Kerr JL. Semaglutide for the treatment of type 2 diabetes mellitus. J Pharm Technol. 2018;34:281-289. doi:10.1177/8755122518790925
Efficacy of Subcutaneous Semaglutide Dose Escalation in Reducing Insulin in Patients With Type 2 Diabetes
Efficacy of Subcutaneous Semaglutide Dose Escalation in Reducing Insulin in Patients With Type 2 Diabetes
Impact of Continuous Glucose Monitoring for American Indian/Alaska Native Adults With Type 2 Diabetes Mellitus Not Using Insulin
Impact of Continuous Glucose Monitoring for American Indian/Alaska Native Adults With Type 2 Diabetes Mellitus Not Using Insulin
Diabetes mellitus (DM) is a national health crisis affecting > 38 million people (11.6%) in the United States.1 American Indian and Alaska Native (AI/AN) adults are disproportionately affected, with a prevalence of 14.5%—the highest among all racial and ethnic groups.1 Type 2 DM (T2DM) accounts for 90% to 95% of all DM cases and is a leading cause of morbidity and mortality due to its association with cardiovascular disease, kidney failure, and other complications.2
Maintaining glycemic control is important for managing T2DM and preventing microvascular and macrovascular complications.3 The cornerstone of diabetes self-management has been patient self-monitored blood glucose (SMBG) using finger-stick glucometers.4 However, SMBG provides measurements from a single point in time and requires frequent, painful, and inconvenient finger pricks, leading to decreased adherence.5,6 These limitations negatively affect patient engagement and overall glycemic control.7
Continuous glucose monitors (CGMs) offer real-time, continuous glucose readings and trends.8 CGMs improve glycemic control and reduce hypoglycemic episodes in patients who are insulin-dependent.9,10 Flash glucose monitors, a type of CGM that requires scanning to obtain glucose readings, provide similar benefits.11 Despite these demonstrated advantages, research has primarily focused on insulin-dependent populations, leaving a significant gap in understanding the effect of CGMs on patients with T2DM who are not insulin-dependent.12
Given the high prevalence of T2DM among AI/AN populations and the potential benefits of CGMs, this study sought to evaluate the effect of CGM use on glycemic control and other health metrics in patients with non–insulin-dependent T2DM in an AI/AN population. This focus addresses a critical knowledge gap and may inform clinical practices and policies to improve diabetes management in this high-risk group.
Methods
A retrospective observational study was conducted using deidentified electronic health records (EHRs) from 2019 to 2024 at a federally operated outpatient Indian Health Service (IHS) clinic serving an AI/AN population in the IHS Portland Area (Oregon, Washington, Idaho). The study protocol was reviewed and deemed exempt by institutional review boards at Washington State University and the Portland Area IHS.
Study Population
This study included patients diagnosed with non–insulin-dependent T2DM, had used a CGM for ≥ 1 year, and had hemoglobin A1c (HbA1c) measurements within 4 months prior to CGM initiation (baseline) and within ± 4 months after 1 year of CGM use. For other health metrics, including blood pressure (BP), weight, low-density lipoprotein cholesterol (LDL-C), and estimated glomerular filtration rate (eGFR), this study required measurements within 6 months before CGM initiation and within 6 months after 1 year of CGM use. The baseline HbA1c in the dataset ranged from 5.3% to > 14%.
Patients were excluded if they used insulin during the study period, had incomplete laboratory or clinical data for the required time frame, or had < 1 year of CGM use. The dataset did not include detailed information on oral DM medications; thus, we could not report or account for the type or number of oral hypoglycemic agents used by the patients. The IHS clinical applications coordinator compiled the dataset from the EHR, identifying patients who were prescribed and received a CGM at the clinic. All patients used the Abbott Freestyle Libre CGM, the only formulary CGM available at the clinic during the study period.
A 1-year follow-up endpoint was selected for several reasons: (1) to capture potential seasonal variations in diet and activity; (2) to align with the clinic’s standard practice of annual comprehensive diabetes evaluations; and (3) to allow sufficient time for patients to adapt to CGM use and reflect any meaningful changes in glycemic control.
All patients received standard DM care according to clinic protocols, which included DM self-management education and training. Patients met with the diabetes educator at least once, during which the educator emphasized making informed decisions using CGM data, such as adjusting dietary choices and physical activity levels to manage blood glucose concentrations effectively.
A total of 302 patients were initially identified. After applying exclusion criteria, 132 were excluded due to insulin use, and 77 were excluded due to incomplete HbA1c data within the specified time frames (Figure 1). The final sample included 93 patients.
Abbreviations: eGFR, estimated glomerular filtration rate; HbA1c, hemoglobin A1c; LDL-C, low-density lipoprotein cholesterol.
Measures
The primary outcome was the change in HbA1c levels from baseline to 1 year after CGM initiation. Secondary outcomes included changes in weight, systolic and diastolic BP, LDL-C concentrations, and eGFR. For the primary outcome, HbA1c values were collected within a grace period of ± 4 months from the baseline and 1-year time points. The laboratory’s upper reporting limit for HbA1c was 14%; values reported as “> 14%” were recorded as 14.1% for data analysis, although the actual values could have been higher.
For secondary outcomes, data were included if measurements were obtained within ± 6 months of the baseline and 1-year time points. Patients who did not have measurements within these time frames for specific metrics were excluded from secondary outcome analysis but remained in the overall study if they met the criteria for HbA1c and CGM use.
Statistical Analysis
Statistical analysis was performed using R statistical software version 4.4.2. Paired t tests were conducted to compare baseline and 1-year follow- up measurements for variables with parametric distributions. Wilcoxon signed-rank test was used for nonparametric data. A linear regression analysis was conducted to examine the relationship between baseline HbA1c levels and the change in HbA1c after 1 year of CGM use. Differences were considered significant at P < .05 set a priori. To guide future research, a posthoc power analysis was performed using Cohen’s d to estimate the required sample sizes for detecting significant effects, assuming a similar population.
Results
The study included 93 patients, with a mean (SD) age of 55 (13) years (range, 29-83 years). Of the participants, 56 were female (60%) and 37 were male (40%). All participants were identified as AI/AN and had non–insulin-dependent T2DM.
Primary Outcomes
A significant reduction in HbA1c levels was observed after 1 year of CGM use. The mean (SD) baseline HbA1c was 9.5% (2.4%), which decreased to 7.6% (2.2%) at 1-year follow-up (Table 1). This difference represents a mean change of -1.86% (2.4%) (95% CI, -2.35 to -1.37; P < .001 [paired t test, -7.53]).
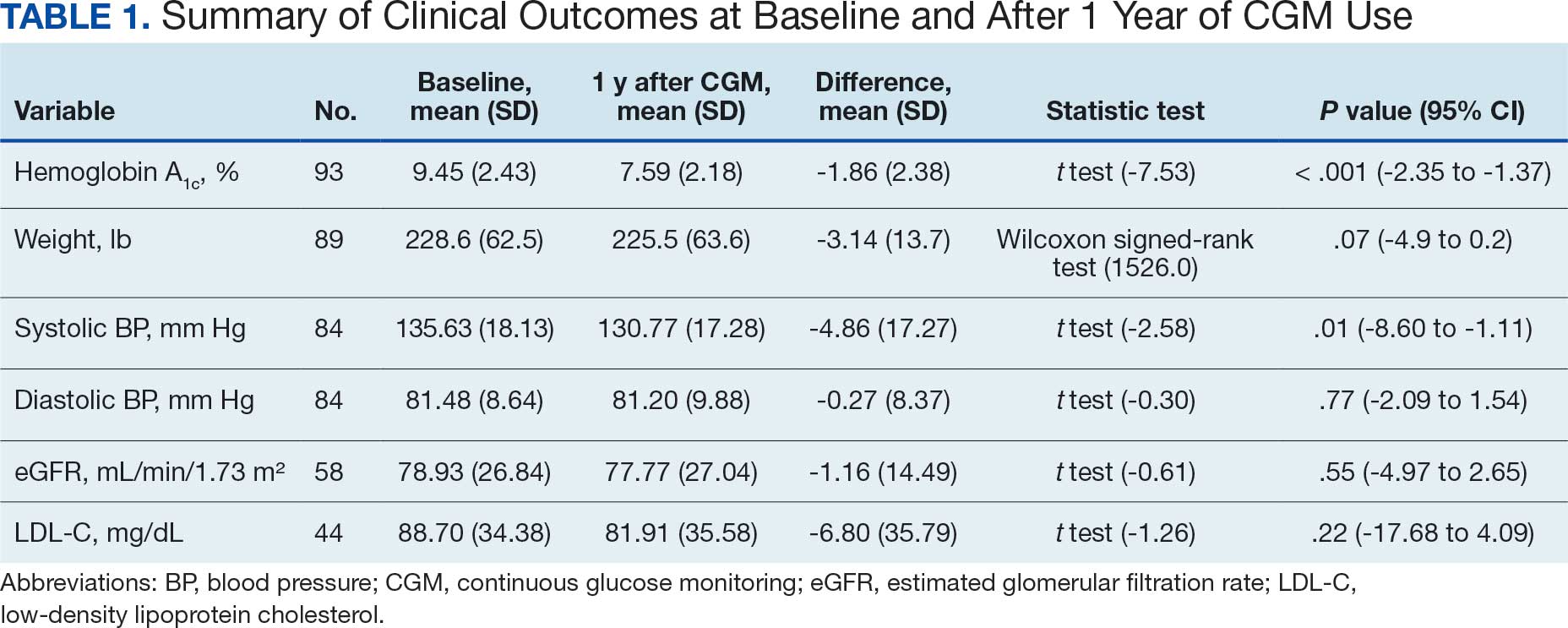
A linear regression model evaluated the relationship between baseline HbA1c (predictor) and the change in HbA1c after 1 year (outcome). The change in HbA1c was calculated as the difference between 1-year follow-up and baseline values. The regression model revealed a significant negative association between baseline HbA1c and the change in HbA1c (Β = -0.576; P < .001), indicating that higher baseline HbA1c values were associated with greater reductions in HbA1c over the year. The regression equation was: Change in HbA1c = 3.587 – 0.576 × Baseline HbA1c
The regression coefficient for baseline HbA1c was -0.576 (standard error, 0.083; t = -6.931; P < .001), indicating that for each 1% increase in baseline HbA1c, the reduction of HbA1c after 1 year increased by approximately 0.576% (Figure 2). The model explained 34.6% of the variance in HbA1c change (R2 = .345; adjusted R2 = .338).
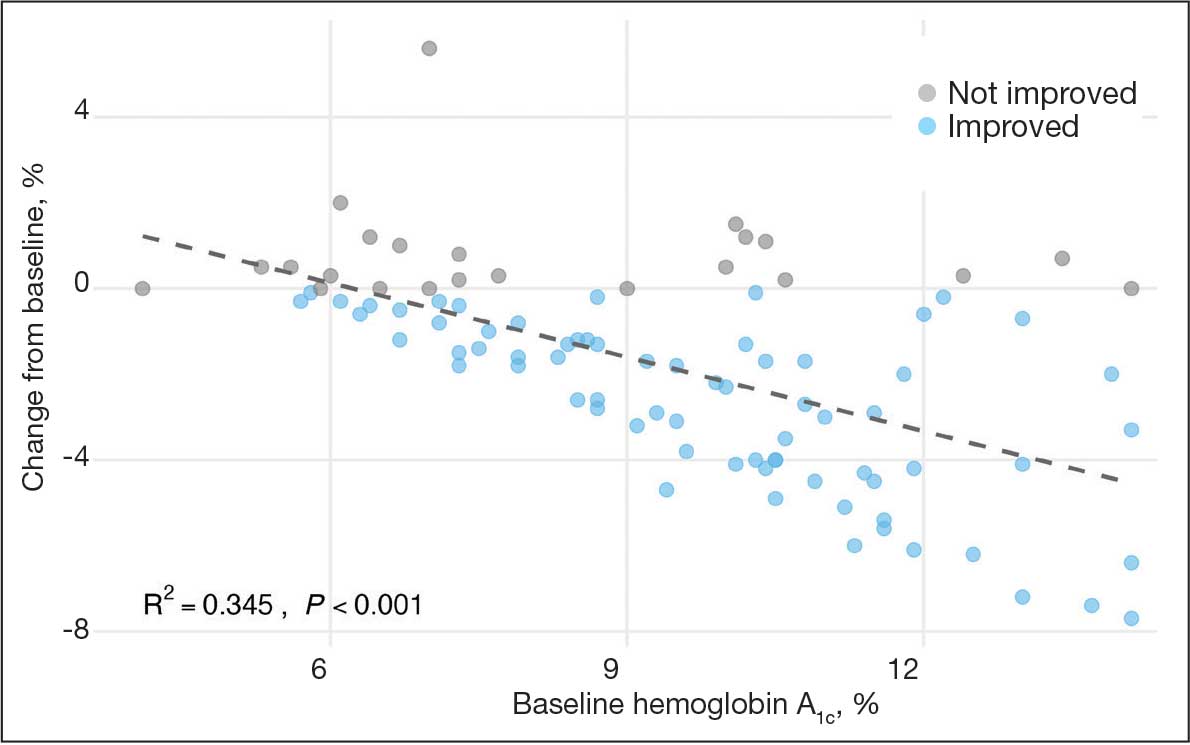
Secondary Outcomes
Systolic BP decreased by a mean (SD) -4.9 (17) mm Hg; 95% CI, -8.6 to -1.11; P = .01, paired t test). However, no significant change was observed for diastolic BP (P = .77, paired t test). Similarly, no significant changes were observed in weight, LDL-C concentrations, or eGFR after 1 year of CGM use. A posthoc power analysis indicated that the study was underpowered to detect smaller effect sizes in secondary outcomes. For example, sample size estimates indicated that detecting significant changes in weight and LDL-C concentrations would require sample sizes of 152 and 220 patients, respectively (Table 2).
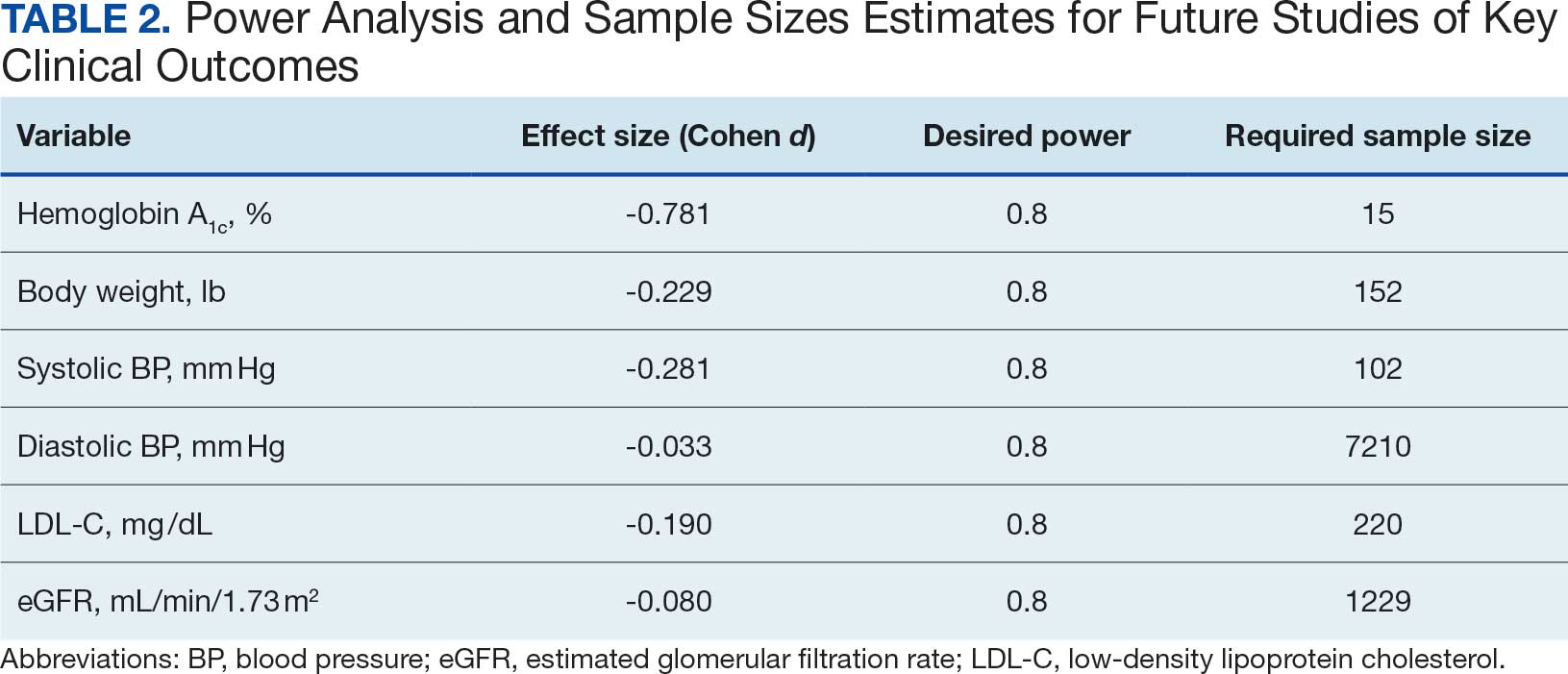
Discussion
This study found a clinically significant reduction in HbA1c levels after 1 year among AI/AN patients with non–insulin-dependent T2DM who used CGMs. The mean HbA1c decreased 1.9%, from 9.5% at baseline to 7.6% after 1 year. This reduction is not only statistically significant (P < .001), it is clinically meaningful—even a 1% decrease in HbA1c is associated with substantial reductions in the risk of microvascular complications.3 The magnitude of the HbA1c reduction observed suggests CGM use may be associated with improved glycemic control in this high-risk population. By achieving lower HbA1c levels, patients may experience improved long-term health outcomes and a reduced burden of DM-related complications.
Changes in oral DM medications during the study period may have contributed to the observed improvements in HbA1c levels. While the dataset lacked detailed information on types or dosages of oral hypoglycemic agents used, adjustments in medication regimens are common in DM management and could significantly affect glycemic control. The inability to account for these changes results in an inability to attribute the improvements in HbA1c solely to CGM use. Future studies should collect comprehensive medication data to better isolate the effects of CGM use from other treatment modifications.
Another factor that may have contributed to the improved glycemic control is the DM self-management education and training patients received as part of standard care. Patients met with diabetes educators at least once and learned how to use the CGM device and interpret the data for self-management decisions. This education may have enhanced patient engagement and empowerment, enabling them to make informed choices about diet, physical activity, and medication adherence. Studies have shown that DM self-management education can significantly improve glycemic control and patient outcomes.13 By combining the CGM technology with targeted education, patients may have been better equipped to manage their condition, contributing to the observed reduction in HbA1c levels. Future studies should consider synergistic effects of CGM use and DM education when evaluating interventions for glycemic control.
The significant reduction in HbA1c indicates CGM use is associated with improved glycemic control in non–insulin-dependent T2DM. The linear regression analysis suggests patients with poorer glycemic control at baseline experienced greater reductions in HbA1c over the course of 1 year. This finding aligns with previous studies that have shown greater HbA1c reductions in patients with higher initial levels when using CGMs. Yaron et al reported similar findings: higher baseline HbA1c levels predicted more substantial improvements with CGM use in patients with T2DM on insulin therapy.14
This study contributes to existing research by examining the association between CGM use and glycemic control in patients with non– insulin-dependent T2DM within an AI/AN population, a group that has been underreported in previous studies. Most prior research has focused on insulin-dependent patients or populations with different ethnic backgrounds.12 By focusing on patients with non–insulin-dependent T2DM, this study highlights the broader applicability of CGMs beyond traditional use, showcasing their potential association with benefits in earlier stages of DM management. Targeting the AI/AN population addresses a critical knowledge gap, given the disproportionately high prevalence of T2DM and associated complications in this group. The findings of this study suggest integrating CGM technology into the standard care of AI/AN patients with non–insulin-dependent T2DM may be associated with improved glycemic control and may help reduce health disparities.
The modest decrease in systolic BP observed in this study may indicate potential cardiovascular benefits associated with CGM use, possibly due to improved glycemic control and increased patient engagement in self-management. However, given the limited sample size and exclusion criteria, the study lacked sufficient power to detect significant associations between CGM use and other secondary outcomes such as BP, weight, LDL-C, and eGFR. Therefore, the significant finding with systolic BP should be interpreted with caution.
The lack of significant changes in secondary outcomes may be attributed to the study’s limited sample size and the relatively short duration for observing changes in these parameters. Larger studies are needed to assess the full impact of CGM on these variables. The required sample sizes for achieving adequate power in future studies were calculated, highlighting the utility of our study as a pilot, providing critical data for the design of larger, adequately powered studies.
Limitations
The retrospective design of this study limits causal inferences. Moreover, potential confounding variables were not controlled, such as changes in medication regimens (other than insulin use), dietary counseling, or physical activity. Additionally, we could not account for the type or number of oral DM medications prescribed to patients. The dataset included only information on insulin use, without detailed records of other antidiabetic medications. This limitation may have influenced the observed change in glycemic control, as variations in medication regimens could affect HbA1c levels.
Because this study lacked a comparator group, the effect of CGM use cannot be definitively isolated from other factors (eg, medication changes, dietary modifications, or physical activity). Moreover, CGM devices can be costly and are not universally covered by all insurance or IHS programs, potentially limiting widespread implementation. Policy-level restrictions and patient-specific barriers may also hinder feasibility in other settings.
The small sample size may limit the generalizability of the findings. Of the initial 302 patients, about 69% were excluded due to insulin use or incomplete laboratory data. A ± 4-month window was selected to balance data quality with real-world practices. Extending this window further (eg, ± 6 months) might have included more participants but risked diluting the 1-year endpoint consistency. The lack of statistical significance in secondary metrics may be due to insufficient power rather than the absence of an effect.
Exclusion of patients due to incomplete data may have introduced selection bias. However, patients were included in the overall analysis if they met the criteria for HbA1c and CGM use, even if they lacked data for secondary outcomes. Additionally, the laboratory’s upper reporting limit for HbA1c was 14%, with values above this reported as “> 14%.” For analysis, these were recorded as 14.1%, which may underestimate the true baseline HbA1c levels and impact of the assessment of change. This occurred for 4 of the 93 patients included.
All patients used the Freestyle Libre CGM, which may limit the generalizability of the findings to other CGM brands or models. Differences in device features, accuracy, scanning frequency, and user experience may influence outcomes, and results might differ with other CGM technologies. The dataset did not include patients’ scanning frequency because this metric was not consistently included in the EHRs.
Conclusions
This study found that CGM use was significantly associated with improved glycemic control in patients with non–insulin-dependent T2DM within an AI/AN population, particularly among patients with higher baseline HbA1c levels. The findings suggest that CGMs may be a valuable tool for managing T2DM beyond insulin-dependent populations.
Additional research with larger sample sizes, control groups, and extended follow-up periods is recommended to explore long-term benefits and impacts on other health metrics. The sample size estimates derived from this study serve as a valuable resource for researchers designing future studies aimed at addressing these gaps. Future research that expands on our findings by including larger, more diverse cohorts, accounting for medication use, and exploring different CGM technologies will enhance understanding and contribute to more effective diabetes management strategies for varied populations.
- National diabetes statistics report. Centers for Disease Control and Prevention. May 15, 2024. Accessed October 7, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- Elsayed NA, Aleppo G, Aroda VR, et al. 2. Classification and diagnosis of diabetes: standards of care in diabetes—2023. Diabetes Care. 2023;46:S19-S40. doi:10.2337/dc23-S002
- Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diabetes. 2011;29:116-122. doi:10.2337/diaclin.29.3.116
- Pleus S, Freckmann G, Schauer S, et al. Self-monitoring of blood glucose as an integral part in the management of people with type 2 diabetes mellitus. Diabetes Ther. 2022;13:829-846. doi:10.1007/s13300-022-01254-8
- Polonsky WH, Fisher L, Schikman CH, et al. Structured self-monitoring of blood glucose significantly reduces A1C levels in poorly controlled, noninsulin-treated type 2 diabetes: results from the Structured Testing Program study. Diabetes Care. 2011;34:262-267. doi:10.2337/dc10-1732
- Tanaka N, Yabe D, Murotani K, et al. Mental distress and health-related quality of life among type 1 and type 2 diabetes patients using self-monitoring of blood glucose: a cross-sectional questionnaire study in Japan. J Diabetes Investig. 2018;9:1203-1211. doi:10.1111/jdi.12827
- Hortensius J, Kars MC, Wierenga WS, et al. Perspectives of patients with type 1 or insulin-treated type 2 diabetes on self-monitoring of blood glucose: a qualitative study. BMC Public Health. 2012;12:167. doi:10.1186/1471-2458-12-167
- Didyuk O, Econom N, Guardia A, Livingston K, Klueh U. Continuous glucose monitoring devices: past, present, and future focus on the history and evolution of technological innovation. J Diabetes Sci Technol. 2021;15:676-683. doi:10.1177/1932296819899394
- Beck RW, Riddlesworth TD, Ruedy K, et al. Effect of continuous glucose monitoring on glycemic control in adults with type 1 diabetes using insulin injections: the DIAMOND randomized clinical trial. JAMA. 2017;317:371-378. doi:10.1001/jama.2016.19975
- Lind M, Polonsky W, Hirsch IB, et al. Continuous glucose monitoring vs conventional therapy for glycemic control in adults with type 1 diabetes treated with multiple daily insulin injections: the GOLD randomized clinical trial. JAMA. 2017;317:379-387. doi:10.1001/jama.2016.19976
- Bolinder J, Antuna R, Geelhoed-Duijvestijn P, et al. Novel glucose-sensing technology and hypoglycemia in type 1 diabetes: a multicenter, non-masked, randomized controlled trial. Lancet. 2016;388:2254-2263. doi:10.1016/S0140-6736(16)31535-5
- Seidu S, Kunutsor SK, Ajjan RA, et al. Efficacy and safety of continuous glucose monitoring and intermittently scanned continuous glucose monitoring in patients with type 2 diabetes: a systematic review and meta-analysis of interventional evidence. Diabetes Care. 2024;47:169-179. doi:10.2337/dc23-1520
- ElSayed NA, Aleppo G, Aroda VR, et al. 5. Facilitating positive health behaviors and well-being to improve health outcomes: standards of care in diabetes-2023. Diabetes Care. 2023;46:S68-S96. doi:10.2337/dc23-S005
- Yaron M, Roitman E, Aharon-Hananel G, et al. Effect of flash glucose monitoring technology on glycemic control and treatment satisfaction in patients with type 2 diabetes. Diabetes Care. 2019;42:1178-1184. doi:10.2337/dc18-0166
Diabetes mellitus (DM) is a national health crisis affecting > 38 million people (11.6%) in the United States.1 American Indian and Alaska Native (AI/AN) adults are disproportionately affected, with a prevalence of 14.5%—the highest among all racial and ethnic groups.1 Type 2 DM (T2DM) accounts for 90% to 95% of all DM cases and is a leading cause of morbidity and mortality due to its association with cardiovascular disease, kidney failure, and other complications.2
Maintaining glycemic control is important for managing T2DM and preventing microvascular and macrovascular complications.3 The cornerstone of diabetes self-management has been patient self-monitored blood glucose (SMBG) using finger-stick glucometers.4 However, SMBG provides measurements from a single point in time and requires frequent, painful, and inconvenient finger pricks, leading to decreased adherence.5,6 These limitations negatively affect patient engagement and overall glycemic control.7
Continuous glucose monitors (CGMs) offer real-time, continuous glucose readings and trends.8 CGMs improve glycemic control and reduce hypoglycemic episodes in patients who are insulin-dependent.9,10 Flash glucose monitors, a type of CGM that requires scanning to obtain glucose readings, provide similar benefits.11 Despite these demonstrated advantages, research has primarily focused on insulin-dependent populations, leaving a significant gap in understanding the effect of CGMs on patients with T2DM who are not insulin-dependent.12
Given the high prevalence of T2DM among AI/AN populations and the potential benefits of CGMs, this study sought to evaluate the effect of CGM use on glycemic control and other health metrics in patients with non–insulin-dependent T2DM in an AI/AN population. This focus addresses a critical knowledge gap and may inform clinical practices and policies to improve diabetes management in this high-risk group.
Methods
A retrospective observational study was conducted using deidentified electronic health records (EHRs) from 2019 to 2024 at a federally operated outpatient Indian Health Service (IHS) clinic serving an AI/AN population in the IHS Portland Area (Oregon, Washington, Idaho). The study protocol was reviewed and deemed exempt by institutional review boards at Washington State University and the Portland Area IHS.
Study Population
This study included patients diagnosed with non–insulin-dependent T2DM, had used a CGM for ≥ 1 year, and had hemoglobin A1c (HbA1c) measurements within 4 months prior to CGM initiation (baseline) and within ± 4 months after 1 year of CGM use. For other health metrics, including blood pressure (BP), weight, low-density lipoprotein cholesterol (LDL-C), and estimated glomerular filtration rate (eGFR), this study required measurements within 6 months before CGM initiation and within 6 months after 1 year of CGM use. The baseline HbA1c in the dataset ranged from 5.3% to > 14%.
Patients were excluded if they used insulin during the study period, had incomplete laboratory or clinical data for the required time frame, or had < 1 year of CGM use. The dataset did not include detailed information on oral DM medications; thus, we could not report or account for the type or number of oral hypoglycemic agents used by the patients. The IHS clinical applications coordinator compiled the dataset from the EHR, identifying patients who were prescribed and received a CGM at the clinic. All patients used the Abbott Freestyle Libre CGM, the only formulary CGM available at the clinic during the study period.
A 1-year follow-up endpoint was selected for several reasons: (1) to capture potential seasonal variations in diet and activity; (2) to align with the clinic’s standard practice of annual comprehensive diabetes evaluations; and (3) to allow sufficient time for patients to adapt to CGM use and reflect any meaningful changes in glycemic control.
All patients received standard DM care according to clinic protocols, which included DM self-management education and training. Patients met with the diabetes educator at least once, during which the educator emphasized making informed decisions using CGM data, such as adjusting dietary choices and physical activity levels to manage blood glucose concentrations effectively.
A total of 302 patients were initially identified. After applying exclusion criteria, 132 were excluded due to insulin use, and 77 were excluded due to incomplete HbA1c data within the specified time frames (Figure 1). The final sample included 93 patients.
Abbreviations: eGFR, estimated glomerular filtration rate; HbA1c, hemoglobin A1c; LDL-C, low-density lipoprotein cholesterol.
Measures
The primary outcome was the change in HbA1c levels from baseline to 1 year after CGM initiation. Secondary outcomes included changes in weight, systolic and diastolic BP, LDL-C concentrations, and eGFR. For the primary outcome, HbA1c values were collected within a grace period of ± 4 months from the baseline and 1-year time points. The laboratory’s upper reporting limit for HbA1c was 14%; values reported as “> 14%” were recorded as 14.1% for data analysis, although the actual values could have been higher.
For secondary outcomes, data were included if measurements were obtained within ± 6 months of the baseline and 1-year time points. Patients who did not have measurements within these time frames for specific metrics were excluded from secondary outcome analysis but remained in the overall study if they met the criteria for HbA1c and CGM use.
Statistical Analysis
Statistical analysis was performed using R statistical software version 4.4.2. Paired t tests were conducted to compare baseline and 1-year follow- up measurements for variables with parametric distributions. Wilcoxon signed-rank test was used for nonparametric data. A linear regression analysis was conducted to examine the relationship between baseline HbA1c levels and the change in HbA1c after 1 year of CGM use. Differences were considered significant at P < .05 set a priori. To guide future research, a posthoc power analysis was performed using Cohen’s d to estimate the required sample sizes for detecting significant effects, assuming a similar population.
Results
The study included 93 patients, with a mean (SD) age of 55 (13) years (range, 29-83 years). Of the participants, 56 were female (60%) and 37 were male (40%). All participants were identified as AI/AN and had non–insulin-dependent T2DM.
Primary Outcomes
A significant reduction in HbA1c levels was observed after 1 year of CGM use. The mean (SD) baseline HbA1c was 9.5% (2.4%), which decreased to 7.6% (2.2%) at 1-year follow-up (Table 1). This difference represents a mean change of -1.86% (2.4%) (95% CI, -2.35 to -1.37; P < .001 [paired t test, -7.53]).
A linear regression model evaluated the relationship between baseline HbA1c (predictor) and the change in HbA1c after 1 year (outcome). The change in HbA1c was calculated as the difference between 1-year follow-up and baseline values. The regression model revealed a significant negative association between baseline HbA1c and the change in HbA1c (Β = -0.576; P < .001), indicating that higher baseline HbA1c values were associated with greater reductions in HbA1c over the year. The regression equation was: Change in HbA1c = 3.587 – 0.576 × Baseline HbA1c
The regression coefficient for baseline HbA1c was -0.576 (standard error, 0.083; t = -6.931; P < .001), indicating that for each 1% increase in baseline HbA1c, the reduction of HbA1c after 1 year increased by approximately 0.576% (Figure 2). The model explained 34.6% of the variance in HbA1c change (R2 = .345; adjusted R2 = .338).
Secondary Outcomes
Systolic BP decreased by a mean (SD) -4.9 (17) mm Hg; 95% CI, -8.6 to -1.11; P = .01, paired t test). However, no significant change was observed for diastolic BP (P = .77, paired t test). Similarly, no significant changes were observed in weight, LDL-C concentrations, or eGFR after 1 year of CGM use. A posthoc power analysis indicated that the study was underpowered to detect smaller effect sizes in secondary outcomes. For example, sample size estimates indicated that detecting significant changes in weight and LDL-C concentrations would require sample sizes of 152 and 220 patients, respectively (Table 2).
Discussion
This study found a clinically significant reduction in HbA1c levels after 1 year among AI/AN patients with non–insulin-dependent T2DM who used CGMs. The mean HbA1c decreased 1.9%, from 9.5% at baseline to 7.6% after 1 year. This reduction is not only statistically significant (P < .001), it is clinically meaningful—even a 1% decrease in HbA1c is associated with substantial reductions in the risk of microvascular complications.3 The magnitude of the HbA1c reduction observed suggests CGM use may be associated with improved glycemic control in this high-risk population. By achieving lower HbA1c levels, patients may experience improved long-term health outcomes and a reduced burden of DM-related complications.
Changes in oral DM medications during the study period may have contributed to the observed improvements in HbA1c levels. While the dataset lacked detailed information on types or dosages of oral hypoglycemic agents used, adjustments in medication regimens are common in DM management and could significantly affect glycemic control. The inability to account for these changes results in an inability to attribute the improvements in HbA1c solely to CGM use. Future studies should collect comprehensive medication data to better isolate the effects of CGM use from other treatment modifications.
Another factor that may have contributed to the improved glycemic control is the DM self-management education and training patients received as part of standard care. Patients met with diabetes educators at least once and learned how to use the CGM device and interpret the data for self-management decisions. This education may have enhanced patient engagement and empowerment, enabling them to make informed choices about diet, physical activity, and medication adherence. Studies have shown that DM self-management education can significantly improve glycemic control and patient outcomes.13 By combining the CGM technology with targeted education, patients may have been better equipped to manage their condition, contributing to the observed reduction in HbA1c levels. Future studies should consider synergistic effects of CGM use and DM education when evaluating interventions for glycemic control.
