User login
Some cardiac devices vulnerable to cybersecurity threats
Implantable cardiac devices made by Medtronic have cybersecurity vulnerabilities, according to a safety communication from the Food and Drug Administration. That said, so far the FDA is unaware of any reports of harm related to these vulnerabilities, and the agency still advises doctors and patients to continue using the devices as intended and in accordance with device labeling.

The Conexus wireless telemetry protocol used with Medtronic’s implantable cardioverter defibrillators and cardiac resynchronization therapy defibrillators, as well as with certain models of Medtronic’s CareLink Programmer and the MyCareLink Monitor, lacks encryption, authentication, or authorization, which leaves the devices open to exploitation. Such exploitation “could allow unauthorized individuals ... to access and potentially manipulate an implantable device, home monitor, or clinic programmer,” the agency said in its safety communication.
The FDA provides several recommendations in the safety communication, including obtaining these devices “directly from the manufacturer to ensure integrity of the system” and operating “the programmers within well-managed networks.”
Implantable cardiac devices made by Medtronic have cybersecurity vulnerabilities, according to a safety communication from the Food and Drug Administration. That said, so far the FDA is unaware of any reports of harm related to these vulnerabilities, and the agency still advises doctors and patients to continue using the devices as intended and in accordance with device labeling.
The Conexus wireless telemetry protocol used with Medtronic’s implantable cardioverter defibrillators and cardiac resynchronization therapy defibrillators, as well as with certain models of Medtronic’s CareLink Programmer and the MyCareLink Monitor, lacks encryption, authentication, or authorization, which leaves the devices open to exploitation. Such exploitation “could allow unauthorized individuals ... to access and potentially manipulate an implantable device, home monitor, or clinic programmer,” the agency said in its safety communication.
The FDA provides several recommendations in the safety communication, including obtaining these devices “directly from the manufacturer to ensure integrity of the system” and operating “the programmers within well-managed networks.”
Implantable cardiac devices made by Medtronic have cybersecurity vulnerabilities, according to a safety communication from the Food and Drug Administration. That said, so far the FDA is unaware of any reports of harm related to these vulnerabilities, and the agency still advises doctors and patients to continue using the devices as intended and in accordance with device labeling.
The Conexus wireless telemetry protocol used with Medtronic’s implantable cardioverter defibrillators and cardiac resynchronization therapy defibrillators, as well as with certain models of Medtronic’s CareLink Programmer and the MyCareLink Monitor, lacks encryption, authentication, or authorization, which leaves the devices open to exploitation. Such exploitation “could allow unauthorized individuals ... to access and potentially manipulate an implantable device, home monitor, or clinic programmer,” the agency said in its safety communication.
The FDA provides several recommendations in the safety communication, including obtaining these devices “directly from the manufacturer to ensure integrity of the system” and operating “the programmers within well-managed networks.”
FDA panel calls for changes to breast implant rupture screening
A Food and Drug Administration advisory panel urged the agency to switch its recommended screening method for silent breast implant ruptures from MRI to ultrasound and to push the first screening examination back from the current 3 years post implant to 5 years.
Members of the FDA’s General and Plastic Surgery Advisory Panel also made suggestions to the FDA regarding how it might improve communication about the risks of breast implants to the public in general and to people considering implants in particular.
The panel also discussed the sort of safety and efficacy assessments the FDA should require for acellular dermal matrix (ADM), also known as mesh, to add the material’s label for use during breast reconstruction or implant augmentation. Surgeons have used mesh routinely as a surgical aid at other body sites, such as the abdomen. Although ADM is now also widely used during breast surgery, it has never undergone testing or labeling for use in that setting.
The FDA convened the advisory committee meeting largely to assess and discuss data and concerns about two recently appreciated complications of breast implant placement – breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL) and a still poorly defined and described constellation of autoimmune and rheumatoid-like symptoms reported anecdotally by some breast implant recipients called Breast Implant Illness (BII). But agency officials asked the panel to also address these other issues related to the safety of breast implants and implant surgery.
The revised screening recommendations were primarily a response to a lack of compliance with current FDA recommendations to screen for breast implant rupture with MRI starting 3 years after placement and then every 2 years.

The problem is that a screening MRI costs about $1,500-$2,000 and is generally not covered by insurance when done for this purpose, although it is often covered when used to investigate a suspected rupture. The result is that less than 5% of implanted patients comply with the recommended screening schedule, noted committee chair Frank R. Lewis Jr., MD, executive director, emeritus, of the American Board of Surgery in Philadelphia.
“Effectively it’s a useless recommendation,” he said. “Ultrasound is far easier, quicker, and cheaper” and seems effective for screening.
The advisory panel recommended starting ultrasound screening 5 years after implantation, based on MRI screening data showing that virtually all ruptures don’t occur until after 5 years, and then following with ultrasound screening every 3 years after that. The panel recommended using MRI when the ultrasound result is equivocal or when the patient has symptoms suggesting rupture.

After hearing testimony during the sessions from several dozen women who told horror stories of the complications they experienced from breast implants, panel member Karen E. Burke, MD, PhD, spoke for many on the panel when she said “no doubt patients feel that the informed consent process failed them, that they were not aware of the risks.”
Dr. Burke suggested that patients must be informed so that they realize that breast implants are not static objects that will always sit unchanged in their body for the rest of their lives, that certain factors such as allergy or family history of tissue disease might predispose them to autoimmune-type reactions and that the diverse symptoms described for BII are possible sequelae.
A black box warning for the potential of developing anaplastic large-cell lymphoma should also go into the label, said Dr. Burke, a dermatologist who practices in New York City.
Dr. Lewis ridiculed the information booklets that implant manufacturers currently provide for patients as too long and dense. “They were not constructed to inform patients in the best way; they were constructed to provide legal protection.” He called for creating a two- or three-page list of potential adverse effects and points to consider.
Other panel members suggested public service advertisements similar to what is used to inform consumers about the risk from cigarettes. Dr. Burke recommended getting the word out about BII to other medical specialties that are more likely to see affected patients first, such as rheumatologists, immunologists, and dermatologists. She vowed to speak about these complications at an upcoming meeting of the American Academy of Dermatology. But other panel members noted that BII right now remains without any official medical definition nor clear causal link to breast implants.

The question of exactly what safety and efficacy data the FDA might require from manufacturers seeking a breast surgery indication for ADM was less clear.
Binita Ashar, MD, director of the FDA’s Division of Surgical Devices, highlighted the agency’s dilemma about considering data for a breast surgery indication. “The challenge for us is that we can’t expect a control arm because everyone today is using” mesh, she explained. “We’re looking for guidance on how to understand the risk-to-benefit profile” of ADM.
A plastic surgeon on the advisory panel, Pierre M. Chevray, MD, PhD, from Houston Methodist Hospital summarized the way ADM mesh reached its current niche in routine, U.S. breast surgery.
About 20 years ago, plastic surgeons began using mesh during implant surgery to improve eventual breast cosmesis. Surgeons began to wrap the implant in mesh and then attached the mesh to the pectoral muscle so that the implant could go on top of the muscle and not beneath it. It greatly diminished capsular contraction around the implant over time, reduced the risk for implant movement, and allowed for more natural positioning of the breast with the implant inside, he said.

Another factor in the growing use of mesh was heavy promotion by manufacturers to a generation of plastic surgeons, Dr. Chevray said. But use of ADM may also lead to a slightly increased rate of seromas and infections.
“The benefit from mesh is hard to prove and is questionable” because it largely depends on a subjective assessment by a surgeon or patient, Dr. Chevray said. “The cost [of ADM] is substantial, but no data have shown that outcomes are better” with its use. Despite that, “nearly every surgeon uses mesh” these days, he noted.
A Food and Drug Administration advisory panel urged the agency to switch its recommended screening method for silent breast implant ruptures from MRI to ultrasound and to push the first screening examination back from the current 3 years post implant to 5 years.
Members of the FDA’s General and Plastic Surgery Advisory Panel also made suggestions to the FDA regarding how it might improve communication about the risks of breast implants to the public in general and to people considering implants in particular.
The panel also discussed the sort of safety and efficacy assessments the FDA should require for acellular dermal matrix (ADM), also known as mesh, to add the material’s label for use during breast reconstruction or implant augmentation. Surgeons have used mesh routinely as a surgical aid at other body sites, such as the abdomen. Although ADM is now also widely used during breast surgery, it has never undergone testing or labeling for use in that setting.
The FDA convened the advisory committee meeting largely to assess and discuss data and concerns about two recently appreciated complications of breast implant placement – breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL) and a still poorly defined and described constellation of autoimmune and rheumatoid-like symptoms reported anecdotally by some breast implant recipients called Breast Implant Illness (BII). But agency officials asked the panel to also address these other issues related to the safety of breast implants and implant surgery.
The revised screening recommendations were primarily a response to a lack of compliance with current FDA recommendations to screen for breast implant rupture with MRI starting 3 years after placement and then every 2 years.
The problem is that a screening MRI costs about $1,500-$2,000 and is generally not covered by insurance when done for this purpose, although it is often covered when used to investigate a suspected rupture. The result is that less than 5% of implanted patients comply with the recommended screening schedule, noted committee chair Frank R. Lewis Jr., MD, executive director, emeritus, of the American Board of Surgery in Philadelphia.
“Effectively it’s a useless recommendation,” he said. “Ultrasound is far easier, quicker, and cheaper” and seems effective for screening.
The advisory panel recommended starting ultrasound screening 5 years after implantation, based on MRI screening data showing that virtually all ruptures don’t occur until after 5 years, and then following with ultrasound screening every 3 years after that. The panel recommended using MRI when the ultrasound result is equivocal or when the patient has symptoms suggesting rupture.
After hearing testimony during the sessions from several dozen women who told horror stories of the complications they experienced from breast implants, panel member Karen E. Burke, MD, PhD, spoke for many on the panel when she said “no doubt patients feel that the informed consent process failed them, that they were not aware of the risks.”
Dr. Burke suggested that patients must be informed so that they realize that breast implants are not static objects that will always sit unchanged in their body for the rest of their lives, that certain factors such as allergy or family history of tissue disease might predispose them to autoimmune-type reactions and that the diverse symptoms described for BII are possible sequelae.
A black box warning for the potential of developing anaplastic large-cell lymphoma should also go into the label, said Dr. Burke, a dermatologist who practices in New York City.
Dr. Lewis ridiculed the information booklets that implant manufacturers currently provide for patients as too long and dense. “They were not constructed to inform patients in the best way; they were constructed to provide legal protection.” He called for creating a two- or three-page list of potential adverse effects and points to consider.
Other panel members suggested public service advertisements similar to what is used to inform consumers about the risk from cigarettes. Dr. Burke recommended getting the word out about BII to other medical specialties that are more likely to see affected patients first, such as rheumatologists, immunologists, and dermatologists. She vowed to speak about these complications at an upcoming meeting of the American Academy of Dermatology. But other panel members noted that BII right now remains without any official medical definition nor clear causal link to breast implants.
The question of exactly what safety and efficacy data the FDA might require from manufacturers seeking a breast surgery indication for ADM was less clear.
Binita Ashar, MD, director of the FDA’s Division of Surgical Devices, highlighted the agency’s dilemma about considering data for a breast surgery indication. “The challenge for us is that we can’t expect a control arm because everyone today is using” mesh, she explained. “We’re looking for guidance on how to understand the risk-to-benefit profile” of ADM.
A plastic surgeon on the advisory panel, Pierre M. Chevray, MD, PhD, from Houston Methodist Hospital summarized the way ADM mesh reached its current niche in routine, U.S. breast surgery.
About 20 years ago, plastic surgeons began using mesh during implant surgery to improve eventual breast cosmesis. Surgeons began to wrap the implant in mesh and then attached the mesh to the pectoral muscle so that the implant could go on top of the muscle and not beneath it. It greatly diminished capsular contraction around the implant over time, reduced the risk for implant movement, and allowed for more natural positioning of the breast with the implant inside, he said.
