User login
Combo may overcome drug resistance in ALL

Credit: Linda Bartlett
Adding the alkylating agent cyclophosphamide to treatment with a monoclonal antibody (mAb) can overcome drug resistance in mice with acute lymphoblastic leukemia (ALL), researchers have reported in Cell.
mAbs such as rituximab and alemtuzumab are designed to bind to proteins found on the surfaces of tumor cells.
Once the mAbs flag the tumor cells, macrophages destroy them. But the drugs have little effect on tumor cells that hide out in the bone marrow.
Experiments in mice with B-cell ALL revealed that cyclophosphamide stimulates the immune response in bone marrow, eliminating the reservoir of cancer cells that can produce new tumors after treatment with a mAb.
Finding hidden ALL cells
Michael Hemann, PhD, of MIT’s Koch Institute for Integrative Cancer Research in Cambridge, Massachusetts, and his colleagues began this research by administering alemtuzumab to the mice.
The drug successfully cleared most ALL cells, but some remained hidden in the bone marrow, which has been identified as a site of drug resistance in many cancers.
The researchers found that, within the bone marrow, alemtuzumab successfully binds to ALL cells. But macrophages do not attack the cells due to the presence of lipid compounds called prostaglandins, which repress macrophage activity.
Scientists believe the bone marrow naturally produces prostaglandins to help protect the immune cells maturing there. Tumor cells that reach the bone marrow can exploit this protective environment to aid their own survival.
The finding is an important contribution to scientists’ understanding of how mAbs act against ALL, according to Ravi Majeti, MD, PhD, of Stanford University in California, who was not involved in this research.
“There clearly has been a lack of understanding about why antibody therapies have been relatively unsuccessful as monotherapies,” Dr Majeti said.
Tricking the immune system
Dr Hemann and his colleagues then tested a variety of anticancer drugs in combination with alemtuzumab. And they discovered that cyclophosphamide can “rewire” the bone marrow microenvironment to make it much more receptive to macrophages, allowing them to destroy the tumor cells hiding there.
“After you treat with cyclophosphamide, you get this flux of macrophages into the bone marrow, and these macrophages are now active and very capable of consuming the targeted tumor cells,” Dr Hemann said.
“Essentially, we are tricking the immune system to suddenly recognize an entity that it wouldn’t typically recognize and aggressively go after antibody-bound tumor cells.”
Following treatment with this combination, the mice survived and remained free of ALL for the duration of the study, which was about 18 months.
However, the researchers found that timing of drug delivery was critical. Alemtuzumab and cyclophosphamide must be administered together so that cyclophosphamide can create the right type of environment for macrophages to become activated in the bone marrow.
The team also obtained good results by combining cyclophosphamide with rituximab.
They now plan to test cyclophosphamide with other mAbs and begin testing the alemtuzumab-cyclophosphamide combination in patients. ![]()
Credit: Linda Bartlett
Adding the alkylating agent cyclophosphamide to treatment with a monoclonal antibody (mAb) can overcome drug resistance in mice with acute lymphoblastic leukemia (ALL), researchers have reported in Cell.
mAbs such as rituximab and alemtuzumab are designed to bind to proteins found on the surfaces of tumor cells.
Once the mAbs flag the tumor cells, macrophages destroy them. But the drugs have little effect on tumor cells that hide out in the bone marrow.
Experiments in mice with B-cell ALL revealed that cyclophosphamide stimulates the immune response in bone marrow, eliminating the reservoir of cancer cells that can produce new tumors after treatment with a mAb.
Finding hidden ALL cells
Michael Hemann, PhD, of MIT’s Koch Institute for Integrative Cancer Research in Cambridge, Massachusetts, and his colleagues began this research by administering alemtuzumab to the mice.
The drug successfully cleared most ALL cells, but some remained hidden in the bone marrow, which has been identified as a site of drug resistance in many cancers.
The researchers found that, within the bone marrow, alemtuzumab successfully binds to ALL cells. But macrophages do not attack the cells due to the presence of lipid compounds called prostaglandins, which repress macrophage activity.
Scientists believe the bone marrow naturally produces prostaglandins to help protect the immune cells maturing there. Tumor cells that reach the bone marrow can exploit this protective environment to aid their own survival.
The finding is an important contribution to scientists’ understanding of how mAbs act against ALL, according to Ravi Majeti, MD, PhD, of Stanford University in California, who was not involved in this research.
“There clearly has been a lack of understanding about why antibody therapies have been relatively unsuccessful as monotherapies,” Dr Majeti said.
Tricking the immune system
Dr Hemann and his colleagues then tested a variety of anticancer drugs in combination with alemtuzumab. And they discovered that cyclophosphamide can “rewire” the bone marrow microenvironment to make it much more receptive to macrophages, allowing them to destroy the tumor cells hiding there.
“After you treat with cyclophosphamide, you get this flux of macrophages into the bone marrow, and these macrophages are now active and very capable of consuming the targeted tumor cells,” Dr Hemann said.
“Essentially, we are tricking the immune system to suddenly recognize an entity that it wouldn’t typically recognize and aggressively go after antibody-bound tumor cells.”
Following treatment with this combination, the mice survived and remained free of ALL for the duration of the study, which was about 18 months.
However, the researchers found that timing of drug delivery was critical. Alemtuzumab and cyclophosphamide must be administered together so that cyclophosphamide can create the right type of environment for macrophages to become activated in the bone marrow.
