User login
A new (old) drug joins the COVID fray, and guess what? It works
This transcript has been edited for clarity.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr F. Perry Wilson of the Yale School of Medicine.
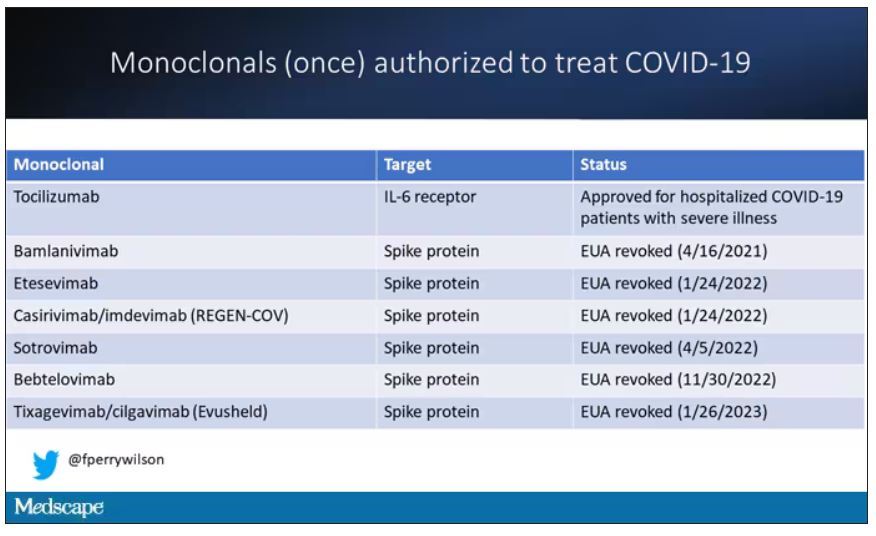
At this point, with the monoclonals found to be essentially useless, we are left with remdesivir with its modest efficacy and Paxlovid, which, for some reason, people don’t seem to be taking.
Part of the reason the monoclonals have failed lately is because of their specificity; they are homogeneous antibodies targeted toward a very specific epitope that may change from variant to variant. We need a broader therapeutic, one that has activity across all variants — maybe even one that has activity against all viruses? We’ve got one. Interferon.
The first mention of interferon as a potential COVID therapy was at the very start of the pandemic, so I’m sort of surprised that the first large, randomized trial is only being reported now in the New England Journal of Medicine.
Before we dig into the results, let’s talk mechanism. This is a trial of interferon-lambda, also known as interleukin-29.
The lambda interferons were only discovered in 2003. They differ from the more familiar interferons only in their cellular receptors; the downstream effects seem quite similar. As opposed to the cellular receptors for interferon alfa, which are widely expressed, the receptors for lambda are restricted to epithelial tissues. This makes it a good choice as a COVID treatment, since the virus also preferentially targets those epithelial cells.
In this study, 1,951 participants from Brazil and Canada, but mostly Brazil, with new COVID infections who were not yet hospitalized were randomized to receive 180 mcg of interferon lambda or placebo.
This was a relatively current COVID trial, as you can see from the participant characteristics. The majority had been vaccinated, and nearly half of the infections were during the Omicron phase of the pandemic.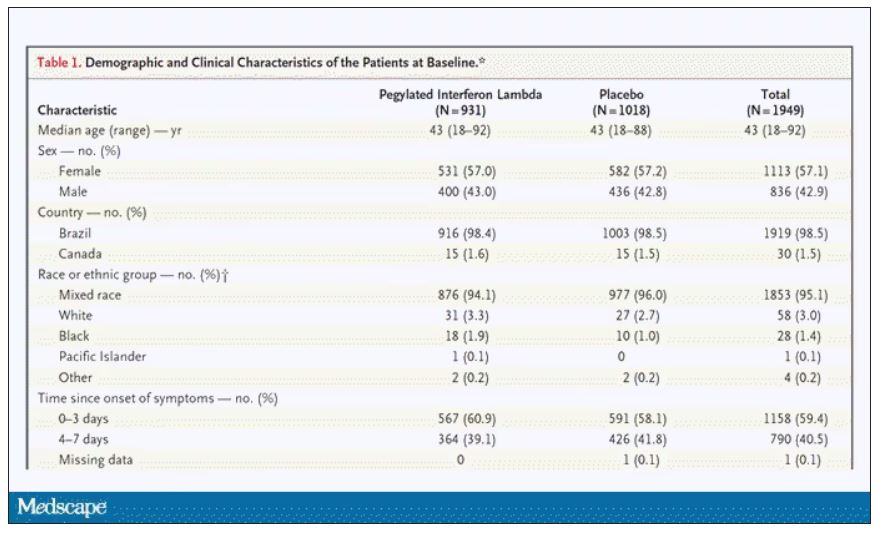
If you just want to cut to the chase, interferon worked.
The primary outcome – hospitalization or a prolonged emergency room visit for COVID – was 50% lower in the interferon group.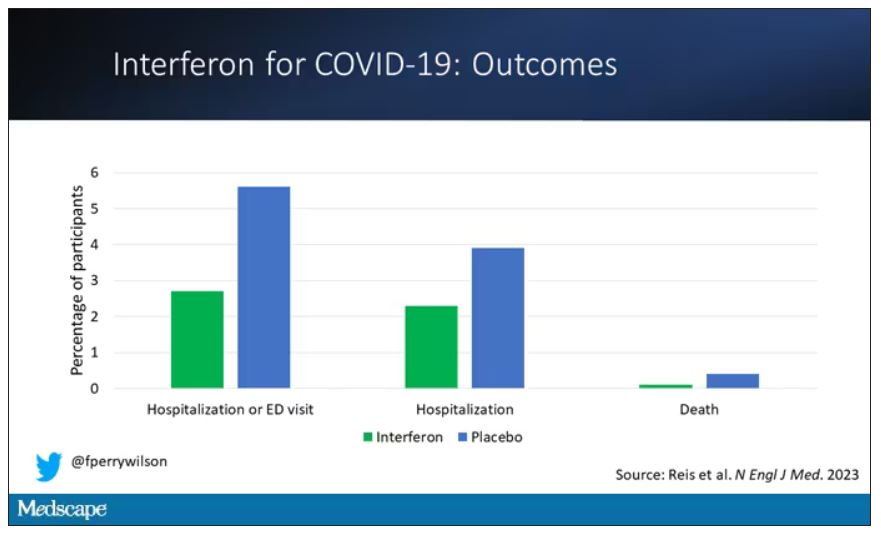
Key secondary outcomes, including death from COVID, were lower in the interferon group as well. These effects persisted across most of the subgroups I was looking out for.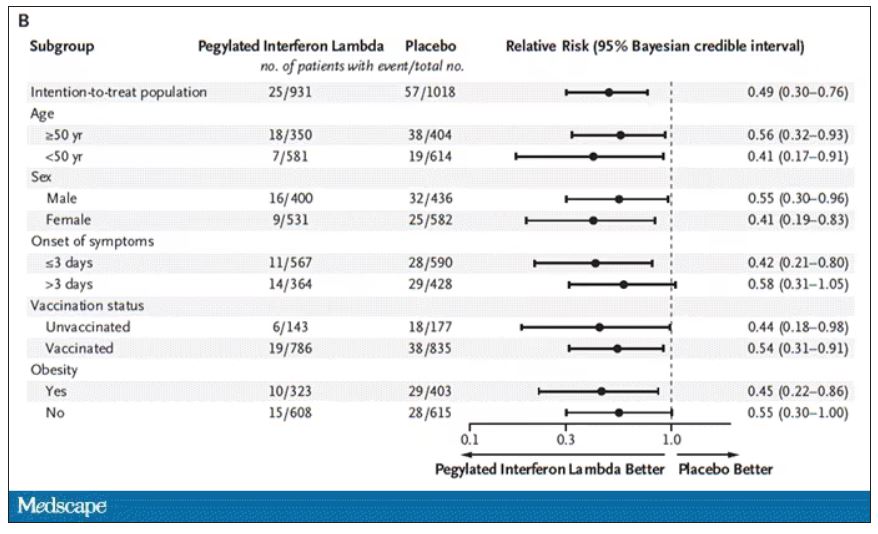
Interferon seemed to help those who were already vaccinated and those who were unvaccinated. There’s a hint that it works better within the first few days of symptoms, which isn’t surprising; we’ve seen this for many of the therapeutics, including Paxlovid. Time is of the essence. Encouragingly, the effect was a bit more pronounced among those infected with Omicron.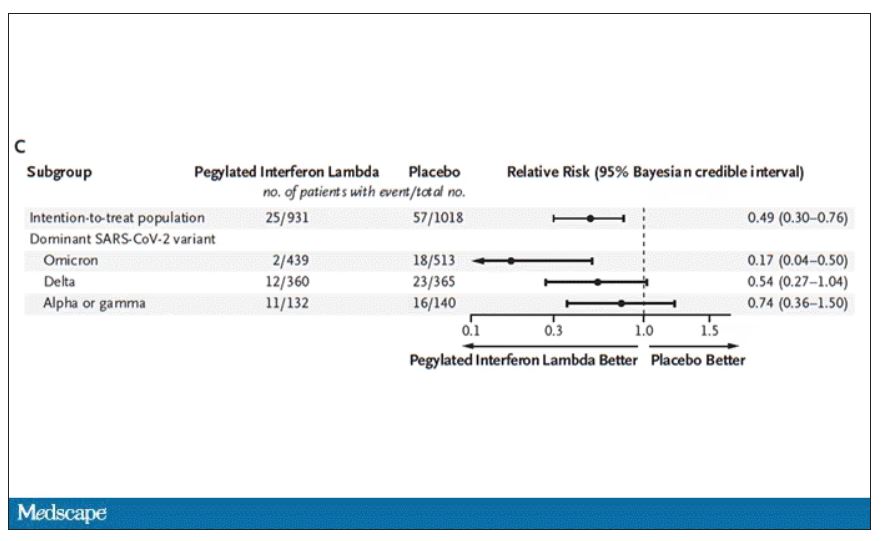
Of course, if you have any experience with interferon, you know that the side effects can be pretty rough. In the bad old days when we treated hepatitis C infection with interferon, patients would get their injections on Friday in anticipation of being essentially out of commission with flu-like symptoms through the weekend. But we don’t see much evidence of adverse events in this trial, maybe due to the greater specificity of interferon lambda.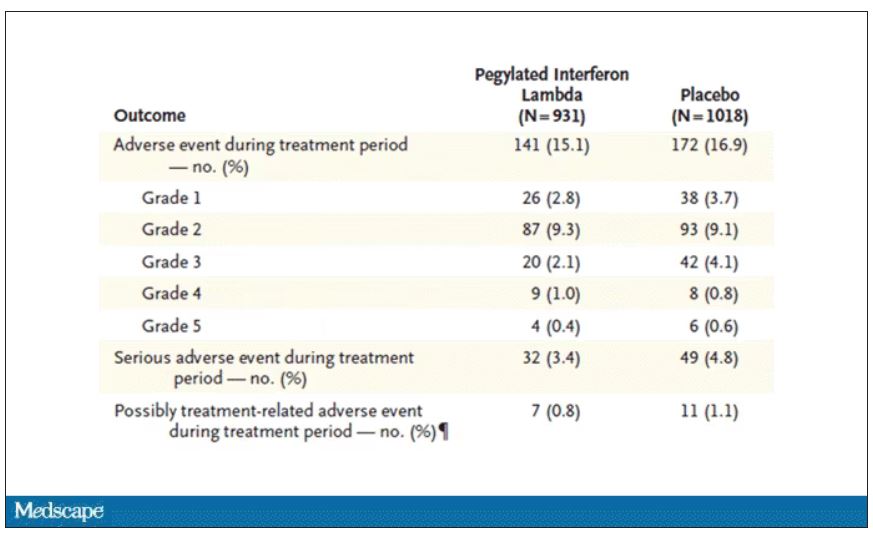
Putting it all together, the state of play for interferons in COVID may be changing. To date, the FDA has not recommended the use of interferon alfa or -beta for COVID-19, citing some data that they are ineffective or even harmful in hospitalized patients with COVID. Interferon lambda is not FDA approved and thus not even available in the United States. But the reason it has not been approved is that there has not been a large, well-conducted interferon lambda trial. Now there is. Will this study be enough to prompt an emergency use authorization? The elephant in the room, of course, is Paxlovid, which at this point has a longer safety track record and, importantly, is oral. I’d love to see a head-to-head trial. Short of that, I tend to be in favor of having more options on the table.
Dr. Perry Wilson is associate professor, department of medicine, and director, Clinical and Translational Research Accelerator, at Yale University, New Haven, Conn. He disclosed no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr F. Perry Wilson of the Yale School of Medicine.
At this point, with the monoclonals found to be essentially useless, we are left with remdesivir with its modest efficacy and Paxlovid, which, for some reason, people don’t seem to be taking.
Part of the reason the monoclonals have failed lately is because of their specificity; they are homogeneous antibodies targeted toward a very specific epitope that may change from variant to variant. We need a broader therapeutic, one that has activity across all variants — maybe even one that has activity against all viruses? We’ve got one. Interferon.
The first mention of interferon as a potential COVID therapy was at the very start of the pandemic, so I’m sort of surprised that the first large, randomized trial is only being reported now in the New England Journal of Medicine.
Before we dig into the results, let’s talk mechanism. This is a trial of interferon-lambda, also known as interleukin-29.
The lambda interferons were only discovered in 2003. They differ from the more familiar interferons only in their cellular receptors; the downstream effects seem quite similar. As opposed to the cellular receptors for interferon alfa, which are widely expressed, the receptors for lambda are restricted to epithelial tissues. This makes it a good choice as a COVID treatment, since the virus also preferentially targets those epithelial cells.
In this study, 1,951 participants from Brazil and Canada, but mostly Brazil, with new COVID infections who were not yet hospitalized were randomized to receive 180 mcg of interferon lambda or placebo.
This was a relatively current COVID trial, as you can see from the participant characteristics. The majority had been vaccinated, and nearly half of the infections were during the Omicron phase of the pandemic.
If you just want to cut to the chase, interferon worked.
The primary outcome – hospitalization or a prolonged emergency room visit for COVID – was 50% lower in the interferon group.
Key secondary outcomes, including death from COVID, were lower in the interferon group as well. These effects persisted across most of the subgroups I was looking out for.
Interferon seemed to help those who were already vaccinated and those who were unvaccinated. There’s a hint that it works better within the first few days of symptoms, which isn’t surprising; we’ve seen this for many of the therapeutics, including Paxlovid. Time is of the essence. Encouragingly, the effect was a bit more pronounced among those infected with Omicron.
Of course, if you have any experience with interferon, you know that the side effects can be pretty rough. In the bad old days when we treated hepatitis C infection with interferon, patients would get their injections on Friday in anticipation of being essentially out of commission with flu-like symptoms through the weekend. But we don’t see much evidence of adverse events in this trial, maybe due to the greater specificity of interferon lambda.
Putting it all together, the state of play for interferons in COVID may be changing. To date, the FDA has not recommended the use of interferon alfa or -beta for COVID-19, citing some data that they are ineffective or even harmful in hospitalized patients with COVID. Interferon lambda is not FDA approved and thus not even available in the United States. But the reason it has not been approved is that there has not been a large, well-conducted interferon lambda trial. Now there is. Will this study be enough to prompt an emergency use authorization? The elephant in the room, of course, is Paxlovid, which at this point has a longer safety track record and, importantly, is oral. I’d love to see a head-to-head trial. Short of that, I tend to be in favor of having more options on the table.
Dr. Perry Wilson is associate professor, department of medicine, and director, Clinical and Translational Research Accelerator, at Yale University, New Haven, Conn. He disclosed no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr F. Perry Wilson of the Yale School of Medicine.
At this point, with the monoclonals found to be essentially useless, we are left with remdesivir with its modest efficacy and Paxlovid, which, for some reason, people don’t seem to be taking.
Part of the reason the monoclonals have failed lately is because of their specificity; they are homogeneous antibodies targeted toward a very specific epitope that may change from variant to variant. We need a broader therapeutic, one that has activity across all variants — maybe even one that has activity against all viruses? We’ve got one. Interferon.
The first mention of interferon as a potential COVID therapy was at the very start of the pandemic, so I’m sort of surprised that the first large, randomized trial is only being reported now in the New England Journal of Medicine.
Before we dig into the results, let’s talk mechanism. This is a trial of interferon-lambda, also known as interleukin-29.
The lambda interferons were only discovered in 2003. They differ from the more familiar interferons only in their cellular receptors; the downstream effects seem quite similar. As opposed to the cellular receptors for interferon alfa, which are widely expressed, the receptors for lambda are restricted to epithelial tissues. This makes it a good choice as a COVID treatment, since the virus also preferentially targets those epithelial cells.
In this study, 1,951 participants from Brazil and Canada, but mostly Brazil, with new COVID infections who were not yet hospitalized were randomized to receive 180 mcg of interferon lambda or placebo.
This was a relatively current COVID trial, as you can see from the participant characteristics. The majority had been vaccinated, and nearly half of the infections were during the Omicron phase of the pandemic.
If you just want to cut to the chase, interferon worked.
The primary outcome – hospitalization or a prolonged emergency room visit for COVID – was 50% lower in the interferon group.
Key secondary outcomes, including death from COVID, were lower in the interferon group as well. These effects persisted across most of the subgroups I was looking out for.
Interferon seemed to help those who were already vaccinated and those who were unvaccinated. There’s a hint that it works better within the first few days of symptoms, which isn’t surprising; we’ve seen this for many of the therapeutics, including Paxlovid. Time is of the essence. Encouragingly, the effect was a bit more pronounced among those infected with Omicron.
Of course, if you have any experience with interferon, you know that the side effects can be pretty rough. In the bad old days when we treated hepatitis C infection with interferon, patients would get their injections on Friday in anticipation of being essentially out of commission with flu-like symptoms through the weekend. But we don’t see much evidence of adverse events in this trial, maybe due to the greater specificity of interferon lambda.
Putting it all together, the state of play for interferons in COVID may be changing. To date, the FDA has not recommended the use of interferon alfa or -beta for COVID-19, citing some data that they are ineffective or even harmful in hospitalized patients with COVID. Interferon lambda is not FDA approved and thus not even available in the United States. But the reason it has not been approved is that there has not been a large, well-conducted interferon lambda trial. Now there is. Will this study be enough to prompt an emergency use authorization? The elephant in the room, of course, is Paxlovid, which at this point has a longer safety track record and, importantly, is oral. I’d love to see a head-to-head trial. Short of that, I tend to be in favor of having more options on the table.
Dr. Perry Wilson is associate professor, department of medicine, and director, Clinical and Translational Research Accelerator, at Yale University, New Haven, Conn. He disclosed no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
Keto for life? Reasons to think twice
Is the ketogenic diet the only way to lose weight? Of course not! Keep track of calories in vs. calories out and almost anyone can lose weight. The problem is keeping it off. To understand that, we need to look at metabolic adaptation and the biology of obesity.
Our bodies have a “set point” that is epigenetically latched onto the environment the brain senses, just as the fetal environment responds to the maternal environment.

If food is plentiful, our hormones force us to eat until our bodies feel that there are enough fat stores to survive. Because of environmental influences such as highly processed food, preservatives, climate change, and regulation of temperature, our brains have decided that we need more adipose tissue than we did 50-100 years ago. It could be that an element in food has caused a dysfunction of the pathways that regulate our body weight, and most of us “defend” a higher body weight in this environment.
How to counteract that? Not easily. The ketogenic diet works temporarily just like any other diet where calorie intake is lower than usual. It seems to be agreeable to many people because they say they feel full after eating protein, fat, and perhaps some vegetables. Protein and fat are certainly more satiating than simple carbohydrates.
If strictly followed, a ketogenic diet will force the body to burn fat and go into ketosis. Without a source for glucose, the brain will burn ketones from fat stores. Owen and colleagues discovered this in 1969 when they did their now-famous studies of fasting in inpatients at Brigham and Women’s hospital, using IV amino acids to protect muscle mass.
Keto for life?
Is the ketogenic diet a healthy diet for the long term? That is a different question.
Of course not – we need high-fiber carbohydrate sources such as whole grains, fruits, and vegetables to keep the colon healthy and obtain the vitamins and minerals needed to make the Krebs cycle, or citric acid cycle, work at its best.
Why, then, are we promoting ketogenic diets for those with obesity and type 2 diabetes? Ketogenic or low-carbohydrate diets are easy to teach and can rapidly help patients lose weight and return their blood glucose, blood pressure, and other metabolic parameters to normal.
The patient will be instructed to avoid all highly processed foods. Studies have shown that highly processed foods, created to maximize flavor, “coerce” people to eat more calories than when presented with the same number of calories in unprocessed foods, a way to fool the brain.
Why are we fooling the brain?
We circumvent the natural satiety mechanisms that start with the gut. When we eat, our gastric fundus and intestinal stretch receptors start the process that informs the hypothalamus about food intake. Highly processed foods are usually devoid of fiber and volume, and pack in the calories in small volumes so that the stretch receptors are not activated until more calories are ingested. The study mentioned above developed two ad lib diets with the same number of calories, sugar, fat, and carbohydrate content – one ultraprocessed and the other unprocessed.
That explanation is just the tip of the iceberg, because a lot more than primitive stretch receptors is informing the brain. There are gut hormones that are secreted before and after meals, such as ghrelin, glucagon-like peptide 1 (GLP-1), glucose-dependent insulinotropic polypeptide (GIP), and cholecystokinin (CCK), among a slew of others. These peptide hormones are all secreted from gut cells into the blood or vagus nerve, or both, and alert the brain that there is or is not enough food to maintain body weight at its set point.
It’s a highly regulated and precise system that regulates body weight for survival of the species in this environment. However, the environment has changed over the past 100 years but our genetic makeup for survival of the fittest has not. The mechanism of action for defense of a higher body weight set point in this new environment has not been elucidated as yet. Most likely, there are many players or instigators involved, such as food-supply changes, sedentary lifestyle, ambient temperature, fetal programming, air quality, and global warming and climate change, to name a few.
The goal of obesity researchers is to investigate the underlying mechanisms of the increased prevalence of obesity over the past 100 years. The goal of obesity medicine specialists is to treat obesity in adults and children, and to prevent obesity as much as possible with lifestyle change and medications that have been shown to help “reverse” the metabolic adaptation to this environment. Our newest GLP-1/GIP receptor agonists have been shown in animal models to hit several pathways that lead to obesity. They are not just appetite suppressants. Yes, they do modulate appetite and satiety, but they also affect energy expenditure. The body’s normal reaction to a lack of calorie intake is to reduce resting energy expenditure until body weight increases back to “set point levels.” These agonists prevent that metabolic adaptation. That is why they are true agents that can treat obesity – the disease.
