User login
HCV testing stagnant among baby boomers
Despite the urging of the United States Preventive Services Task Force and other organizations in 2013, the percentage of baby boomers who underwent testing for hepatitis C (HCV) infection had barely changed 2 years later – from 12.3% in 2013 to 13.8% in 2015.
The numbers are particularly troubling because new and improved antiviral drugs offer cures that could forestall liver cancer, cirrhosis, and other potential complications, with shorter regimens and fewer side effects than older regimens.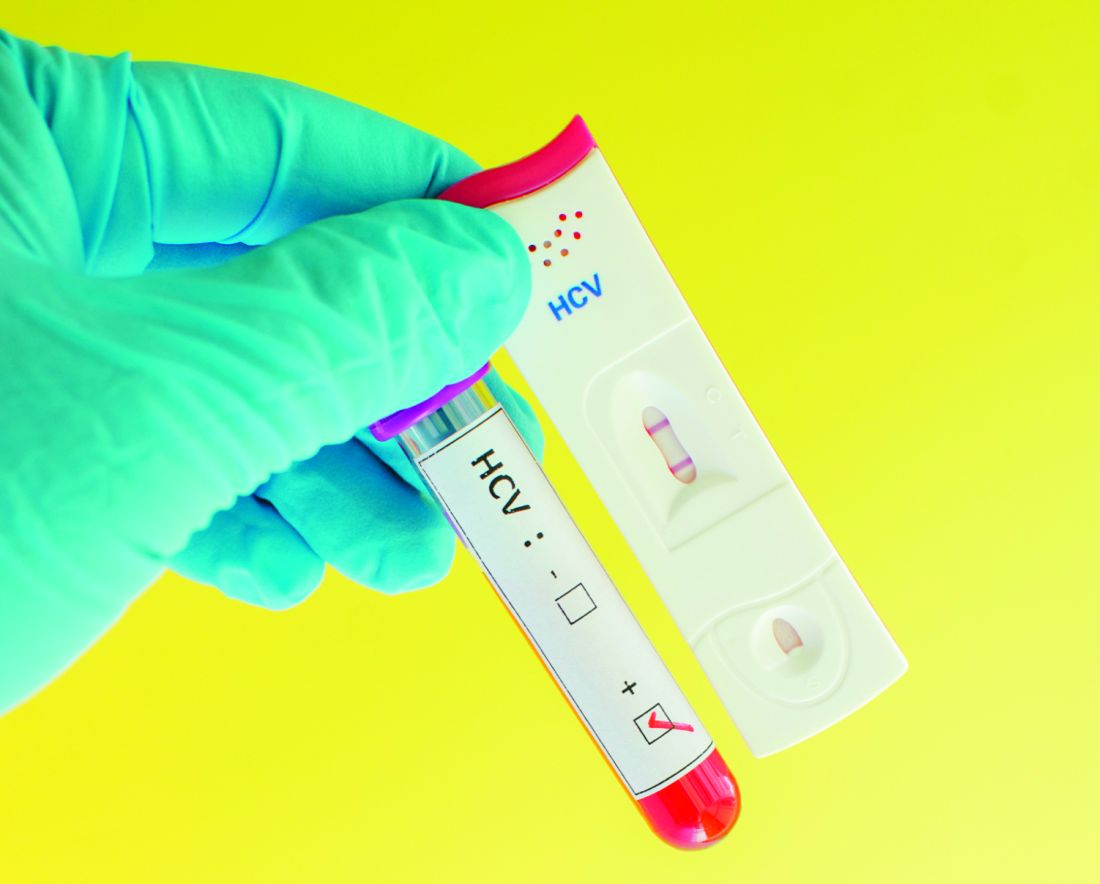
Other reactions were more forceful. “Kind of pathetic, isn’t it?” said John D. Scott, MD, assistant director of the Hepatitis and Liver Clinic at Harborview Medical Center, and an associate professor of medicine at the University of Washington, Seattle.
The researchers analyzed 2013 and 2015 data from the National Health Interview Survey, which included records for 21,827 baby boomers with HCV testing data.
The slight increase overall of 12.3% to 13.8% was small but also statistically significant (P = .013). Some populations fared better: Compared with the privately insured, those with Medicare plus Medicaid were more likely to have been tested (prevalence ratio, 1.83; 95% confidence interval, 1.32-2.53), as were those only on Medicaid (PR, 1.35; 95% CI, 1.04-1.76), and those with military insurance (PR, 1.62; 95% CI, 1.16-2.26).
The study could be subject to recall bias, since it relied on participants’ self-reports.
The authors speculate that the higher prevalence of testing in those with military insurance may reflect efforts by the Veterans Health Administration to reduce the high prevalence of HCV-associated disease among veterans.
It’s entirely possible to increase testing rates, according to Dr. Scott, who has a grant from the Centers for Disease Control and Prevention to study ways to increase uptake. “Probably the easiest thing to do is just incorporate this information into your electronic medical record and make it part of your alerts and standard preventative practices. Try to automate a lot of this rather than remind a very busy primary care doctor of all the things they have to do,” he said.
For example, one strategy that Seattle’s King County has employed is to automatically notify the testing laboratory if an antibody test is positive. “The lab knows to keep that blood and run a second (nucleic acid) test without the patient having to come back. That has helped to get our confirmatory rates up,” said Dr. Scott.
More broadly, the importance of testing needs to be emphasized, according to Paul J. Thuluvath, MD, medical director at the Institute of Digestive Health and Liver Disease at Mercy Medical Center, Baltimore, and a professor of medicine and surgery at the University of Maryland. “We need everybody to buy into this: the primary care physicians, internists, and gynecologists. If they are not convinced of the importance of this, it’s not going to happen. And I don’t think many primary care physicians and internists are convinced yet,” he said.
Despite the urging of the United States Preventive Services Task Force and other organizations in 2013, the percentage of baby boomers who underwent testing for hepatitis C (HCV) infection had barely changed 2 years later – from 12.3% in 2013 to 13.8% in 2015.
The numbers are particularly troubling because new and improved antiviral drugs offer cures that could forestall liver cancer, cirrhosis, and other potential complications, with shorter regimens and fewer side effects than older regimens.
Other reactions were more forceful. “Kind of pathetic, isn’t it?” said John D. Scott, MD, assistant director of the Hepatitis and Liver Clinic at Harborview Medical Center, and an associate professor of medicine at the University of Washington, Seattle.
The researchers analyzed 2013 and 2015 data from the National Health Interview Survey, which included records for 21,827 baby boomers with HCV testing data.
The slight increase overall of 12.3% to 13.8% was small but also statistically significant (P = .013). Some populations fared better: Compared with the privately insured, those with Medicare plus Medicaid were more likely to have been tested (prevalence ratio, 1.83; 95% confidence interval, 1.32-2.53), as were those only on Medicaid (PR, 1.35; 95% CI, 1.04-1.76), and those with military insurance (PR, 1.62; 95% CI, 1.16-2.26).
The study could be subject to recall bias, since it relied on participants’ self-reports.
The authors speculate that the higher prevalence of testing in those with military insurance may reflect efforts by the Veterans Health Administration to reduce the high prevalence of HCV-associated disease among veterans.
It’s entirely possible to increase testing rates, according to Dr. Scott, who has a grant from the Centers for Disease Control and Prevention to study ways to increase uptake. “Probably the easiest thing to do is just incorporate this information into your electronic medical record and make it part of your alerts and standard preventative practices. Try to automate a lot of this rather than remind a very busy primary care doctor of all the things they have to do,” he said.
For example, one strategy that Seattle’s King County has employed is to automatically notify the testing laboratory if an antibody test is positive. “The lab knows to keep that blood and run a second (nucleic acid) test without the patient having to come back. That has helped to get our confirmatory rates up,” said Dr. Scott.
More broadly, the importance of testing needs to be emphasized, according to Paul J. Thuluvath, MD, medical director at the Institute of Digestive Health and Liver Disease at Mercy Medical Center, Baltimore, and a professor of medicine and surgery at the University of Maryland. “We need everybody to buy into this: the primary care physicians, internists, and gynecologists. If they are not convinced of the importance of this, it’s not going to happen. And I don’t think many primary care physicians and internists are convinced yet,” he said.
Despite the urging of the United States Preventive Services Task Force and other organizations in 2013, the percentage of baby boomers who underwent testing for hepatitis C (HCV) infection had barely changed 2 years later – from 12.3% in 2013 to 13.8% in 2015.
The numbers are particularly troubling because new and improved antiviral drugs offer cures that could forestall liver cancer, cirrhosis, and other potential complications, with shorter regimens and fewer side effects than older regimens.
Other reactions were more forceful. “Kind of pathetic, isn’t it?” said John D. Scott, MD, assistant director of the Hepatitis and Liver Clinic at Harborview Medical Center, and an associate professor of medicine at the University of Washington, Seattle.
The researchers analyzed 2013 and 2015 data from the National Health Interview Survey, which included records for 21,827 baby boomers with HCV testing data.
The slight increase overall of 12.3% to 13.8% was small but also statistically significant (P = .013). Some populations fared better: Compared with the privately insured, those with Medicare plus Medicaid were more likely to have been tested (prevalence ratio, 1.83; 95% confidence interval, 1.32-2.53), as were those only on Medicaid (PR, 1.35; 95% CI, 1.04-1.76), and those with military insurance (PR, 1.62; 95% CI, 1.16-2.26).
The study could be subject to recall bias, since it relied on participants’ self-reports.
The authors speculate that the higher prevalence of testing in those with military insurance may reflect efforts by the Veterans Health Administration to reduce the high prevalence of HCV-associated disease among veterans.
It’s entirely possible to increase testing rates, according to Dr. Scott, who has a grant from the Centers for Disease Control and Prevention to study ways to increase uptake. “Probably the easiest thing to do is just incorporate this information into your electronic medical record and make it part of your alerts and standard preventative practices. Try to automate a lot of this rather than remind a very busy primary care doctor of all the things they have to do,” he said.
For example, one strategy that Seattle’s King County has employed is to automatically notify the testing laboratory if an antibody test is positive. “The lab knows to keep that blood and run a second (nucleic acid) test without the patient having to come back. That has helped to get our confirmatory rates up,” said Dr. Scott.
More broadly, the importance of testing needs to be emphasized, according to Paul J. Thuluvath, MD, medical director at the Institute of Digestive Health and Liver Disease at Mercy Medical Center, Baltimore, and a professor of medicine and surgery at the University of Maryland. “We need everybody to buy into this: the primary care physicians, internists, and gynecologists. If they are not convinced of the importance of this, it’s not going to happen. And I don’t think many primary care physicians and internists are convinced yet,” he said.
FROM AMERICAN JOURNAL OF PREVENTIVE MEDICINE
Key clinical point: Primary care physicians are not yet convinced of HCV test’s value.
Major finding: Between 2013 and 2015, HCV testing rates rose from 12.3% to 13.8%.
Data source: Retrospective analysis of 21,827 baby boomers who were part of the National Health Interview Survey.
Disclosures: The study was funded by the American Cancer Society. Dr. Fedewa reported having no financial disclosures. Dr. Scott has received research funding from Merck and serves on the data safety and monitoring board for Tacere Therapeutics. Dr. Thuluvath has received funding from Gilead and has received speaking fees from Gilead and AbbVie.
Federal judge blocks Anthem-Cigna merger
A federal district court judge has blocked health insurer Anthem from acquiring Cigna, ruling the megamerger would violate antitrust laws and stifle competition.
The decision came weeks after another U.S. district court judge barred a merger between health insurance giants Aetna and Humana.
The U.S. Department of Justice praised the latest ruling, calling the decision a victory for patients.
“This merger would have stifled competition, harming consumers by increasing health insurance prices and slowing innovation aimed at lowering the costs of health care,” Acting Assistant Attorney General Brent Snyder said in a statement.
Anthem intends to appeal the decision, said Joseph R. Swedish, Anthem’s chair, president, and chief executive officer. “Anthem is significantly disappointed by the decision, as combining Anthem and Cigna would positively impact the health and well-being of millions of Americans – saving them more than $2 billion in medical costs annually,” Mr. Swedish said in a statement. “If not overturned, the consequences of the decision are far reaching and will hurt American consumers by limiting their access to high-quality affordable care, slowing the industry’s shift to value-based care and improved outcomes for patients, and restricting innovation, which is critical to meeting the evolving needs of health care consumers.”
In a statement, a Cigna official said the company intends to carefully review the opinion and evaluate its options in accordance with the merger agreement.
“Cigna remains focused on helping to improve health care by delivering value to our customers and clients and expanding our business around the world,” the statement said.
The DOJ, 11 states, and the District of Columbia sued Anthem and Cigna in July over their proposed $54 billion consolidation in what would have been the largest merger in history.
The DOJ argued the merger would substantially harm competition and negatively impact the entire insurance industry if allowed to proceed. The consolidation would enhance Anthem’s power to profit at the expense of consumers and the doctors and hospitals who provide their medical care, DOJ attorneys said in their complaint.
Anthem and Cigna argued the proposed acquisition was “procompetitive,” and that the merger would result in efficiencies that would directly benefit consumers via greater access to affordable health care. The benefits of the merger outweigh any alleged anticompetitive effects, according to Anthem.
A trial before Judge Amy Berman Jackson of the U.S. District Court for the District of Columbia ran from November through January.
Judge Berman’s opinion is temporarily under seal to allow parties to review for confidentiality.
The ruling is the second victory for the DOJ in as many weeks. In a Jan. 23 decision, Judge John D. Bates of the U.S. District Court for the District of Columbia denied Aetna’s $37 billion plan to purchase Humana, following a month-long trial that began in early December. Judge Bates ruled the consolidation would violate antitrust laws and reduce competition.
Aetna and Humana did not respond to requests for comment.
On Twitter @legal_med
Michael E. Nelson, MD, FCCP, comments: Any business owner who has been required to absorb yearly double-digit increases in employee health insurance costs cannot help but wonder where Mr. Swedish learned his “new math.” His second statement is even more incogitable – since when were insurers known for expanding access to health care.

Michael E. Nelson, MD, FCCP, comments: Any business owner who has been required to absorb yearly double-digit increases in employee health insurance costs cannot help but wonder where Mr. Swedish learned his “new math.” His second statement is even more incogitable – since when were insurers known for expanding access to health care.
Michael E. Nelson, MD, FCCP, comments: Any business owner who has been required to absorb yearly double-digit increases in employee health insurance costs cannot help but wonder where Mr. Swedish learned his “new math.” His second statement is even more incogitable – since when were insurers known for expanding access to health care.
A federal district court judge has blocked health insurer Anthem from acquiring Cigna, ruling the megamerger would violate antitrust laws and stifle competition.
The decision came weeks after another U.S. district court judge barred a merger between health insurance giants Aetna and Humana.
The U.S. Department of Justice praised the latest ruling, calling the decision a victory for patients.
“This merger would have stifled competition, harming consumers by increasing health insurance prices and slowing innovation aimed at lowering the costs of health care,” Acting Assistant Attorney General Brent Snyder said in a statement.
Anthem intends to appeal the decision, said Joseph R. Swedish, Anthem’s chair, president, and chief executive officer. “Anthem is significantly disappointed by the decision, as combining Anthem and Cigna would positively impact the health and well-being of millions of Americans – saving them more than $2 billion in medical costs annually,” Mr. Swedish said in a statement. “If not overturned, the consequences of the decision are far reaching and will hurt American consumers by limiting their access to high-quality affordable care, slowing the industry’s shift to value-based care and improved outcomes for patients, and restricting innovation, which is critical to meeting the evolving needs of health care consumers.”
In a statement, a Cigna official said the company intends to carefully review the opinion and evaluate its options in accordance with the merger agreement.
“Cigna remains focused on helping to improve health care by delivering value to our customers and clients and expanding our business around the world,” the statement said.
The DOJ, 11 states, and the District of Columbia sued Anthem and Cigna in July over their proposed $54 billion consolidation in what would have been the largest merger in history.
The DOJ argued the merger would substantially harm competition and negatively impact the entire insurance industry if allowed to proceed. The consolidation would enhance Anthem’s power to profit at the expense of consumers and the doctors and hospitals who provide their medical care, DOJ attorneys said in their complaint.
Anthem and Cigna argued the proposed acquisition was “procompetitive,” and that the merger would result in efficiencies that would directly benefit consumers via greater access to affordable health care. The benefits of the merger outweigh any alleged anticompetitive effects, according to Anthem.
