User login
Progressive Axillary Hyperpigmentation
The Diagnosis: Dowling-Degos Disease
Histopathology demonstrated elongation of the epidermal rete ridges with increased basal pigmentation, suprapapillary epithelial thinning, dermal melanophages, and a mild lymphocytic infiltrate (Figure). Given the clinical and histologic findings, a diagnosis of Dowling-Degos disease (DDD) was made. The patient was counseled on the increased risk for her children developing DDD. Treatment with the erbium:YAG (Er:YAG) laser subsequently was initiated.

Dowling-Degos disease (also known as reticulate pigmented anomaly of the flexures) is an uncommon autosomal-dominant condition characterized by reticular hyperpigmentation involving the flexural and intertriginous sites. Classic DDD commonly is caused by lossof-function mutations in the keratin 5 gene, KRT51; however, DDD also may result from loss-of-function mutations in the protein O-fucosyltransferase 1, POFUT1, and protein O-glucosyltransferase 1, POGLUT1, genes.2
Rare cases of DDD associated with hidradenitis suppurativa are caused by mutations in the presenilin enhancer protein 2 gene, PSENEN.3
Of note, a missense mutation in KRT5 is implicated in epidermolysis bullosa simplex with mottled pigmentation. Onset of DDD typically occurs during the third to fourth decades of life. Reticulated hyperpigmented macules initially occur in the axillae and groin and progressively increase over time to involve the neck, inframammary folds, trunk, and flexural surfaces of the arms and thighs. Patients additionally may present with pitted perioral scars, comedolike lesions on the back and neck, epidermoid cysts, and hidradenitis suppurativa. Keratoacanthoma and squamous cell carcinoma rarely have been reported in association with classic DDD.4,5
Dowling-Degos disease usually is asymptomatic, though pruritus seldom may occur in the affected flexural areas. Histologically, the epidermal rete ridges are elongated in a filiform or antlerlike pattern with increased pigmentation of the basal layer and thinning of the suprapapillary epithelium. Dermal melanosis and a mild perivascular lymphohistiocytic infiltrate also are present with no increase in the number of melanocytes.6,7 Galli-Galli disease is a variant of DDD that shares similar clinical and histologic features of DDD but is distinguished from DDD by suprabasilar nondyskeratotic acantholysis on histology.8
Regarding other differential diagnoses for our patient, acanthosis nigricans may be distinguished clinically by the presence of velvety and/or verrucous plaques, commonly in the neck folds and axillae. Histologically, acanthosis nigricans is distinct from DDD and involves hyperkeratosis, acanthosis, and epidermal papillomatosis. Our patient had no history of diabetes mellitus or insulin resistance. Granular parakeratosis presents with hyperpigmented hyperkeratotic papules and plaques classically confined to the axillary region; however, the involvement of other intertriginous areas may occur. Histologically, granular parakeratosis demonstrates compact parakeratosis with small bluish keratohyalin granules within the stratum corneum. Confluent and reticulated papillomatosis presents with red-brown keratotic papules that initially appear in the intermammary region and spread laterally forming a reticulated pattern. Histology is similar to acanthosis nigricans and demonstrates hyperkeratosis, acanthosis, and papillomatosis. Inverse psoriasis presents with symmetric and sharply demarcated, erythematous, nonscaly plaques in the intertriginous areas. The plaques of inverse psoriasis may be pruritic and/or sore and occasionally may become macerated. Inverse psoriasis shares similar histologic findings compared to classic plaque psoriasis but may have less confluent parakeratosis.
Treatment of DDD essentially is reserved for cosmetic reasons. Topical hydroquinone, tretinoin, and corticosteroids have been used with limited to no success.5,9 Beneficial results after treatment with the Er:YAG laser have been reported.10
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-519.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomaldominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135-143.
- Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-508.
- Ujihara M, Kamakura T, Ikeda M, et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-571.
- Weber LA, Kantor GR, Bergfeld WF. Reticulate pigmented anomaly of the flexures (Dowling-Degos disease): a case report associated with hidradenitis suppurativa and squamous cell carcinoma. Cutis. 1990;45:446-450.
- Jones EW, Grice K. Reticulate pigmented anomaly of the flexures. Dowing Degos disease, a new genodermatosis. Arch Dermatol. 1978;114:1150-1157.
- Kim YC, Davis MD, Schanbacher CF, et al. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999; 40:462-467.
- Reisenauer AK, Wordingham SV, York J, et al. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br J Dermatol. 2014;170:1362-1365.
- Oppolzer G, Schwarz T, Duschet P, et al. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids [in German]. Hautarzt. 1987;38:615-618.
- Wenzel G, Petrow W, Tappe K, et al. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatol Surg. 2003;29:1161-1162.
The Diagnosis: Dowling-Degos Disease
Histopathology demonstrated elongation of the epidermal rete ridges with increased basal pigmentation, suprapapillary epithelial thinning, dermal melanophages, and a mild lymphocytic infiltrate (Figure). Given the clinical and histologic findings, a diagnosis of Dowling-Degos disease (DDD) was made. The patient was counseled on the increased risk for her children developing DDD. Treatment with the erbium:YAG (Er:YAG) laser subsequently was initiated.
Dowling-Degos disease (also known as reticulate pigmented anomaly of the flexures) is an uncommon autosomal-dominant condition characterized by reticular hyperpigmentation involving the flexural and intertriginous sites. Classic DDD commonly is caused by lossof-function mutations in the keratin 5 gene, KRT51; however, DDD also may result from loss-of-function mutations in the protein O-fucosyltransferase 1, POFUT1, and protein O-glucosyltransferase 1, POGLUT1, genes.2
Rare cases of DDD associated with hidradenitis suppurativa are caused by mutations in the presenilin enhancer protein 2 gene, PSENEN.3
Of note, a missense mutation in KRT5 is implicated in epidermolysis bullosa simplex with mottled pigmentation. Onset of DDD typically occurs during the third to fourth decades of life. Reticulated hyperpigmented macules initially occur in the axillae and groin and progressively increase over time to involve the neck, inframammary folds, trunk, and flexural surfaces of the arms and thighs. Patients additionally may present with pitted perioral scars, comedolike lesions on the back and neck, epidermoid cysts, and hidradenitis suppurativa. Keratoacanthoma and squamous cell carcinoma rarely have been reported in association with classic DDD.4,5
Dowling-Degos disease usually is asymptomatic, though pruritus seldom may occur in the affected flexural areas. Histologically, the epidermal rete ridges are elongated in a filiform or antlerlike pattern with increased pigmentation of the basal layer and thinning of the suprapapillary epithelium. Dermal melanosis and a mild perivascular lymphohistiocytic infiltrate also are present with no increase in the number of melanocytes.6,7 Galli-Galli disease is a variant of DDD that shares similar clinical and histologic features of DDD but is distinguished from DDD by suprabasilar nondyskeratotic acantholysis on histology.8
Regarding other differential diagnoses for our patient, acanthosis nigricans may be distinguished clinically by the presence of velvety and/or verrucous plaques, commonly in the neck folds and axillae. Histologically, acanthosis nigricans is distinct from DDD and involves hyperkeratosis, acanthosis, and epidermal papillomatosis. Our patient had no history of diabetes mellitus or insulin resistance. Granular parakeratosis presents with hyperpigmented hyperkeratotic papules and plaques classically confined to the axillary region; however, the involvement of other intertriginous areas may occur. Histologically, granular parakeratosis demonstrates compact parakeratosis with small bluish keratohyalin granules within the stratum corneum. Confluent and reticulated papillomatosis presents with red-brown keratotic papules that initially appear in the intermammary region and spread laterally forming a reticulated pattern. Histology is similar to acanthosis nigricans and demonstrates hyperkeratosis, acanthosis, and papillomatosis. Inverse psoriasis presents with symmetric and sharply demarcated, erythematous, nonscaly plaques in the intertriginous areas. The plaques of inverse psoriasis may be pruritic and/or sore and occasionally may become macerated. Inverse psoriasis shares similar histologic findings compared to classic plaque psoriasis but may have less confluent parakeratosis.
Treatment of DDD essentially is reserved for cosmetic reasons. Topical hydroquinone, tretinoin, and corticosteroids have been used with limited to no success.5,9 Beneficial results after treatment with the Er:YAG laser have been reported.10
The Diagnosis: Dowling-Degos Disease
Histopathology demonstrated elongation of the epidermal rete ridges with increased basal pigmentation, suprapapillary epithelial thinning, dermal melanophages, and a mild lymphocytic infiltrate (Figure). Given the clinical and histologic findings, a diagnosis of Dowling-Degos disease (DDD) was made. The patient was counseled on the increased risk for her children developing DDD. Treatment with the erbium:YAG (Er:YAG) laser subsequently was initiated.
Dowling-Degos disease (also known as reticulate pigmented anomaly of the flexures) is an uncommon autosomal-dominant condition characterized by reticular hyperpigmentation involving the flexural and intertriginous sites. Classic DDD commonly is caused by lossof-function mutations in the keratin 5 gene, KRT51; however, DDD also may result from loss-of-function mutations in the protein O-fucosyltransferase 1, POFUT1, and protein O-glucosyltransferase 1, POGLUT1, genes.2
Rare cases of DDD associated with hidradenitis suppurativa are caused by mutations in the presenilin enhancer protein 2 gene, PSENEN.3
Of note, a missense mutation in KRT5 is implicated in epidermolysis bullosa simplex with mottled pigmentation. Onset of DDD typically occurs during the third to fourth decades of life. Reticulated hyperpigmented macules initially occur in the axillae and groin and progressively increase over time to involve the neck, inframammary folds, trunk, and flexural surfaces of the arms and thighs. Patients additionally may present with pitted perioral scars, comedolike lesions on the back and neck, epidermoid cysts, and hidradenitis suppurativa. Keratoacanthoma and squamous cell carcinoma rarely have been reported in association with classic DDD.4,5
Dowling-Degos disease usually is asymptomatic, though pruritus seldom may occur in the affected flexural areas. Histologically, the epidermal rete ridges are elongated in a filiform or antlerlike pattern with increased pigmentation of the basal layer and thinning of the suprapapillary epithelium. Dermal melanosis and a mild perivascular lymphohistiocytic infiltrate also are present with no increase in the number of melanocytes.6,7 Galli-Galli disease is a variant of DDD that shares similar clinical and histologic features of DDD but is distinguished from DDD by suprabasilar nondyskeratotic acantholysis on histology.8
Regarding other differential diagnoses for our patient, acanthosis nigricans may be distinguished clinically by the presence of velvety and/or verrucous plaques, commonly in the neck folds and axillae. Histologically, acanthosis nigricans is distinct from DDD and involves hyperkeratosis, acanthosis, and epidermal papillomatosis. Our patient had no history of diabetes mellitus or insulin resistance. Granular parakeratosis presents with hyperpigmented hyperkeratotic papules and plaques classically confined to the axillary region; however, the involvement of other intertriginous areas may occur. Histologically, granular parakeratosis demonstrates compact parakeratosis with small bluish keratohyalin granules within the stratum corneum. Confluent and reticulated papillomatosis presents with red-brown keratotic papules that initially appear in the intermammary region and spread laterally forming a reticulated pattern. Histology is similar to acanthosis nigricans and demonstrates hyperkeratosis, acanthosis, and papillomatosis. Inverse psoriasis presents with symmetric and sharply demarcated, erythematous, nonscaly plaques in the intertriginous areas. The plaques of inverse psoriasis may be pruritic and/or sore and occasionally may become macerated. Inverse psoriasis shares similar histologic findings compared to classic plaque psoriasis but may have less confluent parakeratosis.
Treatment of DDD essentially is reserved for cosmetic reasons. Topical hydroquinone, tretinoin, and corticosteroids have been used with limited to no success.5,9 Beneficial results after treatment with the Er:YAG laser have been reported.10
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-519.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomaldominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135-143.
- Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-508.
- Ujihara M, Kamakura T, Ikeda M, et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-571.
- Weber LA, Kantor GR, Bergfeld WF. Reticulate pigmented anomaly of the flexures (Dowling-Degos disease): a case report associated with hidradenitis suppurativa and squamous cell carcinoma. Cutis. 1990;45:446-450.
- Jones EW, Grice K. Reticulate pigmented anomaly of the flexures. Dowing Degos disease, a new genodermatosis. Arch Dermatol. 1978;114:1150-1157.
- Kim YC, Davis MD, Schanbacher CF, et al. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999; 40:462-467.
- Reisenauer AK, Wordingham SV, York J, et al. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br J Dermatol. 2014;170:1362-1365.
- Oppolzer G, Schwarz T, Duschet P, et al. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids [in German]. Hautarzt. 1987;38:615-618.
- Wenzel G, Petrow W, Tappe K, et al. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatol Surg. 2003;29:1161-1162.
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-519.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomaldominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135-143.
- Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-508.
- Ujihara M, Kamakura T, Ikeda M, et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-571.
- Weber LA, Kantor GR, Bergfeld WF. Reticulate pigmented anomaly of the flexures (Dowling-Degos disease): a case report associated with hidradenitis suppurativa and squamous cell carcinoma. Cutis. 1990;45:446-450.
- Jones EW, Grice K. Reticulate pigmented anomaly of the flexures. Dowing Degos disease, a new genodermatosis. Arch Dermatol. 1978;114:1150-1157.
- Kim YC, Davis MD, Schanbacher CF, et al. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999; 40:462-467.
- Reisenauer AK, Wordingham SV, York J, et al. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br J Dermatol. 2014;170:1362-1365.
- Oppolzer G, Schwarz T, Duschet P, et al. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids [in German]. Hautarzt. 1987;38:615-618.
- Wenzel G, Petrow W, Tappe K, et al. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatol Surg. 2003;29:1161-1162.

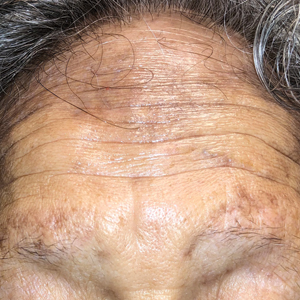
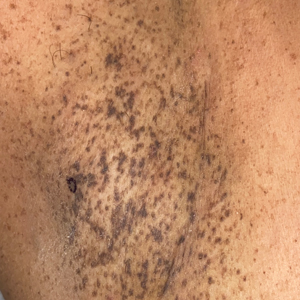
A 50-year-old Hispanic woman presented with asymptomatic, progressive, brown hyperpigmentation involving the axillae, neck, upper back, and inframammary areas of 5 years’ duration. She had no other notable medical history; family history was unremarkable. She had been treated with topical hydroquinone and tretinoin by an outside physician without improvement. Physical examination revealed reticulated hyperpigmented macules and patches involving the inverse regions of the neck, axillae, and inframammary regions. Additionally, acneform pitted scars involving the perioral region were seen. A 4.0-mm punch biopsy of the right axilla was performed.
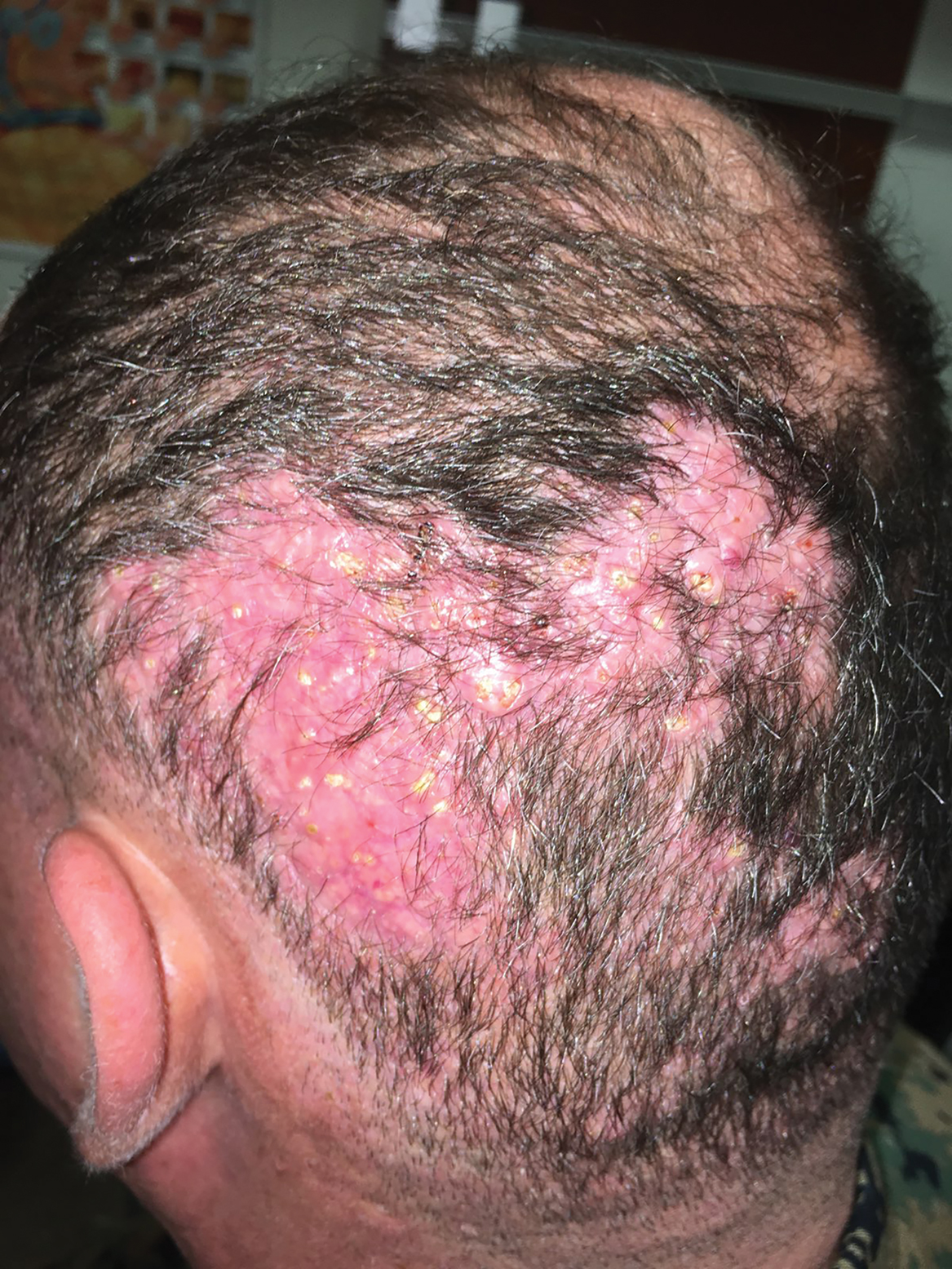
Hard Nodular Plaque on the Scalp
The Diagnosis: Platelike Osteoma Cutis
Histopathologic examination revealed extensive cutaneous ossification in the dermis and subcutis with dermal fibrosis and minimal surrounding inflammation (Figure 1). There was no evidence of infection or neoplasm. Further evaluation did not demonstrate any additional physical dysmorphia, and there were no imbalances of calcium-phosphate metabolism or abnormalities in parathyroid hormone or thyroid hormone function. A diagnosis of platelike osteoma cutis (PLOC) was favored. Computed tomography of the head showed material at the posterior skull of similar density to the adjacent calvarial skull and centered within the dermis, consistent with osteoma cutis (Figure 2).
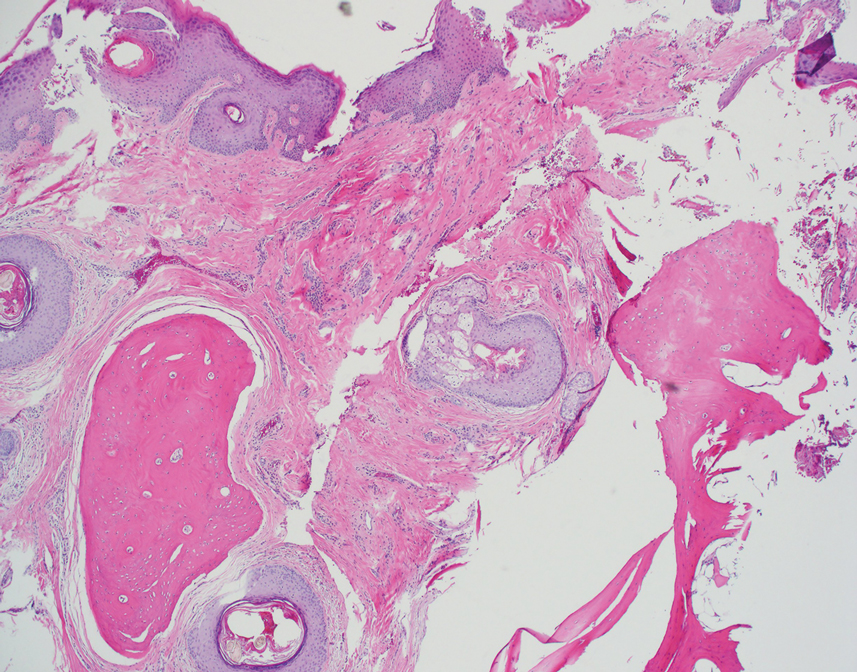
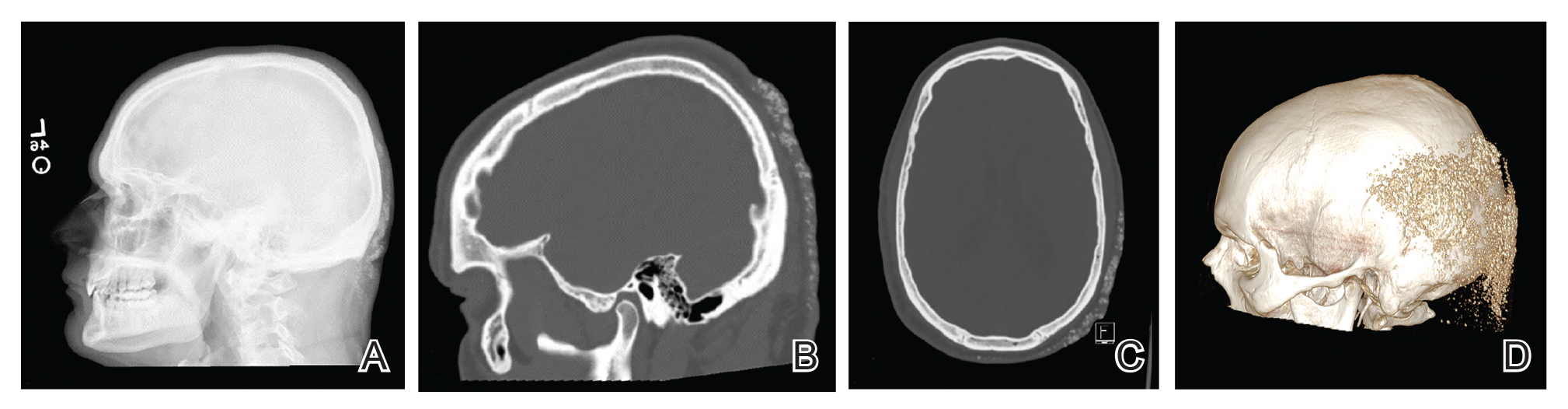
Osteoma cutis describes the formation of bone within the skin. It occurs when hydroxyapatite crystals in a proteinaceous matrix are deposited within the skin, ultimately leading to the formation of bone ultrastructure. Ossification of the skin most often occurs secondary to trauma, inflammation, or neoplasm; however, it rarely may be a primary event.1,2
Platelike osteoma cutis is a rare form of primary cutaneous ossification in which bone forms within the skin in a platelike manner. It most frequently affects the scalp but also has been observed on the trunk and extremities.1 A driving metabolic or endocrine abnormality typically is not identified.2
Platelike osteoma cutis can occur as an isolated finding or as a feature of Albright hereditary osteodystrophy (AHO) or progressive osseous heteroplasia (POH). In addition to cutaneous ossification, AHO involves short stature, endocrinopathy, obesity, shortened fourth and fifth metacarpals, and mental retardation. Progressive osseous heteroplasia is characterized by progressive ossification of the skin and deeper tissues such as muscle and fascia, leading to severe movement restriction; it is believed to be a localized nonprogressive variant of POH.3,4 Mutations in the guanine nucleotide binding protein, alpha stimulating activity polypeptide 1 gene, GNAS1, a key regulatory gene involved in AHO and POH, have been found in several cases of PLOC.3 Our patient lacked any dysmorphic features or laboratory abnormalities suggestive of AHO or POH. Moreover, testing of the tissue and blood for the GNAS1 mutation was negative. Treatment of PLOC often is difficult. Our patient underwent a trial of ablative fractional laser resurfacing, which failed to lead to perceivable improvement.