Back to the ketogenic diet. The ketogenic diet can fool the brain temporarily by using protein and fat to elicit satiety with less food intake in calories. After a while, however, gut hormones and other factors begin to counteract the weight loss with a reduction in resting energy and total energy expenditure, and other metabolic measures, to get the body back to a certain body weight set point.
The ketogenic diet also can help dieters avoid ultra- and highly processed foods. In the end, any type of diet that lowers caloric intake will work for weight loss, but it’s the maintenance of that weight loss that makes a long-term difference, and that involves closing the metabolic gap that the body generates to defend fat mass. Understanding this pathophysiology will allow obesity medicine specialists to assist patients with obesity to lose weight and keep it off.
Dr. Apovian is in the department of medicine, division of endocrinology, diabetes, and hypertension, and codirector, Center for Weight Management and Wellness, Harvard Medical School, Boston. She disclosed ties with Altimmune, Cowen and Company, Currax Pharmaceuticals, EPG Communication Holdings, Gelesis Srl, L-Nutra, NeuroBo Pharmaceuticals, National Institutes of Health, Patient-Centered Outcomes Research Institute, GI Dynamics, and Novo Nordisk. A version of this article first appeared on Medscape.com.
Is the ketogenic diet the only way to lose weight? Of course not! Keep track of calories in vs. calories out and almost anyone can lose weight. The problem is keeping it off. To understand that, we need to look at metabolic adaptation and the biology of obesity.
Our bodies have a “set point” that is epigenetically latched onto the environment the brain senses, just as the fetal environment responds to the maternal environment.
If food is plentiful, our hormones force us to eat until our bodies feel that there are enough fat stores to survive. Because of environmental influences such as highly processed food, preservatives, climate change, and regulation of temperature, our brains have decided that we need more adipose tissue than we did 50-100 years ago. It could be that an element in food has caused a dysfunction of the pathways that regulate our body weight, and most of us “defend” a higher body weight in this environment.
How to counteract that? Not easily. The ketogenic diet works temporarily just like any other diet where calorie intake is lower than usual. It seems to be agreeable to many people because they say they feel full after eating protein, fat, and perhaps some vegetables. Protein and fat are certainly more satiating than simple carbohydrates.
If strictly followed, a ketogenic diet will force the body to burn fat and go into ketosis. Without a source for glucose, the brain will burn ketones from fat stores. Owen and colleagues discovered this in 1969 when they did their now-famous studies of fasting in inpatients at Brigham and Women’s hospital, using IV amino acids to protect muscle mass.
Keto for life?
Is the ketogenic diet a healthy diet for the long term? That is a different question.
Of course not – we need high-fiber carbohydrate sources such as whole grains, fruits, and vegetables to keep the colon healthy and obtain the vitamins and minerals needed to make the Krebs cycle, or citric acid cycle, work at its best.
Why, then, are we promoting ketogenic diets for those with obesity and type 2 diabetes? Ketogenic or low-carbohydrate diets are easy to teach and can rapidly help patients lose weight and return their blood glucose, blood pressure, and other metabolic parameters to normal.
The patient will be instructed to avoid all highly processed foods. Studies have shown that highly processed foods, created to maximize flavor, “coerce” people to eat more calories than when presented with the same number of calories in unprocessed foods, a way to fool the brain.
Why are we fooling the brain?
We circumvent the natural satiety mechanisms that start with the gut. When we eat, our gastric fundus and intestinal stretch receptors start the process that informs the hypothalamus about food intake. Highly processed foods are usually devoid of fiber and volume, and pack in the calories in small volumes so that the stretch receptors are not activated until more calories are ingested. The study mentioned above developed two ad lib diets with the same number of calories, sugar, fat, and carbohydrate content – one ultraprocessed and the other unprocessed.
That explanation is just the tip of the iceberg, because a lot more than primitive stretch receptors is informing the brain. There are gut hormones that are secreted before and after meals, such as ghrelin, glucagon-like peptide 1 (GLP-1), glucose-dependent insulinotropic polypeptide (GIP), and cholecystokinin (CCK), among a slew of others. These peptide hormones are all secreted from gut cells into the blood or vagus nerve, or both, and alert the brain that there is or is not enough food to maintain body weight at its set point.
It’s a highly regulated and precise system that regulates body weight for survival of the species in this environment. However, the environment has changed over the past 100 years but our genetic makeup for survival of the fittest has not. The mechanism of action for defense of a higher body weight set point in this new environment has not been elucidated as yet. Most likely, there are many players or instigators involved, such as food-supply changes, sedentary lifestyle, ambient temperature, fetal programming, air quality, and global warming and climate change, to name a few.
The goal of obesity researchers is to investigate the underlying mechanisms of the increased prevalence of obesity over the past 100 years. The goal of obesity medicine specialists is to treat obesity in adults and children, and to prevent obesity as much as possible with lifestyle change and medications that have been shown to help “reverse” the metabolic adaptation to this environment. Our newest GLP-1/GIP receptor agonists have been shown in animal models to hit several pathways that lead to obesity. They are not just appetite suppressants. Yes, they do modulate appetite and satiety, but they also affect energy expenditure. The body’s normal reaction to a lack of calorie intake is to reduce resting energy expenditure until body weight increases back to “set point levels.” These agonists prevent that metabolic adaptation. That is why they are true agents that can treat obesity – the disease.
Back to the ketogenic diet. The ketogenic diet can fool the brain temporarily by using protein and fat to elicit satiety with less food intake in calories. After a while, however, gut hormones and other factors begin to counteract the weight loss with a reduction in resting energy and total energy expenditure, and other metabolic measures, to get the body back to a certain body weight set point.
The ketogenic diet also can help dieters avoid ultra- and highly processed foods. In the end, any type of diet that lowers caloric intake will work for weight loss, but it’s the maintenance of that weight loss that makes a long-term difference, and that involves closing the metabolic gap that the body generates to defend fat mass. Understanding this pathophysiology will allow obesity medicine specialists to assist patients with obesity to lose weight and keep it off.
Dr. Apovian is in the department of medicine, division of endocrinology, diabetes, and hypertension, and codirector, Center for Weight Management and Wellness, Harvard Medical School, Boston. She disclosed ties with Altimmune, Cowen and Company, Currax Pharmaceuticals, EPG Communication Holdings, Gelesis Srl, L-Nutra, NeuroBo Pharmaceuticals, National Institutes of Health, Patient-Centered Outcomes Research Institute, GI Dynamics, and Novo Nordisk. A version of this article first appeared on Medscape.com.
Is the ketogenic diet the only way to lose weight? Of course not! Keep track of calories in vs. calories out and almost anyone can lose weight. The problem is keeping it off. To understand that, we need to look at metabolic adaptation and the biology of obesity.
Our bodies have a “set point” that is epigenetically latched onto the environment the brain senses, just as the fetal environment responds to the maternal environment.
If food is plentiful, our hormones force us to eat until our bodies feel that there are enough fat stores to survive. Because of environmental influences such as highly processed food, preservatives, climate change, and regulation of temperature, our brains have decided that we need more adipose tissue than we did 50-100 years ago. It could be that an element in food has caused a dysfunction of the pathways that regulate our body weight, and most of us “defend” a higher body weight in this environment.
How to counteract that? Not easily. The ketogenic diet works temporarily just like any other diet where calorie intake is lower than usual. It seems to be agreeable to many people because they say they feel full after eating protein, fat, and perhaps some vegetables. Protein and fat are certainly more satiating than simple carbohydrates.
If strictly followed, a ketogenic diet will force the body to burn fat and go into ketosis. Without a source for glucose, the brain will burn ketones from fat stores. Owen and colleagues discovered this in 1969 when they did their now-famous studies of fasting in inpatients at Brigham and Women’s hospital, using IV amino acids to protect muscle mass.
Keto for life?
Is the ketogenic diet a healthy diet for the long term? That is a different question.
Of course not – we need high-fiber carbohydrate sources such as whole grains, fruits, and vegetables to keep the colon healthy and obtain the vitamins and minerals needed to make the Krebs cycle, or citric acid cycle, work at its best.
Why, then, are we promoting ketogenic diets for those with obesity and type 2 diabetes? Ketogenic or low-carbohydrate diets are easy to teach and can rapidly help patients lose weight and return their blood glucose, blood pressure, and other metabolic parameters to normal.
The patient will be instructed to avoid all highly processed foods. Studies have shown that highly processed foods, created to maximize flavor, “coerce” people to eat more calories than when presented with the same number of calories in unprocessed foods, a way to fool the brain.
Why are we fooling the brain?
We circumvent the natural satiety mechanisms that start with the gut. When we eat, our gastric fundus and intestinal stretch receptors start the process that informs the hypothalamus about food intake. Highly processed foods are usually devoid of fiber and volume, and pack in the calories in small volumes so that the stretch receptors are not activated until more calories are ingested. The study mentioned above developed two ad lib diets with the same number of calories, sugar, fat, and carbohydrate content – one ultraprocessed and the other unprocessed.
That explanation is just the tip of the iceberg, because a lot more than primitive stretch receptors is informing the brain. There are gut hormones that are secreted before and after meals, such as ghrelin, glucagon-like peptide 1 (GLP-1), glucose-dependent insulinotropic polypeptide (GIP), and cholecystokinin (CCK), among a slew of others. These peptide hormones are all secreted from gut cells into the blood or vagus nerve, or both, and alert the brain that there is or is not enough food to maintain body weight at its set point.
It’s a highly regulated and precise system that regulates body weight for survival of the species in this environment. However, the environment has changed over the past 100 years but our genetic makeup for survival of the fittest has not. The mechanism of action for defense of a higher body weight set point in this new environment has not been elucidated as yet. Most likely, there are many players or instigators involved, such as food-supply changes, sedentary lifestyle, ambient temperature, fetal programming, air quality, and global warming and climate change, to name a few.
The goal of obesity researchers is to investigate the underlying mechanisms of the increased prevalence of obesity over the past 100 years. The goal of obesity medicine specialists is to treat obesity in adults and children, and to prevent obesity as much as possible with lifestyle change and medications that have been shown to help “reverse” the metabolic adaptation to this environment. Our newest GLP-1/GIP receptor agonists have been shown in animal models to hit several pathways that lead to obesity. They are not just appetite suppressants. Yes, they do modulate appetite and satiety, but they also affect energy expenditure. The body’s normal reaction to a lack of calorie intake is to reduce resting energy expenditure until body weight increases back to “set point levels.” These agonists prevent that metabolic adaptation. That is why they are true agents that can treat obesity – the disease.
Back to the ketogenic diet. The ketogenic diet can fool the brain temporarily by using protein and fat to elicit satiety with less food intake in calories. After a while, however, gut hormones and other factors begin to counteract the weight loss with a reduction in resting energy and total energy expenditure, and other metabolic measures, to get the body back to a certain body weight set point.
The ketogenic diet also can help dieters avoid ultra- and highly processed foods. In the end, any type of diet that lowers caloric intake will work for weight loss, but it’s the maintenance of that weight loss that makes a long-term difference, and that involves closing the metabolic gap that the body generates to defend fat mass. Understanding this pathophysiology will allow obesity medicine specialists to assist patients with obesity to lose weight and keep it off.
Dr. Apovian is in the department of medicine, division of endocrinology, diabetes, and hypertension, and codirector, Center for Weight Management and Wellness, Harvard Medical School, Boston. She disclosed ties with Altimmune, Cowen and Company, Currax Pharmaceuticals, EPG Communication Holdings, Gelesis Srl, L-Nutra, NeuroBo Pharmaceuticals, National Institutes of Health, Patient-Centered Outcomes Research Institute, GI Dynamics, and Novo Nordisk. A version of this article first appeared on Medscape.com.
‘Ozempic face’: Accepting wrinkles for improved health
This transcript has been edited for clarity.
Last week, a number of patients emailed me regarding their concerns about this phenomenon known as Ozempic face. I went on to read about what this meant. I live in Los Angeles, where most people appear to be on semaglutide (Ozempic). It’s the phenomenon where people lose weight relatively rapidly, making their faces thin out. Then what happens, apparently, is they look older because their face is more wrinkled and baggier. They might have to have further plastic surgery. I say that with slight sarcasm because of where I live.
I want to talk about what I think about this, living here where there’s a great pressure to prescribe semaglutide off label, and what I think about it for my patients with diabetes.

Historically, we haven’t had much in terms of effective medication for treating obesity, and frankly, now we do. We now have agents that are effective, that have relatively few side effects, and that have become part of what’s out there. People now want to use these agents, semaglutide, and there’s been a great need for these agents.
The problem, however, is twofold. One, as we all know, is that it has basically caused a shortage of medication for treating our patients who actually have type 2 diabetes and really need these medications to manage their disease. Then we have people who want these medications who can’t pay for them. Insurance doesn’t cover obesity medications, which is problematic and actually quite frustrating for people who, I think, really would benefit from using these medications.
What I tell people, frankly, is that until I have enough supply for my patients with type 2 diabetes, who need these agents to control their blood sugars, I want to keep this class of drugs available to them. I also hope we’re able to expand it more and more with improving insurance coverage – and that’s a big if, if you ask me – both for people who have prediabetes and for patients who are overweight and obese, because I think it’s really hard for people to lose weight.
It’s frustrating, and for many people, being overweight and obese causes all sorts of other health issues, not only diabetes. I believe that these drugs are both safe and effective and should be more available. I do think we need to be careful in terms of who we prescribe them to, at least at the moment. Hopefully, we’ll be able to expand their use.
Anything that can encourage our population to lose weight and maintain that weight loss is very important. We need to couple weight loss medications with lifestyle interventions. I think people can out-eat any medication; therefore, it’s very important to encourage our patients to eat better, to exercise more, and to do all the other things they need to do to reduce their risks for other comorbidities.
I am incredibly happy to have these newer agents on the market. I tell my patients – at least those who have diabetes – that they have to accept looking a little bit too thin for the benefits that we can see in using these medications.
Thank you.
Dr. Peters is professor of medicine at the University of Southern California, Los Angeles, and director of the USC clinical diabetes programs. She has published more than 200 articles, reviews, and abstracts, and three books, on diabetes, and has been an investigator for more than 40 research studies. She has spoken internationally at over 400 programs and serves on many committees of several professional organizations. She has ties with Abbott Diabetes Care, AstraZeneca Becton Dickinson, Boehringer Ingelheim Pharmaceuticals, Dexcom, Eli Lilly, Lexicon Pharmaceuticals, Livongo, MannKind Corporation, Medscape, Merck, Novo Nordisk, Omada Health, OptumHealth, Sanofi, and Zafgen. A version of this article originally appeared on Medscape.com.
This transcript has been edited for clarity.
Last week, a number of patients emailed me regarding their concerns about this phenomenon known as Ozempic face. I went on to read about what this meant. I live in Los Angeles, where most people appear to be on semaglutide (Ozempic). It’s the phenomenon where people lose weight relatively rapidly, making their faces thin out. Then what happens, apparently, is they look older because their face is more wrinkled and baggier. They might have to have further plastic surgery. I say that with slight sarcasm because of where I live.
I want to talk about what I think about this, living here where there’s a great pressure to prescribe semaglutide off label, and what I think about it for my patients with diabetes.
Historically, we haven’t had much in terms of effective medication for treating obesity, and frankly, now we do. We now have agents that are effective, that have relatively few side effects, and that have become part of what’s out there. People now want to use these agents, semaglutide, and there’s been a great need for these agents.
The problem, however, is twofold. One, as we all know, is that it has basically caused a shortage of medication for treating our patients who actually have type 2 diabetes and really need these medications to manage their disease. Then we have people who want these medications who can’t pay for them. Insurance doesn’t cover obesity medications, which is problematic and actually quite frustrating for people who, I think, really would benefit from using these medications.
What I tell people, frankly, is that until I have enough supply for my patients with type 2 diabetes, who need these agents to control their blood sugars, I want to keep this class of drugs available to them. I also hope we’re able to expand it more and more with improving insurance coverage – and that’s a big if, if you ask me – both for people who have prediabetes and for patients who are overweight and obese, because I think it’s really hard for people to lose weight.
It’s frustrating, and for many people, being overweight and obese causes all sorts of other health issues, not only diabetes. I believe that these drugs are both safe and effective and should be more available. I do think we need to be careful in terms of who we prescribe them to, at least at the moment. Hopefully, we’ll be able to expand their use.
Anything that can encourage our population to lose weight and maintain that weight loss is very important. We need to couple weight loss medications with lifestyle interventions. I think people can out-eat any medication; therefore, it’s very important to encourage our patients to eat better, to exercise more, and to do all the other things they need to do to reduce their risks for other comorbidities.
I am incredibly happy to have these newer agents on the market. I tell my patients – at least those who have diabetes – that they have to accept looking a little bit too thin for the benefits that we can see in using these medications.
Thank you.
Dr. Peters is professor of medicine at the University of Southern California, Los Angeles, and director of the USC clinical diabetes programs. She has published more than 200 articles, reviews, and abstracts, and three books, on diabetes, and has been an investigator for more than 40 research studies. She has spoken internationally at over 400 programs and serves on many committees of several professional organizations. She has ties with Abbott Diabetes Care, AstraZeneca Becton Dickinson, Boehringer Ingelheim Pharmaceuticals, Dexcom, Eli Lilly, Lexicon Pharmaceuticals, Livongo, MannKind Corporation, Medscape, Merck, Novo Nordisk, Omada Health, OptumHealth, Sanofi, and Zafgen. A version of this article originally appeared on Medscape.com.
This transcript has been edited for clarity.
Last week, a number of patients emailed me regarding their concerns about this phenomenon known as Ozempic face. I went on to read about what this meant. I live in Los Angeles, where most people appear to be on semaglutide (Ozempic). It’s the phenomenon where people lose weight relatively rapidly, making their faces thin out. Then what happens, apparently, is they look older because their face is more wrinkled and baggier. They might have to have further plastic surgery. I say that with slight sarcasm because of where I live.
I want to talk about what I think about this, living here where there’s a great pressure to prescribe semaglutide off label, and what I think about it for my patients with diabetes.
Historically, we haven’t had much in terms of effective medication for treating obesity, and frankly, now we do. We now have agents that are effective, that have relatively few side effects, and that have become part of what’s out there. People now want to use these agents, semaglutide, and there’s been a great need for these agents.
The problem, however, is twofold. One, as we all know, is that it has basically caused a shortage of medication for treating our patients who actually have type 2 diabetes and really need these medications to manage their disease. Then we have people who want these medications who can’t pay for them. Insurance doesn’t cover obesity medications, which is problematic and actually quite frustrating for people who, I think, really would benefit from using these medications.
What I tell people, frankly, is that until I have enough supply for my patients with type 2 diabetes, who need these agents to control their blood sugars, I want to keep this class of drugs available to them. I also hope we’re able to expand it more and more with improving insurance coverage – and that’s a big if, if you ask me – both for people who have prediabetes and for patients who are overweight and obese, because I think it’s really hard for people to lose weight.
It’s frustrating, and for many people, being overweight and obese causes all sorts of other health issues, not only diabetes. I believe that these drugs are both safe and effective and should be more available. I do think we need to be careful in terms of who we prescribe them to, at least at the moment. Hopefully, we’ll be able to expand their use.
Anything that can encourage our population to lose weight and maintain that weight loss is very important. We need to couple weight loss medications with lifestyle interventions. I think people can out-eat any medication; therefore, it’s very important to encourage our patients to eat better, to exercise more, and to do all the other things they need to do to reduce their risks for other comorbidities.
I am incredibly happy to have these newer agents on the market. I tell my patients – at least those who have diabetes – that they have to accept looking a little bit too thin for the benefits that we can see in using these medications.
Thank you.
Dr. Peters is professor of medicine at the University of Southern California, Los Angeles, and director of the USC clinical diabetes programs. She has published more than 200 articles, reviews, and abstracts, and three books, on diabetes, and has been an investigator for more than 40 research studies. She has spoken internationally at over 400 programs and serves on many committees of several professional organizations. She has ties with Abbott Diabetes Care, AstraZeneca Becton Dickinson, Boehringer Ingelheim Pharmaceuticals, Dexcom, Eli Lilly, Lexicon Pharmaceuticals, Livongo, MannKind Corporation, Medscape, Merck, Novo Nordisk, Omada Health, OptumHealth, Sanofi, and Zafgen. A version of this article originally appeared on Medscape.com.
Despite ongoing challenges, experts are optimistic about the future of MS therapy
Prior to 1993, a multiple sclerosis (MS) diagnosis could often mean an abbreviated lifespan marked by progressive disability and loss of function. That changed when the Food and Drug Administration approved interferon beta-1b (Betaseron) in 1993, which revolutionized MS therapy and gave hope to the entire MS community.

"The most surprising thing about MS management over the last 30 years is that we’ve been able to treat MS – especially relapsing MS,” said Fred D. Lublin, MD, professor of neurology and director of the Corinne Goldsmith Dickinson Center for Multiple Sclerosis in Mount Sinai in New York. “The approval of interferon was a major therapeutic advancement because it was the first treatment for what was an untreatable disease.”
Mark Gudesblatt, MD, medical director of the Comprehensive MS Care Center of South Shore Neurologic Associates in Patchogue, N.Y., agrees.
“For people with MS, it’s an extraordinarily lucky and amazingly optimistic time,” he said. “Before interferon beta-1b, MS was called ‘the crippler of young adults’ because more than 50% of these people would require a walker 10 years after diagnosis, and a large number of young and middle-age patients with MS were residing in nursing homes.”

According to Dr. Lublin, the emergence of the immunomodulating therapies placed MS at the leading edge of neurotherapeutics. Interferon beta-1b laid the foundation for new therapies such as another interferon (interferon beta-1a; Avonex), glatiramer acetate (Copaxone), and many other effective therapies with different mechanisms of action. Since the emergence of the first therapy, more than 20 oral and infusion agents with moderate to high efficacy have come to market for relapsing MS.
Treatment options, treatment challenges
Dr. Gudesblatt points out that having numerous therapies from which to choose is both a blessing and a problem.
“The good news is that there are so many options for treating relapsing MS today,” he said. “The bad news is there are so many options. Like doctors who are treating high blood pressure, doctors managing patients with MS often struggle to determine which medication is best for individual patients.”
Despite the promise of vastly better outcomes and prolonged lifespan, MS therapy still faces its share of challenges, including effective therapies for progressive MS and reparative-restorative therapies.
“Choice in route of administration and timing of administration allow for larger and broader discussions to try to meet patients’ needs,” Dr. Lublin said. “We’ve been extremely successful at treating relapses, but not as successful in treating progressive disease.”
