User login
Neonatal Sleep Measures Predict Neurodevelopmental Outcomes
VANCOUVER—Among newborns at risk of neurologic dysfunction, measures of neonatal sleep help predict 18-month neurodevelopmental outcomes, according to research presented at the 45th Annual Meeting of the Child Neurology Society.
Studies suggest that abnormal sleep has neurocognitive consequences for older infants and children and that polysomnogram data are associated with brain function in newborns who require neonatal intensive care. “Although sleep is a highly sophisticated brain function, it is not typically included in the newborn clinical neurological assessment,” said Renée A. Shellhaas, MD, MS, Assistant Professor of Pediatrics and Communicable Diseases at the University of Michigan in Ann Arbor, and colleagues.

To evaluate how polysomnography measures may add to standard predictors of neurodevelopmental outcome for newborns who require intensive care and are at risk for neurologic dysfunction, Dr. Shellhaas and colleagues conducted a longitudinal study of 29 newborns. Patients had a gestational age of 35 weeks or more, were cared for in a neonatal intensive care unit, and were clinically determined to be at risk of seizures. Researchers excluded patients with congenital anomalies or syndromes known to affect neurodevelopmental outcome or predispose patients to sleep-disordered breathing. They also excluded patients who had severely abnormal EEG without sleep–wake cycling.
Once a newborn was medically stable, researchers conducted a 12-hour attended, bedside polysomnogram. Polysomnograms were scored by a polysomnography technologist and reviewed by a sleep-medicine physician. Researchers calculated the proportion of each sleep–wake stage, entropy of the sequence of sleep–wake state transitions, and power spectra of the EEG portion of the polysomnogram.
Researchers evaluated neurodevelopmental outcome at 18 months to 22 months using the third edition of the Bayley Scales of Infant Development (BSID). They assessed associations between polysomnogram results and neurodevelopmental outcomes using regression techniques that de-emphasized outliers. Patients’ mean gestational age was 39.6 weeks. Seventeen of the 29 patients were male. Mean birth weight was 3.42 kg, and median five-minute Apgar score was 8.
In univariate analysis, increased time in quiet sleep predicted lower 18-month cognitive, language, and motor BSID scores. Higher entropy of sleep–wake transitions predicted lower motor scores. Increased low-frequency EEG power during quiet sleep predicted higher motor and language BSID scores. Gestational age and illness severity were not predictive of BSID results. A more abnormal neonatal neurologic exam score (ie, Thompson score) predicted lower cognitive and motor BSID scores.
In analyses adjusted for Thompson score, higher EEG power during neonatal quiet sleep was associated with better 18-month motor and language scores. In addition, increased time in neonatal quiet sleep was associated with lower 18-month cognitive and motor scores. “Notably, Thompson score was not an independent predictor of outcome when the sleep data were included in the bivariate models,” Dr. Shellhaas and colleagues said.
“Our results suggest that inefficient neonatal quiet sleep—more time in quiet sleep and lower delta frequency power during that stage—predicts lower 18-month neurodevelopmental outcome scores,” the researchers concluded. “Importantly, these novel measures of brain functional integrity were robust predictors even after adjusting for the neonatal neurologic examination score.”
—Jake Remaly
Suggested Reading
Shellhaas RA, Burns JW, Barks JD, Chervin RD. Quantitative sleep stage analyses as a window to neonatal neurologic function. Neurology. 2014;82(5):390-395.
Shellhaas RA, Burns JW, Wiggins SA, et al. Sleep-wake cycling and cerebral oxygen metabolism among critically ill neonates. J Child Neurol. 2014;29(4):530-533.
VANCOUVER—Among newborns at risk of neurologic dysfunction, measures of neonatal sleep help predict 18-month neurodevelopmental outcomes, according to research presented at the 45th Annual Meeting of the Child Neurology Society.
Studies suggest that abnormal sleep has neurocognitive consequences for older infants and children and that polysomnogram data are associated with brain function in newborns who require neonatal intensive care. “Although sleep is a highly sophisticated brain function, it is not typically included in the newborn clinical neurological assessment,” said Renée A. Shellhaas, MD, MS, Assistant Professor of Pediatrics and Communicable Diseases at the University of Michigan in Ann Arbor, and colleagues.
To evaluate how polysomnography measures may add to standard predictors of neurodevelopmental outcome for newborns who require intensive care and are at risk for neurologic dysfunction, Dr. Shellhaas and colleagues conducted a longitudinal study of 29 newborns. Patients had a gestational age of 35 weeks or more, were cared for in a neonatal intensive care unit, and were clinically determined to be at risk of seizures. Researchers excluded patients with congenital anomalies or syndromes known to affect neurodevelopmental outcome or predispose patients to sleep-disordered breathing. They also excluded patients who had severely abnormal EEG without sleep–wake cycling.
Once a newborn was medically stable, researchers conducted a 12-hour attended, bedside polysomnogram. Polysomnograms were scored by a polysomnography technologist and reviewed by a sleep-medicine physician. Researchers calculated the proportion of each sleep–wake stage, entropy of the sequence of sleep–wake state transitions, and power spectra of the EEG portion of the polysomnogram.
Researchers evaluated neurodevelopmental outcome at 18 months to 22 months using the third edition of the Bayley Scales of Infant Development (BSID). They assessed associations between polysomnogram results and neurodevelopmental outcomes using regression techniques that de-emphasized outliers. Patients’ mean gestational age was 39.6 weeks. Seventeen of the 29 patients were male. Mean birth weight was 3.42 kg, and median five-minute Apgar score was 8.
In univariate analysis, increased time in quiet sleep predicted lower 18-month cognitive, language, and motor BSID scores. Higher entropy of sleep–wake transitions predicted lower motor scores. Increased low-frequency EEG power during quiet sleep predicted higher motor and language BSID scores. Gestational age and illness severity were not predictive of BSID results. A more abnormal neonatal neurologic exam score (ie, Thompson score) predicted lower cognitive and motor BSID scores.
In analyses adjusted for Thompson score, higher EEG power during neonatal quiet sleep was associated with better 18-month motor and language scores. In addition, increased time in neonatal quiet sleep was associated with lower 18-month cognitive and motor scores. “Notably, Thompson score was not an independent predictor of outcome when the sleep data were included in the bivariate models,” Dr. Shellhaas and colleagues said.
“Our results suggest that inefficient neonatal quiet sleep—more time in quiet sleep and lower delta frequency power during that stage—predicts lower 18-month neurodevelopmental outcome scores,” the researchers concluded. “Importantly, these novel measures of brain functional integrity were robust predictors even after adjusting for the neonatal neurologic examination score.”
—Jake Remaly
Suggested Reading
Shellhaas RA, Burns JW, Barks JD, Chervin RD. Quantitative sleep stage analyses as a window to neonatal neurologic function. Neurology. 2014;82(5):390-395.
Shellhaas RA, Burns JW, Wiggins SA, et al. Sleep-wake cycling and cerebral oxygen metabolism among critically ill neonates. J Child Neurol. 2014;29(4):530-533.
VANCOUVER—Among newborns at risk of neurologic dysfunction, measures of neonatal sleep help predict 18-month neurodevelopmental outcomes, according to research presented at the 45th Annual Meeting of the Child Neurology Society.
Studies suggest that abnormal sleep has neurocognitive consequences for older infants and children and that polysomnogram data are associated with brain function in newborns who require neonatal intensive care. “Although sleep is a highly sophisticated brain function, it is not typically included in the newborn clinical neurological assessment,” said Renée A. Shellhaas, MD, MS, Assistant Professor of Pediatrics and Communicable Diseases at the University of Michigan in Ann Arbor, and colleagues.
To evaluate how polysomnography measures may add to standard predictors of neurodevelopmental outcome for newborns who require intensive care and are at risk for neurologic dysfunction, Dr. Shellhaas and colleagues conducted a longitudinal study of 29 newborns. Patients had a gestational age of 35 weeks or more, were cared for in a neonatal intensive care unit, and were clinically determined to be at risk of seizures. Researchers excluded patients with congenital anomalies or syndromes known to affect neurodevelopmental outcome or predispose patients to sleep-disordered breathing. They also excluded patients who had severely abnormal EEG without sleep–wake cycling.
Once a newborn was medically stable, researchers conducted a 12-hour attended, bedside polysomnogram. Polysomnograms were scored by a polysomnography technologist and reviewed by a sleep-medicine physician. Researchers calculated the proportion of each sleep–wake stage, entropy of the sequence of sleep–wake state transitions, and power spectra of the EEG portion of the polysomnogram.
Researchers evaluated neurodevelopmental outcome at 18 months to 22 months using the third edition of the Bayley Scales of Infant Development (BSID). They assessed associations between polysomnogram results and neurodevelopmental outcomes using regression techniques that de-emphasized outliers. Patients’ mean gestational age was 39.6 weeks. Seventeen of the 29 patients were male. Mean birth weight was 3.42 kg, and median five-minute Apgar score was 8.
In univariate analysis, increased time in quiet sleep predicted lower 18-month cognitive, language, and motor BSID scores. Higher entropy of sleep–wake transitions predicted lower motor scores. Increased low-frequency EEG power during quiet sleep predicted higher motor and language BSID scores. Gestational age and illness severity were not predictive of BSID results. A more abnormal neonatal neurologic exam score (ie, Thompson score) predicted lower cognitive and motor BSID scores.
In analyses adjusted for Thompson score, higher EEG power during neonatal quiet sleep was associated with better 18-month motor and language scores. In addition, increased time in neonatal quiet sleep was associated with lower 18-month cognitive and motor scores. “Notably, Thompson score was not an independent predictor of outcome when the sleep data were included in the bivariate models,” Dr. Shellhaas and colleagues said.
“Our results suggest that inefficient neonatal quiet sleep—more time in quiet sleep and lower delta frequency power during that stage—predicts lower 18-month neurodevelopmental outcome scores,” the researchers concluded. “Importantly, these novel measures of brain functional integrity were robust predictors even after adjusting for the neonatal neurologic examination score.”
—Jake Remaly
Suggested Reading
Shellhaas RA, Burns JW, Barks JD, Chervin RD. Quantitative sleep stage analyses as a window to neonatal neurologic function. Neurology. 2014;82(5):390-395.
Shellhaas RA, Burns JW, Wiggins SA, et al. Sleep-wake cycling and cerebral oxygen metabolism among critically ill neonates. J Child Neurol. 2014;29(4):530-533.

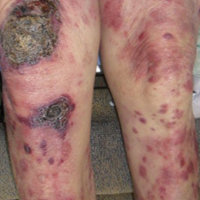
Diffuse Rash With Associated Ulceration
The Diagnosis: Epidermotropic CD8+ T-Cell Lymphoma
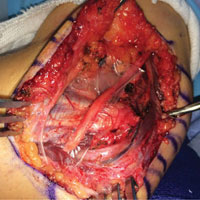
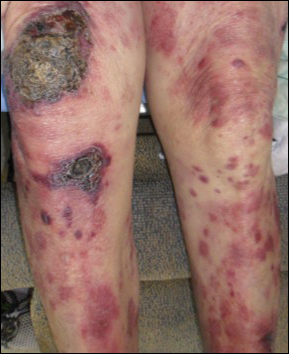
Epidermotropic CD8+ T-cell lymphoma is a rare aggressive form of cutaneous T-cell lymphoma (CTCL), accounting for less than 1% of all cases.1 Since this subtype of CTCL was first described in 1999 by Berti et al,2 approximately 45 cases have been reported in the literature.1 It typically is found in elderly men and presents as disseminated or localized papules, patches, plaques, nodules, and tumors, often with central necrosis, ulceration, crusting, and hemorrhage (Figure 1).1,3 These lesions rapidly progress and can affect any skin site, but acral accentuation and mucosal involvement are common.4 Due to the rapidly progressive nature of this disease, patients typically present with widespread plaque- and tumor-stage disease.3 Frequency of systemic spread is high, with metastasis to the central nervous system, lungs, and testes being most common. Lymph nodes typically are spared, helping to differentiate this form of CTCL from classic mycosis fungoides.
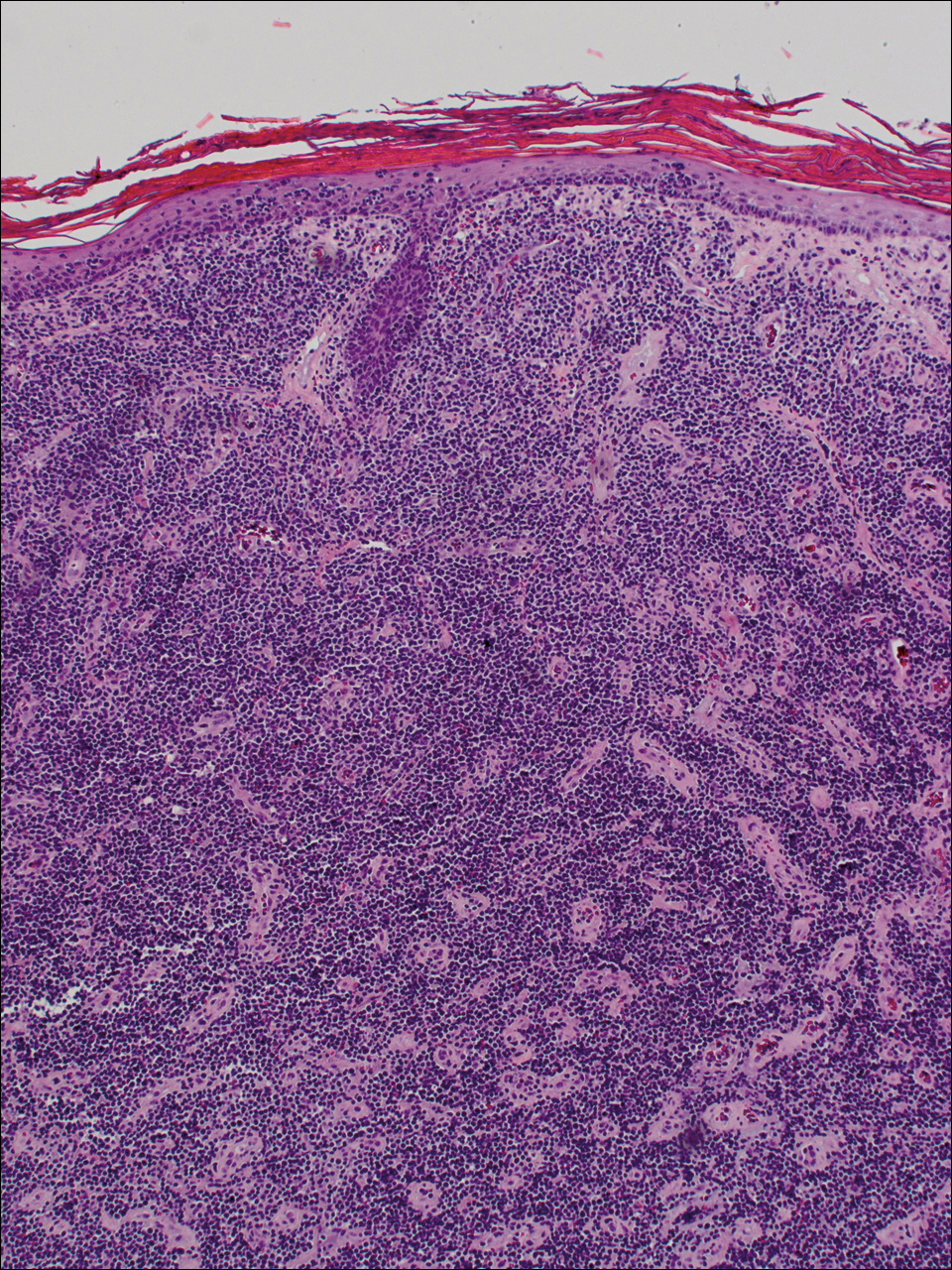
Diagnosis of epidermotropic CD8+ T-cell lymphoma is based on a combination of clinical, histopathologic, and immunohistochemical features. Histopathologic components include epidermotropism, particularly in the basal cell layer, in a pagetoid or linear pattern. A second feature is a dermal infiltrate consisting of a nodular or diffuse pattern of atypical lymphocytes that extend to the subcutaneous fat (Figure 2). All cases of epidermotropic CD8+ T-cell lymphoma express the CD8+ phenotype and most have a high Ki-67 proliferation index and are CD3, CD45RA, and/or T-cell intracellular antigen 1 positive.1
Due to its aggressive nature, epidermotropic CD8+ T-cell lymphoma has a poor prognosis, with an average 5-year survival rate of 18% and median survival of 22.5 months.3 Treatment proves difficult as conventional therapies for CD4+ CTCL have proven ineffective for epidermotropic CD8+ T-cell lymphoma. Partial response has been seen with bexarotene alone and with total skin electron beam therapy combined with oral retinoids.1
- Nofal A, Abdel-Mawla MY, Assaf M, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma: proposed diagnostic criteria and therapeutic evaluation. J Am Acad Dermatol. 2012;67:748-759.
- Berti E, Tomasini D, Vermeer MH, et al. Primary cutaneous CD8-positive epidermotropic cytotoxic T cell lymphomas. a distinct clinicopathological entity with an aggressive clinical behavior. Am J Pathol. 1999;155:483-492.
- Gormley RH, Hess SD, Anand D, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma. J Am Acad Dermatol. 2010;62:300-307.
- Nofal A, Abdel-Mawla MY, Assaf M, et al. Primary cutaneous aggressive epidermotropic CD8+ T cell lymphoma: a diagnostic and therapeutic challenge. Int J Dermatol. 2014;53:76-81.
The Diagnosis: Epidermotropic CD8+ T-Cell Lymphoma
Epidermotropic CD8+ T-cell lymphoma is a rare aggressive form of cutaneous T-cell lymphoma (CTCL), accounting for less than 1% of all cases.1 Since this subtype of CTCL was first described in 1999 by Berti et al,2 approximately 45 cases have been reported in the literature.1 It typically is found in elderly men and presents as disseminated or localized papules, patches, plaques, nodules, and tumors, often with central necrosis, ulceration, crusting, and hemorrhage (Figure 1).1,3 These lesions rapidly progress and can affect any skin site, but acral accentuation and mucosal involvement are common.4 Due to the rapidly progressive nature of this disease, patients typically present with widespread plaque- and tumor-stage disease.3 Frequency of systemic spread is high, with metastasis to the central nervous system, lungs, and testes being most common. Lymph nodes typically are spared, helping to differentiate this form of CTCL from classic mycosis fungoides.
Diagnosis of epidermotropic CD8+ T-cell lymphoma is based on a combination of clinical, histopathologic, and immunohistochemical features. Histopathologic components include epidermotropism, particularly in the basal cell layer, in a pagetoid or linear pattern. A second feature is a dermal infiltrate consisting of a nodular or diffuse pattern of atypical lymphocytes that extend to the subcutaneous fat (Figure 2). All cases of epidermotropic CD8+ T-cell lymphoma express the CD8+ phenotype and most have a high Ki-67 proliferation index and are CD3, CD45RA, and/or T-cell intracellular antigen 1 positive.1
Due to its aggressive nature, epidermotropic CD8+ T-cell lymphoma has a poor prognosis, with an average 5-year survival rate of 18% and median survival of 22.5 months.3 Treatment proves difficult as conventional therapies for CD4+ CTCL have proven ineffective for epidermotropic CD8+ T-cell lymphoma. Partial response has been seen with bexarotene alone and with total skin electron beam therapy combined with oral retinoids.1
The Diagnosis: Epidermotropic CD8+ T-Cell Lymphoma
Epidermotropic CD8+ T-cell lymphoma is a rare aggressive form of cutaneous T-cell lymphoma (CTCL), accounting for less than 1% of all cases.1 Since this subtype of CTCL was first described in 1999 by Berti et al,2 approximately 45 cases have been reported in the literature.1 It typically is found in elderly men and presents as disseminated or localized papules, patches, plaques, nodules, and tumors, often with central necrosis, ulceration, crusting, and hemorrhage (Figure 1).1,3 These lesions rapidly progress and can affect any skin site, but acral accentuation and mucosal involvement are common.4 Due to the rapidly progressive nature of this disease, patients typically present with widespread plaque- and tumor-stage disease.3 Frequency of systemic spread is high, with metastasis to the central nervous system, lungs, and testes being most common. Lymph nodes typically are spared, helping to differentiate this form of CTCL from classic mycosis fungoides.
Diagnosis of epidermotropic CD8+ T-cell lymphoma is based on a combination of clinical, histopathologic, and immunohistochemical features. Histopathologic components include epidermotropism, particularly in the basal cell layer, in a pagetoid or linear pattern. A second feature is a dermal infiltrate consisting of a nodular or diffuse pattern of atypical lymphocytes that extend to the subcutaneous fat (Figure 2). All cases of epidermotropic CD8+ T-cell lymphoma express the CD8+ phenotype and most have a high Ki-67 proliferation index and are CD3, CD45RA, and/or T-cell intracellular antigen 1 positive.1
Due to its aggressive nature, epidermotropic CD8+ T-cell lymphoma has a poor prognosis, with an average 5-year survival rate of 18% and median survival of 22.5 months.3 Treatment proves difficult as conventional therapies for CD4+ CTCL have proven ineffective for epidermotropic CD8+ T-cell lymphoma. Partial response has been seen with bexarotene alone and with total skin electron beam therapy combined with oral retinoids.1
- Nofal A, Abdel-Mawla MY, Assaf M, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma: proposed diagnostic criteria and therapeutic evaluation. J Am Acad Dermatol. 2012;67:748-759.
- Berti E, Tomasini D, Vermeer MH, et al. Primary cutaneous CD8-positive epidermotropic cytotoxic T cell lymphomas. a distinct clinicopathological entity with an aggressive clinical behavior. Am J Pathol. 1999;155:483-492.
- Gormley RH, Hess SD, Anand D, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma. J Am Acad Dermatol. 2010;62:300-307.
- Nofal A, Abdel-Mawla MY, Assaf M, et al. Primary cutaneous aggressive epidermotropic CD8+ T cell lymphoma: a diagnostic and therapeutic challenge. Int J Dermatol. 2014;53:76-81.
- Nofal A, Abdel-Mawla MY, Assaf M, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma: proposed diagnostic criteria and therapeutic evaluation. J Am Acad Dermatol. 2012;67:748-759.
- Berti E, Tomasini D, Vermeer MH, et al. Primary cutaneous CD8-positive epidermotropic cytotoxic T cell lymphomas. a distinct clinicopathological entity with an aggressive clinical behavior. Am J Pathol. 1999;155:483-492.
- Gormley RH, Hess SD, Anand D, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma. J Am Acad Dermatol. 2010;62:300-307.
- Nofal A, Abdel-Mawla MY, Assaf M, et al. Primary cutaneous aggressive epidermotropic CD8+ T cell lymphoma: a diagnostic and therapeutic challenge. Int J Dermatol. 2014;53:76-81.
