User login
-
MM:New Tool Gauges Post–CAR T Relapse Risk
“To our knowledge, this large multicenter study is the first report to identify patients with RRMM at high risk of early relapse after CAR-T,” the authors report in the study, published February 15 in the Journal of Clinical Oncology.
“We saw that early relapse within 5 months from infusion was significantly associated with very poor outcomes, and disease-, treatment-, and inflammation-specific variables were independent predictors of early relapse,” first author Nico Gagelmann, MD, of the University Medical Center Hamburg-Eppendorf, in Hamburg, Germany, explained in presenting the findings at the 6th European CAR T-cell Meeting jointly sponsored by the European Society for Blood and Marrow Transplantation and the European Hematology Association. CAR-T therapy has revolutionized the treatment of RRMM, with the idecabtagene vicleucel (ide-cel) and ciltacabtagene autoleucel (cilta-cel) CAR-T therapies approved for the condition. However, the treatment is far from a cure, with nearly 50% of patients relapsing and having progression of disease within the first year after infusion, prompting a need to better understand the risk factors for who may or may not progress.
With a lack of a universal model to help with those predictions across products and populations, Dr. Gagelmann and colleagues conducted a retrospective observational study utilizing data from 136 patients at seven CAR-T centers in Europe and 133 patients at three centers in the US who had received either commercial or academically produced anti-BCMA CAR-T.
Of the patients, 171 were infused with ide-cel, 38 with cilta-cel, and 60 with an academic CAR-T therapy. The patients had a median age of 63, and extramedullary disease was more common in the US cohort (48%) versus European (35%; P = .04).
Notably, the response rates between the European and US cohorts were similar, despite various differences between the cohorts, including differences in ethnicities and a lower body mass index (BMI) in the European cohort versus US (BMI 25 vs 28, respectively; P < .001). There were also no significant differences in responses between the CAR-T treatments.
The overall response rate was 87% and was comparable between the European and US groups, with complete responses occurring among 48% of patients in Europe and 49% in the US group.
Their measurable residual disease (MRD) negativity rate at any time was 29% and 37%, respectively, and rates of complete response at day 30 were 29% and 26%, respectively. The rate of progression-free survival at 12 months was 40% for the entire cohort, with a rate of 45% in the European group and 34% in the US group (P = .09). Overall survival rates at 12 months were 79% and 65%, respectively (P = .11).
The patients had a median time to relapse of 5 months, and the 5-month incidence of relapse was identical, at 24% in each cohort.
Of those patients, overall survival at 12 months was low, at 30% in the European cohort and 14% in the US group.
“Early relapse within the first 5 months clearly identified patients with poor survival across the cohort,” Dr. Gagelmann said.
Key Risk Factors Identified
Key factors found after multivariate adjustment to be independently predictive of early relapse or progression included extramedullary disease or plasma cell leukemia, being refractory to lenalidomide, having high-risk cytogenetics, and having increased age- and sex-adjusted ferritin at the time of lymphodepletion.
With each of the risk factors valued at 1 point, the MyCARe model ranked scores of 0-1 points as low-risk, 2-3 as intermediate risk, and a score above 4 was considered high-risk.
Based on the model, the risk of early relapse within 5 months among those scored as low risk was 7%, for intermediate risk, 27% (hazard ratio [HR], 3.27 vs low-risk; P < .001), and for high risk, 53% (HR, 7.89 vs low-risk; P < .001), with outcomes overall comparable between the two geographic groups. Importantly, the model maintained utility for patients who did and did not receive salvage therapies; however, “more studies are needed to identify the optimal post–CAR-T approach,” the authors write.
Dr. Gagelmann added that older age was significantly associated with improved progression-free survival in the US cohort, with a 12-month progression-free survival of 27% among patients under 65 versus 43% for those over 65 (P = .03). However, age was not found to be associated with similar outcomes in the European cohort.
The authors note that the MyCARe model outperformed the CAR-HEMATOTOX and more recent disease-specific R2-ISS risk-stratification tools regarding prediction of relapse/progression and progression-free survival.
However, with CAR-HEMATOTOX developed to predict side effects and non-relapse mortality, “our results demonstrate that both scores independently predict different outcomes after anti–BCMA CAR-T in RRMM,” the authors report. Therefore, “they can be used complimentarily to predict complications (CAR-HEMATOTOX) and relapse/progression-free survival (MyCARe model).”
Importantly, the authors add that the tool may help in patient selection for earlier treatment.
“As ide-cel and cilta-cel have shown astonishing efficacy for earlier treatment lines, our model might also be validated for such patients,” the authors note in the study. They conclude that the study provides “the first Euro-American cartography of the efficacy and safety profile of current CAR-T, showing comparable results.”
“We also built the MyCARe model, which can predict early relapse, response, and survival and may facilitate patient selection in this very challenging setting,” the authors report.
Hope for Interventions Based on Patients’ Risk
Commenting on the study, Rahul Banerjee, MD, an assistant professor with the Division of Hematology and Oncology, University of Washington, Seattle, underscored that “we need more cross-border research like this in the myeloma field.”
“Clinically, my hope that this will help us tailor post–CAR-T interventions according to each patient’s risk profile,” he said.
Risk factors such as the presence of extramedullary disease, plasma cell leukemia, or high-risk cytogenetics are expected; however, Dr. Banerjee said the inclusion of increased ferritin before CAR-T was “an interesting new risk factor that we’ve also heard about from our colleagues in the lymphoma space.”
Ferritin perturbations can indicate many things, but high ferritin can be a sign of elevated inflammation at baseline,” he explained. “These patients may have a hyperinflammatory phenotype of their myeloma which can predispose T-cells to exhaustion,” Dr. Banerjee said.
“Exhausted T-cells at collection mean exhausted CAR T-cells at infusion, and so the negative prognostic significance of elevated ferritin — which we don’t always check before CAR-T — makes sense.”
While the authors suggest a potential benefit of the MyCAR3 model in identifying patients who could benefit from other novel therapies at relapse, Dr. Banerjee suggests another possibility. “I’d take this a step further and suggest future studies of this MyCARe model to identify patients who might benefit from post–CAR-T maintenance,” he said.
“The ‘one-and-done’ nature of CAR-T in terms of not requiring further myeloma therapy after infusion is a powerful benefit for patients, but there are some patients who may benefit from low-dose pomalidomide or iberdomide/mezigdomide maintenance to help keep the myeloma at bay and to promote T-cell fitness,” Dr. Banerjee explained. “This risk model may identify patients to prioritize for such types of clinical trials in the future.”
Caveats include that factors beyond the baseline features (used for the risk model) can further influence outcomes,” Dr. Banerjee noted.
“Risk stratification is inherently a dynamic process over time,” he said, questioning, for instance, “what about patients who achieve measurable residual disease negativity [MRD] at day +28 after CAR-T cell? Does the achievement of MRD negativity ‘erase’ a high-risk MyCARe score? We’ll need future studies to tell.”
An overriding take-home message for clinicians should be to simply refer eligible patients to a CAR-T capable center as soon as possible for evaluation.
“In the lymphoma world, they have a nice adage for this: ‘If they recur, you should refer,’ ” he said. “I’d suggest the same here. By no means will we move to CAR-T therapy for every patient at first relapse. However, based on their MyCARe score and other risk factors, there may be patients we prioritize for CAR-T first versus CAR-T with maintenance versus clinical trials.”
Dr. Gagelmann reported relationships with BMS, Pfizer, Stemline, MorphoSys, and Kite. Dr. Banerjee disclosed ties with BMS, Caribou Biosciences, Genentech, Janssen, Karyopharm, Pfizer, Sanofi, SparkCures, Novartis, and Pack Health.
“To our knowledge, this large multicenter study is the first report to identify patients with RRMM at high risk of early relapse after CAR-T,” the authors report in the study, published February 15 in the Journal of Clinical Oncology.
“We saw that early relapse within 5 months from infusion was significantly associated with very poor outcomes, and disease-, treatment-, and inflammation-specific variables were independent predictors of early relapse,” first author Nico Gagelmann, MD, of the University Medical Center Hamburg-Eppendorf, in Hamburg, Germany, explained in presenting the findings at the 6th European CAR T-cell Meeting jointly sponsored by the European Society for Blood and Marrow Transplantation and the European Hematology Association. CAR-T therapy has revolutionized the treatment of RRMM, with the idecabtagene vicleucel (ide-cel) and ciltacabtagene autoleucel (cilta-cel) CAR-T therapies approved for the condition. However, the treatment is far from a cure, with nearly 50% of patients relapsing and having progression of disease within the first year after infusion, prompting a need to better understand the risk factors for who may or may not progress.
With a lack of a universal model to help with those predictions across products and populations, Dr. Gagelmann and colleagues conducted a retrospective observational study utilizing data from 136 patients at seven CAR-T centers in Europe and 133 patients at three centers in the US who had received either commercial or academically produced anti-BCMA CAR-T.
Of the patients, 171 were infused with ide-cel, 38 with cilta-cel, and 60 with an academic CAR-T therapy. The patients had a median age of 63, and extramedullary disease was more common in the US cohort (48%) versus European (35%; P = .04).
Notably, the response rates between the European and US cohorts were similar, despite various differences between the cohorts, including differences in ethnicities and a lower body mass index (BMI) in the European cohort versus US (BMI 25 vs 28, respectively; P < .001). There were also no significant differences in responses between the CAR-T treatments.
The overall response rate was 87% and was comparable between the European and US groups, with complete responses occurring among 48% of patients in Europe and 49% in the US group.
Their measurable residual disease (MRD) negativity rate at any time was 29% and 37%, respectively, and rates of complete response at day 30 were 29% and 26%, respectively. The rate of progression-free survival at 12 months was 40% for the entire cohort, with a rate of 45% in the European group and 34% in the US group (P = .09). Overall survival rates at 12 months were 79% and 65%, respectively (P = .11).
The patients had a median time to relapse of 5 months, and the 5-month incidence of relapse was identical, at 24% in each cohort.
Of those patients, overall survival at 12 months was low, at 30% in the European cohort and 14% in the US group.
“Early relapse within the first 5 months clearly identified patients with poor survival across the cohort,” Dr. Gagelmann said.
Key Risk Factors Identified
Key factors found after multivariate adjustment to be independently predictive of early relapse or progression included extramedullary disease or plasma cell leukemia, being refractory to lenalidomide, having high-risk cytogenetics, and having increased age- and sex-adjusted ferritin at the time of lymphodepletion.
With each of the risk factors valued at 1 point, the MyCARe model ranked scores of 0-1 points as low-risk, 2-3 as intermediate risk, and a score above 4 was considered high-risk.
Based on the model, the risk of early relapse within 5 months among those scored as low risk was 7%, for intermediate risk, 27% (hazard ratio [HR], 3.27 vs low-risk; P < .001), and for high risk, 53% (HR, 7.89 vs low-risk; P < .001), with outcomes overall comparable between the two geographic groups. Importantly, the model maintained utility for patients who did and did not receive salvage therapies; however, “more studies are needed to identify the optimal post–CAR-T approach,” the authors write.
Dr. Gagelmann added that older age was significantly associated with improved progression-free survival in the US cohort, with a 12-month progression-free survival of 27% among patients under 65 versus 43% for those over 65 (P = .03). However, age was not found to be associated with similar outcomes in the European cohort.
The authors note that the MyCARe model outperformed the CAR-HEMATOTOX and more recent disease-specific R2-ISS risk-stratification tools regarding prediction of relapse/progression and progression-free survival.
However, with CAR-HEMATOTOX developed to predict side effects and non-relapse mortality, “our results demonstrate that both scores independently predict different outcomes after anti–BCMA CAR-T in RRMM,” the authors report. Therefore, “they can be used complimentarily to predict complications (CAR-HEMATOTOX) and relapse/progression-free survival (MyCARe model).”
Importantly, the authors add that the tool may help in patient selection for earlier treatment.
“As ide-cel and cilta-cel have shown astonishing efficacy for earlier treatment lines, our model might also be validated for such patients,” the authors note in the study. They conclude that the study provides “the first Euro-American cartography of the efficacy and safety profile of current CAR-T, showing comparable results.”
“We also built the MyCARe model, which can predict early relapse, response, and survival and may facilitate patient selection in this very challenging setting,” the authors report.
Hope for Interventions Based on Patients’ Risk
Commenting on the study, Rahul Banerjee, MD, an assistant professor with the Division of Hematology and Oncology, University of Washington, Seattle, underscored that “we need more cross-border research like this in the myeloma field.”
“Clinically, my hope that this will help us tailor post–CAR-T interventions according to each patient’s risk profile,” he said.
Risk factors such as the presence of extramedullary disease, plasma cell leukemia, or high-risk cytogenetics are expected; however, Dr. Banerjee said the inclusion of increased ferritin before CAR-T was “an interesting new risk factor that we’ve also heard about from our colleagues in the lymphoma space.”
Ferritin perturbations can indicate many things, but high ferritin can be a sign of elevated inflammation at baseline,” he explained. “These patients may have a hyperinflammatory phenotype of their myeloma which can predispose T-cells to exhaustion,” Dr. Banerjee said.
“Exhausted T-cells at collection mean exhausted CAR T-cells at infusion, and so the negative prognostic significance of elevated ferritin — which we don’t always check before CAR-T — makes sense.”
While the authors suggest a potential benefit of the MyCAR3 model in identifying patients who could benefit from other novel therapies at relapse, Dr. Banerjee suggests another possibility. “I’d take this a step further and suggest future studies of this MyCARe model to identify patients who might benefit from post–CAR-T maintenance,” he said.
“The ‘one-and-done’ nature of CAR-T in terms of not requiring further myeloma therapy after infusion is a powerful benefit for patients, but there are some patients who may benefit from low-dose pomalidomide or iberdomide/mezigdomide maintenance to help keep the myeloma at bay and to promote T-cell fitness,” Dr. Banerjee explained. “This risk model may identify patients to prioritize for such types of clinical trials in the future.”
Caveats include that factors beyond the baseline features (used for the risk model) can further influence outcomes,” Dr. Banerjee noted.
“Risk stratification is inherently a dynamic process over time,” he said, questioning, for instance, “what about patients who achieve measurable residual disease negativity [MRD] at day +28 after CAR-T cell? Does the achievement of MRD negativity ‘erase’ a high-risk MyCARe score? We’ll need future studies to tell.”
An overriding take-home message for clinicians should be to simply refer eligible patients to a CAR-T capable center as soon as possible for evaluation.
“In the lymphoma world, they have a nice adage for this: ‘If they recur, you should refer,’ ” he said. “I’d suggest the same here. By no means will we move to CAR-T therapy for every patient at first relapse. However, based on their MyCARe score and other risk factors, there may be patients we prioritize for CAR-T first versus CAR-T with maintenance versus clinical trials.”
Dr. Gagelmann reported relationships with BMS, Pfizer, Stemline, MorphoSys, and Kite. Dr. Banerjee disclosed ties with BMS, Caribou Biosciences, Genentech, Janssen, Karyopharm, Pfizer, Sanofi, SparkCures, Novartis, and Pack Health.
“To our knowledge, this large multicenter study is the first report to identify patients with RRMM at high risk of early relapse after CAR-T,” the authors report in the study, published February 15 in the Journal of Clinical Oncology.
“We saw that early relapse within 5 months from infusion was significantly associated with very poor outcomes, and disease-, treatment-, and inflammation-specific variables were independent predictors of early relapse,” first author Nico Gagelmann, MD, of the University Medical Center Hamburg-Eppendorf, in Hamburg, Germany, explained in presenting the findings at the 6th European CAR T-cell Meeting jointly sponsored by the European Society for Blood and Marrow Transplantation and the European Hematology Association. CAR-T therapy has revolutionized the treatment of RRMM, with the idecabtagene vicleucel (ide-cel) and ciltacabtagene autoleucel (cilta-cel) CAR-T therapies approved for the condition. However, the treatment is far from a cure, with nearly 50% of patients relapsing and having progression of disease within the first year after infusion, prompting a need to better understand the risk factors for who may or may not progress.
With a lack of a universal model to help with those predictions across products and populations, Dr. Gagelmann and colleagues conducted a retrospective observational study utilizing data from 136 patients at seven CAR-T centers in Europe and 133 patients at three centers in the US who had received either commercial or academically produced anti-BCMA CAR-T.
Of the patients, 171 were infused with ide-cel, 38 with cilta-cel, and 60 with an academic CAR-T therapy. The patients had a median age of 63, and extramedullary disease was more common in the US cohort (48%) versus European (35%; P = .04).
Notably, the response rates between the European and US cohorts were similar, despite various differences between the cohorts, including differences in ethnicities and a lower body mass index (BMI) in the European cohort versus US (BMI 25 vs 28, respectively; P < .001). There were also no significant differences in responses between the CAR-T treatments.
The overall response rate was 87% and was comparable between the European and US groups, with complete responses occurring among 48% of patients in Europe and 49% in the US group.
Their measurable residual disease (MRD) negativity rate at any time was 29% and 37%, respectively, and rates of complete response at day 30 were 29% and 26%, respectively. The rate of progression-free survival at 12 months was 40% for the entire cohort, with a rate of 45% in the European group and 34% in the US group (P = .09). Overall survival rates at 12 months were 79% and 65%, respectively (P = .11).
The patients had a median time to relapse of 5 months, and the 5-month incidence of relapse was identical, at 24% in each cohort.
Of those patients, overall survival at 12 months was low, at 30% in the European cohort and 14% in the US group.
“Early relapse within the first 5 months clearly identified patients with poor survival across the cohort,” Dr. Gagelmann said.
Key Risk Factors Identified
Key factors found after multivariate adjustment to be independently predictive of early relapse or progression included extramedullary disease or plasma cell leukemia, being refractory to lenalidomide, having high-risk cytogenetics, and having increased age- and sex-adjusted ferritin at the time of lymphodepletion.
With each of the risk factors valued at 1 point, the MyCARe model ranked scores of 0-1 points as low-risk, 2-3 as intermediate risk, and a score above 4 was considered high-risk.
Based on the model, the risk of early relapse within 5 months among those scored as low risk was 7%, for intermediate risk, 27% (hazard ratio [HR], 3.27 vs low-risk; P < .001), and for high risk, 53% (HR, 7.89 vs low-risk; P < .001), with outcomes overall comparable between the two geographic groups. Importantly, the model maintained utility for patients who did and did not receive salvage therapies; however, “more studies are needed to identify the optimal post–CAR-T approach,” the authors write.
Dr. Gagelmann added that older age was significantly associated with improved progression-free survival in the US cohort, with a 12-month progression-free survival of 27% among patients under 65 versus 43% for those over 65 (P = .03). However, age was not found to be associated with similar outcomes in the European cohort.
The authors note that the MyCARe model outperformed the CAR-HEMATOTOX and more recent disease-specific R2-ISS risk-stratification tools regarding prediction of relapse/progression and progression-free survival.
