User login
Methotrexate users need tuberculosis tests in high-TB areas
People taking even low-dose methotrexate need tuberculosis screening and ongoing clinical care if they live in areas where TB is common, results of a study presented at the virtual annual meeting of the American College of Rheumatology suggest.
Coauthor Carol Hitchon, MD, MSc, a rheumatologist with the University of Manitoba in Winnipeg, who presented the findings, warned that methotrexate (MTX) users who also take corticosteroids or other immunosuppressants are at particular risk and need TB screening.
Current management guidelines for rheumatic disease address TB in relation to biologics, but not in relation to methotrexate, Dr. Hitchon said.
“We know that methotrexate is the foundational DMARD [disease-modifying antirheumatic drug] for many rheumatic diseases, especially rheumatoid arthritis,” Dr. Hitchon noted at a press conference. “It’s safe and effective when dosed properly. However, methotrexate does have the potential for significant liver toxicity as well as infection, particularly for infectious organisms that are targeted by cell-mediated immunity, and TB is one of those agents.”
Using multiple databases, researchers conducted a systematic review of the literature published from 1990 to 2018 on TB rates among people who take less than 30 mg of methotrexate a week. Of the 4,700 studies they examined, 31 fit the criteria for this analysis.
They collected data on tuberculosis incidence or new TB diagnoses vs. reactivation of latent TB infection as well as TB outcomes, such as pulmonary symptoms, dissemination, and mortality.
They found a modest increase in the risk of TB infections in the setting of low-dose methotrexate. In addition, rates of TB in people with rheumatic disease who are treated with either methotrexate or biologics are generally higher than in the general population.
They also found that methotrexate users had higher rates of the type of TB that spreads beyond a patient’s lungs, compared with the general population.
Safety of INH with methotrexate
Researchers also looked at the safety of isoniazid (INH), the antibiotic used to treat TB, and found that isoniazid-related liver toxicity and neutropenia were more common when people took the antibiotic along with methotrexate, but those effects were usually reversible.
TB is endemic in various regions around the world. Historically there hasn’t been much rheumatology capacity in many of these areas, but as that capacity increases more people who are at high risk for developing or reactivating TB will be receiving methotrexate for rheumatic diseases, Dr. Hitchon said.
“It’s prudent for people managing patients who may be at higher risk for TB either from where they live or from where they travel that we should have a high suspicion for TB and consider screening as part of our workup in the course of initiating treatment like methotrexate,” she said.
Narender Annapureddy, MD, a rheumatologist at Vanderbilt University, Nashville, Tenn., who was not involved in the research, pointed out that a limitation of the work is that only 27% of the studies are from developing countries, which are more likely to have endemic TB, and those studies had very few cases.
“This finding needs to be studied in larger populations in TB-endemic areas and in high-risk populations,” he said in an interview.
As for practice implications in the United States, Dr. Annapureddy noted that TB is rare in the United States and most of the cases occur in people born in other countries.
“This population may be at risk for TB and should probably be screened for TB before initiating methotrexate,” he said. “Since biologics are usually the next step, especially in RA after patients fail methotrexate, having information on TB status may also help guide management options after MTX failure.
“Since high-dose steroids are another important risk factor for TB activation,” Dr. Annapureddy continued, “rheumatologists should likely consider screening patients who are going to be on moderate to high doses of steroids with MTX.”
A version of this article originally appeared on Medscape.com.
People taking even low-dose methotrexate need tuberculosis screening and ongoing clinical care if they live in areas where TB is common, results of a study presented at the virtual annual meeting of the American College of Rheumatology suggest.
Coauthor Carol Hitchon, MD, MSc, a rheumatologist with the University of Manitoba in Winnipeg, who presented the findings, warned that methotrexate (MTX) users who also take corticosteroids or other immunosuppressants are at particular risk and need TB screening.
Current management guidelines for rheumatic disease address TB in relation to biologics, but not in relation to methotrexate, Dr. Hitchon said.
“We know that methotrexate is the foundational DMARD [disease-modifying antirheumatic drug] for many rheumatic diseases, especially rheumatoid arthritis,” Dr. Hitchon noted at a press conference. “It’s safe and effective when dosed properly. However, methotrexate does have the potential for significant liver toxicity as well as infection, particularly for infectious organisms that are targeted by cell-mediated immunity, and TB is one of those agents.”
Using multiple databases, researchers conducted a systematic review of the literature published from 1990 to 2018 on TB rates among people who take less than 30 mg of methotrexate a week. Of the 4,700 studies they examined, 31 fit the criteria for this analysis.
They collected data on tuberculosis incidence or new TB diagnoses vs. reactivation of latent TB infection as well as TB outcomes, such as pulmonary symptoms, dissemination, and mortality.
They found a modest increase in the risk of TB infections in the setting of low-dose methotrexate. In addition, rates of TB in people with rheumatic disease who are treated with either methotrexate or biologics are generally higher than in the general population.
They also found that methotrexate users had higher rates of the type of TB that spreads beyond a patient’s lungs, compared with the general population.
Safety of INH with methotrexate
Researchers also looked at the safety of isoniazid (INH), the antibiotic used to treat TB, and found that isoniazid-related liver toxicity and neutropenia were more common when people took the antibiotic along with methotrexate, but those effects were usually reversible.
TB is endemic in various regions around the world. Historically there hasn’t been much rheumatology capacity in many of these areas, but as that capacity increases more people who are at high risk for developing or reactivating TB will be receiving methotrexate for rheumatic diseases, Dr. Hitchon said.
“It’s prudent for people managing patients who may be at higher risk for TB either from where they live or from where they travel that we should have a high suspicion for TB and consider screening as part of our workup in the course of initiating treatment like methotrexate,” she said.
Narender Annapureddy, MD, a rheumatologist at Vanderbilt University, Nashville, Tenn., who was not involved in the research, pointed out that a limitation of the work is that only 27% of the studies are from developing countries, which are more likely to have endemic TB, and those studies had very few cases.
“This finding needs to be studied in larger populations in TB-endemic areas and in high-risk populations,” he said in an interview.
As for practice implications in the United States, Dr. Annapureddy noted that TB is rare in the United States and most of the cases occur in people born in other countries.
“This population may be at risk for TB and should probably be screened for TB before initiating methotrexate,” he said. “Since biologics are usually the next step, especially in RA after patients fail methotrexate, having information on TB status may also help guide management options after MTX failure.
“Since high-dose steroids are another important risk factor for TB activation,” Dr. Annapureddy continued, “rheumatologists should likely consider screening patients who are going to be on moderate to high doses of steroids with MTX.”
A version of this article originally appeared on Medscape.com.
People taking even low-dose methotrexate need tuberculosis screening and ongoing clinical care if they live in areas where TB is common, results of a study presented at the virtual annual meeting of the American College of Rheumatology suggest.
Coauthor Carol Hitchon, MD, MSc, a rheumatologist with the University of Manitoba in Winnipeg, who presented the findings, warned that methotrexate (MTX) users who also take corticosteroids or other immunosuppressants are at particular risk and need TB screening.
Current management guidelines for rheumatic disease address TB in relation to biologics, but not in relation to methotrexate, Dr. Hitchon said.
“We know that methotrexate is the foundational DMARD [disease-modifying antirheumatic drug] for many rheumatic diseases, especially rheumatoid arthritis,” Dr. Hitchon noted at a press conference. “It’s safe and effective when dosed properly. However, methotrexate does have the potential for significant liver toxicity as well as infection, particularly for infectious organisms that are targeted by cell-mediated immunity, and TB is one of those agents.”
Using multiple databases, researchers conducted a systematic review of the literature published from 1990 to 2018 on TB rates among people who take less than 30 mg of methotrexate a week. Of the 4,700 studies they examined, 31 fit the criteria for this analysis.
They collected data on tuberculosis incidence or new TB diagnoses vs. reactivation of latent TB infection as well as TB outcomes, such as pulmonary symptoms, dissemination, and mortality.
They found a modest increase in the risk of TB infections in the setting of low-dose methotrexate. In addition, rates of TB in people with rheumatic disease who are treated with either methotrexate or biologics are generally higher than in the general population.
They also found that methotrexate users had higher rates of the type of TB that spreads beyond a patient’s lungs, compared with the general population.
Safety of INH with methotrexate
Researchers also looked at the safety of isoniazid (INH), the antibiotic used to treat TB, and found that isoniazid-related liver toxicity and neutropenia were more common when people took the antibiotic along with methotrexate, but those effects were usually reversible.
TB is endemic in various regions around the world. Historically there hasn’t been much rheumatology capacity in many of these areas, but as that capacity increases more people who are at high risk for developing or reactivating TB will be receiving methotrexate for rheumatic diseases, Dr. Hitchon said.
“It’s prudent for people managing patients who may be at higher risk for TB either from where they live or from where they travel that we should have a high suspicion for TB and consider screening as part of our workup in the course of initiating treatment like methotrexate,” she said.
Narender Annapureddy, MD, a rheumatologist at Vanderbilt University, Nashville, Tenn., who was not involved in the research, pointed out that a limitation of the work is that only 27% of the studies are from developing countries, which are more likely to have endemic TB, and those studies had very few cases.
“This finding needs to be studied in larger populations in TB-endemic areas and in high-risk populations,” he said in an interview.
As for practice implications in the United States, Dr. Annapureddy noted that TB is rare in the United States and most of the cases occur in people born in other countries.
“This population may be at risk for TB and should probably be screened for TB before initiating methotrexate,” he said. “Since biologics are usually the next step, especially in RA after patients fail methotrexate, having information on TB status may also help guide management options after MTX failure.
“Since high-dose steroids are another important risk factor for TB activation,” Dr. Annapureddy continued, “rheumatologists should likely consider screening patients who are going to be on moderate to high doses of steroids with MTX.”
A version of this article originally appeared on Medscape.com.
Transcutaneous VNS on the ear shows positive effects in lupus pilot trial
during a brief pilot trial.
“Our study population included individuals with significant pain and exemplifies the unmet need for adequate control of pain and fatigue in SLE. Importantly, this was a double-blind, sham-controlled study and neither the subject nor assessor was aware of a subject’s intervention. Objective outcomes, that is, tender and swollen joint counts, were also significantly reduced in subjects receiving taVNS, compared with those receiving [sham stimulation]. The stimulation was well tolerated with no adverse events attributed to the intervention, and, clinical benefits continued after taVNS was stopped,” first author Cynthia Aranow, MD, and her colleagues at the Feinstein Institutes for Medical Research in Manhasset, N.Y., wrote in Annals of the Rheumatic Diseases.
Stimulation of the vagus nerve can be achieved through the ear via its auricular branch, which innervates the cymba concha in the outer ear. Past pilot studies of implanted VNS devices lasting 6 weeks to 6 months in patients with Crohn’s disease or rheumatoid arthritis have shown improvements in measures of disease activity as well as objective markers of inflammation, and a more recent trial testing a transcutaneous devices’s effect in patients with Sjögren’s syndrome found significant reductions in fatigue over a 26-day period, the investigators noted.
In the taVNS device study, the researchers recruited 18 patients with SLE who had musculoskeletal pain rated as 4 or higher on a 10-cm visual analog scale and randomized them in a 2:1 ratio to receive taVNS once per day for 5 minutes for 4 consecutive days versus sham stimulation. Patients were allowed to be on stable doses of disease-modifying antirheumatic drugs, biologics, and/or prednisone ≤ 10 mg/day, with no change of dose within 28 days prior to baseline. The study excluded patients who used tobacco or an anticholinergic medication and those with a diagnosis of fibromyalgia.
The 12 patients who received actual taVNS had a significantly greater reduction in their pain, compared with 6 sham-treated patients (–5.00 vs. 0.10; P = .049), with 10 of 12 and 1 of 6 having a clinical response (a reduction of at least 1.58 on a 10-cm visual analog scale from baseline to day 5). Stimulation-treated patients also reported significantly greater reductions in fatigue, with 10 of 12 achieving a meaningful reduction, defined as a 4-point improvement on the Functional Assessment of Chronic Illness Therapy Fatigue Subscale; none of the sham-treated patients experienced meaningful improvement of fatigue. The patients who received taVNS had resolution of all swollen and tender joints, compared with 5.3% of tender and 9.1% of swollen joints in sham-treated patients. Ex vivo lipopolysaccharide stimulation of whole-blood samples from taVNS-treated patients, however, showed no reductions of inflammatory mediators or chemokines in tests on day 5 and day 12.
The investigators reported that there were no adverse events attributed to taVNS, including no reports of headache, lightheadedness, tinnitus, ear irritation, or changes to the external skin of the outer ear.
The study was supported by a grant from the John and Marcia Goldman Foundation. One author reported a financial relationship with Set Point Medical and My String, and three authors reported having a provisional patent application titled “Auricular stimulation device, system and methods of use.”
SOURCE: Aranow C et al. Ann Rheum Dis. 2020 Nov 3. doi: 10.1136/annrheumdis-2020-217872.
during a brief pilot trial.
“Our study population included individuals with significant pain and exemplifies the unmet need for adequate control of pain and fatigue in SLE. Importantly, this was a double-blind, sham-controlled study and neither the subject nor assessor was aware of a subject’s intervention. Objective outcomes, that is, tender and swollen joint counts, were also significantly reduced in subjects receiving taVNS, compared with those receiving [sham stimulation]. The stimulation was well tolerated with no adverse events attributed to the intervention, and, clinical benefits continued after taVNS was stopped,” first author Cynthia Aranow, MD, and her colleagues at the Feinstein Institutes for Medical Research in Manhasset, N.Y., wrote in Annals of the Rheumatic Diseases.
Stimulation of the vagus nerve can be achieved through the ear via its auricular branch, which innervates the cymba concha in the outer ear. Past pilot studies of implanted VNS devices lasting 6 weeks to 6 months in patients with Crohn’s disease or rheumatoid arthritis have shown improvements in measures of disease activity as well as objective markers of inflammation, and a more recent trial testing a transcutaneous devices’s effect in patients with Sjögren’s syndrome found significant reductions in fatigue over a 26-day period, the investigators noted.
In the taVNS device study, the researchers recruited 18 patients with SLE who had musculoskeletal pain rated as 4 or higher on a 10-cm visual analog scale and randomized them in a 2:1 ratio to receive taVNS once per day for 5 minutes for 4 consecutive days versus sham stimulation. Patients were allowed to be on stable doses of disease-modifying antirheumatic drugs, biologics, and/or prednisone ≤ 10 mg/day, with no change of dose within 28 days prior to baseline. The study excluded patients who used tobacco or an anticholinergic medication and those with a diagnosis of fibromyalgia.
The 12 patients who received actual taVNS had a significantly greater reduction in their pain, compared with 6 sham-treated patients (–5.00 vs. 0.10; P = .049), with 10 of 12 and 1 of 6 having a clinical response (a reduction of at least 1.58 on a 10-cm visual analog scale from baseline to day 5). Stimulation-treated patients also reported significantly greater reductions in fatigue, with 10 of 12 achieving a meaningful reduction, defined as a 4-point improvement on the Functional Assessment of Chronic Illness Therapy Fatigue Subscale; none of the sham-treated patients experienced meaningful improvement of fatigue. The patients who received taVNS had resolution of all swollen and tender joints, compared with 5.3% of tender and 9.1% of swollen joints in sham-treated patients. Ex vivo lipopolysaccharide stimulation of whole-blood samples from taVNS-treated patients, however, showed no reductions of inflammatory mediators or chemokines in tests on day 5 and day 12.
The investigators reported that there were no adverse events attributed to taVNS, including no reports of headache, lightheadedness, tinnitus, ear irritation, or changes to the external skin of the outer ear.
The study was supported by a grant from the John and Marcia Goldman Foundation. One author reported a financial relationship with Set Point Medical and My String, and three authors reported having a provisional patent application titled “Auricular stimulation device, system and methods of use.”
SOURCE: Aranow C et al. Ann Rheum Dis. 2020 Nov 3. doi: 10.1136/annrheumdis-2020-217872.
during a brief pilot trial.
“Our study population included individuals with significant pain and exemplifies the unmet need for adequate control of pain and fatigue in SLE. Importantly, this was a double-blind, sham-controlled study and neither the subject nor assessor was aware of a subject’s intervention. Objective outcomes, that is, tender and swollen joint counts, were also significantly reduced in subjects receiving taVNS, compared with those receiving [sham stimulation]. The stimulation was well tolerated with no adverse events attributed to the intervention, and, clinical benefits continued after taVNS was stopped,” first author Cynthia Aranow, MD, and her colleagues at the Feinstein Institutes for Medical Research in Manhasset, N.Y., wrote in Annals of the Rheumatic Diseases.
Stimulation of the vagus nerve can be achieved through the ear via its auricular branch, which innervates the cymba concha in the outer ear. Past pilot studies of implanted VNS devices lasting 6 weeks to 6 months in patients with Crohn’s disease or rheumatoid arthritis have shown improvements in measures of disease activity as well as objective markers of inflammation, and a more recent trial testing a transcutaneous devices’s effect in patients with Sjögren’s syndrome found significant reductions in fatigue over a 26-day period, the investigators noted.
In the taVNS device study, the researchers recruited 18 patients with SLE who had musculoskeletal pain rated as 4 or higher on a 10-cm visual analog scale and randomized them in a 2:1 ratio to receive taVNS once per day for 5 minutes for 4 consecutive days versus sham stimulation. Patients were allowed to be on stable doses of disease-modifying antirheumatic drugs, biologics, and/or prednisone ≤ 10 mg/day, with no change of dose within 28 days prior to baseline. The study excluded patients who used tobacco or an anticholinergic medication and those with a diagnosis of fibromyalgia.
The 12 patients who received actual taVNS had a significantly greater reduction in their pain, compared with 6 sham-treated patients (–5.00 vs. 0.10; P = .049), with 10 of 12 and 1 of 6 having a clinical response (a reduction of at least 1.58 on a 10-cm visual analog scale from baseline to day 5). Stimulation-treated patients also reported significantly greater reductions in fatigue, with 10 of 12 achieving a meaningful reduction, defined as a 4-point improvement on the Functional Assessment of Chronic Illness Therapy Fatigue Subscale; none of the sham-treated patients experienced meaningful improvement of fatigue. The patients who received taVNS had resolution of all swollen and tender joints, compared with 5.3% of tender and 9.1% of swollen joints in sham-treated patients. Ex vivo lipopolysaccharide stimulation of whole-blood samples from taVNS-treated patients, however, showed no reductions of inflammatory mediators or chemokines in tests on day 5 and day 12.
The investigators reported that there were no adverse events attributed to taVNS, including no reports of headache, lightheadedness, tinnitus, ear irritation, or changes to the external skin of the outer ear.
The study was supported by a grant from the John and Marcia Goldman Foundation. One author reported a financial relationship with Set Point Medical and My String, and three authors reported having a provisional patent application titled “Auricular stimulation device, system and methods of use.”
SOURCE: Aranow C et al. Ann Rheum Dis. 2020 Nov 3. doi: 10.1136/annrheumdis-2020-217872.
FROM ANNALS OF THE RHEUMATIC DISEASES
COVID-19–related HCQ shortages affected rheumatology patients worldwide
New data document the global fallout for rheumatology patients when hydroxychloroquine (HCQ) supplies were being diverted to hospitals for COVID-19 patients.
Demand for HCQ soared on evidence-lacking claims that the drug was effective in treating and preventing SARS-CoV-2 infection. Further research has since shown HCQ to be ineffective for COVID-19 and potentially harmful to patients.
But during the height of the COVID-19-related hype, patients worldwide with autoimmune diseases, particularly lupus and rheumatoid arthritis, had trouble getting the pills at all or couldn’t get as many as they needed for their chronic conditions.
Emily Sirotich, MSc, a PhD student at McMaster University in Hamilton, Ont., presented data at the virtual annual meeting of the American College of Rheumatology demonstrating that the severity of shortages differed widely.
Whereas 26.7% of rheumatology patients in Africa and 21.4% in southeast Asia said their pharmacy ran short of HCQ – which was originally developed as an antimalarial drug but has been found effective in treating some rheumatic diseases – only 6.8% of patients in the Americas and 2.1% in European regions reported the shortages.
“There are large regional disparities in access to antimalarials whether they were caused by the COVID-19 pandemic or already existed,” she said in an interview.
Global survey polled patient experience
Ms. Sirotich’s team analyzed data from the Global Rheumatology Alliance Patient Experience Survey.
They found that from 9,393 respondents (average age 46.1 years and 90% female), 3,872 (41.2%) were taking antimalarials. Of these, 230 (6.2% globally) were unable to keep taking the drugs because their pharmacy ran out.
Researchers evaluated the effect of drug shortages on disease activity, mental health, and physical health by comparing mean values with two-sided independent t-tests to identify significant differences.
They found that patients who were unable to obtain antimalarials had significantly higher levels of rheumatic disease activity as well as poorer mental and physical health (all P < .001).
The survey was distributed online through patient support groups and on social media. Patients with rheumatic diseases or their parents anonymously entered data including their rheumatic disease diagnosis, medications, COVID-19 status, and disease outcomes.
Ms. Sirotich said they are currently gathering new data to see if the gaps in access to HCQ persist and whether the physical and mental consequences of not having the medications continue.
Hospitals stockpiled HCQ in the U.S.
Michael Ganio, PharmD, senior director of pharmacy practice and quality at the American Society of Health-System Pharmacists (ASHP), said in an interview that hospitals in the United States received large amounts of HCQ in late spring and early summer, donated by pharmaceutical companies for COVID-19 before the lack of evidence for efficacy became clear.
Hospitals found themselves sitting on large quantities of HCQ they couldn’t use while prescriptions for rheumatology outpatients were going unfilled.
It is only in recent months that the U.S. Department of Health and Human Services has given clear direction to hospitals on how to redistribute those supplies, Dr. Ganio said.
“There’s no good real good way to move a product from a hospital to a [drug store] down the street,” he said.
The Food and Drug Administration now lists the HCQ shortages as resolved.
Declined prescriptions have frustrated physicians
Brett Smith, DO, a pediatric and adult rheumatologist in Alcoa, Tenn., said he was frustrated by pharmacies declining his prescriptions for HCQ for patients with rheumatoid arthritis.
“I got notes from pharmacies that I should consider alternative agents,” he said in an interview. But the safety profiles of the alternatives were not as good, he said.
