User login
Impact of Rapid Blood Culture Identification on Antibiotic De-escalation at a Veterans Affairs Medical Center
Impact of Rapid Blood Culture Identification on Antibiotic De-escalation at a Veterans Affairs Medical Center
About 530,000 to 628,000 episodes of bloodstream infections (BSI) occur annually in the US.1 Early identification and treatment of bacteremia are essential to improve patient outcomes because it allows for more timely targeted antibiotic therapy.2 Organism identification and susceptibility testing can take 2 to 5 days, prolonging the use of broad-spectrum empiric antibiotics and increasing the risk of adverse events.3,4 The Infectious Disease Society of America recommends the use of rapid diagnostic testing and antimicrobial stewardship programs (ASPs) to improve rates of antibiotic susceptibilities to targeted antibiotics and optimize resource utilization.3 Rapid blood culture identification (BCID) technologies reduce the duration of empiric antibiotics in patients with contaminated blood cultures, resulting in shorter hospital stays and saving money per each patient tested.4
In March 2023, Veteran Health Indiana (VHI) implemented the BioFire FilmArray Blood Culture Identification (BCID2), a BSI panel test that identifies select gram-negative bacteria, gram-positive bacteria, yeast, and antimicrobial resistance genes with an aggregate sensitivity of 99% and a specificity of 99.8%. The BCID2 presents clinically relevant information faster than traditional culture methods, allowing clinicians to make more efficient and educated antibiotic regimen decisions than with previous methods.5
It takes 24 to 48 hours from blood collection for culture incubation, positivity, and gram staining to occur at VHI. If the gram stain is positive, the blood culture is placed on the BioFire BCID2 in addition to traditional culture medium. BioFire BCID2 results are ready in 45 to 60 minutes. Results are uploaded into the electronic health record (EHR) ≤ 2 hours after they are obtained and the primary team is notified if the test is positive for certain critical results. Susceptibility testing of an identified organism typically requires an additional 24 to 48 hours for finalization. VHI Infectious Disease created an evidence-based antibiotic recommendation chart for certain medication(s) and alternate therapies based on the reported organism and its interpreted presence of resistance markers (eg, ceftriaxone for Escherichia coli when extended-spectrum beta lactamases are not detected vs meropenem if extended-spectrum beta lactamases marker are present). These charts optimize the antibiotic regimen while awaiting susceptibility finalizations.
Two previous studies describe the impact of rapid diagnostic testing technology at US Department of Veterans Affairs (VA) medical centers.6,7 In Texas, the ASP reviewed BCID panel results via clinical decision support software for about 1 hour per day.6 A Los Angeles study analyzed the impact of Biofire BCID with an interpretation guide centered on unnecessary vancomycin use and determined that shorter duration of the medication may have been the result of more frequent infectious disease consultation.7
This study assessed the time to optimal antibiotic de-escalation before and after the implementation of BioFire BCID2 with results reviewed by the ASP without active notification or assistance of any clinical decision support technology. The primary objective was to evaluate difference in time to optimal antibiotics from blood culture draw pre- vs postintervention. Secondary objectives included differences in time to organism identification, difference in time on broad-spectrum antibiotics, and difference in time to appropriate antibiotics.
Methods
This quasi-experimental retrospective chart review assessed the impact of BioFire BCID2 use on timely antibiotic de-escalation for patients who experienced a BSI at VHI between March 1, 2022, and October 1, 2023. Microbiology laboratory records identified eligible patients with positive blood cultures within the study time frame. Data were collected from the VHI EHR.
Patients were included if they had a positive bacterial blood culture and received ≥ 1 antibiotic indicated for bacteremia while receiving inpatient care. Patients were excluded if they died prior to blood culture results, transferred out of VHI, left against medical advice, or had untreated contaminants in blood culture results (ie, never received antibiotics aimed at the contaminated culture).
Patient lists were generated for before and after implementation of BioFire BCID2 (pre- and postintervention) using the VHI EHR and microbiology laboratory record system. The pre- and postinterventions groups were different sizes. As a result, a random sampling of the preintervention group was selected and included patients from March 1, 2022, through March 26, 2023. The postintervention group was smaller due to time constraints between initiation of BioFire BCID2 for data collection and included all patients from March 27, 2023, through October 1, 2023.
Optimal antibiotics were defined as escalation from inappropriate therapy to broader agent(s), de-escalation from broad-spectrum therapy to targeted agent(s), discontinuation of therapy due to an organism being identified as a contaminant, or optimization of a regimen to the preferred antimicrobial agent based on evidence-based consensus guidelines. Broad-spectrum antibiotics included: piperacillin/tazobactam, cefepime, ceftazidime, ceftazidime-avibactam, cefiderocol, carbapenems, fluroquinolones, vancomycin, daptomycin, ceftaroline, linezolid, or aztreonam. Appropriate antibiotics were defined as those with activity toward the final identified organism(s).
Deidentified participant data were entered into Microsoft Excel and kept on a secure VA server to complete statistical analyses. Parametric continuous data, such as age, were analyzed using the t-test, while nonparametric continuous data, such as time to optimal antibiotics, were analyzed using the Mann-Whitney U test. Categorical data, like sex and race, were analyzed using either Fisher exact test for small sample sizes or X2 test for a larger sample size. Statistical significance levels was defined as P < .05.
Results
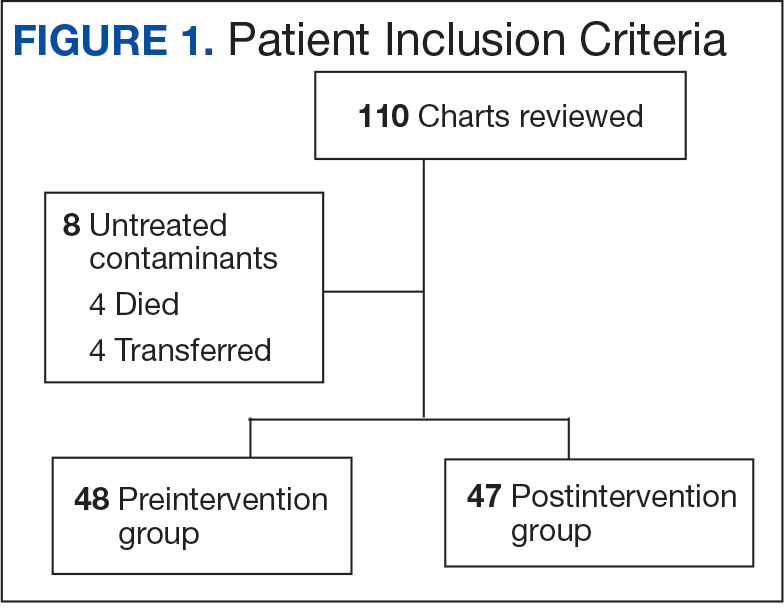
Using patient lists drawn from the EHR and the microbiology laboratory records, 110 electronic charts were randomly selected for review. Fifteen patients were excluded: 8 had untreated contaminants, 4 died, and 3 were transferred out of VHI. Of the 95 patients included, 48 were in the preintervention group and 47 were in the postintervention group (Figure 1).
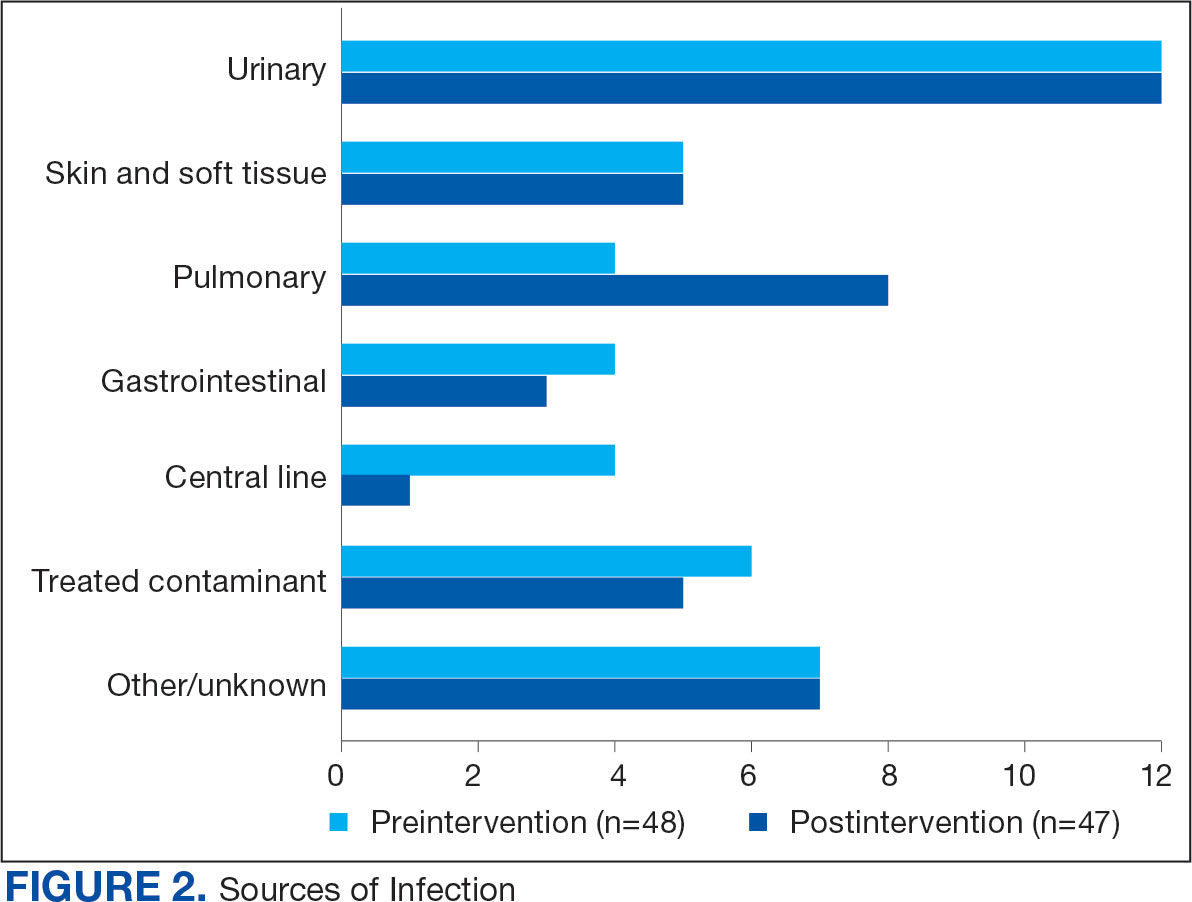
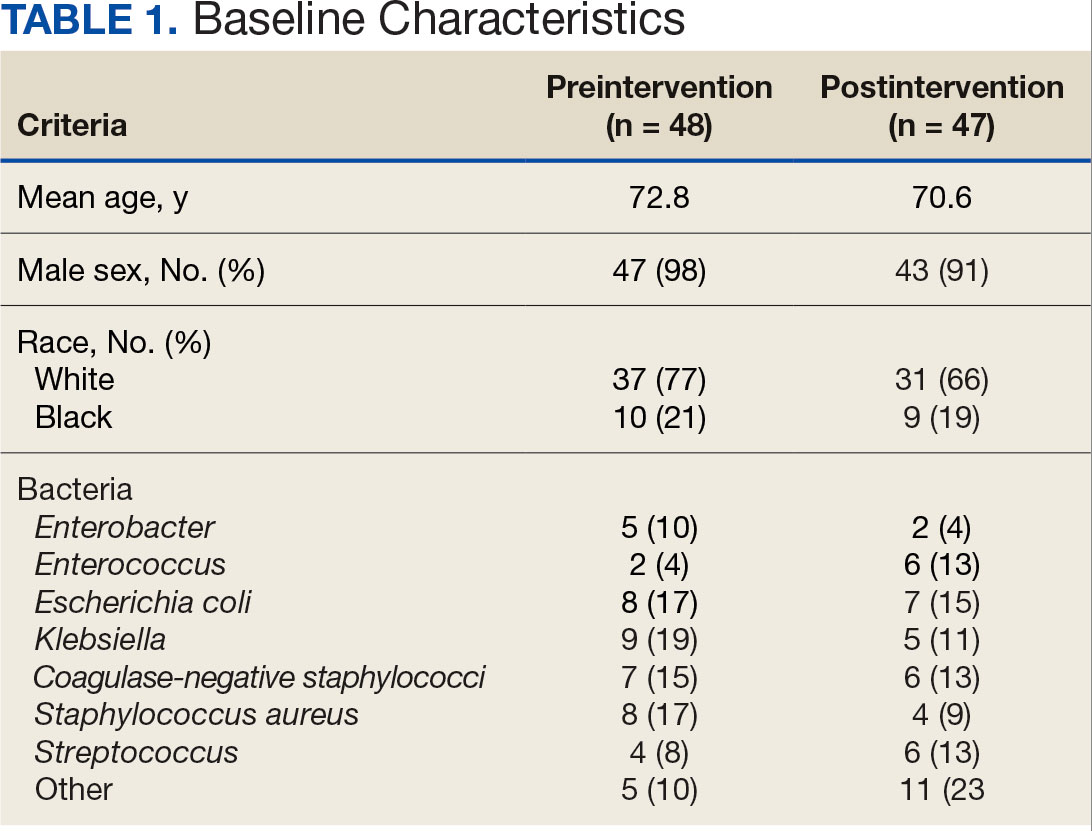
Baseline characteristics were similar between the 2 groups (Table 1). Most patients were White males aged > 70 years in the EHR. The urinary tract was the most common source of infection, impacting 12 patients in each group (Figure 2). Escherichia coli, Klebsiella, Staphylococcus, and Streptococcus were the most common bloodstream isolates identified.
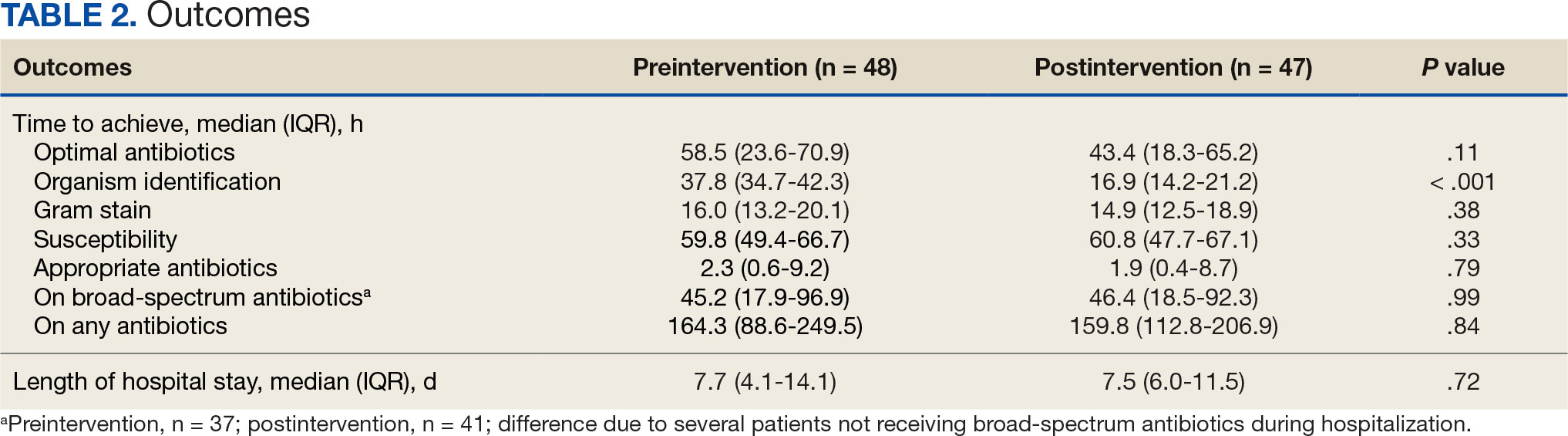
The median time to optimal antibiotics in the preintervention group was 58.5 hours vs 43.4 hours in the postintervention group (P = .11). The median time to organism identification was 37.8 hours in the preintervention group vs 16.9 hours in the postintervention group (P < .001). The median time on broad-spectrum antibiotics was 45.2 hours in the preintervention group vs 46.6 hours in the postintervention group (P = .99). The median time on appropriate antibiotics in the preintervention group was 2.3 hours vs 1.9 hours in the postintervention group (P = .79). Differences in other measured outcomes between the groups were not statistically significant (Table 2).
Although implementation of rapid diagnostic technology reduced the median time to optimal antibiotics, the results were not statistically significant. Shorter time to organism identification in the postintervention group compared to the preintervention group was the lone statistically significant metric (P < .001).
Discussion
A lack of statistical significance in the primary outcome may have been due to nonadherence to facility de-escalation protocols or a suboptimal BioFire BCID2 result notification system. Additionally, use of rapid BCID at VHI may improve over time as clinicians become more familiar with the technology. Gaps in clinical pharmacy coverage during the night shift may have also contributed to delays in antibiotic optimization, particularly if other clinicians are not equipped with the knowledge or training to appropriately deescalate antibiotics based on microorganisms identified. A 2017 study by Donner et al concluded that physician interpretation of BCID results is suboptimal and should be augmented with clinical decision support tools as new technology becomes available.8 Despite the statistically insignificant results of this study, it did highlight potential areas of improvement which can lead to improved patient care.
Previous research has evaluated the impact of rapid BCID technology on antibiotic treatment and clinical outcomes. Chiasson et al found that median time to optimal therapy was 73.8 hours in the pre-BCID arm compared to 34.7 hours in the post- BCID arm (P ≤ .001), emphasizing the importance of combining rapid BCID with clinical decision support tools and pharmacy input.6 Senok et al found that BCID2 implementation led to a significant decrease in median time to culture result, which informed optimal antibiotic therapy and decreased 30-day mortality in the intensive care setting.9 In contrast, the current study did not stratify patients according to medical ward or illness severity even though clinicians may be less likely to de-escalate antibiotic therapy in critically ill patients.
Bae et al reported findings consistent with the current study and concluded that BCID did not affect the clinical outcomes of overall BSIs; however, it contributed to early administration of effective antibiotics in cases of BSIs caused by multidrug-resistant organisms.10 Results of this study were not stratified according to multidrug-resistant organisms because the sample size was too small. The current study also included patients with polymicrobial infections, which may have impacted the results due to a less streamlined approach to antibiotic optimization.
Limitations
This single-center, retrospective study had a small sample size, short time frame, and lacked patient diversity, and therefore may not be generalizable to other health care systems. The sample size was limited by shorter date range and smaller patient list between BioFire BCID2 implementation and data collection, which was used to determine the number of charts selected in each group. Some patients received antibiotics prior to blood cultures being drawn, which may falsely decrease time to optimal/ appropriate antibiotics and falsely increase time on broad spectrum/any antibiotics due to early antibiotic administration. The total number of patients on broad-spectrum antibiotics differed from the total number of patients for other outcomes because several patients never received the defined broad spectrum antibiotics.
Conclusions
When combined with a pre-existing ASP without active notification, the implementation of BioFire BCID2 did not return statistically significant data showing a decrease in time to optimal antibiotics, time to appropriate antibiotics, or time on broad-spectrum antibiotics at VHI. To make this program more successful, pharmacist intervention and clinical decision support tools may be needed.
Additional research is required to determine the optimal integration of antimicrobial stewardship, rapid diagnostic technology, and pharmacy services for maximum benefit. Even though the primary outcome was not statistically significant, the results may be clinically significant from a stewardship perspective. Realigning microbiology workflows to mimic other research, which emphasizes the importance of funneling rapid BCID results through the ASP, may improve outcomes. Future studies may be warranted following the implementation of clinical decision support tools to assess their impact on stewardship practices and patient outcomes.
- Goto M, Al-Hasan MN. Overall burden of bloodstream infection and nosocomial bloodstream infection in North America and Europe. Clin Microbiol Infect. 2013;19(6):501- 509. doi:10.1111/1469-0691.12195
- Pardo J, Klinker KP, Borgert SJ, Butler BM, Giglio PG, Rand KH. Clinical and economic impact of antimicrobial stewardship interventions with the FilmArray blood culture identification panel. Diagn Microbiol Infect Dis. 2016;84(2):159-164. doi:10.1016/j.diagmicrobio.2015.10.023.
- Barlam TF, Cosgrove SE, Abbo LM, et al. Implementing an antibiotic stewardship program: guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America. Clin Infect Dis. 2016;62(10):e51-e77. doi:10.1093/cid/ciw118
- BIOFIRE® Blood Culture Identification 2 (BCID2) Panel. Biomerierux. Updated 2025. Accessed May 10, 2025. https://www.biofiredx.com/products/the-filmarray-panels/filmarraybcid/
- Huang AM, Newton D, Kunapuli A, et al. Impact of rapid organism identification via matrix-assisted laser desorption/ionization time-of-flight combined with antimicrobial stewardship team intervention in adult patients with bacteremia and candidemia. Clin Infect Dis. 2013;57(9):1237-1245. doi:10.1093/cid/cit498
- Chiasson JM, Smith WJ, Jodlowski TZ, Kouma MA, Cutrell JB. Impact of a rapid blood culture diagnostic panel on time to optimal antimicrobial therapy at a veterans affairs medical center. J Pharm Pract. 2022;35(5):722-729. doi:10.1177/08971900211000686
- Wu S, Watson RL, Graber CJ. 2007. Impact of combining rapid diagnostics with an interpretation guide on vancomycin usage for contaminant blood cultures growing coagulase- negative staphylococci (CoNS). Open Forum Infect Dis. 2019;6(Suppl 2):S674. doi:10.1093/ofid/ofz360.1687
- Donner LM, Campbell WS, Lyden E, Van Schooneveld TC. Assessment of rapid-blood-culture-identification result interpretation and antibiotic prescribing practices. J Clin Microbiol. 2017;55(5):1496-1507. doi:10.1128/JCM.02395-16
- Senok A, Dabal LA, Alfaresi M, et al. Clinical impact of the BIOFIRE blood culture identification 2 panel in adult patients with bloodstream infection: a multicentre observational study in the United Arab Emirates. Diagnostics (Basel). 2023;13(14):2433. doi:10.3390/diagnostics13142433
- Bae JY, Bae J, So MK, Choi HJ, Lee M. The impact of the rapid blood culture identification panel on antibiotic treatment and clinical outcomes in bloodstream infections, particularly those associated with multidrug-resistant micro-organisms. Diagnostics (Basel). 2023;13(23):3504. doi:10.3390/diagnostics13233504
About 530,000 to 628,000 episodes of bloodstream infections (BSI) occur annually in the US.1 Early identification and treatment of bacteremia are essential to improve patient outcomes because it allows for more timely targeted antibiotic therapy.2 Organism identification and susceptibility testing can take 2 to 5 days, prolonging the use of broad-spectrum empiric antibiotics and increasing the risk of adverse events.3,4 The Infectious Disease Society of America recommends the use of rapid diagnostic testing and antimicrobial stewardship programs (ASPs) to improve rates of antibiotic susceptibilities to targeted antibiotics and optimize resource utilization.3 Rapid blood culture identification (BCID) technologies reduce the duration of empiric antibiotics in patients with contaminated blood cultures, resulting in shorter hospital stays and saving money per each patient tested.4
In March 2023, Veteran Health Indiana (VHI) implemented the BioFire FilmArray Blood Culture Identification (BCID2), a BSI panel test that identifies select gram-negative bacteria, gram-positive bacteria, yeast, and antimicrobial resistance genes with an aggregate sensitivity of 99% and a specificity of 99.8%. The BCID2 presents clinically relevant information faster than traditional culture methods, allowing clinicians to make more efficient and educated antibiotic regimen decisions than with previous methods.5
It takes 24 to 48 hours from blood collection for culture incubation, positivity, and gram staining to occur at VHI. If the gram stain is positive, the blood culture is placed on the BioFire BCID2 in addition to traditional culture medium. BioFire BCID2 results are ready in 45 to 60 minutes. Results are uploaded into the electronic health record (EHR) ≤ 2 hours after they are obtained and the primary team is notified if the test is positive for certain critical results. Susceptibility testing of an identified organism typically requires an additional 24 to 48 hours for finalization. VHI Infectious Disease created an evidence-based antibiotic recommendation chart for certain medication(s) and alternate therapies based on the reported organism and its interpreted presence of resistance markers (eg, ceftriaxone for Escherichia coli when extended-spectrum beta lactamases are not detected vs meropenem if extended-spectrum beta lactamases marker are present). These charts optimize the antibiotic regimen while awaiting susceptibility finalizations.
Two previous studies describe the impact of rapid diagnostic testing technology at US Department of Veterans Affairs (VA) medical centers.6,7 In Texas, the ASP reviewed BCID panel results via clinical decision support software for about 1 hour per day.6 A Los Angeles study analyzed the impact of Biofire BCID with an interpretation guide centered on unnecessary vancomycin use and determined that shorter duration of the medication may have been the result of more frequent infectious disease consultation.7
This study assessed the time to optimal antibiotic de-escalation before and after the implementation of BioFire BCID2 with results reviewed by the ASP without active notification or assistance of any clinical decision support technology. The primary objective was to evaluate difference in time to optimal antibiotics from blood culture draw pre- vs postintervention. Secondary objectives included differences in time to organism identification, difference in time on broad-spectrum antibiotics, and difference in time to appropriate antibiotics.
Methods
This quasi-experimental retrospective chart review assessed the impact of BioFire BCID2 use on timely antibiotic de-escalation for patients who experienced a BSI at VHI between March 1, 2022, and October 1, 2023. Microbiology laboratory records identified eligible patients with positive blood cultures within the study time frame. Data were collected from the VHI EHR.
Patients were included if they had a positive bacterial blood culture and received ≥ 1 antibiotic indicated for bacteremia while receiving inpatient care. Patients were excluded if they died prior to blood culture results, transferred out of VHI, left against medical advice, or had untreated contaminants in blood culture results (ie, never received antibiotics aimed at the contaminated culture).
Patient lists were generated for before and after implementation of BioFire BCID2 (pre- and postintervention) using the VHI EHR and microbiology laboratory record system. The pre- and postinterventions groups were different sizes. As a result, a random sampling of the preintervention group was selected and included patients from March 1, 2022, through March 26, 2023. The postintervention group was smaller due to time constraints between initiation of BioFire BCID2 for data collection and included all patients from March 27, 2023, through October 1, 2023.
Optimal antibiotics were defined as escalation from inappropriate therapy to broader agent(s), de-escalation from broad-spectrum therapy to targeted agent(s), discontinuation of therapy due to an organism being identified as a contaminant, or optimization of a regimen to the preferred antimicrobial agent based on evidence-based consensus guidelines. Broad-spectrum antibiotics included: piperacillin/tazobactam, cefepime, ceftazidime, ceftazidime-avibactam, cefiderocol, carbapenems, fluroquinolones, vancomycin, daptomycin, ceftaroline, linezolid, or aztreonam. Appropriate antibiotics were defined as those with activity toward the final identified organism(s).
Deidentified participant data were entered into Microsoft Excel and kept on a secure VA server to complete statistical analyses. Parametric continuous data, such as age, were analyzed using the t-test, while nonparametric continuous data, such as time to optimal antibiotics, were analyzed using the Mann-Whitney U test. Categorical data, like sex and race, were analyzed using either Fisher exact test for small sample sizes or X2 test for a larger sample size. Statistical significance levels was defined as P < .05.
Results
Using patient lists drawn from the EHR and the microbiology laboratory records, 110 electronic charts were randomly selected for review. Fifteen patients were excluded: 8 had untreated contaminants, 4 died, and 3 were transferred out of VHI. Of the 95 patients included, 48 were in the preintervention group and 47 were in the postintervention group (Figure 1).
Baseline characteristics were similar between the 2 groups (Table 1). Most patients were White males aged > 70 years in the EHR. The urinary tract was the most common source of infection, impacting 12 patients in each group (Figure 2). Escherichia coli, Klebsiella, Staphylococcus, and Streptococcus were the most common bloodstream isolates identified.
The median time to optimal antibiotics in the preintervention group was 58.5 hours vs 43.4 hours in the postintervention group (P = .11). The median time to organism identification was 37.8 hours in the preintervention group vs 16.9 hours in the postintervention group (P < .001). The median time on broad-spectrum antibiotics was 45.2 hours in the preintervention group vs 46.6 hours in the postintervention group (P = .99). The median time on appropriate antibiotics in the preintervention group was 2.3 hours vs 1.9 hours in the postintervention group (P = .79). Differences in other measured outcomes between the groups were not statistically significant (Table 2).
Although implementation of rapid diagnostic technology reduced the median time to optimal antibiotics, the results were not statistically significant. Shorter time to organism identification in the postintervention group compared to the preintervention group was the lone statistically significant metric (P < .001).
Discussion
A lack of statistical significance in the primary outcome may have been due to nonadherence to facility de-escalation protocols or a suboptimal BioFire BCID2 result notification system. Additionally, use of rapid BCID at VHI may improve over time as clinicians become more familiar with the technology. Gaps in clinical pharmacy coverage during the night shift may have also contributed to delays in antibiotic optimization, particularly if other clinicians are not equipped with the knowledge or training to appropriately deescalate antibiotics based on microorganisms identified. A 2017 study by Donner et al concluded that physician interpretation of BCID results is suboptimal and should be augmented with clinical decision support tools as new technology becomes available.8 Despite the statistically insignificant results of this study, it did highlight potential areas of improvement which can lead to improved patient care.
Previous research has evaluated the impact of rapid BCID technology on antibiotic treatment and clinical outcomes. Chiasson et al found that median time to optimal therapy was 73.8 hours in the pre-BCID arm compared to 34.7 hours in the post- BCID arm (P ≤ .001), emphasizing the importance of combining rapid BCID with clinical decision support tools and pharmacy input.6 Senok et al found that BCID2 implementation led to a significant decrease in median time to culture result, which informed optimal antibiotic therapy and decreased 30-day mortality in the intensive care setting.9 In contrast, the current study did not stratify patients according to medical ward or illness severity even though clinicians may be less likely to de-escalate antibiotic therapy in critically ill patients.
Bae et al reported findings consistent with the current study and concluded that BCID did not affect the clinical outcomes of overall BSIs; however, it contributed to early administration of effective antibiotics in cases of BSIs caused by multidrug-resistant organisms.10 Results of this study were not stratified according to multidrug-resistant organisms because the sample size was too small. The current study also included patients with polymicrobial infections, which may have impacted the results due to a less streamlined approach to antibiotic optimization.
Limitations
This single-center, retrospective study had a small sample size, short time frame, and lacked patient diversity, and therefore may not be generalizable to other health care systems. The sample size was limited by shorter date range and smaller patient list between BioFire BCID2 implementation and data collection, which was used to determine the number of charts selected in each group. Some patients received antibiotics prior to blood cultures being drawn, which may falsely decrease time to optimal/ appropriate antibiotics and falsely increase time on broad spectrum/any antibiotics due to early antibiotic administration. The total number of patients on broad-spectrum antibiotics differed from the total number of patients for other outcomes because several patients never received the defined broad spectrum antibiotics.
Conclusions
When combined with a pre-existing ASP without active notification, the implementation of BioFire BCID2 did not return statistically significant data showing a decrease in time to optimal antibiotics, time to appropriate antibiotics, or time on broad-spectrum antibiotics at VHI. To make this program more successful, pharmacist intervention and clinical decision support tools may be needed.
Additional research is required to determine the optimal integration of antimicrobial stewardship, rapid diagnostic technology, and pharmacy services for maximum benefit. Even though the primary outcome was not statistically significant, the results may be clinically significant from a stewardship perspective. Realigning microbiology workflows to mimic other research, which emphasizes the importance of funneling rapid BCID results through the ASP, may improve outcomes. Future studies may be warranted following the implementation of clinical decision support tools to assess their impact on stewardship practices and patient outcomes.
About 530,000 to 628,000 episodes of bloodstream infections (BSI) occur annually in the US.1 Early identification and treatment of bacteremia are essential to improve patient outcomes because it allows for more timely targeted antibiotic therapy.2 Organism identification and susceptibility testing can take 2 to 5 days, prolonging the use of broad-spectrum empiric antibiotics and increasing the risk of adverse events.3,4 The Infectious Disease Society of America recommends the use of rapid diagnostic testing and antimicrobial stewardship programs (ASPs) to improve rates of antibiotic susceptibilities to targeted antibiotics and optimize resource utilization.3 Rapid blood culture identification (BCID) technologies reduce the duration of empiric antibiotics in patients with contaminated blood cultures, resulting in shorter hospital stays and saving money per each patient tested.4
In March 2023, Veteran Health Indiana (VHI) implemented the BioFire FilmArray Blood Culture Identification (BCID2), a BSI panel test that identifies select gram-negative bacteria, gram-positive bacteria, yeast, and antimicrobial resistance genes with an aggregate sensitivity of 99% and a specificity of 99.8%. The BCID2 presents clinically relevant information faster than traditional culture methods, allowing clinicians to make more efficient and educated antibiotic regimen decisions than with previous methods.5
It takes 24 to 48 hours from blood collection for culture incubation, positivity, and gram staining to occur at VHI. If the gram stain is positive, the blood culture is placed on the BioFire BCID2 in addition to traditional culture medium. BioFire BCID2 results are ready in 45 to 60 minutes. Results are uploaded into the electronic health record (EHR) ≤ 2 hours after they are obtained and the primary team is notified if the test is positive for certain critical results. Susceptibility testing of an identified organism typically requires an additional 24 to 48 hours for finalization. VHI Infectious Disease created an evidence-based antibiotic recommendation chart for certain medication(s) and alternate therapies based on the reported organism and its interpreted presence of resistance markers (eg, ceftriaxone for Escherichia coli when extended-spectrum beta lactamases are not detected vs meropenem if extended-spectrum beta lactamases marker are present). These charts optimize the antibiotic regimen while awaiting susceptibility finalizations.
Two previous studies describe the impact of rapid diagnostic testing technology at US Department of Veterans Affairs (VA) medical centers.6,7 In Texas, the ASP reviewed BCID panel results via clinical decision support software for about 1 hour per day.6 A Los Angeles study analyzed the impact of Biofire BCID with an interpretation guide centered on unnecessary vancomycin use and determined that shorter duration of the medication may have been the result of more frequent infectious disease consultation.7
This study assessed the time to optimal antibiotic de-escalation before and after the implementation of BioFire BCID2 with results reviewed by the ASP without active notification or assistance of any clinical decision support technology. The primary objective was to evaluate difference in time to optimal antibiotics from blood culture draw pre- vs postintervention. Secondary objectives included differences in time to organism identification, difference in time on broad-spectrum antibiotics, and difference in time to appropriate antibiotics.
Methods
This quasi-experimental retrospective chart review assessed the impact of BioFire BCID2 use on timely antibiotic de-escalation for patients who experienced a BSI at VHI between March 1, 2022, and October 1, 2023. Microbiology laboratory records identified eligible patients with positive blood cultures within the study time frame. Data were collected from the VHI EHR.
Patients were included if they had a positive bacterial blood culture and received ≥ 1 antibiotic indicated for bacteremia while receiving inpatient care. Patients were excluded if they died prior to blood culture results, transferred out of VHI, left against medical advice, or had untreated contaminants in blood culture results (ie, never received antibiotics aimed at the contaminated culture).
Patient lists were generated for before and after implementation of BioFire BCID2 (pre- and postintervention) using the VHI EHR and microbiology laboratory record system. The pre- and postinterventions groups were different sizes. As a result, a random sampling of the preintervention group was selected and included patients from March 1, 2022, through March 26, 2023. The postintervention group was smaller due to time constraints between initiation of BioFire BCID2 for data collection and included all patients from March 27, 2023, through October 1, 2023.
Optimal antibiotics were defined as escalation from inappropriate therapy to broader agent(s), de-escalation from broad-spectrum therapy to targeted agent(s), discontinuation of therapy due to an organism being identified as a contaminant, or optimization of a regimen to the preferred antimicrobial agent based on evidence-based consensus guidelines. Broad-spectrum antibiotics included: piperacillin/tazobactam, cefepime, ceftazidime, ceftazidime-avibactam, cefiderocol, carbapenems, fluroquinolones, vancomycin, daptomycin, ceftaroline, linezolid, or aztreonam. Appropriate antibiotics were defined as those with activity toward the final identified organism(s).
Deidentified participant data were entered into Microsoft Excel and kept on a secure VA server to complete statistical analyses. Parametric continuous data, such as age, were analyzed using the t-test, while nonparametric continuous data, such as time to optimal antibiotics, were analyzed using the Mann-Whitney U test. Categorical data, like sex and race, were analyzed using either Fisher exact test for small sample sizes or X2 test for a larger sample size. Statistical significance levels was defined as P < .05.
Results
Using patient lists drawn from the EHR and the microbiology laboratory records, 110 electronic charts were randomly selected for review. Fifteen patients were excluded: 8 had untreated contaminants, 4 died, and 3 were transferred out of VHI. Of the 95 patients included, 48 were in the preintervention group and 47 were in the postintervention group (Figure 1).
Baseline characteristics were similar between the 2 groups (Table 1). Most patients were White males aged > 70 years in the EHR. The urinary tract was the most common source of infection, impacting 12 patients in each group (Figure 2). Escherichia coli, Klebsiella, Staphylococcus, and Streptococcus were the most common bloodstream isolates identified.
The median time to optimal antibiotics in the preintervention group was 58.5 hours vs 43.4 hours in the postintervention group (P = .11). The median time to organism identification was 37.8 hours in the preintervention group vs 16.9 hours in the postintervention group (P < .001). The median time on broad-spectrum antibiotics was 45.2 hours in the preintervention group vs 46.6 hours in the postintervention group (P = .99). The median time on appropriate antibiotics in the preintervention group was 2.3 hours vs 1.9 hours in the postintervention group (P = .79). Differences in other measured outcomes between the groups were not statistically significant (Table 2).
Although implementation of rapid diagnostic technology reduced the median time to optimal antibiotics, the results were not statistically significant. Shorter time to organism identification in the postintervention group compared to the preintervention group was the lone statistically significant metric (P < .001).
Discussion
A lack of statistical significance in the primary outcome may have been due to nonadherence to facility de-escalation protocols or a suboptimal BioFire BCID2 result notification system. Additionally, use of rapid BCID at VHI may improve over time as clinicians become more familiar with the technology. Gaps in clinical pharmacy coverage during the night shift may have also contributed to delays in antibiotic optimization, particularly if other clinicians are not equipped with the knowledge or training to appropriately deescalate antibiotics based on microorganisms identified. A 2017 study by Donner et al concluded that physician interpretation of BCID results is suboptimal and should be augmented with clinical decision support tools as new technology becomes available.8 Despite the statistically insignificant results of this study, it did highlight potential areas of improvement which can lead to improved patient care.
Previous research has evaluated the impact of rapid BCID technology on antibiotic treatment and clinical outcomes. Chiasson et al found that median time to optimal therapy was 73.8 hours in the pre-BCID arm compared to 34.7 hours in the post- BCID arm (P ≤ .001), emphasizing the importance of combining rapid BCID with clinical decision support tools and pharmacy input.6 Senok et al found that BCID2 implementation led to a significant decrease in median time to culture result, which informed optimal antibiotic therapy and decreased 30-day mortality in the intensive care setting.9 In contrast, the current study did not stratify patients according to medical ward or illness severity even though clinicians may be less likely to de-escalate antibiotic therapy in critically ill patients.
Bae et al reported findings consistent with the current study and concluded that BCID did not affect the clinical outcomes of overall BSIs; however, it contributed to early administration of effective antibiotics in cases of BSIs caused by multidrug-resistant organisms.10 Results of this study were not stratified according to multidrug-resistant organisms because the sample size was too small. The current study also included patients with polymicrobial infections, which may have impacted the results due to a less streamlined approach to antibiotic optimization.
Limitations
This single-center, retrospective study had a small sample size, short time frame, and lacked patient diversity, and therefore may not be generalizable to other health care systems. The sample size was limited by shorter date range and smaller patient list between BioFire BCID2 implementation and data collection, which was used to determine the number of charts selected in each group. Some patients received antibiotics prior to blood cultures being drawn, which may falsely decrease time to optimal/ appropriate antibiotics and falsely increase time on broad spectrum/any antibiotics due to early antibiotic administration. The total number of patients on broad-spectrum antibiotics differed from the total number of patients for other outcomes because several patients never received the defined broad spectrum antibiotics.
Conclusions
When combined with a pre-existing ASP without active notification, the implementation of BioFire BCID2 did not return statistically significant data showing a decrease in time to optimal antibiotics, time to appropriate antibiotics, or time on broad-spectrum antibiotics at VHI. To make this program more successful, pharmacist intervention and clinical decision support tools may be needed.
Additional research is required to determine the optimal integration of antimicrobial stewardship, rapid diagnostic technology, and pharmacy services for maximum benefit. Even though the primary outcome was not statistically significant, the results may be clinically significant from a stewardship perspective. Realigning microbiology workflows to mimic other research, which emphasizes the importance of funneling rapid BCID results through the ASP, may improve outcomes. Future studies may be warranted following the implementation of clinical decision support tools to assess their impact on stewardship practices and patient outcomes.
- Goto M, Al-Hasan MN. Overall burden of bloodstream infection and nosocomial bloodstream infection in North America and Europe. Clin Microbiol Infect. 2013;19(6):501- 509. doi:10.1111/1469-0691.12195
- Pardo J, Klinker KP, Borgert SJ, Butler BM, Giglio PG, Rand KH. Clinical and economic impact of antimicrobial stewardship interventions with the FilmArray blood culture identification panel. Diagn Microbiol Infect Dis. 2016;84(2):159-164. doi:10.1016/j.diagmicrobio.2015.10.023.
- Barlam TF, Cosgrove SE, Abbo LM, et al. Implementing an antibiotic stewardship program: guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America. Clin Infect Dis. 2016;62(10):e51-e77. doi:10.1093/cid/ciw118
- BIOFIRE® Blood Culture Identification 2 (BCID2) Panel. Biomerierux. Updated 2025. Accessed May 10, 2025. https://www.biofiredx.com/products/the-filmarray-panels/filmarraybcid/
- Huang AM, Newton D, Kunapuli A, et al. Impact of rapid organism identification via matrix-assisted laser desorption/ionization time-of-flight combined with antimicrobial stewardship team intervention in adult patients with bacteremia and candidemia. Clin Infect Dis. 2013;57(9):1237-1245. doi:10.1093/cid/cit498
- Chiasson JM, Smith WJ, Jodlowski TZ, Kouma MA, Cutrell JB. Impact of a rapid blood culture diagnostic panel on time to optimal antimicrobial therapy at a veterans affairs medical center. J Pharm Pract. 2022;35(5):722-729. doi:10.1177/08971900211000686
- Wu S, Watson RL, Graber CJ. 2007. Impact of combining rapid diagnostics with an interpretation guide on vancomycin usage for contaminant blood cultures growing coagulase- negative staphylococci (CoNS). Open Forum Infect Dis. 2019;6(Suppl 2):S674. doi:10.1093/ofid/ofz360.1687
- Donner LM, Campbell WS, Lyden E, Van Schooneveld TC. Assessment of rapid-blood-culture-identification result interpretation and antibiotic prescribing practices. J Clin Microbiol. 2017;55(5):1496-1507. doi:10.1128/JCM.02395-16
- Senok A, Dabal LA, Alfaresi M, et al. Clinical impact of the BIOFIRE blood culture identification 2 panel in adult patients with bloodstream infection: a multicentre observational study in the United Arab Emirates. Diagnostics (Basel). 2023;13(14):2433. doi:10.3390/diagnostics13142433
- Bae JY, Bae J, So MK, Choi HJ, Lee M. The impact of the rapid blood culture identification panel on antibiotic treatment and clinical outcomes in bloodstream infections, particularly those associated with multidrug-resistant micro-organisms. Diagnostics (Basel). 2023;13(23):3504. doi:10.3390/diagnostics13233504
- Goto M, Al-Hasan MN. Overall burden of bloodstream infection and nosocomial bloodstream infection in North America and Europe. Clin Microbiol Infect. 2013;19(6):501- 509. doi:10.1111/1469-0691.12195
- Pardo J, Klinker KP, Borgert SJ, Butler BM, Giglio PG, Rand KH. Clinical and economic impact of antimicrobial stewardship interventions with the FilmArray blood culture identification panel. Diagn Microbiol Infect Dis. 2016;84(2):159-164. doi:10.1016/j.diagmicrobio.2015.10.023.
- Barlam TF, Cosgrove SE, Abbo LM, et al. Implementing an antibiotic stewardship program: guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America. Clin Infect Dis. 2016;62(10):e51-e77. doi:10.1093/cid/ciw118
- BIOFIRE® Blood Culture Identification 2 (BCID2) Panel. Biomerierux. Updated 2025. Accessed May 10, 2025. https://www.biofiredx.com/products/the-filmarray-panels/filmarraybcid/
- Huang AM, Newton D, Kunapuli A, et al. Impact of rapid organism identification via matrix-assisted laser desorption/ionization time-of-flight combined with antimicrobial stewardship team intervention in adult patients with bacteremia and candidemia. Clin Infect Dis. 2013;57(9):1237-1245. doi:10.1093/cid/cit498
- Chiasson JM, Smith WJ, Jodlowski TZ, Kouma MA, Cutrell JB. Impact of a rapid blood culture diagnostic panel on time to optimal antimicrobial therapy at a veterans affairs medical center. J Pharm Pract. 2022;35(5):722-729. doi:10.1177/08971900211000686
- Wu S, Watson RL, Graber CJ. 2007. Impact of combining rapid diagnostics with an interpretation guide on vancomycin usage for contaminant blood cultures growing coagulase- negative staphylococci (CoNS). Open Forum Infect Dis. 2019;6(Suppl 2):S674. doi:10.1093/ofid/ofz360.1687
- Donner LM, Campbell WS, Lyden E, Van Schooneveld TC. Assessment of rapid-blood-culture-identification result interpretation and antibiotic prescribing practices. J Clin Microbiol. 2017;55(5):1496-1507. doi:10.1128/JCM.02395-16
- Senok A, Dabal LA, Alfaresi M, et al. Clinical impact of the BIOFIRE blood culture identification 2 panel in adult patients with bloodstream infection: a multicentre observational study in the United Arab Emirates. Diagnostics (Basel). 2023;13(14):2433. doi:10.3390/diagnostics13142433
- Bae JY, Bae J, So MK, Choi HJ, Lee M. The impact of the rapid blood culture identification panel on antibiotic treatment and clinical outcomes in bloodstream infections, particularly those associated with multidrug-resistant micro-organisms. Diagnostics (Basel). 2023;13(23):3504. doi:10.3390/diagnostics13233504
Impact of Rapid Blood Culture Identification on Antibiotic De-escalation at a Veterans Affairs Medical Center
Impact of Rapid Blood Culture Identification on Antibiotic De-escalation at a Veterans Affairs Medical Center

Disparate Prednisone Starting Dosages for Systemic Corticosteroid-Naïve Veterans With Active Sarcoidosis
Disparate Prednisone Starting Dosages for Systemic Corticosteroid-Naïve Veterans With Active Sarcoidosis
Sarcoidosis is a multiorgan granulomatous disorder of unknown etiology that impacts many US veterans.1 At diagnosis, clinical manifestations vary and partially depend on the extent and severity of organ involvement, particularly of the lungs, heart, and eyes.2,3 Sarcoidosis may lead to progressive organ dysfunction, long-term disability, and death.1-3 Clinical practice guidelines recommend prednisone 20 to 40 mg daily or equivalent-prednisone dose followed by a slow tapering, as first-line pharmacotherapy for patients with active sarcoidosis who are naïve to systemic corticosteroids.2-4
Use of prolonged, high-dosage prednisone (> 40 mg daily) is discouraged due to a high risk of corticosteroid-related adverse events and associated health care costs.5,6 Research suggests that initial lower prednisone dosage (< 20 mg daily) may be effective in systemic corticosteroid-naïve patients with active sarcoidosis.3
Adherence to this regimen by specialists (eg, pulmonologists, dermatologists, ophthalmologists, rheumatologists, and cardiologists) has not been established. This study sought to determine the starting dosages for prednisone prescribed at the Jesse Brown Department of Veterans Affairs Medical Center (JBVAMC) to patients with active sarcoidosis who were systemic corticosteroid-naïve.
Methods
Patient data were reviewed from the Computerized Patient Record System (CPRS) for individuals diagnosed with sarcoidosis who were corticosteroid-naïve and prescribed initial prednisone dosages by health care practitioners (HCPs) from several specialties between 2014 and 2023 at JBVAMC. This 200-bed acute care facility serves about 62,000 veterans who live in Illinois or Indiana. JBVAMC is affiliated with the University of Illinois College of Medicine at Chicago, Northwestern University Feinberg School of Medicine, and the University of Chicago Pritzker School of Medicine; many JBVAMC HCPs hold academic appointments with these medical schools.
Patient demographics, prescriber specialty, and daily starting dosage were recorded. The decision to initiate prednisone therapy and its dosage were at the discretion of HCPs who diagnosed active sarcoidosis based on compatible clinical and ancillary test findings as documented in CPRS.2-4,6-10 Statistical analyses were conducted using a t test, and a threshold of P < .05 was considered statistically significant. This study was reviewed and determined to be exempt by the JBVAMC institutional review board.
Results
Sixty-eight patients who were systemic corticosteroid- naïve and had sarcoidosis were prescribed prednisone by HCPs at JBVAMC. Fifty-two were Black (76%), 62 were male (91%), and 53 were current or former smokers (78%). The mean (SD) age was 63 (11) years (Table 1). Forty patients (59%) had lung involvement, 6 had eye (9%), 6 had skin (9%), and 5 had musculoskeletal system (7%) involvement.
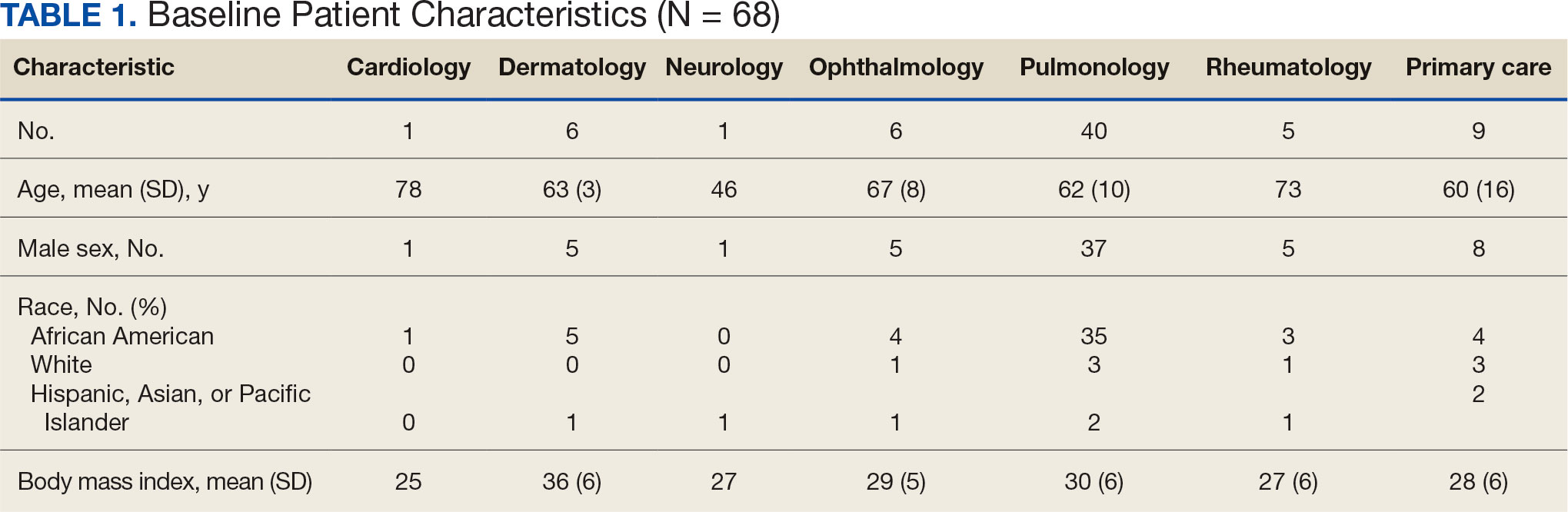
Pulmonologists predominantly prescribed initial dosage of 20 mg to 40 mg (median, 35 mg daily) (Figure). Other HCPs, including primary care, tended to prescribe prednisone < 20 mg (median, 17.5 mg; P < .05) (Table 2). The highest initial prednisone dosage was 80 mg daily, prescribed by a neurologist for a patient with neurosarcoidosis. Voortman et al recommend 20 to 40 mg prednisone daily for neurosarcoidosis.7 Both groups, pulmonologists and nonpulmonologists, had no significant differences in patient characteristics.
Discussion
Disparate prescription patterns of initial prednisone dosages were observed between pulmonologists and nonpulmonologists treating systemic corticosteroid-naïve patients with active sarcoidosis at JBVAMC. This study did not determine the underlying reasons for this phenomenon, nor its impact on patient outcomes.
Clinical practice guidelines have not been independently validated for each organ affected by sarcoidosis.2-4,6-10 Variations in clinical practice for other specialties may account for the variable prednisone starting dosage selection. For example, among 6 patients with active ocular sarcoidosis treated by ophthalmologists, 4 were prescribed an initial prednisone dosage of ≥ 10 mg daily. The American Academy of Ophthalmology recommends an initial short-term course of prednisone at 1 to 1.5 mg/kg daily, tapered down to the lowest effective dosage.10
Limitations
This study used a small, single-center predominantly older Black male patient cohort. The generalizability of these observations is unknown. A larger, multicenter prospective study is warranted to further evaluate these initial observations.
Conclusions
HCPs treating patients who are systemic corticosteroid-naïve with active sarcoidosis for whom prednisone is indicated should adhere to current clinical practice guidelines by prescribing prednisone in the 20 to 40 mg daily range.
- Seedahmed MI, Baugh AD, Albirair MT, et al. Epidemiology of sarcoidosis in U.S. veterans from 2003 to 2019. Ann Am Thorac Soc. 2023;20(6):797-806. doi:10.1513/AnnalsATS.202206-515OC
- Baughman RP, Valeyre D, Korsten P, et al. ERS clinical practice guidelines on treatment of sarcoidosis. Eur Respir J. 2021;58(6):2004079. doi:10.1183/13993003.04079-2020
- Rahaghi FF, Baughman RP, Saketkoo LA, et al. Delphi consensus recommendations for a treatment algorithm in pulmonary sarcoidosis. Eur Respir Rev. 2020;29(155):190146. doi:10.1183/16000617.0146-2019
- Kwon S, Judson MA. Clinical pharmacology in sarcoidosis: how to use and monitor sarcoidosis medications. J Clin Med. 2024;13(5):1250. doi:10.3390/jcm13051250
- Rice JB, White AG, Johnson M, et al. Quantitative characterization of the relationship between levels of extended corticosteroid use and related adverse events in a US population. Curr Med Res Opin. 2018;34(8):1519-1527. doi:10.1080/03007995.2018.1474090
- Rice JB, White AG, Johnson M, Wagh A, Qin Y, Bartels-Peculis L, et al. Healthcare resource use and cost associated with varying dosages of extended corticosteroid exposure in a US population. J Med Econ. 2018;21(9):846-852. doi:10.1080/13696998.2018.1474750
- Voortman M, Drent M, Baughman RP. Management of neurosarcoidosis: a clinical challenge. Curr Opin Neurol. 2019;32(3):475-483. doi:10.1097/WCO.0000000000000684
- Cheng RK, Kittleson MM, Beavers CJ, et al. Diagnosis and management of cardiac sarcoidosis: a scientific statement from the American Heart Association. Circulation. 2024;149(21):e1197-e1216. doi:10.1161/CIR.0000000000001240
- Cohen E, Lheure C, Ingen-Housz-Oro S, et al. Which firstline treatment for cutaneous sarcoidosis? A retrospective study of 120 patients. Eur J Dermatol. 2023;33(6):680-685. doi:10.1684/ejd.2023.4584
- American Academy of Ophthalmology. Ocular manifestations of sarcoidosis. EyeWiki. Accessed June 3, 2025. https://eyewiki.org/Ocular_Manifestations_of_Sarcoidosis
Sarcoidosis is a multiorgan granulomatous disorder of unknown etiology that impacts many US veterans.1 At diagnosis, clinical manifestations vary and partially depend on the extent and severity of organ involvement, particularly of the lungs, heart, and eyes.2,3 Sarcoidosis may lead to progressive organ dysfunction, long-term disability, and death.1-3 Clinical practice guidelines recommend prednisone 20 to 40 mg daily or equivalent-prednisone dose followed by a slow tapering, as first-line pharmacotherapy for patients with active sarcoidosis who are naïve to systemic corticosteroids.2-4
Use of prolonged, high-dosage prednisone (> 40 mg daily) is discouraged due to a high risk of corticosteroid-related adverse events and associated health care costs.5,6 Research suggests that initial lower prednisone dosage (< 20 mg daily) may be effective in systemic corticosteroid-naïve patients with active sarcoidosis.3
Adherence to this regimen by specialists (eg, pulmonologists, dermatologists, ophthalmologists, rheumatologists, and cardiologists) has not been established. This study sought to determine the starting dosages for prednisone prescribed at the Jesse Brown Department of Veterans Affairs Medical Center (JBVAMC) to patients with active sarcoidosis who were systemic corticosteroid-naïve.
Methods
Patient data were reviewed from the Computerized Patient Record System (CPRS) for individuals diagnosed with sarcoidosis who were corticosteroid-naïve and prescribed initial prednisone dosages by health care practitioners (HCPs) from several specialties between 2014 and 2023 at JBVAMC. This 200-bed acute care facility serves about 62,000 veterans who live in Illinois or Indiana. JBVAMC is affiliated with the University of Illinois College of Medicine at Chicago, Northwestern University Feinberg School of Medicine, and the University of Chicago Pritzker School of Medicine; many JBVAMC HCPs hold academic appointments with these medical schools.
