User login
Pedunculated Tumor on the Posterior Neck
The Diagnosis: Nodular Hidradenoma
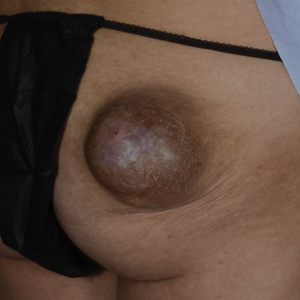
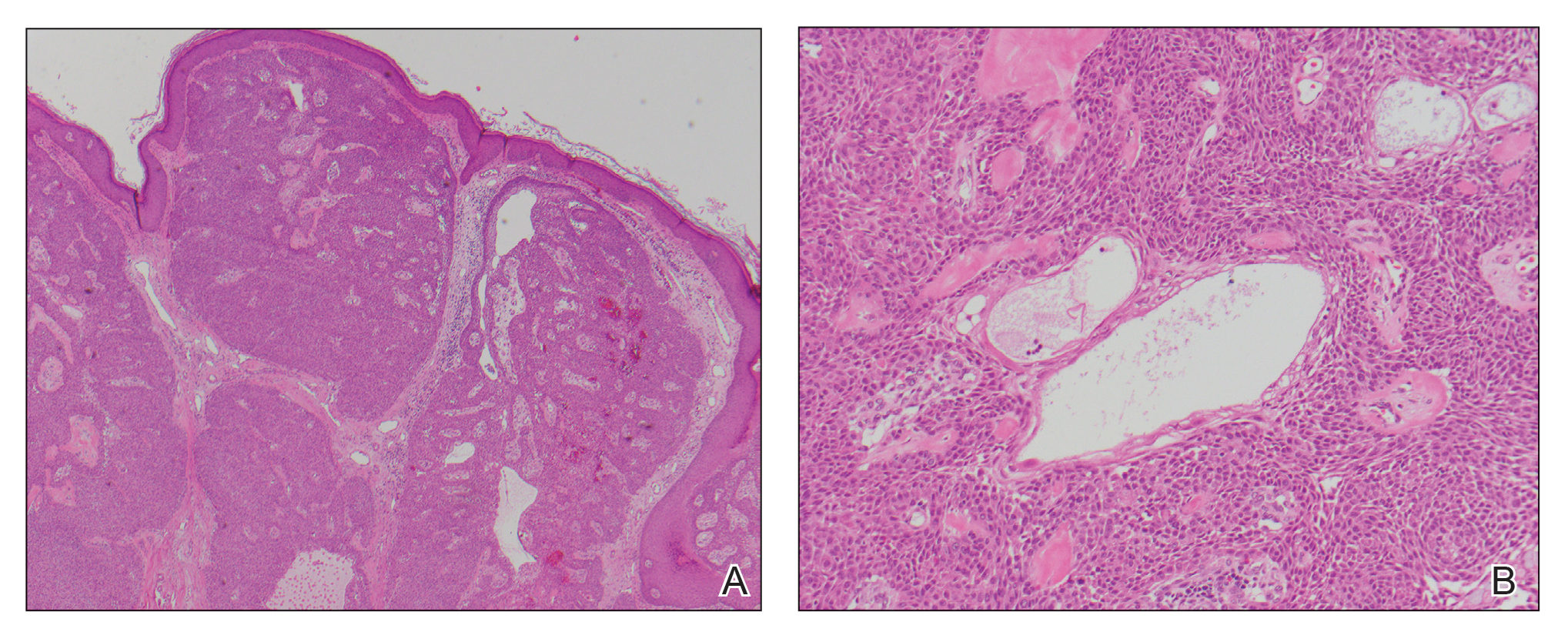
A biopsy of the nodule showed a large, fungating, well-circumscribed, multilobulated neoplasm composed of primarily monotonous eosinophilic cells in a background of keloidal stroma (Figure). There was a minority population of small, monotonous, clear cells within the lobules, and no glandular structures were noted. Neither cytological nor architectural atypia were evident. MART-1/Melan-A and S-100 stains were negative, consistent with a diagnosis of benign nodular hidradenoma.
Nodular hidradenoma (also known as acrospiroma, solid-cystic hidradenoma, clear cell hidradenoma, and eccrine sweat gland adenoma) is a benign adnexal tumor of the apocrine or eccrine glands.1,2 Nodular hidradenoma can arise at any cutaneous site but most commonly arises on the head and anterior portion of the trunk and rarely on the extremities.2 It presents as a solitary nodular, cystic, or pedunculated mass that can reach up to several centimeters in diameter.2,3 Nodular hidradenoma more commonly affects women compared to men with a ratio of 1.7 to 1 and commonly presents between the third and fifth decades of life, with an average age at presentation of 37.2 years.2,4 There can be associated skin changes, including smoothening, thickening, ulceration, and bluish discoloration. Dermoscopy commonly shows a pinkish homogenous area that extends throughout the entire lesion. This homogenous area less commonly can be bluish, brownish, or pink-blue. Most nodular hidradenomas also can exhibit vascularization, with arborizing telangiectases, polymorphous atypical vessels, and linear irregular vessels being most common; however, this is not specific to nodular hidradenoma.3 Occasionally, tumors can drain serous or hemorrhagic fluid. Nodular hidradenoma commonly is a slow-growing tumor.5 Rapid increase in tumor size can be indicative of malignant transformation, hemorrhage into the tumor, or trauma to the area.2
Histologically, nodular hidradenoma consists of a circumscribed, nonencapsulated, multilobular tumor commonly found in the dermis and sometimes extending into the subcutaneous tissue. There usually is no epidermal attachment, and the overlying epidermis largely is normal. The tumor consists of large multilobulated areas of epithelial cells, tubular lamina, and large cystic areas filled with homogenous eosinophilic material.1 It notably is composed of 2 epithelial cell types: (1) fusiform cells with elongated vesicular nuclei and basophilic cytoplasm, and (2) large polygonal cells with round eccentric nuclei and eosinophilic, periodic acid–Schiff–positive cytoplasm that washes away during fixation, giving the appearance of clear cells.5 Both types of cells are small, monotonous, and void of mitosis or dyskeratosis. Although there can be ducts with apocrine secretion present within the lobulated tumor, they are not consistently found. Due to the varying features that are neither mandatory nor consistent to arrive at this diagnosis, some dermatopathologists view the term hidradenoma as a catch-all term that includes several different types of benign sweat gland tumors. Some authors divide the terminology into apocrine hidradenoma and eccrine hidradenoma based on whether the tumor is composed of solely clear mucinous cells, or poroid and cuticular cells, respectively.
Although nodular hidradenoma classically is a benign tumor, total surgical excision is recommended due to the rare risk for malignant transformation. Rarely, longstanding hidradenomas can metastasize to lymph nodes, bone, or viscera; in these instances, metastatic hidradenoma has a 5-year survival rate of 30%. Recurrence may occur in tumors that are inadequately excised, and the rate of recurrence is estimated to be approximately 10% of surgically excised tumors.5 However, utilization of Mohs micrographic surgery for excision of nodular hidradenoma is associated with a reduced recurrence rate.6
Keloids present as painful, sometimes pruritic, raised scars that extend beyond the boundary of the initial injury, commonly arising on the shoulder, upper arm, and chest. Histopathology reveals nodules of thick hyalinized collagen bundles, keloidal collagen with mucinous ground substance, and few fibroblasts.7
Metastatic renal cell carcinoma to the skin most commonly presents on the face and scalp as a nodular, rapidly growing, round to oval lesion that is flesh colored to reddish purple in a patient with history of renal cell carcinoma.8 Histopathology shows clusters of atypical, nucleated clear cells surrounded by chicken wire vasculature.8,9
Verruca vulgaris is caused by human papillomavirus and most commonly occurs on the hands and feet. It presents as a pink to white, sessile lesion with a verrucous surface and exophytic growths. Histopathology shows acanthosis; hypergranulosis; exophytic projections with a fibrovascular core; inward cupping of the rete ridges; and koilocytes, which are cells with an eccentric, raisinlike nucleus and vacuolated cytoplasm in the granular layer of the epidermis.10
Similar to nodular hidradenoma, nodular melanoma most commonly presents on the head and neck as a symmetric, elevated, amelanotic nodule, but in contrast to nodular hidradenoma, it typically is confined to a smaller diameter.11 Histologically, it is characterized by sheets of atypical, commonly epithelioid melanocytes with a lack of maturation and brisk mitotic activity extending through the epidermis and dermis with lateral extension limited to less than 3 rete ridges.12
- Patterson JW, Weedon D. Tumors of cutaneous appendages. In: Patterson JW, Weedon D. Weedon’s Skin Pathology. 5th ed. Elsevier; 2020:951-1016.
- Ngo N, Susa M, Nakagawa T, et al. Malignant transformation of nodular hidradenoma in the lower leg. Case Rep Oncol. 2018;11:298-304. doi:10.1159/000489255
- Zaballos P, Gómez-Martín I, Martin JM, et al. Dermoscopy of adnexal tumors. Dermatol Clin. 2018;36:397-412. doi:10.1016/j .det.2018.05.007
- Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985; 12:15-20. doi:10.1016/s0190-9622(85)70002-3
- Stratigos AJ, Olbricht S, Kwan TH, et al. Nodular hidradenoma. Dermatol Surg. 1998;24:387-391. doi:10.1111/j.1524-4725.1998.tb04173.x
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach. Dermatol Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Lee JY-Y, Yang C-C, Chao S-C, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26:379-384. doi:10.1097/00000372-200410000-00006
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Jaitly V, Jahan-Tigh R, Belousova T, et al. Case report and literature review of nodular hidradenoma, a rare adnexal tumor that mimics breast carcinoma, in a 20-year-old woman. Lab Med. 2019;50:320-325. doi:10.1093/labmed/lmy084
- Betz SJ. HPV-related papillary lesions of the oral mucosa: a review. Head Neck Pathol. 2019;13:80-90. doi:10.1007/s12105-019-01003-7
- Kalkhoran S, Milne O, Zalaudek I, et al. Historical, clinical, and dermoscopic characteristics of thin nodular melanoma. Arch Dermatol. 2010;146:311-318. doi:10.1001/archdermatol.2009.369
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40. doi:10.1038 /modpathol.3800508
The Diagnosis: Nodular Hidradenoma
A biopsy of the nodule showed a large, fungating, well-circumscribed, multilobulated neoplasm composed of primarily monotonous eosinophilic cells in a background of keloidal stroma (Figure). There was a minority population of small, monotonous, clear cells within the lobules, and no glandular structures were noted. Neither cytological nor architectural atypia were evident. MART-1/Melan-A and S-100 stains were negative, consistent with a diagnosis of benign nodular hidradenoma.
Nodular hidradenoma (also known as acrospiroma, solid-cystic hidradenoma, clear cell hidradenoma, and eccrine sweat gland adenoma) is a benign adnexal tumor of the apocrine or eccrine glands.1,2 Nodular hidradenoma can arise at any cutaneous site but most commonly arises on the head and anterior portion of the trunk and rarely on the extremities.2 It presents as a solitary nodular, cystic, or pedunculated mass that can reach up to several centimeters in diameter.2,3 Nodular hidradenoma more commonly affects women compared to men with a ratio of 1.7 to 1 and commonly presents between the third and fifth decades of life, with an average age at presentation of 37.2 years.2,4 There can be associated skin changes, including smoothening, thickening, ulceration, and bluish discoloration. Dermoscopy commonly shows a pinkish homogenous area that extends throughout the entire lesion. This homogenous area less commonly can be bluish, brownish, or pink-blue. Most nodular hidradenomas also can exhibit vascularization, with arborizing telangiectases, polymorphous atypical vessels, and linear irregular vessels being most common; however, this is not specific to nodular hidradenoma.3 Occasionally, tumors can drain serous or hemorrhagic fluid. Nodular hidradenoma commonly is a slow-growing tumor.5 Rapid increase in tumor size can be indicative of malignant transformation, hemorrhage into the tumor, or trauma to the area.2
Histologically, nodular hidradenoma consists of a circumscribed, nonencapsulated, multilobular tumor commonly found in the dermis and sometimes extending into the subcutaneous tissue. There usually is no epidermal attachment, and the overlying epidermis largely is normal. The tumor consists of large multilobulated areas of epithelial cells, tubular lamina, and large cystic areas filled with homogenous eosinophilic material.1 It notably is composed of 2 epithelial cell types: (1) fusiform cells with elongated vesicular nuclei and basophilic cytoplasm, and (2) large polygonal cells with round eccentric nuclei and eosinophilic, periodic acid–Schiff–positive cytoplasm that washes away during fixation, giving the appearance of clear cells.5 Both types of cells are small, monotonous, and void of mitosis or dyskeratosis. Although there can be ducts with apocrine secretion present within the lobulated tumor, they are not consistently found. Due to the varying features that are neither mandatory nor consistent to arrive at this diagnosis, some dermatopathologists view the term hidradenoma as a catch-all term that includes several different types of benign sweat gland tumors. Some authors divide the terminology into apocrine hidradenoma and eccrine hidradenoma based on whether the tumor is composed of solely clear mucinous cells, or poroid and cuticular cells, respectively.
Although nodular hidradenoma classically is a benign tumor, total surgical excision is recommended due to the rare risk for malignant transformation. Rarely, longstanding hidradenomas can metastasize to lymph nodes, bone, or viscera; in these instances, metastatic hidradenoma has a 5-year survival rate of 30%. Recurrence may occur in tumors that are inadequately excised, and the rate of recurrence is estimated to be approximately 10% of surgically excised tumors.5 However, utilization of Mohs micrographic surgery for excision of nodular hidradenoma is associated with a reduced recurrence rate.6
Keloids present as painful, sometimes pruritic, raised scars that extend beyond the boundary of the initial injury, commonly arising on the shoulder, upper arm, and chest. Histopathology reveals nodules of thick hyalinized collagen bundles, keloidal collagen with mucinous ground substance, and few fibroblasts.7
Metastatic renal cell carcinoma to the skin most commonly presents on the face and scalp as a nodular, rapidly growing, round to oval lesion that is flesh colored to reddish purple in a patient with history of renal cell carcinoma.8 Histopathology shows clusters of atypical, nucleated clear cells surrounded by chicken wire vasculature.8,9
Verruca vulgaris is caused by human papillomavirus and most commonly occurs on the hands and feet. It presents as a pink to white, sessile lesion with a verrucous surface and exophytic growths. Histopathology shows acanthosis; hypergranulosis; exophytic projections with a fibrovascular core; inward cupping of the rete ridges; and koilocytes, which are cells with an eccentric, raisinlike nucleus and vacuolated cytoplasm in the granular layer of the epidermis.10
Similar to nodular hidradenoma, nodular melanoma most commonly presents on the head and neck as a symmetric, elevated, amelanotic nodule, but in contrast to nodular hidradenoma, it typically is confined to a smaller diameter.11 Histologically, it is characterized by sheets of atypical, commonly epithelioid melanocytes with a lack of maturation and brisk mitotic activity extending through the epidermis and dermis with lateral extension limited to less than 3 rete ridges.12
The Diagnosis: Nodular Hidradenoma
A biopsy of the nodule showed a large, fungating, well-circumscribed, multilobulated neoplasm composed of primarily monotonous eosinophilic cells in a background of keloidal stroma (Figure). There was a minority population of small, monotonous, clear cells within the lobules, and no glandular structures were noted. Neither cytological nor architectural atypia were evident. MART-1/Melan-A and S-100 stains were negative, consistent with a diagnosis of benign nodular hidradenoma.
Nodular hidradenoma (also known as acrospiroma, solid-cystic hidradenoma, clear cell hidradenoma, and eccrine sweat gland adenoma) is a benign adnexal tumor of the apocrine or eccrine glands.1,2 Nodular hidradenoma can arise at any cutaneous site but most commonly arises on the head and anterior portion of the trunk and rarely on the extremities.2 It presents as a solitary nodular, cystic, or pedunculated mass that can reach up to several centimeters in diameter.2,3 Nodular hidradenoma more commonly affects women compared to men with a ratio of 1.7 to 1 and commonly presents between the third and fifth decades of life, with an average age at presentation of 37.2 years.2,4 There can be associated skin changes, including smoothening, thickening, ulceration, and bluish discoloration. Dermoscopy commonly shows a pinkish homogenous area that extends throughout the entire lesion. This homogenous area less commonly can be bluish, brownish, or pink-blue. Most nodular hidradenomas also can exhibit vascularization, with arborizing telangiectases, polymorphous atypical vessels, and linear irregular vessels being most common; however, this is not specific to nodular hidradenoma.3 Occasionally, tumors can drain serous or hemorrhagic fluid. Nodular hidradenoma commonly is a slow-growing tumor.5 Rapid increase in tumor size can be indicative of malignant transformation, hemorrhage into the tumor, or trauma to the area.2
Histologically, nodular hidradenoma consists of a circumscribed, nonencapsulated, multilobular tumor commonly found in the dermis and sometimes extending into the subcutaneous tissue. There usually is no epidermal attachment, and the overlying epidermis largely is normal. The tumor consists of large multilobulated areas of epithelial cells, tubular lamina, and large cystic areas filled with homogenous eosinophilic material.1 It notably is composed of 2 epithelial cell types: (1) fusiform cells with elongated vesicular nuclei and basophilic cytoplasm, and (2) large polygonal cells with round eccentric nuclei and eosinophilic, periodic acid–Schiff–positive cytoplasm that washes away during fixation, giving the appearance of clear cells.5 Both types of cells are small, monotonous, and void of mitosis or dyskeratosis. Although there can be ducts with apocrine secretion present within the lobulated tumor, they are not consistently found. Due to the varying features that are neither mandatory nor consistent to arrive at this diagnosis, some dermatopathologists view the term hidradenoma as a catch-all term that includes several different types of benign sweat gland tumors. Some authors divide the terminology into apocrine hidradenoma and eccrine hidradenoma based on whether the tumor is composed of solely clear mucinous cells, or poroid and cuticular cells, respectively.
Although nodular hidradenoma classically is a benign tumor, total surgical excision is recommended due to the rare risk for malignant transformation. Rarely, longstanding hidradenomas can metastasize to lymph nodes, bone, or viscera; in these instances, metastatic hidradenoma has a 5-year survival rate of 30%. Recurrence may occur in tumors that are inadequately excised, and the rate of recurrence is estimated to be approximately 10% of surgically excised tumors.5 However, utilization of Mohs micrographic surgery for excision of nodular hidradenoma is associated with a reduced recurrence rate.6
Keloids present as painful, sometimes pruritic, raised scars that extend beyond the boundary of the initial injury, commonly arising on the shoulder, upper arm, and chest. Histopathology reveals nodules of thick hyalinized collagen bundles, keloidal collagen with mucinous ground substance, and few fibroblasts.7
Metastatic renal cell carcinoma to the skin most commonly presents on the face and scalp as a nodular, rapidly growing, round to oval lesion that is flesh colored to reddish purple in a patient with history of renal cell carcinoma.8 Histopathology shows clusters of atypical, nucleated clear cells surrounded by chicken wire vasculature.8,9
Verruca vulgaris is caused by human papillomavirus and most commonly occurs on the hands and feet. It presents as a pink to white, sessile lesion with a verrucous surface and exophytic growths. Histopathology shows acanthosis; hypergranulosis; exophytic projections with a fibrovascular core; inward cupping of the rete ridges; and koilocytes, which are cells with an eccentric, raisinlike nucleus and vacuolated cytoplasm in the granular layer of the epidermis.10
Similar to nodular hidradenoma, nodular melanoma most commonly presents on the head and neck as a symmetric, elevated, amelanotic nodule, but in contrast to nodular hidradenoma, it typically is confined to a smaller diameter.11 Histologically, it is characterized by sheets of atypical, commonly epithelioid melanocytes with a lack of maturation and brisk mitotic activity extending through the epidermis and dermis with lateral extension limited to less than 3 rete ridges.12
- Patterson JW, Weedon D. Tumors of cutaneous appendages. In: Patterson JW, Weedon D. Weedon’s Skin Pathology. 5th ed. Elsevier; 2020:951-1016.
- Ngo N, Susa M, Nakagawa T, et al. Malignant transformation of nodular hidradenoma in the lower leg. Case Rep Oncol. 2018;11:298-304. doi:10.1159/000489255
- Zaballos P, Gómez-Martín I, Martin JM, et al. Dermoscopy of adnexal tumors. Dermatol Clin. 2018;36:397-412. doi:10.1016/j .det.2018.05.007
- Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985; 12:15-20. doi:10.1016/s0190-9622(85)70002-3
- Stratigos AJ, Olbricht S, Kwan TH, et al. Nodular hidradenoma. Dermatol Surg. 1998;24:387-391. doi:10.1111/j.1524-4725.1998.tb04173.x
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach. Dermatol Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Lee JY-Y, Yang C-C, Chao S-C, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26:379-384. doi:10.1097/00000372-200410000-00006
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Jaitly V, Jahan-Tigh R, Belousova T, et al. Case report and literature review of nodular hidradenoma, a rare adnexal tumor that mimics breast carcinoma, in a 20-year-old woman. Lab Med. 2019;50:320-325. doi:10.1093/labmed/lmy084
- Betz SJ. HPV-related papillary lesions of the oral mucosa: a review. Head Neck Pathol. 2019;13:80-90. doi:10.1007/s12105-019-01003-7
- Kalkhoran S, Milne O, Zalaudek I, et al. Historical, clinical, and dermoscopic characteristics of thin nodular melanoma. Arch Dermatol. 2010;146:311-318. doi:10.1001/archdermatol.2009.369
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40. doi:10.1038 /modpathol.3800508
- Patterson JW, Weedon D. Tumors of cutaneous appendages. In: Patterson JW, Weedon D. Weedon’s Skin Pathology. 5th ed. Elsevier; 2020:951-1016.
- Ngo N, Susa M, Nakagawa T, et al. Malignant transformation of nodular hidradenoma in the lower leg. Case Rep Oncol. 2018;11:298-304. doi:10.1159/000489255
- Zaballos P, Gómez-Martín I, Martin JM, et al. Dermoscopy of adnexal tumors. Dermatol Clin. 2018;36:397-412. doi:10.1016/j .det.2018.05.007
- Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985; 12:15-20. doi:10.1016/s0190-9622(85)70002-3
- Stratigos AJ, Olbricht S, Kwan TH, et al. Nodular hidradenoma. Dermatol Surg. 1998;24:387-391. doi:10.1111/j.1524-4725.1998.tb04173.x
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach. Dermatol Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Lee JY-Y, Yang C-C, Chao S-C, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26:379-384. doi:10.1097/00000372-200410000-00006
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Jaitly V, Jahan-Tigh R, Belousova T, et al. Case report and literature review of nodular hidradenoma, a rare adnexal tumor that mimics breast carcinoma, in a 20-year-old woman. Lab Med. 2019;50:320-325. doi:10.1093/labmed/lmy084
- Betz SJ. HPV-related papillary lesions of the oral mucosa: a review. Head Neck Pathol. 2019;13:80-90. doi:10.1007/s12105-019-01003-7
- Kalkhoran S, Milne O, Zalaudek I, et al. Historical, clinical, and dermoscopic characteristics of thin nodular melanoma. Arch Dermatol. 2010;146:311-318. doi:10.1001/archdermatol.2009.369
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40. doi:10.1038 /modpathol.3800508

A 56-year-old man presented with a progressively enlarging lesion on the posterior neck of 8 months’ duration. He reported localized pruritus of the lesion that improved with triamcinolone cream 0.05% and oral hydroxyzine as well as occasional irritation of the mass with oozing of clear fluid and blood. He denied associated pain and constitutional symptoms. Physical examination revealed a 2.5-cm, nodular, pedunculated, rubbery mass with foci of crusting on the central posterior neck. The mass was flesh colored to pink, and no lymphadenopathy was noted on physical examination.
Velvety Plaques on the Abdomen and Extremities
The Diagnosis: Dermatitis Neglecta
A punch biopsy of the abdomen revealed hyperkeratosis and mild papillomatosis (Figure), which can be seen in dermatitis neglecta (DN) and acanthosis nigricans (AN) as well as confluent and reticulated papillomatosis (CARP). Due to the patient’s history of mood and psychotic disorders, collateral information was obtained from the patient’s family, who reported that the patient had a depressed mood in the last few months and was not showering or caring for herself during this period. There was no additional personal or family history of skin disease. Clinical and histopathologic findings led to a diagnosis of DN. Following recommendations for daily cleansing with soap and water along with topical ammonium lactate, near-complete resolution of the rash was achieved in 3 weeks.
Dermatitis neglecta, or unwashed dermatosis, is a skin condition that occurs secondary to poor hygiene, which was first reported in 1995 by Poskitt et al.1 Avoidance of washing in affected areas can be due to physical disability, pain after injury, neurological deficit, or psychologically induced fear or neglect. Sebum, sweat, corneocytes, and bacteria combine into compact adherent crusts of dirt, which appear as hyperkeratotic plaques with cornflakelike scale.2,3 Despite its innate simplicity, DN is a diagnostic challenge, as it clinically and histologically mimics other dermatoses including AN, terra firmaforme dermatosis, and CARP.2,4 Ultimately, the diagnosis of DN can be made when a history of poor hygiene is probable or elicited, and lesions can be removed with soap and water. Treatment of DN includes daily cleansing with soap and water; however, resistant lesions or extensive disease may require keratolytic agents, as in our patient.2-4 In contrast, terra firma-forme dermatosis, which may look similar, is not due to poor hygiene, and the lesions typically are resistant to soap and water, classically requiring isopropyl alcohol for removal. Overall, maintained awareness of DN is imperative, as early diagnosis can avoid unnecessary biopsies and more complex treatment measures as well as facilitate coordination of care when additional medical or psychiatric concerns are present.

Although the diagnoses of DN and terra firma-forme dermatosis can be distinguished based on the patient’s clinical history and response to simple cleansing measures alone, the alternate diagnoses can be excluded based on different clinical distributions and response to other treatment modalities but sometimes may require clinicopathologic correlation for definitive diagnosis. Our patient had a biopsy diagnosis of psoriasiform dermatitis from an outside provider, but neither her clinical disease nor repeated histopathologic findings supported a diagnosis of psoriasis or other classic psoriasiform dermatoses such as contact dermatitis, dermatophyte/ candidal infection, seborrheic dermatitis, pityriasis rubra pilaris, pityriasis rosea, scabies, or syphilis.
It is imperative to exclude alternative diagnoses because they can have systemic implications and can misguide treatment, as was done initially with our patient. Psoriasis vulgaris in its classic form is a chronic inflammatory skin disease that manifests as sharply demarcated, erythematous plaques with overlying thick silvery scale; it has the additional histologic findings of neutrophilic spongiform pustules in the epidermis, tortuous blood vessels in the papillary dermis, and neutrophils and parakeratosis in the stratum corneum. In its benign form, AN is associated with endocrinopathies, most commonly obesity and insulin-resistant diabetes mellitus, and presents as hyperkeratotic, velvety, hyperpigmented plaques typically limited to the neck and axillae. Malignant AN spontaneously arises in association with systemic malignancy and can be extensive and generalized.5 Treatment of AN primarily focuses on resolution of the underlying systemic disease; however, cosmetic treatment with topical or oral retinoids may hasten resolution of cutaneous disease.6 Confluent and reticulated papillomatosis is characterized by reticulated hyperkeratotic plaques with a common distribution over the central and upper trunk. Unlike DN and AN, which may occur at any age, CARP typically is seen in adolescents and young adults.7 There is no evidence-based gold standard for the management of CARP; however, the successful use of various antibiotics, antifungals, and retinoids—alone or in combination—has been reported.8 Overall, compared to the other entities in the differential diagnosis, DN easily can be prevented with consistent use of soap and water and may be underreported given the asymptomatic nature of the disease and the typical patient population.
- Poskitt L, Wayte J, Wojnarowska F, et al. ‘Dermatitis neglecta’: unwashed dermatosis. Br J Dermatol. 1995;132:827-829.
- Perez-Rodriguez IM, Munoz-Garza FZ, Ocampo-Candiani J. An unusually severe case of dermatosis neglecta: a diagnostic challenge. Case Rep Dermatol. 2014;6:194-199.
- Park JM, Roh MR, Kwon JE, et al. A case of generalized dermatitis neglecta mimicking psoriasis vulgaris. Arch Dermatol. 2010;146:1050-1051.
- Lopes S, Vide J, Antunes I, et al. Dermatitis neglecta: a challenging diagnosis in psychodermatology. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:109-110.
- Shah KR, Boland CR, Patel M, et al. Cutaneous manifestations of gastrointestinal disease: part I. J Am Acad Dermatol. 2013;68:189. e1-21; quiz 210.
- Patel NU, Roach C, Alinia H, et al. Current treatment options for acanthosis nigricans. Clin Cosmet Investig Dermatol. 2018; 11:407-413.
- Kurtyka DJ, Burke KT, DeKlotz CMC. Use of topical sirolimus (rapamycin) for treating confluent and reticulated papillomatosis. JAMA Dermatol. 2021;157:121-123.
- Mufti A, Sachdeva M, Maliyar K, et al. Treatment outcomes in confluent and reticulated papillomatosis: a systematic review. J Am Acad Dermatol. 2021;84:825-829.
The Diagnosis: Dermatitis Neglecta
A punch biopsy of the abdomen revealed hyperkeratosis and mild papillomatosis (Figure), which can be seen in dermatitis neglecta (DN) and acanthosis nigricans (AN) as well as confluent and reticulated papillomatosis (CARP). Due to the patient’s history of mood and psychotic disorders, collateral information was obtained from the patient’s family, who reported that the patient had a depressed mood in the last few months and was not showering or caring for herself during this period. There was no additional personal or family history of skin disease. Clinical and histopathologic findings led to a diagnosis of DN. Following recommendations for daily cleansing with soap and water along with topical ammonium lactate, near-complete resolution of the rash was achieved in 3 weeks.