The significant reduction in HbA1c indicates CGM use is associated with improved glycemic control in non–insulin-dependent T2DM. The linear regression analysis suggests patients with poorer glycemic control at baseline experienced greater reductions in HbA1c over the course of 1 year. This finding aligns with previous studies that have shown greater HbA1c reductions in patients with higher initial levels when using CGMs. Yaron et al reported similar findings: higher baseline HbA1c levels predicted more substantial improvements with CGM use in patients with T2DM on insulin therapy.14
This study contributes to existing research by examining the association between CGM use and glycemic control in patients with non– insulin-dependent T2DM within an AI/AN population, a group that has been underreported in previous studies. Most prior research has focused on insulin-dependent patients or populations with different ethnic backgrounds.12 By focusing on patients with non–insulin-dependent T2DM, this study highlights the broader applicability of CGMs beyond traditional use, showcasing their potential association with benefits in earlier stages of DM management. Targeting the AI/AN population addresses a critical knowledge gap, given the disproportionately high prevalence of T2DM and associated complications in this group. The findings of this study suggest integrating CGM technology into the standard care of AI/AN patients with non–insulin-dependent T2DM may be associated with improved glycemic control and may help reduce health disparities.
The modest decrease in systolic BP observed in this study may indicate potential cardiovascular benefits associated with CGM use, possibly due to improved glycemic control and increased patient engagement in self-management. However, given the limited sample size and exclusion criteria, the study lacked sufficient power to detect significant associations between CGM use and other secondary outcomes such as BP, weight, LDL-C, and eGFR. Therefore, the significant finding with systolic BP should be interpreted with caution.
The lack of significant changes in secondary outcomes may be attributed to the study’s limited sample size and the relatively short duration for observing changes in these parameters. Larger studies are needed to assess the full impact of CGM on these variables. The required sample sizes for achieving adequate power in future studies were calculated, highlighting the utility of our study as a pilot, providing critical data for the design of larger, adequately powered studies.
Limitations
The retrospective design of this study limits causal inferences. Moreover, potential confounding variables were not controlled, such as changes in medication regimens (other than insulin use), dietary counseling, or physical activity. Additionally, we could not account for the type or number of oral DM medications prescribed to patients. The dataset included only information on insulin use, without detailed records of other antidiabetic medications. This limitation may have influenced the observed change in glycemic control, as variations in medication regimens could affect HbA1c levels.
Because this study lacked a comparator group, the effect of CGM use cannot be definitively isolated from other factors (eg, medication changes, dietary modifications, or physical activity). Moreover, CGM devices can be costly and are not universally covered by all insurance or IHS programs, potentially limiting widespread implementation. Policy-level restrictions and patient-specific barriers may also hinder feasibility in other settings.
The small sample size may limit the generalizability of the findings. Of the initial 302 patients, about 69% were excluded due to insulin use or incomplete laboratory data. A ± 4-month window was selected to balance data quality with real-world practices. Extending this window further (eg, ± 6 months) might have included more participants but risked diluting the 1-year endpoint consistency. The lack of statistical significance in secondary metrics may be due to insufficient power rather than the absence of an effect.
Exclusion of patients due to incomplete data may have introduced selection bias. However, patients were included in the overall analysis if they met the criteria for HbA1c and CGM use, even if they lacked data for secondary outcomes. Additionally, the laboratory’s upper reporting limit for HbA1c was 14%, with values above this reported as “> 14%.” For analysis, these were recorded as 14.1%, which may underestimate the true baseline HbA1c levels and impact of the assessment of change. This occurred for 4 of the 93 patients included.
All patients used the Freestyle Libre CGM, which may limit the generalizability of the findings to other CGM brands or models. Differences in device features, accuracy, scanning frequency, and user experience may influence outcomes, and results might differ with other CGM technologies. The dataset did not include patients’ scanning frequency because this metric was not consistently included in the EHRs.
Conclusions
This study found that CGM use was significantly associated with improved glycemic control in patients with non–insulin-dependent T2DM within an AI/AN population, particularly among patients with higher baseline HbA1c levels. The findings suggest that CGMs may be a valuable tool for managing T2DM beyond insulin-dependent populations.
Additional research with larger sample sizes, control groups, and extended follow-up periods is recommended to explore long-term benefits and impacts on other health metrics. The sample size estimates derived from this study serve as a valuable resource for researchers designing future studies aimed at addressing these gaps. Future research that expands on our findings by including larger, more diverse cohorts, accounting for medication use, and exploring different CGM technologies will enhance understanding and contribute to more effective diabetes management strategies for varied populations.
Diabetes mellitus (DM) is a national health crisis affecting > 38 million people (11.6%) in the United States.1 American Indian and Alaska Native (AI/AN) adults are disproportionately affected, with a prevalence of 14.5%—the highest among all racial and ethnic groups.1 Type 2 DM (T2DM) accounts for 90% to 95% of all DM cases and is a leading cause of morbidity and mortality due to its association with cardiovascular disease, kidney failure, and other complications.2
Maintaining glycemic control is important for managing T2DM and preventing microvascular and macrovascular complications.3 The cornerstone of diabetes self-management has been patient self-monitored blood glucose (SMBG) using finger-stick glucometers.4 However, SMBG provides measurements from a single point in time and requires frequent, painful, and inconvenient finger pricks, leading to decreased adherence.5,6 These limitations negatively affect patient engagement and overall glycemic control.7
Continuous glucose monitors (CGMs) offer real-time, continuous glucose readings and trends.8 CGMs improve glycemic control and reduce hypoglycemic episodes in patients who are insulin-dependent.9,10 Flash glucose monitors, a type of CGM that requires scanning to obtain glucose readings, provide similar benefits.11 Despite these demonstrated advantages, research has primarily focused on insulin-dependent populations, leaving a significant gap in understanding the effect of CGMs on patients with T2DM who are not insulin-dependent.12
Given the high prevalence of T2DM among AI/AN populations and the potential benefits of CGMs, this study sought to evaluate the effect of CGM use on glycemic control and other health metrics in patients with non–insulin-dependent T2DM in an AI/AN population. This focus addresses a critical knowledge gap and may inform clinical practices and policies to improve diabetes management in this high-risk group.
Methods
A retrospective observational study was conducted using deidentified electronic health records (EHRs) from 2019 to 2024 at a federally operated outpatient Indian Health Service (IHS) clinic serving an AI/AN population in the IHS Portland Area (Oregon, Washington, Idaho). The study protocol was reviewed and deemed exempt by institutional review boards at Washington State University and the Portland Area IHS.
Study Population
This study included patients diagnosed with non–insulin-dependent T2DM, had used a CGM for ≥ 1 year, and had hemoglobin A1c (HbA1c) measurements within 4 months prior to CGM initiation (baseline) and within ± 4 months after 1 year of CGM use. For other health metrics, including blood pressure (BP), weight, low-density lipoprotein cholesterol (LDL-C), and estimated glomerular filtration rate (eGFR), this study required measurements within 6 months before CGM initiation and within 6 months after 1 year of CGM use. The baseline HbA1c in the dataset ranged from 5.3% to > 14%.
Patients were excluded if they used insulin during the study period, had incomplete laboratory or clinical data for the required time frame, or had < 1 year of CGM use. The dataset did not include detailed information on oral DM medications; thus, we could not report or account for the type or number of oral hypoglycemic agents used by the patients. The IHS clinical applications coordinator compiled the dataset from the EHR, identifying patients who were prescribed and received a CGM at the clinic. All patients used the Abbott Freestyle Libre CGM, the only formulary CGM available at the clinic during the study period.
A 1-year follow-up endpoint was selected for several reasons: (1) to capture potential seasonal variations in diet and activity; (2) to align with the clinic’s standard practice of annual comprehensive diabetes evaluations; and (3) to allow sufficient time for patients to adapt to CGM use and reflect any meaningful changes in glycemic control.
All patients received standard DM care according to clinic protocols, which included DM self-management education and training. Patients met with the diabetes educator at least once, during which the educator emphasized making informed decisions using CGM data, such as adjusting dietary choices and physical activity levels to manage blood glucose concentrations effectively.
A total of 302 patients were initially identified. After applying exclusion criteria, 132 were excluded due to insulin use, and 77 were excluded due to incomplete HbA1c data within the specified time frames (Figure 1). The final sample included 93 patients.
Abbreviations: eGFR, estimated glomerular filtration rate; HbA1c, hemoglobin A1c; LDL-C, low-density lipoprotein cholesterol.
Measures
The primary outcome was the change in HbA1c levels from baseline to 1 year after CGM initiation. Secondary outcomes included changes in weight, systolic and diastolic BP, LDL-C concentrations, and eGFR. For the primary outcome, HbA1c values were collected within a grace period of ± 4 months from the baseline and 1-year time points. The laboratory’s upper reporting limit for HbA1c was 14%; values reported as “> 14%” were recorded as 14.1% for data analysis, although the actual values could have been higher.
For secondary outcomes, data were included if measurements were obtained within ± 6 months of the baseline and 1-year time points. Patients who did not have measurements within these time frames for specific metrics were excluded from secondary outcome analysis but remained in the overall study if they met the criteria for HbA1c and CGM use.
Statistical Analysis
Statistical analysis was performed using R statistical software version 4.4.2. Paired t tests were conducted to compare baseline and 1-year follow- up measurements for variables with parametric distributions. Wilcoxon signed-rank test was used for nonparametric data. A linear regression analysis was conducted to examine the relationship between baseline HbA1c levels and the change in HbA1c after 1 year of CGM use. Differences were considered significant at P < .05 set a priori. To guide future research, a posthoc power analysis was performed using Cohen’s d to estimate the required sample sizes for detecting significant effects, assuming a similar population.
Results
The study included 93 patients, with a mean (SD) age of 55 (13) years (range, 29-83 years). Of the participants, 56 were female (60%) and 37 were male (40%). All participants were identified as AI/AN and had non–insulin-dependent T2DM.
Primary Outcomes
A significant reduction in HbA1c levels was observed after 1 year of CGM use. The mean (SD) baseline HbA1c was 9.5% (2.4%), which decreased to 7.6% (2.2%) at 1-year follow-up (Table 1). This difference represents a mean change of -1.86% (2.4%) (95% CI, -2.35 to -1.37; P < .001 [paired t test, -7.53]).
A linear regression model evaluated the relationship between baseline HbA1c (predictor) and the change in HbA1c after 1 year (outcome). The change in HbA1c was calculated as the difference between 1-year follow-up and baseline values. The regression model revealed a significant negative association between baseline HbA1c and the change in HbA1c (Β = -0.576; P < .001), indicating that higher baseline HbA1c values were associated with greater reductions in HbA1c over the year. The regression equation was: Change in HbA1c = 3.587 – 0.576 × Baseline HbA1c
The regression coefficient for baseline HbA1c was -0.576 (standard error, 0.083; t = -6.931; P < .001), indicating that for each 1% increase in baseline HbA1c, the reduction of HbA1c after 1 year increased by approximately 0.576% (Figure 2). The model explained 34.6% of the variance in HbA1c change (R2 = .345; adjusted R2 = .338).
Secondary Outcomes
Systolic BP decreased by a mean (SD) -4.9 (17) mm Hg; 95% CI, -8.6 to -1.11; P = .01, paired t test). However, no significant change was observed for diastolic BP (P = .77, paired t test). Similarly, no significant changes were observed in weight, LDL-C concentrations, or eGFR after 1 year of CGM use. A posthoc power analysis indicated that the study was underpowered to detect smaller effect sizes in secondary outcomes. For example, sample size estimates indicated that detecting significant changes in weight and LDL-C concentrations would require sample sizes of 152 and 220 patients, respectively (Table 2).
Discussion
This study found a clinically significant reduction in HbA1c levels after 1 year among AI/AN patients with non–insulin-dependent T2DM who used CGMs. The mean HbA1c decreased 1.9%, from 9.5% at baseline to 7.6% after 1 year. This reduction is not only statistically significant (P < .001), it is clinically meaningful—even a 1% decrease in HbA1c is associated with substantial reductions in the risk of microvascular complications.3 The magnitude of the HbA1c reduction observed suggests CGM use may be associated with improved glycemic control in this high-risk population. By achieving lower HbA1c levels, patients may experience improved long-term health outcomes and a reduced burden of DM-related complications.
Changes in oral DM medications during the study period may have contributed to the observed improvements in HbA1c levels. While the dataset lacked detailed information on types or dosages of oral hypoglycemic agents used, adjustments in medication regimens are common in DM management and could significantly affect glycemic control. The inability to account for these changes results in an inability to attribute the improvements in HbA1c solely to CGM use. Future studies should collect comprehensive medication data to better isolate the effects of CGM use from other treatment modifications.
Another factor that may have contributed to the improved glycemic control is the DM self-management education and training patients received as part of standard care. Patients met with diabetes educators at least once and learned how to use the CGM device and interpret the data for self-management decisions. This education may have enhanced patient engagement and empowerment, enabling them to make informed choices about diet, physical activity, and medication adherence. Studies have shown that DM self-management education can significantly improve glycemic control and patient outcomes.13 By combining the CGM technology with targeted education, patients may have been better equipped to manage their condition, contributing to the observed reduction in HbA1c levels. Future studies should consider synergistic effects of CGM use and DM education when evaluating interventions for glycemic control.
The significant reduction in HbA1c indicates CGM use is associated with improved glycemic control in non–insulin-dependent T2DM. The linear regression analysis suggests patients with poorer glycemic control at baseline experienced greater reductions in HbA1c over the course of 1 year. This finding aligns with previous studies that have shown greater HbA1c reductions in patients with higher initial levels when using CGMs. Yaron et al reported similar findings: higher baseline HbA1c levels predicted more substantial improvements with CGM use in patients with T2DM on insulin therapy.14
This study contributes to existing research by examining the association between CGM use and glycemic control in patients with non– insulin-dependent T2DM within an AI/AN population, a group that has been underreported in previous studies. Most prior research has focused on insulin-dependent patients or populations with different ethnic backgrounds.12 By focusing on patients with non–insulin-dependent T2DM, this study highlights the broader applicability of CGMs beyond traditional use, showcasing their potential association with benefits in earlier stages of DM management. Targeting the AI/AN population addresses a critical knowledge gap, given the disproportionately high prevalence of T2DM and associated complications in this group. The findings of this study suggest integrating CGM technology into the standard care of AI/AN patients with non–insulin-dependent T2DM may be associated with improved glycemic control and may help reduce health disparities.
The modest decrease in systolic BP observed in this study may indicate potential cardiovascular benefits associated with CGM use, possibly due to improved glycemic control and increased patient engagement in self-management. However, given the limited sample size and exclusion criteria, the study lacked sufficient power to detect significant associations between CGM use and other secondary outcomes such as BP, weight, LDL-C, and eGFR. Therefore, the significant finding with systolic BP should be interpreted with caution.
The lack of significant changes in secondary outcomes may be attributed to the study’s limited sample size and the relatively short duration for observing changes in these parameters. Larger studies are needed to assess the full impact of CGM on these variables. The required sample sizes for achieving adequate power in future studies were calculated, highlighting the utility of our study as a pilot, providing critical data for the design of larger, adequately powered studies.
Limitations
The retrospective design of this study limits causal inferences. Moreover, potential confounding variables were not controlled, such as changes in medication regimens (other than insulin use), dietary counseling, or physical activity. Additionally, we could not account for the type or number of oral DM medications prescribed to patients. The dataset included only information on insulin use, without detailed records of other antidiabetic medications. This limitation may have influenced the observed change in glycemic control, as variations in medication regimens could affect HbA1c levels.
Because this study lacked a comparator group, the effect of CGM use cannot be definitively isolated from other factors (eg, medication changes, dietary modifications, or physical activity). Moreover, CGM devices can be costly and are not universally covered by all insurance or IHS programs, potentially limiting widespread implementation. Policy-level restrictions and patient-specific barriers may also hinder feasibility in other settings.
The small sample size may limit the generalizability of the findings. Of the initial 302 patients, about 69% were excluded due to insulin use or incomplete laboratory data. A ± 4-month window was selected to balance data quality with real-world practices. Extending this window further (eg, ± 6 months) might have included more participants but risked diluting the 1-year endpoint consistency. The lack of statistical significance in secondary metrics may be due to insufficient power rather than the absence of an effect.
Exclusion of patients due to incomplete data may have introduced selection bias. However, patients were included in the overall analysis if they met the criteria for HbA1c and CGM use, even if they lacked data for secondary outcomes. Additionally, the laboratory’s upper reporting limit for HbA1c was 14%, with values above this reported as “> 14%.” For analysis, these were recorded as 14.1%, which may underestimate the true baseline HbA1c levels and impact of the assessment of change. This occurred for 4 of the 93 patients included.
All patients used the Freestyle Libre CGM, which may limit the generalizability of the findings to other CGM brands or models. Differences in device features, accuracy, scanning frequency, and user experience may influence outcomes, and results might differ with other CGM technologies. The dataset did not include patients’ scanning frequency because this metric was not consistently included in the EHRs.
Conclusions
This study found that CGM use was significantly associated with improved glycemic control in patients with non–insulin-dependent T2DM within an AI/AN population, particularly among patients with higher baseline HbA1c levels. The findings suggest that CGMs may be a valuable tool for managing T2DM beyond insulin-dependent populations.
Additional research with larger sample sizes, control groups, and extended follow-up periods is recommended to explore long-term benefits and impacts on other health metrics. The sample size estimates derived from this study serve as a valuable resource for researchers designing future studies aimed at addressing these gaps. Future research that expands on our findings by including larger, more diverse cohorts, accounting for medication use, and exploring different CGM technologies will enhance understanding and contribute to more effective diabetes management strategies for varied populations.
- National diabetes statistics report. Centers for Disease Control and Prevention. May 15, 2024. Accessed October 7, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- Elsayed NA, Aleppo G, Aroda VR, et al. 2. Classification and diagnosis of diabetes: standards of care in diabetes—2023. Diabetes Care. 2023;46:S19-S40. doi:10.2337/dc23-S002
- Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diabetes. 2011;29:116-122. doi:10.2337/diaclin.29.3.116
- Pleus S, Freckmann G, Schauer S, et al. Self-monitoring of blood glucose as an integral part in the management of people with type 2 diabetes mellitus. Diabetes Ther. 2022;13:829-846. doi:10.1007/s13300-022-01254-8
- Polonsky WH, Fisher L, Schikman CH, et al. Structured self-monitoring of blood glucose significantly reduces A1C levels in poorly controlled, noninsulin-treated type 2 diabetes: results from the Structured Testing Program study. Diabetes Care. 2011;34:262-267. doi:10.2337/dc10-1732
- Tanaka N, Yabe D, Murotani K, et al. Mental distress and health-related quality of life among type 1 and type 2 diabetes patients using self-monitoring of blood glucose: a cross-sectional questionnaire study in Japan. J Diabetes Investig. 2018;9:1203-1211. doi:10.1111/jdi.12827
- Hortensius J, Kars MC, Wierenga WS, et al. Perspectives of patients with type 1 or insulin-treated type 2 diabetes on self-monitoring of blood glucose: a qualitative study. BMC Public Health. 2012;12:167. doi:10.1186/1471-2458-12-167
- Didyuk O, Econom N, Guardia A, Livingston K, Klueh U. Continuous glucose monitoring devices: past, present, and future focus on the history and evolution of technological innovation. J Diabetes Sci Technol. 2021;15:676-683. doi:10.1177/1932296819899394
- Beck RW, Riddlesworth TD, Ruedy K, et al. Effect of continuous glucose monitoring on glycemic control in adults with type 1 diabetes using insulin injections: the DIAMOND randomized clinical trial. JAMA. 2017;317:371-378. doi:10.1001/jama.2016.19975
- Lind M, Polonsky W, Hirsch IB, et al. Continuous glucose monitoring vs conventional therapy for glycemic control in adults with type 1 diabetes treated with multiple daily insulin injections: the GOLD randomized clinical trial. JAMA. 2017;317:379-387. doi:10.1001/jama.2016.19976
- Bolinder J, Antuna R, Geelhoed-Duijvestijn P, et al. Novel glucose-sensing technology and hypoglycemia in type 1 diabetes: a multicenter, non-masked, randomized controlled trial. Lancet. 2016;388:2254-2263. doi:10.1016/S0140-6736(16)31535-5
- Seidu S, Kunutsor SK, Ajjan RA, et al. Efficacy and safety of continuous glucose monitoring and intermittently scanned continuous glucose monitoring in patients with type 2 diabetes: a systematic review and meta-analysis of interventional evidence. Diabetes Care. 2024;47:169-179. doi:10.2337/dc23-1520
- ElSayed NA, Aleppo G, Aroda VR, et al. 5. Facilitating positive health behaviors and well-being to improve health outcomes: standards of care in diabetes-2023. Diabetes Care. 2023;46:S68-S96. doi:10.2337/dc23-S005
- Yaron M, Roitman E, Aharon-Hananel G, et al. Effect of flash glucose monitoring technology on glycemic control and treatment satisfaction in patients with type 2 diabetes. Diabetes Care. 2019;42:1178-1184. doi:10.2337/dc18-0166
- National diabetes statistics report. Centers for Disease Control and Prevention. May 15, 2024. Accessed October 7, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- Elsayed NA, Aleppo G, Aroda VR, et al. 2. Classification and diagnosis of diabetes: standards of care in diabetes—2023. Diabetes Care. 2023;46:S19-S40. doi:10.2337/dc23-S002
- Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diabetes. 2011;29:116-122. doi:10.2337/diaclin.29.3.116
- Pleus S, Freckmann G, Schauer S, et al. Self-monitoring of blood glucose as an integral part in the management of people with type 2 diabetes mellitus. Diabetes Ther. 2022;13:829-846. doi:10.1007/s13300-022-01254-8
- Polonsky WH, Fisher L, Schikman CH, et al. Structured self-monitoring of blood glucose significantly reduces A1C levels in poorly controlled, noninsulin-treated type 2 diabetes: results from the Structured Testing Program study. Diabetes Care. 2011;34:262-267. doi:10.2337/dc10-1732
- Tanaka N, Yabe D, Murotani K, et al. Mental distress and health-related quality of life among type 1 and type 2 diabetes patients using self-monitoring of blood glucose: a cross-sectional questionnaire study in Japan. J Diabetes Investig. 2018;9:1203-1211. doi:10.1111/jdi.12827
- Hortensius J, Kars MC, Wierenga WS, et al. Perspectives of patients with type 1 or insulin-treated type 2 diabetes on self-monitoring of blood glucose: a qualitative study. BMC Public Health. 2012;12:167. doi:10.1186/1471-2458-12-167
- Didyuk O, Econom N, Guardia A, Livingston K, Klueh U. Continuous glucose monitoring devices: past, present, and future focus on the history and evolution of technological innovation. J Diabetes Sci Technol. 2021;15:676-683. doi:10.1177/1932296819899394
- Beck RW, Riddlesworth TD, Ruedy K, et al. Effect of continuous glucose monitoring on glycemic control in adults with type 1 diabetes using insulin injections: the DIAMOND randomized clinical trial. JAMA. 2017;317:371-378. doi:10.1001/jama.2016.19975
- Lind M, Polonsky W, Hirsch IB, et al. Continuous glucose monitoring vs conventional therapy for glycemic control in adults with type 1 diabetes treated with multiple daily insulin injections: the GOLD randomized clinical trial. JAMA. 2017;317:379-387. doi:10.1001/jama.2016.19976
- Bolinder J, Antuna R, Geelhoed-Duijvestijn P, et al. Novel glucose-sensing technology and hypoglycemia in type 1 diabetes: a multicenter, non-masked, randomized controlled trial. Lancet. 2016;388:2254-2263. doi:10.1016/S0140-6736(16)31535-5
- Seidu S, Kunutsor SK, Ajjan RA, et al. Efficacy and safety of continuous glucose monitoring and intermittently scanned continuous glucose monitoring in patients with type 2 diabetes: a systematic review and meta-analysis of interventional evidence. Diabetes Care. 2024;47:169-179. doi:10.2337/dc23-1520
- ElSayed NA, Aleppo G, Aroda VR, et al. 5. Facilitating positive health behaviors and well-being to improve health outcomes: standards of care in diabetes-2023. Diabetes Care. 2023;46:S68-S96. doi:10.2337/dc23-S005
- Yaron M, Roitman E, Aharon-Hananel G, et al. Effect of flash glucose monitoring technology on glycemic control and treatment satisfaction in patients with type 2 diabetes. Diabetes Care. 2019;42:1178-1184. doi:10.2337/dc18-0166
Impact of Continuous Glucose Monitoring for American Indian/Alaska Native Adults With Type 2 Diabetes Mellitus Not Using Insulin
Impact of Continuous Glucose Monitoring for American Indian/Alaska Native Adults With Type 2 Diabetes Mellitus Not Using Insulin
Reducing Sex Disparities in Statin Therapy Among Female Veterans With Type 2 Diabetes and/or Cardiovascular Disease
Reducing Sex Disparities in Statin Therapy Among Female Veterans With Type 2 Diabetes and/or Cardiovascular Disease
Cardiovascular disease (CVD) is the leading cause of death among women in the United States.1 Most CVD is due to the buildup of plaque (ie, cholesterol, proteins, calcium, and inflammatory cells) in artery walls.2 The plaque may lead to atherosclerotic cardiovascular disease (ASCVD), which includes coronary heart disease, cerebrovascular disease, peripheral artery disease, and aortic atherosclerotic disease.2,3 Control and reduction of ASCVD risk factors, including high cholesterol levels, elevated blood pressure, insulin resistance, smoking, and a sedentary lifestyle, can contribute to a reduction in ASCVD morbidity and mortality.2 People with type 2 diabetes mellitus (T2DM) have an increased prevalence of lipid abnormalities, contributing to their high risk of ASCVD.4,5
The prescribing of statins (3-hydroxy-3-methyl-glutaryl-coenzmye A reductase inhibitors) is the cornerstone of lipid-lowering therapy and cardiovascular risk reduction for primary and secondary prevention of ASCVD.6 The American Diabetes Association (ADA) and American College of Cardiology/American Heart Association (ACC/AHA) recommend moderate- to high-intensity statins for primary prevention in patients with T2DM and high-intensity statins for secondary prevention in those with or without diabetes when not contraindicated.4,5,7 Despite eligibility according to guideline recommendations, research predominantly shows that women are less likely to receive statin therapy; however, this trend is improving. [6,8-11] To explain the sex differences in statin use, Nanna et al found that there is a combination of women being offered statin therapy less frequently, declining therapy more frequently, and discontinuing treatment more frequently.11 One possibility for discontinuing treatment could be statin-associated muscle symptoms (SAMS), which occur in about 10% of patients.12 The incidence of adverse effects (AEs) may be related to the way statins are metabolized.
Pharmacogenomic testing is free for veterans through the US Department of Veterans Affairs (VA) PHASER program, which offers information and recommendations for a panel of 11 gene variants. The panel includes genes related to common medication classes such as anticoagulants, antiplatelets, proton pump inhibitors, nonsteroidal anti-inflammatory drugs, opioids, antidepressants, and statins. The VA PHASER panel includes the solute carrier organic anion transporter family member 1B1 (SLCO1B1) gene, which is predominantly expressed in the liver and facilitates the hepatic uptake of most statins.13,14 A reduced function of SLCO1B1 can lead to higher statin levels, resulting in increased concentrations that may potentially cause SAMS.13,14 Some alleles associated with reduced function include SLCO1B1*5, *15, *23, *31, and *46 to *49, whereas others are associated with increased function, such as SLCO1B1 *14 and *20 (Appendix).15 Supporting evidence shows the SLCO1B1*5 nucleotide polymorphism increases plasma levels of simvastatin and atorvastatin, affecting effectiveness or toxicity. 13 Females tend to have a lower body weight and higher percentage of body fat compared with males, which might lead to higher concentrations of lipophilic drugs, including atorvastatin and simvastatin, which may be exacerbated by decreased function of SLCO1B1*5.15 With pharmacogenomic testing, therapeutic recommendations can be made to improve the overall safety and efficacy of statins, thus improving adherence using a patient-specific approach.14,15
Methods
Carl Vinson VA Medical Center (CVVAMC) serves about 42,000 veterans in Central and South Georgia, of which about 15% are female. Of the female veterans enrolled in care, 63% identify as Black, 27% White, and 1.5% as Asian, American Indian/Alaska Native, or Native Hawaiian/Other Pacific Islander. The 2020 Veterans Chartbook report showed that female veterans and minority racial and ethnic groups had worse access to health care and higher mortality rates than their male and non-Hispanic White counterparts.16
The Primary Care Equity Dashboard (PCED) was developed to engage the VA health care workforce in the process of identifying and addressing inequities in local patient populations.17 Using electronic quality measure data, the PCED provides Veterans Integrated Service Network-level and facility-level performance on several metrics.18 The PCED had not been previously used at the CVVAMC, and few publications or quality improvement projects regarding its use have been reported by the VA Office of Health Equity. PCED helped identify disparities when comparing female to male patients in the prescribing of statin therapy for patients with CVD and statin therapy for patients with T2DM.
VA PHASER pharmacogenomic analyses provided an opportunity to expand this quality improvement project. Sanford Health and the VA collaborated on the PHASER program to offer free genetic testing for veterans. The program launched in 2019 and expanded to various VA sites, including CVVAMC in March 2023. This program has been extended to December 31, 2025.
The primary objective of this quality improvement project was to increase statin prescribing among female veterans with T2DM and/or CVD to reduce cardiovascular risk. Secondary outcomes included increased pharmacogenomic testing and the assessment of pharmacogenomic results related to statin therapy. This project was approved by the CVVAMC Pharmacy and Therapeutics Committee. The PCED was used to identify female veterans with T2DM and/or CVD without an active prescription for a statin between July and October 2023. A review of Computerized Patient Record System patient charts was completed to screen for prespecified inclusion and exclusion criteria. Veterans were included if they were assigned female at birth, were enrolled in care at CVVAMC, and had a diagnosis of T2DM or CVD (history of myocardial infarction, coronary bypass graft, percutaneous coronary intervention, or other revascularization in any setting).
Veterans were excluded if they were currently pregnant, trying to conceive, breastfeeding, had a T1DM diagnosis, had previously documented hypersensitivity to a statin, active liver failure or decompensated cirrhosis, previously documented statin-associated rhabdomyolysis or autoimmune myopathy, an active prescription for a proprotein convertase subtilisin/kexin type 9 inhibitor, or previously documented statin intolerance (defined as the inability to tolerate ≥ 3 statins, with ≥ 1 prescribed at low intensity or alternate-day dosing). The female veterans were compared to 2 comparators: the facility's male veterans and the VA national average, identified via the PCED.
Once a veteran was screened, they were telephoned between October 2023 and February 2024 and provided education on statin use and pharmacogenomic testing using a standardized note template. An order was placed for participants who provided verbal consent for pharmacogenomic testing. Those who agreed to statin initiation were referred to a clinical pharmacist practitioner (CPP) who contacted them at a later date to prescribe a statin following the recommendations of the 2019 ACC/AHA and 2023 ADA guidelines and pharmacogenomic testing, if applicable.4,5,7 Appropriate monitoring and follow-up occurred at the discretion of each CPP. Data collection included: age, race, diagnoses (T2DM, CVD, or both), baseline lipid panel (total cholesterol, triglycerides, high-density lipoprotein, low-density lipoprotein), hepatic function, name and dose of statin, reasons for declining statin therapy, and pharmacogenomic testing results related to SLCO1B1.
Results
At baseline in July 2023, 77.8% of female veterans with T2DM were prescribed a statin, which exceeded the national VA average (77.0%), but was below the rate for male veterans (78.7%) in the facility comparator group.17 Additionally, 82.2% of females with CVD were prescribed a statin, which was below the national VA average of 86.0% and the 84.9% of male veterans in the facility comparator group.17 The PCED identified 189 female veterans from July 2023 to October 2023 who may benefit from statin therapy. Thirty-three females met the exclusion criteria. Of the 156 included veterans, 129 (82.7%) were successfully contacted and 27 (17.3%) could not be reached by telephone after 3 attempts (Figure 1). The 129 female veterans contacted had a mean age of 59 years and the majority were Black (82.9%) (Table 1).
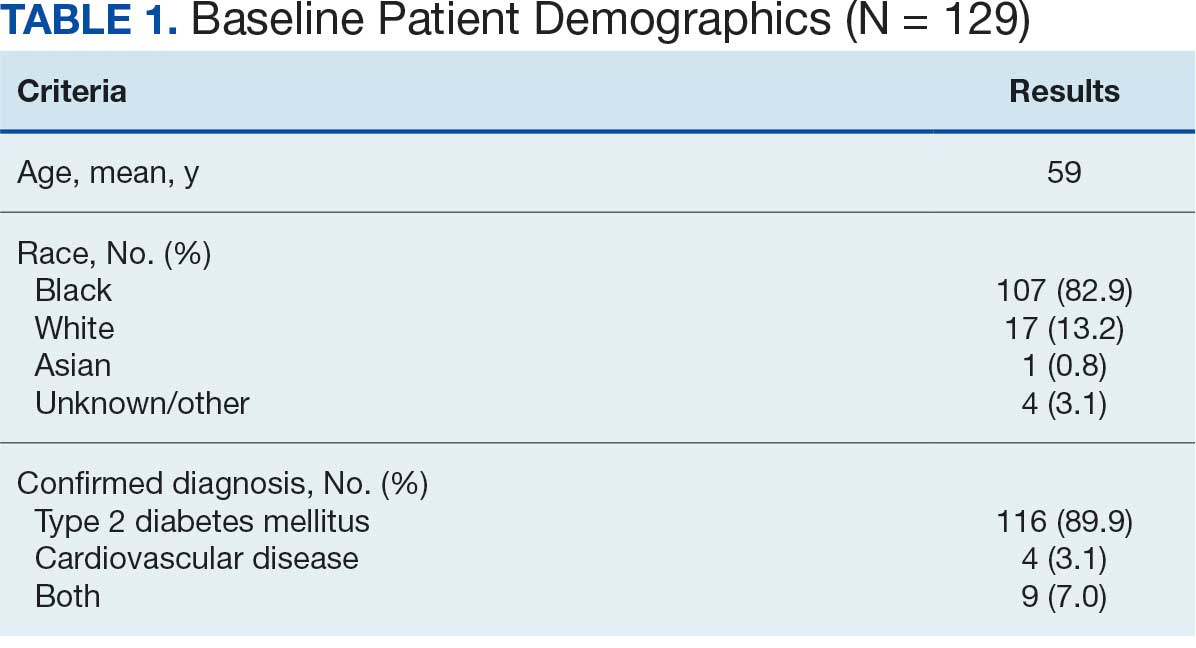
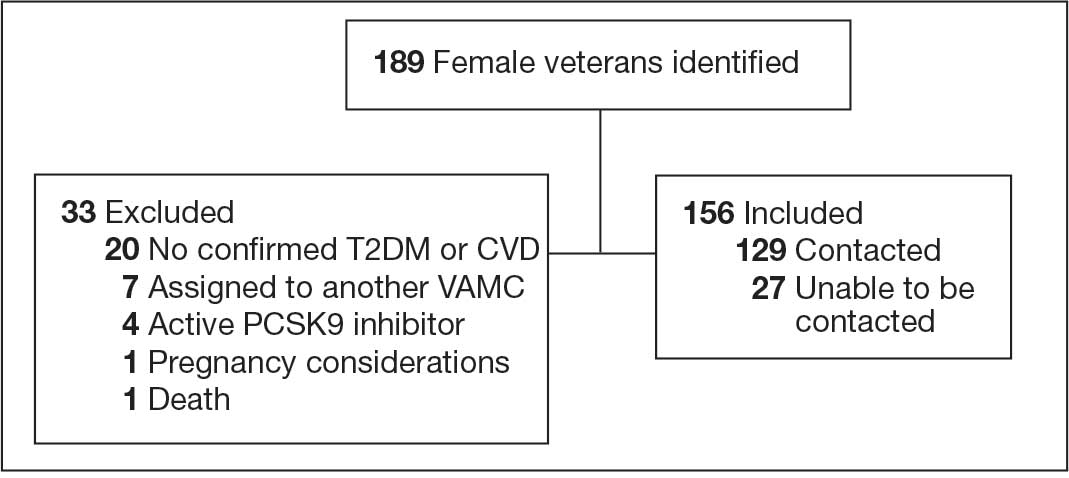
Abbreviations: CVD, cardiovascular disease; PCSK9, proprotein convertase subtilisin/
kexin type 9; T2DM, type 2 diabetes mellitus; VAMC, Veterans Affairs medical center.