The differential diagnoses include a kerion, dissecting cellulitis of the scalp, folliculitis decalvans, and acne keloidalis nuchae. A kerion is a manifestation of tinea capitis characterized by an inflammatory plaque, often with pain or tenderness. Kerions most frequently occur in children aged 5 to 10 years.5 Failure to treat a kerion may result in scarring alopecia. Treatment consists of oral antifungals.
Dissecting cellulitis of the scalp is thought to occur secondary to follicular occlusion. It is characterized by boggy suppurative nodules primarily on the posterior and vertex scalp. Patchy hair loss is present and typically progresses to cicatricial alopecia. Histology characteristically shows areas of dense, predominantly neutrophilic, perifollicular dermal infiltrates.6
Folliculitis decalvans is a primary neutrophilic cicatricial alopecia that primarily occurs in adults. Patients with folliculitis decalvans tend to have multiple pustules on the periphery of confluent areas of scarring alopecia. It is theorized that an immune response to staphylococcal superantigens contributes to this disease process.7
The clinical findings of acne keloidalis nuchae include inflammatory pustules and papules with keloidlike plaques on the posterior neck and scalp. It occurs predominantly in teenaged and adult males of African ancestry.8 Treatment is aimed at reducing inflammation and preventing exacerbating factors. Severe disease courses may lead to scarring alopecia.
- Sanmartín O, Alegre V, Martinez-Aparicio A, et al. Congenital platelike osteoma cutis: case report and review of the literature. Pediatr Dermatol. 1993;10:182-186.
- Talsania N, Jolliffe V, O’Toole EA, et al. Platelike osteoma cutis. J Am Acad Dermatol. 2009;64:613-615.
- Yeh GL, Mathur S, Wivel A, et al. GNAS1 mutation and Cbfa1 misexpression in a child with severe congenital platelike osteoma cutis. J Bone Miner Res. 2000;15:2063-2073.
- Hernandez-Martin A, Perez-Mies B, Torrelo A. Congenital plate-like osteoma cutis in an infant. Pediatr Dermatol. 2009;26:479-481.
- Zaraa I, Hawilo A, Aounallah A, et al. Inflammatory tinea capitis: a 12-year study and a review of the literature. Mycoses. 2013;56:110-116.
- Scheinfeld N. Dissecting cellulitis (perifolliculitis capitis abscedens et suffodiens): a comprehensive review focusing on new treatments and findings of the last decade with commentary comparing the therapies and causes of dissecting cellulitis to hidradenitis suppurativa. Dermatol Online J. 2014;20:22692.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Knable AL Jr, Hanke CW, Gonin R. Prevalence of acne keloidalis nuchae in football players. J Am Acad Dermatol. 1997;37:570-574.
The Diagnosis: Platelike Osteoma Cutis
Histopathologic examination revealed extensive cutaneous ossification in the dermis and subcutis with dermal fibrosis and minimal surrounding inflammation (Figure 1). There was no evidence of infection or neoplasm. Further evaluation did not demonstrate any additional physical dysmorphia, and there were no imbalances of calcium-phosphate metabolism or abnormalities in parathyroid hormone or thyroid hormone function. A diagnosis of platelike osteoma cutis (PLOC) was favored. Computed tomography of the head showed material at the posterior skull of similar density to the adjacent calvarial skull and centered within the dermis, consistent with osteoma cutis (Figure 2).
Osteoma cutis describes the formation of bone within the skin. It occurs when hydroxyapatite crystals in a proteinaceous matrix are deposited within the skin, ultimately leading to the formation of bone ultrastructure. Ossification of the skin most often occurs secondary to trauma, inflammation, or neoplasm; however, it rarely may be a primary event.1,2
Platelike osteoma cutis is a rare form of primary cutaneous ossification in which bone forms within the skin in a platelike manner. It most frequently affects the scalp but also has been observed on the trunk and extremities.1 A driving metabolic or endocrine abnormality typically is not identified.2
Platelike osteoma cutis can occur as an isolated finding or as a feature of Albright hereditary osteodystrophy (AHO) or progressive osseous heteroplasia (POH). In addition to cutaneous ossification, AHO involves short stature, endocrinopathy, obesity, shortened fourth and fifth metacarpals, and mental retardation. Progressive osseous heteroplasia is characterized by progressive ossification of the skin and deeper tissues such as muscle and fascia, leading to severe movement restriction; it is believed to be a localized nonprogressive variant of POH.3,4 Mutations in the guanine nucleotide binding protein, alpha stimulating activity polypeptide 1 gene, GNAS1, a key regulatory gene involved in AHO and POH, have been found in several cases of PLOC.3 Our patient lacked any dysmorphic features or laboratory abnormalities suggestive of AHO or POH. Moreover, testing of the tissue and blood for the GNAS1 mutation was negative. Treatment of PLOC often is difficult. Our patient underwent a trial of ablative fractional laser resurfacing, which failed to lead to perceivable improvement.
The differential diagnoses include a kerion, dissecting cellulitis of the scalp, folliculitis decalvans, and acne keloidalis nuchae. A kerion is a manifestation of tinea capitis characterized by an inflammatory plaque, often with pain or tenderness. Kerions most frequently occur in children aged 5 to 10 years.5 Failure to treat a kerion may result in scarring alopecia. Treatment consists of oral antifungals.
Dissecting cellulitis of the scalp is thought to occur secondary to follicular occlusion. It is characterized by boggy suppurative nodules primarily on the posterior and vertex scalp. Patchy hair loss is present and typically progresses to cicatricial alopecia. Histology characteristically shows areas of dense, predominantly neutrophilic, perifollicular dermal infiltrates.6
Folliculitis decalvans is a primary neutrophilic cicatricial alopecia that primarily occurs in adults. Patients with folliculitis decalvans tend to have multiple pustules on the periphery of confluent areas of scarring alopecia. It is theorized that an immune response to staphylococcal superantigens contributes to this disease process.7
The clinical findings of acne keloidalis nuchae include inflammatory pustules and papules with keloidlike plaques on the posterior neck and scalp. It occurs predominantly in teenaged and adult males of African ancestry.8 Treatment is aimed at reducing inflammation and preventing exacerbating factors. Severe disease courses may lead to scarring alopecia.
The Diagnosis: Platelike Osteoma Cutis
Histopathologic examination revealed extensive cutaneous ossification in the dermis and subcutis with dermal fibrosis and minimal surrounding inflammation (Figure 1). There was no evidence of infection or neoplasm. Further evaluation did not demonstrate any additional physical dysmorphia, and there were no imbalances of calcium-phosphate metabolism or abnormalities in parathyroid hormone or thyroid hormone function. A diagnosis of platelike osteoma cutis (PLOC) was favored. Computed tomography of the head showed material at the posterior skull of similar density to the adjacent calvarial skull and centered within the dermis, consistent with osteoma cutis (Figure 2).
Osteoma cutis describes the formation of bone within the skin. It occurs when hydroxyapatite crystals in a proteinaceous matrix are deposited within the skin, ultimately leading to the formation of bone ultrastructure. Ossification of the skin most often occurs secondary to trauma, inflammation, or neoplasm; however, it rarely may be a primary event.1,2
Platelike osteoma cutis is a rare form of primary cutaneous ossification in which bone forms within the skin in a platelike manner. It most frequently affects the scalp but also has been observed on the trunk and extremities.1 A driving metabolic or endocrine abnormality typically is not identified.2
Platelike osteoma cutis can occur as an isolated finding or as a feature of Albright hereditary osteodystrophy (AHO) or progressive osseous heteroplasia (POH). In addition to cutaneous ossification, AHO involves short stature, endocrinopathy, obesity, shortened fourth and fifth metacarpals, and mental retardation. Progressive osseous heteroplasia is characterized by progressive ossification of the skin and deeper tissues such as muscle and fascia, leading to severe movement restriction; it is believed to be a localized nonprogressive variant of POH.3,4 Mutations in the guanine nucleotide binding protein, alpha stimulating activity polypeptide 1 gene, GNAS1, a key regulatory gene involved in AHO and POH, have been found in several cases of PLOC.3 Our patient lacked any dysmorphic features or laboratory abnormalities suggestive of AHO or POH. Moreover, testing of the tissue and blood for the GNAS1 mutation was negative. Treatment of PLOC often is difficult. Our patient underwent a trial of ablative fractional laser resurfacing, which failed to lead to perceivable improvement.
The differential diagnoses include a kerion, dissecting cellulitis of the scalp, folliculitis decalvans, and acne keloidalis nuchae. A kerion is a manifestation of tinea capitis characterized by an inflammatory plaque, often with pain or tenderness. Kerions most frequently occur in children aged 5 to 10 years.5 Failure to treat a kerion may result in scarring alopecia. Treatment consists of oral antifungals.
Dissecting cellulitis of the scalp is thought to occur secondary to follicular occlusion. It is characterized by boggy suppurative nodules primarily on the posterior and vertex scalp. Patchy hair loss is present and typically progresses to cicatricial alopecia. Histology characteristically shows areas of dense, predominantly neutrophilic, perifollicular dermal infiltrates.6
Folliculitis decalvans is a primary neutrophilic cicatricial alopecia that primarily occurs in adults. Patients with folliculitis decalvans tend to have multiple pustules on the periphery of confluent areas of scarring alopecia. It is theorized that an immune response to staphylococcal superantigens contributes to this disease process.7
The clinical findings of acne keloidalis nuchae include inflammatory pustules and papules with keloidlike plaques on the posterior neck and scalp. It occurs predominantly in teenaged and adult males of African ancestry.8 Treatment is aimed at reducing inflammation and preventing exacerbating factors. Severe disease courses may lead to scarring alopecia.
- Sanmartín O, Alegre V, Martinez-Aparicio A, et al. Congenital platelike osteoma cutis: case report and review of the literature. Pediatr Dermatol. 1993;10:182-186.
- Talsania N, Jolliffe V, O’Toole EA, et al. Platelike osteoma cutis. J Am Acad Dermatol. 2009;64:613-615.
- Yeh GL, Mathur S, Wivel A, et al. GNAS1 mutation and Cbfa1 misexpression in a child with severe congenital platelike osteoma cutis. J Bone Miner Res. 2000;15:2063-2073.
- Hernandez-Martin A, Perez-Mies B, Torrelo A. Congenital plate-like osteoma cutis in an infant. Pediatr Dermatol. 2009;26:479-481.
- Zaraa I, Hawilo A, Aounallah A, et al. Inflammatory tinea capitis: a 12-year study and a review of the literature. Mycoses. 2013;56:110-116.
- Scheinfeld N. Dissecting cellulitis (perifolliculitis capitis abscedens et suffodiens): a comprehensive review focusing on new treatments and findings of the last decade with commentary comparing the therapies and causes of dissecting cellulitis to hidradenitis suppurativa. Dermatol Online J. 2014;20:22692.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Knable AL Jr, Hanke CW, Gonin R. Prevalence of acne keloidalis nuchae in football players. J Am Acad Dermatol. 1997;37:570-574.
- Sanmartín O, Alegre V, Martinez-Aparicio A, et al. Congenital platelike osteoma cutis: case report and review of the literature. Pediatr Dermatol. 1993;10:182-186.
- Talsania N, Jolliffe V, O’Toole EA, et al. Platelike osteoma cutis. J Am Acad Dermatol. 2009;64:613-615.
- Yeh GL, Mathur S, Wivel A, et al. GNAS1 mutation and Cbfa1 misexpression in a child with severe congenital platelike osteoma cutis. J Bone Miner Res. 2000;15:2063-2073.
- Hernandez-Martin A, Perez-Mies B, Torrelo A. Congenital plate-like osteoma cutis in an infant. Pediatr Dermatol. 2009;26:479-481.
- Zaraa I, Hawilo A, Aounallah A, et al. Inflammatory tinea capitis: a 12-year study and a review of the literature. Mycoses. 2013;56:110-116.
- Scheinfeld N. Dissecting cellulitis (perifolliculitis capitis abscedens et suffodiens): a comprehensive review focusing on new treatments and findings of the last decade with commentary comparing the therapies and causes of dissecting cellulitis to hidradenitis suppurativa. Dermatol Online J. 2014;20:22692.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Knable AL Jr, Hanke CW, Gonin R. Prevalence of acne keloidalis nuchae in football players. J Am Acad Dermatol. 1997;37:570-574.
A 35-year-old man presented to the dermatology clinic with a slow-growing plaque on the scalp of 10 years’ duration. The lesion was mildly pruritic and was never associated with any pain or discharge. He denied antecedent trauma or infection. A hard, erythematous, nodular, alopecic plaque with punctate hyperkeratosis on the left posterior temporal and parietal scalp was noted on physical examination. The lesion was slightly tender to palpation.
Oral Verrucous Plaques in a Patient With Urothelial Cancer
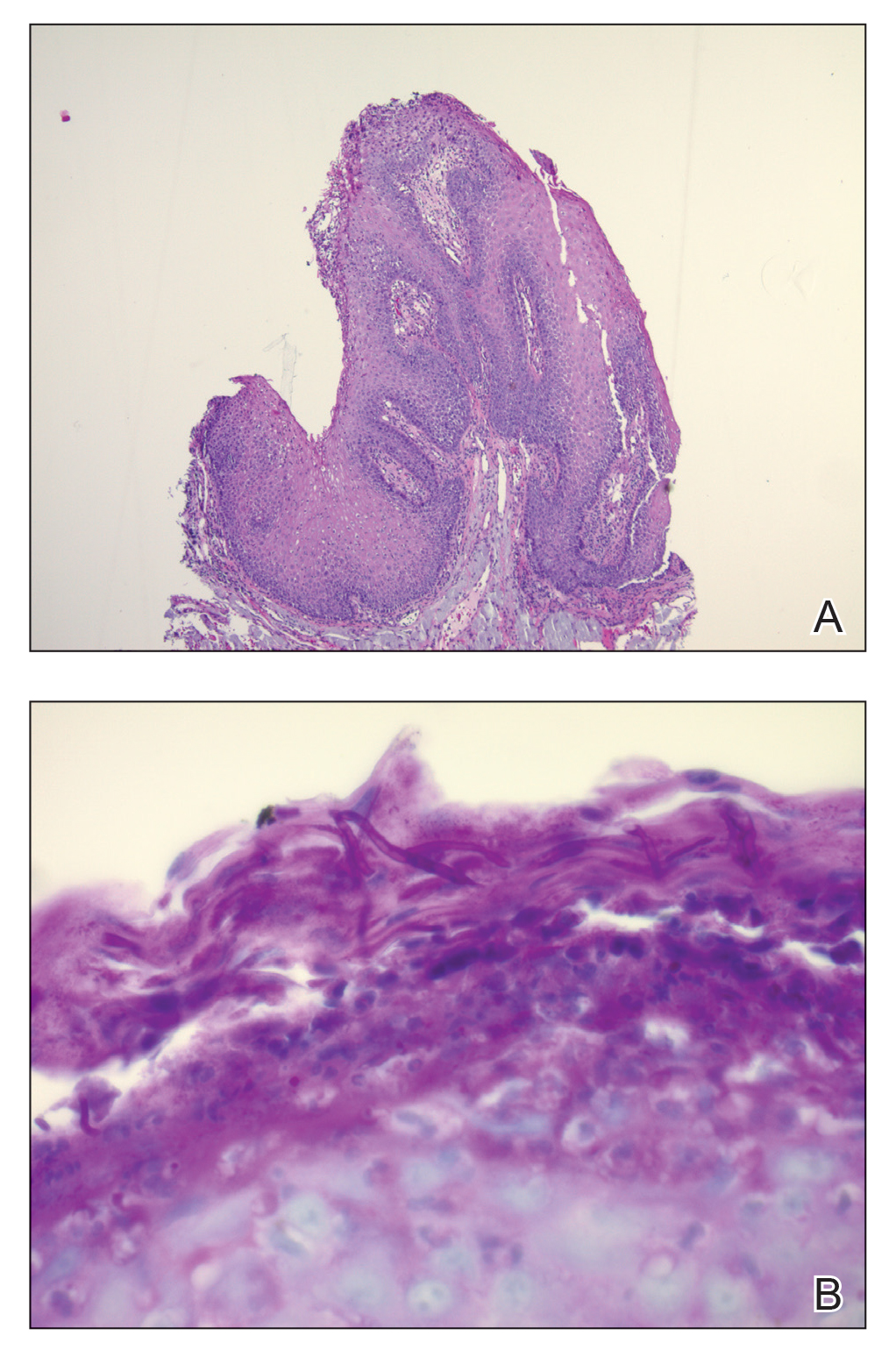
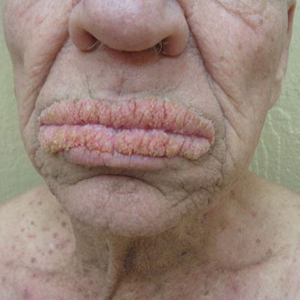
The Diagnosis: Paraneoplastic Acanthosis Nigricans
Histopathologic examination demonstrated verrucous epidermal hyperplasia (Figure, A). Fungal organisms were identified with an Alcian blue and periodic acid-Schiff stain (Figure, B). The organisms demonstrated a vertical orientation in relation to the mucosal surface, which was consistent with candidal organisms.
Given the rapid eruption of these plaques, the distribution on the oral and palmar surfaces (tripe palms), and the minimal improvement with both systemic steroids and antifungal treatment, a diagnosis of paraneoplastic acanthosis nigricans with secondary candidal infection was made. Drug-induced cheilitis was considered; however, improvement with discontinuation of the suspected offending drug would have been expected. Although chronic mucocutaneous candidiasis was possible, more prompt improvement upon initiation of systemic antifungal therapy would have been observed. Oral Crohn disease should be included in the differential, but it was unlikely given the lack of granulomas on pathology and absence of history of gastrointestinal tract symptoms. Melkersson-Rosenthal syndrome also was unlikely given the lack of facial nerve palsy as well as the lack of granulomas on pathology. Furthermore, none of these options would be associated with tripe palms, as seen in our patient.
Acanthosis nigricans is a localized skin disorder characterized by hyperpigmented velvety plaques arising in flexural and intertriginous regions. Although most cases (80%) are associated with idiopathic or benign conditions, the link between acanthosis nigricans and an underlying malignancy has been well documented.1-3 Most commonly associated with an underlying intra-abdominal malignancy (often gastric carcinoma), the lesions of paraneoplastic acanthosis nigricans are indistinguishable from their benign counterparts.1,4 When the condition presents abruptly and extensively in a nonobese patient, prompt workup for malignancy should be initiated. Rapid onset and atypical distribution (ie, palmar, perioral, or mucosal) more commonly is associated with a paraneoplastic etiology.5,6
Histopathology for acanthosis nigricans shows hyperkeratosis and epidermal papillomatosis. Horn pseudocyst formation is possible, but usually no hyperpigmentation is observed. The findings typically are indistinguishable from seborrheic keratoses, epidermal nevi, or lesions of confluent and reticulated papillomatosis of Gougerot and Carteaud.2
The underlying pathogenesis of acanthosis nigricans is poorly understood. In the benign subtype, insulin resistance commonly has been described. In the paraneoplastic subtype, it is proposed that the tumor produces a transforming growth factor that mimics epidermal growth factor and leads to keratinocyte proliferation.7,8 Paraneoplastic acanthosis nigricans has the potential to arise at any point of tumor development, further contributing to the diagnostic challenge. Treatment of the skin lesions involves management of the underlying malignancy. Unfortunately, many such malignancies often are at an advanced stage, and subsequent prognosis is poor.2
- Shah A, Jack A, Liu H, et al. Neoplastic/paraneoplastic dermatitis, fasciitis, and panniculitis. Rheum Dis Clin North Am. 2011;37:573-592.
- Chairatchaneeboon M, Kim EJ. Cutaneous paraneoplastic syndromes. In: Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick's Dermatology. 9th ed. McGraw-Hill Education; 2019:2441-2464.
- Lee HC, Ker KJ, Chong WS. Oral malignant acanthosis nigricans and tripe palms associated with renal urothelial carcinoma. JAMA Dermatol. 2015;151:1381-1383.
- Yu Q, Li XL, Ji G, et al. Malignant acanthosis nigricans: an early diagnostic clue for gastric adenocarcinoma. World J Surg Oncol. 2017;15:208.
- Mohrenschlager M, Vocks E, Wessner DB, et al. Tripe palms and malignant acanthosis nigricans: cutaneous signs of imminent metastasis in bladder cancer? J Urol. 2001;165:1629-1630.
- Cohen PR, Grossman ME, Almeida L, et al. Tripe palms and malignancy. J Clin Oncol. 1989;7:669-678.
- Higgins SP, Freemark M, Prose NS. Acanthosis nigricans: a practical approach to evaluation and management. Dermatol Online J. 2008;14:2.
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101.
The Diagnosis: Paraneoplastic Acanthosis Nigricans
Histopathologic examination demonstrated verrucous epidermal hyperplasia (Figure, A). Fungal organisms were identified with an Alcian blue and periodic acid-Schiff stain (Figure, B). The organisms demonstrated a vertical orientation in relation to the mucosal surface, which was consistent with candidal organisms.
Given the rapid eruption of these plaques, the distribution on the oral and palmar surfaces (tripe palms), and the minimal improvement with both systemic steroids and antifungal treatment, a diagnosis of paraneoplastic acanthosis nigricans with secondary candidal infection was made. Drug-induced cheilitis was considered; however, improvement with discontinuation of the suspected offending drug would have been expected. Although chronic mucocutaneous candidiasis was possible, more prompt improvement upon initiation of systemic antifungal therapy would have been observed. Oral Crohn disease should be included in the differential, but it was unlikely given the lack of granulomas on pathology and absence of history of gastrointestinal tract symptoms. Melkersson-Rosenthal syndrome also was unlikely given the lack of facial nerve palsy as well as the lack of granulomas on pathology. Furthermore, none of these options would be associated with tripe palms, as seen in our patient.