A trial before Judge Amy Berman Jackson of the U.S. District Court for the District of Columbia ran from November through January.
Judge Berman’s opinion is temporarily under seal to allow parties to review for confidentiality.
The ruling is the second victory for the DOJ in as many weeks. In a Jan. 23 decision, Judge John D. Bates of the U.S. District Court for the District of Columbia denied Aetna’s $37 billion plan to purchase Humana, following a month-long trial that began in early December. Judge Bates ruled the consolidation would violate antitrust laws and reduce competition.
Aetna and Humana did not respond to requests for comment.
On Twitter @legal_med
A federal district court judge has blocked health insurer Anthem from acquiring Cigna, ruling the megamerger would violate antitrust laws and stifle competition.
The decision came weeks after another U.S. district court judge barred a merger between health insurance giants Aetna and Humana.
The U.S. Department of Justice praised the latest ruling, calling the decision a victory for patients.
“This merger would have stifled competition, harming consumers by increasing health insurance prices and slowing innovation aimed at lowering the costs of health care,” Acting Assistant Attorney General Brent Snyder said in a statement.
Anthem intends to appeal the decision, said Joseph R. Swedish, Anthem’s chair, president, and chief executive officer. “Anthem is significantly disappointed by the decision, as combining Anthem and Cigna would positively impact the health and well-being of millions of Americans – saving them more than $2 billion in medical costs annually,” Mr. Swedish said in a statement. “If not overturned, the consequences of the decision are far reaching and will hurt American consumers by limiting their access to high-quality affordable care, slowing the industry’s shift to value-based care and improved outcomes for patients, and restricting innovation, which is critical to meeting the evolving needs of health care consumers.”
In a statement, a Cigna official said the company intends to carefully review the opinion and evaluate its options in accordance with the merger agreement.
“Cigna remains focused on helping to improve health care by delivering value to our customers and clients and expanding our business around the world,” the statement said.
The DOJ, 11 states, and the District of Columbia sued Anthem and Cigna in July over their proposed $54 billion consolidation in what would have been the largest merger in history.
The DOJ argued the merger would substantially harm competition and negatively impact the entire insurance industry if allowed to proceed. The consolidation would enhance Anthem’s power to profit at the expense of consumers and the doctors and hospitals who provide their medical care, DOJ attorneys said in their complaint.
Anthem and Cigna argued the proposed acquisition was “procompetitive,” and that the merger would result in efficiencies that would directly benefit consumers via greater access to affordable health care. The benefits of the merger outweigh any alleged anticompetitive effects, according to Anthem.
A trial before Judge Amy Berman Jackson of the U.S. District Court for the District of Columbia ran from November through January.
Judge Berman’s opinion is temporarily under seal to allow parties to review for confidentiality.
The ruling is the second victory for the DOJ in as many weeks. In a Jan. 23 decision, Judge John D. Bates of the U.S. District Court for the District of Columbia denied Aetna’s $37 billion plan to purchase Humana, following a month-long trial that began in early December. Judge Bates ruled the consolidation would violate antitrust laws and reduce competition.
Aetna and Humana did not respond to requests for comment.
On Twitter @legal_med

Trending at the Society of Hospital Medicine
Calling all pediatric hospitalists
Register for Pediatric Hospital Medicine 2017 (PHM17), the premier educational conference for pediatric hospitalists and other clinicians who care for hospitalized children. Re-energize your practice with the latest research, best practices, innovations, and more.
![]()
Register before June 7 to receive the early-bird rates. Visit www.peds2017.org for more information.
SHM can prepare you for MACRA
![]()
Visit www.macraforhm.org for general information and details in the MACRA FAQ and MIPS Tips links.
Don’t miss five new tracks at HM17
![]()
- Learn how to avoid diagnostic and therapeutic overuse, and how to move towards the right care for every hospital medicine patient with the High Value Care Track.
- Don’t miss the Clinical Updates Track, which provides evidence-based updates from recent literature published in medicine subspecialty fields and specific topic areas that all hospitalists need to know.
- Accurate and timely diagnosis are the two cornerstones of high-quality patient care. Find out what topics are in the Diagnostic Reasoning Track.
- Learn from experts during the Health Policy Track who will discuss the most current health care policy issues as they impact hospitalists and what we can expect from a new Presidential administration and changes in Congress.
- The Mini Medical Education Track is for hospitalists who are interested in improving their teaching skills.
Learn more about the HM17 schedule and offerings at www.hospitalmedicine2017.org/schedule.
Prepare for the entire Focused Practice in Hospital Medicine (FPHM) exam with SPARK ONE
![]()
This self-paced study guide engages learners through an open-book format, allowing users to review detailed learning objectives and discussion points and define individual areas of strengths and weaknesses. SHM members Save $150! Learn more at www.hospitalmedicine.org/sparkone.
Improve your treatment of VTE during Blood Clot Awareness Month
March is Blood Clot Awareness Month, and SHM recently introduced a new toolkit and guide surrounding treatment of venous thromboembolism (VTE) in the hospital setting. SHM has a history of providing cutting-edge resources in this space, and Steven B. Deitelzweig, MD, MMM, SFHM, FACP, FACC, system chairman of hospital medicine at Oschner Health System in New Orleans, was integral in editing SHM’s VTE treatment mentored implementation guide and online toolkit.
“SHM has an established track record of implementing evidence-based and guideline-driven learnings successfully, and we continue to see improvement across multiple facilities based on this work with this disease,” Dr. Deitelzweig says. “Whenever possible, I would strongly recommend taking full advantage of SHM’s outstanding programs as they are intensely developed by experts for adoption at hospitals of different sizes, including community and academic centers.”
SHM can help you and your hospital improve treatment of VTE as well – learn how at www.hospitalmedicine.org/vtetreatment.
Share patient experience success stories
Our Patient Experience Committee wants to showcase stories of when care teams or their counterparts in the hospital made a notable shift in a patient’s experience: a special moment or interaction; a successful improvement project; an award for excellence in practice; a memo of commendation; a letter from a patient. Email examples of success to Claudia Stahl at cstahl@hospitalmedicine.org by May 11. Submissions can include photos, letters, or videos. SHM will share these moments that “made all the difference” with members on its website via other channels soon to be announced.
Brett Radler is SHM’s communications specialist.
Not a member? Know someone who should be? Visit www.joinshm.org to learn about the opportunities we can offer hospital medicine professionals.
Calling all pediatric hospitalists
Register for Pediatric Hospital Medicine 2017 (PHM17), the premier educational conference for pediatric hospitalists and other clinicians who care for hospitalized children. Re-energize your practice with the latest research, best practices, innovations, and more.
![]()
Register before June 7 to receive the early-bird rates. Visit www.peds2017.org for more information.
SHM can prepare you for MACRA
![]()
Visit www.macraforhm.org for general information and details in the MACRA FAQ and MIPS Tips links.
Don’t miss five new tracks at HM17
![]()
- Learn how to avoid diagnostic and therapeutic overuse, and how to move towards the right care for every hospital medicine patient with the High Value Care Track.
- Don’t miss the Clinical Updates Track, which provides evidence-based updates from recent literature published in medicine subspecialty fields and specific topic areas that all hospitalists need to know.
- Accurate and timely diagnosis are the two cornerstones of high-quality patient care. Find out what topics are in the Diagnostic Reasoning Track.
- Learn from experts during the Health Policy Track who will discuss the most current health care policy issues as they impact hospitalists and what we can expect from a new Presidential administration and changes in Congress.
- The Mini Medical Education Track is for hospitalists who are interested in improving their teaching skills.
Learn more about the HM17 schedule and offerings at www.hospitalmedicine2017.org/schedule.
Prepare for the entire Focused Practice in Hospital Medicine (FPHM) exam with SPARK ONE
![]()
This self-paced study guide engages learners through an open-book format, allowing users to review detailed learning objectives and discussion points and define individual areas of strengths and weaknesses. SHM members Save $150! Learn more at www.hospitalmedicine.org/sparkone.
Improve your treatment of VTE during Blood Clot Awareness Month
March is Blood Clot Awareness Month, and SHM recently introduced a new toolkit and guide surrounding treatment of venous thromboembolism (VTE) in the hospital setting. SHM has a history of providing cutting-edge resources in this space, and Steven B. Deitelzweig, MD, MMM, SFHM, FACP, FACC, system chairman of hospital medicine at Oschner Health System in New Orleans, was integral in editing SHM’s VTE treatment mentored implementation guide and online toolkit.
“SHM has an established track record of implementing evidence-based and guideline-driven learnings successfully, and we continue to see improvement across multiple facilities based on this work with this disease,” Dr. Deitelzweig says. “Whenever possible, I would strongly recommend taking full advantage of SHM’s outstanding programs as they are intensely developed by experts for adoption at hospitals of different sizes, including community and academic centers.”
SHM can help you and your hospital improve treatment of VTE as well – learn how at www.hospitalmedicine.org/vtetreatment.
Share patient experience success stories
Our Patient Experience Committee wants to showcase stories of when care teams or their counterparts in the hospital made a notable shift in a patient’s experience: a special moment or interaction; a successful improvement project; an award for excellence in practice; a memo of commendation; a letter from a patient. Email examples of success to Claudia Stahl at cstahl@hospitalmedicine.org by May 11. Submissions can include photos, letters, or videos. SHM will share these moments that “made all the difference” with members on its website via other channels soon to be announced.
Brett Radler is SHM’s communications specialist.
Not a member? Know someone who should be? Visit www.joinshm.org to learn about the opportunities we can offer hospital medicine professionals.
Calling all pediatric hospitalists
Register for Pediatric Hospital Medicine 2017 (PHM17), the premier educational conference for pediatric hospitalists and other clinicians who care for hospitalized children. Re-energize your practice with the latest research, best practices, innovations, and more.
![]()
Register before June 7 to receive the early-bird rates. Visit www.peds2017.org for more information.
SHM can prepare you for MACRA
![]()
Visit www.macraforhm.org for general information and details in the MACRA FAQ and MIPS Tips links.
Don’t miss five new tracks at HM17
![]()
- Learn how to avoid diagnostic and therapeutic overuse, and how to move towards the right care for every hospital medicine patient with the High Value Care Track.
- Don’t miss the Clinical Updates Track, which provides evidence-based updates from recent literature published in medicine subspecialty fields and specific topic areas that all hospitalists need to know.
- Accurate and timely diagnosis are the two cornerstones of high-quality patient care. Find out what topics are in the Diagnostic Reasoning Track.
- Learn from experts during the Health Policy Track who will discuss the most current health care policy issues as they impact hospitalists and what we can expect from a new Presidential administration and changes in Congress.
- The Mini Medical Education Track is for hospitalists who are interested in improving their teaching skills.
Learn more about the HM17 schedule and offerings at www.hospitalmedicine2017.org/schedule.
Prepare for the entire Focused Practice in Hospital Medicine (FPHM) exam with SPARK ONE
![]()
This self-paced study guide engages learners through an open-book format, allowing users to review detailed learning objectives and discussion points and define individual areas of strengths and weaknesses. SHM members Save $150! Learn more at www.hospitalmedicine.org/sparkone.
Improve your treatment of VTE during Blood Clot Awareness Month
March is Blood Clot Awareness Month, and SHM recently introduced a new toolkit and guide surrounding treatment of venous thromboembolism (VTE) in the hospital setting. SHM has a history of providing cutting-edge resources in this space, and Steven B. Deitelzweig, MD, MMM, SFHM, FACP, FACC, system chairman of hospital medicine at Oschner Health System in New Orleans, was integral in editing SHM’s VTE treatment mentored implementation guide and online toolkit.
“SHM has an established track record of implementing evidence-based and guideline-driven learnings successfully, and we continue to see improvement across multiple facilities based on this work with this disease,” Dr. Deitelzweig says. “Whenever possible, I would strongly recommend taking full advantage of SHM’s outstanding programs as they are intensely developed by experts for adoption at hospitals of different sizes, including community and academic centers.”
SHM can help you and your hospital improve treatment of VTE as well – learn how at www.hospitalmedicine.org/vtetreatment.
Share patient experience success stories
Our Patient Experience Committee wants to showcase stories of when care teams or their counterparts in the hospital made a notable shift in a patient’s experience: a special moment or interaction; a successful improvement project; an award for excellence in practice; a memo of commendation; a letter from a patient. Email examples of success to Claudia Stahl at cstahl@hospitalmedicine.org by May 11. Submissions can include photos, letters, or videos. SHM will share these moments that “made all the difference” with members on its website via other channels soon to be announced.
Brett Radler is SHM’s communications specialist.
Not a member? Know someone who should be? Visit www.joinshm.org to learn about the opportunities we can offer hospital medicine professionals.
Are you getting the most out of your EHR?
Sparrow Health System in Lansing, Mich., went live with its electronic health record (EHR) system at its main hospital on Dec. 1, 2012. For a year and a half, the system was untapped, innovation-wise. Very few features were turned on, and it sat relatively idle with regard to quality improvement. Hospitalists and others used the EHR, but not ambitiously. Everyone, essentially, used the post-launch period to catch their breath. Some even decided it would be the perfect time to retire, rather than confront the new reality of the EHR.
“It took a good 6 months, probably longer for some, for people to feel comfortable, to start smiling again and really feel like, ‘This isn’t so bad and actually might be working for us,’ ” said Carol Nwelue, MD, medical director of Sparrow’s adult hospitalist service.

Although Sparrow is now probably ahead of the curve when it comes to maximizing its EHR use, its story carries themes that are familiar to hospitalists and to the medical field: The beginning is scary and bumpy; there typically is a long getting-used-to period; and then some hospitalists get ansty and try to get more out of the system, but only gradually – and not without pain.
The bottom line is that most hospitals have a long way to go, said Venkataraman Palabindala, MD, a hospitalist and assistant professor of medicine at the University of Mississippi Medical Center in Jackson.
“We are nowhere close to using the technology to maximum benefit,” said Dr. Palabindala, also a member of the Society of Hospital Medicine’s information technology committee.
How well hospitalists are maximizing their use of EHRs varies from center to center and doctor to doctor. But, for those that are more advanced, Dr. Palabindala and other advocates of better EHR use mention these characteristics that drive the change:
- They have hospitalist leaders with a strong interest in IT who like to tinker and refine – and then share the tricks that work with others at their center.
- They belong to EHR-related committees or work at centers with hospitalists with a big presence in those committees.
- They keep their eyes on what other centers are doing with EHRs and use those projects as models for projects at their own centers.
- They are willing to make changes in their own processes, when feasible, so that they can better dovetail with the EHR.
- They keep their lines of communication open with their EHR vendors.
- They attend user meetings to get questions answered and share information and experiences.
At Sparrow, two committees – one nurse-led and one physician-led – guide EHR enhancement. The committees are a place where, yes, doctors can vent about the EHR (the phrase they use is “pain points”), but also a place where they can get constructive feedback. The committees also keep an eye out for EHR projects elsewhere that they might be able to do themselves.
EHR: a CAUTI example
In 2014, Sparrow doctors and nurses wanted to lower their number of catheter-associated urinary tract infections (CAUTI). With the EHR that had gone live 2 years before, they had the data that they needed. They just had to figure out how to turn the data into a workable plan. Ah, if only things were so simple with EHRs. As any health center that has gone through the great transition from paper to digital can attest, having the data only puts you at the foot of the mountain.
But using a program that Texas Health System had developed as a model, Sparrow got its CAUTI program up and running. The new system included not just a placement order, but the discontinuation order, too. Advisories on best practice were built into the work flow, including alerts on when catheters had been in for 48 hours, and metrics were created to track how well the whole thing worked.

“Once the data [were] obtained and validated, it was quickly shown that more needed to be done within this clinical program to impact our CAUTI numbers,” she said. “With collaboration from end users, the system was tweaked more and BPAs (best practice advisories) were added and removed in certain areas and shifted the focus from physician-facing to nursing-facing in most areas.”
It appears to be working: CAUTI incidence at 836-bed Sparrow Hospital has dropped from a total of 52 in 2014 to 11 over the first 3 quarters of 2016.
Sparrow has also built programs to better use its EHR for sepsis, medical reconciliation, and methicillin-resistant Staphylococcus aureus screening, and one is being developed for heart failure.