Dermatitis neglecta, or unwashed dermatosis, is a skin condition that occurs secondary to poor hygiene, which was first reported in 1995 by Poskitt et al.1 Avoidance of washing in affected areas can be due to physical disability, pain after injury, neurological deficit, or psychologically induced fear or neglect. Sebum, sweat, corneocytes, and bacteria combine into compact adherent crusts of dirt, which appear as hyperkeratotic plaques with cornflakelike scale.2,3 Despite its innate simplicity, DN is a diagnostic challenge, as it clinically and histologically mimics other dermatoses including AN, terra firmaforme dermatosis, and CARP.2,4 Ultimately, the diagnosis of DN can be made when a history of poor hygiene is probable or elicited, and lesions can be removed with soap and water. Treatment of DN includes daily cleansing with soap and water; however, resistant lesions or extensive disease may require keratolytic agents, as in our patient.2-4 In contrast, terra firma-forme dermatosis, which may look similar, is not due to poor hygiene, and the lesions typically are resistant to soap and water, classically requiring isopropyl alcohol for removal. Overall, maintained awareness of DN is imperative, as early diagnosis can avoid unnecessary biopsies and more complex treatment measures as well as facilitate coordination of care when additional medical or psychiatric concerns are present.
Although the diagnoses of DN and terra firma-forme dermatosis can be distinguished based on the patient’s clinical history and response to simple cleansing measures alone, the alternate diagnoses can be excluded based on different clinical distributions and response to other treatment modalities but sometimes may require clinicopathologic correlation for definitive diagnosis. Our patient had a biopsy diagnosis of psoriasiform dermatitis from an outside provider, but neither her clinical disease nor repeated histopathologic findings supported a diagnosis of psoriasis or other classic psoriasiform dermatoses such as contact dermatitis, dermatophyte/ candidal infection, seborrheic dermatitis, pityriasis rubra pilaris, pityriasis rosea, scabies, or syphilis.
It is imperative to exclude alternative diagnoses because they can have systemic implications and can misguide treatment, as was done initially with our patient. Psoriasis vulgaris in its classic form is a chronic inflammatory skin disease that manifests as sharply demarcated, erythematous plaques with overlying thick silvery scale; it has the additional histologic findings of neutrophilic spongiform pustules in the epidermis, tortuous blood vessels in the papillary dermis, and neutrophils and parakeratosis in the stratum corneum. In its benign form, AN is associated with endocrinopathies, most commonly obesity and insulin-resistant diabetes mellitus, and presents as hyperkeratotic, velvety, hyperpigmented plaques typically limited to the neck and axillae. Malignant AN spontaneously arises in association with systemic malignancy and can be extensive and generalized.5 Treatment of AN primarily focuses on resolution of the underlying systemic disease; however, cosmetic treatment with topical or oral retinoids may hasten resolution of cutaneous disease.6 Confluent and reticulated papillomatosis is characterized by reticulated hyperkeratotic plaques with a common distribution over the central and upper trunk. Unlike DN and AN, which may occur at any age, CARP typically is seen in adolescents and young adults.7 There is no evidence-based gold standard for the management of CARP; however, the successful use of various antibiotics, antifungals, and retinoids—alone or in combination—has been reported.8 Overall, compared to the other entities in the differential diagnosis, DN easily can be prevented with consistent use of soap and water and may be underreported given the asymptomatic nature of the disease and the typical patient population.
The Diagnosis: Dermatitis Neglecta
A punch biopsy of the abdomen revealed hyperkeratosis and mild papillomatosis (Figure), which can be seen in dermatitis neglecta (DN) and acanthosis nigricans (AN) as well as confluent and reticulated papillomatosis (CARP). Due to the patient’s history of mood and psychotic disorders, collateral information was obtained from the patient’s family, who reported that the patient had a depressed mood in the last few months and was not showering or caring for herself during this period. There was no additional personal or family history of skin disease. Clinical and histopathologic findings led to a diagnosis of DN. Following recommendations for daily cleansing with soap and water along with topical ammonium lactate, near-complete resolution of the rash was achieved in 3 weeks.
Dermatitis neglecta, or unwashed dermatosis, is a skin condition that occurs secondary to poor hygiene, which was first reported in 1995 by Poskitt et al.1 Avoidance of washing in affected areas can be due to physical disability, pain after injury, neurological deficit, or psychologically induced fear or neglect. Sebum, sweat, corneocytes, and bacteria combine into compact adherent crusts of dirt, which appear as hyperkeratotic plaques with cornflakelike scale.2,3 Despite its innate simplicity, DN is a diagnostic challenge, as it clinically and histologically mimics other dermatoses including AN, terra firmaforme dermatosis, and CARP.2,4 Ultimately, the diagnosis of DN can be made when a history of poor hygiene is probable or elicited, and lesions can be removed with soap and water. Treatment of DN includes daily cleansing with soap and water; however, resistant lesions or extensive disease may require keratolytic agents, as in our patient.2-4 In contrast, terra firma-forme dermatosis, which may look similar, is not due to poor hygiene, and the lesions typically are resistant to soap and water, classically requiring isopropyl alcohol for removal. Overall, maintained awareness of DN is imperative, as early diagnosis can avoid unnecessary biopsies and more complex treatment measures as well as facilitate coordination of care when additional medical or psychiatric concerns are present.
Although the diagnoses of DN and terra firma-forme dermatosis can be distinguished based on the patient’s clinical history and response to simple cleansing measures alone, the alternate diagnoses can be excluded based on different clinical distributions and response to other treatment modalities but sometimes may require clinicopathologic correlation for definitive diagnosis. Our patient had a biopsy diagnosis of psoriasiform dermatitis from an outside provider, but neither her clinical disease nor repeated histopathologic findings supported a diagnosis of psoriasis or other classic psoriasiform dermatoses such as contact dermatitis, dermatophyte/ candidal infection, seborrheic dermatitis, pityriasis rubra pilaris, pityriasis rosea, scabies, or syphilis.
It is imperative to exclude alternative diagnoses because they can have systemic implications and can misguide treatment, as was done initially with our patient. Psoriasis vulgaris in its classic form is a chronic inflammatory skin disease that manifests as sharply demarcated, erythematous plaques with overlying thick silvery scale; it has the additional histologic findings of neutrophilic spongiform pustules in the epidermis, tortuous blood vessels in the papillary dermis, and neutrophils and parakeratosis in the stratum corneum. In its benign form, AN is associated with endocrinopathies, most commonly obesity and insulin-resistant diabetes mellitus, and presents as hyperkeratotic, velvety, hyperpigmented plaques typically limited to the neck and axillae. Malignant AN spontaneously arises in association with systemic malignancy and can be extensive and generalized.5 Treatment of AN primarily focuses on resolution of the underlying systemic disease; however, cosmetic treatment with topical or oral retinoids may hasten resolution of cutaneous disease.6 Confluent and reticulated papillomatosis is characterized by reticulated hyperkeratotic plaques with a common distribution over the central and upper trunk. Unlike DN and AN, which may occur at any age, CARP typically is seen in adolescents and young adults.7 There is no evidence-based gold standard for the management of CARP; however, the successful use of various antibiotics, antifungals, and retinoids—alone or in combination—has been reported.8 Overall, compared to the other entities in the differential diagnosis, DN easily can be prevented with consistent use of soap and water and may be underreported given the asymptomatic nature of the disease and the typical patient population.
- Poskitt L, Wayte J, Wojnarowska F, et al. ‘Dermatitis neglecta’: unwashed dermatosis. Br J Dermatol. 1995;132:827-829.
- Perez-Rodriguez IM, Munoz-Garza FZ, Ocampo-Candiani J. An unusually severe case of dermatosis neglecta: a diagnostic challenge. Case Rep Dermatol. 2014;6:194-199.
- Park JM, Roh MR, Kwon JE, et al. A case of generalized dermatitis neglecta mimicking psoriasis vulgaris. Arch Dermatol. 2010;146:1050-1051.
- Lopes S, Vide J, Antunes I, et al. Dermatitis neglecta: a challenging diagnosis in psychodermatology. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:109-110.
- Shah KR, Boland CR, Patel M, et al. Cutaneous manifestations of gastrointestinal disease: part I. J Am Acad Dermatol. 2013;68:189. e1-21; quiz 210.
- Patel NU, Roach C, Alinia H, et al. Current treatment options for acanthosis nigricans. Clin Cosmet Investig Dermatol. 2018; 11:407-413.
- Kurtyka DJ, Burke KT, DeKlotz CMC. Use of topical sirolimus (rapamycin) for treating confluent and reticulated papillomatosis. JAMA Dermatol. 2021;157:121-123.
- Mufti A, Sachdeva M, Maliyar K, et al. Treatment outcomes in confluent and reticulated papillomatosis: a systematic review. J Am Acad Dermatol. 2021;84:825-829.
- Poskitt L, Wayte J, Wojnarowska F, et al. ‘Dermatitis neglecta’: unwashed dermatosis. Br J Dermatol. 1995;132:827-829.
- Perez-Rodriguez IM, Munoz-Garza FZ, Ocampo-Candiani J. An unusually severe case of dermatosis neglecta: a diagnostic challenge. Case Rep Dermatol. 2014;6:194-199.
- Park JM, Roh MR, Kwon JE, et al. A case of generalized dermatitis neglecta mimicking psoriasis vulgaris. Arch Dermatol. 2010;146:1050-1051.
- Lopes S, Vide J, Antunes I, et al. Dermatitis neglecta: a challenging diagnosis in psychodermatology. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:109-110.
- Shah KR, Boland CR, Patel M, et al. Cutaneous manifestations of gastrointestinal disease: part I. J Am Acad Dermatol. 2013;68:189. e1-21; quiz 210.
- Patel NU, Roach C, Alinia H, et al. Current treatment options for acanthosis nigricans. Clin Cosmet Investig Dermatol. 2018; 11:407-413.
- Kurtyka DJ, Burke KT, DeKlotz CMC. Use of topical sirolimus (rapamycin) for treating confluent and reticulated papillomatosis. JAMA Dermatol. 2021;157:121-123.
- Mufti A, Sachdeva M, Maliyar K, et al. Treatment outcomes in confluent and reticulated papillomatosis: a systematic review. J Am Acad Dermatol. 2021;84:825-829.

A 28-year-old woman was admitted to the medicine service with bilateral pedal numbness and ataxia, as well as an asymptomatic rash on the neck, chest, abdomen, and extremities of a few months’ duration. The patient was seen by an outside dermatologist for the same rash 1 month prior, at which time a punch biopsy of the right forearm was suggestive of psoriasiform dermatitis; however, the rash failed to improve with topical ammonium lactate and corticosteroids. During the current admission, the patient was found to have low methylmalonic acid and vitamin B1 levels; however, vitamin B12, thyroid studies, rapid plasma reagin test, and inflammatory markers, as well as central and peripheral imaging and nerve conduction studies were normal.
Dermatology was consulted. Physical examination revealed retention hyperkeratosis on the neck that was wipeable with 70% isopropyl alcohol, as well as nonwipeable, thin, reticulated plaques on the mid chest and thick velvety plaques on the abdomen and bilateral extremities. There was notable sparing of areas with natural occlusion such as the back and body folds. A punch biopsy of the abdomen was performed.
Spiral Plaque on the Left Ankle
The Diagnosis: Recurrent Cutaneous T-Cell Lymphoma
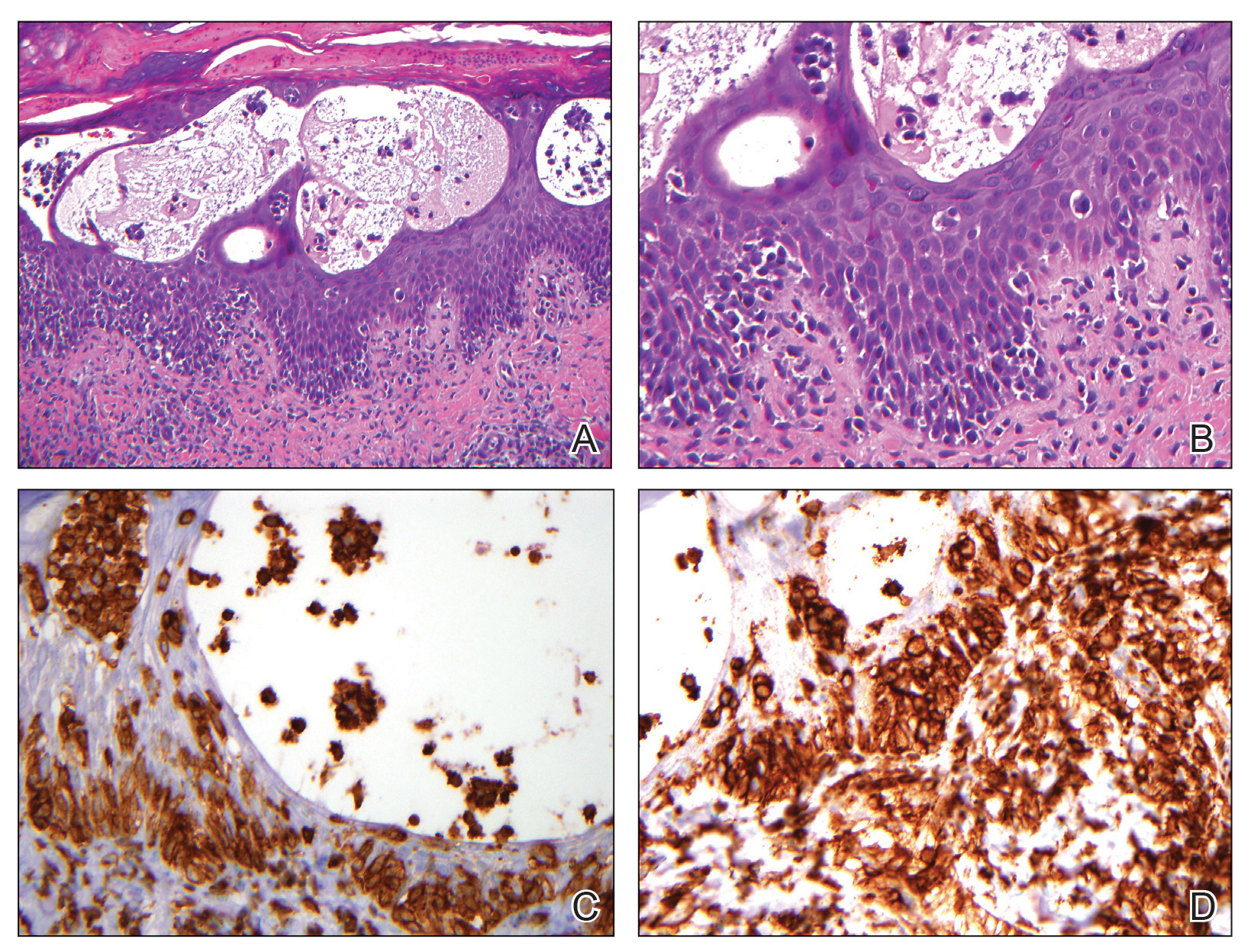
The skin biopsy revealed alternating orthokeratosis and parakeratosis with mild to moderate spongiosis and intraepidermal vesiculation as well as individual and nested atypical mononuclear cells with moderately enlarged hyperchromatic nuclei in the epidermis. There was a superficial interstitial lymphocytic infiltrate with occasional enlarged cells (Figure, A and B), and atypical cells in the epidermis and dermis stained with antibodies against CD3 and CD4 (Figure, C and D) but not against CD20 or CD8. These histopathologic findings were consistent with cutaneous T-cell lymphoma (CTCL), mycosis fungoides (MF) type. Additional application of bexarotene gel on days the patient received narrowband UVB was recommended with noted improvement of the skin.
Cutaneous T-cell lymphomas are a heterogenous group of diseases with monoclonal proliferation of T lymphocytes that largely are confined to the skin at the time of diagnosis.1 The incidence of CTCL rose steadily for more than 25 years, with an annual age-adjusted incidence of 6.4 to 9.6 cases per million individuals in the United States from 1973 to 2002.2 Mycosis fungoides is the most common classification of CTCL. It usually is characterized by patches or plaques of scaly erythema or poikiloderma; however, it also can present with annular, arcuate, concentrative, annular and linear morphologies. Mycosis fungoides tumor cells typically express a mature memory T helper cell phenotype of CD3+, CD4+, and CD8−, but there are different variants that have been discovered.3 Mycosis fungoides distributed in a spiral pattern is a distinctly unusual manifestation. Mechanisms of such dynamic morphologies are unknown but may represent an interplay between malignant cell proliferation and lost immune responses in temporospatial relationships.
The presence of keratotic gyrate lesions on acral surfaces should raise the possibility of pagetoid reticulosis. However, our patient had a history of MF involving areas of the body beyond the extremities, making this diagnosis less likely. Pagetoid reticulosis is categorized as an MF variant under the current World Health Organization– European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas.4 Pagetoid reticulosis clinically presents as a solitary psoriasiform or hyperkeratotic patch or plaque that affects the distal extremities. Variable immunophenotypes have been shown in pagetoid reticulosis, such as CD4−/CD8+ and CD4−/CD8−, while classic MF typically shows CD4+/CD8−, as in our case.5
Tinea pedis is a superficial fungal infection usually caused by anthropophilic dermatophytes, with Trichophyton rubrum being the most common organism. Four common clinical presentations of tinea pedis have been identified: interdigital, moccasin, vesicular, and acute ulcerative. Clinical presentation ranges from macerations, ulcerations, and erosions in the toe web spaces to dry hyperkeratotic scaling and fissures on the plantar foot.6 Tinea pedis primarily affects the plantar and interdigital spaces, sparing the dorsal foot and ankle. Treatment is recommended to alleviate symptoms and limit the spread of infection; topical antifungals for 4 weeks is the treatment of choice. However, recurrence is common, and maintenance therapy often is indicated. Oral antifungals or a combination of both topical and oral medications may be needed in certain cases.7
Erythema annulare centrifugum (EAC) is a rare dermatologic disease described as erythematous or urticarial papules that can enlarge centrifugally to form annular lesions that clear centrally. Thought to be a hypersensitivity reaction to an underlying condition, EAC has been associated with fungal infections, various cutaneous diseases, and even internal malignancies. Clinically, EAC can be divided into 2 forms: deep and superficial. Deep gyrate erythema is characterized by a firm indurated border with rare scaling and pruritus that histologically shows perivascular lymphocytic infiltration in the upper and deep dermis. Superficial gyrate erythema has minimally elevated lesions with an indistinct border and trailing scales and pruritus; histopathologic findings present a dense, perivascular, lymphocytic infiltration restricted to the upper dermis.8 Therapy for EAC is directed at relieving symptoms and treating the underlying condition if there is one associated.
Granuloma annulare (GA) is a common skin disorder classically characterized by ringed erythematous plaques, though many variants have been identified. Localized GA is the most common variant and presents with pink-red, nonscaly, annular patches or plaques, typically affecting the hands and feet. Generalized GA is characterized as diffuse annular patches or plaques classically affecting the trunk and extremities. Histology is notable for mucin with a palisading or interstitial pattern of granulomatous inflammation, which was not evident in our patient.9 Topical or intralesional corticosteroids are the first-line treatment of localized GA; however, localized GA generally is self-limited, and treatment often is not necessary. Treatment with cryosurgery, laser therapy, and topical dapsone and tacrolimus also has been described, but evidence of the efficacy of these agents is limited. For generalized GA, phototherapy currently is the most reliable therapy. Systemic therapies include antimalarials, fumaric acid esters, biologics, antimicrobials, and isotretinoin.10
Erythema gyratum repens (EGR) is a rare dermatologic disease described as erythematous concentric bands arranged in parallel rings that can be annular, figurate, or gyrate, with a fine scale trailing the leading edge. Histopathologic features of EGR are nonspecific but are characterized by a perivascular, superficial, mononuclear dermatitis. Diagnosis is based on its characteristic clinical presentation. Although EGR commonly is associated with internal malignancies such as bronchial carcinoma, it also may be associated with benign conditions.11 Improvement often is seen with successful therapy of the underlying associated malignancy.12
Treatment of MF is based on tumor-node-metastasisblood classification, prognostic factors, and clinical stage at the time of diagnosis. Early-stage MF (IA–IIA) commonly is treated with skin-directed therapies such as topical corticosteroids, topical mechlorethamine, topical retinoids, UV phototherapy, and localized radiotherapy. In late stages (IIB–IV), systemic therapy is indicated and includes systemic retinoids, interferon alfa, chemotherapy, monoclonal antibodies, and psoralen plus UVA.13 In many cases, patients may require combination therapy to achieve remission or better control of their condition, as in our patient.
The Diagnosis: Recurrent Cutaneous T-Cell Lymphoma
The skin biopsy revealed alternating orthokeratosis and parakeratosis with mild to moderate spongiosis and intraepidermal vesiculation as well as individual and nested atypical mononuclear cells with moderately enlarged hyperchromatic nuclei in the epidermis. There was a superficial interstitial lymphocytic infiltrate with occasional enlarged cells (Figure, A and B), and atypical cells in the epidermis and dermis stained with antibodies against CD3 and CD4 (Figure, C and D) but not against CD20 or CD8. These histopathologic findings were consistent with cutaneous T-cell lymphoma (CTCL), mycosis fungoides (MF) type. Additional application of bexarotene gel on days the patient received narrowband UVB was recommended with noted improvement of the skin.
Cutaneous T-cell lymphomas are a heterogenous group of diseases with monoclonal proliferation of T lymphocytes that largely are confined to the skin at the time of diagnosis.1 The incidence of CTCL rose steadily for more than 25 years, with an annual age-adjusted incidence of 6.4 to 9.6 cases per million individuals in the United States from 1973 to 2002.2 Mycosis fungoides is the most common classification of CTCL. It usually is characterized by patches or plaques of scaly erythema or poikiloderma; however, it also can present with annular, arcuate, concentrative, annular and linear morphologies. Mycosis fungoides tumor cells typically express a mature memory T helper cell phenotype of CD3+, CD4+, and CD8−, but there are different variants that have been discovered.3 Mycosis fungoides distributed in a spiral pattern is a distinctly unusual manifestation. Mechanisms of such dynamic morphologies are unknown but may represent an interplay between malignant cell proliferation and lost immune responses in temporospatial relationships.
The presence of keratotic gyrate lesions on acral surfaces should raise the possibility of pagetoid reticulosis. However, our patient had a history of MF involving areas of the body beyond the extremities, making this diagnosis less likely. Pagetoid reticulosis is categorized as an MF variant under the current World Health Organization– European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas.4 Pagetoid reticulosis clinically presents as a solitary psoriasiform or hyperkeratotic patch or plaque that affects the distal extremities. Variable immunophenotypes have been shown in pagetoid reticulosis, such as CD4−/CD8+ and CD4−/CD8−, while classic MF typically shows CD4+/CD8−, as in our case.5
Tinea pedis is a superficial fungal infection usually caused by anthropophilic dermatophytes, with Trichophyton rubrum being the most common organism. Four common clinical presentations of tinea pedis have been identified: interdigital, moccasin, vesicular, and acute ulcerative. Clinical presentation ranges from macerations, ulcerations, and erosions in the toe web spaces to dry hyperkeratotic scaling and fissures on the plantar foot.6 Tinea pedis primarily affects the plantar and interdigital spaces, sparing the dorsal foot and ankle. Treatment is recommended to alleviate symptoms and limit the spread of infection; topical antifungals for 4 weeks is the treatment of choice. However, recurrence is common, and maintenance therapy often is indicated. Oral antifungals or a combination of both topical and oral medications may be needed in certain cases.7
Erythema annulare centrifugum (EAC) is a rare dermatologic disease described as erythematous or urticarial papules that can enlarge centrifugally to form annular lesions that clear centrally. Thought to be a hypersensitivity reaction to an underlying condition, EAC has been associated with fungal infections, various cutaneous diseases, and even internal malignancies. Clinically, EAC can be divided into 2 forms: deep and superficial. Deep gyrate erythema is characterized by a firm indurated border with rare scaling and pruritus that histologically shows perivascular lymphocytic infiltration in the upper and deep dermis. Superficial gyrate erythema has minimally elevated lesions with an indistinct border and trailing scales and pruritus; histopathologic findings present a dense, perivascular, lymphocytic infiltration restricted to the upper dermis.8 Therapy for EAC is directed at relieving symptoms and treating the underlying condition if there is one associated.
Granuloma annulare (GA) is a common skin disorder classically characterized by ringed erythematous plaques, though many variants have been identified. Localized GA is the most common variant and presents with pink-red, nonscaly, annular patches or plaques, typically affecting the hands and feet. Generalized GA is characterized as diffuse annular patches or plaques classically affecting the trunk and extremities. Histology is notable for mucin with a palisading or interstitial pattern of granulomatous inflammation, which was not evident in our patient.9 Topical or intralesional corticosteroids are the first-line treatment of localized GA; however, localized GA generally is self-limited, and treatment often is not necessary. Treatment with cryosurgery, laser therapy, and topical dapsone and tacrolimus also has been described, but evidence of the efficacy of these agents is limited. For generalized GA, phototherapy currently is the most reliable therapy. Systemic therapies include antimalarials, fumaric acid esters, biologics, antimicrobials, and isotretinoin.10
Erythema gyratum repens (EGR) is a rare dermatologic disease described as erythematous concentric bands arranged in parallel rings that can be annular, figurate, or gyrate, with a fine scale trailing the leading edge. Histopathologic features of EGR are nonspecific but are characterized by a perivascular, superficial, mononuclear dermatitis. Diagnosis is based on its characteristic clinical presentation. Although EGR commonly is associated with internal malignancies such as bronchial carcinoma, it also may be associated with benign conditions.11 Improvement often is seen with successful therapy of the underlying associated malignancy.12
Treatment of MF is based on tumor-node-metastasisblood classification, prognostic factors, and clinical stage at the time of diagnosis. Early-stage MF (IA–IIA) commonly is treated with skin-directed therapies such as topical corticosteroids, topical mechlorethamine, topical retinoids, UV phototherapy, and localized radiotherapy. In late stages (IIB–IV), systemic therapy is indicated and includes systemic retinoids, interferon alfa, chemotherapy, monoclonal antibodies, and psoralen plus UVA.13 In many cases, patients may require combination therapy to achieve remission or better control of their condition, as in our patient.
The Diagnosis: Recurrent Cutaneous T-Cell Lymphoma
The skin biopsy revealed alternating orthokeratosis and parakeratosis with mild to moderate spongiosis and intraepidermal vesiculation as well as individual and nested atypical mononuclear cells with moderately enlarged hyperchromatic nuclei in the epidermis. There was a superficial interstitial lymphocytic infiltrate with occasional enlarged cells (Figure, A and B), and atypical cells in the epidermis and dermis stained with antibodies against CD3 and CD4 (Figure, C and D) but not against CD20 or CD8. These histopathologic findings were consistent with cutaneous T-cell lymphoma (CTCL), mycosis fungoides (MF) type. Additional application of bexarotene gel on days the patient received narrowband UVB was recommended with noted improvement of the skin.
Cutaneous T-cell lymphomas are a heterogenous group of diseases with monoclonal proliferation of T lymphocytes that largely are confined to the skin at the time of diagnosis.1 The incidence of CTCL rose steadily for more than 25 years, with an annual age-adjusted incidence of 6.4 to 9.6 cases per million individuals in the United States from 1973 to 2002.2 Mycosis fungoides is the most common classification of CTCL. It usually is characterized by patches or plaques of scaly erythema or poikiloderma; however, it also can present with annular, arcuate, concentrative, annular and linear morphologies. Mycosis fungoides tumor cells typically express a mature memory T helper cell phenotype of CD3+, CD4+, and CD8−, but there are different variants that have been discovered.3 Mycosis fungoides distributed in a spiral pattern is a distinctly unusual manifestation. Mechanisms of such dynamic morphologies are unknown but may represent an interplay between malignant cell proliferation and lost immune responses in temporospatial relationships.
The presence of keratotic gyrate lesions on acral surfaces should raise the possibility of pagetoid reticulosis. However, our patient had a history of MF involving areas of the body beyond the extremities, making this diagnosis less likely. Pagetoid reticulosis is categorized as an MF variant under the current World Health Organization– European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas.4 Pagetoid reticulosis clinically presents as a solitary psoriasiform or hyperkeratotic patch or plaque that affects the distal extremities. Variable immunophenotypes have been shown in pagetoid reticulosis, such as CD4−/CD8+ and CD4−/CD8−, while classic MF typically shows CD4+/CD8−, as in our case.5
Tinea pedis is a superficial fungal infection usually caused by anthropophilic dermatophytes, with Trichophyton rubrum being the most common organism. Four common clinical presentations of tinea pedis have been identified: interdigital, moccasin, vesicular, and acute ulcerative. Clinical presentation ranges from macerations, ulcerations, and erosions in the toe web spaces to dry hyperkeratotic scaling and fissures on the plantar foot.6 Tinea pedis primarily affects the plantar and interdigital spaces, sparing the dorsal foot and ankle. Treatment is recommended to alleviate symptoms and limit the spread of infection; topical antifungals for 4 weeks is the treatment of choice. However, recurrence is common, and maintenance therapy often is indicated. Oral antifungals or a combination of both topical and oral medications may be needed in certain cases.7
Erythema annulare centrifugum (EAC) is a rare dermatologic disease described as erythematous or urticarial papules that can enlarge centrifugally to form annular lesions that clear centrally. Thought to be a hypersensitivity reaction to an underlying condition, EAC has been associated with fungal infections, various cutaneous diseases, and even internal malignancies. Clinically, EAC can be divided into 2 forms: deep and superficial. Deep gyrate erythema is characterized by a firm indurated border with rare scaling and pruritus that histologically shows perivascular lymphocytic infiltration in the upper and deep dermis. Superficial gyrate erythema has minimally elevated lesions with an indistinct border and trailing scales and pruritus; histopathologic findings present a dense, perivascular, lymphocytic infiltration restricted to the upper dermis.8 Therapy for EAC is directed at relieving symptoms and treating the underlying condition if there is one associated.