The team also obtained good results by combining cyclophosphamide with rituximab.
They now plan to test cyclophosphamide with other mAbs and begin testing the alemtuzumab-cyclophosphamide combination in patients. ![]()
Credit: Linda Bartlett
Adding the alkylating agent cyclophosphamide to treatment with a monoclonal antibody (mAb) can overcome drug resistance in mice with acute lymphoblastic leukemia (ALL), researchers have reported in Cell.
mAbs such as rituximab and alemtuzumab are designed to bind to proteins found on the surfaces of tumor cells.
Once the mAbs flag the tumor cells, macrophages destroy them. But the drugs have little effect on tumor cells that hide out in the bone marrow.
Experiments in mice with B-cell ALL revealed that cyclophosphamide stimulates the immune response in bone marrow, eliminating the reservoir of cancer cells that can produce new tumors after treatment with a mAb.
Finding hidden ALL cells
Michael Hemann, PhD, of MIT’s Koch Institute for Integrative Cancer Research in Cambridge, Massachusetts, and his colleagues began this research by administering alemtuzumab to the mice.
The drug successfully cleared most ALL cells, but some remained hidden in the bone marrow, which has been identified as a site of drug resistance in many cancers.
The researchers found that, within the bone marrow, alemtuzumab successfully binds to ALL cells. But macrophages do not attack the cells due to the presence of lipid compounds called prostaglandins, which repress macrophage activity.
Scientists believe the bone marrow naturally produces prostaglandins to help protect the immune cells maturing there. Tumor cells that reach the bone marrow can exploit this protective environment to aid their own survival.
The finding is an important contribution to scientists’ understanding of how mAbs act against ALL, according to Ravi Majeti, MD, PhD, of Stanford University in California, who was not involved in this research.
“There clearly has been a lack of understanding about why antibody therapies have been relatively unsuccessful as monotherapies,” Dr Majeti said.
Tricking the immune system
Dr Hemann and his colleagues then tested a variety of anticancer drugs in combination with alemtuzumab. And they discovered that cyclophosphamide can “rewire” the bone marrow microenvironment to make it much more receptive to macrophages, allowing them to destroy the tumor cells hiding there.
“After you treat with cyclophosphamide, you get this flux of macrophages into the bone marrow, and these macrophages are now active and very capable of consuming the targeted tumor cells,” Dr Hemann said.
“Essentially, we are tricking the immune system to suddenly recognize an entity that it wouldn’t typically recognize and aggressively go after antibody-bound tumor cells.”
Following treatment with this combination, the mice survived and remained free of ALL for the duration of the study, which was about 18 months.
However, the researchers found that timing of drug delivery was critical. Alemtuzumab and cyclophosphamide must be administered together so that cyclophosphamide can create the right type of environment for macrophages to become activated in the bone marrow.
The team also obtained good results by combining cyclophosphamide with rituximab.
They now plan to test cyclophosphamide with other mAbs and begin testing the alemtuzumab-cyclophosphamide combination in patients. ![]()
Experts offer guidance for preventing drug shortages

with chemotherapy drugs
Credit: Bill Branson
A group of healthcare experts has created a “blueprint” for managing and preventing drug shortages.
They devised 6 recommendations for preempting shortages of pediatric oncology drugs, but the suggestions can be applied to other drugs as well.
Some of the recommendations would represent new norms for healthcare practice, such as sharing scarce drugs among healthcare institutions and not giving preferential access to patients participating in clinical trials.
The suggestions appear in a consensus statement published in Pediatrics.
“Although our recommendations were developed with pediatric oncology in mind, and serve as a blueprint for preventing children with cancer from lacking access to essential life-saving medications, we believe that they apply more broadly across medicine to include pediatrics and adult medicine in general,” said statement author Yoram Unguru, MD, of the Johns Hopkins Berman Institute of Bioethics in Baltimore, Maryland.
He and his coauthors developed the following recommendations:
- Support national polices—and develop new measures—to prevent drug shortages
- Use drug supplies efficiently to reduce the likelihood of shortages
- Give equal priority to evidence-based uses of drugs, whether they occur within or outside clinical trials
- Develop an improved clearinghouse for sharing drug shortage information
- Explore the sharing of drug supplies among institutions
- Develop strategies to engage stakeholders in the management of drug shortages.
For each recommendation, the authors included potential barriers to its implementation. A centralized information source of drug supply information, for instance, comes with the risk that such information will encourage hoarding of existing supplies. This includes “gray market” suppliers that sell scarce drugs for inflated prices.
The authors also pointed to the marketplace economy as an obstacle to implementing their recommendations and preventing drug shortages. Drug manufacturers do not like to disclose manufacturing problems that lead to shortages, nor are competitive healthcare institutions accustomed to cooperating to share resources.
Nonetheless, the authors called for an exploration of “ways to facilitate inter-institutional and inter-state transfer of drugs, especially during shortages,” as well as the ethical obligation to patients inside vs outside a healthcare institution when there is a drug shortage.
“The reasons for drug shortages are complex, but we must not lose sight of the fact that, without access to these life-saving drugs, children and adults with cancer will almost certainly die,” Dr Unguru said. “It is untenable for this situation to continue any longer. We have a clear moral obligation to act to address this critical issue.” ![]()
with chemotherapy drugs
Credit: Bill Branson
A group of healthcare experts has created a “blueprint” for managing and preventing drug shortages.
