User login
Urgent recall for Penumbra JET 7 Xtra Flex reperfusion catheters
“All users should stop using this device, and facilities should remove these devices from inventory,” the recall notice, posted on the U.S. Food and Drug Administration website, advises.
The recall covers the JET 7 Xtra Flex catheter, which was cleared for use in June 2019, and the JET 7MAX configuration (which includes the JET 7 Xtra Flex catheter and MAX delivery device), which was cleared in February of this year.
The recall does not apply to the Penumbra JET 7 reperfusion catheter with standard tip.
The FDA says it has received over 200 medical device reports (MDRs) associated with the JET 7 Xtra Flex catheter, including reports of deaths, serious injuries, and malfunctions.
Twenty of these MDRs describe 14 unique patient deaths. Other MDRs describe serious patient injury, such as vessel damage, hemorrhage, and cerebral infarction.
Device malfunctions described in the reports include ballooning, expansion, rupture, breakage or complete separation, and exposure of internal support coils near the distal tip region of the JET 7 Xtra Flex catheter.
According to the FDA, bench testing by the manufacturer, in which the catheter distal tip is plugged and pressurized to failure, indicates that the JET 7 Xtra Flex catheter is not able to withstand the same burst pressures to failure as the manufacturer’s other large-bore aspiration catheters used to remove thrombus for patients with acute ischemic stroke.
Penumbra’s urgent medical device recall letter advises health care providers and facilities to remove and quarantine all unused devices covered by this recall, to complete the product identification and return form, and to return all products to Penumbra in accordance with instructions provided.
For questions regarding this recall, contact Penumbra customer service by phone at 888-272-4606 or by email at notification@penumbrainc.com.
A version of this article first appeared on Medscape.com.
“All users should stop using this device, and facilities should remove these devices from inventory,” the recall notice, posted on the U.S. Food and Drug Administration website, advises.
The recall covers the JET 7 Xtra Flex catheter, which was cleared for use in June 2019, and the JET 7MAX configuration (which includes the JET 7 Xtra Flex catheter and MAX delivery device), which was cleared in February of this year.
The recall does not apply to the Penumbra JET 7 reperfusion catheter with standard tip.
The FDA says it has received over 200 medical device reports (MDRs) associated with the JET 7 Xtra Flex catheter, including reports of deaths, serious injuries, and malfunctions.
Twenty of these MDRs describe 14 unique patient deaths. Other MDRs describe serious patient injury, such as vessel damage, hemorrhage, and cerebral infarction.
Device malfunctions described in the reports include ballooning, expansion, rupture, breakage or complete separation, and exposure of internal support coils near the distal tip region of the JET 7 Xtra Flex catheter.
According to the FDA, bench testing by the manufacturer, in which the catheter distal tip is plugged and pressurized to failure, indicates that the JET 7 Xtra Flex catheter is not able to withstand the same burst pressures to failure as the manufacturer’s other large-bore aspiration catheters used to remove thrombus for patients with acute ischemic stroke.
Penumbra’s urgent medical device recall letter advises health care providers and facilities to remove and quarantine all unused devices covered by this recall, to complete the product identification and return form, and to return all products to Penumbra in accordance with instructions provided.
For questions regarding this recall, contact Penumbra customer service by phone at 888-272-4606 or by email at notification@penumbrainc.com.
A version of this article first appeared on Medscape.com.
“All users should stop using this device, and facilities should remove these devices from inventory,” the recall notice, posted on the U.S. Food and Drug Administration website, advises.
The recall covers the JET 7 Xtra Flex catheter, which was cleared for use in June 2019, and the JET 7MAX configuration (which includes the JET 7 Xtra Flex catheter and MAX delivery device), which was cleared in February of this year.
The recall does not apply to the Penumbra JET 7 reperfusion catheter with standard tip.
The FDA says it has received over 200 medical device reports (MDRs) associated with the JET 7 Xtra Flex catheter, including reports of deaths, serious injuries, and malfunctions.
Twenty of these MDRs describe 14 unique patient deaths. Other MDRs describe serious patient injury, such as vessel damage, hemorrhage, and cerebral infarction.
Device malfunctions described in the reports include ballooning, expansion, rupture, breakage or complete separation, and exposure of internal support coils near the distal tip region of the JET 7 Xtra Flex catheter.
According to the FDA, bench testing by the manufacturer, in which the catheter distal tip is plugged and pressurized to failure, indicates that the JET 7 Xtra Flex catheter is not able to withstand the same burst pressures to failure as the manufacturer’s other large-bore aspiration catheters used to remove thrombus for patients with acute ischemic stroke.
Penumbra’s urgent medical device recall letter advises health care providers and facilities to remove and quarantine all unused devices covered by this recall, to complete the product identification and return form, and to return all products to Penumbra in accordance with instructions provided.
For questions regarding this recall, contact Penumbra customer service by phone at 888-272-4606 or by email at notification@penumbrainc.com.
A version of this article first appeared on Medscape.com.
COVID-19 ranks as a leading cause of death in United States
Adults over age 45 were more likely to die from COVID-19 than car crashes, respiratory diseases, drug overdoses, and suicide. And those over age 55 faced even higher rates of dying because of the coronavirus.
“The current exponential increase in COVID-19 is reaching a calamitous scale in the U.S.,” the authors wrote. “Putting these numbers in perspective may be difficult.”
Population health researchers at Virginia Commonwealth University put COVID-19 deaths into context by comparing this year’s numbers to the leading causes of death for March through October 2018, sorting by age.
By October 2020, COVID-19 had become the third leading cause of death overall for those between the ages of 45 and 84 years, following after heart disease and cancer. For those over age 85, COVID-19 was the second leading cause of death, surpassing cancer and following behind heart disease.
For people aged 35-44 years, COVID-19 surpassed car crashes and respiratory diseases and was slightly lower than suicide, heart disease, and cancer. For those under age 35, drug overdoses, suicide, and car crashes remained the leading causes of death.
Importantly, the authors wrote, death rates for the two leading causes – heart disease and cancer – are about 1,700 and 1,600 per day, respectively. COVID-19 deaths have surpassed these numbers individually throughout December and, on Wednesday, beat them combined. More than 3,400 deaths were reported, according to the COVID Tracking Project, marking an all-time high that continues to increase. Hospitalizations were also at a new high, with more than 113,000 COVID-19 patients in hospitals across the country, and another 232,000 new cases were reported.
“With COVID-19 mortality rates now exceeding these thresholds, this infectious disease has become deadlier than heart disease and cancer,” the authors wrote. “Its lethality may increase further as transmission increases with holiday travel and gatherings and with the intensified indoor exposure that winter brings.”
The reported number of COVID-19 deaths is likely a 20% underestimate, they wrote, attributable to delays in reporting and an increase in non–COVID-19 deaths that were undetected and untreated because of pandemic-related disruptions. Since the coronavirus is communicable and spreads easily, COVID-19 deaths are particularly unique and worrying, they said.
“Individuals who die from homicide or cancer do not transmit the risk of morbidity and mortality to those nearby,” they wrote. “Every COVID-19 death signals the possibility of more deaths among close contacts.”
The fall surge in cases and deaths is widespread nationally, as compared to the spring, with hot spots on both coasts and in rural areas, according to an accompanying editorial in JAMA from public health researchers at the Harvard T.H. Chan School of Public Health, Boston. People of color have faced twice the death rate as well, with one in 875 Black people and one in 925 Indigenous people dying from COVID-19, as compared with one in 1,625 White people.
“The year 2020 ends with COVID-19 massively surging, as it was in the spring, to be the leading cause of death,” they wrote. “The accelerating numbers of deaths fall far short of fully capturing each devastating human story: Every death represents untold loss for countless families.”
Vaccines offer hope, they said, but won’t prevent the upcoming increase in COVID-19 hospitalizations and deaths this winter. In 2021, containing the pandemic will require national coordination, resources to help overwhelmed health care workers, new support for state and local public health officials, a stimulus package for schools and businesses, and financial aid for people on the brink of eviction. The country needs federal coordination of testing, contact tracing, personal protective equipment, travel precautions, and a face mask mandate, they wrote.
“Ending this crisis will require not only further advances in treatment but also unprecedented commitment to all aspects of prevention, vaccination, and public health,” they wrote. “Only by doing so can future years see this illness revert back to the unfamiliar and unknown condition it once was.”
This article first appeared on WebMD.com.
Adults over age 45 were more likely to die from COVID-19 than car crashes, respiratory diseases, drug overdoses, and suicide. And those over age 55 faced even higher rates of dying because of the coronavirus.
“The current exponential increase in COVID-19 is reaching a calamitous scale in the U.S.,” the authors wrote. “Putting these numbers in perspective may be difficult.”
Population health researchers at Virginia Commonwealth University put COVID-19 deaths into context by comparing this year’s numbers to the leading causes of death for March through October 2018, sorting by age.
By October 2020, COVID-19 had become the third leading cause of death overall for those between the ages of 45 and 84 years, following after heart disease and cancer. For those over age 85, COVID-19 was the second leading cause of death, surpassing cancer and following behind heart disease.
For people aged 35-44 years, COVID-19 surpassed car crashes and respiratory diseases and was slightly lower than suicide, heart disease, and cancer. For those under age 35, drug overdoses, suicide, and car crashes remained the leading causes of death.
Importantly, the authors wrote, death rates for the two leading causes – heart disease and cancer – are about 1,700 and 1,600 per day, respectively. COVID-19 deaths have surpassed these numbers individually throughout December and, on Wednesday, beat them combined. More than 3,400 deaths were reported, according to the COVID Tracking Project, marking an all-time high that continues to increase. Hospitalizations were also at a new high, with more than 113,000 COVID-19 patients in hospitals across the country, and another 232,000 new cases were reported.
“With COVID-19 mortality rates now exceeding these thresholds, this infectious disease has become deadlier than heart disease and cancer,” the authors wrote. “Its lethality may increase further as transmission increases with holiday travel and gatherings and with the intensified indoor exposure that winter brings.”
The reported number of COVID-19 deaths is likely a 20% underestimate, they wrote, attributable to delays in reporting and an increase in non–COVID-19 deaths that were undetected and untreated because of pandemic-related disruptions. Since the coronavirus is communicable and spreads easily, COVID-19 deaths are particularly unique and worrying, they said.
“Individuals who die from homicide or cancer do not transmit the risk of morbidity and mortality to those nearby,” they wrote. “Every COVID-19 death signals the possibility of more deaths among close contacts.”
The fall surge in cases and deaths is widespread nationally, as compared to the spring, with hot spots on both coasts and in rural areas, according to an accompanying editorial in JAMA from public health researchers at the Harvard T.H. Chan School of Public Health, Boston. People of color have faced twice the death rate as well, with one in 875 Black people and one in 925 Indigenous people dying from COVID-19, as compared with one in 1,625 White people.
“The year 2020 ends with COVID-19 massively surging, as it was in the spring, to be the leading cause of death,” they wrote. “The accelerating numbers of deaths fall far short of fully capturing each devastating human story: Every death represents untold loss for countless families.”
Vaccines offer hope, they said, but won’t prevent the upcoming increase in COVID-19 hospitalizations and deaths this winter. In 2021, containing the pandemic will require national coordination, resources to help overwhelmed health care workers, new support for state and local public health officials, a stimulus package for schools and businesses, and financial aid for people on the brink of eviction. The country needs federal coordination of testing, contact tracing, personal protective equipment, travel precautions, and a face mask mandate, they wrote.
“Ending this crisis will require not only further advances in treatment but also unprecedented commitment to all aspects of prevention, vaccination, and public health,” they wrote. “Only by doing so can future years see this illness revert back to the unfamiliar and unknown condition it once was.”
This article first appeared on WebMD.com.
Adults over age 45 were more likely to die from COVID-19 than car crashes, respiratory diseases, drug overdoses, and suicide. And those over age 55 faced even higher rates of dying because of the coronavirus.
“The current exponential increase in COVID-19 is reaching a calamitous scale in the U.S.,” the authors wrote. “Putting these numbers in perspective may be difficult.”
Population health researchers at Virginia Commonwealth University put COVID-19 deaths into context by comparing this year’s numbers to the leading causes of death for March through October 2018, sorting by age.
By October 2020, COVID-19 had become the third leading cause of death overall for those between the ages of 45 and 84 years, following after heart disease and cancer. For those over age 85, COVID-19 was the second leading cause of death, surpassing cancer and following behind heart disease.
For people aged 35-44 years, COVID-19 surpassed car crashes and respiratory diseases and was slightly lower than suicide, heart disease, and cancer. For those under age 35, drug overdoses, suicide, and car crashes remained the leading causes of death.
Importantly, the authors wrote, death rates for the two leading causes – heart disease and cancer – are about 1,700 and 1,600 per day, respectively. COVID-19 deaths have surpassed these numbers individually throughout December and, on Wednesday, beat them combined. More than 3,400 deaths were reported, according to the COVID Tracking Project, marking an all-time high that continues to increase. Hospitalizations were also at a new high, with more than 113,000 COVID-19 patients in hospitals across the country, and another 232,000 new cases were reported.
“With COVID-19 mortality rates now exceeding these thresholds, this infectious disease has become deadlier than heart disease and cancer,” the authors wrote. “Its lethality may increase further as transmission increases with holiday travel and gatherings and with the intensified indoor exposure that winter brings.”
The reported number of COVID-19 deaths is likely a 20% underestimate, they wrote, attributable to delays in reporting and an increase in non–COVID-19 deaths that were undetected and untreated because of pandemic-related disruptions. Since the coronavirus is communicable and spreads easily, COVID-19 deaths are particularly unique and worrying, they said.
“Individuals who die from homicide or cancer do not transmit the risk of morbidity and mortality to those nearby,” they wrote. “Every COVID-19 death signals the possibility of more deaths among close contacts.”
The fall surge in cases and deaths is widespread nationally, as compared to the spring, with hot spots on both coasts and in rural areas, according to an accompanying editorial in JAMA from public health researchers at the Harvard T.H. Chan School of Public Health, Boston. People of color have faced twice the death rate as well, with one in 875 Black people and one in 925 Indigenous people dying from COVID-19, as compared with one in 1,625 White people.
“The year 2020 ends with COVID-19 massively surging, as it was in the spring, to be the leading cause of death,” they wrote. “The accelerating numbers of deaths fall far short of fully capturing each devastating human story: Every death represents untold loss for countless families.”
Vaccines offer hope, they said, but won’t prevent the upcoming increase in COVID-19 hospitalizations and deaths this winter. In 2021, containing the pandemic will require national coordination, resources to help overwhelmed health care workers, new support for state and local public health officials, a stimulus package for schools and businesses, and financial aid for people on the brink of eviction. The country needs federal coordination of testing, contact tracing, personal protective equipment, travel precautions, and a face mask mandate, they wrote.
“Ending this crisis will require not only further advances in treatment but also unprecedented commitment to all aspects of prevention, vaccination, and public health,” they wrote. “Only by doing so can future years see this illness revert back to the unfamiliar and unknown condition it once was.”
This article first appeared on WebMD.com.
Encouraging results for new epilepsy drug
Post hoc analyses from an open-label study showed that seizure frequency was significantly reduced and the seizure-freedom rate was significantly improved among 240 adult participants who received cenobamate. The patients’ use of concomitant antiseizure medications was also reduced, with no effect on efficacy.
These results are “fascinating” and “very, very exciting,” said lead author William E. Rosenfeld, MD, director, Comprehensive Epilepsy Care Center for Children and Adults, St. Louis, Mo. Although responder rates were impressive, at 50% or greater and 75% or greater, “what patients really want is to have seizure freedom, or at least a 90% reduction in seizures,” Dr. Rosenfeld said.
The findings were presented at the annual meeting of the American Epilepsy Society, held online this year.
Adverse events
Cenobamate reduces seizures by inhibiting sodium current or affecting the GABAA channel, or potentially through a combination of these two mechanisms, said Dr. Rosenfeld. The drug was approved by the U.S. Food and Drug Administration in November 2019 for the treatment of uncontrolled partial-onset seizures in adults, which represent about 60% of all epileptic seizures. It has been on the market since May 2020.
During the drug’s development, three cases of drug reaction with eosinophilia and systemic symptoms (DRESS) occurred. This condition typically involves a skin rash, fever, swollen lymph nodes, and characteristic blood abnormalities, including a high level of eosinophils. However, an open-label study, published earlier this year in Epilepsia, that assessed safety and pharmacokinetics in 1,347 patients aged 18-70 years who received stable doses of one to three antiseizure medications showed that, with “slow and low titration” of cenobamate, there were no cases of DRESS, Dr. Rosenfeld said.
In that safety study, investigators administered increasing daily doses of cenobamate at 12.5, 25, 50, 100, 150, and 200 mg/day at 2-week intervals. If necessary, the dose could be increased to 400 mg/day via 50-mg/day increments every other week.
The researchers presented post hoc analyses regarding 240 patients from 10 U.S. sites who participated in the safety study. Dr. Rosenfeld noted, “These are all good epilepsy centers, and they all kept seizure records.” Of these participants, 177 continued taking the drug as they had at their last visit for a mean of more than 30 months; for some, it was up to 44 months.