Granuloma annulare (GA) is a common skin disorder classically characterized by ringed erythematous plaques, though many variants have been identified. Localized GA is the most common variant and presents with pink-red, nonscaly, annular patches or plaques, typically affecting the hands and feet. Generalized GA is characterized as diffuse annular patches or plaques classically affecting the trunk and extremities. Histology is notable for mucin with a palisading or interstitial pattern of granulomatous inflammation, which was not evident in our patient.9 Topical or intralesional corticosteroids are the first-line treatment of localized GA; however, localized GA generally is self-limited, and treatment often is not necessary. Treatment with cryosurgery, laser therapy, and topical dapsone and tacrolimus also has been described, but evidence of the efficacy of these agents is limited. For generalized GA, phototherapy currently is the most reliable therapy. Systemic therapies include antimalarials, fumaric acid esters, biologics, antimicrobials, and isotretinoin.10
Erythema gyratum repens (EGR) is a rare dermatologic disease described as erythematous concentric bands arranged in parallel rings that can be annular, figurate, or gyrate, with a fine scale trailing the leading edge. Histopathologic features of EGR are nonspecific but are characterized by a perivascular, superficial, mononuclear dermatitis. Diagnosis is based on its characteristic clinical presentation. Although EGR commonly is associated with internal malignancies such as bronchial carcinoma, it also may be associated with benign conditions.11 Improvement often is seen with successful therapy of the underlying associated malignancy.12
Treatment of MF is based on tumor-node-metastasisblood classification, prognostic factors, and clinical stage at the time of diagnosis. Early-stage MF (IA–IIA) commonly is treated with skin-directed therapies such as topical corticosteroids, topical mechlorethamine, topical retinoids, UV phototherapy, and localized radiotherapy. In late stages (IIB–IV), systemic therapy is indicated and includes systemic retinoids, interferon alfa, chemotherapy, monoclonal antibodies, and psoralen plus UVA.13 In many cases, patients may require combination therapy to achieve remission or better control of their condition, as in our patient.
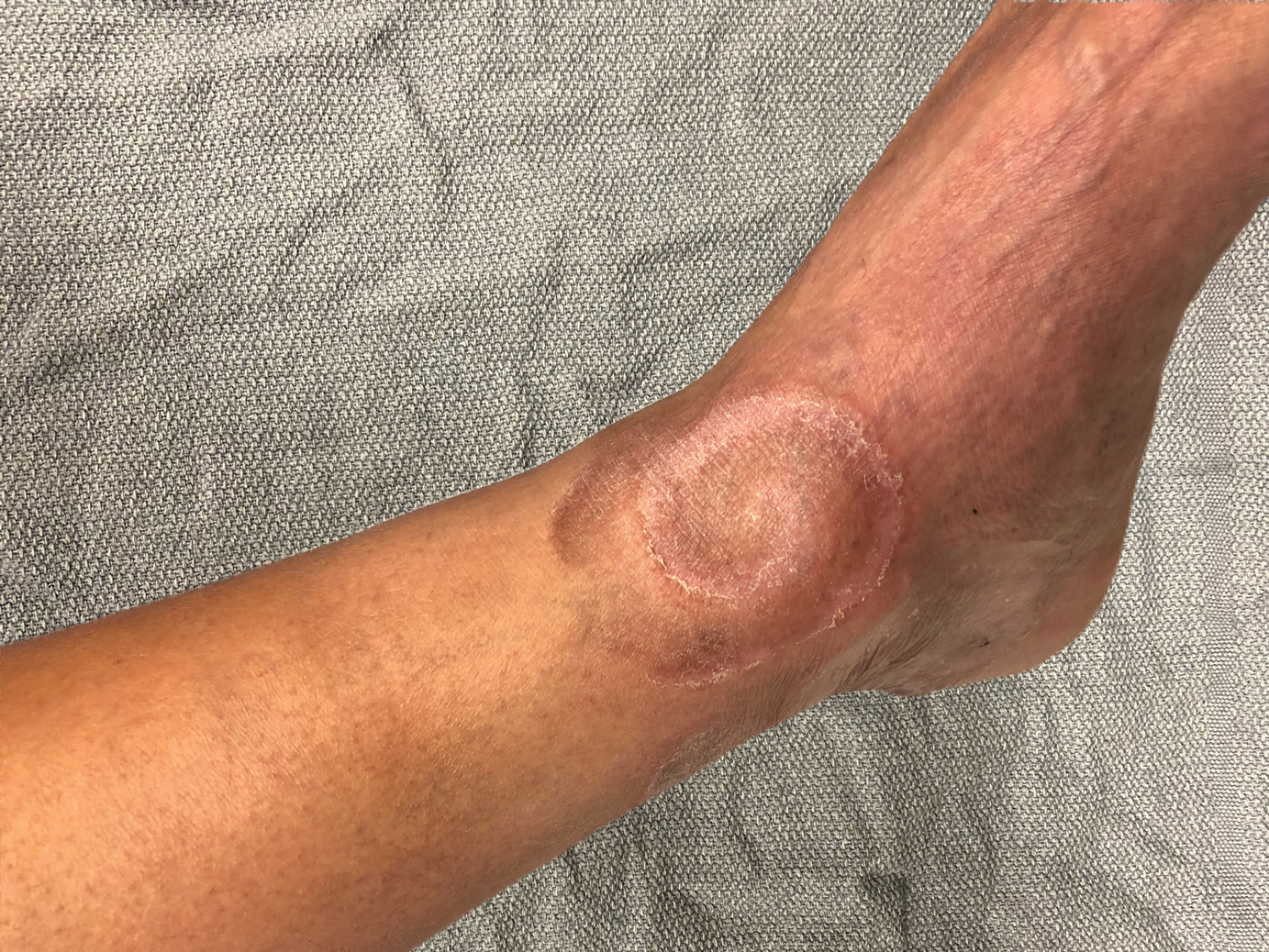
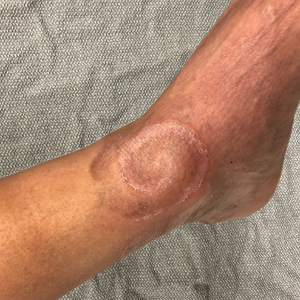
A 60-year-old man presented with a whorl-like plaque on the left ankle that he had noticed while undergoing treatment with narrowband UVB every other week and nitrogen mustard gel daily for stage IB cutaneous T-cell lymphoma, mycosis fungoides type. He denied pain, pruritus, and any other associated symptoms at the site. He denied recent illness, new medications, or changes in diet. His medical history included multiple sclerosis, vascular disease, and stroke. Physical examination revealed an 8×6-cm, welldemarcated, slightly scaly, erythematous plaque with a spiral appearance and peripheral hyperpigmentation involving the left ankle. The remainder of the examination was notable for well-controlled mycosis fungoides with several hyperpigmented patches at sites of prior involvement on the trunk and upper and lower extremities. No cervical, axillary, or inguinal lymphadenopathy was noted. A 4-mm punch biopsy was performed and sent for histopathologic examination.
Lesions on the Thigh After an Organ Transplant
The Diagnosis: Microcystic Lymphatic Malformation
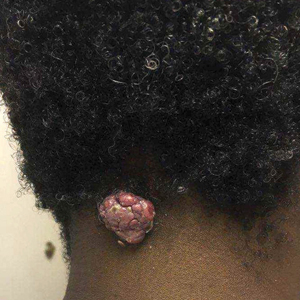
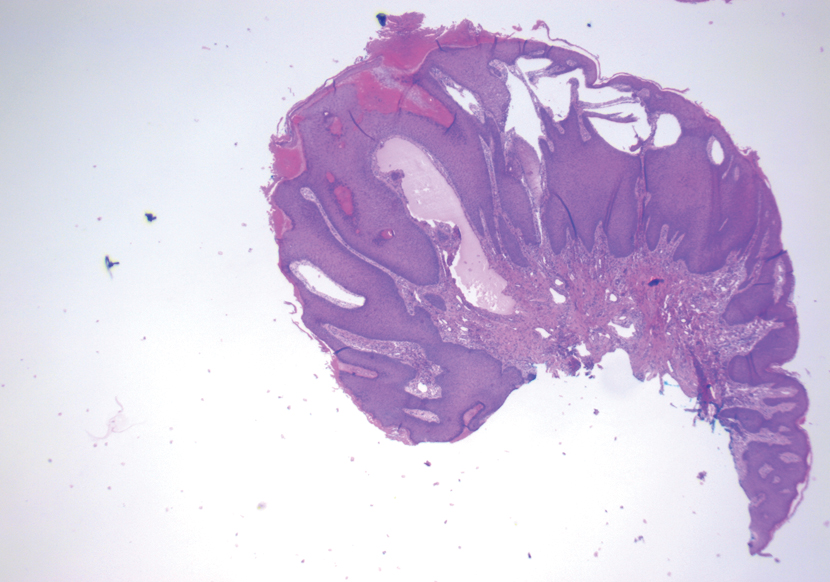
The shave biopsy demonstrated numerous thin-walled vascular spaces filled with lymphatic fluid within the dermis (Figure), consistent with a diagnosis of microcystic lymphatic malformation (LM). Lymphatic malformations represent a class of benign vascular lesions consisting of anomalous or dilated lymphatic vessels, which can be broadly categorized as macrocystic (formerly cavernous lymphangioma or cystic hygroma), microcystic (formerly lymphangioma circumscriptum), or mixed.1 Patients often will present with pruritus, crusting, secondary infection, edema, or oozing.2 The superficial blebs of microcystic LMs resemble frog spawn and range in color from clear to pink, brawny, or deep maroon.3 Although the lymphatic vessels involved in microcystic LMs appear disconnected from the major lymphatic circulation,3 systemic fluid overload could plausibly promote lesional swelling and tenderness; we attributed our patient's worsening symptoms to the cumulative 7.8 L of intravenous fluid he received intraoperatively during his cardiac transplant. The excess fluid allowed communication between lymphatic cisterns and thin-walled vesicles on the skin surface through dilated channels. Overall, LMs represent roughly 26% of pediatric benign vascular tumors and approximately 4% of all vascular tumors.4
Although microcystic LMs may appear especially vascular or verrucous, the differential diagnosis for our patient's LM included condyloma acuminatum,5,6 condyloma lata,7 epidermal nevus, and lymphangiosarcoma. Epidermal nevi are congenital lesions, varying in appearance from velvety to verrucous patches and plaques that often evolve during puberty and become thicker, more verrucous, and hyperpigmented. Keratinocytic epidermal nevus syndromes and other entities such as nevus sebaceous have been associated with somatic mutations affecting proteins in the fibroblast growth factor receptor signaling pathway (eg, FGFR3, HRAS).8 Although the clinical appearance alone may be similar, lymphangiosarcoma can be distinguished from LM via biopsy.
There are several methods to diagnose LM. Duplex sonography is possibly the best noninvasive method to identify the flow between venous valves. Magnetic resonance imaging can detect larger occurrences of LM, and lymphangiography can be utilized to confirm a normal or abnormal lymphatic network.4 Treatment options are broad, including surgical excision, laser ablation, and topical sirolimus. Hypertonic saline sclerotherapy can be injected into the afflicted lymphatic channels to decrease inflammation, erythema, and hyperpigmentation without further treatment or major side effects.4
However, the benefits of sclerotherapy alone in the treatment of LM often come gradually, and radiofrequency ablation may need to be utilized to achieve more immediate results.2 Overall, outcomes are highly variable, but favorable outcomes often can be difficult to obtain due to a high recurrence rate.2,8 Our patient's symptoms improved during his postoperative recovery, and he declined further intervention.
- Elluru RG, Balakrishnan K, Padua HM. Lymphatic malformations: diagnosis and management. Semin Pediatr Surg. 2014;23:178-185. doi:10.1053/j.sempedsurg.2014.07.002
- Niti K, Manish P. Microcystic lymphatic malformation (lymphangioma circumscriptum) treated using a minimally invasive technique of radiofrequency ablation and sclerotherapy. Dermatol Surg. 2010;36:1711-1717. doi:10.1111/j.1524-4725.2010.01723.x
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295. doi:10.1111 /j.1365-4632.2009.04226.x
- Bikowski JB, Dumont AM. Lymphangioma circumscriptum: treatment with hypertonic saline sclerotherapy. J Am Acad Dermatol. 2005;53:442-444. doi:10.1016/j.jaad.2005.04.086
- Costa-Silva M, Fernandes I, Rodrigues AG, et al. Anogenital warts in pediatric population. An Bras Dermatol. 2017;92:675-681. doi:10.1590 /abd1806-4841.201756411
- Darmstadt GL. Perianal lymphangioma circumscriptum mistaken for genital warts. Pediatrics 1996;98;461.
- Bruins FG, van Deudekom FJA, de Vries HJC. Syphilitic condylomata lata mimicking anogenital warts. BMJ. 2015;350:h1259. doi:10.1136 /bmj.h1259
- Asch S, Sugarman JL. Epidermal nevus syndromes: new insights into whorls and swirls. Pediatr Dermatol. 2018;35:21-29. doi:10.1111 /pde.13273
The Diagnosis: Microcystic Lymphatic Malformation
The shave biopsy demonstrated numerous thin-walled vascular spaces filled with lymphatic fluid within the dermis (Figure), consistent with a diagnosis of microcystic lymphatic malformation (LM). Lymphatic malformations represent a class of benign vascular lesions consisting of anomalous or dilated lymphatic vessels, which can be broadly categorized as macrocystic (formerly cavernous lymphangioma or cystic hygroma), microcystic (formerly lymphangioma circumscriptum), or mixed.1 Patients often will present with pruritus, crusting, secondary infection, edema, or oozing.2 The superficial blebs of microcystic LMs resemble frog spawn and range in color from clear to pink, brawny, or deep maroon.3 Although the lymphatic vessels involved in microcystic LMs appear disconnected from the major lymphatic circulation,3 systemic fluid overload could plausibly promote lesional swelling and tenderness; we attributed our patient's worsening symptoms to the cumulative 7.8 L of intravenous fluid he received intraoperatively during his cardiac transplant. The excess fluid allowed communication between lymphatic cisterns and thin-walled vesicles on the skin surface through dilated channels. Overall, LMs represent roughly 26% of pediatric benign vascular tumors and approximately 4% of all vascular tumors.4
Although microcystic LMs may appear especially vascular or verrucous, the differential diagnosis for our patient's LM included condyloma acuminatum,5,6 condyloma lata,7 epidermal nevus, and lymphangiosarcoma. Epidermal nevi are congenital lesions, varying in appearance from velvety to verrucous patches and plaques that often evolve during puberty and become thicker, more verrucous, and hyperpigmented. Keratinocytic epidermal nevus syndromes and other entities such as nevus sebaceous have been associated with somatic mutations affecting proteins in the fibroblast growth factor receptor signaling pathway (eg, FGFR3, HRAS).8 Although the clinical appearance alone may be similar, lymphangiosarcoma can be distinguished from LM via biopsy.
There are several methods to diagnose LM. Duplex sonography is possibly the best noninvasive method to identify the flow between venous valves. Magnetic resonance imaging can detect larger occurrences of LM, and lymphangiography can be utilized to confirm a normal or abnormal lymphatic network.4 Treatment options are broad, including surgical excision, laser ablation, and topical sirolimus. Hypertonic saline sclerotherapy can be injected into the afflicted lymphatic channels to decrease inflammation, erythema, and hyperpigmentation without further treatment or major side effects.4
However, the benefits of sclerotherapy alone in the treatment of LM often come gradually, and radiofrequency ablation may need to be utilized to achieve more immediate results.2 Overall, outcomes are highly variable, but favorable outcomes often can be difficult to obtain due to a high recurrence rate.2,8 Our patient's symptoms improved during his postoperative recovery, and he declined further intervention.
The Diagnosis: Microcystic Lymphatic Malformation
The shave biopsy demonstrated numerous thin-walled vascular spaces filled with lymphatic fluid within the dermis (Figure), consistent with a diagnosis of microcystic lymphatic malformation (LM). Lymphatic malformations represent a class of benign vascular lesions consisting of anomalous or dilated lymphatic vessels, which can be broadly categorized as macrocystic (formerly cavernous lymphangioma or cystic hygroma), microcystic (formerly lymphangioma circumscriptum), or mixed.1 Patients often will present with pruritus, crusting, secondary infection, edema, or oozing.2 The superficial blebs of microcystic LMs resemble frog spawn and range in color from clear to pink, brawny, or deep maroon.3 Although the lymphatic vessels involved in microcystic LMs appear disconnected from the major lymphatic circulation,3 systemic fluid overload could plausibly promote lesional swelling and tenderness; we attributed our patient's worsening symptoms to the cumulative 7.8 L of intravenous fluid he received intraoperatively during his cardiac transplant. The excess fluid allowed communication between lymphatic cisterns and thin-walled vesicles on the skin surface through dilated channels. Overall, LMs represent roughly 26% of pediatric benign vascular tumors and approximately 4% of all vascular tumors.4
Although microcystic LMs may appear especially vascular or verrucous, the differential diagnosis for our patient's LM included condyloma acuminatum,5,6 condyloma lata,7 epidermal nevus, and lymphangiosarcoma. Epidermal nevi are congenital lesions, varying in appearance from velvety to verrucous patches and plaques that often evolve during puberty and become thicker, more verrucous, and hyperpigmented. Keratinocytic epidermal nevus syndromes and other entities such as nevus sebaceous have been associated with somatic mutations affecting proteins in the fibroblast growth factor receptor signaling pathway (eg, FGFR3, HRAS).8 Although the clinical appearance alone may be similar, lymphangiosarcoma can be distinguished from LM via biopsy.
There are several methods to diagnose LM. Duplex sonography is possibly the best noninvasive method to identify the flow between venous valves. Magnetic resonance imaging can detect larger occurrences of LM, and lymphangiography can be utilized to confirm a normal or abnormal lymphatic network.4 Treatment options are broad, including surgical excision, laser ablation, and topical sirolimus. Hypertonic saline sclerotherapy can be injected into the afflicted lymphatic channels to decrease inflammation, erythema, and hyperpigmentation without further treatment or major side effects.4
However, the benefits of sclerotherapy alone in the treatment of LM often come gradually, and radiofrequency ablation may need to be utilized to achieve more immediate results.2 Overall, outcomes are highly variable, but favorable outcomes often can be difficult to obtain due to a high recurrence rate.2,8 Our patient's symptoms improved during his postoperative recovery, and he declined further intervention.
- Elluru RG, Balakrishnan K, Padua HM. Lymphatic malformations: diagnosis and management. Semin Pediatr Surg. 2014;23:178-185. doi:10.1053/j.sempedsurg.2014.07.002
- Niti K, Manish P. Microcystic lymphatic malformation (lymphangioma circumscriptum) treated using a minimally invasive technique of radiofrequency ablation and sclerotherapy. Dermatol Surg. 2010;36:1711-1717. doi:10.1111/j.1524-4725.2010.01723.x
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295. doi:10.1111 /j.1365-4632.2009.04226.x
- Bikowski JB, Dumont AM. Lymphangioma circumscriptum: treatment with hypertonic saline sclerotherapy. J Am Acad Dermatol. 2005;53:442-444. doi:10.1016/j.jaad.2005.04.086
- Costa-Silva M, Fernandes I, Rodrigues AG, et al. Anogenital warts in pediatric population. An Bras Dermatol. 2017;92:675-681. doi:10.1590 /abd1806-4841.201756411
- Darmstadt GL. Perianal lymphangioma circumscriptum mistaken for genital warts. Pediatrics 1996;98;461.
- Bruins FG, van Deudekom FJA, de Vries HJC. Syphilitic condylomata lata mimicking anogenital warts. BMJ. 2015;350:h1259. doi:10.1136 /bmj.h1259
- Asch S, Sugarman JL. Epidermal nevus syndromes: new insights into whorls and swirls. Pediatr Dermatol. 2018;35:21-29. doi:10.1111 /pde.13273
- Elluru RG, Balakrishnan K, Padua HM. Lymphatic malformations: diagnosis and management. Semin Pediatr Surg. 2014;23:178-185. doi:10.1053/j.sempedsurg.2014.07.002
- Niti K, Manish P. Microcystic lymphatic malformation (lymphangioma circumscriptum) treated using a minimally invasive technique of radiofrequency ablation and sclerotherapy. Dermatol Surg. 2010;36:1711-1717. doi:10.1111/j.1524-4725.2010.01723.x
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295. doi:10.1111 /j.1365-4632.2009.04226.x
- Bikowski JB, Dumont AM. Lymphangioma circumscriptum: treatment with hypertonic saline sclerotherapy. J Am Acad Dermatol. 2005;53:442-444. doi:10.1016/j.jaad.2005.04.086
- Costa-Silva M, Fernandes I, Rodrigues AG, et al. Anogenital warts in pediatric population. An Bras Dermatol. 2017;92:675-681. doi:10.1590 /abd1806-4841.201756411
- Darmstadt GL. Perianal lymphangioma circumscriptum mistaken for genital warts. Pediatrics 1996;98;461.
- Bruins FG, van Deudekom FJA, de Vries HJC. Syphilitic condylomata lata mimicking anogenital warts. BMJ. 2015;350:h1259. doi:10.1136 /bmj.h1259
- Asch S, Sugarman JL. Epidermal nevus syndromes: new insights into whorls and swirls. Pediatr Dermatol. 2018;35:21-29. doi:10.1111 /pde.13273
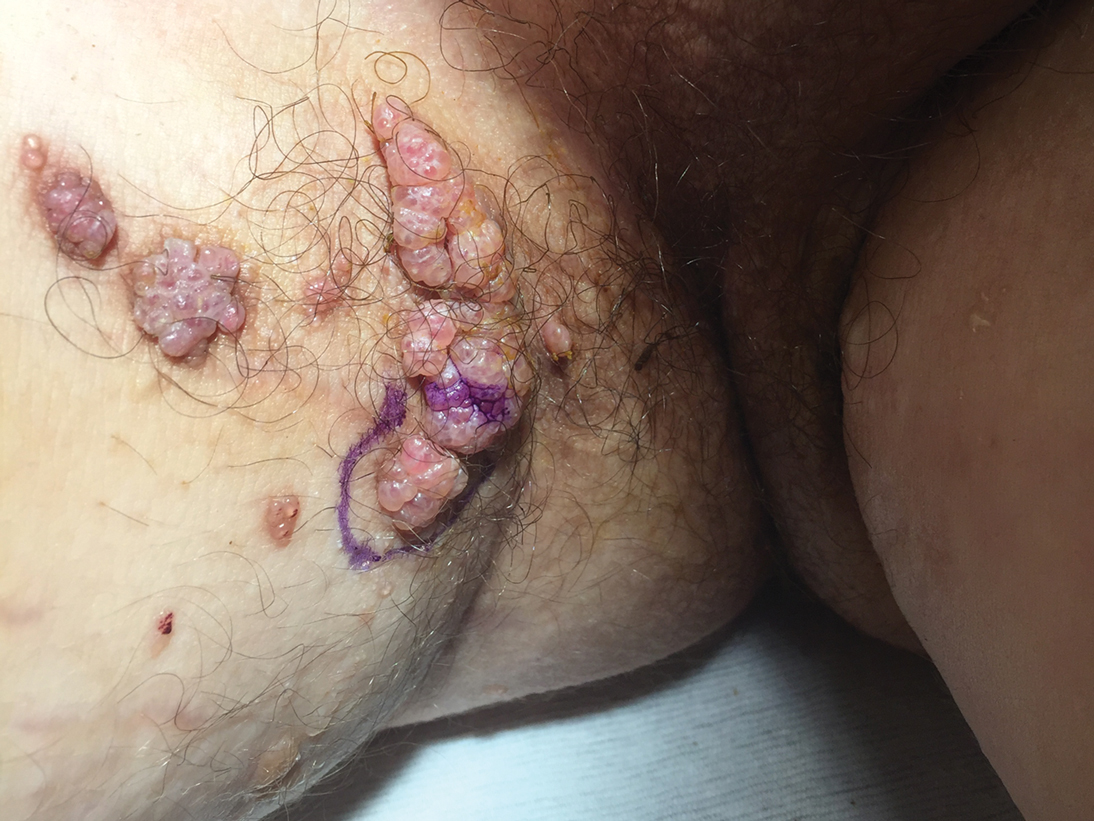
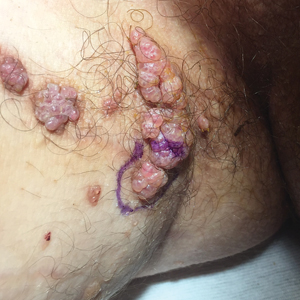
A 17-year-old adolescent boy presented with increasingly painful genital warts on the right thigh, groin, and scrotum that had been present since birth. The patient had a medical history of cardiac transplantation in the months prior to presentation and was on immunosuppressive therapy. The lesions had become more swollen and bothersome in the weeks following the transplantation and now prevented him from ambulating due to discomfort. He denied any history of sexual contact or oral lesions. Physical examination revealed numerous translucent and hemorrhagic vesicles clustered and linearly distributed on the right medial thigh. A shave biopsy of a vesicle was performed.
Verrucous Scalp Plaque and Widespread Eruption
The Diagnosis: Pemphigus Foliaceous
Laboratory workup including a complete blood cell count with differential, comprehensive metabolic panel, antinuclear antibodies, Sjögren syndrome A and B antibodies, hepatitis profile, rapid plasma reagin, HIV screen, aldolase, anti–Jo-1, T-Spot TB test (Quest Diagnostics), and tissue cultures was unremarkable. Two 4-mm punch biopsies were obtained from the left cheek and upper back, both of which demonstrated intragranular acantholysis suggestive of pemphigus foliaceous (Figure 1A). A subsequent punch biopsy from the right lower abdomen sent for direct immunofluorescence demonstrated netlike positivity of IgG and C3 in the upper epidermis (Figure 1B), and serum sent for indirect immunofluorescence demonstrated intercellular IgG antibodies to desmoglein (Dsg) 1 on monkey esophagus and positive Dsg-1 antibodies on enzyme-linked immunosorbent assay, confirming the diagnosis.
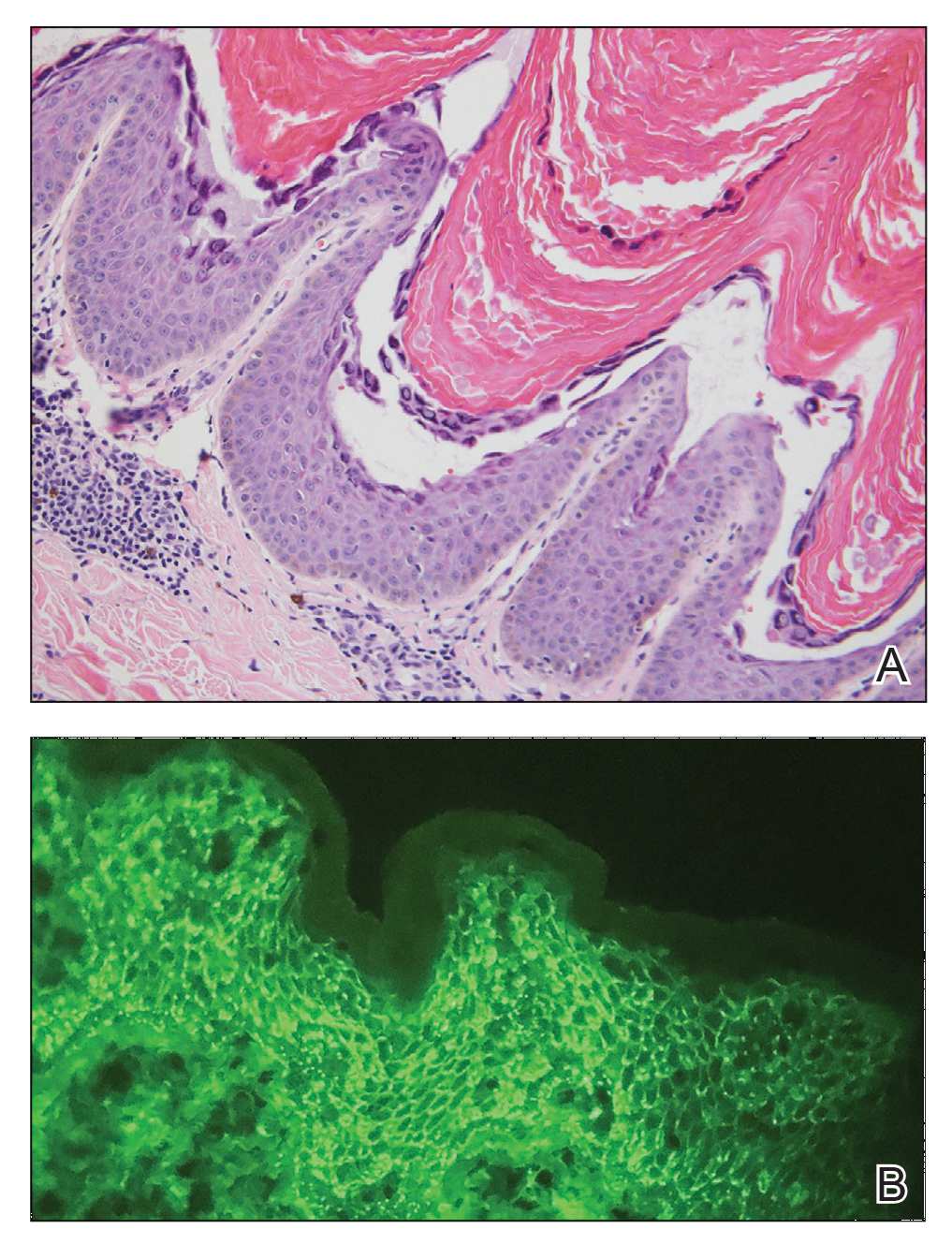
The patient was started on a 60-mg prednisone taper as well as dapsone 50 mg daily; the dapsone was titrated up to 100 mg daily. After tapering down to 10 mg daily of prednisone over 2 months and continuing dapsone with minimal improvement, he was given 2 infusions of rituximab 1000 mg 2 weeks apart. The scalp plaque was dramatically improved at 3-month follow-up (Figure 2), with partial improvement of the cheek plaques (Figure 3). Dapsone was increased to 150 mg daily, and he was encouraged to use triamcinolone acetonide ointment 0.1% twice daily, which led to further improvement.