The unclear mechanism of pathogenesis amplifies the challenges clinicians face in successful management of patients with MS. For example, experts agree that the therapies for progressive MS have only proven moderately effective at best. The paucity of therapies available for progressive MS and the limitations of the current therapies further limit the outcomes.
Looking ahead
Experts expressed optimistic views about the future of MS therapy as a whole. From Dr. Lublin’s perspective, the MS community stands to gain valuable insights from emerging research focused on treating progressive disease along with new testing to understand the underlying mechanism of progressive disease. Enhanced understanding of the underlying pathogenesis of progressive MS coupled with the ability to diagnose MS – such as improved MRI techniques – have facilitated this process.
Among the therapies with novel mechanisms of action in the pipeline include agents that generate myelin sheath repair. Another potential therapeutic class on the horizon, known as TPK inhibitors, addresses the smoldering of the disease. With these and other therapeutic advances, Dr. Lublin hopes to see better control of progressive disease.
An agenda for the future
In addition, barriers such as access to care, cost, insurance coverage, and tolerance remain ongoing stressors that will likely continue weighing on the MS community and its stakeholders into the future.
Dr. Gudesblatt concluded that advancing MS outcomes in the future hinges on several additional factors.
“We need medicines that are better for relapse and progression; medicines that are better tolerated and safer; and better medicine to address the underlying disease as well as its symptoms. But we also need to appreciate, recognize, and address cognitive impairment along the MS continuum and develop effective reparative options,” he said.
Regardless, he emphasized that these “amazing advancements” in MS therapy have renewed hope that research may identify and expand effective treatments for multiple other neurologic conditions such as muscular dystrophies, neurodegenerative and genetic disorders, movement disorders, and dysautonomia-related diseases. Like MS, all of these conditions have limited therapies, some of which have minimal efficacy. But none of these other disorders has disease-modifying therapies currently available.
‘A beacon of hope’
“MS is the beacon of hope for multiple disease states because it’s cracked the door wide open,” Dr. Gudesblatt said. Relapse no longer gauges the prognosis of today’s MS patient – a prognosis both experts think will only continue to improve with forthcoming innovations.
While the challenges for MS still exist, the bright future that lies ahead may eventually eclipse them.
Prior to 1993, a multiple sclerosis (MS) diagnosis could often mean an abbreviated lifespan marked by progressive disability and loss of function. That changed when the Food and Drug Administration approved interferon beta-1b (Betaseron) in 1993, which revolutionized MS therapy and gave hope to the entire MS community.
"The most surprising thing about MS management over the last 30 years is that we’ve been able to treat MS – especially relapsing MS,” said Fred D. Lublin, MD, professor of neurology and director of the Corinne Goldsmith Dickinson Center for Multiple Sclerosis in Mount Sinai in New York. “The approval of interferon was a major therapeutic advancement because it was the first treatment for what was an untreatable disease.”
Mark Gudesblatt, MD, medical director of the Comprehensive MS Care Center of South Shore Neurologic Associates in Patchogue, N.Y., agrees.
“For people with MS, it’s an extraordinarily lucky and amazingly optimistic time,” he said. “Before interferon beta-1b, MS was called ‘the crippler of young adults’ because more than 50% of these people would require a walker 10 years after diagnosis, and a large number of young and middle-age patients with MS were residing in nursing homes.”
According to Dr. Lublin, the emergence of the immunomodulating therapies placed MS at the leading edge of neurotherapeutics. Interferon beta-1b laid the foundation for new therapies such as another interferon (interferon beta-1a; Avonex), glatiramer acetate (Copaxone), and many other effective therapies with different mechanisms of action. Since the emergence of the first therapy, more than 20 oral and infusion agents with moderate to high efficacy have come to market for relapsing MS.
Treatment options, treatment challenges
Dr. Gudesblatt points out that having numerous therapies from which to choose is both a blessing and a problem.
“The good news is that there are so many options for treating relapsing MS today,” he said. “The bad news is there are so many options. Like doctors who are treating high blood pressure, doctors managing patients with MS often struggle to determine which medication is best for individual patients.”
Despite the promise of vastly better outcomes and prolonged lifespan, MS therapy still faces its share of challenges, including effective therapies for progressive MS and reparative-restorative therapies.
“Choice in route of administration and timing of administration allow for larger and broader discussions to try to meet patients’ needs,” Dr. Lublin said. “We’ve been extremely successful at treating relapses, but not as successful in treating progressive disease.”
The unclear mechanism of pathogenesis amplifies the challenges clinicians face in successful management of patients with MS. For example, experts agree that the therapies for progressive MS have only proven moderately effective at best. The paucity of therapies available for progressive MS and the limitations of the current therapies further limit the outcomes.
Looking ahead
Experts expressed optimistic views about the future of MS therapy as a whole. From Dr. Lublin’s perspective, the MS community stands to gain valuable insights from emerging research focused on treating progressive disease along with new testing to understand the underlying mechanism of progressive disease. Enhanced understanding of the underlying pathogenesis of progressive MS coupled with the ability to diagnose MS – such as improved MRI techniques – have facilitated this process.
Among the therapies with novel mechanisms of action in the pipeline include agents that generate myelin sheath repair. Another potential therapeutic class on the horizon, known as TPK inhibitors, addresses the smoldering of the disease. With these and other therapeutic advances, Dr. Lublin hopes to see better control of progressive disease.
An agenda for the future
In addition, barriers such as access to care, cost, insurance coverage, and tolerance remain ongoing stressors that will likely continue weighing on the MS community and its stakeholders into the future.
Dr. Gudesblatt concluded that advancing MS outcomes in the future hinges on several additional factors.
“We need medicines that are better for relapse and progression; medicines that are better tolerated and safer; and better medicine to address the underlying disease as well as its symptoms. But we also need to appreciate, recognize, and address cognitive impairment along the MS continuum and develop effective reparative options,” he said.
Regardless, he emphasized that these “amazing advancements” in MS therapy have renewed hope that research may identify and expand effective treatments for multiple other neurologic conditions such as muscular dystrophies, neurodegenerative and genetic disorders, movement disorders, and dysautonomia-related diseases. Like MS, all of these conditions have limited therapies, some of which have minimal efficacy. But none of these other disorders has disease-modifying therapies currently available.
‘A beacon of hope’
“MS is the beacon of hope for multiple disease states because it’s cracked the door wide open,” Dr. Gudesblatt said. Relapse no longer gauges the prognosis of today’s MS patient – a prognosis both experts think will only continue to improve with forthcoming innovations.
While the challenges for MS still exist, the bright future that lies ahead may eventually eclipse them.
Prior to 1993, a multiple sclerosis (MS) diagnosis could often mean an abbreviated lifespan marked by progressive disability and loss of function. That changed when the Food and Drug Administration approved interferon beta-1b (Betaseron) in 1993, which revolutionized MS therapy and gave hope to the entire MS community.
"The most surprising thing about MS management over the last 30 years is that we’ve been able to treat MS – especially relapsing MS,” said Fred D. Lublin, MD, professor of neurology and director of the Corinne Goldsmith Dickinson Center for Multiple Sclerosis in Mount Sinai in New York. “The approval of interferon was a major therapeutic advancement because it was the first treatment for what was an untreatable disease.”
Mark Gudesblatt, MD, medical director of the Comprehensive MS Care Center of South Shore Neurologic Associates in Patchogue, N.Y., agrees.
“For people with MS, it’s an extraordinarily lucky and amazingly optimistic time,” he said. “Before interferon beta-1b, MS was called ‘the crippler of young adults’ because more than 50% of these people would require a walker 10 years after diagnosis, and a large number of young and middle-age patients with MS were residing in nursing homes.”
According to Dr. Lublin, the emergence of the immunomodulating therapies placed MS at the leading edge of neurotherapeutics. Interferon beta-1b laid the foundation for new therapies such as another interferon (interferon beta-1a; Avonex), glatiramer acetate (Copaxone), and many other effective therapies with different mechanisms of action. Since the emergence of the first therapy, more than 20 oral and infusion agents with moderate to high efficacy have come to market for relapsing MS.
Treatment options, treatment challenges
Dr. Gudesblatt points out that having numerous therapies from which to choose is both a blessing and a problem.
“The good news is that there are so many options for treating relapsing MS today,” he said. “The bad news is there are so many options. Like doctors who are treating high blood pressure, doctors managing patients with MS often struggle to determine which medication is best for individual patients.”
Despite the promise of vastly better outcomes and prolonged lifespan, MS therapy still faces its share of challenges, including effective therapies for progressive MS and reparative-restorative therapies.
“Choice in route of administration and timing of administration allow for larger and broader discussions to try to meet patients’ needs,” Dr. Lublin said. “We’ve been extremely successful at treating relapses, but not as successful in treating progressive disease.”
The unclear mechanism of pathogenesis amplifies the challenges clinicians face in successful management of patients with MS. For example, experts agree that the therapies for progressive MS have only proven moderately effective at best. The paucity of therapies available for progressive MS and the limitations of the current therapies further limit the outcomes.
Looking ahead
Experts expressed optimistic views about the future of MS therapy as a whole. From Dr. Lublin’s perspective, the MS community stands to gain valuable insights from emerging research focused on treating progressive disease along with new testing to understand the underlying mechanism of progressive disease. Enhanced understanding of the underlying pathogenesis of progressive MS coupled with the ability to diagnose MS – such as improved MRI techniques – have facilitated this process.
Among the therapies with novel mechanisms of action in the pipeline include agents that generate myelin sheath repair. Another potential therapeutic class on the horizon, known as TPK inhibitors, addresses the smoldering of the disease. With these and other therapeutic advances, Dr. Lublin hopes to see better control of progressive disease.
An agenda for the future
In addition, barriers such as access to care, cost, insurance coverage, and tolerance remain ongoing stressors that will likely continue weighing on the MS community and its stakeholders into the future.
Dr. Gudesblatt concluded that advancing MS outcomes in the future hinges on several additional factors.
“We need medicines that are better for relapse and progression; medicines that are better tolerated and safer; and better medicine to address the underlying disease as well as its symptoms. But we also need to appreciate, recognize, and address cognitive impairment along the MS continuum and develop effective reparative options,” he said.
Regardless, he emphasized that these “amazing advancements” in MS therapy have renewed hope that research may identify and expand effective treatments for multiple other neurologic conditions such as muscular dystrophies, neurodegenerative and genetic disorders, movement disorders, and dysautonomia-related diseases. Like MS, all of these conditions have limited therapies, some of which have minimal efficacy. But none of these other disorders has disease-modifying therapies currently available.
‘A beacon of hope’
“MS is the beacon of hope for multiple disease states because it’s cracked the door wide open,” Dr. Gudesblatt said. Relapse no longer gauges the prognosis of today’s MS patient – a prognosis both experts think will only continue to improve with forthcoming innovations.
While the challenges for MS still exist, the bright future that lies ahead may eventually eclipse them.
Medication Overuse Headache (MOH): Prevention and Treatment

Medication overuse headache, previously known as rebound headache or medication-induced headache, may be caused by the frequent or excessive use of various acute care medications. When these medications are used too frequently, they can cause headaches rather than relieving them. (Some headache specialists feel that MOH is the result of recurring severe headaches, and the patients’ overuse of medications to relieve them.) These medications, some of which are painkillers or analgesics, include over-the-counter products such as acetaminophen, aspirin, and anti-inflammatories, as well as prescription medications such as triptans, ergots opioids, opioids, and barbiturates. The one category of acute care medication that does not seem to cause MOH is the gepants, such as rimegepant and ubrogepant.
MOH is the fourth most common headache disorder. It is defined by the International Classification of Headache Disorders (ICHD-3) as a headache present 15 days per month, evolving from regular use of strong acute medication (10 or more days of triptans, ergotamines, butalbital medications, opioids, or combination medications or 15 or more days per month of simple analgesics such as aspirin, acetaminophen, or nonsteroidal anti-inflammatories) for 3 months.
Patients are usually not aware they have MOH, and this is the most problematic aspect of the condition. Patients do not realize that the medicine they are taking is making their headaches worse. It can be difficult to explain to the patient exactly what is going on with MOH, and why they are doing the wrong thing by taking the very medication that was prescribed by their doctor to stop a migraine attack. Many doctors do not fully understand MOH either, which can make it difficult to treat patients with this type of headache; therefore, it is imperative to educate both doctors and patients on the causes and treatments of MOH.
One of the most important facets of treating MOH traditionally has been the process of detoxifying patients from their overused medication by gradually or precipitously withdrawing the offending medication. There is variability in how detoxification can be accomplished. Some of my patients stopped medications abruptly and experienced very bad headaches. Others tried reducing dosages on their own and reported experiencing the worst headaches of their lives—some of which lasted for a few weeks. I have found that if patients can endure 2 to 3 weeks of detox, they start to feel better. But because the headaches can worsen before they get better, patients understandably try to avoid the detoxification process.
I start patients on preventive medicine, then slowly increase it to an effective dose, and have them come back in a month for an evaluation. I then have them gradually reduce, but not completely stop, the pain medication before they return. Once I feel their preventive medication is at a therapeutic level, I have them begin a slow detox. After a month of preventive medication, there is a reasonable chance that headaches will start to decrease and be less severe. I tell them that if their headache is less severe try to avoid taking the medicine that they were overusing to prevent perpetuating the MOH.
One plausible physiologic mechanism behind MOH is that chronic exposure to acute care migraine treatment leads to suppression of the serotonergic/norepinephrinergic endogenous antinociceptive system in the upper brain stem, with facilitation of the trigeminal nociceptive process via up-regulation of calcitonin gene-related peptide (CGRP).This increase in CGRP at the end of peripheral nerve terminals in the trigeminovascular system may facilitate pain transmission. An increase in cortical CGRP may cause cortical spreading depression: a wave of excitement traveling through the cortex, followed by a wave of electrical depression seems to cause headache.
Good, effective prevention often helps avoid MOH; medications such as topiramate, nortriptyline, gabapentin, onabotulinumtoxinA, and CGRP monoclonal antibodies or some type of local nerve block have improved MOH in patients, but detoxification is usually necessary is some patients.
Monoclonal antibodies targeting CGRP or its receptor (CGRP-R), given by subcutaneous or intravenous injection or small molecule CGRP receptor antagonists given orally (gepants), seem to be able to treat MOH in some patients without a detoxification. This has been best demonstrated in the monoclonal antibody group, but there is some evidence showing that it may also occur with gepants. These treatments seem to work even when patients are overusing acute care medications; this helps some patients to self-detoxify at their own pace, which is easier for both the patient and the doctor.
Currently, there are 4 monoclonal antibodies against CGRP or the CGRP-R. Erenumab is the only completely human one and the only antibody that blocks the CGRP receptor to prevent the CGRP ligand from docking and exerting its effect. The other 3 (fremanezumab, galcanezumab, and eptinezumab) are humanized monoclonal antibodies that selectively bind to the CGRP ligand, preventing it from docking on its receptor. Patients started on the monoclonal antibodies against CGRP or its receptor usually have fewer headaches in the first week or two of therapy, and this helps make the self-detox easier for the patient.
Further, substantial data have shown that onabotulinumtoxinA reduces the number/frequency of headaches and reduces the need for patients to take acute medication. OnabotulinumtoxinA is currently the only medication approved for preventive treatment of chronic migraine; it has long-term safety data available and has reported efficacy lasting for up to 3 years when given in multiple injection sites every 3 months. Interestingly, although topiramate is used as a preventive medication, a recent study comparing erenumab vs topiramate for reducing monthly migraine days (MMD) showed that erenumab outperformed topiramate with a 50% reduction in MMD, and with fewer reported adverse events.
We are just starting to learn about some other potential cellular mechanisms that could be causing MOH in patients; these data could help create new and improved therapies for treating and possibly preventing MOH in the future. Patient outcomes could also be improved by encouraging the inclusion of MOH as part of a continuing education program for physicians who could potentially be treating new patients presenting with MOH.
Medication overuse headache, previously known as rebound headache or medication-induced headache, may be caused by the frequent or excessive use of various acute care medications. When these medications are used too frequently, they can cause headaches rather than relieving them. (Some headache specialists feel that MOH is the result of recurring severe headaches, and the patients’ overuse of medications to relieve them.) These medications, some of which are painkillers or analgesics, include over-the-counter products such as acetaminophen, aspirin, and anti-inflammatories, as well as prescription medications such as triptans, ergots opioids, opioids, and barbiturates. The one category of acute care medication that does not seem to cause MOH is the gepants, such as rimegepant and ubrogepant.
MOH is the fourth most common headache disorder. It is defined by the International Classification of Headache Disorders (ICHD-3) as a headache present 15 days per month, evolving from regular use of strong acute medication (10 or more days of triptans, ergotamines, butalbital medications, opioids, or combination medications or 15 or more days per month of simple analgesics such as aspirin, acetaminophen, or nonsteroidal anti-inflammatories) for 3 months.
Patients are usually not aware they have MOH, and this is the most problematic aspect of the condition. Patients do not realize that the medicine they are taking is making their headaches worse. It can be difficult to explain to the patient exactly what is going on with MOH, and why they are doing the wrong thing by taking the very medication that was prescribed by their doctor to stop a migraine attack. Many doctors do not fully understand MOH either, which can make it difficult to treat patients with this type of headache; therefore, it is imperative to educate both doctors and patients on the causes and treatments of MOH.
One of the most important facets of treating MOH traditionally has been the process of detoxifying patients from their overused medication by gradually or precipitously withdrawing the offending medication. There is variability in how detoxification can be accomplished. Some of my patients stopped medications abruptly and experienced very bad headaches. Others tried reducing dosages on their own and reported experiencing the worst headaches of their lives—some of which lasted for a few weeks. I have found that if patients can endure 2 to 3 weeks of detox, they start to feel better. But because the headaches can worsen before they get better, patients understandably try to avoid the detoxification process.
I start patients on preventive medicine, then slowly increase it to an effective dose, and have them come back in a month for an evaluation. I then have them gradually reduce, but not completely stop, the pain medication before they return. Once I feel their preventive medication is at a therapeutic level, I have them begin a slow detox. After a month of preventive medication, there is a reasonable chance that headaches will start to decrease and be less severe. I tell them that if their headache is less severe try to avoid taking the medicine that they were overusing to prevent perpetuating the MOH.
One plausible physiologic mechanism behind MOH is that chronic exposure to acute care migraine treatment leads to suppression of the serotonergic/norepinephrinergic endogenous antinociceptive system in the upper brain stem, with facilitation of the trigeminal nociceptive process via up-regulation of calcitonin gene-related peptide (CGRP).This increase in CGRP at the end of peripheral nerve terminals in the trigeminovascular system may facilitate pain transmission. An increase in cortical CGRP may cause cortical spreading depression: a wave of excitement traveling through the cortex, followed by a wave of electrical depression seems to cause headache.
Good, effective prevention often helps avoid MOH; medications such as topiramate, nortriptyline, gabapentin, onabotulinumtoxinA, and CGRP monoclonal antibodies or some type of local nerve block have improved MOH in patients, but detoxification is usually necessary is some patients.
Monoclonal antibodies targeting CGRP or its receptor (CGRP-R), given by subcutaneous or intravenous injection or small molecule CGRP receptor antagonists given orally (gepants), seem to be able to treat MOH in some patients without a detoxification. This has been best demonstrated in the monoclonal antibody group, but there is some evidence showing that it may also occur with gepants. These treatments seem to work even when patients are overusing acute care medications; this helps some patients to self-detoxify at their own pace, which is easier for both the patient and the doctor.
Currently, there are 4 monoclonal antibodies against CGRP or the CGRP-R. Erenumab is the only completely human one and the only antibody that blocks the CGRP receptor to prevent the CGRP ligand from docking and exerting its effect. The other 3 (fremanezumab, galcanezumab, and eptinezumab) are humanized monoclonal antibodies that selectively bind to the CGRP ligand, preventing it from docking on its receptor. Patients started on the monoclonal antibodies against CGRP or its receptor usually have fewer headaches in the first week or two of therapy, and this helps make the self-detox easier for the patient.
Further, substantial data have shown that onabotulinumtoxinA reduces the number/frequency of headaches and reduces the need for patients to take acute medication. OnabotulinumtoxinA is currently the only medication approved for preventive treatment of chronic migraine; it has long-term safety data available and has reported efficacy lasting for up to 3 years when given in multiple injection sites every 3 months. Interestingly, although topiramate is used as a preventive medication, a recent study comparing erenumab vs topiramate for reducing monthly migraine days (MMD) showed that erenumab outperformed topiramate with a 50% reduction in MMD, and with fewer reported adverse events.
We are just starting to learn about some other potential cellular mechanisms that could be causing MOH in patients; these data could help create new and improved therapies for treating and possibly preventing MOH in the future. Patient outcomes could also be improved by encouraging the inclusion of MOH as part of a continuing education program for physicians who could potentially be treating new patients presenting with MOH.
Medication overuse headache, previously known as rebound headache or medication-induced headache, may be caused by the frequent or excessive use of various acute care medications. When these medications are used too frequently, they can cause headaches rather than relieving them. (Some headache specialists feel that MOH is the result of recurring severe headaches, and the patients’ overuse of medications to relieve them.) These medications, some of which are painkillers or analgesics, include over-the-counter products such as acetaminophen, aspirin, and anti-inflammatories, as well as prescription medications such as triptans, ergots opioids, opioids, and barbiturates. The one category of acute care medication that does not seem to cause MOH is the gepants, such as rimegepant and ubrogepant.
MOH is the fourth most common headache disorder. It is defined by the International Classification of Headache Disorders (ICHD-3) as a headache present 15 days per month, evolving from regular use of strong acute medication (10 or more days of triptans, ergotamines, butalbital medications, opioids, or combination medications or 15 or more days per month of simple analgesics such as aspirin, acetaminophen, or nonsteroidal anti-inflammatories) for 3 months.
Patients are usually not aware they have MOH, and this is the most problematic aspect of the condition. Patients do not realize that the medicine they are taking is making their headaches worse. It can be difficult to explain to the patient exactly what is going on with MOH, and why they are doing the wrong thing by taking the very medication that was prescribed by their doctor to stop a migraine attack. Many doctors do not fully understand MOH either, which can make it difficult to treat patients with this type of headache; therefore, it is imperative to educate both doctors and patients on the causes and treatments of MOH.
One of the most important facets of treating MOH traditionally has been the process of detoxifying patients from their overused medication by gradually or precipitously withdrawing the offending medication. There is variability in how detoxification can be accomplished. Some of my patients stopped medications abruptly and experienced very bad headaches. Others tried reducing dosages on their own and reported experiencing the worst headaches of their lives—some of which lasted for a few weeks. I have found that if patients can endure 2 to 3 weeks of detox, they start to feel better. But because the headaches can worsen before they get better, patients understandably try to avoid the detoxification process.
