User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Patient Navigators for Serious Illnesses Can Now Bill Under New Medicare Codes
In a move that acknowledges the gauntlet the US health system poses for people facing serious and fatal illnesses, Medicare will pay for a new class of workers to help patients manage treatments for conditions like cancer and heart failure.
The 2024 Medicare physician fee schedule includes new billing codes, including G0023, to pay for 60 minutes a month of care coordination by certified or trained auxiliary personnel working under the direction of a clinician.
A diagnosis of cancer or another serious illness takes a toll beyond the physical effects of the disease. Patients often scramble to make adjustments in family and work schedules to manage treatment, said Samyukta Mullangi, MD, MBA, medical director of oncology at Thyme Care, a Nashville, Tennessee–based firm that provides navigation and coordination services to oncology practices and insurers.

“It just really does create a bit of a pressure cooker for patients,” Dr. Mullangi told this news organization.
Medicare has for many years paid for medical professionals to help patients cope with the complexities of disease, such as chronic care management (CCM) provided by physicians, nurses, and physician assistants.
The new principal illness navigation (PIN) payments are intended to pay for work that to date typically has been done by people without medical degrees, including those involved in peer support networks and community health programs. The US Centers for Medicare and Medicaid Services(CMS) expects these navigators will undergo training and work under the supervision of clinicians.
The new navigators may coordinate care transitions between medical settings, follow up with patients after emergency department (ED) visits, or communicate with skilled nursing facilities regarding the psychosocial needs and functional deficits of a patient, among other functions.
CMS expects the new navigators may:
- Conduct assessments to understand a patient’s life story, strengths, needs, goals, preferences, and desired outcomes, including understanding cultural and linguistic factors.
- Provide support to accomplish the clinician’s treatment plan.
- Coordinate the receipt of needed services from healthcare facilities, home- and community-based service providers, and caregivers.
Peers as Navigators
The new navigators can be former patients who have undergone similar treatments for serious diseases, CMS said. This approach sets the new program apart from other care management services Medicare already covers, program officials wrote in the 2024 physician fee schedule.
“For some conditions, patients are best able to engage with the healthcare system and access care if they have assistance from a single, dedicated individual who has ‘lived experience,’ ” according to the rule.
The agency has taken a broad initial approach in defining what kinds of illnesses a patient may have to qualify for services. Patients must have a serious condition that is expected to last at least 3 months, such as cancer, heart failure, or substance use disorder.
But those without a definitive diagnosis may also qualify to receive navigator services.
In the rule, CMS cited a case in which a CT scan identified a suspicious mass in a patient’s colon. A clinician might decide this person would benefit from navigation services due to the potential risks for an undiagnosed illness.
“Regardless of the definitive diagnosis of the mass, presence of a colonic mass for that patient may be a serious high-risk condition that could, for example, cause obstruction and lead the patient to present to the emergency department, as well as be potentially indicative of an underlying life-threatening illness such as colon cancer,” CMS wrote in the rule.
Navigators often start their work when cancer patients are screened and guide them through initial diagnosis, potential surgery, radiation, or chemotherapy, said Sharon Gentry, MSN, RN, a former nurse navigator who is now the editor in chief of the Journal of the Academy of Oncology Nurse & Patient Navigators.
The navigators are meant to be a trusted and continual presence for patients, who otherwise might be left to start anew in finding help at each phase of care.
The navigators “see the whole picture. They see the whole journey the patient takes, from pre-diagnosis all the way through diagnosis care out through survival,” Ms. Gentry said.

Gaining a special Medicare payment for these kinds of services will elevate this work, she said.
Many newer drugs can target specific mechanisms and proteins of cancer. Often, oncology treatment involves testing to find out if mutations are allowing the cancer cells to evade a patient’s immune system.
Checking these biomarkers takes time, however. Patients sometimes become frustrated because they are anxious to begin treatment. Patients may receive inaccurate information from friends or family who went through treatment previously. Navigators can provide knowledge on the current state of care for a patient’s disease, helping them better manage anxieties.
“You have to explain to them that things have changed since the guy you drink coffee with was diagnosed with cancer, and there may be a drug that could target that,” Ms. Gentry said.
Potential Challenges
Initial uptake of the new PIN codes may be slow going, however, as clinicians and health systems may already use well-established codes. These include CCM and principal care management services, which may pay higher rates, Mullangi said.
“There might be sensitivity around not wanting to cannibalize existing programs with a new program,” Dr. Mullangi said.
In addition, many patients will have a copay for the services of principal illness navigators, Dr. Mullangi said.
While many patients have additional insurance that would cover the service, not all do. People with traditional Medicare coverage can sometimes pay 20% of the cost of some medical services.
“I think that may give patients pause, particularly if they’re already feeling the financial burden of a cancer treatment journey,” Dr. Mullangi said.
Pay rates for PIN services involve calculations of regional price differences, which are posted publicly by CMS, and potential added fees for services provided by hospital-affiliated organizations.
Consider payments for code G0023, covering 60 minutes of principal navigation services provided in a single month.
A set reimbursement for patients cared for in independent medical practices exists, with variation for local costs. Medicare’s non-facility price for G0023 would be $102.41 in some parts of Silicon Valley in California, including San Jose. In Arkansas, where costs are lower, reimbursement would be $73.14 for this same service.
Patients who get services covered by code G0023 in independent medical practices would have monthly copays of about $15-$20, depending on where they live.
The tab for patients tends to be higher for these same services if delivered through a medical practice owned by a hospital, as this would trigger the addition of facility fees to the payments made to cover the services. Facility fees are difficult for the public to ascertain before getting a treatment or service.
Dr. Mullangi and Ms. Gentry reported no relevant financial disclosures outside of their employers.
A version of this article first appeared on Medscape.com.
In a move that acknowledges the gauntlet the US health system poses for people facing serious and fatal illnesses, Medicare will pay for a new class of workers to help patients manage treatments for conditions like cancer and heart failure.
The 2024 Medicare physician fee schedule includes new billing codes, including G0023, to pay for 60 minutes a month of care coordination by certified or trained auxiliary personnel working under the direction of a clinician.
A diagnosis of cancer or another serious illness takes a toll beyond the physical effects of the disease. Patients often scramble to make adjustments in family and work schedules to manage treatment, said Samyukta Mullangi, MD, MBA, medical director of oncology at Thyme Care, a Nashville, Tennessee–based firm that provides navigation and coordination services to oncology practices and insurers.
“It just really does create a bit of a pressure cooker for patients,” Dr. Mullangi told this news organization.
Medicare has for many years paid for medical professionals to help patients cope with the complexities of disease, such as chronic care management (CCM) provided by physicians, nurses, and physician assistants.
The new principal illness navigation (PIN) payments are intended to pay for work that to date typically has been done by people without medical degrees, including those involved in peer support networks and community health programs. The US Centers for Medicare and Medicaid Services(CMS) expects these navigators will undergo training and work under the supervision of clinicians.
The new navigators may coordinate care transitions between medical settings, follow up with patients after emergency department (ED) visits, or communicate with skilled nursing facilities regarding the psychosocial needs and functional deficits of a patient, among other functions.
CMS expects the new navigators may:
- Conduct assessments to understand a patient’s life story, strengths, needs, goals, preferences, and desired outcomes, including understanding cultural and linguistic factors.
- Provide support to accomplish the clinician’s treatment plan.
- Coordinate the receipt of needed services from healthcare facilities, home- and community-based service providers, and caregivers.
Peers as Navigators
The new navigators can be former patients who have undergone similar treatments for serious diseases, CMS said. This approach sets the new program apart from other care management services Medicare already covers, program officials wrote in the 2024 physician fee schedule.
“For some conditions, patients are best able to engage with the healthcare system and access care if they have assistance from a single, dedicated individual who has ‘lived experience,’ ” according to the rule.
The agency has taken a broad initial approach in defining what kinds of illnesses a patient may have to qualify for services. Patients must have a serious condition that is expected to last at least 3 months, such as cancer, heart failure, or substance use disorder.
But those without a definitive diagnosis may also qualify to receive navigator services.
In the rule, CMS cited a case in which a CT scan identified a suspicious mass in a patient’s colon. A clinician might decide this person would benefit from navigation services due to the potential risks for an undiagnosed illness.
“Regardless of the definitive diagnosis of the mass, presence of a colonic mass for that patient may be a serious high-risk condition that could, for example, cause obstruction and lead the patient to present to the emergency department, as well as be potentially indicative of an underlying life-threatening illness such as colon cancer,” CMS wrote in the rule.
Navigators often start their work when cancer patients are screened and guide them through initial diagnosis, potential surgery, radiation, or chemotherapy, said Sharon Gentry, MSN, RN, a former nurse navigator who is now the editor in chief of the Journal of the Academy of Oncology Nurse & Patient Navigators.
The navigators are meant to be a trusted and continual presence for patients, who otherwise might be left to start anew in finding help at each phase of care.
The navigators “see the whole picture. They see the whole journey the patient takes, from pre-diagnosis all the way through diagnosis care out through survival,” Ms. Gentry said.
Gaining a special Medicare payment for these kinds of services will elevate this work, she said.
Many newer drugs can target specific mechanisms and proteins of cancer. Often, oncology treatment involves testing to find out if mutations are allowing the cancer cells to evade a patient’s immune system.
Checking these biomarkers takes time, however. Patients sometimes become frustrated because they are anxious to begin treatment. Patients may receive inaccurate information from friends or family who went through treatment previously. Navigators can provide knowledge on the current state of care for a patient’s disease, helping them better manage anxieties.
“You have to explain to them that things have changed since the guy you drink coffee with was diagnosed with cancer, and there may be a drug that could target that,” Ms. Gentry said.
Potential Challenges
Initial uptake of the new PIN codes may be slow going, however, as clinicians and health systems may already use well-established codes. These include CCM and principal care management services, which may pay higher rates, Mullangi said.
“There might be sensitivity around not wanting to cannibalize existing programs with a new program,” Dr. Mullangi said.
In addition, many patients will have a copay for the services of principal illness navigators, Dr. Mullangi said.
While many patients have additional insurance that would cover the service, not all do. People with traditional Medicare coverage can sometimes pay 20% of the cost of some medical services.
“I think that may give patients pause, particularly if they’re already feeling the financial burden of a cancer treatment journey,” Dr. Mullangi said.
Pay rates for PIN services involve calculations of regional price differences, which are posted publicly by CMS, and potential added fees for services provided by hospital-affiliated organizations.
Consider payments for code G0023, covering 60 minutes of principal navigation services provided in a single month.
A set reimbursement for patients cared for in independent medical practices exists, with variation for local costs. Medicare’s non-facility price for G0023 would be $102.41 in some parts of Silicon Valley in California, including San Jose. In Arkansas, where costs are lower, reimbursement would be $73.14 for this same service.
Patients who get services covered by code G0023 in independent medical practices would have monthly copays of about $15-$20, depending on where they live.
The tab for patients tends to be higher for these same services if delivered through a medical practice owned by a hospital, as this would trigger the addition of facility fees to the payments made to cover the services. Facility fees are difficult for the public to ascertain before getting a treatment or service.
Dr. Mullangi and Ms. Gentry reported no relevant financial disclosures outside of their employers.
A version of this article first appeared on Medscape.com.
In a move that acknowledges the gauntlet the US health system poses for people facing serious and fatal illnesses, Medicare will pay for a new class of workers to help patients manage treatments for conditions like cancer and heart failure.
The 2024 Medicare physician fee schedule includes new billing codes, including G0023, to pay for 60 minutes a month of care coordination by certified or trained auxiliary personnel working under the direction of a clinician.
A diagnosis of cancer or another serious illness takes a toll beyond the physical effects of the disease. Patients often scramble to make adjustments in family and work schedules to manage treatment, said Samyukta Mullangi, MD, MBA, medical director of oncology at Thyme Care, a Nashville, Tennessee–based firm that provides navigation and coordination services to oncology practices and insurers.
“It just really does create a bit of a pressure cooker for patients,” Dr. Mullangi told this news organization.
Medicare has for many years paid for medical professionals to help patients cope with the complexities of disease, such as chronic care management (CCM) provided by physicians, nurses, and physician assistants.
The new principal illness navigation (PIN) payments are intended to pay for work that to date typically has been done by people without medical degrees, including those involved in peer support networks and community health programs. The US Centers for Medicare and Medicaid Services(CMS) expects these navigators will undergo training and work under the supervision of clinicians.
The new navigators may coordinate care transitions between medical settings, follow up with patients after emergency department (ED) visits, or communicate with skilled nursing facilities regarding the psychosocial needs and functional deficits of a patient, among other functions.
CMS expects the new navigators may:
- Conduct assessments to understand a patient’s life story, strengths, needs, goals, preferences, and desired outcomes, including understanding cultural and linguistic factors.
- Provide support to accomplish the clinician’s treatment plan.
- Coordinate the receipt of needed services from healthcare facilities, home- and community-based service providers, and caregivers.
Peers as Navigators
The new navigators can be former patients who have undergone similar treatments for serious diseases, CMS said. This approach sets the new program apart from other care management services Medicare already covers, program officials wrote in the 2024 physician fee schedule.
“For some conditions, patients are best able to engage with the healthcare system and access care if they have assistance from a single, dedicated individual who has ‘lived experience,’ ” according to the rule.
The agency has taken a broad initial approach in defining what kinds of illnesses a patient may have to qualify for services. Patients must have a serious condition that is expected to last at least 3 months, such as cancer, heart failure, or substance use disorder.
But those without a definitive diagnosis may also qualify to receive navigator services.
In the rule, CMS cited a case in which a CT scan identified a suspicious mass in a patient’s colon. A clinician might decide this person would benefit from navigation services due to the potential risks for an undiagnosed illness.
“Regardless of the definitive diagnosis of the mass, presence of a colonic mass for that patient may be a serious high-risk condition that could, for example, cause obstruction and lead the patient to present to the emergency department, as well as be potentially indicative of an underlying life-threatening illness such as colon cancer,” CMS wrote in the rule.
Navigators often start their work when cancer patients are screened and guide them through initial diagnosis, potential surgery, radiation, or chemotherapy, said Sharon Gentry, MSN, RN, a former nurse navigator who is now the editor in chief of the Journal of the Academy of Oncology Nurse & Patient Navigators.
The navigators are meant to be a trusted and continual presence for patients, who otherwise might be left to start anew in finding help at each phase of care.
The navigators “see the whole picture. They see the whole journey the patient takes, from pre-diagnosis all the way through diagnosis care out through survival,” Ms. Gentry said.
Gaining a special Medicare payment for these kinds of services will elevate this work, she said.
Many newer drugs can target specific mechanisms and proteins of cancer. Often, oncology treatment involves testing to find out if mutations are allowing the cancer cells to evade a patient’s immune system.
Checking these biomarkers takes time, however. Patients sometimes become frustrated because they are anxious to begin treatment. Patients may receive inaccurate information from friends or family who went through treatment previously. Navigators can provide knowledge on the current state of care for a patient’s disease, helping them better manage anxieties.
“You have to explain to them that things have changed since the guy you drink coffee with was diagnosed with cancer, and there may be a drug that could target that,” Ms. Gentry said.
Potential Challenges
Initial uptake of the new PIN codes may be slow going, however, as clinicians and health systems may already use well-established codes. These include CCM and principal care management services, which may pay higher rates, Mullangi said.
“There might be sensitivity around not wanting to cannibalize existing programs with a new program,” Dr. Mullangi said.
In addition, many patients will have a copay for the services of principal illness navigators, Dr. Mullangi said.
While many patients have additional insurance that would cover the service, not all do. People with traditional Medicare coverage can sometimes pay 20% of the cost of some medical services.
“I think that may give patients pause, particularly if they’re already feeling the financial burden of a cancer treatment journey,” Dr. Mullangi said.
Pay rates for PIN services involve calculations of regional price differences, which are posted publicly by CMS, and potential added fees for services provided by hospital-affiliated organizations.
Consider payments for code G0023, covering 60 minutes of principal navigation services provided in a single month.
A set reimbursement for patients cared for in independent medical practices exists, with variation for local costs. Medicare’s non-facility price for G0023 would be $102.41 in some parts of Silicon Valley in California, including San Jose. In Arkansas, where costs are lower, reimbursement would be $73.14 for this same service.
Patients who get services covered by code G0023 in independent medical practices would have monthly copays of about $15-$20, depending on where they live.
The tab for patients tends to be higher for these same services if delivered through a medical practice owned by a hospital, as this would trigger the addition of facility fees to the payments made to cover the services. Facility fees are difficult for the public to ascertain before getting a treatment or service.
Dr. Mullangi and Ms. Gentry reported no relevant financial disclosures outside of their employers.
A version of this article first appeared on Medscape.com.

How to explain physician compounding to legislators
In Ohio, new limits on drug compounding in physicians’ offices went into effect in April and have become a real hindrance to care for dermatology patients. The State of Ohio Board of Pharmacy has defined compounding as combining two or more prescription drugs and has required that physicians who perform this “compounding” must obtain a “Terminal Distributor of Dangerous Drugs” license. Ohio is the “test state,” and these rules, unless vigorously opposed, will be coming to your state.
[polldaddy:9779752]
The rules state that “compounded” drugs used within 6 hours of preparation must be prepared in a designated clean medication area with proper hand hygiene and the use of powder-free gloves. “Compounded” drugs that are used more than 6 hours after preparation, require a designated clean room with access limited to authorized personnel, environmental control devices such as a laminar flow hood, and additional equipment and training of personnel to maintain an aseptic environment. A separate license is required for each office location.
The state pharmacy boards are eager to restrict physicians – as well as dentists and veterinarians – and to collect annual licensing fees. Additionally, according to an article from the Ohio State Medical Association, noncompliant physicians can be fined by the pharmacy board.
We are talking big money, power, and dreams of clinical relevancy (and billable activities) here.
What can dermatologists do to prevent this regulatory overreach? I encourage you to plan a visit to your state representative, where you can demonstrate how these restrictions affect you and your patients – an exercise that should be both fun and compelling. All you need to illustrate your case is a simple kit that includes a syringe (but no needles in the statehouse!), a bottle of lidocaine with epinephrine, a bottle of 8.4% bicarbonate, alcohol pads, and gloves.
First, explain to your audience that there is a skin cancer epidemic with more than 5.4 million new cases a year and that, over the past 20 years, the incidence of skin cancer has doubled and is projected to double again over the next 20 years. Further, explain that dermatologists treat more than 70% of these cases in the office setting, under local anesthesia, at a huge cost savings to the public and government (it costs an average of 12 times as much to remove these cancers in the outpatient department at the hospital). Remember, states foot most of the bill for Medicaid and Medicare gap indigent coverage.
Take the bottle of lidocaine with epinephrine and open the syringe pack (Staffers love this demonstration; everyone is fascinated with shots.). Put on your gloves, wipe the top of the lidocaine bottle with an alcohol swab, and explain that this medicine is the anesthetic preferred for skin cancer surgery. Explain how it not only numbs the skin, but also causes vasoconstriction, so that the cancer can be easily and safely removed in the office.
Then explain that, in order for the epinephrine to be stable, the solution has to be very acidic (a pH of 4.2, in fact). Explain that this makes it burn like hell unless you add 0.1 cc per cc of 8.4% bicarbonate, in which case the perceived pain on a 10-point scale will drop from 8 to 2. Then pick up the bottle of bicarbonate and explain that you will no longer be able to mix these two components anymore without a “Terminal Distributor of Dangerous Drugs” license because your state pharmacy board considers this compounding. Your representative is likely to give you looks of astonishment, disbelief, and then a dawning realization of the absurdity of the situation.
Follow-up questions may include “Why can’t you buy buffered lidocaine with epinephrine from the compounding pharmacy?” Easy answer: because each patient needs an individual prescription, and you may not know in advance which patient will need it, and how much the patient will need, and it becomes unstable once it has been buffered. It also will cost the patient $45 per 5-cc syringe, and it will be degraded by the time the patient returns from the compounding pharmacy. Explain further that it costs you only 84 cents to make a 5-cc syringe of buffered lidocaine; that some patients may need as many as 10 syringes; and that these costs are all included in the surgery (free!) if the physician draws it up in the office.
A simple summary is – less pain, less cost – and no history of infections or complications.
It is an eye-opener when you demonstrate how ridiculous the compounding rules being imposed are for physicians and patients. I’ve used this demonstration at the state and federal legislative level, and more recently, at the Food and Drug Administration.
If you get the chance, when a state legislator is in your office, become an advocate for your patients and fellow physicians. Make sure physician offices are excluded from these definitions of com

This column was updated June 22, 2017.
Dr. Coldiron is in private practice but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of more than 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics. He is a past president of the American Academy of Dermatology. Write to him at dermnews@frontlinemedcom.com.
In Ohio, new limits on drug compounding in physicians’ offices went into effect in April and have become a real hindrance to care for dermatology patients. The State of Ohio Board of Pharmacy has defined compounding as combining two or more prescription drugs and has required that physicians who perform this “compounding” must obtain a “Terminal Distributor of Dangerous Drugs” license. Ohio is the “test state,” and these rules, unless vigorously opposed, will be coming to your state.
[polldaddy:9779752]
The rules state that “compounded” drugs used within 6 hours of preparation must be prepared in a designated clean medication area with proper hand hygiene and the use of powder-free gloves. “Compounded” drugs that are used more than 6 hours after preparation, require a designated clean room with access limited to authorized personnel, environmental control devices such as a laminar flow hood, and additional equipment and training of personnel to maintain an aseptic environment. A separate license is required for each office location.
The state pharmacy boards are eager to restrict physicians – as well as dentists and veterinarians – and to collect annual licensing fees. Additionally, according to an article from the Ohio State Medical Association, noncompliant physicians can be fined by the pharmacy board.
We are talking big money, power, and dreams of clinical relevancy (and billable activities) here.
What can dermatologists do to prevent this regulatory overreach? I encourage you to plan a visit to your state representative, where you can demonstrate how these restrictions affect you and your patients – an exercise that should be both fun and compelling. All you need to illustrate your case is a simple kit that includes a syringe (but no needles in the statehouse!), a bottle of lidocaine with epinephrine, a bottle of 8.4% bicarbonate, alcohol pads, and gloves.
First, explain to your audience that there is a skin cancer epidemic with more than 5.4 million new cases a year and that, over the past 20 years, the incidence of skin cancer has doubled and is projected to double again over the next 20 years. Further, explain that dermatologists treat more than 70% of these cases in the office setting, under local anesthesia, at a huge cost savings to the public and government (it costs an average of 12 times as much to remove these cancers in the outpatient department at the hospital). Remember, states foot most of the bill for Medicaid and Medicare gap indigent coverage.
Take the bottle of lidocaine with epinephrine and open the syringe pack (Staffers love this demonstration; everyone is fascinated with shots.). Put on your gloves, wipe the top of the lidocaine bottle with an alcohol swab, and explain that this medicine is the anesthetic preferred for skin cancer surgery. Explain how it not only numbs the skin, but also causes vasoconstriction, so that the cancer can be easily and safely removed in the office.
Then explain that, in order for the epinephrine to be stable, the solution has to be very acidic (a pH of 4.2, in fact). Explain that this makes it burn like hell unless you add 0.1 cc per cc of 8.4% bicarbonate, in which case the perceived pain on a 10-point scale will drop from 8 to 2. Then pick up the bottle of bicarbonate and explain that you will no longer be able to mix these two components anymore without a “Terminal Distributor of Dangerous Drugs” license because your state pharmacy board considers this compounding. Your representative is likely to give you looks of astonishment, disbelief, and then a dawning realization of the absurdity of the situation.
Follow-up questions may include “Why can’t you buy buffered lidocaine with epinephrine from the compounding pharmacy?” Easy answer: because each patient needs an individual prescription, and you may not know in advance which patient will need it, and how much the patient will need, and it becomes unstable once it has been buffered. It also will cost the patient $45 per 5-cc syringe, and it will be degraded by the time the patient returns from the compounding pharmacy. Explain further that it costs you only 84 cents to make a 5-cc syringe of buffered lidocaine; that some patients may need as many as 10 syringes; and that these costs are all included in the surgery (free!) if the physician draws it up in the office.
A simple summary is – less pain, less cost – and no history of infections or complications.
It is an eye-opener when you demonstrate how ridiculous the compounding rules being imposed are for physicians and patients. I’ve used this demonstration at the state and federal legislative level, and more recently, at the Food and Drug Administration.
If you get the chance, when a state legislator is in your office, become an advocate for your patients and fellow physicians. Make sure physician offices are excluded from these definitions of com
This column was updated June 22, 2017.
Dr. Coldiron is in private practice but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of more than 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics. He is a past president of the American Academy of Dermatology. Write to him at dermnews@frontlinemedcom.com.
In Ohio, new limits on drug compounding in physicians’ offices went into effect in April and have become a real hindrance to care for dermatology patients. The State of Ohio Board of Pharmacy has defined compounding as combining two or more prescription drugs and has required that physicians who perform this “compounding” must obtain a “Terminal Distributor of Dangerous Drugs” license. Ohio is the “test state,” and these rules, unless vigorously opposed, will be coming to your state.
[polldaddy:9779752]
The rules state that “compounded” drugs used within 6 hours of preparation must be prepared in a designated clean medication area with proper hand hygiene and the use of powder-free gloves. “Compounded” drugs that are used more than 6 hours after preparation, require a designated clean room with access limited to authorized personnel, environmental control devices such as a laminar flow hood, and additional equipment and training of personnel to maintain an aseptic environment. A separate license is required for each office location.
The state pharmacy boards are eager to restrict physicians – as well as dentists and veterinarians – and to collect annual licensing fees. Additionally, according to an article from the Ohio State Medical Association, noncompliant physicians can be fined by the pharmacy board.
We are talking big money, power, and dreams of clinical relevancy (and billable activities) here.
What can dermatologists do to prevent this regulatory overreach? I encourage you to plan a visit to your state representative, where you can demonstrate how these restrictions affect you and your patients – an exercise that should be both fun and compelling. All you need to illustrate your case is a simple kit that includes a syringe (but no needles in the statehouse!), a bottle of lidocaine with epinephrine, a bottle of 8.4% bicarbonate, alcohol pads, and gloves.
First, explain to your audience that there is a skin cancer epidemic with more than 5.4 million new cases a year and that, over the past 20 years, the incidence of skin cancer has doubled and is projected to double again over the next 20 years. Further, explain that dermatologists treat more than 70% of these cases in the office setting, under local anesthesia, at a huge cost savings to the public and government (it costs an average of 12 times as much to remove these cancers in the outpatient department at the hospital). Remember, states foot most of the bill for Medicaid and Medicare gap indigent coverage.
Take the bottle of lidocaine with epinephrine and open the syringe pack (Staffers love this demonstration; everyone is fascinated with shots.). Put on your gloves, wipe the top of the lidocaine bottle with an alcohol swab, and explain that this medicine is the anesthetic preferred for skin cancer surgery. Explain how it not only numbs the skin, but also causes vasoconstriction, so that the cancer can be easily and safely removed in the office.
Then explain that, in order for the epinephrine to be stable, the solution has to be very acidic (a pH of 4.2, in fact). Explain that this makes it burn like hell unless you add 0.1 cc per cc of 8.4% bicarbonate, in which case the perceived pain on a 10-point scale will drop from 8 to 2. Then pick up the bottle of bicarbonate and explain that you will no longer be able to mix these two components anymore without a “Terminal Distributor of Dangerous Drugs” license because your state pharmacy board considers this compounding. Your representative is likely to give you looks of astonishment, disbelief, and then a dawning realization of the absurdity of the situation.
Follow-up questions may include “Why can’t you buy buffered lidocaine with epinephrine from the compounding pharmacy?” Easy answer: because each patient needs an individual prescription, and you may not know in advance which patient will need it, and how much the patient will need, and it becomes unstable once it has been buffered. It also will cost the patient $45 per 5-cc syringe, and it will be degraded by the time the patient returns from the compounding pharmacy. Explain further that it costs you only 84 cents to make a 5-cc syringe of buffered lidocaine; that some patients may need as many as 10 syringes; and that these costs are all included in the surgery (free!) if the physician draws it up in the office.
A simple summary is – less pain, less cost – and no history of infections or complications.
It is an eye-opener when you demonstrate how ridiculous the compounding rules being imposed are for physicians and patients. I’ve used this demonstration at the state and federal legislative level, and more recently, at the Food and Drug Administration.
If you get the chance, when a state legislator is in your office, become an advocate for your patients and fellow physicians. Make sure physician offices are excluded from these definitions of com
This column was updated June 22, 2017.
Dr. Coldiron is in private practice but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of more than 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics. He is a past president of the American Academy of Dermatology. Write to him at dermnews@frontlinemedcom.com.