A 72-year-old woman who was admitted for pneumonia and acute hypoxic respiratory failure was seen for an inpatient consultation for a diffuse rash with associated ulceration. She reported a rash of 20 months' duration that began on the legs and then spread to the trunk, arms, head, and neck with minimal pruritus and no pain or photosensitivity. She had been treated with hydroxychloroquine, mycophenolate mofetil, and prednisone without improvement. The patient noted recent ulceration on the rash. Physical examination revealed violaceous patches, plaques, nodules, and tumors with rare ulceration involving the face, trunk, and extremities. Biopsy showed a diffuse infiltration of the dermis with medium-sized atypical lymphocytes with scant cytoplasm and round to irregular hyperchromatic nuclei with clumped chromatin. Epidermotropism with small collections of atypical lymphocytes also was present within the epidermis.
VIDEO: Bariatric surgery may protect against heart failure
NEW ORLEANS – Results of a new 40,000-patient Swedish observational study provide the strongest evidence to date suggesting a causal relationship between bariatric surgery and reduced risk of heart failure, according to Johan Sundström, MD.
The study, which included patients drawn from two large Swedish national registries, demonstrated that bariatric surgery was associated with a 46% reduction in the incidence of heart failure during a median 4.1 years of follow-up, compared with an intensive lifestyle modification program for weight loss.
“These are observational data, but it’s a very large study population – and probably there will never be a large randomized trial of bariatric surgery versus weight loss through intensive lifestyle modification as a means of reducing the risk of heart failure,” Dr. Sundström, professor of epidemiology and a cardiologist at Uppsala (Sweden) University, said at the American Heart Association scientific sessions.
The study included 25,804 bariatric surgery patients in SOReg, the Scandinavian Obesity Surgery Registry, and a matched comparator group of 13,701 participants in a Swedish national registry of obese participants in a commercial Sweden-based intensive structural lifestyle modification program for weight loss called Itrim. The two groups were matched for baseline body mass index, which was a mean of 41.5 kg, and numerous other demographic factors and comorbid conditions. Participants weighed an average of 119 kg at baseline. None of the subjects had a history of heart failure.
The bariatric surgery group lost substantially more weight than did the lifestyle modification group: an average loss of about 35 kg after 1 year, which was 18.8 kg more than in the lifestyle modification group. After 2 years, the bariatric surgery group had an average of 22.6 kg more weight loss than did the comparison group.
The primary outcome was hospitalization for new-onset heart failure during a median 4.1 years of follow-up. Subjects were well below the age range when the incidence of heart failure accelerates – they averaged 41 years of age – but 73 of them did develop heart failure during follow-up. The incidence was 46% lower in the bariatric surgery patients. This supports the study hypothesis that bariatric surgery leads to a low incidence of new-onset heart failure, compared with intensive lifestyle modification because of its larger weight loss effect.
When Dr. Sundström and his coinvestigators combined the two study groups, they found that a 10-kg weight loss at 1 year was associated with a 23% reduction in the risk of heart failure during follow-up, irrespective of whether the weight loss was achieved surgically or through the lifestyle program.
“A great way of studying causality is to take away the exposure and note what happens to the outcome. If there’s a causal link, then if you take away the risk factor – in this case, obesity – the disease should go away,” he explained in a video interview.
The reduced risk of heart failure in the bariatric surgery patients wasn’t because of fewer acute MIs. Indeed, their acute MI rate during follow-up was similar to that of the lifestyle modification group. But bariatric surgery was associated with relative risk reductions of 35%-37% for atrial fibrillation or need for diabetes or blood pressure–lowering medications at 1 year – and atrial fibrillation, diabetes, and hypertension are all established risk factors for heart failure, Dr. Sundström noted.
The Itrim intensive lifestyle modification program entailed an initial very-low-energy diet for the first 3 months in order to achieve massive weight loss, followed by a 9-month maintenance program involving motivational counseling, exercise, behavioral therapy, and a restricted diet.
Dr. Sundström said he and his coinvestigators plan to continue the study and expand it to look at differences in additional cardiovascular endpoints as patients age.
The study was funded by the U.S. National Institute of Diabetes and Digestive and Kidney Diseases, Uppsala University, the Karolinska Institute, and the Swedish Research Council. Dr. Sundström reported serving as a scientific advisor to Itrim.
NEW ORLEANS – Results of a new 40,000-patient Swedish observational study provide the strongest evidence to date suggesting a causal relationship between bariatric surgery and reduced risk of heart failure, according to Johan Sundström, MD.
The study, which included patients drawn from two large Swedish national registries, demonstrated that bariatric surgery was associated with a 46% reduction in the incidence of heart failure during a median 4.1 years of follow-up, compared with an intensive lifestyle modification program for weight loss.
“These are observational data, but it’s a very large study population – and probably there will never be a large randomized trial of bariatric surgery versus weight loss through intensive lifestyle modification as a means of reducing the risk of heart failure,” Dr. Sundström, professor of epidemiology and a cardiologist at Uppsala (Sweden) University, said at the American Heart Association scientific sessions.
The study included 25,804 bariatric surgery patients in SOReg, the Scandinavian Obesity Surgery Registry, and a matched comparator group of 13,701 participants in a Swedish national registry of obese participants in a commercial Sweden-based intensive structural lifestyle modification program for weight loss called Itrim. The two groups were matched for baseline body mass index, which was a mean of 41.5 kg, and numerous other demographic factors and comorbid conditions. Participants weighed an average of 119 kg at baseline. None of the subjects had a history of heart failure.
The bariatric surgery group lost substantially more weight than did the lifestyle modification group: an average loss of about 35 kg after 1 year, which was 18.8 kg more than in the lifestyle modification group. After 2 years, the bariatric surgery group had an average of 22.6 kg more weight loss than did the comparison group.
The primary outcome was hospitalization for new-onset heart failure during a median 4.1 years of follow-up. Subjects were well below the age range when the incidence of heart failure accelerates – they averaged 41 years of age – but 73 of them did develop heart failure during follow-up. The incidence was 46% lower in the bariatric surgery patients. This supports the study hypothesis that bariatric surgery leads to a low incidence of new-onset heart failure, compared with intensive lifestyle modification because of its larger weight loss effect.
When Dr. Sundström and his coinvestigators combined the two study groups, they found that a 10-kg weight loss at 1 year was associated with a 23% reduction in the risk of heart failure during follow-up, irrespective of whether the weight loss was achieved surgically or through the lifestyle program.
“A great way of studying causality is to take away the exposure and note what happens to the outcome. If there’s a causal link, then if you take away the risk factor – in this case, obesity – the disease should go away,” he explained in a video interview.
The reduced risk of heart failure in the bariatric surgery patients wasn’t because of fewer acute MIs. Indeed, their acute MI rate during follow-up was similar to that of the lifestyle modification group. But bariatric surgery was associated with relative risk reductions of 35%-37% for atrial fibrillation or need for diabetes or blood pressure–lowering medications at 1 year – and atrial fibrillation, diabetes, and hypertension are all established risk factors for heart failure, Dr. Sundström noted.
The Itrim intensive lifestyle modification program entailed an initial very-low-energy diet for the first 3 months in order to achieve massive weight loss, followed by a 9-month maintenance program involving motivational counseling, exercise, behavioral therapy, and a restricted diet.
Dr. Sundström said he and his coinvestigators plan to continue the study and expand it to look at differences in additional cardiovascular endpoints as patients age.
The study was funded by the U.S. National Institute of Diabetes and Digestive and Kidney Diseases, Uppsala University, the Karolinska Institute, and the Swedish Research Council. Dr. Sundström reported serving as a scientific advisor to Itrim.
NEW ORLEANS – Results of a new 40,000-patient Swedish observational study provide the strongest evidence to date suggesting a causal relationship between bariatric surgery and reduced risk of heart failure, according to Johan Sundström, MD.
The study, which included patients drawn from two large Swedish national registries, demonstrated that bariatric surgery was associated with a 46% reduction in the incidence of heart failure during a median 4.1 years of follow-up, compared with an intensive lifestyle modification program for weight loss.
“These are observational data, but it’s a very large study population – and probably there will never be a large randomized trial of bariatric surgery versus weight loss through intensive lifestyle modification as a means of reducing the risk of heart failure,” Dr. Sundström, professor of epidemiology and a cardiologist at Uppsala (Sweden) University, said at the American Heart Association scientific sessions.
The study included 25,804 bariatric surgery patients in SOReg, the Scandinavian Obesity Surgery Registry, and a matched comparator group of 13,701 participants in a Swedish national registry of obese participants in a commercial Sweden-based intensive structural lifestyle modification program for weight loss called Itrim. The two groups were matched for baseline body mass index, which was a mean of 41.5 kg, and numerous other demographic factors and comorbid conditions. Participants weighed an average of 119 kg at baseline. None of the subjects had a history of heart failure.
The bariatric surgery group lost substantially more weight than did the lifestyle modification group: an average loss of about 35 kg after 1 year, which was 18.8 kg more than in the lifestyle modification group. After 2 years, the bariatric surgery group had an average of 22.6 kg more weight loss than did the comparison group.
The primary outcome was hospitalization for new-onset heart failure during a median 4.1 years of follow-up. Subjects were well below the age range when the incidence of heart failure accelerates – they averaged 41 years of age – but 73 of them did develop heart failure during follow-up. The incidence was 46% lower in the bariatric surgery patients. This supports the study hypothesis that bariatric surgery leads to a low incidence of new-onset heart failure, compared with intensive lifestyle modification because of its larger weight loss effect.
When Dr. Sundström and his coinvestigators combined the two study groups, they found that a 10-kg weight loss at 1 year was associated with a 23% reduction in the risk of heart failure during follow-up, irrespective of whether the weight loss was achieved surgically or through the lifestyle program.
“A great way of studying causality is to take away the exposure and note what happens to the outcome. If there’s a causal link, then if you take away the risk factor – in this case, obesity – the disease should go away,” he explained in a video interview.
The reduced risk of heart failure in the bariatric surgery patients wasn’t because of fewer acute MIs. Indeed, their acute MI rate during follow-up was similar to that of the lifestyle modification group. But bariatric surgery was associated with relative risk reductions of 35%-37% for atrial fibrillation or need for diabetes or blood pressure–lowering medications at 1 year – and atrial fibrillation, diabetes, and hypertension are all established risk factors for heart failure, Dr. Sundström noted.
The Itrim intensive lifestyle modification program entailed an initial very-low-energy diet for the first 3 months in order to achieve massive weight loss, followed by a 9-month maintenance program involving motivational counseling, exercise, behavioral therapy, and a restricted diet.
Dr. Sundström said he and his coinvestigators plan to continue the study and expand it to look at differences in additional cardiovascular endpoints as patients age.
The study was funded by the U.S. National Institute of Diabetes and Digestive and Kidney Diseases, Uppsala University, the Karolinska Institute, and the Swedish Research Council. Dr. Sundström reported serving as a scientific advisor to Itrim.
AT THE AHA SCIENTIFIC SESSIONS 2016
Key clinical point:
Major finding: The incidence of new-onset heart failure was 46% lower during follow-up after bariatric surgery than among participants in an intensive lifestyle modification program for weight loss.
Data source: This observational registry study followed nearly 26,000 Swedish bariatric surgery patients and 14,000 matched participants in a commercial intensive lifestyle modification program for a median of 4.1 years.
Disclosures: The study was funded by the U.S. National Institutes of Diabetes and Digestive and Kidney Diseases, Uppsala University, the Karolinska Institute, and the Swedish Research Council. The presenter reported serving as a scientific advisor to Itrim.

Mindfulness
How might mindfulness contribute to your mental collapse? Let’s say your work has become tedious. Tottering toward burnout, you decide to try mindfulness meditation to reverse your downward trend. However, you habitually fail to do your daily meditation. Now, “Meditate today” just piles on to your to-do list, a daily reminder of just how weak and disorganized you have become. Voila! Mindfulness is making you more crazy. There are things you can do to avoid this.
There are plenty of things to tip us doctors into burnout. We are not alone in the burnout epidemic, but we are overrepresented. More than 50% of physicians have burnout symptoms according to a recent Mayo Clinic study. Mindfulness training can help.

According to an interview with the program’s director, Douglas Zeidonis, MD, professor and chair of the department of psychiatry at the University of Massachusetts, most of the physicians reported that mindfulness training significantly benefited their work and personal lives. Mindfulness helped them feel more present and engaged with colleagues and patients and made them better clinicians – they reported showing more compassion toward patients.
Like any desirable habit, the key is to do it again and again and again. Here are a few recommendations to help you become more mindful during your workday.
1. Set random alarms (vibrate mode) on your smartphone to remind yourself to take a moment. When it goes off, do this: Breathe (4 seconds in, hold, then 8 seconds out) and be totally present for one minute.
2. Remove deliciously distracting apps from your phone’s home screen. Instead, tuck them away in a folder to reduce the likelihood you’ll click on them when you’re stressed.
3. Put meditation apps where you easily see them. You might try:
• The Mindfulness App: This app offers guided meditations in varying lengths from 3 to 30 minutes, so you can choose the one that’s right for you at any time of the day. Cool features include tracking your progress and setting reminders.
• Headspace: Headspace is known for helping people learn to meditate in just 10 easy minutes a day. Cool features include the ability to track your progress and to buddy up with a friend to help keep you motivated.
• Omvana: This app offers over 500 “transformative” audios to improve all areas of your life from work to personal relationships. Cool features include tracks to improve sleep, something more than a few of us might appreciate.
• Stop, Breathe, & Think: Quicker than Headspace, this app teaches you to meditate in 5 minutes a day and is easy to use at your workplace. Cool features include customizing meditations based upon your mood.
• Take a Break!: Ideal for the workplace, this app will help you carve out time each day to breathe, relax, and focus. Cool features include the ability to choose meditations with voice, music, or nature sounds.
4. Block a 10-minute mindfulness appointment on your schedule in the afternoon. Becoming more resilient will more than offset the short term lost revenue if you avoid retiring too soon due to burnout!
5. If you have an Apple watch, then try the new Breathe app. It reminds you to stop, breathe, and relax and even reports your heart rate afterward.
So unless you are expecting 2017 to be uneventful, I suggest you start building your mindfulness habit today.
Serenity now, serenity now.
Dr. Benabio is a partner physician in the department of dermatology of the Southern California Permanente Group in San Diego. Dr. Benabio is @Dermdoc on Twitter. Write to him at dermnews@frontlinemedcom.com . He has no disclosures related to this column.
How might mindfulness contribute to your mental collapse? Let’s say your work has become tedious. Tottering toward burnout, you decide to try mindfulness meditation to reverse your downward trend. However, you habitually fail to do your daily meditation. Now, “Meditate today” just piles on to your to-do list, a daily reminder of just how weak and disorganized you have become. Voila! Mindfulness is making you more crazy. There are things you can do to avoid this.
There are plenty of things to tip us doctors into burnout. We are not alone in the burnout epidemic, but we are overrepresented. More than 50% of physicians have burnout symptoms according to a recent Mayo Clinic study. Mindfulness training can help.
According to an interview with the program’s director, Douglas Zeidonis, MD, professor and chair of the department of psychiatry at the University of Massachusetts, most of the physicians reported that mindfulness training significantly benefited their work and personal lives. Mindfulness helped them feel more present and engaged with colleagues and patients and made them better clinicians – they reported showing more compassion toward patients.
Like any desirable habit, the key is to do it again and again and again. Here are a few recommendations to help you become more mindful during your workday.
1. Set random alarms (vibrate mode) on your smartphone to remind yourself to take a moment. When it goes off, do this: Breathe (4 seconds in, hold, then 8 seconds out) and be totally present for one minute.
2. Remove deliciously distracting apps from your phone’s home screen. Instead, tuck them away in a folder to reduce the likelihood you’ll click on them when you’re stressed.
3. Put meditation apps where you easily see them. You might try:
• The Mindfulness App: This app offers guided meditations in varying lengths from 3 to 30 minutes, so you can choose the one that’s right for you at any time of the day. Cool features include tracking your progress and setting reminders.
• Headspace: Headspace is known for helping people learn to meditate in just 10 easy minutes a day. Cool features include the ability to track your progress and to buddy up with a friend to help keep you motivated.
• Omvana: This app offers over 500 “transformative” audios to improve all areas of your life from work to personal relationships. Cool features include tracks to improve sleep, something more than a few of us might appreciate.
• Stop, Breathe, & Think: Quicker than Headspace, this app teaches you to meditate in 5 minutes a day and is easy to use at your workplace. Cool features include customizing meditations based upon your mood.
• Take a Break!: Ideal for the workplace, this app will help you carve out time each day to breathe, relax, and focus. Cool features include the ability to choose meditations with voice, music, or nature sounds.
4. Block a 10-minute mindfulness appointment on your schedule in the afternoon. Becoming more resilient will more than offset the short term lost revenue if you avoid retiring too soon due to burnout!
5. If you have an Apple watch, then try the new Breathe app. It reminds you to stop, breathe, and relax and even reports your heart rate afterward.
So unless you are expecting 2017 to be uneventful, I suggest you start building your mindfulness habit today.
Serenity now, serenity now.
Dr. Benabio is a partner physician in the department of dermatology of the Southern California Permanente Group in San Diego. Dr. Benabio is @Dermdoc on Twitter. Write to him at dermnews@frontlinemedcom.com . He has no disclosures related to this column.
How might mindfulness contribute to your mental collapse? Let’s say your work has become tedious. Tottering toward burnout, you decide to try mindfulness meditation to reverse your downward trend. However, you habitually fail to do your daily meditation. Now, “Meditate today” just piles on to your to-do list, a daily reminder of just how weak and disorganized you have become. Voila! Mindfulness is making you more crazy. There are things you can do to avoid this.
There are plenty of things to tip us doctors into burnout. We are not alone in the burnout epidemic, but we are overrepresented. More than 50% of physicians have burnout symptoms according to a recent Mayo Clinic study. Mindfulness training can help.
According to an interview with the program’s director, Douglas Zeidonis, MD, professor and chair of the department of psychiatry at the University of Massachusetts, most of the physicians reported that mindfulness training significantly benefited their work and personal lives. Mindfulness helped them feel more present and engaged with colleagues and patients and made them better clinicians – they reported showing more compassion toward patients.
Like any desirable habit, the key is to do it again and again and again. Here are a few recommendations to help you become more mindful during your workday.
1. Set random alarms (vibrate mode) on your smartphone to remind yourself to take a moment. When it goes off, do this: Breathe (4 seconds in, hold, then 8 seconds out) and be totally present for one minute.
2. Remove deliciously distracting apps from your phone’s home screen. Instead, tuck them away in a folder to reduce the likelihood you’ll click on them when you’re stressed.
3. Put meditation apps where you easily see them. You might try:
• The Mindfulness App: This app offers guided meditations in varying lengths from 3 to 30 minutes, so you can choose the one that’s right for you at any time of the day. Cool features include tracking your progress and setting reminders.
• Headspace: Headspace is known for helping people learn to meditate in just 10 easy minutes a day. Cool features include the ability to track your progress and to buddy up with a friend to help keep you motivated.
• Omvana: This app offers over 500 “transformative” audios to improve all areas of your life from work to personal relationships. Cool features include tracks to improve sleep, something more than a few of us might appreciate.
• Stop, Breathe, & Think: Quicker than Headspace, this app teaches you to meditate in 5 minutes a day and is easy to use at your workplace. Cool features include customizing meditations based upon your mood.
• Take a Break!: Ideal for the workplace, this app will help you carve out time each day to breathe, relax, and focus. Cool features include the ability to choose meditations with voice, music, or nature sounds.
4. Block a 10-minute mindfulness appointment on your schedule in the afternoon. Becoming more resilient will more than offset the short term lost revenue if you avoid retiring too soon due to burnout!
5. If you have an Apple watch, then try the new Breathe app. It reminds you to stop, breathe, and relax and even reports your heart rate afterward.
So unless you are expecting 2017 to be uneventful, I suggest you start building your mindfulness habit today.
Serenity now, serenity now.
Dr. Benabio is a partner physician in the department of dermatology of the Southern California Permanente Group in San Diego. Dr. Benabio is @Dermdoc on Twitter. Write to him at dermnews@frontlinemedcom.com . He has no disclosures related to this column.
How Often Do Children With Epilepsy Have Generalized Tonic–Clonic Seizures?
VANCOUVER—Children with epilepsy have a “remarkable” number of generalized tonic–clonic seizures over 25 years of follow-up, according to a study presented at the 45th Annual Meeting of the Child Neurology Society. Among patients who have more than 20 generalized tonic–clonic seizures, “only half … have normal intelligence, most have focal epilepsy, and the chance of eventual remission is only one-third,” said Carol Camfield, MD, and Peter Camfield, MD, Professors of Neurology at Dalhousie University in Halifax, Canada.

Generalized tonic–clonic seizures, either primarily generalized or focal with secondary generalization, “frighten families, are a risk factor for SUDEP [sudden, unexpected death in epilepsy], and dominate the public’s image of epilepsy,” the researchers said. To study how many children with epilepsy have convulsive seizures with loss of consciousness, how often they occur, and whether they are associated with an increased risk of SUDEP, the investigators analyzed data from 463 patients who had at least 10 years of follow-up in the Nova Scotia population-based childhood epilepsy study. The study population includes patients who had new-onset epilepsy of any kind between 1977 and 1985 and excludes patients with childhood absence epilepsy.
Among the patient characteristics noted in the study were number of generalized tonic–clonic seizures before and after diagnosis, presence of intellectual disability or neurologic abnormality, intractability, presence of terminal remission at the end of follow-up, number of antiepileptic drugs used, and cause of death.
Patients’ average age of epilepsy onset was 6.2, and average follow-up was 25.6 years. Overall, 359 patients (78%) had at least one generalized tonic–clonic seizure. Thirty percent of patients had between one and 10 generalized tonic–clonic seizures, 12% had between 11 and 20, 15% had between 21 and 99, and 21% had more than 100.
Within broad epilepsy syndrome groupings, the proportion of patients with more than 20 generalized tonic–clonic seizures was 40% among patients with focal epilepsy (95 of 235), 52% among patients with juvenile myoclonic epilepsy (11 of 21), and 62% among patients with symptomatic generalized epilepsy (45 of 73).
Forty-eight percent of patients with more than 20 generalized tonic–clonic seizures were intellectually disabled, compared with 12% of patients who had between one and 20 generalized tonic–clonic seizures.
Overall, 62% of patients were in terminal remission and off of antiepileptic drugs at the end of follow-up. Among patients with between one and 20 generalized tonic–clonic seizures, the proportion was 74%. Among patients with more than 20 generalized tonic–clonic seizures, the proportion was 33%.
One patient with intractable epilepsy died from SUDEP at age 23. The patient had more than 100 generalized tonic–clonic seizures.
The researchers noted that patients’ total number of seizures may be over- or underestimated due to caregiver report and the medical record used.
—Jake Remaly
VANCOUVER—Children with epilepsy have a “remarkable” number of generalized tonic–clonic seizures over 25 years of follow-up, according to a study presented at the 45th Annual Meeting of the Child Neurology Society. Among patients who have more than 20 generalized tonic–clonic seizures, “only half … have normal intelligence, most have focal epilepsy, and the chance of eventual remission is only one-third,” said Carol Camfield, MD, and Peter Camfield, MD, Professors of Neurology at Dalhousie University in Halifax, Canada.