I start patients on preventive medicine, then slowly increase it to an effective dose, and have them come back in a month for an evaluation. I then have them gradually reduce, but not completely stop, the pain medication before they return. Once I feel their preventive medication is at a therapeutic level, I have them begin a slow detox. After a month of preventive medication, there is a reasonable chance that headaches will start to decrease and be less severe. I tell them that if their headache is less severe try to avoid taking the medicine that they were overusing to prevent perpetuating the MOH.
One plausible physiologic mechanism behind MOH is that chronic exposure to acute care migraine treatment leads to suppression of the serotonergic/norepinephrinergic endogenous antinociceptive system in the upper brain stem, with facilitation of the trigeminal nociceptive process via up-regulation of calcitonin gene-related peptide (CGRP).This increase in CGRP at the end of peripheral nerve terminals in the trigeminovascular system may facilitate pain transmission. An increase in cortical CGRP may cause cortical spreading depression: a wave of excitement traveling through the cortex, followed by a wave of electrical depression seems to cause headache.
Good, effective prevention often helps avoid MOH; medications such as topiramate, nortriptyline, gabapentin, onabotulinumtoxinA, and CGRP monoclonal antibodies or some type of local nerve block have improved MOH in patients, but detoxification is usually necessary is some patients.
Monoclonal antibodies targeting CGRP or its receptor (CGRP-R), given by subcutaneous or intravenous injection or small molecule CGRP receptor antagonists given orally (gepants), seem to be able to treat MOH in some patients without a detoxification. This has been best demonstrated in the monoclonal antibody group, but there is some evidence showing that it may also occur with gepants. These treatments seem to work even when patients are overusing acute care medications; this helps some patients to self-detoxify at their own pace, which is easier for both the patient and the doctor.
Currently, there are 4 monoclonal antibodies against CGRP or the CGRP-R. Erenumab is the only completely human one and the only antibody that blocks the CGRP receptor to prevent the CGRP ligand from docking and exerting its effect. The other 3 (fremanezumab, galcanezumab, and eptinezumab) are humanized monoclonal antibodies that selectively bind to the CGRP ligand, preventing it from docking on its receptor. Patients started on the monoclonal antibodies against CGRP or its receptor usually have fewer headaches in the first week or two of therapy, and this helps make the self-detox easier for the patient.
Further, substantial data have shown that onabotulinumtoxinA reduces the number/frequency of headaches and reduces the need for patients to take acute medication. OnabotulinumtoxinA is currently the only medication approved for preventive treatment of chronic migraine; it has long-term safety data available and has reported efficacy lasting for up to 3 years when given in multiple injection sites every 3 months. Interestingly, although topiramate is used as a preventive medication, a recent study comparing erenumab vs topiramate for reducing monthly migraine days (MMD) showed that erenumab outperformed topiramate with a 50% reduction in MMD, and with fewer reported adverse events.
We are just starting to learn about some other potential cellular mechanisms that could be causing MOH in patients; these data could help create new and improved therapies for treating and possibly preventing MOH in the future. Patient outcomes could also be improved by encouraging the inclusion of MOH as part of a continuing education program for physicians who could potentially be treating new patients presenting with MOH.
Spikes out: A COVID mystery
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr. F. Perry Wilson of the Yale School of Medicine.
To date, it has been a mystery, like “Glass Onion.” And in the spirit of all the great mysteries, to get to the bottom of this, we’ll need to round up the usual suspects.
Appearing in Circulation, a new study does a great job of systematically evaluating multiple hypotheses linking vaccination to myocarditis, and eliminating them, Poirot-style, one by one until only one remains. We’ll get there.
But first, let’s review the suspects. Why do the mRNA vaccines cause myocarditis in a small subset of people?
There are a few leading candidates.
Number one: antibody responses. There are two flavors here. The quantitative hypothesis suggests that some people simply generate too many antibodies to the vaccine, leading to increased inflammation and heart damage.
The qualitative hypothesis suggests that maybe it’s the nature of the antibodies generated rather than the amount; they might cross-react with some protein on the surface of heart cells for instance.
Or maybe it is driven by T-cell responses, which, of course, are independent of antibody levels.
There’s the idea that myocarditis is due to excessive cytokine release – sort of like what we see in the multisystem inflammatory syndrome in children.
Or it could be due to the viral antigens themselves – the spike protein the mRNA codes for that is generated after vaccination.
To tease all these possibilities apart, researchers led by Lael Yonker at Mass General performed a case-control study. Sixteen children with postvaccine myocarditis were matched by age to 45 control children who had been vaccinated without complications.
The matching was OK, but as you can see here, there were more boys in the myocarditis group, and the time from vaccination was a bit shorter in that group as well. We’ll keep that in mind as we go through the results.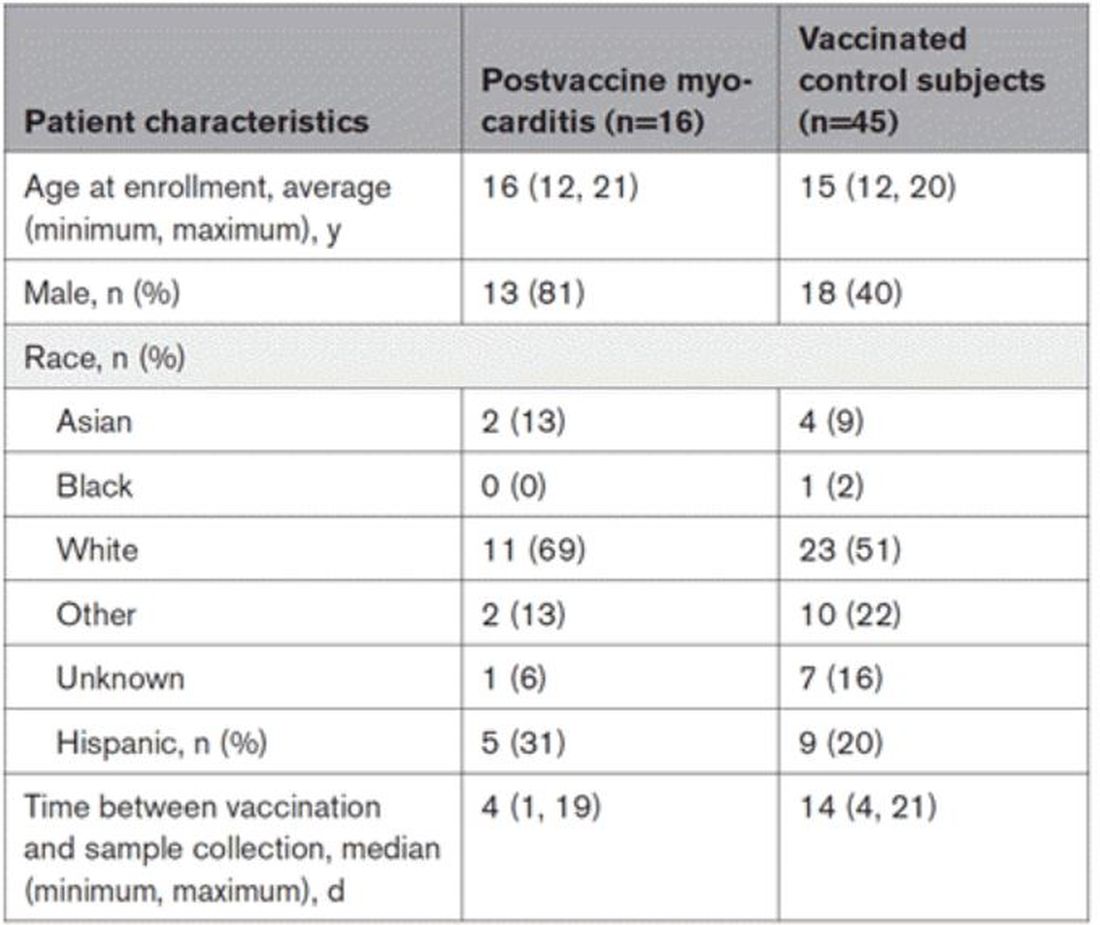
OK, let’s start eliminating suspects.
First, quantitative antibodies. Seems unlikely. Absolute antibody titers were really no different in the myocarditis vs. the control group.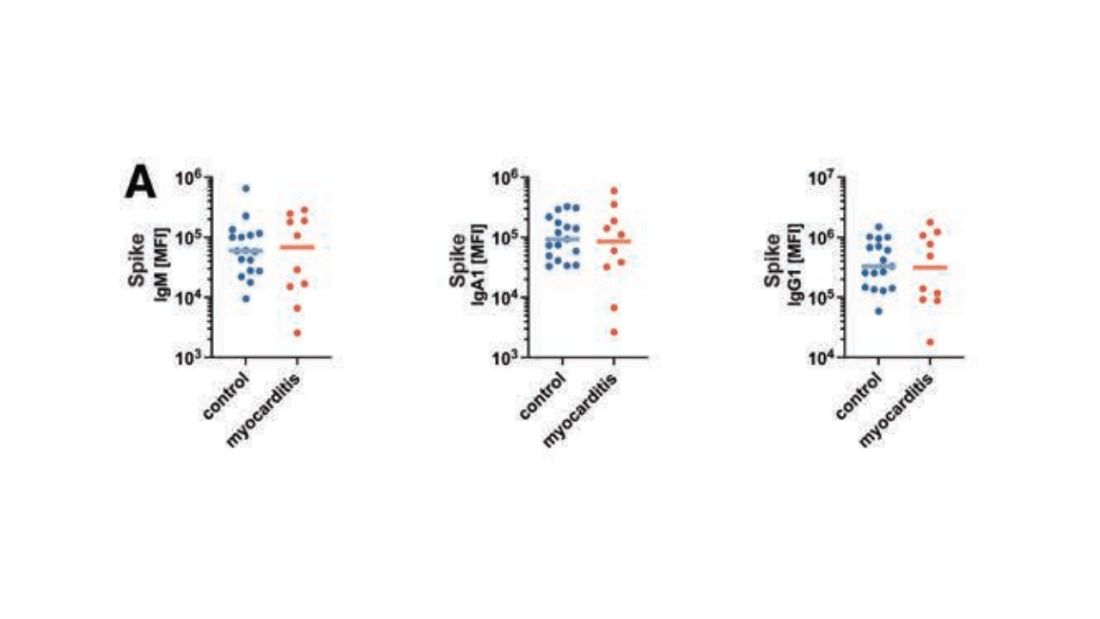
What about the quality of the antibodies? Would the kids with myocarditis have more self-recognizing antibodies present? It doesn’t appear so. Autoantibody levels were similar in the two groups.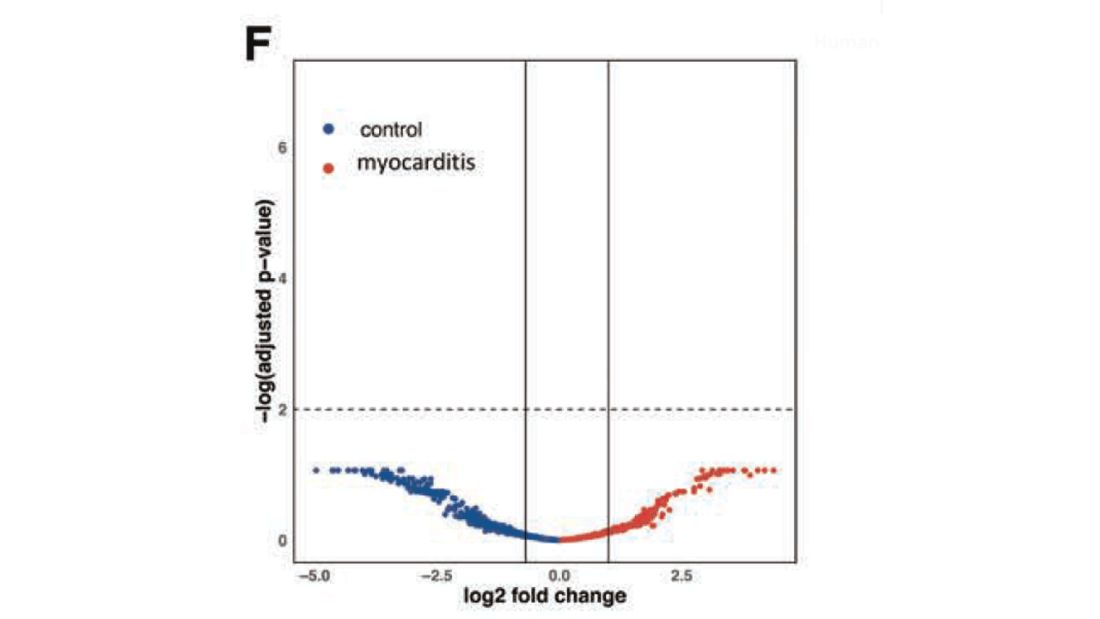
Take antibodies off the list.
T-cell responses come next, and, again, no major differences here, save for one specific T-cell subtype that was moderately elevated in the myocarditis group. Not what I would call a smoking gun, frankly.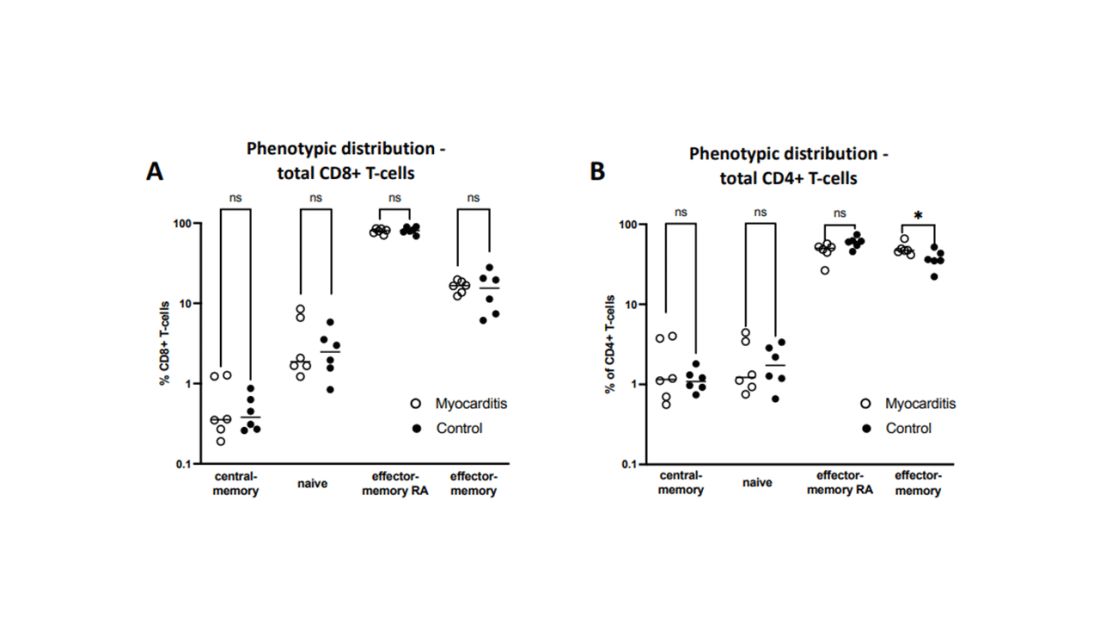
Cytokines give us a bit more to chew on. Levels of interleukin (IL)-8, IL-6, tumor necrosis factor (TNF)-alpha, and IL-10 were all substantially higher in the kids with myocarditis.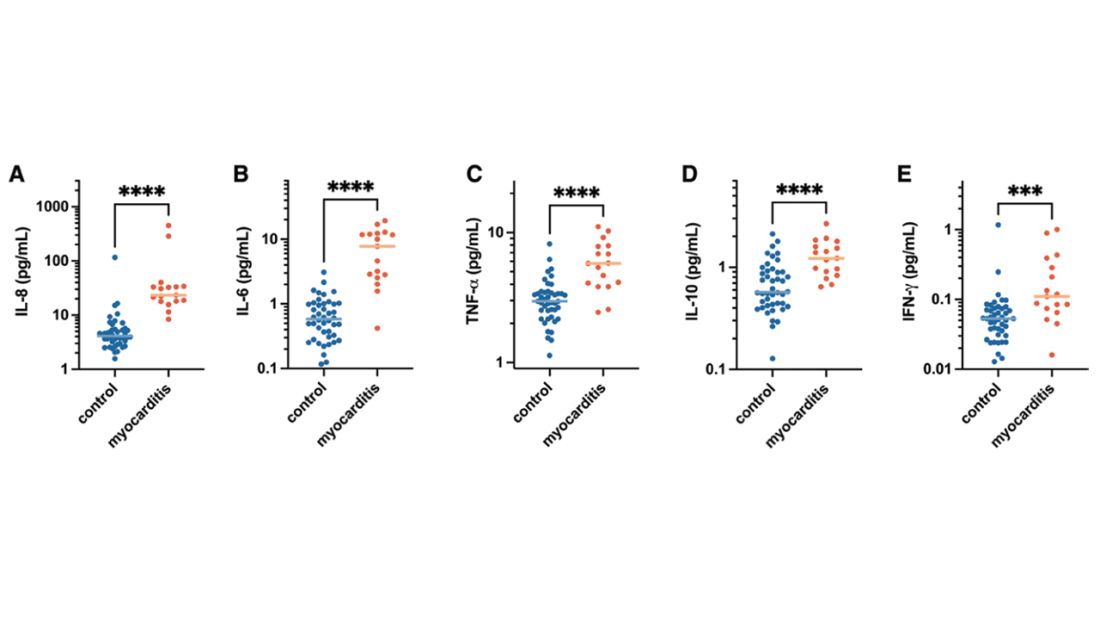
But the thing about cytokines is that they are not particularly specific. OK, kids with myocarditis have more systemic inflammation than kids without; that’s not really surprising. It still leaves us with the question of what is causing all this inflammation? Who is the arch-villain? The kingpin? The don?
It’s the analyses of antigens – the protein products of vaccination – that may hold the key here.
In 12 out of 16 kids with myocarditis, the researchers were able to measure free spike protein in the blood – that is to say spike protein, not bound by antispike antibodies.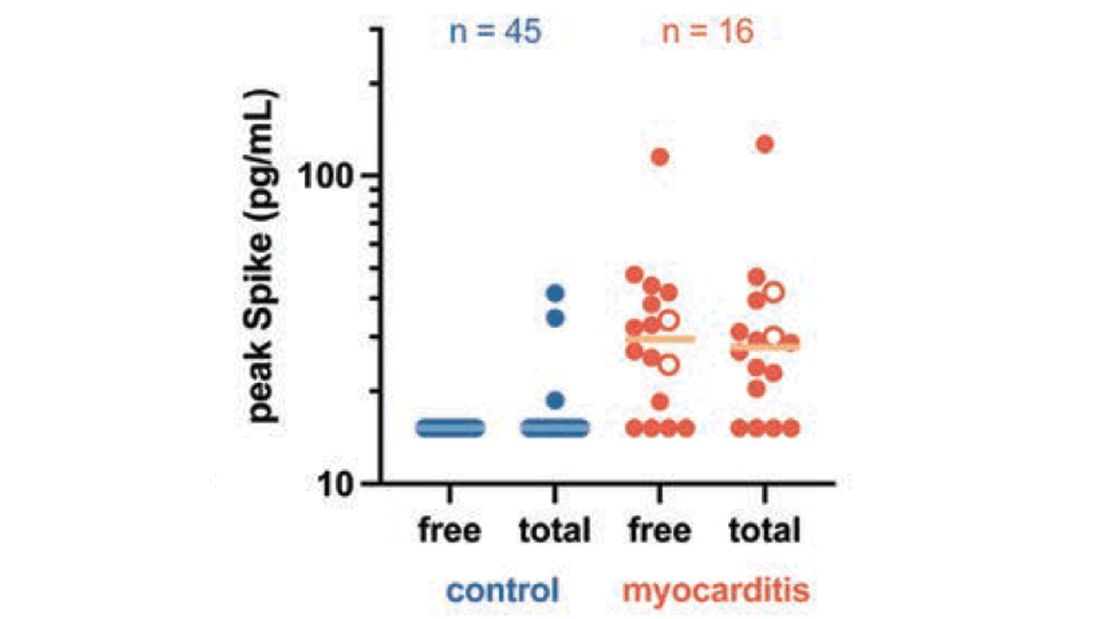
These free spikes were present in – wait for it – zero of the 45 control patients. That makes spike protein itself our prime suspect. J’accuse free spike protein!
Of course, all good detectives need to wrap up the case with a good story: How was it all done?
And here’s where we could use Agatha Christie’s help. How could this all work? The vaccine gets injected; mRNA is taken up into cells, where spike protein is generated and released, generating antibody and T-cell responses all the while. Those responses rapidly clear that spike protein from the system – this has been demonstrated in multiple studies – in adults, at least. But in some small number of people, apparently, spike protein is not cleared. Why? It makes no damn sense. Compels me, though. Some have suggested that inadvertent intravenous injection of vaccine, compared with the appropriate intramuscular route, might distribute the vaccine to sites with less immune surveillance. But that is definitely not proven yet.
We are on the path for sure, but this is, as Benoit Blanc would say, a twisted web – and we are not finished untangling it. Not yet.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here. He tweets @fperrywilson and his new book, “How Medicine Works and When It Doesn’t,” is available for preorder now. He reports no conflicts of interest.
A version of this article first appeared on Medscape.com.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr. F. Perry Wilson of the Yale School of Medicine.
To date, it has been a mystery, like “Glass Onion.” And in the spirit of all the great mysteries, to get to the bottom of this, we’ll need to round up the usual suspects.
Appearing in Circulation, a new study does a great job of systematically evaluating multiple hypotheses linking vaccination to myocarditis, and eliminating them, Poirot-style, one by one until only one remains. We’ll get there.
But first, let’s review the suspects. Why do the mRNA vaccines cause myocarditis in a small subset of people?
There are a few leading candidates.
Number one: antibody responses. There are two flavors here. The quantitative hypothesis suggests that some people simply generate too many antibodies to the vaccine, leading to increased inflammation and heart damage.
The qualitative hypothesis suggests that maybe it’s the nature of the antibodies generated rather than the amount; they might cross-react with some protein on the surface of heart cells for instance.
Or maybe it is driven by T-cell responses, which, of course, are independent of antibody levels.
There’s the idea that myocarditis is due to excessive cytokine release – sort of like what we see in the multisystem inflammatory syndrome in children.
Or it could be due to the viral antigens themselves – the spike protein the mRNA codes for that is generated after vaccination.