Primary Outcomes
Of the 129 contacted veterans, 31 (24.0%) had a non-VA statin prescription, 13 (10.1%) had an active VA statin prescription, and 85 (65.9%) did not have a statin prescription, despite being eligible. Statin adherence was confirmed with participants, and the medication list was updated accordingly.
Of the 85 veterans with no active statin therapy, 37 (43.5%) accepted a new statin prescription and 48 (56.5%) declined. There were various reasons provided for declining statin therapy: 17 participants (35.4%) declined due to concern for AEs (Table 2).

From July 2023 to March 2024, the percentage of female veterans with active statin therapy with T2DM increased from 77.8% to 79.0%. For those with active statin therapy with CVD, usage increased from 82.2% to 90.2%, which exceeded the national VA average and facility male comparator group (Figures 2 and 3).17
Secondary Outcomes
Seventy-one of 129 veterans (55.0%) gave verbal consent, and 47 (66.2%) completed the pharmacogenomic testing; 58 (45.0%) declined. Five veterans (10.6%) had a known SLCO1B1 allele variant present. One veteran required a change in statin therapy based on the results (eAppendix).
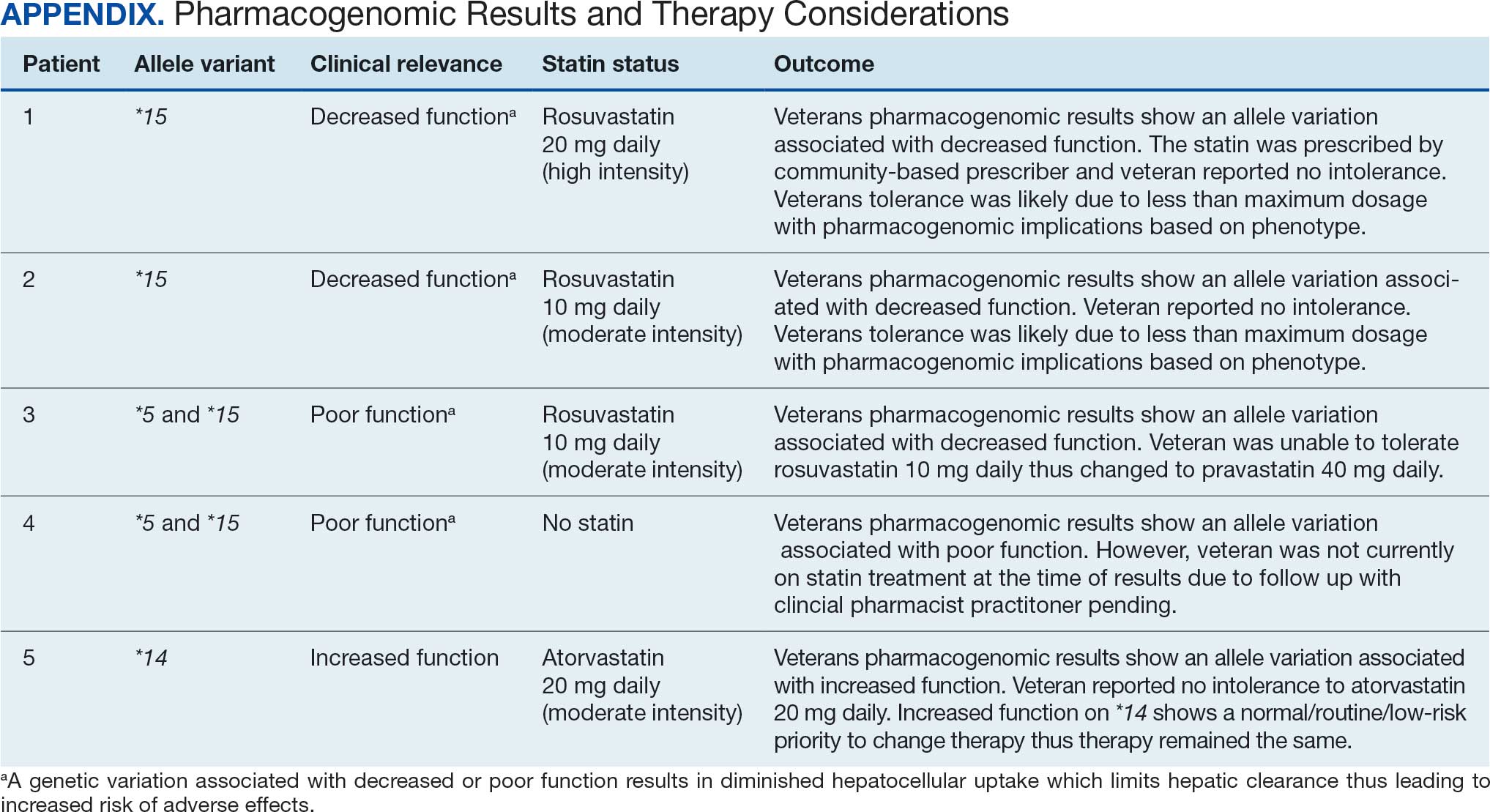
Discussion
This project aimed to increase statin prescribing among female veterans with T2DM and/or CVD to reduce cardiovascular risk and increase pharmacogenomic testing using the PCED and care managed by CPPs. The results of this quality improvement project illustrated that both metrics have improved at CVVAMC as a result of the intervention. The results in both metrics now exceed the PCED national VA average, and the CVD metric also exceeds that of the facility male comparator group. While there was only a 1.2% increase from July 2023 to March 2024 for patients with T2DM, there was an 8.0% increase for patients with CVD. Despite standardized education on statin use, more veterans declined therapy than accepted it, mostly due to concern for AEs. Recording the reasons for declining statin therapy offered valuable insight that can be used in additional discussions with veterans and clinicians.
Pharmacogenomics gives clinicians the unique opportunity to take a proactive approach to better predict drug responses, potentially allowing for less trial and error with medications, fewer AEs, greater trust in the clinician, and improved medication adherence. The CPPs incorporated pharmacogenomic testing into their practice, which led to identifying 5 SLCO1B1 gene abnormalities. The PCED served as a powerful tool for advancing equity-focused quality improvement initiatives on a local level and was crucial in prioritizing the detection of veterans potentially receiving suboptimal care.
Limitations
The nature of “cold calls” made it challenging to establish contact for inclusion in this study. An alternative to increase engagement could have been scheduled phone or face-to-face visits. While the use of the PCED was crucial, data did not account for statins listed in the non-VA medication list. All 31 patients with statins prescribed outside the VA had a start date added to provide the most accurate representation of the data moving forward.
Another limitation in this project was its small sample size and population. CVVAMC serves about 6200 female veterans, with roughly 63% identifying as Black. The preponderance of Black individuals (83%) in this project is typical for the female patient population at CVVAMC but may not reflect the demographics of other populations. Other limitations to this project consisted of scheduling conflicts. Appointments for laboratory draws at community-based outpatient clinics were subject to availability, which resulted in some delay in completion of pharmacogenomic testing.
Conclusions
CPPs can help reduce inequity in health care delivery. Increased incorporation of the PCED into regular practice within the VA is recommended to continue addressing sex disparities in statin use, diabetes control, blood pressure management, cancer screenings, and vaccination needs. CVVAMC plans to expand its use through another quality improvement project focused on reducing sex disparities in blood pressure management. Improving educational resources made available to veterans on the importance of statin therapy and potential to mitigate AEs through use of the VA PHASER program also would be helpful. This project successfully improved CVVAMC metrics for female veterans appropriately prescribed statin therapy and increased access to pharmacogenomic testing. Most importantly, it helped close the sex-based gap in CVD risk reduction care.
- Heron M. Deaths: leading causes for 2018. Nat Vital Stat Rep. 2021;70:1-114.
- US Department of Veterans Affairs, US Department of Defense. VA/DoD Clinical practice guideline for the management of dyslipidemia for cardiovascular risk reduction. Published June 2020. Accessed August 25, 2025. https://www.healthquality.va.gov/guidelines/CD/lipids/VADODDyslipidemiaCPG5087212020.pdf
- Atherosclerotic Cardiovascular Disease (ASCVD). American Heart Association. Accessed August 26, 2025. https:// www.heart.org/en/professional/quality-improvement/ascvd
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular disease and risk management: standards of medical care in diabetes-2022. Diabetes Care. 2022;45(Suppl 1):S144-S174. doi:10.2337/dc22-S010
- American Diabetes Association. Standards of Care in Diabetes— 2023 abridged for primary care providers. Clinical Diabetes. 2022;41(1):4-31. doi:10.2337/cd23-as01
- Virani SS, Woodard LD, Ramsey DJ, et al. Gender disparities in evidence-based statin therapy in patients with cardiovascular disease. Am J Cardiol. 2015;115:21-26. doi:10.1016/j.amjcard.2014.09.041
- Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/ AHA Guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi:10.1161/CIR.0000000000000678
- Buchanan CH, Brown EA, Bishu KG, et al. The magnitude and potential causes of gender disparities in statin therapy in veterans with type 2 diabetes: a 10-year nationwide longitudinal cohort study. Womens Health Issues. 2022;32:274-283. doi:10.1016/j.whi.2021.10.003
- Ahmed F, Lin J, Ahmed T, et al. Health disparities: statin prescribing patterns among patients with diabetes in a family medicine clinic. Health Equity. 2022;6:291-297. doi:10.1089/heq.2021.0144
- Metser G, Bradley C, Moise N, Liyanage-Don N, Kronish I, Ye S. Gaps and disparities in primary prevention statin prescription during outpatient care. Am J Cardiol. 2021;161:36-41. doi:10.1016/j.amjcard.2021.08.070
- Nanna MG, Wang TY, Xiang Q, et al. Sex differences in the use of statins in community practice. Circ Cardiovasc Qual Outcomes. 2019;12(8):e005562. doi:10.1161/CIRCOUTCOMES.118.005562
- Kitzmiller JP, Mikulik EB, Dauki AM, Murkherjee C, Luzum JA. Pharmacogenomics of statins: understanding susceptibility to adverse effects. Pharmgenomics Pers Med. 2016;9:97-106. doi:10.2147/PGPM.S86013
- Türkmen D, Masoli JAH, Kuo CL, Bowden J, Melzer D, Pilling LC. Statin treatment effectiveness and the SLCO1B1*5 reduced function genotype: long-term outcomes in women and men. Br J Clin Pharmacol. 2022;88:3230-3240. doi:10.1111/bcp.15245
- Cooper-DeHoff RM, Niemi M, Ramsey LB, et al. The Clinical Pharmacogenetics Implementation Consortium guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and statin-associated musculoskeletal symptoms. Clin Pharmacol Ther. 2022;111:1007-1021. doi:10.1002/cpt.2557
- Ramsey LB, Gong L, Lee SB, et al. PharmVar GeneFocus: SLCO1B1. Clin Pharmacol Ther. 2023;113:782-793. doi:10.1002/cpt.2705
- National Healthcare Quality and Disparities Report: Chartbook on Healthcare for Veterans. Rockville (MD): Agency for Healthcare Research and Quality (US); November 2020.
- Procario G. Primary Care Equity Dashboard [database online]. Power Bi. 2023. Accessed August 26, 2025. https://app.powerbigov.us
- Hausmann LRM, Lamorte C, Estock JL. Understanding the context for incorporating equity into quality improvement throughout a national health care system. Health Equity. 2023;7(1):312-320. doi:10.1089/heq.2023.0009
Cardiovascular disease (CVD) is the leading cause of death among women in the United States.1 Most CVD is due to the buildup of plaque (ie, cholesterol, proteins, calcium, and inflammatory cells) in artery walls.2 The plaque may lead to atherosclerotic cardiovascular disease (ASCVD), which includes coronary heart disease, cerebrovascular disease, peripheral artery disease, and aortic atherosclerotic disease.2,3 Control and reduction of ASCVD risk factors, including high cholesterol levels, elevated blood pressure, insulin resistance, smoking, and a sedentary lifestyle, can contribute to a reduction in ASCVD morbidity and mortality.2 People with type 2 diabetes mellitus (T2DM) have an increased prevalence of lipid abnormalities, contributing to their high risk of ASCVD.4,5
The prescribing of statins (3-hydroxy-3-methyl-glutaryl-coenzmye A reductase inhibitors) is the cornerstone of lipid-lowering therapy and cardiovascular risk reduction for primary and secondary prevention of ASCVD.6 The American Diabetes Association (ADA) and American College of Cardiology/American Heart Association (ACC/AHA) recommend moderate- to high-intensity statins for primary prevention in patients with T2DM and high-intensity statins for secondary prevention in those with or without diabetes when not contraindicated.4,5,7 Despite eligibility according to guideline recommendations, research predominantly shows that women are less likely to receive statin therapy; however, this trend is improving. [6,8-11] To explain the sex differences in statin use, Nanna et al found that there is a combination of women being offered statin therapy less frequently, declining therapy more frequently, and discontinuing treatment more frequently.11 One possibility for discontinuing treatment could be statin-associated muscle symptoms (SAMS), which occur in about 10% of patients.12 The incidence of adverse effects (AEs) may be related to the way statins are metabolized.
Pharmacogenomic testing is free for veterans through the US Department of Veterans Affairs (VA) PHASER program, which offers information and recommendations for a panel of 11 gene variants. The panel includes genes related to common medication classes such as anticoagulants, antiplatelets, proton pump inhibitors, nonsteroidal anti-inflammatory drugs, opioids, antidepressants, and statins. The VA PHASER panel includes the solute carrier organic anion transporter family member 1B1 (SLCO1B1) gene, which is predominantly expressed in the liver and facilitates the hepatic uptake of most statins.13,14 A reduced function of SLCO1B1 can lead to higher statin levels, resulting in increased concentrations that may potentially cause SAMS.13,14 Some alleles associated with reduced function include SLCO1B1*5, *15, *23, *31, and *46 to *49, whereas others are associated with increased function, such as SLCO1B1 *14 and *20 (Appendix).15 Supporting evidence shows the SLCO1B1*5 nucleotide polymorphism increases plasma levels of simvastatin and atorvastatin, affecting effectiveness or toxicity. 13 Females tend to have a lower body weight and higher percentage of body fat compared with males, which might lead to higher concentrations of lipophilic drugs, including atorvastatin and simvastatin, which may be exacerbated by decreased function of SLCO1B1*5.15 With pharmacogenomic testing, therapeutic recommendations can be made to improve the overall safety and efficacy of statins, thus improving adherence using a patient-specific approach.14,15
Methods
Carl Vinson VA Medical Center (CVVAMC) serves about 42,000 veterans in Central and South Georgia, of which about 15% are female. Of the female veterans enrolled in care, 63% identify as Black, 27% White, and 1.5% as Asian, American Indian/Alaska Native, or Native Hawaiian/Other Pacific Islander. The 2020 Veterans Chartbook report showed that female veterans and minority racial and ethnic groups had worse access to health care and higher mortality rates than their male and non-Hispanic White counterparts.16
The Primary Care Equity Dashboard (PCED) was developed to engage the VA health care workforce in the process of identifying and addressing inequities in local patient populations.17 Using electronic quality measure data, the PCED provides Veterans Integrated Service Network-level and facility-level performance on several metrics.18 The PCED had not been previously used at the CVVAMC, and few publications or quality improvement projects regarding its use have been reported by the VA Office of Health Equity. PCED helped identify disparities when comparing female to male patients in the prescribing of statin therapy for patients with CVD and statin therapy for patients with T2DM.
VA PHASER pharmacogenomic analyses provided an opportunity to expand this quality improvement project. Sanford Health and the VA collaborated on the PHASER program to offer free genetic testing for veterans. The program launched in 2019 and expanded to various VA sites, including CVVAMC in March 2023. This program has been extended to December 31, 2025.
The primary objective of this quality improvement project was to increase statin prescribing among female veterans with T2DM and/or CVD to reduce cardiovascular risk. Secondary outcomes included increased pharmacogenomic testing and the assessment of pharmacogenomic results related to statin therapy. This project was approved by the CVVAMC Pharmacy and Therapeutics Committee. The PCED was used to identify female veterans with T2DM and/or CVD without an active prescription for a statin between July and October 2023. A review of Computerized Patient Record System patient charts was completed to screen for prespecified inclusion and exclusion criteria. Veterans were included if they were assigned female at birth, were enrolled in care at CVVAMC, and had a diagnosis of T2DM or CVD (history of myocardial infarction, coronary bypass graft, percutaneous coronary intervention, or other revascularization in any setting).
Veterans were excluded if they were currently pregnant, trying to conceive, breastfeeding, had a T1DM diagnosis, had previously documented hypersensitivity to a statin, active liver failure or decompensated cirrhosis, previously documented statin-associated rhabdomyolysis or autoimmune myopathy, an active prescription for a proprotein convertase subtilisin/kexin type 9 inhibitor, or previously documented statin intolerance (defined as the inability to tolerate ≥ 3 statins, with ≥ 1 prescribed at low intensity or alternate-day dosing). The female veterans were compared to 2 comparators: the facility's male veterans and the VA national average, identified via the PCED.
Once a veteran was screened, they were telephoned between October 2023 and February 2024 and provided education on statin use and pharmacogenomic testing using a standardized note template. An order was placed for participants who provided verbal consent for pharmacogenomic testing. Those who agreed to statin initiation were referred to a clinical pharmacist practitioner (CPP) who contacted them at a later date to prescribe a statin following the recommendations of the 2019 ACC/AHA and 2023 ADA guidelines and pharmacogenomic testing, if applicable.4,5,7 Appropriate monitoring and follow-up occurred at the discretion of each CPP. Data collection included: age, race, diagnoses (T2DM, CVD, or both), baseline lipid panel (total cholesterol, triglycerides, high-density lipoprotein, low-density lipoprotein), hepatic function, name and dose of statin, reasons for declining statin therapy, and pharmacogenomic testing results related to SLCO1B1.
Results
At baseline in July 2023, 77.8% of female veterans with T2DM were prescribed a statin, which exceeded the national VA average (77.0%), but was below the rate for male veterans (78.7%) in the facility comparator group.17 Additionally, 82.2% of females with CVD were prescribed a statin, which was below the national VA average of 86.0% and the 84.9% of male veterans in the facility comparator group.17 The PCED identified 189 female veterans from July 2023 to October 2023 who may benefit from statin therapy. Thirty-three females met the exclusion criteria. Of the 156 included veterans, 129 (82.7%) were successfully contacted and 27 (17.3%) could not be reached by telephone after 3 attempts (Figure 1). The 129 female veterans contacted had a mean age of 59 years and the majority were Black (82.9%) (Table 1).
Abbreviations: CVD, cardiovascular disease; PCSK9, proprotein convertase subtilisin/
kexin type 9; T2DM, type 2 diabetes mellitus; VAMC, Veterans Affairs medical center.
Primary Outcomes
Of the 129 contacted veterans, 31 (24.0%) had a non-VA statin prescription, 13 (10.1%) had an active VA statin prescription, and 85 (65.9%) did not have a statin prescription, despite being eligible. Statin adherence was confirmed with participants, and the medication list was updated accordingly.
Of the 85 veterans with no active statin therapy, 37 (43.5%) accepted a new statin prescription and 48 (56.5%) declined. There were various reasons provided for declining statin therapy: 17 participants (35.4%) declined due to concern for AEs (Table 2).
From July 2023 to March 2024, the percentage of female veterans with active statin therapy with T2DM increased from 77.8% to 79.0%. For those with active statin therapy with CVD, usage increased from 82.2% to 90.2%, which exceeded the national VA average and facility male comparator group (Figures 2 and 3).17
Secondary Outcomes
Seventy-one of 129 veterans (55.0%) gave verbal consent, and 47 (66.2%) completed the pharmacogenomic testing; 58 (45.0%) declined. Five veterans (10.6%) had a known SLCO1B1 allele variant present. One veteran required a change in statin therapy based on the results (eAppendix).
Discussion
This project aimed to increase statin prescribing among female veterans with T2DM and/or CVD to reduce cardiovascular risk and increase pharmacogenomic testing using the PCED and care managed by CPPs. The results of this quality improvement project illustrated that both metrics have improved at CVVAMC as a result of the intervention. The results in both metrics now exceed the PCED national VA average, and the CVD metric also exceeds that of the facility male comparator group. While there was only a 1.2% increase from July 2023 to March 2024 for patients with T2DM, there was an 8.0% increase for patients with CVD. Despite standardized education on statin use, more veterans declined therapy than accepted it, mostly due to concern for AEs. Recording the reasons for declining statin therapy offered valuable insight that can be used in additional discussions with veterans and clinicians.
Pharmacogenomics gives clinicians the unique opportunity to take a proactive approach to better predict drug responses, potentially allowing for less trial and error with medications, fewer AEs, greater trust in the clinician, and improved medication adherence. The CPPs incorporated pharmacogenomic testing into their practice, which led to identifying 5 SLCO1B1 gene abnormalities. The PCED served as a powerful tool for advancing equity-focused quality improvement initiatives on a local level and was crucial in prioritizing the detection of veterans potentially receiving suboptimal care.
Limitations
The nature of “cold calls” made it challenging to establish contact for inclusion in this study. An alternative to increase engagement could have been scheduled phone or face-to-face visits. While the use of the PCED was crucial, data did not account for statins listed in the non-VA medication list. All 31 patients with statins prescribed outside the VA had a start date added to provide the most accurate representation of the data moving forward.
Another limitation in this project was its small sample size and population. CVVAMC serves about 6200 female veterans, with roughly 63% identifying as Black. The preponderance of Black individuals (83%) in this project is typical for the female patient population at CVVAMC but may not reflect the demographics of other populations. Other limitations to this project consisted of scheduling conflicts. Appointments for laboratory draws at community-based outpatient clinics were subject to availability, which resulted in some delay in completion of pharmacogenomic testing.
Conclusions
CPPs can help reduce inequity in health care delivery. Increased incorporation of the PCED into regular practice within the VA is recommended to continue addressing sex disparities in statin use, diabetes control, blood pressure management, cancer screenings, and vaccination needs. CVVAMC plans to expand its use through another quality improvement project focused on reducing sex disparities in blood pressure management. Improving educational resources made available to veterans on the importance of statin therapy and potential to mitigate AEs through use of the VA PHASER program also would be helpful. This project successfully improved CVVAMC metrics for female veterans appropriately prescribed statin therapy and increased access to pharmacogenomic testing. Most importantly, it helped close the sex-based gap in CVD risk reduction care.
Cardiovascular disease (CVD) is the leading cause of death among women in the United States.1 Most CVD is due to the buildup of plaque (ie, cholesterol, proteins, calcium, and inflammatory cells) in artery walls.2 The plaque may lead to atherosclerotic cardiovascular disease (ASCVD), which includes coronary heart disease, cerebrovascular disease, peripheral artery disease, and aortic atherosclerotic disease.2,3 Control and reduction of ASCVD risk factors, including high cholesterol levels, elevated blood pressure, insulin resistance, smoking, and a sedentary lifestyle, can contribute to a reduction in ASCVD morbidity and mortality.2 People with type 2 diabetes mellitus (T2DM) have an increased prevalence of lipid abnormalities, contributing to their high risk of ASCVD.4,5
The prescribing of statins (3-hydroxy-3-methyl-glutaryl-coenzmye A reductase inhibitors) is the cornerstone of lipid-lowering therapy and cardiovascular risk reduction for primary and secondary prevention of ASCVD.6 The American Diabetes Association (ADA) and American College of Cardiology/American Heart Association (ACC/AHA) recommend moderate- to high-intensity statins for primary prevention in patients with T2DM and high-intensity statins for secondary prevention in those with or without diabetes when not contraindicated.4,5,7 Despite eligibility according to guideline recommendations, research predominantly shows that women are less likely to receive statin therapy; however, this trend is improving. [6,8-11] To explain the sex differences in statin use, Nanna et al found that there is a combination of women being offered statin therapy less frequently, declining therapy more frequently, and discontinuing treatment more frequently.11 One possibility for discontinuing treatment could be statin-associated muscle symptoms (SAMS), which occur in about 10% of patients.12 The incidence of adverse effects (AEs) may be related to the way statins are metabolized.
Pharmacogenomic testing is free for veterans through the US Department of Veterans Affairs (VA) PHASER program, which offers information and recommendations for a panel of 11 gene variants. The panel includes genes related to common medication classes such as anticoagulants, antiplatelets, proton pump inhibitors, nonsteroidal anti-inflammatory drugs, opioids, antidepressants, and statins. The VA PHASER panel includes the solute carrier organic anion transporter family member 1B1 (SLCO1B1) gene, which is predominantly expressed in the liver and facilitates the hepatic uptake of most statins.13,14 A reduced function of SLCO1B1 can lead to higher statin levels, resulting in increased concentrations that may potentially cause SAMS.13,14 Some alleles associated with reduced function include SLCO1B1*5, *15, *23, *31, and *46 to *49, whereas others are associated with increased function, such as SLCO1B1 *14 and *20 (Appendix).15 Supporting evidence shows the SLCO1B1*5 nucleotide polymorphism increases plasma levels of simvastatin and atorvastatin, affecting effectiveness or toxicity. 13 Females tend to have a lower body weight and higher percentage of body fat compared with males, which might lead to higher concentrations of lipophilic drugs, including atorvastatin and simvastatin, which may be exacerbated by decreased function of SLCO1B1*5.15 With pharmacogenomic testing, therapeutic recommendations can be made to improve the overall safety and efficacy of statins, thus improving adherence using a patient-specific approach.14,15
Methods
Carl Vinson VA Medical Center (CVVAMC) serves about 42,000 veterans in Central and South Georgia, of which about 15% are female. Of the female veterans enrolled in care, 63% identify as Black, 27% White, and 1.5% as Asian, American Indian/Alaska Native, or Native Hawaiian/Other Pacific Islander. The 2020 Veterans Chartbook report showed that female veterans and minority racial and ethnic groups had worse access to health care and higher mortality rates than their male and non-Hispanic White counterparts.16
The Primary Care Equity Dashboard (PCED) was developed to engage the VA health care workforce in the process of identifying and addressing inequities in local patient populations.17 Using electronic quality measure data, the PCED provides Veterans Integrated Service Network-level and facility-level performance on several metrics.18 The PCED had not been previously used at the CVVAMC, and few publications or quality improvement projects regarding its use have been reported by the VA Office of Health Equity. PCED helped identify disparities when comparing female to male patients in the prescribing of statin therapy for patients with CVD and statin therapy for patients with T2DM.
VA PHASER pharmacogenomic analyses provided an opportunity to expand this quality improvement project. Sanford Health and the VA collaborated on the PHASER program to offer free genetic testing for veterans. The program launched in 2019 and expanded to various VA sites, including CVVAMC in March 2023. This program has been extended to December 31, 2025.
The primary objective of this quality improvement project was to increase statin prescribing among female veterans with T2DM and/or CVD to reduce cardiovascular risk. Secondary outcomes included increased pharmacogenomic testing and the assessment of pharmacogenomic results related to statin therapy. This project was approved by the CVVAMC Pharmacy and Therapeutics Committee. The PCED was used to identify female veterans with T2DM and/or CVD without an active prescription for a statin between July and October 2023. A review of Computerized Patient Record System patient charts was completed to screen for prespecified inclusion and exclusion criteria. Veterans were included if they were assigned female at birth, were enrolled in care at CVVAMC, and had a diagnosis of T2DM or CVD (history of myocardial infarction, coronary bypass graft, percutaneous coronary intervention, or other revascularization in any setting).
Veterans were excluded if they were currently pregnant, trying to conceive, breastfeeding, had a T1DM diagnosis, had previously documented hypersensitivity to a statin, active liver failure or decompensated cirrhosis, previously documented statin-associated rhabdomyolysis or autoimmune myopathy, an active prescription for a proprotein convertase subtilisin/kexin type 9 inhibitor, or previously documented statin intolerance (defined as the inability to tolerate ≥ 3 statins, with ≥ 1 prescribed at low intensity or alternate-day dosing). The female veterans were compared to 2 comparators: the facility's male veterans and the VA national average, identified via the PCED.
Once a veteran was screened, they were telephoned between October 2023 and February 2024 and provided education on statin use and pharmacogenomic testing using a standardized note template. An order was placed for participants who provided verbal consent for pharmacogenomic testing. Those who agreed to statin initiation were referred to a clinical pharmacist practitioner (CPP) who contacted them at a later date to prescribe a statin following the recommendations of the 2019 ACC/AHA and 2023 ADA guidelines and pharmacogenomic testing, if applicable.4,5,7 Appropriate monitoring and follow-up occurred at the discretion of each CPP. Data collection included: age, race, diagnoses (T2DM, CVD, or both), baseline lipid panel (total cholesterol, triglycerides, high-density lipoprotein, low-density lipoprotein), hepatic function, name and dose of statin, reasons for declining statin therapy, and pharmacogenomic testing results related to SLCO1B1.
Results
At baseline in July 2023, 77.8% of female veterans with T2DM were prescribed a statin, which exceeded the national VA average (77.0%), but was below the rate for male veterans (78.7%) in the facility comparator group.17 Additionally, 82.2% of females with CVD were prescribed a statin, which was below the national VA average of 86.0% and the 84.9% of male veterans in the facility comparator group.17 The PCED identified 189 female veterans from July 2023 to October 2023 who may benefit from statin therapy. Thirty-three females met the exclusion criteria. Of the 156 included veterans, 129 (82.7%) were successfully contacted and 27 (17.3%) could not be reached by telephone after 3 attempts (Figure 1). The 129 female veterans contacted had a mean age of 59 years and the majority were Black (82.9%) (Table 1).
Abbreviations: CVD, cardiovascular disease; PCSK9, proprotein convertase subtilisin/
kexin type 9; T2DM, type 2 diabetes mellitus; VAMC, Veterans Affairs medical center.
Primary Outcomes
Of the 129 contacted veterans, 31 (24.0%) had a non-VA statin prescription, 13 (10.1%) had an active VA statin prescription, and 85 (65.9%) did not have a statin prescription, despite being eligible. Statin adherence was confirmed with participants, and the medication list was updated accordingly.
Of the 85 veterans with no active statin therapy, 37 (43.5%) accepted a new statin prescription and 48 (56.5%) declined. There were various reasons provided for declining statin therapy: 17 participants (35.4%) declined due to concern for AEs (Table 2).
From July 2023 to March 2024, the percentage of female veterans with active statin therapy with T2DM increased from 77.8% to 79.0%. For those with active statin therapy with CVD, usage increased from 82.2% to 90.2%, which exceeded the national VA average and facility male comparator group (Figures 2 and 3).17
Secondary Outcomes
Seventy-one of 129 veterans (55.0%) gave verbal consent, and 47 (66.2%) completed the pharmacogenomic testing; 58 (45.0%) declined. Five veterans (10.6%) had a known SLCO1B1 allele variant present. One veteran required a change in statin therapy based on the results (eAppendix).
Discussion
This project aimed to increase statin prescribing among female veterans with T2DM and/or CVD to reduce cardiovascular risk and increase pharmacogenomic testing using the PCED and care managed by CPPs. The results of this quality improvement project illustrated that both metrics have improved at CVVAMC as a result of the intervention. The results in both metrics now exceed the PCED national VA average, and the CVD metric also exceeds that of the facility male comparator group. While there was only a 1.2% increase from July 2023 to March 2024 for patients with T2DM, there was an 8.0% increase for patients with CVD. Despite standardized education on statin use, more veterans declined therapy than accepted it, mostly due to concern for AEs. Recording the reasons for declining statin therapy offered valuable insight that can be used in additional discussions with veterans and clinicians.
Pharmacogenomics gives clinicians the unique opportunity to take a proactive approach to better predict drug responses, potentially allowing for less trial and error with medications, fewer AEs, greater trust in the clinician, and improved medication adherence. The CPPs incorporated pharmacogenomic testing into their practice, which led to identifying 5 SLCO1B1 gene abnormalities. The PCED served as a powerful tool for advancing equity-focused quality improvement initiatives on a local level and was crucial in prioritizing the detection of veterans potentially receiving suboptimal care.
Limitations
The nature of “cold calls” made it challenging to establish contact for inclusion in this study. An alternative to increase engagement could have been scheduled phone or face-to-face visits. While the use of the PCED was crucial, data did not account for statins listed in the non-VA medication list. All 31 patients with statins prescribed outside the VA had a start date added to provide the most accurate representation of the data moving forward.
Another limitation in this project was its small sample size and population. CVVAMC serves about 6200 female veterans, with roughly 63% identifying as Black. The preponderance of Black individuals (83%) in this project is typical for the female patient population at CVVAMC but may not reflect the demographics of other populations. Other limitations to this project consisted of scheduling conflicts. Appointments for laboratory draws at community-based outpatient clinics were subject to availability, which resulted in some delay in completion of pharmacogenomic testing.
Conclusions
CPPs can help reduce inequity in health care delivery. Increased incorporation of the PCED into regular practice within the VA is recommended to continue addressing sex disparities in statin use, diabetes control, blood pressure management, cancer screenings, and vaccination needs. CVVAMC plans to expand its use through another quality improvement project focused on reducing sex disparities in blood pressure management. Improving educational resources made available to veterans on the importance of statin therapy and potential to mitigate AEs through use of the VA PHASER program also would be helpful. This project successfully improved CVVAMC metrics for female veterans appropriately prescribed statin therapy and increased access to pharmacogenomic testing. Most importantly, it helped close the sex-based gap in CVD risk reduction care.
- Heron M. Deaths: leading causes for 2018. Nat Vital Stat Rep. 2021;70:1-114.
- US Department of Veterans Affairs, US Department of Defense. VA/DoD Clinical practice guideline for the management of dyslipidemia for cardiovascular risk reduction. Published June 2020. Accessed August 25, 2025. https://www.healthquality.va.gov/guidelines/CD/lipids/VADODDyslipidemiaCPG5087212020.pdf
- Atherosclerotic Cardiovascular Disease (ASCVD). American Heart Association. Accessed August 26, 2025. https:// www.heart.org/en/professional/quality-improvement/ascvd
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular disease and risk management: standards of medical care in diabetes-2022. Diabetes Care. 2022;45(Suppl 1):S144-S174. doi:10.2337/dc22-S010
- American Diabetes Association. Standards of Care in Diabetes— 2023 abridged for primary care providers. Clinical Diabetes. 2022;41(1):4-31. doi:10.2337/cd23-as01
- Virani SS, Woodard LD, Ramsey DJ, et al. Gender disparities in evidence-based statin therapy in patients with cardiovascular disease. Am J Cardiol. 2015;115:21-26. doi:10.1016/j.amjcard.2014.09.041
- Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/ AHA Guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi:10.1161/CIR.0000000000000678
- Buchanan CH, Brown EA, Bishu KG, et al. The magnitude and potential causes of gender disparities in statin therapy in veterans with type 2 diabetes: a 10-year nationwide longitudinal cohort study. Womens Health Issues. 2022;32:274-283. doi:10.1016/j.whi.2021.10.003
- Ahmed F, Lin J, Ahmed T, et al. Health disparities: statin prescribing patterns among patients with diabetes in a family medicine clinic. Health Equity. 2022;6:291-297. doi:10.1089/heq.2021.0144
- Metser G, Bradley C, Moise N, Liyanage-Don N, Kronish I, Ye S. Gaps and disparities in primary prevention statin prescription during outpatient care. Am J Cardiol. 2021;161:36-41. doi:10.1016/j.amjcard.2021.08.070
- Nanna MG, Wang TY, Xiang Q, et al. Sex differences in the use of statins in community practice. Circ Cardiovasc Qual Outcomes. 2019;12(8):e005562. doi:10.1161/CIRCOUTCOMES.118.005562
- Kitzmiller JP, Mikulik EB, Dauki AM, Murkherjee C, Luzum JA. Pharmacogenomics of statins: understanding susceptibility to adverse effects. Pharmgenomics Pers Med. 2016;9:97-106. doi:10.2147/PGPM.S86013
- Türkmen D, Masoli JAH, Kuo CL, Bowden J, Melzer D, Pilling LC. Statin treatment effectiveness and the SLCO1B1*5 reduced function genotype: long-term outcomes in women and men. Br J Clin Pharmacol. 2022;88:3230-3240. doi:10.1111/bcp.15245
- Cooper-DeHoff RM, Niemi M, Ramsey LB, et al. The Clinical Pharmacogenetics Implementation Consortium guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and statin-associated musculoskeletal symptoms. Clin Pharmacol Ther. 2022;111:1007-1021. doi:10.1002/cpt.2557
- Ramsey LB, Gong L, Lee SB, et al. PharmVar GeneFocus: SLCO1B1. Clin Pharmacol Ther. 2023;113:782-793. doi:10.1002/cpt.2705
- National Healthcare Quality and Disparities Report: Chartbook on Healthcare for Veterans. Rockville (MD): Agency for Healthcare Research and Quality (US); November 2020.
