User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Debunking Psoriasis Myths: Is Psoriasis Contagious?
Myth: Psoriasis is contagious
One of the challenges patients with psoriasis face is misinformation among their peers, especially about the cause of psoriasis. Results from a survey conducted in France (N=1005) regarding knowledge of psoriasis and perceptions toward psoriasis patients indicated that approximately 16.5% of respondents believed that psoriasis is contagious; 6.8% believed that the disease is related to personal hygiene and 3.2% believed that it affects more people with low personal hygiene (Halioua et al). Moreover, approximately 62.4% of respondents recognized a lack of information about psoriasis and 19.7% noted that they have misconceptions about the disease.
Another study from Poland on the feelings of stigmatization among psoriasis patients (N=102) found that 72 psoriasis patients reported that they felt other people think their skin disease is contagious; only 30 psoriasis patients indicated that they did not feel this way (Hrehorów et al).
These findings underscore the need for more information on psoriasis in the general public worldwide. It is important to spread the message that psoriasis is a chronic inflammatory disease of the immune system that affects approximately 7.5 million individuals in the United States. One cannot "catch" psoriasis; psoriasis starts or worsens because of a variety of triggers (eg, infections, injury to the skin, stress, cold weather, smoking, heavy alcohol consumption, certain medications). The National Institute of Arthritis and Musculoskeletal and Skin Diseases offers a simple but revealing explanation for the cause of psoriasis: "Normally, T cells help protect the body against infection and disease. In the case of psoriasis, T cells are put into action by mistake and become so active that they trigger other immune responses, which lead to inflammation and to rapid turnover of skin cells." Genetics may play a role in the development of psoriasis, along with environmental factors.
With several new therapies available, psoriasis has gained media attention. Dermatologists can circulate more information about the causes of psoriasis, which may work toward helping patients with psoriasis cope with the social impact of the condition.
Halioua B, Sid-Mohand D, Roussel ME, et al. Extent of misconceptions, negative prejudices and discriminatory behaviour to psoriasis patients in France. J Eur Acad Dermatol Venereol. 2016;30:650-654.
Hrehorów E, Salomon J, Matusiak L, et al. Patients with psoriasis feel stigmatized. Acta Derm Venereol. 2012;92:67-72.
Psoriasis. American Academy of Dermatology website. https://www.aad.org/media/stats/conditions/psoriasis. Accessed July 7, 2016.
Psoriasis causes. Mayo Clinic website. http://www.mayoclinic.org/diseases-conditions/psoriasis/basics/causes/con-20030838. Published June 17, 2015. Accessed July 7, 2016.
Questions and answers about psoriasis. National Institute of Arthritis and Musculoskeletal and Skin Diseases website. http://www.niams.nih.gov/Health_Info/Psoriasis/default.asp. Published October 2013. Accessed July 7, 2016.
Myth: Psoriasis is contagious
One of the challenges patients with psoriasis face is misinformation among their peers, especially about the cause of psoriasis. Results from a survey conducted in France (N=1005) regarding knowledge of psoriasis and perceptions toward psoriasis patients indicated that approximately 16.5% of respondents believed that psoriasis is contagious; 6.8% believed that the disease is related to personal hygiene and 3.2% believed that it affects more people with low personal hygiene (Halioua et al). Moreover, approximately 62.4% of respondents recognized a lack of information about psoriasis and 19.7% noted that they have misconceptions about the disease.
Another study from Poland on the feelings of stigmatization among psoriasis patients (N=102) found that 72 psoriasis patients reported that they felt other people think their skin disease is contagious; only 30 psoriasis patients indicated that they did not feel this way (Hrehorów et al).
These findings underscore the need for more information on psoriasis in the general public worldwide. It is important to spread the message that psoriasis is a chronic inflammatory disease of the immune system that affects approximately 7.5 million individuals in the United States. One cannot "catch" psoriasis; psoriasis starts or worsens because of a variety of triggers (eg, infections, injury to the skin, stress, cold weather, smoking, heavy alcohol consumption, certain medications). The National Institute of Arthritis and Musculoskeletal and Skin Diseases offers a simple but revealing explanation for the cause of psoriasis: "Normally, T cells help protect the body against infection and disease. In the case of psoriasis, T cells are put into action by mistake and become so active that they trigger other immune responses, which lead to inflammation and to rapid turnover of skin cells." Genetics may play a role in the development of psoriasis, along with environmental factors.
With several new therapies available, psoriasis has gained media attention. Dermatologists can circulate more information about the causes of psoriasis, which may work toward helping patients with psoriasis cope with the social impact of the condition.
Myth: Psoriasis is contagious
One of the challenges patients with psoriasis face is misinformation among their peers, especially about the cause of psoriasis. Results from a survey conducted in France (N=1005) regarding knowledge of psoriasis and perceptions toward psoriasis patients indicated that approximately 16.5% of respondents believed that psoriasis is contagious; 6.8% believed that the disease is related to personal hygiene and 3.2% believed that it affects more people with low personal hygiene (Halioua et al). Moreover, approximately 62.4% of respondents recognized a lack of information about psoriasis and 19.7% noted that they have misconceptions about the disease.
Another study from Poland on the feelings of stigmatization among psoriasis patients (N=102) found that 72 psoriasis patients reported that they felt other people think their skin disease is contagious; only 30 psoriasis patients indicated that they did not feel this way (Hrehorów et al).
These findings underscore the need for more information on psoriasis in the general public worldwide. It is important to spread the message that psoriasis is a chronic inflammatory disease of the immune system that affects approximately 7.5 million individuals in the United States. One cannot "catch" psoriasis; psoriasis starts or worsens because of a variety of triggers (eg, infections, injury to the skin, stress, cold weather, smoking, heavy alcohol consumption, certain medications). The National Institute of Arthritis and Musculoskeletal and Skin Diseases offers a simple but revealing explanation for the cause of psoriasis: "Normally, T cells help protect the body against infection and disease. In the case of psoriasis, T cells are put into action by mistake and become so active that they trigger other immune responses, which lead to inflammation and to rapid turnover of skin cells." Genetics may play a role in the development of psoriasis, along with environmental factors.
With several new therapies available, psoriasis has gained media attention. Dermatologists can circulate more information about the causes of psoriasis, which may work toward helping patients with psoriasis cope with the social impact of the condition.
Halioua B, Sid-Mohand D, Roussel ME, et al. Extent of misconceptions, negative prejudices and discriminatory behaviour to psoriasis patients in France. J Eur Acad Dermatol Venereol. 2016;30:650-654.