To tease all these possibilities apart, researchers led by Lael Yonker at Mass General performed a case-control study. Sixteen children with postvaccine myocarditis were matched by age to 45 control children who had been vaccinated without complications.
The matching was OK, but as you can see here, there were more boys in the myocarditis group, and the time from vaccination was a bit shorter in that group as well. We’ll keep that in mind as we go through the results.
OK, let’s start eliminating suspects.
First, quantitative antibodies. Seems unlikely. Absolute antibody titers were really no different in the myocarditis vs. the control group.
What about the quality of the antibodies? Would the kids with myocarditis have more self-recognizing antibodies present? It doesn’t appear so. Autoantibody levels were similar in the two groups.
Take antibodies off the list.
T-cell responses come next, and, again, no major differences here, save for one specific T-cell subtype that was moderately elevated in the myocarditis group. Not what I would call a smoking gun, frankly.
Cytokines give us a bit more to chew on. Levels of interleukin (IL)-8, IL-6, tumor necrosis factor (TNF)-alpha, and IL-10 were all substantially higher in the kids with myocarditis.
But the thing about cytokines is that they are not particularly specific. OK, kids with myocarditis have more systemic inflammation than kids without; that’s not really surprising. It still leaves us with the question of what is causing all this inflammation? Who is the arch-villain? The kingpin? The don?
It’s the analyses of antigens – the protein products of vaccination – that may hold the key here.
In 12 out of 16 kids with myocarditis, the researchers were able to measure free spike protein in the blood – that is to say spike protein, not bound by antispike antibodies.
These free spikes were present in – wait for it – zero of the 45 control patients. That makes spike protein itself our prime suspect. J’accuse free spike protein!
Of course, all good detectives need to wrap up the case with a good story: How was it all done?
And here’s where we could use Agatha Christie’s help. How could this all work? The vaccine gets injected; mRNA is taken up into cells, where spike protein is generated and released, generating antibody and T-cell responses all the while. Those responses rapidly clear that spike protein from the system – this has been demonstrated in multiple studies – in adults, at least. But in some small number of people, apparently, spike protein is not cleared. Why? It makes no damn sense. Compels me, though. Some have suggested that inadvertent intravenous injection of vaccine, compared with the appropriate intramuscular route, might distribute the vaccine to sites with less immune surveillance. But that is definitely not proven yet.
We are on the path for sure, but this is, as Benoit Blanc would say, a twisted web – and we are not finished untangling it. Not yet.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here. He tweets @fperrywilson and his new book, “How Medicine Works and When It Doesn’t,” is available for preorder now. He reports no conflicts of interest.
A version of this article first appeared on Medscape.com.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr. F. Perry Wilson of the Yale School of Medicine.
To date, it has been a mystery, like “Glass Onion.” And in the spirit of all the great mysteries, to get to the bottom of this, we’ll need to round up the usual suspects.
Appearing in Circulation, a new study does a great job of systematically evaluating multiple hypotheses linking vaccination to myocarditis, and eliminating them, Poirot-style, one by one until only one remains. We’ll get there.
But first, let’s review the suspects. Why do the mRNA vaccines cause myocarditis in a small subset of people?
There are a few leading candidates.
Number one: antibody responses. There are two flavors here. The quantitative hypothesis suggests that some people simply generate too many antibodies to the vaccine, leading to increased inflammation and heart damage.
The qualitative hypothesis suggests that maybe it’s the nature of the antibodies generated rather than the amount; they might cross-react with some protein on the surface of heart cells for instance.
Or maybe it is driven by T-cell responses, which, of course, are independent of antibody levels.
There’s the idea that myocarditis is due to excessive cytokine release – sort of like what we see in the multisystem inflammatory syndrome in children.
Or it could be due to the viral antigens themselves – the spike protein the mRNA codes for that is generated after vaccination.
To tease all these possibilities apart, researchers led by Lael Yonker at Mass General performed a case-control study. Sixteen children with postvaccine myocarditis were matched by age to 45 control children who had been vaccinated without complications.
The matching was OK, but as you can see here, there were more boys in the myocarditis group, and the time from vaccination was a bit shorter in that group as well. We’ll keep that in mind as we go through the results.
OK, let’s start eliminating suspects.
First, quantitative antibodies. Seems unlikely. Absolute antibody titers were really no different in the myocarditis vs. the control group.
What about the quality of the antibodies? Would the kids with myocarditis have more self-recognizing antibodies present? It doesn’t appear so. Autoantibody levels were similar in the two groups.
Take antibodies off the list.
T-cell responses come next, and, again, no major differences here, save for one specific T-cell subtype that was moderately elevated in the myocarditis group. Not what I would call a smoking gun, frankly.
Cytokines give us a bit more to chew on. Levels of interleukin (IL)-8, IL-6, tumor necrosis factor (TNF)-alpha, and IL-10 were all substantially higher in the kids with myocarditis.
But the thing about cytokines is that they are not particularly specific. OK, kids with myocarditis have more systemic inflammation than kids without; that’s not really surprising. It still leaves us with the question of what is causing all this inflammation? Who is the arch-villain? The kingpin? The don?
It’s the analyses of antigens – the protein products of vaccination – that may hold the key here.
In 12 out of 16 kids with myocarditis, the researchers were able to measure free spike protein in the blood – that is to say spike protein, not bound by antispike antibodies.
These free spikes were present in – wait for it – zero of the 45 control patients. That makes spike protein itself our prime suspect. J’accuse free spike protein!
Of course, all good detectives need to wrap up the case with a good story: How was it all done?
And here’s where we could use Agatha Christie’s help. How could this all work? The vaccine gets injected; mRNA is taken up into cells, where spike protein is generated and released, generating antibody and T-cell responses all the while. Those responses rapidly clear that spike protein from the system – this has been demonstrated in multiple studies – in adults, at least. But in some small number of people, apparently, spike protein is not cleared. Why? It makes no damn sense. Compels me, though. Some have suggested that inadvertent intravenous injection of vaccine, compared with the appropriate intramuscular route, might distribute the vaccine to sites with less immune surveillance. But that is definitely not proven yet.
We are on the path for sure, but this is, as Benoit Blanc would say, a twisted web – and we are not finished untangling it. Not yet.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here. He tweets @fperrywilson and his new book, “How Medicine Works and When It Doesn’t,” is available for preorder now. He reports no conflicts of interest.
A version of this article first appeared on Medscape.com.
Year in Review: How Targeted Drug Therapies Have Expanded Breast Cancer Treatment Options in 2022

Breast cancer (BC) is the most diagnosed cancer and the second leading cause of cancer deaths in women. In 2022, more than 275,000 women were diagnosed with BC, and at least 43,000 of these cases resulted in death. With targeted drug therapy for treating BC now approved by the US Food and Drug Administration (FDA) and available (or in the late phases of clinical trials and pending availability and FDA approval), clinicians are beginning to be able to move away from a “one-size-fits-all” treatment approach that has been used in the past, enhancing prognosis and survival rates in their patients living with BC.
The new targeted drug therapies available are more precise and individualized. They treat patients more effectively because they are based on the patient’s own biology. These therapies open the possibility of having more valuable treatment options, which can be beneficial for the outcome of many patients diagnosed with BC—especially the highly aggressive forms that were previously difficult to treat.
In March 2022, the FDA approved the drug olaparib to treat HER2-, high-risk, early breast cancer (EBC). The approval was made after the conclusion of the phase 3 OlympiA trial. The clinical trial results showed a statistically significant improvement in overall survival rates (by 32%) with adjuvant olaparib compared with placebo for germline BRCA1/2-mutated EBC.
In August 2022, the FDA approved the antibody drug conjugate fam-trastuzumab deruxtecan-nxki (or T-DXd), which is the first FDA-approved therapy targeted to treat patients who have the HER2-low BC subtype. It is also approved for patients with unresectable or metastatic HER2+ breast cancer who have already been treated with 2 or more prior anti-HER2–based therapies in the metastatic setting. The approval of T-DXd was given on the basis of DESTINY-Breast04, a randomized, multicenter, open-label clinical trial that was published in June 2022. T-DXd had been previously approved in HER2+ metastatic breast cancer.
The results from the phase 3 CAPItello-291 clinical trial of capivasertib in combination with fulvestrant suggests the combination could become a new treatment option for patients with hormone receptor–positive, HER2-low, locally advanced or metastatic BC following recurrence or progression after treatment with endocrine therapy and a CDK4/6 inhibitor. Capivasertib is a novel, selective, ATP-competitive, pan-AKT kinase inhibitor. In clinical trials, the drug was shown to successfully block activity of the cancer-driving protein molecule AKT. This research was presented at the 2022 San Antonio Breast Cancer Symposium, and the findings demonstrated a significant improvement in the overall population, as well as the subgroup of patients with PI3K pathway–altered tumors.
The year 2022 was full of exciting discoveries in the field of targeted drug therapies for treating BC, expanding patients’ treatment options and giving hope to people who have been diagnosed with breast cancer and their loved ones. In addition, emerging technologies such as immunotherapy and new antibody-drug conjugates continue to be evaluated as potential treatment options for treating breast cancer in the near future.
Breast cancer (BC) is the most diagnosed cancer and the second leading cause of cancer deaths in women. In 2022, more than 275,000 women were diagnosed with BC, and at least 43,000 of these cases resulted in death. With targeted drug therapy for treating BC now approved by the US Food and Drug Administration (FDA) and available (or in the late phases of clinical trials and pending availability and FDA approval), clinicians are beginning to be able to move away from a “one-size-fits-all” treatment approach that has been used in the past, enhancing prognosis and survival rates in their patients living with BC.
The new targeted drug therapies available are more precise and individualized. They treat patients more effectively because they are based on the patient’s own biology. These therapies open the possibility of having more valuable treatment options, which can be beneficial for the outcome of many patients diagnosed with BC—especially the highly aggressive forms that were previously difficult to treat.
In March 2022, the FDA approved the drug olaparib to treat HER2-, high-risk, early breast cancer (EBC). The approval was made after the conclusion of the phase 3 OlympiA trial. The clinical trial results showed a statistically significant improvement in overall survival rates (by 32%) with adjuvant olaparib compared with placebo for germline BRCA1/2-mutated EBC.
In August 2022, the FDA approved the antibody drug conjugate fam-trastuzumab deruxtecan-nxki (or T-DXd), which is the first FDA-approved therapy targeted to treat patients who have the HER2-low BC subtype. It is also approved for patients with unresectable or metastatic HER2+ breast cancer who have already been treated with 2 or more prior anti-HER2–based therapies in the metastatic setting. The approval of T-DXd was given on the basis of DESTINY-Breast04, a randomized, multicenter, open-label clinical trial that was published in June 2022. T-DXd had been previously approved in HER2+ metastatic breast cancer.
The results from the phase 3 CAPItello-291 clinical trial of capivasertib in combination with fulvestrant suggests the combination could become a new treatment option for patients with hormone receptor–positive, HER2-low, locally advanced or metastatic BC following recurrence or progression after treatment with endocrine therapy and a CDK4/6 inhibitor. Capivasertib is a novel, selective, ATP-competitive, pan-AKT kinase inhibitor. In clinical trials, the drug was shown to successfully block activity of the cancer-driving protein molecule AKT. This research was presented at the 2022 San Antonio Breast Cancer Symposium, and the findings demonstrated a significant improvement in the overall population, as well as the subgroup of patients with PI3K pathway–altered tumors.
The year 2022 was full of exciting discoveries in the field of targeted drug therapies for treating BC, expanding patients’ treatment options and giving hope to people who have been diagnosed with breast cancer and their loved ones. In addition, emerging technologies such as immunotherapy and new antibody-drug conjugates continue to be evaluated as potential treatment options for treating breast cancer in the near future.
Breast cancer (BC) is the most diagnosed cancer and the second leading cause of cancer deaths in women. In 2022, more than 275,000 women were diagnosed with BC, and at least 43,000 of these cases resulted in death. With targeted drug therapy for treating BC now approved by the US Food and Drug Administration (FDA) and available (or in the late phases of clinical trials and pending availability and FDA approval), clinicians are beginning to be able to move away from a “one-size-fits-all” treatment approach that has been used in the past, enhancing prognosis and survival rates in their patients living with BC.
The new targeted drug therapies available are more precise and individualized. They treat patients more effectively because they are based on the patient’s own biology. These therapies open the possibility of having more valuable treatment options, which can be beneficial for the outcome of many patients diagnosed with BC—especially the highly aggressive forms that were previously difficult to treat.
In March 2022, the FDA approved the drug olaparib to treat HER2-, high-risk, early breast cancer (EBC). The approval was made after the conclusion of the phase 3 OlympiA trial. The clinical trial results showed a statistically significant improvement in overall survival rates (by 32%) with adjuvant olaparib compared with placebo for germline BRCA1/2-mutated EBC.
In August 2022, the FDA approved the antibody drug conjugate fam-trastuzumab deruxtecan-nxki (or T-DXd), which is the first FDA-approved therapy targeted to treat patients who have the HER2-low BC subtype. It is also approved for patients with unresectable or metastatic HER2+ breast cancer who have already been treated with 2 or more prior anti-HER2–based therapies in the metastatic setting. The approval of T-DXd was given on the basis of DESTINY-Breast04, a randomized, multicenter, open-label clinical trial that was published in June 2022. T-DXd had been previously approved in HER2+ metastatic breast cancer.
The results from the phase 3 CAPItello-291 clinical trial of capivasertib in combination with fulvestrant suggests the combination could become a new treatment option for patients with hormone receptor–positive, HER2-low, locally advanced or metastatic BC following recurrence or progression after treatment with endocrine therapy and a CDK4/6 inhibitor. Capivasertib is a novel, selective, ATP-competitive, pan-AKT kinase inhibitor. In clinical trials, the drug was shown to successfully block activity of the cancer-driving protein molecule AKT. This research was presented at the 2022 San Antonio Breast Cancer Symposium, and the findings demonstrated a significant improvement in the overall population, as well as the subgroup of patients with PI3K pathway–altered tumors.
The year 2022 was full of exciting discoveries in the field of targeted drug therapies for treating BC, expanding patients’ treatment options and giving hope to people who have been diagnosed with breast cancer and their loved ones. In addition, emerging technologies such as immunotherapy and new antibody-drug conjugates continue to be evaluated as potential treatment options for treating breast cancer in the near future.
How to have a safer and more joyful holiday season
This holiday season, I am looking forward to spending some time with family, as I have in the past. As I have chatted with others, many friends are looking forward to events that are potentially larger and potentially returning to prepandemic type gatherings.

Gathering is important and can bring joy, sense of community, and love to the lives of many. Unfortunately, the risks associated with gathering are not over. as our country faces many cases of respiratory syncytial virus (RSV), COVID-19, and influenza at the same time.
During the first week of December, cases of influenza were rising across the country1 and were rising faster than in previous years. Although getting the vaccine is an important method of influenza prevention and is recommended for everyone over the age of 6 months with rare exception, many have not gotten their vaccine this year.
Influenza
Thus far, “nearly 50% of reported flu-associated hospitalizations in women of childbearing age have been in women who are pregnant.” We are seeing this at a time with lower-than-average uptake of influenza vaccine leaving both the pregnant persons and their babies unprotected. In addition to utilizing vaccines as prevention, isolating when ill, cleaning surfaces, and practicing good hand hygiene can all decrease transmission.
RSV
In addition to rises of influenza, there are currently high rates of RSV in various parts of the country. Prior to 2020, RSV typically started in the fall and peaked in the winter months. However, since the pandemic, the typical seasonal pattern has not returned, and it is unclear when it will. Although RSV hits the very young, the old, and the immunocompromised the most, RSV can infect anyone. Unfortunately, we do not currently have a vaccine for everyone against this virus. Prevention of transmission includes, as with flu, isolating when ill, cleaning surfaces, and washing hands.2
COVID-19
Of course, the effects of the COVID-19 pandemic are also still here as well. During the first week of December, the CDC reported rising cases of COVID across the country. Within the past few months, there have been several developments, though, for protection. There are now bivalent vaccines available as either third doses or booster doses approved for all persons over 6 months of age. As of the first week of December, only 13.5% of those aged 5 and over had received an updated booster.
There is currently wider access to rapid testing, including at-home testing, which can allow individuals to identify if COVID positive. Additionally, there is access to medication to decrease the likelihood of severe disease – though this does not take the place of vaccinations.
If anyone does test positive for COVID, they should follow the most recent quarantine guidelines including wearing a well-fitted mask when they do begin returning to activities.3
With rising cases of all three of these viruses, some may be asking how we can safely gather. There are several things to consider and do to enjoy our events. The first thing everyone can do is to receive updated vaccinations for both influenza and COVID-19 if eligible. Although it may take some time to be effective, vaccination is still one of our most effective methods of disease prevention and is important this winter season. Vaccinations can also help decrease the risk of severe disease.
Although many have stopped masking, as cases rise, it is time to consider masking particularly when community levels of any of these viruses are high. Masks help with preventing and spreading more than just COVID-19. Using them can be especially important for those going places such as stores and to large public gatherings and when riding on buses, planes, or trains.
In summary
Preventing exposure by masking can help keep individuals healthy prior to celebrating the holidays with others. With access to rapid testing, it makes sense to consider testing prior to gathering with friends and family. Most importantly, although we all are looking forward to spending time with our loved ones, it is important to stay home if not feeling well. Following these recommendations will allow us to have a safer and more joyful holiday season.
Dr. Wheat is a family physician at Erie Family Health Center and program director of Northwestern University’s McGaw Family Medicine residency program, both in Chicago. Dr. Wheat serves on the editorial advisory board of Family Practice News. You can contact her at fpnews@mdedge.com.
References
1. Centers for Disease Control and Prevention. Influenza (flu). [Online] Dec. 1, 2022. [Cited: 2022 Dec 10.] https://www.cdc.gov/flu/index.htm.
2. Respiratory syncytial virus. Respiratory syncytial virus infection (RSV). [Online] Oct. 28, 2022. [Cited: 2022 Dec 10.] https://www.cdc.gov/rsv/index.html.
3. COVID-19. [Online] Dec. 7, 2022. [Cited: 2022 Dec 10.] https://www.cdc.gov/coronavirus/2019-ncov/index.html.
This holiday season, I am looking forward to spending some time with family, as I have in the past. As I have chatted with others, many friends are looking forward to events that are potentially larger and potentially returning to prepandemic type gatherings.
Gathering is important and can bring joy, sense of community, and love to the lives of many. Unfortunately, the risks associated with gathering are not over. as our country faces many cases of respiratory syncytial virus (RSV), COVID-19, and influenza at the same time.
During the first week of December, cases of influenza were rising across the country1 and were rising faster than in previous years. Although getting the vaccine is an important method of influenza prevention and is recommended for everyone over the age of 6 months with rare exception, many have not gotten their vaccine this year.
Influenza
Thus far, “nearly 50% of reported flu-associated hospitalizations in women of childbearing age have been in women who are pregnant.” We are seeing this at a time with lower-than-average uptake of influenza vaccine leaving both the pregnant persons and their babies unprotected. In addition to utilizing vaccines as prevention, isolating when ill, cleaning surfaces, and practicing good hand hygiene can all decrease transmission.
RSV
In addition to rises of influenza, there are currently high rates of RSV in various parts of the country. Prior to 2020, RSV typically started in the fall and peaked in the winter months. However, since the pandemic, the typical seasonal pattern has not returned, and it is unclear when it will. Although RSV hits the very young, the old, and the immunocompromised the most, RSV can infect anyone. Unfortunately, we do not currently have a vaccine for everyone against this virus. Prevention of transmission includes, as with flu, isolating when ill, cleaning surfaces, and washing hands.2
COVID-19
Of course, the effects of the COVID-19 pandemic are also still here as well. During the first week of December, the CDC reported rising cases of COVID across the country. Within the past few months, there have been several developments, though, for protection. There are now bivalent vaccines available as either third doses or booster doses approved for all persons over 6 months of age. As of the first week of December, only 13.5% of those aged 5 and over had received an updated booster.
There is currently wider access to rapid testing, including at-home testing, which can allow individuals to identify if COVID positive. Additionally, there is access to medication to decrease the likelihood of severe disease – though this does not take the place of vaccinations.
If anyone does test positive for COVID, they should follow the most recent quarantine guidelines including wearing a well-fitted mask when they do begin returning to activities.3
With rising cases of all three of these viruses, some may be asking how we can safely gather. There are several things to consider and do to enjoy our events. The first thing everyone can do is to receive updated vaccinations for both influenza and COVID-19 if eligible. Although it may take some time to be effective, vaccination is still one of our most effective methods of disease prevention and is important this winter season. Vaccinations can also help decrease the risk of severe disease.
Although many have stopped masking, as cases rise, it is time to consider masking particularly when community levels of any of these viruses are high. Masks help with preventing and spreading more than just COVID-19. Using them can be especially important for those going places such as stores and to large public gatherings and when riding on buses, planes, or trains.
In summary
Preventing exposure by masking can help keep individuals healthy prior to celebrating the holidays with others. With access to rapid testing, it makes sense to consider testing prior to gathering with friends and family. Most importantly, although we all are looking forward to spending time with our loved ones, it is important to stay home if not feeling well. Following these recommendations will allow us to have a safer and more joyful holiday season.
Dr. Wheat is a family physician at Erie Family Health Center and program director of Northwestern University’s McGaw Family Medicine residency program, both in Chicago. Dr. Wheat serves on the editorial advisory board of Family Practice News. You can contact her at fpnews@mdedge.com.
References
1. Centers for Disease Control and Prevention. Influenza (flu). [Online] Dec. 1, 2022. [Cited: 2022 Dec 10.] https://www.cdc.gov/flu/index.htm.
2. Respiratory syncytial virus. Respiratory syncytial virus infection (RSV). [Online] Oct. 28, 2022. [Cited: 2022 Dec 10.] https://www.cdc.gov/rsv/index.html.
3. COVID-19. [Online] Dec. 7, 2022. [Cited: 2022 Dec 10.] https://www.cdc.gov/coronavirus/2019-ncov/index.html.
This holiday season, I am looking forward to spending some time with family, as I have in the past. As I have chatted with others, many friends are looking forward to events that are potentially larger and potentially returning to prepandemic type gatherings.
Gathering is important and can bring joy, sense of community, and love to the lives of many. Unfortunately, the risks associated with gathering are not over. as our country faces many cases of respiratory syncytial virus (RSV), COVID-19, and influenza at the same time.
During the first week of December, cases of influenza were rising across the country1 and were rising faster than in previous years. Although getting the vaccine is an important method of influenza prevention and is recommended for everyone over the age of 6 months with rare exception, many have not gotten their vaccine this year.