Generalized tonic–clonic seizures, either primarily generalized or focal with secondary generalization, “frighten families, are a risk factor for SUDEP [sudden, unexpected death in epilepsy], and dominate the public’s image of epilepsy,” the researchers said. To study how many children with epilepsy have convulsive seizures with loss of consciousness, how often they occur, and whether they are associated with an increased risk of SUDEP, the investigators analyzed data from 463 patients who had at least 10 years of follow-up in the Nova Scotia population-based childhood epilepsy study. The study population includes patients who had new-onset epilepsy of any kind between 1977 and 1985 and excludes patients with childhood absence epilepsy.
Among the patient characteristics noted in the study were number of generalized tonic–clonic seizures before and after diagnosis, presence of intellectual disability or neurologic abnormality, intractability, presence of terminal remission at the end of follow-up, number of antiepileptic drugs used, and cause of death.
Patients’ average age of epilepsy onset was 6.2, and average follow-up was 25.6 years. Overall, 359 patients (78%) had at least one generalized tonic–clonic seizure. Thirty percent of patients had between one and 10 generalized tonic–clonic seizures, 12% had between 11 and 20, 15% had between 21 and 99, and 21% had more than 100.
Within broad epilepsy syndrome groupings, the proportion of patients with more than 20 generalized tonic–clonic seizures was 40% among patients with focal epilepsy (95 of 235), 52% among patients with juvenile myoclonic epilepsy (11 of 21), and 62% among patients with symptomatic generalized epilepsy (45 of 73).
Forty-eight percent of patients with more than 20 generalized tonic–clonic seizures were intellectually disabled, compared with 12% of patients who had between one and 20 generalized tonic–clonic seizures.
Overall, 62% of patients were in terminal remission and off of antiepileptic drugs at the end of follow-up. Among patients with between one and 20 generalized tonic–clonic seizures, the proportion was 74%. Among patients with more than 20 generalized tonic–clonic seizures, the proportion was 33%.
One patient with intractable epilepsy died from SUDEP at age 23. The patient had more than 100 generalized tonic–clonic seizures.
The researchers noted that patients’ total number of seizures may be over- or underestimated due to caregiver report and the medical record used.
—Jake Remaly
VANCOUVER—Children with epilepsy have a “remarkable” number of generalized tonic–clonic seizures over 25 years of follow-up, according to a study presented at the 45th Annual Meeting of the Child Neurology Society. Among patients who have more than 20 generalized tonic–clonic seizures, “only half … have normal intelligence, most have focal epilepsy, and the chance of eventual remission is only one-third,” said Carol Camfield, MD, and Peter Camfield, MD, Professors of Neurology at Dalhousie University in Halifax, Canada.
Generalized tonic–clonic seizures, either primarily generalized or focal with secondary generalization, “frighten families, are a risk factor for SUDEP [sudden, unexpected death in epilepsy], and dominate the public’s image of epilepsy,” the researchers said. To study how many children with epilepsy have convulsive seizures with loss of consciousness, how often they occur, and whether they are associated with an increased risk of SUDEP, the investigators analyzed data from 463 patients who had at least 10 years of follow-up in the Nova Scotia population-based childhood epilepsy study. The study population includes patients who had new-onset epilepsy of any kind between 1977 and 1985 and excludes patients with childhood absence epilepsy.
Among the patient characteristics noted in the study were number of generalized tonic–clonic seizures before and after diagnosis, presence of intellectual disability or neurologic abnormality, intractability, presence of terminal remission at the end of follow-up, number of antiepileptic drugs used, and cause of death.
Patients’ average age of epilepsy onset was 6.2, and average follow-up was 25.6 years. Overall, 359 patients (78%) had at least one generalized tonic–clonic seizure. Thirty percent of patients had between one and 10 generalized tonic–clonic seizures, 12% had between 11 and 20, 15% had between 21 and 99, and 21% had more than 100.
Within broad epilepsy syndrome groupings, the proportion of patients with more than 20 generalized tonic–clonic seizures was 40% among patients with focal epilepsy (95 of 235), 52% among patients with juvenile myoclonic epilepsy (11 of 21), and 62% among patients with symptomatic generalized epilepsy (45 of 73).
Forty-eight percent of patients with more than 20 generalized tonic–clonic seizures were intellectually disabled, compared with 12% of patients who had between one and 20 generalized tonic–clonic seizures.
Overall, 62% of patients were in terminal remission and off of antiepileptic drugs at the end of follow-up. Among patients with between one and 20 generalized tonic–clonic seizures, the proportion was 74%. Among patients with more than 20 generalized tonic–clonic seizures, the proportion was 33%.
One patient with intractable epilepsy died from SUDEP at age 23. The patient had more than 100 generalized tonic–clonic seizures.
The researchers noted that patients’ total number of seizures may be over- or underestimated due to caregiver report and the medical record used.
—Jake Remaly

View of medical cannabis in psychiatry may be changing
Some psychiatrists and other physicians who treat pain have been open for some time to recommending medical cannabis for their patients. The psychiatric community in general, however – largely because of concerns about addiction and psychosis – has been reluctant to do so, despite the drug’s proven benefits for illnesses such as anxiety, posttraumatic stress disorder, and mood disorders.
But this reluctance might be changing, some psychiatrists say.
“Clinicians are increasingly open to this kind of dialogue,” said Christopher G. Fichtner, MD, a Fellow of the American Psychiatric Association (FAPA), and clinical professor of psychiatry at the University of California, Riverside. “As a doctor, I try to be aware of how my patients are using cannabis, what benefits they may attribute to it, and whether they have problems related to its use. At times, it may be possible to advise them about the value, for example, of products rich in cannabidiol (CBD) – known to be anxiolytic, possibly antipsychotic, and potentially mood stabilizing – as opposed to products higher in tetrahydrocannabinol (THC) that are more psychoactive and may aggravate psychotic symptoms for some patients.
“Our ability to advise patients along these lines is helpful to the extent that patients are able to consistently obtain reliable cannabis products,” Dr. Fichtner said, noting that European and Brazilian research supports the view that CBD might have some antipsychotic properties (Schizophr Res. March 2015;162[1-3]:153-61) and (Schizophr Res. 2015 May;164:[1-3]:155-63).
Currently, 25 states and the District of Columbia have legalized cannabis for medical use; and voters in three additional states – Arkansas, Florida, and North Dakota – approved medical cannabis measures on Nov. 8. “Although medical marijuana laws have not made it possible to prescribe cannabis in the strict sense, they allow doctors to recommend it for patients who experience benefit from using it,” Dr. Fichtner said. “Cannabinoid products available in dispensaries are not subject to the same level of regulation and quality control as Food and Drug Administration–approved medicines, but they are often labeled qualitatively and/or quantitatively with the result that they probably are more reliable than marijuana obtained on the streets.”
Traditional vs. counter views
The American Psychiatric Association came out strongly against cannabis use for medicinal purposes in 2013. The APA’s position statement cites the absence of “current scientific evidence” showing that marijuana is in any way beneficial for treating “any psychiatric disorder. In contrast, current evidence supports, at minimum, a strong association of cannabis use with the onset of psychiatric disorders,” the statement says. Likewise, the American Medical Association reaffirmed its opposition to the use of medical cannabis in 2013, citing the drug as a “public health concern.” Another medical group that is in strong opposition to the use of medical cannabis is the American Society of Addiction Medicine (ASAM). In addition to rejecting smoking as a means of delivery, ASAM’s policy statement says “cannabis, cannabis-based products, and cannabis delivery devices should be subject to the same standards that are applicable to other prescription medications and medical devices.”
In a policy revised in 2015, the National Institute on Drug Abuse (NIDA) said that although the FDA has neither recognized nor approved the marijuana plant as medicine, the FDA has approved three medications that contain cannabinoid chemicals. Marinol and Syndros, which include dronabinol, a synthetic delta-9-THC, are approved for treating anorexia tied to weight loss in patients with AIDS. Cesamet, which contains nabilone and has a chemical structure that is similar to THC, also has received FDA approval for nausea and vomiting caused by cancer chemotherapy.
Earlier this year, a new organization founded by David L. Nathan, MD, a Distinguished Fellow of the APA (DFAPA), joined the cannabis debate. The group, known as Doctors for Cannabis Regulation (DFCR), does not advocate for the use of medical cannabis. However, DFCR does support, among other things, “cannabis legalization for adults, preventive education of minors, and regulation of the industry.”

DFCR argues that prohibiting cannabis overburdens the criminal justice system, drains law enforcement, and treats African Americans unfairly, saying that population is “nearly four times more likely than whites to be arrested for cannabis possession, despite similar usage rates between the two groups.” The group, which launched with more than 40 founding members – including 10 psychiatrists – also cites evidence showing that medical cannabis use is correlated with a 25% reduction in deaths from opioid use. In 2014, Marcus A. Bachhuber, MD, of the Philadelphia VA Center, and his colleagues reached those conclusions after comparing three states with medical cannabis laws that were effective before 1999 with 10 states that had enacted medical cannabis laws between 1999 and 2010 (JAMA Intern Med. 2014;174[10]:166-73).
“This is a rapidly changing area; new therapeutics are being developed; and there is great potential for good here” as well as the potential for risks, just as with any other medicine,” said Dan Morhaim, MD, an emergency medicine physician who represents Baltimore County’s 11th legislative district in the Maryland House of Delegates. “It’s not the devil weed; nor is it a panacea,” said Dr. Morhaim, who was active in the process of passing Maryland’s medical cannabis law. “Our job ought to be to see it as another tool in the toolbox to be used responsibly, just as we would with any other medicine.”
Weighing U.S. data
Most of the data on cannabis come from studies of recreational use, Dr. Daviss said. “We know that it can contribute to symptoms of depression, anxiety, psychosis, and addiction. Younger users are particularly at risk for some of these symptoms. We don’t really know how much of this risk applies to monitored medical use,” he said. “And some states have no requirements for monitoring and modulating the dose, so risk data from these states will be different from states where medical use is more regulated and more similar to standard medical care.”
In addition, data on the benefits of marijuana for psychiatric conditions are limited, Dr. Daviss said. “I have certainly heard directly from patients about how marijuana helps them,” he said. “These comments often suggest improvements in sleep, appetite, mood, and anxiety. “A major concern of psychiatric clinicians is that many people asking for medical cannabis are providing contrived reports of symptoms in an attempt to obtain marijuana for nonmedical (recreational) use,” or to share or sell, he said.
Clinicians also remain concerned that “one’s state medical license or federal [Drug Enforcement Administration] license could be at risk for ‘prescribing’ or ‘recommending’ medical cannabis,” Dr. Daviss said. “Being known as a ‘pot doc’ is not viewed as a career booster and could be seen as a risk factor for enhanced regulatory scrutiny of one’s practice.”
However, “there are many who balance those concerns with a harm reduction approach that acknowledges that drug cartels and gangs are fueled by marijuana sales, resulting in preventable violence and deaths, marijuana of unknown quality or adulteration, and negative criminal justice consequences for what should be viewed as a public health problem rather than a criminal problem,” Dr. Daviss said. “Such a harm reduction perspective may increase the likelihood that some physicians will support the use of either medical cannabis or recreational marijuana.”
As more researchers study medical marijuana, the concerns of psychiatrists will evolve along with the science, Dr. Daviss predicted. Relevant outcomes research would include comparative effectiveness studies such as a randomized, double-blind trials of standardized cannabis formulations versus standard treatment for depression or anxiety, he said.
There’s not enough evidence for specific contraindications for medical marijuana use in psychiatry, Dr. Daviss said. However, most clinicians would say that a psychotic disorder such as schizophrenia is a contraindication, because some users of medical marijuana experience paranoia and other psychotic symptoms, he noted.
Dr. Fichtner agreed, and said “studies do find an association between adolescent marijuana use and later development of psychotic disorders, including schizophrenia.
“Even though authors of such studies extrapolate their findings to public health implications in terms of reducing the number of cases of schizophrenia through prevention of marijuana use, such a cause and effect relationship is far from clear – and such conclusions are unwarranted,” he said.
In fact, he said, at least one small case series found prescription molecular THC to be a helpful adjunct to approved antipsychotic medications in schizophrenia patients who reported symptomatic benefit from previous marijuana use (J Clin Psychopharmacol. 2009 Jun;29[3]:255-8). “Even more compelling,” Dr. Fichtner said, “are data on antipsychotic effects of molecular CBD, an important component of herbal cannabis that does not have THC-like psychoactive effects but has been demonstrated to have anxiolytic properties in a number of human studies.”
Dr. Fichtner said authors of research articles on the association between schizophrenia and cannabis use rarely have grappled with the possibility that patients might experience therapeutic benefits through self-medication. In addition, he said, “there are findings in the literature suggesting that among patients with schizophrenia, those using marijuana may have fewer and more selective cognitive deficits than those who do not use marijuana.”
For his part, Dr. Morhaim also supports an open approach. “All health professionals, psychiatrists included, need to consider cannabis as any other medicine,” Dr. Morhaim said. “It has its uses and side effects; reasons to initiate use and reasons to stop; diagnoses for which it is generally accepted that it works (multiple sclerosis, nausea from cancer chemotherapy, and appetite loss) and ones where [effects are] less clear. The medical-scientific potential, however, is expanding rapidly as the shackles that have blocked proper cannabis research are coming off,” he noted.
Several large studies evaluating the use of cannabis for mental illness are underway, Dr. Morhaim said. As the science expands, recent studies have examined the role of cannabis for treating a range of medical conditions, including Alzheimer’s disease, multiple sclerosis, migraine, and even fracture healing and acne, he said. Still, data remain sparse. For example, a literature review that looked at randomized, clinical trials and meta-analyses tied to marijuana and other cannabinoids, and specific diagnoses found no trials that examined marijuana’s efficacy for treating Tourette disorder, PTSD, or Alzheimer’s disease (J Clin Psychiatry. 2016 Aug;77[8]:1050-64). “Given its rapidly changing legal status, there is an urgent need to conduct double-blind, randomized, placebo- and active-controlled studies on the efficacy and safety of marijuana or its constituent cannabinoids for psychiatric conditions,” wrote Samuel T. Wilkinson, MD, of the department of psychiatry at Yale University, New Haven, Conn., and his associates. Meanwhile, a matched case-control, cross-sectional evaluation of veterans with probable PTSD found no significant differences between cases and controls in PTSD scores and frequency of cannabis use (J Affect Disord. 2016 Jan 15;190:439-42).
Nevertheless, “psychiatrists ought to be familiar with medical cannabis, because they may be taking care of patients who are on it for other (somatic) reasons,” Dr. Morhaim said. “This is just like my work as an emergency doctor; I don’t prescribe psych medicines, but I have to know about them, because patients come to the ER who are on them.”
Lessons from patients’ reports
Rachna J. Patel, DO, a psychiatrist who describes herself on her website as “The Medical Marijuana Expert,” treats patients with anxiety and PTSD, and agrees that marijuana is a tool that, when used correctly, can benefit patients. “A visit with me, a medical marijuana doctor, is much like a visit with any other doctor,” said Dr. Patel, who practices in Walnut Creek, Calif.
“I have my patients complete a form that asks about their medical history, as well as the treatments they’ve sought out for their anxiety and/or PTSD, such as prescription medications, therapy, and other methods,” she said. “I then sit down with the patient to go over their history and do a focused physical exam. Once I determine that the patient’s medical condition will benefit with the use of medical marijuana, I spend the remainder of the time step-by-step walking him or her through how to use medical marijuana for the anxiety and/or PTSD,” she added.
Dr. Patel applies some basic considerations when assessing patients for potential medical marijuana treatment, notably the efficacy and side effects of past treatments and patient concerns about past treatments, including the potential for dependency or addiction. She also assesses the impact of anxiety or PTSD on the patient’s quality of life.
Education is key, she said, to addressing patients’ concerns and managing possible risks. “The risks are generally minimized and even eliminated by educating patients on selection of the appropriate combination of cannabinoids, as well as teaching them to use medical marijuana at the lowest effective dose and with the lowest effective frequency,” she said.
“Having practiced in the field of medical marijuana for 4-plus years, I haven’t found marijuana to be a gateway drug,” she said. “The patients I see aren’t looking to ‘get high’ off of medical marijuana. In fact, they’re seeking to get off of the pharmaceutical drugs they’ve been prescribed.”
Dr. Patel walks patients through what she calls “The Patel Protocol for Medical Marijuana,” which includes “teaching patients how to select the appropriate combination of cannabinoids in marijuana products, figuring out how much to use and how often to use it, how to avoid side effects, and what to do in case patients do experience side effects,” she said.
In her experience, “when it comes to dosing, just like with many pharmaceutical medications, it really varies from patient to patient, even more so because cannabinoids are hydrophobic, so their pharmacology is not typical of most pharmaceutical medications.” Dr. Patel follows up with her patients often by phone after the initial office visit, then continues with office visits at least once a year.
“The best way any doctor can help to monitor their patients’ use of medical marijuana is to be as informed about it as any other pharmaceutical drug they’re prescribing,” she said. “Become familiar with the pharmacology [of medical marijuana],” she advised. “Know what scenarios could lead to side effects and how those side effects can be avoided. Know in what scenarios it [marijuana] can exacerbate underlying medical conditions.”
Dr. Fichtner agrees with Dr. Patel’s approach but adds that he generally does not recommend cannabis as a first-line treatment for his patients with serious mental illness. Rather, his approach generally is to prescribe evidence-based treatments.
“But when I do find a history of marijuana use and the patient reports it has been helpful, I work with the patient to assess possible benefits in the areas of symptom management and possible reductions in polypharmacy,” he said. “The latter is of particular concern in PTSD, where patients may be on complex multidrug regimens involving off-label medication prescriptions with only partial symptom relief. It is important to take the patient’s claim seriously, rather than merely jumping to the conclusion that they have a substance use disorder that is undermining their psychiatric care.”
Dr. Patel said a surprising and positive side effect she has observed during her years of recommending medical marijuana is that patients’ use of any anxiety and PTSD medication decreases over time.
“Even though I never expected it, with the use of medical marijuana, many of my patients have been able to significantly reduce or even eliminate the use of their prescription medications for their anxiety and/or PTSD,” she said. “Over time, I find that patients use the medical marijuana on an as-needed basis.”
Dr. Fichtner is the author of book, “Cannabinomics: The Marijuana Policy Tipping Point” (Northbrook, Ill.: Well Mind Books, 2010). Dr. Daviss, Dr. Morhaim, and Dr. Patel had no relevant financial conflicts to disclose.
Some psychiatrists and other physicians who treat pain have been open for some time to recommending medical cannabis for their patients. The psychiatric community in general, however – largely because of concerns about addiction and psychosis – has been reluctant to do so, despite the drug’s proven benefits for illnesses such as anxiety, posttraumatic stress disorder, and mood disorders.
But this reluctance might be changing, some psychiatrists say.
“Clinicians are increasingly open to this kind of dialogue,” said Christopher G. Fichtner, MD, a Fellow of the American Psychiatric Association (FAPA), and clinical professor of psychiatry at the University of California, Riverside. “As a doctor, I try to be aware of how my patients are using cannabis, what benefits they may attribute to it, and whether they have problems related to its use. At times, it may be possible to advise them about the value, for example, of products rich in cannabidiol (CBD) – known to be anxiolytic, possibly antipsychotic, and potentially mood stabilizing – as opposed to products higher in tetrahydrocannabinol (THC) that are more psychoactive and may aggravate psychotic symptoms for some patients.
“Our ability to advise patients along these lines is helpful to the extent that patients are able to consistently obtain reliable cannabis products,” Dr. Fichtner said, noting that European and Brazilian research supports the view that CBD might have some antipsychotic properties (Schizophr Res. March 2015;162[1-3]:153-61) and (Schizophr Res. 2015 May;164:[1-3]:155-63).
Currently, 25 states and the District of Columbia have legalized cannabis for medical use; and voters in three additional states – Arkansas, Florida, and North Dakota – approved medical cannabis measures on Nov. 8. “Although medical marijuana laws have not made it possible to prescribe cannabis in the strict sense, they allow doctors to recommend it for patients who experience benefit from using it,” Dr. Fichtner said. “Cannabinoid products available in dispensaries are not subject to the same level of regulation and quality control as Food and Drug Administration–approved medicines, but they are often labeled qualitatively and/or quantitatively with the result that they probably are more reliable than marijuana obtained on the streets.”
Traditional vs. counter views
The American Psychiatric Association came out strongly against cannabis use for medicinal purposes in 2013. The APA’s position statement cites the absence of “current scientific evidence” showing that marijuana is in any way beneficial for treating “any psychiatric disorder. In contrast, current evidence supports, at minimum, a strong association of cannabis use with the onset of psychiatric disorders,” the statement says. Likewise, the American Medical Association reaffirmed its opposition to the use of medical cannabis in 2013, citing the drug as a “public health concern.” Another medical group that is in strong opposition to the use of medical cannabis is the American Society of Addiction Medicine (ASAM). In addition to rejecting smoking as a means of delivery, ASAM’s policy statement says “cannabis, cannabis-based products, and cannabis delivery devices should be subject to the same standards that are applicable to other prescription medications and medical devices.”
In a policy revised in 2015, the National Institute on Drug Abuse (NIDA) said that although the FDA has neither recognized nor approved the marijuana plant as medicine, the FDA has approved three medications that contain cannabinoid chemicals. Marinol and Syndros, which include dronabinol, a synthetic delta-9-THC, are approved for treating anorexia tied to weight loss in patients with AIDS. Cesamet, which contains nabilone and has a chemical structure that is similar to THC, also has received FDA approval for nausea and vomiting caused by cancer chemotherapy.
Earlier this year, a new organization founded by David L. Nathan, MD, a Distinguished Fellow of the APA (DFAPA), joined the cannabis debate. The group, known as Doctors for Cannabis Regulation (DFCR), does not advocate for the use of medical cannabis. However, DFCR does support, among other things, “cannabis legalization for adults, preventive education of minors, and regulation of the industry.”
DFCR argues that prohibiting cannabis overburdens the criminal justice system, drains law enforcement, and treats African Americans unfairly, saying that population is “nearly four times more likely than whites to be arrested for cannabis possession, despite similar usage rates between the two groups.” The group, which launched with more than 40 founding members – including 10 psychiatrists – also cites evidence showing that medical cannabis use is correlated with a 25% reduction in deaths from opioid use. In 2014, Marcus A. Bachhuber, MD, of the Philadelphia VA Center, and his colleagues reached those conclusions after comparing three states with medical cannabis laws that were effective before 1999 with 10 states that had enacted medical cannabis laws between 1999 and 2010 (JAMA Intern Med. 2014;174[10]:166-73).