“Hydroxychloroquine has no risk of infection and no risk of malignancy, and they were proposing alternative agents that carry those risks,” he said.
“I had some people with RA who couldn’t get [HCQ] who had a substantial increase in swollen joints and pain without it,” he said.
Dr. Smith said some patients who use HCQ for off-label uses such as certain skin disorders still aren’t getting the drug, as off-label use has been discouraged to make sure those with lupus and RA have enough, he said.
Saira Sheikh, MD, director of the University of North Carolina Rheumatology Lupus Clinic in Chapel Hill, said in an interview that during the summer months pharmacists required additional documentation of the diagnosis of autoimmune disease, resulting in unnecessary delays even when patients had been on the medication for many years.
She said emerging research has found patient-reported barriers to filling prescriptions, interruptions in HCQ treatment, and reported emotional stress and anxiety related to medication access during the COVID-19 pandemic.
“This experience with HCQ during the COVID-19 pandemic teaches us that while swift action and progress to address the immediate threats of the pandemic should be commended, it is important that we move forward in a conscious manner, guided by an evidence base that comes from high-quality research, not from rushed judgments based on preliminary studies, or pressure from political leaders,” Dr. Sheikh said.
Ms. Sirotich, Dr. Smith, Dr. Sheikh, and Dr. Ganio have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
New data document the global fallout for rheumatology patients when hydroxychloroquine (HCQ) supplies were being diverted to hospitals for COVID-19 patients.
Demand for HCQ soared on evidence-lacking claims that the drug was effective in treating and preventing SARS-CoV-2 infection. Further research has since shown HCQ to be ineffective for COVID-19 and potentially harmful to patients.
But during the height of the COVID-19-related hype, patients worldwide with autoimmune diseases, particularly lupus and rheumatoid arthritis, had trouble getting the pills at all or couldn’t get as many as they needed for their chronic conditions.
Emily Sirotich, MSc, a PhD student at McMaster University in Hamilton, Ont., presented data at the virtual annual meeting of the American College of Rheumatology demonstrating that the severity of shortages differed widely.
Whereas 26.7% of rheumatology patients in Africa and 21.4% in southeast Asia said their pharmacy ran short of HCQ – which was originally developed as an antimalarial drug but has been found effective in treating some rheumatic diseases – only 6.8% of patients in the Americas and 2.1% in European regions reported the shortages.
“There are large regional disparities in access to antimalarials whether they were caused by the COVID-19 pandemic or already existed,” she said in an interview.
Global survey polled patient experience
Ms. Sirotich’s team analyzed data from the Global Rheumatology Alliance Patient Experience Survey.
They found that from 9,393 respondents (average age 46.1 years and 90% female), 3,872 (41.2%) were taking antimalarials. Of these, 230 (6.2% globally) were unable to keep taking the drugs because their pharmacy ran out.
Researchers evaluated the effect of drug shortages on disease activity, mental health, and physical health by comparing mean values with two-sided independent t-tests to identify significant differences.
They found that patients who were unable to obtain antimalarials had significantly higher levels of rheumatic disease activity as well as poorer mental and physical health (all P < .001).
The survey was distributed online through patient support groups and on social media. Patients with rheumatic diseases or their parents anonymously entered data including their rheumatic disease diagnosis, medications, COVID-19 status, and disease outcomes.
Ms. Sirotich said they are currently gathering new data to see if the gaps in access to HCQ persist and whether the physical and mental consequences of not having the medications continue.
Hospitals stockpiled HCQ in the U.S.
Michael Ganio, PharmD, senior director of pharmacy practice and quality at the American Society of Health-System Pharmacists (ASHP), said in an interview that hospitals in the United States received large amounts of HCQ in late spring and early summer, donated by pharmaceutical companies for COVID-19 before the lack of evidence for efficacy became clear.
Hospitals found themselves sitting on large quantities of HCQ they couldn’t use while prescriptions for rheumatology outpatients were going unfilled.
It is only in recent months that the U.S. Department of Health and Human Services has given clear direction to hospitals on how to redistribute those supplies, Dr. Ganio said.
“There’s no good real good way to move a product from a hospital to a [drug store] down the street,” he said.
The Food and Drug Administration now lists the HCQ shortages as resolved.
Declined prescriptions have frustrated physicians
Brett Smith, DO, a pediatric and adult rheumatologist in Alcoa, Tenn., said he was frustrated by pharmacies declining his prescriptions for HCQ for patients with rheumatoid arthritis.
“I got notes from pharmacies that I should consider alternative agents,” he said in an interview. But the safety profiles of the alternatives were not as good, he said.
“Hydroxychloroquine has no risk of infection and no risk of malignancy, and they were proposing alternative agents that carry those risks,” he said.
“I had some people with RA who couldn’t get [HCQ] who had a substantial increase in swollen joints and pain without it,” he said.
Dr. Smith said some patients who use HCQ for off-label uses such as certain skin disorders still aren’t getting the drug, as off-label use has been discouraged to make sure those with lupus and RA have enough, he said.
Saira Sheikh, MD, director of the University of North Carolina Rheumatology Lupus Clinic in Chapel Hill, said in an interview that during the summer months pharmacists required additional documentation of the diagnosis of autoimmune disease, resulting in unnecessary delays even when patients had been on the medication for many years.
She said emerging research has found patient-reported barriers to filling prescriptions, interruptions in HCQ treatment, and reported emotional stress and anxiety related to medication access during the COVID-19 pandemic.
“This experience with HCQ during the COVID-19 pandemic teaches us that while swift action and progress to address the immediate threats of the pandemic should be commended, it is important that we move forward in a conscious manner, guided by an evidence base that comes from high-quality research, not from rushed judgments based on preliminary studies, or pressure from political leaders,” Dr. Sheikh said.
Ms. Sirotich, Dr. Smith, Dr. Sheikh, and Dr. Ganio have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
New data document the global fallout for rheumatology patients when hydroxychloroquine (HCQ) supplies were being diverted to hospitals for COVID-19 patients.
Demand for HCQ soared on evidence-lacking claims that the drug was effective in treating and preventing SARS-CoV-2 infection. Further research has since shown HCQ to be ineffective for COVID-19 and potentially harmful to patients.
But during the height of the COVID-19-related hype, patients worldwide with autoimmune diseases, particularly lupus and rheumatoid arthritis, had trouble getting the pills at all or couldn’t get as many as they needed for their chronic conditions.
Emily Sirotich, MSc, a PhD student at McMaster University in Hamilton, Ont., presented data at the virtual annual meeting of the American College of Rheumatology demonstrating that the severity of shortages differed widely.
Whereas 26.7% of rheumatology patients in Africa and 21.4% in southeast Asia said their pharmacy ran short of HCQ – which was originally developed as an antimalarial drug but has been found effective in treating some rheumatic diseases – only 6.8% of patients in the Americas and 2.1% in European regions reported the shortages.
“There are large regional disparities in access to antimalarials whether they were caused by the COVID-19 pandemic or already existed,” she said in an interview.
Global survey polled patient experience
Ms. Sirotich’s team analyzed data from the Global Rheumatology Alliance Patient Experience Survey.
They found that from 9,393 respondents (average age 46.1 years and 90% female), 3,872 (41.2%) were taking antimalarials. Of these, 230 (6.2% globally) were unable to keep taking the drugs because their pharmacy ran out.
Researchers evaluated the effect of drug shortages on disease activity, mental health, and physical health by comparing mean values with two-sided independent t-tests to identify significant differences.
They found that patients who were unable to obtain antimalarials had significantly higher levels of rheumatic disease activity as well as poorer mental and physical health (all P < .001).
The survey was distributed online through patient support groups and on social media. Patients with rheumatic diseases or their parents anonymously entered data including their rheumatic disease diagnosis, medications, COVID-19 status, and disease outcomes.
Ms. Sirotich said they are currently gathering new data to see if the gaps in access to HCQ persist and whether the physical and mental consequences of not having the medications continue.
Hospitals stockpiled HCQ in the U.S.
Michael Ganio, PharmD, senior director of pharmacy practice and quality at the American Society of Health-System Pharmacists (ASHP), said in an interview that hospitals in the United States received large amounts of HCQ in late spring and early summer, donated by pharmaceutical companies for COVID-19 before the lack of evidence for efficacy became clear.
Hospitals found themselves sitting on large quantities of HCQ they couldn’t use while prescriptions for rheumatology outpatients were going unfilled.
It is only in recent months that the U.S. Department of Health and Human Services has given clear direction to hospitals on how to redistribute those supplies, Dr. Ganio said.
“There’s no good real good way to move a product from a hospital to a [drug store] down the street,” he said.
The Food and Drug Administration now lists the HCQ shortages as resolved.
Declined prescriptions have frustrated physicians
Brett Smith, DO, a pediatric and adult rheumatologist in Alcoa, Tenn., said he was frustrated by pharmacies declining his prescriptions for HCQ for patients with rheumatoid arthritis.
“I got notes from pharmacies that I should consider alternative agents,” he said in an interview. But the safety profiles of the alternatives were not as good, he said.
“Hydroxychloroquine has no risk of infection and no risk of malignancy, and they were proposing alternative agents that carry those risks,” he said.
“I had some people with RA who couldn’t get [HCQ] who had a substantial increase in swollen joints and pain without it,” he said.
Dr. Smith said some patients who use HCQ for off-label uses such as certain skin disorders still aren’t getting the drug, as off-label use has been discouraged to make sure those with lupus and RA have enough, he said.
Saira Sheikh, MD, director of the University of North Carolina Rheumatology Lupus Clinic in Chapel Hill, said in an interview that during the summer months pharmacists required additional documentation of the diagnosis of autoimmune disease, resulting in unnecessary delays even when patients had been on the medication for many years.
She said emerging research has found patient-reported barriers to filling prescriptions, interruptions in HCQ treatment, and reported emotional stress and anxiety related to medication access during the COVID-19 pandemic.
“This experience with HCQ during the COVID-19 pandemic teaches us that while swift action and progress to address the immediate threats of the pandemic should be commended, it is important that we move forward in a conscious manner, guided by an evidence base that comes from high-quality research, not from rushed judgments based on preliminary studies, or pressure from political leaders,” Dr. Sheikh said.
Ms. Sirotich, Dr. Smith, Dr. Sheikh, and Dr. Ganio have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis Overlap Syndrome in a Patient With Relapsing Polychondritis
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.

Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.
Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.
Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
Practice Points
- The clinical presentation of relapsing polychondritis (RP) may demonstrate cutaneous manifestations other than the typical inflammation of cartilage-rich structures.
- Approximately 30% of patients with RP will have another autoimmune disease.
Systemic sclerosis patients share their perspectives and needs in treatment trials
Patients with systemic sclerosis have variable disease progression but often experience debilitating fatigue, pain, and digestive issues – and they’re extremely concerned about progressive organ damage, according to those who spoke at and provided input at a public meeting on patient-focused drug development for the disease.

The virtual meeting was part of the Food and Drug Administration’s Patient-Focused Drug Development (PFDD) initiative, which began in 2012 and aims to provide a systematic way for patients’ experiences, needs, and priorities to be “captured and meaningfully incorporated” into drug development and evaluation.
Patients rate their most impactful symptoms
Dinesh Khanna, MBBS, MSc, a rheumatologist who directs a scleroderma research program at the University of Michigan, Ann Arbor, attended the meeting after giving an opening presentation on the disease to FDA officials, patients, and other participants. In a later interview, he said that patients’ ratings of their most impactful symptoms was especially striking.

Raynaud’s phenomenon, digestive symptoms, and fatigue were the top three answers to a poll question that asked patients what symptom had the most significant impact on daily life, he noted, “and none of these are being [strongly] addressed right now [in clinical trials] apart from Raynaud’s phenomenon, for which there are some trials ongoing.”
He and other researchers are “struggling with what outcomes measures to use [in their studies],” said Dr. Khanna, the Frederick G.L. Huetwell Professor of Rheumatology at the University. “My takeaway from the meeting as a clinical trialist is that we should be paying close attention to the symptoms that patients tell us are the most important. We should be including these in our trial designs as secondary endpoints, if not primary endpoints. We have not done that [thus far], really.”
Approximately 200,000 patients in the United States have scleroderma, and approximately 75,000-80,000 of these patients have systemic scleroderma, or systemic sclerosis, Dr. Khanna said in his opening presentation. Each year, he estimates, about 6,000 new diagnoses of systemic sclerosis are made.
More than 200 people – patients, FDA officials, and others – participated in the PFDD meeting. Patients participated in one of two panels – one focused on health effects and daily impacts, and the other on treatments – or submitted input electronically. All were invited to answer poll questions.
Raj Nair, MD, one of eight FDA leaders attending the meeting, noted in closing remarks that the pain experienced by patients with systemic sclerosis includes severe pain from Raynaud’s phenomenon and pain caused by digital ulcers and by calcinosis. “We heard about how paralyzing the pain from calcinosis is, and that there are very few options for alleviating this pain,” said Dr. Nair, of the division of rheumatology and transplant medicine.
Another takeaway, he said, is that the “fatigue can be severe and debilitating, leading to days where it is impossible to get out of bed,” and that digestive symptoms can also be severe. “Reflux,” he noted, “requires significant medical intervention.”
Patients describe their experiences
Rosemary Lyons, diagnosed with scleroderma 35 years ago, explained that while her skin is no longer hardened, she is overly sensitive to fabrics and skin care products and has difficulty with sleeping and eating. She moved away from family in the Northeast to live in the South where the climate is warmer, but even on a 90-degree night she needs a blanket and two comforters to curb the cold and attempt to sleep.
Impaired gastrointestinal motility has made food her “biggest problem” for the past 10 years, and because of GI symptoms, she can eat only one meal a day. She also experiences fainting, brain fog, and severe fatigue. On a good day, Ms. Lyons noted, she sometimes opts to do some house chores “knowing that I’ll have 1-3 days of recovery.”
Another patient, Amy Harding, said that 22 years after her scleroderma diagnosis, “the calcinosis I get in my fingers, elbows, toes, and ears tops all the prior symptoms.” The skin tightening and digital ulcers that she experienced in the first 10 years have tapered off, and while Raynaud’s symptoms and heartburn have worsened, they are at least partly manageable with medications, unlike the pain from calcinosis.
Treating symptoms vs. disease may be key in risk-benefit analysis
In questions after patient presentations, FDA officials probed for more perspective on issues such as how fatigue should be assessed, the differences between fatigue and brain fog, the impact of calcinosis on functioning, and how much risk patients would be willing to assume from treatments that have side effects and that may or may not modulate the disease and slow disease progression.
Most patients said in response to an FDA poll question that they definitely (almost 40%) or possibly (almost 50%) would be willing to try a hypothetical new self-injectable medication if it were shown to reduce their most impactful symptoms but had side effects.
“I think what [we’ve been hearing] today is that whether we’re working on the symptoms or the disease itself is [the key]” to patients’ risk-benefit analysis, said meeting moderator Capt. Robyn Bent, RN, MS, of the U.S. Public Health Service, and director of the PFDD.
Anita Devine, diagnosed 13 years ago with systemic sclerosis, was one of several panel members who said she would accept more bothersome treatment side effects and risks “if the gain was control of disease progression and overall quality of life ... and organ preservation.” Ms. Devine, who has needed kidney dialysis and multiple hand surgeries, noted that she previously took anti-neoplastic and anti-inflammatory agents “to try to stem the course of my disease, but unfortunately the disease did not abate.”
Treatments for systemic sclerosis include vasodilators, immunosuppressive medications, antifibrotic therapies, and stem cell transplants, Dr. Khanna said in his opening remarks.
Trials of drugs for scleroderma have focused on early disease that may be amenable to treatment, with the exception of trials for pulmonary arterial hypertension, which affects some patients with systemic sclerosis. There are multiple FDA-approved drugs for pulmonary arterial hypertension and more trials are underway.
Outcomes such as pain and fatigue are included in many of the trials currently underway, but they tend to be lower-level secondary outcomes measures that cannot be incorporated into drug labeling or are more “exploratory in nature,” Dr. Khanna said in the interview.
Dr. Khanna disclosed that he is the chief medical officer (an equity position) for CiVi Biopharma/Eicos Sciences Inc., which is developing a drug for Raynaud’s, and serves as a consultant and grant recipient for numerous companies that make or are developing drugs for systemic sclerosis.
The FDA will accept patient comments until Dec. 15, 2020, at which time comments will be compiled into a summary report, Ms. Bent said.
Patients with systemic sclerosis have variable disease progression but often experience debilitating fatigue, pain, and digestive issues – and they’re extremely concerned about progressive organ damage, according to those who spoke at and provided input at a public meeting on patient-focused drug development for the disease.
The virtual meeting was part of the Food and Drug Administration’s Patient-Focused Drug Development (PFDD) initiative, which began in 2012 and aims to provide a systematic way for patients’ experiences, needs, and priorities to be “captured and meaningfully incorporated” into drug development and evaluation.
Patients rate their most impactful symptoms
Dinesh Khanna, MBBS, MSc, a rheumatologist who directs a scleroderma research program at the University of Michigan, Ann Arbor, attended the meeting after giving an opening presentation on the disease to FDA officials, patients, and other participants. In a later interview, he said that patients’ ratings of their most impactful symptoms was especially striking.
Raynaud’s phenomenon, digestive symptoms, and fatigue were the top three answers to a poll question that asked patients what symptom had the most significant impact on daily life, he noted, “and none of these are being [strongly] addressed right now [in clinical trials] apart from Raynaud’s phenomenon, for which there are some trials ongoing.”
He and other researchers are “struggling with what outcomes measures to use [in their studies],” said Dr. Khanna, the Frederick G.L. Huetwell Professor of Rheumatology at the University. “My takeaway from the meeting as a clinical trialist is that we should be paying close attention to the symptoms that patients tell us are the most important. We should be including these in our trial designs as secondary endpoints, if not primary endpoints. We have not done that [thus far], really.”
Approximately 200,000 patients in the United States have scleroderma, and approximately 75,000-80,000 of these patients have systemic scleroderma, or systemic sclerosis, Dr. Khanna said in his opening presentation. Each year, he estimates, about 6,000 new diagnoses of systemic sclerosis are made.
More than 200 people – patients, FDA officials, and others – participated in the PFDD meeting. Patients participated in one of two panels – one focused on health effects and daily impacts, and the other on treatments – or submitted input electronically. All were invited to answer poll questions.
Raj Nair, MD, one of eight FDA leaders attending the meeting, noted in closing remarks that the pain experienced by patients with systemic sclerosis includes severe pain from Raynaud’s phenomenon and pain caused by digital ulcers and by calcinosis. “We heard about how paralyzing the pain from calcinosis is, and that there are very few options for alleviating this pain,” said Dr. Nair, of the division of rheumatology and transplant medicine.
Another takeaway, he said, is that the “fatigue can be severe and debilitating, leading to days where it is impossible to get out of bed,” and that digestive symptoms can also be severe. “Reflux,” he noted, “requires significant medical intervention.”
Patients describe their experiences
Rosemary Lyons, diagnosed with scleroderma 35 years ago, explained that while her skin is no longer hardened, she is overly sensitive to fabrics and skin care products and has difficulty with sleeping and eating. She moved away from family in the Northeast to live in the South where the climate is warmer, but even on a 90-degree night she needs a blanket and two comforters to curb the cold and attempt to sleep.
Impaired gastrointestinal motility has made food her “biggest problem” for the past 10 years, and because of GI symptoms, she can eat only one meal a day. She also experiences fainting, brain fog, and severe fatigue. On a good day, Ms. Lyons noted, she sometimes opts to do some house chores “knowing that I’ll have 1-3 days of recovery.”
Another patient, Amy Harding, said that 22 years after her scleroderma diagnosis, “the calcinosis I get in my fingers, elbows, toes, and ears tops all the prior symptoms.” The skin tightening and digital ulcers that she experienced in the first 10 years have tapered off, and while Raynaud’s symptoms and heartburn have worsened, they are at least partly manageable with medications, unlike the pain from calcinosis.
Treating symptoms vs. disease may be key in risk-benefit analysis
In questions after patient presentations, FDA officials probed for more perspective on issues such as how fatigue should be assessed, the differences between fatigue and brain fog, the impact of calcinosis on functioning, and how much risk patients would be willing to assume from treatments that have side effects and that may or may not modulate the disease and slow disease progression.
Most patients said in response to an FDA poll question that they definitely (almost 40%) or possibly (almost 50%) would be willing to try a hypothetical new self-injectable medication if it were shown to reduce their most impactful symptoms but had side effects.
“I think what [we’ve been hearing] today is that whether we’re working on the symptoms or the disease itself is [the key]” to patients’ risk-benefit analysis, said meeting moderator Capt. Robyn Bent, RN, MS, of the U.S. Public Health Service, and director of the PFDD.
Anita Devine, diagnosed 13 years ago with systemic sclerosis, was one of several panel members who said she would accept more bothersome treatment side effects and risks “if the gain was control of disease progression and overall quality of life ... and organ preservation.” Ms. Devine, who has needed kidney dialysis and multiple hand surgeries, noted that she previously took anti-neoplastic and anti-inflammatory agents “to try to stem the course of my disease, but unfortunately the disease did not abate.”
Treatments for systemic sclerosis include vasodilators, immunosuppressive medications, antifibrotic therapies, and stem cell transplants, Dr. Khanna said in his opening remarks.
Trials of drugs for scleroderma have focused on early disease that may be amenable to treatment, with the exception of trials for pulmonary arterial hypertension, which affects some patients with systemic sclerosis. There are multiple FDA-approved drugs for pulmonary arterial hypertension and more trials are underway.
Outcomes such as pain and fatigue are included in many of the trials currently underway, but they tend to be lower-level secondary outcomes measures that cannot be incorporated into drug labeling or are more “exploratory in nature,” Dr. Khanna said in the interview.
Dr. Khanna disclosed that he is the chief medical officer (an equity position) for CiVi Biopharma/Eicos Sciences Inc., which is developing a drug for Raynaud’s, and serves as a consultant and grant recipient for numerous companies that make or are developing drugs for systemic sclerosis.
The FDA will accept patient comments until Dec. 15, 2020, at which time comments will be compiled into a summary report, Ms. Bent said.
Patients with systemic sclerosis have variable disease progression but often experience debilitating fatigue, pain, and digestive issues – and they’re extremely concerned about progressive organ damage, according to those who spoke at and provided input at a public meeting on patient-focused drug development for the disease.