Patient demographics, prescriber specialty, and daily starting dosage were recorded. The decision to initiate prednisone therapy and its dosage were at the discretion of HCPs who diagnosed active sarcoidosis based on compatible clinical and ancillary test findings as documented in CPRS.2-4,6-10 Statistical analyses were conducted using a t test, and a threshold of P < .05 was considered statistically significant. This study was reviewed and determined to be exempt by the JBVAMC institutional review board.
Results
Sixty-eight patients who were systemic corticosteroid- naïve and had sarcoidosis were prescribed prednisone by HCPs at JBVAMC. Fifty-two were Black (76%), 62 were male (91%), and 53 were current or former smokers (78%). The mean (SD) age was 63 (11) years (Table 1). Forty patients (59%) had lung involvement, 6 had eye (9%), 6 had skin (9%), and 5 had musculoskeletal system (7%) involvement.
Pulmonologists predominantly prescribed initial dosage of 20 mg to 40 mg (median, 35 mg daily) (Figure). Other HCPs, including primary care, tended to prescribe prednisone < 20 mg (median, 17.5 mg; P < .05) (Table 2). The highest initial prednisone dosage was 80 mg daily, prescribed by a neurologist for a patient with neurosarcoidosis. Voortman et al recommend 20 to 40 mg prednisone daily for neurosarcoidosis.7 Both groups, pulmonologists and nonpulmonologists, had no significant differences in patient characteristics.
Discussion
Disparate prescription patterns of initial prednisone dosages were observed between pulmonologists and nonpulmonologists treating systemic corticosteroid-naïve patients with active sarcoidosis at JBVAMC. This study did not determine the underlying reasons for this phenomenon, nor its impact on patient outcomes.
Clinical practice guidelines have not been independently validated for each organ affected by sarcoidosis.2-4,6-10 Variations in clinical practice for other specialties may account for the variable prednisone starting dosage selection. For example, among 6 patients with active ocular sarcoidosis treated by ophthalmologists, 4 were prescribed an initial prednisone dosage of ≥ 10 mg daily. The American Academy of Ophthalmology recommends an initial short-term course of prednisone at 1 to 1.5 mg/kg daily, tapered down to the lowest effective dosage.10
Limitations
This study used a small, single-center predominantly older Black male patient cohort. The generalizability of these observations is unknown. A larger, multicenter prospective study is warranted to further evaluate these initial observations.
Conclusions
HCPs treating patients who are systemic corticosteroid-naïve with active sarcoidosis for whom prednisone is indicated should adhere to current clinical practice guidelines by prescribing prednisone in the 20 to 40 mg daily range.
Sarcoidosis is a multiorgan granulomatous disorder of unknown etiology that impacts many US veterans.1 At diagnosis, clinical manifestations vary and partially depend on the extent and severity of organ involvement, particularly of the lungs, heart, and eyes.2,3 Sarcoidosis may lead to progressive organ dysfunction, long-term disability, and death.1-3 Clinical practice guidelines recommend prednisone 20 to 40 mg daily or equivalent-prednisone dose followed by a slow tapering, as first-line pharmacotherapy for patients with active sarcoidosis who are naïve to systemic corticosteroids.2-4
Use of prolonged, high-dosage prednisone (> 40 mg daily) is discouraged due to a high risk of corticosteroid-related adverse events and associated health care costs.5,6 Research suggests that initial lower prednisone dosage (< 20 mg daily) may be effective in systemic corticosteroid-naïve patients with active sarcoidosis.3
Adherence to this regimen by specialists (eg, pulmonologists, dermatologists, ophthalmologists, rheumatologists, and cardiologists) has not been established. This study sought to determine the starting dosages for prednisone prescribed at the Jesse Brown Department of Veterans Affairs Medical Center (JBVAMC) to patients with active sarcoidosis who were systemic corticosteroid-naïve.
Methods
Patient data were reviewed from the Computerized Patient Record System (CPRS) for individuals diagnosed with sarcoidosis who were corticosteroid-naïve and prescribed initial prednisone dosages by health care practitioners (HCPs) from several specialties between 2014 and 2023 at JBVAMC. This 200-bed acute care facility serves about 62,000 veterans who live in Illinois or Indiana. JBVAMC is affiliated with the University of Illinois College of Medicine at Chicago, Northwestern University Feinberg School of Medicine, and the University of Chicago Pritzker School of Medicine; many JBVAMC HCPs hold academic appointments with these medical schools.
Patient demographics, prescriber specialty, and daily starting dosage were recorded. The decision to initiate prednisone therapy and its dosage were at the discretion of HCPs who diagnosed active sarcoidosis based on compatible clinical and ancillary test findings as documented in CPRS.2-4,6-10 Statistical analyses were conducted using a t test, and a threshold of P < .05 was considered statistically significant. This study was reviewed and determined to be exempt by the JBVAMC institutional review board.
Results
Sixty-eight patients who were systemic corticosteroid- naïve and had sarcoidosis were prescribed prednisone by HCPs at JBVAMC. Fifty-two were Black (76%), 62 were male (91%), and 53 were current or former smokers (78%). The mean (SD) age was 63 (11) years (Table 1). Forty patients (59%) had lung involvement, 6 had eye (9%), 6 had skin (9%), and 5 had musculoskeletal system (7%) involvement.
Pulmonologists predominantly prescribed initial dosage of 20 mg to 40 mg (median, 35 mg daily) (Figure). Other HCPs, including primary care, tended to prescribe prednisone < 20 mg (median, 17.5 mg; P < .05) (Table 2). The highest initial prednisone dosage was 80 mg daily, prescribed by a neurologist for a patient with neurosarcoidosis. Voortman et al recommend 20 to 40 mg prednisone daily for neurosarcoidosis.7 Both groups, pulmonologists and nonpulmonologists, had no significant differences in patient characteristics.
Discussion
Disparate prescription patterns of initial prednisone dosages were observed between pulmonologists and nonpulmonologists treating systemic corticosteroid-naïve patients with active sarcoidosis at JBVAMC. This study did not determine the underlying reasons for this phenomenon, nor its impact on patient outcomes.
Clinical practice guidelines have not been independently validated for each organ affected by sarcoidosis.2-4,6-10 Variations in clinical practice for other specialties may account for the variable prednisone starting dosage selection. For example, among 6 patients with active ocular sarcoidosis treated by ophthalmologists, 4 were prescribed an initial prednisone dosage of ≥ 10 mg daily. The American Academy of Ophthalmology recommends an initial short-term course of prednisone at 1 to 1.5 mg/kg daily, tapered down to the lowest effective dosage.10
Limitations
This study used a small, single-center predominantly older Black male patient cohort. The generalizability of these observations is unknown. A larger, multicenter prospective study is warranted to further evaluate these initial observations.
Conclusions
HCPs treating patients who are systemic corticosteroid-naïve with active sarcoidosis for whom prednisone is indicated should adhere to current clinical practice guidelines by prescribing prednisone in the 20 to 40 mg daily range.
- Seedahmed MI, Baugh AD, Albirair MT, et al. Epidemiology of sarcoidosis in U.S. veterans from 2003 to 2019. Ann Am Thorac Soc. 2023;20(6):797-806. doi:10.1513/AnnalsATS.202206-515OC
- Baughman RP, Valeyre D, Korsten P, et al. ERS clinical practice guidelines on treatment of sarcoidosis. Eur Respir J. 2021;58(6):2004079. doi:10.1183/13993003.04079-2020
- Rahaghi FF, Baughman RP, Saketkoo LA, et al. Delphi consensus recommendations for a treatment algorithm in pulmonary sarcoidosis. Eur Respir Rev. 2020;29(155):190146. doi:10.1183/16000617.0146-2019
- Kwon S, Judson MA. Clinical pharmacology in sarcoidosis: how to use and monitor sarcoidosis medications. J Clin Med. 2024;13(5):1250. doi:10.3390/jcm13051250
- Rice JB, White AG, Johnson M, et al. Quantitative characterization of the relationship between levels of extended corticosteroid use and related adverse events in a US population. Curr Med Res Opin. 2018;34(8):1519-1527. doi:10.1080/03007995.2018.1474090
- Rice JB, White AG, Johnson M, Wagh A, Qin Y, Bartels-Peculis L, et al. Healthcare resource use and cost associated with varying dosages of extended corticosteroid exposure in a US population. J Med Econ. 2018;21(9):846-852. doi:10.1080/13696998.2018.1474750
- Voortman M, Drent M, Baughman RP. Management of neurosarcoidosis: a clinical challenge. Curr Opin Neurol. 2019;32(3):475-483. doi:10.1097/WCO.0000000000000684
- Cheng RK, Kittleson MM, Beavers CJ, et al. Diagnosis and management of cardiac sarcoidosis: a scientific statement from the American Heart Association. Circulation. 2024;149(21):e1197-e1216. doi:10.1161/CIR.0000000000001240
- Cohen E, Lheure C, Ingen-Housz-Oro S, et al. Which firstline treatment for cutaneous sarcoidosis? A retrospective study of 120 patients. Eur J Dermatol. 2023;33(6):680-685. doi:10.1684/ejd.2023.4584
- American Academy of Ophthalmology. Ocular manifestations of sarcoidosis. EyeWiki. Accessed June 3, 2025. https://eyewiki.org/Ocular_Manifestations_of_Sarcoidosis
- Seedahmed MI, Baugh AD, Albirair MT, et al. Epidemiology of sarcoidosis in U.S. veterans from 2003 to 2019. Ann Am Thorac Soc. 2023;20(6):797-806. doi:10.1513/AnnalsATS.202206-515OC
- Baughman RP, Valeyre D, Korsten P, et al. ERS clinical practice guidelines on treatment of sarcoidosis. Eur Respir J. 2021;58(6):2004079. doi:10.1183/13993003.04079-2020
- Rahaghi FF, Baughman RP, Saketkoo LA, et al. Delphi consensus recommendations for a treatment algorithm in pulmonary sarcoidosis. Eur Respir Rev. 2020;29(155):190146. doi:10.1183/16000617.0146-2019
- Kwon S, Judson MA. Clinical pharmacology in sarcoidosis: how to use and monitor sarcoidosis medications. J Clin Med. 2024;13(5):1250. doi:10.3390/jcm13051250
- Rice JB, White AG, Johnson M, et al. Quantitative characterization of the relationship between levels of extended corticosteroid use and related adverse events in a US population. Curr Med Res Opin. 2018;34(8):1519-1527. doi:10.1080/03007995.2018.1474090
- Rice JB, White AG, Johnson M, Wagh A, Qin Y, Bartels-Peculis L, et al. Healthcare resource use and cost associated with varying dosages of extended corticosteroid exposure in a US population. J Med Econ. 2018;21(9):846-852. doi:10.1080/13696998.2018.1474750
- Voortman M, Drent M, Baughman RP. Management of neurosarcoidosis: a clinical challenge. Curr Opin Neurol. 2019;32(3):475-483. doi:10.1097/WCO.0000000000000684
- Cheng RK, Kittleson MM, Beavers CJ, et al. Diagnosis and management of cardiac sarcoidosis: a scientific statement from the American Heart Association. Circulation. 2024;149(21):e1197-e1216. doi:10.1161/CIR.0000000000001240
- Cohen E, Lheure C, Ingen-Housz-Oro S, et al. Which firstline treatment for cutaneous sarcoidosis? A retrospective study of 120 patients. Eur J Dermatol. 2023;33(6):680-685. doi:10.1684/ejd.2023.4584
- American Academy of Ophthalmology. Ocular manifestations of sarcoidosis. EyeWiki. Accessed June 3, 2025. https://eyewiki.org/Ocular_Manifestations_of_Sarcoidosis
Disparate Prednisone Starting Dosages for Systemic Corticosteroid-Naïve Veterans With Active Sarcoidosis
Disparate Prednisone Starting Dosages for Systemic Corticosteroid-Naïve Veterans With Active Sarcoidosis
Are Oritavancin and Dalbavancin More Cost Effective for Outpatient Parenteral Antimicrobial Therapy at a Veterans Affairs Medical Center?
Are Oritavancin and Dalbavancin More Cost Effective for Outpatient Parenteral Antimicrobial Therapy at a Veterans Affairs Medical Center?
Oritavancin and dalbavancin are long acting lipoglycopeptides indicated for the treatment of acute bacterial skin and skin structure infections (ABSSSI).1,2 Largely due to their long half-lives, prolonged tissue concentrations at sites of infection, tolerability, and minimal requirement for therapeutic drug monitoring, these agents are attractive options in outpatient settings.3,4 A 1- or 2-dose treatment of oritavancin and dalbavancin may be sufficient for conditions traditionally treated with outpatient parenteral antimicrobial therapy (OPAT) via peripherally inserted central catheter (PICC).
Limited research supports the use of dalbavancin and oritavancin for bone and joint infections, infective endocarditis, and bloodstream infections (BSIs). However, the US Food and Drug Administration has approved an indication for the treatment of ABSSSI.3-9 Dosing for these off-label indications varies but typically consists of an initial intravenous (IV) dose (1000 mg, 1200 mg, or 1500 mg), with a subsequent dose 1 to 2 weeks later or administered once weekly.6-10
Due in part to the recent availability of oritavancin and dalbavancin relative to the publication of practice guidelines, their appropriate place in therapy continues to evolve based on emerging literature.11,12 One potential barrier of use for these medications is their cost. Based on the number of doses administered, the 2022 estimated total acquisition cost of therapy for oritavancin and dalbavancin was $1014 to $4397 and $3046 to $7150, respectively (eAppendix). Despite the high acquisition costs, these agents do not require the placement of an indwelling central line, can be administered in outpatient settings, and require minimal therapeutic dose monitoring compared to vancomycin.13-15 This medication use evaluation (MUE) compared the total cost of treatment with oritavancin and dalbavancin vs therapies traditionally used for OPAT or prolonged IV inpatient therapy.
METHODS
This retrospective MUE was conducted at the Boise Veterans Affairs Medical Center (BVAMC), a level 2 facility with an extensive rural catchment area. BVAMC provides many OPAT services, including medications, supplies, and dressing changes after initial clinic or inpatient education. Contracted vendors may also assist with at-home nursing care using supplies provided by the BVAMC. Cases were identified using an internal database of OPAT patients and those who received oritavancin or dalbavancin between September 1, 2017, and November 1, 2022. Patients aged ≥ 18 years who received ≥ 1 dose of oritavancin or dalbavancin for ABSSSI, osteomyelitis/joint infections, endocarditis, and BSI were included. Comparator treatments consisting of ≥ 1 week of vancomycin or daptomycin for ABSSSI, osteomyelitis/joint infections, endocarditis, and BSI were identified through review of OPAT and Infectious Diseases service consults during the same timeframe. Patients were excluded if any antibiotic was prescribed by a non- VA clinician, if medications were not provided by OPAT, or if chart review did not identify an ABSSSI, osteomyelitis/ joint infection, or BSI diagnosis.
Electronic medical record review was conducted using a standardized data collection form (eAppendix). Data collected included demographics, infectious diagnosis, treatment administered, administration procedures and related visits and treatment locations, outcomes including clinical failure, adverse events (AEs), and hospital readmission.
Clinical failure was defined as readmission or death due to worsening infection or readmission secondary to a documented potential AE to the evaluated antibiotics within 90 days after initiation. Clinical failures excluded readmissions not associated with infection including comorbidities or elective procedures. AEs included new onset renal failure (serum creatinine ≥ 0.5 mg/dL), neutropenia (neutrophils ≤ 500), thrombocytopenia (platelets < 100,000), eosinophilia (> 15% eosinophils), or creatine phosphokinase > 10 times the upper limit of normal, and Clostridioides difficile (C. difficile) infection. Line complications included thrombophlebitis, local inflammation, or infection requiring line replacement (eAppendix).
A cost-minimization approach was used to assess the total cost of treatment.16 Patients who received oritavancin or dalbavancin were matched with patients that received vancomycin and daptomycin for the same indication and about 1 month of initiation through the randomization function in Microsoft Excel. This accounted for changes in personnel, nonformulary drug approvals, cost, and changes in practice during the pandemic. Costs were calculated using a decision tree as a base model (Figure 1). In this model, each treatment dyad was assessed for the presence or absence of clinical failure, adverse event (medication and line complications), and treatment setting endpoints. Cost estimates were tabulated for each patient that received treatment using published VA data, literature, pharmacoeconomist guidance, or best faith effort based on workflow. 17-20 All cost estimates were based on 2022 figures or adjusted for inflation if obtained prior to 2022. Secondary endpoints of this analysis included estimated total cost of medication acquisition, administration supplies, laboratory monitoring, and human resources for OPAT visits or receiving home-health services.
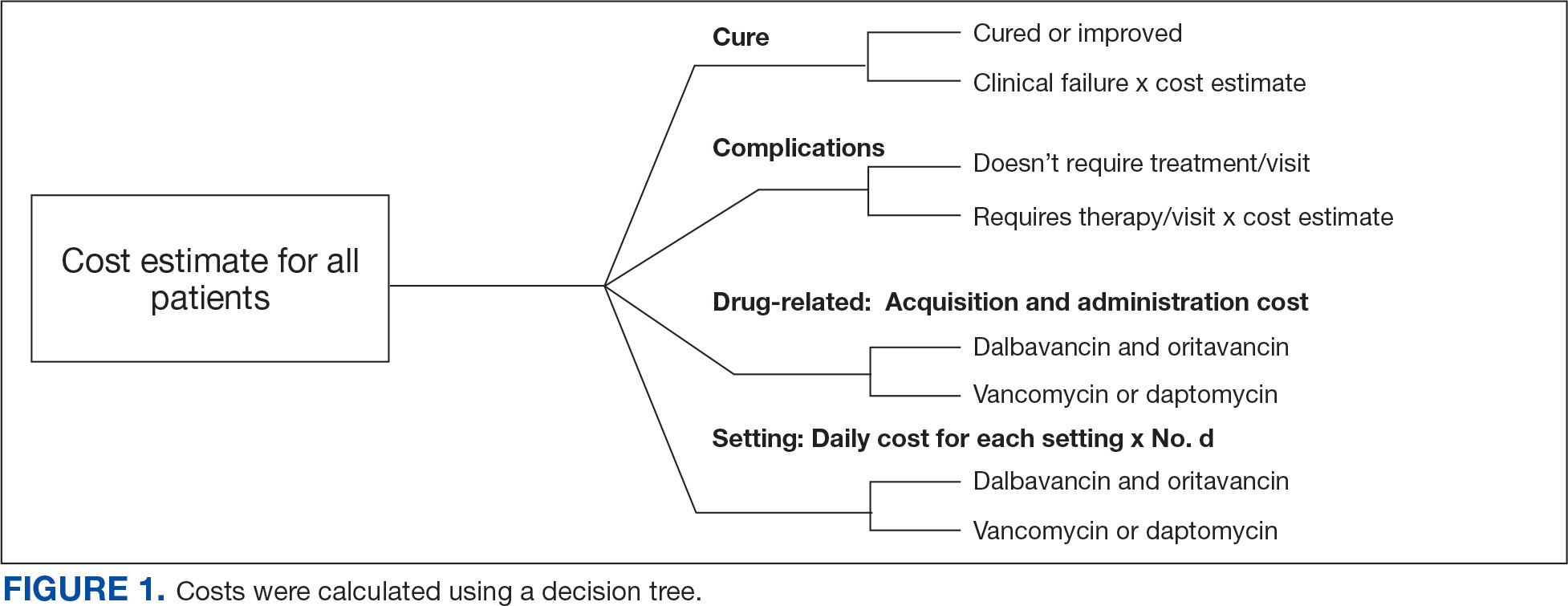
This evaluation was classified by the BVAMC Medication Use Evaluation research determination subcommittee as a quality improvement project and was considered exempt from VA Human Subjects Research requirements based on the VA Policy Handbook guideline 1058.05.
RESULTS
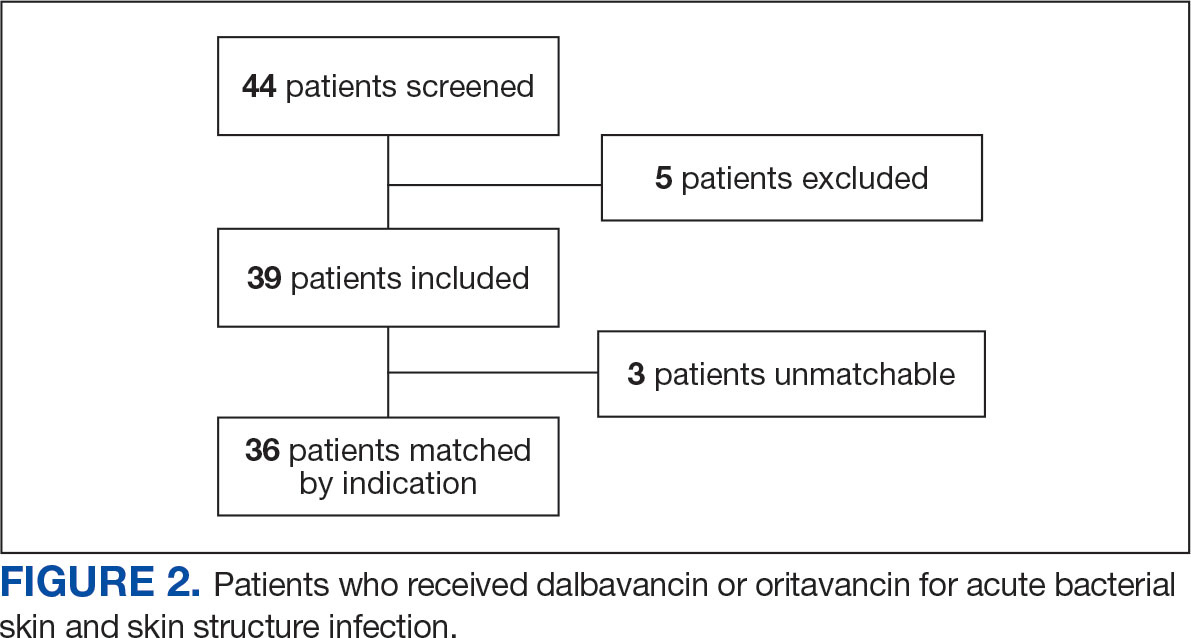
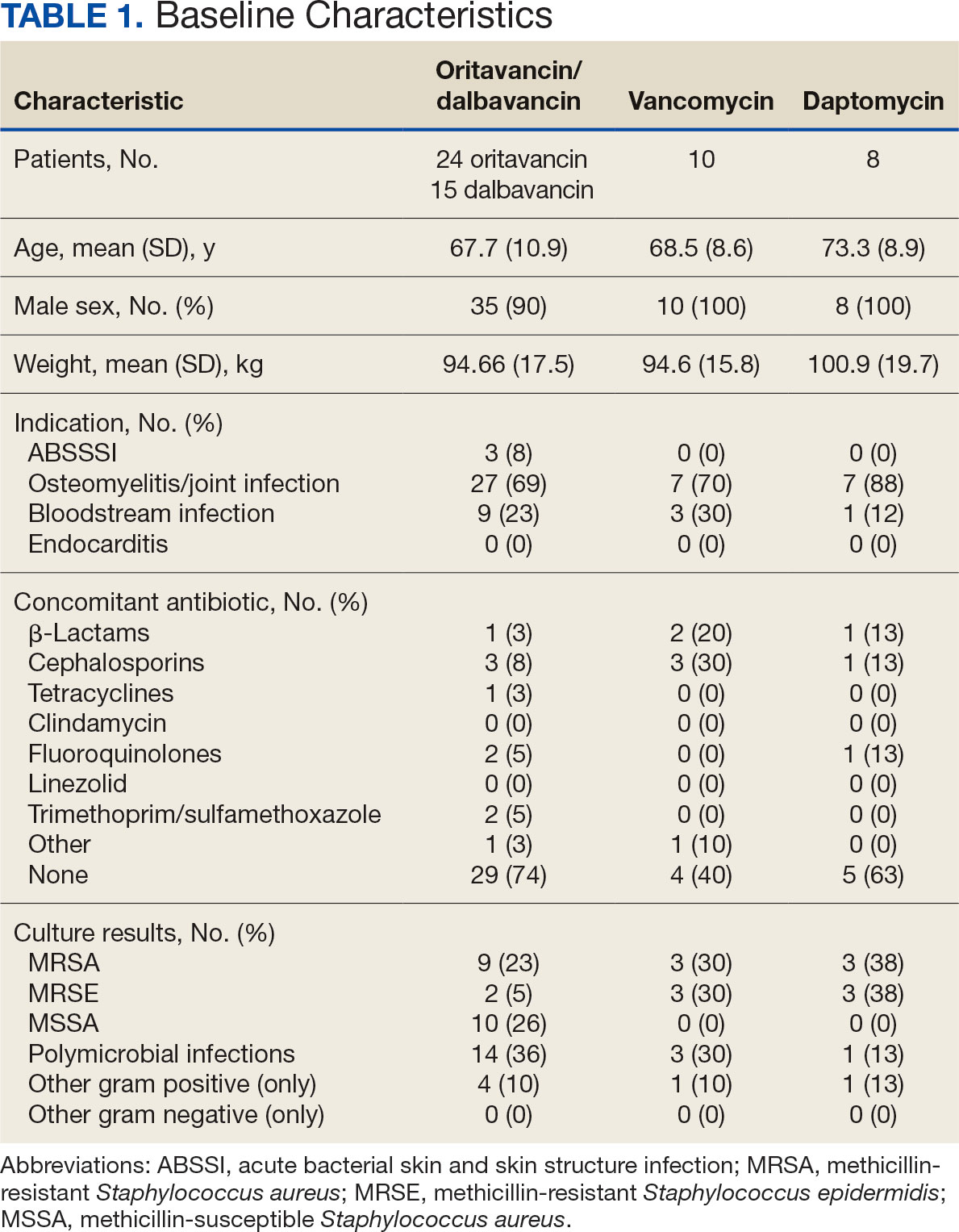
The study identified 44 patients who received dalbavancin or oritavancin between September 1, 2017, and October 31, 2022. Thirty-nine patients were included in the analysis: 24 received oritavancin and 15 received dalbavancin and were matched by indication to 10 patients who received vancomycin and 8 patients who received daptomycin. Three patients could not be matched by indication of ABSSSI (Figure 2). Most patients were male, aged > 65 years, and were treated for osteomyelitis (Table 1). No patients were treated for infective endocarditis. A myriad of concomitant antibiotics were used to treat patients and culture results indicated that most infections treated with oritavancin and dalbavancin were polymicrobial.
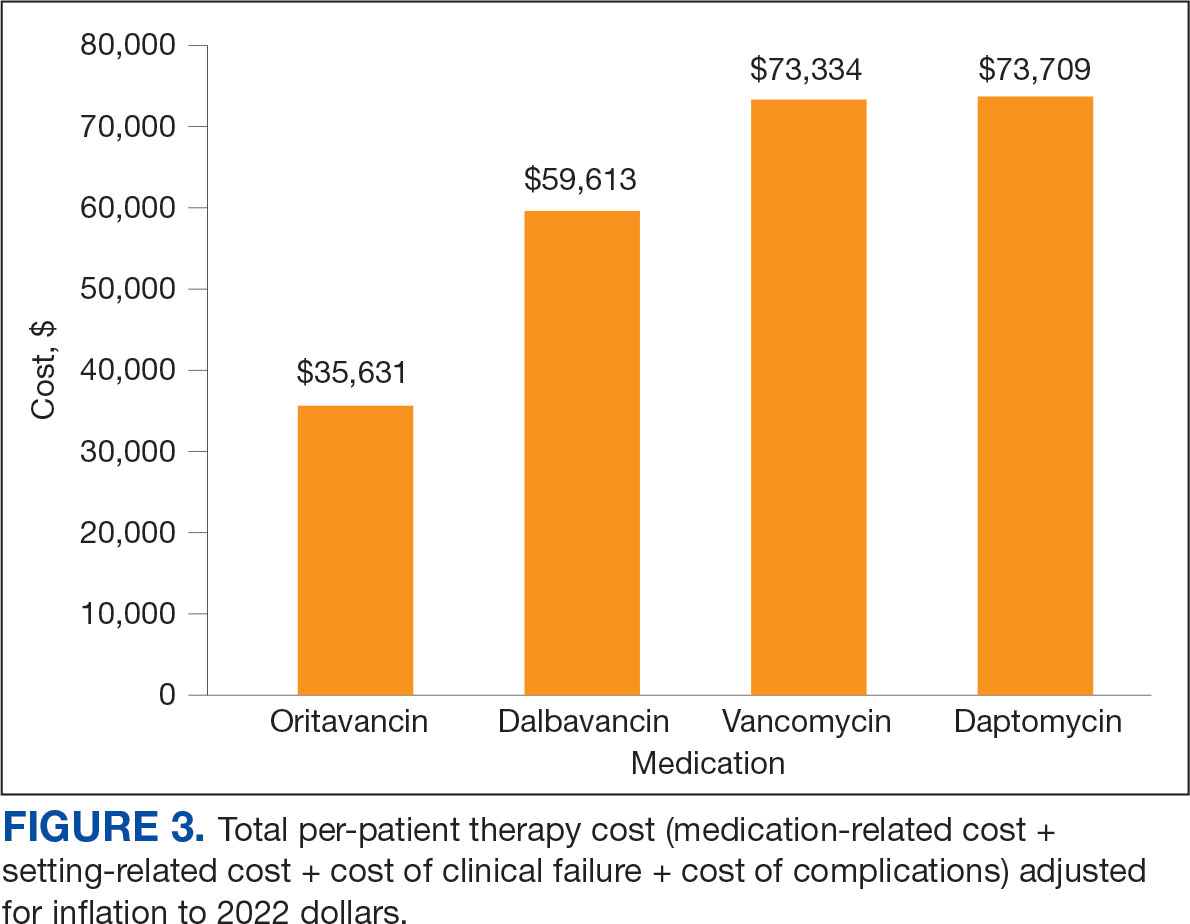
The mean total cost of therapy per patient receiving oritavancin, dalbavancin, vancomycin, and daptomycin was $35,630, $59,612, $73,333, and $73,708, respectively (Figure 3). When stratified by indication, 27 patients (69%) in the oritavancin/dalbavancin group were treated for osteomyelitis/ joint infections (16 oritavancin, 11 dalbavancin), 9 patients (23%) were treated for BSI (6 oritavancin, 3 dalbavancin), and 3 patients (8%) were treated for ABSSSI (2 oritavancin, 1 dalbavancin). The mean cost per patient for osteomyelitis/joint infections with oritavancin, dalbavancin, vancomycin, and daptomycin was $34,678, $54,224, $87,488, and $85,044, respectively. The mean cost per patient for BSI for oritavancin, dalbavancin, vancomycin, and daptomycin was $35,048, $75,349, $40,305, and $68,068, respectively. The mean cost per patient for ABSSSI for oritavancin and dalbavancin was $44,771 and $71,672.51.
Estimated total drug cost represents the cost of drug acquisition, administration supplies, laboratory monitoring, and human resources for OPAT visits or receiving home health services. The mean cost per patient of drug-related therapy for oritavancin, dalbavancin, vancomycin, and daptomycin was $2203, $5924, $3637, and $7146, respectively (Table 2).
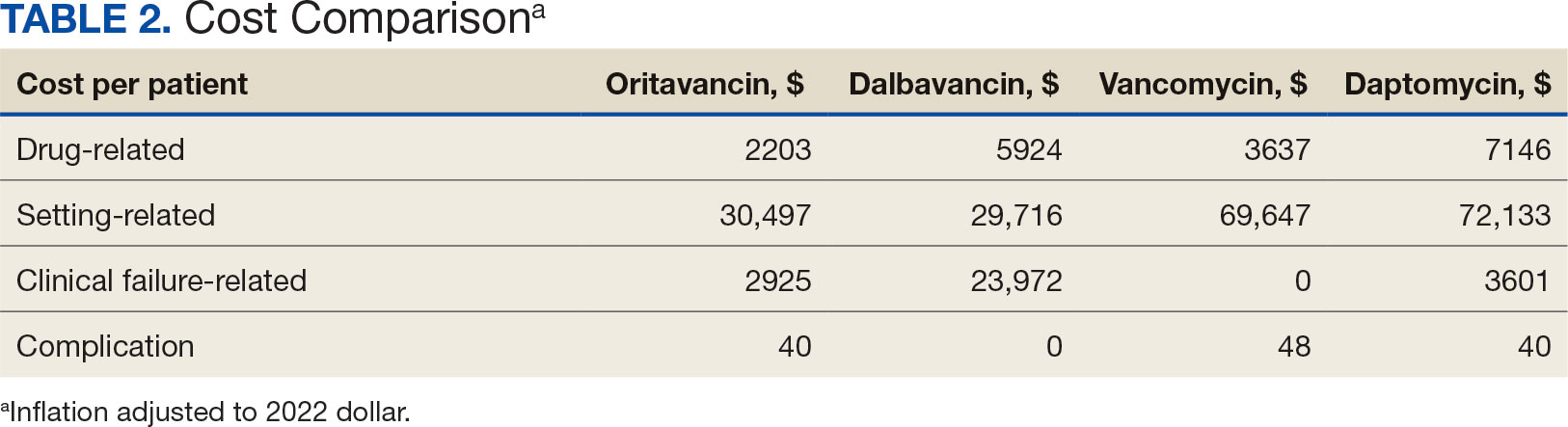
The mean cost per patient for osteomyelitis therapy for oritavancin, dalbavancin, vancomycin, and daptomycin was $2375, $6775, $4164, $8152, respectively. The mean cost of per patient for BSI treatment with oritavancin, dalbavancin, vancomycin, and daptomycin was $1737, $3475, $2409, and $1016, respectively. The mean cost per patient for oritavancin and dalbavancin for ABSSSI treatment, was $1553 and $3910, respectively.
Setting-related costs include expenses from inpatient admissions and postdischarge stays at community living centers (CLCs), skilled nursing facilities (SNFs), or rehabilitation facilities (RFs) for the duration of antimicrobial therapy. The mean setting-related therapy cost for osteomyelitis treatment with oritavancin, dalbavancin, vancomycin, and daptomycin was $27,852, $17,815, $83,324, and $72,856, respectively. The mean setting-related therapy cost per patient for BSI treatment with oritavancin, dalbavancin, vancomycin, and daptomycin was $33,310, $60,668, $37,734, and $67,074, respectively. The mean setting-related therapy cost per patient for ABSSSI treatment for oritavancin and dalbavancin was $43,218 and $67,762.00, respectively.
Six of 39 patients (15%) had clinical failure: 2 patients with oritavancin and 4 patients with dalbavancin. Four patients were readmitted for worsening infection and 2 for AEs. One patient (13%) in the daptomycin group had clinical failure due to readmission for worsening infection. There was no clinical failure with vancomycin. The costs associated with clinical failure per patient for oritavancin, dalbavancin, vancomycin, and daptomycin were $2925, $23,972, $0, and $3601, respectively (Table 3).
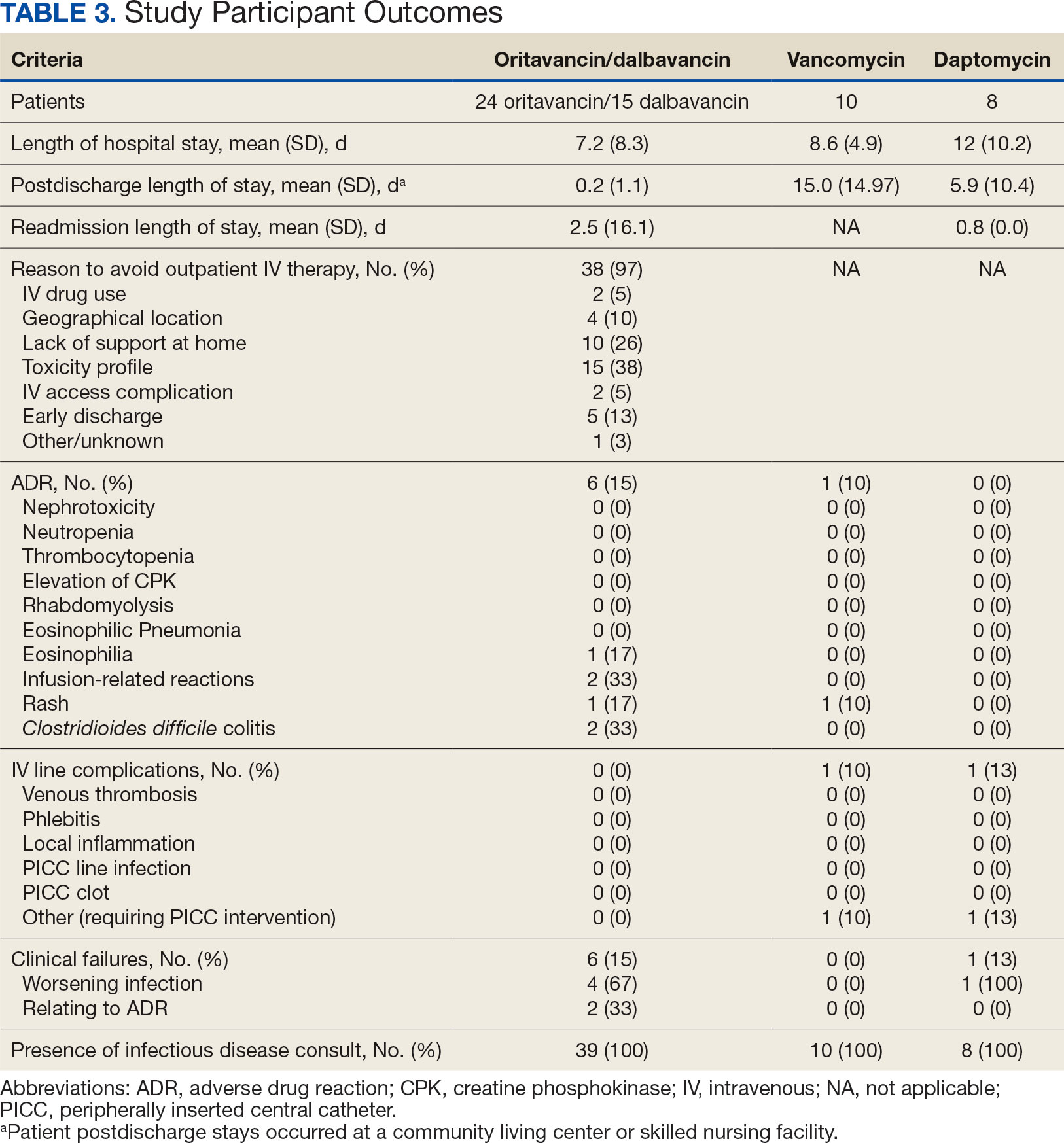
Thirty-eight patients (97%) who received oritavancin or dalbavancin had difficulty adhering to vancomycin or daptomycin OPAT. Oritavancin or dalbavancin was used in 10 patients (26%) who lacked support at home and 15 patients (38%) who had either a contraindication or previous failure with other antimicrobials, which were the most common explanations.
DISCUSSION
Long-acting lipoglycopeptides represent a potential alternative to home IV therapy that can avoid prolonged IV access with traditional OPAT. This offers significant advantages, allowing patients to be discharged from the hospital early, especially in rural areas with little OPAT infrastructure or those with logistic challenges. In this analysis, treatment with oritavancin for osteomyelitis, BSI, or ABSSSI, yielded an estimated cost savings of about $37,000 per patient, compared to treatment of matched indications with vancomycin and daptomycin. For every patient treated with dalbavancin for osteomyelitis, BSI, or ABSSSI, the cost savings was about $13,000 per patient, compared to treatment of matched indications for daptomycin and vancomycin. The estimated cost savings per patient for oritavancin was similar to previously published projections ($30,500 to $55,831).15
Cost savings were primarily driven by setting-related costs. The greatest contrast between the oritavancin and dalbavancin group compared to the vancomycin and daptomycin group was the length of stay in a postdischarge CLC, SNF, or RF setting. This analysis estimated that for every patient treated with oritavancin for osteomyelitis, the setting-related cost savings per patient was about $55,000 compared with vancomycin, and about $45,000 per patient compared with daptomycin. Furthermore, the estimated setting-related cost savings for osteomyelitis treatment with dalbavancin was about $65,000 compared with vancomycin and about $55,000 compared with daptomycin.
Clinical failure occurred with greater frequency in the oritavancin and dalbavancin groups (15%), compared with the vancomycin (0%) and daptomycin (13%) groups. Although the clinical failure rates in patients with osteomyelitis treated with oritavancin and dalbavancin compared with daptomycin were like those in previously published research (10%-30%), the rates of clinical failure for vancomycin in this analysis were lower than those in the oritavancin and dalbavancin group.8,21,22 The discrepancy in clinical failure rates between this analysis and previous research is likely due to selection bias. Based on the percentages of clinical failure found in the analysis, it is not surprising to note that the total clinical failure-related cost per patient was higher for oritavancin and dalbavancin compared to vancomycin, but similar between oritavancin and daptomycin.
This analysis also found that 15% of patients in the oritavancin and dalbavancin group experienced an AE compared to 10% of patients in the vancomycin group and none in the daptomycin group. In the oritavancin and dalbavancin group, the 2 most common AEs were infusion-related reactions and C. difficile colitis. Although infusion related reactions are easier to correspond to oritavancin and dalbavancin, it becomes difficult to definitively attribute the occurrence of C. difficile to these drugs as many patients were receiving concomitant antibiotics. Although not a primary or secondary objective, the rate of IV-line AEs were more prevalent in the vancomycin (10%), and daptomycin (13%) groups, compared to none in the oritavancin and dalbavancin group. This finding was expected; oritavancin and dalbavancin do not require a central IV line for administration.
Pharmacoeconomic literature continues to emerge with long-acting lipoglycopeptides. A 2024 Italian retrospective single-center analysis of 62 patients reported mean cost reductions > €3200 per patient (> $3400) given dalbavancin compared with the standard of care for ABSSSI or more deep-seeded infections such as osteomyelitis.23 A 2023 Spanish observational multicenter analysis of 124 patients with infective endocarditis demonstrated high efficacy, safety and cost-effectiveness with dalbavancin vs conventional treatments, with a mean savings of > €5548 per patient (> $6200).24 An analysis of the implementation of a dalbavancin order pathway for ABSSSI to avert inpatient admissions at 11 US emergency departments found a mean cost savings of $5133 per patient and $1211 per hospitalization day avoided, compared with inpatient usual care.25
Conversely, a multicenter, retrospective study of 209 patients in a community-based health care system failed to show a financial benefit for dalbavancin use when compared to standard of care for ABSSSI with higher readmission rates.26 Turco et al also reported increased cost results for 64 patients who received dalbavancin vs standard of care for ABSSSI.27 These discordant findings in ABSSSI studies may be impacted by the authors' patient selection choices and cost assumptions, especially with significantly cheaper oral alternatives. More data are needed to best identify the optimal therapeutic use for the long-acting lipoglycopeptides.
Limitations
The most significant limitation in this analysis was selection bias: 38 of 39 patients (97%) who received dalbavancin or oritavancin had a documented reason that described why OPAT therapy with traditional medications would not be optimal, including logistics, AEs, or clinical failures. Most patients treated with vancomycin and daptomycin were admitted into a SNF, RF, or CLC for the remainder of their treatment, allowing for closer monitoring and care compared to patients treated with oritavancin and dalbavancin, but at a greater cost. For patients sent to a community based SNF or RF, laboratory data were not available unless internally drawn or documented in the electronic medical record.
Additionally, not all cost data were available from VA sources; some were applied from literature, pharmacoeconomist, or best faith effort based on workflow. The cost data from third party contractors providing OPAT services to some BVAMC patients during the time frame of this analysis were not available. Due to its small sample size, outliers had the potential to affect averages reported and accuracy of the cost analysis. Emerging evidence suggests that daptomycin doses higher than the manufacturer-recommended regimen may be required for select indications, a factor that could affect cost, AEs, and efficacy outcomes.28 The acquisition cost of oritavancin and dalbavancin may vary by institution (ie, VA contract prices vs non- VA contract prices) and change over time. A current assessment of cost is needed to best visualize institutional benefit.
Finally, while the patient demographic of this MUE was highly representative of the demographic treated at the BVAMC (males aged >65 years), it may not be applicable to external patient populations. This analysis evaluated off-label indications for these medications. Consequently, this analysis would likely not be applicable to non-VA institution, as third-party payers (eg, insurance) are unlikely to cover medications for off-label indications.
CONCLUSIONS
This study found cost savings associated with the use of oritavancin and dalbavancin compared with vancomycin and daptomycin, particularly for the treatment of osteomyelitis. As safety and efficacy data continues to emerge, the use of long-acting lipoglycopeptides appears to be an increasingly attractive alternative option compared to traditional outpatient antimicrobial therapy, depending on the structure of the program. Larger, multicenter cost-effectiveness studies are needed to further establish the impact of these novel agents.
- Dalvance. Package insert. AbbVie Inc.; 2025.
- Orbactiv. Package insert. Melinta Therapeutics; 2022.
- Cooper CC, Stein GE, Mitra S, Abubaker A, Havlichek DH. Long-acting lipoglycopeptides for the treatment of bone and joint infections. Surg Infect (Larchmt). 2021;22(8):771- 779. doi:10.1089/sur.2020.413
- Simonetti O, Rizzetto G, Molinelli E, Cirioni O, Offidani A. Review: a safety profile of dalbavancin for on- and offlabel utilization. Ther Clin Risk Manag. 2021;17:223-232. doi:10.2147/TCRM.S271445
- Bloem A, Bax HI, Yusuf E, Verkaik NJ. New-generation antibiotics for treatment of gram-positive infections: a review with focus on endocarditis and osteomyelitis. J Clin Med. 2021;10(8):1743. doi:10.3390/jcm10081743
- Thomas G, Henao-Martínez AF, Franco-Paredes C, Chastain DB. Treatment of osteoarticular, cardiovascular, intravascular-catheter-related and other complicated infections with dalbavancin and oritavancin: a systematic review. Int J Antimicrob Agents. 2020;56(3):106069. doi:10.1016/j.ijantimicag.2020.106069
- Rappo U, Puttagunta S, Shevchenko V, et al. Dalbavancin for the treatment of osteomyelitis in adult patients: a randomized clinical trial of efficacy and safety. Open Forum Infect Dis. 2018;6(1):ofy331. doi:10.1093/ofid/ofy331
- Cain AR, Bremmer DN, Carr DR, et al. Effectiveness of dalbavancin compared with standard of care for the treatment of osteomyelitis: a real-world analysis. Open Forum Infect Dis. 2021;9(2):ofab589. doi:10.1093/ofid/ofab589
- Van Hise NW, Chundi V, Didwania V, et al. Treatment of acute osteomyelitis with once-weekly oritavancin: a two-year, multicenter, retrospective study. Drugs Real World Outcomes. 2020;7(Suppl 1):41-45. doi:10.1007/s40801-020-00195-7
- Cooper MM, Preslaski CR, Shihadeh KC, Hawkins KL, Jenkins TC. Multiple-dose dalbavancin regimens as the predominant treatment of deep-seated or endovascular infections: a scoping review. Open Forum Infect Dis. 2021;8(11):ofab486. doi:10.1093/ofid/ofab486
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132(15):1435-1486. doi:10.1161/CIR.0000000000000296
- Berbari EF, Kanj SS, Kowalski TJ, et al. 2015 Infectious Diseases Society of America (IDSA) Clinical Practice Guidelines for the Diagnosis and Treatment of Native Vertebral Osteomyelitis in Adults. Clin Infect Dis. 2015;61(6):e26-46. doi:10.1093/cid/civ482
- Arrieta-Loitegui M, Caro-Teller JM, Ortiz-Pérez S, López- Medrano F, San Juan-Garrido R, Ferrari-Piquero JM. Effectiveness, safety, and cost analysis of dalbavancin in clinical practice. Eur J Hosp Pharm. 2022;29(1):55-58. doi:10.1136/ejhpharm-2020-002315
- Pascale R, Maccaro A, Mikus E, et al. A retrospective multicentre study on dalbavancin effectiveness and cost-evaluation in sternotomic wound infection treatment: DALBA SWIT study. J Glob Antimicrob Resist. 2022;30:390-394. doi:10.1016/j.jgar.2022.07.018
- Antosz K, Al-Hasan MN, Lu ZK, et at. Clinical utility and cost effectiveness of long-acting lipoglycopeptides used in deep seated infections among patients with social and economic barriers to care. Pharmacy (Basel). 2021;10(1):1. doi:10.3390/pharmacy10010001
- Roberts MS. Economic aspects of evaluation. In: Friedman CP, Wyatt JC, eds. Evaluation Methods in Biomedical Informatics. 2nd ed. Springer; 2006:301-337.
- US Department of Veterans Affairs. HERC inpatient average cost data. Updated May 1, 2025. Accessed May 9, 2025. https://www.herc.research.va.gov/include/page.asp?id=inpatient
- US Department of Veterans Affairs. HERC Outpatient average cost dataset. Updated May 1, 2025. Accessed May 9, 2025. https://www.herc.research.va.gov/include/page.asp?id=outpatient
- Ektare V, Khachatryan A, Xue M, Dunne M, Johnson K, Stephens J. Assessing the economic value of avoiding hospital admissions by shifting the management of gram + acute bacterial skin and skin-structure infections to an outpatient care setting. J Med Econ. 2015;18(12):1092-1101. doi:10.3111/13696998.2015.1078339
- Ruh CA, Parameswaran GI, Wojciechowski AL, Mergenhagen KA. Outcomes and pharmacoeconomic analysis of a home intravenous antibiotic infusion program in veterans. Clin Ther. 2015;37(11):2527-2535. doi:10.1016/j.clinthera.2015.09.009
- Nakrani M, Yu D, Skikka M, et al. Comparison of vancomycin and daptomycin complications and interventions in outpatient parenteral antimicrobial therapy. Open Forum Infect Dis. 2020;7(Suppl 1):S361-S362. doi:10.1093/ofid/ofaa439.791
- Scoble PJ, Reilly J, Tilloston GS. Real-world use of oritavancin for the treatment of osteomyelitis. Drugs Real World Outcomes. 2020;7(Suppl 1):46-54. doi:10.1007/s40801-020-00194-8
- Segala D, Barbieri M, Di Nuzzo M, et al. Clinical, organizational, and pharmacoeconomic perspectives of dalbavancin vs standard of care in the infectious disease network. Glob Reg Health Technol Assess. 2024;11(Suppl 2):5-12. doi:10.33393/grhta.2024.3094
- Gómez A, et al. EN-DALBACEN 2.0 Cohort: real-life study of dalbavancin as sequential/consolidation therapy in patients with infective endocarditis due to Gram-positive cocci. Int J Antimicrob Agents. 2023;62(3):106918. doi:10.1016/j.ijantimicag.2023.106918
- LoVecchio F, McCarthy MW, Ye X, et al. Single intravenous dose dalbavancin pathway for the treatment of acute bacterial skin and skin structure infections: considerations for emergency department implementation and cost savings. J Emerg Med. 2024;67(2):e217-e229. doi:10.1016/j.jemermed.2024.03.003
- Gonzalez J, Andrade DC, Niu J. Cost-consequence analysis of single-dose dalbavancin versus standard of care for the treatment of acute bacterial skin and skin structure infections in a multisite healthcare system. Clin Infect Dis. 2021;73(7):e1436-e1442. doi:10.1093/cid/ciaa1732
- Turco NJ, Kane-Gill SL, Hernandez I, Oleksiuk LM, D’Amico F, Pickering AJ. A cost-minimization analysis of dalbavancin compared to conventional therapy for the outpatient treatment of acute bacterial skin and skin-structure infections. Expert Opin Pharmacother. 2018;19(4):319-325. doi:10.1080/14656566.2018.1442439
- Jones TW, Jun AH, Michal JL, Olney WJ. High-dose daptomycin and clinical applications. Ann Pharmacother. 2021;55(11):1363-1378. doi:10.1177/1060028021991943
Oritavancin and dalbavancin are long acting lipoglycopeptides indicated for the treatment of acute bacterial skin and skin structure infections (ABSSSI).1,2 Largely due to their long half-lives, prolonged tissue concentrations at sites of infection, tolerability, and minimal requirement for therapeutic drug monitoring, these agents are attractive options in outpatient settings.3,4 A 1- or 2-dose treatment of oritavancin and dalbavancin may be sufficient for conditions traditionally treated with outpatient parenteral antimicrobial therapy (OPAT) via peripherally inserted central catheter (PICC).
Limited research supports the use of dalbavancin and oritavancin for bone and joint infections, infective endocarditis, and bloodstream infections (BSIs). However, the US Food and Drug Administration has approved an indication for the treatment of ABSSSI.3-9 Dosing for these off-label indications varies but typically consists of an initial intravenous (IV) dose (1000 mg, 1200 mg, or 1500 mg), with a subsequent dose 1 to 2 weeks later or administered once weekly.6-10
Due in part to the recent availability of oritavancin and dalbavancin relative to the publication of practice guidelines, their appropriate place in therapy continues to evolve based on emerging literature.11,12 One potential barrier of use for these medications is their cost. Based on the number of doses administered, the 2022 estimated total acquisition cost of therapy for oritavancin and dalbavancin was $1014 to $4397 and $3046 to $7150, respectively (eAppendix). Despite the high acquisition costs, these agents do not require the placement of an indwelling central line, can be administered in outpatient settings, and require minimal therapeutic dose monitoring compared to vancomycin.13-15 This medication use evaluation (MUE) compared the total cost of treatment with oritavancin and dalbavancin vs therapies traditionally used for OPAT or prolonged IV inpatient therapy.
METHODS
This retrospective MUE was conducted at the Boise Veterans Affairs Medical Center (BVAMC), a level 2 facility with an extensive rural catchment area. BVAMC provides many OPAT services, including medications, supplies, and dressing changes after initial clinic or inpatient education. Contracted vendors may also assist with at-home nursing care using supplies provided by the BVAMC. Cases were identified using an internal database of OPAT patients and those who received oritavancin or dalbavancin between September 1, 2017, and November 1, 2022. Patients aged ≥ 18 years who received ≥ 1 dose of oritavancin or dalbavancin for ABSSSI, osteomyelitis/joint infections, endocarditis, and BSI were included. Comparator treatments consisting of ≥ 1 week of vancomycin or daptomycin for ABSSSI, osteomyelitis/joint infections, endocarditis, and BSI were identified through review of OPAT and Infectious Diseases service consults during the same timeframe. Patients were excluded if any antibiotic was prescribed by a non- VA clinician, if medications were not provided by OPAT, or if chart review did not identify an ABSSSI, osteomyelitis/ joint infection, or BSI diagnosis.
Electronic medical record review was conducted using a standardized data collection form (eAppendix). Data collected included demographics, infectious diagnosis, treatment administered, administration procedures and related visits and treatment locations, outcomes including clinical failure, adverse events (AEs), and hospital readmission.
Clinical failure was defined as readmission or death due to worsening infection or readmission secondary to a documented potential AE to the evaluated antibiotics within 90 days after initiation. Clinical failures excluded readmissions not associated with infection including comorbidities or elective procedures. AEs included new onset renal failure (serum creatinine ≥ 0.5 mg/dL), neutropenia (neutrophils ≤ 500), thrombocytopenia (platelets < 100,000), eosinophilia (> 15% eosinophils), or creatine phosphokinase > 10 times the upper limit of normal, and Clostridioides difficile (C. difficile) infection. Line complications included thrombophlebitis, local inflammation, or infection requiring line replacement (eAppendix).
A cost-minimization approach was used to assess the total cost of treatment.16 Patients who received oritavancin or dalbavancin were matched with patients that received vancomycin and daptomycin for the same indication and about 1 month of initiation through the randomization function in Microsoft Excel. This accounted for changes in personnel, nonformulary drug approvals, cost, and changes in practice during the pandemic. Costs were calculated using a decision tree as a base model (Figure 1). In this model, each treatment dyad was assessed for the presence or absence of clinical failure, adverse event (medication and line complications), and treatment setting endpoints. Cost estimates were tabulated for each patient that received treatment using published VA data, literature, pharmacoeconomist guidance, or best faith effort based on workflow. 17-20 All cost estimates were based on 2022 figures or adjusted for inflation if obtained prior to 2022. Secondary endpoints of this analysis included estimated total cost of medication acquisition, administration supplies, laboratory monitoring, and human resources for OPAT visits or receiving home-health services.
This evaluation was classified by the BVAMC Medication Use Evaluation research determination subcommittee as a quality improvement project and was considered exempt from VA Human Subjects Research requirements based on the VA Policy Handbook guideline 1058.05.
RESULTS
The study identified 44 patients who received dalbavancin or oritavancin between September 1, 2017, and October 31, 2022. Thirty-nine patients were included in the analysis: 24 received oritavancin and 15 received dalbavancin and were matched by indication to 10 patients who received vancomycin and 8 patients who received daptomycin. Three patients could not be matched by indication of ABSSSI (Figure 2). Most patients were male, aged > 65 years, and were treated for osteomyelitis (Table 1). No patients were treated for infective endocarditis. A myriad of concomitant antibiotics were used to treat patients and culture results indicated that most infections treated with oritavancin and dalbavancin were polymicrobial.
