User login
Enlarging Red Papulonodule on the Chest
The Diagnosis: Metastatic Renal Cell Carcinoma
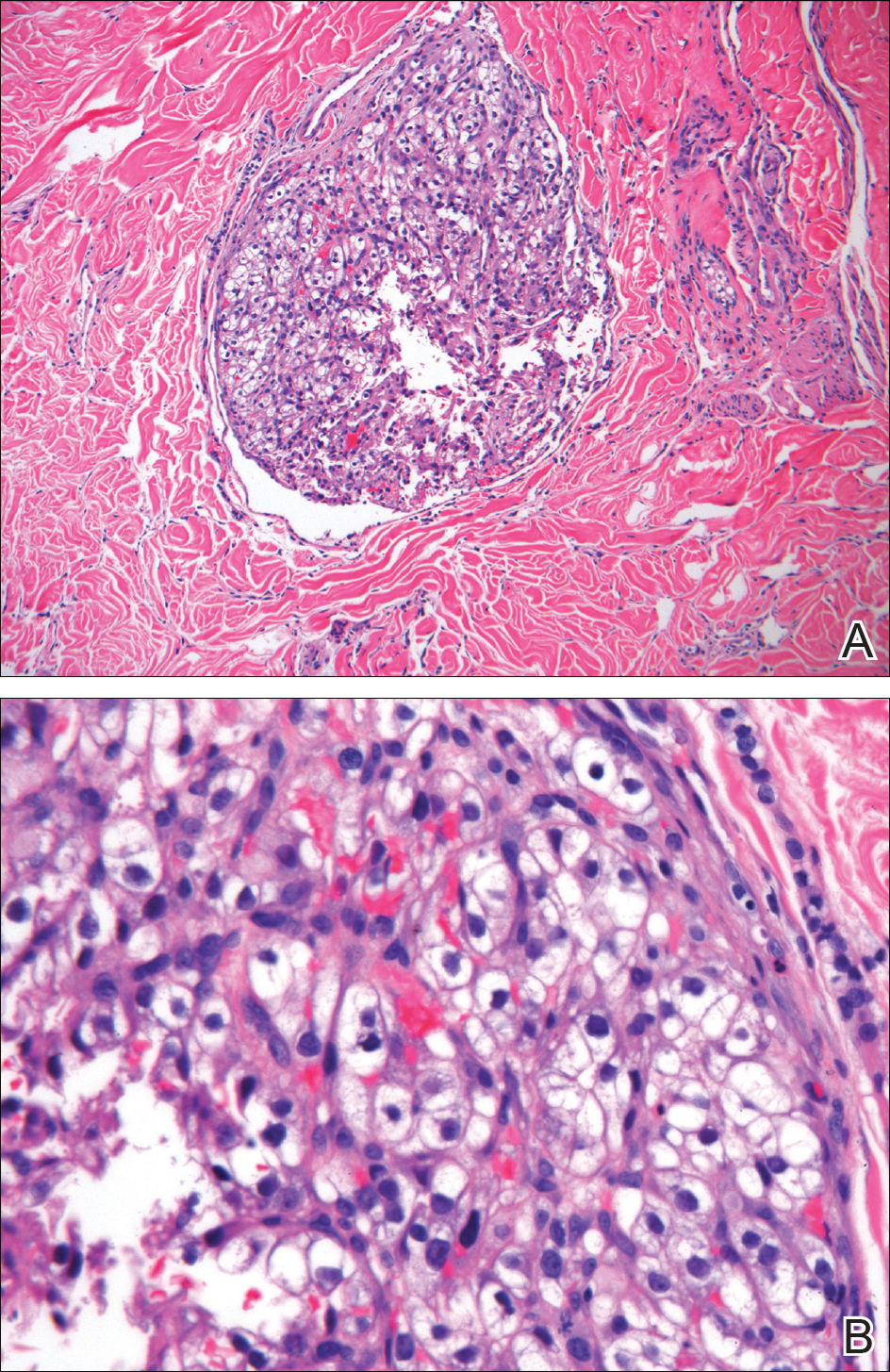
Histopathologic examination of the punch biopsy demonstrated epithelioid cells with abundant clear cytoplasm and numerous chicken wire-like vascular channels consistent with a diagnosis of cutaneous metastasis of renal cell carcinoma (RCC)(Figure). Collateral history revealed that 8 years prior, the patient had been diagnosed with clear cell RCC, stage III (T3aN0M0). At that time, he was treated with radical nephrectomy, which was considered curative. He remained disease free until several months prior to the development of the cutaneous lesion when he was found to have pulmonary and cerebral metastases with biopsies showing metastatic RCC. He was treated with lobectomy and Gamma Knife radiation for the lung and cerebral metastases, respectively. His oncologist planned to initiate therapy with the multikinase inhibitor sunitinib, which inhibits vascular endothelial growth factor (VEGF) signaling. Unfortunately, the patient died prior to treatment due to overwhelming tumor burden.
Clear cell RCC, the most common renal malignancy, presents with metastatic disease at the time of diagnosis in 21% of patients.1 An additional 20% of patients with localized disease develop metastases within several years of receiving a nephrectomy without adjuvant therapy, which is standard treatment for stage I to stage III disease.1,2 Metastatic RCC most frequently targets the lungs, bone, liver, and brain, though virtually any organ can be involved. Cutaneous involvement is estimated to occur in 3.3% of RCC cases,3 accounting for only 1.4% of cutaneous metastases overall.4 The risk for developing cutaneous metastases is greatest within 3 years following nephrectomy.3 However, our patient demonstrates that metastasis of RCC to skin can be long delayed (>5 years) despite an initial diagnosis of localized disease.
Cutaneous RCC classically presents as a painless firm papulonodule with a deep red or purple color due to its high vascularity.4 Several retrospective studies have identified the scalp as the most frequent site of cutaneous involvement, followed by the chest, abdomen, and nephrectomy scar.3,4 The differential diagnosis includes other vascular lesions such as pyogenic granuloma, hemangioma, angiosarcoma, bacillary angiomatosis, and Kaposi sarcoma. Diagnosis usually is easily confirmed histologically. Proliferative nests of epithelioid cells with clear cell morphology are surrounded by delicately branching vessels referred to as chicken wire-like vasculature. Immunohistochemical studies demonstrate positivity for pan-cytokeratin, vimentin, and CD-10, and negativity for p63 and cytokeratins 5 and 6, helping to confirm the diagnosis in more challenging cases, especially when there is no known history of primary RCC.5
If cutaneous metastasis of RCC is diagnosed, a chest and abdominal computed tomography scan as well as serum alkaline phosphatase test are warranted, as up to 90% of patients with RCC in the skin have additional lesions in at least 1 other site such as the lungs, bones, or liver.3 Management of metastatic RCC includes surgical excision if a single metastasis is found and either immunotherapy with high-dose IL-2 or an anti-programmed cell death inhibitor. Patients with progressive disease also may receive targeted anti-VEGF inhibitors (eg, axitinib, pazopanib, sunitinib), which have been shown to increase progression-free survival in metastatic RCC.6-8 Interestingly, some evidence suggests severely delayed recurrence of RCC (>5 years following nephrectomy) may predict better response to systemic therapy.9
This case of severely delayed metastasis of RCC 8 years after nephrectomy raises the question of whether routine surveillance for RCC recurrence should continue beyond 5 years. It also underscores the need for further studies to determine the utility of postsurgical adjuvant therapy for localized disease (stages I-III). A randomized clinical trial showed no significant difference in disease-free survival when the multikinase inhibitors sunitinib and sorafenib were used as adjuvant therapy.10 The randomized, placebo-controlled PROTECT trial showed no significant difference in disease-free survival between the VEGF inhibitor pazopanib and placebo when used as adjuvant therapy.11 However, trials are ongoing to investigate a potential survival advantage of adjuvant therapy with the VEGF receptor inhibitor axitinib and the mammalian target of rapamycin inhibitor everolimus.
- Dabestani S, Thorstenson A, Lindblad P, et al. Renal cell carcinoma recurrences and metastases in primary non-metastatic patients: a population-based study. World J Urol. 2016;34:1081-1086.
- Ljungberg B, Campbell SC, Choi HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615-621.
- Dorairajan LN, Hemal AK, Aron M, et al. Cutaneous metastases in renal cell carcinoma. Urol Int. 1999;63:164-167.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, pt 1):228-236.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061-1068.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584-3590.
- Rini BI, Grunwald V, Fishman MN, et al. Axitinib for first-line metastatic renal cell carcinoma (mRCC): overall efficacy and pharmacokinetic (PK) analyses from a randomized phase II study. J Clin Oncol. 2012;30(suppl). doi:10.1200/jco.2012.30.15_suppl.4503.
- Ficarra V, Novara G. Characterizing late recurrence of renal cell carcinoma. Nat Rev Urol. 2013;10:687-689.
- Haas NB, Manola J, Uzzo RG, et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial [published online March 9, 2016]. Lancet. 2016;387:2008-2016.
- Motzer RJ, Haas NB, Donskov F, et al; PROTECT investigators. Randomized phase III trial of adjuvant pazopanib versus placebo after nephrectomy in patients with localized or locally advanced renal cell carcinoma [published online September 13, 2017]. J Clin Oncol. 2017;35:3916-3923.
The Diagnosis: Metastatic Renal Cell Carcinoma
Histopathologic examination of the punch biopsy demonstrated epithelioid cells with abundant clear cytoplasm and numerous chicken wire-like vascular channels consistent with a diagnosis of cutaneous metastasis of renal cell carcinoma (RCC)(Figure). Collateral history revealed that 8 years prior, the patient had been diagnosed with clear cell RCC, stage III (T3aN0M0). At that time, he was treated with radical nephrectomy, which was considered curative. He remained disease free until several months prior to the development of the cutaneous lesion when he was found to have pulmonary and cerebral metastases with biopsies showing metastatic RCC. He was treated with lobectomy and Gamma Knife radiation for the lung and cerebral metastases, respectively. His oncologist planned to initiate therapy with the multikinase inhibitor sunitinib, which inhibits vascular endothelial growth factor (VEGF) signaling. Unfortunately, the patient died prior to treatment due to overwhelming tumor burden.
Clear cell RCC, the most common renal malignancy, presents with metastatic disease at the time of diagnosis in 21% of patients.1 An additional 20% of patients with localized disease develop metastases within several years of receiving a nephrectomy without adjuvant therapy, which is standard treatment for stage I to stage III disease.1,2 Metastatic RCC most frequently targets the lungs, bone, liver, and brain, though virtually any organ can be involved. Cutaneous involvement is estimated to occur in 3.3% of RCC cases,3 accounting for only 1.4% of cutaneous metastases overall.4 The risk for developing cutaneous metastases is greatest within 3 years following nephrectomy.3 However, our patient demonstrates that metastasis of RCC to skin can be long delayed (>5 years) despite an initial diagnosis of localized disease.
Cutaneous RCC classically presents as a painless firm papulonodule with a deep red or purple color due to its high vascularity.4 Several retrospective studies have identified the scalp as the most frequent site of cutaneous involvement, followed by the chest, abdomen, and nephrectomy scar.3,4 The differential diagnosis includes other vascular lesions such as pyogenic granuloma, hemangioma, angiosarcoma, bacillary angiomatosis, and Kaposi sarcoma. Diagnosis usually is easily confirmed histologically. Proliferative nests of epithelioid cells with clear cell morphology are surrounded by delicately branching vessels referred to as chicken wire-like vasculature. Immunohistochemical studies demonstrate positivity for pan-cytokeratin, vimentin, and CD-10, and negativity for p63 and cytokeratins 5 and 6, helping to confirm the diagnosis in more challenging cases, especially when there is no known history of primary RCC.5
If cutaneous metastasis of RCC is diagnosed, a chest and abdominal computed tomography scan as well as serum alkaline phosphatase test are warranted, as up to 90% of patients with RCC in the skin have additional lesions in at least 1 other site such as the lungs, bones, or liver.3 Management of metastatic RCC includes surgical excision if a single metastasis is found and either immunotherapy with high-dose IL-2 or an anti-programmed cell death inhibitor. Patients with progressive disease also may receive targeted anti-VEGF inhibitors (eg, axitinib, pazopanib, sunitinib), which have been shown to increase progression-free survival in metastatic RCC.6-8 Interestingly, some evidence suggests severely delayed recurrence of RCC (>5 years following nephrectomy) may predict better response to systemic therapy.9
This case of severely delayed metastasis of RCC 8 years after nephrectomy raises the question of whether routine surveillance for RCC recurrence should continue beyond 5 years. It also underscores the need for further studies to determine the utility of postsurgical adjuvant therapy for localized disease (stages I-III). A randomized clinical trial showed no significant difference in disease-free survival when the multikinase inhibitors sunitinib and sorafenib were used as adjuvant therapy.10 The randomized, placebo-controlled PROTECT trial showed no significant difference in disease-free survival between the VEGF inhibitor pazopanib and placebo when used as adjuvant therapy.11 However, trials are ongoing to investigate a potential survival advantage of adjuvant therapy with the VEGF receptor inhibitor axitinib and the mammalian target of rapamycin inhibitor everolimus.
The Diagnosis: Metastatic Renal Cell Carcinoma
Histopathologic examination of the punch biopsy demonstrated epithelioid cells with abundant clear cytoplasm and numerous chicken wire-like vascular channels consistent with a diagnosis of cutaneous metastasis of renal cell carcinoma (RCC)(Figure). Collateral history revealed that 8 years prior, the patient had been diagnosed with clear cell RCC, stage III (T3aN0M0). At that time, he was treated with radical nephrectomy, which was considered curative. He remained disease free until several months prior to the development of the cutaneous lesion when he was found to have pulmonary and cerebral metastases with biopsies showing metastatic RCC. He was treated with lobectomy and Gamma Knife radiation for the lung and cerebral metastases, respectively. His oncologist planned to initiate therapy with the multikinase inhibitor sunitinib, which inhibits vascular endothelial growth factor (VEGF) signaling. Unfortunately, the patient died prior to treatment due to overwhelming tumor burden.
Clear cell RCC, the most common renal malignancy, presents with metastatic disease at the time of diagnosis in 21% of patients.1 An additional 20% of patients with localized disease develop metastases within several years of receiving a nephrectomy without adjuvant therapy, which is standard treatment for stage I to stage III disease.1,2 Metastatic RCC most frequently targets the lungs, bone, liver, and brain, though virtually any organ can be involved. Cutaneous involvement is estimated to occur in 3.3% of RCC cases,3 accounting for only 1.4% of cutaneous metastases overall.4 The risk for developing cutaneous metastases is greatest within 3 years following nephrectomy.3 However, our patient demonstrates that metastasis of RCC to skin can be long delayed (>5 years) despite an initial diagnosis of localized disease.
Cutaneous RCC classically presents as a painless firm papulonodule with a deep red or purple color due to its high vascularity.4 Several retrospective studies have identified the scalp as the most frequent site of cutaneous involvement, followed by the chest, abdomen, and nephrectomy scar.3,4 The differential diagnosis includes other vascular lesions such as pyogenic granuloma, hemangioma, angiosarcoma, bacillary angiomatosis, and Kaposi sarcoma. Diagnosis usually is easily confirmed histologically. Proliferative nests of epithelioid cells with clear cell morphology are surrounded by delicately branching vessels referred to as chicken wire-like vasculature. Immunohistochemical studies demonstrate positivity for pan-cytokeratin, vimentin, and CD-10, and negativity for p63 and cytokeratins 5 and 6, helping to confirm the diagnosis in more challenging cases, especially when there is no known history of primary RCC.5
If cutaneous metastasis of RCC is diagnosed, a chest and abdominal computed tomography scan as well as serum alkaline phosphatase test are warranted, as up to 90% of patients with RCC in the skin have additional lesions in at least 1 other site such as the lungs, bones, or liver.3 Management of metastatic RCC includes surgical excision if a single metastasis is found and either immunotherapy with high-dose IL-2 or an anti-programmed cell death inhibitor. Patients with progressive disease also may receive targeted anti-VEGF inhibitors (eg, axitinib, pazopanib, sunitinib), which have been shown to increase progression-free survival in metastatic RCC.6-8 Interestingly, some evidence suggests severely delayed recurrence of RCC (>5 years following nephrectomy) may predict better response to systemic therapy.9
This case of severely delayed metastasis of RCC 8 years after nephrectomy raises the question of whether routine surveillance for RCC recurrence should continue beyond 5 years. It also underscores the need for further studies to determine the utility of postsurgical adjuvant therapy for localized disease (stages I-III). A randomized clinical trial showed no significant difference in disease-free survival when the multikinase inhibitors sunitinib and sorafenib were used as adjuvant therapy.10 The randomized, placebo-controlled PROTECT trial showed no significant difference in disease-free survival between the VEGF inhibitor pazopanib and placebo when used as adjuvant therapy.11 However, trials are ongoing to investigate a potential survival advantage of adjuvant therapy with the VEGF receptor inhibitor axitinib and the mammalian target of rapamycin inhibitor everolimus.
- Dabestani S, Thorstenson A, Lindblad P, et al. Renal cell carcinoma recurrences and metastases in primary non-metastatic patients: a population-based study. World J Urol. 2016;34:1081-1086.
- Ljungberg B, Campbell SC, Choi HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615-621.
- Dorairajan LN, Hemal AK, Aron M, et al. Cutaneous metastases in renal cell carcinoma. Urol Int. 1999;63:164-167.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, pt 1):228-236.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061-1068.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584-3590.
- Rini BI, Grunwald V, Fishman MN, et al. Axitinib for first-line metastatic renal cell carcinoma (mRCC): overall efficacy and pharmacokinetic (PK) analyses from a randomized phase II study. J Clin Oncol. 2012;30(suppl). doi:10.1200/jco.2012.30.15_suppl.4503.
- Ficarra V, Novara G. Characterizing late recurrence of renal cell carcinoma. Nat Rev Urol. 2013;10:687-689.
- Haas NB, Manola J, Uzzo RG, et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial [published online March 9, 2016]. Lancet. 2016;387:2008-2016.
- Motzer RJ, Haas NB, Donskov F, et al; PROTECT investigators. Randomized phase III trial of adjuvant pazopanib versus placebo after nephrectomy in patients with localized or locally advanced renal cell carcinoma [published online September 13, 2017]. J Clin Oncol. 2017;35:3916-3923.
- Dabestani S, Thorstenson A, Lindblad P, et al. Renal cell carcinoma recurrences and metastases in primary non-metastatic patients: a population-based study. World J Urol. 2016;34:1081-1086.
- Ljungberg B, Campbell SC, Choi HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615-621.
- Dorairajan LN, Hemal AK, Aron M, et al. Cutaneous metastases in renal cell carcinoma. Urol Int. 1999;63:164-167.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, pt 1):228-236.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061-1068.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584-3590.
- Rini BI, Grunwald V, Fishman MN, et al. Axitinib for first-line metastatic renal cell carcinoma (mRCC): overall efficacy and pharmacokinetic (PK) analyses from a randomized phase II study. J Clin Oncol. 2012;30(suppl). doi:10.1200/jco.2012.30.15_suppl.4503.
- Ficarra V, Novara G. Characterizing late recurrence of renal cell carcinoma. Nat Rev Urol. 2013;10:687-689.
- Haas NB, Manola J, Uzzo RG, et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial [published online March 9, 2016]. Lancet. 2016;387:2008-2016.
- Motzer RJ, Haas NB, Donskov F, et al; PROTECT investigators. Randomized phase III trial of adjuvant pazopanib versus placebo after nephrectomy in patients with localized or locally advanced renal cell carcinoma [published online September 13, 2017]. J Clin Oncol. 2017;35:3916-3923.
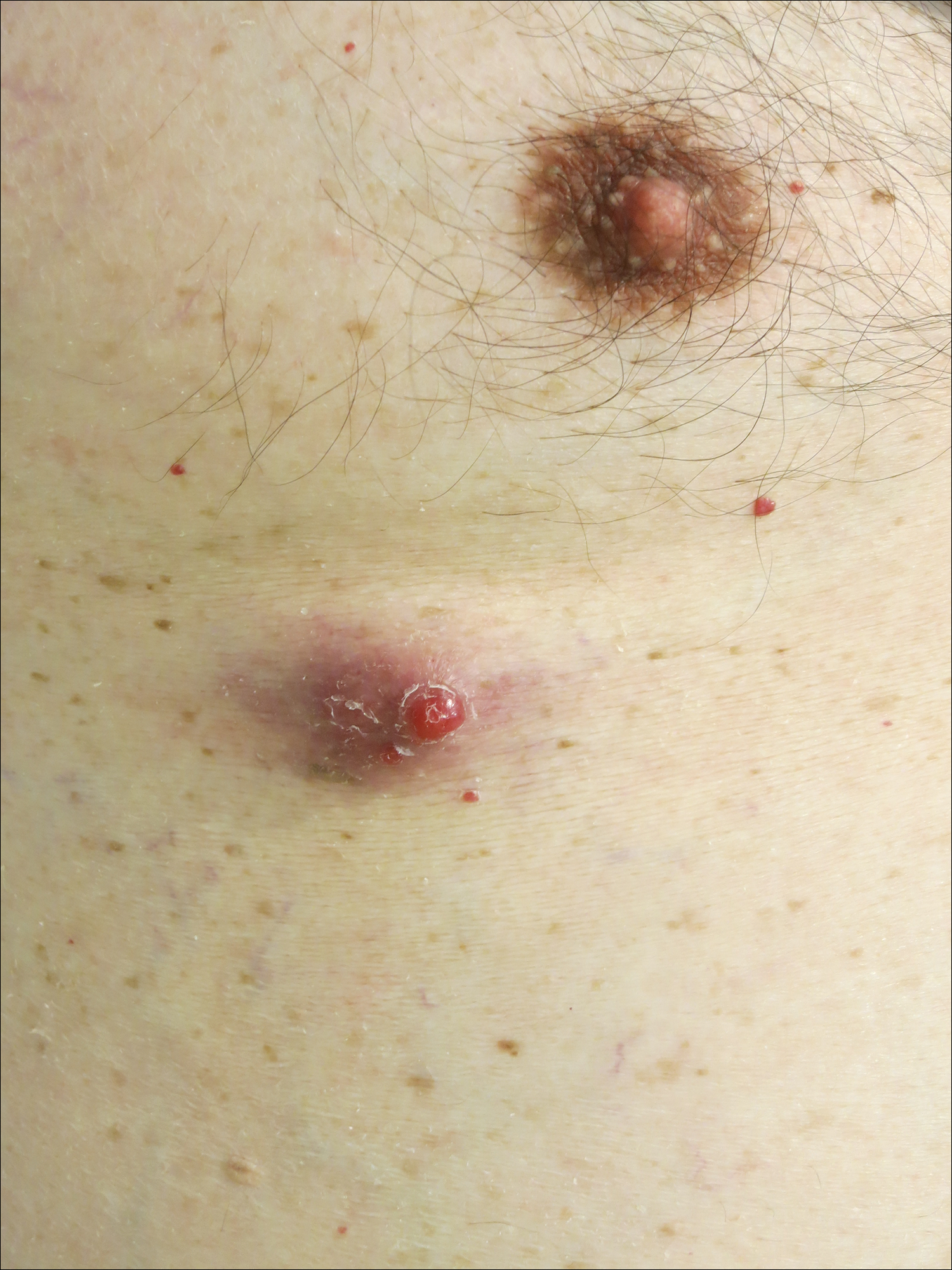
A man in his 60s presented with a subcutaneous nodule on the right side of the chest. Due to impaired mental status, he was unable to describe the precise age of the lesion, but his wife reported it had been present at least several weeks. She recently noted a new, bright red growth on top of the nodule. The lesion was asymptomatic but seemed to be growing in size. Physical examination revealed a 3-cm firm fixed nodule on the right side of the chest with an overlying, exophytic bright red papule. No similar lesions were found elsewhere on physical examination. A punch biopsy of the lesion was performed.
Mobile Medical Apps for Patient Education: A Graded Review of Available Dermatology Apps
According to industry estimates, roughly 64% of US adults were smartphone users in 2015.1 Smartphones enable users to utilize mobile applications (apps) that can perform a variety of functions in many categories, including business, music, photography, entertainment, education, social networking, travel, and lifestyle. The widespread adoption and use of mobile apps has implications for medical practice. Mobile apps have the capability to serve as information sources for patients, educational tools for students, and diagnostic aids for physicians.2 Consequently, a number of medical and health care–oriented apps have already been developed3 and are increasingly utilized by patients and providers.4
Given its visual nature, dermatology is particularly amenable to the integration of mobile medical apps. A study by Brewer et al5 identified more than 229 dermatology-related apps in categories ranging from general dermatology reference, self-surveillance and diagnosis, disease guides, educational aids, sunscreen and UV recommendations, and teledermatology. Patients served as the target audience and principal consumers of more than half of these dermatology apps.5
Mobile medical and health care apps demonstrate great potential for serving as valuable information sources for patients with dermatologic conditions; however, the content, functions, accuracy, and educational value of dermatology mobile apps are not well characterized, making it difficult for patients and health care providers to select and recommend appropriate apps.6 In this study, we created a rubric to objectively grade 44 publicly available mobile dermatology apps with the primary focus of patient education.
Methods
We conducted a search of dermatology-related educational mobile apps that were publicly available via the App Store (Apple Inc) from January 2016 to November 2016. (The pricing, availability, and other features of these apps may have changed since the study period.) The following search terms were used: dermatology, dermoscopy, melanoma, skin cancer, psoriasis, rosacea, acne, eczema, dermal fillers, and Mohs surgery. We excluded apps that were not in English; had a solely commercial focus; were mobile textbooks or scientific journals; were used to provide teledermatology services with no educational purpose; were solely focused on homeopathic, alternative, and/or complementary medicine; or were intended primarily as a reference for students or health care professionals. Our search yielded 44 apps with patient education as a primary objective. The apps were divided into 6 categories based on their focus: general dermatology, cosmetic dermatology, acne, eczema, psoriasis, and skin cancer.
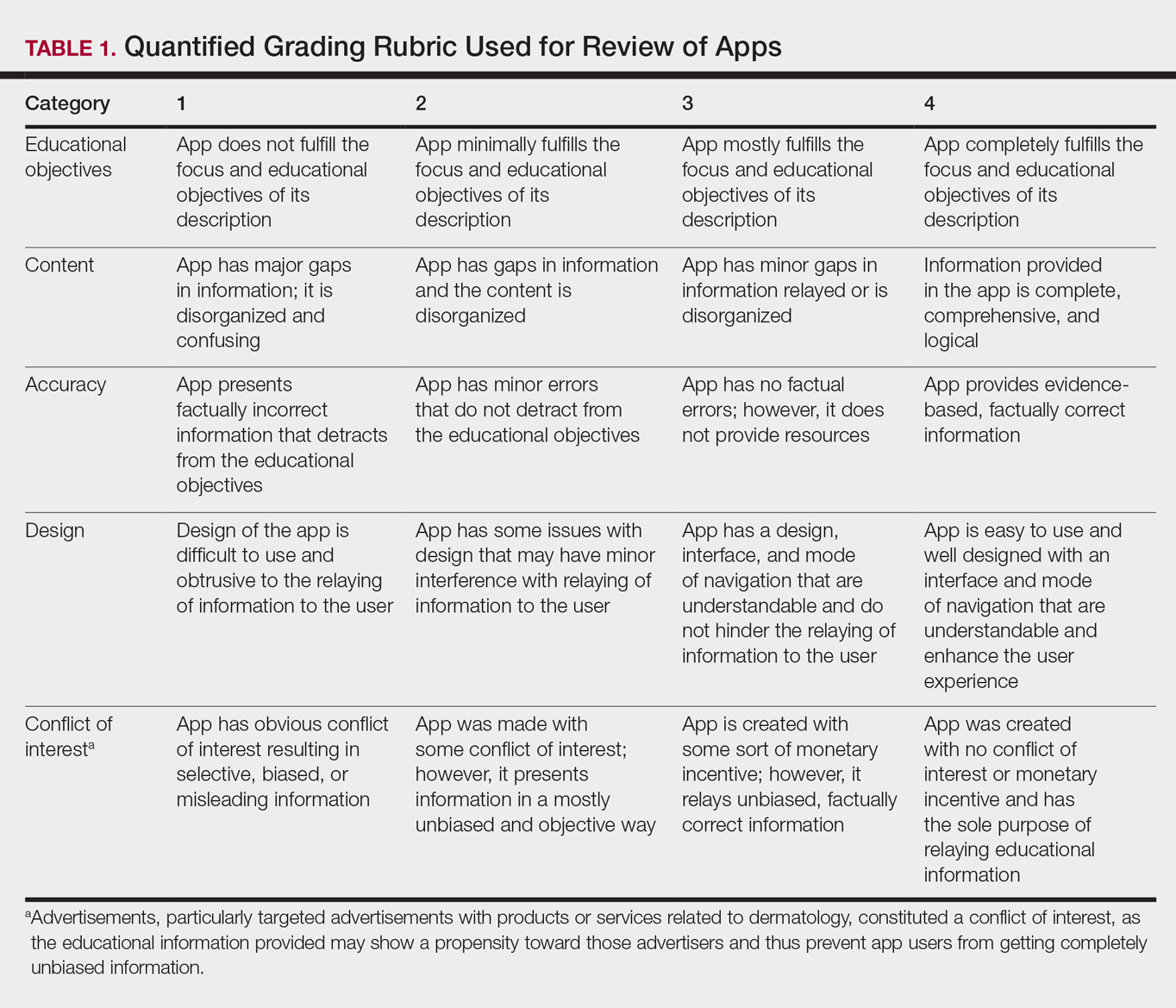
Each app was reviewed using a quantified grading rubric developed by the researchers. In a prior evaluation, Handel7 reviewed 35 health and wellness mobile apps utilizing the categories of ease of use, reliability, quality, scope of information, and aesthetics.4 These criteria were modified and adapted for the purposes of this study, and a 4-point scale was applied to each criterion. The final criteria were (1) educational objectives, (2) content, (3) accuracy, (4) design, and (5) conflict of interest. The quantified grading rubric is described in Table 1.
Results
The possible range of scores based on the grading rubric was 5 to 20. The actual range of scores was 8 to 19 (Table 2). The 44 reviewed apps were categorized by topic as acne, cosmetic dermatology, eczema, general dermatology, psoriasis, or skin cancer. A sample of 15 apps selected to represent the distribution of scores and their grading on the rubric are presented in Table 3.
Comment
The number of dermatology-related apps available to mobile users continues to grow at an increasing rate.8 The apps vary in many aspects, including their purpose, scope, intended audience, and goals of the app publisher. In turn, more individuals are turning to mobile apps for medical information,4 especially in dermatology, thus it is necessary to create a systematic way to evaluate the quality and utility of each app to assist users in making informed decisions about which apps will best meet their needs in the midst of a wide array of choices.
For the purpose of this study, an objective rubric was created that can be used to evaluate the quality of medical apps for patient education in dermatology. An app’s adequacy and usefulness for patient education was thought to depend on 3 possible score ranges into which the app could fall based on the grading rubric. An app with a total score in the range of 5 to 10 was not thought to be useful and may even be detrimental to patients. An app with a total score in the range of 11 to 15 may be used for patient education with some reservations based on shortcomings for certain criteria. An app with a score in the range of 16 to 20 was thought to be valuable and adequate for patient education. For example, the How to Treat Acne app received a total score of 8 and therefore would not be recommended to patients based on the grading rubric used in this study. This particular app provided sparse and sometimes inaccurate information, had a confusing user interface, and contained many obstructive advertisements. In contrast, the Eczema Doc app received a total score of 19, which indicates a quality app deemed to be useful for patient information based on the established rubric. This app met all the objectives that it advertised, contained accurate information with verified citation of sources, and was very easy for users to navigate.
Of the 44 graded apps, only 9 (20.5%) received scores in the highest range of 16 to 20, which indicates a need for improvements in mobile dermatology apps intended for patient education. Adopting the grading rubric developed in this study as a standard in the creation of medical apps could have beneficial implications in disseminating accurate, safe, unbiased, and easy-to-understand information to patients.
- Smith A. U.S. smartphone use in 2015. Pew Research Center website. http://www.pewinternet.org/2015/04/01/us-smartphone-use-in-2015. Published April 1, 2015. Accessed August 29, 2017.
- Nilsen W, Kumar S, Shar A, et al. Advancing the science of mHealth. J Health Commun. 2012;17(suppl 1):5-10.
- West DM. How mobile devices are transforming healthcare issues in technology innovation. Issues Technol Innov. 2012;18:1-14.
- Boudreaux ED, Waring ME, Hayes RB, et al. Evaluating and selecting mobile health apps: strategies for healthcare providers and healthcare organizations. Transl Behav Med. 2014;4:363-371.
- Brewer AC, Endly DC, Henley J, et al. Mobile applications in dermatology. JAMA Dermatol. 2013;149:1300-1304.
- Cummings E, Borycki E, Roehrer E. Issues and considerations for healthcare consumers using mobile applications. Stud Health Technol Inform. 2013;183:227-231.
- Handel MJ. mHealth (mobile health)-using apps for health and wellness. Explore. 2011;7:256-261.
- Boulos MN, Brewer AC, Karimkhani C, et al. Mobile medical and health apps: state of the art, concerns, regulatory control and certification. Online J Public Health Inform. 2014;5:229.