Another factor in the growing use of mesh was heavy promotion by manufacturers to a generation of plastic surgeons, Dr. Chevray said. But use of ADM may also lead to a slightly increased rate of seromas and infections.
“The benefit from mesh is hard to prove and is questionable” because it largely depends on a subjective assessment by a surgeon or patient, Dr. Chevray said. “The cost [of ADM] is substantial, but no data have shown that outcomes are better” with its use. Despite that, “nearly every surgeon uses mesh” these days, he noted.
A Food and Drug Administration advisory panel urged the agency to switch its recommended screening method for silent breast implant ruptures from MRI to ultrasound and to push the first screening examination back from the current 3 years post implant to 5 years.
Members of the FDA’s General and Plastic Surgery Advisory Panel also made suggestions to the FDA regarding how it might improve communication about the risks of breast implants to the public in general and to people considering implants in particular.
The panel also discussed the sort of safety and efficacy assessments the FDA should require for acellular dermal matrix (ADM), also known as mesh, to add the material’s label for use during breast reconstruction or implant augmentation. Surgeons have used mesh routinely as a surgical aid at other body sites, such as the abdomen. Although ADM is now also widely used during breast surgery, it has never undergone testing or labeling for use in that setting.
The FDA convened the advisory committee meeting largely to assess and discuss data and concerns about two recently appreciated complications of breast implant placement – breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL) and a still poorly defined and described constellation of autoimmune and rheumatoid-like symptoms reported anecdotally by some breast implant recipients called Breast Implant Illness (BII). But agency officials asked the panel to also address these other issues related to the safety of breast implants and implant surgery.
The revised screening recommendations were primarily a response to a lack of compliance with current FDA recommendations to screen for breast implant rupture with MRI starting 3 years after placement and then every 2 years.
The problem is that a screening MRI costs about $1,500-$2,000 and is generally not covered by insurance when done for this purpose, although it is often covered when used to investigate a suspected rupture. The result is that less than 5% of implanted patients comply with the recommended screening schedule, noted committee chair Frank R. Lewis Jr., MD, executive director, emeritus, of the American Board of Surgery in Philadelphia.
“Effectively it’s a useless recommendation,” he said. “Ultrasound is far easier, quicker, and cheaper” and seems effective for screening.
The advisory panel recommended starting ultrasound screening 5 years after implantation, based on MRI screening data showing that virtually all ruptures don’t occur until after 5 years, and then following with ultrasound screening every 3 years after that. The panel recommended using MRI when the ultrasound result is equivocal or when the patient has symptoms suggesting rupture.
After hearing testimony during the sessions from several dozen women who told horror stories of the complications they experienced from breast implants, panel member Karen E. Burke, MD, PhD, spoke for many on the panel when she said “no doubt patients feel that the informed consent process failed them, that they were not aware of the risks.”
Dr. Burke suggested that patients must be informed so that they realize that breast implants are not static objects that will always sit unchanged in their body for the rest of their lives, that certain factors such as allergy or family history of tissue disease might predispose them to autoimmune-type reactions and that the diverse symptoms described for BII are possible sequelae.
A black box warning for the potential of developing anaplastic large-cell lymphoma should also go into the label, said Dr. Burke, a dermatologist who practices in New York City.
Dr. Lewis ridiculed the information booklets that implant manufacturers currently provide for patients as too long and dense. “They were not constructed to inform patients in the best way; they were constructed to provide legal protection.” He called for creating a two- or three-page list of potential adverse effects and points to consider.
Other panel members suggested public service advertisements similar to what is used to inform consumers about the risk from cigarettes. Dr. Burke recommended getting the word out about BII to other medical specialties that are more likely to see affected patients first, such as rheumatologists, immunologists, and dermatologists. She vowed to speak about these complications at an upcoming meeting of the American Academy of Dermatology. But other panel members noted that BII right now remains without any official medical definition nor clear causal link to breast implants.
The question of exactly what safety and efficacy data the FDA might require from manufacturers seeking a breast surgery indication for ADM was less clear.
Binita Ashar, MD, director of the FDA’s Division of Surgical Devices, highlighted the agency’s dilemma about considering data for a breast surgery indication. “The challenge for us is that we can’t expect a control arm because everyone today is using” mesh, she explained. “We’re looking for guidance on how to understand the risk-to-benefit profile” of ADM.
A plastic surgeon on the advisory panel, Pierre M. Chevray, MD, PhD, from Houston Methodist Hospital summarized the way ADM mesh reached its current niche in routine, U.S. breast surgery.
About 20 years ago, plastic surgeons began using mesh during implant surgery to improve eventual breast cosmesis. Surgeons began to wrap the implant in mesh and then attached the mesh to the pectoral muscle so that the implant could go on top of the muscle and not beneath it. It greatly diminished capsular contraction around the implant over time, reduced the risk for implant movement, and allowed for more natural positioning of the breast with the implant inside, he said.
Another factor in the growing use of mesh was heavy promotion by manufacturers to a generation of plastic surgeons, Dr. Chevray said. But use of ADM may also lead to a slightly increased rate of seromas and infections.
“The benefit from mesh is hard to prove and is questionable” because it largely depends on a subjective assessment by a surgeon or patient, Dr. Chevray said. “The cost [of ADM] is substantial, but no data have shown that outcomes are better” with its use. Despite that, “nearly every surgeon uses mesh” these days, he noted.
AT AN FDA ADVISORY PANEL MEETING
FDA approves siponimod for relapsing forms of MS
(MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.

Siponimod is a selective sphingosine 1-phosphate (S1P) receptor modulator that binds to S1P1 and S1P5 receptors. Its binding to the S1P1 receptor prevents lymphocytes from leaving the lymph nodes, which contributes to the treatment’s anti-inflammatory effects. Its binding to the S1P5 and S1P1 subreceptors on oligodendrocytes and astrocytes is intended to promote remyelination and prevent inflammation.
The treatment’s approval is based on the results of the phase 3 EXPAND study, according to the agency’s March 26 announcement. This randomized, double-blind study compared siponimod with placebo among 1,651 patients with secondary progressive MS. At baseline, the population’s mean age was 48 years, and mean disease duration was approximately 16 years. More than half the study population had a median Expanded Disability Status Scale score of 6.0 and relied on a walking aid.
Siponimod reduced the risk of 3-month confirmed disability progression (CDP) by 21%, compared with placebo (P = .013). Among participants with relapse activity in the 2 years prior to screening, siponimod reduced the risk of this outcome by 33%, compared with placebo (P = .0100). Siponimod delayed the risk of 6-month CDP by 26%, compared with placebo (P = .0058) and reduced the annualized relapse rate by 55%. In addition, the data suggested beneficial effects of siponimod on cognition, MRI disease activity, and brain volume loss. Siponimod did not provide significant improvements in patients with nonactive secondary progressive MS.
Common adverse events included headache, hypertension, and transaminase increase. The FDA requires siponimod to be dispensed with a medication guide that describes the treatment’s associated risks of infection, macular edema, decreased heart rate, and impaired lung function.
Novartis manufactures the drug. The company expects the drug to be available within 1 week, according to its press release.
(MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.
Siponimod is a selective sphingosine 1-phosphate (S1P) receptor modulator that binds to S1P1 and S1P5 receptors. Its binding to the S1P1 receptor prevents lymphocytes from leaving the lymph nodes, which contributes to the treatment’s anti-inflammatory effects. Its binding to the S1P5 and S1P1 subreceptors on oligodendrocytes and astrocytes is intended to promote remyelination and prevent inflammation.
The treatment’s approval is based on the results of the phase 3 EXPAND study, according to the agency’s March 26 announcement. This randomized, double-blind study compared siponimod with placebo among 1,651 patients with secondary progressive MS. At baseline, the population’s mean age was 48 years, and mean disease duration was approximately 16 years. More than half the study population had a median Expanded Disability Status Scale score of 6.0 and relied on a walking aid.
Siponimod reduced the risk of 3-month confirmed disability progression (CDP) by 21%, compared with placebo (P = .013). Among participants with relapse activity in the 2 years prior to screening, siponimod reduced the risk of this outcome by 33%, compared with placebo (P = .0100). Siponimod delayed the risk of 6-month CDP by 26%, compared with placebo (P = .0058) and reduced the annualized relapse rate by 55%. In addition, the data suggested beneficial effects of siponimod on cognition, MRI disease activity, and brain volume loss. Siponimod did not provide significant improvements in patients with nonactive secondary progressive MS.
Common adverse events included headache, hypertension, and transaminase increase. The FDA requires siponimod to be dispensed with a medication guide that describes the treatment’s associated risks of infection, macular edema, decreased heart rate, and impaired lung function.
Novartis manufactures the drug. The company expects the drug to be available within 1 week, according to its press release.
(MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.
Siponimod is a selective sphingosine 1-phosphate (S1P) receptor modulator that binds to S1P1 and S1P5 receptors. Its binding to the S1P1 receptor prevents lymphocytes from leaving the lymph nodes, which contributes to the treatment’s anti-inflammatory effects. Its binding to the S1P5 and S1P1 subreceptors on oligodendrocytes and astrocytes is intended to promote remyelination and prevent inflammation.
The treatment’s approval is based on the results of the phase 3 EXPAND study, according to the agency’s March 26 announcement. This randomized, double-blind study compared siponimod with placebo among 1,651 patients with secondary progressive MS. At baseline, the population’s mean age was 48 years, and mean disease duration was approximately 16 years. More than half the study population had a median Expanded Disability Status Scale score of 6.0 and relied on a walking aid.
Siponimod reduced the risk of 3-month confirmed disability progression (CDP) by 21%, compared with placebo (P = .013). Among participants with relapse activity in the 2 years prior to screening, siponimod reduced the risk of this outcome by 33%, compared with placebo (P = .0100). Siponimod delayed the risk of 6-month CDP by 26%, compared with placebo (P = .0058) and reduced the annualized relapse rate by 55%. In addition, the data suggested beneficial effects of siponimod on cognition, MRI disease activity, and brain volume loss. Siponimod did not provide significant improvements in patients with nonactive secondary progressive MS.
Common adverse events included headache, hypertension, and transaminase increase. The FDA requires siponimod to be dispensed with a medication guide that describes the treatment’s associated risks of infection, macular edema, decreased heart rate, and impaired lung function.
Novartis manufactures the drug. The company expects the drug to be available within 1 week, according to its press release.
AAP updates 2019-2020 flu vaccine recommendations to include nasal spray
Although the American Academy of Pediatrics had cited a preference for injected flu vaccines for children during the 2018-2019 flu season, this year’s recommendations say either that or the nasal spray formulation are acceptable, according to a press release. The Centers for Disease Control and Prevention has given similar guidance.