“This is a rapidly changing area; new therapeutics are being developed; and there is great potential for good here” as well as the potential for risks, just as with any other medicine,” said Dan Morhaim, MD, an emergency medicine physician who represents Baltimore County’s 11th legislative district in the Maryland House of Delegates. “It’s not the devil weed; nor is it a panacea,” said Dr. Morhaim, who was active in the process of passing Maryland’s medical cannabis law. “Our job ought to be to see it as another tool in the toolbox to be used responsibly, just as we would with any other medicine.”
Weighing U.S. data
Most of the data on cannabis come from studies of recreational use, Dr. Daviss said. “We know that it can contribute to symptoms of depression, anxiety, psychosis, and addiction. Younger users are particularly at risk for some of these symptoms. We don’t really know how much of this risk applies to monitored medical use,” he said. “And some states have no requirements for monitoring and modulating the dose, so risk data from these states will be different from states where medical use is more regulated and more similar to standard medical care.”
In addition, data on the benefits of marijuana for psychiatric conditions are limited, Dr. Daviss said. “I have certainly heard directly from patients about how marijuana helps them,” he said. “These comments often suggest improvements in sleep, appetite, mood, and anxiety. “A major concern of psychiatric clinicians is that many people asking for medical cannabis are providing contrived reports of symptoms in an attempt to obtain marijuana for nonmedical (recreational) use,” or to share or sell, he said.
Clinicians also remain concerned that “one’s state medical license or federal [Drug Enforcement Administration] license could be at risk for ‘prescribing’ or ‘recommending’ medical cannabis,” Dr. Daviss said. “Being known as a ‘pot doc’ is not viewed as a career booster and could be seen as a risk factor for enhanced regulatory scrutiny of one’s practice.”
However, “there are many who balance those concerns with a harm reduction approach that acknowledges that drug cartels and gangs are fueled by marijuana sales, resulting in preventable violence and deaths, marijuana of unknown quality or adulteration, and negative criminal justice consequences for what should be viewed as a public health problem rather than a criminal problem,” Dr. Daviss said. “Such a harm reduction perspective may increase the likelihood that some physicians will support the use of either medical cannabis or recreational marijuana.”
As more researchers study medical marijuana, the concerns of psychiatrists will evolve along with the science, Dr. Daviss predicted. Relevant outcomes research would include comparative effectiveness studies such as a randomized, double-blind trials of standardized cannabis formulations versus standard treatment for depression or anxiety, he said.
There’s not enough evidence for specific contraindications for medical marijuana use in psychiatry, Dr. Daviss said. However, most clinicians would say that a psychotic disorder such as schizophrenia is a contraindication, because some users of medical marijuana experience paranoia and other psychotic symptoms, he noted.
Dr. Fichtner agreed, and said “studies do find an association between adolescent marijuana use and later development of psychotic disorders, including schizophrenia.
“Even though authors of such studies extrapolate their findings to public health implications in terms of reducing the number of cases of schizophrenia through prevention of marijuana use, such a cause and effect relationship is far from clear – and such conclusions are unwarranted,” he said.
In fact, he said, at least one small case series found prescription molecular THC to be a helpful adjunct to approved antipsychotic medications in schizophrenia patients who reported symptomatic benefit from previous marijuana use (J Clin Psychopharmacol. 2009 Jun;29[3]:255-8). “Even more compelling,” Dr. Fichtner said, “are data on antipsychotic effects of molecular CBD, an important component of herbal cannabis that does not have THC-like psychoactive effects but has been demonstrated to have anxiolytic properties in a number of human studies.”
Dr. Fichtner said authors of research articles on the association between schizophrenia and cannabis use rarely have grappled with the possibility that patients might experience therapeutic benefits through self-medication. In addition, he said, “there are findings in the literature suggesting that among patients with schizophrenia, those using marijuana may have fewer and more selective cognitive deficits than those who do not use marijuana.”
For his part, Dr. Morhaim also supports an open approach. “All health professionals, psychiatrists included, need to consider cannabis as any other medicine,” Dr. Morhaim said. “It has its uses and side effects; reasons to initiate use and reasons to stop; diagnoses for which it is generally accepted that it works (multiple sclerosis, nausea from cancer chemotherapy, and appetite loss) and ones where [effects are] less clear. The medical-scientific potential, however, is expanding rapidly as the shackles that have blocked proper cannabis research are coming off,” he noted.
Several large studies evaluating the use of cannabis for mental illness are underway, Dr. Morhaim said. As the science expands, recent studies have examined the role of cannabis for treating a range of medical conditions, including Alzheimer’s disease, multiple sclerosis, migraine, and even fracture healing and acne, he said. Still, data remain sparse. For example, a literature review that looked at randomized, clinical trials and meta-analyses tied to marijuana and other cannabinoids, and specific diagnoses found no trials that examined marijuana’s efficacy for treating Tourette disorder, PTSD, or Alzheimer’s disease (J Clin Psychiatry. 2016 Aug;77[8]:1050-64). “Given its rapidly changing legal status, there is an urgent need to conduct double-blind, randomized, placebo- and active-controlled studies on the efficacy and safety of marijuana or its constituent cannabinoids for psychiatric conditions,” wrote Samuel T. Wilkinson, MD, of the department of psychiatry at Yale University, New Haven, Conn., and his associates. Meanwhile, a matched case-control, cross-sectional evaluation of veterans with probable PTSD found no significant differences between cases and controls in PTSD scores and frequency of cannabis use (J Affect Disord. 2016 Jan 15;190:439-42).
Nevertheless, “psychiatrists ought to be familiar with medical cannabis, because they may be taking care of patients who are on it for other (somatic) reasons,” Dr. Morhaim said. “This is just like my work as an emergency doctor; I don’t prescribe psych medicines, but I have to know about them, because patients come to the ER who are on them.”
Lessons from patients’ reports
Rachna J. Patel, DO, a psychiatrist who describes herself on her website as “The Medical Marijuana Expert,” treats patients with anxiety and PTSD, and agrees that marijuana is a tool that, when used correctly, can benefit patients. “A visit with me, a medical marijuana doctor, is much like a visit with any other doctor,” said Dr. Patel, who practices in Walnut Creek, Calif.
“I have my patients complete a form that asks about their medical history, as well as the treatments they’ve sought out for their anxiety and/or PTSD, such as prescription medications, therapy, and other methods,” she said. “I then sit down with the patient to go over their history and do a focused physical exam. Once I determine that the patient’s medical condition will benefit with the use of medical marijuana, I spend the remainder of the time step-by-step walking him or her through how to use medical marijuana for the anxiety and/or PTSD,” she added.
Dr. Patel applies some basic considerations when assessing patients for potential medical marijuana treatment, notably the efficacy and side effects of past treatments and patient concerns about past treatments, including the potential for dependency or addiction. She also assesses the impact of anxiety or PTSD on the patient’s quality of life.
Education is key, she said, to addressing patients’ concerns and managing possible risks. “The risks are generally minimized and even eliminated by educating patients on selection of the appropriate combination of cannabinoids, as well as teaching them to use medical marijuana at the lowest effective dose and with the lowest effective frequency,” she said.
“Having practiced in the field of medical marijuana for 4-plus years, I haven’t found marijuana to be a gateway drug,” she said. “The patients I see aren’t looking to ‘get high’ off of medical marijuana. In fact, they’re seeking to get off of the pharmaceutical drugs they’ve been prescribed.”
Dr. Patel walks patients through what she calls “The Patel Protocol for Medical Marijuana,” which includes “teaching patients how to select the appropriate combination of cannabinoids in marijuana products, figuring out how much to use and how often to use it, how to avoid side effects, and what to do in case patients do experience side effects,” she said.
In her experience, “when it comes to dosing, just like with many pharmaceutical medications, it really varies from patient to patient, even more so because cannabinoids are hydrophobic, so their pharmacology is not typical of most pharmaceutical medications.” Dr. Patel follows up with her patients often by phone after the initial office visit, then continues with office visits at least once a year.
“The best way any doctor can help to monitor their patients’ use of medical marijuana is to be as informed about it as any other pharmaceutical drug they’re prescribing,” she said. “Become familiar with the pharmacology [of medical marijuana],” she advised. “Know what scenarios could lead to side effects and how those side effects can be avoided. Know in what scenarios it [marijuana] can exacerbate underlying medical conditions.”
Dr. Fichtner agrees with Dr. Patel’s approach but adds that he generally does not recommend cannabis as a first-line treatment for his patients with serious mental illness. Rather, his approach generally is to prescribe evidence-based treatments.
“But when I do find a history of marijuana use and the patient reports it has been helpful, I work with the patient to assess possible benefits in the areas of symptom management and possible reductions in polypharmacy,” he said. “The latter is of particular concern in PTSD, where patients may be on complex multidrug regimens involving off-label medication prescriptions with only partial symptom relief. It is important to take the patient’s claim seriously, rather than merely jumping to the conclusion that they have a substance use disorder that is undermining their psychiatric care.”
Dr. Patel said a surprising and positive side effect she has observed during her years of recommending medical marijuana is that patients’ use of any anxiety and PTSD medication decreases over time.
“Even though I never expected it, with the use of medical marijuana, many of my patients have been able to significantly reduce or even eliminate the use of their prescription medications for their anxiety and/or PTSD,” she said. “Over time, I find that patients use the medical marijuana on an as-needed basis.”
Dr. Fichtner is the author of book, “Cannabinomics: The Marijuana Policy Tipping Point” (Northbrook, Ill.: Well Mind Books, 2010). Dr. Daviss, Dr. Morhaim, and Dr. Patel had no relevant financial conflicts to disclose.
Some psychiatrists and other physicians who treat pain have been open for some time to recommending medical cannabis for their patients. The psychiatric community in general, however – largely because of concerns about addiction and psychosis – has been reluctant to do so, despite the drug’s proven benefits for illnesses such as anxiety, posttraumatic stress disorder, and mood disorders.
But this reluctance might be changing, some psychiatrists say.
“Clinicians are increasingly open to this kind of dialogue,” said Christopher G. Fichtner, MD, a Fellow of the American Psychiatric Association (FAPA), and clinical professor of psychiatry at the University of California, Riverside. “As a doctor, I try to be aware of how my patients are using cannabis, what benefits they may attribute to it, and whether they have problems related to its use. At times, it may be possible to advise them about the value, for example, of products rich in cannabidiol (CBD) – known to be anxiolytic, possibly antipsychotic, and potentially mood stabilizing – as opposed to products higher in tetrahydrocannabinol (THC) that are more psychoactive and may aggravate psychotic symptoms for some patients.
“Our ability to advise patients along these lines is helpful to the extent that patients are able to consistently obtain reliable cannabis products,” Dr. Fichtner said, noting that European and Brazilian research supports the view that CBD might have some antipsychotic properties (Schizophr Res. March 2015;162[1-3]:153-61) and (Schizophr Res. 2015 May;164:[1-3]:155-63).
Currently, 25 states and the District of Columbia have legalized cannabis for medical use; and voters in three additional states – Arkansas, Florida, and North Dakota – approved medical cannabis measures on Nov. 8. “Although medical marijuana laws have not made it possible to prescribe cannabis in the strict sense, they allow doctors to recommend it for patients who experience benefit from using it,” Dr. Fichtner said. “Cannabinoid products available in dispensaries are not subject to the same level of regulation and quality control as Food and Drug Administration–approved medicines, but they are often labeled qualitatively and/or quantitatively with the result that they probably are more reliable than marijuana obtained on the streets.”
Traditional vs. counter views
The American Psychiatric Association came out strongly against cannabis use for medicinal purposes in 2013. The APA’s position statement cites the absence of “current scientific evidence” showing that marijuana is in any way beneficial for treating “any psychiatric disorder. In contrast, current evidence supports, at minimum, a strong association of cannabis use with the onset of psychiatric disorders,” the statement says. Likewise, the American Medical Association reaffirmed its opposition to the use of medical cannabis in 2013, citing the drug as a “public health concern.” Another medical group that is in strong opposition to the use of medical cannabis is the American Society of Addiction Medicine (ASAM). In addition to rejecting smoking as a means of delivery, ASAM’s policy statement says “cannabis, cannabis-based products, and cannabis delivery devices should be subject to the same standards that are applicable to other prescription medications and medical devices.”
In a policy revised in 2015, the National Institute on Drug Abuse (NIDA) said that although the FDA has neither recognized nor approved the marijuana plant as medicine, the FDA has approved three medications that contain cannabinoid chemicals. Marinol and Syndros, which include dronabinol, a synthetic delta-9-THC, are approved for treating anorexia tied to weight loss in patients with AIDS. Cesamet, which contains nabilone and has a chemical structure that is similar to THC, also has received FDA approval for nausea and vomiting caused by cancer chemotherapy.
Earlier this year, a new organization founded by David L. Nathan, MD, a Distinguished Fellow of the APA (DFAPA), joined the cannabis debate. The group, known as Doctors for Cannabis Regulation (DFCR), does not advocate for the use of medical cannabis. However, DFCR does support, among other things, “cannabis legalization for adults, preventive education of minors, and regulation of the industry.”
DFCR argues that prohibiting cannabis overburdens the criminal justice system, drains law enforcement, and treats African Americans unfairly, saying that population is “nearly four times more likely than whites to be arrested for cannabis possession, despite similar usage rates between the two groups.” The group, which launched with more than 40 founding members – including 10 psychiatrists – also cites evidence showing that medical cannabis use is correlated with a 25% reduction in deaths from opioid use. In 2014, Marcus A. Bachhuber, MD, of the Philadelphia VA Center, and his colleagues reached those conclusions after comparing three states with medical cannabis laws that were effective before 1999 with 10 states that had enacted medical cannabis laws between 1999 and 2010 (JAMA Intern Med. 2014;174[10]:166-73).
“This is a rapidly changing area; new therapeutics are being developed; and there is great potential for good here” as well as the potential for risks, just as with any other medicine,” said Dan Morhaim, MD, an emergency medicine physician who represents Baltimore County’s 11th legislative district in the Maryland House of Delegates. “It’s not the devil weed; nor is it a panacea,” said Dr. Morhaim, who was active in the process of passing Maryland’s medical cannabis law. “Our job ought to be to see it as another tool in the toolbox to be used responsibly, just as we would with any other medicine.”
Weighing U.S. data
Most of the data on cannabis come from studies of recreational use, Dr. Daviss said. “We know that it can contribute to symptoms of depression, anxiety, psychosis, and addiction. Younger users are particularly at risk for some of these symptoms. We don’t really know how much of this risk applies to monitored medical use,” he said. “And some states have no requirements for monitoring and modulating the dose, so risk data from these states will be different from states where medical use is more regulated and more similar to standard medical care.”
In addition, data on the benefits of marijuana for psychiatric conditions are limited, Dr. Daviss said. “I have certainly heard directly from patients about how marijuana helps them,” he said. “These comments often suggest improvements in sleep, appetite, mood, and anxiety. “A major concern of psychiatric clinicians is that many people asking for medical cannabis are providing contrived reports of symptoms in an attempt to obtain marijuana for nonmedical (recreational) use,” or to share or sell, he said.
Clinicians also remain concerned that “one’s state medical license or federal [Drug Enforcement Administration] license could be at risk for ‘prescribing’ or ‘recommending’ medical cannabis,” Dr. Daviss said. “Being known as a ‘pot doc’ is not viewed as a career booster and could be seen as a risk factor for enhanced regulatory scrutiny of one’s practice.”
However, “there are many who balance those concerns with a harm reduction approach that acknowledges that drug cartels and gangs are fueled by marijuana sales, resulting in preventable violence and deaths, marijuana of unknown quality or adulteration, and negative criminal justice consequences for what should be viewed as a public health problem rather than a criminal problem,” Dr. Daviss said. “Such a harm reduction perspective may increase the likelihood that some physicians will support the use of either medical cannabis or recreational marijuana.”
As more researchers study medical marijuana, the concerns of psychiatrists will evolve along with the science, Dr. Daviss predicted. Relevant outcomes research would include comparative effectiveness studies such as a randomized, double-blind trials of standardized cannabis formulations versus standard treatment for depression or anxiety, he said.
There’s not enough evidence for specific contraindications for medical marijuana use in psychiatry, Dr. Daviss said. However, most clinicians would say that a psychotic disorder such as schizophrenia is a contraindication, because some users of medical marijuana experience paranoia and other psychotic symptoms, he noted.
Dr. Fichtner agreed, and said “studies do find an association between adolescent marijuana use and later development of psychotic disorders, including schizophrenia.
“Even though authors of such studies extrapolate their findings to public health implications in terms of reducing the number of cases of schizophrenia through prevention of marijuana use, such a cause and effect relationship is far from clear – and such conclusions are unwarranted,” he said.
In fact, he said, at least one small case series found prescription molecular THC to be a helpful adjunct to approved antipsychotic medications in schizophrenia patients who reported symptomatic benefit from previous marijuana use (J Clin Psychopharmacol. 2009 Jun;29[3]:255-8). “Even more compelling,” Dr. Fichtner said, “are data on antipsychotic effects of molecular CBD, an important component of herbal cannabis that does not have THC-like psychoactive effects but has been demonstrated to have anxiolytic properties in a number of human studies.”
Dr. Fichtner said authors of research articles on the association between schizophrenia and cannabis use rarely have grappled with the possibility that patients might experience therapeutic benefits through self-medication. In addition, he said, “there are findings in the literature suggesting that among patients with schizophrenia, those using marijuana may have fewer and more selective cognitive deficits than those who do not use marijuana.”
For his part, Dr. Morhaim also supports an open approach. “All health professionals, psychiatrists included, need to consider cannabis as any other medicine,” Dr. Morhaim said. “It has its uses and side effects; reasons to initiate use and reasons to stop; diagnoses for which it is generally accepted that it works (multiple sclerosis, nausea from cancer chemotherapy, and appetite loss) and ones where [effects are] less clear. The medical-scientific potential, however, is expanding rapidly as the shackles that have blocked proper cannabis research are coming off,” he noted.
Several large studies evaluating the use of cannabis for mental illness are underway, Dr. Morhaim said. As the science expands, recent studies have examined the role of cannabis for treating a range of medical conditions, including Alzheimer’s disease, multiple sclerosis, migraine, and even fracture healing and acne, he said. Still, data remain sparse. For example, a literature review that looked at randomized, clinical trials and meta-analyses tied to marijuana and other cannabinoids, and specific diagnoses found no trials that examined marijuana’s efficacy for treating Tourette disorder, PTSD, or Alzheimer’s disease (J Clin Psychiatry. 2016 Aug;77[8]:1050-64). “Given its rapidly changing legal status, there is an urgent need to conduct double-blind, randomized, placebo- and active-controlled studies on the efficacy and safety of marijuana or its constituent cannabinoids for psychiatric conditions,” wrote Samuel T. Wilkinson, MD, of the department of psychiatry at Yale University, New Haven, Conn., and his associates. Meanwhile, a matched case-control, cross-sectional evaluation of veterans with probable PTSD found no significant differences between cases and controls in PTSD scores and frequency of cannabis use (J Affect Disord. 2016 Jan 15;190:439-42).
Nevertheless, “psychiatrists ought to be familiar with medical cannabis, because they may be taking care of patients who are on it for other (somatic) reasons,” Dr. Morhaim said. “This is just like my work as an emergency doctor; I don’t prescribe psych medicines, but I have to know about them, because patients come to the ER who are on them.”
Lessons from patients’ reports
Rachna J. Patel, DO, a psychiatrist who describes herself on her website as “The Medical Marijuana Expert,” treats patients with anxiety and PTSD, and agrees that marijuana is a tool that, when used correctly, can benefit patients. “A visit with me, a medical marijuana doctor, is much like a visit with any other doctor,” said Dr. Patel, who practices in Walnut Creek, Calif.
“I have my patients complete a form that asks about their medical history, as well as the treatments they’ve sought out for their anxiety and/or PTSD, such as prescription medications, therapy, and other methods,” she said. “I then sit down with the patient to go over their history and do a focused physical exam. Once I determine that the patient’s medical condition will benefit with the use of medical marijuana, I spend the remainder of the time step-by-step walking him or her through how to use medical marijuana for the anxiety and/or PTSD,” she added.
Dr. Patel applies some basic considerations when assessing patients for potential medical marijuana treatment, notably the efficacy and side effects of past treatments and patient concerns about past treatments, including the potential for dependency or addiction. She also assesses the impact of anxiety or PTSD on the patient’s quality of life.
Education is key, she said, to addressing patients’ concerns and managing possible risks. “The risks are generally minimized and even eliminated by educating patients on selection of the appropriate combination of cannabinoids, as well as teaching them to use medical marijuana at the lowest effective dose and with the lowest effective frequency,” she said.
“Having practiced in the field of medical marijuana for 4-plus years, I haven’t found marijuana to be a gateway drug,” she said. “The patients I see aren’t looking to ‘get high’ off of medical marijuana. In fact, they’re seeking to get off of the pharmaceutical drugs they’ve been prescribed.”
Dr. Patel walks patients through what she calls “The Patel Protocol for Medical Marijuana,” which includes “teaching patients how to select the appropriate combination of cannabinoids in marijuana products, figuring out how much to use and how often to use it, how to avoid side effects, and what to do in case patients do experience side effects,” she said.
In her experience, “when it comes to dosing, just like with many pharmaceutical medications, it really varies from patient to patient, even more so because cannabinoids are hydrophobic, so their pharmacology is not typical of most pharmaceutical medications.” Dr. Patel follows up with her patients often by phone after the initial office visit, then continues with office visits at least once a year.
“The best way any doctor can help to monitor their patients’ use of medical marijuana is to be as informed about it as any other pharmaceutical drug they’re prescribing,” she said. “Become familiar with the pharmacology [of medical marijuana],” she advised. “Know what scenarios could lead to side effects and how those side effects can be avoided. Know in what scenarios it [marijuana] can exacerbate underlying medical conditions.”
Dr. Fichtner agrees with Dr. Patel’s approach but adds that he generally does not recommend cannabis as a first-line treatment for his patients with serious mental illness. Rather, his approach generally is to prescribe evidence-based treatments.
“But when I do find a history of marijuana use and the patient reports it has been helpful, I work with the patient to assess possible benefits in the areas of symptom management and possible reductions in polypharmacy,” he said. “The latter is of particular concern in PTSD, where patients may be on complex multidrug regimens involving off-label medication prescriptions with only partial symptom relief. It is important to take the patient’s claim seriously, rather than merely jumping to the conclusion that they have a substance use disorder that is undermining their psychiatric care.”
Dr. Patel said a surprising and positive side effect she has observed during her years of recommending medical marijuana is that patients’ use of any anxiety and PTSD medication decreases over time.
“Even though I never expected it, with the use of medical marijuana, many of my patients have been able to significantly reduce or even eliminate the use of their prescription medications for their anxiety and/or PTSD,” she said. “Over time, I find that patients use the medical marijuana on an as-needed basis.”
Dr. Fichtner is the author of book, “Cannabinomics: The Marijuana Policy Tipping Point” (Northbrook, Ill.: Well Mind Books, 2010). Dr. Daviss, Dr. Morhaim, and Dr. Patel had no relevant financial conflicts to disclose.