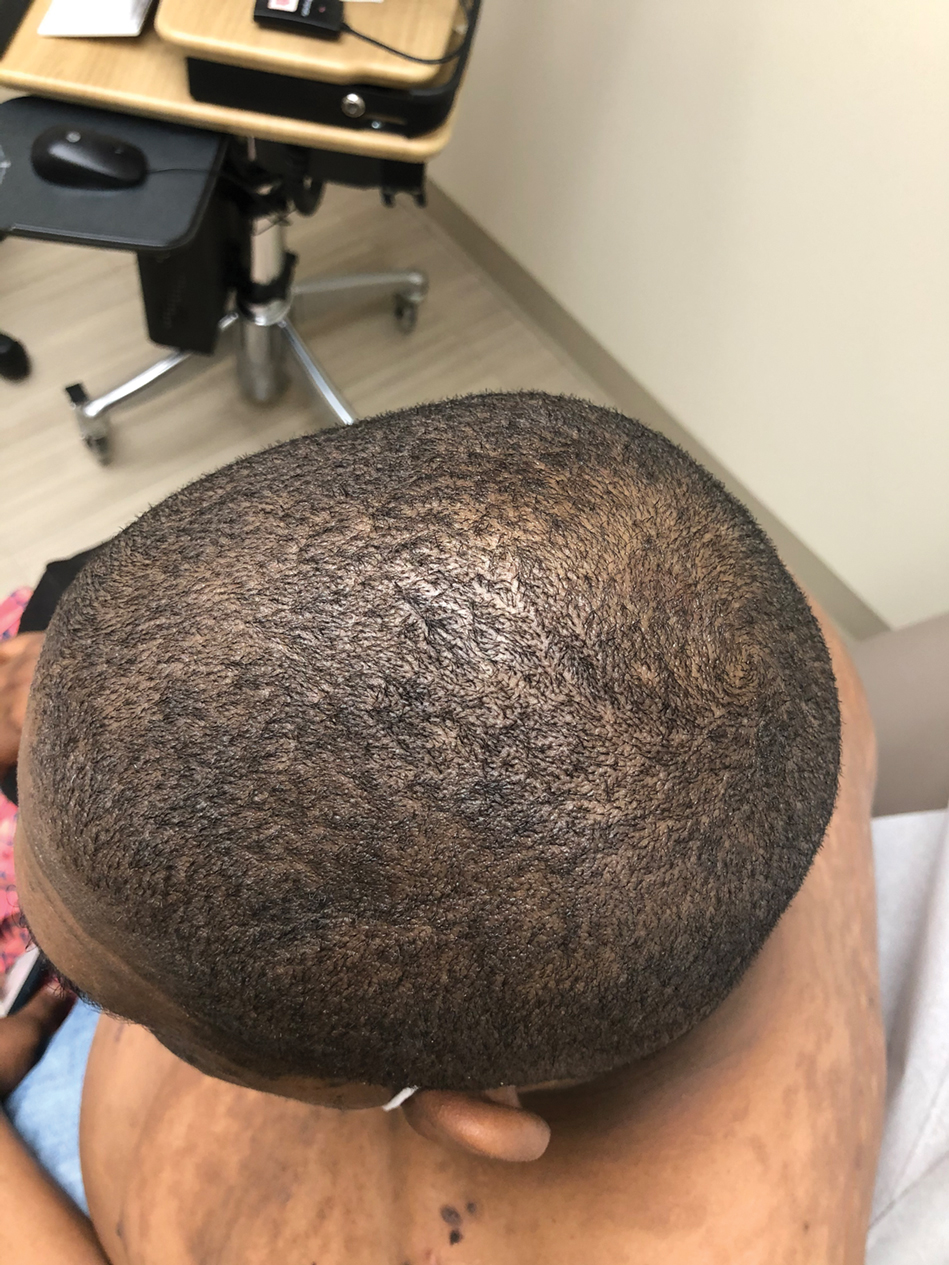
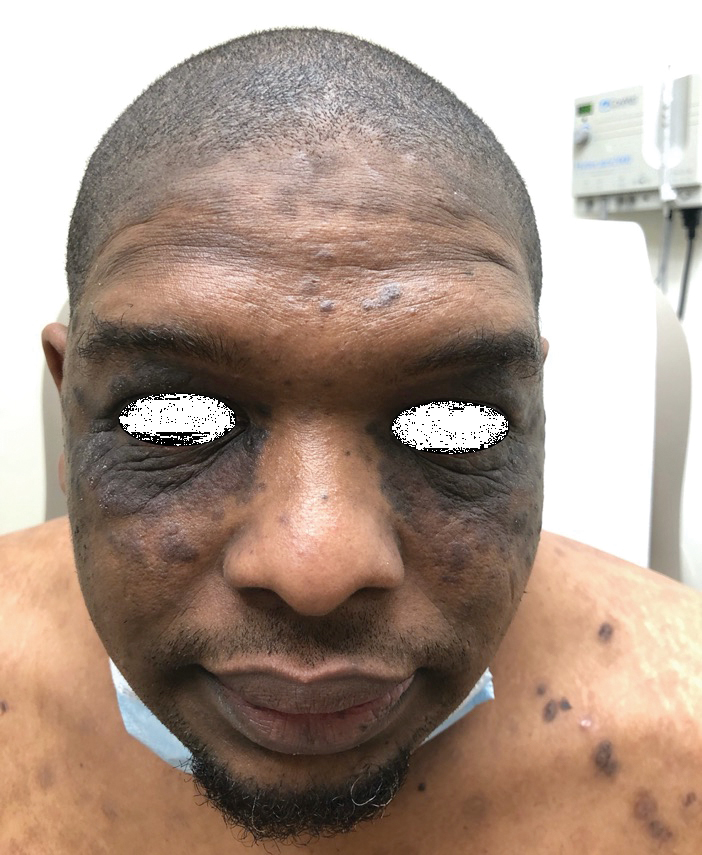
Pemphigus foliaceus is an autoimmune blistering disease that most commonly occurs in middle-aged adults. It generally is less common than pemphigus vulgaris, except in Finland, Tunisia, and Brazil, where there is an endemic condition with an identical clinical and histological presentation known as fogo selvagem.1
The pathogenesis of pemphigus foliaceous is characterized by IgG autoantibodies against Dsg-1, a transmembrane glycoprotein involved in the cellular adhesion of keratinocytes, which is preferentially expressed in the superficial epidermis.2-7 Dysfunction of Dsg-1 results in the separation of superficial epidermal cells, resulting in intraepidermal blisters.2,7 In contrast to pemphigus vulgaris, there typically is a lack of oral mucosal involvement due to compensation by Dsg-3 in the mucosa.4 Potential triggers for pemphigus foliaceous include exposure to UV radiation; radiotherapy; pregnancy; physiologic stress; and drugs, most commonly captopril, penicillamine, and thiols.8
Pemphigus foliaceous lesions clinically appear as eroded and crusted lesions on an erythematous base, commonly in a seborrheic distribution on the face, scalp, and trunk with sparing of the oral mucosa,2,6 but lesions can progress to a widespread and more severe exfoliative dermatitis.7 Lesions also can appear as psoriasiform plaques and often are initially misdiagnosed as psoriasis, particularly in patients with skin of color.9,10
Diagnosis of pemphigus foliaceous typically is made using a combination of histology as well as both direct and indirect immunofluorescence. Histologically, pemphigus foliaceus presents with subcorneal acantholysis, which is most prominent in the granular layer and occasionally the presence of neutrophils and eosinophils in the blister cavity.7 Direct immunofluorescence demonstrates netlike intercellular IgG and C3 in the upper portion of the epidermis.11 Indirect immunofluorescence can help detect circulating IgG antibodies to Dsg-1, with guinea pig esophagus being the ideal substrate.11,12
First-line treatment of pemphigus foliaceus consists of systemic glucocorticoid therapy, often administered with azathioprine, methotrexate, or mycophenolate mofetil.2,6,13 Although first-line treatment is effective in 60% to 80% of patients,2 relapsing cases can be treated with cyclophosphamide, intravenous immunoglobulin, immunoadsorption, plasmapheresis, or rituximab.2
Rituximab is a chimeric monoclonal antibody targeting CD20+ B cells, leading to decreased antibody production, which has been shown to be effective in treating severe and refractory cases of pemphigus foliaceus.6,13Rituximab with short-course prednisone has been found to be more effective in achieving complete remission at 24 months than prednisone alone.14 In patients with contraindications to systemic glucocorticoid therapy, rituximab has been shown as an effective first-line therapy.15 One-quarter of patients treated with rituximab relapsed within 2 years of treatment6 (average time to relapse, 6–26 months).16 High-dose rituximab regimens, along with a higher number of rituximab treatment cycles, have been shown to prolong time to relapse.6 Further, higher baseline levels of Dsg-1 antibody have been correlated to earlier relapse and can be used following rituximab therapy to monitor disease progression.6,16
The differential diagnosis for pemphigus foliaceous includes disseminated blastomycosis, hypertrophic lupus erythematosus, sebopsoriasis, and secondary syphilis. Disseminated blastomycosis presents with cutaneous manifestations such as nodules, papules, or pustules evolving over weeks to months into ulcers with subsequent scarring.17 Hypertrophic lupus erythematosus presents with papules and nodules with associated keratotic scaling on the face, palms, and extensor surfaces of the limbs.18 Sebopsoriasis is characterized by well-defined lesions with an overlying scale distributed on the scalp, face, and chest.19 Secondary syphilis presents as early hyperpigmented macules transitioning to acral papulosquamous lesions involving the palms and soles.20
- Hans-Filho G, Aoki V, Hans Bittner NR, et al. Fogo selvagem: endemic pemphigus foliaceus. An Bras Dermatol. 2018;93:638-650.
- Jenson KK, Burr DM, Edwards BC. Case report: reatment of refractory pemphigus foliaceus with rituximab. Practical Dermatology. February 2016:33-36. Accessed August 27, 2021. https://practicaldermatology.com/articles/2016-feb/case-report -treatment-of-refractory-pemphigus-foliaceus-with-rituximab -financial-matters-aad-asds-resources
- Amagai M, Hashimoto T, Green KJ, et al. Antigen-specific immunoadsorption of pathogenic autoantibodies in pemphigus foliaceus. J Invest Dermatol. 1995;104:895-901.
- Mahoney MG, Wang Z, Rothenberger K, et al. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461-468.
- Oktarina DAM, Sokol E, Kramer D, et al. Endocytosis of IgG, desmoglein 1, and plakoglobin in pemphigus foliaceus patient skin. Front Immunol. 2019;10:1-12.
- Kraft M, Worm M. Pemphigus foliaceus-repeated treatment with rituximab 7 years after initial response: a case report. Front Med. 2018;5:315.
- Hale EK. Pemphigus foliaceous. Dermatol Online J. 2002;8:9.
- Tavakolpour S. Pemphigus trigger factors: special focus on pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol Res. 2018;310:95-106.
- A boobaker J, Morar N, Ramdial PK, et al. Pemphigus in South Africa. Int J Dermatol. 2001;40:115-119.
- Austin E, Millsop JW, Ely H, et al. Psoriasiform pemphigus foliaceus in an African American female: an important clinical manifestation. J Drugs Dermatol. 2018;17:471.
- Arbache ST, Nogueira TG, Delgado L, et al. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-889.
- Sabolinski ML, Beutner EH, Krasny S, et al. Substrate specificity of antiepithelial antibodies of pemphigus vulgaris and pemphigus foliaceus sera in immunofluorescence tests on monkey and guinea pig esophagus sections. J Invest Dermatol. 1987;88:545-549.
- Palacios-Álvarez I, Riquelme-McLoughlin C, Curto-Barredo L, et al. Rituximab treatment of pemphigus foliaceus: a retrospective study of 12 patients. J Am Acad Dermatol. 2021;85:484-486.
- Murrell DF, Sprecher E. Rituximab and short-course prednisone as the new gold standard for new-onset pemphigus vulgaris and pemphigus foliaceus. Br J Dermatol. 2017;177:1143-1144.
- Gregoriou S, Efthymiou O, Stefanaki C, et al. Management of pemphigus vulgaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2015;8:521-527.
- Saleh MA. A prospective study comparing patients with early and late relapsing pemphigus treated with rituximab. J Am Acad Dermatol. 2018;79:97-103.
- Castillo CG, Kauffman CA, Miceli MH. Blastomycosis. Infect Dis Clin North Am. 2016;30:247-264.
- Herzum A, Gasparini G, Emanuele C, et al. Atypical and rare forms of cutaneous lupus erythematosus: the importance of the diagnosis for the best management of patients. Dermatology. 2013;1-10.
- Tull TJ, Noy M, Bunker CB, et al. Sebopsoriasis in patients with HIV: a case series of 20 patients. Br J Dermatol. 2016; 173:813-815.
- Balagula Y, Mattei P, Wisco OJ, et al. The great imitator revised: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
The Diagnosis: Pemphigus Foliaceous
Laboratory workup including a complete blood cell count with differential, comprehensive metabolic panel, antinuclear antibodies, Sjögren syndrome A and B antibodies, hepatitis profile, rapid plasma reagin, HIV screen, aldolase, anti–Jo-1, T-Spot TB test (Quest Diagnostics), and tissue cultures was unremarkable. Two 4-mm punch biopsies were obtained from the left cheek and upper back, both of which demonstrated intragranular acantholysis suggestive of pemphigus foliaceous (Figure 1A). A subsequent punch biopsy from the right lower abdomen sent for direct immunofluorescence demonstrated netlike positivity of IgG and C3 in the upper epidermis (Figure 1B), and serum sent for indirect immunofluorescence demonstrated intercellular IgG antibodies to desmoglein (Dsg) 1 on monkey esophagus and positive Dsg-1 antibodies on enzyme-linked immunosorbent assay, confirming the diagnosis.
The patient was started on a 60-mg prednisone taper as well as dapsone 50 mg daily; the dapsone was titrated up to 100 mg daily. After tapering down to 10 mg daily of prednisone over 2 months and continuing dapsone with minimal improvement, he was given 2 infusions of rituximab 1000 mg 2 weeks apart. The scalp plaque was dramatically improved at 3-month follow-up (Figure 2), with partial improvement of the cheek plaques (Figure 3). Dapsone was increased to 150 mg daily, and he was encouraged to use triamcinolone acetonide ointment 0.1% twice daily, which led to further improvement.
Pemphigus foliaceus is an autoimmune blistering disease that most commonly occurs in middle-aged adults. It generally is less common than pemphigus vulgaris, except in Finland, Tunisia, and Brazil, where there is an endemic condition with an identical clinical and histological presentation known as fogo selvagem.1
The pathogenesis of pemphigus foliaceous is characterized by IgG autoantibodies against Dsg-1, a transmembrane glycoprotein involved in the cellular adhesion of keratinocytes, which is preferentially expressed in the superficial epidermis.2-7 Dysfunction of Dsg-1 results in the separation of superficial epidermal cells, resulting in intraepidermal blisters.2,7 In contrast to pemphigus vulgaris, there typically is a lack of oral mucosal involvement due to compensation by Dsg-3 in the mucosa.4 Potential triggers for pemphigus foliaceous include exposure to UV radiation; radiotherapy; pregnancy; physiologic stress; and drugs, most commonly captopril, penicillamine, and thiols.8
Pemphigus foliaceous lesions clinically appear as eroded and crusted lesions on an erythematous base, commonly in a seborrheic distribution on the face, scalp, and trunk with sparing of the oral mucosa,2,6 but lesions can progress to a widespread and more severe exfoliative dermatitis.7 Lesions also can appear as psoriasiform plaques and often are initially misdiagnosed as psoriasis, particularly in patients with skin of color.9,10
Diagnosis of pemphigus foliaceous typically is made using a combination of histology as well as both direct and indirect immunofluorescence. Histologically, pemphigus foliaceus presents with subcorneal acantholysis, which is most prominent in the granular layer and occasionally the presence of neutrophils and eosinophils in the blister cavity.7 Direct immunofluorescence demonstrates netlike intercellular IgG and C3 in the upper portion of the epidermis.11 Indirect immunofluorescence can help detect circulating IgG antibodies to Dsg-1, with guinea pig esophagus being the ideal substrate.11,12
First-line treatment of pemphigus foliaceus consists of systemic glucocorticoid therapy, often administered with azathioprine, methotrexate, or mycophenolate mofetil.2,6,13 Although first-line treatment is effective in 60% to 80% of patients,2 relapsing cases can be treated with cyclophosphamide, intravenous immunoglobulin, immunoadsorption, plasmapheresis, or rituximab.2
Rituximab is a chimeric monoclonal antibody targeting CD20+ B cells, leading to decreased antibody production, which has been shown to be effective in treating severe and refractory cases of pemphigus foliaceus.6,13Rituximab with short-course prednisone has been found to be more effective in achieving complete remission at 24 months than prednisone alone.14 In patients with contraindications to systemic glucocorticoid therapy, rituximab has been shown as an effective first-line therapy.15 One-quarter of patients treated with rituximab relapsed within 2 years of treatment6 (average time to relapse, 6–26 months).16 High-dose rituximab regimens, along with a higher number of rituximab treatment cycles, have been shown to prolong time to relapse.6 Further, higher baseline levels of Dsg-1 antibody have been correlated to earlier relapse and can be used following rituximab therapy to monitor disease progression.6,16
The differential diagnosis for pemphigus foliaceous includes disseminated blastomycosis, hypertrophic lupus erythematosus, sebopsoriasis, and secondary syphilis. Disseminated blastomycosis presents with cutaneous manifestations such as nodules, papules, or pustules evolving over weeks to months into ulcers with subsequent scarring.17 Hypertrophic lupus erythematosus presents with papules and nodules with associated keratotic scaling on the face, palms, and extensor surfaces of the limbs.18 Sebopsoriasis is characterized by well-defined lesions with an overlying scale distributed on the scalp, face, and chest.19 Secondary syphilis presents as early hyperpigmented macules transitioning to acral papulosquamous lesions involving the palms and soles.20
The Diagnosis: Pemphigus Foliaceous
Laboratory workup including a complete blood cell count with differential, comprehensive metabolic panel, antinuclear antibodies, Sjögren syndrome A and B antibodies, hepatitis profile, rapid plasma reagin, HIV screen, aldolase, anti–Jo-1, T-Spot TB test (Quest Diagnostics), and tissue cultures was unremarkable. Two 4-mm punch biopsies were obtained from the left cheek and upper back, both of which demonstrated intragranular acantholysis suggestive of pemphigus foliaceous (Figure 1A). A subsequent punch biopsy from the right lower abdomen sent for direct immunofluorescence demonstrated netlike positivity of IgG and C3 in the upper epidermis (Figure 1B), and serum sent for indirect immunofluorescence demonstrated intercellular IgG antibodies to desmoglein (Dsg) 1 on monkey esophagus and positive Dsg-1 antibodies on enzyme-linked immunosorbent assay, confirming the diagnosis.
The patient was started on a 60-mg prednisone taper as well as dapsone 50 mg daily; the dapsone was titrated up to 100 mg daily. After tapering down to 10 mg daily of prednisone over 2 months and continuing dapsone with minimal improvement, he was given 2 infusions of rituximab 1000 mg 2 weeks apart. The scalp plaque was dramatically improved at 3-month follow-up (Figure 2), with partial improvement of the cheek plaques (Figure 3). Dapsone was increased to 150 mg daily, and he was encouraged to use triamcinolone acetonide ointment 0.1% twice daily, which led to further improvement.
Pemphigus foliaceus is an autoimmune blistering disease that most commonly occurs in middle-aged adults. It generally is less common than pemphigus vulgaris, except in Finland, Tunisia, and Brazil, where there is an endemic condition with an identical clinical and histological presentation known as fogo selvagem.1
The pathogenesis of pemphigus foliaceous is characterized by IgG autoantibodies against Dsg-1, a transmembrane glycoprotein involved in the cellular adhesion of keratinocytes, which is preferentially expressed in the superficial epidermis.2-7 Dysfunction of Dsg-1 results in the separation of superficial epidermal cells, resulting in intraepidermal blisters.2,7 In contrast to pemphigus vulgaris, there typically is a lack of oral mucosal involvement due to compensation by Dsg-3 in the mucosa.4 Potential triggers for pemphigus foliaceous include exposure to UV radiation; radiotherapy; pregnancy; physiologic stress; and drugs, most commonly captopril, penicillamine, and thiols.8
Pemphigus foliaceous lesions clinically appear as eroded and crusted lesions on an erythematous base, commonly in a seborrheic distribution on the face, scalp, and trunk with sparing of the oral mucosa,2,6 but lesions can progress to a widespread and more severe exfoliative dermatitis.7 Lesions also can appear as psoriasiform plaques and often are initially misdiagnosed as psoriasis, particularly in patients with skin of color.9,10
Diagnosis of pemphigus foliaceous typically is made using a combination of histology as well as both direct and indirect immunofluorescence. Histologically, pemphigus foliaceus presents with subcorneal acantholysis, which is most prominent in the granular layer and occasionally the presence of neutrophils and eosinophils in the blister cavity.7 Direct immunofluorescence demonstrates netlike intercellular IgG and C3 in the upper portion of the epidermis.11 Indirect immunofluorescence can help detect circulating IgG antibodies to Dsg-1, with guinea pig esophagus being the ideal substrate.11,12
First-line treatment of pemphigus foliaceus consists of systemic glucocorticoid therapy, often administered with azathioprine, methotrexate, or mycophenolate mofetil.2,6,13 Although first-line treatment is effective in 60% to 80% of patients,2 relapsing cases can be treated with cyclophosphamide, intravenous immunoglobulin, immunoadsorption, plasmapheresis, or rituximab.2
Rituximab is a chimeric monoclonal antibody targeting CD20+ B cells, leading to decreased antibody production, which has been shown to be effective in treating severe and refractory cases of pemphigus foliaceus.6,13Rituximab with short-course prednisone has been found to be more effective in achieving complete remission at 24 months than prednisone alone.14 In patients with contraindications to systemic glucocorticoid therapy, rituximab has been shown as an effective first-line therapy.15 One-quarter of patients treated with rituximab relapsed within 2 years of treatment6 (average time to relapse, 6–26 months).16 High-dose rituximab regimens, along with a higher number of rituximab treatment cycles, have been shown to prolong time to relapse.6 Further, higher baseline levels of Dsg-1 antibody have been correlated to earlier relapse and can be used following rituximab therapy to monitor disease progression.6,16
The differential diagnosis for pemphigus foliaceous includes disseminated blastomycosis, hypertrophic lupus erythematosus, sebopsoriasis, and secondary syphilis. Disseminated blastomycosis presents with cutaneous manifestations such as nodules, papules, or pustules evolving over weeks to months into ulcers with subsequent scarring.17 Hypertrophic lupus erythematosus presents with papules and nodules with associated keratotic scaling on the face, palms, and extensor surfaces of the limbs.18 Sebopsoriasis is characterized by well-defined lesions with an overlying scale distributed on the scalp, face, and chest.19 Secondary syphilis presents as early hyperpigmented macules transitioning to acral papulosquamous lesions involving the palms and soles.20
- Hans-Filho G, Aoki V, Hans Bittner NR, et al. Fogo selvagem: endemic pemphigus foliaceus. An Bras Dermatol. 2018;93:638-650.
- Jenson KK, Burr DM, Edwards BC. Case report: reatment of refractory pemphigus foliaceus with rituximab. Practical Dermatology. February 2016:33-36. Accessed August 27, 2021. https://practicaldermatology.com/articles/2016-feb/case-report -treatment-of-refractory-pemphigus-foliaceus-with-rituximab -financial-matters-aad-asds-resources
- Amagai M, Hashimoto T, Green KJ, et al. Antigen-specific immunoadsorption of pathogenic autoantibodies in pemphigus foliaceus. J Invest Dermatol. 1995;104:895-901.
- Mahoney MG, Wang Z, Rothenberger K, et al. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461-468.
- Oktarina DAM, Sokol E, Kramer D, et al. Endocytosis of IgG, desmoglein 1, and plakoglobin in pemphigus foliaceus patient skin. Front Immunol. 2019;10:1-12.
- Kraft M, Worm M. Pemphigus foliaceus-repeated treatment with rituximab 7 years after initial response: a case report. Front Med. 2018;5:315.
- Hale EK. Pemphigus foliaceous. Dermatol Online J. 2002;8:9.
- Tavakolpour S. Pemphigus trigger factors: special focus on pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol Res. 2018;310:95-106.
- A boobaker J, Morar N, Ramdial PK, et al. Pemphigus in South Africa. Int J Dermatol. 2001;40:115-119.
- Austin E, Millsop JW, Ely H, et al. Psoriasiform pemphigus foliaceus in an African American female: an important clinical manifestation. J Drugs Dermatol. 2018;17:471.
- Arbache ST, Nogueira TG, Delgado L, et al. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-889.
- Sabolinski ML, Beutner EH, Krasny S, et al. Substrate specificity of antiepithelial antibodies of pemphigus vulgaris and pemphigus foliaceus sera in immunofluorescence tests on monkey and guinea pig esophagus sections. J Invest Dermatol. 1987;88:545-549.
- Palacios-Álvarez I, Riquelme-McLoughlin C, Curto-Barredo L, et al. Rituximab treatment of pemphigus foliaceus: a retrospective study of 12 patients. J Am Acad Dermatol. 2021;85:484-486.
- Murrell DF, Sprecher E. Rituximab and short-course prednisone as the new gold standard for new-onset pemphigus vulgaris and pemphigus foliaceus. Br J Dermatol. 2017;177:1143-1144.
- Gregoriou S, Efthymiou O, Stefanaki C, et al. Management of pemphigus vulgaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2015;8:521-527.
- Saleh MA. A prospective study comparing patients with early and late relapsing pemphigus treated with rituximab. J Am Acad Dermatol. 2018;79:97-103.
- Castillo CG, Kauffman CA, Miceli MH. Blastomycosis. Infect Dis Clin North Am. 2016;30:247-264.
- Herzum A, Gasparini G, Emanuele C, et al. Atypical and rare forms of cutaneous lupus erythematosus: the importance of the diagnosis for the best management of patients. Dermatology. 2013;1-10.
- Tull TJ, Noy M, Bunker CB, et al. Sebopsoriasis in patients with HIV: a case series of 20 patients. Br J Dermatol. 2016; 173:813-815.
- Balagula Y, Mattei P, Wisco OJ, et al. The great imitator revised: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
- Hans-Filho G, Aoki V, Hans Bittner NR, et al. Fogo selvagem: endemic pemphigus foliaceus. An Bras Dermatol. 2018;93:638-650.
- Jenson KK, Burr DM, Edwards BC. Case report: reatment of refractory pemphigus foliaceus with rituximab. Practical Dermatology. February 2016:33-36. Accessed August 27, 2021. https://practicaldermatology.com/articles/2016-feb/case-report -treatment-of-refractory-pemphigus-foliaceus-with-rituximab -financial-matters-aad-asds-resources
- Amagai M, Hashimoto T, Green KJ, et al. Antigen-specific immunoadsorption of pathogenic autoantibodies in pemphigus foliaceus. J Invest Dermatol. 1995;104:895-901.
- Mahoney MG, Wang Z, Rothenberger K, et al. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461-468.
- Oktarina DAM, Sokol E, Kramer D, et al. Endocytosis of IgG, desmoglein 1, and plakoglobin in pemphigus foliaceus patient skin. Front Immunol. 2019;10:1-12.
- Kraft M, Worm M. Pemphigus foliaceus-repeated treatment with rituximab 7 years after initial response: a case report. Front Med. 2018;5:315.
- Hale EK. Pemphigus foliaceous. Dermatol Online J. 2002;8:9.
- Tavakolpour S. Pemphigus trigger factors: special focus on pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol Res. 2018;310:95-106.
- A boobaker J, Morar N, Ramdial PK, et al. Pemphigus in South Africa. Int J Dermatol. 2001;40:115-119.
- Austin E, Millsop JW, Ely H, et al. Psoriasiform pemphigus foliaceus in an African American female: an important clinical manifestation. J Drugs Dermatol. 2018;17:471.
- Arbache ST, Nogueira TG, Delgado L, et al. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-889.
- Sabolinski ML, Beutner EH, Krasny S, et al. Substrate specificity of antiepithelial antibodies of pemphigus vulgaris and pemphigus foliaceus sera in immunofluorescence tests on monkey and guinea pig esophagus sections. J Invest Dermatol. 1987;88:545-549.
- Palacios-Álvarez I, Riquelme-McLoughlin C, Curto-Barredo L, et al. Rituximab treatment of pemphigus foliaceus: a retrospective study of 12 patients. J Am Acad Dermatol. 2021;85:484-486.