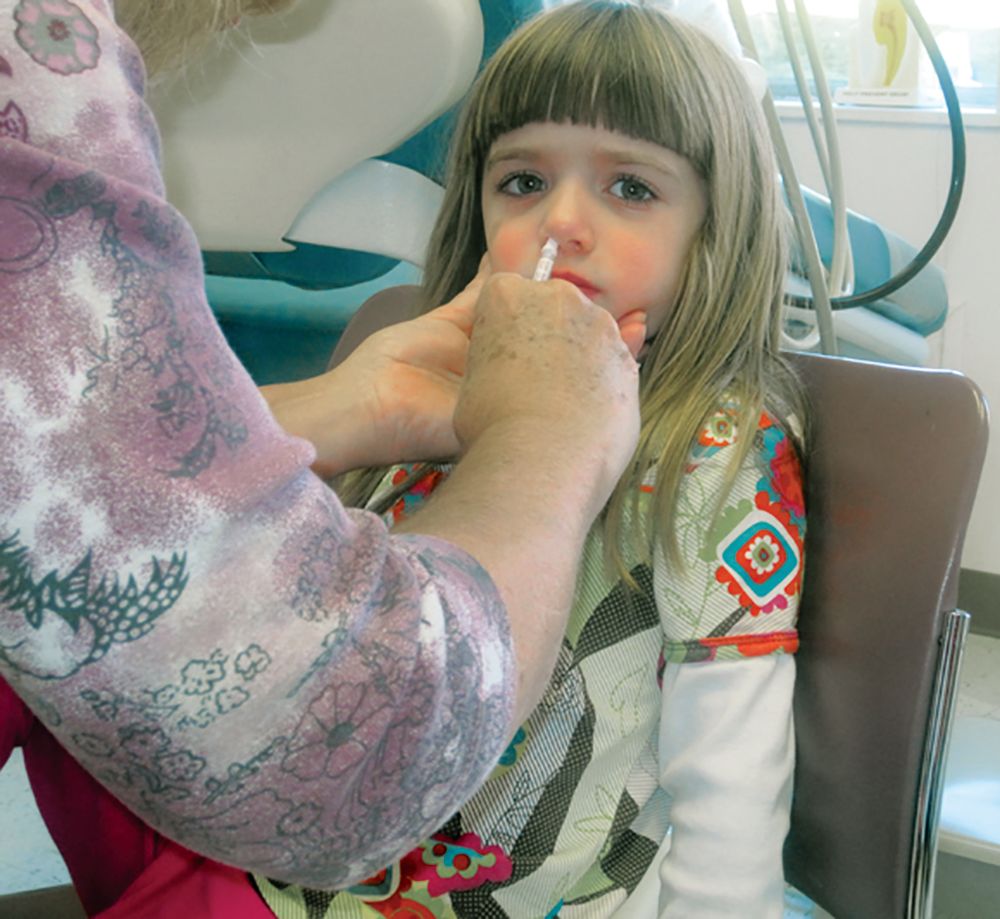
Because the spray did not work as well against A/H1N1 as the injected vaccine had during the 2013-2014 and 2014-2015 seasons, the AAP did not recommend the spray during the 2015-2016 and 2016-2017 seasons. However, in 2017 the spray’s manufacturer included a new strain of A/H1N1, and new data has supported the spray’s effectiveness against some strains.
according to the CDC. That said, the spray is especially appropriate for patients who refuse to receive the injected form, so the choice of formulation is at the pediatrician’s discretion, according to the AAP release.
Although the American Academy of Pediatrics had cited a preference for injected flu vaccines for children during the 2018-2019 flu season, this year’s recommendations say either that or the nasal spray formulation are acceptable, according to a press release. The Centers for Disease Control and Prevention has given similar guidance.
Because the spray did not work as well against A/H1N1 as the injected vaccine had during the 2013-2014 and 2014-2015 seasons, the AAP did not recommend the spray during the 2015-2016 and 2016-2017 seasons. However, in 2017 the spray’s manufacturer included a new strain of A/H1N1, and new data has supported the spray’s effectiveness against some strains.
according to the CDC. That said, the spray is especially appropriate for patients who refuse to receive the injected form, so the choice of formulation is at the pediatrician’s discretion, according to the AAP release.
Although the American Academy of Pediatrics had cited a preference for injected flu vaccines for children during the 2018-2019 flu season, this year’s recommendations say either that or the nasal spray formulation are acceptable, according to a press release. The Centers for Disease Control and Prevention has given similar guidance.
Because the spray did not work as well against A/H1N1 as the injected vaccine had during the 2013-2014 and 2014-2015 seasons, the AAP did not recommend the spray during the 2015-2016 and 2016-2017 seasons. However, in 2017 the spray’s manufacturer included a new strain of A/H1N1, and new data has supported the spray’s effectiveness against some strains.
according to the CDC. That said, the spray is especially appropriate for patients who refuse to receive the injected form, so the choice of formulation is at the pediatrician’s discretion, according to the AAP release.
FDA: Programmable heart failure device approved
. Specifically, these patients are unsuited for other treatments, have marked physical limitations related to their heart failure, and have remained symptomatic despite optimal medical therapy. They also have a regular heart rhythm, are not candidates for resynchronization, and possess a left ventricular ejection fraction of 25%-45%.
The cardiac contractility modulation system is indicated to improve 6-minute hall walk distance, quality of life, and functional status in these patients.
The system is made up of several components, including the implantable pulse generator, a programmer, and software. The pulse generator is connected to three leads that have been implanted in the heart, after which the device is tested and programmed to deliver pulses during normal heartbeats, which improves the heart’s squeezing capability. In randomized, multicenter clinical trials, the system plus optimal medical therapy demonstrated improvements in distance during 6-minute walking tests and standard assessments of heart failure symptoms when compared with optimal medical therapy alone.
The Breakthrough Device designation means this system treats a life-threatening disease and addresses unmet medical needs among some patients. “The FDA recognized the unmet need for these patients and worked with the manufacturer through our Breakthrough Device Program to efficiently bring this product to market, while ensuring it meets our regulatory requirements for safety and effectiveness,” Bram Zuckerman, MD, the director of the division of cardiovascular devices in the FDA’s Center for Devices and Radiological Health said in a news release from the agency.
. Specifically, these patients are unsuited for other treatments, have marked physical limitations related to their heart failure, and have remained symptomatic despite optimal medical therapy. They also have a regular heart rhythm, are not candidates for resynchronization, and possess a left ventricular ejection fraction of 25%-45%.
The cardiac contractility modulation system is indicated to improve 6-minute hall walk distance, quality of life, and functional status in these patients.
The system is made up of several components, including the implantable pulse generator, a programmer, and software. The pulse generator is connected to three leads that have been implanted in the heart, after which the device is tested and programmed to deliver pulses during normal heartbeats, which improves the heart’s squeezing capability. In randomized, multicenter clinical trials, the system plus optimal medical therapy demonstrated improvements in distance during 6-minute walking tests and standard assessments of heart failure symptoms when compared with optimal medical therapy alone.
The Breakthrough Device designation means this system treats a life-threatening disease and addresses unmet medical needs among some patients. “The FDA recognized the unmet need for these patients and worked with the manufacturer through our Breakthrough Device Program to efficiently bring this product to market, while ensuring it meets our regulatory requirements for safety and effectiveness,” Bram Zuckerman, MD, the director of the division of cardiovascular devices in the FDA’s Center for Devices and Radiological Health said in a news release from the agency.
. Specifically, these patients are unsuited for other treatments, have marked physical limitations related to their heart failure, and have remained symptomatic despite optimal medical therapy. They also have a regular heart rhythm, are not candidates for resynchronization, and possess a left ventricular ejection fraction of 25%-45%.
The cardiac contractility modulation system is indicated to improve 6-minute hall walk distance, quality of life, and functional status in these patients.
The system is made up of several components, including the implantable pulse generator, a programmer, and software. The pulse generator is connected to three leads that have been implanted in the heart, after which the device is tested and programmed to deliver pulses during normal heartbeats, which improves the heart’s squeezing capability. In randomized, multicenter clinical trials, the system plus optimal medical therapy demonstrated improvements in distance during 6-minute walking tests and standard assessments of heart failure symptoms when compared with optimal medical therapy alone.
The Breakthrough Device designation means this system treats a life-threatening disease and addresses unmet medical needs among some patients. “The FDA recognized the unmet need for these patients and worked with the manufacturer through our Breakthrough Device Program to efficiently bring this product to market, while ensuring it meets our regulatory requirements for safety and effectiveness,” Bram Zuckerman, MD, the director of the division of cardiovascular devices in the FDA’s Center for Devices and Radiological Health said in a news release from the agency.
FDA chief calls for stricter scrutiny of electronic health records

“What we really need is a much more tailored approach, so that we have appropriate oversight of EHRs when they’re doing things that could create risk for patients,” Dr. Gottlieb said in an interview with Kaiser Health News.
Dr. Gottlieb was responding to “Botched Operation,” a report published March 18 by KHN and Fortune magazine. The investigation found that the federal government has spent more than $36 billion over the past 10 years to switch doctors and hospitals from paper to digital records systems. In that time, thousands of reports of deaths, injuries, and near misses linked to EHRs have piled up in databases – including at least one run by the FDA.
Dr. Gottlieb said Congress would need to enact legislation to define when an EHR would require government oversight. He said that the digital records systems, which store a patient’s medical history, don’t fit neatly under the agency’s existing mandate to regulate items such as drugs and medical devices.
Dr. Gottlieb said the best approach might be to say that an EHR that has a certain capability becomes a medical device. He called EHRs a “unique tool,” noting that the risks posed by their use aren’t the same as for a traditional medical device implanted in a patient. “You need a much different regulatory scheme,” he said.
The 21st Century Cures Act of 2016 excludes the FDA from having oversight over EHRs as a medical device.
Dr. Gottlieb said that health IT companies could add new functions that would improve EHRs, but they have been reluctant to do so because they didn’t want their products to fall under FDA jurisdiction. He added that he was “not calling” for FDA to take over such a duty, however, and suggested that any new approach could be years away. Proponents have long argued that widespread use of EHRs can make medicine safer by alerting doctors to potential medical errors, though critics counter that software glitches and user errors may cause new varieties of medical mistakes.
How closely the FDA should watch over the digital medical record revolution has been controversial for years. The agency’s interest in the issue perked up after Congress decided in February 2009 to spend billions of dollars on digital medical records as part of an economic stimulus program.
At the time, many industry groups argued that FDA regulation would “stifle innovation” and stall the national drive to bring medicine into the modern era. Federal officials responsible for doling out billions in subsidies to doctors and hospitals generally sympathized with that view and were skeptical of allowing the FDA to play a role.
The debate became public in February 2010, when Jeffrey Shuren, MD, an FDA official, testified at a public hearing that the agency had tied 6 deaths and more than 200 injuries to health information technology. In all, the FDA said, it had logged 260 reports in the previous 2 years of “malfunctions with the potential for patient harm.”
The agency said the findings were based largely on reports voluntarily submitted to the FDA and suggested “significant clinical implications and public safety issues.” In one case cited, lab tests done in a hospital emergency department were sent to the wrong patient’s file. Since then, several government and private repositories have associated thousands of injuries, near misses, and deaths to EHR technology.
Dr. Shuren said in 2010 that the agency recognized that health information technology had great potential to improve patient care, but also needed oversight to “assure patient safety.”
While some safety proponents agree that EHRs offer tremendous benefits, they also see a greater opportunities to improve their safety.
Dean Sittig, PhD, a professor of bioinformatics and bioengineering at the University of Texas, Houston, said EHRs have improved safety within the health care system, but they have not eliminated errors to the extent that he would have expected. Federal officials were initially pushing for rapid adoption and ‘there wasn’t a lot of interest in talking about things that could go wrong,’ ” Dr. Sittig told KHN and Fortune.
Earlier in March, Gottlieb announced his resignation from the FDA. His last day is scheduled to be April 5.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente. KHN correspondents Sarah Jane Tribble, Sydney Lupkin, and Julie Rovner contributed to this report.
“What we really need is a much more tailored approach, so that we have appropriate oversight of EHRs when they’re doing things that could create risk for patients,” Dr. Gottlieb said in an interview with Kaiser Health News.
Dr. Gottlieb was responding to “Botched Operation,” a report published March 18 by KHN and Fortune magazine. The investigation found that the federal government has spent more than $36 billion over the past 10 years to switch doctors and hospitals from paper to digital records systems. In that time, thousands of reports of deaths, injuries, and near misses linked to EHRs have piled up in databases – including at least one run by the FDA.
Dr. Gottlieb said Congress would need to enact legislation to define when an EHR would require government oversight. He said that the digital records systems, which store a patient’s medical history, don’t fit neatly under the agency’s existing mandate to regulate items such as drugs and medical devices.
Dr. Gottlieb said the best approach might be to say that an EHR that has a certain capability becomes a medical device. He called EHRs a “unique tool,” noting that the risks posed by their use aren’t the same as for a traditional medical device implanted in a patient. “You need a much different regulatory scheme,” he said.
The 21st Century Cures Act of 2016 excludes the FDA from having oversight over EHRs as a medical device.