- Procario G. Primary Care Equity Dashboard [database online]. Power Bi. 2023. Accessed August 26, 2025. https://app.powerbigov.us
- Hausmann LRM, Lamorte C, Estock JL. Understanding the context for incorporating equity into quality improvement throughout a national health care system. Health Equity. 2023;7(1):312-320. doi:10.1089/heq.2023.0009
- Heron M. Deaths: leading causes for 2018. Nat Vital Stat Rep. 2021;70:1-114.
- US Department of Veterans Affairs, US Department of Defense. VA/DoD Clinical practice guideline for the management of dyslipidemia for cardiovascular risk reduction. Published June 2020. Accessed August 25, 2025. https://www.healthquality.va.gov/guidelines/CD/lipids/VADODDyslipidemiaCPG5087212020.pdf
- Atherosclerotic Cardiovascular Disease (ASCVD). American Heart Association. Accessed August 26, 2025. https:// www.heart.org/en/professional/quality-improvement/ascvd
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular disease and risk management: standards of medical care in diabetes-2022. Diabetes Care. 2022;45(Suppl 1):S144-S174. doi:10.2337/dc22-S010
- American Diabetes Association. Standards of Care in Diabetes— 2023 abridged for primary care providers. Clinical Diabetes. 2022;41(1):4-31. doi:10.2337/cd23-as01
- Virani SS, Woodard LD, Ramsey DJ, et al. Gender disparities in evidence-based statin therapy in patients with cardiovascular disease. Am J Cardiol. 2015;115:21-26. doi:10.1016/j.amjcard.2014.09.041
- Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/ AHA Guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi:10.1161/CIR.0000000000000678
- Buchanan CH, Brown EA, Bishu KG, et al. The magnitude and potential causes of gender disparities in statin therapy in veterans with type 2 diabetes: a 10-year nationwide longitudinal cohort study. Womens Health Issues. 2022;32:274-283. doi:10.1016/j.whi.2021.10.003
- Ahmed F, Lin J, Ahmed T, et al. Health disparities: statin prescribing patterns among patients with diabetes in a family medicine clinic. Health Equity. 2022;6:291-297. doi:10.1089/heq.2021.0144
- Metser G, Bradley C, Moise N, Liyanage-Don N, Kronish I, Ye S. Gaps and disparities in primary prevention statin prescription during outpatient care. Am J Cardiol. 2021;161:36-41. doi:10.1016/j.amjcard.2021.08.070
- Nanna MG, Wang TY, Xiang Q, et al. Sex differences in the use of statins in community practice. Circ Cardiovasc Qual Outcomes. 2019;12(8):e005562. doi:10.1161/CIRCOUTCOMES.118.005562
- Kitzmiller JP, Mikulik EB, Dauki AM, Murkherjee C, Luzum JA. Pharmacogenomics of statins: understanding susceptibility to adverse effects. Pharmgenomics Pers Med. 2016;9:97-106. doi:10.2147/PGPM.S86013
- Türkmen D, Masoli JAH, Kuo CL, Bowden J, Melzer D, Pilling LC. Statin treatment effectiveness and the SLCO1B1*5 reduced function genotype: long-term outcomes in women and men. Br J Clin Pharmacol. 2022;88:3230-3240. doi:10.1111/bcp.15245
- Cooper-DeHoff RM, Niemi M, Ramsey LB, et al. The Clinical Pharmacogenetics Implementation Consortium guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and statin-associated musculoskeletal symptoms. Clin Pharmacol Ther. 2022;111:1007-1021. doi:10.1002/cpt.2557
- Ramsey LB, Gong L, Lee SB, et al. PharmVar GeneFocus: SLCO1B1. Clin Pharmacol Ther. 2023;113:782-793. doi:10.1002/cpt.2705
- National Healthcare Quality and Disparities Report: Chartbook on Healthcare for Veterans. Rockville (MD): Agency for Healthcare Research and Quality (US); November 2020.
- Procario G. Primary Care Equity Dashboard [database online]. Power Bi. 2023. Accessed August 26, 2025. https://app.powerbigov.us
- Hausmann LRM, Lamorte C, Estock JL. Understanding the context for incorporating equity into quality improvement throughout a national health care system. Health Equity. 2023;7(1):312-320. doi:10.1089/heq.2023.0009
Reducing Sex Disparities in Statin Therapy Among Female Veterans With Type 2 Diabetes and/or Cardiovascular Disease
Reducing Sex Disparities in Statin Therapy Among Female Veterans With Type 2 Diabetes and/or Cardiovascular Disease
Implementation of a VHA Virtual Oncology Training Pilot Program for Clinical Pharmacists
Purpose/Background
Oncology clinical pharmacist practitioners (CPP) play a critical role in optimizing drug therapy, managing side effects, and ensuring medication adherence. As a specialized clinical area, specific training is needed to ensure quality of care. Oncology pharmacy training programs are commercially available but pose a financial burden and are not specific to the Veterans Health Administration (VHA). A comprehensive, virtual Oncology Bootcamp series was implemented to upskill new oncology pharmacists (or pharmacists seeking to further their understanding of oncology practice), with didactic materials and clinical tools to enhance and standardize quality care delivery.
Methods
This program was comprised of an online platform of 23 one hour-long continuing education accredited sessions, delivered by leading subject matter experts. Pharmacists from two Veteran Integrated Service Networks (VISNs) were invited for the first year of the bootcamp. The curriculum encompassed fundamentals of oncology practice, patient care assessment, chemotherapy protocol review, practice management, and supportive care. Participants also received in-depth training on managing various cancer types, including but not limited to prostate, lung, gastrointestinal and hematologic malignancies. VHA specific information, including utilization of Oncology Clinical Pathways to promote standardized care was included where applicable. The interactive nature of the virtual sessions provided opportunities for real-time discussion and immediate feedback. To measure the impact of this program, a pre and post program evaluation of participants was conducted.
Results
Over the course of the program, more than 40 pharmacists across two VISNs participated in the bootcamp series. Results of the program evaluation showed an increase in self-reported comfort and skill levels in all criteria that were assessed (oncology pharmacotherapy, solid tumor malignancies, hematologic malignancies and oral anti-cancer therapy management). Additionally, 85% of respondents stated the series met their overall goals and over 90% of respondents stated they were either satisfied or very satisfied with the content, speakers and organization of the course.
Implications/Significance
This initiative has established the viability and significance of a highly accessible, VHA pathway specific and Veteran centric platform for oncology pharmacy professional development. Future directions for the program include a broader nationwide audience, increased content coverage and self-paced learning options.
Purpose/Background
Oncology clinical pharmacist practitioners (CPP) play a critical role in optimizing drug therapy, managing side effects, and ensuring medication adherence. As a specialized clinical area, specific training is needed to ensure quality of care. Oncology pharmacy training programs are commercially available but pose a financial burden and are not specific to the Veterans Health Administration (VHA). A comprehensive, virtual Oncology Bootcamp series was implemented to upskill new oncology pharmacists (or pharmacists seeking to further their understanding of oncology practice), with didactic materials and clinical tools to enhance and standardize quality care delivery.
Methods
This program was comprised of an online platform of 23 one hour-long continuing education accredited sessions, delivered by leading subject matter experts. Pharmacists from two Veteran Integrated Service Networks (VISNs) were invited for the first year of the bootcamp. The curriculum encompassed fundamentals of oncology practice, patient care assessment, chemotherapy protocol review, practice management, and supportive care. Participants also received in-depth training on managing various cancer types, including but not limited to prostate, lung, gastrointestinal and hematologic malignancies. VHA specific information, including utilization of Oncology Clinical Pathways to promote standardized care was included where applicable. The interactive nature of the virtual sessions provided opportunities for real-time discussion and immediate feedback. To measure the impact of this program, a pre and post program evaluation of participants was conducted.
Results
Over the course of the program, more than 40 pharmacists across two VISNs participated in the bootcamp series. Results of the program evaluation showed an increase in self-reported comfort and skill levels in all criteria that were assessed (oncology pharmacotherapy, solid tumor malignancies, hematologic malignancies and oral anti-cancer therapy management). Additionally, 85% of respondents stated the series met their overall goals and over 90% of respondents stated they were either satisfied or very satisfied with the content, speakers and organization of the course.
Implications/Significance
This initiative has established the viability and significance of a highly accessible, VHA pathway specific and Veteran centric platform for oncology pharmacy professional development. Future directions for the program include a broader nationwide audience, increased content coverage and self-paced learning options.
Purpose/Background
Oncology clinical pharmacist practitioners (CPP) play a critical role in optimizing drug therapy, managing side effects, and ensuring medication adherence. As a specialized clinical area, specific training is needed to ensure quality of care. Oncology pharmacy training programs are commercially available but pose a financial burden and are not specific to the Veterans Health Administration (VHA). A comprehensive, virtual Oncology Bootcamp series was implemented to upskill new oncology pharmacists (or pharmacists seeking to further their understanding of oncology practice), with didactic materials and clinical tools to enhance and standardize quality care delivery.
Methods
This program was comprised of an online platform of 23 one hour-long continuing education accredited sessions, delivered by leading subject matter experts. Pharmacists from two Veteran Integrated Service Networks (VISNs) were invited for the first year of the bootcamp. The curriculum encompassed fundamentals of oncology practice, patient care assessment, chemotherapy protocol review, practice management, and supportive care. Participants also received in-depth training on managing various cancer types, including but not limited to prostate, lung, gastrointestinal and hematologic malignancies. VHA specific information, including utilization of Oncology Clinical Pathways to promote standardized care was included where applicable. The interactive nature of the virtual sessions provided opportunities for real-time discussion and immediate feedback. To measure the impact of this program, a pre and post program evaluation of participants was conducted.
Results
Over the course of the program, more than 40 pharmacists across two VISNs participated in the bootcamp series. Results of the program evaluation showed an increase in self-reported comfort and skill levels in all criteria that were assessed (oncology pharmacotherapy, solid tumor malignancies, hematologic malignancies and oral anti-cancer therapy management). Additionally, 85% of respondents stated the series met their overall goals and over 90% of respondents stated they were either satisfied or very satisfied with the content, speakers and organization of the course.
Implications/Significance
This initiative has established the viability and significance of a highly accessible, VHA pathway specific and Veteran centric platform for oncology pharmacy professional development. Future directions for the program include a broader nationwide audience, increased content coverage and self-paced learning options.
Assessing the Impact of Antidepressants on Cancer Treatment: A Retrospective Analysis of 14 Antineoplastic Agents
Assessing the Impact of Antidepressants on Cancer Treatment: A Retrospective Analysis of 14 Antineoplastic Agents
Cancer patients experience depression at rates > 5 times that of the general population.1-11 Despite an increase in palliative care use, depression rates continued to rise.2-4 Between 5% to 16% of outpatients, 4% to 14% of inpatients, and up to 49% of patients receiving palliative care experience depression.5 This issue also impacts families and caregivers.1 A 2021 meta-analysis found that 23% of active military personnel and 20% of veterans experience depression.11
Antidepressants approved by the US Food and Drug Administration (FDA) target the serotonin, norepinephrine, or dopamine systems and include boxed warnings about an increased risk of suicidal thoughts in adults aged 18 to 24 years.12,13 These medications are categorized into several classes: monoamine oxidase inhibitors (MAOIs), tricyclic antidepressants (TCAs), tetracyclic antidepressants (TeCAs), norepinephrine-dopamine reuptake inhibitors (NDRIs), selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), serotonin receptor modulators (SRMs), serotonin-melatonin receptor antagonists (SMRAs), and N—methyl-D-aspartate receptor antagonists (NMDARAs).14,15 The first FDA-approved antidepressants, iproniazid (an MAOI) and imipramine (a TCA) laid the foundation for the development of newer classes like SSRIs and SNRIs.15-17
Older antidepressants such as MAOIs and TCAs are used less due to their adverse effects (AEs) and drug interactions. MAOIs, such as iproniazid, selegiline, moclobemide, tranylcypromine, isocarboxazid, and phenelzine, have numerous AEs and drug interactions, making them unsuitable for first- or second-line treatment of depression.14,18-21 TCAs such as doxepin, amitriptyline, nortriptyline, imipramine, desipramine, clomipramine, trimipramine, protriptyline, maprotiline, and amoxapine have a narrow therapeutic index requiring careful monitoring for signs of toxicity such as QRS widening, tremors, or confusion. Despite the issues, TCAs are generally classified as second-line agents for major depressive disorder (MDD). TCAs have off-label uses for migraine prophylaxis, treatment of obsessive-compulsive disorder (OCD), insomnia, and chronic pain management first-line.14,22-29
Newer antidepressants, including TeCAs and NDRIs, are typically more effective, but also come with safety concerns. TeCAs like mirtazapine interact with several medications, including MAOIs, serotonin-increasing drugs, alcohol, cannabidiol, and marijuana. Mirtazapine is FDA-approved for the treatment of moderate to severe depression in adults. It is also used off-label to treat insomnia, panic disorder, posttraumatic stress disorder (PTSD), generalized anxiety disorder (GAD), social anxiety disorder (SAD), headaches, and migraines. Compared to other antidepressants, mirtazapine is effective for all stages of depression and addresses a broad range of related symptoms.14,30-34 NDRIs, such as bupropion, also interact with various medications, including MAOIs, other antidepressants, stimulants, and alcohol. Bupropion is FDA-approved for smoking cessation and to treat depression and SAD. It is also used off-label for depression- related bipolar disorder or sexual dysfunction, attention-deficit/hyperactivity disorder (ADHD), and obesity.14,35-42
SSRIs, SNRIs, and SRMs should be used with caution. SSRIs such as sertraline, citalopram, escitalopram, fluoxetine, paroxetine, and fluvoxamine are first-line treatments for depression and various psychiatric disorders due to their safety and efficacy. Common AEs of SSRIs include sexual dysfunction, sleep disturbances, weight changes, and gastrointestinal (GI) issues. SSRIs can prolong the QT interval, posing a risk of life-threatening arrhythmia, and may interact with other medications, necessitating treatment adjustments. The FDA approved SSRIs for MDD, GAD, bulimia nervosa, bipolar depression, OCD, panic disorder, premenstrual dysphoric disorder, treatment-resistant depression, PTSD, and SAD. Off-label uses include binge eating disorder, body dysmorphic disorder, fibromyalgia, premature ejaculation, paraphilias, autism, Raynaud phenomenon, and vasomotor symptoms associated with menopause. Among SSRIs, sertraline and escitalopram are noted for their effectiveness and tolerability.14,43-53
SNRIs, including duloxetine, venlafaxine, desvenlafaxine, milnacipran, and levomilnacipran, may increase bleeding risk, especially when taken with blood thinners. They can also elevate blood pressure, which may worsen if combined with stimulants. SNRIs may interact with other medications that affect serotonin levels, increasing the risk of serotonin syndrome when taken with triptans, pain medications, or other antidepressants.14 Desvenlafaxine has been approved by the FDA (but not by the European Medicines Agency).54-56 Duloxetine is FDA-approved for the treatment of depression, neuropathic pain, anxiety disorders, fibromyalgia, and musculoskeletal disorders. It is used off-label to treat chemotherapy-induced peripheral neuropathy and stress urinary incontinence.57-61 Venlafaxine is FDA-approved for depression, SAD, and panic disorder, and is prescribed off-label to treat ADHD, neuropathy, fibromyalgia, cataplexy, and PTSD, either alone or in combination with other medications.62,63 Milnacipran is not approved for MDD; levomilnacipran received approval in 2013.64
SRMs such as trazodone, nefazodone, vilazodone, and vortioxetine also function as serotonin reuptake inhibitors.14,15 Trazodone is FDA-approved for MDD. It has been used off-label to treat anxiety, Alzheimer disease, substance misuse, bulimia nervosa, insomnia, fibromyalgia, and PTSD when first-line SSRIs are ineffective. A notable AE of trazodone is orthostatic hypotension, which can lead to dizziness and increase the risk of falls, especially in geriatric patients.65-70 Nefazodone was discontinued in Europe in 2003 due to rare cases of liver toxicity but remains available in the US.71-74 Vilazodone and vortioxetine are FDA-approved.
The latest classes of antidepressants include SMRAs and NMDARAs.14 Agomelatine, an SMRA, was approved in Europe in 2009 but rejected by the FDA in 2011 due to liver toxicity.75 NMDARAs like esketamine and a combination of dextromethorphan and bupropion received FDA approval in 2019 and 2022, respectively.76,77
This retrospective study analyzes noncancer drugs used during systemic chemotherapy based on a dataset of 14 antineoplastic agents. It sought to identify the most dispensed noncancer drug groups, discuss findings, compare patients with and without antidepressant prescriptions, and examine trends in antidepressant use from 2002 to 2023. This analysis expands on prior research.78-81
Methods
The Walter Reed National Military Medical Center Institutional Review Board approved the study protocol and ensured compliance with the Health Insurance Portability and Accountability Act as an exempt protocol. The Joint Pathology Center (JPC) of the US Department of Defense (DoD) Cancer Registry Program and Military Health System (MHS) data experts from the Comprehensive Ambulatory/Professional Encounter Record (CAPER) and Pharmacy Data Transaction Service (PDTS) provided data for the analysis.
Data Sources
The JPC DoD Cancer Registry Program contains data from 1998 to 2024. CAPER and PDTS are part of the MHS Data Repository/Management Analysis and Reporting Tool database. Each observation in CAPER represents an ambulatory encounter at a military treatment facility (MTF). CAPER records are available from 2003 to 2024. PDTS records are available from 2002 to 2004. Each observation in PDTS represents a prescription filled for an MHS beneficiary, excluding those filled at international civilian pharmacies and inpatient pharmacy prescriptions.
This cross-sectional analysis requested data extraction for specific cancer drugs from the DoD Cancer Registry, focusing on treatment details, diagnosis dates, patient demographics, and physicians’ comments on AEs. After identifying patients, CAPER was used to identify additional health conditions. PDTS was used to compile a list of prescription medications filled during systemic cancer treatment or < 2 years postdiagnosis.
The 2016 Surveillance, Epidemiology, and End Results Program Coding and Staging Manual and International Classification of Diseases for Oncology, 3rd edition, 1st revision, were used to decode disease and cancer types.82,83 Data sorting and analysis were performed using Microsoft Excel. The percentage for the total was calculated by using the number of patients or data available within the subgroup divided by the total number of patients or data variables. To compare the mean number of dispensed antidepressants to those without antidepressants, a 2-tailed, 2-sample z test was used to calculate the P value and determine statistical significance (P < .05) using socscistatistics.com.
Data were extracted 3 times between 2021 and 2023. The initial 2021 protocol focused on erlotinib and gefitinib. A modified protocol in 2022 added paclitaxel, cisplatin, docetaxel, pemetrexed, and crizotinib; further modification in 2023 included 8 new antineoplastic agents and 2 anticoagulants. Sotorasib has not been prescribed in the MHS, and JPC lacks records for noncancer drugs. The 2023 dataset comprised 2210 patients with cancer treated with 14 antineoplastic agents; 2104 had documented diagnoses and 2113 had recorded prescriptions. Data for erlotinib, gefitinib, and paclitaxel have been published previously.78,79
Results
Of 2113 patients with recorded prescriptions, 1297 patients (61.4%) received 109 cancer drugs, including 96 antineoplastics, 7 disease-modifying antirheumatic agents, 4 biologic response modifiers, and 2 calcitonin gene-related peptides. Fourteen antineoplastic agents had complete data from JPC, while others were noted for combination therapies or treatment switches from the PDTS (Table 1). Seventy-six cancer drugs were prescribed with antidepressants in 489 patients (eAppendix).
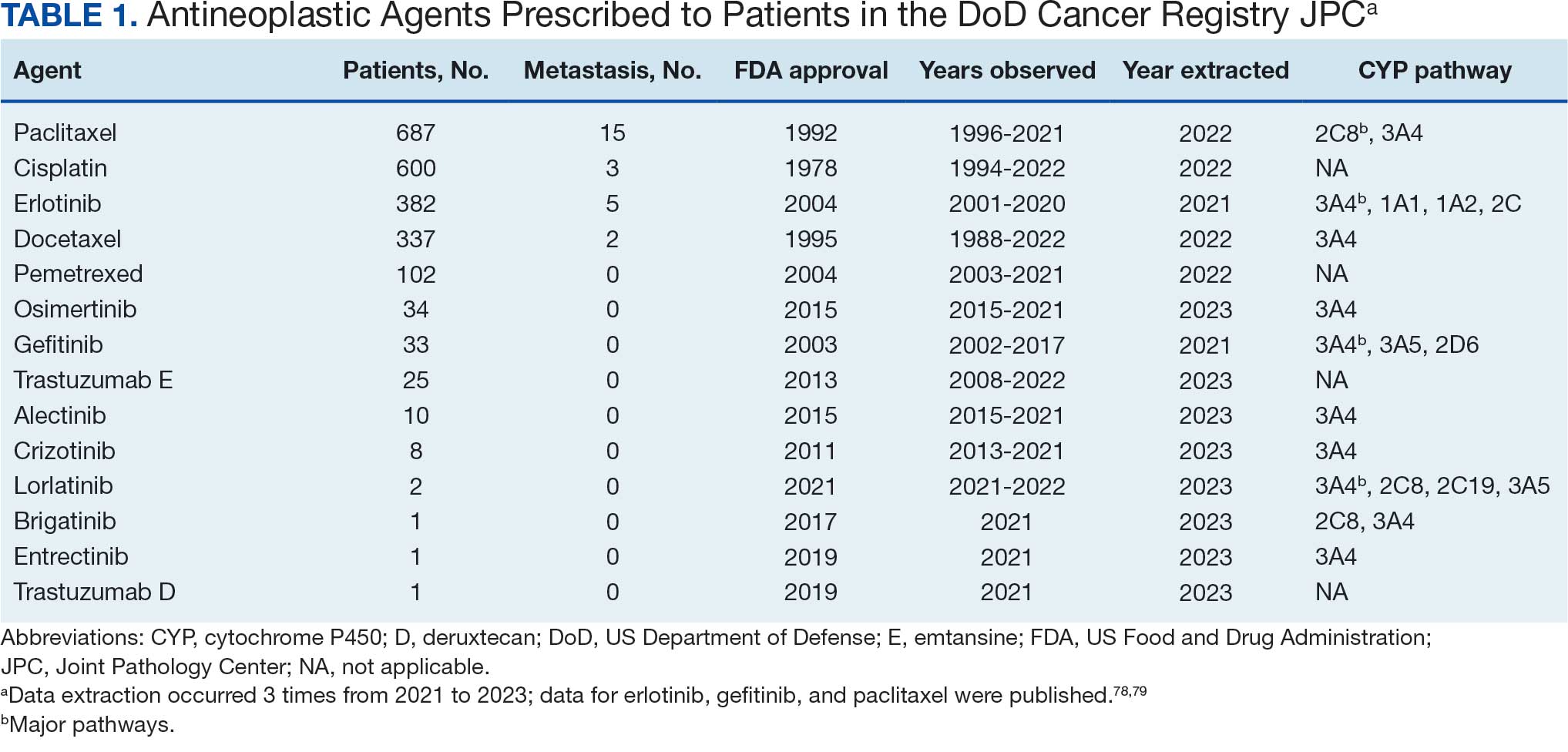
The JPC provided 2242 entries for 2210 patients, ranging in age from 2 months to 88 years (mean, 56 years), documenting treatment from September 1988 to January 2023. Thirty-two patients had duplicate entries due to multiple cancer locations or occurrences. Of the 2242 patients, 1541 (68.7%) were aged > 50 years, 975 patients (43.5%) had cancers that were stage III or IV, and 1267 (56.5%) had cancers that were stage 0, I, II, or not applicable/unknown. There were 51 different types of cancer: breast, lung, testicular, endometrial, and ovarian were most common (n ≥ 100 patients). Forty-two cancer types were documented among 750 patients prescribed antidepressants (Table 2).
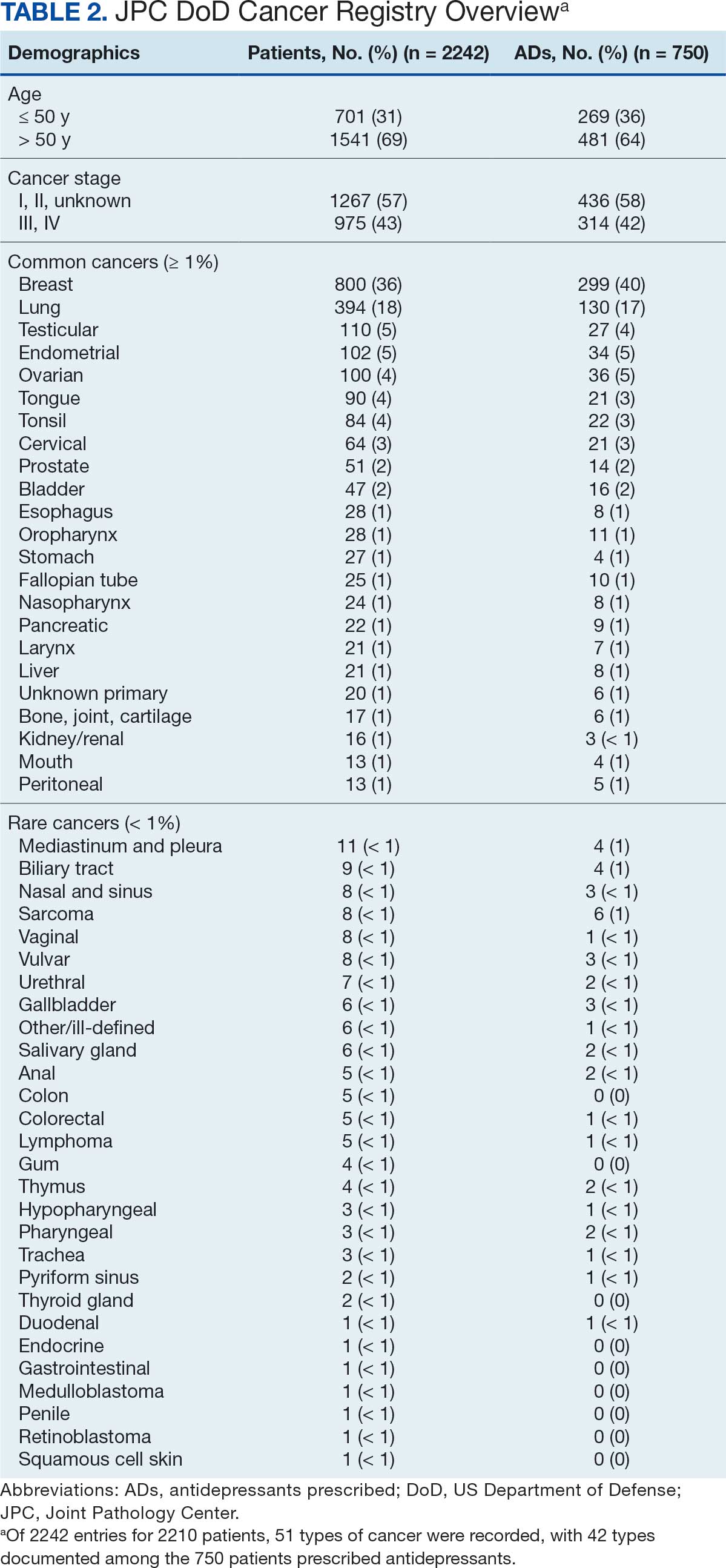
The CAPER database recorded 8882 unique diagnoses for 2104 patients, while PDTS noted 1089 unique prescriptions within 273 therapeutic codes for 2113 patients. Nine therapeutic codes (opiate agonists, adrenals, cathartics-laxatives, nonsteroidal anti-inflammatory agents, antihistamines for GI conditions, 5-HT3 receptor antagonists, analgesics and antipyretic miscellanea, antineoplastic agents, and proton-pump inhibitors) and 8 drugs (dexamethasone, prochlorperazine, ondansetron, docusate, acetaminophen, ibuprofen, oxycodone, and polyethylene glycol 3350) were associated with > 1000 patients (≥ 50%). Patients had between 1 and 275 unique health conditions and filled 1 to 108 prescriptions. The mean (SD) number of diagnoses and prescriptions was 50 (28) and 29 (12), respectively. Of the 273 therapeutic codes, 30 groups were analyzed, with others categorized into miscellaneous groups such as lotions, vaccines, and devices. Significant differences in mean number of prescriptions were found for patients taking antidepressants compared to those not (P < .05), except for anticonvulsants and antipsychotics (P = .12 and .09, respectively) (Table 3).
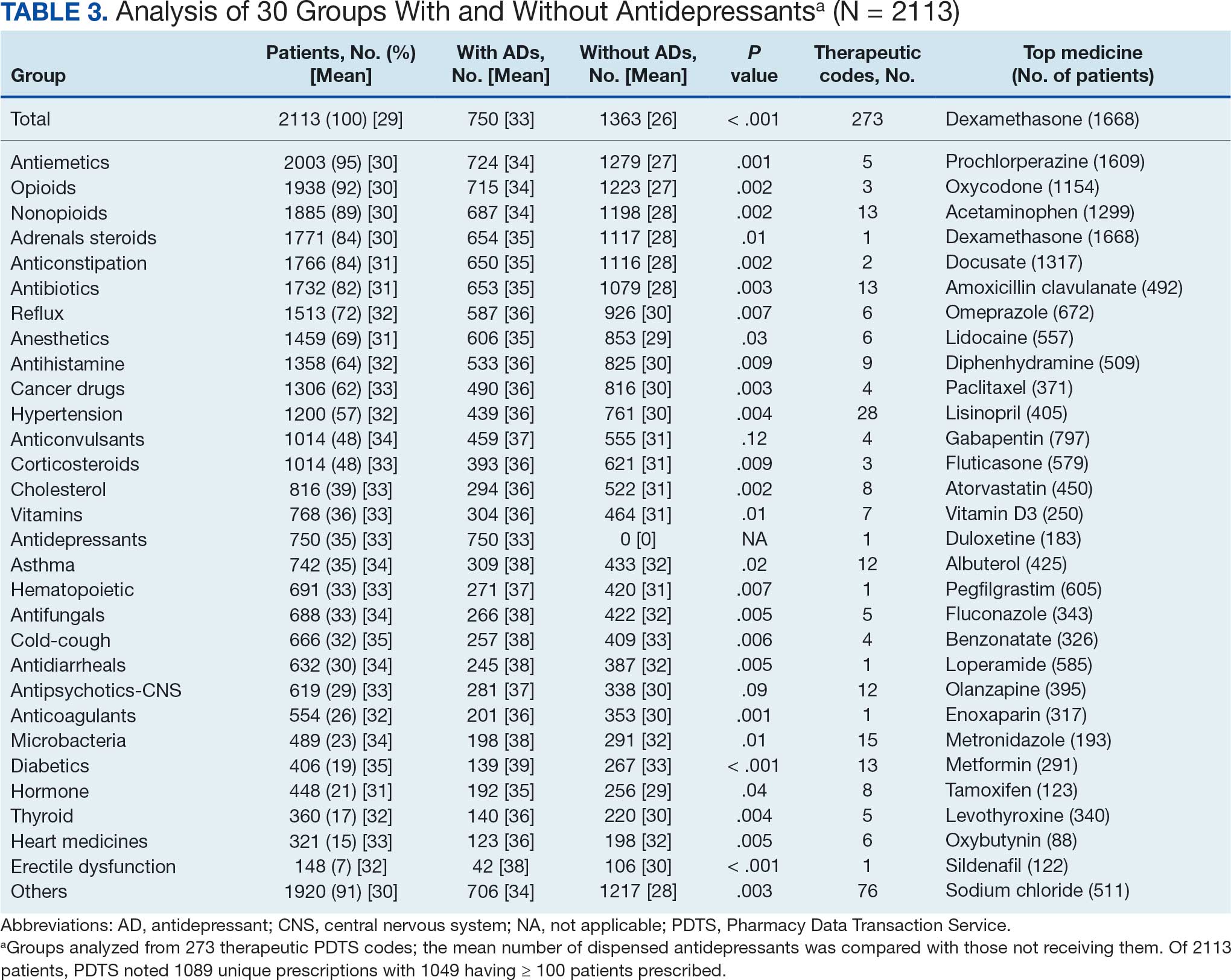
Antidepressants
Of the 2113 patients with recorded prescriptions, 750 (35.5%) were dispensed 17 different antidepressants. Among these 17 antidepressants, 183 (8.7%) patients received duloxetine, 158 (7.5%) received venlafaxine, 118 (5.6%) received trazodone, and 107 (5.1%) received sertraline (Figure 1, Table 4). Of the 750 patients, 509 (67.9%) received 1 antidepressant, 168 (22.4%) received 2, 60 (8.0%) received 3, and 13 (1.7%) received > 3. Combinations varied, but only duloxetine and trazodone were prescribed to > 10 patients.