- Murrell DF, Sprecher E. Rituximab and short-course prednisone as the new gold standard for new-onset pemphigus vulgaris and pemphigus foliaceus. Br J Dermatol. 2017;177:1143-1144.
- Gregoriou S, Efthymiou O, Stefanaki C, et al. Management of pemphigus vulgaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2015;8:521-527.
- Saleh MA. A prospective study comparing patients with early and late relapsing pemphigus treated with rituximab. J Am Acad Dermatol. 2018;79:97-103.
- Castillo CG, Kauffman CA, Miceli MH. Blastomycosis. Infect Dis Clin North Am. 2016;30:247-264.
- Herzum A, Gasparini G, Emanuele C, et al. Atypical and rare forms of cutaneous lupus erythematosus: the importance of the diagnosis for the best management of patients. Dermatology. 2013;1-10.
- Tull TJ, Noy M, Bunker CB, et al. Sebopsoriasis in patients with HIV: a case series of 20 patients. Br J Dermatol. 2016; 173:813-815.
- Balagula Y, Mattei P, Wisco OJ, et al. The great imitator revised: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
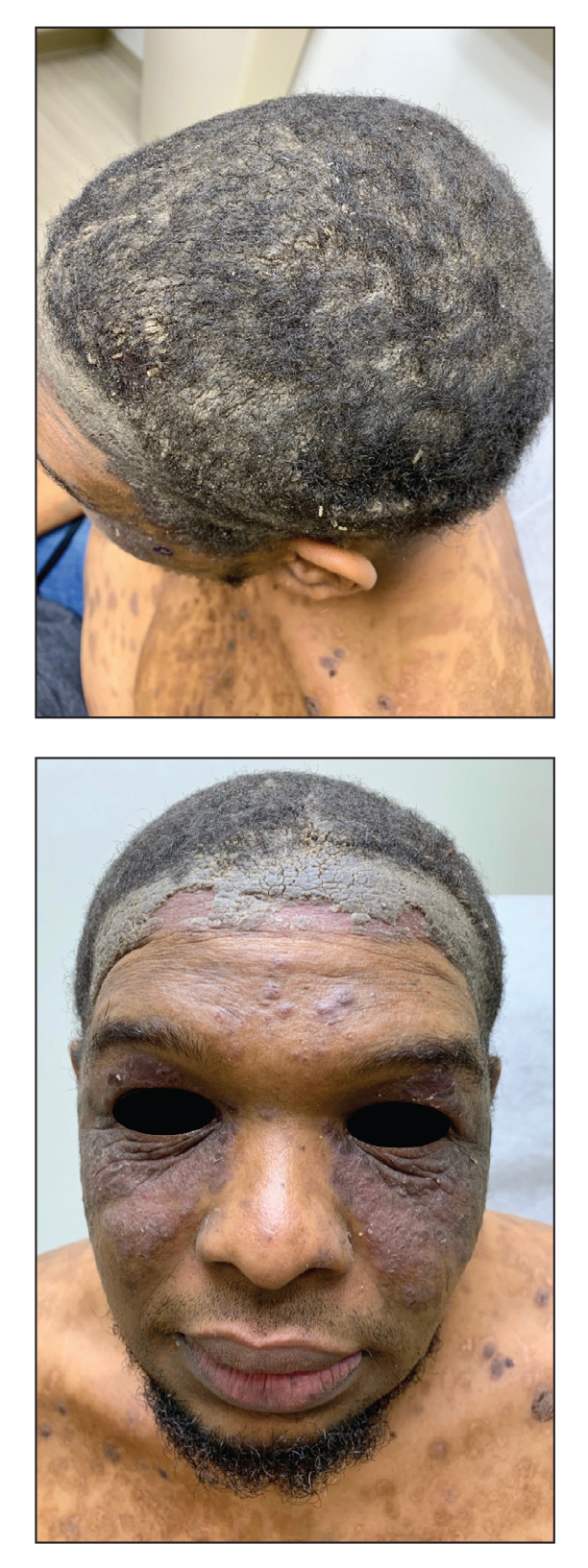
A 40-year-old Black man presented for evaluation of a thick plaque throughout the scalp (top), scaly plaques on the cheeks (bottom), and a spreading rash on the trunk that had progressed over the last few months. He had no relevant medical history, took no medications, and was in a monogamous relationship with a female partner. He previously saw an outside dermatologist who gave him triamcinolone cream, which was mildly helpful. Physical examination revealed a thick verrucous plaque throughout the scalp extending onto the forehead; thick plaques on the cheeks; and numerous, thinly eroded lesions on the trunk. Biopsies and a laboratory workup were performed.
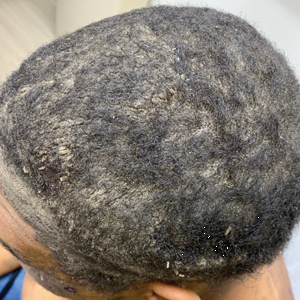
Sudden-Onset Blistering Rash
The Diagnosis: Generalized Bullous Fixed Drug Eruption
A punch biopsy from the left thigh revealed a vacuolar interface dermatitis with full-thickness necrosis of the epidermis and a patchy lichenoid inflammatory cell infiltrate in the superficial dermis consistent with a generalized bullous fixed drug eruption (GBFDE). The patient received supportive care and methylprednisolone with improvement of symptoms.
Generalized bullous fixed drug eruption is a rare, potentially life-threatening form of a fixed drug eruption (FDE), a cutaneous drug reaction that occurs in response to a causative medication. It typically presents with welldemarcated, dusky, erythematous patches or plaques that recur in the same sites with repeat exposure.1 The pathogenesis of FDE has been hypothesized to involve epidermal CD8+ T cells, which are activated by drug exposure and release cytotoxic molecules including Fas, Fas ligand, perforin, and granzyme B, resulting in lysis of the surrounding keratinocytes.1-3 Common eliciting drugs include nonsteroidal anti-inflammatory drugs, antibacterial agents (particularly trimethoprim-sulfamethoxazole), barbiturates, acetaminophen, and antimalarials.1 In addition to the findings seen in FDE, GBFDE is characterized by widespread bullous skin lesions.1-4 Typical histologic patterns seen in GBFDE are dispersed epidermal apoptotic keratinocytes, prominent dermal eosinophilic and lymphocytic infiltrates, and dermal melanophages.3 Discontinuing the causative agent and diligent prevention of re-exposure are the most important steps in management, as additional exposures can increase the number of lesions and overall severity. Symptoms typically resolve 7 to 14 days after drug discontinuation, often with postinflammatory hyperpigmentation.3
Generalized bullous fixed drug eruption presents a diagnostic challenge, as it sometimes involves the oral mucosa and can exhibit the Nikolsky sign. Thus, it often is confused with Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN).1,4 Stevens-Johnson syndrome and TEN are severe cutaneous drug eruptions that also can present with diffuse bullous skin lesions. Stevens-Johnson syndrome and TEN are thought to be a spectrum of the same disease that initially presents with dusky red macules that can coalesce, develop central blistering, and lead to skin detachment.5 Stevens-Johnson syndrome is defined as skin detachment of less than 10% body surface area (BSA); TEN is defined as skin detachment of more than 30% BSA. Stevens-Johnson syndrome/TEN overlap syndrome includes skin detachment of 10% to 30% BSA.5
Causative medications overlap substantially with GBFDE and include anticonvulsants, sulfa-containing drugs, antibiotics, nonsteroidal anti-inflammatory drugs, and uric acid–lowering agents. The histology of SJS/TEN also is quite similar to GBFDE, and these entities may be indistinguishable without clinical information.5 Lee et al1 found that absence of grouped necrotic keratinocytes (fire flag sign), deep inflammatory infiltrates, notable pigment incontinence, and higher eosinophil counts appear to be more common in GBFDE than SJS/TEN. Constitutional symptoms and mucosal involvement also were more frequent in SJS/TEN.
The timing of clinical presentation and medical history can be useful in differentiating between SJS/TEN and GBFDE. In SJS/TEN, drug exposure typically occurs 1 to 3 weeks before onset of symptoms vs 30 minutes to 24 hours in GBFDE.3 Additionally, a history of similar eruption in the same location is pathognomonic for GBFDE. Although GBFDE has been thought to have a better prognosis than SJS/TEN, more recent data suggest mortality rates may be similar.3 A case-control study found a mortality rate of 22% (13/58) in patients with GBFDE compared to 28% (n=170) in SJS/TEN patients.4
Erythema multiforme (EM) is an uncommon immunemediated disorder that typically presents as targetoid lesions with central epidermal necrosis in an acral distribution. Erythema multiforme can arise from a variety of factors, but up to 90% of cases are due to infection, most commonly herpes simplex virus; medications account for less than 10% of cases.6 Previously, EM has been thought to be on the same disease spectrum as SJS and TEN. It is now clear that EM is a separate entity with similar mucosal erosions but different cutaneous findings,6 mainly typical target lesions that differ from the atypical targets seen in SJS.
Staphylococcal scalded skin syndrome is a blistering skin disorder associated with local Staphylococcus aureus infection. It most commonly is seen in children and rarely occurs in adults who are not on dialysis. Some Staphylococcus strains produce exfoliative toxins A and B, which are serine proteases that target and cleave desmoglein 1, a mediator of keratinocyte adhesion. Staphylococcal scalded skin syndrome initially presents with erythema accentuated in the skin folds that becomes generalized. The disruption of keratinocyte adhesion leads to bullae formation in areas of erythema and diffuse sheetlike desquamation. Pathology reveals subcorneal rather than subepidermal blistering, which is seen in GBFDE and SJS/TEN. Treatment involves antistaphylococcal antibiotics and supportive care. With proper treatment, most cases resolve within 2 to 3 weeks.7
Mycoplasma pneumoniae–induced rash and mucositis presents with prominent mucositis and can have cutaneous findings of sparse vesiculobullous or targetoid eruption.8 Mycoplasma pneumoniae typically infects the lungs and is a leading cause of community-acquired pneumonia. However, a subset of patients can have extrapulmonary disease presenting as mucocutaneous eruptions, which is preceded by an approximately weeklong prodrome of fever, cough, and malaise.7 Mycoplasma pneumoniae–induced rash and mucositis also affect children and young patients and is more common in males.8
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15. doi:10.1016/j.dsi.2012.02.002
- Cho Y-T, Lin J-W, Chen Y-C, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548. doi:10.1016/j.jaad.2013.11.015
- Mitre V, Applebaum DS, Albahrani Y, et al. Generalized bullous fixed drug eruption imitating toxic epidermal necrolysis: a case report and literature review. Dermatol Online J. 2017;23: 13030/qt25v009gs.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with StevensJohnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732. doi:10.1111/bjd.12133
- Cho Y-T, Chu C-Y. Treatments for severe cutaneous adverse reactions [published online December 27, 2017]. J Immunol Res. doi:10.1155/2017/1503709
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902. doi:10.1111/j.1365-4632.2011.05348.x
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14:116-120.
- Canavan TN, Mathes EF, Frieden I, et al. Mycoplasma pneumoniae–induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72:239-245. doi:10.1016/j .jaad.2014.06.026
The Diagnosis: Generalized Bullous Fixed Drug Eruption
A punch biopsy from the left thigh revealed a vacuolar interface dermatitis with full-thickness necrosis of the epidermis and a patchy lichenoid inflammatory cell infiltrate in the superficial dermis consistent with a generalized bullous fixed drug eruption (GBFDE). The patient received supportive care and methylprednisolone with improvement of symptoms.
Generalized bullous fixed drug eruption is a rare, potentially life-threatening form of a fixed drug eruption (FDE), a cutaneous drug reaction that occurs in response to a causative medication. It typically presents with welldemarcated, dusky, erythematous patches or plaques that recur in the same sites with repeat exposure.1 The pathogenesis of FDE has been hypothesized to involve epidermal CD8+ T cells, which are activated by drug exposure and release cytotoxic molecules including Fas, Fas ligand, perforin, and granzyme B, resulting in lysis of the surrounding keratinocytes.1-3 Common eliciting drugs include nonsteroidal anti-inflammatory drugs, antibacterial agents (particularly trimethoprim-sulfamethoxazole), barbiturates, acetaminophen, and antimalarials.1 In addition to the findings seen in FDE, GBFDE is characterized by widespread bullous skin lesions.1-4 Typical histologic patterns seen in GBFDE are dispersed epidermal apoptotic keratinocytes, prominent dermal eosinophilic and lymphocytic infiltrates, and dermal melanophages.3 Discontinuing the causative agent and diligent prevention of re-exposure are the most important steps in management, as additional exposures can increase the number of lesions and overall severity. Symptoms typically resolve 7 to 14 days after drug discontinuation, often with postinflammatory hyperpigmentation.3
Generalized bullous fixed drug eruption presents a diagnostic challenge, as it sometimes involves the oral mucosa and can exhibit the Nikolsky sign. Thus, it often is confused with Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN).1,4 Stevens-Johnson syndrome and TEN are severe cutaneous drug eruptions that also can present with diffuse bullous skin lesions. Stevens-Johnson syndrome and TEN are thought to be a spectrum of the same disease that initially presents with dusky red macules that can coalesce, develop central blistering, and lead to skin detachment.5 Stevens-Johnson syndrome is defined as skin detachment of less than 10% body surface area (BSA); TEN is defined as skin detachment of more than 30% BSA. Stevens-Johnson syndrome/TEN overlap syndrome includes skin detachment of 10% to 30% BSA.5
Causative medications overlap substantially with GBFDE and include anticonvulsants, sulfa-containing drugs, antibiotics, nonsteroidal anti-inflammatory drugs, and uric acid–lowering agents. The histology of SJS/TEN also is quite similar to GBFDE, and these entities may be indistinguishable without clinical information.5 Lee et al1 found that absence of grouped necrotic keratinocytes (fire flag sign), deep inflammatory infiltrates, notable pigment incontinence, and higher eosinophil counts appear to be more common in GBFDE than SJS/TEN. Constitutional symptoms and mucosal involvement also were more frequent in SJS/TEN.
The timing of clinical presentation and medical history can be useful in differentiating between SJS/TEN and GBFDE. In SJS/TEN, drug exposure typically occurs 1 to 3 weeks before onset of symptoms vs 30 minutes to 24 hours in GBFDE.3 Additionally, a history of similar eruption in the same location is pathognomonic for GBFDE. Although GBFDE has been thought to have a better prognosis than SJS/TEN, more recent data suggest mortality rates may be similar.3 A case-control study found a mortality rate of 22% (13/58) in patients with GBFDE compared to 28% (n=170) in SJS/TEN patients.4
Erythema multiforme (EM) is an uncommon immunemediated disorder that typically presents as targetoid lesions with central epidermal necrosis in an acral distribution. Erythema multiforme can arise from a variety of factors, but up to 90% of cases are due to infection, most commonly herpes simplex virus; medications account for less than 10% of cases.6 Previously, EM has been thought to be on the same disease spectrum as SJS and TEN. It is now clear that EM is a separate entity with similar mucosal erosions but different cutaneous findings,6 mainly typical target lesions that differ from the atypical targets seen in SJS.
Staphylococcal scalded skin syndrome is a blistering skin disorder associated with local Staphylococcus aureus infection. It most commonly is seen in children and rarely occurs in adults who are not on dialysis. Some Staphylococcus strains produce exfoliative toxins A and B, which are serine proteases that target and cleave desmoglein 1, a mediator of keratinocyte adhesion. Staphylococcal scalded skin syndrome initially presents with erythema accentuated in the skin folds that becomes generalized. The disruption of keratinocyte adhesion leads to bullae formation in areas of erythema and diffuse sheetlike desquamation. Pathology reveals subcorneal rather than subepidermal blistering, which is seen in GBFDE and SJS/TEN. Treatment involves antistaphylococcal antibiotics and supportive care. With proper treatment, most cases resolve within 2 to 3 weeks.7
Mycoplasma pneumoniae–induced rash and mucositis presents with prominent mucositis and can have cutaneous findings of sparse vesiculobullous or targetoid eruption.8 Mycoplasma pneumoniae typically infects the lungs and is a leading cause of community-acquired pneumonia. However, a subset of patients can have extrapulmonary disease presenting as mucocutaneous eruptions, which is preceded by an approximately weeklong prodrome of fever, cough, and malaise.7 Mycoplasma pneumoniae–induced rash and mucositis also affect children and young patients and is more common in males.8
The Diagnosis: Generalized Bullous Fixed Drug Eruption
A punch biopsy from the left thigh revealed a vacuolar interface dermatitis with full-thickness necrosis of the epidermis and a patchy lichenoid inflammatory cell infiltrate in the superficial dermis consistent with a generalized bullous fixed drug eruption (GBFDE). The patient received supportive care and methylprednisolone with improvement of symptoms.
Generalized bullous fixed drug eruption is a rare, potentially life-threatening form of a fixed drug eruption (FDE), a cutaneous drug reaction that occurs in response to a causative medication. It typically presents with welldemarcated, dusky, erythematous patches or plaques that recur in the same sites with repeat exposure.1 The pathogenesis of FDE has been hypothesized to involve epidermal CD8+ T cells, which are activated by drug exposure and release cytotoxic molecules including Fas, Fas ligand, perforin, and granzyme B, resulting in lysis of the surrounding keratinocytes.1-3 Common eliciting drugs include nonsteroidal anti-inflammatory drugs, antibacterial agents (particularly trimethoprim-sulfamethoxazole), barbiturates, acetaminophen, and antimalarials.1 In addition to the findings seen in FDE, GBFDE is characterized by widespread bullous skin lesions.1-4 Typical histologic patterns seen in GBFDE are dispersed epidermal apoptotic keratinocytes, prominent dermal eosinophilic and lymphocytic infiltrates, and dermal melanophages.3 Discontinuing the causative agent and diligent prevention of re-exposure are the most important steps in management, as additional exposures can increase the number of lesions and overall severity. Symptoms typically resolve 7 to 14 days after drug discontinuation, often with postinflammatory hyperpigmentation.3
Generalized bullous fixed drug eruption presents a diagnostic challenge, as it sometimes involves the oral mucosa and can exhibit the Nikolsky sign. Thus, it often is confused with Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN).1,4 Stevens-Johnson syndrome and TEN are severe cutaneous drug eruptions that also can present with diffuse bullous skin lesions. Stevens-Johnson syndrome and TEN are thought to be a spectrum of the same disease that initially presents with dusky red macules that can coalesce, develop central blistering, and lead to skin detachment.5 Stevens-Johnson syndrome is defined as skin detachment of less than 10% body surface area (BSA); TEN is defined as skin detachment of more than 30% BSA. Stevens-Johnson syndrome/TEN overlap syndrome includes skin detachment of 10% to 30% BSA.5
Causative medications overlap substantially with GBFDE and include anticonvulsants, sulfa-containing drugs, antibiotics, nonsteroidal anti-inflammatory drugs, and uric acid–lowering agents. The histology of SJS/TEN also is quite similar to GBFDE, and these entities may be indistinguishable without clinical information.5 Lee et al1 found that absence of grouped necrotic keratinocytes (fire flag sign), deep inflammatory infiltrates, notable pigment incontinence, and higher eosinophil counts appear to be more common in GBFDE than SJS/TEN. Constitutional symptoms and mucosal involvement also were more frequent in SJS/TEN.
The timing of clinical presentation and medical history can be useful in differentiating between SJS/TEN and GBFDE. In SJS/TEN, drug exposure typically occurs 1 to 3 weeks before onset of symptoms vs 30 minutes to 24 hours in GBFDE.3 Additionally, a history of similar eruption in the same location is pathognomonic for GBFDE. Although GBFDE has been thought to have a better prognosis than SJS/TEN, more recent data suggest mortality rates may be similar.3 A case-control study found a mortality rate of 22% (13/58) in patients with GBFDE compared to 28% (n=170) in SJS/TEN patients.4
Erythema multiforme (EM) is an uncommon immunemediated disorder that typically presents as targetoid lesions with central epidermal necrosis in an acral distribution. Erythema multiforme can arise from a variety of factors, but up to 90% of cases are due to infection, most commonly herpes simplex virus; medications account for less than 10% of cases.6 Previously, EM has been thought to be on the same disease spectrum as SJS and TEN. It is now clear that EM is a separate entity with similar mucosal erosions but different cutaneous findings,6 mainly typical target lesions that differ from the atypical targets seen in SJS.
Staphylococcal scalded skin syndrome is a blistering skin disorder associated with local Staphylococcus aureus infection. It most commonly is seen in children and rarely occurs in adults who are not on dialysis. Some Staphylococcus strains produce exfoliative toxins A and B, which are serine proteases that target and cleave desmoglein 1, a mediator of keratinocyte adhesion. Staphylococcal scalded skin syndrome initially presents with erythema accentuated in the skin folds that becomes generalized. The disruption of keratinocyte adhesion leads to bullae formation in areas of erythema and diffuse sheetlike desquamation. Pathology reveals subcorneal rather than subepidermal blistering, which is seen in GBFDE and SJS/TEN. Treatment involves antistaphylococcal antibiotics and supportive care. With proper treatment, most cases resolve within 2 to 3 weeks.7
Mycoplasma pneumoniae–induced rash and mucositis presents with prominent mucositis and can have cutaneous findings of sparse vesiculobullous or targetoid eruption.8 Mycoplasma pneumoniae typically infects the lungs and is a leading cause of community-acquired pneumonia. However, a subset of patients can have extrapulmonary disease presenting as mucocutaneous eruptions, which is preceded by an approximately weeklong prodrome of fever, cough, and malaise.7 Mycoplasma pneumoniae–induced rash and mucositis also affect children and young patients and is more common in males.8
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15. doi:10.1016/j.dsi.2012.02.002
- Cho Y-T, Lin J-W, Chen Y-C, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548. doi:10.1016/j.jaad.2013.11.015
- Mitre V, Applebaum DS, Albahrani Y, et al. Generalized bullous fixed drug eruption imitating toxic epidermal necrolysis: a case report and literature review. Dermatol Online J. 2017;23: 13030/qt25v009gs.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with StevensJohnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732. doi:10.1111/bjd.12133
- Cho Y-T, Chu C-Y. Treatments for severe cutaneous adverse reactions [published online December 27, 2017]. J Immunol Res. doi:10.1155/2017/1503709
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902. doi:10.1111/j.1365-4632.2011.05348.x
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14:116-120.
- Canavan TN, Mathes EF, Frieden I, et al. Mycoplasma pneumoniae–induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72:239-245. doi:10.1016/j .jaad.2014.06.026
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15. doi:10.1016/j.dsi.2012.02.002
- Cho Y-T, Lin J-W, Chen Y-C, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548. doi:10.1016/j.jaad.2013.11.015
- Mitre V, Applebaum DS, Albahrani Y, et al. Generalized bullous fixed drug eruption imitating toxic epidermal necrolysis: a case report and literature review. Dermatol Online J. 2017;23: 13030/qt25v009gs.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with StevensJohnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732. doi:10.1111/bjd.12133
- Cho Y-T, Chu C-Y. Treatments for severe cutaneous adverse reactions [published online December 27, 2017]. J Immunol Res. doi:10.1155/2017/1503709
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902. doi:10.1111/j.1365-4632.2011.05348.x
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14:116-120.
- Canavan TN, Mathes EF, Frieden I, et al. Mycoplasma pneumoniae–induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72:239-245. doi:10.1016/j .jaad.2014.06.026
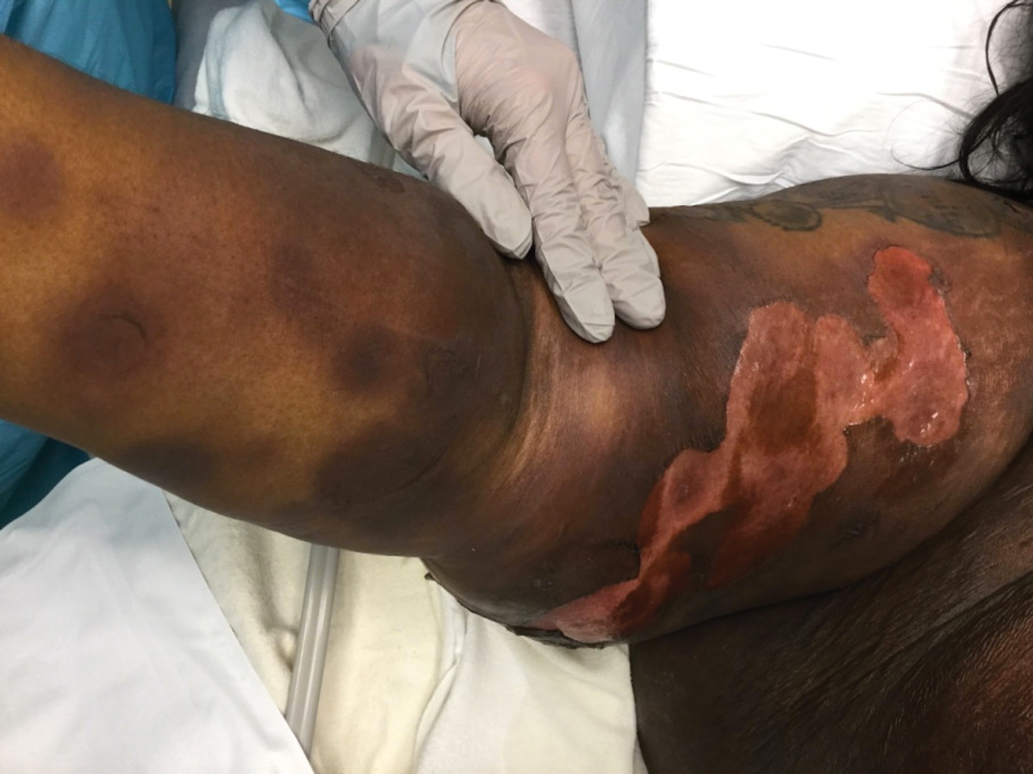
A 45-year-old woman presented with a diffuse rash 2 days after receiving ondansetron. She developed blisters on the arms, legs, trunk, and face 2 hours after exposure. There was no oral or vaginal involvement. She reported a history of leg blisters after prior exposure to ondansetron that were not as severe or numerous as the current episode. Physical examination revealed innumerable coalescing, ovoid and circular, dusky patches, some with central flaccid bullae, along with large areas of denuded skin on the trunk, arms, legs, and face. There were erosions on the lower eyelids without conjunctival or other mucosal involvement.
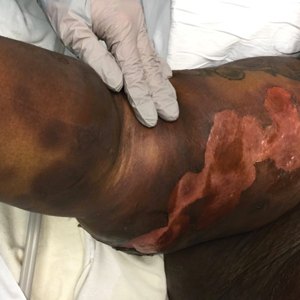
Ulcerated and Verrucous Plaque on the Chest
The Diagnosis: Disseminated Coccidioidomycosis
A6-mm punch biopsy was performed at the periphery of the ulcerated cutaneous lesion on the chest revealing extensive spherules. Serum antibody immunodiffusion for histoplasmosis and blastomycoses both were negative; however, B-D-glucan assay was positive at 364 pg/mL (reference range: <60 pg/mL, negative). Initial HIV-1 and HIV-2 antibody and antigen testing was negative as well as repeat testing at 3 weeks. Immunodiffusion for Coccidioides IgM and IgG was positive, and cocci antibody IgG complement fixation assays were positive at titers of 1:64 (reference range: <1:2, negative). A computed tomography needle-guided biopsy of the paravertebral soft tissue was performed. Gram stains and bacterial cultures of the biopsies were negative; however, fungal cultures were notable for growth of Coccidioides. Given the pertinent testing, a diagnosis of disseminated coccidioidomycosis was made.
