User login
Cardiology: Step One…

Improvements made in safe opioid prescribing practices but crisis far from over
Clinical question: How have national and county-level opioid prescribing practices changed from the years 2006 to 2015?
Background: The opioid epidemic is currently at the forefront of public health crises, with more than 15,000 deaths caused by prescription opioid overdoses in 2015 alone and an estimated 2 million people with an opioid use disorder associated with prescription opioids. The opioid epidemic also has a significant financial burden with the cost of opioid overdose, abuse, and dependence totaling $78.5 billion/year in the United States. As the utilization of opioids to treat noncancer pain quadrupled during 1999-2010, so did the prevalence of opioid misuse disorder and overdose deaths from prescription opioids. This study reviewed prescribing practices at the national and county level during 2006-2015 in hopes of understanding how this affected the opioid crisis.
Setting: The data were summarized from a sample of pharmacies throughout the United States.
Synopsis: Data were obtained via the QuintilesIMS Data Warehouse, which estimated the number of opioid prescriptions, based upon a sample of 59,000 U.S. pharmacies (88% of total prescriptions). The amount of prescriptions peaked in 2010 then decreased yearly through 2015, yet remained about three times as high as prescription rates from 1999. Opioid prescribing practices had significant variation throughout the country, with higher prescription rates associated with smaller cities, larger white population, higher rates of Medicaid and unemployment, and higher prevalence of arthritis and diabetes. Variation in prescribing practices at the county level represents lack of consensus and evidence-based guidelines.
The authors suggest that providers carefully weigh the risks and benefits of opioids and review the Guideline for Prescribing Opioids for Chronic Pain from the Centers for Disease Control and Prevention. At the state and local levels, mandated pain clinic regulations and Physician Drug Monitoring Programs also are needed for continued improvement in opioid-related deaths. Weaknesses of study included lack of clinical outcomes and use of QuintilesIMS to estimate prescriptions that has not been validated.
Bottom line: Although rates of opioid prescriptions have improved since 2010, substantial changes and regulations for prescribing practices are needed at the state and local levels.
Citation: Guy GP Jr. et al. Vital Signs: Changes in opioid prescribing in the United States, 2006-2015. MMWR Morb Mortal Wkly Rep. 2017;66:697704.
Dr. Farber is a medical instructor, Duke University Health System.
Clinical question: How have national and county-level opioid prescribing practices changed from the years 2006 to 2015?
Background: The opioid epidemic is currently at the forefront of public health crises, with more than 15,000 deaths caused by prescription opioid overdoses in 2015 alone and an estimated 2 million people with an opioid use disorder associated with prescription opioids. The opioid epidemic also has a significant financial burden with the cost of opioid overdose, abuse, and dependence totaling $78.5 billion/year in the United States. As the utilization of opioids to treat noncancer pain quadrupled during 1999-2010, so did the prevalence of opioid misuse disorder and overdose deaths from prescription opioids. This study reviewed prescribing practices at the national and county level during 2006-2015 in hopes of understanding how this affected the opioid crisis.
Setting: The data were summarized from a sample of pharmacies throughout the United States.
Synopsis: Data were obtained via the QuintilesIMS Data Warehouse, which estimated the number of opioid prescriptions, based upon a sample of 59,000 U.S. pharmacies (88% of total prescriptions). The amount of prescriptions peaked in 2010 then decreased yearly through 2015, yet remained about three times as high as prescription rates from 1999. Opioid prescribing practices had significant variation throughout the country, with higher prescription rates associated with smaller cities, larger white population, higher rates of Medicaid and unemployment, and higher prevalence of arthritis and diabetes. Variation in prescribing practices at the county level represents lack of consensus and evidence-based guidelines.
The authors suggest that providers carefully weigh the risks and benefits of opioids and review the Guideline for Prescribing Opioids for Chronic Pain from the Centers for Disease Control and Prevention. At the state and local levels, mandated pain clinic regulations and Physician Drug Monitoring Programs also are needed for continued improvement in opioid-related deaths. Weaknesses of study included lack of clinical outcomes and use of QuintilesIMS to estimate prescriptions that has not been validated.
Bottom line: Although rates of opioid prescriptions have improved since 2010, substantial changes and regulations for prescribing practices are needed at the state and local levels.
Citation: Guy GP Jr. et al. Vital Signs: Changes in opioid prescribing in the United States, 2006-2015. MMWR Morb Mortal Wkly Rep. 2017;66:697704.
Dr. Farber is a medical instructor, Duke University Health System.
Clinical question: How have national and county-level opioid prescribing practices changed from the years 2006 to 2015?
Background: The opioid epidemic is currently at the forefront of public health crises, with more than 15,000 deaths caused by prescription opioid overdoses in 2015 alone and an estimated 2 million people with an opioid use disorder associated with prescription opioids. The opioid epidemic also has a significant financial burden with the cost of opioid overdose, abuse, and dependence totaling $78.5 billion/year in the United States. As the utilization of opioids to treat noncancer pain quadrupled during 1999-2010, so did the prevalence of opioid misuse disorder and overdose deaths from prescription opioids. This study reviewed prescribing practices at the national and county level during 2006-2015 in hopes of understanding how this affected the opioid crisis.
Setting: The data were summarized from a sample of pharmacies throughout the United States.
Synopsis: Data were obtained via the QuintilesIMS Data Warehouse, which estimated the number of opioid prescriptions, based upon a sample of 59,000 U.S. pharmacies (88% of total prescriptions). The amount of prescriptions peaked in 2010 then decreased yearly through 2015, yet remained about three times as high as prescription rates from 1999. Opioid prescribing practices had significant variation throughout the country, with higher prescription rates associated with smaller cities, larger white population, higher rates of Medicaid and unemployment, and higher prevalence of arthritis and diabetes. Variation in prescribing practices at the county level represents lack of consensus and evidence-based guidelines.
The authors suggest that providers carefully weigh the risks and benefits of opioids and review the Guideline for Prescribing Opioids for Chronic Pain from the Centers for Disease Control and Prevention. At the state and local levels, mandated pain clinic regulations and Physician Drug Monitoring Programs also are needed for continued improvement in opioid-related deaths. Weaknesses of study included lack of clinical outcomes and use of QuintilesIMS to estimate prescriptions that has not been validated.
Bottom line: Although rates of opioid prescriptions have improved since 2010, substantial changes and regulations for prescribing practices are needed at the state and local levels.
Citation: Guy GP Jr. et al. Vital Signs: Changes in opioid prescribing in the United States, 2006-2015. MMWR Morb Mortal Wkly Rep. 2017;66:697704.
Dr. Farber is a medical instructor, Duke University Health System.
Hydrochlorothiazide use linked to higher skin cancer risk
The common diuretic hydrochlorothiazide is linked to a dose-dependent increased risk of nonmelanoma skin cancer, in particular, squamous cell carcinoma, a case-controlled registry study showed.
This nationwide, case-matched control study examined patients’ cumulative hydrochlorothiazide use between 1995 and 2012 and found a clear dose-response patterns for both basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), with a more than sevenfold increased risk of SCC for a cumulative use of greater than or equal to 200,000 mg of hydrochlorothiazide (HCTZ).
“Assuming causality, the present results suggest that 1 in 10 SCC cases diagnosed during the study period can be attributed to HCTZ use,” wrote the study authors, who were led by Sidsel Arnspang, MD, of the department of neurology at Odense (Denmark) University Hospital, which is affiliated with the University of Southern Denmark.
The authors noted that they previously had reported a sevenfold increased risk of lip squamous cell carcinoma with hydrochlorothiazide. Furthermore, the International Agency for Research on Cancer recently classified the diuretic and antihypertensive as “possibly carcinogenic to humans.”
“As HCTZ is among the most widely used drugs in the U.S. and Western Europe, a carcinogenic effect of HCTZ would have a considerable impact on public health,” they wrote in their paper, published in the Journal of American Academy of Dermatology.
According to the study authors, the few studies that have investigated a potential link between thiazide use and nonmelanoma skin cancer (NMSC) have reported inconsistent results.
They speculated that this could be because HCTZ often is prescribed in combination with other diuretics, and there may have been difficulties with disentangling its effect from those of the other drugs.
Using data from five nationwide data sources, the research team compared HCTZ use among people diagnosed with SCC or BCC of the skin to use among a matched control group without such cancers. People were excluded from the analysis if they had SCC of the lip because they had been evaluated in the research team’s previous study.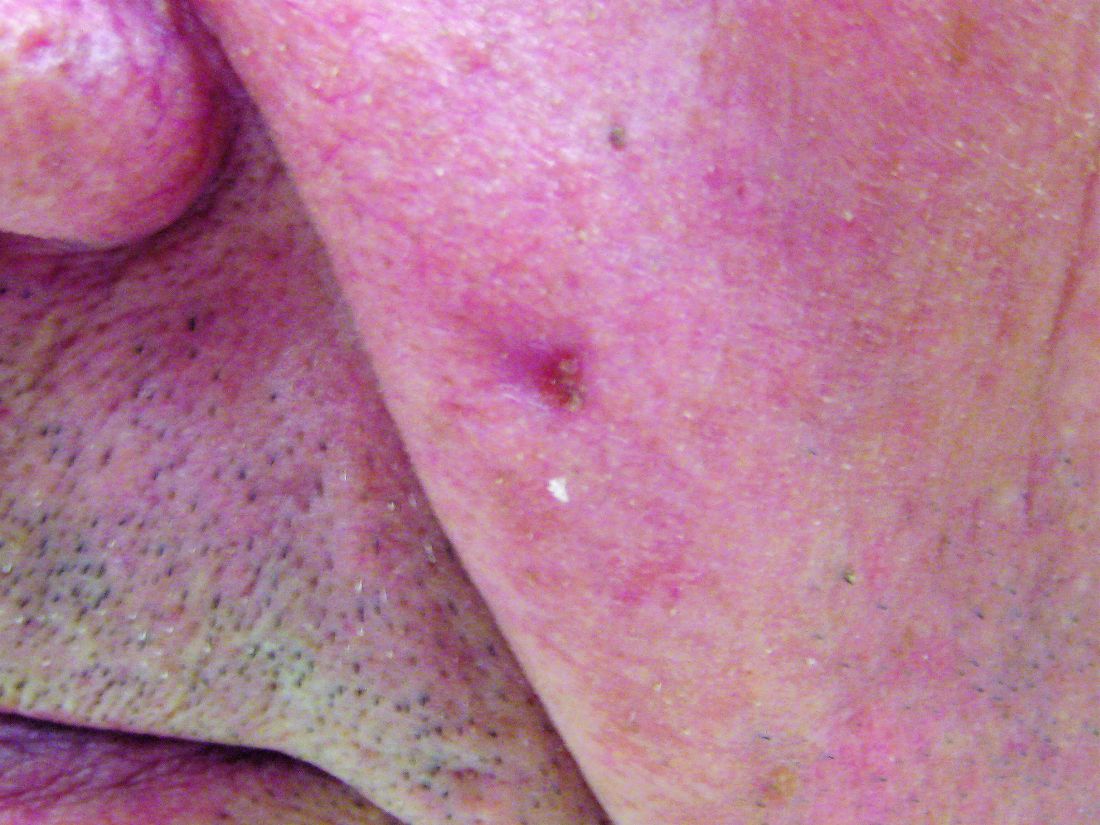
High use of HCTZ was defined as filled prescriptions totaling greater than or equal to 50,000 mg of HCTZ, which corresponds to greater than or equal to 2,000 defined daily doses (for example, approximately 6 years of cumulative use).