Dr. Gottlieb said that health IT companies could add new functions that would improve EHRs, but they have been reluctant to do so because they didn’t want their products to fall under FDA jurisdiction. He added that he was “not calling” for FDA to take over such a duty, however, and suggested that any new approach could be years away. Proponents have long argued that widespread use of EHRs can make medicine safer by alerting doctors to potential medical errors, though critics counter that software glitches and user errors may cause new varieties of medical mistakes.
How closely the FDA should watch over the digital medical record revolution has been controversial for years. The agency’s interest in the issue perked up after Congress decided in February 2009 to spend billions of dollars on digital medical records as part of an economic stimulus program.
At the time, many industry groups argued that FDA regulation would “stifle innovation” and stall the national drive to bring medicine into the modern era. Federal officials responsible for doling out billions in subsidies to doctors and hospitals generally sympathized with that view and were skeptical of allowing the FDA to play a role.
The debate became public in February 2010, when Jeffrey Shuren, MD, an FDA official, testified at a public hearing that the agency had tied 6 deaths and more than 200 injuries to health information technology. In all, the FDA said, it had logged 260 reports in the previous 2 years of “malfunctions with the potential for patient harm.”
The agency said the findings were based largely on reports voluntarily submitted to the FDA and suggested “significant clinical implications and public safety issues.” In one case cited, lab tests done in a hospital emergency department were sent to the wrong patient’s file. Since then, several government and private repositories have associated thousands of injuries, near misses, and deaths to EHR technology.
Dr. Shuren said in 2010 that the agency recognized that health information technology had great potential to improve patient care, but also needed oversight to “assure patient safety.”
While some safety proponents agree that EHRs offer tremendous benefits, they also see a greater opportunities to improve their safety.
Dean Sittig, PhD, a professor of bioinformatics and bioengineering at the University of Texas, Houston, said EHRs have improved safety within the health care system, but they have not eliminated errors to the extent that he would have expected. Federal officials were initially pushing for rapid adoption and ‘there wasn’t a lot of interest in talking about things that could go wrong,’ ” Dr. Sittig told KHN and Fortune.
Earlier in March, Gottlieb announced his resignation from the FDA. His last day is scheduled to be April 5.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente. KHN correspondents Sarah Jane Tribble, Sydney Lupkin, and Julie Rovner contributed to this report.
“What we really need is a much more tailored approach, so that we have appropriate oversight of EHRs when they’re doing things that could create risk for patients,” Dr. Gottlieb said in an interview with Kaiser Health News.
Dr. Gottlieb was responding to “Botched Operation,” a report published March 18 by KHN and Fortune magazine. The investigation found that the federal government has spent more than $36 billion over the past 10 years to switch doctors and hospitals from paper to digital records systems. In that time, thousands of reports of deaths, injuries, and near misses linked to EHRs have piled up in databases – including at least one run by the FDA.
Dr. Gottlieb said Congress would need to enact legislation to define when an EHR would require government oversight. He said that the digital records systems, which store a patient’s medical history, don’t fit neatly under the agency’s existing mandate to regulate items such as drugs and medical devices.
Dr. Gottlieb said the best approach might be to say that an EHR that has a certain capability becomes a medical device. He called EHRs a “unique tool,” noting that the risks posed by their use aren’t the same as for a traditional medical device implanted in a patient. “You need a much different regulatory scheme,” he said.
The 21st Century Cures Act of 2016 excludes the FDA from having oversight over EHRs as a medical device.
Dr. Gottlieb said that health IT companies could add new functions that would improve EHRs, but they have been reluctant to do so because they didn’t want their products to fall under FDA jurisdiction. He added that he was “not calling” for FDA to take over such a duty, however, and suggested that any new approach could be years away. Proponents have long argued that widespread use of EHRs can make medicine safer by alerting doctors to potential medical errors, though critics counter that software glitches and user errors may cause new varieties of medical mistakes.
How closely the FDA should watch over the digital medical record revolution has been controversial for years. The agency’s interest in the issue perked up after Congress decided in February 2009 to spend billions of dollars on digital medical records as part of an economic stimulus program.
At the time, many industry groups argued that FDA regulation would “stifle innovation” and stall the national drive to bring medicine into the modern era. Federal officials responsible for doling out billions in subsidies to doctors and hospitals generally sympathized with that view and were skeptical of allowing the FDA to play a role.
The debate became public in February 2010, when Jeffrey Shuren, MD, an FDA official, testified at a public hearing that the agency had tied 6 deaths and more than 200 injuries to health information technology. In all, the FDA said, it had logged 260 reports in the previous 2 years of “malfunctions with the potential for patient harm.”
The agency said the findings were based largely on reports voluntarily submitted to the FDA and suggested “significant clinical implications and public safety issues.” In one case cited, lab tests done in a hospital emergency department were sent to the wrong patient’s file. Since then, several government and private repositories have associated thousands of injuries, near misses, and deaths to EHR technology.
Dr. Shuren said in 2010 that the agency recognized that health information technology had great potential to improve patient care, but also needed oversight to “assure patient safety.”
While some safety proponents agree that EHRs offer tremendous benefits, they also see a greater opportunities to improve their safety.
Dean Sittig, PhD, a professor of bioinformatics and bioengineering at the University of Texas, Houston, said EHRs have improved safety within the health care system, but they have not eliminated errors to the extent that he would have expected. Federal officials were initially pushing for rapid adoption and ‘there wasn’t a lot of interest in talking about things that could go wrong,’ ” Dr. Sittig told KHN and Fortune.
Earlier in March, Gottlieb announced his resignation from the FDA. His last day is scheduled to be April 5.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente. KHN correspondents Sarah Jane Tribble, Sydney Lupkin, and Julie Rovner contributed to this report.
FDA halts enrollment in trial of venetoclax for multiple myeloma
The Food and Drug Administration has halted enrollment in trials of venetoclax (Venclexta) for multiple myeloma.

The move comes after a review of data from the phase 3 BELLINI trial, which pitted venetoclax against placebo in relapsed and refractory multiple myeloma patients on a background of bortezomib and low-dose dexamethasone. Venetoclax is not approved for the treatment of multiple myeloma; the agency said that patients using the drug for approved indications should continue use of the drug.
There were 41/194 deaths (21.1%) in the venetoclax arm, versus 11/97 (11.3%) in the placebo group; 13 of the deaths in the venetoclax arm (32%) and 1 death in the placebo arm (9%) were treatment related. Sepsis, pneumonia, and cardiac arrest were the most common treatment-related causes of death in the venetoclax group; 8 of the 13 deaths (62%) were due to infection.
The FDA estimated that the drug doubled the risk of death compared to placebo.
The agency warned against off-label use of venetoclax for multiple myeloma, and noted that the drug “is safe and effective for its approved uses,” which include second-line treatment of chronic lymphocytic leukemia and small lymphocytic lymphoma in adults, as well as newly-diagnosed acute myeloid leukemia in adults age 75 years or older or who have contraindications to standard chemotherapy.
There are more than 10 trials in the United States of venetoclax for multiple myeloma, and most of them have been suspended, including BELLINI.
Patients already enrolled in the trial can remain on treatment, but they must re-consent to the trial. The FDA “will be working directly with sponsors of Venclexta, as well as other investigators conducting clinical trials in patients with multiple myeloma, to determine the extent of the safety issue,” the agency said in a statement.
Abbvie, which is developing venetoclax in partnership with Roche, noted in its own press release that the drug otherwise outperformed placebo in BELLINI, both in progression-free survival (22.4 months versus 11.5 months), and in overall (82% versus 68%) and partial (59% versus 36%) response rates.
Severe grade 3-5 toxicity and serious adverse event rates were similar in the two study arms, as was the overall incidence of infections (79.8% versus 77.1%). However, the incidence of pneumonia was 20.7% with venetoclax, versus 15.6% with placebo.
“We will continue working with the FDA and worldwide regulatory agencies to determine appropriate next steps for the multiple myeloma program,” Michael Severino, MD, AbbVie vice chairman and president, said in the press release.
Venetoclax binds and inhibits the B-cell lymphoma-2 protein, which prevents some blood cancer cells from undergoing programmed cell death.
The Food and Drug Administration has halted enrollment in trials of venetoclax (Venclexta) for multiple myeloma.
The move comes after a review of data from the phase 3 BELLINI trial, which pitted venetoclax against placebo in relapsed and refractory multiple myeloma patients on a background of bortezomib and low-dose dexamethasone. Venetoclax is not approved for the treatment of multiple myeloma; the agency said that patients using the drug for approved indications should continue use of the drug.
There were 41/194 deaths (21.1%) in the venetoclax arm, versus 11/97 (11.3%) in the placebo group; 13 of the deaths in the venetoclax arm (32%) and 1 death in the placebo arm (9%) were treatment related. Sepsis, pneumonia, and cardiac arrest were the most common treatment-related causes of death in the venetoclax group; 8 of the 13 deaths (62%) were due to infection.
The FDA estimated that the drug doubled the risk of death compared to placebo.
The agency warned against off-label use of venetoclax for multiple myeloma, and noted that the drug “is safe and effective for its approved uses,” which include second-line treatment of chronic lymphocytic leukemia and small lymphocytic lymphoma in adults, as well as newly-diagnosed acute myeloid leukemia in adults age 75 years or older or who have contraindications to standard chemotherapy.
There are more than 10 trials in the United States of venetoclax for multiple myeloma, and most of them have been suspended, including BELLINI.
Patients already enrolled in the trial can remain on treatment, but they must re-consent to the trial. The FDA “will be working directly with sponsors of Venclexta, as well as other investigators conducting clinical trials in patients with multiple myeloma, to determine the extent of the safety issue,” the agency said in a statement.
Abbvie, which is developing venetoclax in partnership with Roche, noted in its own press release that the drug otherwise outperformed placebo in BELLINI, both in progression-free survival (22.4 months versus 11.5 months), and in overall (82% versus 68%) and partial (59% versus 36%) response rates.
Severe grade 3-5 toxicity and serious adverse event rates were similar in the two study arms, as was the overall incidence of infections (79.8% versus 77.1%). However, the incidence of pneumonia was 20.7% with venetoclax, versus 15.6% with placebo.
“We will continue working with the FDA and worldwide regulatory agencies to determine appropriate next steps for the multiple myeloma program,” Michael Severino, MD, AbbVie vice chairman and president, said in the press release.
Venetoclax binds and inhibits the B-cell lymphoma-2 protein, which prevents some blood cancer cells from undergoing programmed cell death.
The Food and Drug Administration has halted enrollment in trials of venetoclax (Venclexta) for multiple myeloma.
The move comes after a review of data from the phase 3 BELLINI trial, which pitted venetoclax against placebo in relapsed and refractory multiple myeloma patients on a background of bortezomib and low-dose dexamethasone. Venetoclax is not approved for the treatment of multiple myeloma; the agency said that patients using the drug for approved indications should continue use of the drug.
There were 41/194 deaths (21.1%) in the venetoclax arm, versus 11/97 (11.3%) in the placebo group; 13 of the deaths in the venetoclax arm (32%) and 1 death in the placebo arm (9%) were treatment related. Sepsis, pneumonia, and cardiac arrest were the most common treatment-related causes of death in the venetoclax group; 8 of the 13 deaths (62%) were due to infection.
The FDA estimated that the drug doubled the risk of death compared to placebo.
The agency warned against off-label use of venetoclax for multiple myeloma, and noted that the drug “is safe and effective for its approved uses,” which include second-line treatment of chronic lymphocytic leukemia and small lymphocytic lymphoma in adults, as well as newly-diagnosed acute myeloid leukemia in adults age 75 years or older or who have contraindications to standard chemotherapy.
There are more than 10 trials in the United States of venetoclax for multiple myeloma, and most of them have been suspended, including BELLINI.
Patients already enrolled in the trial can remain on treatment, but they must re-consent to the trial. The FDA “will be working directly with sponsors of Venclexta, as well as other investigators conducting clinical trials in patients with multiple myeloma, to determine the extent of the safety issue,” the agency said in a statement.