However, with CAR-HEMATOTOX developed to predict side effects and non-relapse mortality, “our results demonstrate that both scores independently predict different outcomes after anti–BCMA CAR-T in RRMM,” the authors report. Therefore, “they can be used complimentarily to predict complications (CAR-HEMATOTOX) and relapse/progression-free survival (MyCARe model).”
Importantly, the authors add that the tool may help in patient selection for earlier treatment.
“As ide-cel and cilta-cel have shown astonishing efficacy for earlier treatment lines, our model might also be validated for such patients,” the authors note in the study. They conclude that the study provides “the first Euro-American cartography of the efficacy and safety profile of current CAR-T, showing comparable results.”
“We also built the MyCARe model, which can predict early relapse, response, and survival and may facilitate patient selection in this very challenging setting,” the authors report.
Hope for Interventions Based on Patients’ Risk
Commenting on the study, Rahul Banerjee, MD, an assistant professor with the Division of Hematology and Oncology, University of Washington, Seattle, underscored that “we need more cross-border research like this in the myeloma field.”
“Clinically, my hope that this will help us tailor post–CAR-T interventions according to each patient’s risk profile,” he said.
Risk factors such as the presence of extramedullary disease, plasma cell leukemia, or high-risk cytogenetics are expected; however, Dr. Banerjee said the inclusion of increased ferritin before CAR-T was “an interesting new risk factor that we’ve also heard about from our colleagues in the lymphoma space.”
Ferritin perturbations can indicate many things, but high ferritin can be a sign of elevated inflammation at baseline,” he explained. “These patients may have a hyperinflammatory phenotype of their myeloma which can predispose T-cells to exhaustion,” Dr. Banerjee said.
“Exhausted T-cells at collection mean exhausted CAR T-cells at infusion, and so the negative prognostic significance of elevated ferritin — which we don’t always check before CAR-T — makes sense.”
While the authors suggest a potential benefit of the MyCAR3 model in identifying patients who could benefit from other novel therapies at relapse, Dr. Banerjee suggests another possibility. “I’d take this a step further and suggest future studies of this MyCARe model to identify patients who might benefit from post–CAR-T maintenance,” he said.
“The ‘one-and-done’ nature of CAR-T in terms of not requiring further myeloma therapy after infusion is a powerful benefit for patients, but there are some patients who may benefit from low-dose pomalidomide or iberdomide/mezigdomide maintenance to help keep the myeloma at bay and to promote T-cell fitness,” Dr. Banerjee explained. “This risk model may identify patients to prioritize for such types of clinical trials in the future.”
Caveats include that factors beyond the baseline features (used for the risk model) can further influence outcomes,” Dr. Banerjee noted.
“Risk stratification is inherently a dynamic process over time,” he said, questioning, for instance, “what about patients who achieve measurable residual disease negativity [MRD] at day +28 after CAR-T cell? Does the achievement of MRD negativity ‘erase’ a high-risk MyCARe score? We’ll need future studies to tell.”
An overriding take-home message for clinicians should be to simply refer eligible patients to a CAR-T capable center as soon as possible for evaluation.
“In the lymphoma world, they have a nice adage for this: ‘If they recur, you should refer,’ ” he said. “I’d suggest the same here. By no means will we move to CAR-T therapy for every patient at first relapse. However, based on their MyCARe score and other risk factors, there may be patients we prioritize for CAR-T first versus CAR-T with maintenance versus clinical trials.”
Dr. Gagelmann reported relationships with BMS, Pfizer, Stemline, MorphoSys, and Kite. Dr. Banerjee disclosed ties with BMS, Caribou Biosciences, Genentech, Janssen, Karyopharm, Pfizer, Sanofi, SparkCures, Novartis, and Pack Health.
FROM THE 6TH EUROPEAN CAR T-CELL MEETING
FDA Withdraws Melflufen Approval, but EMA Still Allows Its Use
But the European Medicines Agency (EMA) still authorizes the drug’s manufacturer Oncopeptides AB to market the drug, also called Pepaxti, in Europe, Iceland, Lichtenstein, Norway, and the United Kingdom.
Amol Akhade, MBBS, who describes himself as a senior consultant medical and hemato oncologist–bone marrow transplant physician on LinkedIn, raised questions about the inconsistencies between the FDA and EMA’s opinions about these drugs. Dr. Akhad, of Suyog Cancer Clinics in India, posted via the following handle @SuyogCancer on X (Twitter):
“How can one drug and one trial data [have] two diagonally different outcomes from two different drug approval agencies?
Melphalan Flufenamide is finally completely withdrawn by @US_FDA
But approval by @EMA_News stays.
How can be one drug be harmful across one side of Atlantic Ocean and becomes safe and useful on the other side of Atlantic Ocean?
Modern day miracle?”
EMA: Pepaxti’s Benefits Exceed Its Risks
The EMA, which could not be reached for comment regarding why the agency was still allowing patients to use the drug, said the following about Pepaxti on its website:
“The European Medicines Agency decided that Pepaxti’s benefits are greater than its risks and it can be authorised for use in the EU. The Agency noted the unmet medical need for patients with multiple myeloma who no longer improve with the available therapies. Despite some limitations in the studies, the results were considered clinically relevant, with the exception of the subgroup of patients who had an autologous stem cell transplant and whose disease progressed within three years of transplantation.
Regarding safety, although side effects, including severe effects, were seen with treatment involving Pepaxti, these were considered acceptable and manageable,” the agency wrote.
“Recommendations and precautions to be followed by healthcare professionals and patients for the safe and effective use of Pepaxti have been included in the summary of product characteristics and the package leaflet.
As for all medicines, data on the use of Pepaxti are continuously monitored. Suspected side effects reported with Pepaxti are carefully evaluated and any necessary action taken to protect patients,” according to the EMA.
The FDA’s final decision, issued on February 23, 2024, follows its warning in 2021 that meflufen plus dexamethasone exposed patients with multiple myeloma to increased risk for death, and its call for withdrawal of the drug in 2022.
“The grounds for withdrawing approval have been met because: (1) the confirmatory study conducted as a condition of accelerated approval did not confirm Pepaxto’s clinical benefit and (2) the available evidence demonstrates that Pepaxto is not shown to be safe or effective under its conditions of use,” Peter Marks, MD, PhD, Director of the FDA Center for Biologics Evaluation and Research, wrote in the final decision document.
Oncopeptides AB: Drug ‘Caters to a Large Unmet Need’
David Augustsson, Director of Corporate Affairs, Oncopeptides AB, explained in an interview why he thinks the EMA and FDA’s actions regarding the drug differ from each other.

“The European Medicines Agency had the opinion that the OCEAN study met its primary endpoint by demonstrating superior progression-free survival and it agreed that the potential detriment of overall survival was limited to patients progressing less than 36 months after an autologous stem cell transplant,” he said.“The FDA was not willing to acknowledge the observed clinically relevant differences across patient subgroups in the OCEAN study as confirmed.”
Mr. Augustsson added that this decision will deprive US patients of access to “a drug we believe caters to a large unmet need among elderly multiple myeloma patients with few treatment options left.”
“While we remain confident that we have science on our side we are of course disappointed in the decision [to remove Pepaxto from the US market],” Oncopeptides AB CEO Sofia Heigis said in a statement. “At the same time this is no change to our plans and we will continue to focus all our attention on the commercialization in Europe, progression of our pipeline and rest of world opportunities.”
FDA 'Took Swift Action' to Ensure Users of Pepaxto Were Informed of Risks
In February 2021, the FDA used the Accelerated Approval Program to enable certain patients with multiple myeloma to be treated with the peptide conjugated alkylating drug melflufen plus dexamethasone. Under the program, Oncopeptides was required to conduct the phase III randomized, controlled OCEAN clinical trial.
OCEAN enrolled 495 patients with relapsed/refractory multiple myeloma who had 2 to 4 lines of prior therapy and who were refractory to lenalidomide in the last line of therapy. Participants in the multinational study received either melflufen plus dexamethasone or pomalidomide plus dexamethasone until disease progression, unacceptable toxicity, or lack of benefit.
In July 2021, the FDA issued an alert that the study results showed increased risk for death in participants treated with melflufen. In October that year, at FDA request, Oncopeptides removed the drug from the US market but continued to provide it to patients already receiving it. In December 2022, the FDA requested that the company withdraw melflufen’s US marketing authorization.
Responding to questions about the timing of the FDA’s most recent decision about Pepaxto and how the decision will affect patient care in the US, the FDA emailed the following statement to this news organization:
“Since the OCEAN trial results for Pepaxto in 2021, the FDA has responded to safety concerns about Pepaxto by issuing a CDER Alert, communicating concerns to Oncopeptides, holding an Oncologic Drugs Advisory Committee meeting in September 2022, and issuing a letter of notice to Oncopeptides in July 2023, proposing to withdraw Pepaxto (NDA 214383). After receiving the notice, Oncopeptides appealed the withdrawal in August 2023. A meeting was held with the Commissioner’s designee, Dr. Peter Marks, Oncopeptides, and others from FDA in October 2023. Dr. Marks reviewed the record and considered the arguments made on appeal and issued a final decision on February 23, 2024. Prior to reaching a decision, the FDA took swift action to ensure those receiving Pepaxto in the post-confirmatory clinical trial were informed of the risks and that no new patients were enrolled in the trial. We also note that it is our understanding that Pepaxto has not been marketed in the U.S. since October 22, 2021.”
“This is the first time FDA has used the amended procedures for withdrawal of accelerated approval that were enacted in 2023, as part of the Food and Drug Omnibus Report Act of 2022 (FDORA),” the agency wrote in a Feb 23 statement. The agency will also remove melflufen from the Approved Drug Products with Therapeutic Equivalence Evaluations, also called the Orange Book.
But the European Medicines Agency (EMA) still authorizes the drug’s manufacturer Oncopeptides AB to market the drug, also called Pepaxti, in Europe, Iceland, Lichtenstein, Norway, and the United Kingdom.
Amol Akhade, MBBS, who describes himself as a senior consultant medical and hemato oncologist–bone marrow transplant physician on LinkedIn, raised questions about the inconsistencies between the FDA and EMA’s opinions about these drugs. Dr. Akhad, of Suyog Cancer Clinics in India, posted via the following handle @SuyogCancer on X (Twitter):
“How can one drug and one trial data [have] two diagonally different outcomes from two different drug approval agencies?
Melphalan Flufenamide is finally completely withdrawn by @US_FDA
But approval by @EMA_News stays.
How can be one drug be harmful across one side of Atlantic Ocean and becomes safe and useful on the other side of Atlantic Ocean?
Modern day miracle?”
EMA: Pepaxti’s Benefits Exceed Its Risks
The EMA, which could not be reached for comment regarding why the agency was still allowing patients to use the drug, said the following about Pepaxti on its website:
“The European Medicines Agency decided that Pepaxti’s benefits are greater than its risks and it can be authorised for use in the EU. The Agency noted the unmet medical need for patients with multiple myeloma who no longer improve with the available therapies. Despite some limitations in the studies, the results were considered clinically relevant, with the exception of the subgroup of patients who had an autologous stem cell transplant and whose disease progressed within three years of transplantation.
Regarding safety, although side effects, including severe effects, were seen with treatment involving Pepaxti, these were considered acceptable and manageable,” the agency wrote.
“Recommendations and precautions to be followed by healthcare professionals and patients for the safe and effective use of Pepaxti have been included in the summary of product characteristics and the package leaflet.
As for all medicines, data on the use of Pepaxti are continuously monitored. Suspected side effects reported with Pepaxti are carefully evaluated and any necessary action taken to protect patients,” according to the EMA.
The FDA’s final decision, issued on February 23, 2024, follows its warning in 2021 that meflufen plus dexamethasone exposed patients with multiple myeloma to increased risk for death, and its call for withdrawal of the drug in 2022.
“The grounds for withdrawing approval have been met because: (1) the confirmatory study conducted as a condition of accelerated approval did not confirm Pepaxto’s clinical benefit and (2) the available evidence demonstrates that Pepaxto is not shown to be safe or effective under its conditions of use,” Peter Marks, MD, PhD, Director of the FDA Center for Biologics Evaluation and Research, wrote in the final decision document.
Oncopeptides AB: Drug ‘Caters to a Large Unmet Need’
David Augustsson, Director of Corporate Affairs, Oncopeptides AB, explained in an interview why he thinks the EMA and FDA’s actions regarding the drug differ from each other.
“The European Medicines Agency had the opinion that the OCEAN study met its primary endpoint by demonstrating superior progression-free survival and it agreed that the potential detriment of overall survival was limited to patients progressing less than 36 months after an autologous stem cell transplant,” he said.“The FDA was not willing to acknowledge the observed clinically relevant differences across patient subgroups in the OCEAN study as confirmed.”
Mr. Augustsson added that this decision will deprive US patients of access to “a drug we believe caters to a large unmet need among elderly multiple myeloma patients with few treatment options left.”
“While we remain confident that we have science on our side we are of course disappointed in the decision [to remove Pepaxto from the US market],” Oncopeptides AB CEO Sofia Heigis said in a statement. “At the same time this is no change to our plans and we will continue to focus all our attention on the commercialization in Europe, progression of our pipeline and rest of world opportunities.”
FDA 'Took Swift Action' to Ensure Users of Pepaxto Were Informed of Risks
In February 2021, the FDA used the Accelerated Approval Program to enable certain patients with multiple myeloma to be treated with the peptide conjugated alkylating drug melflufen plus dexamethasone. Under the program, Oncopeptides was required to conduct the phase III randomized, controlled OCEAN clinical trial.
OCEAN enrolled 495 patients with relapsed/refractory multiple myeloma who had 2 to 4 lines of prior therapy and who were refractory to lenalidomide in the last line of therapy. Participants in the multinational study received either melflufen plus dexamethasone or pomalidomide plus dexamethasone until disease progression, unacceptable toxicity, or lack of benefit.
In July 2021, the FDA issued an alert that the study results showed increased risk for death in participants treated with melflufen. In October that year, at FDA request, Oncopeptides removed the drug from the US market but continued to provide it to patients already receiving it. In December 2022, the FDA requested that the company withdraw melflufen’s US marketing authorization.
Responding to questions about the timing of the FDA’s most recent decision about Pepaxto and how the decision will affect patient care in the US, the FDA emailed the following statement to this news organization:
“Since the OCEAN trial results for Pepaxto in 2021, the FDA has responded to safety concerns about Pepaxto by issuing a CDER Alert, communicating concerns to Oncopeptides, holding an Oncologic Drugs Advisory Committee meeting in September 2022, and issuing a letter of notice to Oncopeptides in July 2023, proposing to withdraw Pepaxto (NDA 214383). After receiving the notice, Oncopeptides appealed the withdrawal in August 2023. A meeting was held with the Commissioner’s designee, Dr. Peter Marks, Oncopeptides, and others from FDA in October 2023. Dr. Marks reviewed the record and considered the arguments made on appeal and issued a final decision on February 23, 2024. Prior to reaching a decision, the FDA took swift action to ensure those receiving Pepaxto in the post-confirmatory clinical trial were informed of the risks and that no new patients were enrolled in the trial. We also note that it is our understanding that Pepaxto has not been marketed in the U.S. since October 22, 2021.”
“This is the first time FDA has used the amended procedures for withdrawal of accelerated approval that were enacted in 2023, as part of the Food and Drug Omnibus Report Act of 2022 (FDORA),” the agency wrote in a Feb 23 statement. The agency will also remove melflufen from the Approved Drug Products with Therapeutic Equivalence Evaluations, also called the Orange Book.
But the European Medicines Agency (EMA) still authorizes the drug’s manufacturer Oncopeptides AB to market the drug, also called Pepaxti, in Europe, Iceland, Lichtenstein, Norway, and the United Kingdom.
Amol Akhade, MBBS, who describes himself as a senior consultant medical and hemato oncologist–bone marrow transplant physician on LinkedIn, raised questions about the inconsistencies between the FDA and EMA’s opinions about these drugs. Dr. Akhad, of Suyog Cancer Clinics in India, posted via the following handle @SuyogCancer on X (Twitter):
“How can one drug and one trial data [have] two diagonally different outcomes from two different drug approval agencies?
Melphalan Flufenamide is finally completely withdrawn by @US_FDA
But approval by @EMA_News stays.
How can be one drug be harmful across one side of Atlantic Ocean and becomes safe and useful on the other side of Atlantic Ocean?
Modern day miracle?”
EMA: Pepaxti’s Benefits Exceed Its Risks
The EMA, which could not be reached for comment regarding why the agency was still allowing patients to use the drug, said the following about Pepaxti on its website:
“The European Medicines Agency decided that Pepaxti’s benefits are greater than its risks and it can be authorised for use in the EU. The Agency noted the unmet medical need for patients with multiple myeloma who no longer improve with the available therapies. Despite some limitations in the studies, the results were considered clinically relevant, with the exception of the subgroup of patients who had an autologous stem cell transplant and whose disease progressed within three years of transplantation.
Regarding safety, although side effects, including severe effects, were seen with treatment involving Pepaxti, these were considered acceptable and manageable,” the agency wrote.
“Recommendations and precautions to be followed by healthcare professionals and patients for the safe and effective use of Pepaxti have been included in the summary of product characteristics and the package leaflet.
As for all medicines, data on the use of Pepaxti are continuously monitored. Suspected side effects reported with Pepaxti are carefully evaluated and any necessary action taken to protect patients,” according to the EMA.
The FDA’s final decision, issued on February 23, 2024, follows its warning in 2021 that meflufen plus dexamethasone exposed patients with multiple myeloma to increased risk for death, and its call for withdrawal of the drug in 2022.
“The grounds for withdrawing approval have been met because: (1) the confirmatory study conducted as a condition of accelerated approval did not confirm Pepaxto’s clinical benefit and (2) the available evidence demonstrates that Pepaxto is not shown to be safe or effective under its conditions of use,” Peter Marks, MD, PhD, Director of the FDA Center for Biologics Evaluation and Research, wrote in the final decision document.
Oncopeptides AB: Drug ‘Caters to a Large Unmet Need’
David Augustsson, Director of Corporate Affairs, Oncopeptides AB, explained in an interview why he thinks the EMA and FDA’s actions regarding the drug differ from each other.
“The European Medicines Agency had the opinion that the OCEAN study met its primary endpoint by demonstrating superior progression-free survival and it agreed that the potential detriment of overall survival was limited to patients progressing less than 36 months after an autologous stem cell transplant,” he said.“The FDA was not willing to acknowledge the observed clinically relevant differences across patient subgroups in the OCEAN study as confirmed.”
Mr. Augustsson added that this decision will deprive US patients of access to “a drug we believe caters to a large unmet need among elderly multiple myeloma patients with few treatment options left.”
“While we remain confident that we have science on our side we are of course disappointed in the decision [to remove Pepaxto from the US market],” Oncopeptides AB CEO Sofia Heigis said in a statement. “At the same time this is no change to our plans and we will continue to focus all our attention on the commercialization in Europe, progression of our pipeline and rest of world opportunities.”