According to industry estimates, roughly 64% of US adults were smartphone users in 2015.1 Smartphones enable users to utilize mobile applications (apps) that can perform a variety of functions in many categories, including business, music, photography, entertainment, education, social networking, travel, and lifestyle. The widespread adoption and use of mobile apps has implications for medical practice. Mobile apps have the capability to serve as information sources for patients, educational tools for students, and diagnostic aids for physicians.2 Consequently, a number of medical and health care–oriented apps have already been developed3 and are increasingly utilized by patients and providers.4
Given its visual nature, dermatology is particularly amenable to the integration of mobile medical apps. A study by Brewer et al5 identified more than 229 dermatology-related apps in categories ranging from general dermatology reference, self-surveillance and diagnosis, disease guides, educational aids, sunscreen and UV recommendations, and teledermatology. Patients served as the target audience and principal consumers of more than half of these dermatology apps.5
Mobile medical and health care apps demonstrate great potential for serving as valuable information sources for patients with dermatologic conditions; however, the content, functions, accuracy, and educational value of dermatology mobile apps are not well characterized, making it difficult for patients and health care providers to select and recommend appropriate apps.6 In this study, we created a rubric to objectively grade 44 publicly available mobile dermatology apps with the primary focus of patient education.
Methods
We conducted a search of dermatology-related educational mobile apps that were publicly available via the App Store (Apple Inc) from January 2016 to November 2016. (The pricing, availability, and other features of these apps may have changed since the study period.) The following search terms were used: dermatology, dermoscopy, melanoma, skin cancer, psoriasis, rosacea, acne, eczema, dermal fillers, and Mohs surgery. We excluded apps that were not in English; had a solely commercial focus; were mobile textbooks or scientific journals; were used to provide teledermatology services with no educational purpose; were solely focused on homeopathic, alternative, and/or complementary medicine; or were intended primarily as a reference for students or health care professionals. Our search yielded 44 apps with patient education as a primary objective. The apps were divided into 6 categories based on their focus: general dermatology, cosmetic dermatology, acne, eczema, psoriasis, and skin cancer.
Each app was reviewed using a quantified grading rubric developed by the researchers. In a prior evaluation, Handel7 reviewed 35 health and wellness mobile apps utilizing the categories of ease of use, reliability, quality, scope of information, and aesthetics.4 These criteria were modified and adapted for the purposes of this study, and a 4-point scale was applied to each criterion. The final criteria were (1) educational objectives, (2) content, (3) accuracy, (4) design, and (5) conflict of interest. The quantified grading rubric is described in Table 1.
Results
The possible range of scores based on the grading rubric was 5 to 20. The actual range of scores was 8 to 19 (Table 2). The 44 reviewed apps were categorized by topic as acne, cosmetic dermatology, eczema, general dermatology, psoriasis, or skin cancer. A sample of 15 apps selected to represent the distribution of scores and their grading on the rubric are presented in Table 3.
Comment
The number of dermatology-related apps available to mobile users continues to grow at an increasing rate.8 The apps vary in many aspects, including their purpose, scope, intended audience, and goals of the app publisher. In turn, more individuals are turning to mobile apps for medical information,4 especially in dermatology, thus it is necessary to create a systematic way to evaluate the quality and utility of each app to assist users in making informed decisions about which apps will best meet their needs in the midst of a wide array of choices.
For the purpose of this study, an objective rubric was created that can be used to evaluate the quality of medical apps for patient education in dermatology. An app’s adequacy and usefulness for patient education was thought to depend on 3 possible score ranges into which the app could fall based on the grading rubric. An app with a total score in the range of 5 to 10 was not thought to be useful and may even be detrimental to patients. An app with a total score in the range of 11 to 15 may be used for patient education with some reservations based on shortcomings for certain criteria. An app with a score in the range of 16 to 20 was thought to be valuable and adequate for patient education. For example, the How to Treat Acne app received a total score of 8 and therefore would not be recommended to patients based on the grading rubric used in this study. This particular app provided sparse and sometimes inaccurate information, had a confusing user interface, and contained many obstructive advertisements. In contrast, the Eczema Doc app received a total score of 19, which indicates a quality app deemed to be useful for patient information based on the established rubric. This app met all the objectives that it advertised, contained accurate information with verified citation of sources, and was very easy for users to navigate.
Of the 44 graded apps, only 9 (20.5%) received scores in the highest range of 16 to 20, which indicates a need for improvements in mobile dermatology apps intended for patient education. Adopting the grading rubric developed in this study as a standard in the creation of medical apps could have beneficial implications in disseminating accurate, safe, unbiased, and easy-to-understand information to patients.
According to industry estimates, roughly 64% of US adults were smartphone users in 2015.1 Smartphones enable users to utilize mobile applications (apps) that can perform a variety of functions in many categories, including business, music, photography, entertainment, education, social networking, travel, and lifestyle. The widespread adoption and use of mobile apps has implications for medical practice. Mobile apps have the capability to serve as information sources for patients, educational tools for students, and diagnostic aids for physicians.2 Consequently, a number of medical and health care–oriented apps have already been developed3 and are increasingly utilized by patients and providers.4
Given its visual nature, dermatology is particularly amenable to the integration of mobile medical apps. A study by Brewer et al5 identified more than 229 dermatology-related apps in categories ranging from general dermatology reference, self-surveillance and diagnosis, disease guides, educational aids, sunscreen and UV recommendations, and teledermatology. Patients served as the target audience and principal consumers of more than half of these dermatology apps.5
Mobile medical and health care apps demonstrate great potential for serving as valuable information sources for patients with dermatologic conditions; however, the content, functions, accuracy, and educational value of dermatology mobile apps are not well characterized, making it difficult for patients and health care providers to select and recommend appropriate apps.6 In this study, we created a rubric to objectively grade 44 publicly available mobile dermatology apps with the primary focus of patient education.
Methods
We conducted a search of dermatology-related educational mobile apps that were publicly available via the App Store (Apple Inc) from January 2016 to November 2016. (The pricing, availability, and other features of these apps may have changed since the study period.) The following search terms were used: dermatology, dermoscopy, melanoma, skin cancer, psoriasis, rosacea, acne, eczema, dermal fillers, and Mohs surgery. We excluded apps that were not in English; had a solely commercial focus; were mobile textbooks or scientific journals; were used to provide teledermatology services with no educational purpose; were solely focused on homeopathic, alternative, and/or complementary medicine; or were intended primarily as a reference for students or health care professionals. Our search yielded 44 apps with patient education as a primary objective. The apps were divided into 6 categories based on their focus: general dermatology, cosmetic dermatology, acne, eczema, psoriasis, and skin cancer.
Each app was reviewed using a quantified grading rubric developed by the researchers. In a prior evaluation, Handel7 reviewed 35 health and wellness mobile apps utilizing the categories of ease of use, reliability, quality, scope of information, and aesthetics.4 These criteria were modified and adapted for the purposes of this study, and a 4-point scale was applied to each criterion. The final criteria were (1) educational objectives, (2) content, (3) accuracy, (4) design, and (5) conflict of interest. The quantified grading rubric is described in Table 1.
Results
The possible range of scores based on the grading rubric was 5 to 20. The actual range of scores was 8 to 19 (Table 2). The 44 reviewed apps were categorized by topic as acne, cosmetic dermatology, eczema, general dermatology, psoriasis, or skin cancer. A sample of 15 apps selected to represent the distribution of scores and their grading on the rubric are presented in Table 3.
Comment
The number of dermatology-related apps available to mobile users continues to grow at an increasing rate.8 The apps vary in many aspects, including their purpose, scope, intended audience, and goals of the app publisher. In turn, more individuals are turning to mobile apps for medical information,4 especially in dermatology, thus it is necessary to create a systematic way to evaluate the quality and utility of each app to assist users in making informed decisions about which apps will best meet their needs in the midst of a wide array of choices.
For the purpose of this study, an objective rubric was created that can be used to evaluate the quality of medical apps for patient education in dermatology. An app’s adequacy and usefulness for patient education was thought to depend on 3 possible score ranges into which the app could fall based on the grading rubric. An app with a total score in the range of 5 to 10 was not thought to be useful and may even be detrimental to patients. An app with a total score in the range of 11 to 15 may be used for patient education with some reservations based on shortcomings for certain criteria. An app with a score in the range of 16 to 20 was thought to be valuable and adequate for patient education. For example, the How to Treat Acne app received a total score of 8 and therefore would not be recommended to patients based on the grading rubric used in this study. This particular app provided sparse and sometimes inaccurate information, had a confusing user interface, and contained many obstructive advertisements. In contrast, the Eczema Doc app received a total score of 19, which indicates a quality app deemed to be useful for patient information based on the established rubric. This app met all the objectives that it advertised, contained accurate information with verified citation of sources, and was very easy for users to navigate.
Of the 44 graded apps, only 9 (20.5%) received scores in the highest range of 16 to 20, which indicates a need for improvements in mobile dermatology apps intended for patient education. Adopting the grading rubric developed in this study as a standard in the creation of medical apps could have beneficial implications in disseminating accurate, safe, unbiased, and easy-to-understand information to patients.
- Smith A. U.S. smartphone use in 2015. Pew Research Center website. http://www.pewinternet.org/2015/04/01/us-smartphone-use-in-2015. Published April 1, 2015. Accessed August 29, 2017.
- Nilsen W, Kumar S, Shar A, et al. Advancing the science of mHealth. J Health Commun. 2012;17(suppl 1):5-10.
- West DM. How mobile devices are transforming healthcare issues in technology innovation. Issues Technol Innov. 2012;18:1-14.
- Boudreaux ED, Waring ME, Hayes RB, et al. Evaluating and selecting mobile health apps: strategies for healthcare providers and healthcare organizations. Transl Behav Med. 2014;4:363-371.
- Brewer AC, Endly DC, Henley J, et al. Mobile applications in dermatology. JAMA Dermatol. 2013;149:1300-1304.
- Cummings E, Borycki E, Roehrer E. Issues and considerations for healthcare consumers using mobile applications. Stud Health Technol Inform. 2013;183:227-231.
- Handel MJ. mHealth (mobile health)-using apps for health and wellness. Explore. 2011;7:256-261.
- Boulos MN, Brewer AC, Karimkhani C, et al. Mobile medical and health apps: state of the art, concerns, regulatory control and certification. Online J Public Health Inform. 2014;5:229.
- Smith A. U.S. smartphone use in 2015. Pew Research Center website. http://www.pewinternet.org/2015/04/01/us-smartphone-use-in-2015. Published April 1, 2015. Accessed August 29, 2017.
- Nilsen W, Kumar S, Shar A, et al. Advancing the science of mHealth. J Health Commun. 2012;17(suppl 1):5-10.
- West DM. How mobile devices are transforming healthcare issues in technology innovation. Issues Technol Innov. 2012;18:1-14.
- Boudreaux ED, Waring ME, Hayes RB, et al. Evaluating and selecting mobile health apps: strategies for healthcare providers and healthcare organizations. Transl Behav Med. 2014;4:363-371.
- Brewer AC, Endly DC, Henley J, et al. Mobile applications in dermatology. JAMA Dermatol. 2013;149:1300-1304.
- Cummings E, Borycki E, Roehrer E. Issues and considerations for healthcare consumers using mobile applications. Stud Health Technol Inform. 2013;183:227-231.
- Handel MJ. mHealth (mobile health)-using apps for health and wellness. Explore. 2011;7:256-261.
- Boulos MN, Brewer AC, Karimkhani C, et al. Mobile medical and health apps: state of the art, concerns, regulatory control and certification. Online J Public Health Inform. 2014;5:229.
Practice Points
- Mobile dermatology apps for educational purposes should be objectively reviewed before being used by patients.
- In our study, only 9 (20.5%) of the 44 dermatology apps evaluated were considered adequate for patient information based on our grading criteria.
Primary Cutaneous Follicle Center Lymphoma Mimicking Folliculitis
The 2008 World Health Organization and European Organization for Treatment of Cancer joint classification has distinguished 3 categories of primary cutaneous B-cell lymphomas: primary cutaneous follicle center lymphoma (PCFCL), primary cutaneous diffuse large B-cell lymphoma, and primary cutaneous marginal zone lymphoma.1-3 Primary cutaneous follicle center lymphoma is the most common type of cutaneous B-cell lymphoma, accounting for approximately 60% of cases worldwide.4 The median age at diagnosis is 60 years, and most lesions are located on the scalp, forehead, neck, and trunk.5 Histologically, PCFCL is characterized by dermal proliferation of centrocytes and centroblasts derived from germinal center B cells that are arranged in either a follicular, diffuse, or mixed growth pattern.1 The cutaneous manifestations of PCFCL include solitary erythematous or violaceous plaques, nodules, or tumors of varying sizes.4 Grouped lesions also may be observed, but multifocal disease is rare.1 We report a rare presentation of PCFCL mimicking folliculitis with multiple multifocal papules on the back.
Case Report
A 54-year-old woman presented with fever and leukocytosis of 4 days’ duration and was admitted to the hospital for presumed sepsis. She had a history of mastectomy for treatment of ductal carcinoma in situ of the right breast 5 years prior to the current presentation and endocrine therapy with tamoxifen. Her symptoms were thought to be a complication from a surgery for implantation of a tissue expander in the right breast 5 years prior to presentation.
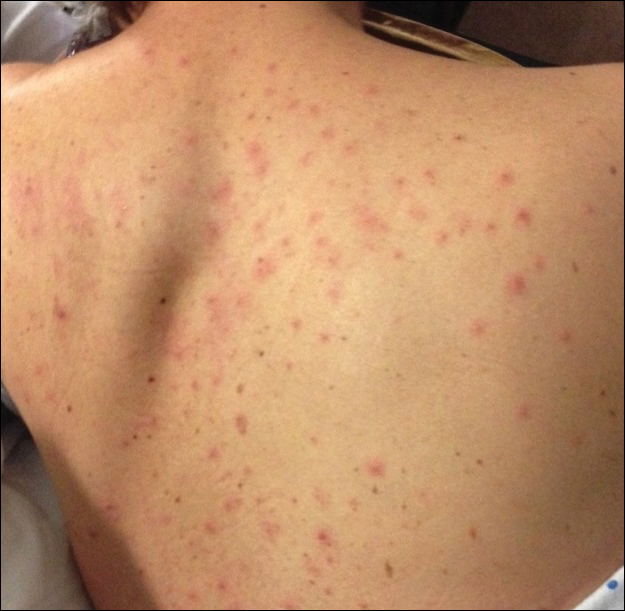
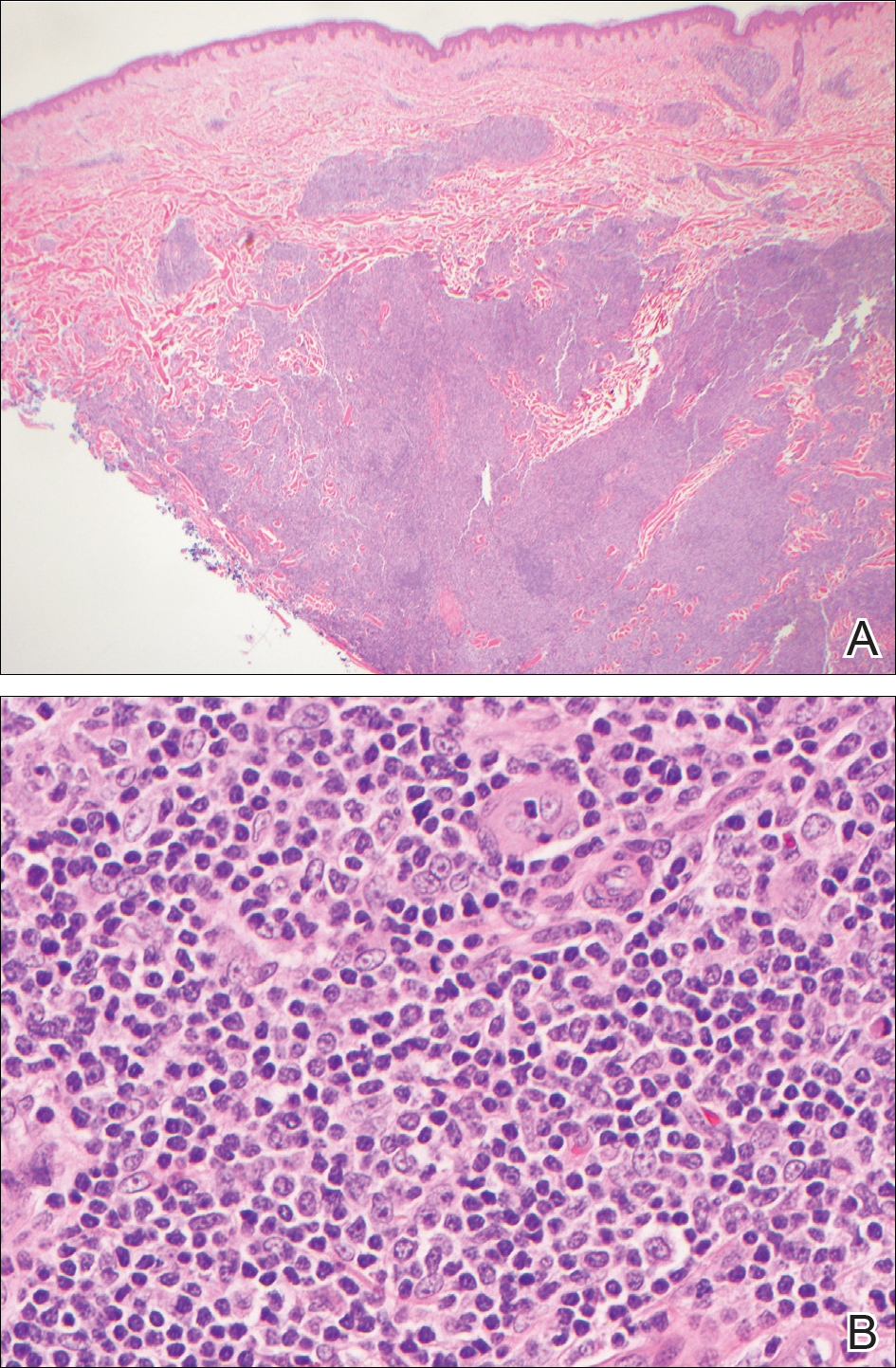
During her hospital admission, she developed a papular and cystic eruption on the back that was clinically suggestive of folliculitis, transient acantholytic dermatosis (Grover disease), or miliaria rubra (Figure 1). This papular and cystic eruption initially was managed conservatively with observation as she recovered from an occult infection. Due to the persistent nature of the eruption on the back, an excisional biopsy of the cystic component was performed 2 months after her discharge from the hospital. Histologic studies showed a dense infiltrate of lymphocytes, which expanded into the deep dermis in a nodular and diffuse growth pattern that was accentuated in the periadnexal areas. The B lymphocytes were small and hyperchromatic with few scattered centroblasts (Figure 2). Further immunohistochemical studies demonstrated that the neoplastic cells were positive for CD20, CD79a, BCL-2, and BCL-6; CD3, CD5, and cyclin D1 were negative. Staining for antigen Ki-67 revealed a proliferation index of 15% to 20% among the neoplastic cells (Figure 3). These findings were consistent with either PCFCL or secondary cutaneous follicle center lymphoma.
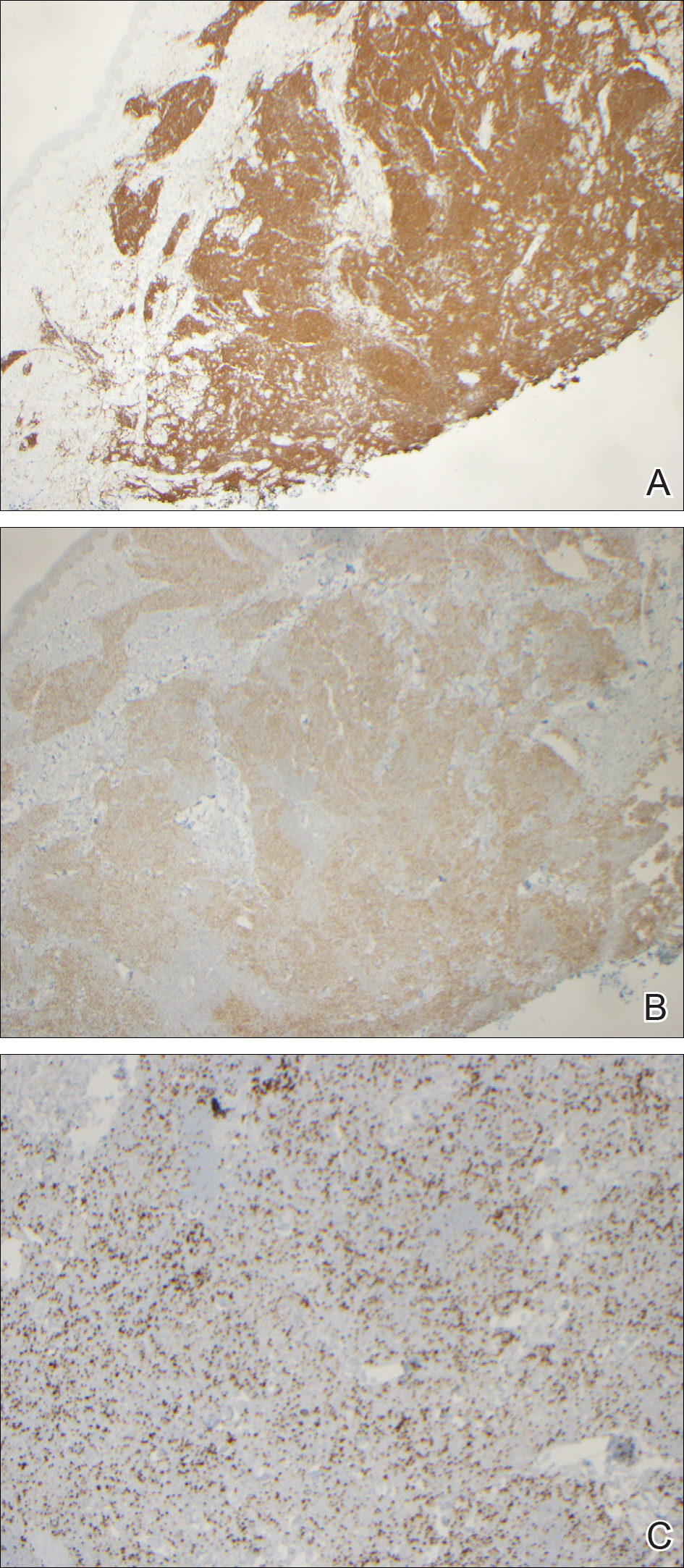
Further evaluation for systemic disease was unremarkable. Positron emission tomography–computed tomography revealed no evidence of nodal lymphoma, and a bone marrow biopsy was negative. Other laboratory studies including lactate dehydrogenase were within reference range, which conferred a diagnosis of PCFCL. The patient was treated with localized electron beam radiation therapy to the skin of the mid back for a total dose of 24 Gy in 12 fractions at 2 Gy per fraction once daily over a 12-day period. She tolerated the treatment well and has remained clinically and radiographically without evidence of disease for more than 3 years.
Comment
Because the incidence of cutaneous B-cell lymphomas has been increasing, especially among males, non-Hispanic whites, and adults older than 50 years,1 it is important for clinicians to have a high index of suspicion for this entity. In our patient, the clinical findings of a papular, largely asymptomatic eruption on the back with acute onset were initially thought to be consistent with folliculitis; the differential diagnosis included transient acantholytic dermatosis and miliaria rubra. Lymphoma was not in the initial clinical differential, and we only arrived at this diagnosis based on histopathologic evaluation.
The neoplastic cells typically are positive for CD20, CD79a, and BCL-6, and negative for BCL-2.4 Most cases of PCFCL do not express the t(14;18) translocation involving the BCL-2 locus, in contrast to systemic follicular lymphoma.1 Systemic imaging and evaluation is needed to definitively differentiate PCFCL from systemic lymphoma with cutaneous involvement. Our patient was unusual in that BCL-2 was strongly staining in the setting of a negative systemic workup.
With regard to treatment of PCFCL, electron beam radiation therapy is highly effective and safe in patients with solitary lesions, as the remission rate is close to 100%.1 For patients with multiple lesions confined to one area, electron beam radiation therapy also can be helpful, as in our patient. In patients with more extensive skin involvement, rituximab therapy may be preferable. Relapse following treatment with either radiation or rituximab occurs in approximately one-third of patients, but these relapses generally are limited to the skin.1 The International Extranodal Lymphoma Study Group has noted that elevated lactate dehydrogenase, presence of more than 2 skin lesions, and presence of nodular lesions are negative prognostic factors in patients with PCFCL6; however, PCFCL has an excellent prognosis overall with a 5-year survival rate of 95%.1
Other rare heterogeneous presentations of PCFCL have been reported in the literature. A large multinodular mass on the scalp with multifocal facial lesions has been described in a patient with essential thrombocytopenia.7 Another report identified a variant of PCFCL characterized by multiple erythematous firm papules that were distributed in a miliary pattern, predominantly on the forehead and cheeks.8 Barzilai et al9 described 4 patients with PCFCL who developed lesions that were clinically similar to rosacea or rhinophyma, including papulonodular eruptions on the cheeks; infiltrated erythematous nasal plaques; and small flesh-colored to erythematous papules on the cheeks, nose, helices, and upper back. Hodak et al10 identified 2 cases of PCFCL that manifested as anetoderma, a condition characterized by the focal loss of elastic tissue. In the setting of chronic lymphocytic leukemia, PCFCL has been observed as a red or violaceous nodule with a centrally depressed scar on the legs.11 In one case, PCFCL manifested as recurrent episodes of extraorbital swelling and a multifocal red-blue macular lesion that extended from the inferior orbital rim to the nasojugal fold.12 An interesting presentation of PCFCL was noted as a small, recurring, blood-filled blister on the cheek with perineural spread of the tumor along cranial nerves V2, V3, VII, and VIII.13 In the pediatric literature, PCFCL has been reported to present as an erythematous nodule with a smooth surface and a hard elastic consistency that appeared on the nose and nasolabial fold and spread to the ipsilateral cheek, maxillary sinus, and soft palate.14 In many of these unusual cases, the diagnosis of PCFCL was made after treatment with topical or systemic anti-inflammatory therapies failed.
Increased recognition of anomalous presentations of PCFCL among dermatologists can lead to more timely diagnoses and treatment. Based on our experience with this patient, we recommend considering biopsy for histopathologic evaluation when treating patients with presumed folliculitis or transient acantholytic dermatosis that does not improve with routine treatment or is accompanied by systemic symptoms.
- Wilcox RA. Cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- World Health Organization. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: World Health Organization; 2008: 227.
- Dilly M, Ben-Rejeb H, Vergier B, et al. Primary cutaneous follicle center lymphoma with Hodgkin and Reed-Sternberg-like cells: a new histopathologic variant. J Cutan Pathol. 2014;41:797-801.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 341-342.
- Mian M, Marcheselli L, Luminari S, et al. CLIPI: a new prognostic index for indolent cutaneous B cell lymphoma proposed by the International Extranodal Lymphoma Study Group (IELSG 11) [published online September 25, 2010]. Ann Hematol. 2011;90:401-408.
- Tirefort Y, Pham XC, Ibrahim YL, et al. A rare case of primary cutaneous follicle centre lymphoma presenting as a giant tumour of the scalp and combined with JAK2V617F positive essential thrombocythaemia. Biomark Res. 2014;2:7.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: report of 18 cases.J Am Acad Dermatol. 2011;65:749-755.
- Barzilai A, Feuerman H, Quaglino P, et al. Cutaneous B-cell neoplasms mimicking granulomatous rosacea or rhinophyma. Arch Dermatol. 2012;148:824-831.
- Hodak E, Feuerman H, Barzilai A, et al. Anetodermic primary cutaneous B-cell lymphoma: a unique clinicopathological presentation of lymphoma possibly associated with antiphospholipid antibodies. Arch Dermatol. 2010;146:175-182.
- Konda S, Beckford A, Demierre MF, et al. Primary cutaneous follicle center lymphoma in the setting of chronic lymphocytic leukemia. Indian J Dermatol Venereol Leprol. 2011;77:314-317.
- Pandya VB, Conway RM, Taylor SF. Primary cutaneous B cell lymphoma presenting as recurrent eyelid swelling. Clin Exp Ophthalmol. 2008;36:672-674.
- Buda-Okreglak EM, Walden MJ, Brissette MD. Perineural CNS invasion in primary cutaneous follicular center lymphoma. J Clin Oncol. 2007;25:4684-4686.
- Ghislanzoni M, Gambini D, Perrone T, et al. Primary cutaneous follicular center cell lymphoma of the nose with maxillary sinus involvement in a pediatric patient. J Am Acad Dermatol. 2005;52(5 suppl 1):S73-S75.
The 2008 World Health Organization and European Organization for Treatment of Cancer joint classification has distinguished 3 categories of primary cutaneous B-cell lymphomas: primary cutaneous follicle center lymphoma (PCFCL), primary cutaneous diffuse large B-cell lymphoma, and primary cutaneous marginal zone lymphoma.1-3 Primary cutaneous follicle center lymphoma is the most common type of cutaneous B-cell lymphoma, accounting for approximately 60% of cases worldwide.4 The median age at diagnosis is 60 years, and most lesions are located on the scalp, forehead, neck, and trunk.5 Histologically, PCFCL is characterized by dermal proliferation of centrocytes and centroblasts derived from germinal center B cells that are arranged in either a follicular, diffuse, or mixed growth pattern.1 The cutaneous manifestations of PCFCL include solitary erythematous or violaceous plaques, nodules, or tumors of varying sizes.4 Grouped lesions also may be observed, but multifocal disease is rare.1 We report a rare presentation of PCFCL mimicking folliculitis with multiple multifocal papules on the back.
Case Report
A 54-year-old woman presented with fever and leukocytosis of 4 days’ duration and was admitted to the hospital for presumed sepsis. She had a history of mastectomy for treatment of ductal carcinoma in situ of the right breast 5 years prior to the current presentation and endocrine therapy with tamoxifen. Her symptoms were thought to be a complication from a surgery for implantation of a tissue expander in the right breast 5 years prior to presentation.
During her hospital admission, she developed a papular and cystic eruption on the back that was clinically suggestive of folliculitis, transient acantholytic dermatosis (Grover disease), or miliaria rubra (Figure 1). This papular and cystic eruption initially was managed conservatively with observation as she recovered from an occult infection. Due to the persistent nature of the eruption on the back, an excisional biopsy of the cystic component was performed 2 months after her discharge from the hospital. Histologic studies showed a dense infiltrate of lymphocytes, which expanded into the deep dermis in a nodular and diffuse growth pattern that was accentuated in the periadnexal areas. The B lymphocytes were small and hyperchromatic with few scattered centroblasts (Figure 2). Further immunohistochemical studies demonstrated that the neoplastic cells were positive for CD20, CD79a, BCL-2, and BCL-6; CD3, CD5, and cyclin D1 were negative. Staining for antigen Ki-67 revealed a proliferation index of 15% to 20% among the neoplastic cells (Figure 3). These findings were consistent with either PCFCL or secondary cutaneous follicle center lymphoma.
Further evaluation for systemic disease was unremarkable. Positron emission tomography–computed tomography revealed no evidence of nodal lymphoma, and a bone marrow biopsy was negative. Other laboratory studies including lactate dehydrogenase were within reference range, which conferred a diagnosis of PCFCL. The patient was treated with localized electron beam radiation therapy to the skin of the mid back for a total dose of 24 Gy in 12 fractions at 2 Gy per fraction once daily over a 12-day period. She tolerated the treatment well and has remained clinically and radiographically without evidence of disease for more than 3 years.
Comment
Because the incidence of cutaneous B-cell lymphomas has been increasing, especially among males, non-Hispanic whites, and adults older than 50 years,1 it is important for clinicians to have a high index of suspicion for this entity. In our patient, the clinical findings of a papular, largely asymptomatic eruption on the back with acute onset were initially thought to be consistent with folliculitis; the differential diagnosis included transient acantholytic dermatosis and miliaria rubra. Lymphoma was not in the initial clinical differential, and we only arrived at this diagnosis based on histopathologic evaluation.
The neoplastic cells typically are positive for CD20, CD79a, and BCL-6, and negative for BCL-2.4 Most cases of PCFCL do not express the t(14;18) translocation involving the BCL-2 locus, in contrast to systemic follicular lymphoma.1 Systemic imaging and evaluation is needed to definitively differentiate PCFCL from systemic lymphoma with cutaneous involvement. Our patient was unusual in that BCL-2 was strongly staining in the setting of a negative systemic workup.