Vendor engagement = QI opportunity
Sparrow and many other health systems are motivated to use more of Epic’s features and to innovate through an Epic rewards program that gives rebates for advanced use that can total hundreds of thousands of dollars. That innovation helps Epic problem solve and it can then point to that innovation in its marketing.
Almost all hospitals, and their hospitalists, are using the EHR for such basics as reducing unnecessary testing, medical reconciliation, and to document more accurately, said Eric Helsher, vice president of client success at Epic, whose job is to foster the spread of new and better ways to use the EHR. Most hospitals use the EHR, to at least some degree, for targeted quality improvement (QI) and patient safety programs, he said.
Dr. Palabindala pointed to record-sharing features as a way clinicians can share records within minutes without having to bother with faxing or emailing. Integrating smart-paging into the EHR is another way for doctors to communicate – it may not be as good as a phone call, but it’s less disruptive during a workday, he notes.
Epic is just now rolling out a secure text-messaging system hospitalists and others can use to communicate with one another – the header of the text thread clearly shows the patient it is referencing, Mr. Helsher said. Other EHR uses, such as telemedicine, are being used around the country but are far less widespread. But users are generally becoming more ambitious, he said.
“For the last 5-10 years, we’ve been in such an implementation rush,” Mr. Helsher explained. “ Now, at much more of a macro scale, the mentality has changed to ‘OK, we have these systems, let’s go from the implementation era to the value era.’ ”
Corinne Boudreau, senior marketing manager of physician experience at Meditech, said their sepsis tool has been very popular, while messaging features and shortcut commands for simpler charting are gradually coming into wider use. Meditech also expects their Web-based EHR – designed to give patients access on their mobile devices – will give doctors the mobility they want.
Still, there’s a wide range in how much hospitalists and other doctors are using even the fundamental tools that are available to them.
“I think that between implementation and maximization there is a period of adoption, and I think that that’s where a lot of folks are these days,” she said.
As “physician engagement” has become a buzzword in the industry, Meditech has worked with physician leaders on how to get doctors to absorb the message that the EHR really can help them do their jobs better.
“If you get [doctors] at the right time, you show them how it can make things easier or take time off their workload,” Ms. Boudreau said. “For some physicians that time to get them might be first thing in the morning before they see patients. Another physician might want to do it in the evening. If you hit that evening physician in the morning, you’ve missed that window of opportunity.”
Given the demands on doctors’ time and either an inability or unwillingness to put the time in that’s needed to learn about all functions the EHR can offer, there’s a growing acknowledgment that doctors often can’t simply do this on their own.
“There’s more recognition that this is a project that needs to be resourced,” Ms. Boudreau said. “They’re already strapped for time; to put something additional on top of it needs to be accommodated for. It needs to be resourced in terms of time, it needs to be resourced in terms of compensation. There need to be governance and support of that.”
Early adopters vs. late bloomers
Many hospitalists and HM groups have advanced, but some places have lagged behind, said John Nelson, MD, MHM, a veteran hospitalist, practice management consultant, and longtime columnist with The Hospitalist.
“We find it’s reasonably common to go to a place where they’re still keeping their census in an Excel spreadsheet,” he said. “Last year, we found people who do billing on paper and index cards.”
He said that often, a failure to adopt new EHR functionality isn’t because hospitals and HM groups are avoiding it. He said he sees IT shortcomings as a major blocker.
“They want to use it,” he said. “Inertia might be part of the reason people are failing to fully capture the benefit the EHR could offer, [but] the bigger reason is local IT configurations and support.”
As an example, Dr. Nelson explained that at some of the centers he has worked with the name of the attending physician is not always reflected in the EHR. That’s a big no-no, he said. The problem, he’s sometimes found, isn’t really the EHR, but quirks in the hospital system: The EHR is locked down for that information and can be changed only by a person in the admitting department.
“It would require the hospitalist to call down [to admissions] and get someone else to make that change – and that’s tedious a big headache. They give up and don’t do it anymore,” he said. “Ideally, you’d want to make it so the hospitalists can make the change themselves.”
At his center, Overlake Hospital Medical Center in Bellevue, Wash., a go-to hospitalist is David Chu, MD, who has gone through Epic training and shares tips with colleagues. He is one of a relatively few physicians there who has taken the time to use the drop-down menu feature for putting information into a chart.
That might sound like a fairly basic use for a multimillion-dollar EHR system. But it still can take hours and hours to get it right.
“The way to do it is a little bit of a programmer’s way of looking at things,” Dr. Chu said, noting it involves programming-style language with double colons, commas, and quotations marks.
“For me, I think it took a good 10, 12, 15 hours on my part to get things going,” he said. “It was a good time investment up front to help me on that end, but it’s just hard getting people to want to commit that time, especially if they’re not that savvy with computers.”
His hospitalist colleague, Ryan Chew, MD, is more advanced – he has a taxonomy-like shorthand he uses to give him the right set of basic fields for a given type of case. For someone admitted with pneumonia, he’d want to know certain things all the time. Were they short of breath? Did they have chest pain? What were their vital signs? What about inflammatory markers?
Dr. Chew can get all of those fields to pop up by typing “.rchppneumonia.” The “.” means that a special code is to follow. The “rc” is for Ryan Chew, the “hp” is for history and physical, and “pneumonia,” is the type of case. For cases that require other information to be entered, he can add that as needed.
Hospitalists might try to write shortcut phrases, but unless they have a well-defined system, it won’t be helpful over the long run, he said.
“If you don’t have a good organization system … you’ll never remember it,” Dr. Chew said.
But even he hasn’t created the drop-down menus. He said he just hasn’t been willing to take the time, especially since he feels his own way of doing things seems to be working just fine.
Effort is essential
Expanding the functionalities of the EHR takes effort, no doubt. As a result, some physicians and hospitalist groups have not been open-minded to the idea – and opportunities – of the EHR as a database.
“I think for some people, even still, working with the EHR, it’s become more something they’ve learned to get used to rather than something that they sought to take advantage of, in terms of helping things,” Dr. Chew said. “They’re still working against the EHR a little bit.”
Dr. Palabindala agreed, and said that regardless of resistance or complaint, EHRs work.
“No matter how much we argue, it is proven in multiple studies that EHRs showed increased patient safety and better documentation and better transfer of the data,” he said.
He suggests hospitalists make more of an effort.
“I strongly encourage hospitalists to be part of the every EHR-related committee, including CPOE [computerized physician order entry], analytics, and utilization-review committees,” he said. “Learning about the upgrades and learning about all the possible options, exploring clinical informatics on a regular basis is important. I also encourage [hospitalists] to participate in online, EHR-related surveys to learn more about the EHR utility and what is missing in their home institution.”
He acknowledges that it’s “hard to develop a passion.” Then he put it in terms he thought might resonate: “Think of it like a new version of smart phone. Show the enthusiasm as if you are ready for next version of iPhone or Pixel.” TH
Is hospitalists’ EHR efficiency taken advantage of?
Even though their level of EHR use can be hit or miss, hospitalists tend to be ahead of the game, many agree. But that can come with some drawbacks. They’re often the go-to people everyone else in the hospital relies on to handle the system that some think is too unwieldy to bother with.
“One thing that really distinguishes hospitalists from many other providers, particularly on the inpatient side, is just the frequency with which they use the EHR,” said Eric Helsher of Epic. Many hospitalists are chosen by administrators to test pilot projects for that reason, he adds. “They want to get it out there with a group who they know will have a lot of exposure to the system and may be more willing to make those changes for long-term gain.”
Sometimes that expertise leads to situations that go beyond the hospitalist simply being leaders of change – they’re doing work they were never really intended to do.
John Nelson, MD, MHM, a hospitalist consultant based in Seattle, said hospitalists tell him that a subspecialist might handle a case but will not want to be the attending physician specifically so they don’t have to deal with the EHR. He said the specialist in such cases will say something along the lines of, “You can call me, I’ll help you, and I’ll come by and say hello to the patient and make the care decisions, but I need you to be the attending so you can document in the chart and you can do the med rec because ‘I can’t figure out how to do those buttons right.’ ”
Some will ask hospitalists “for a hand” with a case when really all they want is for the hospitalist to enter information into the system. It’s a tricky situation for the hospitalist, Dr. Nelson said.
“Some will be transparent and say I don’t really have a medical question – I just can’t figure out how to do the med rec and the discharge, so would you do it?” he said, adding the systems issues are largely because of new rounding patterns sparked by HM’s expanding role in-hospital. “I think it meaningfully contributes to what I perceive to be a decline in hospitalist morale in the last 2 or 3 years.”
Tom Collins is a freelance writer in South Florida.
Sparrow Health System in Lansing, Mich., went live with its electronic health record (EHR) system at its main hospital on Dec. 1, 2012. For a year and a half, the system was untapped, innovation-wise. Very few features were turned on, and it sat relatively idle with regard to quality improvement. Hospitalists and others used the EHR, but not ambitiously. Everyone, essentially, used the post-launch period to catch their breath. Some even decided it would be the perfect time to retire, rather than confront the new reality of the EHR.
“It took a good 6 months, probably longer for some, for people to feel comfortable, to start smiling again and really feel like, ‘This isn’t so bad and actually might be working for us,’ ” said Carol Nwelue, MD, medical director of Sparrow’s adult hospitalist service.
Although Sparrow is now probably ahead of the curve when it comes to maximizing its EHR use, its story carries themes that are familiar to hospitalists and to the medical field: The beginning is scary and bumpy; there typically is a long getting-used-to period; and then some hospitalists get ansty and try to get more out of the system, but only gradually – and not without pain.
The bottom line is that most hospitals have a long way to go, said Venkataraman Palabindala, MD, a hospitalist and assistant professor of medicine at the University of Mississippi Medical Center in Jackson.
“We are nowhere close to using the technology to maximum benefit,” said Dr. Palabindala, also a member of the Society of Hospital Medicine’s information technology committee.
How well hospitalists are maximizing their use of EHRs varies from center to center and doctor to doctor. But, for those that are more advanced, Dr. Palabindala and other advocates of better EHR use mention these characteristics that drive the change:
- They have hospitalist leaders with a strong interest in IT who like to tinker and refine – and then share the tricks that work with others at their center.
- They belong to EHR-related committees or work at centers with hospitalists with a big presence in those committees.
- They keep their eyes on what other centers are doing with EHRs and use those projects as models for projects at their own centers.
- They are willing to make changes in their own processes, when feasible, so that they can better dovetail with the EHR.
- They keep their lines of communication open with their EHR vendors.
- They attend user meetings to get questions answered and share information and experiences.
At Sparrow, two committees – one nurse-led and one physician-led – guide EHR enhancement. The committees are a place where, yes, doctors can vent about the EHR (the phrase they use is “pain points”), but also a place where they can get constructive feedback. The committees also keep an eye out for EHR projects elsewhere that they might be able to do themselves.
EHR: a CAUTI example
In 2014, Sparrow doctors and nurses wanted to lower their number of catheter-associated urinary tract infections (CAUTI). With the EHR that had gone live 2 years before, they had the data that they needed. They just had to figure out how to turn the data into a workable plan. Ah, if only things were so simple with EHRs. As any health center that has gone through the great transition from paper to digital can attest, having the data only puts you at the foot of the mountain.
But using a program that Texas Health System had developed as a model, Sparrow got its CAUTI program up and running. The new system included not just a placement order, but the discontinuation order, too. Advisories on best practice were built into the work flow, including alerts on when catheters had been in for 48 hours, and metrics were created to track how well the whole thing worked.
“Once the data [were] obtained and validated, it was quickly shown that more needed to be done within this clinical program to impact our CAUTI numbers,” she said. “With collaboration from end users, the system was tweaked more and BPAs (best practice advisories) were added and removed in certain areas and shifted the focus from physician-facing to nursing-facing in most areas.”
It appears to be working: CAUTI incidence at 836-bed Sparrow Hospital has dropped from a total of 52 in 2014 to 11 over the first 3 quarters of 2016.
Sparrow has also built programs to better use its EHR for sepsis, medical reconciliation, and methicillin-resistant Staphylococcus aureus screening, and one is being developed for heart failure.
Vendor engagement = QI opportunity
Sparrow and many other health systems are motivated to use more of Epic’s features and to innovate through an Epic rewards program that gives rebates for advanced use that can total hundreds of thousands of dollars. That innovation helps Epic problem solve and it can then point to that innovation in its marketing.
Almost all hospitals, and their hospitalists, are using the EHR for such basics as reducing unnecessary testing, medical reconciliation, and to document more accurately, said Eric Helsher, vice president of client success at Epic, whose job is to foster the spread of new and better ways to use the EHR. Most hospitals use the EHR, to at least some degree, for targeted quality improvement (QI) and patient safety programs, he said.
Dr. Palabindala pointed to record-sharing features as a way clinicians can share records within minutes without having to bother with faxing or emailing. Integrating smart-paging into the EHR is another way for doctors to communicate – it may not be as good as a phone call, but it’s less disruptive during a workday, he notes.
Epic is just now rolling out a secure text-messaging system hospitalists and others can use to communicate with one another – the header of the text thread clearly shows the patient it is referencing, Mr. Helsher said. Other EHR uses, such as telemedicine, are being used around the country but are far less widespread. But users are generally becoming more ambitious, he said.
“For the last 5-10 years, we’ve been in such an implementation rush,” Mr. Helsher explained. “ Now, at much more of a macro scale, the mentality has changed to ‘OK, we have these systems, let’s go from the implementation era to the value era.’ ”
Corinne Boudreau, senior marketing manager of physician experience at Meditech, said their sepsis tool has been very popular, while messaging features and shortcut commands for simpler charting are gradually coming into wider use. Meditech also expects their Web-based EHR – designed to give patients access on their mobile devices – will give doctors the mobility they want.
Still, there’s a wide range in how much hospitalists and other doctors are using even the fundamental tools that are available to them.
“I think that between implementation and maximization there is a period of adoption, and I think that that’s where a lot of folks are these days,” she said.
As “physician engagement” has become a buzzword in the industry, Meditech has worked with physician leaders on how to get doctors to absorb the message that the EHR really can help them do their jobs better.
“If you get [doctors] at the right time, you show them how it can make things easier or take time off their workload,” Ms. Boudreau said. “For some physicians that time to get them might be first thing in the morning before they see patients. Another physician might want to do it in the evening. If you hit that evening physician in the morning, you’ve missed that window of opportunity.”
Given the demands on doctors’ time and either an inability or unwillingness to put the time in that’s needed to learn about all functions the EHR can offer, there’s a growing acknowledgment that doctors often can’t simply do this on their own.
“There’s more recognition that this is a project that needs to be resourced,” Ms. Boudreau said. “They’re already strapped for time; to put something additional on top of it needs to be accommodated for. It needs to be resourced in terms of time, it needs to be resourced in terms of compensation. There need to be governance and support of that.”
Early adopters vs. late bloomers
Many hospitalists and HM groups have advanced, but some places have lagged behind, said John Nelson, MD, MHM, a veteran hospitalist, practice management consultant, and longtime columnist with The Hospitalist.
“We find it’s reasonably common to go to a place where they’re still keeping their census in an Excel spreadsheet,” he said. “Last year, we found people who do billing on paper and index cards.”
He said that often, a failure to adopt new EHR functionality isn’t because hospitals and HM groups are avoiding it. He said he sees IT shortcomings as a major blocker.
“They want to use it,” he said. “Inertia might be part of the reason people are failing to fully capture the benefit the EHR could offer, [but] the bigger reason is local IT configurations and support.”
As an example, Dr. Nelson explained that at some of the centers he has worked with the name of the attending physician is not always reflected in the EHR. That’s a big no-no, he said. The problem, he’s sometimes found, isn’t really the EHR, but quirks in the hospital system: The EHR is locked down for that information and can be changed only by a person in the admitting department.
“It would require the hospitalist to call down [to admissions] and get someone else to make that change – and that’s tedious a big headache. They give up and don’t do it anymore,” he said. “Ideally, you’d want to make it so the hospitalists can make the change themselves.”
At his center, Overlake Hospital Medical Center in Bellevue, Wash., a go-to hospitalist is David Chu, MD, who has gone through Epic training and shares tips with colleagues. He is one of a relatively few physicians there who has taken the time to use the drop-down menu feature for putting information into a chart.
That might sound like a fairly basic use for a multimillion-dollar EHR system. But it still can take hours and hours to get it right.
“The way to do it is a little bit of a programmer’s way of looking at things,” Dr. Chu said, noting it involves programming-style language with double colons, commas, and quotations marks.