FDA 'Took Swift Action' to Ensure Users of Pepaxto Were Informed of Risks
In February 2021, the FDA used the Accelerated Approval Program to enable certain patients with multiple myeloma to be treated with the peptide conjugated alkylating drug melflufen plus dexamethasone. Under the program, Oncopeptides was required to conduct the phase III randomized, controlled OCEAN clinical trial.
OCEAN enrolled 495 patients with relapsed/refractory multiple myeloma who had 2 to 4 lines of prior therapy and who were refractory to lenalidomide in the last line of therapy. Participants in the multinational study received either melflufen plus dexamethasone or pomalidomide plus dexamethasone until disease progression, unacceptable toxicity, or lack of benefit.
In July 2021, the FDA issued an alert that the study results showed increased risk for death in participants treated with melflufen. In October that year, at FDA request, Oncopeptides removed the drug from the US market but continued to provide it to patients already receiving it. In December 2022, the FDA requested that the company withdraw melflufen’s US marketing authorization.
Responding to questions about the timing of the FDA’s most recent decision about Pepaxto and how the decision will affect patient care in the US, the FDA emailed the following statement to this news organization:
“Since the OCEAN trial results for Pepaxto in 2021, the FDA has responded to safety concerns about Pepaxto by issuing a CDER Alert, communicating concerns to Oncopeptides, holding an Oncologic Drugs Advisory Committee meeting in September 2022, and issuing a letter of notice to Oncopeptides in July 2023, proposing to withdraw Pepaxto (NDA 214383). After receiving the notice, Oncopeptides appealed the withdrawal in August 2023. A meeting was held with the Commissioner’s designee, Dr. Peter Marks, Oncopeptides, and others from FDA in October 2023. Dr. Marks reviewed the record and considered the arguments made on appeal and issued a final decision on February 23, 2024. Prior to reaching a decision, the FDA took swift action to ensure those receiving Pepaxto in the post-confirmatory clinical trial were informed of the risks and that no new patients were enrolled in the trial. We also note that it is our understanding that Pepaxto has not been marketed in the U.S. since October 22, 2021.”
“This is the first time FDA has used the amended procedures for withdrawal of accelerated approval that were enacted in 2023, as part of the Food and Drug Omnibus Report Act of 2022 (FDORA),” the agency wrote in a Feb 23 statement. The agency will also remove melflufen from the Approved Drug Products with Therapeutic Equivalence Evaluations, also called the Orange Book.
New Trials in Leukemia and Lymphoma: Could Your Patient Benefit?
Several clinical trials in leukemia and lymphoma have started enrolling recently. Maybe one of your patients could benefit from taking part?
run by the Center for International Blood and Bone Marrow Transplant Research.
The purpose of the study is to test whether cyclophosphamide, which is given to prevent a dreaded complication of stem cell transplantation called graft-versus-host disease, can be safely reduced without increasing infection or reducing protection. All participants will receive cyclophosphamide on days 3 and 4 post transplant. One group will receive a reduced dose of cyclophosphamide (25 mg/kg per dose), and the other will be given a usual dose (37.5 mg/kg).
Sites in Michigan, Missouri, Oregon, Virginia, and Washington started recruiting for 190 participants in December 2023. Study centers in Florida, Massachusetts, New York, and Wisconsin are also planned. Infection-free survival is the primary endpoint, and overall survival is a secondary measure. Quality of life (QoL) is not recorded. More details at clinicaltrials.gov.
Untreated chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). Adults who are newly diagnosed with this type of cancer and have active disease may wish to consider a randomized, open-label, phase 3 trial testing an experimental Bruton tyrosine kinase (BTK) inhibitor, nemtabrutinib (from Merck Sharp & Dohme), against standard-of-care BTK inhibitors ibrutinib (Imbruvica) and acalabrutinib (Calquence).
BTK inhibitors target B-cell proliferation in B-cell cancers such as CLL/SLL and allow for chemotherapy-free treatment of some hematological malignancies. In this study, until disease progression, unacceptable toxicity, or another reason for discontinuation occurs, participants will take daily oral nemtabrutinib, ibrutinib, or acalabrutinib.
The study opened in December 2023 in Pennsylvania, Washington, Taiwan, Israel, and the United Kingdom seeking 1200 participants. The primary outcomes are objective response rate and progression-free survival. Overall survival is a secondary outcome, and QoL is not measured. More details at clinicaltrials.gov.
Relapsed or refractory leukemia with a KMT2A-gene rearrangement (KMT2A-r). Children aged 1 month to younger than 6 years with this diagnosis may be able to join an open-label, nonrandomized, Children’s Oncology Group phase 2 study to determine the most tolerable and/or effective dose of an experimental oral drug called revumenib when added to chemotherapy.
KMT2A-gene alterations are associated with a poor prognosis in leukemia. These alterations cause blood cells to dedifferentiate and start proliferating uncontrollably as leukemia cells. The expression of the damaged KMT2A gene relies on a protein called menin. Revumenib, from Syndax Pharmaceuticals, blocks menin and prevents expression of KMT2A.
Children in the study will receive two different regimens of revumenib in combination with chemotherapy for up to a year, or until disease progression or unacceptable toxicity, and will then be followed for up to 5 years. Trial centers in 12 US states opened their doors in January 2024 looking for 78 participants. Toxicities and minimal residual disease are the primary outcomes; overall survival is a secondary outcome, and QoL is not assessed. More details at clinicaltrials.gov.
Previously untreated follicular lymphoma or diffuse large B-cell lymphoma. Adults with one of these types of lymphoma may be eligible for one of three open-label, randomized, phase 3 trials testing odronextamab (from Regeneron). This bispecific antibody is designed to ‘lock together’ CD20 on cancer cells with CD3-expressing cancer-killing T cells. It has shown anti-lymphoma activity in heavily pretreated patients.
Late in 2023, three phase 3 trials turned the spotlight on treatment-naive patients and started recruiting 2115 participants to assess odronextamab in this setting. The trial OLYMPIA-1 will compare odronextamab with standard-of-care rituximab (Rituxan) plus chemotherapy in follicular lymphoma. OLYMPIA-2 will test the drug in combination with chemotherapy, also in follicular lymphoma. OLYMPIA-3 will evaluate odronextamab plus chemotherapy against rituximab and chemotherapy in people with large B-cell lymphoma.
All study drugs, including odronextamab, will be administered by intravenous infusion, and participants will be followed for up to 5 years. Research centers across eight US states and Australia, Czechia, France, Italy, Poland, Spain, Turkey, and Thailand are currently accepting participants for the three trials. The primary outcomes are various measures of toxicity and complete response at 30 months in the follicular lymphoma studies and toxicity and progression-free survival in large B-cell lymphoma. All three trials are measuring overall survival and QoL as secondary endpoints.
Previously untreated stage II, III, or IV follicular lymphoma. Adults with this type of cancer may be eligible to participate in a randomized, open-label, phase 3 study testing whether an experimental therapy called epcoritamab (from AbbVie) improves disease response and is tolerable when added to standard therapy. For up to 120 weeks, one group of participants will receive a combination of intravenous rituximab and oral lenalidomide (Revlimid), while a second group will also receive subcutaneous injections of epcoritamab. Some participants may be offered investigators’ choice of chemotherapy as well.
Sites across Iowa, Maryland, Missouri, Ohio, Washington, and Montana started welcoming their 900 participants in February 2024. The primary outcome is complete response at 30 months. Overall survival and QoL are secondary outcomes. More details at clinicaltrials.gov.
Relapsed or refractory mantle cell lymphoma. Adults facing one of these clinical scenarios can join an Academic and Community Cancer Research United open label, phase 2 trial examining the effectiveness of combining tafasitamab (Monjuvi), lenalidomide, and venetoclax (Venclexta) for such patients.
Frontline therapy does not cure mantle cell lymphoma, and continued relapses are common. In this situation, treatments can include acalabrutinib, ibrutinib, stem cell transplantation, venetoclax, lenalidomide, and rituximab.
In this study, participants will take venetoclax and lenalidomide daily and receive intravenous tafasitamab every 2 weeks after an initial ramp-up period as per clinic standards. Participants will be followed for 5 years after entering the trial. The Mayo Clinic in Rochester, Minnesota, began recruiting the planned 100 trial participants in January 2024. The primary outcome is objective response rate; overall survival is a secondary outcome, and QoL will not be tracked. More details at clinicaltrials.gov.
All trial information is from the National Institutes of Health US National Library of Medicine (online at clinicaltrials.gov).
A version of this article appeared on Medscape.com .
Several clinical trials in leukemia and lymphoma have started enrolling recently. Maybe one of your patients could benefit from taking part?
run by the Center for International Blood and Bone Marrow Transplant Research.
The purpose of the study is to test whether cyclophosphamide, which is given to prevent a dreaded complication of stem cell transplantation called graft-versus-host disease, can be safely reduced without increasing infection or reducing protection. All participants will receive cyclophosphamide on days 3 and 4 post transplant. One group will receive a reduced dose of cyclophosphamide (25 mg/kg per dose), and the other will be given a usual dose (37.5 mg/kg).
Sites in Michigan, Missouri, Oregon, Virginia, and Washington started recruiting for 190 participants in December 2023. Study centers in Florida, Massachusetts, New York, and Wisconsin are also planned. Infection-free survival is the primary endpoint, and overall survival is a secondary measure. Quality of life (QoL) is not recorded. More details at clinicaltrials.gov.
Untreated chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). Adults who are newly diagnosed with this type of cancer and have active disease may wish to consider a randomized, open-label, phase 3 trial testing an experimental Bruton tyrosine kinase (BTK) inhibitor, nemtabrutinib (from Merck Sharp & Dohme), against standard-of-care BTK inhibitors ibrutinib (Imbruvica) and acalabrutinib (Calquence).
BTK inhibitors target B-cell proliferation in B-cell cancers such as CLL/SLL and allow for chemotherapy-free treatment of some hematological malignancies. In this study, until disease progression, unacceptable toxicity, or another reason for discontinuation occurs, participants will take daily oral nemtabrutinib, ibrutinib, or acalabrutinib.
The study opened in December 2023 in Pennsylvania, Washington, Taiwan, Israel, and the United Kingdom seeking 1200 participants. The primary outcomes are objective response rate and progression-free survival. Overall survival is a secondary outcome, and QoL is not measured. More details at clinicaltrials.gov.
Relapsed or refractory leukemia with a KMT2A-gene rearrangement (KMT2A-r). Children aged 1 month to younger than 6 years with this diagnosis may be able to join an open-label, nonrandomized, Children’s Oncology Group phase 2 study to determine the most tolerable and/or effective dose of an experimental oral drug called revumenib when added to chemotherapy.
KMT2A-gene alterations are associated with a poor prognosis in leukemia. These alterations cause blood cells to dedifferentiate and start proliferating uncontrollably as leukemia cells. The expression of the damaged KMT2A gene relies on a protein called menin. Revumenib, from Syndax Pharmaceuticals, blocks menin and prevents expression of KMT2A.
Children in the study will receive two different regimens of revumenib in combination with chemotherapy for up to a year, or until disease progression or unacceptable toxicity, and will then be followed for up to 5 years. Trial centers in 12 US states opened their doors in January 2024 looking for 78 participants. Toxicities and minimal residual disease are the primary outcomes; overall survival is a secondary outcome, and QoL is not assessed. More details at clinicaltrials.gov.
Previously untreated follicular lymphoma or diffuse large B-cell lymphoma. Adults with one of these types of lymphoma may be eligible for one of three open-label, randomized, phase 3 trials testing odronextamab (from Regeneron). This bispecific antibody is designed to ‘lock together’ CD20 on cancer cells with CD3-expressing cancer-killing T cells. It has shown anti-lymphoma activity in heavily pretreated patients.
Late in 2023, three phase 3 trials turned the spotlight on treatment-naive patients and started recruiting 2115 participants to assess odronextamab in this setting. The trial OLYMPIA-1 will compare odronextamab with standard-of-care rituximab (Rituxan) plus chemotherapy in follicular lymphoma. OLYMPIA-2 will test the drug in combination with chemotherapy, also in follicular lymphoma. OLYMPIA-3 will evaluate odronextamab plus chemotherapy against rituximab and chemotherapy in people with large B-cell lymphoma.
All study drugs, including odronextamab, will be administered by intravenous infusion, and participants will be followed for up to 5 years. Research centers across eight US states and Australia, Czechia, France, Italy, Poland, Spain, Turkey, and Thailand are currently accepting participants for the three trials. The primary outcomes are various measures of toxicity and complete response at 30 months in the follicular lymphoma studies and toxicity and progression-free survival in large B-cell lymphoma. All three trials are measuring overall survival and QoL as secondary endpoints.
Previously untreated stage II, III, or IV follicular lymphoma. Adults with this type of cancer may be eligible to participate in a randomized, open-label, phase 3 study testing whether an experimental therapy called epcoritamab (from AbbVie) improves disease response and is tolerable when added to standard therapy. For up to 120 weeks, one group of participants will receive a combination of intravenous rituximab and oral lenalidomide (Revlimid), while a second group will also receive subcutaneous injections of epcoritamab. Some participants may be offered investigators’ choice of chemotherapy as well.
Sites across Iowa, Maryland, Missouri, Ohio, Washington, and Montana started welcoming their 900 participants in February 2024. The primary outcome is complete response at 30 months. Overall survival and QoL are secondary outcomes. More details at clinicaltrials.gov.
Relapsed or refractory mantle cell lymphoma. Adults facing one of these clinical scenarios can join an Academic and Community Cancer Research United open label, phase 2 trial examining the effectiveness of combining tafasitamab (Monjuvi), lenalidomide, and venetoclax (Venclexta) for such patients.
Frontline therapy does not cure mantle cell lymphoma, and continued relapses are common. In this situation, treatments can include acalabrutinib, ibrutinib, stem cell transplantation, venetoclax, lenalidomide, and rituximab.
In this study, participants will take venetoclax and lenalidomide daily and receive intravenous tafasitamab every 2 weeks after an initial ramp-up period as per clinic standards. Participants will be followed for 5 years after entering the trial. The Mayo Clinic in Rochester, Minnesota, began recruiting the planned 100 trial participants in January 2024. The primary outcome is objective response rate; overall survival is a secondary outcome, and QoL will not be tracked. More details at clinicaltrials.gov.
All trial information is from the National Institutes of Health US National Library of Medicine (online at clinicaltrials.gov).
A version of this article appeared on Medscape.com .
Several clinical trials in leukemia and lymphoma have started enrolling recently. Maybe one of your patients could benefit from taking part?
run by the Center for International Blood and Bone Marrow Transplant Research.
The purpose of the study is to test whether cyclophosphamide, which is given to prevent a dreaded complication of stem cell transplantation called graft-versus-host disease, can be safely reduced without increasing infection or reducing protection. All participants will receive cyclophosphamide on days 3 and 4 post transplant. One group will receive a reduced dose of cyclophosphamide (25 mg/kg per dose), and the other will be given a usual dose (37.5 mg/kg).
Sites in Michigan, Missouri, Oregon, Virginia, and Washington started recruiting for 190 participants in December 2023. Study centers in Florida, Massachusetts, New York, and Wisconsin are also planned. Infection-free survival is the primary endpoint, and overall survival is a secondary measure. Quality of life (QoL) is not recorded. More details at clinicaltrials.gov.
Untreated chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). Adults who are newly diagnosed with this type of cancer and have active disease may wish to consider a randomized, open-label, phase 3 trial testing an experimental Bruton tyrosine kinase (BTK) inhibitor, nemtabrutinib (from Merck Sharp & Dohme), against standard-of-care BTK inhibitors ibrutinib (Imbruvica) and acalabrutinib (Calquence).
BTK inhibitors target B-cell proliferation in B-cell cancers such as CLL/SLL and allow for chemotherapy-free treatment of some hematological malignancies. In this study, until disease progression, unacceptable toxicity, or another reason for discontinuation occurs, participants will take daily oral nemtabrutinib, ibrutinib, or acalabrutinib.
The study opened in December 2023 in Pennsylvania, Washington, Taiwan, Israel, and the United Kingdom seeking 1200 participants. The primary outcomes are objective response rate and progression-free survival. Overall survival is a secondary outcome, and QoL is not measured. More details at clinicaltrials.gov.
Relapsed or refractory leukemia with a KMT2A-gene rearrangement (KMT2A-r). Children aged 1 month to younger than 6 years with this diagnosis may be able to join an open-label, nonrandomized, Children’s Oncology Group phase 2 study to determine the most tolerable and/or effective dose of an experimental oral drug called revumenib when added to chemotherapy.
KMT2A-gene alterations are associated with a poor prognosis in leukemia. These alterations cause blood cells to dedifferentiate and start proliferating uncontrollably as leukemia cells. The expression of the damaged KMT2A gene relies on a protein called menin. Revumenib, from Syndax Pharmaceuticals, blocks menin and prevents expression of KMT2A.
Children in the study will receive two different regimens of revumenib in combination with chemotherapy for up to a year, or until disease progression or unacceptable toxicity, and will then be followed for up to 5 years. Trial centers in 12 US states opened their doors in January 2024 looking for 78 participants. Toxicities and minimal residual disease are the primary outcomes; overall survival is a secondary outcome, and QoL is not assessed. More details at clinicaltrials.gov.
Previously untreated follicular lymphoma or diffuse large B-cell lymphoma. Adults with one of these types of lymphoma may be eligible for one of three open-label, randomized, phase 3 trials testing odronextamab (from Regeneron). This bispecific antibody is designed to ‘lock together’ CD20 on cancer cells with CD3-expressing cancer-killing T cells. It has shown anti-lymphoma activity in heavily pretreated patients.
Late in 2023, three phase 3 trials turned the spotlight on treatment-naive patients and started recruiting 2115 participants to assess odronextamab in this setting. The trial OLYMPIA-1 will compare odronextamab with standard-of-care rituximab (Rituxan) plus chemotherapy in follicular lymphoma. OLYMPIA-2 will test the drug in combination with chemotherapy, also in follicular lymphoma. OLYMPIA-3 will evaluate odronextamab plus chemotherapy against rituximab and chemotherapy in people with large B-cell lymphoma.
All study drugs, including odronextamab, will be administered by intravenous infusion, and participants will be followed for up to 5 years. Research centers across eight US states and Australia, Czechia, France, Italy, Poland, Spain, Turkey, and Thailand are currently accepting participants for the three trials. The primary outcomes are various measures of toxicity and complete response at 30 months in the follicular lymphoma studies and toxicity and progression-free survival in large B-cell lymphoma. All three trials are measuring overall survival and QoL as secondary endpoints.
Previously untreated stage II, III, or IV follicular lymphoma. Adults with this type of cancer may be eligible to participate in a randomized, open-label, phase 3 study testing whether an experimental therapy called epcoritamab (from AbbVie) improves disease response and is tolerable when added to standard therapy. For up to 120 weeks, one group of participants will receive a combination of intravenous rituximab and oral lenalidomide (Revlimid), while a second group will also receive subcutaneous injections of epcoritamab. Some participants may be offered investigators’ choice of chemotherapy as well.