Acanthosis nigricans is a localized skin disorder characterized by hyperpigmented velvety plaques arising in flexural and intertriginous regions. Although most cases (80%) are associated with idiopathic or benign conditions, the link between acanthosis nigricans and an underlying malignancy has been well documented.1-3 Most commonly associated with an underlying intra-abdominal malignancy (often gastric carcinoma), the lesions of paraneoplastic acanthosis nigricans are indistinguishable from their benign counterparts.1,4 When the condition presents abruptly and extensively in a nonobese patient, prompt workup for malignancy should be initiated. Rapid onset and atypical distribution (ie, palmar, perioral, or mucosal) more commonly is associated with a paraneoplastic etiology.5,6
Histopathology for acanthosis nigricans shows hyperkeratosis and epidermal papillomatosis. Horn pseudocyst formation is possible, but usually no hyperpigmentation is observed. The findings typically are indistinguishable from seborrheic keratoses, epidermal nevi, or lesions of confluent and reticulated papillomatosis of Gougerot and Carteaud.2
The underlying pathogenesis of acanthosis nigricans is poorly understood. In the benign subtype, insulin resistance commonly has been described. In the paraneoplastic subtype, it is proposed that the tumor produces a transforming growth factor that mimics epidermal growth factor and leads to keratinocyte proliferation.7,8 Paraneoplastic acanthosis nigricans has the potential to arise at any point of tumor development, further contributing to the diagnostic challenge. Treatment of the skin lesions involves management of the underlying malignancy. Unfortunately, many such malignancies often are at an advanced stage, and subsequent prognosis is poor.2
The Diagnosis: Paraneoplastic Acanthosis Nigricans
Histopathologic examination demonstrated verrucous epidermal hyperplasia (Figure, A). Fungal organisms were identified with an Alcian blue and periodic acid-Schiff stain (Figure, B). The organisms demonstrated a vertical orientation in relation to the mucosal surface, which was consistent with candidal organisms.
Given the rapid eruption of these plaques, the distribution on the oral and palmar surfaces (tripe palms), and the minimal improvement with both systemic steroids and antifungal treatment, a diagnosis of paraneoplastic acanthosis nigricans with secondary candidal infection was made. Drug-induced cheilitis was considered; however, improvement with discontinuation of the suspected offending drug would have been expected. Although chronic mucocutaneous candidiasis was possible, more prompt improvement upon initiation of systemic antifungal therapy would have been observed. Oral Crohn disease should be included in the differential, but it was unlikely given the lack of granulomas on pathology and absence of history of gastrointestinal tract symptoms. Melkersson-Rosenthal syndrome also was unlikely given the lack of facial nerve palsy as well as the lack of granulomas on pathology. Furthermore, none of these options would be associated with tripe palms, as seen in our patient.
Acanthosis nigricans is a localized skin disorder characterized by hyperpigmented velvety plaques arising in flexural and intertriginous regions. Although most cases (80%) are associated with idiopathic or benign conditions, the link between acanthosis nigricans and an underlying malignancy has been well documented.1-3 Most commonly associated with an underlying intra-abdominal malignancy (often gastric carcinoma), the lesions of paraneoplastic acanthosis nigricans are indistinguishable from their benign counterparts.1,4 When the condition presents abruptly and extensively in a nonobese patient, prompt workup for malignancy should be initiated. Rapid onset and atypical distribution (ie, palmar, perioral, or mucosal) more commonly is associated with a paraneoplastic etiology.5,6
Histopathology for acanthosis nigricans shows hyperkeratosis and epidermal papillomatosis. Horn pseudocyst formation is possible, but usually no hyperpigmentation is observed. The findings typically are indistinguishable from seborrheic keratoses, epidermal nevi, or lesions of confluent and reticulated papillomatosis of Gougerot and Carteaud.2
The underlying pathogenesis of acanthosis nigricans is poorly understood. In the benign subtype, insulin resistance commonly has been described. In the paraneoplastic subtype, it is proposed that the tumor produces a transforming growth factor that mimics epidermal growth factor and leads to keratinocyte proliferation.7,8 Paraneoplastic acanthosis nigricans has the potential to arise at any point of tumor development, further contributing to the diagnostic challenge. Treatment of the skin lesions involves management of the underlying malignancy. Unfortunately, many such malignancies often are at an advanced stage, and subsequent prognosis is poor.2
- Shah A, Jack A, Liu H, et al. Neoplastic/paraneoplastic dermatitis, fasciitis, and panniculitis. Rheum Dis Clin North Am. 2011;37:573-592.
- Chairatchaneeboon M, Kim EJ. Cutaneous paraneoplastic syndromes. In: Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick's Dermatology. 9th ed. McGraw-Hill Education; 2019:2441-2464.
- Lee HC, Ker KJ, Chong WS. Oral malignant acanthosis nigricans and tripe palms associated with renal urothelial carcinoma. JAMA Dermatol. 2015;151:1381-1383.
- Yu Q, Li XL, Ji G, et al. Malignant acanthosis nigricans: an early diagnostic clue for gastric adenocarcinoma. World J Surg Oncol. 2017;15:208.
- Mohrenschlager M, Vocks E, Wessner DB, et al. Tripe palms and malignant acanthosis nigricans: cutaneous signs of imminent metastasis in bladder cancer? J Urol. 2001;165:1629-1630.
- Cohen PR, Grossman ME, Almeida L, et al. Tripe palms and malignancy. J Clin Oncol. 1989;7:669-678.
- Higgins SP, Freemark M, Prose NS. Acanthosis nigricans: a practical approach to evaluation and management. Dermatol Online J. 2008;14:2.
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101.
- Shah A, Jack A, Liu H, et al. Neoplastic/paraneoplastic dermatitis, fasciitis, and panniculitis. Rheum Dis Clin North Am. 2011;37:573-592.
- Chairatchaneeboon M, Kim EJ. Cutaneous paraneoplastic syndromes. In: Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick's Dermatology. 9th ed. McGraw-Hill Education; 2019:2441-2464.
- Lee HC, Ker KJ, Chong WS. Oral malignant acanthosis nigricans and tripe palms associated with renal urothelial carcinoma. JAMA Dermatol. 2015;151:1381-1383.
- Yu Q, Li XL, Ji G, et al. Malignant acanthosis nigricans: an early diagnostic clue for gastric adenocarcinoma. World J Surg Oncol. 2017;15:208.
- Mohrenschlager M, Vocks E, Wessner DB, et al. Tripe palms and malignant acanthosis nigricans: cutaneous signs of imminent metastasis in bladder cancer? J Urol. 2001;165:1629-1630.
- Cohen PR, Grossman ME, Almeida L, et al. Tripe palms and malignancy. J Clin Oncol. 1989;7:669-678.
- Higgins SP, Freemark M, Prose NS. Acanthosis nigricans: a practical approach to evaluation and management. Dermatol Online J. 2008;14:2.
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101.
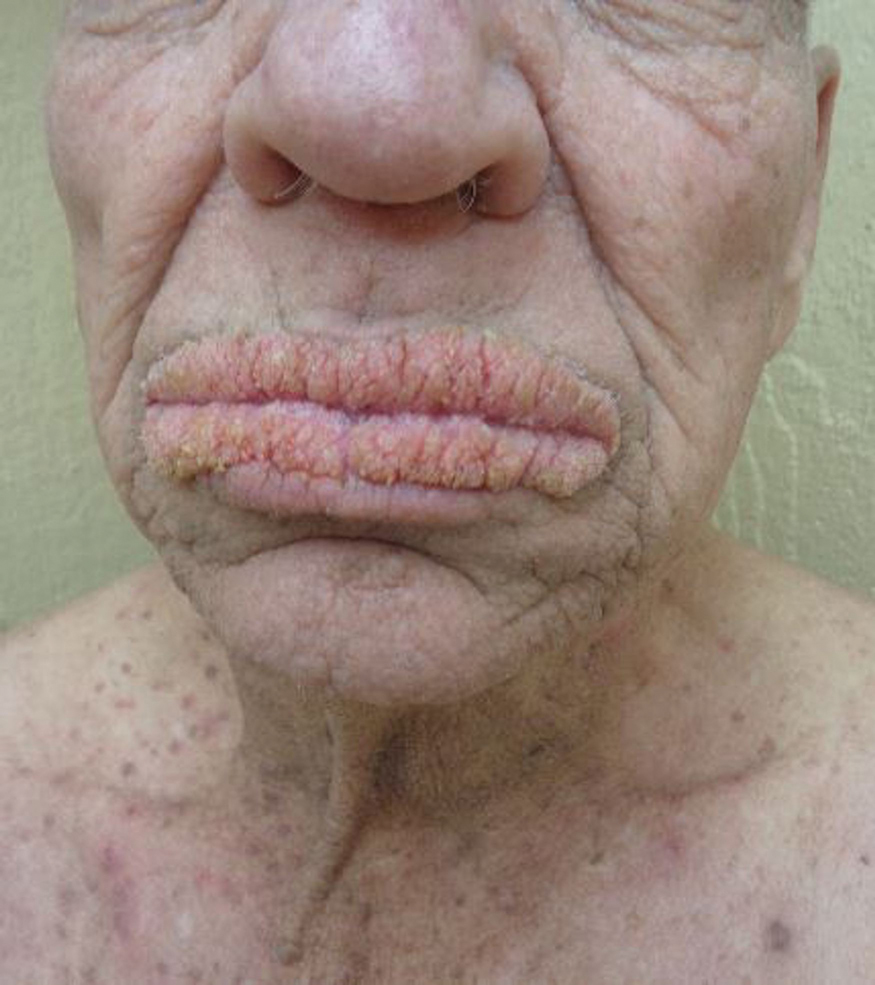
A 75-year-old nonobese man with metastatic urothelial carcinoma presented for evaluation and treatment of swollen lips. The patient stated that his lips began to swell and crack shortly after beginning pembrolizumab approximately 5 months prior. The swelling had progressively worsened, prompting discontinuation of the pembrolizumab by oncology about 2 months prior to presentation to our dermatology clinic. He reported slight improvement after the discontinuation of pembrolizumab, and he had since been started on carboplatin and gemcitabine. He previously was treated with oral corticosteroids without improvement. His oncologist started him on oral fluconazole for treatment of oral thrush on the day of presentation to our clinic. Physical examination revealed diffuse papillomatous and verrucous plaques of the upper and lower lips with involvement of the buccal mucosa. He also had deep fissures and white plaques on the tongue. Velvety hyperpigmented plaques were noted in the axillae, and he had confluent thickening of the palms. A 3-mm punch biopsy from the lower lip was performed. The patient subsequently was evaluated 2 weeks after the initial appointment, and minor improvement in the oral verrucous hyperplasia was noted following antifungal therapy, with resolution of the candidiasis.
Periorbital and Tragal Cutaneous Lesions
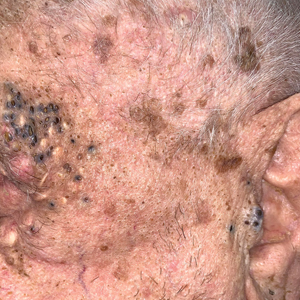
The Diagnosis: Favre-Racouchot Syndrome
Favre-Racouchot syndrome, also known as nodular elastosis with cysts and comedones, is seen in approximately 6% of adults aged 40 to 60 years and predominantly is observed in White males.1 Typically, patients have a history of prolonged recreational or occupational UV exposure and tobacco use. The diagnosis can be made clinically; no biopsy is necessary. If a biopsy is performed, histologic findings typically consist of notable actinic elastosis, epidermal atrophy, and comedones. The differential diagnosis includes acne comedones, colloid milium, milia, chloracne, and trichoepithelioma.2 Associated conditions that have been found concurrently include cutis rhomboidalis nuchae, actinic keratosis, erosive pustular dermatosis, actinic granuloma, and basal and squamous cell carcinomas.2
The pathogenesis, while not fully understood, seems to involve a combination of chronic UV radiation exposure and heavy cigarette smoking that eventually leads to cutaneous atrophy and keratinization of the pilosebaceous follicles as well as the formation of comedones.2 Radiation therapy also has been implicated as a possible causative agent of Favre-Racouchot syndrome.1 Clinically, symmetric distribution of large black comedones over the temporal and periorbital areas is seen surrounded by distinct signs of UV-damaged skin, including wrinkles and atrophic skin.3 Although there seems to be a synergistic effect between cigarette smoking and chronic UV exposure, evidence favors smoking as the major cause of this condition,4,5 which causes striking visual changes but is a benign process. UV protection and smoking cessation are the most important factors for prevention and limiting progression.
Treatment consists of typical comedonal therapies such as tretinoin or comedone extraction. Procedural options in conjunction with medical therapy include dermabrasion or laser therapy. Newer studies have shown promising results for both CO2 laser treatment and plasma exeresis.6 Plasma exeresis is a noninvasive technique that causes ionization of atmospheric gas between the device and tissue, ultimately causing sublimation of the target tissue.7 It is important to carefully evaluate and follow up with these patients due to their history of extensive UV exposure. Both short-term and long-term follow-up is recommended due to high rates of reoccurrence within 10 to 12 months and the dangers of chronic UV exposure– related malignancies.6
- Lewis KG, Bercovitch L, Dill SW, et al. Acquired disorders of elastic tissue: part i. increased elastic tissue and solar elastotic syndromes. J Am Acad Dermatol. 2004;51:1-21; quiz 22-24. doi:10.1016/j.jaad.2004.03.013
- Patterson WM, Fox MD, Schwartz RA. Favre-Racouchot disease. Int J Dermatol. 2004;43:167-169. doi:10.1111/j.1365-4632.2004.01546.x
- Sonthalia S, Arora R, Chhabra N, et al. Favre-Racouchot syndrome. Indian Dermatol Online J. 2014;5(suppl 2):S128-S129. doi:10.4103/2229-5178.146192
- Keough GC, Laws RA, Elston DM. Favre-Racouchot syndrome: a case for smokers’ comedones. Arch Dermatol. 1997;133:796-797. doi:10.1001/archderm.133.6.796
- Muto H, Takizawa Y. Dioxins in cigarette smoke. Arch Environ Health. 1989;44:171-174. doi:10.1080/00039896.1989.9935882
- Paganelli A, Mandel VD, Kaleci S, et al. Favre-Racouchot disease: systematic review and possible therapeutic strategies. J Eur Acad Dermatol Venereol. 2018;33:32-41. doi:10.1111/jdv.15184
- Rossi E, Paganelli A, Mandel VD, et al. Plasma exeresis treatment for epidermoid cysts: a minimal scarring technique. Dermatol Surg. 2018;44:1509-1515. doi:10.1097/dss.0000000000001604
The Diagnosis: Favre-Racouchot Syndrome
Favre-Racouchot syndrome, also known as nodular elastosis with cysts and comedones, is seen in approximately 6% of adults aged 40 to 60 years and predominantly is observed in White males.1 Typically, patients have a history of prolonged recreational or occupational UV exposure and tobacco use. The diagnosis can be made clinically; no biopsy is necessary. If a biopsy is performed, histologic findings typically consist of notable actinic elastosis, epidermal atrophy, and comedones. The differential diagnosis includes acne comedones, colloid milium, milia, chloracne, and trichoepithelioma.2 Associated conditions that have been found concurrently include cutis rhomboidalis nuchae, actinic keratosis, erosive pustular dermatosis, actinic granuloma, and basal and squamous cell carcinomas.2
The pathogenesis, while not fully understood, seems to involve a combination of chronic UV radiation exposure and heavy cigarette smoking that eventually leads to cutaneous atrophy and keratinization of the pilosebaceous follicles as well as the formation of comedones.2 Radiation therapy also has been implicated as a possible causative agent of Favre-Racouchot syndrome.1 Clinically, symmetric distribution of large black comedones over the temporal and periorbital areas is seen surrounded by distinct signs of UV-damaged skin, including wrinkles and atrophic skin.3 Although there seems to be a synergistic effect between cigarette smoking and chronic UV exposure, evidence favors smoking as the major cause of this condition,4,5 which causes striking visual changes but is a benign process. UV protection and smoking cessation are the most important factors for prevention and limiting progression.
Treatment consists of typical comedonal therapies such as tretinoin or comedone extraction. Procedural options in conjunction with medical therapy include dermabrasion or laser therapy. Newer studies have shown promising results for both CO2 laser treatment and plasma exeresis.6 Plasma exeresis is a noninvasive technique that causes ionization of atmospheric gas between the device and tissue, ultimately causing sublimation of the target tissue.7 It is important to carefully evaluate and follow up with these patients due to their history of extensive UV exposure. Both short-term and long-term follow-up is recommended due to high rates of reoccurrence within 10 to 12 months and the dangers of chronic UV exposure– related malignancies.6
The Diagnosis: Favre-Racouchot Syndrome
Favre-Racouchot syndrome, also known as nodular elastosis with cysts and comedones, is seen in approximately 6% of adults aged 40 to 60 years and predominantly is observed in White males.1 Typically, patients have a history of prolonged recreational or occupational UV exposure and tobacco use. The diagnosis can be made clinically; no biopsy is necessary. If a biopsy is performed, histologic findings typically consist of notable actinic elastosis, epidermal atrophy, and comedones. The differential diagnosis includes acne comedones, colloid milium, milia, chloracne, and trichoepithelioma.2 Associated conditions that have been found concurrently include cutis rhomboidalis nuchae, actinic keratosis, erosive pustular dermatosis, actinic granuloma, and basal and squamous cell carcinomas.2
The pathogenesis, while not fully understood, seems to involve a combination of chronic UV radiation exposure and heavy cigarette smoking that eventually leads to cutaneous atrophy and keratinization of the pilosebaceous follicles as well as the formation of comedones.2 Radiation therapy also has been implicated as a possible causative agent of Favre-Racouchot syndrome.1 Clinically, symmetric distribution of large black comedones over the temporal and periorbital areas is seen surrounded by distinct signs of UV-damaged skin, including wrinkles and atrophic skin.3 Although there seems to be a synergistic effect between cigarette smoking and chronic UV exposure, evidence favors smoking as the major cause of this condition,4,5 which causes striking visual changes but is a benign process. UV protection and smoking cessation are the most important factors for prevention and limiting progression.
Treatment consists of typical comedonal therapies such as tretinoin or comedone extraction. Procedural options in conjunction with medical therapy include dermabrasion or laser therapy. Newer studies have shown promising results for both CO2 laser treatment and plasma exeresis.6 Plasma exeresis is a noninvasive technique that causes ionization of atmospheric gas between the device and tissue, ultimately causing sublimation of the target tissue.7 It is important to carefully evaluate and follow up with these patients due to their history of extensive UV exposure. Both short-term and long-term follow-up is recommended due to high rates of reoccurrence within 10 to 12 months and the dangers of chronic UV exposure– related malignancies.6
- Lewis KG, Bercovitch L, Dill SW, et al. Acquired disorders of elastic tissue: part i. increased elastic tissue and solar elastotic syndromes. J Am Acad Dermatol. 2004;51:1-21; quiz 22-24. doi:10.1016/j.jaad.2004.03.013
- Patterson WM, Fox MD, Schwartz RA. Favre-Racouchot disease. Int J Dermatol. 2004;43:167-169. doi:10.1111/j.1365-4632.2004.01546.x
- Sonthalia S, Arora R, Chhabra N, et al. Favre-Racouchot syndrome. Indian Dermatol Online J. 2014;5(suppl 2):S128-S129. doi:10.4103/2229-5178.146192
- Keough GC, Laws RA, Elston DM. Favre-Racouchot syndrome: a case for smokers’ comedones. Arch Dermatol. 1997;133:796-797. doi:10.1001/archderm.133.6.796
- Muto H, Takizawa Y. Dioxins in cigarette smoke. Arch Environ Health. 1989;44:171-174. doi:10.1080/00039896.1989.9935882
- Paganelli A, Mandel VD, Kaleci S, et al. Favre-Racouchot disease: systematic review and possible therapeutic strategies. J Eur Acad Dermatol Venereol. 2018;33:32-41. doi:10.1111/jdv.15184
- Rossi E, Paganelli A, Mandel VD, et al. Plasma exeresis treatment for epidermoid cysts: a minimal scarring technique. Dermatol Surg. 2018;44:1509-1515. doi:10.1097/dss.0000000000001604
- Lewis KG, Bercovitch L, Dill SW, et al. Acquired disorders of elastic tissue: part i. increased elastic tissue and solar elastotic syndromes. J Am Acad Dermatol. 2004;51:1-21; quiz 22-24. doi:10.1016/j.jaad.2004.03.013
- Patterson WM, Fox MD, Schwartz RA. Favre-Racouchot disease. Int J Dermatol. 2004;43:167-169. doi:10.1111/j.1365-4632.2004.01546.x
- Sonthalia S, Arora R, Chhabra N, et al. Favre-Racouchot syndrome. Indian Dermatol Online J. 2014;5(suppl 2):S128-S129. doi:10.4103/2229-5178.146192
- Keough GC, Laws RA, Elston DM. Favre-Racouchot syndrome: a case for smokers’ comedones. Arch Dermatol. 1997;133:796-797. doi:10.1001/archderm.133.6.796
- Muto H, Takizawa Y. Dioxins in cigarette smoke. Arch Environ Health. 1989;44:171-174. doi:10.1080/00039896.1989.9935882
- Paganelli A, Mandel VD, Kaleci S, et al. Favre-Racouchot disease: systematic review and possible therapeutic strategies. J Eur Acad Dermatol Venereol. 2018;33:32-41. doi:10.1111/jdv.15184
- Rossi E, Paganelli A, Mandel VD, et al. Plasma exeresis treatment for epidermoid cysts: a minimal scarring technique. Dermatol Surg. 2018;44:1509-1515. doi:10.1097/dss.0000000000001604

A 91-year-old White man with no personal or family history of skin cancer presented to the dermatology clinic for a total-body skin examination. A 6×5-cm grouped cluster of open comedones in the periorbital region and on the left tragus as well as surrounding actinic damaged skin with coarse rhytides, dyschromia, and lentigines were seen. He had a history of excessive UV exposure and noted that the lesions had been present for approximately 10 years. They were asymptomatic and remained unchanged since their onset.
Multiple Crusted Swellings on the Chin
The Diagnosis: Cutaneous Cryptococcosis
Histologic examination revealed infiltration of the dermis and subcutaneous tissue with rounded basophilic cells on low magnification (Figure 1A). On higher magnification, encapsulated yeast cells (cryptococci) of varying size accompanied by chronic granulomatous inflammatory infiltration with occasional giant cells were seen (Figure 1B). Alcian blue stain showed mucinous capsular material (Figure 1C). There was no history of diabetes mellitus, tuberculosis, steroid therapy, or immunosuppression. Moreover, systemic involvement or systemic focus of infection was ruled out after computed tomography of the head, chest, and abdomen. Therefore, the diagnosis of primary cutaneous cryptococcosis (PCC) was established. The patient was started on oral itraconazole 100 mg twice daily along with 5 drops of a saturated solution of potassium iodide 3 times daily that later was increased to 20 drops 3 times daily at a weekly interval. The lesions started improving after 1 month and healed completely after 9 months of treatment (Figure 2).