Hrehorów E, Salomon J, Matusiak L, et al. Patients with psoriasis feel stigmatized. Acta Derm Venereol. 2012;92:67-72.
Psoriasis. American Academy of Dermatology website. https://www.aad.org/media/stats/conditions/psoriasis. Accessed July 7, 2016.
Psoriasis causes. Mayo Clinic website. http://www.mayoclinic.org/diseases-conditions/psoriasis/basics/causes/con-20030838. Published June 17, 2015. Accessed July 7, 2016.
Questions and answers about psoriasis. National Institute of Arthritis and Musculoskeletal and Skin Diseases website. http://www.niams.nih.gov/Health_Info/Psoriasis/default.asp. Published October 2013. Accessed July 7, 2016.
Halioua B, Sid-Mohand D, Roussel ME, et al. Extent of misconceptions, negative prejudices and discriminatory behaviour to psoriasis patients in France. J Eur Acad Dermatol Venereol. 2016;30:650-654.
Hrehorów E, Salomon J, Matusiak L, et al. Patients with psoriasis feel stigmatized. Acta Derm Venereol. 2012;92:67-72.
Psoriasis. American Academy of Dermatology website. https://www.aad.org/media/stats/conditions/psoriasis. Accessed July 7, 2016.
Psoriasis causes. Mayo Clinic website. http://www.mayoclinic.org/diseases-conditions/psoriasis/basics/causes/con-20030838. Published June 17, 2015. Accessed July 7, 2016.
Questions and answers about psoriasis. National Institute of Arthritis and Musculoskeletal and Skin Diseases website. http://www.niams.nih.gov/Health_Info/Psoriasis/default.asp. Published October 2013. Accessed July 7, 2016.

Cognitive Biases in Dermatology Training
As young physicians, we are taught to be as objective as possible when evaluating a patient; however, cognitive biases are regularly encountered in day-to-day patient experiences and can unfortunately influence our clinical decision-making skills to be subpar.
Consider the following case: An overweight 74-year-old man with diabetes mellitus and a nonhealing ulceration on the left lower extremity presented to the emergency department for repeat evaluation. He previously had been treated by an outside dermatologist for stasis dermatitis and was being managed with compression, elevation, and lubrication of both lower extremities. Often, the initial reaction is to conclude that the patient does in fact have an ulceration associated with stasis dermatitis and changing the management strategy or performing a biopsy would not change the outcome. However, this response limits the potential to provide the patient with a thorough examination. If the patient is treated with the same management strategies that previously failed rather than delving into all the causes for nonhealing ulceration on the left lower extremity, a vital diagnosis could be missed. In this scenario, when the patient was ultimately biopsied, the diagnosis was an ulcerative squamous cell carcinoma.
These subconscious predetermined decisions regarding difficult patient encounters come from the physician’s heuristics, a process of decision-making wherein a snap judgment about a patient occurs because it is similar to prior patient encounters or a set of views from prior knowledge of the disease.1,2
A recent article by Cohen and Burgin3 elucidated a set of cognitive biases that often are encountered in dermatology practices, including affective, anchoring, availability, and confirmation biases; zebra retreat; and Sutton’s slip.
Affective Bias
Affective bias is a process in which emotions regarding a patient interaction alter the objective prospective and reasoning of a patient. For example, consider the case of a pemphigus vulgaris patient who does not want to be on prednisone due to weight gain and persistently presents to the dermatology clinic insisting that the physician taper the dosage. To avoid the constant frustration and upsetting the patient further, the dermatologist tapers the dosage of prednisone prematurely and the patient has a flare.
Anchoring Bias
Anchoring bias occurs when initial information regarding a patient causes one to jump to a conclusion rather than developing a thorough history. An example may be if an infant presents with a mole on the nasal dorsum that the patient’s father reports has only been present for a short while. Without performing imaging studies or asking for further history, the physician decides to biopsy the lesion. The biopsy results show a neural mass, such that a nasal glioma cannot be ruled out. In this bias, magnetic resonance imaging would have been prudent prior to biopsy and premature action.
Availability Bias
Availability bias refers to a diagnosis that immediately comes to mind, as it is common or recently encountered, such as in the example presented at the beginning of this column about the patient with squamous cell carcinoma.
Confirmation Bias
Confirmation bias caters to elucidating information that confirms your own clinical suspicion as opposed to determining the true cause of the disease etiology. Consider the following example: An obese patient presents with a history of painful sores on the bilateral lower extremities. The physician asks specifically about diabetes mellitus and mobility. When the patient answers yes to poor mobility and diabetes mellitus, the physician asks questions confirming an initial suspected diagnosis of stasis dermatitis. Unfortunately, as the patient continues to get worse, it is revealed that his medication history indicates he has been taking sulfasalazine for several years, and it is eventually determined that the patient has cutaneous Crohn disease.
Zebra Retreat
This bias describes a physician’s unwillingness to consider a diagnosis because it is very obscure, even if it is correct. For example, the case of the patient described in the previous example with a diagnosis of cutaneous Crohn disease also can be considered as an example of zebra retreat. Because the clinician may rarely think of this diagnosis due to its infrequent presentation, he/she may not consider doing a biopsy or investigate further.
Sutton’s Slip
This bias describes a situation in which a physician disregards a problem because a thorough examination is not performed, which is classically noted when physicians treat their family and friends. If asked about a mole or lesion regarding its questionable nature, a dermatologist may either disregard it or not evaluate it carefully, as the person is in a casual setting.
Final Thoughts
Although there are several other types of cognitive biases, those described here show that on several occasions, dermatologists can be swayed toward an incorrect diagnosis simply because of a subconscious thought process. Often times, such as in multiple-choice examinations, initial guesses are usually the best answers, but care has to be taken when in a clinical setting. Our patients rarely are good historians and do not present in well-written question stems. The biases emphasize that dermatologists in training should keep their minds open, focus on getting a clear and concise history, and use their knowledge as a tool to derive a well thought-out answer.
1. Croskerry P. The importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med. 2003;78:775-780.
2. Hicks EP, Kluemper GT. Heuristic reasoning and cognitive biases: are they hindrances to judgments and decision making in orthodontics? Am J Orthod Dentofacial Orthop. 2011;139:297-304.
3. Cohen JM, Burgin S. Cognitive biases in clinical decision making: a primer for the practicing dermatologist. JAMA Dermatol. 2016;152:253-254.
As young physicians, we are taught to be as objective as possible when evaluating a patient; however, cognitive biases are regularly encountered in day-to-day patient experiences and can unfortunately influence our clinical decision-making skills to be subpar.