The mean total cost of therapy per patient receiving oritavancin, dalbavancin, vancomycin, and daptomycin was $35,630, $59,612, $73,333, and $73,708, respectively (Figure 3). When stratified by indication, 27 patients (69%) in the oritavancin/dalbavancin group were treated for osteomyelitis/ joint infections (16 oritavancin, 11 dalbavancin), 9 patients (23%) were treated for BSI (6 oritavancin, 3 dalbavancin), and 3 patients (8%) were treated for ABSSSI (2 oritavancin, 1 dalbavancin). The mean cost per patient for osteomyelitis/joint infections with oritavancin, dalbavancin, vancomycin, and daptomycin was $34,678, $54,224, $87,488, and $85,044, respectively. The mean cost per patient for BSI for oritavancin, dalbavancin, vancomycin, and daptomycin was $35,048, $75,349, $40,305, and $68,068, respectively. The mean cost per patient for ABSSSI for oritavancin and dalbavancin was $44,771 and $71,672.51.
Estimated total drug cost represents the cost of drug acquisition, administration supplies, laboratory monitoring, and human resources for OPAT visits or receiving home health services. The mean cost per patient of drug-related therapy for oritavancin, dalbavancin, vancomycin, and daptomycin was $2203, $5924, $3637, and $7146, respectively (Table 2).
The mean cost per patient for osteomyelitis therapy for oritavancin, dalbavancin, vancomycin, and daptomycin was $2375, $6775, $4164, $8152, respectively. The mean cost of per patient for BSI treatment with oritavancin, dalbavancin, vancomycin, and daptomycin was $1737, $3475, $2409, and $1016, respectively. The mean cost per patient for oritavancin and dalbavancin for ABSSSI treatment, was $1553 and $3910, respectively.
Setting-related costs include expenses from inpatient admissions and postdischarge stays at community living centers (CLCs), skilled nursing facilities (SNFs), or rehabilitation facilities (RFs) for the duration of antimicrobial therapy. The mean setting-related therapy cost for osteomyelitis treatment with oritavancin, dalbavancin, vancomycin, and daptomycin was $27,852, $17,815, $83,324, and $72,856, respectively. The mean setting-related therapy cost per patient for BSI treatment with oritavancin, dalbavancin, vancomycin, and daptomycin was $33,310, $60,668, $37,734, and $67,074, respectively. The mean setting-related therapy cost per patient for ABSSSI treatment for oritavancin and dalbavancin was $43,218 and $67,762.00, respectively.
Six of 39 patients (15%) had clinical failure: 2 patients with oritavancin and 4 patients with dalbavancin. Four patients were readmitted for worsening infection and 2 for AEs. One patient (13%) in the daptomycin group had clinical failure due to readmission for worsening infection. There was no clinical failure with vancomycin. The costs associated with clinical failure per patient for oritavancin, dalbavancin, vancomycin, and daptomycin were $2925, $23,972, $0, and $3601, respectively (Table 3).
Thirty-eight patients (97%) who received oritavancin or dalbavancin had difficulty adhering to vancomycin or daptomycin OPAT. Oritavancin or dalbavancin was used in 10 patients (26%) who lacked support at home and 15 patients (38%) who had either a contraindication or previous failure with other antimicrobials, which were the most common explanations.
DISCUSSION
Long-acting lipoglycopeptides represent a potential alternative to home IV therapy that can avoid prolonged IV access with traditional OPAT. This offers significant advantages, allowing patients to be discharged from the hospital early, especially in rural areas with little OPAT infrastructure or those with logistic challenges. In this analysis, treatment with oritavancin for osteomyelitis, BSI, or ABSSSI, yielded an estimated cost savings of about $37,000 per patient, compared to treatment of matched indications with vancomycin and daptomycin. For every patient treated with dalbavancin for osteomyelitis, BSI, or ABSSSI, the cost savings was about $13,000 per patient, compared to treatment of matched indications for daptomycin and vancomycin. The estimated cost savings per patient for oritavancin was similar to previously published projections ($30,500 to $55,831).15
Cost savings were primarily driven by setting-related costs. The greatest contrast between the oritavancin and dalbavancin group compared to the vancomycin and daptomycin group was the length of stay in a postdischarge CLC, SNF, or RF setting. This analysis estimated that for every patient treated with oritavancin for osteomyelitis, the setting-related cost savings per patient was about $55,000 compared with vancomycin, and about $45,000 per patient compared with daptomycin. Furthermore, the estimated setting-related cost savings for osteomyelitis treatment with dalbavancin was about $65,000 compared with vancomycin and about $55,000 compared with daptomycin.
Clinical failure occurred with greater frequency in the oritavancin and dalbavancin groups (15%), compared with the vancomycin (0%) and daptomycin (13%) groups. Although the clinical failure rates in patients with osteomyelitis treated with oritavancin and dalbavancin compared with daptomycin were like those in previously published research (10%-30%), the rates of clinical failure for vancomycin in this analysis were lower than those in the oritavancin and dalbavancin group.8,21,22 The discrepancy in clinical failure rates between this analysis and previous research is likely due to selection bias. Based on the percentages of clinical failure found in the analysis, it is not surprising to note that the total clinical failure-related cost per patient was higher for oritavancin and dalbavancin compared to vancomycin, but similar between oritavancin and daptomycin.
This analysis also found that 15% of patients in the oritavancin and dalbavancin group experienced an AE compared to 10% of patients in the vancomycin group and none in the daptomycin group. In the oritavancin and dalbavancin group, the 2 most common AEs were infusion-related reactions and C. difficile colitis. Although infusion related reactions are easier to correspond to oritavancin and dalbavancin, it becomes difficult to definitively attribute the occurrence of C. difficile to these drugs as many patients were receiving concomitant antibiotics. Although not a primary or secondary objective, the rate of IV-line AEs were more prevalent in the vancomycin (10%), and daptomycin (13%) groups, compared to none in the oritavancin and dalbavancin group. This finding was expected; oritavancin and dalbavancin do not require a central IV line for administration.
Pharmacoeconomic literature continues to emerge with long-acting lipoglycopeptides. A 2024 Italian retrospective single-center analysis of 62 patients reported mean cost reductions > €3200 per patient (> $3400) given dalbavancin compared with the standard of care for ABSSSI or more deep-seeded infections such as osteomyelitis.23 A 2023 Spanish observational multicenter analysis of 124 patients with infective endocarditis demonstrated high efficacy, safety and cost-effectiveness with dalbavancin vs conventional treatments, with a mean savings of > €5548 per patient (> $6200).24 An analysis of the implementation of a dalbavancin order pathway for ABSSSI to avert inpatient admissions at 11 US emergency departments found a mean cost savings of $5133 per patient and $1211 per hospitalization day avoided, compared with inpatient usual care.25
Conversely, a multicenter, retrospective study of 209 patients in a community-based health care system failed to show a financial benefit for dalbavancin use when compared to standard of care for ABSSSI with higher readmission rates.26 Turco et al also reported increased cost results for 64 patients who received dalbavancin vs standard of care for ABSSSI.27 These discordant findings in ABSSSI studies may be impacted by the authors' patient selection choices and cost assumptions, especially with significantly cheaper oral alternatives. More data are needed to best identify the optimal therapeutic use for the long-acting lipoglycopeptides.
Limitations
The most significant limitation in this analysis was selection bias: 38 of 39 patients (97%) who received dalbavancin or oritavancin had a documented reason that described why OPAT therapy with traditional medications would not be optimal, including logistics, AEs, or clinical failures. Most patients treated with vancomycin and daptomycin were admitted into a SNF, RF, or CLC for the remainder of their treatment, allowing for closer monitoring and care compared to patients treated with oritavancin and dalbavancin, but at a greater cost. For patients sent to a community based SNF or RF, laboratory data were not available unless internally drawn or documented in the electronic medical record.
Additionally, not all cost data were available from VA sources; some were applied from literature, pharmacoeconomist, or best faith effort based on workflow. The cost data from third party contractors providing OPAT services to some BVAMC patients during the time frame of this analysis were not available. Due to its small sample size, outliers had the potential to affect averages reported and accuracy of the cost analysis. Emerging evidence suggests that daptomycin doses higher than the manufacturer-recommended regimen may be required for select indications, a factor that could affect cost, AEs, and efficacy outcomes.28 The acquisition cost of oritavancin and dalbavancin may vary by institution (ie, VA contract prices vs non- VA contract prices) and change over time. A current assessment of cost is needed to best visualize institutional benefit.
Finally, while the patient demographic of this MUE was highly representative of the demographic treated at the BVAMC (males aged >65 years), it may not be applicable to external patient populations. This analysis evaluated off-label indications for these medications. Consequently, this analysis would likely not be applicable to non-VA institution, as third-party payers (eg, insurance) are unlikely to cover medications for off-label indications.
CONCLUSIONS
This study found cost savings associated with the use of oritavancin and dalbavancin compared with vancomycin and daptomycin, particularly for the treatment of osteomyelitis. As safety and efficacy data continues to emerge, the use of long-acting lipoglycopeptides appears to be an increasingly attractive alternative option compared to traditional outpatient antimicrobial therapy, depending on the structure of the program. Larger, multicenter cost-effectiveness studies are needed to further establish the impact of these novel agents.
Oritavancin and dalbavancin are long acting lipoglycopeptides indicated for the treatment of acute bacterial skin and skin structure infections (ABSSSI).1,2 Largely due to their long half-lives, prolonged tissue concentrations at sites of infection, tolerability, and minimal requirement for therapeutic drug monitoring, these agents are attractive options in outpatient settings.3,4 A 1- or 2-dose treatment of oritavancin and dalbavancin may be sufficient for conditions traditionally treated with outpatient parenteral antimicrobial therapy (OPAT) via peripherally inserted central catheter (PICC).
Limited research supports the use of dalbavancin and oritavancin for bone and joint infections, infective endocarditis, and bloodstream infections (BSIs). However, the US Food and Drug Administration has approved an indication for the treatment of ABSSSI.3-9 Dosing for these off-label indications varies but typically consists of an initial intravenous (IV) dose (1000 mg, 1200 mg, or 1500 mg), with a subsequent dose 1 to 2 weeks later or administered once weekly.6-10
Due in part to the recent availability of oritavancin and dalbavancin relative to the publication of practice guidelines, their appropriate place in therapy continues to evolve based on emerging literature.11,12 One potential barrier of use for these medications is their cost. Based on the number of doses administered, the 2022 estimated total acquisition cost of therapy for oritavancin and dalbavancin was $1014 to $4397 and $3046 to $7150, respectively (eAppendix). Despite the high acquisition costs, these agents do not require the placement of an indwelling central line, can be administered in outpatient settings, and require minimal therapeutic dose monitoring compared to vancomycin.13-15 This medication use evaluation (MUE) compared the total cost of treatment with oritavancin and dalbavancin vs therapies traditionally used for OPAT or prolonged IV inpatient therapy.
METHODS
This retrospective MUE was conducted at the Boise Veterans Affairs Medical Center (BVAMC), a level 2 facility with an extensive rural catchment area. BVAMC provides many OPAT services, including medications, supplies, and dressing changes after initial clinic or inpatient education. Contracted vendors may also assist with at-home nursing care using supplies provided by the BVAMC. Cases were identified using an internal database of OPAT patients and those who received oritavancin or dalbavancin between September 1, 2017, and November 1, 2022. Patients aged ≥ 18 years who received ≥ 1 dose of oritavancin or dalbavancin for ABSSSI, osteomyelitis/joint infections, endocarditis, and BSI were included. Comparator treatments consisting of ≥ 1 week of vancomycin or daptomycin for ABSSSI, osteomyelitis/joint infections, endocarditis, and BSI were identified through review of OPAT and Infectious Diseases service consults during the same timeframe. Patients were excluded if any antibiotic was prescribed by a non- VA clinician, if medications were not provided by OPAT, or if chart review did not identify an ABSSSI, osteomyelitis/ joint infection, or BSI diagnosis.
Electronic medical record review was conducted using a standardized data collection form (eAppendix). Data collected included demographics, infectious diagnosis, treatment administered, administration procedures and related visits and treatment locations, outcomes including clinical failure, adverse events (AEs), and hospital readmission.
Clinical failure was defined as readmission or death due to worsening infection or readmission secondary to a documented potential AE to the evaluated antibiotics within 90 days after initiation. Clinical failures excluded readmissions not associated with infection including comorbidities or elective procedures. AEs included new onset renal failure (serum creatinine ≥ 0.5 mg/dL), neutropenia (neutrophils ≤ 500), thrombocytopenia (platelets < 100,000), eosinophilia (> 15% eosinophils), or creatine phosphokinase > 10 times the upper limit of normal, and Clostridioides difficile (C. difficile) infection. Line complications included thrombophlebitis, local inflammation, or infection requiring line replacement (eAppendix).
A cost-minimization approach was used to assess the total cost of treatment.16 Patients who received oritavancin or dalbavancin were matched with patients that received vancomycin and daptomycin for the same indication and about 1 month of initiation through the randomization function in Microsoft Excel. This accounted for changes in personnel, nonformulary drug approvals, cost, and changes in practice during the pandemic. Costs were calculated using a decision tree as a base model (Figure 1). In this model, each treatment dyad was assessed for the presence or absence of clinical failure, adverse event (medication and line complications), and treatment setting endpoints. Cost estimates were tabulated for each patient that received treatment using published VA data, literature, pharmacoeconomist guidance, or best faith effort based on workflow. 17-20 All cost estimates were based on 2022 figures or adjusted for inflation if obtained prior to 2022. Secondary endpoints of this analysis included estimated total cost of medication acquisition, administration supplies, laboratory monitoring, and human resources for OPAT visits or receiving home-health services.
This evaluation was classified by the BVAMC Medication Use Evaluation research determination subcommittee as a quality improvement project and was considered exempt from VA Human Subjects Research requirements based on the VA Policy Handbook guideline 1058.05.
RESULTS
The study identified 44 patients who received dalbavancin or oritavancin between September 1, 2017, and October 31, 2022. Thirty-nine patients were included in the analysis: 24 received oritavancin and 15 received dalbavancin and were matched by indication to 10 patients who received vancomycin and 8 patients who received daptomycin. Three patients could not be matched by indication of ABSSSI (Figure 2). Most patients were male, aged > 65 years, and were treated for osteomyelitis (Table 1). No patients were treated for infective endocarditis. A myriad of concomitant antibiotics were used to treat patients and culture results indicated that most infections treated with oritavancin and dalbavancin were polymicrobial.
The mean total cost of therapy per patient receiving oritavancin, dalbavancin, vancomycin, and daptomycin was $35,630, $59,612, $73,333, and $73,708, respectively (Figure 3). When stratified by indication, 27 patients (69%) in the oritavancin/dalbavancin group were treated for osteomyelitis/ joint infections (16 oritavancin, 11 dalbavancin), 9 patients (23%) were treated for BSI (6 oritavancin, 3 dalbavancin), and 3 patients (8%) were treated for ABSSSI (2 oritavancin, 1 dalbavancin). The mean cost per patient for osteomyelitis/joint infections with oritavancin, dalbavancin, vancomycin, and daptomycin was $34,678, $54,224, $87,488, and $85,044, respectively. The mean cost per patient for BSI for oritavancin, dalbavancin, vancomycin, and daptomycin was $35,048, $75,349, $40,305, and $68,068, respectively. The mean cost per patient for ABSSSI for oritavancin and dalbavancin was $44,771 and $71,672.51.
Estimated total drug cost represents the cost of drug acquisition, administration supplies, laboratory monitoring, and human resources for OPAT visits or receiving home health services. The mean cost per patient of drug-related therapy for oritavancin, dalbavancin, vancomycin, and daptomycin was $2203, $5924, $3637, and $7146, respectively (Table 2).
The mean cost per patient for osteomyelitis therapy for oritavancin, dalbavancin, vancomycin, and daptomycin was $2375, $6775, $4164, $8152, respectively. The mean cost of per patient for BSI treatment with oritavancin, dalbavancin, vancomycin, and daptomycin was $1737, $3475, $2409, and $1016, respectively. The mean cost per patient for oritavancin and dalbavancin for ABSSSI treatment, was $1553 and $3910, respectively.
Setting-related costs include expenses from inpatient admissions and postdischarge stays at community living centers (CLCs), skilled nursing facilities (SNFs), or rehabilitation facilities (RFs) for the duration of antimicrobial therapy. The mean setting-related therapy cost for osteomyelitis treatment with oritavancin, dalbavancin, vancomycin, and daptomycin was $27,852, $17,815, $83,324, and $72,856, respectively. The mean setting-related therapy cost per patient for BSI treatment with oritavancin, dalbavancin, vancomycin, and daptomycin was $33,310, $60,668, $37,734, and $67,074, respectively. The mean setting-related therapy cost per patient for ABSSSI treatment for oritavancin and dalbavancin was $43,218 and $67,762.00, respectively.
Six of 39 patients (15%) had clinical failure: 2 patients with oritavancin and 4 patients with dalbavancin. Four patients were readmitted for worsening infection and 2 for AEs. One patient (13%) in the daptomycin group had clinical failure due to readmission for worsening infection. There was no clinical failure with vancomycin. The costs associated with clinical failure per patient for oritavancin, dalbavancin, vancomycin, and daptomycin were $2925, $23,972, $0, and $3601, respectively (Table 3).
Thirty-eight patients (97%) who received oritavancin or dalbavancin had difficulty adhering to vancomycin or daptomycin OPAT. Oritavancin or dalbavancin was used in 10 patients (26%) who lacked support at home and 15 patients (38%) who had either a contraindication or previous failure with other antimicrobials, which were the most common explanations.
DISCUSSION
Long-acting lipoglycopeptides represent a potential alternative to home IV therapy that can avoid prolonged IV access with traditional OPAT. This offers significant advantages, allowing patients to be discharged from the hospital early, especially in rural areas with little OPAT infrastructure or those with logistic challenges. In this analysis, treatment with oritavancin for osteomyelitis, BSI, or ABSSSI, yielded an estimated cost savings of about $37,000 per patient, compared to treatment of matched indications with vancomycin and daptomycin. For every patient treated with dalbavancin for osteomyelitis, BSI, or ABSSSI, the cost savings was about $13,000 per patient, compared to treatment of matched indications for daptomycin and vancomycin. The estimated cost savings per patient for oritavancin was similar to previously published projections ($30,500 to $55,831).15
Cost savings were primarily driven by setting-related costs. The greatest contrast between the oritavancin and dalbavancin group compared to the vancomycin and daptomycin group was the length of stay in a postdischarge CLC, SNF, or RF setting. This analysis estimated that for every patient treated with oritavancin for osteomyelitis, the setting-related cost savings per patient was about $55,000 compared with vancomycin, and about $45,000 per patient compared with daptomycin. Furthermore, the estimated setting-related cost savings for osteomyelitis treatment with dalbavancin was about $65,000 compared with vancomycin and about $55,000 compared with daptomycin.
Clinical failure occurred with greater frequency in the oritavancin and dalbavancin groups (15%), compared with the vancomycin (0%) and daptomycin (13%) groups. Although the clinical failure rates in patients with osteomyelitis treated with oritavancin and dalbavancin compared with daptomycin were like those in previously published research (10%-30%), the rates of clinical failure for vancomycin in this analysis were lower than those in the oritavancin and dalbavancin group.8,21,22 The discrepancy in clinical failure rates between this analysis and previous research is likely due to selection bias. Based on the percentages of clinical failure found in the analysis, it is not surprising to note that the total clinical failure-related cost per patient was higher for oritavancin and dalbavancin compared to vancomycin, but similar between oritavancin and daptomycin.
This analysis also found that 15% of patients in the oritavancin and dalbavancin group experienced an AE compared to 10% of patients in the vancomycin group and none in the daptomycin group. In the oritavancin and dalbavancin group, the 2 most common AEs were infusion-related reactions and C. difficile colitis. Although infusion related reactions are easier to correspond to oritavancin and dalbavancin, it becomes difficult to definitively attribute the occurrence of C. difficile to these drugs as many patients were receiving concomitant antibiotics. Although not a primary or secondary objective, the rate of IV-line AEs were more prevalent in the vancomycin (10%), and daptomycin (13%) groups, compared to none in the oritavancin and dalbavancin group. This finding was expected; oritavancin and dalbavancin do not require a central IV line for administration.
Pharmacoeconomic literature continues to emerge with long-acting lipoglycopeptides. A 2024 Italian retrospective single-center analysis of 62 patients reported mean cost reductions > €3200 per patient (> $3400) given dalbavancin compared with the standard of care for ABSSSI or more deep-seeded infections such as osteomyelitis.23 A 2023 Spanish observational multicenter analysis of 124 patients with infective endocarditis demonstrated high efficacy, safety and cost-effectiveness with dalbavancin vs conventional treatments, with a mean savings of > €5548 per patient (> $6200).24 An analysis of the implementation of a dalbavancin order pathway for ABSSSI to avert inpatient admissions at 11 US emergency departments found a mean cost savings of $5133 per patient and $1211 per hospitalization day avoided, compared with inpatient usual care.25
Conversely, a multicenter, retrospective study of 209 patients in a community-based health care system failed to show a financial benefit for dalbavancin use when compared to standard of care for ABSSSI with higher readmission rates.26 Turco et al also reported increased cost results for 64 patients who received dalbavancin vs standard of care for ABSSSI.27 These discordant findings in ABSSSI studies may be impacted by the authors' patient selection choices and cost assumptions, especially with significantly cheaper oral alternatives. More data are needed to best identify the optimal therapeutic use for the long-acting lipoglycopeptides.
Limitations
The most significant limitation in this analysis was selection bias: 38 of 39 patients (97%) who received dalbavancin or oritavancin had a documented reason that described why OPAT therapy with traditional medications would not be optimal, including logistics, AEs, or clinical failures. Most patients treated with vancomycin and daptomycin were admitted into a SNF, RF, or CLC for the remainder of their treatment, allowing for closer monitoring and care compared to patients treated with oritavancin and dalbavancin, but at a greater cost. For patients sent to a community based SNF or RF, laboratory data were not available unless internally drawn or documented in the electronic medical record.
Additionally, not all cost data were available from VA sources; some were applied from literature, pharmacoeconomist, or best faith effort based on workflow. The cost data from third party contractors providing OPAT services to some BVAMC patients during the time frame of this analysis were not available. Due to its small sample size, outliers had the potential to affect averages reported and accuracy of the cost analysis. Emerging evidence suggests that daptomycin doses higher than the manufacturer-recommended regimen may be required for select indications, a factor that could affect cost, AEs, and efficacy outcomes.28 The acquisition cost of oritavancin and dalbavancin may vary by institution (ie, VA contract prices vs non- VA contract prices) and change over time. A current assessment of cost is needed to best visualize institutional benefit.
Finally, while the patient demographic of this MUE was highly representative of the demographic treated at the BVAMC (males aged >65 years), it may not be applicable to external patient populations. This analysis evaluated off-label indications for these medications. Consequently, this analysis would likely not be applicable to non-VA institution, as third-party payers (eg, insurance) are unlikely to cover medications for off-label indications.
CONCLUSIONS
This study found cost savings associated with the use of oritavancin and dalbavancin compared with vancomycin and daptomycin, particularly for the treatment of osteomyelitis. As safety and efficacy data continues to emerge, the use of long-acting lipoglycopeptides appears to be an increasingly attractive alternative option compared to traditional outpatient antimicrobial therapy, depending on the structure of the program. Larger, multicenter cost-effectiveness studies are needed to further establish the impact of these novel agents.
- Dalvance. Package insert. AbbVie Inc.; 2025.
- Orbactiv. Package insert. Melinta Therapeutics; 2022.
- Cooper CC, Stein GE, Mitra S, Abubaker A, Havlichek DH. Long-acting lipoglycopeptides for the treatment of bone and joint infections. Surg Infect (Larchmt). 2021;22(8):771- 779. doi:10.1089/sur.2020.413
- Simonetti O, Rizzetto G, Molinelli E, Cirioni O, Offidani A. Review: a safety profile of dalbavancin for on- and offlabel utilization. Ther Clin Risk Manag. 2021;17:223-232. doi:10.2147/TCRM.S271445
- Bloem A, Bax HI, Yusuf E, Verkaik NJ. New-generation antibiotics for treatment of gram-positive infections: a review with focus on endocarditis and osteomyelitis. J Clin Med. 2021;10(8):1743. doi:10.3390/jcm10081743
- Thomas G, Henao-Martínez AF, Franco-Paredes C, Chastain DB. Treatment of osteoarticular, cardiovascular, intravascular-catheter-related and other complicated infections with dalbavancin and oritavancin: a systematic review. Int J Antimicrob Agents. 2020;56(3):106069. doi:10.1016/j.ijantimicag.2020.106069
- Rappo U, Puttagunta S, Shevchenko V, et al. Dalbavancin for the treatment of osteomyelitis in adult patients: a randomized clinical trial of efficacy and safety. Open Forum Infect Dis. 2018;6(1):ofy331. doi:10.1093/ofid/ofy331
- Cain AR, Bremmer DN, Carr DR, et al. Effectiveness of dalbavancin compared with standard of care for the treatment of osteomyelitis: a real-world analysis. Open Forum Infect Dis. 2021;9(2):ofab589. doi:10.1093/ofid/ofab589
- Van Hise NW, Chundi V, Didwania V, et al. Treatment of acute osteomyelitis with once-weekly oritavancin: a two-year, multicenter, retrospective study. Drugs Real World Outcomes. 2020;7(Suppl 1):41-45. doi:10.1007/s40801-020-00195-7
- Cooper MM, Preslaski CR, Shihadeh KC, Hawkins KL, Jenkins TC. Multiple-dose dalbavancin regimens as the predominant treatment of deep-seated or endovascular infections: a scoping review. Open Forum Infect Dis. 2021;8(11):ofab486. doi:10.1093/ofid/ofab486
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132(15):1435-1486. doi:10.1161/CIR.0000000000000296
- Berbari EF, Kanj SS, Kowalski TJ, et al. 2015 Infectious Diseases Society of America (IDSA) Clinical Practice Guidelines for the Diagnosis and Treatment of Native Vertebral Osteomyelitis in Adults. Clin Infect Dis. 2015;61(6):e26-46. doi:10.1093/cid/civ482
- Arrieta-Loitegui M, Caro-Teller JM, Ortiz-Pérez S, López- Medrano F, San Juan-Garrido R, Ferrari-Piquero JM. Effectiveness, safety, and cost analysis of dalbavancin in clinical practice. Eur J Hosp Pharm. 2022;29(1):55-58. doi:10.1136/ejhpharm-2020-002315
- Pascale R, Maccaro A, Mikus E, et al. A retrospective multicentre study on dalbavancin effectiveness and cost-evaluation in sternotomic wound infection treatment: DALBA SWIT study. J Glob Antimicrob Resist. 2022;30:390-394. doi:10.1016/j.jgar.2022.07.018
- Antosz K, Al-Hasan MN, Lu ZK, et at. Clinical utility and cost effectiveness of long-acting lipoglycopeptides used in deep seated infections among patients with social and economic barriers to care. Pharmacy (Basel). 2021;10(1):1. doi:10.3390/pharmacy10010001
- Roberts MS. Economic aspects of evaluation. In: Friedman CP, Wyatt JC, eds. Evaluation Methods in Biomedical Informatics. 2nd ed. Springer; 2006:301-337.
- US Department of Veterans Affairs. HERC inpatient average cost data. Updated May 1, 2025. Accessed May 9, 2025. https://www.herc.research.va.gov/include/page.asp?id=inpatient
- US Department of Veterans Affairs. HERC Outpatient average cost dataset. Updated May 1, 2025. Accessed May 9, 2025. https://www.herc.research.va.gov/include/page.asp?id=outpatient
- Ektare V, Khachatryan A, Xue M, Dunne M, Johnson K, Stephens J. Assessing the economic value of avoiding hospital admissions by shifting the management of gram + acute bacterial skin and skin-structure infections to an outpatient care setting. J Med Econ. 2015;18(12):1092-1101. doi:10.3111/13696998.2015.1078339
- Ruh CA, Parameswaran GI, Wojciechowski AL, Mergenhagen KA. Outcomes and pharmacoeconomic analysis of a home intravenous antibiotic infusion program in veterans. Clin Ther. 2015;37(11):2527-2535. doi:10.1016/j.clinthera.2015.09.009
- Nakrani M, Yu D, Skikka M, et al. Comparison of vancomycin and daptomycin complications and interventions in outpatient parenteral antimicrobial therapy. Open Forum Infect Dis. 2020;7(Suppl 1):S361-S362. doi:10.1093/ofid/ofaa439.791
- Scoble PJ, Reilly J, Tilloston GS. Real-world use of oritavancin for the treatment of osteomyelitis. Drugs Real World Outcomes. 2020;7(Suppl 1):46-54. doi:10.1007/s40801-020-00194-8
- Segala D, Barbieri M, Di Nuzzo M, et al. Clinical, organizational, and pharmacoeconomic perspectives of dalbavancin vs standard of care in the infectious disease network. Glob Reg Health Technol Assess. 2024;11(Suppl 2):5-12. doi:10.33393/grhta.2024.3094
- Gómez A, et al. EN-DALBACEN 2.0 Cohort: real-life study of dalbavancin as sequential/consolidation therapy in patients with infective endocarditis due to Gram-positive cocci. Int J Antimicrob Agents. 2023;62(3):106918. doi:10.1016/j.ijantimicag.2023.106918
- LoVecchio F, McCarthy MW, Ye X, et al. Single intravenous dose dalbavancin pathway for the treatment of acute bacterial skin and skin structure infections: considerations for emergency department implementation and cost savings. J Emerg Med. 2024;67(2):e217-e229. doi:10.1016/j.jemermed.2024.03.003
- Gonzalez J, Andrade DC, Niu J. Cost-consequence analysis of single-dose dalbavancin versus standard of care for the treatment of acute bacterial skin and skin structure infections in a multisite healthcare system. Clin Infect Dis. 2021;73(7):e1436-e1442. doi:10.1093/cid/ciaa1732
- Turco NJ, Kane-Gill SL, Hernandez I, Oleksiuk LM, D’Amico F, Pickering AJ. A cost-minimization analysis of dalbavancin compared to conventional therapy for the outpatient treatment of acute bacterial skin and skin-structure infections. Expert Opin Pharmacother. 2018;19(4):319-325. doi:10.1080/14656566.2018.1442439
- Jones TW, Jun AH, Michal JL, Olney WJ. High-dose daptomycin and clinical applications. Ann Pharmacother. 2021;55(11):1363-1378. doi:10.1177/1060028021991943
- Dalvance. Package insert. AbbVie Inc.; 2025.
- Orbactiv. Package insert. Melinta Therapeutics; 2022.
- Cooper CC, Stein GE, Mitra S, Abubaker A, Havlichek DH. Long-acting lipoglycopeptides for the treatment of bone and joint infections. Surg Infect (Larchmt). 2021;22(8):771- 779. doi:10.1089/sur.2020.413
- Simonetti O, Rizzetto G, Molinelli E, Cirioni O, Offidani A. Review: a safety profile of dalbavancin for on- and offlabel utilization. Ther Clin Risk Manag. 2021;17:223-232. doi:10.2147/TCRM.S271445
- Bloem A, Bax HI, Yusuf E, Verkaik NJ. New-generation antibiotics for treatment of gram-positive infections: a review with focus on endocarditis and osteomyelitis. J Clin Med. 2021;10(8):1743. doi:10.3390/jcm10081743
- Thomas G, Henao-Martínez AF, Franco-Paredes C, Chastain DB. Treatment of osteoarticular, cardiovascular, intravascular-catheter-related and other complicated infections with dalbavancin and oritavancin: a systematic review. Int J Antimicrob Agents. 2020;56(3):106069. doi:10.1016/j.ijantimicag.2020.106069
- Rappo U, Puttagunta S, Shevchenko V, et al. Dalbavancin for the treatment of osteomyelitis in adult patients: a randomized clinical trial of efficacy and safety. Open Forum Infect Dis. 2018;6(1):ofy331. doi:10.1093/ofid/ofy331
- Cain AR, Bremmer DN, Carr DR, et al. Effectiveness of dalbavancin compared with standard of care for the treatment of osteomyelitis: a real-world analysis. Open Forum Infect Dis. 2021;9(2):ofab589. doi:10.1093/ofid/ofab589
- Van Hise NW, Chundi V, Didwania V, et al. Treatment of acute osteomyelitis with once-weekly oritavancin: a two-year, multicenter, retrospective study. Drugs Real World Outcomes. 2020;7(Suppl 1):41-45. doi:10.1007/s40801-020-00195-7
- Cooper MM, Preslaski CR, Shihadeh KC, Hawkins KL, Jenkins TC. Multiple-dose dalbavancin regimens as the predominant treatment of deep-seated or endovascular infections: a scoping review. Open Forum Infect Dis. 2021;8(11):ofab486. doi:10.1093/ofid/ofab486
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132(15):1435-1486. doi:10.1161/CIR.0000000000000296
- Berbari EF, Kanj SS, Kowalski TJ, et al. 2015 Infectious Diseases Society of America (IDSA) Clinical Practice Guidelines for the Diagnosis and Treatment of Native Vertebral Osteomyelitis in Adults. Clin Infect Dis. 2015;61(6):e26-46. doi:10.1093/cid/civ482
- Arrieta-Loitegui M, Caro-Teller JM, Ortiz-Pérez S, López- Medrano F, San Juan-Garrido R, Ferrari-Piquero JM. Effectiveness, safety, and cost analysis of dalbavancin in clinical practice. Eur J Hosp Pharm. 2022;29(1):55-58. doi:10.1136/ejhpharm-2020-002315
- Pascale R, Maccaro A, Mikus E, et al. A retrospective multicentre study on dalbavancin effectiveness and cost-evaluation in sternotomic wound infection treatment: DALBA SWIT study. J Glob Antimicrob Resist. 2022;30:390-394. doi:10.1016/j.jgar.2022.07.018
- Antosz K, Al-Hasan MN, Lu ZK, et at. Clinical utility and cost effectiveness of long-acting lipoglycopeptides used in deep seated infections among patients with social and economic barriers to care. Pharmacy (Basel). 2021;10(1):1. doi:10.3390/pharmacy10010001
- Roberts MS. Economic aspects of evaluation. In: Friedman CP, Wyatt JC, eds. Evaluation Methods in Biomedical Informatics. 2nd ed. Springer; 2006:301-337.
- US Department of Veterans Affairs. HERC inpatient average cost data. Updated May 1, 2025. Accessed May 9, 2025. https://www.herc.research.va.gov/include/page.asp?id=inpatient
- US Department of Veterans Affairs. HERC Outpatient average cost dataset. Updated May 1, 2025. Accessed May 9, 2025. https://www.herc.research.va.gov/include/page.asp?id=outpatient
- Ektare V, Khachatryan A, Xue M, Dunne M, Johnson K, Stephens J. Assessing the economic value of avoiding hospital admissions by shifting the management of gram + acute bacterial skin and skin-structure infections to an outpatient care setting. J Med Econ. 2015;18(12):1092-1101. doi:10.3111/13696998.2015.1078339
- Ruh CA, Parameswaran GI, Wojciechowski AL, Mergenhagen KA. Outcomes and pharmacoeconomic analysis of a home intravenous antibiotic infusion program in veterans. Clin Ther. 2015;37(11):2527-2535. doi:10.1016/j.clinthera.2015.09.009
- Nakrani M, Yu D, Skikka M, et al. Comparison of vancomycin and daptomycin complications and interventions in outpatient parenteral antimicrobial therapy. Open Forum Infect Dis. 2020;7(Suppl 1):S361-S362. doi:10.1093/ofid/ofaa439.791
- Scoble PJ, Reilly J, Tilloston GS. Real-world use of oritavancin for the treatment of osteomyelitis. Drugs Real World Outcomes. 2020;7(Suppl 1):46-54. doi:10.1007/s40801-020-00194-8
- Segala D, Barbieri M, Di Nuzzo M, et al. Clinical, organizational, and pharmacoeconomic perspectives of dalbavancin vs standard of care in the infectious disease network. Glob Reg Health Technol Assess. 2024;11(Suppl 2):5-12. doi:10.33393/grhta.2024.3094
- Gómez A, et al. EN-DALBACEN 2.0 Cohort: real-life study of dalbavancin as sequential/consolidation therapy in patients with infective endocarditis due to Gram-positive cocci. Int J Antimicrob Agents. 2023;62(3):106918. doi:10.1016/j.ijantimicag.2023.106918
- LoVecchio F, McCarthy MW, Ye X, et al. Single intravenous dose dalbavancin pathway for the treatment of acute bacterial skin and skin structure infections: considerations for emergency department implementation and cost savings. J Emerg Med. 2024;67(2):e217-e229. doi:10.1016/j.jemermed.2024.03.003
- Gonzalez J, Andrade DC, Niu J. Cost-consequence analysis of single-dose dalbavancin versus standard of care for the treatment of acute bacterial skin and skin structure infections in a multisite healthcare system. Clin Infect Dis. 2021;73(7):e1436-e1442. doi:10.1093/cid/ciaa1732
- Turco NJ, Kane-Gill SL, Hernandez I, Oleksiuk LM, D’Amico F, Pickering AJ. A cost-minimization analysis of dalbavancin compared to conventional therapy for the outpatient treatment of acute bacterial skin and skin-structure infections. Expert Opin Pharmacother. 2018;19(4):319-325. doi:10.1080/14656566.2018.1442439
- Jones TW, Jun AH, Michal JL, Olney WJ. High-dose daptomycin and clinical applications. Ann Pharmacother. 2021;55(11):1363-1378. doi:10.1177/1060028021991943
Are Oritavancin and Dalbavancin More Cost Effective for Outpatient Parenteral Antimicrobial Therapy at a Veterans Affairs Medical Center?
Are Oritavancin and Dalbavancin More Cost Effective for Outpatient Parenteral Antimicrobial Therapy at a Veterans Affairs Medical Center?
Impact of Multisite Patient Education on Pharmacotherapy for Veterans With Alcohol Use Disorder
Impact of Multisite Patient Education on Pharmacotherapy for Veterans With Alcohol Use Disorder
Excessive alcohol use is one of the leading preventable causes of death in the United States, responsible for about 178,000 deaths annually and an average of 488 daily deaths in 2020 and 2021.1Alcohol-related deaths increased by 49% between 2006 and 2019.2 This trend continued during the COVID-19 pandemic, with death certificates that listed alcohol increasing by > 25% from 2019 to 2020, and another 10% in 2021.3 This increase of alcohol-related deaths includes those as a direct result of chronic alcohol use, such as alcoholic cardiomyopathy, alcoholic hepatitis and cirrhosis, and alcohol-induced pancreatitis, as well as a result of acute use such as alcohol poisoning, suicide by exposure to alcohol, and alcohol-impaired driving fatalities.4
Excessive alcohol consumption poses other serious risks, including cases when intake is abruptly reduced without proper management. Alcohol withdrawal syndrome (AWS) can vary in severity, with potentially life-threatening complications such as hallucinations, seizures, and delirium tremens.5
These risks highlight the importance of professional intervention and support, not only to mitigate risks associated with AWS, but provide a pathway towards recovery from alcohol use disorder (AUD).
According to the 2022 National Survey on Drug Use and Health, 28.8 million US adults had AUD in the prior year, yet only 7.6% of these individuals received treatment and an even smaller group (2.2%) received medication-assisted treatment for alcohol.6,7 This is despite American Psychiatric Association guidelines for the pharmacological treatment of patients with AUD, including the use of naltrexone, acamprosate, disulfiram, topiramate, or gabapentin, depending on therapy goals, past medication trials, medication contraindications, and patient preference.8 Several of these medications are approved by the US Food and Drug Administration (FDA) for the treatment of AUD and have support for effectiveness from randomized controlled trials and meta-analyses.9-11
Clinical practice guidelines for the management of substance use disorders (SUDs) from the US Department of Veterans Affairs (VA) and US Department of Defense have strong recommendations for naltrexone and topiramate as first-line pharmacotherapies for moderate to severe AUD. Acamprosate and disulfiram are weak recommendations as alternative options. Gabapentin is a weak recommendation for cases where first-line treatments are contraindicated or ineffective. The guidelines emphasize the importance of a comprehensive approach to AUD treatment, including psychosocial interventions in addition to pharmacotherapy.12
A 2023 national survey found veterans reported higher alcohol consumption than nonveterans.13 At the end of fiscal year 2023, > 4.4 million veterans—6% of Veterans Health Administration patients—had been diagnosed with AUD.14 However, > 87% of these patients nationally, and 88% of Veterans Integrated Service Network (VISN) 21 patients, were not receiving naltrexone, acamprosate, disulfiram, or topiramate as part of their treatment. The VA Academic Detailing Service (ADS) now includes AUD pharmacotherapy as a campaign focus, highlighting its importance. The ADS is a pharmacy educational outreach program that uses unbiased clinical guidelines to promote aligning prescribing behavior with best practices. Academic detailing methods include speaking with health care practitioners (HCPs), and direct-to-consumer (DTC) patient education.
ADS campaigns include DTC educational handouts. Past ADS projects and research using DTC have demonstrated a significant improvement in outcomes and positively influencing patients’ pharmacotherapy treatment. 15,16 A VA quality improvement project found a positive correlation between the initiation of AUD pharmacotherapy and engagement with mental health care following the distribution of AUD DTC patient education. 17 This project aimed to apply the same principles of prior research to explore the use of DTC across multiple facilities within VISN 21 to increase AUD pharmacotherapy. VISN 21 includes VA facilities and clinics across the Pacific Islands, Nevada, and California and serves about 350,000 veterans.
METHODS
A prospective cohort of VISN 21 veterans with or at high risk for AUD was identified using the VA ADS AUD Dashboard. The cohort included those not on acamprosate, disulfiram, naltrexone, topiramate, or gabapentin for treatment of AUD and had an elevated Alcohol Use Disorder Identification Test-Consumption (AUDIT-C) score of ≥ 6 (high risk) with an AUD diagnosis or ≥ 8 (severe risk) without a diagnosis. The AUDIT-C scores used in the dashboard are supported by the VA AUD clinician guide as the minimum scores when AUD pharmacotherapy should be offered to patients.18 Prescriptions filled outside the VA were not included in this dashboard.
Data and patient information were collected using the VA Corporate Data Warehouse. To be eligible, veterans needed a valid mailing address within the VISN 21 region and a primary care, mental health, or SUD clinician prescriber visit scheduled between October 1, 2023, and January 31, 2024. Veterans were excluded if they were in hospice, had a 1-year mortality risk score > 50% based on their Care Assessment Need (CAN) score, or facility leadership opted out of project involvement. Patients with both severe renal and hepatic impairments were excluded because they were ineligible for AUD pharmacotherapy. However, veterans with either renal or hepatic impairment (but not both) were included, as they could be potential candidates for ≥ 1 AUD pharmacotherapy option.
Initial correspondence with facilities was initiated through local academic detailers. A local champion was identified for the 1 facility without an academic detailer. Facilities could opt in or out of the project. Approval was provided by the local pharmacy and therapeutics committee, pharmacy, primary care, or psychiatry leadership. Approval process and clinician involvement varied by site.
Education
The selected AUD patient education was designed and approved by the national VA ADS (eappendix). The DTC patient education provided general knowledge about alcohol, including what constitutes a standard amount of alcohol, what is considered heavy drinking, risks of heavy drinking, creating a plan with a clinician to reduce and manage withdrawal symptoms, and additional resources. The DTC was accompanied by a cover letter that included a local facility contact number.
A centralized mailing facility was used for all materials. VA Northern California Health Care System provided the funding to cover the cost of postage. The list of veterans to be contacted was updated on a rolling basis and DTC education was mailed 2 weeks prior to their scheduled prescriber visit.
The eligible cohort of 1260 veterans received DTC education. A comparator group of 2048 veterans that did not receive DTC education was obtained retrospectively by using the same inclusion and exclusion criteria with a scheduled primary care, mental health, or SUD HCP visit from October 1, 2022, to January 31, 2023. The outcomes assessed were within 30 days of the scheduled visit, with the primary outcome as the initiation of AUD-related pharmacotherapy and the secondary outcome as the placement of a consultation for mental health or SUD services. Any consultations sent to Behavioral Health, Addiction, Mental Health, Psychiatric, and SUD services following the HCP visit, within the specified time frame, were used for the secondary outcome.
Matching and Analysis
A 1-to-1 nearest neighbor propensity score (PS) matching without replacement was used to pair the 1260 veterans from the intervention group with similarly scored comparator group veterans for a PS-matched final dataset of 2520 veterans. The PS model was a multivariate logistic regression with the outcome being exposure and comparator group status. Baseline characteristics used in the PS model were age, birth sex, race, facility of care, baseline AUDIT-C score, and days between project start and scheduled appointment. Covariate imbalance for the PS-matched sample was assessed to ensure the standardized mean difference for all covariates fell under a 0.1 threshold (Figure).19
A frequency table was provided to compare the discrete distributions of the baseline characteristics in the intervention and comparator groups. Logistic regression analysis was performed to evaluate the association between DTC education exposure and pharmacotherapy initiation, while controlling for potential confounders. Univariate and multivariate P value results for each variable included in the model were reported along with the multivariate odds ratios (ORs) and their associated 95% CIs. Logistic regression analyses were run for both outcomes. Each model included the exposure and comparator group status as well as the baseline characteristics included in the PS model. Statistical significance was set at P < .05. All statistical analyses were performed with R version 4.2.1.
RESULTS
Two of 7 VISN 21 sites did not participate, and 3 had restrictions on participation. DTC education was mailed about 2 weeks prior to scheduled visit for 1260 veterans; 53.6% identified as White, 37.6% were aged 41 to 60 years, and 79.2% had an AUDIT-C ≥ 8 (Table 1). Of those mailed education, there were 173 no-show appointments (13.7%). Thirty-two veterans (2.5%) in the DTC group and 33 veterans (2.6%) in the comparator group received an AUD-related pharmacotherapy prescription (P = .88) (Table 2). One hundred seventy-one veterans (13.6%) in the DTC group and 160 veterans (12.7%) in the comparator group had a consult placed for mental health or SUD services within 30 days of their appointment (P = .59) (Table 3).
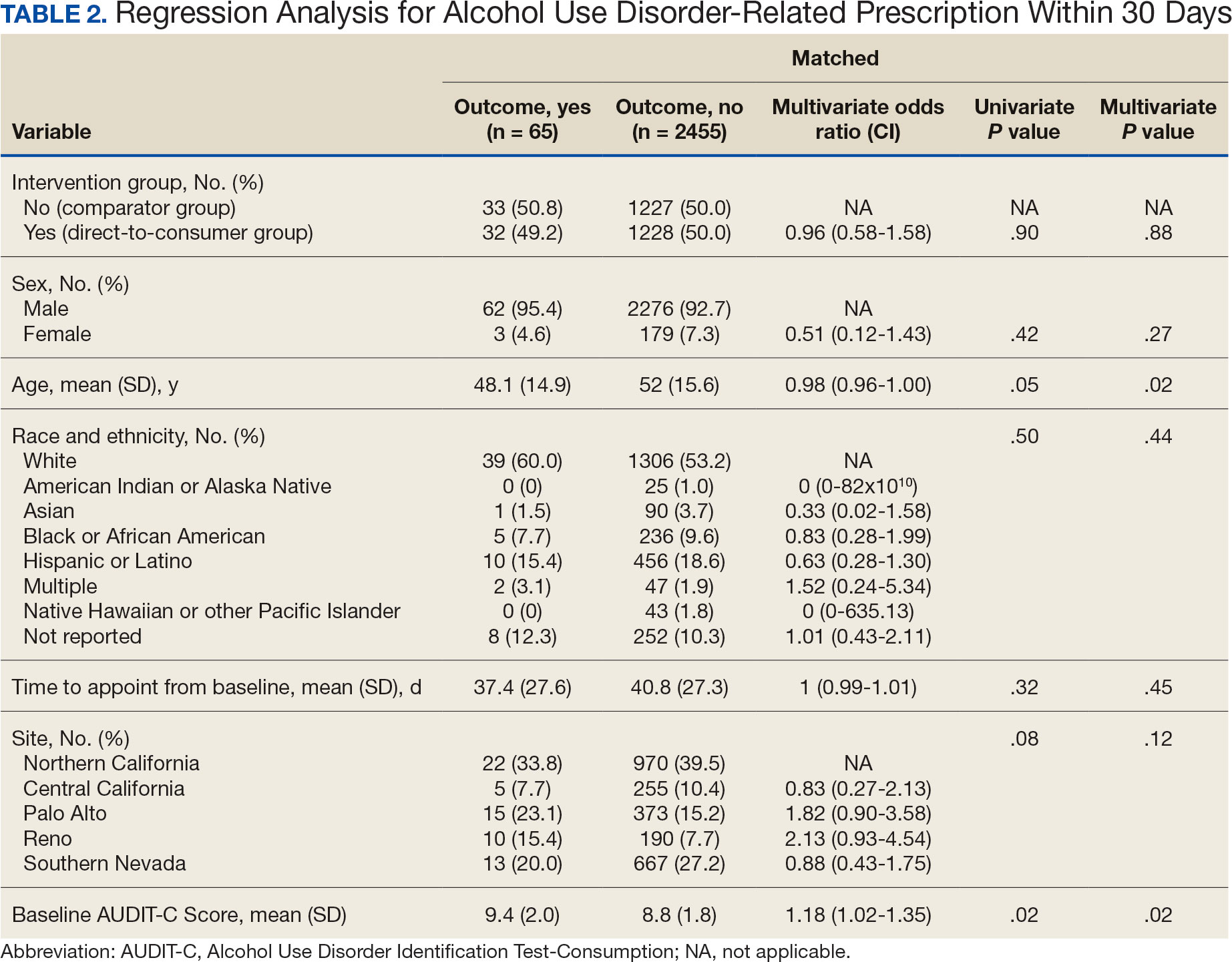
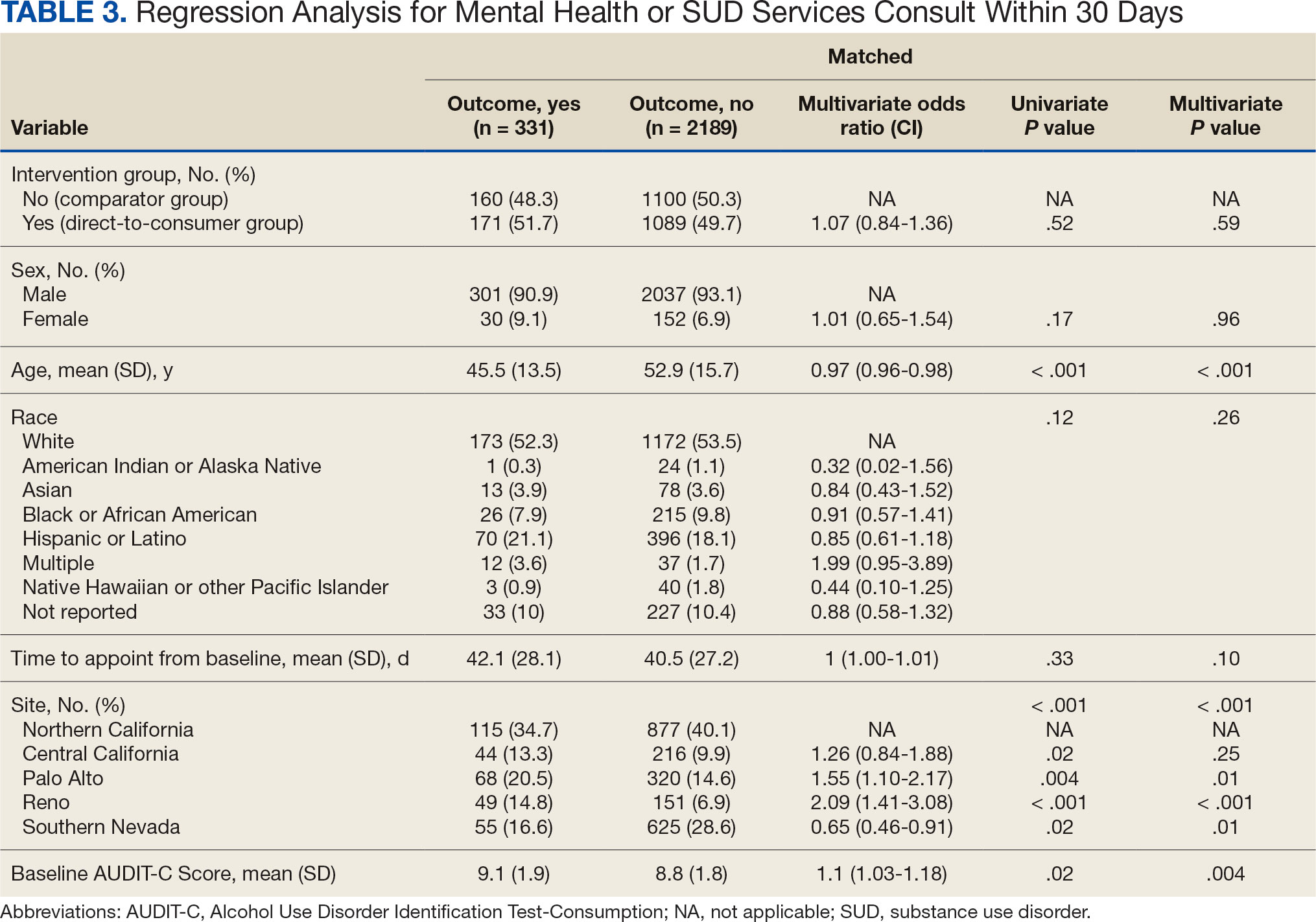
DISCUSSION
This project did not yield statistically significant differences in either the primary or secondary outcomes within the 30-day follow-up window and found limited impact from the DTC educational outreach to veterans. The percentage of veterans that received AUD-related pharmacotherapy or consultations for mental health or SUD services was similarly low in the DTC and comparator groups. These findings suggest that although DTC education may raise awareness, it may not be sufficient on its own to drive changes in prescribing behavior or referral patterns without system-level support.
Addiction is a complex disease faced with stigma and requiring readiness by both the HCP and patient to move forward in support and treatment. The consequences of stigma can be severe: the more stigma perceived by a person with AUD, the less likely they are to seek treatment.20 Stigma may exist even within HCPs and may lead to compromised care including shortened visits, less engagement, and less empathy.19 Cultural attitude towards alcohol use and intoxication can also be influenced through a wide range of sources including social media, movies, music, and television. Studies have shown targeted alcohol marketing may result in the development of positive beliefs about drinking and expand environments where alcohol use is socially acceptable and encouraged.21 These factors can impact drinking behavior, including the onset of drinking, binge drinking, and increased alcohol consumption.22
Three VISN 21 sites in this study had restrictions on or excluded primary care from participation. Leadership at some of these facilities were concerned that primary care teams did not have the bandwidth to take on additional items and/or there was variable primary care readiness for initiating AUD pharmacotherapy. Further attempts should be made to integrate primary care into the process of initiating AUD treatment as significant research suggests that integrated care models for AUD may be associated with improved process and outcome measures of care.23
There are several differences between this quality improvement project and prior research investigating the impact of DTC education for other conditions, such as the EMPOWER randomized controlled trial and VISN 22 project, which both demonstrated effectiveness of DTC education for reducing benzodiazepine use in geriatric veterans. 15,16 These studies focused on reducing or stopping pharmacotherapy use, whereas this project sought to promote the initiation of AUD pharmacotherapy. These studies evaluated outcomes at least 6 months postindex date, whereas this project evaluated outcomes within 30 days postappointment. Furthermore, the educational content varied significantly. Other projects provided patients with information focused on specific medications and interventions, such as benzodiazepine tapering, while this project mailed general information on heavy drinking, its risks, and strategies for cutting back, without mentioning pharmacotherapy. The DTC material used in this project was chosen because it was a preapproved national VA ADS resource, which expedited the project timeline by avoiding the need for additional approvals at each participating site. These differences may impact the observed effectiveness of DTC education in this project, especially regarding the primary outcome.
Strengths and Limitations
This quality improvement project sent a large sample of veterans DTC education in a clinical setting across multiple sites. Additionally, PS matching methods were used to balance covariates between the comparator and DTC education group, thereby simulating a randomized controlled trial and reducing selection bias. The project brought attention to the VISN 21 AUD treatment rates, stimulated conversation across sites about available treatments and resources for AUD, and sparked collaboration between academic detailing, mental health, and primary care services. The time frame for visits was selected during the winter; the National Institute on Alcohol Abuse and Alcoholism notes this is a time when people may be more likely to engage in excessive alcohol consumption than at other times of the year.24
The 30-day time frame for outcomes may have been too short to observe changes in prescribing or referral patterns. Additionally, the comparator group was comprised of veterans seen from October 1, 2022, to January 31, 2023, where seasonal timing may have influenced alcohol consumption behaviors and skewed the results. There were also no-show appointments in the DTC education group (13.7%), though it is likely some patients rescheduled and still received AUD pharmacotherapy within 30 days of the original appointment. Finally, it was not possible to confirm whether a patient opened and read the education that was mailed to them. This may be another reason to explore electronic distribution of DTC education. This all may have contributed to the lack of statistically significant differences in both the primary and secondary outcomes.
There was a high level of variability between facility participation in the project. Two of 7 sites did not participate, and 3 sites restricted primary care engagement. This represents a significant limitation, particularly for the secondary outcome of placing consultations for MH or SUD services. Facilities that only included mental health or SUD HCPs may have resulted in lower consultation rates due to their inherent specialization, reducing the likelihood of self-referrals.
The project may overestimate prescribed AUD pharmacotherapy in the primary outcome due to potential misclassification of medications. While the project adhered to the national VA ADS AUD dashboard’s definition of AUD pharmacotherapy, including acamprosate, disulfiram, naltrexone, topiramate, and gabapentin, some of these medications have multiple indications. For example, gabapentin is commonly prescribed for peripheral neuropathy, and topiramate is used to treat migraines and seizures. The multipurpose use adds uncertainty about whether they were prescribed specifically for AUD treatment, especially in cases where the HCP is responsible for treating a broad range of disease states, as in primary care.