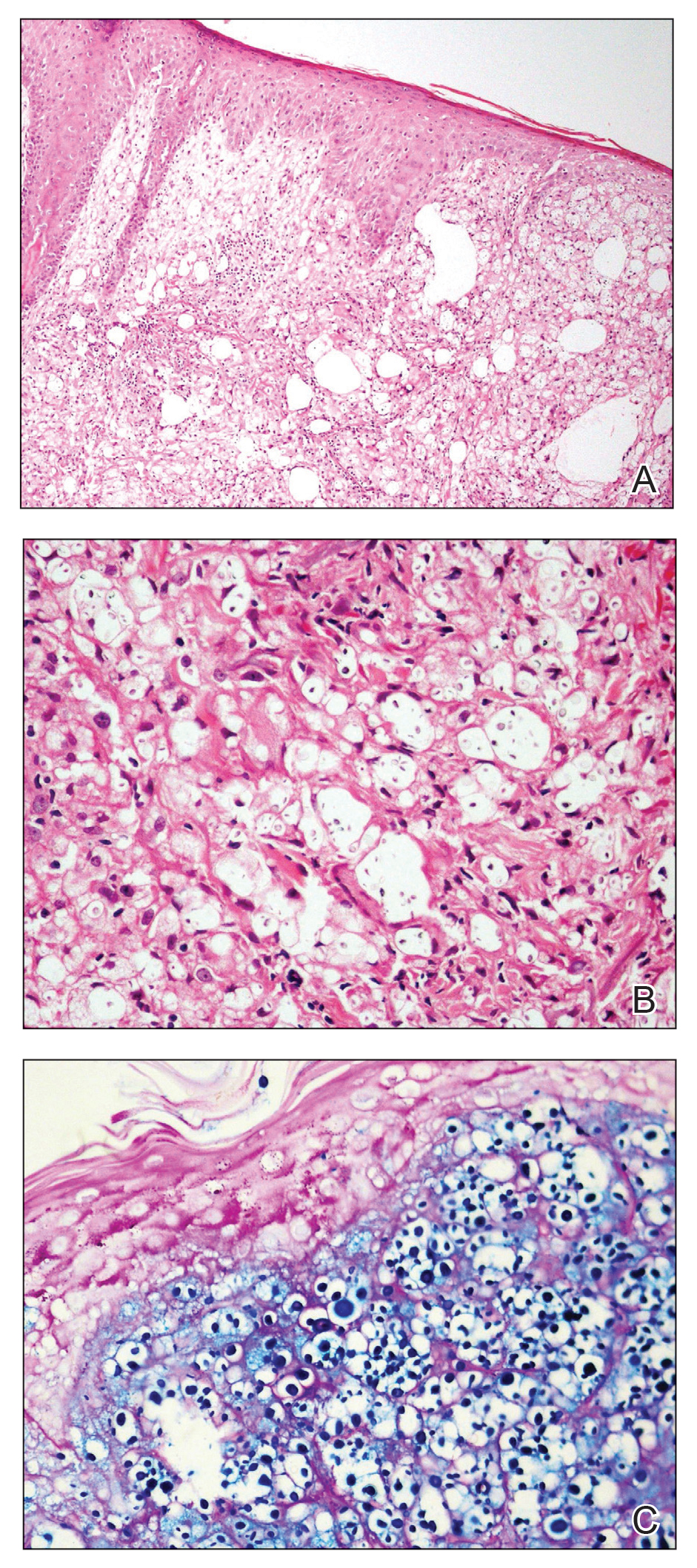
Primary cutaneous cryptococcosis is the identification of Cryptococcus neoformans in a skin lesion without evidence of simultaneous disseminated disease. Neuville et al1 observed that skin lesions resemble cellulitis, ulcerations, or whitlows and were located on unclothed areas. In contrast, lesions from disseminated disease presented as scattered umbilicated papules resembling molluscum contagiosum. Diagnosis of PCC is based on the observation of encapsulated yeasts by direct microscopic examination, isolation of C neoformans or Cryptococcus gattii in culture, and by the demonstration of capsular antigen in various fluids, including serum and cerebrospinal fluid by latex particle agglutination or enzyme-linked immunosorbent assay. Histologically, Cryptococcus species produce a proliferative inflammatory reaction in immunocompetent hosts with the formation of compact epithelioid granulomas, with giant cells and a peripheral layer of lymphocytes. Treatment options for PCC infection range from antifungal medications and surgical debridement to observation.
The differential diagnosis may include cutaneous leishmaniasis, cutaneous tuberculosis, cutaneous histoplasmosis, and basal cell carcinoma. These entities may have similar presentations and can only be confidently differentiated on direct microscopy and histopathologic examination. The characteristic Leishmania donovani bodies on microscopy in cutaneous leishmaniasis and tubercular granuloma with central necrosis on histology in cutaneous tuberculosis can differentiate these conditions from cryptococcosis. In some patients with cryptococcosis, the yeast may produce a less characteristic polysaccharide capsule and thus may be confused with histoplasmosis. Fontana-Masson staining may show melanin-producing yeast, which is characteristic of cryptococci.2 Ulcerated basal cell carcinoma may present similar clinically; however, histopathology will rule it out.
Cutaneous cryptococcal infection should be presumed to be disseminated until proven otherwise, and a search for other sites of involvement must immediately be undertaken. Cutaneous signs may be the first indication of infection, preceding the diagnosis of disseminated disease by 2 to 8 months, making its recognition crucial to early treatment. It is not possible to diagnose PCC on a specific clinical manifestation because a diverse range of skin lesions may be present. Therefore, culture and histology are the gold standards for diagnosis of cryptococcosis.
- Neuville S, Dromer F, Morin O, et al. Primary cutaneous cryptococcosis: a distinct clinical entity. Clin Infect Dis. 2003;36:337-347.
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280.
The Diagnosis: Cutaneous Cryptococcosis
Histologic examination revealed infiltration of the dermis and subcutaneous tissue with rounded basophilic cells on low magnification (Figure 1A). On higher magnification, encapsulated yeast cells (cryptococci) of varying size accompanied by chronic granulomatous inflammatory infiltration with occasional giant cells were seen (Figure 1B). Alcian blue stain showed mucinous capsular material (Figure 1C). There was no history of diabetes mellitus, tuberculosis, steroid therapy, or immunosuppression. Moreover, systemic involvement or systemic focus of infection was ruled out after computed tomography of the head, chest, and abdomen. Therefore, the diagnosis of primary cutaneous cryptococcosis (PCC) was established. The patient was started on oral itraconazole 100 mg twice daily along with 5 drops of a saturated solution of potassium iodide 3 times daily that later was increased to 20 drops 3 times daily at a weekly interval. The lesions started improving after 1 month and healed completely after 9 months of treatment (Figure 2).
Primary cutaneous cryptococcosis is the identification of Cryptococcus neoformans in a skin lesion without evidence of simultaneous disseminated disease. Neuville et al1 observed that skin lesions resemble cellulitis, ulcerations, or whitlows and were located on unclothed areas. In contrast, lesions from disseminated disease presented as scattered umbilicated papules resembling molluscum contagiosum. Diagnosis of PCC is based on the observation of encapsulated yeasts by direct microscopic examination, isolation of C neoformans or Cryptococcus gattii in culture, and by the demonstration of capsular antigen in various fluids, including serum and cerebrospinal fluid by latex particle agglutination or enzyme-linked immunosorbent assay. Histologically, Cryptococcus species produce a proliferative inflammatory reaction in immunocompetent hosts with the formation of compact epithelioid granulomas, with giant cells and a peripheral layer of lymphocytes. Treatment options for PCC infection range from antifungal medications and surgical debridement to observation.
The differential diagnosis may include cutaneous leishmaniasis, cutaneous tuberculosis, cutaneous histoplasmosis, and basal cell carcinoma. These entities may have similar presentations and can only be confidently differentiated on direct microscopy and histopathologic examination. The characteristic Leishmania donovani bodies on microscopy in cutaneous leishmaniasis and tubercular granuloma with central necrosis on histology in cutaneous tuberculosis can differentiate these conditions from cryptococcosis. In some patients with cryptococcosis, the yeast may produce a less characteristic polysaccharide capsule and thus may be confused with histoplasmosis. Fontana-Masson staining may show melanin-producing yeast, which is characteristic of cryptococci.2 Ulcerated basal cell carcinoma may present similar clinically; however, histopathology will rule it out.
Cutaneous cryptococcal infection should be presumed to be disseminated until proven otherwise, and a search for other sites of involvement must immediately be undertaken. Cutaneous signs may be the first indication of infection, preceding the diagnosis of disseminated disease by 2 to 8 months, making its recognition crucial to early treatment. It is not possible to diagnose PCC on a specific clinical manifestation because a diverse range of skin lesions may be present. Therefore, culture and histology are the gold standards for diagnosis of cryptococcosis.
The Diagnosis: Cutaneous Cryptococcosis
Histologic examination revealed infiltration of the dermis and subcutaneous tissue with rounded basophilic cells on low magnification (Figure 1A). On higher magnification, encapsulated yeast cells (cryptococci) of varying size accompanied by chronic granulomatous inflammatory infiltration with occasional giant cells were seen (Figure 1B). Alcian blue stain showed mucinous capsular material (Figure 1C). There was no history of diabetes mellitus, tuberculosis, steroid therapy, or immunosuppression. Moreover, systemic involvement or systemic focus of infection was ruled out after computed tomography of the head, chest, and abdomen. Therefore, the diagnosis of primary cutaneous cryptococcosis (PCC) was established. The patient was started on oral itraconazole 100 mg twice daily along with 5 drops of a saturated solution of potassium iodide 3 times daily that later was increased to 20 drops 3 times daily at a weekly interval. The lesions started improving after 1 month and healed completely after 9 months of treatment (Figure 2).
Primary cutaneous cryptococcosis is the identification of Cryptococcus neoformans in a skin lesion without evidence of simultaneous disseminated disease. Neuville et al1 observed that skin lesions resemble cellulitis, ulcerations, or whitlows and were located on unclothed areas. In contrast, lesions from disseminated disease presented as scattered umbilicated papules resembling molluscum contagiosum. Diagnosis of PCC is based on the observation of encapsulated yeasts by direct microscopic examination, isolation of C neoformans or Cryptococcus gattii in culture, and by the demonstration of capsular antigen in various fluids, including serum and cerebrospinal fluid by latex particle agglutination or enzyme-linked immunosorbent assay. Histologically, Cryptococcus species produce a proliferative inflammatory reaction in immunocompetent hosts with the formation of compact epithelioid granulomas, with giant cells and a peripheral layer of lymphocytes. Treatment options for PCC infection range from antifungal medications and surgical debridement to observation.
The differential diagnosis may include cutaneous leishmaniasis, cutaneous tuberculosis, cutaneous histoplasmosis, and basal cell carcinoma. These entities may have similar presentations and can only be confidently differentiated on direct microscopy and histopathologic examination. The characteristic Leishmania donovani bodies on microscopy in cutaneous leishmaniasis and tubercular granuloma with central necrosis on histology in cutaneous tuberculosis can differentiate these conditions from cryptococcosis. In some patients with cryptococcosis, the yeast may produce a less characteristic polysaccharide capsule and thus may be confused with histoplasmosis. Fontana-Masson staining may show melanin-producing yeast, which is characteristic of cryptococci.2 Ulcerated basal cell carcinoma may present similar clinically; however, histopathology will rule it out.
Cutaneous cryptococcal infection should be presumed to be disseminated until proven otherwise, and a search for other sites of involvement must immediately be undertaken. Cutaneous signs may be the first indication of infection, preceding the diagnosis of disseminated disease by 2 to 8 months, making its recognition crucial to early treatment. It is not possible to diagnose PCC on a specific clinical manifestation because a diverse range of skin lesions may be present. Therefore, culture and histology are the gold standards for diagnosis of cryptococcosis.
- Neuville S, Dromer F, Morin O, et al. Primary cutaneous cryptococcosis: a distinct clinical entity. Clin Infect Dis. 2003;36:337-347.
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280.
- Neuville S, Dromer F, Morin O, et al. Primary cutaneous cryptococcosis: a distinct clinical entity. Clin Infect Dis. 2003;36:337-347.
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280.
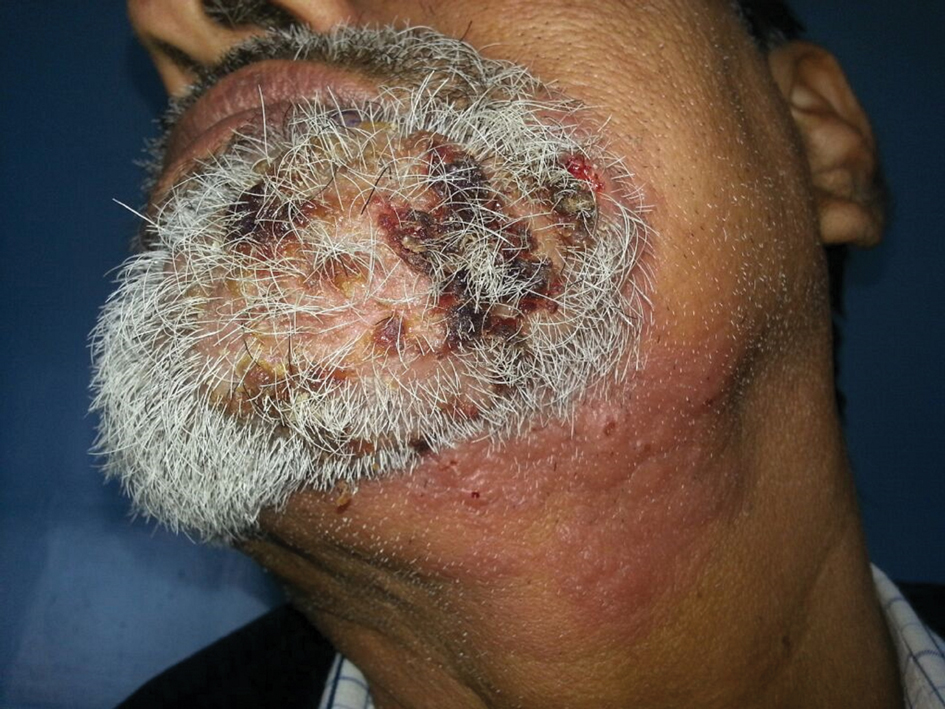
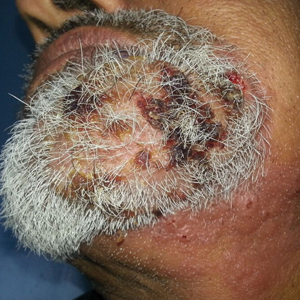
A 54-year-old man with no comorbidities presented with multiple painless swellings on the left side of the chin of 1 month’s duration that progressively were increasing, both in size and number. He denied any discharge of pus or grains from the lesion, facial trauma, insect bites, or dental procedures. The patient was treated with oral antibiotics for 15 days with no relief at an outside hospital. All routine blood and serologic investigations including viral markers and chest radiography were normal. Bacterial and fungal cultures as well as an acid-fast bacilli culture were negative. Systemic examination was normal, and vitals were within reference range. Mucocutaneous examination revealed multiple nontender small nodules and plaques with yellow-brown to dark brown hemorrhagic crusts with mild perilesional erythema on the left side of the chin extending to the adjacent neck. All mucosal sites were normal, and a biopsy was performed.
Widespread Hyperkeratotic Papules in a Transplant Recipient
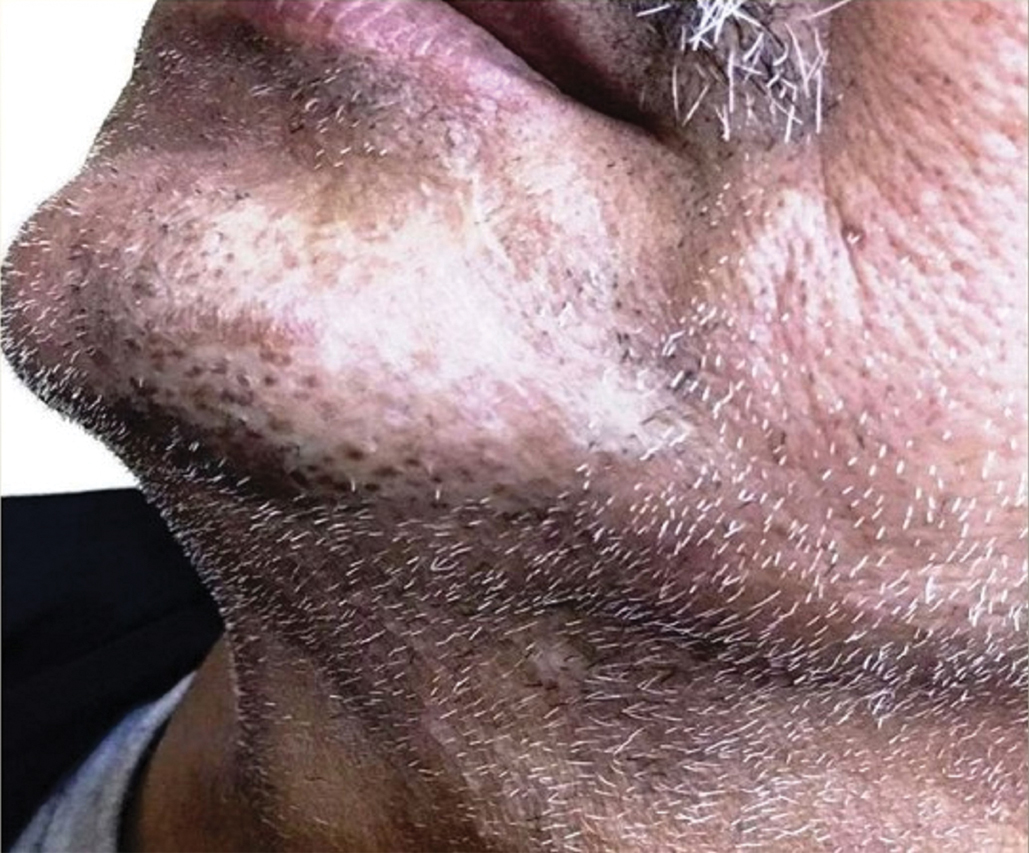
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
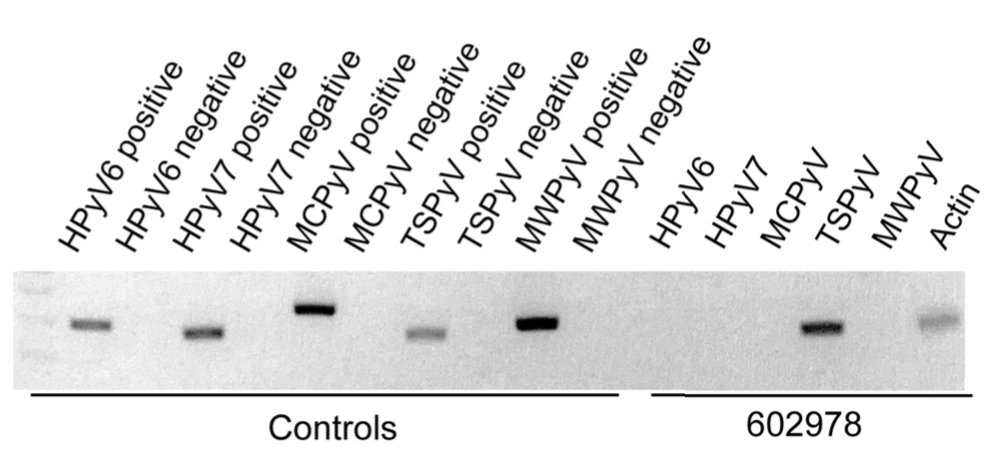
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
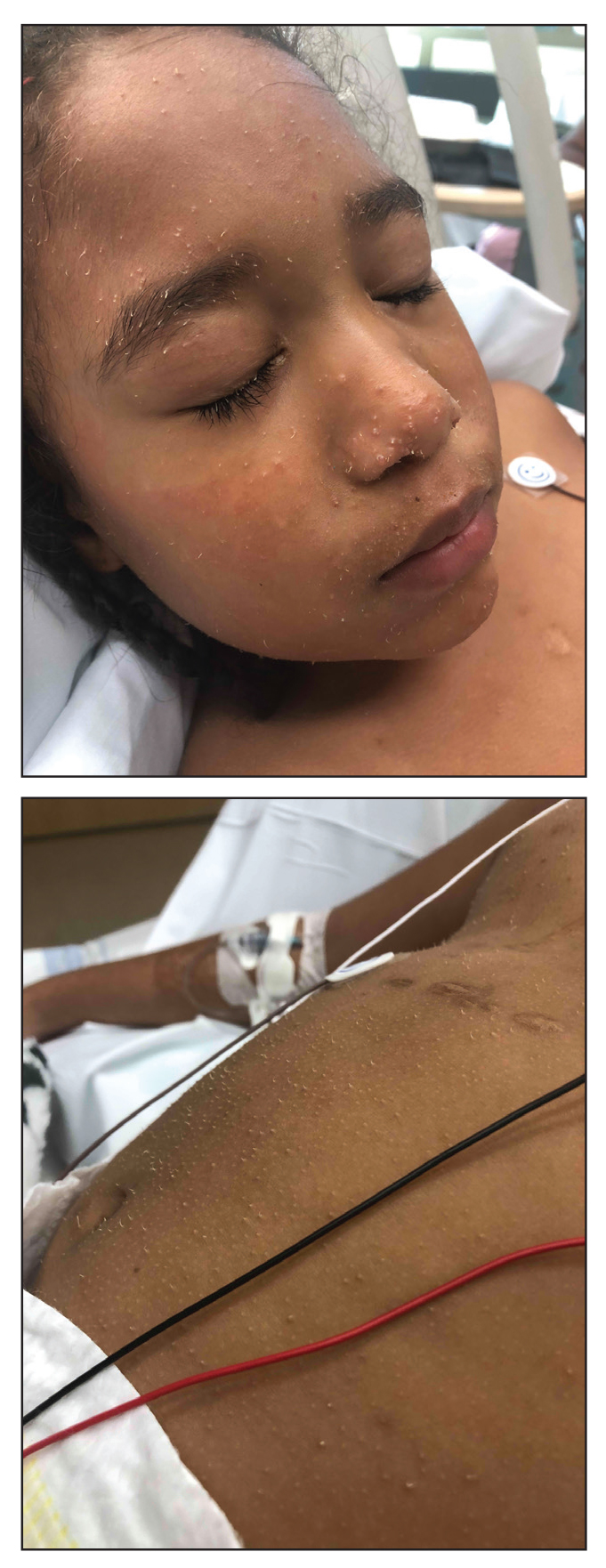
A 4-year-old girl with a history of cardiac transplantation 1 year prior for dilated cardiomyopathy presented to the dermatology consultation service with widespread hyperkeratotic papules of 2 months’ duration. The eruption initially had appeared on the face with subsequent involvement of the trunk and extremities. Her immunosuppressive medications included oral tacrolimus and mycophenolate mofetil. No over-the-counter or prescription treatments had been used for the eruption; the patient’s mother had been manually extracting the spicules from the nose, cheeks, and forehead with tweezers. The lesions were asymptomatic with only mild follicular erythema. Physical examination revealed multiple folliculocentric keratinous spicules on the nose, cheeks, forehead (top), trunk (bottom), arms, and legs.