With regard to treatment of PCFCL, electron beam radiation therapy is highly effective and safe in patients with solitary lesions, as the remission rate is close to 100%.1 For patients with multiple lesions confined to one area, electron beam radiation therapy also can be helpful, as in our patient. In patients with more extensive skin involvement, rituximab therapy may be preferable. Relapse following treatment with either radiation or rituximab occurs in approximately one-third of patients, but these relapses generally are limited to the skin.1 The International Extranodal Lymphoma Study Group has noted that elevated lactate dehydrogenase, presence of more than 2 skin lesions, and presence of nodular lesions are negative prognostic factors in patients with PCFCL6; however, PCFCL has an excellent prognosis overall with a 5-year survival rate of 95%.1
Other rare heterogeneous presentations of PCFCL have been reported in the literature. A large multinodular mass on the scalp with multifocal facial lesions has been described in a patient with essential thrombocytopenia.7 Another report identified a variant of PCFCL characterized by multiple erythematous firm papules that were distributed in a miliary pattern, predominantly on the forehead and cheeks.8 Barzilai et al9 described 4 patients with PCFCL who developed lesions that were clinically similar to rosacea or rhinophyma, including papulonodular eruptions on the cheeks; infiltrated erythematous nasal plaques; and small flesh-colored to erythematous papules on the cheeks, nose, helices, and upper back. Hodak et al10 identified 2 cases of PCFCL that manifested as anetoderma, a condition characterized by the focal loss of elastic tissue. In the setting of chronic lymphocytic leukemia, PCFCL has been observed as a red or violaceous nodule with a centrally depressed scar on the legs.11 In one case, PCFCL manifested as recurrent episodes of extraorbital swelling and a multifocal red-blue macular lesion that extended from the inferior orbital rim to the nasojugal fold.12 An interesting presentation of PCFCL was noted as a small, recurring, blood-filled blister on the cheek with perineural spread of the tumor along cranial nerves V2, V3, VII, and VIII.13 In the pediatric literature, PCFCL has been reported to present as an erythematous nodule with a smooth surface and a hard elastic consistency that appeared on the nose and nasolabial fold and spread to the ipsilateral cheek, maxillary sinus, and soft palate.14 In many of these unusual cases, the diagnosis of PCFCL was made after treatment with topical or systemic anti-inflammatory therapies failed.
Increased recognition of anomalous presentations of PCFCL among dermatologists can lead to more timely diagnoses and treatment. Based on our experience with this patient, we recommend considering biopsy for histopathologic evaluation when treating patients with presumed folliculitis or transient acantholytic dermatosis that does not improve with routine treatment or is accompanied by systemic symptoms.
The 2008 World Health Organization and European Organization for Treatment of Cancer joint classification has distinguished 3 categories of primary cutaneous B-cell lymphomas: primary cutaneous follicle center lymphoma (PCFCL), primary cutaneous diffuse large B-cell lymphoma, and primary cutaneous marginal zone lymphoma.1-3 Primary cutaneous follicle center lymphoma is the most common type of cutaneous B-cell lymphoma, accounting for approximately 60% of cases worldwide.4 The median age at diagnosis is 60 years, and most lesions are located on the scalp, forehead, neck, and trunk.5 Histologically, PCFCL is characterized by dermal proliferation of centrocytes and centroblasts derived from germinal center B cells that are arranged in either a follicular, diffuse, or mixed growth pattern.1 The cutaneous manifestations of PCFCL include solitary erythematous or violaceous plaques, nodules, or tumors of varying sizes.4 Grouped lesions also may be observed, but multifocal disease is rare.1 We report a rare presentation of PCFCL mimicking folliculitis with multiple multifocal papules on the back.
Case Report
A 54-year-old woman presented with fever and leukocytosis of 4 days’ duration and was admitted to the hospital for presumed sepsis. She had a history of mastectomy for treatment of ductal carcinoma in situ of the right breast 5 years prior to the current presentation and endocrine therapy with tamoxifen. Her symptoms were thought to be a complication from a surgery for implantation of a tissue expander in the right breast 5 years prior to presentation.
During her hospital admission, she developed a papular and cystic eruption on the back that was clinically suggestive of folliculitis, transient acantholytic dermatosis (Grover disease), or miliaria rubra (Figure 1). This papular and cystic eruption initially was managed conservatively with observation as she recovered from an occult infection. Due to the persistent nature of the eruption on the back, an excisional biopsy of the cystic component was performed 2 months after her discharge from the hospital. Histologic studies showed a dense infiltrate of lymphocytes, which expanded into the deep dermis in a nodular and diffuse growth pattern that was accentuated in the periadnexal areas. The B lymphocytes were small and hyperchromatic with few scattered centroblasts (Figure 2). Further immunohistochemical studies demonstrated that the neoplastic cells were positive for CD20, CD79a, BCL-2, and BCL-6; CD3, CD5, and cyclin D1 were negative. Staining for antigen Ki-67 revealed a proliferation index of 15% to 20% among the neoplastic cells (Figure 3). These findings were consistent with either PCFCL or secondary cutaneous follicle center lymphoma.
Further evaluation for systemic disease was unremarkable. Positron emission tomography–computed tomography revealed no evidence of nodal lymphoma, and a bone marrow biopsy was negative. Other laboratory studies including lactate dehydrogenase were within reference range, which conferred a diagnosis of PCFCL. The patient was treated with localized electron beam radiation therapy to the skin of the mid back for a total dose of 24 Gy in 12 fractions at 2 Gy per fraction once daily over a 12-day period. She tolerated the treatment well and has remained clinically and radiographically without evidence of disease for more than 3 years.
Comment
Because the incidence of cutaneous B-cell lymphomas has been increasing, especially among males, non-Hispanic whites, and adults older than 50 years,1 it is important for clinicians to have a high index of suspicion for this entity. In our patient, the clinical findings of a papular, largely asymptomatic eruption on the back with acute onset were initially thought to be consistent with folliculitis; the differential diagnosis included transient acantholytic dermatosis and miliaria rubra. Lymphoma was not in the initial clinical differential, and we only arrived at this diagnosis based on histopathologic evaluation.
The neoplastic cells typically are positive for CD20, CD79a, and BCL-6, and negative for BCL-2.4 Most cases of PCFCL do not express the t(14;18) translocation involving the BCL-2 locus, in contrast to systemic follicular lymphoma.1 Systemic imaging and evaluation is needed to definitively differentiate PCFCL from systemic lymphoma with cutaneous involvement. Our patient was unusual in that BCL-2 was strongly staining in the setting of a negative systemic workup.
With regard to treatment of PCFCL, electron beam radiation therapy is highly effective and safe in patients with solitary lesions, as the remission rate is close to 100%.1 For patients with multiple lesions confined to one area, electron beam radiation therapy also can be helpful, as in our patient. In patients with more extensive skin involvement, rituximab therapy may be preferable. Relapse following treatment with either radiation or rituximab occurs in approximately one-third of patients, but these relapses generally are limited to the skin.1 The International Extranodal Lymphoma Study Group has noted that elevated lactate dehydrogenase, presence of more than 2 skin lesions, and presence of nodular lesions are negative prognostic factors in patients with PCFCL6; however, PCFCL has an excellent prognosis overall with a 5-year survival rate of 95%.1
Other rare heterogeneous presentations of PCFCL have been reported in the literature. A large multinodular mass on the scalp with multifocal facial lesions has been described in a patient with essential thrombocytopenia.7 Another report identified a variant of PCFCL characterized by multiple erythematous firm papules that were distributed in a miliary pattern, predominantly on the forehead and cheeks.8 Barzilai et al9 described 4 patients with PCFCL who developed lesions that were clinically similar to rosacea or rhinophyma, including papulonodular eruptions on the cheeks; infiltrated erythematous nasal plaques; and small flesh-colored to erythematous papules on the cheeks, nose, helices, and upper back. Hodak et al10 identified 2 cases of PCFCL that manifested as anetoderma, a condition characterized by the focal loss of elastic tissue. In the setting of chronic lymphocytic leukemia, PCFCL has been observed as a red or violaceous nodule with a centrally depressed scar on the legs.11 In one case, PCFCL manifested as recurrent episodes of extraorbital swelling and a multifocal red-blue macular lesion that extended from the inferior orbital rim to the nasojugal fold.12 An interesting presentation of PCFCL was noted as a small, recurring, blood-filled blister on the cheek with perineural spread of the tumor along cranial nerves V2, V3, VII, and VIII.13 In the pediatric literature, PCFCL has been reported to present as an erythematous nodule with a smooth surface and a hard elastic consistency that appeared on the nose and nasolabial fold and spread to the ipsilateral cheek, maxillary sinus, and soft palate.14 In many of these unusual cases, the diagnosis of PCFCL was made after treatment with topical or systemic anti-inflammatory therapies failed.
Increased recognition of anomalous presentations of PCFCL among dermatologists can lead to more timely diagnoses and treatment. Based on our experience with this patient, we recommend considering biopsy for histopathologic evaluation when treating patients with presumed folliculitis or transient acantholytic dermatosis that does not improve with routine treatment or is accompanied by systemic symptoms.
- Wilcox RA. Cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- World Health Organization. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: World Health Organization; 2008: 227.
- Dilly M, Ben-Rejeb H, Vergier B, et al. Primary cutaneous follicle center lymphoma with Hodgkin and Reed-Sternberg-like cells: a new histopathologic variant. J Cutan Pathol. 2014;41:797-801.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 341-342.
- Mian M, Marcheselli L, Luminari S, et al. CLIPI: a new prognostic index for indolent cutaneous B cell lymphoma proposed by the International Extranodal Lymphoma Study Group (IELSG 11) [published online September 25, 2010]. Ann Hematol. 2011;90:401-408.
- Tirefort Y, Pham XC, Ibrahim YL, et al. A rare case of primary cutaneous follicle centre lymphoma presenting as a giant tumour of the scalp and combined with JAK2V617F positive essential thrombocythaemia. Biomark Res. 2014;2:7.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: report of 18 cases.J Am Acad Dermatol. 2011;65:749-755.
- Barzilai A, Feuerman H, Quaglino P, et al. Cutaneous B-cell neoplasms mimicking granulomatous rosacea or rhinophyma. Arch Dermatol. 2012;148:824-831.
- Hodak E, Feuerman H, Barzilai A, et al. Anetodermic primary cutaneous B-cell lymphoma: a unique clinicopathological presentation of lymphoma possibly associated with antiphospholipid antibodies. Arch Dermatol. 2010;146:175-182.
- Konda S, Beckford A, Demierre MF, et al. Primary cutaneous follicle center lymphoma in the setting of chronic lymphocytic leukemia. Indian J Dermatol Venereol Leprol. 2011;77:314-317.
- Pandya VB, Conway RM, Taylor SF. Primary cutaneous B cell lymphoma presenting as recurrent eyelid swelling. Clin Exp Ophthalmol. 2008;36:672-674.
- Buda-Okreglak EM, Walden MJ, Brissette MD. Perineural CNS invasion in primary cutaneous follicular center lymphoma. J Clin Oncol. 2007;25:4684-4686.
- Ghislanzoni M, Gambini D, Perrone T, et al. Primary cutaneous follicular center cell lymphoma of the nose with maxillary sinus involvement in a pediatric patient. J Am Acad Dermatol. 2005;52(5 suppl 1):S73-S75.
- Wilcox RA. Cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- World Health Organization. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: World Health Organization; 2008: 227.
- Dilly M, Ben-Rejeb H, Vergier B, et al. Primary cutaneous follicle center lymphoma with Hodgkin and Reed-Sternberg-like cells: a new histopathologic variant. J Cutan Pathol. 2014;41:797-801.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 341-342.
- Mian M, Marcheselli L, Luminari S, et al. CLIPI: a new prognostic index for indolent cutaneous B cell lymphoma proposed by the International Extranodal Lymphoma Study Group (IELSG 11) [published online September 25, 2010]. Ann Hematol. 2011;90:401-408.
- Tirefort Y, Pham XC, Ibrahim YL, et al. A rare case of primary cutaneous follicle centre lymphoma presenting as a giant tumour of the scalp and combined with JAK2V617F positive essential thrombocythaemia. Biomark Res. 2014;2:7.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: report of 18 cases.J Am Acad Dermatol. 2011;65:749-755.
- Barzilai A, Feuerman H, Quaglino P, et al. Cutaneous B-cell neoplasms mimicking granulomatous rosacea or rhinophyma. Arch Dermatol. 2012;148:824-831.
- Hodak E, Feuerman H, Barzilai A, et al. Anetodermic primary cutaneous B-cell lymphoma: a unique clinicopathological presentation of lymphoma possibly associated with antiphospholipid antibodies. Arch Dermatol. 2010;146:175-182.
- Konda S, Beckford A, Demierre MF, et al. Primary cutaneous follicle center lymphoma in the setting of chronic lymphocytic leukemia. Indian J Dermatol Venereol Leprol. 2011;77:314-317.
- Pandya VB, Conway RM, Taylor SF. Primary cutaneous B cell lymphoma presenting as recurrent eyelid swelling. Clin Exp Ophthalmol. 2008;36:672-674.
- Buda-Okreglak EM, Walden MJ, Brissette MD. Perineural CNS invasion in primary cutaneous follicular center lymphoma. J Clin Oncol. 2007;25:4684-4686.
- Ghislanzoni M, Gambini D, Perrone T, et al. Primary cutaneous follicular center cell lymphoma of the nose with maxillary sinus involvement in a pediatric patient. J Am Acad Dermatol. 2005;52(5 suppl 1):S73-S75.
Practice Points
- Atypical or unresponsive folliculitis should be biopsied.
- Primary cutaneous follicle center lymphoma can mimic folliculitis or Grover disease.

Complete Remission of Metastatic Merkel Cell Carcinoma in a Patient With Severe Psoriasis
To the Editor:
A 69-year-old white man presented with a skin lesion on the back of 1 to 2 weeks’ duration. The patient stated he was unaware of it, but his wife had recently noticed the new spot. He denied any bleeding, pain, pruritus, or other associated symptoms with the lesion. He also denied any prior treatment to the area. The patient’s medical history was remarkable for severe psoriasis involving more than 80% body surface area, psoriatic arthritis, rheumatoid arthritis, ankylosing spondylitis, coronary artery disease, squamous cell carcinoma, and actinic keratoses. He had been on multiple treatment regimens over the last 20 years for control of psoriasis including topical corticosteroids, psoralen plus UVA and UVB phototherapy, gold injections, acitretin, prednisone, efalizumab, ustekinumab, and alefacept upon evaluation of this new skin lesion. Utilization of immunosuppressive agents also provided an additional benefit of controlling the patient’s inflammatory arthritic disease.
On physical examination a 0.6×0.7-cm, pink to erythematous, pearly papule with superficial telangiectases was noted on the right side of the dorsal thorax (Figure 1). Multiple well-demarcated erythematous plaques with silvery scale and areas of secondary excoriation were noted on the trunk and both legs consistent with the patient’s history of psoriasis.
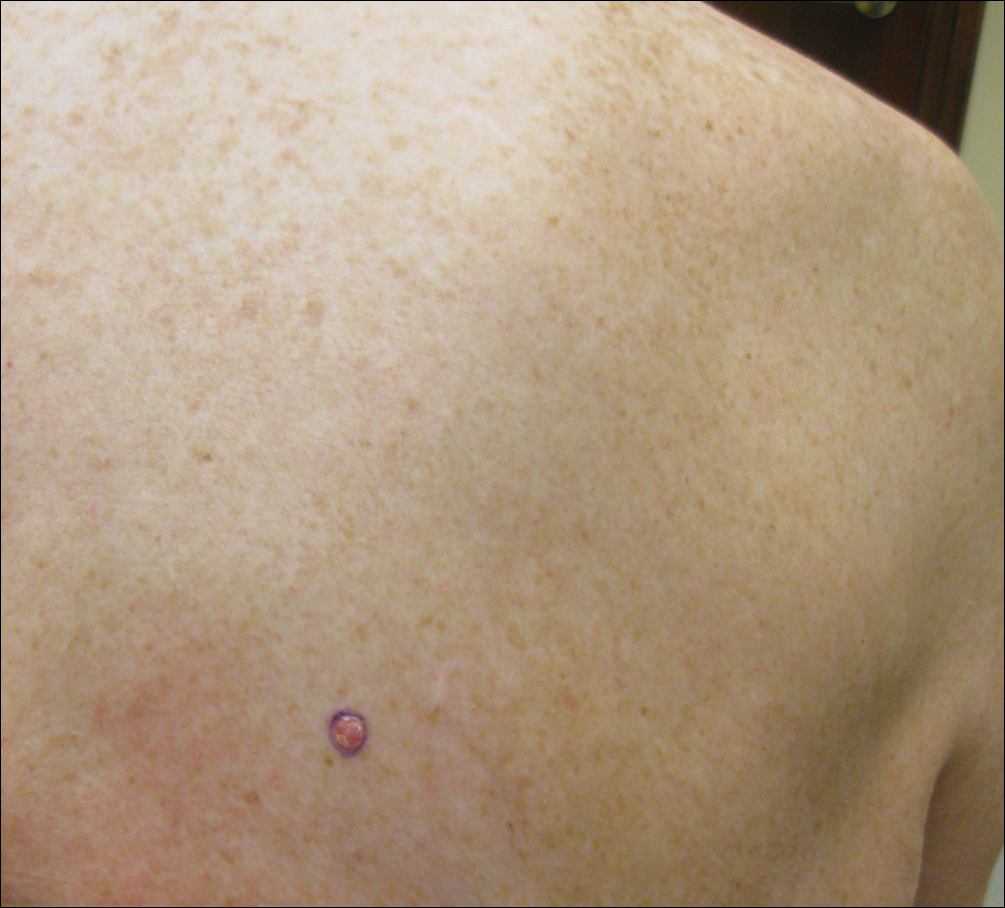
A shave biopsy was performed on the skin lesion on the right side of the dorsal thorax with a suspected clinical diagnosis of basal cell carcinoma. Two weeks later the patient returned for a discussion of the pathology report, which revealed nodules of basaloid cells with tightly packed vesicular nuclei and scant cytoplasm in sheets within the superficial dermis, as well as areas of nuclear molding, numerous mitotic figures, and areas of focal necrosis (Figure 2). In addition, immunostaining was positive for cytokeratin (CK) 20 antibodies with a characteristic paranuclear dot uptake of the antibody. These findings were consistent with a diagnosis of Merkel cell carcinoma (MCC). At that time, alefacept was discontinued and he was referred to a tertiary referral center for further evaluation and treatment.
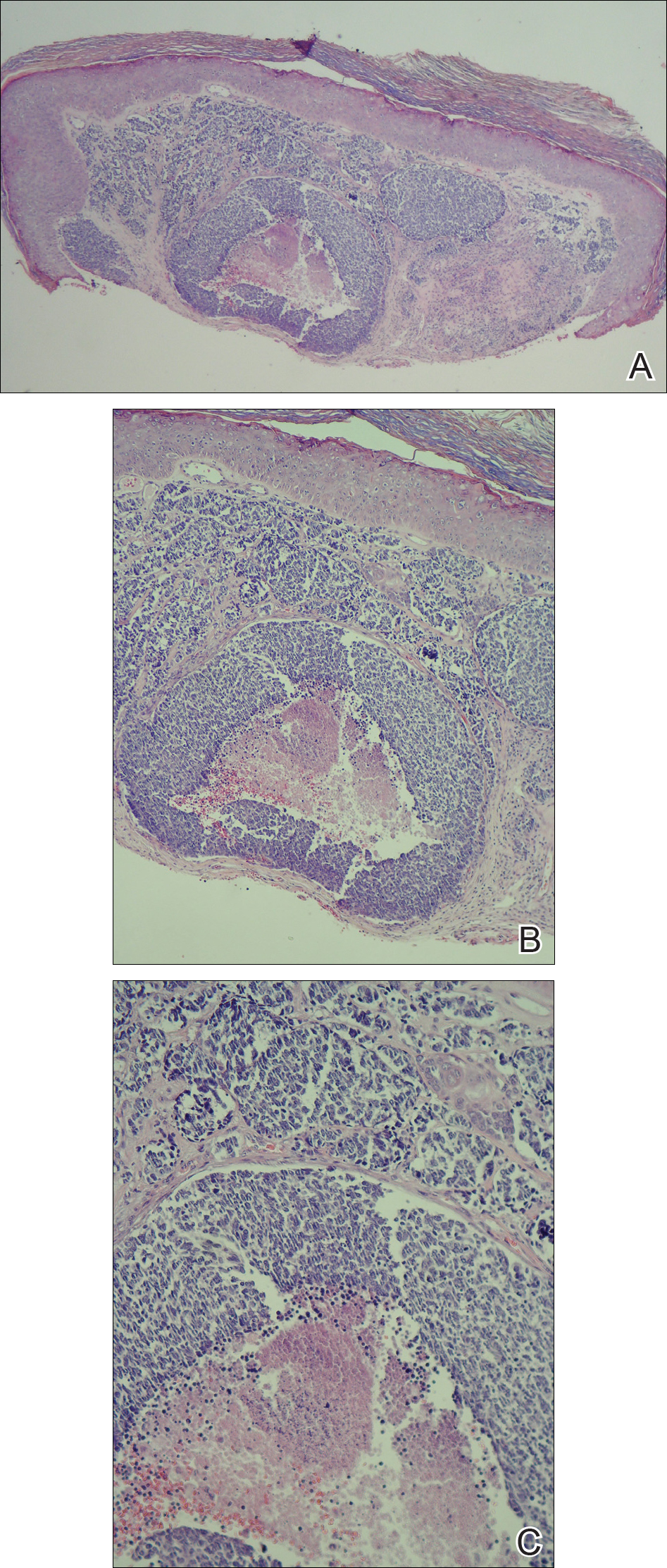
The patient subsequently underwent wide excision with 1-cm margins of the MCC, with intraoperative lymphatic mapping/sentinel lymph node biopsy (SLNB) of the right axillary nodal basin 1 month later, which he tolerated well without any associated complications. Further histopathologic examination revealed the deep, medial, and lateral surgical margins to be negative of residual neoplasm. However, one sentinel lymph node indicated positivity for micrometastatic MCC, consistent with stage IIIA disease progression.
He underwent a second procedure the following month for complete right axillary lymph node dissection. Histopathologic examination of the right axillary contents included 28 lymph nodes, which were negative for carcinoma. He continued to do well without any signs of clinical recurrence or distant metastasis at subsequent follow-up visits.
Approximately 2.5 years after the second procedure, the patient began to develop right upper quadrant abdominal pain of an unclear etiology. Computed tomography of the abdomen and pelvis was performed, revealing areas of calcification and findings consistent with malignant lymphadenopathy. Multiple hepatic lesions also were noted including a 9-cm lesion in the posterior right hepatic lobe. Computed tomography–guided biopsy of the liver lesion was performed and the findings were consistent with metastatic MCC, indicating progression to stage IV disease.
The patient was subsequently started on combination chemotherapeutic treatment with carboplatin and VP-16, with a planned treatment course of 4 to 6 cycles. He was able to complete a total of 6 cycles over a 4-month period, tolerating the treatment regimen fairly well. Follow-up positron emission tomography–computed tomography was within normal limits with no evidence of any hypermetabolic activity noted, indicating a complete radiographic remission of MCC. He was seen approximately 1 month after completion of treatment for clinical follow-up and monthly thereafter.
While on chemotherapy, the patient experienced a notable improvement in the psoriasis and psoriatic joint disease. Upon completion of chemotherapy, he was restarted on the same treatment plan that was utilized prior to surgery including topical corticosteroids, calcitriol, intramuscular steroid injections, and UVB phototherapy, which provided substantial control of psoriasis and arthritic joint disease. The patient later died, likely due to his multiple comorbidities.
Merkel cells are slow-responding mechanoreceptors located within the basal layer of the epidermis and are the source of a rare aggressive cutaneous malignancy.1 Merkel cell carcinoma was first noted in 1972 and termed trabecular carcinoma of the skin, and it accounts for less than 1% of all nonmelanoma skin cancer.2,3 This primary neuroendocrine carcinoma has remarkable metastatic potential (34%–75%) and can invade regional lymph nodes, as well as distant metastasis most commonly to the liver, lungs, bones, and brain.2 Approximately 25% of patients present with palpable lymphadenopathy and 5% with distant metastasis at the time of diagnosis. This frequency of metastasis at diagnosis as well as the recurrence after treatment contributes to the poor prognosis of MCC. Local recurrence rates have been reported at 25% with lymph node involvement in 52% and metastasis in 34%, with most recurrences occurring within 2 years of diagnosis. Patient mortality is dependent on the aggressiveness of the tumor, with 5-year survival rates of 83.3% without lymph node involvement, 58.3% with lymph node involvement, and 31.3% in those with metastatic disease.4
The tumor classically presents as a red to violaceous, painless nodule with a smooth shiny surface most often on the head and neck region.4-6 Approximately 50% of MCC cases present in the head and neck region, 32% to 38% on the extremities, and 12% to 14% on the trunk.1 This nonspecific presentation may lead to diagnostic uncertainty and a consequent delay in treatment. Definitive diagnosis of MCC is achieved with a skin biopsy and allows for distinction from other clinically similar–appearing neoplasms. Merkel cell carcinoma presents histologically as small round basophilic cells penetrating through the dermis in 3 histologic patterns: the trabecular, intermediate (80% of cases), and small cell type.5 It may be differentiated immunohistochemically from other neoplasms, as it displays CK20 positivity (showing paranuclear dotlike depositions in the cytoplasm or cell membrane) and is negative for CK7. Chromagranin and synaptophysin positivity also may provide further histologic confirmation. In addition, absence of peripheral palisading, retraction artifact, and a fibromyxoid stroma allow for distinction from cutaneous basal cell carcinoma, which may display these features histologically. Other immunohistochemical markers that may be of value include thyroid transcription factor 1, which is typically positive in cutaneous metastasis of neuroendocrine carcinoma of the lung; S-100 and human melanoma black 45, which are positive in melanoma; and leukocyte common antigen (CD45), which can be positive in lymphoma. These stains are classically negative in MCC.3
Merkel cell carcinoma is commonly associated with the presence of Merkel cell polyomavirus (MCPyV) in tumor specimens, with a prevalence of 70% to 80% in all cases. Merkel cell polyomavirus is a class 2A carcinogen (ie, a probable carcinogen to humans) and is classified among a group of viruses that encode T antigens (ie, an antigen coded by a viral genome associated with transformation of infected cells by tumor viruses), which can lead to initiation of tumorigenesis through interference with cellular tumor suppressing proteins such as p53.5 In addition, several risk factors have been associated with the development of MCC including immunosuppression, older age (>50 years), and UV-exposed fair skin.7 One explanation for this phenomenon is the increase in MCPyV small T antigen transcripts induced by UV irradiation.5 In addition, as with other cancers induced by viruses, host immunity can impede tumor progression and development. Therefore, impairment of normal immune function likely creates a higher risk for MCC development and potential for a worse prognosis.3Although the exact incidence of MCC in immunosuppressed patients appears unclear, chronic immunosuppressive therapy may play a notable role in the pathogenesis of the tumor.3
Although each of these factors was observed in our patient, it also was possible that his associated comorbidities further contributed to disease presentation. In particular, rheumatoid arthritis has been shown to carry an increased risk for the development of MCC.8 In addition, inflammatory monocytes infected with MCPyV, as evidenced in a patient with a history of chronic psoriasis prior to diagnosis of MCC, also may contribute to the pathogenesis of MCC by traveling to inflammatory skin lesions, such as those seen in psoriasis, releasing MCPyV locally and infecting Merkel cells.9 Although MCPyV testing was never performed in our patient, it certainly would be prudent as well as further studies determining the correlation of MCC to these disease processes.
Although regression is rare, multiple cases have documented spontaneous regression of MCC after biopsy of these lesions.4,6,10 The exact mechanism is unclear, but apoptosis induced by T-cell immunity is suspected to play a role. Programmed cell death 1 protein (PD-1)–positive cells play a role. The PD-1 receptor is an inhibitory receptor expressed by T cells and in approximately half of tumor-infiltrating cells in MCC. It was found that in a regressed case of MCC there was a notably lower percentage of PD-1 positivity compared to cases with no apparent regression, suggesting that PD-1–positive cells suppress tumor immunity to MCC and that significant reduction in these cells may induce clinical regression.10 Additional investigation would be beneficial to examine the relationship of this phenomenon to tumor regression.
Initial evaluation of these patients should include a meticulous clinical examination with an emphasis on detection of cutaneous, lymph node, and distant metastasis. Due to the risk of metastatic potential, regional lymph node ultrasonography and computed tomography of the chest, abdomen, and pelvis typically are recommended at baseline. Other imaging modalities may be warranted based on clinical findings.3 Treatment modalities include various approaches, with surgical excision of the primary tumor with more than 1-cm margin to the fascial plane being the primary modality for uncomplicated cases.1,3,7 In addition, SLNB also should be performed at the time of the procedure. In the case of a positive SLNB or suspected regional lymph node involvement upon initial examination, radical regional lymph node dissection also is recommended.3 Although some authorities advocate postsurgical radiation therapy to minimize the risk of local recurrence, there does not appear to be a clear benefit in survival rate.3,5 However, radiation treatment as monotherapy has been advocated in certain instances, particularly in cases of unresectable tumors or patients who are poor surgical candidates.5,7 Cases of distant metastasis (stage IV disease) may include management with surgery, radiation, and/or chemotherapy. Although none of these modalities have consistently shown to improve survival, there appears to be up to a 60% response with chemotherapy in these patients.3
Because MCC tends to affect an older population, often with other notable comorbidities, important considerations involving a treatment plan include the cost, side effects, and convenience for patients. The combination of carboplatin and VP-16 (etoposide) was utilized and tolerated well in our patient, and it has been successful in achieving complete radiologic and clinical remission of his metastatic disease. This combination appears to prolong survival in patients with distant metastasis, as compared to those patients not receiving chemotherapy.1 Our patient has since died, but in these high-risk patients, close clinical monitoring is essential to help optimize their prognosis.
Merkel cell carcinoma is a rare aggressive cutaneous neoplasm that most commonly affects the elderly, immunosuppressed, and those with chronic UV sun damage. An association between the oncogenesis of MCC and infection with MCPyV has been documented, but other underlying diseases also may play a role in this process including rheumatoid arthritis and psoriasis. Although these risk factors were associated with our patient, his history of chronic immunosuppressive therapy for treatment of his psoriasis and inflammatory joint disease likely played a role in the pathogenesis of the tumor and should be an important point of discussion with any patient requiring this type of long-term management for disease control. Our unique clinical case highlights a patient with substantial comorbidities who developed metastatic MCC and achieved complete clinical and radiologic remission after treatment with surgery and chemotherapy.
- Timmer FC, Klop WM, Relyveld GN, et al. Merkel cell carcinoma of the head and neck: emphasizing the risk of undertreatment [published online March 11, 2015]. Eur Arch Otorhinolaryngol. 2016;273:1243-1252.
- Açıkalın A, Paydas¸ S, Güleç ÜK, et al. A unique case of Merkel cell carcinoma with ovarian metastasis [published online December 1, 2014]. Balkan Med J. 2014;31:356-359.
- Samimi M, Gardair C, Nicol JT, et al. Merkel cell polyomavirus in Merkel cell carcinoma: clinical and therapeutic perspectives [published online Dec 31, 2014]. Semin Oncol. 2015;42:347-358.
- Grandhaye M, Teixeira PG, Henrot P, et al. Focus on Merkel cell carcinoma: diagnosis and staging [published online January 30, 2015]. Skeletal Radiol. 2015;44:777-786.
- Chatzinasiou F, Papadavid E, Korkolopoulou P, et al. An unusual case of diffuse Merkel cell carcinoma successfully treated with low dose radiotherapy [published online May 14, 2015]. Dermatol Ther. 2015;28:282-286.
- Pang C, Sharma D, Sankar T. Spontaneous regression of Merkel cell carcinoma: a case report and review of the literature [published online November 13, 2014]. Int J Surg Case Rep. 2015;7C:104-108.
- Kitamura N, Tomita R, Yamamoto M, et al. Complete remission of Merkel cell carcinoma on the upper lip treated with radiation monotherapy and a literature review of Japanese cases. World J Surg Oncol. 2015;13:152.
- Lanoy E, Engels EA. Skin cancers associated with autoimmune conditions among elderly adults [published online June 15, 2010]. Br J Cancer. 2010;103:112-114.
- Mertz KD, Junt T, Schmid M, et al. Inflammatory monocytes are a reservoir for Merkel cell polyomavirus [published online December 17, 2009]. J Invest Dermatol. 2009;130:1146-1151.
- Fujimoto N, Nakanishi G, Kabuto M, et al. Merkel cell carcinoma showing regression after biopsy: evaluation of programmed cell death 1-positive cells [published online February 24, 2015]. J Dermatol. 2015;42:496-499.
To the Editor:
A 69-year-old white man presented with a skin lesion on the back of 1 to 2 weeks’ duration. The patient stated he was unaware of it, but his wife had recently noticed the new spot. He denied any bleeding, pain, pruritus, or other associated symptoms with the lesion. He also denied any prior treatment to the area. The patient’s medical history was remarkable for severe psoriasis involving more than 80% body surface area, psoriatic arthritis, rheumatoid arthritis, ankylosing spondylitis, coronary artery disease, squamous cell carcinoma, and actinic keratoses. He had been on multiple treatment regimens over the last 20 years for control of psoriasis including topical corticosteroids, psoralen plus UVA and UVB phototherapy, gold injections, acitretin, prednisone, efalizumab, ustekinumab, and alefacept upon evaluation of this new skin lesion. Utilization of immunosuppressive agents also provided an additional benefit of controlling the patient’s inflammatory arthritic disease.
On physical examination a 0.6×0.7-cm, pink to erythematous, pearly papule with superficial telangiectases was noted on the right side of the dorsal thorax (Figure 1). Multiple well-demarcated erythematous plaques with silvery scale and areas of secondary excoriation were noted on the trunk and both legs consistent with the patient’s history of psoriasis.
A shave biopsy was performed on the skin lesion on the right side of the dorsal thorax with a suspected clinical diagnosis of basal cell carcinoma. Two weeks later the patient returned for a discussion of the pathology report, which revealed nodules of basaloid cells with tightly packed vesicular nuclei and scant cytoplasm in sheets within the superficial dermis, as well as areas of nuclear molding, numerous mitotic figures, and areas of focal necrosis (Figure 2). In addition, immunostaining was positive for cytokeratin (CK) 20 antibodies with a characteristic paranuclear dot uptake of the antibody. These findings were consistent with a diagnosis of Merkel cell carcinoma (MCC). At that time, alefacept was discontinued and he was referred to a tertiary referral center for further evaluation and treatment.
