User login
Periungual Papules in an Elderly Woman
The Diagnosis: Multicentric Reticulohistiocytosis
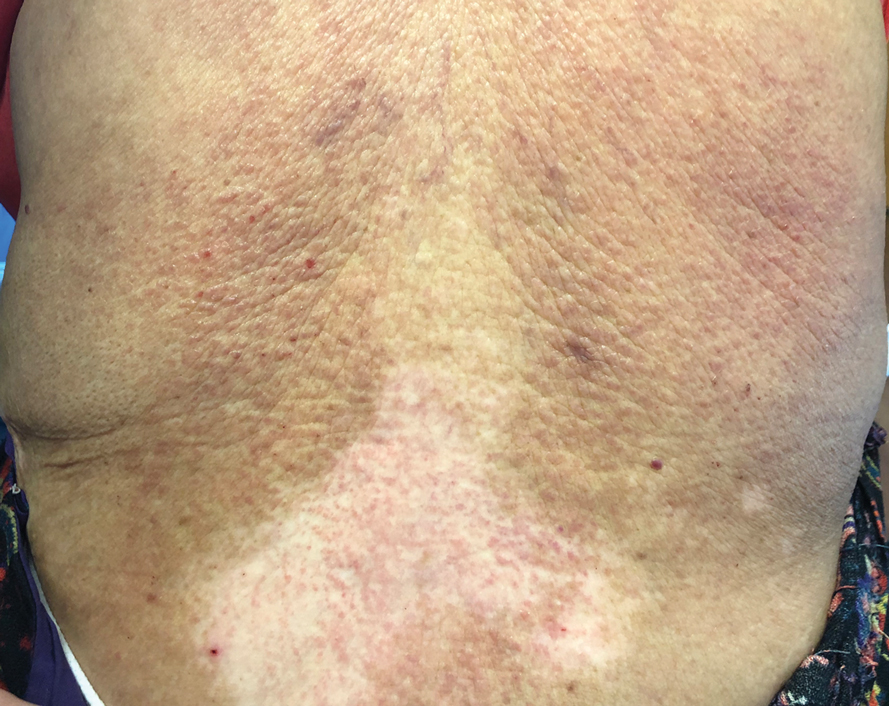
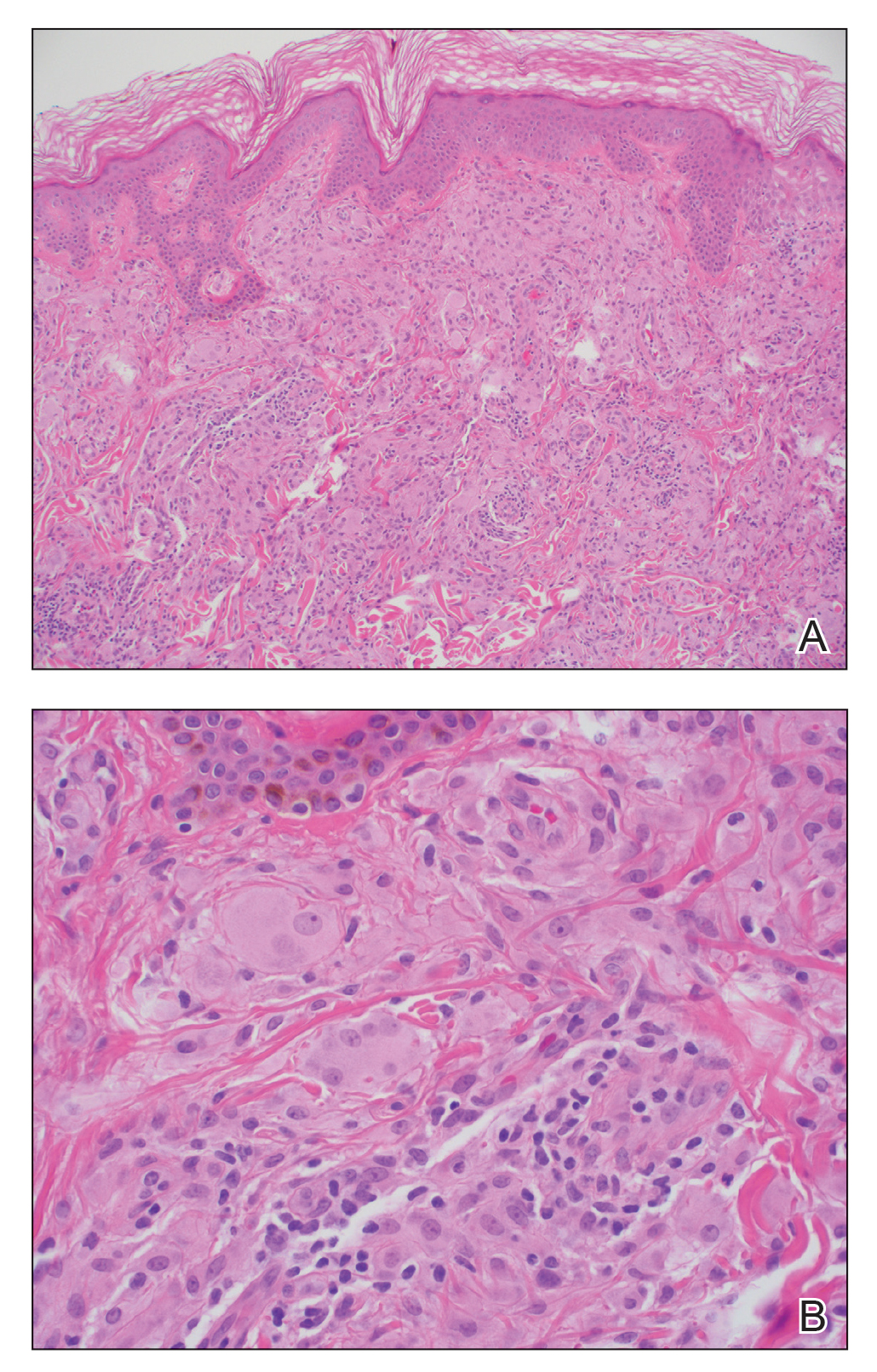
Te patient presented with pink papules coalescing into plaques on the upper chest and lower back (Figure 1) as well as a characteristic finding of periungual papules with a coral bead appearance. Histopathologic examination revealed a dense infiltrate of epithelioid histiocytes with amphophilic ground-glass cytoplasm in a nodular configuration (Figure 2). This pattern in conjunction with the clinical features seen in our patient was consistent with a diagnosis of multicentric reticulohistiocytosis (MRH).1-3 The cutaneous symptoms were managed with triamcinolone ointment 0.1% twice daily and oral hydroxyzine 10 mg 3 times daily as needed for itching with moderate improvement. She was referred to rheumatology for arthritis management, and the initial cancer screening was negative.
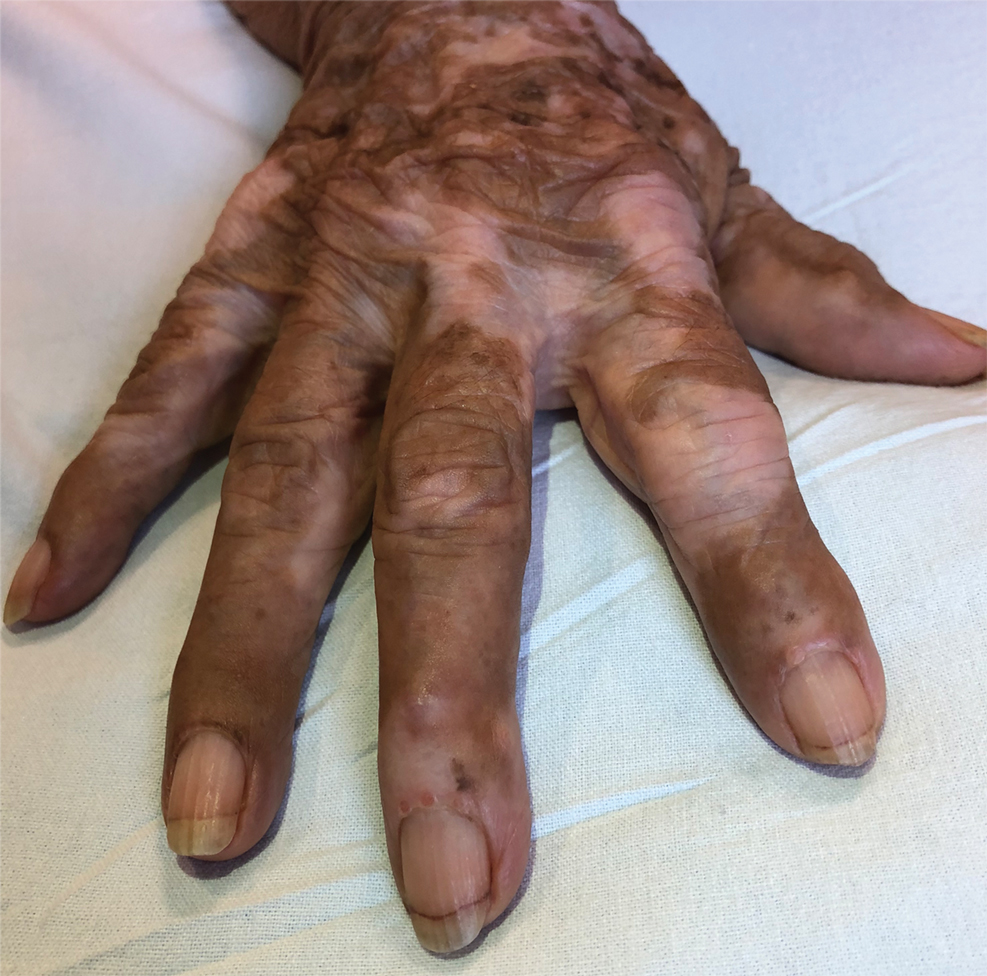
Multicentric reticulohistiocytosis is a rare granulomatous disease characterized by papulonodular cutaneous lesions and severe erosive arthritis. It has an insidious onset and most commonly affects middle-aged women.1 Multicentric reticulohistiocytosis typically presents as rounded pruritic papules or nodules that may be pink, red, or brown primarily affecting the face and distal upper extremities.1,3 Mucosal involvement occurs in more than half of patients and is characterized by multiple erythematous papules and nodules on the oral and nasopharyngeal mucosae that rarely can produce leonine facies.2 A hallmark feature of MRH is the presence of multiple shiny erythematous papules along the proximal and lateral nail folds that take on a coral bead appearance.1,3,4 Furthermore, nail changes such as atrophy, longitudinal ridging, brittleness, and hyperpigmentation can occur secondary to a synovial reaction that disturbs the nail matrix.4,5
Joint involvement precedes cutaneous involvement in most cases of MRH.1,5 Multicentric reticulohistiocytosis is associated with a symmetric destructive arthritis affecting the hands, knees, shoulders, and hips that often is associated with pain, stiffness, and swelling.1,3 The arthritis rapidly progresses in the early stages of the disease but then becomes less active over the subsequent 8 to 10 years.1 It has the potential to develop into arthritis mutilans, an end-stage form of arthritis also seen in psoriatic and rheumatoid arthritis that leads to severe joint deformity and debilitation.1,2
The etiology of MRH still is unknown, but it has an association with underlying malignancy in up to 25% of patients.6 Multicentric reticulohistiocytosis has been reported in the context of a wide variety of malignancies including melanoma; sarcoma; lymphoma; leukemia; and carcinomas of the breast, colon, and lung. In some cases, the diagnosis of MRH may even precede the diagnosis of cancer.3 Multicentric reticulohistiocytosis also may be associated with autoimmune conditions,3 as seen in our patient who had a history of both hypothyroidism and vitiligo.
Histopathologic examination is essential in distinguishing MRH from other autoimmune disorders associated with hand lesions, rash, and arthralgia. Erythema elevatum diutinum is associated with symmetric, violaceous, red or brown papules and plaques located on the extensor surfaces of the extremities and hands; however, histology reveals a leukocytoclastic vasculitis with a mixture of polymorphonuclear leukocytes and lymphocytes.7 Dermatomyositis may present with arthralgia, flattopped, erythematous (Gottron) papules localized over the proximal interphalangeal and distal interphalangeal joints, as well as proximal nail findings. The latter generally presents with periungual erythema associated with dilated capillary loops rather than the discrete orderly papules seen in MRH. Histologic examination of dermatomyositis shows mild epidermal atrophy, vacuolar changes in the basal keratinocyte layer, and a dermal perivascular lymphocytic infiltrate.8 Because MRH initially can present with joint symptoms and hand nodules, it may be confused with rheumatoid arthritis. However, rheumatoid arthritis typically is associated with severe osteopenia and tends to affect the metacarpophalangeal and proximal interphalangeal joints rather than the distal interphalangeal joints that most often are affected in MRH.1 Histologic examination of rheumatoid nodules reveals palisading granulomas surrounding a central area of fibrinoid necrosis.9 Sarcoidosis is a multisystem disease that can present with cutaneous involvement including erythema nodosum, skin plaques, subcutaneous nodules, and papular eruptions in addition to joint lesions.10 Sarcoidosis most frequently involves the lungs, manifesting as diffuse interstitial lung disease with bilateral hilar lymphadenopathy. Furthermore, histologic examination of lesions demonstrates classic noncaseating granulomas containing epithelioid cells, multinucleated giant cells with inclusion bodies, and lymphocytes.11
A skin biopsy is required to establish the diagnosis of MRH. In general, patients with MRH and no underlying malignancy have a good prognosis and respond to anti-inflammatory therapies such as nonsteroidal antiinflammatory drugs and corticosteroids. Other agents including methotrexate, cyclophosphamide, and tumor necrosis factor α inhibitors also have been effective in more severe cases.1,3,12 Finally, in addition to treating the cutaneous manifestations of MRH, it is important to screen patients for underlying malignancies and other autoimmune conditions.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492.
- Gold RH, Metzger AL, Mirra JM, et al. Multicentric reticulohistiocytosis (lipoid dermato-arthritis). an erosive polyarthritis with distinctive clinical, roentgenographic and pathologic features. Am J Roentgenol Radium Ther Nucl Med. 1975;124:610-624.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, et al. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol. 2001;15:524-531.
- Barrow MV. The nails in multicentric reticulohistiocytosis. (lipoid dermato-arthritis). Arch Dermatol. 1967;95:200-201.
- Barrow MV, Holubar K. Multicentric reticulohistiocytosis. a review of 33 patients. Medicine (Baltimore). 1969;48:287-305.
- Snow JL, Muller SA. Malignancy-associated multicentric reticulohistiocytosis: a clinical, histological and immunophenotypic study. Br J Dermatol. 1995;133:71-76.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Smith ES, Hallman JR, DeLuca AM, et al. Dermatomyositis: a clinicopathological study of 40 patients. Am J Dermatopathol. 2009; 31:61-67.
- Athanasou NA, Quinn J, Woods CG, et al. Immunohistology of rheumatoid nodules and rheumatoid synovium. Ann Rheum Dis. 1988;47:398-403.
- Yanardag H, Pamuk ON, Karayel T. Cutaneous involvement in sarcoidosis: analysis of the features in 170 patients. Respir Med. 2003;97:978-982.
- Ma Y, Gal A, Koss MN. The pathology of pulmonary sarcoidosis: update. Semin Diagn Pathol. 2007;24:150-161.
- Kovach BT, Calamia KT, Walsh JS, et al. Treatment of multicentric reticulohistiocytosis with etanercept. Arch Dermatol. 2004;140:919-921.
The Diagnosis: Multicentric Reticulohistiocytosis
Te patient presented with pink papules coalescing into plaques on the upper chest and lower back (Figure 1) as well as a characteristic finding of periungual papules with a coral bead appearance. Histopathologic examination revealed a dense infiltrate of epithelioid histiocytes with amphophilic ground-glass cytoplasm in a nodular configuration (Figure 2). This pattern in conjunction with the clinical features seen in our patient was consistent with a diagnosis of multicentric reticulohistiocytosis (MRH).1-3 The cutaneous symptoms were managed with triamcinolone ointment 0.1% twice daily and oral hydroxyzine 10 mg 3 times daily as needed for itching with moderate improvement. She was referred to rheumatology for arthritis management, and the initial cancer screening was negative.
Multicentric reticulohistiocytosis is a rare granulomatous disease characterized by papulonodular cutaneous lesions and severe erosive arthritis. It has an insidious onset and most commonly affects middle-aged women.1 Multicentric reticulohistiocytosis typically presents as rounded pruritic papules or nodules that may be pink, red, or brown primarily affecting the face and distal upper extremities.1,3 Mucosal involvement occurs in more than half of patients and is characterized by multiple erythematous papules and nodules on the oral and nasopharyngeal mucosae that rarely can produce leonine facies.2 A hallmark feature of MRH is the presence of multiple shiny erythematous papules along the proximal and lateral nail folds that take on a coral bead appearance.1,3,4 Furthermore, nail changes such as atrophy, longitudinal ridging, brittleness, and hyperpigmentation can occur secondary to a synovial reaction that disturbs the nail matrix.4,5
Joint involvement precedes cutaneous involvement in most cases of MRH.1,5 Multicentric reticulohistiocytosis is associated with a symmetric destructive arthritis affecting the hands, knees, shoulders, and hips that often is associated with pain, stiffness, and swelling.1,3 The arthritis rapidly progresses in the early stages of the disease but then becomes less active over the subsequent 8 to 10 years.1 It has the potential to develop into arthritis mutilans, an end-stage form of arthritis also seen in psoriatic and rheumatoid arthritis that leads to severe joint deformity and debilitation.1,2
The etiology of MRH still is unknown, but it has an association with underlying malignancy in up to 25% of patients.6 Multicentric reticulohistiocytosis has been reported in the context of a wide variety of malignancies including melanoma; sarcoma; lymphoma; leukemia; and carcinomas of the breast, colon, and lung. In some cases, the diagnosis of MRH may even precede the diagnosis of cancer.3 Multicentric reticulohistiocytosis also may be associated with autoimmune conditions,3 as seen in our patient who had a history of both hypothyroidism and vitiligo.
Histopathologic examination is essential in distinguishing MRH from other autoimmune disorders associated with hand lesions, rash, and arthralgia. Erythema elevatum diutinum is associated with symmetric, violaceous, red or brown papules and plaques located on the extensor surfaces of the extremities and hands; however, histology reveals a leukocytoclastic vasculitis with a mixture of polymorphonuclear leukocytes and lymphocytes.7 Dermatomyositis may present with arthralgia, flattopped, erythematous (Gottron) papules localized over the proximal interphalangeal and distal interphalangeal joints, as well as proximal nail findings. The latter generally presents with periungual erythema associated with dilated capillary loops rather than the discrete orderly papules seen in MRH. Histologic examination of dermatomyositis shows mild epidermal atrophy, vacuolar changes in the basal keratinocyte layer, and a dermal perivascular lymphocytic infiltrate.8 Because MRH initially can present with joint symptoms and hand nodules, it may be confused with rheumatoid arthritis. However, rheumatoid arthritis typically is associated with severe osteopenia and tends to affect the metacarpophalangeal and proximal interphalangeal joints rather than the distal interphalangeal joints that most often are affected in MRH.1 Histologic examination of rheumatoid nodules reveals palisading granulomas surrounding a central area of fibrinoid necrosis.9 Sarcoidosis is a multisystem disease that can present with cutaneous involvement including erythema nodosum, skin plaques, subcutaneous nodules, and papular eruptions in addition to joint lesions.10 Sarcoidosis most frequently involves the lungs, manifesting as diffuse interstitial lung disease with bilateral hilar lymphadenopathy. Furthermore, histologic examination of lesions demonstrates classic noncaseating granulomas containing epithelioid cells, multinucleated giant cells with inclusion bodies, and lymphocytes.11
A skin biopsy is required to establish the diagnosis of MRH. In general, patients with MRH and no underlying malignancy have a good prognosis and respond to anti-inflammatory therapies such as nonsteroidal antiinflammatory drugs and corticosteroids. Other agents including methotrexate, cyclophosphamide, and tumor necrosis factor α inhibitors also have been effective in more severe cases.1,3,12 Finally, in addition to treating the cutaneous manifestations of MRH, it is important to screen patients for underlying malignancies and other autoimmune conditions.
The Diagnosis: Multicentric Reticulohistiocytosis
Te patient presented with pink papules coalescing into plaques on the upper chest and lower back (Figure 1) as well as a characteristic finding of periungual papules with a coral bead appearance. Histopathologic examination revealed a dense infiltrate of epithelioid histiocytes with amphophilic ground-glass cytoplasm in a nodular configuration (Figure 2). This pattern in conjunction with the clinical features seen in our patient was consistent with a diagnosis of multicentric reticulohistiocytosis (MRH).1-3 The cutaneous symptoms were managed with triamcinolone ointment 0.1% twice daily and oral hydroxyzine 10 mg 3 times daily as needed for itching with moderate improvement. She was referred to rheumatology for arthritis management, and the initial cancer screening was negative.
Multicentric reticulohistiocytosis is a rare granulomatous disease characterized by papulonodular cutaneous lesions and severe erosive arthritis. It has an insidious onset and most commonly affects middle-aged women.1 Multicentric reticulohistiocytosis typically presents as rounded pruritic papules or nodules that may be pink, red, or brown primarily affecting the face and distal upper extremities.1,3 Mucosal involvement occurs in more than half of patients and is characterized by multiple erythematous papules and nodules on the oral and nasopharyngeal mucosae that rarely can produce leonine facies.2 A hallmark feature of MRH is the presence of multiple shiny erythematous papules along the proximal and lateral nail folds that take on a coral bead appearance.1,3,4 Furthermore, nail changes such as atrophy, longitudinal ridging, brittleness, and hyperpigmentation can occur secondary to a synovial reaction that disturbs the nail matrix.4,5
Joint involvement precedes cutaneous involvement in most cases of MRH.1,5 Multicentric reticulohistiocytosis is associated with a symmetric destructive arthritis affecting the hands, knees, shoulders, and hips that often is associated with pain, stiffness, and swelling.1,3 The arthritis rapidly progresses in the early stages of the disease but then becomes less active over the subsequent 8 to 10 years.1 It has the potential to develop into arthritis mutilans, an end-stage form of arthritis also seen in psoriatic and rheumatoid arthritis that leads to severe joint deformity and debilitation.1,2
The etiology of MRH still is unknown, but it has an association with underlying malignancy in up to 25% of patients.6 Multicentric reticulohistiocytosis has been reported in the context of a wide variety of malignancies including melanoma; sarcoma; lymphoma; leukemia; and carcinomas of the breast, colon, and lung. In some cases, the diagnosis of MRH may even precede the diagnosis of cancer.3 Multicentric reticulohistiocytosis also may be associated with autoimmune conditions,3 as seen in our patient who had a history of both hypothyroidism and vitiligo.
Histopathologic examination is essential in distinguishing MRH from other autoimmune disorders associated with hand lesions, rash, and arthralgia. Erythema elevatum diutinum is associated with symmetric, violaceous, red or brown papules and plaques located on the extensor surfaces of the extremities and hands; however, histology reveals a leukocytoclastic vasculitis with a mixture of polymorphonuclear leukocytes and lymphocytes.7 Dermatomyositis may present with arthralgia, flattopped, erythematous (Gottron) papules localized over the proximal interphalangeal and distal interphalangeal joints, as well as proximal nail findings. The latter generally presents with periungual erythema associated with dilated capillary loops rather than the discrete orderly papules seen in MRH. Histologic examination of dermatomyositis shows mild epidermal atrophy, vacuolar changes in the basal keratinocyte layer, and a dermal perivascular lymphocytic infiltrate.8 Because MRH initially can present with joint symptoms and hand nodules, it may be confused with rheumatoid arthritis. However, rheumatoid arthritis typically is associated with severe osteopenia and tends to affect the metacarpophalangeal and proximal interphalangeal joints rather than the distal interphalangeal joints that most often are affected in MRH.1 Histologic examination of rheumatoid nodules reveals palisading granulomas surrounding a central area of fibrinoid necrosis.9 Sarcoidosis is a multisystem disease that can present with cutaneous involvement including erythema nodosum, skin plaques, subcutaneous nodules, and papular eruptions in addition to joint lesions.10 Sarcoidosis most frequently involves the lungs, manifesting as diffuse interstitial lung disease with bilateral hilar lymphadenopathy. Furthermore, histologic examination of lesions demonstrates classic noncaseating granulomas containing epithelioid cells, multinucleated giant cells with inclusion bodies, and lymphocytes.11
A skin biopsy is required to establish the diagnosis of MRH. In general, patients with MRH and no underlying malignancy have a good prognosis and respond to anti-inflammatory therapies such as nonsteroidal antiinflammatory drugs and corticosteroids. Other agents including methotrexate, cyclophosphamide, and tumor necrosis factor α inhibitors also have been effective in more severe cases.1,3,12 Finally, in addition to treating the cutaneous manifestations of MRH, it is important to screen patients for underlying malignancies and other autoimmune conditions.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492.
- Gold RH, Metzger AL, Mirra JM, et al. Multicentric reticulohistiocytosis (lipoid dermato-arthritis). an erosive polyarthritis with distinctive clinical, roentgenographic and pathologic features. Am J Roentgenol Radium Ther Nucl Med. 1975;124:610-624.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, et al. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol. 2001;15:524-531.
- Barrow MV. The nails in multicentric reticulohistiocytosis. (lipoid dermato-arthritis). Arch Dermatol. 1967;95:200-201.
- Barrow MV, Holubar K. Multicentric reticulohistiocytosis. a review of 33 patients. Medicine (Baltimore). 1969;48:287-305.
- Snow JL, Muller SA. Malignancy-associated multicentric reticulohistiocytosis: a clinical, histological and immunophenotypic study. Br J Dermatol. 1995;133:71-76.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Smith ES, Hallman JR, DeLuca AM, et al. Dermatomyositis: a clinicopathological study of 40 patients. Am J Dermatopathol. 2009; 31:61-67.
- Athanasou NA, Quinn J, Woods CG, et al. Immunohistology of rheumatoid nodules and rheumatoid synovium. Ann Rheum Dis. 1988;47:398-403.
- Yanardag H, Pamuk ON, Karayel T. Cutaneous involvement in sarcoidosis: analysis of the features in 170 patients. Respir Med. 2003;97:978-982.
- Ma Y, Gal A, Koss MN. The pathology of pulmonary sarcoidosis: update. Semin Diagn Pathol. 2007;24:150-161.
- Kovach BT, Calamia KT, Walsh JS, et al. Treatment of multicentric reticulohistiocytosis with etanercept. Arch Dermatol. 2004;140:919-921.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492.
- Gold RH, Metzger AL, Mirra JM, et al. Multicentric reticulohistiocytosis (lipoid dermato-arthritis). an erosive polyarthritis with distinctive clinical, roentgenographic and pathologic features. Am J Roentgenol Radium Ther Nucl Med. 1975;124:610-624.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, et al. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol. 2001;15:524-531.
- Barrow MV. The nails in multicentric reticulohistiocytosis. (lipoid dermato-arthritis). Arch Dermatol. 1967;95:200-201.
- Barrow MV, Holubar K. Multicentric reticulohistiocytosis. a review of 33 patients. Medicine (Baltimore). 1969;48:287-305.
- Snow JL, Muller SA. Malignancy-associated multicentric reticulohistiocytosis: a clinical, histological and immunophenotypic study. Br J Dermatol. 1995;133:71-76.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Smith ES, Hallman JR, DeLuca AM, et al. Dermatomyositis: a clinicopathological study of 40 patients. Am J Dermatopathol. 2009; 31:61-67.
- Athanasou NA, Quinn J, Woods CG, et al. Immunohistology of rheumatoid nodules and rheumatoid synovium. Ann Rheum Dis. 1988;47:398-403.
- Yanardag H, Pamuk ON, Karayel T. Cutaneous involvement in sarcoidosis: analysis of the features in 170 patients. Respir Med. 2003;97:978-982.
- Ma Y, Gal A, Koss MN. The pathology of pulmonary sarcoidosis: update. Semin Diagn Pathol. 2007;24:150-161.
- Kovach BT, Calamia KT, Walsh JS, et al. Treatment of multicentric reticulohistiocytosis with etanercept. Arch Dermatol. 2004;140:919-921.
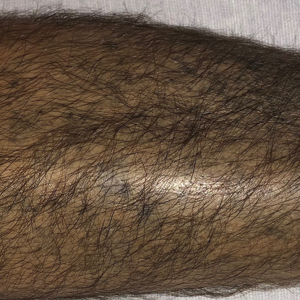
A 79-year-old woman presented with pruritic papules and plaques on the chest, back, arms, hands, legs, and feet of 1 year’s duration. She reported a history of hypothyroidism, arthritis, and vitiligo but denied a history of cancer. Physical examination showed pink papules coalescing into plaques on the upper chest and lower back as well as lichenified plaques on the forearms and knees. Erythematous papules on the proximal nail folds of the right first and second digits also were noted. Multiple depigmented patches on the hands, wrists, arms, and lower back also were present, and deformities of the hands and bulbous-appearing knees were observed. Results from a complete blood cell count and blood chemistry analyses showed mild anemia but were otherwise normal. Radiography of the right knee showed degenerative changes and periarticular radiolucencies consistent with an inflammatory arthropathy. A 4-mm punch biopsy specimen from the back was obtained for histopathologic examination.
Multiple Lesions With Recurrent Bleeding
The Diagnosis: Nevoid Basal Cell Carcinoma Syndrome
Nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin syndrome, is a rare autosomal-dominant disorder that increases the risk for developing various carcinomas and affects multiple organ systems. Nevoid basal cell carcinoma syndrome is estimated at 1 per 40,000 to 60,000 individuals with no sexual predilection.1,2 Pathogenesis of NBCCS occurs through molecular alterations in the dormant hedgehog signaling pathway, causing constitutive signaling activity and a loss of function in the tumor suppressor patched 1 gene, PTCH1. As a result, the inhibition of smoothened oncogenes is released, Gli proteins are activated, and the hedgehog signaling pathway is no longer quiescent.2 Additional loss of function in the suppressor of fused homolog protein, a negative regulator of the hedgehog pathway, allows for further tumor proliferation. The crucial role these genes play in the hedgehog signaling pathway and their mutation association with NBCCS allows for molecular confirmation in the diagnosis of NBCCS. Allelic losses at the PTCH1 gene site are thought to occur in approximately 70% of NBCCS patients.2
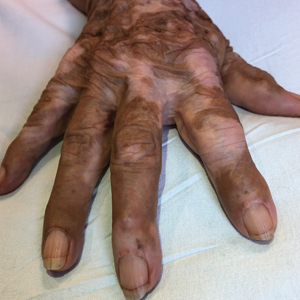
Diagnosis of NBCCS is based on genetic testing to examine pathogenic gene variants, notably in the PTCH1 gene, and identification of characteristic clinical findings.2 Diagnosis of NBCCS requires either 2 minor suggestive criteria and 1 major suggestive criterion, 2 major suggestive criteria and 1 minor suggestive criterion, or 1 major suggestive criterion with molecular confirmation. The presence of basal cell carcinomas (BCCs) before 20 years of age or an excessive numbers of BCCs, keratocystic odontogenic tumors (KOTs), palmar or plantar pitting, and first-degree relatives with NBCCS are classified as major suggestive criteria.2 Nevoid basal cell carcinoma syndrome patients typically have BCCs that crust, ulcerate, or bleed. Minor suggestive criteria for NBCCS are rib abnormalities, skeletal malformations, macrocephaly, cleft lip or palate, and desmoplastic medulloblastoma.2-4 Suppressor of fused homolog protein mutations may increase the risk for desmoplastic medulloblastoma in NBCCS patients. Our patient had 4 of the major suggestive criteria, including a history of KOTs, multiple BCCs, first-degree relatives with NBCCS, and palmar or plantar pitting (bottom quiz image), while having 1 minor suggestive criterion of frontal bossing.