“So we had a 73.8% retention rate over the course of the open label, which is the maintenance phase of the study,” Dr. Rosenfeld said.
Among the entire group of 240 patients, 25.8% had been seizure free for more than 12 months at their last visit. Of the 177 who continued to take cenobamate, 33.9% were seizure free for an average of 23.5 months.
“We have never seen those kinds of numbers in the past,” said Dr. Rosenfeld, adding, “it’s so important for patients to get seizure freedom.” These promising results may be related to the fact that the drug works on more than one mechanism of seizure, he speculated.
For some patients, the drug will “make a big difference” by providing them with the best quality of life and allow them to resume normal activities, Dr. Rosenfeld noted. In addition, the drug was well tolerated. The most common adverse events were dizziness/diplopia and sleepiness/drowsiness.
Concomitant drug reductions
Another post hoc analysis of the 240 patients showed that many patients were able to reduce use of other antiseizure medications. At study outset, about 41% were taking lacosamide, 35.7% were taking levetiracetam, and 27.7% were taking lamotrigine. Among patients who continued to take cenobamate, 22.7% of concomitant baseline antiseizure medications were discontinued. Carbamazepine was discontinued by 31.3%, oxcarbazepine by 26.7%, lacosamide by 23.4%, eslicarbazepine by 23.1%, clobazam by 26.7%, lamotrigine by 14.6%, and levetiracetam by 20.3%.
“We found that the patients who stayed in the study the longest had greater reductions in their concomitant antiepileptic mediation,” said Dr. Rosenfeld. Lowering concomitant medications did not reduce efficacy at a target dose of 200 mg/day.
The investigators hope to test the drug in children and in patients with different seizure types.
Promising, with caveats
Commenting on the research, Jong Woo Lee, MD, PhD, associate professor of neurology, the Edward B. Bromfield Epilepsy Program, Brigham and Women’s Hospital, Boston, said cenobamate “has certainly given new hope” to some of his patients. He noted that a few of these patients had been experiencing daily or nearly daily seizures and had been taking three or more medications for many years.
“The chances of another medication being effective for these patients is very low,” said Dr. Lee, who was not involved with the research. “But several of these patients responded to cenobamate, and some of them achieved complete seizure freedom.”
However, as with all new promising medications, there are some caveats. “The concern is for long-term efficacy for more than 5 years and, of course, unforeseen side effects,” Dr. Lee said.
The studies were funded by SK Life Science. Dr. Rosenfeld has been a consultant for SK Life Science. Dr. Lee has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Post hoc analyses from an open-label study showed that seizure frequency was significantly reduced and the seizure-freedom rate was significantly improved among 240 adult participants who received cenobamate. The patients’ use of concomitant antiseizure medications was also reduced, with no effect on efficacy.
These results are “fascinating” and “very, very exciting,” said lead author William E. Rosenfeld, MD, director, Comprehensive Epilepsy Care Center for Children and Adults, St. Louis, Mo. Although responder rates were impressive, at 50% or greater and 75% or greater, “what patients really want is to have seizure freedom, or at least a 90% reduction in seizures,” Dr. Rosenfeld said.
The findings were presented at the annual meeting of the American Epilepsy Society, held online this year.
Adverse events
Cenobamate reduces seizures by inhibiting sodium current or affecting the GABAA channel, or potentially through a combination of these two mechanisms, said Dr. Rosenfeld. The drug was approved by the U.S. Food and Drug Administration in November 2019 for the treatment of uncontrolled partial-onset seizures in adults, which represent about 60% of all epileptic seizures. It has been on the market since May 2020.
During the drug’s development, three cases of drug reaction with eosinophilia and systemic symptoms (DRESS) occurred. This condition typically involves a skin rash, fever, swollen lymph nodes, and characteristic blood abnormalities, including a high level of eosinophils. However, an open-label study, published earlier this year in Epilepsia, that assessed safety and pharmacokinetics in 1,347 patients aged 18-70 years who received stable doses of one to three antiseizure medications showed that, with “slow and low titration” of cenobamate, there were no cases of DRESS, Dr. Rosenfeld said.
In that safety study, investigators administered increasing daily doses of cenobamate at 12.5, 25, 50, 100, 150, and 200 mg/day at 2-week intervals. If necessary, the dose could be increased to 400 mg/day via 50-mg/day increments every other week.
The researchers presented post hoc analyses regarding 240 patients from 10 U.S. sites who participated in the safety study. Dr. Rosenfeld noted, “These are all good epilepsy centers, and they all kept seizure records.” Of these participants, 177 continued taking the drug as they had at their last visit for a mean of more than 30 months; for some, it was up to 44 months.
“So we had a 73.8% retention rate over the course of the open label, which is the maintenance phase of the study,” Dr. Rosenfeld said.
Among the entire group of 240 patients, 25.8% had been seizure free for more than 12 months at their last visit. Of the 177 who continued to take cenobamate, 33.9% were seizure free for an average of 23.5 months.
“We have never seen those kinds of numbers in the past,” said Dr. Rosenfeld, adding, “it’s so important for patients to get seizure freedom.” These promising results may be related to the fact that the drug works on more than one mechanism of seizure, he speculated.
For some patients, the drug will “make a big difference” by providing them with the best quality of life and allow them to resume normal activities, Dr. Rosenfeld noted. In addition, the drug was well tolerated. The most common adverse events were dizziness/diplopia and sleepiness/drowsiness.
Concomitant drug reductions
Another post hoc analysis of the 240 patients showed that many patients were able to reduce use of other antiseizure medications. At study outset, about 41% were taking lacosamide, 35.7% were taking levetiracetam, and 27.7% were taking lamotrigine. Among patients who continued to take cenobamate, 22.7% of concomitant baseline antiseizure medications were discontinued. Carbamazepine was discontinued by 31.3%, oxcarbazepine by 26.7%, lacosamide by 23.4%, eslicarbazepine by 23.1%, clobazam by 26.7%, lamotrigine by 14.6%, and levetiracetam by 20.3%.
“We found that the patients who stayed in the study the longest had greater reductions in their concomitant antiepileptic mediation,” said Dr. Rosenfeld. Lowering concomitant medications did not reduce efficacy at a target dose of 200 mg/day.
The investigators hope to test the drug in children and in patients with different seizure types.
Promising, with caveats
Commenting on the research, Jong Woo Lee, MD, PhD, associate professor of neurology, the Edward B. Bromfield Epilepsy Program, Brigham and Women’s Hospital, Boston, said cenobamate “has certainly given new hope” to some of his patients. He noted that a few of these patients had been experiencing daily or nearly daily seizures and had been taking three or more medications for many years.
“The chances of another medication being effective for these patients is very low,” said Dr. Lee, who was not involved with the research. “But several of these patients responded to cenobamate, and some of them achieved complete seizure freedom.”
However, as with all new promising medications, there are some caveats. “The concern is for long-term efficacy for more than 5 years and, of course, unforeseen side effects,” Dr. Lee said.
The studies were funded by SK Life Science. Dr. Rosenfeld has been a consultant for SK Life Science. Dr. Lee has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Post hoc analyses from an open-label study showed that seizure frequency was significantly reduced and the seizure-freedom rate was significantly improved among 240 adult participants who received cenobamate. The patients’ use of concomitant antiseizure medications was also reduced, with no effect on efficacy.
These results are “fascinating” and “very, very exciting,” said lead author William E. Rosenfeld, MD, director, Comprehensive Epilepsy Care Center for Children and Adults, St. Louis, Mo. Although responder rates were impressive, at 50% or greater and 75% or greater, “what patients really want is to have seizure freedom, or at least a 90% reduction in seizures,” Dr. Rosenfeld said.
The findings were presented at the annual meeting of the American Epilepsy Society, held online this year.
Adverse events
Cenobamate reduces seizures by inhibiting sodium current or affecting the GABAA channel, or potentially through a combination of these two mechanisms, said Dr. Rosenfeld. The drug was approved by the U.S. Food and Drug Administration in November 2019 for the treatment of uncontrolled partial-onset seizures in adults, which represent about 60% of all epileptic seizures. It has been on the market since May 2020.
During the drug’s development, three cases of drug reaction with eosinophilia and systemic symptoms (DRESS) occurred. This condition typically involves a skin rash, fever, swollen lymph nodes, and characteristic blood abnormalities, including a high level of eosinophils. However, an open-label study, published earlier this year in Epilepsia, that assessed safety and pharmacokinetics in 1,347 patients aged 18-70 years who received stable doses of one to three antiseizure medications showed that, with “slow and low titration” of cenobamate, there were no cases of DRESS, Dr. Rosenfeld said.
In that safety study, investigators administered increasing daily doses of cenobamate at 12.5, 25, 50, 100, 150, and 200 mg/day at 2-week intervals. If necessary, the dose could be increased to 400 mg/day via 50-mg/day increments every other week.
The researchers presented post hoc analyses regarding 240 patients from 10 U.S. sites who participated in the safety study. Dr. Rosenfeld noted, “These are all good epilepsy centers, and they all kept seizure records.” Of these participants, 177 continued taking the drug as they had at their last visit for a mean of more than 30 months; for some, it was up to 44 months.
“So we had a 73.8% retention rate over the course of the open label, which is the maintenance phase of the study,” Dr. Rosenfeld said.
Among the entire group of 240 patients, 25.8% had been seizure free for more than 12 months at their last visit. Of the 177 who continued to take cenobamate, 33.9% were seizure free for an average of 23.5 months.
“We have never seen those kinds of numbers in the past,” said Dr. Rosenfeld, adding, “it’s so important for patients to get seizure freedom.” These promising results may be related to the fact that the drug works on more than one mechanism of seizure, he speculated.
For some patients, the drug will “make a big difference” by providing them with the best quality of life and allow them to resume normal activities, Dr. Rosenfeld noted. In addition, the drug was well tolerated. The most common adverse events were dizziness/diplopia and sleepiness/drowsiness.
Concomitant drug reductions
Another post hoc analysis of the 240 patients showed that many patients were able to reduce use of other antiseizure medications. At study outset, about 41% were taking lacosamide, 35.7% were taking levetiracetam, and 27.7% were taking lamotrigine. Among patients who continued to take cenobamate, 22.7% of concomitant baseline antiseizure medications were discontinued. Carbamazepine was discontinued by 31.3%, oxcarbazepine by 26.7%, lacosamide by 23.4%, eslicarbazepine by 23.1%, clobazam by 26.7%, lamotrigine by 14.6%, and levetiracetam by 20.3%.
“We found that the patients who stayed in the study the longest had greater reductions in their concomitant antiepileptic mediation,” said Dr. Rosenfeld. Lowering concomitant medications did not reduce efficacy at a target dose of 200 mg/day.
The investigators hope to test the drug in children and in patients with different seizure types.
Promising, with caveats
Commenting on the research, Jong Woo Lee, MD, PhD, associate professor of neurology, the Edward B. Bromfield Epilepsy Program, Brigham and Women’s Hospital, Boston, said cenobamate “has certainly given new hope” to some of his patients. He noted that a few of these patients had been experiencing daily or nearly daily seizures and had been taking three or more medications for many years.
“The chances of another medication being effective for these patients is very low,” said Dr. Lee, who was not involved with the research. “But several of these patients responded to cenobamate, and some of them achieved complete seizure freedom.”
However, as with all new promising medications, there are some caveats. “The concern is for long-term efficacy for more than 5 years and, of course, unforeseen side effects,” Dr. Lee said.
The studies were funded by SK Life Science. Dr. Rosenfeld has been a consultant for SK Life Science. Dr. Lee has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM AES 2020
13 best practices to increase hospitalist billing efficiency
As an aspiring physician, I like learning about how things work. Since medical students learn very little about the “business” of medicine in school, this led me to pioneer a project on missed billing by hospitalists at a medium-sized hospital in the northeastern US. Although hospitalists do a tremendous amount of work, they do not always bill for what they are doing. The question became: Why are hospitalists missing charges and what can we do to stop it?

Shortly into my study, I recognized there was little daily communication between the administrators and the hospitalists; neither the hospitalists nor administrators understood the different dynamics that the others faced in their own workplace. It became apparent that administrators needed to learn what was important to hospitalists and to address them at their level in order to bring about change.
Some trending themes emerged as I started shadowing the hospitalists. Many of them asked how this project would benefit them. They argued that administrative needs should be dealt with at the administrative level. A major point was made that current incentives, such as the bonuses given for exceeding a certain number of RVUs, were not the motivating force behind their work ethics. From my observations, the motivating factors were the quality of their patient care, the needs of their patients, and teaching. The hospitalists also were eager to teach and continually instructed me on clinical skills and how to be a better medical student.
Bonuses or notoriety didn’t seem to be the main incentives for them. However, efficiency – especially in rounding – was important, and that became the focal point of the project. I found several studies that showed that improvements in aspects of rounding led to increased quality of patient care, decreased burnout, increased patient satisfaction, and decreased workload and discussed some of those findings with the hospitalists.1-10 When the hospitalists felt that their concerns were being heard, they became even more involved in the project, and the administrators and hospitalists started working together as a team.
One hospitalist spent two hours helping me design the platform that would be used for hospitalists to report barriers in their rounding process that may cause them to miss a charge. Once we identified those barriers, we discussed the possibility of standardizing their workflow based off these data. Many hospitalists argued that each physician has unique skills and practices that make them successful; therefore, the disruption of an already established workflow may cause a decrease in efficiency.
The hospitalists and I talked a lot about the importance of them rounding more efficiently and how that could positively affect the time that they have with their patients and themselves. We discussed that due to the additional work missed billing causes, minimizing this burden can possibly help decrease burnout. As a result, seven hospitalists, the administrative staff, and I met and created thirteen best practices, six of which they were able to get approved to use immediately. To note, hospitalists bill differently; some use a software company, fill out paper forms still or have integration within their EMR. Although these solutions were made for a program which has the ability to bill within the EMR, many of the principles will apply to your program too.

The 13 best practices that the seven hospitalists agreed upon are the following:
When a doctor signs a note, it opens a charge option or there is a hard stop.
Charge delinquencies are sent via email to the hospitalist.
Standardize that hospitalists charge directly after writing a note consistently as part of their workflow.*
Prioritize discharges before rounding.*
Standardize the use of the “my prof charges” column, a feature of this hospital’s EMR system that tells them if they had made a charge to a patient or not, in order to remind them to/confirm billing a patient.*
Create reports by the EMR system to provide charge data for individual providers.
Create a report for bill vs note to help providers self-audit. At this hospital, this feature was offered to the administrators as a way to audit their providers and doctors.
Ensure that when a patient is seen by a physician hospitalist as well as an NP/PA hospitalist, the appropriate charge for the physician is entered.
Notifications get sent to the physician hospitalist if a charge gets deleted by another person (e.g., NP/PA hospitalist).
Handoff of daily rounding sheets, or a paper copy of the patients assigned to a hospitalist for his/her shift, at the end of the shift to the project specialist.*
To keep the rounding sheets a complete and accurate account of the patients seen by the hospitalist.*
Hospitalists are to complete and check all billing at the end of their shift at the latest.*
Hospitalists are to participate on Provider Efficiency Training to optimize workflow, by creating more efficient note-writing behavior using Dragon.
*Indicates the practices the hospitalists were able to implement immediately. Practices 1, 2, 6, 7, and 9 request EMR changes. Practice 8 was already an established practice the hospitalists wished to continue. Practice 13 was suggested by the Lean Director for the continuation of a previous project.
Six of the best practices were easier to implement right away because they were at the discretion of the hospitalists. We found that the hospitalists who had the highest billing performances were more likely to start writing notes and charge earlier while rounding. Those who had poorer billing performances were more likely to leave all note writing and billing towards the end of their shift. The few exceptions (hospitalists who left all note writing and charging to the end of their shift yet had high billing performances) were found to have a consistent and standardized workflow. This was unlike the hospitalists who had the lowest billing performances. Having practices that help remind hospitalists to bill will surely help prevent missed billing, but because of the findings from this project, it was important to have consistent and standardized practices to additionally improve missed billing.

When we followed up with the hospitalist division two months later, we learned they were making great progress. Not only were hospitalists using their best practices, but in working with the administrators, they were designing sessions to further educate fellow hospitalists to prevent further missed billing. These sessions outlined shortcuts, resources and ways hospitalists may modify their personal EMR accounts to prevent missed billing. None of the progress could have been made without first understanding and addressing what is truly important to the hospitalists.
In summary, we noted these general observations in this project:
- Hospitalists favor solutions that benefit them or their patients.
- Hospitalists want to be part of the solution process.
- Hospitalists were more likely to accept ideas to improve their rounding if it meant they could keep their routine.
Obstacles exist in our health care system that prevent administrators and hospitalists from working together as a team. The more we are able to communicate and collaborate to fix problems in the health system, the more we can use the system to our mutual advantage. With the ongoing changes in medicine, especially during uncertain times, better communication needs be a major priority to affect positive change.