Sites across Iowa, Maryland, Missouri, Ohio, Washington, and Montana started welcoming their 900 participants in February 2024. The primary outcome is complete response at 30 months. Overall survival and QoL are secondary outcomes. More details at clinicaltrials.gov.
Relapsed or refractory mantle cell lymphoma. Adults facing one of these clinical scenarios can join an Academic and Community Cancer Research United open label, phase 2 trial examining the effectiveness of combining tafasitamab (Monjuvi), lenalidomide, and venetoclax (Venclexta) for such patients.
Frontline therapy does not cure mantle cell lymphoma, and continued relapses are common. In this situation, treatments can include acalabrutinib, ibrutinib, stem cell transplantation, venetoclax, lenalidomide, and rituximab.
In this study, participants will take venetoclax and lenalidomide daily and receive intravenous tafasitamab every 2 weeks after an initial ramp-up period as per clinic standards. Participants will be followed for 5 years after entering the trial. The Mayo Clinic in Rochester, Minnesota, began recruiting the planned 100 trial participants in January 2024. The primary outcome is objective response rate; overall survival is a secondary outcome, and QoL will not be tracked. More details at clinicaltrials.gov.
All trial information is from the National Institutes of Health US National Library of Medicine (online at clinicaltrials.gov).
A version of this article appeared on Medscape.com .
AML: Genetic Testing Unlocks Hope
For adult patients, “we’ve seen a series of remarkable and well-overdue advances in a space that had not changed much over the prior decades,” hematologist/oncologist Thomas William LeBlanc, MD, associate professor of medicine at Duke University School of Medicine, Durham, North Carolina, said in an interview.
According to the National Cancer Institute, AML will be newly diagnosed in 20,800 patients in 2024, at a median age of 69, and will cause 11,220 deaths. As many as 70% of adult patients will reach complete remission, and 45% of those will live for more than 3 years and potentially be cured. As for children, the Leukemia & Lymphoma Society says the 5-year survival rate from 2012-2018 was 69% for those under 15 years old.
As the American Cancer Society notes, the goal of AML treatment “is to put the leukemia into complete remission (the bone marrow and blood cell counts return to normal), preferably a complete molecular remission (no signs of leukemia in the bone marrow, even using sensitive lab tests), and to keep it that way.”
Chemotherapy Strategies Shift Over Time
In terms of the treatment of adults with AML, “targeted therapies, in addition to the expanding role of venetoclax, has really altered our approach to AML from diagnosis, including after relapse, and later in the disease,” hematologist/oncologist Andrew M. Brunner, MD, of Harvard Medical School and Massachusetts General Hospital, Boston, said in an interview. “The ability to explore these options as monotherapy and in novel combinations has dramatically expanded our treatment options.”
Much depends on the underlying genetic profile of the disease, he said. “There certainly have been gains in patient survival in AML, but those improvements remain fairly heterogeneous and dependent on the underlying genetic profile of the disease. For instance, advances in FLT3- and IDH1/2-mutated AML are a direct result of the improvements in targeted therapies directed at these mutations. Similarly, some molecular and cytogenetic subtypes of AML are particularly responsive to venetoclax-based regimens, and these regimens have been expanded to previously undertreated populations, particularly those over age 60.”
Specifically, Dr. LeBlanc said, the Food and Drug Administration has approved “3 different FLT3 inhibitors, 2 IDH1 inhibitors, 1 IDH2 inhibitor, a BCL-2 inhibitor, a smoothened/hedgehog pathway inhibitor, an oral maintenance chemotherapy/hypomethylating agent (CC-486/oral azacitidine), a CD33-targeting antibody-drug conjugate, and even a novel formulation of two older chemotherapies that improves efficacy in a poor prognosis subgroup (CPX-351/liposomal daunorubicin and cytarabine).”
There’s also been a shift in treatment protocols for patients who were not fit for intensive chemotherapy. In the past, he said, it was standard “to give single-agent hypomethylating chemotherapy with azacitidine or decitabine, or in some contexts, low-dose chemotherapy with cytarabine. Today, many patients who are older and/or more frail are receiving novel therapies either alone or in combination, with greater efficacy and longer duration of response than previously seen with chemotherapy alone.”
Outcomes Improve but Remain Grim in High-Risk Cases
As a result, Dr. LeBlanc said, “we’re definitely seeing much better outcomes in AML overall. It takes some time to prove this via outcomes data assessments in a large population, but I expect that registries will show significant improvements in overall survival in the coming years, owing to the many new FDA approvals in AML”
Dr. LeBlanc highlighted national data from 2013-2019 showing that the 5-year relative survival rate from AML is 31.7%. That’s up from 26% just a few years ago, and the numbers “always lag several years behind the current year of practice,” he said. However, “the major area where we still have relatively poor outcomes and significant unmet needs remains the ‘adverse risk’ group of patients, particularly those who are older and/or not candidates for hematopoietic stem cell transplantation, which generally is the only potentially curative option for adverse-risk AML.”
He went on to say that “this risk grouping includes those with TP53 mutations, most of which confer a particularly poor prognosis. Exciting therapies that many of us were hoping would prove effective in this subgroup have unfortunately failed in recent clinical trials. We still have a lot of work to do in adverse-risk AML particularly, and also for those whose leukemia has relapsed.”
Mikkael Sekeres, MD, MS, chief of the Division of Hematology at the University of Miami Miller School of Medicine/Sylvester Comprehensive Cancer Center, agreed that more progress is needed, since survival rates are low even as lifespans improve. One key will be “better identifying subtypes of acute myeloid leukemia, and identifying the therapies that will benefit those people most,” he said in an interview. On the other side, it’s important to identify “when aggressive therapies aren’t going to work in somebody and maybe turn toward less-aggressive approaches so we can maximize that person’s quality of life.”
What advice do AML experts have for their colleagues? Dr. LeBlanc said “older patients are not often enough considered for allogeneic stem cell transplantation, which could potentially cure their AML when given as a consolidation treatment for those in remission. I have several patients who are healthy and in their 70s who have enormously benefited from transplants and are now being several years out from transplant with adverse risk AML and without relapse. They’ve had no significant impairments of their quality of life, including no significant graft vs. host disease.”
Dr. Sekeres highlighted the American Society of Hematology’s guidelines for treating older adults with AML, which are currently being updated. It’s crucial to order genetic testing “up front,” he said. “I’m often pleasantly surprised when genetic testing returns and reveals that I have other treatment options.”
However, it’s crucial to understand a patient’s priorities. “I’ve had patients who are 75 who say to me, ‘Do everything under the sun to get rid of my leukemia, I want to live as long as possible.’ And I’ve had patients who say, ‘I want to see as little of doctors and nurses as I can. I want you to maximize my quality of life and keep me out of the hospital.’ ”
Dr. Sekeres also noted that insurers may not cover some pill-based AML treatments such as venetoclax. “We work with our patients and assistance programs. For the most part, we’re pretty successful at getting these drugs for our patients,” he said.
In Pediatrics, Clinical Trials Are Crucial
AML in children is less well-known than in adults, since the number of cases is so small. The disease is diagnosed in about 500 children a year in the United States, according to St. Jude Children’s Research Hospital, adding, however, that AML is “the most common second cancer among children treated for other cancers.”
AML in children gained attention earlier this year when the 2-year-old daughter of a Boston Herald NFL reporter died of the disease following a bone marrow transplant and chemotherapy. Despite the agonies of her treatment, reporter Doug Kyed told a reporter that his daughter Hallie “was still able to find joy every day.”
In an interview, hematologist/oncologist Sarah K. Tasian, MD, of Children’s Hospital of Philadelphia, said researchers are discovering that pediatric AML is significantly different on from a biological perspective from adult AML. “We’ve come to understand a lot more about who these patients are, what makes these leukemias tick, and what their Achilles’ heels are. Then we can align that with the clinical trials outcome data that we have.”
About 80%-90% of pediatric patients with AML nationwide are enrolled in clinical trials, Dr. Tasian said, and an international consortium called the Children’s Oncology Group gathers data about genetics. About 60%-70% of patients will be cured, she added.
However, “we’ve kind of been stuck for about the last 20 years,” she said. “A lot of improving the survival of patients has not been because we’ve been better at chemotherapy or using new chemo, but because we’ve gotten better at supportive care, at treating infections that can be fatal.”
There haven’t been major conflicts with insurers over coverage, she said, although drug shortages are a problem, especially in relapsed AML.
As for advice to colleagues, Dr. Tasian counseled them to understand the importance of genetic testing and the expanding role of stem cell transplants. “We are now transplanting somewhere between 30% and 50% of children with AML, which is a higher rate than we used to do,” she said. The number is up thanks to genetic testing that reveals which patients are most likely to benefit.
Also, she noted, “the chemotherapy that we get to these patients is really strong, and patients have a lot of complications. Really pay attention to supportive care.”
Dr. LeBlanc reported ties with AbbVie, Agios/Servier, Astellas, BMS/Celgene, Genentech, Pfizer, Incyte, Rige, Deverra, GSK, Jazz, and Seattle Genetics. Dr. Sekeres discloses relationships with BMS and Kurome. Dr. Tasian serves as the Leukemia & Lymphoma Society Pediatric Acute Leukemia consortium clinical trials leader and works with pharmaceutical companies on clinical trials under confidentiality agreements. Dr. Brunner has no disclosures.
For adult patients, “we’ve seen a series of remarkable and well-overdue advances in a space that had not changed much over the prior decades,” hematologist/oncologist Thomas William LeBlanc, MD, associate professor of medicine at Duke University School of Medicine, Durham, North Carolina, said in an interview.
According to the National Cancer Institute, AML will be newly diagnosed in 20,800 patients in 2024, at a median age of 69, and will cause 11,220 deaths. As many as 70% of adult patients will reach complete remission, and 45% of those will live for more than 3 years and potentially be cured. As for children, the Leukemia & Lymphoma Society says the 5-year survival rate from 2012-2018 was 69% for those under 15 years old.
As the American Cancer Society notes, the goal of AML treatment “is to put the leukemia into complete remission (the bone marrow and blood cell counts return to normal), preferably a complete molecular remission (no signs of leukemia in the bone marrow, even using sensitive lab tests), and to keep it that way.”
Chemotherapy Strategies Shift Over Time
In terms of the treatment of adults with AML, “targeted therapies, in addition to the expanding role of venetoclax, has really altered our approach to AML from diagnosis, including after relapse, and later in the disease,” hematologist/oncologist Andrew M. Brunner, MD, of Harvard Medical School and Massachusetts General Hospital, Boston, said in an interview. “The ability to explore these options as monotherapy and in novel combinations has dramatically expanded our treatment options.”
Much depends on the underlying genetic profile of the disease, he said. “There certainly have been gains in patient survival in AML, but those improvements remain fairly heterogeneous and dependent on the underlying genetic profile of the disease. For instance, advances in FLT3- and IDH1/2-mutated AML are a direct result of the improvements in targeted therapies directed at these mutations. Similarly, some molecular and cytogenetic subtypes of AML are particularly responsive to venetoclax-based regimens, and these regimens have been expanded to previously undertreated populations, particularly those over age 60.”
Specifically, Dr. LeBlanc said, the Food and Drug Administration has approved “3 different FLT3 inhibitors, 2 IDH1 inhibitors, 1 IDH2 inhibitor, a BCL-2 inhibitor, a smoothened/hedgehog pathway inhibitor, an oral maintenance chemotherapy/hypomethylating agent (CC-486/oral azacitidine), a CD33-targeting antibody-drug conjugate, and even a novel formulation of two older chemotherapies that improves efficacy in a poor prognosis subgroup (CPX-351/liposomal daunorubicin and cytarabine).”
There’s also been a shift in treatment protocols for patients who were not fit for intensive chemotherapy. In the past, he said, it was standard “to give single-agent hypomethylating chemotherapy with azacitidine or decitabine, or in some contexts, low-dose chemotherapy with cytarabine. Today, many patients who are older and/or more frail are receiving novel therapies either alone or in combination, with greater efficacy and longer duration of response than previously seen with chemotherapy alone.”
Outcomes Improve but Remain Grim in High-Risk Cases
As a result, Dr. LeBlanc said, “we’re definitely seeing much better outcomes in AML overall. It takes some time to prove this via outcomes data assessments in a large population, but I expect that registries will show significant improvements in overall survival in the coming years, owing to the many new FDA approvals in AML”
Dr. LeBlanc highlighted national data from 2013-2019 showing that the 5-year relative survival rate from AML is 31.7%. That’s up from 26% just a few years ago, and the numbers “always lag several years behind the current year of practice,” he said. However, “the major area where we still have relatively poor outcomes and significant unmet needs remains the ‘adverse risk’ group of patients, particularly those who are older and/or not candidates for hematopoietic stem cell transplantation, which generally is the only potentially curative option for adverse-risk AML.”
He went on to say that “this risk grouping includes those with TP53 mutations, most of which confer a particularly poor prognosis. Exciting therapies that many of us were hoping would prove effective in this subgroup have unfortunately failed in recent clinical trials. We still have a lot of work to do in adverse-risk AML particularly, and also for those whose leukemia has relapsed.”
Mikkael Sekeres, MD, MS, chief of the Division of Hematology at the University of Miami Miller School of Medicine/Sylvester Comprehensive Cancer Center, agreed that more progress is needed, since survival rates are low even as lifespans improve. One key will be “better identifying subtypes of acute myeloid leukemia, and identifying the therapies that will benefit those people most,” he said in an interview. On the other side, it’s important to identify “when aggressive therapies aren’t going to work in somebody and maybe turn toward less-aggressive approaches so we can maximize that person’s quality of life.”
What advice do AML experts have for their colleagues? Dr. LeBlanc said “older patients are not often enough considered for allogeneic stem cell transplantation, which could potentially cure their AML when given as a consolidation treatment for those in remission. I have several patients who are healthy and in their 70s who have enormously benefited from transplants and are now being several years out from transplant with adverse risk AML and without relapse. They’ve had no significant impairments of their quality of life, including no significant graft vs. host disease.”
Dr. Sekeres highlighted the American Society of Hematology’s guidelines for treating older adults with AML, which are currently being updated. It’s crucial to order genetic testing “up front,” he said. “I’m often pleasantly surprised when genetic testing returns and reveals that I have other treatment options.”
However, it’s crucial to understand a patient’s priorities. “I’ve had patients who are 75 who say to me, ‘Do everything under the sun to get rid of my leukemia, I want to live as long as possible.’ And I’ve had patients who say, ‘I want to see as little of doctors and nurses as I can. I want you to maximize my quality of life and keep me out of the hospital.’ ”
Dr. Sekeres also noted that insurers may not cover some pill-based AML treatments such as venetoclax. “We work with our patients and assistance programs. For the most part, we’re pretty successful at getting these drugs for our patients,” he said.
In Pediatrics, Clinical Trials Are Crucial
AML in children is less well-known than in adults, since the number of cases is so small. The disease is diagnosed in about 500 children a year in the United States, according to St. Jude Children’s Research Hospital, adding, however, that AML is “the most common second cancer among children treated for other cancers.”
AML in children gained attention earlier this year when the 2-year-old daughter of a Boston Herald NFL reporter died of the disease following a bone marrow transplant and chemotherapy. Despite the agonies of her treatment, reporter Doug Kyed told a reporter that his daughter Hallie “was still able to find joy every day.”
In an interview, hematologist/oncologist Sarah K. Tasian, MD, of Children’s Hospital of Philadelphia, said researchers are discovering that pediatric AML is significantly different on from a biological perspective from adult AML. “We’ve come to understand a lot more about who these patients are, what makes these leukemias tick, and what their Achilles’ heels are. Then we can align that with the clinical trials outcome data that we have.”
About 80%-90% of pediatric patients with AML nationwide are enrolled in clinical trials, Dr. Tasian said, and an international consortium called the Children’s Oncology Group gathers data about genetics. About 60%-70% of patients will be cured, she added.
However, “we’ve kind of been stuck for about the last 20 years,” she said. “A lot of improving the survival of patients has not been because we’ve been better at chemotherapy or using new chemo, but because we’ve gotten better at supportive care, at treating infections that can be fatal.”
There haven’t been major conflicts with insurers over coverage, she said, although drug shortages are a problem, especially in relapsed AML.
As for advice to colleagues, Dr. Tasian counseled them to understand the importance of genetic testing and the expanding role of stem cell transplants. “We are now transplanting somewhere between 30% and 50% of children with AML, which is a higher rate than we used to do,” she said. The number is up thanks to genetic testing that reveals which patients are most likely to benefit.
Also, she noted, “the chemotherapy that we get to these patients is really strong, and patients have a lot of complications. Really pay attention to supportive care.”
Dr. LeBlanc reported ties with AbbVie, Agios/Servier, Astellas, BMS/Celgene, Genentech, Pfizer, Incyte, Rige, Deverra, GSK, Jazz, and Seattle Genetics. Dr. Sekeres discloses relationships with BMS and Kurome. Dr. Tasian serves as the Leukemia & Lymphoma Society Pediatric Acute Leukemia consortium clinical trials leader and works with pharmaceutical companies on clinical trials under confidentiality agreements. Dr. Brunner has no disclosures.
For adult patients, “we’ve seen a series of remarkable and well-overdue advances in a space that had not changed much over the prior decades,” hematologist/oncologist Thomas William LeBlanc, MD, associate professor of medicine at Duke University School of Medicine, Durham, North Carolina, said in an interview.
According to the National Cancer Institute, AML will be newly diagnosed in 20,800 patients in 2024, at a median age of 69, and will cause 11,220 deaths. As many as 70% of adult patients will reach complete remission, and 45% of those will live for more than 3 years and potentially be cured. As for children, the Leukemia & Lymphoma Society says the 5-year survival rate from 2012-2018 was 69% for those under 15 years old.
As the American Cancer Society notes, the goal of AML treatment “is to put the leukemia into complete remission (the bone marrow and blood cell counts return to normal), preferably a complete molecular remission (no signs of leukemia in the bone marrow, even using sensitive lab tests), and to keep it that way.”
Chemotherapy Strategies Shift Over Time
In terms of the treatment of adults with AML, “targeted therapies, in addition to the expanding role of venetoclax, has really altered our approach to AML from diagnosis, including after relapse, and later in the disease,” hematologist/oncologist Andrew M. Brunner, MD, of Harvard Medical School and Massachusetts General Hospital, Boston, said in an interview. “The ability to explore these options as monotherapy and in novel combinations has dramatically expanded our treatment options.”
Much depends on the underlying genetic profile of the disease, he said. “There certainly have been gains in patient survival in AML, but those improvements remain fairly heterogeneous and dependent on the underlying genetic profile of the disease. For instance, advances in FLT3- and IDH1/2-mutated AML are a direct result of the improvements in targeted therapies directed at these mutations. Similarly, some molecular and cytogenetic subtypes of AML are particularly responsive to venetoclax-based regimens, and these regimens have been expanded to previously undertreated populations, particularly those over age 60.”