Patients with NBCCS have high phenotypic variability, as their skin carcinomas do not have the classic features of pearly surfaces or corkscrew telangiectasia that typically are associated with BCCs.1 Basal cell carcinomas in NBCCS-affected individuals usually are indistinguishable from sporadic lesions that arise in sun-exposed areas, making NBCCS difficult to diagnose. These sporadic lesions often are misdiagnosed as psoriatic or eczematous lesions, and additional subsequent examination is required. The findings of multiple papules and plaques spanning the body as well as lesions with rolled borders and ulcerated bases, indicative of BCCs, aid dermatologists in distinguishing benign lesions from those of NBCCS.1
Additional differential diagnoses are required to distinguish NBCCS from other similar inherited skin disorders that are characterized by BCCs. The presence of multiple incidental BCCs early in life remains a histopathologic clue for NBCCS diagnosis, as opposed to Rombo syndrome, in which BCCs often develop in adulthood.2,4 In addition, although both Bazex syndrome and Muir-Torre syndrome are characterized by the early onset of BCCs, the lack of skeletal abnormalities and palmar and plantar pitting distinguish these entities from NBCCS.2,4 Furthermore, though psoriasis also can present on the scalp, clinical presentation often includes well-demarcated and symmetric plaques that are erythematous and silvery, all of which were not present in our patient and typically are not seen in NBCCS.5
The recommended treatment of NBCCS is vismodegib, a specific oncogene inhibitor. This medication suppresses the hedgehog signaling pathway by inhibiting smoothened oncogenes and downstream target molecules, thereby decreasing tumor proliferation.6 In doing so, vismodegib inhibits the development of new BCCs while reducing the burden of present ones. Additionally, vismodegib appears to effectively treat KOTs. If successful, this medication may be able to suppress KOTs in patients with NBCCS and thus facilitate surgery.6 Additional hedgehog inhibitors include patidegib, sonidegib, and itraconazole. Patidegib gel 2% currently is in phase 3 clinical trials for evaluation of efficacy and safety in treatment of NBCCS. Sonidegib is approved for the treatment of locally advanced BCCs in the United States and the European Union and for both locally advanced BCCs and metastatic BCCs in Switzerland and Australia.7 Further research is needed before recommending antifungal itraconazole for NBCCS clinical use.8 Other medications for localized areas include topical application of 5-fluorouracil and imiquimod.2
- Sangehra R, Grewal P. Gorlin syndrome presentation and the importance of differential diagnosis of skin cancer: a case report. J Pharm Pharm Sci. 2018;21:222-224.
- Bresler S, Padwa B, Granter S. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Head Neck Pathol. 2016;10:119-124.
- Fujii K, Miyashita T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014;56:667-674.
- Evans G, Farndon P. Nevoid basal cell carcinoma syndrome. GeneReviews [Internet]. University of Washington; 1993-2020.
- Kim WB, Jerome D, Yeung J. Diagnosis and management of psoriasis. Can Fam Physician. 2017;63:278-285.
- Booms P, Harth M, Sader R, et al. Vismodegib hedgehog-signaling inhibition and treatment of basal cell carcinomas as well as keratocystic odontogenic tumors in Gorlin syndrome. Ann Maxillofac Surg. 2015;5:14-19.
- Gutzmer R, Soloon J. Hedgehog pathway inhibition for the treatment of basal cell carcinoma. Target Oncol. 2019;14:253-267.
- Leavitt E, Lask G, Martin S. Sonic hedgehog pathway inhibition in the treatment of advanced basal cell carcinoma. Curr Treat Options Oncol. 2019;20:84.
The Diagnosis: Nevoid Basal Cell Carcinoma Syndrome
Nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin syndrome, is a rare autosomal-dominant disorder that increases the risk for developing various carcinomas and affects multiple organ systems. Nevoid basal cell carcinoma syndrome is estimated at 1 per 40,000 to 60,000 individuals with no sexual predilection.1,2 Pathogenesis of NBCCS occurs through molecular alterations in the dormant hedgehog signaling pathway, causing constitutive signaling activity and a loss of function in the tumor suppressor patched 1 gene, PTCH1. As a result, the inhibition of smoothened oncogenes is released, Gli proteins are activated, and the hedgehog signaling pathway is no longer quiescent.2 Additional loss of function in the suppressor of fused homolog protein, a negative regulator of the hedgehog pathway, allows for further tumor proliferation. The crucial role these genes play in the hedgehog signaling pathway and their mutation association with NBCCS allows for molecular confirmation in the diagnosis of NBCCS. Allelic losses at the PTCH1 gene site are thought to occur in approximately 70% of NBCCS patients.2
Diagnosis of NBCCS is based on genetic testing to examine pathogenic gene variants, notably in the PTCH1 gene, and identification of characteristic clinical findings.2 Diagnosis of NBCCS requires either 2 minor suggestive criteria and 1 major suggestive criterion, 2 major suggestive criteria and 1 minor suggestive criterion, or 1 major suggestive criterion with molecular confirmation. The presence of basal cell carcinomas (BCCs) before 20 years of age or an excessive numbers of BCCs, keratocystic odontogenic tumors (KOTs), palmar or plantar pitting, and first-degree relatives with NBCCS are classified as major suggestive criteria.2 Nevoid basal cell carcinoma syndrome patients typically have BCCs that crust, ulcerate, or bleed. Minor suggestive criteria for NBCCS are rib abnormalities, skeletal malformations, macrocephaly, cleft lip or palate, and desmoplastic medulloblastoma.2-4 Suppressor of fused homolog protein mutations may increase the risk for desmoplastic medulloblastoma in NBCCS patients. Our patient had 4 of the major suggestive criteria, including a history of KOTs, multiple BCCs, first-degree relatives with NBCCS, and palmar or plantar pitting (bottom quiz image), while having 1 minor suggestive criterion of frontal bossing.
Patients with NBCCS have high phenotypic variability, as their skin carcinomas do not have the classic features of pearly surfaces or corkscrew telangiectasia that typically are associated with BCCs.1 Basal cell carcinomas in NBCCS-affected individuals usually are indistinguishable from sporadic lesions that arise in sun-exposed areas, making NBCCS difficult to diagnose. These sporadic lesions often are misdiagnosed as psoriatic or eczematous lesions, and additional subsequent examination is required. The findings of multiple papules and plaques spanning the body as well as lesions with rolled borders and ulcerated bases, indicative of BCCs, aid dermatologists in distinguishing benign lesions from those of NBCCS.1
Additional differential diagnoses are required to distinguish NBCCS from other similar inherited skin disorders that are characterized by BCCs. The presence of multiple incidental BCCs early in life remains a histopathologic clue for NBCCS diagnosis, as opposed to Rombo syndrome, in which BCCs often develop in adulthood.2,4 In addition, although both Bazex syndrome and Muir-Torre syndrome are characterized by the early onset of BCCs, the lack of skeletal abnormalities and palmar and plantar pitting distinguish these entities from NBCCS.2,4 Furthermore, though psoriasis also can present on the scalp, clinical presentation often includes well-demarcated and symmetric plaques that are erythematous and silvery, all of which were not present in our patient and typically are not seen in NBCCS.5
The recommended treatment of NBCCS is vismodegib, a specific oncogene inhibitor. This medication suppresses the hedgehog signaling pathway by inhibiting smoothened oncogenes and downstream target molecules, thereby decreasing tumor proliferation.6 In doing so, vismodegib inhibits the development of new BCCs while reducing the burden of present ones. Additionally, vismodegib appears to effectively treat KOTs. If successful, this medication may be able to suppress KOTs in patients with NBCCS and thus facilitate surgery.6 Additional hedgehog inhibitors include patidegib, sonidegib, and itraconazole. Patidegib gel 2% currently is in phase 3 clinical trials for evaluation of efficacy and safety in treatment of NBCCS. Sonidegib is approved for the treatment of locally advanced BCCs in the United States and the European Union and for both locally advanced BCCs and metastatic BCCs in Switzerland and Australia.7 Further research is needed before recommending antifungal itraconazole for NBCCS clinical use.8 Other medications for localized areas include topical application of 5-fluorouracil and imiquimod.2
The Diagnosis: Nevoid Basal Cell Carcinoma Syndrome
Nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin syndrome, is a rare autosomal-dominant disorder that increases the risk for developing various carcinomas and affects multiple organ systems. Nevoid basal cell carcinoma syndrome is estimated at 1 per 40,000 to 60,000 individuals with no sexual predilection.1,2 Pathogenesis of NBCCS occurs through molecular alterations in the dormant hedgehog signaling pathway, causing constitutive signaling activity and a loss of function in the tumor suppressor patched 1 gene, PTCH1. As a result, the inhibition of smoothened oncogenes is released, Gli proteins are activated, and the hedgehog signaling pathway is no longer quiescent.2 Additional loss of function in the suppressor of fused homolog protein, a negative regulator of the hedgehog pathway, allows for further tumor proliferation. The crucial role these genes play in the hedgehog signaling pathway and their mutation association with NBCCS allows for molecular confirmation in the diagnosis of NBCCS. Allelic losses at the PTCH1 gene site are thought to occur in approximately 70% of NBCCS patients.2
Diagnosis of NBCCS is based on genetic testing to examine pathogenic gene variants, notably in the PTCH1 gene, and identification of characteristic clinical findings.2 Diagnosis of NBCCS requires either 2 minor suggestive criteria and 1 major suggestive criterion, 2 major suggestive criteria and 1 minor suggestive criterion, or 1 major suggestive criterion with molecular confirmation. The presence of basal cell carcinomas (BCCs) before 20 years of age or an excessive numbers of BCCs, keratocystic odontogenic tumors (KOTs), palmar or plantar pitting, and first-degree relatives with NBCCS are classified as major suggestive criteria.2 Nevoid basal cell carcinoma syndrome patients typically have BCCs that crust, ulcerate, or bleed. Minor suggestive criteria for NBCCS are rib abnormalities, skeletal malformations, macrocephaly, cleft lip or palate, and desmoplastic medulloblastoma.2-4 Suppressor of fused homolog protein mutations may increase the risk for desmoplastic medulloblastoma in NBCCS patients. Our patient had 4 of the major suggestive criteria, including a history of KOTs, multiple BCCs, first-degree relatives with NBCCS, and palmar or plantar pitting (bottom quiz image), while having 1 minor suggestive criterion of frontal bossing.
Patients with NBCCS have high phenotypic variability, as their skin carcinomas do not have the classic features of pearly surfaces or corkscrew telangiectasia that typically are associated with BCCs.1 Basal cell carcinomas in NBCCS-affected individuals usually are indistinguishable from sporadic lesions that arise in sun-exposed areas, making NBCCS difficult to diagnose. These sporadic lesions often are misdiagnosed as psoriatic or eczematous lesions, and additional subsequent examination is required. The findings of multiple papules and plaques spanning the body as well as lesions with rolled borders and ulcerated bases, indicative of BCCs, aid dermatologists in distinguishing benign lesions from those of NBCCS.1
Additional differential diagnoses are required to distinguish NBCCS from other similar inherited skin disorders that are characterized by BCCs. The presence of multiple incidental BCCs early in life remains a histopathologic clue for NBCCS diagnosis, as opposed to Rombo syndrome, in which BCCs often develop in adulthood.2,4 In addition, although both Bazex syndrome and Muir-Torre syndrome are characterized by the early onset of BCCs, the lack of skeletal abnormalities and palmar and plantar pitting distinguish these entities from NBCCS.2,4 Furthermore, though psoriasis also can present on the scalp, clinical presentation often includes well-demarcated and symmetric plaques that are erythematous and silvery, all of which were not present in our patient and typically are not seen in NBCCS.5
The recommended treatment of NBCCS is vismodegib, a specific oncogene inhibitor. This medication suppresses the hedgehog signaling pathway by inhibiting smoothened oncogenes and downstream target molecules, thereby decreasing tumor proliferation.6 In doing so, vismodegib inhibits the development of new BCCs while reducing the burden of present ones. Additionally, vismodegib appears to effectively treat KOTs. If successful, this medication may be able to suppress KOTs in patients with NBCCS and thus facilitate surgery.6 Additional hedgehog inhibitors include patidegib, sonidegib, and itraconazole. Patidegib gel 2% currently is in phase 3 clinical trials for evaluation of efficacy and safety in treatment of NBCCS. Sonidegib is approved for the treatment of locally advanced BCCs in the United States and the European Union and for both locally advanced BCCs and metastatic BCCs in Switzerland and Australia.7 Further research is needed before recommending antifungal itraconazole for NBCCS clinical use.8 Other medications for localized areas include topical application of 5-fluorouracil and imiquimod.2
- Sangehra R, Grewal P. Gorlin syndrome presentation and the importance of differential diagnosis of skin cancer: a case report. J Pharm Pharm Sci. 2018;21:222-224.
- Bresler S, Padwa B, Granter S. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Head Neck Pathol. 2016;10:119-124.
- Fujii K, Miyashita T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014;56:667-674.
- Evans G, Farndon P. Nevoid basal cell carcinoma syndrome. GeneReviews [Internet]. University of Washington; 1993-2020.
- Kim WB, Jerome D, Yeung J. Diagnosis and management of psoriasis. Can Fam Physician. 2017;63:278-285.
- Booms P, Harth M, Sader R, et al. Vismodegib hedgehog-signaling inhibition and treatment of basal cell carcinomas as well as keratocystic odontogenic tumors in Gorlin syndrome. Ann Maxillofac Surg. 2015;5:14-19.
- Gutzmer R, Soloon J. Hedgehog pathway inhibition for the treatment of basal cell carcinoma. Target Oncol. 2019;14:253-267.
- Leavitt E, Lask G, Martin S. Sonic hedgehog pathway inhibition in the treatment of advanced basal cell carcinoma. Curr Treat Options Oncol. 2019;20:84.
- Sangehra R, Grewal P. Gorlin syndrome presentation and the importance of differential diagnosis of skin cancer: a case report. J Pharm Pharm Sci. 2018;21:222-224.
- Bresler S, Padwa B, Granter S. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Head Neck Pathol. 2016;10:119-124.
- Fujii K, Miyashita T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014;56:667-674.
- Evans G, Farndon P. Nevoid basal cell carcinoma syndrome. GeneReviews [Internet]. University of Washington; 1993-2020.
- Kim WB, Jerome D, Yeung J. Diagnosis and management of psoriasis. Can Fam Physician. 2017;63:278-285.
- Booms P, Harth M, Sader R, et al. Vismodegib hedgehog-signaling inhibition and treatment of basal cell carcinomas as well as keratocystic odontogenic tumors in Gorlin syndrome. Ann Maxillofac Surg. 2015;5:14-19.
- Gutzmer R, Soloon J. Hedgehog pathway inhibition for the treatment of basal cell carcinoma. Target Oncol. 2019;14:253-267.
- Leavitt E, Lask G, Martin S. Sonic hedgehog pathway inhibition in the treatment of advanced basal cell carcinoma. Curr Treat Options Oncol. 2019;20:84.

A 63-year-old man with frontal bossing and a history of jaw cysts presented with numerous lesions on the scalp, trunk, and legs with recurrent bleeding. Both of his siblings had similar findings. Many lesions had been present for at least 40 years. Physical examination revealed a large, irregular, atrophic, hyperpigmented plaque on the central scalp with multiple translucent hyperpigmented papules at the periphery (top). Similar papules and plaques were present at the temples, around the waist, and on the distal lower extremities, leading to surgical excision of the largest leg lesions. In addition, there were many pinpoint pits on both palms (bottom). A biopsy was submitted for review, which confirmed basal cell carcinomas on the scalp.
Large Leg Ulcers After Swimming in the Ocean
The Diagnosis: Vibrio vulnificus Infection
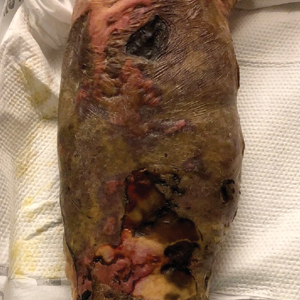
At the initial presentation, the differential diagnosis included infectious processes such as bacterial or angioinvasive fungal infections or an inflammatory process such as pyoderma gangrenosum. Blood cultures were found to be positive for pansensitive Vibrio vulnificus. He initially was treated with piperacillin-tazobactam and received surgical debridement of the affected tissues. Pathologic interpretation of the wound tissues revealed a diagnosis of necrotizing softtissue infection and positive Candida albicans growth. He received topical bacitracin on discharge as well as a 7-day course of amoxicillin-clavulanate and fluconazole. He continued to receive debridement procedures and skin grafts, followed by topical mupirocin treatment and silver sulfadiazine. He was seen 6 weeks after discharge with healing wounds and healthy-appearing granulation tissue at the base.
Our patient’s presentation of retiform purpura with stellate necrosis was consistent with a wide range of serious pathologies ranging from medium-vessel vasculitis to thromboembolic phenomena and angioinvasive fungal infections.1 Although Vibrio infection rarely is the first explanation that comes to mind when observing necrotic retiform purpura, the chronic nonhealing injury on the leg combined with the recent history of ocean swimming made V vulnificus stand out as a likely culprit. Although V vulnificus infection traditionally presents with cellulitis, edema, and hemorrhagic bulla,2 necrosis also has been observed.3 Vibrio vulnificus produces multiple virulence factors, and it is believed that these severe cutaneous symptoms are attributable to the production of a specific metalloprotease that enhances vascular permeability, thereby inducing hemorrhage within the vascular basement membrane zone.2
Vibrio vulnificus is an opportunistic bacterial pathogen associated with consumption of contaminated seafood or swimming in ocean waters with open wounds. Infections are rare, with only approximately 100 cases reported annually in the United States.4 However, V vulnificus infections have demonstrated increasing incidence in recent years, especially infections of pre-existing wounds.4,5 Risk factors for their development include age over 40 years and underlying conditions including liver disease, diabetes mellitus, and immune dysfunction.4 Vibrio vulnificus infections also demonstrate a strong male predilection, with almost 90% of infections occurring in males.4 Although the precise etiology of this sex discrepancy remains unknown, estrogen has been suggested to be a protective factor.6 Alternatively, behavioral differences also have been proposed as possible explanations for this discrepancy, with women less likely to consume seafood or go swimming. However, epidemiologic data reveal strong correlations between male sex and liver cirrhosis, a primary risk factor for V vulnificus infections, suggesting that male sex may simply be a confounding variable.7
Infections with V vulnificus are notable for their short incubation periods, with onset of symptoms occurring within 24 hours of exposure, making prompt diagnosis and treatment of high importance.8 Although rare, V vulnificus infections are associated with high mortality rates. From 1988 to 2010, nearly 600 deaths were reported secondary to V vulnificus infections.4 Wound infections carry a 17.6% fatality rate,4 while bloodborne V vulnificus infections exceed 50% fatality.8 Although sepsis secondary to V vulnificus usually is caused by ingestion of raw or undercooked shellfish, primarily oysters,4 our case highlights a rarer instance of both sepsis and localized infection stemming from ocean water exposure.
Vibrio vulnificus is an obligate halophile and therefore is found in marine environments rather than freshwater bodies. However, it rarely is isolated from bodies of water with salinities over 25 parts per thousand, such as the Mediterranean Sea; it usually is found in warmer waters, making it more common in the summer months from May to October.4 Given this proclivity for warmer environments, climate change has contributed to both a greater incidence and global distribution of V vulnificus. 9,10
Treatment of V vulnificus infections centers on antibiotic treatment, with Vibrio species generally demonstrating susceptibility to most antibiotics of human significance.11 However, some Vibrio isolates within the United States have demonstrated antibiotic resistance; 45% of a variety of clinical and environmental samples from South Carolina and Georgia demonstrated resistance to at least 3 antibiotic classes, and 17.3% resisted 8 or more classes of antibiotics.12 These included medications such as doxycycline, tetracycline, aminoglycosides, and cephalosporins—agents that normally are prescribed for V vulnificus infections. Although tetracyclines have long been touted as the preferred treatment of V vulnificus infections, the spread of antibiotic resistance may require greater reliance on alternative regimens such as combinations of cephalosporins and doxycycline or a single fluoroquinolone.13 Although rare, Vibrio infections can have rapidly fatal consequences and should be given serious consideration when evaluating patients with relevant risk factors.
The differential diagnosis included angioinvasive mucormycosis, calciphylaxis, pyoderma gangrenosum, and Stevens-Johnson syndrome/toxic epidermal necrolysis. Mucormycosis is a fungal infection caused by Mucorales fungi that most commonly is seen in patients with diabetes mellitus, hematologic malignancies, neutropenia, and immunocompromise.14 Calciphylaxis is a condition involving microvascular occlusion due to diffuse calcium deposition in cutaneous blood vessels. It typically presents as violaceous retiform patches and plaques commonly seen on areas such as the thighs, buttocks, or abdomen and usually is associated with chronic renal failure, hemodialysis, and/or secondary hyperparathyroidism.15 Pyoderma gangrenosum is an inflammatory condition involving neutrophilic ulceration of the skin that typically presents as ulceration with a classically undermined border. It frequently is considered a diagnosis of exclusion and therefore requires that providers rule out other causes of ulceration prior to diagnosis.16 Stevens-Johnson syndrome/toxic epidermal necrolysis is a rare drug reaction involving mucosal erosions and cutaneous detachment.17 This diagnosis is less likely given that our patient lacked mucosal involvement and did not have any notable medication exposures prior to symptom onset.
- Wysong A, Venkatesan P. An approach to the patient with retiform purpura. Dermatol Ther. 2011;24:151-172. doi:10.1111/j .1529-8019.2011.01392.x
- Miyoshi S-I. Vibrio vulnificus infection and metalloprotease. J Dermatol. 2006;33:589-595. doi:10.1111/j.1346-8138.2006.00139.x
- Patel VJ, Gardner E, Burton CS. Vibrio vulnificus septicemia and leg ulcer. J Am Acad Dermatol. 2002;46(5 suppl):S144-S145. doi:10.1067 /mjd.2002.107778
- Baker-Austin C, Oliver JD. Vibrio vulnificus: new insights into a deadly opportunistic pathogen. Environ Microbiol. 2018;20:423-430. doi:10.1111/1462-2920.13955
- Preliminary FoodNet data on the incidence of infection with pathogens transmitted commonly through food —10 states, 2009. CDC website. Published April 16, 2010. Accessed November 3, 2021. https://www.cdc .gov/mmwr/preview/mmwrhtml/mm5914a2.htm
- Merkel SM, Alexander S, Zufall E, et al. Essential role for estrogen in protection against Vibrio vulnificus-induced endotoxic shock. Infect Immun. 2001;69:6119-6122. doi:10.1128/IAI.69.10.6119 -6122.2001
- Scaglione S, Kliethermes S, Cao G, et al. The epidemiology of cirrhosis in the United States: a population-based study. J Clin Gastroenterol. 2015;49:690-696. doi:10.1097/MCG.0000000000000208
- Jones M, Oliver J. Vibrio vulnificus: disease and pathogenesis [published online December 20, 2020]. Infect Immun. https://doi.org/10.1128 /IAI.01046-08
- Paz S, Bisharat N, Paz E, et al. Climate change and the emergence of Vibrio vulnificus disease in Israel. Environ Res. 2007;103:390-396. doi:10.1016/j.envres.2006.07.002
- Martinez-Urtaza J, Bowers JC, Trinanes J, et al. Climate anomalies and the increasing risk of Vibrio parahaemolyticus and Vibrio vulnificus illnesses. Food Res Int. 2010;43:1780-1790. doi:10.1016/j. foodres.2010.04.001
- Oliver JD. Vibrio vulnificus. In: Thompson FL, Austin B, Swings J, eds. The Biology of Vibrios. ASM Press; 2006:349-366.
- Baker-Austin C, McArthur JV, Lindell AH, et al. Multi-site analysis reveals widespread antibiotic resistance in the marine pathogen Vibrio vulnificus. Microb Ecol. 2009;57:151-159. doi:10.1007 /s00248-008-9413-8
- Elmahdi S, DaSilva LV, Parveen S. Antibiotic resistance of Vibrio parahaemolyticus and Vibrio vulnificus in various countries: a review. Food Microbiol. 2016;57:128-134. doi:10.1016/j.fm.2016.02.008
- Prasad P, Wong V, Burgin S, et al. Mucormycosis. VisualDx website. Accessed November 13, 2021. https://www-visualdx-com.proxy.lib.ohio-state.edu/visualdx/diagnosis/mucormycosis?diagnosisId=51981 &moduleId=101
- Blum A, Song P, Tan B, et al. Calciphylaxis. VisualDx website. Accessed November 13, 2021. https://www-visualdx-com.proxy.lib.ohio-state.edu/visualdx/diagnosis/calciphylaxis?diagnosisId=51241&moduleId=101
- Cohen J, Wong V, Burgin S. Pyoderma gangrenosum. VisualDx website. Accessed November 13, 2021. https://www-visualdx-com.proxy.lib.ohio-state.edu/visualdx/diagnosis/pyoderma+gangrenosum?diagnosis Id=52242&moduleId=101
- Walls A, Burgin S. Stevens-Johnson syndrome. VisualDx website. Accessed November 13, 2021. https://www-visualdx-com.proxy.lib.ohio-state.edu/visualdx/diagnosis/stevens-johnson+syndrome?diagnosisId=52342&moduleId=101
The Diagnosis: Vibrio vulnificus Infection
At the initial presentation, the differential diagnosis included infectious processes such as bacterial or angioinvasive fungal infections or an inflammatory process such as pyoderma gangrenosum. Blood cultures were found to be positive for pansensitive Vibrio vulnificus. He initially was treated with piperacillin-tazobactam and received surgical debridement of the affected tissues. Pathologic interpretation of the wound tissues revealed a diagnosis of necrotizing softtissue infection and positive Candida albicans growth. He received topical bacitracin on discharge as well as a 7-day course of amoxicillin-clavulanate and fluconazole. He continued to receive debridement procedures and skin grafts, followed by topical mupirocin treatment and silver sulfadiazine. He was seen 6 weeks after discharge with healing wounds and healthy-appearing granulation tissue at the base.
Our patient’s presentation of retiform purpura with stellate necrosis was consistent with a wide range of serious pathologies ranging from medium-vessel vasculitis to thromboembolic phenomena and angioinvasive fungal infections.1 Although Vibrio infection rarely is the first explanation that comes to mind when observing necrotic retiform purpura, the chronic nonhealing injury on the leg combined with the recent history of ocean swimming made V vulnificus stand out as a likely culprit. Although V vulnificus infection traditionally presents with cellulitis, edema, and hemorrhagic bulla,2 necrosis also has been observed.3 Vibrio vulnificus produces multiple virulence factors, and it is believed that these severe cutaneous symptoms are attributable to the production of a specific metalloprotease that enhances vascular permeability, thereby inducing hemorrhage within the vascular basement membrane zone.2
Vibrio vulnificus is an opportunistic bacterial pathogen associated with consumption of contaminated seafood or swimming in ocean waters with open wounds. Infections are rare, with only approximately 100 cases reported annually in the United States.4 However, V vulnificus infections have demonstrated increasing incidence in recent years, especially infections of pre-existing wounds.4,5 Risk factors for their development include age over 40 years and underlying conditions including liver disease, diabetes mellitus, and immune dysfunction.4 Vibrio vulnificus infections also demonstrate a strong male predilection, with almost 90% of infections occurring in males.4 Although the precise etiology of this sex discrepancy remains unknown, estrogen has been suggested to be a protective factor.6 Alternatively, behavioral differences also have been proposed as possible explanations for this discrepancy, with women less likely to consume seafood or go swimming. However, epidemiologic data reveal strong correlations between male sex and liver cirrhosis, a primary risk factor for V vulnificus infections, suggesting that male sex may simply be a confounding variable.7
Infections with V vulnificus are notable for their short incubation periods, with onset of symptoms occurring within 24 hours of exposure, making prompt diagnosis and treatment of high importance.8 Although rare, V vulnificus infections are associated with high mortality rates. From 1988 to 2010, nearly 600 deaths were reported secondary to V vulnificus infections.4 Wound infections carry a 17.6% fatality rate,4 while bloodborne V vulnificus infections exceed 50% fatality.8 Although sepsis secondary to V vulnificus usually is caused by ingestion of raw or undercooked shellfish, primarily oysters,4 our case highlights a rarer instance of both sepsis and localized infection stemming from ocean water exposure.
Vibrio vulnificus is an obligate halophile and therefore is found in marine environments rather than freshwater bodies. However, it rarely is isolated from bodies of water with salinities over 25 parts per thousand, such as the Mediterranean Sea; it usually is found in warmer waters, making it more common in the summer months from May to October.4 Given this proclivity for warmer environments, climate change has contributed to both a greater incidence and global distribution of V vulnificus. 9,10
Treatment of V vulnificus infections centers on antibiotic treatment, with Vibrio species generally demonstrating susceptibility to most antibiotics of human significance.11 However, some Vibrio isolates within the United States have demonstrated antibiotic resistance; 45% of a variety of clinical and environmental samples from South Carolina and Georgia demonstrated resistance to at least 3 antibiotic classes, and 17.3% resisted 8 or more classes of antibiotics.12 These included medications such as doxycycline, tetracycline, aminoglycosides, and cephalosporins—agents that normally are prescribed for V vulnificus infections. Although tetracyclines have long been touted as the preferred treatment of V vulnificus infections, the spread of antibiotic resistance may require greater reliance on alternative regimens such as combinations of cephalosporins and doxycycline or a single fluoroquinolone.13 Although rare, Vibrio infections can have rapidly fatal consequences and should be given serious consideration when evaluating patients with relevant risk factors.
The differential diagnosis included angioinvasive mucormycosis, calciphylaxis, pyoderma gangrenosum, and Stevens-Johnson syndrome/toxic epidermal necrolysis. Mucormycosis is a fungal infection caused by Mucorales fungi that most commonly is seen in patients with diabetes mellitus, hematologic malignancies, neutropenia, and immunocompromise.14 Calciphylaxis is a condition involving microvascular occlusion due to diffuse calcium deposition in cutaneous blood vessels. It typically presents as violaceous retiform patches and plaques commonly seen on areas such as the thighs, buttocks, or abdomen and usually is associated with chronic renal failure, hemodialysis, and/or secondary hyperparathyroidism.15 Pyoderma gangrenosum is an inflammatory condition involving neutrophilic ulceration of the skin that typically presents as ulceration with a classically undermined border. It frequently is considered a diagnosis of exclusion and therefore requires that providers rule out other causes of ulceration prior to diagnosis.16 Stevens-Johnson syndrome/toxic epidermal necrolysis is a rare drug reaction involving mucosal erosions and cutaneous detachment.17 This diagnosis is less likely given that our patient lacked mucosal involvement and did not have any notable medication exposures prior to symptom onset.