CONCLUSIONS
Results of this quality improvement project did not show a statistically significant difference between patients sent DTC education and the comparator group for the initiation of AUD pharmacotherapy or placement of a consult to mental health or SUD services within 30 days of their scheduled visit. Future studies may seek to implement stricter criteria to confirm the intended use of topiramate and gabapentin, such as looking for keywords in the prescription instructions for use, performing chart reviews, and/or only including these medications if prescribed by a mental health or SUD HCP. Alternatively, future studies may consider limiting the analysis to only FDA-approved AUD medications: acamprosate, disulfiram, and naltrexone. It is vital to continue to enhance primary care HCP readiness to treat AUD, given the existing relationships and trust they often have with patients. Electronic methods for distributing DTC education could also be advantageous, as these methods may have the ability to track whether a message has been opened and read. Despite a lack of statistical significance, this project sparked crucial conversations and collaboration around AUD, available treatments, and addressing potential barriers to connecting patients to care within VISN 21.
- Centers for Disease Control and Prevention. Facts about U.S. deaths from excessive alcohol use. August 6, 2024. Accessed February 5, 2025. https://www.cdc.gov/alcohol/facts-stats/
- State Health Access Data Assistance Center. Escalating alcohol-involved death rates: trends and variation across the nation and in the states from 2006 to 2019. April 19, 2021. Accessed February 5, 2025. https://www.shadac.org/escalating-alcohol-involved-death-rates-trends-and-variation-across-nation-and-states-2006-2019
- National Institute on Alcohol Abuse and Alcoholism. Alcohol- related emergencies and deaths in the United States. Updated November 2024. Accessed February 5, 2025. https://www.niaaa.nih.gov/alcohols-effects-health/alcohol-topics/alcohol-facts-and-statistics/alcohol-related-emergencies-and-deaths-united-states
- Esser MB, Sherk A, Liu Y, Naimi TS. Deaths from excessive alcohol use - United States, 2016- 2021. MMWR Morb Mortal Wkly Rep. 2024;73(8):154-161. doi:10.15585/mmwr.mm7308a1
- Canver BR, Newman RK, Gomez AE. Alcohol Withdrawal Syndrome. In: StatPearls. StatPearls Publishing; 2024.
- National Institute on Alcohol Abuse and Alcoholism. Alcohol treatment in the United States. Updated January 2025. Accessed February 5, 2025. https://www.niaaa.nih.gov/alcohols-effects-health/alcohol-topics/alcohol-facts-and-statistics/alcohol-treatment-united-states
- National Institute on Alcohol Abuse and Alcoholism. Alcohol use disorder (AUD) in the United States: age groups and demographic characteristics. Updated September 2024. Accessed February 5, 2025. https://www.niaaa.nih.gov/alcohols-effects-health/alcohol-topics/alcohol-facts-and-statistics/alcohol-use-disorder-aud-united-states-age-groups-and-demographic-characteristics
- Reus VI, Fochtmann LJ, Bukstein O, et al. The American Psychiatric Association practice guideline for the pharmacological treatment of patients with alcohol use disorder. Am J Psychiatry. 2018;175(1):86-90. doi:10.1176/appi.ajp.2017.1750101
- Blodgett JC, Del Re AC, Maisel NC, Finney JW. A meta-analysis of topiramate’s effects for individuals with alcohol use disorders. Alcohol Clin Exp Res. 2014;38(6):1481-1488. doi:10.1111/acer.12411
- Maisel NC, Blodgett JC, Wilbourne PL, Humphreys K, Finney JW. Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: when are these medications most helpful? Addiction. 2013;108(2):275-293. doi:10.1111/j.1360-0443.2012.04054.x
- Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA. 2014;311(18):1889-1900. doi:10.1001/jama.2014.3628
- US Department of Veterans Affairs, Department of Defense. VA/DoD clinical practice guideline for the management of substance use disorders. August 2021. Accessed February 5, 2025. https://www.healthquality.va.gov/guidelines/MH/sud/VADODSUDCPG.pdf
- Ranney RM, Bernhard PA, Vogt D, et al. Alcohol use and treatment utilization in a national sample of veterans and nonveterans. J Subst Use Addict Treat. 2023;146:208964. doi:10.1016/j.josat.2023.208964
- US Department of Veterans Affairs, Pharmacy Benefit Management Service, Academic Detailing Service. AUD Trend Report. https://vaww.pbi.cdw.va.gov/PBIRS/Pages/ReportViewer.aspx?/GPE/PBM_AD/SSRS/AUD/AUD_TrendReport
- Mendes MA, Smith JP, Marin JK, et al. Reducing benzodiazepine prescribing in older veterans: a direct-to-consumer educational brochure. Fed Pract. 2018;35(9):36-43.
- Tannenbaum C, Martin P, Tamblyn R, Benedetti A, Ahmed S. Reduction of inappropriate benzodiazepine prescriptions among older adults through direct patient education: the EMPOWER cluster randomized trial. JAMA Intern Med. 2014;174(6):890-898. doi:10.1001/jamainternmed.2014.949
- Maloney R, Funmilayo M. Acting on the AUDIT-C: implementation of direct-to-consumer education on unhealth alcohol use. Presented on March 31, 2023; Central Virginia Veterans Affairs Health Care System, Richmond, Virginia.
- US Department of Veterans Affairs, Pharmacy Benefit Management Service. Alcohol use disorder (AUD) – leading the charge in the treatment of AUD: a VA clinician’s guide. February 2022. Accessed February 5, 2025. https://www.pbm.va.gov/PBM/AcademicDetailingService/Documents/508/10-1530_AUD_ClinicianGuide_508Conformant.pdf
- Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res. 2011;46(3):399-424. doi:10.1080/00273171.2011.568786
- National Institute on Alcohol Abuse and Alcoholism. Stigma: overcoming a pervasive barrier to optimal care. Updated January 6, 2025. Accessed February 5, 2025. https://www.niaaa.nih.gov/health-professionals-communities/core-resource-on-alcohol/stigma-overcoming-pervasive-barrier-optimal-care
- Sudhinaraset M, Wigglesworth C, Takeuchi DT. Social and cultural contexts of alcohol use: influences in a socialecological framework. Alcohol Res. 2016;38(1):35-45.
- Tanski SE, McClure AC, Li Z, et al. Cued recall of alcohol advertising on television and underage drinking behavior. JAMA Pediatr. 2015;169(3):264-271. doi:10.1001/jamapediatrics.2014.3345
- Hyland CJ, McDowell MJ, Bain PA, Huskamp HA, Busch AB. Integration of pharmacotherapy for alcohol use disorder treatment in primary care settings: a scoping review. J Subst Abuse Treat. 2023;144:108919. doi:10.1016/j.jsat.2022.108919
- National Institute on Alcohol Abuse and Alcoholism. The truth about holiday spirits. Updated November 2023. Accessed February 5, 2025. ,a href="https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/truth-about-holiday-spirits">https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/truth-about-holiday-spirits
Excessive alcohol use is one of the leading preventable causes of death in the United States, responsible for about 178,000 deaths annually and an average of 488 daily deaths in 2020 and 2021.1Alcohol-related deaths increased by 49% between 2006 and 2019.2 This trend continued during the COVID-19 pandemic, with death certificates that listed alcohol increasing by > 25% from 2019 to 2020, and another 10% in 2021.3 This increase of alcohol-related deaths includes those as a direct result of chronic alcohol use, such as alcoholic cardiomyopathy, alcoholic hepatitis and cirrhosis, and alcohol-induced pancreatitis, as well as a result of acute use such as alcohol poisoning, suicide by exposure to alcohol, and alcohol-impaired driving fatalities.4
Excessive alcohol consumption poses other serious risks, including cases when intake is abruptly reduced without proper management. Alcohol withdrawal syndrome (AWS) can vary in severity, with potentially life-threatening complications such as hallucinations, seizures, and delirium tremens.5
These risks highlight the importance of professional intervention and support, not only to mitigate risks associated with AWS, but provide a pathway towards recovery from alcohol use disorder (AUD).
According to the 2022 National Survey on Drug Use and Health, 28.8 million US adults had AUD in the prior year, yet only 7.6% of these individuals received treatment and an even smaller group (2.2%) received medication-assisted treatment for alcohol.6,7 This is despite American Psychiatric Association guidelines for the pharmacological treatment of patients with AUD, including the use of naltrexone, acamprosate, disulfiram, topiramate, or gabapentin, depending on therapy goals, past medication trials, medication contraindications, and patient preference.8 Several of these medications are approved by the US Food and Drug Administration (FDA) for the treatment of AUD and have support for effectiveness from randomized controlled trials and meta-analyses.9-11
Clinical practice guidelines for the management of substance use disorders (SUDs) from the US Department of Veterans Affairs (VA) and US Department of Defense have strong recommendations for naltrexone and topiramate as first-line pharmacotherapies for moderate to severe AUD. Acamprosate and disulfiram are weak recommendations as alternative options. Gabapentin is a weak recommendation for cases where first-line treatments are contraindicated or ineffective. The guidelines emphasize the importance of a comprehensive approach to AUD treatment, including psychosocial interventions in addition to pharmacotherapy.12
A 2023 national survey found veterans reported higher alcohol consumption than nonveterans.13 At the end of fiscal year 2023, > 4.4 million veterans—6% of Veterans Health Administration patients—had been diagnosed with AUD.14 However, > 87% of these patients nationally, and 88% of Veterans Integrated Service Network (VISN) 21 patients, were not receiving naltrexone, acamprosate, disulfiram, or topiramate as part of their treatment. The VA Academic Detailing Service (ADS) now includes AUD pharmacotherapy as a campaign focus, highlighting its importance. The ADS is a pharmacy educational outreach program that uses unbiased clinical guidelines to promote aligning prescribing behavior with best practices. Academic detailing methods include speaking with health care practitioners (HCPs), and direct-to-consumer (DTC) patient education.
ADS campaigns include DTC educational handouts. Past ADS projects and research using DTC have demonstrated a significant improvement in outcomes and positively influencing patients’ pharmacotherapy treatment. 15,16 A VA quality improvement project found a positive correlation between the initiation of AUD pharmacotherapy and engagement with mental health care following the distribution of AUD DTC patient education. 17 This project aimed to apply the same principles of prior research to explore the use of DTC across multiple facilities within VISN 21 to increase AUD pharmacotherapy. VISN 21 includes VA facilities and clinics across the Pacific Islands, Nevada, and California and serves about 350,000 veterans.
METHODS
A prospective cohort of VISN 21 veterans with or at high risk for AUD was identified using the VA ADS AUD Dashboard. The cohort included those not on acamprosate, disulfiram, naltrexone, topiramate, or gabapentin for treatment of AUD and had an elevated Alcohol Use Disorder Identification Test-Consumption (AUDIT-C) score of ≥ 6 (high risk) with an AUD diagnosis or ≥ 8 (severe risk) without a diagnosis. The AUDIT-C scores used in the dashboard are supported by the VA AUD clinician guide as the minimum scores when AUD pharmacotherapy should be offered to patients.18 Prescriptions filled outside the VA were not included in this dashboard.
Data and patient information were collected using the VA Corporate Data Warehouse. To be eligible, veterans needed a valid mailing address within the VISN 21 region and a primary care, mental health, or SUD clinician prescriber visit scheduled between October 1, 2023, and January 31, 2024. Veterans were excluded if they were in hospice, had a 1-year mortality risk score > 50% based on their Care Assessment Need (CAN) score, or facility leadership opted out of project involvement. Patients with both severe renal and hepatic impairments were excluded because they were ineligible for AUD pharmacotherapy. However, veterans with either renal or hepatic impairment (but not both) were included, as they could be potential candidates for ≥ 1 AUD pharmacotherapy option.
Initial correspondence with facilities was initiated through local academic detailers. A local champion was identified for the 1 facility without an academic detailer. Facilities could opt in or out of the project. Approval was provided by the local pharmacy and therapeutics committee, pharmacy, primary care, or psychiatry leadership. Approval process and clinician involvement varied by site.
Education
The selected AUD patient education was designed and approved by the national VA ADS (eappendix). The DTC patient education provided general knowledge about alcohol, including what constitutes a standard amount of alcohol, what is considered heavy drinking, risks of heavy drinking, creating a plan with a clinician to reduce and manage withdrawal symptoms, and additional resources. The DTC was accompanied by a cover letter that included a local facility contact number.
A centralized mailing facility was used for all materials. VA Northern California Health Care System provided the funding to cover the cost of postage. The list of veterans to be contacted was updated on a rolling basis and DTC education was mailed 2 weeks prior to their scheduled prescriber visit.
The eligible cohort of 1260 veterans received DTC education. A comparator group of 2048 veterans that did not receive DTC education was obtained retrospectively by using the same inclusion and exclusion criteria with a scheduled primary care, mental health, or SUD HCP visit from October 1, 2022, to January 31, 2023. The outcomes assessed were within 30 days of the scheduled visit, with the primary outcome as the initiation of AUD-related pharmacotherapy and the secondary outcome as the placement of a consultation for mental health or SUD services. Any consultations sent to Behavioral Health, Addiction, Mental Health, Psychiatric, and SUD services following the HCP visit, within the specified time frame, were used for the secondary outcome.
Matching and Analysis
A 1-to-1 nearest neighbor propensity score (PS) matching without replacement was used to pair the 1260 veterans from the intervention group with similarly scored comparator group veterans for a PS-matched final dataset of 2520 veterans. The PS model was a multivariate logistic regression with the outcome being exposure and comparator group status. Baseline characteristics used in the PS model were age, birth sex, race, facility of care, baseline AUDIT-C score, and days between project start and scheduled appointment. Covariate imbalance for the PS-matched sample was assessed to ensure the standardized mean difference for all covariates fell under a 0.1 threshold (Figure).19
A frequency table was provided to compare the discrete distributions of the baseline characteristics in the intervention and comparator groups. Logistic regression analysis was performed to evaluate the association between DTC education exposure and pharmacotherapy initiation, while controlling for potential confounders. Univariate and multivariate P value results for each variable included in the model were reported along with the multivariate odds ratios (ORs) and their associated 95% CIs. Logistic regression analyses were run for both outcomes. Each model included the exposure and comparator group status as well as the baseline characteristics included in the PS model. Statistical significance was set at P < .05. All statistical analyses were performed with R version 4.2.1.
RESULTS
Two of 7 VISN 21 sites did not participate, and 3 had restrictions on participation. DTC education was mailed about 2 weeks prior to scheduled visit for 1260 veterans; 53.6% identified as White, 37.6% were aged 41 to 60 years, and 79.2% had an AUDIT-C ≥ 8 (Table 1). Of those mailed education, there were 173 no-show appointments (13.7%). Thirty-two veterans (2.5%) in the DTC group and 33 veterans (2.6%) in the comparator group received an AUD-related pharmacotherapy prescription (P = .88) (Table 2). One hundred seventy-one veterans (13.6%) in the DTC group and 160 veterans (12.7%) in the comparator group had a consult placed for mental health or SUD services within 30 days of their appointment (P = .59) (Table 3).
DISCUSSION
This project did not yield statistically significant differences in either the primary or secondary outcomes within the 30-day follow-up window and found limited impact from the DTC educational outreach to veterans. The percentage of veterans that received AUD-related pharmacotherapy or consultations for mental health or SUD services was similarly low in the DTC and comparator groups. These findings suggest that although DTC education may raise awareness, it may not be sufficient on its own to drive changes in prescribing behavior or referral patterns without system-level support.
Addiction is a complex disease faced with stigma and requiring readiness by both the HCP and patient to move forward in support and treatment. The consequences of stigma can be severe: the more stigma perceived by a person with AUD, the less likely they are to seek treatment.20 Stigma may exist even within HCPs and may lead to compromised care including shortened visits, less engagement, and less empathy.19 Cultural attitude towards alcohol use and intoxication can also be influenced through a wide range of sources including social media, movies, music, and television. Studies have shown targeted alcohol marketing may result in the development of positive beliefs about drinking and expand environments where alcohol use is socially acceptable and encouraged.21 These factors can impact drinking behavior, including the onset of drinking, binge drinking, and increased alcohol consumption.22
Three VISN 21 sites in this study had restrictions on or excluded primary care from participation. Leadership at some of these facilities were concerned that primary care teams did not have the bandwidth to take on additional items and/or there was variable primary care readiness for initiating AUD pharmacotherapy. Further attempts should be made to integrate primary care into the process of initiating AUD treatment as significant research suggests that integrated care models for AUD may be associated with improved process and outcome measures of care.23
There are several differences between this quality improvement project and prior research investigating the impact of DTC education for other conditions, such as the EMPOWER randomized controlled trial and VISN 22 project, which both demonstrated effectiveness of DTC education for reducing benzodiazepine use in geriatric veterans. 15,16 These studies focused on reducing or stopping pharmacotherapy use, whereas this project sought to promote the initiation of AUD pharmacotherapy. These studies evaluated outcomes at least 6 months postindex date, whereas this project evaluated outcomes within 30 days postappointment. Furthermore, the educational content varied significantly. Other projects provided patients with information focused on specific medications and interventions, such as benzodiazepine tapering, while this project mailed general information on heavy drinking, its risks, and strategies for cutting back, without mentioning pharmacotherapy. The DTC material used in this project was chosen because it was a preapproved national VA ADS resource, which expedited the project timeline by avoiding the need for additional approvals at each participating site. These differences may impact the observed effectiveness of DTC education in this project, especially regarding the primary outcome.
Strengths and Limitations
This quality improvement project sent a large sample of veterans DTC education in a clinical setting across multiple sites. Additionally, PS matching methods were used to balance covariates between the comparator and DTC education group, thereby simulating a randomized controlled trial and reducing selection bias. The project brought attention to the VISN 21 AUD treatment rates, stimulated conversation across sites about available treatments and resources for AUD, and sparked collaboration between academic detailing, mental health, and primary care services. The time frame for visits was selected during the winter; the National Institute on Alcohol Abuse and Alcoholism notes this is a time when people may be more likely to engage in excessive alcohol consumption than at other times of the year.24
The 30-day time frame for outcomes may have been too short to observe changes in prescribing or referral patterns. Additionally, the comparator group was comprised of veterans seen from October 1, 2022, to January 31, 2023, where seasonal timing may have influenced alcohol consumption behaviors and skewed the results. There were also no-show appointments in the DTC education group (13.7%), though it is likely some patients rescheduled and still received AUD pharmacotherapy within 30 days of the original appointment. Finally, it was not possible to confirm whether a patient opened and read the education that was mailed to them. This may be another reason to explore electronic distribution of DTC education. This all may have contributed to the lack of statistically significant differences in both the primary and secondary outcomes.
There was a high level of variability between facility participation in the project. Two of 7 sites did not participate, and 3 sites restricted primary care engagement. This represents a significant limitation, particularly for the secondary outcome of placing consultations for MH or SUD services. Facilities that only included mental health or SUD HCPs may have resulted in lower consultation rates due to their inherent specialization, reducing the likelihood of self-referrals.
The project may overestimate prescribed AUD pharmacotherapy in the primary outcome due to potential misclassification of medications. While the project adhered to the national VA ADS AUD dashboard’s definition of AUD pharmacotherapy, including acamprosate, disulfiram, naltrexone, topiramate, and gabapentin, some of these medications have multiple indications. For example, gabapentin is commonly prescribed for peripheral neuropathy, and topiramate is used to treat migraines and seizures. The multipurpose use adds uncertainty about whether they were prescribed specifically for AUD treatment, especially in cases where the HCP is responsible for treating a broad range of disease states, as in primary care.
CONCLUSIONS
Results of this quality improvement project did not show a statistically significant difference between patients sent DTC education and the comparator group for the initiation of AUD pharmacotherapy or placement of a consult to mental health or SUD services within 30 days of their scheduled visit. Future studies may seek to implement stricter criteria to confirm the intended use of topiramate and gabapentin, such as looking for keywords in the prescription instructions for use, performing chart reviews, and/or only including these medications if prescribed by a mental health or SUD HCP. Alternatively, future studies may consider limiting the analysis to only FDA-approved AUD medications: acamprosate, disulfiram, and naltrexone. It is vital to continue to enhance primary care HCP readiness to treat AUD, given the existing relationships and trust they often have with patients. Electronic methods for distributing DTC education could also be advantageous, as these methods may have the ability to track whether a message has been opened and read. Despite a lack of statistical significance, this project sparked crucial conversations and collaboration around AUD, available treatments, and addressing potential barriers to connecting patients to care within VISN 21.
Excessive alcohol use is one of the leading preventable causes of death in the United States, responsible for about 178,000 deaths annually and an average of 488 daily deaths in 2020 and 2021.1Alcohol-related deaths increased by 49% between 2006 and 2019.2 This trend continued during the COVID-19 pandemic, with death certificates that listed alcohol increasing by > 25% from 2019 to 2020, and another 10% in 2021.3 This increase of alcohol-related deaths includes those as a direct result of chronic alcohol use, such as alcoholic cardiomyopathy, alcoholic hepatitis and cirrhosis, and alcohol-induced pancreatitis, as well as a result of acute use such as alcohol poisoning, suicide by exposure to alcohol, and alcohol-impaired driving fatalities.4
Excessive alcohol consumption poses other serious risks, including cases when intake is abruptly reduced without proper management. Alcohol withdrawal syndrome (AWS) can vary in severity, with potentially life-threatening complications such as hallucinations, seizures, and delirium tremens.5
These risks highlight the importance of professional intervention and support, not only to mitigate risks associated with AWS, but provide a pathway towards recovery from alcohol use disorder (AUD).
According to the 2022 National Survey on Drug Use and Health, 28.8 million US adults had AUD in the prior year, yet only 7.6% of these individuals received treatment and an even smaller group (2.2%) received medication-assisted treatment for alcohol.6,7 This is despite American Psychiatric Association guidelines for the pharmacological treatment of patients with AUD, including the use of naltrexone, acamprosate, disulfiram, topiramate, or gabapentin, depending on therapy goals, past medication trials, medication contraindications, and patient preference.8 Several of these medications are approved by the US Food and Drug Administration (FDA) for the treatment of AUD and have support for effectiveness from randomized controlled trials and meta-analyses.9-11
Clinical practice guidelines for the management of substance use disorders (SUDs) from the US Department of Veterans Affairs (VA) and US Department of Defense have strong recommendations for naltrexone and topiramate as first-line pharmacotherapies for moderate to severe AUD. Acamprosate and disulfiram are weak recommendations as alternative options. Gabapentin is a weak recommendation for cases where first-line treatments are contraindicated or ineffective. The guidelines emphasize the importance of a comprehensive approach to AUD treatment, including psychosocial interventions in addition to pharmacotherapy.12
A 2023 national survey found veterans reported higher alcohol consumption than nonveterans.13 At the end of fiscal year 2023, > 4.4 million veterans—6% of Veterans Health Administration patients—had been diagnosed with AUD.14 However, > 87% of these patients nationally, and 88% of Veterans Integrated Service Network (VISN) 21 patients, were not receiving naltrexone, acamprosate, disulfiram, or topiramate as part of their treatment. The VA Academic Detailing Service (ADS) now includes AUD pharmacotherapy as a campaign focus, highlighting its importance. The ADS is a pharmacy educational outreach program that uses unbiased clinical guidelines to promote aligning prescribing behavior with best practices. Academic detailing methods include speaking with health care practitioners (HCPs), and direct-to-consumer (DTC) patient education.
ADS campaigns include DTC educational handouts. Past ADS projects and research using DTC have demonstrated a significant improvement in outcomes and positively influencing patients’ pharmacotherapy treatment. 15,16 A VA quality improvement project found a positive correlation between the initiation of AUD pharmacotherapy and engagement with mental health care following the distribution of AUD DTC patient education. 17 This project aimed to apply the same principles of prior research to explore the use of DTC across multiple facilities within VISN 21 to increase AUD pharmacotherapy. VISN 21 includes VA facilities and clinics across the Pacific Islands, Nevada, and California and serves about 350,000 veterans.
METHODS
A prospective cohort of VISN 21 veterans with or at high risk for AUD was identified using the VA ADS AUD Dashboard. The cohort included those not on acamprosate, disulfiram, naltrexone, topiramate, or gabapentin for treatment of AUD and had an elevated Alcohol Use Disorder Identification Test-Consumption (AUDIT-C) score of ≥ 6 (high risk) with an AUD diagnosis or ≥ 8 (severe risk) without a diagnosis. The AUDIT-C scores used in the dashboard are supported by the VA AUD clinician guide as the minimum scores when AUD pharmacotherapy should be offered to patients.18 Prescriptions filled outside the VA were not included in this dashboard.
Data and patient information were collected using the VA Corporate Data Warehouse. To be eligible, veterans needed a valid mailing address within the VISN 21 region and a primary care, mental health, or SUD clinician prescriber visit scheduled between October 1, 2023, and January 31, 2024. Veterans were excluded if they were in hospice, had a 1-year mortality risk score > 50% based on their Care Assessment Need (CAN) score, or facility leadership opted out of project involvement. Patients with both severe renal and hepatic impairments were excluded because they were ineligible for AUD pharmacotherapy. However, veterans with either renal or hepatic impairment (but not both) were included, as they could be potential candidates for ≥ 1 AUD pharmacotherapy option.
Initial correspondence with facilities was initiated through local academic detailers. A local champion was identified for the 1 facility without an academic detailer. Facilities could opt in or out of the project. Approval was provided by the local pharmacy and therapeutics committee, pharmacy, primary care, or psychiatry leadership. Approval process and clinician involvement varied by site.
Education
The selected AUD patient education was designed and approved by the national VA ADS (eappendix). The DTC patient education provided general knowledge about alcohol, including what constitutes a standard amount of alcohol, what is considered heavy drinking, risks of heavy drinking, creating a plan with a clinician to reduce and manage withdrawal symptoms, and additional resources. The DTC was accompanied by a cover letter that included a local facility contact number.
A centralized mailing facility was used for all materials. VA Northern California Health Care System provided the funding to cover the cost of postage. The list of veterans to be contacted was updated on a rolling basis and DTC education was mailed 2 weeks prior to their scheduled prescriber visit.
The eligible cohort of 1260 veterans received DTC education. A comparator group of 2048 veterans that did not receive DTC education was obtained retrospectively by using the same inclusion and exclusion criteria with a scheduled primary care, mental health, or SUD HCP visit from October 1, 2022, to January 31, 2023. The outcomes assessed were within 30 days of the scheduled visit, with the primary outcome as the initiation of AUD-related pharmacotherapy and the secondary outcome as the placement of a consultation for mental health or SUD services. Any consultations sent to Behavioral Health, Addiction, Mental Health, Psychiatric, and SUD services following the HCP visit, within the specified time frame, were used for the secondary outcome.
Matching and Analysis
A 1-to-1 nearest neighbor propensity score (PS) matching without replacement was used to pair the 1260 veterans from the intervention group with similarly scored comparator group veterans for a PS-matched final dataset of 2520 veterans. The PS model was a multivariate logistic regression with the outcome being exposure and comparator group status. Baseline characteristics used in the PS model were age, birth sex, race, facility of care, baseline AUDIT-C score, and days between project start and scheduled appointment. Covariate imbalance for the PS-matched sample was assessed to ensure the standardized mean difference for all covariates fell under a 0.1 threshold (Figure).19
A frequency table was provided to compare the discrete distributions of the baseline characteristics in the intervention and comparator groups. Logistic regression analysis was performed to evaluate the association between DTC education exposure and pharmacotherapy initiation, while controlling for potential confounders. Univariate and multivariate P value results for each variable included in the model were reported along with the multivariate odds ratios (ORs) and their associated 95% CIs. Logistic regression analyses were run for both outcomes. Each model included the exposure and comparator group status as well as the baseline characteristics included in the PS model. Statistical significance was set at P < .05. All statistical analyses were performed with R version 4.2.1.
RESULTS
Two of 7 VISN 21 sites did not participate, and 3 had restrictions on participation. DTC education was mailed about 2 weeks prior to scheduled visit for 1260 veterans; 53.6% identified as White, 37.6% were aged 41 to 60 years, and 79.2% had an AUDIT-C ≥ 8 (Table 1). Of those mailed education, there were 173 no-show appointments (13.7%). Thirty-two veterans (2.5%) in the DTC group and 33 veterans (2.6%) in the comparator group received an AUD-related pharmacotherapy prescription (P = .88) (Table 2). One hundred seventy-one veterans (13.6%) in the DTC group and 160 veterans (12.7%) in the comparator group had a consult placed for mental health or SUD services within 30 days of their appointment (P = .59) (Table 3).
DISCUSSION
This project did not yield statistically significant differences in either the primary or secondary outcomes within the 30-day follow-up window and found limited impact from the DTC educational outreach to veterans. The percentage of veterans that received AUD-related pharmacotherapy or consultations for mental health or SUD services was similarly low in the DTC and comparator groups. These findings suggest that although DTC education may raise awareness, it may not be sufficient on its own to drive changes in prescribing behavior or referral patterns without system-level support.
Addiction is a complex disease faced with stigma and requiring readiness by both the HCP and patient to move forward in support and treatment. The consequences of stigma can be severe: the more stigma perceived by a person with AUD, the less likely they are to seek treatment.20 Stigma may exist even within HCPs and may lead to compromised care including shortened visits, less engagement, and less empathy.19 Cultural attitude towards alcohol use and intoxication can also be influenced through a wide range of sources including social media, movies, music, and television. Studies have shown targeted alcohol marketing may result in the development of positive beliefs about drinking and expand environments where alcohol use is socially acceptable and encouraged.21 These factors can impact drinking behavior, including the onset of drinking, binge drinking, and increased alcohol consumption.22
Three VISN 21 sites in this study had restrictions on or excluded primary care from participation. Leadership at some of these facilities were concerned that primary care teams did not have the bandwidth to take on additional items and/or there was variable primary care readiness for initiating AUD pharmacotherapy. Further attempts should be made to integrate primary care into the process of initiating AUD treatment as significant research suggests that integrated care models for AUD may be associated with improved process and outcome measures of care.23
There are several differences between this quality improvement project and prior research investigating the impact of DTC education for other conditions, such as the EMPOWER randomized controlled trial and VISN 22 project, which both demonstrated effectiveness of DTC education for reducing benzodiazepine use in geriatric veterans. 15,16 These studies focused on reducing or stopping pharmacotherapy use, whereas this project sought to promote the initiation of AUD pharmacotherapy. These studies evaluated outcomes at least 6 months postindex date, whereas this project evaluated outcomes within 30 days postappointment. Furthermore, the educational content varied significantly. Other projects provided patients with information focused on specific medications and interventions, such as benzodiazepine tapering, while this project mailed general information on heavy drinking, its risks, and strategies for cutting back, without mentioning pharmacotherapy. The DTC material used in this project was chosen because it was a preapproved national VA ADS resource, which expedited the project timeline by avoiding the need for additional approvals at each participating site. These differences may impact the observed effectiveness of DTC education in this project, especially regarding the primary outcome.
Strengths and Limitations
This quality improvement project sent a large sample of veterans DTC education in a clinical setting across multiple sites. Additionally, PS matching methods were used to balance covariates between the comparator and DTC education group, thereby simulating a randomized controlled trial and reducing selection bias. The project brought attention to the VISN 21 AUD treatment rates, stimulated conversation across sites about available treatments and resources for AUD, and sparked collaboration between academic detailing, mental health, and primary care services. The time frame for visits was selected during the winter; the National Institute on Alcohol Abuse and Alcoholism notes this is a time when people may be more likely to engage in excessive alcohol consumption than at other times of the year.24
The 30-day time frame for outcomes may have been too short to observe changes in prescribing or referral patterns. Additionally, the comparator group was comprised of veterans seen from October 1, 2022, to January 31, 2023, where seasonal timing may have influenced alcohol consumption behaviors and skewed the results. There were also no-show appointments in the DTC education group (13.7%), though it is likely some patients rescheduled and still received AUD pharmacotherapy within 30 days of the original appointment. Finally, it was not possible to confirm whether a patient opened and read the education that was mailed to them. This may be another reason to explore electronic distribution of DTC education. This all may have contributed to the lack of statistically significant differences in both the primary and secondary outcomes.
There was a high level of variability between facility participation in the project. Two of 7 sites did not participate, and 3 sites restricted primary care engagement. This represents a significant limitation, particularly for the secondary outcome of placing consultations for MH or SUD services. Facilities that only included mental health or SUD HCPs may have resulted in lower consultation rates due to their inherent specialization, reducing the likelihood of self-referrals.
The project may overestimate prescribed AUD pharmacotherapy in the primary outcome due to potential misclassification of medications. While the project adhered to the national VA ADS AUD dashboard’s definition of AUD pharmacotherapy, including acamprosate, disulfiram, naltrexone, topiramate, and gabapentin, some of these medications have multiple indications. For example, gabapentin is commonly prescribed for peripheral neuropathy, and topiramate is used to treat migraines and seizures. The multipurpose use adds uncertainty about whether they were prescribed specifically for AUD treatment, especially in cases where the HCP is responsible for treating a broad range of disease states, as in primary care.
CONCLUSIONS
Results of this quality improvement project did not show a statistically significant difference between patients sent DTC education and the comparator group for the initiation of AUD pharmacotherapy or placement of a consult to mental health or SUD services within 30 days of their scheduled visit. Future studies may seek to implement stricter criteria to confirm the intended use of topiramate and gabapentin, such as looking for keywords in the prescription instructions for use, performing chart reviews, and/or only including these medications if prescribed by a mental health or SUD HCP. Alternatively, future studies may consider limiting the analysis to only FDA-approved AUD medications: acamprosate, disulfiram, and naltrexone. It is vital to continue to enhance primary care HCP readiness to treat AUD, given the existing relationships and trust they often have with patients. Electronic methods for distributing DTC education could also be advantageous, as these methods may have the ability to track whether a message has been opened and read. Despite a lack of statistical significance, this project sparked crucial conversations and collaboration around AUD, available treatments, and addressing potential barriers to connecting patients to care within VISN 21.
- Centers for Disease Control and Prevention. Facts about U.S. deaths from excessive alcohol use. August 6, 2024. Accessed February 5, 2025. https://www.cdc.gov/alcohol/facts-stats/
- State Health Access Data Assistance Center. Escalating alcohol-involved death rates: trends and variation across the nation and in the states from 2006 to 2019. April 19, 2021. Accessed February 5, 2025. https://www.shadac.org/escalating-alcohol-involved-death-rates-trends-and-variation-across-nation-and-states-2006-2019
- National Institute on Alcohol Abuse and Alcoholism. Alcohol- related emergencies and deaths in the United States. Updated November 2024. Accessed February 5, 2025. https://www.niaaa.nih.gov/alcohols-effects-health/alcohol-topics/alcohol-facts-and-statistics/alcohol-related-emergencies-and-deaths-united-states
- Esser MB, Sherk A, Liu Y, Naimi TS. Deaths from excessive alcohol use - United States, 2016- 2021. MMWR Morb Mortal Wkly Rep. 2024;73(8):154-161. doi:10.15585/mmwr.mm7308a1
- Canver BR, Newman RK, Gomez AE. Alcohol Withdrawal Syndrome. In: StatPearls. StatPearls Publishing; 2024.
- National Institute on Alcohol Abuse and Alcoholism. Alcohol treatment in the United States. Updated January 2025. Accessed February 5, 2025. https://www.niaaa.nih.gov/alcohols-effects-health/alcohol-topics/alcohol-facts-and-statistics/alcohol-treatment-united-states
- National Institute on Alcohol Abuse and Alcoholism. Alcohol use disorder (AUD) in the United States: age groups and demographic characteristics. Updated September 2024. Accessed February 5, 2025. https://www.niaaa.nih.gov/alcohols-effects-health/alcohol-topics/alcohol-facts-and-statistics/alcohol-use-disorder-aud-united-states-age-groups-and-demographic-characteristics
- Reus VI, Fochtmann LJ, Bukstein O, et al. The American Psychiatric Association practice guideline for the pharmacological treatment of patients with alcohol use disorder. Am J Psychiatry. 2018;175(1):86-90. doi:10.1176/appi.ajp.2017.1750101
- Blodgett JC, Del Re AC, Maisel NC, Finney JW. A meta-analysis of topiramate’s effects for individuals with alcohol use disorders. Alcohol Clin Exp Res. 2014;38(6):1481-1488. doi:10.1111/acer.12411
- Maisel NC, Blodgett JC, Wilbourne PL, Humphreys K, Finney JW. Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: when are these medications most helpful? Addiction. 2013;108(2):275-293. doi:10.1111/j.1360-0443.2012.04054.x
- Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA. 2014;311(18):1889-1900. doi:10.1001/jama.2014.3628
- US Department of Veterans Affairs, Department of Defense. VA/DoD clinical practice guideline for the management of substance use disorders. August 2021. Accessed February 5, 2025. https://www.healthquality.va.gov/guidelines/MH/sud/VADODSUDCPG.pdf
- Ranney RM, Bernhard PA, Vogt D, et al. Alcohol use and treatment utilization in a national sample of veterans and nonveterans. J Subst Use Addict Treat. 2023;146:208964. doi:10.1016/j.josat.2023.208964
- US Department of Veterans Affairs, Pharmacy Benefit Management Service, Academic Detailing Service. AUD Trend Report. https://vaww.pbi.cdw.va.gov/PBIRS/Pages/ReportViewer.aspx?/GPE/PBM_AD/SSRS/AUD/AUD_TrendReport
- Mendes MA, Smith JP, Marin JK, et al. Reducing benzodiazepine prescribing in older veterans: a direct-to-consumer educational brochure. Fed Pract. 2018;35(9):36-43.
- Tannenbaum C, Martin P, Tamblyn R, Benedetti A, Ahmed S. Reduction of inappropriate benzodiazepine prescriptions among older adults through direct patient education: the EMPOWER cluster randomized trial. JAMA Intern Med. 2014;174(6):890-898. doi:10.1001/jamainternmed.2014.949
- Maloney R, Funmilayo M. Acting on the AUDIT-C: implementation of direct-to-consumer education on unhealth alcohol use. Presented on March 31, 2023; Central Virginia Veterans Affairs Health Care System, Richmond, Virginia.
- US Department of Veterans Affairs, Pharmacy Benefit Management Service. Alcohol use disorder (AUD) – leading the charge in the treatment of AUD: a VA clinician’s guide. February 2022. Accessed February 5, 2025. https://www.pbm.va.gov/PBM/AcademicDetailingService/Documents/508/10-1530_AUD_ClinicianGuide_508Conformant.pdf
- Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res. 2011;46(3):399-424. doi:10.1080/00273171.2011.568786
- National Institute on Alcohol Abuse and Alcoholism. Stigma: overcoming a pervasive barrier to optimal care. Updated January 6, 2025. Accessed February 5, 2025. https://www.niaaa.nih.gov/health-professionals-communities/core-resource-on-alcohol/stigma-overcoming-pervasive-barrier-optimal-care
- Sudhinaraset M, Wigglesworth C, Takeuchi DT. Social and cultural contexts of alcohol use: influences in a socialecological framework. Alcohol Res. 2016;38(1):35-45.
- Tanski SE, McClure AC, Li Z, et al. Cued recall of alcohol advertising on television and underage drinking behavior. JAMA Pediatr. 2015;169(3):264-271. doi:10.1001/jamapediatrics.2014.3345
- Hyland CJ, McDowell MJ, Bain PA, Huskamp HA, Busch AB. Integration of pharmacotherapy for alcohol use disorder treatment in primary care settings: a scoping review. J Subst Abuse Treat. 2023;144:108919. doi:10.1016/j.jsat.2022.108919
- National Institute on Alcohol Abuse and Alcoholism. The truth about holiday spirits. Updated November 2023. Accessed February 5, 2025. ,a href="https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/truth-about-holiday-spirits">https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/truth-about-holiday-spirits
- Centers for Disease Control and Prevention. Facts about U.S. deaths from excessive alcohol use. August 6, 2024. Accessed February 5, 2025. https://www.cdc.gov/alcohol/facts-stats/
- State Health Access Data Assistance Center. Escalating alcohol-involved death rates: trends and variation across the nation and in the states from 2006 to 2019. April 19, 2021. Accessed February 5, 2025. https://www.shadac.org/escalating-alcohol-involved-death-rates-trends-and-variation-across-nation-and-states-2006-2019
- National Institute on Alcohol Abuse and Alcoholism. Alcohol- related emergencies and deaths in the United States. Updated November 2024. Accessed February 5, 2025. https://www.niaaa.nih.gov/alcohols-effects-health/alcohol-topics/alcohol-facts-and-statistics/alcohol-related-emergencies-and-deaths-united-states
- Esser MB, Sherk A, Liu Y, Naimi TS. Deaths from excessive alcohol use - United States, 2016- 2021. MMWR Morb Mortal Wkly Rep. 2024;73(8):154-161. doi:10.15585/mmwr.mm7308a1
- Canver BR, Newman RK, Gomez AE. Alcohol Withdrawal Syndrome. In: StatPearls. StatPearls Publishing; 2024.
- National Institute on Alcohol Abuse and Alcoholism. Alcohol treatment in the United States. Updated January 2025. Accessed February 5, 2025. https://www.niaaa.nih.gov/alcohols-effects-health/alcohol-topics/alcohol-facts-and-statistics/alcohol-treatment-united-states
- National Institute on Alcohol Abuse and Alcoholism. Alcohol use disorder (AUD) in the United States: age groups and demographic characteristics. Updated September 2024. Accessed February 5, 2025. https://www.niaaa.nih.gov/alcohols-effects-health/alcohol-topics/alcohol-facts-and-statistics/alcohol-use-disorder-aud-united-states-age-groups-and-demographic-characteristics
- Reus VI, Fochtmann LJ, Bukstein O, et al. The American Psychiatric Association practice guideline for the pharmacological treatment of patients with alcohol use disorder. Am J Psychiatry. 2018;175(1):86-90. doi:10.1176/appi.ajp.2017.1750101
- Blodgett JC, Del Re AC, Maisel NC, Finney JW. A meta-analysis of topiramate’s effects for individuals with alcohol use disorders. Alcohol Clin Exp Res. 2014;38(6):1481-1488. doi:10.1111/acer.12411
- Maisel NC, Blodgett JC, Wilbourne PL, Humphreys K, Finney JW. Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: when are these medications most helpful? Addiction. 2013;108(2):275-293. doi:10.1111/j.1360-0443.2012.04054.x
- Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA. 2014;311(18):1889-1900. doi:10.1001/jama.2014.3628
- US Department of Veterans Affairs, Department of Defense. VA/DoD clinical practice guideline for the management of substance use disorders. August 2021. Accessed February 5, 2025. https://www.healthquality.va.gov/guidelines/MH/sud/VADODSUDCPG.pdf
- Ranney RM, Bernhard PA, Vogt D, et al. Alcohol use and treatment utilization in a national sample of veterans and nonveterans. J Subst Use Addict Treat. 2023;146:208964. doi:10.1016/j.josat.2023.208964
- US Department of Veterans Affairs, Pharmacy Benefit Management Service, Academic Detailing Service. AUD Trend Report. https://vaww.pbi.cdw.va.gov/PBIRS/Pages/ReportViewer.aspx?/GPE/PBM_AD/SSRS/AUD/AUD_TrendReport
- Mendes MA, Smith JP, Marin JK, et al. Reducing benzodiazepine prescribing in older veterans: a direct-to-consumer educational brochure. Fed Pract. 2018;35(9):36-43.
- Tannenbaum C, Martin P, Tamblyn R, Benedetti A, Ahmed S. Reduction of inappropriate benzodiazepine prescriptions among older adults through direct patient education: the EMPOWER cluster randomized trial. JAMA Intern Med. 2014;174(6):890-898. doi:10.1001/jamainternmed.2014.949
- Maloney R, Funmilayo M. Acting on the AUDIT-C: implementation of direct-to-consumer education on unhealth alcohol use. Presented on March 31, 2023; Central Virginia Veterans Affairs Health Care System, Richmond, Virginia.
- US Department of Veterans Affairs, Pharmacy Benefit Management Service. Alcohol use disorder (AUD) – leading the charge in the treatment of AUD: a VA clinician’s guide. February 2022. Accessed February 5, 2025. https://www.pbm.va.gov/PBM/AcademicDetailingService/Documents/508/10-1530_AUD_ClinicianGuide_508Conformant.pdf
- Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res. 2011;46(3):399-424. doi:10.1080/00273171.2011.568786
- National Institute on Alcohol Abuse and Alcoholism. Stigma: overcoming a pervasive barrier to optimal care. Updated January 6, 2025. Accessed February 5, 2025. https://www.niaaa.nih.gov/health-professionals-communities/core-resource-on-alcohol/stigma-overcoming-pervasive-barrier-optimal-care
- Sudhinaraset M, Wigglesworth C, Takeuchi DT. Social and cultural contexts of alcohol use: influences in a socialecological framework. Alcohol Res. 2016;38(1):35-45.
- Tanski SE, McClure AC, Li Z, et al. Cued recall of alcohol advertising on television and underage drinking behavior. JAMA Pediatr. 2015;169(3):264-271. doi:10.1001/jamapediatrics.2014.3345
- Hyland CJ, McDowell MJ, Bain PA, Huskamp HA, Busch AB. Integration of pharmacotherapy for alcohol use disorder treatment in primary care settings: a scoping review. J Subst Abuse Treat. 2023;144:108919. doi:10.1016/j.jsat.2022.108919
- National Institute on Alcohol Abuse and Alcoholism. The truth about holiday spirits. Updated November 2023. Accessed February 5, 2025. ,a href="https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/truth-about-holiday-spirits">https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/truth-about-holiday-spirits
Impact of Multisite Patient Education on Pharmacotherapy for Veterans With Alcohol Use Disorder
Impact of Multisite Patient Education on Pharmacotherapy for Veterans With Alcohol Use Disorder
Improved Pharmacogenomic Testing Process for Veterans in Outpatient Settings by Clinical Pharmacist Practitioners
Peer-review, evidence-based, detailed gene/drug clinical practice guidelines suggest that genetic variations can impact how individuals metabolize medications, which is sometimes included in medication prescribing information.1-3 Pharmacogenomic testing identifies genetic markers so medication selection and dosing can be tailored to each individual by identifying whether a specific medication is likely to be safe and effective prior to prescribing.4
Pharmacogenomics can be a valuable tool for personalizing medicine but has had suboptimal implementation since its discovery. The US Department of Veterans Affairs (VA) health care system reviewed the implementation of the Pharmacogenomic Testing for Veterans (PHASER) program. This review identified clinician barriers pre- and post-PHASER program implementation; staffing issues, competing clinical priorities, and inadequate PHASER program resources were the most frequently reported barriers to implementation of pharmacogenomic testing.5
Another evaluation of the implementation of the PHASER program that surveyed VA patients found that patients could be separated into 3 groups. Acceptors of pharmacogenomic testing emphasized potential health benefits of testing. Patients that declined testing often cited concerns for genetic information affecting insurance coverage, being misused, or being susceptible to data breach. The third group—identified as contemplators—reported the need for clinician outreach to impact their decision on whether or not to receive pharmacogenomic testing.6 These studies suggest that removing barriers by providing ample pharmacogenomics resources to clinicians, in addition to detailed training on how to offer and follow up with patients regarding pharmacogenomic testing, is crucial to successful implementation of the PHASER program.
PHASER
In 2019, the VA began working with Sanford Health to establish the PHASER program and offer pharmacogenomic testing. PHASER has since expanded to 25 VA medical centers, including the VA Central Ohio Healthcare System (VACOHCS).7,8 Pharmacogenomic testing through PHASER is conducted using a standardized laboratory panel that includes 12 different medication classes.9 The drug classes include certain anti-infective, anticoagulant, antiplatelet, cardiovascular, cholesterol, gastrointestinal, mental health, neurological, oncology, pain, transplant, and other miscellaneous medications. Medications are correlated to each class and assessed for therapeutic impacts based on gene panel results.
Clinical recommendations for medication-gene interactions can range from monitoring for increased risk of adverse effects or therapeutic failure to recommending avoiding a medication. For example, patients who test positive for the HLA-B gene have significantly increased risk of hypersensitivity to abacavir, an HIV treatment.10
Similarly, patients who cannot adequately metabolize cytochrome P450 2C19 should consider avoiding clopidogrel as they are unlikely to convert clopidogrel to its active prodrug, which reduces its effectiveness.11 Pharmacists can play a critical role educating patients about pharmacogenomic testing, especially within hematology and oncology.12 Patients can benefit from this testing even if they are not currently taking medications with known concerns as they could be prescribed in the future. The SLCO1B1 gene-drug test, for example, can identify risk for statin-associated muscle symptoms.13
Clinical pharmacist practitioners (CPPs) can increase access to genetic testing because they interact with patients in a variety of settings and can order this laboratory test.12,14 Recent research has demonstrated that most VA patients carry ≥ 1 genetic variant that may influence medication decisions and that half of veterans are prescribed a medication with known gene-drug interactions.15 CPP ordering of pharmacogenomic tests at the VACOHCS outpatient clinic was evaluated through collection of baseline data from March 8, 2023, to September 8, 2023. A goal was identified to increase orders by 50% for a patient care quality improvement initiative and use CPPs to increase access to pharmacogenomic testing. The purpose of this quality improvement initiative was to expand access to pharmacogenomic testing through process implementation and improvement within CPP-led clinic settings.
Gap Analysis
Lean Six Sigma A3 methodology was used to identify ways to increase the use of pharmacogenomic testing for veterans at VACOHCS and develop an improved process for increased ordering of pharmacogenomic testing. Lean Six Sigma A3 methodology is a stepwise approach to process improvement that helps identify gaps in efficiency, sustainable changes, and eliminate waste.16 Baseline data were collected from March 8, 2023, to September 8, 2023, to determine the frequency of CPPs ordering pharmacogenomic laboratory panels during clinic appointments. The ordering of pharmacogenomic panels was monitored by the VACOHCS PHASER coordinator.
CPPs were surveyed to identify perceived barriers to PHASER implementation. A gap analysis was conducted using Lean Six Sigma A3 methodology. Gap analyses use lean tools such as a Fishbone Diagram to illustrate and identify the gap between current state and ideal state. (Figure 1).The following barriers were identified: lack of clinician education materials, lack of a standardized patient screening process, time constraints on patient education and ordering, higher priority clinical needs, forgetting to order, lack of comfort with pharmacogenomics ordering and education, lack of support for the initiative, and increased workload and burnout. Among these perceived barriers, higher priority clinical needs, forgetting to order, and time constraints ranked highest in importance among CPPs.
In line with Lean Six Sigma A3 methodology, several tests of change were used to improve pharmacogenomic testing ordering. These changes focused on increasing patient and clinician awareness, facilitating discussion, educating clinicians, and simplifying documentation to ease time constraints. Several strategies were employed postimplementation (Figure 2). Prefilled templates simplified documentation. These templates helped identify patients without pharmacogenomic testing, provided reminders, and saved documentation time during visits. CPPs also received training and materials on PHASER ordering and documentation within encounter notes. Additionally, patient-directed advertisements were displayed in CPP examination rooms to help inspire and facilitate discussion between veterans and CPPs.
Process Improvement Data
The quality improvement project goal was to increase PHASER orders by 50% after 3 months. PHASER orders increased from 87 at baseline (March 8, 2023, to September 8, 2023) to 196 during the intervention (November 16, 2023, to February 16, 2024), a 125% increase. Changes were consistent and sustained with 65 orders the first month, 67 orders the second month, and 64 orders the third month.
Discussion
Using Lean Six Sigma A3 methodology for a quality improvement process to increase PHASER orders by CPPs revealed barriers and guided potential solutions to overcome these barriers. Interventions included additional CPP training and ordering, tools for easier identification of potential patients, documentation best practices, patient-directed advertisements to facilitate conversations. These interventions required about 8 hours for preparation, distribution, development, and interpretation of surveys, education, and documentation materials. The financial impact of these interventions was already included in allotted office materials budgeted and provided. Additional funding was not needed to provide patient-directed advertisements or education materials. The VACOHCS pharmacogenomics CPP discusses PHASER test results with patients at a separate appointment.
Future directions include educating other CPPs to assist in discussing results with veterans. Overall, the changes implemented to improve the PHASER ordering process were low effort and exemplify the ease of streamlining future initiatives, allowing for sustained optimal implementation of pharmacogenomic testing.
Conclusions
A quality improvement initiative resulted in increased PHASER orders and a clearly defined process, allowing for a continued increase and sustained support. Perceived barriers were identified, and the changes implemented were often low effort but exhibited a sustained impact. The insights gleaned from this process will shape future process development initiatives and continue to sustain pharmacogenomic testing ordering by CPPs. This process will be extended to other VACOHCS clinical departments to further support increased access to pharmacogenomic testing, reduce medication trial and error, and reduce hospitalizations from adverse effects for veterans.
Cecchin E, Stocco G. Pharmacogenomics and personalized medicine. Genes (Basel). 2020;11(6):679. doi:10.3390/genes11060679
Guidelines. CPIC. Accessed April 16, 2025. https://cpicpgx.org/guidelines/
PharmGKB. PharmGKB. 2025. Accessed April 16, 2025. https://www.pharmgkb.org
Centers for Disease Control and Prevention. Pharmacogenomics. Updated November 13, 2024. Accessed April 16, 2024. https://www.cdc.gov/genomics-and-health/pharmacogenomics/
Dong OM, Roberts MC, Wu RR, et al. Evaluation of the Veterans Affairs Pharmacogenomic Testing for Veterans (PHASER) clinical program at initial test sites. Pharmacogenomics. 2021;22(17):1121-1133. doi:10.2217/pgs-2021-0089
Melendez K, Gutierrez-Meza D, Gavin KL, et al. Patient perspectives of barriers and facilitators for the uptake of pharmacogenomic testing in Veterans Affairs’ pharmacogenomic testing for the veterans (PHASER) program. J Pers Med. 2023;13(9):1367. doi:10.3390/jpm13091367
Sanford Health Imagenetics. FREQUENTLY ASKED QUESTIONS (FAQs) about the “Pharmacogenomic Teting for Vetans” (PHASER) Program. US Department of Veterans Affairs. December 20, 2019. Accessed April 16, 2025. https://www.va.gov/opa/publications/factsheets/PHASER-FLYER-VA-Patient-FAQ.pdf
Peterson H. PHASER program testing informs how you respond to medicines. VA News. September 6, 2022. Accessed April 16, 2025. https://news.va.gov/108091/phaser-program-testing-respond-medicines/
Pharmacogenomics (PGx). Sanford Health Imagenetics. 2025. Accessed April 16, 2025. https://imagenetics.sanfordhealth.org/pharmacogenomics/
Martin MA, Hoffman JM, Freimuth RR, et al. Clinical pharmacogenetics implementation consortium guidelines for HLA-B genotype and abacavir dosing: 2014 update. Clin Pharmacol Ther. 2014;95(5):499-500. doi:10.1038/clpt.2014.38
Lee CR, Luzum JA, Sangkuhl K, et al. Clinical pharmacogenetics implementation consortium guideline for CYP2C19 genotype and clopidogrel therapy: 2022 update. Clin Pharmacol Ther. 2022;112(5):959-967. doi:10.1002/cpt.2526
Dreischmeier E, Hecht H, Crocker E, et al. Integration of a clinical pharmacist practitioner-led pharmacogenomics service in a Veterans Affairs hematology/oncology clinic. Am J Health Syst Pharm. 2024;81(19):e634-e639. doi:10.1093/ajhp/zxae122
Tomcsanyi KM, Tran KA, Bates J, et al. Veterans Health Administration: implementation of pharmacogenomic clinical decision support with statin medications and the SLCO1B1 gene as an exemplar. Am J Health Syst Pharm. 2023;80(16):1082-1089. doi:10.1093/ajhp/zxad111
Gammal RS, Lee YM, Petry NJ, et al. Pharmacists leading the way to precision medicine: updates to the core pharmacist competencies in genomics. Am J Pharm Educ. 2022;86(4):8634. doi:10.5688/ajpe8634
Chanfreau-Coffinier C, Hull LE, Lynch JA, et al. Projected prevalence of actionable pharmacogenetic variants and level A drugs prescribed among US Veterans Health Administration pharmacy users. JAMA Netw Open. 2019;2(6):e195345. doi:10.1001/jamanetworkopen.2019.5345
Shaffie S, Shahbazi S. The McGraw-Hill 36-Hour Course: Lean Six Sigma. McGraw-Hill; 2012.
Peer-review, evidence-based, detailed gene/drug clinical practice guidelines suggest that genetic variations can impact how individuals metabolize medications, which is sometimes included in medication prescribing information.1-3 Pharmacogenomic testing identifies genetic markers so medication selection and dosing can be tailored to each individual by identifying whether a specific medication is likely to be safe and effective prior to prescribing.4
Pharmacogenomics can be a valuable tool for personalizing medicine but has had suboptimal implementation since its discovery. The US Department of Veterans Affairs (VA) health care system reviewed the implementation of the Pharmacogenomic Testing for Veterans (PHASER) program. This review identified clinician barriers pre- and post-PHASER program implementation; staffing issues, competing clinical priorities, and inadequate PHASER program resources were the most frequently reported barriers to implementation of pharmacogenomic testing.5
Another evaluation of the implementation of the PHASER program that surveyed VA patients found that patients could be separated into 3 groups. Acceptors of pharmacogenomic testing emphasized potential health benefits of testing. Patients that declined testing often cited concerns for genetic information affecting insurance coverage, being misused, or being susceptible to data breach. The third group—identified as contemplators—reported the need for clinician outreach to impact their decision on whether or not to receive pharmacogenomic testing.6 These studies suggest that removing barriers by providing ample pharmacogenomics resources to clinicians, in addition to detailed training on how to offer and follow up with patients regarding pharmacogenomic testing, is crucial to successful implementation of the PHASER program.