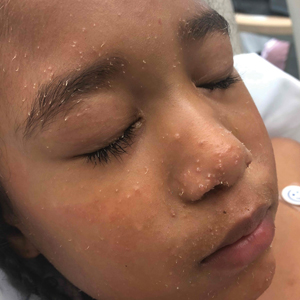
Nodule on the Neck
The Diagnosis: Primary Cutaneous Anaplastic Large Cell Lymphoma
Microscopic analysis showed a dense proliferation of mononuclear cells filling and expanding the dermis with focal epidermotropism (Figure 1). Immunohistochemistry demonstrated strong and diffuse staining for CD3, CD4, and CD30 (Figure 2) and lack of staining for anaplastic lymphoma kinase (ALK). Workup to exclude systemic disease was initiated and included unremarkable computed tomography (CT) of the neck, chest, abdomen, and pelvis along with no abnormal cells on bone marrow biopsy. Complete blood cell count, basic metabolic panel, and lactate dehydrogenase were within reference range. Given the lack of evidence for systemic involvement, a diagnosis of primary cutaneous anaplastic large cell lymphoma (PC-ALCL) was made. The treatment plan for our patient with a solitary lesion was localized radiation therapy.
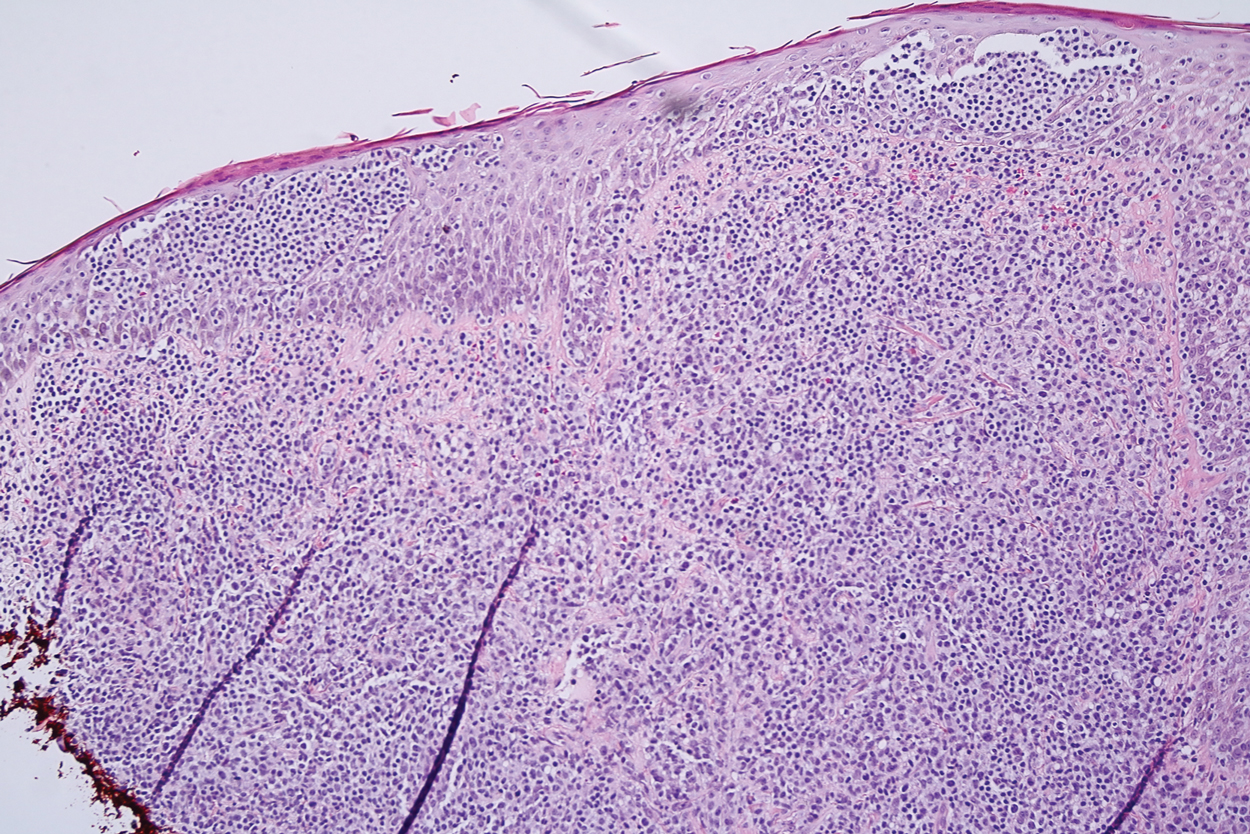
Primary cutaneous CD30+ lymphoproliferative disorders encompass a spectrum of conditions, with premalignant lymphomatoid papulosis (LyP) at one extreme and the malignant PC-ALCL on the other.1 The diagnosis of PC-ALCL is made by clinicopathologic correlation, and lesions typically present abruptly as solitary or grouped nodules with a tendency to ulcerate over time. Spontaneous regression has been reported, but relapse in the skin is frequent.2
A representative, typically excisional, biopsy should be performed if the clinician suspects PC-ALCL. Histologic criteria include a dense dermal infiltrate of large pleomorphic cells and the expression of CD30 in at least 75% of tumor cells.3 Primary cutaneous anaplastic large cell lymphoma typically lacks the ALK gene translocation with the nucleophosmin gene, NPM, that is common in systemic disease; however, a small subset of PC-ALCL may be ALK positive and indicate a higher chance of transformation into systemic disease.2
The extent of the lymphoma should be staged to exclude the possibility of systemic disease. This assessment includes a complete physical examination; laboratory investigation, including complete blood cell count with differential and blood chemistries; and radiography. A positron emission tomography-CT scan of the neck, chest, abdomen, and pelvis, or a whole-body integrated positron emission tomography-CT are sufficient for the radiographic examination.3
The initial choice of treatment for solitary or localized PC-ALCL is localized radiation therapy or low-dose methotrexate. Targeted therapy such as brentuximab has been shown to be effective for those with multifocal systemic involvement or refractory disease.2 Cure rates from radiation therapy alone approach 95%.3 It is important to highlight radiation therapy as the initial management plan to increase awareness and to avoid inappropriate treatment of PC-ALCL with traditional chemotherapy.
Large lesions of LyP may appear similar to PC-ALCL on histopathology, making the two entities difficult to distinguish. However, in contrast to PC-ALCL, LyP classically has a different clinical course characterized by waxing and waning crops of lesions that typically are smaller (<1 cm) than those of PC-ALCL.2 Large cell transformation of mycosis fungoides is another entity to consider, but these patients usually have a known history of mycosis fungoides.4
Keratoacanthomas, considered to be a variant of a well-differentiated squamous cell carcinoma, present as rapidly enlarging crateriform nodules with a keratotic core. They usually are found on the head and neck or sun-exposed areas of the extremities and may regress spontaneously.5 Histology will show atypical, highly differentiated squamous epithelia. Merkel cell carcinoma also has a predilection for the head and neck in older patients and may present as a rapidly growing nodule. However, histology will show an aggressive tumor with small round blue cells, and immunohistochemistry will show the characteristic paranuclear dot staining for CK20 along with staining for various neuroendocrine markers. Similarly, atypical fibroxanthoma is a low-grade sarcoma that also presents on the head and neck of elderly sun-damaged patients.5 Histology will show dermal proliferation of spindle cells that often extend up against the epidermis along with pleomorphism and atypical mitoses. Basal cell carcinoma is a common tumor that can present on the head and neck in sun-damaged patients. Nodular basal cell carcinomas can enlarge and ulcerate, but growth is seen over years rather than weeks.5 Histology characteristically will show tumor islands composed of basaloid cells with peripheral palisading and clefting between the tumor islands and the stroma.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Brown RA, Fernandez-Pol S, Kim J. Primary cutaneous anaplastic large cell lymphoma. J Cutan Pathol. 2017;44:570-577.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014;70:223.e1-17.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Saunders Elsevier; 2015:475-489.
The Diagnosis: Primary Cutaneous Anaplastic Large Cell Lymphoma
Microscopic analysis showed a dense proliferation of mononuclear cells filling and expanding the dermis with focal epidermotropism (Figure 1). Immunohistochemistry demonstrated strong and diffuse staining for CD3, CD4, and CD30 (Figure 2) and lack of staining for anaplastic lymphoma kinase (ALK). Workup to exclude systemic disease was initiated and included unremarkable computed tomography (CT) of the neck, chest, abdomen, and pelvis along with no abnormal cells on bone marrow biopsy. Complete blood cell count, basic metabolic panel, and lactate dehydrogenase were within reference range. Given the lack of evidence for systemic involvement, a diagnosis of primary cutaneous anaplastic large cell lymphoma (PC-ALCL) was made. The treatment plan for our patient with a solitary lesion was localized radiation therapy.
Primary cutaneous CD30+ lymphoproliferative disorders encompass a spectrum of conditions, with premalignant lymphomatoid papulosis (LyP) at one extreme and the malignant PC-ALCL on the other.1 The diagnosis of PC-ALCL is made by clinicopathologic correlation, and lesions typically present abruptly as solitary or grouped nodules with a tendency to ulcerate over time. Spontaneous regression has been reported, but relapse in the skin is frequent.2
A representative, typically excisional, biopsy should be performed if the clinician suspects PC-ALCL. Histologic criteria include a dense dermal infiltrate of large pleomorphic cells and the expression of CD30 in at least 75% of tumor cells.3 Primary cutaneous anaplastic large cell lymphoma typically lacks the ALK gene translocation with the nucleophosmin gene, NPM, that is common in systemic disease; however, a small subset of PC-ALCL may be ALK positive and indicate a higher chance of transformation into systemic disease.2
The extent of the lymphoma should be staged to exclude the possibility of systemic disease. This assessment includes a complete physical examination; laboratory investigation, including complete blood cell count with differential and blood chemistries; and radiography. A positron emission tomography-CT scan of the neck, chest, abdomen, and pelvis, or a whole-body integrated positron emission tomography-CT are sufficient for the radiographic examination.3
The initial choice of treatment for solitary or localized PC-ALCL is localized radiation therapy or low-dose methotrexate. Targeted therapy such as brentuximab has been shown to be effective for those with multifocal systemic involvement or refractory disease.2 Cure rates from radiation therapy alone approach 95%.3 It is important to highlight radiation therapy as the initial management plan to increase awareness and to avoid inappropriate treatment of PC-ALCL with traditional chemotherapy.
Large lesions of LyP may appear similar to PC-ALCL on histopathology, making the two entities difficult to distinguish. However, in contrast to PC-ALCL, LyP classically has a different clinical course characterized by waxing and waning crops of lesions that typically are smaller (<1 cm) than those of PC-ALCL.2 Large cell transformation of mycosis fungoides is another entity to consider, but these patients usually have a known history of mycosis fungoides.4
Keratoacanthomas, considered to be a variant of a well-differentiated squamous cell carcinoma, present as rapidly enlarging crateriform nodules with a keratotic core. They usually are found on the head and neck or sun-exposed areas of the extremities and may regress spontaneously.5 Histology will show atypical, highly differentiated squamous epithelia. Merkel cell carcinoma also has a predilection for the head and neck in older patients and may present as a rapidly growing nodule. However, histology will show an aggressive tumor with small round blue cells, and immunohistochemistry will show the characteristic paranuclear dot staining for CK20 along with staining for various neuroendocrine markers. Similarly, atypical fibroxanthoma is a low-grade sarcoma that also presents on the head and neck of elderly sun-damaged patients.5 Histology will show dermal proliferation of spindle cells that often extend up against the epidermis along with pleomorphism and atypical mitoses. Basal cell carcinoma is a common tumor that can present on the head and neck in sun-damaged patients. Nodular basal cell carcinomas can enlarge and ulcerate, but growth is seen over years rather than weeks.5 Histology characteristically will show tumor islands composed of basaloid cells with peripheral palisading and clefting between the tumor islands and the stroma.
The Diagnosis: Primary Cutaneous Anaplastic Large Cell Lymphoma
Microscopic analysis showed a dense proliferation of mononuclear cells filling and expanding the dermis with focal epidermotropism (Figure 1). Immunohistochemistry demonstrated strong and diffuse staining for CD3, CD4, and CD30 (Figure 2) and lack of staining for anaplastic lymphoma kinase (ALK). Workup to exclude systemic disease was initiated and included unremarkable computed tomography (CT) of the neck, chest, abdomen, and pelvis along with no abnormal cells on bone marrow biopsy. Complete blood cell count, basic metabolic panel, and lactate dehydrogenase were within reference range. Given the lack of evidence for systemic involvement, a diagnosis of primary cutaneous anaplastic large cell lymphoma (PC-ALCL) was made. The treatment plan for our patient with a solitary lesion was localized radiation therapy.
Primary cutaneous CD30+ lymphoproliferative disorders encompass a spectrum of conditions, with premalignant lymphomatoid papulosis (LyP) at one extreme and the malignant PC-ALCL on the other.1 The diagnosis of PC-ALCL is made by clinicopathologic correlation, and lesions typically present abruptly as solitary or grouped nodules with a tendency to ulcerate over time. Spontaneous regression has been reported, but relapse in the skin is frequent.2
A representative, typically excisional, biopsy should be performed if the clinician suspects PC-ALCL. Histologic criteria include a dense dermal infiltrate of large pleomorphic cells and the expression of CD30 in at least 75% of tumor cells.3 Primary cutaneous anaplastic large cell lymphoma typically lacks the ALK gene translocation with the nucleophosmin gene, NPM, that is common in systemic disease; however, a small subset of PC-ALCL may be ALK positive and indicate a higher chance of transformation into systemic disease.2
The extent of the lymphoma should be staged to exclude the possibility of systemic disease. This assessment includes a complete physical examination; laboratory investigation, including complete blood cell count with differential and blood chemistries; and radiography. A positron emission tomography-CT scan of the neck, chest, abdomen, and pelvis, or a whole-body integrated positron emission tomography-CT are sufficient for the radiographic examination.3
The initial choice of treatment for solitary or localized PC-ALCL is localized radiation therapy or low-dose methotrexate. Targeted therapy such as brentuximab has been shown to be effective for those with multifocal systemic involvement or refractory disease.2 Cure rates from radiation therapy alone approach 95%.3 It is important to highlight radiation therapy as the initial management plan to increase awareness and to avoid inappropriate treatment of PC-ALCL with traditional chemotherapy.
Large lesions of LyP may appear similar to PC-ALCL on histopathology, making the two entities difficult to distinguish. However, in contrast to PC-ALCL, LyP classically has a different clinical course characterized by waxing and waning crops of lesions that typically are smaller (<1 cm) than those of PC-ALCL.2 Large cell transformation of mycosis fungoides is another entity to consider, but these patients usually have a known history of mycosis fungoides.4
Keratoacanthomas, considered to be a variant of a well-differentiated squamous cell carcinoma, present as rapidly enlarging crateriform nodules with a keratotic core. They usually are found on the head and neck or sun-exposed areas of the extremities and may regress spontaneously.5 Histology will show atypical, highly differentiated squamous epithelia. Merkel cell carcinoma also has a predilection for the head and neck in older patients and may present as a rapidly growing nodule. However, histology will show an aggressive tumor with small round blue cells, and immunohistochemistry will show the characteristic paranuclear dot staining for CK20 along with staining for various neuroendocrine markers. Similarly, atypical fibroxanthoma is a low-grade sarcoma that also presents on the head and neck of elderly sun-damaged patients.5 Histology will show dermal proliferation of spindle cells that often extend up against the epidermis along with pleomorphism and atypical mitoses. Basal cell carcinoma is a common tumor that can present on the head and neck in sun-damaged patients. Nodular basal cell carcinomas can enlarge and ulcerate, but growth is seen over years rather than weeks.5 Histology characteristically will show tumor islands composed of basaloid cells with peripheral palisading and clefting between the tumor islands and the stroma.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Brown RA, Fernandez-Pol S, Kim J. Primary cutaneous anaplastic large cell lymphoma. J Cutan Pathol. 2017;44:570-577.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014;70:223.e1-17.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Saunders Elsevier; 2015:475-489.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Brown RA, Fernandez-Pol S, Kim J. Primary cutaneous anaplastic large cell lymphoma. J Cutan Pathol. 2017;44:570-577.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014;70:223.e1-17.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Saunders Elsevier; 2015:475-489.
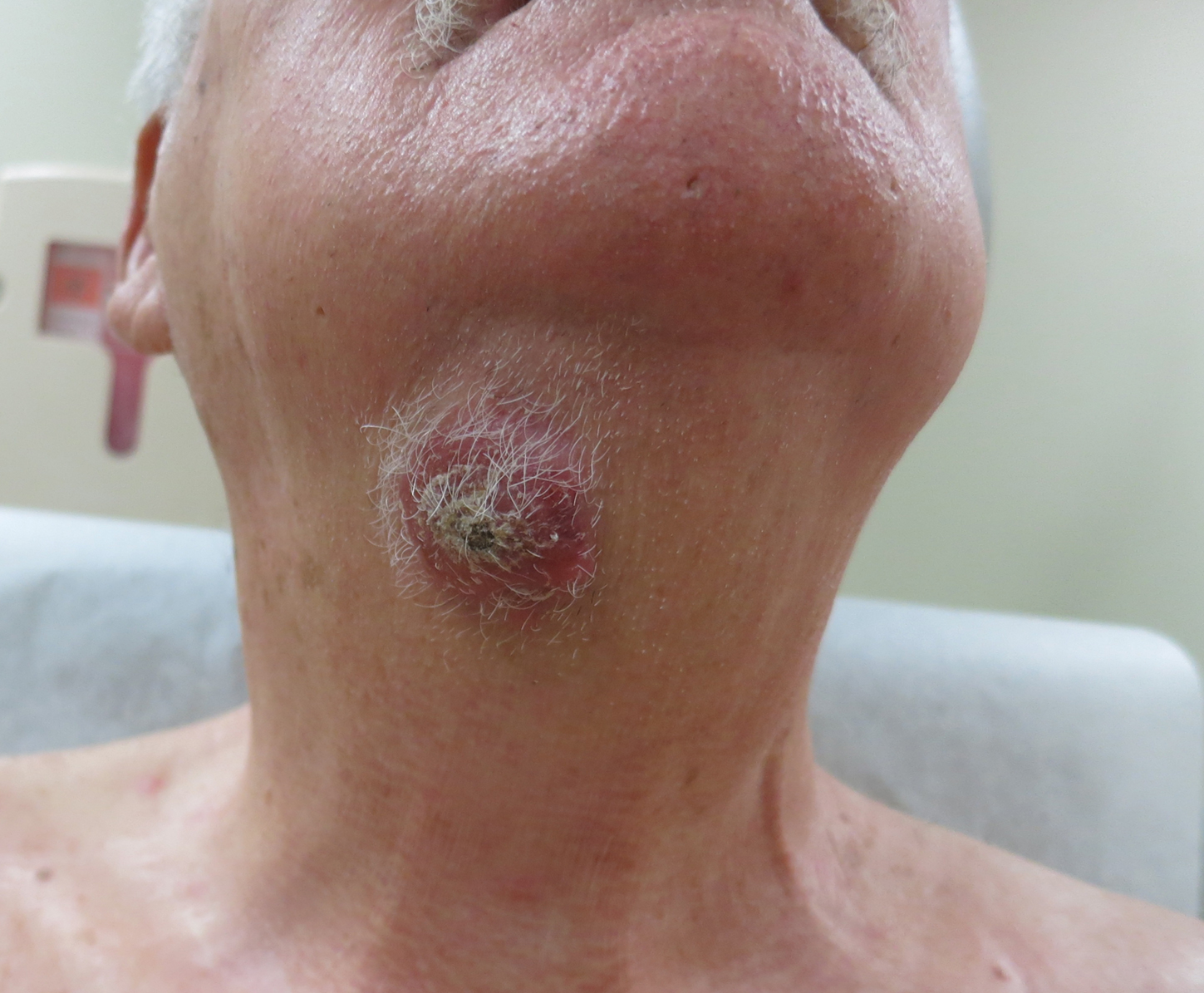
An 80-year-old man presented to our clinic with a large lesion on the right upper neck of approximately 4 weeks’ duration. He reported that it was rapidly increasing in size and had bled on several occasions. No treatments were attempted prior to the initial visit. He denied any constitutional symptoms. The patient had a history of nonmelanoma skin cancers but no other chronic medical problems. Physical examination revealed a large, 35×40-mm, erythematous nodule with central ulceration and overlying hyperkeratosis on the right upper neck. No palpable cervical, supraclavicular, or axillary lymphadenopathy was observed. An excisional biopsy of the lesion was obtained.
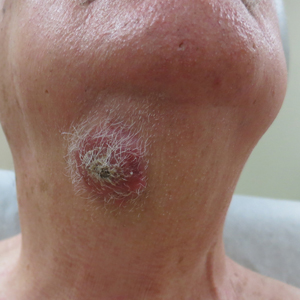
Pruritic and Pustular Eruption on the Face
The Diagnosis: Demodicosis
A clinical diagnosis of facial demodicosis triggered by topical corticosteroid therapy was suspected in our patient. A 3-mm punch biopsy of the right forehead demonstrated a follicular infundibulum rich in Demodex folliculorum with a discrete mononuclear infiltrate in the dermis (Figure 1), confirming the diagnosis. The patient was prescribed metronidazole gel 2% twice daily. Complete clearance of the facial rash was observed at 1-week follow-up (Figure 2).