Specifically, Dr. LeBlanc said, the Food and Drug Administration has approved “3 different FLT3 inhibitors, 2 IDH1 inhibitors, 1 IDH2 inhibitor, a BCL-2 inhibitor, a smoothened/hedgehog pathway inhibitor, an oral maintenance chemotherapy/hypomethylating agent (CC-486/oral azacitidine), a CD33-targeting antibody-drug conjugate, and even a novel formulation of two older chemotherapies that improves efficacy in a poor prognosis subgroup (CPX-351/liposomal daunorubicin and cytarabine).”
There’s also been a shift in treatment protocols for patients who were not fit for intensive chemotherapy. In the past, he said, it was standard “to give single-agent hypomethylating chemotherapy with azacitidine or decitabine, or in some contexts, low-dose chemotherapy with cytarabine. Today, many patients who are older and/or more frail are receiving novel therapies either alone or in combination, with greater efficacy and longer duration of response than previously seen with chemotherapy alone.”
Outcomes Improve but Remain Grim in High-Risk Cases
As a result, Dr. LeBlanc said, “we’re definitely seeing much better outcomes in AML overall. It takes some time to prove this via outcomes data assessments in a large population, but I expect that registries will show significant improvements in overall survival in the coming years, owing to the many new FDA approvals in AML”
Dr. LeBlanc highlighted national data from 2013-2019 showing that the 5-year relative survival rate from AML is 31.7%. That’s up from 26% just a few years ago, and the numbers “always lag several years behind the current year of practice,” he said. However, “the major area where we still have relatively poor outcomes and significant unmet needs remains the ‘adverse risk’ group of patients, particularly those who are older and/or not candidates for hematopoietic stem cell transplantation, which generally is the only potentially curative option for adverse-risk AML.”
He went on to say that “this risk grouping includes those with TP53 mutations, most of which confer a particularly poor prognosis. Exciting therapies that many of us were hoping would prove effective in this subgroup have unfortunately failed in recent clinical trials. We still have a lot of work to do in adverse-risk AML particularly, and also for those whose leukemia has relapsed.”
Mikkael Sekeres, MD, MS, chief of the Division of Hematology at the University of Miami Miller School of Medicine/Sylvester Comprehensive Cancer Center, agreed that more progress is needed, since survival rates are low even as lifespans improve. One key will be “better identifying subtypes of acute myeloid leukemia, and identifying the therapies that will benefit those people most,” he said in an interview. On the other side, it’s important to identify “when aggressive therapies aren’t going to work in somebody and maybe turn toward less-aggressive approaches so we can maximize that person’s quality of life.”
What advice do AML experts have for their colleagues? Dr. LeBlanc said “older patients are not often enough considered for allogeneic stem cell transplantation, which could potentially cure their AML when given as a consolidation treatment for those in remission. I have several patients who are healthy and in their 70s who have enormously benefited from transplants and are now being several years out from transplant with adverse risk AML and without relapse. They’ve had no significant impairments of their quality of life, including no significant graft vs. host disease.”
Dr. Sekeres highlighted the American Society of Hematology’s guidelines for treating older adults with AML, which are currently being updated. It’s crucial to order genetic testing “up front,” he said. “I’m often pleasantly surprised when genetic testing returns and reveals that I have other treatment options.”
However, it’s crucial to understand a patient’s priorities. “I’ve had patients who are 75 who say to me, ‘Do everything under the sun to get rid of my leukemia, I want to live as long as possible.’ And I’ve had patients who say, ‘I want to see as little of doctors and nurses as I can. I want you to maximize my quality of life and keep me out of the hospital.’ ”
Dr. Sekeres also noted that insurers may not cover some pill-based AML treatments such as venetoclax. “We work with our patients and assistance programs. For the most part, we’re pretty successful at getting these drugs for our patients,” he said.
In Pediatrics, Clinical Trials Are Crucial
AML in children is less well-known than in adults, since the number of cases is so small. The disease is diagnosed in about 500 children a year in the United States, according to St. Jude Children’s Research Hospital, adding, however, that AML is “the most common second cancer among children treated for other cancers.”
AML in children gained attention earlier this year when the 2-year-old daughter of a Boston Herald NFL reporter died of the disease following a bone marrow transplant and chemotherapy. Despite the agonies of her treatment, reporter Doug Kyed told a reporter that his daughter Hallie “was still able to find joy every day.”
In an interview, hematologist/oncologist Sarah K. Tasian, MD, of Children’s Hospital of Philadelphia, said researchers are discovering that pediatric AML is significantly different on from a biological perspective from adult AML. “We’ve come to understand a lot more about who these patients are, what makes these leukemias tick, and what their Achilles’ heels are. Then we can align that with the clinical trials outcome data that we have.”
About 80%-90% of pediatric patients with AML nationwide are enrolled in clinical trials, Dr. Tasian said, and an international consortium called the Children’s Oncology Group gathers data about genetics. About 60%-70% of patients will be cured, she added.
However, “we’ve kind of been stuck for about the last 20 years,” she said. “A lot of improving the survival of patients has not been because we’ve been better at chemotherapy or using new chemo, but because we’ve gotten better at supportive care, at treating infections that can be fatal.”
There haven’t been major conflicts with insurers over coverage, she said, although drug shortages are a problem, especially in relapsed AML.
As for advice to colleagues, Dr. Tasian counseled them to understand the importance of genetic testing and the expanding role of stem cell transplants. “We are now transplanting somewhere between 30% and 50% of children with AML, which is a higher rate than we used to do,” she said. The number is up thanks to genetic testing that reveals which patients are most likely to benefit.
Also, she noted, “the chemotherapy that we get to these patients is really strong, and patients have a lot of complications. Really pay attention to supportive care.”
Dr. LeBlanc reported ties with AbbVie, Agios/Servier, Astellas, BMS/Celgene, Genentech, Pfizer, Incyte, Rige, Deverra, GSK, Jazz, and Seattle Genetics. Dr. Sekeres discloses relationships with BMS and Kurome. Dr. Tasian serves as the Leukemia & Lymphoma Society Pediatric Acute Leukemia consortium clinical trials leader and works with pharmaceutical companies on clinical trials under confidentiality agreements. Dr. Brunner has no disclosures.
Doxorubicin Increases Breast Cancer Risk in Women With Hodgkin Lymphoma
TOPLINE:
METHODOLOGY:
- Doxorubicin is a mainstay of Hodgkin lymphoma treatment.
- Studies suggest that girls with Hodgkin lymphoma who receive doxorubicin have a higher risk for breast cancer later in life, but it is unclear if women treated as adults face that same risk.
- To find out, investigators reviewed breast cancer incidence in 1964 Dutch women, ages 15-50, who were treated for Hodgkin lymphoma from 1975 to 2008.
- Patients had survived for at least 5 years, and 57% received doxorubicin.
TAKEAWAY:
- Women treated with doxorubicin had a 40% higher risk for breast cancer, and that risk was independent of age of treatment, receipt of chest radiation, and the use of gonadotoxic agents.
- The risk for breast cancer with doxorubicin was dose-dependent, with each 100 mg/m2 dose increment increasing the risk by 18%.
- The findings held whether women were treated years ago or more recently, despite the evolution of treatment strategies for Hodgkin lymphoma.
- After 30 years of follow-up, nearly one in five survivors (20.8%) developed breast cancer. It took 20 years for the elevated risk for breast cancer following treatment with doxorubicin to emerge.
IN PRACTICE:
The study suggests that adolescent and adult women survivors of Hodgkin lymphoma who received doxorubicin have an increased risk for breast cancer, and this risk is independent of age at first Hodgkin lymphoma treatment, receipt of chest radiotherapy, and gonadotoxic treatment, the authors concluded. “Our results have implications for [breast cancer] surveillance guidelines for [Hodgkin lymphoma] survivors and treatment strategies for patients with newly diagnosed” Hodgkin lymphoma.
SOURCE:
The study, led by Suzanne Neppelenbroek of the Netherlands Cancer Institute, Amsterdam, was published February 15 in the Journal of Clinical Oncology.
LIMITATIONS:
Recruitment ended in 2008 before the advent of newer treatments such as antibody-drug conjugates and immune checkpoint inhibitors.
DISCLOSURES:
The work was funded by the Dutch Cancer Society. Several authors reported ties to Lilly, AbbVie, Amgen, and other companies.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Doxorubicin is a mainstay of Hodgkin lymphoma treatment.
- Studies suggest that girls with Hodgkin lymphoma who receive doxorubicin have a higher risk for breast cancer later in life, but it is unclear if women treated as adults face that same risk.
- To find out, investigators reviewed breast cancer incidence in 1964 Dutch women, ages 15-50, who were treated for Hodgkin lymphoma from 1975 to 2008.
- Patients had survived for at least 5 years, and 57% received doxorubicin.
TAKEAWAY:
- Women treated with doxorubicin had a 40% higher risk for breast cancer, and that risk was independent of age of treatment, receipt of chest radiation, and the use of gonadotoxic agents.
- The risk for breast cancer with doxorubicin was dose-dependent, with each 100 mg/m2 dose increment increasing the risk by 18%.
- The findings held whether women were treated years ago or more recently, despite the evolution of treatment strategies for Hodgkin lymphoma.
- After 30 years of follow-up, nearly one in five survivors (20.8%) developed breast cancer. It took 20 years for the elevated risk for breast cancer following treatment with doxorubicin to emerge.
IN PRACTICE:
The study suggests that adolescent and adult women survivors of Hodgkin lymphoma who received doxorubicin have an increased risk for breast cancer, and this risk is independent of age at first Hodgkin lymphoma treatment, receipt of chest radiotherapy, and gonadotoxic treatment, the authors concluded. “Our results have implications for [breast cancer] surveillance guidelines for [Hodgkin lymphoma] survivors and treatment strategies for patients with newly diagnosed” Hodgkin lymphoma.
SOURCE:
The study, led by Suzanne Neppelenbroek of the Netherlands Cancer Institute, Amsterdam, was published February 15 in the Journal of Clinical Oncology.
LIMITATIONS:
Recruitment ended in 2008 before the advent of newer treatments such as antibody-drug conjugates and immune checkpoint inhibitors.
DISCLOSURES:
The work was funded by the Dutch Cancer Society. Several authors reported ties to Lilly, AbbVie, Amgen, and other companies.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Doxorubicin is a mainstay of Hodgkin lymphoma treatment.
- Studies suggest that girls with Hodgkin lymphoma who receive doxorubicin have a higher risk for breast cancer later in life, but it is unclear if women treated as adults face that same risk.
- To find out, investigators reviewed breast cancer incidence in 1964 Dutch women, ages 15-50, who were treated for Hodgkin lymphoma from 1975 to 2008.
- Patients had survived for at least 5 years, and 57% received doxorubicin.
TAKEAWAY:
- Women treated with doxorubicin had a 40% higher risk for breast cancer, and that risk was independent of age of treatment, receipt of chest radiation, and the use of gonadotoxic agents.
- The risk for breast cancer with doxorubicin was dose-dependent, with each 100 mg/m2 dose increment increasing the risk by 18%.
- The findings held whether women were treated years ago or more recently, despite the evolution of treatment strategies for Hodgkin lymphoma.
- After 30 years of follow-up, nearly one in five survivors (20.8%) developed breast cancer. It took 20 years for the elevated risk for breast cancer following treatment with doxorubicin to emerge.
IN PRACTICE:
The study suggests that adolescent and adult women survivors of Hodgkin lymphoma who received doxorubicin have an increased risk for breast cancer, and this risk is independent of age at first Hodgkin lymphoma treatment, receipt of chest radiotherapy, and gonadotoxic treatment, the authors concluded. “Our results have implications for [breast cancer] surveillance guidelines for [Hodgkin lymphoma] survivors and treatment strategies for patients with newly diagnosed” Hodgkin lymphoma.
SOURCE:
The study, led by Suzanne Neppelenbroek of the Netherlands Cancer Institute, Amsterdam, was published February 15 in the Journal of Clinical Oncology.
LIMITATIONS:
Recruitment ended in 2008 before the advent of newer treatments such as antibody-drug conjugates and immune checkpoint inhibitors.
DISCLOSURES:
The work was funded by the Dutch Cancer Society. Several authors reported ties to Lilly, AbbVie, Amgen, and other companies.
A version of this article appeared on Medscape.com.
Are Food Emulsifiers Associated With Increased Cancer Risk?
Food emulsifiers are among the most widespread food additives.
Ultraprocessed foods constitute a significant part of our diet, representing approximately 30% of energy intake in France.
Large epidemiologic studies have already linked diets rich in ultraprocessed products to an increased risk for cardiovascular diseases, diabetes, obesity, and mortality. Possible explanations for this association include the presence of additives, particularly emulsifiers. These additives are intended to improve the texture and shelf life of foods.
Recent experimental studies have shown that emulsifiers alter the gut microbiota and may lead to low-grade inflammation. Dysbiosis and chronic inflammation not only increase the risk for inflammatory bowel diseases but are also implicated in the etiology of several other chronic pathologies and certain extraintestinal cancers.
The NutriNet-Santé study provided extensive information on the dietary habits of > 100,000 French participants. A new analysis was conducted, examining the possible link between the presence of emulsifiers in the diet and cancer occurrence. Data from 92,000 participants (78.8% women) were utilized. They covered an average follow-up of 6.7 years, during which 2604 cancer cases were diagnosed, including 750 breast cancers, 322 prostate cancers, and 207 colorectal cancers.
In this cohort, the risk for cancer increased with a higher presence in the diet of products containing certain emulsifiers widely used in industrial food in Europe: Carrageenans (E407), mono- and diglycerides of fatty acids (E471), pectins (E440), and sodium carbonate (E500).
Notably, the highest consumption of mono- and diglycerides of fatty acids (E471) was associated with a 15% increase in the risk for all types of cancer, a 24% increase in breast cancer risk, and a 46% increase in prostate cancer risk. The highest consumption of carrageenans (E407) was associated with a 28% increase in breast cancer risk.
In an analysis by menopausal status, the risk for breast cancer before menopause was associated with high consumption of diphosphates (E450; 45% increase), pectins (E440; 55% increase), and sodium bicarbonate (E500; 48% increase). No link was found between emulsifier consumption and colorectal cancer risk. While some associations were observed for other emulsifiers, they did not persist in sensitivity analyses.
The European Food Safety Agency recently evaluated the risks of emulsifiers, however, and found no safety issues or need to limit daily consumption of several of them, notably E471.
It is certain that cancer is multifactorial, and a single factor (here, exposure to emulsifiers) will not significantly increase the risk. However, while not essential to human health, emulsifiers are widely prevalent in the global market. Therefore, if causality is established, the increased risk could translate into a significant number of preventable cancers at the population level. Confirmation of this causal link will need to be obtained through experimental and epidemiological studies.
This story was translated from JIM, which is part of the Medscape professional network, using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Food emulsifiers are among the most widespread food additives.
Ultraprocessed foods constitute a significant part of our diet, representing approximately 30% of energy intake in France.
Large epidemiologic studies have already linked diets rich in ultraprocessed products to an increased risk for cardiovascular diseases, diabetes, obesity, and mortality. Possible explanations for this association include the presence of additives, particularly emulsifiers. These additives are intended to improve the texture and shelf life of foods.
Recent experimental studies have shown that emulsifiers alter the gut microbiota and may lead to low-grade inflammation. Dysbiosis and chronic inflammation not only increase the risk for inflammatory bowel diseases but are also implicated in the etiology of several other chronic pathologies and certain extraintestinal cancers.
The NutriNet-Santé study provided extensive information on the dietary habits of > 100,000 French participants. A new analysis was conducted, examining the possible link between the presence of emulsifiers in the diet and cancer occurrence. Data from 92,000 participants (78.8% women) were utilized. They covered an average follow-up of 6.7 years, during which 2604 cancer cases were diagnosed, including 750 breast cancers, 322 prostate cancers, and 207 colorectal cancers.
In this cohort, the risk for cancer increased with a higher presence in the diet of products containing certain emulsifiers widely used in industrial food in Europe: Carrageenans (E407), mono- and diglycerides of fatty acids (E471), pectins (E440), and sodium carbonate (E500).
Notably, the highest consumption of mono- and diglycerides of fatty acids (E471) was associated with a 15% increase in the risk for all types of cancer, a 24% increase in breast cancer risk, and a 46% increase in prostate cancer risk. The highest consumption of carrageenans (E407) was associated with a 28% increase in breast cancer risk.
In an analysis by menopausal status, the risk for breast cancer before menopause was associated with high consumption of diphosphates (E450; 45% increase), pectins (E440; 55% increase), and sodium bicarbonate (E500; 48% increase). No link was found between emulsifier consumption and colorectal cancer risk. While some associations were observed for other emulsifiers, they did not persist in sensitivity analyses.
The European Food Safety Agency recently evaluated the risks of emulsifiers, however, and found no safety issues or need to limit daily consumption of several of them, notably E471.
It is certain that cancer is multifactorial, and a single factor (here, exposure to emulsifiers) will not significantly increase the risk. However, while not essential to human health, emulsifiers are widely prevalent in the global market. Therefore, if causality is established, the increased risk could translate into a significant number of preventable cancers at the population level. Confirmation of this causal link will need to be obtained through experimental and epidemiological studies.
This story was translated from JIM, which is part of the Medscape professional network, using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Food emulsifiers are among the most widespread food additives.
Ultraprocessed foods constitute a significant part of our diet, representing approximately 30% of energy intake in France.
Large epidemiologic studies have already linked diets rich in ultraprocessed products to an increased risk for cardiovascular diseases, diabetes, obesity, and mortality. Possible explanations for this association include the presence of additives, particularly emulsifiers. These additives are intended to improve the texture and shelf life of foods.
Recent experimental studies have shown that emulsifiers alter the gut microbiota and may lead to low-grade inflammation. Dysbiosis and chronic inflammation not only increase the risk for inflammatory bowel diseases but are also implicated in the etiology of several other chronic pathologies and certain extraintestinal cancers.
The NutriNet-Santé study provided extensive information on the dietary habits of > 100,000 French participants. A new analysis was conducted, examining the possible link between the presence of emulsifiers in the diet and cancer occurrence. Data from 92,000 participants (78.8% women) were utilized. They covered an average follow-up of 6.7 years, during which 2604 cancer cases were diagnosed, including 750 breast cancers, 322 prostate cancers, and 207 colorectal cancers.
In this cohort, the risk for cancer increased with a higher presence in the diet of products containing certain emulsifiers widely used in industrial food in Europe: Carrageenans (E407), mono- and diglycerides of fatty acids (E471), pectins (E440), and sodium carbonate (E500).
Notably, the highest consumption of mono- and diglycerides of fatty acids (E471) was associated with a 15% increase in the risk for all types of cancer, a 24% increase in breast cancer risk, and a 46% increase in prostate cancer risk. The highest consumption of carrageenans (E407) was associated with a 28% increase in breast cancer risk.