The Diagnosis: Vibrio vulnificus Infection
At the initial presentation, the differential diagnosis included infectious processes such as bacterial or angioinvasive fungal infections or an inflammatory process such as pyoderma gangrenosum. Blood cultures were found to be positive for pansensitive Vibrio vulnificus. He initially was treated with piperacillin-tazobactam and received surgical debridement of the affected tissues. Pathologic interpretation of the wound tissues revealed a diagnosis of necrotizing softtissue infection and positive Candida albicans growth. He received topical bacitracin on discharge as well as a 7-day course of amoxicillin-clavulanate and fluconazole. He continued to receive debridement procedures and skin grafts, followed by topical mupirocin treatment and silver sulfadiazine. He was seen 6 weeks after discharge with healing wounds and healthy-appearing granulation tissue at the base.
Our patient’s presentation of retiform purpura with stellate necrosis was consistent with a wide range of serious pathologies ranging from medium-vessel vasculitis to thromboembolic phenomena and angioinvasive fungal infections.1 Although Vibrio infection rarely is the first explanation that comes to mind when observing necrotic retiform purpura, the chronic nonhealing injury on the leg combined with the recent history of ocean swimming made V vulnificus stand out as a likely culprit. Although V vulnificus infection traditionally presents with cellulitis, edema, and hemorrhagic bulla,2 necrosis also has been observed.3 Vibrio vulnificus produces multiple virulence factors, and it is believed that these severe cutaneous symptoms are attributable to the production of a specific metalloprotease that enhances vascular permeability, thereby inducing hemorrhage within the vascular basement membrane zone.2
Vibrio vulnificus is an opportunistic bacterial pathogen associated with consumption of contaminated seafood or swimming in ocean waters with open wounds. Infections are rare, with only approximately 100 cases reported annually in the United States.4 However, V vulnificus infections have demonstrated increasing incidence in recent years, especially infections of pre-existing wounds.4,5 Risk factors for their development include age over 40 years and underlying conditions including liver disease, diabetes mellitus, and immune dysfunction.4 Vibrio vulnificus infections also demonstrate a strong male predilection, with almost 90% of infections occurring in males.4 Although the precise etiology of this sex discrepancy remains unknown, estrogen has been suggested to be a protective factor.6 Alternatively, behavioral differences also have been proposed as possible explanations for this discrepancy, with women less likely to consume seafood or go swimming. However, epidemiologic data reveal strong correlations between male sex and liver cirrhosis, a primary risk factor for V vulnificus infections, suggesting that male sex may simply be a confounding variable.7
Infections with V vulnificus are notable for their short incubation periods, with onset of symptoms occurring within 24 hours of exposure, making prompt diagnosis and treatment of high importance.8 Although rare, V vulnificus infections are associated with high mortality rates. From 1988 to 2010, nearly 600 deaths were reported secondary to V vulnificus infections.4 Wound infections carry a 17.6% fatality rate,4 while bloodborne V vulnificus infections exceed 50% fatality.8 Although sepsis secondary to V vulnificus usually is caused by ingestion of raw or undercooked shellfish, primarily oysters,4 our case highlights a rarer instance of both sepsis and localized infection stemming from ocean water exposure.
Vibrio vulnificus is an obligate halophile and therefore is found in marine environments rather than freshwater bodies. However, it rarely is isolated from bodies of water with salinities over 25 parts per thousand, such as the Mediterranean Sea; it usually is found in warmer waters, making it more common in the summer months from May to October.4 Given this proclivity for warmer environments, climate change has contributed to both a greater incidence and global distribution of V vulnificus. 9,10
Treatment of V vulnificus infections centers on antibiotic treatment, with Vibrio species generally demonstrating susceptibility to most antibiotics of human significance.11 However, some Vibrio isolates within the United States have demonstrated antibiotic resistance; 45% of a variety of clinical and environmental samples from South Carolina and Georgia demonstrated resistance to at least 3 antibiotic classes, and 17.3% resisted 8 or more classes of antibiotics.12 These included medications such as doxycycline, tetracycline, aminoglycosides, and cephalosporins—agents that normally are prescribed for V vulnificus infections. Although tetracyclines have long been touted as the preferred treatment of V vulnificus infections, the spread of antibiotic resistance may require greater reliance on alternative regimens such as combinations of cephalosporins and doxycycline or a single fluoroquinolone.13 Although rare, Vibrio infections can have rapidly fatal consequences and should be given serious consideration when evaluating patients with relevant risk factors.
The differential diagnosis included angioinvasive mucormycosis, calciphylaxis, pyoderma gangrenosum, and Stevens-Johnson syndrome/toxic epidermal necrolysis. Mucormycosis is a fungal infection caused by Mucorales fungi that most commonly is seen in patients with diabetes mellitus, hematologic malignancies, neutropenia, and immunocompromise.14 Calciphylaxis is a condition involving microvascular occlusion due to diffuse calcium deposition in cutaneous blood vessels. It typically presents as violaceous retiform patches and plaques commonly seen on areas such as the thighs, buttocks, or abdomen and usually is associated with chronic renal failure, hemodialysis, and/or secondary hyperparathyroidism.15 Pyoderma gangrenosum is an inflammatory condition involving neutrophilic ulceration of the skin that typically presents as ulceration with a classically undermined border. It frequently is considered a diagnosis of exclusion and therefore requires that providers rule out other causes of ulceration prior to diagnosis.16 Stevens-Johnson syndrome/toxic epidermal necrolysis is a rare drug reaction involving mucosal erosions and cutaneous detachment.17 This diagnosis is less likely given that our patient lacked mucosal involvement and did not have any notable medication exposures prior to symptom onset.
- Wysong A, Venkatesan P. An approach to the patient with retiform purpura. Dermatol Ther. 2011;24:151-172. doi:10.1111/j .1529-8019.2011.01392.x
- Miyoshi S-I. Vibrio vulnificus infection and metalloprotease. J Dermatol. 2006;33:589-595. doi:10.1111/j.1346-8138.2006.00139.x
- Patel VJ, Gardner E, Burton CS. Vibrio vulnificus septicemia and leg ulcer. J Am Acad Dermatol. 2002;46(5 suppl):S144-S145. doi:10.1067 /mjd.2002.107778
- Baker-Austin C, Oliver JD. Vibrio vulnificus: new insights into a deadly opportunistic pathogen. Environ Microbiol. 2018;20:423-430. doi:10.1111/1462-2920.13955
- Preliminary FoodNet data on the incidence of infection with pathogens transmitted commonly through food —10 states, 2009. CDC website. Published April 16, 2010. Accessed November 3, 2021. https://www.cdc .gov/mmwr/preview/mmwrhtml/mm5914a2.htm
- Merkel SM, Alexander S, Zufall E, et al. Essential role for estrogen in protection against Vibrio vulnificus-induced endotoxic shock. Infect Immun. 2001;69:6119-6122. doi:10.1128/IAI.69.10.6119 -6122.2001
- Scaglione S, Kliethermes S, Cao G, et al. The epidemiology of cirrhosis in the United States: a population-based study. J Clin Gastroenterol. 2015;49:690-696. doi:10.1097/MCG.0000000000000208
- Jones M, Oliver J. Vibrio vulnificus: disease and pathogenesis [published online December 20, 2020]. Infect Immun. https://doi.org/10.1128 /IAI.01046-08
- Paz S, Bisharat N, Paz E, et al. Climate change and the emergence of Vibrio vulnificus disease in Israel. Environ Res. 2007;103:390-396. doi:10.1016/j.envres.2006.07.002
- Martinez-Urtaza J, Bowers JC, Trinanes J, et al. Climate anomalies and the increasing risk of Vibrio parahaemolyticus and Vibrio vulnificus illnesses. Food Res Int. 2010;43:1780-1790. doi:10.1016/j. foodres.2010.04.001
- Oliver JD. Vibrio vulnificus. In: Thompson FL, Austin B, Swings J, eds. The Biology of Vibrios. ASM Press; 2006:349-366.
- Baker-Austin C, McArthur JV, Lindell AH, et al. Multi-site analysis reveals widespread antibiotic resistance in the marine pathogen Vibrio vulnificus. Microb Ecol. 2009;57:151-159. doi:10.1007 /s00248-008-9413-8
- Elmahdi S, DaSilva LV, Parveen S. Antibiotic resistance of Vibrio parahaemolyticus and Vibrio vulnificus in various countries: a review. Food Microbiol. 2016;57:128-134. doi:10.1016/j.fm.2016.02.008
- Prasad P, Wong V, Burgin S, et al. Mucormycosis. VisualDx website. Accessed November 13, 2021. https://www-visualdx-com.proxy.lib.ohio-state.edu/visualdx/diagnosis/mucormycosis?diagnosisId=51981 &moduleId=101
- Blum A, Song P, Tan B, et al. Calciphylaxis. VisualDx website. Accessed November 13, 2021. https://www-visualdx-com.proxy.lib.ohio-state.edu/visualdx/diagnosis/calciphylaxis?diagnosisId=51241&moduleId=101
- Cohen J, Wong V, Burgin S. Pyoderma gangrenosum. VisualDx website. Accessed November 13, 2021. https://www-visualdx-com.proxy.lib.ohio-state.edu/visualdx/diagnosis/pyoderma+gangrenosum?diagnosis Id=52242&moduleId=101
- Walls A, Burgin S. Stevens-Johnson syndrome. VisualDx website. Accessed November 13, 2021. https://www-visualdx-com.proxy.lib.ohio-state.edu/visualdx/diagnosis/stevens-johnson+syndrome?diagnosisId=52342&moduleId=101
- Wysong A, Venkatesan P. An approach to the patient with retiform purpura. Dermatol Ther. 2011;24:151-172. doi:10.1111/j .1529-8019.2011.01392.x
- Miyoshi S-I. Vibrio vulnificus infection and metalloprotease. J Dermatol. 2006;33:589-595. doi:10.1111/j.1346-8138.2006.00139.x
- Patel VJ, Gardner E, Burton CS. Vibrio vulnificus septicemia and leg ulcer. J Am Acad Dermatol. 2002;46(5 suppl):S144-S145. doi:10.1067 /mjd.2002.107778
- Baker-Austin C, Oliver JD. Vibrio vulnificus: new insights into a deadly opportunistic pathogen. Environ Microbiol. 2018;20:423-430. doi:10.1111/1462-2920.13955
- Preliminary FoodNet data on the incidence of infection with pathogens transmitted commonly through food —10 states, 2009. CDC website. Published April 16, 2010. Accessed November 3, 2021. https://www.cdc .gov/mmwr/preview/mmwrhtml/mm5914a2.htm
- Merkel SM, Alexander S, Zufall E, et al. Essential role for estrogen in protection against Vibrio vulnificus-induced endotoxic shock. Infect Immun. 2001;69:6119-6122. doi:10.1128/IAI.69.10.6119 -6122.2001
- Scaglione S, Kliethermes S, Cao G, et al. The epidemiology of cirrhosis in the United States: a population-based study. J Clin Gastroenterol. 2015;49:690-696. doi:10.1097/MCG.0000000000000208
- Jones M, Oliver J. Vibrio vulnificus: disease and pathogenesis [published online December 20, 2020]. Infect Immun. https://doi.org/10.1128 /IAI.01046-08
- Paz S, Bisharat N, Paz E, et al. Climate change and the emergence of Vibrio vulnificus disease in Israel. Environ Res. 2007;103:390-396. doi:10.1016/j.envres.2006.07.002
- Martinez-Urtaza J, Bowers JC, Trinanes J, et al. Climate anomalies and the increasing risk of Vibrio parahaemolyticus and Vibrio vulnificus illnesses. Food Res Int. 2010;43:1780-1790. doi:10.1016/j. foodres.2010.04.001
- Oliver JD. Vibrio vulnificus. In: Thompson FL, Austin B, Swings J, eds. The Biology of Vibrios. ASM Press; 2006:349-366.
- Baker-Austin C, McArthur JV, Lindell AH, et al. Multi-site analysis reveals widespread antibiotic resistance in the marine pathogen Vibrio vulnificus. Microb Ecol. 2009;57:151-159. doi:10.1007 /s00248-008-9413-8
- Elmahdi S, DaSilva LV, Parveen S. Antibiotic resistance of Vibrio parahaemolyticus and Vibrio vulnificus in various countries: a review. Food Microbiol. 2016;57:128-134. doi:10.1016/j.fm.2016.02.008
- Prasad P, Wong V, Burgin S, et al. Mucormycosis. VisualDx website. Accessed November 13, 2021. https://www-visualdx-com.proxy.lib.ohio-state.edu/visualdx/diagnosis/mucormycosis?diagnosisId=51981 &moduleId=101
- Blum A, Song P, Tan B, et al. Calciphylaxis. VisualDx website. Accessed November 13, 2021. https://www-visualdx-com.proxy.lib.ohio-state.edu/visualdx/diagnosis/calciphylaxis?diagnosisId=51241&moduleId=101
- Cohen J, Wong V, Burgin S. Pyoderma gangrenosum. VisualDx website. Accessed November 13, 2021. https://www-visualdx-com.proxy.lib.ohio-state.edu/visualdx/diagnosis/pyoderma+gangrenosum?diagnosis Id=52242&moduleId=101
- Walls A, Burgin S. Stevens-Johnson syndrome. VisualDx website. Accessed November 13, 2021. https://www-visualdx-com.proxy.lib.ohio-state.edu/visualdx/diagnosis/stevens-johnson+syndrome?diagnosisId=52342&moduleId=101
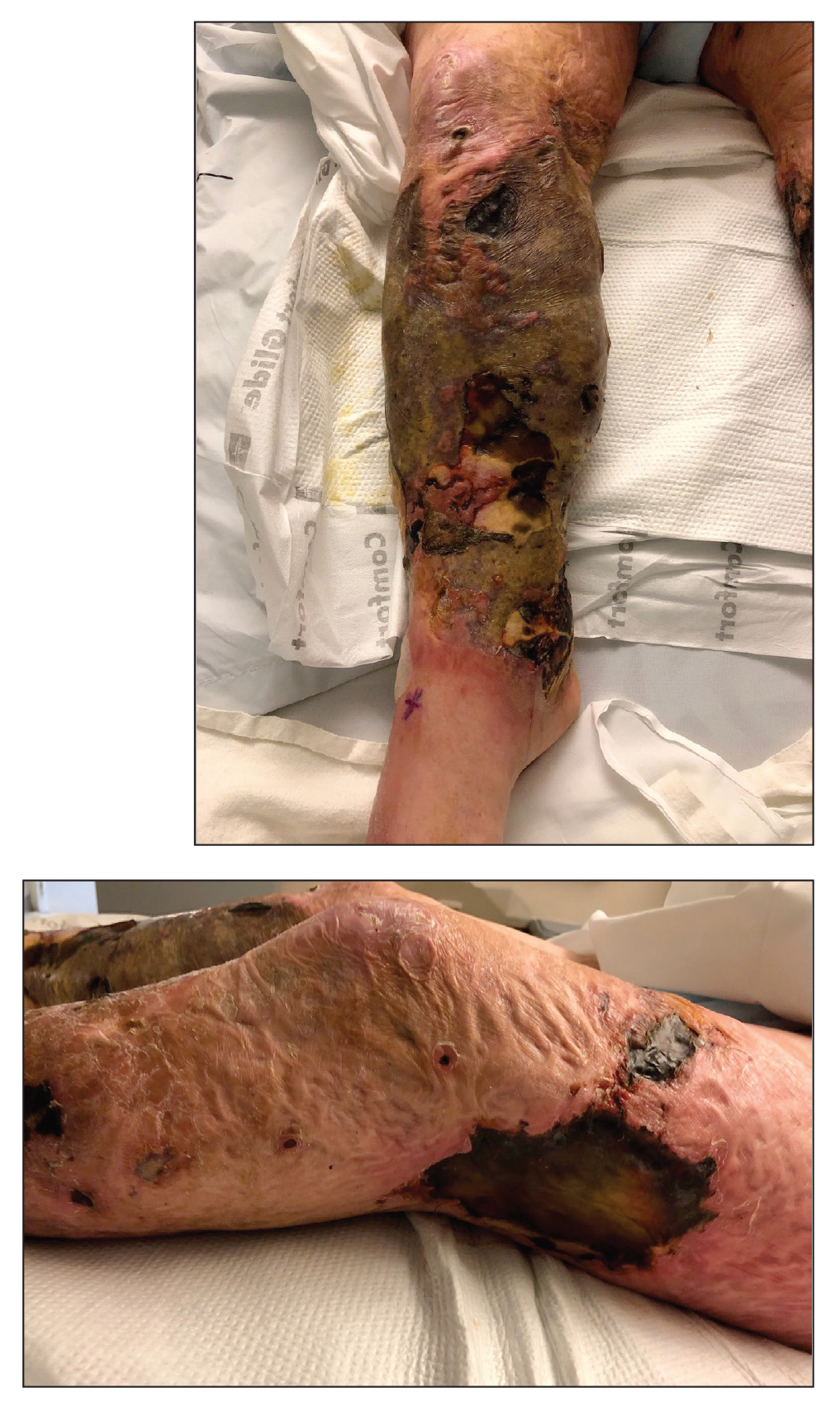
A 48-year-old man presented to the emergency department with pain in both legs after swimming in the ocean surrounding Florida 1 month prior to presentation. His medical history included skin graft treatment of burns during childhood and a chronic lower extremity ulcer that developed after trauma. He received hemodialysis for acute renal failure approximately 1 month prior to the current presentation. At the current presentation he was found to be septic and quickly developed rapidly expanding regions of retiform purpura with stellate necrosis on the legs.
Erythematous Nodule With Central Erosions on the Calf
The Diagnosis: Osteoma Cutis
Osteoma cutis is the heterotopic development of cutaneous ossifications in the dermis or subcutaneous fat and presents as plaquelike, stony, hard nodules. It can manifest as either a primary or secondary condition based on the presence or absence of a prior skin insult at the lesion site. Primary osteoma cutis occurs in 15% of patients and arises either de novo or in association with any of several inflammatory, neoplastic, and metabolic diseases that provide a favorable environment for abnormal mesenchymal stem cell commitment to osteoid,1 including Albright hereditary osteodystrophy, myositis ossificans progressiva, and progressive osseous heteroplasia, which are all associated with mutations in the heterotrimeric G-protein alpha subunit encoding gene, GNAS. 1,2 It is suggested that an insufficiency of Gsα leads to uncontrolled negative regulation of nonosseous connective tissue differentiation, forming osteoid.3 Additionally, diseases involving gain-of-function mutations in the activin A receptor type 1 encoding gene, ACVR1, such as fibrodysplasia ossificans progressiva, have been associated with osteoma cutis.4 These mutations lead to decreased receptor affinity to molecular safeguards of bone morphogenetic protein signaling, ultimately contributing to progressive ectopic bone formation.5 Secondary osteoma cutis occurs in 85% of patients and develops at the site of prior skin damage due to inflammation, neoplasm, or trauma.6 It is believed that tissue damage and degeneration lead to mesenchymal stem cell proliferation and skeletogenicinducing factor recruitment forming cartilaginous tissue, later replaced by bone through endochondral ossification.7
Although osteoma cutis previously was believed to be rare, more recent radiologic studies suggest otherwise, detecting cutaneous osteomas in up to 42.1% of patients.8 Consequently, it is likely that osteoma cutis is underdiagnosed due to its subclinical nature. Our patient, however, presented with a solitary osteoma cutis with perforation of the epidermis, a rare phenomenon.9-12
A shave biopsy in our patient revealed moderate to focally marked, irregular epidermal hyperplasia with a large focus of moderate, compact, parakeratotic crust overlying the epidermis in the center of the specimen. The papillary dermis in the center of the specimen revealed large foci of dark pink to purple bone fragments surrounded by moderate lymphocytic infiltrate with few foci perforating through the overlying epidermis (Figure, A). These findings were characteristic of osteoma cutis with perforation through the overlying epidermis.
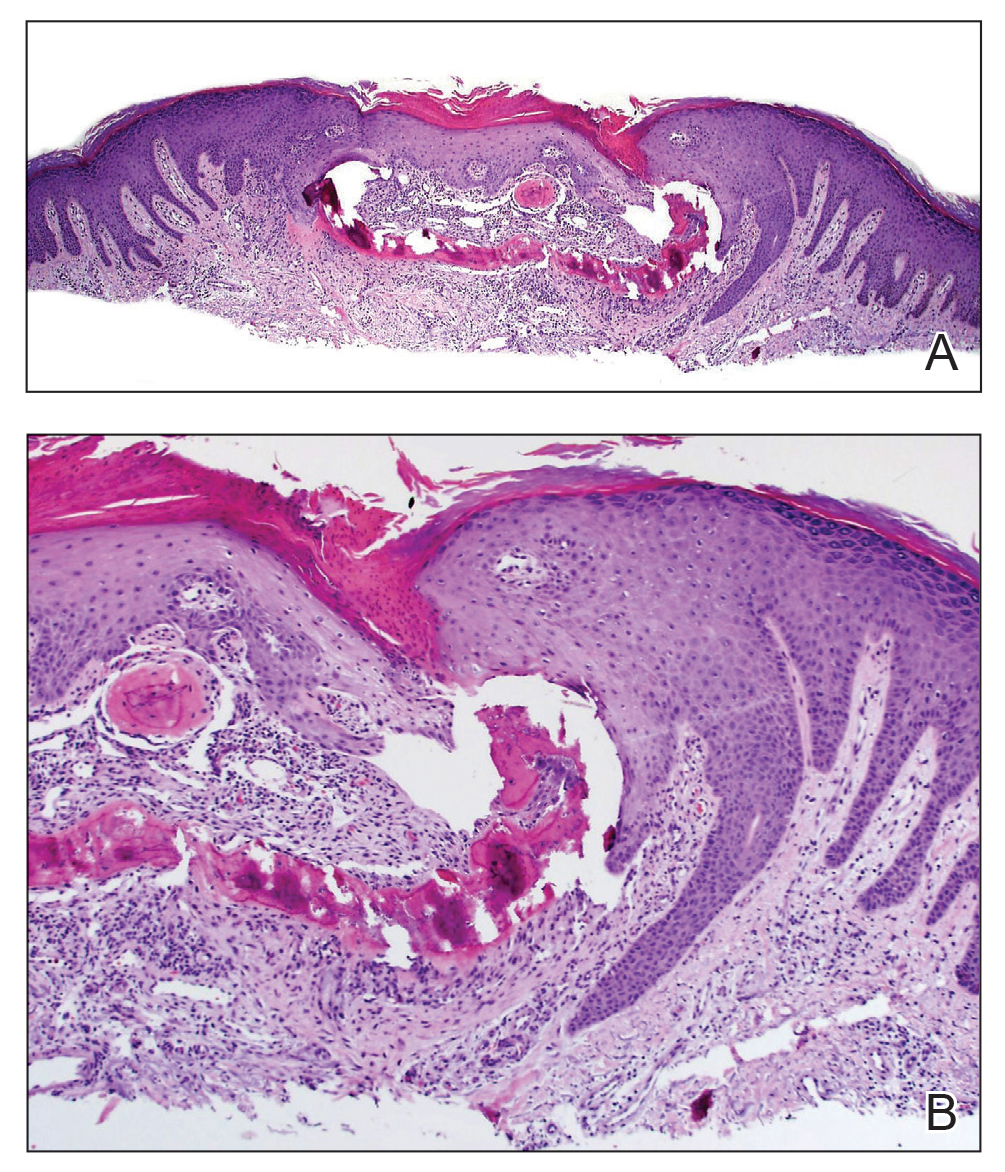
The diagnosis of osteoma cutis at the age of 62 years suggested that the lesion was not primary in association with previously described diseases. Furthermore, the lack of phenotypic features of these diseases including obesity, developmental disability, and high parathyroid hormone levels essentially excluded this possibility. The presence of the lesion on the lower extremities initially may have suggested osteoma cutis secondary to chronic venous insufficiency13; however, the absence of visible varicose veins or obvious signs of stasis disease made this unlikely. No further cutaneous disorders at or around the lesion site clinically and histologically suggested that our patient’s lesion was primary and of idiopathic nature. Dermatofibroma can present similarly in appearance but would characteristically dimple centrally when pinched. Keratoacanthoma presents with central ulceration and keratin plugging. Pilomatricoma more commonly presents on the head and neck and less frequently as a firm nodule. Lastly, prurigo nodularis more commonly presents as a symmetrically diffuse rash compared to an isolated nodule.
Osteoma cutis is a cutaneous ossification that may be primary or secondary in nature and less rare than originally thought. Workup for potentially associated inflammatory, neoplastic, and metabolic diseases should be considered in patients with this condition. Perforating osteoma cutis is a rare variant that presents as solitary or multiple nodules with central erosion and crust. The mechanism of transepidermal elimination leading to skin perforation is hypothesized to involve epidermal hyperproliferation leading to upward movement.14 Shave biopsy establishes a definitive histopathologic diagnosis and often is curative. Given that lesions of osteoma cutis themselves are benign, removal may not be necessary.
- Falsey RR, Ackerman L. Eruptive, hard cutaneous nodules in a 61-yearold woman. osteoma cutis in a patient with Albright hereditary osteodystrophy (AHO). JAMA Dermatol. 2013;149:975-976.
- Martin J, Tucker M, Browning JC. Infantile osteoma cutis as a presentation of a GNAS mutation. Pediatr Dermatol. 2012;29:483-484.
- Shore EM, Ahn J, de Beur SJ, et al. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med. 2002;346:99-106.
- Kaplan FS, Le Merrer M, Glaser DL, et al. Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol. 2008;22:191-205.
- Song GA, Kim HJ, Woo KM, et al. Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J Biol Chem. 2010;285:22542-22553.
- Roth SI, Stowell RE, Helwig EB, et al. Cutaneous ossification. report of 120 cases and review of the literature. Arch Pathol. 1963;76:44-54.
- Shimono K, Uchibe K, Kuboki T, et al. The pathophysiology of heterotopic ossification: current treatment considerations in dentistry. Japanese Dental Science Review. 2014;50:1-8.
- Kim D, Franco GA, Shigehara H, et al. Benign miliary osteoma cutis of the face: a common incidental CT finding. AJNR Am J Neuroradiol. 2017;38:789-794.
- Basu P, Erickson CP, Calame A, et al. Osteoma cutis: an adverse event following tattoo placement. Cureus. 2019;11:E4323.
- Cohen PR. Perforating osteoma cutis: case report and literature review of patients with a solitary perforating osteoma cutis lesion. Dermatol Online J. 2018;24:13030/qt6kt5n92w.
- Hong SH, Kang HY. A case of perforating osteoma cutis. Ann Dermatol. 2003;15:153-155.
- Kim BK, Ahn SK. Acquired perforating osteoma cutis. Ann Dermatol. 2015;27:452-453.
- Lippmann HI, Goldin RR. Subcutaneous ossification of the legs in chronic venous insufficiency. Radiology. 1960;74:279-288.
- Haro R, Revelles JM, Angulo J, et al. Plaque-like osteoma cutis with transepidermal elimination. J Cutan Pathol. 2009;36:591-593.
The Diagnosis: Osteoma Cutis
Osteoma cutis is the heterotopic development of cutaneous ossifications in the dermis or subcutaneous fat and presents as plaquelike, stony, hard nodules. It can manifest as either a primary or secondary condition based on the presence or absence of a prior skin insult at the lesion site. Primary osteoma cutis occurs in 15% of patients and arises either de novo or in association with any of several inflammatory, neoplastic, and metabolic diseases that provide a favorable environment for abnormal mesenchymal stem cell commitment to osteoid,1 including Albright hereditary osteodystrophy, myositis ossificans progressiva, and progressive osseous heteroplasia, which are all associated with mutations in the heterotrimeric G-protein alpha subunit encoding gene, GNAS. 1,2 It is suggested that an insufficiency of Gsα leads to uncontrolled negative regulation of nonosseous connective tissue differentiation, forming osteoid.3 Additionally, diseases involving gain-of-function mutations in the activin A receptor type 1 encoding gene, ACVR1, such as fibrodysplasia ossificans progressiva, have been associated with osteoma cutis.4 These mutations lead to decreased receptor affinity to molecular safeguards of bone morphogenetic protein signaling, ultimately contributing to progressive ectopic bone formation.5 Secondary osteoma cutis occurs in 85% of patients and develops at the site of prior skin damage due to inflammation, neoplasm, or trauma.6 It is believed that tissue damage and degeneration lead to mesenchymal stem cell proliferation and skeletogenicinducing factor recruitment forming cartilaginous tissue, later replaced by bone through endochondral ossification.7
Although osteoma cutis previously was believed to be rare, more recent radiologic studies suggest otherwise, detecting cutaneous osteomas in up to 42.1% of patients.8 Consequently, it is likely that osteoma cutis is underdiagnosed due to its subclinical nature. Our patient, however, presented with a solitary osteoma cutis with perforation of the epidermis, a rare phenomenon.9-12
A shave biopsy in our patient revealed moderate to focally marked, irregular epidermal hyperplasia with a large focus of moderate, compact, parakeratotic crust overlying the epidermis in the center of the specimen. The papillary dermis in the center of the specimen revealed large foci of dark pink to purple bone fragments surrounded by moderate lymphocytic infiltrate with few foci perforating through the overlying epidermis (Figure, A). These findings were characteristic of osteoma cutis with perforation through the overlying epidermis.
The diagnosis of osteoma cutis at the age of 62 years suggested that the lesion was not primary in association with previously described diseases. Furthermore, the lack of phenotypic features of these diseases including obesity, developmental disability, and high parathyroid hormone levels essentially excluded this possibility. The presence of the lesion on the lower extremities initially may have suggested osteoma cutis secondary to chronic venous insufficiency13; however, the absence of visible varicose veins or obvious signs of stasis disease made this unlikely. No further cutaneous disorders at or around the lesion site clinically and histologically suggested that our patient’s lesion was primary and of idiopathic nature. Dermatofibroma can present similarly in appearance but would characteristically dimple centrally when pinched. Keratoacanthoma presents with central ulceration and keratin plugging. Pilomatricoma more commonly presents on the head and neck and less frequently as a firm nodule. Lastly, prurigo nodularis more commonly presents as a symmetrically diffuse rash compared to an isolated nodule.
Osteoma cutis is a cutaneous ossification that may be primary or secondary in nature and less rare than originally thought. Workup for potentially associated inflammatory, neoplastic, and metabolic diseases should be considered in patients with this condition. Perforating osteoma cutis is a rare variant that presents as solitary or multiple nodules with central erosion and crust. The mechanism of transepidermal elimination leading to skin perforation is hypothesized to involve epidermal hyperproliferation leading to upward movement.14 Shave biopsy establishes a definitive histopathologic diagnosis and often is curative. Given that lesions of osteoma cutis themselves are benign, removal may not be necessary.