The virtual meeting was part of the Food and Drug Administration’s Patient-Focused Drug Development (PFDD) initiative, which began in 2012 and aims to provide a systematic way for patients’ experiences, needs, and priorities to be “captured and meaningfully incorporated” into drug development and evaluation.
Patients rate their most impactful symptoms
Dinesh Khanna, MBBS, MSc, a rheumatologist who directs a scleroderma research program at the University of Michigan, Ann Arbor, attended the meeting after giving an opening presentation on the disease to FDA officials, patients, and other participants. In a later interview, he said that patients’ ratings of their most impactful symptoms was especially striking.
Raynaud’s phenomenon, digestive symptoms, and fatigue were the top three answers to a poll question that asked patients what symptom had the most significant impact on daily life, he noted, “and none of these are being [strongly] addressed right now [in clinical trials] apart from Raynaud’s phenomenon, for which there are some trials ongoing.”
He and other researchers are “struggling with what outcomes measures to use [in their studies],” said Dr. Khanna, the Frederick G.L. Huetwell Professor of Rheumatology at the University. “My takeaway from the meeting as a clinical trialist is that we should be paying close attention to the symptoms that patients tell us are the most important. We should be including these in our trial designs as secondary endpoints, if not primary endpoints. We have not done that [thus far], really.”
Approximately 200,000 patients in the United States have scleroderma, and approximately 75,000-80,000 of these patients have systemic scleroderma, or systemic sclerosis, Dr. Khanna said in his opening presentation. Each year, he estimates, about 6,000 new diagnoses of systemic sclerosis are made.
More than 200 people – patients, FDA officials, and others – participated in the PFDD meeting. Patients participated in one of two panels – one focused on health effects and daily impacts, and the other on treatments – or submitted input electronically. All were invited to answer poll questions.
Raj Nair, MD, one of eight FDA leaders attending the meeting, noted in closing remarks that the pain experienced by patients with systemic sclerosis includes severe pain from Raynaud’s phenomenon and pain caused by digital ulcers and by calcinosis. “We heard about how paralyzing the pain from calcinosis is, and that there are very few options for alleviating this pain,” said Dr. Nair, of the division of rheumatology and transplant medicine.
Another takeaway, he said, is that the “fatigue can be severe and debilitating, leading to days where it is impossible to get out of bed,” and that digestive symptoms can also be severe. “Reflux,” he noted, “requires significant medical intervention.”
Patients describe their experiences
Rosemary Lyons, diagnosed with scleroderma 35 years ago, explained that while her skin is no longer hardened, she is overly sensitive to fabrics and skin care products and has difficulty with sleeping and eating. She moved away from family in the Northeast to live in the South where the climate is warmer, but even on a 90-degree night she needs a blanket and two comforters to curb the cold and attempt to sleep.
Impaired gastrointestinal motility has made food her “biggest problem” for the past 10 years, and because of GI symptoms, she can eat only one meal a day. She also experiences fainting, brain fog, and severe fatigue. On a good day, Ms. Lyons noted, she sometimes opts to do some house chores “knowing that I’ll have 1-3 days of recovery.”
Another patient, Amy Harding, said that 22 years after her scleroderma diagnosis, “the calcinosis I get in my fingers, elbows, toes, and ears tops all the prior symptoms.” The skin tightening and digital ulcers that she experienced in the first 10 years have tapered off, and while Raynaud’s symptoms and heartburn have worsened, they are at least partly manageable with medications, unlike the pain from calcinosis.
Treating symptoms vs. disease may be key in risk-benefit analysis
In questions after patient presentations, FDA officials probed for more perspective on issues such as how fatigue should be assessed, the differences between fatigue and brain fog, the impact of calcinosis on functioning, and how much risk patients would be willing to assume from treatments that have side effects and that may or may not modulate the disease and slow disease progression.
Most patients said in response to an FDA poll question that they definitely (almost 40%) or possibly (almost 50%) would be willing to try a hypothetical new self-injectable medication if it were shown to reduce their most impactful symptoms but had side effects.
“I think what [we’ve been hearing] today is that whether we’re working on the symptoms or the disease itself is [the key]” to patients’ risk-benefit analysis, said meeting moderator Capt. Robyn Bent, RN, MS, of the U.S. Public Health Service, and director of the PFDD.
Anita Devine, diagnosed 13 years ago with systemic sclerosis, was one of several panel members who said she would accept more bothersome treatment side effects and risks “if the gain was control of disease progression and overall quality of life ... and organ preservation.” Ms. Devine, who has needed kidney dialysis and multiple hand surgeries, noted that she previously took anti-neoplastic and anti-inflammatory agents “to try to stem the course of my disease, but unfortunately the disease did not abate.”
Treatments for systemic sclerosis include vasodilators, immunosuppressive medications, antifibrotic therapies, and stem cell transplants, Dr. Khanna said in his opening remarks.
Trials of drugs for scleroderma have focused on early disease that may be amenable to treatment, with the exception of trials for pulmonary arterial hypertension, which affects some patients with systemic sclerosis. There are multiple FDA-approved drugs for pulmonary arterial hypertension and more trials are underway.
Outcomes such as pain and fatigue are included in many of the trials currently underway, but they tend to be lower-level secondary outcomes measures that cannot be incorporated into drug labeling or are more “exploratory in nature,” Dr. Khanna said in the interview.
Dr. Khanna disclosed that he is the chief medical officer (an equity position) for CiVi Biopharma/Eicos Sciences Inc., which is developing a drug for Raynaud’s, and serves as a consultant and grant recipient for numerous companies that make or are developing drugs for systemic sclerosis.
The FDA will accept patient comments until Dec. 15, 2020, at which time comments will be compiled into a summary report, Ms. Bent said.
FROM AN FDA PATIENT-FOCUSED DRUG DEVELOPMENT MEETING
Low IgA levels associated with increased infection risk in SLE patients
A new study of immunoglobulin levels in adult patients with systemic lupus erythematosus (SLE) has found that acquired low levels of IgA are associated with a higher risk of infection.

To the knowledge of first author Ibrahim Almaghlouth, MD, of the division of rheumatology at the University of Toronto, and colleagues, “this is the first dedicated study to examine the relationship between acquired low immunoglobulins and infection risk in adult patients with SLE.” But as to whether there may be a “protective role for immunoglobulins and the potential effect of immunoglobulin replacement in a setting of recurrent or severe infection among SLE patients requires further study.”
To determine if the risk of infection was tied to acquired low immunoglobulin levels, the researchers launched a retrospective analysis of data from a prospective cohort study of adult SLE patients from a Toronto lupus cohort that was established in 1970. The study was published in Rheumatology.
A total of 448 patients with at least two low immunoglobulin tests were matched with 656 SLE patients with no low immunoglobulins according to enrollment decade. The average age of the low-immunoglobulin group was 41.8 years, compared with 39.3 years in the control group. Average disease duration was 11.2 years in the low-immunoglobulin group and 7.6 years in the control group.
Of the patients in the low-immunoglobulin group, 221 had consecutive low tests and 227 had nonconsecutive low tests. Overall, 98 of those patients had low IgG, 251 patients had low IgM, and 51 patients had low IgA. Only 48 patients had overlapping low levels, including 5 with all three.
Average levels among the low-immunoglobulin group at baseline were 11.5 (standard deviation, 6.1) g/L of IgG, 0.8 (1.1) g/L of IgM, and 2.4 (1.6) g/L of IgA, while average levels among the control group were 16.3 (6.4) g/L of IgG, 1.8 (1.2) g/L of IgM, and 3.2 (1.5) g/L of IgA. In the primary analysis, after adjustment using propensity scoring, there were 97 infections: 47 in the low-immunoglobulin group and 50 in the control group. The most common types were respiratory and urinary tract infections, and the rate of infection was higher in patients with low IgA. The IgA level associated with risk of infection was less than 0.75 g/L.
After Cox regression analysis, the only variable that significantly increased infection risk was a low IgA level (hazard ratio, 3.19; 95% confidence interval, 1.17-8.71), not a low IgG level (HR, 1.87; 95% CI, 0.77-4.54) or low IgM level (HR, 0.63; 95% CI, 0.34-1.17). In regard to recovery among the low-immunoglobulin group, 11 patients (2.5%) recovered from low immunoglobulins within in the first year, followed by 36 (8.2%) in the second year, 44 (10.1%) in the third year, and 80 (18.4%) in the fourth year. All told, 60% (263) of patients with acquired hypogammaglobulinemia recovered over a 4-year period.
Is there clinical relevance to low IgA?
“I don’t see us using this clinically immediately,” Karen Costenbader, MD, a rheumatologist at Brigham and Women’s Hospital and professor of medicine at Harvard Medical School, both in Boston, said in an interview. “We do test immunoglobulins often, especially in patients who’ve had biologic therapy. Will we start thinking about their IgA levels? It’s not clear, and the researchers leave it up in the air as to what this means, beyond them being at high risk.”
That said, she added, “IgA levels are interesting, especially in a time of COVID, because they’re associated with mucosal immunity. Is this subset of patients going to be at particularly high risk for the coronavirus?”
She also noted that, though immunoglobulin replacement has been helpful in her patients, it’s an expensive therapy to recommend for low IgA levels without knowing exactly what is causing these deficiencies. “My question is, would it be useful to follow these levels in lupus patients, even we don’t know what to do about them?” she asked. “We know there are a lot of risk factors for infections, so is the IgA going to be useful above and beyond that, and then what can we do about it?”
The authors acknowledged their study’s potential limitations, including low infection rates and yearly measurements of immunoglobulin levels, which could’ve led to misclassifying a lab error as true low immunoglobulin. They also highlighted its strengths, including using various methods to reduce selection and confounding bias while also reporting consistent results after examining multiple definitions of low immunoglobulins and outcomes.
The study received no specific funding, and the authors reported no potential conflicts of interest.
SOURCE: Almaghlouth I et al. Rheumatology. 2020 Oct 2. doi: 10.1093/rheumatology/keaa641.
A new study of immunoglobulin levels in adult patients with systemic lupus erythematosus (SLE) has found that acquired low levels of IgA are associated with a higher risk of infection.
To the knowledge of first author Ibrahim Almaghlouth, MD, of the division of rheumatology at the University of Toronto, and colleagues, “this is the first dedicated study to examine the relationship between acquired low immunoglobulins and infection risk in adult patients with SLE.” But as to whether there may be a “protective role for immunoglobulins and the potential effect of immunoglobulin replacement in a setting of recurrent or severe infection among SLE patients requires further study.”
To determine if the risk of infection was tied to acquired low immunoglobulin levels, the researchers launched a retrospective analysis of data from a prospective cohort study of adult SLE patients from a Toronto lupus cohort that was established in 1970. The study was published in Rheumatology.
A total of 448 patients with at least two low immunoglobulin tests were matched with 656 SLE patients with no low immunoglobulins according to enrollment decade. The average age of the low-immunoglobulin group was 41.8 years, compared with 39.3 years in the control group. Average disease duration was 11.2 years in the low-immunoglobulin group and 7.6 years in the control group.
Of the patients in the low-immunoglobulin group, 221 had consecutive low tests and 227 had nonconsecutive low tests. Overall, 98 of those patients had low IgG, 251 patients had low IgM, and 51 patients had low IgA. Only 48 patients had overlapping low levels, including 5 with all three.
Average levels among the low-immunoglobulin group at baseline were 11.5 (standard deviation, 6.1) g/L of IgG, 0.8 (1.1) g/L of IgM, and 2.4 (1.6) g/L of IgA, while average levels among the control group were 16.3 (6.4) g/L of IgG, 1.8 (1.2) g/L of IgM, and 3.2 (1.5) g/L of IgA. In the primary analysis, after adjustment using propensity scoring, there were 97 infections: 47 in the low-immunoglobulin group and 50 in the control group. The most common types were respiratory and urinary tract infections, and the rate of infection was higher in patients with low IgA. The IgA level associated with risk of infection was less than 0.75 g/L.
After Cox regression analysis, the only variable that significantly increased infection risk was a low IgA level (hazard ratio, 3.19; 95% confidence interval, 1.17-8.71), not a low IgG level (HR, 1.87; 95% CI, 0.77-4.54) or low IgM level (HR, 0.63; 95% CI, 0.34-1.17). In regard to recovery among the low-immunoglobulin group, 11 patients (2.5%) recovered from low immunoglobulins within in the first year, followed by 36 (8.2%) in the second year, 44 (10.1%) in the third year, and 80 (18.4%) in the fourth year. All told, 60% (263) of patients with acquired hypogammaglobulinemia recovered over a 4-year period.
Is there clinical relevance to low IgA?
“I don’t see us using this clinically immediately,” Karen Costenbader, MD, a rheumatologist at Brigham and Women’s Hospital and professor of medicine at Harvard Medical School, both in Boston, said in an interview. “We do test immunoglobulins often, especially in patients who’ve had biologic therapy. Will we start thinking about their IgA levels? It’s not clear, and the researchers leave it up in the air as to what this means, beyond them being at high risk.”
That said, she added, “IgA levels are interesting, especially in a time of COVID, because they’re associated with mucosal immunity. Is this subset of patients going to be at particularly high risk for the coronavirus?”
She also noted that, though immunoglobulin replacement has been helpful in her patients, it’s an expensive therapy to recommend for low IgA levels without knowing exactly what is causing these deficiencies. “My question is, would it be useful to follow these levels in lupus patients, even we don’t know what to do about them?” she asked. “We know there are a lot of risk factors for infections, so is the IgA going to be useful above and beyond that, and then what can we do about it?”
The authors acknowledged their study’s potential limitations, including low infection rates and yearly measurements of immunoglobulin levels, which could’ve led to misclassifying a lab error as true low immunoglobulin. They also highlighted its strengths, including using various methods to reduce selection and confounding bias while also reporting consistent results after examining multiple definitions of low immunoglobulins and outcomes.
The study received no specific funding, and the authors reported no potential conflicts of interest.
SOURCE: Almaghlouth I et al. Rheumatology. 2020 Oct 2. doi: 10.1093/rheumatology/keaa641.
A new study of immunoglobulin levels in adult patients with systemic lupus erythematosus (SLE) has found that acquired low levels of IgA are associated with a higher risk of infection.
To the knowledge of first author Ibrahim Almaghlouth, MD, of the division of rheumatology at the University of Toronto, and colleagues, “this is the first dedicated study to examine the relationship between acquired low immunoglobulins and infection risk in adult patients with SLE.” But as to whether there may be a “protective role for immunoglobulins and the potential effect of immunoglobulin replacement in a setting of recurrent or severe infection among SLE patients requires further study.”
To determine if the risk of infection was tied to acquired low immunoglobulin levels, the researchers launched a retrospective analysis of data from a prospective cohort study of adult SLE patients from a Toronto lupus cohort that was established in 1970. The study was published in Rheumatology.
A total of 448 patients with at least two low immunoglobulin tests were matched with 656 SLE patients with no low immunoglobulins according to enrollment decade. The average age of the low-immunoglobulin group was 41.8 years, compared with 39.3 years in the control group. Average disease duration was 11.2 years in the low-immunoglobulin group and 7.6 years in the control group.
Of the patients in the low-immunoglobulin group, 221 had consecutive low tests and 227 had nonconsecutive low tests. Overall, 98 of those patients had low IgG, 251 patients had low IgM, and 51 patients had low IgA. Only 48 patients had overlapping low levels, including 5 with all three.
Average levels among the low-immunoglobulin group at baseline were 11.5 (standard deviation, 6.1) g/L of IgG, 0.8 (1.1) g/L of IgM, and 2.4 (1.6) g/L of IgA, while average levels among the control group were 16.3 (6.4) g/L of IgG, 1.8 (1.2) g/L of IgM, and 3.2 (1.5) g/L of IgA. In the primary analysis, after adjustment using propensity scoring, there were 97 infections: 47 in the low-immunoglobulin group and 50 in the control group. The most common types were respiratory and urinary tract infections, and the rate of infection was higher in patients with low IgA. The IgA level associated with risk of infection was less than 0.75 g/L.
After Cox regression analysis, the only variable that significantly increased infection risk was a low IgA level (hazard ratio, 3.19; 95% confidence interval, 1.17-8.71), not a low IgG level (HR, 1.87; 95% CI, 0.77-4.54) or low IgM level (HR, 0.63; 95% CI, 0.34-1.17). In regard to recovery among the low-immunoglobulin group, 11 patients (2.5%) recovered from low immunoglobulins within in the first year, followed by 36 (8.2%) in the second year, 44 (10.1%) in the third year, and 80 (18.4%) in the fourth year. All told, 60% (263) of patients with acquired hypogammaglobulinemia recovered over a 4-year period.
Is there clinical relevance to low IgA?
“I don’t see us using this clinically immediately,” Karen Costenbader, MD, a rheumatologist at Brigham and Women’s Hospital and professor of medicine at Harvard Medical School, both in Boston, said in an interview. “We do test immunoglobulins often, especially in patients who’ve had biologic therapy. Will we start thinking about their IgA levels? It’s not clear, and the researchers leave it up in the air as to what this means, beyond them being at high risk.”
That said, she added, “IgA levels are interesting, especially in a time of COVID, because they’re associated with mucosal immunity. Is this subset of patients going to be at particularly high risk for the coronavirus?”
She also noted that, though immunoglobulin replacement has been helpful in her patients, it’s an expensive therapy to recommend for low IgA levels without knowing exactly what is causing these deficiencies. “My question is, would it be useful to follow these levels in lupus patients, even we don’t know what to do about them?” she asked. “We know there are a lot of risk factors for infections, so is the IgA going to be useful above and beyond that, and then what can we do about it?”
The authors acknowledged their study’s potential limitations, including low infection rates and yearly measurements of immunoglobulin levels, which could’ve led to misclassifying a lab error as true low immunoglobulin. They also highlighted its strengths, including using various methods to reduce selection and confounding bias while also reporting consistent results after examining multiple definitions of low immunoglobulins and outcomes.
The study received no specific funding, and the authors reported no potential conflicts of interest.
SOURCE: Almaghlouth I et al. Rheumatology. 2020 Oct 2. doi: 10.1093/rheumatology/keaa641.
FROM RHEUMATOLOGY
Hidradenitis Suppurativa in the Military
Case Report
A 19-year-old female marine with a 10-year history of hidradenitis suppurativa (HS) presented with hyperpigmented nodules in the inguinal folds and a recurrent cyst in the right groin area of 2 to 3 weeks’ duration. She denied axillary or inframammary involvement. She underwent several incision and drainage procedures 1 year prior to her enlistment in the US Marine Corps at 18 years of age. She previously had been treated by dermatology with doxycycline 100-mg tablets twice daily, benzoyl peroxide wash 5% applied to affected areas and rinsed daily, and clindamycin solution 1% with minimal improvement. She denied smoking or alcohol intake and said she typically wore a loose-fitting uniform to work. As a marine, she was expected to participate in daily physical training and exercises with her military unit, during which she wore a standardized physical training uniform, including nylon shorts and a cotton T-shirt. She requested light duty—military duty status with physical limitations or restrictions—to avoid physical training that would cause further friction and irritation to the inguinal region.
Physical examination demonstrated a woman with Fitzpatrick skin type III and normal body mass index. There were hyperpigmented nodules and scarring in the inguinal folds, most consistent with Hurley stage 2. A single, 0.5-cm, draining lesion was visualized. No hyperhidrosis was noted. The patient was placed on light duty for 7 days, with physical training only at her own pace and discretion. Moreover, she was restricted from field training, rifle range training, and other situations where she may excessively sweat or not be able to adequately maintain personal hygiene. She was encouraged to continue clindamycin solution 1% to the affected area twice daily and was prescribed chlorhexidine solution 4% to use when washing affected areas in the shower. The patient also was referred to the dermatology department at the Naval Hospital Camp Pendleton (Oceanside, California), where she was treated with laser hair removal in the inguinal region, thus avoiding waxing and further aggravation of HS flares. Due to the combination of topical therapies along with laser hair removal and duty restrictions, the patient had a dramatic decrease in development of severe nodular lesions.
Comment
Presentation
Historically, the incidence of HS is estimated at 0.5% to 4% of the general population with female predominance.1 Predisposing factors include obesity, smoking, genetic predisposition to acne, apocrine duct obstruction, and secondary bacterial infection.2 During acute flares, patients generally present with tender subcutaneous nodules that drain malodorous purulent material.3,4 Acute flares are unpredictable, and patients deal with chronic, recurrent, draining wounds, leading to a poor quality of life with resulting physical, psychological, financial, social, and emotional distress.3-5 The negative impact of HS on a patient’s quality of life has been reported to be greater than other dermatologic conditions.6 Lesions can be particularly painful and can cause disfiguration to the surface of the skin.7 Lesion severity is described using the Hurley staging system. Patient quality of life is directly correlated with disease severity and Hurley stage. In stage 1, abscesses develop, but no sinus tracts or cicatrization is present. In stage 2, recurrent abscesses will form tracts and cicatrization. In stage 3, the abscesses become diffuse or near diffuse, with multiple interconnected tracts and abscesses across the entire area of the body.8,9
Severe or refractory HS within the physically active military population may require consideration of light or limited duty or even separation from service. Similarly, severe HS may pose challenges with other physically demanding occupations, such as the police force and firefighters.
Prevention Focus
Prevention of flares is key for patients with HS; secondary prevention aims to reduce impact of the disease or injury that has already occurred,10,11 which includes prevention of the infundibulofolliculitis from becoming a deep folliculitis, nodule, or fistula, as well as Hurley stage progression. Prompt diagnosis with appropriate treatment can decrease the severity of lesions, pain, and scarring. Globally, HS patients continue to experience considerable diagnostic delays of 8 to 12 years after onset of initial symptoms.11,12 Earlier accurate diagnosis and initiation of treatment from the primary care provider or general medical officer is imperative. Initial accurate management may help keep symptoms from progressing to more severe painful lesions. Similarly, patients should be educated on how to prevent HS flares. Patients should avoid known triggers, including smoking, obesity, sweating, mechanical irritation, stress, and poor hygiene.11
Shaving for hair reduction creates ingrown hair shafts, which may lead to folliculitis in mechanically stressed areas in skin folds, thus initiating the inflammatory cascade of HS.11,13 Therefore, shaving along with any other mechanical stress should be avoided in patients with HS. Laser hair removal has been shown to be quite helpful in both the prevention and treatment of HS. In one study, 22 patients with Hurley stage 2 to 3 disease were treated with an Nd:YAG laser once monthly. Results demonstrated a 65% decrease in disease severity after 3 monthly treatments.11 Similarly, other lasers have been used with success in several small case series; an 800-nm diode laser, intense pulsed light therapy, and a ruby laser have each demonstrated efficacy.14 Given these results, hair removal should be recommended to patients with HS. Military servicemembers (SMs) with certain conditions, such as polycystic ovary syndrome, pseudofolliculitis barbae, and HS, are eligible for laser hair removal when available at local military treatment facilities. Primary care providers for military SMs must have a working understanding of the disease process of HS and awareness of what resources are available for treatment, which allows for more streamlined care and improved outcomes.