Continue DMARDs, biologics in RA surgery patients
WASHINGTON – The perioperative use of disease-modifying antirheumatic drug monotherapy or combined therapy with methotrexate and a tumor necrosis factor (TNF) inhibitor is not associated with increased rates of postoperative infectious complications or wound infections in patients with rheumatoid arthritis, according to findings from a retrospective review of more than 9,000 surgeries.
With respect to monotherapy, treatment was continued in 1,951 of 2,601 surgeries among patients receiving methotrexate, in 1,496 of 2,012 surgeries among patients receiving hydroxychloroquine, and in 508 of 652 surgeries among patient receiving leflunomide. The odds ratios for postoperative infection (including urinary tract, pneumonia, or sepsis) and postoperative wound infection, respectively, were 0.79 and 0.77 with methotrexate continuation, 0.93 and 0.86 with hydroxychloroquine continuation, and 0.78 and 0.87 with leflunomide continuation, Hsin-Hsuan Juo, MD, reported at the annual meeting of the American College of Rheumatology.
Data for this study were derived from the U.S. Department of Veterans Affairs administrative database and surgical quality registry. Rheumatoid arthritis patients who underwent a surgical procedure and who were on at least one disease-modifying antirheumatic drug (DMARD) or one biologic agent in the perioperative period during the study period of Oct. 1, 1999, through Sept. 30, 2009, were included. Subjects had a mean age of 67 years, and 91% were men.
The finding that the continuation of DMARD monotherapy or the combination of methotrexate and TNF inhibitor therapy for RA in the perioperative setting was not associated with increased rates of overall postoperative infectious complications and wound infections is important, because many patients are advised to stop taking these drugs prior to surgery because of concerns about increased susceptibility to infection. Discontinuing RA medication can increase the risk of disease flares requiring treatment with prednisone, which can further increase the risk of postsurgical complications, Dr. Juo said.
Clear, consistent guidance on the continuation of treatment among RA patients undergoing surgery has been lacking, she said, noting that guidelines over the years from the ACR, the British Society for Rheumatology, and the Canadian Rheumatology Association have differed in their recommendations.
A new draft guideline reported the morning of Dr. Juo’s presentation at the ACR annual meeting recommended continuing DMARDs but discontinuing biologics prior to surgery, but that guideline is limited to orthopedic surgery among patients with various rheumatic diseases.
“With literature review, the results are conflicting as well; some recommend continuing medication, and others recommend discontinuing medications prior to surgery,” she said.
The current findings, though limited by the study’s observational design and generally older, male population, suggest that continuing antirheumatic medications during the perioperative period is not associated with increased rates of postoperative complications.
“Our study results suggest that discontinuing DMARDs and biologic agents prior to surgery may not be necessary. Therefore, being on DMARDs or biologic agents should not preclude patients from receiving urgent surgeries,” Dr. Juo concluded.
Dr. Juo reported having no disclosures.
WASHINGTON – The perioperative use of disease-modifying antirheumatic drug monotherapy or combined therapy with methotrexate and a tumor necrosis factor (TNF) inhibitor is not associated with increased rates of postoperative infectious complications or wound infections in patients with rheumatoid arthritis, according to findings from a retrospective review of more than 9,000 surgeries.
With respect to monotherapy, treatment was continued in 1,951 of 2,601 surgeries among patients receiving methotrexate, in 1,496 of 2,012 surgeries among patients receiving hydroxychloroquine, and in 508 of 652 surgeries among patient receiving leflunomide. The odds ratios for postoperative infection (including urinary tract, pneumonia, or sepsis) and postoperative wound infection, respectively, were 0.79 and 0.77 with methotrexate continuation, 0.93 and 0.86 with hydroxychloroquine continuation, and 0.78 and 0.87 with leflunomide continuation, Hsin-Hsuan Juo, MD, reported at the annual meeting of the American College of Rheumatology.
Data for this study were derived from the U.S. Department of Veterans Affairs administrative database and surgical quality registry. Rheumatoid arthritis patients who underwent a surgical procedure and who were on at least one disease-modifying antirheumatic drug (DMARD) or one biologic agent in the perioperative period during the study period of Oct. 1, 1999, through Sept. 30, 2009, were included. Subjects had a mean age of 67 years, and 91% were men.
The finding that the continuation of DMARD monotherapy or the combination of methotrexate and TNF inhibitor therapy for RA in the perioperative setting was not associated with increased rates of overall postoperative infectious complications and wound infections is important, because many patients are advised to stop taking these drugs prior to surgery because of concerns about increased susceptibility to infection. Discontinuing RA medication can increase the risk of disease flares requiring treatment with prednisone, which can further increase the risk of postsurgical complications, Dr. Juo said.
Clear, consistent guidance on the continuation of treatment among RA patients undergoing surgery has been lacking, she said, noting that guidelines over the years from the ACR, the British Society for Rheumatology, and the Canadian Rheumatology Association have differed in their recommendations.
A new draft guideline reported the morning of Dr. Juo’s presentation at the ACR annual meeting recommended continuing DMARDs but discontinuing biologics prior to surgery, but that guideline is limited to orthopedic surgery among patients with various rheumatic diseases.
“With literature review, the results are conflicting as well; some recommend continuing medication, and others recommend discontinuing medications prior to surgery,” she said.
The current findings, though limited by the study’s observational design and generally older, male population, suggest that continuing antirheumatic medications during the perioperative period is not associated with increased rates of postoperative complications.
“Our study results suggest that discontinuing DMARDs and biologic agents prior to surgery may not be necessary. Therefore, being on DMARDs or biologic agents should not preclude patients from receiving urgent surgeries,” Dr. Juo concluded.
Dr. Juo reported having no disclosures.
WASHINGTON – The perioperative use of disease-modifying antirheumatic drug monotherapy or combined therapy with methotrexate and a tumor necrosis factor (TNF) inhibitor is not associated with increased rates of postoperative infectious complications or wound infections in patients with rheumatoid arthritis, according to findings from a retrospective review of more than 9,000 surgeries.
With respect to monotherapy, treatment was continued in 1,951 of 2,601 surgeries among patients receiving methotrexate, in 1,496 of 2,012 surgeries among patients receiving hydroxychloroquine, and in 508 of 652 surgeries among patient receiving leflunomide. The odds ratios for postoperative infection (including urinary tract, pneumonia, or sepsis) and postoperative wound infection, respectively, were 0.79 and 0.77 with methotrexate continuation, 0.93 and 0.86 with hydroxychloroquine continuation, and 0.78 and 0.87 with leflunomide continuation, Hsin-Hsuan Juo, MD, reported at the annual meeting of the American College of Rheumatology.
Data for this study were derived from the U.S. Department of Veterans Affairs administrative database and surgical quality registry. Rheumatoid arthritis patients who underwent a surgical procedure and who were on at least one disease-modifying antirheumatic drug (DMARD) or one biologic agent in the perioperative period during the study period of Oct. 1, 1999, through Sept. 30, 2009, were included. Subjects had a mean age of 67 years, and 91% were men.
The finding that the continuation of DMARD monotherapy or the combination of methotrexate and TNF inhibitor therapy for RA in the perioperative setting was not associated with increased rates of overall postoperative infectious complications and wound infections is important, because many patients are advised to stop taking these drugs prior to surgery because of concerns about increased susceptibility to infection. Discontinuing RA medication can increase the risk of disease flares requiring treatment with prednisone, which can further increase the risk of postsurgical complications, Dr. Juo said.
Clear, consistent guidance on the continuation of treatment among RA patients undergoing surgery has been lacking, she said, noting that guidelines over the years from the ACR, the British Society for Rheumatology, and the Canadian Rheumatology Association have differed in their recommendations.
A new draft guideline reported the morning of Dr. Juo’s presentation at the ACR annual meeting recommended continuing DMARDs but discontinuing biologics prior to surgery, but that guideline is limited to orthopedic surgery among patients with various rheumatic diseases.
“With literature review, the results are conflicting as well; some recommend continuing medication, and others recommend discontinuing medications prior to surgery,” she said.
The current findings, though limited by the study’s observational design and generally older, male population, suggest that continuing antirheumatic medications during the perioperative period is not associated with increased rates of postoperative complications.
“Our study results suggest that discontinuing DMARDs and biologic agents prior to surgery may not be necessary. Therefore, being on DMARDs or biologic agents should not preclude patients from receiving urgent surgeries,” Dr. Juo concluded.
Dr. Juo reported having no disclosures.
AT THE ACR ANNUAL MEETING
Key clinical point:
Major finding: Odds ratios for postoperative infection and postoperative wound infection, respectively, were 0.79 and 0.77 with methotrexate continuation, 0.93 and 0.86 with hydroxychloroquine continuation, 0.78 and 0.87 with leflunomide continuation, and 0.35 and 0.38 with combined methotrexate/TNF inhibitor continuation.
Data source: A retrospective review of more than 9,000 surgeries.
Disclosures: Dr. Juo reported having no disclosures.
CSL112 enhances cholesterol efflux capacity after acute MI
CSL112, a plasma-derived apolipoprotein A-1 (apoA-1) that enhances cholesterol efflux capacity, was found safe for use after acute MI in a manufacturer-sponsored phase IIb trial, Michael Gibson, MD, reported at the American Heart Association scientific sessions.
Cholesterol efflux capacity is a measure of HDL cholesterol’s ability to remove excess cholesterol from atherosclerotic plaque for transport to the liver. Drugs that improve this capacity rather than simply raising HDL cholesterol are expected to reduce plaque burden and stabilize vulnerable plaque immediately following acute MI, when cholesterol efflux is significantly impaired. But they have not been tested in this patient population until now, said Dr. Gibson of Beth Israel Deaconess Medical Center and Harvard, both in Boston.
An earlier prototype formulation of CSL112 was discontinued because of concerns about transient elevations in liver enzymes and the potential for renal toxicity due to the agent’s high sucrose content. Dr. Gibson and his associates in the AEGIS-1 trial (ApoA-1 Event Reducing in Ischemic Syndromes 1) now report their findings for the current formulation of CSL112, which contains lower phosphatidylcholine and lower sucrose levels, in 1,258 patients who had MI during the preceding week and who had normal or only mildly impaired renal function. These results were reported at the meeting and simultaneously published online in Circulation (2016, Nov 15. doi: 10.1161/CIRCULATION AHA.116.025687).
The study participants were enrolled in 16 countries during an 11-month period. They were randomly assigned to receive low-dose (2 g) CSL112 (419 patients), high-dose (6 g) CSL112 (421 patients), or a matching placebo (418 patients) via IV infusion every week for 4 consecutive weeks. The median duration of follow-up at the time of this report was 7.5 months.
The two primary safety end points – the rate of hepatic impairment and the rate of renal impairment during treatment – were not significantly different across the three study groups. Hepatic impairment developed in 1.0% of the low-dose group, 0.5% of the high-dose group, and 0.0% of the placebo group. Renal impairment developed in 0.0% of the low-dose group, 0.7% of the high-dose group, and 0.2% of the placebo group.
Similarly, rates of major adverse cardiovascular events were comparable across the three study groups, as were rates of any grade of bleeding, rates of serious and life-threatening adverse events, and rates of adverse events leading to drug discontinuation.
This study focused on safety and tolerability and was not designed or powered to assess efficacy. Nevertheless, CSL112 caused a substantial and dose-dependent increase in both apoA-1 and cholesterol efflux capacity. The low-dose drug raised total cholesterol efflux capacity by 1.87-fold, and the high-dose drug raised it 2.45-fold.
As the first study to establish the safety and feasibility of adding CSL112 to standard care for acute MI, this trial demonstrates that an adequately powered, multicenter, phase III trial is warranted, Dr. Gibson said.
The current formulation of CSL112 didn’t provoke hepatic toxicity, and even though it was given shortly after contrast studies in MI patients, it also didn’t provoke renal toxicity. “This demonstrates the feasibility of administering CSL112 to patients with MI who have normal renal function or mild renal impairment shortly after angiography. A study in MI patients who have moderate renal impairment is now under way,” he noted.
This study was sponsored by CSL Behring, maker of CSL112. Dr. Gibson and his associates reported financial ties to Behring and numerous other industry sources.
CSL112, a plasma-derived apolipoprotein A-1 (apoA-1) that enhances cholesterol efflux capacity, was found safe for use after acute MI in a manufacturer-sponsored phase IIb trial, Michael Gibson, MD, reported at the American Heart Association scientific sessions.
Cholesterol efflux capacity is a measure of HDL cholesterol’s ability to remove excess cholesterol from atherosclerotic plaque for transport to the liver. Drugs that improve this capacity rather than simply raising HDL cholesterol are expected to reduce plaque burden and stabilize vulnerable plaque immediately following acute MI, when cholesterol efflux is significantly impaired. But they have not been tested in this patient population until now, said Dr. Gibson of Beth Israel Deaconess Medical Center and Harvard, both in Boston.
An earlier prototype formulation of CSL112 was discontinued because of concerns about transient elevations in liver enzymes and the potential for renal toxicity due to the agent’s high sucrose content. Dr. Gibson and his associates in the AEGIS-1 trial (ApoA-1 Event Reducing in Ischemic Syndromes 1) now report their findings for the current formulation of CSL112, which contains lower phosphatidylcholine and lower sucrose levels, in 1,258 patients who had MI during the preceding week and who had normal or only mildly impaired renal function. These results were reported at the meeting and simultaneously published online in Circulation (2016, Nov 15. doi: 10.1161/CIRCULATION AHA.116.025687).
The study participants were enrolled in 16 countries during an 11-month period. They were randomly assigned to receive low-dose (2 g) CSL112 (419 patients), high-dose (6 g) CSL112 (421 patients), or a matching placebo (418 patients) via IV infusion every week for 4 consecutive weeks. The median duration of follow-up at the time of this report was 7.5 months.
The two primary safety end points – the rate of hepatic impairment and the rate of renal impairment during treatment – were not significantly different across the three study groups. Hepatic impairment developed in 1.0% of the low-dose group, 0.5% of the high-dose group, and 0.0% of the placebo group. Renal impairment developed in 0.0% of the low-dose group, 0.7% of the high-dose group, and 0.2% of the placebo group.
Similarly, rates of major adverse cardiovascular events were comparable across the three study groups, as were rates of any grade of bleeding, rates of serious and life-threatening adverse events, and rates of adverse events leading to drug discontinuation.
This study focused on safety and tolerability and was not designed or powered to assess efficacy. Nevertheless, CSL112 caused a substantial and dose-dependent increase in both apoA-1 and cholesterol efflux capacity. The low-dose drug raised total cholesterol efflux capacity by 1.87-fold, and the high-dose drug raised it 2.45-fold.
As the first study to establish the safety and feasibility of adding CSL112 to standard care for acute MI, this trial demonstrates that an adequately powered, multicenter, phase III trial is warranted, Dr. Gibson said.
The current formulation of CSL112 didn’t provoke hepatic toxicity, and even though it was given shortly after contrast studies in MI patients, it also didn’t provoke renal toxicity. “This demonstrates the feasibility of administering CSL112 to patients with MI who have normal renal function or mild renal impairment shortly after angiography. A study in MI patients who have moderate renal impairment is now under way,” he noted.
This study was sponsored by CSL Behring, maker of CSL112. Dr. Gibson and his associates reported financial ties to Behring and numerous other industry sources.
CSL112, a plasma-derived apolipoprotein A-1 (apoA-1) that enhances cholesterol efflux capacity, was found safe for use after acute MI in a manufacturer-sponsored phase IIb trial, Michael Gibson, MD, reported at the American Heart Association scientific sessions.
Cholesterol efflux capacity is a measure of HDL cholesterol’s ability to remove excess cholesterol from atherosclerotic plaque for transport to the liver. Drugs that improve this capacity rather than simply raising HDL cholesterol are expected to reduce plaque burden and stabilize vulnerable plaque immediately following acute MI, when cholesterol efflux is significantly impaired. But they have not been tested in this patient population until now, said Dr. Gibson of Beth Israel Deaconess Medical Center and Harvard, both in Boston.
An earlier prototype formulation of CSL112 was discontinued because of concerns about transient elevations in liver enzymes and the potential for renal toxicity due to the agent’s high sucrose content. Dr. Gibson and his associates in the AEGIS-1 trial (ApoA-1 Event Reducing in Ischemic Syndromes 1) now report their findings for the current formulation of CSL112, which contains lower phosphatidylcholine and lower sucrose levels, in 1,258 patients who had MI during the preceding week and who had normal or only mildly impaired renal function. These results were reported at the meeting and simultaneously published online in Circulation (2016, Nov 15. doi: 10.1161/CIRCULATION AHA.116.025687).
The study participants were enrolled in 16 countries during an 11-month period. They were randomly assigned to receive low-dose (2 g) CSL112 (419 patients), high-dose (6 g) CSL112 (421 patients), or a matching placebo (418 patients) via IV infusion every week for 4 consecutive weeks. The median duration of follow-up at the time of this report was 7.5 months.
The two primary safety end points – the rate of hepatic impairment and the rate of renal impairment during treatment – were not significantly different across the three study groups. Hepatic impairment developed in 1.0% of the low-dose group, 0.5% of the high-dose group, and 0.0% of the placebo group. Renal impairment developed in 0.0% of the low-dose group, 0.7% of the high-dose group, and 0.2% of the placebo group.
Similarly, rates of major adverse cardiovascular events were comparable across the three study groups, as were rates of any grade of bleeding, rates of serious and life-threatening adverse events, and rates of adverse events leading to drug discontinuation.
This study focused on safety and tolerability and was not designed or powered to assess efficacy. Nevertheless, CSL112 caused a substantial and dose-dependent increase in both apoA-1 and cholesterol efflux capacity. The low-dose drug raised total cholesterol efflux capacity by 1.87-fold, and the high-dose drug raised it 2.45-fold.
As the first study to establish the safety and feasibility of adding CSL112 to standard care for acute MI, this trial demonstrates that an adequately powered, multicenter, phase III trial is warranted, Dr. Gibson said.
The current formulation of CSL112 didn’t provoke hepatic toxicity, and even though it was given shortly after contrast studies in MI patients, it also didn’t provoke renal toxicity. “This demonstrates the feasibility of administering CSL112 to patients with MI who have normal renal function or mild renal impairment shortly after angiography. A study in MI patients who have moderate renal impairment is now under way,” he noted.
This study was sponsored by CSL Behring, maker of CSL112. Dr. Gibson and his associates reported financial ties to Behring and numerous other industry sources.
FROM THE AHA SCIENTIFIC SESSIONS
Key clinical point: CSL112, a plasma-derived apolipoprotein A-1 that enhances cholesterol efflux capacity, was found safe for use after acute MI in an international phase IIb trial.
Major finding: Hepatic impairment developed in 1.0% of the low-dose group, 0.5% of the high-dose group, and 0.0% of the placebo group, while renal impairment developed in 0.0% of the low-dose group, 0.7% of the high-dose group, and 0.2% of the placebo group – all nonsignificant differences.
Data source: A manufacturer-sponsored randomized double-blind placebo-controlled phase IIb trial involving 1,258 patients in 16 countries.
Disclosures: This study was sponsored by CSL Behring, maker of CSL112. Dr. Gibson and his associates reported financial ties to Behring and numerous other industry sources.
Heart failure readmission metric not linked to care quality
Metrics used by the Centers for Medicare & Medicaid Services to determine penalties for heart failure hospital readmissions are not associated with quality of care or overall clinical outcomes, according to data presented at the annual scientific sessions of the American Heart Association.
Ambarish Pandey, MD, of the University of Texas Southwestern Medical Center in Dallas, and his colleagues analyzed data from centers participating in the American Heart Association’s Get With The Guidelines-Heart Failure (GWTG-HF) registry linked to Medicare claims from July 2008 to June 2011. Centers were stratified as having low risk-adjusted readmission rates and high risk-adjusted readmission rates based on publicly available data from 2013.
The study included 171 centers with 43,143 patients. Centers were almost evenly split between low- and high-risk–adjusted 30-day readmission rates, with just a few more (51%) falling in the low-risk–adjusted category.
Performance was nearly equal (95.7% for centers with a low risk-adjusted readmission rate vs. 96.5% for those with high risk-adjusted rate) for median adherence to all performance measures, as was the case for median percentage of defect-free care (90.0% vs. 91.1%, respectively) and composite 1-year outcome of death or all-cause readmission rates (median 62.9% vs. 65.3%, respectively). The higher readmission group had higher 1-year all-cause readmission rates (median, 59.1% vs. 54.7%), Dr. Pandey and his colleagues reported in the study that was published simultaneously in JACC: Heart Failure (2016 Nov 15. doi. org/10.1016/j.jchf.2016). One-year mortality rates were lower in the higher readmission group with a trend toward statistical significance (median, 28.2% vs. 31.7%; P = 0.07).
Taken together, the findings suggest the 30-day readmission metrics currently used by CMS to determine readmission penalties are not associated with quality of care or overall clinical outcomes, Dr. Pandey and his colleagues wrote. Results showing higher 30-day readmissions do not necessarily reflect poor quality of care and may be related to other factors.
“These findings question the usefulness of the [hospital readmission reduction program] metric in identifying and penalizing hospitals with low quality of care,” Dr. Pandey wrote, adding that the findings were consistent with previous studies that have demonstrated a lack of association between in-hospital quality of care and 30-day readmission rates.
CMS implemented the federal Hospital Readmissions Reduction Program (HRRP) in 2012 to provide financial incentives for hospitals to reduce readmissions. Under the program, CMS uses claims data to determine whether readmission rates for heart failure, acute myocardial infarction, and pneumonia at eligible hospitals are higher than would be predicted by CMS models. Centers with higher than expected readmission rates face up to a 3% reimbursement penalty.
agallegos@frontlinemedcom.com
On Twitter @legal_med
These authors add to a chorus of voices expressing concern regarding the appropriateness and validity of the 30-day readmission metric. Arguably, this metric has driven our entire provider workforce to construct machinery designed to reduce short-term posthospitalization utilization, while doing little to improve quality for the 5.7 million (and counting) Americans with heart failure.

Marvin A. Konstam, MD, of Tufts University, Boston, made these comments in an accompanying editorial (JACC: Heart Fail. 2016 Nov 15. doi: 10.1016/j.jchf.2016.10.004). He reported no relevant disclosures.
These authors add to a chorus of voices expressing concern regarding the appropriateness and validity of the 30-day readmission metric. Arguably, this metric has driven our entire provider workforce to construct machinery designed to reduce short-term posthospitalization utilization, while doing little to improve quality for the 5.7 million (and counting) Americans with heart failure.
Marvin A. Konstam, MD, of Tufts University, Boston, made these comments in an accompanying editorial (JACC: Heart Fail. 2016 Nov 15. doi: 10.1016/j.jchf.2016.10.004). He reported no relevant disclosures.
These authors add to a chorus of voices expressing concern regarding the appropriateness and validity of the 30-day readmission metric. Arguably, this metric has driven our entire provider workforce to construct machinery designed to reduce short-term posthospitalization utilization, while doing little to improve quality for the 5.7 million (and counting) Americans with heart failure.