Ms. Mirabella attends the Frank H. Netter MD School of Medicine at Quinnipiac University, Hamden, Conn., in the class of 2022. She has interests in internal/hospital medicine, primary care, and health management and leadership. Dr. Rosenberg is associate professor at the Frank H. Netter MD School of Medicine at Quinnipiac University where she is director of clinical skills coaching. Dr. Kiassat is associate dean of the School of Engineering and associate clinical professor at Frank H. Netter MD School of Medicine, at Quinnipiac University. His research interests are in process improvement in health care, using Lean Six Sigma.
References
1. Burdick K, et al. Bedside interprofessional rounding. J Patient Exp. 2017;4(1):22-27. doi: 10.1177/2374373517692910.
2. Patel CR. Improving communication between hospitalists and consultants. The Hospital Leader. 2018. https://thehospitalleader.org/improving-communication-between-hospitalists-and-consultants/.
3. Adams TN, et al. Hospitalist perspective of interactions with medicine subspecialty consult services. J Hosp Med. 2018;13(5):318-323. doi: 10.12788/jhm.2882.
4. Michtalik HJ, et al. Impact of attending physician workload on patient care: A survey of hospitalists. JAMA Intern Med. 2013;173(5):375-377. doi: 10.1001/jamainternmed.2013.1864.
5. Chandra R, et al. How hospitalists can improve efficiency on inpatient wards. The Hospitalist. 2014. https://www.the-hospitalist.org/hospitalist/article/126231/how-hospitalists-can-improve-efficiency-inpatient-wards.
6. Chand DV. Observational study using the tools of lean six sigma to improve the efficiency of the resident rounding process. J Grad Med Educ. 2011;3(2):144-150. doi: 10.4300/JGME-D-10-00116.1.
7. O’Leary KJ, et al. How hospitalists spend their time: Insights on efficiency and safety. J Hosp Med. 2006;1(2):88-93. doi: 10.1002/jhm.88.
8. Wachter RM. Hospitalist workload: The search for the magic number. JAMA Intern Med. 2014;174(5):794-795. doi: 10.1001/jamainternmed.2014.18.
9. Bryson C, et al. Geographical assignment of hospitalists in an urban teaching hospital: Feasibility and impact on efficiency and provider satisfaction. Hospital Practice. 2017;45(4):135-142. doi: 10.1080/21548331.2017.1353884.
10. Calderon AS, et al. Transforming ward rounds through rounding-in-flow. J Grad Med Educ. 2014 Dec;6(4):750-5. doi: 10.4300/JGME-D-13-00324.1.
As an aspiring physician, I like learning about how things work. Since medical students learn very little about the “business” of medicine in school, this led me to pioneer a project on missed billing by hospitalists at a medium-sized hospital in the northeastern US. Although hospitalists do a tremendous amount of work, they do not always bill for what they are doing. The question became: Why are hospitalists missing charges and what can we do to stop it?
Shortly into my study, I recognized there was little daily communication between the administrators and the hospitalists; neither the hospitalists nor administrators understood the different dynamics that the others faced in their own workplace. It became apparent that administrators needed to learn what was important to hospitalists and to address them at their level in order to bring about change.
Some trending themes emerged as I started shadowing the hospitalists. Many of them asked how this project would benefit them. They argued that administrative needs should be dealt with at the administrative level. A major point was made that current incentives, such as the bonuses given for exceeding a certain number of RVUs, were not the motivating force behind their work ethics. From my observations, the motivating factors were the quality of their patient care, the needs of their patients, and teaching. The hospitalists also were eager to teach and continually instructed me on clinical skills and how to be a better medical student.
Bonuses or notoriety didn’t seem to be the main incentives for them. However, efficiency – especially in rounding – was important, and that became the focal point of the project. I found several studies that showed that improvements in aspects of rounding led to increased quality of patient care, decreased burnout, increased patient satisfaction, and decreased workload and discussed some of those findings with the hospitalists.1-10 When the hospitalists felt that their concerns were being heard, they became even more involved in the project, and the administrators and hospitalists started working together as a team.
One hospitalist spent two hours helping me design the platform that would be used for hospitalists to report barriers in their rounding process that may cause them to miss a charge. Once we identified those barriers, we discussed the possibility of standardizing their workflow based off these data. Many hospitalists argued that each physician has unique skills and practices that make them successful; therefore, the disruption of an already established workflow may cause a decrease in efficiency.
The hospitalists and I talked a lot about the importance of them rounding more efficiently and how that could positively affect the time that they have with their patients and themselves. We discussed that due to the additional work missed billing causes, minimizing this burden can possibly help decrease burnout. As a result, seven hospitalists, the administrative staff, and I met and created thirteen best practices, six of which they were able to get approved to use immediately. To note, hospitalists bill differently; some use a software company, fill out paper forms still or have integration within their EMR. Although these solutions were made for a program which has the ability to bill within the EMR, many of the principles will apply to your program too.
The 13 best practices that the seven hospitalists agreed upon are the following:
When a doctor signs a note, it opens a charge option or there is a hard stop.
Charge delinquencies are sent via email to the hospitalist.
Standardize that hospitalists charge directly after writing a note consistently as part of their workflow.*
Prioritize discharges before rounding.*
Standardize the use of the “my prof charges” column, a feature of this hospital’s EMR system that tells them if they had made a charge to a patient or not, in order to remind them to/confirm billing a patient.*
Create reports by the EMR system to provide charge data for individual providers.
Create a report for bill vs note to help providers self-audit. At this hospital, this feature was offered to the administrators as a way to audit their providers and doctors.
Ensure that when a patient is seen by a physician hospitalist as well as an NP/PA hospitalist, the appropriate charge for the physician is entered.
Notifications get sent to the physician hospitalist if a charge gets deleted by another person (e.g., NP/PA hospitalist).
Handoff of daily rounding sheets, or a paper copy of the patients assigned to a hospitalist for his/her shift, at the end of the shift to the project specialist.*
To keep the rounding sheets a complete and accurate account of the patients seen by the hospitalist.*
Hospitalists are to complete and check all billing at the end of their shift at the latest.*
Hospitalists are to participate on Provider Efficiency Training to optimize workflow, by creating more efficient note-writing behavior using Dragon.
*Indicates the practices the hospitalists were able to implement immediately. Practices 1, 2, 6, 7, and 9 request EMR changes. Practice 8 was already an established practice the hospitalists wished to continue. Practice 13 was suggested by the Lean Director for the continuation of a previous project.
Six of the best practices were easier to implement right away because they were at the discretion of the hospitalists. We found that the hospitalists who had the highest billing performances were more likely to start writing notes and charge earlier while rounding. Those who had poorer billing performances were more likely to leave all note writing and billing towards the end of their shift. The few exceptions (hospitalists who left all note writing and charging to the end of their shift yet had high billing performances) were found to have a consistent and standardized workflow. This was unlike the hospitalists who had the lowest billing performances. Having practices that help remind hospitalists to bill will surely help prevent missed billing, but because of the findings from this project, it was important to have consistent and standardized practices to additionally improve missed billing.
When we followed up with the hospitalist division two months later, we learned they were making great progress. Not only were hospitalists using their best practices, but in working with the administrators, they were designing sessions to further educate fellow hospitalists to prevent further missed billing. These sessions outlined shortcuts, resources and ways hospitalists may modify their personal EMR accounts to prevent missed billing. None of the progress could have been made without first understanding and addressing what is truly important to the hospitalists.
In summary, we noted these general observations in this project:
- Hospitalists favor solutions that benefit them or their patients.
- Hospitalists want to be part of the solution process.
- Hospitalists were more likely to accept ideas to improve their rounding if it meant they could keep their routine.
Obstacles exist in our health care system that prevent administrators and hospitalists from working together as a team. The more we are able to communicate and collaborate to fix problems in the health system, the more we can use the system to our mutual advantage. With the ongoing changes in medicine, especially during uncertain times, better communication needs be a major priority to affect positive change.
Ms. Mirabella attends the Frank H. Netter MD School of Medicine at Quinnipiac University, Hamden, Conn., in the class of 2022. She has interests in internal/hospital medicine, primary care, and health management and leadership. Dr. Rosenberg is associate professor at the Frank H. Netter MD School of Medicine at Quinnipiac University where she is director of clinical skills coaching. Dr. Kiassat is associate dean of the School of Engineering and associate clinical professor at Frank H. Netter MD School of Medicine, at Quinnipiac University. His research interests are in process improvement in health care, using Lean Six Sigma.
References
1. Burdick K, et al. Bedside interprofessional rounding. J Patient Exp. 2017;4(1):22-27. doi: 10.1177/2374373517692910.
2. Patel CR. Improving communication between hospitalists and consultants. The Hospital Leader. 2018. https://thehospitalleader.org/improving-communication-between-hospitalists-and-consultants/.
3. Adams TN, et al. Hospitalist perspective of interactions with medicine subspecialty consult services. J Hosp Med. 2018;13(5):318-323. doi: 10.12788/jhm.2882.
4. Michtalik HJ, et al. Impact of attending physician workload on patient care: A survey of hospitalists. JAMA Intern Med. 2013;173(5):375-377. doi: 10.1001/jamainternmed.2013.1864.
5. Chandra R, et al. How hospitalists can improve efficiency on inpatient wards. The Hospitalist. 2014. https://www.the-hospitalist.org/hospitalist/article/126231/how-hospitalists-can-improve-efficiency-inpatient-wards.
6. Chand DV. Observational study using the tools of lean six sigma to improve the efficiency of the resident rounding process. J Grad Med Educ. 2011;3(2):144-150. doi: 10.4300/JGME-D-10-00116.1.
7. O’Leary KJ, et al. How hospitalists spend their time: Insights on efficiency and safety. J Hosp Med. 2006;1(2):88-93. doi: 10.1002/jhm.88.
8. Wachter RM. Hospitalist workload: The search for the magic number. JAMA Intern Med. 2014;174(5):794-795. doi: 10.1001/jamainternmed.2014.18.
9. Bryson C, et al. Geographical assignment of hospitalists in an urban teaching hospital: Feasibility and impact on efficiency and provider satisfaction. Hospital Practice. 2017;45(4):135-142. doi: 10.1080/21548331.2017.1353884.
10. Calderon AS, et al. Transforming ward rounds through rounding-in-flow. J Grad Med Educ. 2014 Dec;6(4):750-5. doi: 10.4300/JGME-D-13-00324.1.
As an aspiring physician, I like learning about how things work. Since medical students learn very little about the “business” of medicine in school, this led me to pioneer a project on missed billing by hospitalists at a medium-sized hospital in the northeastern US. Although hospitalists do a tremendous amount of work, they do not always bill for what they are doing. The question became: Why are hospitalists missing charges and what can we do to stop it?
Shortly into my study, I recognized there was little daily communication between the administrators and the hospitalists; neither the hospitalists nor administrators understood the different dynamics that the others faced in their own workplace. It became apparent that administrators needed to learn what was important to hospitalists and to address them at their level in order to bring about change.
Some trending themes emerged as I started shadowing the hospitalists. Many of them asked how this project would benefit them. They argued that administrative needs should be dealt with at the administrative level. A major point was made that current incentives, such as the bonuses given for exceeding a certain number of RVUs, were not the motivating force behind their work ethics. From my observations, the motivating factors were the quality of their patient care, the needs of their patients, and teaching. The hospitalists also were eager to teach and continually instructed me on clinical skills and how to be a better medical student.
Bonuses or notoriety didn’t seem to be the main incentives for them. However, efficiency – especially in rounding – was important, and that became the focal point of the project. I found several studies that showed that improvements in aspects of rounding led to increased quality of patient care, decreased burnout, increased patient satisfaction, and decreased workload and discussed some of those findings with the hospitalists.1-10 When the hospitalists felt that their concerns were being heard, they became even more involved in the project, and the administrators and hospitalists started working together as a team.
One hospitalist spent two hours helping me design the platform that would be used for hospitalists to report barriers in their rounding process that may cause them to miss a charge. Once we identified those barriers, we discussed the possibility of standardizing their workflow based off these data. Many hospitalists argued that each physician has unique skills and practices that make them successful; therefore, the disruption of an already established workflow may cause a decrease in efficiency.
The hospitalists and I talked a lot about the importance of them rounding more efficiently and how that could positively affect the time that they have with their patients and themselves. We discussed that due to the additional work missed billing causes, minimizing this burden can possibly help decrease burnout. As a result, seven hospitalists, the administrative staff, and I met and created thirteen best practices, six of which they were able to get approved to use immediately. To note, hospitalists bill differently; some use a software company, fill out paper forms still or have integration within their EMR. Although these solutions were made for a program which has the ability to bill within the EMR, many of the principles will apply to your program too.
The 13 best practices that the seven hospitalists agreed upon are the following:
When a doctor signs a note, it opens a charge option or there is a hard stop.
Charge delinquencies are sent via email to the hospitalist.
Standardize that hospitalists charge directly after writing a note consistently as part of their workflow.*
Prioritize discharges before rounding.*
Standardize the use of the “my prof charges” column, a feature of this hospital’s EMR system that tells them if they had made a charge to a patient or not, in order to remind them to/confirm billing a patient.*
Create reports by the EMR system to provide charge data for individual providers.
Create a report for bill vs note to help providers self-audit. At this hospital, this feature was offered to the administrators as a way to audit their providers and doctors.
Ensure that when a patient is seen by a physician hospitalist as well as an NP/PA hospitalist, the appropriate charge for the physician is entered.
Notifications get sent to the physician hospitalist if a charge gets deleted by another person (e.g., NP/PA hospitalist).
Handoff of daily rounding sheets, or a paper copy of the patients assigned to a hospitalist for his/her shift, at the end of the shift to the project specialist.*
To keep the rounding sheets a complete and accurate account of the patients seen by the hospitalist.*
Hospitalists are to complete and check all billing at the end of their shift at the latest.*
Hospitalists are to participate on Provider Efficiency Training to optimize workflow, by creating more efficient note-writing behavior using Dragon.
*Indicates the practices the hospitalists were able to implement immediately. Practices 1, 2, 6, 7, and 9 request EMR changes. Practice 8 was already an established practice the hospitalists wished to continue. Practice 13 was suggested by the Lean Director for the continuation of a previous project.
Six of the best practices were easier to implement right away because they were at the discretion of the hospitalists. We found that the hospitalists who had the highest billing performances were more likely to start writing notes and charge earlier while rounding. Those who had poorer billing performances were more likely to leave all note writing and billing towards the end of their shift. The few exceptions (hospitalists who left all note writing and charging to the end of their shift yet had high billing performances) were found to have a consistent and standardized workflow. This was unlike the hospitalists who had the lowest billing performances. Having practices that help remind hospitalists to bill will surely help prevent missed billing, but because of the findings from this project, it was important to have consistent and standardized practices to additionally improve missed billing.
When we followed up with the hospitalist division two months later, we learned they were making great progress. Not only were hospitalists using their best practices, but in working with the administrators, they were designing sessions to further educate fellow hospitalists to prevent further missed billing. These sessions outlined shortcuts, resources and ways hospitalists may modify their personal EMR accounts to prevent missed billing. None of the progress could have been made without first understanding and addressing what is truly important to the hospitalists.
In summary, we noted these general observations in this project:
- Hospitalists favor solutions that benefit them or their patients.
- Hospitalists want to be part of the solution process.
- Hospitalists were more likely to accept ideas to improve their rounding if it meant they could keep their routine.
Obstacles exist in our health care system that prevent administrators and hospitalists from working together as a team. The more we are able to communicate and collaborate to fix problems in the health system, the more we can use the system to our mutual advantage. With the ongoing changes in medicine, especially during uncertain times, better communication needs be a major priority to affect positive change.
Ms. Mirabella attends the Frank H. Netter MD School of Medicine at Quinnipiac University, Hamden, Conn., in the class of 2022. She has interests in internal/hospital medicine, primary care, and health management and leadership. Dr. Rosenberg is associate professor at the Frank H. Netter MD School of Medicine at Quinnipiac University where she is director of clinical skills coaching. Dr. Kiassat is associate dean of the School of Engineering and associate clinical professor at Frank H. Netter MD School of Medicine, at Quinnipiac University. His research interests are in process improvement in health care, using Lean Six Sigma.
References
1. Burdick K, et al. Bedside interprofessional rounding. J Patient Exp. 2017;4(1):22-27. doi: 10.1177/2374373517692910.
2. Patel CR. Improving communication between hospitalists and consultants. The Hospital Leader. 2018. https://thehospitalleader.org/improving-communication-between-hospitalists-and-consultants/.
3. Adams TN, et al. Hospitalist perspective of interactions with medicine subspecialty consult services. J Hosp Med. 2018;13(5):318-323. doi: 10.12788/jhm.2882.
4. Michtalik HJ, et al. Impact of attending physician workload on patient care: A survey of hospitalists. JAMA Intern Med. 2013;173(5):375-377. doi: 10.1001/jamainternmed.2013.1864.
5. Chandra R, et al. How hospitalists can improve efficiency on inpatient wards. The Hospitalist. 2014. https://www.the-hospitalist.org/hospitalist/article/126231/how-hospitalists-can-improve-efficiency-inpatient-wards.
6. Chand DV. Observational study using the tools of lean six sigma to improve the efficiency of the resident rounding process. J Grad Med Educ. 2011;3(2):144-150. doi: 10.4300/JGME-D-10-00116.1.