“For me, I think it took a good 10, 12, 15 hours on my part to get things going,” he said. “It was a good time investment up front to help me on that end, but it’s just hard getting people to want to commit that time, especially if they’re not that savvy with computers.”
His hospitalist colleague, Ryan Chew, MD, is more advanced – he has a taxonomy-like shorthand he uses to give him the right set of basic fields for a given type of case. For someone admitted with pneumonia, he’d want to know certain things all the time. Were they short of breath? Did they have chest pain? What were their vital signs? What about inflammatory markers?
Dr. Chew can get all of those fields to pop up by typing “.rchppneumonia.” The “.” means that a special code is to follow. The “rc” is for Ryan Chew, the “hp” is for history and physical, and “pneumonia,” is the type of case. For cases that require other information to be entered, he can add that as needed.
Hospitalists might try to write shortcut phrases, but unless they have a well-defined system, it won’t be helpful over the long run, he said.
“If you don’t have a good organization system … you’ll never remember it,” Dr. Chew said.
But even he hasn’t created the drop-down menus. He said he just hasn’t been willing to take the time, especially since he feels his own way of doing things seems to be working just fine.
Effort is essential
Expanding the functionalities of the EHR takes effort, no doubt. As a result, some physicians and hospitalist groups have not been open-minded to the idea – and opportunities – of the EHR as a database.
“I think for some people, even still, working with the EHR, it’s become more something they’ve learned to get used to rather than something that they sought to take advantage of, in terms of helping things,” Dr. Chew said. “They’re still working against the EHR a little bit.”
Dr. Palabindala agreed, and said that regardless of resistance or complaint, EHRs work.
“No matter how much we argue, it is proven in multiple studies that EHRs showed increased patient safety and better documentation and better transfer of the data,” he said.
He suggests hospitalists make more of an effort.
“I strongly encourage hospitalists to be part of the every EHR-related committee, including CPOE [computerized physician order entry], analytics, and utilization-review committees,” he said. “Learning about the upgrades and learning about all the possible options, exploring clinical informatics on a regular basis is important. I also encourage [hospitalists] to participate in online, EHR-related surveys to learn more about the EHR utility and what is missing in their home institution.”
He acknowledges that it’s “hard to develop a passion.” Then he put it in terms he thought might resonate: “Think of it like a new version of smart phone. Show the enthusiasm as if you are ready for next version of iPhone or Pixel.” TH
Is hospitalists’ EHR efficiency taken advantage of?
Even though their level of EHR use can be hit or miss, hospitalists tend to be ahead of the game, many agree. But that can come with some drawbacks. They’re often the go-to people everyone else in the hospital relies on to handle the system that some think is too unwieldy to bother with.
“One thing that really distinguishes hospitalists from many other providers, particularly on the inpatient side, is just the frequency with which they use the EHR,” said Eric Helsher of Epic. Many hospitalists are chosen by administrators to test pilot projects for that reason, he adds. “They want to get it out there with a group who they know will have a lot of exposure to the system and may be more willing to make those changes for long-term gain.”
Sometimes that expertise leads to situations that go beyond the hospitalist simply being leaders of change – they’re doing work they were never really intended to do.
John Nelson, MD, MHM, a hospitalist consultant based in Seattle, said hospitalists tell him that a subspecialist might handle a case but will not want to be the attending physician specifically so they don’t have to deal with the EHR. He said the specialist in such cases will say something along the lines of, “You can call me, I’ll help you, and I’ll come by and say hello to the patient and make the care decisions, but I need you to be the attending so you can document in the chart and you can do the med rec because ‘I can’t figure out how to do those buttons right.’ ”
Some will ask hospitalists “for a hand” with a case when really all they want is for the hospitalist to enter information into the system. It’s a tricky situation for the hospitalist, Dr. Nelson said.
“Some will be transparent and say I don’t really have a medical question – I just can’t figure out how to do the med rec and the discharge, so would you do it?” he said, adding the systems issues are largely because of new rounding patterns sparked by HM’s expanding role in-hospital. “I think it meaningfully contributes to what I perceive to be a decline in hospitalist morale in the last 2 or 3 years.”
Tom Collins is a freelance writer in South Florida.
Sparrow Health System in Lansing, Mich., went live with its electronic health record (EHR) system at its main hospital on Dec. 1, 2012. For a year and a half, the system was untapped, innovation-wise. Very few features were turned on, and it sat relatively idle with regard to quality improvement. Hospitalists and others used the EHR, but not ambitiously. Everyone, essentially, used the post-launch period to catch their breath. Some even decided it would be the perfect time to retire, rather than confront the new reality of the EHR.
“It took a good 6 months, probably longer for some, for people to feel comfortable, to start smiling again and really feel like, ‘This isn’t so bad and actually might be working for us,’ ” said Carol Nwelue, MD, medical director of Sparrow’s adult hospitalist service.
Although Sparrow is now probably ahead of the curve when it comes to maximizing its EHR use, its story carries themes that are familiar to hospitalists and to the medical field: The beginning is scary and bumpy; there typically is a long getting-used-to period; and then some hospitalists get ansty and try to get more out of the system, but only gradually – and not without pain.
The bottom line is that most hospitals have a long way to go, said Venkataraman Palabindala, MD, a hospitalist and assistant professor of medicine at the University of Mississippi Medical Center in Jackson.
“We are nowhere close to using the technology to maximum benefit,” said Dr. Palabindala, also a member of the Society of Hospital Medicine’s information technology committee.
How well hospitalists are maximizing their use of EHRs varies from center to center and doctor to doctor. But, for those that are more advanced, Dr. Palabindala and other advocates of better EHR use mention these characteristics that drive the change:
- They have hospitalist leaders with a strong interest in IT who like to tinker and refine – and then share the tricks that work with others at their center.
- They belong to EHR-related committees or work at centers with hospitalists with a big presence in those committees.
- They keep their eyes on what other centers are doing with EHRs and use those projects as models for projects at their own centers.
- They are willing to make changes in their own processes, when feasible, so that they can better dovetail with the EHR.
- They keep their lines of communication open with their EHR vendors.
- They attend user meetings to get questions answered and share information and experiences.
At Sparrow, two committees – one nurse-led and one physician-led – guide EHR enhancement. The committees are a place where, yes, doctors can vent about the EHR (the phrase they use is “pain points”), but also a place where they can get constructive feedback. The committees also keep an eye out for EHR projects elsewhere that they might be able to do themselves.
EHR: a CAUTI example
In 2014, Sparrow doctors and nurses wanted to lower their number of catheter-associated urinary tract infections (CAUTI). With the EHR that had gone live 2 years before, they had the data that they needed. They just had to figure out how to turn the data into a workable plan. Ah, if only things were so simple with EHRs. As any health center that has gone through the great transition from paper to digital can attest, having the data only puts you at the foot of the mountain.
But using a program that Texas Health System had developed as a model, Sparrow got its CAUTI program up and running. The new system included not just a placement order, but the discontinuation order, too. Advisories on best practice were built into the work flow, including alerts on when catheters had been in for 48 hours, and metrics were created to track how well the whole thing worked.
“Once the data [were] obtained and validated, it was quickly shown that more needed to be done within this clinical program to impact our CAUTI numbers,” she said. “With collaboration from end users, the system was tweaked more and BPAs (best practice advisories) were added and removed in certain areas and shifted the focus from physician-facing to nursing-facing in most areas.”
It appears to be working: CAUTI incidence at 836-bed Sparrow Hospital has dropped from a total of 52 in 2014 to 11 over the first 3 quarters of 2016.
Sparrow has also built programs to better use its EHR for sepsis, medical reconciliation, and methicillin-resistant Staphylococcus aureus screening, and one is being developed for heart failure.
Vendor engagement = QI opportunity
Sparrow and many other health systems are motivated to use more of Epic’s features and to innovate through an Epic rewards program that gives rebates for advanced use that can total hundreds of thousands of dollars. That innovation helps Epic problem solve and it can then point to that innovation in its marketing.
Almost all hospitals, and their hospitalists, are using the EHR for such basics as reducing unnecessary testing, medical reconciliation, and to document more accurately, said Eric Helsher, vice president of client success at Epic, whose job is to foster the spread of new and better ways to use the EHR. Most hospitals use the EHR, to at least some degree, for targeted quality improvement (QI) and patient safety programs, he said.
Dr. Palabindala pointed to record-sharing features as a way clinicians can share records within minutes without having to bother with faxing or emailing. Integrating smart-paging into the EHR is another way for doctors to communicate – it may not be as good as a phone call, but it’s less disruptive during a workday, he notes.
Epic is just now rolling out a secure text-messaging system hospitalists and others can use to communicate with one another – the header of the text thread clearly shows the patient it is referencing, Mr. Helsher said. Other EHR uses, such as telemedicine, are being used around the country but are far less widespread. But users are generally becoming more ambitious, he said.
“For the last 5-10 years, we’ve been in such an implementation rush,” Mr. Helsher explained. “ Now, at much more of a macro scale, the mentality has changed to ‘OK, we have these systems, let’s go from the implementation era to the value era.’ ”
Corinne Boudreau, senior marketing manager of physician experience at Meditech, said their sepsis tool has been very popular, while messaging features and shortcut commands for simpler charting are gradually coming into wider use. Meditech also expects their Web-based EHR – designed to give patients access on their mobile devices – will give doctors the mobility they want.
Still, there’s a wide range in how much hospitalists and other doctors are using even the fundamental tools that are available to them.
“I think that between implementation and maximization there is a period of adoption, and I think that that’s where a lot of folks are these days,” she said.
As “physician engagement” has become a buzzword in the industry, Meditech has worked with physician leaders on how to get doctors to absorb the message that the EHR really can help them do their jobs better.
“If you get [doctors] at the right time, you show them how it can make things easier or take time off their workload,” Ms. Boudreau said. “For some physicians that time to get them might be first thing in the morning before they see patients. Another physician might want to do it in the evening. If you hit that evening physician in the morning, you’ve missed that window of opportunity.”
Given the demands on doctors’ time and either an inability or unwillingness to put the time in that’s needed to learn about all functions the EHR can offer, there’s a growing acknowledgment that doctors often can’t simply do this on their own.
“There’s more recognition that this is a project that needs to be resourced,” Ms. Boudreau said. “They’re already strapped for time; to put something additional on top of it needs to be accommodated for. It needs to be resourced in terms of time, it needs to be resourced in terms of compensation. There need to be governance and support of that.”
Early adopters vs. late bloomers
Many hospitalists and HM groups have advanced, but some places have lagged behind, said John Nelson, MD, MHM, a veteran hospitalist, practice management consultant, and longtime columnist with The Hospitalist.
“We find it’s reasonably common to go to a place where they’re still keeping their census in an Excel spreadsheet,” he said. “Last year, we found people who do billing on paper and index cards.”
He said that often, a failure to adopt new EHR functionality isn’t because hospitals and HM groups are avoiding it. He said he sees IT shortcomings as a major blocker.
“They want to use it,” he said. “Inertia might be part of the reason people are failing to fully capture the benefit the EHR could offer, [but] the bigger reason is local IT configurations and support.”
As an example, Dr. Nelson explained that at some of the centers he has worked with the name of the attending physician is not always reflected in the EHR. That’s a big no-no, he said. The problem, he’s sometimes found, isn’t really the EHR, but quirks in the hospital system: The EHR is locked down for that information and can be changed only by a person in the admitting department.
“It would require the hospitalist to call down [to admissions] and get someone else to make that change – and that’s tedious a big headache. They give up and don’t do it anymore,” he said. “Ideally, you’d want to make it so the hospitalists can make the change themselves.”
At his center, Overlake Hospital Medical Center in Bellevue, Wash., a go-to hospitalist is David Chu, MD, who has gone through Epic training and shares tips with colleagues. He is one of a relatively few physicians there who has taken the time to use the drop-down menu feature for putting information into a chart.
That might sound like a fairly basic use for a multimillion-dollar EHR system. But it still can take hours and hours to get it right.
“The way to do it is a little bit of a programmer’s way of looking at things,” Dr. Chu said, noting it involves programming-style language with double colons, commas, and quotations marks.
“For me, I think it took a good 10, 12, 15 hours on my part to get things going,” he said. “It was a good time investment up front to help me on that end, but it’s just hard getting people to want to commit that time, especially if they’re not that savvy with computers.”
His hospitalist colleague, Ryan Chew, MD, is more advanced – he has a taxonomy-like shorthand he uses to give him the right set of basic fields for a given type of case. For someone admitted with pneumonia, he’d want to know certain things all the time. Were they short of breath? Did they have chest pain? What were their vital signs? What about inflammatory markers?
Dr. Chew can get all of those fields to pop up by typing “.rchppneumonia.” The “.” means that a special code is to follow. The “rc” is for Ryan Chew, the “hp” is for history and physical, and “pneumonia,” is the type of case. For cases that require other information to be entered, he can add that as needed.
Hospitalists might try to write shortcut phrases, but unless they have a well-defined system, it won’t be helpful over the long run, he said.
“If you don’t have a good organization system … you’ll never remember it,” Dr. Chew said.
But even he hasn’t created the drop-down menus. He said he just hasn’t been willing to take the time, especially since he feels his own way of doing things seems to be working just fine.
Effort is essential
Expanding the functionalities of the EHR takes effort, no doubt. As a result, some physicians and hospitalist groups have not been open-minded to the idea – and opportunities – of the EHR as a database.
“I think for some people, even still, working with the EHR, it’s become more something they’ve learned to get used to rather than something that they sought to take advantage of, in terms of helping things,” Dr. Chew said. “They’re still working against the EHR a little bit.”
Dr. Palabindala agreed, and said that regardless of resistance or complaint, EHRs work.
“No matter how much we argue, it is proven in multiple studies that EHRs showed increased patient safety and better documentation and better transfer of the data,” he said.
He suggests hospitalists make more of an effort.
“I strongly encourage hospitalists to be part of the every EHR-related committee, including CPOE [computerized physician order entry], analytics, and utilization-review committees,” he said. “Learning about the upgrades and learning about all the possible options, exploring clinical informatics on a regular basis is important. I also encourage [hospitalists] to participate in online, EHR-related surveys to learn more about the EHR utility and what is missing in their home institution.”
He acknowledges that it’s “hard to develop a passion.” Then he put it in terms he thought might resonate: “Think of it like a new version of smart phone. Show the enthusiasm as if you are ready for next version of iPhone or Pixel.” TH
Is hospitalists’ EHR efficiency taken advantage of?
Even though their level of EHR use can be hit or miss, hospitalists tend to be ahead of the game, many agree. But that can come with some drawbacks. They’re often the go-to people everyone else in the hospital relies on to handle the system that some think is too unwieldy to bother with.
“One thing that really distinguishes hospitalists from many other providers, particularly on the inpatient side, is just the frequency with which they use the EHR,” said Eric Helsher of Epic. Many hospitalists are chosen by administrators to test pilot projects for that reason, he adds. “They want to get it out there with a group who they know will have a lot of exposure to the system and may be more willing to make those changes for long-term gain.”
Sometimes that expertise leads to situations that go beyond the hospitalist simply being leaders of change – they’re doing work they were never really intended to do.
John Nelson, MD, MHM, a hospitalist consultant based in Seattle, said hospitalists tell him that a subspecialist might handle a case but will not want to be the attending physician specifically so they don’t have to deal with the EHR. He said the specialist in such cases will say something along the lines of, “You can call me, I’ll help you, and I’ll come by and say hello to the patient and make the care decisions, but I need you to be the attending so you can document in the chart and you can do the med rec because ‘I can’t figure out how to do those buttons right.’ ”
Some will ask hospitalists “for a hand” with a case when really all they want is for the hospitalist to enter information into the system. It’s a tricky situation for the hospitalist, Dr. Nelson said.
“Some will be transparent and say I don’t really have a medical question – I just can’t figure out how to do the med rec and the discharge, so would you do it?” he said, adding the systems issues are largely because of new rounding patterns sparked by HM’s expanding role in-hospital. “I think it meaningfully contributes to what I perceive to be a decline in hospitalist morale in the last 2 or 3 years.”
Tom Collins is a freelance writer in South Florida.