Marvin A. Konstam, MD, of Tufts University, Boston, made these comments in an accompanying editorial (JACC: Heart Fail. 2016 Nov 15. doi: 10.1016/j.jchf.2016.10.004). He reported no relevant disclosures.
Metrics used by the Centers for Medicare & Medicaid Services to determine penalties for heart failure hospital readmissions are not associated with quality of care or overall clinical outcomes, according to data presented at the annual scientific sessions of the American Heart Association.
Ambarish Pandey, MD, of the University of Texas Southwestern Medical Center in Dallas, and his colleagues analyzed data from centers participating in the American Heart Association’s Get With The Guidelines-Heart Failure (GWTG-HF) registry linked to Medicare claims from July 2008 to June 2011. Centers were stratified as having low risk-adjusted readmission rates and high risk-adjusted readmission rates based on publicly available data from 2013.
The study included 171 centers with 43,143 patients. Centers were almost evenly split between low- and high-risk–adjusted 30-day readmission rates, with just a few more (51%) falling in the low-risk–adjusted category.
Performance was nearly equal (95.7% for centers with a low risk-adjusted readmission rate vs. 96.5% for those with high risk-adjusted rate) for median adherence to all performance measures, as was the case for median percentage of defect-free care (90.0% vs. 91.1%, respectively) and composite 1-year outcome of death or all-cause readmission rates (median 62.9% vs. 65.3%, respectively). The higher readmission group had higher 1-year all-cause readmission rates (median, 59.1% vs. 54.7%), Dr. Pandey and his colleagues reported in the study that was published simultaneously in JACC: Heart Failure (2016 Nov 15. doi. org/10.1016/j.jchf.2016). One-year mortality rates were lower in the higher readmission group with a trend toward statistical significance (median, 28.2% vs. 31.7%; P = 0.07).
Taken together, the findings suggest the 30-day readmission metrics currently used by CMS to determine readmission penalties are not associated with quality of care or overall clinical outcomes, Dr. Pandey and his colleagues wrote. Results showing higher 30-day readmissions do not necessarily reflect poor quality of care and may be related to other factors.
“These findings question the usefulness of the [hospital readmission reduction program] metric in identifying and penalizing hospitals with low quality of care,” Dr. Pandey wrote, adding that the findings were consistent with previous studies that have demonstrated a lack of association between in-hospital quality of care and 30-day readmission rates.
CMS implemented the federal Hospital Readmissions Reduction Program (HRRP) in 2012 to provide financial incentives for hospitals to reduce readmissions. Under the program, CMS uses claims data to determine whether readmission rates for heart failure, acute myocardial infarction, and pneumonia at eligible hospitals are higher than would be predicted by CMS models. Centers with higher than expected readmission rates face up to a 3% reimbursement penalty.
agallegos@frontlinemedcom.com
On Twitter @legal_med
Metrics used by the Centers for Medicare & Medicaid Services to determine penalties for heart failure hospital readmissions are not associated with quality of care or overall clinical outcomes, according to data presented at the annual scientific sessions of the American Heart Association.
Ambarish Pandey, MD, of the University of Texas Southwestern Medical Center in Dallas, and his colleagues analyzed data from centers participating in the American Heart Association’s Get With The Guidelines-Heart Failure (GWTG-HF) registry linked to Medicare claims from July 2008 to June 2011. Centers were stratified as having low risk-adjusted readmission rates and high risk-adjusted readmission rates based on publicly available data from 2013.
The study included 171 centers with 43,143 patients. Centers were almost evenly split between low- and high-risk–adjusted 30-day readmission rates, with just a few more (51%) falling in the low-risk–adjusted category.
Performance was nearly equal (95.7% for centers with a low risk-adjusted readmission rate vs. 96.5% for those with high risk-adjusted rate) for median adherence to all performance measures, as was the case for median percentage of defect-free care (90.0% vs. 91.1%, respectively) and composite 1-year outcome of death or all-cause readmission rates (median 62.9% vs. 65.3%, respectively). The higher readmission group had higher 1-year all-cause readmission rates (median, 59.1% vs. 54.7%), Dr. Pandey and his colleagues reported in the study that was published simultaneously in JACC: Heart Failure (2016 Nov 15. doi. org/10.1016/j.jchf.2016). One-year mortality rates were lower in the higher readmission group with a trend toward statistical significance (median, 28.2% vs. 31.7%; P = 0.07).
Taken together, the findings suggest the 30-day readmission metrics currently used by CMS to determine readmission penalties are not associated with quality of care or overall clinical outcomes, Dr. Pandey and his colleagues wrote. Results showing higher 30-day readmissions do not necessarily reflect poor quality of care and may be related to other factors.
“These findings question the usefulness of the [hospital readmission reduction program] metric in identifying and penalizing hospitals with low quality of care,” Dr. Pandey wrote, adding that the findings were consistent with previous studies that have demonstrated a lack of association between in-hospital quality of care and 30-day readmission rates.
CMS implemented the federal Hospital Readmissions Reduction Program (HRRP) in 2012 to provide financial incentives for hospitals to reduce readmissions. Under the program, CMS uses claims data to determine whether readmission rates for heart failure, acute myocardial infarction, and pneumonia at eligible hospitals are higher than would be predicted by CMS models. Centers with higher than expected readmission rates face up to a 3% reimbursement penalty.
agallegos@frontlinemedcom.com
On Twitter @legal_med
FROM THE AHA SCIENTIFIC SESSIONS
Key clinical point:
Major finding: Performance was nearly equal (95.7% for centers with a low risk-adjusted readmission rate vs. 96.5% for those with high risk-adjusted rate) for median adherence to all performance measures.
Data source: Analysis of publicly available data reported to the CMS Hospital Readmission Reduction program.
Disclosures: No relevant conflicts of interest.
Application of Amniotic Tissue in Orthopedic Surgery
The amniotic membrane is a multilayer tissue forming the innermost layer of the amniotic sac that surrounds the developing fetus. It is comprised of 5 layers, from the inside out: a single layer of epithelial cells, a thick basement membrane, a compact layer, a fibroblast layer, and a spongy layer that abuts the surrounding chorion (Figure 1).1 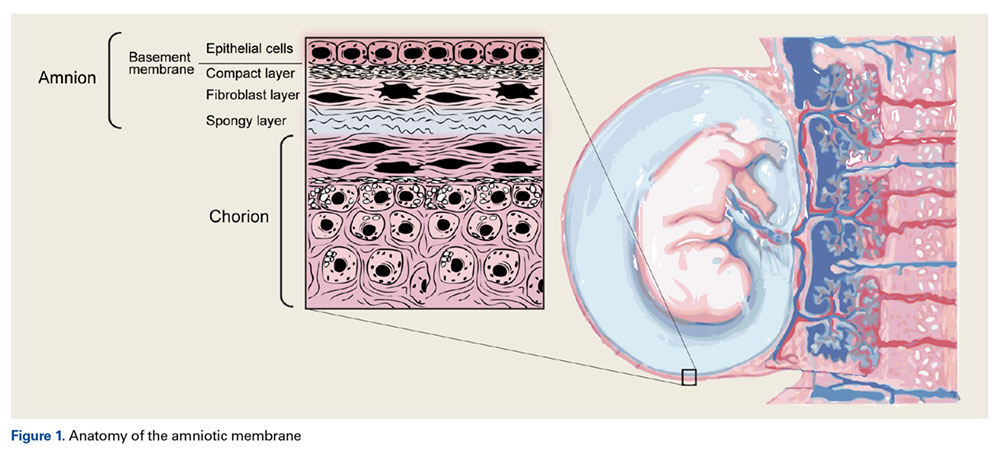
Amniotic epithelial cells are derived from the pluripotent epiblast at approximately day 8 of gestation. This is well before gastrulation occurs at days 15 to 17, considered the “tipping point” when pluripotent cells differentiate into ectoderm, mesoderm, and endoderm.3 These cells express Oct-4 and Nanog, 2 molecular markers that are indicative of pluripotency.3 Two cell types have been identified in amniotic tissues that possess stem cell-like characteristics: human amniotic epithelial cells and human amniotic mesenchymal stromal cells.4 Both of these cell types have demonstrated the ability to differentiate into various cell lineages, including endothelial cells, adipocytes, myogenic cells, neurogenic cells, chondrocytes, tenocytes, and osteogenic cells.5-7 These previously reported findings indicate that amniotic cells and tissue have the capability to generate mesenchymal tissues.
FDA Classification and Available Forms
The US Food and Drug Administration (FDA) classifies amnion as an allograft tissue under Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps) 361. To meet criteria, the tissue needs to be minimally manipulated. It is to be for homologous use and cannot be combined with other cells or tissues. There can be no systemic effect or dependence on the metabolic activity of living cells to achieve its primary function. The tissue has to have a localized effect in vivo. Therefore, amnion allograft tissue can be commercialized, provided it is not marketed as a stem cell product or to contain viable cells.
Amniotic tissue is commercially available in several forms. 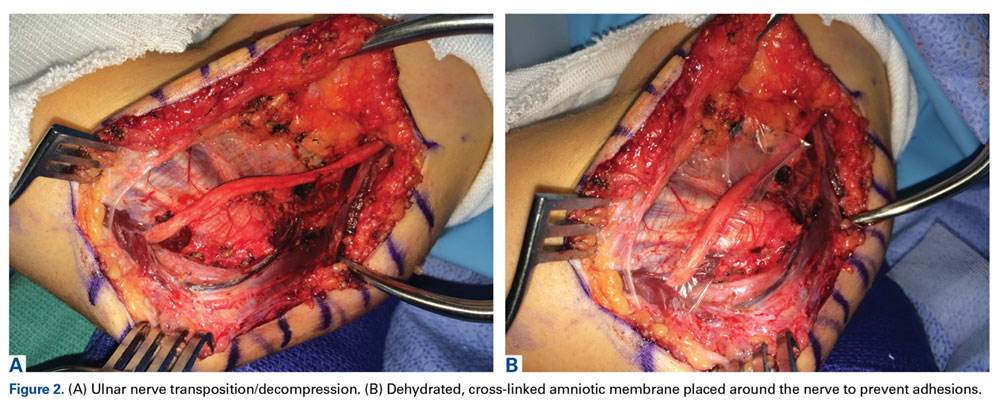
Safety
Amniotic tissue has been used for over 100 years in burn, ophthalmology, and chronic wound patients with favorable outcomes and no adverse effects reported in the literature. Unlike embryonic stem cells, which may be tumorigenic,8 amniotic cells do not possess any known tumorigenicity.9 In one study, 50 immunodeficient mice were injected with 1 to 2 million amniotic epithelial cells and observed for a maximum of 516 days with no tumorigenicity observed in any of the animals.10 In another study, amniotic epithelial cells were implanted into the forearms of healthy volunteers and no immunologic response was observed in any of the recipients.11 Furthermore, viable amniotic cells were recovered via biopsy 7 weeks following transplantation, demonstrating viability of the transplanted cells.11 The lack of tumorigenicity and immunologic response in hosts is due in part to the fact that amniotic cells do not express human leukocyte antigen class II antigens and only express class I antigens in small amounts.3
Advantages of Amnion Tissue
Amniotic tissue is readily available, as it is often discarded after childbirth. The use of this tissue poses no added risk to the fetus or mother, eliminating the ethical concerns associated with obtaining embryonic stem cells. Amniotic tissue is comprised of an extracellular matrix, which acts as a natural scaffold for cellular attachment and structural support for cells as well as collagen types I, III, IV, V, and VI, hyaluronic acid, and a host of growth factors.12 In addition, it possesses antimicrobial properties, including beta-defensins.13
Amniotic tissue has been shown to exert an anti-inflammatory effect by inhibiting the inflammatory cascade. Specifically, it has been shown to inhibit cytokines such as tumor necrosis factor-alpha in the presence of dendritic cells,14 as well as inhibiting transforming growth factor-beta, interleukin-8, and fibroblast proliferation.15 These findings indicate that amniotic tissue has the ability to dampen the “cytokine storm” that occurs after an injury in an adult, which would lead to beneficial impacts on healing and scar formation in patients.16
Basic Science and Animal Studies
Several studies have demonstrated promising outcomes for orthopedic applications in vitro. A comparison of osteogenic potential found that amniotic fluid-derived cells were able to produce approximately 5 times more mineralized matrix than bone marrow-derived mesenchymal stem cells.17 More recently, Si and colleagues18 compared the osteogenic potential of human amniotic epithelial cells, amniotic cells, and human bone marrow-derived mesenchymal stem cells. They found that all 3 cell lines were osteogenic, though the amniotic epithelial cells had better immunomodulatory properties and marginally less osteogenic potential than the other 2 cell types. Furthermore, several in vivo animal studies have demonstrated the ability of human amniotic cells to stimulate bone growth in rats,19,20 rabbits,21 and sheep.22
Amniotic tissue also possesses potential for chondrogenesis. Cryopreserved human amniotic membrane cells used for in vitro human osteoarthritis tissue scaffolds did not differentiate in culture, and they integrated and repaired damaged articular cartilage.23 Various in vitro24,25 and animal in vivo26,27 studies have reported similar supportive findings. Kunisaki and colleagues28 used sheep amniotic fluid mesenchymal stem cells to reconstruct lamb tracheal cartilage in utero, concluding that cells obtained from the amniotic fluid possess chondrogenic capabilities. Further in utero lamb studies of cartilage artificial defects, given 7 days to settle before adding a hypocellular matrix as a scaffold, showed chondrocyte density and cell architecture was restored at the defect site after 28 days without the formation of an inflammatory response or scar tissue.29
Amniotic tissue has had similar success in tendon repair studies in vivo.9,30,31 Barboni and colleagues32 implanted amniotic epithelial cells (AECs) into artificially created sheep Achilles tendon defects in situ, inducing superior structural and mechanical recovery in the defects at a faster rate compared to controls not receiving AECs. Healing via AECs started at the healthy tissue around the borders of the defect and progressed centrally, suggesting recruitment of native progenitor cells to the lesion.32 Kueckelhaus and colleagues33 investigated the role of amnion-derived cellular cytokine solution in the healing of transections of rat Achilles tendons, reporting improved mechanical properties of healing tendons at early time points compared to controls. Beredjiklian and colleagues34 compared the healing of transected extensor tendons of pregnant ewes and of their fetus in utero, reporting a reparative form of healing with scar formation in adult subjects and regenerative form of healing without scar formation or inflammation in fetal subjects.
Amniotic tissue has properties that prevent adhesion formation around tendons following injury and reconstruction.35 Ozgenel36 investigated the effects of hyaluronic acid and amniotic membrane alone and in combination on the presence of adhesions and the rate of healing following chicken flexor tendon repair. The study found amniotic membrane wrapped around the repaired tendon was superior in preventing adhesion formation. Kim and colleagues37 report a similar reduction in fibrosis and adhesion following application of a human amniotic membrane wrap to rabbit ulnar neurorrhaphy sites.
This barrier function of amniotic tissue has also been investigated in the prevention of surgical scarring and peridural fibrosis in animal models following spinal discectomy. A study in canine models showed a reduction of scarring following the application of cross-linked amniotic membrane compared to freeze dried amniotic membrane.38 Similar reductions in scarring in rat models with the application of freeze-dried amniotic membrane compared to negative controls have been reported.39
Human Studies
A randomized trial investigated the outcomes of prenatal vs postnatal repair of myelomeningocele in humans, finding a reduced need for implanted shunts and improved functional outcomes at 30 months of life in the prenatal intervention group compared to the postnatal group.40 This study was concluded early due to the efficacy of prenatal surgery and the benefit of nervous system repair in utero in the presence of amniotic growth factors.
Vines and colleagues41 performed a 6-patient feasibility study using amnion injections to treat symptomatic knee osteoarthritis. Each patient received a single intra-articular cryopreserved amniotic suspension allograft (ASA) injection and was followed for 1 year. No adverse outcomes were reported, with the only abnormal finding being a small increase in serum immunoglobulin G and immunoglobulin E levels. Intra-articular ASA injection was found to be safe, but a large-scale trial investigating symptomatic relief was recommended.41
Most of the human studies using amnion pertain to foot and ankle surgery. Its use as a treatment for diabetic foot ulcers and recalcitrant plantar fasciitis was one of the early-recognized successes.42-45 Zelen and colleagues46 investigated the applications of injectable micronized dehydrated human amniotic/chorionic membrane as an alternative to surgical intervention in the treatment of refractory plantar fasciitis. This prospective, randomized trial with 45 patients showed significant improvement in plantar fasciitis symptoms at 8 weeks compared to controls (saline injections). A similar study compared the use of cryopreserved human amniotic membrane (c-hAM) injections to corticosteroid injections in plantar fasciitis patients.47 The results indicated that c-hAM is safe and comparable to corticosteroids, with the authors noting that pain improvement was greatest in patients receiving 2 injections of c-hAM at 18 weeks.
Tendon wrapping, in which the amniotic membrane is laid over a tendon repair, has been reported with success. Amniotic membrane is superior to collagen for tendon wrapping as it actively contributes to healing while minimizing adhesions, which collagen alone cannot do.48 The membrane serves as a protective sheath around repaired tendons with anti-inflammatory, anti-adhesive, immunomodulatory, and antimicrobial benefits. A 124-patient study demonstrated the safety of using amnion in this manner, and the authors reported a decreased rate of complication compared to previously published data.49 Another study of 14 patients undergoing foot and ankle surgery with tendon wrapping reported clinical improvement with reduced pain and greater functional outcomes postoperatively compared to preoperative measurements.50
Conclusion
Amniotic membrane-derived tissues are safe and non-tumorigenic, producing an abundance of growth factors that have shown promise as tissue scaffolds and as aids in the regeneration of human bone and soft tissues. Amnion applications in orthopedic surgery may be numerous, but development is ongoing. Given the vast array of in vitro and in vivo animal data supporting the benefits of amnion in tissue regeneration, orthopedic surgeons and researchers should place emphasis on conducting clinical studies to validate the safety and efficacy of amniotic cells in the treatment of orthopedic conditions.
Am J Orthop. 2016;45(7):E421-E425. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Benirschke K, Kaufman P. Anatomy and pathology of the placental membranes. In: Pathology of the Human Placent., 4th ed. New York, NY: Springer-Verlag; 2000:281-334.
2. Mamede AC, Carvalho MJ, Abrantes AM, Laranjo M, Maia CJ, Botelho MF. Amniotic membrane: from structure and functions to clinical applications. Cell Tissue Res. 2012;349(2):447-458.
3. Miki T, Strom SC. Amnion-derived pluripotent/multipotent stem cells. Stem Cell Rev. 2006;2(2):133-142.
4. Parolini O, Alviano F, Bagnara GP, et al. Concise review: isolation and characterization of cells from human term placenta: outcome of the first international workshop on placenta derived stem cells. Stem Cells. 2008;26(2):300-311.
5. Ilancheran S, Michalska A, Peh G, Wallace EM, Pera M, Manuelpillai U. Stem cells derived from human fetal membranes display multilineage differentiation potential. Biol Reprod. 2007;77(3):577-588.
6. Alviano F, Fossati V, Marchionni C, et al. Term amniotic membrane is a high throughput source for multipotent mesenchymal stem cells with the ability to differentiate into endothelial cells in vitro. BMC Dev Biol. 2007;7:11.
7. Barboni B, Curini V, Russo V, et al. Indirect co-culture with tendons or tenocytes can program amniotic epithelial cells towards stepwise tenogenic differentiation. PLoS One. 2012;7(2):e30974.
8. Ben-David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nature Reviews Cancer. 2011;11(4):268-277.
9. Lange-Consiglio A, Rossi D, Tassan S, Perego R, Cremonesi F, Parolini O. Conditioned medium from horse amniotic membrane-derived multipotent progenitor cells: immunomodulatory activity in vitro and first clinical application in tendon and ligament injuries in vivo. Stem Cells Dev. 2013;22(22):3015-3024.
10. Miki T. Amnion-derived stem cells: in quest of clinical applications. Stem Cell Res Ther. 2011;2(3):25.
11. Akle CA, Adinolfi M, Welsh KI, Leibowitz S, McColl I. Immunogenicity of human amniotic epithelial cells after transplantation into volunteers. Lancet. 1981;2(8254):1003-1035.
12. Gupta A, Kedige SD, Jain K. Amnion and chorion membranes: potential stem cell reservoir with wide applications in periodontics. Int J Biomater. 2015;2015:274082.
13. Buhimschi IA, Jabr M, Buhimschi CS, Petkova AP, Weiner CP, Saed GM. The novel antimicrobial peptide beta3-defensin is produced by the amnion: a possible role of the fetal membranes in innate immunity of the amniotic cavity. Am J Obstet Gynecol. 2004;191(5):1678-1687.
14. Magatti M, De Munari S, Vertua E, et al. Amniotic mesenchymal tissue cells inhibit dendritic cell differentiation of peripheral blood and amnion resident monocytes. Cell Transplant. 2009;18(8):899-914.
15. Solomon A, Wajngarten M, Alviano F, et al. Suppression of inflammatory and fibrotic responses in allergic inflammation by the amniotic membrane stromal matrix. Clin Exp Allergy. 2005;35(7):941-948.
16. Silini A, Parolini O, Huppertz B, Lang I. Soluble factors of amnion-derived cells in treatment of inflammatory and fibrotic pathologies. Curr Stem Cell Res Ther. 2013;8(1):6-14.
17. Peister A, Woodruff MA, Prince JJ, Gray DP, Hutmacher DW, Guldberg RE. Cell sourcing for bone tissue engineering: amniotic fluid stem cells have a delayed, robust differentiation compared to mesenchymal stem cells. Stem Cell Res. 2011;7(1):17-27.
18. Si J, Dai J, Zhang J, et al. Comparative investigation of human amniotic epithelial cells and mesenchymal stem cells for application in bone tissue engineering. Stem Cells Int. 2015;2015:565732.
19. Starecki M, Schwartz JA, Grande DA. Evaluation of amniotic-derived membrane biomaterial as an adjunct for repair of critical sized bone defects. Advances in Orthopedic Surgery. 2014;2014:572586.
20. Kerimoglu S, Livaoglu M, Sönmez B, et al. Effects of human amniotic fluid on fracture healing in rat tibia. J Surg Res. 2009;152(2):281-287.
21. Karaçal N, Kocucu P, Cobanglu U, Kutlu N. Effect of human amniotic fluid on bone healing. J Surg Res. 2005;129(2):283-287.
22. Barboni B, Mangano C, Valbonetti L, et al. Synthetic bone substitute engineered with amniotic epithelial cells enhances bone regeneration after maxillary sinus augmentation. PLoS One. 2013;8(5):e63256.
23. Díaz-Prado S, Rendal-Vázquez ME, Muiños-Lopez E, et al. Potential use of the human amniotic membrane as a scaffold in human articular cartilage repair. Cell Tissue Bank. 2010;11(2):183-195.
24. Krishnamurithy G, Shilpa PN, Ahmad RE, Sulaiman S, Ng CL, Kamarul T. Human amniotic membrane as a chondrocyte carrier vehicle/substrate: in vitro study. J Biomed Mater Res A. 2011;99(3):500-506.