Best Practices: Protecting Dry Vulnerable Skin with CeraVe® Healing Ointment
A supplement to Dermatology News. This advertising supplement is sponsored by Valeant Pharmaceuticals.

- Reinforcing the Skin Barrier
- NEA Seal of Acceptance
- A Preventative Approach to Dry, Cracked Skin
- CeraVe Ointment in the Clinical Setting
Faculty/Faculty Disclosure
Sheila Fallon Friedlander, MD
Professor of Clinical Dermatology & Pediatrics
Director, Pediatric Dermatology Fellowship Training Program
University of California at San Diego School of Medicine
Rady Children’s Hospital,
San Diego, California
Dr. Friedlander was compensated for her participation in the development of this article.
CeraVe is a registered trademark of Valeant Pharmaceuticals International, Inc. or its affiliates.
A supplement to Dermatology News. This advertising supplement is sponsored by Valeant Pharmaceuticals.
- Reinforcing the Skin Barrier
- NEA Seal of Acceptance
- A Preventative Approach to Dry, Cracked Skin
- CeraVe Ointment in the Clinical Setting
Faculty/Faculty Disclosure
Sheila Fallon Friedlander, MD
Professor of Clinical Dermatology & Pediatrics
Director, Pediatric Dermatology Fellowship Training Program
University of California at San Diego School of Medicine
Rady Children’s Hospital,
San Diego, California
Dr. Friedlander was compensated for her participation in the development of this article.
CeraVe is a registered trademark of Valeant Pharmaceuticals International, Inc. or its affiliates.
A supplement to Dermatology News. This advertising supplement is sponsored by Valeant Pharmaceuticals.
- Reinforcing the Skin Barrier
- NEA Seal of Acceptance
- A Preventative Approach to Dry, Cracked Skin
- CeraVe Ointment in the Clinical Setting
Faculty/Faculty Disclosure
Sheila Fallon Friedlander, MD
Professor of Clinical Dermatology & Pediatrics
Director, Pediatric Dermatology Fellowship Training Program
University of California at San Diego School of Medicine
Rady Children’s Hospital,
San Diego, California
Dr. Friedlander was compensated for her participation in the development of this article.
CeraVe is a registered trademark of Valeant Pharmaceuticals International, Inc. or its affiliates.

Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
A 72-year-old man with a history of multiple cancers, including melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC), presented to the dermatology clinic for a regularly scheduled full-body skin examination. His family history was negative for malignancy, but due to his personal history of both primary internal cancers and skin cancers, the patient previously had been referred by dermatology to a medical geneticist for evaluation. He tested positive for a pathogenic POT1 (protection of telomeres 1) variant associated with tumor predisposition, which most often is associated with cutaneous melanoma, chronic lymphocytic leukemia (CLL), angiosarcoma, and gliomas.1
At the current presentation, physical examination revealed a small, asymmetric, pink papule on the superior thoracic spine. A biopsy of the lesion was performed (Figure 1). Pathology demonstrated cornifying cystic structures with a granulomatous response at the surface of the tumor, ductal differentiation with depth, and infiltrative strands and cords of hyperchromatic cells within a collagenous stroma at the base of the specimen (Figures 2A and 2B). One unusual finding was the presence of prominent clear-cell change within the superficial portion of the neoplasm (Figure 2C). Immunohistochemical stains revealed strong p63 and p40 positivity. Epithelial membrane antigen staining was positive in the hyperchromatic strands and cords with depth but not in the clear-cell superficial portion. Similarly, periodic acid–Schiff–positive material increased within tumor cells in proportion to depth of infiltration. Additional immunohistochemical staining showed carcinoembryonic antigen was largely negative (with rare positivity in a few ductal lumina), with negative results for S100, SOX10, CD117, BerEP4, factor XIIIa, CD34, and cytokeratin 7 (Figures 2D and 2E).
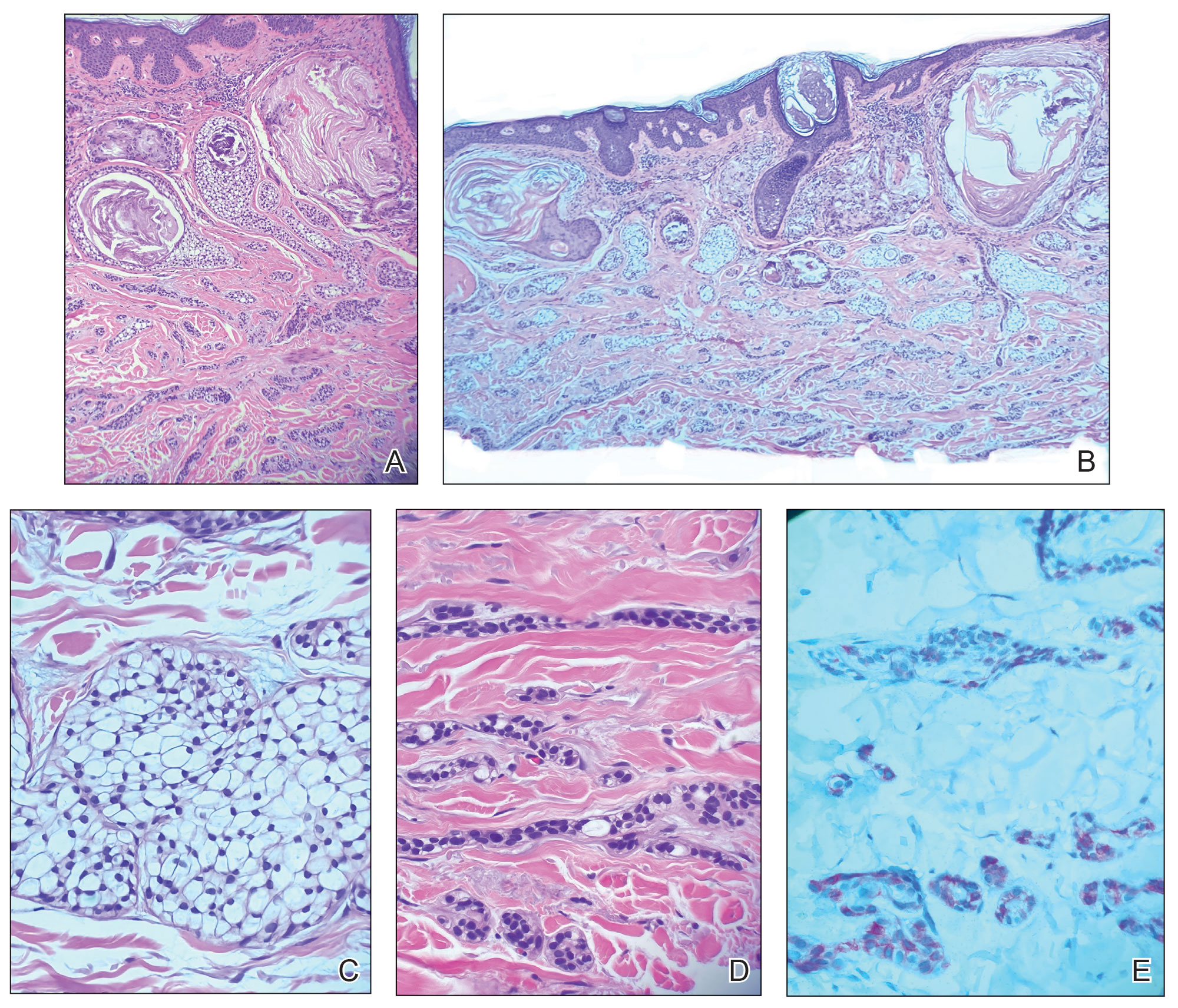
The differential diagnoses included trichilemmal carcinoma (which may manifest with CD34 expression),2 clear cell BCC, adenoid cystic carcinoma (tubular variant), sebaceous carcinoma, and eccrine carcinoma. Importantly, the patient was under continuous oncologic surveillance, with no evidence of a primary internal tumor to suggest metastasis. Despite negative carcinoembryonic antigen staining, the immunohistochemical and histopathologic findings fit best with a primary cutaneous malignant eccrine tumor, specifically microcystic adnexal carcinoma (MAC), in which p63 typically stains peripheral cells but solid variants have been described.3
Eccrine carcinoma is exceedingly rare, reported in 0.01% of diagnosed cutaneous malignancies, and demonstrates overlapping features to other malignant eccrine tumors. It possesses an inconsistent immunohistochemical staining profile, making the distinction from other malignant sweat gland tumors challenging.4 Given that the morphologic features were otherwise classic for MAC in our patient, we favored a clear-cell variant.
Sixteen years prior to the current presentation, our patient presented to urology with a history of prostatitis and increasing prostate-specific antigen levels. Biopsies were negative until prostate-specific antigen reached 13 ng/mL, confirming stage 1A prostate cancer. The patient subsequently underwent a robot-assisted radical prostatectomy. At age 63 years, dysphagia that was unresponsive to antibiotics led to a tonsillar biopsy revealing T2N2bM0 stage IVA SCC of the right tonsil with confirmed HPV type 16 with extracapsular extension. The patient underwent transoral robotic radical tonsillectomy and right neck dissection, followed by adjuvant chemoradiation consisting of intensity-modulated radiation therapy (IMRT) to a total dose of 63 Gy in 33 fractions, with concurrent weekly cisplatin. At age 67 years, dyspepsia, dysphagia, pyrosis, and gastroesophageal reflux prompted endoscopy, revealing T1aNxMx esophageal adenocarcinoma. Three months later, the patient underwent laparoscopic-assisted esophagectomy, with no recurrence. At age 68 years, an atypical intramelanocytic proliferation was found on the left cheek and was treated with Mohs micrographic surgery.
At age 71 years, acral lentiginous malignant melanoma (Breslow thickness 0.8 mm; Clark level IV; American Joint Committee on Cancer T1b) was diagnosed on the left plantar foot and treated with Mohs micrographic surgery. Sentinel lymph node biopsy was negative. Squamous cell carcinoma in situ on the frontal scalp and nodular BCC on the right upper back also were diagnosed.
While there are no guidelines for surveillance of individuals with POT1, recommendations were given in consensus from a medical genetics team,1 including comprehensive monitoring—specifically baseline imaging utilizing brain and full-body magnetic resonance imaging. Furthermore, considering the crucial role of POT1 in maintaining telomeres, it was advised to measure telomere length as part of the surveillance process. Given the patient’s susceptibility to CLL, routine complete blood count assessments were recommended. Additionally, we advised close monitoring for seizures and consideration of genetic testing in first-degree relatives.
Literature Review
Given our patient’s history of multiple skin cancers, including the most recent MAC, we sought to conduct a review of the literature to evaluate existing skin cancer associations and reports for patients with known POT1 mutations to guide recommendations for dermatologic surveillance (Table). A search of PubMed articles indexed for MEDLINE through April 2023 using the terms microcystic adnexal carcinoma, POT1, melanoma, basal cell carcinoma, squamous cell carcinoma, and skin cancer yielded no reported cases of MAC associated with POT1 mutations. POT1 is one of 6 proteins (TERF1, TERF2, RAP1, TIN2, TPP1, and POT1) belonging to the shelterin complex, which plays a crucial role in telomeric DNA remodeling and regulation of telomere length.5 Mutation in the POT1 gene disrupts the shelterin complex, causing telomeres to become elongated and unstable, resulting in chromosomal abnormalities and promoting cancer development.5
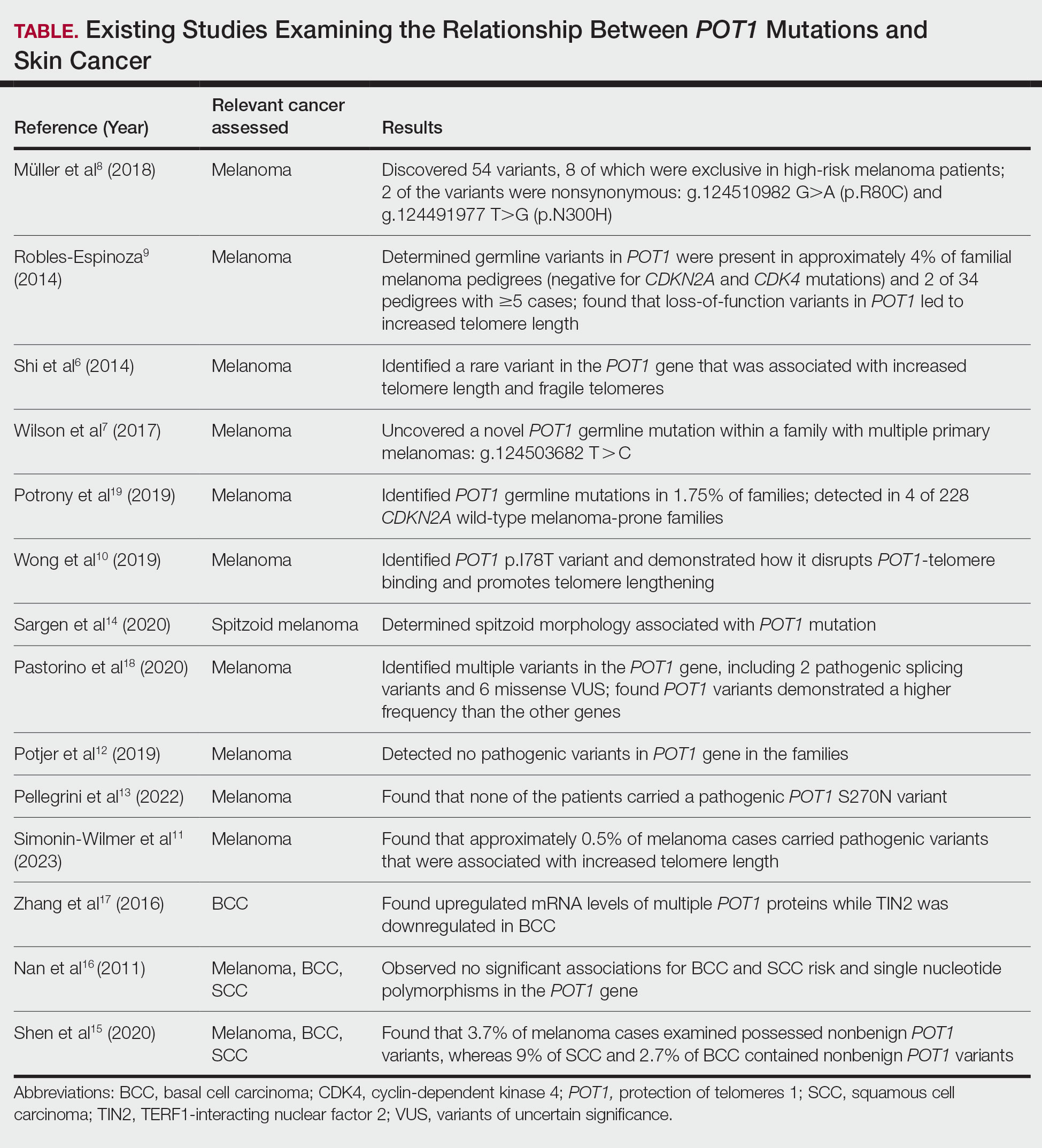
While our literature review did not reveal any associations between the shelterin complex genes and MAC, mutations in the POT1 gene have been studied in other types of skin cancer, particularly melanoma.1 One of the earliest studies was conducted in 2014 by Shi et al,6 in which whole-exome sequencing was performed on families with a history of melanoma. Multiple POT1 gene pathogenic variants associated with increased telomere length and fragility were identified in unrelated families. Subsequent studies have confirmed POT1 variants in melanoma-prone families,7 supporting an association between increased telomere length and melanoma risk8-11; however, other studies have yielded nonsignificant findings.12,13 Further investigation also has identified morphologic characteristics consistent with POT1 mutation, including spitzoid morphology.14
The association between POT1 mutations and nonmelanoma skin cancers has been relatively understudied. While a few studies have explored this link, results have shown mixed findings. Some studies have suggested a potential role for POT1 mutations in cutaneous SCC risk,15 while other studies have shown no significant associations for both BCC and SCC risk and telomere gene mutations.16 Additionally, mRNA levels of POT1 were upregulated in BCC cases compared to normal tissue in a gene expression.17
Comment
In the literature, POT1 mutations are well established as high-penetrance alterations associated with melanoma.9,18,19 However, the correlation between POT1 and other forms of skin cancer is not yet delineated. Recent insights suggest that POT1 mutations play a major role in promoting melanoma progression through telomere elongation, an established driver of melanoma progression, thereby extending the proliferative capacity of incipient cancer cells.20 This notion is supported by observations of increased telomere length in melanomaprone families with POT1 mutations. Given this association, research has focused on examining the relationship between telomere length and skin cancer.
Several studies have examined the relationship between telomere length and the risk for various types of skin cancer, including melanoma, BCC, and SCC. Prior investigations have suggested that shorter telomere length is associated with a decreased risk for melanoma and an increased risk for BCC, while no significant association has been observed for SCC.16 However, subsequent reports analyzing POT1 variants have failed to reveal any conclusive associations between BCC and SCC and telomere length.16,21
In contrast, other genetic variants associated with melanoma susceptibility have demonstrated notable associations with BCC and SCC; for instance, the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, which is the first gene linked to high-risk familial melanoma, exhibits an increased presence of mutations in individuals with BCC and SCC.22 Similarly, the MC1R (melanocortin 1 receptor) variant, a gene involved in human pigmentation and known to increase the risk for melanoma, carries a statistically significantly higher risk for BCC (summary odds ratio, 1.39; 95% CI, 1.15-1.69) and SCC (summary odds ratio, 1.61; 95% CI, 1.35-1.91) when at least one variant is present and an even greater risk with 2 or more variants.23
Considering the potential importance of POT1 mutations and their association with melanoma, as well as the inconsistencies surrounding POT1 mutations and their associations with BCC and SCC, further research may clarify the impact of POT1 mutations on the development and progression of different types of skin cancers and improve understanding of the complex interplay among telomere length, genetic variants, and skin cancer susceptibility. Given the established risk for melanoma with POT1 mutations, regular dermatology surveillance seems prudent. Dermatologists should consider referring patients with multiple skin cancers (especially melanoma) and any strong family history of internal malignancies to genetic testing for POT1. Though melanoma, CLL, angiosarcoma, and gliomas are the most commonly associated malignancies with POT1 mutations, as our case demonstrates, presentations can be heterogeneous, and the spectrum of malignancies associated with POT1 may be more expansive than previously thought.
For our patient, the current surveillance plan is fullbody skin examinations every 3 months. Given no prior family history of malignancies, presumably our patient’s case was a spontaneous mutation. Interestingly, despite his many primary cancer diagnoses and metastases, our patient has responded well to all treatments without recurrence. It is unclear if these characteristics and treatment successes are features of POT1associated cancers. Further research is needed to refine recommendations for screening and management of patients with identified POT1 mutations.
Conclusion
This case report highlights a rare occurrence of MAC in a patient with a POT1 mutation. Given the limited research conducted on investigating POT1 mutations and skin cancer, it is important to consider various forms of skin cancer, in addition to melanoma, when treating patients with a POT1 mutation.
- Accardo ML, Osborne J, Else T. POT1 tumor predisposition. GeneReviews®. October 29, 2020. Updated December 4, 2025. University of Washington.
- Chaichamnan K, Satayasoontorn K, Puttanupaab S, et al. Malignant proliferating trichilemmal tumors with CD34 expression. J Med Assoc Thai. 2010;93(suppl 6):S28-S34.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Kaseb H, Babiker HM. Eccrine carcinoma. StatPearls [Internet]. Updated June 26, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK541042
- Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649-1654. doi:10.1101/gad.1215404
- Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482-486. doi:10.1038/ng.2941
- Wilson TL, Hattangady N, Lerario AM, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. 2017;16:561-566. doi:10.1007/s10689-017-9984-y
- Müller C, Krunic M, Wendt J, et al. Germline variants in the POT1- gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8:1475-1480. doi:10.1534/g3.117.300394
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-offunction variants predispose to familial melanoma. Nat Genet. 2014;46:478-481. doi:10.1038/ng.2947
- Wong K, Robles-Espinoza CD, Rodriguez D, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. 2019;155:604-609. doi:10.1001/jamadermatol.2018.3662
- Simonin-Wilmer I, Ossio R, Leddin EM, et al. Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet. 2023;60:692-696. doi:10.1136/jmg-2022-108776
- Potjer TP, Bollen S, Grimbergen AJEM, et al; Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144:2453- 2464. doi:10.1002/ijc.31984
- Pellegrini C, Raimondi S, Di Nardo L, et al; Italian Melanoma Intergroup (IMI). Melanoma in children and adolescents: analysis of susceptibility genes in 123 Italian patients. J Eur Acad Dermatol Venereol. 2022;36:213-221. doi:10.1111/jdv.17735
- Sargen MR, Calista D, Elder DE, et al. Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol. 2020;83:860-869. doi:10.1016/j.jaad.2020.03.100
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J Med Genet. 2020;57:664-670. doi:10.1136 /jmedgenet-2019-106657
- Nan H, Qureshi AA, Prescott J, et al. Genetic variants in telomere-maintaining genes and skin cancer risk. Hum Genet. 2011;129:247-253. doi:10.1007/s00439-010-0921-5
- Zhang L, Huang X, Zhu X, et al. Differential senescence capacities in meibomian gland carcinoma and basal cell carcinoma. Int J Cancer. 2016;138:1442-1452. doi:10.1002/ijc.29882
- Pastorino L, Andreotti V, Dalmasso B, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers (Basel). 2020;12:1007. doi:10.3390/cancers12041007
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. 2019;181:105-113. doi:10.1111/bjd.17443
- Kim WT, Hennick K, Johnson J, et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021;40:e107346.
- Ventura A, Pellegrini C, Cardelli L, et al. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int J Mol Sci. 2019;20:1333. doi:10.3390/ijms20061333
- Helgadottir H, Höiom V, Jönsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51:545-552. doi:10.1136/jmedgenet-2014-102320
- Tagliabue E, Fargnoli MC, Gandini S, et al; M-SKIP Study Group. MC1R gene variants and non-melanoma skin cancer: a pooledanalysis from the M-SKIP project. Br J Cancer. 2015;113:354-363. doi:10.1038/bjc.2015.231
A 72-year-old man with a history of multiple cancers, including melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC), presented to the dermatology clinic for a regularly scheduled full-body skin examination. His family history was negative for malignancy, but due to his personal history of both primary internal cancers and skin cancers, the patient previously had been referred by dermatology to a medical geneticist for evaluation. He tested positive for a pathogenic POT1 (protection of telomeres 1) variant associated with tumor predisposition, which most often is associated with cutaneous melanoma, chronic lymphocytic leukemia (CLL), angiosarcoma, and gliomas.1
At the current presentation, physical examination revealed a small, asymmetric, pink papule on the superior thoracic spine. A biopsy of the lesion was performed (Figure 1). Pathology demonstrated cornifying cystic structures with a granulomatous response at the surface of the tumor, ductal differentiation with depth, and infiltrative strands and cords of hyperchromatic cells within a collagenous stroma at the base of the specimen (Figures 2A and 2B). One unusual finding was the presence of prominent clear-cell change within the superficial portion of the neoplasm (Figure 2C). Immunohistochemical stains revealed strong p63 and p40 positivity. Epithelial membrane antigen staining was positive in the hyperchromatic strands and cords with depth but not in the clear-cell superficial portion. Similarly, periodic acid–Schiff–positive material increased within tumor cells in proportion to depth of infiltration. Additional immunohistochemical staining showed carcinoembryonic antigen was largely negative (with rare positivity in a few ductal lumina), with negative results for S100, SOX10, CD117, BerEP4, factor XIIIa, CD34, and cytokeratin 7 (Figures 2D and 2E).
The differential diagnoses included trichilemmal carcinoma (which may manifest with CD34 expression),2 clear cell BCC, adenoid cystic carcinoma (tubular variant), sebaceous carcinoma, and eccrine carcinoma. Importantly, the patient was under continuous oncologic surveillance, with no evidence of a primary internal tumor to suggest metastasis. Despite negative carcinoembryonic antigen staining, the immunohistochemical and histopathologic findings fit best with a primary cutaneous malignant eccrine tumor, specifically microcystic adnexal carcinoma (MAC), in which p63 typically stains peripheral cells but solid variants have been described.3
Eccrine carcinoma is exceedingly rare, reported in 0.01% of diagnosed cutaneous malignancies, and demonstrates overlapping features to other malignant eccrine tumors. It possesses an inconsistent immunohistochemical staining profile, making the distinction from other malignant sweat gland tumors challenging.4 Given that the morphologic features were otherwise classic for MAC in our patient, we favored a clear-cell variant.
Sixteen years prior to the current presentation, our patient presented to urology with a history of prostatitis and increasing prostate-specific antigen levels. Biopsies were negative until prostate-specific antigen reached 13 ng/mL, confirming stage 1A prostate cancer. The patient subsequently underwent a robot-assisted radical prostatectomy. At age 63 years, dysphagia that was unresponsive to antibiotics led to a tonsillar biopsy revealing T2N2bM0 stage IVA SCC of the right tonsil with confirmed HPV type 16 with extracapsular extension. The patient underwent transoral robotic radical tonsillectomy and right neck dissection, followed by adjuvant chemoradiation consisting of intensity-modulated radiation therapy (IMRT) to a total dose of 63 Gy in 33 fractions, with concurrent weekly cisplatin. At age 67 years, dyspepsia, dysphagia, pyrosis, and gastroesophageal reflux prompted endoscopy, revealing T1aNxMx esophageal adenocarcinoma. Three months later, the patient underwent laparoscopic-assisted esophagectomy, with no recurrence. At age 68 years, an atypical intramelanocytic proliferation was found on the left cheek and was treated with Mohs micrographic surgery.
At age 71 years, acral lentiginous malignant melanoma (Breslow thickness 0.8 mm; Clark level IV; American Joint Committee on Cancer T1b) was diagnosed on the left plantar foot and treated with Mohs micrographic surgery. Sentinel lymph node biopsy was negative. Squamous cell carcinoma in situ on the frontal scalp and nodular BCC on the right upper back also were diagnosed.
While there are no guidelines for surveillance of individuals with POT1, recommendations were given in consensus from a medical genetics team,1 including comprehensive monitoring—specifically baseline imaging utilizing brain and full-body magnetic resonance imaging. Furthermore, considering the crucial role of POT1 in maintaining telomeres, it was advised to measure telomere length as part of the surveillance process. Given the patient’s susceptibility to CLL, routine complete blood count assessments were recommended. Additionally, we advised close monitoring for seizures and consideration of genetic testing in first-degree relatives.
Literature Review
Given our patient’s history of multiple skin cancers, including the most recent MAC, we sought to conduct a review of the literature to evaluate existing skin cancer associations and reports for patients with known POT1 mutations to guide recommendations for dermatologic surveillance (Table). A search of PubMed articles indexed for MEDLINE through April 2023 using the terms microcystic adnexal carcinoma, POT1, melanoma, basal cell carcinoma, squamous cell carcinoma, and skin cancer yielded no reported cases of MAC associated with POT1 mutations. POT1 is one of 6 proteins (TERF1, TERF2, RAP1, TIN2, TPP1, and POT1) belonging to the shelterin complex, which plays a crucial role in telomeric DNA remodeling and regulation of telomere length.5 Mutation in the POT1 gene disrupts the shelterin complex, causing telomeres to become elongated and unstable, resulting in chromosomal abnormalities and promoting cancer development.5
While our literature review did not reveal any associations between the shelterin complex genes and MAC, mutations in the POT1 gene have been studied in other types of skin cancer, particularly melanoma.1 One of the earliest studies was conducted in 2014 by Shi et al,6 in which whole-exome sequencing was performed on families with a history of melanoma. Multiple POT1 gene pathogenic variants associated with increased telomere length and fragility were identified in unrelated families. Subsequent studies have confirmed POT1 variants in melanoma-prone families,7 supporting an association between increased telomere length and melanoma risk8-11; however, other studies have yielded nonsignificant findings.12,13 Further investigation also has identified morphologic characteristics consistent with POT1 mutation, including spitzoid morphology.14
The association between POT1 mutations and nonmelanoma skin cancers has been relatively understudied. While a few studies have explored this link, results have shown mixed findings. Some studies have suggested a potential role for POT1 mutations in cutaneous SCC risk,15 while other studies have shown no significant associations for both BCC and SCC risk and telomere gene mutations.16 Additionally, mRNA levels of POT1 were upregulated in BCC cases compared to normal tissue in a gene expression.17
Comment
In the literature, POT1 mutations are well established as high-penetrance alterations associated with melanoma.9,18,19 However, the correlation between POT1 and other forms of skin cancer is not yet delineated. Recent insights suggest that POT1 mutations play a major role in promoting melanoma progression through telomere elongation, an established driver of melanoma progression, thereby extending the proliferative capacity of incipient cancer cells.20 This notion is supported by observations of increased telomere length in melanomaprone families with POT1 mutations. Given this association, research has focused on examining the relationship between telomere length and skin cancer.