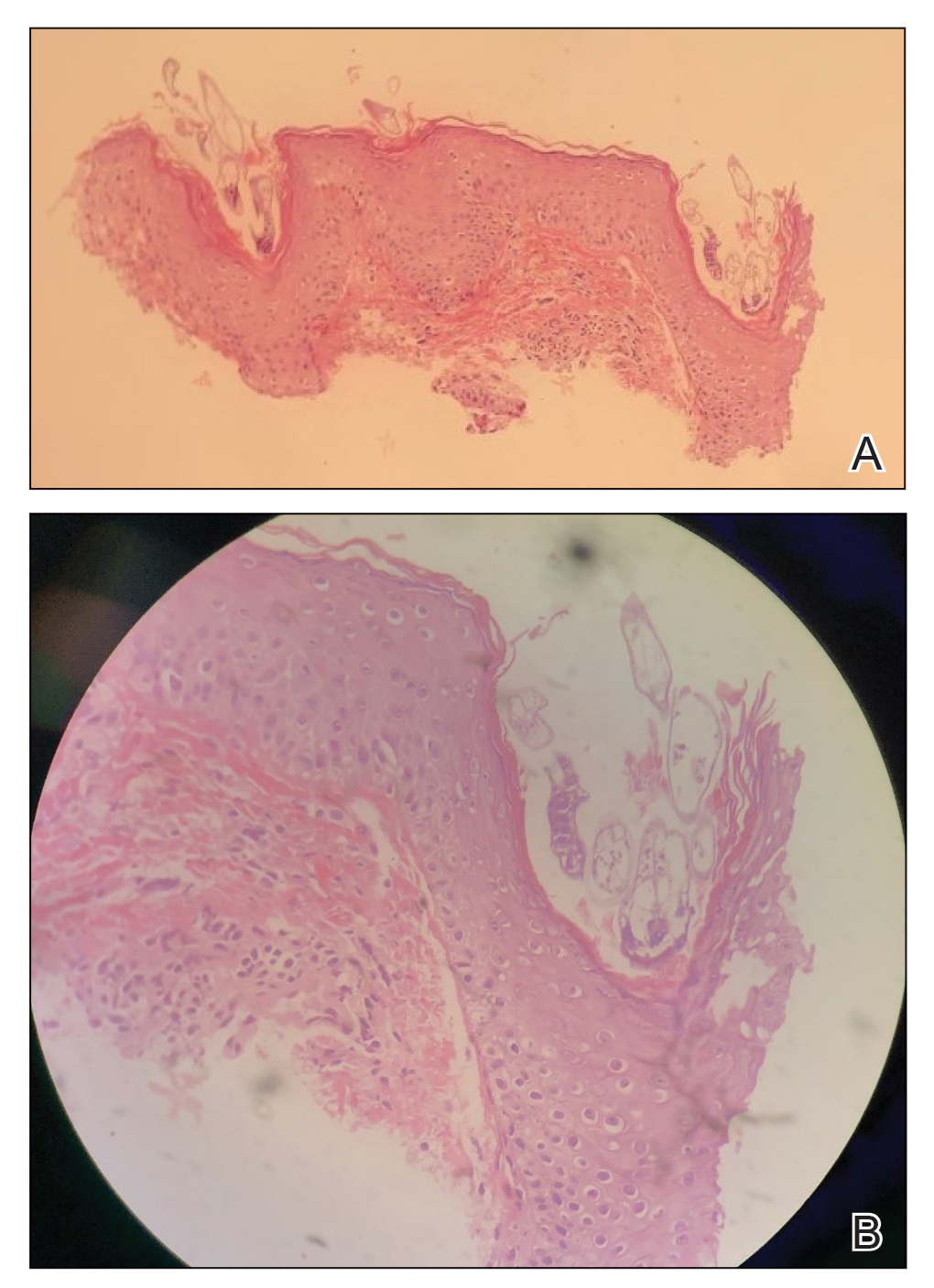
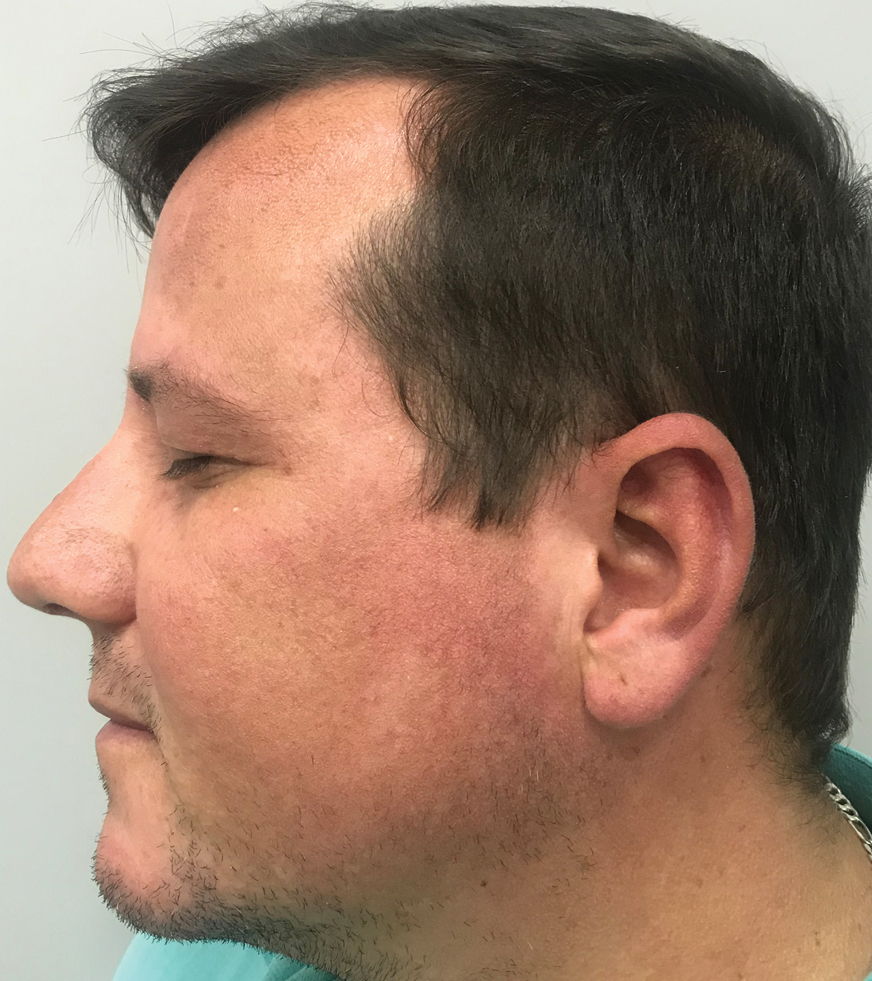
Demodex folliculorum is a mite that parasitizes the pilosebaceous follicles of human skin, becoming pathogenic with excessive colonization.1-3 Facial demodicosis presents as pruritic papules and pustules in a pilosebaceous distribution on the face.2,3 The diagnosis of facial demodicosis can be difficult due to similarities with many other common facial rashes such as acne, rosacea, contact dermatitis, and folliculitis.
Similar to facial demodicosis, rosacea can present with erythema, papules, and pustules on the face. Patients with rosacea also have a higher prevalence and degree of Demodex mite infestation.4 However, these facial rashes can be differentiated when symptom acuity and personal and family histories are considered. In patients with a long-standing personal and/or family history of erythema, papules, and pustules, the chronicity of disease implies a diagnosis of rosacea. The acute onset of symptomatology and absence of personal or family history of rosacea in our patient favored a diagnosis of demodicosis.
Pityrosporum folliculitis is an eruption caused by Malassezia furfur, the organism implicated in tinea versicolor. It can present similarly to Demodex folliculitis with follicular papules and pustules on the forehead and back. Features that help to differentiate Pityrosporum folliculitis from Demodex folliculitis include involvement of the upper trunk and onset after antibiotic usage.5 Diagnosis of Pityrosporum folliculitis can be confirmed by potassium hydroxide preparation, which demonstrates spaghetti-and-meatball-like hyphae and spores.
Eosinophilic folliculitis is a chronic sterile folliculitis that usually presents as intensely pruritic papules and pustules on the face with an eruptive onset. There are 3 variants of disease: the classic form (also known as Ofuji disease), immunosuppression-associated disease, and infancy-associated disease.6 Eosinophilic folliculitis also may present with annular plaques and peripheral blood eosinophilia, and it is more prevalent in patients of Japanese descent. Differentiation from Demodex folliculitis can be done by histologic examination, which demonstrates spongiosis with exocytosis of eosinophils into the epithelium and a clear deficiency of infiltrating mites.6
Miliaria, also known as sweat rash, is a common condition that occurs due to occlusion of eccrine sweat glands.7 Clinically, miliaria is characterized by erythematous papules ranging from 2 to 4 mm in size that may be vesicular or pustular. Miliaria and demodicosis may have similar clinical presentations; however, several characteristic differences can be noted. Based on the pathophysiology, miliaria will not be folliculocentric, which differs from demodicosis. Additionally, miliaria most commonly occurs in areas of occlusion, such as in skin folds or on the trunk under tight clothing. Miliaria rarely can appear confluent and sunburnlike.8 Furthermore, miliaria is less common on the face, while demodicosis almost exclusively is found on the face.
We present the case of an otherwise healthy patient with acute onset of pruritic papules and pustules involving the superior face following topical corticosteroid use. Similar cases frequently are encountered by dermatologists and require broad differential diagnoses.
- Aylesworth R, Vance JC. Demodex folliculorum and Demodex brevis in cutaneous biopsies. J Am Acad Dermatol. 1982;7:583-589.
- Kligman AM, Christensen MS. Demodex folliculorum: requirements for understanding its role in human skin disease. J Invest Dermatol. 2011;131:8-10.
- Zomorodian K, Geramishoar M, Saadat F, et al. Facial demodicosis. Eur J Dermatol. 2004;14:121-122.
- Chang YS, Huang YC. Role of Demodex mite infestation in rosacea: a systematic review and meta-analysis. J Am Acad Dermatol. 2017;77:441-447.
- Prindaville B, Belazarian L, Levin NA, et al. Pityrosporum folliculitis: a retrospective review of 110 cases. J Am Acad Dermatol. 2018;78:511-514.
- Lankerani L, Thompson R. Eosinophilic pustular folliculitis: case report and review of the literature. Cutis. 2010;86:190-194.
- Hölzle ER, Kligman AM. The pathogenesis of miliaria rubra. role of the resident microflora. Br J Dermatol. 1978;99:117-137.
- Al-Hilo MM, Al-Saedy SJ, Alwan AI. Atypical presentation of miliaria in Iraqi patients attending Al-Kindy Teaching Hospital in Baghdad: a clinical descriptive study. Am J Dermatol Venereol. 2012;1:41-46.
The Diagnosis: Demodicosis
A clinical diagnosis of facial demodicosis triggered by topical corticosteroid therapy was suspected in our patient. A 3-mm punch biopsy of the right forehead demonstrated a follicular infundibulum rich in Demodex folliculorum with a discrete mononuclear infiltrate in the dermis (Figure 1), confirming the diagnosis. The patient was prescribed metronidazole gel 2% twice daily. Complete clearance of the facial rash was observed at 1-week follow-up (Figure 2).
Demodex folliculorum is a mite that parasitizes the pilosebaceous follicles of human skin, becoming pathogenic with excessive colonization.1-3 Facial demodicosis presents as pruritic papules and pustules in a pilosebaceous distribution on the face.2,3 The diagnosis of facial demodicosis can be difficult due to similarities with many other common facial rashes such as acne, rosacea, contact dermatitis, and folliculitis.
Similar to facial demodicosis, rosacea can present with erythema, papules, and pustules on the face. Patients with rosacea also have a higher prevalence and degree of Demodex mite infestation.4 However, these facial rashes can be differentiated when symptom acuity and personal and family histories are considered. In patients with a long-standing personal and/or family history of erythema, papules, and pustules, the chronicity of disease implies a diagnosis of rosacea. The acute onset of symptomatology and absence of personal or family history of rosacea in our patient favored a diagnosis of demodicosis.
Pityrosporum folliculitis is an eruption caused by Malassezia furfur, the organism implicated in tinea versicolor. It can present similarly to Demodex folliculitis with follicular papules and pustules on the forehead and back. Features that help to differentiate Pityrosporum folliculitis from Demodex folliculitis include involvement of the upper trunk and onset after antibiotic usage.5 Diagnosis of Pityrosporum folliculitis can be confirmed by potassium hydroxide preparation, which demonstrates spaghetti-and-meatball-like hyphae and spores.
Eosinophilic folliculitis is a chronic sterile folliculitis that usually presents as intensely pruritic papules and pustules on the face with an eruptive onset. There are 3 variants of disease: the classic form (also known as Ofuji disease), immunosuppression-associated disease, and infancy-associated disease.6 Eosinophilic folliculitis also may present with annular plaques and peripheral blood eosinophilia, and it is more prevalent in patients of Japanese descent. Differentiation from Demodex folliculitis can be done by histologic examination, which demonstrates spongiosis with exocytosis of eosinophils into the epithelium and a clear deficiency of infiltrating mites.6
Miliaria, also known as sweat rash, is a common condition that occurs due to occlusion of eccrine sweat glands.7 Clinically, miliaria is characterized by erythematous papules ranging from 2 to 4 mm in size that may be vesicular or pustular. Miliaria and demodicosis may have similar clinical presentations; however, several characteristic differences can be noted. Based on the pathophysiology, miliaria will not be folliculocentric, which differs from demodicosis. Additionally, miliaria most commonly occurs in areas of occlusion, such as in skin folds or on the trunk under tight clothing. Miliaria rarely can appear confluent and sunburnlike.8 Furthermore, miliaria is less common on the face, while demodicosis almost exclusively is found on the face.
We present the case of an otherwise healthy patient with acute onset of pruritic papules and pustules involving the superior face following topical corticosteroid use. Similar cases frequently are encountered by dermatologists and require broad differential diagnoses.
The Diagnosis: Demodicosis
A clinical diagnosis of facial demodicosis triggered by topical corticosteroid therapy was suspected in our patient. A 3-mm punch biopsy of the right forehead demonstrated a follicular infundibulum rich in Demodex folliculorum with a discrete mononuclear infiltrate in the dermis (Figure 1), confirming the diagnosis. The patient was prescribed metronidazole gel 2% twice daily. Complete clearance of the facial rash was observed at 1-week follow-up (Figure 2).
Demodex folliculorum is a mite that parasitizes the pilosebaceous follicles of human skin, becoming pathogenic with excessive colonization.1-3 Facial demodicosis presents as pruritic papules and pustules in a pilosebaceous distribution on the face.2,3 The diagnosis of facial demodicosis can be difficult due to similarities with many other common facial rashes such as acne, rosacea, contact dermatitis, and folliculitis.
Similar to facial demodicosis, rosacea can present with erythema, papules, and pustules on the face. Patients with rosacea also have a higher prevalence and degree of Demodex mite infestation.4 However, these facial rashes can be differentiated when symptom acuity and personal and family histories are considered. In patients with a long-standing personal and/or family history of erythema, papules, and pustules, the chronicity of disease implies a diagnosis of rosacea. The acute onset of symptomatology and absence of personal or family history of rosacea in our patient favored a diagnosis of demodicosis.
Pityrosporum folliculitis is an eruption caused by Malassezia furfur, the organism implicated in tinea versicolor. It can present similarly to Demodex folliculitis with follicular papules and pustules on the forehead and back. Features that help to differentiate Pityrosporum folliculitis from Demodex folliculitis include involvement of the upper trunk and onset after antibiotic usage.5 Diagnosis of Pityrosporum folliculitis can be confirmed by potassium hydroxide preparation, which demonstrates spaghetti-and-meatball-like hyphae and spores.
Eosinophilic folliculitis is a chronic sterile folliculitis that usually presents as intensely pruritic papules and pustules on the face with an eruptive onset. There are 3 variants of disease: the classic form (also known as Ofuji disease), immunosuppression-associated disease, and infancy-associated disease.6 Eosinophilic folliculitis also may present with annular plaques and peripheral blood eosinophilia, and it is more prevalent in patients of Japanese descent. Differentiation from Demodex folliculitis can be done by histologic examination, which demonstrates spongiosis with exocytosis of eosinophils into the epithelium and a clear deficiency of infiltrating mites.6
Miliaria, also known as sweat rash, is a common condition that occurs due to occlusion of eccrine sweat glands.7 Clinically, miliaria is characterized by erythematous papules ranging from 2 to 4 mm in size that may be vesicular or pustular. Miliaria and demodicosis may have similar clinical presentations; however, several characteristic differences can be noted. Based on the pathophysiology, miliaria will not be folliculocentric, which differs from demodicosis. Additionally, miliaria most commonly occurs in areas of occlusion, such as in skin folds or on the trunk under tight clothing. Miliaria rarely can appear confluent and sunburnlike.8 Furthermore, miliaria is less common on the face, while demodicosis almost exclusively is found on the face.
We present the case of an otherwise healthy patient with acute onset of pruritic papules and pustules involving the superior face following topical corticosteroid use. Similar cases frequently are encountered by dermatologists and require broad differential diagnoses.
- Aylesworth R, Vance JC. Demodex folliculorum and Demodex brevis in cutaneous biopsies. J Am Acad Dermatol. 1982;7:583-589.
- Kligman AM, Christensen MS. Demodex folliculorum: requirements for understanding its role in human skin disease. J Invest Dermatol. 2011;131:8-10.
- Zomorodian K, Geramishoar M, Saadat F, et al. Facial demodicosis. Eur J Dermatol. 2004;14:121-122.
- Chang YS, Huang YC. Role of Demodex mite infestation in rosacea: a systematic review and meta-analysis. J Am Acad Dermatol. 2017;77:441-447.
- Prindaville B, Belazarian L, Levin NA, et al. Pityrosporum folliculitis: a retrospective review of 110 cases. J Am Acad Dermatol. 2018;78:511-514.
- Lankerani L, Thompson R. Eosinophilic pustular folliculitis: case report and review of the literature. Cutis. 2010;86:190-194.
- Hölzle ER, Kligman AM. The pathogenesis of miliaria rubra. role of the resident microflora. Br J Dermatol. 1978;99:117-137.
- Al-Hilo MM, Al-Saedy SJ, Alwan AI. Atypical presentation of miliaria in Iraqi patients attending Al-Kindy Teaching Hospital in Baghdad: a clinical descriptive study. Am J Dermatol Venereol. 2012;1:41-46.
- Aylesworth R, Vance JC. Demodex folliculorum and Demodex brevis in cutaneous biopsies. J Am Acad Dermatol. 1982;7:583-589.
- Kligman AM, Christensen MS. Demodex folliculorum: requirements for understanding its role in human skin disease. J Invest Dermatol. 2011;131:8-10.
- Zomorodian K, Geramishoar M, Saadat F, et al. Facial demodicosis. Eur J Dermatol. 2004;14:121-122.
- Chang YS, Huang YC. Role of Demodex mite infestation in rosacea: a systematic review and meta-analysis. J Am Acad Dermatol. 2017;77:441-447.
- Prindaville B, Belazarian L, Levin NA, et al. Pityrosporum folliculitis: a retrospective review of 110 cases. J Am Acad Dermatol. 2018;78:511-514.
- Lankerani L, Thompson R. Eosinophilic pustular folliculitis: case report and review of the literature. Cutis. 2010;86:190-194.
- Hölzle ER, Kligman AM. The pathogenesis of miliaria rubra. role of the resident microflora. Br J Dermatol. 1978;99:117-137.
- Al-Hilo MM, Al-Saedy SJ, Alwan AI. Atypical presentation of miliaria in Iraqi patients attending Al-Kindy Teaching Hospital in Baghdad: a clinical descriptive study. Am J Dermatol Venereol. 2012;1:41-46.
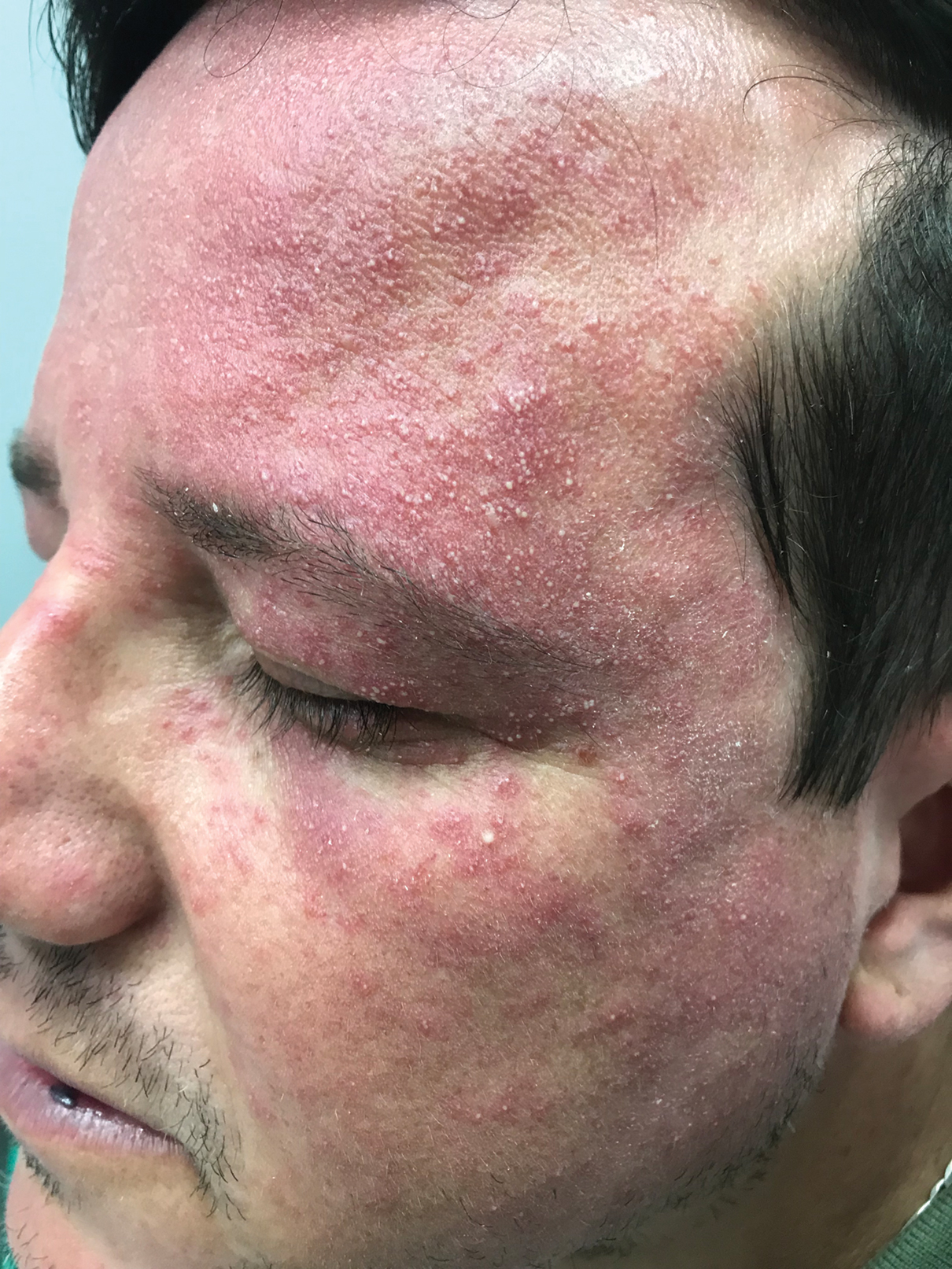
A 37-year-old man presented with a progressively pruritic and pustular eruption on the face of 2 weeks’ duration. Twenty days prior to the rash onset, he began treatment for scalp psoriasis with a mixture of salicylic acid 20 mg/mL and betamethasone dipropionate 0.5 mg/mL, which inadvertently extended to the facial area. One week after rash onset, he presented to the emergency department at a local hospital, where he was given intravenous hydrocortisone 100 mg with no improvement of the rash. He had no history of skin cancer, and his family history was negative for dermatologic disease. At the time of examination, he had an erythematous eruption of follicular micropustules on the forehead, upper and lower eyelids, temples, cheeks, and mandible.