Overall, the study population involved 71,533 BCC and 8,629 SCC cases that were matched to 1,430,883 and 172,462 population controls, respectively.
Baseline characteristics of the cases and controls were similar, except BCC cases were slightly more educated than controls. Results showed that high use of hydrochlorothiazide was associated with odds ratios of 1.29 (95% confidence interval, 1.23-1.35) for BCC and 3.98 (95% CI, 3.68-4.31) for SCC.
A clear dose-response relationship was observed with HCTZ use for both BCC and SCC, with the highest ORs observed in the upper exposure category (greater than or equal to 200,000 mg): The OR for BCC in this category was 1.54 (95% CI, 1.38-1.71) and 7.38 for SCC (95% CI, 6.32-8.60).
The researchers observed no associations for BCC or SCC risk with use of other diuretics and other hypertensives, a finding that they said supported a potential causal association between HCTZ and NMSC risk.
Little variation was seen in the association between HCTZ use and BCC or SCC risk in the subgroup analyses, except for notably stronger associations among younger individuals and females.
In analyses stratified according to tumor localization, the authors saw stronger associations for cancers at sun-exposed skin sites, especially the skin of the lower limbs.
“Given the considerable use of HCTZ worldwide and the morbidity associated with NMSC, a causal association between HCTZ use and NMSC risk would have significant public health implications,” Dr. Arnspang and associates concluded. “The use of HCTZ should be carefully considered as several other antihypertensive agents with similar indications and efficiency are available but without known associations with skin cancer.”
The investigators cited several limitations. For example, information on ethnicity and skin type was not available. This information would have been useful in evaluating participants’ photosensitivity as a possible mechanism for a higher skin cancer risk with the use of HCTZ.
The study was funded by a grant from the Danish Cancer Society and the Danish Council of Independent Research. Several of the authors reported receiving grants and or honoraria from pharmaceutical companies.
SOURCE: Arnspang S et al. JAAD. 2017. doi: 10.1016/j.jaad.2017.11.042.
The common diuretic hydrochlorothiazide is linked to a dose-dependent increased risk of nonmelanoma skin cancer, in particular, squamous cell carcinoma, a case-controlled registry study showed.
This nationwide, case-matched control study examined patients’ cumulative hydrochlorothiazide use between 1995 and 2012 and found a clear dose-response patterns for both basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), with a more than sevenfold increased risk of SCC for a cumulative use of greater than or equal to 200,000 mg of hydrochlorothiazide (HCTZ).
“Assuming causality, the present results suggest that 1 in 10 SCC cases diagnosed during the study period can be attributed to HCTZ use,” wrote the study authors, who were led by Sidsel Arnspang, MD, of the department of neurology at Odense (Denmark) University Hospital, which is affiliated with the University of Southern Denmark.
The authors noted that they previously had reported a sevenfold increased risk of lip squamous cell carcinoma with hydrochlorothiazide. Furthermore, the International Agency for Research on Cancer recently classified the diuretic and antihypertensive as “possibly carcinogenic to humans.”
“As HCTZ is among the most widely used drugs in the U.S. and Western Europe, a carcinogenic effect of HCTZ would have a considerable impact on public health,” they wrote in their paper, published in the Journal of American Academy of Dermatology.
According to the study authors, the few studies that have investigated a potential link between thiazide use and nonmelanoma skin cancer (NMSC) have reported inconsistent results.
They speculated that this could be because HCTZ often is prescribed in combination with other diuretics, and there may have been difficulties with disentangling its effect from those of the other drugs.
Using data from five nationwide data sources, the research team compared HCTZ use among people diagnosed with SCC or BCC of the skin to use among a matched control group without such cancers. People were excluded from the analysis if they had SCC of the lip because they had been evaluated in the research team’s previous study.
High use of HCTZ was defined as filled prescriptions totaling greater than or equal to 50,000 mg of HCTZ, which corresponds to greater than or equal to 2,000 defined daily doses (for example, approximately 6 years of cumulative use).
Overall, the study population involved 71,533 BCC and 8,629 SCC cases that were matched to 1,430,883 and 172,462 population controls, respectively.
Baseline characteristics of the cases and controls were similar, except BCC cases were slightly more educated than controls. Results showed that high use of hydrochlorothiazide was associated with odds ratios of 1.29 (95% confidence interval, 1.23-1.35) for BCC and 3.98 (95% CI, 3.68-4.31) for SCC.
A clear dose-response relationship was observed with HCTZ use for both BCC and SCC, with the highest ORs observed in the upper exposure category (greater than or equal to 200,000 mg): The OR for BCC in this category was 1.54 (95% CI, 1.38-1.71) and 7.38 for SCC (95% CI, 6.32-8.60).
The researchers observed no associations for BCC or SCC risk with use of other diuretics and other hypertensives, a finding that they said supported a potential causal association between HCTZ and NMSC risk.
Little variation was seen in the association between HCTZ use and BCC or SCC risk in the subgroup analyses, except for notably stronger associations among younger individuals and females.
In analyses stratified according to tumor localization, the authors saw stronger associations for cancers at sun-exposed skin sites, especially the skin of the lower limbs.
“Given the considerable use of HCTZ worldwide and the morbidity associated with NMSC, a causal association between HCTZ use and NMSC risk would have significant public health implications,” Dr. Arnspang and associates concluded. “The use of HCTZ should be carefully considered as several other antihypertensive agents with similar indications and efficiency are available but without known associations with skin cancer.”
The investigators cited several limitations. For example, information on ethnicity and skin type was not available. This information would have been useful in evaluating participants’ photosensitivity as a possible mechanism for a higher skin cancer risk with the use of HCTZ.
The study was funded by a grant from the Danish Cancer Society and the Danish Council of Independent Research. Several of the authors reported receiving grants and or honoraria from pharmaceutical companies.
SOURCE: Arnspang S et al. JAAD. 2017. doi: 10.1016/j.jaad.2017.11.042.
The common diuretic hydrochlorothiazide is linked to a dose-dependent increased risk of nonmelanoma skin cancer, in particular, squamous cell carcinoma, a case-controlled registry study showed.
This nationwide, case-matched control study examined patients’ cumulative hydrochlorothiazide use between 1995 and 2012 and found a clear dose-response patterns for both basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), with a more than sevenfold increased risk of SCC for a cumulative use of greater than or equal to 200,000 mg of hydrochlorothiazide (HCTZ).
“Assuming causality, the present results suggest that 1 in 10 SCC cases diagnosed during the study period can be attributed to HCTZ use,” wrote the study authors, who were led by Sidsel Arnspang, MD, of the department of neurology at Odense (Denmark) University Hospital, which is affiliated with the University of Southern Denmark.
The authors noted that they previously had reported a sevenfold increased risk of lip squamous cell carcinoma with hydrochlorothiazide. Furthermore, the International Agency for Research on Cancer recently classified the diuretic and antihypertensive as “possibly carcinogenic to humans.”
“As HCTZ is among the most widely used drugs in the U.S. and Western Europe, a carcinogenic effect of HCTZ would have a considerable impact on public health,” they wrote in their paper, published in the Journal of American Academy of Dermatology.
According to the study authors, the few studies that have investigated a potential link between thiazide use and nonmelanoma skin cancer (NMSC) have reported inconsistent results.
They speculated that this could be because HCTZ often is prescribed in combination with other diuretics, and there may have been difficulties with disentangling its effect from those of the other drugs.
Using data from five nationwide data sources, the research team compared HCTZ use among people diagnosed with SCC or BCC of the skin to use among a matched control group without such cancers. People were excluded from the analysis if they had SCC of the lip because they had been evaluated in the research team’s previous study.
High use of HCTZ was defined as filled prescriptions totaling greater than or equal to 50,000 mg of HCTZ, which corresponds to greater than or equal to 2,000 defined daily doses (for example, approximately 6 years of cumulative use).
Overall, the study population involved 71,533 BCC and 8,629 SCC cases that were matched to 1,430,883 and 172,462 population controls, respectively.
Baseline characteristics of the cases and controls were similar, except BCC cases were slightly more educated than controls. Results showed that high use of hydrochlorothiazide was associated with odds ratios of 1.29 (95% confidence interval, 1.23-1.35) for BCC and 3.98 (95% CI, 3.68-4.31) for SCC.
A clear dose-response relationship was observed with HCTZ use for both BCC and SCC, with the highest ORs observed in the upper exposure category (greater than or equal to 200,000 mg): The OR for BCC in this category was 1.54 (95% CI, 1.38-1.71) and 7.38 for SCC (95% CI, 6.32-8.60).
The researchers observed no associations for BCC or SCC risk with use of other diuretics and other hypertensives, a finding that they said supported a potential causal association between HCTZ and NMSC risk.
Little variation was seen in the association between HCTZ use and BCC or SCC risk in the subgroup analyses, except for notably stronger associations among younger individuals and females.
In analyses stratified according to tumor localization, the authors saw stronger associations for cancers at sun-exposed skin sites, especially the skin of the lower limbs.
“Given the considerable use of HCTZ worldwide and the morbidity associated with NMSC, a causal association between HCTZ use and NMSC risk would have significant public health implications,” Dr. Arnspang and associates concluded. “The use of HCTZ should be carefully considered as several other antihypertensive agents with similar indications and efficiency are available but without known associations with skin cancer.”