Influenza
Thus far, “nearly 50% of reported flu-associated hospitalizations in women of childbearing age have been in women who are pregnant.” We are seeing this at a time with lower-than-average uptake of influenza vaccine leaving both the pregnant persons and their babies unprotected. In addition to utilizing vaccines as prevention, isolating when ill, cleaning surfaces, and practicing good hand hygiene can all decrease transmission.
RSV
In addition to rises of influenza, there are currently high rates of RSV in various parts of the country. Prior to 2020, RSV typically started in the fall and peaked in the winter months. However, since the pandemic, the typical seasonal pattern has not returned, and it is unclear when it will. Although RSV hits the very young, the old, and the immunocompromised the most, RSV can infect anyone. Unfortunately, we do not currently have a vaccine for everyone against this virus. Prevention of transmission includes, as with flu, isolating when ill, cleaning surfaces, and washing hands.2
COVID-19
Of course, the effects of the COVID-19 pandemic are also still here as well. During the first week of December, the CDC reported rising cases of COVID across the country. Within the past few months, there have been several developments, though, for protection. There are now bivalent vaccines available as either third doses or booster doses approved for all persons over 6 months of age. As of the first week of December, only 13.5% of those aged 5 and over had received an updated booster.
There is currently wider access to rapid testing, including at-home testing, which can allow individuals to identify if COVID positive. Additionally, there is access to medication to decrease the likelihood of severe disease – though this does not take the place of vaccinations.
If anyone does test positive for COVID, they should follow the most recent quarantine guidelines including wearing a well-fitted mask when they do begin returning to activities.3
With rising cases of all three of these viruses, some may be asking how we can safely gather. There are several things to consider and do to enjoy our events. The first thing everyone can do is to receive updated vaccinations for both influenza and COVID-19 if eligible. Although it may take some time to be effective, vaccination is still one of our most effective methods of disease prevention and is important this winter season. Vaccinations can also help decrease the risk of severe disease.
Although many have stopped masking, as cases rise, it is time to consider masking particularly when community levels of any of these viruses are high. Masks help with preventing and spreading more than just COVID-19. Using them can be especially important for those going places such as stores and to large public gatherings and when riding on buses, planes, or trains.
In summary
Preventing exposure by masking can help keep individuals healthy prior to celebrating the holidays with others. With access to rapid testing, it makes sense to consider testing prior to gathering with friends and family. Most importantly, although we all are looking forward to spending time with our loved ones, it is important to stay home if not feeling well. Following these recommendations will allow us to have a safer and more joyful holiday season.
Dr. Wheat is a family physician at Erie Family Health Center and program director of Northwestern University’s McGaw Family Medicine residency program, both in Chicago. Dr. Wheat serves on the editorial advisory board of Family Practice News. You can contact her at fpnews@mdedge.com.
References
1. Centers for Disease Control and Prevention. Influenza (flu). [Online] Dec. 1, 2022. [Cited: 2022 Dec 10.] https://www.cdc.gov/flu/index.htm.
2. Respiratory syncytial virus. Respiratory syncytial virus infection (RSV). [Online] Oct. 28, 2022. [Cited: 2022 Dec 10.] https://www.cdc.gov/rsv/index.html.
3. COVID-19. [Online] Dec. 7, 2022. [Cited: 2022 Dec 10.] https://www.cdc.gov/coronavirus/2019-ncov/index.html.
Principles and Process for Reducing the Need for Insulin in Patients With Type 2 Diabetes

For people living with type 2 diabetes mellitus (T2D), exogenous insulin, whether given early or later in T2D diagnosis, can provide many pharmacologically desirable effects. But it has always been clear, and is now more widely recognized, that insulin treatments are not completely risk-free for the patient. There are now newer, non-insulin therapy options that could be used, along with certain patient lifestyle changes in diet and activity levels, that have been shown to achieve desired glucose control—without the associated risks that insulin can bring.
But is it possible to markedly reduce the need for insulin in some 90% of T2D patients and to reduce the doses in the others? Yes—if patients have sufficient beta-cell function and are willing to change their lifestyle. This mode of treatment has been slowly gaining momentum as of late in the medical community because of the benefits it ultimately provides for the patient. In my practice, I personally have done this by using an evidence-based approach that includes thinking inside a larger box. It is a 2-way street, and each should drive the other: the right drugs (in the right doses), and in the right patients.
Why avoid early insulin therapy?
Is the requirement of early insulin therapy in many or most patients a myth?
Yes. It resulted from “old logic,” which was to use insulin early to reduce glucotoxicity and lipotoxicity. The American Diabetes Association guidelines recommend that glycated hemoglobin (HbA1c) should not exceed 8.0% and consider a fasting blood glucose level >250 mg/dL as high, with a need to start insulin treatment right away; other guidelines recommend initiating insulin immediately in patients with HbA1c >9% and postprandial glucose 300 mg/dL. But this was at a time when oral agents were not as effective and took time to titrate or engender good control. We now have agents that are more effective and start working right away.
However, the main problem in early insulin treatment is the significant risk of over-insulinization—a vicious cycle of insulin-caused increased appetite, hypoglycemia-resultant increased weight gain, insulin resistance (poorer control), increased circulating insulin, etc. Moreover, weight gain and individual hypoglycemic events can cause an increase in the risk of cardiovascular (CV) events.
I believe clinicians must start as early as possible in the natural history of T2D to prevent progressive beta-cell failure. Do not believe in “first-, second-, or third-line”; in other words, do not prioritize, so there is no competition between classes. The goal I have for my patients is to provide therapies that aim for the lowest HbA1c possible without hypoglycemia, provide the greatest CV benefit, and assist in weight reduction.
My protocol, “the egregious eleven,” involves using the least number of agents in combinations that treat the greatest number of mechanisms of hyperglycemia—without the use of sulfonylureas (which cause beta-cell apoptosis, hypoglycemia, and weight gain). Fortunately, newer agents, such as glucagon-like peptide 1 receptor agonist (GLP-1 RA) and sodium-glucose cotransporter 1 (SGLT-2) inhibitors, work right away, cause weight reduction, and have side benefits of CV risk reduction—as well as preserve beta-cell function. Metformin remains a valuable agent and has its own potential side benefits, and bromocriptine-QR and pioglitazone have CV side benefits. So, there is really no need for early insulin in true T2D patients (ie, those that are non-ketosis prone and have sufficient beta-cell reserve).
Why reduce insulin in patients who are already on insulin?
Prior protocols resulted in 40%-50% of T2D patients being placed on insulin unnecessarily. As discussed, the side effects of insulin are many; they include weight gain, insulin resistance, hypoglycemia, and CV complications—all of which have been associated with a decline in quality of life.
What is your approach to reduce or eliminate insulin in those already on it (unnecessarily)?
First, I pick the right patient. Physicians should use sound clinical judgment to identify patients with likely residual beta-cell function. It is not just the “insulin-resistant patient," as 30%-50% of type 1 diabetes mellitus patients also have insulin resistance.
It needs to be a definite T2D patient: not ketosis prone, a family history T2D, no islet cell antibodies (if one has any concerns, check for them). They were often started on insulin in the emergency department with no ketosis and never received non-insulin therapy.
Patients need to be willing to commit to my strict, no-concentrated-sweets diet, to perform careful glucose monitoring, and to check their ketones. Patients should be willing to contact me if their sugar level is >250 mg/dL for 2 measurements in a row, while testing 4 times a day or using a continuous glucose-monitoring (CGM) device.
First, estimate a patient’s “current insulin need” (CIN), or the dose they might be on if they had not been subject to over-insulinization (ie, if they had not been subject to the “vicious cycle” discussed above). I do this by taking their total basal and bolus insulin dose, then reducing it by ~25% as the patient changes to a no-concentrated-sweets diet with an additional up-to-25% dose reduction if the patient has been experiencing symptomatic or asymptomatic hypoglycemia.
Next, I reduce this CIN number by ~25% upon starting a rapid-acting subcutaneous GLP-1 RA (liraglutide or oral semaglutide) and reduce the CIN another 20% as they start the SGLT-2 inhibitor. If patients come into my office on <40 U/d, I stop insulin as I start a GLP-1 RA and an SGLT-2 inhibitor and have them monitor home glucose levels to assure reasonable results as they go off the insulin and on their new therapy.
If patients come into my office on >40 U/d, they go home on a GLP-1 RA and an SGLT-2 inhibitor and ~30% of their presenting dose, apportioned between basal/bolus dosing based on when they are currently getting hypoglycemic.
The rapid initial reduction in their insulin doses, with initial adjustments in estimated insulin doses as needed based on home glucose monitoring, and rapid stabilization of glycemic levels by the effectiveness of these 2 agents give patients great motivation to keep up with the diet/program.
Then, as patients lose weight, they are told to report any glucose measurements <80 mg/dL, so that further reduction in insulin doses can be made. When patients achieve a new steady state of glycemia, weight, and GLP-1 RA and SGLT-2 inhibitor doses, you can add bromocriptine-QR, pioglitazone, and/or metformin as needed to allow for a further reduction of insulin. And, as you see the delayed effects of subsequently adding these new agents (eg, glucose <80 mg/dL), you can ultimately stop insulin when they get to <10-12 U/d. The process works very well, even in those starting on up to 300 units of insulin daily. Patients love the outcome and will greatly appreciate your care.
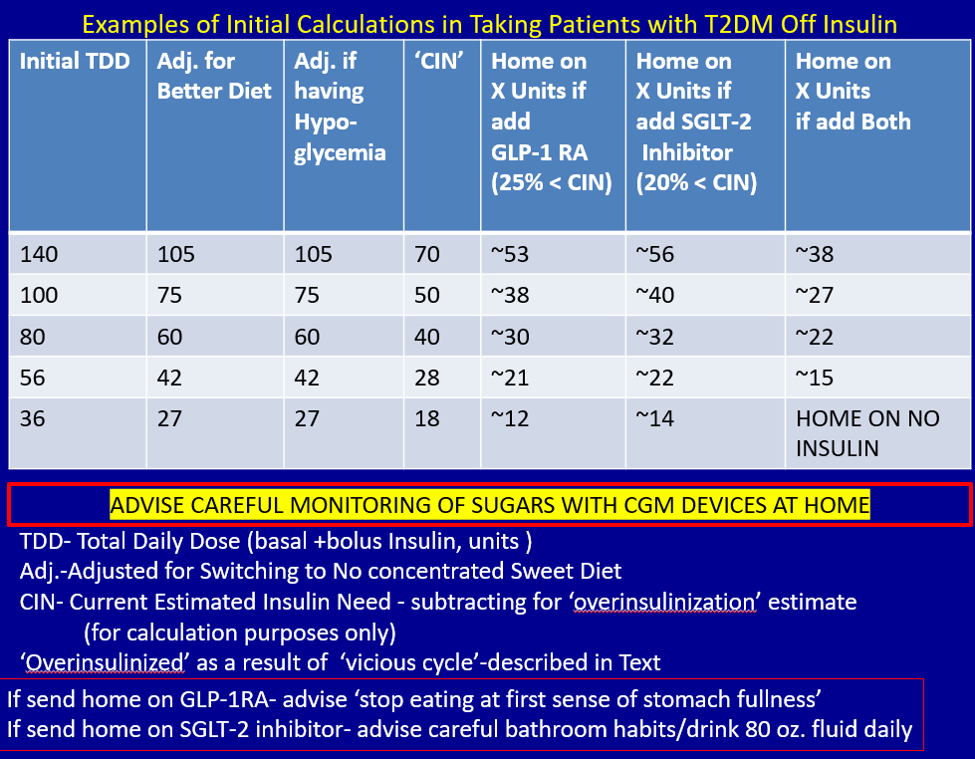
Feel free to contact Dr. Schwartz at stschwar@gmail.com with any questions about his protocol or diet.
For people living with type 2 diabetes mellitus (T2D), exogenous insulin, whether given early or later in T2D diagnosis, can provide many pharmacologically desirable effects. But it has always been clear, and is now more widely recognized, that insulin treatments are not completely risk-free for the patient. There are now newer, non-insulin therapy options that could be used, along with certain patient lifestyle changes in diet and activity levels, that have been shown to achieve desired glucose control—without the associated risks that insulin can bring.
But is it possible to markedly reduce the need for insulin in some 90% of T2D patients and to reduce the doses in the others? Yes—if patients have sufficient beta-cell function and are willing to change their lifestyle. This mode of treatment has been slowly gaining momentum as of late in the medical community because of the benefits it ultimately provides for the patient. In my practice, I personally have done this by using an evidence-based approach that includes thinking inside a larger box. It is a 2-way street, and each should drive the other: the right drugs (in the right doses), and in the right patients.
Why avoid early insulin therapy?
Is the requirement of early insulin therapy in many or most patients a myth?
Yes. It resulted from “old logic,” which was to use insulin early to reduce glucotoxicity and lipotoxicity. The American Diabetes Association guidelines recommend that glycated hemoglobin (HbA1c) should not exceed 8.0% and consider a fasting blood glucose level >250 mg/dL as high, with a need to start insulin treatment right away; other guidelines recommend initiating insulin immediately in patients with HbA1c >9% and postprandial glucose 300 mg/dL. But this was at a time when oral agents were not as effective and took time to titrate or engender good control. We now have agents that are more effective and start working right away.
However, the main problem in early insulin treatment is the significant risk of over-insulinization—a vicious cycle of insulin-caused increased appetite, hypoglycemia-resultant increased weight gain, insulin resistance (poorer control), increased circulating insulin, etc. Moreover, weight gain and individual hypoglycemic events can cause an increase in the risk of cardiovascular (CV) events.
I believe clinicians must start as early as possible in the natural history of T2D to prevent progressive beta-cell failure. Do not believe in “first-, second-, or third-line”; in other words, do not prioritize, so there is no competition between classes. The goal I have for my patients is to provide therapies that aim for the lowest HbA1c possible without hypoglycemia, provide the greatest CV benefit, and assist in weight reduction.
My protocol, “the egregious eleven,” involves using the least number of agents in combinations that treat the greatest number of mechanisms of hyperglycemia—without the use of sulfonylureas (which cause beta-cell apoptosis, hypoglycemia, and weight gain). Fortunately, newer agents, such as glucagon-like peptide 1 receptor agonist (GLP-1 RA) and sodium-glucose cotransporter 1 (SGLT-2) inhibitors, work right away, cause weight reduction, and have side benefits of CV risk reduction—as well as preserve beta-cell function. Metformin remains a valuable agent and has its own potential side benefits, and bromocriptine-QR and pioglitazone have CV side benefits. So, there is really no need for early insulin in true T2D patients (ie, those that are non-ketosis prone and have sufficient beta-cell reserve).
Why reduce insulin in patients who are already on insulin?
Prior protocols resulted in 40%-50% of T2D patients being placed on insulin unnecessarily. As discussed, the side effects of insulin are many; they include weight gain, insulin resistance, hypoglycemia, and CV complications—all of which have been associated with a decline in quality of life.
What is your approach to reduce or eliminate insulin in those already on it (unnecessarily)?
First, I pick the right patient. Physicians should use sound clinical judgment to identify patients with likely residual beta-cell function. It is not just the “insulin-resistant patient," as 30%-50% of type 1 diabetes mellitus patients also have insulin resistance.
It needs to be a definite T2D patient: not ketosis prone, a family history T2D, no islet cell antibodies (if one has any concerns, check for them). They were often started on insulin in the emergency department with no ketosis and never received non-insulin therapy.
Patients need to be willing to commit to my strict, no-concentrated-sweets diet, to perform careful glucose monitoring, and to check their ketones. Patients should be willing to contact me if their sugar level is >250 mg/dL for 2 measurements in a row, while testing 4 times a day or using a continuous glucose-monitoring (CGM) device.
First, estimate a patient’s “current insulin need” (CIN), or the dose they might be on if they had not been subject to over-insulinization (ie, if they had not been subject to the “vicious cycle” discussed above). I do this by taking their total basal and bolus insulin dose, then reducing it by ~25% as the patient changes to a no-concentrated-sweets diet with an additional up-to-25% dose reduction if the patient has been experiencing symptomatic or asymptomatic hypoglycemia.
Next, I reduce this CIN number by ~25% upon starting a rapid-acting subcutaneous GLP-1 RA (liraglutide or oral semaglutide) and reduce the CIN another 20% as they start the SGLT-2 inhibitor. If patients come into my office on <40 U/d, I stop insulin as I start a GLP-1 RA and an SGLT-2 inhibitor and have them monitor home glucose levels to assure reasonable results as they go off the insulin and on their new therapy.
If patients come into my office on >40 U/d, they go home on a GLP-1 RA and an SGLT-2 inhibitor and ~30% of their presenting dose, apportioned between basal/bolus dosing based on when they are currently getting hypoglycemic.
The rapid initial reduction in their insulin doses, with initial adjustments in estimated insulin doses as needed based on home glucose monitoring, and rapid stabilization of glycemic levels by the effectiveness of these 2 agents give patients great motivation to keep up with the diet/program.
Then, as patients lose weight, they are told to report any glucose measurements <80 mg/dL, so that further reduction in insulin doses can be made. When patients achieve a new steady state of glycemia, weight, and GLP-1 RA and SGLT-2 inhibitor doses, you can add bromocriptine-QR, pioglitazone, and/or metformin as needed to allow for a further reduction of insulin. And, as you see the delayed effects of subsequently adding these new agents (eg, glucose <80 mg/dL), you can ultimately stop insulin when they get to <10-12 U/d. The process works very well, even in those starting on up to 300 units of insulin daily. Patients love the outcome and will greatly appreciate your care.
Feel free to contact Dr. Schwartz at stschwar@gmail.com with any questions about his protocol or diet.
For people living with type 2 diabetes mellitus (T2D), exogenous insulin, whether given early or later in T2D diagnosis, can provide many pharmacologically desirable effects. But it has always been clear, and is now more widely recognized, that insulin treatments are not completely risk-free for the patient. There are now newer, non-insulin therapy options that could be used, along with certain patient lifestyle changes in diet and activity levels, that have been shown to achieve desired glucose control—without the associated risks that insulin can bring.
But is it possible to markedly reduce the need for insulin in some 90% of T2D patients and to reduce the doses in the others? Yes—if patients have sufficient beta-cell function and are willing to change their lifestyle. This mode of treatment has been slowly gaining momentum as of late in the medical community because of the benefits it ultimately provides for the patient. In my practice, I personally have done this by using an evidence-based approach that includes thinking inside a larger box. It is a 2-way street, and each should drive the other: the right drugs (in the right doses), and in the right patients.
Why avoid early insulin therapy?
Is the requirement of early insulin therapy in many or most patients a myth?
Yes. It resulted from “old logic,” which was to use insulin early to reduce glucotoxicity and lipotoxicity. The American Diabetes Association guidelines recommend that glycated hemoglobin (HbA1c) should not exceed 8.0% and consider a fasting blood glucose level >250 mg/dL as high, with a need to start insulin treatment right away; other guidelines recommend initiating insulin immediately in patients with HbA1c >9% and postprandial glucose 300 mg/dL. But this was at a time when oral agents were not as effective and took time to titrate or engender good control. We now have agents that are more effective and start working right away.
However, the main problem in early insulin treatment is the significant risk of over-insulinization—a vicious cycle of insulin-caused increased appetite, hypoglycemia-resultant increased weight gain, insulin resistance (poorer control), increased circulating insulin, etc. Moreover, weight gain and individual hypoglycemic events can cause an increase in the risk of cardiovascular (CV) events.
I believe clinicians must start as early as possible in the natural history of T2D to prevent progressive beta-cell failure. Do not believe in “first-, second-, or third-line”; in other words, do not prioritize, so there is no competition between classes. The goal I have for my patients is to provide therapies that aim for the lowest HbA1c possible without hypoglycemia, provide the greatest CV benefit, and assist in weight reduction.
My protocol, “the egregious eleven,” involves using the least number of agents in combinations that treat the greatest number of mechanisms of hyperglycemia—without the use of sulfonylureas (which cause beta-cell apoptosis, hypoglycemia, and weight gain). Fortunately, newer agents, such as glucagon-like peptide 1 receptor agonist (GLP-1 RA) and sodium-glucose cotransporter 1 (SGLT-2) inhibitors, work right away, cause weight reduction, and have side benefits of CV risk reduction—as well as preserve beta-cell function. Metformin remains a valuable agent and has its own potential side benefits, and bromocriptine-QR and pioglitazone have CV side benefits. So, there is really no need for early insulin in true T2D patients (ie, those that are non-ketosis prone and have sufficient beta-cell reserve).
Why reduce insulin in patients who are already on insulin?
Prior protocols resulted in 40%-50% of T2D patients being placed on insulin unnecessarily. As discussed, the side effects of insulin are many; they include weight gain, insulin resistance, hypoglycemia, and CV complications—all of which have been associated with a decline in quality of life.
What is your approach to reduce or eliminate insulin in those already on it (unnecessarily)?
First, I pick the right patient. Physicians should use sound clinical judgment to identify patients with likely residual beta-cell function. It is not just the “insulin-resistant patient," as 30%-50% of type 1 diabetes mellitus patients also have insulin resistance.
It needs to be a definite T2D patient: not ketosis prone, a family history T2D, no islet cell antibodies (if one has any concerns, check for them). They were often started on insulin in the emergency department with no ketosis and never received non-insulin therapy.
Patients need to be willing to commit to my strict, no-concentrated-sweets diet, to perform careful glucose monitoring, and to check their ketones. Patients should be willing to contact me if their sugar level is >250 mg/dL for 2 measurements in a row, while testing 4 times a day or using a continuous glucose-monitoring (CGM) device.
First, estimate a patient’s “current insulin need” (CIN), or the dose they might be on if they had not been subject to over-insulinization (ie, if they had not been subject to the “vicious cycle” discussed above). I do this by taking their total basal and bolus insulin dose, then reducing it by ~25% as the patient changes to a no-concentrated-sweets diet with an additional up-to-25% dose reduction if the patient has been experiencing symptomatic or asymptomatic hypoglycemia.
Next, I reduce this CIN number by ~25% upon starting a rapid-acting subcutaneous GLP-1 RA (liraglutide or oral semaglutide) and reduce the CIN another 20% as they start the SGLT-2 inhibitor. If patients come into my office on <40 U/d, I stop insulin as I start a GLP-1 RA and an SGLT-2 inhibitor and have them monitor home glucose levels to assure reasonable results as they go off the insulin and on their new therapy.
If patients come into my office on >40 U/d, they go home on a GLP-1 RA and an SGLT-2 inhibitor and ~30% of their presenting dose, apportioned between basal/bolus dosing based on when they are currently getting hypoglycemic.
The rapid initial reduction in their insulin doses, with initial adjustments in estimated insulin doses as needed based on home glucose monitoring, and rapid stabilization of glycemic levels by the effectiveness of these 2 agents give patients great motivation to keep up with the diet/program.