Several studies have examined the relationship between telomere length and the risk for various types of skin cancer, including melanoma, BCC, and SCC. Prior investigations have suggested that shorter telomere length is associated with a decreased risk for melanoma and an increased risk for BCC, while no significant association has been observed for SCC.16 However, subsequent reports analyzing POT1 variants have failed to reveal any conclusive associations between BCC and SCC and telomere length.16,21
In contrast, other genetic variants associated with melanoma susceptibility have demonstrated notable associations with BCC and SCC; for instance, the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, which is the first gene linked to high-risk familial melanoma, exhibits an increased presence of mutations in individuals with BCC and SCC.22 Similarly, the MC1R (melanocortin 1 receptor) variant, a gene involved in human pigmentation and known to increase the risk for melanoma, carries a statistically significantly higher risk for BCC (summary odds ratio, 1.39; 95% CI, 1.15-1.69) and SCC (summary odds ratio, 1.61; 95% CI, 1.35-1.91) when at least one variant is present and an even greater risk with 2 or more variants.23
Considering the potential importance of POT1 mutations and their association with melanoma, as well as the inconsistencies surrounding POT1 mutations and their associations with BCC and SCC, further research may clarify the impact of POT1 mutations on the development and progression of different types of skin cancers and improve understanding of the complex interplay among telomere length, genetic variants, and skin cancer susceptibility. Given the established risk for melanoma with POT1 mutations, regular dermatology surveillance seems prudent. Dermatologists should consider referring patients with multiple skin cancers (especially melanoma) and any strong family history of internal malignancies to genetic testing for POT1. Though melanoma, CLL, angiosarcoma, and gliomas are the most commonly associated malignancies with POT1 mutations, as our case demonstrates, presentations can be heterogeneous, and the spectrum of malignancies associated with POT1 may be more expansive than previously thought.
For our patient, the current surveillance plan is fullbody skin examinations every 3 months. Given no prior family history of malignancies, presumably our patient’s case was a spontaneous mutation. Interestingly, despite his many primary cancer diagnoses and metastases, our patient has responded well to all treatments without recurrence. It is unclear if these characteristics and treatment successes are features of POT1associated cancers. Further research is needed to refine recommendations for screening and management of patients with identified POT1 mutations.
Conclusion
This case report highlights a rare occurrence of MAC in a patient with a POT1 mutation. Given the limited research conducted on investigating POT1 mutations and skin cancer, it is important to consider various forms of skin cancer, in addition to melanoma, when treating patients with a POT1 mutation.
A 72-year-old man with a history of multiple cancers, including melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC), presented to the dermatology clinic for a regularly scheduled full-body skin examination. His family history was negative for malignancy, but due to his personal history of both primary internal cancers and skin cancers, the patient previously had been referred by dermatology to a medical geneticist for evaluation. He tested positive for a pathogenic POT1 (protection of telomeres 1) variant associated with tumor predisposition, which most often is associated with cutaneous melanoma, chronic lymphocytic leukemia (CLL), angiosarcoma, and gliomas.1
At the current presentation, physical examination revealed a small, asymmetric, pink papule on the superior thoracic spine. A biopsy of the lesion was performed (Figure 1). Pathology demonstrated cornifying cystic structures with a granulomatous response at the surface of the tumor, ductal differentiation with depth, and infiltrative strands and cords of hyperchromatic cells within a collagenous stroma at the base of the specimen (Figures 2A and 2B). One unusual finding was the presence of prominent clear-cell change within the superficial portion of the neoplasm (Figure 2C). Immunohistochemical stains revealed strong p63 and p40 positivity. Epithelial membrane antigen staining was positive in the hyperchromatic strands and cords with depth but not in the clear-cell superficial portion. Similarly, periodic acid–Schiff–positive material increased within tumor cells in proportion to depth of infiltration. Additional immunohistochemical staining showed carcinoembryonic antigen was largely negative (with rare positivity in a few ductal lumina), with negative results for S100, SOX10, CD117, BerEP4, factor XIIIa, CD34, and cytokeratin 7 (Figures 2D and 2E).
The differential diagnoses included trichilemmal carcinoma (which may manifest with CD34 expression),2 clear cell BCC, adenoid cystic carcinoma (tubular variant), sebaceous carcinoma, and eccrine carcinoma. Importantly, the patient was under continuous oncologic surveillance, with no evidence of a primary internal tumor to suggest metastasis. Despite negative carcinoembryonic antigen staining, the immunohistochemical and histopathologic findings fit best with a primary cutaneous malignant eccrine tumor, specifically microcystic adnexal carcinoma (MAC), in which p63 typically stains peripheral cells but solid variants have been described.3
Eccrine carcinoma is exceedingly rare, reported in 0.01% of diagnosed cutaneous malignancies, and demonstrates overlapping features to other malignant eccrine tumors. It possesses an inconsistent immunohistochemical staining profile, making the distinction from other malignant sweat gland tumors challenging.4 Given that the morphologic features were otherwise classic for MAC in our patient, we favored a clear-cell variant.
Sixteen years prior to the current presentation, our patient presented to urology with a history of prostatitis and increasing prostate-specific antigen levels. Biopsies were negative until prostate-specific antigen reached 13 ng/mL, confirming stage 1A prostate cancer. The patient subsequently underwent a robot-assisted radical prostatectomy. At age 63 years, dysphagia that was unresponsive to antibiotics led to a tonsillar biopsy revealing T2N2bM0 stage IVA SCC of the right tonsil with confirmed HPV type 16 with extracapsular extension. The patient underwent transoral robotic radical tonsillectomy and right neck dissection, followed by adjuvant chemoradiation consisting of intensity-modulated radiation therapy (IMRT) to a total dose of 63 Gy in 33 fractions, with concurrent weekly cisplatin. At age 67 years, dyspepsia, dysphagia, pyrosis, and gastroesophageal reflux prompted endoscopy, revealing T1aNxMx esophageal adenocarcinoma. Three months later, the patient underwent laparoscopic-assisted esophagectomy, with no recurrence. At age 68 years, an atypical intramelanocytic proliferation was found on the left cheek and was treated with Mohs micrographic surgery.
At age 71 years, acral lentiginous malignant melanoma (Breslow thickness 0.8 mm; Clark level IV; American Joint Committee on Cancer T1b) was diagnosed on the left plantar foot and treated with Mohs micrographic surgery. Sentinel lymph node biopsy was negative. Squamous cell carcinoma in situ on the frontal scalp and nodular BCC on the right upper back also were diagnosed.
While there are no guidelines for surveillance of individuals with POT1, recommendations were given in consensus from a medical genetics team,1 including comprehensive monitoring—specifically baseline imaging utilizing brain and full-body magnetic resonance imaging. Furthermore, considering the crucial role of POT1 in maintaining telomeres, it was advised to measure telomere length as part of the surveillance process. Given the patient’s susceptibility to CLL, routine complete blood count assessments were recommended. Additionally, we advised close monitoring for seizures and consideration of genetic testing in first-degree relatives.
Literature Review
Given our patient’s history of multiple skin cancers, including the most recent MAC, we sought to conduct a review of the literature to evaluate existing skin cancer associations and reports for patients with known POT1 mutations to guide recommendations for dermatologic surveillance (Table). A search of PubMed articles indexed for MEDLINE through April 2023 using the terms microcystic adnexal carcinoma, POT1, melanoma, basal cell carcinoma, squamous cell carcinoma, and skin cancer yielded no reported cases of MAC associated with POT1 mutations. POT1 is one of 6 proteins (TERF1, TERF2, RAP1, TIN2, TPP1, and POT1) belonging to the shelterin complex, which plays a crucial role in telomeric DNA remodeling and regulation of telomere length.5 Mutation in the POT1 gene disrupts the shelterin complex, causing telomeres to become elongated and unstable, resulting in chromosomal abnormalities and promoting cancer development.5
While our literature review did not reveal any associations between the shelterin complex genes and MAC, mutations in the POT1 gene have been studied in other types of skin cancer, particularly melanoma.1 One of the earliest studies was conducted in 2014 by Shi et al,6 in which whole-exome sequencing was performed on families with a history of melanoma. Multiple POT1 gene pathogenic variants associated with increased telomere length and fragility were identified in unrelated families. Subsequent studies have confirmed POT1 variants in melanoma-prone families,7 supporting an association between increased telomere length and melanoma risk8-11; however, other studies have yielded nonsignificant findings.12,13 Further investigation also has identified morphologic characteristics consistent with POT1 mutation, including spitzoid morphology.14
The association between POT1 mutations and nonmelanoma skin cancers has been relatively understudied. While a few studies have explored this link, results have shown mixed findings. Some studies have suggested a potential role for POT1 mutations in cutaneous SCC risk,15 while other studies have shown no significant associations for both BCC and SCC risk and telomere gene mutations.16 Additionally, mRNA levels of POT1 were upregulated in BCC cases compared to normal tissue in a gene expression.17
Comment
In the literature, POT1 mutations are well established as high-penetrance alterations associated with melanoma.9,18,19 However, the correlation between POT1 and other forms of skin cancer is not yet delineated. Recent insights suggest that POT1 mutations play a major role in promoting melanoma progression through telomere elongation, an established driver of melanoma progression, thereby extending the proliferative capacity of incipient cancer cells.20 This notion is supported by observations of increased telomere length in melanomaprone families with POT1 mutations. Given this association, research has focused on examining the relationship between telomere length and skin cancer.
Several studies have examined the relationship between telomere length and the risk for various types of skin cancer, including melanoma, BCC, and SCC. Prior investigations have suggested that shorter telomere length is associated with a decreased risk for melanoma and an increased risk for BCC, while no significant association has been observed for SCC.16 However, subsequent reports analyzing POT1 variants have failed to reveal any conclusive associations between BCC and SCC and telomere length.16,21
In contrast, other genetic variants associated with melanoma susceptibility have demonstrated notable associations with BCC and SCC; for instance, the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, which is the first gene linked to high-risk familial melanoma, exhibits an increased presence of mutations in individuals with BCC and SCC.22 Similarly, the MC1R (melanocortin 1 receptor) variant, a gene involved in human pigmentation and known to increase the risk for melanoma, carries a statistically significantly higher risk for BCC (summary odds ratio, 1.39; 95% CI, 1.15-1.69) and SCC (summary odds ratio, 1.61; 95% CI, 1.35-1.91) when at least one variant is present and an even greater risk with 2 or more variants.23
Considering the potential importance of POT1 mutations and their association with melanoma, as well as the inconsistencies surrounding POT1 mutations and their associations with BCC and SCC, further research may clarify the impact of POT1 mutations on the development and progression of different types of skin cancers and improve understanding of the complex interplay among telomere length, genetic variants, and skin cancer susceptibility. Given the established risk for melanoma with POT1 mutations, regular dermatology surveillance seems prudent. Dermatologists should consider referring patients with multiple skin cancers (especially melanoma) and any strong family history of internal malignancies to genetic testing for POT1. Though melanoma, CLL, angiosarcoma, and gliomas are the most commonly associated malignancies with POT1 mutations, as our case demonstrates, presentations can be heterogeneous, and the spectrum of malignancies associated with POT1 may be more expansive than previously thought.
For our patient, the current surveillance plan is fullbody skin examinations every 3 months. Given no prior family history of malignancies, presumably our patient’s case was a spontaneous mutation. Interestingly, despite his many primary cancer diagnoses and metastases, our patient has responded well to all treatments without recurrence. It is unclear if these characteristics and treatment successes are features of POT1associated cancers. Further research is needed to refine recommendations for screening and management of patients with identified POT1 mutations.
Conclusion
This case report highlights a rare occurrence of MAC in a patient with a POT1 mutation. Given the limited research conducted on investigating POT1 mutations and skin cancer, it is important to consider various forms of skin cancer, in addition to melanoma, when treating patients with a POT1 mutation.
- Accardo ML, Osborne J, Else T. POT1 tumor predisposition. GeneReviews®. October 29, 2020. Updated December 4, 2025. University of Washington.
- Chaichamnan K, Satayasoontorn K, Puttanupaab S, et al. Malignant proliferating trichilemmal tumors with CD34 expression. J Med Assoc Thai. 2010;93(suppl 6):S28-S34.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Kaseb H, Babiker HM. Eccrine carcinoma. StatPearls [Internet]. Updated June 26, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK541042
- Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649-1654. doi:10.1101/gad.1215404
- Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482-486. doi:10.1038/ng.2941
- Wilson TL, Hattangady N, Lerario AM, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. 2017;16:561-566. doi:10.1007/s10689-017-9984-y
- Müller C, Krunic M, Wendt J, et al. Germline variants in the POT1- gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8:1475-1480. doi:10.1534/g3.117.300394
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-offunction variants predispose to familial melanoma. Nat Genet. 2014;46:478-481. doi:10.1038/ng.2947
- Wong K, Robles-Espinoza CD, Rodriguez D, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. 2019;155:604-609. doi:10.1001/jamadermatol.2018.3662
- Simonin-Wilmer I, Ossio R, Leddin EM, et al. Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet. 2023;60:692-696. doi:10.1136/jmg-2022-108776
- Potjer TP, Bollen S, Grimbergen AJEM, et al; Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144:2453- 2464. doi:10.1002/ijc.31984
- Pellegrini C, Raimondi S, Di Nardo L, et al; Italian Melanoma Intergroup (IMI). Melanoma in children and adolescents: analysis of susceptibility genes in 123 Italian patients. J Eur Acad Dermatol Venereol. 2022;36:213-221. doi:10.1111/jdv.17735
- Sargen MR, Calista D, Elder DE, et al. Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol. 2020;83:860-869. doi:10.1016/j.jaad.2020.03.100
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J Med Genet. 2020;57:664-670. doi:10.1136 /jmedgenet-2019-106657
- Nan H, Qureshi AA, Prescott J, et al. Genetic variants in telomere-maintaining genes and skin cancer risk. Hum Genet. 2011;129:247-253. doi:10.1007/s00439-010-0921-5
- Zhang L, Huang X, Zhu X, et al. Differential senescence capacities in meibomian gland carcinoma and basal cell carcinoma. Int J Cancer. 2016;138:1442-1452. doi:10.1002/ijc.29882
- Pastorino L, Andreotti V, Dalmasso B, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers (Basel). 2020;12:1007. doi:10.3390/cancers12041007
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. 2019;181:105-113. doi:10.1111/bjd.17443
- Kim WT, Hennick K, Johnson J, et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021;40:e107346.
- Ventura A, Pellegrini C, Cardelli L, et al. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int J Mol Sci. 2019;20:1333. doi:10.3390/ijms20061333
- Helgadottir H, Höiom V, Jönsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51:545-552. doi:10.1136/jmedgenet-2014-102320
- Tagliabue E, Fargnoli MC, Gandini S, et al; M-SKIP Study Group. MC1R gene variants and non-melanoma skin cancer: a pooledanalysis from the M-SKIP project. Br J Cancer. 2015;113:354-363. doi:10.1038/bjc.2015.231
- Accardo ML, Osborne J, Else T. POT1 tumor predisposition. GeneReviews®. October 29, 2020. Updated December 4, 2025. University of Washington.
- Chaichamnan K, Satayasoontorn K, Puttanupaab S, et al. Malignant proliferating trichilemmal tumors with CD34 expression. J Med Assoc Thai. 2010;93(suppl 6):S28-S34.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Kaseb H, Babiker HM. Eccrine carcinoma. StatPearls [Internet]. Updated June 26, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK541042
- Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649-1654. doi:10.1101/gad.1215404
- Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482-486. doi:10.1038/ng.2941
- Wilson TL, Hattangady N, Lerario AM, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. 2017;16:561-566. doi:10.1007/s10689-017-9984-y
- Müller C, Krunic M, Wendt J, et al. Germline variants in the POT1- gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8:1475-1480. doi:10.1534/g3.117.300394
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-offunction variants predispose to familial melanoma. Nat Genet. 2014;46:478-481. doi:10.1038/ng.2947
- Wong K, Robles-Espinoza CD, Rodriguez D, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. 2019;155:604-609. doi:10.1001/jamadermatol.2018.3662
- Simonin-Wilmer I, Ossio R, Leddin EM, et al. Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet. 2023;60:692-696. doi:10.1136/jmg-2022-108776
- Potjer TP, Bollen S, Grimbergen AJEM, et al; Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144:2453- 2464. doi:10.1002/ijc.31984
- Pellegrini C, Raimondi S, Di Nardo L, et al; Italian Melanoma Intergroup (IMI). Melanoma in children and adolescents: analysis of susceptibility genes in 123 Italian patients. J Eur Acad Dermatol Venereol. 2022;36:213-221. doi:10.1111/jdv.17735
- Sargen MR, Calista D, Elder DE, et al. Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol. 2020;83:860-869. doi:10.1016/j.jaad.2020.03.100
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J Med Genet. 2020;57:664-670. doi:10.1136 /jmedgenet-2019-106657
- Nan H, Qureshi AA, Prescott J, et al. Genetic variants in telomere-maintaining genes and skin cancer risk. Hum Genet. 2011;129:247-253. doi:10.1007/s00439-010-0921-5
- Zhang L, Huang X, Zhu X, et al. Differential senescence capacities in meibomian gland carcinoma and basal cell carcinoma. Int J Cancer. 2016;138:1442-1452. doi:10.1002/ijc.29882
- Pastorino L, Andreotti V, Dalmasso B, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers (Basel). 2020;12:1007. doi:10.3390/cancers12041007
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. 2019;181:105-113. doi:10.1111/bjd.17443
- Kim WT, Hennick K, Johnson J, et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021;40:e107346.
- Ventura A, Pellegrini C, Cardelli L, et al. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int J Mol Sci. 2019;20:1333. doi:10.3390/ijms20061333
- Helgadottir H, Höiom V, Jönsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51:545-552. doi:10.1136/jmedgenet-2014-102320
- Tagliabue E, Fargnoli MC, Gandini S, et al; M-SKIP Study Group. MC1R gene variants and non-melanoma skin cancer: a pooledanalysis from the M-SKIP project. Br J Cancer. 2015;113:354-363. doi:10.1038/bjc.2015.231
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
PRACTICE POINTS
- Dermatologists should consider referring patients with both a history of skin cancer and a strong family history of internal malignancy for genetic testing for POT1 (protection of telomeres 1) mutations.
- Although melanoma, chronic lymphocytic leukemia, angiosarcoma, and gliomas are most commonly associated with POT1 mutations, this case suggests a broader and more heterogeneous malignancy spectrum than previously recognized.

Ulcerated Lesions on the Right Leg
Ulcerated Lesions on the Right Leg
THE DIAGNOSIS: Mycobacteria infection
Despite the initial biopsy for tissue culture showing no growth, a subsequent biopsy performed 1 month later yielded a positive result. Mycobacterium marinum was identified through organism genome sequencing. The patient was further treated by infectious disease with clarithromycin and ethambutol, with complete resolution of the lesions.
Although initial staining with acid-fast bacilli and tissue culture were negative, we suspected a diagnosis of mycobacterial infection with sporotrichoid spread of multiple nodular and ulcerated lesions that was unresponsive to antibiotics. Performing a tissue culture is crucial for diagnosing mycobacterial skin and soft-tissue infections, as an acid-fast bacilli stain alone cannot distinguish between different mycobacterial species. Lowenstein-Jensen agar is a selective medium specifically used for the culture and isolation of Mycobacterium species. The strict temperature requirement of 30 °C to 32 °C (86-89.6 °F) for the growth of this organism suggests that the infection predominantly affects the limbs, which tend to have a slightly lower temperature compared to the core of the body.1 In our case, the histologic findings and clinical history suggested granulomatous involvement due to fungi or mycobacteria.
Cutaneous leishmaniasis is characterized by ulcers with possible accompanying nodular lymphangitis; however, the patient did not have relevant travel history. Leishmaniasis results from a parasite transmitted by a sandfly, with most cases occurring in Afghanistan, Algeria, Brazil, Iran, Pakistan, Peru, Saudi Arabia, and Syria.2
Ecthyma gangrenosum is characterized by tender necrotic plaques seen predominantly in immunocompromised patients and is associated with Pseudomonas aeruginosa bacteremia.3 Our patient had lesions present for a duration of 5 months, which is inconsistent with the more rapidly progressing course of ecthyma gangrenosum.
Leukocytoclastic vasculitis may manifest with palpable purpura of the lower extremities. An infectious trigger, such as Mycobacterium, may lead to a leukocytoclastic vasculitis. The histopathologic findings classically demonstrate neutrophil deposition in vessel walls, deposition of fibrin in the vessel lumen, and nuclear debris.4
Despite the presence of granulomatous changes in our patient, the presentation of ulcerated nodules in a sporotrichoid pattern on one extremity suggests a diagnosis of infectious etiology rather than sarcoidosis.
- Gonçalves IC, Furtado I, Gonçalves MJ, et al. Mycobacterium marinum cutaneous infection: a series of three cases and literature review. Cureus. 2022;14:E31787. doi:10.7759/cureus.31787
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007 /s40257-022-00726-8
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. Updated August 8, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482159/
THE DIAGNOSIS: Mycobacteria infection
Despite the initial biopsy for tissue culture showing no growth, a subsequent biopsy performed 1 month later yielded a positive result. Mycobacterium marinum was identified through organism genome sequencing. The patient was further treated by infectious disease with clarithromycin and ethambutol, with complete resolution of the lesions.
Although initial staining with acid-fast bacilli and tissue culture were negative, we suspected a diagnosis of mycobacterial infection with sporotrichoid spread of multiple nodular and ulcerated lesions that was unresponsive to antibiotics. Performing a tissue culture is crucial for diagnosing mycobacterial skin and soft-tissue infections, as an acid-fast bacilli stain alone cannot distinguish between different mycobacterial species. Lowenstein-Jensen agar is a selective medium specifically used for the culture and isolation of Mycobacterium species. The strict temperature requirement of 30 °C to 32 °C (86-89.6 °F) for the growth of this organism suggests that the infection predominantly affects the limbs, which tend to have a slightly lower temperature compared to the core of the body.1 In our case, the histologic findings and clinical history suggested granulomatous involvement due to fungi or mycobacteria.
Cutaneous leishmaniasis is characterized by ulcers with possible accompanying nodular lymphangitis; however, the patient did not have relevant travel history. Leishmaniasis results from a parasite transmitted by a sandfly, with most cases occurring in Afghanistan, Algeria, Brazil, Iran, Pakistan, Peru, Saudi Arabia, and Syria.2
Ecthyma gangrenosum is characterized by tender necrotic plaques seen predominantly in immunocompromised patients and is associated with Pseudomonas aeruginosa bacteremia.3 Our patient had lesions present for a duration of 5 months, which is inconsistent with the more rapidly progressing course of ecthyma gangrenosum.
Leukocytoclastic vasculitis may manifest with palpable purpura of the lower extremities. An infectious trigger, such as Mycobacterium, may lead to a leukocytoclastic vasculitis. The histopathologic findings classically demonstrate neutrophil deposition in vessel walls, deposition of fibrin in the vessel lumen, and nuclear debris.4
Despite the presence of granulomatous changes in our patient, the presentation of ulcerated nodules in a sporotrichoid pattern on one extremity suggests a diagnosis of infectious etiology rather than sarcoidosis.
THE DIAGNOSIS: Mycobacteria infection
Despite the initial biopsy for tissue culture showing no growth, a subsequent biopsy performed 1 month later yielded a positive result. Mycobacterium marinum was identified through organism genome sequencing. The patient was further treated by infectious disease with clarithromycin and ethambutol, with complete resolution of the lesions.
Although initial staining with acid-fast bacilli and tissue culture were negative, we suspected a diagnosis of mycobacterial infection with sporotrichoid spread of multiple nodular and ulcerated lesions that was unresponsive to antibiotics. Performing a tissue culture is crucial for diagnosing mycobacterial skin and soft-tissue infections, as an acid-fast bacilli stain alone cannot distinguish between different mycobacterial species. Lowenstein-Jensen agar is a selective medium specifically used for the culture and isolation of Mycobacterium species. The strict temperature requirement of 30 °C to 32 °C (86-89.6 °F) for the growth of this organism suggests that the infection predominantly affects the limbs, which tend to have a slightly lower temperature compared to the core of the body.1 In our case, the histologic findings and clinical history suggested granulomatous involvement due to fungi or mycobacteria.
Cutaneous leishmaniasis is characterized by ulcers with possible accompanying nodular lymphangitis; however, the patient did not have relevant travel history. Leishmaniasis results from a parasite transmitted by a sandfly, with most cases occurring in Afghanistan, Algeria, Brazil, Iran, Pakistan, Peru, Saudi Arabia, and Syria.2
Ecthyma gangrenosum is characterized by tender necrotic plaques seen predominantly in immunocompromised patients and is associated with Pseudomonas aeruginosa bacteremia.3 Our patient had lesions present for a duration of 5 months, which is inconsistent with the more rapidly progressing course of ecthyma gangrenosum.
Leukocytoclastic vasculitis may manifest with palpable purpura of the lower extremities. An infectious trigger, such as Mycobacterium, may lead to a leukocytoclastic vasculitis. The histopathologic findings classically demonstrate neutrophil deposition in vessel walls, deposition of fibrin in the vessel lumen, and nuclear debris.4
Despite the presence of granulomatous changes in our patient, the presentation of ulcerated nodules in a sporotrichoid pattern on one extremity suggests a diagnosis of infectious etiology rather than sarcoidosis.
- Gonçalves IC, Furtado I, Gonçalves MJ, et al. Mycobacterium marinum cutaneous infection: a series of three cases and literature review. Cureus. 2022;14:E31787. doi:10.7759/cureus.31787
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007 /s40257-022-00726-8
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. Updated August 8, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482159/
- Gonçalves IC, Furtado I, Gonçalves MJ, et al. Mycobacterium marinum cutaneous infection: a series of three cases and literature review. Cureus. 2022;14:E31787. doi:10.7759/cureus.31787
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007 /s40257-022-00726-8
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. Updated August 8, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482159/
Ulcerated Lesions on the Right Leg
Ulcerated Lesions on the Right Leg
A 78-year-old man was referred to our dermatology clinic for evaluation of nontender erythematous plaques and nodules with central ulceration on the right leg of 5 months’ duration. The patient’s medical history was remarkable for hyperlipidemia, gastroesophageal reflux disease, prostate cancer, and colon cancer status post resection. He denied any relevant travel history but noted that he was an avid hiker and suspected he may have obtained a puncture wound from a bush or a mosquito bite prior to the appearance of the lesions. Previous therapies prescribed by outside physicians and our practice included trimethoprim/sulfamethoxazole, ceftriaxone, levofloxacin, mupirocin, and topical corticosteroids, all with minimal benefit. Clinical examination on initial presentation revealed multiple ulcerations of the lower extremities present for more than 2 months. Punch biopsy of a sample lesion at the current presentation revealed granulomatous change, focal necrosis, and a mixed inflammatory cell infiltrate. Grocott-Gomori methenamine silver and periodic acid–Schiff stains were negative for fungal organisms. The initial acid-fast bacilli stain was negative for mycobacteria, and tissue culture showed no growth.