Recalcitrant Hyperkeratotic Plaques
The Diagnosis: Hypertrophic Lupus Erythematosus
Physical examination at initial presentation revealed well-demarcated, 2- to 3-cm plaques with scale distributed most extensively on the elbows and shins with lesser involvement of the chest and abdomen. After treatment with topical steroids, adalimumab, methotrexate, and narrowband UVB phototherapy, new annular, erythematous, and edematous lesions began to appear on the chest and abdomen (Figure 1). These new lesions appeared less hyperkeratotic than the older ones.
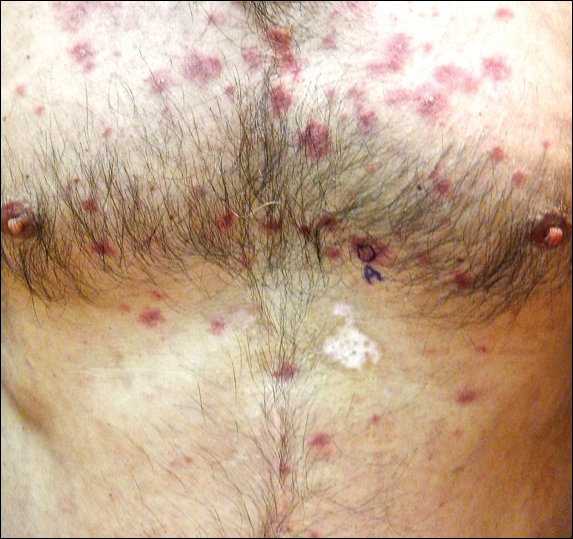
Biopsy of a hyperkeratotic lesion from the patient's arm revealed marked hyperkeratosis, parakeratosis, epidermal hyperplasia, focal vacuolar change, solar elastosis, and transepidermal elastotic elimination (Figure 2A). A second biopsy performed on a newer chest lesion revealed interface changes, degeneration of the basal layer, follicular plugging, and dermal mucin (Figure 2B). Serology revealed an antinuclear antibody (ANA) titer of 1:1280 (reference range, <1:40 dilution) and hemoglobin of 11.5 g/dL (reference range, 14.0-17.5 g/dL). On the basis of clinical, histologic, and serologic findings, hypertrophic lupus erythematosus (LE) was diagnosed. The patient was treated with oral prednisone, which resulted in rapid improvement.
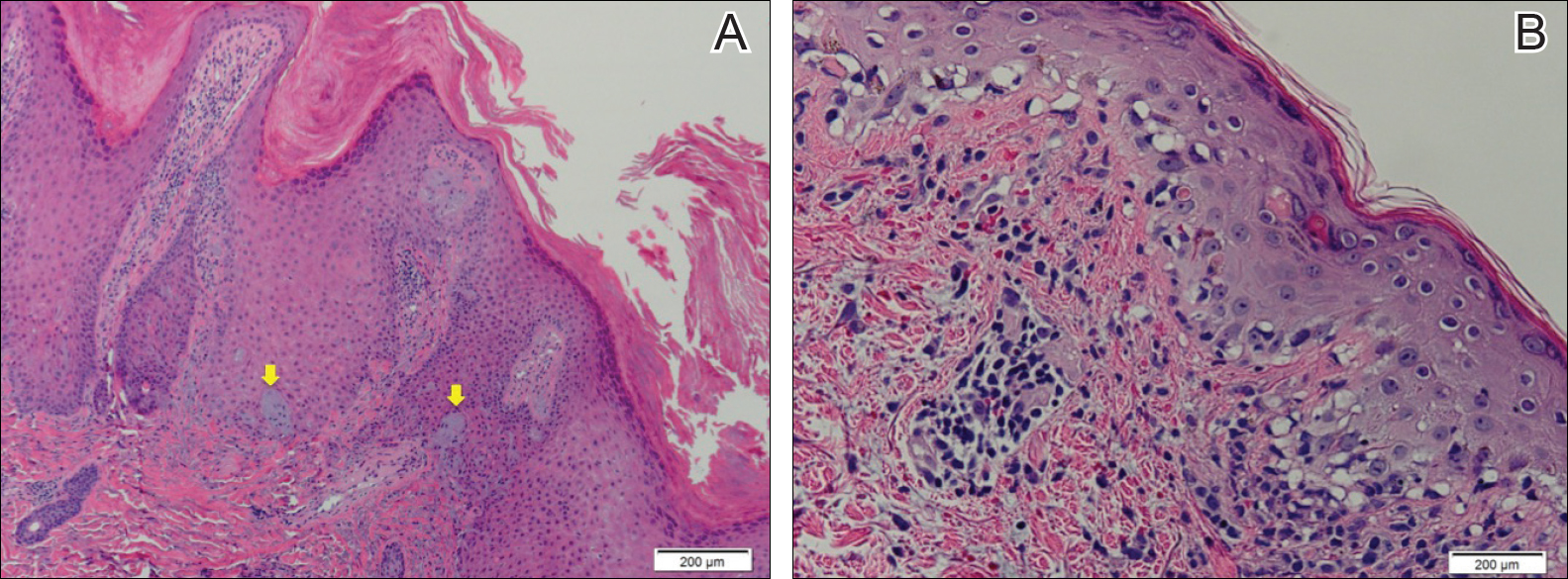
Hypertrophic LE is a rare subset of chronic cutaneous lupus first described by Behcet1 in 1942. Lesions are identified as verrucous keratotic plaques with a characteristic erythematous indurated border.2 Patients predominantly are middle-aged women with lesions distributed on sun-exposed areas. Most often, hypertrophic LE is seen in association with the classic lesions of discoid LE; however, patients may present exclusively with the cutaneous manifestations of hypertrophic LE. More rarely, as seen in this case, hypertrophic LE may present in conjunction with systemic features.3 The diagnosis of systemic LE requires 4 of the following criteria be fulfilled: malar rash; discoid rash; photosensitivity; oral ulcers; arthritis; cardiopulmonary serositis; renal involvement; positive ANA titer; and neurologic, hematologic, or immunologic disorders.4 Our patient qualified for discoid rash, photosensitivity, cardiopulmonary involvement with mitral valve defects and pulmonary pleuritis, hematologic disorder (anemia), and a positive ANA titer. Furthermore, in patients with only cutaneous discoid LE, serology generally reveals negative or low-titer ANA and negative anti-Ro antibodies.5
Hypertrophic LE is characterized histologically by irregular epidermal hyperplasia in association with features of classic cutaneous LE. Distinctive features of cutaneous LE include interface changes, follicular plugging, dermal mucin, and angiocentric lymphocytic inflammation.6 Notably, additional biopsies of the less hyperkeratotic lesions on our patient's chest and abdomen were performed, which revealed classic cutaneous LE features (Figure 2B).
Hypertrophic LE has 2 histological variants: lichen planus-like and keratoacanthoma (KA)-like patterns. Most cases are described as lichen planus-like, with a dense bandlike infiltrate in association with irregular epidermal hyperplasia, vacuolar interface changes, and reactive squamous atypia.5 In contrast, the less common KA-like lesions consist of a keratinous center with vigorous squamous epithelial proliferation.6
Clinically, hypertrophic LE may resemble hypertrophic psoriasis, lichen planus, KA, or squamous cell carcinoma (SCC). Due to the presence of pseudocarcinomatous hyperplasia, the histopathologic differential includes hypertrophic lichen planus, SCC, KA, and deep fungal infections. However, these other diseases lack the classic features of cutaneous LE, which include interface changes, follicular plugging, dermal mucin, and perivascular lymphocytic inflammation. Additionally, transepidermal elastotic elimination (Figure 2A) helps distinguish hypertrophic LE from other diagnoses.7 One of the most important tasks is distinguishing hypertrophic LE from SCC. Hypertrophic LE does not typically display eosinophil infiltrates, which differentiates it from SCC and KA. Additionally, studies report that CD123 positivity can be useful.6 Positive plasmacytoid dendritic cells are abundant at the dermoepidermal junction in hypertrophic LE, while only single or rare clusters of CD123+ cells are seen in SCC.8 Also, SCC has been found to arise in long-standing cutaneous LE lesions including both discoid and hypertrophic LE. Therefore, clinical and sometimes histological follow-up is required.
Hypertrophic LE often is challenging to treat and frequently is resistant to antimalarial drugs. The primary goals of treatment involve reducing inflammatory infiltrate and minimizing hyperkeratinization. Topical corticosteroids and calcineurin inhibitors often are inadequate as monotherapy due to reduced penetrance through the thick lesions; however, intralesional corticosteroids may be beneficial in patients with localized disease.9 Unfortunately, topical or intralesional treatments are impractical in patients with extensive lesions, as seen in our patient, in which case systemic corticosteroids can be beneficial.
Topical retinoids also have been found to be highly effective.10 Specifically, retinoids such as acitretin and isotretinoin, in some cases combined with antimalarial drugs, are effective in reducing the keratinization of these lesions. Successful treatment also has been reported with ustekinumab, thalidomide, mycophenolate mofetil, and pulsed dye laser.11 As in other types of cutaneous LE, hyperkeratotic LE is photosensitive; avoidance of prolonged sun exposure should be advised.8
- Bechet PE. Lupus erythematosus hypertrophicus et profundus. Arch Derm Syphilol. 1942;45:33-39.
- Bernardi M, Bahrami S, Callen JP. Hypertrophic lupus erythematous complicating long-standing systemic lupus erythematous. Lupus. 2011;20:549-550.
- Spann CR, Callen JP, Klein JB, et al. Clinical, serologic and immunogenetic studies in patients with chronic cutaneous (discoid) lupus erythematosus who have verrucous and/or hypertrophic skin lesions. J Rheumatol. 1988;15:256-261.
- Yu C, Gershwin E, Chang C. Diagnostic criteria for systemic lupus erythematosus: a critical review [published online January 21, 2014]. J Autoimmun. 2014;48-49:10-13.
- Provost TT. The relationship between discoid and systemic lupus erythematous. Arch Dermatol. 1994;130:1308-1310.
- Arps DP, Patel RM. Cutaneous hypertrophic lupus erythematous: a challenging histopathologic diagnosis in the absence of clinical information. Arch Pathol Lab Med. 2013;137:1205-1210.
- Daldon PE, De Souza EM, Cintra ML. Hypertrophic lupus erythematous: a clinicopathological study of 14 cases. J Cutan Pathol. 2003;30:443-448.
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophiclupus erythematous: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. issues in diagnosis and treatment. Am J Clin Dermatol. 2009;10:366-381.
- Al-Mutairi N, Rijhwani M, Nour-Eldin O. Hypertrophic lupus erythematosus treated successfully with acitretin as monotherapy. J Dermatol. 2005;32:482-486.
- Winchester D, Duffin KC, Hansen C. Response to ustekinumab in a patient with both severe psoriasis and hypertrophic cutaneous lupus. Lupus. 2012;12:1007-1010.
The Diagnosis: Hypertrophic Lupus Erythematosus
Physical examination at initial presentation revealed well-demarcated, 2- to 3-cm plaques with scale distributed most extensively on the elbows and shins with lesser involvement of the chest and abdomen. After treatment with topical steroids, adalimumab, methotrexate, and narrowband UVB phototherapy, new annular, erythematous, and edematous lesions began to appear on the chest and abdomen (Figure 1). These new lesions appeared less hyperkeratotic than the older ones.
Biopsy of a hyperkeratotic lesion from the patient's arm revealed marked hyperkeratosis, parakeratosis, epidermal hyperplasia, focal vacuolar change, solar elastosis, and transepidermal elastotic elimination (Figure 2A). A second biopsy performed on a newer chest lesion revealed interface changes, degeneration of the basal layer, follicular plugging, and dermal mucin (Figure 2B). Serology revealed an antinuclear antibody (ANA) titer of 1:1280 (reference range, <1:40 dilution) and hemoglobin of 11.5 g/dL (reference range, 14.0-17.5 g/dL). On the basis of clinical, histologic, and serologic findings, hypertrophic lupus erythematosus (LE) was diagnosed. The patient was treated with oral prednisone, which resulted in rapid improvement.
Hypertrophic LE is a rare subset of chronic cutaneous lupus first described by Behcet1 in 1942. Lesions are identified as verrucous keratotic plaques with a characteristic erythematous indurated border.2 Patients predominantly are middle-aged women with lesions distributed on sun-exposed areas. Most often, hypertrophic LE is seen in association with the classic lesions of discoid LE; however, patients may present exclusively with the cutaneous manifestations of hypertrophic LE. More rarely, as seen in this case, hypertrophic LE may present in conjunction with systemic features.3 The diagnosis of systemic LE requires 4 of the following criteria be fulfilled: malar rash; discoid rash; photosensitivity; oral ulcers; arthritis; cardiopulmonary serositis; renal involvement; positive ANA titer; and neurologic, hematologic, or immunologic disorders.4 Our patient qualified for discoid rash, photosensitivity, cardiopulmonary involvement with mitral valve defects and pulmonary pleuritis, hematologic disorder (anemia), and a positive ANA titer. Furthermore, in patients with only cutaneous discoid LE, serology generally reveals negative or low-titer ANA and negative anti-Ro antibodies.5
Hypertrophic LE is characterized histologically by irregular epidermal hyperplasia in association with features of classic cutaneous LE. Distinctive features of cutaneous LE include interface changes, follicular plugging, dermal mucin, and angiocentric lymphocytic inflammation.6 Notably, additional biopsies of the less hyperkeratotic lesions on our patient's chest and abdomen were performed, which revealed classic cutaneous LE features (Figure 2B).
Hypertrophic LE has 2 histological variants: lichen planus-like and keratoacanthoma (KA)-like patterns. Most cases are described as lichen planus-like, with a dense bandlike infiltrate in association with irregular epidermal hyperplasia, vacuolar interface changes, and reactive squamous atypia.5 In contrast, the less common KA-like lesions consist of a keratinous center with vigorous squamous epithelial proliferation.6
Clinically, hypertrophic LE may resemble hypertrophic psoriasis, lichen planus, KA, or squamous cell carcinoma (SCC). Due to the presence of pseudocarcinomatous hyperplasia, the histopathologic differential includes hypertrophic lichen planus, SCC, KA, and deep fungal infections. However, these other diseases lack the classic features of cutaneous LE, which include interface changes, follicular plugging, dermal mucin, and perivascular lymphocytic inflammation. Additionally, transepidermal elastotic elimination (Figure 2A) helps distinguish hypertrophic LE from other diagnoses.7 One of the most important tasks is distinguishing hypertrophic LE from SCC. Hypertrophic LE does not typically display eosinophil infiltrates, which differentiates it from SCC and KA. Additionally, studies report that CD123 positivity can be useful.6 Positive plasmacytoid dendritic cells are abundant at the dermoepidermal junction in hypertrophic LE, while only single or rare clusters of CD123+ cells are seen in SCC.8 Also, SCC has been found to arise in long-standing cutaneous LE lesions including both discoid and hypertrophic LE. Therefore, clinical and sometimes histological follow-up is required.
Hypertrophic LE often is challenging to treat and frequently is resistant to antimalarial drugs. The primary goals of treatment involve reducing inflammatory infiltrate and minimizing hyperkeratinization. Topical corticosteroids and calcineurin inhibitors often are inadequate as monotherapy due to reduced penetrance through the thick lesions; however, intralesional corticosteroids may be beneficial in patients with localized disease.9 Unfortunately, topical or intralesional treatments are impractical in patients with extensive lesions, as seen in our patient, in which case systemic corticosteroids can be beneficial.
Topical retinoids also have been found to be highly effective.10 Specifically, retinoids such as acitretin and isotretinoin, in some cases combined with antimalarial drugs, are effective in reducing the keratinization of these lesions. Successful treatment also has been reported with ustekinumab, thalidomide, mycophenolate mofetil, and pulsed dye laser.11 As in other types of cutaneous LE, hyperkeratotic LE is photosensitive; avoidance of prolonged sun exposure should be advised.8
The Diagnosis: Hypertrophic Lupus Erythematosus
Physical examination at initial presentation revealed well-demarcated, 2- to 3-cm plaques with scale distributed most extensively on the elbows and shins with lesser involvement of the chest and abdomen. After treatment with topical steroids, adalimumab, methotrexate, and narrowband UVB phototherapy, new annular, erythematous, and edematous lesions began to appear on the chest and abdomen (Figure 1). These new lesions appeared less hyperkeratotic than the older ones.