Consider the following case: An overweight 74-year-old man with diabetes mellitus and a nonhealing ulceration on the left lower extremity presented to the emergency department for repeat evaluation. He previously had been treated by an outside dermatologist for stasis dermatitis and was being managed with compression, elevation, and lubrication of both lower extremities. Often, the initial reaction is to conclude that the patient does in fact have an ulceration associated with stasis dermatitis and changing the management strategy or performing a biopsy would not change the outcome. However, this response limits the potential to provide the patient with a thorough examination. If the patient is treated with the same management strategies that previously failed rather than delving into all the causes for nonhealing ulceration on the left lower extremity, a vital diagnosis could be missed. In this scenario, when the patient was ultimately biopsied, the diagnosis was an ulcerative squamous cell carcinoma.
These subconscious predetermined decisions regarding difficult patient encounters come from the physician’s heuristics, a process of decision-making wherein a snap judgment about a patient occurs because it is similar to prior patient encounters or a set of views from prior knowledge of the disease.1,2
A recent article by Cohen and Burgin3 elucidated a set of cognitive biases that often are encountered in dermatology practices, including affective, anchoring, availability, and confirmation biases; zebra retreat; and Sutton’s slip.
Affective Bias
Affective bias is a process in which emotions regarding a patient interaction alter the objective prospective and reasoning of a patient. For example, consider the case of a pemphigus vulgaris patient who does not want to be on prednisone due to weight gain and persistently presents to the dermatology clinic insisting that the physician taper the dosage. To avoid the constant frustration and upsetting the patient further, the dermatologist tapers the dosage of prednisone prematurely and the patient has a flare.
Anchoring Bias
Anchoring bias occurs when initial information regarding a patient causes one to jump to a conclusion rather than developing a thorough history. An example may be if an infant presents with a mole on the nasal dorsum that the patient’s father reports has only been present for a short while. Without performing imaging studies or asking for further history, the physician decides to biopsy the lesion. The biopsy results show a neural mass, such that a nasal glioma cannot be ruled out. In this bias, magnetic resonance imaging would have been prudent prior to biopsy and premature action.
Availability Bias
Availability bias refers to a diagnosis that immediately comes to mind, as it is common or recently encountered, such as in the example presented at the beginning of this column about the patient with squamous cell carcinoma.
Confirmation Bias
Confirmation bias caters to elucidating information that confirms your own clinical suspicion as opposed to determining the true cause of the disease etiology. Consider the following example: An obese patient presents with a history of painful sores on the bilateral lower extremities. The physician asks specifically about diabetes mellitus and mobility. When the patient answers yes to poor mobility and diabetes mellitus, the physician asks questions confirming an initial suspected diagnosis of stasis dermatitis. Unfortunately, as the patient continues to get worse, it is revealed that his medication history indicates he has been taking sulfasalazine for several years, and it is eventually determined that the patient has cutaneous Crohn disease.
Zebra Retreat
This bias describes a physician’s unwillingness to consider a diagnosis because it is very obscure, even if it is correct. For example, the case of the patient described in the previous example with a diagnosis of cutaneous Crohn disease also can be considered as an example of zebra retreat. Because the clinician may rarely think of this diagnosis due to its infrequent presentation, he/she may not consider doing a biopsy or investigate further.
Sutton’s Slip
This bias describes a situation in which a physician disregards a problem because a thorough examination is not performed, which is classically noted when physicians treat their family and friends. If asked about a mole or lesion regarding its questionable nature, a dermatologist may either disregard it or not evaluate it carefully, as the person is in a casual setting.
Final Thoughts
Although there are several other types of cognitive biases, those described here show that on several occasions, dermatologists can be swayed toward an incorrect diagnosis simply because of a subconscious thought process. Often times, such as in multiple-choice examinations, initial guesses are usually the best answers, but care has to be taken when in a clinical setting. Our patients rarely are good historians and do not present in well-written question stems. The biases emphasize that dermatologists in training should keep their minds open, focus on getting a clear and concise history, and use their knowledge as a tool to derive a well thought-out answer.
As young physicians, we are taught to be as objective as possible when evaluating a patient; however, cognitive biases are regularly encountered in day-to-day patient experiences and can unfortunately influence our clinical decision-making skills to be subpar.
Consider the following case: An overweight 74-year-old man with diabetes mellitus and a nonhealing ulceration on the left lower extremity presented to the emergency department for repeat evaluation. He previously had been treated by an outside dermatologist for stasis dermatitis and was being managed with compression, elevation, and lubrication of both lower extremities. Often, the initial reaction is to conclude that the patient does in fact have an ulceration associated with stasis dermatitis and changing the management strategy or performing a biopsy would not change the outcome. However, this response limits the potential to provide the patient with a thorough examination. If the patient is treated with the same management strategies that previously failed rather than delving into all the causes for nonhealing ulceration on the left lower extremity, a vital diagnosis could be missed. In this scenario, when the patient was ultimately biopsied, the diagnosis was an ulcerative squamous cell carcinoma.
These subconscious predetermined decisions regarding difficult patient encounters come from the physician’s heuristics, a process of decision-making wherein a snap judgment about a patient occurs because it is similar to prior patient encounters or a set of views from prior knowledge of the disease.1,2
A recent article by Cohen and Burgin3 elucidated a set of cognitive biases that often are encountered in dermatology practices, including affective, anchoring, availability, and confirmation biases; zebra retreat; and Sutton’s slip.
Affective Bias
Affective bias is a process in which emotions regarding a patient interaction alter the objective prospective and reasoning of a patient. For example, consider the case of a pemphigus vulgaris patient who does not want to be on prednisone due to weight gain and persistently presents to the dermatology clinic insisting that the physician taper the dosage. To avoid the constant frustration and upsetting the patient further, the dermatologist tapers the dosage of prednisone prematurely and the patient has a flare.
Anchoring Bias
Anchoring bias occurs when initial information regarding a patient causes one to jump to a conclusion rather than developing a thorough history. An example may be if an infant presents with a mole on the nasal dorsum that the patient’s father reports has only been present for a short while. Without performing imaging studies or asking for further history, the physician decides to biopsy the lesion. The biopsy results show a neural mass, such that a nasal glioma cannot be ruled out. In this bias, magnetic resonance imaging would have been prudent prior to biopsy and premature action.
Availability Bias
Availability bias refers to a diagnosis that immediately comes to mind, as it is common or recently encountered, such as in the example presented at the beginning of this column about the patient with squamous cell carcinoma.