25. Tan SL, Sulaiman S, Pingguan-Murphy B, Selvaratnam L, Tai CC, Kamarul T. Human amnion as a novel cell delivery vehicle for chondrogenic mesenchymal stem cells. Cell Tissue Bank. 2011;12(1):59-70.
26. Jin CZ, Park SR, Choi BH, Lee KY, Kang CK, Min BH. Human amniotic membrane as a delivery matrix for articular cartilage repair. Tissue Eng. 2007;13(4):693-702.
27. Garcia D, Longo UG, Vaquero J, et al. Amniotic membrane transplant for articular cartilage repair: an experimental study in sheep. Curr Stem Cell Res Ther. 2014;10(1):77-83.
28. Kunisaki SM, Freedman DA, Fauza DO. Fetal tracheal reconstruction with cartilaginous grafts engineered from mesenchymal amniocytes. J Pediatr Surg. 2006;41(4):675-682.
29. Namba RS, Meuli M, Sullivan KM, Le AX, Adzick NS. Spontaneous repair of superficial defects in articular cartilage in a fetal lamb model. J Bone Joint Surg Am. 1998;80(1):4-10.
30. Philip J, Hackl F, Canseco JA, et al. Amnion-derived multipotent progenitor cells improve achilles tendon repair in rats. Eplasty. 2013;13:e31.
31. Lange-Consiglio A, Tassan S, Corradetti B, et al. Investigating the efficacy of amnion-derived compared with bone marrow–derived mesenchymal stromal cells in equine tendon and ligament injuries. Cytotherapy. 2013;15(8):1011-1020.
32. Barboni B, Russo V, Curini V, et al. Achilles tendon regeneration can be improved by amniotic epithelial cell allotransplantation. Cell Transplant. 2012;21(11):2377-2395.
33. Kueckelhaus M, Philip J, Kamel RA, et al. Sustained release of amnion-derived cellular cytokine solution facilitates achilles tendon healing in rats. Eplasty. 2014;14:e29.
34. Beredjiklian PK, Favata M, Cartmell JS, Flanagan CL, Crombleholme TM, Soslowski LJ. Regenerative versus reparative healing in tendon: a study of biomechanical and histological properties in fetal sheep. Ann Biomed Eng. 2003;31(10):1143-1152.
35. Demirkan F, Colakoglu N, Herek O, Erkula G. The use of amniotic membrane in flexor tendon repair: an experimental model. Arch Orthop Trauma Surg. 2002;122(7):396-369.
36. Ozgenel GY. The effects of a combination of hyaluronic and amniotic membrane on the formation of peritendinous adhesions after flexor tendon surgery in chickens. J Bone Joint Surg Br. 2004;86(2):301-307.
37. Kim SS, Sohn SK, Lee KY, Lee MJ, Roh MS, Kim CH. Use of human amniotic membrane wrap in reducing perineural adhesions in a rabbit model of ulnar nerve neurorrhaphy. J Hand Surg Eur Vol. 2010;35(3):214-219.
38. Tao H, Fan H. Implantation of amniotic membrane to reduce postlaminectomy epidural adhesions. Eur Spine J. 2009;18(8):1202-1212.
39. Choi HJ, Kim KB, Kwon YM. Effect of amniotic membrane to reduce postlaminectomy epidural adhesion on a rat model. J Korean Neurosurg Soc. 2011;49(6):323-328.
40. Adzick NS, Thom EA, Spong CY, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011;364(11):993-1004.
41. Vines JB, Aliprantis AO, Gomoll AH, Farr J. Cryopreserved amniotic suspension for the treatment of knee osteoarthritis. J Knee Surg. 2016;29(6):443-450.
42. Zelen CM. An evaluation of dehydrated human amniotic membrane allografts in patients with DFUs. J Wound Care. 2013;22(7):347-348,350-351.
43. Zelen CM, Serena TE, Denoziere G, Fetterolf DE. A prospective randomised comparative parallel study of amniotic membrane wound graft in the management of diabetic foot ulcers. Int Wound J. 2013;10(5):502-507.
44. Zelen CM, Serena TE, Snyder RJ. A prospective, randomised comparative study of weekly versus biweekly application of dehydrated human amnion/chorion membrane allograft in the management of diabetic foot ulcers. Int Wound J. 2014;11(2):122-128.
45. Zelen CM, Snyder RJ, Serena TE, Li WW. The use of human amnion/chorion membrane in the clinical setting for lower extremity repair: a review. Clin Podiatr Med Surg. 2015;32(1):135-146.
46. Zelen CM, Poka A, Andrews J. Prospective, randomized, blinded, comparative study of injectable micronized dehydrated amniotic/chorionic membrane allograft for plantar fasciitis: a feasibility study. Foot Ankle Int. 2013;34(10):1332-1339.
47. Hanselman AE, Tidwell JE, Santrock RD. Cryopreserved human amniotic membrane injection for plantar fasciitis: a randomized, controlled, double-blind pilot study. Foot Ankle Int. 2015;36(2):151-158.
48. Jay RM. Initial clinical experience with the use of human amniotic membrane tissue during repair of posterior tibial and achilles tendons. 2009. http://encompassbiologics.com/wp-content/uploads/2015/07/DrJayClinicalExperience.pdf. Accessed September 29, 2016.
49. DeMill SL, Granata JD, McAlister JE, Berlet GC, Hyer CF. Safety analysis of cryopreserved amniotic membrane/umbilical cord tissue in foot and ankle surgery: a consecutive case series of 124 patients. Surg Technol Int. 2014;25:257-261.
50. Warner M, Lasyone L. An open-label, single-center, retrospective study of cryopreserved amniotic membrane and umbilical cord tissue as an adjunct for foot and ankle surgery. Surg Technol Int. 2014;25:251-255.
The amniotic membrane is a multilayer tissue forming the innermost layer of the amniotic sac that surrounds the developing fetus. It is comprised of 5 layers, from the inside out: a single layer of epithelial cells, a thick basement membrane, a compact layer, a fibroblast layer, and a spongy layer that abuts the surrounding chorion (Figure 1).1
Amniotic epithelial cells are derived from the pluripotent epiblast at approximately day 8 of gestation. This is well before gastrulation occurs at days 15 to 17, considered the “tipping point” when pluripotent cells differentiate into ectoderm, mesoderm, and endoderm.3 These cells express Oct-4 and Nanog, 2 molecular markers that are indicative of pluripotency.3 Two cell types have been identified in amniotic tissues that possess stem cell-like characteristics: human amniotic epithelial cells and human amniotic mesenchymal stromal cells.4 Both of these cell types have demonstrated the ability to differentiate into various cell lineages, including endothelial cells, adipocytes, myogenic cells, neurogenic cells, chondrocytes, tenocytes, and osteogenic cells.5-7 These previously reported findings indicate that amniotic cells and tissue have the capability to generate mesenchymal tissues.
FDA Classification and Available Forms
The US Food and Drug Administration (FDA) classifies amnion as an allograft tissue under Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps) 361. To meet criteria, the tissue needs to be minimally manipulated. It is to be for homologous use and cannot be combined with other cells or tissues. There can be no systemic effect or dependence on the metabolic activity of living cells to achieve its primary function. The tissue has to have a localized effect in vivo. Therefore, amnion allograft tissue can be commercialized, provided it is not marketed as a stem cell product or to contain viable cells.
Amniotic tissue is commercially available in several forms.
Safety
Amniotic tissue has been used for over 100 years in burn, ophthalmology, and chronic wound patients with favorable outcomes and no adverse effects reported in the literature. Unlike embryonic stem cells, which may be tumorigenic,8 amniotic cells do not possess any known tumorigenicity.9 In one study, 50 immunodeficient mice were injected with 1 to 2 million amniotic epithelial cells and observed for a maximum of 516 days with no tumorigenicity observed in any of the animals.10 In another study, amniotic epithelial cells were implanted into the forearms of healthy volunteers and no immunologic response was observed in any of the recipients.11 Furthermore, viable amniotic cells were recovered via biopsy 7 weeks following transplantation, demonstrating viability of the transplanted cells.11 The lack of tumorigenicity and immunologic response in hosts is due in part to the fact that amniotic cells do not express human leukocyte antigen class II antigens and only express class I antigens in small amounts.3
Advantages of Amnion Tissue
Amniotic tissue is readily available, as it is often discarded after childbirth. The use of this tissue poses no added risk to the fetus or mother, eliminating the ethical concerns associated with obtaining embryonic stem cells. Amniotic tissue is comprised of an extracellular matrix, which acts as a natural scaffold for cellular attachment and structural support for cells as well as collagen types I, III, IV, V, and VI, hyaluronic acid, and a host of growth factors.12 In addition, it possesses antimicrobial properties, including beta-defensins.13
Amniotic tissue has been shown to exert an anti-inflammatory effect by inhibiting the inflammatory cascade. Specifically, it has been shown to inhibit cytokines such as tumor necrosis factor-alpha in the presence of dendritic cells,14 as well as inhibiting transforming growth factor-beta, interleukin-8, and fibroblast proliferation.15 These findings indicate that amniotic tissue has the ability to dampen the “cytokine storm” that occurs after an injury in an adult, which would lead to beneficial impacts on healing and scar formation in patients.16
Basic Science and Animal Studies
Several studies have demonstrated promising outcomes for orthopedic applications in vitro. A comparison of osteogenic potential found that amniotic fluid-derived cells were able to produce approximately 5 times more mineralized matrix than bone marrow-derived mesenchymal stem cells.17 More recently, Si and colleagues18 compared the osteogenic potential of human amniotic epithelial cells, amniotic cells, and human bone marrow-derived mesenchymal stem cells. They found that all 3 cell lines were osteogenic, though the amniotic epithelial cells had better immunomodulatory properties and marginally less osteogenic potential than the other 2 cell types. Furthermore, several in vivo animal studies have demonstrated the ability of human amniotic cells to stimulate bone growth in rats,19,20 rabbits,21 and sheep.22
Amniotic tissue also possesses potential for chondrogenesis. Cryopreserved human amniotic membrane cells used for in vitro human osteoarthritis tissue scaffolds did not differentiate in culture, and they integrated and repaired damaged articular cartilage.23 Various in vitro24,25 and animal in vivo26,27 studies have reported similar supportive findings. Kunisaki and colleagues28 used sheep amniotic fluid mesenchymal stem cells to reconstruct lamb tracheal cartilage in utero, concluding that cells obtained from the amniotic fluid possess chondrogenic capabilities. Further in utero lamb studies of cartilage artificial defects, given 7 days to settle before adding a hypocellular matrix as a scaffold, showed chondrocyte density and cell architecture was restored at the defect site after 28 days without the formation of an inflammatory response or scar tissue.29
Amniotic tissue has had similar success in tendon repair studies in vivo.9,30,31 Barboni and colleagues32 implanted amniotic epithelial cells (AECs) into artificially created sheep Achilles tendon defects in situ, inducing superior structural and mechanical recovery in the defects at a faster rate compared to controls not receiving AECs. Healing via AECs started at the healthy tissue around the borders of the defect and progressed centrally, suggesting recruitment of native progenitor cells to the lesion.32 Kueckelhaus and colleagues33 investigated the role of amnion-derived cellular cytokine solution in the healing of transections of rat Achilles tendons, reporting improved mechanical properties of healing tendons at early time points compared to controls. Beredjiklian and colleagues34 compared the healing of transected extensor tendons of pregnant ewes and of their fetus in utero, reporting a reparative form of healing with scar formation in adult subjects and regenerative form of healing without scar formation or inflammation in fetal subjects.
Amniotic tissue has properties that prevent adhesion formation around tendons following injury and reconstruction.35 Ozgenel36 investigated the effects of hyaluronic acid and amniotic membrane alone and in combination on the presence of adhesions and the rate of healing following chicken flexor tendon repair. The study found amniotic membrane wrapped around the repaired tendon was superior in preventing adhesion formation. Kim and colleagues37 report a similar reduction in fibrosis and adhesion following application of a human amniotic membrane wrap to rabbit ulnar neurorrhaphy sites.
This barrier function of amniotic tissue has also been investigated in the prevention of surgical scarring and peridural fibrosis in animal models following spinal discectomy. A study in canine models showed a reduction of scarring following the application of cross-linked amniotic membrane compared to freeze dried amniotic membrane.38 Similar reductions in scarring in rat models with the application of freeze-dried amniotic membrane compared to negative controls have been reported.39
Human Studies
A randomized trial investigated the outcomes of prenatal vs postnatal repair of myelomeningocele in humans, finding a reduced need for implanted shunts and improved functional outcomes at 30 months of life in the prenatal intervention group compared to the postnatal group.40 This study was concluded early due to the efficacy of prenatal surgery and the benefit of nervous system repair in utero in the presence of amniotic growth factors.
Vines and colleagues41 performed a 6-patient feasibility study using amnion injections to treat symptomatic knee osteoarthritis. Each patient received a single intra-articular cryopreserved amniotic suspension allograft (ASA) injection and was followed for 1 year. No adverse outcomes were reported, with the only abnormal finding being a small increase in serum immunoglobulin G and immunoglobulin E levels. Intra-articular ASA injection was found to be safe, but a large-scale trial investigating symptomatic relief was recommended.41
Most of the human studies using amnion pertain to foot and ankle surgery. Its use as a treatment for diabetic foot ulcers and recalcitrant plantar fasciitis was one of the early-recognized successes.42-45 Zelen and colleagues46 investigated the applications of injectable micronized dehydrated human amniotic/chorionic membrane as an alternative to surgical intervention in the treatment of refractory plantar fasciitis. This prospective, randomized trial with 45 patients showed significant improvement in plantar fasciitis symptoms at 8 weeks compared to controls (saline injections). A similar study compared the use of cryopreserved human amniotic membrane (c-hAM) injections to corticosteroid injections in plantar fasciitis patients.47 The results indicated that c-hAM is safe and comparable to corticosteroids, with the authors noting that pain improvement was greatest in patients receiving 2 injections of c-hAM at 18 weeks.
Tendon wrapping, in which the amniotic membrane is laid over a tendon repair, has been reported with success. Amniotic membrane is superior to collagen for tendon wrapping as it actively contributes to healing while minimizing adhesions, which collagen alone cannot do.48 The membrane serves as a protective sheath around repaired tendons with anti-inflammatory, anti-adhesive, immunomodulatory, and antimicrobial benefits. A 124-patient study demonstrated the safety of using amnion in this manner, and the authors reported a decreased rate of complication compared to previously published data.49 Another study of 14 patients undergoing foot and ankle surgery with tendon wrapping reported clinical improvement with reduced pain and greater functional outcomes postoperatively compared to preoperative measurements.50
Conclusion
Amniotic membrane-derived tissues are safe and non-tumorigenic, producing an abundance of growth factors that have shown promise as tissue scaffolds and as aids in the regeneration of human bone and soft tissues. Amnion applications in orthopedic surgery may be numerous, but development is ongoing. Given the vast array of in vitro and in vivo animal data supporting the benefits of amnion in tissue regeneration, orthopedic surgeons and researchers should place emphasis on conducting clinical studies to validate the safety and efficacy of amniotic cells in the treatment of orthopedic conditions.
Am J Orthop. 2016;45(7):E421-E425. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
The amniotic membrane is a multilayer tissue forming the innermost layer of the amniotic sac that surrounds the developing fetus. It is comprised of 5 layers, from the inside out: a single layer of epithelial cells, a thick basement membrane, a compact layer, a fibroblast layer, and a spongy layer that abuts the surrounding chorion (Figure 1).1
Amniotic epithelial cells are derived from the pluripotent epiblast at approximately day 8 of gestation. This is well before gastrulation occurs at days 15 to 17, considered the “tipping point” when pluripotent cells differentiate into ectoderm, mesoderm, and endoderm.3 These cells express Oct-4 and Nanog, 2 molecular markers that are indicative of pluripotency.3 Two cell types have been identified in amniotic tissues that possess stem cell-like characteristics: human amniotic epithelial cells and human amniotic mesenchymal stromal cells.4 Both of these cell types have demonstrated the ability to differentiate into various cell lineages, including endothelial cells, adipocytes, myogenic cells, neurogenic cells, chondrocytes, tenocytes, and osteogenic cells.5-7 These previously reported findings indicate that amniotic cells and tissue have the capability to generate mesenchymal tissues.
FDA Classification and Available Forms
The US Food and Drug Administration (FDA) classifies amnion as an allograft tissue under Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps) 361. To meet criteria, the tissue needs to be minimally manipulated. It is to be for homologous use and cannot be combined with other cells or tissues. There can be no systemic effect or dependence on the metabolic activity of living cells to achieve its primary function. The tissue has to have a localized effect in vivo. Therefore, amnion allograft tissue can be commercialized, provided it is not marketed as a stem cell product or to contain viable cells.
Amniotic tissue is commercially available in several forms.
Safety
Amniotic tissue has been used for over 100 years in burn, ophthalmology, and chronic wound patients with favorable outcomes and no adverse effects reported in the literature. Unlike embryonic stem cells, which may be tumorigenic,8 amniotic cells do not possess any known tumorigenicity.9 In one study, 50 immunodeficient mice were injected with 1 to 2 million amniotic epithelial cells and observed for a maximum of 516 days with no tumorigenicity observed in any of the animals.10 In another study, amniotic epithelial cells were implanted into the forearms of healthy volunteers and no immunologic response was observed in any of the recipients.11 Furthermore, viable amniotic cells were recovered via biopsy 7 weeks following transplantation, demonstrating viability of the transplanted cells.11 The lack of tumorigenicity and immunologic response in hosts is due in part to the fact that amniotic cells do not express human leukocyte antigen class II antigens and only express class I antigens in small amounts.3
Advantages of Amnion Tissue
Amniotic tissue is readily available, as it is often discarded after childbirth. The use of this tissue poses no added risk to the fetus or mother, eliminating the ethical concerns associated with obtaining embryonic stem cells. Amniotic tissue is comprised of an extracellular matrix, which acts as a natural scaffold for cellular attachment and structural support for cells as well as collagen types I, III, IV, V, and VI, hyaluronic acid, and a host of growth factors.12 In addition, it possesses antimicrobial properties, including beta-defensins.13
Amniotic tissue has been shown to exert an anti-inflammatory effect by inhibiting the inflammatory cascade. Specifically, it has been shown to inhibit cytokines such as tumor necrosis factor-alpha in the presence of dendritic cells,14 as well as inhibiting transforming growth factor-beta, interleukin-8, and fibroblast proliferation.15 These findings indicate that amniotic tissue has the ability to dampen the “cytokine storm” that occurs after an injury in an adult, which would lead to beneficial impacts on healing and scar formation in patients.16
Basic Science and Animal Studies
Several studies have demonstrated promising outcomes for orthopedic applications in vitro. A comparison of osteogenic potential found that amniotic fluid-derived cells were able to produce approximately 5 times more mineralized matrix than bone marrow-derived mesenchymal stem cells.17 More recently, Si and colleagues18 compared the osteogenic potential of human amniotic epithelial cells, amniotic cells, and human bone marrow-derived mesenchymal stem cells. They found that all 3 cell lines were osteogenic, though the amniotic epithelial cells had better immunomodulatory properties and marginally less osteogenic potential than the other 2 cell types. Furthermore, several in vivo animal studies have demonstrated the ability of human amniotic cells to stimulate bone growth in rats,19,20 rabbits,21 and sheep.22
Amniotic tissue also possesses potential for chondrogenesis. Cryopreserved human amniotic membrane cells used for in vitro human osteoarthritis tissue scaffolds did not differentiate in culture, and they integrated and repaired damaged articular cartilage.23 Various in vitro24,25 and animal in vivo26,27 studies have reported similar supportive findings. Kunisaki and colleagues28 used sheep amniotic fluid mesenchymal stem cells to reconstruct lamb tracheal cartilage in utero, concluding that cells obtained from the amniotic fluid possess chondrogenic capabilities. Further in utero lamb studies of cartilage artificial defects, given 7 days to settle before adding a hypocellular matrix as a scaffold, showed chondrocyte density and cell architecture was restored at the defect site after 28 days without the formation of an inflammatory response or scar tissue.29
Amniotic tissue has had similar success in tendon repair studies in vivo.9,30,31 Barboni and colleagues32 implanted amniotic epithelial cells (AECs) into artificially created sheep Achilles tendon defects in situ, inducing superior structural and mechanical recovery in the defects at a faster rate compared to controls not receiving AECs. Healing via AECs started at the healthy tissue around the borders of the defect and progressed centrally, suggesting recruitment of native progenitor cells to the lesion.32 Kueckelhaus and colleagues33 investigated the role of amnion-derived cellular cytokine solution in the healing of transections of rat Achilles tendons, reporting improved mechanical properties of healing tendons at early time points compared to controls. Beredjiklian and colleagues34 compared the healing of transected extensor tendons of pregnant ewes and of their fetus in utero, reporting a reparative form of healing with scar formation in adult subjects and regenerative form of healing without scar formation or inflammation in fetal subjects.
Amniotic tissue has properties that prevent adhesion formation around tendons following injury and reconstruction.35 Ozgenel36 investigated the effects of hyaluronic acid and amniotic membrane alone and in combination on the presence of adhesions and the rate of healing following chicken flexor tendon repair. The study found amniotic membrane wrapped around the repaired tendon was superior in preventing adhesion formation. Kim and colleagues37 report a similar reduction in fibrosis and adhesion following application of a human amniotic membrane wrap to rabbit ulnar neurorrhaphy sites.
This barrier function of amniotic tissue has also been investigated in the prevention of surgical scarring and peridural fibrosis in animal models following spinal discectomy. A study in canine models showed a reduction of scarring following the application of cross-linked amniotic membrane compared to freeze dried amniotic membrane.38 Similar reductions in scarring in rat models with the application of freeze-dried amniotic membrane compared to negative controls have been reported.39
Human Studies
A randomized trial investigated the outcomes of prenatal vs postnatal repair of myelomeningocele in humans, finding a reduced need for implanted shunts and improved functional outcomes at 30 months of life in the prenatal intervention group compared to the postnatal group.40 This study was concluded early due to the efficacy of prenatal surgery and the benefit of nervous system repair in utero in the presence of amniotic growth factors.
Vines and colleagues41 performed a 6-patient feasibility study using amnion injections to treat symptomatic knee osteoarthritis. Each patient received a single intra-articular cryopreserved amniotic suspension allograft (ASA) injection and was followed for 1 year. No adverse outcomes were reported, with the only abnormal finding being a small increase in serum immunoglobulin G and immunoglobulin E levels. Intra-articular ASA injection was found to be safe, but a large-scale trial investigating symptomatic relief was recommended.41
Most of the human studies using amnion pertain to foot and ankle surgery. Its use as a treatment for diabetic foot ulcers and recalcitrant plantar fasciitis was one of the early-recognized successes.42-45 Zelen and colleagues46 investigated the applications of injectable micronized dehydrated human amniotic/chorionic membrane as an alternative to surgical intervention in the treatment of refractory plantar fasciitis. This prospective, randomized trial with 45 patients showed significant improvement in plantar fasciitis symptoms at 8 weeks compared to controls (saline injections). A similar study compared the use of cryopreserved human amniotic membrane (c-hAM) injections to corticosteroid injections in plantar fasciitis patients.47 The results indicated that c-hAM is safe and comparable to corticosteroids, with the authors noting that pain improvement was greatest in patients receiving 2 injections of c-hAM at 18 weeks.