Abbvie, which is developing venetoclax in partnership with Roche, noted in its own press release that the drug otherwise outperformed placebo in BELLINI, both in progression-free survival (22.4 months versus 11.5 months), and in overall (82% versus 68%) and partial (59% versus 36%) response rates.
Severe grade 3-5 toxicity and serious adverse event rates were similar in the two study arms, as was the overall incidence of infections (79.8% versus 77.1%). However, the incidence of pneumonia was 20.7% with venetoclax, versus 15.6% with placebo.
“We will continue working with the FDA and worldwide regulatory agencies to determine appropriate next steps for the multiple myeloma program,” Michael Severino, MD, AbbVie vice chairman and president, said in the press release.
Venetoclax binds and inhibits the B-cell lymphoma-2 protein, which prevents some blood cancer cells from undergoing programmed cell death.
FDA examines changing donation policies for men who have sex with men
The
At a meeting of the FDA’s Blood Products Advisory Committee, the agency shared the content of the 5-item questionnaire and reviewed the proposed study design with committee members, who were asked to comment – but not vote – on the best path forward for MSM donation policies.
The FDA is “committed to ongoing evaluation of the MSM deferral policy” and remains open to adjusting the policy based on the best available scientific evidence, said Barbee Whitaker, PhD, a lead scientist in the agency’s Office of Emerging and Transfusion Transmitted Disease
After recruiting 2,000 men who have had sex with men at least once during the past 3 months, the study will aim to identify individuals who have very recently become HIV infected, in order to assess the discriminant function of the set of behavioral questions that are proposed in the questionnaire.
The crux of the problem currently, noted Dr. Whitaker, is identifying those individuals who are very recently infected with HIV. Nucleic acid testing has tightened the window of undetectability considerably, but the current 12-month deferral window after men have had sexual contact with other men is designed to ensure safety of the blood supply.
Social justice concerns have been raised about the blanket deferral, said Dr. Whitaker; the behavioral questions in the pilot study will ask about the number of different sexual partners men have had within the past 1, 3, and 12 months and ask about the type of sexual contact (oral sex, or anal penetrative or receptive intercourse). The questionnaire also asks about sex with a partner known to be HIV positive, condom use, and use of pre-exposure prophylaxis (PrEP).
The FDA will ask for proposals to conduct the study with an eye to having sites in such cities as Washington, Atlanta, and Miami, which have high incidences of HIV, to improve chances of early detection.
The behavioral questionnaire is not seen as an immediate replacement for the 12-month deferral policy, the FDA made clear in its briefing documents and in discussion with the committee. Instead, its utility will be in the information gleaned from the pilot study and a follow-on that may include several hundred thousand individuals. These data should provide “population-based evidence upon which to base regulatory decisions to ensure blood safety,” she said.
Donation policies outside the United States
Whether a change in blood donation deferral policies for MSM would be a shortened window or a move toward a behavioral questionnaire is currently not known. Globally, a variety of practices are used for blood screening, said Mindy Goldman, MD, medical director of Canadian Blood Services, who reviewed international perspectives on blood donation for MSM.
“There’s no general consensus on donation deferrals internationally,” she said. Factors influencing policy can include epidemiology, risk analysis, modeling, and history of response to threats in the past.
However, “there’s basically a couple of main approaches” to handling deferrals for MSM, Dr. Goldman said. One is time-based deferral – the strategy used in the United States, as well as Canada, the United Kingdom, Japan, and Australia.
Japan and the U.K. have recently moved to 3-month deferral periods, a figure arrived at by doubling the window period for nucleic acid testing for HIV, roughly, Dr. Goldman said. Early data from the U.K. experience has not shown an increase in HIV rates among donors, or an increase in NAT-only positive donors, she said. An application to move from a 12-month to a 3-month deferral period is pending in Canada.
A strong advantage of time-based deferral as a risk management strategy, Dr. Goldman said, is standardization. “For us, standardization is close to godliness.”
However, she added, “another major limitation is that you’re still deferring all sexually active MSM, including those who are in a stable monogamous relationship from donating. From a justice perspective for the lowest risk population of MSM – they are still being deferred using this type of approach.”
Some nations, such as Spain and Italy, use individual risk assessment via physician-led interviews. These approaches are often not standardized. “There’s no national uniform questionnaire, so there’s less standardization, and more variability between blood centers,” Dr. Goldman said. “So you wind up trying to compare apples with oranges.”
This means the results are harder to evaluate on a national level. However, there appears to be higher residual risk, with HIV rates among first-time donors approaching those of the general population, Dr. Goldman said.
Another strategy, used in France, is a test-retest model, where blood from first-time MSM that initially tests negative for HIV is held until the individual returns for re-testing or an additional donation, with a second negative test. This approach increases operational complexity and cost, noted Dr. Goldman, and because of the short shelf life of platelets, it’s not practical for this blood component.
In general questioning and discussion after this and other background presentations, the committee could agree on one point: this isn’t an easy question.
“I’m increasingly struck by how difficult this problem is,” said committee member Roger Lewis, MD, PhD, professor at the University of California, Los Angeles, and chair of the department of emergency medicine at Harbor-UCLA Medical Center. Regarding just the problem of completing the pilot study, Dr. Lewis commented, “It sounds like it’s going to be impossible to get the data that directly answers the questions.”
Peter Marx, MD, PhD, who directs the FDA’s Center for Biologics Evaluation and Research (CBER), which oversees blood products safety, joined the discussion to acknowledge the difficulty, but underscore the social importance of a careful examination of the current MSM donation policy.
“We understand the issues here…. With all due respect to our European colleagues, there’s not enough data. That’s the point of this study; we also know that the U.S. has a very different epidemiology of HIV than the U.K. and a lot of other places,” Dr. Marx said. “The pilot study is a way to get some data where we might be able to get away from a time-based deferral. The LGBT community finds any time-based deferral discriminatory.”
Pathogen reduction technology
The committee heard a proposal for a completely different strategy during its afternoon session: pathogen reduction technology (PRT) holds promise to achieve virtual elimination of HIV and other pathogens from donated blood products.
The FDA is reviewing a variance request from the nonprofit blood donation organization Bloodworks Northwest organization to use PRT for apheresis platelet donations from MSM who would otherwise be deferred because of sexual activity within the 12-month deferral window.
James AuBuchon, MD, president of Bloodworks Northwest, explained that his organization takes in about 225,000 donations annually. The variance sought would use the FDA-approved INTERCEPT device to achieve pathogen reduction for donations that meet all requirements except the MSM deferral, and that would still undergo all relevant transfusion transmitted infection testing.
The INTERCEPT device uses amotosalen, which intercalates with DNA and RNA, inactivating it after exposure to ultraviolet A light. Amotosalen is then removed from the blood product before administration. The pathogen reduction activity doesn’t interfere with platelets or plasma, and is active against a wide range of viruses, bacteria, and fungal pathogens, explained Dr. AuBuchon, who is also a professor of hematology at the University of Washington, Seattle.
Dr. AuBuchon walked the committee through procedures designed to flag donors for PRT platelet apheresis, and to ensure these donations receive the intended PRT treatment. Platelets were chosen for this variance request, he explained, because demand outstrips supply. “We are all spending additional time and resources in recruiting a new framework and demographic, and it is exceedingly difficult to keep enough donors coming through the door,” he said. “Our platelet utilization climbs continually – it’s up 15% in the last 4 years.”
Committee members circled around the idea that all risk can’t be eliminated, even with the highly effective PRT technology. But the risk is exceedingly low, said committee chair Richard Kaufman, MD, medical director of the adult transfusion service at Brigham and Women’s Hospital, Boston. “It’s not possible to get rid of the window. We can kind of hammer down the risk by shrinking down the window by using incredibly sensitive tests. But that risk continues to exist. Pathogen reduction can take care of that residual risk…. So what’s left is really quite a low risk,” Dr. Kaufman said.
Susan Stramer, PhD, vice president of scientific affairs for the American Red Cross, concurred, noting that pathogen reduction techniques are already in use for many other blood products, particularly within the plasma industry.
Wrapping up, Dr. Kaufman asked individual committee members to summarize their position on the variance request, though the FDA had not placed a voting question before the committee. Consensus in the room was that this real-world examination of PRT could point to a path to expanding the donor pool while maintaining patient safety – a concern all agreed was paramount.
The FDA usually follows the recommendations of its committees.
The
At a meeting of the FDA’s Blood Products Advisory Committee, the agency shared the content of the 5-item questionnaire and reviewed the proposed study design with committee members, who were asked to comment – but not vote – on the best path forward for MSM donation policies.
The FDA is “committed to ongoing evaluation of the MSM deferral policy” and remains open to adjusting the policy based on the best available scientific evidence, said Barbee Whitaker, PhD, a lead scientist in the agency’s Office of Emerging and Transfusion Transmitted Disease
After recruiting 2,000 men who have had sex with men at least once during the past 3 months, the study will aim to identify individuals who have very recently become HIV infected, in order to assess the discriminant function of the set of behavioral questions that are proposed in the questionnaire.
The crux of the problem currently, noted Dr. Whitaker, is identifying those individuals who are very recently infected with HIV. Nucleic acid testing has tightened the window of undetectability considerably, but the current 12-month deferral window after men have had sexual contact with other men is designed to ensure safety of the blood supply.
Social justice concerns have been raised about the blanket deferral, said Dr. Whitaker; the behavioral questions in the pilot study will ask about the number of different sexual partners men have had within the past 1, 3, and 12 months and ask about the type of sexual contact (oral sex, or anal penetrative or receptive intercourse). The questionnaire also asks about sex with a partner known to be HIV positive, condom use, and use of pre-exposure prophylaxis (PrEP).
The FDA will ask for proposals to conduct the study with an eye to having sites in such cities as Washington, Atlanta, and Miami, which have high incidences of HIV, to improve chances of early detection.
The behavioral questionnaire is not seen as an immediate replacement for the 12-month deferral policy, the FDA made clear in its briefing documents and in discussion with the committee. Instead, its utility will be in the information gleaned from the pilot study and a follow-on that may include several hundred thousand individuals. These data should provide “population-based evidence upon which to base regulatory decisions to ensure blood safety,” she said.
Donation policies outside the United States
Whether a change in blood donation deferral policies for MSM would be a shortened window or a move toward a behavioral questionnaire is currently not known. Globally, a variety of practices are used for blood screening, said Mindy Goldman, MD, medical director of Canadian Blood Services, who reviewed international perspectives on blood donation for MSM.
“There’s no general consensus on donation deferrals internationally,” she said. Factors influencing policy can include epidemiology, risk analysis, modeling, and history of response to threats in the past.
However, “there’s basically a couple of main approaches” to handling deferrals for MSM, Dr. Goldman said. One is time-based deferral – the strategy used in the United States, as well as Canada, the United Kingdom, Japan, and Australia.
Japan and the U.K. have recently moved to 3-month deferral periods, a figure arrived at by doubling the window period for nucleic acid testing for HIV, roughly, Dr. Goldman said. Early data from the U.K. experience has not shown an increase in HIV rates among donors, or an increase in NAT-only positive donors, she said. An application to move from a 12-month to a 3-month deferral period is pending in Canada.
A strong advantage of time-based deferral as a risk management strategy, Dr. Goldman said, is standardization. “For us, standardization is close to godliness.”
However, she added, “another major limitation is that you’re still deferring all sexually active MSM, including those who are in a stable monogamous relationship from donating. From a justice perspective for the lowest risk population of MSM – they are still being deferred using this type of approach.”
Some nations, such as Spain and Italy, use individual risk assessment via physician-led interviews. These approaches are often not standardized. “There’s no national uniform questionnaire, so there’s less standardization, and more variability between blood centers,” Dr. Goldman said. “So you wind up trying to compare apples with oranges.”
This means the results are harder to evaluate on a national level. However, there appears to be higher residual risk, with HIV rates among first-time donors approaching those of the general population, Dr. Goldman said.