Confirmation Bias
Confirmation bias caters to elucidating information that confirms your own clinical suspicion as opposed to determining the true cause of the disease etiology. Consider the following example: An obese patient presents with a history of painful sores on the bilateral lower extremities. The physician asks specifically about diabetes mellitus and mobility. When the patient answers yes to poor mobility and diabetes mellitus, the physician asks questions confirming an initial suspected diagnosis of stasis dermatitis. Unfortunately, as the patient continues to get worse, it is revealed that his medication history indicates he has been taking sulfasalazine for several years, and it is eventually determined that the patient has cutaneous Crohn disease.
Zebra Retreat
This bias describes a physician’s unwillingness to consider a diagnosis because it is very obscure, even if it is correct. For example, the case of the patient described in the previous example with a diagnosis of cutaneous Crohn disease also can be considered as an example of zebra retreat. Because the clinician may rarely think of this diagnosis due to its infrequent presentation, he/she may not consider doing a biopsy or investigate further.
Sutton’s Slip
This bias describes a situation in which a physician disregards a problem because a thorough examination is not performed, which is classically noted when physicians treat their family and friends. If asked about a mole or lesion regarding its questionable nature, a dermatologist may either disregard it or not evaluate it carefully, as the person is in a casual setting.
Final Thoughts
Although there are several other types of cognitive biases, those described here show that on several occasions, dermatologists can be swayed toward an incorrect diagnosis simply because of a subconscious thought process. Often times, such as in multiple-choice examinations, initial guesses are usually the best answers, but care has to be taken when in a clinical setting. Our patients rarely are good historians and do not present in well-written question stems. The biases emphasize that dermatologists in training should keep their minds open, focus on getting a clear and concise history, and use their knowledge as a tool to derive a well thought-out answer.
1. Croskerry P. The importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med. 2003;78:775-780.
2. Hicks EP, Kluemper GT. Heuristic reasoning and cognitive biases: are they hindrances to judgments and decision making in orthodontics? Am J Orthod Dentofacial Orthop. 2011;139:297-304.
3. Cohen JM, Burgin S. Cognitive biases in clinical decision making: a primer for the practicing dermatologist. JAMA Dermatol. 2016;152:253-254.
1. Croskerry P. The importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med. 2003;78:775-780.
2. Hicks EP, Kluemper GT. Heuristic reasoning and cognitive biases: are they hindrances to judgments and decision making in orthodontics? Am J Orthod Dentofacial Orthop. 2011;139:297-304.
3. Cohen JM, Burgin S. Cognitive biases in clinical decision making: a primer for the practicing dermatologist. JAMA Dermatol. 2016;152:253-254.

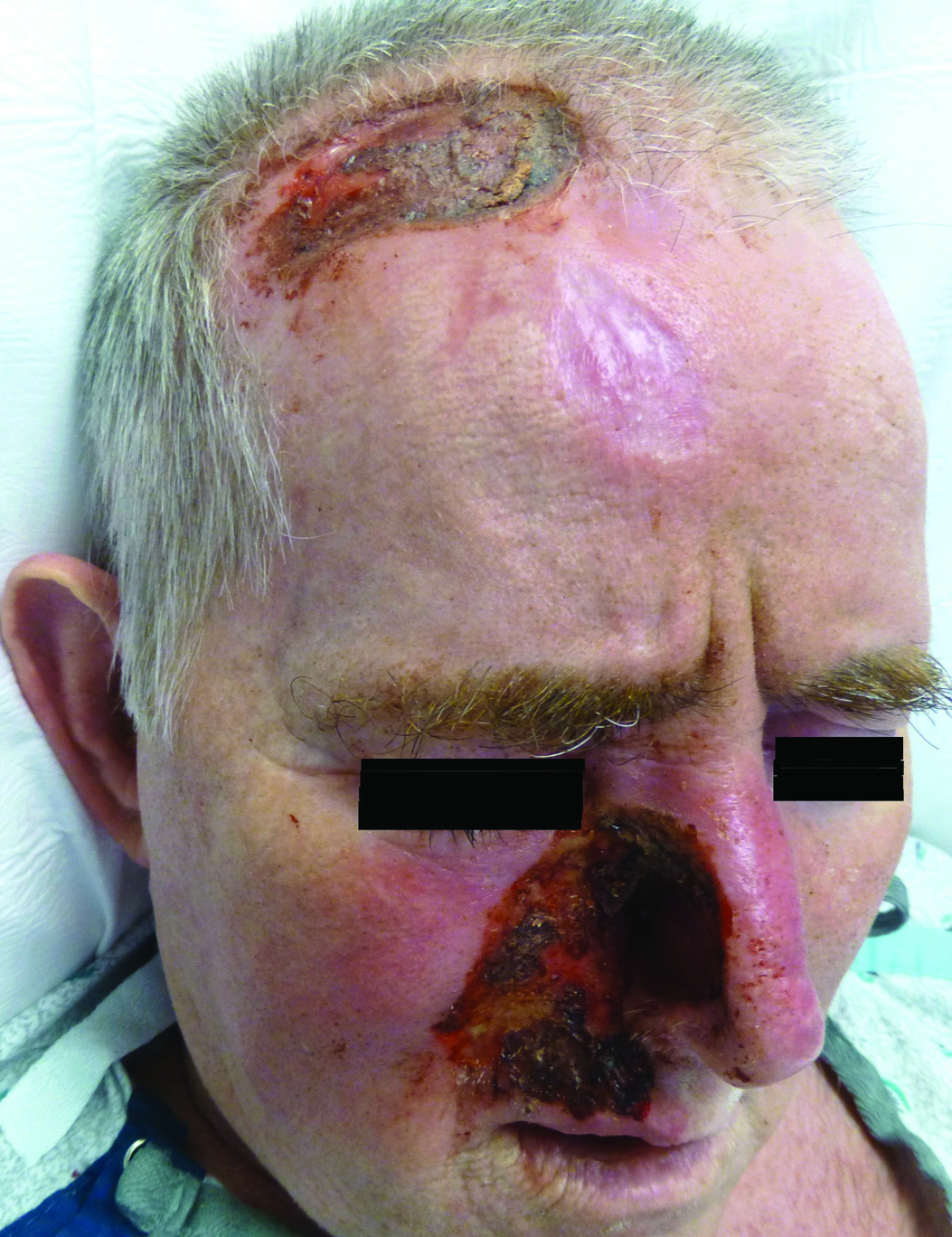
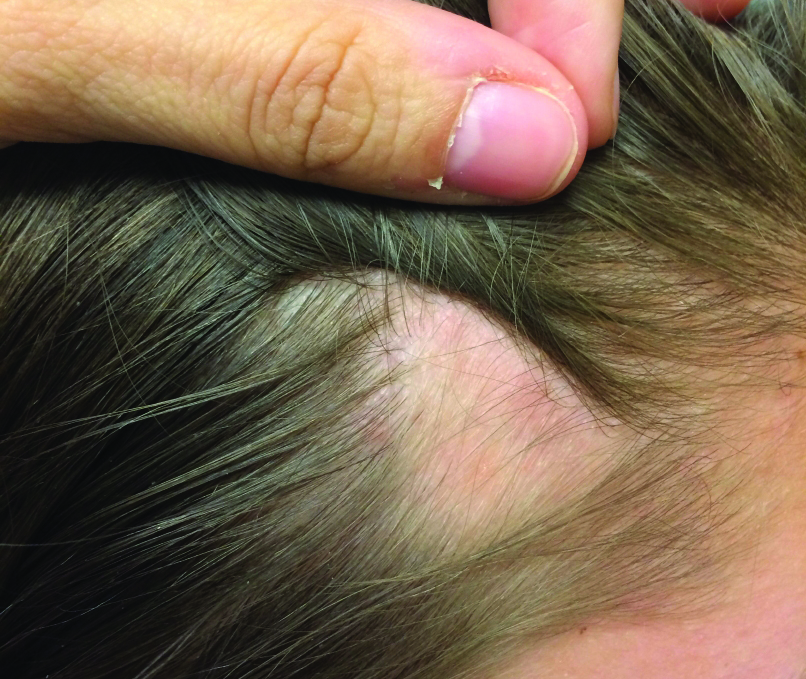
Patch of Hair Loss on the Scalp
The Diagnosis: Temporal Triangular Alopecia
Temporal triangular alopecia (TTA), also known as congenital triangular alopecia, was first described in the early 1900s.1 It presents clinically as a triangular-shaped area of nonscarring alopecia either unilaterally or bilaterally. Limited clinical data suggest that most unilateral cases are on the left frontotemporal region of the scalp. In bilateral cases, there may be asymmetry in size of the area involved.2 Dermatoscopically, TTA is characterized by decreased terminal hair follicle density as well as the presence of vellus hairs with an absence of inflammation.3 The majority of TTA is noted between birth and 6 years of life with the areas staying stable thereafter. Large areas of TTA may suggest cerebello-trigeminal-dermal dysplasia (Gomez-Lopez-Hernandez syndrome), a rare neurocutaneous syndrome characterized by rhombencephalosynapsis, trigeminal anesthesia, and parietooccipital alopecia (Online Mendelian Inheritance in Man 601853).4 Although TTA is largely idiopathic, it has been suggested that the trait may be paradominant, whereby a postzygotic loss of the wild-type allele in a heterozygotic state causes triangular alopecia and reflects hamartomatous mosaicism.5 It also is an important mimicker of alopecia areata. Correct identification prevents unnecessary treatment to the areas of the scalp. Hair restoration surgery has been reported as a tool to treat this disorder.6