PHASER
In 2019, the VA began working with Sanford Health to establish the PHASER program and offer pharmacogenomic testing. PHASER has since expanded to 25 VA medical centers, including the VA Central Ohio Healthcare System (VACOHCS).7,8 Pharmacogenomic testing through PHASER is conducted using a standardized laboratory panel that includes 12 different medication classes.9 The drug classes include certain anti-infective, anticoagulant, antiplatelet, cardiovascular, cholesterol, gastrointestinal, mental health, neurological, oncology, pain, transplant, and other miscellaneous medications. Medications are correlated to each class and assessed for therapeutic impacts based on gene panel results.
Clinical recommendations for medication-gene interactions can range from monitoring for increased risk of adverse effects or therapeutic failure to recommending avoiding a medication. For example, patients who test positive for the HLA-B gene have significantly increased risk of hypersensitivity to abacavir, an HIV treatment.10
Similarly, patients who cannot adequately metabolize cytochrome P450 2C19 should consider avoiding clopidogrel as they are unlikely to convert clopidogrel to its active prodrug, which reduces its effectiveness.11 Pharmacists can play a critical role educating patients about pharmacogenomic testing, especially within hematology and oncology.12 Patients can benefit from this testing even if they are not currently taking medications with known concerns as they could be prescribed in the future. The SLCO1B1 gene-drug test, for example, can identify risk for statin-associated muscle symptoms.13
Clinical pharmacist practitioners (CPPs) can increase access to genetic testing because they interact with patients in a variety of settings and can order this laboratory test.12,14 Recent research has demonstrated that most VA patients carry ≥ 1 genetic variant that may influence medication decisions and that half of veterans are prescribed a medication with known gene-drug interactions.15 CPP ordering of pharmacogenomic tests at the VACOHCS outpatient clinic was evaluated through collection of baseline data from March 8, 2023, to September 8, 2023. A goal was identified to increase orders by 50% for a patient care quality improvement initiative and use CPPs to increase access to pharmacogenomic testing. The purpose of this quality improvement initiative was to expand access to pharmacogenomic testing through process implementation and improvement within CPP-led clinic settings.
Gap Analysis
Lean Six Sigma A3 methodology was used to identify ways to increase the use of pharmacogenomic testing for veterans at VACOHCS and develop an improved process for increased ordering of pharmacogenomic testing. Lean Six Sigma A3 methodology is a stepwise approach to process improvement that helps identify gaps in efficiency, sustainable changes, and eliminate waste.16 Baseline data were collected from March 8, 2023, to September 8, 2023, to determine the frequency of CPPs ordering pharmacogenomic laboratory panels during clinic appointments. The ordering of pharmacogenomic panels was monitored by the VACOHCS PHASER coordinator.
CPPs were surveyed to identify perceived barriers to PHASER implementation. A gap analysis was conducted using Lean Six Sigma A3 methodology. Gap analyses use lean tools such as a Fishbone Diagram to illustrate and identify the gap between current state and ideal state. (Figure 1).The following barriers were identified: lack of clinician education materials, lack of a standardized patient screening process, time constraints on patient education and ordering, higher priority clinical needs, forgetting to order, lack of comfort with pharmacogenomics ordering and education, lack of support for the initiative, and increased workload and burnout. Among these perceived barriers, higher priority clinical needs, forgetting to order, and time constraints ranked highest in importance among CPPs.
In line with Lean Six Sigma A3 methodology, several tests of change were used to improve pharmacogenomic testing ordering. These changes focused on increasing patient and clinician awareness, facilitating discussion, educating clinicians, and simplifying documentation to ease time constraints. Several strategies were employed postimplementation (Figure 2). Prefilled templates simplified documentation. These templates helped identify patients without pharmacogenomic testing, provided reminders, and saved documentation time during visits. CPPs also received training and materials on PHASER ordering and documentation within encounter notes. Additionally, patient-directed advertisements were displayed in CPP examination rooms to help inspire and facilitate discussion between veterans and CPPs.
Process Improvement Data
The quality improvement project goal was to increase PHASER orders by 50% after 3 months. PHASER orders increased from 87 at baseline (March 8, 2023, to September 8, 2023) to 196 during the intervention (November 16, 2023, to February 16, 2024), a 125% increase. Changes were consistent and sustained with 65 orders the first month, 67 orders the second month, and 64 orders the third month.
Discussion
Using Lean Six Sigma A3 methodology for a quality improvement process to increase PHASER orders by CPPs revealed barriers and guided potential solutions to overcome these barriers. Interventions included additional CPP training and ordering, tools for easier identification of potential patients, documentation best practices, patient-directed advertisements to facilitate conversations. These interventions required about 8 hours for preparation, distribution, development, and interpretation of surveys, education, and documentation materials. The financial impact of these interventions was already included in allotted office materials budgeted and provided. Additional funding was not needed to provide patient-directed advertisements or education materials. The VACOHCS pharmacogenomics CPP discusses PHASER test results with patients at a separate appointment.
Future directions include educating other CPPs to assist in discussing results with veterans. Overall, the changes implemented to improve the PHASER ordering process were low effort and exemplify the ease of streamlining future initiatives, allowing for sustained optimal implementation of pharmacogenomic testing.
Conclusions
A quality improvement initiative resulted in increased PHASER orders and a clearly defined process, allowing for a continued increase and sustained support. Perceived barriers were identified, and the changes implemented were often low effort but exhibited a sustained impact. The insights gleaned from this process will shape future process development initiatives and continue to sustain pharmacogenomic testing ordering by CPPs. This process will be extended to other VACOHCS clinical departments to further support increased access to pharmacogenomic testing, reduce medication trial and error, and reduce hospitalizations from adverse effects for veterans.
Peer-review, evidence-based, detailed gene/drug clinical practice guidelines suggest that genetic variations can impact how individuals metabolize medications, which is sometimes included in medication prescribing information.1-3 Pharmacogenomic testing identifies genetic markers so medication selection and dosing can be tailored to each individual by identifying whether a specific medication is likely to be safe and effective prior to prescribing.4
Pharmacogenomics can be a valuable tool for personalizing medicine but has had suboptimal implementation since its discovery. The US Department of Veterans Affairs (VA) health care system reviewed the implementation of the Pharmacogenomic Testing for Veterans (PHASER) program. This review identified clinician barriers pre- and post-PHASER program implementation; staffing issues, competing clinical priorities, and inadequate PHASER program resources were the most frequently reported barriers to implementation of pharmacogenomic testing.5
Another evaluation of the implementation of the PHASER program that surveyed VA patients found that patients could be separated into 3 groups. Acceptors of pharmacogenomic testing emphasized potential health benefits of testing. Patients that declined testing often cited concerns for genetic information affecting insurance coverage, being misused, or being susceptible to data breach. The third group—identified as contemplators—reported the need for clinician outreach to impact their decision on whether or not to receive pharmacogenomic testing.6 These studies suggest that removing barriers by providing ample pharmacogenomics resources to clinicians, in addition to detailed training on how to offer and follow up with patients regarding pharmacogenomic testing, is crucial to successful implementation of the PHASER program.
PHASER
In 2019, the VA began working with Sanford Health to establish the PHASER program and offer pharmacogenomic testing. PHASER has since expanded to 25 VA medical centers, including the VA Central Ohio Healthcare System (VACOHCS).7,8 Pharmacogenomic testing through PHASER is conducted using a standardized laboratory panel that includes 12 different medication classes.9 The drug classes include certain anti-infective, anticoagulant, antiplatelet, cardiovascular, cholesterol, gastrointestinal, mental health, neurological, oncology, pain, transplant, and other miscellaneous medications. Medications are correlated to each class and assessed for therapeutic impacts based on gene panel results.
Clinical recommendations for medication-gene interactions can range from monitoring for increased risk of adverse effects or therapeutic failure to recommending avoiding a medication. For example, patients who test positive for the HLA-B gene have significantly increased risk of hypersensitivity to abacavir, an HIV treatment.10
Similarly, patients who cannot adequately metabolize cytochrome P450 2C19 should consider avoiding clopidogrel as they are unlikely to convert clopidogrel to its active prodrug, which reduces its effectiveness.11 Pharmacists can play a critical role educating patients about pharmacogenomic testing, especially within hematology and oncology.12 Patients can benefit from this testing even if they are not currently taking medications with known concerns as they could be prescribed in the future. The SLCO1B1 gene-drug test, for example, can identify risk for statin-associated muscle symptoms.13
Clinical pharmacist practitioners (CPPs) can increase access to genetic testing because they interact with patients in a variety of settings and can order this laboratory test.12,14 Recent research has demonstrated that most VA patients carry ≥ 1 genetic variant that may influence medication decisions and that half of veterans are prescribed a medication with known gene-drug interactions.15 CPP ordering of pharmacogenomic tests at the VACOHCS outpatient clinic was evaluated through collection of baseline data from March 8, 2023, to September 8, 2023. A goal was identified to increase orders by 50% for a patient care quality improvement initiative and use CPPs to increase access to pharmacogenomic testing. The purpose of this quality improvement initiative was to expand access to pharmacogenomic testing through process implementation and improvement within CPP-led clinic settings.
Gap Analysis
Lean Six Sigma A3 methodology was used to identify ways to increase the use of pharmacogenomic testing for veterans at VACOHCS and develop an improved process for increased ordering of pharmacogenomic testing. Lean Six Sigma A3 methodology is a stepwise approach to process improvement that helps identify gaps in efficiency, sustainable changes, and eliminate waste.16 Baseline data were collected from March 8, 2023, to September 8, 2023, to determine the frequency of CPPs ordering pharmacogenomic laboratory panels during clinic appointments. The ordering of pharmacogenomic panels was monitored by the VACOHCS PHASER coordinator.
CPPs were surveyed to identify perceived barriers to PHASER implementation. A gap analysis was conducted using Lean Six Sigma A3 methodology. Gap analyses use lean tools such as a Fishbone Diagram to illustrate and identify the gap between current state and ideal state. (Figure 1).The following barriers were identified: lack of clinician education materials, lack of a standardized patient screening process, time constraints on patient education and ordering, higher priority clinical needs, forgetting to order, lack of comfort with pharmacogenomics ordering and education, lack of support for the initiative, and increased workload and burnout. Among these perceived barriers, higher priority clinical needs, forgetting to order, and time constraints ranked highest in importance among CPPs.
In line with Lean Six Sigma A3 methodology, several tests of change were used to improve pharmacogenomic testing ordering. These changes focused on increasing patient and clinician awareness, facilitating discussion, educating clinicians, and simplifying documentation to ease time constraints. Several strategies were employed postimplementation (Figure 2). Prefilled templates simplified documentation. These templates helped identify patients without pharmacogenomic testing, provided reminders, and saved documentation time during visits. CPPs also received training and materials on PHASER ordering and documentation within encounter notes. Additionally, patient-directed advertisements were displayed in CPP examination rooms to help inspire and facilitate discussion between veterans and CPPs.
Process Improvement Data
The quality improvement project goal was to increase PHASER orders by 50% after 3 months. PHASER orders increased from 87 at baseline (March 8, 2023, to September 8, 2023) to 196 during the intervention (November 16, 2023, to February 16, 2024), a 125% increase. Changes were consistent and sustained with 65 orders the first month, 67 orders the second month, and 64 orders the third month.
Discussion
Using Lean Six Sigma A3 methodology for a quality improvement process to increase PHASER orders by CPPs revealed barriers and guided potential solutions to overcome these barriers. Interventions included additional CPP training and ordering, tools for easier identification of potential patients, documentation best practices, patient-directed advertisements to facilitate conversations. These interventions required about 8 hours for preparation, distribution, development, and interpretation of surveys, education, and documentation materials. The financial impact of these interventions was already included in allotted office materials budgeted and provided. Additional funding was not needed to provide patient-directed advertisements or education materials. The VACOHCS pharmacogenomics CPP discusses PHASER test results with patients at a separate appointment.
Future directions include educating other CPPs to assist in discussing results with veterans. Overall, the changes implemented to improve the PHASER ordering process were low effort and exemplify the ease of streamlining future initiatives, allowing for sustained optimal implementation of pharmacogenomic testing.
Conclusions
A quality improvement initiative resulted in increased PHASER orders and a clearly defined process, allowing for a continued increase and sustained support. Perceived barriers were identified, and the changes implemented were often low effort but exhibited a sustained impact. The insights gleaned from this process will shape future process development initiatives and continue to sustain pharmacogenomic testing ordering by CPPs. This process will be extended to other VACOHCS clinical departments to further support increased access to pharmacogenomic testing, reduce medication trial and error, and reduce hospitalizations from adverse effects for veterans.
Cecchin E, Stocco G. Pharmacogenomics and personalized medicine. Genes (Basel). 2020;11(6):679. doi:10.3390/genes11060679
Guidelines. CPIC. Accessed April 16, 2025. https://cpicpgx.org/guidelines/
PharmGKB. PharmGKB. 2025. Accessed April 16, 2025. https://www.pharmgkb.org
Centers for Disease Control and Prevention. Pharmacogenomics. Updated November 13, 2024. Accessed April 16, 2024. https://www.cdc.gov/genomics-and-health/pharmacogenomics/
Dong OM, Roberts MC, Wu RR, et al. Evaluation of the Veterans Affairs Pharmacogenomic Testing for Veterans (PHASER) clinical program at initial test sites. Pharmacogenomics. 2021;22(17):1121-1133. doi:10.2217/pgs-2021-0089
Melendez K, Gutierrez-Meza D, Gavin KL, et al. Patient perspectives of barriers and facilitators for the uptake of pharmacogenomic testing in Veterans Affairs’ pharmacogenomic testing for the veterans (PHASER) program. J Pers Med. 2023;13(9):1367. doi:10.3390/jpm13091367
Sanford Health Imagenetics. FREQUENTLY ASKED QUESTIONS (FAQs) about the “Pharmacogenomic Teting for Vetans” (PHASER) Program. US Department of Veterans Affairs. December 20, 2019. Accessed April 16, 2025. https://www.va.gov/opa/publications/factsheets/PHASER-FLYER-VA-Patient-FAQ.pdf
Peterson H. PHASER program testing informs how you respond to medicines. VA News. September 6, 2022. Accessed April 16, 2025. https://news.va.gov/108091/phaser-program-testing-respond-medicines/
Pharmacogenomics (PGx). Sanford Health Imagenetics. 2025. Accessed April 16, 2025. https://imagenetics.sanfordhealth.org/pharmacogenomics/
Martin MA, Hoffman JM, Freimuth RR, et al. Clinical pharmacogenetics implementation consortium guidelines for HLA-B genotype and abacavir dosing: 2014 update. Clin Pharmacol Ther. 2014;95(5):499-500. doi:10.1038/clpt.2014.38
Lee CR, Luzum JA, Sangkuhl K, et al. Clinical pharmacogenetics implementation consortium guideline for CYP2C19 genotype and clopidogrel therapy: 2022 update. Clin Pharmacol Ther. 2022;112(5):959-967. doi:10.1002/cpt.2526
Dreischmeier E, Hecht H, Crocker E, et al. Integration of a clinical pharmacist practitioner-led pharmacogenomics service in a Veterans Affairs hematology/oncology clinic. Am J Health Syst Pharm. 2024;81(19):e634-e639. doi:10.1093/ajhp/zxae122
Tomcsanyi KM, Tran KA, Bates J, et al. Veterans Health Administration: implementation of pharmacogenomic clinical decision support with statin medications and the SLCO1B1 gene as an exemplar. Am J Health Syst Pharm. 2023;80(16):1082-1089. doi:10.1093/ajhp/zxad111
Gammal RS, Lee YM, Petry NJ, et al. Pharmacists leading the way to precision medicine: updates to the core pharmacist competencies in genomics. Am J Pharm Educ. 2022;86(4):8634. doi:10.5688/ajpe8634
Chanfreau-Coffinier C, Hull LE, Lynch JA, et al. Projected prevalence of actionable pharmacogenetic variants and level A drugs prescribed among US Veterans Health Administration pharmacy users. JAMA Netw Open. 2019;2(6):e195345. doi:10.1001/jamanetworkopen.2019.5345
Shaffie S, Shahbazi S. The McGraw-Hill 36-Hour Course: Lean Six Sigma. McGraw-Hill; 2012.
Cecchin E, Stocco G. Pharmacogenomics and personalized medicine. Genes (Basel). 2020;11(6):679. doi:10.3390/genes11060679
Guidelines. CPIC. Accessed April 16, 2025. https://cpicpgx.org/guidelines/
PharmGKB. PharmGKB. 2025. Accessed April 16, 2025. https://www.pharmgkb.org
Centers for Disease Control and Prevention. Pharmacogenomics. Updated November 13, 2024. Accessed April 16, 2024. https://www.cdc.gov/genomics-and-health/pharmacogenomics/
Dong OM, Roberts MC, Wu RR, et al. Evaluation of the Veterans Affairs Pharmacogenomic Testing for Veterans (PHASER) clinical program at initial test sites. Pharmacogenomics. 2021;22(17):1121-1133. doi:10.2217/pgs-2021-0089
Melendez K, Gutierrez-Meza D, Gavin KL, et al. Patient perspectives of barriers and facilitators for the uptake of pharmacogenomic testing in Veterans Affairs’ pharmacogenomic testing for the veterans (PHASER) program. J Pers Med. 2023;13(9):1367. doi:10.3390/jpm13091367
Sanford Health Imagenetics. FREQUENTLY ASKED QUESTIONS (FAQs) about the “Pharmacogenomic Teting for Vetans” (PHASER) Program. US Department of Veterans Affairs. December 20, 2019. Accessed April 16, 2025. https://www.va.gov/opa/publications/factsheets/PHASER-FLYER-VA-Patient-FAQ.pdf
Peterson H. PHASER program testing informs how you respond to medicines. VA News. September 6, 2022. Accessed April 16, 2025. https://news.va.gov/108091/phaser-program-testing-respond-medicines/
Pharmacogenomics (PGx). Sanford Health Imagenetics. 2025. Accessed April 16, 2025. https://imagenetics.sanfordhealth.org/pharmacogenomics/
Martin MA, Hoffman JM, Freimuth RR, et al. Clinical pharmacogenetics implementation consortium guidelines for HLA-B genotype and abacavir dosing: 2014 update. Clin Pharmacol Ther. 2014;95(5):499-500. doi:10.1038/clpt.2014.38
Lee CR, Luzum JA, Sangkuhl K, et al. Clinical pharmacogenetics implementation consortium guideline for CYP2C19 genotype and clopidogrel therapy: 2022 update. Clin Pharmacol Ther. 2022;112(5):959-967. doi:10.1002/cpt.2526
Dreischmeier E, Hecht H, Crocker E, et al. Integration of a clinical pharmacist practitioner-led pharmacogenomics service in a Veterans Affairs hematology/oncology clinic. Am J Health Syst Pharm. 2024;81(19):e634-e639. doi:10.1093/ajhp/zxae122
Tomcsanyi KM, Tran KA, Bates J, et al. Veterans Health Administration: implementation of pharmacogenomic clinical decision support with statin medications and the SLCO1B1 gene as an exemplar. Am J Health Syst Pharm. 2023;80(16):1082-1089. doi:10.1093/ajhp/zxad111
Gammal RS, Lee YM, Petry NJ, et al. Pharmacists leading the way to precision medicine: updates to the core pharmacist competencies in genomics. Am J Pharm Educ. 2022;86(4):8634. doi:10.5688/ajpe8634
Chanfreau-Coffinier C, Hull LE, Lynch JA, et al. Projected prevalence of actionable pharmacogenetic variants and level A drugs prescribed among US Veterans Health Administration pharmacy users. JAMA Netw Open. 2019;2(6):e195345. doi:10.1001/jamanetworkopen.2019.5345
Shaffie S, Shahbazi S. The McGraw-Hill 36-Hour Course: Lean Six Sigma. McGraw-Hill; 2012.
Comparison of Prescribing Patterns of Intranasal Naloxone in a Veteran Population
Comparison of Prescribing Patterns of Intranasal Naloxone in a Veteran Population
Since 1999, annual deaths attributed to opioid overdose in the United States have increased from about 10,000 to about 50,000 in 2019.1 During the COVID-19 pandemic > 74,000 opioid overdose deaths occurred in the US from April 2020 to April 2021.2,3 Opioid-related overdoses now account for about 75% of all drug-related overdose deaths.1 In 2017, the cost of opioid overdose deaths and opioid use disorder (OUD) reached $1.02 trillion in the United States and $26 million in Indiana.4 The total deaths and costs would likely be higher if it were not for naloxone.
Naloxone hydrochloride was first patented in the 1960s and approved by the US Food and Drug Administration (FDA) in 1971 to treat opioid-related toxicity.1 It is the most frequently prescribed antidote for opioid toxicity due to its activity as a pure υ-opioid receptor competitive antagonist. Naloxone formulations include intramuscular, intravenous, subcutaneous, and intranasal delivery methods.5 According to the Centers for Disease Control and Prevention, clinicians should offer naloxone to patients at high risk for opioid-related adverse events. Risk factors include a history of overdose, opioid dosages of ≥ 50 morphine mg equivalents/day, and concurrent use of opioids with benzodiazepines.6
Intranasal naloxone 4 mg has become more accessible following the classification of opioid use as a public health emergency in 2017 and its over-the-counter availability since 2023. Intranasal naloxone 4 mg was approved by the FDA in 2015 for the prevention of opioid overdoses (accidental or intentional), which can be caused by heroin, fentanyl, carfentanil, hydrocodone, oxycodone, methadone, and other substances. 7 Fentanyl has most recently been associated with xylazine, a nonopioid tranquilizer linked to increased opioid overdose deaths.8 Recent data suggest that 34% of opioid overdose reversals involved ≥ 2 doses of intranasal naloxone 4 mg, which led to FDA approval of an intranasal naloxone 8 mg spray in April 2021.9-11
Veteran Health Indiana (VHI) has implemented several initiatives to promote naloxone prescribing. Established in 2020, the Opioid Overdose Education and Naloxone Distribution (OEND) program sought to prevent opioid-related deaths through education and product distribution. These criteria included an opioid prescription for ≥ 30 days. In 2021, the Stratification Tool for Opioid Risk Mitigation (STORM) was created to identify patients at high risk of opioid overdose and allowing pharmacists to prescribe naloxone for at-risk patients without restrictions, increasing accessibility.12
Recent cases of fentanyl-related overdoses involving stronger fentanyl analogues highlight the need for higher naloxone dosing to prevent overdose. A pharmacokinetic comparison of intranasal naloxone 8 mg vs 4 mg demonstrated maximum plasma concentrations of 10.3 ng/mL and 5.3 ng/mL, respectively. 13 Patients may be at an increased risk of precipitated opioid withdrawal when using intranasal naloxone 8 mg over 4 mg; however, some patients may benefit from achieving higher serum concentrations and therefore require larger doses of naloxone.
No clinical trials have demonstrated a difference in reversal rates between naloxone doses. No clinical practice guidelines support a specific naloxone formulation, and limited US Department of Veterans Affairs (VA)-specific guidance exists. VA Naloxone Rescue: Recommendations for Use states that selection of naloxone 8 mg should be based on shared decision-making between the patient and clinician and based on individual risk factors.12 The purpose of this study is to analyze data to determine if there is a difference in prescribing patterns of intranasal naloxone 4 mg and intranasal naloxone 8 mg.
METHODS
A retrospective chart reviews using the VA Computerized Patient Record System (CPRS) analyzed patients prescribed intranasal naloxone 4 mg or intranasal naloxone 8 mg at VHI. A patient list was generated based on active naloxone prescriptions between April 1, 2022, and April 1, 2023. Data were obtained exclusively through CPRS and patients were not contacted. This study was reviewed and deemed exempt by the Indiana University Health Institutional Review Board and the VHI Research and Development Committee.
Patients were included if they were aged ≥ 18 years and had an active prescription for intranasal naloxone 4 mg or intranasal naloxone 8 mg during the trial period. Patients were excluded if their naloxone prescription was written by a non-VHI clinician, if the dose was not 4 mg or 8 mg, or if the dosage form was other than intranasal spray.
The primary endpoint was the comparison for prescribing patterns for intranasal naloxone 4 mg and intranasal naloxone 8 mg during the study period. Secondary endpoints included total naloxone prescriptions; monthly prescriptions; number of patients with repeated naloxone prescriptions; prescriber type by naloxone dose; clinic type by naloxone dose; and documented indication for naloxone use by dose.
Demographic data collected included baseline age, sex, race, comorbid mental health conditions, and active central nervous system depressant medications on patient profile (ie, opioids, gabapentinoids, benzodiazepines, antidepressants, antipsychotics). Opioid prescriptions that were active or discontinued within the last 3 months were also recorded. Comorbid mental health conditions were collected based on the most recent clinical note before initiating medication.
Prescription-related data included strength of medication prescribed (4 mg, 8 mg, or both), documented use of medication, prescriber name, prescriber discipline, prescription entered by, number of times naloxone was filled or refilled during the study period, indication, clinic location, and clinic name. If > 1 prescription was active during the study period, the number of refills, prescriber name and clinic location of the first prescription in the study period was recorded. Additionally, the indication of OUD was differentiated from substance use disorder (SUD) if the patient was only dependent on opioids, excluding tobacco or alcohol. Patients with SUDs may include opioid dependence in addition to other substance dependence (eg, cannabis, stimulants, gabapentinoids, or benzodiazepines).
Basic descriptive statistics, including mean, ranges, and percentages were used to characterize the study subjects. For nominal data, X2 tests were used. A 2-sided 5% significance level was used for all statistical tests.
RESULTS
A total of 1952 active naloxone prescriptions from 1739 patients met the inclusion criteria; none were eliminated based on the exclusion criteria and some were included multiple times because data were collected for each active prescription during the study period. One hundred one patients were randomized and included in the final analysis (Figure). Most patients identified as White (81%), male (90%), and had a mean (SD) age of 60.9 (14.2) years. Common mental health comorbidities included 59 patients with depression, 50 with tobacco use disorder, and 31 with anxiety. Eighty-four patients had opioid and 60 had antidepressants/antianxiety, and 40 had gabapentinoids prescriptions. Forty-three patients had ≥ 3 mental health comorbidities. Thirty-four patients had 2 active central nervous system depressant prescriptions, 30 had 3 active prescriptions, and 9 had ≥ 4 active prescriptions. Most patients (n = 83) had an active or recently discontinued opioid prescription (Table 1).
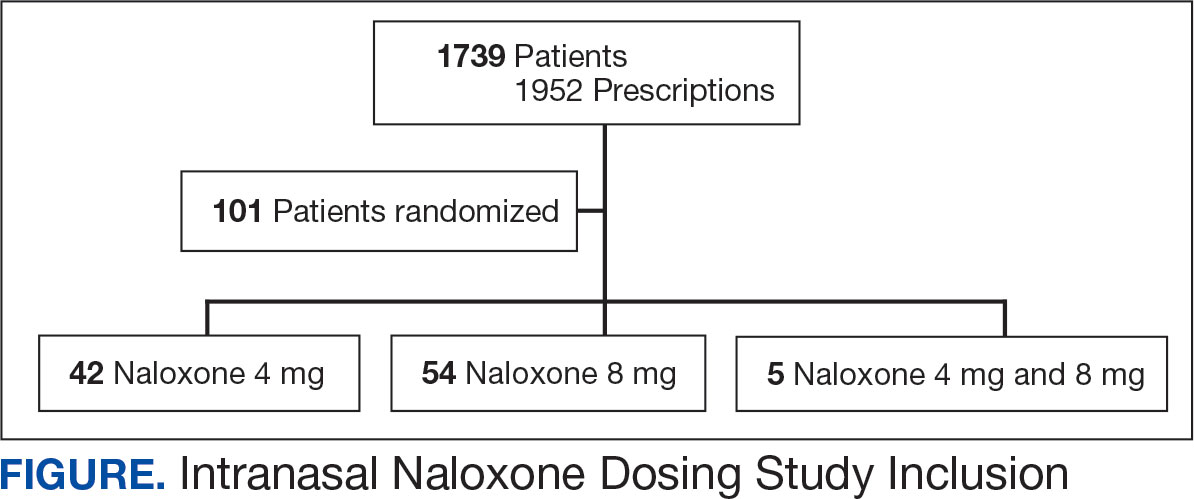
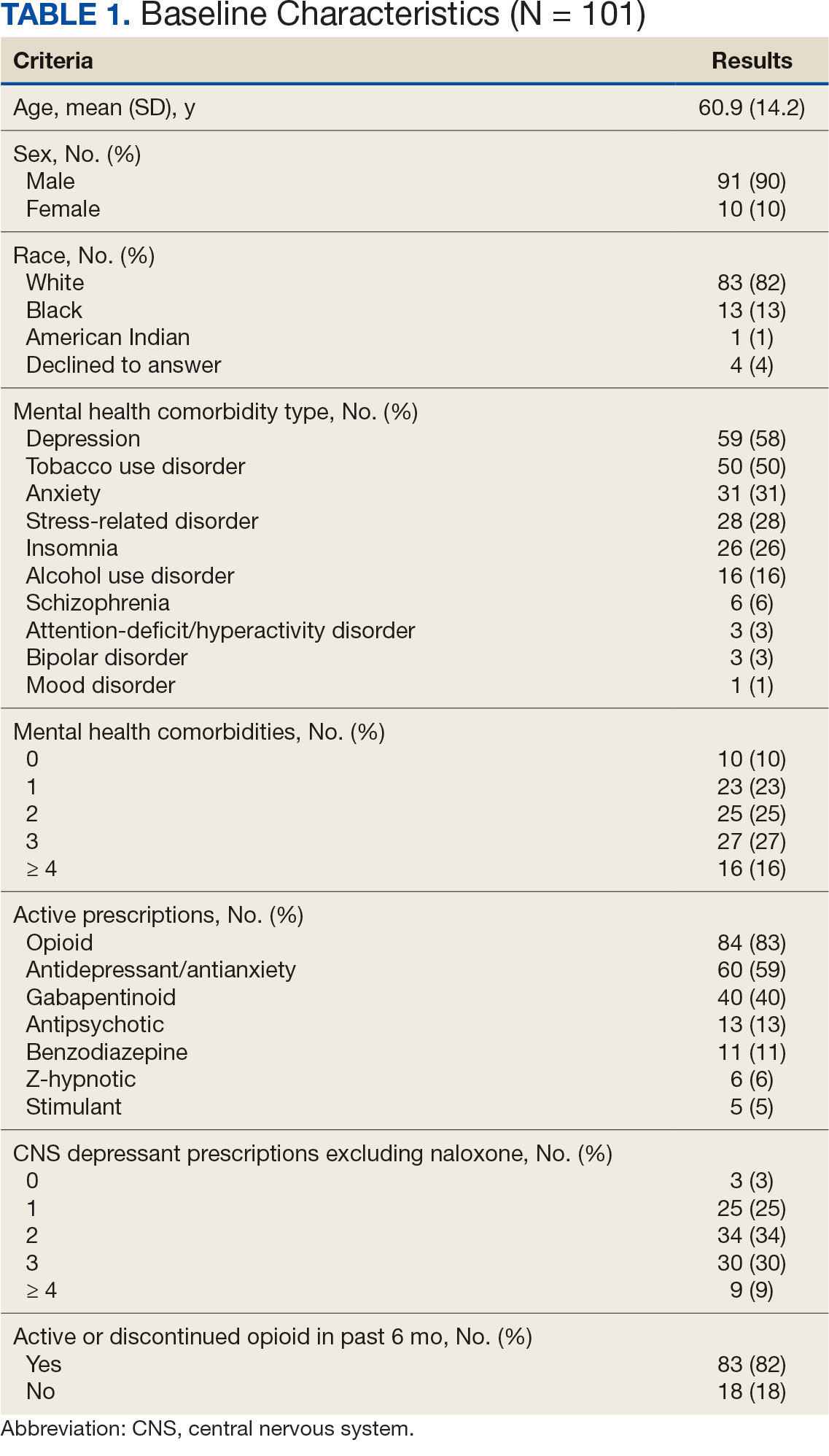
The 101 patients received 54 prescriptions for naloxone 8 mg and 47 for 4 mg (Table 2). Five patients received prescriptions for both the 4 mg and 8 mg intranasal naloxone formulations. Sixty-six patients had naloxone filled once (66%) during the study period. Intranasal naloxone 4 mg was prescribed to 30 patients by nurse practitioners, 17 patients by physicians, and not prescribed by pharmacists. Intranasal naloxone 8 mg was prescribed to 40 patients by pharmacists, 13 patients by physicians, and 6 patients by nurses. Patients who received prescriptions for both intranasal naloxone 4 mg and 8 mg were most routinely ordered by physicians (n = 3; 60%) in primary care (n = 2; 40%) for chronic opioid use (n = 2; 40%).
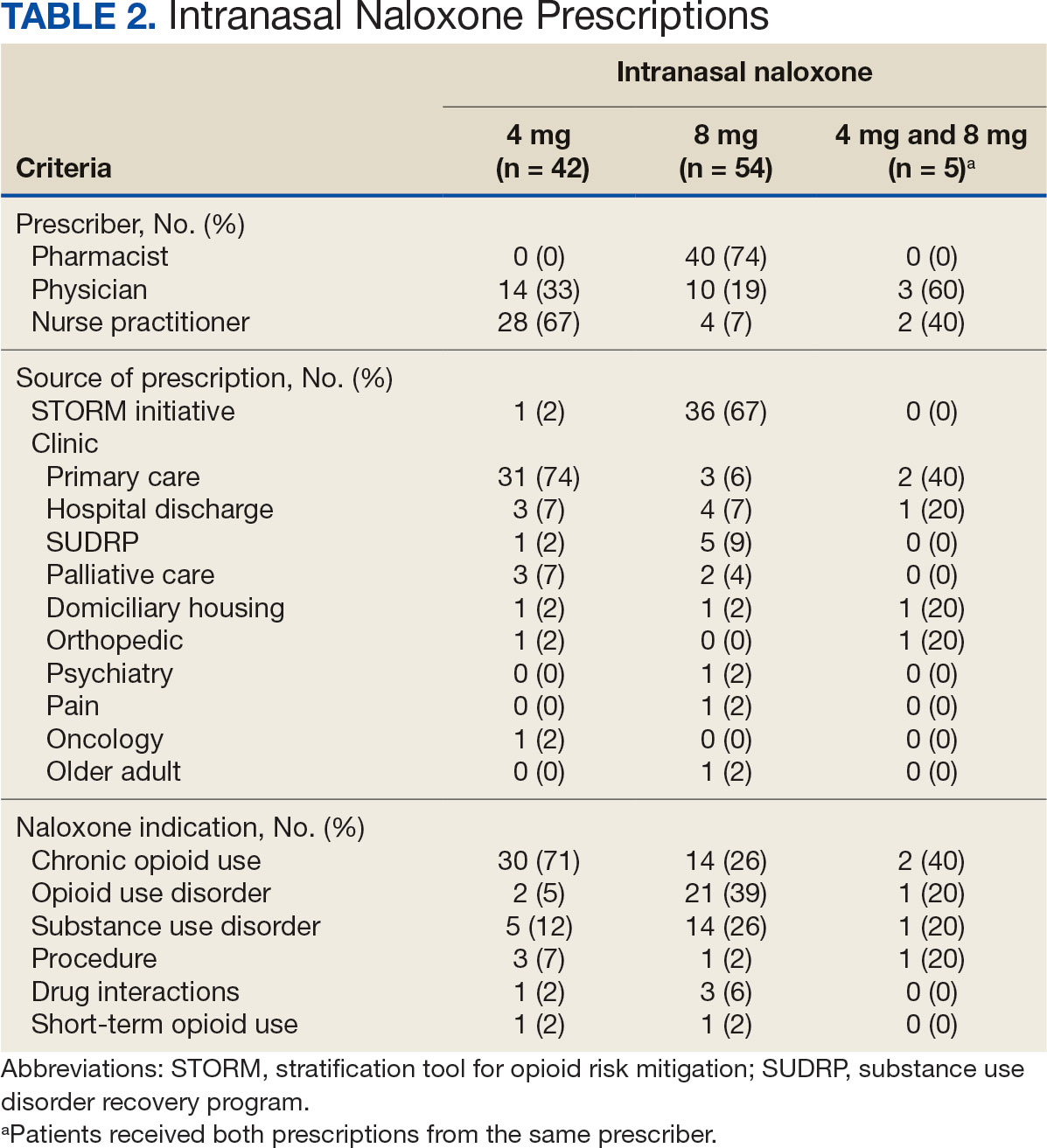
Patients access naloxone from many different VHI clinics. Primary care clinics prescribed the 4 mg formulation to 31 patients, 8 mg to 3 patients, and both to 2 patients. The STORM initiative was used for 37 of 106 prescriptions (35%): 4 mg intranasal naloxone was prescribed to 1 patient, 8 mg to 36 patients, and no patients received both formulations. Chronic opioid use was the most common indication (46%) with 30 patients prescribed intranasal naloxone 4 mg, 14 patients prescribed 8 mg, and 2 patients prescribed both. OUD was the indication for 24% of patients: 2 patients prescribed intranasal naloxone 4 mg, 21 patients prescribed 8 mg, and 1 patient prescribed both.
The 106 intranasal naloxone prescriptions were equally distributed across each month from April 1, 2022, to April 1, 2023. Of the 101 patients, 34 had multiple naloxone prescriptions filled during the study period. Pharmacists wrote 40 of 106 naloxone prescriptions (38%), all for the 8 mg formulation. Nurse practitioners prescribed naloxone 4 mg 30 times and 8 mg 6 times for 36 of 106 prescriptions (34%). Physicians prescribed 30 of 106 prescriptions (28%), including intranasal naloxone 4 mg 17 times and 8 mg 13 times.
Statistics were analyzed using a X2 test; however, it was determined that the expected frequencies made the tests inappropriate. Differences in prescribing patterns between naloxone doses, prescriber disciplines, source of the prescription, or indications were not statistically significant.
DISCUSSION
Many pharmacists possess a scope of practice under state law and/or institution policy to prescribe naloxone. In this study, pharmacists prescribed the most naloxone prescriptions compared to physicians and nurse practitioners. Initiatives such as OEND and STORM have given pharmacists at VHI an avenue to combat the growing opioid epidemic while expanding their scope of practice. A systematic review of 67 studies found that pharmacist-led OEND programs showed a statistically significant increase in naloxone orders. A statistical significance was likely met given the large sample sizes ranging from 10 to 217,000 individuals, whereas this study only assessed a small portion of patients.14 This study contributes to the overwhelming amount of data that highlights pharmacists’ impact on overall naloxone distribution.
The STORM initiative and primary care clinics were responsible for large portions of naloxone prescriptions in this study. STORM was used by pharmacists and contributed to more than half of the higher dose naloxone prescriptions. Following a discussion with members of the pain management team, pharmacists involved in STORM prescribing were revealed to exclusively prescribe intranasal naloxone 8 mg as opposed to 4 mg. At the risk of precipitating withdrawal from higher doses of naloxone, it was agreed that this risk was heavily outweighed by the benefit of successful opioid reversal. In this context, it is expected for this avenue of prescribing to influence naloxone prescribing patterns at VHI.
Prescribing in primary care clinics was shown to be equally as substantial. Primary care-based multidisciplinary transition clinics have been reported to be associated with increased access to OUD treatment.15 Primary care clinics at VHI, or patient aligned care teams (PACT), largely consist of multidisciplinary health care teams. PACT clinicians are heavily involved in transitions of care because one system provides patients with comprehensive acute and chronic care. Continuing to encourage naloxone distribution through primary care and using STORM affords various patient populations access to high-level care.
Notable differences were observed between indications for naloxone use and the corresponding dose. Patients with OUD or SUD were more likely to receive intranasal naloxone 8 mg as opposed to patients receiving intranasal naloxone for chronic opioid use, who were more likely to receive the 4 mg dose. This may be due to a rationale to provide a higher dose of naloxone to combat overdoses in the case of ingesting substances mixed with fentanyl or xylazine.12,13 Without standard of care guidelines, concerns remain for varying outcomes in opioid overdose prevention within vulnerable populations.
Limitations
Chart data were dependent on documentation, which may have omitted pertinent baseline characteristics and risk factors. Additional data collection could have further assessed a patient’s specific risk factors (eg, opioid dose in morphine equivalents) to draw conclusions to the dose of naloxone prescribed. The sample size was small, and the patient population was largely White and male, which minimized the generalizability of the results.
CONCLUSIONS
This study evaluated the differences in intranasal naloxone prescribing patterns within a veteran population at VHI over 12 months. Findings revealed that most prescriptions were written for intranasal naloxone 8 mg, by a pharmacist, in a primary care setting, and for chronic opioid use. The results revealed evidence of differing naloxone prescribing practices, which emphasize the need for clinical guidelines and better defined recommendations in relation to naloxone dosing.
The most evident gap in patient care could be addressed by urging the VA Pharmacy Benefits Management group to update naloxone recommendations for use to include more concrete dosing recommendations. Furthermore, it would be beneficial to re-educate clinicians on naloxone prescribing to increase awareness of different doses and the importance of equipping patients with the correct amount of naloxone in an emergency. Additional research assessing change in prescribing patterns is warranted as the use of higher dose naloxone becomes more routine.
- Britch SC, Walsh SL. Treatment of opioid overdose: current approaches and recent advances. Psychopharmacology (Berl). 2022;239(7):2063-2081. doi:10.1007/s00213-022-06125-5
- Ahmad FB, Cisewski JA, Rossen LM, Sutton P. Provisional Drug Overdose Death Counts. National Center for Health Statistics, Centers for Disease Control and Prevention; 2023. Accessed April 10, 2025. https://www.cdc.gov/nchs/nvss/vsrr/drug-overdose-data.htm
- O’Donnell J, Tanz LJ, Gladden RM, Davis NL, Bitting J. Trends in and characteristics of drug overdose deaths involving illicitly manufactured fentanyls — United States, 2019–2020. MMWR Morb Mortal Wkly Rep. 2021;70:1740-1746. doi:10.15585/mmwr.mm7050e3
- Luo F, Li M, Florence C. State-level economic costs of opioid use disorder and fatal opioid overdose — United States, 2017. MMWR Morb Mortal Wkly Rep. 2021;70:541-546. doi:10.15585/mmwr.mm7015a1
- Lexicomp. Lexicomp Online. Accessed April 10, 2025. http://online.lexi.com
- Dowell D, Ragan KR, Jones CM, Baldwin GT, Chou R. CDC Clinical practice guideline for prescribing opioids for pain — United States, 2022. MMWR Recomm Rep. 2022;71(3):1-95. doi:10.15585/mmwr.rr7103a1
- Narcan (naloxone) FDA approval history. Drugs.com. Accessed April 10, 2025. https://www.drugs.com/history/narcan.html
- Centers for Disease Control and Prevention. What you should know about xylazine. May 16, 2024. Accessed April 10, 2025. https://www.cdc.gov/overdose-prevention/about/what-you-should-know-about-xylazine.html
- Avetian GK, Fiuty P, Mazzella S, Koppa D, Heye V, Hebbar P. Use of naloxone nasal spray 4 mg in the community setting: a survey of use by community organizations. Curr Med Res Opin. 2018;34(4):573-576. doi:10.1080/03007995.2017.1334637
- Kloxxado [package insert]. Hikma Pharmaceuticals USA Inc; 2021.
- FDA approves higher dosage of naloxone nasal spray to treat opioid overdose. News release. FDA. April 30, 2021. Accessed April 10, 2025. https://www.fda.gov/news-events/press-announcements/fda-approves-higher-dosage-naloxone-nasal-spray-treat-opioid-overdose
- US Department of Veterans Affairs, Pharmacy Benefits Management Services and National Formulary Committee in Collaboration with the VA National Harm Reduction Support & Development Workgroup. Naloxone Rescue: Recommendations for Use. June 2014. Updated March 2024. Accessed April 10, 2025. https://www.va.gov/formularyadvisor/DOC_PDF/CRE_Naloxone_Rescue_Guidance_March_2024.pdf
- Krieter P, Chiang N, Gyaw S, et al. Pharmacokinetic properties and human use characteristics of an FDA-approved intranasal naloxone product for the treatment of opioid overdose. J Clin Pharmacol. 2016;56(10):1243-1253. doi:10.1002/jcph.759
- Rawal S, Osae SP, Cobran EK, Albert A, Young HN. Pharmacists’ naloxone services beyond community pharmacy settings: a systematic review. Res Social Adm Pharm. 2023;19(2):243-265. doi:10.1016/j.sapharm.2022.09.002
- Incze MA, Sehgal SL, Hansen A, Garcia L, Stolebarger L. Evaluation of a primary care-based multidisciplinary transition clinic for patients newly initiated on buprenorphine in the emergency department. Subst Abus. 2023;44(3):220-225. doi:10.1177/08897077231188592
Since 1999, annual deaths attributed to opioid overdose in the United States have increased from about 10,000 to about 50,000 in 2019.1 During the COVID-19 pandemic > 74,000 opioid overdose deaths occurred in the US from April 2020 to April 2021.2,3 Opioid-related overdoses now account for about 75% of all drug-related overdose deaths.1 In 2017, the cost of opioid overdose deaths and opioid use disorder (OUD) reached $1.02 trillion in the United States and $26 million in Indiana.4 The total deaths and costs would likely be higher if it were not for naloxone.
Naloxone hydrochloride was first patented in the 1960s and approved by the US Food and Drug Administration (FDA) in 1971 to treat opioid-related toxicity.1 It is the most frequently prescribed antidote for opioid toxicity due to its activity as a pure υ-opioid receptor competitive antagonist. Naloxone formulations include intramuscular, intravenous, subcutaneous, and intranasal delivery methods.5 According to the Centers for Disease Control and Prevention, clinicians should offer naloxone to patients at high risk for opioid-related adverse events. Risk factors include a history of overdose, opioid dosages of ≥ 50 morphine mg equivalents/day, and concurrent use of opioids with benzodiazepines.6
Intranasal naloxone 4 mg has become more accessible following the classification of opioid use as a public health emergency in 2017 and its over-the-counter availability since 2023. Intranasal naloxone 4 mg was approved by the FDA in 2015 for the prevention of opioid overdoses (accidental or intentional), which can be caused by heroin, fentanyl, carfentanil, hydrocodone, oxycodone, methadone, and other substances. 7 Fentanyl has most recently been associated with xylazine, a nonopioid tranquilizer linked to increased opioid overdose deaths.8 Recent data suggest that 34% of opioid overdose reversals involved ≥ 2 doses of intranasal naloxone 4 mg, which led to FDA approval of an intranasal naloxone 8 mg spray in April 2021.9-11
Veteran Health Indiana (VHI) has implemented several initiatives to promote naloxone prescribing. Established in 2020, the Opioid Overdose Education and Naloxone Distribution (OEND) program sought to prevent opioid-related deaths through education and product distribution. These criteria included an opioid prescription for ≥ 30 days. In 2021, the Stratification Tool for Opioid Risk Mitigation (STORM) was created to identify patients at high risk of opioid overdose and allowing pharmacists to prescribe naloxone for at-risk patients without restrictions, increasing accessibility.12
Recent cases of fentanyl-related overdoses involving stronger fentanyl analogues highlight the need for higher naloxone dosing to prevent overdose. A pharmacokinetic comparison of intranasal naloxone 8 mg vs 4 mg demonstrated maximum plasma concentrations of 10.3 ng/mL and 5.3 ng/mL, respectively. 13 Patients may be at an increased risk of precipitated opioid withdrawal when using intranasal naloxone 8 mg over 4 mg; however, some patients may benefit from achieving higher serum concentrations and therefore require larger doses of naloxone.
No clinical trials have demonstrated a difference in reversal rates between naloxone doses. No clinical practice guidelines support a specific naloxone formulation, and limited US Department of Veterans Affairs (VA)-specific guidance exists. VA Naloxone Rescue: Recommendations for Use states that selection of naloxone 8 mg should be based on shared decision-making between the patient and clinician and based on individual risk factors.12 The purpose of this study is to analyze data to determine if there is a difference in prescribing patterns of intranasal naloxone 4 mg and intranasal naloxone 8 mg.
METHODS
A retrospective chart reviews using the VA Computerized Patient Record System (CPRS) analyzed patients prescribed intranasal naloxone 4 mg or intranasal naloxone 8 mg at VHI. A patient list was generated based on active naloxone prescriptions between April 1, 2022, and April 1, 2023. Data were obtained exclusively through CPRS and patients were not contacted. This study was reviewed and deemed exempt by the Indiana University Health Institutional Review Board and the VHI Research and Development Committee.
Patients were included if they were aged ≥ 18 years and had an active prescription for intranasal naloxone 4 mg or intranasal naloxone 8 mg during the trial period. Patients were excluded if their naloxone prescription was written by a non-VHI clinician, if the dose was not 4 mg or 8 mg, or if the dosage form was other than intranasal spray.
The primary endpoint was the comparison for prescribing patterns for intranasal naloxone 4 mg and intranasal naloxone 8 mg during the study period. Secondary endpoints included total naloxone prescriptions; monthly prescriptions; number of patients with repeated naloxone prescriptions; prescriber type by naloxone dose; clinic type by naloxone dose; and documented indication for naloxone use by dose.
Demographic data collected included baseline age, sex, race, comorbid mental health conditions, and active central nervous system depressant medications on patient profile (ie, opioids, gabapentinoids, benzodiazepines, antidepressants, antipsychotics). Opioid prescriptions that were active or discontinued within the last 3 months were also recorded. Comorbid mental health conditions were collected based on the most recent clinical note before initiating medication.
Prescription-related data included strength of medication prescribed (4 mg, 8 mg, or both), documented use of medication, prescriber name, prescriber discipline, prescription entered by, number of times naloxone was filled or refilled during the study period, indication, clinic location, and clinic name. If > 1 prescription was active during the study period, the number of refills, prescriber name and clinic location of the first prescription in the study period was recorded. Additionally, the indication of OUD was differentiated from substance use disorder (SUD) if the patient was only dependent on opioids, excluding tobacco or alcohol. Patients with SUDs may include opioid dependence in addition to other substance dependence (eg, cannabis, stimulants, gabapentinoids, or benzodiazepines).
Basic descriptive statistics, including mean, ranges, and percentages were used to characterize the study subjects. For nominal data, X2 tests were used. A 2-sided 5% significance level was used for all statistical tests.
RESULTS
A total of 1952 active naloxone prescriptions from 1739 patients met the inclusion criteria; none were eliminated based on the exclusion criteria and some were included multiple times because data were collected for each active prescription during the study period. One hundred one patients were randomized and included in the final analysis (Figure). Most patients identified as White (81%), male (90%), and had a mean (SD) age of 60.9 (14.2) years. Common mental health comorbidities included 59 patients with depression, 50 with tobacco use disorder, and 31 with anxiety. Eighty-four patients had opioid and 60 had antidepressants/antianxiety, and 40 had gabapentinoids prescriptions. Forty-three patients had ≥ 3 mental health comorbidities. Thirty-four patients had 2 active central nervous system depressant prescriptions, 30 had 3 active prescriptions, and 9 had ≥ 4 active prescriptions. Most patients (n = 83) had an active or recently discontinued opioid prescription (Table 1).
The 101 patients received 54 prescriptions for naloxone 8 mg and 47 for 4 mg (Table 2). Five patients received prescriptions for both the 4 mg and 8 mg intranasal naloxone formulations. Sixty-six patients had naloxone filled once (66%) during the study period. Intranasal naloxone 4 mg was prescribed to 30 patients by nurse practitioners, 17 patients by physicians, and not prescribed by pharmacists. Intranasal naloxone 8 mg was prescribed to 40 patients by pharmacists, 13 patients by physicians, and 6 patients by nurses. Patients who received prescriptions for both intranasal naloxone 4 mg and 8 mg were most routinely ordered by physicians (n = 3; 60%) in primary care (n = 2; 40%) for chronic opioid use (n = 2; 40%).
Patients access naloxone from many different VHI clinics. Primary care clinics prescribed the 4 mg formulation to 31 patients, 8 mg to 3 patients, and both to 2 patients. The STORM initiative was used for 37 of 106 prescriptions (35%): 4 mg intranasal naloxone was prescribed to 1 patient, 8 mg to 36 patients, and no patients received both formulations. Chronic opioid use was the most common indication (46%) with 30 patients prescribed intranasal naloxone 4 mg, 14 patients prescribed 8 mg, and 2 patients prescribed both. OUD was the indication for 24% of patients: 2 patients prescribed intranasal naloxone 4 mg, 21 patients prescribed 8 mg, and 1 patient prescribed both.
The 106 intranasal naloxone prescriptions were equally distributed across each month from April 1, 2022, to April 1, 2023. Of the 101 patients, 34 had multiple naloxone prescriptions filled during the study period. Pharmacists wrote 40 of 106 naloxone prescriptions (38%), all for the 8 mg formulation. Nurse practitioners prescribed naloxone 4 mg 30 times and 8 mg 6 times for 36 of 106 prescriptions (34%). Physicians prescribed 30 of 106 prescriptions (28%), including intranasal naloxone 4 mg 17 times and 8 mg 13 times.
Statistics were analyzed using a X2 test; however, it was determined that the expected frequencies made the tests inappropriate. Differences in prescribing patterns between naloxone doses, prescriber disciplines, source of the prescription, or indications were not statistically significant.
DISCUSSION
Many pharmacists possess a scope of practice under state law and/or institution policy to prescribe naloxone. In this study, pharmacists prescribed the most naloxone prescriptions compared to physicians and nurse practitioners. Initiatives such as OEND and STORM have given pharmacists at VHI an avenue to combat the growing opioid epidemic while expanding their scope of practice. A systematic review of 67 studies found that pharmacist-led OEND programs showed a statistically significant increase in naloxone orders. A statistical significance was likely met given the large sample sizes ranging from 10 to 217,000 individuals, whereas this study only assessed a small portion of patients.14 This study contributes to the overwhelming amount of data that highlights pharmacists’ impact on overall naloxone distribution.
The STORM initiative and primary care clinics were responsible for large portions of naloxone prescriptions in this study. STORM was used by pharmacists and contributed to more than half of the higher dose naloxone prescriptions. Following a discussion with members of the pain management team, pharmacists involved in STORM prescribing were revealed to exclusively prescribe intranasal naloxone 8 mg as opposed to 4 mg. At the risk of precipitating withdrawal from higher doses of naloxone, it was agreed that this risk was heavily outweighed by the benefit of successful opioid reversal. In this context, it is expected for this avenue of prescribing to influence naloxone prescribing patterns at VHI.
Prescribing in primary care clinics was shown to be equally as substantial. Primary care-based multidisciplinary transition clinics have been reported to be associated with increased access to OUD treatment.15 Primary care clinics at VHI, or patient aligned care teams (PACT), largely consist of multidisciplinary health care teams. PACT clinicians are heavily involved in transitions of care because one system provides patients with comprehensive acute and chronic care. Continuing to encourage naloxone distribution through primary care and using STORM affords various patient populations access to high-level care.
Notable differences were observed between indications for naloxone use and the corresponding dose. Patients with OUD or SUD were more likely to receive intranasal naloxone 8 mg as opposed to patients receiving intranasal naloxone for chronic opioid use, who were more likely to receive the 4 mg dose. This may be due to a rationale to provide a higher dose of naloxone to combat overdoses in the case of ingesting substances mixed with fentanyl or xylazine.12,13 Without standard of care guidelines, concerns remain for varying outcomes in opioid overdose prevention within vulnerable populations.
Limitations
Chart data were dependent on documentation, which may have omitted pertinent baseline characteristics and risk factors. Additional data collection could have further assessed a patient’s specific risk factors (eg, opioid dose in morphine equivalents) to draw conclusions to the dose of naloxone prescribed. The sample size was small, and the patient population was largely White and male, which minimized the generalizability of the results.
CONCLUSIONS
This study evaluated the differences in intranasal naloxone prescribing patterns within a veteran population at VHI over 12 months. Findings revealed that most prescriptions were written for intranasal naloxone 8 mg, by a pharmacist, in a primary care setting, and for chronic opioid use. The results revealed evidence of differing naloxone prescribing practices, which emphasize the need for clinical guidelines and better defined recommendations in relation to naloxone dosing.
The most evident gap in patient care could be addressed by urging the VA Pharmacy Benefits Management group to update naloxone recommendations for use to include more concrete dosing recommendations. Furthermore, it would be beneficial to re-educate clinicians on naloxone prescribing to increase awareness of different doses and the importance of equipping patients with the correct amount of naloxone in an emergency. Additional research assessing change in prescribing patterns is warranted as the use of higher dose naloxone becomes more routine.
Since 1999, annual deaths attributed to opioid overdose in the United States have increased from about 10,000 to about 50,000 in 2019.1 During the COVID-19 pandemic > 74,000 opioid overdose deaths occurred in the US from April 2020 to April 2021.2,3 Opioid-related overdoses now account for about 75% of all drug-related overdose deaths.1 In 2017, the cost of opioid overdose deaths and opioid use disorder (OUD) reached $1.02 trillion in the United States and $26 million in Indiana.4 The total deaths and costs would likely be higher if it were not for naloxone.