Treatment Options
Treatment options are diverse and depend on the severity of HS. Typically, treatment begins with medical therapy followed by escalation to surgical intervention. Medical therapies often include antibiotics, acne treatments, antiandrogen therapy, immunosuppressive agents, and biologic therapy.15,16 If first-line medical interventions fail to control HS, surgical interventions should be considered. Surgical intervention in conjunction with medical therapy decreases the chance for recurrence.3,15,16
Although HS is internationally recognized as an inflammatory disease and not an infectious process, topical antibiotics can help to prevent and improve formation of abscesses, nodules, and pustules.11 Agents such as clindamycin and chlorhexidine wash have proven effective in preventing flares.11,16 Other antibiotics used alone or in combination also are efficacious. Tetracyclines are recommended as monotherapy for mild stages of HS.17-19 Doxycycline is the most commonly used tetracycline in HS patients and has been demonstrated to penetrate Staphylococcus aureus biofilm in high enough concentrations to maintain its antibacterial activity.20 Moreover, doxycycline, as with other tetracyclines, has a multitude of anti-inflammatory and immunomodulatory properties21 and can reduce the production of IL-1, IL-6, tumor necrosis factor α, and IL-8; downregulate chemotaxis; and promote lipo-oxygenase, matrix metalloproteinase, and nuclear factor κB (NF-κB) signaling inhibition.17
Clindamycin is the only known agent that has been studied for topical treatment and utilization in milder cases of HS.17,22 Systemic combination of clindamycin and rifampicin is the most studied, with well-established efficacy in managing HS.17,23,24 Clindamycin has bacteriostatic activity toward both aerobic and anaerobic gram-positive bacteria by binding irreversibly to the 50S ribosomal subunit, thereby inhibiting bacterial protein synthesis. Rifampicin binds to the beta subunit of DNA-dependent RNA polymerase, inhibiting bacterial DNA-dependent RNA synthesis. Rifampicin has broad-spectrum activity, mostly against gram-positive as well as some gram-negative bacteria. Moreover, rifampicin has anti-inflammatory and immunomodulatory properties, including evidence that it inhibits excessive helper T cell (TH17) responses by reducing inducible nitric oxide synthase transcription and NF-κB activity.25,26
Metronidazole, moxifloxacin, and rifampicin as triple combination therapy has been shown to be effective in reducing HS activity in moderate to severe cases that were refractory to other treatments.27 Research suggests that moxifloxacin has anti-inflammatory properties, mainly by reducing IL-1β, IL-8, and tumor necrosis factor α; stabilizing IXb protein; suppressing NF-κB signaling; and reducing IL-17A.28,29
Ertapenem can be utilized as a single 6-week antibiotic course during surgical planning or rescue therapy.18 Moreover, ertapenem can be used to treat complicated skin and soft tissue infections and has been shown to substantially improve clinical aspects of severe HS.17,27
Disease-modifying antirheumatic drugs are effective in the treatment of moderate to severe HS.17-19 In 2 phase 3 trials (PIONEER I and II), adalimumab was used as monotherapy or in conjunction with antibiotics in patients with moderate to severe HS compared to placebo.30 Results demonstrated a disease burden reduction of greater than 50%. Antibiotic dual therapy was not noted to significantly affect disease burden.30 Of note, use of immunosuppressants in the military affects an SM’s availability for worldwide deployment and duty station assignment.
Antiandrogen therapies have demonstrated some reduction in HS flares. Although recommendations for use in HS is based on limited evidence, one randomized controlled trial compared ethinyl estradiol–norgestrel to ethinyl estradiol and cyproterone acetate. Both therapies resulted in similar efficacy, with 12 of 24 (50%) patients reporting HS symptoms improving or completely resolved.31 In another retrospective study of women treated with antiandrogen therapies, including ethinyl estriol, cyproterone acetate, and spironolactone, 16 of 29 (55%) patients reported improvement.32 In another study, daily doses of 100 to 150 mg of spironolactone resulted in improvement in 17 of 20 (85%) patients, including complete remission in 11 of 20 (55%) patients. Of the 3 patients with severe HS, none had complete clearing of disease burden.33 Patients with polycystic ovary syndrome or HS flares that occur around menstruation are more likely to benefit from treatment with spironolactone.18,32,34
Retinoids frequently have been utilized in the management of HS. In some retrospective studies and other prospective studies with 5 or more patients, isotretinoin monotherapy was utilized for a 4- to 10-month period.18,35-38 In the Alikhan et al18 study, 85 of 207 patients demonstrated improvement of HS symptoms, with more remarkable improvements in milder cases. Isotretinoin for management of patients with HS who have concomitant nodulocystic acne would have two-fold benefits.18
Wound Care
Given the purulent nodular formation in HS, adequate wound care management is vital. There is an abundance of HS wound care management strategies utilized by clinicians and patients. When selecting the appropriate dressing, consideration for the type of HS wound, cost, ease of application, patient comfort, absorbency, and odor management is important.3 However, living arrangements for military SMs can create difficulties applying and maintaining HS dressings, especially if deployed or in a field setting. Active-duty SMs often find themselves in austere living conditions in the field, aboard ships, or in other scenarios where they may or may not have running water or showers. Maintaining adequate hygiene may be difficult, and additional education about how to keep wounds clean must be imparted. Ideal dressings for HS should be highly absorbent, comfortable when applied to the anatomic locations of the HS lesions, and easily self-applied. Ideally, dressings would have atraumatic adhesion and antimicrobial properties.3 Cost-effective dressing options that have good absorption capability include sanitary napkins, adult briefs, infant diapers, and gauze.3 These dressings help to wick moisture, thus protecting the wound from maceration, which is a common patient concern. Although gauze dressings are easier to obtain, they are not as absorbent. Abdominal pads can be utilized, but they are moderately absorbent, bulky, and more challenging to obtain over-the-counter. Hydrofiber and calcium alginate dressings with silver are not accessible to the common consumer and are more expensive than the aforementioned dressings, but they do have some antimicrobial activity. Silver-impregnated foam dressings are moldable to intertriginous areas, easy to self-apply, and have moderate-heavy absorption abilities.
Final Thoughts
Hidradenitis suppurativa poses cumbersome and uncomfortable symptoms for all patients and may pose additional hardships for military SMs or those with physically demanding occupations who work in austere environments. Severe HS can restrict a military SM from certain duty stations, positions, or deployments. Early identification of HS can help reduce HS flares, disfigurement, and placement on limited duty status, therefore rendering the SM more able to engage in his/her operational responsibilities. Hidradenitis suppurativa should be discussed with the patient, with the goal to prevent flares for SMs that will be in the field, placed in austere environments, or be deployed. Use of immunosuppressants in active-duty SMs may affect their deployability, duty assignment, and retention.
For a military SM with HS, all aspects of prevention and treatment need to be balanced with his/her ability to remain deployable and complete his/her daily duties. Military SMs are not guaranteed the ideal scenario for treatment and prevention of HS. Unsanitary environments and occlusive uniforms undoubtedly contribute to disease process and make treatment more challenging. If a military SM is in a field setting or deployed, frequent daily dressing changes should still be attempted.
- Dufour DN, Emtestam L, Jemec GB. Hidradenitis suppurativa: a common and burdensome, yet under-recognised, inflammatory skin disease. Postgrad Med J. 2014;90:216-221.
- Beshara MA. Hidradenitis suppurativa: a clinician’s tool for early diagnosis and treatment. Nurse Pract. 2010;35:24-28.
- Kazemi A, Carnaggio K, Clark M, et al. Optimal wound care management in hidradenitis suppurativa. J Dermatolog Treat. 2017;29:165-167.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003:49:96-98.
- Blattner C, Polley DC, Ferrito F, et al. Central centrifugal cicatricial alopecia. Indian Dermatol Online J. 2013:4:50.
- Wolkenstein P, Loundou A, Barrau K, et al. Quality of life impairment in hidradenitis suppurativa: a study of 61 cases. J Am Acad Dermatol. 2007;56:621-623.
- Smith HS, Chao JD, Teitelbaum J. Painful hidradenitis suppurativa. Clin J Pain. 2010;26:435-444.
- Alavi A, Anooshirvani N, Kim WB, et al. Quality-of-life impairment in patients with hidradenitis suppurativa: a Canadian study. Am J Clin Dermatol. 2015;16:61-65.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- Kligman AM. Welcome letter. 2nd International Conference on the Sebaceous Gland, Acne, Rosacea and Related Disorders; September 13-16, 2008; Rome Italy.
- Kurzen H, Kurzen M. Secondary prevention of hidradenitis suppurativa. Dermatol Reports. 2019;11:8243.
- Sabat R, Tsaousi A, Rossbacher J, et al. Acne inversa/hidradenitis suppurativa: an update [in German]. Hautarzt. 2017;68:999-1006.
- Boer J, Nazary M, Riis PT. The role of mechanical stress in hidradenitis suppurativa. Dermatol Clin. 2016;34:37-43.
- Hamzavi IH, Griffith JL, Riyaz F, et al. Laser and light-based treatment options for hidradenitis suppurativa. J Am Acad Dermatol. 2015;73(5 suppl 1):S78-S81.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Michel C, DiBianco JM, Sabarwal V, et al. The treatment of genitoperineal hidradenitis suppurativa: a review of the literature. Urology. 2019;124:1-5.
- Constantinou CA, Fragoulis GE, Nikiphorou E. Hidradenitis suppurativa: infection, autoimmunity, or both [published online December 30, 2019]? Ther Adv Musculoskelet Dis. doi:10.1177/1759720x19895488.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Zouboulis CC, Desai N, Emtestam, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29:619-644.
- Mandell JB, Orr S, Koch J, et al. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J Orthop Res. 2019;37:1604-1609.
- Sun J, Shigemi H, Tanaka Y, et al. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397-404.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22:325-328.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219:148-154.
- Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154:977-978.
- Ma K, Chen X, Chen J-C, et al. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151-1162.
- Yuhas Y, Berent E, Ovadiah H, et al. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob Agents Chemother. 2006;50:396-398.
- Join-Lambert O, Coignard H, Jais J-P, et al. Efficacy of rifampin-moxifloxacin-metronidazole combination therapy in hidradenitis suppurativa. Dermatology. 2011;222:49-58.
- Choi J-H, Song M-J, Kim S-H, et al. Effect of moxifloxacin on production of proinflammatory cytokines from human peripheral blood mononuclear cells. Antimicrob Agents Chemother. 2003;47:3704-3707.
- Weiss T, Shalit I, Blau H, et al. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines.” Antimicrob Agents Chemother. 2004;48:1974-1982.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Mortimer PS, Dawber RP, Gales MA, et al. A double-blind controlled cross-over trial of cyproterone acetate in females with hidradenitis suppurativa. Br J Dermatol. 1986;115:263-268.
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. J Cutan Med Surg. 2007;11:125-131.
- Lee A, Fischer G. A case series of 20 women with hidradenitis suppurativa treated with spironolactone. Australas J Dermatol. 2015;56:192-196.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a “male” therapy for a predominantly “female” disease. J Clin Aesthet Dermatol. 2016;9:44-50.
- Dicken CH, Powell ST, Spear KL. Evaluation of isotretinoin treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1984;11:500-502.
- Huang CM, Kirchof MG. A new perspective on isotretinoin treatment of hidradenitis suppurativa: a retrospective chart review of patient outcomes. Dermatology. 2017;233:120-125.
- Norris JF, Cunliffe WJ. Failure of treatment of familial widespread hidradenitis suppurativa with isotretinoin. Clin Exp Dermatol. 1986;11:579-583.
- Soria A, Canoui-Poitrine F, Wolkenstein P, et al. Absence of efficacy of oral isotretinoin in hidradenitis suppurativa: a retrospective study based on patients’ outcome assessment. Dermatology. 2009;218:134-135.
Case Report
A 19-year-old female marine with a 10-year history of hidradenitis suppurativa (HS) presented with hyperpigmented nodules in the inguinal folds and a recurrent cyst in the right groin area of 2 to 3 weeks’ duration. She denied axillary or inframammary involvement. She underwent several incision and drainage procedures 1 year prior to her enlistment in the US Marine Corps at 18 years of age. She previously had been treated by dermatology with doxycycline 100-mg tablets twice daily, benzoyl peroxide wash 5% applied to affected areas and rinsed daily, and clindamycin solution 1% with minimal improvement. She denied smoking or alcohol intake and said she typically wore a loose-fitting uniform to work. As a marine, she was expected to participate in daily physical training and exercises with her military unit, during which she wore a standardized physical training uniform, including nylon shorts and a cotton T-shirt. She requested light duty—military duty status with physical limitations or restrictions—to avoid physical training that would cause further friction and irritation to the inguinal region.
Physical examination demonstrated a woman with Fitzpatrick skin type III and normal body mass index. There were hyperpigmented nodules and scarring in the inguinal folds, most consistent with Hurley stage 2. A single, 0.5-cm, draining lesion was visualized. No hyperhidrosis was noted. The patient was placed on light duty for 7 days, with physical training only at her own pace and discretion. Moreover, she was restricted from field training, rifle range training, and other situations where she may excessively sweat or not be able to adequately maintain personal hygiene. She was encouraged to continue clindamycin solution 1% to the affected area twice daily and was prescribed chlorhexidine solution 4% to use when washing affected areas in the shower. The patient also was referred to the dermatology department at the Naval Hospital Camp Pendleton (Oceanside, California), where she was treated with laser hair removal in the inguinal region, thus avoiding waxing and further aggravation of HS flares. Due to the combination of topical therapies along with laser hair removal and duty restrictions, the patient had a dramatic decrease in development of severe nodular lesions.
Comment
Presentation
Historically, the incidence of HS is estimated at 0.5% to 4% of the general population with female predominance.1 Predisposing factors include obesity, smoking, genetic predisposition to acne, apocrine duct obstruction, and secondary bacterial infection.2 During acute flares, patients generally present with tender subcutaneous nodules that drain malodorous purulent material.3,4 Acute flares are unpredictable, and patients deal with chronic, recurrent, draining wounds, leading to a poor quality of life with resulting physical, psychological, financial, social, and emotional distress.3-5 The negative impact of HS on a patient’s quality of life has been reported to be greater than other dermatologic conditions.6 Lesions can be particularly painful and can cause disfiguration to the surface of the skin.7 Lesion severity is described using the Hurley staging system. Patient quality of life is directly correlated with disease severity and Hurley stage. In stage 1, abscesses develop, but no sinus tracts or cicatrization is present. In stage 2, recurrent abscesses will form tracts and cicatrization. In stage 3, the abscesses become diffuse or near diffuse, with multiple interconnected tracts and abscesses across the entire area of the body.8,9
Severe or refractory HS within the physically active military population may require consideration of light or limited duty or even separation from service. Similarly, severe HS may pose challenges with other physically demanding occupations, such as the police force and firefighters.
Prevention Focus
Prevention of flares is key for patients with HS; secondary prevention aims to reduce impact of the disease or injury that has already occurred,10,11 which includes prevention of the infundibulofolliculitis from becoming a deep folliculitis, nodule, or fistula, as well as Hurley stage progression. Prompt diagnosis with appropriate treatment can decrease the severity of lesions, pain, and scarring. Globally, HS patients continue to experience considerable diagnostic delays of 8 to 12 years after onset of initial symptoms.11,12 Earlier accurate diagnosis and initiation of treatment from the primary care provider or general medical officer is imperative. Initial accurate management may help keep symptoms from progressing to more severe painful lesions. Similarly, patients should be educated on how to prevent HS flares. Patients should avoid known triggers, including smoking, obesity, sweating, mechanical irritation, stress, and poor hygiene.11
Shaving for hair reduction creates ingrown hair shafts, which may lead to folliculitis in mechanically stressed areas in skin folds, thus initiating the inflammatory cascade of HS.11,13 Therefore, shaving along with any other mechanical stress should be avoided in patients with HS. Laser hair removal has been shown to be quite helpful in both the prevention and treatment of HS. In one study, 22 patients with Hurley stage 2 to 3 disease were treated with an Nd:YAG laser once monthly. Results demonstrated a 65% decrease in disease severity after 3 monthly treatments.11 Similarly, other lasers have been used with success in several small case series; an 800-nm diode laser, intense pulsed light therapy, and a ruby laser have each demonstrated efficacy.14 Given these results, hair removal should be recommended to patients with HS. Military servicemembers (SMs) with certain conditions, such as polycystic ovary syndrome, pseudofolliculitis barbae, and HS, are eligible for laser hair removal when available at local military treatment facilities. Primary care providers for military SMs must have a working understanding of the disease process of HS and awareness of what resources are available for treatment, which allows for more streamlined care and improved outcomes.
Treatment Options
Treatment options are diverse and depend on the severity of HS. Typically, treatment begins with medical therapy followed by escalation to surgical intervention. Medical therapies often include antibiotics, acne treatments, antiandrogen therapy, immunosuppressive agents, and biologic therapy.15,16 If first-line medical interventions fail to control HS, surgical interventions should be considered. Surgical intervention in conjunction with medical therapy decreases the chance for recurrence.3,15,16
Although HS is internationally recognized as an inflammatory disease and not an infectious process, topical antibiotics can help to prevent and improve formation of abscesses, nodules, and pustules.11 Agents such as clindamycin and chlorhexidine wash have proven effective in preventing flares.11,16 Other antibiotics used alone or in combination also are efficacious. Tetracyclines are recommended as monotherapy for mild stages of HS.17-19 Doxycycline is the most commonly used tetracycline in HS patients and has been demonstrated to penetrate Staphylococcus aureus biofilm in high enough concentrations to maintain its antibacterial activity.20 Moreover, doxycycline, as with other tetracyclines, has a multitude of anti-inflammatory and immunomodulatory properties21 and can reduce the production of IL-1, IL-6, tumor necrosis factor α, and IL-8; downregulate chemotaxis; and promote lipo-oxygenase, matrix metalloproteinase, and nuclear factor κB (NF-κB) signaling inhibition.17
Clindamycin is the only known agent that has been studied for topical treatment and utilization in milder cases of HS.17,22 Systemic combination of clindamycin and rifampicin is the most studied, with well-established efficacy in managing HS.17,23,24 Clindamycin has bacteriostatic activity toward both aerobic and anaerobic gram-positive bacteria by binding irreversibly to the 50S ribosomal subunit, thereby inhibiting bacterial protein synthesis. Rifampicin binds to the beta subunit of DNA-dependent RNA polymerase, inhibiting bacterial DNA-dependent RNA synthesis. Rifampicin has broad-spectrum activity, mostly against gram-positive as well as some gram-negative bacteria. Moreover, rifampicin has anti-inflammatory and immunomodulatory properties, including evidence that it inhibits excessive helper T cell (TH17) responses by reducing inducible nitric oxide synthase transcription and NF-κB activity.25,26
Metronidazole, moxifloxacin, and rifampicin as triple combination therapy has been shown to be effective in reducing HS activity in moderate to severe cases that were refractory to other treatments.27 Research suggests that moxifloxacin has anti-inflammatory properties, mainly by reducing IL-1β, IL-8, and tumor necrosis factor α; stabilizing IXb protein; suppressing NF-κB signaling; and reducing IL-17A.28,29
Ertapenem can be utilized as a single 6-week antibiotic course during surgical planning or rescue therapy.18 Moreover, ertapenem can be used to treat complicated skin and soft tissue infections and has been shown to substantially improve clinical aspects of severe HS.17,27
Disease-modifying antirheumatic drugs are effective in the treatment of moderate to severe HS.17-19 In 2 phase 3 trials (PIONEER I and II), adalimumab was used as monotherapy or in conjunction with antibiotics in patients with moderate to severe HS compared to placebo.30 Results demonstrated a disease burden reduction of greater than 50%. Antibiotic dual therapy was not noted to significantly affect disease burden.30 Of note, use of immunosuppressants in the military affects an SM’s availability for worldwide deployment and duty station assignment.
Antiandrogen therapies have demonstrated some reduction in HS flares. Although recommendations for use in HS is based on limited evidence, one randomized controlled trial compared ethinyl estradiol–norgestrel to ethinyl estradiol and cyproterone acetate. Both therapies resulted in similar efficacy, with 12 of 24 (50%) patients reporting HS symptoms improving or completely resolved.31 In another retrospective study of women treated with antiandrogen therapies, including ethinyl estriol, cyproterone acetate, and spironolactone, 16 of 29 (55%) patients reported improvement.32 In another study, daily doses of 100 to 150 mg of spironolactone resulted in improvement in 17 of 20 (85%) patients, including complete remission in 11 of 20 (55%) patients. Of the 3 patients with severe HS, none had complete clearing of disease burden.33 Patients with polycystic ovary syndrome or HS flares that occur around menstruation are more likely to benefit from treatment with spironolactone.18,32,34
Retinoids frequently have been utilized in the management of HS. In some retrospective studies and other prospective studies with 5 or more patients, isotretinoin monotherapy was utilized for a 4- to 10-month period.18,35-38 In the Alikhan et al18 study, 85 of 207 patients demonstrated improvement of HS symptoms, with more remarkable improvements in milder cases. Isotretinoin for management of patients with HS who have concomitant nodulocystic acne would have two-fold benefits.18
Wound Care
Given the purulent nodular formation in HS, adequate wound care management is vital. There is an abundance of HS wound care management strategies utilized by clinicians and patients. When selecting the appropriate dressing, consideration for the type of HS wound, cost, ease of application, patient comfort, absorbency, and odor management is important.3 However, living arrangements for military SMs can create difficulties applying and maintaining HS dressings, especially if deployed or in a field setting. Active-duty SMs often find themselves in austere living conditions in the field, aboard ships, or in other scenarios where they may or may not have running water or showers. Maintaining adequate hygiene may be difficult, and additional education about how to keep wounds clean must be imparted. Ideal dressings for HS should be highly absorbent, comfortable when applied to the anatomic locations of the HS lesions, and easily self-applied. Ideally, dressings would have atraumatic adhesion and antimicrobial properties.3 Cost-effective dressing options that have good absorption capability include sanitary napkins, adult briefs, infant diapers, and gauze.3 These dressings help to wick moisture, thus protecting the wound from maceration, which is a common patient concern. Although gauze dressings are easier to obtain, they are not as absorbent. Abdominal pads can be utilized, but they are moderately absorbent, bulky, and more challenging to obtain over-the-counter. Hydrofiber and calcium alginate dressings with silver are not accessible to the common consumer and are more expensive than the aforementioned dressings, but they do have some antimicrobial activity. Silver-impregnated foam dressings are moldable to intertriginous areas, easy to self-apply, and have moderate-heavy absorption abilities.