The Diagnosis: Osteoma Cutis
Osteoma cutis is the heterotopic development of cutaneous ossifications in the dermis or subcutaneous fat and presents as plaquelike, stony, hard nodules. It can manifest as either a primary or secondary condition based on the presence or absence of a prior skin insult at the lesion site. Primary osteoma cutis occurs in 15% of patients and arises either de novo or in association with any of several inflammatory, neoplastic, and metabolic diseases that provide a favorable environment for abnormal mesenchymal stem cell commitment to osteoid,1 including Albright hereditary osteodystrophy, myositis ossificans progressiva, and progressive osseous heteroplasia, which are all associated with mutations in the heterotrimeric G-protein alpha subunit encoding gene, GNAS. 1,2 It is suggested that an insufficiency of Gsα leads to uncontrolled negative regulation of nonosseous connective tissue differentiation, forming osteoid.3 Additionally, diseases involving gain-of-function mutations in the activin A receptor type 1 encoding gene, ACVR1, such as fibrodysplasia ossificans progressiva, have been associated with osteoma cutis.4 These mutations lead to decreased receptor affinity to molecular safeguards of bone morphogenetic protein signaling, ultimately contributing to progressive ectopic bone formation.5 Secondary osteoma cutis occurs in 85% of patients and develops at the site of prior skin damage due to inflammation, neoplasm, or trauma.6 It is believed that tissue damage and degeneration lead to mesenchymal stem cell proliferation and skeletogenicinducing factor recruitment forming cartilaginous tissue, later replaced by bone through endochondral ossification.7
Although osteoma cutis previously was believed to be rare, more recent radiologic studies suggest otherwise, detecting cutaneous osteomas in up to 42.1% of patients.8 Consequently, it is likely that osteoma cutis is underdiagnosed due to its subclinical nature. Our patient, however, presented with a solitary osteoma cutis with perforation of the epidermis, a rare phenomenon.9-12
A shave biopsy in our patient revealed moderate to focally marked, irregular epidermal hyperplasia with a large focus of moderate, compact, parakeratotic crust overlying the epidermis in the center of the specimen. The papillary dermis in the center of the specimen revealed large foci of dark pink to purple bone fragments surrounded by moderate lymphocytic infiltrate with few foci perforating through the overlying epidermis (Figure, A). These findings were characteristic of osteoma cutis with perforation through the overlying epidermis.
The diagnosis of osteoma cutis at the age of 62 years suggested that the lesion was not primary in association with previously described diseases. Furthermore, the lack of phenotypic features of these diseases including obesity, developmental disability, and high parathyroid hormone levels essentially excluded this possibility. The presence of the lesion on the lower extremities initially may have suggested osteoma cutis secondary to chronic venous insufficiency13; however, the absence of visible varicose veins or obvious signs of stasis disease made this unlikely. No further cutaneous disorders at or around the lesion site clinically and histologically suggested that our patient’s lesion was primary and of idiopathic nature. Dermatofibroma can present similarly in appearance but would characteristically dimple centrally when pinched. Keratoacanthoma presents with central ulceration and keratin plugging. Pilomatricoma more commonly presents on the head and neck and less frequently as a firm nodule. Lastly, prurigo nodularis more commonly presents as a symmetrically diffuse rash compared to an isolated nodule.
Osteoma cutis is a cutaneous ossification that may be primary or secondary in nature and less rare than originally thought. Workup for potentially associated inflammatory, neoplastic, and metabolic diseases should be considered in patients with this condition. Perforating osteoma cutis is a rare variant that presents as solitary or multiple nodules with central erosion and crust. The mechanism of transepidermal elimination leading to skin perforation is hypothesized to involve epidermal hyperproliferation leading to upward movement.14 Shave biopsy establishes a definitive histopathologic diagnosis and often is curative. Given that lesions of osteoma cutis themselves are benign, removal may not be necessary.
- Falsey RR, Ackerman L. Eruptive, hard cutaneous nodules in a 61-yearold woman. osteoma cutis in a patient with Albright hereditary osteodystrophy (AHO). JAMA Dermatol. 2013;149:975-976.
- Martin J, Tucker M, Browning JC. Infantile osteoma cutis as a presentation of a GNAS mutation. Pediatr Dermatol. 2012;29:483-484.
- Shore EM, Ahn J, de Beur SJ, et al. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med. 2002;346:99-106.
- Kaplan FS, Le Merrer M, Glaser DL, et al. Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol. 2008;22:191-205.
- Song GA, Kim HJ, Woo KM, et al. Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J Biol Chem. 2010;285:22542-22553.
- Roth SI, Stowell RE, Helwig EB, et al. Cutaneous ossification. report of 120 cases and review of the literature. Arch Pathol. 1963;76:44-54.
- Shimono K, Uchibe K, Kuboki T, et al. The pathophysiology of heterotopic ossification: current treatment considerations in dentistry. Japanese Dental Science Review. 2014;50:1-8.
- Kim D, Franco GA, Shigehara H, et al. Benign miliary osteoma cutis of the face: a common incidental CT finding. AJNR Am J Neuroradiol. 2017;38:789-794.
- Basu P, Erickson CP, Calame A, et al. Osteoma cutis: an adverse event following tattoo placement. Cureus. 2019;11:E4323.
- Cohen PR. Perforating osteoma cutis: case report and literature review of patients with a solitary perforating osteoma cutis lesion. Dermatol Online J. 2018;24:13030/qt6kt5n92w.
- Hong SH, Kang HY. A case of perforating osteoma cutis. Ann Dermatol. 2003;15:153-155.
- Kim BK, Ahn SK. Acquired perforating osteoma cutis. Ann Dermatol. 2015;27:452-453.
- Lippmann HI, Goldin RR. Subcutaneous ossification of the legs in chronic venous insufficiency. Radiology. 1960;74:279-288.
- Haro R, Revelles JM, Angulo J, et al. Plaque-like osteoma cutis with transepidermal elimination. J Cutan Pathol. 2009;36:591-593.
- Falsey RR, Ackerman L. Eruptive, hard cutaneous nodules in a 61-yearold woman. osteoma cutis in a patient with Albright hereditary osteodystrophy (AHO). JAMA Dermatol. 2013;149:975-976.
- Martin J, Tucker M, Browning JC. Infantile osteoma cutis as a presentation of a GNAS mutation. Pediatr Dermatol. 2012;29:483-484.
- Shore EM, Ahn J, de Beur SJ, et al. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med. 2002;346:99-106.
- Kaplan FS, Le Merrer M, Glaser DL, et al. Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol. 2008;22:191-205.
- Song GA, Kim HJ, Woo KM, et al. Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J Biol Chem. 2010;285:22542-22553.
- Roth SI, Stowell RE, Helwig EB, et al. Cutaneous ossification. report of 120 cases and review of the literature. Arch Pathol. 1963;76:44-54.
- Shimono K, Uchibe K, Kuboki T, et al. The pathophysiology of heterotopic ossification: current treatment considerations in dentistry. Japanese Dental Science Review. 2014;50:1-8.
- Kim D, Franco GA, Shigehara H, et al. Benign miliary osteoma cutis of the face: a common incidental CT finding. AJNR Am J Neuroradiol. 2017;38:789-794.
- Basu P, Erickson CP, Calame A, et al. Osteoma cutis: an adverse event following tattoo placement. Cureus. 2019;11:E4323.
- Cohen PR. Perforating osteoma cutis: case report and literature review of patients with a solitary perforating osteoma cutis lesion. Dermatol Online J. 2018;24:13030/qt6kt5n92w.
- Hong SH, Kang HY. A case of perforating osteoma cutis. Ann Dermatol. 2003;15:153-155.
- Kim BK, Ahn SK. Acquired perforating osteoma cutis. Ann Dermatol. 2015;27:452-453.
- Lippmann HI, Goldin RR. Subcutaneous ossification of the legs in chronic venous insufficiency. Radiology. 1960;74:279-288.
- Haro R, Revelles JM, Angulo J, et al. Plaque-like osteoma cutis with transepidermal elimination. J Cutan Pathol. 2009;36:591-593.
![]()
A 62-year-old woman presented with an irregular, erythematous, 4-mm nodule with central erosions on the left proximal calf of 2 months’ duration. The patient had a history of actinic keratoses and dysplastic nevi. She had no other notable medical history. She was not taking any medications and reported no history of trauma to the area. A shave biopsy of the lesion (encircled by black ink) was performed.
Linear Violaceous Papules in a Child
The Diagnosis: Linear Lichen Planus
The patient was clinically diagnosed with linear lichen planus and was started on betamethasone dipropionate ointment 0.05% applied once daily with improvement in both the pruritus and appearance at 4-month follow-up. A biopsy was deferred based on the parents’ wishes.
Lichen planus is an inflammatory disorder involving the skin and oral mucosa. Cutaneous lichen planus classically presents as flat-topped, violaceous, pruritic, polygonal papules with overlying fine white or grey lines known as Wickham striae.1 Postinflammatory hyperpigmentation is common, especially in patients with darker skin tones. Expected histologic findings include orthokeratosis, apoptotic keratinocytes, and bandlike lymphocytic infiltration at the dermoepidermal junction.1
An estimated 5% of cases of cutaneous lichen planus occur in children.2 A study of 316 children with lichen planus demonstrated that the classic morphology remained the most common presentation, while the linear variant was present in only 6.9% of pediatric cases.3 Linear lichen planus appears to be more common among children than adults. A study of 36 pediatric cases showed a greater representation of lichen planus in Black children (67% affected vs 21% cohort).2
Cutaneous lichen planus often clears spontaneously in approximately 1 year.4 Treatment in children primarily is focused on shortening the time to resolution and relieving pruritus, with topical corticosteroids as firstline therapy.3,4 Oral corticosteroids have a faster clinical response; greater efficacy; and more effectively prevent residual hyperpigmentation, which is especially relevant in individuals with darker skin.3 Nonetheless, oral corticosteroids are considered a second-line treatment due to their unfavorable side-effect profile. Additional treatment options include oral aromatic retinoids (acitretin) and phototherapy.3
Incontinentia pigmenti is characterized by a defect in the inhibitor of nuclear factor–κB kinase regulatory subunit gamma, IKBKG, gene on the X chromosome. Incontinentia pigmenti usually is lethal in males; in females, it leads to ectodermal dysplasia associated with skin findings in a blaschkoid distribution occurring in 4 stages.5 The verrucous stage is preceded by the vesicular stage and expected to occur within the first few months of life, making it unlikely in our 5-year-old patient. Inflammatory linear verrucous epidermal nevus usually occurs in children younger than 5 years and is characterized by psoriasiform papules coalescing into a plaque with substantial scale instead of Wickham striae, as seen in our patient.6 Lichen striatus consists of smaller, pink to flesh-colored papules that rarely are pruritic.7 It is more common among atopic individuals and is associated with postinflammatory hypopigmentation.8 Linear psoriasis presents similarly to inflammatory linear verrucous epidermal nevus, with greater erythema and scale compared to the fine lacy Wickham striae that were seen in our patient.8
- Tziotzios C, Lee JYW, Brier T, et al. Lichen planus and lichenoid dermatoses: clinical overview and molecular basis. J Am Acad Dermatol. 2018;79:789-804.
- Walton KE, Bowers EV, Drolet BA, et al. Childhood lichen planus: demographics of a U.S. population. Pediatr Dermatol. 2010;27:34-38.
- Pandhi D, Singal A, Bhattacharya SN. Lichen planus in childhood: a series of 316 patients. Pediatr Dermatol. 2014;31:59-67.
- Le Cleach L, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;366:723-732.
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:E45-E52.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1894-1916.
- Payette MJ, Weston G, Humphrey S, et al. Lichen planus and other lichenoid dermatoses: kids are not just little people. Clin Dermatol. 2015;33:631-643.
- Moss C, Browne F. Mosaicism and linear lesions. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1894-1916.
The Diagnosis: Linear Lichen Planus
The patient was clinically diagnosed with linear lichen planus and was started on betamethasone dipropionate ointment 0.05% applied once daily with improvement in both the pruritus and appearance at 4-month follow-up. A biopsy was deferred based on the parents’ wishes.
Lichen planus is an inflammatory disorder involving the skin and oral mucosa. Cutaneous lichen planus classically presents as flat-topped, violaceous, pruritic, polygonal papules with overlying fine white or grey lines known as Wickham striae.1 Postinflammatory hyperpigmentation is common, especially in patients with darker skin tones. Expected histologic findings include orthokeratosis, apoptotic keratinocytes, and bandlike lymphocytic infiltration at the dermoepidermal junction.1
An estimated 5% of cases of cutaneous lichen planus occur in children.2 A study of 316 children with lichen planus demonstrated that the classic morphology remained the most common presentation, while the linear variant was present in only 6.9% of pediatric cases.3 Linear lichen planus appears to be more common among children than adults. A study of 36 pediatric cases showed a greater representation of lichen planus in Black children (67% affected vs 21% cohort).2
Cutaneous lichen planus often clears spontaneously in approximately 1 year.4 Treatment in children primarily is focused on shortening the time to resolution and relieving pruritus, with topical corticosteroids as firstline therapy.3,4 Oral corticosteroids have a faster clinical response; greater efficacy; and more effectively prevent residual hyperpigmentation, which is especially relevant in individuals with darker skin.3 Nonetheless, oral corticosteroids are considered a second-line treatment due to their unfavorable side-effect profile. Additional treatment options include oral aromatic retinoids (acitretin) and phototherapy.3
Incontinentia pigmenti is characterized by a defect in the inhibitor of nuclear factor–κB kinase regulatory subunit gamma, IKBKG, gene on the X chromosome. Incontinentia pigmenti usually is lethal in males; in females, it leads to ectodermal dysplasia associated with skin findings in a blaschkoid distribution occurring in 4 stages.5 The verrucous stage is preceded by the vesicular stage and expected to occur within the first few months of life, making it unlikely in our 5-year-old patient. Inflammatory linear verrucous epidermal nevus usually occurs in children younger than 5 years and is characterized by psoriasiform papules coalescing into a plaque with substantial scale instead of Wickham striae, as seen in our patient.6 Lichen striatus consists of smaller, pink to flesh-colored papules that rarely are pruritic.7 It is more common among atopic individuals and is associated with postinflammatory hypopigmentation.8 Linear psoriasis presents similarly to inflammatory linear verrucous epidermal nevus, with greater erythema and scale compared to the fine lacy Wickham striae that were seen in our patient.8
The Diagnosis: Linear Lichen Planus
The patient was clinically diagnosed with linear lichen planus and was started on betamethasone dipropionate ointment 0.05% applied once daily with improvement in both the pruritus and appearance at 4-month follow-up. A biopsy was deferred based on the parents’ wishes.
Lichen planus is an inflammatory disorder involving the skin and oral mucosa. Cutaneous lichen planus classically presents as flat-topped, violaceous, pruritic, polygonal papules with overlying fine white or grey lines known as Wickham striae.1 Postinflammatory hyperpigmentation is common, especially in patients with darker skin tones. Expected histologic findings include orthokeratosis, apoptotic keratinocytes, and bandlike lymphocytic infiltration at the dermoepidermal junction.1
An estimated 5% of cases of cutaneous lichen planus occur in children.2 A study of 316 children with lichen planus demonstrated that the classic morphology remained the most common presentation, while the linear variant was present in only 6.9% of pediatric cases.3 Linear lichen planus appears to be more common among children than adults. A study of 36 pediatric cases showed a greater representation of lichen planus in Black children (67% affected vs 21% cohort).2
Cutaneous lichen planus often clears spontaneously in approximately 1 year.4 Treatment in children primarily is focused on shortening the time to resolution and relieving pruritus, with topical corticosteroids as firstline therapy.3,4 Oral corticosteroids have a faster clinical response; greater efficacy; and more effectively prevent residual hyperpigmentation, which is especially relevant in individuals with darker skin.3 Nonetheless, oral corticosteroids are considered a second-line treatment due to their unfavorable side-effect profile. Additional treatment options include oral aromatic retinoids (acitretin) and phototherapy.3
Incontinentia pigmenti is characterized by a defect in the inhibitor of nuclear factor–κB kinase regulatory subunit gamma, IKBKG, gene on the X chromosome. Incontinentia pigmenti usually is lethal in males; in females, it leads to ectodermal dysplasia associated with skin findings in a blaschkoid distribution occurring in 4 stages.5 The verrucous stage is preceded by the vesicular stage and expected to occur within the first few months of life, making it unlikely in our 5-year-old patient. Inflammatory linear verrucous epidermal nevus usually occurs in children younger than 5 years and is characterized by psoriasiform papules coalescing into a plaque with substantial scale instead of Wickham striae, as seen in our patient.6 Lichen striatus consists of smaller, pink to flesh-colored papules that rarely are pruritic.7 It is more common among atopic individuals and is associated with postinflammatory hypopigmentation.8 Linear psoriasis presents similarly to inflammatory linear verrucous epidermal nevus, with greater erythema and scale compared to the fine lacy Wickham striae that were seen in our patient.8
- Tziotzios C, Lee JYW, Brier T, et al. Lichen planus and lichenoid dermatoses: clinical overview and molecular basis. J Am Acad Dermatol. 2018;79:789-804.
- Walton KE, Bowers EV, Drolet BA, et al. Childhood lichen planus: demographics of a U.S. population. Pediatr Dermatol. 2010;27:34-38.
- Pandhi D, Singal A, Bhattacharya SN. Lichen planus in childhood: a series of 316 patients. Pediatr Dermatol. 2014;31:59-67.
- Le Cleach L, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;366:723-732.
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:E45-E52.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1894-1916.
- Payette MJ, Weston G, Humphrey S, et al. Lichen planus and other lichenoid dermatoses: kids are not just little people. Clin Dermatol. 2015;33:631-643.
- Moss C, Browne F. Mosaicism and linear lesions. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1894-1916.
- Tziotzios C, Lee JYW, Brier T, et al. Lichen planus and lichenoid dermatoses: clinical overview and molecular basis. J Am Acad Dermatol. 2018;79:789-804.
- Walton KE, Bowers EV, Drolet BA, et al. Childhood lichen planus: demographics of a U.S. population. Pediatr Dermatol. 2010;27:34-38.
- Pandhi D, Singal A, Bhattacharya SN. Lichen planus in childhood: a series of 316 patients. Pediatr Dermatol. 2014;31:59-67.
- Le Cleach L, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;366:723-732.
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:E45-E52.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1894-1916.
- Payette MJ, Weston G, Humphrey S, et al. Lichen planus and other lichenoid dermatoses: kids are not just little people. Clin Dermatol. 2015;33:631-643.
- Moss C, Browne F. Mosaicism and linear lesions. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1894-1916.
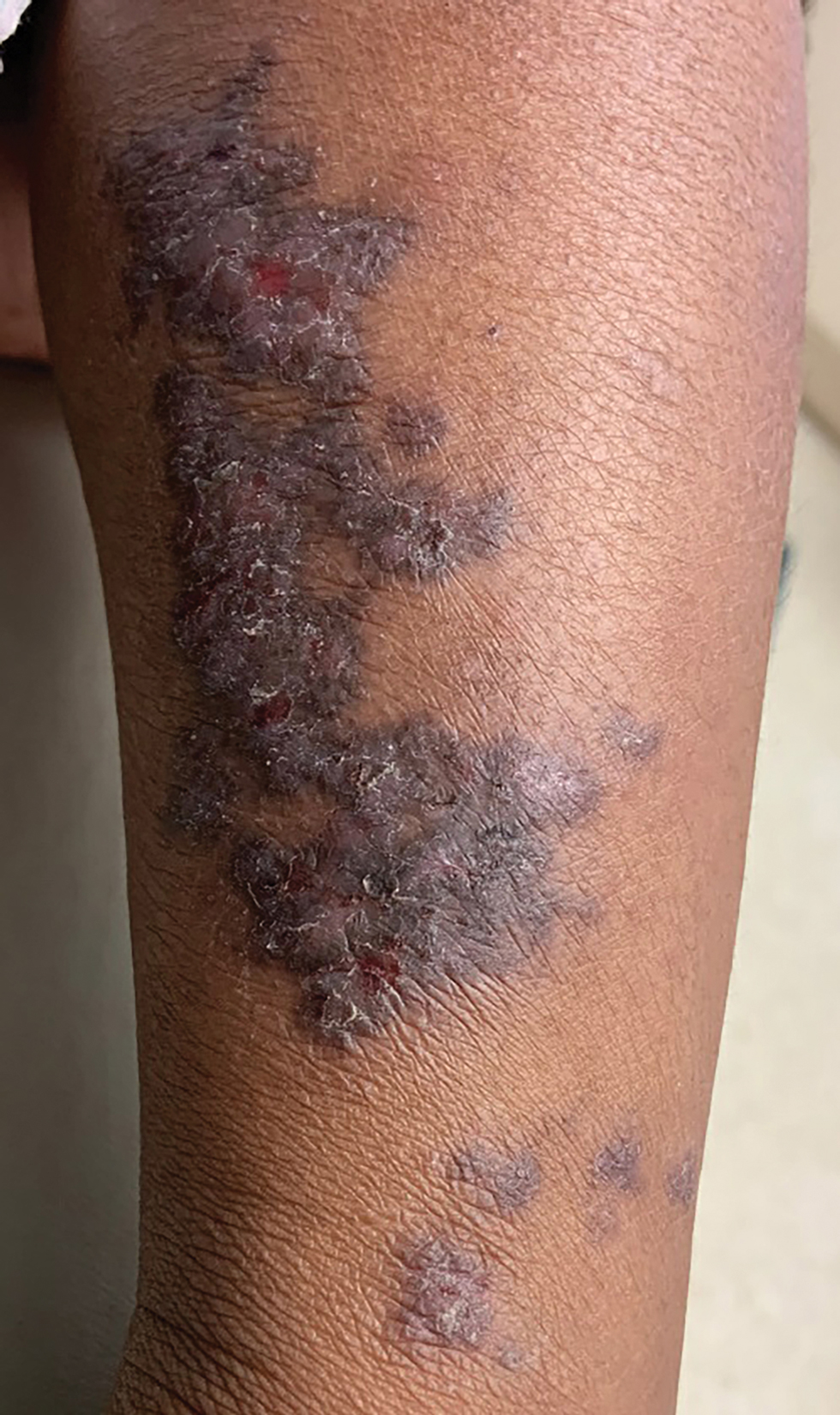
A 5-year-old Black girl presented to the dermatology clinic with a stable pruritic eruption on the right leg of 1 month’s duration. Over-the-counter hydrocortisone cream was applied for 3 days with no response. Physical examination revealed grouped, flat-topped, violaceous papules coalescing into plaques with overlying lacy white striae along the right lower leg, wrapping around to the right dorsal foot in a blaschkoid distribution. The patient was otherwise healthy and up-to-date on immunizations and had an unremarkable birth history.
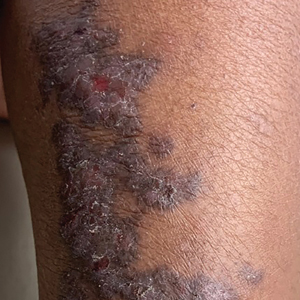
Ulcer on the Leg
The Diagnosis: Calcinosis Cutis Due to Systemic Sclerosis Sine Scleroderma
Laboratory evaluation was notable for high titers of antinuclear antibodies (>1/320; reference range, 0–1/80) and positive anticentromere antibodies. There were no other relevant laboratory findings; phosphocalcic metabolism was within normal limits, and urinary sediment was normal. Biopsy of the edge of the ulcer revealed basophilic material compatible with calcium deposits. In a 3D volume rendering reconstruction from the lower limb scanner, grouped calcifications were observed in subcutaneous cellular tissue near the ulcer (Figure). The patient had a restrictive ventilatory pattern observed in a pulmonary function test. An esophageal motility study was normal.
The patient was diagnosed with systemic sclerosis sine scleroderma (ssSSc) type II because she met the 4 criteria established by Poormoghim et al1 : (1) Raynaud phenomenon or a peripheral vascular equivalent (ie, digital pitting scars, digital-tip ulcers, digital-tip gangrene, abnormal nail fold capillaries); (2) positive antinuclear antibodies; (3) distal esophageal hypomotility, small bowel hypomotility, pulmonary interstitial fibrosis, primary pulmonary arterial hypertension (without fibrosis), cardiac involvement typical of scleroderma, or renal failure; and (4) no other defined connective tissue or other disease as a cause of the prior conditions.
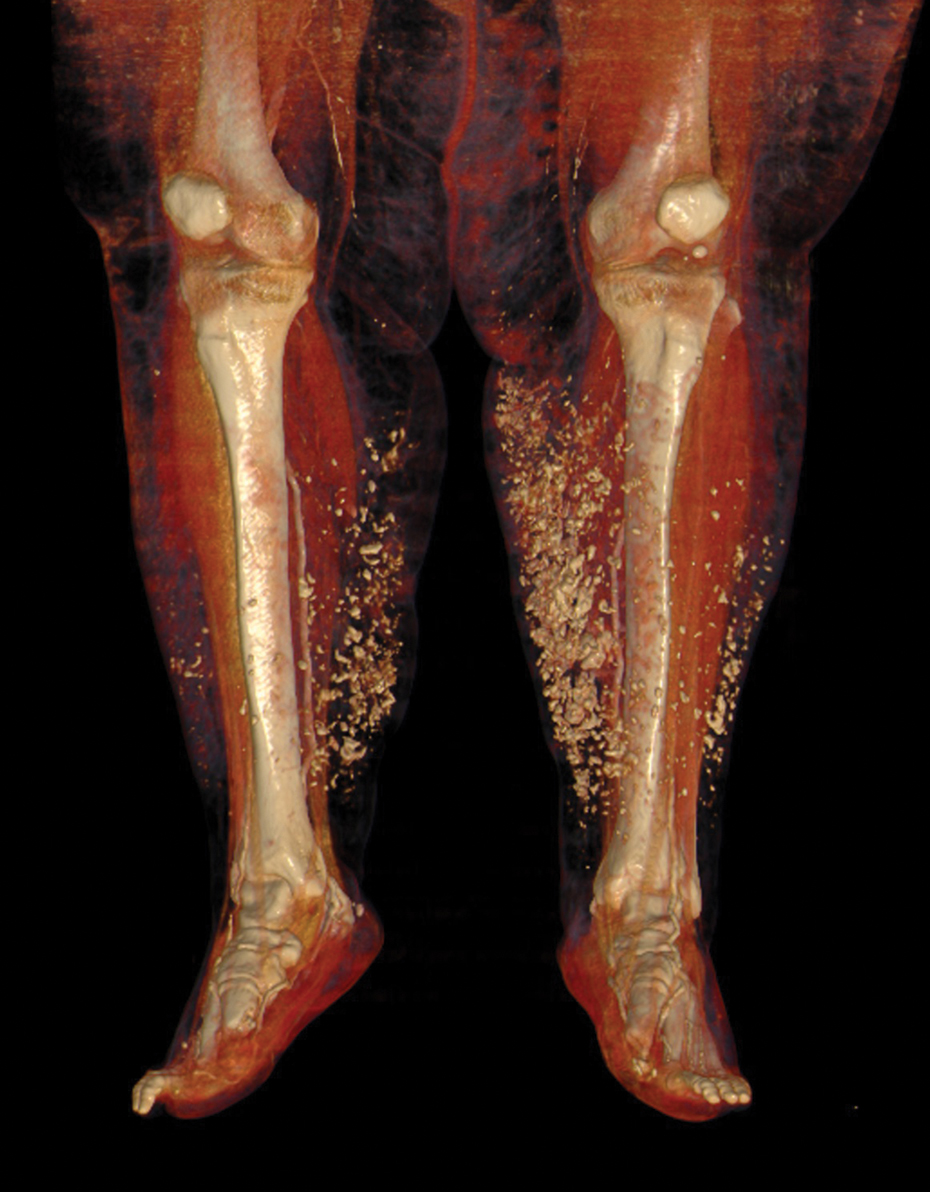
Systemic sclerosis is a chronic disease characterized by progressive fibrosis of the skin and other internal organs—especially the lungs, kidneys, digestive tract, and heart—as well as generalized vascular dysfunction. Cutaneous induration is its hallmark; however, up to 10% of affected patients have ssSSc.2 This entity is characterized by the total or partial absence of cutaneous manifestations of systemic sclerosis with the occurrence of internal organ involvement and serologic abnormalities. There are 3 types of ssSSc depending on the grade of skin involvement. Type I is characterized by the lack of any typical cutaneous stigmata of the disease. Type II is without sclerodactyly but can coexist with other cutaneous findings such as calcifications, telangiectases, or pitting scars. Type III is characterized clinically by internal organ involvement, typical of systemic sclerosis, that has appeared before skin changes.2
An abnormal deposit of calcium in the cutaneous and subcutaneous tissue is called calcinosis cutis. There are 5 subtypes of calcinosis cutis: dystrophic, metastatic, idiopathic, iatrogenic, and calciphylaxis. Dystrophic skin calcifications may appear in patients with connective tissue diseases such as dermatomyositis or systemic sclerosis.3 Up to 25% of patients with systemic sclerosis can develop calcinosis cutis due to local tissue damage, with normal phosphocalcic metabolism.3
Calcinosis cutis is more common in patients with systemic sclerosis and positive anticentromere antibodies.4 The calcifications usually are located in areas that are subject to repeated trauma, such as the fingers or arms, though other locations have been described such as cervical, paraspinal, or on the hips.5,6 Our patient developed calcifications on both legs, which represent atypical areas for this process.
Dermatomyositis also can present with calcinosis cutis. There are 4 patterns of calcification: superficial nodulelike calcified masses; deep calcified masses; deep sheetlike calcifications within the fascial planes; and a rare, diffuse, superficial lacy and reticular calcification that involves almost the entire body surface area.7 Patients with calcinosis cutis secondary to dermatomyositis usually develop proximal muscle weakness, high titers of creatine kinase, heliotrope rash, or interstitial lung disease with specific antibodies.
Calciphylaxis is a serious disorder involving the calcification of dermal and subcutaneous arterioles and capillaries. It presents with painful cutaneous areas of necrosis.
Venous ulcers also can present with secondary dystrophic calcification due to local tissue damage. These patients usually have cutaneous signs of chronic venous insufficiency. Our patient denied prior trauma to the area; therefore, a traumatic ulcer with secondary calcification was ruled out.
The most concerning complication of calcinosis cutis is the development of ulcers, which occurred in 154 of 316 calcinoses (48.7%) in patients with systemic sclerosis and secondary calcifications.8 These ulcers can cause disabling pain or become superinfected, as in our patient.
There currently is no drug capable of removing dystrophic calcifications, but diltiazem, minocycline, or colchicine can reduce their size and prevent their progression. In the event of neurologic compromise or intractable pain, the treatment of choice is surgical removal of the calcification.9 Curettage, intralesional sodium thiosulfate, and intravenous sodium thiosulfate also have been suggested as therapeutic options.10 Antibiotic treatment was carried out in our patient, which controlled the superinfection of the ulcers. Diltiazem also was started, with stabilization of the calcium deposits without a reduction in their size.
There are few studies evaluating the presence of nondigital ulcers in patients with systemic sclerosis. Shanmugam et al11 calculated a 4% (N=249) prevalence of ulcers in the lower limbs of systemic sclerosis patients. In a study by Bohelay et al12 of 45 patients, the estimated prevalence of lower limb ulcers was 12.8%, and the etiologies consisted of 22 cases of venous insufficiency (49%), 21 cases of ischemic causes (47%), and 2 cases of other causes (4%).
We present the case of a woman with ssSSc who developed dystrophic calcinosis cutis in atypical areas with secondary ulceration and superinfection. The skin usually plays a key role in the diagnosis of systemic sclerosis, as sclerodactyly and the characteristic generalized skin induration stand out in affected individuals. Although our patient was diagnosed with ssSSc, her skin manifestations also were crucial for the diagnosis, as she had ulcers on the lower limbs.
- Poormoghim H, Lucas M, Fertig N, et al. Systemic sclerosis sine scleroderma: demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum. 2000;43:444-451.
- Kucharz EJ, Kopec´-Me˛ drek M. Systemic sclerosis sine scleroderma. Adv Clin Exp Med. 2017;26:875-880.