Another strategy, used in France, is a test-retest model, where blood from first-time MSM that initially tests negative for HIV is held until the individual returns for re-testing or an additional donation, with a second negative test. This approach increases operational complexity and cost, noted Dr. Goldman, and because of the short shelf life of platelets, it’s not practical for this blood component.
In general questioning and discussion after this and other background presentations, the committee could agree on one point: this isn’t an easy question.
“I’m increasingly struck by how difficult this problem is,” said committee member Roger Lewis, MD, PhD, professor at the University of California, Los Angeles, and chair of the department of emergency medicine at Harbor-UCLA Medical Center. Regarding just the problem of completing the pilot study, Dr. Lewis commented, “It sounds like it’s going to be impossible to get the data that directly answers the questions.”
Peter Marx, MD, PhD, who directs the FDA’s Center for Biologics Evaluation and Research (CBER), which oversees blood products safety, joined the discussion to acknowledge the difficulty, but underscore the social importance of a careful examination of the current MSM donation policy.
“We understand the issues here…. With all due respect to our European colleagues, there’s not enough data. That’s the point of this study; we also know that the U.S. has a very different epidemiology of HIV than the U.K. and a lot of other places,” Dr. Marx said. “The pilot study is a way to get some data where we might be able to get away from a time-based deferral. The LGBT community finds any time-based deferral discriminatory.”
Pathogen reduction technology
The committee heard a proposal for a completely different strategy during its afternoon session: pathogen reduction technology (PRT) holds promise to achieve virtual elimination of HIV and other pathogens from donated blood products.
The FDA is reviewing a variance request from the nonprofit blood donation organization Bloodworks Northwest organization to use PRT for apheresis platelet donations from MSM who would otherwise be deferred because of sexual activity within the 12-month deferral window.
James AuBuchon, MD, president of Bloodworks Northwest, explained that his organization takes in about 225,000 donations annually. The variance sought would use the FDA-approved INTERCEPT device to achieve pathogen reduction for donations that meet all requirements except the MSM deferral, and that would still undergo all relevant transfusion transmitted infection testing.
The INTERCEPT device uses amotosalen, which intercalates with DNA and RNA, inactivating it after exposure to ultraviolet A light. Amotosalen is then removed from the blood product before administration. The pathogen reduction activity doesn’t interfere with platelets or plasma, and is active against a wide range of viruses, bacteria, and fungal pathogens, explained Dr. AuBuchon, who is also a professor of hematology at the University of Washington, Seattle.
Dr. AuBuchon walked the committee through procedures designed to flag donors for PRT platelet apheresis, and to ensure these donations receive the intended PRT treatment. Platelets were chosen for this variance request, he explained, because demand outstrips supply. “We are all spending additional time and resources in recruiting a new framework and demographic, and it is exceedingly difficult to keep enough donors coming through the door,” he said. “Our platelet utilization climbs continually – it’s up 15% in the last 4 years.”
Committee members circled around the idea that all risk can’t be eliminated, even with the highly effective PRT technology. But the risk is exceedingly low, said committee chair Richard Kaufman, MD, medical director of the adult transfusion service at Brigham and Women’s Hospital, Boston. “It’s not possible to get rid of the window. We can kind of hammer down the risk by shrinking down the window by using incredibly sensitive tests. But that risk continues to exist. Pathogen reduction can take care of that residual risk…. So what’s left is really quite a low risk,” Dr. Kaufman said.
Susan Stramer, PhD, vice president of scientific affairs for the American Red Cross, concurred, noting that pathogen reduction techniques are already in use for many other blood products, particularly within the plasma industry.
Wrapping up, Dr. Kaufman asked individual committee members to summarize their position on the variance request, though the FDA had not placed a voting question before the committee. Consensus in the room was that this real-world examination of PRT could point to a path to expanding the donor pool while maintaining patient safety – a concern all agreed was paramount.
The FDA usually follows the recommendations of its committees.
The
At a meeting of the FDA’s Blood Products Advisory Committee, the agency shared the content of the 5-item questionnaire and reviewed the proposed study design with committee members, who were asked to comment – but not vote – on the best path forward for MSM donation policies.
The FDA is “committed to ongoing evaluation of the MSM deferral policy” and remains open to adjusting the policy based on the best available scientific evidence, said Barbee Whitaker, PhD, a lead scientist in the agency’s Office of Emerging and Transfusion Transmitted Disease
After recruiting 2,000 men who have had sex with men at least once during the past 3 months, the study will aim to identify individuals who have very recently become HIV infected, in order to assess the discriminant function of the set of behavioral questions that are proposed in the questionnaire.
The crux of the problem currently, noted Dr. Whitaker, is identifying those individuals who are very recently infected with HIV. Nucleic acid testing has tightened the window of undetectability considerably, but the current 12-month deferral window after men have had sexual contact with other men is designed to ensure safety of the blood supply.
Social justice concerns have been raised about the blanket deferral, said Dr. Whitaker; the behavioral questions in the pilot study will ask about the number of different sexual partners men have had within the past 1, 3, and 12 months and ask about the type of sexual contact (oral sex, or anal penetrative or receptive intercourse). The questionnaire also asks about sex with a partner known to be HIV positive, condom use, and use of pre-exposure prophylaxis (PrEP).
The FDA will ask for proposals to conduct the study with an eye to having sites in such cities as Washington, Atlanta, and Miami, which have high incidences of HIV, to improve chances of early detection.
The behavioral questionnaire is not seen as an immediate replacement for the 12-month deferral policy, the FDA made clear in its briefing documents and in discussion with the committee. Instead, its utility will be in the information gleaned from the pilot study and a follow-on that may include several hundred thousand individuals. These data should provide “population-based evidence upon which to base regulatory decisions to ensure blood safety,” she said.
Donation policies outside the United States
Whether a change in blood donation deferral policies for MSM would be a shortened window or a move toward a behavioral questionnaire is currently not known. Globally, a variety of practices are used for blood screening, said Mindy Goldman, MD, medical director of Canadian Blood Services, who reviewed international perspectives on blood donation for MSM.
“There’s no general consensus on donation deferrals internationally,” she said. Factors influencing policy can include epidemiology, risk analysis, modeling, and history of response to threats in the past.
However, “there’s basically a couple of main approaches” to handling deferrals for MSM, Dr. Goldman said. One is time-based deferral – the strategy used in the United States, as well as Canada, the United Kingdom, Japan, and Australia.
Japan and the U.K. have recently moved to 3-month deferral periods, a figure arrived at by doubling the window period for nucleic acid testing for HIV, roughly, Dr. Goldman said. Early data from the U.K. experience has not shown an increase in HIV rates among donors, or an increase in NAT-only positive donors, she said. An application to move from a 12-month to a 3-month deferral period is pending in Canada.
A strong advantage of time-based deferral as a risk management strategy, Dr. Goldman said, is standardization. “For us, standardization is close to godliness.”
However, she added, “another major limitation is that you’re still deferring all sexually active MSM, including those who are in a stable monogamous relationship from donating. From a justice perspective for the lowest risk population of MSM – they are still being deferred using this type of approach.”
Some nations, such as Spain and Italy, use individual risk assessment via physician-led interviews. These approaches are often not standardized. “There’s no national uniform questionnaire, so there’s less standardization, and more variability between blood centers,” Dr. Goldman said. “So you wind up trying to compare apples with oranges.”
This means the results are harder to evaluate on a national level. However, there appears to be higher residual risk, with HIV rates among first-time donors approaching those of the general population, Dr. Goldman said.
Another strategy, used in France, is a test-retest model, where blood from first-time MSM that initially tests negative for HIV is held until the individual returns for re-testing or an additional donation, with a second negative test. This approach increases operational complexity and cost, noted Dr. Goldman, and because of the short shelf life of platelets, it’s not practical for this blood component.
In general questioning and discussion after this and other background presentations, the committee could agree on one point: this isn’t an easy question.
“I’m increasingly struck by how difficult this problem is,” said committee member Roger Lewis, MD, PhD, professor at the University of California, Los Angeles, and chair of the department of emergency medicine at Harbor-UCLA Medical Center. Regarding just the problem of completing the pilot study, Dr. Lewis commented, “It sounds like it’s going to be impossible to get the data that directly answers the questions.”
Peter Marx, MD, PhD, who directs the FDA’s Center for Biologics Evaluation and Research (CBER), which oversees blood products safety, joined the discussion to acknowledge the difficulty, but underscore the social importance of a careful examination of the current MSM donation policy.
“We understand the issues here…. With all due respect to our European colleagues, there’s not enough data. That’s the point of this study; we also know that the U.S. has a very different epidemiology of HIV than the U.K. and a lot of other places,” Dr. Marx said. “The pilot study is a way to get some data where we might be able to get away from a time-based deferral. The LGBT community finds any time-based deferral discriminatory.”
Pathogen reduction technology
The committee heard a proposal for a completely different strategy during its afternoon session: pathogen reduction technology (PRT) holds promise to achieve virtual elimination of HIV and other pathogens from donated blood products.
The FDA is reviewing a variance request from the nonprofit blood donation organization Bloodworks Northwest organization to use PRT for apheresis platelet donations from MSM who would otherwise be deferred because of sexual activity within the 12-month deferral window.
James AuBuchon, MD, president of Bloodworks Northwest, explained that his organization takes in about 225,000 donations annually. The variance sought would use the FDA-approved INTERCEPT device to achieve pathogen reduction for donations that meet all requirements except the MSM deferral, and that would still undergo all relevant transfusion transmitted infection testing.
The INTERCEPT device uses amotosalen, which intercalates with DNA and RNA, inactivating it after exposure to ultraviolet A light. Amotosalen is then removed from the blood product before administration. The pathogen reduction activity doesn’t interfere with platelets or plasma, and is active against a wide range of viruses, bacteria, and fungal pathogens, explained Dr. AuBuchon, who is also a professor of hematology at the University of Washington, Seattle.
Dr. AuBuchon walked the committee through procedures designed to flag donors for PRT platelet apheresis, and to ensure these donations receive the intended PRT treatment. Platelets were chosen for this variance request, he explained, because demand outstrips supply. “We are all spending additional time and resources in recruiting a new framework and demographic, and it is exceedingly difficult to keep enough donors coming through the door,” he said. “Our platelet utilization climbs continually – it’s up 15% in the last 4 years.”
Committee members circled around the idea that all risk can’t be eliminated, even with the highly effective PRT technology. But the risk is exceedingly low, said committee chair Richard Kaufman, MD, medical director of the adult transfusion service at Brigham and Women’s Hospital, Boston. “It’s not possible to get rid of the window. We can kind of hammer down the risk by shrinking down the window by using incredibly sensitive tests. But that risk continues to exist. Pathogen reduction can take care of that residual risk…. So what’s left is really quite a low risk,” Dr. Kaufman said.
Susan Stramer, PhD, vice president of scientific affairs for the American Red Cross, concurred, noting that pathogen reduction techniques are already in use for many other blood products, particularly within the plasma industry.
Wrapping up, Dr. Kaufman asked individual committee members to summarize their position on the variance request, though the FDA had not placed a voting question before the committee. Consensus in the room was that this real-world examination of PRT could point to a path to expanding the donor pool while maintaining patient safety – a concern all agreed was paramount.
The FDA usually follows the recommendations of its committees.
FROM AN FDA ADVISORY COMMITTEE MEETING
FDA committee advises status quo for blood supply Zika testing
Most members of a Food and Drug Administration advisory committee considered that data support maintaining current testing protocols for Zika virus in the blood donor pool. However, committee discussion entertained the idea of revisiting testing strategies after another year or 2 of Zika virus epidemiological data are available.
In its last guidance regarding Zika virus testing, issued in July 2018, the FDA recommended that either minipool nucleic acid testing (MP NAT) or individual donor (ID) NAT be used to screen for Zika virus. Current guidance still requires conversion to all-ID NAT “when certain threshold conditions are met that indicate an increased risk of suspected mosquito-borne transmission in a defined geographic collection area.”