They devised 6 recommendations for preempting shortages of pediatric oncology drugs, but the suggestions can be applied to other drugs as well.
Some of the recommendations would represent new norms for healthcare practice, such as sharing scarce drugs among healthcare institutions and not giving preferential access to patients participating in clinical trials.
The suggestions appear in a consensus statement published in Pediatrics.
“Although our recommendations were developed with pediatric oncology in mind, and serve as a blueprint for preventing children with cancer from lacking access to essential life-saving medications, we believe that they apply more broadly across medicine to include pediatrics and adult medicine in general,” said statement author Yoram Unguru, MD, of the Johns Hopkins Berman Institute of Bioethics in Baltimore, Maryland.
He and his coauthors developed the following recommendations:
- Support national polices—and develop new measures—to prevent drug shortages
- Use drug supplies efficiently to reduce the likelihood of shortages
- Give equal priority to evidence-based uses of drugs, whether they occur within or outside clinical trials
- Develop an improved clearinghouse for sharing drug shortage information
- Explore the sharing of drug supplies among institutions
- Develop strategies to engage stakeholders in the management of drug shortages.
For each recommendation, the authors included potential barriers to its implementation. A centralized information source of drug supply information, for instance, comes with the risk that such information will encourage hoarding of existing supplies. This includes “gray market” suppliers that sell scarce drugs for inflated prices.
The authors also pointed to the marketplace economy as an obstacle to implementing their recommendations and preventing drug shortages. Drug manufacturers do not like to disclose manufacturing problems that lead to shortages, nor are competitive healthcare institutions accustomed to cooperating to share resources.
Nonetheless, the authors called for an exploration of “ways to facilitate inter-institutional and inter-state transfer of drugs, especially during shortages,” as well as the ethical obligation to patients inside vs outside a healthcare institution when there is a drug shortage.
“The reasons for drug shortages are complex, but we must not lose sight of the fact that, without access to these life-saving drugs, children and adults with cancer will almost certainly die,” Dr Unguru said. “It is untenable for this situation to continue any longer. We have a clear moral obligation to act to address this critical issue.” ![]()
with chemotherapy drugs
Credit: Bill Branson
A group of healthcare experts has created a “blueprint” for managing and preventing drug shortages.
They devised 6 recommendations for preempting shortages of pediatric oncology drugs, but the suggestions can be applied to other drugs as well.
Some of the recommendations would represent new norms for healthcare practice, such as sharing scarce drugs among healthcare institutions and not giving preferential access to patients participating in clinical trials.
The suggestions appear in a consensus statement published in Pediatrics.
“Although our recommendations were developed with pediatric oncology in mind, and serve as a blueprint for preventing children with cancer from lacking access to essential life-saving medications, we believe that they apply more broadly across medicine to include pediatrics and adult medicine in general,” said statement author Yoram Unguru, MD, of the Johns Hopkins Berman Institute of Bioethics in Baltimore, Maryland.
He and his coauthors developed the following recommendations:
- Support national polices—and develop new measures—to prevent drug shortages
- Use drug supplies efficiently to reduce the likelihood of shortages
- Give equal priority to evidence-based uses of drugs, whether they occur within or outside clinical trials
- Develop an improved clearinghouse for sharing drug shortage information
- Explore the sharing of drug supplies among institutions
- Develop strategies to engage stakeholders in the management of drug shortages.
For each recommendation, the authors included potential barriers to its implementation. A centralized information source of drug supply information, for instance, comes with the risk that such information will encourage hoarding of existing supplies. This includes “gray market” suppliers that sell scarce drugs for inflated prices.
The authors also pointed to the marketplace economy as an obstacle to implementing their recommendations and preventing drug shortages. Drug manufacturers do not like to disclose manufacturing problems that lead to shortages, nor are competitive healthcare institutions accustomed to cooperating to share resources.
Nonetheless, the authors called for an exploration of “ways to facilitate inter-institutional and inter-state transfer of drugs, especially during shortages,” as well as the ethical obligation to patients inside vs outside a healthcare institution when there is a drug shortage.
“The reasons for drug shortages are complex, but we must not lose sight of the fact that, without access to these life-saving drugs, children and adults with cancer will almost certainly die,” Dr Unguru said. “It is untenable for this situation to continue any longer. We have a clear moral obligation to act to address this critical issue.” ![]()
Antibody prevents thrombosis in rabbits

oxygenator membranes
Credit: Kjell Hultenby
A newly developed antibody can prevent thrombosis without increasing the risk of bleeding in rabbits, according to research published in Science Translational Medicine.
The antibody, called 3F7, works by blocking a protein that’s active in the coagulation system factor XII (FXII).
Scientists have long known that humans deficient in FXII do not bleed excessively.
And in 2005, Thomas Renné, MD, PhD, of the Karolinska Institutet in Stockholm, Sweden, and his colleagues discovered that mice lacking FXII could have neither a stroke nor a pulmonary embolism, even though they had normal bleeding patterns.
“Since then, our goal has been to find an effective way to block FXII,” Dr Renné said. “Now, we have developed an antibody that blocks FXII in human blood, mice, and rabbits. This provides protection against thrombosis without increasing the risk of bleeding.”