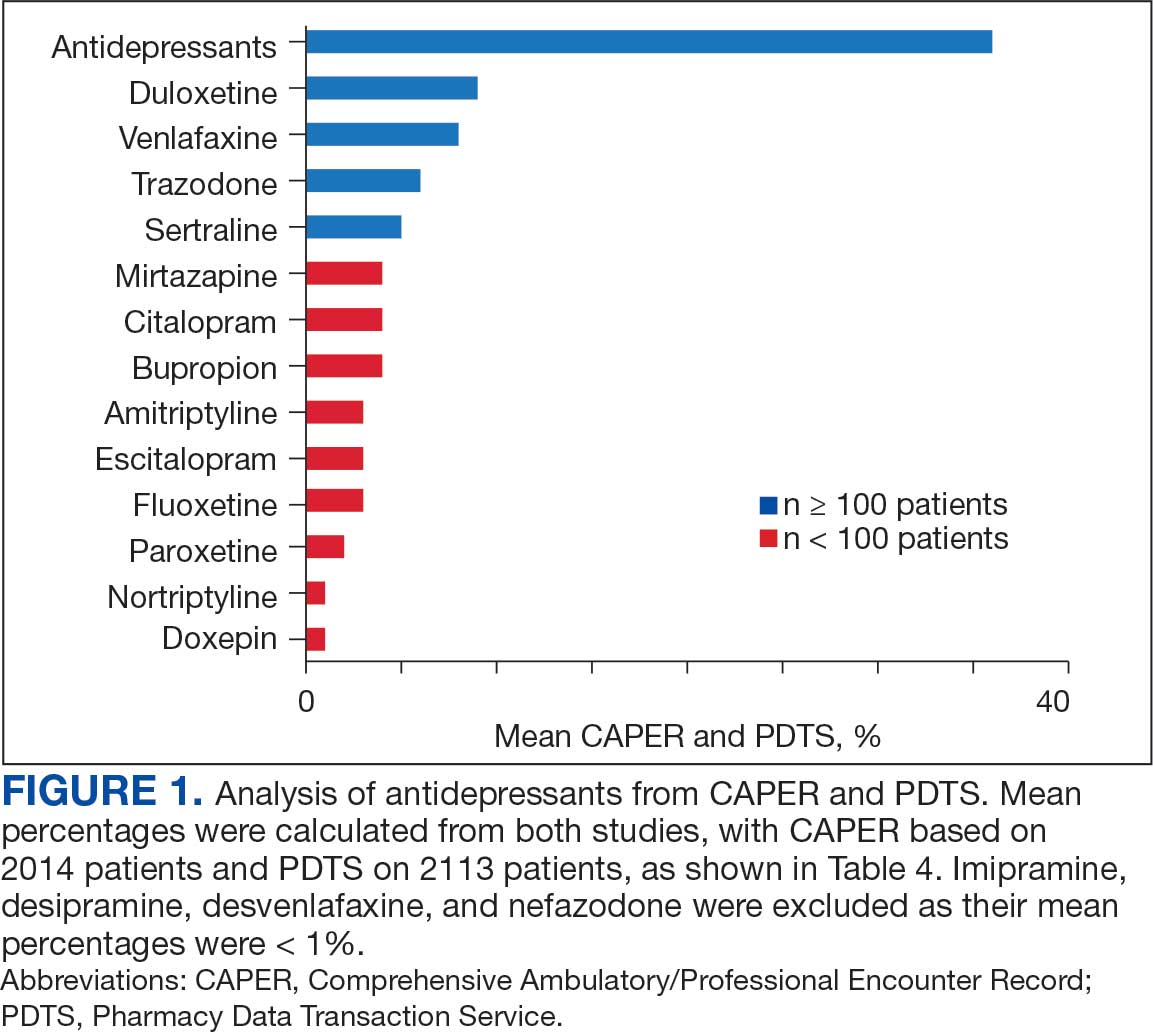
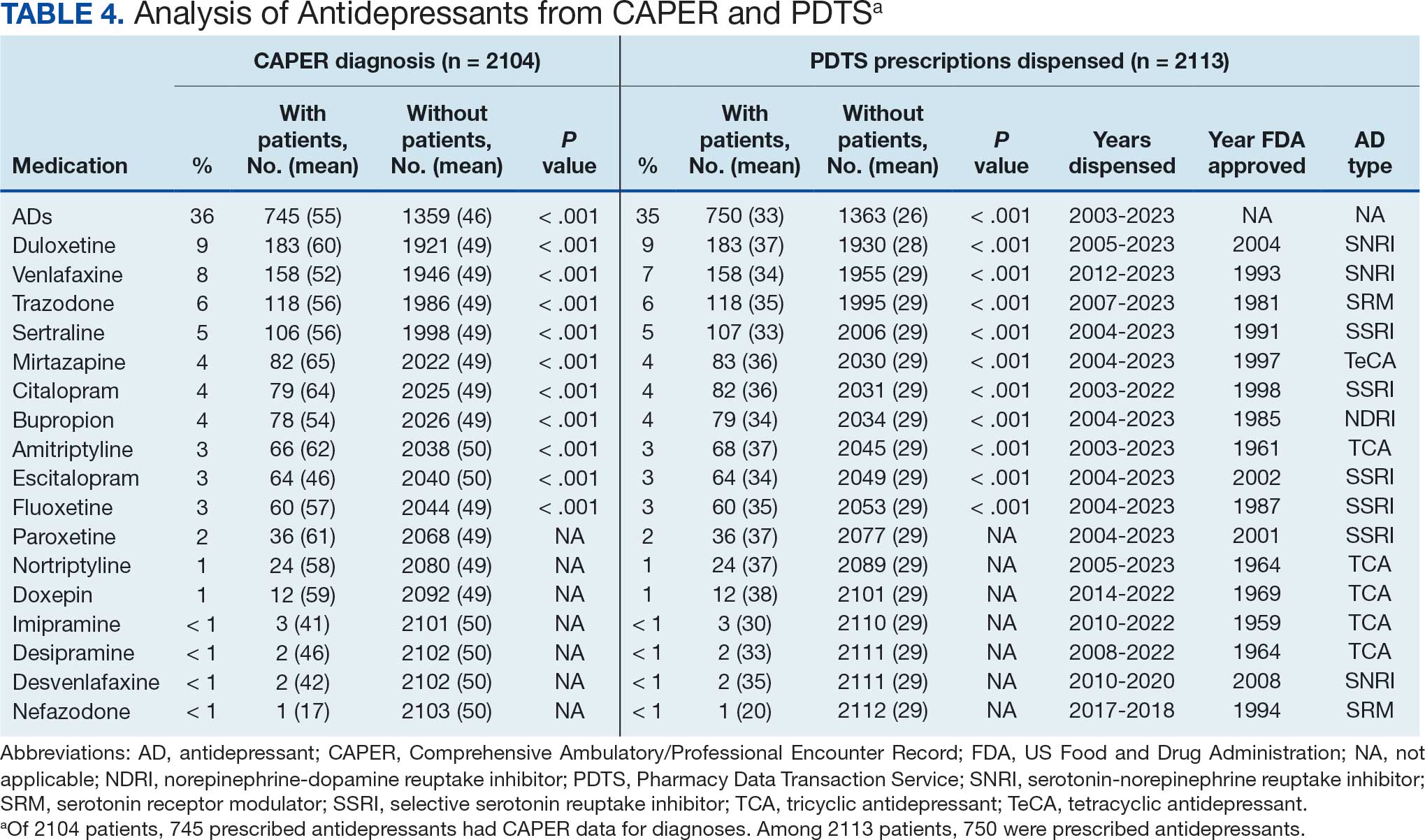
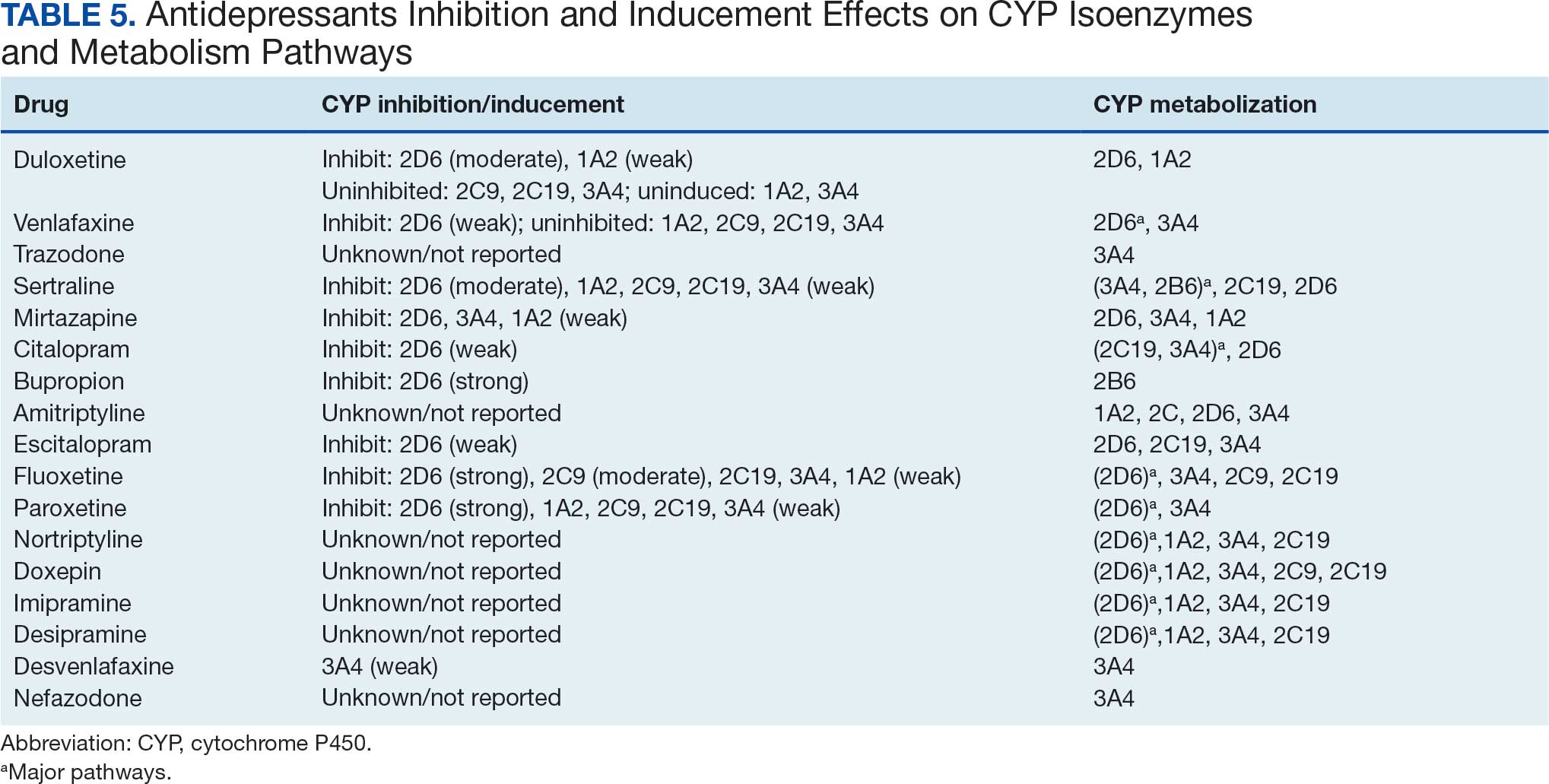
Antidepressants were prescribed annually at an overall mean (SD) rate of 23% (5%) from 2003 to 2022 (Figure 2). Patients on antidepressants during systemic therapy had a greater number of diagnosed medical conditions and received more prescription medications compared to those not taking antidepressants (P < .001) (Figure 3). The 745 patients taking antidepressants in CAPER data had between 1 and 275 diagnosed medical issues, with a mean (SD) of 55 (31) vs a range of 1 to 209 and a mean (SD) of 46 (26) for the 1359 patients not taking antidepressants. The 750 patients on antidepressants in PDTS data had between 8 and 108 prescriptions dispensed, with a mean (SD) of 32 (12), vs a range of 1 to 65 prescriptions and a mean (SD) of 29 (12) for 1363 patients not taking antidepressants.
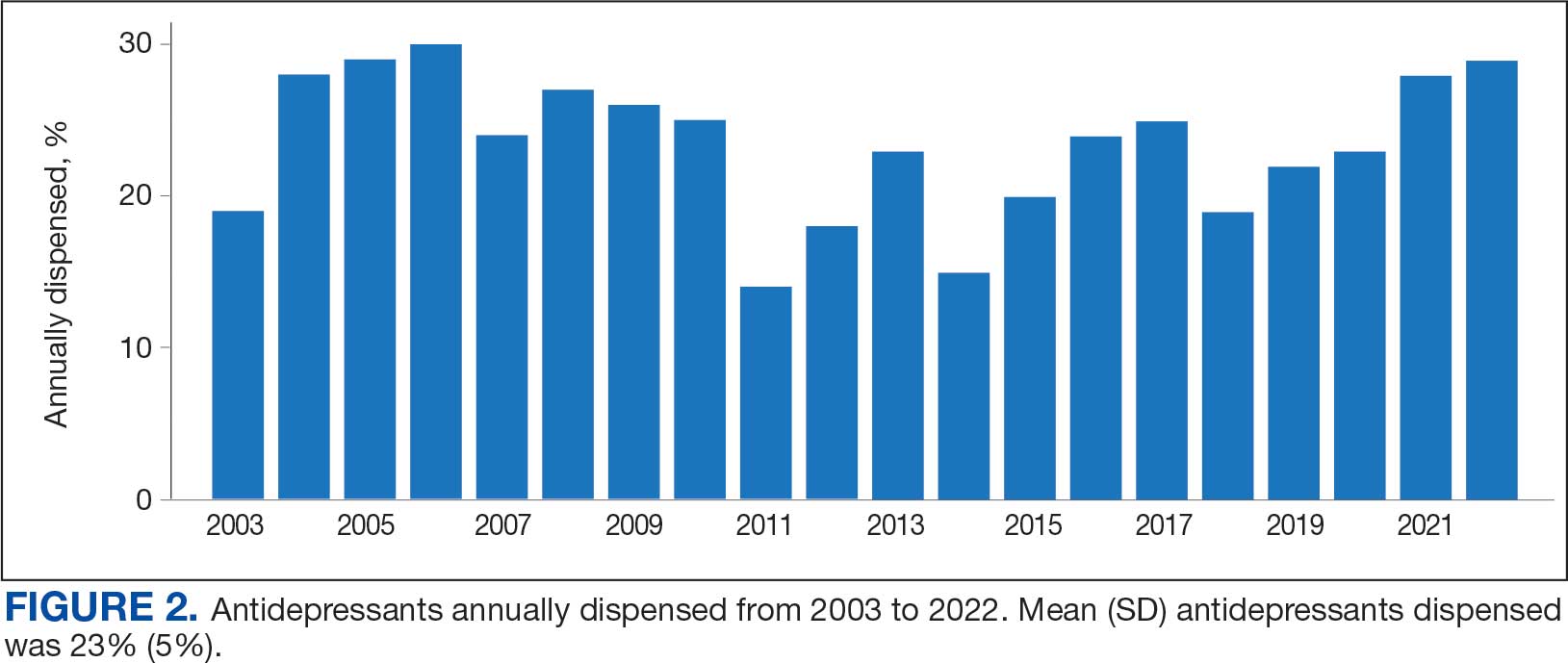
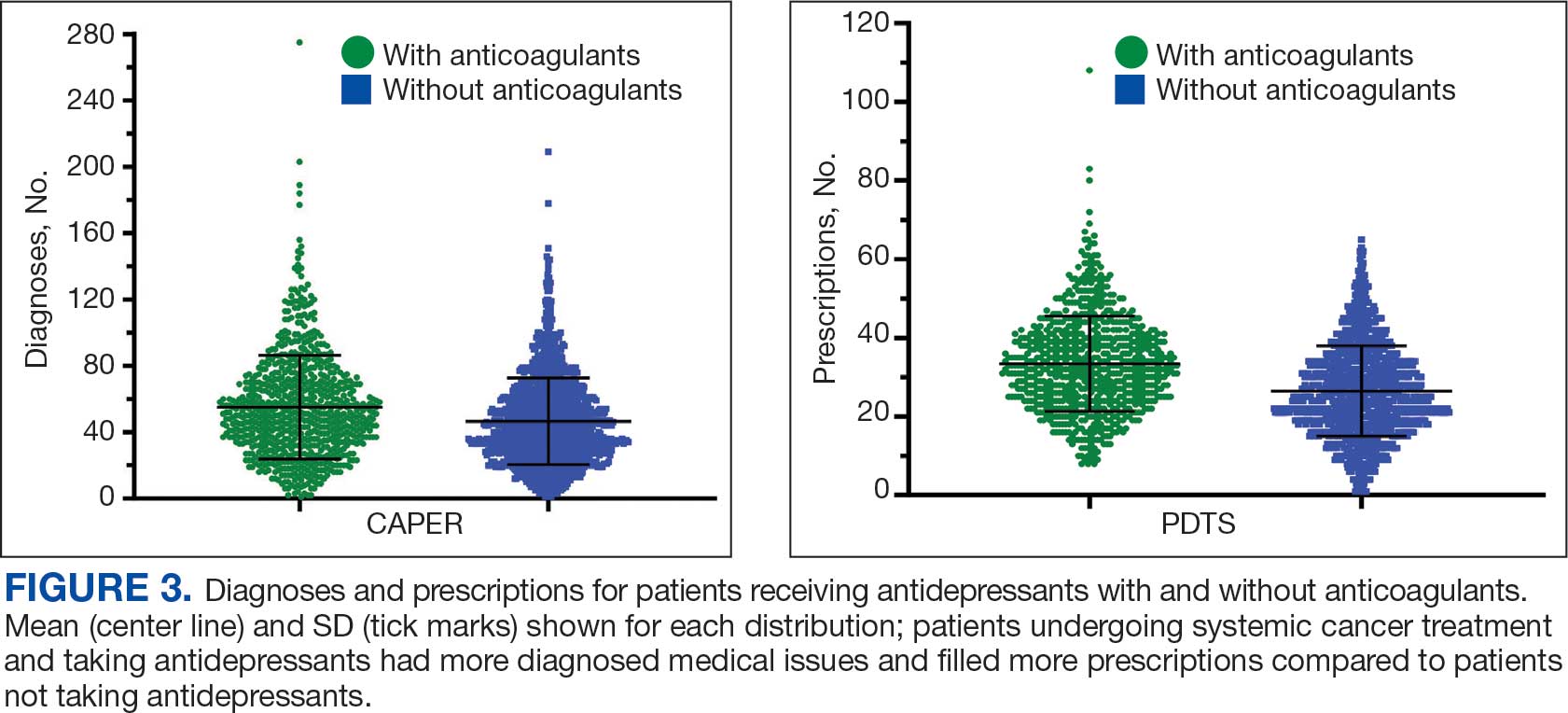
Discussion
The JPC DoD Cancer Registry includes information on cancer types, stages, treatment regimens, and physicians’ notes, while noncancer drugs are sourced from the PDTS database. The pharmacy uses a different documentation system, leading to varied classifications.
Database reliance has its drawbacks. For example, megestrol is coded as a cancer drug, although it’s primarily used for endometrial or gynecologic cancers. Many drugs have multiple therapeutic codes assigned to them, including 10 antineoplastic agents: diclofenac, Bacillus Calmette-Guérin (BCG), megestrol acetate, tamoxifen, anastrozole, letrozole, leuprolide, goserelin, degarelix, and fluorouracil. Diclofenac, BCG, and mitomycin have been repurposed for cancer treatment.84-87 From 2003 to 2023, diclofenac was prescribed to 350 patients for mild-to-moderate pain, with only 2 patients receiving it for cancer in 2018. FDA-approved for bladder cancer in 1990, BCG was prescribed for cancer treatment for 1 patient in 2021 after being used for vaccines between 2003 and 2018. Tamoxifen, used for hormone receptor-positive breast cancer from 2004 to 2017 with 53 patients, switched to estrogen agonist-antagonists from 2017 to 2023 with 123 patients. Only a few of the 168 patients were prescribed tamoxifen using both codes.88-91 Anastrozole and letrozole were coded as antiestrogens for 7 and 18 patients, respectively, while leuprolide and goserelin were coded as gonadotropins for 59 and 18 patients. Degarelix was coded as antigonadotropins, fluorouracil as skin and mucous membrane agents miscellaneous, and megestrol acetate as progestins for 7, 6, and 3 patients, respectively. Duloxetine was given to 186 patients, primarily for depression from 2005 to 2023, with 7 patients treated for fibromyalgia from 2022 to 2023.
Antidepressants Observed
Tables 1 and 5 provide insight into the FDA approval of 14 antineoplastics and antidepressants and their CYP metabolic pathways.92-122 In Table 4, the most prescribed antidepressant classes are SNRIs, SRMs, SSRIs, TeCAs, NDRIs, and TCAs. This trend highlights a preference for newer medications with weak CYP inhibition. A total of 349 patients were prescribed SSRIs, 343 SNRIs, 119 SRMs, 109 TCAs, 83 TeCAs, and 79 NDRIs. MAOIs, SMRAs, and NMDARAs were not observed in this dataset. While there are instances of dextromethorphan-bupropion and sertraline-escitalopram being dispensed together, it remains unclear whether these were NMDARA combinations.
Among the 14 specific antineoplastic agents, 10 are metabolized by CYP isoenzymes, primarily CYP3A4. Duloxetine neither inhibits nor is metabolized by CYP3A4, a reason it is often recommended, following venlafaxine.
Both duloxetine and venlafaxine are used off-label for chemotherapy-induced peripheral neuropathy related to paclitaxel and docetaxel. According to the CYP metabolized pathway, duloxetine tends to have more favorable DDIs than venlafaxine. In PDTS data, 371 patients were treated with paclitaxel and 180 with docetaxel, with respective antidepressant prescriptions of 156 and 70. Of the 156 patients dispensed paclitaxel, 62 (40%) were dispensed with duloxetine compared to 43 (28%) with venlafaxine. Of the 70 patients dispensed docetaxel, 23 (33%) received duloxetine vs 24 (34%) with venlafaxine.
Of 85 patients prescribed duloxetine, 75 received it with either paclitaxel or docetaxel (5 received both). Five patients had documented AEs (1 neuropathy related). Of 67 patients prescribed venlafaxine, 66 received it with either paclitaxel or docetaxel. Two patients had documented AEs (1 was neuropathy related, the same patient who received duloxetine). Of the 687 patients treated with paclitaxel and 337 with docetaxel in all databases, 4 experienced neuropathic AEs from both medications.79
Antidepressants can increase the risk of bleeding, especially when combined with blood thinners, and may elevate blood pressure, particularly alongside stimulants. Of the 554 patients prescribed 9 different anticoagulants, enoxaparin, apixaban, and rivaroxaban were the most common (each > 100 patients). Among these, 201 patients (36%) received both anticoagulants and antidepressants: duloxetine for 64 patients, venlafaxine for 30, trazodone for 35, and sertraline for 26. There were no data available to assess bleeding rates related to the evaluation of DDIs between these medication classes.
Antidepressants can be prescribed for erectile dysfunction. Of the 148 patients prescribed an antidepressant for erectile dysfunction, duloxetine, trazodone, and mirtazapine were the most common. Antidepressant preferences varied by cancer type. Duloxetine was the only antidepressant used for all types of cancer. Venlafaxine, duloxetine, trazodone, sertraline, and escitalopram were the most prescribed antidepressants for breast cancer, while duloxetine, mirtazapine, citalopram, sertraline, and trazodone were the most prescribed for lung cancer. Sertraline, duloxetine, trazodone, amitriptyline, and escitalopram were most common for testicular cancer. Duloxetine, venlafaxine, trazodone, amitriptyline, and sertraline were the most prescribed for endometrial cancer, while duloxetine, venlafaxine, amitriptyline, citalopram, and sertraline were most prescribed for ovarian cancer.
The broadness of International Statistical Classification of Diseases, Tenth Revision codes made it challenging to identify nondepression diagnoses in the analyzed population. However, if all antidepressants were prescribed to treat depression, service members with cancer exhibited a higher depression rate (35%) than the general population (25%). Of 2104 patients, 191 (9.1%) had mood disorders, and 706 (33.6%) had mental disorders: 346 (49.0%) had 1 diagnosis, and 360 (51.0%) had multiple diagnoses. The percentage of diagnoses varied yearly, with notable drops in 2003, 2007, 2011, 2014, and 2018, and peaks in 2006, 2008, 2013, 2017, and 2022. This fluctuation was influenced by events like the establishment of PDTS in 2002, the 2008 economic recession, a hospital relocation in 2011, the 2014 Ebola outbreak, and the COVID-19 pandemic. Although the number of patients receiving antidepressants increased from 2019 to 2022, the overall percentage of patients receiving them did not significantly change from 2003 to 2022, aligning with previous research.5,125
Many medications have potential uses beyond what is detailed in the prescribing information. Antidepressants can relieve pain, while pain medications may help with depression. Opioids were once thought to effectively treat depression, but this perspective has changed with a greater understanding of their risks, including misuse.126-131 Pain is a severe and often unbearable AE of cancer. Of 2113 patients, 92% received opioids; 34% received both opioids and antidepressants; 2% received only antidepressants; and 7% received neither. This study didn’t clarify whether those on opioids alone recognized their depression or if those on both were aware of their dependence. While SSRIs are generally not addictive, they can lead to physical dependence, and any medication can be abused if not managed properly.132-134
Conclusions
This retrospective study analyzes data from antineoplastic agents used in systemic cancer treatment between 1988 and 2023, with a particular focus on the use of antidepressants. Data on antidepressant prescriptions are incomplete and specific to these agents, which means the findings cannot be generalized to all antidepressants. Hence, the results indicate that patients taking antidepressants had more diagnosed health issues and received more medications compared to patients who were not on these drugs.
This study underscores the need for further research into the effects of antidepressants on cancer treatment, utilizing all data from the DoD Cancer Registry. Future research should explore DDIs between antidepressants and other cancer and noncancer medications, as this study did not assess AE documentation, unlike in studies involving erlotinib, gefitinib, and paclitaxel.78,79 Further investigation is needed to evaluate the impact of discontinuing antidepressant use during cancer treatment. This comprehensive overview provides insights for clinicians to help them make informed decisions regarding the prescription of antidepressants in the context of cancer treatment.
- National Cancer Institute. Depression (PDQ)-Health Professional Version. Updated July 25, 2024. Accessed April 4, 2025. https://www.cancer.gov/about-cancer/coping/feelings/depression-hp-pdq
- Krebber AM, Buffart LM, Kleijn G, et al. Prevalence of depression in cancer patients: a meta-analysis of diagnostic interviews and self-report instruments. Psychooncology. 2014;23(2):121-130. doi:10.1002/pon.3409
- Xiang X, An R, Gehlert S. Trends in antidepressant use among U.S. cancer survivors, 1999-2012. Psychiatr Serv. 2015;66(6):564. doi:10.1176/appi.ps.201500007
- Hartung TJ, Brähler E, Faller H, et al. The risk of being depressed is significantly higher in cancer patients than in the general population: Prevalence and severity of depressive symptoms across major cancer types. Eur J Cancer. 2017;72:46-53. doi:10.1016/j.ejca.2016.11.017
- Walker J, Holm Hansen C, Martin P, et al. Prevalence of depression in adults with cancer: a systematic review. Ann Oncol. 2013;24(4):895-900. doi:10.1093/annonc/mds575
- Pitman A, Suleman S, Hyde N, Hodgkiss A. Depression and anxiety in patients with cancer. BMJ. 2018;361:k1415. doi:10.1136/bmj.k1415
- Kisely S, Alotiby MKN, Protani MM, Soole R, Arnautovska U, Siskind D. Breast cancer treatment disparities in patients with severe mental illness: a systematic review and meta-analysis. Psychooncology. 2023;32(5):651-662. doi:10.1002/pon.6120
- Massie MJ. Prevalence of depression in patients with cancer. J Natl Cancer Inst Monogr. 2004;(32):57-71. doi:10.1093/jncimonographs/lgh014
- Rodin G, Katz M, Lloyd N, Green E, Mackay JA, Wong RK. Treatment of depression in cancer patients. Curr Oncol. 2007;14(5):180-188. doi:10.3747/co.2007.146
- Wilson KG, Chochinov HM, Skirko MG, et al. Depression and anxiety disorders in palliative cancer care. J Pain Symptom Manage. 2007;33(2):118-129. doi:10.1016/j.jpainsymman.2006.07.016
- Moradi Y, Dowran B, Sepandi M. The global prevalence of depression, suicide ideation, and attempts in the military forces: a systematic review and meta-analysis of cross sectional studies. BMC Psychiatry. 2021;21(1):510. doi:10.1186/s12888-021-03526-2
- Lu CY, Zhang F, Lakoma MD, et al. Changes in antidepressant use by young people and suicidal behavior after FDA warnings and media coverage: quasi-experimental study. BMJ. 2014;348:g3596. doi:10.1136/bmj.g3596
- Friedman RA. Antidepressants’ black-box warning--10 years later. N Engl J Med. 2014;371(18):1666-1668. doi:10.1056/NEJMp1408480
- Sheffler ZM, Patel P, Abdijadid S. Antidepressants. In: StatPearls. StatPearls Publishing. Updated May 26, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK538182/
- Miller JJ. Antidepressants, Part 1: 100 Years and Counting. Accessed April 4, 2025. Psychiatric Times. https:// www.psychiatrictimes.com/view/antidepressants-part-1-100-years-and-counting
- Hillhouse TM, Porter JH. A brief history of the development of antidepressant drugs: from monoamines to glutamate. Exp Clin Psychopharmacol. 2015;23(1):1-21. doi:10.1037/a0038550
- Mitchell PB, Mitchell MS. The management of depression. Part 2. The place of the new antidepressants. Aust Fam Physician. 1994;23(9):1771-1781.
- Sabri MA, Saber-Ayad MM. MAO Inhibitors. In: Stat- Pearls. StatPearls Publishing. Updated June 5, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557395/
- Sub Laban T, Saadabadi A. Monoamine Oxidase Inhibitors (MAOI). In: StatPearls. StatPearls Publishing. Updated July 17, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539848/
- Fiedorowicz JG, Swartz KL. The role of monoamine oxidase inhibitors in current psychiatric practice. J Psychiatr Pract. 2004;10(4):239-248. doi:10.1097/00131746-200407000-00005
- Flockhart DA. Dietary restrictions and drug interactions with monoamine oxidase inhibitors: an update. J Clin Psychiatry. 2012;73 Suppl 1:17-24. doi:10.4088/JCP.11096su1c.03
- Moraczewski J, Awosika AO, Aedma KK. Tricyclic Antidepressants. In: StatPearls. StatPearls Publishing. Updated August 17, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557791/
- Almasi A, Patel P, Meza CE. Doxepin. In: StatPearls. StatPearls Publishing. Updated February 14, 2024. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK542306/
- Thour A, Marwaha R. Amitriptyline. In: StatPearls. Stat- Pearls Publishing. Updated July 18, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK537225/
- Radley DC, Finkelstein SN, Stafford RS. Off-label prescribing among office-based physicians. Arch Intern Med. 2006;166(9):1021-1026. doi:10.1001/archinte.166.9.1021
- Tesfaye S, Sloan G, Petrie J, et al. Comparison of amitriptyline supplemented with pregabalin, pregabalin supplemented with amitriptyline, and duloxetine supplemented with pregabalin for the treatment of diabetic peripheral neuropathic pain (OPTION-DM): a multicentre, double-blind, randomised crossover trial. Lancet. 2022;400(10353):680- 690. doi:10.1016/S0140-6736(22)01472-6
- Farag HM, Yunusa I, Goswami H, Sultan I, Doucette JA, Eguale T. Comparison of amitriptyline and US Food and Drug Administration-approved treatments for fibromyalgia: a systematic review and network metaanalysis. JAMA Netw Open. 2022;5(5):e2212939. doi:10.1001/jamanetworkopen.2022.12939
- Merwar G, Gibbons JR, Hosseini SA, Saadabadi A. Nortriptyline. In: StatPearls. StatPearls Publishing. Updated June 5, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK482214/
- Fayez R, Gupta V. Imipramine. In: StatPearls. StatPearls Publishing. Updated May 22, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557656/
- Jilani TN, Gibbons JR, Faizy RM, Saadabadi A. Mirtazapine. In: StatPearls. StatPearls Publishing. Updated August 28, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK519059/
- Nutt DJ. Tolerability and safety aspects of mirtazapine. Hum Psychopharmacol. 2002;17 Suppl 1:S37-S41. doi:10.1002/hup.388
- Gandotra K, Chen P, Jaskiw GE, Konicki PE, Strohl KP. Effective treatment of insomnia with mirtazapine attenuates concomitant suicidal ideation. J Clin Sleep Med. 2018;14(5):901-902. doi:10.5664/jcsm.7142
- Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264. doi:10.1111/j.1527-3458.2001.tb00198.x
- Wang SM, Han C, Bahk WM, et al. Addressing the side effects of contemporary antidepressant drugs: a comprehensive review. Chonnam Med J. 2018;54(2):101-112. doi:10.4068/cmj.2018.54.2.101
- Huecker MR, Smiley A, Saadabadi A. Bupropion. In: StatPearls. StatPearls Publishing. Updated April 9, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK470212/
- Hsieh MT, Tseng PT, Wu YC, et al. Effects of different pharmacologic smoking cessation treatments on body weight changes and success rates in patients with nicotine dependence: a network meta-analysis. Obes Rev. 2019;20(6):895- 905. doi:10.1111/obr.12835
- Hankosky ER, Bush HM, Dwoskin LP, et al. Retrospective analysis of health claims to evaluate pharmacotherapies with potential for repurposing: association of bupropion and stimulant use disorder remission. AMIA Annu Symp Proc. 2018;2018:1292-1299.
- Livingstone-Banks J, Norris E, Hartmann-Boyce J, West R, Jarvis M, Hajek P. Relapse prevention interventions for smoking cessation. Cochrane Database Syst Rev. 2019; 2(2):CD003999. doi:10.1002/14651858.CD003999.pub5
- Clayton AH, Kingsberg SA, Goldstein I. Evaluation and management of hypoactive sexual desire disorder. Sex Med. 2018;6(2):59-74. doi:10.1016/j.esxm.2018.01.004
- Verbeeck W, Bekkering GE, Van den Noortgate W, Kramers C. Bupropion for attention deficit hyperactivity disorder (ADHD) in adults. Cochrane Database Syst Rev. 2017;10(10):CD009504. doi:10.1002/14651858.CD009504.pub2
- Ng QX. A systematic review of the use of bupropion for attention-deficit/hyperactivity disorder in children and adolescents. J Child Adolesc Psychopharmacol. 2017;27(2):112-116. doi:10.1089/cap.2016.0124
- Fava M, Rush AJ, Thase ME, et al. 15 years of clinical experience with bupropion HCl: from bupropion to bupropion SR to bupropion XL. Prim Care Companion J Clin Psychiatry. 2005;7(3):106-113. doi:10.4088/pcc.v07n0305
- Chu A, Wadhwa R. Selective Serotonin Reuptake Inhibitors. In: StatPearls. StatPearls Publishing. Updated May 1, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK554406/
- Singh HK, Saadabadi A. Sertraline. In: StatPearls. Stat- Pearls Publishing. Updated February 13, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK547689/
- MacQueen G, Born L, Steiner M. The selective serotonin reuptake inhibitor sertraline: its profile and use in psychiatric disorders. CNS Drug Rev. 2001;7(1):1-24. doi:10.1111/j.1527-3458.2001.tb00188.x
- Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373(9665):746-758. doi:10.1016/S0140-6736(09)60046-5
- Nelson JC. The STAR*D study: a four-course meal that leaves us wanting more. Am J Psychiatry. 2006;163(11):1864-1866. doi:10.1176/ajp.2006.163.11.1864
- Sharbaf Shoar N, Fariba KA, Padhy RK. Citalopram. In: StatPearls. StatPearls Publishing. Updated November 7, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK482222/
- Landy K, Rosani A, Estevez R. Escitalopram. In: Stat- Pearls. StatPearls Publishing. Updated November 10, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557734/
- Cavanah LR, Ray P, Goldhirsh JL, Huey LY, Piper BJ. Rise of escitalopram and the fall of citalopram. medRxiv. Preprint published online May 8, 2023. doi:10.1101/2023.05.07.23289632
- Sohel AJ, Shutter MC, Patel P, Molla M. Fluoxetine. In: StatPearls. StatPearls Publishing. Updated July 4, 2022. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK459223/
- Wong DT, Perry KW, Bymaster FP. Case history: the discovery of fluoxetine hydrochloride (Prozac). Nat Rev Drug Discov. 2005;4(9):764-774. doi:10.1038/nrd1821
- Shrestha P, Fariba KA, Abdijadid S. Paroxetine. In: Stat- Pearls. StatPearls Publishing. Updated July 17, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK526022/
- Naseeruddin R, Rosani A, Marwaha R. Desvenlafaxine. In: StatPearls. StatPearls Publishing. Updated July 10, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK534829
- Lieberman DZ, Massey SH. Desvenlafaxine in major depressive disorder: an evidence-based review of its place in therapy. Core Evid. 2010;4:67-82. doi:10.2147/ce.s5998
- Withdrawal assessment report for Ellefore (desvenlafaxine). European Medicine Agency. 2009. Accessed April 4, 2025. https://www.ema.europa.eu/en/documents/withdrawal-report/withdrawal-assessment-report-ellefore_en.pdf
- Dhaliwal JS, Spurling BC, Molla M. Duloxetine. In: Stat- Pearls. Statpearls Publishing. Updated May 29, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK549806/
- Hershman DL, Lacchetti C, Dworkin RH, et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2014;32(18):1941-1967. doi:10.1200/JCO.2013.54.0914
- Sommer C, Häuser W, Alten R, et al. Medikamentöse Therapie des Fibromyalgiesyndroms. Systematische Übersicht und Metaanalyse [Drug therapy of fibromyalgia syndrome. Systematic review, meta-analysis and guideline]. Schmerz. 2012;26(3):297-310. doi:10.1007/s00482-012-1172-2
- Bril V, England J, Franklin GM, et al. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2011;76(20):1758-1765. doi:10.1212/WNL.0b013e3182166ebe
- Attal N, Cruccu G, Baron R, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol. 2010;17(9):1113-e88. doi:10.1111/j.1468-1331.2010.02999.x
- Singh D, Saadabadi A. Venlafaxine. In: StatPearls. StatePearls Publishing. Updated February 26, 2024. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK535363/
- Sertraline and venlafaxine: new indication. Prevention of recurrent depression: no advance. Prescrire Int. 2005;14(75):19-20.