Tendon wrapping, in which the amniotic membrane is laid over a tendon repair, has been reported with success. Amniotic membrane is superior to collagen for tendon wrapping as it actively contributes to healing while minimizing adhesions, which collagen alone cannot do.48 The membrane serves as a protective sheath around repaired tendons with anti-inflammatory, anti-adhesive, immunomodulatory, and antimicrobial benefits. A 124-patient study demonstrated the safety of using amnion in this manner, and the authors reported a decreased rate of complication compared to previously published data.49 Another study of 14 patients undergoing foot and ankle surgery with tendon wrapping reported clinical improvement with reduced pain and greater functional outcomes postoperatively compared to preoperative measurements.50
Conclusion
Amniotic membrane-derived tissues are safe and non-tumorigenic, producing an abundance of growth factors that have shown promise as tissue scaffolds and as aids in the regeneration of human bone and soft tissues. Amnion applications in orthopedic surgery may be numerous, but development is ongoing. Given the vast array of in vitro and in vivo animal data supporting the benefits of amnion in tissue regeneration, orthopedic surgeons and researchers should place emphasis on conducting clinical studies to validate the safety and efficacy of amniotic cells in the treatment of orthopedic conditions.
Am J Orthop. 2016;45(7):E421-E425. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Benirschke K, Kaufman P. Anatomy and pathology of the placental membranes. In: Pathology of the Human Placent., 4th ed. New York, NY: Springer-Verlag; 2000:281-334.
2. Mamede AC, Carvalho MJ, Abrantes AM, Laranjo M, Maia CJ, Botelho MF. Amniotic membrane: from structure and functions to clinical applications. Cell Tissue Res. 2012;349(2):447-458.
3. Miki T, Strom SC. Amnion-derived pluripotent/multipotent stem cells. Stem Cell Rev. 2006;2(2):133-142.
4. Parolini O, Alviano F, Bagnara GP, et al. Concise review: isolation and characterization of cells from human term placenta: outcome of the first international workshop on placenta derived stem cells. Stem Cells. 2008;26(2):300-311.
5. Ilancheran S, Michalska A, Peh G, Wallace EM, Pera M, Manuelpillai U. Stem cells derived from human fetal membranes display multilineage differentiation potential. Biol Reprod. 2007;77(3):577-588.
6. Alviano F, Fossati V, Marchionni C, et al. Term amniotic membrane is a high throughput source for multipotent mesenchymal stem cells with the ability to differentiate into endothelial cells in vitro. BMC Dev Biol. 2007;7:11.
7. Barboni B, Curini V, Russo V, et al. Indirect co-culture with tendons or tenocytes can program amniotic epithelial cells towards stepwise tenogenic differentiation. PLoS One. 2012;7(2):e30974.
8. Ben-David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nature Reviews Cancer. 2011;11(4):268-277.
9. Lange-Consiglio A, Rossi D, Tassan S, Perego R, Cremonesi F, Parolini O. Conditioned medium from horse amniotic membrane-derived multipotent progenitor cells: immunomodulatory activity in vitro and first clinical application in tendon and ligament injuries in vivo. Stem Cells Dev. 2013;22(22):3015-3024.
10. Miki T. Amnion-derived stem cells: in quest of clinical applications. Stem Cell Res Ther. 2011;2(3):25.
11. Akle CA, Adinolfi M, Welsh KI, Leibowitz S, McColl I. Immunogenicity of human amniotic epithelial cells after transplantation into volunteers. Lancet. 1981;2(8254):1003-1035.
12. Gupta A, Kedige SD, Jain K. Amnion and chorion membranes: potential stem cell reservoir with wide applications in periodontics. Int J Biomater. 2015;2015:274082.
13. Buhimschi IA, Jabr M, Buhimschi CS, Petkova AP, Weiner CP, Saed GM. The novel antimicrobial peptide beta3-defensin is produced by the amnion: a possible role of the fetal membranes in innate immunity of the amniotic cavity. Am J Obstet Gynecol. 2004;191(5):1678-1687.
14. Magatti M, De Munari S, Vertua E, et al. Amniotic mesenchymal tissue cells inhibit dendritic cell differentiation of peripheral blood and amnion resident monocytes. Cell Transplant. 2009;18(8):899-914.
15. Solomon A, Wajngarten M, Alviano F, et al. Suppression of inflammatory and fibrotic responses in allergic inflammation by the amniotic membrane stromal matrix. Clin Exp Allergy. 2005;35(7):941-948.
16. Silini A, Parolini O, Huppertz B, Lang I. Soluble factors of amnion-derived cells in treatment of inflammatory and fibrotic pathologies. Curr Stem Cell Res Ther. 2013;8(1):6-14.
17. Peister A, Woodruff MA, Prince JJ, Gray DP, Hutmacher DW, Guldberg RE. Cell sourcing for bone tissue engineering: amniotic fluid stem cells have a delayed, robust differentiation compared to mesenchymal stem cells. Stem Cell Res. 2011;7(1):17-27.
18. Si J, Dai J, Zhang J, et al. Comparative investigation of human amniotic epithelial cells and mesenchymal stem cells for application in bone tissue engineering. Stem Cells Int. 2015;2015:565732.
19. Starecki M, Schwartz JA, Grande DA. Evaluation of amniotic-derived membrane biomaterial as an adjunct for repair of critical sized bone defects. Advances in Orthopedic Surgery. 2014;2014:572586.
20. Kerimoglu S, Livaoglu M, Sönmez B, et al. Effects of human amniotic fluid on fracture healing in rat tibia. J Surg Res. 2009;152(2):281-287.
21. Karaçal N, Kocucu P, Cobanglu U, Kutlu N. Effect of human amniotic fluid on bone healing. J Surg Res. 2005;129(2):283-287.
22. Barboni B, Mangano C, Valbonetti L, et al. Synthetic bone substitute engineered with amniotic epithelial cells enhances bone regeneration after maxillary sinus augmentation. PLoS One. 2013;8(5):e63256.
23. Díaz-Prado S, Rendal-Vázquez ME, Muiños-Lopez E, et al. Potential use of the human amniotic membrane as a scaffold in human articular cartilage repair. Cell Tissue Bank. 2010;11(2):183-195.
24. Krishnamurithy G, Shilpa PN, Ahmad RE, Sulaiman S, Ng CL, Kamarul T. Human amniotic membrane as a chondrocyte carrier vehicle/substrate: in vitro study. J Biomed Mater Res A. 2011;99(3):500-506.
25. Tan SL, Sulaiman S, Pingguan-Murphy B, Selvaratnam L, Tai CC, Kamarul T. Human amnion as a novel cell delivery vehicle for chondrogenic mesenchymal stem cells. Cell Tissue Bank. 2011;12(1):59-70.
26. Jin CZ, Park SR, Choi BH, Lee KY, Kang CK, Min BH. Human amniotic membrane as a delivery matrix for articular cartilage repair. Tissue Eng. 2007;13(4):693-702.
27. Garcia D, Longo UG, Vaquero J, et al. Amniotic membrane transplant for articular cartilage repair: an experimental study in sheep. Curr Stem Cell Res Ther. 2014;10(1):77-83.
28. Kunisaki SM, Freedman DA, Fauza DO. Fetal tracheal reconstruction with cartilaginous grafts engineered from mesenchymal amniocytes. J Pediatr Surg. 2006;41(4):675-682.
29. Namba RS, Meuli M, Sullivan KM, Le AX, Adzick NS. Spontaneous repair of superficial defects in articular cartilage in a fetal lamb model. J Bone Joint Surg Am. 1998;80(1):4-10.
30. Philip J, Hackl F, Canseco JA, et al. Amnion-derived multipotent progenitor cells improve achilles tendon repair in rats. Eplasty. 2013;13:e31.
31. Lange-Consiglio A, Tassan S, Corradetti B, et al. Investigating the efficacy of amnion-derived compared with bone marrow–derived mesenchymal stromal cells in equine tendon and ligament injuries. Cytotherapy. 2013;15(8):1011-1020.
32. Barboni B, Russo V, Curini V, et al. Achilles tendon regeneration can be improved by amniotic epithelial cell allotransplantation. Cell Transplant. 2012;21(11):2377-2395.
33. Kueckelhaus M, Philip J, Kamel RA, et al. Sustained release of amnion-derived cellular cytokine solution facilitates achilles tendon healing in rats. Eplasty. 2014;14:e29.
34. Beredjiklian PK, Favata M, Cartmell JS, Flanagan CL, Crombleholme TM, Soslowski LJ. Regenerative versus reparative healing in tendon: a study of biomechanical and histological properties in fetal sheep. Ann Biomed Eng. 2003;31(10):1143-1152.
35. Demirkan F, Colakoglu N, Herek O, Erkula G. The use of amniotic membrane in flexor tendon repair: an experimental model. Arch Orthop Trauma Surg. 2002;122(7):396-369.
36. Ozgenel GY. The effects of a combination of hyaluronic and amniotic membrane on the formation of peritendinous adhesions after flexor tendon surgery in chickens. J Bone Joint Surg Br. 2004;86(2):301-307.
37. Kim SS, Sohn SK, Lee KY, Lee MJ, Roh MS, Kim CH. Use of human amniotic membrane wrap in reducing perineural adhesions in a rabbit model of ulnar nerve neurorrhaphy. J Hand Surg Eur Vol. 2010;35(3):214-219.
38. Tao H, Fan H. Implantation of amniotic membrane to reduce postlaminectomy epidural adhesions. Eur Spine J. 2009;18(8):1202-1212.
39. Choi HJ, Kim KB, Kwon YM. Effect of amniotic membrane to reduce postlaminectomy epidural adhesion on a rat model. J Korean Neurosurg Soc. 2011;49(6):323-328.
40. Adzick NS, Thom EA, Spong CY, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011;364(11):993-1004.
41. Vines JB, Aliprantis AO, Gomoll AH, Farr J. Cryopreserved amniotic suspension for the treatment of knee osteoarthritis. J Knee Surg. 2016;29(6):443-450.
42. Zelen CM. An evaluation of dehydrated human amniotic membrane allografts in patients with DFUs. J Wound Care. 2013;22(7):347-348,350-351.
43. Zelen CM, Serena TE, Denoziere G, Fetterolf DE. A prospective randomised comparative parallel study of amniotic membrane wound graft in the management of diabetic foot ulcers. Int Wound J. 2013;10(5):502-507.
44. Zelen CM, Serena TE, Snyder RJ. A prospective, randomised comparative study of weekly versus biweekly application of dehydrated human amnion/chorion membrane allograft in the management of diabetic foot ulcers. Int Wound J. 2014;11(2):122-128.
45. Zelen CM, Snyder RJ, Serena TE, Li WW. The use of human amnion/chorion membrane in the clinical setting for lower extremity repair: a review. Clin Podiatr Med Surg. 2015;32(1):135-146.
46. Zelen CM, Poka A, Andrews J. Prospective, randomized, blinded, comparative study of injectable micronized dehydrated amniotic/chorionic membrane allograft for plantar fasciitis: a feasibility study. Foot Ankle Int. 2013;34(10):1332-1339.
47. Hanselman AE, Tidwell JE, Santrock RD. Cryopreserved human amniotic membrane injection for plantar fasciitis: a randomized, controlled, double-blind pilot study. Foot Ankle Int. 2015;36(2):151-158.
48. Jay RM. Initial clinical experience with the use of human amniotic membrane tissue during repair of posterior tibial and achilles tendons. 2009. http://encompassbiologics.com/wp-content/uploads/2015/07/DrJayClinicalExperience.pdf. Accessed September 29, 2016.
49. DeMill SL, Granata JD, McAlister JE, Berlet GC, Hyer CF. Safety analysis of cryopreserved amniotic membrane/umbilical cord tissue in foot and ankle surgery: a consecutive case series of 124 patients. Surg Technol Int. 2014;25:257-261.
50. Warner M, Lasyone L. An open-label, single-center, retrospective study of cryopreserved amniotic membrane and umbilical cord tissue as an adjunct for foot and ankle surgery. Surg Technol Int. 2014;25:251-255.
1. Benirschke K, Kaufman P. Anatomy and pathology of the placental membranes. In: Pathology of the Human Placent., 4th ed. New York, NY: Springer-Verlag; 2000:281-334.
2. Mamede AC, Carvalho MJ, Abrantes AM, Laranjo M, Maia CJ, Botelho MF. Amniotic membrane: from structure and functions to clinical applications. Cell Tissue Res. 2012;349(2):447-458.
3. Miki T, Strom SC. Amnion-derived pluripotent/multipotent stem cells. Stem Cell Rev. 2006;2(2):133-142.
4. Parolini O, Alviano F, Bagnara GP, et al. Concise review: isolation and characterization of cells from human term placenta: outcome of the first international workshop on placenta derived stem cells. Stem Cells. 2008;26(2):300-311.
5. Ilancheran S, Michalska A, Peh G, Wallace EM, Pera M, Manuelpillai U. Stem cells derived from human fetal membranes display multilineage differentiation potential. Biol Reprod. 2007;77(3):577-588.
6. Alviano F, Fossati V, Marchionni C, et al. Term amniotic membrane is a high throughput source for multipotent mesenchymal stem cells with the ability to differentiate into endothelial cells in vitro. BMC Dev Biol. 2007;7:11.
7. Barboni B, Curini V, Russo V, et al. Indirect co-culture with tendons or tenocytes can program amniotic epithelial cells towards stepwise tenogenic differentiation. PLoS One. 2012;7(2):e30974.
8. Ben-David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nature Reviews Cancer. 2011;11(4):268-277.
9. Lange-Consiglio A, Rossi D, Tassan S, Perego R, Cremonesi F, Parolini O. Conditioned medium from horse amniotic membrane-derived multipotent progenitor cells: immunomodulatory activity in vitro and first clinical application in tendon and ligament injuries in vivo. Stem Cells Dev. 2013;22(22):3015-3024.
10. Miki T. Amnion-derived stem cells: in quest of clinical applications. Stem Cell Res Ther. 2011;2(3):25.
11. Akle CA, Adinolfi M, Welsh KI, Leibowitz S, McColl I. Immunogenicity of human amniotic epithelial cells after transplantation into volunteers. Lancet. 1981;2(8254):1003-1035.
12. Gupta A, Kedige SD, Jain K. Amnion and chorion membranes: potential stem cell reservoir with wide applications in periodontics. Int J Biomater. 2015;2015:274082.
13. Buhimschi IA, Jabr M, Buhimschi CS, Petkova AP, Weiner CP, Saed GM. The novel antimicrobial peptide beta3-defensin is produced by the amnion: a possible role of the fetal membranes in innate immunity of the amniotic cavity. Am J Obstet Gynecol. 2004;191(5):1678-1687.
14. Magatti M, De Munari S, Vertua E, et al. Amniotic mesenchymal tissue cells inhibit dendritic cell differentiation of peripheral blood and amnion resident monocytes. Cell Transplant. 2009;18(8):899-914.
15. Solomon A, Wajngarten M, Alviano F, et al. Suppression of inflammatory and fibrotic responses in allergic inflammation by the amniotic membrane stromal matrix. Clin Exp Allergy. 2005;35(7):941-948.
16. Silini A, Parolini O, Huppertz B, Lang I. Soluble factors of amnion-derived cells in treatment of inflammatory and fibrotic pathologies. Curr Stem Cell Res Ther. 2013;8(1):6-14.
17. Peister A, Woodruff MA, Prince JJ, Gray DP, Hutmacher DW, Guldberg RE. Cell sourcing for bone tissue engineering: amniotic fluid stem cells have a delayed, robust differentiation compared to mesenchymal stem cells. Stem Cell Res. 2011;7(1):17-27.
18. Si J, Dai J, Zhang J, et al. Comparative investigation of human amniotic epithelial cells and mesenchymal stem cells for application in bone tissue engineering. Stem Cells Int. 2015;2015:565732.
19. Starecki M, Schwartz JA, Grande DA. Evaluation of amniotic-derived membrane biomaterial as an adjunct for repair of critical sized bone defects. Advances in Orthopedic Surgery. 2014;2014:572586.
20. Kerimoglu S, Livaoglu M, Sönmez B, et al. Effects of human amniotic fluid on fracture healing in rat tibia. J Surg Res. 2009;152(2):281-287.
21. Karaçal N, Kocucu P, Cobanglu U, Kutlu N. Effect of human amniotic fluid on bone healing. J Surg Res. 2005;129(2):283-287.
22. Barboni B, Mangano C, Valbonetti L, et al. Synthetic bone substitute engineered with amniotic epithelial cells enhances bone regeneration after maxillary sinus augmentation. PLoS One. 2013;8(5):e63256.
23. Díaz-Prado S, Rendal-Vázquez ME, Muiños-Lopez E, et al. Potential use of the human amniotic membrane as a scaffold in human articular cartilage repair. Cell Tissue Bank. 2010;11(2):183-195.
24. Krishnamurithy G, Shilpa PN, Ahmad RE, Sulaiman S, Ng CL, Kamarul T. Human amniotic membrane as a chondrocyte carrier vehicle/substrate: in vitro study. J Biomed Mater Res A. 2011;99(3):500-506.
25. Tan SL, Sulaiman S, Pingguan-Murphy B, Selvaratnam L, Tai CC, Kamarul T. Human amnion as a novel cell delivery vehicle for chondrogenic mesenchymal stem cells. Cell Tissue Bank. 2011;12(1):59-70.
26. Jin CZ, Park SR, Choi BH, Lee KY, Kang CK, Min BH. Human amniotic membrane as a delivery matrix for articular cartilage repair. Tissue Eng. 2007;13(4):693-702.
27. Garcia D, Longo UG, Vaquero J, et al. Amniotic membrane transplant for articular cartilage repair: an experimental study in sheep. Curr Stem Cell Res Ther. 2014;10(1):77-83.
28. Kunisaki SM, Freedman DA, Fauza DO. Fetal tracheal reconstruction with cartilaginous grafts engineered from mesenchymal amniocytes. J Pediatr Surg. 2006;41(4):675-682.
29. Namba RS, Meuli M, Sullivan KM, Le AX, Adzick NS. Spontaneous repair of superficial defects in articular cartilage in a fetal lamb model. J Bone Joint Surg Am. 1998;80(1):4-10.
30. Philip J, Hackl F, Canseco JA, et al. Amnion-derived multipotent progenitor cells improve achilles tendon repair in rats. Eplasty. 2013;13:e31.
31. Lange-Consiglio A, Tassan S, Corradetti B, et al. Investigating the efficacy of amnion-derived compared with bone marrow–derived mesenchymal stromal cells in equine tendon and ligament injuries. Cytotherapy. 2013;15(8):1011-1020.
32. Barboni B, Russo V, Curini V, et al. Achilles tendon regeneration can be improved by amniotic epithelial cell allotransplantation. Cell Transplant. 2012;21(11):2377-2395.
33. Kueckelhaus M, Philip J, Kamel RA, et al. Sustained release of amnion-derived cellular cytokine solution facilitates achilles tendon healing in rats. Eplasty. 2014;14:e29.
34. Beredjiklian PK, Favata M, Cartmell JS, Flanagan CL, Crombleholme TM, Soslowski LJ. Regenerative versus reparative healing in tendon: a study of biomechanical and histological properties in fetal sheep. Ann Biomed Eng. 2003;31(10):1143-1152.
35. Demirkan F, Colakoglu N, Herek O, Erkula G. The use of amniotic membrane in flexor tendon repair: an experimental model. Arch Orthop Trauma Surg. 2002;122(7):396-369.
36. Ozgenel GY. The effects of a combination of hyaluronic and amniotic membrane on the formation of peritendinous adhesions after flexor tendon surgery in chickens. J Bone Joint Surg Br. 2004;86(2):301-307.
37. Kim SS, Sohn SK, Lee KY, Lee MJ, Roh MS, Kim CH. Use of human amniotic membrane wrap in reducing perineural adhesions in a rabbit model of ulnar nerve neurorrhaphy. J Hand Surg Eur Vol. 2010;35(3):214-219.
38. Tao H, Fan H. Implantation of amniotic membrane to reduce postlaminectomy epidural adhesions. Eur Spine J. 2009;18(8):1202-1212.
39. Choi HJ, Kim KB, Kwon YM. Effect of amniotic membrane to reduce postlaminectomy epidural adhesion on a rat model. J Korean Neurosurg Soc. 2011;49(6):323-328.
40. Adzick NS, Thom EA, Spong CY, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011;364(11):993-1004.
41. Vines JB, Aliprantis AO, Gomoll AH, Farr J. Cryopreserved amniotic suspension for the treatment of knee osteoarthritis. J Knee Surg. 2016;29(6):443-450.
42. Zelen CM. An evaluation of dehydrated human amniotic membrane allografts in patients with DFUs. J Wound Care. 2013;22(7):347-348,350-351.
43. Zelen CM, Serena TE, Denoziere G, Fetterolf DE. A prospective randomised comparative parallel study of amniotic membrane wound graft in the management of diabetic foot ulcers. Int Wound J. 2013;10(5):502-507.
44. Zelen CM, Serena TE, Snyder RJ. A prospective, randomised comparative study of weekly versus biweekly application of dehydrated human amnion/chorion membrane allograft in the management of diabetic foot ulcers. Int Wound J. 2014;11(2):122-128.
45. Zelen CM, Snyder RJ, Serena TE, Li WW. The use of human amnion/chorion membrane in the clinical setting for lower extremity repair: a review. Clin Podiatr Med Surg. 2015;32(1):135-146.
46. Zelen CM, Poka A, Andrews J. Prospective, randomized, blinded, comparative study of injectable micronized dehydrated amniotic/chorionic membrane allograft for plantar fasciitis: a feasibility study. Foot Ankle Int. 2013;34(10):1332-1339.
47. Hanselman AE, Tidwell JE, Santrock RD. Cryopreserved human amniotic membrane injection for plantar fasciitis: a randomized, controlled, double-blind pilot study. Foot Ankle Int. 2015;36(2):151-158.
48. Jay RM. Initial clinical experience with the use of human amniotic membrane tissue during repair of posterior tibial and achilles tendons. 2009. http://encompassbiologics.com/wp-content/uploads/2015/07/DrJayClinicalExperience.pdf. Accessed September 29, 2016.
49. DeMill SL, Granata JD, McAlister JE, Berlet GC, Hyer CF. Safety analysis of cryopreserved amniotic membrane/umbilical cord tissue in foot and ankle surgery: a consecutive case series of 124 patients. Surg Technol Int. 2014;25:257-261.
50. Warner M, Lasyone L. An open-label, single-center, retrospective study of cryopreserved amniotic membrane and umbilical cord tissue as an adjunct for foot and ankle surgery. Surg Technol Int. 2014;25:251-255.