The researchers tested 3F7 in rabbits during extracorporeal membrane oxygenation (ECMO) treatment. ECMO is an advanced heart-lung machine used in life-threatening conditions, but contact with the plastic tubing causes the blood to clot.
The incidence of thrombosis in rabbits on ECMO receiving 3F7 was as low as in rabbits receiving heparin. There were significantly fewer fibrin clots in the oxygenators of 3F7-treated rabbits and heparin-treated rabbits than in saline-treated controls—4 ± 3%, 8 ± 6%, and 100 ± 19%, respectively.
However, bleeding was more severe in the heparin-treated rabbits than in 3F7-treated rabbits and controls. Incision-provoked skin bleeding time was more than 600 seconds in the heparin group, 160 ± 40 s in the 3F7 group, and 130 ± 15 s in controls.
Cuticle bleeding time was more than 600 s in the heparin group, 165 ± 40 s in the 3F7 group, and 120 ± 30 s in controls. The mean blood loss from cuticle wounds was 5.1 ± 1.1 mL/10 min, 0.3 ± 0.1 mL/10 min, and 0.2 ± 0.05 mL/10 min, respectively.
“Blocking FXII appears to be an effective strategy against thrombus formation, and we have shown this in experiments on rabbits in a clinically relevant context,” Dr Renné said in conclusion.
“We plan to test the antibody in a phase 1 study. It is possible that the antibody also blocks inflammation mediated by FXII, an interesting area for future studies.” ![]()
oxygenator membranes
Credit: Kjell Hultenby
A newly developed antibody can prevent thrombosis without increasing the risk of bleeding in rabbits, according to research published in Science Translational Medicine.
The antibody, called 3F7, works by blocking a protein that’s active in the coagulation system factor XII (FXII).
Scientists have long known that humans deficient in FXII do not bleed excessively.
And in 2005, Thomas Renné, MD, PhD, of the Karolinska Institutet in Stockholm, Sweden, and his colleagues discovered that mice lacking FXII could have neither a stroke nor a pulmonary embolism, even though they had normal bleeding patterns.
“Since then, our goal has been to find an effective way to block FXII,” Dr Renné said. “Now, we have developed an antibody that blocks FXII in human blood, mice, and rabbits. This provides protection against thrombosis without increasing the risk of bleeding.”
The researchers tested 3F7 in rabbits during extracorporeal membrane oxygenation (ECMO) treatment. ECMO is an advanced heart-lung machine used in life-threatening conditions, but contact with the plastic tubing causes the blood to clot.
The incidence of thrombosis in rabbits on ECMO receiving 3F7 was as low as in rabbits receiving heparin. There were significantly fewer fibrin clots in the oxygenators of 3F7-treated rabbits and heparin-treated rabbits than in saline-treated controls—4 ± 3%, 8 ± 6%, and 100 ± 19%, respectively.
However, bleeding was more severe in the heparin-treated rabbits than in 3F7-treated rabbits and controls. Incision-provoked skin bleeding time was more than 600 seconds in the heparin group, 160 ± 40 s in the 3F7 group, and 130 ± 15 s in controls.
Cuticle bleeding time was more than 600 s in the heparin group, 165 ± 40 s in the 3F7 group, and 120 ± 30 s in controls. The mean blood loss from cuticle wounds was 5.1 ± 1.1 mL/10 min, 0.3 ± 0.1 mL/10 min, and 0.2 ± 0.05 mL/10 min, respectively.
“Blocking FXII appears to be an effective strategy against thrombus formation, and we have shown this in experiments on rabbits in a clinically relevant context,” Dr Renné said in conclusion.
“We plan to test the antibody in a phase 1 study. It is possible that the antibody also blocks inflammation mediated by FXII, an interesting area for future studies.” ![]()
oxygenator membranes
Credit: Kjell Hultenby
A newly developed antibody can prevent thrombosis without increasing the risk of bleeding in rabbits, according to research published in Science Translational Medicine.
The antibody, called 3F7, works by blocking a protein that’s active in the coagulation system factor XII (FXII).
Scientists have long known that humans deficient in FXII do not bleed excessively.
And in 2005, Thomas Renné, MD, PhD, of the Karolinska Institutet in Stockholm, Sweden, and his colleagues discovered that mice lacking FXII could have neither a stroke nor a pulmonary embolism, even though they had normal bleeding patterns.
“Since then, our goal has been to find an effective way to block FXII,” Dr Renné said. “Now, we have developed an antibody that blocks FXII in human blood, mice, and rabbits. This provides protection against thrombosis without increasing the risk of bleeding.”
The researchers tested 3F7 in rabbits during extracorporeal membrane oxygenation (ECMO) treatment. ECMO is an advanced heart-lung machine used in life-threatening conditions, but contact with the plastic tubing causes the blood to clot.
The incidence of thrombosis in rabbits on ECMO receiving 3F7 was as low as in rabbits receiving heparin. There were significantly fewer fibrin clots in the oxygenators of 3F7-treated rabbits and heparin-treated rabbits than in saline-treated controls—4 ± 3%, 8 ± 6%, and 100 ± 19%, respectively.
However, bleeding was more severe in the heparin-treated rabbits than in 3F7-treated rabbits and controls. Incision-provoked skin bleeding time was more than 600 seconds in the heparin group, 160 ± 40 s in the 3F7 group, and 130 ± 15 s in controls.