Naloxone hydrochloride was first patented in the 1960s and approved by the US Food and Drug Administration (FDA) in 1971 to treat opioid-related toxicity.1 It is the most frequently prescribed antidote for opioid toxicity due to its activity as a pure υ-opioid receptor competitive antagonist. Naloxone formulations include intramuscular, intravenous, subcutaneous, and intranasal delivery methods.5 According to the Centers for Disease Control and Prevention, clinicians should offer naloxone to patients at high risk for opioid-related adverse events. Risk factors include a history of overdose, opioid dosages of ≥ 50 morphine mg equivalents/day, and concurrent use of opioids with benzodiazepines.6
Intranasal naloxone 4 mg has become more accessible following the classification of opioid use as a public health emergency in 2017 and its over-the-counter availability since 2023. Intranasal naloxone 4 mg was approved by the FDA in 2015 for the prevention of opioid overdoses (accidental or intentional), which can be caused by heroin, fentanyl, carfentanil, hydrocodone, oxycodone, methadone, and other substances. 7 Fentanyl has most recently been associated with xylazine, a nonopioid tranquilizer linked to increased opioid overdose deaths.8 Recent data suggest that 34% of opioid overdose reversals involved ≥ 2 doses of intranasal naloxone 4 mg, which led to FDA approval of an intranasal naloxone 8 mg spray in April 2021.9-11
Veteran Health Indiana (VHI) has implemented several initiatives to promote naloxone prescribing. Established in 2020, the Opioid Overdose Education and Naloxone Distribution (OEND) program sought to prevent opioid-related deaths through education and product distribution. These criteria included an opioid prescription for ≥ 30 days. In 2021, the Stratification Tool for Opioid Risk Mitigation (STORM) was created to identify patients at high risk of opioid overdose and allowing pharmacists to prescribe naloxone for at-risk patients without restrictions, increasing accessibility.12
Recent cases of fentanyl-related overdoses involving stronger fentanyl analogues highlight the need for higher naloxone dosing to prevent overdose. A pharmacokinetic comparison of intranasal naloxone 8 mg vs 4 mg demonstrated maximum plasma concentrations of 10.3 ng/mL and 5.3 ng/mL, respectively. 13 Patients may be at an increased risk of precipitated opioid withdrawal when using intranasal naloxone 8 mg over 4 mg; however, some patients may benefit from achieving higher serum concentrations and therefore require larger doses of naloxone.
No clinical trials have demonstrated a difference in reversal rates between naloxone doses. No clinical practice guidelines support a specific naloxone formulation, and limited US Department of Veterans Affairs (VA)-specific guidance exists. VA Naloxone Rescue: Recommendations for Use states that selection of naloxone 8 mg should be based on shared decision-making between the patient and clinician and based on individual risk factors.12 The purpose of this study is to analyze data to determine if there is a difference in prescribing patterns of intranasal naloxone 4 mg and intranasal naloxone 8 mg.
METHODS
A retrospective chart reviews using the VA Computerized Patient Record System (CPRS) analyzed patients prescribed intranasal naloxone 4 mg or intranasal naloxone 8 mg at VHI. A patient list was generated based on active naloxone prescriptions between April 1, 2022, and April 1, 2023. Data were obtained exclusively through CPRS and patients were not contacted. This study was reviewed and deemed exempt by the Indiana University Health Institutional Review Board and the VHI Research and Development Committee.
Patients were included if they were aged ≥ 18 years and had an active prescription for intranasal naloxone 4 mg or intranasal naloxone 8 mg during the trial period. Patients were excluded if their naloxone prescription was written by a non-VHI clinician, if the dose was not 4 mg or 8 mg, or if the dosage form was other than intranasal spray.
The primary endpoint was the comparison for prescribing patterns for intranasal naloxone 4 mg and intranasal naloxone 8 mg during the study period. Secondary endpoints included total naloxone prescriptions; monthly prescriptions; number of patients with repeated naloxone prescriptions; prescriber type by naloxone dose; clinic type by naloxone dose; and documented indication for naloxone use by dose.
Demographic data collected included baseline age, sex, race, comorbid mental health conditions, and active central nervous system depressant medications on patient profile (ie, opioids, gabapentinoids, benzodiazepines, antidepressants, antipsychotics). Opioid prescriptions that were active or discontinued within the last 3 months were also recorded. Comorbid mental health conditions were collected based on the most recent clinical note before initiating medication.
Prescription-related data included strength of medication prescribed (4 mg, 8 mg, or both), documented use of medication, prescriber name, prescriber discipline, prescription entered by, number of times naloxone was filled or refilled during the study period, indication, clinic location, and clinic name. If > 1 prescription was active during the study period, the number of refills, prescriber name and clinic location of the first prescription in the study period was recorded. Additionally, the indication of OUD was differentiated from substance use disorder (SUD) if the patient was only dependent on opioids, excluding tobacco or alcohol. Patients with SUDs may include opioid dependence in addition to other substance dependence (eg, cannabis, stimulants, gabapentinoids, or benzodiazepines).
Basic descriptive statistics, including mean, ranges, and percentages were used to characterize the study subjects. For nominal data, X2 tests were used. A 2-sided 5% significance level was used for all statistical tests.
RESULTS
A total of 1952 active naloxone prescriptions from 1739 patients met the inclusion criteria; none were eliminated based on the exclusion criteria and some were included multiple times because data were collected for each active prescription during the study period. One hundred one patients were randomized and included in the final analysis (Figure). Most patients identified as White (81%), male (90%), and had a mean (SD) age of 60.9 (14.2) years. Common mental health comorbidities included 59 patients with depression, 50 with tobacco use disorder, and 31 with anxiety. Eighty-four patients had opioid and 60 had antidepressants/antianxiety, and 40 had gabapentinoids prescriptions. Forty-three patients had ≥ 3 mental health comorbidities. Thirty-four patients had 2 active central nervous system depressant prescriptions, 30 had 3 active prescriptions, and 9 had ≥ 4 active prescriptions. Most patients (n = 83) had an active or recently discontinued opioid prescription (Table 1).
The 101 patients received 54 prescriptions for naloxone 8 mg and 47 for 4 mg (Table 2). Five patients received prescriptions for both the 4 mg and 8 mg intranasal naloxone formulations. Sixty-six patients had naloxone filled once (66%) during the study period. Intranasal naloxone 4 mg was prescribed to 30 patients by nurse practitioners, 17 patients by physicians, and not prescribed by pharmacists. Intranasal naloxone 8 mg was prescribed to 40 patients by pharmacists, 13 patients by physicians, and 6 patients by nurses. Patients who received prescriptions for both intranasal naloxone 4 mg and 8 mg were most routinely ordered by physicians (n = 3; 60%) in primary care (n = 2; 40%) for chronic opioid use (n = 2; 40%).
Patients access naloxone from many different VHI clinics. Primary care clinics prescribed the 4 mg formulation to 31 patients, 8 mg to 3 patients, and both to 2 patients. The STORM initiative was used for 37 of 106 prescriptions (35%): 4 mg intranasal naloxone was prescribed to 1 patient, 8 mg to 36 patients, and no patients received both formulations. Chronic opioid use was the most common indication (46%) with 30 patients prescribed intranasal naloxone 4 mg, 14 patients prescribed 8 mg, and 2 patients prescribed both. OUD was the indication for 24% of patients: 2 patients prescribed intranasal naloxone 4 mg, 21 patients prescribed 8 mg, and 1 patient prescribed both.
The 106 intranasal naloxone prescriptions were equally distributed across each month from April 1, 2022, to April 1, 2023. Of the 101 patients, 34 had multiple naloxone prescriptions filled during the study period. Pharmacists wrote 40 of 106 naloxone prescriptions (38%), all for the 8 mg formulation. Nurse practitioners prescribed naloxone 4 mg 30 times and 8 mg 6 times for 36 of 106 prescriptions (34%). Physicians prescribed 30 of 106 prescriptions (28%), including intranasal naloxone 4 mg 17 times and 8 mg 13 times.
Statistics were analyzed using a X2 test; however, it was determined that the expected frequencies made the tests inappropriate. Differences in prescribing patterns between naloxone doses, prescriber disciplines, source of the prescription, or indications were not statistically significant.
DISCUSSION
Many pharmacists possess a scope of practice under state law and/or institution policy to prescribe naloxone. In this study, pharmacists prescribed the most naloxone prescriptions compared to physicians and nurse practitioners. Initiatives such as OEND and STORM have given pharmacists at VHI an avenue to combat the growing opioid epidemic while expanding their scope of practice. A systematic review of 67 studies found that pharmacist-led OEND programs showed a statistically significant increase in naloxone orders. A statistical significance was likely met given the large sample sizes ranging from 10 to 217,000 individuals, whereas this study only assessed a small portion of patients.14 This study contributes to the overwhelming amount of data that highlights pharmacists’ impact on overall naloxone distribution.
The STORM initiative and primary care clinics were responsible for large portions of naloxone prescriptions in this study. STORM was used by pharmacists and contributed to more than half of the higher dose naloxone prescriptions. Following a discussion with members of the pain management team, pharmacists involved in STORM prescribing were revealed to exclusively prescribe intranasal naloxone 8 mg as opposed to 4 mg. At the risk of precipitating withdrawal from higher doses of naloxone, it was agreed that this risk was heavily outweighed by the benefit of successful opioid reversal. In this context, it is expected for this avenue of prescribing to influence naloxone prescribing patterns at VHI.
Prescribing in primary care clinics was shown to be equally as substantial. Primary care-based multidisciplinary transition clinics have been reported to be associated with increased access to OUD treatment.15 Primary care clinics at VHI, or patient aligned care teams (PACT), largely consist of multidisciplinary health care teams. PACT clinicians are heavily involved in transitions of care because one system provides patients with comprehensive acute and chronic care. Continuing to encourage naloxone distribution through primary care and using STORM affords various patient populations access to high-level care.
Notable differences were observed between indications for naloxone use and the corresponding dose. Patients with OUD or SUD were more likely to receive intranasal naloxone 8 mg as opposed to patients receiving intranasal naloxone for chronic opioid use, who were more likely to receive the 4 mg dose. This may be due to a rationale to provide a higher dose of naloxone to combat overdoses in the case of ingesting substances mixed with fentanyl or xylazine.12,13 Without standard of care guidelines, concerns remain for varying outcomes in opioid overdose prevention within vulnerable populations.
Limitations
Chart data were dependent on documentation, which may have omitted pertinent baseline characteristics and risk factors. Additional data collection could have further assessed a patient’s specific risk factors (eg, opioid dose in morphine equivalents) to draw conclusions to the dose of naloxone prescribed. The sample size was small, and the patient population was largely White and male, which minimized the generalizability of the results.
CONCLUSIONS
This study evaluated the differences in intranasal naloxone prescribing patterns within a veteran population at VHI over 12 months. Findings revealed that most prescriptions were written for intranasal naloxone 8 mg, by a pharmacist, in a primary care setting, and for chronic opioid use. The results revealed evidence of differing naloxone prescribing practices, which emphasize the need for clinical guidelines and better defined recommendations in relation to naloxone dosing.
The most evident gap in patient care could be addressed by urging the VA Pharmacy Benefits Management group to update naloxone recommendations for use to include more concrete dosing recommendations. Furthermore, it would be beneficial to re-educate clinicians on naloxone prescribing to increase awareness of different doses and the importance of equipping patients with the correct amount of naloxone in an emergency. Additional research assessing change in prescribing patterns is warranted as the use of higher dose naloxone becomes more routine.
- Britch SC, Walsh SL. Treatment of opioid overdose: current approaches and recent advances. Psychopharmacology (Berl). 2022;239(7):2063-2081. doi:10.1007/s00213-022-06125-5
- Ahmad FB, Cisewski JA, Rossen LM, Sutton P. Provisional Drug Overdose Death Counts. National Center for Health Statistics, Centers for Disease Control and Prevention; 2023. Accessed April 10, 2025. https://www.cdc.gov/nchs/nvss/vsrr/drug-overdose-data.htm
- O’Donnell J, Tanz LJ, Gladden RM, Davis NL, Bitting J. Trends in and characteristics of drug overdose deaths involving illicitly manufactured fentanyls — United States, 2019–2020. MMWR Morb Mortal Wkly Rep. 2021;70:1740-1746. doi:10.15585/mmwr.mm7050e3
- Luo F, Li M, Florence C. State-level economic costs of opioid use disorder and fatal opioid overdose — United States, 2017. MMWR Morb Mortal Wkly Rep. 2021;70:541-546. doi:10.15585/mmwr.mm7015a1
- Lexicomp. Lexicomp Online. Accessed April 10, 2025. http://online.lexi.com
- Dowell D, Ragan KR, Jones CM, Baldwin GT, Chou R. CDC Clinical practice guideline for prescribing opioids for pain — United States, 2022. MMWR Recomm Rep. 2022;71(3):1-95. doi:10.15585/mmwr.rr7103a1
- Narcan (naloxone) FDA approval history. Drugs.com. Accessed April 10, 2025. https://www.drugs.com/history/narcan.html
- Centers for Disease Control and Prevention. What you should know about xylazine. May 16, 2024. Accessed April 10, 2025. https://www.cdc.gov/overdose-prevention/about/what-you-should-know-about-xylazine.html
- Avetian GK, Fiuty P, Mazzella S, Koppa D, Heye V, Hebbar P. Use of naloxone nasal spray 4 mg in the community setting: a survey of use by community organizations. Curr Med Res Opin. 2018;34(4):573-576. doi:10.1080/03007995.2017.1334637
- Kloxxado [package insert]. Hikma Pharmaceuticals USA Inc; 2021.
- FDA approves higher dosage of naloxone nasal spray to treat opioid overdose. News release. FDA. April 30, 2021. Accessed April 10, 2025. https://www.fda.gov/news-events/press-announcements/fda-approves-higher-dosage-naloxone-nasal-spray-treat-opioid-overdose
- US Department of Veterans Affairs, Pharmacy Benefits Management Services and National Formulary Committee in Collaboration with the VA National Harm Reduction Support & Development Workgroup. Naloxone Rescue: Recommendations for Use. June 2014. Updated March 2024. Accessed April 10, 2025. https://www.va.gov/formularyadvisor/DOC_PDF/CRE_Naloxone_Rescue_Guidance_March_2024.pdf
- Krieter P, Chiang N, Gyaw S, et al. Pharmacokinetic properties and human use characteristics of an FDA-approved intranasal naloxone product for the treatment of opioid overdose. J Clin Pharmacol. 2016;56(10):1243-1253. doi:10.1002/jcph.759
- Rawal S, Osae SP, Cobran EK, Albert A, Young HN. Pharmacists’ naloxone services beyond community pharmacy settings: a systematic review. Res Social Adm Pharm. 2023;19(2):243-265. doi:10.1016/j.sapharm.2022.09.002
- Incze MA, Sehgal SL, Hansen A, Garcia L, Stolebarger L. Evaluation of a primary care-based multidisciplinary transition clinic for patients newly initiated on buprenorphine in the emergency department. Subst Abus. 2023;44(3):220-225. doi:10.1177/08897077231188592
- Britch SC, Walsh SL. Treatment of opioid overdose: current approaches and recent advances. Psychopharmacology (Berl). 2022;239(7):2063-2081. doi:10.1007/s00213-022-06125-5
- Ahmad FB, Cisewski JA, Rossen LM, Sutton P. Provisional Drug Overdose Death Counts. National Center for Health Statistics, Centers for Disease Control and Prevention; 2023. Accessed April 10, 2025. https://www.cdc.gov/nchs/nvss/vsrr/drug-overdose-data.htm
- O’Donnell J, Tanz LJ, Gladden RM, Davis NL, Bitting J. Trends in and characteristics of drug overdose deaths involving illicitly manufactured fentanyls — United States, 2019–2020. MMWR Morb Mortal Wkly Rep. 2021;70:1740-1746. doi:10.15585/mmwr.mm7050e3
- Luo F, Li M, Florence C. State-level economic costs of opioid use disorder and fatal opioid overdose — United States, 2017. MMWR Morb Mortal Wkly Rep. 2021;70:541-546. doi:10.15585/mmwr.mm7015a1
- Lexicomp. Lexicomp Online. Accessed April 10, 2025. http://online.lexi.com
- Dowell D, Ragan KR, Jones CM, Baldwin GT, Chou R. CDC Clinical practice guideline for prescribing opioids for pain — United States, 2022. MMWR Recomm Rep. 2022;71(3):1-95. doi:10.15585/mmwr.rr7103a1
- Narcan (naloxone) FDA approval history. Drugs.com. Accessed April 10, 2025. https://www.drugs.com/history/narcan.html
- Centers for Disease Control and Prevention. What you should know about xylazine. May 16, 2024. Accessed April 10, 2025. https://www.cdc.gov/overdose-prevention/about/what-you-should-know-about-xylazine.html
- Avetian GK, Fiuty P, Mazzella S, Koppa D, Heye V, Hebbar P. Use of naloxone nasal spray 4 mg in the community setting: a survey of use by community organizations. Curr Med Res Opin. 2018;34(4):573-576. doi:10.1080/03007995.2017.1334637
- Kloxxado [package insert]. Hikma Pharmaceuticals USA Inc; 2021.
- FDA approves higher dosage of naloxone nasal spray to treat opioid overdose. News release. FDA. April 30, 2021. Accessed April 10, 2025. https://www.fda.gov/news-events/press-announcements/fda-approves-higher-dosage-naloxone-nasal-spray-treat-opioid-overdose
- US Department of Veterans Affairs, Pharmacy Benefits Management Services and National Formulary Committee in Collaboration with the VA National Harm Reduction Support & Development Workgroup. Naloxone Rescue: Recommendations for Use. June 2014. Updated March 2024. Accessed April 10, 2025. https://www.va.gov/formularyadvisor/DOC_PDF/CRE_Naloxone_Rescue_Guidance_March_2024.pdf
- Krieter P, Chiang N, Gyaw S, et al. Pharmacokinetic properties and human use characteristics of an FDA-approved intranasal naloxone product for the treatment of opioid overdose. J Clin Pharmacol. 2016;56(10):1243-1253. doi:10.1002/jcph.759
- Rawal S, Osae SP, Cobran EK, Albert A, Young HN. Pharmacists’ naloxone services beyond community pharmacy settings: a systematic review. Res Social Adm Pharm. 2023;19(2):243-265. doi:10.1016/j.sapharm.2022.09.002
- Incze MA, Sehgal SL, Hansen A, Garcia L, Stolebarger L. Evaluation of a primary care-based multidisciplinary transition clinic for patients newly initiated on buprenorphine in the emergency department. Subst Abus. 2023;44(3):220-225. doi:10.1177/08897077231188592
Comparison of Prescribing Patterns of Intranasal Naloxone in a Veteran Population
Comparison of Prescribing Patterns of Intranasal Naloxone in a Veteran Population

The Unholy Trinity: Unlawful Prescriptions, False Claims, and Dangerous Drugs
The Unholy Trinity: Unlawful Prescriptions, False Claims, and Dangerous Drugs
Express Scripts, the contractor that manages the pharmacy benefit for Tricare, the military health insurance program, announced in 2021 that after a 5-year absence, CVS Pharmacy was once more in the network. In 2023, CVS had the largest profits of any pharmacy chain in the United States, about $159 billion, and generated a quarter of the overall revenue of the US pharmacy industry.1 Tricare officials heralded the return of CVS as a move that would offer US Department of Defense (DoD) beneficiaries more competitive prices, convenient access, and overall quality.2
DOJ Files Lawsuit Against CVS
In December 2024, the US Department of Justice (DoJ) filed a lawsuit alleging that CVS violated both the Controlled Substances Act (CSA) and the False Claims Act (FCA).3,4 The United States ex rel. Estright v Health Corporation, et al, filed in Rhode Island, charged that CVS “routinely” and “knowingly” filled invalid prescriptions for controlled substances violating the CSA and then billed federal health care programs for payment for these prescriptions, a breach of the FCA.5 The DoJ alleged that CVS pharmacies and pharmacists filled prescriptions for controlled substances that (1) lacked a legitimate medical purpose; (2) were not legally valid; and/or (3) were not issued in the usual course of medical practice. 6 CVS contests the charges and issued an official response, stating that it disputes the allegations as false, plans to disprove them in litigation, and has nonetheless fully cooperated with the investigation.7
The allegations involved prescriptions for drugs like opioids and benzodiazepines, primary culprits in the American overdose epidemic.8 The complaint notes that the prescriptions were early refills in excessive quantities and included what has been called the “holy trinity” of dangerous medications: opioids, benzodiazepines, and muscle relaxants. 5,8 Even worse (if that is possible), as the complaint outlines, CVS had access to data from both inside and outside the company that these prescriptions came from notorious pill mills and were hence unlawful and yet continued to fill them, leading the DoJ to file the more serious charge that the corporation “knowingly” violated the CSA and “prioritized profits over safety in dispensing controlled substances.”5,6
The Unholy Trinity
The infamous members of what I prefer to call the “unholy trinity” are a benzodiazepine, often alprazolam, an opioid, and the muscle relaxant carisoprodol. The combination amplifies each agent’s independent risk of respiratory depression. The latter is a schedule IV medication with an active metabolite, meprobamate, that also has this adverse effect. All 3 drugs have high abuse potential and, when combined, increase the risk of fatal overdose. The colloquial name holy trinity derives from the synergistic euphoria experienced when taking this triple cocktail of sedative agents.9 This pharmacological recipe for disaster is the house specialty of pill mills: infamous storefront practices that generate high profits and exploit persons with chronic pain and addiction by handing out controlled substances with little clinical assessment and even less oversight.10
When the Means Become the End
The DoJ allegations suggest that the violations resulted from “corporate-mandated performance metrics, incentive compensation, and staffing policies that prioritized corporate profits over patient safety.”6 If the allegations are true, why would a company reinvited by Tricare to serve the nation’s heroes seemingly engage in illegal practices? While CVS has not responded in court, their statement argued that “too often, we have seen government agencies and trial lawyers question the good-faith decisions made by pharmacists while a patient waits at the pharmacy counter, often in pain.”6
The DoJ complaint offers a cautionary warning for the US health care system, which is increasingly being micromanaged in the pursuit of efficiency. Like many practitioners in and out of the federal system, I get a cold chill when I read the word productivity. “CVS pharmacists described working at CVS as ‘soul crushing’ because it was impossible to meet the company’s expectations,” the complaint alleges, because “CVS set staffing levels so low that it was impossible for pharmacists to comply with their legal obligations and meet CVS’s demanding metrics.”5 Did top-down mandates drive the alleged activities by imposing unattainable performance metrics on pharmacists, offering incentives that encouraged and rewarded corner-cutting, and refusing to fund sufficient staffing to ensure patient safety? This may be what happens when the means (efficiency) become the end rather than a mechanism to achieve the goal of more accessible, affordable, high-quality health care.
Ethically, what is most concerning is that leadership intentionally “deprived its pharmacists of crucial information” about specific practitioners known to engage in illegal prescribing practices.6 CVS did not provide pharmacists with “information about prescribers’ prescribing habits that CVS routinely collected and reviewed at the corporate level,” and even removed prescriber blocks that were implemented at Target pharmacies before it was acquired by CVS.5 The first element of informed consent is providing patients with adequate information upon which to decide whether to accept or decline treatment. 11 In this situation, however, CVS allegedly prevented “pharmacists from warning one another about certain prescribers.”6
If true, the company deprived frontline pharmacists of the information they needed to safely and responsibly dispense medications: “The practices alleged contributed to the opioid crisis and opioid-related deaths, and today’s complaint seeks to hold CVS accountable for its misconduct.”6 Though the cost in human life that may have resulted from CSA violations must absolutely and always outweigh financial considerations, the economic damage to Tricare from fraudulent billing and the betrayal of its fiduciary responsinility cannot be underestimated.
A Corporate Morality Play
CVS is not the only company, nor is pharmacy the only industry in health care, that has been the subject of watchdog agency lawsuits or variegated forms of wrongdoing, including violations of the CSA and FCA.10,12 As of this writing, the DoJ case against CVS has not been heard, much less adjudicated in a court of law. It is ironic that both the DoJ claims and the CVS rebuttal describe the manifest conflict of obligation that pharmacists confront between protecting their livelihood and safeguarding patients’ lives as suggested in the epigraph that has been attributed to the 19th-century British physician and medical educator Peter Mere Latham. It is a dilemma that a growing number of health care practitioners face daily in a vocation becoming increasingly commercialized. It is all too easy for an individual physician, nurse, or pharmacist to feel hopeless and helpless before the behemoth might of a large and looming entity. Yet, it was a whistleblower whose moral courage led to the DoJ investigation and subsequent charges.13 We must all never doubt the power of a committed person of conscience to withstand the pressure to mutate medications into poison and stand up for the principles of our professions and inspire a community of colleagues to follow their example.
- Fein AJ. The Top U.S. pharmacy markets of 2023: market shares and revenues at the biggest chains and PBMs. Drug Channels. March 12, 2024. Accessed February 24, 2025. https://www.drugchannels.net/2024/03/the-top-15-us-pharmacies-of-2023-market.html
- Jowers K. CVS returns to the military Tricare network. Walmart’s out. Military Times. October 18, 2021. Accessed February 24, 2025. https://www.militarytimes.com/pay-benefits/mil-money/2021/10/28/cvs-returns-to-the-military-tricare-pharmacy-network-walmarts-out/
- False Claims, 31 USC § 3729 (2009). Accessed February 24, 2025. https://www.govinfo.gov/content/pkg/USCODE-2011-title31/pdf/USCODE-2011-title31-subtitleIII-chap37-subchapIII-sec3729.pdf
- Drug Abuse Prevention and Control, Control and Enforcement, 21 USC 13 § 801 (2022). Accessed February 24, 2025. https://www.govinfo.gov/app/details/USCODE-2021-title21/USCODE-2021-title21-chap13-subchapI-partA-sec801
- United States ex rel. Estright v Health Corporation, et al. Accessed February 26, 2025. https://www.justice.gov/archives/opa/media/1381111/dl
- US Department of Justice. Justice Department files nationwide lawsuit alleging CVS knowingly dispensed controlled substances in violation of the Controlled Substances ACT and the False Claims Act. News release. December 18, 2024. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/justice-department-files-nationwide-lawsuit-alleging-cvs-knowingly-dispensed-controlled
- CVS Health. CVS Health statement regarding the U.S. Department of Justice’s lawsuit against CVS pharmacy. News release. December 18, 2024. Accessed February 24, 2025. https://www.cvshealth.com/impact/healthy-community/our-opioid-response.html
- Park TW, Saitz R, Ganoczy D, Ilgen MA, Bohnert AS. Benzodiazepine prescribing patterns and deaths from drug overdose among US veterans receiving opioid analgesics: case-cohort study. BMJ. 2015;350:h2698. doi:10.1136/bmj.h2698
- Wang Y, Delcher C, Li Y, Goldberger BA, Reisfield GM. Overlapping prescriptions of opioids, benzodiazepines, and carisoprodol: “Holy Trinity” prescribing in the state of Florida. Drug Alcohol Depend. 2019;205:107693. doi:10.1016/j.drugalcdep.2019.107693
- Wolf AA. The perfect storm: opioid risks and ‘The Holy Trinity’. Pharmacy Times. September 24, 2014. Accessed February 24, 2025. https://www.pharmacytimes.com/view/the-perfect-storm-opioid-risks-and-the-holy-trinity
- The meaning and justification of informed consent. In: Beauchamp TL, Childress JF. Principles of Biomedical Ethics. Eighth Edition. Oxford University Press; 2019:118-123.
- US Department of Justice. OptumRX agrees to pay $20M to resolve allegations that it filled certain opioid prescriptions in violation of the Controlled Substances Act. News release. June 27, 2024. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/optumrx-agrees-pay-20m-resolve-allegations-it-filled-certain-opioid-prescriptions-violation
- US Department of Justice. False Claims Act settlements and judgments exceed $2.9B in fiscal year 2024. News release. January 15, 2025. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/false-claims-act-settlements-and-judgments-exceed-29b-fiscal-year-2024
Express Scripts, the contractor that manages the pharmacy benefit for Tricare, the military health insurance program, announced in 2021 that after a 5-year absence, CVS Pharmacy was once more in the network. In 2023, CVS had the largest profits of any pharmacy chain in the United States, about $159 billion, and generated a quarter of the overall revenue of the US pharmacy industry.1 Tricare officials heralded the return of CVS as a move that would offer US Department of Defense (DoD) beneficiaries more competitive prices, convenient access, and overall quality.2
DOJ Files Lawsuit Against CVS
In December 2024, the US Department of Justice (DoJ) filed a lawsuit alleging that CVS violated both the Controlled Substances Act (CSA) and the False Claims Act (FCA).3,4 The United States ex rel. Estright v Health Corporation, et al, filed in Rhode Island, charged that CVS “routinely” and “knowingly” filled invalid prescriptions for controlled substances violating the CSA and then billed federal health care programs for payment for these prescriptions, a breach of the FCA.5 The DoJ alleged that CVS pharmacies and pharmacists filled prescriptions for controlled substances that (1) lacked a legitimate medical purpose; (2) were not legally valid; and/or (3) were not issued in the usual course of medical practice. 6 CVS contests the charges and issued an official response, stating that it disputes the allegations as false, plans to disprove them in litigation, and has nonetheless fully cooperated with the investigation.7
The allegations involved prescriptions for drugs like opioids and benzodiazepines, primary culprits in the American overdose epidemic.8 The complaint notes that the prescriptions were early refills in excessive quantities and included what has been called the “holy trinity” of dangerous medications: opioids, benzodiazepines, and muscle relaxants. 5,8 Even worse (if that is possible), as the complaint outlines, CVS had access to data from both inside and outside the company that these prescriptions came from notorious pill mills and were hence unlawful and yet continued to fill them, leading the DoJ to file the more serious charge that the corporation “knowingly” violated the CSA and “prioritized profits over safety in dispensing controlled substances.”5,6
The Unholy Trinity
The infamous members of what I prefer to call the “unholy trinity” are a benzodiazepine, often alprazolam, an opioid, and the muscle relaxant carisoprodol. The combination amplifies each agent’s independent risk of respiratory depression. The latter is a schedule IV medication with an active metabolite, meprobamate, that also has this adverse effect. All 3 drugs have high abuse potential and, when combined, increase the risk of fatal overdose. The colloquial name holy trinity derives from the synergistic euphoria experienced when taking this triple cocktail of sedative agents.9 This pharmacological recipe for disaster is the house specialty of pill mills: infamous storefront practices that generate high profits and exploit persons with chronic pain and addiction by handing out controlled substances with little clinical assessment and even less oversight.10
When the Means Become the End
The DoJ allegations suggest that the violations resulted from “corporate-mandated performance metrics, incentive compensation, and staffing policies that prioritized corporate profits over patient safety.”6 If the allegations are true, why would a company reinvited by Tricare to serve the nation’s heroes seemingly engage in illegal practices? While CVS has not responded in court, their statement argued that “too often, we have seen government agencies and trial lawyers question the good-faith decisions made by pharmacists while a patient waits at the pharmacy counter, often in pain.”6
The DoJ complaint offers a cautionary warning for the US health care system, which is increasingly being micromanaged in the pursuit of efficiency. Like many practitioners in and out of the federal system, I get a cold chill when I read the word productivity. “CVS pharmacists described working at CVS as ‘soul crushing’ because it was impossible to meet the company’s expectations,” the complaint alleges, because “CVS set staffing levels so low that it was impossible for pharmacists to comply with their legal obligations and meet CVS’s demanding metrics.”5 Did top-down mandates drive the alleged activities by imposing unattainable performance metrics on pharmacists, offering incentives that encouraged and rewarded corner-cutting, and refusing to fund sufficient staffing to ensure patient safety? This may be what happens when the means (efficiency) become the end rather than a mechanism to achieve the goal of more accessible, affordable, high-quality health care.
Ethically, what is most concerning is that leadership intentionally “deprived its pharmacists of crucial information” about specific practitioners known to engage in illegal prescribing practices.6 CVS did not provide pharmacists with “information about prescribers’ prescribing habits that CVS routinely collected and reviewed at the corporate level,” and even removed prescriber blocks that were implemented at Target pharmacies before it was acquired by CVS.5 The first element of informed consent is providing patients with adequate information upon which to decide whether to accept or decline treatment. 11 In this situation, however, CVS allegedly prevented “pharmacists from warning one another about certain prescribers.”6
If true, the company deprived frontline pharmacists of the information they needed to safely and responsibly dispense medications: “The practices alleged contributed to the opioid crisis and opioid-related deaths, and today’s complaint seeks to hold CVS accountable for its misconduct.”6 Though the cost in human life that may have resulted from CSA violations must absolutely and always outweigh financial considerations, the economic damage to Tricare from fraudulent billing and the betrayal of its fiduciary responsinility cannot be underestimated.
A Corporate Morality Play
CVS is not the only company, nor is pharmacy the only industry in health care, that has been the subject of watchdog agency lawsuits or variegated forms of wrongdoing, including violations of the CSA and FCA.10,12 As of this writing, the DoJ case against CVS has not been heard, much less adjudicated in a court of law. It is ironic that both the DoJ claims and the CVS rebuttal describe the manifest conflict of obligation that pharmacists confront between protecting their livelihood and safeguarding patients’ lives as suggested in the epigraph that has been attributed to the 19th-century British physician and medical educator Peter Mere Latham. It is a dilemma that a growing number of health care practitioners face daily in a vocation becoming increasingly commercialized. It is all too easy for an individual physician, nurse, or pharmacist to feel hopeless and helpless before the behemoth might of a large and looming entity. Yet, it was a whistleblower whose moral courage led to the DoJ investigation and subsequent charges.13 We must all never doubt the power of a committed person of conscience to withstand the pressure to mutate medications into poison and stand up for the principles of our professions and inspire a community of colleagues to follow their example.
Express Scripts, the contractor that manages the pharmacy benefit for Tricare, the military health insurance program, announced in 2021 that after a 5-year absence, CVS Pharmacy was once more in the network. In 2023, CVS had the largest profits of any pharmacy chain in the United States, about $159 billion, and generated a quarter of the overall revenue of the US pharmacy industry.1 Tricare officials heralded the return of CVS as a move that would offer US Department of Defense (DoD) beneficiaries more competitive prices, convenient access, and overall quality.2
DOJ Files Lawsuit Against CVS
In December 2024, the US Department of Justice (DoJ) filed a lawsuit alleging that CVS violated both the Controlled Substances Act (CSA) and the False Claims Act (FCA).3,4 The United States ex rel. Estright v Health Corporation, et al, filed in Rhode Island, charged that CVS “routinely” and “knowingly” filled invalid prescriptions for controlled substances violating the CSA and then billed federal health care programs for payment for these prescriptions, a breach of the FCA.5 The DoJ alleged that CVS pharmacies and pharmacists filled prescriptions for controlled substances that (1) lacked a legitimate medical purpose; (2) were not legally valid; and/or (3) were not issued in the usual course of medical practice. 6 CVS contests the charges and issued an official response, stating that it disputes the allegations as false, plans to disprove them in litigation, and has nonetheless fully cooperated with the investigation.7
The allegations involved prescriptions for drugs like opioids and benzodiazepines, primary culprits in the American overdose epidemic.8 The complaint notes that the prescriptions were early refills in excessive quantities and included what has been called the “holy trinity” of dangerous medications: opioids, benzodiazepines, and muscle relaxants. 5,8 Even worse (if that is possible), as the complaint outlines, CVS had access to data from both inside and outside the company that these prescriptions came from notorious pill mills and were hence unlawful and yet continued to fill them, leading the DoJ to file the more serious charge that the corporation “knowingly” violated the CSA and “prioritized profits over safety in dispensing controlled substances.”5,6
The Unholy Trinity
The infamous members of what I prefer to call the “unholy trinity” are a benzodiazepine, often alprazolam, an opioid, and the muscle relaxant carisoprodol. The combination amplifies each agent’s independent risk of respiratory depression. The latter is a schedule IV medication with an active metabolite, meprobamate, that also has this adverse effect. All 3 drugs have high abuse potential and, when combined, increase the risk of fatal overdose. The colloquial name holy trinity derives from the synergistic euphoria experienced when taking this triple cocktail of sedative agents.9 This pharmacological recipe for disaster is the house specialty of pill mills: infamous storefront practices that generate high profits and exploit persons with chronic pain and addiction by handing out controlled substances with little clinical assessment and even less oversight.10
When the Means Become the End
The DoJ allegations suggest that the violations resulted from “corporate-mandated performance metrics, incentive compensation, and staffing policies that prioritized corporate profits over patient safety.”6 If the allegations are true, why would a company reinvited by Tricare to serve the nation’s heroes seemingly engage in illegal practices? While CVS has not responded in court, their statement argued that “too often, we have seen government agencies and trial lawyers question the good-faith decisions made by pharmacists while a patient waits at the pharmacy counter, often in pain.”6
The DoJ complaint offers a cautionary warning for the US health care system, which is increasingly being micromanaged in the pursuit of efficiency. Like many practitioners in and out of the federal system, I get a cold chill when I read the word productivity. “CVS pharmacists described working at CVS as ‘soul crushing’ because it was impossible to meet the company’s expectations,” the complaint alleges, because “CVS set staffing levels so low that it was impossible for pharmacists to comply with their legal obligations and meet CVS’s demanding metrics.”5 Did top-down mandates drive the alleged activities by imposing unattainable performance metrics on pharmacists, offering incentives that encouraged and rewarded corner-cutting, and refusing to fund sufficient staffing to ensure patient safety? This may be what happens when the means (efficiency) become the end rather than a mechanism to achieve the goal of more accessible, affordable, high-quality health care.
Ethically, what is most concerning is that leadership intentionally “deprived its pharmacists of crucial information” about specific practitioners known to engage in illegal prescribing practices.6 CVS did not provide pharmacists with “information about prescribers’ prescribing habits that CVS routinely collected and reviewed at the corporate level,” and even removed prescriber blocks that were implemented at Target pharmacies before it was acquired by CVS.5 The first element of informed consent is providing patients with adequate information upon which to decide whether to accept or decline treatment. 11 In this situation, however, CVS allegedly prevented “pharmacists from warning one another about certain prescribers.”6
If true, the company deprived frontline pharmacists of the information they needed to safely and responsibly dispense medications: “The practices alleged contributed to the opioid crisis and opioid-related deaths, and today’s complaint seeks to hold CVS accountable for its misconduct.”6 Though the cost in human life that may have resulted from CSA violations must absolutely and always outweigh financial considerations, the economic damage to Tricare from fraudulent billing and the betrayal of its fiduciary responsinility cannot be underestimated.
A Corporate Morality Play
CVS is not the only company, nor is pharmacy the only industry in health care, that has been the subject of watchdog agency lawsuits or variegated forms of wrongdoing, including violations of the CSA and FCA.10,12 As of this writing, the DoJ case against CVS has not been heard, much less adjudicated in a court of law. It is ironic that both the DoJ claims and the CVS rebuttal describe the manifest conflict of obligation that pharmacists confront between protecting their livelihood and safeguarding patients’ lives as suggested in the epigraph that has been attributed to the 19th-century British physician and medical educator Peter Mere Latham. It is a dilemma that a growing number of health care practitioners face daily in a vocation becoming increasingly commercialized. It is all too easy for an individual physician, nurse, or pharmacist to feel hopeless and helpless before the behemoth might of a large and looming entity. Yet, it was a whistleblower whose moral courage led to the DoJ investigation and subsequent charges.13 We must all never doubt the power of a committed person of conscience to withstand the pressure to mutate medications into poison and stand up for the principles of our professions and inspire a community of colleagues to follow their example.
- Fein AJ. The Top U.S. pharmacy markets of 2023: market shares and revenues at the biggest chains and PBMs. Drug Channels. March 12, 2024. Accessed February 24, 2025. https://www.drugchannels.net/2024/03/the-top-15-us-pharmacies-of-2023-market.html
- Jowers K. CVS returns to the military Tricare network. Walmart’s out. Military Times. October 18, 2021. Accessed February 24, 2025. https://www.militarytimes.com/pay-benefits/mil-money/2021/10/28/cvs-returns-to-the-military-tricare-pharmacy-network-walmarts-out/
- False Claims, 31 USC § 3729 (2009). Accessed February 24, 2025. https://www.govinfo.gov/content/pkg/USCODE-2011-title31/pdf/USCODE-2011-title31-subtitleIII-chap37-subchapIII-sec3729.pdf
- Drug Abuse Prevention and Control, Control and Enforcement, 21 USC 13 § 801 (2022). Accessed February 24, 2025. https://www.govinfo.gov/app/details/USCODE-2021-title21/USCODE-2021-title21-chap13-subchapI-partA-sec801
- United States ex rel. Estright v Health Corporation, et al. Accessed February 26, 2025. https://www.justice.gov/archives/opa/media/1381111/dl
- US Department of Justice. Justice Department files nationwide lawsuit alleging CVS knowingly dispensed controlled substances in violation of the Controlled Substances ACT and the False Claims Act. News release. December 18, 2024. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/justice-department-files-nationwide-lawsuit-alleging-cvs-knowingly-dispensed-controlled
- CVS Health. CVS Health statement regarding the U.S. Department of Justice’s lawsuit against CVS pharmacy. News release. December 18, 2024. Accessed February 24, 2025. https://www.cvshealth.com/impact/healthy-community/our-opioid-response.html
- Park TW, Saitz R, Ganoczy D, Ilgen MA, Bohnert AS. Benzodiazepine prescribing patterns and deaths from drug overdose among US veterans receiving opioid analgesics: case-cohort study. BMJ. 2015;350:h2698. doi:10.1136/bmj.h2698
- Wang Y, Delcher C, Li Y, Goldberger BA, Reisfield GM. Overlapping prescriptions of opioids, benzodiazepines, and carisoprodol: “Holy Trinity” prescribing in the state of Florida. Drug Alcohol Depend. 2019;205:107693. doi:10.1016/j.drugalcdep.2019.107693
- Wolf AA. The perfect storm: opioid risks and ‘The Holy Trinity’. Pharmacy Times. September 24, 2014. Accessed February 24, 2025. https://www.pharmacytimes.com/view/the-perfect-storm-opioid-risks-and-the-holy-trinity
- The meaning and justification of informed consent. In: Beauchamp TL, Childress JF. Principles of Biomedical Ethics. Eighth Edition. Oxford University Press; 2019:118-123.
- US Department of Justice. OptumRX agrees to pay $20M to resolve allegations that it filled certain opioid prescriptions in violation of the Controlled Substances Act. News release. June 27, 2024. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/optumrx-agrees-pay-20m-resolve-allegations-it-filled-certain-opioid-prescriptions-violation
- US Department of Justice. False Claims Act settlements and judgments exceed $2.9B in fiscal year 2024. News release. January 15, 2025. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/false-claims-act-settlements-and-judgments-exceed-29b-fiscal-year-2024
- Fein AJ. The Top U.S. pharmacy markets of 2023: market shares and revenues at the biggest chains and PBMs. Drug Channels. March 12, 2024. Accessed February 24, 2025. https://www.drugchannels.net/2024/03/the-top-15-us-pharmacies-of-2023-market.html
- Jowers K. CVS returns to the military Tricare network. Walmart’s out. Military Times. October 18, 2021. Accessed February 24, 2025. https://www.militarytimes.com/pay-benefits/mil-money/2021/10/28/cvs-returns-to-the-military-tricare-pharmacy-network-walmarts-out/
- False Claims, 31 USC § 3729 (2009). Accessed February 24, 2025. https://www.govinfo.gov/content/pkg/USCODE-2011-title31/pdf/USCODE-2011-title31-subtitleIII-chap37-subchapIII-sec3729.pdf
- Drug Abuse Prevention and Control, Control and Enforcement, 21 USC 13 § 801 (2022). Accessed February 24, 2025. https://www.govinfo.gov/app/details/USCODE-2021-title21/USCODE-2021-title21-chap13-subchapI-partA-sec801
- United States ex rel. Estright v Health Corporation, et al. Accessed February 26, 2025. https://www.justice.gov/archives/opa/media/1381111/dl
- US Department of Justice. Justice Department files nationwide lawsuit alleging CVS knowingly dispensed controlled substances in violation of the Controlled Substances ACT and the False Claims Act. News release. December 18, 2024. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/justice-department-files-nationwide-lawsuit-alleging-cvs-knowingly-dispensed-controlled
- CVS Health. CVS Health statement regarding the U.S. Department of Justice’s lawsuit against CVS pharmacy. News release. December 18, 2024. Accessed February 24, 2025. https://www.cvshealth.com/impact/healthy-community/our-opioid-response.html
- Park TW, Saitz R, Ganoczy D, Ilgen MA, Bohnert AS. Benzodiazepine prescribing patterns and deaths from drug overdose among US veterans receiving opioid analgesics: case-cohort study. BMJ. 2015;350:h2698. doi:10.1136/bmj.h2698
- Wang Y, Delcher C, Li Y, Goldberger BA, Reisfield GM. Overlapping prescriptions of opioids, benzodiazepines, and carisoprodol: “Holy Trinity” prescribing in the state of Florida. Drug Alcohol Depend. 2019;205:107693. doi:10.1016/j.drugalcdep.2019.107693
- Wolf AA. The perfect storm: opioid risks and ‘The Holy Trinity’. Pharmacy Times. September 24, 2014. Accessed February 24, 2025. https://www.pharmacytimes.com/view/the-perfect-storm-opioid-risks-and-the-holy-trinity
- The meaning and justification of informed consent. In: Beauchamp TL, Childress JF. Principles of Biomedical Ethics. Eighth Edition. Oxford University Press; 2019:118-123.
- US Department of Justice. OptumRX agrees to pay $20M to resolve allegations that it filled certain opioid prescriptions in violation of the Controlled Substances Act. News release. June 27, 2024. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/optumrx-agrees-pay-20m-resolve-allegations-it-filled-certain-opioid-prescriptions-violation
- US Department of Justice. False Claims Act settlements and judgments exceed $2.9B in fiscal year 2024. News release. January 15, 2025. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/false-claims-act-settlements-and-judgments-exceed-29b-fiscal-year-2024
The Unholy Trinity: Unlawful Prescriptions, False Claims, and Dangerous Drugs
The Unholy Trinity: Unlawful Prescriptions, False Claims, and Dangerous Drugs
Improving High-Risk Osteoporosis Medication Adherence and Safety With an Automated Dashboard
Improving High-Risk Osteoporosis Medication Adherence and Safety With an Automated Dashboard
Osteoporotic fragility fractures constitute a significant public health concern, with 1 in 2 women and 1 in 5 men aged > 50 years sustaining an osteoporotic fracture.1 Osteoporotic fractures are costly and associated with reduced quality of life and impaired survival.2-6 Many interventions including fall mitigation, calcium, vitamin D supplementation, and osteoporosis—specific medications reduce fracture risk.7 New medications for treating osteoporosis, including anabolic therapies, are costly and require clinical oversight to ensure safe delivery. This includes laboratory monitoring, timing of in-clinic dosing and provision of sequence therapy.8,9 COVID-19 introduced numerous barriers to osteoporosis care, raising concerns for medication interruption and patients lost to follow-up, which made monitoring these high risk and costly medications even more important.
The US Department of Veterans Affairs (VA) was an early adopter of using the electronic health record to analyze and implement system-wide processes for population management and quality improvement.10 This enabled the creation of clinical dashboards to display key performance indicator data that support quality improvement and patient care initiatives.11-15 The VA Puget Sound Health Care System (VAPSHCS) has a dedicated osteoporosis clinic focused on preventing and treating veterans at high risk for fracture. Considering the growing utilization of osteoporosis medications, particularly those requiring timed sequential therapy to prevent bone mineral density loss and rebound osteoporotic fractures, close monitoring and follow-up is required. The COVID-19 pandemic made clear the need for proactive osteoporosis management. This article describes the creation and use of an automated clinic dashboard to identify and contact veterans with osteoporosis-related care needs, such as prescription refills, laboratory tests, and clinical visits.
Methods
An automated dashboard was created in partnership with VA pharmacy clinical informatics to display the osteoporosis medication prescription (including last refill), monitoring laboratory test values and most recent osteoporosis clinic visit for each clinic patient. Data from the VA Corporate Data Warehouse were extracted. The resulting tables were used to create a patient cohort with ≥ 1 active medication for alendronate, zoledronic acid, the parathyroid hormone analogues (PTH) teriparatide or abaloparatide, denosumab, or romosozumab. Notably, alendronate was the only oral bisphosphonate prescribed in the clinic. These data were formatted and displayed using Microsoft SQL Server Reporting Services. The secure and encrypted dashboard alerts the clinic staff when prescriptions, appointments, or laboratory tests, such as estimated glomerular filtration rate, 25-hydroxy vitamin D, calcium, and PTH are overdue or out of reference range. The dashboard tracked the most recent clinic visit or dual-energy X-ray absorptiometry (DXA) scan if performed within the VA. Overdue laboratory test alerts for bisphosphonates were flagged if delayed 12 months and 6 months for all other medications.
On March 20, 2021, the VAPSHCS osteoporosis clinic was staffed by 1 endocrinologist, 1 geriatrician, 1 rheumatologist, and 1 registered nurse (RN) coordinator. Overdue or out-of-range alerts were reviewed weekly by the RN coordinator, who addressed alerts. For any overdue laboratory work or prescription refills, the RN coordinator alerted the primary osteoporosis physician via the electronic health record for updated orders. Patients were contacted by phone to schedule a clinic visit, complete ordered laboratory work, or discuss osteoporosis medication refills based on the need identified by the dashboard. A letter was mailed to the patient requesting they contact the osteoporosis clinic for patients who could not be reached by phone after 2 attempts. If 3 attempts (2 phone calls and a letter) were unsuccessful, the osteoporosis physician was alerted so they could either call the patient, alert the primary referring clinician, or discontinue the osteoporosis medication.
Results
As of March 20, 2021, 139 patients were included on the dashboard. Ninety-two patients (66%) had unmet care needs and 29% were female. Ages ranged from 40 to 100 years (Table). The dashboard alerted the team to 3 patients lost to follow-up, all of whom had transferred to care outside the clinic. Twenty-three patients (17%) had overdue medications, including 2 (9%) who had not refilled oral bisphosphonate and 18 (78%) who were overdue for intravenous bisphosphonate treatment. One veteran flagged as overdue for their denosumab injection was unable to receive it due to a significant change in health status. Two veterans were overdue for a PTH analogue refill, 1 of whom had completed their course and transitioned to bisphosphonate.
The most common alert was 40 patients (29%) with overdue laboratory tests, 37 of which were receiving bisphosphonates. One patient included on the dashboard was taking romosozumab and all their monitoring parameters were up to date, thus their data were not included in the Table to prevent possible identification.
Discussion
A dashboard alerted the osteoporosis clinic team to veterans who were overdue for visits, laboratory work, and prescription renewals. Overall, 92 patients (66%) had unmet care needs identified by the dashboard, all of which were addressed with phone calls and/or letters. Most of the overdue medication refills and laboratory tests were for patients taking bisphosphonates avoiding VAPSHCS during the COVID-19 pandemic. The dashboard enabled the RN coordinator to promptly contact the patient, facilitate coordination of care requirements, and guarantee the safe and efficient delivery of osteoporosis care.
The VA has historically been a leader in the creation of clinical dashboards to support health campaigns.11,12 These dashboards have successfully improved quality metrics towards the treatment of hepatitis C virus, heart failure, and highrisk opioid prescribing.13-15 Data have shown that successful clinical dashboard implementation must be done in conjunction with protected time or staff to support care improvements.16 Additionally, the time required for clinical dashboards can limit their sustainability and feasibility.17 A study aimed at improving osteoporosis care for patients with Parkinson disease found that weekly multidisciplinary review of at-risk patients resulted in all new patients and 91% of follow-up patients receiving evidence- based osteoporosis treatments.17 However, despite the benefits, the intervention required significant time and resources. In contrast, the osteoporosis dashboard implemented at VAPSHCS was not time or resource intensive, requiring about 1 hour per week for the RN coordinator to review the dashboard and coordinate patient care needs.
Limitations
This study setting is unique from other health care organizations or VA health care systems. Implementation of a similar dashboard in other clinical settings where patients receive medical care in multiple health care systems may differ. The VA dedicates resources to support veteran population health management, which may not be available in other health care systems.11,12 These issues may pose a barrier to implementing a similar osteoporosis dashboard in non-VA facilities. In addition, it is significant that while the dashboard can be reconfigured and adapted to track veterans across different VA facilities, certain complexities arise if essential data, such as laboratory tests and DXA imaging, are conducted outside of VA facilities. In such cases, manual entry of this information into the dashboard would be necessary. Because the dashboard was quickly developed during the COVID-19 pandemic, this study lacked preimplementation data on laboratory testing, medication refills, and DXA imaging, which would have enabled a comparison of adherence before and after dashboard implementation. Finally, we acknowledge the delay in publishing these findings; however, we believe sharing innovative approaches to providing care for high-risk populations is essential, as demonstrated during the COVID-19 pandemic.
Conclusions
An osteoporosis clinic dashboard served as a valuable clinical support tool to ensure safe and effective osteoporosis medication delivery at VAPSHCS. Considering the growing utilization of osteoporosis medications, this dashboard plays a vital role in facilitating care coordination for patients receiving these high-risk treatments.18 Use of the dashboard supported the effective use of high-cost osteoporosis medications and is likely to improve clinical osteoporosis outcomes.
Despite the known fracture risk reduction, osteoporosis medication adherence is low.19,20 Maintaining consistent pharmacotherapy for osteoporosis is essential not only for fracture prevention but also reducing health care costs related to osteoporosis and preserving patient independence and functionality.21-24 While initially developed in response to the COVID-19 pandemic, the dashboard remains useful. The VAPSHCS osteoporosis clinic is now staffed by 2 physicians (endocrine and rheumatology) and the dashboard is still in use. The RN coordinator spends about 15 minutes per week using the dashboard and managing the 67 veterans on osteoporosis therapy. This dashboard represents a sustainable clinical tool with the capacity to minimize osteoporosis care gaps and improve outcomes.
- Johnell O, Kanis J. Epidemiology of osteoporotic fractures. Osteoporos Int. 2005;16(suppl 2):S3-S7. doi:10.1007/s00198-004-1702-6
- van Staa TP, Dennison EM, Leufkens HG, Cooper C. Epidemiology of fractures in England and Wales. Bone. 2001;29:517-522. doi:10.1016/s8756-3282(01)00614-7
- Dennison E, Cooper C. Epidemiology of osteoporotic fractures. Horm Res. 2000;54(suppl 1):58-63. doi:10.1159/000063449
- Cooper C. Epidemiology and public health impact of osteoporosis. Baillieres Clin Rheumatol. 1993;7:459-477. doi:10.1016/s0950-3579(05)80073-1
- Dolan P, Torgerson DJ. The cost of treating osteoporotic fractures in the United Kingdom female population. Osteoporos Int. 1998;8:611-617. doi:10.1007/s001980050107
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475. doi:10.1359/jbmr.061113
- Palacios S. Medical treatment of osteoporosis. Climacteric. 2022;25:43-49. doi:10.1080/13697137.2021.1951697
- Eastell R, Rosen CJ, Black DM, Cheung AM, Murad MH, Shoback D. Pharmacological management of osteoporosis in postmenopausal women: an Endocrine Society* clinical practice guideline. J Clin Endocrinol Metab. 2019;104:1595-1622. doi:10.1210/jc.2019-00221
- Watts NB, Adler RA, Bilezikian JP, et al. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:1802-1822. doi:10.1210/jc.2011-3045
- Lau MK, Bounthavong M, Kay CL, Harvey MA, Christopher MLD. Clinical dashboard development and use for academic detailing in the U.S. Department of Veterans Affairs. J Am Pharm Assoc (2003). 2019;59(2S):S96-S103.e3. doi:10.1016/j.japh.2018.12.006
- Mould DR, D’Haens G, Upton RN. Clinical decision support tools: the evolution of a revolution. Clin Pharmacol Ther. 2016;99:405-418. doi:10.1002/cpt.334
- Kizer KW, Fonseca ML, Long LM. The veterans healthcare system: preparing for the twenty-first century. Hosp Health Serv Adm. 1997;42:283-298.
- Park A, Gonzalez R, Chartier M, et al. Screening and treating hepatitis c in the VA: achieving excellence using lean and system redesign. Fed Pract. 2018;35:24-29.
- Brownell N, Kay C, Parra D, et al. Development and optimization of the Veterans Affairs’ national heart failure dashboard for population health management. J Card Fail. 2024;30:452-459. doi:10.1016/j.cardfail.2023.08.024
- Lin LA, Bohnert ASB, Kerns RD, Clay MA, Ganoczy D, Ilgen MA. Impact of the opioid safety initiative on opioidrelated prescribing in veterans. Pain. 2017;158:833-839. doi:10.1097/j.pain.0000000000000837
- Twohig PA, Rivington JR, Gunzler D, Daprano J, Margolius D. Clinician dashboard views and improvement in preventative health outcome measures: a retrospective analysis. BMC Health Serv Res. 2019;19:475. doi:10.1186/s12913-019-4327-3
- Singh I, Fletcher R, Scanlon L, Tyler M, Aithal S. A quality improvement initiative on the management of osteoporosis in older people with Parkinsonism. BMJ Qual Improv Rep. 2016;5:u210921.w5756. doi:10.1136/bmjquality.u210921.w5756
- Anastasilakis AD, Makras P, Yavropoulou MP, Tabacco G, Naciu AM, Palermo A. Denosumab discontinuation and the rebound phenomenon: a narrative review. J Clin Med. 2021;10:152. doi:10.3390/jcm10010152
- Sharman Moser S, Yu J, Goldshtein I, et al. Cost and consequences of nonadherence with oral bisphosphonate therapy: findings from a real-world data analysis. Ann Pharmacother. 2016;50:262-269. doi:10.1177/1060028015626935
- Olsen KR, Hansen C, Abrahamsen B. Association between refill compliance to oral bisphosphonate treatment, incident fractures, and health care costs--an analysis using national health databases. Osteoporos Int. 2013;24:2639-2647. doi:10.1007/s00198-013-2365-y
- Blouin J, Dragomir A, Fredette M, Ste-Marie LG, Fernandes JC, Perreault S. Comparison of direct health care costs related to the pharmacological treatment of osteoporosis and to the management of osteoporotic fractures among compliant and noncompliant users of alendronate and risedronate: a population-based study. Osteoporos Int. 2009;20:1571-1581. doi:10.1007/s00198-008-0818-5
- Cotté F-E, De Pouvourville G. Cost of non-persistence with oral bisphosphonates in post-menopausal osteoporosis treatment in France. BMC Health Serv Res. 2011;11:151. doi:10.1186/1472-6963-11-151
- Cho H, Byun J-H, Song I, et al. Effect of improved medication adherence on health care costs in osteoporosis patients. Medicine (Baltimore). 2018;97:e11470. doi:10.1097/MD.0000000000011470
- Li N, Cornelissen D, Silverman S, et al. An updated systematic review of cost-effectiveness analyses of drugs for osteoporosis. Pharmacoeconomics. 2021;39:181-209. doi:10.1007/s40273-020-00965-9
Osteoporotic fragility fractures constitute a significant public health concern, with 1 in 2 women and 1 in 5 men aged > 50 years sustaining an osteoporotic fracture.1 Osteoporotic fractures are costly and associated with reduced quality of life and impaired survival.2-6 Many interventions including fall mitigation, calcium, vitamin D supplementation, and osteoporosis—specific medications reduce fracture risk.7 New medications for treating osteoporosis, including anabolic therapies, are costly and require clinical oversight to ensure safe delivery. This includes laboratory monitoring, timing of in-clinic dosing and provision of sequence therapy.8,9 COVID-19 introduced numerous barriers to osteoporosis care, raising concerns for medication interruption and patients lost to follow-up, which made monitoring these high risk and costly medications even more important.