7. O’Leary KJ, et al. How hospitalists spend their time: Insights on efficiency and safety. J Hosp Med. 2006;1(2):88-93. doi: 10.1002/jhm.88.
8. Wachter RM. Hospitalist workload: The search for the magic number. JAMA Intern Med. 2014;174(5):794-795. doi: 10.1001/jamainternmed.2014.18.
9. Bryson C, et al. Geographical assignment of hospitalists in an urban teaching hospital: Feasibility and impact on efficiency and provider satisfaction. Hospital Practice. 2017;45(4):135-142. doi: 10.1080/21548331.2017.1353884.
10. Calderon AS, et al. Transforming ward rounds through rounding-in-flow. J Grad Med Educ. 2014 Dec;6(4):750-5. doi: 10.4300/JGME-D-13-00324.1.
Should we use antibiotics to treat sore throats?
The use of antibiotics to treat a sore throat remains contentious, with guidelines from around the world providing contradictory advice. This topic generated a lively debate at the annual meeting of the European Society for Paediatric Infectious Diseases, held virtually this year.

Lauri Ivaska, MD, of the department of pediatrics and adolescent medicine at Turku (Finland) University Hospital, argued for the use of antibiotics, while Borbála Zsigmond, MD, of Heim Pál Children’s Hospital in Budapest, made the case against their use. Interestingly, this debate occurred against the background of a poll conducted before the debate, which found that only 11% of the audience voted in favor of using antibiotics to treat sore throats.
Both speakers began by exploring their approach to the treatment of a recent clinical case involving a 4-year-old girl presenting with sore throat. Dr. Ivaska stressed the difference between a sore throat, pharyngitis, and tonsillitis: the latter two refer to a physical finding, while the former is a subjective symptom.
International guidelines differ on the subject
The debate moved to discussing the international guidelines for treating pharyngitis and tonsillitis. Dr. Zsigmond believes that these are flawed and unhelpful, arguing that they differ depending on what part of the world a physician is practicing in. For example, the 2012 Infectious Diseases Society of America guidelines recommend using best clinical judgment and then backing this up by testing. If testing proves positive for group A Streptococcus pyogenes (GAS), the physician should universally treat. By comparison, the European Society of Clinical Microbiology and Infectious Diseases Sore Throat Guideline Group focuses on severity rather than the cause of the infection. If the case is deemed to be serious, antibiotics can be prescribed without a positive test.

Sore throat is frequently associated with a common cold. In a recent study, more that 80% of students with an acute viral respiratory tract infection had soreness at the beginning of their illness.
Reporting from his own research, Dr. Ivaska argued that viruses can be detected in almost two-thirds of children with pharyngitis using polymerase chain reaction analysis. He thinks antibiotics should be reserved for those 30%-40% of patients with a confirmed GAS infection. The potential role of Fusobacterium necrophorum was raised, but there is no evidence of the benefits of antibiotic treatment in such cases.
There are diagnostic aids for GAS infection
It was suggested that, instead of concentrating on sore throat, the debate should be about whether to use antibiotics to treat GAS infection. But how can the diagnosis be confirmed simply in a clinical setting? Dr. Ivaska recommended adopting diagnostic aids such as Centor, McIsaac, and FeverPAIN, which award scores for several common disease features – the higher the score, the more likely a patient is to be suffering from a GAS infection.
Dr. Zsigmond also likes scoring symptoms but believes they are often inaccurate, especially in young children. She pointed to a report that examined the use of the Centor tool among 441 children attending a pediatric ED. The authors concluded that the Centor criteria were ineffective in predicting a positive GAS culture in throat swabs taken from symptomatic patients.
When are antibiotics warranted?
It is widely accepted that antibiotics should be avoided for viral infections. Returning to the case described at the start of this debate, Dr. Zsigmond calculated that her patient with a 2-day history of sore throat, elevated temperature, pussy tonsils, and enlarged cervical lymph glands but no cough or rhinitis had a FeverPAIN score of 4-5 and a Centor score of 4, meaning that, according to the European guidelines, she should receive antibiotic treatment. However, viral swabs proved positive for adenovirus.
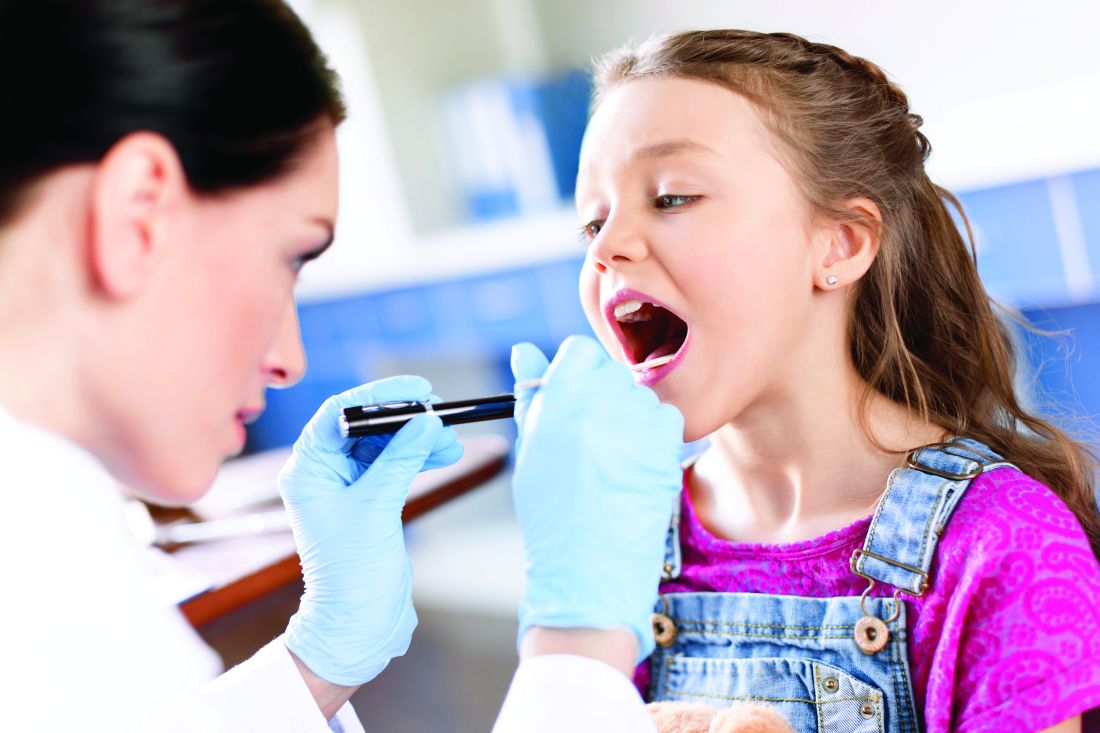
Dr. Ivaska responded with his recent experiences of a similar case, where a 5-year-old boy had a FeverPAIN score of 4-5 and Centor score of 3. Cultures from his throat were GAS positive, illustrating the problem of differentiating between bacterial and viral infections.
But does a GAS-positive pharyngeal culture necessarily mean that antibiotic treatment is indicated? Dr. Ivaska believes it does, citing the importance of preventing serious complications such as rheumatic fever. Dr. Zsigmind countered by pointing out the low levels of acute rheumatic fever in developed nations. In her own country, Hungary, there has not been a case in the last 30 years. Giving antibiotics for historical reasons cannot, in her view, be justified.
Dr. Ivaska responded that perhaps this is because of early treatment in children with sore throats.
Another complication of tonsillitis is quinsy. Dr. Zsigmond cited a study showing that there is no statistically significant evidence demonstrating that antibiotics prevent quinsy. She attributed this to quinsy appearing quickly, typically within 2 days. Delay in seeking help means that the window to treat is often missed. However, should symptoms present early, there is no statistical evidence that prior antibiotic use can prevent quinsy. Also, given the rarity of this condition, prevention would mean excessive use of antibiotics.
Are there other possible benefits of antibiotic treatment in patients with a sore throat? Dr. Ivaska referred to a Cochrane review that found a shortening in duration of throat soreness and fever. Furthermore, compared with placebo, antibiotics reduced the incidence of suppurative complications such as acute otitis media and sinusitis following a sore throat. Other studies have also pointed to the potential benefits of reduced transmission in families where one member with pharyngitis was GAS positive.
As the debate ended, Dr. Zsigmond reported evidence of global antibiotic overprescribing for sore throat ranging from 53% in Europe to 94% in Australia. She also highlighted risks such as altered gut flora, drug resistance, and rashes.
Robin Marlow from the University of Bristol (England), PhD, MBBS, commented that “one of the most enjoyable parts of the ESPID meeting is hearing different viewpoints rationally explained from across the world. As [antibiotic prescription for a sore throat is] a clinical conundrum that faces pediatricians every day, I thought this debate was a really great example of how, despite our different health care systems and ways of working, we are all striving together to improve children’s health using the best evidence available.”
The presenters had no financial conflicts of interest to declare.
The use of antibiotics to treat a sore throat remains contentious, with guidelines from around the world providing contradictory advice. This topic generated a lively debate at the annual meeting of the European Society for Paediatric Infectious Diseases, held virtually this year.
Lauri Ivaska, MD, of the department of pediatrics and adolescent medicine at Turku (Finland) University Hospital, argued for the use of antibiotics, while Borbála Zsigmond, MD, of Heim Pál Children’s Hospital in Budapest, made the case against their use. Interestingly, this debate occurred against the background of a poll conducted before the debate, which found that only 11% of the audience voted in favor of using antibiotics to treat sore throats.
Both speakers began by exploring their approach to the treatment of a recent clinical case involving a 4-year-old girl presenting with sore throat. Dr. Ivaska stressed the difference between a sore throat, pharyngitis, and tonsillitis: the latter two refer to a physical finding, while the former is a subjective symptom.
International guidelines differ on the subject
The debate moved to discussing the international guidelines for treating pharyngitis and tonsillitis. Dr. Zsigmond believes that these are flawed and unhelpful, arguing that they differ depending on what part of the world a physician is practicing in. For example, the 2012 Infectious Diseases Society of America guidelines recommend using best clinical judgment and then backing this up by testing. If testing proves positive for group A Streptococcus pyogenes (GAS), the physician should universally treat. By comparison, the European Society of Clinical Microbiology and Infectious Diseases Sore Throat Guideline Group focuses on severity rather than the cause of the infection. If the case is deemed to be serious, antibiotics can be prescribed without a positive test.
Sore throat is frequently associated with a common cold. In a recent study, more that 80% of students with an acute viral respiratory tract infection had soreness at the beginning of their illness.
Reporting from his own research, Dr. Ivaska argued that viruses can be detected in almost two-thirds of children with pharyngitis using polymerase chain reaction analysis. He thinks antibiotics should be reserved for those 30%-40% of patients with a confirmed GAS infection. The potential role of Fusobacterium necrophorum was raised, but there is no evidence of the benefits of antibiotic treatment in such cases.
There are diagnostic aids for GAS infection
It was suggested that, instead of concentrating on sore throat, the debate should be about whether to use antibiotics to treat GAS infection. But how can the diagnosis be confirmed simply in a clinical setting? Dr. Ivaska recommended adopting diagnostic aids such as Centor, McIsaac, and FeverPAIN, which award scores for several common disease features – the higher the score, the more likely a patient is to be suffering from a GAS infection.
Dr. Zsigmond also likes scoring symptoms but believes they are often inaccurate, especially in young children. She pointed to a report that examined the use of the Centor tool among 441 children attending a pediatric ED. The authors concluded that the Centor criteria were ineffective in predicting a positive GAS culture in throat swabs taken from symptomatic patients.
When are antibiotics warranted?
It is widely accepted that antibiotics should be avoided for viral infections. Returning to the case described at the start of this debate, Dr. Zsigmond calculated that her patient with a 2-day history of sore throat, elevated temperature, pussy tonsils, and enlarged cervical lymph glands but no cough or rhinitis had a FeverPAIN score of 4-5 and a Centor score of 4, meaning that, according to the European guidelines, she should receive antibiotic treatment. However, viral swabs proved positive for adenovirus.
Dr. Ivaska responded with his recent experiences of a similar case, where a 5-year-old boy had a FeverPAIN score of 4-5 and Centor score of 3. Cultures from his throat were GAS positive, illustrating the problem of differentiating between bacterial and viral infections.
But does a GAS-positive pharyngeal culture necessarily mean that antibiotic treatment is indicated? Dr. Ivaska believes it does, citing the importance of preventing serious complications such as rheumatic fever. Dr. Zsigmind countered by pointing out the low levels of acute rheumatic fever in developed nations. In her own country, Hungary, there has not been a case in the last 30 years. Giving antibiotics for historical reasons cannot, in her view, be justified.
Dr. Ivaska responded that perhaps this is because of early treatment in children with sore throats.
Another complication of tonsillitis is quinsy. Dr. Zsigmond cited a study showing that there is no statistically significant evidence demonstrating that antibiotics prevent quinsy. She attributed this to quinsy appearing quickly, typically within 2 days. Delay in seeking help means that the window to treat is often missed. However, should symptoms present early, there is no statistical evidence that prior antibiotic use can prevent quinsy. Also, given the rarity of this condition, prevention would mean excessive use of antibiotics.
Are there other possible benefits of antibiotic treatment in patients with a sore throat? Dr. Ivaska referred to a Cochrane review that found a shortening in duration of throat soreness and fever. Furthermore, compared with placebo, antibiotics reduced the incidence of suppurative complications such as acute otitis media and sinusitis following a sore throat. Other studies have also pointed to the potential benefits of reduced transmission in families where one member with pharyngitis was GAS positive.
As the debate ended, Dr. Zsigmond reported evidence of global antibiotic overprescribing for sore throat ranging from 53% in Europe to 94% in Australia. She also highlighted risks such as altered gut flora, drug resistance, and rashes.
Robin Marlow from the University of Bristol (England), PhD, MBBS, commented that “one of the most enjoyable parts of the ESPID meeting is hearing different viewpoints rationally explained from across the world. As [antibiotic prescription for a sore throat is] a clinical conundrum that faces pediatricians every day, I thought this debate was a really great example of how, despite our different health care systems and ways of working, we are all striving together to improve children’s health using the best evidence available.”
The presenters had no financial conflicts of interest to declare.
The use of antibiotics to treat a sore throat remains contentious, with guidelines from around the world providing contradictory advice. This topic generated a lively debate at the annual meeting of the European Society for Paediatric Infectious Diseases, held virtually this year.
Lauri Ivaska, MD, of the department of pediatrics and adolescent medicine at Turku (Finland) University Hospital, argued for the use of antibiotics, while Borbála Zsigmond, MD, of Heim Pál Children’s Hospital in Budapest, made the case against their use. Interestingly, this debate occurred against the background of a poll conducted before the debate, which found that only 11% of the audience voted in favor of using antibiotics to treat sore throats.
Both speakers began by exploring their approach to the treatment of a recent clinical case involving a 4-year-old girl presenting with sore throat. Dr. Ivaska stressed the difference between a sore throat, pharyngitis, and tonsillitis: the latter two refer to a physical finding, while the former is a subjective symptom.
International guidelines differ on the subject
The debate moved to discussing the international guidelines for treating pharyngitis and tonsillitis. Dr. Zsigmond believes that these are flawed and unhelpful, arguing that they differ depending on what part of the world a physician is practicing in. For example, the 2012 Infectious Diseases Society of America guidelines recommend using best clinical judgment and then backing this up by testing. If testing proves positive for group A Streptococcus pyogenes (GAS), the physician should universally treat. By comparison, the European Society of Clinical Microbiology and Infectious Diseases Sore Throat Guideline Group focuses on severity rather than the cause of the infection. If the case is deemed to be serious, antibiotics can be prescribed without a positive test.
Sore throat is frequently associated with a common cold. In a recent study, more that 80% of students with an acute viral respiratory tract infection had soreness at the beginning of their illness.
Reporting from his own research, Dr. Ivaska argued that viruses can be detected in almost two-thirds of children with pharyngitis using polymerase chain reaction analysis. He thinks antibiotics should be reserved for those 30%-40% of patients with a confirmed GAS infection. The potential role of Fusobacterium necrophorum was raised, but there is no evidence of the benefits of antibiotic treatment in such cases.
There are diagnostic aids for GAS infection
It was suggested that, instead of concentrating on sore throat, the debate should be about whether to use antibiotics to treat GAS infection. But how can the diagnosis be confirmed simply in a clinical setting? Dr. Ivaska recommended adopting diagnostic aids such as Centor, McIsaac, and FeverPAIN, which award scores for several common disease features – the higher the score, the more likely a patient is to be suffering from a GAS infection.
Dr. Zsigmond also likes scoring symptoms but believes they are often inaccurate, especially in young children. She pointed to a report that examined the use of the Centor tool among 441 children attending a pediatric ED. The authors concluded that the Centor criteria were ineffective in predicting a positive GAS culture in throat swabs taken from symptomatic patients.
When are antibiotics warranted?