The investigators cited several limitations. For example, information on ethnicity and skin type was not available. This information would have been useful in evaluating participants’ photosensitivity as a possible mechanism for a higher skin cancer risk with the use of HCTZ.
The study was funded by a grant from the Danish Cancer Society and the Danish Council of Independent Research. Several of the authors reported receiving grants and or honoraria from pharmaceutical companies.
SOURCE: Arnspang S et al. JAAD. 2017. doi: 10.1016/j.jaad.2017.11.042.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Key clinical point: The use of hydrochlorothiazide should be carefully considered because it is linked to a substantially increased risk of nonmelanoma skin cancer.
Major finding: A clear, dose-response pattern was found for both basal cell carcinoma and squamous cell carcinoma, with a more than sevenfold increased risk of SCC for a cumulative use of greater than or equal to 200,000 mg of the diuretic and antihypertensive.
Study details: The study population involved 71,533 BCC and 8,629 SCC cases that were matched to 1,430,883 and 172,462 population controls, respectively.
Disclosures: The study was funded by a grant from the Danish Cancer Society and the Danish Council of Independent Research. Several of the authors reported receiving grants and or honoraria from pharmaceutical companies.
Source: Arnspang S et al. JAAD. 2017. doi: 10.1016/j.jaad.2017.11.042
More evidence backs adjunctive intranasal esketamine for refractory depression
Esketamine – the S-enantiomer of the anesthetic ketamine – continues to look promising as an adjunctive treatment for refractory depression, phase 2 results of a four-phase multicenter trial suggested.
“We observed a significant and clinically meaningful treatment effect (vs. placebo) with 28-mg, 56-mg, and 84-mg doses of esketamine,” reported Ella J. Daly, MD, of Janssen, and her associates. The results were apparent 1 week after treatment, and they persisted over the follow-up phase, which lasted 8 weeks.
“,” Dr. Daly and her associates wrote in JAMA Psychiatry (2017 Dec 27. doi: 10.1001/jamapsychiatry.2017.3739).
In the study, patients aged 20-64 years with a diagnosis of major depressive disorder were recruited from several outpatient referral centers. All of the participants had treatment-resistant depression, defined by the study as an inadequate response despite the use of two or more antidepressants. Overall, 67 patients were randomized to receive one of the three doses of intranasal esketamine or a placebo nasal spray. In addition, participants continued to take oral antidepressants during the study period. People with a history of psychotic symptoms, use of substances such as alcohol and cannabis, or significant medical comorbidities were excluded.
Among participants in the treatment groups, the mean total score changes on the Montgomery-Åsberg Depression Rating Scale (MADRS) surpassed the MADRS score changes among those on placebo. Specifically, the mean MADRS score change for those on the 28-mg dose was –4.2 (P = 0.2), on the 56-mg dose was –6.3 (P = .001), and on the 84-mg dose was –9 (P less than .001).
The most common side effects among participants treated with esketamine were dizziness, headache, and dissociative symptoms. However, most adverse events were transient and “either mild or moderate in severity,” the investigators reported.
Dr. Daly and her associates cited several limitations, including the small sample size and the study’s exclusion criteria. Despite those limitations, Dr. Daly and her associates said, the results support further investigation of intranasal esketamine for treatment-resistant depression. They said a phase 3 study aimed at evaluating the frequency needed for dosing and duration of effect is underway.
Janssen funded the study. Dr. Daly and several of the other investigators are Janssen employees.
ghenderson@frontlinemedcom.com
SOURCE: Daly et al. JAMA Psychiatry. 2017 Dec 27. doi: 10.1001/jamapsychiatry.2017.3739
The study by Daly et al. is of interest for two key reasons, wrote Daniel S. Quintana, PhD, and his associates in an accompanying editorial (JAMA Psychiatry. 2017 Dec 27. doi: 10.1001/jamapsychiatry.2017.3738). First, of interest is the drug’s impact on depressive symptoms as measured on the Montgomery-Åsberg Depression Rating Scale. Second, the delivery mechanism is of interest, particularly in light of the increased bioavailability made possible by intranasal delivery. However, esketamine should be used with caution for psychiatric patients, he said.
“One of the three most common adverse effects was perceptual changes or dissociative symptoms, which fits with the known effect of ketamine and should be further clarified before starting routine in clinical practice,” he wrote. “Moreover, several issues related to long-term use, including the potential for addiction and adverse effects (somatic and cognitive) need to be carefully assessed in forthcoming studies.”
Dr. Quintana is affiliated with Oslo University Hospital & Institute of Clinical Medicine at the University of Oslo.
The study by Daly et al. is of interest for two key reasons, wrote Daniel S. Quintana, PhD, and his associates in an accompanying editorial (JAMA Psychiatry. 2017 Dec 27. doi: 10.1001/jamapsychiatry.2017.3738). First, of interest is the drug’s impact on depressive symptoms as measured on the Montgomery-Åsberg Depression Rating Scale. Second, the delivery mechanism is of interest, particularly in light of the increased bioavailability made possible by intranasal delivery. However, esketamine should be used with caution for psychiatric patients, he said.
“One of the three most common adverse effects was perceptual changes or dissociative symptoms, which fits with the known effect of ketamine and should be further clarified before starting routine in clinical practice,” he wrote. “Moreover, several issues related to long-term use, including the potential for addiction and adverse effects (somatic and cognitive) need to be carefully assessed in forthcoming studies.”
Dr. Quintana is affiliated with Oslo University Hospital & Institute of Clinical Medicine at the University of Oslo.
The study by Daly et al. is of interest for two key reasons, wrote Daniel S. Quintana, PhD, and his associates in an accompanying editorial (JAMA Psychiatry. 2017 Dec 27. doi: 10.1001/jamapsychiatry.2017.3738). First, of interest is the drug’s impact on depressive symptoms as measured on the Montgomery-Åsberg Depression Rating Scale. Second, the delivery mechanism is of interest, particularly in light of the increased bioavailability made possible by intranasal delivery. However, esketamine should be used with caution for psychiatric patients, he said.
“One of the three most common adverse effects was perceptual changes or dissociative symptoms, which fits with the known effect of ketamine and should be further clarified before starting routine in clinical practice,” he wrote. “Moreover, several issues related to long-term use, including the potential for addiction and adverse effects (somatic and cognitive) need to be carefully assessed in forthcoming studies.”
Dr. Quintana is affiliated with Oslo University Hospital & Institute of Clinical Medicine at the University of Oslo.
Esketamine – the S-enantiomer of the anesthetic ketamine – continues to look promising as an adjunctive treatment for refractory depression, phase 2 results of a four-phase multicenter trial suggested.
“We observed a significant and clinically meaningful treatment effect (vs. placebo) with 28-mg, 56-mg, and 84-mg doses of esketamine,” reported Ella J. Daly, MD, of Janssen, and her associates. The results were apparent 1 week after treatment, and they persisted over the follow-up phase, which lasted 8 weeks.
“,” Dr. Daly and her associates wrote in JAMA Psychiatry (2017 Dec 27. doi: 10.1001/jamapsychiatry.2017.3739).
In the study, patients aged 20-64 years with a diagnosis of major depressive disorder were recruited from several outpatient referral centers. All of the participants had treatment-resistant depression, defined by the study as an inadequate response despite the use of two or more antidepressants. Overall, 67 patients were randomized to receive one of the three doses of intranasal esketamine or a placebo nasal spray. In addition, participants continued to take oral antidepressants during the study period. People with a history of psychotic symptoms, use of substances such as alcohol and cannabis, or significant medical comorbidities were excluded.
Among participants in the treatment groups, the mean total score changes on the Montgomery-Åsberg Depression Rating Scale (MADRS) surpassed the MADRS score changes among those on placebo. Specifically, the mean MADRS score change for those on the 28-mg dose was –4.2 (P = 0.2), on the 56-mg dose was –6.3 (P = .001), and on the 84-mg dose was –9 (P less than .001).
The most common side effects among participants treated with esketamine were dizziness, headache, and dissociative symptoms. However, most adverse events were transient and “either mild or moderate in severity,” the investigators reported.
Dr. Daly and her associates cited several limitations, including the small sample size and the study’s exclusion criteria. Despite those limitations, Dr. Daly and her associates said, the results support further investigation of intranasal esketamine for treatment-resistant depression. They said a phase 3 study aimed at evaluating the frequency needed for dosing and duration of effect is underway.
Janssen funded the study. Dr. Daly and several of the other investigators are Janssen employees.
ghenderson@frontlinemedcom.com
SOURCE: Daly et al. JAMA Psychiatry. 2017 Dec 27. doi: 10.1001/jamapsychiatry.2017.3739
Esketamine – the S-enantiomer of the anesthetic ketamine – continues to look promising as an adjunctive treatment for refractory depression, phase 2 results of a four-phase multicenter trial suggested.
“We observed a significant and clinically meaningful treatment effect (vs. placebo) with 28-mg, 56-mg, and 84-mg doses of esketamine,” reported Ella J. Daly, MD, of Janssen, and her associates. The results were apparent 1 week after treatment, and they persisted over the follow-up phase, which lasted 8 weeks.
“,” Dr. Daly and her associates wrote in JAMA Psychiatry (2017 Dec 27. doi: 10.1001/jamapsychiatry.2017.3739).
In the study, patients aged 20-64 years with a diagnosis of major depressive disorder were recruited from several outpatient referral centers. All of the participants had treatment-resistant depression, defined by the study as an inadequate response despite the use of two or more antidepressants. Overall, 67 patients were randomized to receive one of the three doses of intranasal esketamine or a placebo nasal spray. In addition, participants continued to take oral antidepressants during the study period. People with a history of psychotic symptoms, use of substances such as alcohol and cannabis, or significant medical comorbidities were excluded.
Among participants in the treatment groups, the mean total score changes on the Montgomery-Åsberg Depression Rating Scale (MADRS) surpassed the MADRS score changes among those on placebo. Specifically, the mean MADRS score change for those on the 28-mg dose was –4.2 (P = 0.2), on the 56-mg dose was –6.3 (P = .001), and on the 84-mg dose was –9 (P less than .001).
The most common side effects among participants treated with esketamine were dizziness, headache, and dissociative symptoms. However, most adverse events were transient and “either mild or moderate in severity,” the investigators reported.