Cuticle bleeding time was more than 600 s in the heparin group, 165 ± 40 s in the 3F7 group, and 120 ± 30 s in controls. The mean blood loss from cuticle wounds was 5.1 ± 1.1 mL/10 min, 0.3 ± 0.1 mL/10 min, and 0.2 ± 0.05 mL/10 min, respectively.
“Blocking FXII appears to be an effective strategy against thrombus formation, and we have shown this in experiments on rabbits in a clinically relevant context,” Dr Renné said in conclusion.
“We plan to test the antibody in a phase 1 study. It is possible that the antibody also blocks inflammation mediated by FXII, an interesting area for future studies.” ![]()
Minnesota-based Hospital Readmissions Reduction Campaign Earns Prestigious Award
How's this for a quality-improvement success story? In 2011, three Minnesota-based institutions—the Institute for Clinical Systems Improvement (ICSI), the Minnesota Hospital Association, and Stratis Health—launched the Reducing Avoidable Readmissions Effectively (RARE) campaign to reduce avoidable hospital readmissions across the state. So far, the campaign's 82 participating hospitals and 100 community partners have prevented 6,211 readmissions between Jan. 1, 2011 and June 30, 2013.
The efforts were recognized last month when RARE was named a recipient of the 2013 John M. Eisenberg Patient Safety and Quality Award for Innovation in Patient Safety and Quality.
Launched in 2002 by National Quality Forum and Joint Commission, the award honors John M. Eisenberg, MD, MBA, a former administrator of the Agency for Healthcare Research and Quality and an advocate for patient safety and healthcare quality. SHM won the John M. Eisenberg Innovation in Patient Safety and Quality award in 2011 for its mentored-implementation program.
According to ICSI project manager Kathy Cummins, RN, MA, hospitals involved in RARE aren't given specific instructions on how to reduce readmissions, but are encouraged to focus their efforts on these areas:
- Comprehensive discharge planning;
- Medication management;
- Patient and family engagement;
- Transition-care support; and
- Transition communications.
While the campaign provides guidance and technical support, each hospital comes up with its own strategies for achieving these goals. Ms. Cummins, for example, describes how one hospital that was tasked with reducing readmissions without adding staff had paramedics use their downtime to visit recently discharged patients. The paramedics now check in to see if patients are exhibiting warning signs of illness and make sure they’re taking their prescribed medications.
SHM board member Howard Epstein, MD, FHM, ICSI's chief health systems officer, says the RARE campaign targets issues hospitalists have long struggled with.
"Hospitalists don't want to see their patients readmitted to the hospital," Dr. Epstein says. "It doesn't look good on their part, and it's not the best thing for their patients. The [RARE] campaign galvanized the system to support what hospitalists have been demanding for many years."
Stephanie C. Mackiewicz is a freelance author in California.
Visit our website for more information about the RARE campaign.
How's this for a quality-improvement success story? In 2011, three Minnesota-based institutions—the Institute for Clinical Systems Improvement (ICSI), the Minnesota Hospital Association, and Stratis Health—launched the Reducing Avoidable Readmissions Effectively (RARE) campaign to reduce avoidable hospital readmissions across the state. So far, the campaign's 82 participating hospitals and 100 community partners have prevented 6,211 readmissions between Jan. 1, 2011 and June 30, 2013.
The efforts were recognized last month when RARE was named a recipient of the 2013 John M. Eisenberg Patient Safety and Quality Award for Innovation in Patient Safety and Quality.
Launched in 2002 by National Quality Forum and Joint Commission, the award honors John M. Eisenberg, MD, MBA, a former administrator of the Agency for Healthcare Research and Quality and an advocate for patient safety and healthcare quality. SHM won the John M. Eisenberg Innovation in Patient Safety and Quality award in 2011 for its mentored-implementation program.
According to ICSI project manager Kathy Cummins, RN, MA, hospitals involved in RARE aren't given specific instructions on how to reduce readmissions, but are encouraged to focus their efforts on these areas:
- Comprehensive discharge planning;
- Medication management;
- Patient and family engagement;
- Transition-care support; and
- Transition communications.
While the campaign provides guidance and technical support, each hospital comes up with its own strategies for achieving these goals. Ms. Cummins, for example, describes how one hospital that was tasked with reducing readmissions without adding staff had paramedics use their downtime to visit recently discharged patients. The paramedics now check in to see if patients are exhibiting warning signs of illness and make sure they’re taking their prescribed medications.
SHM board member Howard Epstein, MD, FHM, ICSI's chief health systems officer, says the RARE campaign targets issues hospitalists have long struggled with.
"Hospitalists don't want to see their patients readmitted to the hospital," Dr. Epstein says. "It doesn't look good on their part, and it's not the best thing for their patients. The [RARE] campaign galvanized the system to support what hospitalists have been demanding for many years."
Stephanie C. Mackiewicz is a freelance author in California.
Visit our website for more information about the RARE campaign.
How's this for a quality-improvement success story? In 2011, three Minnesota-based institutions—the Institute for Clinical Systems Improvement (ICSI), the Minnesota Hospital Association, and Stratis Health—launched the Reducing Avoidable Readmissions Effectively (RARE) campaign to reduce avoidable hospital readmissions across the state. So far, the campaign's 82 participating hospitals and 100 community partners have prevented 6,211 readmissions between Jan. 1, 2011 and June 30, 2013.