Cutaneous coccidioidomycosis can occur in 3 situations: direct inoculation (primary cutaneous coccidioidomycosis), disseminated infection (disseminated cutaneous coccidioidomycosis), or as a reactive component of pulmonary infection.1,2 Of them, primary and disseminated cutaneous coccidioidomycosis are organism specific and display characteristic spherules and fungus on histopathology and cultures, respectively. Reactive coccidioidomycosis differs from organism-specific disease, as it does not contain spherules in histopathologic sections of tissue biopsies.1 Reactive skin manifestations occur in 12% to 50% of patients with primary pulmonary infection and include erythema nodosum, erythema multiforme, acute generalized exanthema, reactive interstitial granulomatous dermatitis, and Sweet syndrome.3
Organism-specific cutaneous coccidioidomycosis most often is correlated with hematogenous dissemination of primary pulmonary disease rather than direct inoculation of skin.1 The skin is the most common site of extrapulmonary involvement in disseminated coccidioidomycosis, and cutaneous lesions have been reported in 15% to 67% of cases of disseminated disease.1,4 In cutaneous disseminated disease, nodules, papules, macules, and verrucous plaques have been described. In a case series of disseminated cutaneous coccidioidomycosis, nodules were the most common cutaneous presentation and occurred in 39% (7/18) of patients, while verrucous plaques were the rarest and occurred in only 6% (1/18) of patients.5
The rate of coccidioidomycosis dissemination varies based on exposure and patient characteristics. Increased rates of dissemination have been reported in patients of African and Filipino descent, along with individuals that are immunosuppressed due to disease or medical therapy. Dissemination is clinically significant, as patients with multifocal dissemination have a greater than 50% risk for mortality.6
Disseminated coccidioidomycosis is a relatively rare manifestation of Coccidioides infection; approximately 1.6% of patients exposed to and infected with Coccidioides ultimately will develop systemic or disseminated disease.7,8 Although the rates of primary pulmonary infection are similar between patients of varying ethnicities, the rates of dissemination are higher in patients of African and Filipino ethnicity.8 In population studies of coccidioidomycosis (N=332), Black patients represented 33.3% (4/12) of disseminated cases but only 8.7% of Coccidioides cases overall.7
Population studies of Black patients with coccidioidomycosis have shown a 4-fold higher predisposition for severe disease compared to mild disease.9 Spondylitis and meningitis also are disproportionately more common in Black patients.8 Black patients comprised 75% of all spondylitis cases in a cohort where only 25% of patients were Black. Additionally, 33% of all meningitis cases occurred in Black patients in a cohort where 8% of total cases were Black patients.8 Within the United States, the highest rates of coccidioidomycosis meningitis are seen in Black patients.10
The pathophysiology underlying the increased susceptibility of individuals of African or Filipino descent to disseminated and severe coccidioidomycosis remains unknown.8 The level of vulnerability within this patient population has no association with increased environmental exposure or poor immunologic response to Coccidioides, as demonstrated by the ability of these populations to respond to experimental vaccination and skin testing (spherulin, coccidioidin) to a similar extent as other ethnicities.8 Class II HLA-DRB1*1301 alleles have been associated with an increased risk for severe disseminated Coccidioides infection regardless of ethnicity; however, these alleles are not overrepresented in these patient populations.8
In patients with primary pulmonary coccidioidomycosis with no evidence of dissemination, guidelines generally recommend offering treatment to groups at high risk of dissemination, such as pregnant women and patients with diabetes mellitus. Given the high incidence of disseminated and severe disease in Black and Filipino patients, some guidelines endorse treatment of all cases of coccidioidomycosis in this patient population.8 No current data are available to help determine whether this broad treatment approach reduces the development of disseminated infection in these populations. Frequent monitoring for disease progression and/or dissemination involving clinical and laboratory reevaluation every 3 months for 2 years is highly recommended.8
Treatment generally is based on location and severity of infection, with disseminated nonmeningeal infection being treated with oral azole therapy (ketoconazole, itraconazole, or fluconazole).11 If there is involvement of the central nervous system structures or rapidly worsening disease despite azole therapy, amphotericin B is recommended at 0.5 to 0.7 mg/kg daily. In patients with disseminated meningeal infection, oral fluconazole (800–1000 mg/d) or a combination of an azole with intrathecal amphotericin B (0.01–1.5 mg/dose, interval ranging from daily to 1 week) is recommended to improve response.11
The differential diagnosis of cutaneous disseminated coccidioidomycosis is broad and includes other systemic endemic mycoses (histoplasmosis, blastomycosis) and infections (mycobacteria, leishmania). Lupus vulgaris, a form of cutaneous tuberculosis, presents as a palpable tubercular lesion that may coalesce into erythematous plaques, which may mimic endemic mycoses, especially in patients with risk factors for both infectious etiologies such as our patient.12 Disseminated histoplasmosis may present as polymorphic plaques, pustules, nodules, and ulcerated skin lesions, whereas disseminated blastomycosis characteristically presents as a crusted verrucous lesion with raised borders and painful ulcers, both of which may mimic coccidioidomycosis.13 Biopsy would reveal the characteristic intracellular yeast in Histoplasma capsulatum and broad-based budding yeast form of Blastomyces dermatitidis in histoplasmosis and blastomycosis, respectively, in contrast to the spherules seen in our patient’s biopsy.13 Localized cutaneous leishmaniasis initially develops as a nodular or papular lesion and can progress to open ulcerations with raised borders. Biopsy and histopathology would reveal round protozoal amastigotes.14 Other diagnoses that should be considered include mycetoma, nocardiosis, and sporotrichosis.15 As the cutaneous manifestations of Coccidioides infections are varied, a broad differential diagnosis should be maintained, and probable environmental and infectious exposures should be considered prior to ordering diagnostic studies.
- Garcia Garcia SC, Salas Alanis JC, Flores MG, et al. Coccidioidomycosis and the skin: a comprehensive review. An Bras Dermatol. 2015; 90:610-619.
- DiCaudo DJ. Coccidioidomycosis: a review and update. J Am Acad Dermatol. 2006;55:929-942; quiz 943-925.
- DiCaudo DJ, Yiannias JA, Laman SD, et al. The exanthem of acute pulmonary coccidioidomycosis: clinical and histopathologic features of 3 cases and review of the literature. Arch Dermatol. 2006;142:744-746.
- Blair JE. State-of-the-art treatment of coccidioidomycosis: skin and soft-tissue infections. Ann N Y Acad Sci. 2007;1111:411-421.
- Crum NF, Lederman ER, Stafford CM, et al. Coccidioidomycosis: a descriptive survey of a reemerging disease. clinical characteristics and current controversies. Medicine (Baltimore). 2004;83:149-175.
- Borchers AT, Gershwin ME. The immune response in coccidioidomycosis. Autoimmun Rev. 2010;10:94-102.
- Smith CE, Beard RR. Varieties of coccidioidal infection in relation to the epidemiology and control of the diseases. Am J Public Health Nations Health. 1946;36:1394-1402.
- Ruddy BE, Mayer AP, Ko MG, et al. Coccidioidomycosis in African Americans. Mayo Clin Proc. 2011;86:63-69.
- Louie L, Ng S, Hajjeh R, et al. Influence of host genetics on the severity of coccidioidomycosis. Emerg Infect Dis. 1999;5:672-680.
- McCotter OZ, Benedict K, Engelthaler DM, et al. Update on the epidemiology of coccidioidomycosis in the United States. Med Mycol. 2019;57(suppl 1):S30-S40.
- Galgiani JN, Ampel NM, Catanzaro A, et al. Practice guideline for the treatment of coccidioidomycosis. Infectious Diseases Society of America. Clin Infect Dis. 2000;30:658-661.
- Khadka P, Koirala S, Thapaliya J. Cutaneous tuberculosis: clinicopathologic arrays and diagnostic challenges. Dermatol Res Pract. 2018;2018:7201973.
- Smith JA, Riddell JT, Kauffman CA. Cutaneous manifestations of endemic mycoses. Curr Infect Dis Rep. 2013;15:440-449.
- Scorza BM, Carvalho EM, Wilson ME. Cutaneous manifestations of human and murine leishmaniasis. Int J Mol Sci. 2017;18:1296.
The Diagnosis: Disseminated Coccidioidomycosis
A6-mm punch biopsy was performed at the periphery of the ulcerated cutaneous lesion on the chest revealing extensive spherules. Serum antibody immunodiffusion for histoplasmosis and blastomycoses both were negative; however, B-D-glucan assay was positive at 364 pg/mL (reference range: <60 pg/mL, negative). Initial HIV-1 and HIV-2 antibody and antigen testing was negative as well as repeat testing at 3 weeks. Immunodiffusion for Coccidioides IgM and IgG was positive, and cocci antibody IgG complement fixation assays were positive at titers of 1:64 (reference range: <1:2, negative). A computed tomography needle-guided biopsy of the paravertebral soft tissue was performed. Gram stains and bacterial cultures of the biopsies were negative; however, fungal cultures were notable for growth of Coccidioides. Given the pertinent testing, a diagnosis of disseminated coccidioidomycosis was made.
Cutaneous coccidioidomycosis can occur in 3 situations: direct inoculation (primary cutaneous coccidioidomycosis), disseminated infection (disseminated cutaneous coccidioidomycosis), or as a reactive component of pulmonary infection.1,2 Of them, primary and disseminated cutaneous coccidioidomycosis are organism specific and display characteristic spherules and fungus on histopathology and cultures, respectively. Reactive coccidioidomycosis differs from organism-specific disease, as it does not contain spherules in histopathologic sections of tissue biopsies.1 Reactive skin manifestations occur in 12% to 50% of patients with primary pulmonary infection and include erythema nodosum, erythema multiforme, acute generalized exanthema, reactive interstitial granulomatous dermatitis, and Sweet syndrome.3
Organism-specific cutaneous coccidioidomycosis most often is correlated with hematogenous dissemination of primary pulmonary disease rather than direct inoculation of skin.1 The skin is the most common site of extrapulmonary involvement in disseminated coccidioidomycosis, and cutaneous lesions have been reported in 15% to 67% of cases of disseminated disease.1,4 In cutaneous disseminated disease, nodules, papules, macules, and verrucous plaques have been described. In a case series of disseminated cutaneous coccidioidomycosis, nodules were the most common cutaneous presentation and occurred in 39% (7/18) of patients, while verrucous plaques were the rarest and occurred in only 6% (1/18) of patients.5
The rate of coccidioidomycosis dissemination varies based on exposure and patient characteristics. Increased rates of dissemination have been reported in patients of African and Filipino descent, along with individuals that are immunosuppressed due to disease or medical therapy. Dissemination is clinically significant, as patients with multifocal dissemination have a greater than 50% risk for mortality.6
Disseminated coccidioidomycosis is a relatively rare manifestation of Coccidioides infection; approximately 1.6% of patients exposed to and infected with Coccidioides ultimately will develop systemic or disseminated disease.7,8 Although the rates of primary pulmonary infection are similar between patients of varying ethnicities, the rates of dissemination are higher in patients of African and Filipino ethnicity.8 In population studies of coccidioidomycosis (N=332), Black patients represented 33.3% (4/12) of disseminated cases but only 8.7% of Coccidioides cases overall.7
Population studies of Black patients with coccidioidomycosis have shown a 4-fold higher predisposition for severe disease compared to mild disease.9 Spondylitis and meningitis also are disproportionately more common in Black patients.8 Black patients comprised 75% of all spondylitis cases in a cohort where only 25% of patients were Black. Additionally, 33% of all meningitis cases occurred in Black patients in a cohort where 8% of total cases were Black patients.8 Within the United States, the highest rates of coccidioidomycosis meningitis are seen in Black patients.10
The pathophysiology underlying the increased susceptibility of individuals of African or Filipino descent to disseminated and severe coccidioidomycosis remains unknown.8 The level of vulnerability within this patient population has no association with increased environmental exposure or poor immunologic response to Coccidioides, as demonstrated by the ability of these populations to respond to experimental vaccination and skin testing (spherulin, coccidioidin) to a similar extent as other ethnicities.8 Class II HLA-DRB1*1301 alleles have been associated with an increased risk for severe disseminated Coccidioides infection regardless of ethnicity; however, these alleles are not overrepresented in these patient populations.8
In patients with primary pulmonary coccidioidomycosis with no evidence of dissemination, guidelines generally recommend offering treatment to groups at high risk of dissemination, such as pregnant women and patients with diabetes mellitus. Given the high incidence of disseminated and severe disease in Black and Filipino patients, some guidelines endorse treatment of all cases of coccidioidomycosis in this patient population.8 No current data are available to help determine whether this broad treatment approach reduces the development of disseminated infection in these populations. Frequent monitoring for disease progression and/or dissemination involving clinical and laboratory reevaluation every 3 months for 2 years is highly recommended.8
Treatment generally is based on location and severity of infection, with disseminated nonmeningeal infection being treated with oral azole therapy (ketoconazole, itraconazole, or fluconazole).11 If there is involvement of the central nervous system structures or rapidly worsening disease despite azole therapy, amphotericin B is recommended at 0.5 to 0.7 mg/kg daily. In patients with disseminated meningeal infection, oral fluconazole (800–1000 mg/d) or a combination of an azole with intrathecal amphotericin B (0.01–1.5 mg/dose, interval ranging from daily to 1 week) is recommended to improve response.11
The differential diagnosis of cutaneous disseminated coccidioidomycosis is broad and includes other systemic endemic mycoses (histoplasmosis, blastomycosis) and infections (mycobacteria, leishmania). Lupus vulgaris, a form of cutaneous tuberculosis, presents as a palpable tubercular lesion that may coalesce into erythematous plaques, which may mimic endemic mycoses, especially in patients with risk factors for both infectious etiologies such as our patient.12 Disseminated histoplasmosis may present as polymorphic plaques, pustules, nodules, and ulcerated skin lesions, whereas disseminated blastomycosis characteristically presents as a crusted verrucous lesion with raised borders and painful ulcers, both of which may mimic coccidioidomycosis.13 Biopsy would reveal the characteristic intracellular yeast in Histoplasma capsulatum and broad-based budding yeast form of Blastomyces dermatitidis in histoplasmosis and blastomycosis, respectively, in contrast to the spherules seen in our patient’s biopsy.13 Localized cutaneous leishmaniasis initially develops as a nodular or papular lesion and can progress to open ulcerations with raised borders. Biopsy and histopathology would reveal round protozoal amastigotes.14 Other diagnoses that should be considered include mycetoma, nocardiosis, and sporotrichosis.15 As the cutaneous manifestations of Coccidioides infections are varied, a broad differential diagnosis should be maintained, and probable environmental and infectious exposures should be considered prior to ordering diagnostic studies.
The Diagnosis: Disseminated Coccidioidomycosis
A6-mm punch biopsy was performed at the periphery of the ulcerated cutaneous lesion on the chest revealing extensive spherules. Serum antibody immunodiffusion for histoplasmosis and blastomycoses both were negative; however, B-D-glucan assay was positive at 364 pg/mL (reference range: <60 pg/mL, negative). Initial HIV-1 and HIV-2 antibody and antigen testing was negative as well as repeat testing at 3 weeks. Immunodiffusion for Coccidioides IgM and IgG was positive, and cocci antibody IgG complement fixation assays were positive at titers of 1:64 (reference range: <1:2, negative). A computed tomography needle-guided biopsy of the paravertebral soft tissue was performed. Gram stains and bacterial cultures of the biopsies were negative; however, fungal cultures were notable for growth of Coccidioides. Given the pertinent testing, a diagnosis of disseminated coccidioidomycosis was made.
Cutaneous coccidioidomycosis can occur in 3 situations: direct inoculation (primary cutaneous coccidioidomycosis), disseminated infection (disseminated cutaneous coccidioidomycosis), or as a reactive component of pulmonary infection.1,2 Of them, primary and disseminated cutaneous coccidioidomycosis are organism specific and display characteristic spherules and fungus on histopathology and cultures, respectively. Reactive coccidioidomycosis differs from organism-specific disease, as it does not contain spherules in histopathologic sections of tissue biopsies.1 Reactive skin manifestations occur in 12% to 50% of patients with primary pulmonary infection and include erythema nodosum, erythema multiforme, acute generalized exanthema, reactive interstitial granulomatous dermatitis, and Sweet syndrome.3
Organism-specific cutaneous coccidioidomycosis most often is correlated with hematogenous dissemination of primary pulmonary disease rather than direct inoculation of skin.1 The skin is the most common site of extrapulmonary involvement in disseminated coccidioidomycosis, and cutaneous lesions have been reported in 15% to 67% of cases of disseminated disease.1,4 In cutaneous disseminated disease, nodules, papules, macules, and verrucous plaques have been described. In a case series of disseminated cutaneous coccidioidomycosis, nodules were the most common cutaneous presentation and occurred in 39% (7/18) of patients, while verrucous plaques were the rarest and occurred in only 6% (1/18) of patients.5
The rate of coccidioidomycosis dissemination varies based on exposure and patient characteristics. Increased rates of dissemination have been reported in patients of African and Filipino descent, along with individuals that are immunosuppressed due to disease or medical therapy. Dissemination is clinically significant, as patients with multifocal dissemination have a greater than 50% risk for mortality.6
Disseminated coccidioidomycosis is a relatively rare manifestation of Coccidioides infection; approximately 1.6% of patients exposed to and infected with Coccidioides ultimately will develop systemic or disseminated disease.7,8 Although the rates of primary pulmonary infection are similar between patients of varying ethnicities, the rates of dissemination are higher in patients of African and Filipino ethnicity.8 In population studies of coccidioidomycosis (N=332), Black patients represented 33.3% (4/12) of disseminated cases but only 8.7% of Coccidioides cases overall.7
Population studies of Black patients with coccidioidomycosis have shown a 4-fold higher predisposition for severe disease compared to mild disease.9 Spondylitis and meningitis also are disproportionately more common in Black patients.8 Black patients comprised 75% of all spondylitis cases in a cohort where only 25% of patients were Black. Additionally, 33% of all meningitis cases occurred in Black patients in a cohort where 8% of total cases were Black patients.8 Within the United States, the highest rates of coccidioidomycosis meningitis are seen in Black patients.10
The pathophysiology underlying the increased susceptibility of individuals of African or Filipino descent to disseminated and severe coccidioidomycosis remains unknown.8 The level of vulnerability within this patient population has no association with increased environmental exposure or poor immunologic response to Coccidioides, as demonstrated by the ability of these populations to respond to experimental vaccination and skin testing (spherulin, coccidioidin) to a similar extent as other ethnicities.8 Class II HLA-DRB1*1301 alleles have been associated with an increased risk for severe disseminated Coccidioides infection regardless of ethnicity; however, these alleles are not overrepresented in these patient populations.8
In patients with primary pulmonary coccidioidomycosis with no evidence of dissemination, guidelines generally recommend offering treatment to groups at high risk of dissemination, such as pregnant women and patients with diabetes mellitus. Given the high incidence of disseminated and severe disease in Black and Filipino patients, some guidelines endorse treatment of all cases of coccidioidomycosis in this patient population.8 No current data are available to help determine whether this broad treatment approach reduces the development of disseminated infection in these populations. Frequent monitoring for disease progression and/or dissemination involving clinical and laboratory reevaluation every 3 months for 2 years is highly recommended.8
Treatment generally is based on location and severity of infection, with disseminated nonmeningeal infection being treated with oral azole therapy (ketoconazole, itraconazole, or fluconazole).11 If there is involvement of the central nervous system structures or rapidly worsening disease despite azole therapy, amphotericin B is recommended at 0.5 to 0.7 mg/kg daily. In patients with disseminated meningeal infection, oral fluconazole (800–1000 mg/d) or a combination of an azole with intrathecal amphotericin B (0.01–1.5 mg/dose, interval ranging from daily to 1 week) is recommended to improve response.11
The differential diagnosis of cutaneous disseminated coccidioidomycosis is broad and includes other systemic endemic mycoses (histoplasmosis, blastomycosis) and infections (mycobacteria, leishmania). Lupus vulgaris, a form of cutaneous tuberculosis, presents as a palpable tubercular lesion that may coalesce into erythematous plaques, which may mimic endemic mycoses, especially in patients with risk factors for both infectious etiologies such as our patient.12 Disseminated histoplasmosis may present as polymorphic plaques, pustules, nodules, and ulcerated skin lesions, whereas disseminated blastomycosis characteristically presents as a crusted verrucous lesion with raised borders and painful ulcers, both of which may mimic coccidioidomycosis.13 Biopsy would reveal the characteristic intracellular yeast in Histoplasma capsulatum and broad-based budding yeast form of Blastomyces dermatitidis in histoplasmosis and blastomycosis, respectively, in contrast to the spherules seen in our patient’s biopsy.13 Localized cutaneous leishmaniasis initially develops as a nodular or papular lesion and can progress to open ulcerations with raised borders. Biopsy and histopathology would reveal round protozoal amastigotes.14 Other diagnoses that should be considered include mycetoma, nocardiosis, and sporotrichosis.15 As the cutaneous manifestations of Coccidioides infections are varied, a broad differential diagnosis should be maintained, and probable environmental and infectious exposures should be considered prior to ordering diagnostic studies.
- Garcia Garcia SC, Salas Alanis JC, Flores MG, et al. Coccidioidomycosis and the skin: a comprehensive review. An Bras Dermatol. 2015; 90:610-619.
- DiCaudo DJ. Coccidioidomycosis: a review and update. J Am Acad Dermatol. 2006;55:929-942; quiz 943-925.
- DiCaudo DJ, Yiannias JA, Laman SD, et al. The exanthem of acute pulmonary coccidioidomycosis: clinical and histopathologic features of 3 cases and review of the literature. Arch Dermatol. 2006;142:744-746.
- Blair JE. State-of-the-art treatment of coccidioidomycosis: skin and soft-tissue infections. Ann N Y Acad Sci. 2007;1111:411-421.
- Crum NF, Lederman ER, Stafford CM, et al. Coccidioidomycosis: a descriptive survey of a reemerging disease. clinical characteristics and current controversies. Medicine (Baltimore). 2004;83:149-175.
- Borchers AT, Gershwin ME. The immune response in coccidioidomycosis. Autoimmun Rev. 2010;10:94-102.
- Smith CE, Beard RR. Varieties of coccidioidal infection in relation to the epidemiology and control of the diseases. Am J Public Health Nations Health. 1946;36:1394-1402.
- Ruddy BE, Mayer AP, Ko MG, et al. Coccidioidomycosis in African Americans. Mayo Clin Proc. 2011;86:63-69.
- Louie L, Ng S, Hajjeh R, et al. Influence of host genetics on the severity of coccidioidomycosis. Emerg Infect Dis. 1999;5:672-680.
- McCotter OZ, Benedict K, Engelthaler DM, et al. Update on the epidemiology of coccidioidomycosis in the United States. Med Mycol. 2019;57(suppl 1):S30-S40.
- Galgiani JN, Ampel NM, Catanzaro A, et al. Practice guideline for the treatment of coccidioidomycosis. Infectious Diseases Society of America. Clin Infect Dis. 2000;30:658-661.
- Khadka P, Koirala S, Thapaliya J. Cutaneous tuberculosis: clinicopathologic arrays and diagnostic challenges. Dermatol Res Pract. 2018;2018:7201973.
- Smith JA, Riddell JT, Kauffman CA. Cutaneous manifestations of endemic mycoses. Curr Infect Dis Rep. 2013;15:440-449.
- Scorza BM, Carvalho EM, Wilson ME. Cutaneous manifestations of human and murine leishmaniasis. Int J Mol Sci. 2017;18:1296.
- Garcia Garcia SC, Salas Alanis JC, Flores MG, et al. Coccidioidomycosis and the skin: a comprehensive review. An Bras Dermatol. 2015; 90:610-619.
- DiCaudo DJ. Coccidioidomycosis: a review and update. J Am Acad Dermatol. 2006;55:929-942; quiz 943-925.
- DiCaudo DJ, Yiannias JA, Laman SD, et al. The exanthem of acute pulmonary coccidioidomycosis: clinical and histopathologic features of 3 cases and review of the literature. Arch Dermatol. 2006;142:744-746.
- Blair JE. State-of-the-art treatment of coccidioidomycosis: skin and soft-tissue infections. Ann N Y Acad Sci. 2007;1111:411-421.
- Crum NF, Lederman ER, Stafford CM, et al. Coccidioidomycosis: a descriptive survey of a reemerging disease. clinical characteristics and current controversies. Medicine (Baltimore). 2004;83:149-175.
- Borchers AT, Gershwin ME. The immune response in coccidioidomycosis. Autoimmun Rev. 2010;10:94-102.
- Smith CE, Beard RR. Varieties of coccidioidal infection in relation to the epidemiology and control of the diseases. Am J Public Health Nations Health. 1946;36:1394-1402.
- Ruddy BE, Mayer AP, Ko MG, et al. Coccidioidomycosis in African Americans. Mayo Clin Proc. 2011;86:63-69.
- Louie L, Ng S, Hajjeh R, et al. Influence of host genetics on the severity of coccidioidomycosis. Emerg Infect Dis. 1999;5:672-680.
- McCotter OZ, Benedict K, Engelthaler DM, et al. Update on the epidemiology of coccidioidomycosis in the United States. Med Mycol. 2019;57(suppl 1):S30-S40.
- Galgiani JN, Ampel NM, Catanzaro A, et al. Practice guideline for the treatment of coccidioidomycosis. Infectious Diseases Society of America. Clin Infect Dis. 2000;30:658-661.
- Khadka P, Koirala S, Thapaliya J. Cutaneous tuberculosis: clinicopathologic arrays and diagnostic challenges. Dermatol Res Pract. 2018;2018:7201973.
- Smith JA, Riddell JT, Kauffman CA. Cutaneous manifestations of endemic mycoses. Curr Infect Dis Rep. 2013;15:440-449.
- Scorza BM, Carvalho EM, Wilson ME. Cutaneous manifestations of human and murine leishmaniasis. Int J Mol Sci. 2017;18:1296.

A 36-year-old man presented to an emergency department in the southwestern United States with a cough, fatigue, and worsening back pain associated with night sweats of 1 month’s duration. He experienced a 9.07-kg weight loss, as well as development of a rough, nontender, nonpruritic rash along the left upper chest over the prior month. The patient was born in West Africa and reported that he had moved to the southwestern United States from the eastern United States approximately 6 years prior to presentation. Physical examination on admission revealed a 5×3-cm, purple-brown, verrucous plaque with a central pink cobblestone appearance and ulceration. Chest radiography was notable for perihilar adenopathy with no focal infiltrates or cavitary lesions. Computed tomography and magnetic resonance imaging of the chest were notable for miliary nodules throughout the lungs; extensive lytic spine lesions of cervical, thoracic, and lumbar vertebral bodies and left twelfth rib; and a left paraspinal thoracic epidural soft tissue phlegmon. Initial laboratory investigations revealed peripheral eosinophilia without absolute leukocytosis and a microcytic anemia.
Reticular Rash on the Chest
The Diagnosis: Erythema Ab Igne
Based on the clinical findings and history, a diagnosis of erythema ab igne (EAI), a skin reaction to chronic infrared radiation exposure, was made. The name of this condition translates from Latin as “redness from fire”; other names include toasted skin syndrome and fire stains. The most common presentation is reticulated hyperpigmentation, erythema, and cutaneous atrophy, as well as possible crusting, scaling, or telangiectasia. The rash also typically presents in areas of heat exposure—from heated blankets, heating pads, or the use of infrared heaters or lamps.1,2 The patient usually will have pain and pruritus over the affected areas. The diagnosis of EAI largely is clinical and based on the patient’s history of exposure; it rarely requires biopsy and histologic analysis. However, some of the common histopathologic findings include hyperkeratosis, a hyperpigmented basal layer, hemosiderin deposits, prominent melanophages, basal cell degeneration, course collagen, and elastosis.2,3 These changes are common with UV radiation exposure and thermal damage. The primary treatment in all cases is to remove or reduce the source of infrared radiation. However, EAI has been reported to be successfully treated with removal of the insult as well as topical agents such as imiquimod and 5-fluorouracil.4 Possible complications include increased risk for malignancies such as squamous cell carcinoma in the affected area.1
The possible differential for EAI includes livedo reticularis, livedo racemosa, cutis marmorata, and cutis marmorata telangiectatica congenita. All of these conditions are related to dysfunction of the cutaneous vasculature that creates a reticular, mottled, reddish purple rash. When the livedo is reversible and idiopathic, it is referred to as livedo reticularis, but when it is generalized and permanent it is referred to as livedo racemosa. Livedo racemosa can be caused by a variety of conditions, including systemic lupus erythematosus and antiphospholipid syndrome.1 Physiologic livedo reticularis that is more transient and can be reversed by warming is referred to as cutis marmorata. Finally, cutis marmorata telangiectatica congenita primarily is found in neonates, and although persistent, it usually improves with age. Erythema ab igne also is a type of livedo with a known heat exposure and localized distribution.
Our patient was educated on the etiology of the rash, specifically related to heating pad usage for multiple years, and the risk for cutaneous malignancy after longstanding EAI. It was recommended that she discontinue use of a heating pad on the affected areas to allow them to properly heal. If she found that heating pad usage was necessary, she was advised to limit use to 5 to 10 minutes with 2 to 3 hours in between applications. In addition, she was advised to apply petroleum jelly daily for assistance with wound healing as well as anti-itch sensitive lotion twice daily on the arms and back to alleviate some of the tingling pain. We explained that areas of hyperpigmentation may improve with time; however, areas of erythema/ atrophy may be long-lasting.
- Aria AB, Chen L, Silapunt S. Erythema ab igne from heating pad use: a report of three clinical cases and a differential diagnosis. Cureus. 2018;10:E2635.
- Dellavalle RP, Gillum P. Erythema ab igne following heating/cooling blanket use in the intensive care unit. Cutis. 2000;66:136-138.
- Finlayson GR, Sams WM Jr, Smith JG Jr. Erythema ab igne: a histopathological study. J Invest Dermatol. 1966;46:104-108.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
The Diagnosis: Erythema Ab Igne
Based on the clinical findings and history, a diagnosis of erythema ab igne (EAI), a skin reaction to chronic infrared radiation exposure, was made. The name of this condition translates from Latin as “redness from fire”; other names include toasted skin syndrome and fire stains. The most common presentation is reticulated hyperpigmentation, erythema, and cutaneous atrophy, as well as possible crusting, scaling, or telangiectasia. The rash also typically presents in areas of heat exposure—from heated blankets, heating pads, or the use of infrared heaters or lamps.1,2 The patient usually will have pain and pruritus over the affected areas. The diagnosis of EAI largely is clinical and based on the patient’s history of exposure; it rarely requires biopsy and histologic analysis. However, some of the common histopathologic findings include hyperkeratosis, a hyperpigmented basal layer, hemosiderin deposits, prominent melanophages, basal cell degeneration, course collagen, and elastosis.2,3 These changes are common with UV radiation exposure and thermal damage. The primary treatment in all cases is to remove or reduce the source of infrared radiation. However, EAI has been reported to be successfully treated with removal of the insult as well as topical agents such as imiquimod and 5-fluorouracil.4 Possible complications include increased risk for malignancies such as squamous cell carcinoma in the affected area.1
The possible differential for EAI includes livedo reticularis, livedo racemosa, cutis marmorata, and cutis marmorata telangiectatica congenita. All of these conditions are related to dysfunction of the cutaneous vasculature that creates a reticular, mottled, reddish purple rash. When the livedo is reversible and idiopathic, it is referred to as livedo reticularis, but when it is generalized and permanent it is referred to as livedo racemosa. Livedo racemosa can be caused by a variety of conditions, including systemic lupus erythematosus and antiphospholipid syndrome.1 Physiologic livedo reticularis that is more transient and can be reversed by warming is referred to as cutis marmorata. Finally, cutis marmorata telangiectatica congenita primarily is found in neonates, and although persistent, it usually improves with age. Erythema ab igne also is a type of livedo with a known heat exposure and localized distribution.