The patient subsequently underwent wide excision with 1-cm margins of the MCC, with intraoperative lymphatic mapping/sentinel lymph node biopsy (SLNB) of the right axillary nodal basin 1 month later, which he tolerated well without any associated complications. Further histopathologic examination revealed the deep, medial, and lateral surgical margins to be negative of residual neoplasm. However, one sentinel lymph node indicated positivity for micrometastatic MCC, consistent with stage IIIA disease progression.
He underwent a second procedure the following month for complete right axillary lymph node dissection. Histopathologic examination of the right axillary contents included 28 lymph nodes, which were negative for carcinoma. He continued to do well without any signs of clinical recurrence or distant metastasis at subsequent follow-up visits.
Approximately 2.5 years after the second procedure, the patient began to develop right upper quadrant abdominal pain of an unclear etiology. Computed tomography of the abdomen and pelvis was performed, revealing areas of calcification and findings consistent with malignant lymphadenopathy. Multiple hepatic lesions also were noted including a 9-cm lesion in the posterior right hepatic lobe. Computed tomography–guided biopsy of the liver lesion was performed and the findings were consistent with metastatic MCC, indicating progression to stage IV disease.
The patient was subsequently started on combination chemotherapeutic treatment with carboplatin and VP-16, with a planned treatment course of 4 to 6 cycles. He was able to complete a total of 6 cycles over a 4-month period, tolerating the treatment regimen fairly well. Follow-up positron emission tomography–computed tomography was within normal limits with no evidence of any hypermetabolic activity noted, indicating a complete radiographic remission of MCC. He was seen approximately 1 month after completion of treatment for clinical follow-up and monthly thereafter.
While on chemotherapy, the patient experienced a notable improvement in the psoriasis and psoriatic joint disease. Upon completion of chemotherapy, he was restarted on the same treatment plan that was utilized prior to surgery including topical corticosteroids, calcitriol, intramuscular steroid injections, and UVB phototherapy, which provided substantial control of psoriasis and arthritic joint disease. The patient later died, likely due to his multiple comorbidities.
Merkel cells are slow-responding mechanoreceptors located within the basal layer of the epidermis and are the source of a rare aggressive cutaneous malignancy.1 Merkel cell carcinoma was first noted in 1972 and termed trabecular carcinoma of the skin, and it accounts for less than 1% of all nonmelanoma skin cancer.2,3 This primary neuroendocrine carcinoma has remarkable metastatic potential (34%–75%) and can invade regional lymph nodes, as well as distant metastasis most commonly to the liver, lungs, bones, and brain.2 Approximately 25% of patients present with palpable lymphadenopathy and 5% with distant metastasis at the time of diagnosis. This frequency of metastasis at diagnosis as well as the recurrence after treatment contributes to the poor prognosis of MCC. Local recurrence rates have been reported at 25% with lymph node involvement in 52% and metastasis in 34%, with most recurrences occurring within 2 years of diagnosis. Patient mortality is dependent on the aggressiveness of the tumor, with 5-year survival rates of 83.3% without lymph node involvement, 58.3% with lymph node involvement, and 31.3% in those with metastatic disease.4
The tumor classically presents as a red to violaceous, painless nodule with a smooth shiny surface most often on the head and neck region.4-6 Approximately 50% of MCC cases present in the head and neck region, 32% to 38% on the extremities, and 12% to 14% on the trunk.1 This nonspecific presentation may lead to diagnostic uncertainty and a consequent delay in treatment. Definitive diagnosis of MCC is achieved with a skin biopsy and allows for distinction from other clinically similar–appearing neoplasms. Merkel cell carcinoma presents histologically as small round basophilic cells penetrating through the dermis in 3 histologic patterns: the trabecular, intermediate (80% of cases), and small cell type.5 It may be differentiated immunohistochemically from other neoplasms, as it displays CK20 positivity (showing paranuclear dotlike depositions in the cytoplasm or cell membrane) and is negative for CK7. Chromagranin and synaptophysin positivity also may provide further histologic confirmation. In addition, absence of peripheral palisading, retraction artifact, and a fibromyxoid stroma allow for distinction from cutaneous basal cell carcinoma, which may display these features histologically. Other immunohistochemical markers that may be of value include thyroid transcription factor 1, which is typically positive in cutaneous metastasis of neuroendocrine carcinoma of the lung; S-100 and human melanoma black 45, which are positive in melanoma; and leukocyte common antigen (CD45), which can be positive in lymphoma. These stains are classically negative in MCC.3
Merkel cell carcinoma is commonly associated with the presence of Merkel cell polyomavirus (MCPyV) in tumor specimens, with a prevalence of 70% to 80% in all cases. Merkel cell polyomavirus is a class 2A carcinogen (ie, a probable carcinogen to humans) and is classified among a group of viruses that encode T antigens (ie, an antigen coded by a viral genome associated with transformation of infected cells by tumor viruses), which can lead to initiation of tumorigenesis through interference with cellular tumor suppressing proteins such as p53.5 In addition, several risk factors have been associated with the development of MCC including immunosuppression, older age (>50 years), and UV-exposed fair skin.7 One explanation for this phenomenon is the increase in MCPyV small T antigen transcripts induced by UV irradiation.5 In addition, as with other cancers induced by viruses, host immunity can impede tumor progression and development. Therefore, impairment of normal immune function likely creates a higher risk for MCC development and potential for a worse prognosis.3Although the exact incidence of MCC in immunosuppressed patients appears unclear, chronic immunosuppressive therapy may play a notable role in the pathogenesis of the tumor.3
Although each of these factors was observed in our patient, it also was possible that his associated comorbidities further contributed to disease presentation. In particular, rheumatoid arthritis has been shown to carry an increased risk for the development of MCC.8 In addition, inflammatory monocytes infected with MCPyV, as evidenced in a patient with a history of chronic psoriasis prior to diagnosis of MCC, also may contribute to the pathogenesis of MCC by traveling to inflammatory skin lesions, such as those seen in psoriasis, releasing MCPyV locally and infecting Merkel cells.9 Although MCPyV testing was never performed in our patient, it certainly would be prudent as well as further studies determining the correlation of MCC to these disease processes.
Although regression is rare, multiple cases have documented spontaneous regression of MCC after biopsy of these lesions.4,6,10 The exact mechanism is unclear, but apoptosis induced by T-cell immunity is suspected to play a role. Programmed cell death 1 protein (PD-1)–positive cells play a role. The PD-1 receptor is an inhibitory receptor expressed by T cells and in approximately half of tumor-infiltrating cells in MCC. It was found that in a regressed case of MCC there was a notably lower percentage of PD-1 positivity compared to cases with no apparent regression, suggesting that PD-1–positive cells suppress tumor immunity to MCC and that significant reduction in these cells may induce clinical regression.10 Additional investigation would be beneficial to examine the relationship of this phenomenon to tumor regression.
Initial evaluation of these patients should include a meticulous clinical examination with an emphasis on detection of cutaneous, lymph node, and distant metastasis. Due to the risk of metastatic potential, regional lymph node ultrasonography and computed tomography of the chest, abdomen, and pelvis typically are recommended at baseline. Other imaging modalities may be warranted based on clinical findings.3 Treatment modalities include various approaches, with surgical excision of the primary tumor with more than 1-cm margin to the fascial plane being the primary modality for uncomplicated cases.1,3,7 In addition, SLNB also should be performed at the time of the procedure. In the case of a positive SLNB or suspected regional lymph node involvement upon initial examination, radical regional lymph node dissection also is recommended.3 Although some authorities advocate postsurgical radiation therapy to minimize the risk of local recurrence, there does not appear to be a clear benefit in survival rate.3,5 However, radiation treatment as monotherapy has been advocated in certain instances, particularly in cases of unresectable tumors or patients who are poor surgical candidates.5,7 Cases of distant metastasis (stage IV disease) may include management with surgery, radiation, and/or chemotherapy. Although none of these modalities have consistently shown to improve survival, there appears to be up to a 60% response with chemotherapy in these patients.3
Because MCC tends to affect an older population, often with other notable comorbidities, important considerations involving a treatment plan include the cost, side effects, and convenience for patients. The combination of carboplatin and VP-16 (etoposide) was utilized and tolerated well in our patient, and it has been successful in achieving complete radiologic and clinical remission of his metastatic disease. This combination appears to prolong survival in patients with distant metastasis, as compared to those patients not receiving chemotherapy.1 Our patient has since died, but in these high-risk patients, close clinical monitoring is essential to help optimize their prognosis.
Merkel cell carcinoma is a rare aggressive cutaneous neoplasm that most commonly affects the elderly, immunosuppressed, and those with chronic UV sun damage. An association between the oncogenesis of MCC and infection with MCPyV has been documented, but other underlying diseases also may play a role in this process including rheumatoid arthritis and psoriasis. Although these risk factors were associated with our patient, his history of chronic immunosuppressive therapy for treatment of his psoriasis and inflammatory joint disease likely played a role in the pathogenesis of the tumor and should be an important point of discussion with any patient requiring this type of long-term management for disease control. Our unique clinical case highlights a patient with substantial comorbidities who developed metastatic MCC and achieved complete clinical and radiologic remission after treatment with surgery and chemotherapy.
To the Editor:
A 69-year-old white man presented with a skin lesion on the back of 1 to 2 weeks’ duration. The patient stated he was unaware of it, but his wife had recently noticed the new spot. He denied any bleeding, pain, pruritus, or other associated symptoms with the lesion. He also denied any prior treatment to the area. The patient’s medical history was remarkable for severe psoriasis involving more than 80% body surface area, psoriatic arthritis, rheumatoid arthritis, ankylosing spondylitis, coronary artery disease, squamous cell carcinoma, and actinic keratoses. He had been on multiple treatment regimens over the last 20 years for control of psoriasis including topical corticosteroids, psoralen plus UVA and UVB phototherapy, gold injections, acitretin, prednisone, efalizumab, ustekinumab, and alefacept upon evaluation of this new skin lesion. Utilization of immunosuppressive agents also provided an additional benefit of controlling the patient’s inflammatory arthritic disease.
On physical examination a 0.6×0.7-cm, pink to erythematous, pearly papule with superficial telangiectases was noted on the right side of the dorsal thorax (Figure 1). Multiple well-demarcated erythematous plaques with silvery scale and areas of secondary excoriation were noted on the trunk and both legs consistent with the patient’s history of psoriasis.
A shave biopsy was performed on the skin lesion on the right side of the dorsal thorax with a suspected clinical diagnosis of basal cell carcinoma. Two weeks later the patient returned for a discussion of the pathology report, which revealed nodules of basaloid cells with tightly packed vesicular nuclei and scant cytoplasm in sheets within the superficial dermis, as well as areas of nuclear molding, numerous mitotic figures, and areas of focal necrosis (Figure 2). In addition, immunostaining was positive for cytokeratin (CK) 20 antibodies with a characteristic paranuclear dot uptake of the antibody. These findings were consistent with a diagnosis of Merkel cell carcinoma (MCC). At that time, alefacept was discontinued and he was referred to a tertiary referral center for further evaluation and treatment.
The patient subsequently underwent wide excision with 1-cm margins of the MCC, with intraoperative lymphatic mapping/sentinel lymph node biopsy (SLNB) of the right axillary nodal basin 1 month later, which he tolerated well without any associated complications. Further histopathologic examination revealed the deep, medial, and lateral surgical margins to be negative of residual neoplasm. However, one sentinel lymph node indicated positivity for micrometastatic MCC, consistent with stage IIIA disease progression.
He underwent a second procedure the following month for complete right axillary lymph node dissection. Histopathologic examination of the right axillary contents included 28 lymph nodes, which were negative for carcinoma. He continued to do well without any signs of clinical recurrence or distant metastasis at subsequent follow-up visits.
Approximately 2.5 years after the second procedure, the patient began to develop right upper quadrant abdominal pain of an unclear etiology. Computed tomography of the abdomen and pelvis was performed, revealing areas of calcification and findings consistent with malignant lymphadenopathy. Multiple hepatic lesions also were noted including a 9-cm lesion in the posterior right hepatic lobe. Computed tomography–guided biopsy of the liver lesion was performed and the findings were consistent with metastatic MCC, indicating progression to stage IV disease.
The patient was subsequently started on combination chemotherapeutic treatment with carboplatin and VP-16, with a planned treatment course of 4 to 6 cycles. He was able to complete a total of 6 cycles over a 4-month period, tolerating the treatment regimen fairly well. Follow-up positron emission tomography–computed tomography was within normal limits with no evidence of any hypermetabolic activity noted, indicating a complete radiographic remission of MCC. He was seen approximately 1 month after completion of treatment for clinical follow-up and monthly thereafter.
While on chemotherapy, the patient experienced a notable improvement in the psoriasis and psoriatic joint disease. Upon completion of chemotherapy, he was restarted on the same treatment plan that was utilized prior to surgery including topical corticosteroids, calcitriol, intramuscular steroid injections, and UVB phototherapy, which provided substantial control of psoriasis and arthritic joint disease. The patient later died, likely due to his multiple comorbidities.
Merkel cells are slow-responding mechanoreceptors located within the basal layer of the epidermis and are the source of a rare aggressive cutaneous malignancy.1 Merkel cell carcinoma was first noted in 1972 and termed trabecular carcinoma of the skin, and it accounts for less than 1% of all nonmelanoma skin cancer.2,3 This primary neuroendocrine carcinoma has remarkable metastatic potential (34%–75%) and can invade regional lymph nodes, as well as distant metastasis most commonly to the liver, lungs, bones, and brain.2 Approximately 25% of patients present with palpable lymphadenopathy and 5% with distant metastasis at the time of diagnosis. This frequency of metastasis at diagnosis as well as the recurrence after treatment contributes to the poor prognosis of MCC. Local recurrence rates have been reported at 25% with lymph node involvement in 52% and metastasis in 34%, with most recurrences occurring within 2 years of diagnosis. Patient mortality is dependent on the aggressiveness of the tumor, with 5-year survival rates of 83.3% without lymph node involvement, 58.3% with lymph node involvement, and 31.3% in those with metastatic disease.4
The tumor classically presents as a red to violaceous, painless nodule with a smooth shiny surface most often on the head and neck region.4-6 Approximately 50% of MCC cases present in the head and neck region, 32% to 38% on the extremities, and 12% to 14% on the trunk.1 This nonspecific presentation may lead to diagnostic uncertainty and a consequent delay in treatment. Definitive diagnosis of MCC is achieved with a skin biopsy and allows for distinction from other clinically similar–appearing neoplasms. Merkel cell carcinoma presents histologically as small round basophilic cells penetrating through the dermis in 3 histologic patterns: the trabecular, intermediate (80% of cases), and small cell type.5 It may be differentiated immunohistochemically from other neoplasms, as it displays CK20 positivity (showing paranuclear dotlike depositions in the cytoplasm or cell membrane) and is negative for CK7. Chromagranin and synaptophysin positivity also may provide further histologic confirmation. In addition, absence of peripheral palisading, retraction artifact, and a fibromyxoid stroma allow for distinction from cutaneous basal cell carcinoma, which may display these features histologically. Other immunohistochemical markers that may be of value include thyroid transcription factor 1, which is typically positive in cutaneous metastasis of neuroendocrine carcinoma of the lung; S-100 and human melanoma black 45, which are positive in melanoma; and leukocyte common antigen (CD45), which can be positive in lymphoma. These stains are classically negative in MCC.3
Merkel cell carcinoma is commonly associated with the presence of Merkel cell polyomavirus (MCPyV) in tumor specimens, with a prevalence of 70% to 80% in all cases. Merkel cell polyomavirus is a class 2A carcinogen (ie, a probable carcinogen to humans) and is classified among a group of viruses that encode T antigens (ie, an antigen coded by a viral genome associated with transformation of infected cells by tumor viruses), which can lead to initiation of tumorigenesis through interference with cellular tumor suppressing proteins such as p53.5 In addition, several risk factors have been associated with the development of MCC including immunosuppression, older age (>50 years), and UV-exposed fair skin.7 One explanation for this phenomenon is the increase in MCPyV small T antigen transcripts induced by UV irradiation.5 In addition, as with other cancers induced by viruses, host immunity can impede tumor progression and development. Therefore, impairment of normal immune function likely creates a higher risk for MCC development and potential for a worse prognosis.3Although the exact incidence of MCC in immunosuppressed patients appears unclear, chronic immunosuppressive therapy may play a notable role in the pathogenesis of the tumor.3
Although each of these factors was observed in our patient, it also was possible that his associated comorbidities further contributed to disease presentation. In particular, rheumatoid arthritis has been shown to carry an increased risk for the development of MCC.8 In addition, inflammatory monocytes infected with MCPyV, as evidenced in a patient with a history of chronic psoriasis prior to diagnosis of MCC, also may contribute to the pathogenesis of MCC by traveling to inflammatory skin lesions, such as those seen in psoriasis, releasing MCPyV locally and infecting Merkel cells.9 Although MCPyV testing was never performed in our patient, it certainly would be prudent as well as further studies determining the correlation of MCC to these disease processes.
Although regression is rare, multiple cases have documented spontaneous regression of MCC after biopsy of these lesions.4,6,10 The exact mechanism is unclear, but apoptosis induced by T-cell immunity is suspected to play a role. Programmed cell death 1 protein (PD-1)–positive cells play a role. The PD-1 receptor is an inhibitory receptor expressed by T cells and in approximately half of tumor-infiltrating cells in MCC. It was found that in a regressed case of MCC there was a notably lower percentage of PD-1 positivity compared to cases with no apparent regression, suggesting that PD-1–positive cells suppress tumor immunity to MCC and that significant reduction in these cells may induce clinical regression.10 Additional investigation would be beneficial to examine the relationship of this phenomenon to tumor regression.
Initial evaluation of these patients should include a meticulous clinical examination with an emphasis on detection of cutaneous, lymph node, and distant metastasis. Due to the risk of metastatic potential, regional lymph node ultrasonography and computed tomography of the chest, abdomen, and pelvis typically are recommended at baseline. Other imaging modalities may be warranted based on clinical findings.3 Treatment modalities include various approaches, with surgical excision of the primary tumor with more than 1-cm margin to the fascial plane being the primary modality for uncomplicated cases.1,3,7 In addition, SLNB also should be performed at the time of the procedure. In the case of a positive SLNB or suspected regional lymph node involvement upon initial examination, radical regional lymph node dissection also is recommended.3 Although some authorities advocate postsurgical radiation therapy to minimize the risk of local recurrence, there does not appear to be a clear benefit in survival rate.3,5 However, radiation treatment as monotherapy has been advocated in certain instances, particularly in cases of unresectable tumors or patients who are poor surgical candidates.5,7 Cases of distant metastasis (stage IV disease) may include management with surgery, radiation, and/or chemotherapy. Although none of these modalities have consistently shown to improve survival, there appears to be up to a 60% response with chemotherapy in these patients.3
Because MCC tends to affect an older population, often with other notable comorbidities, important considerations involving a treatment plan include the cost, side effects, and convenience for patients. The combination of carboplatin and VP-16 (etoposide) was utilized and tolerated well in our patient, and it has been successful in achieving complete radiologic and clinical remission of his metastatic disease. This combination appears to prolong survival in patients with distant metastasis, as compared to those patients not receiving chemotherapy.1 Our patient has since died, but in these high-risk patients, close clinical monitoring is essential to help optimize their prognosis.
Merkel cell carcinoma is a rare aggressive cutaneous neoplasm that most commonly affects the elderly, immunosuppressed, and those with chronic UV sun damage. An association between the oncogenesis of MCC and infection with MCPyV has been documented, but other underlying diseases also may play a role in this process including rheumatoid arthritis and psoriasis. Although these risk factors were associated with our patient, his history of chronic immunosuppressive therapy for treatment of his psoriasis and inflammatory joint disease likely played a role in the pathogenesis of the tumor and should be an important point of discussion with any patient requiring this type of long-term management for disease control. Our unique clinical case highlights a patient with substantial comorbidities who developed metastatic MCC and achieved complete clinical and radiologic remission after treatment with surgery and chemotherapy.
- Timmer FC, Klop WM, Relyveld GN, et al. Merkel cell carcinoma of the head and neck: emphasizing the risk of undertreatment [published online March 11, 2015]. Eur Arch Otorhinolaryngol. 2016;273:1243-1252.
- Açıkalın A, Paydas¸ S, Güleç ÜK, et al. A unique case of Merkel cell carcinoma with ovarian metastasis [published online December 1, 2014]. Balkan Med J. 2014;31:356-359.
- Samimi M, Gardair C, Nicol JT, et al. Merkel cell polyomavirus in Merkel cell carcinoma: clinical and therapeutic perspectives [published online Dec 31, 2014]. Semin Oncol. 2015;42:347-358.
- Grandhaye M, Teixeira PG, Henrot P, et al. Focus on Merkel cell carcinoma: diagnosis and staging [published online January 30, 2015]. Skeletal Radiol. 2015;44:777-786.
- Chatzinasiou F, Papadavid E, Korkolopoulou P, et al. An unusual case of diffuse Merkel cell carcinoma successfully treated with low dose radiotherapy [published online May 14, 2015]. Dermatol Ther. 2015;28:282-286.
- Pang C, Sharma D, Sankar T. Spontaneous regression of Merkel cell carcinoma: a case report and review of the literature [published online November 13, 2014]. Int J Surg Case Rep. 2015;7C:104-108.
- Kitamura N, Tomita R, Yamamoto M, et al. Complete remission of Merkel cell carcinoma on the upper lip treated with radiation monotherapy and a literature review of Japanese cases. World J Surg Oncol. 2015;13:152.
- Lanoy E, Engels EA. Skin cancers associated with autoimmune conditions among elderly adults [published online June 15, 2010]. Br J Cancer. 2010;103:112-114.
- Mertz KD, Junt T, Schmid M, et al. Inflammatory monocytes are a reservoir for Merkel cell polyomavirus [published online December 17, 2009]. J Invest Dermatol. 2009;130:1146-1151.
- Fujimoto N, Nakanishi G, Kabuto M, et al. Merkel cell carcinoma showing regression after biopsy: evaluation of programmed cell death 1-positive cells [published online February 24, 2015]. J Dermatol. 2015;42:496-499.
- Timmer FC, Klop WM, Relyveld GN, et al. Merkel cell carcinoma of the head and neck: emphasizing the risk of undertreatment [published online March 11, 2015]. Eur Arch Otorhinolaryngol. 2016;273:1243-1252.
- Açıkalın A, Paydas¸ S, Güleç ÜK, et al. A unique case of Merkel cell carcinoma with ovarian metastasis [published online December 1, 2014]. Balkan Med J. 2014;31:356-359.
- Samimi M, Gardair C, Nicol JT, et al. Merkel cell polyomavirus in Merkel cell carcinoma: clinical and therapeutic perspectives [published online Dec 31, 2014]. Semin Oncol. 2015;42:347-358.
- Grandhaye M, Teixeira PG, Henrot P, et al. Focus on Merkel cell carcinoma: diagnosis and staging [published online January 30, 2015]. Skeletal Radiol. 2015;44:777-786.
- Chatzinasiou F, Papadavid E, Korkolopoulou P, et al. An unusual case of diffuse Merkel cell carcinoma successfully treated with low dose radiotherapy [published online May 14, 2015]. Dermatol Ther. 2015;28:282-286.
- Pang C, Sharma D, Sankar T. Spontaneous regression of Merkel cell carcinoma: a case report and review of the literature [published online November 13, 2014]. Int J Surg Case Rep. 2015;7C:104-108.
- Kitamura N, Tomita R, Yamamoto M, et al. Complete remission of Merkel cell carcinoma on the upper lip treated with radiation monotherapy and a literature review of Japanese cases. World J Surg Oncol. 2015;13:152.
- Lanoy E, Engels EA. Skin cancers associated with autoimmune conditions among elderly adults [published online June 15, 2010]. Br J Cancer. 2010;103:112-114.
- Mertz KD, Junt T, Schmid M, et al. Inflammatory monocytes are a reservoir for Merkel cell polyomavirus [published online December 17, 2009]. J Invest Dermatol. 2009;130:1146-1151.
- Fujimoto N, Nakanishi G, Kabuto M, et al. Merkel cell carcinoma showing regression after biopsy: evaluation of programmed cell death 1-positive cells [published online February 24, 2015]. J Dermatol. 2015;42:496-499.
Practice Points
- Merkel cell carcinoma (MCC) has remarkable metastatic potential.
- Initial evaluation of patients with MCC should include clinical examination to detect cutaneous, lymph node, and distant metastasis.
- Risk factors associated with the development of MCC include immunosuppression, older age, and UV-exposed fair skin.
Best practices address latest trends in PDT, skin cancer treatment
MIAMI – Pearls for providers of photodynamic therapy (PDT) include tips on skin preparation, eye protection, and use of three new codes to maximize reimbursement. Also trending in medical dermatology are best practices for intralesional injections of 5-FU to treat the often challenging isomorphic squamous cell carcinomas (SCCs) or keratoacanthomas on the lower leg, as well as use of neoadjuvant hedgehog inhibitors to shrink large skin cancer lesions, according to Glenn David Goldman, MD.
“This talk is about what you can do medically as a dermatologic surgeon,” Dr. Goldman said at the Orlando Dermatology Aesthetic and Clinical Conference.
Use new billing codes for photodynamic therapy
There are now three new PDT billing codes. “Make sure your coders are using these properly. They are active now, and if you don’t use them, you won’t get paid properly,” said Dr. Goldman, professor and medical director of dermatology at the University of Vermont, Burlington. Specifically, 96567 is for standard PDT applied by staff; 96573 is for PDT applied by a physician; and 96574 is for PDT and curettage performed by a physician.
“Be involved, don’t delegate,” Dr. Goldman added. “If you do, you will get paid half as much as you used to, which means you will lose money on every single patient you treat.”
What type of PDT physicians choose to use in their practice remains controversial. “Do you do short-contact PDT, do you do daylight PDT? We’ve gone back and forth in our practice,” Dr. Goldman said. “I’m not impressed with daylight PDT. I know this is at odds with some of the people here, but at least in Vermont, it doesn’t work very well.”
The way PDT was described in the original trials (a photosensitizer applied in the office followed by PDT) “works the best, with one caveat,” Dr. Goldman said. The caveat is that dermatologists should aim for a PDT clearance that approaches the efficacy of 5-fluorouracil (5-FU). “If you can get to that – which is difficult by the way – I think your patients will really appreciate this.”
An additional PDT pearl Dr. Goldman shared involves skin preparation: the use of acetone to defat the skin, even in patients with very thick lesions. Apply acetone with gauze to the site for 5 minutes and “all of that hyperkeratosis just wipes away,” curette off any residual hyperkeratosis – and consider a ring anesthetic block to control pain for the patient with severe disease, he advised.
Another tip is to forgo the goggles that come with most PDT kits. Instead, purchase smaller, disposable laser eye shields for PDT patients, Dr. Goldman said. “They work better. You can get closer to the eye … and they are more comfortable for the patient.”
Dr. Goldman’s practice is providing more PDT and much less 5-FU for patient convenience. “I believe if someone is willing to go through 3 weeks of 5-FU or 12-16 weeks of imiquimod, they get the best results. However, most people don’t want to do that if they can sit in front of a light for 15 minutes.”
Consider intralesional injections for SCCs and KAs on the legs
An ongoing challenge in medical dermatology is preventing rapid recurrence of SCCs and/or keratoacanthomas (KAs) near sites of previous excision on the legs. “We all see this quite a bit. Often you get lesions on the leg, you cut them out, and they come right back” close to the excision site, Dr. Goldman said.
He does not recommend methotrexate injections for these lesions. “Methotrexate does not work. It doesn’t hurt, but I’ve injected methotrexate into squamous cell carcinomas many times and they’ve never gone away.” In contrast, 5-FU “works incredibly well. They go away, I’ve had tremendous success. This has changed the way we treat these lesions.” 5-FU is inexpensive and can be obtained from oncology pharmacies. One caveat is 5-FU injections can be painful and patients require anesthesia prior to injection.
Using a 25-gauge or 27-gauge needle, Dr. Goldman injects 5-FU “exactly as I would a hypertrophic scar. I inject a squamous cell carcinoma carefully and ‘expand’ the tumor.” He typically injects a lesion every 2 weeks until it resolves completely, which typically takes two or three sessions.
“I want to emphasize that that’s really true about intralesional 5-FU for those KAs and scars on the legs,” said session moderator James Spencer, MD, a dermatologist in private practice in St. Petersburg, Florida. “Otherwise, you’re just chasing your tail trying to cut them out. You’ll do much better with the intralesional 5-FU; it’s easy to get, it’s affordable, it comes as 50 mg/mL … just keep it in the office.”
A recommended role for hedgehog inhibitors
Hedgehog inhibitors work best as neoadjuvant therapy to shrink large skin cancer tumors prior to excision, Dr. Goldman said. “Hedgehog inhibitors don’t cure anything … except for rare cases of small basal cell carcinomas.” For most lesions, however, the strategy is not curative.
“I don’t believe in hedgehog inhibitors for things that are readily resectable. We use them to shrink things down,” he added.
Dr. Goldman recommended treating patients with neoadjuvant hedgehog inhibitors to achieve the maximum tumor shrinking effect. Adverse effects tend to develop slowly over time, typically after a 6-week “grace period.” Nighttime leg muscle cramps, loss of taste, hair loss, and weight loss can occur. Also, electrolyte imbalances can occur, particularly in older patients with renal clearance issues. When the patient can no longer reasonably tolerate the adverse effects, which is usually the case, “then you do the surgery,” Dr. Goldman said.
“The benefits of neoadjuvant hedgehog inhibitors include predictable shrinkage of tumors,” and manufacturers have been helpful with financial issues, he noted.
Dr. Goldman had no relevant financial disclosures. Dr. Spencer has served on the speakers bureau for Genentech and Leo Pharma.
MIAMI – Pearls for providers of photodynamic therapy (PDT) include tips on skin preparation, eye protection, and use of three new codes to maximize reimbursement. Also trending in medical dermatology are best practices for intralesional injections of 5-FU to treat the often challenging isomorphic squamous cell carcinomas (SCCs) or keratoacanthomas on the lower leg, as well as use of neoadjuvant hedgehog inhibitors to shrink large skin cancer lesions, according to Glenn David Goldman, MD.
“This talk is about what you can do medically as a dermatologic surgeon,” Dr. Goldman said at the Orlando Dermatology Aesthetic and Clinical Conference.
Use new billing codes for photodynamic therapy
There are now three new PDT billing codes. “Make sure your coders are using these properly. They are active now, and if you don’t use them, you won’t get paid properly,” said Dr. Goldman, professor and medical director of dermatology at the University of Vermont, Burlington. Specifically, 96567 is for standard PDT applied by staff; 96573 is for PDT applied by a physician; and 96574 is for PDT and curettage performed by a physician.
“Be involved, don’t delegate,” Dr. Goldman added. “If you do, you will get paid half as much as you used to, which means you will lose money on every single patient you treat.”
What type of PDT physicians choose to use in their practice remains controversial. “Do you do short-contact PDT, do you do daylight PDT? We’ve gone back and forth in our practice,” Dr. Goldman said. “I’m not impressed with daylight PDT. I know this is at odds with some of the people here, but at least in Vermont, it doesn’t work very well.”
The way PDT was described in the original trials (a photosensitizer applied in the office followed by PDT) “works the best, with one caveat,” Dr. Goldman said. The caveat is that dermatologists should aim for a PDT clearance that approaches the efficacy of 5-fluorouracil (5-FU). “If you can get to that – which is difficult by the way – I think your patients will really appreciate this.”
An additional PDT pearl Dr. Goldman shared involves skin preparation: the use of acetone to defat the skin, even in patients with very thick lesions. Apply acetone with gauze to the site for 5 minutes and “all of that hyperkeratosis just wipes away,” curette off any residual hyperkeratosis – and consider a ring anesthetic block to control pain for the patient with severe disease, he advised.
Another tip is to forgo the goggles that come with most PDT kits. Instead, purchase smaller, disposable laser eye shields for PDT patients, Dr. Goldman said. “They work better. You can get closer to the eye … and they are more comfortable for the patient.”
Dr. Goldman’s practice is providing more PDT and much less 5-FU for patient convenience. “I believe if someone is willing to go through 3 weeks of 5-FU or 12-16 weeks of imiquimod, they get the best results. However, most people don’t want to do that if they can sit in front of a light for 15 minutes.”
Consider intralesional injections for SCCs and KAs on the legs
An ongoing challenge in medical dermatology is preventing rapid recurrence of SCCs and/or keratoacanthomas (KAs) near sites of previous excision on the legs. “We all see this quite a bit. Often you get lesions on the leg, you cut them out, and they come right back” close to the excision site, Dr. Goldman said.
He does not recommend methotrexate injections for these lesions. “Methotrexate does not work. It doesn’t hurt, but I’ve injected methotrexate into squamous cell carcinomas many times and they’ve never gone away.” In contrast, 5-FU “works incredibly well. They go away, I’ve had tremendous success. This has changed the way we treat these lesions.” 5-FU is inexpensive and can be obtained from oncology pharmacies. One caveat is 5-FU injections can be painful and patients require anesthesia prior to injection.
Using a 25-gauge or 27-gauge needle, Dr. Goldman injects 5-FU “exactly as I would a hypertrophic scar. I inject a squamous cell carcinoma carefully and ‘expand’ the tumor.” He typically injects a lesion every 2 weeks until it resolves completely, which typically takes two or three sessions.