In an analysis by menopausal status, the risk for breast cancer before menopause was associated with high consumption of diphosphates (E450; 45% increase), pectins (E440; 55% increase), and sodium bicarbonate (E500; 48% increase). No link was found between emulsifier consumption and colorectal cancer risk. While some associations were observed for other emulsifiers, they did not persist in sensitivity analyses.
The European Food Safety Agency recently evaluated the risks of emulsifiers, however, and found no safety issues or need to limit daily consumption of several of them, notably E471.
It is certain that cancer is multifactorial, and a single factor (here, exposure to emulsifiers) will not significantly increase the risk. However, while not essential to human health, emulsifiers are widely prevalent in the global market. Therefore, if causality is established, the increased risk could translate into a significant number of preventable cancers at the population level. Confirmation of this causal link will need to be obtained through experimental and epidemiological studies.
This story was translated from JIM, which is part of the Medscape professional network, using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Democratic Lawmakers Press Pfizer on Chemotherapy Drug Shortages
In a statement about their February 21 action, the legislators, led by Rep. Jamie Raskin (D-Md.), the committee’s ranking minority member, described their work as a follow up to an earlier investigation into price hikes of generic drugs. While the committee members queried Pfizer over the three oncology medications only, they also sent letters to drugmakers Teva and Sandoz with respect to shortages in other drug classes.
A representative for Pfizer confirmed to MDedge Oncology that the company had received the representatives’ letter but said “we have no further details to provide at this time.”
What is the basis for concern?
All three generic chemotherapy drugs are mainstay treatments used across a broad array of cancers. Though shortages have been reported for several years, they became especially acute after December 2022, when an inspection by the US Food and Drug Administration (FDA) led to regulatory action against an Indian manufacturer, Intas, that produced up to half of the platinum-based therapies supplied globally. The National Comprehensive Cancer Care Network reported in October 2023 that more than 90% of its member centers were struggling to maintain adequate supplies of carboplatin, and 70% had trouble obtaining cisplatin, while the American Society of Clinical Oncology published clinical guidance on alternative treatment strategies.
What has the government done in response to the recent shortages?
The White House and the FDA announced in September that they were working with several manufacturers to help increase supplies of the platinum-based chemotherapies and of methotrexate, and taking measures that included relaxing rules on imports. Recent guidance under a pandemic-era federal law, the 2020 CARES Act, strengthened manufacturer reporting requirements related to drug shortages, and other measures have been proposed. While federal regulators have many tools with which to address drug shortages, they cannot legally oblige a manufacturer to increase production of a drug.
What can the lawmakers expect to achieve with their letter?
By pressuring Pfizer publicly, the lawmakers may be able to nudge the company to take measures to assure more consistent supplies of the three drugs. The lawmakers also said they hoped to glean from Pfizer more insight into the root causes of the shortages and potential remedies. They noted that, in a May 2023 letter by Pfizer to customers, the company had warned of depleted and limited supplies of the three drugs and said it was “working diligently” to increase output. However, the lawmakers wrote, “the root cause is not yet resolved and carboplatin, cisplatin, and methotrexate continue to experience residual delays.”
Why did the committee target Pfizer specifically?
Pfizer and its subsidiaries are among the major manufacturers of the three generic chemotherapy agents mentioned in the letter. The legislators noted that “pharmaceutical companies may not be motivated to produce generic drugs like carboplatin, cisplatin, and methotrexate, because they are not as lucrative as producing patented brand name drugs,” and that “as a principal supplier of carboplatin, cisplatin, and methotrexate, it is critical that Pfizer continues to increase production of these life-sustaining cancer medications, even amidst potential lower profitability.”
The committee members also made reference to news reports of price-gouging with these medications, as smaller hospitals or oncology centers are forced to turn to unscrupulous third-party suppliers.
What is being demanded of Pfizer?
Pfizer was given until March 6 to respond, in writing and in a briefing with committee staff, to a six questions. These queries concern what specific steps the company has taken to increase supplies of the three generic oncology drugs, what Pfizer is doing to help avert price-gouging, whether further oncology drug shortages are anticipated, and how the company is working with the FDA on the matter.
In a statement about their February 21 action, the legislators, led by Rep. Jamie Raskin (D-Md.), the committee’s ranking minority member, described their work as a follow up to an earlier investigation into price hikes of generic drugs. While the committee members queried Pfizer over the three oncology medications only, they also sent letters to drugmakers Teva and Sandoz with respect to shortages in other drug classes.
A representative for Pfizer confirmed to MDedge Oncology that the company had received the representatives’ letter but said “we have no further details to provide at this time.”
What is the basis for concern?
All three generic chemotherapy drugs are mainstay treatments used across a broad array of cancers. Though shortages have been reported for several years, they became especially acute after December 2022, when an inspection by the US Food and Drug Administration (FDA) led to regulatory action against an Indian manufacturer, Intas, that produced up to half of the platinum-based therapies supplied globally. The National Comprehensive Cancer Care Network reported in October 2023 that more than 90% of its member centers were struggling to maintain adequate supplies of carboplatin, and 70% had trouble obtaining cisplatin, while the American Society of Clinical Oncology published clinical guidance on alternative treatment strategies.
What has the government done in response to the recent shortages?
The White House and the FDA announced in September that they were working with several manufacturers to help increase supplies of the platinum-based chemotherapies and of methotrexate, and taking measures that included relaxing rules on imports. Recent guidance under a pandemic-era federal law, the 2020 CARES Act, strengthened manufacturer reporting requirements related to drug shortages, and other measures have been proposed. While federal regulators have many tools with which to address drug shortages, they cannot legally oblige a manufacturer to increase production of a drug.
What can the lawmakers expect to achieve with their letter?
By pressuring Pfizer publicly, the lawmakers may be able to nudge the company to take measures to assure more consistent supplies of the three drugs. The lawmakers also said they hoped to glean from Pfizer more insight into the root causes of the shortages and potential remedies. They noted that, in a May 2023 letter by Pfizer to customers, the company had warned of depleted and limited supplies of the three drugs and said it was “working diligently” to increase output. However, the lawmakers wrote, “the root cause is not yet resolved and carboplatin, cisplatin, and methotrexate continue to experience residual delays.”
Why did the committee target Pfizer specifically?
Pfizer and its subsidiaries are among the major manufacturers of the three generic chemotherapy agents mentioned in the letter. The legislators noted that “pharmaceutical companies may not be motivated to produce generic drugs like carboplatin, cisplatin, and methotrexate, because they are not as lucrative as producing patented brand name drugs,” and that “as a principal supplier of carboplatin, cisplatin, and methotrexate, it is critical that Pfizer continues to increase production of these life-sustaining cancer medications, even amidst potential lower profitability.”
The committee members also made reference to news reports of price-gouging with these medications, as smaller hospitals or oncology centers are forced to turn to unscrupulous third-party suppliers.
What is being demanded of Pfizer?
Pfizer was given until March 6 to respond, in writing and in a briefing with committee staff, to a six questions. These queries concern what specific steps the company has taken to increase supplies of the three generic oncology drugs, what Pfizer is doing to help avert price-gouging, whether further oncology drug shortages are anticipated, and how the company is working with the FDA on the matter.
In a statement about their February 21 action, the legislators, led by Rep. Jamie Raskin (D-Md.), the committee’s ranking minority member, described their work as a follow up to an earlier investigation into price hikes of generic drugs. While the committee members queried Pfizer over the three oncology medications only, they also sent letters to drugmakers Teva and Sandoz with respect to shortages in other drug classes.
A representative for Pfizer confirmed to MDedge Oncology that the company had received the representatives’ letter but said “we have no further details to provide at this time.”
What is the basis for concern?
All three generic chemotherapy drugs are mainstay treatments used across a broad array of cancers. Though shortages have been reported for several years, they became especially acute after December 2022, when an inspection by the US Food and Drug Administration (FDA) led to regulatory action against an Indian manufacturer, Intas, that produced up to half of the platinum-based therapies supplied globally. The National Comprehensive Cancer Care Network reported in October 2023 that more than 90% of its member centers were struggling to maintain adequate supplies of carboplatin, and 70% had trouble obtaining cisplatin, while the American Society of Clinical Oncology published clinical guidance on alternative treatment strategies.
What has the government done in response to the recent shortages?
The White House and the FDA announced in September that they were working with several manufacturers to help increase supplies of the platinum-based chemotherapies and of methotrexate, and taking measures that included relaxing rules on imports. Recent guidance under a pandemic-era federal law, the 2020 CARES Act, strengthened manufacturer reporting requirements related to drug shortages, and other measures have been proposed. While federal regulators have many tools with which to address drug shortages, they cannot legally oblige a manufacturer to increase production of a drug.
What can the lawmakers expect to achieve with their letter?
By pressuring Pfizer publicly, the lawmakers may be able to nudge the company to take measures to assure more consistent supplies of the three drugs. The lawmakers also said they hoped to glean from Pfizer more insight into the root causes of the shortages and potential remedies. They noted that, in a May 2023 letter by Pfizer to customers, the company had warned of depleted and limited supplies of the three drugs and said it was “working diligently” to increase output. However, the lawmakers wrote, “the root cause is not yet resolved and carboplatin, cisplatin, and methotrexate continue to experience residual delays.”
Why did the committee target Pfizer specifically?
Pfizer and its subsidiaries are among the major manufacturers of the three generic chemotherapy agents mentioned in the letter. The legislators noted that “pharmaceutical companies may not be motivated to produce generic drugs like carboplatin, cisplatin, and methotrexate, because they are not as lucrative as producing patented brand name drugs,” and that “as a principal supplier of carboplatin, cisplatin, and methotrexate, it is critical that Pfizer continues to increase production of these life-sustaining cancer medications, even amidst potential lower profitability.”
The committee members also made reference to news reports of price-gouging with these medications, as smaller hospitals or oncology centers are forced to turn to unscrupulous third-party suppliers.
What is being demanded of Pfizer?
Pfizer was given until March 6 to respond, in writing and in a briefing with committee staff, to a six questions. These queries concern what specific steps the company has taken to increase supplies of the three generic oncology drugs, what Pfizer is doing to help avert price-gouging, whether further oncology drug shortages are anticipated, and how the company is working with the FDA on the matter.
Asparaginase in ALL: Innovative Ways to Manage Toxicity
The good news, hematologists note, is that new strategies have been developed to address side effects. “We’ve gotten better at managing them,” pediatric oncologist Birte Wistinghausen, MD, of Children’s National Hospital in Washington, DC, said in an interview.
According to her, key approaches include sensitivity testing and “pre-medication” to prevent adverse effects from appearing in the first place.
The American Cancer Society estimates that 6,550 new cases of ALL appear in the United States each year, and 1,330 people die from the disease.
“Most cases of ALL occur in children, but most deaths from ALL (about 4 out of 5) occur in adults,” the organization reports. Indeed, the 5-year survival rate in children is now at about 90%, a number that hematologists partially attribute to the power of asparaginase.
Researchers believe that asparaginase, an enzyme, works by breaking down a substance called asparagine, which ALL cells use to reproduce. The drug is “universally used throughout the treatment of ALL in children and adolescents,” Luke Maese, DO, associate professor of pediatrics at the University of Utah–Huntsman Cancer Institute, Salt Lake City, and director of Leukemia/Lymphoma at Primary Children’s Hospital, said in an interview. “It has become more and more adopted in the treatment of young adults as well.”
The formulations of available asparaginase have evolved over the years, Dr. Maese said. “Currently, the first-line asparaginase products delivered in the majority of patients throughout the world are pegylated, meaning they have an extended duration of action. There are non-pegylated asparaginase products that are used as well.”
The pegylated drugs are much easier on patients since they don’t require frequent injections, according to experts.
Treatment protocols vary, Dr. Maese said. “Some use the drug intermittently intermixed throughout therapy, and others have periods of continuous asparaginase use — i.e. 10-20 weeks of repeated doses of the drug.”
All patients are likely to experience side effects, he said, and about 5%-10% of standard-risk and 20%-25% of high-risk patients will experience clinically significant problems.
When asparaginase is given by IV, its rapid onset can lead to a condition called acute hyperammonemia, in which ammonia levels rise and patients develop flushing, anxiety, and low blood pressure, said Dr. Wistinghausen of Children’s National Hospital. “But that is not a reason to abandon asparaginase.”
It can be difficult to differentiate this effect from hypersensitivity — allergic reactions — which can range from hives to full anaphylactic shock that requires treatment with epinephrine, she said.
According to Dr. Maese, other major side effects other than hypersensitivity include pancreatitis, hepatotoxicity, and thrombosis. The most dangerous of these side effects are hypersensitivity and pancreatitis, which can lead to discontinuation of treatment, he said. Indeed, a 2017 study found that 2% of 465 patients with ALL who developed asparaginase-associated pancreatitis died, and 8% needed mechanical ventilation.
There’s no way to predict which patients may be susceptible to pancreatitis, Michael J. Burke, MD, professor of pediatrics and director of leukemia/lymphoma director at Children’s Wisconsin and Medical College of Wisconsin, said in an interview.
As for therapy options if pancreatitis develops, a 2022 review cowritten by Dr. Maese reported that clinicians have been leaning toward re-treating patients with asparaginase since it’s so crucial to treatment. This has worked about 50% of the time, the review reported, and “many groups consider it in the setting of all grade 2 pancreatitis and grade 3 pancreatitis without prolonged illness or severe complications.”
As for hypersensitivity, the most prevalent adverse effect, clinicians frequently administer anti-allergy medications prior to infusion. This approach, known as “pre-medication,” is controversial. Research has produced conflicting results, with a 2022 study in the journal Blood finding that pre-medication had no effect on hypersensitivity in children with ALL.
“Although there is mixed data, most institutions utilize this,” Dr. Maese said. “At our institution, we continue to use pre-medication prior to pegylated asparaginase but do not use it with non-pegylated asparaginase.”
Specifically, most institutions administer H2 and H1 blockers, Dr. Wistinghausen said. “Some institutions also use hydrocortisone” — a steroid — “but our institution only uses it if patients have a reaction.”
Other potential adverse effects to treatment include infusion reactions, which can mimic allergic reactions such as nausea, vomiting, abdominal pain, and flushing, Dr. Maese said.
Asked how to treat patients who cannot tolerate first-line treatment with asparaginase, Dr. Burke responded, “There are second-generation asparaginase formulations for once a patient develops an allergy.”
Dr. Maese said his institution switches patients when necessary to asparaginase Erwinia chrysanthemi (recombinant)-rywn, also known as Rylaze.
Another recent development in ALL treatment is the widespread use of drug monitoring to make sure asparaginase is reaching therapeutic levels. “Asparagine itself is difficult to measure so we use a surrogate of asparaginase levels to demonstrate efficacy of the drug,” he said. “There is conflicting literature as to what constitutes a therapeutic level, but the internationally accepted standard is a level of ≥ 0.1 IU/mL. We monitor asparaginase levels routinely with pegylated asparaginase but not with non-pegylated asparaginase.”
Tests can turn up “silent inactivation,” a term that refers to when the drug is “inactivated” and is not effective, Dr. Maese said. “There are several guidelines that have defined inactivation.” According to the 2022 report cowritten by Dr. Maese, Rylaze can be an alternative option if initial asparaginase treatment isn’t working.
With regard to cost, treatment with asparaginase can cost tens of thousands of dollars. However, insurers routinely pay for treatment plus pre-medication and testing, Dr. Burke said. “There’s no pushback. It seems to be accepted.”
What’s next on the horizon? “We need to understand better those patients who are at risk for toxicity,” Dr. Maese noted. “We understand obesity causes risk for certain toxicities, but have little else to go on. There has been some work with genomics and its relationship to risk of toxicity. However, it has been difficult to translate what has been found to patients.”
There’s work in progress that is exploring other preventive approaches to decrease toxicity, he said. Also, “optimizing the dosing of asparaginase has been explored more in Europe and within a smaller consortium in North America.”
In addition, he said, “as we begin to increase use of immunotherapy within our chemotherapy backbones, we need to understand the relationship these drugs have with asparaginase treatment.”
Dr. Burke and Dr. Wistinghausen have no disclosures. Dr. Maese discloses relationships with Jazz (advisory board, consultant, speakers bureau) and Servier (advisory board).
The good news, hematologists note, is that new strategies have been developed to address side effects. “We’ve gotten better at managing them,” pediatric oncologist Birte Wistinghausen, MD, of Children’s National Hospital in Washington, DC, said in an interview.
According to her, key approaches include sensitivity testing and “pre-medication” to prevent adverse effects from appearing in the first place.
The American Cancer Society estimates that 6,550 new cases of ALL appear in the United States each year, and 1,330 people die from the disease.
“Most cases of ALL occur in children, but most deaths from ALL (about 4 out of 5) occur in adults,” the organization reports. Indeed, the 5-year survival rate in children is now at about 90%, a number that hematologists partially attribute to the power of asparaginase.
Researchers believe that asparaginase, an enzyme, works by breaking down a substance called asparagine, which ALL cells use to reproduce. The drug is “universally used throughout the treatment of ALL in children and adolescents,” Luke Maese, DO, associate professor of pediatrics at the University of Utah–Huntsman Cancer Institute, Salt Lake City, and director of Leukemia/Lymphoma at Primary Children’s Hospital, said in an interview. “It has become more and more adopted in the treatment of young adults as well.”
The formulations of available asparaginase have evolved over the years, Dr. Maese said. “Currently, the first-line asparaginase products delivered in the majority of patients throughout the world are pegylated, meaning they have an extended duration of action. There are non-pegylated asparaginase products that are used as well.”
The pegylated drugs are much easier on patients since they don’t require frequent injections, according to experts.
Treatment protocols vary, Dr. Maese said. “Some use the drug intermittently intermixed throughout therapy, and others have periods of continuous asparaginase use — i.e. 10-20 weeks of repeated doses of the drug.”
All patients are likely to experience side effects, he said, and about 5%-10% of standard-risk and 20%-25% of high-risk patients will experience clinically significant problems.
When asparaginase is given by IV, its rapid onset can lead to a condition called acute hyperammonemia, in which ammonia levels rise and patients develop flushing, anxiety, and low blood pressure, said Dr. Wistinghausen of Children’s National Hospital. “But that is not a reason to abandon asparaginase.”
It can be difficult to differentiate this effect from hypersensitivity — allergic reactions — which can range from hives to full anaphylactic shock that requires treatment with epinephrine, she said.
According to Dr. Maese, other major side effects other than hypersensitivity include pancreatitis, hepatotoxicity, and thrombosis. The most dangerous of these side effects are hypersensitivity and pancreatitis, which can lead to discontinuation of treatment, he said. Indeed, a 2017 study found that 2% of 465 patients with ALL who developed asparaginase-associated pancreatitis died, and 8% needed mechanical ventilation.