The efforts were recognized last month when RARE was named a recipient of the 2013 John M. Eisenberg Patient Safety and Quality Award for Innovation in Patient Safety and Quality.
Launched in 2002 by National Quality Forum and Joint Commission, the award honors John M. Eisenberg, MD, MBA, a former administrator of the Agency for Healthcare Research and Quality and an advocate for patient safety and healthcare quality. SHM won the John M. Eisenberg Innovation in Patient Safety and Quality award in 2011 for its mentored-implementation program.
According to ICSI project manager Kathy Cummins, RN, MA, hospitals involved in RARE aren't given specific instructions on how to reduce readmissions, but are encouraged to focus their efforts on these areas:
- Comprehensive discharge planning;
- Medication management;
- Patient and family engagement;
- Transition-care support; and
- Transition communications.
While the campaign provides guidance and technical support, each hospital comes up with its own strategies for achieving these goals. Ms. Cummins, for example, describes how one hospital that was tasked with reducing readmissions without adding staff had paramedics use their downtime to visit recently discharged patients. The paramedics now check in to see if patients are exhibiting warning signs of illness and make sure they’re taking their prescribed medications.
SHM board member Howard Epstein, MD, FHM, ICSI's chief health systems officer, says the RARE campaign targets issues hospitalists have long struggled with.
"Hospitalists don't want to see their patients readmitted to the hospital," Dr. Epstein says. "It doesn't look good on their part, and it's not the best thing for their patients. The [RARE] campaign galvanized the system to support what hospitalists have been demanding for many years."
Stephanie C. Mackiewicz is a freelance author in California.
Visit our website for more information about the RARE campaign.
Report Offers Practice Management Roadmap for Hospital Medicine Groups
A new white paper from the Society of Hospital Medicine (SHM) is the specialty's first formal blueprint on best practices for running a hospital medicine group (HMG).
"The Key Principles and Characteristics of an Effective Hospital Medicine Group: An Assessment Guide for Hospitals and Hospitalists" report published this week in the Journal of Hospital Medicine, is the culmination of a two-year effort to give group leaders and hospital executives a self-assessment tool, says former SHM President John Nelson, MD, MHM, a practicing hospitalist and principal in Nelson Flores Hospital Medicine Consultants in Bellevue, Wash.
Dr. Nelson, one of the report's authors, says the 10 guiding principles and 47 individual characteristics are a launching point for group leaders, C-suite administrators, and others to discuss what ideals apply to their respective practices.
"It's like reading any other thing about how to get fit, how to eat a healthy diet, how to lead a good life," he adds. "It's pretty hard to rigidly pursue everything that an expert or well—considered document might recommend, but you tend to adapt it to your own circumstances and some things resonate."
Culled from more than 200 stakeholders, the report's 10 principles focus on:
Dr. Nelson says it's important for group leaders and hospital executives not to negatively view their practice in light of the report. Because characteristics recommended by the paper won’t necessarily apply everywhere, he suggests instead that readers pick out a handful of principles that apply most specifically to them.
"Two or three might be the right number to zero in on, but those are likely to be different for different groups," he adds. "I don't think there's any way for this to be used in the same manner at each group."
Likewise, Patrick Cawley, MD, MHM, chief executive officer at the Medical University of South Carolina (MUSC) Medical Center in Charleston, S.C. and author of the paper, says it shouldn't be viewed as a scientific conclusion, but as an aspirational approach to improvement.
"We feel that hospital medicine can be better than it is today," says Dr. Cawley, a former SHM president, "by laying out a road map not only [for HMGs] but [also for] hospital medicine leaders and hospital leaders about the things they should concentrate on raises the bar for everybody."
Dr. Cawley says the report is a first step for physicians and executives looking to benchmark their practices. In the future, SHM could follow up with other assessment tools that help groups improve themselves further.
"There are leaders and hospitals trying to improve their group. They're looking for something to measure themselves against," he adds. "This is instantly available for them, and we think this will be self-fulfilling. If people aren't using it, we haven't done our job right."
Richard Quinn is a freelance author in New Jersey.
Visit our website for more information on hospital medicine improvement initiatives.
A new white paper from the Society of Hospital Medicine (SHM) is the specialty's first formal blueprint on best practices for running a hospital medicine group (HMG).
"The Key Principles and Characteristics of an Effective Hospital Medicine Group: An Assessment Guide for Hospitals and Hospitalists" report published this week in the Journal of Hospital Medicine, is the culmination of a two-year effort to give group leaders and hospital executives a self-assessment tool, says former SHM President John Nelson, MD, MHM, a practicing hospitalist and principal in Nelson Flores Hospital Medicine Consultants in Bellevue, Wash.
Dr. Nelson, one of the report's authors, says the 10 guiding principles and 47 individual characteristics are a launching point for group leaders, C-suite administrators, and others to discuss what ideals apply to their respective practices.
"It's like reading any other thing about how to get fit, how to eat a healthy diet, how to lead a good life," he adds. "It's pretty hard to rigidly pursue everything that an expert or well—considered document might recommend, but you tend to adapt it to your own circumstances and some things resonate."