- Tosti A. Congenital triangular alopecia. report of fourteen cases. J Am Acad Dermatol. 1987;16:991-993.
- Armstrong DK, Burrows D. Congenital triangular alopecia. Pediatr Dermatol. 1996;13:394-396.
- Iorizzo M, Pazzaglia M, Starace M, et al. Videodermoscopy: a useful tool for diagnosing congenital triangular alopecia. Pediatr Dermatol. 2008;25:652-654.
- Assoly P, Happle R. A hairy paradox: congenital triangular alopecia with a central hair tuft. Dermatology. 2010;221:107-109.
- Happle R. Congenital triangular alopecia may be categorized as a paradominant trait. Eur J Dermatol. 2003;13:346-347.
- Wu WY, Otberg N, Kang H, et al. Successful treatment of temporal triangular alopecia by hair restoration surgery using follicular unit transplantation. Dermatol Surg. 2009;35:1307-1310.
The Diagnosis: Temporal Triangular Alopecia
Temporal triangular alopecia (TTA), also known as congenital triangular alopecia, was first described in the early 1900s.1 It presents clinically as a triangular-shaped area of nonscarring alopecia either unilaterally or bilaterally. Limited clinical data suggest that most unilateral cases are on the left frontotemporal region of the scalp. In bilateral cases, there may be asymmetry in size of the area involved.2 Dermatoscopically, TTA is characterized by decreased terminal hair follicle density as well as the presence of vellus hairs with an absence of inflammation.3 The majority of TTA is noted between birth and 6 years of life with the areas staying stable thereafter. Large areas of TTA may suggest cerebello-trigeminal-dermal dysplasia (Gomez-Lopez-Hernandez syndrome), a rare neurocutaneous syndrome characterized by rhombencephalosynapsis, trigeminal anesthesia, and parietooccipital alopecia (Online Mendelian Inheritance in Man 601853).4 Although TTA is largely idiopathic, it has been suggested that the trait may be paradominant, whereby a postzygotic loss of the wild-type allele in a heterozygotic state causes triangular alopecia and reflects hamartomatous mosaicism.5 It also is an important mimicker of alopecia areata. Correct identification prevents unnecessary treatment to the areas of the scalp. Hair restoration surgery has been reported as a tool to treat this disorder.6
The Diagnosis: Temporal Triangular Alopecia
Temporal triangular alopecia (TTA), also known as congenital triangular alopecia, was first described in the early 1900s.1 It presents clinically as a triangular-shaped area of nonscarring alopecia either unilaterally or bilaterally. Limited clinical data suggest that most unilateral cases are on the left frontotemporal region of the scalp. In bilateral cases, there may be asymmetry in size of the area involved.2 Dermatoscopically, TTA is characterized by decreased terminal hair follicle density as well as the presence of vellus hairs with an absence of inflammation.3 The majority of TTA is noted between birth and 6 years of life with the areas staying stable thereafter. Large areas of TTA may suggest cerebello-trigeminal-dermal dysplasia (Gomez-Lopez-Hernandez syndrome), a rare neurocutaneous syndrome characterized by rhombencephalosynapsis, trigeminal anesthesia, and parietooccipital alopecia (Online Mendelian Inheritance in Man 601853).4 Although TTA is largely idiopathic, it has been suggested that the trait may be paradominant, whereby a postzygotic loss of the wild-type allele in a heterozygotic state causes triangular alopecia and reflects hamartomatous mosaicism.5 It also is an important mimicker of alopecia areata. Correct identification prevents unnecessary treatment to the areas of the scalp. Hair restoration surgery has been reported as a tool to treat this disorder.6
- Tosti A. Congenital triangular alopecia. report of fourteen cases. J Am Acad Dermatol. 1987;16:991-993.