- Valenzuela A, Baron M, Herrick AL, et al. Calcinosis is associated with digital ulcers and osteoporosis in patients with systemic sclerosis: a scleroderma clinical trials consortium study. Semin Arthritis Rheum. 2016;46:344-349.
- D’Aoust J, Hudson M, Tatibouet S, et al. Clinical and serologic correlates of antiPM/Scl antibodies in systemic sclerosis: a multicenter study of 763 patients. Arthritis Rheum. 2014;66:1608-1615.
- Contreras I, Sallés M, Mínguez S, et al. Hard paracervical tumor in a patient with limited systemic sclerosis. Rheumatol Clin. 2014; 10:336-337.
- Meriglier E, Lafourcade F, Gombert B, et al. Giant calcinosis revealing systemic sclerosis. Int J Rheum Dis. 2019;22:1787-1788.
- Chung CH. Calcinosis universalis in juvenile dermatomyositis [published online September 24, 2020]. Chonnam Med J. 2020;56:212-213.
- Bartoli F, Fiori G, Braschi F, et al. Calcinosis in systemic sclerosis: subsets, distribution and complications [published online May 30, 2016]. Rheumatology (Oxford). 2016;55:1610-1614.
- Jung H, Lee D, Cho J, et al. Surgical treatment of extensive tumoral calcinosis associated with systemic sclerosis. Korean J Thorac Cardiovasc Surg. 2015;48:151-154.
- Badawi AH, Patel V, Warner AE, et al. Dystrophic calcinosis cutis: treatment with intravenous sodium thiosulfate. Cutis. 2020;106:E15-E17.
- Shanmugam V, Price P, Attinger C, et al. Lower extremity ulcers in systemic sclerosis: features and response to therapy [published online August 18, 2010]. Int J Rheumatol. doi:10.1155/2010/747946
- Bohelay G, Blaise S, Levy P, et al. Lower-limb ulcers in systemic sclerosis: a multicentre retrospective case-control study. Acta Derm Venereol. 2018;98:677-682.
The Diagnosis: Calcinosis Cutis Due to Systemic Sclerosis Sine Scleroderma
Laboratory evaluation was notable for high titers of antinuclear antibodies (>1/320; reference range, 0–1/80) and positive anticentromere antibodies. There were no other relevant laboratory findings; phosphocalcic metabolism was within normal limits, and urinary sediment was normal. Biopsy of the edge of the ulcer revealed basophilic material compatible with calcium deposits. In a 3D volume rendering reconstruction from the lower limb scanner, grouped calcifications were observed in subcutaneous cellular tissue near the ulcer (Figure). The patient had a restrictive ventilatory pattern observed in a pulmonary function test. An esophageal motility study was normal.
The patient was diagnosed with systemic sclerosis sine scleroderma (ssSSc) type II because she met the 4 criteria established by Poormoghim et al1 : (1) Raynaud phenomenon or a peripheral vascular equivalent (ie, digital pitting scars, digital-tip ulcers, digital-tip gangrene, abnormal nail fold capillaries); (2) positive antinuclear antibodies; (3) distal esophageal hypomotility, small bowel hypomotility, pulmonary interstitial fibrosis, primary pulmonary arterial hypertension (without fibrosis), cardiac involvement typical of scleroderma, or renal failure; and (4) no other defined connective tissue or other disease as a cause of the prior conditions.
Systemic sclerosis is a chronic disease characterized by progressive fibrosis of the skin and other internal organs—especially the lungs, kidneys, digestive tract, and heart—as well as generalized vascular dysfunction. Cutaneous induration is its hallmark; however, up to 10% of affected patients have ssSSc.2 This entity is characterized by the total or partial absence of cutaneous manifestations of systemic sclerosis with the occurrence of internal organ involvement and serologic abnormalities. There are 3 types of ssSSc depending on the grade of skin involvement. Type I is characterized by the lack of any typical cutaneous stigmata of the disease. Type II is without sclerodactyly but can coexist with other cutaneous findings such as calcifications, telangiectases, or pitting scars. Type III is characterized clinically by internal organ involvement, typical of systemic sclerosis, that has appeared before skin changes.2
An abnormal deposit of calcium in the cutaneous and subcutaneous tissue is called calcinosis cutis. There are 5 subtypes of calcinosis cutis: dystrophic, metastatic, idiopathic, iatrogenic, and calciphylaxis. Dystrophic skin calcifications may appear in patients with connective tissue diseases such as dermatomyositis or systemic sclerosis.3 Up to 25% of patients with systemic sclerosis can develop calcinosis cutis due to local tissue damage, with normal phosphocalcic metabolism.3
Calcinosis cutis is more common in patients with systemic sclerosis and positive anticentromere antibodies.4 The calcifications usually are located in areas that are subject to repeated trauma, such as the fingers or arms, though other locations have been described such as cervical, paraspinal, or on the hips.5,6 Our patient developed calcifications on both legs, which represent atypical areas for this process.
Dermatomyositis also can present with calcinosis cutis. There are 4 patterns of calcification: superficial nodulelike calcified masses; deep calcified masses; deep sheetlike calcifications within the fascial planes; and a rare, diffuse, superficial lacy and reticular calcification that involves almost the entire body surface area.7 Patients with calcinosis cutis secondary to dermatomyositis usually develop proximal muscle weakness, high titers of creatine kinase, heliotrope rash, or interstitial lung disease with specific antibodies.
Calciphylaxis is a serious disorder involving the calcification of dermal and subcutaneous arterioles and capillaries. It presents with painful cutaneous areas of necrosis.
Venous ulcers also can present with secondary dystrophic calcification due to local tissue damage. These patients usually have cutaneous signs of chronic venous insufficiency. Our patient denied prior trauma to the area; therefore, a traumatic ulcer with secondary calcification was ruled out.
The most concerning complication of calcinosis cutis is the development of ulcers, which occurred in 154 of 316 calcinoses (48.7%) in patients with systemic sclerosis and secondary calcifications.8 These ulcers can cause disabling pain or become superinfected, as in our patient.
There currently is no drug capable of removing dystrophic calcifications, but diltiazem, minocycline, or colchicine can reduce their size and prevent their progression. In the event of neurologic compromise or intractable pain, the treatment of choice is surgical removal of the calcification.9 Curettage, intralesional sodium thiosulfate, and intravenous sodium thiosulfate also have been suggested as therapeutic options.10 Antibiotic treatment was carried out in our patient, which controlled the superinfection of the ulcers. Diltiazem also was started, with stabilization of the calcium deposits without a reduction in their size.
There are few studies evaluating the presence of nondigital ulcers in patients with systemic sclerosis. Shanmugam et al11 calculated a 4% (N=249) prevalence of ulcers in the lower limbs of systemic sclerosis patients. In a study by Bohelay et al12 of 45 patients, the estimated prevalence of lower limb ulcers was 12.8%, and the etiologies consisted of 22 cases of venous insufficiency (49%), 21 cases of ischemic causes (47%), and 2 cases of other causes (4%).
We present the case of a woman with ssSSc who developed dystrophic calcinosis cutis in atypical areas with secondary ulceration and superinfection. The skin usually plays a key role in the diagnosis of systemic sclerosis, as sclerodactyly and the characteristic generalized skin induration stand out in affected individuals. Although our patient was diagnosed with ssSSc, her skin manifestations also were crucial for the diagnosis, as she had ulcers on the lower limbs.
The Diagnosis: Calcinosis Cutis Due to Systemic Sclerosis Sine Scleroderma
Laboratory evaluation was notable for high titers of antinuclear antibodies (>1/320; reference range, 0–1/80) and positive anticentromere antibodies. There were no other relevant laboratory findings; phosphocalcic metabolism was within normal limits, and urinary sediment was normal. Biopsy of the edge of the ulcer revealed basophilic material compatible with calcium deposits. In a 3D volume rendering reconstruction from the lower limb scanner, grouped calcifications were observed in subcutaneous cellular tissue near the ulcer (Figure). The patient had a restrictive ventilatory pattern observed in a pulmonary function test. An esophageal motility study was normal.
The patient was diagnosed with systemic sclerosis sine scleroderma (ssSSc) type II because she met the 4 criteria established by Poormoghim et al1 : (1) Raynaud phenomenon or a peripheral vascular equivalent (ie, digital pitting scars, digital-tip ulcers, digital-tip gangrene, abnormal nail fold capillaries); (2) positive antinuclear antibodies; (3) distal esophageal hypomotility, small bowel hypomotility, pulmonary interstitial fibrosis, primary pulmonary arterial hypertension (without fibrosis), cardiac involvement typical of scleroderma, or renal failure; and (4) no other defined connective tissue or other disease as a cause of the prior conditions.
Systemic sclerosis is a chronic disease characterized by progressive fibrosis of the skin and other internal organs—especially the lungs, kidneys, digestive tract, and heart—as well as generalized vascular dysfunction. Cutaneous induration is its hallmark; however, up to 10% of affected patients have ssSSc.2 This entity is characterized by the total or partial absence of cutaneous manifestations of systemic sclerosis with the occurrence of internal organ involvement and serologic abnormalities. There are 3 types of ssSSc depending on the grade of skin involvement. Type I is characterized by the lack of any typical cutaneous stigmata of the disease. Type II is without sclerodactyly but can coexist with other cutaneous findings such as calcifications, telangiectases, or pitting scars. Type III is characterized clinically by internal organ involvement, typical of systemic sclerosis, that has appeared before skin changes.2
An abnormal deposit of calcium in the cutaneous and subcutaneous tissue is called calcinosis cutis. There are 5 subtypes of calcinosis cutis: dystrophic, metastatic, idiopathic, iatrogenic, and calciphylaxis. Dystrophic skin calcifications may appear in patients with connective tissue diseases such as dermatomyositis or systemic sclerosis.3 Up to 25% of patients with systemic sclerosis can develop calcinosis cutis due to local tissue damage, with normal phosphocalcic metabolism.3
Calcinosis cutis is more common in patients with systemic sclerosis and positive anticentromere antibodies.4 The calcifications usually are located in areas that are subject to repeated trauma, such as the fingers or arms, though other locations have been described such as cervical, paraspinal, or on the hips.5,6 Our patient developed calcifications on both legs, which represent atypical areas for this process.
Dermatomyositis also can present with calcinosis cutis. There are 4 patterns of calcification: superficial nodulelike calcified masses; deep calcified masses; deep sheetlike calcifications within the fascial planes; and a rare, diffuse, superficial lacy and reticular calcification that involves almost the entire body surface area.7 Patients with calcinosis cutis secondary to dermatomyositis usually develop proximal muscle weakness, high titers of creatine kinase, heliotrope rash, or interstitial lung disease with specific antibodies.
Calciphylaxis is a serious disorder involving the calcification of dermal and subcutaneous arterioles and capillaries. It presents with painful cutaneous areas of necrosis.
Venous ulcers also can present with secondary dystrophic calcification due to local tissue damage. These patients usually have cutaneous signs of chronic venous insufficiency. Our patient denied prior trauma to the area; therefore, a traumatic ulcer with secondary calcification was ruled out.
The most concerning complication of calcinosis cutis is the development of ulcers, which occurred in 154 of 316 calcinoses (48.7%) in patients with systemic sclerosis and secondary calcifications.8 These ulcers can cause disabling pain or become superinfected, as in our patient.
There currently is no drug capable of removing dystrophic calcifications, but diltiazem, minocycline, or colchicine can reduce their size and prevent their progression. In the event of neurologic compromise or intractable pain, the treatment of choice is surgical removal of the calcification.9 Curettage, intralesional sodium thiosulfate, and intravenous sodium thiosulfate also have been suggested as therapeutic options.10 Antibiotic treatment was carried out in our patient, which controlled the superinfection of the ulcers. Diltiazem also was started, with stabilization of the calcium deposits without a reduction in their size.
There are few studies evaluating the presence of nondigital ulcers in patients with systemic sclerosis. Shanmugam et al11 calculated a 4% (N=249) prevalence of ulcers in the lower limbs of systemic sclerosis patients. In a study by Bohelay et al12 of 45 patients, the estimated prevalence of lower limb ulcers was 12.8%, and the etiologies consisted of 22 cases of venous insufficiency (49%), 21 cases of ischemic causes (47%), and 2 cases of other causes (4%).
We present the case of a woman with ssSSc who developed dystrophic calcinosis cutis in atypical areas with secondary ulceration and superinfection. The skin usually plays a key role in the diagnosis of systemic sclerosis, as sclerodactyly and the characteristic generalized skin induration stand out in affected individuals. Although our patient was diagnosed with ssSSc, her skin manifestations also were crucial for the diagnosis, as she had ulcers on the lower limbs.
- Poormoghim H, Lucas M, Fertig N, et al. Systemic sclerosis sine scleroderma: demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum. 2000;43:444-451.
- Kucharz EJ, Kopec´-Me˛ drek M. Systemic sclerosis sine scleroderma. Adv Clin Exp Med. 2017;26:875-880.
- Valenzuela A, Baron M, Herrick AL, et al. Calcinosis is associated with digital ulcers and osteoporosis in patients with systemic sclerosis: a scleroderma clinical trials consortium study. Semin Arthritis Rheum. 2016;46:344-349.
- D’Aoust J, Hudson M, Tatibouet S, et al. Clinical and serologic correlates of antiPM/Scl antibodies in systemic sclerosis: a multicenter study of 763 patients. Arthritis Rheum. 2014;66:1608-1615.
- Contreras I, Sallés M, Mínguez S, et al. Hard paracervical tumor in a patient with limited systemic sclerosis. Rheumatol Clin. 2014; 10:336-337.
- Meriglier E, Lafourcade F, Gombert B, et al. Giant calcinosis revealing systemic sclerosis. Int J Rheum Dis. 2019;22:1787-1788.
- Chung CH. Calcinosis universalis in juvenile dermatomyositis [published online September 24, 2020]. Chonnam Med J. 2020;56:212-213.
- Bartoli F, Fiori G, Braschi F, et al. Calcinosis in systemic sclerosis: subsets, distribution and complications [published online May 30, 2016]. Rheumatology (Oxford). 2016;55:1610-1614.
- Jung H, Lee D, Cho J, et al. Surgical treatment of extensive tumoral calcinosis associated with systemic sclerosis. Korean J Thorac Cardiovasc Surg. 2015;48:151-154.
- Badawi AH, Patel V, Warner AE, et al. Dystrophic calcinosis cutis: treatment with intravenous sodium thiosulfate. Cutis. 2020;106:E15-E17.
- Shanmugam V, Price P, Attinger C, et al. Lower extremity ulcers in systemic sclerosis: features and response to therapy [published online August 18, 2010]. Int J Rheumatol. doi:10.1155/2010/747946
- Bohelay G, Blaise S, Levy P, et al. Lower-limb ulcers in systemic sclerosis: a multicentre retrospective case-control study. Acta Derm Venereol. 2018;98:677-682.
- Poormoghim H, Lucas M, Fertig N, et al. Systemic sclerosis sine scleroderma: demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum. 2000;43:444-451.
- Kucharz EJ, Kopec´-Me˛ drek M. Systemic sclerosis sine scleroderma. Adv Clin Exp Med. 2017;26:875-880.
- Valenzuela A, Baron M, Herrick AL, et al. Calcinosis is associated with digital ulcers and osteoporosis in patients with systemic sclerosis: a scleroderma clinical trials consortium study. Semin Arthritis Rheum. 2016;46:344-349.
- D’Aoust J, Hudson M, Tatibouet S, et al. Clinical and serologic correlates of antiPM/Scl antibodies in systemic sclerosis: a multicenter study of 763 patients. Arthritis Rheum. 2014;66:1608-1615.
- Contreras I, Sallés M, Mínguez S, et al. Hard paracervical tumor in a patient with limited systemic sclerosis. Rheumatol Clin. 2014; 10:336-337.
- Meriglier E, Lafourcade F, Gombert B, et al. Giant calcinosis revealing systemic sclerosis. Int J Rheum Dis. 2019;22:1787-1788.
- Chung CH. Calcinosis universalis in juvenile dermatomyositis [published online September 24, 2020]. Chonnam Med J. 2020;56:212-213.
- Bartoli F, Fiori G, Braschi F, et al. Calcinosis in systemic sclerosis: subsets, distribution and complications [published online May 30, 2016]. Rheumatology (Oxford). 2016;55:1610-1614.
- Jung H, Lee D, Cho J, et al. Surgical treatment of extensive tumoral calcinosis associated with systemic sclerosis. Korean J Thorac Cardiovasc Surg. 2015;48:151-154.
- Badawi AH, Patel V, Warner AE, et al. Dystrophic calcinosis cutis: treatment with intravenous sodium thiosulfate. Cutis. 2020;106:E15-E17.
- Shanmugam V, Price P, Attinger C, et al. Lower extremity ulcers in systemic sclerosis: features and response to therapy [published online August 18, 2010]. Int J Rheumatol. doi:10.1155/2010/747946
- Bohelay G, Blaise S, Levy P, et al. Lower-limb ulcers in systemic sclerosis: a multicentre retrospective case-control study. Acta Derm Venereol. 2018;98:677-682.
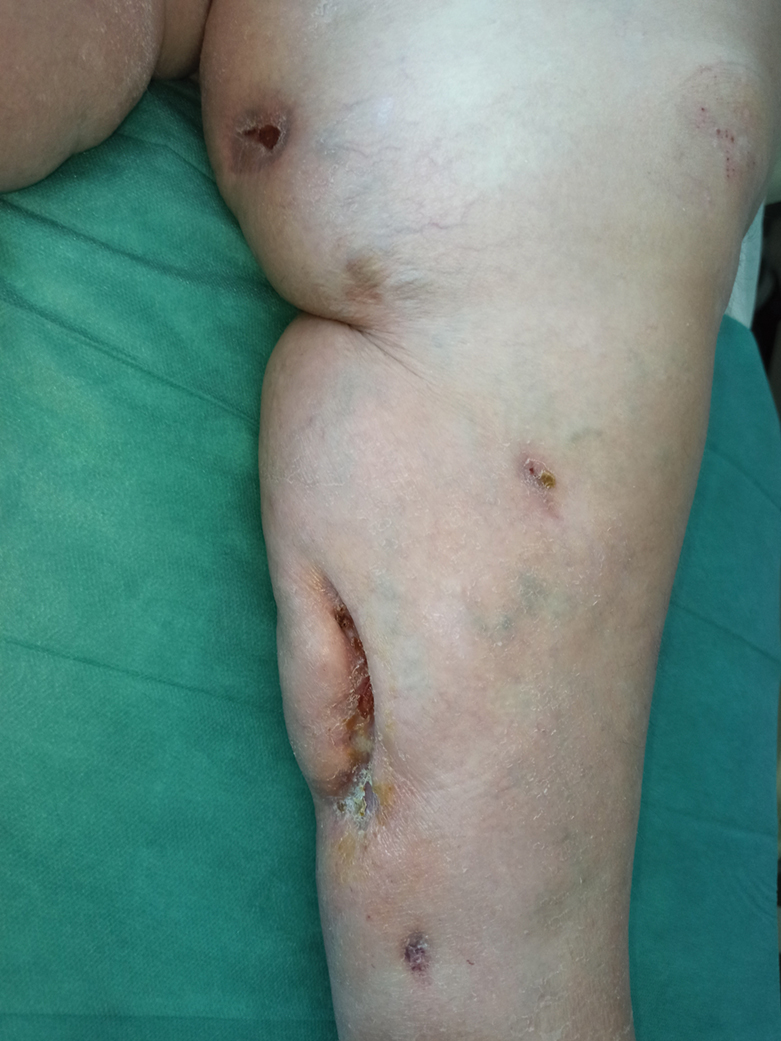
A 49-year-old woman with type 2 diabetes mellitus, morbid obesity, pulmonary fibrosis, and pulmonary arterial hypertension presented to our hospital with an ulcer on the left leg of unknown etiology that was superinfected by multidrug-resistant Klebsiella according to bacterial culture. She had an axillary temperature of 38.6 °C. She underwent amputation of the second and third toes on the left foot 5 years prior to presentation due to distal necrotic ulcers of ischemic origin. Physical examination revealed an 8×2-cm deep ulcer with abrupt edges on the left leg with fibrin and a purulent exudate. Deep palpation of the perilesional skin revealed indurated subcutaneous nodules. She also had scars on the fingertips of both hands with no induration on the rest of the skin surface. Capillaroscopy showed no pathologic findings. Blood cultures were performed, and she was admitted to the hospital for intravenous antibiotic therapy. During ulcer debridement, some solid whitish material was released.
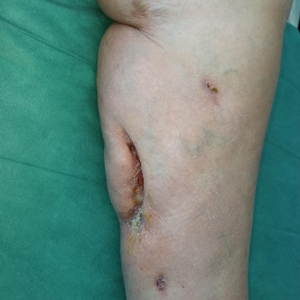
Flesh-Colored Papule in the Nose of a Child
The Diagnosis: Striated Muscle Hamartoma
Histopathologic evaluation revealed a dome-shaped papule with a center composed of mature striated muscle bundles, vellus hairs, sebaceous lobules, and nerve twigs (Figure) consistent with a diagnosis of striated muscle hamartoma (SMH).
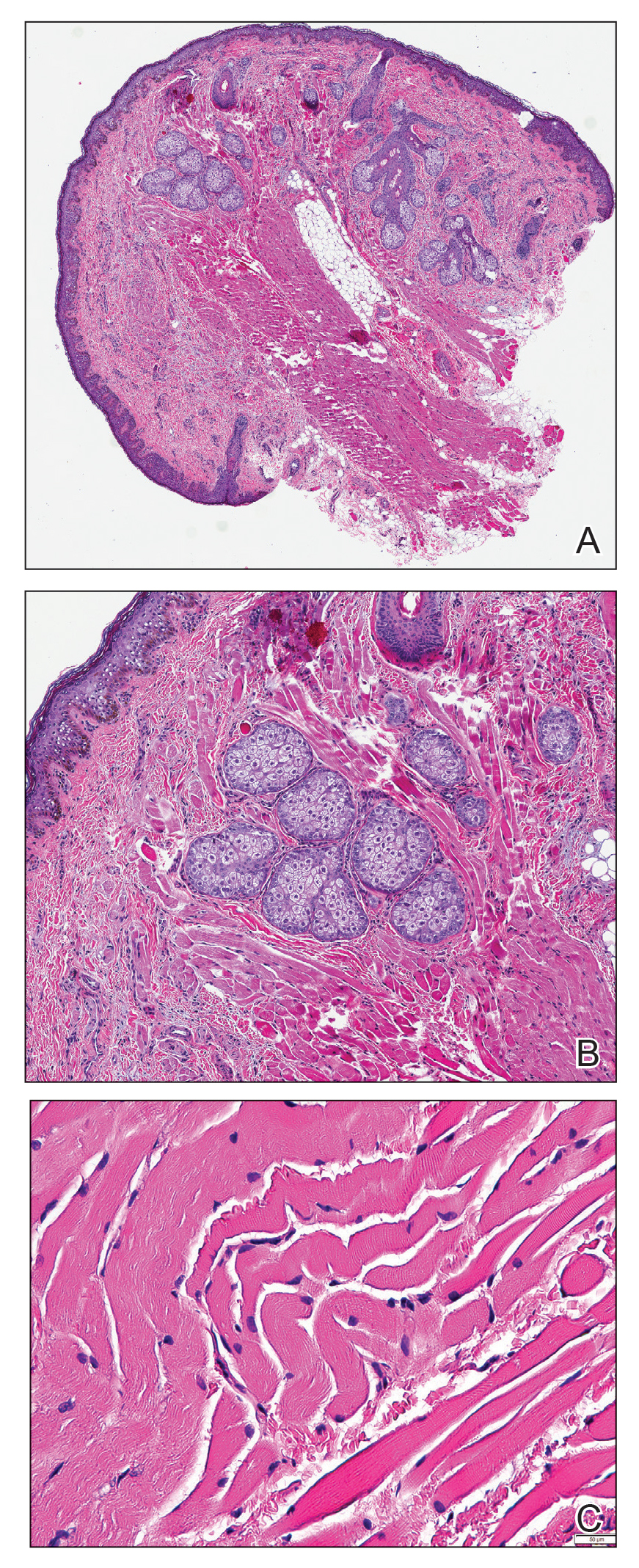
Striated muscle hamartoma was first described in 1986 by Hendrick et al1 with 2 cases in neonates. Biopsies of the lesions taken from the upper lip and sternum showed a characteristic histology consisting of dermal striated muscle fibers and nerve bundles in the central core of the papules associated with a marked number of adnexa. In 1989, the diagnosis of rhabdomyomatous mesenchymal hamartoma was described, which showed similar findings.2 Cases reported since these entities were discovered have used the terms striated muscle hamartoma and rhabdomyomatous mesenchymal hamartoma interchangeably.3
Most commonly found on the head and neck, SMH has now been observed in diverse locations including the sternum, hallux, vagina, and oral cavity.1-15 Many reported cases describe lesions around or in the nose.4,7,8 Multiple congenital anomalies have been described alongside SMH and may be associated with this entity including amniotic bands, cleft lip and palate, coloboma, and Delleman syndrome.1,3,4 Almost all of the lesions present as a sessile or pedunculated papule, polyp, nodule, or plaque measuring from 0.3 cm up to 4.9 cm and typically are present since birth.3,5,15 However, there are a few cases of lesions presenting in adults with no prior history.5,6,15
Microscopically, SMH is defined by a dermal lesion with a core comprised of mature skeletal muscle admixed with adipose tissue, adnexa, nerve bundles, and fibrovascular tissue.1 There are other entities that should be considered before making the diagnosis of SMH. Other hamartomas such as accessory tragus, connective tissue nevus, fibrous hamartoma of infancy, and nevus lipomatosis may present similarly; however, these lesions classically lack skeletal muscle. Benign triton tumors, or neuromuscular hamartomas, are rare lesions composed of skeletal muscle and abundant, intimately associated neural tissue. Neuromuscular hamartomas frequently involve large nerves.16 Rhabdomyomas also should be considered. Adult rhabdomyomas are composed of eosinophilic polygonal cells with granular cytoplasm and occasional cross-striations. Fetal rhabdomyomas have multiple histologic types and are defined by a variable myxoid stroma, eosinophilic spindled cells, and rhabdomyocytes in various stages of maturity. Genital rhabdomyomas histopathologically appear similar to fetal rhabdomyomas but are confined to the genital region. The skeletal muscle present in rhabdomyomas typically is less differentiated.17 TMature skeletal bundles should be a dominant component of the lesion before diagnosing SMH.
Typically presenting as congenital lesions in the head and neck region, papules with a dermal core of mature skeletal muscle associated with adnexa and nerve twigs should prompt consideration of a diagnosis of SMH or rhabdomyomatous mesenchymal hamartoma. These lesions are benign and usually are cured with complete excision.
- Hendrick SJ, Sanchez RL, Blackwell SJ, et al. Striated muscle hamartoma: description of two cases. Pediatr Dermatol. 1986;3:153-157.
- Mills AE. Rhabdomyomatous mesenchymal hamartoma of skin. Am J Dermatopathol. 1989;1:58-63.
- Rosenberg AS, Kirk J, Morgan MB. Rhabdomyomatous mesenchymal hamartoma: an unusual dermal entity with a report of two cases and a review of the literature. J Cutan Pathol. 2002;29:238-243.
- Sánchez RL, Raimer SS. Clinical and histologic features of striated muscle hamartoma: possible relationship to Delleman’s syndrome. J Cutan Pathol. 1994;21:40-46.
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005;21:185-188.
- Harris MA, Dutton JJ, Proia AD. Striated muscle hamartoma of the eyelid in an adult woman. Ophthalmic Plast Reconstr Surg. 2008;24:492-494.
- Nakanishi H, Hashimoto I, Takiwaki H, et al. Striated muscle hamartoma of the nostril. J Dermatol. 1995;22:504-507.
- Farris PE, Manning S, Veatch F. Rhabdomyomatous mesenchymal hamartoma. Am J Dermatopathol. 1994;16:73-75.
- Grilli R, Escalonilla P, Soriano ML, et al. The so-called striated muscle hamartoma is a hamartoma of cutaneous adnexa and mesenchyme, but not of striated muscle. Acta Derm Venereol. 1998;78:390.
- Sampat K, Cheesman E, Siminas S. Perianal rhabdomyomatous mesenchymal hamartoma. Ann R Coll Surg Engl. 2017;99:E193-E195.
- Brinster NK, Farmer ER. Rhabdomyomatous mesenchymal hamartoma presenting on a digit. J Cutan Pathol. 2009;36:61-63.
- Han SH, Song HJ, Hong WK, et al. Rhabdomyomatous mesenchymal hamartoma of the vagina. Pediatr Dermatol. 2009;26:753-755.
- De la Sotta P, Salomone C, González S. Rhabdomyomatous (mesenchymal) hamartoma of the tongue: report of a case. J Oral Pathol Med. 2007;36:58-59.
- Magro G, Di Benedetto A, Sanges G, et al. Rhabdomyomatous mesenchymal hamartoma of oral cavity: an unusual location for such a rare lesion. Virchows Arch. 2005;446:346-347.
- Wang Y, Zhao H, Yue X, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a big subcutaneous mass on the neck: a case report. J Med Case Rep. 2014;8:410.
- Amita K, Shankar SV, Nischal KC, et al. Benign triton tumor: a rare entity in head and neck region. Korean J Pathol. 2013;47:74-76.
- Walsh S, Hurt M. Cutaneous fetal rhabdomyoma: a case report and historical review of the literature. Am J Surg Pathol. 2008;32:485-491.
The Diagnosis: Striated Muscle Hamartoma
Histopathologic evaluation revealed a dome-shaped papule with a center composed of mature striated muscle bundles, vellus hairs, sebaceous lobules, and nerve twigs (Figure) consistent with a diagnosis of striated muscle hamartoma (SMH).
Striated muscle hamartoma was first described in 1986 by Hendrick et al1 with 2 cases in neonates. Biopsies of the lesions taken from the upper lip and sternum showed a characteristic histology consisting of dermal striated muscle fibers and nerve bundles in the central core of the papules associated with a marked number of adnexa. In 1989, the diagnosis of rhabdomyomatous mesenchymal hamartoma was described, which showed similar findings.2 Cases reported since these entities were discovered have used the terms striated muscle hamartoma and rhabdomyomatous mesenchymal hamartoma interchangeably.3
Most commonly found on the head and neck, SMH has now been observed in diverse locations including the sternum, hallux, vagina, and oral cavity.1-15 Many reported cases describe lesions around or in the nose.4,7,8 Multiple congenital anomalies have been described alongside SMH and may be associated with this entity including amniotic bands, cleft lip and palate, coloboma, and Delleman syndrome.1,3,4 Almost all of the lesions present as a sessile or pedunculated papule, polyp, nodule, or plaque measuring from 0.3 cm up to 4.9 cm and typically are present since birth.3,5,15 However, there are a few cases of lesions presenting in adults with no prior history.5,6,15
Microscopically, SMH is defined by a dermal lesion with a core comprised of mature skeletal muscle admixed with adipose tissue, adnexa, nerve bundles, and fibrovascular tissue.1 There are other entities that should be considered before making the diagnosis of SMH. Other hamartomas such as accessory tragus, connective tissue nevus, fibrous hamartoma of infancy, and nevus lipomatosis may present similarly; however, these lesions classically lack skeletal muscle. Benign triton tumors, or neuromuscular hamartomas, are rare lesions composed of skeletal muscle and abundant, intimately associated neural tissue. Neuromuscular hamartomas frequently involve large nerves.16 Rhabdomyomas also should be considered. Adult rhabdomyomas are composed of eosinophilic polygonal cells with granular cytoplasm and occasional cross-striations. Fetal rhabdomyomas have multiple histologic types and are defined by a variable myxoid stroma, eosinophilic spindled cells, and rhabdomyocytes in various stages of maturity. Genital rhabdomyomas histopathologically appear similar to fetal rhabdomyomas but are confined to the genital region. The skeletal muscle present in rhabdomyomas typically is less differentiated.17 TMature skeletal bundles should be a dominant component of the lesion before diagnosing SMH.