There’s no way to predict which patients may be susceptible to pancreatitis, Michael J. Burke, MD, professor of pediatrics and director of leukemia/lymphoma director at Children’s Wisconsin and Medical College of Wisconsin, said in an interview.
As for therapy options if pancreatitis develops, a 2022 review cowritten by Dr. Maese reported that clinicians have been leaning toward re-treating patients with asparaginase since it’s so crucial to treatment. This has worked about 50% of the time, the review reported, and “many groups consider it in the setting of all grade 2 pancreatitis and grade 3 pancreatitis without prolonged illness or severe complications.”
As for hypersensitivity, the most prevalent adverse effect, clinicians frequently administer anti-allergy medications prior to infusion. This approach, known as “pre-medication,” is controversial. Research has produced conflicting results, with a 2022 study in the journal Blood finding that pre-medication had no effect on hypersensitivity in children with ALL.
“Although there is mixed data, most institutions utilize this,” Dr. Maese said. “At our institution, we continue to use pre-medication prior to pegylated asparaginase but do not use it with non-pegylated asparaginase.”
Specifically, most institutions administer H2 and H1 blockers, Dr. Wistinghausen said. “Some institutions also use hydrocortisone” — a steroid — “but our institution only uses it if patients have a reaction.”
Other potential adverse effects to treatment include infusion reactions, which can mimic allergic reactions such as nausea, vomiting, abdominal pain, and flushing, Dr. Maese said.
Asked how to treat patients who cannot tolerate first-line treatment with asparaginase, Dr. Burke responded, “There are second-generation asparaginase formulations for once a patient develops an allergy.”
Dr. Maese said his institution switches patients when necessary to asparaginase Erwinia chrysanthemi (recombinant)-rywn, also known as Rylaze.
Another recent development in ALL treatment is the widespread use of drug monitoring to make sure asparaginase is reaching therapeutic levels. “Asparagine itself is difficult to measure so we use a surrogate of asparaginase levels to demonstrate efficacy of the drug,” he said. “There is conflicting literature as to what constitutes a therapeutic level, but the internationally accepted standard is a level of ≥ 0.1 IU/mL. We monitor asparaginase levels routinely with pegylated asparaginase but not with non-pegylated asparaginase.”
Tests can turn up “silent inactivation,” a term that refers to when the drug is “inactivated” and is not effective, Dr. Maese said. “There are several guidelines that have defined inactivation.” According to the 2022 report cowritten by Dr. Maese, Rylaze can be an alternative option if initial asparaginase treatment isn’t working.
With regard to cost, treatment with asparaginase can cost tens of thousands of dollars. However, insurers routinely pay for treatment plus pre-medication and testing, Dr. Burke said. “There’s no pushback. It seems to be accepted.”
What’s next on the horizon? “We need to understand better those patients who are at risk for toxicity,” Dr. Maese noted. “We understand obesity causes risk for certain toxicities, but have little else to go on. There has been some work with genomics and its relationship to risk of toxicity. However, it has been difficult to translate what has been found to patients.”
There’s work in progress that is exploring other preventive approaches to decrease toxicity, he said. Also, “optimizing the dosing of asparaginase has been explored more in Europe and within a smaller consortium in North America.”
In addition, he said, “as we begin to increase use of immunotherapy within our chemotherapy backbones, we need to understand the relationship these drugs have with asparaginase treatment.”
Dr. Burke and Dr. Wistinghausen have no disclosures. Dr. Maese discloses relationships with Jazz (advisory board, consultant, speakers bureau) and Servier (advisory board).
The good news, hematologists note, is that new strategies have been developed to address side effects. “We’ve gotten better at managing them,” pediatric oncologist Birte Wistinghausen, MD, of Children’s National Hospital in Washington, DC, said in an interview.
According to her, key approaches include sensitivity testing and “pre-medication” to prevent adverse effects from appearing in the first place.
The American Cancer Society estimates that 6,550 new cases of ALL appear in the United States each year, and 1,330 people die from the disease.
“Most cases of ALL occur in children, but most deaths from ALL (about 4 out of 5) occur in adults,” the organization reports. Indeed, the 5-year survival rate in children is now at about 90%, a number that hematologists partially attribute to the power of asparaginase.
Researchers believe that asparaginase, an enzyme, works by breaking down a substance called asparagine, which ALL cells use to reproduce. The drug is “universally used throughout the treatment of ALL in children and adolescents,” Luke Maese, DO, associate professor of pediatrics at the University of Utah–Huntsman Cancer Institute, Salt Lake City, and director of Leukemia/Lymphoma at Primary Children’s Hospital, said in an interview. “It has become more and more adopted in the treatment of young adults as well.”
The formulations of available asparaginase have evolved over the years, Dr. Maese said. “Currently, the first-line asparaginase products delivered in the majority of patients throughout the world are pegylated, meaning they have an extended duration of action. There are non-pegylated asparaginase products that are used as well.”
The pegylated drugs are much easier on patients since they don’t require frequent injections, according to experts.
Treatment protocols vary, Dr. Maese said. “Some use the drug intermittently intermixed throughout therapy, and others have periods of continuous asparaginase use — i.e. 10-20 weeks of repeated doses of the drug.”
All patients are likely to experience side effects, he said, and about 5%-10% of standard-risk and 20%-25% of high-risk patients will experience clinically significant problems.
When asparaginase is given by IV, its rapid onset can lead to a condition called acute hyperammonemia, in which ammonia levels rise and patients develop flushing, anxiety, and low blood pressure, said Dr. Wistinghausen of Children’s National Hospital. “But that is not a reason to abandon asparaginase.”
It can be difficult to differentiate this effect from hypersensitivity — allergic reactions — which can range from hives to full anaphylactic shock that requires treatment with epinephrine, she said.
According to Dr. Maese, other major side effects other than hypersensitivity include pancreatitis, hepatotoxicity, and thrombosis. The most dangerous of these side effects are hypersensitivity and pancreatitis, which can lead to discontinuation of treatment, he said. Indeed, a 2017 study found that 2% of 465 patients with ALL who developed asparaginase-associated pancreatitis died, and 8% needed mechanical ventilation.
There’s no way to predict which patients may be susceptible to pancreatitis, Michael J. Burke, MD, professor of pediatrics and director of leukemia/lymphoma director at Children’s Wisconsin and Medical College of Wisconsin, said in an interview.
As for therapy options if pancreatitis develops, a 2022 review cowritten by Dr. Maese reported that clinicians have been leaning toward re-treating patients with asparaginase since it’s so crucial to treatment. This has worked about 50% of the time, the review reported, and “many groups consider it in the setting of all grade 2 pancreatitis and grade 3 pancreatitis without prolonged illness or severe complications.”
As for hypersensitivity, the most prevalent adverse effect, clinicians frequently administer anti-allergy medications prior to infusion. This approach, known as “pre-medication,” is controversial. Research has produced conflicting results, with a 2022 study in the journal Blood finding that pre-medication had no effect on hypersensitivity in children with ALL.
“Although there is mixed data, most institutions utilize this,” Dr. Maese said. “At our institution, we continue to use pre-medication prior to pegylated asparaginase but do not use it with non-pegylated asparaginase.”
Specifically, most institutions administer H2 and H1 blockers, Dr. Wistinghausen said. “Some institutions also use hydrocortisone” — a steroid — “but our institution only uses it if patients have a reaction.”
Other potential adverse effects to treatment include infusion reactions, which can mimic allergic reactions such as nausea, vomiting, abdominal pain, and flushing, Dr. Maese said.
Asked how to treat patients who cannot tolerate first-line treatment with asparaginase, Dr. Burke responded, “There are second-generation asparaginase formulations for once a patient develops an allergy.”
Dr. Maese said his institution switches patients when necessary to asparaginase Erwinia chrysanthemi (recombinant)-rywn, also known as Rylaze.
Another recent development in ALL treatment is the widespread use of drug monitoring to make sure asparaginase is reaching therapeutic levels. “Asparagine itself is difficult to measure so we use a surrogate of asparaginase levels to demonstrate efficacy of the drug,” he said. “There is conflicting literature as to what constitutes a therapeutic level, but the internationally accepted standard is a level of ≥ 0.1 IU/mL. We monitor asparaginase levels routinely with pegylated asparaginase but not with non-pegylated asparaginase.”
Tests can turn up “silent inactivation,” a term that refers to when the drug is “inactivated” and is not effective, Dr. Maese said. “There are several guidelines that have defined inactivation.” According to the 2022 report cowritten by Dr. Maese, Rylaze can be an alternative option if initial asparaginase treatment isn’t working.
With regard to cost, treatment with asparaginase can cost tens of thousands of dollars. However, insurers routinely pay for treatment plus pre-medication and testing, Dr. Burke said. “There’s no pushback. It seems to be accepted.”
What’s next on the horizon? “We need to understand better those patients who are at risk for toxicity,” Dr. Maese noted. “We understand obesity causes risk for certain toxicities, but have little else to go on. There has been some work with genomics and its relationship to risk of toxicity. However, it has been difficult to translate what has been found to patients.”
There’s work in progress that is exploring other preventive approaches to decrease toxicity, he said. Also, “optimizing the dosing of asparaginase has been explored more in Europe and within a smaller consortium in North America.”
In addition, he said, “as we begin to increase use of immunotherapy within our chemotherapy backbones, we need to understand the relationship these drugs have with asparaginase treatment.”
Dr. Burke and Dr. Wistinghausen have no disclosures. Dr. Maese discloses relationships with Jazz (advisory board, consultant, speakers bureau) and Servier (advisory board).
Unleashing Our Immune Response to Quash Cancer
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
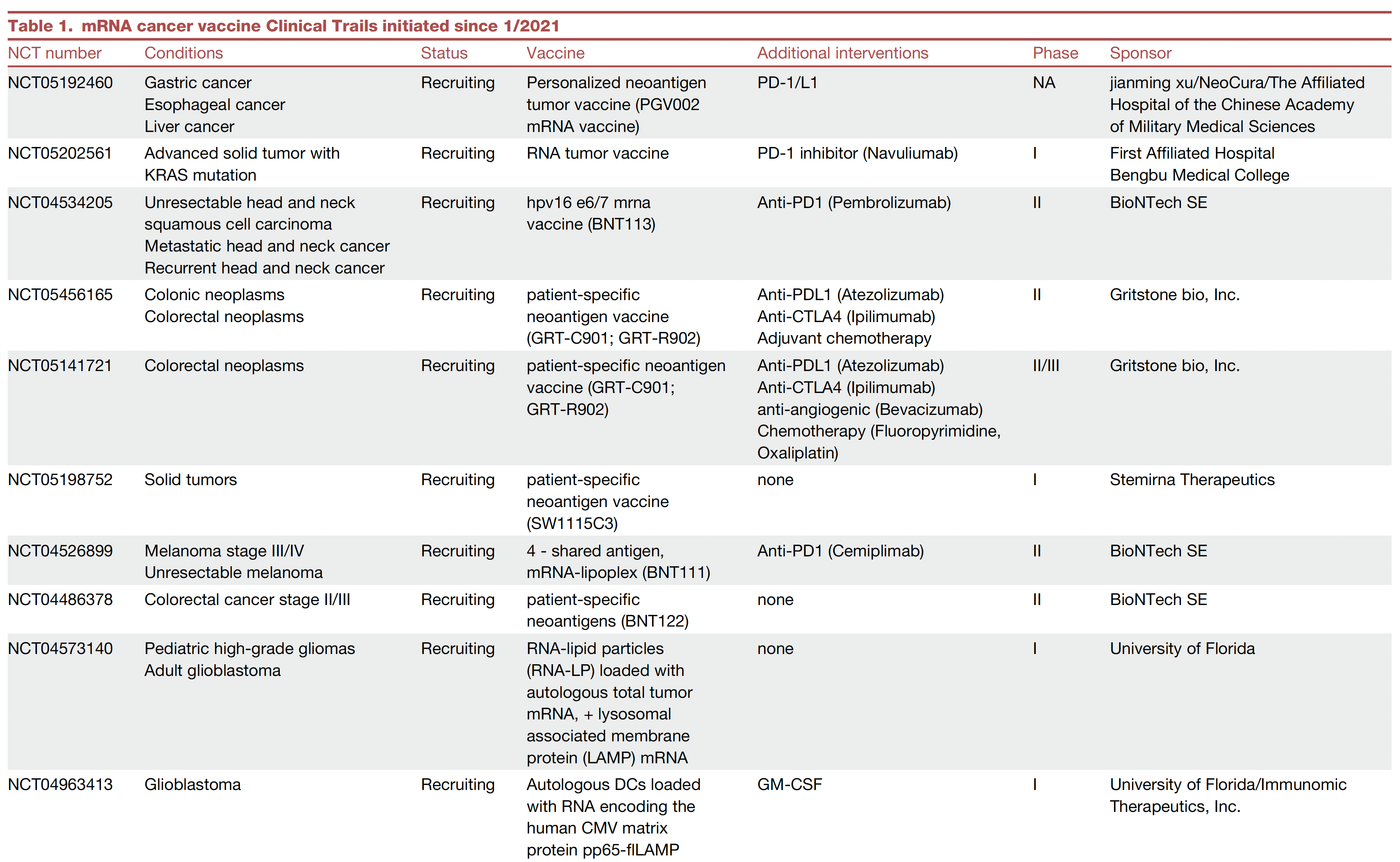{kind=link}
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
{kind=link}
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
CAR T-Cell: Do Benefits Still Outweigh Risks?
Importantly, most specialists agree, so far the risk appears no greater than the known risk of secondary primary malignancies that is well established with other cancer therapies.
“The data that we have so far suggest that the risk of secondary T-cell lymphoma in patients treated with CAR T-cells is similar to [that] of patients treated with other cancer therapies, [including] chemotherapy, radiation, transplantation,” Marco Ruella, MD, said in an interview. He reported on a case of a T-cell lymphoma occurring following CAR-T therapy at the University of Pennsylvania.
While his team is still investigating the development of such malignancies, “the FDA notice does not change our clinical practice and patients should be reassured that the benefit of CAR-T therapy significantly outweighs the potential risk of secondary malignancies including T-cell lymphoma,” said Dr. Ruella, scientific director of the Lymphoma Program, Division of Hematology and Oncology and Center for Cellular Immunotherapies, at the University of Pennsylvania, Philadelphia.
FDA: 28 Reports of Malignancies; 3 with Evidence of ‘Likely’ CAR T Involvement
Concerns were raised last November when the FDA announced in a safety communication that it was investigating the “serious risk of T-cell malignancy” following B-cell maturation antigen (BCMA)-directed or CD19-directed CAR T-cell immunotherapies, citing reports from clinical trials and/or postmarketing adverse event data sources. Subsequently, in January, the FDA called for the boxed warning on all approved BCMA- and CD19-targeted genetically modified autologous T-cell immunotherapies, which include: Abecma (idecabtagene vicleucel); Breyanzi (lisocabtagene maraleucel); Carvykti (ciltacabtagene autoleucel); Kymriah (tisagenlecleucel); Tecartus (brexucabtagene autoleucel); and Yescarta (axicabtagene ciloleucel).
“Although the overall benefits of these products continue to outweigh their potential risks for their approved uses, the FDA continues to investigate the identified risk of T-cell malignancy with serious outcomes, including hospitalization and death,” the FDA reported in discussing the safety warnings.
The cases were detailed in a report from FDA researchers published in the New England Journal of Medicine, noting that as of December 31, 2023, the FDA had become aware of 22 cases of T-cell cancers occurring following CAR T-cell treatment, including T-cell lymphoma, T-cell large granular lymphocytosis, peripheral T-cell lymphoma, and cutaneous T-cell lymphoma.
Report coauthor Peter Marks, MD, PhD, of the FDA’s Center for Biologics Evaluation and Research in Silver Spring, Maryland, said in an interview that since the publication of their report, six new cases have emerged.
“As reported in the NEJM Perspective, there were 22 cases of T-cell malignancy after treatment with CAR T-cell immunotherapies as of December 31, 2023, but we have received additional reports and, as of February 9, 2024, FDA has now received 28 reports,” he said. “Note that as new cases are being reported, there will be updates to the total number of cases under ongoing review by FDA.”
The initial 22 cases all occurred relatively soon after treatment. Of 14 cases with sufficient data, all developed within 2 years of the CAR-T therapy, ranging from 1 to 19 months, with about half occurring in the first year after administration.
The cases involved five of the six FDA-approved CAR-T products, with the numbers too low to suggest an association with any particular product.
In three of the cases, the lymphoma was found in genetic testing to contain the CAR construction, “indicating that the CAR-T product was most likely involved in the development of the T-cell cancer,” according to the FDA researchers.
With inadequate genetic sampling in most of the remaining 19 cases, the association is less clear, however “the timing of several of the cases makes association a possibility,” Dr. Marks said. In their report, Dr. Marks and colleagues added that “determination of whether the T-cell cancer is associated with the CAR construct ... most likely won’t be possible for every case reported to date.”
Even if all the reported cases are assumed to be related to CAR-T treatment, the numbers still represent a very small proportion of the more than 27,000 doses of the six CAR-T therapies approved in the United States, the authors noted, but they cautioned that the numbers could indeed be higher than reported.
“Relying on postmarketing reporting may lead to underestimates of such cases,” they said.
Life-Long Monitoring Recommended
In response to the reports, the FDA is urging that clinicians’ monitoring of patients treated with CAR-T therapy should be lifelong.
“Patients and clinical trial participants receiving treatment with these products should be monitored lifelong for new malignancies,” Dr. Marks said.
“In the event that a new malignancy occurs following treatment with these products, contact the manufacturer to report the event and obtain instructions on collection of patient samples for testing for the presence of the CAR transgene.”
In addition, cases should be reported to the FDA, either by calling or through the FDA’s medical product safety reporting program.
T-Cell Malignancy Case Report
In describing the case at their medical center in the report in Nature Medicine, Dr. Ruella and colleagues said a T-cell lymphoma occurred in a patient with non-Hodgkin B-cell lymphoma 3 months after an anti-CD19 CAR T-cell treatment.
As a result, the team conducted a subsequent analysis of 449 patients treated with CAR-T therapy at the University of Pennsylvania center, and with a median follow-up of 10.3 months, 16 patients (3.6%) had developed a secondary primary malignancy, with a median onset time of 26.4 months for solid and 9.7 months for hematological malignancies.
The patient who had developed a T-cell lymphoma tested negative for CAR integration upon diagnosis, and regarding the other cancers, Dr. Ruella noted that “we have no indication that the secondary malignancies are directly caused by the CAR-T therapy.
“We have many patients with a very long follow-up beyond 5 and even 10 years,” he said. “In these patients, we don’t see an increased risk of T-cell lymphoma.”