It is widely accepted that antibiotics should be avoided for viral infections. Returning to the case described at the start of this debate, Dr. Zsigmond calculated that her patient with a 2-day history of sore throat, elevated temperature, pussy tonsils, and enlarged cervical lymph glands but no cough or rhinitis had a FeverPAIN score of 4-5 and a Centor score of 4, meaning that, according to the European guidelines, she should receive antibiotic treatment. However, viral swabs proved positive for adenovirus.
Dr. Ivaska responded with his recent experiences of a similar case, where a 5-year-old boy had a FeverPAIN score of 4-5 and Centor score of 3. Cultures from his throat were GAS positive, illustrating the problem of differentiating between bacterial and viral infections.
But does a GAS-positive pharyngeal culture necessarily mean that antibiotic treatment is indicated? Dr. Ivaska believes it does, citing the importance of preventing serious complications such as rheumatic fever. Dr. Zsigmind countered by pointing out the low levels of acute rheumatic fever in developed nations. In her own country, Hungary, there has not been a case in the last 30 years. Giving antibiotics for historical reasons cannot, in her view, be justified.
Dr. Ivaska responded that perhaps this is because of early treatment in children with sore throats.
Another complication of tonsillitis is quinsy. Dr. Zsigmond cited a study showing that there is no statistically significant evidence demonstrating that antibiotics prevent quinsy. She attributed this to quinsy appearing quickly, typically within 2 days. Delay in seeking help means that the window to treat is often missed. However, should symptoms present early, there is no statistical evidence that prior antibiotic use can prevent quinsy. Also, given the rarity of this condition, prevention would mean excessive use of antibiotics.
Are there other possible benefits of antibiotic treatment in patients with a sore throat? Dr. Ivaska referred to a Cochrane review that found a shortening in duration of throat soreness and fever. Furthermore, compared with placebo, antibiotics reduced the incidence of suppurative complications such as acute otitis media and sinusitis following a sore throat. Other studies have also pointed to the potential benefits of reduced transmission in families where one member with pharyngitis was GAS positive.
As the debate ended, Dr. Zsigmond reported evidence of global antibiotic overprescribing for sore throat ranging from 53% in Europe to 94% in Australia. She also highlighted risks such as altered gut flora, drug resistance, and rashes.
Robin Marlow from the University of Bristol (England), PhD, MBBS, commented that “one of the most enjoyable parts of the ESPID meeting is hearing different viewpoints rationally explained from across the world. As [antibiotic prescription for a sore throat is] a clinical conundrum that faces pediatricians every day, I thought this debate was a really great example of how, despite our different health care systems and ways of working, we are all striving together to improve children’s health using the best evidence available.”
The presenters had no financial conflicts of interest to declare.
FROM ESPID 2020
E-cigarette use tied to increased COPD, asthma risk

Results from a large national prospective cohort study of adults demonstrated that the use of electronic cigarettes is associated with an increased risk of asthma, chronic obstructive pulmonary disease (COPD), emphysema, and chronic bronchitis – independent of cigarette smoking and other combustible tobacco product use.
“Our longitudinal results are consistent with the findings of prior population studies,” researchers led by Wubin Xie, DrPH, MPH, wrote in a study published online in JAMA Network Open. “With a more refined study design assessing multiple respiratory conditions and extensive sensitivity checks to mitigate bias from reverse causation and residual confounding by cigarette smoking and other tobacco product use, our results strengthen the evidence of the potential role of e-cigarette use in pulmonary disease pathogenesis. The findings may be used to inform counseling of patients on the potential risks of e-cigarette use.”
Dr. Xie of Boston University, and colleagues used data from the Population Assessment of Tobacco and Health (PATH) study waves 1-4 to examine the association of e-cigarette use with incident respiratory conditions, including COPD, emphysema, chronic bronchitis, and asthma. An earlier analysis of PATH data found an association between e-cigarette use with a composite respiratory disease outcome, but it did not consider the timing of respiratory events over follow-up and was underpowered to evaluate specific respiratory conditions.
The current analysis included data from 21,618 U.S. adults who were surveyed in four waves of PATH between 2013 and 2018. Of these, 49% were men, 65% were non-Hispanic White, 12% were non-Hispanic Black, 16% were Hispanic, and the remainder were non-Hispanic other. Their mean pack-years was 6.7 at baseline, 26% had self-reported hypertension, and their mean body mass index was 27.8 kg/m2. The analysis was limited to data from the wave 1 cohort of adults and the prospective follow-up at waves 2-4 from public use files. It excluded adults who reported a history of a respiratory condition such as COPD, emphysema, chronic bronchitis, or asthma at wave 1 (baseline).
Two-thirds of respondents (66%) were never e-cigarette users, 12% were former e-cigarette users, and 5% were current e-cigarette users. After the researchers adjusted for cigarette and other combustible tobacco product use, demographic characteristics, and chronic health conditions, they observed an increased risk of respiratory disease among former e-cigarette users (incidence rate ratio, 1.28) and current e-cigarette users (IRR, 1.31). Among respondents with good self-reported health, the IRR for former e-cigarette users was 1.21 and the IRR for current e-cigarette users was 1.43. As for specific respiratory diseases among current e-cigarette users, the IRR was 1.33 for chronic bronchitis, 1.69 for emphysema, 1.57 for COPD, and 1.31 for asthma.
“Our findings on clinical outcome were consistent with studies assessing in vivo biomarkers of e-cigarette exposure in animal subjects, human participants, and population studies,” the authors wrote. “Studies have documented that exclusive e-cigarette use may be associated with higher exposure to harmful and potentially harmful constituents, compared with tobacco nonuse. The potential mechanisms of the association of e-cigarette exposure with pulmonary diseases include pulmonary inflammation, increased oxidative stress, and inhibited immune response. Animal studies have generated substantial evidence on e-cigarette exposure and emphysematous lung destruction, loss of pulmonary capillaries, reduced small airway function, and airway hyperresponsiveness, suggesting the plausibility of e-cigarettes causing chronic lung diseases.”
They acknowledged certain limitations of the study, including its reliance on self-reported measures of e-cigarette and other tobacco product use and its reliance on self-reported diagnoses of respiratory diseases.
The study was supported by grants from the National Heart, Lung, and Blood Institute; the Food and Drug Administration Center for Tobacco Products; and the American Lung Association Public Policy Research Award. Dr. Xie reported having no financial disclosures. His coauthors reported having received research grants and personal fees from a variety of sources.
SOURCE: Xie W et al. JAMA Netw Open. 2020 Nov 12. doi: 10.1001/jamanetworkopen.2020.20816
Results from a large national prospective cohort study of adults demonstrated that the use of electronic cigarettes is associated with an increased risk of asthma, chronic obstructive pulmonary disease (COPD), emphysema, and chronic bronchitis – independent of cigarette smoking and other combustible tobacco product use.
“Our longitudinal results are consistent with the findings of prior population studies,” researchers led by Wubin Xie, DrPH, MPH, wrote in a study published online in JAMA Network Open. “With a more refined study design assessing multiple respiratory conditions and extensive sensitivity checks to mitigate bias from reverse causation and residual confounding by cigarette smoking and other tobacco product use, our results strengthen the evidence of the potential role of e-cigarette use in pulmonary disease pathogenesis. The findings may be used to inform counseling of patients on the potential risks of e-cigarette use.”
Dr. Xie of Boston University, and colleagues used data from the Population Assessment of Tobacco and Health (PATH) study waves 1-4 to examine the association of e-cigarette use with incident respiratory conditions, including COPD, emphysema, chronic bronchitis, and asthma. An earlier analysis of PATH data found an association between e-cigarette use with a composite respiratory disease outcome, but it did not consider the timing of respiratory events over follow-up and was underpowered to evaluate specific respiratory conditions.
The current analysis included data from 21,618 U.S. adults who were surveyed in four waves of PATH between 2013 and 2018. Of these, 49% were men, 65% were non-Hispanic White, 12% were non-Hispanic Black, 16% were Hispanic, and the remainder were non-Hispanic other. Their mean pack-years was 6.7 at baseline, 26% had self-reported hypertension, and their mean body mass index was 27.8 kg/m2. The analysis was limited to data from the wave 1 cohort of adults and the prospective follow-up at waves 2-4 from public use files. It excluded adults who reported a history of a respiratory condition such as COPD, emphysema, chronic bronchitis, or asthma at wave 1 (baseline).
Two-thirds of respondents (66%) were never e-cigarette users, 12% were former e-cigarette users, and 5% were current e-cigarette users. After the researchers adjusted for cigarette and other combustible tobacco product use, demographic characteristics, and chronic health conditions, they observed an increased risk of respiratory disease among former e-cigarette users (incidence rate ratio, 1.28) and current e-cigarette users (IRR, 1.31). Among respondents with good self-reported health, the IRR for former e-cigarette users was 1.21 and the IRR for current e-cigarette users was 1.43. As for specific respiratory diseases among current e-cigarette users, the IRR was 1.33 for chronic bronchitis, 1.69 for emphysema, 1.57 for COPD, and 1.31 for asthma.
“Our findings on clinical outcome were consistent with studies assessing in vivo biomarkers of e-cigarette exposure in animal subjects, human participants, and population studies,” the authors wrote. “Studies have documented that exclusive e-cigarette use may be associated with higher exposure to harmful and potentially harmful constituents, compared with tobacco nonuse. The potential mechanisms of the association of e-cigarette exposure with pulmonary diseases include pulmonary inflammation, increased oxidative stress, and inhibited immune response. Animal studies have generated substantial evidence on e-cigarette exposure and emphysematous lung destruction, loss of pulmonary capillaries, reduced small airway function, and airway hyperresponsiveness, suggesting the plausibility of e-cigarettes causing chronic lung diseases.”
They acknowledged certain limitations of the study, including its reliance on self-reported measures of e-cigarette and other tobacco product use and its reliance on self-reported diagnoses of respiratory diseases.
The study was supported by grants from the National Heart, Lung, and Blood Institute; the Food and Drug Administration Center for Tobacco Products; and the American Lung Association Public Policy Research Award. Dr. Xie reported having no financial disclosures. His coauthors reported having received research grants and personal fees from a variety of sources.
SOURCE: Xie W et al. JAMA Netw Open. 2020 Nov 12. doi: 10.1001/jamanetworkopen.2020.20816
Results from a large national prospective cohort study of adults demonstrated that the use of electronic cigarettes is associated with an increased risk of asthma, chronic obstructive pulmonary disease (COPD), emphysema, and chronic bronchitis – independent of cigarette smoking and other combustible tobacco product use.
“Our longitudinal results are consistent with the findings of prior population studies,” researchers led by Wubin Xie, DrPH, MPH, wrote in a study published online in JAMA Network Open. “With a more refined study design assessing multiple respiratory conditions and extensive sensitivity checks to mitigate bias from reverse causation and residual confounding by cigarette smoking and other tobacco product use, our results strengthen the evidence of the potential role of e-cigarette use in pulmonary disease pathogenesis. The findings may be used to inform counseling of patients on the potential risks of e-cigarette use.”
Dr. Xie of Boston University, and colleagues used data from the Population Assessment of Tobacco and Health (PATH) study waves 1-4 to examine the association of e-cigarette use with incident respiratory conditions, including COPD, emphysema, chronic bronchitis, and asthma. An earlier analysis of PATH data found an association between e-cigarette use with a composite respiratory disease outcome, but it did not consider the timing of respiratory events over follow-up and was underpowered to evaluate specific respiratory conditions.
The current analysis included data from 21,618 U.S. adults who were surveyed in four waves of PATH between 2013 and 2018. Of these, 49% were men, 65% were non-Hispanic White, 12% were non-Hispanic Black, 16% were Hispanic, and the remainder were non-Hispanic other. Their mean pack-years was 6.7 at baseline, 26% had self-reported hypertension, and their mean body mass index was 27.8 kg/m2. The analysis was limited to data from the wave 1 cohort of adults and the prospective follow-up at waves 2-4 from public use files. It excluded adults who reported a history of a respiratory condition such as COPD, emphysema, chronic bronchitis, or asthma at wave 1 (baseline).
Two-thirds of respondents (66%) were never e-cigarette users, 12% were former e-cigarette users, and 5% were current e-cigarette users. After the researchers adjusted for cigarette and other combustible tobacco product use, demographic characteristics, and chronic health conditions, they observed an increased risk of respiratory disease among former e-cigarette users (incidence rate ratio, 1.28) and current e-cigarette users (IRR, 1.31). Among respondents with good self-reported health, the IRR for former e-cigarette users was 1.21 and the IRR for current e-cigarette users was 1.43. As for specific respiratory diseases among current e-cigarette users, the IRR was 1.33 for chronic bronchitis, 1.69 for emphysema, 1.57 for COPD, and 1.31 for asthma.
“Our findings on clinical outcome were consistent with studies assessing in vivo biomarkers of e-cigarette exposure in animal subjects, human participants, and population studies,” the authors wrote. “Studies have documented that exclusive e-cigarette use may be associated with higher exposure to harmful and potentially harmful constituents, compared with tobacco nonuse. The potential mechanisms of the association of e-cigarette exposure with pulmonary diseases include pulmonary inflammation, increased oxidative stress, and inhibited immune response. Animal studies have generated substantial evidence on e-cigarette exposure and emphysematous lung destruction, loss of pulmonary capillaries, reduced small airway function, and airway hyperresponsiveness, suggesting the plausibility of e-cigarettes causing chronic lung diseases.”
They acknowledged certain limitations of the study, including its reliance on self-reported measures of e-cigarette and other tobacco product use and its reliance on self-reported diagnoses of respiratory diseases.
The study was supported by grants from the National Heart, Lung, and Blood Institute; the Food and Drug Administration Center for Tobacco Products; and the American Lung Association Public Policy Research Award. Dr. Xie reported having no financial disclosures. His coauthors reported having received research grants and personal fees from a variety of sources.
SOURCE: Xie W et al. JAMA Netw Open. 2020 Nov 12. doi: 10.1001/jamanetworkopen.2020.20816
FROM JAMA NETWORK OPEN
Pediatricians want kids to be part of COVID vaccine trials
If clinical trials for COVID-19 vaccines aren’t expanded soon to include children, it’s unlikely that even kids in their teens will be vaccinated in time for the next school year.
The hurdle is that COVID vaccine makers are only in the early stages of testing their products on children. The Pfizer vaccine authorized for use by the Food and Drug Administration on Friday was greenlighted only for people aged 16 years and older. Moderna just started trials for 12- to 17-year-olds for its vaccine, likely to be authorized later this month.
It will take months to approve use of the vaccines for middle- and high school–aged kids, and months more to test them in younger children. But some pediatricians say that concerns about the safety of the front-runner vaccines make the wait worthwhile.
Although most pediatricians believe the eventual vaccination of children will be crucial to subduing the COVID virus, they’re split on how fast to move toward that, says James Campbell, MD, professor of pediatrics at the University of Maryland’s Center for Vaccine Development and Global Health in Baltimore. Dr. Campbell and colleagues said it’s a matter of urgency to get the vaccines tested in kids, while others want to hold off on those trials until millions of adults have been safely vaccinated.
Much of the debate centers on two issues: the degree of harm COVID-19 causes children and the extent to which children are spreading the virus to their friends, teachers, parents and grandparents.
COVID-19’s impact on children represents a tiny fraction of the suffering and death experienced by vulnerable adults. Yet it would qualify as a pretty serious childhood disease, having caused 154 deaths and more than 7,500 hospitalizations as of Dec. 3 among aged people 19 years and younger in the United States. Those numbers rank it as worse than a typical year of influenza, and worse than diseases like mumps or hepatitis B in children before the vaccination era.
Studies thus far show that 1%-2% of children infected with the virus end up requiring intensive care, Stanley Plotkin, MD, professor emeritus of pediatrics at the University of Pennsylvania, Philadelphia told a federal panel. That’s in line with the percentage who become gravely ill as result of infections like Haemophilus influenzae type B, for which doctors have vaccinated children since the 1980s, he pointed out.
Dr. Campbell, who with colleagues has developed a plan for how to run pediatric COVID vaccine trials, pointed out that, “in a universe where COVID mainly affected children the way it’s affecting them now, and we had potential vaccines, people would be clamoring for them.”
The evidence that teens can transmit the disease is pretty clear, and transmission has been documented in children as young as 8. Fear of spread by children has been enough to close schools, and led the American Academy of Pediatrics to demand that children be quickly included in vaccine testing.
“The longer we take to start kids in trials, the longer it will take them to get vaccinated and to break the chains of transmission,” said Yvonne Maldonado, MD, a professor of pediatrics at Stanford (Calif.) University who chairs the AAP’s infectious disease committee. “If you want kids to go back to school and not have the teachers union terrified, you have to make sure they aren’t a risk.”
Other pediatricians worry that early pediatric trials could backfire. Cody Meissner, MD, chief of pediatric infectious diseases at Tufts Medical Center and a member of the FDA’s advisory committee on vaccines, is worried that whatever causes multisystem inflammatory syndrome in children, a rare but frightening COVID-related disorder, might also be triggered, however rarely, by vaccination.
Dr. Meissner abstained from the committee’s vote Thursday that supported, by a 17-4 vote, an emergency authorization of the Pfizer vaccine for people aged 16 years and older.