Dr. Daly and her associates cited several limitations, including the small sample size and the study’s exclusion criteria. Despite those limitations, Dr. Daly and her associates said, the results support further investigation of intranasal esketamine for treatment-resistant depression. They said a phase 3 study aimed at evaluating the frequency needed for dosing and duration of effect is underway.
Janssen funded the study. Dr. Daly and several of the other investigators are Janssen employees.
ghenderson@frontlinemedcom.com
SOURCE: Daly et al. JAMA Psychiatry. 2017 Dec 27. doi: 10.1001/jamapsychiatry.2017.3739
FROM JAMA PSYCHIATRY
Key clinical point: Adjunctive intranasal esketamine appears to lift treatment-resistant depression symptoms quickly, and the results last for more than 2 months “with a lower dosing frequency.”
Major finding: The MADRS mean score change for participants on the 28-mg dose was –4.2 (P = .2), –6.3 (P = .001) for those on the 56-mg dose, and –.9.0 (P greater than .001) for those on the 84-mg dose.
Study details: A phase 2 placebo-controlled study of 67 participants with treatment-resistant depression conducted in four phases at several outpatient treatment centers.
Disclosures: Janssen funded the study, and Dr. Daly and several of the other investigators are Janssen employees.
Source: Daly et al. JAMA Psychiatry. 2017 Dec. 27. doi: 10.1001/jamapsychiatry.2017.3739.
Survival improvements lag for young Hispanic patients with myeloma
ATLANTA –
Among U.S. adults diagnosed with multiple myeloma by age 40 years, 5-year and 10-year survival improved significantly (P less than .0001) for non-Hispanic blacks and whites, but not for Hispanics (5-year survival, P = .08; 10-year survival, P = .13), Abdel-Ghani Azzouqa, MD, and colleagues reported in a poster at the annual meeting of the American Society of Hematology.

Other population-based studies have uncovered racial and ethnic disparities in myeloma outcomes but had not honed in on the experience of young adult patients, who make up a growing proportion of diagnosed patients, said Dr. Azzouqa.
He and his associates analyzed Surveillance Epidemiology and End Results (SEER) data on patients diagnosed between ages 18 and 40 years with histologically confirmed multiple myeloma. The dataset spanned 1973-2014 and included 1,460 patients, of whom about 60% were male. Median age at diagnosis was 37 years; 47% of patients were non-Hispanic white, 28% were non-Hispanic black, 18% were Hispanic, 5.5% were Asian, and about 1% were of other ethnicities.
For young Hispanic patients with myeloma, 5-year survival improved from 39% before 1996, when stem cell transplants and novel therapies became available, to 56% from 2002 onward. This change was not statistically significant (P = .08), and 10-year survival rates also did not change significantly (from 21% to 33%; P = .13).
Five-year and 10-year survival did improve significantly for both genders (P = .0001) and among non-Hispanic blacks (P = .0001) and non-Hispanic whites (P = .0001).
Racial/ethnic subgroups did not differ significantly by median age at diagnosis, gender distribution, or listed cause of death, Dr. Azzouqa noted. Thus, reasons for the difference in survival for Hispanic patients remain unclear. Perhaps they reflect differences in disease biology, treatment response, or access or use of effective novel therapies, he said.
The researchers had no external funding sources. Dr. Azzouqa had no conflicts of interest. Lead author Dr. Sikander Ailawadhi disclosed ties to funding Pharmacyclics, Amgen, Novartis, and Takeda.
SOURCE: Ailawadhi S et al. ASH Abstract 2149
ATLANTA –
Among U.S. adults diagnosed with multiple myeloma by age 40 years, 5-year and 10-year survival improved significantly (P less than .0001) for non-Hispanic blacks and whites, but not for Hispanics (5-year survival, P = .08; 10-year survival, P = .13), Abdel-Ghani Azzouqa, MD, and colleagues reported in a poster at the annual meeting of the American Society of Hematology.
Other population-based studies have uncovered racial and ethnic disparities in myeloma outcomes but had not honed in on the experience of young adult patients, who make up a growing proportion of diagnosed patients, said Dr. Azzouqa.
He and his associates analyzed Surveillance Epidemiology and End Results (SEER) data on patients diagnosed between ages 18 and 40 years with histologically confirmed multiple myeloma. The dataset spanned 1973-2014 and included 1,460 patients, of whom about 60% were male. Median age at diagnosis was 37 years; 47% of patients were non-Hispanic white, 28% were non-Hispanic black, 18% were Hispanic, 5.5% were Asian, and about 1% were of other ethnicities.
For young Hispanic patients with myeloma, 5-year survival improved from 39% before 1996, when stem cell transplants and novel therapies became available, to 56% from 2002 onward. This change was not statistically significant (P = .08), and 10-year survival rates also did not change significantly (from 21% to 33%; P = .13).
Five-year and 10-year survival did improve significantly for both genders (P = .0001) and among non-Hispanic blacks (P = .0001) and non-Hispanic whites (P = .0001).
Racial/ethnic subgroups did not differ significantly by median age at diagnosis, gender distribution, or listed cause of death, Dr. Azzouqa noted. Thus, reasons for the difference in survival for Hispanic patients remain unclear. Perhaps they reflect differences in disease biology, treatment response, or access or use of effective novel therapies, he said.
The researchers had no external funding sources. Dr. Azzouqa had no conflicts of interest. Lead author Dr. Sikander Ailawadhi disclosed ties to funding Pharmacyclics, Amgen, Novartis, and Takeda.
SOURCE: Ailawadhi S et al. ASH Abstract 2149
ATLANTA –
Among U.S. adults diagnosed with multiple myeloma by age 40 years, 5-year and 10-year survival improved significantly (P less than .0001) for non-Hispanic blacks and whites, but not for Hispanics (5-year survival, P = .08; 10-year survival, P = .13), Abdel-Ghani Azzouqa, MD, and colleagues reported in a poster at the annual meeting of the American Society of Hematology.
Other population-based studies have uncovered racial and ethnic disparities in myeloma outcomes but had not honed in on the experience of young adult patients, who make up a growing proportion of diagnosed patients, said Dr. Azzouqa.
He and his associates analyzed Surveillance Epidemiology and End Results (SEER) data on patients diagnosed between ages 18 and 40 years with histologically confirmed multiple myeloma. The dataset spanned 1973-2014 and included 1,460 patients, of whom about 60% were male. Median age at diagnosis was 37 years; 47% of patients were non-Hispanic white, 28% were non-Hispanic black, 18% were Hispanic, 5.5% were Asian, and about 1% were of other ethnicities.
For young Hispanic patients with myeloma, 5-year survival improved from 39% before 1996, when stem cell transplants and novel therapies became available, to 56% from 2002 onward. This change was not statistically significant (P = .08), and 10-year survival rates also did not change significantly (from 21% to 33%; P = .13).
Five-year and 10-year survival did improve significantly for both genders (P = .0001) and among non-Hispanic blacks (P = .0001) and non-Hispanic whites (P = .0001).
Racial/ethnic subgroups did not differ significantly by median age at diagnosis, gender distribution, or listed cause of death, Dr. Azzouqa noted. Thus, reasons for the difference in survival for Hispanic patients remain unclear. Perhaps they reflect differences in disease biology, treatment response, or access or use of effective novel therapies, he said.
The researchers had no external funding sources. Dr. Azzouqa had no conflicts of interest. Lead author Dr. Sikander Ailawadhi disclosed ties to funding Pharmacyclics, Amgen, Novartis, and Takeda.
SOURCE: Ailawadhi S et al. ASH Abstract 2149
REPORTING FROM ASH 2017
Key clinical point: Recent improvements in multiple myeloma survival have left young Hispanics behind.
Major finding: Five-year and 10-year survival have improved significantly among young blacks and non-Hispanic whites with multiple myeloma (P less than .0001 for all comparisons) but not Hispanics (5-year survival P = .08; 10-year survival P = .13).
Data source: Surveillance Epidemiology and End Results (SEER) data for 1,460 adults up to 40 years old when diagnosed with multiple myeloma.
Disclosures: The researchers had no external funding sources. Dr. Azzouqa had no conflicts of interest. Lead author Dr. Sikander Ailawadhi disclosed funding from Pharmacyclics, Amgen, Novartis, and Takeda.
Source: Ailawadhi S et al. ASH Abstract 2149.
FDA expands approval of nivolumab for melanoma treatment
The Food and Drug Administration has approved nivolumab for the adjuvant treatment of patients with melanoma with involvement of lymph nodes or in patients with metastatic disease who have undergone complete resection.
Nivolumab was previously approved for the treatment of patients with unresectable or metastatic melanoma, the FDA said in a press statement.
Approval was based on results from the CHECKMATE-238 trial, where 906 patients with completely resected stage IIIB/C or stage IV melanoma received either nivolumab or ipilimumab for up to 1 year. Recurrence-free survival was superior in patients who received nivolumab, with 34% of patients in the nivolumab group experiencing recurrence/death, compared to 45.5% in the ipilimumab group.
The recommended dose and schedule of nivolumab in adjuvant melanoma is 240 mg administered as an IV infusion over 60 minutes every 2 weeks until disease recurrence or unacceptable toxicity, for a maximum of 1 year, according to the FDA.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb Company.
Find the full press release on the FDA website.
The Food and Drug Administration has approved nivolumab for the adjuvant treatment of patients with melanoma with involvement of lymph nodes or in patients with metastatic disease who have undergone complete resection.
Nivolumab was previously approved for the treatment of patients with unresectable or metastatic melanoma, the FDA said in a press statement.
Approval was based on results from the CHECKMATE-238 trial, where 906 patients with completely resected stage IIIB/C or stage IV melanoma received either nivolumab or ipilimumab for up to 1 year. Recurrence-free survival was superior in patients who received nivolumab, with 34% of patients in the nivolumab group experiencing recurrence/death, compared to 45.5% in the ipilimumab group.
The recommended dose and schedule of nivolumab in adjuvant melanoma is 240 mg administered as an IV infusion over 60 minutes every 2 weeks until disease recurrence or unacceptable toxicity, for a maximum of 1 year, according to the FDA.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb Company.
Find the full press release on the FDA website.
The Food and Drug Administration has approved nivolumab for the adjuvant treatment of patients with melanoma with involvement of lymph nodes or in patients with metastatic disease who have undergone complete resection.