‘Cautious Reassurance’ Urged in Discussion with Patients
With alarming headlines on the findings suggesting that CAR-T therapy may cause cancer, Rahul Banerjee, MD, and colleagues at the University of Washington, Seattle, recommend the use of “cautious reassurance” in discussing the issue with patients. In a paper published in January in Blood Advances, they suggest a three-part response: underscoring that the benefits of CAR T “far outweigh” the risks in relapsed/refractory malignancies, that the ‘one-and-done’ nature of CAR-T infusions provide meaningful improvements in quality of life, and that the active cancer at hand is “a much larger threat than a hypothetical cancer years later.”
In many cases, patients may only have months to live without CAR-T therapy and will have already had multiple prior lines of therapy, therefore the CAR-T treatment itself may provide time for the secondary primary cancers from any of the treatments to emerge, as experts have noted.
“One has to be alive to be diagnosed with a secondary primary malignancy, and it’s thus very possible that CAR-T may be creating a type of ‘immortal time bias’ wherein patients live long enough to experience the unfortunate sequelae of their previous therapies,” Dr. Banerjee explained in an interview.
Nevertheless, the potential for substantial improvements in quality of life with CAR-T therapy compared with traditional treatments addresses a top priority for patients, he added.
“For most patients with [for instance], myeloma, the ability of CAR-T to put them rapidly into a deep remission without the need for maintenance is an unheard-of potential for them,” Dr. Banerjee said.
“In multiple myeloma, no CAR-T therapy has (yet) demonstrated an overall survival benefit — but I think the substantial quality-of-life benefit stands by itself as a big reason why patients continue to prefer CAR-T.”
Keep Patients In Touch with CAR T Centers
In light of the concerns regarding the secondary malignancies, Dr. Banerjee underscored that CAR-T patients should be kept in close touch with centers that have CAR-T treatment expertise.
With most patients followed primarily at community practices where CAR-T therapy is not administered, “I’d strongly encourage my colleagues in community practices to refer all eligible patients to a CAR-T-capable center for evaluation regardless of what their risk of post-CAR-T secondary primary malignancies may be,” Dr. Banerjee urged.
“Based on the evidence we have currently, which includes the FDA’s updated information, there are many more unknowns about this potential secondary primary malignancy risk than knowns,” he said. “This is of course a much more nuanced issue than any one package insert can convey, and CAR-T experts at treating centers can have these conversations at length with eligible patients who are nervous about these recent updates.”
Dr. Ruella disclosed that he holds patents related to CD19 CAR T cells, as well as relationships with NanoString, Bristol Myers Squibb, GlaxoSmithKline, Scailyte, Bayer, AbClon, Oxford NanoImaging, CURIOX, and Beckman Coulter, and he was the scientific founder of viTToria Biotherapeutics. Dr. Banerjee reported ties with BMS, Caribou Biosciences, Genentech, Janssen, Karyopharm, Pfizer, Sanofi, SparkCures, Novartis, and Pack Health.
Importantly, most specialists agree, so far the risk appears no greater than the known risk of secondary primary malignancies that is well established with other cancer therapies.
“The data that we have so far suggest that the risk of secondary T-cell lymphoma in patients treated with CAR T-cells is similar to [that] of patients treated with other cancer therapies, [including] chemotherapy, radiation, transplantation,” Marco Ruella, MD, said in an interview. He reported on a case of a T-cell lymphoma occurring following CAR-T therapy at the University of Pennsylvania.
While his team is still investigating the development of such malignancies, “the FDA notice does not change our clinical practice and patients should be reassured that the benefit of CAR-T therapy significantly outweighs the potential risk of secondary malignancies including T-cell lymphoma,” said Dr. Ruella, scientific director of the Lymphoma Program, Division of Hematology and Oncology and Center for Cellular Immunotherapies, at the University of Pennsylvania, Philadelphia.
FDA: 28 Reports of Malignancies; 3 with Evidence of ‘Likely’ CAR T Involvement
Concerns were raised last November when the FDA announced in a safety communication that it was investigating the “serious risk of T-cell malignancy” following B-cell maturation antigen (BCMA)-directed or CD19-directed CAR T-cell immunotherapies, citing reports from clinical trials and/or postmarketing adverse event data sources. Subsequently, in January, the FDA called for the boxed warning on all approved BCMA- and CD19-targeted genetically modified autologous T-cell immunotherapies, which include: Abecma (idecabtagene vicleucel); Breyanzi (lisocabtagene maraleucel); Carvykti (ciltacabtagene autoleucel); Kymriah (tisagenlecleucel); Tecartus (brexucabtagene autoleucel); and Yescarta (axicabtagene ciloleucel).
“Although the overall benefits of these products continue to outweigh their potential risks for their approved uses, the FDA continues to investigate the identified risk of T-cell malignancy with serious outcomes, including hospitalization and death,” the FDA reported in discussing the safety warnings.
The cases were detailed in a report from FDA researchers published in the New England Journal of Medicine, noting that as of December 31, 2023, the FDA had become aware of 22 cases of T-cell cancers occurring following CAR T-cell treatment, including T-cell lymphoma, T-cell large granular lymphocytosis, peripheral T-cell lymphoma, and cutaneous T-cell lymphoma.
Report coauthor Peter Marks, MD, PhD, of the FDA’s Center for Biologics Evaluation and Research in Silver Spring, Maryland, said in an interview that since the publication of their report, six new cases have emerged.
“As reported in the NEJM Perspective, there were 22 cases of T-cell malignancy after treatment with CAR T-cell immunotherapies as of December 31, 2023, but we have received additional reports and, as of February 9, 2024, FDA has now received 28 reports,” he said. “Note that as new cases are being reported, there will be updates to the total number of cases under ongoing review by FDA.”
The initial 22 cases all occurred relatively soon after treatment. Of 14 cases with sufficient data, all developed within 2 years of the CAR-T therapy, ranging from 1 to 19 months, with about half occurring in the first year after administration.
The cases involved five of the six FDA-approved CAR-T products, with the numbers too low to suggest an association with any particular product.
In three of the cases, the lymphoma was found in genetic testing to contain the CAR construction, “indicating that the CAR-T product was most likely involved in the development of the T-cell cancer,” according to the FDA researchers.
With inadequate genetic sampling in most of the remaining 19 cases, the association is less clear, however “the timing of several of the cases makes association a possibility,” Dr. Marks said. In their report, Dr. Marks and colleagues added that “determination of whether the T-cell cancer is associated with the CAR construct ... most likely won’t be possible for every case reported to date.”
Even if all the reported cases are assumed to be related to CAR-T treatment, the numbers still represent a very small proportion of the more than 27,000 doses of the six CAR-T therapies approved in the United States, the authors noted, but they cautioned that the numbers could indeed be higher than reported.
“Relying on postmarketing reporting may lead to underestimates of such cases,” they said.
Life-Long Monitoring Recommended
In response to the reports, the FDA is urging that clinicians’ monitoring of patients treated with CAR-T therapy should be lifelong.
“Patients and clinical trial participants receiving treatment with these products should be monitored lifelong for new malignancies,” Dr. Marks said.
“In the event that a new malignancy occurs following treatment with these products, contact the manufacturer to report the event and obtain instructions on collection of patient samples for testing for the presence of the CAR transgene.”
In addition, cases should be reported to the FDA, either by calling or through the FDA’s medical product safety reporting program.
T-Cell Malignancy Case Report
In describing the case at their medical center in the report in Nature Medicine, Dr. Ruella and colleagues said a T-cell lymphoma occurred in a patient with non-Hodgkin B-cell lymphoma 3 months after an anti-CD19 CAR T-cell treatment.
As a result, the team conducted a subsequent analysis of 449 patients treated with CAR-T therapy at the University of Pennsylvania center, and with a median follow-up of 10.3 months, 16 patients (3.6%) had developed a secondary primary malignancy, with a median onset time of 26.4 months for solid and 9.7 months for hematological malignancies.
The patient who had developed a T-cell lymphoma tested negative for CAR integration upon diagnosis, and regarding the other cancers, Dr. Ruella noted that “we have no indication that the secondary malignancies are directly caused by the CAR-T therapy.
“We have many patients with a very long follow-up beyond 5 and even 10 years,” he said. “In these patients, we don’t see an increased risk of T-cell lymphoma.”
‘Cautious Reassurance’ Urged in Discussion with Patients
With alarming headlines on the findings suggesting that CAR-T therapy may cause cancer, Rahul Banerjee, MD, and colleagues at the University of Washington, Seattle, recommend the use of “cautious reassurance” in discussing the issue with patients. In a paper published in January in Blood Advances, they suggest a three-part response: underscoring that the benefits of CAR T “far outweigh” the risks in relapsed/refractory malignancies, that the ‘one-and-done’ nature of CAR-T infusions provide meaningful improvements in quality of life, and that the active cancer at hand is “a much larger threat than a hypothetical cancer years later.”
In many cases, patients may only have months to live without CAR-T therapy and will have already had multiple prior lines of therapy, therefore the CAR-T treatment itself may provide time for the secondary primary cancers from any of the treatments to emerge, as experts have noted.
“One has to be alive to be diagnosed with a secondary primary malignancy, and it’s thus very possible that CAR-T may be creating a type of ‘immortal time bias’ wherein patients live long enough to experience the unfortunate sequelae of their previous therapies,” Dr. Banerjee explained in an interview.
Nevertheless, the potential for substantial improvements in quality of life with CAR-T therapy compared with traditional treatments addresses a top priority for patients, he added.
“For most patients with [for instance], myeloma, the ability of CAR-T to put them rapidly into a deep remission without the need for maintenance is an unheard-of potential for them,” Dr. Banerjee said.
“In multiple myeloma, no CAR-T therapy has (yet) demonstrated an overall survival benefit — but I think the substantial quality-of-life benefit stands by itself as a big reason why patients continue to prefer CAR-T.”
Keep Patients In Touch with CAR T Centers
In light of the concerns regarding the secondary malignancies, Dr. Banerjee underscored that CAR-T patients should be kept in close touch with centers that have CAR-T treatment expertise.
With most patients followed primarily at community practices where CAR-T therapy is not administered, “I’d strongly encourage my colleagues in community practices to refer all eligible patients to a CAR-T-capable center for evaluation regardless of what their risk of post-CAR-T secondary primary malignancies may be,” Dr. Banerjee urged.
“Based on the evidence we have currently, which includes the FDA’s updated information, there are many more unknowns about this potential secondary primary malignancy risk than knowns,” he said. “This is of course a much more nuanced issue than any one package insert can convey, and CAR-T experts at treating centers can have these conversations at length with eligible patients who are nervous about these recent updates.”
Dr. Ruella disclosed that he holds patents related to CD19 CAR T cells, as well as relationships with NanoString, Bristol Myers Squibb, GlaxoSmithKline, Scailyte, Bayer, AbClon, Oxford NanoImaging, CURIOX, and Beckman Coulter, and he was the scientific founder of viTToria Biotherapeutics. Dr. Banerjee reported ties with BMS, Caribou Biosciences, Genentech, Janssen, Karyopharm, Pfizer, Sanofi, SparkCures, Novartis, and Pack Health.
Importantly, most specialists agree, so far the risk appears no greater than the known risk of secondary primary malignancies that is well established with other cancer therapies.
“The data that we have so far suggest that the risk of secondary T-cell lymphoma in patients treated with CAR T-cells is similar to [that] of patients treated with other cancer therapies, [including] chemotherapy, radiation, transplantation,” Marco Ruella, MD, said in an interview. He reported on a case of a T-cell lymphoma occurring following CAR-T therapy at the University of Pennsylvania.
While his team is still investigating the development of such malignancies, “the FDA notice does not change our clinical practice and patients should be reassured that the benefit of CAR-T therapy significantly outweighs the potential risk of secondary malignancies including T-cell lymphoma,” said Dr. Ruella, scientific director of the Lymphoma Program, Division of Hematology and Oncology and Center for Cellular Immunotherapies, at the University of Pennsylvania, Philadelphia.
FDA: 28 Reports of Malignancies; 3 with Evidence of ‘Likely’ CAR T Involvement
Concerns were raised last November when the FDA announced in a safety communication that it was investigating the “serious risk of T-cell malignancy” following B-cell maturation antigen (BCMA)-directed or CD19-directed CAR T-cell immunotherapies, citing reports from clinical trials and/or postmarketing adverse event data sources. Subsequently, in January, the FDA called for the boxed warning on all approved BCMA- and CD19-targeted genetically modified autologous T-cell immunotherapies, which include: Abecma (idecabtagene vicleucel); Breyanzi (lisocabtagene maraleucel); Carvykti (ciltacabtagene autoleucel); Kymriah (tisagenlecleucel); Tecartus (brexucabtagene autoleucel); and Yescarta (axicabtagene ciloleucel).
“Although the overall benefits of these products continue to outweigh their potential risks for their approved uses, the FDA continues to investigate the identified risk of T-cell malignancy with serious outcomes, including hospitalization and death,” the FDA reported in discussing the safety warnings.
The cases were detailed in a report from FDA researchers published in the New England Journal of Medicine, noting that as of December 31, 2023, the FDA had become aware of 22 cases of T-cell cancers occurring following CAR T-cell treatment, including T-cell lymphoma, T-cell large granular lymphocytosis, peripheral T-cell lymphoma, and cutaneous T-cell lymphoma.
Report coauthor Peter Marks, MD, PhD, of the FDA’s Center for Biologics Evaluation and Research in Silver Spring, Maryland, said in an interview that since the publication of their report, six new cases have emerged.
“As reported in the NEJM Perspective, there were 22 cases of T-cell malignancy after treatment with CAR T-cell immunotherapies as of December 31, 2023, but we have received additional reports and, as of February 9, 2024, FDA has now received 28 reports,” he said. “Note that as new cases are being reported, there will be updates to the total number of cases under ongoing review by FDA.”
The initial 22 cases all occurred relatively soon after treatment. Of 14 cases with sufficient data, all developed within 2 years of the CAR-T therapy, ranging from 1 to 19 months, with about half occurring in the first year after administration.
The cases involved five of the six FDA-approved CAR-T products, with the numbers too low to suggest an association with any particular product.
In three of the cases, the lymphoma was found in genetic testing to contain the CAR construction, “indicating that the CAR-T product was most likely involved in the development of the T-cell cancer,” according to the FDA researchers.
With inadequate genetic sampling in most of the remaining 19 cases, the association is less clear, however “the timing of several of the cases makes association a possibility,” Dr. Marks said. In their report, Dr. Marks and colleagues added that “determination of whether the T-cell cancer is associated with the CAR construct ... most likely won’t be possible for every case reported to date.”
Even if all the reported cases are assumed to be related to CAR-T treatment, the numbers still represent a very small proportion of the more than 27,000 doses of the six CAR-T therapies approved in the United States, the authors noted, but they cautioned that the numbers could indeed be higher than reported.
“Relying on postmarketing reporting may lead to underestimates of such cases,” they said.
Life-Long Monitoring Recommended
In response to the reports, the FDA is urging that clinicians’ monitoring of patients treated with CAR-T therapy should be lifelong.
“Patients and clinical trial participants receiving treatment with these products should be monitored lifelong for new malignancies,” Dr. Marks said.
“In the event that a new malignancy occurs following treatment with these products, contact the manufacturer to report the event and obtain instructions on collection of patient samples for testing for the presence of the CAR transgene.”
In addition, cases should be reported to the FDA, either by calling or through the FDA’s medical product safety reporting program.
T-Cell Malignancy Case Report
In describing the case at their medical center in the report in Nature Medicine, Dr. Ruella and colleagues said a T-cell lymphoma occurred in a patient with non-Hodgkin B-cell lymphoma 3 months after an anti-CD19 CAR T-cell treatment.
As a result, the team conducted a subsequent analysis of 449 patients treated with CAR-T therapy at the University of Pennsylvania center, and with a median follow-up of 10.3 months, 16 patients (3.6%) had developed a secondary primary malignancy, with a median onset time of 26.4 months for solid and 9.7 months for hematological malignancies.
The patient who had developed a T-cell lymphoma tested negative for CAR integration upon diagnosis, and regarding the other cancers, Dr. Ruella noted that “we have no indication that the secondary malignancies are directly caused by the CAR-T therapy.
“We have many patients with a very long follow-up beyond 5 and even 10 years,” he said. “In these patients, we don’t see an increased risk of T-cell lymphoma.”
‘Cautious Reassurance’ Urged in Discussion with Patients
With alarming headlines on the findings suggesting that CAR-T therapy may cause cancer, Rahul Banerjee, MD, and colleagues at the University of Washington, Seattle, recommend the use of “cautious reassurance” in discussing the issue with patients. In a paper published in January in Blood Advances, they suggest a three-part response: underscoring that the benefits of CAR T “far outweigh” the risks in relapsed/refractory malignancies, that the ‘one-and-done’ nature of CAR-T infusions provide meaningful improvements in quality of life, and that the active cancer at hand is “a much larger threat than a hypothetical cancer years later.”
In many cases, patients may only have months to live without CAR-T therapy and will have already had multiple prior lines of therapy, therefore the CAR-T treatment itself may provide time for the secondary primary cancers from any of the treatments to emerge, as experts have noted.
“One has to be alive to be diagnosed with a secondary primary malignancy, and it’s thus very possible that CAR-T may be creating a type of ‘immortal time bias’ wherein patients live long enough to experience the unfortunate sequelae of their previous therapies,” Dr. Banerjee explained in an interview.
Nevertheless, the potential for substantial improvements in quality of life with CAR-T therapy compared with traditional treatments addresses a top priority for patients, he added.
“For most patients with [for instance], myeloma, the ability of CAR-T to put them rapidly into a deep remission without the need for maintenance is an unheard-of potential for them,” Dr. Banerjee said.
“In multiple myeloma, no CAR-T therapy has (yet) demonstrated an overall survival benefit — but I think the substantial quality-of-life benefit stands by itself as a big reason why patients continue to prefer CAR-T.”
Keep Patients In Touch with CAR T Centers
In light of the concerns regarding the secondary malignancies, Dr. Banerjee underscored that CAR-T patients should be kept in close touch with centers that have CAR-T treatment expertise.
With most patients followed primarily at community practices where CAR-T therapy is not administered, “I’d strongly encourage my colleagues in community practices to refer all eligible patients to a CAR-T-capable center for evaluation regardless of what their risk of post-CAR-T secondary primary malignancies may be,” Dr. Banerjee urged.
“Based on the evidence we have currently, which includes the FDA’s updated information, there are many more unknowns about this potential secondary primary malignancy risk than knowns,” he said. “This is of course a much more nuanced issue than any one package insert can convey, and CAR-T experts at treating centers can have these conversations at length with eligible patients who are nervous about these recent updates.”
Dr. Ruella disclosed that he holds patents related to CD19 CAR T cells, as well as relationships with NanoString, Bristol Myers Squibb, GlaxoSmithKline, Scailyte, Bayer, AbClon, Oxford NanoImaging, CURIOX, and Beckman Coulter, and he was the scientific founder of viTToria Biotherapeutics. Dr. Banerjee reported ties with BMS, Caribou Biosciences, Genentech, Janssen, Karyopharm, Pfizer, Sanofi, SparkCures, Novartis, and Pack Health.