- Bruno A, Morabito P, S p i n a E , M u s c a t ello MRl. The role of levomilnacipran in the management of major depressive disorder: a comprehensive review. Curr Neuro pharmacol. 2016;14(2):191-199. doi:10.2174/1570159x14666151117122458
- Shin JJ, Saadabadi A. Trazodone. In: StatPearls. StatPearls Publishing. Updated February 29, 2024. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK470560/
- Khouzam HR. A review of trazodone use in psychiatric and medical conditions. Postgrad Med. 2017;129(1):140-148. doi:10.1080/00325481.2017.1249265
- Smales ET, Edwards BA, Deyoung PN, et al. Trazodone effects on obstructive sleep apnea and non-REM arousal threshold. Ann Am Thorac Soc. 2015;12(5):758-764. doi:10.1513/AnnalsATS.201408-399OC
- Eckert DJ, Malhotra A, Wellman A, White DP. Trazodone increases the respiratory arousal threshold in patients with obstructive sleep apnea and a low arousal threshold. Sleep. 2014;37(4):811-819. doi:10.5665/sleep.3596
- Schatzberg AF, Nemeroff CB, eds. The American Psychiat ric Association Publishing Textbook of Psychopharmacology. 4th ed. American Psychiatric Publishing; 2009.
- Ruxton K, Woodman RJ, Mangoni AA. Drugs with anticholinergic effects and cognitive impairment, falls and allcause mortality in older adults: a systematic review and meta-analysis. Br J Clin Pharmacol. 2015;80(2):209-220. doi:10.1111/bcp.12617
- Nefazodone. In: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. National Institute of Diabetes and Digestive and Kidney Diseases. Updated March 6, 2020. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK548179/
- Drugs of Current Interest: Nefazodone. WHO Pharmaceuticals Newsletter:(1). 2003(1):7. https://web.archive.org/web/20150403165029/http:/apps.who.int/medicinedocs/en/d/Js4944e/3.html
- Choi S. Nefazodone (serzone) withdrawn because of hepatotoxicity. CMAJ. 2003;169(11):1187.
- Teva Nefazodone Statement. News release. Teva USA. December 20, 2021. Accessed April 4, 2025. https:// www.tevausa.com/news-and-media/press-releases/teva-nefazodone-statement/
- Levitan MN, Papelbaum M, Nardi AE. Profile of agomelatine and its potential in the treatment of generalized anxiety disorder. Neuropsychiatr Dis Treat. 2015;11:1149- 1155. doi:10.2147/NDT.S67470
- Fu DJ, Ionescu DF, Li X, et al. Esketamine nasal spray for rapid reduction of major depressive disorder symptoms in patients who have active suicidal ideation with intent: double-blind, randomized study (ASPIRE I). J Clin Psychiatry. 2020;81(3):19m13191. doi:10.4088/JCP.19m13191
- Iosifescu DV, Jones A, O’Gorman C, et al. Efficacy and safety of AXS-05 (dextromethorphan-bupropion) in patients with major depressive disorder: a phase 3 randomized clinical trial (GEMINI). J Clin Psychiatry. 2022;83(4): 21m14345. doi:10.4088/JCP.21m14345
- Luong TT, Powers CN, Reinhardt BJ, et al. Retrospective evaluation of drug-drug interactions with erlotinib and gefitinib use in the Military Health System. Fed Pract. 2023;40(Suppl 3):S24-S34. doi:10.12788/fp.0401
- Luong TT, Shou KJ, Reinhard BJ, Kigelman OF, Greenfield KM. Paclitaxel drug-drug interactions in the Military Health System. Fed Pract. 2024;41(Suppl 3):S70-S82. doi:10.12788/fp.0499
- Luong TT, Powers CN, Reinhardt BJ, Weina PJ. Preclinical drug-drug interactions (DDIs) of gefitinib with/without losartan and selective serotonin reuptake inhibitors (SSRIs): citalopram, fluoxetine, fluvoxamine, paroxetine, sertraline, and venlafaxine. Curr Res Pharmacol Drug Discov. 2022;3:100112. doi:10.1016/j.crphar.2022.100112
- Luong TT, McAnulty MJ, Evers DL, Reinhardt BJ, Weina PJ. Pre-clinical drug-drug interaction (DDI) of gefitinib or erlotinib with cytochrome P450 (CYP) inhibiting drugs, fluoxetine and/or losartan. Curr Res Toxicol. 2021;2:217- 224. doi:10.1016/j.crtox.2021.05.006,
- Adamo M, Dickie L, Ruhl J. SEER program coding and staging manual 2016. National Cancer Institute; 2016. Accessed April 4, 2025. https://seer.cancer.gov/archive/manuals/2016/SPCSM_2016_maindoc.pdf
- World Health Organization. International classification of diseases for oncology (ICD-O). 3rd ed, 1st revision. World Health Organization; 2013. Accessed April 4, 2025. https://apps.who.int/iris/handle/10665/96612
- Simon S. FDA approves Jelmyto (mitomycin gel) for urothelial cancer. American Cancer Society. April 20, 2020. Accessed April 4, 2025. https://www.cancer.org/cancer/latest-news/fda-approves-jelmyto-mitomycin-gel-for-urothelial-cancer.html
- Pantziarka P, Sukhatme V, Bouche G, Meheus L, Sukhatme VP. Repurposing drugs in oncology (ReDO)- diclofenac as an anti-cancer agent. Ecancermedicalscience. 2016;10:610. doi:10.3332/ecancer.2016.610
- Choi S, Kim S, Park J, Lee SE, Kim C, Kang D. Diclofenac: a nonsteroidal anti-inflammatory drug inducing cancer cell death by inhibiting microtubule polymerization and autophagy flux. Antioxidants (Basel). 2022;11(5):1009. doi:10.3390/antiox11051009
- Tontonoz M. The ABCs of BCG: oldest approved immunotherapy gets new explanation. Memorial Sloan Kettering Cancer Center. July 17, 2020. Accessed April 4, 2025. https://www.mskcc.org/news/oldest-approved-immunotherapy-gets-new-explanation
- Jordan VC. Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocr Relat Cancer. 2014;21(3):R235-R246. doi:10.1530/ERC-14-0092
- Cole MP, Jones CT, Todd ID. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br J Cancer. 1971;25(2):270-275. doi:10.1038/bjc.1971.33
- Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2(3):205-213. doi:10.1038/nrd1031
- Maximov PY, McDaniel RE, Jordan VC. Tamoxifen: Pioneering Medicine in Breast Cancer. Springer Basel; 2013. Accessed April 4, 2025. https://link.springer.com/book/10.1007/978-3-0348-0664-0
- Taxol (paclitaxel). Prescribing information. Bristol-Myers Squibb Company; 2011. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020262s049lbl.pdf
- Abraxane (paclitaxel). Prescribing information. Celgene Corporation; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/021660s047lbl.pdf
- Tarceva (erlotinib). Prescribing information. OSI Pharmaceuticals, LLC; 2016. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021743s025lbl.pdf
- Docetaxel. Prescribing information. Sichuan Hyiyu Pharmaceutical Co.; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215813s000lbl.pdf
- Alimta (pemetrexed). Prescribing information. Teva Pharmaceuticals; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/208419s004lbl.pdf
- Tagrisso (osimertinib). Prescribing information. Astra- Zeneca Pharmaceuticals; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208065s021lbl.pdf
- Iressa (gefitinib). Prescribing information. AstraZeneca Pharmaceuticals; 2018. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/206995s003lbl.pdf
- Kadcyla (ado-trastuzumab emtansine). Prescribing information. Genentech, Inc.; 2013. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125427lbl.pdf
- Alecensa (alectinib). Prescribing information. Genetech, Inc.; 2017. Accessed April 4, 2025. https:// www.accessdata.fda.gov/drugsatfda_docs/label/2017/208434s003lbl.pdf
- Xalkori (crizotinib). Prescribing information. Pfizer Laboratories; 2022. Accessed April 4, 2025. https:// www.accessdata.fda.gov/drugsatfda_docs/label/2022/202570s033lbl.pdf
- Lorbrena (lorlatinib). Prescribing information. Pfizer Laboratories; 2018. Accessed April 14, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210868s000lbl.pdf
- Alunbrig (brigatinib). Prescribing information. Takeda Pharmaceutical Company; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208772s008lbl.pdf
- Rozlytrek (entrectinib). Prescribing information. Genentech, Inc.; 2019. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212725s000lbl.pdf
- Herceptin (trastuzumab). Prescribing information. Genentech, Inc.; 2010. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/103792s5250lbl.pdf
- Cybalta (duloxetine). Prescribing information. Eli Lilly and Company; 2017. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021427s049lbl.pdf
- Effexor XR (venlafaxine). Prescribing information. Pfizer Wyeth Pharmaceuticals Inc; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/020699s112lbl.pdf
- Desyrel (trazodone hydrochloride). Prescribing information. Pragma Pharmaceuticals; 2017. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/018207s032lbl.pdf
- Sertraline hydrochloride. Prescribing information. Almatica Pharma LLC; 2021. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215133s000lbl.pdf
- Remeron (mirtazapine). Prescribing information. Merck & Co. Inc; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/020415s029,%20021208s019lbl.pdf
- Celexa (citalopram). Prescribing information. Allergan USA Inc; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/020822s041lbl.pdf
- information. GlaxoSmithKline; 2019. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/020358s066lbl.pdf
- Amitriptyline hydrochloride tablet. Prescribing information. Quality Care Products LLC; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/spl/data/0f12f50f-7087-46e7-a2e6-356b4c566c9f/0f12f50f-7087-46e7-a2e6-356b4c566c9f.xml
- Lexapro (escitalopram). Prescribing information. AbbVie Inc; 2023. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/021323s055,021365s039lbl.pdf
- Fluoxetine. Prescribing information. Edgemont Pharmaceutical, LLC; 2017. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/202133s004s005lbl.pdf
- Paxil (paroxetine). Prescribing Information. Apotex Inc; 2021. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/020031s077lbl.pdf
- Pamelor (nortriptyline HCl). Prescribing information. Mallinckrodt, Inc; 2012. Accessed April 4, 2025. https:// www.accessdata.fda.gov/drugsatfda_docs/label/2012/018012s029,018013s061lbl.pdf
- Silenor (doxepin). Prescribing information. Currax Pharmaceuticals; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/022036s006lbl.pdf
- Tofranil-PM (imipramine pamote). Prescribing information. Mallinckrodt, Inc; 2014. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/017090s078lbl.pdf
- Norpramin (desipramine hydrochloride). Prescribing information. Sanofi-aventis U.S. LLC; 2014. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/014399s069lbl.pdf
- Khedezla (desvenlafaxine). Prescribing information. Osmotical Pharmaceutical US LLC; 2019. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/204683s006lbl.pdf
- Nefazodone hydrochloride. Prescribing information. Bryant Ranch Prepack; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/spl/data/0bd4c34a-4f43-4c84-8b98-1d074cba97d5/0bd4c34a-4f43-4c84-8b98-1d074cba97d5.xml
- Grassi L, Nanni MG, Rodin G, Li M, Caruso R. The use of antidepressants in oncology: a review and practical tips for oncologists. Ann Oncol. 2018;29(1):101-111. doi:10.1093/annonc/mdx526
- Lee E, Park Y, Li D, Rodriguez-Fuguet A, Wang X, Zhang WC. Antidepressant use and lung cancer risk and survival: a meta-analysis of observational studies. Cancer Res Commun. 2023;3(6):1013-1025. doi:10.1158/2767-9764.CRC-23-0003
- Olfson M, Marcus SC. National patterns in antidepressant medication treatment. Arch Gen Psychiatry. 2009;66(8):848 -856. doi:10.1001/archgenpsychiatry.2009.81
- Grattan A, Sullivan MD, Saunders KW, Campbell CI, Von Korff MR. Depression and prescription opioid misuse among chronic opioid therapy recipients with no history of substance abuse. Ann Fam Med. 2012;10(4):304-311. doi:10.1370/afm.1371
- Cowan DT, Wilson-Barnett J, Griffiths P, Allan LG. A survey of chronic noncancer pain patients prescribed opioid analgesics. Pain Med. 2003;4(4):340-351. doi:10.1111/j.1526-4637.2003.03038.x
- Breckenridge J, Clark JD. Patient characteristics associated with opioid versus nonsteroidal anti-inflammatory drug management of chronic low back pain. J Pain. 2003;4(6):344-350. doi:10.1016/s1526-5900(03)00638-2
- Edlund MJ, Martin BC, Devries A, Fan MY, Braden JB, Sullivan MD. Trends in use of opioids for chronic noncancer pain among individuals with mental health and substance use disorders: the TROUP study. Clin J Pain. 2010;26(1):1-8. doi:10.1097/AJP.0b013e3181b99f35
- Sullivan MD, Edlund MJ, Fan MY, DeVries A, Braden JB, Martin BC. Risks for possible and probable opioid misuse among recipients of chronic opioid therapy in commercial and medicaid insurance plans: the TROUP study. Pain. 2010;150(2):332-339. doi:10.1016/j.pain.2010.05.020
- Dunn KM, Saunders KW, Rutter CM, et al. Opioid prescriptions for chronic pain and overdose: a cohort study. Ann Intern Med. 2010;152(2):85-92. doi:10.7326/0003-4819-152-2-201001190-00006
- Haddad P. Do antidepressants have any potential to cause addiction? J Psychopharmacol. 1999;13(3):300- 307. doi:10.1177/026988119901300321
- Lakeview Health Staff. America’s most abused antidepressants. Lakeview Health. January 24, 2004. Accessed April 4, 2025. https://www.lakeviewhealth.com/blog/us-most-abused-antidepressants/
- Greenhouse Treatment Center Editorial Staff. Addiction to antidepressants: is it possible? America Addiction Centers: Greenhouse Treatment Center. Updated April 23, 2024. Accessed April 4, 2025. https://greenhousetreatment.com/prescription-medication/antidepressants/
Cancer patients experience depression at rates > 5 times that of the general population.1-11 Despite an increase in palliative care use, depression rates continued to rise.2-4 Between 5% to 16% of outpatients, 4% to 14% of inpatients, and up to 49% of patients receiving palliative care experience depression.5 This issue also impacts families and caregivers.1 A 2021 meta-analysis found that 23% of active military personnel and 20% of veterans experience depression.11
Antidepressants approved by the US Food and Drug Administration (FDA) target the serotonin, norepinephrine, or dopamine systems and include boxed warnings about an increased risk of suicidal thoughts in adults aged 18 to 24 years.12,13 These medications are categorized into several classes: monoamine oxidase inhibitors (MAOIs), tricyclic antidepressants (TCAs), tetracyclic antidepressants (TeCAs), norepinephrine-dopamine reuptake inhibitors (NDRIs), selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), serotonin receptor modulators (SRMs), serotonin-melatonin receptor antagonists (SMRAs), and N—methyl-D-aspartate receptor antagonists (NMDARAs).14,15 The first FDA-approved antidepressants, iproniazid (an MAOI) and imipramine (a TCA) laid the foundation for the development of newer classes like SSRIs and SNRIs.15-17
Older antidepressants such as MAOIs and TCAs are used less due to their adverse effects (AEs) and drug interactions. MAOIs, such as iproniazid, selegiline, moclobemide, tranylcypromine, isocarboxazid, and phenelzine, have numerous AEs and drug interactions, making them unsuitable for first- or second-line treatment of depression.14,18-21 TCAs such as doxepin, amitriptyline, nortriptyline, imipramine, desipramine, clomipramine, trimipramine, protriptyline, maprotiline, and amoxapine have a narrow therapeutic index requiring careful monitoring for signs of toxicity such as QRS widening, tremors, or confusion. Despite the issues, TCAs are generally classified as second-line agents for major depressive disorder (MDD). TCAs have off-label uses for migraine prophylaxis, treatment of obsessive-compulsive disorder (OCD), insomnia, and chronic pain management first-line.14,22-29
Newer antidepressants, including TeCAs and NDRIs, are typically more effective, but also come with safety concerns. TeCAs like mirtazapine interact with several medications, including MAOIs, serotonin-increasing drugs, alcohol, cannabidiol, and marijuana. Mirtazapine is FDA-approved for the treatment of moderate to severe depression in adults. It is also used off-label to treat insomnia, panic disorder, posttraumatic stress disorder (PTSD), generalized anxiety disorder (GAD), social anxiety disorder (SAD), headaches, and migraines. Compared to other antidepressants, mirtazapine is effective for all stages of depression and addresses a broad range of related symptoms.14,30-34 NDRIs, such as bupropion, also interact with various medications, including MAOIs, other antidepressants, stimulants, and alcohol. Bupropion is FDA-approved for smoking cessation and to treat depression and SAD. It is also used off-label for depression- related bipolar disorder or sexual dysfunction, attention-deficit/hyperactivity disorder (ADHD), and obesity.14,35-42
SSRIs, SNRIs, and SRMs should be used with caution. SSRIs such as sertraline, citalopram, escitalopram, fluoxetine, paroxetine, and fluvoxamine are first-line treatments for depression and various psychiatric disorders due to their safety and efficacy. Common AEs of SSRIs include sexual dysfunction, sleep disturbances, weight changes, and gastrointestinal (GI) issues. SSRIs can prolong the QT interval, posing a risk of life-threatening arrhythmia, and may interact with other medications, necessitating treatment adjustments. The FDA approved SSRIs for MDD, GAD, bulimia nervosa, bipolar depression, OCD, panic disorder, premenstrual dysphoric disorder, treatment-resistant depression, PTSD, and SAD. Off-label uses include binge eating disorder, body dysmorphic disorder, fibromyalgia, premature ejaculation, paraphilias, autism, Raynaud phenomenon, and vasomotor symptoms associated with menopause. Among SSRIs, sertraline and escitalopram are noted for their effectiveness and tolerability.14,43-53
SNRIs, including duloxetine, venlafaxine, desvenlafaxine, milnacipran, and levomilnacipran, may increase bleeding risk, especially when taken with blood thinners. They can also elevate blood pressure, which may worsen if combined with stimulants. SNRIs may interact with other medications that affect serotonin levels, increasing the risk of serotonin syndrome when taken with triptans, pain medications, or other antidepressants.14 Desvenlafaxine has been approved by the FDA (but not by the European Medicines Agency).54-56 Duloxetine is FDA-approved for the treatment of depression, neuropathic pain, anxiety disorders, fibromyalgia, and musculoskeletal disorders. It is used off-label to treat chemotherapy-induced peripheral neuropathy and stress urinary incontinence.57-61 Venlafaxine is FDA-approved for depression, SAD, and panic disorder, and is prescribed off-label to treat ADHD, neuropathy, fibromyalgia, cataplexy, and PTSD, either alone or in combination with other medications.62,63 Milnacipran is not approved for MDD; levomilnacipran received approval in 2013.64
SRMs such as trazodone, nefazodone, vilazodone, and vortioxetine also function as serotonin reuptake inhibitors.14,15 Trazodone is FDA-approved for MDD. It has been used off-label to treat anxiety, Alzheimer disease, substance misuse, bulimia nervosa, insomnia, fibromyalgia, and PTSD when first-line SSRIs are ineffective. A notable AE of trazodone is orthostatic hypotension, which can lead to dizziness and increase the risk of falls, especially in geriatric patients.65-70 Nefazodone was discontinued in Europe in 2003 due to rare cases of liver toxicity but remains available in the US.71-74 Vilazodone and vortioxetine are FDA-approved.
The latest classes of antidepressants include SMRAs and NMDARAs.14 Agomelatine, an SMRA, was approved in Europe in 2009 but rejected by the FDA in 2011 due to liver toxicity.75 NMDARAs like esketamine and a combination of dextromethorphan and bupropion received FDA approval in 2019 and 2022, respectively.76,77
This retrospective study analyzes noncancer drugs used during systemic chemotherapy based on a dataset of 14 antineoplastic agents. It sought to identify the most dispensed noncancer drug groups, discuss findings, compare patients with and without antidepressant prescriptions, and examine trends in antidepressant use from 2002 to 2023. This analysis expands on prior research.78-81
Methods
The Walter Reed National Military Medical Center Institutional Review Board approved the study protocol and ensured compliance with the Health Insurance Portability and Accountability Act as an exempt protocol. The Joint Pathology Center (JPC) of the US Department of Defense (DoD) Cancer Registry Program and Military Health System (MHS) data experts from the Comprehensive Ambulatory/Professional Encounter Record (CAPER) and Pharmacy Data Transaction Service (PDTS) provided data for the analysis.
Data Sources
The JPC DoD Cancer Registry Program contains data from 1998 to 2024. CAPER and PDTS are part of the MHS Data Repository/Management Analysis and Reporting Tool database. Each observation in CAPER represents an ambulatory encounter at a military treatment facility (MTF). CAPER records are available from 2003 to 2024. PDTS records are available from 2002 to 2004. Each observation in PDTS represents a prescription filled for an MHS beneficiary, excluding those filled at international civilian pharmacies and inpatient pharmacy prescriptions.
This cross-sectional analysis requested data extraction for specific cancer drugs from the DoD Cancer Registry, focusing on treatment details, diagnosis dates, patient demographics, and physicians’ comments on AEs. After identifying patients, CAPER was used to identify additional health conditions. PDTS was used to compile a list of prescription medications filled during systemic cancer treatment or < 2 years postdiagnosis.
The 2016 Surveillance, Epidemiology, and End Results Program Coding and Staging Manual and International Classification of Diseases for Oncology, 3rd edition, 1st revision, were used to decode disease and cancer types.82,83 Data sorting and analysis were performed using Microsoft Excel. The percentage for the total was calculated by using the number of patients or data available within the subgroup divided by the total number of patients or data variables. To compare the mean number of dispensed antidepressants to those without antidepressants, a 2-tailed, 2-sample z test was used to calculate the P value and determine statistical significance (P < .05) using socscistatistics.com.
Data were extracted 3 times between 2021 and 2023. The initial 2021 protocol focused on erlotinib and gefitinib. A modified protocol in 2022 added paclitaxel, cisplatin, docetaxel, pemetrexed, and crizotinib; further modification in 2023 included 8 new antineoplastic agents and 2 anticoagulants. Sotorasib has not been prescribed in the MHS, and JPC lacks records for noncancer drugs. The 2023 dataset comprised 2210 patients with cancer treated with 14 antineoplastic agents; 2104 had documented diagnoses and 2113 had recorded prescriptions. Data for erlotinib, gefitinib, and paclitaxel have been published previously.78,79
Results
Of 2113 patients with recorded prescriptions, 1297 patients (61.4%) received 109 cancer drugs, including 96 antineoplastics, 7 disease-modifying antirheumatic agents, 4 biologic response modifiers, and 2 calcitonin gene-related peptides. Fourteen antineoplastic agents had complete data from JPC, while others were noted for combination therapies or treatment switches from the PDTS (Table 1). Seventy-six cancer drugs were prescribed with antidepressants in 489 patients (eAppendix).
The JPC provided 2242 entries for 2210 patients, ranging in age from 2 months to 88 years (mean, 56 years), documenting treatment from September 1988 to January 2023. Thirty-two patients had duplicate entries due to multiple cancer locations or occurrences. Of the 2242 patients, 1541 (68.7%) were aged > 50 years, 975 patients (43.5%) had cancers that were stage III or IV, and 1267 (56.5%) had cancers that were stage 0, I, II, or not applicable/unknown. There were 51 different types of cancer: breast, lung, testicular, endometrial, and ovarian were most common (n ≥ 100 patients). Forty-two cancer types were documented among 750 patients prescribed antidepressants (Table 2).
The CAPER database recorded 8882 unique diagnoses for 2104 patients, while PDTS noted 1089 unique prescriptions within 273 therapeutic codes for 2113 patients. Nine therapeutic codes (opiate agonists, adrenals, cathartics-laxatives, nonsteroidal anti-inflammatory agents, antihistamines for GI conditions, 5-HT3 receptor antagonists, analgesics and antipyretic miscellanea, antineoplastic agents, and proton-pump inhibitors) and 8 drugs (dexamethasone, prochlorperazine, ondansetron, docusate, acetaminophen, ibuprofen, oxycodone, and polyethylene glycol 3350) were associated with > 1000 patients (≥ 50%). Patients had between 1 and 275 unique health conditions and filled 1 to 108 prescriptions. The mean (SD) number of diagnoses and prescriptions was 50 (28) and 29 (12), respectively. Of the 273 therapeutic codes, 30 groups were analyzed, with others categorized into miscellaneous groups such as lotions, vaccines, and devices. Significant differences in mean number of prescriptions were found for patients taking antidepressants compared to those not (P < .05), except for anticonvulsants and antipsychotics (P = .12 and .09, respectively) (Table 3).
Antidepressants
Of the 2113 patients with recorded prescriptions, 750 (35.5%) were dispensed 17 different antidepressants. Among these 17 antidepressants, 183 (8.7%) patients received duloxetine, 158 (7.5%) received venlafaxine, 118 (5.6%) received trazodone, and 107 (5.1%) received sertraline (Figure 1, Table 4). Of the 750 patients, 509 (67.9%) received 1 antidepressant, 168 (22.4%) received 2, 60 (8.0%) received 3, and 13 (1.7%) received > 3. Combinations varied, but only duloxetine and trazodone were prescribed to > 10 patients.
Antidepressants were prescribed annually at an overall mean (SD) rate of 23% (5%) from 2003 to 2022 (Figure 2). Patients on antidepressants during systemic therapy had a greater number of diagnosed medical conditions and received more prescription medications compared to those not taking antidepressants (P < .001) (Figure 3). The 745 patients taking antidepressants in CAPER data had between 1 and 275 diagnosed medical issues, with a mean (SD) of 55 (31) vs a range of 1 to 209 and a mean (SD) of 46 (26) for the 1359 patients not taking antidepressants. The 750 patients on antidepressants in PDTS data had between 8 and 108 prescriptions dispensed, with a mean (SD) of 32 (12), vs a range of 1 to 65 prescriptions and a mean (SD) of 29 (12) for 1363 patients not taking antidepressants.
Discussion
The JPC DoD Cancer Registry includes information on cancer types, stages, treatment regimens, and physicians’ notes, while noncancer drugs are sourced from the PDTS database. The pharmacy uses a different documentation system, leading to varied classifications.
Database reliance has its drawbacks. For example, megestrol is coded as a cancer drug, although it’s primarily used for endometrial or gynecologic cancers. Many drugs have multiple therapeutic codes assigned to them, including 10 antineoplastic agents: diclofenac, Bacillus Calmette-Guérin (BCG), megestrol acetate, tamoxifen, anastrozole, letrozole, leuprolide, goserelin, degarelix, and fluorouracil. Diclofenac, BCG, and mitomycin have been repurposed for cancer treatment.84-87 From 2003 to 2023, diclofenac was prescribed to 350 patients for mild-to-moderate pain, with only 2 patients receiving it for cancer in 2018. FDA-approved for bladder cancer in 1990, BCG was prescribed for cancer treatment for 1 patient in 2021 after being used for vaccines between 2003 and 2018. Tamoxifen, used for hormone receptor-positive breast cancer from 2004 to 2017 with 53 patients, switched to estrogen agonist-antagonists from 2017 to 2023 with 123 patients. Only a few of the 168 patients were prescribed tamoxifen using both codes.88-91 Anastrozole and letrozole were coded as antiestrogens for 7 and 18 patients, respectively, while leuprolide and goserelin were coded as gonadotropins for 59 and 18 patients. Degarelix was coded as antigonadotropins, fluorouracil as skin and mucous membrane agents miscellaneous, and megestrol acetate as progestins for 7, 6, and 3 patients, respectively. Duloxetine was given to 186 patients, primarily for depression from 2005 to 2023, with 7 patients treated for fibromyalgia from 2022 to 2023.
Antidepressants Observed
Tables 1 and 5 provide insight into the FDA approval of 14 antineoplastics and antidepressants and their CYP metabolic pathways.92-122 In Table 4, the most prescribed antidepressant classes are SNRIs, SRMs, SSRIs, TeCAs, NDRIs, and TCAs. This trend highlights a preference for newer medications with weak CYP inhibition. A total of 349 patients were prescribed SSRIs, 343 SNRIs, 119 SRMs, 109 TCAs, 83 TeCAs, and 79 NDRIs. MAOIs, SMRAs, and NMDARAs were not observed in this dataset. While there are instances of dextromethorphan-bupropion and sertraline-escitalopram being dispensed together, it remains unclear whether these were NMDARA combinations.
Among the 14 specific antineoplastic agents, 10 are metabolized by CYP isoenzymes, primarily CYP3A4. Duloxetine neither inhibits nor is metabolized by CYP3A4, a reason it is often recommended, following venlafaxine.
Both duloxetine and venlafaxine are used off-label for chemotherapy-induced peripheral neuropathy related to paclitaxel and docetaxel. According to the CYP metabolized pathway, duloxetine tends to have more favorable DDIs than venlafaxine. In PDTS data, 371 patients were treated with paclitaxel and 180 with docetaxel, with respective antidepressant prescriptions of 156 and 70. Of the 156 patients dispensed paclitaxel, 62 (40%) were dispensed with duloxetine compared to 43 (28%) with venlafaxine. Of the 70 patients dispensed docetaxel, 23 (33%) received duloxetine vs 24 (34%) with venlafaxine.
Of 85 patients prescribed duloxetine, 75 received it with either paclitaxel or docetaxel (5 received both). Five patients had documented AEs (1 neuropathy related). Of 67 patients prescribed venlafaxine, 66 received it with either paclitaxel or docetaxel. Two patients had documented AEs (1 was neuropathy related, the same patient who received duloxetine). Of the 687 patients treated with paclitaxel and 337 with docetaxel in all databases, 4 experienced neuropathic AEs from both medications.79
Antidepressants can increase the risk of bleeding, especially when combined with blood thinners, and may elevate blood pressure, particularly alongside stimulants. Of the 554 patients prescribed 9 different anticoagulants, enoxaparin, apixaban, and rivaroxaban were the most common (each > 100 patients). Among these, 201 patients (36%) received both anticoagulants and antidepressants: duloxetine for 64 patients, venlafaxine for 30, trazodone for 35, and sertraline for 26. There were no data available to assess bleeding rates related to the evaluation of DDIs between these medication classes.
Antidepressants can be prescribed for erectile dysfunction. Of the 148 patients prescribed an antidepressant for erectile dysfunction, duloxetine, trazodone, and mirtazapine were the most common. Antidepressant preferences varied by cancer type. Duloxetine was the only antidepressant used for all types of cancer. Venlafaxine, duloxetine, trazodone, sertraline, and escitalopram were the most prescribed antidepressants for breast cancer, while duloxetine, mirtazapine, citalopram, sertraline, and trazodone were the most prescribed for lung cancer. Sertraline, duloxetine, trazodone, amitriptyline, and escitalopram were most common for testicular cancer. Duloxetine, venlafaxine, trazodone, amitriptyline, and sertraline were the most prescribed for endometrial cancer, while duloxetine, venlafaxine, amitriptyline, citalopram, and sertraline were most prescribed for ovarian cancer.
The broadness of International Statistical Classification of Diseases, Tenth Revision codes made it challenging to identify nondepression diagnoses in the analyzed population. However, if all antidepressants were prescribed to treat depression, service members with cancer exhibited a higher depression rate (35%) than the general population (25%). Of 2104 patients, 191 (9.1%) had mood disorders, and 706 (33.6%) had mental disorders: 346 (49.0%) had 1 diagnosis, and 360 (51.0%) had multiple diagnoses. The percentage of diagnoses varied yearly, with notable drops in 2003, 2007, 2011, 2014, and 2018, and peaks in 2006, 2008, 2013, 2017, and 2022. This fluctuation was influenced by events like the establishment of PDTS in 2002, the 2008 economic recession, a hospital relocation in 2011, the 2014 Ebola outbreak, and the COVID-19 pandemic. Although the number of patients receiving antidepressants increased from 2019 to 2022, the overall percentage of patients receiving them did not significantly change from 2003 to 2022, aligning with previous research.5,125
Many medications have potential uses beyond what is detailed in the prescribing information. Antidepressants can relieve pain, while pain medications may help with depression. Opioids were once thought to effectively treat depression, but this perspective has changed with a greater understanding of their risks, including misuse.126-131 Pain is a severe and often unbearable AE of cancer. Of 2113 patients, 92% received opioids; 34% received both opioids and antidepressants; 2% received only antidepressants; and 7% received neither. This study didn’t clarify whether those on opioids alone recognized their depression or if those on both were aware of their dependence. While SSRIs are generally not addictive, they can lead to physical dependence, and any medication can be abused if not managed properly.132-134
Conclusions
This retrospective study analyzes data from antineoplastic agents used in systemic cancer treatment between 1988 and 2023, with a particular focus on the use of antidepressants. Data on antidepressant prescriptions are incomplete and specific to these agents, which means the findings cannot be generalized to all antidepressants. Hence, the results indicate that patients taking antidepressants had more diagnosed health issues and received more medications compared to patients who were not on these drugs.
This study underscores the need for further research into the effects of antidepressants on cancer treatment, utilizing all data from the DoD Cancer Registry. Future research should explore DDIs between antidepressants and other cancer and noncancer medications, as this study did not assess AE documentation, unlike in studies involving erlotinib, gefitinib, and paclitaxel.78,79 Further investigation is needed to evaluate the impact of discontinuing antidepressant use during cancer treatment. This comprehensive overview provides insights for clinicians to help them make informed decisions regarding the prescription of antidepressants in the context of cancer treatment.