“I have trouble justifying it for children so unlikely to get the disease,” he said during debate on the measure.
But panel member Ofer Levy, MD, PhD, director of the Precision Vaccines Program at Boston Children’s Hospital, said the 16-years-and-up authorization would speed the vaccine’s testing in and approval for younger children. That is vital for the world’s protection from COVID-19, he said, since in the United States and most places “most vaccines are delivered early in life.”
While vaccines given to tens of thousands of people so far appear to be safe, the lack of understanding of the inflammatory syndrome means that children in any trials should be followed closely, said Emily Erbelding, MD, MPH, director of the division of microbiology and infectious diseases at the National Institute of Allergy and Infectious Diseases.
Under a 2003 law, vaccine companies are required eventually to test all their products on children. By late November, Pfizer had vaccinated approximately 100 children aged 12-15 years, said spokesperson Jerica Pitts.
Moderna has started enrolling 3,000 children aged 12 years and over in another clinical trial, and other companies have similar plans. Assuming the trials show the vaccines are safe and provide a good immune response, future tests could include progressively younger children, moving to, say, 6- to 12-year-olds next, then 2- to 6-year-olds. Eventually, trials could include younger toddlers and infants.
Similar step-down approaches were taken to test vaccines against human papillomavirus (HPV), influenza and other diseases in the past, Dr. Erbelding noted. Such trials are easiest to conduct when researchers know that a measurable immune response, like antibody levels in the blood, translates to effective protection against disease. Armed with such knowledge, they can see whether children were protected without them having to be exposed to the virus. Federal scientists hope to get that data from the Moderna and Pfizer adult vaccine trials, she said.
Vaccine trials geared to tweens or younger children may involve testing half-doses, which, if protective, would require less vaccine and might cause fewer incidents of sore arms and fevers that afflicted many who’ve received the Pfizer and Moderna vaccines, Dr. Campbell said.
But unless additional studies begin quickly, the window for having an FDA-authorized vaccine available before the next school year “will be closed even for our oldest children,” said Evan Anderson, MD, a pediatrics professor at Emory University, Atlanta. “Our younger children are almost certainly going into next school year without a vaccine option available for them.”
In the meantime, teachers are likely to be high on the priority list for vaccination. Protecting school staff could allow more schools to reopen even if most children can’t be vaccinated, Dr. Erbelding said.
Eventually, if the SARS-CoV-2 virus remains in circulation, governments may want to mandate childhood vaccination against the virus to protect them as they grow up and protect society as a whole, Dr. Plotkin said.
In the 1960s, Dr. Plotkin invented the rubella vaccine that has been given to hundreds of millions of children since. Like COVID-19, rubella – or German measles – is not usually a serious illness for children. But congenital rubella syndrome afflicted babies in the womb with blindness, deafness, developmental delays, and autism. Immunizing toddlers, which, in turn, protects their pregnant mothers, has indirectly prevented hundreds of thousands of such cases.
“We don’t want to use children to protect everyone in the community,” said Dr. Campbell. “But when you can protect both children and their community, that’s important.”
And while a coronavirus infection may not be bad for most children, missed school, absent friends, and distanced families have caused them immense suffering, he said.
“It’s a huge burden on a child to have their entire world flipped around,” Dr. Campbell said. “If vaccinating could help to flip it back, we should begin testing to see if that’s possible.”
Kaiser Health News is a nonprofit news service covering health issues. It is an editorially independent program of KFF (Kaiser Family Foundation), which is not affiliated with Kaiser Permanente.
If clinical trials for COVID-19 vaccines aren’t expanded soon to include children, it’s unlikely that even kids in their teens will be vaccinated in time for the next school year.
The hurdle is that COVID vaccine makers are only in the early stages of testing their products on children. The Pfizer vaccine authorized for use by the Food and Drug Administration on Friday was greenlighted only for people aged 16 years and older. Moderna just started trials for 12- to 17-year-olds for its vaccine, likely to be authorized later this month.
It will take months to approve use of the vaccines for middle- and high school–aged kids, and months more to test them in younger children. But some pediatricians say that concerns about the safety of the front-runner vaccines make the wait worthwhile.
Although most pediatricians believe the eventual vaccination of children will be crucial to subduing the COVID virus, they’re split on how fast to move toward that, says James Campbell, MD, professor of pediatrics at the University of Maryland’s Center for Vaccine Development and Global Health in Baltimore. Dr. Campbell and colleagues said it’s a matter of urgency to get the vaccines tested in kids, while others want to hold off on those trials until millions of adults have been safely vaccinated.
Much of the debate centers on two issues: the degree of harm COVID-19 causes children and the extent to which children are spreading the virus to their friends, teachers, parents and grandparents.
COVID-19’s impact on children represents a tiny fraction of the suffering and death experienced by vulnerable adults. Yet it would qualify as a pretty serious childhood disease, having caused 154 deaths and more than 7,500 hospitalizations as of Dec. 3 among aged people 19 years and younger in the United States. Those numbers rank it as worse than a typical year of influenza, and worse than diseases like mumps or hepatitis B in children before the vaccination era.
Studies thus far show that 1%-2% of children infected with the virus end up requiring intensive care, Stanley Plotkin, MD, professor emeritus of pediatrics at the University of Pennsylvania, Philadelphia told a federal panel. That’s in line with the percentage who become gravely ill as result of infections like Haemophilus influenzae type B, for which doctors have vaccinated children since the 1980s, he pointed out.
Dr. Campbell, who with colleagues has developed a plan for how to run pediatric COVID vaccine trials, pointed out that, “in a universe where COVID mainly affected children the way it’s affecting them now, and we had potential vaccines, people would be clamoring for them.”
The evidence that teens can transmit the disease is pretty clear, and transmission has been documented in children as young as 8. Fear of spread by children has been enough to close schools, and led the American Academy of Pediatrics to demand that children be quickly included in vaccine testing.
“The longer we take to start kids in trials, the longer it will take them to get vaccinated and to break the chains of transmission,” said Yvonne Maldonado, MD, a professor of pediatrics at Stanford (Calif.) University who chairs the AAP’s infectious disease committee. “If you want kids to go back to school and not have the teachers union terrified, you have to make sure they aren’t a risk.”
Other pediatricians worry that early pediatric trials could backfire. Cody Meissner, MD, chief of pediatric infectious diseases at Tufts Medical Center and a member of the FDA’s advisory committee on vaccines, is worried that whatever causes multisystem inflammatory syndrome in children, a rare but frightening COVID-related disorder, might also be triggered, however rarely, by vaccination.
Dr. Meissner abstained from the committee’s vote Thursday that supported, by a 17-4 vote, an emergency authorization of the Pfizer vaccine for people aged 16 years and older.
“I have trouble justifying it for children so unlikely to get the disease,” he said during debate on the measure.
But panel member Ofer Levy, MD, PhD, director of the Precision Vaccines Program at Boston Children’s Hospital, said the 16-years-and-up authorization would speed the vaccine’s testing in and approval for younger children. That is vital for the world’s protection from COVID-19, he said, since in the United States and most places “most vaccines are delivered early in life.”
While vaccines given to tens of thousands of people so far appear to be safe, the lack of understanding of the inflammatory syndrome means that children in any trials should be followed closely, said Emily Erbelding, MD, MPH, director of the division of microbiology and infectious diseases at the National Institute of Allergy and Infectious Diseases.
Under a 2003 law, vaccine companies are required eventually to test all their products on children. By late November, Pfizer had vaccinated approximately 100 children aged 12-15 years, said spokesperson Jerica Pitts.
Moderna has started enrolling 3,000 children aged 12 years and over in another clinical trial, and other companies have similar plans. Assuming the trials show the vaccines are safe and provide a good immune response, future tests could include progressively younger children, moving to, say, 6- to 12-year-olds next, then 2- to 6-year-olds. Eventually, trials could include younger toddlers and infants.
Similar step-down approaches were taken to test vaccines against human papillomavirus (HPV), influenza and other diseases in the past, Dr. Erbelding noted. Such trials are easiest to conduct when researchers know that a measurable immune response, like antibody levels in the blood, translates to effective protection against disease. Armed with such knowledge, they can see whether children were protected without them having to be exposed to the virus. Federal scientists hope to get that data from the Moderna and Pfizer adult vaccine trials, she said.
Vaccine trials geared to tweens or younger children may involve testing half-doses, which, if protective, would require less vaccine and might cause fewer incidents of sore arms and fevers that afflicted many who’ve received the Pfizer and Moderna vaccines, Dr. Campbell said.
But unless additional studies begin quickly, the window for having an FDA-authorized vaccine available before the next school year “will be closed even for our oldest children,” said Evan Anderson, MD, a pediatrics professor at Emory University, Atlanta. “Our younger children are almost certainly going into next school year without a vaccine option available for them.”
In the meantime, teachers are likely to be high on the priority list for vaccination. Protecting school staff could allow more schools to reopen even if most children can’t be vaccinated, Dr. Erbelding said.
Eventually, if the SARS-CoV-2 virus remains in circulation, governments may want to mandate childhood vaccination against the virus to protect them as they grow up and protect society as a whole, Dr. Plotkin said.
In the 1960s, Dr. Plotkin invented the rubella vaccine that has been given to hundreds of millions of children since. Like COVID-19, rubella – or German measles – is not usually a serious illness for children. But congenital rubella syndrome afflicted babies in the womb with blindness, deafness, developmental delays, and autism. Immunizing toddlers, which, in turn, protects their pregnant mothers, has indirectly prevented hundreds of thousands of such cases.
“We don’t want to use children to protect everyone in the community,” said Dr. Campbell. “But when you can protect both children and their community, that’s important.”
And while a coronavirus infection may not be bad for most children, missed school, absent friends, and distanced families have caused them immense suffering, he said.
“It’s a huge burden on a child to have their entire world flipped around,” Dr. Campbell said. “If vaccinating could help to flip it back, we should begin testing to see if that’s possible.”
Kaiser Health News is a nonprofit news service covering health issues. It is an editorially independent program of KFF (Kaiser Family Foundation), which is not affiliated with Kaiser Permanente.
If clinical trials for COVID-19 vaccines aren’t expanded soon to include children, it’s unlikely that even kids in their teens will be vaccinated in time for the next school year.
The hurdle is that COVID vaccine makers are only in the early stages of testing their products on children. The Pfizer vaccine authorized for use by the Food and Drug Administration on Friday was greenlighted only for people aged 16 years and older. Moderna just started trials for 12- to 17-year-olds for its vaccine, likely to be authorized later this month.
It will take months to approve use of the vaccines for middle- and high school–aged kids, and months more to test them in younger children. But some pediatricians say that concerns about the safety of the front-runner vaccines make the wait worthwhile.
Although most pediatricians believe the eventual vaccination of children will be crucial to subduing the COVID virus, they’re split on how fast to move toward that, says James Campbell, MD, professor of pediatrics at the University of Maryland’s Center for Vaccine Development and Global Health in Baltimore. Dr. Campbell and colleagues said it’s a matter of urgency to get the vaccines tested in kids, while others want to hold off on those trials until millions of adults have been safely vaccinated.
Much of the debate centers on two issues: the degree of harm COVID-19 causes children and the extent to which children are spreading the virus to their friends, teachers, parents and grandparents.
COVID-19’s impact on children represents a tiny fraction of the suffering and death experienced by vulnerable adults. Yet it would qualify as a pretty serious childhood disease, having caused 154 deaths and more than 7,500 hospitalizations as of Dec. 3 among aged people 19 years and younger in the United States. Those numbers rank it as worse than a typical year of influenza, and worse than diseases like mumps or hepatitis B in children before the vaccination era.
Studies thus far show that 1%-2% of children infected with the virus end up requiring intensive care, Stanley Plotkin, MD, professor emeritus of pediatrics at the University of Pennsylvania, Philadelphia told a federal panel. That’s in line with the percentage who become gravely ill as result of infections like Haemophilus influenzae type B, for which doctors have vaccinated children since the 1980s, he pointed out.
Dr. Campbell, who with colleagues has developed a plan for how to run pediatric COVID vaccine trials, pointed out that, “in a universe where COVID mainly affected children the way it’s affecting them now, and we had potential vaccines, people would be clamoring for them.”
The evidence that teens can transmit the disease is pretty clear, and transmission has been documented in children as young as 8. Fear of spread by children has been enough to close schools, and led the American Academy of Pediatrics to demand that children be quickly included in vaccine testing.
“The longer we take to start kids in trials, the longer it will take them to get vaccinated and to break the chains of transmission,” said Yvonne Maldonado, MD, a professor of pediatrics at Stanford (Calif.) University who chairs the AAP’s infectious disease committee. “If you want kids to go back to school and not have the teachers union terrified, you have to make sure they aren’t a risk.”
Other pediatricians worry that early pediatric trials could backfire. Cody Meissner, MD, chief of pediatric infectious diseases at Tufts Medical Center and a member of the FDA’s advisory committee on vaccines, is worried that whatever causes multisystem inflammatory syndrome in children, a rare but frightening COVID-related disorder, might also be triggered, however rarely, by vaccination.
Dr. Meissner abstained from the committee’s vote Thursday that supported, by a 17-4 vote, an emergency authorization of the Pfizer vaccine for people aged 16 years and older.
“I have trouble justifying it for children so unlikely to get the disease,” he said during debate on the measure.
But panel member Ofer Levy, MD, PhD, director of the Precision Vaccines Program at Boston Children’s Hospital, said the 16-years-and-up authorization would speed the vaccine’s testing in and approval for younger children. That is vital for the world’s protection from COVID-19, he said, since in the United States and most places “most vaccines are delivered early in life.”
While vaccines given to tens of thousands of people so far appear to be safe, the lack of understanding of the inflammatory syndrome means that children in any trials should be followed closely, said Emily Erbelding, MD, MPH, director of the division of microbiology and infectious diseases at the National Institute of Allergy and Infectious Diseases.
Under a 2003 law, vaccine companies are required eventually to test all their products on children. By late November, Pfizer had vaccinated approximately 100 children aged 12-15 years, said spokesperson Jerica Pitts.
Moderna has started enrolling 3,000 children aged 12 years and over in another clinical trial, and other companies have similar plans. Assuming the trials show the vaccines are safe and provide a good immune response, future tests could include progressively younger children, moving to, say, 6- to 12-year-olds next, then 2- to 6-year-olds. Eventually, trials could include younger toddlers and infants.
Similar step-down approaches were taken to test vaccines against human papillomavirus (HPV), influenza and other diseases in the past, Dr. Erbelding noted. Such trials are easiest to conduct when researchers know that a measurable immune response, like antibody levels in the blood, translates to effective protection against disease. Armed with such knowledge, they can see whether children were protected without them having to be exposed to the virus. Federal scientists hope to get that data from the Moderna and Pfizer adult vaccine trials, she said.
Vaccine trials geared to tweens or younger children may involve testing half-doses, which, if protective, would require less vaccine and might cause fewer incidents of sore arms and fevers that afflicted many who’ve received the Pfizer and Moderna vaccines, Dr. Campbell said.
But unless additional studies begin quickly, the window for having an FDA-authorized vaccine available before the next school year “will be closed even for our oldest children,” said Evan Anderson, MD, a pediatrics professor at Emory University, Atlanta. “Our younger children are almost certainly going into next school year without a vaccine option available for them.”
In the meantime, teachers are likely to be high on the priority list for vaccination. Protecting school staff could allow more schools to reopen even if most children can’t be vaccinated, Dr. Erbelding said.
Eventually, if the SARS-CoV-2 virus remains in circulation, governments may want to mandate childhood vaccination against the virus to protect them as they grow up and protect society as a whole, Dr. Plotkin said.
In the 1960s, Dr. Plotkin invented the rubella vaccine that has been given to hundreds of millions of children since. Like COVID-19, rubella – or German measles – is not usually a serious illness for children. But congenital rubella syndrome afflicted babies in the womb with blindness, deafness, developmental delays, and autism. Immunizing toddlers, which, in turn, protects their pregnant mothers, has indirectly prevented hundreds of thousands of such cases.
“We don’t want to use children to protect everyone in the community,” said Dr. Campbell. “But when you can protect both children and their community, that’s important.”
And while a coronavirus infection may not be bad for most children, missed school, absent friends, and distanced families have caused them immense suffering, he said.
“It’s a huge burden on a child to have their entire world flipped around,” Dr. Campbell said. “If vaccinating could help to flip it back, we should begin testing to see if that’s possible.”
Kaiser Health News is a nonprofit news service covering health issues. It is an editorially independent program of KFF (Kaiser Family Foundation), which is not affiliated with Kaiser Permanente.
Teenage bone density declines following sleeve gastrectomy
Adolescents who undergo sleeve gastrectomy have lower bone density and higher bone marrow fat at 1 year following surgery, new research shows.

“It’s almost paradoxical,” Miriam Bredella, MD, of Massachusetts General Hospital in Boston, told Medscape Medical News. “Despite marked loss of body fat, these children have more fat in their bones and decreased bone density.”
She explained that the dissected part of the stomach is filled with anabolic cells that are important for building bone mass. “When those cells are cut out, the body cannot produce the hormones for building up bone.” It’s a malabsorption problem, she added. “Cutting out parts of the stomach or gut leads to less absorption.”