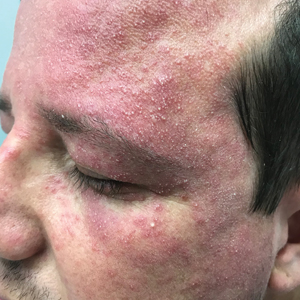
Dark Brown Hyperkeratotic Nodule on the Back
The Diagnosis: Seborrheic Keratosis-like Melanoma
Seborrheic keratosis (SK) is a benign neoplasm commonly encountered on the skin and frequently diagnosed by clinical examination alone. Seborrheic keratosis-like melanomas are melanomas that clinically or dermatoscopically resemble SKs and thus can be challenging to accurately diagnose. Melanomas can have a hyperkeratotic or verrucous appearance1-3 and can even exhibit dermatoscopic and microscopic features that are found in SKs such as comedolike openings and milialike cysts as well as acanthosis and pseudohorn cysts, respectively.2
In our patient, histopathology revealed SK-like architecture with hyperorthokeratosis, papillomatosis, pseudohorn cyst formation, and basaloid acanthosis (Figure). However, within the lesion was an asymmetric proliferation of nested atypical melanocytes with melanin pigment production. The atypical melanocytes filled and expanded papillomatous projections without notable pagetoid growth and extended into the dermis. There was a background congenital nevus component. These findings were diagnostic of invasive malignant melanoma, extending to a Breslow depth of 5.5 mm. A follow-up sentinel lymph node biopsy was negative for metastatic melanoma. The clinical and histologic findings did not show melanoma in the surrounding skin to suggest colonization of an SK by an adjacent melanoma. The clinical history of a long-standing lesion in conjunction with a congenital nevus component on histology favored a diagnosis of melanoma arising in association with a congenital nevus with an SK-like architecture rather than arising in a preexisting SK or de novo melanoma.
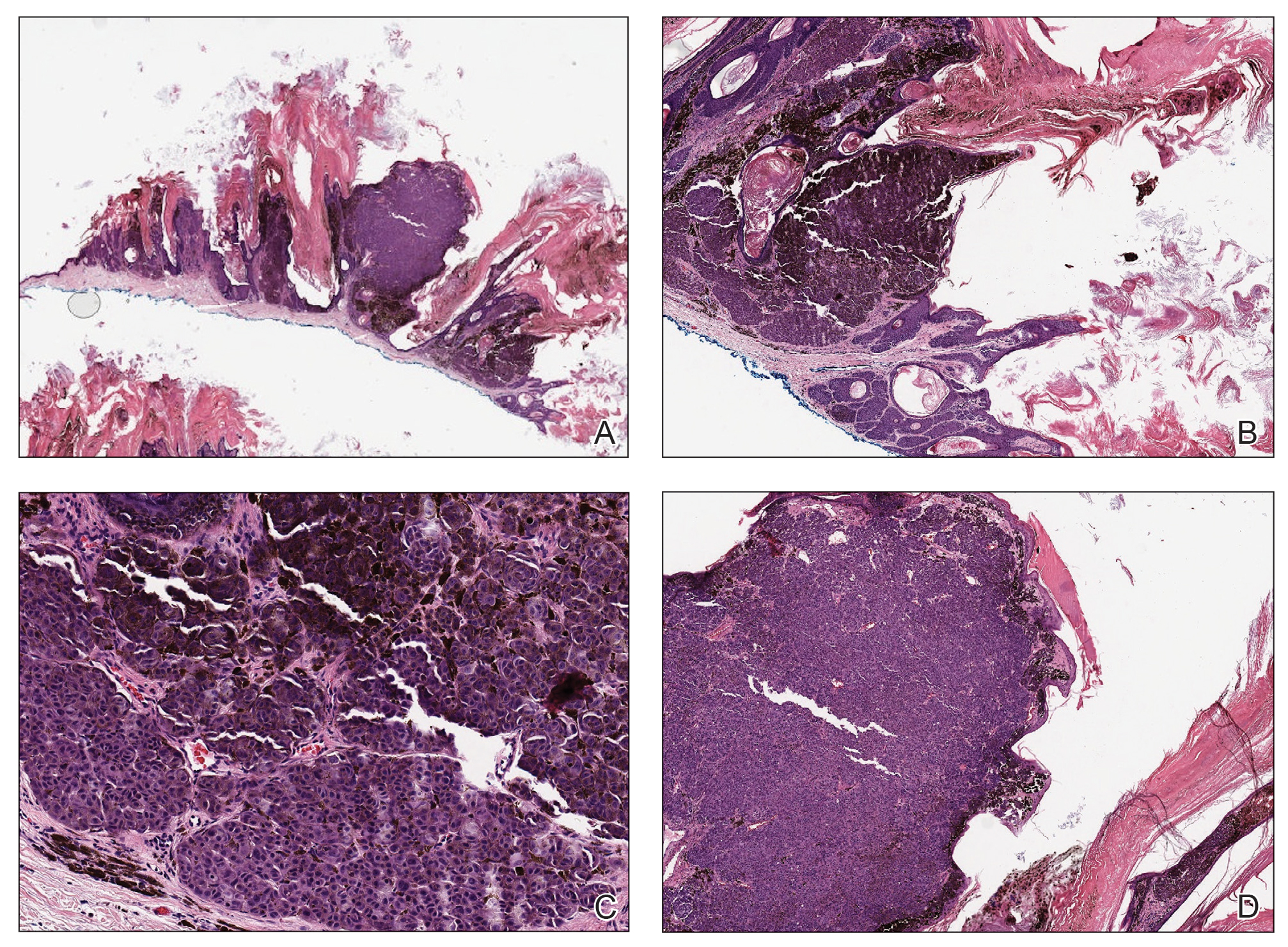
Because our patient did not have multiple widespread SKs and reported rapid growth in the lesion in the last 6 months, there was concern for a malignant neoplasm. However, in patients with numerous SKs or areas of chronically sun-damaged skin, it can be difficult to identify suspicious lesions. It is important for clinicians to remain aware of SK-like melanomas and have a lower threshold for biopsy of any changing or symptomatic lesion that clinically resembles an SK. In our case, the history of change and the markedly different clinical appearance of the lesion in comparison to our patient's SKs prompted the biopsy. Criteria have been proposed to help differentiate these entities under dermoscopy, with melanoma showing the presence of the blue-black sign, pigment network, pseudopods or streaks, and/or the blue-white veil.4
Cutaneous metastases classically present as dermal nodules, plaques, or ulcers.5,6 A rare pigmented case of metastatic breast adenocarcinoma clinically mimicking melanoma has been reported.7 There is limited literature on the dermoscopic features of cutaneous metastases, but it appears that polymorphic vascular patterns are most common.5,8 The possibility of a metastatic melanoma involving an SK is a theoretical consideration, but there was no prior history of melanoma in our patient, and the histologic findings were consistent with primary melanoma. There was no histologic evidence of pigmented metastatic breast carcinoma or metastatic lung carcinoma.
Pigmented malignant hidroacanthoma simplex and pigmented porocarcinomas are rare malignant sweat gland tumors.9-11 Their benign counterparts are the more commonly encountered hidroacanthoma simplex (intraepidermal poroma) and poroma. Pigmented malignant hidroacanthoma simplex has been reported to clinically mimic an irritated SK.10 The histopathology of our case did not have features of malignant hidroacanthoma simplex or porocarcinoma. Pigmented squamous cell carcinoma is an uncommon variant of squamous cell carcinoma, and histopathology would reveal proliferation of atypical keratinocytes.12
- Saggini A, Cota C, Lora V, et al. Uncommon histopathological variants of malignant melanoma. part 2. Am J Dermatopathol. 2019;41:321-342.
- Klebanov N, Gunasekera N, Lin WM, et al. The clinical spectrum of cutaneous melanoma morphology. J Am Acad Dermatol. 2019;80:178-188.
- Tran PT, Truong AK, Munday W, et al. Verrucous melanoma masquerading as a seborrheic keratosis. Dermatol Online J. 2019;25:13030/qt1m07k7fm.
- Carrera C, Segura S, Aguilera P. Dermoscopic clues for diagnosing melanomas that resemble seborrheic keratosis. JAMA Dermatol. 2017;153:544-551.
- Strickley JD, Jenson AB, Jung JY. Cutaneous metastasis. Hematol Oncol Clin North Am. 2019;33:173-197.
- Chernoff KA, Marghoob AA, Lacouture ME. Dermoscopic findings in cutaneous metastases. JAMA Dermatol. 2014;150:429-433.
- Marti N, Molina I, Monteagudo C, et al. Cutaneous metastasis of breast carcinoma mimicking malignant melanoma in scalp. Dermatol Online J. 2008;14:12.
- Kelati A, Gallouj S. Dermoscopy of skin metastases from breast cancer: two case reports. J Med Case Rep. 2018;12:273.
- Ishida M, Hotta M, Kushima R, et al. A case of porocarcinoma arising in pigmented hidroacanthoma simplex with multiple lymph node, liver and bone metastases. J Cutan Pathol. 2011;38:227-231.
- Lee JY, Lin MH. Pigmented malignant hidroacanthoma simplex mimicking irritated seborrheic keratosis. J Cutan Pathol. 2006;33:705-708.
- Ueo T, Kashima K, Daa T, et al. Porocarcinoma arising in pigmented hidroacanthoma simplex. Am J Dermatopathol. 2005;27:500-503.
- Motta de Morais P, Schettini A, Rocha J, et al. Pigmented squamous cell carcinoma: case report and importance of differential diagnosis. An Bras Dermatol. 2018;93:96-98.
The Diagnosis: Seborrheic Keratosis-like Melanoma
Seborrheic keratosis (SK) is a benign neoplasm commonly encountered on the skin and frequently diagnosed by clinical examination alone. Seborrheic keratosis-like melanomas are melanomas that clinically or dermatoscopically resemble SKs and thus can be challenging to accurately diagnose. Melanomas can have a hyperkeratotic or verrucous appearance1-3 and can even exhibit dermatoscopic and microscopic features that are found in SKs such as comedolike openings and milialike cysts as well as acanthosis and pseudohorn cysts, respectively.2
In our patient, histopathology revealed SK-like architecture with hyperorthokeratosis, papillomatosis, pseudohorn cyst formation, and basaloid acanthosis (Figure). However, within the lesion was an asymmetric proliferation of nested atypical melanocytes with melanin pigment production. The atypical melanocytes filled and expanded papillomatous projections without notable pagetoid growth and extended into the dermis. There was a background congenital nevus component. These findings were diagnostic of invasive malignant melanoma, extending to a Breslow depth of 5.5 mm. A follow-up sentinel lymph node biopsy was negative for metastatic melanoma. The clinical and histologic findings did not show melanoma in the surrounding skin to suggest colonization of an SK by an adjacent melanoma. The clinical history of a long-standing lesion in conjunction with a congenital nevus component on histology favored a diagnosis of melanoma arising in association with a congenital nevus with an SK-like architecture rather than arising in a preexisting SK or de novo melanoma.
Because our patient did not have multiple widespread SKs and reported rapid growth in the lesion in the last 6 months, there was concern for a malignant neoplasm. However, in patients with numerous SKs or areas of chronically sun-damaged skin, it can be difficult to identify suspicious lesions. It is important for clinicians to remain aware of SK-like melanomas and have a lower threshold for biopsy of any changing or symptomatic lesion that clinically resembles an SK. In our case, the history of change and the markedly different clinical appearance of the lesion in comparison to our patient's SKs prompted the biopsy. Criteria have been proposed to help differentiate these entities under dermoscopy, with melanoma showing the presence of the blue-black sign, pigment network, pseudopods or streaks, and/or the blue-white veil.4
Cutaneous metastases classically present as dermal nodules, plaques, or ulcers.5,6 A rare pigmented case of metastatic breast adenocarcinoma clinically mimicking melanoma has been reported.7 There is limited literature on the dermoscopic features of cutaneous metastases, but it appears that polymorphic vascular patterns are most common.5,8 The possibility of a metastatic melanoma involving an SK is a theoretical consideration, but there was no prior history of melanoma in our patient, and the histologic findings were consistent with primary melanoma. There was no histologic evidence of pigmented metastatic breast carcinoma or metastatic lung carcinoma.
Pigmented malignant hidroacanthoma simplex and pigmented porocarcinomas are rare malignant sweat gland tumors.9-11 Their benign counterparts are the more commonly encountered hidroacanthoma simplex (intraepidermal poroma) and poroma. Pigmented malignant hidroacanthoma simplex has been reported to clinically mimic an irritated SK.10 The histopathology of our case did not have features of malignant hidroacanthoma simplex or porocarcinoma. Pigmented squamous cell carcinoma is an uncommon variant of squamous cell carcinoma, and histopathology would reveal proliferation of atypical keratinocytes.12
The Diagnosis: Seborrheic Keratosis-like Melanoma
Seborrheic keratosis (SK) is a benign neoplasm commonly encountered on the skin and frequently diagnosed by clinical examination alone. Seborrheic keratosis-like melanomas are melanomas that clinically or dermatoscopically resemble SKs and thus can be challenging to accurately diagnose. Melanomas can have a hyperkeratotic or verrucous appearance1-3 and can even exhibit dermatoscopic and microscopic features that are found in SKs such as comedolike openings and milialike cysts as well as acanthosis and pseudohorn cysts, respectively.2
In our patient, histopathology revealed SK-like architecture with hyperorthokeratosis, papillomatosis, pseudohorn cyst formation, and basaloid acanthosis (Figure). However, within the lesion was an asymmetric proliferation of nested atypical melanocytes with melanin pigment production. The atypical melanocytes filled and expanded papillomatous projections without notable pagetoid growth and extended into the dermis. There was a background congenital nevus component. These findings were diagnostic of invasive malignant melanoma, extending to a Breslow depth of 5.5 mm. A follow-up sentinel lymph node biopsy was negative for metastatic melanoma. The clinical and histologic findings did not show melanoma in the surrounding skin to suggest colonization of an SK by an adjacent melanoma. The clinical history of a long-standing lesion in conjunction with a congenital nevus component on histology favored a diagnosis of melanoma arising in association with a congenital nevus with an SK-like architecture rather than arising in a preexisting SK or de novo melanoma.
Because our patient did not have multiple widespread SKs and reported rapid growth in the lesion in the last 6 months, there was concern for a malignant neoplasm. However, in patients with numerous SKs or areas of chronically sun-damaged skin, it can be difficult to identify suspicious lesions. It is important for clinicians to remain aware of SK-like melanomas and have a lower threshold for biopsy of any changing or symptomatic lesion that clinically resembles an SK. In our case, the history of change and the markedly different clinical appearance of the lesion in comparison to our patient's SKs prompted the biopsy. Criteria have been proposed to help differentiate these entities under dermoscopy, with melanoma showing the presence of the blue-black sign, pigment network, pseudopods or streaks, and/or the blue-white veil.4
Cutaneous metastases classically present as dermal nodules, plaques, or ulcers.5,6 A rare pigmented case of metastatic breast adenocarcinoma clinically mimicking melanoma has been reported.7 There is limited literature on the dermoscopic features of cutaneous metastases, but it appears that polymorphic vascular patterns are most common.5,8 The possibility of a metastatic melanoma involving an SK is a theoretical consideration, but there was no prior history of melanoma in our patient, and the histologic findings were consistent with primary melanoma. There was no histologic evidence of pigmented metastatic breast carcinoma or metastatic lung carcinoma.
Pigmented malignant hidroacanthoma simplex and pigmented porocarcinomas are rare malignant sweat gland tumors.9-11 Their benign counterparts are the more commonly encountered hidroacanthoma simplex (intraepidermal poroma) and poroma. Pigmented malignant hidroacanthoma simplex has been reported to clinically mimic an irritated SK.10 The histopathology of our case did not have features of malignant hidroacanthoma simplex or porocarcinoma. Pigmented squamous cell carcinoma is an uncommon variant of squamous cell carcinoma, and histopathology would reveal proliferation of atypical keratinocytes.12
- Saggini A, Cota C, Lora V, et al. Uncommon histopathological variants of malignant melanoma. part 2. Am J Dermatopathol. 2019;41:321-342.
- Klebanov N, Gunasekera N, Lin WM, et al. The clinical spectrum of cutaneous melanoma morphology. J Am Acad Dermatol. 2019;80:178-188.
- Tran PT, Truong AK, Munday W, et al. Verrucous melanoma masquerading as a seborrheic keratosis. Dermatol Online J. 2019;25:13030/qt1m07k7fm.
- Carrera C, Segura S, Aguilera P. Dermoscopic clues for diagnosing melanomas that resemble seborrheic keratosis. JAMA Dermatol. 2017;153:544-551.
- Strickley JD, Jenson AB, Jung JY. Cutaneous metastasis. Hematol Oncol Clin North Am. 2019;33:173-197.
- Chernoff KA, Marghoob AA, Lacouture ME. Dermoscopic findings in cutaneous metastases. JAMA Dermatol. 2014;150:429-433.
- Marti N, Molina I, Monteagudo C, et al. Cutaneous metastasis of breast carcinoma mimicking malignant melanoma in scalp. Dermatol Online J. 2008;14:12.
- Kelati A, Gallouj S. Dermoscopy of skin metastases from breast cancer: two case reports. J Med Case Rep. 2018;12:273.
- Ishida M, Hotta M, Kushima R, et al. A case of porocarcinoma arising in pigmented hidroacanthoma simplex with multiple lymph node, liver and bone metastases. J Cutan Pathol. 2011;38:227-231.
- Lee JY, Lin MH. Pigmented malignant hidroacanthoma simplex mimicking irritated seborrheic keratosis. J Cutan Pathol. 2006;33:705-708.
- Ueo T, Kashima K, Daa T, et al. Porocarcinoma arising in pigmented hidroacanthoma simplex. Am J Dermatopathol. 2005;27:500-503.
- Motta de Morais P, Schettini A, Rocha J, et al. Pigmented squamous cell carcinoma: case report and importance of differential diagnosis. An Bras Dermatol. 2018;93:96-98.
- Saggini A, Cota C, Lora V, et al. Uncommon histopathological variants of malignant melanoma. part 2. Am J Dermatopathol. 2019;41:321-342.
- Klebanov N, Gunasekera N, Lin WM, et al. The clinical spectrum of cutaneous melanoma morphology. J Am Acad Dermatol. 2019;80:178-188.
- Tran PT, Truong AK, Munday W, et al. Verrucous melanoma masquerading as a seborrheic keratosis. Dermatol Online J. 2019;25:13030/qt1m07k7fm.
- Carrera C, Segura S, Aguilera P. Dermoscopic clues for diagnosing melanomas that resemble seborrheic keratosis. JAMA Dermatol. 2017;153:544-551.
- Strickley JD, Jenson AB, Jung JY. Cutaneous metastasis. Hematol Oncol Clin North Am. 2019;33:173-197.
- Chernoff KA, Marghoob AA, Lacouture ME. Dermoscopic findings in cutaneous metastases. JAMA Dermatol. 2014;150:429-433.
- Marti N, Molina I, Monteagudo C, et al. Cutaneous metastasis of breast carcinoma mimicking malignant melanoma in scalp. Dermatol Online J. 2008;14:12.
- Kelati A, Gallouj S. Dermoscopy of skin metastases from breast cancer: two case reports. J Med Case Rep. 2018;12:273.
- Ishida M, Hotta M, Kushima R, et al. A case of porocarcinoma arising in pigmented hidroacanthoma simplex with multiple lymph node, liver and bone metastases. J Cutan Pathol. 2011;38:227-231.
- Lee JY, Lin MH. Pigmented malignant hidroacanthoma simplex mimicking irritated seborrheic keratosis. J Cutan Pathol. 2006;33:705-708.
- Ueo T, Kashima K, Daa T, et al. Porocarcinoma arising in pigmented hidroacanthoma simplex. Am J Dermatopathol. 2005;27:500-503.
- Motta de Morais P, Schettini A, Rocha J, et al. Pigmented squamous cell carcinoma: case report and importance of differential diagnosis. An Bras Dermatol. 2018;93:96-98.
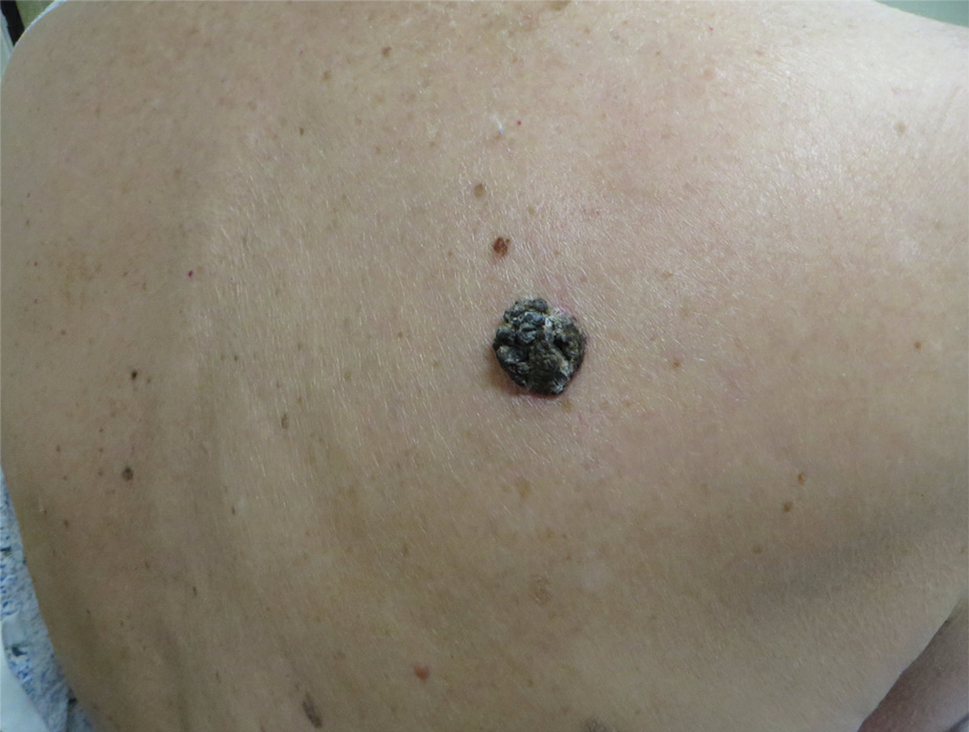
A 71-year-old woman presented with a persistent asymptomatic lesion on the right upper back that had recently increased in size and changed in color, shape, and texture. The lesion had been present for many years. Physical examination revealed a 1.5-cm, dark brown, hyperkeratotic nodule with no identifiable pigment network on dermatoscopy. The patient had no personal history of melanoma but did have a history of stage I non–small cell lung cancer. A review of systems was noncontributory. A shave biopsy of the lesion was performed.