Final Thoughts
Hidradenitis suppurativa poses cumbersome and uncomfortable symptoms for all patients and may pose additional hardships for military SMs or those with physically demanding occupations who work in austere environments. Severe HS can restrict a military SM from certain duty stations, positions, or deployments. Early identification of HS can help reduce HS flares, disfigurement, and placement on limited duty status, therefore rendering the SM more able to engage in his/her operational responsibilities. Hidradenitis suppurativa should be discussed with the patient, with the goal to prevent flares for SMs that will be in the field, placed in austere environments, or be deployed. Use of immunosuppressants in active-duty SMs may affect their deployability, duty assignment, and retention.
For a military SM with HS, all aspects of prevention and treatment need to be balanced with his/her ability to remain deployable and complete his/her daily duties. Military SMs are not guaranteed the ideal scenario for treatment and prevention of HS. Unsanitary environments and occlusive uniforms undoubtedly contribute to disease process and make treatment more challenging. If a military SM is in a field setting or deployed, frequent daily dressing changes should still be attempted.
Case Report
A 19-year-old female marine with a 10-year history of hidradenitis suppurativa (HS) presented with hyperpigmented nodules in the inguinal folds and a recurrent cyst in the right groin area of 2 to 3 weeks’ duration. She denied axillary or inframammary involvement. She underwent several incision and drainage procedures 1 year prior to her enlistment in the US Marine Corps at 18 years of age. She previously had been treated by dermatology with doxycycline 100-mg tablets twice daily, benzoyl peroxide wash 5% applied to affected areas and rinsed daily, and clindamycin solution 1% with minimal improvement. She denied smoking or alcohol intake and said she typically wore a loose-fitting uniform to work. As a marine, she was expected to participate in daily physical training and exercises with her military unit, during which she wore a standardized physical training uniform, including nylon shorts and a cotton T-shirt. She requested light duty—military duty status with physical limitations or restrictions—to avoid physical training that would cause further friction and irritation to the inguinal region.
Physical examination demonstrated a woman with Fitzpatrick skin type III and normal body mass index. There were hyperpigmented nodules and scarring in the inguinal folds, most consistent with Hurley stage 2. A single, 0.5-cm, draining lesion was visualized. No hyperhidrosis was noted. The patient was placed on light duty for 7 days, with physical training only at her own pace and discretion. Moreover, she was restricted from field training, rifle range training, and other situations where she may excessively sweat or not be able to adequately maintain personal hygiene. She was encouraged to continue clindamycin solution 1% to the affected area twice daily and was prescribed chlorhexidine solution 4% to use when washing affected areas in the shower. The patient also was referred to the dermatology department at the Naval Hospital Camp Pendleton (Oceanside, California), where she was treated with laser hair removal in the inguinal region, thus avoiding waxing and further aggravation of HS flares. Due to the combination of topical therapies along with laser hair removal and duty restrictions, the patient had a dramatic decrease in development of severe nodular lesions.
Comment
Presentation
Historically, the incidence of HS is estimated at 0.5% to 4% of the general population with female predominance.1 Predisposing factors include obesity, smoking, genetic predisposition to acne, apocrine duct obstruction, and secondary bacterial infection.2 During acute flares, patients generally present with tender subcutaneous nodules that drain malodorous purulent material.3,4 Acute flares are unpredictable, and patients deal with chronic, recurrent, draining wounds, leading to a poor quality of life with resulting physical, psychological, financial, social, and emotional distress.3-5 The negative impact of HS on a patient’s quality of life has been reported to be greater than other dermatologic conditions.6 Lesions can be particularly painful and can cause disfiguration to the surface of the skin.7 Lesion severity is described using the Hurley staging system. Patient quality of life is directly correlated with disease severity and Hurley stage. In stage 1, abscesses develop, but no sinus tracts or cicatrization is present. In stage 2, recurrent abscesses will form tracts and cicatrization. In stage 3, the abscesses become diffuse or near diffuse, with multiple interconnected tracts and abscesses across the entire area of the body.8,9
Severe or refractory HS within the physically active military population may require consideration of light or limited duty or even separation from service. Similarly, severe HS may pose challenges with other physically demanding occupations, such as the police force and firefighters.
Prevention Focus
Prevention of flares is key for patients with HS; secondary prevention aims to reduce impact of the disease or injury that has already occurred,10,11 which includes prevention of the infundibulofolliculitis from becoming a deep folliculitis, nodule, or fistula, as well as Hurley stage progression. Prompt diagnosis with appropriate treatment can decrease the severity of lesions, pain, and scarring. Globally, HS patients continue to experience considerable diagnostic delays of 8 to 12 years after onset of initial symptoms.11,12 Earlier accurate diagnosis and initiation of treatment from the primary care provider or general medical officer is imperative. Initial accurate management may help keep symptoms from progressing to more severe painful lesions. Similarly, patients should be educated on how to prevent HS flares. Patients should avoid known triggers, including smoking, obesity, sweating, mechanical irritation, stress, and poor hygiene.11
Shaving for hair reduction creates ingrown hair shafts, which may lead to folliculitis in mechanically stressed areas in skin folds, thus initiating the inflammatory cascade of HS.11,13 Therefore, shaving along with any other mechanical stress should be avoided in patients with HS. Laser hair removal has been shown to be quite helpful in both the prevention and treatment of HS. In one study, 22 patients with Hurley stage 2 to 3 disease were treated with an Nd:YAG laser once monthly. Results demonstrated a 65% decrease in disease severity after 3 monthly treatments.11 Similarly, other lasers have been used with success in several small case series; an 800-nm diode laser, intense pulsed light therapy, and a ruby laser have each demonstrated efficacy.14 Given these results, hair removal should be recommended to patients with HS. Military servicemembers (SMs) with certain conditions, such as polycystic ovary syndrome, pseudofolliculitis barbae, and HS, are eligible for laser hair removal when available at local military treatment facilities. Primary care providers for military SMs must have a working understanding of the disease process of HS and awareness of what resources are available for treatment, which allows for more streamlined care and improved outcomes.
Treatment Options
Treatment options are diverse and depend on the severity of HS. Typically, treatment begins with medical therapy followed by escalation to surgical intervention. Medical therapies often include antibiotics, acne treatments, antiandrogen therapy, immunosuppressive agents, and biologic therapy.15,16 If first-line medical interventions fail to control HS, surgical interventions should be considered. Surgical intervention in conjunction with medical therapy decreases the chance for recurrence.3,15,16
Although HS is internationally recognized as an inflammatory disease and not an infectious process, topical antibiotics can help to prevent and improve formation of abscesses, nodules, and pustules.11 Agents such as clindamycin and chlorhexidine wash have proven effective in preventing flares.11,16 Other antibiotics used alone or in combination also are efficacious. Tetracyclines are recommended as monotherapy for mild stages of HS.17-19 Doxycycline is the most commonly used tetracycline in HS patients and has been demonstrated to penetrate Staphylococcus aureus biofilm in high enough concentrations to maintain its antibacterial activity.20 Moreover, doxycycline, as with other tetracyclines, has a multitude of anti-inflammatory and immunomodulatory properties21 and can reduce the production of IL-1, IL-6, tumor necrosis factor α, and IL-8; downregulate chemotaxis; and promote lipo-oxygenase, matrix metalloproteinase, and nuclear factor κB (NF-κB) signaling inhibition.17
Clindamycin is the only known agent that has been studied for topical treatment and utilization in milder cases of HS.17,22 Systemic combination of clindamycin and rifampicin is the most studied, with well-established efficacy in managing HS.17,23,24 Clindamycin has bacteriostatic activity toward both aerobic and anaerobic gram-positive bacteria by binding irreversibly to the 50S ribosomal subunit, thereby inhibiting bacterial protein synthesis. Rifampicin binds to the beta subunit of DNA-dependent RNA polymerase, inhibiting bacterial DNA-dependent RNA synthesis. Rifampicin has broad-spectrum activity, mostly against gram-positive as well as some gram-negative bacteria. Moreover, rifampicin has anti-inflammatory and immunomodulatory properties, including evidence that it inhibits excessive helper T cell (TH17) responses by reducing inducible nitric oxide synthase transcription and NF-κB activity.25,26
Metronidazole, moxifloxacin, and rifampicin as triple combination therapy has been shown to be effective in reducing HS activity in moderate to severe cases that were refractory to other treatments.27 Research suggests that moxifloxacin has anti-inflammatory properties, mainly by reducing IL-1β, IL-8, and tumor necrosis factor α; stabilizing IXb protein; suppressing NF-κB signaling; and reducing IL-17A.28,29
Ertapenem can be utilized as a single 6-week antibiotic course during surgical planning or rescue therapy.18 Moreover, ertapenem can be used to treat complicated skin and soft tissue infections and has been shown to substantially improve clinical aspects of severe HS.17,27
Disease-modifying antirheumatic drugs are effective in the treatment of moderate to severe HS.17-19 In 2 phase 3 trials (PIONEER I and II), adalimumab was used as monotherapy or in conjunction with antibiotics in patients with moderate to severe HS compared to placebo.30 Results demonstrated a disease burden reduction of greater than 50%. Antibiotic dual therapy was not noted to significantly affect disease burden.30 Of note, use of immunosuppressants in the military affects an SM’s availability for worldwide deployment and duty station assignment.
Antiandrogen therapies have demonstrated some reduction in HS flares. Although recommendations for use in HS is based on limited evidence, one randomized controlled trial compared ethinyl estradiol–norgestrel to ethinyl estradiol and cyproterone acetate. Both therapies resulted in similar efficacy, with 12 of 24 (50%) patients reporting HS symptoms improving or completely resolved.31 In another retrospective study of women treated with antiandrogen therapies, including ethinyl estriol, cyproterone acetate, and spironolactone, 16 of 29 (55%) patients reported improvement.32 In another study, daily doses of 100 to 150 mg of spironolactone resulted in improvement in 17 of 20 (85%) patients, including complete remission in 11 of 20 (55%) patients. Of the 3 patients with severe HS, none had complete clearing of disease burden.33 Patients with polycystic ovary syndrome or HS flares that occur around menstruation are more likely to benefit from treatment with spironolactone.18,32,34
Retinoids frequently have been utilized in the management of HS. In some retrospective studies and other prospective studies with 5 or more patients, isotretinoin monotherapy was utilized for a 4- to 10-month period.18,35-38 In the Alikhan et al18 study, 85 of 207 patients demonstrated improvement of HS symptoms, with more remarkable improvements in milder cases. Isotretinoin for management of patients with HS who have concomitant nodulocystic acne would have two-fold benefits.18
Wound Care
Given the purulent nodular formation in HS, adequate wound care management is vital. There is an abundance of HS wound care management strategies utilized by clinicians and patients. When selecting the appropriate dressing, consideration for the type of HS wound, cost, ease of application, patient comfort, absorbency, and odor management is important.3 However, living arrangements for military SMs can create difficulties applying and maintaining HS dressings, especially if deployed or in a field setting. Active-duty SMs often find themselves in austere living conditions in the field, aboard ships, or in other scenarios where they may or may not have running water or showers. Maintaining adequate hygiene may be difficult, and additional education about how to keep wounds clean must be imparted. Ideal dressings for HS should be highly absorbent, comfortable when applied to the anatomic locations of the HS lesions, and easily self-applied. Ideally, dressings would have atraumatic adhesion and antimicrobial properties.3 Cost-effective dressing options that have good absorption capability include sanitary napkins, adult briefs, infant diapers, and gauze.3 These dressings help to wick moisture, thus protecting the wound from maceration, which is a common patient concern. Although gauze dressings are easier to obtain, they are not as absorbent. Abdominal pads can be utilized, but they are moderately absorbent, bulky, and more challenging to obtain over-the-counter. Hydrofiber and calcium alginate dressings with silver are not accessible to the common consumer and are more expensive than the aforementioned dressings, but they do have some antimicrobial activity. Silver-impregnated foam dressings are moldable to intertriginous areas, easy to self-apply, and have moderate-heavy absorption abilities.
Final Thoughts
Hidradenitis suppurativa poses cumbersome and uncomfortable symptoms for all patients and may pose additional hardships for military SMs or those with physically demanding occupations who work in austere environments. Severe HS can restrict a military SM from certain duty stations, positions, or deployments. Early identification of HS can help reduce HS flares, disfigurement, and placement on limited duty status, therefore rendering the SM more able to engage in his/her operational responsibilities. Hidradenitis suppurativa should be discussed with the patient, with the goal to prevent flares for SMs that will be in the field, placed in austere environments, or be deployed. Use of immunosuppressants in active-duty SMs may affect their deployability, duty assignment, and retention.
For a military SM with HS, all aspects of prevention and treatment need to be balanced with his/her ability to remain deployable and complete his/her daily duties. Military SMs are not guaranteed the ideal scenario for treatment and prevention of HS. Unsanitary environments and occlusive uniforms undoubtedly contribute to disease process and make treatment more challenging. If a military SM is in a field setting or deployed, frequent daily dressing changes should still be attempted.
- Dufour DN, Emtestam L, Jemec GB. Hidradenitis suppurativa: a common and burdensome, yet under-recognised, inflammatory skin disease. Postgrad Med J. 2014;90:216-221.
- Beshara MA. Hidradenitis suppurativa: a clinician’s tool for early diagnosis and treatment. Nurse Pract. 2010;35:24-28.
- Kazemi A, Carnaggio K, Clark M, et al. Optimal wound care management in hidradenitis suppurativa. J Dermatolog Treat. 2017;29:165-167.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003:49:96-98.
- Blattner C, Polley DC, Ferrito F, et al. Central centrifugal cicatricial alopecia. Indian Dermatol Online J. 2013:4:50.
- Wolkenstein P, Loundou A, Barrau K, et al. Quality of life impairment in hidradenitis suppurativa: a study of 61 cases. J Am Acad Dermatol. 2007;56:621-623.
- Smith HS, Chao JD, Teitelbaum J. Painful hidradenitis suppurativa. Clin J Pain. 2010;26:435-444.
- Alavi A, Anooshirvani N, Kim WB, et al. Quality-of-life impairment in patients with hidradenitis suppurativa: a Canadian study. Am J Clin Dermatol. 2015;16:61-65.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- Kligman AM. Welcome letter. 2nd International Conference on the Sebaceous Gland, Acne, Rosacea and Related Disorders; September 13-16, 2008; Rome Italy.
- Kurzen H, Kurzen M. Secondary prevention of hidradenitis suppurativa. Dermatol Reports. 2019;11:8243.
- Sabat R, Tsaousi A, Rossbacher J, et al. Acne inversa/hidradenitis suppurativa: an update [in German]. Hautarzt. 2017;68:999-1006.
- Boer J, Nazary M, Riis PT. The role of mechanical stress in hidradenitis suppurativa. Dermatol Clin. 2016;34:37-43.
- Hamzavi IH, Griffith JL, Riyaz F, et al. Laser and light-based treatment options for hidradenitis suppurativa. J Am Acad Dermatol. 2015;73(5 suppl 1):S78-S81.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Michel C, DiBianco JM, Sabarwal V, et al. The treatment of genitoperineal hidradenitis suppurativa: a review of the literature. Urology. 2019;124:1-5.
- Constantinou CA, Fragoulis GE, Nikiphorou E. Hidradenitis suppurativa: infection, autoimmunity, or both [published online December 30, 2019]? Ther Adv Musculoskelet Dis. doi:10.1177/1759720x19895488.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Zouboulis CC, Desai N, Emtestam, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29:619-644.
- Mandell JB, Orr S, Koch J, et al. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J Orthop Res. 2019;37:1604-1609.
- Sun J, Shigemi H, Tanaka Y, et al. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397-404.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22:325-328.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219:148-154.
- Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154:977-978.
- Ma K, Chen X, Chen J-C, et al. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151-1162.
- Yuhas Y, Berent E, Ovadiah H, et al. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob Agents Chemother. 2006;50:396-398.
- Join-Lambert O, Coignard H, Jais J-P, et al. Efficacy of rifampin-moxifloxacin-metronidazole combination therapy in hidradenitis suppurativa. Dermatology. 2011;222:49-58.
- Choi J-H, Song M-J, Kim S-H, et al. Effect of moxifloxacin on production of proinflammatory cytokines from human peripheral blood mononuclear cells. Antimicrob Agents Chemother. 2003;47:3704-3707.
- Weiss T, Shalit I, Blau H, et al. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines.” Antimicrob Agents Chemother. 2004;48:1974-1982.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Mortimer PS, Dawber RP, Gales MA, et al. A double-blind controlled cross-over trial of cyproterone acetate in females with hidradenitis suppurativa. Br J Dermatol. 1986;115:263-268.
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. J Cutan Med Surg. 2007;11:125-131.
- Lee A, Fischer G. A case series of 20 women with hidradenitis suppurativa treated with spironolactone. Australas J Dermatol. 2015;56:192-196.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a “male” therapy for a predominantly “female” disease. J Clin Aesthet Dermatol. 2016;9:44-50.
- Dicken CH, Powell ST, Spear KL. Evaluation of isotretinoin treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1984;11:500-502.
- Huang CM, Kirchof MG. A new perspective on isotretinoin treatment of hidradenitis suppurativa: a retrospective chart review of patient outcomes. Dermatology. 2017;233:120-125.
- Norris JF, Cunliffe WJ. Failure of treatment of familial widespread hidradenitis suppurativa with isotretinoin. Clin Exp Dermatol. 1986;11:579-583.
- Soria A, Canoui-Poitrine F, Wolkenstein P, et al. Absence of efficacy of oral isotretinoin in hidradenitis suppurativa: a retrospective study based on patients’ outcome assessment. Dermatology. 2009;218:134-135.
- Dufour DN, Emtestam L, Jemec GB. Hidradenitis suppurativa: a common and burdensome, yet under-recognised, inflammatory skin disease. Postgrad Med J. 2014;90:216-221.
- Beshara MA. Hidradenitis suppurativa: a clinician’s tool for early diagnosis and treatment. Nurse Pract. 2010;35:24-28.
- Kazemi A, Carnaggio K, Clark M, et al. Optimal wound care management in hidradenitis suppurativa. J Dermatolog Treat. 2017;29:165-167.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003:49:96-98.
- Blattner C, Polley DC, Ferrito F, et al. Central centrifugal cicatricial alopecia. Indian Dermatol Online J. 2013:4:50.
- Wolkenstein P, Loundou A, Barrau K, et al. Quality of life impairment in hidradenitis suppurativa: a study of 61 cases. J Am Acad Dermatol. 2007;56:621-623.
- Smith HS, Chao JD, Teitelbaum J. Painful hidradenitis suppurativa. Clin J Pain. 2010;26:435-444.
- Alavi A, Anooshirvani N, Kim WB, et al. Quality-of-life impairment in patients with hidradenitis suppurativa: a Canadian study. Am J Clin Dermatol. 2015;16:61-65.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- Kligman AM. Welcome letter. 2nd International Conference on the Sebaceous Gland, Acne, Rosacea and Related Disorders; September 13-16, 2008; Rome Italy.
- Kurzen H, Kurzen M. Secondary prevention of hidradenitis suppurativa. Dermatol Reports. 2019;11:8243.
- Sabat R, Tsaousi A, Rossbacher J, et al. Acne inversa/hidradenitis suppurativa: an update [in German]. Hautarzt. 2017;68:999-1006.
- Boer J, Nazary M, Riis PT. The role of mechanical stress in hidradenitis suppurativa. Dermatol Clin. 2016;34:37-43.
- Hamzavi IH, Griffith JL, Riyaz F, et al. Laser and light-based treatment options for hidradenitis suppurativa. J Am Acad Dermatol. 2015;73(5 suppl 1):S78-S81.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Michel C, DiBianco JM, Sabarwal V, et al. The treatment of genitoperineal hidradenitis suppurativa: a review of the literature. Urology. 2019;124:1-5.
- Constantinou CA, Fragoulis GE, Nikiphorou E. Hidradenitis suppurativa: infection, autoimmunity, or both [published online December 30, 2019]? Ther Adv Musculoskelet Dis. doi:10.1177/1759720x19895488.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Zouboulis CC, Desai N, Emtestam, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29:619-644.
- Mandell JB, Orr S, Koch J, et al. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J Orthop Res. 2019;37:1604-1609.
- Sun J, Shigemi H, Tanaka Y, et al. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397-404.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22:325-328.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219:148-154.
- Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154:977-978.
- Ma K, Chen X, Chen J-C, et al. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151-1162.
- Yuhas Y, Berent E, Ovadiah H, et al. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob Agents Chemother. 2006;50:396-398.
- Join-Lambert O, Coignard H, Jais J-P, et al. Efficacy of rifampin-moxifloxacin-metronidazole combination therapy in hidradenitis suppurativa. Dermatology. 2011;222:49-58.
- Choi J-H, Song M-J, Kim S-H, et al. Effect of moxifloxacin on production of proinflammatory cytokines from human peripheral blood mononuclear cells. Antimicrob Agents Chemother. 2003;47:3704-3707.
- Weiss T, Shalit I, Blau H, et al. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines.” Antimicrob Agents Chemother. 2004;48:1974-1982.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Mortimer PS, Dawber RP, Gales MA, et al. A double-blind controlled cross-over trial of cyproterone acetate in females with hidradenitis suppurativa. Br J Dermatol. 1986;115:263-268.
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. J Cutan Med Surg. 2007;11:125-131.