Cancer patients experience depression at rates > 5 times that of the general population.1-11 Despite an increase in palliative care use, depression rates continued to rise.2-4 Between 5% to 16% of outpatients, 4% to 14% of inpatients, and up to 49% of patients receiving palliative care experience depression.5 This issue also impacts families and caregivers.1 A 2021 meta-analysis found that 23% of active military personnel and 20% of veterans experience depression.11
Antidepressants approved by the US Food and Drug Administration (FDA) target the serotonin, norepinephrine, or dopamine systems and include boxed warnings about an increased risk of suicidal thoughts in adults aged 18 to 24 years.12,13 These medications are categorized into several classes: monoamine oxidase inhibitors (MAOIs), tricyclic antidepressants (TCAs), tetracyclic antidepressants (TeCAs), norepinephrine-dopamine reuptake inhibitors (NDRIs), selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), serotonin receptor modulators (SRMs), serotonin-melatonin receptor antagonists (SMRAs), and N—methyl-D-aspartate receptor antagonists (NMDARAs).14,15 The first FDA-approved antidepressants, iproniazid (an MAOI) and imipramine (a TCA) laid the foundation for the development of newer classes like SSRIs and SNRIs.15-17
Older antidepressants such as MAOIs and TCAs are used less due to their adverse effects (AEs) and drug interactions. MAOIs, such as iproniazid, selegiline, moclobemide, tranylcypromine, isocarboxazid, and phenelzine, have numerous AEs and drug interactions, making them unsuitable for first- or second-line treatment of depression.14,18-21 TCAs such as doxepin, amitriptyline, nortriptyline, imipramine, desipramine, clomipramine, trimipramine, protriptyline, maprotiline, and amoxapine have a narrow therapeutic index requiring careful monitoring for signs of toxicity such as QRS widening, tremors, or confusion. Despite the issues, TCAs are generally classified as second-line agents for major depressive disorder (MDD). TCAs have off-label uses for migraine prophylaxis, treatment of obsessive-compulsive disorder (OCD), insomnia, and chronic pain management first-line.14,22-29
Newer antidepressants, including TeCAs and NDRIs, are typically more effective, but also come with safety concerns. TeCAs like mirtazapine interact with several medications, including MAOIs, serotonin-increasing drugs, alcohol, cannabidiol, and marijuana. Mirtazapine is FDA-approved for the treatment of moderate to severe depression in adults. It is also used off-label to treat insomnia, panic disorder, posttraumatic stress disorder (PTSD), generalized anxiety disorder (GAD), social anxiety disorder (SAD), headaches, and migraines. Compared to other antidepressants, mirtazapine is effective for all stages of depression and addresses a broad range of related symptoms.14,30-34 NDRIs, such as bupropion, also interact with various medications, including MAOIs, other antidepressants, stimulants, and alcohol. Bupropion is FDA-approved for smoking cessation and to treat depression and SAD. It is also used off-label for depression- related bipolar disorder or sexual dysfunction, attention-deficit/hyperactivity disorder (ADHD), and obesity.14,35-42
SSRIs, SNRIs, and SRMs should be used with caution. SSRIs such as sertraline, citalopram, escitalopram, fluoxetine, paroxetine, and fluvoxamine are first-line treatments for depression and various psychiatric disorders due to their safety and efficacy. Common AEs of SSRIs include sexual dysfunction, sleep disturbances, weight changes, and gastrointestinal (GI) issues. SSRIs can prolong the QT interval, posing a risk of life-threatening arrhythmia, and may interact with other medications, necessitating treatment adjustments. The FDA approved SSRIs for MDD, GAD, bulimia nervosa, bipolar depression, OCD, panic disorder, premenstrual dysphoric disorder, treatment-resistant depression, PTSD, and SAD. Off-label uses include binge eating disorder, body dysmorphic disorder, fibromyalgia, premature ejaculation, paraphilias, autism, Raynaud phenomenon, and vasomotor symptoms associated with menopause. Among SSRIs, sertraline and escitalopram are noted for their effectiveness and tolerability.14,43-53
SNRIs, including duloxetine, venlafaxine, desvenlafaxine, milnacipran, and levomilnacipran, may increase bleeding risk, especially when taken with blood thinners. They can also elevate blood pressure, which may worsen if combined with stimulants. SNRIs may interact with other medications that affect serotonin levels, increasing the risk of serotonin syndrome when taken with triptans, pain medications, or other antidepressants.14 Desvenlafaxine has been approved by the FDA (but not by the European Medicines Agency).54-56 Duloxetine is FDA-approved for the treatment of depression, neuropathic pain, anxiety disorders, fibromyalgia, and musculoskeletal disorders. It is used off-label to treat chemotherapy-induced peripheral neuropathy and stress urinary incontinence.57-61 Venlafaxine is FDA-approved for depression, SAD, and panic disorder, and is prescribed off-label to treat ADHD, neuropathy, fibromyalgia, cataplexy, and PTSD, either alone or in combination with other medications.62,63 Milnacipran is not approved for MDD; levomilnacipran received approval in 2013.64
SRMs such as trazodone, nefazodone, vilazodone, and vortioxetine also function as serotonin reuptake inhibitors.14,15 Trazodone is FDA-approved for MDD. It has been used off-label to treat anxiety, Alzheimer disease, substance misuse, bulimia nervosa, insomnia, fibromyalgia, and PTSD when first-line SSRIs are ineffective. A notable AE of trazodone is orthostatic hypotension, which can lead to dizziness and increase the risk of falls, especially in geriatric patients.65-70 Nefazodone was discontinued in Europe in 2003 due to rare cases of liver toxicity but remains available in the US.71-74 Vilazodone and vortioxetine are FDA-approved.
The latest classes of antidepressants include SMRAs and NMDARAs.14 Agomelatine, an SMRA, was approved in Europe in 2009 but rejected by the FDA in 2011 due to liver toxicity.75 NMDARAs like esketamine and a combination of dextromethorphan and bupropion received FDA approval in 2019 and 2022, respectively.76,77
This retrospective study analyzes noncancer drugs used during systemic chemotherapy based on a dataset of 14 antineoplastic agents. It sought to identify the most dispensed noncancer drug groups, discuss findings, compare patients with and without antidepressant prescriptions, and examine trends in antidepressant use from 2002 to 2023. This analysis expands on prior research.78-81
Methods
The Walter Reed National Military Medical Center Institutional Review Board approved the study protocol and ensured compliance with the Health Insurance Portability and Accountability Act as an exempt protocol. The Joint Pathology Center (JPC) of the US Department of Defense (DoD) Cancer Registry Program and Military Health System (MHS) data experts from the Comprehensive Ambulatory/Professional Encounter Record (CAPER) and Pharmacy Data Transaction Service (PDTS) provided data for the analysis.
Data Sources
The JPC DoD Cancer Registry Program contains data from 1998 to 2024. CAPER and PDTS are part of the MHS Data Repository/Management Analysis and Reporting Tool database. Each observation in CAPER represents an ambulatory encounter at a military treatment facility (MTF). CAPER records are available from 2003 to 2024. PDTS records are available from 2002 to 2004. Each observation in PDTS represents a prescription filled for an MHS beneficiary, excluding those filled at international civilian pharmacies and inpatient pharmacy prescriptions.
This cross-sectional analysis requested data extraction for specific cancer drugs from the DoD Cancer Registry, focusing on treatment details, diagnosis dates, patient demographics, and physicians’ comments on AEs. After identifying patients, CAPER was used to identify additional health conditions. PDTS was used to compile a list of prescription medications filled during systemic cancer treatment or < 2 years postdiagnosis.
The 2016 Surveillance, Epidemiology, and End Results Program Coding and Staging Manual and International Classification of Diseases for Oncology, 3rd edition, 1st revision, were used to decode disease and cancer types.82,83 Data sorting and analysis were performed using Microsoft Excel. The percentage for the total was calculated by using the number of patients or data available within the subgroup divided by the total number of patients or data variables. To compare the mean number of dispensed antidepressants to those without antidepressants, a 2-tailed, 2-sample z test was used to calculate the P value and determine statistical significance (P < .05) using socscistatistics.com.
Data were extracted 3 times between 2021 and 2023. The initial 2021 protocol focused on erlotinib and gefitinib. A modified protocol in 2022 added paclitaxel, cisplatin, docetaxel, pemetrexed, and crizotinib; further modification in 2023 included 8 new antineoplastic agents and 2 anticoagulants. Sotorasib has not been prescribed in the MHS, and JPC lacks records for noncancer drugs. The 2023 dataset comprised 2210 patients with cancer treated with 14 antineoplastic agents; 2104 had documented diagnoses and 2113 had recorded prescriptions. Data for erlotinib, gefitinib, and paclitaxel have been published previously.78,79
Results
Of 2113 patients with recorded prescriptions, 1297 patients (61.4%) received 109 cancer drugs, including 96 antineoplastics, 7 disease-modifying antirheumatic agents, 4 biologic response modifiers, and 2 calcitonin gene-related peptides. Fourteen antineoplastic agents had complete data from JPC, while others were noted for combination therapies or treatment switches from the PDTS (Table 1). Seventy-six cancer drugs were prescribed with antidepressants in 489 patients (eAppendix).
The JPC provided 2242 entries for 2210 patients, ranging in age from 2 months to 88 years (mean, 56 years), documenting treatment from September 1988 to January 2023. Thirty-two patients had duplicate entries due to multiple cancer locations or occurrences. Of the 2242 patients, 1541 (68.7%) were aged > 50 years, 975 patients (43.5%) had cancers that were stage III or IV, and 1267 (56.5%) had cancers that were stage 0, I, II, or not applicable/unknown. There were 51 different types of cancer: breast, lung, testicular, endometrial, and ovarian were most common (n ≥ 100 patients). Forty-two cancer types were documented among 750 patients prescribed antidepressants (Table 2).
The CAPER database recorded 8882 unique diagnoses for 2104 patients, while PDTS noted 1089 unique prescriptions within 273 therapeutic codes for 2113 patients. Nine therapeutic codes (opiate agonists, adrenals, cathartics-laxatives, nonsteroidal anti-inflammatory agents, antihistamines for GI conditions, 5-HT3 receptor antagonists, analgesics and antipyretic miscellanea, antineoplastic agents, and proton-pump inhibitors) and 8 drugs (dexamethasone, prochlorperazine, ondansetron, docusate, acetaminophen, ibuprofen, oxycodone, and polyethylene glycol 3350) were associated with > 1000 patients (≥ 50%). Patients had between 1 and 275 unique health conditions and filled 1 to 108 prescriptions. The mean (SD) number of diagnoses and prescriptions was 50 (28) and 29 (12), respectively. Of the 273 therapeutic codes, 30 groups were analyzed, with others categorized into miscellaneous groups such as lotions, vaccines, and devices. Significant differences in mean number of prescriptions were found for patients taking antidepressants compared to those not (P < .05), except for anticonvulsants and antipsychotics (P = .12 and .09, respectively) (Table 3).
Antidepressants
Of the 2113 patients with recorded prescriptions, 750 (35.5%) were dispensed 17 different antidepressants. Among these 17 antidepressants, 183 (8.7%) patients received duloxetine, 158 (7.5%) received venlafaxine, 118 (5.6%) received trazodone, and 107 (5.1%) received sertraline (Figure 1, Table 4). Of the 750 patients, 509 (67.9%) received 1 antidepressant, 168 (22.4%) received 2, 60 (8.0%) received 3, and 13 (1.7%) received > 3. Combinations varied, but only duloxetine and trazodone were prescribed to > 10 patients.
Antidepressants were prescribed annually at an overall mean (SD) rate of 23% (5%) from 2003 to 2022 (Figure 2). Patients on antidepressants during systemic therapy had a greater number of diagnosed medical conditions and received more prescription medications compared to those not taking antidepressants (P < .001) (Figure 3). The 745 patients taking antidepressants in CAPER data had between 1 and 275 diagnosed medical issues, with a mean (SD) of 55 (31) vs a range of 1 to 209 and a mean (SD) of 46 (26) for the 1359 patients not taking antidepressants. The 750 patients on antidepressants in PDTS data had between 8 and 108 prescriptions dispensed, with a mean (SD) of 32 (12), vs a range of 1 to 65 prescriptions and a mean (SD) of 29 (12) for 1363 patients not taking antidepressants.
Discussion
The JPC DoD Cancer Registry includes information on cancer types, stages, treatment regimens, and physicians’ notes, while noncancer drugs are sourced from the PDTS database. The pharmacy uses a different documentation system, leading to varied classifications.
Database reliance has its drawbacks. For example, megestrol is coded as a cancer drug, although it’s primarily used for endometrial or gynecologic cancers. Many drugs have multiple therapeutic codes assigned to them, including 10 antineoplastic agents: diclofenac, Bacillus Calmette-Guérin (BCG), megestrol acetate, tamoxifen, anastrozole, letrozole, leuprolide, goserelin, degarelix, and fluorouracil. Diclofenac, BCG, and mitomycin have been repurposed for cancer treatment.84-87 From 2003 to 2023, diclofenac was prescribed to 350 patients for mild-to-moderate pain, with only 2 patients receiving it for cancer in 2018. FDA-approved for bladder cancer in 1990, BCG was prescribed for cancer treatment for 1 patient in 2021 after being used for vaccines between 2003 and 2018. Tamoxifen, used for hormone receptor-positive breast cancer from 2004 to 2017 with 53 patients, switched to estrogen agonist-antagonists from 2017 to 2023 with 123 patients. Only a few of the 168 patients were prescribed tamoxifen using both codes.88-91 Anastrozole and letrozole were coded as antiestrogens for 7 and 18 patients, respectively, while leuprolide and goserelin were coded as gonadotropins for 59 and 18 patients. Degarelix was coded as antigonadotropins, fluorouracil as skin and mucous membrane agents miscellaneous, and megestrol acetate as progestins for 7, 6, and 3 patients, respectively. Duloxetine was given to 186 patients, primarily for depression from 2005 to 2023, with 7 patients treated for fibromyalgia from 2022 to 2023.
Antidepressants Observed
Tables 1 and 5 provide insight into the FDA approval of 14 antineoplastics and antidepressants and their CYP metabolic pathways.92-122 In Table 4, the most prescribed antidepressant classes are SNRIs, SRMs, SSRIs, TeCAs, NDRIs, and TCAs. This trend highlights a preference for newer medications with weak CYP inhibition. A total of 349 patients were prescribed SSRIs, 343 SNRIs, 119 SRMs, 109 TCAs, 83 TeCAs, and 79 NDRIs. MAOIs, SMRAs, and NMDARAs were not observed in this dataset. While there are instances of dextromethorphan-bupropion and sertraline-escitalopram being dispensed together, it remains unclear whether these were NMDARA combinations.
Among the 14 specific antineoplastic agents, 10 are metabolized by CYP isoenzymes, primarily CYP3A4. Duloxetine neither inhibits nor is metabolized by CYP3A4, a reason it is often recommended, following venlafaxine.
Both duloxetine and venlafaxine are used off-label for chemotherapy-induced peripheral neuropathy related to paclitaxel and docetaxel. According to the CYP metabolized pathway, duloxetine tends to have more favorable DDIs than venlafaxine. In PDTS data, 371 patients were treated with paclitaxel and 180 with docetaxel, with respective antidepressant prescriptions of 156 and 70. Of the 156 patients dispensed paclitaxel, 62 (40%) were dispensed with duloxetine compared to 43 (28%) with venlafaxine. Of the 70 patients dispensed docetaxel, 23 (33%) received duloxetine vs 24 (34%) with venlafaxine.
Of 85 patients prescribed duloxetine, 75 received it with either paclitaxel or docetaxel (5 received both). Five patients had documented AEs (1 neuropathy related). Of 67 patients prescribed venlafaxine, 66 received it with either paclitaxel or docetaxel. Two patients had documented AEs (1 was neuropathy related, the same patient who received duloxetine). Of the 687 patients treated with paclitaxel and 337 with docetaxel in all databases, 4 experienced neuropathic AEs from both medications.79
Antidepressants can increase the risk of bleeding, especially when combined with blood thinners, and may elevate blood pressure, particularly alongside stimulants. Of the 554 patients prescribed 9 different anticoagulants, enoxaparin, apixaban, and rivaroxaban were the most common (each > 100 patients). Among these, 201 patients (36%) received both anticoagulants and antidepressants: duloxetine for 64 patients, venlafaxine for 30, trazodone for 35, and sertraline for 26. There were no data available to assess bleeding rates related to the evaluation of DDIs between these medication classes.
Antidepressants can be prescribed for erectile dysfunction. Of the 148 patients prescribed an antidepressant for erectile dysfunction, duloxetine, trazodone, and mirtazapine were the most common. Antidepressant preferences varied by cancer type. Duloxetine was the only antidepressant used for all types of cancer. Venlafaxine, duloxetine, trazodone, sertraline, and escitalopram were the most prescribed antidepressants for breast cancer, while duloxetine, mirtazapine, citalopram, sertraline, and trazodone were the most prescribed for lung cancer. Sertraline, duloxetine, trazodone, amitriptyline, and escitalopram were most common for testicular cancer. Duloxetine, venlafaxine, trazodone, amitriptyline, and sertraline were the most prescribed for endometrial cancer, while duloxetine, venlafaxine, amitriptyline, citalopram, and sertraline were most prescribed for ovarian cancer.
The broadness of International Statistical Classification of Diseases, Tenth Revision codes made it challenging to identify nondepression diagnoses in the analyzed population. However, if all antidepressants were prescribed to treat depression, service members with cancer exhibited a higher depression rate (35%) than the general population (25%). Of 2104 patients, 191 (9.1%) had mood disorders, and 706 (33.6%) had mental disorders: 346 (49.0%) had 1 diagnosis, and 360 (51.0%) had multiple diagnoses. The percentage of diagnoses varied yearly, with notable drops in 2003, 2007, 2011, 2014, and 2018, and peaks in 2006, 2008, 2013, 2017, and 2022. This fluctuation was influenced by events like the establishment of PDTS in 2002, the 2008 economic recession, a hospital relocation in 2011, the 2014 Ebola outbreak, and the COVID-19 pandemic. Although the number of patients receiving antidepressants increased from 2019 to 2022, the overall percentage of patients receiving them did not significantly change from 2003 to 2022, aligning with previous research.5,125
Many medications have potential uses beyond what is detailed in the prescribing information. Antidepressants can relieve pain, while pain medications may help with depression. Opioids were once thought to effectively treat depression, but this perspective has changed with a greater understanding of their risks, including misuse.126-131 Pain is a severe and often unbearable AE of cancer. Of 2113 patients, 92% received opioids; 34% received both opioids and antidepressants; 2% received only antidepressants; and 7% received neither. This study didn’t clarify whether those on opioids alone recognized their depression or if those on both were aware of their dependence. While SSRIs are generally not addictive, they can lead to physical dependence, and any medication can be abused if not managed properly.132-134
Conclusions
This retrospective study analyzes data from antineoplastic agents used in systemic cancer treatment between 1988 and 2023, with a particular focus on the use of antidepressants. Data on antidepressant prescriptions are incomplete and specific to these agents, which means the findings cannot be generalized to all antidepressants. Hence, the results indicate that patients taking antidepressants had more diagnosed health issues and received more medications compared to patients who were not on these drugs.
This study underscores the need for further research into the effects of antidepressants on cancer treatment, utilizing all data from the DoD Cancer Registry. Future research should explore DDIs between antidepressants and other cancer and noncancer medications, as this study did not assess AE documentation, unlike in studies involving erlotinib, gefitinib, and paclitaxel.78,79 Further investigation is needed to evaluate the impact of discontinuing antidepressant use during cancer treatment. This comprehensive overview provides insights for clinicians to help them make informed decisions regarding the prescription of antidepressants in the context of cancer treatment.
- National Cancer Institute. Depression (PDQ)-Health Professional Version. Updated July 25, 2024. Accessed April 4, 2025. https://www.cancer.gov/about-cancer/coping/feelings/depression-hp-pdq
- Krebber AM, Buffart LM, Kleijn G, et al. Prevalence of depression in cancer patients: a meta-analysis of diagnostic interviews and self-report instruments. Psychooncology. 2014;23(2):121-130. doi:10.1002/pon.3409
- Xiang X, An R, Gehlert S. Trends in antidepressant use among U.S. cancer survivors, 1999-2012. Psychiatr Serv. 2015;66(6):564. doi:10.1176/appi.ps.201500007
- Hartung TJ, Brähler E, Faller H, et al. The risk of being depressed is significantly higher in cancer patients than in the general population: Prevalence and severity of depressive symptoms across major cancer types. Eur J Cancer. 2017;72:46-53. doi:10.1016/j.ejca.2016.11.017
- Walker J, Holm Hansen C, Martin P, et al. Prevalence of depression in adults with cancer: a systematic review. Ann Oncol. 2013;24(4):895-900. doi:10.1093/annonc/mds575
- Pitman A, Suleman S, Hyde N, Hodgkiss A. Depression and anxiety in patients with cancer. BMJ. 2018;361:k1415. doi:10.1136/bmj.k1415
- Kisely S, Alotiby MKN, Protani MM, Soole R, Arnautovska U, Siskind D. Breast cancer treatment disparities in patients with severe mental illness: a systematic review and meta-analysis. Psychooncology. 2023;32(5):651-662. doi:10.1002/pon.6120
- Massie MJ. Prevalence of depression in patients with cancer. J Natl Cancer Inst Monogr. 2004;(32):57-71. doi:10.1093/jncimonographs/lgh014
- Rodin G, Katz M, Lloyd N, Green E, Mackay JA, Wong RK. Treatment of depression in cancer patients. Curr Oncol. 2007;14(5):180-188. doi:10.3747/co.2007.146
- Wilson KG, Chochinov HM, Skirko MG, et al. Depression and anxiety disorders in palliative cancer care. J Pain Symptom Manage. 2007;33(2):118-129. doi:10.1016/j.jpainsymman.2006.07.016
- Moradi Y, Dowran B, Sepandi M. The global prevalence of depression, suicide ideation, and attempts in the military forces: a systematic review and meta-analysis of cross sectional studies. BMC Psychiatry. 2021;21(1):510. doi:10.1186/s12888-021-03526-2
- Lu CY, Zhang F, Lakoma MD, et al. Changes in antidepressant use by young people and suicidal behavior after FDA warnings and media coverage: quasi-experimental study. BMJ. 2014;348:g3596. doi:10.1136/bmj.g3596
- Friedman RA. Antidepressants’ black-box warning--10 years later. N Engl J Med. 2014;371(18):1666-1668. doi:10.1056/NEJMp1408480
- Sheffler ZM, Patel P, Abdijadid S. Antidepressants. In: StatPearls. StatPearls Publishing. Updated May 26, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK538182/
- Miller JJ. Antidepressants, Part 1: 100 Years and Counting. Accessed April 4, 2025. Psychiatric Times. https:// www.psychiatrictimes.com/view/antidepressants-part-1-100-years-and-counting
- Hillhouse TM, Porter JH. A brief history of the development of antidepressant drugs: from monoamines to glutamate. Exp Clin Psychopharmacol. 2015;23(1):1-21. doi:10.1037/a0038550
- Mitchell PB, Mitchell MS. The management of depression. Part 2. The place of the new antidepressants. Aust Fam Physician. 1994;23(9):1771-1781.
- Sabri MA, Saber-Ayad MM. MAO Inhibitors. In: Stat- Pearls. StatPearls Publishing. Updated June 5, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557395/
- Sub Laban T, Saadabadi A. Monoamine Oxidase Inhibitors (MAOI). In: StatPearls. StatPearls Publishing. Updated July 17, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539848/
- Fiedorowicz JG, Swartz KL. The role of monoamine oxidase inhibitors in current psychiatric practice. J Psychiatr Pract. 2004;10(4):239-248. doi:10.1097/00131746-200407000-00005
- Flockhart DA. Dietary restrictions and drug interactions with monoamine oxidase inhibitors: an update. J Clin Psychiatry. 2012;73 Suppl 1:17-24. doi:10.4088/JCP.11096su1c.03
- Moraczewski J, Awosika AO, Aedma KK. Tricyclic Antidepressants. In: StatPearls. StatPearls Publishing. Updated August 17, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557791/
- Almasi A, Patel P, Meza CE. Doxepin. In: StatPearls. StatPearls Publishing. Updated February 14, 2024. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK542306/
- Thour A, Marwaha R. Amitriptyline. In: StatPearls. Stat- Pearls Publishing. Updated July 18, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK537225/
- Radley DC, Finkelstein SN, Stafford RS. Off-label prescribing among office-based physicians. Arch Intern Med. 2006;166(9):1021-1026. doi:10.1001/archinte.166.9.1021
- Tesfaye S, Sloan G, Petrie J, et al. Comparison of amitriptyline supplemented with pregabalin, pregabalin supplemented with amitriptyline, and duloxetine supplemented with pregabalin for the treatment of diabetic peripheral neuropathic pain (OPTION-DM): a multicentre, double-blind, randomised crossover trial. Lancet. 2022;400(10353):680- 690. doi:10.1016/S0140-6736(22)01472-6
- Farag HM, Yunusa I, Goswami H, Sultan I, Doucette JA, Eguale T. Comparison of amitriptyline and US Food and Drug Administration-approved treatments for fibromyalgia: a systematic review and network metaanalysis. JAMA Netw Open. 2022;5(5):e2212939. doi:10.1001/jamanetworkopen.2022.12939
- Merwar G, Gibbons JR, Hosseini SA, Saadabadi A. Nortriptyline. In: StatPearls. StatPearls Publishing. Updated June 5, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK482214/
- Fayez R, Gupta V. Imipramine. In: StatPearls. StatPearls Publishing. Updated May 22, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557656/
- Jilani TN, Gibbons JR, Faizy RM, Saadabadi A. Mirtazapine. In: StatPearls. StatPearls Publishing. Updated August 28, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK519059/
- Nutt DJ. Tolerability and safety aspects of mirtazapine. Hum Psychopharmacol. 2002;17 Suppl 1:S37-S41. doi:10.1002/hup.388
- Gandotra K, Chen P, Jaskiw GE, Konicki PE, Strohl KP. Effective treatment of insomnia with mirtazapine attenuates concomitant suicidal ideation. J Clin Sleep Med. 2018;14(5):901-902. doi:10.5664/jcsm.7142
- Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264. doi:10.1111/j.1527-3458.2001.tb00198.x
- Wang SM, Han C, Bahk WM, et al. Addressing the side effects of contemporary antidepressant drugs: a comprehensive review. Chonnam Med J. 2018;54(2):101-112. doi:10.4068/cmj.2018.54.2.101
- Huecker MR, Smiley A, Saadabadi A. Bupropion. In: StatPearls. StatPearls Publishing. Updated April 9, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK470212/
- Hsieh MT, Tseng PT, Wu YC, et al. Effects of different pharmacologic smoking cessation treatments on body weight changes and success rates in patients with nicotine dependence: a network meta-analysis. Obes Rev. 2019;20(6):895- 905. doi:10.1111/obr.12835
- Hankosky ER, Bush HM, Dwoskin LP, et al. Retrospective analysis of health claims to evaluate pharmacotherapies with potential for repurposing: association of bupropion and stimulant use disorder remission. AMIA Annu Symp Proc. 2018;2018:1292-1299.
- Livingstone-Banks J, Norris E, Hartmann-Boyce J, West R, Jarvis M, Hajek P. Relapse prevention interventions for smoking cessation. Cochrane Database Syst Rev. 2019; 2(2):CD003999. doi:10.1002/14651858.CD003999.pub5
- Clayton AH, Kingsberg SA, Goldstein I. Evaluation and management of hypoactive sexual desire disorder. Sex Med. 2018;6(2):59-74. doi:10.1016/j.esxm.2018.01.004
- Verbeeck W, Bekkering GE, Van den Noortgate W, Kramers C. Bupropion for attention deficit hyperactivity disorder (ADHD) in adults. Cochrane Database Syst Rev. 2017;10(10):CD009504. doi:10.1002/14651858.CD009504.pub2
- Ng QX. A systematic review of the use of bupropion for attention-deficit/hyperactivity disorder in children and adolescents. J Child Adolesc Psychopharmacol. 2017;27(2):112-116. doi:10.1089/cap.2016.0124
- Fava M, Rush AJ, Thase ME, et al. 15 years of clinical experience with bupropion HCl: from bupropion to bupropion SR to bupropion XL. Prim Care Companion J Clin Psychiatry. 2005;7(3):106-113. doi:10.4088/pcc.v07n0305
- Chu A, Wadhwa R. Selective Serotonin Reuptake Inhibitors. In: StatPearls. StatPearls Publishing. Updated May 1, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK554406/
- Singh HK, Saadabadi A. Sertraline. In: StatPearls. Stat- Pearls Publishing. Updated February 13, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK547689/
- MacQueen G, Born L, Steiner M. The selective serotonin reuptake inhibitor sertraline: its profile and use in psychiatric disorders. CNS Drug Rev. 2001;7(1):1-24. doi:10.1111/j.1527-3458.2001.tb00188.x
- Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373(9665):746-758. doi:10.1016/S0140-6736(09)60046-5
- Nelson JC. The STAR*D study: a four-course meal that leaves us wanting more. Am J Psychiatry. 2006;163(11):1864-1866. doi:10.1176/ajp.2006.163.11.1864
- Sharbaf Shoar N, Fariba KA, Padhy RK. Citalopram. In: StatPearls. StatPearls Publishing. Updated November 7, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK482222/
- Landy K, Rosani A, Estevez R. Escitalopram. In: Stat- Pearls. StatPearls Publishing. Updated November 10, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557734/
- Cavanah LR, Ray P, Goldhirsh JL, Huey LY, Piper BJ. Rise of escitalopram and the fall of citalopram. medRxiv. Preprint published online May 8, 2023. doi:10.1101/2023.05.07.23289632
- Sohel AJ, Shutter MC, Patel P, Molla M. Fluoxetine. In: StatPearls. StatPearls Publishing. Updated July 4, 2022. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK459223/
- Wong DT, Perry KW, Bymaster FP. Case history: the discovery of fluoxetine hydrochloride (Prozac). Nat Rev Drug Discov. 2005;4(9):764-774. doi:10.1038/nrd1821
- Shrestha P, Fariba KA, Abdijadid S. Paroxetine. In: Stat- Pearls. StatPearls Publishing. Updated July 17, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK526022/
- Naseeruddin R, Rosani A, Marwaha R. Desvenlafaxine. In: StatPearls. StatPearls Publishing. Updated July 10, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK534829
- Lieberman DZ, Massey SH. Desvenlafaxine in major depressive disorder: an evidence-based review of its place in therapy. Core Evid. 2010;4:67-82. doi:10.2147/ce.s5998
- Withdrawal assessment report for Ellefore (desvenlafaxine). European Medicine Agency. 2009. Accessed April 4, 2025. https://www.ema.europa.eu/en/documents/withdrawal-report/withdrawal-assessment-report-ellefore_en.pdf
- Dhaliwal JS, Spurling BC, Molla M. Duloxetine. In: Stat- Pearls. Statpearls Publishing. Updated May 29, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK549806/
- Hershman DL, Lacchetti C, Dworkin RH, et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2014;32(18):1941-1967. doi:10.1200/JCO.2013.54.0914
- Sommer C, Häuser W, Alten R, et al. Medikamentöse Therapie des Fibromyalgiesyndroms. Systematische Übersicht und Metaanalyse [Drug therapy of fibromyalgia syndrome. Systematic review, meta-analysis and guideline]. Schmerz. 2012;26(3):297-310. doi:10.1007/s00482-012-1172-2
- Bril V, England J, Franklin GM, et al. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2011;76(20):1758-1765. doi:10.1212/WNL.0b013e3182166ebe
- Attal N, Cruccu G, Baron R, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol. 2010;17(9):1113-e88. doi:10.1111/j.1468-1331.2010.02999.x
- Singh D, Saadabadi A. Venlafaxine. In: StatPearls. StatePearls Publishing. Updated February 26, 2024. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK535363/
- Sertraline and venlafaxine: new indication. Prevention of recurrent depression: no advance. Prescrire Int. 2005;14(75):19-20.
- Bruno A, Morabito P, S p i n a E , M u s c a t ello MRl. The role of levomilnacipran in the management of major depressive disorder: a comprehensive review. Curr Neuro pharmacol. 2016;14(2):191-199. doi:10.2174/1570159x14666151117122458
- Shin JJ, Saadabadi A. Trazodone. In: StatPearls. StatPearls Publishing. Updated February 29, 2024. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK470560/
- Khouzam HR. A review of trazodone use in psychiatric and medical conditions. Postgrad Med. 2017;129(1):140-148. doi:10.1080/00325481.2017.1249265
- Smales ET, Edwards BA, Deyoung PN, et al. Trazodone effects on obstructive sleep apnea and non-REM arousal threshold. Ann Am Thorac Soc. 2015;12(5):758-764. doi:10.1513/AnnalsATS.201408-399OC
- Eckert DJ, Malhotra A, Wellman A, White DP. Trazodone increases the respiratory arousal threshold in patients with obstructive sleep apnea and a low arousal threshold. Sleep. 2014;37(4):811-819. doi:10.5665/sleep.3596
- Schatzberg AF, Nemeroff CB, eds. The American Psychiat ric Association Publishing Textbook of Psychopharmacology. 4th ed. American Psychiatric Publishing; 2009.
- Ruxton K, Woodman RJ, Mangoni AA. Drugs with anticholinergic effects and cognitive impairment, falls and allcause mortality in older adults: a systematic review and meta-analysis. Br J Clin Pharmacol. 2015;80(2):209-220. doi:10.1111/bcp.12617
- Nefazodone. In: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. National Institute of Diabetes and Digestive and Kidney Diseases. Updated March 6, 2020. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK548179/
- Drugs of Current Interest: Nefazodone. WHO Pharmaceuticals Newsletter:(1). 2003(1):7. https://web.archive.org/web/20150403165029/http:/apps.who.int/medicinedocs/en/d/Js4944e/3.html
- Choi S. Nefazodone (serzone) withdrawn because of hepatotoxicity. CMAJ. 2003;169(11):1187.