Then, as patients lose weight, they are told to report any glucose measurements <80 mg/dL, so that further reduction in insulin doses can be made. When patients achieve a new steady state of glycemia, weight, and GLP-1 RA and SGLT-2 inhibitor doses, you can add bromocriptine-QR, pioglitazone, and/or metformin as needed to allow for a further reduction of insulin. And, as you see the delayed effects of subsequently adding these new agents (eg, glucose <80 mg/dL), you can ultimately stop insulin when they get to <10-12 U/d. The process works very well, even in those starting on up to 300 units of insulin daily. Patients love the outcome and will greatly appreciate your care.
Feel free to contact Dr. Schwartz at stschwar@gmail.com with any questions about his protocol or diet.
MS and Emotional Stress: Is There a Relation?

Sir Augustus d’Este (1794-1848) described the circumstances preceding his development of neurological symptoms as follows:1 “I travelled from Ramsgate to the Highlands of Scotland for the purpose of passing some days with a Relation for whom I had the affection of a Son. On my arrival I found him dead. Shortly after the funeral I was obliged to have my letters read to me, and their answers written for me, as my eyes were so attacked that when fixed upon minute objects indistinctness of vision was the consequence: Soon after I went to Ireland, and without any thing having been done to my eyes, they completely recovered their strength and distinctness of vision…" He then described a clinical course of relapsing-remitting neurologic symptoms merging into a progressive stage of unrelenting illness, most fitting with what we know today as multiple sclerosis (MS).1 Why did Sir Augustus d'Este connect the event of the unexpected death to the onset of a lifelong neurologic disease?
Jean-Martin Charcot first described MS in a way close to what we know it as today. Charcot considered stress a factor in MS. He linked grief, vexation, and adverse changes in social circumstances to the onset of MS at that time.2 I, as a practicing MS specialist, am surprised neither by Sir Augustus d'Este's diary nor by Charcot's earlier assessments of MS triggers.3 As I write this narrative, I think of the many times I heard from people diagnosed with MS. "It happened to me because of stress" is a statement not estranged from my daily clinical practice
MS as a multifactorial disease
It is tempting to make a case for emotional stress as a cause of MS, but one must remember that MS is a very complex disease with unclear etiologies. MS, a treatable but not yet curable disease, is the interplay between the genetics of the host and numerous environmental factors that exploit a susceptible immune system leading to unrelenting immune dysregulation.4 Recent studies have brought some pieces of this intricate puzzle together. The role of Epstein-Barr virus (EBV) in the pathogenesis of MS is being dissected.5 The possible synergy between vitamin D deficiency, EBV, and certain genetic variations is being studied.6 The roles of smoking, environmental toxins, obesity, diet, Western lifestyle, and the gut microbiome are some of the top areas of clinical, translational, and basic research.7-11 But what about emotional stress? Where does it fit, if anywhere, in the current research paradigm?
Emotional stress and MS—Causality or not?
In the scientific method, several criteria must be proven for an element to be suspected in the etiology of a disease.12 First, the suspect element must be present before the disease starts—i.e., a temporal association. Second, there must be a plausible biological explanation of how the suspect element acts in the disease's causation. Third, other variables that could confound the picture must be controlled for or dismissed. It is clear that no single factor is the cause of MS. By now, MS is agreed upon as a disease caused by multiple factors, some of which remain to be unraveled.9 The term "cause" has been utilized more recently by many authors when referring to EBV in relation to MS development, reasoning that in one study, in a small number of individuals with MS, EBV infection preceded the MS clinical diagnosis.13 Thus, the temporal association was provided. But does MS start at the onset of clinical symptoms?
For Sir Augustus d'Este, the disease may have started years before he visited the Highlands of Scotland, but only at that visit did MS become clinically apparent. So, the emotional trauma may have acted as a "trigger" for an MS flare-up rather than being a "cause" of MS. This might be a more plausible explanation of the association between emotional trauma and MS development. However, MS pathogenesis is complex, and one could argue that the disease starts many years before the first clinical symptoms that lead to diagnosis.
The MS prodrome has been demonstrated by several studies that suggest that MS may start many years before the clinical diagnosis.14 Radiologically isolated syndrome (RIS) further argues that MS may be clinically dormant for years, and clinical symptoms may not appear until later in the disease process.15 One may think that immune attacks on the optic nerves, spinal cord, or areas of the brainstem might be readily symptomatic compared to attacks on other structures of the central nervous system (e.g., periventricular or juxtacortical brain areas) that may be clinically silent. So, while for Sir Augustus d'Este it seemed that the disease started at the time of his visit to the Highlands of Scotland, it is equally plausible that it started years before the first clinical attack. Nevertheless, how could emotional stress play a role in the pathophysiology of MS?
Stress and the Immune System
At times of chronic stress, one may become more susceptible to infections. Reactivation of certain viruses can lead to oral ulcers, increased common cold symptoms, or other illnesses. For example, stress can reactivate herpes simplex type 1 and interestingly, EBV.16,17 In MS, the immune system is dysregulated and has an autoimmune component. The effect of acute emotional stress differs from that of chronic stress.18 Several studies have examined the immune responses to both forms of stress.19-21
Interestingly, acute stress activates cell-mediated immunity, increases immune cell trafficking to areas of injury, and, importantly, increases blood-brain barrier (BBB) permeability by activating resident mast cells in the brain and other areas, including the optic nerves.22,23 Mast cell activation leads to BBB disruption, which is a key early step in the pathogenesis of MS. Thus, it is plausible that the proinflammatory changes associated with acute stress could be implicated in the pathogenesis of MS. This contrasts with chronic stress, which attenuates various immune responses, including suppressing cell-mediated immunity, but also dysregulate the immune system.
One could establish a biological plausibility for stress playing a role in the proinflammatory responses in MS. Whether it is causal or not, scientists can further explore the potential biologic explanations. While studying the association between acute stress and MS development or disease activity is difficult, several groups have examined the potential association. Many studies, however, have limitations due to the difficult nature of studying such an association, especially in quantifying or defining acute stress in general.
A limited number of studies on MS and stress: What do we know? And what are the challenges?
Rare studies have reported a potential association between MS development and stressful life events, while others reported no association.24-26 Also, some studies observed an increase in MS relapses or the development of new magnetic resonance imaging (MRI) lesions following stressful life events or wartime, while others failed to show such an association.26-30 There are few studies directly addressing the potential association between acute emotional stress and MS. The results of published studies are variable, and limitations are numerous. Limitations include the difficulty in measuring acute emotional stress, difficulty in its prediction, and ethical challenges of experimental design and recruiting participants. So, studies have focused on observational aspects, retrospective reviews, and surveys of memories prone to various biases. Rarely was the design of these clinical studies prospective. A few prospective studies reported an association between stressful life events and increased MS relapses and increased number of brain lesions.27,31,32 Rare clinical trials have attempted to test stress reduction strategies and reported on the modest improvement of patient-reported outcomes and, in one study, a modest improvement in new MRI lesions.33-35
Overall, several lines of evidence support a potential association between acute emotional stress and MS. Yet, the association is challenging to study, and future research might focus on stress-mitigation strategies and improving coping mechanisms in persons living with MS. It is important to note that it will be very difficult to design prospective studies to examine the potential association between acute emotional trauma and the development of de novo MS. Such studies will require a large number of participants (e.g., hundreds of thousands), long durations of follow-up (e.g., decades), and ways to classify repeated stressful events. An alternative approach is to ask persons newly diagnosed with MS at the time of initial diagnosis about any temporal association between their first symptom and stressful life events. However, this approach would provide some information on any association between the two, but not on causality of the disease itself.
Conclusion
The potential association between acute emotional stress and MS dates to the times of early descriptions of MS. Yet, research has been very limited and challenging. To date, the potential association remains elusive. Lines of evidence, while with limitations, have provided possible biologic explanations for the relationship between MS symptom onset and acute emotional stress. Although avoiding acute emotional stress is nearly impossible, incorporating global stress-coping strategies in early childhood education and secondary education might theoretically have potential beneficial effects on the subsequent risk of MS development or symptom flare-up, depending on a variety of factors.
But for now, when patients and colleagues ask me, “Can acute emotional stress be a ‘trigger’ for MS symptomology?,” my answer will remain, “Potentially, until proven otherwise.”
- Firth D. The case of Augustus d'Este (1794-1848): the first account of disseminated sclerosis: (section of the History of Medicine). Proc R Soc Med. 1941;34(7):381-384.
- Lectures on the diseases of the nervous system. Br Foreign Med Chir Rev. 1877;60(119):180-181.
- Obeidat, A, Cope T. Stressful life events and multiple sclerosis: a call for re-evaluation. Paper presented at: Fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers; May 13, 2013; Orlando, FL.
- Waubant E, Lucas R, Mowry E, et al. Environmental and genetic risk factors for MS: an integrated review. Ann Clin Transl Neurol. 2019;6(9):1905-1922. doi:10.1002/acn3.50862
- Soldan SS, Lieberman PM. Epstein-Barr virus and multiple sclerosis. Nat Rev Microbiol. 2022;1-14. doi:10.1038/s41579-022-00770-5
- Marcucci SB, Obeidat AZ. EBNA1, EBNA2, and EBNA3 link Epstein-Barr virus and hypovitaminosis D in multiple sclerosis pathogenesis. J Neuroimmunol. 2020;339:57711 doi:10.1016/j.jneuroim.2019.577116
- Alfredsson L, Olsson T. Lifestyle and environmental factors in multiple sclerosis. Cold Spring Harb Perspect Med. 2019;9(4):a028944. doi:10.1101/cshperspect.a028944
- Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622-1636. doi:10.1016/S0140-6736(18)30481-1
- Dobson R, Giovannoni G. Multiple sclerosis – a review. Eur J Neurol. 2019;26(1):27-40. doi:10.1111/ene.13819
- Arneth B. Multiple sclerosis and smoking. Am J Med. 2020;133(7):783-788. doi:1016/j.amjmed.2020.03.008
- Correale J, Hohlfeld R, Baranzini SE. The role of the gut microbiota in multiple sclerosis. Nat Rev Neurol. 2022;18(9):544-558. doi:10.1038/s41582-022-00697-8
- Gianicolo EAL, Eichler M, Muensterer O, Strauch K, Blettner M. Methods for evaluating causality in observational studies. Dtsch Arztebl Int. 2020;116(7):101-107. doi:10.3238/arztebl.2020.0101
- Bjornevik K, Cortese M, Healy BC, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. 2022;375(6578):296-301. doi:10.1126/science.abj8222
- Makhani N, Tremlett H. The multiple sclerosis prodrome. Nat Rev Neurol. 2021;17(8):515-521. doi:10.1038/s41582-021-00519-3
- Hosseiny M, Newsome SD, Yousem DM. Radiologically isolated syndrome: a review for neuroradiologists. AJNR Am J Neuroradiol. 2020;41(9):1542-1549. doi:10.3174/ajnr.A6649
- Padgett DA, Sheridan JF, Dorne J, Berntson GG, Candelora J, Glaser R. Social stress and the reactivation of latent herpes simplex virus type 1 [published correction appears in Proc Natl Acad Sci U S A. 1998;95(20):12070]. Proc Natl Acad Sci U S A. 1998;95(12):7231-7235. doi:10.1073/pnas.95.12.7231
- Glaser R, Pearson GR, Jones JF, et al. Stress-related activation of Epstein-Barr virus. Brain Behav Immun. 1991;5(2):219-232. doi:10.1016/0889-1591(91)90018-6
- Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16(5):300-317. doi:10.1159/000216188
- Musazzi L, Tornese P, Sala N, Popoli M. Acute or chronic? A stressful question. Trends Neurosci. 2017;40(9):525-535. doi:10.1016/j.tins.2017.07.002
- Dhabhar FS, McEwen BS. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: a potential role for leukocyte trafficking. Brain Behav Immun. 1997;11(4):286-306. doi:10.1006/brbi.1997.0508
- Maydych V, Claus M, Dychus N, et al. Impact of chronic and acute academic stress on lymphocyte subsets and monocyte function. PLoS One. 2017;12(11):e0188108. Published 2017 Nov 16. doi:10.1371/journal.pone.0188108
- Esposito P, Gheorghe D, Kandere K, et al. Acute stress increases permeability of the blood-brain-barrier through activation of brain mast cells. Brain Res. 2001;888(1):117-127. doi:10.1016/s0006-8993(00)03026-2
- Kempuraj D, Mentor S, Thangavel R, et al. Mast cells in stress, pain, blood-brain barrier, neuroinflammation and Alzheimer's disease. Front Cell Neurosci. 2019;13:54. doi:10.3389/fncel.2019.00054
- Karagkouni A, Alevizos M, Theoharides TC. Effect of stress on brain inflammation and multiple sclerosis. Autoimmun Rev. 2013;12(10):947-953. doi:10.1016/j.autrev.2013.02.006
- Briones-Buixassa L, Milà R, Mª Aragonès J, Bufill E, Olaya B, Arrufat FX. Stress and multiple sclerosis: a systematic review considering potential moderating and mediating factors and methods of assessing stress. Health Psychol Open. 2015;2(2):2055102915612271. doi:10.1177/2055102915612271
- Riise T, Mohr DC, Munger KL, Rich-Edwards JW, Kawachi I, Ascherio A. Stress and the risk of multiple sclerosis. Neurology. 2011;76(22):1866-1871. doi:10.1212/WNL.0b013e31821d74c5
- Burns MN, Nawacki E, Kwasny MJ, Pelletier D, Mohr DC. Do positive or negative stressful events predict the development of new brain lesions in people with multiple sclerosis? Psychol Med. 2014;44(2):349-359. doi:10.1017/S0033291713000755
- Mohr DC, Goodkin DE, Bacchetti P, et al. Psychological stress and the subsequent appearance of new brain MRI lesions in MS. Neurology. 2000;55(1):55-61. doi:10.1212/wnl.55.1.55
- Yamout B, Itani S, Hourany R, Sibaii AM, Yaghi S. The effect of war stress on multiple sclerosis exacerbations and radiological disease activity. J Neurol Sci. 2010;288(1-2):42-44. doi:10.1016/j.jns.2009.10.012
- Artemiadis AK, Anagnostouli MC, Alexopoulos EC. Stress as a risk factor for multiple sclerosis onset or relapse: a systematic review. Neuroepidemiology. 2011;36(2):109-120. doi:10.1159/000323953
- Brown RF, Tennant CC, Sharrock M, Hodgkinson S, Dunn SM, Pollard JD. Relationship between stress and relapse in multiple sclerosis: Part I. Important features. Mult Scler. 2006;12(4):453-464. doi:10.1191/1352458506ms1295oa
- Buljevac D, Hop WCJ, Reedeker W, et al. Self-reported stressful life events and exacerbations in multiple sclerosis: prospective study. BMJ. 2003;327(7416):646. doi:10.1136/bmj.327.7416.646
- Senders A, Hanes D, Bourdette D, Carson K, Marshall LM, Shinto L. Impact of mindfulness-based stress reduction for people with multiple sclerosis at 8 weeks and 12 months: A randomized clinical trial. Mult Scler. 2019;25(8):1178-1188. doi:10.1177/1352458518786650
- Morrow SA, Riccio P, Vording N, Rosehart H, Casserly C, MacDougall A. A mindfulness group intervention in newly diagnosed persons with multiple sclerosis: A pilot study. Mult Scler Relat Disord. 2021;52:103016. doi:10.1016/j.msard.2021.103016
- Mohr DC, Lovera J, Brown T, et al. A randomized trial of stress management for the prevention of new brain lesions in MS. Neurology. 2012;79(5):412-419. doi:10.1212/WNL.0b013e3182616ff9
Sir Augustus d’Este (1794-1848) described the circumstances preceding his development of neurological symptoms as follows:1 “I travelled from Ramsgate to the Highlands of Scotland for the purpose of passing some days with a Relation for whom I had the affection of a Son. On my arrival I found him dead. Shortly after the funeral I was obliged to have my letters read to me, and their answers written for me, as my eyes were so attacked that when fixed upon minute objects indistinctness of vision was the consequence: Soon after I went to Ireland, and without any thing having been done to my eyes, they completely recovered their strength and distinctness of vision…" He then described a clinical course of relapsing-remitting neurologic symptoms merging into a progressive stage of unrelenting illness, most fitting with what we know today as multiple sclerosis (MS).1 Why did Sir Augustus d'Este connect the event of the unexpected death to the onset of a lifelong neurologic disease?
Jean-Martin Charcot first described MS in a way close to what we know it as today. Charcot considered stress a factor in MS. He linked grief, vexation, and adverse changes in social circumstances to the onset of MS at that time.2 I, as a practicing MS specialist, am surprised neither by Sir Augustus d'Este's diary nor by Charcot's earlier assessments of MS triggers.3 As I write this narrative, I think of the many times I heard from people diagnosed with MS. "It happened to me because of stress" is a statement not estranged from my daily clinical practice
MS as a multifactorial disease
It is tempting to make a case for emotional stress as a cause of MS, but one must remember that MS is a very complex disease with unclear etiologies. MS, a treatable but not yet curable disease, is the interplay between the genetics of the host and numerous environmental factors that exploit a susceptible immune system leading to unrelenting immune dysregulation.4 Recent studies have brought some pieces of this intricate puzzle together. The role of Epstein-Barr virus (EBV) in the pathogenesis of MS is being dissected.5 The possible synergy between vitamin D deficiency, EBV, and certain genetic variations is being studied.6 The roles of smoking, environmental toxins, obesity, diet, Western lifestyle, and the gut microbiome are some of the top areas of clinical, translational, and basic research.7-11 But what about emotional stress? Where does it fit, if anywhere, in the current research paradigm?
Emotional stress and MS—Causality or not?
In the scientific method, several criteria must be proven for an element to be suspected in the etiology of a disease.12 First, the suspect element must be present before the disease starts—i.e., a temporal association. Second, there must be a plausible biological explanation of how the suspect element acts in the disease's causation. Third, other variables that could confound the picture must be controlled for or dismissed. It is clear that no single factor is the cause of MS. By now, MS is agreed upon as a disease caused by multiple factors, some of which remain to be unraveled.9 The term "cause" has been utilized more recently by many authors when referring to EBV in relation to MS development, reasoning that in one study, in a small number of individuals with MS, EBV infection preceded the MS clinical diagnosis.13 Thus, the temporal association was provided. But does MS start at the onset of clinical symptoms?
For Sir Augustus d'Este, the disease may have started years before he visited the Highlands of Scotland, but only at that visit did MS become clinically apparent. So, the emotional trauma may have acted as a "trigger" for an MS flare-up rather than being a "cause" of MS. This might be a more plausible explanation of the association between emotional trauma and MS development. However, MS pathogenesis is complex, and one could argue that the disease starts many years before the first clinical symptoms that lead to diagnosis.
The MS prodrome has been demonstrated by several studies that suggest that MS may start many years before the clinical diagnosis.14 Radiologically isolated syndrome (RIS) further argues that MS may be clinically dormant for years, and clinical symptoms may not appear until later in the disease process.15 One may think that immune attacks on the optic nerves, spinal cord, or areas of the brainstem might be readily symptomatic compared to attacks on other structures of the central nervous system (e.g., periventricular or juxtacortical brain areas) that may be clinically silent. So, while for Sir Augustus d'Este it seemed that the disease started at the time of his visit to the Highlands of Scotland, it is equally plausible that it started years before the first clinical attack. Nevertheless, how could emotional stress play a role in the pathophysiology of MS?
Stress and the Immune System
At times of chronic stress, one may become more susceptible to infections. Reactivation of certain viruses can lead to oral ulcers, increased common cold symptoms, or other illnesses. For example, stress can reactivate herpes simplex type 1 and interestingly, EBV.16,17 In MS, the immune system is dysregulated and has an autoimmune component. The effect of acute emotional stress differs from that of chronic stress.18 Several studies have examined the immune responses to both forms of stress.19-21
Interestingly, acute stress activates cell-mediated immunity, increases immune cell trafficking to areas of injury, and, importantly, increases blood-brain barrier (BBB) permeability by activating resident mast cells in the brain and other areas, including the optic nerves.22,23 Mast cell activation leads to BBB disruption, which is a key early step in the pathogenesis of MS. Thus, it is plausible that the proinflammatory changes associated with acute stress could be implicated in the pathogenesis of MS. This contrasts with chronic stress, which attenuates various immune responses, including suppressing cell-mediated immunity, but also dysregulate the immune system.
One could establish a biological plausibility for stress playing a role in the proinflammatory responses in MS. Whether it is causal or not, scientists can further explore the potential biologic explanations. While studying the association between acute stress and MS development or disease activity is difficult, several groups have examined the potential association. Many studies, however, have limitations due to the difficult nature of studying such an association, especially in quantifying or defining acute stress in general.
A limited number of studies on MS and stress: What do we know? And what are the challenges?
Rare studies have reported a potential association between MS development and stressful life events, while others reported no association.24-26 Also, some studies observed an increase in MS relapses or the development of new magnetic resonance imaging (MRI) lesions following stressful life events or wartime, while others failed to show such an association.26-30 There are few studies directly addressing the potential association between acute emotional stress and MS. The results of published studies are variable, and limitations are numerous. Limitations include the difficulty in measuring acute emotional stress, difficulty in its prediction, and ethical challenges of experimental design and recruiting participants. So, studies have focused on observational aspects, retrospective reviews, and surveys of memories prone to various biases. Rarely was the design of these clinical studies prospective. A few prospective studies reported an association between stressful life events and increased MS relapses and increased number of brain lesions.27,31,32 Rare clinical trials have attempted to test stress reduction strategies and reported on the modest improvement of patient-reported outcomes and, in one study, a modest improvement in new MRI lesions.33-35
Overall, several lines of evidence support a potential association between acute emotional stress and MS. Yet, the association is challenging to study, and future research might focus on stress-mitigation strategies and improving coping mechanisms in persons living with MS. It is important to note that it will be very difficult to design prospective studies to examine the potential association between acute emotional trauma and the development of de novo MS. Such studies will require a large number of participants (e.g., hundreds of thousands), long durations of follow-up (e.g., decades), and ways to classify repeated stressful events. An alternative approach is to ask persons newly diagnosed with MS at the time of initial diagnosis about any temporal association between their first symptom and stressful life events. However, this approach would provide some information on any association between the two, but not on causality of the disease itself.
Conclusion
The potential association between acute emotional stress and MS dates to the times of early descriptions of MS. Yet, research has been very limited and challenging. To date, the potential association remains elusive. Lines of evidence, while with limitations, have provided possible biologic explanations for the relationship between MS symptom onset and acute emotional stress. Although avoiding acute emotional stress is nearly impossible, incorporating global stress-coping strategies in early childhood education and secondary education might theoretically have potential beneficial effects on the subsequent risk of MS development or symptom flare-up, depending on a variety of factors.