- Lee A, Fischer G. A case series of 20 women with hidradenitis suppurativa treated with spironolactone. Australas J Dermatol. 2015;56:192-196.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a “male” therapy for a predominantly “female” disease. J Clin Aesthet Dermatol. 2016;9:44-50.
- Dicken CH, Powell ST, Spear KL. Evaluation of isotretinoin treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1984;11:500-502.
- Huang CM, Kirchof MG. A new perspective on isotretinoin treatment of hidradenitis suppurativa: a retrospective chart review of patient outcomes. Dermatology. 2017;233:120-125.
- Norris JF, Cunliffe WJ. Failure of treatment of familial widespread hidradenitis suppurativa with isotretinoin. Clin Exp Dermatol. 1986;11:579-583.
- Soria A, Canoui-Poitrine F, Wolkenstein P, et al. Absence of efficacy of oral isotretinoin in hidradenitis suppurativa: a retrospective study based on patients’ outcome assessment. Dermatology. 2009;218:134-135.
Practice Points
- Hidradenitis suppurativa (HS) can be more difficult to treat in physically active military servicemembers (SMs).
- Patient education and primary care physician awareness of HS is critical to initial diagnosis and long-term management.
- Primary care physician knowledge of HS as well as an understanding of the capabilities at local military medical facilities is important for optimal treatment of HS in military SMs.

New lupus classification criteria perform well in children, young adults
, according to results from a single-center, retrospective study.
However, the 2019 criteria, which were developed using cohorts of adult patients with SLE, were statistically no better than the 1997 ACR criteria at identifying those without the disease, first author Najla Aljaberi, MBBS, of the Cincinnati Children’s Hospital Medical Center, and colleagues reported in Arthritis Care & Research.
The 2019 criteria were especially good at correctly classifying SLE in non-White youths, but the two sets of criteria performed equally well among male and female youths with SLE and across age groups.
“Our study confirms superior sensitivity of the new criteria over the 1997-ACR criteria in youths with SLE. The difference in sensitivity estimates between the two criteria sets (2019-EULAR/ACR vs. 1997-ACR) may be explained by a higher weight being assigned to immunologic criteria, less strict hematologic criteria (not requiring >2 occurrences), and the inclusion of subjective features of arthritis. Notably, our estimates of the sensitivity of the 2019-EULAR/ACR criteria were similar to those reported from a Brazilian pediatric study by Fonseca et al. (87.7%) that also used physician diagnosis as reference standard,” the researchers wrote.
Dr. Aljaberi and colleagues reviewed electronic medical records of 112 patients with SLE aged 2-21 years and 105 controls aged 1-19 years at Cincinnati Children’s Hospital Medical Center during 2008-2019. Patients identified in the records at the center were considered to have SLE based on ICD-10 codes assigned by experienced pediatric rheumatologists. The control patients included 69 (66%) with juvenile dermatomyositis and 36 with juvenile scleroderma/systemic sclerosis, based on corresponding ICD-10 codes.
Among the SLE cases, 57% were White and 81% were female, while Whites represented 83% and females 71% of control patients. Young adults aged 18-21 years represented a minority of SLE cases (18%) and controls (7%).
The 2019 criteria had significantly higher sensitivity than did the 1997 criteria (85% vs. 72%, respectively; P = .023) but similar specificity (83% vs. 87%; P = .456). A total of 17 out of the 112 SLE cases failed to meet the 2019 criteria, 13 (76%) of whom were White. Overall, 31 SLE cases did not meet the 1997 criteria, but 15 of those fulfilled the 2019 criteria. While there was no statistically significant difference in the sensitivity of the 2019 criteria between non-White and White cases (92% vs. 80%, respectively; P = .08), the difference in sensitivity was significant with the 1997 criteria (83% vs. 64%; P < .02).
The 2019 criteria had similar sensitivity in males and females (86% vs. 81%, respectively), as well as specificity (81% vs. 87%). The 1997 criteria also provided similar sensitivity between males and females (71% vs. 76%) as well as specificity (85% vs. 90%).
In only four instances did SLE cases meet 2019 criteria before ICD-10 diagnosis of SLE, whereas in the other 108 cases the ICD-10 diagnosis coincided with reaching the threshold for meeting 2019 criteria.
There was no funding secured for the study, and the authors had no conflicts of interest to disclose.
SOURCE: Aljaberi N et al. Arthritis Care Res. 2020 Aug 25. doi: 10.1002/acr.24430.
, according to results from a single-center, retrospective study.
However, the 2019 criteria, which were developed using cohorts of adult patients with SLE, were statistically no better than the 1997 ACR criteria at identifying those without the disease, first author Najla Aljaberi, MBBS, of the Cincinnati Children’s Hospital Medical Center, and colleagues reported in Arthritis Care & Research.
The 2019 criteria were especially good at correctly classifying SLE in non-White youths, but the two sets of criteria performed equally well among male and female youths with SLE and across age groups.
“Our study confirms superior sensitivity of the new criteria over the 1997-ACR criteria in youths with SLE. The difference in sensitivity estimates between the two criteria sets (2019-EULAR/ACR vs. 1997-ACR) may be explained by a higher weight being assigned to immunologic criteria, less strict hematologic criteria (not requiring >2 occurrences), and the inclusion of subjective features of arthritis. Notably, our estimates of the sensitivity of the 2019-EULAR/ACR criteria were similar to those reported from a Brazilian pediatric study by Fonseca et al. (87.7%) that also used physician diagnosis as reference standard,” the researchers wrote.
Dr. Aljaberi and colleagues reviewed electronic medical records of 112 patients with SLE aged 2-21 years and 105 controls aged 1-19 years at Cincinnati Children’s Hospital Medical Center during 2008-2019. Patients identified in the records at the center were considered to have SLE based on ICD-10 codes assigned by experienced pediatric rheumatologists. The control patients included 69 (66%) with juvenile dermatomyositis and 36 with juvenile scleroderma/systemic sclerosis, based on corresponding ICD-10 codes.
Among the SLE cases, 57% were White and 81% were female, while Whites represented 83% and females 71% of control patients. Young adults aged 18-21 years represented a minority of SLE cases (18%) and controls (7%).
The 2019 criteria had significantly higher sensitivity than did the 1997 criteria (85% vs. 72%, respectively; P = .023) but similar specificity (83% vs. 87%; P = .456). A total of 17 out of the 112 SLE cases failed to meet the 2019 criteria, 13 (76%) of whom were White. Overall, 31 SLE cases did not meet the 1997 criteria, but 15 of those fulfilled the 2019 criteria. While there was no statistically significant difference in the sensitivity of the 2019 criteria between non-White and White cases (92% vs. 80%, respectively; P = .08), the difference in sensitivity was significant with the 1997 criteria (83% vs. 64%; P < .02).
The 2019 criteria had similar sensitivity in males and females (86% vs. 81%, respectively), as well as specificity (81% vs. 87%). The 1997 criteria also provided similar sensitivity between males and females (71% vs. 76%) as well as specificity (85% vs. 90%).
In only four instances did SLE cases meet 2019 criteria before ICD-10 diagnosis of SLE, whereas in the other 108 cases the ICD-10 diagnosis coincided with reaching the threshold for meeting 2019 criteria.
There was no funding secured for the study, and the authors had no conflicts of interest to disclose.
SOURCE: Aljaberi N et al. Arthritis Care Res. 2020 Aug 25. doi: 10.1002/acr.24430.
, according to results from a single-center, retrospective study.
However, the 2019 criteria, which were developed using cohorts of adult patients with SLE, were statistically no better than the 1997 ACR criteria at identifying those without the disease, first author Najla Aljaberi, MBBS, of the Cincinnati Children’s Hospital Medical Center, and colleagues reported in Arthritis Care & Research.
The 2019 criteria were especially good at correctly classifying SLE in non-White youths, but the two sets of criteria performed equally well among male and female youths with SLE and across age groups.
“Our study confirms superior sensitivity of the new criteria over the 1997-ACR criteria in youths with SLE. The difference in sensitivity estimates between the two criteria sets (2019-EULAR/ACR vs. 1997-ACR) may be explained by a higher weight being assigned to immunologic criteria, less strict hematologic criteria (not requiring >2 occurrences), and the inclusion of subjective features of arthritis. Notably, our estimates of the sensitivity of the 2019-EULAR/ACR criteria were similar to those reported from a Brazilian pediatric study by Fonseca et al. (87.7%) that also used physician diagnosis as reference standard,” the researchers wrote.
Dr. Aljaberi and colleagues reviewed electronic medical records of 112 patients with SLE aged 2-21 years and 105 controls aged 1-19 years at Cincinnati Children’s Hospital Medical Center during 2008-2019. Patients identified in the records at the center were considered to have SLE based on ICD-10 codes assigned by experienced pediatric rheumatologists. The control patients included 69 (66%) with juvenile dermatomyositis and 36 with juvenile scleroderma/systemic sclerosis, based on corresponding ICD-10 codes.
Among the SLE cases, 57% were White and 81% were female, while Whites represented 83% and females 71% of control patients. Young adults aged 18-21 years represented a minority of SLE cases (18%) and controls (7%).
The 2019 criteria had significantly higher sensitivity than did the 1997 criteria (85% vs. 72%, respectively; P = .023) but similar specificity (83% vs. 87%; P = .456). A total of 17 out of the 112 SLE cases failed to meet the 2019 criteria, 13 (76%) of whom were White. Overall, 31 SLE cases did not meet the 1997 criteria, but 15 of those fulfilled the 2019 criteria. While there was no statistically significant difference in the sensitivity of the 2019 criteria between non-White and White cases (92% vs. 80%, respectively; P = .08), the difference in sensitivity was significant with the 1997 criteria (83% vs. 64%; P < .02).
The 2019 criteria had similar sensitivity in males and females (86% vs. 81%, respectively), as well as specificity (81% vs. 87%). The 1997 criteria also provided similar sensitivity between males and females (71% vs. 76%) as well as specificity (85% vs. 90%).
In only four instances did SLE cases meet 2019 criteria before ICD-10 diagnosis of SLE, whereas in the other 108 cases the ICD-10 diagnosis coincided with reaching the threshold for meeting 2019 criteria.
There was no funding secured for the study, and the authors had no conflicts of interest to disclose.
SOURCE: Aljaberi N et al. Arthritis Care Res. 2020 Aug 25. doi: 10.1002/acr.24430.
FROM ARTHRITIS CARE & RESEARCH
Promising drugs line up for lupus treatment
Systemic lupus erythematosus remains a treatment challenge, but a variety of drugs in the pipeline are set to target type I interferons, cytokines, and B cells, according to Richard Furie, MD, chief of the division of rheumatology at Northwell Health and professor of medicine at Hofstra University, Hempstead, N.Y.

In general, when treating patients with systemic lupus erythematosus (SLE), “we just don’t see satisfactory response rates,” Dr. Furie said in an online presentation at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education.
“I think the greatest unmet need is in lupus nephritis,” he said. The data show that not even one-third of patients are adequately responding to standard of care treatment. “We need to do better in lupus nephritis but also for those patients with moderate-severe manifestations outside the kidney.”
Patients with SLE have elevated levels of interferon-alpha, Dr. Furie noted. Data from recent studies show that interferon inhibitors can reduce clinical activity in SLE patients, he said.
“About two-thirds to three-quarters of lupus patients have evidence of interferon pathway activation,” he said. There are three types of interferons, and five subtypes of type I interferon, and all five subtypes of type I interferon bind to the same receptor, which is an important strategy for drug development.
In particular, recent phase 2 and 3 trials have focused on targeting type I interferons with anifrolumab, which blocks all five subtypes.
Dr. Furie cited “very robust results” from a phase 2 study. Results of two phase 3 trials of anifrolumab led to a split decision, but the totality of the data collected across the phase 2 and 3 studies points to a drug that is effective for patients with SLE. The two phase 3 studies were published in Lancet Rheumatology and the New England Journal of Medicine.
Dr. Furie also identified recent studies of baricitinib (Olumiant), which has the ability to target several different cytokines. A phase 2 study in 2018 showed a significant difference in SLE Responder Index between lupus patients who received 4 mg of baricitinib or placebo, and a phase 3 study is underway, he said.
For lupus nephritis, Dr. Furie cited the BLISS-LN trial, a 104-week, randomized trial of patients with active lupus nephritis. The group of patients who received belimumab (Benlysta), a monoclonal antibody that targets B-cell activating factor, in addition to standard therapy had significant improvements in renal responses, compared with standard therapy alone (43.0% vs. 32.3%). The outcome measure was Primary Efficacy Renal Response, defined as urinary protein/creatinine ratio <0.7, eGFR ≥60 mL/min per 1.73 m2, confirmation on consecutive visits, and required tapering of background glucocorticoids.
Although belimumab was approved for SLE in 2011, the BLISS-LN study focused on SLE patients with active kidney disease. “Neutralizing B-cell activating factor and down-regulating autoreactive B-cell function in kidneys represented a compelling therapeutic approach to lupus nephritis,” he explained.
Voclosporin, distinct from cyclosporine, has also been studied in lupus nephritis, Dr. Furie said. Voclosporin offers several benefits over cyclosporine, including greater potency and a lower drug and metabolite load, as well as a more consistent pharmacokinetic and pharmacodynamic relationship, he said. In the phase 3 AURORA study, presented at this year’s EULAR congress, 40% of patients with lupus nephritis met the primary endpoint of a renal response at 52 weeks, compared with 22.5% of placebo patients.
Looking ahead to the treatment of SLE in 2021, “I feel very strongly that voclosporin will be approved for lupus nephritis,” he said. He also predicted that the use of belimumab will be officially extended for lupus nephritis and that anifrolumab will receive an approval for SLE patients.
In addition, the future may witness the increased use of biomarkers and development of more individualized therapy. These breakthroughs will yield better outcomes for all lupus patients, he said.
Dr. Furie disclosed relationships with GlaxoSmithKline, Genentech/Roche, Aurinia Pharmaceuticals, AstraZeneca/MedImmune, and Eli Lilly. Global Academy for Medical Education and this news organization are owned by the same parent company.
Systemic lupus erythematosus remains a treatment challenge, but a variety of drugs in the pipeline are set to target type I interferons, cytokines, and B cells, according to Richard Furie, MD, chief of the division of rheumatology at Northwell Health and professor of medicine at Hofstra University, Hempstead, N.Y.
In general, when treating patients with systemic lupus erythematosus (SLE), “we just don’t see satisfactory response rates,” Dr. Furie said in an online presentation at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education.
“I think the greatest unmet need is in lupus nephritis,” he said. The data show that not even one-third of patients are adequately responding to standard of care treatment. “We need to do better in lupus nephritis but also for those patients with moderate-severe manifestations outside the kidney.”
Patients with SLE have elevated levels of interferon-alpha, Dr. Furie noted. Data from recent studies show that interferon inhibitors can reduce clinical activity in SLE patients, he said.
“About two-thirds to three-quarters of lupus patients have evidence of interferon pathway activation,” he said. There are three types of interferons, and five subtypes of type I interferon, and all five subtypes of type I interferon bind to the same receptor, which is an important strategy for drug development.
In particular, recent phase 2 and 3 trials have focused on targeting type I interferons with anifrolumab, which blocks all five subtypes.
Dr. Furie cited “very robust results” from a phase 2 study. Results of two phase 3 trials of anifrolumab led to a split decision, but the totality of the data collected across the phase 2 and 3 studies points to a drug that is effective for patients with SLE. The two phase 3 studies were published in Lancet Rheumatology and the New England Journal of Medicine.
Dr. Furie also identified recent studies of baricitinib (Olumiant), which has the ability to target several different cytokines. A phase 2 study in 2018 showed a significant difference in SLE Responder Index between lupus patients who received 4 mg of baricitinib or placebo, and a phase 3 study is underway, he said.
For lupus nephritis, Dr. Furie cited the BLISS-LN trial, a 104-week, randomized trial of patients with active lupus nephritis. The group of patients who received belimumab (Benlysta), a monoclonal antibody that targets B-cell activating factor, in addition to standard therapy had significant improvements in renal responses, compared with standard therapy alone (43.0% vs. 32.3%). The outcome measure was Primary Efficacy Renal Response, defined as urinary protein/creatinine ratio <0.7, eGFR ≥60 mL/min per 1.73 m2, confirmation on consecutive visits, and required tapering of background glucocorticoids.
Although belimumab was approved for SLE in 2011, the BLISS-LN study focused on SLE patients with active kidney disease. “Neutralizing B-cell activating factor and down-regulating autoreactive B-cell function in kidneys represented a compelling therapeutic approach to lupus nephritis,” he explained.
Voclosporin, distinct from cyclosporine, has also been studied in lupus nephritis, Dr. Furie said. Voclosporin offers several benefits over cyclosporine, including greater potency and a lower drug and metabolite load, as well as a more consistent pharmacokinetic and pharmacodynamic relationship, he said. In the phase 3 AURORA study, presented at this year’s EULAR congress, 40% of patients with lupus nephritis met the primary endpoint of a renal response at 52 weeks, compared with 22.5% of placebo patients.
Looking ahead to the treatment of SLE in 2021, “I feel very strongly that voclosporin will be approved for lupus nephritis,” he said. He also predicted that the use of belimumab will be officially extended for lupus nephritis and that anifrolumab will receive an approval for SLE patients.
In addition, the future may witness the increased use of biomarkers and development of more individualized therapy. These breakthroughs will yield better outcomes for all lupus patients, he said.
Dr. Furie disclosed relationships with GlaxoSmithKline, Genentech/Roche, Aurinia Pharmaceuticals, AstraZeneca/MedImmune, and Eli Lilly. Global Academy for Medical Education and this news organization are owned by the same parent company.
Systemic lupus erythematosus remains a treatment challenge, but a variety of drugs in the pipeline are set to target type I interferons, cytokines, and B cells, according to Richard Furie, MD, chief of the division of rheumatology at Northwell Health and professor of medicine at Hofstra University, Hempstead, N.Y.
In general, when treating patients with systemic lupus erythematosus (SLE), “we just don’t see satisfactory response rates,” Dr. Furie said in an online presentation at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education.
“I think the greatest unmet need is in lupus nephritis,” he said. The data show that not even one-third of patients are adequately responding to standard of care treatment. “We need to do better in lupus nephritis but also for those patients with moderate-severe manifestations outside the kidney.”
Patients with SLE have elevated levels of interferon-alpha, Dr. Furie noted. Data from recent studies show that interferon inhibitors can reduce clinical activity in SLE patients, he said.
“About two-thirds to three-quarters of lupus patients have evidence of interferon pathway activation,” he said. There are three types of interferons, and five subtypes of type I interferon, and all five subtypes of type I interferon bind to the same receptor, which is an important strategy for drug development.
In particular, recent phase 2 and 3 trials have focused on targeting type I interferons with anifrolumab, which blocks all five subtypes.
Dr. Furie cited “very robust results” from a phase 2 study. Results of two phase 3 trials of anifrolumab led to a split decision, but the totality of the data collected across the phase 2 and 3 studies points to a drug that is effective for patients with SLE. The two phase 3 studies were published in Lancet Rheumatology and the New England Journal of Medicine.
Dr. Furie also identified recent studies of baricitinib (Olumiant), which has the ability to target several different cytokines. A phase 2 study in 2018 showed a significant difference in SLE Responder Index between lupus patients who received 4 mg of baricitinib or placebo, and a phase 3 study is underway, he said.
For lupus nephritis, Dr. Furie cited the BLISS-LN trial, a 104-week, randomized trial of patients with active lupus nephritis. The group of patients who received belimumab (Benlysta), a monoclonal antibody that targets B-cell activating factor, in addition to standard therapy had significant improvements in renal responses, compared with standard therapy alone (43.0% vs. 32.3%). The outcome measure was Primary Efficacy Renal Response, defined as urinary protein/creatinine ratio <0.7, eGFR ≥60 mL/min per 1.73 m2, confirmation on consecutive visits, and required tapering of background glucocorticoids.
Although belimumab was approved for SLE in 2011, the BLISS-LN study focused on SLE patients with active kidney disease. “Neutralizing B-cell activating factor and down-regulating autoreactive B-cell function in kidneys represented a compelling therapeutic approach to lupus nephritis,” he explained.
Voclosporin, distinct from cyclosporine, has also been studied in lupus nephritis, Dr. Furie said. Voclosporin offers several benefits over cyclosporine, including greater potency and a lower drug and metabolite load, as well as a more consistent pharmacokinetic and pharmacodynamic relationship, he said. In the phase 3 AURORA study, presented at this year’s EULAR congress, 40% of patients with lupus nephritis met the primary endpoint of a renal response at 52 weeks, compared with 22.5% of placebo patients.
Looking ahead to the treatment of SLE in 2021, “I feel very strongly that voclosporin will be approved for lupus nephritis,” he said. He also predicted that the use of belimumab will be officially extended for lupus nephritis and that anifrolumab will receive an approval for SLE patients.
In addition, the future may witness the increased use of biomarkers and development of more individualized therapy. These breakthroughs will yield better outcomes for all lupus patients, he said.
Dr. Furie disclosed relationships with GlaxoSmithKline, Genentech/Roche, Aurinia Pharmaceuticals, AstraZeneca/MedImmune, and Eli Lilly. Global Academy for Medical Education and this news organization are owned by the same parent company.
FROM PRD 2020
Management of Classic Ulcerative Pyoderma Gangrenosum
Pyoderma gangrenosum (PG) is a rare, chronic, ulcerative, neutrophilic dermatosis of unclear etiology. Large, multicentered, randomized controlled trials (RCTs) are challenging due to the rarity of PG and the lack of a diagnostic confirmatory test; therefore, evidence-based guidelines for diagnosis and treatment are not well established. Current management of PG primarily is guided by case series, small clinical trials, and expert opinion.1-4 We conducted a survey of expert medical dermatologists to highlight best practices in diagnostic and therapeutic approaches to PG.
Methods
The Society of Dermatology Hospitalists (SDH) Scientific Task Force gathered expert opinions from members of the SDH and Rheumatologic Dermatology Society (RDS) regarding PG workup and treatment through an online survey of 15 items (eTable 1). Subscribers of the SDH and RDS LISTSERVs were invited via email to participate in the survey from January 2016 to February 2016. Anonymous survey responses were collected and collated using SurveyMonkey. The survey results identified expert recommendations for evaluation, diagnosis, and treatment of PG and are reported as the sum of the percentage of respondents who answered always (almost 100% of the time) or often (more than half the time) following a particular course of action. A subanalysis was performed defining 2 groups of respondents based on the number of cases of PG treated per year (≥10 vs <10). Survey responses between each group were compared using χ2 analysis with statistical significance set at P=.05.