- Teva Nefazodone Statement. News release. Teva USA. December 20, 2021. Accessed April 4, 2025. https:// www.tevausa.com/news-and-media/press-releases/teva-nefazodone-statement/
- Levitan MN, Papelbaum M, Nardi AE. Profile of agomelatine and its potential in the treatment of generalized anxiety disorder. Neuropsychiatr Dis Treat. 2015;11:1149- 1155. doi:10.2147/NDT.S67470
- Fu DJ, Ionescu DF, Li X, et al. Esketamine nasal spray for rapid reduction of major depressive disorder symptoms in patients who have active suicidal ideation with intent: double-blind, randomized study (ASPIRE I). J Clin Psychiatry. 2020;81(3):19m13191. doi:10.4088/JCP.19m13191
- Iosifescu DV, Jones A, O’Gorman C, et al. Efficacy and safety of AXS-05 (dextromethorphan-bupropion) in patients with major depressive disorder: a phase 3 randomized clinical trial (GEMINI). J Clin Psychiatry. 2022;83(4): 21m14345. doi:10.4088/JCP.21m14345
- Luong TT, Powers CN, Reinhardt BJ, et al. Retrospective evaluation of drug-drug interactions with erlotinib and gefitinib use in the Military Health System. Fed Pract. 2023;40(Suppl 3):S24-S34. doi:10.12788/fp.0401
- Luong TT, Shou KJ, Reinhard BJ, Kigelman OF, Greenfield KM. Paclitaxel drug-drug interactions in the Military Health System. Fed Pract. 2024;41(Suppl 3):S70-S82. doi:10.12788/fp.0499
- Luong TT, Powers CN, Reinhardt BJ, Weina PJ. Preclinical drug-drug interactions (DDIs) of gefitinib with/without losartan and selective serotonin reuptake inhibitors (SSRIs): citalopram, fluoxetine, fluvoxamine, paroxetine, sertraline, and venlafaxine. Curr Res Pharmacol Drug Discov. 2022;3:100112. doi:10.1016/j.crphar.2022.100112
- Luong TT, McAnulty MJ, Evers DL, Reinhardt BJ, Weina PJ. Pre-clinical drug-drug interaction (DDI) of gefitinib or erlotinib with cytochrome P450 (CYP) inhibiting drugs, fluoxetine and/or losartan. Curr Res Toxicol. 2021;2:217- 224. doi:10.1016/j.crtox.2021.05.006,
- Adamo M, Dickie L, Ruhl J. SEER program coding and staging manual 2016. National Cancer Institute; 2016. Accessed April 4, 2025. https://seer.cancer.gov/archive/manuals/2016/SPCSM_2016_maindoc.pdf
- World Health Organization. International classification of diseases for oncology (ICD-O). 3rd ed, 1st revision. World Health Organization; 2013. Accessed April 4, 2025. https://apps.who.int/iris/handle/10665/96612
- Simon S. FDA approves Jelmyto (mitomycin gel) for urothelial cancer. American Cancer Society. April 20, 2020. Accessed April 4, 2025. https://www.cancer.org/cancer/latest-news/fda-approves-jelmyto-mitomycin-gel-for-urothelial-cancer.html
- Pantziarka P, Sukhatme V, Bouche G, Meheus L, Sukhatme VP. Repurposing drugs in oncology (ReDO)- diclofenac as an anti-cancer agent. Ecancermedicalscience. 2016;10:610. doi:10.3332/ecancer.2016.610
- Choi S, Kim S, Park J, Lee SE, Kim C, Kang D. Diclofenac: a nonsteroidal anti-inflammatory drug inducing cancer cell death by inhibiting microtubule polymerization and autophagy flux. Antioxidants (Basel). 2022;11(5):1009. doi:10.3390/antiox11051009
- Tontonoz M. The ABCs of BCG: oldest approved immunotherapy gets new explanation. Memorial Sloan Kettering Cancer Center. July 17, 2020. Accessed April 4, 2025. https://www.mskcc.org/news/oldest-approved-immunotherapy-gets-new-explanation
- Jordan VC. Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocr Relat Cancer. 2014;21(3):R235-R246. doi:10.1530/ERC-14-0092
- Cole MP, Jones CT, Todd ID. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br J Cancer. 1971;25(2):270-275. doi:10.1038/bjc.1971.33
- Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2(3):205-213. doi:10.1038/nrd1031
- Maximov PY, McDaniel RE, Jordan VC. Tamoxifen: Pioneering Medicine in Breast Cancer. Springer Basel; 2013. Accessed April 4, 2025. https://link.springer.com/book/10.1007/978-3-0348-0664-0
- Taxol (paclitaxel). Prescribing information. Bristol-Myers Squibb Company; 2011. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020262s049lbl.pdf
- Abraxane (paclitaxel). Prescribing information. Celgene Corporation; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/021660s047lbl.pdf
- Tarceva (erlotinib). Prescribing information. OSI Pharmaceuticals, LLC; 2016. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021743s025lbl.pdf
- Docetaxel. Prescribing information. Sichuan Hyiyu Pharmaceutical Co.; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215813s000lbl.pdf
- Alimta (pemetrexed). Prescribing information. Teva Pharmaceuticals; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/208419s004lbl.pdf
- Tagrisso (osimertinib). Prescribing information. Astra- Zeneca Pharmaceuticals; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208065s021lbl.pdf
- Iressa (gefitinib). Prescribing information. AstraZeneca Pharmaceuticals; 2018. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/206995s003lbl.pdf
- Kadcyla (ado-trastuzumab emtansine). Prescribing information. Genentech, Inc.; 2013. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125427lbl.pdf
- Alecensa (alectinib). Prescribing information. Genetech, Inc.; 2017. Accessed April 4, 2025. https:// www.accessdata.fda.gov/drugsatfda_docs/label/2017/208434s003lbl.pdf
- Xalkori (crizotinib). Prescribing information. Pfizer Laboratories; 2022. Accessed April 4, 2025. https:// www.accessdata.fda.gov/drugsatfda_docs/label/2022/202570s033lbl.pdf
- Lorbrena (lorlatinib). Prescribing information. Pfizer Laboratories; 2018. Accessed April 14, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210868s000lbl.pdf
- Alunbrig (brigatinib). Prescribing information. Takeda Pharmaceutical Company; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208772s008lbl.pdf
- Rozlytrek (entrectinib). Prescribing information. Genentech, Inc.; 2019. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212725s000lbl.pdf
- Herceptin (trastuzumab). Prescribing information. Genentech, Inc.; 2010. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/103792s5250lbl.pdf
- Cybalta (duloxetine). Prescribing information. Eli Lilly and Company; 2017. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021427s049lbl.pdf
- Effexor XR (venlafaxine). Prescribing information. Pfizer Wyeth Pharmaceuticals Inc; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/020699s112lbl.pdf
- Desyrel (trazodone hydrochloride). Prescribing information. Pragma Pharmaceuticals; 2017. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/018207s032lbl.pdf
- Sertraline hydrochloride. Prescribing information. Almatica Pharma LLC; 2021. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215133s000lbl.pdf
- Remeron (mirtazapine). Prescribing information. Merck & Co. Inc; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/020415s029,%20021208s019lbl.pdf
- Celexa (citalopram). Prescribing information. Allergan USA Inc; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/020822s041lbl.pdf
- information. GlaxoSmithKline; 2019. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/020358s066lbl.pdf
- Amitriptyline hydrochloride tablet. Prescribing information. Quality Care Products LLC; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/spl/data/0f12f50f-7087-46e7-a2e6-356b4c566c9f/0f12f50f-7087-46e7-a2e6-356b4c566c9f.xml
- Lexapro (escitalopram). Prescribing information. AbbVie Inc; 2023. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/021323s055,021365s039lbl.pdf
- Fluoxetine. Prescribing information. Edgemont Pharmaceutical, LLC; 2017. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/202133s004s005lbl.pdf
- Paxil (paroxetine). Prescribing Information. Apotex Inc; 2021. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/020031s077lbl.pdf
- Pamelor (nortriptyline HCl). Prescribing information. Mallinckrodt, Inc; 2012. Accessed April 4, 2025. https:// www.accessdata.fda.gov/drugsatfda_docs/label/2012/018012s029,018013s061lbl.pdf
- Silenor (doxepin). Prescribing information. Currax Pharmaceuticals; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/022036s006lbl.pdf
- Tofranil-PM (imipramine pamote). Prescribing information. Mallinckrodt, Inc; 2014. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/017090s078lbl.pdf
- Norpramin (desipramine hydrochloride). Prescribing information. Sanofi-aventis U.S. LLC; 2014. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/014399s069lbl.pdf
- Khedezla (desvenlafaxine). Prescribing information. Osmotical Pharmaceutical US LLC; 2019. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/204683s006lbl.pdf
- Nefazodone hydrochloride. Prescribing information. Bryant Ranch Prepack; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/spl/data/0bd4c34a-4f43-4c84-8b98-1d074cba97d5/0bd4c34a-4f43-4c84-8b98-1d074cba97d5.xml
- Grassi L, Nanni MG, Rodin G, Li M, Caruso R. The use of antidepressants in oncology: a review and practical tips for oncologists. Ann Oncol. 2018;29(1):101-111. doi:10.1093/annonc/mdx526
- Lee E, Park Y, Li D, Rodriguez-Fuguet A, Wang X, Zhang WC. Antidepressant use and lung cancer risk and survival: a meta-analysis of observational studies. Cancer Res Commun. 2023;3(6):1013-1025. doi:10.1158/2767-9764.CRC-23-0003
- Olfson M, Marcus SC. National patterns in antidepressant medication treatment. Arch Gen Psychiatry. 2009;66(8):848 -856. doi:10.1001/archgenpsychiatry.2009.81
- Grattan A, Sullivan MD, Saunders KW, Campbell CI, Von Korff MR. Depression and prescription opioid misuse among chronic opioid therapy recipients with no history of substance abuse. Ann Fam Med. 2012;10(4):304-311. doi:10.1370/afm.1371
- Cowan DT, Wilson-Barnett J, Griffiths P, Allan LG. A survey of chronic noncancer pain patients prescribed opioid analgesics. Pain Med. 2003;4(4):340-351. doi:10.1111/j.1526-4637.2003.03038.x
- Breckenridge J, Clark JD. Patient characteristics associated with opioid versus nonsteroidal anti-inflammatory drug management of chronic low back pain. J Pain. 2003;4(6):344-350. doi:10.1016/s1526-5900(03)00638-2
- Edlund MJ, Martin BC, Devries A, Fan MY, Braden JB, Sullivan MD. Trends in use of opioids for chronic noncancer pain among individuals with mental health and substance use disorders: the TROUP study. Clin J Pain. 2010;26(1):1-8. doi:10.1097/AJP.0b013e3181b99f35
- Sullivan MD, Edlund MJ, Fan MY, DeVries A, Braden JB, Martin BC. Risks for possible and probable opioid misuse among recipients of chronic opioid therapy in commercial and medicaid insurance plans: the TROUP study. Pain. 2010;150(2):332-339. doi:10.1016/j.pain.2010.05.020
- Dunn KM, Saunders KW, Rutter CM, et al. Opioid prescriptions for chronic pain and overdose: a cohort study. Ann Intern Med. 2010;152(2):85-92. doi:10.7326/0003-4819-152-2-201001190-00006
- Haddad P. Do antidepressants have any potential to cause addiction? J Psychopharmacol. 1999;13(3):300- 307. doi:10.1177/026988119901300321
- Lakeview Health Staff. America’s most abused antidepressants. Lakeview Health. January 24, 2004. Accessed April 4, 2025. https://www.lakeviewhealth.com/blog/us-most-abused-antidepressants/
- Greenhouse Treatment Center Editorial Staff. Addiction to antidepressants: is it possible? America Addiction Centers: Greenhouse Treatment Center. Updated April 23, 2024. Accessed April 4, 2025. https://greenhousetreatment.com/prescription-medication/antidepressants/
- National Cancer Institute. Depression (PDQ)-Health Professional Version. Updated July 25, 2024. Accessed April 4, 2025. https://www.cancer.gov/about-cancer/coping/feelings/depression-hp-pdq
- Krebber AM, Buffart LM, Kleijn G, et al. Prevalence of depression in cancer patients: a meta-analysis of diagnostic interviews and self-report instruments. Psychooncology. 2014;23(2):121-130. doi:10.1002/pon.3409
- Xiang X, An R, Gehlert S. Trends in antidepressant use among U.S. cancer survivors, 1999-2012. Psychiatr Serv. 2015;66(6):564. doi:10.1176/appi.ps.201500007
- Hartung TJ, Brähler E, Faller H, et al. The risk of being depressed is significantly higher in cancer patients than in the general population: Prevalence and severity of depressive symptoms across major cancer types. Eur J Cancer. 2017;72:46-53. doi:10.1016/j.ejca.2016.11.017
- Walker J, Holm Hansen C, Martin P, et al. Prevalence of depression in adults with cancer: a systematic review. Ann Oncol. 2013;24(4):895-900. doi:10.1093/annonc/mds575
- Pitman A, Suleman S, Hyde N, Hodgkiss A. Depression and anxiety in patients with cancer. BMJ. 2018;361:k1415. doi:10.1136/bmj.k1415
- Kisely S, Alotiby MKN, Protani MM, Soole R, Arnautovska U, Siskind D. Breast cancer treatment disparities in patients with severe mental illness: a systematic review and meta-analysis. Psychooncology. 2023;32(5):651-662. doi:10.1002/pon.6120
- Massie MJ. Prevalence of depression in patients with cancer. J Natl Cancer Inst Monogr. 2004;(32):57-71. doi:10.1093/jncimonographs/lgh014
- Rodin G, Katz M, Lloyd N, Green E, Mackay JA, Wong RK. Treatment of depression in cancer patients. Curr Oncol. 2007;14(5):180-188. doi:10.3747/co.2007.146
- Wilson KG, Chochinov HM, Skirko MG, et al. Depression and anxiety disorders in palliative cancer care. J Pain Symptom Manage. 2007;33(2):118-129. doi:10.1016/j.jpainsymman.2006.07.016
- Moradi Y, Dowran B, Sepandi M. The global prevalence of depression, suicide ideation, and attempts in the military forces: a systematic review and meta-analysis of cross sectional studies. BMC Psychiatry. 2021;21(1):510. doi:10.1186/s12888-021-03526-2
- Lu CY, Zhang F, Lakoma MD, et al. Changes in antidepressant use by young people and suicidal behavior after FDA warnings and media coverage: quasi-experimental study. BMJ. 2014;348:g3596. doi:10.1136/bmj.g3596
- Friedman RA. Antidepressants’ black-box warning--10 years later. N Engl J Med. 2014;371(18):1666-1668. doi:10.1056/NEJMp1408480
- Sheffler ZM, Patel P, Abdijadid S. Antidepressants. In: StatPearls. StatPearls Publishing. Updated May 26, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK538182/
- Miller JJ. Antidepressants, Part 1: 100 Years and Counting. Accessed April 4, 2025. Psychiatric Times. https:// www.psychiatrictimes.com/view/antidepressants-part-1-100-years-and-counting
- Hillhouse TM, Porter JH. A brief history of the development of antidepressant drugs: from monoamines to glutamate. Exp Clin Psychopharmacol. 2015;23(1):1-21. doi:10.1037/a0038550
- Mitchell PB, Mitchell MS. The management of depression. Part 2. The place of the new antidepressants. Aust Fam Physician. 1994;23(9):1771-1781.
- Sabri MA, Saber-Ayad MM. MAO Inhibitors. In: Stat- Pearls. StatPearls Publishing. Updated June 5, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557395/
- Sub Laban T, Saadabadi A. Monoamine Oxidase Inhibitors (MAOI). In: StatPearls. StatPearls Publishing. Updated July 17, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539848/
- Fiedorowicz JG, Swartz KL. The role of monoamine oxidase inhibitors in current psychiatric practice. J Psychiatr Pract. 2004;10(4):239-248. doi:10.1097/00131746-200407000-00005
- Flockhart DA. Dietary restrictions and drug interactions with monoamine oxidase inhibitors: an update. J Clin Psychiatry. 2012;73 Suppl 1:17-24. doi:10.4088/JCP.11096su1c.03
- Moraczewski J, Awosika AO, Aedma KK. Tricyclic Antidepressants. In: StatPearls. StatPearls Publishing. Updated August 17, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557791/
- Almasi A, Patel P, Meza CE. Doxepin. In: StatPearls. StatPearls Publishing. Updated February 14, 2024. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK542306/
- Thour A, Marwaha R. Amitriptyline. In: StatPearls. Stat- Pearls Publishing. Updated July 18, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK537225/
- Radley DC, Finkelstein SN, Stafford RS. Off-label prescribing among office-based physicians. Arch Intern Med. 2006;166(9):1021-1026. doi:10.1001/archinte.166.9.1021
- Tesfaye S, Sloan G, Petrie J, et al. Comparison of amitriptyline supplemented with pregabalin, pregabalin supplemented with amitriptyline, and duloxetine supplemented with pregabalin for the treatment of diabetic peripheral neuropathic pain (OPTION-DM): a multicentre, double-blind, randomised crossover trial. Lancet. 2022;400(10353):680- 690. doi:10.1016/S0140-6736(22)01472-6
- Farag HM, Yunusa I, Goswami H, Sultan I, Doucette JA, Eguale T. Comparison of amitriptyline and US Food and Drug Administration-approved treatments for fibromyalgia: a systematic review and network metaanalysis. JAMA Netw Open. 2022;5(5):e2212939. doi:10.1001/jamanetworkopen.2022.12939
- Merwar G, Gibbons JR, Hosseini SA, Saadabadi A. Nortriptyline. In: StatPearls. StatPearls Publishing. Updated June 5, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK482214/
- Fayez R, Gupta V. Imipramine. In: StatPearls. StatPearls Publishing. Updated May 22, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557656/
- Jilani TN, Gibbons JR, Faizy RM, Saadabadi A. Mirtazapine. In: StatPearls. StatPearls Publishing. Updated August 28, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK519059/
- Nutt DJ. Tolerability and safety aspects of mirtazapine. Hum Psychopharmacol. 2002;17 Suppl 1:S37-S41. doi:10.1002/hup.388
- Gandotra K, Chen P, Jaskiw GE, Konicki PE, Strohl KP. Effective treatment of insomnia with mirtazapine attenuates concomitant suicidal ideation. J Clin Sleep Med. 2018;14(5):901-902. doi:10.5664/jcsm.7142
- Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264. doi:10.1111/j.1527-3458.2001.tb00198.x
- Wang SM, Han C, Bahk WM, et al. Addressing the side effects of contemporary antidepressant drugs: a comprehensive review. Chonnam Med J. 2018;54(2):101-112. doi:10.4068/cmj.2018.54.2.101
- Huecker MR, Smiley A, Saadabadi A. Bupropion. In: StatPearls. StatPearls Publishing. Updated April 9, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK470212/
- Hsieh MT, Tseng PT, Wu YC, et al. Effects of different pharmacologic smoking cessation treatments on body weight changes and success rates in patients with nicotine dependence: a network meta-analysis. Obes Rev. 2019;20(6):895- 905. doi:10.1111/obr.12835
- Hankosky ER, Bush HM, Dwoskin LP, et al. Retrospective analysis of health claims to evaluate pharmacotherapies with potential for repurposing: association of bupropion and stimulant use disorder remission. AMIA Annu Symp Proc. 2018;2018:1292-1299.
- Livingstone-Banks J, Norris E, Hartmann-Boyce J, West R, Jarvis M, Hajek P. Relapse prevention interventions for smoking cessation. Cochrane Database Syst Rev. 2019; 2(2):CD003999. doi:10.1002/14651858.CD003999.pub5
- Clayton AH, Kingsberg SA, Goldstein I. Evaluation and management of hypoactive sexual desire disorder. Sex Med. 2018;6(2):59-74. doi:10.1016/j.esxm.2018.01.004
- Verbeeck W, Bekkering GE, Van den Noortgate W, Kramers C. Bupropion for attention deficit hyperactivity disorder (ADHD) in adults. Cochrane Database Syst Rev. 2017;10(10):CD009504. doi:10.1002/14651858.CD009504.pub2
- Ng QX. A systematic review of the use of bupropion for attention-deficit/hyperactivity disorder in children and adolescents. J Child Adolesc Psychopharmacol. 2017;27(2):112-116. doi:10.1089/cap.2016.0124
- Fava M, Rush AJ, Thase ME, et al. 15 years of clinical experience with bupropion HCl: from bupropion to bupropion SR to bupropion XL. Prim Care Companion J Clin Psychiatry. 2005;7(3):106-113. doi:10.4088/pcc.v07n0305
- Chu A, Wadhwa R. Selective Serotonin Reuptake Inhibitors. In: StatPearls. StatPearls Publishing. Updated May 1, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK554406/
- Singh HK, Saadabadi A. Sertraline. In: StatPearls. Stat- Pearls Publishing. Updated February 13, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK547689/
- MacQueen G, Born L, Steiner M. The selective serotonin reuptake inhibitor sertraline: its profile and use in psychiatric disorders. CNS Drug Rev. 2001;7(1):1-24. doi:10.1111/j.1527-3458.2001.tb00188.x
- Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373(9665):746-758. doi:10.1016/S0140-6736(09)60046-5
- Nelson JC. The STAR*D study: a four-course meal that leaves us wanting more. Am J Psychiatry. 2006;163(11):1864-1866. doi:10.1176/ajp.2006.163.11.1864
- Sharbaf Shoar N, Fariba KA, Padhy RK. Citalopram. In: StatPearls. StatPearls Publishing. Updated November 7, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK482222/
- Landy K, Rosani A, Estevez R. Escitalopram. In: Stat- Pearls. StatPearls Publishing. Updated November 10, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557734/
- Cavanah LR, Ray P, Goldhirsh JL, Huey LY, Piper BJ. Rise of escitalopram and the fall of citalopram. medRxiv. Preprint published online May 8, 2023. doi:10.1101/2023.05.07.23289632
- Sohel AJ, Shutter MC, Patel P, Molla M. Fluoxetine. In: StatPearls. StatPearls Publishing. Updated July 4, 2022. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK459223/
- Wong DT, Perry KW, Bymaster FP. Case history: the discovery of fluoxetine hydrochloride (Prozac). Nat Rev Drug Discov. 2005;4(9):764-774. doi:10.1038/nrd1821
- Shrestha P, Fariba KA, Abdijadid S. Paroxetine. In: Stat- Pearls. StatPearls Publishing. Updated July 17, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK526022/
- Naseeruddin R, Rosani A, Marwaha R. Desvenlafaxine. In: StatPearls. StatPearls Publishing. Updated July 10, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK534829
- Lieberman DZ, Massey SH. Desvenlafaxine in major depressive disorder: an evidence-based review of its place in therapy. Core Evid. 2010;4:67-82. doi:10.2147/ce.s5998
- Withdrawal assessment report for Ellefore (desvenlafaxine). European Medicine Agency. 2009. Accessed April 4, 2025. https://www.ema.europa.eu/en/documents/withdrawal-report/withdrawal-assessment-report-ellefore_en.pdf
- Dhaliwal JS, Spurling BC, Molla M. Duloxetine. In: Stat- Pearls. Statpearls Publishing. Updated May 29, 2023. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK549806/
- Hershman DL, Lacchetti C, Dworkin RH, et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2014;32(18):1941-1967. doi:10.1200/JCO.2013.54.0914
- Sommer C, Häuser W, Alten R, et al. Medikamentöse Therapie des Fibromyalgiesyndroms. Systematische Übersicht und Metaanalyse [Drug therapy of fibromyalgia syndrome. Systematic review, meta-analysis and guideline]. Schmerz. 2012;26(3):297-310. doi:10.1007/s00482-012-1172-2
- Bril V, England J, Franklin GM, et al. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2011;76(20):1758-1765. doi:10.1212/WNL.0b013e3182166ebe
- Attal N, Cruccu G, Baron R, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol. 2010;17(9):1113-e88. doi:10.1111/j.1468-1331.2010.02999.x
- Singh D, Saadabadi A. Venlafaxine. In: StatPearls. StatePearls Publishing. Updated February 26, 2024. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK535363/
- Sertraline and venlafaxine: new indication. Prevention of recurrent depression: no advance. Prescrire Int. 2005;14(75):19-20.
- Bruno A, Morabito P, S p i n a E , M u s c a t ello MRl. The role of levomilnacipran in the management of major depressive disorder: a comprehensive review. Curr Neuro pharmacol. 2016;14(2):191-199. doi:10.2174/1570159x14666151117122458
- Shin JJ, Saadabadi A. Trazodone. In: StatPearls. StatPearls Publishing. Updated February 29, 2024. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK470560/
- Khouzam HR. A review of trazodone use in psychiatric and medical conditions. Postgrad Med. 2017;129(1):140-148. doi:10.1080/00325481.2017.1249265
- Smales ET, Edwards BA, Deyoung PN, et al. Trazodone effects on obstructive sleep apnea and non-REM arousal threshold. Ann Am Thorac Soc. 2015;12(5):758-764. doi:10.1513/AnnalsATS.201408-399OC
- Eckert DJ, Malhotra A, Wellman A, White DP. Trazodone increases the respiratory arousal threshold in patients with obstructive sleep apnea and a low arousal threshold. Sleep. 2014;37(4):811-819. doi:10.5665/sleep.3596
- Schatzberg AF, Nemeroff CB, eds. The American Psychiat ric Association Publishing Textbook of Psychopharmacology. 4th ed. American Psychiatric Publishing; 2009.
- Ruxton K, Woodman RJ, Mangoni AA. Drugs with anticholinergic effects and cognitive impairment, falls and allcause mortality in older adults: a systematic review and meta-analysis. Br J Clin Pharmacol. 2015;80(2):209-220. doi:10.1111/bcp.12617
- Nefazodone. In: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. National Institute of Diabetes and Digestive and Kidney Diseases. Updated March 6, 2020. Accessed April 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK548179/
- Drugs of Current Interest: Nefazodone. WHO Pharmaceuticals Newsletter:(1). 2003(1):7. https://web.archive.org/web/20150403165029/http:/apps.who.int/medicinedocs/en/d/Js4944e/3.html
- Choi S. Nefazodone (serzone) withdrawn because of hepatotoxicity. CMAJ. 2003;169(11):1187.
- Teva Nefazodone Statement. News release. Teva USA. December 20, 2021. Accessed April 4, 2025. https:// www.tevausa.com/news-and-media/press-releases/teva-nefazodone-statement/
- Levitan MN, Papelbaum M, Nardi AE. Profile of agomelatine and its potential in the treatment of generalized anxiety disorder. Neuropsychiatr Dis Treat. 2015;11:1149- 1155. doi:10.2147/NDT.S67470
- Fu DJ, Ionescu DF, Li X, et al. Esketamine nasal spray for rapid reduction of major depressive disorder symptoms in patients who have active suicidal ideation with intent: double-blind, randomized study (ASPIRE I). J Clin Psychiatry. 2020;81(3):19m13191. doi:10.4088/JCP.19m13191
- Iosifescu DV, Jones A, O’Gorman C, et al. Efficacy and safety of AXS-05 (dextromethorphan-bupropion) in patients with major depressive disorder: a phase 3 randomized clinical trial (GEMINI). J Clin Psychiatry. 2022;83(4): 21m14345. doi:10.4088/JCP.21m14345
- Luong TT, Powers CN, Reinhardt BJ, et al. Retrospective evaluation of drug-drug interactions with erlotinib and gefitinib use in the Military Health System. Fed Pract. 2023;40(Suppl 3):S24-S34. doi:10.12788/fp.0401
- Luong TT, Shou KJ, Reinhard BJ, Kigelman OF, Greenfield KM. Paclitaxel drug-drug interactions in the Military Health System. Fed Pract. 2024;41(Suppl 3):S70-S82. doi:10.12788/fp.0499
- Luong TT, Powers CN, Reinhardt BJ, Weina PJ. Preclinical drug-drug interactions (DDIs) of gefitinib with/without losartan and selective serotonin reuptake inhibitors (SSRIs): citalopram, fluoxetine, fluvoxamine, paroxetine, sertraline, and venlafaxine. Curr Res Pharmacol Drug Discov. 2022;3:100112. doi:10.1016/j.crphar.2022.100112
- Luong TT, McAnulty MJ, Evers DL, Reinhardt BJ, Weina PJ. Pre-clinical drug-drug interaction (DDI) of gefitinib or erlotinib with cytochrome P450 (CYP) inhibiting drugs, fluoxetine and/or losartan. Curr Res Toxicol. 2021;2:217- 224. doi:10.1016/j.crtox.2021.05.006,
- Adamo M, Dickie L, Ruhl J. SEER program coding and staging manual 2016. National Cancer Institute; 2016. Accessed April 4, 2025. https://seer.cancer.gov/archive/manuals/2016/SPCSM_2016_maindoc.pdf
- World Health Organization. International classification of diseases for oncology (ICD-O). 3rd ed, 1st revision. World Health Organization; 2013. Accessed April 4, 2025. https://apps.who.int/iris/handle/10665/96612
- Simon S. FDA approves Jelmyto (mitomycin gel) for urothelial cancer. American Cancer Society. April 20, 2020. Accessed April 4, 2025. https://www.cancer.org/cancer/latest-news/fda-approves-jelmyto-mitomycin-gel-for-urothelial-cancer.html
- Pantziarka P, Sukhatme V, Bouche G, Meheus L, Sukhatme VP. Repurposing drugs in oncology (ReDO)- diclofenac as an anti-cancer agent. Ecancermedicalscience. 2016;10:610. doi:10.3332/ecancer.2016.610
- Choi S, Kim S, Park J, Lee SE, Kim C, Kang D. Diclofenac: a nonsteroidal anti-inflammatory drug inducing cancer cell death by inhibiting microtubule polymerization and autophagy flux. Antioxidants (Basel). 2022;11(5):1009. doi:10.3390/antiox11051009
- Tontonoz M. The ABCs of BCG: oldest approved immunotherapy gets new explanation. Memorial Sloan Kettering Cancer Center. July 17, 2020. Accessed April 4, 2025. https://www.mskcc.org/news/oldest-approved-immunotherapy-gets-new-explanation
- Jordan VC. Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocr Relat Cancer. 2014;21(3):R235-R246. doi:10.1530/ERC-14-0092
- Cole MP, Jones CT, Todd ID. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br J Cancer. 1971;25(2):270-275. doi:10.1038/bjc.1971.33
- Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2(3):205-213. doi:10.1038/nrd1031
- Maximov PY, McDaniel RE, Jordan VC. Tamoxifen: Pioneering Medicine in Breast Cancer. Springer Basel; 2013. Accessed April 4, 2025. https://link.springer.com/book/10.1007/978-3-0348-0664-0
- Taxol (paclitaxel). Prescribing information. Bristol-Myers Squibb Company; 2011. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020262s049lbl.pdf
- Abraxane (paclitaxel). Prescribing information. Celgene Corporation; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/021660s047lbl.pdf
- Tarceva (erlotinib). Prescribing information. OSI Pharmaceuticals, LLC; 2016. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021743s025lbl.pdf
- Docetaxel. Prescribing information. Sichuan Hyiyu Pharmaceutical Co.; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215813s000lbl.pdf
- Alimta (pemetrexed). Prescribing information. Teva Pharmaceuticals; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/208419s004lbl.pdf
- Tagrisso (osimertinib). Prescribing information. Astra- Zeneca Pharmaceuticals; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208065s021lbl.pdf
- Iressa (gefitinib). Prescribing information. AstraZeneca Pharmaceuticals; 2018. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/206995s003lbl.pdf
- Kadcyla (ado-trastuzumab emtansine). Prescribing information. Genentech, Inc.; 2013. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125427lbl.pdf
- Alecensa (alectinib). Prescribing information. Genetech, Inc.; 2017. Accessed April 4, 2025. https:// www.accessdata.fda.gov/drugsatfda_docs/label/2017/208434s003lbl.pdf
- Xalkori (crizotinib). Prescribing information. Pfizer Laboratories; 2022. Accessed April 4, 2025. https:// www.accessdata.fda.gov/drugsatfda_docs/label/2022/202570s033lbl.pdf
- Lorbrena (lorlatinib). Prescribing information. Pfizer Laboratories; 2018. Accessed April 14, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210868s000lbl.pdf
- Alunbrig (brigatinib). Prescribing information. Takeda Pharmaceutical Company; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208772s008lbl.pdf
- Rozlytrek (entrectinib). Prescribing information. Genentech, Inc.; 2019. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212725s000lbl.pdf
- Herceptin (trastuzumab). Prescribing information. Genentech, Inc.; 2010. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/103792s5250lbl.pdf
- Cybalta (duloxetine). Prescribing information. Eli Lilly and Company; 2017. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021427s049lbl.pdf
- Effexor XR (venlafaxine). Prescribing information. Pfizer Wyeth Pharmaceuticals Inc; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/020699s112lbl.pdf
- Desyrel (trazodone hydrochloride). Prescribing information. Pragma Pharmaceuticals; 2017. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/018207s032lbl.pdf
- Sertraline hydrochloride. Prescribing information. Almatica Pharma LLC; 2021. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215133s000lbl.pdf
- Remeron (mirtazapine). Prescribing information. Merck & Co. Inc; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/020415s029,%20021208s019lbl.pdf
- Celexa (citalopram). Prescribing information. Allergan USA Inc; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/020822s041lbl.pdf
- information. GlaxoSmithKline; 2019. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/020358s066lbl.pdf
- Amitriptyline hydrochloride tablet. Prescribing information. Quality Care Products LLC; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/spl/data/0f12f50f-7087-46e7-a2e6-356b4c566c9f/0f12f50f-7087-46e7-a2e6-356b4c566c9f.xml
- Lexapro (escitalopram). Prescribing information. AbbVie Inc; 2023. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/021323s055,021365s039lbl.pdf
- Fluoxetine. Prescribing information. Edgemont Pharmaceutical, LLC; 2017. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/202133s004s005lbl.pdf
- Paxil (paroxetine). Prescribing Information. Apotex Inc; 2021. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/020031s077lbl.pdf
- Pamelor (nortriptyline HCl). Prescribing information. Mallinckrodt, Inc; 2012. Accessed April 4, 2025. https:// www.accessdata.fda.gov/drugsatfda_docs/label/2012/018012s029,018013s061lbl.pdf
- Silenor (doxepin). Prescribing information. Currax Pharmaceuticals; 2020. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/022036s006lbl.pdf
- Tofranil-PM (imipramine pamote). Prescribing information. Mallinckrodt, Inc; 2014. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/017090s078lbl.pdf
- Norpramin (desipramine hydrochloride). Prescribing information. Sanofi-aventis U.S. LLC; 2014. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/014399s069lbl.pdf
- Khedezla (desvenlafaxine). Prescribing information. Osmotical Pharmaceutical US LLC; 2019. Accessed April 4, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/204683s006lbl.pdf
- Nefazodone hydrochloride. Prescribing information. Bryant Ranch Prepack; 2022. Accessed April 4, 2025. https://www.accessdata.fda.gov/spl/data/0bd4c34a-4f43-4c84-8b98-1d074cba97d5/0bd4c34a-4f43-4c84-8b98-1d074cba97d5.xml
- Grassi L, Nanni MG, Rodin G, Li M, Caruso R. The use of antidepressants in oncology: a review and practical tips for oncologists. Ann Oncol. 2018;29(1):101-111. doi:10.1093/annonc/mdx526
- Lee E, Park Y, Li D, Rodriguez-Fuguet A, Wang X, Zhang WC. Antidepressant use and lung cancer risk and survival: a meta-analysis of observational studies. Cancer Res Commun. 2023;3(6):1013-1025. doi:10.1158/2767-9764.CRC-23-0003
- Olfson M, Marcus SC. National patterns in antidepressant medication treatment. Arch Gen Psychiatry. 2009;66(8):848 -856. doi:10.1001/archgenpsychiatry.2009.81
- Grattan A, Sullivan MD, Saunders KW, Campbell CI, Von Korff MR. Depression and prescription opioid misuse among chronic opioid therapy recipients with no history of substance abuse. Ann Fam Med. 2012;10(4):304-311. doi:10.1370/afm.1371
- Cowan DT, Wilson-Barnett J, Griffiths P, Allan LG. A survey of chronic noncancer pain patients prescribed opioid analgesics. Pain Med. 2003;4(4):340-351. doi:10.1111/j.1526-4637.2003.03038.x
- Breckenridge J, Clark JD. Patient characteristics associated with opioid versus nonsteroidal anti-inflammatory drug management of chronic low back pain. J Pain. 2003;4(6):344-350. doi:10.1016/s1526-5900(03)00638-2
- Edlund MJ, Martin BC, Devries A, Fan MY, Braden JB, Sullivan MD. Trends in use of opioids for chronic noncancer pain among individuals with mental health and substance use disorders: the TROUP study. Clin J Pain. 2010;26(1):1-8. doi:10.1097/AJP.0b013e3181b99f35
- Sullivan MD, Edlund MJ, Fan MY, DeVries A, Braden JB, Martin BC. Risks for possible and probable opioid misuse among recipients of chronic opioid therapy in commercial and medicaid insurance plans: the TROUP study. Pain. 2010;150(2):332-339. doi:10.1016/j.pain.2010.05.020
- Dunn KM, Saunders KW, Rutter CM, et al. Opioid prescriptions for chronic pain and overdose: a cohort study. Ann Intern Med. 2010;152(2):85-92. doi:10.7326/0003-4819-152-2-201001190-00006
- Haddad P. Do antidepressants have any potential to cause addiction? J Psychopharmacol. 1999;13(3):300- 307. doi:10.1177/026988119901300321
- Lakeview Health Staff. America’s most abused antidepressants. Lakeview Health. January 24, 2004. Accessed April 4, 2025. https://www.lakeviewhealth.com/blog/us-most-abused-antidepressants/
- Greenhouse Treatment Center Editorial Staff. Addiction to antidepressants: is it possible? America Addiction Centers: Greenhouse Treatment Center. Updated April 23, 2024. Accessed April 4, 2025. https://greenhousetreatment.com/prescription-medication/antidepressants/
Assessing the Impact of Antidepressants on Cancer Treatment: A Retrospective Analysis of 14 Antineoplastic Agents
Assessing the Impact of Antidepressants on Cancer Treatment: A Retrospective Analysis of 14 Antineoplastic Agents