The US Department of Veterans Affairs (VA) was an early adopter of using the electronic health record to analyze and implement system-wide processes for population management and quality improvement.10 This enabled the creation of clinical dashboards to display key performance indicator data that support quality improvement and patient care initiatives.11-15 The VA Puget Sound Health Care System (VAPSHCS) has a dedicated osteoporosis clinic focused on preventing and treating veterans at high risk for fracture. Considering the growing utilization of osteoporosis medications, particularly those requiring timed sequential therapy to prevent bone mineral density loss and rebound osteoporotic fractures, close monitoring and follow-up is required. The COVID-19 pandemic made clear the need for proactive osteoporosis management. This article describes the creation and use of an automated clinic dashboard to identify and contact veterans with osteoporosis-related care needs, such as prescription refills, laboratory tests, and clinical visits.
Methods
An automated dashboard was created in partnership with VA pharmacy clinical informatics to display the osteoporosis medication prescription (including last refill), monitoring laboratory test values and most recent osteoporosis clinic visit for each clinic patient. Data from the VA Corporate Data Warehouse were extracted. The resulting tables were used to create a patient cohort with ≥ 1 active medication for alendronate, zoledronic acid, the parathyroid hormone analogues (PTH) teriparatide or abaloparatide, denosumab, or romosozumab. Notably, alendronate was the only oral bisphosphonate prescribed in the clinic. These data were formatted and displayed using Microsoft SQL Server Reporting Services. The secure and encrypted dashboard alerts the clinic staff when prescriptions, appointments, or laboratory tests, such as estimated glomerular filtration rate, 25-hydroxy vitamin D, calcium, and PTH are overdue or out of reference range. The dashboard tracked the most recent clinic visit or dual-energy X-ray absorptiometry (DXA) scan if performed within the VA. Overdue laboratory test alerts for bisphosphonates were flagged if delayed 12 months and 6 months for all other medications.
On March 20, 2021, the VAPSHCS osteoporosis clinic was staffed by 1 endocrinologist, 1 geriatrician, 1 rheumatologist, and 1 registered nurse (RN) coordinator. Overdue or out-of-range alerts were reviewed weekly by the RN coordinator, who addressed alerts. For any overdue laboratory work or prescription refills, the RN coordinator alerted the primary osteoporosis physician via the electronic health record for updated orders. Patients were contacted by phone to schedule a clinic visit, complete ordered laboratory work, or discuss osteoporosis medication refills based on the need identified by the dashboard. A letter was mailed to the patient requesting they contact the osteoporosis clinic for patients who could not be reached by phone after 2 attempts. If 3 attempts (2 phone calls and a letter) were unsuccessful, the osteoporosis physician was alerted so they could either call the patient, alert the primary referring clinician, or discontinue the osteoporosis medication.
Results
As of March 20, 2021, 139 patients were included on the dashboard. Ninety-two patients (66%) had unmet care needs and 29% were female. Ages ranged from 40 to 100 years (Table). The dashboard alerted the team to 3 patients lost to follow-up, all of whom had transferred to care outside the clinic. Twenty-three patients (17%) had overdue medications, including 2 (9%) who had not refilled oral bisphosphonate and 18 (78%) who were overdue for intravenous bisphosphonate treatment. One veteran flagged as overdue for their denosumab injection was unable to receive it due to a significant change in health status. Two veterans were overdue for a PTH analogue refill, 1 of whom had completed their course and transitioned to bisphosphonate.
The most common alert was 40 patients (29%) with overdue laboratory tests, 37 of which were receiving bisphosphonates. One patient included on the dashboard was taking romosozumab and all their monitoring parameters were up to date, thus their data were not included in the Table to prevent possible identification.
Discussion
A dashboard alerted the osteoporosis clinic team to veterans who were overdue for visits, laboratory work, and prescription renewals. Overall, 92 patients (66%) had unmet care needs identified by the dashboard, all of which were addressed with phone calls and/or letters. Most of the overdue medication refills and laboratory tests were for patients taking bisphosphonates avoiding VAPSHCS during the COVID-19 pandemic. The dashboard enabled the RN coordinator to promptly contact the patient, facilitate coordination of care requirements, and guarantee the safe and efficient delivery of osteoporosis care.
The VA has historically been a leader in the creation of clinical dashboards to support health campaigns.11,12 These dashboards have successfully improved quality metrics towards the treatment of hepatitis C virus, heart failure, and highrisk opioid prescribing.13-15 Data have shown that successful clinical dashboard implementation must be done in conjunction with protected time or staff to support care improvements.16 Additionally, the time required for clinical dashboards can limit their sustainability and feasibility.17 A study aimed at improving osteoporosis care for patients with Parkinson disease found that weekly multidisciplinary review of at-risk patients resulted in all new patients and 91% of follow-up patients receiving evidence- based osteoporosis treatments.17 However, despite the benefits, the intervention required significant time and resources. In contrast, the osteoporosis dashboard implemented at VAPSHCS was not time or resource intensive, requiring about 1 hour per week for the RN coordinator to review the dashboard and coordinate patient care needs.
Limitations
This study setting is unique from other health care organizations or VA health care systems. Implementation of a similar dashboard in other clinical settings where patients receive medical care in multiple health care systems may differ. The VA dedicates resources to support veteran population health management, which may not be available in other health care systems.11,12 These issues may pose a barrier to implementing a similar osteoporosis dashboard in non-VA facilities. In addition, it is significant that while the dashboard can be reconfigured and adapted to track veterans across different VA facilities, certain complexities arise if essential data, such as laboratory tests and DXA imaging, are conducted outside of VA facilities. In such cases, manual entry of this information into the dashboard would be necessary. Because the dashboard was quickly developed during the COVID-19 pandemic, this study lacked preimplementation data on laboratory testing, medication refills, and DXA imaging, which would have enabled a comparison of adherence before and after dashboard implementation. Finally, we acknowledge the delay in publishing these findings; however, we believe sharing innovative approaches to providing care for high-risk populations is essential, as demonstrated during the COVID-19 pandemic.
Conclusions
An osteoporosis clinic dashboard served as a valuable clinical support tool to ensure safe and effective osteoporosis medication delivery at VAPSHCS. Considering the growing utilization of osteoporosis medications, this dashboard plays a vital role in facilitating care coordination for patients receiving these high-risk treatments.18 Use of the dashboard supported the effective use of high-cost osteoporosis medications and is likely to improve clinical osteoporosis outcomes.
Despite the known fracture risk reduction, osteoporosis medication adherence is low.19,20 Maintaining consistent pharmacotherapy for osteoporosis is essential not only for fracture prevention but also reducing health care costs related to osteoporosis and preserving patient independence and functionality.21-24 While initially developed in response to the COVID-19 pandemic, the dashboard remains useful. The VAPSHCS osteoporosis clinic is now staffed by 2 physicians (endocrine and rheumatology) and the dashboard is still in use. The RN coordinator spends about 15 minutes per week using the dashboard and managing the 67 veterans on osteoporosis therapy. This dashboard represents a sustainable clinical tool with the capacity to minimize osteoporosis care gaps and improve outcomes.
Osteoporotic fragility fractures constitute a significant public health concern, with 1 in 2 women and 1 in 5 men aged > 50 years sustaining an osteoporotic fracture.1 Osteoporotic fractures are costly and associated with reduced quality of life and impaired survival.2-6 Many interventions including fall mitigation, calcium, vitamin D supplementation, and osteoporosis—specific medications reduce fracture risk.7 New medications for treating osteoporosis, including anabolic therapies, are costly and require clinical oversight to ensure safe delivery. This includes laboratory monitoring, timing of in-clinic dosing and provision of sequence therapy.8,9 COVID-19 introduced numerous barriers to osteoporosis care, raising concerns for medication interruption and patients lost to follow-up, which made monitoring these high risk and costly medications even more important.
The US Department of Veterans Affairs (VA) was an early adopter of using the electronic health record to analyze and implement system-wide processes for population management and quality improvement.10 This enabled the creation of clinical dashboards to display key performance indicator data that support quality improvement and patient care initiatives.11-15 The VA Puget Sound Health Care System (VAPSHCS) has a dedicated osteoporosis clinic focused on preventing and treating veterans at high risk for fracture. Considering the growing utilization of osteoporosis medications, particularly those requiring timed sequential therapy to prevent bone mineral density loss and rebound osteoporotic fractures, close monitoring and follow-up is required. The COVID-19 pandemic made clear the need for proactive osteoporosis management. This article describes the creation and use of an automated clinic dashboard to identify and contact veterans with osteoporosis-related care needs, such as prescription refills, laboratory tests, and clinical visits.
Methods
An automated dashboard was created in partnership with VA pharmacy clinical informatics to display the osteoporosis medication prescription (including last refill), monitoring laboratory test values and most recent osteoporosis clinic visit for each clinic patient. Data from the VA Corporate Data Warehouse were extracted. The resulting tables were used to create a patient cohort with ≥ 1 active medication for alendronate, zoledronic acid, the parathyroid hormone analogues (PTH) teriparatide or abaloparatide, denosumab, or romosozumab. Notably, alendronate was the only oral bisphosphonate prescribed in the clinic. These data were formatted and displayed using Microsoft SQL Server Reporting Services. The secure and encrypted dashboard alerts the clinic staff when prescriptions, appointments, or laboratory tests, such as estimated glomerular filtration rate, 25-hydroxy vitamin D, calcium, and PTH are overdue or out of reference range. The dashboard tracked the most recent clinic visit or dual-energy X-ray absorptiometry (DXA) scan if performed within the VA. Overdue laboratory test alerts for bisphosphonates were flagged if delayed 12 months and 6 months for all other medications.
On March 20, 2021, the VAPSHCS osteoporosis clinic was staffed by 1 endocrinologist, 1 geriatrician, 1 rheumatologist, and 1 registered nurse (RN) coordinator. Overdue or out-of-range alerts were reviewed weekly by the RN coordinator, who addressed alerts. For any overdue laboratory work or prescription refills, the RN coordinator alerted the primary osteoporosis physician via the electronic health record for updated orders. Patients were contacted by phone to schedule a clinic visit, complete ordered laboratory work, or discuss osteoporosis medication refills based on the need identified by the dashboard. A letter was mailed to the patient requesting they contact the osteoporosis clinic for patients who could not be reached by phone after 2 attempts. If 3 attempts (2 phone calls and a letter) were unsuccessful, the osteoporosis physician was alerted so they could either call the patient, alert the primary referring clinician, or discontinue the osteoporosis medication.
Results
As of March 20, 2021, 139 patients were included on the dashboard. Ninety-two patients (66%) had unmet care needs and 29% were female. Ages ranged from 40 to 100 years (Table). The dashboard alerted the team to 3 patients lost to follow-up, all of whom had transferred to care outside the clinic. Twenty-three patients (17%) had overdue medications, including 2 (9%) who had not refilled oral bisphosphonate and 18 (78%) who were overdue for intravenous bisphosphonate treatment. One veteran flagged as overdue for their denosumab injection was unable to receive it due to a significant change in health status. Two veterans were overdue for a PTH analogue refill, 1 of whom had completed their course and transitioned to bisphosphonate.
The most common alert was 40 patients (29%) with overdue laboratory tests, 37 of which were receiving bisphosphonates. One patient included on the dashboard was taking romosozumab and all their monitoring parameters were up to date, thus their data were not included in the Table to prevent possible identification.
Discussion
A dashboard alerted the osteoporosis clinic team to veterans who were overdue for visits, laboratory work, and prescription renewals. Overall, 92 patients (66%) had unmet care needs identified by the dashboard, all of which were addressed with phone calls and/or letters. Most of the overdue medication refills and laboratory tests were for patients taking bisphosphonates avoiding VAPSHCS during the COVID-19 pandemic. The dashboard enabled the RN coordinator to promptly contact the patient, facilitate coordination of care requirements, and guarantee the safe and efficient delivery of osteoporosis care.
The VA has historically been a leader in the creation of clinical dashboards to support health campaigns.11,12 These dashboards have successfully improved quality metrics towards the treatment of hepatitis C virus, heart failure, and highrisk opioid prescribing.13-15 Data have shown that successful clinical dashboard implementation must be done in conjunction with protected time or staff to support care improvements.16 Additionally, the time required for clinical dashboards can limit their sustainability and feasibility.17 A study aimed at improving osteoporosis care for patients with Parkinson disease found that weekly multidisciplinary review of at-risk patients resulted in all new patients and 91% of follow-up patients receiving evidence- based osteoporosis treatments.17 However, despite the benefits, the intervention required significant time and resources. In contrast, the osteoporosis dashboard implemented at VAPSHCS was not time or resource intensive, requiring about 1 hour per week for the RN coordinator to review the dashboard and coordinate patient care needs.
Limitations
This study setting is unique from other health care organizations or VA health care systems. Implementation of a similar dashboard in other clinical settings where patients receive medical care in multiple health care systems may differ. The VA dedicates resources to support veteran population health management, which may not be available in other health care systems.11,12 These issues may pose a barrier to implementing a similar osteoporosis dashboard in non-VA facilities. In addition, it is significant that while the dashboard can be reconfigured and adapted to track veterans across different VA facilities, certain complexities arise if essential data, such as laboratory tests and DXA imaging, are conducted outside of VA facilities. In such cases, manual entry of this information into the dashboard would be necessary. Because the dashboard was quickly developed during the COVID-19 pandemic, this study lacked preimplementation data on laboratory testing, medication refills, and DXA imaging, which would have enabled a comparison of adherence before and after dashboard implementation. Finally, we acknowledge the delay in publishing these findings; however, we believe sharing innovative approaches to providing care for high-risk populations is essential, as demonstrated during the COVID-19 pandemic.
Conclusions
An osteoporosis clinic dashboard served as a valuable clinical support tool to ensure safe and effective osteoporosis medication delivery at VAPSHCS. Considering the growing utilization of osteoporosis medications, this dashboard plays a vital role in facilitating care coordination for patients receiving these high-risk treatments.18 Use of the dashboard supported the effective use of high-cost osteoporosis medications and is likely to improve clinical osteoporosis outcomes.
Despite the known fracture risk reduction, osteoporosis medication adherence is low.19,20 Maintaining consistent pharmacotherapy for osteoporosis is essential not only for fracture prevention but also reducing health care costs related to osteoporosis and preserving patient independence and functionality.21-24 While initially developed in response to the COVID-19 pandemic, the dashboard remains useful. The VAPSHCS osteoporosis clinic is now staffed by 2 physicians (endocrine and rheumatology) and the dashboard is still in use. The RN coordinator spends about 15 minutes per week using the dashboard and managing the 67 veterans on osteoporosis therapy. This dashboard represents a sustainable clinical tool with the capacity to minimize osteoporosis care gaps and improve outcomes.
- Johnell O, Kanis J. Epidemiology of osteoporotic fractures. Osteoporos Int. 2005;16(suppl 2):S3-S7. doi:10.1007/s00198-004-1702-6
- van Staa TP, Dennison EM, Leufkens HG, Cooper C. Epidemiology of fractures in England and Wales. Bone. 2001;29:517-522. doi:10.1016/s8756-3282(01)00614-7
- Dennison E, Cooper C. Epidemiology of osteoporotic fractures. Horm Res. 2000;54(suppl 1):58-63. doi:10.1159/000063449
- Cooper C. Epidemiology and public health impact of osteoporosis. Baillieres Clin Rheumatol. 1993;7:459-477. doi:10.1016/s0950-3579(05)80073-1
- Dolan P, Torgerson DJ. The cost of treating osteoporotic fractures in the United Kingdom female population. Osteoporos Int. 1998;8:611-617. doi:10.1007/s001980050107
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475. doi:10.1359/jbmr.061113
- Palacios S. Medical treatment of osteoporosis. Climacteric. 2022;25:43-49. doi:10.1080/13697137.2021.1951697
- Eastell R, Rosen CJ, Black DM, Cheung AM, Murad MH, Shoback D. Pharmacological management of osteoporosis in postmenopausal women: an Endocrine Society* clinical practice guideline. J Clin Endocrinol Metab. 2019;104:1595-1622. doi:10.1210/jc.2019-00221
- Watts NB, Adler RA, Bilezikian JP, et al. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:1802-1822. doi:10.1210/jc.2011-3045
- Lau MK, Bounthavong M, Kay CL, Harvey MA, Christopher MLD. Clinical dashboard development and use for academic detailing in the U.S. Department of Veterans Affairs. J Am Pharm Assoc (2003). 2019;59(2S):S96-S103.e3. doi:10.1016/j.japh.2018.12.006
- Mould DR, D’Haens G, Upton RN. Clinical decision support tools: the evolution of a revolution. Clin Pharmacol Ther. 2016;99:405-418. doi:10.1002/cpt.334
- Kizer KW, Fonseca ML, Long LM. The veterans healthcare system: preparing for the twenty-first century. Hosp Health Serv Adm. 1997;42:283-298.
- Park A, Gonzalez R, Chartier M, et al. Screening and treating hepatitis c in the VA: achieving excellence using lean and system redesign. Fed Pract. 2018;35:24-29.
- Brownell N, Kay C, Parra D, et al. Development and optimization of the Veterans Affairs’ national heart failure dashboard for population health management. J Card Fail. 2024;30:452-459. doi:10.1016/j.cardfail.2023.08.024
- Lin LA, Bohnert ASB, Kerns RD, Clay MA, Ganoczy D, Ilgen MA. Impact of the opioid safety initiative on opioidrelated prescribing in veterans. Pain. 2017;158:833-839. doi:10.1097/j.pain.0000000000000837
- Twohig PA, Rivington JR, Gunzler D, Daprano J, Margolius D. Clinician dashboard views and improvement in preventative health outcome measures: a retrospective analysis. BMC Health Serv Res. 2019;19:475. doi:10.1186/s12913-019-4327-3
- Singh I, Fletcher R, Scanlon L, Tyler M, Aithal S. A quality improvement initiative on the management of osteoporosis in older people with Parkinsonism. BMJ Qual Improv Rep. 2016;5:u210921.w5756. doi:10.1136/bmjquality.u210921.w5756
- Anastasilakis AD, Makras P, Yavropoulou MP, Tabacco G, Naciu AM, Palermo A. Denosumab discontinuation and the rebound phenomenon: a narrative review. J Clin Med. 2021;10:152. doi:10.3390/jcm10010152
- Sharman Moser S, Yu J, Goldshtein I, et al. Cost and consequences of nonadherence with oral bisphosphonate therapy: findings from a real-world data analysis. Ann Pharmacother. 2016;50:262-269. doi:10.1177/1060028015626935
- Olsen KR, Hansen C, Abrahamsen B. Association between refill compliance to oral bisphosphonate treatment, incident fractures, and health care costs--an analysis using national health databases. Osteoporos Int. 2013;24:2639-2647. doi:10.1007/s00198-013-2365-y
- Blouin J, Dragomir A, Fredette M, Ste-Marie LG, Fernandes JC, Perreault S. Comparison of direct health care costs related to the pharmacological treatment of osteoporosis and to the management of osteoporotic fractures among compliant and noncompliant users of alendronate and risedronate: a population-based study. Osteoporos Int. 2009;20:1571-1581. doi:10.1007/s00198-008-0818-5
- Cotté F-E, De Pouvourville G. Cost of non-persistence with oral bisphosphonates in post-menopausal osteoporosis treatment in France. BMC Health Serv Res. 2011;11:151. doi:10.1186/1472-6963-11-151
- Cho H, Byun J-H, Song I, et al. Effect of improved medication adherence on health care costs in osteoporosis patients. Medicine (Baltimore). 2018;97:e11470. doi:10.1097/MD.0000000000011470
- Li N, Cornelissen D, Silverman S, et al. An updated systematic review of cost-effectiveness analyses of drugs for osteoporosis. Pharmacoeconomics. 2021;39:181-209. doi:10.1007/s40273-020-00965-9
- Johnell O, Kanis J. Epidemiology of osteoporotic fractures. Osteoporos Int. 2005;16(suppl 2):S3-S7. doi:10.1007/s00198-004-1702-6
- van Staa TP, Dennison EM, Leufkens HG, Cooper C. Epidemiology of fractures in England and Wales. Bone. 2001;29:517-522. doi:10.1016/s8756-3282(01)00614-7
- Dennison E, Cooper C. Epidemiology of osteoporotic fractures. Horm Res. 2000;54(suppl 1):58-63. doi:10.1159/000063449
- Cooper C. Epidemiology and public health impact of osteoporosis. Baillieres Clin Rheumatol. 1993;7:459-477. doi:10.1016/s0950-3579(05)80073-1
- Dolan P, Torgerson DJ. The cost of treating osteoporotic fractures in the United Kingdom female population. Osteoporos Int. 1998;8:611-617. doi:10.1007/s001980050107
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475. doi:10.1359/jbmr.061113
- Palacios S. Medical treatment of osteoporosis. Climacteric. 2022;25:43-49. doi:10.1080/13697137.2021.1951697
- Eastell R, Rosen CJ, Black DM, Cheung AM, Murad MH, Shoback D. Pharmacological management of osteoporosis in postmenopausal women: an Endocrine Society* clinical practice guideline. J Clin Endocrinol Metab. 2019;104:1595-1622. doi:10.1210/jc.2019-00221
- Watts NB, Adler RA, Bilezikian JP, et al. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:1802-1822. doi:10.1210/jc.2011-3045
- Lau MK, Bounthavong M, Kay CL, Harvey MA, Christopher MLD. Clinical dashboard development and use for academic detailing in the U.S. Department of Veterans Affairs. J Am Pharm Assoc (2003). 2019;59(2S):S96-S103.e3. doi:10.1016/j.japh.2018.12.006
- Mould DR, D’Haens G, Upton RN. Clinical decision support tools: the evolution of a revolution. Clin Pharmacol Ther. 2016;99:405-418. doi:10.1002/cpt.334
- Kizer KW, Fonseca ML, Long LM. The veterans healthcare system: preparing for the twenty-first century. Hosp Health Serv Adm. 1997;42:283-298.
- Park A, Gonzalez R, Chartier M, et al. Screening and treating hepatitis c in the VA: achieving excellence using lean and system redesign. Fed Pract. 2018;35:24-29.
- Brownell N, Kay C, Parra D, et al. Development and optimization of the Veterans Affairs’ national heart failure dashboard for population health management. J Card Fail. 2024;30:452-459. doi:10.1016/j.cardfail.2023.08.024
- Lin LA, Bohnert ASB, Kerns RD, Clay MA, Ganoczy D, Ilgen MA. Impact of the opioid safety initiative on opioidrelated prescribing in veterans. Pain. 2017;158:833-839. doi:10.1097/j.pain.0000000000000837
- Twohig PA, Rivington JR, Gunzler D, Daprano J, Margolius D. Clinician dashboard views and improvement in preventative health outcome measures: a retrospective analysis. BMC Health Serv Res. 2019;19:475. doi:10.1186/s12913-019-4327-3
- Singh I, Fletcher R, Scanlon L, Tyler M, Aithal S. A quality improvement initiative on the management of osteoporosis in older people with Parkinsonism. BMJ Qual Improv Rep. 2016;5:u210921.w5756. doi:10.1136/bmjquality.u210921.w5756
- Anastasilakis AD, Makras P, Yavropoulou MP, Tabacco G, Naciu AM, Palermo A. Denosumab discontinuation and the rebound phenomenon: a narrative review. J Clin Med. 2021;10:152. doi:10.3390/jcm10010152
- Sharman Moser S, Yu J, Goldshtein I, et al. Cost and consequences of nonadherence with oral bisphosphonate therapy: findings from a real-world data analysis. Ann Pharmacother. 2016;50:262-269. doi:10.1177/1060028015626935
- Olsen KR, Hansen C, Abrahamsen B. Association between refill compliance to oral bisphosphonate treatment, incident fractures, and health care costs--an analysis using national health databases. Osteoporos Int. 2013;24:2639-2647. doi:10.1007/s00198-013-2365-y
- Blouin J, Dragomir A, Fredette M, Ste-Marie LG, Fernandes JC, Perreault S. Comparison of direct health care costs related to the pharmacological treatment of osteoporosis and to the management of osteoporotic fractures among compliant and noncompliant users of alendronate and risedronate: a population-based study. Osteoporos Int. 2009;20:1571-1581. doi:10.1007/s00198-008-0818-5
- Cotté F-E, De Pouvourville G. Cost of non-persistence with oral bisphosphonates in post-menopausal osteoporosis treatment in France. BMC Health Serv Res. 2011;11:151. doi:10.1186/1472-6963-11-151
- Cho H, Byun J-H, Song I, et al. Effect of improved medication adherence on health care costs in osteoporosis patients. Medicine (Baltimore). 2018;97:e11470. doi:10.1097/MD.0000000000011470
- Li N, Cornelissen D, Silverman S, et al. An updated systematic review of cost-effectiveness analyses of drugs for osteoporosis. Pharmacoeconomics. 2021;39:181-209. doi:10.1007/s40273-020-00965-9
Improving High-Risk Osteoporosis Medication Adherence and Safety With an Automated Dashboard
Improving High-Risk Osteoporosis Medication Adherence and Safety With an Automated Dashboard
Efficacy of Anti-Obesity Medications in Adult and Older Adult Veteran Populations
Efficacy of Anti-Obesity Medications in Adult and Older Adult Veteran Populations
The impact of obesity in the United States is significant. Between August 2021 and August 2023, the prevalence of obesity (body mass index ≥ 30) in US adults was 40.3%.1 The prevalence of obesity in adults aged 40 to 59 years was 46.4%, higher than the prevalence in adults aged 20 to 39 years (35.5%) and those aged ≥ 60 years (38.9%).1 The excess annual medical costs associated with obesity in the US are estimated at nearly $173 billion.2
The first-line treatment for obesity is lifestyle modifications, including a healthy diet and exercise. When lifestyle modifications are not enough to achieve weight-loss goals, bariatric surgery and anti-obesity medications (AOMs) are often considered. Five medications were approved for the long-term tretament of obesity by the US Food and Drug Administration (FDA) between 2021 and 2023, when this study was conducted: semaglutide (Wegovy), liraglutide (Saxenda), phentermine and topiramate, naltrexone and bupropion, and orlistat. The clinically meaningful (and commonly accepted) weight-loss target for these medications is ≥ 5% from baseline by week 12 of the maximally tolerated dose of therapy. A 5% weight loss has been shown to be clinically significant in improving cardiometabolic risk factors.3,4 These medications are intended to be used as an adjunct to healthy diet and exercise. Of note, semaglutide and liraglutide carry brand names, which are associated with different dosing for the treatment of type 2 diabetes mellitus (T2DM).
All 5 FDA-approved AOMs were available at the Veterans Affairs Sioux Falls Health Care System (VASFHCS) for the treatment of obesity at the time of the study. To qualify for an AOM, a veteran at VASFHCS must first work with a dietitian or be enrolled in the MOVE! clinic to participate in the weight management program, which focuses on dietary, exercise, and behavioral changes. At VASFHCS, AOMs are prescribed by primary care practitioners, clinical pharmacy providers, and advanced practitioners within the MOVE! program.
Ample data exist for the efficacy of AOMs. However, no published research has reported on AOM efficacy by age group (Appendix).5-11 While most of the AOM clinical trials included older adults, the average age of participants was typically between 40 and 50 years. It is well-known that pharmacokinetic and pharmacodynamic changes occur as age increases. Renal and hepatic clearance is reduced while the volume distribution and sensitivities to some medications may increase. 12 Although this study did not focus on specific pharmacokinetic and pharmacodynamic changes with respect to AOM, it is important to recognize that this may play a role in the efficacy and safety of AOMs in older adults.
Methods
This retrospective single-center chart review was performed using the VASFHCS Computerized Patient Record System to compare the efficacy of AOMs in older adults (aged ≥ 65 years) vs adults (aged < 65 years). The primary endpoint was the percent change in body weight from baseline to 6 and 12 months after initiation of AOM therapy in the older adult vs adult population. Secondary endpoints included changes in low-density lipoprotein (LDL), hemoglobin A1c (HbA1c), and blood pressure (BP) from baseline compared to 12 months on AOM therapy. HbA1c was assessed in patients with T2DM or prediabetes at the time of AOM initiation. Two safety endpoints were also explored to determine the incidence of medication adverse events (AEs) and subsequent discontinuation of AOM. A subset analysis was performed to determine whether there was a difference in percent change in body weight between patients in 3 age groups: 18 to 40 years, 41 to 64 years, and ≥ 65 years.
The study population included patients who were prescribed an AOM between January 1, 2021, and June 30, 2023. Patients were excluded if they did not continue AOM therapy for ≥ 6 months after initiation or if they underwent gastric bypass surgery while undergoing AOM therapy. Patients taking semaglutide (Ozempic) or liraglutide (Victoza) for both T2DM and weight loss who were eventually switched to the weight loss formulations (Wegovy or Saxenda) were included. Patients who switched between semaglutide and liraglutide for weight loss were also included. Those taking semaglutide or liraglutide solely for T2DM treatment were excluded because they are dosed differently.
Collected data included age, gender, race, weight (baseline, 6 and 12 months after initiation of AOM), metabolic laboratory values/vital signs (HbA1c, LDL, and BP at baseline and 12 months after initiation of AOM), diagnosis of T2DM or prediabetes, reported AEs associated with AOM therapy, and date of AOM initiation and discontinuation (if applicable). Baseline values were defined at the time of medication initiation or values documented within 6 months prior to medication initiation if true baseline data were not reported. If values were not recorded at months 6 and 12 after AOM initiation, values documented closest to those targets were used. Weights were used for baseline, 6-, and 12-month data unless they were unavailable due to use of virtual care modalities. In these cases, patient-reported weights were used. Patients were included in the 6-month data, but not the 12-month data, if they were taking AOMs for > 6 months but not for 12 months. If patients had been on multiple AOMs, baseline data were recorded at the start of the first medication that was used for 6 months or longer. Twelve-month data were recorded after subsequent medication change. Twelve-month metabolic laboratory values/vital signs were recorded for patients included in the study even if they did not complete ≥ 12 months of AOM therapy.
Statistical Analysis
Data from patients who were prescribed an AOM from January 2021 to June 2023 and who remained on the medication for ≥ 6 months were analyzed. Baseline characteristics were analyzed using descriptive statistics. The primary and secondary endpoints were evaluated using the t test. The safety endpoints were analyzed using descriptive statistics. An analysis of variance test was used for the subset analysis. Results with P < .05 were statistically significant.
Results
A total of 144 participants were included in this study, 116 in the adult group (aged < 65 years) and 28 in the older adult group (aged ≥ 65 years). Sixty-seven patients were excluded due to prespecified inclusion and exclusion criteria.
Other than the predetermined mean age differences (48 years vs 71 years), there were multiple differences in patient baseline characteristics. When comparing older adults and adults, average weight (283 lb vs 269 lb) and White race (89% vs 87%) were slightly higher in the older adult group. Also, a higher prevalence of T2DM (54% and 18%) and a lower prevalence of prediabetes (21% and 33%) was noted in the older adult group. HbA1c and BP were similar between both groups at baseline, while LDL was slightly lower in the older adult group (Table 1).
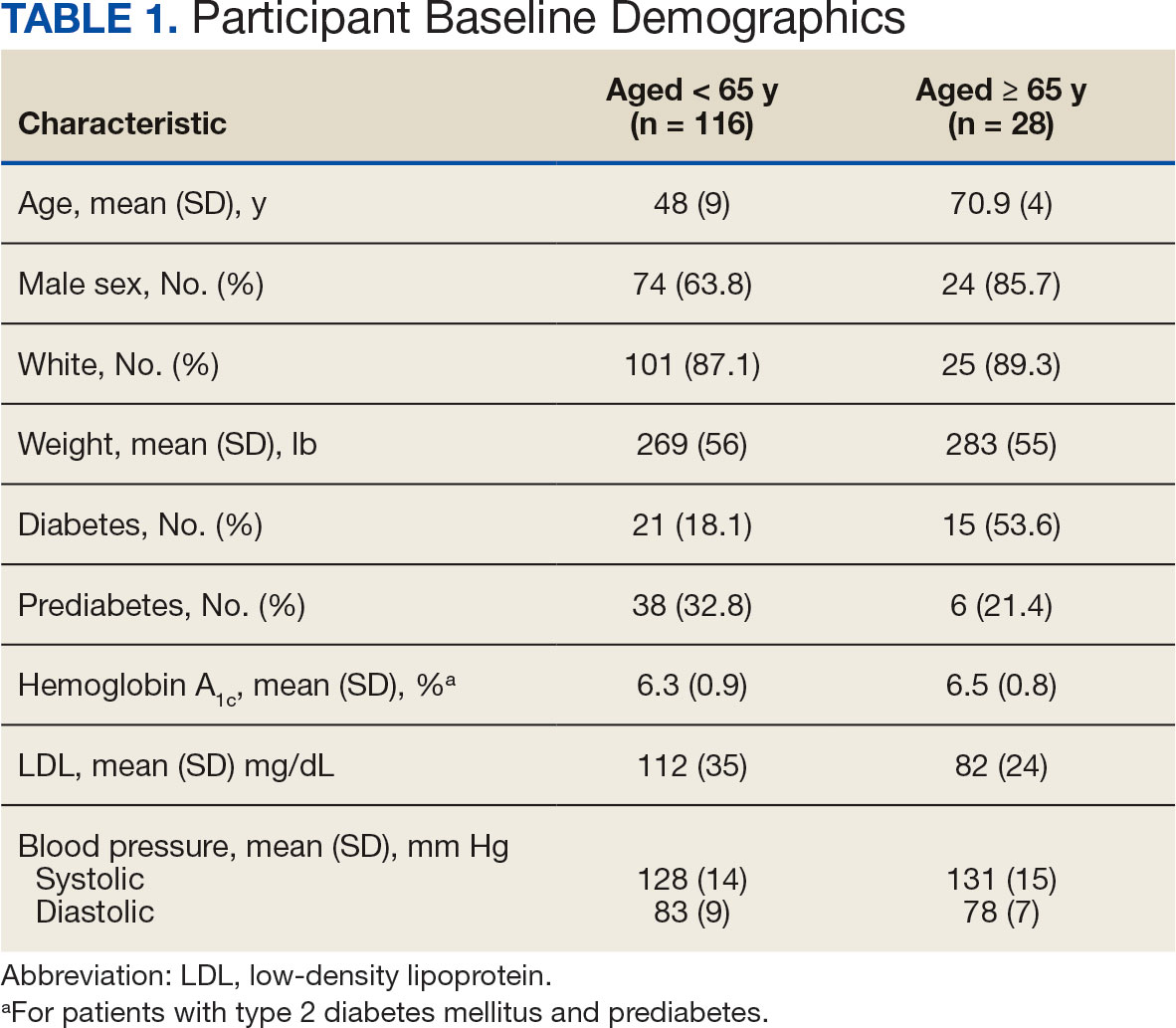
Patients in the adult group lost a mean 7.0% and 8.7% of body weight at 6 and 12 months, respectively, while the older adult group lost 5.0% and 6.6% body weight at 6 and 12 months, respectively. The difference in percent change in body weight was not statistically different at 6 (P = .08) or 12 (P = .26) months between patients in the adult group vs the older adult group or in the specific age groups (18-40 years, 41-64 years, ≥ 65 years) at 6 months (P = .24) or 12 months (P = .53) (Figure).
At 12 months, the difference between the adult group vs the older adult group was not statistically significant for HbA1c in patients with T2DM or prediabetes (P = .73), LDL (P = .95), systolic BP (P = .58), or diastolic BP (P = .51) (Table 2).
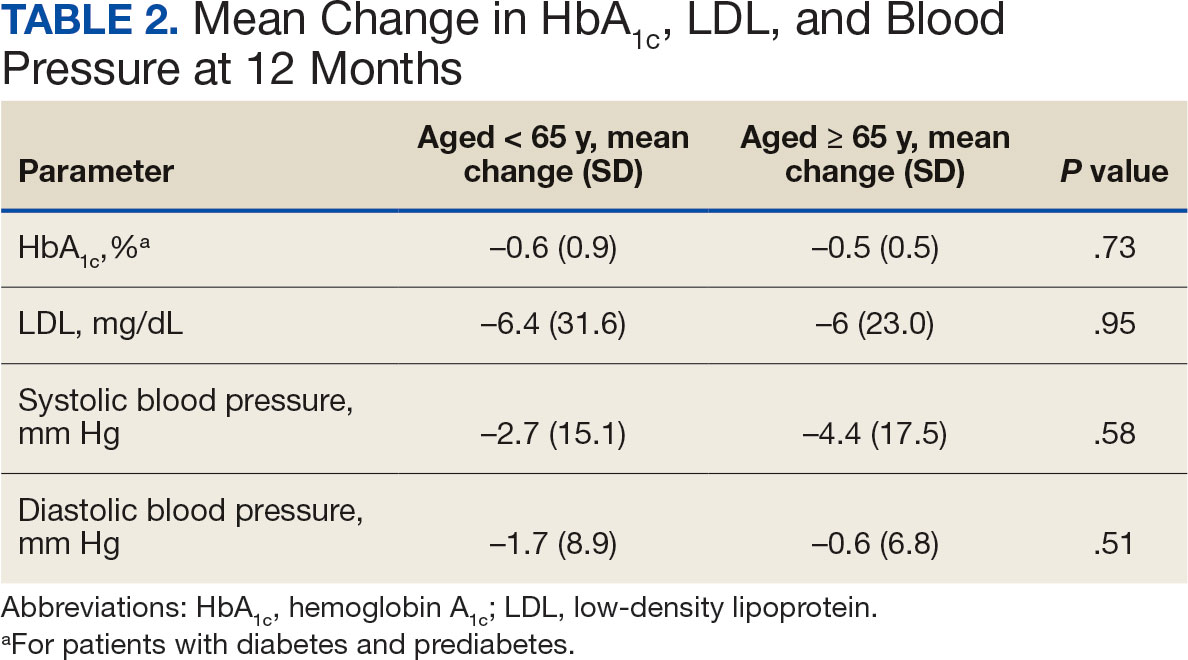
For the safety endpoint, the incidence of AEs was found to be different between groups. There were more reported AEs (61.2% vs 39.3%) and a greater increase in therapy discontinuation due to AEs (6.0% vs 0%) in the adult group compared to the older adult group (Table 3).

Discussion
Patients taking AOMs revealed no statistically significant difference in percent change in body weight at 6 or 12 months between adults aged < 65 years and older adults aged ≥ 65 years. The subset analysis also showed no statistically significant difference in change in percent body weight between more narrowly defined age groups of 18 to 40 years, 41 to 64 years, and ≥ 65 years. This suggests that AOM may have similar efficacy for weight loss in all ages of adults.
Secondary endpoint findings showed no statistically significant difference in HbA1c (in patients with T2DM or prediabetes), LDL, or BP at 12 months between the 2 groups. Although this study did not differentiate secondary outcomes based on the individual AOM, the change in HbA1c in both groups was expected, given that 70% of the patients included in this study were taking a glucagon-like peptide-1 agonist (liraglutide and semaglutide) at some point during the study. It’s also worth noting that secondary endpoints were collected for patients who discontinued the AOM between 6 and 12 months. Therefore, the patients’ HbA1c, LDL, and BP may not have accurately reflected the change that could have been expected if they had continued AOM therapy beyond the 12-month period.
Due to the different mechanisms and range in efficacy that AOMs have in regard to weight loss, changes in all outcomes, including weight, HbA1c, LDL, and BP were expected to vary as patients were included even after switching AOM (collection of data started after ≥ 6 months on a single AOM). Switching of AOM after the first 6 months of therapy was recorded in 25% of the patients in the ≥ 65 years group and 330% of the patients in the < 65 years group.
The incidence of AEs and subsequent discontinuation of AOMs in this study was higher in the adult group. This study excluded patients who did not continue taking an AOM for at least 6 months. As a result, the incidence of AEs between the 2 groups within the first 6 months of AOM therapy remains unknown. It is possible that during the first 6 months of therapy, patients aged < 65 years were more willing to tolerate or had fewer severe AEs compared with the older adult group. It’s also possible that the smaller number of patients in the older adult group was due to increased AEs that led them to discontinue early (before completion of 6 months of therapy) and/or prescriber discomfort in using AOMs in the older adult population. In addition, because the specific medication(s) taken by patients in each group were not detailed, it is unknown whether the adult group was taking AOMs associated with a greater number of AEs.
Limitations
This was a retrospective study with a relatively small sample size. A larger sample size may have shown more precise differences between age groups and may be more representative of the general population. Additionally, data were reliant on appropriate documentation, and adherence to AOM therapy was not assessed due to the retrospective nature of this study. At times, the study relied on patient reported data points, such as weight, if a clinic weight was not available. Also, this study did not account for many potential confounding factors such as other medications taken by the patient, which can affect outcomes including weight, HbA1c, LDL, blood pressure, and AEs.
Conclusions
This retrospective study of patients taking AOMs showed no statistically significant difference in weight loss at 6 or 12 months between adults aged < 65 years and older adults aged ≥ 65 years. A subset analysis found no statistically significant difference in change in body weight between specific age groups (18-40 years, 41-64 years, and ≥ 65 years). There was also no statistically significant difference in secondary outcomes, including change in HbA1c (in patients with T2DM or prediabetes), LDL or BP between age groups. The safety endpoints showed a higher incidence of medication AEs in the adult group, with more of these adults discontinuing therapy due to AEs. This study indicates that AOM may have similar outcomes for weight loss and metabolic laboratory values/vital sign changes between adults and older adults. Also, our findings suggest that patients aged < 65 years may experience more AEs than patients aged ≥ 65 years after ≥ 6 months of AOM therapy. Larger studies are needed to further evaluate these age-specific findings.
- Emmerich SD, Fryar CD, Stierman B, Ogden CL. Obesity and severe obesity prevalence in adults: United States, August 2021-August 2023. NCHS Data Brief No. 508. National Center for Health Statistics; 2024. Accessed December 11, 2024. https://www.cdc.gov/nchs/products/databriefs/db508.htm
- Ward ZJ, Bleich SN, Long MW, Gortmaker SL. Association of body mass index with health care expenditures in the United States by age and sex. PLoS One. 2021;16(3):e0247307. doi:10.1371/journal.pone.0247307
- Horn DB, Almandoz JP, Look M. What is clinically relevant weight loss for your patients and how can it be achieved? A narrative review. Postgrad Med. 2022;134(4):359-375. doi:10.1080/00325481.2022.2051366
- American Diabetes Association (ADA). Standards of care in diabetes–2023. Diabetes Care. 2023;46(suppl 1):S128- S2139. doi:10.2337/dc23-S008
- Wilding JPH, Batterham RL, Calanna S, et al. Onceweekly semaglutide in adults with overweight or obesity. N Engl J Med. 2021;384(11):989-1002. doi:10.1056/NEJMoa2032183
- Pi-Sunyer X, Astrup A, Fujioka K, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373(1):11-22. doi:10.1056/NEJMoa1411892
- Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20(2):330-342. doi:10.1038/oby.2011.330
- Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9774):1341-1352. doi:10.1016/S0140-6736(11)60205-5
- Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95(2):297-308. doi:10.3945/ajcn.111.024927
- Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605. doi:10.1016/S0140-6736(10)60888-4
- Sjöström L, Rissanen A, Andersen T, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet. 1998;352(9123):167-172. doi:10.1016s0140-6736(97)11509-4
- Mangoni AA, Jackson SHD. Age-related changes in pharmacokinetics and pharmacodynamics: basic principles and practical applications. Br J Clin Pharmacol. 2004;57(1):6-14. doi:10.1046/j.1365-2125.2003.02007.x
The impact of obesity in the United States is significant. Between August 2021 and August 2023, the prevalence of obesity (body mass index ≥ 30) in US adults was 40.3%.1 The prevalence of obesity in adults aged 40 to 59 years was 46.4%, higher than the prevalence in adults aged 20 to 39 years (35.5%) and those aged ≥ 60 years (38.9%).1 The excess annual medical costs associated with obesity in the US are estimated at nearly $173 billion.2
The first-line treatment for obesity is lifestyle modifications, including a healthy diet and exercise. When lifestyle modifications are not enough to achieve weight-loss goals, bariatric surgery and anti-obesity medications (AOMs) are often considered. Five medications were approved for the long-term tretament of obesity by the US Food and Drug Administration (FDA) between 2021 and 2023, when this study was conducted: semaglutide (Wegovy), liraglutide (Saxenda), phentermine and topiramate, naltrexone and bupropion, and orlistat. The clinically meaningful (and commonly accepted) weight-loss target for these medications is ≥ 5% from baseline by week 12 of the maximally tolerated dose of therapy. A 5% weight loss has been shown to be clinically significant in improving cardiometabolic risk factors.3,4 These medications are intended to be used as an adjunct to healthy diet and exercise. Of note, semaglutide and liraglutide carry brand names, which are associated with different dosing for the treatment of type 2 diabetes mellitus (T2DM).
All 5 FDA-approved AOMs were available at the Veterans Affairs Sioux Falls Health Care System (VASFHCS) for the treatment of obesity at the time of the study. To qualify for an AOM, a veteran at VASFHCS must first work with a dietitian or be enrolled in the MOVE! clinic to participate in the weight management program, which focuses on dietary, exercise, and behavioral changes. At VASFHCS, AOMs are prescribed by primary care practitioners, clinical pharmacy providers, and advanced practitioners within the MOVE! program.
Ample data exist for the efficacy of AOMs. However, no published research has reported on AOM efficacy by age group (Appendix).5-11 While most of the AOM clinical trials included older adults, the average age of participants was typically between 40 and 50 years. It is well-known that pharmacokinetic and pharmacodynamic changes occur as age increases. Renal and hepatic clearance is reduced while the volume distribution and sensitivities to some medications may increase. 12 Although this study did not focus on specific pharmacokinetic and pharmacodynamic changes with respect to AOM, it is important to recognize that this may play a role in the efficacy and safety of AOMs in older adults.
Methods
This retrospective single-center chart review was performed using the VASFHCS Computerized Patient Record System to compare the efficacy of AOMs in older adults (aged ≥ 65 years) vs adults (aged < 65 years). The primary endpoint was the percent change in body weight from baseline to 6 and 12 months after initiation of AOM therapy in the older adult vs adult population. Secondary endpoints included changes in low-density lipoprotein (LDL), hemoglobin A1c (HbA1c), and blood pressure (BP) from baseline compared to 12 months on AOM therapy. HbA1c was assessed in patients with T2DM or prediabetes at the time of AOM initiation. Two safety endpoints were also explored to determine the incidence of medication adverse events (AEs) and subsequent discontinuation of AOM. A subset analysis was performed to determine whether there was a difference in percent change in body weight between patients in 3 age groups: 18 to 40 years, 41 to 64 years, and ≥ 65 years.
The study population included patients who were prescribed an AOM between January 1, 2021, and June 30, 2023. Patients were excluded if they did not continue AOM therapy for ≥ 6 months after initiation or if they underwent gastric bypass surgery while undergoing AOM therapy. Patients taking semaglutide (Ozempic) or liraglutide (Victoza) for both T2DM and weight loss who were eventually switched to the weight loss formulations (Wegovy or Saxenda) were included. Patients who switched between semaglutide and liraglutide for weight loss were also included. Those taking semaglutide or liraglutide solely for T2DM treatment were excluded because they are dosed differently.
Collected data included age, gender, race, weight (baseline, 6 and 12 months after initiation of AOM), metabolic laboratory values/vital signs (HbA1c, LDL, and BP at baseline and 12 months after initiation of AOM), diagnosis of T2DM or prediabetes, reported AEs associated with AOM therapy, and date of AOM initiation and discontinuation (if applicable). Baseline values were defined at the time of medication initiation or values documented within 6 months prior to medication initiation if true baseline data were not reported. If values were not recorded at months 6 and 12 after AOM initiation, values documented closest to those targets were used. Weights were used for baseline, 6-, and 12-month data unless they were unavailable due to use of virtual care modalities. In these cases, patient-reported weights were used. Patients were included in the 6-month data, but not the 12-month data, if they were taking AOMs for > 6 months but not for 12 months. If patients had been on multiple AOMs, baseline data were recorded at the start of the first medication that was used for 6 months or longer. Twelve-month data were recorded after subsequent medication change. Twelve-month metabolic laboratory values/vital signs were recorded for patients included in the study even if they did not complete ≥ 12 months of AOM therapy.
Statistical Analysis
Data from patients who were prescribed an AOM from January 2021 to June 2023 and who remained on the medication for ≥ 6 months were analyzed. Baseline characteristics were analyzed using descriptive statistics. The primary and secondary endpoints were evaluated using the t test. The safety endpoints were analyzed using descriptive statistics. An analysis of variance test was used for the subset analysis. Results with P < .05 were statistically significant.
Results
A total of 144 participants were included in this study, 116 in the adult group (aged < 65 years) and 28 in the older adult group (aged ≥ 65 years). Sixty-seven patients were excluded due to prespecified inclusion and exclusion criteria.
Other than the predetermined mean age differences (48 years vs 71 years), there were multiple differences in patient baseline characteristics. When comparing older adults and adults, average weight (283 lb vs 269 lb) and White race (89% vs 87%) were slightly higher in the older adult group. Also, a higher prevalence of T2DM (54% and 18%) and a lower prevalence of prediabetes (21% and 33%) was noted in the older adult group. HbA1c and BP were similar between both groups at baseline, while LDL was slightly lower in the older adult group (Table 1).
Patients in the adult group lost a mean 7.0% and 8.7% of body weight at 6 and 12 months, respectively, while the older adult group lost 5.0% and 6.6% body weight at 6 and 12 months, respectively. The difference in percent change in body weight was not statistically different at 6 (P = .08) or 12 (P = .26) months between patients in the adult group vs the older adult group or in the specific age groups (18-40 years, 41-64 years, ≥ 65 years) at 6 months (P = .24) or 12 months (P = .53) (Figure).
At 12 months, the difference between the adult group vs the older adult group was not statistically significant for HbA1c in patients with T2DM or prediabetes (P = .73), LDL (P = .95), systolic BP (P = .58), or diastolic BP (P = .51) (Table 2).
For the safety endpoint, the incidence of AEs was found to be different between groups. There were more reported AEs (61.2% vs 39.3%) and a greater increase in therapy discontinuation due to AEs (6.0% vs 0%) in the adult group compared to the older adult group (Table 3).
Discussion
Patients taking AOMs revealed no statistically significant difference in percent change in body weight at 6 or 12 months between adults aged < 65 years and older adults aged ≥ 65 years. The subset analysis also showed no statistically significant difference in change in percent body weight between more narrowly defined age groups of 18 to 40 years, 41 to 64 years, and ≥ 65 years. This suggests that AOM may have similar efficacy for weight loss in all ages of adults.
Secondary endpoint findings showed no statistically significant difference in HbA1c (in patients with T2DM or prediabetes), LDL, or BP at 12 months between the 2 groups. Although this study did not differentiate secondary outcomes based on the individual AOM, the change in HbA1c in both groups was expected, given that 70% of the patients included in this study were taking a glucagon-like peptide-1 agonist (liraglutide and semaglutide) at some point during the study. It’s also worth noting that secondary endpoints were collected for patients who discontinued the AOM between 6 and 12 months. Therefore, the patients’ HbA1c, LDL, and BP may not have accurately reflected the change that could have been expected if they had continued AOM therapy beyond the 12-month period.
Due to the different mechanisms and range in efficacy that AOMs have in regard to weight loss, changes in all outcomes, including weight, HbA1c, LDL, and BP were expected to vary as patients were included even after switching AOM (collection of data started after ≥ 6 months on a single AOM). Switching of AOM after the first 6 months of therapy was recorded in 25% of the patients in the ≥ 65 years group and 330% of the patients in the < 65 years group.
The incidence of AEs and subsequent discontinuation of AOMs in this study was higher in the adult group. This study excluded patients who did not continue taking an AOM for at least 6 months. As a result, the incidence of AEs between the 2 groups within the first 6 months of AOM therapy remains unknown. It is possible that during the first 6 months of therapy, patients aged < 65 years were more willing to tolerate or had fewer severe AEs compared with the older adult group. It’s also possible that the smaller number of patients in the older adult group was due to increased AEs that led them to discontinue early (before completion of 6 months of therapy) and/or prescriber discomfort in using AOMs in the older adult population. In addition, because the specific medication(s) taken by patients in each group were not detailed, it is unknown whether the adult group was taking AOMs associated with a greater number of AEs.
Limitations
This was a retrospective study with a relatively small sample size. A larger sample size may have shown more precise differences between age groups and may be more representative of the general population. Additionally, data were reliant on appropriate documentation, and adherence to AOM therapy was not assessed due to the retrospective nature of this study. At times, the study relied on patient reported data points, such as weight, if a clinic weight was not available. Also, this study did not account for many potential confounding factors such as other medications taken by the patient, which can affect outcomes including weight, HbA1c, LDL, blood pressure, and AEs.
Conclusions
This retrospective study of patients taking AOMs showed no statistically significant difference in weight loss at 6 or 12 months between adults aged < 65 years and older adults aged ≥ 65 years. A subset analysis found no statistically significant difference in change in body weight between specific age groups (18-40 years, 41-64 years, and ≥ 65 years). There was also no statistically significant difference in secondary outcomes, including change in HbA1c (in patients with T2DM or prediabetes), LDL or BP between age groups. The safety endpoints showed a higher incidence of medication AEs in the adult group, with more of these adults discontinuing therapy due to AEs. This study indicates that AOM may have similar outcomes for weight loss and metabolic laboratory values/vital sign changes between adults and older adults. Also, our findings suggest that patients aged < 65 years may experience more AEs than patients aged ≥ 65 years after ≥ 6 months of AOM therapy. Larger studies are needed to further evaluate these age-specific findings.
The impact of obesity in the United States is significant. Between August 2021 and August 2023, the prevalence of obesity (body mass index ≥ 30) in US adults was 40.3%.1 The prevalence of obesity in adults aged 40 to 59 years was 46.4%, higher than the prevalence in adults aged 20 to 39 years (35.5%) and those aged ≥ 60 years (38.9%).1 The excess annual medical costs associated with obesity in the US are estimated at nearly $173 billion.2
The first-line treatment for obesity is lifestyle modifications, including a healthy diet and exercise. When lifestyle modifications are not enough to achieve weight-loss goals, bariatric surgery and anti-obesity medications (AOMs) are often considered. Five medications were approved for the long-term tretament of obesity by the US Food and Drug Administration (FDA) between 2021 and 2023, when this study was conducted: semaglutide (Wegovy), liraglutide (Saxenda), phentermine and topiramate, naltrexone and bupropion, and orlistat. The clinically meaningful (and commonly accepted) weight-loss target for these medications is ≥ 5% from baseline by week 12 of the maximally tolerated dose of therapy. A 5% weight loss has been shown to be clinically significant in improving cardiometabolic risk factors.3,4 These medications are intended to be used as an adjunct to healthy diet and exercise. Of note, semaglutide and liraglutide carry brand names, which are associated with different dosing for the treatment of type 2 diabetes mellitus (T2DM).
All 5 FDA-approved AOMs were available at the Veterans Affairs Sioux Falls Health Care System (VASFHCS) for the treatment of obesity at the time of the study. To qualify for an AOM, a veteran at VASFHCS must first work with a dietitian or be enrolled in the MOVE! clinic to participate in the weight management program, which focuses on dietary, exercise, and behavioral changes. At VASFHCS, AOMs are prescribed by primary care practitioners, clinical pharmacy providers, and advanced practitioners within the MOVE! program.
Ample data exist for the efficacy of AOMs. However, no published research has reported on AOM efficacy by age group (Appendix).5-11 While most of the AOM clinical trials included older adults, the average age of participants was typically between 40 and 50 years. It is well-known that pharmacokinetic and pharmacodynamic changes occur as age increases. Renal and hepatic clearance is reduced while the volume distribution and sensitivities to some medications may increase. 12 Although this study did not focus on specific pharmacokinetic and pharmacodynamic changes with respect to AOM, it is important to recognize that this may play a role in the efficacy and safety of AOMs in older adults.