Multiple Grouped Erythematous to Violaceous Preauricular Papules
Multiple Grouped Erythematous to Violaceous Preauricular Papules
THE DIAGNOSIS: Angiolymphoid Hyperplasia With Eosinophilia
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare, benign, inflammatory vascular proliferation with lymphocytic and eosinophilic infiltration. Bleeding and pruritus associated with ALHE can substantially affect a patient’s quality of life, necessitating correct diagnosis and effective treatment.1 The etiopathogenesis of ALHE is poorly understood, and it often is attributed to an underlying vascular malformation or local trauma. Vascular proliferation due to hyperestrogenemia could explain why pregnancy is considered a predisposing factor for ALHE.1,2
Angiolymphoid hyperplasia with eosinophilia typically manifests with solitary or multiple pink to red-brown, dome-shaped papules or nodules occurring most frequently on the head and neck. Lesions may be either asymptomatic or associated with pruritus, pain, and spontaneous bleeding.1 Dermoscopy is crucial to diagnosis. The most frequent dermoscopic findings include a polymorphic vascular pattern such as dotted and linear irregular vessels over a pink background, white lines, white dots, white structureless areas, and red-purple lacunae.2,3 Histopathology will demonstrate a vascular proliferation with plump epithelioid endothelial cells showing abundant eosinophilic cytoplasm, accompanied by a variable lymphocytic and eosinophilic inflammatory infiltrate (Figure 1).1
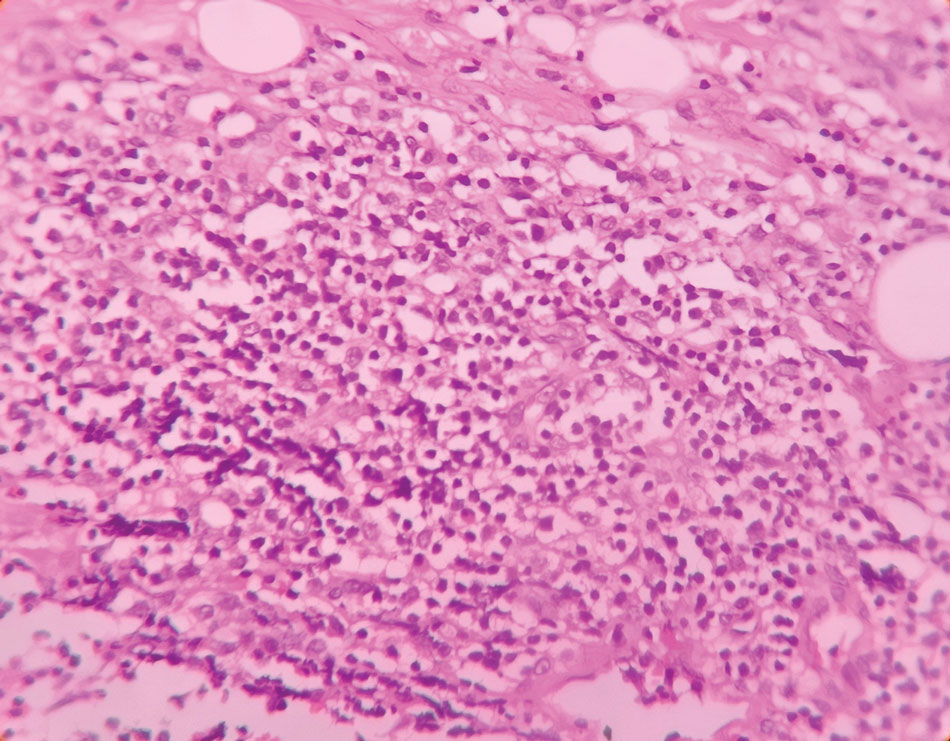
In our case, dermoscopic-histopathologic correlation suggested that the polymorphic vascular pattern and clods on a pink background corresponded to thin- and thick-walled vessels containing plump endothelial cells and intraluminal erythrocytes within the superficial and deep dermis. White structures could represent underlying fibrosis and altered dermal collagen due to vascular proliferation. The brown pigment network and peripheral brownish pigmentation were most likely secondary to increased melanin and accentuation of the pigment network in the setting of Fitzpatrick skin types IV to V, although pruritic trauma with postinflammatory hyperpigmentation may also have contributed, making dermoscopic-histopathologic correlation challenging.
Surgical excision is considered the primary treatment modality for ALHE, with the lowest recurrence rates.1 Alternative therapeutic options include intralesional steroids, cryotherapy, sclerotherapy, radiofrequency, pulsed dye laser, and carbon dioxide laser, with varying efficacy reported.1 Our patient was treated with a combination of a long-pulse Nd:YAG laser (pulse width of 30 ms) to target the vascular component, followed by a single session with an ablative Er:YAG laser. After 4 weeks, healing with good cosmetic results was observed (Figure 2). At 6-month follow-up, there was no recurrence of the lesions.
Kimura disease, often considered the closest differential diagnosis for ALHE, is a rare lymphoproliferative fibroinflammatory condition. Patients present with subcutaneous nodules on the head and neck, often associated with lymphadenopathy. Elevated serum IgE levels and peripheral blood eosinophilia are common.1 Another consideration in the differential diagnosis is cutaneous bacillary angiomatosis caused by Bartonella species, a vascular proliferative condition that mostly affects individuals with HIV, transplant recipients, and those taking immunosuppressive medications.4 Pyogenic granuloma, also known as lobular capillary haemangioma, is another benign vascular proliferation that resembles ALHE. Clinically, it manifests as a solitary, painless, flesh-colored to erythematous papulonodule; however, multiple grouped lesions also can occur. The lesions often are associated with bleeding and erosions.5 Epithelioid hemangioendothelioma is a rare vascular tumor most frequently manifesting in the liver, lungs, or bones, and very rarely is limited to skin. Cutaneous epithelioid hemangioendothelioma mimics ALHE and may manifest as a solitary erythematous mass, multiple dome-shaped masses, or dermal nodules.6
- Brahs A, Sledge B, Mullen H, et al. Angiolymphoid hyperplasia with eosinophilia: many syllables, many unanswered questions. J Clin Aesthet Dermatol. 2021;14:49-54.
- Kalantri M, Khopkar U. Spectrum of dermoscopic pattern in a patient with angiolymphoid hyperplasia with tissue eosinophilia. Indian J Dermatol. 2020;65:556-558.
- Chauhan P, Vinay K, Jindal R, et al. Dermoscopic characterisation of angiolymphoid hyperplasia in skin of colour: a case series of six patients with review of literature. Indian J Dermatol Venereol Leprol. 2024;90:848.
- Ramírez Ramírez CR, Saavedra S, Ramírez Ronda CH. Bacillary angiomatosis: microbiology, histopathology, clinical presentation, diagnosis and management. Bol Asoc Med PR. 1996;88:46-51.
- Leung AKC, Barankin B, Hon KL. Pyogenic granuloma. Clinics Mother Child Health. 2014;11:E106. doi:10.4172/2090-7214.1000e106
- Kumar V, Kachhawa D, Rekha S, et al. Cutaneous epithelioid hemangioendothelioma: a rare presentation. Indian J Dermatol Venereol Leprol. 2018;84:739-742.
THE DIAGNOSIS: Angiolymphoid Hyperplasia With Eosinophilia
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare, benign, inflammatory vascular proliferation with lymphocytic and eosinophilic infiltration. Bleeding and pruritus associated with ALHE can substantially affect a patient’s quality of life, necessitating correct diagnosis and effective treatment.1 The etiopathogenesis of ALHE is poorly understood, and it often is attributed to an underlying vascular malformation or local trauma. Vascular proliferation due to hyperestrogenemia could explain why pregnancy is considered a predisposing factor for ALHE.1,2
Angiolymphoid hyperplasia with eosinophilia typically manifests with solitary or multiple pink to red-brown, dome-shaped papules or nodules occurring most frequently on the head and neck. Lesions may be either asymptomatic or associated with pruritus, pain, and spontaneous bleeding.1 Dermoscopy is crucial to diagnosis. The most frequent dermoscopic findings include a polymorphic vascular pattern such as dotted and linear irregular vessels over a pink background, white lines, white dots, white structureless areas, and red-purple lacunae.2,3 Histopathology will demonstrate a vascular proliferation with plump epithelioid endothelial cells showing abundant eosinophilic cytoplasm, accompanied by a variable lymphocytic and eosinophilic inflammatory infiltrate (Figure 1).1
In our case, dermoscopic-histopathologic correlation suggested that the polymorphic vascular pattern and clods on a pink background corresponded to thin- and thick-walled vessels containing plump endothelial cells and intraluminal erythrocytes within the superficial and deep dermis. White structures could represent underlying fibrosis and altered dermal collagen due to vascular proliferation. The brown pigment network and peripheral brownish pigmentation were most likely secondary to increased melanin and accentuation of the pigment network in the setting of Fitzpatrick skin types IV to V, although pruritic trauma with postinflammatory hyperpigmentation may also have contributed, making dermoscopic-histopathologic correlation challenging.
Surgical excision is considered the primary treatment modality for ALHE, with the lowest recurrence rates.1 Alternative therapeutic options include intralesional steroids, cryotherapy, sclerotherapy, radiofrequency, pulsed dye laser, and carbon dioxide laser, with varying efficacy reported.1 Our patient was treated with a combination of a long-pulse Nd:YAG laser (pulse width of 30 ms) to target the vascular component, followed by a single session with an ablative Er:YAG laser. After 4 weeks, healing with good cosmetic results was observed (Figure 2). At 6-month follow-up, there was no recurrence of the lesions.
Kimura disease, often considered the closest differential diagnosis for ALHE, is a rare lymphoproliferative fibroinflammatory condition. Patients present with subcutaneous nodules on the head and neck, often associated with lymphadenopathy. Elevated serum IgE levels and peripheral blood eosinophilia are common.1 Another consideration in the differential diagnosis is cutaneous bacillary angiomatosis caused by Bartonella species, a vascular proliferative condition that mostly affects individuals with HIV, transplant recipients, and those taking immunosuppressive medications.4 Pyogenic granuloma, also known as lobular capillary haemangioma, is another benign vascular proliferation that resembles ALHE. Clinically, it manifests as a solitary, painless, flesh-colored to erythematous papulonodule; however, multiple grouped lesions also can occur. The lesions often are associated with bleeding and erosions.5 Epithelioid hemangioendothelioma is a rare vascular tumor most frequently manifesting in the liver, lungs, or bones, and very rarely is limited to skin. Cutaneous epithelioid hemangioendothelioma mimics ALHE and may manifest as a solitary erythematous mass, multiple dome-shaped masses, or dermal nodules.6
THE DIAGNOSIS: Angiolymphoid Hyperplasia With Eosinophilia
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare, benign, inflammatory vascular proliferation with lymphocytic and eosinophilic infiltration. Bleeding and pruritus associated with ALHE can substantially affect a patient’s quality of life, necessitating correct diagnosis and effective treatment.1 The etiopathogenesis of ALHE is poorly understood, and it often is attributed to an underlying vascular malformation or local trauma. Vascular proliferation due to hyperestrogenemia could explain why pregnancy is considered a predisposing factor for ALHE.1,2
Angiolymphoid hyperplasia with eosinophilia typically manifests with solitary or multiple pink to red-brown, dome-shaped papules or nodules occurring most frequently on the head and neck. Lesions may be either asymptomatic or associated with pruritus, pain, and spontaneous bleeding.1 Dermoscopy is crucial to diagnosis. The most frequent dermoscopic findings include a polymorphic vascular pattern such as dotted and linear irregular vessels over a pink background, white lines, white dots, white structureless areas, and red-purple lacunae.2,3 Histopathology will demonstrate a vascular proliferation with plump epithelioid endothelial cells showing abundant eosinophilic cytoplasm, accompanied by a variable lymphocytic and eosinophilic inflammatory infiltrate (Figure 1).1
In our case, dermoscopic-histopathologic correlation suggested that the polymorphic vascular pattern and clods on a pink background corresponded to thin- and thick-walled vessels containing plump endothelial cells and intraluminal erythrocytes within the superficial and deep dermis. White structures could represent underlying fibrosis and altered dermal collagen due to vascular proliferation. The brown pigment network and peripheral brownish pigmentation were most likely secondary to increased melanin and accentuation of the pigment network in the setting of Fitzpatrick skin types IV to V, although pruritic trauma with postinflammatory hyperpigmentation may also have contributed, making dermoscopic-histopathologic correlation challenging.
Surgical excision is considered the primary treatment modality for ALHE, with the lowest recurrence rates.1 Alternative therapeutic options include intralesional steroids, cryotherapy, sclerotherapy, radiofrequency, pulsed dye laser, and carbon dioxide laser, with varying efficacy reported.1 Our patient was treated with a combination of a long-pulse Nd:YAG laser (pulse width of 30 ms) to target the vascular component, followed by a single session with an ablative Er:YAG laser. After 4 weeks, healing with good cosmetic results was observed (Figure 2). At 6-month follow-up, there was no recurrence of the lesions.
Kimura disease, often considered the closest differential diagnosis for ALHE, is a rare lymphoproliferative fibroinflammatory condition. Patients present with subcutaneous nodules on the head and neck, often associated with lymphadenopathy. Elevated serum IgE levels and peripheral blood eosinophilia are common.1 Another consideration in the differential diagnosis is cutaneous bacillary angiomatosis caused by Bartonella species, a vascular proliferative condition that mostly affects individuals with HIV, transplant recipients, and those taking immunosuppressive medications.4 Pyogenic granuloma, also known as lobular capillary haemangioma, is another benign vascular proliferation that resembles ALHE. Clinically, it manifests as a solitary, painless, flesh-colored to erythematous papulonodule; however, multiple grouped lesions also can occur. The lesions often are associated with bleeding and erosions.5 Epithelioid hemangioendothelioma is a rare vascular tumor most frequently manifesting in the liver, lungs, or bones, and very rarely is limited to skin. Cutaneous epithelioid hemangioendothelioma mimics ALHE and may manifest as a solitary erythematous mass, multiple dome-shaped masses, or dermal nodules.6
- Brahs A, Sledge B, Mullen H, et al. Angiolymphoid hyperplasia with eosinophilia: many syllables, many unanswered questions. J Clin Aesthet Dermatol. 2021;14:49-54.
- Kalantri M, Khopkar U. Spectrum of dermoscopic pattern in a patient with angiolymphoid hyperplasia with tissue eosinophilia. Indian J Dermatol. 2020;65:556-558.
- Chauhan P, Vinay K, Jindal R, et al. Dermoscopic characterisation of angiolymphoid hyperplasia in skin of colour: a case series of six patients with review of literature. Indian J Dermatol Venereol Leprol. 2024;90:848.
- Ramírez Ramírez CR, Saavedra S, Ramírez Ronda CH. Bacillary angiomatosis: microbiology, histopathology, clinical presentation, diagnosis and management. Bol Asoc Med PR. 1996;88:46-51.
- Leung AKC, Barankin B, Hon KL. Pyogenic granuloma. Clinics Mother Child Health. 2014;11:E106. doi:10.4172/2090-7214.1000e106
- Kumar V, Kachhawa D, Rekha S, et al. Cutaneous epithelioid hemangioendothelioma: a rare presentation. Indian J Dermatol Venereol Leprol. 2018;84:739-742.
- Brahs A, Sledge B, Mullen H, et al. Angiolymphoid hyperplasia with eosinophilia: many syllables, many unanswered questions. J Clin Aesthet Dermatol. 2021;14:49-54.
- Kalantri M, Khopkar U. Spectrum of dermoscopic pattern in a patient with angiolymphoid hyperplasia with tissue eosinophilia. Indian J Dermatol. 2020;65:556-558.
- Chauhan P, Vinay K, Jindal R, et al. Dermoscopic characterisation of angiolymphoid hyperplasia in skin of colour: a case series of six patients with review of literature. Indian J Dermatol Venereol Leprol. 2024;90:848.
- Ramírez Ramírez CR, Saavedra S, Ramírez Ronda CH. Bacillary angiomatosis: microbiology, histopathology, clinical presentation, diagnosis and management. Bol Asoc Med PR. 1996;88:46-51.
- Leung AKC, Barankin B, Hon KL. Pyogenic granuloma. Clinics Mother Child Health. 2014;11:E106. doi:10.4172/2090-7214.1000e106
- Kumar V, Kachhawa D, Rekha S, et al. Cutaneous epithelioid hemangioendothelioma: a rare presentation. Indian J Dermatol Venereol Leprol. 2018;84:739-742.
Multiple Grouped Erythematous to Violaceous Preauricular Papules
Multiple Grouped Erythematous to Violaceous Preauricular Papules
A 35-year-old woman presented with an insidious onset of multiple grouped erythematous to violaceous papules over the left preauricular area of 3 months’ duration (top quiz image). The lesions were soft, itchy, nontender, and friable and were associated with bleeding on excoriation and preauricular lymphadenopathy. Serology for HIV was nonreactive, and Gram staining revealed no bacilli. Laboratory assessment including a complete blood count, urinalysis, and liver and renal function tests was normal.
On dermoscopy (middle quiz image), multiple linear and dotted vessels (circle), reddish lacunae (clods), hemorrhagic crusting (blue arrow), white scaling (black arrow), a brown pigment network (square), white structureless areas (yellow arrow), and white lines were seen over a pale-pink background (green arrow). Scaling and crusting over some lesions, along with a peripheral rim of scaling and brownish pigmentation, also was appreciated. Histopathology revealed a proliferation of vascular channels admixed with lymphocytes, plasma cells, and eosinophils along with a proliferation of thin- and thick-walled blood vessels in the superficial as well as deep dermis (bottom quiz image).
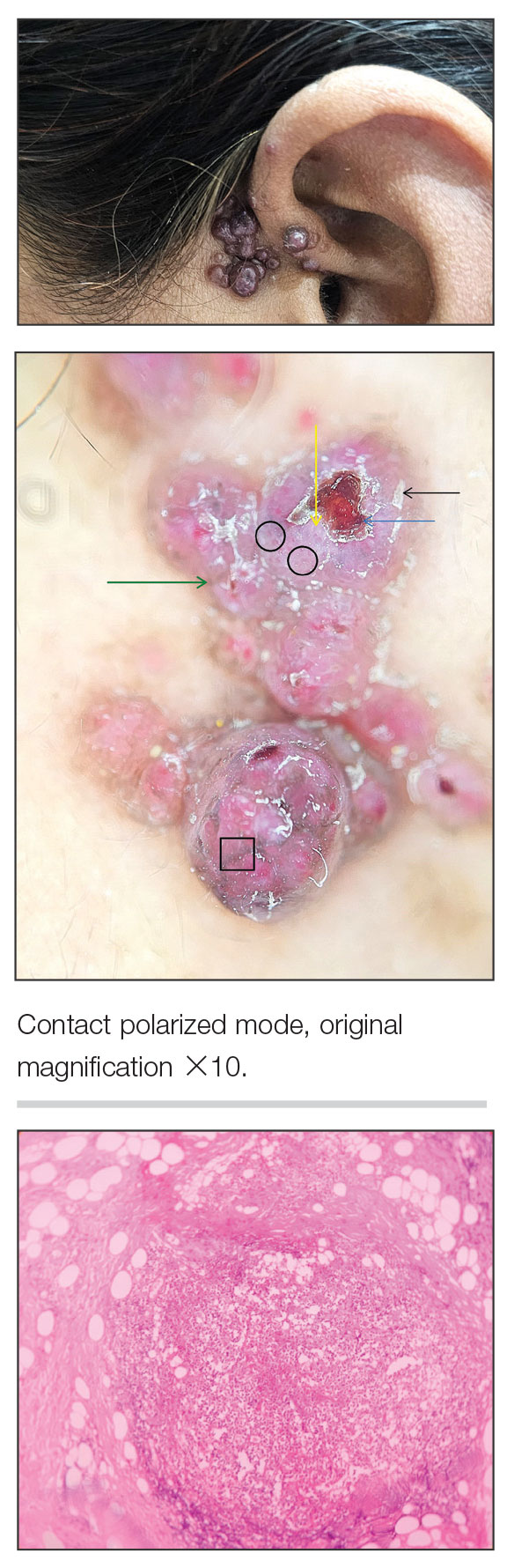

Horse Flies: Identification, Bite Reactions, and Clinical Management
Horse Flies: Identification, Bite Reactions, and Clinical Management
Horse flies (Tabanidae) are hematophagous dipteran insects that feed on the blood of their hosts, including humans.1 Their bites can cause minor cutaneous reactions (eg, urticaria) or, rarely, severe reactions such as anaphylaxis. They also are vectors of tularemia, which may manifest with cutaneous ulcers and systemic illness. In this article, we discuss identifying features of horse flies as well as clinical manifestations from bite reactions, symptomatic and emergency management, and strategies for prevention and control.
Morphology and Geographic Distribution
Horse flies, which can grow as large as 30 mm, can be identified by their brown or black bodies and characteristic large heads and proboscises, wing venation, large calypters, pulvilliform empodium between large pulvilli, and lack of bristles on the body.2 Occasionally, their bodies may be gray, yellow, green, or blue, but this is less likely than in the other species of the Tabanidae family. Short hairs are present on the head and thorax. The eyes are large and often patterned, multicolored, and bright, though they also can exhibit shades of dark brown, gray, or black. There is variation in the appearance of male vs female horse flies: females have eyes that are widely spaced apart, while males have eyes that are closer together.2 It is important to note the difference between male and female horseflies, as hematophagy is exhibited only by females.1
Horse flies are found worldwide, with the exception of Hawaii, Greenland, and Iceland.3,4 They are especially prevalent in warm and moist regions, as these conditions are optimal for breeding.3-5 They tend to be active during the day and inactive at night due to a preference for sunlight and warmth.6 Due to this preference, horse flies’ seasonal activity depends on the climate; for many regions, activity persists from summer to early autumn.7
Clinical Manifestations and Treatment
Female horse flies use their mouthparts to pierce the host’s skin, inject saliva, and suck blood. The saliva contains anticoagulant properties. The bites are painful for the host, and various reactions can occur, including large urticarial wheals or papules at the site of the bite. Treatment for these minor cutaneous reactions is largely symptomatic. The bite site should be washed with soap and water; ice can be applied to help reduce inflammation.8 Oral antihistamines may be administered to reduce pruritus and treat urticaria. Topical steroids also can be prescribed for symptomatic relief. Acetaminophen and nonsteroidal anti-inflammatory drugs can be administered for pain control.8
While most cases of horse fly bites are minor, there have been reports of anaphylaxis.9 Horse fly bite–induced anaphylaxis can manifest as generalized itching, urticaria, and angioedema within minutes of being bitten. This may be followed by pharyngeal constriction, shortness of breath, nausea, vomiting, shivers, perspiration, and loss of consciousness.9 Anaphylaxis symptoms should be treated with immediate administration of intramuscular epinephrine.10
Pathogen Transmission, Prevention, and Control
Although horse flies have been found to carry numerous viruses, bacteria, and protozoa that affect other mammals, there is not enough evidence to suggest that they are vectors of transmission for humans for most diseases.11,12 In particular, West Nile virus and Borrelia burgdorferi both have been found in horse flies, but there are no reports of transmission of these diseases to humans through their bites.12
Horse flies, their close cousins deer flies (specifically Chrysops discalis), and ticks are known vectors of Francisella tularensis.13 These bacteria cause tularemia, which can manifest with symptoms such as fever, headache, and malaise. Ulceroglandular tularemia is the most common manifestation, in which the patient develops a cutaneous ulceration at the site of the horse fly bite and exhibits associated tender regional lymphadenopathy.14 Exudative conjunctivitis, exudative pharyngitis, abdominal pain, diarrhea, vomiting, and severe bilateral pneumonia also are common symptoms. The most severe form of tularemia is systemic or typhoidal tularemia, which can manifest with fever, septic shock, and hepatosplenomegaly.14 The current treatment of choice for all forms of tularemia is intravenous gentamicin, with a recommended dosage of 5 mg/kg/d for 7 to 14 days; streptomycin is an acceptable alternative.14-16 Ciprofloxacin is used less commonly and is reserved for milder disease. Incision and drainage of the affected lymph nodes also may be necessary.14 It is important to promptly identify and treat tularemia, as the mortality rate can be as high as 50% for untreated disease, especially in patients with systemic symptoms. Even after treatment, many patients exhibit residual scarring at the site of the ulcer, as well as lung, kidney, and muscle damage.14
It is advised to avoid contact with horse flies due to the range of symptom severity caused by their bites, but avoidance and control can be difficult. Malaise traps, consisting of a tent and polyester netting, can be used to capture the insects.17 Octenol has been shown to be effective for attracting horse flies and can be applied to the trap in order to increase its effectiveness.18 A Manitoba horse fly trap is a modified version of the Malaise trap that contains a suspended dark sphere to further attract horse flies.19 Patients also should be instructed to wear long-sleeved shirts and pants when outdoors in areas with horse flies to avoid contact, and application of DEET (N,N-diethylmeta-toluamide), picaridin, citronella, or geraniol-based repellents also can be effective in reducing exposure.20
Final Thoughts
Horse flies are large, blood‑feeding dipteran insects whose bites usually produce painful local reactions. Although most bites are benign, they rarely can cause anaphylaxis, and certain Tabanidae insects can transmit Francisella tularensis; therefore, clinicians should consider the risk for tularemia infection in patients presenting with horse fly bites and start appropriate antibiotic therapy when indicated. Due to the risks, prevention of bites and reduction of contact with horse flies via protective clothing, repellents, and trapping methods is recommended. Patients should be advised on bite care and to seek urgent care for systemic symptoms or rapidly progressive local signs.
- Lucas M, Krolow TK, Riet-Correa F, et al. Diversity and seasonality of horse flies (Diptera: Tabanidae) in Uruguay. Sci Rep. 2020;10:401.
- Chainey JE. Horse‑flies, deer‑flies and clegs (Tabanidae). In: Lane RP, Crosskey RW, eds. Medical Insects and Arachnids. Springer; 1993:310‑332.
- Downes JA. The post‑glacial colonization of the North Atlantic islands. Memoirs of the Entomological Society of Canada. 1988;120(S144):55‑92.
- Squitier JM. Deer flies, yellow flies and horse flies. Featured Creatures. University of Florida; April 1, 2014. Accessed September 15, 2023.
- Middlekauff WW, Lane RS. Adult and immature Tabanidae (Diptera) of California. University of California Press. 1980:1‑2.
- Horse flies and deer flies. University of Kentucky. Accessed September 15, 2023. https://entomology.mgcafe.uky.edu/ef511
- Hoover J. Horse flies. LSU College of Agriculture. May 28, 2020. Accessed May 20, 2026. https://www.lsuagcenter.com/profiles/jhoover/articles/page1590683239678
- Powers J, Syed HA, McDowell RH. Insect bites. StatPearls [Internet]. Updated February 15, 2026. Accessed May 12, 2026. https://www.ncbi.nlm.nih.gov/books/NBK537235/
- Hemmer W, Focke M, Vieluf D, et al. Anaphylaxis induced by horsefly bites: identification of a 69 kd IgE-binding salivary gland protein from Chrysops spp. (Diptera, Tabanidae) by Western blot analysis. J Allergy Clin Immunol. 1998;101:134-136.
- McLendon K, Sternard BT. Anaphylaxis. StatPearls [Internet]. Updated January 26, 2023. Accessed May 12, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482124/
- Cheng TC. General Parasitology. Elsevier Science; 2012:660.
- Purdue Medical Entomology. Horse and deer flies. Purdue University. Accessed April 28, 2026. https://extension.entm.purdue.edu/publichealth/diseases/tabanid.html
- US Geological Survey. Tularemia. USGS Publications Warehouse. Accessed April 28, 2026. https://pubs.usgs.gov/circ/1297/report.pdf
- Snowden J, Simonsen KA. Tularemia. StatPearls [Internet]. Updated July 17, 2023. Accessed May 12, 2026. https://www.ncbi.nlm.nih.gov/books/NBK430905/
- Enderlin G, Morales L, Jacobs RF, et al. Streptomycin and alternative agents for the treatment of tularemia: review of the literature. Clin Infect Dis. 1994;19:42-47.
- Balestra A, Bytyci H, Guillod C, et al. A case of ulceroglandular tularemia presenting with lymphadenopathy and an ulcer on a linear morphoea lesion surrounded by erysipelas. Int Med Case Rep J. 2018;11:313-318.
- Malaise R. A new insect‑trap. Entomologisk Tidskrift. 1937;58:148‑160.
- French F, Kline D. l-Octen-3-ol, an effective attractant for Tabanidae (Diptera). J Med Entomol. 1989;26:459-461
- Axtell RC, Edwards TD, Dukes JC. Rigid canopy trap for Tabanidae (Diptera). J Georgia Entomol Soc. 1975;10: 64-67.
- Squitier JM. Deer flies, yellow flies and horse flies. Featured Creatures. University of Florida. April 1, 2014. Accessed May 12, 2026. https://ask.ifas.ufl.edu/publication/IN155
Horse flies (Tabanidae) are hematophagous dipteran insects that feed on the blood of their hosts, including humans.1 Their bites can cause minor cutaneous reactions (eg, urticaria) or, rarely, severe reactions such as anaphylaxis. They also are vectors of tularemia, which may manifest with cutaneous ulcers and systemic illness. In this article, we discuss identifying features of horse flies as well as clinical manifestations from bite reactions, symptomatic and emergency management, and strategies for prevention and control.