Biopsy of a hyperkeratotic lesion from the patient's arm revealed marked hyperkeratosis, parakeratosis, epidermal hyperplasia, focal vacuolar change, solar elastosis, and transepidermal elastotic elimination (Figure 2A). A second biopsy performed on a newer chest lesion revealed interface changes, degeneration of the basal layer, follicular plugging, and dermal mucin (Figure 2B). Serology revealed an antinuclear antibody (ANA) titer of 1:1280 (reference range, <1:40 dilution) and hemoglobin of 11.5 g/dL (reference range, 14.0-17.5 g/dL). On the basis of clinical, histologic, and serologic findings, hypertrophic lupus erythematosus (LE) was diagnosed. The patient was treated with oral prednisone, which resulted in rapid improvement.
Hypertrophic LE is a rare subset of chronic cutaneous lupus first described by Behcet1 in 1942. Lesions are identified as verrucous keratotic plaques with a characteristic erythematous indurated border.2 Patients predominantly are middle-aged women with lesions distributed on sun-exposed areas. Most often, hypertrophic LE is seen in association with the classic lesions of discoid LE; however, patients may present exclusively with the cutaneous manifestations of hypertrophic LE. More rarely, as seen in this case, hypertrophic LE may present in conjunction with systemic features.3 The diagnosis of systemic LE requires 4 of the following criteria be fulfilled: malar rash; discoid rash; photosensitivity; oral ulcers; arthritis; cardiopulmonary serositis; renal involvement; positive ANA titer; and neurologic, hematologic, or immunologic disorders.4 Our patient qualified for discoid rash, photosensitivity, cardiopulmonary involvement with mitral valve defects and pulmonary pleuritis, hematologic disorder (anemia), and a positive ANA titer. Furthermore, in patients with only cutaneous discoid LE, serology generally reveals negative or low-titer ANA and negative anti-Ro antibodies.5
Hypertrophic LE is characterized histologically by irregular epidermal hyperplasia in association with features of classic cutaneous LE. Distinctive features of cutaneous LE include interface changes, follicular plugging, dermal mucin, and angiocentric lymphocytic inflammation.6 Notably, additional biopsies of the less hyperkeratotic lesions on our patient's chest and abdomen were performed, which revealed classic cutaneous LE features (Figure 2B).
Hypertrophic LE has 2 histological variants: lichen planus-like and keratoacanthoma (KA)-like patterns. Most cases are described as lichen planus-like, with a dense bandlike infiltrate in association with irregular epidermal hyperplasia, vacuolar interface changes, and reactive squamous atypia.5 In contrast, the less common KA-like lesions consist of a keratinous center with vigorous squamous epithelial proliferation.6
Clinically, hypertrophic LE may resemble hypertrophic psoriasis, lichen planus, KA, or squamous cell carcinoma (SCC). Due to the presence of pseudocarcinomatous hyperplasia, the histopathologic differential includes hypertrophic lichen planus, SCC, KA, and deep fungal infections. However, these other diseases lack the classic features of cutaneous LE, which include interface changes, follicular plugging, dermal mucin, and perivascular lymphocytic inflammation. Additionally, transepidermal elastotic elimination (Figure 2A) helps distinguish hypertrophic LE from other diagnoses.7 One of the most important tasks is distinguishing hypertrophic LE from SCC. Hypertrophic LE does not typically display eosinophil infiltrates, which differentiates it from SCC and KA. Additionally, studies report that CD123 positivity can be useful.6 Positive plasmacytoid dendritic cells are abundant at the dermoepidermal junction in hypertrophic LE, while only single or rare clusters of CD123+ cells are seen in SCC.8 Also, SCC has been found to arise in long-standing cutaneous LE lesions including both discoid and hypertrophic LE. Therefore, clinical and sometimes histological follow-up is required.
Hypertrophic LE often is challenging to treat and frequently is resistant to antimalarial drugs. The primary goals of treatment involve reducing inflammatory infiltrate and minimizing hyperkeratinization. Topical corticosteroids and calcineurin inhibitors often are inadequate as monotherapy due to reduced penetrance through the thick lesions; however, intralesional corticosteroids may be beneficial in patients with localized disease.9 Unfortunately, topical or intralesional treatments are impractical in patients with extensive lesions, as seen in our patient, in which case systemic corticosteroids can be beneficial.
Topical retinoids also have been found to be highly effective.10 Specifically, retinoids such as acitretin and isotretinoin, in some cases combined with antimalarial drugs, are effective in reducing the keratinization of these lesions. Successful treatment also has been reported with ustekinumab, thalidomide, mycophenolate mofetil, and pulsed dye laser.11 As in other types of cutaneous LE, hyperkeratotic LE is photosensitive; avoidance of prolonged sun exposure should be advised.8
- Bechet PE. Lupus erythematosus hypertrophicus et profundus. Arch Derm Syphilol. 1942;45:33-39.
- Bernardi M, Bahrami S, Callen JP. Hypertrophic lupus erythematous complicating long-standing systemic lupus erythematous. Lupus. 2011;20:549-550.
- Spann CR, Callen JP, Klein JB, et al. Clinical, serologic and immunogenetic studies in patients with chronic cutaneous (discoid) lupus erythematosus who have verrucous and/or hypertrophic skin lesions. J Rheumatol. 1988;15:256-261.
- Yu C, Gershwin E, Chang C. Diagnostic criteria for systemic lupus erythematosus: a critical review [published online January 21, 2014]. J Autoimmun. 2014;48-49:10-13.
- Provost TT. The relationship between discoid and systemic lupus erythematous. Arch Dermatol. 1994;130:1308-1310.
- Arps DP, Patel RM. Cutaneous hypertrophic lupus erythematous: a challenging histopathologic diagnosis in the absence of clinical information. Arch Pathol Lab Med. 2013;137:1205-1210.
- Daldon PE, De Souza EM, Cintra ML. Hypertrophic lupus erythematous: a clinicopathological study of 14 cases. J Cutan Pathol. 2003;30:443-448.
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophiclupus erythematous: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. issues in diagnosis and treatment. Am J Clin Dermatol. 2009;10:366-381.
- Al-Mutairi N, Rijhwani M, Nour-Eldin O. Hypertrophic lupus erythematosus treated successfully with acitretin as monotherapy. J Dermatol. 2005;32:482-486.
- Winchester D, Duffin KC, Hansen C. Response to ustekinumab in a patient with both severe psoriasis and hypertrophic cutaneous lupus. Lupus. 2012;12:1007-1010.
- Bechet PE. Lupus erythematosus hypertrophicus et profundus. Arch Derm Syphilol. 1942;45:33-39.
- Bernardi M, Bahrami S, Callen JP. Hypertrophic lupus erythematous complicating long-standing systemic lupus erythematous. Lupus. 2011;20:549-550.
- Spann CR, Callen JP, Klein JB, et al. Clinical, serologic and immunogenetic studies in patients with chronic cutaneous (discoid) lupus erythematosus who have verrucous and/or hypertrophic skin lesions. J Rheumatol. 1988;15:256-261.
- Yu C, Gershwin E, Chang C. Diagnostic criteria for systemic lupus erythematosus: a critical review [published online January 21, 2014]. J Autoimmun. 2014;48-49:10-13.
- Provost TT. The relationship between discoid and systemic lupus erythematous. Arch Dermatol. 1994;130:1308-1310.
- Arps DP, Patel RM. Cutaneous hypertrophic lupus erythematous: a challenging histopathologic diagnosis in the absence of clinical information. Arch Pathol Lab Med. 2013;137:1205-1210.
- Daldon PE, De Souza EM, Cintra ML. Hypertrophic lupus erythematous: a clinicopathological study of 14 cases. J Cutan Pathol. 2003;30:443-448.
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophiclupus erythematous: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. issues in diagnosis and treatment. Am J Clin Dermatol. 2009;10:366-381.
- Al-Mutairi N, Rijhwani M, Nour-Eldin O. Hypertrophic lupus erythematosus treated successfully with acitretin as monotherapy. J Dermatol. 2005;32:482-486.
- Winchester D, Duffin KC, Hansen C. Response to ustekinumab in a patient with both severe psoriasis and hypertrophic cutaneous lupus. Lupus. 2012;12:1007-1010.
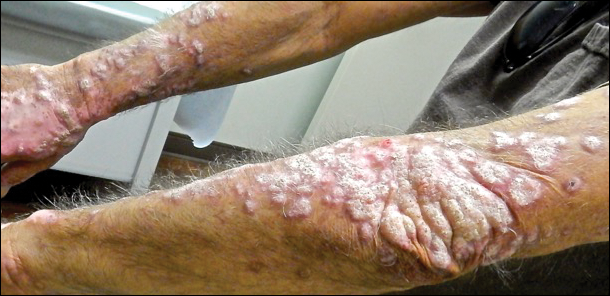
A 53-year-old man presented with a persistent, hyperkeratotic, pruritic rash on the arms, chest, and abdomen. The patient was treated for presumed psoriasis for 9 months by a primary care physician. However, despite an extensive treatment history, which included topical steroids, adalimumab, methotrexate, and narrowband UVB phototherapy, his condition worsened, and new erythematous and edematous lesions with no scale appeared on the back and chest. The patient's history also was notable for splenic rupture and mitral valve defects for which he was maintained on warfarin. In addition, he was evaluated by an allergist for new-onset dyspnea and treated with prednisone, which subsequently resulted in partial resolution of the skin lesions.
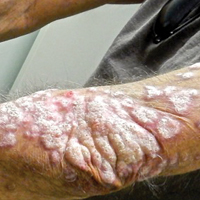
Biomarker score not predictive of successful TNF inhibitor tapering in RA
A multi-biomarker disease activity score in patients with longstanding rheumatoid arthritis and low disease activity prior to tapering adalimumab or etanercept did not predict flare-related outcomes in an 18-month, open-label, randomized clinical trial.
These findings seemed “robust and valid, at least in this specific context,” said investigators led by Chantal A. M. Bouman, MD, of Sint Maartenskliniek Nijmegen (the Netherlands), because multiple other outcomes measured in the trial, including discontinuation of biologic and radiographic progression, were not predicted by the multi-biomarker disease activity (MBDA) score, which is derived from an algorithm using a biomarker panel of 12 serum proteins and is marketed under the name Vectra DA.
The researchers sought to determine if measurement of disease activity using biomarkers with the MBDA score has a potential for smaller measurement error, compared with clinical decision making, in predicting the effects of dose tapering of biologic disease-modifying antirheumatic drugs by examining blood samples taken from 171 people with longstanding RA with low disease activity who were participating in the Dutch Dose Reduction Strategies of Subcutaneous TNF inhibitors (DRESS) trial (Rheumatology [Oxford]. 2017 Feb 22. doi: 10.1093/rheumatology/kex003).
The use of biomarkers could potentially overcome some of the drawbacks of the most widely used and extensively validated measure of RA disease activity, the Disease Activity Score in 28 joints (DAS28), which relies on “clinical assessments [that] are subject to interobserver variability, resulting in measurement error and suboptimal precision,” the investigators wrote. “Also, DAS28 can be influenced by factors other than RA disease activity (e.g., OA, [fibromyalgia], or other causes of inflammation, such an infection), resulting in clinical misclassification of disease activity state.”
The randomized DRESS trial investigated the noninferiority of a dose reduction strategy of adalimumab or etanercept (n = 115), compared with usual care (n = 56), on the rate of flare, defined as a DAS28 (using C-reactive protein) increase of more than 1.2 or 0.6 if the current DAS was more than or equal to 3.2. A major flare was one lasting more than 3 months despite treatment intervention.
The baseline MBDA score did not predict successful tapering based on an area under the receiver operating characteristic (AUROC) of 0.53 (95% confidence interval, 0.41-0.66), nor did it predict the discontinuation of either biologic (AUROC = 0.51; 95% CI, 0.36-0.66) or the occurrence of flare (AUROC = 0.50; 95% CI, 0.41-0.59 for both groups combined) or major flare (AUROC = 0.46; 95% CI, 0.32-0.65 for both groups combined).
Although the authors found a borderline positive predictive value of baseline MBDA score for major flare in the usual care group (AUROC = 0.72; 95% CI, 0.56-0.88), they said the finding should be “interpreted cautiously” as multiple testing may have resulted in false-positive findings.
The researchers also discovered that, in contrast to findings from five studies in four cohorts of patients with established or early RA, the MBDA did not predict radiographic progression (AUROC = 0.53 for predicting radiographic progression of more than 0.5 Sharp–van der Heijde points; 95% CI, 0.43-0.63 for both groups combined).
The inability of the MBDA score to predict radiographic outcomes “might be attributable to the low frequency and severity of radiographic progression in our study, with only a small difference in favor of the [usual care] group. It might reflect the strict tight control that was applied to patients who were already in low disease activity or remission,” the investigators suggested.
They also said that, in spite of the findings, the MBDA score may have predictive value in other groups of patients, such as those with early rheumatoid arthritis, higher disease activity, or suboptimal disease control, and suggested that the study’s findings be validated in further studies.
The study received no specific funding. However, one author is an employee of Crescendo Bioscience and reported receiving stock grants from its parent company, Myriad Genetics. Several authors reported relationships with industry.
A multi-biomarker disease activity score in patients with longstanding rheumatoid arthritis and low disease activity prior to tapering adalimumab or etanercept did not predict flare-related outcomes in an 18-month, open-label, randomized clinical trial.
These findings seemed “robust and valid, at least in this specific context,” said investigators led by Chantal A. M. Bouman, MD, of Sint Maartenskliniek Nijmegen (the Netherlands), because multiple other outcomes measured in the trial, including discontinuation of biologic and radiographic progression, were not predicted by the multi-biomarker disease activity (MBDA) score, which is derived from an algorithm using a biomarker panel of 12 serum proteins and is marketed under the name Vectra DA.
The researchers sought to determine if measurement of disease activity using biomarkers with the MBDA score has a potential for smaller measurement error, compared with clinical decision making, in predicting the effects of dose tapering of biologic disease-modifying antirheumatic drugs by examining blood samples taken from 171 people with longstanding RA with low disease activity who were participating in the Dutch Dose Reduction Strategies of Subcutaneous TNF inhibitors (DRESS) trial (Rheumatology [Oxford]. 2017 Feb 22. doi: 10.1093/rheumatology/kex003).
The use of biomarkers could potentially overcome some of the drawbacks of the most widely used and extensively validated measure of RA disease activity, the Disease Activity Score in 28 joints (DAS28), which relies on “clinical assessments [that] are subject to interobserver variability, resulting in measurement error and suboptimal precision,” the investigators wrote. “Also, DAS28 can be influenced by factors other than RA disease activity (e.g., OA, [fibromyalgia], or other causes of inflammation, such an infection), resulting in clinical misclassification of disease activity state.”
The randomized DRESS trial investigated the noninferiority of a dose reduction strategy of adalimumab or etanercept (n = 115), compared with usual care (n = 56), on the rate of flare, defined as a DAS28 (using C-reactive protein) increase of more than 1.2 or 0.6 if the current DAS was more than or equal to 3.2. A major flare was one lasting more than 3 months despite treatment intervention.
The baseline MBDA score did not predict successful tapering based on an area under the receiver operating characteristic (AUROC) of 0.53 (95% confidence interval, 0.41-0.66), nor did it predict the discontinuation of either biologic (AUROC = 0.51; 95% CI, 0.36-0.66) or the occurrence of flare (AUROC = 0.50; 95% CI, 0.41-0.59 for both groups combined) or major flare (AUROC = 0.46; 95% CI, 0.32-0.65 for both groups combined).
Although the authors found a borderline positive predictive value of baseline MBDA score for major flare in the usual care group (AUROC = 0.72; 95% CI, 0.56-0.88), they said the finding should be “interpreted cautiously” as multiple testing may have resulted in false-positive findings.
The researchers also discovered that, in contrast to findings from five studies in four cohorts of patients with established or early RA, the MBDA did not predict radiographic progression (AUROC = 0.53 for predicting radiographic progression of more than 0.5 Sharp–van der Heijde points; 95% CI, 0.43-0.63 for both groups combined).
The inability of the MBDA score to predict radiographic outcomes “might be attributable to the low frequency and severity of radiographic progression in our study, with only a small difference in favor of the [usual care] group. It might reflect the strict tight control that was applied to patients who were already in low disease activity or remission,” the investigators suggested.
They also said that, in spite of the findings, the MBDA score may have predictive value in other groups of patients, such as those with early rheumatoid arthritis, higher disease activity, or suboptimal disease control, and suggested that the study’s findings be validated in further studies.