It is well known that bariatric surgery in adults has long-term effects on bone, she said, but this is the first time it has been studied in children.
“Nobody thinks about bone loss in children, but it’s extremely important,” Bredella reports. “The adolescent years up to age 25 are when we accrue bone density, so if something happens during this critical time, it can lead to weak bones later in life.” In the case of these adolescents, peak bone mass is never reached.
To investigate the effects of sleeve gastrectomy on bone density and marrow adipose tissue in extremely obese teenagers, researchers at Massachusetts General Hospital and Harvard Medical School recruited 52 adolescents with a mean body mass index (BMI) of 45. They measured volumetric bone mineral density using quantitative computer tomography (QCT) of the lumbar spine.
“We used QCT instead of DEXA [dual energy x-ray absorptiometry] scan because it isn’t affected by changes in soft tissue; it’s less susceptible to extreme changes in body weight,” Bredella said. “With DEXA scan there are too many artifacts.”
Half of the group (n = 26) underwent surgery. At 1 year, those who underwent surgery lost an average of 34 kg (75 lb). Adolescents in the control group lost an average of 0.2 kg (0.5 lb) (P < .0001).
Both groups repeated the QCT scan at the 1-year follow-up. Researchers found a decrease in bone density in those who underwent sleeve gastrectomy vs. controls (P = .046).
In her presentation, Bredella showed the QCT of the L2 spine in a 17-year old female before surgery and 12 months later. Her volumetric bone mineral density decreased from 183 mg/cm3 to 146 mg/cm3.
“Sleeve gastrectomy in children is bad for bones,” Bradella said. “You have to take care of your bones. This is something people are not thinking about and it probably won’t be a problem when they’re young but will likely affect these patients with osteoporosis when they are older.”
Patients need to be aware of this, she warns, and take steps to combat the bone loss. “Drinking milk, taking vitamin D, and doing weight-bearing exercise may help increase the bone density,” she said.
The increased fat in the bone is also concerning, she said. “Increased fat in the bone is a phenomenon that we see in anorexic patients,” Bredella explained.
The body appears to store the fat in bone in case of need later on, she explained. “We know that in severe states of malnutrition the body has the ability to metabolize the fat in the bones.”
The obesity epidemic in America has given way to a 100-fold increase in sleeve gastrectomy procedures in teenagers between 2005 and 2014. “These patients need this surgery so they don›t die of cardiac arrest or diabetes,” she said. “But we need to make sure they get their bone mineral density checked frequently.”
“The results of this study are important,” Marc Michalsky, MD, Nationwide Children’s Hospital, Columbus, Ohio, told Medscape Medical News. “But they need to be put into context.”
“There is an impetus and argument to support bariatric surgery as it offers a significant reduction in BMI and an associated reversal and complete amelioration of obesity related diseases.”
What this study doesn’t address, he said, is whether this population will experience an increase in bone density-related fractures down the road.
“These results are a snapshot in time — a picture of one postoperative time point,” Michalsky pointed out. “Are we seeing a process that represents continued change in bone mineralization? It’s not unreasonable to assume that the radiological findings here may lead to real clinical impact, but we don’t know.”
Bredella and Michalsky have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Adolescents who undergo sleeve gastrectomy have lower bone density and higher bone marrow fat at 1 year following surgery, new research shows.
“It’s almost paradoxical,” Miriam Bredella, MD, of Massachusetts General Hospital in Boston, told Medscape Medical News. “Despite marked loss of body fat, these children have more fat in their bones and decreased bone density.”
She explained that the dissected part of the stomach is filled with anabolic cells that are important for building bone mass. “When those cells are cut out, the body cannot produce the hormones for building up bone.” It’s a malabsorption problem, she added. “Cutting out parts of the stomach or gut leads to less absorption.”
It is well known that bariatric surgery in adults has long-term effects on bone, she said, but this is the first time it has been studied in children.
“Nobody thinks about bone loss in children, but it’s extremely important,” Bredella reports. “The adolescent years up to age 25 are when we accrue bone density, so if something happens during this critical time, it can lead to weak bones later in life.” In the case of these adolescents, peak bone mass is never reached.
To investigate the effects of sleeve gastrectomy on bone density and marrow adipose tissue in extremely obese teenagers, researchers at Massachusetts General Hospital and Harvard Medical School recruited 52 adolescents with a mean body mass index (BMI) of 45. They measured volumetric bone mineral density using quantitative computer tomography (QCT) of the lumbar spine.
“We used QCT instead of DEXA [dual energy x-ray absorptiometry] scan because it isn’t affected by changes in soft tissue; it’s less susceptible to extreme changes in body weight,” Bredella said. “With DEXA scan there are too many artifacts.”
Half of the group (n = 26) underwent surgery. At 1 year, those who underwent surgery lost an average of 34 kg (75 lb). Adolescents in the control group lost an average of 0.2 kg (0.5 lb) (P < .0001).
Both groups repeated the QCT scan at the 1-year follow-up. Researchers found a decrease in bone density in those who underwent sleeve gastrectomy vs. controls (P = .046).
In her presentation, Bredella showed the QCT of the L2 spine in a 17-year old female before surgery and 12 months later. Her volumetric bone mineral density decreased from 183 mg/cm3 to 146 mg/cm3.
“Sleeve gastrectomy in children is bad for bones,” Bradella said. “You have to take care of your bones. This is something people are not thinking about and it probably won’t be a problem when they’re young but will likely affect these patients with osteoporosis when they are older.”
Patients need to be aware of this, she warns, and take steps to combat the bone loss. “Drinking milk, taking vitamin D, and doing weight-bearing exercise may help increase the bone density,” she said.
The increased fat in the bone is also concerning, she said. “Increased fat in the bone is a phenomenon that we see in anorexic patients,” Bredella explained.
The body appears to store the fat in bone in case of need later on, she explained. “We know that in severe states of malnutrition the body has the ability to metabolize the fat in the bones.”
The obesity epidemic in America has given way to a 100-fold increase in sleeve gastrectomy procedures in teenagers between 2005 and 2014. “These patients need this surgery so they don›t die of cardiac arrest or diabetes,” she said. “But we need to make sure they get their bone mineral density checked frequently.”
“The results of this study are important,” Marc Michalsky, MD, Nationwide Children’s Hospital, Columbus, Ohio, told Medscape Medical News. “But they need to be put into context.”
“There is an impetus and argument to support bariatric surgery as it offers a significant reduction in BMI and an associated reversal and complete amelioration of obesity related diseases.”
What this study doesn’t address, he said, is whether this population will experience an increase in bone density-related fractures down the road.
“These results are a snapshot in time — a picture of one postoperative time point,” Michalsky pointed out. “Are we seeing a process that represents continued change in bone mineralization? It’s not unreasonable to assume that the radiological findings here may lead to real clinical impact, but we don’t know.”
Bredella and Michalsky have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Adolescents who undergo sleeve gastrectomy have lower bone density and higher bone marrow fat at 1 year following surgery, new research shows.
“It’s almost paradoxical,” Miriam Bredella, MD, of Massachusetts General Hospital in Boston, told Medscape Medical News. “Despite marked loss of body fat, these children have more fat in their bones and decreased bone density.”
She explained that the dissected part of the stomach is filled with anabolic cells that are important for building bone mass. “When those cells are cut out, the body cannot produce the hormones for building up bone.” It’s a malabsorption problem, she added. “Cutting out parts of the stomach or gut leads to less absorption.”
It is well known that bariatric surgery in adults has long-term effects on bone, she said, but this is the first time it has been studied in children.
“Nobody thinks about bone loss in children, but it’s extremely important,” Bredella reports. “The adolescent years up to age 25 are when we accrue bone density, so if something happens during this critical time, it can lead to weak bones later in life.” In the case of these adolescents, peak bone mass is never reached.
To investigate the effects of sleeve gastrectomy on bone density and marrow adipose tissue in extremely obese teenagers, researchers at Massachusetts General Hospital and Harvard Medical School recruited 52 adolescents with a mean body mass index (BMI) of 45. They measured volumetric bone mineral density using quantitative computer tomography (QCT) of the lumbar spine.
“We used QCT instead of DEXA [dual energy x-ray absorptiometry] scan because it isn’t affected by changes in soft tissue; it’s less susceptible to extreme changes in body weight,” Bredella said. “With DEXA scan there are too many artifacts.”
Half of the group (n = 26) underwent surgery. At 1 year, those who underwent surgery lost an average of 34 kg (75 lb). Adolescents in the control group lost an average of 0.2 kg (0.5 lb) (P < .0001).
Both groups repeated the QCT scan at the 1-year follow-up. Researchers found a decrease in bone density in those who underwent sleeve gastrectomy vs. controls (P = .046).
In her presentation, Bredella showed the QCT of the L2 spine in a 17-year old female before surgery and 12 months later. Her volumetric bone mineral density decreased from 183 mg/cm3 to 146 mg/cm3.
“Sleeve gastrectomy in children is bad for bones,” Bradella said. “You have to take care of your bones. This is something people are not thinking about and it probably won’t be a problem when they’re young but will likely affect these patients with osteoporosis when they are older.”
Patients need to be aware of this, she warns, and take steps to combat the bone loss. “Drinking milk, taking vitamin D, and doing weight-bearing exercise may help increase the bone density,” she said.
The increased fat in the bone is also concerning, she said. “Increased fat in the bone is a phenomenon that we see in anorexic patients,” Bredella explained.
The body appears to store the fat in bone in case of need later on, she explained. “We know that in severe states of malnutrition the body has the ability to metabolize the fat in the bones.”
The obesity epidemic in America has given way to a 100-fold increase in sleeve gastrectomy procedures in teenagers between 2005 and 2014. “These patients need this surgery so they don›t die of cardiac arrest or diabetes,” she said. “But we need to make sure they get their bone mineral density checked frequently.”
“The results of this study are important,” Marc Michalsky, MD, Nationwide Children’s Hospital, Columbus, Ohio, told Medscape Medical News. “But they need to be put into context.”
“There is an impetus and argument to support bariatric surgery as it offers a significant reduction in BMI and an associated reversal and complete amelioration of obesity related diseases.”
What this study doesn’t address, he said, is whether this population will experience an increase in bone density-related fractures down the road.
“These results are a snapshot in time — a picture of one postoperative time point,” Michalsky pointed out. “Are we seeing a process that represents continued change in bone mineralization? It’s not unreasonable to assume that the radiological findings here may lead to real clinical impact, but we don’t know.”
Bredella and Michalsky have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
An all-oral option for advanced HR+, HER2– breast cancer?
These results suggest tesetaxel plus capecitabine is “a potential new treatment option” for this patient population, said study investigator Joyce O’Shaughnessy, MD, of Baylor University Medical Center and Texas Oncology, both in Dallas.

“This should launch an oral taxane into the clinical space, which will be a nice addition to the toolbox for treating advanced breast cancer, with real upsides for patients,” said Hal Burstein, MD, PhD, of Dana-Farber Cancer Institute and Harvard Medical School, both in Boston, who was not involved in the trial but commented on the results in an interview.
Another commenter was more critical of CONTESSA’s results, which were presented at the 2020 San Antonio Breast Cancer Symposium.
“Three months’ difference in PFS in this setting is meaningless without overall survival [OS] results,” Fatima Cardoso, MD, of Champalimaud Clinical Center in Lisbon, Portugal, said in a question submitted through the virtual meeting’s chat system.
At this point, the OS data are immature, and mature data won’t be available for another couple of years at least, according to the study’s protocol.
Dr. O’Shaughnessy defended the PFS result as being significant, however, saying it was comparable with outcomes seen previously with docetaxel-capecitabine and paclitaxel-gemcitabine combinations.
Other meeting attendees questioned why the waters had been muddied by testing the effects of tesetaxel in combination with capecitabine, albeit at a reduced dose, versus the approved full dose of capecitabine as monotherapy, particularly as a phase 2 trial had shown that tesetaxel demonstrated “significant activity” as monotherapy.
“The reason for the combination versus a monotherapy is because it was designed as a registration trial,” Dr. O’Shaughnessy explained. The trial was designed to be very similar to early taxane studies where docetaxel was assessed with or without capecitabine, or paclitaxel with or without gemcitabine.
“Probably we’re going to be using a doublet for patients who have virulent disease who really need a response,” Dr. O’Shaughnessy explained. She noted that the objective response rate was much higher with the tesetaxel-capecitabine combination than with capecitabine alone, and that result alone is “probably enough that we would utilize a doublet.”
The key thing is that it now gives patients an all-oral option, Dr. O’Shaughnessy said.
“The data are exciting because it would be terrific to have an orally available taxane chemotherapy,” agreed Dr. Burstein. “It is far more convenient for patients and opens access globally in places that do not have adequate resources for administration of IV therapeutics. Also, the data suggest that tesetaxel has a different side effect profile than IV taxane, with less neuropathy and less alopecia.”
Trial design
CONTESSA is an ongoing randomized, controlled trial that started in 2017 and is projected to end in early 2023. It is investigating the use of tesetaxel plus a reduced dose of capecitabine versus the approved dose of capecitabine alone in 685 women with hormone receptor–positive, HER2-negative locally advanced or metastatic breast cancer who had previously been treated with a taxane.
Being intrinsically orally bioavailable and more soluble than the other taxanes means that tesetaxel has a much longer half-life that allows for a “more convenient treatment experience for patients,” Dr. O’Shaughnessy observed.
Indeed, because tesetaxel only needs to be dosed once every 3 weeks, patients in the trial received tesetaxel at 27 mg/m2 only on the first day of a 21-day treatment cycle. This was combined with a reduced, 825-mg/m2 dose of capecitabine, given orally twice-daily on days 2-14 but once daily on the evening of day 1 and on the morning of day 15.
The combination regimen was compared with the recommended full dose of capecitabine alone, 1,250 mg/m2 given orally twice daily on days 2-14 but once daily on the evening of day 1 and on the morning of day 15.
Efficacy and safety
PFS was 9.8 months with tesetaxel plus capecitabine and 6.9 months with capecitabine alone, representing a 2.9-month improvement with the combination (hazard ratio, 0.716; P = .003).
A similar PFS benefit was seen regardless of multiple predefined subgroups, such as age, baseline performance status, duration of disease-free interval before study entry, and the use of CDK4/6 inhibitors.
The objective response rate was 57% with tesetaxel plus capecitabine and 41% with capecitabine alone (P = .0002). The 24-week disease control rate was 67% and 50%, respectively (P < .0001).
The most frequent treatment-emergent adverse event seen with the tesetaxel-capecitabine combination was neutropenia, occurring in 76.9% of patients, compared with 22.6% of patients in the monotherapy arm. Rates of grade 3-4 neutropenia were much higher in the combination arm (32.6% and 38.3%, respectively) than in the monotherapy arm (7.4% and 0.9%, respectively).
The neutropenia seen was “generally manageable,” Dr. O’Shaughnessy said, primarily with dose reductions and granulocyte colony–stimulating factor as needed.
She pointed out that rates of grade 3 or higher neuropathy and grade 2 alopecia were low, a respective 5.9% and 8%, with the combination.
The dose of capecitabine used in the control arm was noted to be higher than that used in usual practice.
“This was because of the global nature of the study and the regulatory requirements globally,” Dr. O’Shaughnessy said.
“The dose-modification scheme was that patients could have a dose reduction at the first sign of grade 2 toxicity,” she added, giving investigators the flexibility to reduce the dose as soon as possible.
This study was sponsored by Odonate Therapeutics. Dr. O’Shaughnessy disclosed consulting fees from AbbVie, Agendia, AstraZeneca, Bristol-Myers Squibb, Celgene, Eisai, Genentech/Roche, Genomic Health, GRAIL, Heron, Immunomedics, Ipsen, Jounce, Lilly, Novartis, Odonate, Pfizer, Puma, and Seagen. Dr. Burstein and Dr. Cardoso had no relevant disclosures.
SOURCE: O’Shaughnessy J et al. SABCS 2020, Abstract GS4-01.
These results suggest tesetaxel plus capecitabine is “a potential new treatment option” for this patient population, said study investigator Joyce O’Shaughnessy, MD, of Baylor University Medical Center and Texas Oncology, both in Dallas.
“This should launch an oral taxane into the clinical space, which will be a nice addition to the toolbox for treating advanced breast cancer, with real upsides for patients,” said Hal Burstein, MD, PhD, of Dana-Farber Cancer Institute and Harvard Medical School, both in Boston, who was not involved in the trial but commented on the results in an interview.
Another commenter was more critical of CONTESSA’s results, which were presented at the 2020 San Antonio Breast Cancer Symposium.
“Three months’ difference in PFS in this setting is meaningless without overall survival [OS] results,” Fatima Cardoso, MD, of Champalimaud Clinical Center in Lisbon, Portugal, said in a question submitted through the virtual meeting’s chat system.
At this point, the OS data are immature, and mature data won’t be available for another couple of years at least, according to the study’s protocol.
Dr. O’Shaughnessy defended the PFS result as being significant, however, saying it was comparable with outcomes seen previously with docetaxel-capecitabine and paclitaxel-gemcitabine combinations.