Culled from more than 200 stakeholders, the report's 10 principles focus on:
Dr. Nelson says it's important for group leaders and hospital executives not to negatively view their practice in light of the report. Because characteristics recommended by the paper won’t necessarily apply everywhere, he suggests instead that readers pick out a handful of principles that apply most specifically to them.
"Two or three might be the right number to zero in on, but those are likely to be different for different groups," he adds. "I don't think there's any way for this to be used in the same manner at each group."
Likewise, Patrick Cawley, MD, MHM, chief executive officer at the Medical University of South Carolina (MUSC) Medical Center in Charleston, S.C. and author of the paper, says it shouldn't be viewed as a scientific conclusion, but as an aspirational approach to improvement.
"We feel that hospital medicine can be better than it is today," says Dr. Cawley, a former SHM president, "by laying out a road map not only [for HMGs] but [also for] hospital medicine leaders and hospital leaders about the things they should concentrate on raises the bar for everybody."
Dr. Cawley says the report is a first step for physicians and executives looking to benchmark their practices. In the future, SHM could follow up with other assessment tools that help groups improve themselves further.
"There are leaders and hospitals trying to improve their group. They're looking for something to measure themselves against," he adds. "This is instantly available for them, and we think this will be self-fulfilling. If people aren't using it, we haven't done our job right."
Richard Quinn is a freelance author in New Jersey.
Visit our website for more information on hospital medicine improvement initiatives.
A new white paper from the Society of Hospital Medicine (SHM) is the specialty's first formal blueprint on best practices for running a hospital medicine group (HMG).
"The Key Principles and Characteristics of an Effective Hospital Medicine Group: An Assessment Guide for Hospitals and Hospitalists" report published this week in the Journal of Hospital Medicine, is the culmination of a two-year effort to give group leaders and hospital executives a self-assessment tool, says former SHM President John Nelson, MD, MHM, a practicing hospitalist and principal in Nelson Flores Hospital Medicine Consultants in Bellevue, Wash.
Dr. Nelson, one of the report's authors, says the 10 guiding principles and 47 individual characteristics are a launching point for group leaders, C-suite administrators, and others to discuss what ideals apply to their respective practices.
"It's like reading any other thing about how to get fit, how to eat a healthy diet, how to lead a good life," he adds. "It's pretty hard to rigidly pursue everything that an expert or well—considered document might recommend, but you tend to adapt it to your own circumstances and some things resonate."
Culled from more than 200 stakeholders, the report's 10 principles focus on:
Dr. Nelson says it's important for group leaders and hospital executives not to negatively view their practice in light of the report. Because characteristics recommended by the paper won’t necessarily apply everywhere, he suggests instead that readers pick out a handful of principles that apply most specifically to them.
"Two or three might be the right number to zero in on, but those are likely to be different for different groups," he adds. "I don't think there's any way for this to be used in the same manner at each group."
Likewise, Patrick Cawley, MD, MHM, chief executive officer at the Medical University of South Carolina (MUSC) Medical Center in Charleston, S.C. and author of the paper, says it shouldn't be viewed as a scientific conclusion, but as an aspirational approach to improvement.
"We feel that hospital medicine can be better than it is today," says Dr. Cawley, a former SHM president, "by laying out a road map not only [for HMGs] but [also for] hospital medicine leaders and hospital leaders about the things they should concentrate on raises the bar for everybody."
Dr. Cawley says the report is a first step for physicians and executives looking to benchmark their practices. In the future, SHM could follow up with other assessment tools that help groups improve themselves further.
"There are leaders and hospitals trying to improve their group. They're looking for something to measure themselves against," he adds. "This is instantly available for them, and we think this will be self-fulfilling. If people aren't using it, we haven't done our job right."
Richard Quinn is a freelance author in New Jersey.
Visit our website for more information on hospital medicine improvement initiatives.
Ulerythema Ophryogenes: Updates and Insights
Test your knowledge on ulerythema ophryogenes with MD-IQ: the medical intelligence quiz. Click here to answer 5 questions.
Consensus Recommendations From the American Acne & Rosacea Society on the Management of Rosacea, Part 4: A Status Report on Physical Modalities and Devices
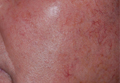
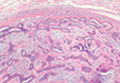
Chondroid Syringoma
What's Eating You? Turkey Mite and Lone Star Tick (Amblyomma americanum)

Polymer could improve blood cryopreservation
![]()
Credit: UAB Hospital
Scientists have found that a common polymer can help red blood cells (RBCs) survive storage at freezing temperatures, and it may offer benefits over current methods of cryopreservation.
The polymer, polyvinyl alcohol, mimics antifreeze properties found in cold-acclimatized fish, such as arctic cod.
And experiments revealed that using polyvinyl alcohol in blood cryopreservation can inhibit the growth of ice crystals, which damage RBCs and make them unusable.
Matthew Gibson, PhD, of the University of Warwick in the UK, and his colleagues conducted these experiments and reported the results in Nature Communications.
“We know that certain types of fish survive perfectly well in sub-zero sea temperatures without their blood freezing,” Dr Gibson said. “We used this as a starting point to search for synthetic substances which reflect what nature already does so well.”
“On closer examination, it turns out that polyvinyl alcohol, which is actually a derivative of wood glue, mimics the properties of the antifreeze proteins found in these kinds of fish.”
So Dr Gibson and his colleagues decided to see how polyvinyl alcohol fared in blood cryopreservation.