In the first of three separate votes, 11 of 15 voting members of the FDA’s Blood Products Advisory Committee (BPAC) answered in the affirmative to the question of whether available data support continuing the status quo for Zika testing. Committee members then were asked to weigh whether current data support scaling back to a regional testing strategy targeting at-risk areas. Here, six committee members answered in the affirmative, and nine in the negative.
Just one committee member, F. Blaine Hollinger, MD, voted in favor of the third option, elimination of all Zika virus testing without reintroducing donor screening for risk factors in risk-free areas pending another outbreak in the United States. Dr. Hollinger is a professor of virology and microbiology at Baylor College of Medicine, Houston.
The committee as whole wasn’t swayed by a line of questioning put forward by chairman Richard Kaufman, MD. “I will be the devil’s advocate a little bit: We learned that there have been zero confirmed positives from blood donors for the past year. Would anyone be comfortable with just stopping screening of donors?” asked Dr. Kaufman, medical director of the adult transfusion service at Brigham and Women’s Hospital, Boston.
A wide-ranging morning of presentations put data regarding historical trends and current global Zika hot spots in front of the committee. Current upticks in infection rates in northwest Mexico and in some states in India were areas of concern, given North American travel patterns, noted speaker Marc Fisher, MD, of the Center for Disease Control and Prevention’s Arboviral Disease Branch (Fort Collins, Colo.) “We’re going to see sporadic outbreaks; it’s hard to predict the future,” he said. “The new outbreak in India raises concerns.”
Briefing information from the FDA explained that Zika virus local transmission peaked in the United States in late summer of 2016. More than 5,000 cases were reported in the United States and over 36,000 in Puerto Rico. This has plummeted to 220 in 2018, with about two-thirds of these cases occurring in the territories, mostly (97%) from Puerto Rico across all 3 years.
Zika viremic blood donors dropped by an order of magnitude yearly, totaling 363 in 2016, 38 in 2017, and just 3 in 2018. Of the 363 detected in 2016, 96% came from Puerto Rico or Florida, noted Dr. Fisher.
The number of suspected and confirmed cases in the Americas overall has also dropped from over 650,000 in 2016 to under 30,000 in 2018, with most cases in 2018 being suspected rather than laboratory confirmed. In contrast to testing conducted in North America, few cases in much of Central and South America were laboratory confirmed.
Asymptomatic infections have occurred in blood donors, said the FDA, with 1.8% of blood donations in Puerto Rico testing positive for Zika virus during the peak of the outbreak. Transmission by transfusion is thought to have occurred in Brazil.
Although Zika virus infections have plummeted in the United States and worldwide, prevalence and rates of local transmission are unpredictable, said the FDA, which pointed to sporadic increases in autochthonous transmission of viruses such as dengue and chikungunya that are carried by the same mosquito vector as Zika.
Some of the committee’s discussion centered around finding a way to carve out protection for those most harmed by Zika virus – pregnant women and their fetuses. Martin Schreiber, MD, professor of surgery at Oregon Health and Sciences University, Portland, proposed a point-of-care testing strategy in which only blood destined for pregnant women would be tested for Zika virus. Dr. Schreiber, a trauma surgeon, put forward the rationale that Zika virus causes harm almost exclusively to fetuses, except for rare cases of Guillain-Barré syndrome.
In response, Dr. Kaufman pointed out that with rare exceptions for some bacterial testing, all testing is done from samples taken at the point of donation. The supply chain for donor blood is not set up to accommodate point-of-care testing, he said.
Answering questions about another targeted strategy – maintaining a separate, Zika-tested supply of blood for pregnant women – Susan Stramer, PhD, vice president of scientific affairs for the American Red Cross, said, “Most hospitals do not want, and are very adamant against, carrying a dual inventory.”
Ultimately, the committee’s discussion swung toward the realization that it may be too soon after the recent spike in U.S. Zika cases to plot the best course for ongoing testing strategies. “We are at the tail end of a waning epidemic. ... I think it would probably be a pretty easy question for the committee and for the agency if we actually had some way of having a crystal ball and knowing that the current trend was likely to continue,” said Roger Lewis, MD, PhD, professor at the University of California, Los Angeles, and chair of the department of emergency medicine at Harbor-UCLA Medical Center.
“I think that is not the question,” he went on. “I think the question is, What is the optimal strategy if we have no idea if that tail is going to continue in this current trend. ... And that maybe the committee ought to be thinking about what is the right strategy for the next 2 years – with an underlying assumption that this is a question that can be brought back as we learn more about how this disease behaves.”
The FDA usually follows the recommendations of its advisory committees.
Most members of a Food and Drug Administration advisory committee considered that data support maintaining current testing protocols for Zika virus in the blood donor pool. However, committee discussion entertained the idea of revisiting testing strategies after another year or 2 of Zika virus epidemiological data are available.
In its last guidance regarding Zika virus testing, issued in July 2018, the FDA recommended that either minipool nucleic acid testing (MP NAT) or individual donor (ID) NAT be used to screen for Zika virus. Current guidance still requires conversion to all-ID NAT “when certain threshold conditions are met that indicate an increased risk of suspected mosquito-borne transmission in a defined geographic collection area.”
In the first of three separate votes, 11 of 15 voting members of the FDA’s Blood Products Advisory Committee (BPAC) answered in the affirmative to the question of whether available data support continuing the status quo for Zika testing. Committee members then were asked to weigh whether current data support scaling back to a regional testing strategy targeting at-risk areas. Here, six committee members answered in the affirmative, and nine in the negative.
Just one committee member, F. Blaine Hollinger, MD, voted in favor of the third option, elimination of all Zika virus testing without reintroducing donor screening for risk factors in risk-free areas pending another outbreak in the United States. Dr. Hollinger is a professor of virology and microbiology at Baylor College of Medicine, Houston.
The committee as whole wasn’t swayed by a line of questioning put forward by chairman Richard Kaufman, MD. “I will be the devil’s advocate a little bit: We learned that there have been zero confirmed positives from blood donors for the past year. Would anyone be comfortable with just stopping screening of donors?” asked Dr. Kaufman, medical director of the adult transfusion service at Brigham and Women’s Hospital, Boston.
A wide-ranging morning of presentations put data regarding historical trends and current global Zika hot spots in front of the committee. Current upticks in infection rates in northwest Mexico and in some states in India were areas of concern, given North American travel patterns, noted speaker Marc Fisher, MD, of the Center for Disease Control and Prevention’s Arboviral Disease Branch (Fort Collins, Colo.) “We’re going to see sporadic outbreaks; it’s hard to predict the future,” he said. “The new outbreak in India raises concerns.”
Briefing information from the FDA explained that Zika virus local transmission peaked in the United States in late summer of 2016. More than 5,000 cases were reported in the United States and over 36,000 in Puerto Rico. This has plummeted to 220 in 2018, with about two-thirds of these cases occurring in the territories, mostly (97%) from Puerto Rico across all 3 years.
Zika viremic blood donors dropped by an order of magnitude yearly, totaling 363 in 2016, 38 in 2017, and just 3 in 2018. Of the 363 detected in 2016, 96% came from Puerto Rico or Florida, noted Dr. Fisher.
The number of suspected and confirmed cases in the Americas overall has also dropped from over 650,000 in 2016 to under 30,000 in 2018, with most cases in 2018 being suspected rather than laboratory confirmed. In contrast to testing conducted in North America, few cases in much of Central and South America were laboratory confirmed.
Asymptomatic infections have occurred in blood donors, said the FDA, with 1.8% of blood donations in Puerto Rico testing positive for Zika virus during the peak of the outbreak. Transmission by transfusion is thought to have occurred in Brazil.
Although Zika virus infections have plummeted in the United States and worldwide, prevalence and rates of local transmission are unpredictable, said the FDA, which pointed to sporadic increases in autochthonous transmission of viruses such as dengue and chikungunya that are carried by the same mosquito vector as Zika.
Some of the committee’s discussion centered around finding a way to carve out protection for those most harmed by Zika virus – pregnant women and their fetuses. Martin Schreiber, MD, professor of surgery at Oregon Health and Sciences University, Portland, proposed a point-of-care testing strategy in which only blood destined for pregnant women would be tested for Zika virus. Dr. Schreiber, a trauma surgeon, put forward the rationale that Zika virus causes harm almost exclusively to fetuses, except for rare cases of Guillain-Barré syndrome.
In response, Dr. Kaufman pointed out that with rare exceptions for some bacterial testing, all testing is done from samples taken at the point of donation. The supply chain for donor blood is not set up to accommodate point-of-care testing, he said.
Answering questions about another targeted strategy – maintaining a separate, Zika-tested supply of blood for pregnant women – Susan Stramer, PhD, vice president of scientific affairs for the American Red Cross, said, “Most hospitals do not want, and are very adamant against, carrying a dual inventory.”
Ultimately, the committee’s discussion swung toward the realization that it may be too soon after the recent spike in U.S. Zika cases to plot the best course for ongoing testing strategies. “We are at the tail end of a waning epidemic. ... I think it would probably be a pretty easy question for the committee and for the agency if we actually had some way of having a crystal ball and knowing that the current trend was likely to continue,” said Roger Lewis, MD, PhD, professor at the University of California, Los Angeles, and chair of the department of emergency medicine at Harbor-UCLA Medical Center.
“I think that is not the question,” he went on. “I think the question is, What is the optimal strategy if we have no idea if that tail is going to continue in this current trend. ... And that maybe the committee ought to be thinking about what is the right strategy for the next 2 years – with an underlying assumption that this is a question that can be brought back as we learn more about how this disease behaves.”
The FDA usually follows the recommendations of its advisory committees.
Most members of a Food and Drug Administration advisory committee considered that data support maintaining current testing protocols for Zika virus in the blood donor pool. However, committee discussion entertained the idea of revisiting testing strategies after another year or 2 of Zika virus epidemiological data are available.
In its last guidance regarding Zika virus testing, issued in July 2018, the FDA recommended that either minipool nucleic acid testing (MP NAT) or individual donor (ID) NAT be used to screen for Zika virus. Current guidance still requires conversion to all-ID NAT “when certain threshold conditions are met that indicate an increased risk of suspected mosquito-borne transmission in a defined geographic collection area.”
In the first of three separate votes, 11 of 15 voting members of the FDA’s Blood Products Advisory Committee (BPAC) answered in the affirmative to the question of whether available data support continuing the status quo for Zika testing. Committee members then were asked to weigh whether current data support scaling back to a regional testing strategy targeting at-risk areas. Here, six committee members answered in the affirmative, and nine in the negative.
Just one committee member, F. Blaine Hollinger, MD, voted in favor of the third option, elimination of all Zika virus testing without reintroducing donor screening for risk factors in risk-free areas pending another outbreak in the United States. Dr. Hollinger is a professor of virology and microbiology at Baylor College of Medicine, Houston.
The committee as whole wasn’t swayed by a line of questioning put forward by chairman Richard Kaufman, MD. “I will be the devil’s advocate a little bit: We learned that there have been zero confirmed positives from blood donors for the past year. Would anyone be comfortable with just stopping screening of donors?” asked Dr. Kaufman, medical director of the adult transfusion service at Brigham and Women’s Hospital, Boston.
A wide-ranging morning of presentations put data regarding historical trends and current global Zika hot spots in front of the committee. Current upticks in infection rates in northwest Mexico and in some states in India were areas of concern, given North American travel patterns, noted speaker Marc Fisher, MD, of the Center for Disease Control and Prevention’s Arboviral Disease Branch (Fort Collins, Colo.) “We’re going to see sporadic outbreaks; it’s hard to predict the future,” he said. “The new outbreak in India raises concerns.”
Briefing information from the FDA explained that Zika virus local transmission peaked in the United States in late summer of 2016. More than 5,000 cases were reported in the United States and over 36,000 in Puerto Rico. This has plummeted to 220 in 2018, with about two-thirds of these cases occurring in the territories, mostly (97%) from Puerto Rico across all 3 years.