Morphology and Geographic Distribution
Horse flies, which can grow as large as 30 mm, can be identified by their brown or black bodies and characteristic large heads and proboscises, wing venation, large calypters, pulvilliform empodium between large pulvilli, and lack of bristles on the body.2 Occasionally, their bodies may be gray, yellow, green, or blue, but this is less likely than in the other species of the Tabanidae family. Short hairs are present on the head and thorax. The eyes are large and often patterned, multicolored, and bright, though they also can exhibit shades of dark brown, gray, or black. There is variation in the appearance of male vs female horse flies: females have eyes that are widely spaced apart, while males have eyes that are closer together.2 It is important to note the difference between male and female horseflies, as hematophagy is exhibited only by females.1
Horse flies are found worldwide, with the exception of Hawaii, Greenland, and Iceland.3,4 They are especially prevalent in warm and moist regions, as these conditions are optimal for breeding.3-5 They tend to be active during the day and inactive at night due to a preference for sunlight and warmth.6 Due to this preference, horse flies’ seasonal activity depends on the climate; for many regions, activity persists from summer to early autumn.7
Clinical Manifestations and Treatment
Female horse flies use their mouthparts to pierce the host’s skin, inject saliva, and suck blood. The saliva contains anticoagulant properties. The bites are painful for the host, and various reactions can occur, including large urticarial wheals or papules at the site of the bite. Treatment for these minor cutaneous reactions is largely symptomatic. The bite site should be washed with soap and water; ice can be applied to help reduce inflammation.8 Oral antihistamines may be administered to reduce pruritus and treat urticaria. Topical steroids also can be prescribed for symptomatic relief. Acetaminophen and nonsteroidal anti-inflammatory drugs can be administered for pain control.8
While most cases of horse fly bites are minor, there have been reports of anaphylaxis.9 Horse fly bite–induced anaphylaxis can manifest as generalized itching, urticaria, and angioedema within minutes of being bitten. This may be followed by pharyngeal constriction, shortness of breath, nausea, vomiting, shivers, perspiration, and loss of consciousness.9 Anaphylaxis symptoms should be treated with immediate administration of intramuscular epinephrine.10
Pathogen Transmission, Prevention, and Control
Although horse flies have been found to carry numerous viruses, bacteria, and protozoa that affect other mammals, there is not enough evidence to suggest that they are vectors of transmission for humans for most diseases.11,12 In particular, West Nile virus and Borrelia burgdorferi both have been found in horse flies, but there are no reports of transmission of these diseases to humans through their bites.12
Horse flies, their close cousins deer flies (specifically Chrysops discalis), and ticks are known vectors of Francisella tularensis.13 These bacteria cause tularemia, which can manifest with symptoms such as fever, headache, and malaise. Ulceroglandular tularemia is the most common manifestation, in which the patient develops a cutaneous ulceration at the site of the horse fly bite and exhibits associated tender regional lymphadenopathy.14 Exudative conjunctivitis, exudative pharyngitis, abdominal pain, diarrhea, vomiting, and severe bilateral pneumonia also are common symptoms. The most severe form of tularemia is systemic or typhoidal tularemia, which can manifest with fever, septic shock, and hepatosplenomegaly.14 The current treatment of choice for all forms of tularemia is intravenous gentamicin, with a recommended dosage of 5 mg/kg/d for 7 to 14 days; streptomycin is an acceptable alternative.14-16 Ciprofloxacin is used less commonly and is reserved for milder disease. Incision and drainage of the affected lymph nodes also may be necessary.14 It is important to promptly identify and treat tularemia, as the mortality rate can be as high as 50% for untreated disease, especially in patients with systemic symptoms. Even after treatment, many patients exhibit residual scarring at the site of the ulcer, as well as lung, kidney, and muscle damage.14
It is advised to avoid contact with horse flies due to the range of symptom severity caused by their bites, but avoidance and control can be difficult. Malaise traps, consisting of a tent and polyester netting, can be used to capture the insects.17 Octenol has been shown to be effective for attracting horse flies and can be applied to the trap in order to increase its effectiveness.18 A Manitoba horse fly trap is a modified version of the Malaise trap that contains a suspended dark sphere to further attract horse flies.19 Patients also should be instructed to wear long-sleeved shirts and pants when outdoors in areas with horse flies to avoid contact, and application of DEET (N,N-diethylmeta-toluamide), picaridin, citronella, or geraniol-based repellents also can be effective in reducing exposure.20
Final Thoughts
Horse flies are large, blood‑feeding dipteran insects whose bites usually produce painful local reactions. Although most bites are benign, they rarely can cause anaphylaxis, and certain Tabanidae insects can transmit Francisella tularensis; therefore, clinicians should consider the risk for tularemia infection in patients presenting with horse fly bites and start appropriate antibiotic therapy when indicated. Due to the risks, prevention of bites and reduction of contact with horse flies via protective clothing, repellents, and trapping methods is recommended. Patients should be advised on bite care and to seek urgent care for systemic symptoms or rapidly progressive local signs.
Horse flies (Tabanidae) are hematophagous dipteran insects that feed on the blood of their hosts, including humans.1 Their bites can cause minor cutaneous reactions (eg, urticaria) or, rarely, severe reactions such as anaphylaxis. They also are vectors of tularemia, which may manifest with cutaneous ulcers and systemic illness. In this article, we discuss identifying features of horse flies as well as clinical manifestations from bite reactions, symptomatic and emergency management, and strategies for prevention and control.
Morphology and Geographic Distribution
Horse flies, which can grow as large as 30 mm, can be identified by their brown or black bodies and characteristic large heads and proboscises, wing venation, large calypters, pulvilliform empodium between large pulvilli, and lack of bristles on the body.2 Occasionally, their bodies may be gray, yellow, green, or blue, but this is less likely than in the other species of the Tabanidae family. Short hairs are present on the head and thorax. The eyes are large and often patterned, multicolored, and bright, though they also can exhibit shades of dark brown, gray, or black. There is variation in the appearance of male vs female horse flies: females have eyes that are widely spaced apart, while males have eyes that are closer together.2 It is important to note the difference between male and female horseflies, as hematophagy is exhibited only by females.1
Horse flies are found worldwide, with the exception of Hawaii, Greenland, and Iceland.3,4 They are especially prevalent in warm and moist regions, as these conditions are optimal for breeding.3-5 They tend to be active during the day and inactive at night due to a preference for sunlight and warmth.6 Due to this preference, horse flies’ seasonal activity depends on the climate; for many regions, activity persists from summer to early autumn.7
Clinical Manifestations and Treatment
Female horse flies use their mouthparts to pierce the host’s skin, inject saliva, and suck blood. The saliva contains anticoagulant properties. The bites are painful for the host, and various reactions can occur, including large urticarial wheals or papules at the site of the bite. Treatment for these minor cutaneous reactions is largely symptomatic. The bite site should be washed with soap and water; ice can be applied to help reduce inflammation.8 Oral antihistamines may be administered to reduce pruritus and treat urticaria. Topical steroids also can be prescribed for symptomatic relief. Acetaminophen and nonsteroidal anti-inflammatory drugs can be administered for pain control.8
While most cases of horse fly bites are minor, there have been reports of anaphylaxis.9 Horse fly bite–induced anaphylaxis can manifest as generalized itching, urticaria, and angioedema within minutes of being bitten. This may be followed by pharyngeal constriction, shortness of breath, nausea, vomiting, shivers, perspiration, and loss of consciousness.9 Anaphylaxis symptoms should be treated with immediate administration of intramuscular epinephrine.10
Pathogen Transmission, Prevention, and Control
Although horse flies have been found to carry numerous viruses, bacteria, and protozoa that affect other mammals, there is not enough evidence to suggest that they are vectors of transmission for humans for most diseases.11,12 In particular, West Nile virus and Borrelia burgdorferi both have been found in horse flies, but there are no reports of transmission of these diseases to humans through their bites.12
Horse flies, their close cousins deer flies (specifically Chrysops discalis), and ticks are known vectors of Francisella tularensis.13 These bacteria cause tularemia, which can manifest with symptoms such as fever, headache, and malaise. Ulceroglandular tularemia is the most common manifestation, in which the patient develops a cutaneous ulceration at the site of the horse fly bite and exhibits associated tender regional lymphadenopathy.14 Exudative conjunctivitis, exudative pharyngitis, abdominal pain, diarrhea, vomiting, and severe bilateral pneumonia also are common symptoms. The most severe form of tularemia is systemic or typhoidal tularemia, which can manifest with fever, septic shock, and hepatosplenomegaly.14 The current treatment of choice for all forms of tularemia is intravenous gentamicin, with a recommended dosage of 5 mg/kg/d for 7 to 14 days; streptomycin is an acceptable alternative.14-16 Ciprofloxacin is used less commonly and is reserved for milder disease. Incision and drainage of the affected lymph nodes also may be necessary.14 It is important to promptly identify and treat tularemia, as the mortality rate can be as high as 50% for untreated disease, especially in patients with systemic symptoms. Even after treatment, many patients exhibit residual scarring at the site of the ulcer, as well as lung, kidney, and muscle damage.14
It is advised to avoid contact with horse flies due to the range of symptom severity caused by their bites, but avoidance and control can be difficult. Malaise traps, consisting of a tent and polyester netting, can be used to capture the insects.17 Octenol has been shown to be effective for attracting horse flies and can be applied to the trap in order to increase its effectiveness.18 A Manitoba horse fly trap is a modified version of the Malaise trap that contains a suspended dark sphere to further attract horse flies.19 Patients also should be instructed to wear long-sleeved shirts and pants when outdoors in areas with horse flies to avoid contact, and application of DEET (N,N-diethylmeta-toluamide), picaridin, citronella, or geraniol-based repellents also can be effective in reducing exposure.20
Final Thoughts
Horse flies are large, blood‑feeding dipteran insects whose bites usually produce painful local reactions. Although most bites are benign, they rarely can cause anaphylaxis, and certain Tabanidae insects can transmit Francisella tularensis; therefore, clinicians should consider the risk for tularemia infection in patients presenting with horse fly bites and start appropriate antibiotic therapy when indicated. Due to the risks, prevention of bites and reduction of contact with horse flies via protective clothing, repellents, and trapping methods is recommended. Patients should be advised on bite care and to seek urgent care for systemic symptoms or rapidly progressive local signs.
- Lucas M, Krolow TK, Riet-Correa F, et al. Diversity and seasonality of horse flies (Diptera: Tabanidae) in Uruguay. Sci Rep. 2020;10:401.
- Chainey JE. Horse‑flies, deer‑flies and clegs (Tabanidae). In: Lane RP, Crosskey RW, eds. Medical Insects and Arachnids. Springer; 1993:310‑332.
- Downes JA. The post‑glacial colonization of the North Atlantic islands. Memoirs of the Entomological Society of Canada. 1988;120(S144):55‑92.
- Squitier JM. Deer flies, yellow flies and horse flies. Featured Creatures. University of Florida; April 1, 2014. Accessed September 15, 2023.
- Middlekauff WW, Lane RS. Adult and immature Tabanidae (Diptera) of California. University of California Press. 1980:1‑2.
- Horse flies and deer flies. University of Kentucky. Accessed September 15, 2023. https://entomology.mgcafe.uky.edu/ef511
- Hoover J. Horse flies. LSU College of Agriculture. May 28, 2020. Accessed May 20, 2026. https://www.lsuagcenter.com/profiles/jhoover/articles/page1590683239678
- Powers J, Syed HA, McDowell RH. Insect bites. StatPearls [Internet]. Updated February 15, 2026. Accessed May 12, 2026. https://www.ncbi.nlm.nih.gov/books/NBK537235/
- Hemmer W, Focke M, Vieluf D, et al. Anaphylaxis induced by horsefly bites: identification of a 69 kd IgE-binding salivary gland protein from Chrysops spp. (Diptera, Tabanidae) by Western blot analysis. J Allergy Clin Immunol. 1998;101:134-136.
- McLendon K, Sternard BT. Anaphylaxis. StatPearls [Internet]. Updated January 26, 2023. Accessed May 12, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482124/
- Cheng TC. General Parasitology. Elsevier Science; 2012:660.
- Purdue Medical Entomology. Horse and deer flies. Purdue University. Accessed April 28, 2026. https://extension.entm.purdue.edu/publichealth/diseases/tabanid.html
- US Geological Survey. Tularemia. USGS Publications Warehouse. Accessed April 28, 2026. https://pubs.usgs.gov/circ/1297/report.pdf
- Snowden J, Simonsen KA. Tularemia. StatPearls [Internet]. Updated July 17, 2023. Accessed May 12, 2026. https://www.ncbi.nlm.nih.gov/books/NBK430905/
- Enderlin G, Morales L, Jacobs RF, et al. Streptomycin and alternative agents for the treatment of tularemia: review of the literature. Clin Infect Dis. 1994;19:42-47.
- Balestra A, Bytyci H, Guillod C, et al. A case of ulceroglandular tularemia presenting with lymphadenopathy and an ulcer on a linear morphoea lesion surrounded by erysipelas. Int Med Case Rep J. 2018;11:313-318.
- Malaise R. A new insect‑trap. Entomologisk Tidskrift. 1937;58:148‑160.
- French F, Kline D. l-Octen-3-ol, an effective attractant for Tabanidae (Diptera). J Med Entomol. 1989;26:459-461
- Axtell RC, Edwards TD, Dukes JC. Rigid canopy trap for Tabanidae (Diptera). J Georgia Entomol Soc. 1975;10: 64-67.
- Squitier JM. Deer flies, yellow flies and horse flies. Featured Creatures. University of Florida. April 1, 2014. Accessed May 12, 2026. https://ask.ifas.ufl.edu/publication/IN155
- Lucas M, Krolow TK, Riet-Correa F, et al. Diversity and seasonality of horse flies (Diptera: Tabanidae) in Uruguay. Sci Rep. 2020;10:401.
- Chainey JE. Horse‑flies, deer‑flies and clegs (Tabanidae). In: Lane RP, Crosskey RW, eds. Medical Insects and Arachnids. Springer; 1993:310‑332.
- Downes JA. The post‑glacial colonization of the North Atlantic islands. Memoirs of the Entomological Society of Canada. 1988;120(S144):55‑92.
- Squitier JM. Deer flies, yellow flies and horse flies. Featured Creatures. University of Florida; April 1, 2014. Accessed September 15, 2023.
- Middlekauff WW, Lane RS. Adult and immature Tabanidae (Diptera) of California. University of California Press. 1980:1‑2.
- Horse flies and deer flies. University of Kentucky. Accessed September 15, 2023. https://entomology.mgcafe.uky.edu/ef511
- Hoover J. Horse flies. LSU College of Agriculture. May 28, 2020. Accessed May 20, 2026. https://www.lsuagcenter.com/profiles/jhoover/articles/page1590683239678
- Powers J, Syed HA, McDowell RH. Insect bites. StatPearls [Internet]. Updated February 15, 2026. Accessed May 12, 2026. https://www.ncbi.nlm.nih.gov/books/NBK537235/
- Hemmer W, Focke M, Vieluf D, et al. Anaphylaxis induced by horsefly bites: identification of a 69 kd IgE-binding salivary gland protein from Chrysops spp. (Diptera, Tabanidae) by Western blot analysis. J Allergy Clin Immunol. 1998;101:134-136.
- McLendon K, Sternard BT. Anaphylaxis. StatPearls [Internet]. Updated January 26, 2023. Accessed May 12, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482124/
- Cheng TC. General Parasitology. Elsevier Science; 2012:660.
- Purdue Medical Entomology. Horse and deer flies. Purdue University. Accessed April 28, 2026. https://extension.entm.purdue.edu/publichealth/diseases/tabanid.html
- US Geological Survey. Tularemia. USGS Publications Warehouse. Accessed April 28, 2026. https://pubs.usgs.gov/circ/1297/report.pdf
- Snowden J, Simonsen KA. Tularemia. StatPearls [Internet]. Updated July 17, 2023. Accessed May 12, 2026. https://www.ncbi.nlm.nih.gov/books/NBK430905/
- Enderlin G, Morales L, Jacobs RF, et al. Streptomycin and alternative agents for the treatment of tularemia: review of the literature. Clin Infect Dis. 1994;19:42-47.
- Balestra A, Bytyci H, Guillod C, et al. A case of ulceroglandular tularemia presenting with lymphadenopathy and an ulcer on a linear morphoea lesion surrounded by erysipelas. Int Med Case Rep J. 2018;11:313-318.
- Malaise R. A new insect‑trap. Entomologisk Tidskrift. 1937;58:148‑160.
- French F, Kline D. l-Octen-3-ol, an effective attractant for Tabanidae (Diptera). J Med Entomol. 1989;26:459-461
- Axtell RC, Edwards TD, Dukes JC. Rigid canopy trap for Tabanidae (Diptera). J Georgia Entomol Soc. 1975;10: 64-67.
- Squitier JM. Deer flies, yellow flies and horse flies. Featured Creatures. University of Florida. April 1, 2014. Accessed May 12, 2026. https://ask.ifas.ufl.edu/publication/IN155
Horse Flies: Identification, Bite Reactions, and Clinical Management
Horse Flies: Identification, Bite Reactions, and Clinical Management
PRACTICE POINTS
- Horse flies (Tabanidae) are hematophagous insects that can cause minor cutaneous reactions (eg, urticaria) or, rarely, severe reactions such as anaphylaxis. They also are vectors of tularemia, which may manifest with cutaneous ulcers or systemic illness.
- Mild reactions are managed symptomatically; anaphylaxis requires epinephrine, and tularemia requires systemic antibiotics such as gentamicin.
- Patients should be counseled on avoidance strategies, including wearing protective clothing and using topical repellents and environmental traps.

Getting a Grip on Occupational Hand Dermatitis: Key Considerations for Evaluation and Management
Getting a Grip on Occupational Hand Dermatitis: Key Considerations for Evaluation and Management
Hand dermatitis (HD) is a common dermatologic concern that can impair quality of life, work productivity, and daily functioning.1 Occupational HD is defined as hand eczema caused or worsened by workplace exposures. When caused by work, HD may lead to reduced productivity and even job loss. Subtypes of HD include irritant contact dermatitis (ICD), allergic contact dermatitis (ACD), protein contact dermatitis (PCD), atopic dermatitis (AD), hyperkeratotic HD, and dyshidrotic eczema.2,3
Often caused by wet work, ICD is the most common subtype, whereas PCD—which is caused by immediate hypersensitivity to protein—is less common and usually seen in food service workers.3,4 When HD does not improve with standard treatment, particularly in occupational cases, patch testing is prudent to evaluate for contact allergens. In this article, we review practical considerations for evaluation and management of occupational irritant and allergic HD, highlighting relevant exposures and pearls on workup and management.
Epidemiology of Hand Dermatitis
A 2021 systematic review and meta-analysis of European studies reported a 1-year HD prevalence of 9.1% and a lifetime prevalence of 14.5%.5 Hand dermatitis is most common in women; individuals aged 30 to 39 years; and those who are employed, underscoring the role of workplace exposure.6 High-risk occupations are those involving substantial wet work, such as hairdressers, beauticians, cleaners, and health care and construction workers.7 Individuals with a history of AD also are at high risk for HD.8
Hand Dermatitis Subtypes
Irritant Contact Dermatitis—Irritant contact dermatitis, the most common form of occupational HD, is caused by repeated exposure to irritants (eg, water, detergents, cleansers, and soaps) that disrupt the skin barrier.9 Occupations that involve wet work are a major risk factor, associated with a 56% higher likelihood of ICD.8 Wet work involves frequent handwashing, prolonged contact with liquids, or occlusive glove use.9 As a ubiquitous skin irritant, water can penetrate the stratum corneum, impair the skin barrier, and increase sensitization risk. The dorsal hands usually are affected by ICD due to the thinner stratum corneum in this area.9
Allergic Contact Dermatitis—Allergic contact dermatitis should be considered in cases of chronic, recurrent, or treatment-resistant disease. Clinical clues include dermatitis beyond irritant contact sites, recurrent pruritic and vesicular HD, and flares with occupational exposures or materials; however, it can be difficult to distinguish ACD from ICD on clinical presentation alone, as they have many overlapping features. When ACD is suspected, patch testing remains the gold standard for identifying allergens and guiding avoidance strategies, product alternatives, and workplace modifications.
Unique Occupational Considerations
Hairdressers—Hairdressers have an increased risk for HD due to wet work and exposure to sensitizers, with a pooled lifetime prevalence of 38.2% (including ICD, ACD, and occupational cases).10 Notably, frequent shampooing, rinsing, cutting wet hair, handwashing, and glove use increase the risk for ICD. Hairdressers also are exposed to allergens in hair products, including p-phenylenediamine, toluene-2,5-diamine, persulfate salts, glyceryl thioglycolate, preservatives, and fragrances. Occupational exposure to the preservative methylisothiazolinone is high among hairdressers, with a sensitization rate of 10.5% in HD cases.11
It has been reported that hyperkeratotic fissured eczema of the dorsal hands caused by wet work often indicates ICD, whereas pruritic dyshidrotic eczema involving the lateral fingers or palms suggests ACD; however, these conditions can share overlapping features.7 If ACD is suspected, broad patch testing with baseline and hairdresser series, along with specific chemicals that may be encountered in the workplace, is necessary. Management includes allergen avoidance, reduced wet work tasks, use of nitrile gloves with glove changes to mitigate occlusive effects, and skin barrier protection with emollients.
Health Care Workers—Health care workers are vulnerable to HD due to intensive hand hygiene, prolonged glove use, and allergen exposures, with a lifetime prevalence of self-reported HD of 33.4%.12 Common allergens among health care workers include rubber accelerators, most often from rubber gloves.13 Frequent handwashing and glove use can further impair the skin barrier, increasing irritant and sensitization risks.14 In contrast, alcohol-based hand sanitizers containing emollients are less irritating, with prior analyses showing no significant association with HD risk.15,16 Conversely, handwashing 8 to 10 times daily increased HD risk, with a relative risk of 1.51.15
Surgeons and proceduralists face unique risks for HD from preoperative scrubbing with products that can contain potential allergens such as chlorhexidine gluconate, chloroxylenol, povidone-iodine, fragrance, cocamide diethanolamine, lanolin, alkyl glucosides, sodium benzoate, sorbic acid, tocopherol, and propylene glycol.17,18 Subsequent occlusion under glove layers drives ICD and ACD risks, highlighting the importance of patch testing in affected individuals. While patch testing, exposure avoidance, and limited glove use can mitigate HD risk, frequent handwashing can contribute to refractory HD.
Food Service Workers—Food service workers have an increased risk for HD from allergens and irritants. In a retrospective study of patients with occupational food-related HD (N=372), 57% were diagnosed with ICD, 22% with PCD, and 1.8% with ACD.19 Skin barrier disruption from wet work, occlusion from glove use, and contact with food proteins increase HD risk, especially in bakers exposed to flours and grains, which can cause IgE–mediated PCD manifesting with contact urticaria. Protein contact dermatitis is confirmed by prick testing with suspected foods.20 Additionally, exposure to garlic can cause ICD and ACD due to sulfur-containing compounds, particularly allicin and diallyl disulfide.21,22 Pineapple also can trigger ICD associated with bromelain, a proteolytic enzyme that can break down the skin.23 Nickel exposure is another concern, as steel utensils and cookware can release nickel onto the skin of sensitized individuals.24 Rubber accelerator exposure from gloves also contributes to contact allergy and HD among food service workers; vinyl gloves usually are a good alternative in this setting.25 Management of food-related HD involves exposure avoidance, which may affect occupational viability
Construction Workers—Construction workers are at risk for occupational HD due to contact with irritants and sensitizers such as paints, adhesives, asphalt, cement, solvents, and gloves.26 A retrospective analysis of North American Contact Dermatitis Group data identified HD in 37.2% (253/681) of patch-tested construction workers. The most common occupational allergens include potassium dichromate, which can be present in cement and leather items; bisphenol A epoxy resin; cobalt chloride hexahydrate; and the rubber accelerators carba mix and thiuram mix.26 A thorough occupational history should assess materials handled, and patch testing should include common construction-related allergens to inform avoidance strategies. Workplace task modification can reduce exposure, as certain managerial roles in construction work may involve less contact with irritants and sensitizers.26
Nail Technicians—Nail technicians are at risk for HD, especially ACD from acrylate monomers used in nail gels, dips, and acrylics. In a 10-year analysis, around 87.5% (14/16) of nail technicians with contact allergy to methacrylate demonstrated hand involvement.27 Common acrylate monomers include 2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate, and ethyl cyanoacrylate. 28 Evaluation requires a detailed occupational history, assessing HD onset relative to exposure, services performed, glove-use practices, and whether symptoms improve away from work. While gloves may appear to reduce exposure, a glove-penetration study showed that acrylate-containing nail products can penetrate commonly used disposable gloves from within seconds to approximately 20 minutes, depending on glove and product type.29 Among available options, nitrile gloves may provide dexterity and allergen avoidance when acrylate exposure is brief, with glove changes required every 15 to 30 minutes.30 Patch testing with 2-hydroxyethyl methacrylate and ethyl cyanoacrylate can identify nail acrylate allergy; however, avoidance can be challenging for nail technicians, as these products often are ubiquitous in their work.
Florists—Florists can develop HD from plant allergens and irritants, particularly tulipalin A and calcium oxalate, with a lifetime prevalence of 19.6%.31 Tulipalin A is a well-documented sensitizer causing ACD among florists exposed to tulip bulbs and other Alstroemeria flowers.32 The term tulip fingers actually was coined to describe ACD caused by tulip bulbs in the European tulip industry.33 Patch testing involves testing for tulipalin A, which may be commercially limited, or tulip plant materials; however, fresh tulips require open testing with small amounts due to higher allergen concentration.32 Additionally, the term daffodil itch describes a type of ICD caused by calcium oxalate crystals in daffodil bulbs and tulip sap.32,34 Diagnosis of plant-related HD requires an occupational history and targeted patch testing, while glove protection and exposure avoidance are essential for improvement.
Evaluation and Management
The workup for HD involves physical examination and medical history, including disease onset, course, and history of AD, along with occupational and exposure history to identify allergens and irritants. Understanding the patient’s tasks and responsibilities and workplace practices along with the materials they handle allows the dermatologist to anticipate relevant allergens for patch testing.
Patch testing should be comprehensive, as baseline screening series alone may miss between 26.3% and 50% of occupationally relevant allergens.35,36 Comprehensive patch testing also should include specialty series and supplemental allergens based on the patient’s clinical history and exposures. Specialty series may include hairdressing, bakery, cosmetics, dental, machinists, and adhesives.37 Gloves also warrant attention, as they may be overlooked as a sensitizer following repeated contact and occlusion. In persistent HD associated with glove use, patch testing should include a rubber accelerator series with relevant allergens, such as thiurams, carbamates, mercaptobenzothiazole, diphenylguanidine, and the patient’s own gloves.38 Latex allergy also should be considered, particularly in immediate-type reactions, and can be evaluated with latex-specific IgE testing.39
Management of HD relies on accurate diagnosis and allergen avoidance, which can be challenging in occupational settings. Structured tools, such as the American Contact Dermatitis Society’s Contact Allergen Management Program (https://www.contactderm.org/ resources/acds-camp), can help identify safe alternatives.
In occupational HD, risk assessment should identify occupational exposures and determine appropriate personal protective equipment while minimizing the risk for HD associated with such equipment. Protective gloves are advised to prevent contact with allergens and irritants. When glove use lasts more than 10 minutes, cotton glove liners may be worn to avoid occlusion and moisture retention.40 For wet work, vinyl gloves are recommended, with regular emollient use to support skin-barrier repair. Overall, gloves should be used when possible, changed regularly, and worn for limited periods of time to prevent ICD.
Work modification may be required to reduce exposure and flares, including task reassignment or substitution of materials containing allergens and irritants. Occupational HD may necessitate workplace accommodations, disability evaluation, medical leave, or even permanent job change. Dermatologists play a crucial role in the medical determination of work relatedness and functional impairment, guiding patients through occupational health, disability, and workers’ compensation when warranted.
Treatments for Occupational HD
Treatment of occupational HD depends on disease severity, chronicity, and avoidance of allergens and irritants in ACD, ICD, and PCD. Foundational management includes regular emollient use, which can even serve as monotherapy in mild occupational HD.40 Corticosteroids are the cornerstone of topical therapy, while calcineurin inhibitors can be used as a steroid-sparing option in milder disease.41 Off-label topical calcipotriol and AD-approved therapies crisaborole and ruxolitinib may be effective. For refractory disease after topical treatments, phototherapy can be considered.40 Biologic and targeted therapies also have emerged as potential treatments. Dupilumab is effective for atopic chronic HD and has demonstrated promise for nonatopic chronic HD.42 Recently, delgocitinib, a topical pan–Janus kinase inhibitor cream, showed clinical efficacy for chronic hand eczema and was approved by the US Food and Drug Administration.43 Off-label use of alternative systemic therapies, including acitretin, cyclosporine, methotrexate, and azathioprine, and other biologics and systemic Janus kinase inhibitors also may treat HD, but larger studies are lacking.40
Our Final Interpretation
Occupational HD is a common skin condition with multiple etiologies. It is important for clinicians to gather a thorough occupational and exposure history to narrow the differential diagnosis, inform patch testing, and guide effective management. In practice, successful treatment depends on screening for and diagnosis of workplace exposures driving disease.