“I want to emphasize that that’s really true about intralesional 5-FU for those KAs and scars on the legs,” said session moderator James Spencer, MD, a dermatologist in private practice in St. Petersburg, Florida. “Otherwise, you’re just chasing your tail trying to cut them out. You’ll do much better with the intralesional 5-FU; it’s easy to get, it’s affordable, it comes as 50 mg/mL … just keep it in the office.”
A recommended role for hedgehog inhibitors
Hedgehog inhibitors work best as neoadjuvant therapy to shrink large skin cancer tumors prior to excision, Dr. Goldman said. “Hedgehog inhibitors don’t cure anything … except for rare cases of small basal cell carcinomas.” For most lesions, however, the strategy is not curative.
“I don’t believe in hedgehog inhibitors for things that are readily resectable. We use them to shrink things down,” he added.
Dr. Goldman recommended treating patients with neoadjuvant hedgehog inhibitors to achieve the maximum tumor shrinking effect. Adverse effects tend to develop slowly over time, typically after a 6-week “grace period.” Nighttime leg muscle cramps, loss of taste, hair loss, and weight loss can occur. Also, electrolyte imbalances can occur, particularly in older patients with renal clearance issues. When the patient can no longer reasonably tolerate the adverse effects, which is usually the case, “then you do the surgery,” Dr. Goldman said.
“The benefits of neoadjuvant hedgehog inhibitors include predictable shrinkage of tumors,” and manufacturers have been helpful with financial issues, he noted.
Dr. Goldman had no relevant financial disclosures. Dr. Spencer has served on the speakers bureau for Genentech and Leo Pharma.
MIAMI – Pearls for providers of photodynamic therapy (PDT) include tips on skin preparation, eye protection, and use of three new codes to maximize reimbursement. Also trending in medical dermatology are best practices for intralesional injections of 5-FU to treat the often challenging isomorphic squamous cell carcinomas (SCCs) or keratoacanthomas on the lower leg, as well as use of neoadjuvant hedgehog inhibitors to shrink large skin cancer lesions, according to Glenn David Goldman, MD.
“This talk is about what you can do medically as a dermatologic surgeon,” Dr. Goldman said at the Orlando Dermatology Aesthetic and Clinical Conference.
Use new billing codes for photodynamic therapy
There are now three new PDT billing codes. “Make sure your coders are using these properly. They are active now, and if you don’t use them, you won’t get paid properly,” said Dr. Goldman, professor and medical director of dermatology at the University of Vermont, Burlington. Specifically, 96567 is for standard PDT applied by staff; 96573 is for PDT applied by a physician; and 96574 is for PDT and curettage performed by a physician.
“Be involved, don’t delegate,” Dr. Goldman added. “If you do, you will get paid half as much as you used to, which means you will lose money on every single patient you treat.”
What type of PDT physicians choose to use in their practice remains controversial. “Do you do short-contact PDT, do you do daylight PDT? We’ve gone back and forth in our practice,” Dr. Goldman said. “I’m not impressed with daylight PDT. I know this is at odds with some of the people here, but at least in Vermont, it doesn’t work very well.”
The way PDT was described in the original trials (a photosensitizer applied in the office followed by PDT) “works the best, with one caveat,” Dr. Goldman said. The caveat is that dermatologists should aim for a PDT clearance that approaches the efficacy of 5-fluorouracil (5-FU). “If you can get to that – which is difficult by the way – I think your patients will really appreciate this.”
An additional PDT pearl Dr. Goldman shared involves skin preparation: the use of acetone to defat the skin, even in patients with very thick lesions. Apply acetone with gauze to the site for 5 minutes and “all of that hyperkeratosis just wipes away,” curette off any residual hyperkeratosis – and consider a ring anesthetic block to control pain for the patient with severe disease, he advised.
Another tip is to forgo the goggles that come with most PDT kits. Instead, purchase smaller, disposable laser eye shields for PDT patients, Dr. Goldman said. “They work better. You can get closer to the eye … and they are more comfortable for the patient.”
Dr. Goldman’s practice is providing more PDT and much less 5-FU for patient convenience. “I believe if someone is willing to go through 3 weeks of 5-FU or 12-16 weeks of imiquimod, they get the best results. However, most people don’t want to do that if they can sit in front of a light for 15 minutes.”
Consider intralesional injections for SCCs and KAs on the legs
An ongoing challenge in medical dermatology is preventing rapid recurrence of SCCs and/or keratoacanthomas (KAs) near sites of previous excision on the legs. “We all see this quite a bit. Often you get lesions on the leg, you cut them out, and they come right back” close to the excision site, Dr. Goldman said.
He does not recommend methotrexate injections for these lesions. “Methotrexate does not work. It doesn’t hurt, but I’ve injected methotrexate into squamous cell carcinomas many times and they’ve never gone away.” In contrast, 5-FU “works incredibly well. They go away, I’ve had tremendous success. This has changed the way we treat these lesions.” 5-FU is inexpensive and can be obtained from oncology pharmacies. One caveat is 5-FU injections can be painful and patients require anesthesia prior to injection.
Using a 25-gauge or 27-gauge needle, Dr. Goldman injects 5-FU “exactly as I would a hypertrophic scar. I inject a squamous cell carcinoma carefully and ‘expand’ the tumor.” He typically injects a lesion every 2 weeks until it resolves completely, which typically takes two or three sessions.
“I want to emphasize that that’s really true about intralesional 5-FU for those KAs and scars on the legs,” said session moderator James Spencer, MD, a dermatologist in private practice in St. Petersburg, Florida. “Otherwise, you’re just chasing your tail trying to cut them out. You’ll do much better with the intralesional 5-FU; it’s easy to get, it’s affordable, it comes as 50 mg/mL … just keep it in the office.”
A recommended role for hedgehog inhibitors
Hedgehog inhibitors work best as neoadjuvant therapy to shrink large skin cancer tumors prior to excision, Dr. Goldman said. “Hedgehog inhibitors don’t cure anything … except for rare cases of small basal cell carcinomas.” For most lesions, however, the strategy is not curative.
“I don’t believe in hedgehog inhibitors for things that are readily resectable. We use them to shrink things down,” he added.
Dr. Goldman recommended treating patients with neoadjuvant hedgehog inhibitors to achieve the maximum tumor shrinking effect. Adverse effects tend to develop slowly over time, typically after a 6-week “grace period.” Nighttime leg muscle cramps, loss of taste, hair loss, and weight loss can occur. Also, electrolyte imbalances can occur, particularly in older patients with renal clearance issues. When the patient can no longer reasonably tolerate the adverse effects, which is usually the case, “then you do the surgery,” Dr. Goldman said.
“The benefits of neoadjuvant hedgehog inhibitors include predictable shrinkage of tumors,” and manufacturers have been helpful with financial issues, he noted.
Dr. Goldman had no relevant financial disclosures. Dr. Spencer has served on the speakers bureau for Genentech and Leo Pharma.
REPORTING FROM ODAC 2018
Study IDs predictors of nonmelanoma skin cancer in IBD
LAS VEGAS – , a large national analysis showed.
“Some studies have shown that patients with IBD may be at increased risk for nonmelanoma skin cancer (NMSC) because of the immunomodulators that they take for the management of their disease,” Zubair Khan, MD, said in an interview at the Crohn’s & Colitis Congress, a partnership of the Crohn’s & Colitis Foundation and the American Gastroenterological Association. “But these are mostly small, single-center studies.” In an effort to determine the epidemiology of NMSC in patients hospitalized with IBD, Dr. Khan, associate chief resident in the internal medicine department at the University of Toledo (Ohio) Medical Center and his associates analyzed the National Inpatient Sample (NIS) database for all subjects who had a primary or secondary discharge diagnosis of IBD during 2002-2014. Next, they used ICD-9 codes to identify the rate of NMSC in this population.

Compared with IBD patients without NMSC, most of the IBD patients with NMSC were males (54% vs. 42%; P less than 0.001), covered by Medicare (65% vs. 37%), were white (96% vs. 81%; P less than .001), lived in the Midwest or Western United States (27% and 26% vs. 22% and 17%), were admitted to urban teaching hospitals (57% vs. 51%; P less than .001), were discharged to skilled nursing facilities (16% vs. 10%; P less than .001), required home health care (17% vs. 11%), and were admitted electively (27% vs. 20%). The researchers observed no significant difference in mortality among IBD patients with and without NMSC (1.61% vs. 1.53%; P = .22).
Multivariate analysis revealed that the following factors were predictive of NMSC in IBD: comorbid diagnosis of rheumatoid arthritis, collagen vasculature diseases, male sex, and white race. “Patients with those risk factors should be made more aware of their risk for developing NMSC,” Dr. Khan said. “They shouldn’t be taken lightly.”
Dr. Khan reported having no financial disclosures.
*This story was updated on 3/26.
SOURCE: Khan Z et al. Crohn’s & Colitis Congress, Poster 209.
LAS VEGAS – , a large national analysis showed.
“Some studies have shown that patients with IBD may be at increased risk for nonmelanoma skin cancer (NMSC) because of the immunomodulators that they take for the management of their disease,” Zubair Khan, MD, said in an interview at the Crohn’s & Colitis Congress, a partnership of the Crohn’s & Colitis Foundation and the American Gastroenterological Association. “But these are mostly small, single-center studies.” In an effort to determine the epidemiology of NMSC in patients hospitalized with IBD, Dr. Khan, associate chief resident in the internal medicine department at the University of Toledo (Ohio) Medical Center and his associates analyzed the National Inpatient Sample (NIS) database for all subjects who had a primary or secondary discharge diagnosis of IBD during 2002-2014. Next, they used ICD-9 codes to identify the rate of NMSC in this population.
Compared with IBD patients without NMSC, most of the IBD patients with NMSC were males (54% vs. 42%; P less than 0.001), covered by Medicare (65% vs. 37%), were white (96% vs. 81%; P less than .001), lived in the Midwest or Western United States (27% and 26% vs. 22% and 17%), were admitted to urban teaching hospitals (57% vs. 51%; P less than .001), were discharged to skilled nursing facilities (16% vs. 10%; P less than .001), required home health care (17% vs. 11%), and were admitted electively (27% vs. 20%). The researchers observed no significant difference in mortality among IBD patients with and without NMSC (1.61% vs. 1.53%; P = .22).
Multivariate analysis revealed that the following factors were predictive of NMSC in IBD: comorbid diagnosis of rheumatoid arthritis, collagen vasculature diseases, male sex, and white race. “Patients with those risk factors should be made more aware of their risk for developing NMSC,” Dr. Khan said. “They shouldn’t be taken lightly.”
Dr. Khan reported having no financial disclosures.
*This story was updated on 3/26.
SOURCE: Khan Z et al. Crohn’s & Colitis Congress, Poster 209.
LAS VEGAS – , a large national analysis showed.
“Some studies have shown that patients with IBD may be at increased risk for nonmelanoma skin cancer (NMSC) because of the immunomodulators that they take for the management of their disease,” Zubair Khan, MD, said in an interview at the Crohn’s & Colitis Congress, a partnership of the Crohn’s & Colitis Foundation and the American Gastroenterological Association. “But these are mostly small, single-center studies.” In an effort to determine the epidemiology of NMSC in patients hospitalized with IBD, Dr. Khan, associate chief resident in the internal medicine department at the University of Toledo (Ohio) Medical Center and his associates analyzed the National Inpatient Sample (NIS) database for all subjects who had a primary or secondary discharge diagnosis of IBD during 2002-2014. Next, they used ICD-9 codes to identify the rate of NMSC in this population.
Compared with IBD patients without NMSC, most of the IBD patients with NMSC were males (54% vs. 42%; P less than 0.001), covered by Medicare (65% vs. 37%), were white (96% vs. 81%; P less than .001), lived in the Midwest or Western United States (27% and 26% vs. 22% and 17%), were admitted to urban teaching hospitals (57% vs. 51%; P less than .001), were discharged to skilled nursing facilities (16% vs. 10%; P less than .001), required home health care (17% vs. 11%), and were admitted electively (27% vs. 20%). The researchers observed no significant difference in mortality among IBD patients with and without NMSC (1.61% vs. 1.53%; P = .22).
Multivariate analysis revealed that the following factors were predictive of NMSC in IBD: comorbid diagnosis of rheumatoid arthritis, collagen vasculature diseases, male sex, and white race. “Patients with those risk factors should be made more aware of their risk for developing NMSC,” Dr. Khan said. “They shouldn’t be taken lightly.”
Dr. Khan reported having no financial disclosures.
*This story was updated on 3/26.
SOURCE: Khan Z et al. Crohn’s & Colitis Congress, Poster 209.
REPORTING FROM THE CROHN’S & COLITIS CONGRESS
Key clinical point: Many factors predict which IBD patients are at risk for developing nonmelanoma skin cancer.
Major finding: Compared with IBD patients without NMSC, most of the IBD patients with NMSC were males (54% vs. 42%; P less than .001) and white (96% vs. 81%; P less than .001).
Study details: An analysis of 22,620 patients who had a primary or secondary discharge diagnosis of IBD during 2002-2014.
Disclosures: Dr. Khan reported having no financial disclosures.
Source: Khan Z et al. Crohn’s & Colitis Congress, Poster 209.
AAD guidelines favor surgery for nonmelanoma skin cancers
, according to new practice guidelines issued by the American Academy of Dermatology.
Nonsurgical approaches such as cryotherapy, photodynamic therapy, and radiation may be considered for low-risk cancers if surgery is contraindicated, but these methods have lower cure rates, according to the guidelines. Christopher K. Bichakjian, MD, professor of dermatology, University of Michigan, Ann Arbor, and Murad Alam, MD, professor of dermatology, Northwestern University, Chicago, cochaired the work groups that developed the guidelines.

The guidelines for BCC and cSCC, published online in two separate papers (J Am Acad Dermatol. 2018 Jan. 10. doi: 10.1016/j.jaad.2017.10.006; J Am Acad Dermatol. 2018 Jan. 10. doi: 10.1016/j.jaad.2017.10.007), also discuss biopsy techniques, tumor staging, and prevention of recurrence of nonmelanoma skin cancers.
The most suitable stratification for localized BCC and cSCC is the framework provided by the National Comprehensive Cancer Network, the authors said in the guidelines.
For suspected BCC and cSCC, recommended biopsy techniques are punch biopsy, shave biopsy, and excisional biopsy. Biopsy technique is “contingent on the clinical characteristics of the suspected tumor, including morphology, expected histologic subtype and depth, natural history, and anatomic location; patient-specific factors, such as bleeding and wound healing diatheses; and patient preference and physician judgment,” the guidelines state. If the initial biopsy proves insufficient for diagnosis, a repeat biopsy may be considered.
For surgical treatment of BCC, curettage and electrodessication may be considered for low-risk tumors in nonterminal hair-bearing locations. Surgical excision with 4-mm clinical margins and histologic margin assessment is recommended for low-risk primary BCC. For high-risk BCC, Mohs micrographic surgery is recommended, the authors said.
Surgical options for cSCC also include curettage and electrodessication and standard excision for low-risk disease, and Mohs micrographic surgery for high-risk cSCC. In both BCC and cSCC, standard excision may be considered for high-risk tumors in some cases, but “strong caution is advised when selecting a treatment modality” for high-risk tumors “without complete margin assessment,” the guidelines state.

Nonsurgical therapies are generally not recommended as first-line treatment, especially in cSCC because of possible recurrence and metastasis. In cases where nonsurgical therapies are preferred, options may include cryosurgery, topical therapy, photodynamic therapy, radiation, or laser therapy, “with the understanding that the cure rate may be lower,” the authors wrote.
Patients with diagnosed nonmelanoma skin cancer should continue to undergo screening for new primary skin cancers (including BCC, cSCC, and melanoma) at least once per year, the guideline states. They should also be counseled on sun protection, tanning bed avoidance, and regular use of broad-spectrum sunscreen.
Although the new guidelines mainly “reaffirm common knowledge and current practice,” they offer a reminder of “alternative therapeutic or preventive options when insufficient evidence is available to support new therapies or previously dogmatic practice patterns,” the authors said.
These are the first guidelines of care for BCC and cSCC published by the AAD. Commonly used guidelines for the management of BCC and cSCC are published by the National Comprehensive Cancer Network, which are frequently referenced throughout the new AAD guidelines, Dr. Bichakjian said in an interview. While the aim of the cancer network is to develop multidisciplinary guidelines, reflected by the composition of the panel members, “AAD guidelines of care are established primarily by dermatologists for dermatologists,” he pointed out. “However, the work group recognizes that a variety of health care providers outside of dermatology take care of patients with BCC and cSCC, and acknowledges the importance of multidisciplinary care,” he added. “With these considerations in mind, reviewers from specialties outside of dermatology, including plastic surgery, otolaryngology/head and neck surgery, medical oncology, radiation oncology, and family medicine, were invited to critically review the current guidelines.”
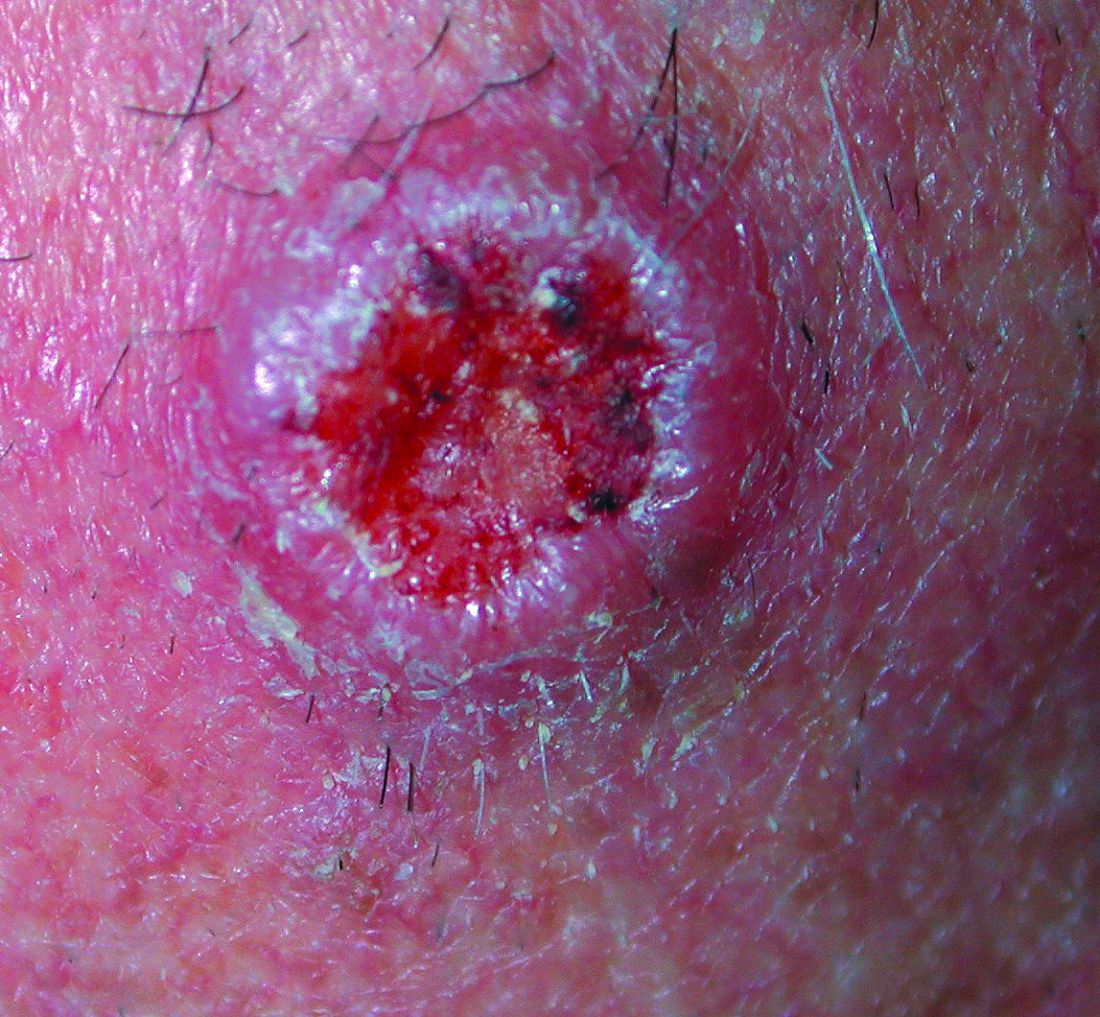
The guidelines do not cover the management of actinic keratosis and cSCC in situ, he said. “The work group acknowledges the importance of appropriate management of premalignant and in situ lesions in the prevention of their potential progression to cSCC. However, additional data to provide comprehensive evidence-based recommendations were deemed too extensive to include in the current guidelines and will need to addressed separately.”
In an interview, David J. Leffell, MD, who was not an author of the guidelines, said that the new guidelines do an effective job of “highlighting where valid outcomes data exist and areas where they do not” for a wide range of therapies. They also “attempt to standardize approaches to diagnosis and care of nonmelanoma skin cancer and in general are consistent with established practice patterns,” he added. “Those contemporary approaches have developed in largely empirical fashion over many decades, but bear clarification and reinforcement,” said Dr. Leffell, professor of dermatology and surgery and chief of the section of dermatologic surgery and cutaneous oncology at Yale University, New Haven, Conn.
The guidelines “thoroughly summarize evidence-based recommendations for the entire spectrum of disease management,” Daniel D. Bennett, MD, of the department of dermatology at the University of Wisconsin – Madison, said in an interview. “While surgery remains the mainstay of treatment for BCC and cutaneous SCC, these guidelines include excellent reviews of nonsurgical management options,” he said.
Dr. Bichakjian, who is also chief of the division of cutaneous surgery and oncology at the University of Michigan, had no relevant financial disclosures to report. Dr. Alam, who is also chief of cutaneous and aesthetic surgery in the department of dermatology at Northwestern, disclosed relationships with Amway, OptMed, and 3M. Dr. Bennett and Dr. Leffell had no relevant disclosures.
, according to new practice guidelines issued by the American Academy of Dermatology.
Nonsurgical approaches such as cryotherapy, photodynamic therapy, and radiation may be considered for low-risk cancers if surgery is contraindicated, but these methods have lower cure rates, according to the guidelines. Christopher K. Bichakjian, MD, professor of dermatology, University of Michigan, Ann Arbor, and Murad Alam, MD, professor of dermatology, Northwestern University, Chicago, cochaired the work groups that developed the guidelines.
The guidelines for BCC and cSCC, published online in two separate papers (J Am Acad Dermatol. 2018 Jan. 10. doi: 10.1016/j.jaad.2017.10.006; J Am Acad Dermatol. 2018 Jan. 10. doi: 10.1016/j.jaad.2017.10.007), also discuss biopsy techniques, tumor staging, and prevention of recurrence of nonmelanoma skin cancers.
The most suitable stratification for localized BCC and cSCC is the framework provided by the National Comprehensive Cancer Network, the authors said in the guidelines.
For suspected BCC and cSCC, recommended biopsy techniques are punch biopsy, shave biopsy, and excisional biopsy. Biopsy technique is “contingent on the clinical characteristics of the suspected tumor, including morphology, expected histologic subtype and depth, natural history, and anatomic location; patient-specific factors, such as bleeding and wound healing diatheses; and patient preference and physician judgment,” the guidelines state. If the initial biopsy proves insufficient for diagnosis, a repeat biopsy may be considered.
For surgical treatment of BCC, curettage and electrodessication may be considered for low-risk tumors in nonterminal hair-bearing locations. Surgical excision with 4-mm clinical margins and histologic margin assessment is recommended for low-risk primary BCC. For high-risk BCC, Mohs micrographic surgery is recommended, the authors said.
Surgical options for cSCC also include curettage and electrodessication and standard excision for low-risk disease, and Mohs micrographic surgery for high-risk cSCC. In both BCC and cSCC, standard excision may be considered for high-risk tumors in some cases, but “strong caution is advised when selecting a treatment modality” for high-risk tumors “without complete margin assessment,” the guidelines state.
Nonsurgical therapies are generally not recommended as first-line treatment, especially in cSCC because of possible recurrence and metastasis. In cases where nonsurgical therapies are preferred, options may include cryosurgery, topical therapy, photodynamic therapy, radiation, or laser therapy, “with the understanding that the cure rate may be lower,” the authors wrote.
Patients with diagnosed nonmelanoma skin cancer should continue to undergo screening for new primary skin cancers (including BCC, cSCC, and melanoma) at least once per year, the guideline states. They should also be counseled on sun protection, tanning bed avoidance, and regular use of broad-spectrum sunscreen.
Although the new guidelines mainly “reaffirm common knowledge and current practice,” they offer a reminder of “alternative therapeutic or preventive options when insufficient evidence is available to support new therapies or previously dogmatic practice patterns,” the authors said.
These are the first guidelines of care for BCC and cSCC published by the AAD. Commonly used guidelines for the management of BCC and cSCC are published by the National Comprehensive Cancer Network, which are frequently referenced throughout the new AAD guidelines, Dr. Bichakjian said in an interview. While the aim of the cancer network is to develop multidisciplinary guidelines, reflected by the composition of the panel members, “AAD guidelines of care are established primarily by dermatologists for dermatologists,” he pointed out. “However, the work group recognizes that a variety of health care providers outside of dermatology take care of patients with BCC and cSCC, and acknowledges the importance of multidisciplinary care,” he added. “With these considerations in mind, reviewers from specialties outside of dermatology, including plastic surgery, otolaryngology/head and neck surgery, medical oncology, radiation oncology, and family medicine, were invited to critically review the current guidelines.”
The guidelines do not cover the management of actinic keratosis and cSCC in situ, he said. “The work group acknowledges the importance of appropriate management of premalignant and in situ lesions in the prevention of their potential progression to cSCC. However, additional data to provide comprehensive evidence-based recommendations were deemed too extensive to include in the current guidelines and will need to addressed separately.”
In an interview, David J. Leffell, MD, who was not an author of the guidelines, said that the new guidelines do an effective job of “highlighting where valid outcomes data exist and areas where they do not” for a wide range of therapies. They also “attempt to standardize approaches to diagnosis and care of nonmelanoma skin cancer and in general are consistent with established practice patterns,” he added. “Those contemporary approaches have developed in largely empirical fashion over many decades, but bear clarification and reinforcement,” said Dr. Leffell, professor of dermatology and surgery and chief of the section of dermatologic surgery and cutaneous oncology at Yale University, New Haven, Conn.
The guidelines “thoroughly summarize evidence-based recommendations for the entire spectrum of disease management,” Daniel D. Bennett, MD, of the department of dermatology at the University of Wisconsin – Madison, said in an interview. “While surgery remains the mainstay of treatment for BCC and cutaneous SCC, these guidelines include excellent reviews of nonsurgical management options,” he said.
Dr. Bichakjian, who is also chief of the division of cutaneous surgery and oncology at the University of Michigan, had no relevant financial disclosures to report. Dr. Alam, who is also chief of cutaneous and aesthetic surgery in the department of dermatology at Northwestern, disclosed relationships with Amway, OptMed, and 3M. Dr. Bennett and Dr. Leffell had no relevant disclosures.
, according to new practice guidelines issued by the American Academy of Dermatology.
Nonsurgical approaches such as cryotherapy, photodynamic therapy, and radiation may be considered for low-risk cancers if surgery is contraindicated, but these methods have lower cure rates, according to the guidelines. Christopher K. Bichakjian, MD, professor of dermatology, University of Michigan, Ann Arbor, and Murad Alam, MD, professor of dermatology, Northwestern University, Chicago, cochaired the work groups that developed the guidelines.
The guidelines for BCC and cSCC, published online in two separate papers (J Am Acad Dermatol. 2018 Jan. 10. doi: 10.1016/j.jaad.2017.10.006; J Am Acad Dermatol. 2018 Jan. 10. doi: 10.1016/j.jaad.2017.10.007), also discuss biopsy techniques, tumor staging, and prevention of recurrence of nonmelanoma skin cancers.
The most suitable stratification for localized BCC and cSCC is the framework provided by the National Comprehensive Cancer Network, the authors said in the guidelines.
For suspected BCC and cSCC, recommended biopsy techniques are punch biopsy, shave biopsy, and excisional biopsy. Biopsy technique is “contingent on the clinical characteristics of the suspected tumor, including morphology, expected histologic subtype and depth, natural history, and anatomic location; patient-specific factors, such as bleeding and wound healing diatheses; and patient preference and physician judgment,” the guidelines state. If the initial biopsy proves insufficient for diagnosis, a repeat biopsy may be considered.
For surgical treatment of BCC, curettage and electrodessication may be considered for low-risk tumors in nonterminal hair-bearing locations. Surgical excision with 4-mm clinical margins and histologic margin assessment is recommended for low-risk primary BCC. For high-risk BCC, Mohs micrographic surgery is recommended, the authors said.
Surgical options for cSCC also include curettage and electrodessication and standard excision for low-risk disease, and Mohs micrographic surgery for high-risk cSCC. In both BCC and cSCC, standard excision may be considered for high-risk tumors in some cases, but “strong caution is advised when selecting a treatment modality” for high-risk tumors “without complete margin assessment,” the guidelines state.
Nonsurgical therapies are generally not recommended as first-line treatment, especially in cSCC because of possible recurrence and metastasis. In cases where nonsurgical therapies are preferred, options may include cryosurgery, topical therapy, photodynamic therapy, radiation, or laser therapy, “with the understanding that the cure rate may be lower,” the authors wrote.
Patients with diagnosed nonmelanoma skin cancer should continue to undergo screening for new primary skin cancers (including BCC, cSCC, and melanoma) at least once per year, the guideline states. They should also be counseled on sun protection, tanning bed avoidance, and regular use of broad-spectrum sunscreen.
Although the new guidelines mainly “reaffirm common knowledge and current practice,” they offer a reminder of “alternative therapeutic or preventive options when insufficient evidence is available to support new therapies or previously dogmatic practice patterns,” the authors said.
These are the first guidelines of care for BCC and cSCC published by the AAD. Commonly used guidelines for the management of BCC and cSCC are published by the National Comprehensive Cancer Network, which are frequently referenced throughout the new AAD guidelines, Dr. Bichakjian said in an interview. While the aim of the cancer network is to develop multidisciplinary guidelines, reflected by the composition of the panel members, “AAD guidelines of care are established primarily by dermatologists for dermatologists,” he pointed out. “However, the work group recognizes that a variety of health care providers outside of dermatology take care of patients with BCC and cSCC, and acknowledges the importance of multidisciplinary care,” he added. “With these considerations in mind, reviewers from specialties outside of dermatology, including plastic surgery, otolaryngology/head and neck surgery, medical oncology, radiation oncology, and family medicine, were invited to critically review the current guidelines.”
The guidelines do not cover the management of actinic keratosis and cSCC in situ, he said. “The work group acknowledges the importance of appropriate management of premalignant and in situ lesions in the prevention of their potential progression to cSCC. However, additional data to provide comprehensive evidence-based recommendations were deemed too extensive to include in the current guidelines and will need to addressed separately.”
In an interview, David J. Leffell, MD, who was not an author of the guidelines, said that the new guidelines do an effective job of “highlighting where valid outcomes data exist and areas where they do not” for a wide range of therapies. They also “attempt to standardize approaches to diagnosis and care of nonmelanoma skin cancer and in general are consistent with established practice patterns,” he added. “Those contemporary approaches have developed in largely empirical fashion over many decades, but bear clarification and reinforcement,” said Dr. Leffell, professor of dermatology and surgery and chief of the section of dermatologic surgery and cutaneous oncology at Yale University, New Haven, Conn.
The guidelines “thoroughly summarize evidence-based recommendations for the entire spectrum of disease management,” Daniel D. Bennett, MD, of the department of dermatology at the University of Wisconsin – Madison, said in an interview. “While surgery remains the mainstay of treatment for BCC and cutaneous SCC, these guidelines include excellent reviews of nonsurgical management options,” he said.
Dr. Bichakjian, who is also chief of the division of cutaneous surgery and oncology at the University of Michigan, had no relevant financial disclosures to report. Dr. Alam, who is also chief of cutaneous and aesthetic surgery in the department of dermatology at Northwestern, disclosed relationships with Amway, OptMed, and 3M. Dr. Bennett and Dr. Leffell had no relevant disclosures.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Nonmalignant Cutaneous Findings Associated With Vemurafenib
To the Editor:
A 53-year-old woman was referred by her oncologist to our dermatology office with lesions on the face and body that presented 8 days after starting vemurafenib 960 mg twice daily for metastatic melanoma. The patient denied any symptoms from the lesions but was concerned they would spread to cover her entire face and body.
The patient's medical history included a diagnosis of metastatic melanoma 6 years prior to presentation. She stated that the primary cutaneous melanoma site was unknown. The patient had endured numerous surgeries to excise lymph node tumors, with some lesions up to 3 cm. The patient recently started vemurafenib, a treatment for BRAF V600E mutation-positive metastatic melanoma. The patient's personal history was notable for hepatitis A, B, and C, and her family history revealed her mother had metastatic lung cancer.
Physical examination revealed numerous 2- to 3-mm, round-oval, flesh-colored to light-brown papules on the cheeks, chest, abdomen (Figure 1), back, and both arms and legs. Some papules were inflamed and some had a stuck-on appearance. Lesions on the chest between the breasts and inframammary region were slightly inflamed. Two skin biopsies were performed. Biopsy of the lesion on the right lateral back revealed solar lentigo, early macular seborrheic keratosis, and a focus of inflamed mild solar keratosis. The dermis showed a mild superficial perivascular and interstitial inflammatory infiltrate composed mostly of lymphocytes, histiocytes, and eosinophils. There were occasional melanophages present (Figure 2). Biopsy of the lesion between the breasts revealed inflamed verrucous seborrheic keratosis (Figure 3).