Hyperpigmentation on the Head and Neck
The Diagnosis: Frontal Fibrosing Alopecia Overlapping With Lichen Planus Pigmentosus
Microscopic examination revealed focal dermal pigmentation, papillary fibrosis, and epidermal atrophy. These clinical and histologic findings indicated a diagnosis of fully developed lichen planus pigmentosus (LPP) overlapping with frontal fibrosing alopecia (FFA). Other cases have demonstrated an association between LPP and FFA.1,2
Lichen planus pigmentosus is considered an uncommon variant of lichen planus, as it has similar histopathologic findings and occasional coexistence.3,4 It is characterized by hyperpigmented macules primarily located in sun-exposed and flexural areas of the skin. First described in India,5 this disease has a predilection for darker skin (Fitzpatrick skin types III-V),6,7 and it has been reported in other racial and ethnic groups including Latin Americans, Middle Eastern populations, Japanese, and Koreans.4,8 Typically, lesions initially appear as ill-defined, blue-grey, round to oval macules that coalesce into hyperpigmented patches. Involvement most commonly begins at the forehead and temples, which are affected in nearly all patients. Infrequently, LPP can be generalized or affect the oral mucosa; involvement of the palms, soles, and nails does not occur. Patients may be asymptomatic, but some experience mild pruritus and burning. The disease course is chronic and insidious, with new lesions appearing over time and old lesions progressively darkening and expanding.6,7,9
Although the pathogenesis of LPP is unknown, several exposures have been implicated, such as amla oil, mustard oil, henna, hair dye, and environmental pollutants.7 Because lesions characteristically occur in sun-exposed areas, UV light also may be involved. In addition, studies have suggested that LPP is associated with endocrinopathies such as diabetes mellitus and dyslipidemias, as in our patient, as well as autoimmune conditions such as vitiligo and systemic lupus erythematosus.10,11
Histopathologic findings are characterized by vacuolar degeneration of the basal layer in the epidermis as well as perivascular lymphohistiocytic infiltration and the presence of melanophages in the dermis.3,9 Lichen planus pigmentosus is difficult to treat, as no consistently effective modality has been established. Topical tacrolimus, topical corticosteroids, oral retinoids, lasers, and sun protection have been implemented with underwhelming results.12
Frontal fibrosing alopecia is a variant of lichen planopilaris that predominantly affects postmenopausal women and presents with frontotemporal hair loss in a bandlike distribution.5,13 Both terminal and vellus hairs are affected. Involvement of multiple hair-bearing sites of the skin have been reported, including the entire scalp, eyebrows, and eyelashes. Affected areas may display hypopigmentation and be accompanied by pruritus and trichodynia.14,15 The pathogenesis currently is under investigation, with studies demonstrating autoimmune, genetic, and possibly even endocrine predispositions.16-18 Biopsies of lesions are indistinguishable from lichen planopilaris, which shows follicular lymphocytic infiltration, perifollicular fibrosis, interface dermatitis of the follicular infundibulum and isthmus, and vertical fibrous tracks.5 Patients with FFA have demonstrated variable responses to treatments, with one study showing improvement with oral finasteride or dutasteride.14 Topical and intralesional corticosteroids have yielded suboptimal effects. Other modalities include hydroxychloroquine and mycophenolate mofetil.15,19
Co-occurrence of LPP and FFA primarily is seen in postmenopausal women with darker skin,14,15 as in our patient, though premenopausal cases have been reported. Lichen planus pigmentosus may serve as a harbinger in most patients.1,2 In a similar fashion, our patient presented with hyperpigmented macular lesions prior to the onset of frontotemporal hair loss.
Our patient was started on finasteride 2.5 mg daily, minoxidil foam 5%, clobetasol solution 0.05%, triamcinolone ointment 0.1%, and hydrocortisone ointment 2.5%. She was instructed to commence treatment and follow up in 6 months.
The differential diagnosis includes dermatologic conditions that mimic both LPP and FFA. Postinflammatory hyperpigmentation and fixed drug reaction were unlikely based on the patient's history. The lesions of ashy dermatosis are characteristically gray erythematous macules on the trunk and limbs. Riehl melanosis is a rare pigmented contact dermatitis that is associated with a history of repeated contact with sensitizing allergens. Although Hori nevus is characterized by small, blue-gray or brown macules on the face, lesions predominantly occur on the bony prominences of the cheeks. Melasma also presents with dark to gray macules that affect the face and less commonly the neck, as in our patient.2
Early discoid lupus erythematosus presents with round erythematous plaques with overlying scale extending into the hair follicles. In pseudopalade of Brocq, an idiopathic cicatricial alopecia, lesions typically are flesh colored. Biopsy also shows epidermal atrophy with additional dermal sclerosis and fibrosis. Folliculitis decalvans is a scarring form of alopecia associated with erythema and pustules, findings that were not present in our patient. Keratosis follicularis spinulosa decalvans is a rare, X-linked inherited ichthyosis manifesting as scarring alopecia with follicular depressions and papules on the scalp in younger males. Photophobia and other manifestations may be present. Alopecia mucinosa is a nonscarring alopecia with grouped follicular erythematous patches or plaques. Mucin sometimes can be squeezed from affected areas, and histopathologic examination shows mucin accumulation.4
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-442.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Rieder E, Kaplan J, Kamino H, et al. Lichen planus pigmentosus. Dermatol Online J. 2013;19:20713.
- Kashima A, Tajiri A, Yamashita A, et al. Two Japanese cases of lichen planus pigmentosus-inversus. Int J Dermatol. 2007;46:740-742.
- Bhutani L, Bedi T, Pandhi R. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Kanwa AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514.
- Torres J, Guadalupe A, Reyes E, et al. Lichen planus pigmentosus in patients with endocrinopathies and hepatitis C. J Am Acad Dermatol. 2013;68:AB139.
- Kim JE, Won CH, Chang S, et al. Linear lichen planus pigmentosus of the forehead treated by neodymium:yttrium-aluminum-garnet laser and topical tacrolimus. J Dermatol. 2012;39:189-191.
- Kossard S. Postmenopausal frontal fibrosing alopecia: scarring alopecia in a pattern distribution. Arch Dermatol. 1994;130:770-774.
- Vano-Galvan S, Molina-Ruiz AM, Serrano-Falcon C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol. 2012;67:955-961.
- Harries MJ, Meyer K, Chaudhry I, et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle's epithelial stem cell niche. J Pathol. 2013;231:236-247.
- Karnik P, Tekeste Z, McCormick TS, et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243-1257.
- Rodriguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Rácz E, Gho C, Moorman PW, et al. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol. 2013;27:1461-1470.
The Diagnosis: Frontal Fibrosing Alopecia Overlapping With Lichen Planus Pigmentosus
Microscopic examination revealed focal dermal pigmentation, papillary fibrosis, and epidermal atrophy. These clinical and histologic findings indicated a diagnosis of fully developed lichen planus pigmentosus (LPP) overlapping with frontal fibrosing alopecia (FFA). Other cases have demonstrated an association between LPP and FFA.1,2
Lichen planus pigmentosus is considered an uncommon variant of lichen planus, as it has similar histopathologic findings and occasional coexistence.3,4 It is characterized by hyperpigmented macules primarily located in sun-exposed and flexural areas of the skin. First described in India,5 this disease has a predilection for darker skin (Fitzpatrick skin types III-V),6,7 and it has been reported in other racial and ethnic groups including Latin Americans, Middle Eastern populations, Japanese, and Koreans.4,8 Typically, lesions initially appear as ill-defined, blue-grey, round to oval macules that coalesce into hyperpigmented patches. Involvement most commonly begins at the forehead and temples, which are affected in nearly all patients. Infrequently, LPP can be generalized or affect the oral mucosa; involvement of the palms, soles, and nails does not occur. Patients may be asymptomatic, but some experience mild pruritus and burning. The disease course is chronic and insidious, with new lesions appearing over time and old lesions progressively darkening and expanding.6,7,9
Although the pathogenesis of LPP is unknown, several exposures have been implicated, such as amla oil, mustard oil, henna, hair dye, and environmental pollutants.7 Because lesions characteristically occur in sun-exposed areas, UV light also may be involved. In addition, studies have suggested that LPP is associated with endocrinopathies such as diabetes mellitus and dyslipidemias, as in our patient, as well as autoimmune conditions such as vitiligo and systemic lupus erythematosus.10,11
Histopathologic findings are characterized by vacuolar degeneration of the basal layer in the epidermis as well as perivascular lymphohistiocytic infiltration and the presence of melanophages in the dermis.3,9 Lichen planus pigmentosus is difficult to treat, as no consistently effective modality has been established. Topical tacrolimus, topical corticosteroids, oral retinoids, lasers, and sun protection have been implemented with underwhelming results.12
Frontal fibrosing alopecia is a variant of lichen planopilaris that predominantly affects postmenopausal women and presents with frontotemporal hair loss in a bandlike distribution.5,13 Both terminal and vellus hairs are affected. Involvement of multiple hair-bearing sites of the skin have been reported, including the entire scalp, eyebrows, and eyelashes. Affected areas may display hypopigmentation and be accompanied by pruritus and trichodynia.14,15 The pathogenesis currently is under investigation, with studies demonstrating autoimmune, genetic, and possibly even endocrine predispositions.16-18 Biopsies of lesions are indistinguishable from lichen planopilaris, which shows follicular lymphocytic infiltration, perifollicular fibrosis, interface dermatitis of the follicular infundibulum and isthmus, and vertical fibrous tracks.5 Patients with FFA have demonstrated variable responses to treatments, with one study showing improvement with oral finasteride or dutasteride.14 Topical and intralesional corticosteroids have yielded suboptimal effects. Other modalities include hydroxychloroquine and mycophenolate mofetil.15,19
Co-occurrence of LPP and FFA primarily is seen in postmenopausal women with darker skin,14,15 as in our patient, though premenopausal cases have been reported. Lichen planus pigmentosus may serve as a harbinger in most patients.1,2 In a similar fashion, our patient presented with hyperpigmented macular lesions prior to the onset of frontotemporal hair loss.
Our patient was started on finasteride 2.5 mg daily, minoxidil foam 5%, clobetasol solution 0.05%, triamcinolone ointment 0.1%, and hydrocortisone ointment 2.5%. She was instructed to commence treatment and follow up in 6 months.
The differential diagnosis includes dermatologic conditions that mimic both LPP and FFA. Postinflammatory hyperpigmentation and fixed drug reaction were unlikely based on the patient's history. The lesions of ashy dermatosis are characteristically gray erythematous macules on the trunk and limbs. Riehl melanosis is a rare pigmented contact dermatitis that is associated with a history of repeated contact with sensitizing allergens. Although Hori nevus is characterized by small, blue-gray or brown macules on the face, lesions predominantly occur on the bony prominences of the cheeks. Melasma also presents with dark to gray macules that affect the face and less commonly the neck, as in our patient.2
Early discoid lupus erythematosus presents with round erythematous plaques with overlying scale extending into the hair follicles. In pseudopalade of Brocq, an idiopathic cicatricial alopecia, lesions typically are flesh colored. Biopsy also shows epidermal atrophy with additional dermal sclerosis and fibrosis. Folliculitis decalvans is a scarring form of alopecia associated with erythema and pustules, findings that were not present in our patient. Keratosis follicularis spinulosa decalvans is a rare, X-linked inherited ichthyosis manifesting as scarring alopecia with follicular depressions and papules on the scalp in younger males. Photophobia and other manifestations may be present. Alopecia mucinosa is a nonscarring alopecia with grouped follicular erythematous patches or plaques. Mucin sometimes can be squeezed from affected areas, and histopathologic examination shows mucin accumulation.4
The Diagnosis: Frontal Fibrosing Alopecia Overlapping With Lichen Planus Pigmentosus
Microscopic examination revealed focal dermal pigmentation, papillary fibrosis, and epidermal atrophy. These clinical and histologic findings indicated a diagnosis of fully developed lichen planus pigmentosus (LPP) overlapping with frontal fibrosing alopecia (FFA). Other cases have demonstrated an association between LPP and FFA.1,2
Lichen planus pigmentosus is considered an uncommon variant of lichen planus, as it has similar histopathologic findings and occasional coexistence.3,4 It is characterized by hyperpigmented macules primarily located in sun-exposed and flexural areas of the skin. First described in India,5 this disease has a predilection for darker skin (Fitzpatrick skin types III-V),6,7 and it has been reported in other racial and ethnic groups including Latin Americans, Middle Eastern populations, Japanese, and Koreans.4,8 Typically, lesions initially appear as ill-defined, blue-grey, round to oval macules that coalesce into hyperpigmented patches. Involvement most commonly begins at the forehead and temples, which are affected in nearly all patients. Infrequently, LPP can be generalized or affect the oral mucosa; involvement of the palms, soles, and nails does not occur. Patients may be asymptomatic, but some experience mild pruritus and burning. The disease course is chronic and insidious, with new lesions appearing over time and old lesions progressively darkening and expanding.6,7,9
Although the pathogenesis of LPP is unknown, several exposures have been implicated, such as amla oil, mustard oil, henna, hair dye, and environmental pollutants.7 Because lesions characteristically occur in sun-exposed areas, UV light also may be involved. In addition, studies have suggested that LPP is associated with endocrinopathies such as diabetes mellitus and dyslipidemias, as in our patient, as well as autoimmune conditions such as vitiligo and systemic lupus erythematosus.10,11
Histopathologic findings are characterized by vacuolar degeneration of the basal layer in the epidermis as well as perivascular lymphohistiocytic infiltration and the presence of melanophages in the dermis.3,9 Lichen planus pigmentosus is difficult to treat, as no consistently effective modality has been established. Topical tacrolimus, topical corticosteroids, oral retinoids, lasers, and sun protection have been implemented with underwhelming results.12
Frontal fibrosing alopecia is a variant of lichen planopilaris that predominantly affects postmenopausal women and presents with frontotemporal hair loss in a bandlike distribution.5,13 Both terminal and vellus hairs are affected. Involvement of multiple hair-bearing sites of the skin have been reported, including the entire scalp, eyebrows, and eyelashes. Affected areas may display hypopigmentation and be accompanied by pruritus and trichodynia.14,15 The pathogenesis currently is under investigation, with studies demonstrating autoimmune, genetic, and possibly even endocrine predispositions.16-18 Biopsies of lesions are indistinguishable from lichen planopilaris, which shows follicular lymphocytic infiltration, perifollicular fibrosis, interface dermatitis of the follicular infundibulum and isthmus, and vertical fibrous tracks.5 Patients with FFA have demonstrated variable responses to treatments, with one study showing improvement with oral finasteride or dutasteride.14 Topical and intralesional corticosteroids have yielded suboptimal effects. Other modalities include hydroxychloroquine and mycophenolate mofetil.15,19
Co-occurrence of LPP and FFA primarily is seen in postmenopausal women with darker skin,14,15 as in our patient, though premenopausal cases have been reported. Lichen planus pigmentosus may serve as a harbinger in most patients.1,2 In a similar fashion, our patient presented with hyperpigmented macular lesions prior to the onset of frontotemporal hair loss.
Our patient was started on finasteride 2.5 mg daily, minoxidil foam 5%, clobetasol solution 0.05%, triamcinolone ointment 0.1%, and hydrocortisone ointment 2.5%. She was instructed to commence treatment and follow up in 6 months.
The differential diagnosis includes dermatologic conditions that mimic both LPP and FFA. Postinflammatory hyperpigmentation and fixed drug reaction were unlikely based on the patient's history. The lesions of ashy dermatosis are characteristically gray erythematous macules on the trunk and limbs. Riehl melanosis is a rare pigmented contact dermatitis that is associated with a history of repeated contact with sensitizing allergens. Although Hori nevus is characterized by small, blue-gray or brown macules on the face, lesions predominantly occur on the bony prominences of the cheeks. Melasma also presents with dark to gray macules that affect the face and less commonly the neck, as in our patient.2
Early discoid lupus erythematosus presents with round erythematous plaques with overlying scale extending into the hair follicles. In pseudopalade of Brocq, an idiopathic cicatricial alopecia, lesions typically are flesh colored. Biopsy also shows epidermal atrophy with additional dermal sclerosis and fibrosis. Folliculitis decalvans is a scarring form of alopecia associated with erythema and pustules, findings that were not present in our patient. Keratosis follicularis spinulosa decalvans is a rare, X-linked inherited ichthyosis manifesting as scarring alopecia with follicular depressions and papules on the scalp in younger males. Photophobia and other manifestations may be present. Alopecia mucinosa is a nonscarring alopecia with grouped follicular erythematous patches or plaques. Mucin sometimes can be squeezed from affected areas, and histopathologic examination shows mucin accumulation.4
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-442.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Rieder E, Kaplan J, Kamino H, et al. Lichen planus pigmentosus. Dermatol Online J. 2013;19:20713.
- Kashima A, Tajiri A, Yamashita A, et al. Two Japanese cases of lichen planus pigmentosus-inversus. Int J Dermatol. 2007;46:740-742.
- Bhutani L, Bedi T, Pandhi R. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Kanwa AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514.
- Torres J, Guadalupe A, Reyes E, et al. Lichen planus pigmentosus in patients with endocrinopathies and hepatitis C. J Am Acad Dermatol. 2013;68:AB139.
- Kim JE, Won CH, Chang S, et al. Linear lichen planus pigmentosus of the forehead treated by neodymium:yttrium-aluminum-garnet laser and topical tacrolimus. J Dermatol. 2012;39:189-191.
- Kossard S. Postmenopausal frontal fibrosing alopecia: scarring alopecia in a pattern distribution. Arch Dermatol. 1994;130:770-774.
- Vano-Galvan S, Molina-Ruiz AM, Serrano-Falcon C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol. 2012;67:955-961.
- Harries MJ, Meyer K, Chaudhry I, et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle's epithelial stem cell niche. J Pathol. 2013;231:236-247.
- Karnik P, Tekeste Z, McCormick TS, et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243-1257.
- Rodriguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Rácz E, Gho C, Moorman PW, et al. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol. 2013;27:1461-1470.
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-442.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Rieder E, Kaplan J, Kamino H, et al. Lichen planus pigmentosus. Dermatol Online J. 2013;19:20713.
- Kashima A, Tajiri A, Yamashita A, et al. Two Japanese cases of lichen planus pigmentosus-inversus. Int J Dermatol. 2007;46:740-742.
- Bhutani L, Bedi T, Pandhi R. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Kanwa AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514.
- Torres J, Guadalupe A, Reyes E, et al. Lichen planus pigmentosus in patients with endocrinopathies and hepatitis C. J Am Acad Dermatol. 2013;68:AB139.
- Kim JE, Won CH, Chang S, et al. Linear lichen planus pigmentosus of the forehead treated by neodymium:yttrium-aluminum-garnet laser and topical tacrolimus. J Dermatol. 2012;39:189-191.
- Kossard S. Postmenopausal frontal fibrosing alopecia: scarring alopecia in a pattern distribution. Arch Dermatol. 1994;130:770-774.
- Vano-Galvan S, Molina-Ruiz AM, Serrano-Falcon C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol. 2012;67:955-961.
- Harries MJ, Meyer K, Chaudhry I, et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle's epithelial stem cell niche. J Pathol. 2013;231:236-247.
- Karnik P, Tekeste Z, McCormick TS, et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243-1257.
- Rodriguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Rácz E, Gho C, Moorman PW, et al. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol. 2013;27:1461-1470.
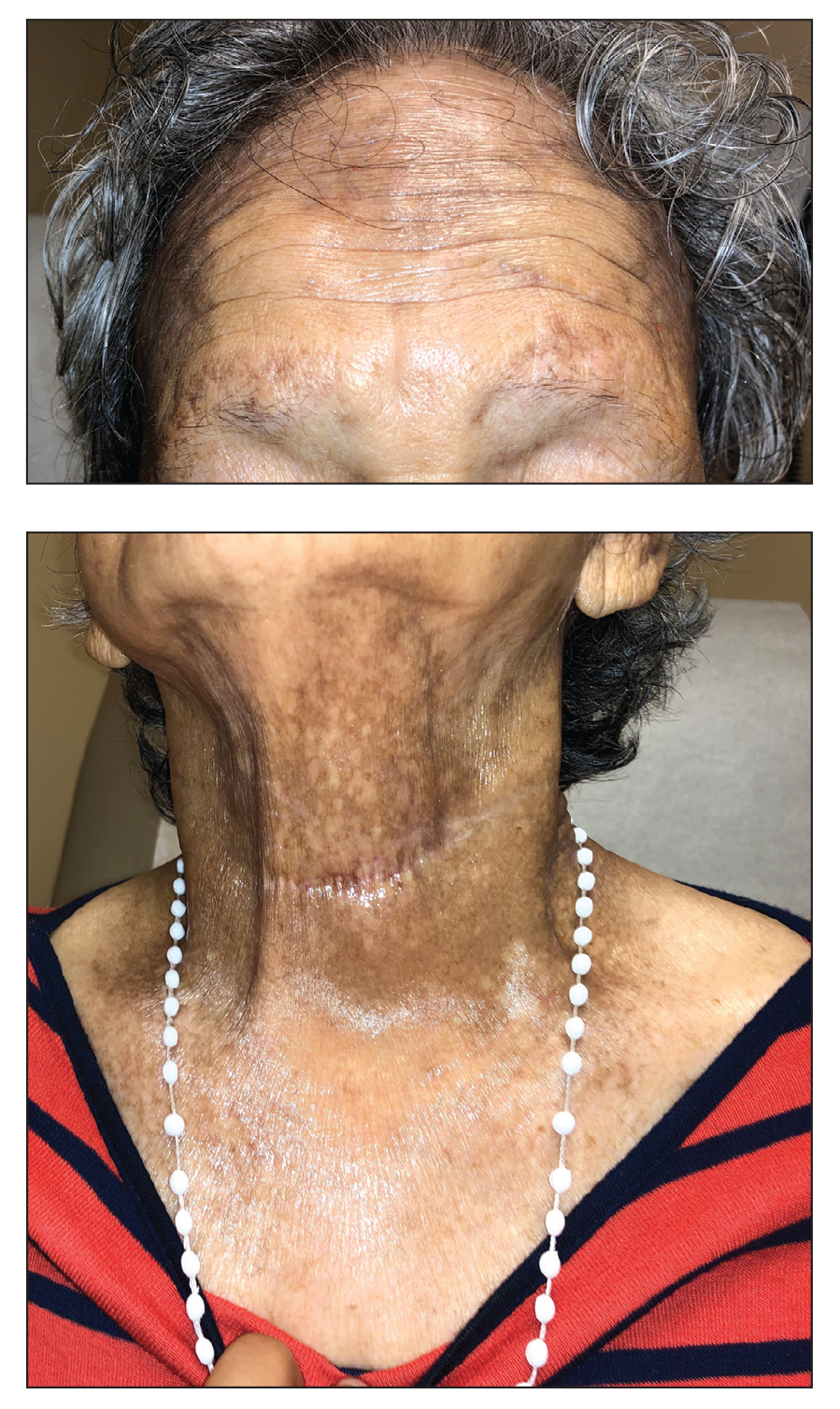
A 78-year-old Asian woman presented to the dermatology clinic with progressively worsening dark spots on the forehead and neck of 3 months’ duration. She noted mild pruritis and hair loss involving the eyebrows and anterior scalp. Her medical history was notable for type 2 diabetes mellitus. She denied any new medical conditions or medications and had no prior history of similar symptoms. Physical examination showed hyperpigmented brown macules and patches on the forehead (top) and anterior neck (bottom) with sparing of the posterior neck and lower face. Alopecia with areas of perifollicular erythema and hyperpigmentation with reduced follicular openings were present on the eyebrows and anterior forehead. Two punch biopsies of head and neck lesions were performed.