Our patient was educated on the etiology of the rash, specifically related to heating pad usage for multiple years, and the risk for cutaneous malignancy after longstanding EAI. It was recommended that she discontinue use of a heating pad on the affected areas to allow them to properly heal. If she found that heating pad usage was necessary, she was advised to limit use to 5 to 10 minutes with 2 to 3 hours in between applications. In addition, she was advised to apply petroleum jelly daily for assistance with wound healing as well as anti-itch sensitive lotion twice daily on the arms and back to alleviate some of the tingling pain. We explained that areas of hyperpigmentation may improve with time; however, areas of erythema/ atrophy may be long-lasting.
The Diagnosis: Erythema Ab Igne
Based on the clinical findings and history, a diagnosis of erythema ab igne (EAI), a skin reaction to chronic infrared radiation exposure, was made. The name of this condition translates from Latin as “redness from fire”; other names include toasted skin syndrome and fire stains. The most common presentation is reticulated hyperpigmentation, erythema, and cutaneous atrophy, as well as possible crusting, scaling, or telangiectasia. The rash also typically presents in areas of heat exposure—from heated blankets, heating pads, or the use of infrared heaters or lamps.1,2 The patient usually will have pain and pruritus over the affected areas. The diagnosis of EAI largely is clinical and based on the patient’s history of exposure; it rarely requires biopsy and histologic analysis. However, some of the common histopathologic findings include hyperkeratosis, a hyperpigmented basal layer, hemosiderin deposits, prominent melanophages, basal cell degeneration, course collagen, and elastosis.2,3 These changes are common with UV radiation exposure and thermal damage. The primary treatment in all cases is to remove or reduce the source of infrared radiation. However, EAI has been reported to be successfully treated with removal of the insult as well as topical agents such as imiquimod and 5-fluorouracil.4 Possible complications include increased risk for malignancies such as squamous cell carcinoma in the affected area.1
The possible differential for EAI includes livedo reticularis, livedo racemosa, cutis marmorata, and cutis marmorata telangiectatica congenita. All of these conditions are related to dysfunction of the cutaneous vasculature that creates a reticular, mottled, reddish purple rash. When the livedo is reversible and idiopathic, it is referred to as livedo reticularis, but when it is generalized and permanent it is referred to as livedo racemosa. Livedo racemosa can be caused by a variety of conditions, including systemic lupus erythematosus and antiphospholipid syndrome.1 Physiologic livedo reticularis that is more transient and can be reversed by warming is referred to as cutis marmorata. Finally, cutis marmorata telangiectatica congenita primarily is found in neonates, and although persistent, it usually improves with age. Erythema ab igne also is a type of livedo with a known heat exposure and localized distribution.
Our patient was educated on the etiology of the rash, specifically related to heating pad usage for multiple years, and the risk for cutaneous malignancy after longstanding EAI. It was recommended that she discontinue use of a heating pad on the affected areas to allow them to properly heal. If she found that heating pad usage was necessary, she was advised to limit use to 5 to 10 minutes with 2 to 3 hours in between applications. In addition, she was advised to apply petroleum jelly daily for assistance with wound healing as well as anti-itch sensitive lotion twice daily on the arms and back to alleviate some of the tingling pain. We explained that areas of hyperpigmentation may improve with time; however, areas of erythema/ atrophy may be long-lasting.
- Aria AB, Chen L, Silapunt S. Erythema ab igne from heating pad use: a report of three clinical cases and a differential diagnosis. Cureus. 2018;10:E2635.
- Dellavalle RP, Gillum P. Erythema ab igne following heating/cooling blanket use in the intensive care unit. Cutis. 2000;66:136-138.
- Finlayson GR, Sams WM Jr, Smith JG Jr. Erythema ab igne: a histopathological study. J Invest Dermatol. 1966;46:104-108.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Aria AB, Chen L, Silapunt S. Erythema ab igne from heating pad use: a report of three clinical cases and a differential diagnosis. Cureus. 2018;10:E2635.
- Dellavalle RP, Gillum P. Erythema ab igne following heating/cooling blanket use in the intensive care unit. Cutis. 2000;66:136-138.
- Finlayson GR, Sams WM Jr, Smith JG Jr. Erythema ab igne: a histopathological study. J Invest Dermatol. 1966;46:104-108.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
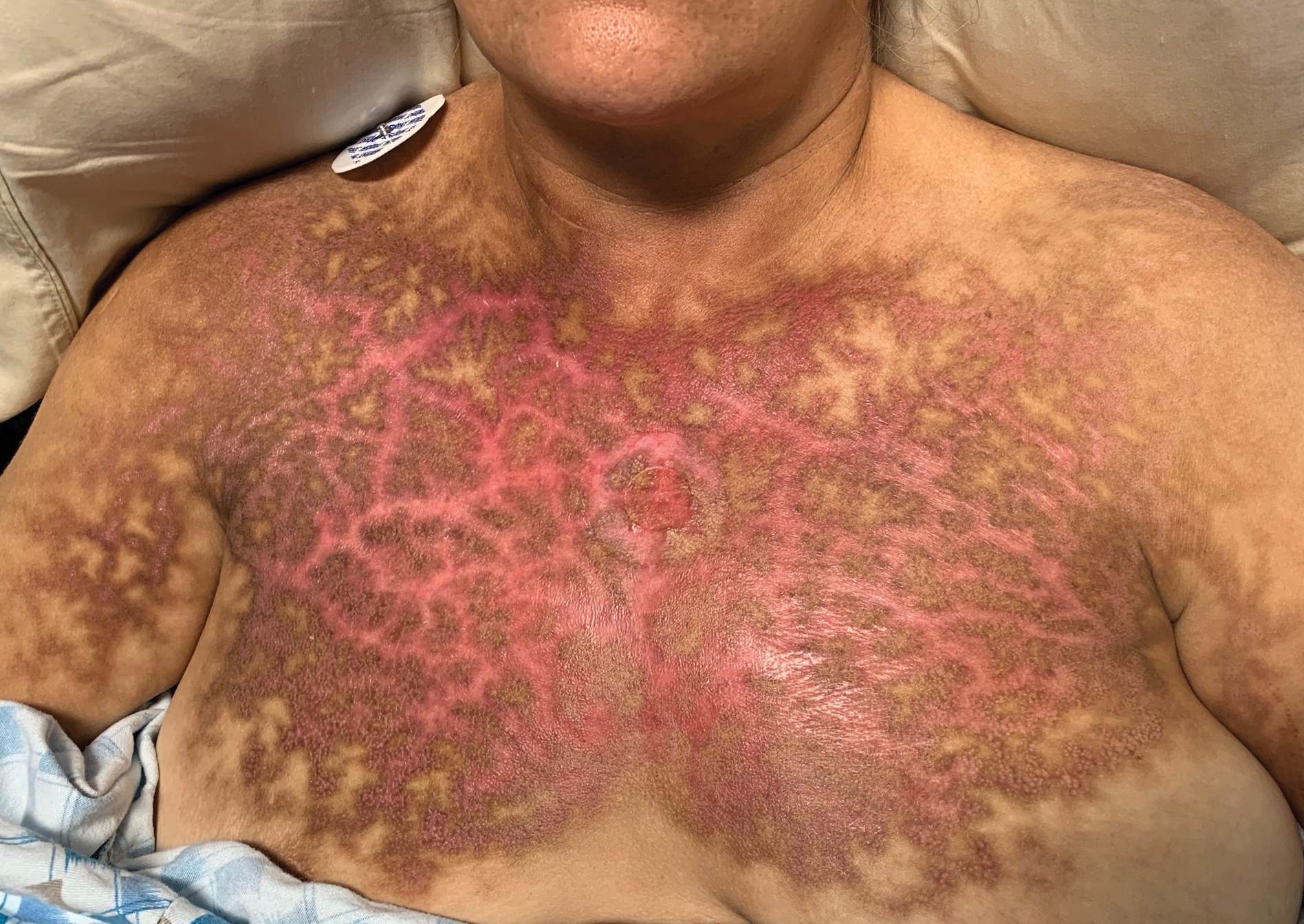
A 53-year-old woman with a history of diabetes mellitus, hypertension, chronic complex regional pain syndrome type 1, and chronic prescription opiate use presented to the hospital with a pruritic rash on the chest of 15 years’ duration that started a few weeks after a left shoulder repair. The patient was using fentanyl patches and acetaminophen with oxycodone as well as a heating pad for 20 to 22 hours per day for many years to help with her chronic pain. She also described similar lesions on the abdomen and back when she used the heating pad on those areas for weeks at a time. Vital signs were within normal limits. Physical examination revealed a lacy, reticular, eroded, well-demarcated rash on the chest along with areas of cracking. Laboratory evaluation did not reveal any abnormalities.
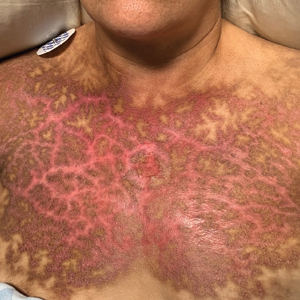
Pink, Scaly, Annular Plaques in Concentric Rings Localized to Vitiliginous Patches
The Diagnosis: Tinea Pseudoimbricata
Tinea pseudoimbricata and tinea indecisiva are synonyms describing cases of tinea corporis that manifest in scaly plaques in concentric rings evocative of those present in tinea imbricata. However, in contrast to tinea imbricata, cases of tinea pseudoimbricata are caused by dermatophytes other than Trichophyton concentricum. 1 Tinea pseudoimbricata usually presents in association with immunosuppression, either systemic or local, and can be produced by application of topical medications such as corticosteroids.2 Mask-Bull et al3 reported the case of a 21-year-old man in the United States with no history of immunosuppressive conditions who presented with scaly erythematous annular plaques on the lateral neck that resolved with 2 pulsed doses of terbinafine. Potassium hydroxide preparation and fungal culture were both consistent with Trichophyton tonsurans.3
Trichophyton concentricum is an anthropophilic species of dermatophyte endemic to areas within the South Pacific, Southeast Asia, and Central and South America. Infection with T concentricum produces tinea imbricata, which presents with concentric, scaly, annular rings. Cutaneous lesions of tinea imbricata caused by T concentricum have a more generalized distribution and more densely grouped, concentric circles than the cutaneous findings seen in patients with tinea pseudoimbricata.4 Affected patients typically demonstrate negative delayed-type hypersensitivity to T concentricum cytoplasmic antigen and T-lymphocyte hyporeactivity, which may contribute to the development of sequential waves of scaling observed in tinea imbricata.5
Trichophyton rubrum, the most common cause of tinea corporis, has been reported to cause some cases of tinea pseudoimbricata (indecisiva).1,2 It utilizes keratinases such as subtilisins (Sub3 and Sub4), leucine aminopeptidases (Lap1 and Lap2), and dipeptidyl peptidases (DppIV and DppV) to invade the skin. Once inside, mannans, glycoprotein constituents of the cell wall, are released and bind to the cell surface of mononuclear phagocytes, subsequently moving into the cell by phagocytosis, thereafter interfering with RNA synthesis that is necessary for presentation of antigens to appropriate T cells and allowing for initiation of chronic infection.6,7 The cytotoxic response to superficial dermatophyte infection is triggered by major histocompatibility complex class I molecule activation of CD8+ cells.6,8
Our case is of interest given the localization of the superficial dermatophyte infection to only vitiliginous skin. This distribution and appearance while undergoing narrowband UVB (NB-UVB) treatment is rare. We postulate that our patient likely represents a case of locus minoris resistentiae, a phenomenon in which an area of skin exhibits a compromised immune microenvironment that predisposes it to disease.9
In vitiligo, NB-UVB modulates the immune response by increasing IL-10, thereby promoting regulatory T-cell differentiation with suppression of autoreactive T cells and induction of direct T-lymphocyte apoptosis.10,11 Although the mechanism accounting for our patient’s presentation is unknown, we suspect NB-UVB–induced immunosuppression enabled persistence of the dermatophyte infection. The localization of the infection to the vitiliginous patches may result from the greater penetration of the UV light relative to the surrounding, normally pigmented skin. This relative difference in UV penetration would be expected to result in increased immunosuppression in the vitiliginous lesions and enhanced susceptibility to the fungal organisms.
Erythema annulare centrifugum is characterized by annular lesions with a trailing scale instead of the concentric rings seen in tinea pseudoimbricata. Erythema marginatum is seen in acute rheumatic fever and presents with a transient nonpruritic rash, usually on the trunk or extremities. Erythema migrans presents with fewer lesions that are less circinate in shape, and the patient often has a history of a tick bite. Tinea imbricata is caused by T concentricum, while tinea pseudoimbricata is caused by T tonsurans and other dermatophytes.
With the increasing use of immunosuppressant drugs, the prevalence of tinea pseudoimbricata is hypothesized to increase.1 The presence of tinea pseudoimbricata should alert dermatologists to the possible overuse of topical corticosteroids, and other forms of immunosuppression also should be considered.
- Lim SP, Smith AG. “Tinea pseudoimbricata”: tinea corporis in a renal transplant recipient mimicking the concentric rings of tinea imbricata. Clin Exp Dermatol. 2003;28:332-333.
- Batta K, Ramlogan D, Smith AG, et al. ‘Tinea indecisiva’ may mimic the concentric rings of tinea imbricata. Br J Dermatol. 2002;147:384.
- Mask-Bull L, Patel R, Tarbox MB. America’s first case of tinea pseudoimbricata. Am J Dermatol Venereol. 2015;4:15-17.
- Meena M, Mittal A. Tinea pseudo-imbricata. J Assoc Physicians India. 2018;66:79.
- Hay RJ, Reid S, Talwat E, et al. Immune responses of patients with tinea imbricata. Br J Dermatol. 1983;108:581-586.
- Dahl MV. Suppression of immunity and inflammation by products produced by dermatophytes. J Am Acad Dermatol. 1993;28(5 pt 1):S19-S23.
- Blutfield MS, Lohre JM, Pawich DA, et al. The immunologic response to Trichophyton rubrum in lower extremity fungal infections. J Fungi (Basel). 2015;1:130-137.
- De Hoog S, Monod M, Dawson T, et al. Skin fungi from colonization to infection [published online July 2017]. Microbiol Spectr. doi:10.1128/ microbiolspec.FUNK-0049-2016
- Lo Schiavo A, Ruocco E, Russo T, et al. Locus minoris resistentiae: an old but still valid way of thinking in medicine. Clin Dermatol. 2014;32:553-556.
- Ponsonby AL, Lucas RM, van der Mei IA. UVR, vitamin D and three autoimmune diseases—multiple sclerosis, type 1 diabetes, rheumatoid arthritis. Photochem Photobiol. 2005;81:1267-1275.
- Yazdani Abyaneh M, Griffith RD, Falto-Aizpurua L, et al. Narrowband ultraviolet B phototherapy in combination with other therapies for vitiligo: mechanisms and efficacies. J Eur Acad Dermatol Venereol. 2014;28:1610-1622.
The Diagnosis: Tinea Pseudoimbricata
Tinea pseudoimbricata and tinea indecisiva are synonyms describing cases of tinea corporis that manifest in scaly plaques in concentric rings evocative of those present in tinea imbricata. However, in contrast to tinea imbricata, cases of tinea pseudoimbricata are caused by dermatophytes other than Trichophyton concentricum. 1 Tinea pseudoimbricata usually presents in association with immunosuppression, either systemic or local, and can be produced by application of topical medications such as corticosteroids.2 Mask-Bull et al3 reported the case of a 21-year-old man in the United States with no history of immunosuppressive conditions who presented with scaly erythematous annular plaques on the lateral neck that resolved with 2 pulsed doses of terbinafine. Potassium hydroxide preparation and fungal culture were both consistent with Trichophyton tonsurans.3
Trichophyton concentricum is an anthropophilic species of dermatophyte endemic to areas within the South Pacific, Southeast Asia, and Central and South America. Infection with T concentricum produces tinea imbricata, which presents with concentric, scaly, annular rings. Cutaneous lesions of tinea imbricata caused by T concentricum have a more generalized distribution and more densely grouped, concentric circles than the cutaneous findings seen in patients with tinea pseudoimbricata.4 Affected patients typically demonstrate negative delayed-type hypersensitivity to T concentricum cytoplasmic antigen and T-lymphocyte hyporeactivity, which may contribute to the development of sequential waves of scaling observed in tinea imbricata.5
Trichophyton rubrum, the most common cause of tinea corporis, has been reported to cause some cases of tinea pseudoimbricata (indecisiva).1,2 It utilizes keratinases such as subtilisins (Sub3 and Sub4), leucine aminopeptidases (Lap1 and Lap2), and dipeptidyl peptidases (DppIV and DppV) to invade the skin. Once inside, mannans, glycoprotein constituents of the cell wall, are released and bind to the cell surface of mononuclear phagocytes, subsequently moving into the cell by phagocytosis, thereafter interfering with RNA synthesis that is necessary for presentation of antigens to appropriate T cells and allowing for initiation of chronic infection.6,7 The cytotoxic response to superficial dermatophyte infection is triggered by major histocompatibility complex class I molecule activation of CD8+ cells.6,8
Our case is of interest given the localization of the superficial dermatophyte infection to only vitiliginous skin. This distribution and appearance while undergoing narrowband UVB (NB-UVB) treatment is rare. We postulate that our patient likely represents a case of locus minoris resistentiae, a phenomenon in which an area of skin exhibits a compromised immune microenvironment that predisposes it to disease.9
In vitiligo, NB-UVB modulates the immune response by increasing IL-10, thereby promoting regulatory T-cell differentiation with suppression of autoreactive T cells and induction of direct T-lymphocyte apoptosis.10,11 Although the mechanism accounting for our patient’s presentation is unknown, we suspect NB-UVB–induced immunosuppression enabled persistence of the dermatophyte infection. The localization of the infection to the vitiliginous patches may result from the greater penetration of the UV light relative to the surrounding, normally pigmented skin. This relative difference in UV penetration would be expected to result in increased immunosuppression in the vitiliginous lesions and enhanced susceptibility to the fungal organisms.
Erythema annulare centrifugum is characterized by annular lesions with a trailing scale instead of the concentric rings seen in tinea pseudoimbricata. Erythema marginatum is seen in acute rheumatic fever and presents with a transient nonpruritic rash, usually on the trunk or extremities. Erythema migrans presents with fewer lesions that are less circinate in shape, and the patient often has a history of a tick bite. Tinea imbricata is caused by T concentricum, while tinea pseudoimbricata is caused by T tonsurans and other dermatophytes.
With the increasing use of immunosuppressant drugs, the prevalence of tinea pseudoimbricata is hypothesized to increase.1 The presence of tinea pseudoimbricata should alert dermatologists to the possible overuse of topical corticosteroids, and other forms of immunosuppression also should be considered.
The Diagnosis: Tinea Pseudoimbricata
Tinea pseudoimbricata and tinea indecisiva are synonyms describing cases of tinea corporis that manifest in scaly plaques in concentric rings evocative of those present in tinea imbricata. However, in contrast to tinea imbricata, cases of tinea pseudoimbricata are caused by dermatophytes other than Trichophyton concentricum. 1 Tinea pseudoimbricata usually presents in association with immunosuppression, either systemic or local, and can be produced by application of topical medications such as corticosteroids.2 Mask-Bull et al3 reported the case of a 21-year-old man in the United States with no history of immunosuppressive conditions who presented with scaly erythematous annular plaques on the lateral neck that resolved with 2 pulsed doses of terbinafine. Potassium hydroxide preparation and fungal culture were both consistent with Trichophyton tonsurans.3
Trichophyton concentricum is an anthropophilic species of dermatophyte endemic to areas within the South Pacific, Southeast Asia, and Central and South America. Infection with T concentricum produces tinea imbricata, which presents with concentric, scaly, annular rings. Cutaneous lesions of tinea imbricata caused by T concentricum have a more generalized distribution and more densely grouped, concentric circles than the cutaneous findings seen in patients with tinea pseudoimbricata.4 Affected patients typically demonstrate negative delayed-type hypersensitivity to T concentricum cytoplasmic antigen and T-lymphocyte hyporeactivity, which may contribute to the development of sequential waves of scaling observed in tinea imbricata.5
Trichophyton rubrum, the most common cause of tinea corporis, has been reported to cause some cases of tinea pseudoimbricata (indecisiva).1,2 It utilizes keratinases such as subtilisins (Sub3 and Sub4), leucine aminopeptidases (Lap1 and Lap2), and dipeptidyl peptidases (DppIV and DppV) to invade the skin. Once inside, mannans, glycoprotein constituents of the cell wall, are released and bind to the cell surface of mononuclear phagocytes, subsequently moving into the cell by phagocytosis, thereafter interfering with RNA synthesis that is necessary for presentation of antigens to appropriate T cells and allowing for initiation of chronic infection.6,7 The cytotoxic response to superficial dermatophyte infection is triggered by major histocompatibility complex class I molecule activation of CD8+ cells.6,8
Our case is of interest given the localization of the superficial dermatophyte infection to only vitiliginous skin. This distribution and appearance while undergoing narrowband UVB (NB-UVB) treatment is rare. We postulate that our patient likely represents a case of locus minoris resistentiae, a phenomenon in which an area of skin exhibits a compromised immune microenvironment that predisposes it to disease.9
In vitiligo, NB-UVB modulates the immune response by increasing IL-10, thereby promoting regulatory T-cell differentiation with suppression of autoreactive T cells and induction of direct T-lymphocyte apoptosis.10,11 Although the mechanism accounting for our patient’s presentation is unknown, we suspect NB-UVB–induced immunosuppression enabled persistence of the dermatophyte infection. The localization of the infection to the vitiliginous patches may result from the greater penetration of the UV light relative to the surrounding, normally pigmented skin. This relative difference in UV penetration would be expected to result in increased immunosuppression in the vitiliginous lesions and enhanced susceptibility to the fungal organisms.
Erythema annulare centrifugum is characterized by annular lesions with a trailing scale instead of the concentric rings seen in tinea pseudoimbricata. Erythema marginatum is seen in acute rheumatic fever and presents with a transient nonpruritic rash, usually on the trunk or extremities. Erythema migrans presents with fewer lesions that are less circinate in shape, and the patient often has a history of a tick bite. Tinea imbricata is caused by T concentricum, while tinea pseudoimbricata is caused by T tonsurans and other dermatophytes.
With the increasing use of immunosuppressant drugs, the prevalence of tinea pseudoimbricata is hypothesized to increase.1 The presence of tinea pseudoimbricata should alert dermatologists to the possible overuse of topical corticosteroids, and other forms of immunosuppression also should be considered.
- Lim SP, Smith AG. “Tinea pseudoimbricata”: tinea corporis in a renal transplant recipient mimicking the concentric rings of tinea imbricata. Clin Exp Dermatol. 2003;28:332-333.
- Batta K, Ramlogan D, Smith AG, et al. ‘Tinea indecisiva’ may mimic the concentric rings of tinea imbricata. Br J Dermatol. 2002;147:384.
- Mask-Bull L, Patel R, Tarbox MB. America’s first case of tinea pseudoimbricata. Am J Dermatol Venereol. 2015;4:15-17.
- Meena M, Mittal A. Tinea pseudo-imbricata. J Assoc Physicians India. 2018;66:79.
- Hay RJ, Reid S, Talwat E, et al. Immune responses of patients with tinea imbricata. Br J Dermatol. 1983;108:581-586.
- Dahl MV. Suppression of immunity and inflammation by products produced by dermatophytes. J Am Acad Dermatol. 1993;28(5 pt 1):S19-S23.
- Blutfield MS, Lohre JM, Pawich DA, et al. The immunologic response to Trichophyton rubrum in lower extremity fungal infections. J Fungi (Basel). 2015;1:130-137.
- De Hoog S, Monod M, Dawson T, et al. Skin fungi from colonization to infection [published online July 2017]. Microbiol Spectr. doi:10.1128/ microbiolspec.FUNK-0049-2016
- Lo Schiavo A, Ruocco E, Russo T, et al. Locus minoris resistentiae: an old but still valid way of thinking in medicine. Clin Dermatol. 2014;32:553-556.
- Ponsonby AL, Lucas RM, van der Mei IA. UVR, vitamin D and three autoimmune diseases—multiple sclerosis, type 1 diabetes, rheumatoid arthritis. Photochem Photobiol. 2005;81:1267-1275.
- Yazdani Abyaneh M, Griffith RD, Falto-Aizpurua L, et al. Narrowband ultraviolet B phototherapy in combination with other therapies for vitiligo: mechanisms and efficacies. J Eur Acad Dermatol Venereol. 2014;28:1610-1622.
- Lim SP, Smith AG. “Tinea pseudoimbricata”: tinea corporis in a renal transplant recipient mimicking the concentric rings of tinea imbricata. Clin Exp Dermatol. 2003;28:332-333.
- Batta K, Ramlogan D, Smith AG, et al. ‘Tinea indecisiva’ may mimic the concentric rings of tinea imbricata. Br J Dermatol. 2002;147:384.
- Mask-Bull L, Patel R, Tarbox MB. America’s first case of tinea pseudoimbricata. Am J Dermatol Venereol. 2015;4:15-17.
- Meena M, Mittal A. Tinea pseudo-imbricata. J Assoc Physicians India. 2018;66:79.
- Hay RJ, Reid S, Talwat E, et al. Immune responses of patients with tinea imbricata. Br J Dermatol. 1983;108:581-586.
- Dahl MV. Suppression of immunity and inflammation by products produced by dermatophytes. J Am Acad Dermatol. 1993;28(5 pt 1):S19-S23.
- Blutfield MS, Lohre JM, Pawich DA, et al. The immunologic response to Trichophyton rubrum in lower extremity fungal infections. J Fungi (Basel). 2015;1:130-137.
- De Hoog S, Monod M, Dawson T, et al. Skin fungi from colonization to infection [published online July 2017]. Microbiol Spectr. doi:10.1128/ microbiolspec.FUNK-0049-2016
- Lo Schiavo A, Ruocco E, Russo T, et al. Locus minoris resistentiae: an old but still valid way of thinking in medicine. Clin Dermatol. 2014;32:553-556.
- Ponsonby AL, Lucas RM, van der Mei IA. UVR, vitamin D and three autoimmune diseases—multiple sclerosis, type 1 diabetes, rheumatoid arthritis. Photochem Photobiol. 2005;81:1267-1275.
- Yazdani Abyaneh M, Griffith RD, Falto-Aizpurua L, et al. Narrowband ultraviolet B phototherapy in combination with other therapies for vitiligo: mechanisms and efficacies. J Eur Acad Dermatol Venereol. 2014;28:1610-1622.
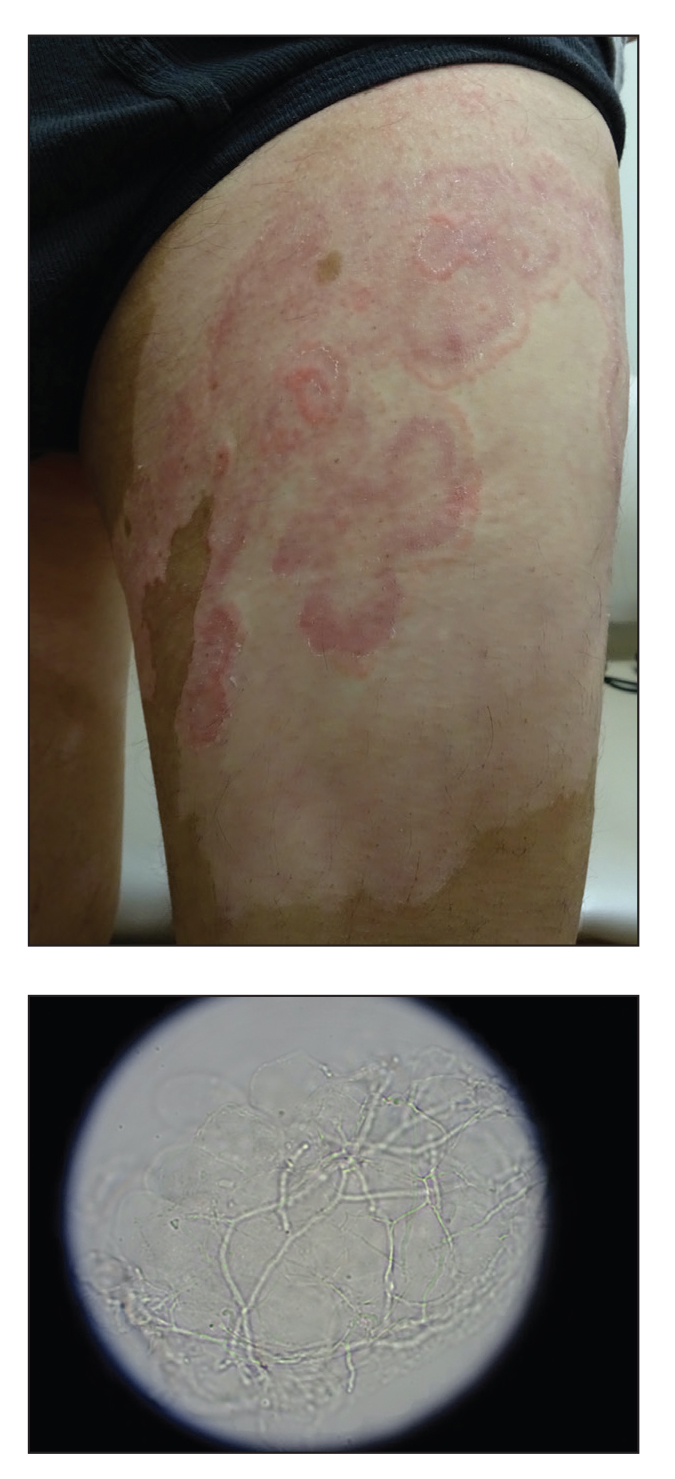
A 64-year-old man presented with generalized vitiligo. In addition to extensive depigmented macules, physical examination revealed the presence of onychomycosis and tinea corporis confirmed by microscopic examination of potassium hydroxide–treated superficial skin scrapings. Vitiligo treatment was postponed, and a 3-month course of oral terbinafine and naftifine cream was undertaken for the dermatophyte infections. Subsequent examination revealed that the patient’s tinea corporis had improved, though there were localized areas of persistence. Given the patient’s eagerness to treat his vitiligo, narrowband UVB phototherapy was started along with tolnaftate cream 1% for treatment of the residual tinea corporis. After 2 months of narrowband UVB, partial repigmentation of the vitiligo was observed; however, he had developed extensive pink, scaly, annular plaques in concentric rings within residual vitiliginous patches on the lower extremities (top). Repeat examination of potassium hydroxide–treated skin scrapings revealed numerous hyphae (bottom). A fungal culture identified Trichophyton rubrum.