Typically presenting as congenital lesions in the head and neck region, papules with a dermal core of mature skeletal muscle associated with adnexa and nerve twigs should prompt consideration of a diagnosis of SMH or rhabdomyomatous mesenchymal hamartoma. These lesions are benign and usually are cured with complete excision.
The Diagnosis: Striated Muscle Hamartoma
Histopathologic evaluation revealed a dome-shaped papule with a center composed of mature striated muscle bundles, vellus hairs, sebaceous lobules, and nerve twigs (Figure) consistent with a diagnosis of striated muscle hamartoma (SMH).
Striated muscle hamartoma was first described in 1986 by Hendrick et al1 with 2 cases in neonates. Biopsies of the lesions taken from the upper lip and sternum showed a characteristic histology consisting of dermal striated muscle fibers and nerve bundles in the central core of the papules associated with a marked number of adnexa. In 1989, the diagnosis of rhabdomyomatous mesenchymal hamartoma was described, which showed similar findings.2 Cases reported since these entities were discovered have used the terms striated muscle hamartoma and rhabdomyomatous mesenchymal hamartoma interchangeably.3
Most commonly found on the head and neck, SMH has now been observed in diverse locations including the sternum, hallux, vagina, and oral cavity.1-15 Many reported cases describe lesions around or in the nose.4,7,8 Multiple congenital anomalies have been described alongside SMH and may be associated with this entity including amniotic bands, cleft lip and palate, coloboma, and Delleman syndrome.1,3,4 Almost all of the lesions present as a sessile or pedunculated papule, polyp, nodule, or plaque measuring from 0.3 cm up to 4.9 cm and typically are present since birth.3,5,15 However, there are a few cases of lesions presenting in adults with no prior history.5,6,15
Microscopically, SMH is defined by a dermal lesion with a core comprised of mature skeletal muscle admixed with adipose tissue, adnexa, nerve bundles, and fibrovascular tissue.1 There are other entities that should be considered before making the diagnosis of SMH. Other hamartomas such as accessory tragus, connective tissue nevus, fibrous hamartoma of infancy, and nevus lipomatosis may present similarly; however, these lesions classically lack skeletal muscle. Benign triton tumors, or neuromuscular hamartomas, are rare lesions composed of skeletal muscle and abundant, intimately associated neural tissue. Neuromuscular hamartomas frequently involve large nerves.16 Rhabdomyomas also should be considered. Adult rhabdomyomas are composed of eosinophilic polygonal cells with granular cytoplasm and occasional cross-striations. Fetal rhabdomyomas have multiple histologic types and are defined by a variable myxoid stroma, eosinophilic spindled cells, and rhabdomyocytes in various stages of maturity. Genital rhabdomyomas histopathologically appear similar to fetal rhabdomyomas but are confined to the genital region. The skeletal muscle present in rhabdomyomas typically is less differentiated.17 TMature skeletal bundles should be a dominant component of the lesion before diagnosing SMH.
Typically presenting as congenital lesions in the head and neck region, papules with a dermal core of mature skeletal muscle associated with adnexa and nerve twigs should prompt consideration of a diagnosis of SMH or rhabdomyomatous mesenchymal hamartoma. These lesions are benign and usually are cured with complete excision.
- Hendrick SJ, Sanchez RL, Blackwell SJ, et al. Striated muscle hamartoma: description of two cases. Pediatr Dermatol. 1986;3:153-157.
- Mills AE. Rhabdomyomatous mesenchymal hamartoma of skin. Am J Dermatopathol. 1989;1:58-63.
- Rosenberg AS, Kirk J, Morgan MB. Rhabdomyomatous mesenchymal hamartoma: an unusual dermal entity with a report of two cases and a review of the literature. J Cutan Pathol. 2002;29:238-243.
- Sánchez RL, Raimer SS. Clinical and histologic features of striated muscle hamartoma: possible relationship to Delleman’s syndrome. J Cutan Pathol. 1994;21:40-46.
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005;21:185-188.
- Harris MA, Dutton JJ, Proia AD. Striated muscle hamartoma of the eyelid in an adult woman. Ophthalmic Plast Reconstr Surg. 2008;24:492-494.
- Nakanishi H, Hashimoto I, Takiwaki H, et al. Striated muscle hamartoma of the nostril. J Dermatol. 1995;22:504-507.
- Farris PE, Manning S, Veatch F. Rhabdomyomatous mesenchymal hamartoma. Am J Dermatopathol. 1994;16:73-75.
- Grilli R, Escalonilla P, Soriano ML, et al. The so-called striated muscle hamartoma is a hamartoma of cutaneous adnexa and mesenchyme, but not of striated muscle. Acta Derm Venereol. 1998;78:390.
- Sampat K, Cheesman E, Siminas S. Perianal rhabdomyomatous mesenchymal hamartoma. Ann R Coll Surg Engl. 2017;99:E193-E195.
- Brinster NK, Farmer ER. Rhabdomyomatous mesenchymal hamartoma presenting on a digit. J Cutan Pathol. 2009;36:61-63.
- Han SH, Song HJ, Hong WK, et al. Rhabdomyomatous mesenchymal hamartoma of the vagina. Pediatr Dermatol. 2009;26:753-755.
- De la Sotta P, Salomone C, González S. Rhabdomyomatous (mesenchymal) hamartoma of the tongue: report of a case. J Oral Pathol Med. 2007;36:58-59.
- Magro G, Di Benedetto A, Sanges G, et al. Rhabdomyomatous mesenchymal hamartoma of oral cavity: an unusual location for such a rare lesion. Virchows Arch. 2005;446:346-347.
- Wang Y, Zhao H, Yue X, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a big subcutaneous mass on the neck: a case report. J Med Case Rep. 2014;8:410.
- Amita K, Shankar SV, Nischal KC, et al. Benign triton tumor: a rare entity in head and neck region. Korean J Pathol. 2013;47:74-76.
- Walsh S, Hurt M. Cutaneous fetal rhabdomyoma: a case report and historical review of the literature. Am J Surg Pathol. 2008;32:485-491.
- Hendrick SJ, Sanchez RL, Blackwell SJ, et al. Striated muscle hamartoma: description of two cases. Pediatr Dermatol. 1986;3:153-157.
- Mills AE. Rhabdomyomatous mesenchymal hamartoma of skin. Am J Dermatopathol. 1989;1:58-63.
- Rosenberg AS, Kirk J, Morgan MB. Rhabdomyomatous mesenchymal hamartoma: an unusual dermal entity with a report of two cases and a review of the literature. J Cutan Pathol. 2002;29:238-243.
- Sánchez RL, Raimer SS. Clinical and histologic features of striated muscle hamartoma: possible relationship to Delleman’s syndrome. J Cutan Pathol. 1994;21:40-46.
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005;21:185-188.
- Harris MA, Dutton JJ, Proia AD. Striated muscle hamartoma of the eyelid in an adult woman. Ophthalmic Plast Reconstr Surg. 2008;24:492-494.
- Nakanishi H, Hashimoto I, Takiwaki H, et al. Striated muscle hamartoma of the nostril. J Dermatol. 1995;22:504-507.
- Farris PE, Manning S, Veatch F. Rhabdomyomatous mesenchymal hamartoma. Am J Dermatopathol. 1994;16:73-75.
- Grilli R, Escalonilla P, Soriano ML, et al. The so-called striated muscle hamartoma is a hamartoma of cutaneous adnexa and mesenchyme, but not of striated muscle. Acta Derm Venereol. 1998;78:390.
- Sampat K, Cheesman E, Siminas S. Perianal rhabdomyomatous mesenchymal hamartoma. Ann R Coll Surg Engl. 2017;99:E193-E195.
- Brinster NK, Farmer ER. Rhabdomyomatous mesenchymal hamartoma presenting on a digit. J Cutan Pathol. 2009;36:61-63.
- Han SH, Song HJ, Hong WK, et al. Rhabdomyomatous mesenchymal hamartoma of the vagina. Pediatr Dermatol. 2009;26:753-755.
- De la Sotta P, Salomone C, González S. Rhabdomyomatous (mesenchymal) hamartoma of the tongue: report of a case. J Oral Pathol Med. 2007;36:58-59.
- Magro G, Di Benedetto A, Sanges G, et al. Rhabdomyomatous mesenchymal hamartoma of oral cavity: an unusual location for such a rare lesion. Virchows Arch. 2005;446:346-347.
- Wang Y, Zhao H, Yue X, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a big subcutaneous mass on the neck: a case report. J Med Case Rep. 2014;8:410.
- Amita K, Shankar SV, Nischal KC, et al. Benign triton tumor: a rare entity in head and neck region. Korean J Pathol. 2013;47:74-76.
- Walsh S, Hurt M. Cutaneous fetal rhabdomyoma: a case report and historical review of the literature. Am J Surg Pathol. 2008;32:485-491.
A 4-year-old girl presented to our clinic with an asymptomatic flesh-colored papule in the left nostril. The lesion had been present since birth and grew in relation to the patient with no rapid changes. There had been no pigmentation changes and no bleeding, pain, or itching. The patient’s birth and developmental history were normal. Physical examination revealed a singular, 10×5-mm, flesh-colored, pedunculated mass on the left nasal sill. There were no additional lesions present. An excisional biopsy was performed and submitted for pathologic diagnosis.

Tender Annular Plaque on the Thigh
The Diagnosis: Ecthyma Gangrenosum
Histopathology revealed basophilic bacterial rods around necrotic vessels with thrombosis and edema (Figure). Blood and tissue cultures grew Pseudomonas aeruginosa. Based on the histopathology and clinical presentation, a diagnosis of P aeruginosa–associated ecthyma gangrenosum (EG) was made. The patient’s symptoms resolved with intravenous cefepime, and he later was transitioned to oral levofloxacin for outpatient treatment.

Ecthyma gangrenosum is an uncommon cutaneous manifestation of bacteremia that most commonly occurs secondary to P aeruginosa in immunocompromised patients, particularly patients with severe neutropenia in the setting of recent chemotherapy.1,2 Ecthyma gangrenosum can occur anywhere on the body, predominantly in moist areas such as the axillae and groin; the arms and legs, such as in our patient, as well as the trunk and face also may be involved.3 Other causes of EG skin lesions include methicillin-resistant Staphylococcus aureus, Citrobacter freundii, Escherichia coli, fungi such as Candida, and viruses such as herpes simplex virus.2,4-6 Common predisposing conditions associated with EG include neutropenia, leukemia, HIV, diabetes mellitus, extensive burn wounds, and a history of immunosuppressive medications. It also has been known to occur in otherwise healthy, immunocompetent individuals with no difference in clinical manifestation.2
The diagnosis is clinicopathologic, with initial evaluation including blood and wound cultures as well as a complete blood cell count once EG is suspected. An excisional or punch biopsy is performed for confirmation, showing many gram-negative, rod-shaped bacteria in cases of pseudomonal EG.7 Histopathology is characterized by bacterial perivascular invasion that then leads to secondary arteriole thrombosis, tissue edema, and separation of the epidermis.7,8 Resultant ischemic necrosis results in the classic macroscopic appearance of an erythematous macule that rapidly progresses into a central necrotic lesion surrounded by an erythematous or violaceous halo after undergoing a hemorrhagic bullous stage.1,9 A Wood lamp can be used to expedite the diagnosis, as Pseudomonas bacteria excretes a pigment (pyoverdine) that fluoresces yellowish green.10
Ecthyma gangrenosum can be classified as a primary skin lesion that may or may not be followed by bacteremia or as a lesion secondary to pseudomonal bacteremia.11 Bacteremia has been reported in half of cases, with hematogenous metastasis of the infection, likely in manifestations with multiple bilateral lesions.2 Our patient’s presentation of a single lesion revealed a positive blood culture result. Lesions also can develop by direct inoculation of the epidermis causing local destruction of the surrounding tissue. The nonbacteremic form of EG has been associated with a lower mortality rate of around 15% compared to patients with bacteremia ranging from 38% to 96%.12 The presence of neutropenia is the most important prognostic factor for mortality at the time of diagnosis.13
Prompt empiric therapy should be initiated after obtaining wound and blood cultures in those with infection until the causative organism and its susceptibility are identified. Pseudomonal infections account for 4% of all cases of hospital-acquired bacteremia and are the third leading cause of gram-negative bloodstream infection.7 Initial broad-spectrum antibiotics include antipseudomonal β-lactams (piperacillin-tazobactam), cephalosporins (cefepime), fluoroquinolones (levofloxacin), and carbapenems (imipenem).1,7 Medical therapy alone may be sufficient without requiring extensive surgical debridement to remove necrotic tissue in some patients. Surgical debridement usually is warranted for lesions larger than 10 cm in diameter.3 Our patient was treated with intravenous cefepime with resolution and was followed with outpatient oral levofloxacin as appropriate. A high index of suspicion should be maintained for relapsing pseudomonal EG infection among patients with AIDS, as the reported recurrence rate is 57%.14
Clinically, the differential diagnosis of EG presenting in immunocompromised patients or individuals with underlying malignancy includes pyoderma gangrenosum, papulonecrotic tuberculid, and leukemia cutis. An erythematous rash with central necrosis presenting in a patient with systemic symptoms is pathognomonic for erythema migrans and should be considered as a diagnostic possibility in areas endemic for Lyme disease in the United States, including the northeastern, mid-Atlantic, and north-central regions.15 A thorough history, physical examination, basic laboratory studies, and histopathology are critical to differentiate between these entities with similar macroscopic features. Pyoderma gangrenosum histologically manifests as a noninfectious, deep, suppurative folliculitis with leukocytoclastic vasculitis in 40% of cases.16 Although papulonecrotic tuberculid can present with dermal necrosis resulting from a hypersensitivity reaction to antigenic components of mycobacteria, there typically are granulomatous infiltrates present and a lack of observed organisms on histopathology.17 Although leukemia cutis infrequently occurs in patients diagnosed with leukemia, its salient features on pathology are nodular or diffuse infiltrates of leukemic cells in the dermis and subcutis with a high nuclear-to-cytoplasmic ratio, often with prominent nucleoli.18 Lyme disease can present in various ways; however, cutaneous involvement in the primary lesion is histologically characterized by a perivascular lymphohistiocytic infiltrate containing plasma cells at the periphery of the expanding annular lesion and eosinophils present at the center.19
- Abdou A, Hassam B. Ecthyma gangrenosum [in French]. Pan Afr Med J. 2018;30:95. doi:10.11604/pamj.2018.30.95.6244
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639. doi:10.1007/s10096-014-2277-6
- Vaiman M, Lasarovitch T, Heller L, et al. Ecthyma gangrenosum versus ecthyma-like lesions: should we separate these conditions? Acta Dermatovenerol Alp Pannonica Adriat. 2015;24:69-72. doi:10.15570 /actaapa.2015.18
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl): S114-S117. doi:10.1016/j.jaad.2003.09.019
- Hawkley T, Chang D, Pollard W, et al. Ecthyma gangrenosum caused by Citrobacter freundii [published online July 27, 2017]. BMJ Case Rep. doi:10.1136/bcr-2017-220996
- Santhaseelan RG, Muralidhar V. Non-pseudomonal ecthyma gangrenosum caused by methicillin-resistant Staphylococcus aureus (MRSA) in a chronic alcoholic patient [published online August 3, 2017]. BMJ Case Rep. doi:10.1136/bcr-2017-220983m
- Bassetti M, Vena A, Croxatto A, et al. How to manage Pseudomonas aeruginosa infections [published online May 29, 2018]. Drugs Context. 2018;7:212527. doi:10.7573/dic.212527
- Llamas-Velasco M, Alegría V, Santos-Briz Á, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662. doi:10.1097/DAD.0000000000000766
- Sarkar S, Patra AK, Mondal M. Ecthyma gangrenosum in the periorbital region in a previously healthy immunocompetent woman without bacteremia. Indian Dermatol Online J. 2016;7:36-39. doi:10.4103/2229-5178.174326
- Ponka D, Baddar F. Wood lamp examination. Can Fam Physician. 2012;58:976.
- Van den Broek PJ, Van der Meer JWM, Kunst MW. The pathogenesis of ecthyma gangrenosum. J Infect. 1979;1:263-267. doi:10.1016 /S0163-4453(79)91329-X
- Downey DM, O’Bryan MC, Burdette SD, et al. Ecthyma gangrenosum in a patient with toxic epidermal necrolysis. J Burn Care Res. 2007;28:198-202. doi:10.1097/BCR.0B013E31802CA481
- Martínez-Longoria CA, Rosales-Solis GM, Ocampo-Garza J, et al. Ecthyma gangrenosum: a report of eight cases. An Bras Dermatol. 2017;92:698-700. doi:10.1590/abd1806-4841.20175580
- Khan MO, Montecalvo MA, Davis I, et al. Ecthyma gangrenosum in patients with acquired immunodeficiency syndrome. Cutis. 2000;66:121-123.
- Nadelman RB, Wormser GP. Lyme borreliosis. Lancet. 1998; 352:557-565.
- Su WP, Schroeter AL, Perry HO, et al. Histopathologic and immunopathologic study of pyoderma gangrenosum. J Cutan Pathol. 1986;13:323-330. doi:10.1111/j.1600-0560.1986.tb00466.x
- Tirumalae R, Yeliur IK, Antony M, et al. Papulonecrotic tuberculidclinicopathologic and molecular features of 12 Indian patients. Dermatol Pract Concept. 2014;4:17-22. doi:10.5826/dpc.0402a03
- Obiozor C, Ganguly S, Fraga GR. Leukemia cutis with lymphoglandular bodies: a clue to acute lymphoblastic leukemia cutis [published online August 15, 2015]. Dermatol Online J. 2015;21:13030/qt6m18g35f
- Vasudevan B, Chatterjee M. Lyme borreliosis and skin. Indian J Dermatol. 2013;58:167-174. doi:10.4103/0019-5154.110822
The Diagnosis: Ecthyma Gangrenosum
Histopathology revealed basophilic bacterial rods around necrotic vessels with thrombosis and edema (Figure). Blood and tissue cultures grew Pseudomonas aeruginosa. Based on the histopathology and clinical presentation, a diagnosis of P aeruginosa–associated ecthyma gangrenosum (EG) was made. The patient’s symptoms resolved with intravenous cefepime, and he later was transitioned to oral levofloxacin for outpatient treatment.
Ecthyma gangrenosum is an uncommon cutaneous manifestation of bacteremia that most commonly occurs secondary to P aeruginosa in immunocompromised patients, particularly patients with severe neutropenia in the setting of recent chemotherapy.1,2 Ecthyma gangrenosum can occur anywhere on the body, predominantly in moist areas such as the axillae and groin; the arms and legs, such as in our patient, as well as the trunk and face also may be involved.3 Other causes of EG skin lesions include methicillin-resistant Staphylococcus aureus, Citrobacter freundii, Escherichia coli, fungi such as Candida, and viruses such as herpes simplex virus.2,4-6 Common predisposing conditions associated with EG include neutropenia, leukemia, HIV, diabetes mellitus, extensive burn wounds, and a history of immunosuppressive medications. It also has been known to occur in otherwise healthy, immunocompetent individuals with no difference in clinical manifestation.2
The diagnosis is clinicopathologic, with initial evaluation including blood and wound cultures as well as a complete blood cell count once EG is suspected. An excisional or punch biopsy is performed for confirmation, showing many gram-negative, rod-shaped bacteria in cases of pseudomonal EG.7 Histopathology is characterized by bacterial perivascular invasion that then leads to secondary arteriole thrombosis, tissue edema, and separation of the epidermis.7,8 Resultant ischemic necrosis results in the classic macroscopic appearance of an erythematous macule that rapidly progresses into a central necrotic lesion surrounded by an erythematous or violaceous halo after undergoing a hemorrhagic bullous stage.1,9 A Wood lamp can be used to expedite the diagnosis, as Pseudomonas bacteria excretes a pigment (pyoverdine) that fluoresces yellowish green.10
Ecthyma gangrenosum can be classified as a primary skin lesion that may or may not be followed by bacteremia or as a lesion secondary to pseudomonal bacteremia.11 Bacteremia has been reported in half of cases, with hematogenous metastasis of the infection, likely in manifestations with multiple bilateral lesions.2 Our patient’s presentation of a single lesion revealed a positive blood culture result. Lesions also can develop by direct inoculation of the epidermis causing local destruction of the surrounding tissue. The nonbacteremic form of EG has been associated with a lower mortality rate of around 15% compared to patients with bacteremia ranging from 38% to 96%.12 The presence of neutropenia is the most important prognostic factor for mortality at the time of diagnosis.13
Prompt empiric therapy should be initiated after obtaining wound and blood cultures in those with infection until the causative organism and its susceptibility are identified. Pseudomonal infections account for 4% of all cases of hospital-acquired bacteremia and are the third leading cause of gram-negative bloodstream infection.7 Initial broad-spectrum antibiotics include antipseudomonal β-lactams (piperacillin-tazobactam), cephalosporins (cefepime), fluoroquinolones (levofloxacin), and carbapenems (imipenem).1,7 Medical therapy alone may be sufficient without requiring extensive surgical debridement to remove necrotic tissue in some patients. Surgical debridement usually is warranted for lesions larger than 10 cm in diameter.3 Our patient was treated with intravenous cefepime with resolution and was followed with outpatient oral levofloxacin as appropriate. A high index of suspicion should be maintained for relapsing pseudomonal EG infection among patients with AIDS, as the reported recurrence rate is 57%.14
Clinically, the differential diagnosis of EG presenting in immunocompromised patients or individuals with underlying malignancy includes pyoderma gangrenosum, papulonecrotic tuberculid, and leukemia cutis. An erythematous rash with central necrosis presenting in a patient with systemic symptoms is pathognomonic for erythema migrans and should be considered as a diagnostic possibility in areas endemic for Lyme disease in the United States, including the northeastern, mid-Atlantic, and north-central regions.15 A thorough history, physical examination, basic laboratory studies, and histopathology are critical to differentiate between these entities with similar macroscopic features. Pyoderma gangrenosum histologically manifests as a noninfectious, deep, suppurative folliculitis with leukocytoclastic vasculitis in 40% of cases.16 Although papulonecrotic tuberculid can present with dermal necrosis resulting from a hypersensitivity reaction to antigenic components of mycobacteria, there typically are granulomatous infiltrates present and a lack of observed organisms on histopathology.17 Although leukemia cutis infrequently occurs in patients diagnosed with leukemia, its salient features on pathology are nodular or diffuse infiltrates of leukemic cells in the dermis and subcutis with a high nuclear-to-cytoplasmic ratio, often with prominent nucleoli.18 Lyme disease can present in various ways; however, cutaneous involvement in the primary lesion is histologically characterized by a perivascular lymphohistiocytic infiltrate containing plasma cells at the periphery of the expanding annular lesion and eosinophils present at the center.19
The Diagnosis: Ecthyma Gangrenosum
Histopathology revealed basophilic bacterial rods around necrotic vessels with thrombosis and edema (Figure). Blood and tissue cultures grew Pseudomonas aeruginosa. Based on the histopathology and clinical presentation, a diagnosis of P aeruginosa–associated ecthyma gangrenosum (EG) was made. The patient’s symptoms resolved with intravenous cefepime, and he later was transitioned to oral levofloxacin for outpatient treatment.
Ecthyma gangrenosum is an uncommon cutaneous manifestation of bacteremia that most commonly occurs secondary to P aeruginosa in immunocompromised patients, particularly patients with severe neutropenia in the setting of recent chemotherapy.1,2 Ecthyma gangrenosum can occur anywhere on the body, predominantly in moist areas such as the axillae and groin; the arms and legs, such as in our patient, as well as the trunk and face also may be involved.3 Other causes of EG skin lesions include methicillin-resistant Staphylococcus aureus, Citrobacter freundii, Escherichia coli, fungi such as Candida, and viruses such as herpes simplex virus.2,4-6 Common predisposing conditions associated with EG include neutropenia, leukemia, HIV, diabetes mellitus, extensive burn wounds, and a history of immunosuppressive medications. It also has been known to occur in otherwise healthy, immunocompetent individuals with no difference in clinical manifestation.2
The diagnosis is clinicopathologic, with initial evaluation including blood and wound cultures as well as a complete blood cell count once EG is suspected. An excisional or punch biopsy is performed for confirmation, showing many gram-negative, rod-shaped bacteria in cases of pseudomonal EG.7 Histopathology is characterized by bacterial perivascular invasion that then leads to secondary arteriole thrombosis, tissue edema, and separation of the epidermis.7,8 Resultant ischemic necrosis results in the classic macroscopic appearance of an erythematous macule that rapidly progresses into a central necrotic lesion surrounded by an erythematous or violaceous halo after undergoing a hemorrhagic bullous stage.1,9 A Wood lamp can be used to expedite the diagnosis, as Pseudomonas bacteria excretes a pigment (pyoverdine) that fluoresces yellowish green.10
Ecthyma gangrenosum can be classified as a primary skin lesion that may or may not be followed by bacteremia or as a lesion secondary to pseudomonal bacteremia.11 Bacteremia has been reported in half of cases, with hematogenous metastasis of the infection, likely in manifestations with multiple bilateral lesions.2 Our patient’s presentation of a single lesion revealed a positive blood culture result. Lesions also can develop by direct inoculation of the epidermis causing local destruction of the surrounding tissue. The nonbacteremic form of EG has been associated with a lower mortality rate of around 15% compared to patients with bacteremia ranging from 38% to 96%.12 The presence of neutropenia is the most important prognostic factor for mortality at the time of diagnosis.13
Prompt empiric therapy should be initiated after obtaining wound and blood cultures in those with infection until the causative organism and its susceptibility are identified. Pseudomonal infections account for 4% of all cases of hospital-acquired bacteremia and are the third leading cause of gram-negative bloodstream infection.7 Initial broad-spectrum antibiotics include antipseudomonal β-lactams (piperacillin-tazobactam), cephalosporins (cefepime), fluoroquinolones (levofloxacin), and carbapenems (imipenem).1,7 Medical therapy alone may be sufficient without requiring extensive surgical debridement to remove necrotic tissue in some patients. Surgical debridement usually is warranted for lesions larger than 10 cm in diameter.3 Our patient was treated with intravenous cefepime with resolution and was followed with outpatient oral levofloxacin as appropriate. A high index of suspicion should be maintained for relapsing pseudomonal EG infection among patients with AIDS, as the reported recurrence rate is 57%.14
Clinically, the differential diagnosis of EG presenting in immunocompromised patients or individuals with underlying malignancy includes pyoderma gangrenosum, papulonecrotic tuberculid, and leukemia cutis. An erythematous rash with central necrosis presenting in a patient with systemic symptoms is pathognomonic for erythema migrans and should be considered as a diagnostic possibility in areas endemic for Lyme disease in the United States, including the northeastern, mid-Atlantic, and north-central regions.15 A thorough history, physical examination, basic laboratory studies, and histopathology are critical to differentiate between these entities with similar macroscopic features. Pyoderma gangrenosum histologically manifests as a noninfectious, deep, suppurative folliculitis with leukocytoclastic vasculitis in 40% of cases.16 Although papulonecrotic tuberculid can present with dermal necrosis resulting from a hypersensitivity reaction to antigenic components of mycobacteria, there typically are granulomatous infiltrates present and a lack of observed organisms on histopathology.17 Although leukemia cutis infrequently occurs in patients diagnosed with leukemia, its salient features on pathology are nodular or diffuse infiltrates of leukemic cells in the dermis and subcutis with a high nuclear-to-cytoplasmic ratio, often with prominent nucleoli.18 Lyme disease can present in various ways; however, cutaneous involvement in the primary lesion is histologically characterized by a perivascular lymphohistiocytic infiltrate containing plasma cells at the periphery of the expanding annular lesion and eosinophils present at the center.19
- Abdou A, Hassam B. Ecthyma gangrenosum [in French]. Pan Afr Med J. 2018;30:95. doi:10.11604/pamj.2018.30.95.6244
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639. doi:10.1007/s10096-014-2277-6
- Vaiman M, Lasarovitch T, Heller L, et al. Ecthyma gangrenosum versus ecthyma-like lesions: should we separate these conditions? Acta Dermatovenerol Alp Pannonica Adriat. 2015;24:69-72. doi:10.15570 /actaapa.2015.18
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl): S114-S117. doi:10.1016/j.jaad.2003.09.019
- Hawkley T, Chang D, Pollard W, et al. Ecthyma gangrenosum caused by Citrobacter freundii [published online July 27, 2017]. BMJ Case Rep. doi:10.1136/bcr-2017-220996
- Santhaseelan RG, Muralidhar V. Non-pseudomonal ecthyma gangrenosum caused by methicillin-resistant Staphylococcus aureus (MRSA) in a chronic alcoholic patient [published online August 3, 2017]. BMJ Case Rep. doi:10.1136/bcr-2017-220983m
- Bassetti M, Vena A, Croxatto A, et al. How to manage Pseudomonas aeruginosa infections [published online May 29, 2018]. Drugs Context. 2018;7:212527. doi:10.7573/dic.212527
- Llamas-Velasco M, Alegría V, Santos-Briz Á, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662. doi:10.1097/DAD.0000000000000766
- Sarkar S, Patra AK, Mondal M. Ecthyma gangrenosum in the periorbital region in a previously healthy immunocompetent woman without bacteremia. Indian Dermatol Online J. 2016;7:36-39. doi:10.4103/2229-5178.174326
- Ponka D, Baddar F. Wood lamp examination. Can Fam Physician. 2012;58:976.
- Van den Broek PJ, Van der Meer JWM, Kunst MW. The pathogenesis of ecthyma gangrenosum. J Infect. 1979;1:263-267. doi:10.1016 /S0163-4453(79)91329-X
- Downey DM, O’Bryan MC, Burdette SD, et al. Ecthyma gangrenosum in a patient with toxic epidermal necrolysis. J Burn Care Res. 2007;28:198-202. doi:10.1097/BCR.0B013E31802CA481
- Martínez-Longoria CA, Rosales-Solis GM, Ocampo-Garza J, et al. Ecthyma gangrenosum: a report of eight cases. An Bras Dermatol. 2017;92:698-700. doi:10.1590/abd1806-4841.20175580
- Khan MO, Montecalvo MA, Davis I, et al. Ecthyma gangrenosum in patients with acquired immunodeficiency syndrome. Cutis. 2000;66:121-123.
- Nadelman RB, Wormser GP. Lyme borreliosis. Lancet. 1998; 352:557-565.