- Agner T, Andersen KE, Brandao FM, et al. Hand eczema severity and quality of life: a cross-sectional, multicentre study of hand eczema patients. Contact Dermatitis. 2008;59:43-47. doi:10.1111 /j.1600-0536.2008.01362.x
- Agner T, Aalto-Korte K, Andersen KE, et al. Classification of hand eczema. J Eur Acad Dermatol Venereol. 2015;29:2417-2422. doi:10.1111 /jdv.13308
- Bissonnette R, Agner T, Molin S, et al. Hand eczema—part 1: epidemiology, pathogenesis, diagnosis, and work-up. J Am Acad Dermatol. 2025;93:1201-1210. doi:10.1016/j.jaad.2024.09.048
- Barbaud A. Mechanism and diagnosis of protein contact dermatitis. Curr Opin Allergy Clin Immunol. 2020;20:117-121. doi:10.1097/ACI.0000000000000621
- Quaade AS, Simonsen AB, Halling AS, et al. Prevalence, incidence, and severity of hand eczema in the general population - a systematic review and meta-analysis. Contact Dermatitis. 2021;84:361-374. doi:10.1111/cod.13804
- Apfelbacher C, Bewley A, Molin S, et al. Prevalence of chronic hand eczema in adults: a cross-sectional survey of over 60 000 respondents from the general population of Canada, France, Germany, Italy, Spain and the UK. Br J Dermatol. 2025;192:1047-1054. doi:10.1093 /bjd/ljaf020
- Weidinger S, Novak N. Hand eczema. Lancet. 2024;404:2476-2486. doi:10.1016/S0140-6736(24)01810-5
- Schütte MG, Tamminga SJ, de Groene GJ, et al. Work-related and personal risk factors for occupational contact dermatitis: a systematic review of the literature with meta-analysis. Contact Dermatitis. 2023;88:171-187. doi:10.1111/cod.14253
- Behroozy A, Keegel TG. Wet-work exposure: a main risk factor for occupational hand dermatitis. Saf Health Work. 2014;5:175-180. doi:10.1016/j.shaw.2014.08.001
- Havmose MS, Kezic S, Uter W, et al. Prevalence and incidence of hand eczema in hairdressers-a systematic review and meta-analysis of the published literature from 2000-2021. Contact Dermatitis. 2022;86:254-265. doi:10.1111/cod.14048
- Uter W, Hallmann S, Gefeller O, et al. Contact allergy to ingredients of hair cosmetics in female hairdressers and female consumers—an update based on IVDK data 2013–2020. Contact Dermatitis. 2023;89:161-170. doi:10.1111/cod.14363
- Yüksel YT, Symanzik C, Christensen MO, et al. Prevalence and incidence of hand eczema in healthcare workers: a systematic review and meta-analysis. Contact Dermatitis. 2024;90:331-342. doi:10.1111 /cod.14489
- Warshaw EM, Schram SE, Maibach HI, et al. Occupation-related contact dermatitis in North American health care workers referred for patch testing: cross-sectional data, 1998 to 2004. Dermatitis. 2008;19:261-274. doi:10.2310/6620.2008.07059
- Hamnerius N, Svedman C, Bergendorff O, et al. Wet work exposure and hand eczema among healthcare workers: a cross-sectional study. Br J Dermatol. 2018;178:452-461. doi:10.1111 /bjd.15813
- Loh EDW, Yew YW. Hand hygiene and hand eczema: a systematic review and meta-analysis. Contact Dermatitis. 2022;87:303-314. doi:10.1111/cod.14133
- Lotfinejad N, Peters A, Tartari E, et al. Hand hygiene in health care: 20 years of ongoing advances and perspectives. Lancet Infect Dis. 2021;21:e209-e221. doi:10.1016/S1473-3099(21)00383-2
- Schlarbaum JP, Hylwa SA. Allergic contact dermatitis to operating room scrubs and disinfectants. Dermat Contact Atopic Occup Drug. 2019;30:363-370. doi:10.1097/DER.0000000000000525
- Rodriguez-Homs LG, Atwater AR. Allergens in medical hand skin cleansers. Dermat Contact Atopic Occup Drug. 2019;30:336-341. doi:10.1097/DER.0000000000000504
- Vester L, Thyssen JP, Menné T, et al. Occupational food-related hand dermatoses seen over a 10-year period. Contact Dermatitis. 2012;66:264-270. doi:10.1111/j.1600-0536.2011.02048.x
- Pesonen M, Koskela K, Aalto-Korte K. Contact urticaria and protein contact dermatitis in the Finnish Register of Occupational Diseases in a period of 12 years. Contact Dermatitis. 2020;83:1-7. doi:10.1111/cod.13547
- McFadden JP, White JML, Basketter DA, et al. Reduced allergy rates in atopic eczema to contact allergens used in both skin products and foods: atopy and the “hapten-atopy hypothesis.” Contact Dermatitis. 2008;58:156-158. doi:10.1111/j.1600-0536.2007.01291.x
- Kao SH, Hsu CH, Su SN, et al. Identification and immunologic characterization of an allergen, alliin lyase, from garlic (Allium sativum). J Allergy Clin Immunol. 2004;113:161-168. doi:10.1016/j.jaci.2003.10.040
- Reddy VB, Lerner EA. Plant cysteine proteases that evoke itch activate protease-activated receptors. Br J Dermatol. 2010;163:532-535. doi:10.1111/j.1365-2133.2010.09862.x
- Silverberg NB, Pelletier JL, Jacob SE, et al; Section on Dermatology, Section on Allergy and Immunology. Nickel allergic contact dermatitis: identification, treatment, and prevention. Pediatrics. 2020;145:e20200628. doi:10.1542/peds.2020-0628
- Clément A, Ferrier le Bouëdec MC, Crépy MN, et al. Hand eczema in glove-wearing patients. Contact Dermatitis. 2023;89:143-152. doi:10.1111/cod.14357
- Reeder MJ, Idrogo-Lam A, Aravamuthan SR, et al. Occupational contact dermatitis in construction workers: a retrospective analysis of the North American Contact Dermatitis Group Data, 2001-2020. Dermat Contact Atopic Occup Drug. 2024;35:467-475. doi:10.1089/derm.2024.0018
- Fisch A, Hamnerius N, Isaksson M. Dermatitis and occupational (meth)acrylate contact allergy in nail technicians-a 10-year study. Contact Dermatitis. 2019;81:58-60. doi:10.1111/cod.13216
- Atwater AR, Reeder M. Trends in nail services may cause dermatitis: not your mother’s nail polish. Cutis. 2019;103:315-317.
- Suuronen K, Ylinen K, Heikkilä J, et al. Acrylates in artificial nails— results of product analyses and glove penetration studies. Contact Dermatitis. 2024;90:266-272. doi:10.1111/cod.14474
- Morgado F, Batista M, Gonçalo M. Short exposures and glove protection against (meth)acrylates in nail beauticians-thoughts on a rising concern. Contact Dermatitis. 2019;81:62-63. doi:10.1111 /cod.13222
- Paulsen E, Søgaard J, Andersen KE. Occupational dermatitis in Danish gardeners and greenhouse workers (I). prevalence and possible risk factors. Contact Dermatitis. 1997;37:263-270. doi:10.1111/j.1600-0536.1997.tb02462.x
- Fonacier L, Bernstein DI, Pacheco K, et al. Contact dermatitis: a practice parameter–update 2015. J Allergy Clin Immunol Pract. 2015; 3(3 suppl):S1-S39. doi:10.1016/j.jaip.2015.02.009
- Gette MT, Marks JE. Tulip fingers. Arch Dermatol. 1990;126:203-205.
- Bruynzeel DP. Bulb dermatitis. Dermatological problems in the flower bulb industries. Contact Dermatitis. 1997;37:70-77. doi:10.1111/j.1600-0536.1997.tb00042.x
- Nettis E, Marcandrea M, Colanardi MC, et al. Results of standard series patch testing in patients with occupational allergic contact dermatitis. Allergy. 2003;58:1304-1307. doi:10.1046/j.1398-9995.2003.00346.x
- Saripalli YV, Achen F, Belsito DV. The detection of clinically relevant contact allergens using a standard screening tray of twenty-three allergens. J Am Acad Dermatol. 2003;49:65-69. doi:10.1067/mjd.2003.489
- Warshaw EM, Buonomo M, DeKoven JG, et al. Importance of supplemental patch testing beyond a screening series for patients with dermatitis: the North American Contact Dermatitis Group experience. JAMA Dermatol. 2021;157:1456-1465. doi:10.1001/jamadermatol.2021.4314
- Geier J, Lessmann H, Mahler V, et al. Occupational contact allergy caused by rubber gloves--nothing has changed. Contact Dermatitis. 2012;67:149-156. doi:10.1111/j.1600-0536.2012.02139.x
- Toraason M, Sussman G, Biagini R, et al. Latex allergy in the workplace. Toxicol Sci Off J Soc Toxicol. 2000;58:5-14. doi:10.1093/toxsci/58.1.5
- Bissonnette R, Agner T, Taylor JS, et al. Hand eczema-part 2: prevention, management, and treatment. J Am Acad Dermatol. 2025;93:1213-1224. doi:10.1016/j.jaad.2024.09.049
- Schliemann S, Kelterer D, Bauer A, et al. Tacrolimus ointment in the treatment of occupationally induced chronic hand dermatitis. Contact Dermatitis. 2008;58:299-306. doi:10.1111/j.1600-0536.2007.01314.x
- Voorberg AN, Kamphuis E, Christoffers WA, et al. Efficacy and safety of dupilumab in patients with severe chronic hand eczema with inadequate response or intolerance to alitretinoin: a randomized, double-blind, placebo-controlled phase IIb proof-of-concept study. Br J Dermatol. 2023;189:400-409. doi:10.1093/bjd/ljad156
- Gooderham M, Molin S, Bissonnette R, et al. Long-term safety and efficacy of delgocitinib cream for up to 52 weeks in adults with chronic hand eczema: results of the phase 3 open-label extension DELTA 3 trial following the DELTA 1 and 2 trials. J Am Acad Dermatol. 2025;93:95-103. doi:10.1016/j.jaad.2025.03.008
Hand dermatitis (HD) is a common dermatologic concern that can impair quality of life, work productivity, and daily functioning.1 Occupational HD is defined as hand eczema caused or worsened by workplace exposures. When caused by work, HD may lead to reduced productivity and even job loss. Subtypes of HD include irritant contact dermatitis (ICD), allergic contact dermatitis (ACD), protein contact dermatitis (PCD), atopic dermatitis (AD), hyperkeratotic HD, and dyshidrotic eczema.2,3
Often caused by wet work, ICD is the most common subtype, whereas PCD—which is caused by immediate hypersensitivity to protein—is less common and usually seen in food service workers.3,4 When HD does not improve with standard treatment, particularly in occupational cases, patch testing is prudent to evaluate for contact allergens. In this article, we review practical considerations for evaluation and management of occupational irritant and allergic HD, highlighting relevant exposures and pearls on workup and management.
Epidemiology of Hand Dermatitis
A 2021 systematic review and meta-analysis of European studies reported a 1-year HD prevalence of 9.1% and a lifetime prevalence of 14.5%.5 Hand dermatitis is most common in women; individuals aged 30 to 39 years; and those who are employed, underscoring the role of workplace exposure.6 High-risk occupations are those involving substantial wet work, such as hairdressers, beauticians, cleaners, and health care and construction workers.7 Individuals with a history of AD also are at high risk for HD.8
Hand Dermatitis Subtypes
Irritant Contact Dermatitis—Irritant contact dermatitis, the most common form of occupational HD, is caused by repeated exposure to irritants (eg, water, detergents, cleansers, and soaps) that disrupt the skin barrier.9 Occupations that involve wet work are a major risk factor, associated with a 56% higher likelihood of ICD.8 Wet work involves frequent handwashing, prolonged contact with liquids, or occlusive glove use.9 As a ubiquitous skin irritant, water can penetrate the stratum corneum, impair the skin barrier, and increase sensitization risk. The dorsal hands usually are affected by ICD due to the thinner stratum corneum in this area.9
Allergic Contact Dermatitis—Allergic contact dermatitis should be considered in cases of chronic, recurrent, or treatment-resistant disease. Clinical clues include dermatitis beyond irritant contact sites, recurrent pruritic and vesicular HD, and flares with occupational exposures or materials; however, it can be difficult to distinguish ACD from ICD on clinical presentation alone, as they have many overlapping features. When ACD is suspected, patch testing remains the gold standard for identifying allergens and guiding avoidance strategies, product alternatives, and workplace modifications.
Unique Occupational Considerations
Hairdressers—Hairdressers have an increased risk for HD due to wet work and exposure to sensitizers, with a pooled lifetime prevalence of 38.2% (including ICD, ACD, and occupational cases).10 Notably, frequent shampooing, rinsing, cutting wet hair, handwashing, and glove use increase the risk for ICD. Hairdressers also are exposed to allergens in hair products, including p-phenylenediamine, toluene-2,5-diamine, persulfate salts, glyceryl thioglycolate, preservatives, and fragrances. Occupational exposure to the preservative methylisothiazolinone is high among hairdressers, with a sensitization rate of 10.5% in HD cases.11
It has been reported that hyperkeratotic fissured eczema of the dorsal hands caused by wet work often indicates ICD, whereas pruritic dyshidrotic eczema involving the lateral fingers or palms suggests ACD; however, these conditions can share overlapping features.7 If ACD is suspected, broad patch testing with baseline and hairdresser series, along with specific chemicals that may be encountered in the workplace, is necessary. Management includes allergen avoidance, reduced wet work tasks, use of nitrile gloves with glove changes to mitigate occlusive effects, and skin barrier protection with emollients.
Health Care Workers—Health care workers are vulnerable to HD due to intensive hand hygiene, prolonged glove use, and allergen exposures, with a lifetime prevalence of self-reported HD of 33.4%.12 Common allergens among health care workers include rubber accelerators, most often from rubber gloves.13 Frequent handwashing and glove use can further impair the skin barrier, increasing irritant and sensitization risks.14 In contrast, alcohol-based hand sanitizers containing emollients are less irritating, with prior analyses showing no significant association with HD risk.15,16 Conversely, handwashing 8 to 10 times daily increased HD risk, with a relative risk of 1.51.15
Surgeons and proceduralists face unique risks for HD from preoperative scrubbing with products that can contain potential allergens such as chlorhexidine gluconate, chloroxylenol, povidone-iodine, fragrance, cocamide diethanolamine, lanolin, alkyl glucosides, sodium benzoate, sorbic acid, tocopherol, and propylene glycol.17,18 Subsequent occlusion under glove layers drives ICD and ACD risks, highlighting the importance of patch testing in affected individuals. While patch testing, exposure avoidance, and limited glove use can mitigate HD risk, frequent handwashing can contribute to refractory HD.
Food Service Workers—Food service workers have an increased risk for HD from allergens and irritants. In a retrospective study of patients with occupational food-related HD (N=372), 57% were diagnosed with ICD, 22% with PCD, and 1.8% with ACD.19 Skin barrier disruption from wet work, occlusion from glove use, and contact with food proteins increase HD risk, especially in bakers exposed to flours and grains, which can cause IgE–mediated PCD manifesting with contact urticaria. Protein contact dermatitis is confirmed by prick testing with suspected foods.20 Additionally, exposure to garlic can cause ICD and ACD due to sulfur-containing compounds, particularly allicin and diallyl disulfide.21,22 Pineapple also can trigger ICD associated with bromelain, a proteolytic enzyme that can break down the skin.23 Nickel exposure is another concern, as steel utensils and cookware can release nickel onto the skin of sensitized individuals.24 Rubber accelerator exposure from gloves also contributes to contact allergy and HD among food service workers; vinyl gloves usually are a good alternative in this setting.25 Management of food-related HD involves exposure avoidance, which may affect occupational viability
Construction Workers—Construction workers are at risk for occupational HD due to contact with irritants and sensitizers such as paints, adhesives, asphalt, cement, solvents, and gloves.26 A retrospective analysis of North American Contact Dermatitis Group data identified HD in 37.2% (253/681) of patch-tested construction workers. The most common occupational allergens include potassium dichromate, which can be present in cement and leather items; bisphenol A epoxy resin; cobalt chloride hexahydrate; and the rubber accelerators carba mix and thiuram mix.26 A thorough occupational history should assess materials handled, and patch testing should include common construction-related allergens to inform avoidance strategies. Workplace task modification can reduce exposure, as certain managerial roles in construction work may involve less contact with irritants and sensitizers.26
Nail Technicians—Nail technicians are at risk for HD, especially ACD from acrylate monomers used in nail gels, dips, and acrylics. In a 10-year analysis, around 87.5% (14/16) of nail technicians with contact allergy to methacrylate demonstrated hand involvement.27 Common acrylate monomers include 2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate, and ethyl cyanoacrylate. 28 Evaluation requires a detailed occupational history, assessing HD onset relative to exposure, services performed, glove-use practices, and whether symptoms improve away from work. While gloves may appear to reduce exposure, a glove-penetration study showed that acrylate-containing nail products can penetrate commonly used disposable gloves from within seconds to approximately 20 minutes, depending on glove and product type.29 Among available options, nitrile gloves may provide dexterity and allergen avoidance when acrylate exposure is brief, with glove changes required every 15 to 30 minutes.30 Patch testing with 2-hydroxyethyl methacrylate and ethyl cyanoacrylate can identify nail acrylate allergy; however, avoidance can be challenging for nail technicians, as these products often are ubiquitous in their work.
Florists—Florists can develop HD from plant allergens and irritants, particularly tulipalin A and calcium oxalate, with a lifetime prevalence of 19.6%.31 Tulipalin A is a well-documented sensitizer causing ACD among florists exposed to tulip bulbs and other Alstroemeria flowers.32 The term tulip fingers actually was coined to describe ACD caused by tulip bulbs in the European tulip industry.33 Patch testing involves testing for tulipalin A, which may be commercially limited, or tulip plant materials; however, fresh tulips require open testing with small amounts due to higher allergen concentration.32 Additionally, the term daffodil itch describes a type of ICD caused by calcium oxalate crystals in daffodil bulbs and tulip sap.32,34 Diagnosis of plant-related HD requires an occupational history and targeted patch testing, while glove protection and exposure avoidance are essential for improvement.
Evaluation and Management
The workup for HD involves physical examination and medical history, including disease onset, course, and history of AD, along with occupational and exposure history to identify allergens and irritants. Understanding the patient’s tasks and responsibilities and workplace practices along with the materials they handle allows the dermatologist to anticipate relevant allergens for patch testing.
Patch testing should be comprehensive, as baseline screening series alone may miss between 26.3% and 50% of occupationally relevant allergens.35,36 Comprehensive patch testing also should include specialty series and supplemental allergens based on the patient’s clinical history and exposures. Specialty series may include hairdressing, bakery, cosmetics, dental, machinists, and adhesives.37 Gloves also warrant attention, as they may be overlooked as a sensitizer following repeated contact and occlusion. In persistent HD associated with glove use, patch testing should include a rubber accelerator series with relevant allergens, such as thiurams, carbamates, mercaptobenzothiazole, diphenylguanidine, and the patient’s own gloves.38 Latex allergy also should be considered, particularly in immediate-type reactions, and can be evaluated with latex-specific IgE testing.39
Management of HD relies on accurate diagnosis and allergen avoidance, which can be challenging in occupational settings. Structured tools, such as the American Contact Dermatitis Society’s Contact Allergen Management Program (https://www.contactderm.org/ resources/acds-camp), can help identify safe alternatives.
In occupational HD, risk assessment should identify occupational exposures and determine appropriate personal protective equipment while minimizing the risk for HD associated with such equipment. Protective gloves are advised to prevent contact with allergens and irritants. When glove use lasts more than 10 minutes, cotton glove liners may be worn to avoid occlusion and moisture retention.40 For wet work, vinyl gloves are recommended, with regular emollient use to support skin-barrier repair. Overall, gloves should be used when possible, changed regularly, and worn for limited periods of time to prevent ICD.
Work modification may be required to reduce exposure and flares, including task reassignment or substitution of materials containing allergens and irritants. Occupational HD may necessitate workplace accommodations, disability evaluation, medical leave, or even permanent job change. Dermatologists play a crucial role in the medical determination of work relatedness and functional impairment, guiding patients through occupational health, disability, and workers’ compensation when warranted.
Treatments for Occupational HD
Treatment of occupational HD depends on disease severity, chronicity, and avoidance of allergens and irritants in ACD, ICD, and PCD. Foundational management includes regular emollient use, which can even serve as monotherapy in mild occupational HD.40 Corticosteroids are the cornerstone of topical therapy, while calcineurin inhibitors can be used as a steroid-sparing option in milder disease.41 Off-label topical calcipotriol and AD-approved therapies crisaborole and ruxolitinib may be effective. For refractory disease after topical treatments, phototherapy can be considered.40 Biologic and targeted therapies also have emerged as potential treatments. Dupilumab is effective for atopic chronic HD and has demonstrated promise for nonatopic chronic HD.42 Recently, delgocitinib, a topical pan–Janus kinase inhibitor cream, showed clinical efficacy for chronic hand eczema and was approved by the US Food and Drug Administration.43 Off-label use of alternative systemic therapies, including acitretin, cyclosporine, methotrexate, and azathioprine, and other biologics and systemic Janus kinase inhibitors also may treat HD, but larger studies are lacking.40
Our Final Interpretation
Occupational HD is a common skin condition with multiple etiologies. It is important for clinicians to gather a thorough occupational and exposure history to narrow the differential diagnosis, inform patch testing, and guide effective management. In practice, successful treatment depends on screening for and diagnosis of workplace exposures driving disease.
Hand dermatitis (HD) is a common dermatologic concern that can impair quality of life, work productivity, and daily functioning.1 Occupational HD is defined as hand eczema caused or worsened by workplace exposures. When caused by work, HD may lead to reduced productivity and even job loss. Subtypes of HD include irritant contact dermatitis (ICD), allergic contact dermatitis (ACD), protein contact dermatitis (PCD), atopic dermatitis (AD), hyperkeratotic HD, and dyshidrotic eczema.2,3
Often caused by wet work, ICD is the most common subtype, whereas PCD—which is caused by immediate hypersensitivity to protein—is less common and usually seen in food service workers.3,4 When HD does not improve with standard treatment, particularly in occupational cases, patch testing is prudent to evaluate for contact allergens. In this article, we review practical considerations for evaluation and management of occupational irritant and allergic HD, highlighting relevant exposures and pearls on workup and management.
Epidemiology of Hand Dermatitis
A 2021 systematic review and meta-analysis of European studies reported a 1-year HD prevalence of 9.1% and a lifetime prevalence of 14.5%.5 Hand dermatitis is most common in women; individuals aged 30 to 39 years; and those who are employed, underscoring the role of workplace exposure.6 High-risk occupations are those involving substantial wet work, such as hairdressers, beauticians, cleaners, and health care and construction workers.7 Individuals with a history of AD also are at high risk for HD.8
Hand Dermatitis Subtypes
Irritant Contact Dermatitis—Irritant contact dermatitis, the most common form of occupational HD, is caused by repeated exposure to irritants (eg, water, detergents, cleansers, and soaps) that disrupt the skin barrier.9 Occupations that involve wet work are a major risk factor, associated with a 56% higher likelihood of ICD.8 Wet work involves frequent handwashing, prolonged contact with liquids, or occlusive glove use.9 As a ubiquitous skin irritant, water can penetrate the stratum corneum, impair the skin barrier, and increase sensitization risk. The dorsal hands usually are affected by ICD due to the thinner stratum corneum in this area.9
Allergic Contact Dermatitis—Allergic contact dermatitis should be considered in cases of chronic, recurrent, or treatment-resistant disease. Clinical clues include dermatitis beyond irritant contact sites, recurrent pruritic and vesicular HD, and flares with occupational exposures or materials; however, it can be difficult to distinguish ACD from ICD on clinical presentation alone, as they have many overlapping features. When ACD is suspected, patch testing remains the gold standard for identifying allergens and guiding avoidance strategies, product alternatives, and workplace modifications.
Unique Occupational Considerations
Hairdressers—Hairdressers have an increased risk for HD due to wet work and exposure to sensitizers, with a pooled lifetime prevalence of 38.2% (including ICD, ACD, and occupational cases).10 Notably, frequent shampooing, rinsing, cutting wet hair, handwashing, and glove use increase the risk for ICD. Hairdressers also are exposed to allergens in hair products, including p-phenylenediamine, toluene-2,5-diamine, persulfate salts, glyceryl thioglycolate, preservatives, and fragrances. Occupational exposure to the preservative methylisothiazolinone is high among hairdressers, with a sensitization rate of 10.5% in HD cases.11
It has been reported that hyperkeratotic fissured eczema of the dorsal hands caused by wet work often indicates ICD, whereas pruritic dyshidrotic eczema involving the lateral fingers or palms suggests ACD; however, these conditions can share overlapping features.7 If ACD is suspected, broad patch testing with baseline and hairdresser series, along with specific chemicals that may be encountered in the workplace, is necessary. Management includes allergen avoidance, reduced wet work tasks, use of nitrile gloves with glove changes to mitigate occlusive effects, and skin barrier protection with emollients.
Health Care Workers—Health care workers are vulnerable to HD due to intensive hand hygiene, prolonged glove use, and allergen exposures, with a lifetime prevalence of self-reported HD of 33.4%.12 Common allergens among health care workers include rubber accelerators, most often from rubber gloves.13 Frequent handwashing and glove use can further impair the skin barrier, increasing irritant and sensitization risks.14 In contrast, alcohol-based hand sanitizers containing emollients are less irritating, with prior analyses showing no significant association with HD risk.15,16 Conversely, handwashing 8 to 10 times daily increased HD risk, with a relative risk of 1.51.15
Surgeons and proceduralists face unique risks for HD from preoperative scrubbing with products that can contain potential allergens such as chlorhexidine gluconate, chloroxylenol, povidone-iodine, fragrance, cocamide diethanolamine, lanolin, alkyl glucosides, sodium benzoate, sorbic acid, tocopherol, and propylene glycol.17,18 Subsequent occlusion under glove layers drives ICD and ACD risks, highlighting the importance of patch testing in affected individuals. While patch testing, exposure avoidance, and limited glove use can mitigate HD risk, frequent handwashing can contribute to refractory HD.
Food Service Workers—Food service workers have an increased risk for HD from allergens and irritants. In a retrospective study of patients with occupational food-related HD (N=372), 57% were diagnosed with ICD, 22% with PCD, and 1.8% with ACD.19 Skin barrier disruption from wet work, occlusion from glove use, and contact with food proteins increase HD risk, especially in bakers exposed to flours and grains, which can cause IgE–mediated PCD manifesting with contact urticaria. Protein contact dermatitis is confirmed by prick testing with suspected foods.20 Additionally, exposure to garlic can cause ICD and ACD due to sulfur-containing compounds, particularly allicin and diallyl disulfide.21,22 Pineapple also can trigger ICD associated with bromelain, a proteolytic enzyme that can break down the skin.23 Nickel exposure is another concern, as steel utensils and cookware can release nickel onto the skin of sensitized individuals.24 Rubber accelerator exposure from gloves also contributes to contact allergy and HD among food service workers; vinyl gloves usually are a good alternative in this setting.25 Management of food-related HD involves exposure avoidance, which may affect occupational viability
Construction Workers—Construction workers are at risk for occupational HD due to contact with irritants and sensitizers such as paints, adhesives, asphalt, cement, solvents, and gloves.26 A retrospective analysis of North American Contact Dermatitis Group data identified HD in 37.2% (253/681) of patch-tested construction workers. The most common occupational allergens include potassium dichromate, which can be present in cement and leather items; bisphenol A epoxy resin; cobalt chloride hexahydrate; and the rubber accelerators carba mix and thiuram mix.26 A thorough occupational history should assess materials handled, and patch testing should include common construction-related allergens to inform avoidance strategies. Workplace task modification can reduce exposure, as certain managerial roles in construction work may involve less contact with irritants and sensitizers.26
Nail Technicians—Nail technicians are at risk for HD, especially ACD from acrylate monomers used in nail gels, dips, and acrylics. In a 10-year analysis, around 87.5% (14/16) of nail technicians with contact allergy to methacrylate demonstrated hand involvement.27 Common acrylate monomers include 2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate, and ethyl cyanoacrylate. 28 Evaluation requires a detailed occupational history, assessing HD onset relative to exposure, services performed, glove-use practices, and whether symptoms improve away from work. While gloves may appear to reduce exposure, a glove-penetration study showed that acrylate-containing nail products can penetrate commonly used disposable gloves from within seconds to approximately 20 minutes, depending on glove and product type.29 Among available options, nitrile gloves may provide dexterity and allergen avoidance when acrylate exposure is brief, with glove changes required every 15 to 30 minutes.30 Patch testing with 2-hydroxyethyl methacrylate and ethyl cyanoacrylate can identify nail acrylate allergy; however, avoidance can be challenging for nail technicians, as these products often are ubiquitous in their work.