- Armstrong DK, Burrows D. Congenital triangular alopecia. Pediatr Dermatol. 1996;13:394-396.
- Iorizzo M, Pazzaglia M, Starace M, et al. Videodermoscopy: a useful tool for diagnosing congenital triangular alopecia. Pediatr Dermatol. 2008;25:652-654.
- Assoly P, Happle R. A hairy paradox: congenital triangular alopecia with a central hair tuft. Dermatology. 2010;221:107-109.
- Happle R. Congenital triangular alopecia may be categorized as a paradominant trait. Eur J Dermatol. 2003;13:346-347.
- Wu WY, Otberg N, Kang H, et al. Successful treatment of temporal triangular alopecia by hair restoration surgery using follicular unit transplantation. Dermatol Surg. 2009;35:1307-1310.
- Tosti A. Congenital triangular alopecia. report of fourteen cases. J Am Acad Dermatol. 1987;16:991-993.
- Armstrong DK, Burrows D. Congenital triangular alopecia. Pediatr Dermatol. 1996;13:394-396.
- Iorizzo M, Pazzaglia M, Starace M, et al. Videodermoscopy: a useful tool for diagnosing congenital triangular alopecia. Pediatr Dermatol. 2008;25:652-654.
- Assoly P, Happle R. A hairy paradox: congenital triangular alopecia with a central hair tuft. Dermatology. 2010;221:107-109.
- Happle R. Congenital triangular alopecia may be categorized as a paradominant trait. Eur J Dermatol. 2003;13:346-347.
- Wu WY, Otberg N, Kang H, et al. Successful treatment of temporal triangular alopecia by hair restoration surgery using follicular unit transplantation. Dermatol Surg. 2009;35:1307-1310.
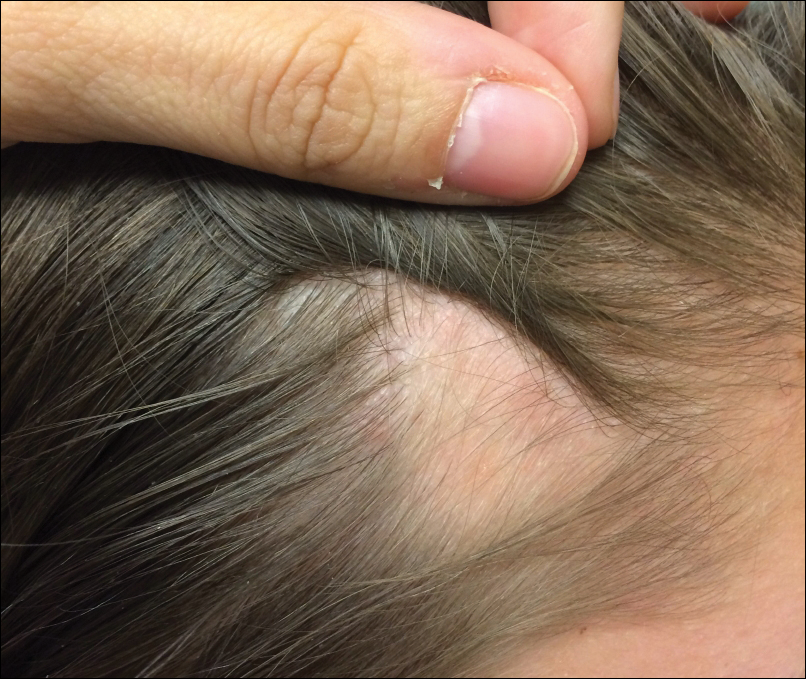
An 11-year-old girl presented for evaluation of a patch of hair loss on the right parietal scalp that had been present and stable for 2.5 years. Physical examination revealed a unilateral area of hair loss that was triangular in shape on the right parietal/temporal region, measuring 2.1×2.2 cm. Dermatoscope examination showed vellus hairs throughout. A hair-pull test was negative and the patient confirmed that the area had never been completely smooth. There were no associated symptoms and no family history of autoimmune disease or hair loss. Prior to presentation, the patient underwent a trial of intralesional steroids and topical steroids to the area without effect.
Azathioprine Hypersensitivity Presenting as Neutrophilic Dermatosis and Erythema Nodosum
To the Editor:
Azathioprine (AZA) hypersensitivity is an immunologically mediated reaction that presents within 1 to 4 weeks of drug initiation.1 Its cutaneous manifestations include Sweet syndrome, erythema nodosum (EN), and acute generalized exanthematous pustulosis, with 88% of cases presenting as neutrophilic dermatoses.2 Confirmation with cutaneous biopsy and cessation of medication is essential to prevent life-threatening anaphylactoid reactions.
A 58-year-old man with a history of Crohn disease was admitted with high fevers (>38.9°C); abdominal pain; diarrhea; and a nonpruritic “pimplelike” rash on the face, chest, and back with a tender nodule on the right leg of 5 days’ duration. Eight days prior to admission, he had started AZA for treatment of Crohn disease. In the hospital he received intravenous metronidazole for a presumed bowel infection; however, the lesions and symptoms did not resolve. Other medical history included psoriatic arthritis for which he was taking oral prednisone 50 mg daily; prednisone was continued during hospitalization.
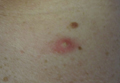
Physical examination showed that the patient was alert and well appearing. On the face, upper chest and back (Figure 1), shoulders, and knees were fewer than 20 sparsely distributed, nontender, 3- to 4-mm pustules. The patient’s scalp, lower back, abdomen, arms, and feet were spared. There also was a solitary 3.5-cm, tender, erythematous nodule on the right lower leg (Figure 2). Blood tests revealed leukocytosis (15,000/mm3 [reference range, 4300–10,300/mm3]) with neutrophilia (90%) and an elevated C-reactive protein level of 173 mg/L (reference range, <10 mg/L). Liver function tests were normal. Thiopurine methyltransferase (TPMT) was on the low end of the reference range. Tissue culture of a shoulder pustule grew only Staphylococcus non-aureus. Blood cultures were negative. A 4-mm punch biopsy specimen from the right leg nodule revealed septal panniculitis with neutrophilic and granulomatous infiltrate consistent with EN.