- Su WP, Schroeter AL, Perry HO, et al. Histopathologic and immunopathologic study of pyoderma gangrenosum. J Cutan Pathol. 1986;13:323-330. doi:10.1111/j.1600-0560.1986.tb00466.x
- Tirumalae R, Yeliur IK, Antony M, et al. Papulonecrotic tuberculidclinicopathologic and molecular features of 12 Indian patients. Dermatol Pract Concept. 2014;4:17-22. doi:10.5826/dpc.0402a03
- Obiozor C, Ganguly S, Fraga GR. Leukemia cutis with lymphoglandular bodies: a clue to acute lymphoblastic leukemia cutis [published online August 15, 2015]. Dermatol Online J. 2015;21:13030/qt6m18g35f
- Vasudevan B, Chatterjee M. Lyme borreliosis and skin. Indian J Dermatol. 2013;58:167-174. doi:10.4103/0019-5154.110822
- Abdou A, Hassam B. Ecthyma gangrenosum [in French]. Pan Afr Med J. 2018;30:95. doi:10.11604/pamj.2018.30.95.6244
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639. doi:10.1007/s10096-014-2277-6
- Vaiman M, Lasarovitch T, Heller L, et al. Ecthyma gangrenosum versus ecthyma-like lesions: should we separate these conditions? Acta Dermatovenerol Alp Pannonica Adriat. 2015;24:69-72. doi:10.15570 /actaapa.2015.18
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl): S114-S117. doi:10.1016/j.jaad.2003.09.019
- Hawkley T, Chang D, Pollard W, et al. Ecthyma gangrenosum caused by Citrobacter freundii [published online July 27, 2017]. BMJ Case Rep. doi:10.1136/bcr-2017-220996
- Santhaseelan RG, Muralidhar V. Non-pseudomonal ecthyma gangrenosum caused by methicillin-resistant Staphylococcus aureus (MRSA) in a chronic alcoholic patient [published online August 3, 2017]. BMJ Case Rep. doi:10.1136/bcr-2017-220983m
- Bassetti M, Vena A, Croxatto A, et al. How to manage Pseudomonas aeruginosa infections [published online May 29, 2018]. Drugs Context. 2018;7:212527. doi:10.7573/dic.212527
- Llamas-Velasco M, Alegría V, Santos-Briz Á, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662. doi:10.1097/DAD.0000000000000766
- Sarkar S, Patra AK, Mondal M. Ecthyma gangrenosum in the periorbital region in a previously healthy immunocompetent woman without bacteremia. Indian Dermatol Online J. 2016;7:36-39. doi:10.4103/2229-5178.174326
- Ponka D, Baddar F. Wood lamp examination. Can Fam Physician. 2012;58:976.
- Van den Broek PJ, Van der Meer JWM, Kunst MW. The pathogenesis of ecthyma gangrenosum. J Infect. 1979;1:263-267. doi:10.1016 /S0163-4453(79)91329-X
- Downey DM, O’Bryan MC, Burdette SD, et al. Ecthyma gangrenosum in a patient with toxic epidermal necrolysis. J Burn Care Res. 2007;28:198-202. doi:10.1097/BCR.0B013E31802CA481
- Martínez-Longoria CA, Rosales-Solis GM, Ocampo-Garza J, et al. Ecthyma gangrenosum: a report of eight cases. An Bras Dermatol. 2017;92:698-700. doi:10.1590/abd1806-4841.20175580
- Khan MO, Montecalvo MA, Davis I, et al. Ecthyma gangrenosum in patients with acquired immunodeficiency syndrome. Cutis. 2000;66:121-123.
- Nadelman RB, Wormser GP. Lyme borreliosis. Lancet. 1998; 352:557-565.
- Su WP, Schroeter AL, Perry HO, et al. Histopathologic and immunopathologic study of pyoderma gangrenosum. J Cutan Pathol. 1986;13:323-330. doi:10.1111/j.1600-0560.1986.tb00466.x
- Tirumalae R, Yeliur IK, Antony M, et al. Papulonecrotic tuberculidclinicopathologic and molecular features of 12 Indian patients. Dermatol Pract Concept. 2014;4:17-22. doi:10.5826/dpc.0402a03
- Obiozor C, Ganguly S, Fraga GR. Leukemia cutis with lymphoglandular bodies: a clue to acute lymphoblastic leukemia cutis [published online August 15, 2015]. Dermatol Online J. 2015;21:13030/qt6m18g35f
- Vasudevan B, Chatterjee M. Lyme borreliosis and skin. Indian J Dermatol. 2013;58:167-174. doi:10.4103/0019-5154.110822
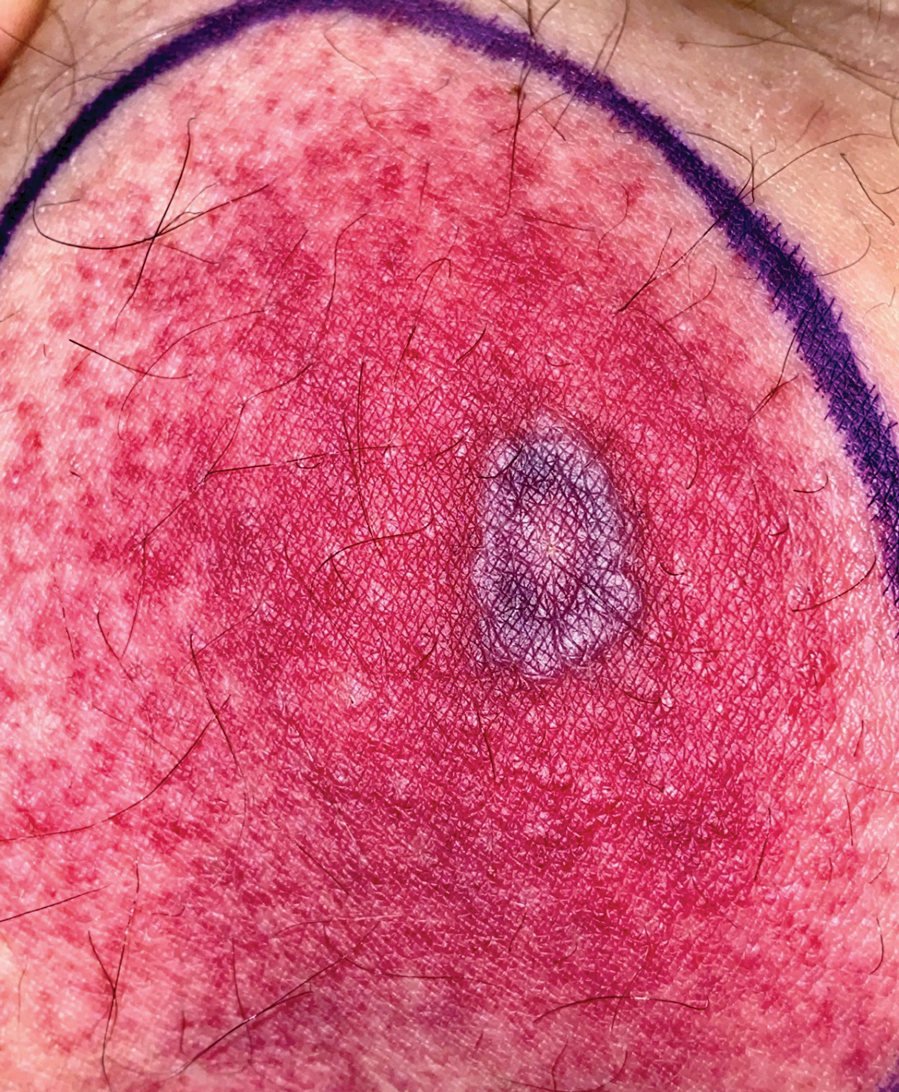
A 58-year-old man who was receiving gilteritinib therapy for relapsed acute myeloid leukemia presented to the emergency department with a painful, rapidly enlarging lesion on the right medial thigh of 2 days’ duration that was accompanied by fever (temperature, 39.2 °C) and body aches. Physical examination revealed a tender annular plaque with a dark violaceous halo overlying a larger area of erythema and induration. Laboratory evaluation revealed a white blood cell count of 600/μL (reference range, 4500–11,000/μL) and an absolute neutrophil count of 200/μL (reference range, 1800–7000/μL). A biopsy was performed.
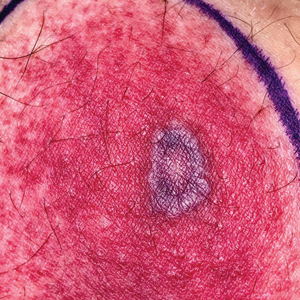
Painful Psoriasiform Plaques
The Diagnosis: Acquired Acrodermatitis Enteropathica
A punch biopsy of an elevated scaly border of the rash on the thigh revealed parakeratosis, absence of the granular layer, and epidermal pallor with psoriasiform and spongiotic dermatitis (Figure). Serum zinc levels were 60.1 μg/dL (reference range, 75.0–120.0 μg/dL), suggestive of a nutritional deficiency dermatitis. Laboratory and histopathologic findings were most consistent with a diagnosis of acquired acrodermatitis enteropathica (AE).
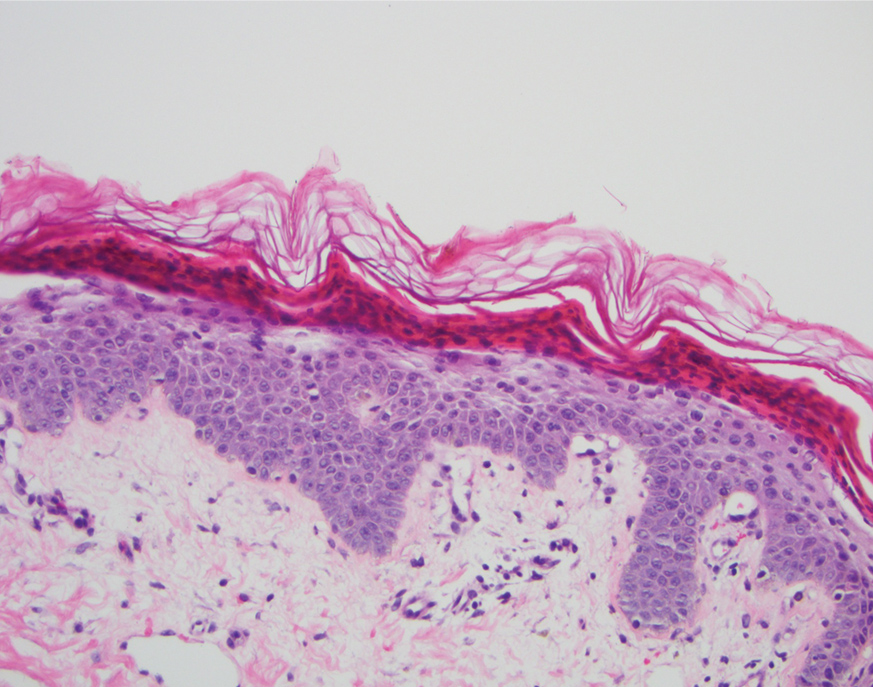
Acrodermatitis enteropathica has been associated with Roux-en-Y gastric bypass and alcohol use disorder working synergistically to cause malabsorption and malnutrition, respectively.1 Zinc functions in the structural integrity, wound healing, and anti-inflammatory properties of the skin. There is a 17.3% risk for hypozincemia worldwide; in developed nations there is an estimated 3% to 10% occurrence rate.2 Acrodermatitis enteropathica can be classified as either acquired or hereditary. Both classically present as a triad of acral dermatitis, diarrhea, and alopecia, though the complete triad is seen in 20% of cases.3,4
Hereditary AE is an autosomal-recessive disorder presenting in infancy that results in the loss of a zinc transporter. In contrast, acquired AE occurs later in life and usually is seen in patients who have decreased intake, malabsorption, or excessive loss of zinc.4 Acrodermatitis enteropathica is observed in individuals with conditions such as anorexia nervosa, pancreatic insufficiency, celiac disease, Crohn disease, or gastric bypass surgery (as in our case) and alcohol recidivism. In early disease, AE often presents with angular cheilitis and paronychia, but if left untreated, it can progress to mental status changes, hypogonadism, and depression.4 Acrodermatitis enteropathica presents as erythematous, erosive, scaly plaques or a papulosquamous psoriasiform rash with well-demarcated borders typically involving the orificial, acral, and intertriginous areas of the body.1,4
Acrodermatitis enteropathica belongs to a family of deficiency dermatoses that includes pellagra, necrolytic acral erythema (NAE), and necrolytic migratory erythema (NME).5 It is important to distinguish AE from NAE, as they can present similarly with well-defined and tender psoriasiform lesions peripherally. Histologically, NAE mimics AE with psoriasiform hyperplasia with parakeratosis.6 Necrolytic acral erythema characteristically is associated with active hepatitis C infection, which was absent in our patient.7
Similar to AE, NME affects the perineal and intertriginous surfaces.8 However, necrolytic migratory erythema has cutaneous manifestations in up to 70% of patients with glucagonoma syndrome, which classically presents as a triad of NME, weight loss, and diabetes mellitus.5 Laboratory studies show marked hyperglucagonemia, and imaging reveals enteropancreatic neoplasia. Necrolytic migratory erythema will rapidly resolve once the glucagonoma has been surgically removed.5 Bazex syndrome, or acrokeratosis paraneoplastica, is a paraneoplastic skin disease that is linked to underlying aerodigestive tract malignancies.
Bazex syndrome clinically is characterized by hyperkeratotic and psoriasiform lesions favoring the ears, nails, and nose.9
Psoriasis vulgaris is a common chronic inflammatory skin condition that usually presents as well-demarcated plaques with silvery scale and observed pinpoint bleeding when layers of scale are removed (Auspitz sign). Lesions typically are found on the extensor surfaces of the body in addition to the neck, feet, hands, and trunk. Treatment of psoriasis vulgaris ranges from topical steroids for mild cases to systemic biologics for moderate to severe circumstances.10 In our patient, topical triamcinolone offered little relief.
Acrodermatitis enteropathica displays clinical and histologic characteristics analogous to many deficiency dermatoses and may represent a spectrum of disease. Because the clinicopathologic findings are nonspecific, it is critical to obtain a comprehensive history and maintain a high index of suspicion in patients with risk factors for malnutrition. The treatment for AE is supplemental oral zinc usually initiated at 0.5 to 1 mg/kg daily in children and 30 to 45 mg daily in adults.3 Our patient initially was prescribed oral zinc supplementation; however, at 1-month follow-up, the rash had not improved. Failure of zinc monotherapy supports a multifactorial nutritional deficiency, which necessitated comprehensive nutritional appraisal and supplementation in our patient. Due to the steatorrhea, fecal pancreatic elastase levels were evaluated and were less than 15 μg/g (reference range, ≥201 μg/g), confirming pancreatic exocrine insufficiency, a known complication of Roux-en-Y gastric bypass.11 Pancrelipase 500 U/kg per meal was added in addition to zinc oxide 40% paste to apply to the rash twice daily, with more frequent applications to the anogenital regions after bowel movements. The patient had substantial clinical improvement after 2 months.
- Shahsavari D, Ahmed Z, Karikkineth A, et al. Zinc-deficiency acrodermatitis in a patient with chronic alcoholism and gastric bypass: a case report. J Community Hosp Intern Med Perspect. 2014. doi:10.3402/jchimp.v4.24707
- Kelly S, Stelzer JW, Esplin N, et al. Acquired acrodermatitis enteropathica: a case study. Cureus. 2017;9:E1667.
- Guliani A, Bishnoi A. Acquired acrodermatitis enteropathica. JAMA Dermatol. 2019;155:1305.
- Baruch D, Naga L, Driscoll M, et al. Acrodermatitis enteropathica from zinc-deficient total parenteral nutrition. Cutis. 2018;101:450-453.
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537.
- Botelho LF, Enokihara MM, Enokihara MY. Necrolytic acral erythema: a rare skin disease associated with hepatitis C virus infection. An Bras Dermatol. 2016;91:649-651.
- Abdallah MA, Ghozzi MY, Monib HA, et al. Necrolytic acral erythema: a cutaneous sign of hepatitis C virus infection. J Am Acad Dermatol. 2005;53:247-251.
- Tolliver S, Graham J, Kaffenberger BH. A review of cutaneous manifestations within glucagonoma syndrome: necrolytic migratory erythema. Int J Dermatol. 2018;57:642-645.
- Poligone B, Christensen SR, Lazova R, et al. Bazex syndrome (acrokeratosis paraneoplastica). Lancet. 2007;369:530. 10. Kupetsky EA, Keller M. Psoriasis vulgaris: an evidencebased guide for primary care. J Am Board Fam Med. 2013; 26:787-801.
- Borbély Y, Plebani A, Kröll D, et al. Exocrine pancreatic insufficiency after Roux-en-Y gastric bypass. Surg Obes Relat Dis. 2016;12:790-794.
The Diagnosis: Acquired Acrodermatitis Enteropathica
A punch biopsy of an elevated scaly border of the rash on the thigh revealed parakeratosis, absence of the granular layer, and epidermal pallor with psoriasiform and spongiotic dermatitis (Figure). Serum zinc levels were 60.1 μg/dL (reference range, 75.0–120.0 μg/dL), suggestive of a nutritional deficiency dermatitis. Laboratory and histopathologic findings were most consistent with a diagnosis of acquired acrodermatitis enteropathica (AE).
Acrodermatitis enteropathica has been associated with Roux-en-Y gastric bypass and alcohol use disorder working synergistically to cause malabsorption and malnutrition, respectively.1 Zinc functions in the structural integrity, wound healing, and anti-inflammatory properties of the skin. There is a 17.3% risk for hypozincemia worldwide; in developed nations there is an estimated 3% to 10% occurrence rate.2 Acrodermatitis enteropathica can be classified as either acquired or hereditary. Both classically present as a triad of acral dermatitis, diarrhea, and alopecia, though the complete triad is seen in 20% of cases.3,4
Hereditary AE is an autosomal-recessive disorder presenting in infancy that results in the loss of a zinc transporter. In contrast, acquired AE occurs later in life and usually is seen in patients who have decreased intake, malabsorption, or excessive loss of zinc.4 Acrodermatitis enteropathica is observed in individuals with conditions such as anorexia nervosa, pancreatic insufficiency, celiac disease, Crohn disease, or gastric bypass surgery (as in our case) and alcohol recidivism. In early disease, AE often presents with angular cheilitis and paronychia, but if left untreated, it can progress to mental status changes, hypogonadism, and depression.4 Acrodermatitis enteropathica presents as erythematous, erosive, scaly plaques or a papulosquamous psoriasiform rash with well-demarcated borders typically involving the orificial, acral, and intertriginous areas of the body.1,4
Acrodermatitis enteropathica belongs to a family of deficiency dermatoses that includes pellagra, necrolytic acral erythema (NAE), and necrolytic migratory erythema (NME).5 It is important to distinguish AE from NAE, as they can present similarly with well-defined and tender psoriasiform lesions peripherally. Histologically, NAE mimics AE with psoriasiform hyperplasia with parakeratosis.6 Necrolytic acral erythema characteristically is associated with active hepatitis C infection, which was absent in our patient.7
Similar to AE, NME affects the perineal and intertriginous surfaces.8 However, necrolytic migratory erythema has cutaneous manifestations in up to 70% of patients with glucagonoma syndrome, which classically presents as a triad of NME, weight loss, and diabetes mellitus.5 Laboratory studies show marked hyperglucagonemia, and imaging reveals enteropancreatic neoplasia. Necrolytic migratory erythema will rapidly resolve once the glucagonoma has been surgically removed.5 Bazex syndrome, or acrokeratosis paraneoplastica, is a paraneoplastic skin disease that is linked to underlying aerodigestive tract malignancies.
Bazex syndrome clinically is characterized by hyperkeratotic and psoriasiform lesions favoring the ears, nails, and nose.9
Psoriasis vulgaris is a common chronic inflammatory skin condition that usually presents as well-demarcated plaques with silvery scale and observed pinpoint bleeding when layers of scale are removed (Auspitz sign). Lesions typically are found on the extensor surfaces of the body in addition to the neck, feet, hands, and trunk. Treatment of psoriasis vulgaris ranges from topical steroids for mild cases to systemic biologics for moderate to severe circumstances.10 In our patient, topical triamcinolone offered little relief.
Acrodermatitis enteropathica displays clinical and histologic characteristics analogous to many deficiency dermatoses and may represent a spectrum of disease. Because the clinicopathologic findings are nonspecific, it is critical to obtain a comprehensive history and maintain a high index of suspicion in patients with risk factors for malnutrition. The treatment for AE is supplemental oral zinc usually initiated at 0.5 to 1 mg/kg daily in children and 30 to 45 mg daily in adults.3 Our patient initially was prescribed oral zinc supplementation; however, at 1-month follow-up, the rash had not improved. Failure of zinc monotherapy supports a multifactorial nutritional deficiency, which necessitated comprehensive nutritional appraisal and supplementation in our patient. Due to the steatorrhea, fecal pancreatic elastase levels were evaluated and were less than 15 μg/g (reference range, ≥201 μg/g), confirming pancreatic exocrine insufficiency, a known complication of Roux-en-Y gastric bypass.11 Pancrelipase 500 U/kg per meal was added in addition to zinc oxide 40% paste to apply to the rash twice daily, with more frequent applications to the anogenital regions after bowel movements. The patient had substantial clinical improvement after 2 months.
The Diagnosis: Acquired Acrodermatitis Enteropathica
A punch biopsy of an elevated scaly border of the rash on the thigh revealed parakeratosis, absence of the granular layer, and epidermal pallor with psoriasiform and spongiotic dermatitis (Figure). Serum zinc levels were 60.1 μg/dL (reference range, 75.0–120.0 μg/dL), suggestive of a nutritional deficiency dermatitis. Laboratory and histopathologic findings were most consistent with a diagnosis of acquired acrodermatitis enteropathica (AE).
Acrodermatitis enteropathica has been associated with Roux-en-Y gastric bypass and alcohol use disorder working synergistically to cause malabsorption and malnutrition, respectively.1 Zinc functions in the structural integrity, wound healing, and anti-inflammatory properties of the skin. There is a 17.3% risk for hypozincemia worldwide; in developed nations there is an estimated 3% to 10% occurrence rate.2 Acrodermatitis enteropathica can be classified as either acquired or hereditary. Both classically present as a triad of acral dermatitis, diarrhea, and alopecia, though the complete triad is seen in 20% of cases.3,4
Hereditary AE is an autosomal-recessive disorder presenting in infancy that results in the loss of a zinc transporter. In contrast, acquired AE occurs later in life and usually is seen in patients who have decreased intake, malabsorption, or excessive loss of zinc.4 Acrodermatitis enteropathica is observed in individuals with conditions such as anorexia nervosa, pancreatic insufficiency, celiac disease, Crohn disease, or gastric bypass surgery (as in our case) and alcohol recidivism. In early disease, AE often presents with angular cheilitis and paronychia, but if left untreated, it can progress to mental status changes, hypogonadism, and depression.4 Acrodermatitis enteropathica presents as erythematous, erosive, scaly plaques or a papulosquamous psoriasiform rash with well-demarcated borders typically involving the orificial, acral, and intertriginous areas of the body.1,4
Acrodermatitis enteropathica belongs to a family of deficiency dermatoses that includes pellagra, necrolytic acral erythema (NAE), and necrolytic migratory erythema (NME).5 It is important to distinguish AE from NAE, as they can present similarly with well-defined and tender psoriasiform lesions peripherally. Histologically, NAE mimics AE with psoriasiform hyperplasia with parakeratosis.6 Necrolytic acral erythema characteristically is associated with active hepatitis C infection, which was absent in our patient.7
Similar to AE, NME affects the perineal and intertriginous surfaces.8 However, necrolytic migratory erythema has cutaneous manifestations in up to 70% of patients with glucagonoma syndrome, which classically presents as a triad of NME, weight loss, and diabetes mellitus.5 Laboratory studies show marked hyperglucagonemia, and imaging reveals enteropancreatic neoplasia. Necrolytic migratory erythema will rapidly resolve once the glucagonoma has been surgically removed.5 Bazex syndrome, or acrokeratosis paraneoplastica, is a paraneoplastic skin disease that is linked to underlying aerodigestive tract malignancies.
Bazex syndrome clinically is characterized by hyperkeratotic and psoriasiform lesions favoring the ears, nails, and nose.9
Psoriasis vulgaris is a common chronic inflammatory skin condition that usually presents as well-demarcated plaques with silvery scale and observed pinpoint bleeding when layers of scale are removed (Auspitz sign). Lesions typically are found on the extensor surfaces of the body in addition to the neck, feet, hands, and trunk. Treatment of psoriasis vulgaris ranges from topical steroids for mild cases to systemic biologics for moderate to severe circumstances.10 In our patient, topical triamcinolone offered little relief.
Acrodermatitis enteropathica displays clinical and histologic characteristics analogous to many deficiency dermatoses and may represent a spectrum of disease. Because the clinicopathologic findings are nonspecific, it is critical to obtain a comprehensive history and maintain a high index of suspicion in patients with risk factors for malnutrition. The treatment for AE is supplemental oral zinc usually initiated at 0.5 to 1 mg/kg daily in children and 30 to 45 mg daily in adults.3 Our patient initially was prescribed oral zinc supplementation; however, at 1-month follow-up, the rash had not improved. Failure of zinc monotherapy supports a multifactorial nutritional deficiency, which necessitated comprehensive nutritional appraisal and supplementation in our patient. Due to the steatorrhea, fecal pancreatic elastase levels were evaluated and were less than 15 μg/g (reference range, ≥201 μg/g), confirming pancreatic exocrine insufficiency, a known complication of Roux-en-Y gastric bypass.11 Pancrelipase 500 U/kg per meal was added in addition to zinc oxide 40% paste to apply to the rash twice daily, with more frequent applications to the anogenital regions after bowel movements. The patient had substantial clinical improvement after 2 months.
- Shahsavari D, Ahmed Z, Karikkineth A, et al. Zinc-deficiency acrodermatitis in a patient with chronic alcoholism and gastric bypass: a case report. J Community Hosp Intern Med Perspect. 2014. doi:10.3402/jchimp.v4.24707
- Kelly S, Stelzer JW, Esplin N, et al. Acquired acrodermatitis enteropathica: a case study. Cureus. 2017;9:E1667.
- Guliani A, Bishnoi A. Acquired acrodermatitis enteropathica. JAMA Dermatol. 2019;155:1305.
- Baruch D, Naga L, Driscoll M, et al. Acrodermatitis enteropathica from zinc-deficient total parenteral nutrition. Cutis. 2018;101:450-453.
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537.
- Botelho LF, Enokihara MM, Enokihara MY. Necrolytic acral erythema: a rare skin disease associated with hepatitis C virus infection. An Bras Dermatol. 2016;91:649-651.
- Abdallah MA, Ghozzi MY, Monib HA, et al. Necrolytic acral erythema: a cutaneous sign of hepatitis C virus infection. J Am Acad Dermatol. 2005;53:247-251.
- Tolliver S, Graham J, Kaffenberger BH. A review of cutaneous manifestations within glucagonoma syndrome: necrolytic migratory erythema. Int J Dermatol. 2018;57:642-645.
- Poligone B, Christensen SR, Lazova R, et al. Bazex syndrome (acrokeratosis paraneoplastica). Lancet. 2007;369:530. 10. Kupetsky EA, Keller M. Psoriasis vulgaris: an evidencebased guide for primary care. J Am Board Fam Med. 2013; 26:787-801.
- Borbély Y, Plebani A, Kröll D, et al. Exocrine pancreatic insufficiency after Roux-en-Y gastric bypass. Surg Obes Relat Dis. 2016;12:790-794.
- Shahsavari D, Ahmed Z, Karikkineth A, et al. Zinc-deficiency acrodermatitis in a patient with chronic alcoholism and gastric bypass: a case report. J Community Hosp Intern Med Perspect. 2014. doi:10.3402/jchimp.v4.24707
- Kelly S, Stelzer JW, Esplin N, et al. Acquired acrodermatitis enteropathica: a case study. Cureus. 2017;9:E1667.
- Guliani A, Bishnoi A. Acquired acrodermatitis enteropathica. JAMA Dermatol. 2019;155:1305.
- Baruch D, Naga L, Driscoll M, et al. Acrodermatitis enteropathica from zinc-deficient total parenteral nutrition. Cutis. 2018;101:450-453.
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537.
- Botelho LF, Enokihara MM, Enokihara MY. Necrolytic acral erythema: a rare skin disease associated with hepatitis C virus infection. An Bras Dermatol. 2016;91:649-651.
- Abdallah MA, Ghozzi MY, Monib HA, et al. Necrolytic acral erythema: a cutaneous sign of hepatitis C virus infection. J Am Acad Dermatol. 2005;53:247-251.
- Tolliver S, Graham J, Kaffenberger BH. A review of cutaneous manifestations within glucagonoma syndrome: necrolytic migratory erythema. Int J Dermatol. 2018;57:642-645.
- Poligone B, Christensen SR, Lazova R, et al. Bazex syndrome (acrokeratosis paraneoplastica). Lancet. 2007;369:530. 10. Kupetsky EA, Keller M. Psoriasis vulgaris: an evidencebased guide for primary care. J Am Board Fam Med. 2013; 26:787-801.
- Borbély Y, Plebani A, Kröll D, et al. Exocrine pancreatic insufficiency after Roux-en-Y gastric bypass. Surg Obes Relat Dis. 2016;12:790-794.
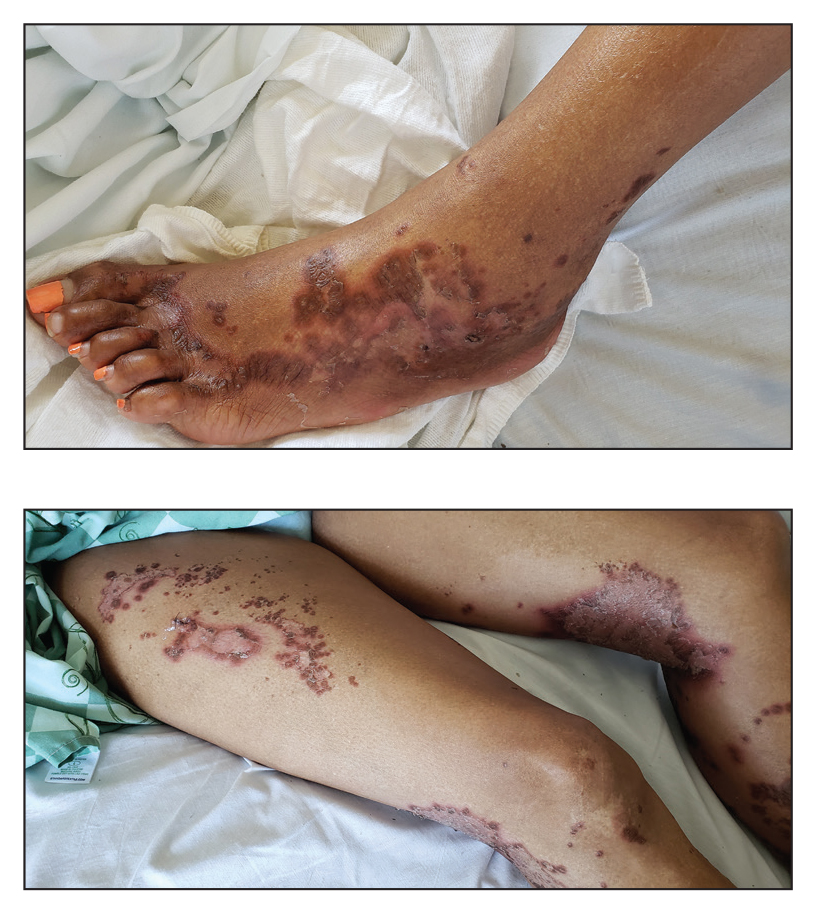
A 45-year-old woman presented to the emergency department with a painful skin eruption and malaise of 5 weeks’ duration. She had an orthotopic liver transplant 5 years prior for end-stage liver disease due to mixed nonalcoholic and alcoholic steatohepatitis and was on mycophenolate mofetil and tacrolimus for graft rejection prophylaxis. Her medical history also included Roux-en-Y gastric bypass 15 years prior, alcohol use disorder, hypothyroidism, and depression.