Nivolumab was previously approved for the treatment of patients with unresectable or metastatic melanoma, the FDA said in a press statement.
Approval was based on results from the CHECKMATE-238 trial, where 906 patients with completely resected stage IIIB/C or stage IV melanoma received either nivolumab or ipilimumab for up to 1 year. Recurrence-free survival was superior in patients who received nivolumab, with 34% of patients in the nivolumab group experiencing recurrence/death, compared to 45.5% in the ipilimumab group.
The recommended dose and schedule of nivolumab in adjuvant melanoma is 240 mg administered as an IV infusion over 60 minutes every 2 weeks until disease recurrence or unacceptable toxicity, for a maximum of 1 year, according to the FDA.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb Company.
Find the full press release on the FDA website.
Venetoclax/rituximab boosts PFS in relapsed/refractory CLL
ATLANTA – In patients with relapsed/refractory chronic lymphocytic leukemia (CLL), a combination of venetoclax (Venclexta) and rituximab was superior to bendamustine (Treanda) and rituximab for prolonging progression-free survival (PFS), with effects consistent across subgroups, regardless of mutational status, and a clinically meaningful improvement in overall survival.
An interim analysis from the phase 3 MURANO trial showed that after a median follow-up of 23.8 months, the median PFS for patients randomized to venetoclax/rituximab had not been reached, compared with 17 months for patients assigned to bendamustine/rituximab, reported John F. Seymour, MBBS, PhD, of the Peter MacCallum Cancer Centre at the University of Melbourne.

Relapsed/refractory CLL often has a suboptimal response to conventional chemotherapy because of adverse biological features that can accumulate in cells, he said.
The combination of bendamustine and rituximab has been associated with about 60% overall responses rates, a median PFS of approximately 15 months, and overall survival of nearly 3 years in patients with CLL, he noted.
The rationale for pairing venetoclax with rituximab in this population comes from evidence showing efficacy of the monoclonal antibody, an oral B-cell lymphoma–2 (BCL-2) inhibitor, as monotherapy in patients with relapsed/refractory CLL, including those with poor prognostic features such as the 17p deletion (del17p).
Dr. Seymour and his colleagues recently published results from a phase 1b trial of venetoclax/rituximab in patients with relapsed/refractory CLL. The combination was associated with a 51% complete response rate, and a 28% rate of negative marrow minimal residual disease (MRD) (Lancet Oncol. 2017 Feb;18[2]:230-40)
In the MURANO study (NCT02005471), the investigators evaluated whether time-limited therapy with venetoclax/rituximab could improve PFS over bendamustine/rituximab.
Patients 18 and older with CLL who had been treated with one to three prior lines of therapy, including at least one chemotherapy-containing regimen, were enrolled. Prior treatment with bendamustine was allowed only if patients had had a duration of response of at least 24 months.
After stratification by del17p status, responsiveness to prior therapy, and geographic region, 389 patients were randomly assigned to receive rituximab 375 mg/m2 on day 1 of cycle 1 and 500 mg/m2 on day 1 of cycles 2 through 6, plus either bendamustine 70 mg/m2 on days 1 and 2 of each of six cycles, or venetoclax 400 mg orally once daily until disease progression, cessation for toxicity, or up to a maximum of 2 years starting from day 1 of cycle 1.
As noted,
The respective 1- and 2-year PFS rates with venetoclax were 91.2% and 82.8%, compared with 74.1% and 37.4% with bendamustine.
The venetoclax/rituximab combination was also significantly superior across all subgroups, regardless of the number of prior therapies, refractory vs. relapsed after most recent prior therapy, del17p status, TP53 mutational status, or baseline immunoglobulin heavy chain variable (IGHV) mutated or unmutated status
Response rates assessed by both investigators and independent reviewers were also better with venetoclax. The investigator-assessed overall response rate (ORR) was 93.3%, compared with 67.7% for bendamustine/rituximab, including 26.8% complete responses (CR), compared with 8.2%. Independent reviewers decreed an ORR of 92.3% for venentoclax, vs. 72.3% for bendamustine, including respective CR rates of 8.2% and 3.6%.
The investigators also found that the percentage of MRD negativity was higher with venetoclax/rituximab, with 62% of patients in this group being MRD negative at 9 months. This rate remained fairly constant at 12-, 15- and 18-month follow-ups (60%, 57%, and 60%, respectively).
In contrast, 13% of patients treated with bendamustine were MRD negative at 9 months, and the rates gradually declined over time to 10%, 9%, and 5%.
Investigators also saw a clinically meaningful improvement in overall survival with the venetoclax/rituximab duo, although survival data are still not mature in this ongoing trial. The median OS had not been reached in either group at the time of data cutoff.
Respective 1- and 2-year OS rates with venetoclax were 95.9% and 91.9%, and with bendamustine were 91.1% and 86.6%.
At the time of this interim analysis, the hazard ratio favoring venetoclax/rituximab was 0.48 (P = .0186).
Drug discontinuation was more frequent with venetoclax/rituximab (25% vs, 17%), with disease progression and adverse events without progression being the most frequent reasons for stopping in each arm.
Serious adverse events occurred in 46% of patients on venetoclax/rituximab and 43% on bendamustine/rituximab. A higher percentage of patients on venetoclax/rituximab had grade 3 or 4 adverse events (82% vs, 70%). Ten patients (5%) in the venetoclax/rituximab arm died, and 11 patients (6%) on bendamustine/rituximab died.
Events with a greater than 2% difference included more frequent neutropenia, tumor lysis syndrome, hyperglycemia and hypogammaglobulinema with venetoclax/rituximab, and more frequent anemia, thrombocytopenia, febrile neutropenia, pneumonia, infusion-related reactions, and hypotension with bendamustine/rituximab.
In the question-and-response portion following Dr. Seymour’s presentation, an audience member commented that the continuation of venetoclax/rituximab beyond the initial treatment cycles amounted to a maintenance strategy, and that patients in the experimental arm were in treatment longer, which likely influenced the results.
“You’re absolutely correct that the treatment duration differed, although, of course, the capacity to deliver more than six cycles of bendamustine/rituximab would have been problematic,” Dr. Seymour replied.
“There are some data that antibody treatment may prolong progression-free survival. However, when this study was designed in 2013 that data was certainly not available, and I believe currently even maintenance antibodies are not an accepted standard of treatment,” he added.
The MURANO trial was funded by AbbVie and Genentech. Dr. Seymour disclosed honoraria, speakers bureau, research funding, and advisory activities with AbbVie and other companies.
SOURCE: Seymour J et al. ASH 2017 LBA-2.
ATLANTA – In patients with relapsed/refractory chronic lymphocytic leukemia (CLL), a combination of venetoclax (Venclexta) and rituximab was superior to bendamustine (Treanda) and rituximab for prolonging progression-free survival (PFS), with effects consistent across subgroups, regardless of mutational status, and a clinically meaningful improvement in overall survival.
An interim analysis from the phase 3 MURANO trial showed that after a median follow-up of 23.8 months, the median PFS for patients randomized to venetoclax/rituximab had not been reached, compared with 17 months for patients assigned to bendamustine/rituximab, reported John F. Seymour, MBBS, PhD, of the Peter MacCallum Cancer Centre at the University of Melbourne.
Relapsed/refractory CLL often has a suboptimal response to conventional chemotherapy because of adverse biological features that can accumulate in cells, he said.
The combination of bendamustine and rituximab has been associated with about 60% overall responses rates, a median PFS of approximately 15 months, and overall survival of nearly 3 years in patients with CLL, he noted.
The rationale for pairing venetoclax with rituximab in this population comes from evidence showing efficacy of the monoclonal antibody, an oral B-cell lymphoma–2 (BCL-2) inhibitor, as monotherapy in patients with relapsed/refractory CLL, including those with poor prognostic features such as the 17p deletion (del17p).
Dr. Seymour and his colleagues recently published results from a phase 1b trial of venetoclax/rituximab in patients with relapsed/refractory CLL. The combination was associated with a 51% complete response rate, and a 28% rate of negative marrow minimal residual disease (MRD) (Lancet Oncol. 2017 Feb;18[2]:230-40)
In the MURANO study (NCT02005471), the investigators evaluated whether time-limited therapy with venetoclax/rituximab could improve PFS over bendamustine/rituximab.
Patients 18 and older with CLL who had been treated with one to three prior lines of therapy, including at least one chemotherapy-containing regimen, were enrolled. Prior treatment with bendamustine was allowed only if patients had had a duration of response of at least 24 months.
After stratification by del17p status, responsiveness to prior therapy, and geographic region, 389 patients were randomly assigned to receive rituximab 375 mg/m2 on day 1 of cycle 1 and 500 mg/m2 on day 1 of cycles 2 through 6, plus either bendamustine 70 mg/m2 on days 1 and 2 of each of six cycles, or venetoclax 400 mg orally once daily until disease progression, cessation for toxicity, or up to a maximum of 2 years starting from day 1 of cycle 1.
As noted,
The respective 1- and 2-year PFS rates with venetoclax were 91.2% and 82.8%, compared with 74.1% and 37.4% with bendamustine.
The venetoclax/rituximab combination was also significantly superior across all subgroups, regardless of the number of prior therapies, refractory vs. relapsed after most recent prior therapy, del17p status, TP53 mutational status, or baseline immunoglobulin heavy chain variable (IGHV) mutated or unmutated status
Response rates assessed by both investigators and independent reviewers were also better with venetoclax. The investigator-assessed overall response rate (ORR) was 93.3%, compared with 67.7% for bendamustine/rituximab, including 26.8% complete responses (CR), compared with 8.2%. Independent reviewers decreed an ORR of 92.3% for venentoclax, vs. 72.3% for bendamustine, including respective CR rates of 8.2% and 3.6%.
The investigators also found that the percentage of MRD negativity was higher with venetoclax/rituximab, with 62% of patients in this group being MRD negative at 9 months. This rate remained fairly constant at 12-, 15- and 18-month follow-ups (60%, 57%, and 60%, respectively).
In contrast, 13% of patients treated with bendamustine were MRD negative at 9 months, and the rates gradually declined over time to 10%, 9%, and 5%.
Investigators also saw a clinically meaningful improvement in overall survival with the venetoclax/rituximab duo, although survival data are still not mature in this ongoing trial. The median OS had not been reached in either group at the time of data cutoff.