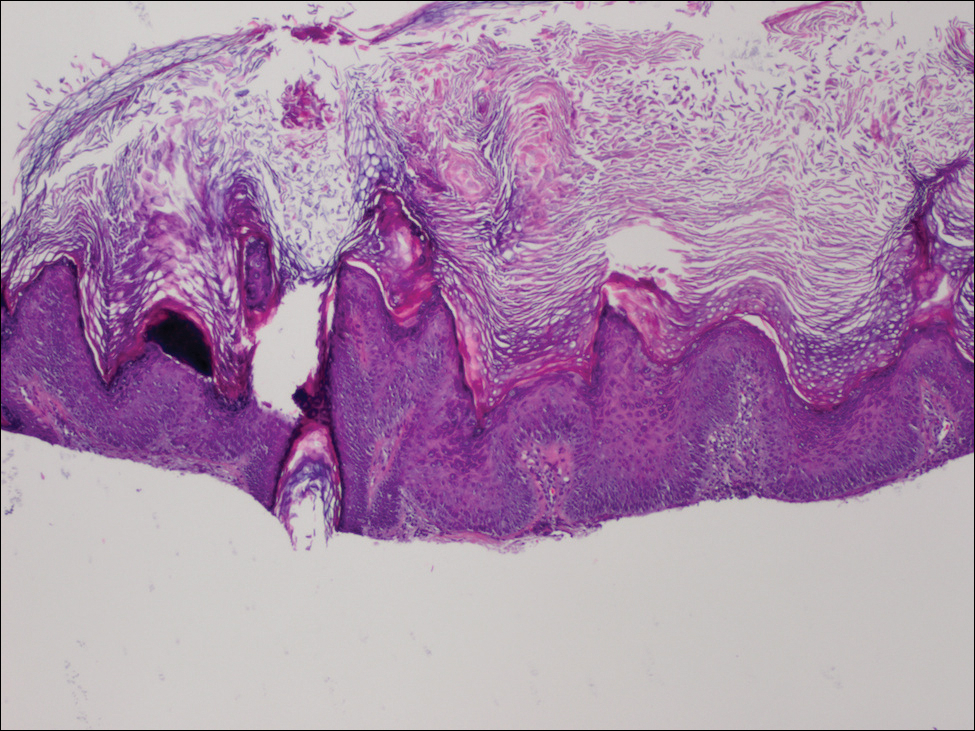
We treated the lesion on the right lateral back with cycles of cryotherapy and explained to the patient that the lesion between the breasts was benign. We also reiterated to the patient the importance of wearing sun-protective clothing and UVA/UVB sunblock with a sun protection factor of 30 or higher.
Our patient was diagnosed with pneumonia and subsequently had to discontinue vemurafenib. During the period of nontreatment, the keratotic lesions cleared with postinflammatory hyperpigmentation and no epidermal changes, which showed a possible inference of a direct relationship between the vemurafenib and the appearance of the nonmalignant cutaneous lesions. Although this report only represents 1 patient, other patients possibly can benefit from a modified dose of vemurafenib, which either would resolve or lessen the quantity of these lesions.
Vemurafenib is the first US Food and Drug Administration-approved treatment for nonresectable metastatic melanoma with the BRAF V600E mutation as detected by a US Food and Drug Administration-approved test.1,2 Mutated BRAF is present in approximately 60% of cutaneous melanomas.3 Vemurafenib targets the oncogenic BRAF V600E making the protein inactive, thus inhibiting cell proliferation and leading to apoptosis and shrinkage of the metastatic tumors.3-5 Vemurafenib has a response rate of more than 50% and is associated with rapid improvement in quality of life.3
Cutaneous side effects include increased incidence of squamous cell carcinoma and keratoacanthomas, appearing approximately 7 to 8 weeks after starting vemurafenib.4 The incidence of these lesions increases in patients 65 years and older and in patients with prior skin cancer and chronic sun exposure. The paradoxical activation of the mitogen-activated protein kinase pathway by mutant BRAF-selective inhibitors provides an explanation of the induction of squamous cell carcinomas.4 Prior to the initiation of vemurafenib, all patients should receive a total-body skin examination and every 2 months thereafter while on treatment. After discontinuation of the medicine, the patient should continue to receive total-body skin evaluations every 6 months indefinitely.
Patients should be aware of the potential for mild to severe photosensitivity reactions. They should be advised to limit their sun exposure time and to wear sun-protective clothing when outdoors. The use of broad-spectrum UVA/UVB sunscreen and lip protectant with a sun protection factor of 30 or higher also should be stressed.6,7 Patients should be aware that UVA rays penetrate glass; therefore, UV-protective clothing should be worn throughout the day and during all seasons.7
In clinical trials of vemurafenib, Stevens-Johnson syndrome and toxic epidermal necrolysis was reported in 2 patients.8,9 Clinical trials also reported patients developing new primary malignant melanoma lesions.10 These findings further emphasize the need for patients to undergo total-body skin examinations during and after treatment.
Other possible dermatologic reactions include a generalized rash, erythema, alopecia, and pruritus.2,3 The development of benign growths associated with patients on vemurafenib include follicular plugging seen in keratosis pilaris, palmar and plantar hyperkeratosis, seborrheic dermatitis-like rashes, verrucous keratosis, and acantholytic dyskeratosis.8,11,12
We report a case of nonmalignant growths occurring 8 days after starting vemurafenib. This case illustrates potential cutaneous adverse reactions that were benign yet still of great concern to our patient. Many of these nonmalignant cutaneous findings are associated with abnormal follicular keratinization thought to be secondary to abnormal signaling of the mitogen-activated protein kinase pathway that occurs with the use of BRAF inhibitors.8 Although in this case malignant lesions were not discovered, the need for total-body skin examinations exists during all stages of treatment. Supportive care and reassurance should be given to patients along with local treatments including topical therapies (steroids, retinoids), cryotherapy, and biopsies or excisions when necessary.13,14
- Holstein S, Hohl R. Therapeutic additions and possible deletions in oncology in 2011. Clin Pharmacol Ther. 2011;91:15-17.
- Zambon A, Niculescu-Dovaz I, Niculescu-Dovaz D, et al. Small molecule inhibitors of BRAF in clinical trials. Bioorg Med Chem Lett. 2012;22:789-792.
- Luke JJ, Hodi FS. Vemurafenib and BRAF inhibition: a new class of treatment for metastatic melanoma [published online November 14, 2011]. Clin Cancer Res. 2012;18:9-14.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363:809-819.
- Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041-3046.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Dummer R, Rinderknecht J, Goldinger SM. Ultraviolet A and photosensitivity during vemuranefib therapy. N Engl J Med. 2012;366:480-481.
- Bovd KP, Vincent B, Andrea A, et al. Nonmalignant cutaneous findings associated with vemurafenib use in patients with metastatic melanoma. J Am Acad Dermatol. 2012;67:1375-1379.
- Wang CM, Fleming KF Hsu S. A case of vemurafenib-induced keratosis pilaris-like eruption. Dermatol Online J. 2012;18:7.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Huang V, Hepper D, Anadkat M, et al. Cutaneous toxic effects associated with vemurafenib and inhibition of the BRAF pathway. Arch Dermatol. 2012;148:628-633.
- Gupta M, Huang V, Linette G, et al. Unusual complication of vemurafenib treatment of metastatic melanoma: exacerbation of acantholytic dyskeratosis complicated by Kaposi varicelliform eruption. Arch Dermatol. 2012;148:966-968;
- Sinha R, Edmonds K, Newton-Bishop JA, et al. Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: practical advice on diagnosis, preventions and management of the main treatment related skin toxicities. Br J Dermatol. 2012;167:987-994.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
To the Editor:
A 53-year-old woman was referred by her oncologist to our dermatology office with lesions on the face and body that presented 8 days after starting vemurafenib 960 mg twice daily for metastatic melanoma. The patient denied any symptoms from the lesions but was concerned they would spread to cover her entire face and body.
The patient's medical history included a diagnosis of metastatic melanoma 6 years prior to presentation. She stated that the primary cutaneous melanoma site was unknown. The patient had endured numerous surgeries to excise lymph node tumors, with some lesions up to 3 cm. The patient recently started vemurafenib, a treatment for BRAF V600E mutation-positive metastatic melanoma. The patient's personal history was notable for hepatitis A, B, and C, and her family history revealed her mother had metastatic lung cancer.
Physical examination revealed numerous 2- to 3-mm, round-oval, flesh-colored to light-brown papules on the cheeks, chest, abdomen (Figure 1), back, and both arms and legs. Some papules were inflamed and some had a stuck-on appearance. Lesions on the chest between the breasts and inframammary region were slightly inflamed. Two skin biopsies were performed. Biopsy of the lesion on the right lateral back revealed solar lentigo, early macular seborrheic keratosis, and a focus of inflamed mild solar keratosis. The dermis showed a mild superficial perivascular and interstitial inflammatory infiltrate composed mostly of lymphocytes, histiocytes, and eosinophils. There were occasional melanophages present (Figure 2). Biopsy of the lesion between the breasts revealed inflamed verrucous seborrheic keratosis (Figure 3).
We treated the lesion on the right lateral back with cycles of cryotherapy and explained to the patient that the lesion between the breasts was benign. We also reiterated to the patient the importance of wearing sun-protective clothing and UVA/UVB sunblock with a sun protection factor of 30 or higher.
Our patient was diagnosed with pneumonia and subsequently had to discontinue vemurafenib. During the period of nontreatment, the keratotic lesions cleared with postinflammatory hyperpigmentation and no epidermal changes, which showed a possible inference of a direct relationship between the vemurafenib and the appearance of the nonmalignant cutaneous lesions. Although this report only represents 1 patient, other patients possibly can benefit from a modified dose of vemurafenib, which either would resolve or lessen the quantity of these lesions.
Vemurafenib is the first US Food and Drug Administration-approved treatment for nonresectable metastatic melanoma with the BRAF V600E mutation as detected by a US Food and Drug Administration-approved test.1,2 Mutated BRAF is present in approximately 60% of cutaneous melanomas.3 Vemurafenib targets the oncogenic BRAF V600E making the protein inactive, thus inhibiting cell proliferation and leading to apoptosis and shrinkage of the metastatic tumors.3-5 Vemurafenib has a response rate of more than 50% and is associated with rapid improvement in quality of life.3
Cutaneous side effects include increased incidence of squamous cell carcinoma and keratoacanthomas, appearing approximately 7 to 8 weeks after starting vemurafenib.4 The incidence of these lesions increases in patients 65 years and older and in patients with prior skin cancer and chronic sun exposure. The paradoxical activation of the mitogen-activated protein kinase pathway by mutant BRAF-selective inhibitors provides an explanation of the induction of squamous cell carcinomas.4 Prior to the initiation of vemurafenib, all patients should receive a total-body skin examination and every 2 months thereafter while on treatment. After discontinuation of the medicine, the patient should continue to receive total-body skin evaluations every 6 months indefinitely.
Patients should be aware of the potential for mild to severe photosensitivity reactions. They should be advised to limit their sun exposure time and to wear sun-protective clothing when outdoors. The use of broad-spectrum UVA/UVB sunscreen and lip protectant with a sun protection factor of 30 or higher also should be stressed.6,7 Patients should be aware that UVA rays penetrate glass; therefore, UV-protective clothing should be worn throughout the day and during all seasons.7
In clinical trials of vemurafenib, Stevens-Johnson syndrome and toxic epidermal necrolysis was reported in 2 patients.8,9 Clinical trials also reported patients developing new primary malignant melanoma lesions.10 These findings further emphasize the need for patients to undergo total-body skin examinations during and after treatment.
Other possible dermatologic reactions include a generalized rash, erythema, alopecia, and pruritus.2,3 The development of benign growths associated with patients on vemurafenib include follicular plugging seen in keratosis pilaris, palmar and plantar hyperkeratosis, seborrheic dermatitis-like rashes, verrucous keratosis, and acantholytic dyskeratosis.8,11,12
We report a case of nonmalignant growths occurring 8 days after starting vemurafenib. This case illustrates potential cutaneous adverse reactions that were benign yet still of great concern to our patient. Many of these nonmalignant cutaneous findings are associated with abnormal follicular keratinization thought to be secondary to abnormal signaling of the mitogen-activated protein kinase pathway that occurs with the use of BRAF inhibitors.8 Although in this case malignant lesions were not discovered, the need for total-body skin examinations exists during all stages of treatment. Supportive care and reassurance should be given to patients along with local treatments including topical therapies (steroids, retinoids), cryotherapy, and biopsies or excisions when necessary.13,14
To the Editor:
A 53-year-old woman was referred by her oncologist to our dermatology office with lesions on the face and body that presented 8 days after starting vemurafenib 960 mg twice daily for metastatic melanoma. The patient denied any symptoms from the lesions but was concerned they would spread to cover her entire face and body.
The patient's medical history included a diagnosis of metastatic melanoma 6 years prior to presentation. She stated that the primary cutaneous melanoma site was unknown. The patient had endured numerous surgeries to excise lymph node tumors, with some lesions up to 3 cm. The patient recently started vemurafenib, a treatment for BRAF V600E mutation-positive metastatic melanoma. The patient's personal history was notable for hepatitis A, B, and C, and her family history revealed her mother had metastatic lung cancer.
Physical examination revealed numerous 2- to 3-mm, round-oval, flesh-colored to light-brown papules on the cheeks, chest, abdomen (Figure 1), back, and both arms and legs. Some papules were inflamed and some had a stuck-on appearance. Lesions on the chest between the breasts and inframammary region were slightly inflamed. Two skin biopsies were performed. Biopsy of the lesion on the right lateral back revealed solar lentigo, early macular seborrheic keratosis, and a focus of inflamed mild solar keratosis. The dermis showed a mild superficial perivascular and interstitial inflammatory infiltrate composed mostly of lymphocytes, histiocytes, and eosinophils. There were occasional melanophages present (Figure 2). Biopsy of the lesion between the breasts revealed inflamed verrucous seborrheic keratosis (Figure 3).
We treated the lesion on the right lateral back with cycles of cryotherapy and explained to the patient that the lesion between the breasts was benign. We also reiterated to the patient the importance of wearing sun-protective clothing and UVA/UVB sunblock with a sun protection factor of 30 or higher.
Our patient was diagnosed with pneumonia and subsequently had to discontinue vemurafenib. During the period of nontreatment, the keratotic lesions cleared with postinflammatory hyperpigmentation and no epidermal changes, which showed a possible inference of a direct relationship between the vemurafenib and the appearance of the nonmalignant cutaneous lesions. Although this report only represents 1 patient, other patients possibly can benefit from a modified dose of vemurafenib, which either would resolve or lessen the quantity of these lesions.
Vemurafenib is the first US Food and Drug Administration-approved treatment for nonresectable metastatic melanoma with the BRAF V600E mutation as detected by a US Food and Drug Administration-approved test.1,2 Mutated BRAF is present in approximately 60% of cutaneous melanomas.3 Vemurafenib targets the oncogenic BRAF V600E making the protein inactive, thus inhibiting cell proliferation and leading to apoptosis and shrinkage of the metastatic tumors.3-5 Vemurafenib has a response rate of more than 50% and is associated with rapid improvement in quality of life.3
Cutaneous side effects include increased incidence of squamous cell carcinoma and keratoacanthomas, appearing approximately 7 to 8 weeks after starting vemurafenib.4 The incidence of these lesions increases in patients 65 years and older and in patients with prior skin cancer and chronic sun exposure. The paradoxical activation of the mitogen-activated protein kinase pathway by mutant BRAF-selective inhibitors provides an explanation of the induction of squamous cell carcinomas.4 Prior to the initiation of vemurafenib, all patients should receive a total-body skin examination and every 2 months thereafter while on treatment. After discontinuation of the medicine, the patient should continue to receive total-body skin evaluations every 6 months indefinitely.
Patients should be aware of the potential for mild to severe photosensitivity reactions. They should be advised to limit their sun exposure time and to wear sun-protective clothing when outdoors. The use of broad-spectrum UVA/UVB sunscreen and lip protectant with a sun protection factor of 30 or higher also should be stressed.6,7 Patients should be aware that UVA rays penetrate glass; therefore, UV-protective clothing should be worn throughout the day and during all seasons.7
In clinical trials of vemurafenib, Stevens-Johnson syndrome and toxic epidermal necrolysis was reported in 2 patients.8,9 Clinical trials also reported patients developing new primary malignant melanoma lesions.10 These findings further emphasize the need for patients to undergo total-body skin examinations during and after treatment.
Other possible dermatologic reactions include a generalized rash, erythema, alopecia, and pruritus.2,3 The development of benign growths associated with patients on vemurafenib include follicular plugging seen in keratosis pilaris, palmar and plantar hyperkeratosis, seborrheic dermatitis-like rashes, verrucous keratosis, and acantholytic dyskeratosis.8,11,12
We report a case of nonmalignant growths occurring 8 days after starting vemurafenib. This case illustrates potential cutaneous adverse reactions that were benign yet still of great concern to our patient. Many of these nonmalignant cutaneous findings are associated with abnormal follicular keratinization thought to be secondary to abnormal signaling of the mitogen-activated protein kinase pathway that occurs with the use of BRAF inhibitors.8 Although in this case malignant lesions were not discovered, the need for total-body skin examinations exists during all stages of treatment. Supportive care and reassurance should be given to patients along with local treatments including topical therapies (steroids, retinoids), cryotherapy, and biopsies or excisions when necessary.13,14
- Holstein S, Hohl R. Therapeutic additions and possible deletions in oncology in 2011. Clin Pharmacol Ther. 2011;91:15-17.
- Zambon A, Niculescu-Dovaz I, Niculescu-Dovaz D, et al. Small molecule inhibitors of BRAF in clinical trials. Bioorg Med Chem Lett. 2012;22:789-792.
- Luke JJ, Hodi FS. Vemurafenib and BRAF inhibition: a new class of treatment for metastatic melanoma [published online November 14, 2011]. Clin Cancer Res. 2012;18:9-14.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363:809-819.
- Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041-3046.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Dummer R, Rinderknecht J, Goldinger SM. Ultraviolet A and photosensitivity during vemuranefib therapy. N Engl J Med. 2012;366:480-481.
- Bovd KP, Vincent B, Andrea A, et al. Nonmalignant cutaneous findings associated with vemurafenib use in patients with metastatic melanoma. J Am Acad Dermatol. 2012;67:1375-1379.
- Wang CM, Fleming KF Hsu S. A case of vemurafenib-induced keratosis pilaris-like eruption. Dermatol Online J. 2012;18:7.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Huang V, Hepper D, Anadkat M, et al. Cutaneous toxic effects associated with vemurafenib and inhibition of the BRAF pathway. Arch Dermatol. 2012;148:628-633.
- Gupta M, Huang V, Linette G, et al. Unusual complication of vemurafenib treatment of metastatic melanoma: exacerbation of acantholytic dyskeratosis complicated by Kaposi varicelliform eruption. Arch Dermatol. 2012;148:966-968;
- Sinha R, Edmonds K, Newton-Bishop JA, et al. Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: practical advice on diagnosis, preventions and management of the main treatment related skin toxicities. Br J Dermatol. 2012;167:987-994.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Holstein S, Hohl R. Therapeutic additions and possible deletions in oncology in 2011. Clin Pharmacol Ther. 2011;91:15-17.
- Zambon A, Niculescu-Dovaz I, Niculescu-Dovaz D, et al. Small molecule inhibitors of BRAF in clinical trials. Bioorg Med Chem Lett. 2012;22:789-792.
- Luke JJ, Hodi FS. Vemurafenib and BRAF inhibition: a new class of treatment for metastatic melanoma [published online November 14, 2011]. Clin Cancer Res. 2012;18:9-14.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363:809-819.
- Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041-3046.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Dummer R, Rinderknecht J, Goldinger SM. Ultraviolet A and photosensitivity during vemuranefib therapy. N Engl J Med. 2012;366:480-481.
- Bovd KP, Vincent B, Andrea A, et al. Nonmalignant cutaneous findings associated with vemurafenib use in patients with metastatic melanoma. J Am Acad Dermatol. 2012;67:1375-1379.
- Wang CM, Fleming KF Hsu S. A case of vemurafenib-induced keratosis pilaris-like eruption. Dermatol Online J. 2012;18:7.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Huang V, Hepper D, Anadkat M, et al. Cutaneous toxic effects associated with vemurafenib and inhibition of the BRAF pathway. Arch Dermatol. 2012;148:628-633.
- Gupta M, Huang V, Linette G, et al. Unusual complication of vemurafenib treatment of metastatic melanoma: exacerbation of acantholytic dyskeratosis complicated by Kaposi varicelliform eruption. Arch Dermatol. 2012;148:966-968;
- Sinha R, Edmonds K, Newton-Bishop JA, et al. Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: practical advice on diagnosis, preventions and management of the main treatment related skin toxicities. Br J Dermatol. 2012;167:987-994.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
Practice Points
- Prior to starting a BRAF inhibitor, clinicians should perform a baseline total-body skin examination and follow-up every 2 months.
- Take photographs of the patient's entire body on initial total-body skin examination.
- Encourage sun protection for exposed areas on the body in all seasons.
Scaly Pink Patches: Differentiating Psoriasis From Basal Cell Carcinoma
Dermoscopy increases diagnostic accuracy in the analysis of skin growths.1,2 Recently the use of dermoscopy has broadened to include inflammatory dermatoses and skin infections.3 To substantiate the value of dermoscopy in assessing psoriasis, we performed a systematic review of the literature and briefly reviewed 31 articles. We also report a case that highlights the differences between psoriasis and basal cell carcinoma (BCC) under dermoscopic examination, and we discuss the literature on the dermoscopic findings of psoriasis with an emphasis on the relative sensitivities and specificities of dermoscopic findings for psoriasis and for BCC.
Case Report
A 63-year-old man with psoriasis and a history of BCC presented for follow-up of psoriasis, which was well-controlled on etanercept. The physical examination was remarkable for scaly pink papules scattered on the trunk and extremities. A new larger red-pink patch was located on the left lower back (Figure 1). Dermoscopic evaluation of the new patch revealed shiny white lines and branching blood vessels (Figure 2).
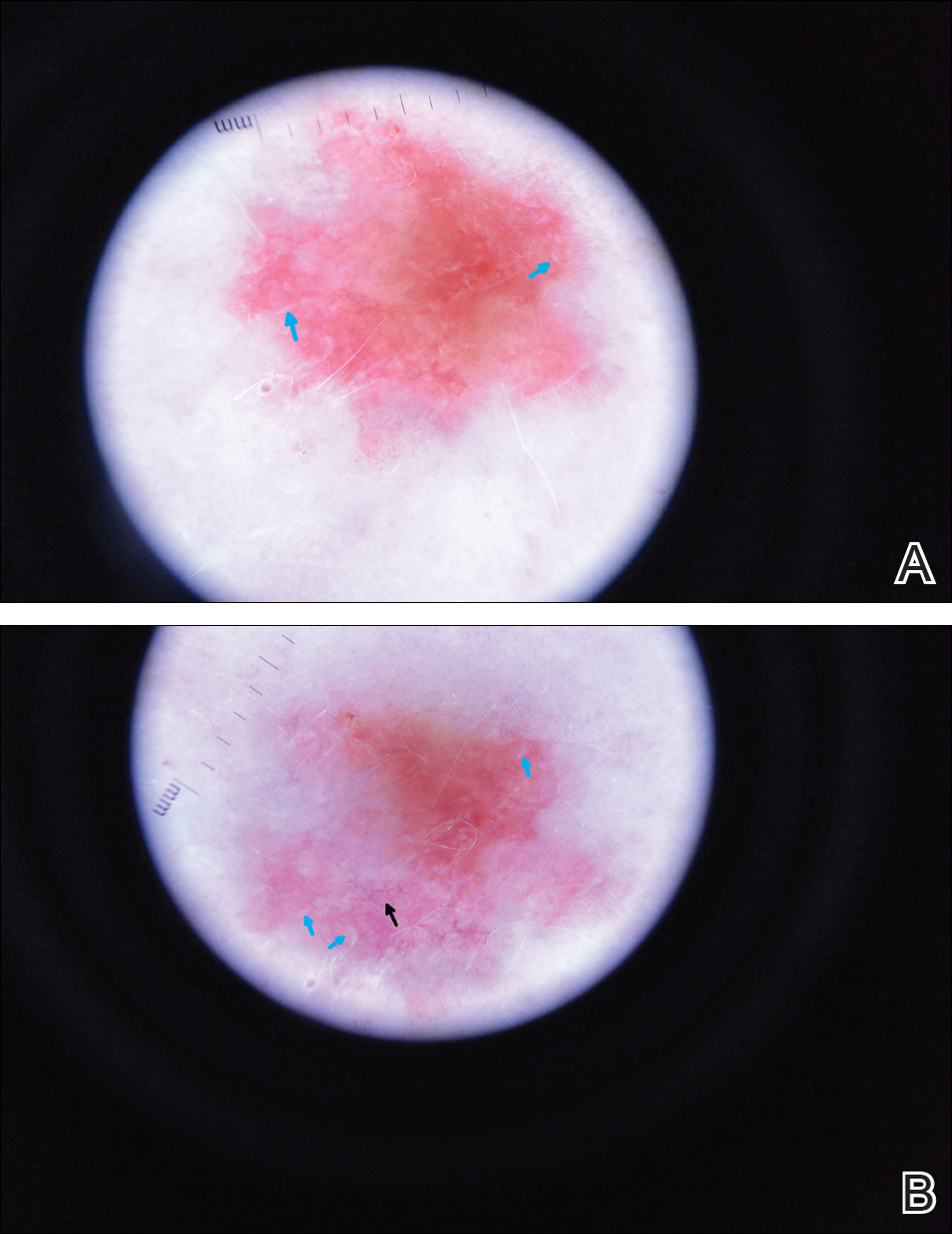
Comment
The clinical morphology of psoriasis and BCC can be similar, and dermoscopy can help in differentiating between the 2 conditions.
Literature Search on Dermoscopy and Psoriasis
We performed a PubMed search of articles indexed for MEDLINE to review the published literature on dermoscopy and psoriasis. Two reviewers (C.H. and L.C.) searched for psoriasis paired with the terms dermoscopy or dermatoscopy or epiluminescence microscopy. Only English-language articles published between 1996 and 2016 were included in the search. Articles that focused solely on confocal microscopy were excluded. Article titles and abstracts were evaluated and articles that omitted mention of dermoscopy and psoriasis were excluded, yielding a total of 31 articles. Of these articles, only 2 discussed the specificity or sensitivity of the dermoscopic findings of psoriasis.4,5 Most of the articles were case reports and descriptive cross-sectional studies. The reports addressed multiple subtypes of psoriasis, but reports on psoriasis vulgaris and scalp psoriasis were most common (Table). Lallas et al6 provided a comprehensive descriptive review of the main findings on dermoscopy for psoriasis and other inflammatory skin conditions, but it lacked a comparison between psoriasis and BCC or data on the sensitivity and specificity of the findings. Two studies reported sensitivity and specificity values for the dermoscopic findings of psoriasis.4,5 Pan et al5 reported a 98% diagnostic probability of psoriasis if red dots, homogeneous vascular pattern, and a light red background are all present. Additionally, they reported that the presence of 4 of 6 criteria for BCC—scattered vascular pattern, arborizing microvessels, telangiectatic or atypical vessels, milky-pink background, and brown dots⁄globules—yielded a diagnostic probability of 99%.5 Similarly, Lallas et al6 demonstrated that the presence of dotted vessels alone is not sufficient to presume a diagnosis of psoriasis, as this finding can be seen in other inflammatory skin conditions. However, “the combination of regularly distributed dotted vessels over a light red background associated with diffuse white scales was highly predictive of [plaque psoriasis] and allowed a correct diagnosis with 88.0% specificity and 84.9% sensitivity.”4 Figure 3 shows a dermoscopic image of plaque psoriasis that demonstrates these findings. The remaining literature corroborated this evidence, with the most commonly reported dermoscopic findings of psoriasis being red dots, red globules, glomerular vessels (also known as twisted capillary loops), red globular ring
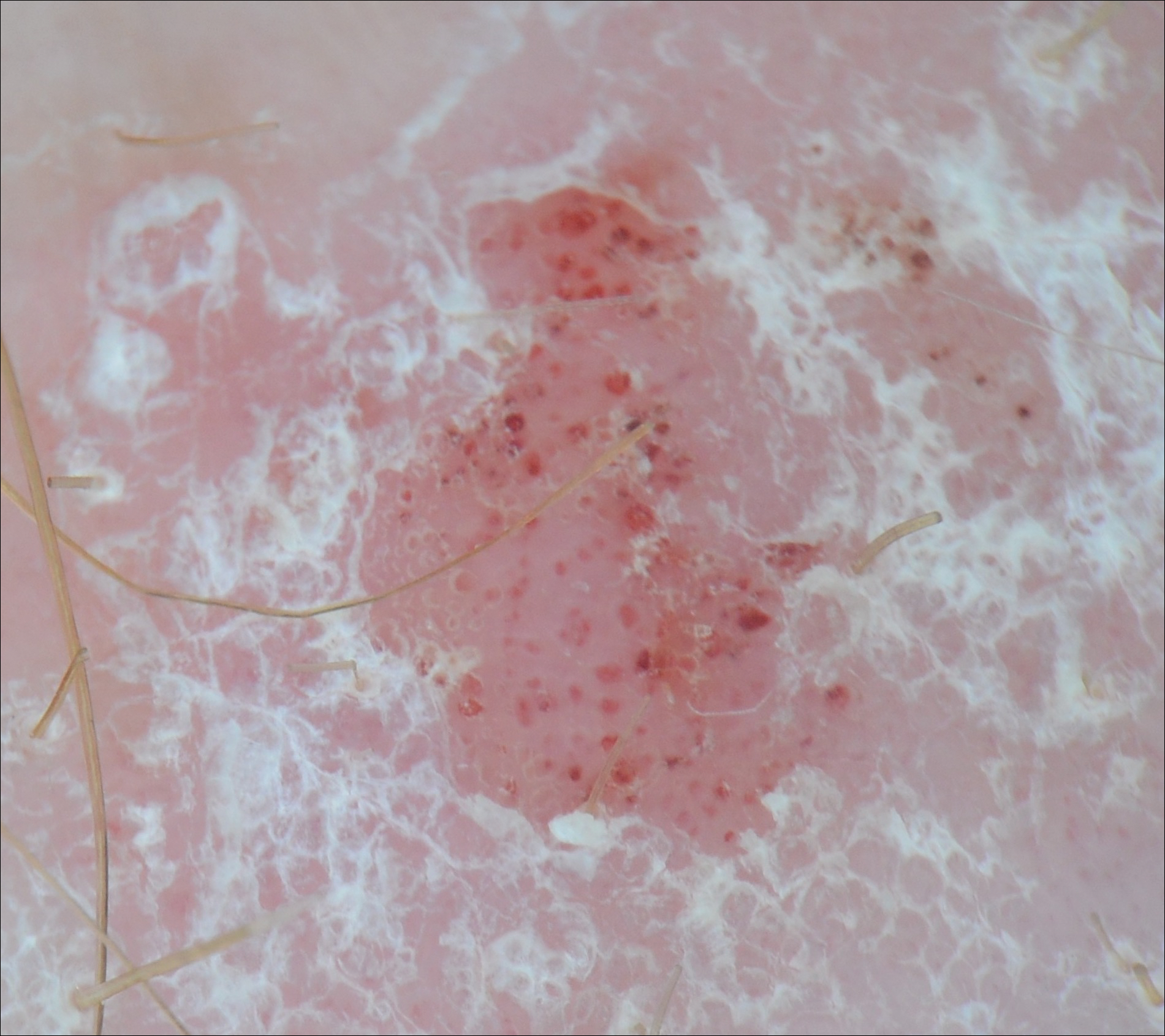
Dermoscopy and BCC
Much has been published on the dermoscopic findings of BCC.5,13-15 The dermoscopic findings of BCC include large blue-gray ovoid nests, leaflike areas, spoke-wheel–like areas, arborizing vessels (telangiectasia), and ulceration.15 Superficial BCC is characterized by short fine or arborizing telangiectasia, shallow erosions, and shiny white areas.15 The positive predictive value of dermoscopy in BCC is as high as 97%.16 Additionally, multiple studies report a sensitivity of 95% to 99%5,13,14 and a specificity of 79% to 99% in the use of dermoscopy for identifying BCC. According to Pan et al,5 the most sensitive finding for BCC is a scattered vascular pattern (97%), while the most specific finding is arborizing microvessels (99%).
Utility of Dermoscopy
Our case of a 63-year-old man with a history of psoriasis and BCC highlights the usefulness of dermoscopy in accurately determining the features of each condition. Additionally, dermoscopy aids in differentiating between psoriasis and squamous cell carcinoma. In contrast to the dotted vessels seen in psoriasis, squamous cell carcinomas often have peripheral hairpin (glomerular) vessels.17
If future reports confirm dermoscopy’s utility in accurately diagnosing psoriasis, fewer biopsies may be needed when evaluating patients with new rashes. Furthermore, dermoscopy may expedite treatment of psoriasis (as it can for malignant conditions) by obviating the wait for pathology results currently needed to initiate systemic treatment. For patients with psoriasis who also have sun-damaged skin, dermoscopy may assist in differentiating pink patches and plaques of psoriasis from skin cancer, such as superficial BCCs, which often have shiny white lines not seen in psoriasis.15
- Kittler H, Pehamberger H, Wolff K, et al. Diagnostic accuracy of dermoscopy. Lancet Oncol. 2002;3:159-165.
- Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676.
- Lallas A, Giacomel J, Argenziano G, et al. Dermoscopy in general dermatology: practical tips for the clinician. Br J Dermatol. 2014;170:514-526.
- Lallas A, Kyrgidis A, Tzellos TG, et al. Accuracy of dermoscopic criteria for the diagnosis of psoriasis, dermatitis, lichen planus and pityriasis rosea. Br J Dermatol. 2012;166:1198-1205.
- Pan Y, Chamberlain AJ, Bailey M, et al. Dermatoscopy aids in the diagnosis of the solitary red scaly patch or plaque–features distinguishing superficial basal cell carcinoma, intraepidermal carcinoma, and psoriasis. J Am Acad Dermatol. 2008;59:268-274.
- Lallas A, Apalla Z, Argenziano G, et al. Dermoscopic pattern of psoriatic lesions on specific body sites. Dermatology. 2014;228:250-254.
- Almeida MC, Romiti R, Doche I, et al. Psoriatic scarring alopecia. An Bras Dermatol. 2013;88:29-31.
- Zalaudek I, Argenziano G. Dermoscopy subpatterns of inflammatory skin disorders. Arch Dermatol. 2006;142:808.
- Miteva M, Tosti A. Hair and scalp dermatoscopy. J Am Acad Dermatol. 2012;67:1040-1048.
- Vázquez-López F, Zaballos P, Fueyo-Casado A, et al. A dermoscopy subpattern of plaque-type psoriasis: red globular rings. Arch Dermatol. 2007;143:1612.
- Lacarrubba F, Nasca MR, Micali G. Videodermatoscopy enhances diagnostic capability in psoriatic balanitis. J Am Acad Dermatol. 2009;61:1084-1086.
- Liebman TN, Wang SQ. Detection of early basal cell carcinoma with dermoscopy in a patient with psoriasis. Dermatol Online J. 2011;17:12.
- Menzies SW, Westerhoff K, Rabinovitz H, et al. Surface microscopy of pigmented basal cell carcinoma. Arch Dermatol. 2000;136:1012-1016.
- Altamura D, Menzies SW, Argenziano G, et al. Dermatoscopy of basal cell carcinoma: morphologic variability of global and local features and accuracy of diagnosis. J Am Acad Dermatol. 2010;62:67-75.
- Marghoob AA, Malvehy J, Braun RP, eds. An Atlas of Dermoscopy. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Nelson SA, Scope A, Rishpon A, et al. Accuracy and confidence in the clinical diagnosis of basal cell cancer using dermoscopy and reflex confocal microscopy. Int J Dermatol. 2016;55:1351-1356.
- Zalaudek I, Kreusch J, Giacomel J, et al. How to diagnose nonpigmented skin tumors: a review of vascular structures seen with dermoscopy: part I. melanocytic skin tumors. J Am Acad Dermatol. 2010;63:361-374.
Dermoscopy increases diagnostic accuracy in the analysis of skin growths.1,2 Recently the use of dermoscopy has broadened to include inflammatory dermatoses and skin infections.3 To substantiate the value of dermoscopy in assessing psoriasis, we performed a systematic review of the literature and briefly reviewed 31 articles. We also report a case that highlights the differences between psoriasis and basal cell carcinoma (BCC) under dermoscopic examination, and we discuss the literature on the dermoscopic findings of psoriasis with an emphasis on the relative sensitivities and specificities of dermoscopic findings for psoriasis and for BCC.
Case Report
A 63-year-old man with psoriasis and a history of BCC presented for follow-up of psoriasis, which was well-controlled on etanercept. The physical examination was remarkable for scaly pink papules scattered on the trunk and extremities. A new larger red-pink patch was located on the left lower back (Figure 1). Dermoscopic evaluation of the new patch revealed shiny white lines and branching blood vessels (Figure 2).
Comment
The clinical morphology of psoriasis and BCC can be similar, and dermoscopy can help in differentiating between the 2 conditions.
Literature Search on Dermoscopy and Psoriasis
We performed a PubMed search of articles indexed for MEDLINE to review the published literature on dermoscopy and psoriasis. Two reviewers (C.H. and L.C.) searched for psoriasis paired with the terms dermoscopy or dermatoscopy or epiluminescence microscopy. Only English-language articles published between 1996 and 2016 were included in the search. Articles that focused solely on confocal microscopy were excluded. Article titles and abstracts were evaluated and articles that omitted mention of dermoscopy and psoriasis were excluded, yielding a total of 31 articles. Of these articles, only 2 discussed the specificity or sensitivity of the dermoscopic findings of psoriasis.4,5 Most of the articles were case reports and descriptive cross-sectional studies. The reports addressed multiple subtypes of psoriasis, but reports on psoriasis vulgaris and scalp psoriasis were most common (Table). Lallas et al6 provided a comprehensive descriptive review of the main findings on dermoscopy for psoriasis and other inflammatory skin conditions, but it lacked a comparison between psoriasis and BCC or data on the sensitivity and specificity of the findings. Two studies reported sensitivity and specificity values for the dermoscopic findings of psoriasis.4,5 Pan et al5 reported a 98% diagnostic probability of psoriasis if red dots, homogeneous vascular pattern, and a light red background are all present. Additionally, they reported that the presence of 4 of 6 criteria for BCC—scattered vascular pattern, arborizing microvessels, telangiectatic or atypical vessels, milky-pink background, and brown dots⁄globules—yielded a diagnostic probability of 99%.5 Similarly, Lallas et al6 demonstrated that the presence of dotted vessels alone is not sufficient to presume a diagnosis of psoriasis, as this finding can be seen in other inflammatory skin conditions. However, “the combination of regularly distributed dotted vessels over a light red background associated with diffuse white scales was highly predictive of [plaque psoriasis] and allowed a correct diagnosis with 88.0% specificity and 84.9% sensitivity.”4 Figure 3 shows a dermoscopic image of plaque psoriasis that demonstrates these findings. The remaining literature corroborated this evidence, with the most commonly reported dermoscopic findings of psoriasis being red dots, red globules, glomerular vessels (also known as twisted capillary loops), red globular ring
Dermoscopy and BCC
Much has been published on the dermoscopic findings of BCC.5,13-15 The dermoscopic findings of BCC include large blue-gray ovoid nests, leaflike areas, spoke-wheel–like areas, arborizing vessels (telangiectasia), and ulceration.15 Superficial BCC is characterized by short fine or arborizing telangiectasia, shallow erosions, and shiny white areas.15 The positive predictive value of dermoscopy in BCC is as high as 97%.16 Additionally, multiple studies report a sensitivity of 95% to 99%5,13,14 and a specificity of 79% to 99% in the use of dermoscopy for identifying BCC. According to Pan et al,5 the most sensitive finding for BCC is a scattered vascular pattern (97%), while the most specific finding is arborizing microvessels (99%).
Utility of Dermoscopy
Our case of a 63-year-old man with a history of psoriasis and BCC highlights the usefulness of dermoscopy in accurately determining the features of each condition. Additionally, dermoscopy aids in differentiating between psoriasis and squamous cell carcinoma. In contrast to the dotted vessels seen in psoriasis, squamous cell carcinomas often have peripheral hairpin (glomerular) vessels.17
If future reports confirm dermoscopy’s utility in accurately diagnosing psoriasis, fewer biopsies may be needed when evaluating patients with new rashes. Furthermore, dermoscopy may expedite treatment of psoriasis (as it can for malignant conditions) by obviating the wait for pathology results currently needed to initiate systemic treatment. For patients with psoriasis who also have sun-damaged skin, dermoscopy may assist in differentiating pink patches and plaques of psoriasis from skin cancer, such as superficial BCCs, which often have shiny white lines not seen in psoriasis.15
Dermoscopy increases diagnostic accuracy in the analysis of skin growths.1,2 Recently the use of dermoscopy has broadened to include inflammatory dermatoses and skin infections.3 To substantiate the value of dermoscopy in assessing psoriasis, we performed a systematic review of the literature and briefly reviewed 31 articles. We also report a case that highlights the differences between psoriasis and basal cell carcinoma (BCC) under dermoscopic examination, and we discuss the literature on the dermoscopic findings of psoriasis with an emphasis on the relative sensitivities and specificities of dermoscopic findings for psoriasis and for BCC.
Case Report
A 63-year-old man with psoriasis and a history of BCC presented for follow-up of psoriasis, which was well-controlled on etanercept. The physical examination was remarkable for scaly pink papules scattered on the trunk and extremities. A new larger red-pink patch was located on the left lower back (Figure 1). Dermoscopic evaluation of the new patch revealed shiny white lines and branching blood vessels (Figure 2).
Comment
The clinical morphology of psoriasis and BCC can be similar, and dermoscopy can help in differentiating between the 2 conditions.
Literature Search on Dermoscopy and Psoriasis
We performed a PubMed search of articles indexed for MEDLINE to review the published literature on dermoscopy and psoriasis. Two reviewers (C.H. and L.C.) searched for psoriasis paired with the terms dermoscopy or dermatoscopy or epiluminescence microscopy. Only English-language articles published between 1996 and 2016 were included in the search. Articles that focused solely on confocal microscopy were excluded. Article titles and abstracts were evaluated and articles that omitted mention of dermoscopy and psoriasis were excluded, yielding a total of 31 articles. Of these articles, only 2 discussed the specificity or sensitivity of the dermoscopic findings of psoriasis.4,5 Most of the articles were case reports and descriptive cross-sectional studies. The reports addressed multiple subtypes of psoriasis, but reports on psoriasis vulgaris and scalp psoriasis were most common (Table). Lallas et al6 provided a comprehensive descriptive review of the main findings on dermoscopy for psoriasis and other inflammatory skin conditions, but it lacked a comparison between psoriasis and BCC or data on the sensitivity and specificity of the findings. Two studies reported sensitivity and specificity values for the dermoscopic findings of psoriasis.4,5 Pan et al5 reported a 98% diagnostic probability of psoriasis if red dots, homogeneous vascular pattern, and a light red background are all present. Additionally, they reported that the presence of 4 of 6 criteria for BCC—scattered vascular pattern, arborizing microvessels, telangiectatic or atypical vessels, milky-pink background, and brown dots⁄globules—yielded a diagnostic probability of 99%.5 Similarly, Lallas et al6 demonstrated that the presence of dotted vessels alone is not sufficient to presume a diagnosis of psoriasis, as this finding can be seen in other inflammatory skin conditions. However, “the combination of regularly distributed dotted vessels over a light red background associated with diffuse white scales was highly predictive of [plaque psoriasis] and allowed a correct diagnosis with 88.0% specificity and 84.9% sensitivity.”4 Figure 3 shows a dermoscopic image of plaque psoriasis that demonstrates these findings. The remaining literature corroborated this evidence, with the most commonly reported dermoscopic findings of psoriasis being red dots, red globules, glomerular vessels (also known as twisted capillary loops), red globular ring
Dermoscopy and BCC
Much has been published on the dermoscopic findings of BCC.5,13-15 The dermoscopic findings of BCC include large blue-gray ovoid nests, leaflike areas, spoke-wheel–like areas, arborizing vessels (telangiectasia), and ulceration.15 Superficial BCC is characterized by short fine or arborizing telangiectasia, shallow erosions, and shiny white areas.15 The positive predictive value of dermoscopy in BCC is as high as 97%.16 Additionally, multiple studies report a sensitivity of 95% to 99%5,13,14 and a specificity of 79% to 99% in the use of dermoscopy for identifying BCC. According to Pan et al,5 the most sensitive finding for BCC is a scattered vascular pattern (97%), while the most specific finding is arborizing microvessels (99%).
Utility of Dermoscopy
Our case of a 63-year-old man with a history of psoriasis and BCC highlights the usefulness of dermoscopy in accurately determining the features of each condition. Additionally, dermoscopy aids in differentiating between psoriasis and squamous cell carcinoma. In contrast to the dotted vessels seen in psoriasis, squamous cell carcinomas often have peripheral hairpin (glomerular) vessels.17
If future reports confirm dermoscopy’s utility in accurately diagnosing psoriasis, fewer biopsies may be needed when evaluating patients with new rashes. Furthermore, dermoscopy may expedite treatment of psoriasis (as it can for malignant conditions) by obviating the wait for pathology results currently needed to initiate systemic treatment. For patients with psoriasis who also have sun-damaged skin, dermoscopy may assist in differentiating pink patches and plaques of psoriasis from skin cancer, such as superficial BCCs, which often have shiny white lines not seen in psoriasis.15
- Kittler H, Pehamberger H, Wolff K, et al. Diagnostic accuracy of dermoscopy. Lancet Oncol. 2002;3:159-165.
- Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676.
- Lallas A, Giacomel J, Argenziano G, et al. Dermoscopy in general dermatology: practical tips for the clinician. Br J Dermatol. 2014;170:514-526.
- Lallas A, Kyrgidis A, Tzellos TG, et al. Accuracy of dermoscopic criteria for the diagnosis of psoriasis, dermatitis, lichen planus and pityriasis rosea. Br J Dermatol. 2012;166:1198-1205.
- Pan Y, Chamberlain AJ, Bailey M, et al. Dermatoscopy aids in the diagnosis of the solitary red scaly patch or plaque–features distinguishing superficial basal cell carcinoma, intraepidermal carcinoma, and psoriasis. J Am Acad Dermatol. 2008;59:268-274.
- Lallas A, Apalla Z, Argenziano G, et al. Dermoscopic pattern of psoriatic lesions on specific body sites. Dermatology. 2014;228:250-254.
- Almeida MC, Romiti R, Doche I, et al. Psoriatic scarring alopecia. An Bras Dermatol. 2013;88:29-31.
- Zalaudek I, Argenziano G. Dermoscopy subpatterns of inflammatory skin disorders. Arch Dermatol. 2006;142:808.
- Miteva M, Tosti A. Hair and scalp dermatoscopy. J Am Acad Dermatol. 2012;67:1040-1048.
- Vázquez-López F, Zaballos P, Fueyo-Casado A, et al. A dermoscopy subpattern of plaque-type psoriasis: red globular rings. Arch Dermatol. 2007;143:1612.
- Lacarrubba F, Nasca MR, Micali G. Videodermatoscopy enhances diagnostic capability in psoriatic balanitis. J Am Acad Dermatol. 2009;61:1084-1086.
- Liebman TN, Wang SQ. Detection of early basal cell carcinoma with dermoscopy in a patient with psoriasis. Dermatol Online J. 2011;17:12.
- Menzies SW, Westerhoff K, Rabinovitz H, et al. Surface microscopy of pigmented basal cell carcinoma. Arch Dermatol. 2000;136:1012-1016.
- Altamura D, Menzies SW, Argenziano G, et al. Dermatoscopy of basal cell carcinoma: morphologic variability of global and local features and accuracy of diagnosis. J Am Acad Dermatol. 2010;62:67-75.
- Marghoob AA, Malvehy J, Braun RP, eds. An Atlas of Dermoscopy. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Nelson SA, Scope A, Rishpon A, et al. Accuracy and confidence in the clinical diagnosis of basal cell cancer using dermoscopy and reflex confocal microscopy. Int J Dermatol. 2016;55:1351-1356.
- Zalaudek I, Kreusch J, Giacomel J, et al. How to diagnose nonpigmented skin tumors: a review of vascular structures seen with dermoscopy: part I. melanocytic skin tumors. J Am Acad Dermatol. 2010;63:361-374.
- Kittler H, Pehamberger H, Wolff K, et al. Diagnostic accuracy of dermoscopy. Lancet Oncol. 2002;3:159-165.
- Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676.
- Lallas A, Giacomel J, Argenziano G, et al. Dermoscopy in general dermatology: practical tips for the clinician. Br J Dermatol. 2014;170:514-526.
- Lallas A, Kyrgidis A, Tzellos TG, et al. Accuracy of dermoscopic criteria for the diagnosis of psoriasis, dermatitis, lichen planus and pityriasis rosea. Br J Dermatol. 2012;166:1198-1205.
- Pan Y, Chamberlain AJ, Bailey M, et al. Dermatoscopy aids in the diagnosis of the solitary red scaly patch or plaque–features distinguishing superficial basal cell carcinoma, intraepidermal carcinoma, and psoriasis. J Am Acad Dermatol. 2008;59:268-274.
- Lallas A, Apalla Z, Argenziano G, et al. Dermoscopic pattern of psoriatic lesions on specific body sites. Dermatology. 2014;228:250-254.
- Almeida MC, Romiti R, Doche I, et al. Psoriatic scarring alopecia. An Bras Dermatol. 2013;88:29-31.
- Zalaudek I, Argenziano G. Dermoscopy subpatterns of inflammatory skin disorders. Arch Dermatol. 2006;142:808.
- Miteva M, Tosti A. Hair and scalp dermatoscopy. J Am Acad Dermatol. 2012;67:1040-1048.
- Vázquez-López F, Zaballos P, Fueyo-Casado A, et al. A dermoscopy subpattern of plaque-type psoriasis: red globular rings. Arch Dermatol. 2007;143:1612.
- Lacarrubba F, Nasca MR, Micali G. Videodermatoscopy enhances diagnostic capability in psoriatic balanitis. J Am Acad Dermatol. 2009;61:1084-1086.
- Liebman TN, Wang SQ. Detection of early basal cell carcinoma with dermoscopy in a patient with psoriasis. Dermatol Online J. 2011;17:12.
- Menzies SW, Westerhoff K, Rabinovitz H, et al. Surface microscopy of pigmented basal cell carcinoma. Arch Dermatol. 2000;136:1012-1016.
- Altamura D, Menzies SW, Argenziano G, et al. Dermatoscopy of basal cell carcinoma: morphologic variability of global and local features and accuracy of diagnosis. J Am Acad Dermatol. 2010;62:67-75.
- Marghoob AA, Malvehy J, Braun RP, eds. An Atlas of Dermoscopy. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Nelson SA, Scope A, Rishpon A, et al. Accuracy and confidence in the clinical diagnosis of basal cell cancer using dermoscopy and reflex confocal microscopy. Int J Dermatol. 2016;55:1351-1356.
- Zalaudek I, Kreusch J, Giacomel J, et al. How to diagnose nonpigmented skin tumors: a review of vascular structures seen with dermoscopy: part I. melanocytic skin tumors. J Am Acad Dermatol. 2010;63:361-374.
Practice Points
- Dermoscopy has been largely utilized for the evaluation of malignant lesions. It also is gaining traction in the evaluation of inflammatory dermatoses.
- Early distinction between basal cell carcinoma and psoriasis is important for both treatment options and health care costs.
Perceptions of Tanning Risk Among Melanoma Patients With a History of Indoor Tanning
The incidence of melanoma is increasing at a rate greater than any other cancer,1 possibly due to the increasing use of indoor tanning devices. These devices emit unnaturally high levels of UVA and low levels of UVA and UVB rays.2 The risks of using these devices include increased incidence of melanoma (3438 cases attributed to indoor tanning in 2008) and keratinocytes cancer (increased risk of squamous cell carcinoma by 67% and basal cell carcinoma by 29%), severe sunburns (61.1% of female users and 44.6% of male users have reported sunburns), and aggravation of underlying disorders such as systemic lupus erythematosus.3-5
The literature varies in its explanation of how indoor tanning increases the risk of developing melanoma. Some authors suggest it is due to increased frequency of use, duration of sessions, and years of using tanning devices.1,6 Others suggest the increased cancer risk is the result of starting to tan at an earlier age.2,3,6-10 There is conflicting literature on the level of increased risk of melanoma in those who tan indoors at a young age (<35 years). Although the estimated rate of increased skin cancer risk varies, with rates up to 75% compared to nonusers, nearly all sources support an increased rate.6 Despite the growing body of knowledge that indoor tanning is dangerous, as well as the academic publication of these risks (eg, carcinogenesis, short-term and long-term eye injury, burns, UV sensitivity when combined with certain medications), teenagers in the United States and affluent countries appear to disregard the risks of tanning.11
Tanning companies have promoted the misconception that only UVB rays cause cell damage and UVA rays, which the devices emit, result in “damage-free” or “safe” tans.2,3 Until 2013, indoor tanning devices were classified by the US Food and Drug Administration (FDA) as class I, indicating that they are safe in terms of electrical shock. Many indoor tanning facilities have promoted the FDA “safe” label without clarifying that the safety indications only referred to electrical-shock potential. Nonetheless, it is known now that these devices, which emit high UVA and low UVB rays, promote melanoma, nonmelanoma skin cancers, and severe sunburns, as well as aggravate existing conditions (eg, systemic lupus erythematosus).4 As a result of an unacceptably high incidence of these disease complications, a 2014 FDA regulation categorized tanning beds as class II, requiring that tanning bed users be informed of the risk of skin cancer in an effort to reverse the growing trend of indoor tanning.12 Despite these regulatory interventions, it is not clear if this knowledge of cancer risk deters patients from indoor tanning.
The purpose of this study was to investigate the patients’ perspective on indoor tanning behaviors as associated with the severity of their melanoma and the time frame in which they were diagnosed as well as their perceived views on the safety of indoor tanning and the frequency in which they continue to tan indoors. This information is highly relevant in helping to determine if requiring a warning of the risk of skin cancer will deter patients from this unhealthy habit, especially given recent reclassification of sunbeds as class II by the FDA. Additional insights from these data may clarify if indoor tanning decreases the time frame in which melanoma is diagnosed or increases the severity of the resulting melanoma. Moreover, it will help elucidate whether or not the age at which indoor tanning is initiated affects the time frame to melanoma onset and corresponding severity.
Methods
An original unvalidated online survey was conducted worldwide via a link distributed to the following supporting institutions: Advanced Dermatology & Cosmetic Surgery, Ameriderm Research, Melanoma Research Foundation (a melanoma patient advocacy group), Florida State University Department of Dermatology, Moffitt Cancer Center Cutaneous Oncology Program, Cleveland Clinic, Ohio State University Division of Medical Oncology, Harvard Medical School Department of Dermatology, The University of Texas MD Anderson Cancer Center Department of Dermatology, University of Colorado Department of Dermatology, and Northwestern University Department of Dermatology. However, there was not confirmation that all of these institutions promoted the survey. Additionally, respondents were recruited through patient advocacy groups and social media sites including Facebook, Twitter, LinkedIn, Tumblr, and Instagram. The patient advocacy groups and social media sites invited participation through recruitment announcements, including DermNetNZ (a global dermatology patient information site), with additional help from the International Federation of Dermatology Clinical Trial Network.
The survey was restricted to those who were self-identified as 18 years or older and who self-reported a diagnosis of melanoma following the use of indoor tanning devices. The survey was hosted by SurveyMonkey, which allowed consent to be obtained and responses to remain anonymous. Access to the survey was sponsored by the Basal Cell Carcinoma Nevus Syndrome Life Support Network. The University of Central Florida (Orlando, Florida) institutional review board reviewed and approved this study as exempt human research.
Survey responses collected from January 2014 to June 2015 were analyzed herein. The survey contained 58 questions and was divided into different topics including indoor tanning background (eg, states/countries in which participants tanned indoors, age when they first tanned, frequency of tanning), consenting process (eg, length, did someone review the consent with participants, what was contained in the consent), indoor tanning and melanoma (eg, how long after tanning did melanoma develop, age at development, location of melanoma), indoor tanning postmelanoma (eg, did participants tan after diagnosis and why), and other risk factors (eg, did participants smoke or drink pre- or postmelanoma).
Statistical Analysis
The data consist of both categorical and continuous variables. The categorical variables included age (<35 years or ≥35 years), frequency of indoor tanning (≤1 time weekly or >1 time weekly), and onset of melanoma diagnosis (within or after 5 years
Difference in proportions among groups, age, frequency of tanning, onset of melanoma diagnosis within or after 5 years of starting indoor tanning, and knowledge of cancer risks was tested for significance using the χ² test. Reported P values were 2-tailed, corresponding with a significance level of P<.05. All data were analyzed using SPSS (version 21.0). All statistical analyses were conducted independent of the participants’ sex.
Results
Of the 454 participants who accessed the survey, 448 were analyzed in this study; 6 participants did not complete the questionnaire. Both males and females were analyzed: 289 females, 12 males, and 153 who did not report gender. The age range of participants was 18 to 69 years. The age at start of indoor tanning ranged from 8 to 54 years, with a mean of 22 years. Additional participant characteristics are described in Table 1. The mean frequency of indoor tanning was reported as 2 times weekly. When participants were asked if they were warned of the risk of skin cancer, 21.5% reported yes while 78.4% reported not being told of the risk. This knowledge was compared to their frequency of indoor tanning. Having the knowledge of the risk of skin cancer had no influence on their frequency of indoor tanning (Table 2).
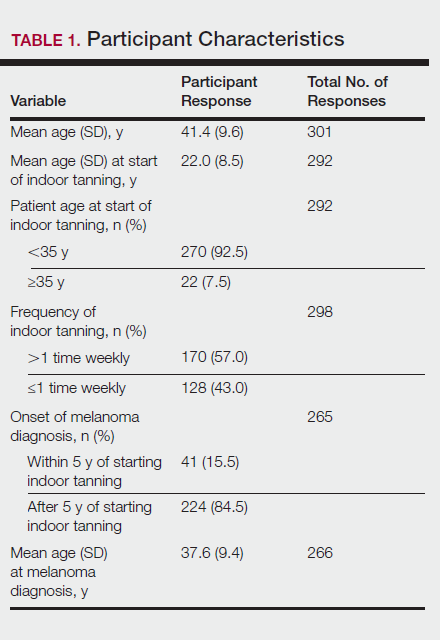
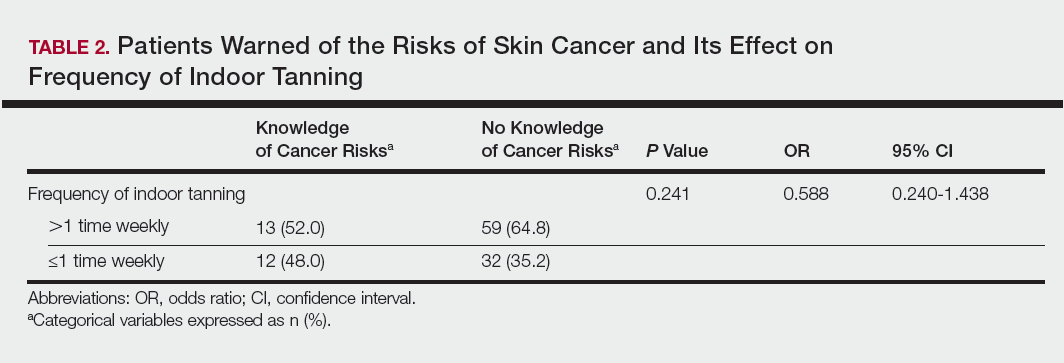
Among responders, those who perceived indoor tanning as safer than outdoor tanning tanned indoors more frequently than those who do not (Spearman r=−0.224; P<.05)(Table 3). The frequency of indoor tanning was divided into those who tanned indoors more than once weekly and those who tanned indoors once a week or less. This study showed that the frequency of indoor tanning had no effect on the latency time between the commencement of indoor tanning and diagnosis of melanoma (Table 4). The time frame from the onset of melanoma diagnosis also was compared to the age at which the participants started to tan indoors. Age was divided into those younger than 35 years and those 35 years and older. There was no correlation between the age when indoor tanning began and the time frame in which the melanoma was diagnosed (eTable).
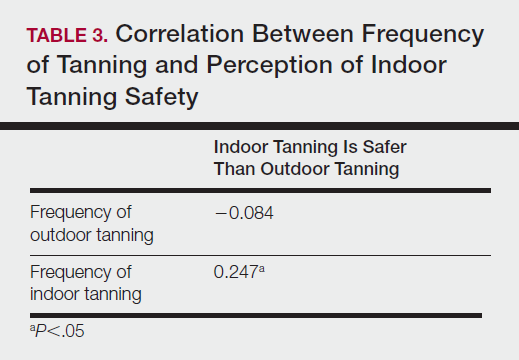

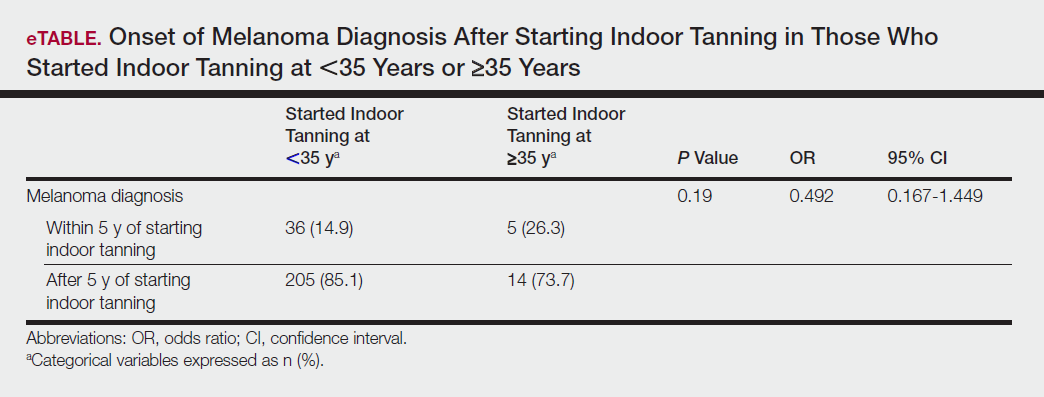
Table 5 shows the correlations between indoor tanning behaviors and melanoma characteristics. Those who started indoor tanning at an earlier age were diagnosed with melanoma at an earlier age compared to those who started indoor tanning later in life (r=0.549; P<.01). Moreover, those who started indoor tanning at a later age reported being diagnosed with a melanoma of greater Breslow depth (r=0.173; P<.01). Those who reported being diagnosed with a greater Breslow depth also reported a higher Clark level (r=0.608; P<.01). Among responders, those who more frequently tanned indoors also reported greater frequency of outdoor tanning (r=0.197; P<.01). This study showed no correlation between the age at melanoma diagnosis and the frequency of indoor (r=0.004; P>.05 not significant) or outdoor (r=0.093; P>.05 not significant) tanning. Having the knowledge of the risk of skin cancer had no relationship on the frequency of indoor tanning (r=−0.04; P>.05 not significant).
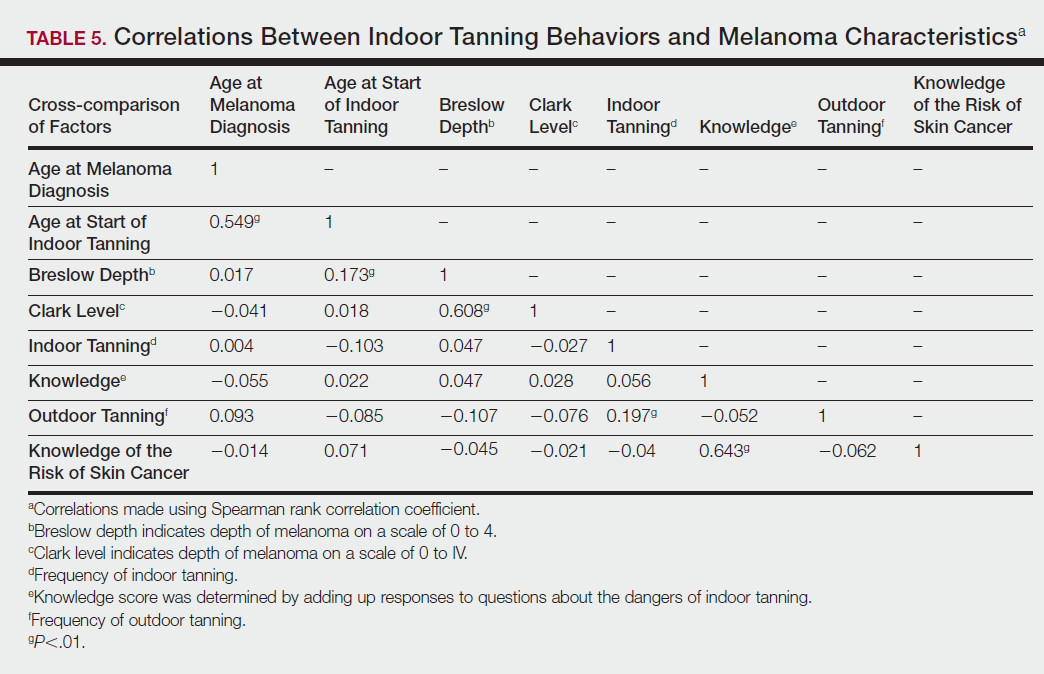
Comment
Thirty million Americans utilize indoor tanning devices at least once a year.13 UVA light comprises the majority of the spectrum used by indoor tanning devices, with a fraction (<5%) being UVB light. Until recently, UVB light was the only solar spectrum considered carcinogenic. In 2009, the International Agency for Research on Cancer classified the whole spectrum as carcinogenic to humans.5,11 Despite this evidence, indoor tanning facilities have promoted indoor tanning as damage free.3 The goal of this study was to collect the patient perspective on the safety of indoor tanning, indoor tanning behaviors, time frame of onset of melanoma, and the severity (ie, Breslow depth) of those melanomas.