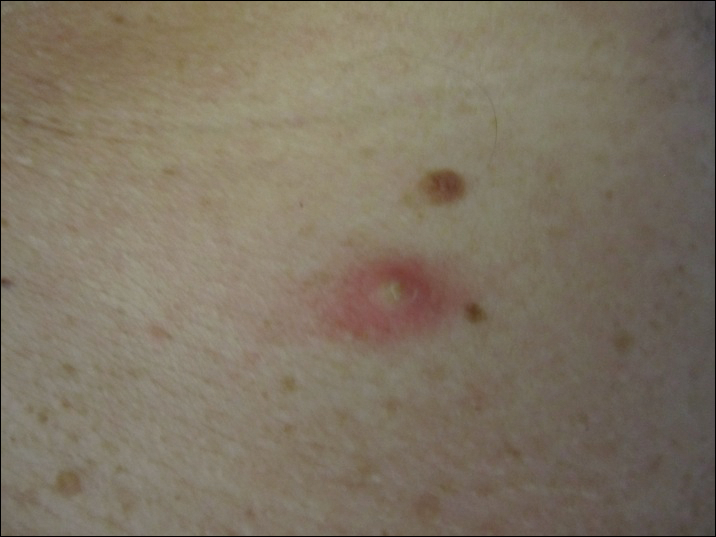
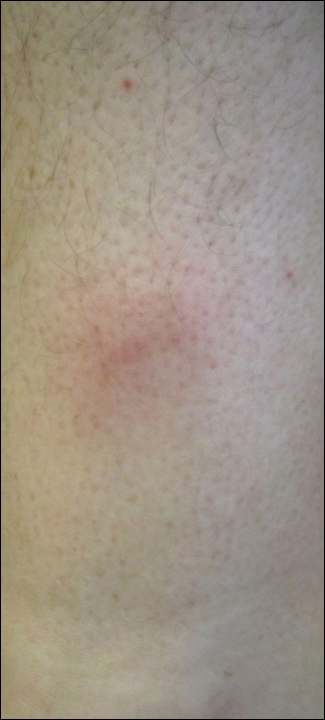
A clinical diagnosis of AZA hypersensitivity was made. Antibiotics and AZA were discontinued and the patient’s lesions resolved within 6 days. Medication rechallenge was not attempted and the patient is now managed with infliximab.
Azathioprine is a well-known and commonly used drug for inflammatory bowel diseases, rheumatoid arthritis, and prevention of transplant rejection. Hypersensitivity is a lesser-known complication of AZA therapy, with most reactions occurring within 4 weeks of treatment initiation. A PubMed search of articles indexed for MEDLINE using the search terms azathioprine and hypersensitivity found only 67 documented cases of AZA hypersensitivity between 1986 and 2009.2 Common findings include fever, malaise, arthralgia, nausea, vomiting, diarrhea, headache, and neutrophilic dermatoses.
Previously reported cases of AZA hypersensitivity with cutaneous manifestations include Sweet syndrome (17.9%), small vessel vasculitis (10.4%), EN (4.4%), acute generalized exanthematous pustulosis (4.4%), and nonspecific cutaneous findings (11.9%).2 One other case reported AZA hypersensitivity presenting as EN with a neutrophilic pustular dermatosis.3 Although Sweet syndrome–like lesions, EN, and acute generalized exanthematous pustulosis have been reported in the context of inflammatory bowel disease, in this case the appearance of these symptoms within 1 week of AZA initiation and resolution after AZA discontinuation is highly suggestive of AZA hypersensitivity. Also, several reports have documented rapid (within a few hours) recurrence of symptoms on rechallenge with AZA.4-6 Moreover, cases of cutaneous AZA hypersensitivity reactions in patients with no history of inflammatory bowel diseases have been reported.6-8
As in this case, cutaneous AZA hypersensitivity can occur even in the setting of normal TPMT levels, suggesting that this phenomenon is a dose-independent reaction.2 Abnormal metabolism of AZA does not appear to be related to previously reported neutrophilic pustular dermatosis3,4 or EN.4 Although the mechanism of hypersensitivity is unclear, there is a report of a patient who developed AZA hypersensitivity but was able to tolerate 6-mercaptopurine, a metabolite of AZA. The authors suggested that the imidazole component of AZA might be responsible for hypersensitivity reactions.9
The differential diagnosis of a patient with these findings includes infectious, rheumatologic, neurologic, or autoimmune diseases, as well as septic shock. Hence, negative cultures and a failure to respond to antibiotics make infection less likely. An appropriate time course of AZA initiation, the development of rash, and a cutaneous biopsy can lead to prompt diagnosis and cessation of AZA.
Once AZA hypersensitivity is suspected, the drug should be discontinued and the reaction should resolve within 2 to 3 days2 and the skin lesions within 5 to 6 days.2,10 Medication rechallenge is contraindicated because AZA rarely has been associated with shock syndrome and hypotension.11-19
Azathioprine hypersensitivity is a serious yet still underrecognized condition in the dermatologic community. In our case, symptoms appeared rapidly and resolved quickly after AZA was discontinued. Azathioprine-induced neutrophilic dermatosis presenting with EN should be recognized as a potential dermatologic manifestation of AZA hypersensitivity, which is a dose-dependent reaction even with normal TPMT levels. Rechallenge with AZA is not recommended due to the risk of a life-threatening anaphylactoid reaction.
- Meggitt SJ, Anstey AV, Mohd Mustapa MF, et al. British Association of Dermatologists’ guidelines for the safe and effective prescribing of azathioprine 2011. Br J Dermatol. 2011;165:711-734.
- Bidinger JJ, Sky K, Battafarano DF, et al. The cutaneous and systemic manifestations of azathioprine hypersensitivity syndrome. J Am Acad Dermatol. 2011;65:184-191.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- De Fonclare AL, Khosrotehrani K, Aractingi S, et al. Erythema nodosum-like eruption as a manifestation of azathioprine hypersensitivity in patients with inflammatory bowel disease. Arch Dermatol. 2007;143:744-748.
- Jeurissen ME, Boerbooms AM, van de Putte LB, et al. Azathioprine induced fever, chills, rash, and hepatotoxicity in rheumatoid arthritis. Ann Rheum Dis. 1990;49:25-27.
- Goldenberg DL, Stor RA. Azathioprine hypersensitivity mimicking an acute exacerbation of dermatomyositis. J Rheumatol. 1975;2:346-349.
- Watts GF, Corston R. Hypersensitivity to azathioprine in myasthenia gravis. Postgrad Med J. 1984;60:362-363.