Respective 1- and 2-year OS rates with venetoclax were 95.9% and 91.9%, and with bendamustine were 91.1% and 86.6%.
At the time of this interim analysis, the hazard ratio favoring venetoclax/rituximab was 0.48 (P = .0186).
Drug discontinuation was more frequent with venetoclax/rituximab (25% vs, 17%), with disease progression and adverse events without progression being the most frequent reasons for stopping in each arm.
Serious adverse events occurred in 46% of patients on venetoclax/rituximab and 43% on bendamustine/rituximab. A higher percentage of patients on venetoclax/rituximab had grade 3 or 4 adverse events (82% vs, 70%). Ten patients (5%) in the venetoclax/rituximab arm died, and 11 patients (6%) on bendamustine/rituximab died.
Events with a greater than 2% difference included more frequent neutropenia, tumor lysis syndrome, hyperglycemia and hypogammaglobulinema with venetoclax/rituximab, and more frequent anemia, thrombocytopenia, febrile neutropenia, pneumonia, infusion-related reactions, and hypotension with bendamustine/rituximab.
In the question-and-response portion following Dr. Seymour’s presentation, an audience member commented that the continuation of venetoclax/rituximab beyond the initial treatment cycles amounted to a maintenance strategy, and that patients in the experimental arm were in treatment longer, which likely influenced the results.
“You’re absolutely correct that the treatment duration differed, although, of course, the capacity to deliver more than six cycles of bendamustine/rituximab would have been problematic,” Dr. Seymour replied.
“There are some data that antibody treatment may prolong progression-free survival. However, when this study was designed in 2013 that data was certainly not available, and I believe currently even maintenance antibodies are not an accepted standard of treatment,” he added.
The MURANO trial was funded by AbbVie and Genentech. Dr. Seymour disclosed honoraria, speakers bureau, research funding, and advisory activities with AbbVie and other companies.
SOURCE: Seymour J et al. ASH 2017 LBA-2.
ATLANTA – In patients with relapsed/refractory chronic lymphocytic leukemia (CLL), a combination of venetoclax (Venclexta) and rituximab was superior to bendamustine (Treanda) and rituximab for prolonging progression-free survival (PFS), with effects consistent across subgroups, regardless of mutational status, and a clinically meaningful improvement in overall survival.
An interim analysis from the phase 3 MURANO trial showed that after a median follow-up of 23.8 months, the median PFS for patients randomized to venetoclax/rituximab had not been reached, compared with 17 months for patients assigned to bendamustine/rituximab, reported John F. Seymour, MBBS, PhD, of the Peter MacCallum Cancer Centre at the University of Melbourne.
Relapsed/refractory CLL often has a suboptimal response to conventional chemotherapy because of adverse biological features that can accumulate in cells, he said.
The combination of bendamustine and rituximab has been associated with about 60% overall responses rates, a median PFS of approximately 15 months, and overall survival of nearly 3 years in patients with CLL, he noted.
The rationale for pairing venetoclax with rituximab in this population comes from evidence showing efficacy of the monoclonal antibody, an oral B-cell lymphoma–2 (BCL-2) inhibitor, as monotherapy in patients with relapsed/refractory CLL, including those with poor prognostic features such as the 17p deletion (del17p).
Dr. Seymour and his colleagues recently published results from a phase 1b trial of venetoclax/rituximab in patients with relapsed/refractory CLL. The combination was associated with a 51% complete response rate, and a 28% rate of negative marrow minimal residual disease (MRD) (Lancet Oncol. 2017 Feb;18[2]:230-40)
In the MURANO study (NCT02005471), the investigators evaluated whether time-limited therapy with venetoclax/rituximab could improve PFS over bendamustine/rituximab.
Patients 18 and older with CLL who had been treated with one to three prior lines of therapy, including at least one chemotherapy-containing regimen, were enrolled. Prior treatment with bendamustine was allowed only if patients had had a duration of response of at least 24 months.
After stratification by del17p status, responsiveness to prior therapy, and geographic region, 389 patients were randomly assigned to receive rituximab 375 mg/m2 on day 1 of cycle 1 and 500 mg/m2 on day 1 of cycles 2 through 6, plus either bendamustine 70 mg/m2 on days 1 and 2 of each of six cycles, or venetoclax 400 mg orally once daily until disease progression, cessation for toxicity, or up to a maximum of 2 years starting from day 1 of cycle 1.
As noted,
The respective 1- and 2-year PFS rates with venetoclax were 91.2% and 82.8%, compared with 74.1% and 37.4% with bendamustine.
The venetoclax/rituximab combination was also significantly superior across all subgroups, regardless of the number of prior therapies, refractory vs. relapsed after most recent prior therapy, del17p status, TP53 mutational status, or baseline immunoglobulin heavy chain variable (IGHV) mutated or unmutated status
Response rates assessed by both investigators and independent reviewers were also better with venetoclax. The investigator-assessed overall response rate (ORR) was 93.3%, compared with 67.7% for bendamustine/rituximab, including 26.8% complete responses (CR), compared with 8.2%. Independent reviewers decreed an ORR of 92.3% for venentoclax, vs. 72.3% for bendamustine, including respective CR rates of 8.2% and 3.6%.
The investigators also found that the percentage of MRD negativity was higher with venetoclax/rituximab, with 62% of patients in this group being MRD negative at 9 months. This rate remained fairly constant at 12-, 15- and 18-month follow-ups (60%, 57%, and 60%, respectively).
In contrast, 13% of patients treated with bendamustine were MRD negative at 9 months, and the rates gradually declined over time to 10%, 9%, and 5%.
Investigators also saw a clinically meaningful improvement in overall survival with the venetoclax/rituximab duo, although survival data are still not mature in this ongoing trial. The median OS had not been reached in either group at the time of data cutoff.
Respective 1- and 2-year OS rates with venetoclax were 95.9% and 91.9%, and with bendamustine were 91.1% and 86.6%.
At the time of this interim analysis, the hazard ratio favoring venetoclax/rituximab was 0.48 (P = .0186).
Drug discontinuation was more frequent with venetoclax/rituximab (25% vs, 17%), with disease progression and adverse events without progression being the most frequent reasons for stopping in each arm.
Serious adverse events occurred in 46% of patients on venetoclax/rituximab and 43% on bendamustine/rituximab. A higher percentage of patients on venetoclax/rituximab had grade 3 or 4 adverse events (82% vs, 70%). Ten patients (5%) in the venetoclax/rituximab arm died, and 11 patients (6%) on bendamustine/rituximab died.
Events with a greater than 2% difference included more frequent neutropenia, tumor lysis syndrome, hyperglycemia and hypogammaglobulinema with venetoclax/rituximab, and more frequent anemia, thrombocytopenia, febrile neutropenia, pneumonia, infusion-related reactions, and hypotension with bendamustine/rituximab.
In the question-and-response portion following Dr. Seymour’s presentation, an audience member commented that the continuation of venetoclax/rituximab beyond the initial treatment cycles amounted to a maintenance strategy, and that patients in the experimental arm were in treatment longer, which likely influenced the results.
“You’re absolutely correct that the treatment duration differed, although, of course, the capacity to deliver more than six cycles of bendamustine/rituximab would have been problematic,” Dr. Seymour replied.
“There are some data that antibody treatment may prolong progression-free survival. However, when this study was designed in 2013 that data was certainly not available, and I believe currently even maintenance antibodies are not an accepted standard of treatment,” he added.
The MURANO trial was funded by AbbVie and Genentech. Dr. Seymour disclosed honoraria, speakers bureau, research funding, and advisory activities with AbbVie and other companies.
SOURCE: Seymour J et al. ASH 2017 LBA-2.
REPORTING FROM ASH 2017
Key clinical point: Compared with bendamustine/rituximab, venetoclax/rituximab was associated with significantly superior progression-free survival of relapsed/refractory chronic lymphocytic leukemia.
Major finding: The hazard ratio for PFS with venetoclax/rituximab was 0.17 (P less than .001).
Data source: A randomized phase 3, open-label trial in 389 patients with relapsed/refractory CLL.
Disclosures: The MURANO trial was funded by AbbVie and Genetech. Dr. Seymour disclosed honoraria, speakers bureau, research funding, and advisory activities with AbbVie and other companies.
Source: Seymour J et al. ASH 2017 LBA-2.
Lapatinib plus trastuzumab improves outcomes in HER2+ breast cancer
SAN ANTONIO – Dual HER2 targeting with lapatinib added to 16 weeks of trastuzumab significantly improved event-free survival (EFS), compared with trastuzumab alone, among women with HER2-positive breast cancer.
EFS was significantly longer in the combination arm versus single-blockade therapy (hazard ratio, 0.35; 95% confidence interval, 0.15-0.84; P = .013).
“Overall survival was also increased, but the number of events was very small,” said study author Ian E. Krop, MD, of the Dana-Farber Cancer Institute in Boston, who presented the study results at the San Antonio Breast Cancer Symposium.
As expected, and consistent with other studies, the pathologic complete response (pCR) rate with dual blockade was associated with favorable long-term outcomes, and the effect was most pronounced in HR- and HER2 enriched cancers.
However, Dr. Krop cautioned that these results need to be seen in the context of two larger trials that failed to find a benefit for dual-blockade HER2 therapy in either the adjuvant or neoadjuvant setting.
Previous research has shown that trastuzumab and lapatinib are synergistic, potently inhibiting HER2 signaling in preclinical trials. The combination of both agents is also active in both untreated and heavily pretreated HER2+ advanced breast cancer. The addition of lapatinib to trastuzumab plus chemotherapy has also been associated with a pCR rate that was statistically significant in many studies.
But despite this “wealth of evidence,” the largest of these studies, the phase 3 ALTTO study, failed to show a benefit for dual HER2 blockade. The same was true for the neoadjuvant Neo ALTTO study. Both studies failed to show an improvement in EFS or overall survival.
The current trial, CALGB 40601, a randomized phase 3 trial, assessed the effect of dual HER2 blockade consisting of trastuzumab and lapatinib added to paclitaxel on rates of pCR, and the researchers also looked at tumor and microenvironment molecular features. Dr. Krop noted that they had previously found that rates of pCR were numerically but not significantly increased with combination-blockade therapy; they had also found that the tumor molecular subtype and evidence of immune activation were independent factors affecting rates of pCR.