Methods
This retrospective single-center chart review was performed using the VASFHCS Computerized Patient Record System to compare the efficacy of AOMs in older adults (aged ≥ 65 years) vs adults (aged < 65 years). The primary endpoint was the percent change in body weight from baseline to 6 and 12 months after initiation of AOM therapy in the older adult vs adult population. Secondary endpoints included changes in low-density lipoprotein (LDL), hemoglobin A1c (HbA1c), and blood pressure (BP) from baseline compared to 12 months on AOM therapy. HbA1c was assessed in patients with T2DM or prediabetes at the time of AOM initiation. Two safety endpoints were also explored to determine the incidence of medication adverse events (AEs) and subsequent discontinuation of AOM. A subset analysis was performed to determine whether there was a difference in percent change in body weight between patients in 3 age groups: 18 to 40 years, 41 to 64 years, and ≥ 65 years.
The study population included patients who were prescribed an AOM between January 1, 2021, and June 30, 2023. Patients were excluded if they did not continue AOM therapy for ≥ 6 months after initiation or if they underwent gastric bypass surgery while undergoing AOM therapy. Patients taking semaglutide (Ozempic) or liraglutide (Victoza) for both T2DM and weight loss who were eventually switched to the weight loss formulations (Wegovy or Saxenda) were included. Patients who switched between semaglutide and liraglutide for weight loss were also included. Those taking semaglutide or liraglutide solely for T2DM treatment were excluded because they are dosed differently.
Collected data included age, gender, race, weight (baseline, 6 and 12 months after initiation of AOM), metabolic laboratory values/vital signs (HbA1c, LDL, and BP at baseline and 12 months after initiation of AOM), diagnosis of T2DM or prediabetes, reported AEs associated with AOM therapy, and date of AOM initiation and discontinuation (if applicable). Baseline values were defined at the time of medication initiation or values documented within 6 months prior to medication initiation if true baseline data were not reported. If values were not recorded at months 6 and 12 after AOM initiation, values documented closest to those targets were used. Weights were used for baseline, 6-, and 12-month data unless they were unavailable due to use of virtual care modalities. In these cases, patient-reported weights were used. Patients were included in the 6-month data, but not the 12-month data, if they were taking AOMs for > 6 months but not for 12 months. If patients had been on multiple AOMs, baseline data were recorded at the start of the first medication that was used for 6 months or longer. Twelve-month data were recorded after subsequent medication change. Twelve-month metabolic laboratory values/vital signs were recorded for patients included in the study even if they did not complete ≥ 12 months of AOM therapy.
Statistical Analysis
Data from patients who were prescribed an AOM from January 2021 to June 2023 and who remained on the medication for ≥ 6 months were analyzed. Baseline characteristics were analyzed using descriptive statistics. The primary and secondary endpoints were evaluated using the t test. The safety endpoints were analyzed using descriptive statistics. An analysis of variance test was used for the subset analysis. Results with P < .05 were statistically significant.
Results
A total of 144 participants were included in this study, 116 in the adult group (aged < 65 years) and 28 in the older adult group (aged ≥ 65 years). Sixty-seven patients were excluded due to prespecified inclusion and exclusion criteria.
Other than the predetermined mean age differences (48 years vs 71 years), there were multiple differences in patient baseline characteristics. When comparing older adults and adults, average weight (283 lb vs 269 lb) and White race (89% vs 87%) were slightly higher in the older adult group. Also, a higher prevalence of T2DM (54% and 18%) and a lower prevalence of prediabetes (21% and 33%) was noted in the older adult group. HbA1c and BP were similar between both groups at baseline, while LDL was slightly lower in the older adult group (Table 1).
Patients in the adult group lost a mean 7.0% and 8.7% of body weight at 6 and 12 months, respectively, while the older adult group lost 5.0% and 6.6% body weight at 6 and 12 months, respectively. The difference in percent change in body weight was not statistically different at 6 (P = .08) or 12 (P = .26) months between patients in the adult group vs the older adult group or in the specific age groups (18-40 years, 41-64 years, ≥ 65 years) at 6 months (P = .24) or 12 months (P = .53) (Figure).
At 12 months, the difference between the adult group vs the older adult group was not statistically significant for HbA1c in patients with T2DM or prediabetes (P = .73), LDL (P = .95), systolic BP (P = .58), or diastolic BP (P = .51) (Table 2).
For the safety endpoint, the incidence of AEs was found to be different between groups. There were more reported AEs (61.2% vs 39.3%) and a greater increase in therapy discontinuation due to AEs (6.0% vs 0%) in the adult group compared to the older adult group (Table 3).
Discussion
Patients taking AOMs revealed no statistically significant difference in percent change in body weight at 6 or 12 months between adults aged < 65 years and older adults aged ≥ 65 years. The subset analysis also showed no statistically significant difference in change in percent body weight between more narrowly defined age groups of 18 to 40 years, 41 to 64 years, and ≥ 65 years. This suggests that AOM may have similar efficacy for weight loss in all ages of adults.
Secondary endpoint findings showed no statistically significant difference in HbA1c (in patients with T2DM or prediabetes), LDL, or BP at 12 months between the 2 groups. Although this study did not differentiate secondary outcomes based on the individual AOM, the change in HbA1c in both groups was expected, given that 70% of the patients included in this study were taking a glucagon-like peptide-1 agonist (liraglutide and semaglutide) at some point during the study. It’s also worth noting that secondary endpoints were collected for patients who discontinued the AOM between 6 and 12 months. Therefore, the patients’ HbA1c, LDL, and BP may not have accurately reflected the change that could have been expected if they had continued AOM therapy beyond the 12-month period.
Due to the different mechanisms and range in efficacy that AOMs have in regard to weight loss, changes in all outcomes, including weight, HbA1c, LDL, and BP were expected to vary as patients were included even after switching AOM (collection of data started after ≥ 6 months on a single AOM). Switching of AOM after the first 6 months of therapy was recorded in 25% of the patients in the ≥ 65 years group and 330% of the patients in the < 65 years group.
The incidence of AEs and subsequent discontinuation of AOMs in this study was higher in the adult group. This study excluded patients who did not continue taking an AOM for at least 6 months. As a result, the incidence of AEs between the 2 groups within the first 6 months of AOM therapy remains unknown. It is possible that during the first 6 months of therapy, patients aged < 65 years were more willing to tolerate or had fewer severe AEs compared with the older adult group. It’s also possible that the smaller number of patients in the older adult group was due to increased AEs that led them to discontinue early (before completion of 6 months of therapy) and/or prescriber discomfort in using AOMs in the older adult population. In addition, because the specific medication(s) taken by patients in each group were not detailed, it is unknown whether the adult group was taking AOMs associated with a greater number of AEs.
Limitations
This was a retrospective study with a relatively small sample size. A larger sample size may have shown more precise differences between age groups and may be more representative of the general population. Additionally, data were reliant on appropriate documentation, and adherence to AOM therapy was not assessed due to the retrospective nature of this study. At times, the study relied on patient reported data points, such as weight, if a clinic weight was not available. Also, this study did not account for many potential confounding factors such as other medications taken by the patient, which can affect outcomes including weight, HbA1c, LDL, blood pressure, and AEs.
Conclusions
This retrospective study of patients taking AOMs showed no statistically significant difference in weight loss at 6 or 12 months between adults aged < 65 years and older adults aged ≥ 65 years. A subset analysis found no statistically significant difference in change in body weight between specific age groups (18-40 years, 41-64 years, and ≥ 65 years). There was also no statistically significant difference in secondary outcomes, including change in HbA1c (in patients with T2DM or prediabetes), LDL or BP between age groups. The safety endpoints showed a higher incidence of medication AEs in the adult group, with more of these adults discontinuing therapy due to AEs. This study indicates that AOM may have similar outcomes for weight loss and metabolic laboratory values/vital sign changes between adults and older adults. Also, our findings suggest that patients aged < 65 years may experience more AEs than patients aged ≥ 65 years after ≥ 6 months of AOM therapy. Larger studies are needed to further evaluate these age-specific findings.
- Emmerich SD, Fryar CD, Stierman B, Ogden CL. Obesity and severe obesity prevalence in adults: United States, August 2021-August 2023. NCHS Data Brief No. 508. National Center for Health Statistics; 2024. Accessed December 11, 2024. https://www.cdc.gov/nchs/products/databriefs/db508.htm
- Ward ZJ, Bleich SN, Long MW, Gortmaker SL. Association of body mass index with health care expenditures in the United States by age and sex. PLoS One. 2021;16(3):e0247307. doi:10.1371/journal.pone.0247307
- Horn DB, Almandoz JP, Look M. What is clinically relevant weight loss for your patients and how can it be achieved? A narrative review. Postgrad Med. 2022;134(4):359-375. doi:10.1080/00325481.2022.2051366
- American Diabetes Association (ADA). Standards of care in diabetes–2023. Diabetes Care. 2023;46(suppl 1):S128- S2139. doi:10.2337/dc23-S008
- Wilding JPH, Batterham RL, Calanna S, et al. Onceweekly semaglutide in adults with overweight or obesity. N Engl J Med. 2021;384(11):989-1002. doi:10.1056/NEJMoa2032183
- Pi-Sunyer X, Astrup A, Fujioka K, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373(1):11-22. doi:10.1056/NEJMoa1411892
- Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20(2):330-342. doi:10.1038/oby.2011.330
- Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9774):1341-1352. doi:10.1016/S0140-6736(11)60205-5
- Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95(2):297-308. doi:10.3945/ajcn.111.024927
- Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605. doi:10.1016/S0140-6736(10)60888-4
- Sjöström L, Rissanen A, Andersen T, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet. 1998;352(9123):167-172. doi:10.1016s0140-6736(97)11509-4
- Mangoni AA, Jackson SHD. Age-related changes in pharmacokinetics and pharmacodynamics: basic principles and practical applications. Br J Clin Pharmacol. 2004;57(1):6-14. doi:10.1046/j.1365-2125.2003.02007.x
- Emmerich SD, Fryar CD, Stierman B, Ogden CL. Obesity and severe obesity prevalence in adults: United States, August 2021-August 2023. NCHS Data Brief No. 508. National Center for Health Statistics; 2024. Accessed December 11, 2024. https://www.cdc.gov/nchs/products/databriefs/db508.htm
- Ward ZJ, Bleich SN, Long MW, Gortmaker SL. Association of body mass index with health care expenditures in the United States by age and sex. PLoS One. 2021;16(3):e0247307. doi:10.1371/journal.pone.0247307
- Horn DB, Almandoz JP, Look M. What is clinically relevant weight loss for your patients and how can it be achieved? A narrative review. Postgrad Med. 2022;134(4):359-375. doi:10.1080/00325481.2022.2051366
- American Diabetes Association (ADA). Standards of care in diabetes–2023. Diabetes Care. 2023;46(suppl 1):S128- S2139. doi:10.2337/dc23-S008
- Wilding JPH, Batterham RL, Calanna S, et al. Onceweekly semaglutide in adults with overweight or obesity. N Engl J Med. 2021;384(11):989-1002. doi:10.1056/NEJMoa2032183
- Pi-Sunyer X, Astrup A, Fujioka K, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373(1):11-22. doi:10.1056/NEJMoa1411892
- Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20(2):330-342. doi:10.1038/oby.2011.330
- Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9774):1341-1352. doi:10.1016/S0140-6736(11)60205-5
- Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95(2):297-308. doi:10.3945/ajcn.111.024927
- Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605. doi:10.1016/S0140-6736(10)60888-4
- Sjöström L, Rissanen A, Andersen T, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet. 1998;352(9123):167-172. doi:10.1016s0140-6736(97)11509-4
- Mangoni AA, Jackson SHD. Age-related changes in pharmacokinetics and pharmacodynamics: basic principles and practical applications. Br J Clin Pharmacol. 2004;57(1):6-14. doi:10.1046/j.1365-2125.2003.02007.x
Efficacy of Anti-Obesity Medications in Adult and Older Adult Veteran Populations
Efficacy of Anti-Obesity Medications in Adult and Older Adult Veteran Populations
Pharmacist-Driven Deprescribing to Reduce Anticholinergic Burden in Veterans With Dementia
Pharmacist-Driven Deprescribing to Reduce Anticholinergic Burden in Veterans With Dementia
Anticholinergic medications block the activity of the neurotransmitter acetylcholine by binding to either muscarinic or nicotinic receptors in both the peripheral and central nervous system. Anticholinergic medications typically refer to antimuscarinic medications and have been prescribed to treat a variety of conditions common in older adults, including overactive bladder, allergies, muscle spasms, and sleep disorders.1,2 Since muscarinic receptors are present throughout the body, anticholinergic medications are associated with many adverse effects (AEs), including constipation, urinary retention, xerostomia, and delirium. Older adults are more sensitive to these AEs due to physiological changes associated with aging.1
The American Geriatric Society Beers Criteria for Potentially Inappropriate Medications Use in Older Adults identifies drugs with strong anticholinergic properties. The Beers Criteria strongly recommends avoiding these medications in patients with dementia or cognitive impairment due to the risk of central nervous system AEs. In the updated 2023 Beers Criteria, the rationale was expanded to recognize the risks of the cumulative anticholinergic burden associated with concurrent anticholinergic use.3,4
Given the prevalent use of anticholinergic medications in older adults, there has been significant research demonstrating their AEs, specifically delirium and cognitive impairment in geriatric patients. A systematic review of 14 articles conducted in 7 different countries of patients with median age of 76.4 to 86.1 years reviewed clinical outcomes of anticholinergic use in patients with dementia. Five studies found anticholinergics were associated with increased all-cause mortality in patients with dementia, and 3 studies found anticholinergics were associated with longer hospital stays. Other studies found that anticholinergics were associated with delirium and reduced health-related quality of life.5
About 35% of veterans with dementia have been prescribed a medication regimen with a high anticholinergic burden.6 In 2018, the US Department of Veterans Affairs (VA) Pharmacy Benfits Management Center for Medical Safety completed a centrally aggregated medication use evaluation (CAMUE) to assess the appropriateness of anticholinergic medication use in patients with dementia. The retrospective chart review included 1094 veterans from 19 sites. Overall, about 15% of the veterans experienced new falls, delirium, or worsening dementia within 30 days of starting an anticholinergic medication. Furthermore, < 40% had documentation of a nonanticholinergic alternative medication trial, and < 20% had documented nonpharmacologic therapy. The documentation of risk-benefit assessment acknowledging the risks of anticholinergic medication use in veterans with dementia occurred only about 13% of the time. The CAMUE concluded that the risks of initiating an anticholinergic medication in veterans with dementia are likely underdocumented and possibly under considered by prescribers.7
Developed within the Veterans Health Administration (VHA), VIONE (Vital, Important, Optional, Not Indicated, Every medication has an indication) is a medication management methodology that aims to reduce polypharmacy and improve patient safety consistent with high-reliability organizations. Since it launched in 2016, VIONE has gradually been implemented at many VHA facilities. The VIONE deprescribing dashboard had not been used at the VA Louisville Healthcare System prior to this quality improvement project.
This dashboard uses the Beers Criteria to identify potentially inappropriate anticholinergic medications. It uses the Anticholinergic Cognitive Burden (ACB) scale to calculate the cumulative anticholinergic risk for each patient. Medications with an ACB score of 2 or 3 have clinically relevant cognitive effects such as delirium and dementia (Table 1). For each point increase in total ACB score, a decline in mini-mental state examination score of 0.33 points over 2 years has been shown. Each point increase has also been correlated with a 26% increase in risk of death.8-10
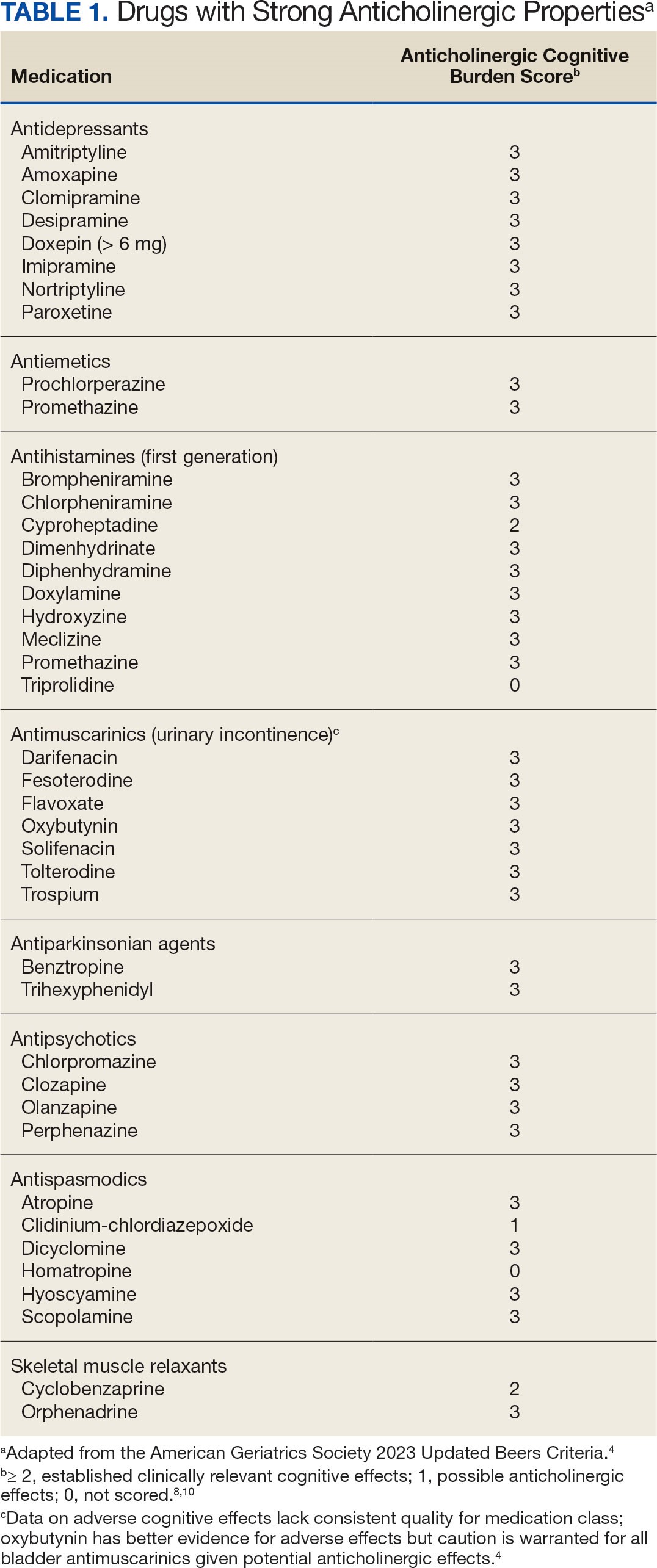
Methods
The purpose of this quality improvement project was to determine the impact of pharmacist-driven deprescribing on the anticholinergic burden in veterans with dementia at VA Louisville Healthcare System. Data were obtained through the Computerized Patient Record System (CPRS) and VIONE deprescribing dashboard and entered in a secure Microsoft Excel spreadsheet. Pharmacist deprescribing steps were entered as CPRS progress notes. A deprescribing note template was created, and 11 templates with indication-specific recommendations were created for each anticholinergic indication identified (contact authors for deprescribing note template examples). Usage of anticholinergic medications was reexamined 3 months after the deprescribing note was entered.
Eligible patients identified in the VIONE deprescribing dashboard had an outpatient order for a medication with strong anticholinergic properties as identified using the Beers Criteria and were aged ≥ 65 years. Patients also had to be diagnosed with dementia or cognitive impairment. Patients were excluded if they were receiving hospice care or if the anticholinergic medication was from a non-VA prescriber or filled at a non-VA pharmacy. The VIONE deprescribing dashboard also excluded skeletal muscle relaxants if the patient had a spinal cord-related visit in the previous 2 years, first-generation antihistamines if the patient had a vertigo diagnosis, hydroxyzine if the indication was for anxiety, trospium if the indication was for overactive bladder, and antipsychotics if the patient had been diagnosed with schizophrenia or bipolar disorder. The following were included in the deprescribing recommendations if the dashboard identified the patient due to receiving a second strongly anticholinergic medication: first generation antihistamines if the patient was diagnosed with vertigo and hydroxyzine if the indication is for anxiety.
Each eligible patient received a focused medication review by a pharmacist via electronic chart review and a templated CPRS progress note with patient-specific recommendations. The prescriber and the patient’s primary care practitioner were recommended to perform a patient-specific risk-benefit assessment, deprescribe potentially inappropriate anticholinergic medications, and consider nonanticholinergic alternatives (both pharmacologic and nonpharmacologic). Data collected included baseline age, sex, prespecified comorbidities (type of dementia, cognitive impairment, delirium, benign prostatic hyperplasia/lower urinary tract symptoms), duration of prescribed anticholinergic medication, indication and deprescribing rate for each anticholinergic agent, and concurrent dementia medications (acetylcholinesterase inhibitors, memantine, or both).
The primary outcome was the number of patients that had = 1 medication with strong anticholinergic properties deprescribed. Deprescribing was defined as medication discontinuation or reduction of total daily dose. Secondary outcomes were the mean change in ACB scale, the number of patients with dose tapering, documented patient-specific risk-benefit assessment, and initiated nonanticholinergic alternative per pharmacist recommendation.
Results
The VIONE deprescribing dashboard identified 121 patients; 45 were excluded for non-VA prescriber or pharmacy, and 8 patients were excluded for other reasons. Sixty-eight patients were included in the deprescribing initiative. The mean age was 73.4 years (range, 67-93), 65 (96%) were male, and 34 (50%) had unspecified dementia (Table 2). Thirty-one patients (46%) had concurrent cholinesterase inhibitor prescriptions for dementia. The median duration of use of a strong anticholinergic medication was 11 months.
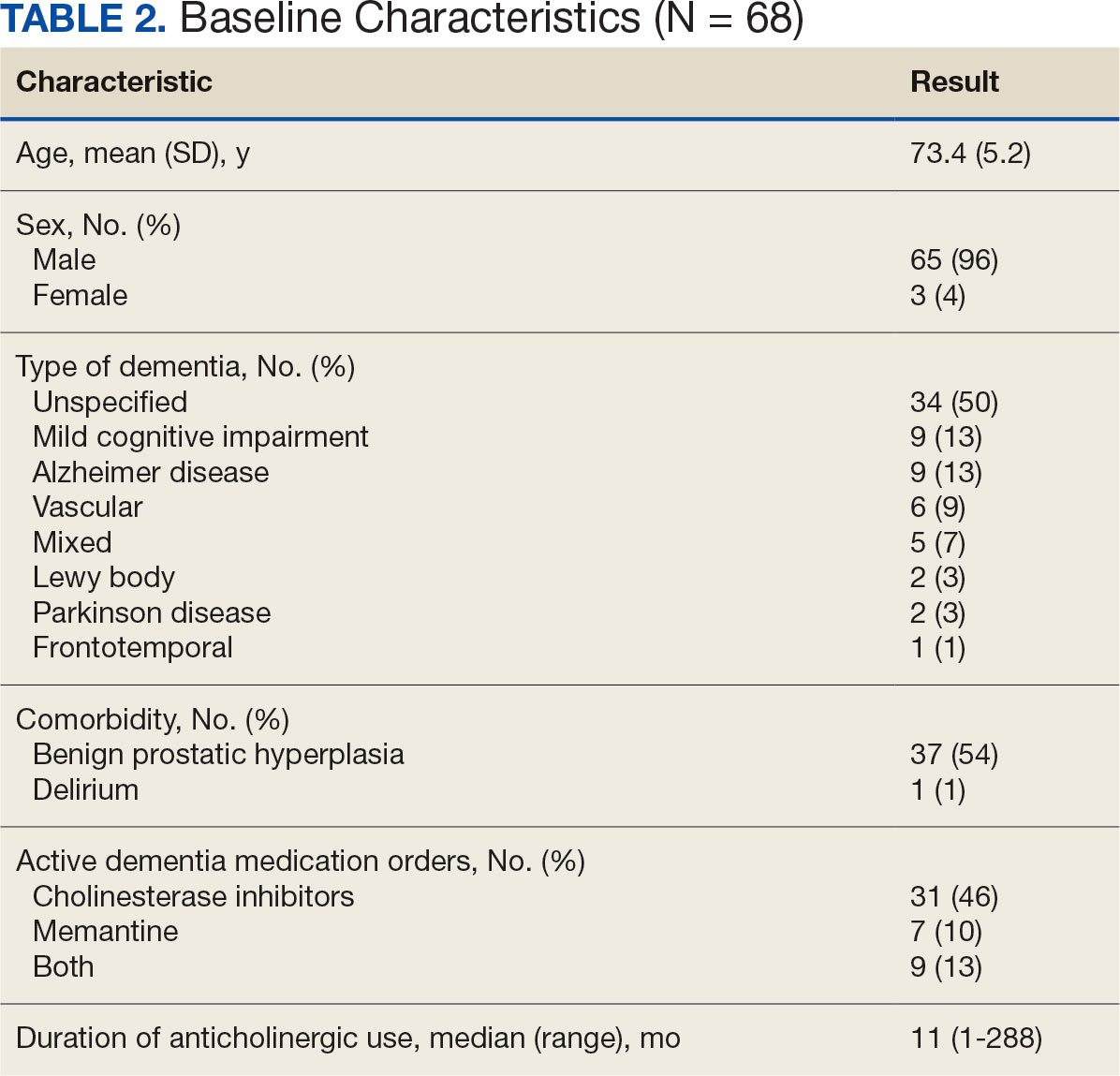
Twenty-nine patients (43%) had ≥ 1 medication with strong anticholinergic properties deprescribed. Anticholinergic medication was discontinued for 26 patients, and the dose was decreased for 3 patients. ACB score fell by a mean of 1.1 per patient. There was an increase in the documented risk-benefit assessment for anticholinergic medications from a baseline of 4 (6%) to 19 (28%) 3 months after the deprescribing note. Cyclobenzaprine, paroxetine, and oxybutynin were deprescribed the most, and amitriptyline had the lowest rate of deprescribing (Table 3). Thirty patients (44%) had a pharmacologic, nonanticholinergic alternative initiated per pharmacist recommendation, and 6 patients (9%) had a nonpharmacologic alternative initiated per pharmacist recommendation.
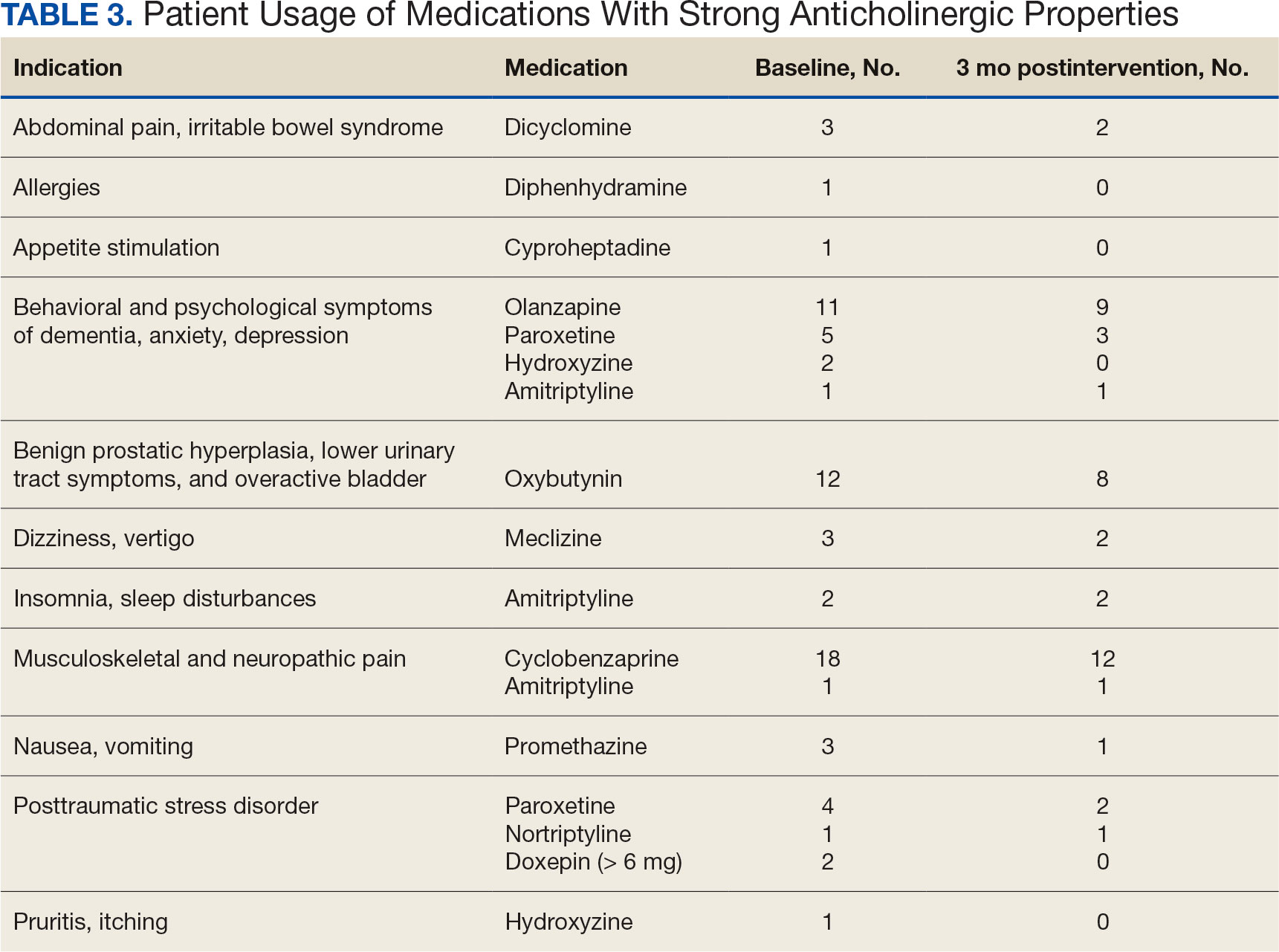
Discussion
This quality improvement project suggests that with the use of population health management tools such as the VIONE deprescribing dashboard, pharmacists can help identify and deprescribe strong anticholinergic medications in patients with cognitive impairment or dementia. Pharmacists can also aid in deprescribing through evidence-based recommendations to guide risk-benefit discussion and consider safer, nonanticholinergic alternatives. The authors were able to help reduce anticholinergic cognitive burden in 43% of patients in this sample. The mean 1.1 ACB score reduction was considered clinically significant based on prior studies that found that each 1-point increase in ACB score correlated with declined cognition and increased mortality.8,10 The VIONE deprescribing dashboard provided real-time patient data and helped target patients at the highest risk of anticholinergic AEs. The creation of the note templates based on the indication helped streamline recommendations. Typically, the prescriber addressed the recommendations at a routine follow-up appointment. The deprescribing method used in this project was time-efficient and could be easily replicated once the CPRS note templates were created. Future deprescribing projects could consider more direct pharmacist intervention and medication management.
Limitations
There was no direct assessment of clinical outcomes such as change in cognition using cognitive function tests. However, multiple studies have demonstrated AEs associated with strong anticholinergic medication use and additive anticholinergic burden in patients with dementia or cognitive impairment.1,5 Also, the 3-month follow-up period was relatively short. The pharmacist’s deprescribing recommendations may have been accepted after 3 months, or patients could have restarted their anticholinergic medications. Longer follow-up time could provide more robust results and conclusions. Thirdly, there was no formal definition of what constituted a risk-benefit assessment of anticholinergic medications. The risk-benefit assessment was determined at the discretion of the authors, which was subjective and allowed for bias. Finally, 6 patients died during the 3-month follow-up. The data for these patients were included in the baseline characteristics but not in the study outcomes. If these patients had been excluded from the results, a higher percentage of patients (47%) would have had ≥ 1 anticholinergic medication deprescribed.
Conclusions
In collaboration with the interdisciplinary team, pharmacist recommendations resulted in deprescribing of anticholinergic medications in veterans with dementia or cognitive impairment. The VIONE deprescribing dashboard, an easily accessible population health management tool, can identify patients prescribed potentially inappropriate medications and help target patients at the highest risk of anticholinergic AEs. To prevent worsening cognitive impairment, delirium, falls, and other AEs, this deprescribing initiative can be replicated at other VHA facilities. Future projects could have a longer follow-up period, incorporate more direct pharmacist intervention, and assess clinical outcomes of deprescribing.
- Gray SL, Hanlon JT. Anticholinergic medication use and dementia: latest evidence and clinical implications. Ther Adv Drug Saf. 2016;7(5):217-224. doi:10.1177/2042098616658399
- Kersten H, Wyller TB. Anticholinergic drug burden in older people’s brain - how well is it measured? Basic Clin Pharmacol Toxicol. 2014;114(2):151-159. doi:10.1111/bcpt.12140
- By the 2019 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2019 updated AGS beers criteria® for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2019;67(4):674-694. doi:10.1111/jgs.15767
- By the 2023 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2023 updated AGS Beers Criteria® for potentially inappropriate medication use in older adults J Am Geriatr Soc. 2023;71(7):2052-2081. doi:10.1111/jgs.18372
- Wang K, Alan J, Page AT, Dimopoulos E, Etherton-Beer C. Anticholinergics and clinical outcomes amongst people with pre-existing dementia: a systematic review. Maturitas. 2021;151:1-14. doi:10.1016/j.maturitas.2021.06.004
- Thorpe JM, Thorpe CT, Gellad WF, et al. Dual health care system use and high-risk prescribing in patients with dementia: a national cohort study. Ann Intern Med. 2017;166(3):157-163. doi:10.7326/M16-0551
- McCarren M, Burk M, Carico R, Glassman P, Good CB, Cunningham F. Design of a centrally aggregated medication use evaluation (CAMUE): anticholinergics in dementia. Presented at: 2019 HSR&D/QUERI National Conference; October 29-31, 2019; Washington, DC. https://www.hsrd.research.va.gov/meetings/2019/abstract-display.cfm?AbsNum=4027
- Boustani, M, Campbell, N, Munger S, et al. Impact of anticholinergics on the aging brain: a review and practical application. Aging Health. 2008;4(3):311-320. doi:10.2217/1745509.x
- Constantino-Corpuz JK, Alonso MTD. Assessment of a medication deprescribing tool on polypharmacy and cost avoidance. Fed Pract. 2021;38(7):332-336. doi:10.12788/fp.0146
- Fox C, Richardson K, Maidment ID, et al. Anticholinergic medication use and cognitive impairment in the older population: the medical research council cognitive function and ageing study. J Am Geriatr Soc. 2011;59(8):1477-1483. doi:10.1111/j.1532-5415.2011.03491.x
Anticholinergic medications block the activity of the neurotransmitter acetylcholine by binding to either muscarinic or nicotinic receptors in both the peripheral and central nervous system. Anticholinergic medications typically refer to antimuscarinic medications and have been prescribed to treat a variety of conditions common in older adults, including overactive bladder, allergies, muscle spasms, and sleep disorders.1,2 Since muscarinic receptors are present throughout the body, anticholinergic medications are associated with many adverse effects (AEs), including constipation, urinary retention, xerostomia, and delirium. Older adults are more sensitive to these AEs due to physiological changes associated with aging.1
The American Geriatric Society Beers Criteria for Potentially Inappropriate Medications Use in Older Adults identifies drugs with strong anticholinergic properties. The Beers Criteria strongly recommends avoiding these medications in patients with dementia or cognitive impairment due to the risk of central nervous system AEs. In the updated 2023 Beers Criteria, the rationale was expanded to recognize the risks of the cumulative anticholinergic burden associated with concurrent anticholinergic use.3,4
Given the prevalent use of anticholinergic medications in older adults, there has been significant research demonstrating their AEs, specifically delirium and cognitive impairment in geriatric patients. A systematic review of 14 articles conducted in 7 different countries of patients with median age of 76.4 to 86.1 years reviewed clinical outcomes of anticholinergic use in patients with dementia. Five studies found anticholinergics were associated with increased all-cause mortality in patients with dementia, and 3 studies found anticholinergics were associated with longer hospital stays. Other studies found that anticholinergics were associated with delirium and reduced health-related quality of life.5
About 35% of veterans with dementia have been prescribed a medication regimen with a high anticholinergic burden.6 In 2018, the US Department of Veterans Affairs (VA) Pharmacy Benfits Management Center for Medical Safety completed a centrally aggregated medication use evaluation (CAMUE) to assess the appropriateness of anticholinergic medication use in patients with dementia. The retrospective chart review included 1094 veterans from 19 sites. Overall, about 15% of the veterans experienced new falls, delirium, or worsening dementia within 30 days of starting an anticholinergic medication. Furthermore, < 40% had documentation of a nonanticholinergic alternative medication trial, and < 20% had documented nonpharmacologic therapy. The documentation of risk-benefit assessment acknowledging the risks of anticholinergic medication use in veterans with dementia occurred only about 13% of the time. The CAMUE concluded that the risks of initiating an anticholinergic medication in veterans with dementia are likely underdocumented and possibly under considered by prescribers.7
Developed within the Veterans Health Administration (VHA), VIONE (Vital, Important, Optional, Not Indicated, Every medication has an indication) is a medication management methodology that aims to reduce polypharmacy and improve patient safety consistent with high-reliability organizations. Since it launched in 2016, VIONE has gradually been implemented at many VHA facilities. The VIONE deprescribing dashboard had not been used at the VA Louisville Healthcare System prior to this quality improvement project.
This dashboard uses the Beers Criteria to identify potentially inappropriate anticholinergic medications. It uses the Anticholinergic Cognitive Burden (ACB) scale to calculate the cumulative anticholinergic risk for each patient. Medications with an ACB score of 2 or 3 have clinically relevant cognitive effects such as delirium and dementia (Table 1). For each point increase in total ACB score, a decline in mini-mental state examination score of 0.33 points over 2 years has been shown. Each point increase has also been correlated with a 26% increase in risk of death.8-10
Methods
The purpose of this quality improvement project was to determine the impact of pharmacist-driven deprescribing on the anticholinergic burden in veterans with dementia at VA Louisville Healthcare System. Data were obtained through the Computerized Patient Record System (CPRS) and VIONE deprescribing dashboard and entered in a secure Microsoft Excel spreadsheet. Pharmacist deprescribing steps were entered as CPRS progress notes. A deprescribing note template was created, and 11 templates with indication-specific recommendations were created for each anticholinergic indication identified (contact authors for deprescribing note template examples). Usage of anticholinergic medications was reexamined 3 months after the deprescribing note was entered.
Eligible patients identified in the VIONE deprescribing dashboard had an outpatient order for a medication with strong anticholinergic properties as identified using the Beers Criteria and were aged ≥ 65 years. Patients also had to be diagnosed with dementia or cognitive impairment. Patients were excluded if they were receiving hospice care or if the anticholinergic medication was from a non-VA prescriber or filled at a non-VA pharmacy. The VIONE deprescribing dashboard also excluded skeletal muscle relaxants if the patient had a spinal cord-related visit in the previous 2 years, first-generation antihistamines if the patient had a vertigo diagnosis, hydroxyzine if the indication was for anxiety, trospium if the indication was for overactive bladder, and antipsychotics if the patient had been diagnosed with schizophrenia or bipolar disorder. The following were included in the deprescribing recommendations if the dashboard identified the patient due to receiving a second strongly anticholinergic medication: first generation antihistamines if the patient was diagnosed with vertigo and hydroxyzine if the indication is for anxiety.
Each eligible patient received a focused medication review by a pharmacist via electronic chart review and a templated CPRS progress note with patient-specific recommendations. The prescriber and the patient’s primary care practitioner were recommended to perform a patient-specific risk-benefit assessment, deprescribe potentially inappropriate anticholinergic medications, and consider nonanticholinergic alternatives (both pharmacologic and nonpharmacologic). Data collected included baseline age, sex, prespecified comorbidities (type of dementia, cognitive impairment, delirium, benign prostatic hyperplasia/lower urinary tract symptoms), duration of prescribed anticholinergic medication, indication and deprescribing rate for each anticholinergic agent, and concurrent dementia medications (acetylcholinesterase inhibitors, memantine, or both).
The primary outcome was the number of patients that had = 1 medication with strong anticholinergic properties deprescribed. Deprescribing was defined as medication discontinuation or reduction of total daily dose. Secondary outcomes were the mean change in ACB scale, the number of patients with dose tapering, documented patient-specific risk-benefit assessment, and initiated nonanticholinergic alternative per pharmacist recommendation.
Results
The VIONE deprescribing dashboard identified 121 patients; 45 were excluded for non-VA prescriber or pharmacy, and 8 patients were excluded for other reasons. Sixty-eight patients were included in the deprescribing initiative. The mean age was 73.4 years (range, 67-93), 65 (96%) were male, and 34 (50%) had unspecified dementia (Table 2). Thirty-one patients (46%) had concurrent cholinesterase inhibitor prescriptions for dementia. The median duration of use of a strong anticholinergic medication was 11 months.
Twenty-nine patients (43%) had ≥ 1 medication with strong anticholinergic properties deprescribed. Anticholinergic medication was discontinued for 26 patients, and the dose was decreased for 3 patients. ACB score fell by a mean of 1.1 per patient. There was an increase in the documented risk-benefit assessment for anticholinergic medications from a baseline of 4 (6%) to 19 (28%) 3 months after the deprescribing note. Cyclobenzaprine, paroxetine, and oxybutynin were deprescribed the most, and amitriptyline had the lowest rate of deprescribing (Table 3). Thirty patients (44%) had a pharmacologic, nonanticholinergic alternative initiated per pharmacist recommendation, and 6 patients (9%) had a nonpharmacologic alternative initiated per pharmacist recommendation.
Discussion
This quality improvement project suggests that with the use of population health management tools such as the VIONE deprescribing dashboard, pharmacists can help identify and deprescribe strong anticholinergic medications in patients with cognitive impairment or dementia. Pharmacists can also aid in deprescribing through evidence-based recommendations to guide risk-benefit discussion and consider safer, nonanticholinergic alternatives. The authors were able to help reduce anticholinergic cognitive burden in 43% of patients in this sample. The mean 1.1 ACB score reduction was considered clinically significant based on prior studies that found that each 1-point increase in ACB score correlated with declined cognition and increased mortality.8,10 The VIONE deprescribing dashboard provided real-time patient data and helped target patients at the highest risk of anticholinergic AEs. The creation of the note templates based on the indication helped streamline recommendations. Typically, the prescriber addressed the recommendations at a routine follow-up appointment. The deprescribing method used in this project was time-efficient and could be easily replicated once the CPRS note templates were created. Future deprescribing projects could consider more direct pharmacist intervention and medication management.
Limitations
There was no direct assessment of clinical outcomes such as change in cognition using cognitive function tests. However, multiple studies have demonstrated AEs associated with strong anticholinergic medication use and additive anticholinergic burden in patients with dementia or cognitive impairment.1,5 Also, the 3-month follow-up period was relatively short. The pharmacist’s deprescribing recommendations may have been accepted after 3 months, or patients could have restarted their anticholinergic medications. Longer follow-up time could provide more robust results and conclusions. Thirdly, there was no formal definition of what constituted a risk-benefit assessment of anticholinergic medications. The risk-benefit assessment was determined at the discretion of the authors, which was subjective and allowed for bias. Finally, 6 patients died during the 3-month follow-up. The data for these patients were included in the baseline characteristics but not in the study outcomes. If these patients had been excluded from the results, a higher percentage of patients (47%) would have had ≥ 1 anticholinergic medication deprescribed.
Conclusions
In collaboration with the interdisciplinary team, pharmacist recommendations resulted in deprescribing of anticholinergic medications in veterans with dementia or cognitive impairment. The VIONE deprescribing dashboard, an easily accessible population health management tool, can identify patients prescribed potentially inappropriate medications and help target patients at the highest risk of anticholinergic AEs. To prevent worsening cognitive impairment, delirium, falls, and other AEs, this deprescribing initiative can be replicated at other VHA facilities. Future projects could have a longer follow-up period, incorporate more direct pharmacist intervention, and assess clinical outcomes of deprescribing.
Anticholinergic medications block the activity of the neurotransmitter acetylcholine by binding to either muscarinic or nicotinic receptors in both the peripheral and central nervous system. Anticholinergic medications typically refer to antimuscarinic medications and have been prescribed to treat a variety of conditions common in older adults, including overactive bladder, allergies, muscle spasms, and sleep disorders.1,2 Since muscarinic receptors are present throughout the body, anticholinergic medications are associated with many adverse effects (AEs), including constipation, urinary retention, xerostomia, and delirium. Older adults are more sensitive to these AEs due to physiological changes associated with aging.1
The American Geriatric Society Beers Criteria for Potentially Inappropriate Medications Use in Older Adults identifies drugs with strong anticholinergic properties. The Beers Criteria strongly recommends avoiding these medications in patients with dementia or cognitive impairment due to the risk of central nervous system AEs. In the updated 2023 Beers Criteria, the rationale was expanded to recognize the risks of the cumulative anticholinergic burden associated with concurrent anticholinergic use.3,4
Given the prevalent use of anticholinergic medications in older adults, there has been significant research demonstrating their AEs, specifically delirium and cognitive impairment in geriatric patients. A systematic review of 14 articles conducted in 7 different countries of patients with median age of 76.4 to 86.1 years reviewed clinical outcomes of anticholinergic use in patients with dementia. Five studies found anticholinergics were associated with increased all-cause mortality in patients with dementia, and 3 studies found anticholinergics were associated with longer hospital stays. Other studies found that anticholinergics were associated with delirium and reduced health-related quality of life.5
About 35% of veterans with dementia have been prescribed a medication regimen with a high anticholinergic burden.6 In 2018, the US Department of Veterans Affairs (VA) Pharmacy Benfits Management Center for Medical Safety completed a centrally aggregated medication use evaluation (CAMUE) to assess the appropriateness of anticholinergic medication use in patients with dementia. The retrospective chart review included 1094 veterans from 19 sites. Overall, about 15% of the veterans experienced new falls, delirium, or worsening dementia within 30 days of starting an anticholinergic medication. Furthermore, < 40% had documentation of a nonanticholinergic alternative medication trial, and < 20% had documented nonpharmacologic therapy. The documentation of risk-benefit assessment acknowledging the risks of anticholinergic medication use in veterans with dementia occurred only about 13% of the time. The CAMUE concluded that the risks of initiating an anticholinergic medication in veterans with dementia are likely underdocumented and possibly under considered by prescribers.7
Developed within the Veterans Health Administration (VHA), VIONE (Vital, Important, Optional, Not Indicated, Every medication has an indication) is a medication management methodology that aims to reduce polypharmacy and improve patient safety consistent with high-reliability organizations. Since it launched in 2016, VIONE has gradually been implemented at many VHA facilities. The VIONE deprescribing dashboard had not been used at the VA Louisville Healthcare System prior to this quality improvement project.
This dashboard uses the Beers Criteria to identify potentially inappropriate anticholinergic medications. It uses the Anticholinergic Cognitive Burden (ACB) scale to calculate the cumulative anticholinergic risk for each patient. Medications with an ACB score of 2 or 3 have clinically relevant cognitive effects such as delirium and dementia (Table 1). For each point increase in total ACB score, a decline in mini-mental state examination score of 0.33 points over 2 years has been shown. Each point increase has also been correlated with a 26% increase in risk of death.8-10
Methods
The purpose of this quality improvement project was to determine the impact of pharmacist-driven deprescribing on the anticholinergic burden in veterans with dementia at VA Louisville Healthcare System. Data were obtained through the Computerized Patient Record System (CPRS) and VIONE deprescribing dashboard and entered in a secure Microsoft Excel spreadsheet. Pharmacist deprescribing steps were entered as CPRS progress notes. A deprescribing note template was created, and 11 templates with indication-specific recommendations were created for each anticholinergic indication identified (contact authors for deprescribing note template examples). Usage of anticholinergic medications was reexamined 3 months after the deprescribing note was entered.
Eligible patients identified in the VIONE deprescribing dashboard had an outpatient order for a medication with strong anticholinergic properties as identified using the Beers Criteria and were aged ≥ 65 years. Patients also had to be diagnosed with dementia or cognitive impairment. Patients were excluded if they were receiving hospice care or if the anticholinergic medication was from a non-VA prescriber or filled at a non-VA pharmacy. The VIONE deprescribing dashboard also excluded skeletal muscle relaxants if the patient had a spinal cord-related visit in the previous 2 years, first-generation antihistamines if the patient had a vertigo diagnosis, hydroxyzine if the indication was for anxiety, trospium if the indication was for overactive bladder, and antipsychotics if the patient had been diagnosed with schizophrenia or bipolar disorder. The following were included in the deprescribing recommendations if the dashboard identified the patient due to receiving a second strongly anticholinergic medication: first generation antihistamines if the patient was diagnosed with vertigo and hydroxyzine if the indication is for anxiety.
Each eligible patient received a focused medication review by a pharmacist via electronic chart review and a templated CPRS progress note with patient-specific recommendations. The prescriber and the patient’s primary care practitioner were recommended to perform a patient-specific risk-benefit assessment, deprescribe potentially inappropriate anticholinergic medications, and consider nonanticholinergic alternatives (both pharmacologic and nonpharmacologic). Data collected included baseline age, sex, prespecified comorbidities (type of dementia, cognitive impairment, delirium, benign prostatic hyperplasia/lower urinary tract symptoms), duration of prescribed anticholinergic medication, indication and deprescribing rate for each anticholinergic agent, and concurrent dementia medications (acetylcholinesterase inhibitors, memantine, or both).
The primary outcome was the number of patients that had = 1 medication with strong anticholinergic properties deprescribed. Deprescribing was defined as medication discontinuation or reduction of total daily dose. Secondary outcomes were the mean change in ACB scale, the number of patients with dose tapering, documented patient-specific risk-benefit assessment, and initiated nonanticholinergic alternative per pharmacist recommendation.
Results
The VIONE deprescribing dashboard identified 121 patients; 45 were excluded for non-VA prescriber or pharmacy, and 8 patients were excluded for other reasons. Sixty-eight patients were included in the deprescribing initiative. The mean age was 73.4 years (range, 67-93), 65 (96%) were male, and 34 (50%) had unspecified dementia (Table 2). Thirty-one patients (46%) had concurrent cholinesterase inhibitor prescriptions for dementia. The median duration of use of a strong anticholinergic medication was 11 months.
Twenty-nine patients (43%) had ≥ 1 medication with strong anticholinergic properties deprescribed. Anticholinergic medication was discontinued for 26 patients, and the dose was decreased for 3 patients. ACB score fell by a mean of 1.1 per patient. There was an increase in the documented risk-benefit assessment for anticholinergic medications from a baseline of 4 (6%) to 19 (28%) 3 months after the deprescribing note. Cyclobenzaprine, paroxetine, and oxybutynin were deprescribed the most, and amitriptyline had the lowest rate of deprescribing (Table 3). Thirty patients (44%) had a pharmacologic, nonanticholinergic alternative initiated per pharmacist recommendation, and 6 patients (9%) had a nonpharmacologic alternative initiated per pharmacist recommendation.
Discussion
This quality improvement project suggests that with the use of population health management tools such as the VIONE deprescribing dashboard, pharmacists can help identify and deprescribe strong anticholinergic medications in patients with cognitive impairment or dementia. Pharmacists can also aid in deprescribing through evidence-based recommendations to guide risk-benefit discussion and consider safer, nonanticholinergic alternatives. The authors were able to help reduce anticholinergic cognitive burden in 43% of patients in this sample. The mean 1.1 ACB score reduction was considered clinically significant based on prior studies that found that each 1-point increase in ACB score correlated with declined cognition and increased mortality.8,10 The VIONE deprescribing dashboard provided real-time patient data and helped target patients at the highest risk of anticholinergic AEs. The creation of the note templates based on the indication helped streamline recommendations. Typically, the prescriber addressed the recommendations at a routine follow-up appointment. The deprescribing method used in this project was time-efficient and could be easily replicated once the CPRS note templates were created. Future deprescribing projects could consider more direct pharmacist intervention and medication management.
Limitations
There was no direct assessment of clinical outcomes such as change in cognition using cognitive function tests. However, multiple studies have demonstrated AEs associated with strong anticholinergic medication use and additive anticholinergic burden in patients with dementia or cognitive impairment.1,5 Also, the 3-month follow-up period was relatively short. The pharmacist’s deprescribing recommendations may have been accepted after 3 months, or patients could have restarted their anticholinergic medications. Longer follow-up time could provide more robust results and conclusions. Thirdly, there was no formal definition of what constituted a risk-benefit assessment of anticholinergic medications. The risk-benefit assessment was determined at the discretion of the authors, which was subjective and allowed for bias. Finally, 6 patients died during the 3-month follow-up. The data for these patients were included in the baseline characteristics but not in the study outcomes. If these patients had been excluded from the results, a higher percentage of patients (47%) would have had ≥ 1 anticholinergic medication deprescribed.
Conclusions
In collaboration with the interdisciplinary team, pharmacist recommendations resulted in deprescribing of anticholinergic medications in veterans with dementia or cognitive impairment. The VIONE deprescribing dashboard, an easily accessible population health management tool, can identify patients prescribed potentially inappropriate medications and help target patients at the highest risk of anticholinergic AEs. To prevent worsening cognitive impairment, delirium, falls, and other AEs, this deprescribing initiative can be replicated at other VHA facilities. Future projects could have a longer follow-up period, incorporate more direct pharmacist intervention, and assess clinical outcomes of deprescribing.
- Gray SL, Hanlon JT. Anticholinergic medication use and dementia: latest evidence and clinical implications. Ther Adv Drug Saf. 2016;7(5):217-224. doi:10.1177/2042098616658399
- Kersten H, Wyller TB. Anticholinergic drug burden in older people’s brain - how well is it measured? Basic Clin Pharmacol Toxicol. 2014;114(2):151-159. doi:10.1111/bcpt.12140
- By the 2019 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2019 updated AGS beers criteria® for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2019;67(4):674-694. doi:10.1111/jgs.15767
- By the 2023 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2023 updated AGS Beers Criteria® for potentially inappropriate medication use in older adults J Am Geriatr Soc. 2023;71(7):2052-2081. doi:10.1111/jgs.18372
- Wang K, Alan J, Page AT, Dimopoulos E, Etherton-Beer C. Anticholinergics and clinical outcomes amongst people with pre-existing dementia: a systematic review. Maturitas. 2021;151:1-14. doi:10.1016/j.maturitas.2021.06.004
- Thorpe JM, Thorpe CT, Gellad WF, et al. Dual health care system use and high-risk prescribing in patients with dementia: a national cohort study. Ann Intern Med. 2017;166(3):157-163. doi:10.7326/M16-0551
- McCarren M, Burk M, Carico R, Glassman P, Good CB, Cunningham F. Design of a centrally aggregated medication use evaluation (CAMUE): anticholinergics in dementia. Presented at: 2019 HSR&D/QUERI National Conference; October 29-31, 2019; Washington, DC. https://www.hsrd.research.va.gov/meetings/2019/abstract-display.cfm?AbsNum=4027
- Boustani, M, Campbell, N, Munger S, et al. Impact of anticholinergics on the aging brain: a review and practical application. Aging Health. 2008;4(3):311-320. doi:10.2217/1745509.x
- Constantino-Corpuz JK, Alonso MTD. Assessment of a medication deprescribing tool on polypharmacy and cost avoidance. Fed Pract. 2021;38(7):332-336. doi:10.12788/fp.0146
- Fox C, Richardson K, Maidment ID, et al. Anticholinergic medication use and cognitive impairment in the older population: the medical research council cognitive function and ageing study. J Am Geriatr Soc. 2011;59(8):1477-1483. doi:10.1111/j.1532-5415.2011.03491.x
- Gray SL, Hanlon JT. Anticholinergic medication use and dementia: latest evidence and clinical implications. Ther Adv Drug Saf. 2016;7(5):217-224. doi:10.1177/2042098616658399
- Kersten H, Wyller TB. Anticholinergic drug burden in older people’s brain - how well is it measured? Basic Clin Pharmacol Toxicol. 2014;114(2):151-159. doi:10.1111/bcpt.12140
- By the 2019 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2019 updated AGS beers criteria® for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2019;67(4):674-694. doi:10.1111/jgs.15767
- By the 2023 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2023 updated AGS Beers Criteria® for potentially inappropriate medication use in older adults J Am Geriatr Soc. 2023;71(7):2052-2081. doi:10.1111/jgs.18372
- Wang K, Alan J, Page AT, Dimopoulos E, Etherton-Beer C. Anticholinergics and clinical outcomes amongst people with pre-existing dementia: a systematic review. Maturitas. 2021;151:1-14. doi:10.1016/j.maturitas.2021.06.004
- Thorpe JM, Thorpe CT, Gellad WF, et al. Dual health care system use and high-risk prescribing in patients with dementia: a national cohort study. Ann Intern Med. 2017;166(3):157-163. doi:10.7326/M16-0551
- McCarren M, Burk M, Carico R, Glassman P, Good CB, Cunningham F. Design of a centrally aggregated medication use evaluation (CAMUE): anticholinergics in dementia. Presented at: 2019 HSR&D/QUERI National Conference; October 29-31, 2019; Washington, DC. https://www.hsrd.research.va.gov/meetings/2019/abstract-display.cfm?AbsNum=4027
- Boustani, M, Campbell, N, Munger S, et al. Impact of anticholinergics on the aging brain: a review and practical application. Aging Health. 2008;4(3):311-320. doi:10.2217/1745509.x
- Constantino-Corpuz JK, Alonso MTD. Assessment of a medication deprescribing tool on polypharmacy and cost avoidance. Fed Pract. 2021;38(7):332-336. doi:10.12788/fp.0146
- Fox C, Richardson K, Maidment ID, et al. Anticholinergic medication use and cognitive impairment in the older population: the medical research council cognitive function and ageing study. J Am Geriatr Soc. 2011;59(8):1477-1483. doi:10.1111/j.1532-5415.2011.03491.x
Pharmacist-Driven Deprescribing to Reduce Anticholinergic Burden in Veterans With Dementia
Pharmacist-Driven Deprescribing to Reduce Anticholinergic Burden in Veterans With Dementia