Florists—Florists can develop HD from plant allergens and irritants, particularly tulipalin A and calcium oxalate, with a lifetime prevalence of 19.6%.31 Tulipalin A is a well-documented sensitizer causing ACD among florists exposed to tulip bulbs and other Alstroemeria flowers.32 The term tulip fingers actually was coined to describe ACD caused by tulip bulbs in the European tulip industry.33 Patch testing involves testing for tulipalin A, which may be commercially limited, or tulip plant materials; however, fresh tulips require open testing with small amounts due to higher allergen concentration.32 Additionally, the term daffodil itch describes a type of ICD caused by calcium oxalate crystals in daffodil bulbs and tulip sap.32,34 Diagnosis of plant-related HD requires an occupational history and targeted patch testing, while glove protection and exposure avoidance are essential for improvement.
Evaluation and Management
The workup for HD involves physical examination and medical history, including disease onset, course, and history of AD, along with occupational and exposure history to identify allergens and irritants. Understanding the patient’s tasks and responsibilities and workplace practices along with the materials they handle allows the dermatologist to anticipate relevant allergens for patch testing.
Patch testing should be comprehensive, as baseline screening series alone may miss between 26.3% and 50% of occupationally relevant allergens.35,36 Comprehensive patch testing also should include specialty series and supplemental allergens based on the patient’s clinical history and exposures. Specialty series may include hairdressing, bakery, cosmetics, dental, machinists, and adhesives.37 Gloves also warrant attention, as they may be overlooked as a sensitizer following repeated contact and occlusion. In persistent HD associated with glove use, patch testing should include a rubber accelerator series with relevant allergens, such as thiurams, carbamates, mercaptobenzothiazole, diphenylguanidine, and the patient’s own gloves.38 Latex allergy also should be considered, particularly in immediate-type reactions, and can be evaluated with latex-specific IgE testing.39
Management of HD relies on accurate diagnosis and allergen avoidance, which can be challenging in occupational settings. Structured tools, such as the American Contact Dermatitis Society’s Contact Allergen Management Program (https://www.contactderm.org/ resources/acds-camp), can help identify safe alternatives.
In occupational HD, risk assessment should identify occupational exposures and determine appropriate personal protective equipment while minimizing the risk for HD associated with such equipment. Protective gloves are advised to prevent contact with allergens and irritants. When glove use lasts more than 10 minutes, cotton glove liners may be worn to avoid occlusion and moisture retention.40 For wet work, vinyl gloves are recommended, with regular emollient use to support skin-barrier repair. Overall, gloves should be used when possible, changed regularly, and worn for limited periods of time to prevent ICD.
Work modification may be required to reduce exposure and flares, including task reassignment or substitution of materials containing allergens and irritants. Occupational HD may necessitate workplace accommodations, disability evaluation, medical leave, or even permanent job change. Dermatologists play a crucial role in the medical determination of work relatedness and functional impairment, guiding patients through occupational health, disability, and workers’ compensation when warranted.
Treatments for Occupational HD
Treatment of occupational HD depends on disease severity, chronicity, and avoidance of allergens and irritants in ACD, ICD, and PCD. Foundational management includes regular emollient use, which can even serve as monotherapy in mild occupational HD.40 Corticosteroids are the cornerstone of topical therapy, while calcineurin inhibitors can be used as a steroid-sparing option in milder disease.41 Off-label topical calcipotriol and AD-approved therapies crisaborole and ruxolitinib may be effective. For refractory disease after topical treatments, phototherapy can be considered.40 Biologic and targeted therapies also have emerged as potential treatments. Dupilumab is effective for atopic chronic HD and has demonstrated promise for nonatopic chronic HD.42 Recently, delgocitinib, a topical pan–Janus kinase inhibitor cream, showed clinical efficacy for chronic hand eczema and was approved by the US Food and Drug Administration.43 Off-label use of alternative systemic therapies, including acitretin, cyclosporine, methotrexate, and azathioprine, and other biologics and systemic Janus kinase inhibitors also may treat HD, but larger studies are lacking.40
Our Final Interpretation
Occupational HD is a common skin condition with multiple etiologies. It is important for clinicians to gather a thorough occupational and exposure history to narrow the differential diagnosis, inform patch testing, and guide effective management. In practice, successful treatment depends on screening for and diagnosis of workplace exposures driving disease.
- Agner T, Andersen KE, Brandao FM, et al. Hand eczema severity and quality of life: a cross-sectional, multicentre study of hand eczema patients. Contact Dermatitis. 2008;59:43-47. doi:10.1111 /j.1600-0536.2008.01362.x
- Agner T, Aalto-Korte K, Andersen KE, et al. Classification of hand eczema. J Eur Acad Dermatol Venereol. 2015;29:2417-2422. doi:10.1111 /jdv.13308
- Bissonnette R, Agner T, Molin S, et al. Hand eczema—part 1: epidemiology, pathogenesis, diagnosis, and work-up. J Am Acad Dermatol. 2025;93:1201-1210. doi:10.1016/j.jaad.2024.09.048
- Barbaud A. Mechanism and diagnosis of protein contact dermatitis. Curr Opin Allergy Clin Immunol. 2020;20:117-121. doi:10.1097/ACI.0000000000000621
- Quaade AS, Simonsen AB, Halling AS, et al. Prevalence, incidence, and severity of hand eczema in the general population - a systematic review and meta-analysis. Contact Dermatitis. 2021;84:361-374. doi:10.1111/cod.13804
- Apfelbacher C, Bewley A, Molin S, et al. Prevalence of chronic hand eczema in adults: a cross-sectional survey of over 60 000 respondents from the general population of Canada, France, Germany, Italy, Spain and the UK. Br J Dermatol. 2025;192:1047-1054. doi:10.1093 /bjd/ljaf020
- Weidinger S, Novak N. Hand eczema. Lancet. 2024;404:2476-2486. doi:10.1016/S0140-6736(24)01810-5
- Schütte MG, Tamminga SJ, de Groene GJ, et al. Work-related and personal risk factors for occupational contact dermatitis: a systematic review of the literature with meta-analysis. Contact Dermatitis. 2023;88:171-187. doi:10.1111/cod.14253
- Behroozy A, Keegel TG. Wet-work exposure: a main risk factor for occupational hand dermatitis. Saf Health Work. 2014;5:175-180. doi:10.1016/j.shaw.2014.08.001
- Havmose MS, Kezic S, Uter W, et al. Prevalence and incidence of hand eczema in hairdressers-a systematic review and meta-analysis of the published literature from 2000-2021. Contact Dermatitis. 2022;86:254-265. doi:10.1111/cod.14048
- Uter W, Hallmann S, Gefeller O, et al. Contact allergy to ingredients of hair cosmetics in female hairdressers and female consumers—an update based on IVDK data 2013–2020. Contact Dermatitis. 2023;89:161-170. doi:10.1111/cod.14363
- Yüksel YT, Symanzik C, Christensen MO, et al. Prevalence and incidence of hand eczema in healthcare workers: a systematic review and meta-analysis. Contact Dermatitis. 2024;90:331-342. doi:10.1111 /cod.14489
- Warshaw EM, Schram SE, Maibach HI, et al. Occupation-related contact dermatitis in North American health care workers referred for patch testing: cross-sectional data, 1998 to 2004. Dermatitis. 2008;19:261-274. doi:10.2310/6620.2008.07059
- Hamnerius N, Svedman C, Bergendorff O, et al. Wet work exposure and hand eczema among healthcare workers: a cross-sectional study. Br J Dermatol. 2018;178:452-461. doi:10.1111 /bjd.15813
- Loh EDW, Yew YW. Hand hygiene and hand eczema: a systematic review and meta-analysis. Contact Dermatitis. 2022;87:303-314. doi:10.1111/cod.14133
- Lotfinejad N, Peters A, Tartari E, et al. Hand hygiene in health care: 20 years of ongoing advances and perspectives. Lancet Infect Dis. 2021;21:e209-e221. doi:10.1016/S1473-3099(21)00383-2
- Schlarbaum JP, Hylwa SA. Allergic contact dermatitis to operating room scrubs and disinfectants. Dermat Contact Atopic Occup Drug. 2019;30:363-370. doi:10.1097/DER.0000000000000525
- Rodriguez-Homs LG, Atwater AR. Allergens in medical hand skin cleansers. Dermat Contact Atopic Occup Drug. 2019;30:336-341. doi:10.1097/DER.0000000000000504
- Vester L, Thyssen JP, Menné T, et al. Occupational food-related hand dermatoses seen over a 10-year period. Contact Dermatitis. 2012;66:264-270. doi:10.1111/j.1600-0536.2011.02048.x
- Pesonen M, Koskela K, Aalto-Korte K. Contact urticaria and protein contact dermatitis in the Finnish Register of Occupational Diseases in a period of 12 years. Contact Dermatitis. 2020;83:1-7. doi:10.1111/cod.13547
- McFadden JP, White JML, Basketter DA, et al. Reduced allergy rates in atopic eczema to contact allergens used in both skin products and foods: atopy and the “hapten-atopy hypothesis.” Contact Dermatitis. 2008;58:156-158. doi:10.1111/j.1600-0536.2007.01291.x
- Kao SH, Hsu CH, Su SN, et al. Identification and immunologic characterization of an allergen, alliin lyase, from garlic (Allium sativum). J Allergy Clin Immunol. 2004;113:161-168. doi:10.1016/j.jaci.2003.10.040
- Reddy VB, Lerner EA. Plant cysteine proteases that evoke itch activate protease-activated receptors. Br J Dermatol. 2010;163:532-535. doi:10.1111/j.1365-2133.2010.09862.x
- Silverberg NB, Pelletier JL, Jacob SE, et al; Section on Dermatology, Section on Allergy and Immunology. Nickel allergic contact dermatitis: identification, treatment, and prevention. Pediatrics. 2020;145:e20200628. doi:10.1542/peds.2020-0628
- Clément A, Ferrier le Bouëdec MC, Crépy MN, et al. Hand eczema in glove-wearing patients. Contact Dermatitis. 2023;89:143-152. doi:10.1111/cod.14357
- Reeder MJ, Idrogo-Lam A, Aravamuthan SR, et al. Occupational contact dermatitis in construction workers: a retrospective analysis of the North American Contact Dermatitis Group Data, 2001-2020. Dermat Contact Atopic Occup Drug. 2024;35:467-475. doi:10.1089/derm.2024.0018
- Fisch A, Hamnerius N, Isaksson M. Dermatitis and occupational (meth)acrylate contact allergy in nail technicians-a 10-year study. Contact Dermatitis. 2019;81:58-60. doi:10.1111/cod.13216
- Atwater AR, Reeder M. Trends in nail services may cause dermatitis: not your mother’s nail polish. Cutis. 2019;103:315-317.
- Suuronen K, Ylinen K, Heikkilä J, et al. Acrylates in artificial nails— results of product analyses and glove penetration studies. Contact Dermatitis. 2024;90:266-272. doi:10.1111/cod.14474
- Morgado F, Batista M, Gonçalo M. Short exposures and glove protection against (meth)acrylates in nail beauticians-thoughts on a rising concern. Contact Dermatitis. 2019;81:62-63. doi:10.1111 /cod.13222
- Paulsen E, Søgaard J, Andersen KE. Occupational dermatitis in Danish gardeners and greenhouse workers (I). prevalence and possible risk factors. Contact Dermatitis. 1997;37:263-270. doi:10.1111/j.1600-0536.1997.tb02462.x
- Fonacier L, Bernstein DI, Pacheco K, et al. Contact dermatitis: a practice parameter–update 2015. J Allergy Clin Immunol Pract. 2015; 3(3 suppl):S1-S39. doi:10.1016/j.jaip.2015.02.009
- Gette MT, Marks JE. Tulip fingers. Arch Dermatol. 1990;126:203-205.
- Bruynzeel DP. Bulb dermatitis. Dermatological problems in the flower bulb industries. Contact Dermatitis. 1997;37:70-77. doi:10.1111/j.1600-0536.1997.tb00042.x
- Nettis E, Marcandrea M, Colanardi MC, et al. Results of standard series patch testing in patients with occupational allergic contact dermatitis. Allergy. 2003;58:1304-1307. doi:10.1046/j.1398-9995.2003.00346.x
- Saripalli YV, Achen F, Belsito DV. The detection of clinically relevant contact allergens using a standard screening tray of twenty-three allergens. J Am Acad Dermatol. 2003;49:65-69. doi:10.1067/mjd.2003.489
- Warshaw EM, Buonomo M, DeKoven JG, et al. Importance of supplemental patch testing beyond a screening series for patients with dermatitis: the North American Contact Dermatitis Group experience. JAMA Dermatol. 2021;157:1456-1465. doi:10.1001/jamadermatol.2021.4314
- Geier J, Lessmann H, Mahler V, et al. Occupational contact allergy caused by rubber gloves--nothing has changed. Contact Dermatitis. 2012;67:149-156. doi:10.1111/j.1600-0536.2012.02139.x
- Toraason M, Sussman G, Biagini R, et al. Latex allergy in the workplace. Toxicol Sci Off J Soc Toxicol. 2000;58:5-14. doi:10.1093/toxsci/58.1.5
- Bissonnette R, Agner T, Taylor JS, et al. Hand eczema-part 2: prevention, management, and treatment. J Am Acad Dermatol. 2025;93:1213-1224. doi:10.1016/j.jaad.2024.09.049
- Schliemann S, Kelterer D, Bauer A, et al. Tacrolimus ointment in the treatment of occupationally induced chronic hand dermatitis. Contact Dermatitis. 2008;58:299-306. doi:10.1111/j.1600-0536.2007.01314.x
- Voorberg AN, Kamphuis E, Christoffers WA, et al. Efficacy and safety of dupilumab in patients with severe chronic hand eczema with inadequate response or intolerance to alitretinoin: a randomized, double-blind, placebo-controlled phase IIb proof-of-concept study. Br J Dermatol. 2023;189:400-409. doi:10.1093/bjd/ljad156
- Gooderham M, Molin S, Bissonnette R, et al. Long-term safety and efficacy of delgocitinib cream for up to 52 weeks in adults with chronic hand eczema: results of the phase 3 open-label extension DELTA 3 trial following the DELTA 1 and 2 trials. J Am Acad Dermatol. 2025;93:95-103. doi:10.1016/j.jaad.2025.03.008
- Agner T, Andersen KE, Brandao FM, et al. Hand eczema severity and quality of life: a cross-sectional, multicentre study of hand eczema patients. Contact Dermatitis. 2008;59:43-47. doi:10.1111 /j.1600-0536.2008.01362.x
- Agner T, Aalto-Korte K, Andersen KE, et al. Classification of hand eczema. J Eur Acad Dermatol Venereol. 2015;29:2417-2422. doi:10.1111 /jdv.13308
- Bissonnette R, Agner T, Molin S, et al. Hand eczema—part 1: epidemiology, pathogenesis, diagnosis, and work-up. J Am Acad Dermatol. 2025;93:1201-1210. doi:10.1016/j.jaad.2024.09.048
- Barbaud A. Mechanism and diagnosis of protein contact dermatitis. Curr Opin Allergy Clin Immunol. 2020;20:117-121. doi:10.1097/ACI.0000000000000621
- Quaade AS, Simonsen AB, Halling AS, et al. Prevalence, incidence, and severity of hand eczema in the general population - a systematic review and meta-analysis. Contact Dermatitis. 2021;84:361-374. doi:10.1111/cod.13804
- Apfelbacher C, Bewley A, Molin S, et al. Prevalence of chronic hand eczema in adults: a cross-sectional survey of over 60 000 respondents from the general population of Canada, France, Germany, Italy, Spain and the UK. Br J Dermatol. 2025;192:1047-1054. doi:10.1093 /bjd/ljaf020
- Weidinger S, Novak N. Hand eczema. Lancet. 2024;404:2476-2486. doi:10.1016/S0140-6736(24)01810-5
- Schütte MG, Tamminga SJ, de Groene GJ, et al. Work-related and personal risk factors for occupational contact dermatitis: a systematic review of the literature with meta-analysis. Contact Dermatitis. 2023;88:171-187. doi:10.1111/cod.14253
- Behroozy A, Keegel TG. Wet-work exposure: a main risk factor for occupational hand dermatitis. Saf Health Work. 2014;5:175-180. doi:10.1016/j.shaw.2014.08.001
- Havmose MS, Kezic S, Uter W, et al. Prevalence and incidence of hand eczema in hairdressers-a systematic review and meta-analysis of the published literature from 2000-2021. Contact Dermatitis. 2022;86:254-265. doi:10.1111/cod.14048
- Uter W, Hallmann S, Gefeller O, et al. Contact allergy to ingredients of hair cosmetics in female hairdressers and female consumers—an update based on IVDK data 2013–2020. Contact Dermatitis. 2023;89:161-170. doi:10.1111/cod.14363
- Yüksel YT, Symanzik C, Christensen MO, et al. Prevalence and incidence of hand eczema in healthcare workers: a systematic review and meta-analysis. Contact Dermatitis. 2024;90:331-342. doi:10.1111 /cod.14489
- Warshaw EM, Schram SE, Maibach HI, et al. Occupation-related contact dermatitis in North American health care workers referred for patch testing: cross-sectional data, 1998 to 2004. Dermatitis. 2008;19:261-274. doi:10.2310/6620.2008.07059
- Hamnerius N, Svedman C, Bergendorff O, et al. Wet work exposure and hand eczema among healthcare workers: a cross-sectional study. Br J Dermatol. 2018;178:452-461. doi:10.1111 /bjd.15813
- Loh EDW, Yew YW. Hand hygiene and hand eczema: a systematic review and meta-analysis. Contact Dermatitis. 2022;87:303-314. doi:10.1111/cod.14133
- Lotfinejad N, Peters A, Tartari E, et al. Hand hygiene in health care: 20 years of ongoing advances and perspectives. Lancet Infect Dis. 2021;21:e209-e221. doi:10.1016/S1473-3099(21)00383-2
- Schlarbaum JP, Hylwa SA. Allergic contact dermatitis to operating room scrubs and disinfectants. Dermat Contact Atopic Occup Drug. 2019;30:363-370. doi:10.1097/DER.0000000000000525
- Rodriguez-Homs LG, Atwater AR. Allergens in medical hand skin cleansers. Dermat Contact Atopic Occup Drug. 2019;30:336-341. doi:10.1097/DER.0000000000000504
- Vester L, Thyssen JP, Menné T, et al. Occupational food-related hand dermatoses seen over a 10-year period. Contact Dermatitis. 2012;66:264-270. doi:10.1111/j.1600-0536.2011.02048.x
- Pesonen M, Koskela K, Aalto-Korte K. Contact urticaria and protein contact dermatitis in the Finnish Register of Occupational Diseases in a period of 12 years. Contact Dermatitis. 2020;83:1-7. doi:10.1111/cod.13547
- McFadden JP, White JML, Basketter DA, et al. Reduced allergy rates in atopic eczema to contact allergens used in both skin products and foods: atopy and the “hapten-atopy hypothesis.” Contact Dermatitis. 2008;58:156-158. doi:10.1111/j.1600-0536.2007.01291.x
- Kao SH, Hsu CH, Su SN, et al. Identification and immunologic characterization of an allergen, alliin lyase, from garlic (Allium sativum). J Allergy Clin Immunol. 2004;113:161-168. doi:10.1016/j.jaci.2003.10.040
- Reddy VB, Lerner EA. Plant cysteine proteases that evoke itch activate protease-activated receptors. Br J Dermatol. 2010;163:532-535. doi:10.1111/j.1365-2133.2010.09862.x
- Silverberg NB, Pelletier JL, Jacob SE, et al; Section on Dermatology, Section on Allergy and Immunology. Nickel allergic contact dermatitis: identification, treatment, and prevention. Pediatrics. 2020;145:e20200628. doi:10.1542/peds.2020-0628
- Clément A, Ferrier le Bouëdec MC, Crépy MN, et al. Hand eczema in glove-wearing patients. Contact Dermatitis. 2023;89:143-152. doi:10.1111/cod.14357
- Reeder MJ, Idrogo-Lam A, Aravamuthan SR, et al. Occupational contact dermatitis in construction workers: a retrospective analysis of the North American Contact Dermatitis Group Data, 2001-2020. Dermat Contact Atopic Occup Drug. 2024;35:467-475. doi:10.1089/derm.2024.0018
- Fisch A, Hamnerius N, Isaksson M. Dermatitis and occupational (meth)acrylate contact allergy in nail technicians-a 10-year study. Contact Dermatitis. 2019;81:58-60. doi:10.1111/cod.13216
- Atwater AR, Reeder M. Trends in nail services may cause dermatitis: not your mother’s nail polish. Cutis. 2019;103:315-317.
- Suuronen K, Ylinen K, Heikkilä J, et al. Acrylates in artificial nails— results of product analyses and glove penetration studies. Contact Dermatitis. 2024;90:266-272. doi:10.1111/cod.14474
- Morgado F, Batista M, Gonçalo M. Short exposures and glove protection against (meth)acrylates in nail beauticians-thoughts on a rising concern. Contact Dermatitis. 2019;81:62-63. doi:10.1111 /cod.13222
- Paulsen E, Søgaard J, Andersen KE. Occupational dermatitis in Danish gardeners and greenhouse workers (I). prevalence and possible risk factors. Contact Dermatitis. 1997;37:263-270. doi:10.1111/j.1600-0536.1997.tb02462.x
- Fonacier L, Bernstein DI, Pacheco K, et al. Contact dermatitis: a practice parameter–update 2015. J Allergy Clin Immunol Pract. 2015; 3(3 suppl):S1-S39. doi:10.1016/j.jaip.2015.02.009
- Gette MT, Marks JE. Tulip fingers. Arch Dermatol. 1990;126:203-205.
- Bruynzeel DP. Bulb dermatitis. Dermatological problems in the flower bulb industries. Contact Dermatitis. 1997;37:70-77. doi:10.1111/j.1600-0536.1997.tb00042.x
- Nettis E, Marcandrea M, Colanardi MC, et al. Results of standard series patch testing in patients with occupational allergic contact dermatitis. Allergy. 2003;58:1304-1307. doi:10.1046/j.1398-9995.2003.00346.x
- Saripalli YV, Achen F, Belsito DV. The detection of clinically relevant contact allergens using a standard screening tray of twenty-three allergens. J Am Acad Dermatol. 2003;49:65-69. doi:10.1067/mjd.2003.489
- Warshaw EM, Buonomo M, DeKoven JG, et al. Importance of supplemental patch testing beyond a screening series for patients with dermatitis: the North American Contact Dermatitis Group experience. JAMA Dermatol. 2021;157:1456-1465. doi:10.1001/jamadermatol.2021.4314
- Geier J, Lessmann H, Mahler V, et al. Occupational contact allergy caused by rubber gloves--nothing has changed. Contact Dermatitis. 2012;67:149-156. doi:10.1111/j.1600-0536.2012.02139.x
- Toraason M, Sussman G, Biagini R, et al. Latex allergy in the workplace. Toxicol Sci Off J Soc Toxicol. 2000;58:5-14. doi:10.1093/toxsci/58.1.5
- Bissonnette R, Agner T, Taylor JS, et al. Hand eczema-part 2: prevention, management, and treatment. J Am Acad Dermatol. 2025;93:1213-1224. doi:10.1016/j.jaad.2024.09.049
- Schliemann S, Kelterer D, Bauer A, et al. Tacrolimus ointment in the treatment of occupationally induced chronic hand dermatitis. Contact Dermatitis. 2008;58:299-306. doi:10.1111/j.1600-0536.2007.01314.x
- Voorberg AN, Kamphuis E, Christoffers WA, et al. Efficacy and safety of dupilumab in patients with severe chronic hand eczema with inadequate response or intolerance to alitretinoin: a randomized, double-blind, placebo-controlled phase IIb proof-of-concept study. Br J Dermatol. 2023;189:400-409. doi:10.1093/bjd/ljad156
- Gooderham M, Molin S, Bissonnette R, et al. Long-term safety and efficacy of delgocitinib cream for up to 52 weeks in adults with chronic hand eczema: results of the phase 3 open-label extension DELTA 3 trial following the DELTA 1 and 2 trials. J Am Acad Dermatol. 2025;93:95-103. doi:10.1016/j.jaad.2025.03.008
Getting a Grip on Occupational Hand Dermatitis: Key Considerations for Evaluation and Management
Getting a Grip on Occupational Hand Dermatitis: Key Considerations for Evaluation and Management
PRACTICE POINTS
- Occupational hand dermatitis (HD) is a common multifactorial disease with a major impact on quality of life, worker safety, and productivity.
- High-risk occupations include those involving wet work, such as hairdressers, beauticians, cleaners, and health care and construction workers.
- A detailed occupational and exposure history is essential for managing occupational HD.

Consumer Trends Driving Contact Dermatitis: Insights from JiaDe Yu, MD, MS
Consumer Trends Driving Contact Dermatitis: Insights from JiaDe Yu, MD, MS
How do social media trends and influencer driven product fads affect the patterns of contact dermatitis you are seeing?
DR. YU: Social media and influencers are huge marketing opportunities for cosmetic and personal care companies and drive consumer demand. One example from a few years ago is slime as a toy for kids. For a period of time, every kid was making slime at home, resulting in high numbers of hand allergic contact dermatitis. Making slime requires a combination of borax (irritant), glue (irritant and allergen), laundry detergent or dish soap (irritant and allergen), and fragrances (irritant and allergen). This fad has been slowing since I cowrote an article on it (doi:10.1111 /pde.13792). More recently, the rise of “Sephora kids” (preteens and adolescents influenced by social media trends promoting multistep skin care and anti-aging products) has raised concerns about contact dermatitis, as many of these products contain ingredients that can disrupt the skin barrier or trigger sensitization in younger patients.
How can products labeled free of fragrances or preservatives still trigger allergic contact dermatitis?
DR. YU: Fragrances are frequently in the top 10 ingredients that cause allergic contact dermatitis in adults and children. For people with sensitive skin, we almost unequivocally recommend fragrance-free products. Now, not all fragrance-free products are truly free of fragrance allergens. Some fragrance chemicals may be used for another purpose (benzyl alcohol as a preservative, for example), so the product can still be fragrance free even though benzyl alcohol has a fragrance. Most products cannot truly be preservative free if they are expected to have a shelf life. One-time-use products do exist and can be preservative free, but they are very rare and very expensive to manufacture and maintain.
Have you seen spikes in reactions from trendy products like CBD-infused creams, botanical serums, or exfoliating acids?
DR. YU: Not yet, but I would not be surprised that this is rising in prevalence. The issue might not be CBD itself; it’s really the other additives in these CBD products that will cause problems. Looking at some CBD products for sale from major retailers, many contain fragrances such as lemongrass oil and botanical extracts such as calendula that have been noted to cause allergic contact dermatitis.
Do certain patient behaviors (eg, layering multiple natural products, frequent product switching, prolonged leave-on use) increase the risk for ACD?
DR. YU: Absolutely possible. The more products you use, the more likely you will develop allergic contact dermatitis due to increased exposure to potential allergens. We know that leave-on products are higher risk than rinse-offs in general. Furthermore, more products used also increase the risk for irritant dermatitis that might break the skin barrier, increasing the odds that someone will develop allergic contact dermatitis. We see this often with facial skin care products where some people might layer on glycolic acid with retinoid acid with vitamin C oil with kojic acid, etc, all leading to irritation on the face.
How do emerging consumer product trends influence your patch-testing approach?
DR. YU: We try to customize our patch-tested allergens to the patient’s rash and symptoms. If it’s a patient with facial dermatitis, for example, we would patch test the patient to a core allergen series (eg, American Contact Dermatitis Society 90, North American Comprehensive 80, North American Contact Dermatitis Group 80) and add on other supplemental panels including cosmetic series if applicable. It is also preferable to patch test for products that are used and/or suspected of causing the rash. For example, if a blush is a suspected cause of dermatitis, we would certainly patch test to that as well. We generally try to encourage the patient to bring in all their products so we can evaluate them for appropriateness for patch testing.
Which consumer-driven ingredients do you now consider high-yield targets for testing?
DR. YU: Fragrances, preservatives, and botanical extracts are all likely causes of allergic contact dermatitis. We are uncovering new allergens all the time, so testing directly to patient products is also important. Just because something has not been reported to be a contact allergen doesn’t mean it can’t become one.
Have you observed any demographic or cultural trends in patients with allergic contact dermatitis related to consumer products?
DR. YU: There are various papers that outline different allergens in adults vs children vs older adults. However, in general, the prevalence of contact dermatitis is very similar across all age groups and distributions. I do think there are definitely gender and cultural variations. Women are more likely to be allergic to nickel, for example, which is more often found in jewelry. However, there really aren’t studies that demonstrate one population is more likely to develop allergic contact dermatitis than others. It really comes down to exposure. For example, neomycin, which is contained in triple antibiotics in the United States and is sold over the counter, is a common allergen here. However, it’s not readily available in other countries, and therefore, neomycin is a rare allergen in those countries.