Dr. Krop presented results from a secondary analysis in which they evaluated the effects of treatment arm and gene expression–defined subgroups on EFS.
A total of 305 patients with stage II and III HER2+ breast cancer were randomized to receive 16 weeks of either paclitaxel plus trastuzumab alone or that combination with lapatinib as well before undergoing surgery. Evaluable information regarding EFS and RNA-sequencing gene expression was available for 265 patients.
“Despite the lack of significant benefits in other studies, we did demonstrate an improvement in EFS,” said Dr. Krop, noting that it was more pronounced in the subset of HR- patients (HR, 0.12; P = .0160).
EFS was also significantly longer among those who achieved pCR than those who didn’t (HR, 0.34; P = .0032).
In an exploratory analysis, Dr. Krop and his colleagues assessed whether there was a difference in rates of pCR by subtype, and they found that luminal A and luminal B did not have much of an effect on rates of pCR. However, patients with HER2-enriched disease who had a pCR had significantly better EFS than those who did not (HR, 0.14; 95% CI, 0.04-0.44; P less than .0001).
Conversely, patients with HER2-enriched disease who did not achieve pCR had a substantial risk of recurrence.
When looking at EFS by intrinsic subtype, the addition of lapatinib had the most effect on luminal A cancers. “This shows that there was some discordance between analyses by pCR and by long-term endpoint,” Dr. Krop said.
Finally, among gene expression signatures, only immune activation measured by an immunoglobulin G signature was associated with an improvement in EFS (HR, 0.70; 95% CI, 0.50-0.98; P = .04).
“These data are hypothesis generating and require validation,” Dr. Krop concluded. He added that a combined analysis of this trial, along with the Neo ALTTO and others, is planned.
SOURCE: Krop IE et al. SABCS 2017 Abstract GS3-02.
SAN ANTONIO – Dual HER2 targeting with lapatinib added to 16 weeks of trastuzumab significantly improved event-free survival (EFS), compared with trastuzumab alone, among women with HER2-positive breast cancer.
EFS was significantly longer in the combination arm versus single-blockade therapy (hazard ratio, 0.35; 95% confidence interval, 0.15-0.84; P = .013).
“Overall survival was also increased, but the number of events was very small,” said study author Ian E. Krop, MD, of the Dana-Farber Cancer Institute in Boston, who presented the study results at the San Antonio Breast Cancer Symposium.
As expected, and consistent with other studies, the pathologic complete response (pCR) rate with dual blockade was associated with favorable long-term outcomes, and the effect was most pronounced in HR- and HER2 enriched cancers.
However, Dr. Krop cautioned that these results need to be seen in the context of two larger trials that failed to find a benefit for dual-blockade HER2 therapy in either the adjuvant or neoadjuvant setting.
Previous research has shown that trastuzumab and lapatinib are synergistic, potently inhibiting HER2 signaling in preclinical trials. The combination of both agents is also active in both untreated and heavily pretreated HER2+ advanced breast cancer. The addition of lapatinib to trastuzumab plus chemotherapy has also been associated with a pCR rate that was statistically significant in many studies.
But despite this “wealth of evidence,” the largest of these studies, the phase 3 ALTTO study, failed to show a benefit for dual HER2 blockade. The same was true for the neoadjuvant Neo ALTTO study. Both studies failed to show an improvement in EFS or overall survival.
The current trial, CALGB 40601, a randomized phase 3 trial, assessed the effect of dual HER2 blockade consisting of trastuzumab and lapatinib added to paclitaxel on rates of pCR, and the researchers also looked at tumor and microenvironment molecular features. Dr. Krop noted that they had previously found that rates of pCR were numerically but not significantly increased with combination-blockade therapy; they had also found that the tumor molecular subtype and evidence of immune activation were independent factors affecting rates of pCR.
Dr. Krop presented results from a secondary analysis in which they evaluated the effects of treatment arm and gene expression–defined subgroups on EFS.
A total of 305 patients with stage II and III HER2+ breast cancer were randomized to receive 16 weeks of either paclitaxel plus trastuzumab alone or that combination with lapatinib as well before undergoing surgery. Evaluable information regarding EFS and RNA-sequencing gene expression was available for 265 patients.
“Despite the lack of significant benefits in other studies, we did demonstrate an improvement in EFS,” said Dr. Krop, noting that it was more pronounced in the subset of HR- patients (HR, 0.12; P = .0160).
EFS was also significantly longer among those who achieved pCR than those who didn’t (HR, 0.34; P = .0032).
In an exploratory analysis, Dr. Krop and his colleagues assessed whether there was a difference in rates of pCR by subtype, and they found that luminal A and luminal B did not have much of an effect on rates of pCR. However, patients with HER2-enriched disease who had a pCR had significantly better EFS than those who did not (HR, 0.14; 95% CI, 0.04-0.44; P less than .0001).
Conversely, patients with HER2-enriched disease who did not achieve pCR had a substantial risk of recurrence.
When looking at EFS by intrinsic subtype, the addition of lapatinib had the most effect on luminal A cancers. “This shows that there was some discordance between analyses by pCR and by long-term endpoint,” Dr. Krop said.
Finally, among gene expression signatures, only immune activation measured by an immunoglobulin G signature was associated with an improvement in EFS (HR, 0.70; 95% CI, 0.50-0.98; P = .04).
“These data are hypothesis generating and require validation,” Dr. Krop concluded. He added that a combined analysis of this trial, along with the Neo ALTTO and others, is planned.
SOURCE: Krop IE et al. SABCS 2017 Abstract GS3-02.
SAN ANTONIO – Dual HER2 targeting with lapatinib added to 16 weeks of trastuzumab significantly improved event-free survival (EFS), compared with trastuzumab alone, among women with HER2-positive breast cancer.
EFS was significantly longer in the combination arm versus single-blockade therapy (hazard ratio, 0.35; 95% confidence interval, 0.15-0.84; P = .013).
“Overall survival was also increased, but the number of events was very small,” said study author Ian E. Krop, MD, of the Dana-Farber Cancer Institute in Boston, who presented the study results at the San Antonio Breast Cancer Symposium.
As expected, and consistent with other studies, the pathologic complete response (pCR) rate with dual blockade was associated with favorable long-term outcomes, and the effect was most pronounced in HR- and HER2 enriched cancers.
However, Dr. Krop cautioned that these results need to be seen in the context of two larger trials that failed to find a benefit for dual-blockade HER2 therapy in either the adjuvant or neoadjuvant setting.
Previous research has shown that trastuzumab and lapatinib are synergistic, potently inhibiting HER2 signaling in preclinical trials. The combination of both agents is also active in both untreated and heavily pretreated HER2+ advanced breast cancer. The addition of lapatinib to trastuzumab plus chemotherapy has also been associated with a pCR rate that was statistically significant in many studies.
But despite this “wealth of evidence,” the largest of these studies, the phase 3 ALTTO study, failed to show a benefit for dual HER2 blockade. The same was true for the neoadjuvant Neo ALTTO study. Both studies failed to show an improvement in EFS or overall survival.
The current trial, CALGB 40601, a randomized phase 3 trial, assessed the effect of dual HER2 blockade consisting of trastuzumab and lapatinib added to paclitaxel on rates of pCR, and the researchers also looked at tumor and microenvironment molecular features. Dr. Krop noted that they had previously found that rates of pCR were numerically but not significantly increased with combination-blockade therapy; they had also found that the tumor molecular subtype and evidence of immune activation were independent factors affecting rates of pCR.
Dr. Krop presented results from a secondary analysis in which they evaluated the effects of treatment arm and gene expression–defined subgroups on EFS.
A total of 305 patients with stage II and III HER2+ breast cancer were randomized to receive 16 weeks of either paclitaxel plus trastuzumab alone or that combination with lapatinib as well before undergoing surgery. Evaluable information regarding EFS and RNA-sequencing gene expression was available for 265 patients.
“Despite the lack of significant benefits in other studies, we did demonstrate an improvement in EFS,” said Dr. Krop, noting that it was more pronounced in the subset of HR- patients (HR, 0.12; P = .0160).
EFS was also significantly longer among those who achieved pCR than those who didn’t (HR, 0.34; P = .0032).
In an exploratory analysis, Dr. Krop and his colleagues assessed whether there was a difference in rates of pCR by subtype, and they found that luminal A and luminal B did not have much of an effect on rates of pCR. However, patients with HER2-enriched disease who had a pCR had significantly better EFS than those who did not (HR, 0.14; 95% CI, 0.04-0.44; P less than .0001).
Conversely, patients with HER2-enriched disease who did not achieve pCR had a substantial risk of recurrence.
When looking at EFS by intrinsic subtype, the addition of lapatinib had the most effect on luminal A cancers. “This shows that there was some discordance between analyses by pCR and by long-term endpoint,” Dr. Krop said.
Finally, among gene expression signatures, only immune activation measured by an immunoglobulin G signature was associated with an improvement in EFS (HR, 0.70; 95% CI, 0.50-0.98; P = .04).
“These data are hypothesis generating and require validation,” Dr. Krop concluded. He added that a combined analysis of this trial, along with the Neo ALTTO and others, is planned.
SOURCE: Krop IE et al. SABCS 2017 Abstract GS3-02.
REPORTING FROM SABCS 2017
Key clinical point: In contrast to previous trials,
Major finding: Event-free survival was significantly longer in the dual HER2 blockade arm versus trastuzumab alone (HR, 0.35; 95% CI, 0.15-0.84; P = .013).
Data source: Phase 3 randomized trial that included 305 women with HER2+ breast cancer.
Disclosures: The study was sponsored by the Alliance for Clinical Trials in Oncology. Dr. Krop reports relationships with Genentech/Roche and MacroGenics.
Source: Krop IE et al. SABCS 2017 Abstract GS3-02.
FDA updates nilotinib product label outlining criteria for discontinuation
for patients who meet certain criteria. Nilotinib, a kinase inhibitor that blocks the BCR-ABL protein that promotes abnormal cell growth, was originally approved in 2007 and was indicated for use in patients with Philadelphia chromosome positive (Ph+) chronic myeloid leukemia (CML). In accordance with the new label update, patients who have early phase CML, have been using nilotinib for 3 years or more, and whose leukemia has responded to treatment according to a test that has received FDA marketing authorization, may be eligible to discontinue use of nilotinib.