The team tested RBCs from sheep and humans and found that polyvinyl alcohol could inhibit ice crystal growth, even when used at concentrations as low as 0.01wt%.
The polymer was most effective at 0.1wt%, which allowed for 40% RBC recovery. Higher concentrations of polyvinyl alcohol did reduce the growth of ice crystals, but the benefits were counteracted by the secondary effects of ice shaping, which can pierce cell membranes.
The researchers noted that the current method of cryopreservation typically requires more than 20wt% of organic solvents to prevent ice formation. And the solvents must be removed before the blood can be used.
“Polyvinyl alcohol has 3 things in its favor when applied to freezing blood,” Dr Gibson said. “Firstly, it reduces the growth of ice crystals during thawing. Secondly, it reduces the need for organic solvents, and, crucially, it reduces the time between defrosting and having transfusion-ready blood by eliminating the need to remove solvent.”
Dr Gibson pointed out that, although polyvinyl alcohol appears to be a promising option for cryopreservation, additional research is needed. But if the polymer proves effective in subsequent studies, it could be used on other cell types as well. ![]()
![]()
Credit: UAB Hospital
Scientists have found that a common polymer can help red blood cells (RBCs) survive storage at freezing temperatures, and it may offer benefits over current methods of cryopreservation.
The polymer, polyvinyl alcohol, mimics antifreeze properties found in cold-acclimatized fish, such as arctic cod.
And experiments revealed that using polyvinyl alcohol in blood cryopreservation can inhibit the growth of ice crystals, which damage RBCs and make them unusable.
Matthew Gibson, PhD, of the University of Warwick in the UK, and his colleagues conducted these experiments and reported the results in Nature Communications.
“We know that certain types of fish survive perfectly well in sub-zero sea temperatures without their blood freezing,” Dr Gibson said. “We used this as a starting point to search for synthetic substances which reflect what nature already does so well.”
“On closer examination, it turns out that polyvinyl alcohol, which is actually a derivative of wood glue, mimics the properties of the antifreeze proteins found in these kinds of fish.”
So Dr Gibson and his colleagues decided to see how polyvinyl alcohol fared in blood cryopreservation.
The team tested RBCs from sheep and humans and found that polyvinyl alcohol could inhibit ice crystal growth, even when used at concentrations as low as 0.01wt%.
The polymer was most effective at 0.1wt%, which allowed for 40% RBC recovery. Higher concentrations of polyvinyl alcohol did reduce the growth of ice crystals, but the benefits were counteracted by the secondary effects of ice shaping, which can pierce cell membranes.
The researchers noted that the current method of cryopreservation typically requires more than 20wt% of organic solvents to prevent ice formation. And the solvents must be removed before the blood can be used.
“Polyvinyl alcohol has 3 things in its favor when applied to freezing blood,” Dr Gibson said. “Firstly, it reduces the growth of ice crystals during thawing. Secondly, it reduces the need for organic solvents, and, crucially, it reduces the time between defrosting and having transfusion-ready blood by eliminating the need to remove solvent.”
Dr Gibson pointed out that, although polyvinyl alcohol appears to be a promising option for cryopreservation, additional research is needed. But if the polymer proves effective in subsequent studies, it could be used on other cell types as well. ![]()
![]()
Credit: UAB Hospital
Scientists have found that a common polymer can help red blood cells (RBCs) survive storage at freezing temperatures, and it may offer benefits over current methods of cryopreservation.
The polymer, polyvinyl alcohol, mimics antifreeze properties found in cold-acclimatized fish, such as arctic cod.
And experiments revealed that using polyvinyl alcohol in blood cryopreservation can inhibit the growth of ice crystals, which damage RBCs and make them unusable.
Matthew Gibson, PhD, of the University of Warwick in the UK, and his colleagues conducted these experiments and reported the results in Nature Communications.
“We know that certain types of fish survive perfectly well in sub-zero sea temperatures without their blood freezing,” Dr Gibson said. “We used this as a starting point to search for synthetic substances which reflect what nature already does so well.”
“On closer examination, it turns out that polyvinyl alcohol, which is actually a derivative of wood glue, mimics the properties of the antifreeze proteins found in these kinds of fish.”
So Dr Gibson and his colleagues decided to see how polyvinyl alcohol fared in blood cryopreservation.
The team tested RBCs from sheep and humans and found that polyvinyl alcohol could inhibit ice crystal growth, even when used at concentrations as low as 0.01wt%.
The polymer was most effective at 0.1wt%, which allowed for 40% RBC recovery. Higher concentrations of polyvinyl alcohol did reduce the growth of ice crystals, but the benefits were counteracted by the secondary effects of ice shaping, which can pierce cell membranes.
The researchers noted that the current method of cryopreservation typically requires more than 20wt% of organic solvents to prevent ice formation. And the solvents must be removed before the blood can be used.
“Polyvinyl alcohol has 3 things in its favor when applied to freezing blood,” Dr Gibson said. “Firstly, it reduces the growth of ice crystals during thawing. Secondly, it reduces the need for organic solvents, and, crucially, it reduces the time between defrosting and having transfusion-ready blood by eliminating the need to remove solvent.”
Dr Gibson pointed out that, although polyvinyl alcohol appears to be a promising option for cryopreservation, additional research is needed. But if the polymer proves effective in subsequent studies, it could be used on other cell types as well. ![]()