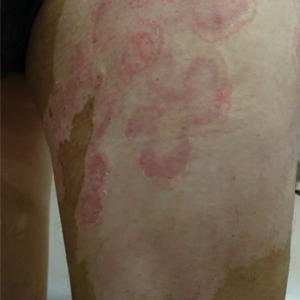
Exophytic Tumor on the Buttock
The Diagnosis: Hidradenocarcinoma
An excisional biopsy revealed a neoplasm in the dermis with focal invasion into the adjacent soft tissue (Figure 1). The tumor consisted of sheets of cells with cytoplasmic vacuoles and ductal differentiation (Figure 2), as well as cells with mild atypia, mild pleomorphism, rare mitotic figures, and abundant pale cytoplasm. Immunohistochemical staining was positive for cytokeratin (CK) 5, CK7, CK20, CK AE1/AE3, and p63 (Figure 3). The culmination of features including the large tumor size, immunohistochemical staining pattern, and mild pleomorphism with focal invasion into the soft tissue supported the diagnosis of hidradenocarcinoma.

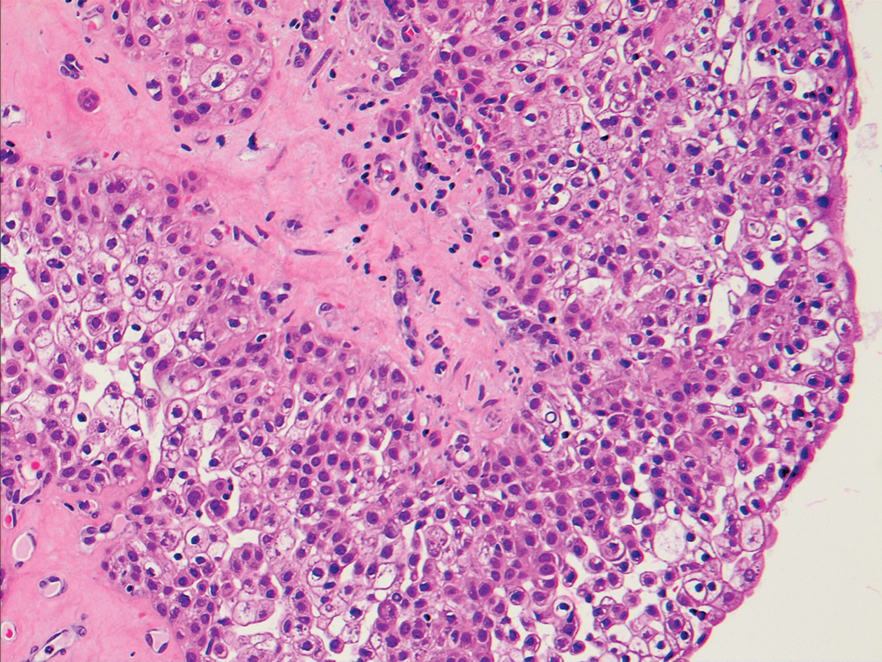
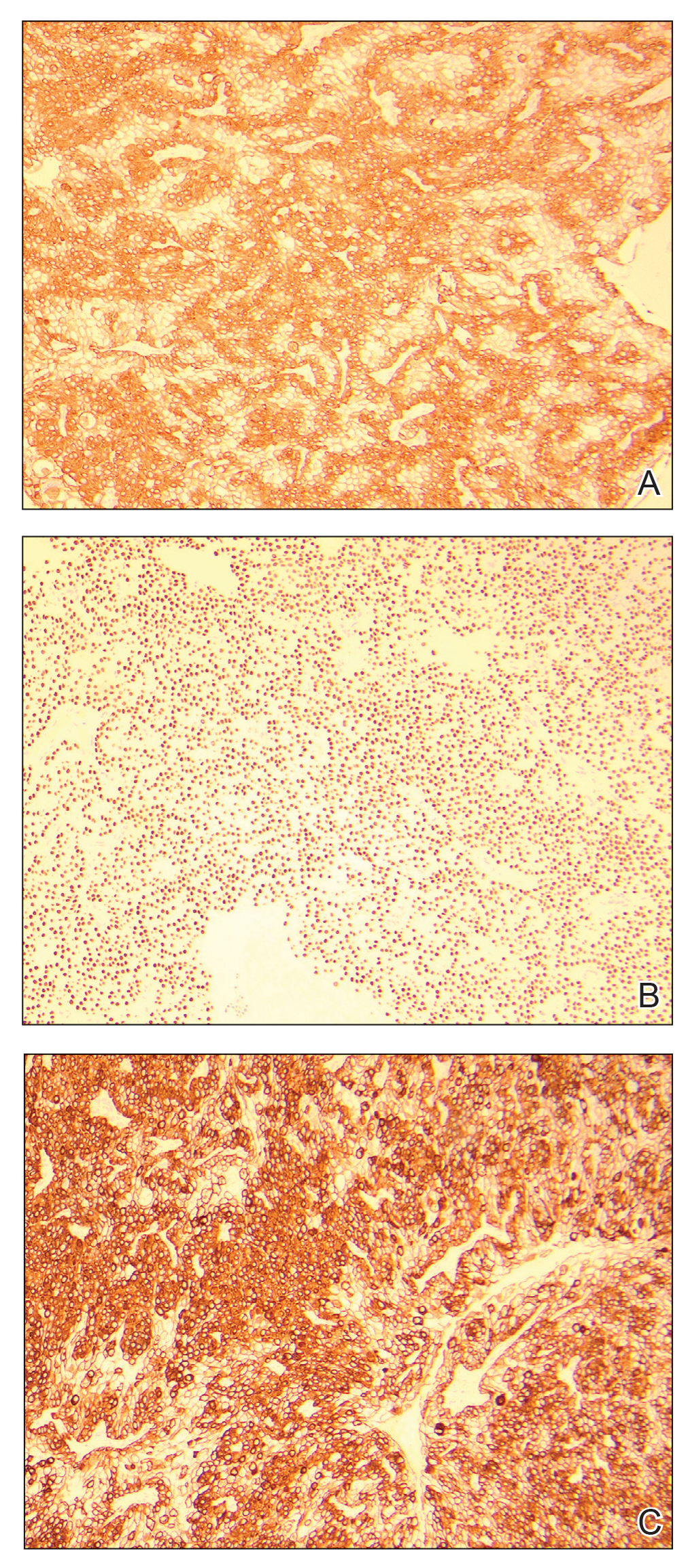
Hidradenocarcinoma is an exceedingly rare malignant tumor of eccrine and/or apocrine origin.1 It accounts for less than 0.001% of all tumors and 1 of 13,000 skin biopsies.2 It usually arises in the head and neck region and most commonly affects older adults aged 50 to 70 years.3 The size of hidradenocarcinomas can vary; however, they typically are large, often growing to be greater than 5 cm in diameter.2 It tends to be an aggressive tumor that generally spreads to regional lymph nodes and distant viscera.4 Although it most commonly arises de novo, it may occasionally derive from a benign hidradenoma.1 The diagnosis of hidradenocarcinoma is made based on the tumor’s morphologic and pathologic characteristics. Histologically, it is characterized by an infiltrative and invasive proliferation of lobules made of large clear cells with atypical mitotic figures and nuclear pleomorphism as well as immunohistochemical features displaying various positive markers, such as carcinoembryonic antigen, epithelial membrane antigen, S-100 protein, and CKs AE1/AE3 and 5/6.2 Invasion of the adjacent soft tissue can be present and helps to confirm the diagnosis.
The differential diagnosis for hidradenocarcinoma primarily is the benign hidradenoma, which is similar both clinically and histologically with a few important differences. Hidradenocarcinomas often are larger and ulcerated. Histologically, they usually are more pleomorphic with the presence of mitotic figures in clear cells and tend to invade locally into the surrounding soft tissue. Other similar lesions such as spiradenoma, Merkel cell carcinoma, lymphangioma, cutaneous Crohn disease, tumors metastatic to the skin, and metastatic clear cell carcinomas originating from other organs also are included in the differential diagnosis.2
Spiradenomas are dermal tumors originating from the sweat glands. They typically present as bluish, painful, solitary nodules on the ventral surfaces of the upper body, though multiple nodules also are reported.5 Spiradenomas manifest as a central constellation of pale large cells surrounded by small, dark, basaloid cells containing hyperchromatic nuclei. The microscopic appearance of the blue basaloid cells contrasts with the clear cells seen in hidradenoma.5
Merkel cell carcinoma is a cutaneous neuroendocrine tumor affecting elderly or immunosuppressed individuals. It arises in sun-exposed areas and often is associated with Merkel cell polyomavirus infection. The histologic features display small and round cells that stain positive for CK8, CK18, CK19, and CK20 but stain negative for CK7, a marker that often is positive in hidradenocarcinoma.6
Lymphangioma, particularly cavernous lymphangioma, may resemble the gross appearance of hidradenoma/ hidradenocarcinoma. It usually presents as irregular clear blue papules and nodules in the skin and subcutaneous tissue.7 The key histopathologic finding in this tumor is the endothelium-lined channels that stain positive for D2-40, a lymphatic endothelium marker.7,8
Cutaneous Crohn disease is classified as noncaseating granulomatous skin lesions that are noncontinuous with the gastrointestinal tract.9 Clinical presentations in addition to skin edema include erythematous plaques, ulcerations, and erosions. Histopathology reveals sterile noncaseating granulomas made of Langerhans giant cells, epithelioid histocytes, and plasma cells.9
Metastatic clear cell carcinomas, such as renal cell carcinoma, can be differentiated by a history of primary carcinoma, demonstration of histologic vascular stroma, and other features related to metastatic clear cell carcinoma.2
There are no well-established therapeutic guidelines for hidradenocarcinoma. Wide local excision with margins greater than 2 cm is the preferred initial treatment and often is performed in conjunction with sentinel lymph node biopsy. External beam radiotherapy and adjunctive chemotherapy have been used for tumors that could not be surgically cleared. However, the efficacy of these treatments has not been well established.2 Targeted therapies recently have emerged as an alternative treatment choice for hidradenocarcinoma due to the utilization of immunohistochemical and genomic testing. The discovery of specific gene mutations or the expression of hormonal receptors in this tumor have paved the way for targeting HER2-expressing hidradenocarcinomas with trastuzumab and those expressing estrogen receptor with the estrogen receptor inhibitor tamoxifen.1 Epidermal growth factor receptor inhibitors and PI3K/Akt/mTOR (phosphatidylinositol-3-kinase/AKT/mammalian target of rapamycin) pathway inhibitors also have been used to target various signal transduction pathways.2
Wide excision with 2.5-cm margins was performed on our patient, and a positron emission tomography– computed tomography scan revealed no metastatic disease. She declined sentinel lymph node biopsy and additional treatment. Due to the risk for recurrence, she was monitored closely with skin examinations and positron emission tomography–computed tomography every 3 months for the first year and every 6 months thereafter. Thus far, she has had no evidence of local or regional recurrence.
- Miller DH, Peterson JL, Buskirk SJ, et al. Management of metastatic apocrine hidradenocarcinoma with chemotherapy and radiation. Rare Tumors. 2015;7:6082.
- Soni A, Bansal N, Kaushal V, et al. Current management approach to hidradenocarcinoma: a comprehensive review of the literature. Ecancermedicalscience. 2015;9:517.
- Jinnah AH, Emory CL, Mai NH, et al. Hidradenocarcinoma presenting as soft tissue mass: case report with cytomorphologic description, histologic correlation, and differential diagnosis. Diagn Cytopathol. 2016;44:438-441.
- Khan BM, Mansha MA, Ali N, et al. Hidradenocarcinoma: five years of local and systemic control of a rare sweat gland neoplasm with nodal metastasis. Cureus. 2018;10:E2884.
- Miceli A, Ferrer-Bruker SJ. Spiradenoma. StatPearls. StatPearls Publishing; 2019.
- Banks PD, Sandhu S, Gyorki DE, et al. Recent insights and advances in the management of Merkel cell carcinoma. J Oncol Pract. 2016; 12:637-646.
- Flanagan BP, Helwig EB. Cutaneous lymphangioma. Arch Dermatol. 1977;113:24-30.
- Kalof AN, Cooper K. D2-40 immunohistochemistry—so far! Adv Anat Pathol. 2009;16:62-64.
- Schneider SL, Foster K, Patel D, et al. Cutaneous manifestations of metastatic Crohn’s disease. Pediatr Dermatol. 2018;35:566-574.
The Diagnosis: Hidradenocarcinoma
An excisional biopsy revealed a neoplasm in the dermis with focal invasion into the adjacent soft tissue (Figure 1). The tumor consisted of sheets of cells with cytoplasmic vacuoles and ductal differentiation (Figure 2), as well as cells with mild atypia, mild pleomorphism, rare mitotic figures, and abundant pale cytoplasm. Immunohistochemical staining was positive for cytokeratin (CK) 5, CK7, CK20, CK AE1/AE3, and p63 (Figure 3). The culmination of features including the large tumor size, immunohistochemical staining pattern, and mild pleomorphism with focal invasion into the soft tissue supported the diagnosis of hidradenocarcinoma.
Hidradenocarcinoma is an exceedingly rare malignant tumor of eccrine and/or apocrine origin.1 It accounts for less than 0.001% of all tumors and 1 of 13,000 skin biopsies.2 It usually arises in the head and neck region and most commonly affects older adults aged 50 to 70 years.3 The size of hidradenocarcinomas can vary; however, they typically are large, often growing to be greater than 5 cm in diameter.2 It tends to be an aggressive tumor that generally spreads to regional lymph nodes and distant viscera.4 Although it most commonly arises de novo, it may occasionally derive from a benign hidradenoma.1 The diagnosis of hidradenocarcinoma is made based on the tumor’s morphologic and pathologic characteristics. Histologically, it is characterized by an infiltrative and invasive proliferation of lobules made of large clear cells with atypical mitotic figures and nuclear pleomorphism as well as immunohistochemical features displaying various positive markers, such as carcinoembryonic antigen, epithelial membrane antigen, S-100 protein, and CKs AE1/AE3 and 5/6.2 Invasion of the adjacent soft tissue can be present and helps to confirm the diagnosis.
The differential diagnosis for hidradenocarcinoma primarily is the benign hidradenoma, which is similar both clinically and histologically with a few important differences. Hidradenocarcinomas often are larger and ulcerated. Histologically, they usually are more pleomorphic with the presence of mitotic figures in clear cells and tend to invade locally into the surrounding soft tissue. Other similar lesions such as spiradenoma, Merkel cell carcinoma, lymphangioma, cutaneous Crohn disease, tumors metastatic to the skin, and metastatic clear cell carcinomas originating from other organs also are included in the differential diagnosis.2
Spiradenomas are dermal tumors originating from the sweat glands. They typically present as bluish, painful, solitary nodules on the ventral surfaces of the upper body, though multiple nodules also are reported.5 Spiradenomas manifest as a central constellation of pale large cells surrounded by small, dark, basaloid cells containing hyperchromatic nuclei. The microscopic appearance of the blue basaloid cells contrasts with the clear cells seen in hidradenoma.5
Merkel cell carcinoma is a cutaneous neuroendocrine tumor affecting elderly or immunosuppressed individuals. It arises in sun-exposed areas and often is associated with Merkel cell polyomavirus infection. The histologic features display small and round cells that stain positive for CK8, CK18, CK19, and CK20 but stain negative for CK7, a marker that often is positive in hidradenocarcinoma.6
Lymphangioma, particularly cavernous lymphangioma, may resemble the gross appearance of hidradenoma/ hidradenocarcinoma. It usually presents as irregular clear blue papules and nodules in the skin and subcutaneous tissue.7 The key histopathologic finding in this tumor is the endothelium-lined channels that stain positive for D2-40, a lymphatic endothelium marker.7,8
Cutaneous Crohn disease is classified as noncaseating granulomatous skin lesions that are noncontinuous with the gastrointestinal tract.9 Clinical presentations in addition to skin edema include erythematous plaques, ulcerations, and erosions. Histopathology reveals sterile noncaseating granulomas made of Langerhans giant cells, epithelioid histocytes, and plasma cells.9
Metastatic clear cell carcinomas, such as renal cell carcinoma, can be differentiated by a history of primary carcinoma, demonstration of histologic vascular stroma, and other features related to metastatic clear cell carcinoma.2
There are no well-established therapeutic guidelines for hidradenocarcinoma. Wide local excision with margins greater than 2 cm is the preferred initial treatment and often is performed in conjunction with sentinel lymph node biopsy. External beam radiotherapy and adjunctive chemotherapy have been used for tumors that could not be surgically cleared. However, the efficacy of these treatments has not been well established.2 Targeted therapies recently have emerged as an alternative treatment choice for hidradenocarcinoma due to the utilization of immunohistochemical and genomic testing. The discovery of specific gene mutations or the expression of hormonal receptors in this tumor have paved the way for targeting HER2-expressing hidradenocarcinomas with trastuzumab and those expressing estrogen receptor with the estrogen receptor inhibitor tamoxifen.1 Epidermal growth factor receptor inhibitors and PI3K/Akt/mTOR (phosphatidylinositol-3-kinase/AKT/mammalian target of rapamycin) pathway inhibitors also have been used to target various signal transduction pathways.2
Wide excision with 2.5-cm margins was performed on our patient, and a positron emission tomography– computed tomography scan revealed no metastatic disease. She declined sentinel lymph node biopsy and additional treatment. Due to the risk for recurrence, she was monitored closely with skin examinations and positron emission tomography–computed tomography every 3 months for the first year and every 6 months thereafter. Thus far, she has had no evidence of local or regional recurrence.
The Diagnosis: Hidradenocarcinoma
An excisional biopsy revealed a neoplasm in the dermis with focal invasion into the adjacent soft tissue (Figure 1). The tumor consisted of sheets of cells with cytoplasmic vacuoles and ductal differentiation (Figure 2), as well as cells with mild atypia, mild pleomorphism, rare mitotic figures, and abundant pale cytoplasm. Immunohistochemical staining was positive for cytokeratin (CK) 5, CK7, CK20, CK AE1/AE3, and p63 (Figure 3). The culmination of features including the large tumor size, immunohistochemical staining pattern, and mild pleomorphism with focal invasion into the soft tissue supported the diagnosis of hidradenocarcinoma.
Hidradenocarcinoma is an exceedingly rare malignant tumor of eccrine and/or apocrine origin.1 It accounts for less than 0.001% of all tumors and 1 of 13,000 skin biopsies.2 It usually arises in the head and neck region and most commonly affects older adults aged 50 to 70 years.3 The size of hidradenocarcinomas can vary; however, they typically are large, often growing to be greater than 5 cm in diameter.2 It tends to be an aggressive tumor that generally spreads to regional lymph nodes and distant viscera.4 Although it most commonly arises de novo, it may occasionally derive from a benign hidradenoma.1 The diagnosis of hidradenocarcinoma is made based on the tumor’s morphologic and pathologic characteristics. Histologically, it is characterized by an infiltrative and invasive proliferation of lobules made of large clear cells with atypical mitotic figures and nuclear pleomorphism as well as immunohistochemical features displaying various positive markers, such as carcinoembryonic antigen, epithelial membrane antigen, S-100 protein, and CKs AE1/AE3 and 5/6.2 Invasion of the adjacent soft tissue can be present and helps to confirm the diagnosis.
The differential diagnosis for hidradenocarcinoma primarily is the benign hidradenoma, which is similar both clinically and histologically with a few important differences. Hidradenocarcinomas often are larger and ulcerated. Histologically, they usually are more pleomorphic with the presence of mitotic figures in clear cells and tend to invade locally into the surrounding soft tissue. Other similar lesions such as spiradenoma, Merkel cell carcinoma, lymphangioma, cutaneous Crohn disease, tumors metastatic to the skin, and metastatic clear cell carcinomas originating from other organs also are included in the differential diagnosis.2
Spiradenomas are dermal tumors originating from the sweat glands. They typically present as bluish, painful, solitary nodules on the ventral surfaces of the upper body, though multiple nodules also are reported.5 Spiradenomas manifest as a central constellation of pale large cells surrounded by small, dark, basaloid cells containing hyperchromatic nuclei. The microscopic appearance of the blue basaloid cells contrasts with the clear cells seen in hidradenoma.5
Merkel cell carcinoma is a cutaneous neuroendocrine tumor affecting elderly or immunosuppressed individuals. It arises in sun-exposed areas and often is associated with Merkel cell polyomavirus infection. The histologic features display small and round cells that stain positive for CK8, CK18, CK19, and CK20 but stain negative for CK7, a marker that often is positive in hidradenocarcinoma.6
Lymphangioma, particularly cavernous lymphangioma, may resemble the gross appearance of hidradenoma/ hidradenocarcinoma. It usually presents as irregular clear blue papules and nodules in the skin and subcutaneous tissue.7 The key histopathologic finding in this tumor is the endothelium-lined channels that stain positive for D2-40, a lymphatic endothelium marker.7,8
Cutaneous Crohn disease is classified as noncaseating granulomatous skin lesions that are noncontinuous with the gastrointestinal tract.9 Clinical presentations in addition to skin edema include erythematous plaques, ulcerations, and erosions. Histopathology reveals sterile noncaseating granulomas made of Langerhans giant cells, epithelioid histocytes, and plasma cells.9
Metastatic clear cell carcinomas, such as renal cell carcinoma, can be differentiated by a history of primary carcinoma, demonstration of histologic vascular stroma, and other features related to metastatic clear cell carcinoma.2
There are no well-established therapeutic guidelines for hidradenocarcinoma. Wide local excision with margins greater than 2 cm is the preferred initial treatment and often is performed in conjunction with sentinel lymph node biopsy. External beam radiotherapy and adjunctive chemotherapy have been used for tumors that could not be surgically cleared. However, the efficacy of these treatments has not been well established.2 Targeted therapies recently have emerged as an alternative treatment choice for hidradenocarcinoma due to the utilization of immunohistochemical and genomic testing. The discovery of specific gene mutations or the expression of hormonal receptors in this tumor have paved the way for targeting HER2-expressing hidradenocarcinomas with trastuzumab and those expressing estrogen receptor with the estrogen receptor inhibitor tamoxifen.1 Epidermal growth factor receptor inhibitors and PI3K/Akt/mTOR (phosphatidylinositol-3-kinase/AKT/mammalian target of rapamycin) pathway inhibitors also have been used to target various signal transduction pathways.2
Wide excision with 2.5-cm margins was performed on our patient, and a positron emission tomography– computed tomography scan revealed no metastatic disease. She declined sentinel lymph node biopsy and additional treatment. Due to the risk for recurrence, she was monitored closely with skin examinations and positron emission tomography–computed tomography every 3 months for the first year and every 6 months thereafter. Thus far, she has had no evidence of local or regional recurrence.
- Miller DH, Peterson JL, Buskirk SJ, et al. Management of metastatic apocrine hidradenocarcinoma with chemotherapy and radiation. Rare Tumors. 2015;7:6082.
- Soni A, Bansal N, Kaushal V, et al. Current management approach to hidradenocarcinoma: a comprehensive review of the literature. Ecancermedicalscience. 2015;9:517.
- Jinnah AH, Emory CL, Mai NH, et al. Hidradenocarcinoma presenting as soft tissue mass: case report with cytomorphologic description, histologic correlation, and differential diagnosis. Diagn Cytopathol. 2016;44:438-441.
- Khan BM, Mansha MA, Ali N, et al. Hidradenocarcinoma: five years of local and systemic control of a rare sweat gland neoplasm with nodal metastasis. Cureus. 2018;10:E2884.
- Miceli A, Ferrer-Bruker SJ. Spiradenoma. StatPearls. StatPearls Publishing; 2019.
- Banks PD, Sandhu S, Gyorki DE, et al. Recent insights and advances in the management of Merkel cell carcinoma. J Oncol Pract. 2016; 12:637-646.
- Flanagan BP, Helwig EB. Cutaneous lymphangioma. Arch Dermatol. 1977;113:24-30.
- Kalof AN, Cooper K. D2-40 immunohistochemistry—so far! Adv Anat Pathol. 2009;16:62-64.
- Schneider SL, Foster K, Patel D, et al. Cutaneous manifestations of metastatic Crohn’s disease. Pediatr Dermatol. 2018;35:566-574.
- Miller DH, Peterson JL, Buskirk SJ, et al. Management of metastatic apocrine hidradenocarcinoma with chemotherapy and radiation. Rare Tumors. 2015;7:6082.
- Soni A, Bansal N, Kaushal V, et al. Current management approach to hidradenocarcinoma: a comprehensive review of the literature. Ecancermedicalscience. 2015;9:517.
- Jinnah AH, Emory CL, Mai NH, et al. Hidradenocarcinoma presenting as soft tissue mass: case report with cytomorphologic description, histologic correlation, and differential diagnosis. Diagn Cytopathol. 2016;44:438-441.
- Khan BM, Mansha MA, Ali N, et al. Hidradenocarcinoma: five years of local and systemic control of a rare sweat gland neoplasm with nodal metastasis. Cureus. 2018;10:E2884.
- Miceli A, Ferrer-Bruker SJ. Spiradenoma. StatPearls. StatPearls Publishing; 2019.
- Banks PD, Sandhu S, Gyorki DE, et al. Recent insights and advances in the management of Merkel cell carcinoma. J Oncol Pract. 2016; 12:637-646.
- Flanagan BP, Helwig EB. Cutaneous lymphangioma. Arch Dermatol. 1977;113:24-30.
- Kalof AN, Cooper K. D2-40 immunohistochemistry—so far! Adv Anat Pathol. 2009;16:62-64.
- Schneider SL, Foster K, Patel D, et al. Cutaneous manifestations of metastatic Crohn’s disease. Pediatr Dermatol. 2018;35:566-574.
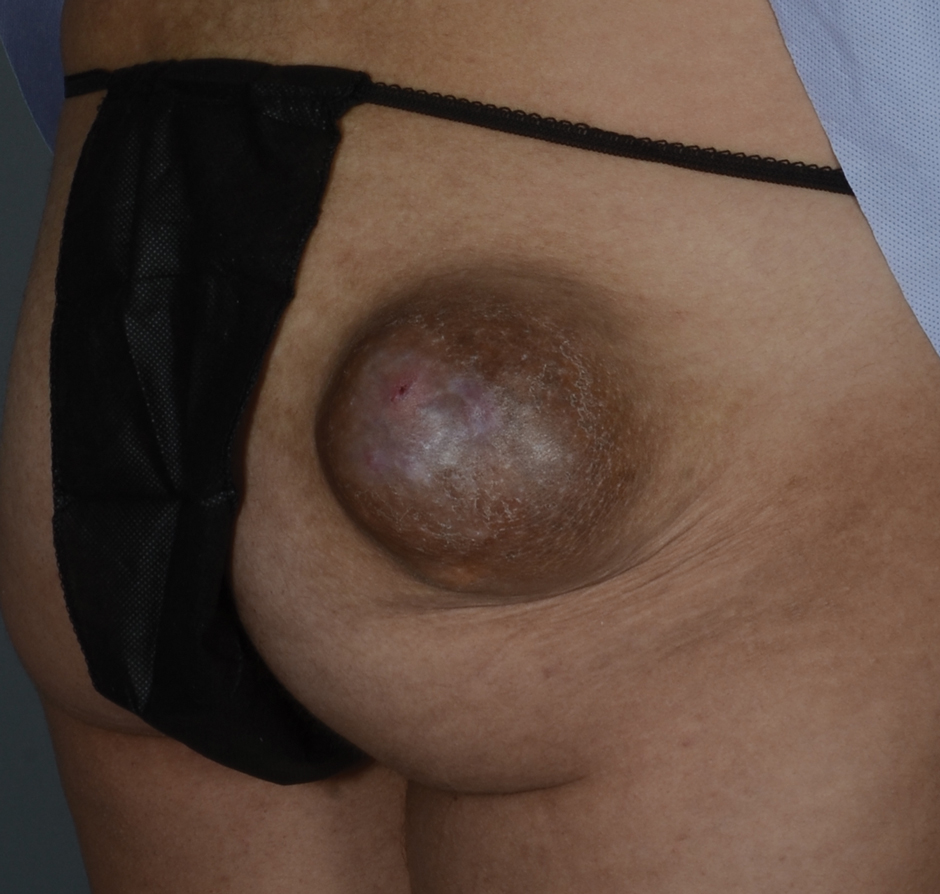
A 20-year-old woman with no notable medical history presented to the dermatology clinic with an enlarging mass on the right buttock that had been growing over the course of several years. The mass progressed from a small, mildly tender nodule to a 10×10-cm, hyperpigmented, exophytic tumor. There were no other abnormal findings on physical examination, and the patient denied any systemic symptoms.