The information that led to the FDA approved label update was based on two single-arm trials of patients with chronic phase Ph+ CML. The trial measured the length of time patients were able to discontinue use of nilotinib without leukemia returning, and who had entered treatment-free remission (TFR). In the first trial, among 190 newly diagnosed CML patients who discontinued taking nilotinib after using the drug for 3 or more years, 51.6% were still in TFR after about 1 year (48 weeks) and 48.9% were still in TFR after nearly 2 years (96 weeks). Similar results were seen in the second trial, among 126 patients, with 57.9% in TFR after about a year (48 weeks) and 53.2% in TFR after approximately 2 years (96 weeks).
An important element of these trials was regular monitoring of specific RNA information that specifies the level of BCR-ABL protein in the blood using a diagnostic test that has received FDA marketing authorization. Monitoring with a test that accurately detects the reductions of RNA information in the blood with accuracy and precision is critical in discontinuing the use of nilotinib. This monitoring will allow physicians to detect the first signs of relapse.
“Patients diagnosed with CML generally face a lifetime of treatment to keep their leukemia from growing or recurring,” said Richard Pazdur, MD, acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval shows that some patients may be able to stop treatment with Tasigna altogether if they are showing a strong response to therapy. While we welcome this progress in patient care, it’s important to note that any discontinuation of treatment still means patients must be regularly monitored for disease recurrence,” Dr. Pazdur said in the FDA statement.
Common side effects after discontinuing use of nilotinib include body aches and pain in the bones and extremities. Severe side effects of taking nilotinib can include myelosuppression, blockages in the heart and arteries, and inflammation of the pancreas. Severe liver damage can also occur.
Severe side effects typically associated with nilotinib administration occurred less frequently in patients who discontinued the drug. However, the long-term outcomes of patients discontinuing versus continuing treatment are unknown at this time, the FDA noted.
for patients who meet certain criteria. Nilotinib, a kinase inhibitor that blocks the BCR-ABL protein that promotes abnormal cell growth, was originally approved in 2007 and was indicated for use in patients with Philadelphia chromosome positive (Ph+) chronic myeloid leukemia (CML). In accordance with the new label update, patients who have early phase CML, have been using nilotinib for 3 years or more, and whose leukemia has responded to treatment according to a test that has received FDA marketing authorization, may be eligible to discontinue use of nilotinib.
The information that led to the FDA approved label update was based on two single-arm trials of patients with chronic phase Ph+ CML. The trial measured the length of time patients were able to discontinue use of nilotinib without leukemia returning, and who had entered treatment-free remission (TFR). In the first trial, among 190 newly diagnosed CML patients who discontinued taking nilotinib after using the drug for 3 or more years, 51.6% were still in TFR after about 1 year (48 weeks) and 48.9% were still in TFR after nearly 2 years (96 weeks). Similar results were seen in the second trial, among 126 patients, with 57.9% in TFR after about a year (48 weeks) and 53.2% in TFR after approximately 2 years (96 weeks).
An important element of these trials was regular monitoring of specific RNA information that specifies the level of BCR-ABL protein in the blood using a diagnostic test that has received FDA marketing authorization. Monitoring with a test that accurately detects the reductions of RNA information in the blood with accuracy and precision is critical in discontinuing the use of nilotinib. This monitoring will allow physicians to detect the first signs of relapse.
“Patients diagnosed with CML generally face a lifetime of treatment to keep their leukemia from growing or recurring,” said Richard Pazdur, MD, acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval shows that some patients may be able to stop treatment with Tasigna altogether if they are showing a strong response to therapy. While we welcome this progress in patient care, it’s important to note that any discontinuation of treatment still means patients must be regularly monitored for disease recurrence,” Dr. Pazdur said in the FDA statement.
Common side effects after discontinuing use of nilotinib include body aches and pain in the bones and extremities. Severe side effects of taking nilotinib can include myelosuppression, blockages in the heart and arteries, and inflammation of the pancreas. Severe liver damage can also occur.
Severe side effects typically associated with nilotinib administration occurred less frequently in patients who discontinued the drug. However, the long-term outcomes of patients discontinuing versus continuing treatment are unknown at this time, the FDA noted.
for patients who meet certain criteria. Nilotinib, a kinase inhibitor that blocks the BCR-ABL protein that promotes abnormal cell growth, was originally approved in 2007 and was indicated for use in patients with Philadelphia chromosome positive (Ph+) chronic myeloid leukemia (CML). In accordance with the new label update, patients who have early phase CML, have been using nilotinib for 3 years or more, and whose leukemia has responded to treatment according to a test that has received FDA marketing authorization, may be eligible to discontinue use of nilotinib.
The information that led to the FDA approved label update was based on two single-arm trials of patients with chronic phase Ph+ CML. The trial measured the length of time patients were able to discontinue use of nilotinib without leukemia returning, and who had entered treatment-free remission (TFR). In the first trial, among 190 newly diagnosed CML patients who discontinued taking nilotinib after using the drug for 3 or more years, 51.6% were still in TFR after about 1 year (48 weeks) and 48.9% were still in TFR after nearly 2 years (96 weeks). Similar results were seen in the second trial, among 126 patients, with 57.9% in TFR after about a year (48 weeks) and 53.2% in TFR after approximately 2 years (96 weeks).
An important element of these trials was regular monitoring of specific RNA information that specifies the level of BCR-ABL protein in the blood using a diagnostic test that has received FDA marketing authorization. Monitoring with a test that accurately detects the reductions of RNA information in the blood with accuracy and precision is critical in discontinuing the use of nilotinib. This monitoring will allow physicians to detect the first signs of relapse.
“Patients diagnosed with CML generally face a lifetime of treatment to keep their leukemia from growing or recurring,” said Richard Pazdur, MD, acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval shows that some patients may be able to stop treatment with Tasigna altogether if they are showing a strong response to therapy. While we welcome this progress in patient care, it’s important to note that any discontinuation of treatment still means patients must be regularly monitored for disease recurrence,” Dr. Pazdur said in the FDA statement.
Common side effects after discontinuing use of nilotinib include body aches and pain in the bones and extremities. Severe side effects of taking nilotinib can include myelosuppression, blockages in the heart and arteries, and inflammation of the pancreas. Severe liver damage can also occur.
Severe side effects typically associated with nilotinib administration occurred less frequently in patients who discontinued the drug. However, the long-term outcomes of patients discontinuing versus continuing treatment are unknown at this time, the FDA noted.
FDA approves raltegravir for newborns at risk for HIV-1 infection
The Food and Drug Administration has approved the use of raltegravir (Isentress), in combination with other antiretrovirals, for HIV-1 exposed newborns who weigh at least 2 kg and are at high risk for acquiring HIV-1 infection from their mothers, according to a press release from Merck, the manufacturer of Isentress.
“With this FDA approval, Isentress becomes the only integrase inhibitor approved in the U.S. for the treatment of HIV-1, in combination with other antiretroviral agents, for neonates weighing at least 2 kg,” Eliav Barr, MD, senior vice president, global clinical development, infectious diseases and vaccines, Merck Research Laboratories, said in a statement.

The study, IMPAACT P1110, was composed of two cohorts receiving different doses of raltegravir at different time intervals. The first cohort, of 16 newborns, received two single doses of raltegravir: the first within 48 hours of birth and the second between 7 and 10 days of age. The second cohort of 26 newborns received daily doses based on weight for 6 weeks. After observation for 24 weeks, all infants were HIV-1 negative.
Raltegravir does not cure HIV-1 infection or AIDS and severe side effects have been reported, including hypersensitivity reaction and toxic epidermal necrolysis, according to the Merck release.
Raltegravir should not be used in newborns and infants weighing under 2 kg. If raltegravir has been administered to the mother 2-24 hours before delivery, the first dose for the newborn should be given between 24 and 48 hours after birth.
The Food and Drug Administration has approved the use of raltegravir (Isentress), in combination with other antiretrovirals, for HIV-1 exposed newborns who weigh at least 2 kg and are at high risk for acquiring HIV-1 infection from their mothers, according to a press release from Merck, the manufacturer of Isentress.
“With this FDA approval, Isentress becomes the only integrase inhibitor approved in the U.S. for the treatment of HIV-1, in combination with other antiretroviral agents, for neonates weighing at least 2 kg,” Eliav Barr, MD, senior vice president, global clinical development, infectious diseases and vaccines, Merck Research Laboratories, said in a statement.
The study, IMPAACT P1110, was composed of two cohorts receiving different doses of raltegravir at different time intervals. The first cohort, of 16 newborns, received two single doses of raltegravir: the first within 48 hours of birth and the second between 7 and 10 days of age. The second cohort of 26 newborns received daily doses based on weight for 6 weeks. After observation for 24 weeks, all infants were HIV-1 negative.
Raltegravir does not cure HIV-1 infection or AIDS and severe side effects have been reported, including hypersensitivity reaction and toxic epidermal necrolysis, according to the Merck release.
Raltegravir should not be used in newborns and infants weighing under 2 kg. If raltegravir has been administered to the mother 2-24 hours before delivery, the first dose for the newborn should be given between 24 and 48 hours after birth.
The Food and Drug Administration has approved the use of raltegravir (Isentress), in combination with other antiretrovirals, for HIV-1 exposed newborns who weigh at least 2 kg and are at high risk for acquiring HIV-1 infection from their mothers, according to a press release from Merck, the manufacturer of Isentress.
“With this FDA approval, Isentress becomes the only integrase inhibitor approved in the U.S. for the treatment of HIV-1, in combination with other antiretroviral agents, for neonates weighing at least 2 kg,” Eliav Barr, MD, senior vice president, global clinical development, infectious diseases and vaccines, Merck Research Laboratories, said in a statement.
The study, IMPAACT P1110, was composed of two cohorts receiving different doses of raltegravir at different time intervals. The first cohort, of 16 newborns, received two single doses of raltegravir: the first within 48 hours of birth and the second between 7 and 10 days of age. The second cohort of 26 newborns received daily doses based on weight for 6 weeks. After observation for 24 weeks, all infants were HIV-1 negative.
Raltegravir does not cure HIV-1 infection or AIDS and severe side effects have been reported, including hypersensitivity reaction and toxic epidermal necrolysis, according to the Merck release.
Raltegravir should not be used in newborns and infants weighing under 2 kg. If raltegravir has been administered to the mother 2-24 hours before delivery, the first dose for the newborn should be given between 24 and 48 hours after birth.