But for now, when patients and colleagues ask me, “Can acute emotional stress be a ‘trigger’ for MS symptomology?,” my answer will remain, “Potentially, until proven otherwise.”
Sir Augustus d’Este (1794-1848) described the circumstances preceding his development of neurological symptoms as follows:1 “I travelled from Ramsgate to the Highlands of Scotland for the purpose of passing some days with a Relation for whom I had the affection of a Son. On my arrival I found him dead. Shortly after the funeral I was obliged to have my letters read to me, and their answers written for me, as my eyes were so attacked that when fixed upon minute objects indistinctness of vision was the consequence: Soon after I went to Ireland, and without any thing having been done to my eyes, they completely recovered their strength and distinctness of vision…" He then described a clinical course of relapsing-remitting neurologic symptoms merging into a progressive stage of unrelenting illness, most fitting with what we know today as multiple sclerosis (MS).1 Why did Sir Augustus d'Este connect the event of the unexpected death to the onset of a lifelong neurologic disease?
Jean-Martin Charcot first described MS in a way close to what we know it as today. Charcot considered stress a factor in MS. He linked grief, vexation, and adverse changes in social circumstances to the onset of MS at that time.2 I, as a practicing MS specialist, am surprised neither by Sir Augustus d'Este's diary nor by Charcot's earlier assessments of MS triggers.3 As I write this narrative, I think of the many times I heard from people diagnosed with MS. "It happened to me because of stress" is a statement not estranged from my daily clinical practice
MS as a multifactorial disease
It is tempting to make a case for emotional stress as a cause of MS, but one must remember that MS is a very complex disease with unclear etiologies. MS, a treatable but not yet curable disease, is the interplay between the genetics of the host and numerous environmental factors that exploit a susceptible immune system leading to unrelenting immune dysregulation.4 Recent studies have brought some pieces of this intricate puzzle together. The role of Epstein-Barr virus (EBV) in the pathogenesis of MS is being dissected.5 The possible synergy between vitamin D deficiency, EBV, and certain genetic variations is being studied.6 The roles of smoking, environmental toxins, obesity, diet, Western lifestyle, and the gut microbiome are some of the top areas of clinical, translational, and basic research.7-11 But what about emotional stress? Where does it fit, if anywhere, in the current research paradigm?
Emotional stress and MS—Causality or not?
In the scientific method, several criteria must be proven for an element to be suspected in the etiology of a disease.12 First, the suspect element must be present before the disease starts—i.e., a temporal association. Second, there must be a plausible biological explanation of how the suspect element acts in the disease's causation. Third, other variables that could confound the picture must be controlled for or dismissed. It is clear that no single factor is the cause of MS. By now, MS is agreed upon as a disease caused by multiple factors, some of which remain to be unraveled.9 The term "cause" has been utilized more recently by many authors when referring to EBV in relation to MS development, reasoning that in one study, in a small number of individuals with MS, EBV infection preceded the MS clinical diagnosis.13 Thus, the temporal association was provided. But does MS start at the onset of clinical symptoms?
For Sir Augustus d'Este, the disease may have started years before he visited the Highlands of Scotland, but only at that visit did MS become clinically apparent. So, the emotional trauma may have acted as a "trigger" for an MS flare-up rather than being a "cause" of MS. This might be a more plausible explanation of the association between emotional trauma and MS development. However, MS pathogenesis is complex, and one could argue that the disease starts many years before the first clinical symptoms that lead to diagnosis.
The MS prodrome has been demonstrated by several studies that suggest that MS may start many years before the clinical diagnosis.14 Radiologically isolated syndrome (RIS) further argues that MS may be clinically dormant for years, and clinical symptoms may not appear until later in the disease process.15 One may think that immune attacks on the optic nerves, spinal cord, or areas of the brainstem might be readily symptomatic compared to attacks on other structures of the central nervous system (e.g., periventricular or juxtacortical brain areas) that may be clinically silent. So, while for Sir Augustus d'Este it seemed that the disease started at the time of his visit to the Highlands of Scotland, it is equally plausible that it started years before the first clinical attack. Nevertheless, how could emotional stress play a role in the pathophysiology of MS?
Stress and the Immune System
At times of chronic stress, one may become more susceptible to infections. Reactivation of certain viruses can lead to oral ulcers, increased common cold symptoms, or other illnesses. For example, stress can reactivate herpes simplex type 1 and interestingly, EBV.16,17 In MS, the immune system is dysregulated and has an autoimmune component. The effect of acute emotional stress differs from that of chronic stress.18 Several studies have examined the immune responses to both forms of stress.19-21
Interestingly, acute stress activates cell-mediated immunity, increases immune cell trafficking to areas of injury, and, importantly, increases blood-brain barrier (BBB) permeability by activating resident mast cells in the brain and other areas, including the optic nerves.22,23 Mast cell activation leads to BBB disruption, which is a key early step in the pathogenesis of MS. Thus, it is plausible that the proinflammatory changes associated with acute stress could be implicated in the pathogenesis of MS. This contrasts with chronic stress, which attenuates various immune responses, including suppressing cell-mediated immunity, but also dysregulate the immune system.
One could establish a biological plausibility for stress playing a role in the proinflammatory responses in MS. Whether it is causal or not, scientists can further explore the potential biologic explanations. While studying the association between acute stress and MS development or disease activity is difficult, several groups have examined the potential association. Many studies, however, have limitations due to the difficult nature of studying such an association, especially in quantifying or defining acute stress in general.
A limited number of studies on MS and stress: What do we know? And what are the challenges?
Rare studies have reported a potential association between MS development and stressful life events, while others reported no association.24-26 Also, some studies observed an increase in MS relapses or the development of new magnetic resonance imaging (MRI) lesions following stressful life events or wartime, while others failed to show such an association.26-30 There are few studies directly addressing the potential association between acute emotional stress and MS. The results of published studies are variable, and limitations are numerous. Limitations include the difficulty in measuring acute emotional stress, difficulty in its prediction, and ethical challenges of experimental design and recruiting participants. So, studies have focused on observational aspects, retrospective reviews, and surveys of memories prone to various biases. Rarely was the design of these clinical studies prospective. A few prospective studies reported an association between stressful life events and increased MS relapses and increased number of brain lesions.27,31,32 Rare clinical trials have attempted to test stress reduction strategies and reported on the modest improvement of patient-reported outcomes and, in one study, a modest improvement in new MRI lesions.33-35
Overall, several lines of evidence support a potential association between acute emotional stress and MS. Yet, the association is challenging to study, and future research might focus on stress-mitigation strategies and improving coping mechanisms in persons living with MS. It is important to note that it will be very difficult to design prospective studies to examine the potential association between acute emotional trauma and the development of de novo MS. Such studies will require a large number of participants (e.g., hundreds of thousands), long durations of follow-up (e.g., decades), and ways to classify repeated stressful events. An alternative approach is to ask persons newly diagnosed with MS at the time of initial diagnosis about any temporal association between their first symptom and stressful life events. However, this approach would provide some information on any association between the two, but not on causality of the disease itself.
Conclusion
The potential association between acute emotional stress and MS dates to the times of early descriptions of MS. Yet, research has been very limited and challenging. To date, the potential association remains elusive. Lines of evidence, while with limitations, have provided possible biologic explanations for the relationship between MS symptom onset and acute emotional stress. Although avoiding acute emotional stress is nearly impossible, incorporating global stress-coping strategies in early childhood education and secondary education might theoretically have potential beneficial effects on the subsequent risk of MS development or symptom flare-up, depending on a variety of factors.
But for now, when patients and colleagues ask me, “Can acute emotional stress be a ‘trigger’ for MS symptomology?,” my answer will remain, “Potentially, until proven otherwise.”
- Firth D. The case of Augustus d'Este (1794-1848): the first account of disseminated sclerosis: (section of the History of Medicine). Proc R Soc Med. 1941;34(7):381-384.
- Lectures on the diseases of the nervous system. Br Foreign Med Chir Rev. 1877;60(119):180-181.
- Obeidat, A, Cope T. Stressful life events and multiple sclerosis: a call for re-evaluation. Paper presented at: Fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers; May 13, 2013; Orlando, FL.
- Waubant E, Lucas R, Mowry E, et al. Environmental and genetic risk factors for MS: an integrated review. Ann Clin Transl Neurol. 2019;6(9):1905-1922. doi:10.1002/acn3.50862
- Soldan SS, Lieberman PM. Epstein-Barr virus and multiple sclerosis. Nat Rev Microbiol. 2022;1-14. doi:10.1038/s41579-022-00770-5
- Marcucci SB, Obeidat AZ. EBNA1, EBNA2, and EBNA3 link Epstein-Barr virus and hypovitaminosis D in multiple sclerosis pathogenesis. J Neuroimmunol. 2020;339:57711 doi:10.1016/j.jneuroim.2019.577116
- Alfredsson L, Olsson T. Lifestyle and environmental factors in multiple sclerosis. Cold Spring Harb Perspect Med. 2019;9(4):a028944. doi:10.1101/cshperspect.a028944
- Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622-1636. doi:10.1016/S0140-6736(18)30481-1
- Dobson R, Giovannoni G. Multiple sclerosis – a review. Eur J Neurol. 2019;26(1):27-40. doi:10.1111/ene.13819
- Arneth B. Multiple sclerosis and smoking. Am J Med. 2020;133(7):783-788. doi:1016/j.amjmed.2020.03.008
- Correale J, Hohlfeld R, Baranzini SE. The role of the gut microbiota in multiple sclerosis. Nat Rev Neurol. 2022;18(9):544-558. doi:10.1038/s41582-022-00697-8
- Gianicolo EAL, Eichler M, Muensterer O, Strauch K, Blettner M. Methods for evaluating causality in observational studies. Dtsch Arztebl Int. 2020;116(7):101-107. doi:10.3238/arztebl.2020.0101
- Bjornevik K, Cortese M, Healy BC, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. 2022;375(6578):296-301. doi:10.1126/science.abj8222
- Makhani N, Tremlett H. The multiple sclerosis prodrome. Nat Rev Neurol. 2021;17(8):515-521. doi:10.1038/s41582-021-00519-3
- Hosseiny M, Newsome SD, Yousem DM. Radiologically isolated syndrome: a review for neuroradiologists. AJNR Am J Neuroradiol. 2020;41(9):1542-1549. doi:10.3174/ajnr.A6649
- Padgett DA, Sheridan JF, Dorne J, Berntson GG, Candelora J, Glaser R. Social stress and the reactivation of latent herpes simplex virus type 1 [published correction appears in Proc Natl Acad Sci U S A. 1998;95(20):12070]. Proc Natl Acad Sci U S A. 1998;95(12):7231-7235. doi:10.1073/pnas.95.12.7231
- Glaser R, Pearson GR, Jones JF, et al. Stress-related activation of Epstein-Barr virus. Brain Behav Immun. 1991;5(2):219-232. doi:10.1016/0889-1591(91)90018-6
- Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16(5):300-317. doi:10.1159/000216188
- Musazzi L, Tornese P, Sala N, Popoli M. Acute or chronic? A stressful question. Trends Neurosci. 2017;40(9):525-535. doi:10.1016/j.tins.2017.07.002
- Dhabhar FS, McEwen BS. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: a potential role for leukocyte trafficking. Brain Behav Immun. 1997;11(4):286-306. doi:10.1006/brbi.1997.0508
- Maydych V, Claus M, Dychus N, et al. Impact of chronic and acute academic stress on lymphocyte subsets and monocyte function. PLoS One. 2017;12(11):e0188108. Published 2017 Nov 16. doi:10.1371/journal.pone.0188108
- Esposito P, Gheorghe D, Kandere K, et al. Acute stress increases permeability of the blood-brain-barrier through activation of brain mast cells. Brain Res. 2001;888(1):117-127. doi:10.1016/s0006-8993(00)03026-2
- Kempuraj D, Mentor S, Thangavel R, et al. Mast cells in stress, pain, blood-brain barrier, neuroinflammation and Alzheimer's disease. Front Cell Neurosci. 2019;13:54. doi:10.3389/fncel.2019.00054
- Karagkouni A, Alevizos M, Theoharides TC. Effect of stress on brain inflammation and multiple sclerosis. Autoimmun Rev. 2013;12(10):947-953. doi:10.1016/j.autrev.2013.02.006
- Briones-Buixassa L, Milà R, Mª Aragonès J, Bufill E, Olaya B, Arrufat FX. Stress and multiple sclerosis: a systematic review considering potential moderating and mediating factors and methods of assessing stress. Health Psychol Open. 2015;2(2):2055102915612271. doi:10.1177/2055102915612271
- Riise T, Mohr DC, Munger KL, Rich-Edwards JW, Kawachi I, Ascherio A. Stress and the risk of multiple sclerosis. Neurology. 2011;76(22):1866-1871. doi:10.1212/WNL.0b013e31821d74c5
- Burns MN, Nawacki E, Kwasny MJ, Pelletier D, Mohr DC. Do positive or negative stressful events predict the development of new brain lesions in people with multiple sclerosis? Psychol Med. 2014;44(2):349-359. doi:10.1017/S0033291713000755
- Mohr DC, Goodkin DE, Bacchetti P, et al. Psychological stress and the subsequent appearance of new brain MRI lesions in MS. Neurology. 2000;55(1):55-61. doi:10.1212/wnl.55.1.55
- Yamout B, Itani S, Hourany R, Sibaii AM, Yaghi S. The effect of war stress on multiple sclerosis exacerbations and radiological disease activity. J Neurol Sci. 2010;288(1-2):42-44. doi:10.1016/j.jns.2009.10.012
- Artemiadis AK, Anagnostouli MC, Alexopoulos EC. Stress as a risk factor for multiple sclerosis onset or relapse: a systematic review. Neuroepidemiology. 2011;36(2):109-120. doi:10.1159/000323953
- Brown RF, Tennant CC, Sharrock M, Hodgkinson S, Dunn SM, Pollard JD. Relationship between stress and relapse in multiple sclerosis: Part I. Important features. Mult Scler. 2006;12(4):453-464. doi:10.1191/1352458506ms1295oa
- Buljevac D, Hop WCJ, Reedeker W, et al. Self-reported stressful life events and exacerbations in multiple sclerosis: prospective study. BMJ. 2003;327(7416):646. doi:10.1136/bmj.327.7416.646
- Senders A, Hanes D, Bourdette D, Carson K, Marshall LM, Shinto L. Impact of mindfulness-based stress reduction for people with multiple sclerosis at 8 weeks and 12 months: A randomized clinical trial. Mult Scler. 2019;25(8):1178-1188. doi:10.1177/1352458518786650
- Morrow SA, Riccio P, Vording N, Rosehart H, Casserly C, MacDougall A. A mindfulness group intervention in newly diagnosed persons with multiple sclerosis: A pilot study. Mult Scler Relat Disord. 2021;52:103016. doi:10.1016/j.msard.2021.103016
- Mohr DC, Lovera J, Brown T, et al. A randomized trial of stress management for the prevention of new brain lesions in MS. Neurology. 2012;79(5):412-419. doi:10.1212/WNL.0b013e3182616ff9
- Firth D. The case of Augustus d'Este (1794-1848): the first account of disseminated sclerosis: (section of the History of Medicine). Proc R Soc Med. 1941;34(7):381-384.
- Lectures on the diseases of the nervous system. Br Foreign Med Chir Rev. 1877;60(119):180-181.
- Obeidat, A, Cope T. Stressful life events and multiple sclerosis: a call for re-evaluation. Paper presented at: Fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers; May 13, 2013; Orlando, FL.
- Waubant E, Lucas R, Mowry E, et al. Environmental and genetic risk factors for MS: an integrated review. Ann Clin Transl Neurol. 2019;6(9):1905-1922. doi:10.1002/acn3.50862
- Soldan SS, Lieberman PM. Epstein-Barr virus and multiple sclerosis. Nat Rev Microbiol. 2022;1-14. doi:10.1038/s41579-022-00770-5
- Marcucci SB, Obeidat AZ. EBNA1, EBNA2, and EBNA3 link Epstein-Barr virus and hypovitaminosis D in multiple sclerosis pathogenesis. J Neuroimmunol. 2020;339:57711 doi:10.1016/j.jneuroim.2019.577116
- Alfredsson L, Olsson T. Lifestyle and environmental factors in multiple sclerosis. Cold Spring Harb Perspect Med. 2019;9(4):a028944. doi:10.1101/cshperspect.a028944
- Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622-1636. doi:10.1016/S0140-6736(18)30481-1
- Dobson R, Giovannoni G. Multiple sclerosis – a review. Eur J Neurol. 2019;26(1):27-40. doi:10.1111/ene.13819
- Arneth B. Multiple sclerosis and smoking. Am J Med. 2020;133(7):783-788. doi:1016/j.amjmed.2020.03.008
- Correale J, Hohlfeld R, Baranzini SE. The role of the gut microbiota in multiple sclerosis. Nat Rev Neurol. 2022;18(9):544-558. doi:10.1038/s41582-022-00697-8
- Gianicolo EAL, Eichler M, Muensterer O, Strauch K, Blettner M. Methods for evaluating causality in observational studies. Dtsch Arztebl Int. 2020;116(7):101-107. doi:10.3238/arztebl.2020.0101
- Bjornevik K, Cortese M, Healy BC, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. 2022;375(6578):296-301. doi:10.1126/science.abj8222
- Makhani N, Tremlett H. The multiple sclerosis prodrome. Nat Rev Neurol. 2021;17(8):515-521. doi:10.1038/s41582-021-00519-3
- Hosseiny M, Newsome SD, Yousem DM. Radiologically isolated syndrome: a review for neuroradiologists. AJNR Am J Neuroradiol. 2020;41(9):1542-1549. doi:10.3174/ajnr.A6649
- Padgett DA, Sheridan JF, Dorne J, Berntson GG, Candelora J, Glaser R. Social stress and the reactivation of latent herpes simplex virus type 1 [published correction appears in Proc Natl Acad Sci U S A. 1998;95(20):12070]. Proc Natl Acad Sci U S A. 1998;95(12):7231-7235. doi:10.1073/pnas.95.12.7231
- Glaser R, Pearson GR, Jones JF, et al. Stress-related activation of Epstein-Barr virus. Brain Behav Immun. 1991;5(2):219-232. doi:10.1016/0889-1591(91)90018-6
- Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16(5):300-317. doi:10.1159/000216188
- Musazzi L, Tornese P, Sala N, Popoli M. Acute or chronic? A stressful question. Trends Neurosci. 2017;40(9):525-535. doi:10.1016/j.tins.2017.07.002
- Dhabhar FS, McEwen BS. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: a potential role for leukocyte trafficking. Brain Behav Immun. 1997;11(4):286-306. doi:10.1006/brbi.1997.0508
- Maydych V, Claus M, Dychus N, et al. Impact of chronic and acute academic stress on lymphocyte subsets and monocyte function. PLoS One. 2017;12(11):e0188108. Published 2017 Nov 16. doi:10.1371/journal.pone.0188108
- Esposito P, Gheorghe D, Kandere K, et al. Acute stress increases permeability of the blood-brain-barrier through activation of brain mast cells. Brain Res. 2001;888(1):117-127. doi:10.1016/s0006-8993(00)03026-2
- Kempuraj D, Mentor S, Thangavel R, et al. Mast cells in stress, pain, blood-brain barrier, neuroinflammation and Alzheimer's disease. Front Cell Neurosci. 2019;13:54. doi:10.3389/fncel.2019.00054
- Karagkouni A, Alevizos M, Theoharides TC. Effect of stress on brain inflammation and multiple sclerosis. Autoimmun Rev. 2013;12(10):947-953. doi:10.1016/j.autrev.2013.02.006
- Briones-Buixassa L, Milà R, Mª Aragonès J, Bufill E, Olaya B, Arrufat FX. Stress and multiple sclerosis: a systematic review considering potential moderating and mediating factors and methods of assessing stress. Health Psychol Open. 2015;2(2):2055102915612271. doi:10.1177/2055102915612271
- Riise T, Mohr DC, Munger KL, Rich-Edwards JW, Kawachi I, Ascherio A. Stress and the risk of multiple sclerosis. Neurology. 2011;76(22):1866-1871. doi:10.1212/WNL.0b013e31821d74c5
- Burns MN, Nawacki E, Kwasny MJ, Pelletier D, Mohr DC. Do positive or negative stressful events predict the development of new brain lesions in people with multiple sclerosis? Psychol Med. 2014;44(2):349-359. doi:10.1017/S0033291713000755
- Mohr DC, Goodkin DE, Bacchetti P, et al. Psychological stress and the subsequent appearance of new brain MRI lesions in MS. Neurology. 2000;55(1):55-61. doi:10.1212/wnl.55.1.55
- Yamout B, Itani S, Hourany R, Sibaii AM, Yaghi S. The effect of war stress on multiple sclerosis exacerbations and radiological disease activity. J Neurol Sci. 2010;288(1-2):42-44. doi:10.1016/j.jns.2009.10.012
- Artemiadis AK, Anagnostouli MC, Alexopoulos EC. Stress as a risk factor for multiple sclerosis onset or relapse: a systematic review. Neuroepidemiology. 2011;36(2):109-120. doi:10.1159/000323953
- Brown RF, Tennant CC, Sharrock M, Hodgkinson S, Dunn SM, Pollard JD. Relationship between stress and relapse in multiple sclerosis: Part I. Important features. Mult Scler. 2006;12(4):453-464. doi:10.1191/1352458506ms1295oa
- Buljevac D, Hop WCJ, Reedeker W, et al. Self-reported stressful life events and exacerbations in multiple sclerosis: prospective study. BMJ. 2003;327(7416):646. doi:10.1136/bmj.327.7416.646
- Senders A, Hanes D, Bourdette D, Carson K, Marshall LM, Shinto L. Impact of mindfulness-based stress reduction for people with multiple sclerosis at 8 weeks and 12 months: A randomized clinical trial. Mult Scler. 2019;25(8):1178-1188. doi:10.1177/1352458518786650
- Morrow SA, Riccio P, Vording N, Rosehart H, Casserly C, MacDougall A. A mindfulness group intervention in newly diagnosed persons with multiple sclerosis: A pilot study. Mult Scler Relat Disord. 2021;52:103016. doi:10.1016/j.msard.2021.103016
- Mohr DC, Lovera J, Brown T, et al. A randomized trial of stress management for the prevention of new brain lesions in MS. Neurology. 2012;79(5):412-419. doi:10.1212/WNL.0b013e3182616ff9