Melanoma is the most prevalent cancer in females aged 25 to 29 years.3 The median age of diagnosis of melanoma (with and without the use of indoor tanning devices) is approximately 60 years14 versus our study, which found the average age at diagnosis was 37.6 years. Our findings are consistent with other literature in that those who start indoor tanning earlier (<35 years of age) develop melanoma at an earlier age.14,15 Cust et al14 also promoted the idea that patients develop melanoma earlier because younger individuals are more biologically susceptible to the carcinogenic effects of artificial UV light. However, our study found that those who started indoor tanning at an older age reported being diagnosed with a melanoma of greater Breslow depth, seemingly incongruent with the aforementioned hypothesis. One limitation is the age range for this research sample (18–69 years). The young age range may be attributable to the recruitment through social media, which is geared toward a younger population. Additionally, indoor tanning is a relatively new phenomenon practiced since the 1980s,2 which may contribute to the younger sample size. However, 2.7 billion individuals use social media worldwide with 40% of those older than 65 years on social media.16
Prior research has shown that those who start indoor tanning before the age of 35 years have a 75% increased risk of developing melanoma.14 Another study also has suggested that UVA-rich sunlamps may shorten the latency period for induction of melanoma and nonmelanoma skin cancers.3 Our study used similar age cutoffs in concluding that there was no earlier onset of melanoma diagnosis between those who started indoor tanning before the age of 35 years and those who started at the age of 35 years or older. Limitations include that our study is cross-sectional, and therefore time course cannot be established. Also, survey responses were self-reported, allowing the possibility of recall bias.
A plethora of research has been conducted to determine if there is a connection between the use of indoor tanning devices and developing melanoma. Cust et al14 suggested the risk of melanoma was 41% higher for those who had ever used a sunbed in comparison to those who had not. Other studies describe the difficulty in making the connection between indoor tanning and melanoma, as those who more frequently tan indoors also more frequently tan outdoors,11 as suggested by this study. However, there is a paucity of literature on the patients’ perspectives on the safety of indoor tanning. This study determined that those who more frequently tan indoors believed that indoor tanning is safer than outdoor tanning. With this altered perception promoted by the indoor tanning industry, the FDA has added a warning label to all indoor tanning devices about the risk of skin cancer. Our study revealed that having the knowledge of the risk of skin cancer had no influence on the frequency of indoor tanning. This concerning finding highlights a pressing need for an alternative approach to increase awareness of the harmful consequences that accompany indoor tanning. Further studies may elaborate on potential effective methods and messages to relate to an indoor tanning population comprised mostly of young females.
Acknowledgments
Supported and funded by the Basal Cell Carcinoma Nevus Syndrome Life Support Network. This research project was completed as part of the FIRE Module at the University of Central Florida, College of Medicine. We thank the FIRE Module faculty and staff for their assistance with this project.
- Fisher DE, James WD. Indoor tanning—science, behavior, and policy. N Engl J Med. 2010;363:901-903.
- Boniol M, Autier P, Boyle P, et al. Cutaneous melanoma attributable to sunbed use: systematic review and meta-analysis. BMJ. 2012;345:e4757.
- Coelho SG, Hearing VJ. UVA tanning is involved in the increased incidence of skin cancers in fair-skinned young women. Pigment Cell Melanoma Res. 2010;23:57-63.
- Klein RS, Sayre RM, Dowdy JC, et al. The risk of ultraviolet radiation exposure from indoor lamps in lupus erythematosus. Autoimmun Rev. 2009;8:320-324.
- O’Sullivan NA, Tait CP. Tanning bed and nail lamp use and the risk of cutaneous malignancy: a review of the literature. Australas J Dermatol. 2014;55:99-106.
- Schmidt CW. UV radiation and skin cancer: the science behind age restrictions for tanning beds. Environ Health Perspect. 2012;120:a308-a313.
- Lazovich D, Vogel RI, Berwick M, et al. Indoor tanning and risk of melanoma: a case-control study in a highly exposed population. Cancer Epidemiol Biomarkers Prev. 2010;19:1557-1568.
- Centers for Disease Control and Prevention (CDC). Use of indoor tanning devices by adults—United States, 2010. MMWR Morb Mortal Wkly Rep. 2012;61:323-326.
- Nielsen K, Masback A, Olsson H, et al. A prospective, population-based study of 40,000 women regarding host factors, UV exposure and sunbed use in relation to risk and anatomic site of cutaneous melanoma. Int J Cancer. 2012;131:706-715.
- Gandini S, Autier P, Boniol M. Reviews on sun exposure and artificial light and melanoma. Prog Biophys Mol Biol. 2011;107:362-366.
- Indoor tanning: the risks of ultraviolet rays. US Food and Drug Administration website. http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm186687.htm. Updated September 11, 2017. Accessed November 2, 2017.
- Food and Drug Administration, HHS. General and plastic surgery devices: reclassification of ultraviolet lamps for tanning, henceforth to be known as sunlamp products and ultraviolet lamps intended for use in sunlamp products. Fed Regist. 2014;79:31205-31214.
- Brady MS. Public health and the tanning bed controversy. J Clin Oncol. 2012;30:1571-1573.
- Cust AE, Armstrong BK, Goumas C, et al. Sunbed use during adolescence and early adulthood is associated with increased risk of early-onset melanoma. Int J Cancer. 2011;128:2425-2435.
- International Agency for Research on Cancer Working Group on artificial ultraviolet (UV) light and skin cancer. The association of use of sunbeds with cutaneous malignant melanoma and other skin cancers: a systematic review. Int J Cancer. 2007;120:1116-1122.
- Greenwood S, Perrin A, Duggan M. Social media update 2016. Pew Research Center website. http://www.pewinternet.org/2016/11/11/social-media-update-2016/. Published November 11, 2016. Accessed December 12, 2017.
The incidence of melanoma is increasing at a rate greater than any other cancer,1 possibly due to the increasing use of indoor tanning devices. These devices emit unnaturally high levels of UVA and low levels of UVA and UVB rays.2 The risks of using these devices include increased incidence of melanoma (3438 cases attributed to indoor tanning in 2008) and keratinocytes cancer (increased risk of squamous cell carcinoma by 67% and basal cell carcinoma by 29%), severe sunburns (61.1% of female users and 44.6% of male users have reported sunburns), and aggravation of underlying disorders such as systemic lupus erythematosus.3-5
The literature varies in its explanation of how indoor tanning increases the risk of developing melanoma. Some authors suggest it is due to increased frequency of use, duration of sessions, and years of using tanning devices.1,6 Others suggest the increased cancer risk is the result of starting to tan at an earlier age.2,3,6-10 There is conflicting literature on the level of increased risk of melanoma in those who tan indoors at a young age (<35 years). Although the estimated rate of increased skin cancer risk varies, with rates up to 75% compared to nonusers, nearly all sources support an increased rate.6 Despite the growing body of knowledge that indoor tanning is dangerous, as well as the academic publication of these risks (eg, carcinogenesis, short-term and long-term eye injury, burns, UV sensitivity when combined with certain medications), teenagers in the United States and affluent countries appear to disregard the risks of tanning.11
Tanning companies have promoted the misconception that only UVB rays cause cell damage and UVA rays, which the devices emit, result in “damage-free” or “safe” tans.2,3 Until 2013, indoor tanning devices were classified by the US Food and Drug Administration (FDA) as class I, indicating that they are safe in terms of electrical shock. Many indoor tanning facilities have promoted the FDA “safe” label without clarifying that the safety indications only referred to electrical-shock potential. Nonetheless, it is known now that these devices, which emit high UVA and low UVB rays, promote melanoma, nonmelanoma skin cancers, and severe sunburns, as well as aggravate existing conditions (eg, systemic lupus erythematosus).4 As a result of an unacceptably high incidence of these disease complications, a 2014 FDA regulation categorized tanning beds as class II, requiring that tanning bed users be informed of the risk of skin cancer in an effort to reverse the growing trend of indoor tanning.12 Despite these regulatory interventions, it is not clear if this knowledge of cancer risk deters patients from indoor tanning.
The purpose of this study was to investigate the patients’ perspective on indoor tanning behaviors as associated with the severity of their melanoma and the time frame in which they were diagnosed as well as their perceived views on the safety of indoor tanning and the frequency in which they continue to tan indoors. This information is highly relevant in helping to determine if requiring a warning of the risk of skin cancer will deter patients from this unhealthy habit, especially given recent reclassification of sunbeds as class II by the FDA. Additional insights from these data may clarify if indoor tanning decreases the time frame in which melanoma is diagnosed or increases the severity of the resulting melanoma. Moreover, it will help elucidate whether or not the age at which indoor tanning is initiated affects the time frame to melanoma onset and corresponding severity.
Methods
An original unvalidated online survey was conducted worldwide via a link distributed to the following supporting institutions: Advanced Dermatology & Cosmetic Surgery, Ameriderm Research, Melanoma Research Foundation (a melanoma patient advocacy group), Florida State University Department of Dermatology, Moffitt Cancer Center Cutaneous Oncology Program, Cleveland Clinic, Ohio State University Division of Medical Oncology, Harvard Medical School Department of Dermatology, The University of Texas MD Anderson Cancer Center Department of Dermatology, University of Colorado Department of Dermatology, and Northwestern University Department of Dermatology. However, there was not confirmation that all of these institutions promoted the survey. Additionally, respondents were recruited through patient advocacy groups and social media sites including Facebook, Twitter, LinkedIn, Tumblr, and Instagram. The patient advocacy groups and social media sites invited participation through recruitment announcements, including DermNetNZ (a global dermatology patient information site), with additional help from the International Federation of Dermatology Clinical Trial Network.
The survey was restricted to those who were self-identified as 18 years or older and who self-reported a diagnosis of melanoma following the use of indoor tanning devices. The survey was hosted by SurveyMonkey, which allowed consent to be obtained and responses to remain anonymous. Access to the survey was sponsored by the Basal Cell Carcinoma Nevus Syndrome Life Support Network. The University of Central Florida (Orlando, Florida) institutional review board reviewed and approved this study as exempt human research.
Survey responses collected from January 2014 to June 2015 were analyzed herein. The survey contained 58 questions and was divided into different topics including indoor tanning background (eg, states/countries in which participants tanned indoors, age when they first tanned, frequency of tanning), consenting process (eg, length, did someone review the consent with participants, what was contained in the consent), indoor tanning and melanoma (eg, how long after tanning did melanoma develop, age at development, location of melanoma), indoor tanning postmelanoma (eg, did participants tan after diagnosis and why), and other risk factors (eg, did participants smoke or drink pre- or postmelanoma).
Statistical Analysis
The data consist of both categorical and continuous variables. The categorical variables included age (<35 years or ≥35 years), frequency of indoor tanning (≤1 time weekly or >1 time weekly), and onset of melanoma diagnosis (within or after 5 years
Difference in proportions among groups, age, frequency of tanning, onset of melanoma diagnosis within or after 5 years of starting indoor tanning, and knowledge of cancer risks was tested for significance using the χ² test. Reported P values were 2-tailed, corresponding with a significance level of P<.05. All data were analyzed using SPSS (version 21.0). All statistical analyses were conducted independent of the participants’ sex.
Results
Of the 454 participants who accessed the survey, 448 were analyzed in this study; 6 participants did not complete the questionnaire. Both males and females were analyzed: 289 females, 12 males, and 153 who did not report gender. The age range of participants was 18 to 69 years. The age at start of indoor tanning ranged from 8 to 54 years, with a mean of 22 years. Additional participant characteristics are described in Table 1. The mean frequency of indoor tanning was reported as 2 times weekly. When participants were asked if they were warned of the risk of skin cancer, 21.5% reported yes while 78.4% reported not being told of the risk. This knowledge was compared to their frequency of indoor tanning. Having the knowledge of the risk of skin cancer had no influence on their frequency of indoor tanning (Table 2).
Among responders, those who perceived indoor tanning as safer than outdoor tanning tanned indoors more frequently than those who do not (Spearman r=−0.224; P<.05)(Table 3). The frequency of indoor tanning was divided into those who tanned indoors more than once weekly and those who tanned indoors once a week or less. This study showed that the frequency of indoor tanning had no effect on the latency time between the commencement of indoor tanning and diagnosis of melanoma (Table 4). The time frame from the onset of melanoma diagnosis also was compared to the age at which the participants started to tan indoors. Age was divided into those younger than 35 years and those 35 years and older. There was no correlation between the age when indoor tanning began and the time frame in which the melanoma was diagnosed (eTable).
Table 5 shows the correlations between indoor tanning behaviors and melanoma characteristics. Those who started indoor tanning at an earlier age were diagnosed with melanoma at an earlier age compared to those who started indoor tanning later in life (r=0.549; P<.01). Moreover, those who started indoor tanning at a later age reported being diagnosed with a melanoma of greater Breslow depth (r=0.173; P<.01). Those who reported being diagnosed with a greater Breslow depth also reported a higher Clark level (r=0.608; P<.01). Among responders, those who more frequently tanned indoors also reported greater frequency of outdoor tanning (r=0.197; P<.01). This study showed no correlation between the age at melanoma diagnosis and the frequency of indoor (r=0.004; P>.05 not significant) or outdoor (r=0.093; P>.05 not significant) tanning. Having the knowledge of the risk of skin cancer had no relationship on the frequency of indoor tanning (r=−0.04; P>.05 not significant).
Comment
Thirty million Americans utilize indoor tanning devices at least once a year.13 UVA light comprises the majority of the spectrum used by indoor tanning devices, with a fraction (<5%) being UVB light. Until recently, UVB light was the only solar spectrum considered carcinogenic. In 2009, the International Agency for Research on Cancer classified the whole spectrum as carcinogenic to humans.5,11 Despite this evidence, indoor tanning facilities have promoted indoor tanning as damage free.3 The goal of this study was to collect the patient perspective on the safety of indoor tanning, indoor tanning behaviors, time frame of onset of melanoma, and the severity (ie, Breslow depth) of those melanomas.
Melanoma is the most prevalent cancer in females aged 25 to 29 years.3 The median age of diagnosis of melanoma (with and without the use of indoor tanning devices) is approximately 60 years14 versus our study, which found the average age at diagnosis was 37.6 years. Our findings are consistent with other literature in that those who start indoor tanning earlier (<35 years of age) develop melanoma at an earlier age.14,15 Cust et al14 also promoted the idea that patients develop melanoma earlier because younger individuals are more biologically susceptible to the carcinogenic effects of artificial UV light. However, our study found that those who started indoor tanning at an older age reported being diagnosed with a melanoma of greater Breslow depth, seemingly incongruent with the aforementioned hypothesis. One limitation is the age range for this research sample (18–69 years). The young age range may be attributable to the recruitment through social media, which is geared toward a younger population. Additionally, indoor tanning is a relatively new phenomenon practiced since the 1980s,2 which may contribute to the younger sample size. However, 2.7 billion individuals use social media worldwide with 40% of those older than 65 years on social media.16
Prior research has shown that those who start indoor tanning before the age of 35 years have a 75% increased risk of developing melanoma.14 Another study also has suggested that UVA-rich sunlamps may shorten the latency period for induction of melanoma and nonmelanoma skin cancers.3 Our study used similar age cutoffs in concluding that there was no earlier onset of melanoma diagnosis between those who started indoor tanning before the age of 35 years and those who started at the age of 35 years or older. Limitations include that our study is cross-sectional, and therefore time course cannot be established. Also, survey responses were self-reported, allowing the possibility of recall bias.
A plethora of research has been conducted to determine if there is a connection between the use of indoor tanning devices and developing melanoma. Cust et al14 suggested the risk of melanoma was 41% higher for those who had ever used a sunbed in comparison to those who had not. Other studies describe the difficulty in making the connection between indoor tanning and melanoma, as those who more frequently tan indoors also more frequently tan outdoors,11 as suggested by this study. However, there is a paucity of literature on the patients’ perspectives on the safety of indoor tanning. This study determined that those who more frequently tan indoors believed that indoor tanning is safer than outdoor tanning. With this altered perception promoted by the indoor tanning industry, the FDA has added a warning label to all indoor tanning devices about the risk of skin cancer. Our study revealed that having the knowledge of the risk of skin cancer had no influence on the frequency of indoor tanning. This concerning finding highlights a pressing need for an alternative approach to increase awareness of the harmful consequences that accompany indoor tanning. Further studies may elaborate on potential effective methods and messages to relate to an indoor tanning population comprised mostly of young females.
Acknowledgments
Supported and funded by the Basal Cell Carcinoma Nevus Syndrome Life Support Network. This research project was completed as part of the FIRE Module at the University of Central Florida, College of Medicine. We thank the FIRE Module faculty and staff for their assistance with this project.
The incidence of melanoma is increasing at a rate greater than any other cancer,1 possibly due to the increasing use of indoor tanning devices. These devices emit unnaturally high levels of UVA and low levels of UVA and UVB rays.2 The risks of using these devices include increased incidence of melanoma (3438 cases attributed to indoor tanning in 2008) and keratinocytes cancer (increased risk of squamous cell carcinoma by 67% and basal cell carcinoma by 29%), severe sunburns (61.1% of female users and 44.6% of male users have reported sunburns), and aggravation of underlying disorders such as systemic lupus erythematosus.3-5
The literature varies in its explanation of how indoor tanning increases the risk of developing melanoma. Some authors suggest it is due to increased frequency of use, duration of sessions, and years of using tanning devices.1,6 Others suggest the increased cancer risk is the result of starting to tan at an earlier age.2,3,6-10 There is conflicting literature on the level of increased risk of melanoma in those who tan indoors at a young age (<35 years). Although the estimated rate of increased skin cancer risk varies, with rates up to 75% compared to nonusers, nearly all sources support an increased rate.6 Despite the growing body of knowledge that indoor tanning is dangerous, as well as the academic publication of these risks (eg, carcinogenesis, short-term and long-term eye injury, burns, UV sensitivity when combined with certain medications), teenagers in the United States and affluent countries appear to disregard the risks of tanning.11
Tanning companies have promoted the misconception that only UVB rays cause cell damage and UVA rays, which the devices emit, result in “damage-free” or “safe” tans.2,3 Until 2013, indoor tanning devices were classified by the US Food and Drug Administration (FDA) as class I, indicating that they are safe in terms of electrical shock. Many indoor tanning facilities have promoted the FDA “safe” label without clarifying that the safety indications only referred to electrical-shock potential. Nonetheless, it is known now that these devices, which emit high UVA and low UVB rays, promote melanoma, nonmelanoma skin cancers, and severe sunburns, as well as aggravate existing conditions (eg, systemic lupus erythematosus).4 As a result of an unacceptably high incidence of these disease complications, a 2014 FDA regulation categorized tanning beds as class II, requiring that tanning bed users be informed of the risk of skin cancer in an effort to reverse the growing trend of indoor tanning.12 Despite these regulatory interventions, it is not clear if this knowledge of cancer risk deters patients from indoor tanning.
The purpose of this study was to investigate the patients’ perspective on indoor tanning behaviors as associated with the severity of their melanoma and the time frame in which they were diagnosed as well as their perceived views on the safety of indoor tanning and the frequency in which they continue to tan indoors. This information is highly relevant in helping to determine if requiring a warning of the risk of skin cancer will deter patients from this unhealthy habit, especially given recent reclassification of sunbeds as class II by the FDA. Additional insights from these data may clarify if indoor tanning decreases the time frame in which melanoma is diagnosed or increases the severity of the resulting melanoma. Moreover, it will help elucidate whether or not the age at which indoor tanning is initiated affects the time frame to melanoma onset and corresponding severity.
Methods
An original unvalidated online survey was conducted worldwide via a link distributed to the following supporting institutions: Advanced Dermatology & Cosmetic Surgery, Ameriderm Research, Melanoma Research Foundation (a melanoma patient advocacy group), Florida State University Department of Dermatology, Moffitt Cancer Center Cutaneous Oncology Program, Cleveland Clinic, Ohio State University Division of Medical Oncology, Harvard Medical School Department of Dermatology, The University of Texas MD Anderson Cancer Center Department of Dermatology, University of Colorado Department of Dermatology, and Northwestern University Department of Dermatology. However, there was not confirmation that all of these institutions promoted the survey. Additionally, respondents were recruited through patient advocacy groups and social media sites including Facebook, Twitter, LinkedIn, Tumblr, and Instagram. The patient advocacy groups and social media sites invited participation through recruitment announcements, including DermNetNZ (a global dermatology patient information site), with additional help from the International Federation of Dermatology Clinical Trial Network.
The survey was restricted to those who were self-identified as 18 years or older and who self-reported a diagnosis of melanoma following the use of indoor tanning devices. The survey was hosted by SurveyMonkey, which allowed consent to be obtained and responses to remain anonymous. Access to the survey was sponsored by the Basal Cell Carcinoma Nevus Syndrome Life Support Network. The University of Central Florida (Orlando, Florida) institutional review board reviewed and approved this study as exempt human research.
Survey responses collected from January 2014 to June 2015 were analyzed herein. The survey contained 58 questions and was divided into different topics including indoor tanning background (eg, states/countries in which participants tanned indoors, age when they first tanned, frequency of tanning), consenting process (eg, length, did someone review the consent with participants, what was contained in the consent), indoor tanning and melanoma (eg, how long after tanning did melanoma develop, age at development, location of melanoma), indoor tanning postmelanoma (eg, did participants tan after diagnosis and why), and other risk factors (eg, did participants smoke or drink pre- or postmelanoma).
Statistical Analysis
The data consist of both categorical and continuous variables. The categorical variables included age (<35 years or ≥35 years), frequency of indoor tanning (≤1 time weekly or >1 time weekly), and onset of melanoma diagnosis (within or after 5 years
Difference in proportions among groups, age, frequency of tanning, onset of melanoma diagnosis within or after 5 years of starting indoor tanning, and knowledge of cancer risks was tested for significance using the χ² test. Reported P values were 2-tailed, corresponding with a significance level of P<.05. All data were analyzed using SPSS (version 21.0). All statistical analyses were conducted independent of the participants’ sex.
Results
Of the 454 participants who accessed the survey, 448 were analyzed in this study; 6 participants did not complete the questionnaire. Both males and females were analyzed: 289 females, 12 males, and 153 who did not report gender. The age range of participants was 18 to 69 years. The age at start of indoor tanning ranged from 8 to 54 years, with a mean of 22 years. Additional participant characteristics are described in Table 1. The mean frequency of indoor tanning was reported as 2 times weekly. When participants were asked if they were warned of the risk of skin cancer, 21.5% reported yes while 78.4% reported not being told of the risk. This knowledge was compared to their frequency of indoor tanning. Having the knowledge of the risk of skin cancer had no influence on their frequency of indoor tanning (Table 2).
Among responders, those who perceived indoor tanning as safer than outdoor tanning tanned indoors more frequently than those who do not (Spearman r=−0.224; P<.05)(Table 3). The frequency of indoor tanning was divided into those who tanned indoors more than once weekly and those who tanned indoors once a week or less. This study showed that the frequency of indoor tanning had no effect on the latency time between the commencement of indoor tanning and diagnosis of melanoma (Table 4). The time frame from the onset of melanoma diagnosis also was compared to the age at which the participants started to tan indoors. Age was divided into those younger than 35 years and those 35 years and older. There was no correlation between the age when indoor tanning began and the time frame in which the melanoma was diagnosed (eTable).
Table 5 shows the correlations between indoor tanning behaviors and melanoma characteristics. Those who started indoor tanning at an earlier age were diagnosed with melanoma at an earlier age compared to those who started indoor tanning later in life (r=0.549; P<.01). Moreover, those who started indoor tanning at a later age reported being diagnosed with a melanoma of greater Breslow depth (r=0.173; P<.01). Those who reported being diagnosed with a greater Breslow depth also reported a higher Clark level (r=0.608; P<.01). Among responders, those who more frequently tanned indoors also reported greater frequency of outdoor tanning (r=0.197; P<.01). This study showed no correlation between the age at melanoma diagnosis and the frequency of indoor (r=0.004; P>.05 not significant) or outdoor (r=0.093; P>.05 not significant) tanning. Having the knowledge of the risk of skin cancer had no relationship on the frequency of indoor tanning (r=−0.04; P>.05 not significant).
Comment
Thirty million Americans utilize indoor tanning devices at least once a year.13 UVA light comprises the majority of the spectrum used by indoor tanning devices, with a fraction (<5%) being UVB light. Until recently, UVB light was the only solar spectrum considered carcinogenic. In 2009, the International Agency for Research on Cancer classified the whole spectrum as carcinogenic to humans.5,11 Despite this evidence, indoor tanning facilities have promoted indoor tanning as damage free.3 The goal of this study was to collect the patient perspective on the safety of indoor tanning, indoor tanning behaviors, time frame of onset of melanoma, and the severity (ie, Breslow depth) of those melanomas.
Melanoma is the most prevalent cancer in females aged 25 to 29 years.3 The median age of diagnosis of melanoma (with and without the use of indoor tanning devices) is approximately 60 years14 versus our study, which found the average age at diagnosis was 37.6 years. Our findings are consistent with other literature in that those who start indoor tanning earlier (<35 years of age) develop melanoma at an earlier age.14,15 Cust et al14 also promoted the idea that patients develop melanoma earlier because younger individuals are more biologically susceptible to the carcinogenic effects of artificial UV light. However, our study found that those who started indoor tanning at an older age reported being diagnosed with a melanoma of greater Breslow depth, seemingly incongruent with the aforementioned hypothesis. One limitation is the age range for this research sample (18–69 years). The young age range may be attributable to the recruitment through social media, which is geared toward a younger population. Additionally, indoor tanning is a relatively new phenomenon practiced since the 1980s,2 which may contribute to the younger sample size. However, 2.7 billion individuals use social media worldwide with 40% of those older than 65 years on social media.16
Prior research has shown that those who start indoor tanning before the age of 35 years have a 75% increased risk of developing melanoma.14 Another study also has suggested that UVA-rich sunlamps may shorten the latency period for induction of melanoma and nonmelanoma skin cancers.3 Our study used similar age cutoffs in concluding that there was no earlier onset of melanoma diagnosis between those who started indoor tanning before the age of 35 years and those who started at the age of 35 years or older. Limitations include that our study is cross-sectional, and therefore time course cannot be established. Also, survey responses were self-reported, allowing the possibility of recall bias.
A plethora of research has been conducted to determine if there is a connection between the use of indoor tanning devices and developing melanoma. Cust et al14 suggested the risk of melanoma was 41% higher for those who had ever used a sunbed in comparison to those who had not. Other studies describe the difficulty in making the connection between indoor tanning and melanoma, as those who more frequently tan indoors also more frequently tan outdoors,11 as suggested by this study. However, there is a paucity of literature on the patients’ perspectives on the safety of indoor tanning. This study determined that those who more frequently tan indoors believed that indoor tanning is safer than outdoor tanning. With this altered perception promoted by the indoor tanning industry, the FDA has added a warning label to all indoor tanning devices about the risk of skin cancer. Our study revealed that having the knowledge of the risk of skin cancer had no influence on the frequency of indoor tanning. This concerning finding highlights a pressing need for an alternative approach to increase awareness of the harmful consequences that accompany indoor tanning. Further studies may elaborate on potential effective methods and messages to relate to an indoor tanning population comprised mostly of young females.
Acknowledgments
Supported and funded by the Basal Cell Carcinoma Nevus Syndrome Life Support Network. This research project was completed as part of the FIRE Module at the University of Central Florida, College of Medicine. We thank the FIRE Module faculty and staff for their assistance with this project.
- Fisher DE, James WD. Indoor tanning—science, behavior, and policy. N Engl J Med. 2010;363:901-903.
- Boniol M, Autier P, Boyle P, et al. Cutaneous melanoma attributable to sunbed use: systematic review and meta-analysis. BMJ. 2012;345:e4757.
- Coelho SG, Hearing VJ. UVA tanning is involved in the increased incidence of skin cancers in fair-skinned young women. Pigment Cell Melanoma Res. 2010;23:57-63.
- Klein RS, Sayre RM, Dowdy JC, et al. The risk of ultraviolet radiation exposure from indoor lamps in lupus erythematosus. Autoimmun Rev. 2009;8:320-324.
- O’Sullivan NA, Tait CP. Tanning bed and nail lamp use and the risk of cutaneous malignancy: a review of the literature. Australas J Dermatol. 2014;55:99-106.
- Schmidt CW. UV radiation and skin cancer: the science behind age restrictions for tanning beds. Environ Health Perspect. 2012;120:a308-a313.
- Lazovich D, Vogel RI, Berwick M, et al. Indoor tanning and risk of melanoma: a case-control study in a highly exposed population. Cancer Epidemiol Biomarkers Prev. 2010;19:1557-1568.
- Centers for Disease Control and Prevention (CDC). Use of indoor tanning devices by adults—United States, 2010. MMWR Morb Mortal Wkly Rep. 2012;61:323-326.
- Nielsen K, Masback A, Olsson H, et al. A prospective, population-based study of 40,000 women regarding host factors, UV exposure and sunbed use in relation to risk and anatomic site of cutaneous melanoma. Int J Cancer. 2012;131:706-715.
- Gandini S, Autier P, Boniol M. Reviews on sun exposure and artificial light and melanoma. Prog Biophys Mol Biol. 2011;107:362-366.
- Indoor tanning: the risks of ultraviolet rays. US Food and Drug Administration website. http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm186687.htm. Updated September 11, 2017. Accessed November 2, 2017.
- Food and Drug Administration, HHS. General and plastic surgery devices: reclassification of ultraviolet lamps for tanning, henceforth to be known as sunlamp products and ultraviolet lamps intended for use in sunlamp products. Fed Regist. 2014;79:31205-31214.
- Brady MS. Public health and the tanning bed controversy. J Clin Oncol. 2012;30:1571-1573.
- Cust AE, Armstrong BK, Goumas C, et al. Sunbed use during adolescence and early adulthood is associated with increased risk of early-onset melanoma. Int J Cancer. 2011;128:2425-2435.
- International Agency for Research on Cancer Working Group on artificial ultraviolet (UV) light and skin cancer. The association of use of sunbeds with cutaneous malignant melanoma and other skin cancers: a systematic review. Int J Cancer. 2007;120:1116-1122.
- Greenwood S, Perrin A, Duggan M. Social media update 2016. Pew Research Center website. http://www.pewinternet.org/2016/11/11/social-media-update-2016/. Published November 11, 2016. Accessed December 12, 2017.
- Fisher DE, James WD. Indoor tanning—science, behavior, and policy. N Engl J Med. 2010;363:901-903.
- Boniol M, Autier P, Boyle P, et al. Cutaneous melanoma attributable to sunbed use: systematic review and meta-analysis. BMJ. 2012;345:e4757.
- Coelho SG, Hearing VJ. UVA tanning is involved in the increased incidence of skin cancers in fair-skinned young women. Pigment Cell Melanoma Res. 2010;23:57-63.
- Klein RS, Sayre RM, Dowdy JC, et al. The risk of ultraviolet radiation exposure from indoor lamps in lupus erythematosus. Autoimmun Rev. 2009;8:320-324.
- O’Sullivan NA, Tait CP. Tanning bed and nail lamp use and the risk of cutaneous malignancy: a review of the literature. Australas J Dermatol. 2014;55:99-106.
- Schmidt CW. UV radiation and skin cancer: the science behind age restrictions for tanning beds. Environ Health Perspect. 2012;120:a308-a313.
- Lazovich D, Vogel RI, Berwick M, et al. Indoor tanning and risk of melanoma: a case-control study in a highly exposed population. Cancer Epidemiol Biomarkers Prev. 2010;19:1557-1568.
- Centers for Disease Control and Prevention (CDC). Use of indoor tanning devices by adults—United States, 2010. MMWR Morb Mortal Wkly Rep. 2012;61:323-326.
- Nielsen K, Masback A, Olsson H, et al. A prospective, population-based study of 40,000 women regarding host factors, UV exposure and sunbed use in relation to risk and anatomic site of cutaneous melanoma. Int J Cancer. 2012;131:706-715.
- Gandini S, Autier P, Boniol M. Reviews on sun exposure and artificial light and melanoma. Prog Biophys Mol Biol. 2011;107:362-366.
- Indoor tanning: the risks of ultraviolet rays. US Food and Drug Administration website. http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm186687.htm. Updated September 11, 2017. Accessed November 2, 2017.
- Food and Drug Administration, HHS. General and plastic surgery devices: reclassification of ultraviolet lamps for tanning, henceforth to be known as sunlamp products and ultraviolet lamps intended for use in sunlamp products. Fed Regist. 2014;79:31205-31214.
- Brady MS. Public health and the tanning bed controversy. J Clin Oncol. 2012;30:1571-1573.
- Cust AE, Armstrong BK, Goumas C, et al. Sunbed use during adolescence and early adulthood is associated with increased risk of early-onset melanoma. Int J Cancer. 2011;128:2425-2435.
- International Agency for Research on Cancer Working Group on artificial ultraviolet (UV) light and skin cancer. The association of use of sunbeds with cutaneous malignant melanoma and other skin cancers: a systematic review. Int J Cancer. 2007;120:1116-1122.
- Greenwood S, Perrin A, Duggan M. Social media update 2016. Pew Research Center website. http://www.pewinternet.org/2016/11/11/social-media-update-2016/. Published November 11, 2016. Accessed December 12, 2017.
Practice Points
- Despite US Food and Drug Administration reclassification and publicity of the risks of skin cancer, many patients continue to use sunbeds.
- It is important to assess how patients are obtaining information regarding sunbed safety, as indoor tanning companies are promoting sunbeds as “safe” tans.
- The increased combination of sunbed use and outdoor tanning is putting people at greater risk for the development of melanoma and nonmelanoma skin cancer.