Other meeting attendees questioned why the waters had been muddied by testing the effects of tesetaxel in combination with capecitabine, albeit at a reduced dose, versus the approved full dose of capecitabine as monotherapy, particularly as a phase 2 trial had shown that tesetaxel demonstrated “significant activity” as monotherapy.
“The reason for the combination versus a monotherapy is because it was designed as a registration trial,” Dr. O’Shaughnessy explained. The trial was designed to be very similar to early taxane studies where docetaxel was assessed with or without capecitabine, or paclitaxel with or without gemcitabine.
“Probably we’re going to be using a doublet for patients who have virulent disease who really need a response,” Dr. O’Shaughnessy explained. She noted that the objective response rate was much higher with the tesetaxel-capecitabine combination than with capecitabine alone, and that result alone is “probably enough that we would utilize a doublet.”
The key thing is that it now gives patients an all-oral option, Dr. O’Shaughnessy said.
“The data are exciting because it would be terrific to have an orally available taxane chemotherapy,” agreed Dr. Burstein. “It is far more convenient for patients and opens access globally in places that do not have adequate resources for administration of IV therapeutics. Also, the data suggest that tesetaxel has a different side effect profile than IV taxane, with less neuropathy and less alopecia.”
Trial design
CONTESSA is an ongoing randomized, controlled trial that started in 2017 and is projected to end in early 2023. It is investigating the use of tesetaxel plus a reduced dose of capecitabine versus the approved dose of capecitabine alone in 685 women with hormone receptor–positive, HER2-negative locally advanced or metastatic breast cancer who had previously been treated with a taxane.
Being intrinsically orally bioavailable and more soluble than the other taxanes means that tesetaxel has a much longer half-life that allows for a “more convenient treatment experience for patients,” Dr. O’Shaughnessy observed.
Indeed, because tesetaxel only needs to be dosed once every 3 weeks, patients in the trial received tesetaxel at 27 mg/m2 only on the first day of a 21-day treatment cycle. This was combined with a reduced, 825-mg/m2 dose of capecitabine, given orally twice-daily on days 2-14 but once daily on the evening of day 1 and on the morning of day 15.
The combination regimen was compared with the recommended full dose of capecitabine alone, 1,250 mg/m2 given orally twice daily on days 2-14 but once daily on the evening of day 1 and on the morning of day 15.
Efficacy and safety
PFS was 9.8 months with tesetaxel plus capecitabine and 6.9 months with capecitabine alone, representing a 2.9-month improvement with the combination (hazard ratio, 0.716; P = .003).
A similar PFS benefit was seen regardless of multiple predefined subgroups, such as age, baseline performance status, duration of disease-free interval before study entry, and the use of CDK4/6 inhibitors.
The objective response rate was 57% with tesetaxel plus capecitabine and 41% with capecitabine alone (P = .0002). The 24-week disease control rate was 67% and 50%, respectively (P < .0001).
The most frequent treatment-emergent adverse event seen with the tesetaxel-capecitabine combination was neutropenia, occurring in 76.9% of patients, compared with 22.6% of patients in the monotherapy arm. Rates of grade 3-4 neutropenia were much higher in the combination arm (32.6% and 38.3%, respectively) than in the monotherapy arm (7.4% and 0.9%, respectively).
The neutropenia seen was “generally manageable,” Dr. O’Shaughnessy said, primarily with dose reductions and granulocyte colony–stimulating factor as needed.
She pointed out that rates of grade 3 or higher neuropathy and grade 2 alopecia were low, a respective 5.9% and 8%, with the combination.
The dose of capecitabine used in the control arm was noted to be higher than that used in usual practice.
“This was because of the global nature of the study and the regulatory requirements globally,” Dr. O’Shaughnessy said.
“The dose-modification scheme was that patients could have a dose reduction at the first sign of grade 2 toxicity,” she added, giving investigators the flexibility to reduce the dose as soon as possible.
This study was sponsored by Odonate Therapeutics. Dr. O’Shaughnessy disclosed consulting fees from AbbVie, Agendia, AstraZeneca, Bristol-Myers Squibb, Celgene, Eisai, Genentech/Roche, Genomic Health, GRAIL, Heron, Immunomedics, Ipsen, Jounce, Lilly, Novartis, Odonate, Pfizer, Puma, and Seagen. Dr. Burstein and Dr. Cardoso had no relevant disclosures.
SOURCE: O’Shaughnessy J et al. SABCS 2020, Abstract GS4-01.
These results suggest tesetaxel plus capecitabine is “a potential new treatment option” for this patient population, said study investigator Joyce O’Shaughnessy, MD, of Baylor University Medical Center and Texas Oncology, both in Dallas.
“This should launch an oral taxane into the clinical space, which will be a nice addition to the toolbox for treating advanced breast cancer, with real upsides for patients,” said Hal Burstein, MD, PhD, of Dana-Farber Cancer Institute and Harvard Medical School, both in Boston, who was not involved in the trial but commented on the results in an interview.
Another commenter was more critical of CONTESSA’s results, which were presented at the 2020 San Antonio Breast Cancer Symposium.
“Three months’ difference in PFS in this setting is meaningless without overall survival [OS] results,” Fatima Cardoso, MD, of Champalimaud Clinical Center in Lisbon, Portugal, said in a question submitted through the virtual meeting’s chat system.
At this point, the OS data are immature, and mature data won’t be available for another couple of years at least, according to the study’s protocol.
Dr. O’Shaughnessy defended the PFS result as being significant, however, saying it was comparable with outcomes seen previously with docetaxel-capecitabine and paclitaxel-gemcitabine combinations.
Other meeting attendees questioned why the waters had been muddied by testing the effects of tesetaxel in combination with capecitabine, albeit at a reduced dose, versus the approved full dose of capecitabine as monotherapy, particularly as a phase 2 trial had shown that tesetaxel demonstrated “significant activity” as monotherapy.
“The reason for the combination versus a monotherapy is because it was designed as a registration trial,” Dr. O’Shaughnessy explained. The trial was designed to be very similar to early taxane studies where docetaxel was assessed with or without capecitabine, or paclitaxel with or without gemcitabine.
“Probably we’re going to be using a doublet for patients who have virulent disease who really need a response,” Dr. O’Shaughnessy explained. She noted that the objective response rate was much higher with the tesetaxel-capecitabine combination than with capecitabine alone, and that result alone is “probably enough that we would utilize a doublet.”
The key thing is that it now gives patients an all-oral option, Dr. O’Shaughnessy said.
“The data are exciting because it would be terrific to have an orally available taxane chemotherapy,” agreed Dr. Burstein. “It is far more convenient for patients and opens access globally in places that do not have adequate resources for administration of IV therapeutics. Also, the data suggest that tesetaxel has a different side effect profile than IV taxane, with less neuropathy and less alopecia.”
Trial design
CONTESSA is an ongoing randomized, controlled trial that started in 2017 and is projected to end in early 2023. It is investigating the use of tesetaxel plus a reduced dose of capecitabine versus the approved dose of capecitabine alone in 685 women with hormone receptor–positive, HER2-negative locally advanced or metastatic breast cancer who had previously been treated with a taxane.
Being intrinsically orally bioavailable and more soluble than the other taxanes means that tesetaxel has a much longer half-life that allows for a “more convenient treatment experience for patients,” Dr. O’Shaughnessy observed.
Indeed, because tesetaxel only needs to be dosed once every 3 weeks, patients in the trial received tesetaxel at 27 mg/m2 only on the first day of a 21-day treatment cycle. This was combined with a reduced, 825-mg/m2 dose of capecitabine, given orally twice-daily on days 2-14 but once daily on the evening of day 1 and on the morning of day 15.
The combination regimen was compared with the recommended full dose of capecitabine alone, 1,250 mg/m2 given orally twice daily on days 2-14 but once daily on the evening of day 1 and on the morning of day 15.
Efficacy and safety
PFS was 9.8 months with tesetaxel plus capecitabine and 6.9 months with capecitabine alone, representing a 2.9-month improvement with the combination (hazard ratio, 0.716; P = .003).
A similar PFS benefit was seen regardless of multiple predefined subgroups, such as age, baseline performance status, duration of disease-free interval before study entry, and the use of CDK4/6 inhibitors.
The objective response rate was 57% with tesetaxel plus capecitabine and 41% with capecitabine alone (P = .0002). The 24-week disease control rate was 67% and 50%, respectively (P < .0001).
The most frequent treatment-emergent adverse event seen with the tesetaxel-capecitabine combination was neutropenia, occurring in 76.9% of patients, compared with 22.6% of patients in the monotherapy arm. Rates of grade 3-4 neutropenia were much higher in the combination arm (32.6% and 38.3%, respectively) than in the monotherapy arm (7.4% and 0.9%, respectively).
The neutropenia seen was “generally manageable,” Dr. O’Shaughnessy said, primarily with dose reductions and granulocyte colony–stimulating factor as needed.
She pointed out that rates of grade 3 or higher neuropathy and grade 2 alopecia were low, a respective 5.9% and 8%, with the combination.
The dose of capecitabine used in the control arm was noted to be higher than that used in usual practice.
“This was because of the global nature of the study and the regulatory requirements globally,” Dr. O’Shaughnessy said.
“The dose-modification scheme was that patients could have a dose reduction at the first sign of grade 2 toxicity,” she added, giving investigators the flexibility to reduce the dose as soon as possible.
This study was sponsored by Odonate Therapeutics. Dr. O’Shaughnessy disclosed consulting fees from AbbVie, Agendia, AstraZeneca, Bristol-Myers Squibb, Celgene, Eisai, Genentech/Roche, Genomic Health, GRAIL, Heron, Immunomedics, Ipsen, Jounce, Lilly, Novartis, Odonate, Pfizer, Puma, and Seagen. Dr. Burstein and Dr. Cardoso had no relevant disclosures.
SOURCE: O’Shaughnessy J et al. SABCS 2020, Abstract GS4-01.
FROM SABCS 2020
Vaccine-preventable infection risk high for pediatric hematopoietic cell transplantation recipients
Vaccine-preventable infections (VPIs) in pediatric hematopoietic cell transplantation (HCT) recipients cause significant morbidity, health care burden, and mortality.
Dana Danino, MD, and colleagues presented their evaluation of the prevalence and epidemiology of pediatric VPI-associated hospitalizations occurring within 5 years post HCT at the annual meeting of the European Society for Paediatric Infectious Diseases, held virtually this year.
“Pediatric HCT recipients are at increased risk of VPIs, and HCT recipients have poor outcomes from VPIs, compared with the general population,” explained Dr. Danino, of the department of pediatrics, and divisions of infectious diseases and host defense at the Ohio State University, Columbus. “However, the contemporary prevalence, risk factors, morbidity and mortality resulting from VPIs in children post HCT are not well known.”
Their epidemiological study, using the Pediatric Health Information System (PHIS) database, identified all children under 18 years that underwent allogeneic or autologous HCT in an 8-year period. A total of 9,591 unique HCT recipients were identified.
The researchers demonstrated that 7.1% of this cohort were hospitalized for a VPI in the first 5 years post HCT. Dr. Danino explained that 67% of VPI hospitalizations occurred during the first year, at a median of 222 days, and 22% of VPIs occurred during the initial HCT admission.
As to the type of infection, Dr. Danino and colleagues found that, the prevalence of VPI hospitalizations were highest for influenza, followed by varicella and invasive pneumococcal infections. They identified no hospitalizations due to measles or rubella during the study period.
The study findings revealed that the influenza infections occurred a median 231 days post HCT; varicella infections occurred a median 190 days; and invasive pneumococcal infections occurred a median 311 days post HCT.
“When we did a multivariate analysis by time post HCT, we found that age at transplantation, primary immune deficiency as an indication for transplantation, and graft versus host disease were independent predictors of VPIs during the initial HCT admission,” said Dr. Danino.
Children with a VPI who spent longer in hospital were more likely to be admitted to an ICU and have higher mortality, compared with children without a VPI diagnosis.
“VPIs led to longer duration of hospitalization, higher rates of ICU admission, and higher mortality, compared to HCT recipients without VPIs,” Dr. Danino explained. It was not possible in this retrospective study to determine whether increased mortality was VPI related.
These results underline the seriousness of infections in vulnerable children after HCT. Dr. Danino concluded by saying that “efforts to optimize vaccination strategies early post HCT are warranted to decrease VPIs.”
Dr. Danino had nothing to disclose.
Vaccine-preventable infections (VPIs) in pediatric hematopoietic cell transplantation (HCT) recipients cause significant morbidity, health care burden, and mortality.
Dana Danino, MD, and colleagues presented their evaluation of the prevalence and epidemiology of pediatric VPI-associated hospitalizations occurring within 5 years post HCT at the annual meeting of the European Society for Paediatric Infectious Diseases, held virtually this year.
“Pediatric HCT recipients are at increased risk of VPIs, and HCT recipients have poor outcomes from VPIs, compared with the general population,” explained Dr. Danino, of the department of pediatrics, and divisions of infectious diseases and host defense at the Ohio State University, Columbus. “However, the contemporary prevalence, risk factors, morbidity and mortality resulting from VPIs in children post HCT are not well known.”
Their epidemiological study, using the Pediatric Health Information System (PHIS) database, identified all children under 18 years that underwent allogeneic or autologous HCT in an 8-year period. A total of 9,591 unique HCT recipients were identified.
The researchers demonstrated that 7.1% of this cohort were hospitalized for a VPI in the first 5 years post HCT. Dr. Danino explained that 67% of VPI hospitalizations occurred during the first year, at a median of 222 days, and 22% of VPIs occurred during the initial HCT admission.
As to the type of infection, Dr. Danino and colleagues found that, the prevalence of VPI hospitalizations were highest for influenza, followed by varicella and invasive pneumococcal infections. They identified no hospitalizations due to measles or rubella during the study period.
The study findings revealed that the influenza infections occurred a median 231 days post HCT; varicella infections occurred a median 190 days; and invasive pneumococcal infections occurred a median 311 days post HCT.
“When we did a multivariate analysis by time post HCT, we found that age at transplantation, primary immune deficiency as an indication for transplantation, and graft versus host disease were independent predictors of VPIs during the initial HCT admission,” said Dr. Danino.
Children with a VPI who spent longer in hospital were more likely to be admitted to an ICU and have higher mortality, compared with children without a VPI diagnosis.
“VPIs led to longer duration of hospitalization, higher rates of ICU admission, and higher mortality, compared to HCT recipients without VPIs,” Dr. Danino explained. It was not possible in this retrospective study to determine whether increased mortality was VPI related.
These results underline the seriousness of infections in vulnerable children after HCT. Dr. Danino concluded by saying that “efforts to optimize vaccination strategies early post HCT are warranted to decrease VPIs.”
Dr. Danino had nothing to disclose.
Vaccine-preventable infections (VPIs) in pediatric hematopoietic cell transplantation (HCT) recipients cause significant morbidity, health care burden, and mortality.
Dana Danino, MD, and colleagues presented their evaluation of the prevalence and epidemiology of pediatric VPI-associated hospitalizations occurring within 5 years post HCT at the annual meeting of the European Society for Paediatric Infectious Diseases, held virtually this year.
“Pediatric HCT recipients are at increased risk of VPIs, and HCT recipients have poor outcomes from VPIs, compared with the general population,” explained Dr. Danino, of the department of pediatrics, and divisions of infectious diseases and host defense at the Ohio State University, Columbus. “However, the contemporary prevalence, risk factors, morbidity and mortality resulting from VPIs in children post HCT are not well known.”
Their epidemiological study, using the Pediatric Health Information System (PHIS) database, identified all children under 18 years that underwent allogeneic or autologous HCT in an 8-year period. A total of 9,591 unique HCT recipients were identified.
The researchers demonstrated that 7.1% of this cohort were hospitalized for a VPI in the first 5 years post HCT. Dr. Danino explained that 67% of VPI hospitalizations occurred during the first year, at a median of 222 days, and 22% of VPIs occurred during the initial HCT admission.
As to the type of infection, Dr. Danino and colleagues found that, the prevalence of VPI hospitalizations were highest for influenza, followed by varicella and invasive pneumococcal infections. They identified no hospitalizations due to measles or rubella during the study period.
The study findings revealed that the influenza infections occurred a median 231 days post HCT; varicella infections occurred a median 190 days; and invasive pneumococcal infections occurred a median 311 days post HCT.
“When we did a multivariate analysis by time post HCT, we found that age at transplantation, primary immune deficiency as an indication for transplantation, and graft versus host disease were independent predictors of VPIs during the initial HCT admission,” said Dr. Danino.
Children with a VPI who spent longer in hospital were more likely to be admitted to an ICU and have higher mortality, compared with children without a VPI diagnosis.
“VPIs led to longer duration of hospitalization, higher rates of ICU admission, and higher mortality, compared to HCT recipients without VPIs,” Dr. Danino explained. It was not possible in this retrospective study to determine whether increased mortality was VPI related.
These results underline the seriousness of infections in vulnerable children after HCT. Dr. Danino concluded by saying that “efforts to optimize vaccination strategies early post HCT are warranted to decrease VPIs.”
Dr. Danino had nothing to disclose.
FROM ESPID 2020