The study received no specific funding. However, one author is an employee of Crescendo Bioscience and reported receiving stock grants from its parent company, Myriad Genetics. Several authors reported relationships with industry.
A multi-biomarker disease activity score in patients with longstanding rheumatoid arthritis and low disease activity prior to tapering adalimumab or etanercept did not predict flare-related outcomes in an 18-month, open-label, randomized clinical trial.
These findings seemed “robust and valid, at least in this specific context,” said investigators led by Chantal A. M. Bouman, MD, of Sint Maartenskliniek Nijmegen (the Netherlands), because multiple other outcomes measured in the trial, including discontinuation of biologic and radiographic progression, were not predicted by the multi-biomarker disease activity (MBDA) score, which is derived from an algorithm using a biomarker panel of 12 serum proteins and is marketed under the name Vectra DA.
The researchers sought to determine if measurement of disease activity using biomarkers with the MBDA score has a potential for smaller measurement error, compared with clinical decision making, in predicting the effects of dose tapering of biologic disease-modifying antirheumatic drugs by examining blood samples taken from 171 people with longstanding RA with low disease activity who were participating in the Dutch Dose Reduction Strategies of Subcutaneous TNF inhibitors (DRESS) trial (Rheumatology [Oxford]. 2017 Feb 22. doi: 10.1093/rheumatology/kex003).
The use of biomarkers could potentially overcome some of the drawbacks of the most widely used and extensively validated measure of RA disease activity, the Disease Activity Score in 28 joints (DAS28), which relies on “clinical assessments [that] are subject to interobserver variability, resulting in measurement error and suboptimal precision,” the investigators wrote. “Also, DAS28 can be influenced by factors other than RA disease activity (e.g., OA, [fibromyalgia], or other causes of inflammation, such an infection), resulting in clinical misclassification of disease activity state.”
The randomized DRESS trial investigated the noninferiority of a dose reduction strategy of adalimumab or etanercept (n = 115), compared with usual care (n = 56), on the rate of flare, defined as a DAS28 (using C-reactive protein) increase of more than 1.2 or 0.6 if the current DAS was more than or equal to 3.2. A major flare was one lasting more than 3 months despite treatment intervention.
The baseline MBDA score did not predict successful tapering based on an area under the receiver operating characteristic (AUROC) of 0.53 (95% confidence interval, 0.41-0.66), nor did it predict the discontinuation of either biologic (AUROC = 0.51; 95% CI, 0.36-0.66) or the occurrence of flare (AUROC = 0.50; 95% CI, 0.41-0.59 for both groups combined) or major flare (AUROC = 0.46; 95% CI, 0.32-0.65 for both groups combined).
Although the authors found a borderline positive predictive value of baseline MBDA score for major flare in the usual care group (AUROC = 0.72; 95% CI, 0.56-0.88), they said the finding should be “interpreted cautiously” as multiple testing may have resulted in false-positive findings.
The researchers also discovered that, in contrast to findings from five studies in four cohorts of patients with established or early RA, the MBDA did not predict radiographic progression (AUROC = 0.53 for predicting radiographic progression of more than 0.5 Sharp–van der Heijde points; 95% CI, 0.43-0.63 for both groups combined).
The inability of the MBDA score to predict radiographic outcomes “might be attributable to the low frequency and severity of radiographic progression in our study, with only a small difference in favor of the [usual care] group. It might reflect the strict tight control that was applied to patients who were already in low disease activity or remission,” the investigators suggested.
They also said that, in spite of the findings, the MBDA score may have predictive value in other groups of patients, such as those with early rheumatoid arthritis, higher disease activity, or suboptimal disease control, and suggested that the study’s findings be validated in further studies.
The study received no specific funding. However, one author is an employee of Crescendo Bioscience and reported receiving stock grants from its parent company, Myriad Genetics. Several authors reported relationships with industry.
FROM RHEUMATOLOGY
Key clinical point:
Main finding: The baseline MBDA score did not predict successful tapering based on an area under the receiver operating characteristic of 0.53 (95% CI, 0.41-0.66).
Data source: Analysis of serum samples from 171 RA patients taking part in the noninferiority, randomized, open-label, controlled Dose Reduction Strategies of Subcutaneous TNF inhibitors (DRESS) trial.
Disclosures: The study received no specific funding. However, one author is an employee of Crescendo Bioscience and reported receiving stock grants from its parent company, Myriad Genetics. Several authors reported relationships with industry.
VAM Registration, Housing Now Open
Registration and housing for the 2017 Vascular Annual Meeting are now open. VAM will be held May 31-June 3 in San Diego, with plenaries and exhibits open June 1-3. Register here and make housing reservations here. Already, more than 150 people have registered; look who’s coming here.
Registration and housing for the 2017 Vascular Annual Meeting are now open. VAM will be held May 31-June 3 in San Diego, with plenaries and exhibits open June 1-3. Register here and make housing reservations here. Already, more than 150 people have registered; look who’s coming here.
Registration and housing for the 2017 Vascular Annual Meeting are now open. VAM will be held May 31-June 3 in San Diego, with plenaries and exhibits open June 1-3. Register here and make housing reservations here. Already, more than 150 people have registered; look who’s coming here.

Plan to attend CHEST 2017 in Toronto
Oct 28 – Nov 1
Toronto, Ontario, Canada
Join us in wonderful Toronto for CHEST 2017, where we’ll connect a global community in clinical chest medicine. Our program will deliver current pulmonary, critical care, and sleep medicine topics presented by world-renowned faculty in a variety of innovative instruction formats. Take advantage of these opportunities to get involved now:
Submit Abstracts and Case Reports
Submission deadline: March 31![]()
- Fellow Case Reports.
- Medical Student/Resident Case Reports.
- Global Case Reports.
- Clinical Case Puzzlers.
Learn more and submit at chest2017.abstractcentral.com.
Apply for 2017 CHEST Foundation Grants
Application deadline: March 31
The CHEST Foundation has started accepting applications for its clinical research, distinguished scholar, and community service grants. Every year, the CHEST Foundation awards more than a half-million dollars to the next generation of lung health champions.
The grants available are:
- GlaxoSmithKline Distinguished Scholar Research Grant in Respiratory Health: $150,000 over 3 years
- CHEST Foundation Research Grant in Lung Cancer: $50,000-$100,000* over 2 years
- CHEST Foundation Research Grant in Pulmonary Arterial Hypertension: $25,000 1-year grant
- CHEST Foundation and Alpha-1 Foundation Research Grant in Alpha-1 Antitrypsin Deficiency: $25,000 1-year grant
- CHEST Foundation Research Grant in Nontuberculous Mycobacteria: $10,000-$30,000* 1-year grant
- CHEST Foundation Research Grant in Venous Thromboembolism: $30,000 1-year grant
- CHEST Foundation Research Grant in Pulmonary Fibrosis: $30,000 1-year grant
- CHEST Foundation Research Grant in Chronic Obstructive Pulmonary Disease: $50,000 1-year grant
- CHEST Foundation Research Grant in Women’s Lung Health: $10,000 1-year grant
- CHEST Foundation Research Grant in Asthma: $15,000 - $30,000* 1-year grant
- CHEST Foundation Research Grant in Cystic Fibrosis: $30,000 1-year grant
- Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP: multiple awards up to $15,000 per 1-year grant
*Amount contingent on funding.Apply for grants at chestfoundation.org/grants.
Oct 28 – Nov 1
Toronto, Ontario, Canada
Join us in wonderful Toronto for CHEST 2017, where we’ll connect a global community in clinical chest medicine. Our program will deliver current pulmonary, critical care, and sleep medicine topics presented by world-renowned faculty in a variety of innovative instruction formats. Take advantage of these opportunities to get involved now:
Submit Abstracts and Case Reports
Submission deadline: March 31![]()
- Fellow Case Reports.
- Medical Student/Resident Case Reports.
- Global Case Reports.
- Clinical Case Puzzlers.
Learn more and submit at chest2017.abstractcentral.com.
Apply for 2017 CHEST Foundation Grants
Application deadline: March 31
The CHEST Foundation has started accepting applications for its clinical research, distinguished scholar, and community service grants. Every year, the CHEST Foundation awards more than a half-million dollars to the next generation of lung health champions.
The grants available are:
- GlaxoSmithKline Distinguished Scholar Research Grant in Respiratory Health: $150,000 over 3 years
- CHEST Foundation Research Grant in Lung Cancer: $50,000-$100,000* over 2 years
- CHEST Foundation Research Grant in Pulmonary Arterial Hypertension: $25,000 1-year grant
- CHEST Foundation and Alpha-1 Foundation Research Grant in Alpha-1 Antitrypsin Deficiency: $25,000 1-year grant
- CHEST Foundation Research Grant in Nontuberculous Mycobacteria: $10,000-$30,000* 1-year grant
- CHEST Foundation Research Grant in Venous Thromboembolism: $30,000 1-year grant
- CHEST Foundation Research Grant in Pulmonary Fibrosis: $30,000 1-year grant
- CHEST Foundation Research Grant in Chronic Obstructive Pulmonary Disease: $50,000 1-year grant
- CHEST Foundation Research Grant in Women’s Lung Health: $10,000 1-year grant
- CHEST Foundation Research Grant in Asthma: $15,000 - $30,000* 1-year grant
- CHEST Foundation Research Grant in Cystic Fibrosis: $30,000 1-year grant
- Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP: multiple awards up to $15,000 per 1-year grant
*Amount contingent on funding.Apply for grants at chestfoundation.org/grants.
Oct 28 – Nov 1
Toronto, Ontario, Canada
Join us in wonderful Toronto for CHEST 2017, where we’ll connect a global community in clinical chest medicine. Our program will deliver current pulmonary, critical care, and sleep medicine topics presented by world-renowned faculty in a variety of innovative instruction formats. Take advantage of these opportunities to get involved now:
Submit Abstracts and Case Reports
Submission deadline: March 31![]()
- Fellow Case Reports.
- Medical Student/Resident Case Reports.
- Global Case Reports.
- Clinical Case Puzzlers.
Learn more and submit at chest2017.abstractcentral.com.
Apply for 2017 CHEST Foundation Grants
Application deadline: March 31
The CHEST Foundation has started accepting applications for its clinical research, distinguished scholar, and community service grants. Every year, the CHEST Foundation awards more than a half-million dollars to the next generation of lung health champions.
The grants available are:
- GlaxoSmithKline Distinguished Scholar Research Grant in Respiratory Health: $150,000 over 3 years
- CHEST Foundation Research Grant in Lung Cancer: $50,000-$100,000* over 2 years
- CHEST Foundation Research Grant in Pulmonary Arterial Hypertension: $25,000 1-year grant
- CHEST Foundation and Alpha-1 Foundation Research Grant in Alpha-1 Antitrypsin Deficiency: $25,000 1-year grant
- CHEST Foundation Research Grant in Nontuberculous Mycobacteria: $10,000-$30,000* 1-year grant
- CHEST Foundation Research Grant in Venous Thromboembolism: $30,000 1-year grant
- CHEST Foundation Research Grant in Pulmonary Fibrosis: $30,000 1-year grant
- CHEST Foundation Research Grant in Chronic Obstructive Pulmonary Disease: $50,000 1-year grant
- CHEST Foundation Research Grant in Women’s Lung Health: $10,000 1-year grant
- CHEST Foundation Research Grant in Asthma: $15,000 - $30,000* 1-year grant
- CHEST Foundation Research Grant in Cystic Fibrosis: $30,000 1-year grant
- Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP: multiple awards up to $15,000 per 1-year grant
*Amount contingent on funding.Apply for grants at chestfoundation.org/grants.
Household air pollution: Foundation grantee champions lung health
In 2016, Catherine Oberg, MD, was awarded the CHEST Foundation Research Grant in Women’s Lung Health for her project on household air pollution in Ghana. In this recent interview with Dr. Oberg, she describes how she is championing lung health.
How I got involved
In medical school, I was very interested in international medicine and took a trip to Tanzania to do primary care work when I was in my fourth year. I saw firsthand how the people, women especially, sleep, cook, eat, and take care of their children and animals all in one house. I saw how direct smoke exposure from cooking caused symptoms of cough, phlegm, and shortness of breath. I knew this was an area where I could make an impact.

Fortunately, I learned about CHEST Foundation grants through my mentor, Alison Lee, MD, who was a CHEST Foundation grant recipient early in her career. With the help of the grant, I was able to furnish my own supplies, get everything to Ghana, train native health-care providers, and start doing assessments. I received the CHEST Foundation grant at the perfect time. I am so appreciative and honored to be a CHEST Foundation grant recipient. It’s such a humbling experience to be able to act on these things that I’ve been looking into for so many months. I’m just excited and thankful, and can’t wait to see what we’re able to show.
Tackling a leading cause of lung disease
In rural areas around the world, people cook with ineffective fuels, such as animal dung, that cause damaging household air pollution. This is a leading cause of asthma, COPD, and lung cancer worldwide, and it preferentially affects women and children because of their roles in the household. My project focuses on household air pollution with a goal to measure the effectiveness of utilizing a clean burning stove as an intervention.
We have a cohort of women in Ghana and have had randomized clusters using either a liquefied petroleum gas (LPG) clean burning stove or a traditional cook stove for 18 months now. We’re going to look at their lung function, inflammatory markers, and respiratory symptoms and compare the groups to see if the intervention has made a difference.
The impact
Being able to breathe is a function many of us take for granted. The ability to impact something this vital to everyday life is a really exciting and important challenge. It’s an area where I think we can make a big impact.

The future
This project could bring about further research and hopefully provide evidence supporting these types of interventions. The impact could affect millions of people around the world. The CHEST Foundation grant is providing materials that are the foundation of our project. This grant allows us to design better studies in the future, to educate patients in a more effective manner, and to prevent these life-threatening diseases.
The next CHEST Foundation grants cycle is open from February 1 to March 31, 2017. How will you champion lung health? Learn more about foundation grants and how you can apply at https://chest.realmagnet.land/chest-foundation-grants.
In 2016, Catherine Oberg, MD, was awarded the CHEST Foundation Research Grant in Women’s Lung Health for her project on household air pollution in Ghana. In this recent interview with Dr. Oberg, she describes how she is championing lung health.
How I got involved
In medical school, I was very interested in international medicine and took a trip to Tanzania to do primary care work when I was in my fourth year. I saw firsthand how the people, women especially, sleep, cook, eat, and take care of their children and animals all in one house. I saw how direct smoke exposure from cooking caused symptoms of cough, phlegm, and shortness of breath. I knew this was an area where I could make an impact.
Fortunately, I learned about CHEST Foundation grants through my mentor, Alison Lee, MD, who was a CHEST Foundation grant recipient early in her career. With the help of the grant, I was able to furnish my own supplies, get everything to Ghana, train native health-care providers, and start doing assessments. I received the CHEST Foundation grant at the perfect time. I am so appreciative and honored to be a CHEST Foundation grant recipient. It’s such a humbling experience to be able to act on these things that I’ve been looking into for so many months. I’m just excited and thankful, and can’t wait to see what we’re able to show.
Tackling a leading cause of lung disease
In rural areas around the world, people cook with ineffective fuels, such as animal dung, that cause damaging household air pollution. This is a leading cause of asthma, COPD, and lung cancer worldwide, and it preferentially affects women and children because of their roles in the household. My project focuses on household air pollution with a goal to measure the effectiveness of utilizing a clean burning stove as an intervention.
We have a cohort of women in Ghana and have had randomized clusters using either a liquefied petroleum gas (LPG) clean burning stove or a traditional cook stove for 18 months now. We’re going to look at their lung function, inflammatory markers, and respiratory symptoms and compare the groups to see if the intervention has made a difference.
The impact
Being able to breathe is a function many of us take for granted. The ability to impact something this vital to everyday life is a really exciting and important challenge. It’s an area where I think we can make a big impact.
The future
This project could bring about further research and hopefully provide evidence supporting these types of interventions. The impact could affect millions of people around the world. The CHEST Foundation grant is providing materials that are the foundation of our project. This grant allows us to design better studies in the future, to educate patients in a more effective manner, and to prevent these life-threatening diseases.
The next CHEST Foundation grants cycle is open from February 1 to March 31, 2017. How will you champion lung health? Learn more about foundation grants and how you can apply at https://chest.realmagnet.land/chest-foundation-grants.