Looking forward, which emerging consumer trends do you anticipate will create the next wave of contact dermatitis cases?
DR. YU: We have seen an increase in allergic contact dermatitis in the wearables industry, especially in continuous glucose monitors. They are now being sold over the counter so people without diabetes and without a prescription will be able to purchase them from retailers like Amazon or CVS. The adhesives in these glucose monitors have been shown to cause allergic contact dermatitis in a sizeable number of kids and adults. I suspect this problem will continue to increase with increased exposure to the allergens in these adhesives.
How do social media trends and influencer driven product fads affect the patterns of contact dermatitis you are seeing?
DR. YU: Social media and influencers are huge marketing opportunities for cosmetic and personal care companies and drive consumer demand. One example from a few years ago is slime as a toy for kids. For a period of time, every kid was making slime at home, resulting in high numbers of hand allergic contact dermatitis. Making slime requires a combination of borax (irritant), glue (irritant and allergen), laundry detergent or dish soap (irritant and allergen), and fragrances (irritant and allergen). This fad has been slowing since I cowrote an article on it (doi:10.1111 /pde.13792). More recently, the rise of “Sephora kids” (preteens and adolescents influenced by social media trends promoting multistep skin care and anti-aging products) has raised concerns about contact dermatitis, as many of these products contain ingredients that can disrupt the skin barrier or trigger sensitization in younger patients.
How can products labeled free of fragrances or preservatives still trigger allergic contact dermatitis?
DR. YU: Fragrances are frequently in the top 10 ingredients that cause allergic contact dermatitis in adults and children. For people with sensitive skin, we almost unequivocally recommend fragrance-free products. Now, not all fragrance-free products are truly free of fragrance allergens. Some fragrance chemicals may be used for another purpose (benzyl alcohol as a preservative, for example), so the product can still be fragrance free even though benzyl alcohol has a fragrance. Most products cannot truly be preservative free if they are expected to have a shelf life. One-time-use products do exist and can be preservative free, but they are very rare and very expensive to manufacture and maintain.
Have you seen spikes in reactions from trendy products like CBD-infused creams, botanical serums, or exfoliating acids?
DR. YU: Not yet, but I would not be surprised that this is rising in prevalence. The issue might not be CBD itself; it’s really the other additives in these CBD products that will cause problems. Looking at some CBD products for sale from major retailers, many contain fragrances such as lemongrass oil and botanical extracts such as calendula that have been noted to cause allergic contact dermatitis.
Do certain patient behaviors (eg, layering multiple natural products, frequent product switching, prolonged leave-on use) increase the risk for ACD?
DR. YU: Absolutely possible. The more products you use, the more likely you will develop allergic contact dermatitis due to increased exposure to potential allergens. We know that leave-on products are higher risk than rinse-offs in general. Furthermore, more products used also increase the risk for irritant dermatitis that might break the skin barrier, increasing the odds that someone will develop allergic contact dermatitis. We see this often with facial skin care products where some people might layer on glycolic acid with retinoid acid with vitamin C oil with kojic acid, etc, all leading to irritation on the face.
How do emerging consumer product trends influence your patch-testing approach?
DR. YU: We try to customize our patch-tested allergens to the patient’s rash and symptoms. If it’s a patient with facial dermatitis, for example, we would patch test the patient to a core allergen series (eg, American Contact Dermatitis Society 90, North American Comprehensive 80, North American Contact Dermatitis Group 80) and add on other supplemental panels including cosmetic series if applicable. It is also preferable to patch test for products that are used and/or suspected of causing the rash. For example, if a blush is a suspected cause of dermatitis, we would certainly patch test to that as well. We generally try to encourage the patient to bring in all their products so we can evaluate them for appropriateness for patch testing.
Which consumer-driven ingredients do you now consider high-yield targets for testing?
DR. YU: Fragrances, preservatives, and botanical extracts are all likely causes of allergic contact dermatitis. We are uncovering new allergens all the time, so testing directly to patient products is also important. Just because something has not been reported to be a contact allergen doesn’t mean it can’t become one.
Have you observed any demographic or cultural trends in patients with allergic contact dermatitis related to consumer products?
DR. YU: There are various papers that outline different allergens in adults vs children vs older adults. However, in general, the prevalence of contact dermatitis is very similar across all age groups and distributions. I do think there are definitely gender and cultural variations. Women are more likely to be allergic to nickel, for example, which is more often found in jewelry. However, there really aren’t studies that demonstrate one population is more likely to develop allergic contact dermatitis than others. It really comes down to exposure. For example, neomycin, which is contained in triple antibiotics in the United States and is sold over the counter, is a common allergen here. However, it’s not readily available in other countries, and therefore, neomycin is a rare allergen in those countries.
Looking forward, which emerging consumer trends do you anticipate will create the next wave of contact dermatitis cases?
DR. YU: We have seen an increase in allergic contact dermatitis in the wearables industry, especially in continuous glucose monitors. They are now being sold over the counter so people without diabetes and without a prescription will be able to purchase them from retailers like Amazon or CVS. The adhesives in these glucose monitors have been shown to cause allergic contact dermatitis in a sizeable number of kids and adults. I suspect this problem will continue to increase with increased exposure to the allergens in these adhesives.
How do social media trends and influencer driven product fads affect the patterns of contact dermatitis you are seeing?
DR. YU: Social media and influencers are huge marketing opportunities for cosmetic and personal care companies and drive consumer demand. One example from a few years ago is slime as a toy for kids. For a period of time, every kid was making slime at home, resulting in high numbers of hand allergic contact dermatitis. Making slime requires a combination of borax (irritant), glue (irritant and allergen), laundry detergent or dish soap (irritant and allergen), and fragrances (irritant and allergen). This fad has been slowing since I cowrote an article on it (doi:10.1111 /pde.13792). More recently, the rise of “Sephora kids” (preteens and adolescents influenced by social media trends promoting multistep skin care and anti-aging products) has raised concerns about contact dermatitis, as many of these products contain ingredients that can disrupt the skin barrier or trigger sensitization in younger patients.
How can products labeled free of fragrances or preservatives still trigger allergic contact dermatitis?
DR. YU: Fragrances are frequently in the top 10 ingredients that cause allergic contact dermatitis in adults and children. For people with sensitive skin, we almost unequivocally recommend fragrance-free products. Now, not all fragrance-free products are truly free of fragrance allergens. Some fragrance chemicals may be used for another purpose (benzyl alcohol as a preservative, for example), so the product can still be fragrance free even though benzyl alcohol has a fragrance. Most products cannot truly be preservative free if they are expected to have a shelf life. One-time-use products do exist and can be preservative free, but they are very rare and very expensive to manufacture and maintain.
Have you seen spikes in reactions from trendy products like CBD-infused creams, botanical serums, or exfoliating acids?
DR. YU: Not yet, but I would not be surprised that this is rising in prevalence. The issue might not be CBD itself; it’s really the other additives in these CBD products that will cause problems. Looking at some CBD products for sale from major retailers, many contain fragrances such as lemongrass oil and botanical extracts such as calendula that have been noted to cause allergic contact dermatitis.
Do certain patient behaviors (eg, layering multiple natural products, frequent product switching, prolonged leave-on use) increase the risk for ACD?
DR. YU: Absolutely possible. The more products you use, the more likely you will develop allergic contact dermatitis due to increased exposure to potential allergens. We know that leave-on products are higher risk than rinse-offs in general. Furthermore, more products used also increase the risk for irritant dermatitis that might break the skin barrier, increasing the odds that someone will develop allergic contact dermatitis. We see this often with facial skin care products where some people might layer on glycolic acid with retinoid acid with vitamin C oil with kojic acid, etc, all leading to irritation on the face.
How do emerging consumer product trends influence your patch-testing approach?
DR. YU: We try to customize our patch-tested allergens to the patient’s rash and symptoms. If it’s a patient with facial dermatitis, for example, we would patch test the patient to a core allergen series (eg, American Contact Dermatitis Society 90, North American Comprehensive 80, North American Contact Dermatitis Group 80) and add on other supplemental panels including cosmetic series if applicable. It is also preferable to patch test for products that are used and/or suspected of causing the rash. For example, if a blush is a suspected cause of dermatitis, we would certainly patch test to that as well. We generally try to encourage the patient to bring in all their products so we can evaluate them for appropriateness for patch testing.
Which consumer-driven ingredients do you now consider high-yield targets for testing?
DR. YU: Fragrances, preservatives, and botanical extracts are all likely causes of allergic contact dermatitis. We are uncovering new allergens all the time, so testing directly to patient products is also important. Just because something has not been reported to be a contact allergen doesn’t mean it can’t become one.
Have you observed any demographic or cultural trends in patients with allergic contact dermatitis related to consumer products?
DR. YU: There are various papers that outline different allergens in adults vs children vs older adults. However, in general, the prevalence of contact dermatitis is very similar across all age groups and distributions. I do think there are definitely gender and cultural variations. Women are more likely to be allergic to nickel, for example, which is more often found in jewelry. However, there really aren’t studies that demonstrate one population is more likely to develop allergic contact dermatitis than others. It really comes down to exposure. For example, neomycin, which is contained in triple antibiotics in the United States and is sold over the counter, is a common allergen here. However, it’s not readily available in other countries, and therefore, neomycin is a rare allergen in those countries.
Looking forward, which emerging consumer trends do you anticipate will create the next wave of contact dermatitis cases?
DR. YU: We have seen an increase in allergic contact dermatitis in the wearables industry, especially in continuous glucose monitors. They are now being sold over the counter so people without diabetes and without a prescription will be able to purchase them from retailers like Amazon or CVS. The adhesives in these glucose monitors have been shown to cause allergic contact dermatitis in a sizeable number of kids and adults. I suspect this problem will continue to increase with increased exposure to the allergens in these adhesives.
Consumer Trends Driving Contact Dermatitis: Insights from JiaDe Yu, MD, MS
Consumer Trends Driving Contact Dermatitis: Insights from JiaDe Yu, MD, MS

Using Intralesional Adalimumab for Chronic Refractory Cutaneous Granulomatous Inflammation
Using Intralesional Adalimumab for Chronic Refractory Cutaneous Granulomatous Inflammation
Practice Gap
Chronic localized granulomatous inflammation can be difficult to manage, particularly when manifesting on the face. Intralesional corticosteroids may lead to atrophy and dyspigmentation and therefore must be used cautiously in cosmetically sensitive areas.1 Surgical removal can lead to recurrence, and systemic agents may carry risks disproportionate to disease burden. Although tumor necrosis factor (TNF) α inhibitors are effective systemically, their localized use in cutaneous granulomatous dermatoses remains underreported.1-3 We describe a technique using intralesional injection of adalimumab to treat chronic refractory cutaneous granulomatous inflammation.
The Technique
A 69-year-old woman presented with a crusted erythematous papule with surrounding inflammation on the left nasal ala of 5 years’ duration (Figure 1). Histopathology demonstrated a localized cutaneous granulomatous process. There was no clinical, radiographic, or laboratory evidence of systemic sarcoidosis. Infectious causes were excluded through negative tissue cultures and special stains, including auramine-rhodamine. Over a 3-month period following initial presentation, the lesion proved refractory to intralesional 5-fluorouracil, intralesional triamcinolone acetonide, pentoxifylline, N-acetylcysteine, and shave excision (Figure 2).
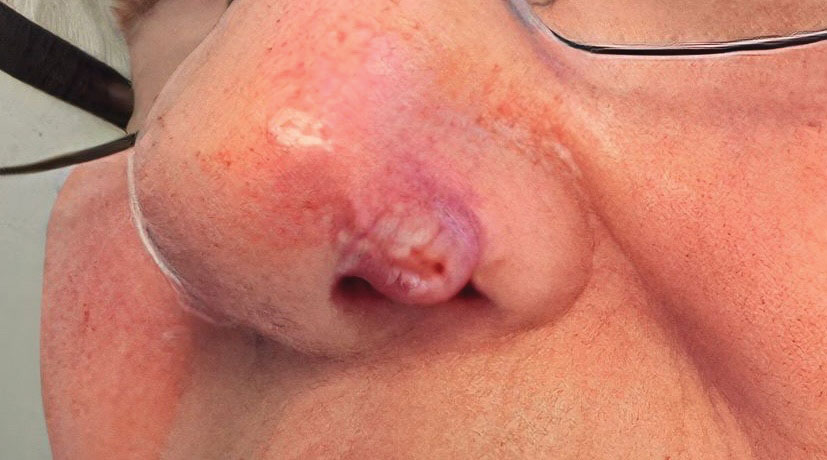
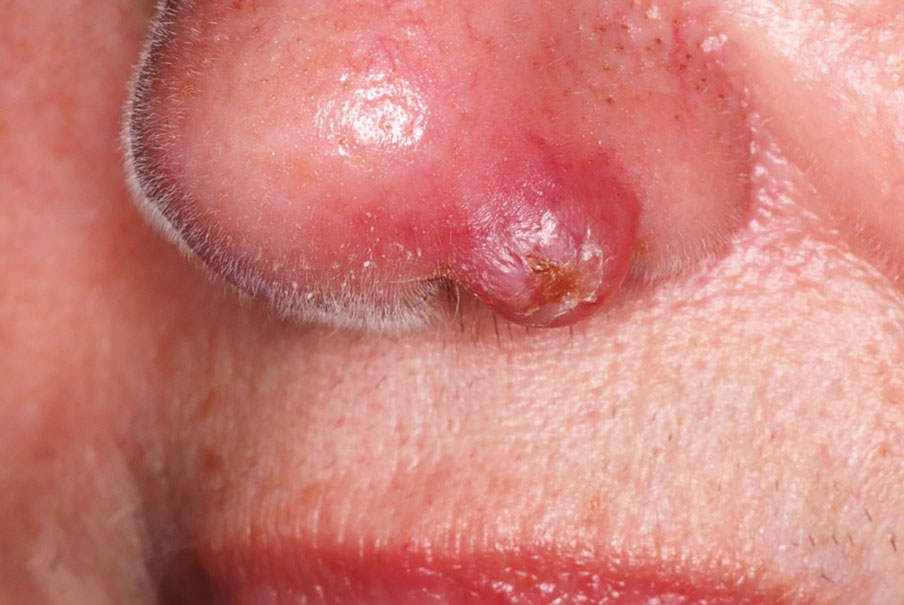
At 3-month follow-up, given the lesion’s persistence despite local and systemic anti-inflammatory approaches and our intent to avoid repeated corticosteroid exposure or more aggressive surgery in a cosmetically sensitive facial site, we attempted treatment with intralesional adalimumab. A 40-mg/0.4-mL dose of adalimumab was withdrawn directly from a prefilled autoinjector and placed into a sterile container, then transferred to a syringe fitted with a 30-gauge needle. Finally, the full 0.4 mL was injected intralesionally (Figure 3) until complete blanching of the lesion was achieved.
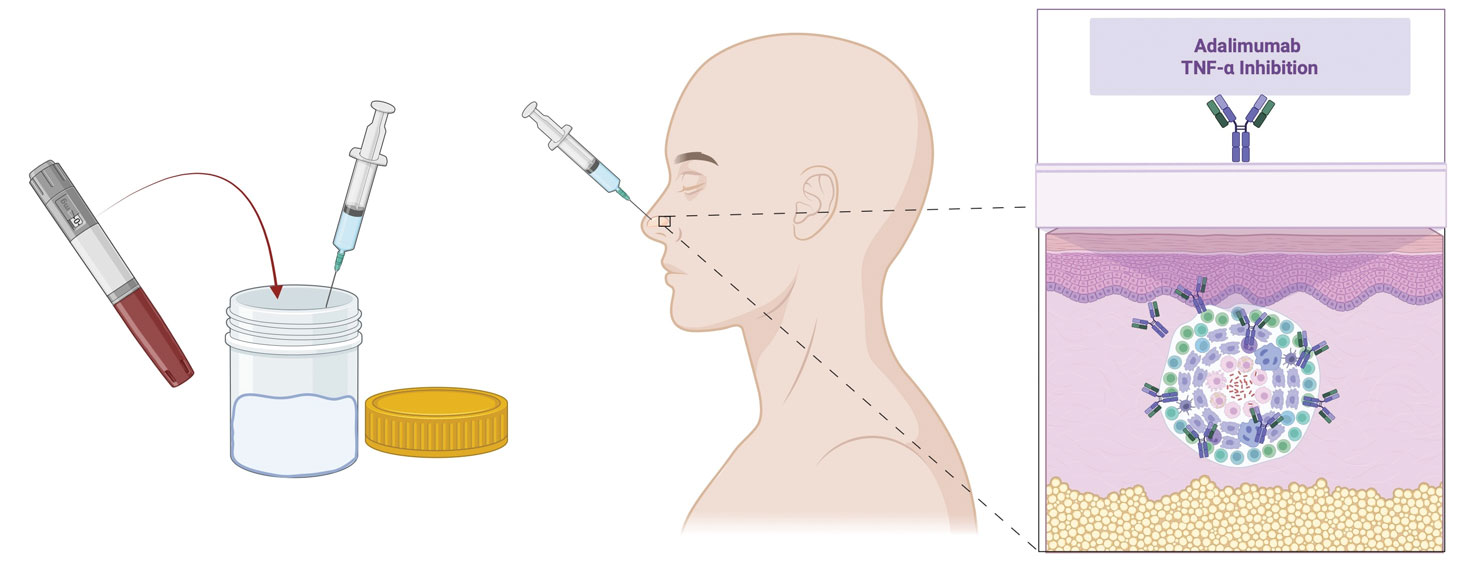
At 1-month follow-up, the lesion demonstrated decreased erythema and crusting (Figure 4A). The patient subsequently underwent 12 adalimumab injections over an 18-month period with marked reduction in size and erythema of the lesion without complications (Figure 4B). In addition, doxycycline 100 mg/d was started 11 months after the first adalimumab injection to address mild residual inflammation (Figure 4C); after 4 months, the dose was reduced to 50 mg/d due to gastrointestinal adverse effects. Doxycycline was maintained for 3 additional months with persistent improvement of the lesion.
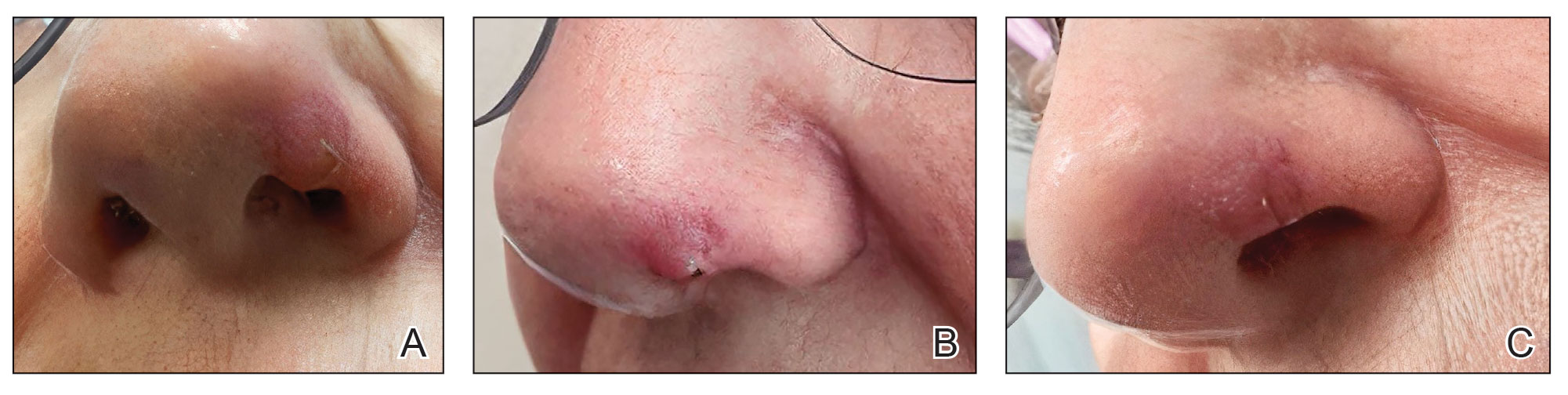
Practice Implication
Intralesional administration of adalimumab may represent a useful therapeutic option for localized refractory granulomatous inflammation, particularly in sensitive areas such as the face, where conventional therapies may be limited by adverse effects or suboptimal response. Localized delivery of TNF-α inhibition directly to the site of inflammation may allow for clinical improvement while minimizing systemic exposure associated with biologic therapy.2 This approach may be particularly advantageous in cases in which repeated intralesional corticosteroid injections raise concern for atrophy or dyspigmentation, or when surgical intervention carries a risk for recurrence or cosmetic morbidity.1,2 Given the established role of TNF-α in granuloma formation and maintenance, intralesional adalimumab provides a biologically plausible targeted therapeutic strategy. Further studies are needed to evaluate the potential applications in other cutaneous granulomatous dermatoses.2,3
- Philips MA, Lynch J, Azmi FH. Ulcerative cutaneous sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917. doi:10.1016/j.jaad.2005.02.023
- Balan K, Sagut P, Ederle AC, et al. Cutaneous sarcoidosis treated with intralesional adalimumab. Int J Dermatol. 2025;64:1120-1121. doi:10.1111/ijd.17549
- Dunn C, Whitney Z, Foss M, et al. Intralesional certolizumab for refractory lupus pernio. JAMA Dermatol. 2023;159:890-891. doi:10.1001 /jamadermatol.2023.0987
Practice Gap
Chronic localized granulomatous inflammation can be difficult to manage, particularly when manifesting on the face. Intralesional corticosteroids may lead to atrophy and dyspigmentation and therefore must be used cautiously in cosmetically sensitive areas.1 Surgical removal can lead to recurrence, and systemic agents may carry risks disproportionate to disease burden. Although tumor necrosis factor (TNF) α inhibitors are effective systemically, their localized use in cutaneous granulomatous dermatoses remains underreported.1-3 We describe a technique using intralesional injection of adalimumab to treat chronic refractory cutaneous granulomatous inflammation.
The Technique
A 69-year-old woman presented with a crusted erythematous papule with surrounding inflammation on the left nasal ala of 5 years’ duration (Figure 1). Histopathology demonstrated a localized cutaneous granulomatous process. There was no clinical, radiographic, or laboratory evidence of systemic sarcoidosis. Infectious causes were excluded through negative tissue cultures and special stains, including auramine-rhodamine. Over a 3-month period following initial presentation, the lesion proved refractory to intralesional 5-fluorouracil, intralesional triamcinolone acetonide, pentoxifylline, N-acetylcysteine, and shave excision (Figure 2).
At 3-month follow-up, given the lesion’s persistence despite local and systemic anti-inflammatory approaches and our intent to avoid repeated corticosteroid exposure or more aggressive surgery in a cosmetically sensitive facial site, we attempted treatment with intralesional adalimumab. A 40-mg/0.4-mL dose of adalimumab was withdrawn directly from a prefilled autoinjector and placed into a sterile container, then transferred to a syringe fitted with a 30-gauge needle. Finally, the full 0.4 mL was injected intralesionally (Figure 3) until complete blanching of the lesion was achieved.
At 1-month follow-up, the lesion demonstrated decreased erythema and crusting (Figure 4A). The patient subsequently underwent 12 adalimumab injections over an 18-month period with marked reduction in size and erythema of the lesion without complications (Figure 4B). In addition, doxycycline 100 mg/d was started 11 months after the first adalimumab injection to address mild residual inflammation (Figure 4C); after 4 months, the dose was reduced to 50 mg/d due to gastrointestinal adverse effects. Doxycycline was maintained for 3 additional months with persistent improvement of the lesion.
Practice Implication
Intralesional administration of adalimumab may represent a useful therapeutic option for localized refractory granulomatous inflammation, particularly in sensitive areas such as the face, where conventional therapies may be limited by adverse effects or suboptimal response. Localized delivery of TNF-α inhibition directly to the site of inflammation may allow for clinical improvement while minimizing systemic exposure associated with biologic therapy.2 This approach may be particularly advantageous in cases in which repeated intralesional corticosteroid injections raise concern for atrophy or dyspigmentation, or when surgical intervention carries a risk for recurrence or cosmetic morbidity.1,2 Given the established role of TNF-α in granuloma formation and maintenance, intralesional adalimumab provides a biologically plausible targeted therapeutic strategy. Further studies are needed to evaluate the potential applications in other cutaneous granulomatous dermatoses.2,3
Practice Gap
Chronic localized granulomatous inflammation can be difficult to manage, particularly when manifesting on the face. Intralesional corticosteroids may lead to atrophy and dyspigmentation and therefore must be used cautiously in cosmetically sensitive areas.1 Surgical removal can lead to recurrence, and systemic agents may carry risks disproportionate to disease burden. Although tumor necrosis factor (TNF) α inhibitors are effective systemically, their localized use in cutaneous granulomatous dermatoses remains underreported.1-3 We describe a technique using intralesional injection of adalimumab to treat chronic refractory cutaneous granulomatous inflammation.
The Technique
A 69-year-old woman presented with a crusted erythematous papule with surrounding inflammation on the left nasal ala of 5 years’ duration (Figure 1). Histopathology demonstrated a localized cutaneous granulomatous process. There was no clinical, radiographic, or laboratory evidence of systemic sarcoidosis. Infectious causes were excluded through negative tissue cultures and special stains, including auramine-rhodamine. Over a 3-month period following initial presentation, the lesion proved refractory to intralesional 5-fluorouracil, intralesional triamcinolone acetonide, pentoxifylline, N-acetylcysteine, and shave excision (Figure 2).
At 3-month follow-up, given the lesion’s persistence despite local and systemic anti-inflammatory approaches and our intent to avoid repeated corticosteroid exposure or more aggressive surgery in a cosmetically sensitive facial site, we attempted treatment with intralesional adalimumab. A 40-mg/0.4-mL dose of adalimumab was withdrawn directly from a prefilled autoinjector and placed into a sterile container, then transferred to a syringe fitted with a 30-gauge needle. Finally, the full 0.4 mL was injected intralesionally (Figure 3) until complete blanching of the lesion was achieved.
At 1-month follow-up, the lesion demonstrated decreased erythema and crusting (Figure 4A). The patient subsequently underwent 12 adalimumab injections over an 18-month period with marked reduction in size and erythema of the lesion without complications (Figure 4B). In addition, doxycycline 100 mg/d was started 11 months after the first adalimumab injection to address mild residual inflammation (Figure 4C); after 4 months, the dose was reduced to 50 mg/d due to gastrointestinal adverse effects. Doxycycline was maintained for 3 additional months with persistent improvement of the lesion.
Practice Implication
Intralesional administration of adalimumab may represent a useful therapeutic option for localized refractory granulomatous inflammation, particularly in sensitive areas such as the face, where conventional therapies may be limited by adverse effects or suboptimal response. Localized delivery of TNF-α inhibition directly to the site of inflammation may allow for clinical improvement while minimizing systemic exposure associated with biologic therapy.2 This approach may be particularly advantageous in cases in which repeated intralesional corticosteroid injections raise concern for atrophy or dyspigmentation, or when surgical intervention carries a risk for recurrence or cosmetic morbidity.1,2 Given the established role of TNF-α in granuloma formation and maintenance, intralesional adalimumab provides a biologically plausible targeted therapeutic strategy. Further studies are needed to evaluate the potential applications in other cutaneous granulomatous dermatoses.2,3
- Philips MA, Lynch J, Azmi FH. Ulcerative cutaneous sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917. doi:10.1016/j.jaad.2005.02.023
- Balan K, Sagut P, Ederle AC, et al. Cutaneous sarcoidosis treated with intralesional adalimumab. Int J Dermatol. 2025;64:1120-1121. doi:10.1111/ijd.17549
- Dunn C, Whitney Z, Foss M, et al. Intralesional certolizumab for refractory lupus pernio. JAMA Dermatol. 2023;159:890-891. doi:10.1001 /jamadermatol.2023.0987
- Philips MA, Lynch J, Azmi FH. Ulcerative cutaneous sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917. doi:10.1016/j.jaad.2005.02.023
- Balan K, Sagut P, Ederle AC, et al. Cutaneous sarcoidosis treated with intralesional adalimumab. Int J Dermatol. 2025;64:1120-1121. doi:10.1111/ijd.17549
- Dunn C, Whitney Z, Foss M, et al. Intralesional certolizumab for refractory lupus pernio. JAMA Dermatol. 2023;159:890-891. doi:10.1001 /jamadermatol.2023.0987
Using Intralesional Adalimumab for Chronic Refractory Cutaneous Granulomatous Inflammation
Using Intralesional Adalimumab for Chronic Refractory Cutaneous Granulomatous Inflammation