Results
Fifty-one respondents completed the survey out of 140 surveyed (36% response rate). All respondents were dermatologists, and 96% (49/51) were affiliated with an academic institution. Among the respondents, the number of PG cases managed per year ranged from 2 to 35.
Respondents consistently ordered skin biopsies (92% [47/51]) and tissue cultures (90% [46/51]), as well as certain ancillary tests, including complete blood cell count (96% [49/51]), complete metabolic panel (86% [44/51]), serum protein electrophoresis (76% [39/51]), and hepatitis panel (71% [36/51]). Other frequently ordered studies were rheumatoid factor (69% [35/51]), antinuclear antibodies (67% [34/51]), and antineutrophilic antibodies (65% [33/51]). Respondents frequently ordered erythrocyte sedimentation rate (59% [30/51]), C-reactive protein (55% [28/51]), cryoglobulins (53% [27/51]), urine protein electrophoresis (53% [27/51]), hypercoagulability workup (49% [25/51]), and serum immunofixation test (49% [25/51]). Human immunodeficiency virus testing (43% [22/51]), chest radiograph (41% [21/51]), colonoscopy (41% [21/51]) and referral to other specialties for workup—gastroenterology (38% [19/51]), hematology/oncology (14% [7/51]), and rheumatology (10% [5/51])—were less frequently ordered (eTable 2).
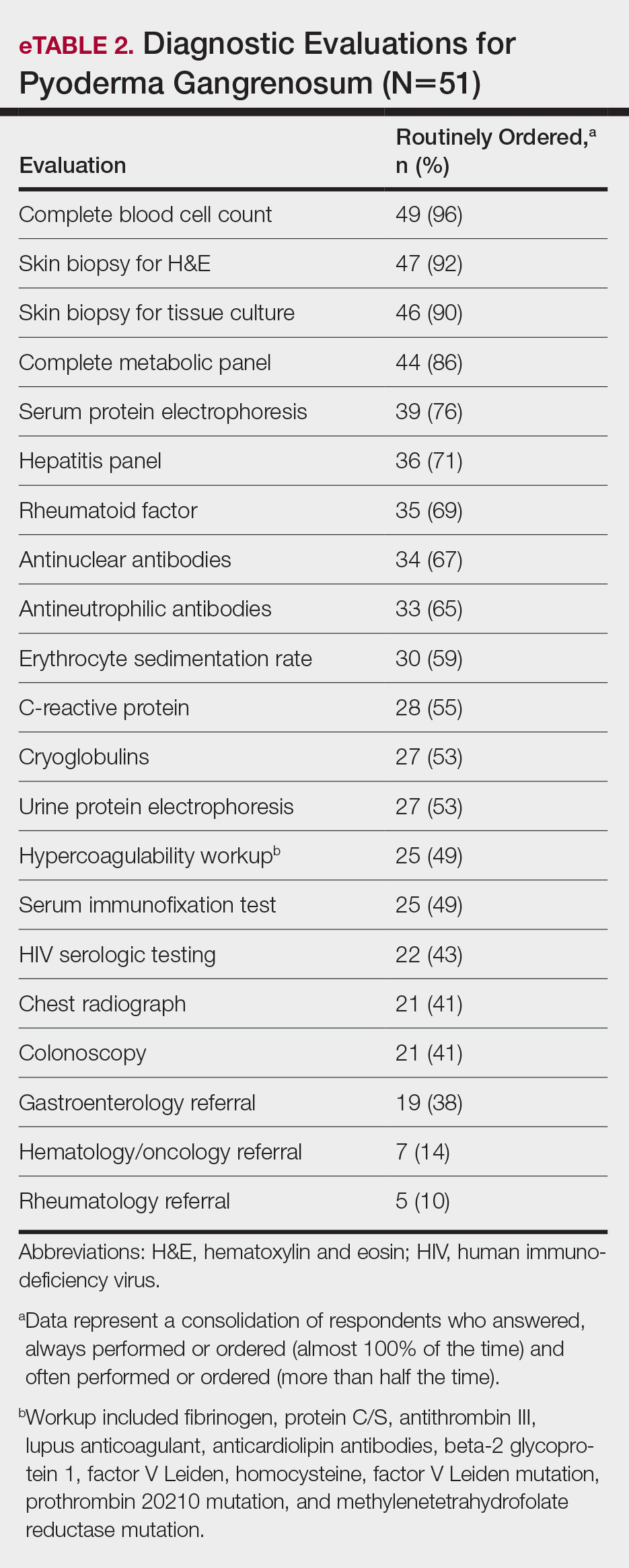
Systemic corticosteroids were reported as first-line therapy by most respondents (94% [48/51]), followed by topical immunomodulatory therapies (63% [32/51]). Topical corticosteroids (75% [38/51]) were the most common first-line topical agents. Thirty-nine percent of respondents (20/51) prescribed topical calcineurin inhibitors as first-line topical therapy. Additional therapies frequently used included systemic cyclosporine (47% [24/51]), antineutrophilic agents (41% [21/51]), and biologic agents (37% [19/51]). Fifty-seven percent of respondents (29/51) supported using combination topical and systemic therapy (Table).
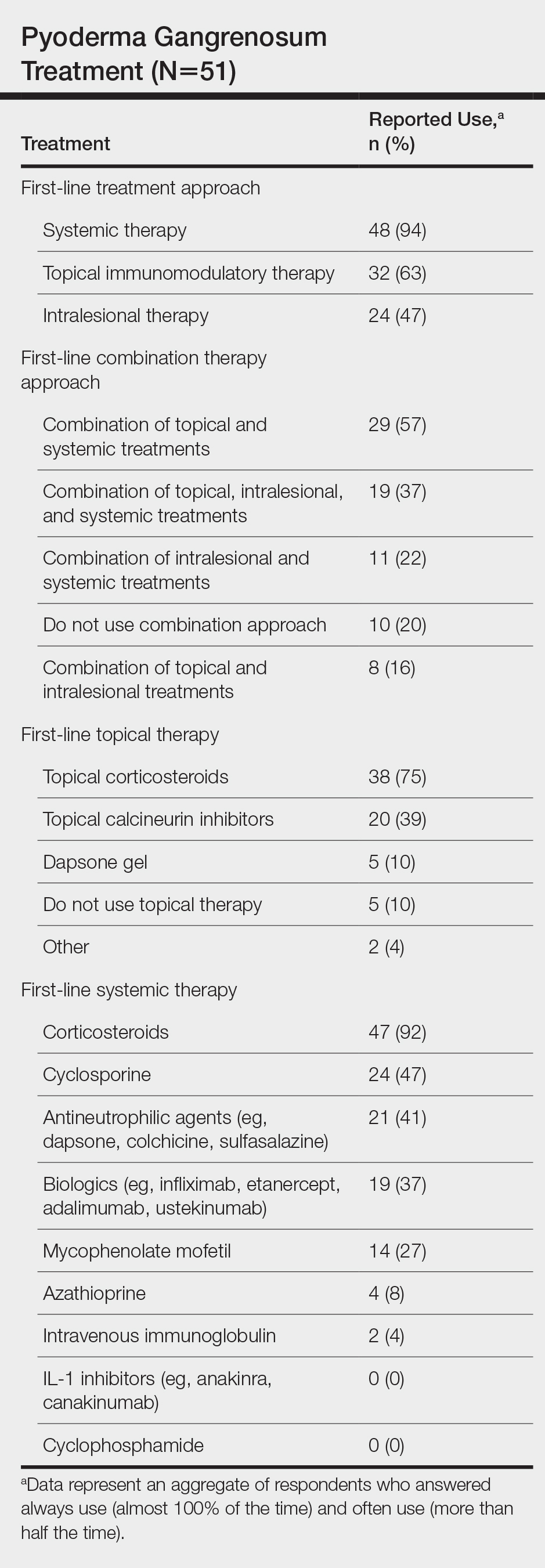
A wide variety of wound care practices were reported in the management of PG. Seventy-six percent of respondents (39/51) favored petroleum-impregnated gauze, 69% (35/51) used nonadhesive dressings, and 43% (22/51) added antimicrobial therapy for PG wound care (eTable 3). In the subanalysis, there were no significant differences in the majority of answer responses in patients treating 10 or more PG cases per year vs fewer than 10 PG cases, except with regard to the practice of combination therapy. Those treating more than 10 cases of PG per year more frequently reported use of combination therapies compared to respondents treating fewer than 10 cases (P=.04).
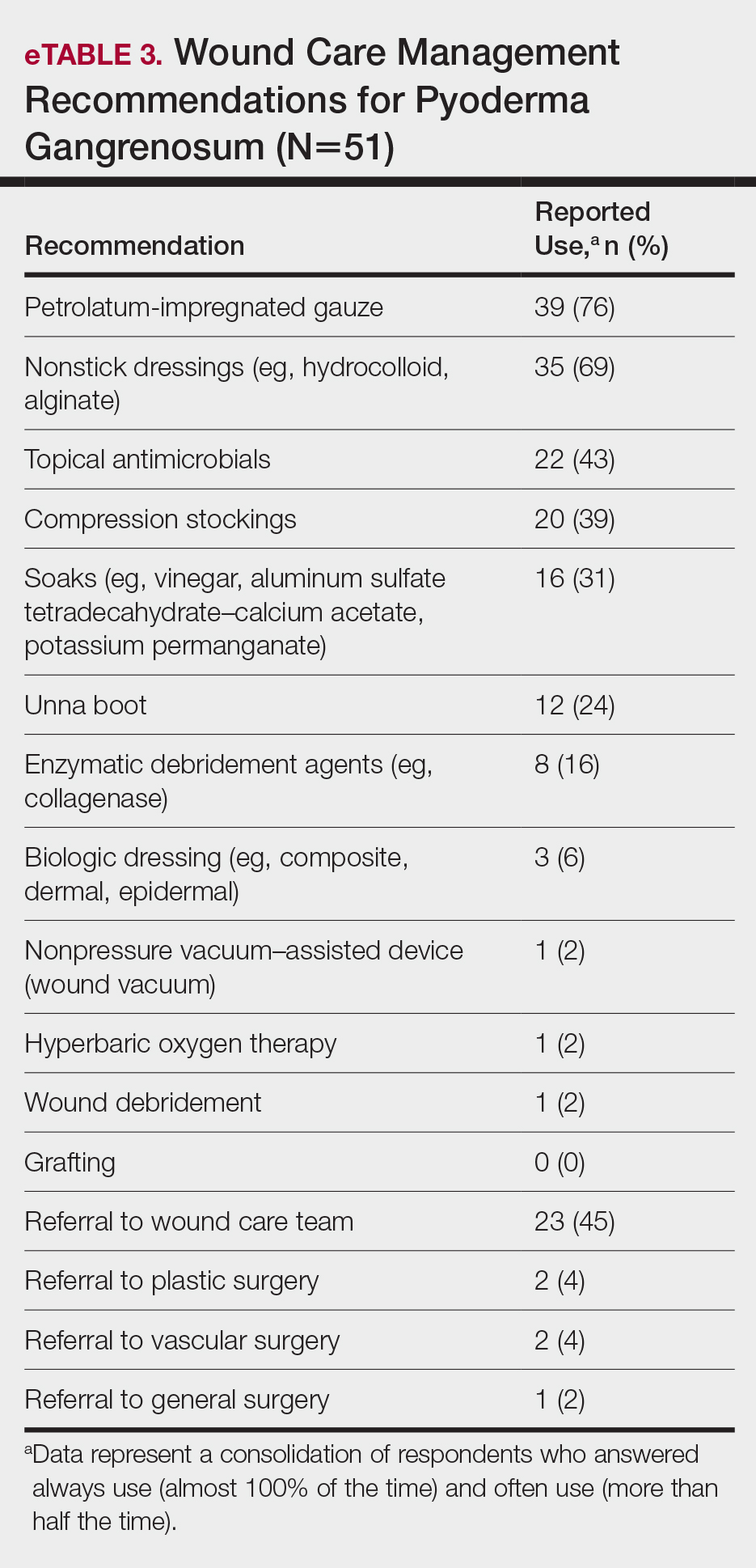
Comment
Skin biopsies and tissue cultures were strongly recommended (>90% survey respondents) for the initial evaluation of lesions suspected to be PG to evaluate for typical histopathologic changes that appear early in the disease, to rule out PG mimickers such as infectious or vascular causes, and to prevent the detrimental effects of inappropriate treatment and delayed diagnosis.5
Suspected PG warrants a reasonable search for related conditions because more than 50% of PG cases are associated with comorbidities such as rheumatoid arthritis, inflammatory bowel disease, and hematologic disease/malignancy.6,7 A complete blood cell count and comprehensive metabolic panel were recommended by most respondents, aiding in the preliminary screening for hematologic and infectious causes as well as detecting liver and kidney dysfunction associated with systemic conditions. Additionally, exclusion of infection or malignancy may be particularly important if the patient will undergo systemic immunosuppression. In challenging PG cases when initial findings are inconclusive and the clinical presentation does not direct workup (eg, colonoscopy to evaluate gastrointestinal tract symptoms), serum protein electrophoresis, hepatitis panel, rheumatoid factor, antinuclear antibodies, and antineutrophilic antibody tests also were frequently ordered by respondents to further evaluate for underlying or associated conditions.
This consensus regarding skin biopsies and certain ancillary tests is consistent with the proposed diagnostic criteria for classic ulcerative PG in which the absence or exclusion of other relevant causes of cutaneous ulcers is required based on the criteria.8 The importance of ensuring an accurate diagnosis is paramount, as a 10% misdiagnosis rate has been documented in the literature.5
Importantly, a stepwise diagnostic workup for PG is proposed based on survey results, which may limit unnecessary testing and the associated costs to the health care system (Figure 1). Selection of additional testing is guided by initial test results and features of the patient’s clinical presentation, including age, review of systems, and associated comorbidities. Available data suggest that underlying inflammatory bowel disease is more frequent in PG patients who are younger than 65 years, whereas those who are 65 years and older are more likely to have inflammatory arthritis, cancer, or an underlying hematologic disorder.9
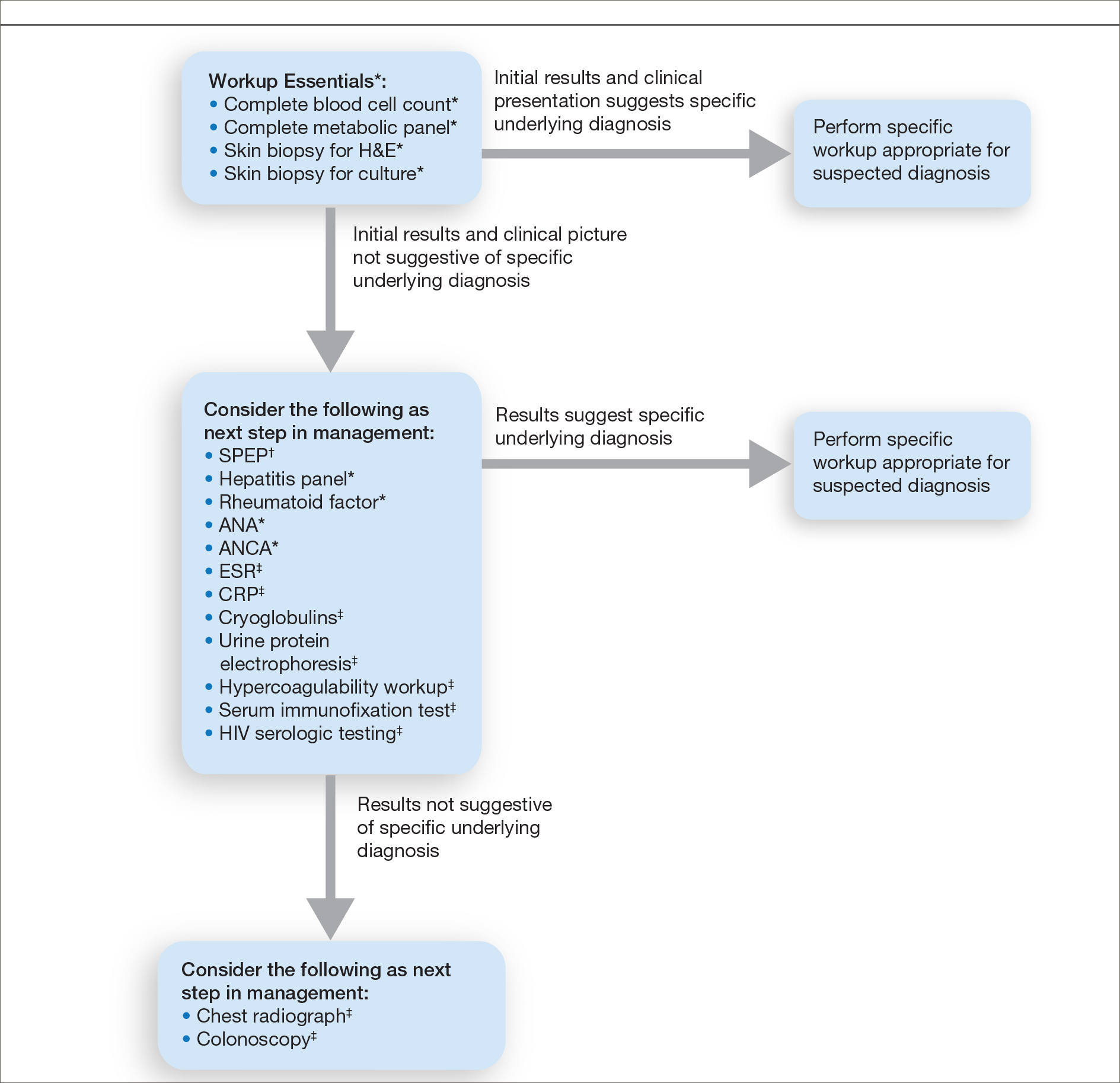
Treatment of PG should address both the inflammatory and wound components of the disease (Figure 2).7 In our survey results, systemic corticosteroids were identified as an important first-line therapy supported by reasonable evidence and were favored for their rapid response and minimal cost.1,10,11 Many respondents endorsed the use of systemic therapy in combination with topical steroids or calcineurin inhibitors. Combination therapy may provide more immediate control of rapidly progressing disease while minimizing adverse effects of long-term systemic corticosteroid use. A survey of German wound experts similarly endorsed frequent use of topical calcineurin inhibitors and combination systemic and topical glucocorticoid therapy as common therapeutic approaches.1
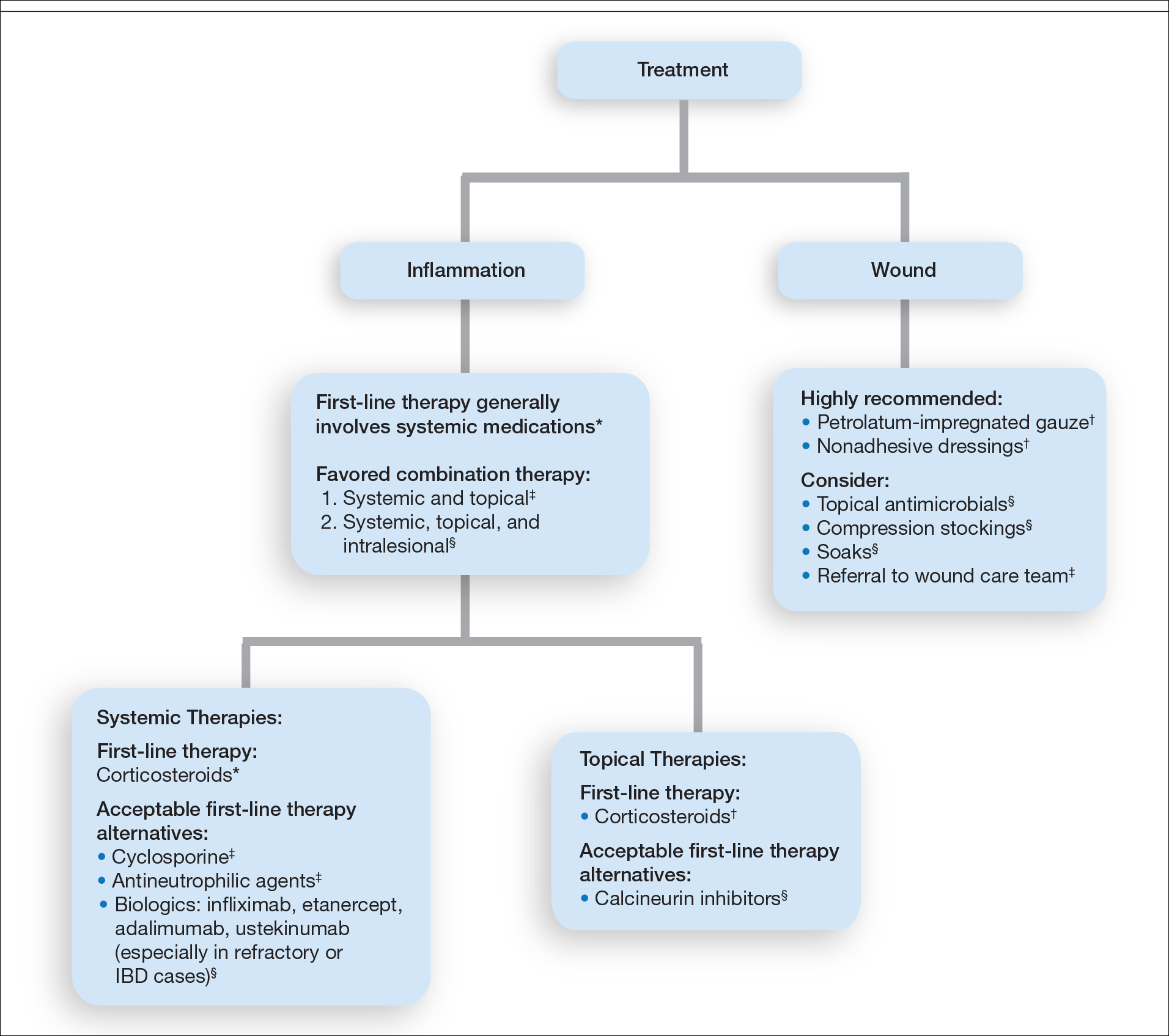
Importantly, treatments may vary depending on patient characteristics, comorbidities, and underlying disease, which underscores the need for individualized treatment approaches. Alternative first-line systemic treatments favored by respondents were cyclosporine, biologic medications, and antineutrophilic agents such as dapsone. Cyclosporine has demonstrated comparable efficacy to systemic glucocorticoids in one RCT and is considered an important steroid-sparing alternative for PG treatment.2 Biologic agents, especially tumor necrosis factor inhibitors, may be effective in treating cases of refractory PG or for concomitant inflammatory bowel disease management, as demonstrated by a small RCT documenting improvement of PG following infliximab infusion.3
Respondents strongly recommended petrolatum-impregnated gauze and other nonadhesive dressings, including alginate and hydrocolloid dressings, as part of PG wound care. Topical antimicrobials and compression stockings also were recommended by respondents. These practices aim to promote moist environments for healing, avoid maceration, prevent superinfection, optimize wound healing, and minimize damage from adhesive injury.12 Wound debridement and grafting generally were not recommended. However, pathergy is not a universal phenomenon in PG, and wounds that are no longer in the inflammatory phase may benefit from gentle debridement of necrotic tissue and/or grafting in select cases.10
Conclusion
An approach to modifying PG management based on clinical presentation and the practice of combination therapy with multiple systemic agents in refractory PG cases was not addressed in our survey. The low response rate is a limitation; however, the opinions of 51 medical dermatologist experts who regularly manage PG (in contrast to papers based on individualized clinical experience) can provide important clinical guidance until more scientific evidence is established.