In 2016, Catherine Oberg, MD, was awarded the CHEST Foundation Research Grant in Women’s Lung Health for her project on household air pollution in Ghana. In this recent interview with Dr. Oberg, she describes how she is championing lung health.
How I got involved
In medical school, I was very interested in international medicine and took a trip to Tanzania to do primary care work when I was in my fourth year. I saw firsthand how the people, women especially, sleep, cook, eat, and take care of their children and animals all in one house. I saw how direct smoke exposure from cooking caused symptoms of cough, phlegm, and shortness of breath. I knew this was an area where I could make an impact.
Fortunately, I learned about CHEST Foundation grants through my mentor, Alison Lee, MD, who was a CHEST Foundation grant recipient early in her career. With the help of the grant, I was able to furnish my own supplies, get everything to Ghana, train native health-care providers, and start doing assessments. I received the CHEST Foundation grant at the perfect time. I am so appreciative and honored to be a CHEST Foundation grant recipient. It’s such a humbling experience to be able to act on these things that I’ve been looking into for so many months. I’m just excited and thankful, and can’t wait to see what we’re able to show.
Tackling a leading cause of lung disease
In rural areas around the world, people cook with ineffective fuels, such as animal dung, that cause damaging household air pollution. This is a leading cause of asthma, COPD, and lung cancer worldwide, and it preferentially affects women and children because of their roles in the household. My project focuses on household air pollution with a goal to measure the effectiveness of utilizing a clean burning stove as an intervention.
We have a cohort of women in Ghana and have had randomized clusters using either a liquefied petroleum gas (LPG) clean burning stove or a traditional cook stove for 18 months now. We’re going to look at their lung function, inflammatory markers, and respiratory symptoms and compare the groups to see if the intervention has made a difference.
The impact
Being able to breathe is a function many of us take for granted. The ability to impact something this vital to everyday life is a really exciting and important challenge. It’s an area where I think we can make a big impact.
The future
This project could bring about further research and hopefully provide evidence supporting these types of interventions. The impact could affect millions of people around the world. The CHEST Foundation grant is providing materials that are the foundation of our project. This grant allows us to design better studies in the future, to educate patients in a more effective manner, and to prevent these life-threatening diseases.
The next CHEST Foundation grants cycle is open from February 1 to March 31, 2017. How will you champion lung health? Learn more about foundation grants and how you can apply at https://chest.realmagnet.land/chest-foundation-grants.
To save on drug costs, insurer wants to steer you to ‘preferred’ pharmacies
One of California’s largest insurers has proposed a change in the benefits of commercial plans next year that would require consumers to pay more for drugs at pharmacies outside an established network.
Blue Shield of California wants to create “a tiered pharmacy network” in its 2018 small- and large-group plans, according to preliminary proposals the company submitted to the California Department of Managed Health Care (DMHC), a state health insurance regulator.
If the proposal is approved by the department before the end of the year, it would affect the coverage of more than 1.8 million consumers, based on 2015 numbers from the regulator.
Under Blue Shield’s proposal, consumers still would have a broad selection of pharmacies, but they would have to choose a “preferred” pharmacy to maintain this year’s copayment amount. Outside of that network, consumers could pay up to $50 more for the same prescription, the company document says.
The move is part of a larger trend among insurers and other health care payers to narrow networks of providers to keep costs down, experts say. Consumers already are familiar with insurers steering them toward certain physicians and hospitals to save money. And narrower pharmacy networks are increasingly common in Medicare and employer-sponsored health coverage.
The Blue Shield proposal would expand pharmacy networks to commercial plans – something California regulators say insurers under their purview aren’t currently doing.
Nationally, insurers and their pharmaceutical benefit managers have been known to selectively contract with pharmacies, although it’s difficult to say how common that practice is.
Pharmacy chain CVS Health expects to dispense tens of millions fewer prescriptions in 2017 as a result of being excluded from health plan pharmacy networks around the nation, according to news media reports. And late last year, because CVS was not included in a Blue Cross of Alabama network, hundreds of thousands of customers were notified that they should switch to a preferred pharmacy if they wanted lower prices.
Raymond Brown, a clinical pharmacy leader at Mercer, a New York-based employee benefits consulting firm, says every health care payer – government, employers, and insurers – scrutinizes the pharmaceutical supply chain to find places to control rising drug costs.
“They’re saying, ‘What else can we do?’ ” he said. “To say ‘Can we do something on the distribution side?’ … I think is a natural additional step.”
Blue Shield of California spokeswoman Molly Weedn said the preferred pharmacy network proposal is an effort to “stabilize the increasingly high cost of drugs for our members.” Blue Shield of California already has preferred pharmacies in its Medicare plans, including CVS/Target, Walmart, Costco, and Safeway/Albertsons and some independent pharmacies. The insurer’s latest proposal would use that same network of pharmacies in its state-regulated commercial market.
But some patient advocates and pharmacists say pharmacy networks can confuse consumers and interfere with patient care.
Another major California insurer, Anthem Blue Cross, joined Blue Shield of California in proposing to create preferred pharmacy networks in Covered California policies for 2018.
But in a letter in December to the health insurance marketplace, attorneys for low-income consumers and other advocacy groups objected. “We reject the allowance of tiered pharmacies as the right solution to our shared concerns about the ever-escalating prices of prescription drugs,” the letter said.
Covered California staff decided not to allow the change in the 2018 health insurance marketplace, spokeswoman Amy Palmer said, because it wouldn’t have considerably brought down health care costs.
Advocates with Consumers Union, which hasn’t taken a position on the most recent Blue Shield proposal, say pharmacy networks could create more complexity for lower-income people in an already complicated health insurance system, one that faces more uncertainty under an Obamacare repeal.
“It’s really not a good time to add one more layer of information that [patients] have to deal with,” said Betsy Imholz, special projects director at Consumers Union. “People will be befuddled.”
Imholz said creating economic incentives to steer patients toward network pharmacies could inconvenience the most vulnerable patients. If the preferred pharmacy is farther away, or in a rural area, lower-income patients dependent on public transit could have a harder time reaching the preferred pharmacy, she said.
“It’s going to add to your costs and your time, and if you have a medical condition, to get to … the cheaper tier of pharmacy, that can be a real burden,” Imholz said.
Many pharmacists don’t like narrow pharmacy networks, either.
Jon Roth, CEO of the California Pharmacists Association, says creating preferred pharmacies can sever long-term relationships that pharmacists have with patients.
Pharmacists “may know everything about [a patient’s] health history, their medication use” and help patients “adhere to their medication instructions,” Roth said. “It’s very distressing.”
But Blue Shield, in its proposals to the DMHC, said its pharmacy network idea is intended to make “health care affordable for all Californians.” And the company suggests that almost all of its commercial plan members live close to a preferred pharmacy.
Still, Katie Keith, a policy consultant and contributor to a recent report on pharmacy access, says the preferred pharmacy approach is not a panacea for rising drug prices.
“It’s not going to solve everything or even get close to solving the whole problem,” Keith said. “It’s a trend to keep an eye on, but I’m not sure how far this will go.”
This story was produced by Kaiser Health News, which publishes California Healthline, an editorially independent service of the California Health Care Foundation. Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
One of California’s largest insurers has proposed a change in the benefits of commercial plans next year that would require consumers to pay more for drugs at pharmacies outside an established network.
Blue Shield of California wants to create “a tiered pharmacy network” in its 2018 small- and large-group plans, according to preliminary proposals the company submitted to the California Department of Managed Health Care (DMHC), a state health insurance regulator.
If the proposal is approved by the department before the end of the year, it would affect the coverage of more than 1.8 million consumers, based on 2015 numbers from the regulator.
Under Blue Shield’s proposal, consumers still would have a broad selection of pharmacies, but they would have to choose a “preferred” pharmacy to maintain this year’s copayment amount. Outside of that network, consumers could pay up to $50 more for the same prescription, the company document says.
The move is part of a larger trend among insurers and other health care payers to narrow networks of providers to keep costs down, experts say. Consumers already are familiar with insurers steering them toward certain physicians and hospitals to save money. And narrower pharmacy networks are increasingly common in Medicare and employer-sponsored health coverage.
The Blue Shield proposal would expand pharmacy networks to commercial plans – something California regulators say insurers under their purview aren’t currently doing.
Nationally, insurers and their pharmaceutical benefit managers have been known to selectively contract with pharmacies, although it’s difficult to say how common that practice is.
Pharmacy chain CVS Health expects to dispense tens of millions fewer prescriptions in 2017 as a result of being excluded from health plan pharmacy networks around the nation, according to news media reports. And late last year, because CVS was not included in a Blue Cross of Alabama network, hundreds of thousands of customers were notified that they should switch to a preferred pharmacy if they wanted lower prices.
Raymond Brown, a clinical pharmacy leader at Mercer, a New York-based employee benefits consulting firm, says every health care payer – government, employers, and insurers – scrutinizes the pharmaceutical supply chain to find places to control rising drug costs.
“They’re saying, ‘What else can we do?’ ” he said. “To say ‘Can we do something on the distribution side?’ … I think is a natural additional step.”
Blue Shield of California spokeswoman Molly Weedn said the preferred pharmacy network proposal is an effort to “stabilize the increasingly high cost of drugs for our members.” Blue Shield of California already has preferred pharmacies in its Medicare plans, including CVS/Target, Walmart, Costco, and Safeway/Albertsons and some independent pharmacies. The insurer’s latest proposal would use that same network of pharmacies in its state-regulated commercial market.
But some patient advocates and pharmacists say pharmacy networks can confuse consumers and interfere with patient care.
Another major California insurer, Anthem Blue Cross, joined Blue Shield of California in proposing to create preferred pharmacy networks in Covered California policies for 2018.
But in a letter in December to the health insurance marketplace, attorneys for low-income consumers and other advocacy groups objected. “We reject the allowance of tiered pharmacies as the right solution to our shared concerns about the ever-escalating prices of prescription drugs,” the letter said.
Covered California staff decided not to allow the change in the 2018 health insurance marketplace, spokeswoman Amy Palmer said, because it wouldn’t have considerably brought down health care costs.
Advocates with Consumers Union, which hasn’t taken a position on the most recent Blue Shield proposal, say pharmacy networks could create more complexity for lower-income people in an already complicated health insurance system, one that faces more uncertainty under an Obamacare repeal.
“It’s really not a good time to add one more layer of information that [patients] have to deal with,” said Betsy Imholz, special projects director at Consumers Union. “People will be befuddled.”
Imholz said creating economic incentives to steer patients toward network pharmacies could inconvenience the most vulnerable patients. If the preferred pharmacy is farther away, or in a rural area, lower-income patients dependent on public transit could have a harder time reaching the preferred pharmacy, she said.
“It’s going to add to your costs and your time, and if you have a medical condition, to get to … the cheaper tier of pharmacy, that can be a real burden,” Imholz said.
Many pharmacists don’t like narrow pharmacy networks, either.
Jon Roth, CEO of the California Pharmacists Association, says creating preferred pharmacies can sever long-term relationships that pharmacists have with patients.
Pharmacists “may know everything about [a patient’s] health history, their medication use” and help patients “adhere to their medication instructions,” Roth said. “It’s very distressing.”
But Blue Shield, in its proposals to the DMHC, said its pharmacy network idea is intended to make “health care affordable for all Californians.” And the company suggests that almost all of its commercial plan members live close to a preferred pharmacy.
Still, Katie Keith, a policy consultant and contributor to a recent report on pharmacy access, says the preferred pharmacy approach is not a panacea for rising drug prices.
“It’s not going to solve everything or even get close to solving the whole problem,” Keith said. “It’s a trend to keep an eye on, but I’m not sure how far this will go.”
This story was produced by Kaiser Health News, which publishes California Healthline, an editorially independent service of the California Health Care Foundation. Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
One of California’s largest insurers has proposed a change in the benefits of commercial plans next year that would require consumers to pay more for drugs at pharmacies outside an established network.
Blue Shield of California wants to create “a tiered pharmacy network” in its 2018 small- and large-group plans, according to preliminary proposals the company submitted to the California Department of Managed Health Care (DMHC), a state health insurance regulator.
If the proposal is approved by the department before the end of the year, it would affect the coverage of more than 1.8 million consumers, based on 2015 numbers from the regulator.
Under Blue Shield’s proposal, consumers still would have a broad selection of pharmacies, but they would have to choose a “preferred” pharmacy to maintain this year’s copayment amount. Outside of that network, consumers could pay up to $50 more for the same prescription, the company document says.
The move is part of a larger trend among insurers and other health care payers to narrow networks of providers to keep costs down, experts say. Consumers already are familiar with insurers steering them toward certain physicians and hospitals to save money. And narrower pharmacy networks are increasingly common in Medicare and employer-sponsored health coverage.
The Blue Shield proposal would expand pharmacy networks to commercial plans – something California regulators say insurers under their purview aren’t currently doing.
Nationally, insurers and their pharmaceutical benefit managers have been known to selectively contract with pharmacies, although it’s difficult to say how common that practice is.
Pharmacy chain CVS Health expects to dispense tens of millions fewer prescriptions in 2017 as a result of being excluded from health plan pharmacy networks around the nation, according to news media reports. And late last year, because CVS was not included in a Blue Cross of Alabama network, hundreds of thousands of customers were notified that they should switch to a preferred pharmacy if they wanted lower prices.
Raymond Brown, a clinical pharmacy leader at Mercer, a New York-based employee benefits consulting firm, says every health care payer – government, employers, and insurers – scrutinizes the pharmaceutical supply chain to find places to control rising drug costs.
“They’re saying, ‘What else can we do?’ ” he said. “To say ‘Can we do something on the distribution side?’ … I think is a natural additional step.”
Blue Shield of California spokeswoman Molly Weedn said the preferred pharmacy network proposal is an effort to “stabilize the increasingly high cost of drugs for our members.” Blue Shield of California already has preferred pharmacies in its Medicare plans, including CVS/Target, Walmart, Costco, and Safeway/Albertsons and some independent pharmacies. The insurer’s latest proposal would use that same network of pharmacies in its state-regulated commercial market.
But some patient advocates and pharmacists say pharmacy networks can confuse consumers and interfere with patient care.
Another major California insurer, Anthem Blue Cross, joined Blue Shield of California in proposing to create preferred pharmacy networks in Covered California policies for 2018.
But in a letter in December to the health insurance marketplace, attorneys for low-income consumers and other advocacy groups objected. “We reject the allowance of tiered pharmacies as the right solution to our shared concerns about the ever-escalating prices of prescription drugs,” the letter said.
Covered California staff decided not to allow the change in the 2018 health insurance marketplace, spokeswoman Amy Palmer said, because it wouldn’t have considerably brought down health care costs.
Advocates with Consumers Union, which hasn’t taken a position on the most recent Blue Shield proposal, say pharmacy networks could create more complexity for lower-income people in an already complicated health insurance system, one that faces more uncertainty under an Obamacare repeal.
“It’s really not a good time to add one more layer of information that [patients] have to deal with,” said Betsy Imholz, special projects director at Consumers Union. “People will be befuddled.”
Imholz said creating economic incentives to steer patients toward network pharmacies could inconvenience the most vulnerable patients. If the preferred pharmacy is farther away, or in a rural area, lower-income patients dependent on public transit could have a harder time reaching the preferred pharmacy, she said.
“It’s going to add to your costs and your time, and if you have a medical condition, to get to … the cheaper tier of pharmacy, that can be a real burden,” Imholz said.
Many pharmacists don’t like narrow pharmacy networks, either.
Jon Roth, CEO of the California Pharmacists Association, says creating preferred pharmacies can sever long-term relationships that pharmacists have with patients.
Pharmacists “may know everything about [a patient’s] health history, their medication use” and help patients “adhere to their medication instructions,” Roth said. “It’s very distressing.”
But Blue Shield, in its proposals to the DMHC, said its pharmacy network idea is intended to make “health care affordable for all Californians.” And the company suggests that almost all of its commercial plan members live close to a preferred pharmacy.
Still, Katie Keith, a policy consultant and contributor to a recent report on pharmacy access, says the preferred pharmacy approach is not a panacea for rising drug prices.
“It’s not going to solve everything or even get close to solving the whole problem,” Keith said. “It’s a trend to keep an eye on, but I’m not sure how far this will go.”
This story was produced by Kaiser Health News, which publishes California Healthline, an editorially independent service of the California Health Care Foundation. Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.