The exanthem began on the legs as pruritic, red, raised, exudative lesions that gradually crusted. Over the 2 weeks prior to the current presentation, the rash became tender as it spread to the feet, thighs, perianal skin, buttocks, and elbows. Triamcinolone ointment prescribed for a presumed nummular dermatitis effected marginal benefit. A review of systems was notable for a 15-pound weight loss over several weeks; lowgrade fever of 3 days’ duration; epigastric abdominal pain; and long-standing, frequent defecation of oily, foul-smelling feces.
Physical examination revealed a combination of flat-topped, violaceous papules and serpiginous, polycyclic, annular plaques coalescing to form larger psoriasiform plaques with hyperkeratotic rims and dusky borders on the dorsal aspect of the feet (top), lateral ankles, legs (bottom), lateral thighs, buttocks, perianal skin, and elbows. Bilateral angular cheilitis, a smooth and fissured tongue, and pitting of all fingernails were noted.
Chronic Hyperpigmented Patches on the Legs
The Diagnosis: Drug-Induced Hyperpigmentation
Additional history provided by the patient’s caretaker elucidated an extensive list of medications including chlorpromazine and minocycline, among several others. The caretaker revealed that the patient began treatment for acne vulgaris 2 years prior; despite the acne resolving, therapy was not discontinued. The blue-gray and brown pigmentation on our patient’s shins likely was attributed to a medication he was taking.
Both chlorpromazine and minocycline, among many other medications, are known to cause abnormal pigmentation of the skin.1 Minocycline is a tetracycline antibiotic prescribed for acne and other inflammatory cutaneous conditions. It is highly lipophilic, allowing it to reach high drug concentrations in the skin and nail unit.2 Patients taking minocycline long term and at high doses are at greatest risk for pigment deposition.3,4
Minocycline-induced hyperpigmentation is classified into 3 types. Type I describes blue-black deposition of pigment in acne scars and areas of inflammation, typically on facial skin.1,5 Histologically, type I stains positive for Perls Prussian blue, indicating an increased deposition of iron as hemosiderin,1 which likely occurs because minocycline is thought to play a role in defective clearance of hemosiderin from the dermis of injured tissue.5 Type II hyperpigmentation presents as bluegray pigment on the lower legs and occasionally the arms.6,7 Type II stains positive for both Perls Prussian blue and Fontana-Masson, demonstrating hemosiderin and melanin, respectively.6 The third form of hyperpigmentation results in diffuse, dark brown to gray pigmentation with a predilection for sun-exposed areas.8 Histology of type III shows increased pigment in the basal portion of the epidermis and brown-black pigment in macrophages of the dermis. Type III stains positive for Fontana-Masson and negative for Perls Prussian blue. The etiology of hyperpigmentation has been suspected to be caused by minocycline stimulating melanin production and/or deposition of minocycline-melanin complexes in dermal macrophages after a certain drug level; this largely is seen in patients receiving 100 to 200 mg daily as early as 1 year into treatment.8
Chlorpromazine is a typical antipsychotic that causes abnormal skin pigmentation in sun-exposed areas due to increased melanogenesis.9 Similar to type III minocyclineinduced hyperpigmentation, a histologic specimen may stain positive for Fontana-Masson yet negative for Perls Prussian blue. Lal et al10 demonstrated complete resolution of abnormal skin pigmentation within 5 years after stopping chlorpromazine. In contrast, minocyclineinduced hyperpigmentation may be permanent in some cases. There is substantial clinical and histologic overlap for drug-induced hyperpigmentation etiologies; it would behoove the clinician to focus on the most common locations affected and the generalized coloration.
Treatment of minocycline-induced hyperpigmentation includes the use of Q-switched lasers, specifically Q-switched ruby and Q-switched alexandrite.11 The use of the Q-switched Nd:YAG laser appears to be ineffective at clearing minocycline-induced pigmentation.7,11 In our patient, minocycline was discontinued immediately. Due to the patient’s critical condition, he deferred all other therapy. Erythema dyschromicum perstans, also referred to as ashy dermatosis, is an idiopathic form of hyperpigmentation.12 Lesions start as blue-gray to ashy gray macules, occasionally surrounded by a slightly erythematous, raised border.
Erythema dyschromicum perstans typically presents on the trunk, face, and arms of patients with Fitzpatrick skin types III and IV; it is considered a variant of lichen planus actinicus.12 Histologically, erythema dyschromicum perstans may mimic lichen planus pigmentosus (LPP); however, subtle differences exist to distinguish the 2 conditions. Erythema dyschromicum perstans demonstrates a mild lichenoid infiltrate, focal basal vacuolization at the dermoepidermal junction, and melanophage deposition.13 In contrast, LPP demonstrates pigmentary incontinence and a more severe inflammatory infiltrate. A perifollicular infiltrate and fibrosis also can be seen in LPP, which may explain the frontal fibrosing alopecia that often precedes LPP.13
Addison disease, also known as primary adrenal insufficiency, can cause diffuse hyperpigmentation in the skin, mucosae, and nail beds. The pigmentation is prominent in regions of naturally increased pigmentation, such as the flexural surfaces and intertriginous areas.14 Patients with adrenal insufficiency will have accompanying weight loss, hypotension, and fatigue, among other symptoms related to deficiency of cortisol and aldosterone. Skin biopsy shows acanthosis, hyperkeratosis, focal parakeratosis, spongiosis, superficial perivascular lymphocytic infiltrate, basal melanin deposition, and superficial dermal macrophages.15
Confluent and reticulated papillomatosis is an uncommon dermatosis that presents with multiple hyperpigmented macules and papules that coalesce to form patches and plaques centrally with reticulation in the periphery.16 Confluent and reticulated papillomatosis commonly presents on the upper trunk, axillae, and neck, though involvement can include flexural surfaces as well as the lower trunk and legs.16,17 Biopsy demonstrates undulating hyperkeratosis, papillomatosis, acanthosis, and negative fungal staining.16
Pretibial myxedema most commonly is associated with Graves disease and presents as well-defined thickening and induration with overlying pink or purple-brown papules in the pretibial region.18 An acral surface and mucin deposition within the entire dermis may be appreciated on histology with staining for colloidal iron or Alcian blue.
- Fenske NA, Millns JL, Greer KE. Minocycline-induced pigmentation at sites of cutaneous inflammation. JAMA. 1980;244:1103-1106. doi:10.1001/jama.1980.03310100021021
- Snodgrass A, Motaparthi K. Systemic antibacterial agents. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2020:69-98.
- Eisen D, Hakim MD. Minocycline-induced pigmentation. incidence, prevention and management. Drug Saf. 1998;18:431-440. doi:10.2165/00002018-199818060-00004
- Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. 1996;134:693-695. doi:10.1111/j.1365-2133.1996.tb06972.x
- Basler RS, Kohnen PW. Localized hemosiderosis as a sequela of acne. Arch Dermatol. 1978;114:1695-1697.
- Ridgway HA, Sonnex TS, Kennedy CT, et al. Hyperpigmentation associated with oral minocycline. Br J Dermatol. 1982;107:95-102. doi:10.1111/j.1365-2133.1982.tb00296.x
- Nisar MS, Iyer K, Brodell RT, et al. Minocycline-induced hyperpigmentation: comparison of 3 Q-switched lasers to reverse its effects. Clin Cosmet Investig Dermatol. 2013;6:159-162. doi:10.2147/CCID.S42166
- Simons JJ, Morales A. Minocycline and generalized cutaneous pigmentation. J Am Acad Dermatol. 1980;3:244-247. doi:10.1016/s0190 -9622(80)80186-1
- Perry TL, Culling CF, Berry K, et al. 7-Hydroxychlorpromazine: potential toxic drug metabolite in psychiatric patients. Science. 1964;146:81-83. doi:10.1126/science.146.3640.81
- Lal S, Bloom D, Silver B, et al. Replacement of chlorpromazine with other neuroleptics: effect on abnormal skin pigmentation and ocular changes. J Psychiatry Neurosci. 1993;18:173-177.
- Tsao H, Busam K, Barnhill RL, et al. Treatment of minocycline-induced hyperpigmentation with the Q-switched ruby laser. Arch Dermatol. 1996;132:1250-1251.
- Knox JM, Dodge BG, Freeman RG. Erythema dyschromicum perstans. Arch Dermatol. 1968;97:262-272. doi:10.1001 /archderm.1968.01610090034006
- Rutnin S, Udompanich S, Pratumchart N, et al. Ashy dermatosis and lichen planus pigmentosus: the histopathological differences. Biomed Res Int. 2019;2019:5829185. doi:10.1155/2019/5829185
- Montgomery H, O’Leary PA. Pigmentation of the skin in Addison’s disease, acanthosis nigricans and hemochromatosis. Arch Derm Syphilol. 1930;21:970-984. doi:10.1001 /archderm.1930.01440120072005
- Fernandez-Flores A, Cassarino DS. Histopathologic findings of cutaneous hyperpigmentation in Addison disease and immunostain of the melanocytic population. Am J Dermatopathol. 2017;39:924-927. doi:10.1097/DAD.0000000000000937
- Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): a minocycline-responsive dermatosis without evidence for yeast in pathogenesis. a study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol. 2006;154:287-293. doi:10.1111/j.1365-2133.2005.06955.x
- Jo S, Park HS, Cho S, et al. Updated diagnosis criteria for confluent and reticulated papillomatosis: a case report. Ann Dermatol. 2014; 26:409-410. doi:10.5021/ad.2014.26.3.409
- Lause M, Kamboj A, Fernandez Faith E. Dermatologic manifestations of endocrine disorders. Transl Pediatr. 2017;6:300-312. doi:10.21037 /tp.2017.09.08
The Diagnosis: Drug-Induced Hyperpigmentation
Additional history provided by the patient’s caretaker elucidated an extensive list of medications including chlorpromazine and minocycline, among several others. The caretaker revealed that the patient began treatment for acne vulgaris 2 years prior; despite the acne resolving, therapy was not discontinued. The blue-gray and brown pigmentation on our patient’s shins likely was attributed to a medication he was taking.
Both chlorpromazine and minocycline, among many other medications, are known to cause abnormal pigmentation of the skin.1 Minocycline is a tetracycline antibiotic prescribed for acne and other inflammatory cutaneous conditions. It is highly lipophilic, allowing it to reach high drug concentrations in the skin and nail unit.2 Patients taking minocycline long term and at high doses are at greatest risk for pigment deposition.3,4
Minocycline-induced hyperpigmentation is classified into 3 types. Type I describes blue-black deposition of pigment in acne scars and areas of inflammation, typically on facial skin.1,5 Histologically, type I stains positive for Perls Prussian blue, indicating an increased deposition of iron as hemosiderin,1 which likely occurs because minocycline is thought to play a role in defective clearance of hemosiderin from the dermis of injured tissue.5 Type II hyperpigmentation presents as bluegray pigment on the lower legs and occasionally the arms.6,7 Type II stains positive for both Perls Prussian blue and Fontana-Masson, demonstrating hemosiderin and melanin, respectively.6 The third form of hyperpigmentation results in diffuse, dark brown to gray pigmentation with a predilection for sun-exposed areas.8 Histology of type III shows increased pigment in the basal portion of the epidermis and brown-black pigment in macrophages of the dermis. Type III stains positive for Fontana-Masson and negative for Perls Prussian blue. The etiology of hyperpigmentation has been suspected to be caused by minocycline stimulating melanin production and/or deposition of minocycline-melanin complexes in dermal macrophages after a certain drug level; this largely is seen in patients receiving 100 to 200 mg daily as early as 1 year into treatment.8
Chlorpromazine is a typical antipsychotic that causes abnormal skin pigmentation in sun-exposed areas due to increased melanogenesis.9 Similar to type III minocyclineinduced hyperpigmentation, a histologic specimen may stain positive for Fontana-Masson yet negative for Perls Prussian blue. Lal et al10 demonstrated complete resolution of abnormal skin pigmentation within 5 years after stopping chlorpromazine. In contrast, minocyclineinduced hyperpigmentation may be permanent in some cases. There is substantial clinical and histologic overlap for drug-induced hyperpigmentation etiologies; it would behoove the clinician to focus on the most common locations affected and the generalized coloration.
Treatment of minocycline-induced hyperpigmentation includes the use of Q-switched lasers, specifically Q-switched ruby and Q-switched alexandrite.11 The use of the Q-switched Nd:YAG laser appears to be ineffective at clearing minocycline-induced pigmentation.7,11 In our patient, minocycline was discontinued immediately. Due to the patient’s critical condition, he deferred all other therapy. Erythema dyschromicum perstans, also referred to as ashy dermatosis, is an idiopathic form of hyperpigmentation.12 Lesions start as blue-gray to ashy gray macules, occasionally surrounded by a slightly erythematous, raised border.
Erythema dyschromicum perstans typically presents on the trunk, face, and arms of patients with Fitzpatrick skin types III and IV; it is considered a variant of lichen planus actinicus.12 Histologically, erythema dyschromicum perstans may mimic lichen planus pigmentosus (LPP); however, subtle differences exist to distinguish the 2 conditions. Erythema dyschromicum perstans demonstrates a mild lichenoid infiltrate, focal basal vacuolization at the dermoepidermal junction, and melanophage deposition.13 In contrast, LPP demonstrates pigmentary incontinence and a more severe inflammatory infiltrate. A perifollicular infiltrate and fibrosis also can be seen in LPP, which may explain the frontal fibrosing alopecia that often precedes LPP.13
Addison disease, also known as primary adrenal insufficiency, can cause diffuse hyperpigmentation in the skin, mucosae, and nail beds. The pigmentation is prominent in regions of naturally increased pigmentation, such as the flexural surfaces and intertriginous areas.14 Patients with adrenal insufficiency will have accompanying weight loss, hypotension, and fatigue, among other symptoms related to deficiency of cortisol and aldosterone. Skin biopsy shows acanthosis, hyperkeratosis, focal parakeratosis, spongiosis, superficial perivascular lymphocytic infiltrate, basal melanin deposition, and superficial dermal macrophages.15
Confluent and reticulated papillomatosis is an uncommon dermatosis that presents with multiple hyperpigmented macules and papules that coalesce to form patches and plaques centrally with reticulation in the periphery.16 Confluent and reticulated papillomatosis commonly presents on the upper trunk, axillae, and neck, though involvement can include flexural surfaces as well as the lower trunk and legs.16,17 Biopsy demonstrates undulating hyperkeratosis, papillomatosis, acanthosis, and negative fungal staining.16
Pretibial myxedema most commonly is associated with Graves disease and presents as well-defined thickening and induration with overlying pink or purple-brown papules in the pretibial region.18 An acral surface and mucin deposition within the entire dermis may be appreciated on histology with staining for colloidal iron or Alcian blue.
The Diagnosis: Drug-Induced Hyperpigmentation
Additional history provided by the patient’s caretaker elucidated an extensive list of medications including chlorpromazine and minocycline, among several others. The caretaker revealed that the patient began treatment for acne vulgaris 2 years prior; despite the acne resolving, therapy was not discontinued. The blue-gray and brown pigmentation on our patient’s shins likely was attributed to a medication he was taking.
Both chlorpromazine and minocycline, among many other medications, are known to cause abnormal pigmentation of the skin.1 Minocycline is a tetracycline antibiotic prescribed for acne and other inflammatory cutaneous conditions. It is highly lipophilic, allowing it to reach high drug concentrations in the skin and nail unit.2 Patients taking minocycline long term and at high doses are at greatest risk for pigment deposition.3,4
Minocycline-induced hyperpigmentation is classified into 3 types. Type I describes blue-black deposition of pigment in acne scars and areas of inflammation, typically on facial skin.1,5 Histologically, type I stains positive for Perls Prussian blue, indicating an increased deposition of iron as hemosiderin,1 which likely occurs because minocycline is thought to play a role in defective clearance of hemosiderin from the dermis of injured tissue.5 Type II hyperpigmentation presents as bluegray pigment on the lower legs and occasionally the arms.6,7 Type II stains positive for both Perls Prussian blue and Fontana-Masson, demonstrating hemosiderin and melanin, respectively.6 The third form of hyperpigmentation results in diffuse, dark brown to gray pigmentation with a predilection for sun-exposed areas.8 Histology of type III shows increased pigment in the basal portion of the epidermis and brown-black pigment in macrophages of the dermis. Type III stains positive for Fontana-Masson and negative for Perls Prussian blue. The etiology of hyperpigmentation has been suspected to be caused by minocycline stimulating melanin production and/or deposition of minocycline-melanin complexes in dermal macrophages after a certain drug level; this largely is seen in patients receiving 100 to 200 mg daily as early as 1 year into treatment.8
Chlorpromazine is a typical antipsychotic that causes abnormal skin pigmentation in sun-exposed areas due to increased melanogenesis.9 Similar to type III minocyclineinduced hyperpigmentation, a histologic specimen may stain positive for Fontana-Masson yet negative for Perls Prussian blue. Lal et al10 demonstrated complete resolution of abnormal skin pigmentation within 5 years after stopping chlorpromazine. In contrast, minocyclineinduced hyperpigmentation may be permanent in some cases. There is substantial clinical and histologic overlap for drug-induced hyperpigmentation etiologies; it would behoove the clinician to focus on the most common locations affected and the generalized coloration.
Treatment of minocycline-induced hyperpigmentation includes the use of Q-switched lasers, specifically Q-switched ruby and Q-switched alexandrite.11 The use of the Q-switched Nd:YAG laser appears to be ineffective at clearing minocycline-induced pigmentation.7,11 In our patient, minocycline was discontinued immediately. Due to the patient’s critical condition, he deferred all other therapy. Erythema dyschromicum perstans, also referred to as ashy dermatosis, is an idiopathic form of hyperpigmentation.12 Lesions start as blue-gray to ashy gray macules, occasionally surrounded by a slightly erythematous, raised border.
Erythema dyschromicum perstans typically presents on the trunk, face, and arms of patients with Fitzpatrick skin types III and IV; it is considered a variant of lichen planus actinicus.12 Histologically, erythema dyschromicum perstans may mimic lichen planus pigmentosus (LPP); however, subtle differences exist to distinguish the 2 conditions. Erythema dyschromicum perstans demonstrates a mild lichenoid infiltrate, focal basal vacuolization at the dermoepidermal junction, and melanophage deposition.13 In contrast, LPP demonstrates pigmentary incontinence and a more severe inflammatory infiltrate. A perifollicular infiltrate and fibrosis also can be seen in LPP, which may explain the frontal fibrosing alopecia that often precedes LPP.13
Addison disease, also known as primary adrenal insufficiency, can cause diffuse hyperpigmentation in the skin, mucosae, and nail beds. The pigmentation is prominent in regions of naturally increased pigmentation, such as the flexural surfaces and intertriginous areas.14 Patients with adrenal insufficiency will have accompanying weight loss, hypotension, and fatigue, among other symptoms related to deficiency of cortisol and aldosterone. Skin biopsy shows acanthosis, hyperkeratosis, focal parakeratosis, spongiosis, superficial perivascular lymphocytic infiltrate, basal melanin deposition, and superficial dermal macrophages.15
Confluent and reticulated papillomatosis is an uncommon dermatosis that presents with multiple hyperpigmented macules and papules that coalesce to form patches and plaques centrally with reticulation in the periphery.16 Confluent and reticulated papillomatosis commonly presents on the upper trunk, axillae, and neck, though involvement can include flexural surfaces as well as the lower trunk and legs.16,17 Biopsy demonstrates undulating hyperkeratosis, papillomatosis, acanthosis, and negative fungal staining.16
Pretibial myxedema most commonly is associated with Graves disease and presents as well-defined thickening and induration with overlying pink or purple-brown papules in the pretibial region.18 An acral surface and mucin deposition within the entire dermis may be appreciated on histology with staining for colloidal iron or Alcian blue.
- Fenske NA, Millns JL, Greer KE. Minocycline-induced pigmentation at sites of cutaneous inflammation. JAMA. 1980;244:1103-1106. doi:10.1001/jama.1980.03310100021021
- Snodgrass A, Motaparthi K. Systemic antibacterial agents. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2020:69-98.
- Eisen D, Hakim MD. Minocycline-induced pigmentation. incidence, prevention and management. Drug Saf. 1998;18:431-440. doi:10.2165/00002018-199818060-00004
- Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. 1996;134:693-695. doi:10.1111/j.1365-2133.1996.tb06972.x
- Basler RS, Kohnen PW. Localized hemosiderosis as a sequela of acne. Arch Dermatol. 1978;114:1695-1697.
- Ridgway HA, Sonnex TS, Kennedy CT, et al. Hyperpigmentation associated with oral minocycline. Br J Dermatol. 1982;107:95-102. doi:10.1111/j.1365-2133.1982.tb00296.x
- Nisar MS, Iyer K, Brodell RT, et al. Minocycline-induced hyperpigmentation: comparison of 3 Q-switched lasers to reverse its effects. Clin Cosmet Investig Dermatol. 2013;6:159-162. doi:10.2147/CCID.S42166
- Simons JJ, Morales A. Minocycline and generalized cutaneous pigmentation. J Am Acad Dermatol. 1980;3:244-247. doi:10.1016/s0190 -9622(80)80186-1
- Perry TL, Culling CF, Berry K, et al. 7-Hydroxychlorpromazine: potential toxic drug metabolite in psychiatric patients. Science. 1964;146:81-83. doi:10.1126/science.146.3640.81
- Lal S, Bloom D, Silver B, et al. Replacement of chlorpromazine with other neuroleptics: effect on abnormal skin pigmentation and ocular changes. J Psychiatry Neurosci. 1993;18:173-177.
- Tsao H, Busam K, Barnhill RL, et al. Treatment of minocycline-induced hyperpigmentation with the Q-switched ruby laser. Arch Dermatol. 1996;132:1250-1251.
- Knox JM, Dodge BG, Freeman RG. Erythema dyschromicum perstans. Arch Dermatol. 1968;97:262-272. doi:10.1001 /archderm.1968.01610090034006
- Rutnin S, Udompanich S, Pratumchart N, et al. Ashy dermatosis and lichen planus pigmentosus: the histopathological differences. Biomed Res Int. 2019;2019:5829185. doi:10.1155/2019/5829185
- Montgomery H, O’Leary PA. Pigmentation of the skin in Addison’s disease, acanthosis nigricans and hemochromatosis. Arch Derm Syphilol. 1930;21:970-984. doi:10.1001 /archderm.1930.01440120072005
- Fernandez-Flores A, Cassarino DS. Histopathologic findings of cutaneous hyperpigmentation in Addison disease and immunostain of the melanocytic population. Am J Dermatopathol. 2017;39:924-927. doi:10.1097/DAD.0000000000000937
- Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): a minocycline-responsive dermatosis without evidence for yeast in pathogenesis. a study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol. 2006;154:287-293. doi:10.1111/j.1365-2133.2005.06955.x
- Jo S, Park HS, Cho S, et al. Updated diagnosis criteria for confluent and reticulated papillomatosis: a case report. Ann Dermatol. 2014; 26:409-410. doi:10.5021/ad.2014.26.3.409
- Lause M, Kamboj A, Fernandez Faith E. Dermatologic manifestations of endocrine disorders. Transl Pediatr. 2017;6:300-312. doi:10.21037 /tp.2017.09.08
- Fenske NA, Millns JL, Greer KE. Minocycline-induced pigmentation at sites of cutaneous inflammation. JAMA. 1980;244:1103-1106. doi:10.1001/jama.1980.03310100021021
- Snodgrass A, Motaparthi K. Systemic antibacterial agents. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2020:69-98.
- Eisen D, Hakim MD. Minocycline-induced pigmentation. incidence, prevention and management. Drug Saf. 1998;18:431-440. doi:10.2165/00002018-199818060-00004
- Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. 1996;134:693-695. doi:10.1111/j.1365-2133.1996.tb06972.x
- Basler RS, Kohnen PW. Localized hemosiderosis as a sequela of acne. Arch Dermatol. 1978;114:1695-1697.
- Ridgway HA, Sonnex TS, Kennedy CT, et al. Hyperpigmentation associated with oral minocycline. Br J Dermatol. 1982;107:95-102. doi:10.1111/j.1365-2133.1982.tb00296.x
- Nisar MS, Iyer K, Brodell RT, et al. Minocycline-induced hyperpigmentation: comparison of 3 Q-switched lasers to reverse its effects. Clin Cosmet Investig Dermatol. 2013;6:159-162. doi:10.2147/CCID.S42166
- Simons JJ, Morales A. Minocycline and generalized cutaneous pigmentation. J Am Acad Dermatol. 1980;3:244-247. doi:10.1016/s0190 -9622(80)80186-1
- Perry TL, Culling CF, Berry K, et al. 7-Hydroxychlorpromazine: potential toxic drug metabolite in psychiatric patients. Science. 1964;146:81-83. doi:10.1126/science.146.3640.81
- Lal S, Bloom D, Silver B, et al. Replacement of chlorpromazine with other neuroleptics: effect on abnormal skin pigmentation and ocular changes. J Psychiatry Neurosci. 1993;18:173-177.
- Tsao H, Busam K, Barnhill RL, et al. Treatment of minocycline-induced hyperpigmentation with the Q-switched ruby laser. Arch Dermatol. 1996;132:1250-1251.
- Knox JM, Dodge BG, Freeman RG. Erythema dyschromicum perstans. Arch Dermatol. 1968;97:262-272. doi:10.1001 /archderm.1968.01610090034006
- Rutnin S, Udompanich S, Pratumchart N, et al. Ashy dermatosis and lichen planus pigmentosus: the histopathological differences. Biomed Res Int. 2019;2019:5829185. doi:10.1155/2019/5829185
- Montgomery H, O’Leary PA. Pigmentation of the skin in Addison’s disease, acanthosis nigricans and hemochromatosis. Arch Derm Syphilol. 1930;21:970-984. doi:10.1001 /archderm.1930.01440120072005
- Fernandez-Flores A, Cassarino DS. Histopathologic findings of cutaneous hyperpigmentation in Addison disease and immunostain of the melanocytic population. Am J Dermatopathol. 2017;39:924-927. doi:10.1097/DAD.0000000000000937
- Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): a minocycline-responsive dermatosis without evidence for yeast in pathogenesis. a study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol. 2006;154:287-293. doi:10.1111/j.1365-2133.2005.06955.x
- Jo S, Park HS, Cho S, et al. Updated diagnosis criteria for confluent and reticulated papillomatosis: a case report. Ann Dermatol. 2014; 26:409-410. doi:10.5021/ad.2014.26.3.409
- Lause M, Kamboj A, Fernandez Faith E. Dermatologic manifestations of endocrine disorders. Transl Pediatr. 2017;6:300-312. doi:10.21037 /tp.2017.09.08
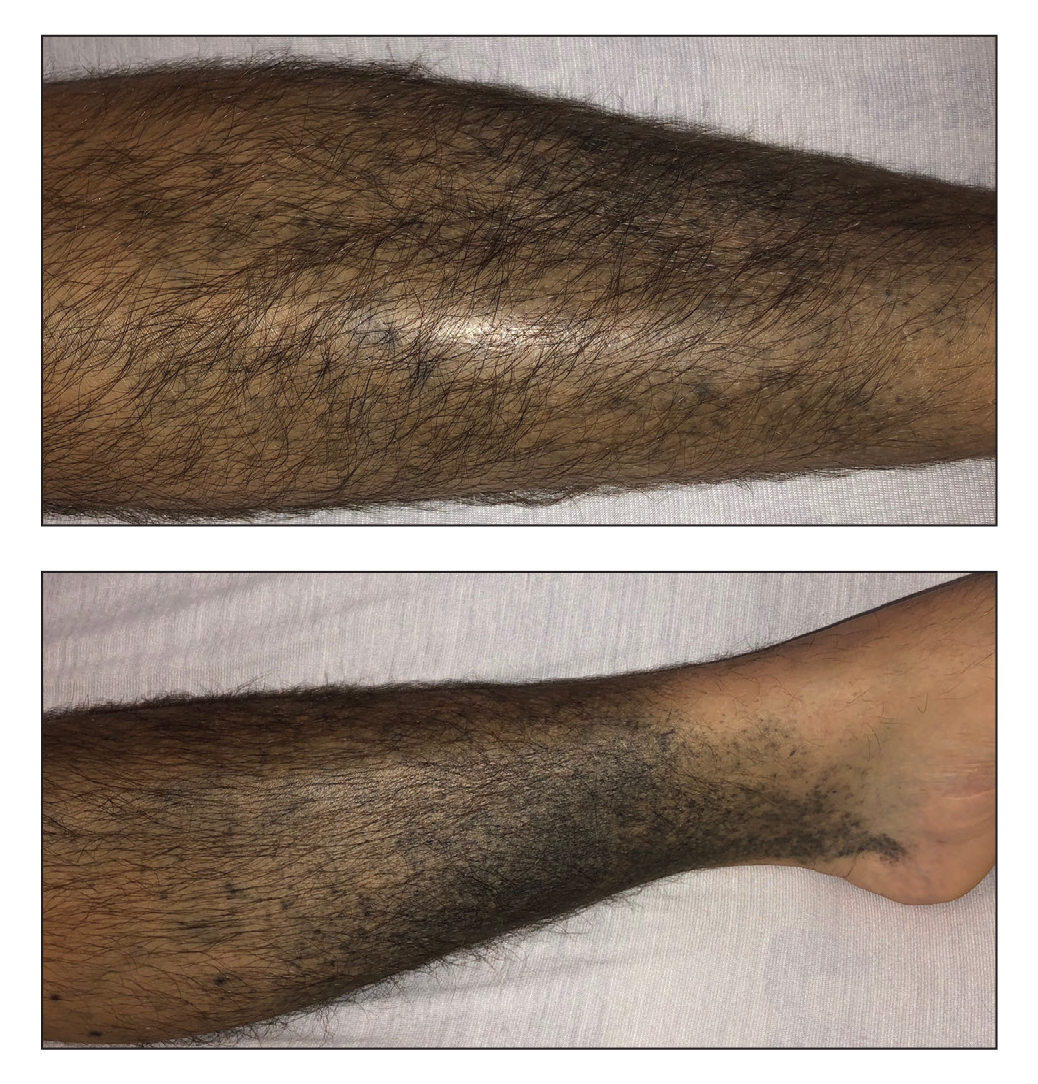
A 37-year-old man with a history of cerebral palsy, bipolar disorder, and impulse control disorder presented to the emergency department with breathing difficulty and worsening malaise. The patient subsequently was intubated due to hypoxic respiratory failure and was found to be positive for SARS-CoV-2. He was admitted to the intensive care unit, and dermatology was consulted due to concern that the cutaneous findings were demonstrative of a vasculitic process. Physical examination revealed diffuse, symmetric, dark brown to blue-gray macules coalescing into patches on the anterior tibia (top) and covering the entire lower leg (bottom). The patches were mottled and did not blanch with pressure. According to the patient’s caretaker, the leg hyperpigmentation had been present for 2 years.