Acknowledgments
We would like to thank the SDH and RDS membership for their participation in this survey. We especially acknowledge the other members of the SDH Scientific Task Force for their feedback: Misha Rosenbach, MD (Philadelphia, Pennsylvania); Robert G. Micheletti, MD (Philadelphia, Pennsylvania); Karolyn Wanat, MD (Milwaukee, Wisconsin); Amy Chen, MD (Cromwell, Connecticut); and A. Rambi Cardones, MD (Durham, North Carolina).
- Al Ghazal P, Dissemond J. Therapy of pyoderma gangrenosum in Germany: results of a survey among wound experts. J Dtsch Dermatol Ges . 2015;13:317-324.
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958.
- Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Al Ghazal P, Klode J, Dissemond J. Diagnostic criteria for pyoderma gangrenosum: results of a survey among dermatologic wound experts in Germany. J Dtsch Dermatol Ges. 2014;12:1129-1131.
- Weenig RH, Davis MD, Dahl PR, et al. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347:1412-1418.
- Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395-409.
- Bennett ML, Jackson JM, Jorizzo JL, et al. Pyoderma gangrenosum: a comparison of typical and atypical forms with an emphasis on time to remission. case review of 86 patients from 2 institutions. Medicine. 2000;79:37-46.
- Su WP, Davis MD, Weening RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Aschyan H, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Binus AM, Qureshi AA, Li VW, et al. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol. 2011;165:1244-1250.
- Reichrath J, Bens G, Bonowitz A, et al. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005;53:273-283.
- Miller J, Yentzer BA, Clark A, et al. Pyoderma gangrenosum: a review and update on new therapies. J Am Acad Dermatol. 2010;62:646-654.
Pyoderma gangrenosum (PG) is a rare, chronic, ulcerative, neutrophilic dermatosis of unclear etiology. Large, multicentered, randomized controlled trials (RCTs) are challenging due to the rarity of PG and the lack of a diagnostic confirmatory test; therefore, evidence-based guidelines for diagnosis and treatment are not well established. Current management of PG primarily is guided by case series, small clinical trials, and expert opinion.1-4 We conducted a survey of expert medical dermatologists to highlight best practices in diagnostic and therapeutic approaches to PG.
Methods
The Society of Dermatology Hospitalists (SDH) Scientific Task Force gathered expert opinions from members of the SDH and Rheumatologic Dermatology Society (RDS) regarding PG workup and treatment through an online survey of 15 items (eTable 1). Subscribers of the SDH and RDS LISTSERVs were invited via email to participate in the survey from January 2016 to February 2016. Anonymous survey responses were collected and collated using SurveyMonkey. The survey results identified expert recommendations for evaluation, diagnosis, and treatment of PG and are reported as the sum of the percentage of respondents who answered always (almost 100% of the time) or often (more than half the time) following a particular course of action. A subanalysis was performed defining 2 groups of respondents based on the number of cases of PG treated per year (≥10 vs <10). Survey responses between each group were compared using χ2 analysis with statistical significance set at P=.05.
Results
Fifty-one respondents completed the survey out of 140 surveyed (36% response rate). All respondents were dermatologists, and 96% (49/51) were affiliated with an academic institution. Among the respondents, the number of PG cases managed per year ranged from 2 to 35.
Respondents consistently ordered skin biopsies (92% [47/51]) and tissue cultures (90% [46/51]), as well as certain ancillary tests, including complete blood cell count (96% [49/51]), complete metabolic panel (86% [44/51]), serum protein electrophoresis (76% [39/51]), and hepatitis panel (71% [36/51]). Other frequently ordered studies were rheumatoid factor (69% [35/51]), antinuclear antibodies (67% [34/51]), and antineutrophilic antibodies (65% [33/51]). Respondents frequently ordered erythrocyte sedimentation rate (59% [30/51]), C-reactive protein (55% [28/51]), cryoglobulins (53% [27/51]), urine protein electrophoresis (53% [27/51]), hypercoagulability workup (49% [25/51]), and serum immunofixation test (49% [25/51]). Human immunodeficiency virus testing (43% [22/51]), chest radiograph (41% [21/51]), colonoscopy (41% [21/51]) and referral to other specialties for workup—gastroenterology (38% [19/51]), hematology/oncology (14% [7/51]), and rheumatology (10% [5/51])—were less frequently ordered (eTable 2).
Systemic corticosteroids were reported as first-line therapy by most respondents (94% [48/51]), followed by topical immunomodulatory therapies (63% [32/51]). Topical corticosteroids (75% [38/51]) were the most common first-line topical agents. Thirty-nine percent of respondents (20/51) prescribed topical calcineurin inhibitors as first-line topical therapy. Additional therapies frequently used included systemic cyclosporine (47% [24/51]), antineutrophilic agents (41% [21/51]), and biologic agents (37% [19/51]). Fifty-seven percent of respondents (29/51) supported using combination topical and systemic therapy (Table).
A wide variety of wound care practices were reported in the management of PG. Seventy-six percent of respondents (39/51) favored petroleum-impregnated gauze, 69% (35/51) used nonadhesive dressings, and 43% (22/51) added antimicrobial therapy for PG wound care (eTable 3). In the subanalysis, there were no significant differences in the majority of answer responses in patients treating 10 or more PG cases per year vs fewer than 10 PG cases, except with regard to the practice of combination therapy. Those treating more than 10 cases of PG per year more frequently reported use of combination therapies compared to respondents treating fewer than 10 cases (P=.04).
Comment
Skin biopsies and tissue cultures were strongly recommended (>90% survey respondents) for the initial evaluation of lesions suspected to be PG to evaluate for typical histopathologic changes that appear early in the disease, to rule out PG mimickers such as infectious or vascular causes, and to prevent the detrimental effects of inappropriate treatment and delayed diagnosis.5
Suspected PG warrants a reasonable search for related conditions because more than 50% of PG cases are associated with comorbidities such as rheumatoid arthritis, inflammatory bowel disease, and hematologic disease/malignancy.6,7 A complete blood cell count and comprehensive metabolic panel were recommended by most respondents, aiding in the preliminary screening for hematologic and infectious causes as well as detecting liver and kidney dysfunction associated with systemic conditions. Additionally, exclusion of infection or malignancy may be particularly important if the patient will undergo systemic immunosuppression. In challenging PG cases when initial findings are inconclusive and the clinical presentation does not direct workup (eg, colonoscopy to evaluate gastrointestinal tract symptoms), serum protein electrophoresis, hepatitis panel, rheumatoid factor, antinuclear antibodies, and antineutrophilic antibody tests also were frequently ordered by respondents to further evaluate for underlying or associated conditions.
This consensus regarding skin biopsies and certain ancillary tests is consistent with the proposed diagnostic criteria for classic ulcerative PG in which the absence or exclusion of other relevant causes of cutaneous ulcers is required based on the criteria.8 The importance of ensuring an accurate diagnosis is paramount, as a 10% misdiagnosis rate has been documented in the literature.5
Importantly, a stepwise diagnostic workup for PG is proposed based on survey results, which may limit unnecessary testing and the associated costs to the health care system (Figure 1). Selection of additional testing is guided by initial test results and features of the patient’s clinical presentation, including age, review of systems, and associated comorbidities. Available data suggest that underlying inflammatory bowel disease is more frequent in PG patients who are younger than 65 years, whereas those who are 65 years and older are more likely to have inflammatory arthritis, cancer, or an underlying hematologic disorder.9
Treatment of PG should address both the inflammatory and wound components of the disease (Figure 2).7 In our survey results, systemic corticosteroids were identified as an important first-line therapy supported by reasonable evidence and were favored for their rapid response and minimal cost.1,10,11 Many respondents endorsed the use of systemic therapy in combination with topical steroids or calcineurin inhibitors. Combination therapy may provide more immediate control of rapidly progressing disease while minimizing adverse effects of long-term systemic corticosteroid use. A survey of German wound experts similarly endorsed frequent use of topical calcineurin inhibitors and combination systemic and topical glucocorticoid therapy as common therapeutic approaches.1
Importantly, treatments may vary depending on patient characteristics, comorbidities, and underlying disease, which underscores the need for individualized treatment approaches. Alternative first-line systemic treatments favored by respondents were cyclosporine, biologic medications, and antineutrophilic agents such as dapsone. Cyclosporine has demonstrated comparable efficacy to systemic glucocorticoids in one RCT and is considered an important steroid-sparing alternative for PG treatment.2 Biologic agents, especially tumor necrosis factor inhibitors, may be effective in treating cases of refractory PG or for concomitant inflammatory bowel disease management, as demonstrated by a small RCT documenting improvement of PG following infliximab infusion.3
Respondents strongly recommended petrolatum-impregnated gauze and other nonadhesive dressings, including alginate and hydrocolloid dressings, as part of PG wound care. Topical antimicrobials and compression stockings also were recommended by respondents. These practices aim to promote moist environments for healing, avoid maceration, prevent superinfection, optimize wound healing, and minimize damage from adhesive injury.12 Wound debridement and grafting generally were not recommended. However, pathergy is not a universal phenomenon in PG, and wounds that are no longer in the inflammatory phase may benefit from gentle debridement of necrotic tissue and/or grafting in select cases.10
Conclusion
An approach to modifying PG management based on clinical presentation and the practice of combination therapy with multiple systemic agents in refractory PG cases was not addressed in our survey. The low response rate is a limitation; however, the opinions of 51 medical dermatologist experts who regularly manage PG (in contrast to papers based on individualized clinical experience) can provide important clinical guidance until more scientific evidence is established.
Acknowledgments
We would like to thank the SDH and RDS membership for their participation in this survey. We especially acknowledge the other members of the SDH Scientific Task Force for their feedback: Misha Rosenbach, MD (Philadelphia, Pennsylvania); Robert G. Micheletti, MD (Philadelphia, Pennsylvania); Karolyn Wanat, MD (Milwaukee, Wisconsin); Amy Chen, MD (Cromwell, Connecticut); and A. Rambi Cardones, MD (Durham, North Carolina).
Pyoderma gangrenosum (PG) is a rare, chronic, ulcerative, neutrophilic dermatosis of unclear etiology. Large, multicentered, randomized controlled trials (RCTs) are challenging due to the rarity of PG and the lack of a diagnostic confirmatory test; therefore, evidence-based guidelines for diagnosis and treatment are not well established. Current management of PG primarily is guided by case series, small clinical trials, and expert opinion.1-4 We conducted a survey of expert medical dermatologists to highlight best practices in diagnostic and therapeutic approaches to PG.
Methods
The Society of Dermatology Hospitalists (SDH) Scientific Task Force gathered expert opinions from members of the SDH and Rheumatologic Dermatology Society (RDS) regarding PG workup and treatment through an online survey of 15 items (eTable 1). Subscribers of the SDH and RDS LISTSERVs were invited via email to participate in the survey from January 2016 to February 2016. Anonymous survey responses were collected and collated using SurveyMonkey. The survey results identified expert recommendations for evaluation, diagnosis, and treatment of PG and are reported as the sum of the percentage of respondents who answered always (almost 100% of the time) or often (more than half the time) following a particular course of action. A subanalysis was performed defining 2 groups of respondents based on the number of cases of PG treated per year (≥10 vs <10). Survey responses between each group were compared using χ2 analysis with statistical significance set at P=.05.
Results
Fifty-one respondents completed the survey out of 140 surveyed (36% response rate). All respondents were dermatologists, and 96% (49/51) were affiliated with an academic institution. Among the respondents, the number of PG cases managed per year ranged from 2 to 35.
Respondents consistently ordered skin biopsies (92% [47/51]) and tissue cultures (90% [46/51]), as well as certain ancillary tests, including complete blood cell count (96% [49/51]), complete metabolic panel (86% [44/51]), serum protein electrophoresis (76% [39/51]), and hepatitis panel (71% [36/51]). Other frequently ordered studies were rheumatoid factor (69% [35/51]), antinuclear antibodies (67% [34/51]), and antineutrophilic antibodies (65% [33/51]). Respondents frequently ordered erythrocyte sedimentation rate (59% [30/51]), C-reactive protein (55% [28/51]), cryoglobulins (53% [27/51]), urine protein electrophoresis (53% [27/51]), hypercoagulability workup (49% [25/51]), and serum immunofixation test (49% [25/51]). Human immunodeficiency virus testing (43% [22/51]), chest radiograph (41% [21/51]), colonoscopy (41% [21/51]) and referral to other specialties for workup—gastroenterology (38% [19/51]), hematology/oncology (14% [7/51]), and rheumatology (10% [5/51])—were less frequently ordered (eTable 2).
Systemic corticosteroids were reported as first-line therapy by most respondents (94% [48/51]), followed by topical immunomodulatory therapies (63% [32/51]). Topical corticosteroids (75% [38/51]) were the most common first-line topical agents. Thirty-nine percent of respondents (20/51) prescribed topical calcineurin inhibitors as first-line topical therapy. Additional therapies frequently used included systemic cyclosporine (47% [24/51]), antineutrophilic agents (41% [21/51]), and biologic agents (37% [19/51]). Fifty-seven percent of respondents (29/51) supported using combination topical and systemic therapy (Table).
A wide variety of wound care practices were reported in the management of PG. Seventy-six percent of respondents (39/51) favored petroleum-impregnated gauze, 69% (35/51) used nonadhesive dressings, and 43% (22/51) added antimicrobial therapy for PG wound care (eTable 3). In the subanalysis, there were no significant differences in the majority of answer responses in patients treating 10 or more PG cases per year vs fewer than 10 PG cases, except with regard to the practice of combination therapy. Those treating more than 10 cases of PG per year more frequently reported use of combination therapies compared to respondents treating fewer than 10 cases (P=.04).
Comment
Skin biopsies and tissue cultures were strongly recommended (>90% survey respondents) for the initial evaluation of lesions suspected to be PG to evaluate for typical histopathologic changes that appear early in the disease, to rule out PG mimickers such as infectious or vascular causes, and to prevent the detrimental effects of inappropriate treatment and delayed diagnosis.5
Suspected PG warrants a reasonable search for related conditions because more than 50% of PG cases are associated with comorbidities such as rheumatoid arthritis, inflammatory bowel disease, and hematologic disease/malignancy.6,7 A complete blood cell count and comprehensive metabolic panel were recommended by most respondents, aiding in the preliminary screening for hematologic and infectious causes as well as detecting liver and kidney dysfunction associated with systemic conditions. Additionally, exclusion of infection or malignancy may be particularly important if the patient will undergo systemic immunosuppression. In challenging PG cases when initial findings are inconclusive and the clinical presentation does not direct workup (eg, colonoscopy to evaluate gastrointestinal tract symptoms), serum protein electrophoresis, hepatitis panel, rheumatoid factor, antinuclear antibodies, and antineutrophilic antibody tests also were frequently ordered by respondents to further evaluate for underlying or associated conditions.
This consensus regarding skin biopsies and certain ancillary tests is consistent with the proposed diagnostic criteria for classic ulcerative PG in which the absence or exclusion of other relevant causes of cutaneous ulcers is required based on the criteria.8 The importance of ensuring an accurate diagnosis is paramount, as a 10% misdiagnosis rate has been documented in the literature.5
Importantly, a stepwise diagnostic workup for PG is proposed based on survey results, which may limit unnecessary testing and the associated costs to the health care system (Figure 1). Selection of additional testing is guided by initial test results and features of the patient’s clinical presentation, including age, review of systems, and associated comorbidities. Available data suggest that underlying inflammatory bowel disease is more frequent in PG patients who are younger than 65 years, whereas those who are 65 years and older are more likely to have inflammatory arthritis, cancer, or an underlying hematologic disorder.9
Treatment of PG should address both the inflammatory and wound components of the disease (Figure 2).7 In our survey results, systemic corticosteroids were identified as an important first-line therapy supported by reasonable evidence and were favored for their rapid response and minimal cost.1,10,11 Many respondents endorsed the use of systemic therapy in combination with topical steroids or calcineurin inhibitors. Combination therapy may provide more immediate control of rapidly progressing disease while minimizing adverse effects of long-term systemic corticosteroid use. A survey of German wound experts similarly endorsed frequent use of topical calcineurin inhibitors and combination systemic and topical glucocorticoid therapy as common therapeutic approaches.1
Importantly, treatments may vary depending on patient characteristics, comorbidities, and underlying disease, which underscores the need for individualized treatment approaches. Alternative first-line systemic treatments favored by respondents were cyclosporine, biologic medications, and antineutrophilic agents such as dapsone. Cyclosporine has demonstrated comparable efficacy to systemic glucocorticoids in one RCT and is considered an important steroid-sparing alternative for PG treatment.2 Biologic agents, especially tumor necrosis factor inhibitors, may be effective in treating cases of refractory PG or for concomitant inflammatory bowel disease management, as demonstrated by a small RCT documenting improvement of PG following infliximab infusion.3
Respondents strongly recommended petrolatum-impregnated gauze and other nonadhesive dressings, including alginate and hydrocolloid dressings, as part of PG wound care. Topical antimicrobials and compression stockings also were recommended by respondents. These practices aim to promote moist environments for healing, avoid maceration, prevent superinfection, optimize wound healing, and minimize damage from adhesive injury.12 Wound debridement and grafting generally were not recommended. However, pathergy is not a universal phenomenon in PG, and wounds that are no longer in the inflammatory phase may benefit from gentle debridement of necrotic tissue and/or grafting in select cases.10
Conclusion
An approach to modifying PG management based on clinical presentation and the practice of combination therapy with multiple systemic agents in refractory PG cases was not addressed in our survey. The low response rate is a limitation; however, the opinions of 51 medical dermatologist experts who regularly manage PG (in contrast to papers based on individualized clinical experience) can provide important clinical guidance until more scientific evidence is established.
Acknowledgments
We would like to thank the SDH and RDS membership for their participation in this survey. We especially acknowledge the other members of the SDH Scientific Task Force for their feedback: Misha Rosenbach, MD (Philadelphia, Pennsylvania); Robert G. Micheletti, MD (Philadelphia, Pennsylvania); Karolyn Wanat, MD (Milwaukee, Wisconsin); Amy Chen, MD (Cromwell, Connecticut); and A. Rambi Cardones, MD (Durham, North Carolina).
- Al Ghazal P, Dissemond J. Therapy of pyoderma gangrenosum in Germany: results of a survey among wound experts. J Dtsch Dermatol Ges . 2015;13:317-324.
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958.
- Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Al Ghazal P, Klode J, Dissemond J. Diagnostic criteria for pyoderma gangrenosum: results of a survey among dermatologic wound experts in Germany. J Dtsch Dermatol Ges. 2014;12:1129-1131.
- Weenig RH, Davis MD, Dahl PR, et al. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347:1412-1418.
- Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395-409.
- Bennett ML, Jackson JM, Jorizzo JL, et al. Pyoderma gangrenosum: a comparison of typical and atypical forms with an emphasis on time to remission. case review of 86 patients from 2 institutions. Medicine. 2000;79:37-46.
- Su WP, Davis MD, Weening RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Aschyan H, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Binus AM, Qureshi AA, Li VW, et al. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol. 2011;165:1244-1250.
- Reichrath J, Bens G, Bonowitz A, et al. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005;53:273-283.
- Miller J, Yentzer BA, Clark A, et al. Pyoderma gangrenosum: a review and update on new therapies. J Am Acad Dermatol. 2010;62:646-654.
- Al Ghazal P, Dissemond J. Therapy of pyoderma gangrenosum in Germany: results of a survey among wound experts. J Dtsch Dermatol Ges . 2015;13:317-324.
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958.
- Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Al Ghazal P, Klode J, Dissemond J. Diagnostic criteria for pyoderma gangrenosum: results of a survey among dermatologic wound experts in Germany. J Dtsch Dermatol Ges. 2014;12:1129-1131.
- Weenig RH, Davis MD, Dahl PR, et al. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347:1412-1418.
- Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395-409.
- Bennett ML, Jackson JM, Jorizzo JL, et al. Pyoderma gangrenosum: a comparison of typical and atypical forms with an emphasis on time to remission. case review of 86 patients from 2 institutions. Medicine. 2000;79:37-46.
- Su WP, Davis MD, Weening RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Aschyan H, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Binus AM, Qureshi AA, Li VW, et al. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol. 2011;165:1244-1250.
- Reichrath J, Bens G, Bonowitz A, et al. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005;53:273-283.
- Miller J, Yentzer BA, Clark A, et al. Pyoderma gangrenosum: a review and update on new therapies. J Am Acad Dermatol. 2010;62:646-654.
Practice Points
- The diagnosis of pyoderma gangrenosum (PG) poses a challenge in clinical practice that could be minimized by following a stepwise algorithm based on initial test results (including skin biopsies) and features of the patient’s clinical presentation.
- As there is no US Food and Drug Administration–approved treatment for PG, a stepwise algorithm approach in combination with the clinical experience addressing inflammation and wound care is essential to reach control and remission of PG.