Zika viremic blood donors dropped by an order of magnitude yearly, totaling 363 in 2016, 38 in 2017, and just 3 in 2018. Of the 363 detected in 2016, 96% came from Puerto Rico or Florida, noted Dr. Fisher.
The number of suspected and confirmed cases in the Americas overall has also dropped from over 650,000 in 2016 to under 30,000 in 2018, with most cases in 2018 being suspected rather than laboratory confirmed. In contrast to testing conducted in North America, few cases in much of Central and South America were laboratory confirmed.
Asymptomatic infections have occurred in blood donors, said the FDA, with 1.8% of blood donations in Puerto Rico testing positive for Zika virus during the peak of the outbreak. Transmission by transfusion is thought to have occurred in Brazil.
Although Zika virus infections have plummeted in the United States and worldwide, prevalence and rates of local transmission are unpredictable, said the FDA, which pointed to sporadic increases in autochthonous transmission of viruses such as dengue and chikungunya that are carried by the same mosquito vector as Zika.
Some of the committee’s discussion centered around finding a way to carve out protection for those most harmed by Zika virus – pregnant women and their fetuses. Martin Schreiber, MD, professor of surgery at Oregon Health and Sciences University, Portland, proposed a point-of-care testing strategy in which only blood destined for pregnant women would be tested for Zika virus. Dr. Schreiber, a trauma surgeon, put forward the rationale that Zika virus causes harm almost exclusively to fetuses, except for rare cases of Guillain-Barré syndrome.
In response, Dr. Kaufman pointed out that with rare exceptions for some bacterial testing, all testing is done from samples taken at the point of donation. The supply chain for donor blood is not set up to accommodate point-of-care testing, he said.
Answering questions about another targeted strategy – maintaining a separate, Zika-tested supply of blood for pregnant women – Susan Stramer, PhD, vice president of scientific affairs for the American Red Cross, said, “Most hospitals do not want, and are very adamant against, carrying a dual inventory.”
Ultimately, the committee’s discussion swung toward the realization that it may be too soon after the recent spike in U.S. Zika cases to plot the best course for ongoing testing strategies. “We are at the tail end of a waning epidemic. ... I think it would probably be a pretty easy question for the committee and for the agency if we actually had some way of having a crystal ball and knowing that the current trend was likely to continue,” said Roger Lewis, MD, PhD, professor at the University of California, Los Angeles, and chair of the department of emergency medicine at Harbor-UCLA Medical Center.
“I think that is not the question,” he went on. “I think the question is, What is the optimal strategy if we have no idea if that tail is going to continue in this current trend. ... And that maybe the committee ought to be thinking about what is the right strategy for the next 2 years – with an underlying assumption that this is a question that can be brought back as we learn more about how this disease behaves.”
The FDA usually follows the recommendations of its advisory committees.
FDA approves brexanolone for postpartum depression
The Food and Drug Administration on March 19 approved the first medication specifically for the treatment of postpartum depression.
The drug, brexanolone (Zulresso), is to be administered as a single continuous 60-hour infusion for each episode of postpartum depression. . By binding to GABA A receptors, brexanolone increases receptor functionality. The recommended maximum dose of brexanolone is 90 µg/kg/h, and the infusion includes three dosing phases.
Brexanolone provides “an important new treatment option,” said Tiffany Farchione, MD, acting director of the division of psychiatry products in the FDA’s Center for Drug Evaluation and Research, in a press release. “Because of concerns about serious risks, including excessive sedation or sudden loss of consciousness during administration, Zulresso has been approved with a Risk Evaluation and Mitigation Strategy (REMS) and is only available to patients through a restricted distribution program at certified health care facilities where the health care provider can carefully monitor the patient.”
The approval was based on results of three phase 3 trials, which were double-blind, randomized, and placebo-controlled studies in which the primary efficacy endpoint was a change in baseline 60 hours after the start of the infusion on the Hamilton Depression Rating Scale (HAM-D). In all two of the trials, known as Hummingbird 202B and 202C, brexanolone’s impact on the patients’ HAM-D scores was greater than that of placebo, the FDA reported in briefing document released late last year. In addition, the impact of brexanolone on postpartum depression proved both rapid and durable.
Side effects observed in about 3% of the brexanolone patients included dizziness, dry mouth, fatigue, headache, infusion site pain, somnolence, and loss of consciousness. The FDA’s concern about loss of consciousness led the agency to recommend a REMS protocol before a hearing of its Psychopharmacologic Drugs Advisory and Drug Safety and Risk Management Advisory panels late last year. The Zulresso REMS Program will require that the drug be administered by a clinician in a health care facility that is certified. Patients will have to be monitored for excessive sedation and “sudden loss of consciousness and have continuous pulse oximetry monitoring (monitors oxygen levels in the blood),” the FDA said. Another requirement is that patients who receive the infusion will have to be accompanied while interacting with their children. Patients will be advised not to drive, operate machinery or engage in other dangerous activities until they feel totally alert. Those requirements will be addressed in a boxed warning.
The drug should be either adjusted or discontinued for patients whose postpartum depression becomes worse or for those experience suicidal thoughts and behaviors after taking brexanolone, the agency said.
Some physicians use antidepressants to treat postpartum depression, but their effectiveness is limited, according to the FDA. Interventions such as electroconvulsive therapy and psychotherapy also are used, but getting results can several weeks.
The symptoms of postpartum depression are indistinguishable from major depressive disorder, but “the timing of its onset has led to its recognition as potentially unique illness,” the FDA said. Postpartum depression in the United States affects up to 12% of births. In the developed world, suicide is the most common cause of maternal death after childbirth. This suicide risk makes postpartum depression a condition that is life-threatening. In addition, the condition has “profound negative effects on the maternal-infant bond and later infant development,” the FDA said.
SAGE Therapeutics, developer of brexanolone, secured the approval through the FDA’s breakthrough therapy designation process.
Heidi Splete contributed to this article.
The Food and Drug Administration on March 19 approved the first medication specifically for the treatment of postpartum depression.
The drug, brexanolone (Zulresso), is to be administered as a single continuous 60-hour infusion for each episode of postpartum depression. . By binding to GABA A receptors, brexanolone increases receptor functionality. The recommended maximum dose of brexanolone is 90 µg/kg/h, and the infusion includes three dosing phases.
Brexanolone provides “an important new treatment option,” said Tiffany Farchione, MD, acting director of the division of psychiatry products in the FDA’s Center for Drug Evaluation and Research, in a press release. “Because of concerns about serious risks, including excessive sedation or sudden loss of consciousness during administration, Zulresso has been approved with a Risk Evaluation and Mitigation Strategy (REMS) and is only available to patients through a restricted distribution program at certified health care facilities where the health care provider can carefully monitor the patient.”
The approval was based on results of three phase 3 trials, which were double-blind, randomized, and placebo-controlled studies in which the primary efficacy endpoint was a change in baseline 60 hours after the start of the infusion on the Hamilton Depression Rating Scale (HAM-D). In all two of the trials, known as Hummingbird 202B and 202C, brexanolone’s impact on the patients’ HAM-D scores was greater than that of placebo, the FDA reported in briefing document released late last year. In addition, the impact of brexanolone on postpartum depression proved both rapid and durable.
Side effects observed in about 3% of the brexanolone patients included dizziness, dry mouth, fatigue, headache, infusion site pain, somnolence, and loss of consciousness. The FDA’s concern about loss of consciousness led the agency to recommend a REMS protocol before a hearing of its Psychopharmacologic Drugs Advisory and Drug Safety and Risk Management Advisory panels late last year. The Zulresso REMS Program will require that the drug be administered by a clinician in a health care facility that is certified. Patients will have to be monitored for excessive sedation and “sudden loss of consciousness and have continuous pulse oximetry monitoring (monitors oxygen levels in the blood),” the FDA said. Another requirement is that patients who receive the infusion will have to be accompanied while interacting with their children. Patients will be advised not to drive, operate machinery or engage in other dangerous activities until they feel totally alert. Those requirements will be addressed in a boxed warning.
The drug should be either adjusted or discontinued for patients whose postpartum depression becomes worse or for those experience suicidal thoughts and behaviors after taking brexanolone, the agency said.
Some physicians use antidepressants to treat postpartum depression, but their effectiveness is limited, according to the FDA. Interventions such as electroconvulsive therapy and psychotherapy also are used, but getting results can several weeks.
The symptoms of postpartum depression are indistinguishable from major depressive disorder, but “the timing of its onset has led to its recognition as potentially unique illness,” the FDA said. Postpartum depression in the United States affects up to 12% of births. In the developed world, suicide is the most common cause of maternal death after childbirth. This suicide risk makes postpartum depression a condition that is life-threatening. In addition, the condition has “profound negative effects on the maternal-infant bond and later infant development,” the FDA said.
SAGE Therapeutics, developer of brexanolone, secured the approval through the FDA’s breakthrough therapy designation process.
Heidi Splete contributed to this article.
The Food and Drug Administration on March 19 approved the first medication specifically for the treatment of postpartum depression.
The drug, brexanolone (Zulresso), is to be administered as a single continuous 60-hour infusion for each episode of postpartum depression. . By binding to GABA A receptors, brexanolone increases receptor functionality. The recommended maximum dose of brexanolone is 90 µg/kg/h, and the infusion includes three dosing phases.
Brexanolone provides “an important new treatment option,” said Tiffany Farchione, MD, acting director of the division of psychiatry products in the FDA’s Center for Drug Evaluation and Research, in a press release. “Because of concerns about serious risks, including excessive sedation or sudden loss of consciousness during administration, Zulresso has been approved with a Risk Evaluation and Mitigation Strategy (REMS) and is only available to patients through a restricted distribution program at certified health care facilities where the health care provider can carefully monitor the patient.”
The approval was based on results of three phase 3 trials, which were double-blind, randomized, and placebo-controlled studies in which the primary efficacy endpoint was a change in baseline 60 hours after the start of the infusion on the Hamilton Depression Rating Scale (HAM-D). In all two of the trials, known as Hummingbird 202B and 202C, brexanolone’s impact on the patients’ HAM-D scores was greater than that of placebo, the FDA reported in briefing document released late last year. In addition, the impact of brexanolone on postpartum depression proved both rapid and durable.
Side effects observed in about 3% of the brexanolone patients included dizziness, dry mouth, fatigue, headache, infusion site pain, somnolence, and loss of consciousness. The FDA’s concern about loss of consciousness led the agency to recommend a REMS protocol before a hearing of its Psychopharmacologic Drugs Advisory and Drug Safety and Risk Management Advisory panels late last year. The Zulresso REMS Program will require that the drug be administered by a clinician in a health care facility that is certified. Patients will have to be monitored for excessive sedation and “sudden loss of consciousness and have continuous pulse oximetry monitoring (monitors oxygen levels in the blood),” the FDA said. Another requirement is that patients who receive the infusion will have to be accompanied while interacting with their children. Patients will be advised not to drive, operate machinery or engage in other dangerous activities until they feel totally alert. Those requirements will be addressed in a boxed warning.
The drug should be either adjusted or discontinued for patients whose postpartum depression becomes worse or for those experience suicidal thoughts and behaviors after taking brexanolone, the agency said.
Some physicians use antidepressants to treat postpartum depression, but their effectiveness is limited, according to the FDA. Interventions such as electroconvulsive therapy and psychotherapy also are used, but getting results can several weeks.
The symptoms of postpartum depression are indistinguishable from major depressive disorder, but “the timing of its onset has led to its recognition as potentially unique illness,” the FDA said. Postpartum depression in the United States affects up to 12% of births. In the developed world, suicide is the most common cause of maternal death after childbirth. This suicide risk makes postpartum depression a condition that is life-threatening. In addition, the condition has “profound negative effects on the maternal-infant bond and later infant development,” the FDA said.
SAGE Therapeutics, developer of brexanolone, secured the approval through the FDA’s breakthrough therapy designation process.
Heidi Splete contributed to this article.