- El-Azhary RA, Brunner KL, Gibson LE. Sweet syndrome as a manifestation of azathioprine hypersensitivity. Mayo Clin Proc. 2008;83:1026-1030.
- Stetter M, Schmidl M, Krapf R. Azathioprine hypersensitivity mimicking Goodpasture’s syndrome. Am J Kidney Dis. 1994;23:874-877.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Cunningham T, Barraclough D, Muirdin K. Azathioprine induced shock. Br Med J. 1981;283:823-824.
- Elston GE, Johnston GA, Mortimer NJ, et al. Acute generalized exanthematous pustulosis associated with azathioprine hypersensitivity. Clin Exp Dermatol. 2007;32:52-53.
- Fields CL, Robinson JW, Roy TM, et al. Hypersensitivity reaction to azathioprine. South Med J. 1998;91:471-474.
- Keystone E, Schabas R. Hypotension with oliguria: a side effect of azathioprine. Arthritis Rheum. 1981;24:1453-1454.
- Rosenthal E. Azathioprine shock. Postgrad Med J. 1986;62:677-678.
- Sofat N, Houghton J, McHale J, et al. Azathioprine hypersensitivity. Ann Rheum Dis. 2001;60:719-720.
- Knowles SR, Gupta AK, Shear NH, et al. Azathioprine hypersensitivity-like reactions—a case report and a review of the literature. Clin Exp Dermatol. 1995;20:353-356.
- Demirtaş-Ertan G, Rowshani AT, ten Berge IJ. Azathioprine-induced shock in a patient suffering from undifferentiated erosive oligoarthritis. Neth J Med. 2006;64:124-126.
- Zaltzman M, Kallenbach J, Shapiro T, et al. Life-threatening hypotension associated with azathioprine therapy. a case report. S Afr Med J. 1984;65:306.
To the Editor:
Azathioprine (AZA) hypersensitivity is an immunologically mediated reaction that presents within 1 to 4 weeks of drug initiation.1 Its cutaneous manifestations include Sweet syndrome, erythema nodosum (EN), and acute generalized exanthematous pustulosis, with 88% of cases presenting as neutrophilic dermatoses.2 Confirmation with cutaneous biopsy and cessation of medication is essential to prevent life-threatening anaphylactoid reactions.
A 58-year-old man with a history of Crohn disease was admitted with high fevers (>38.9°C); abdominal pain; diarrhea; and a nonpruritic “pimplelike” rash on the face, chest, and back with a tender nodule on the right leg of 5 days’ duration. Eight days prior to admission, he had started AZA for treatment of Crohn disease. In the hospital he received intravenous metronidazole for a presumed bowel infection; however, the lesions and symptoms did not resolve. Other medical history included psoriatic arthritis for which he was taking oral prednisone 50 mg daily; prednisone was continued during hospitalization.
Physical examination showed that the patient was alert and well appearing. On the face, upper chest and back (Figure 1), shoulders, and knees were fewer than 20 sparsely distributed, nontender, 3- to 4-mm pustules. The patient’s scalp, lower back, abdomen, arms, and feet were spared. There also was a solitary 3.5-cm, tender, erythematous nodule on the right lower leg (Figure 2). Blood tests revealed leukocytosis (15,000/mm3 [reference range, 4300–10,300/mm3]) with neutrophilia (90%) and an elevated C-reactive protein level of 173 mg/L (reference range, <10 mg/L). Liver function tests were normal. Thiopurine methyltransferase (TPMT) was on the low end of the reference range. Tissue culture of a shoulder pustule grew only Staphylococcus non-aureus. Blood cultures were negative. A 4-mm punch biopsy specimen from the right leg nodule revealed septal panniculitis with neutrophilic and granulomatous infiltrate consistent with EN.
A clinical diagnosis of AZA hypersensitivity was made. Antibiotics and AZA were discontinued and the patient’s lesions resolved within 6 days. Medication rechallenge was not attempted and the patient is now managed with infliximab.
Azathioprine is a well-known and commonly used drug for inflammatory bowel diseases, rheumatoid arthritis, and prevention of transplant rejection. Hypersensitivity is a lesser-known complication of AZA therapy, with most reactions occurring within 4 weeks of treatment initiation. A PubMed search of articles indexed for MEDLINE using the search terms azathioprine and hypersensitivity found only 67 documented cases of AZA hypersensitivity between 1986 and 2009.2 Common findings include fever, malaise, arthralgia, nausea, vomiting, diarrhea, headache, and neutrophilic dermatoses.
Previously reported cases of AZA hypersensitivity with cutaneous manifestations include Sweet syndrome (17.9%), small vessel vasculitis (10.4%), EN (4.4%), acute generalized exanthematous pustulosis (4.4%), and nonspecific cutaneous findings (11.9%).2 One other case reported AZA hypersensitivity presenting as EN with a neutrophilic pustular dermatosis.3 Although Sweet syndrome–like lesions, EN, and acute generalized exanthematous pustulosis have been reported in the context of inflammatory bowel disease, in this case the appearance of these symptoms within 1 week of AZA initiation and resolution after AZA discontinuation is highly suggestive of AZA hypersensitivity. Also, several reports have documented rapid (within a few hours) recurrence of symptoms on rechallenge with AZA.4-6 Moreover, cases of cutaneous AZA hypersensitivity reactions in patients with no history of inflammatory bowel diseases have been reported.6-8
As in this case, cutaneous AZA hypersensitivity can occur even in the setting of normal TPMT levels, suggesting that this phenomenon is a dose-independent reaction.2 Abnormal metabolism of AZA does not appear to be related to previously reported neutrophilic pustular dermatosis3,4 or EN.4 Although the mechanism of hypersensitivity is unclear, there is a report of a patient who developed AZA hypersensitivity but was able to tolerate 6-mercaptopurine, a metabolite of AZA. The authors suggested that the imidazole component of AZA might be responsible for hypersensitivity reactions.9
The differential diagnosis of a patient with these findings includes infectious, rheumatologic, neurologic, or autoimmune diseases, as well as septic shock. Hence, negative cultures and a failure to respond to antibiotics make infection less likely. An appropriate time course of AZA initiation, the development of rash, and a cutaneous biopsy can lead to prompt diagnosis and cessation of AZA.
Once AZA hypersensitivity is suspected, the drug should be discontinued and the reaction should resolve within 2 to 3 days2 and the skin lesions within 5 to 6 days.2,10 Medication rechallenge is contraindicated because AZA rarely has been associated with shock syndrome and hypotension.11-19
Azathioprine hypersensitivity is a serious yet still underrecognized condition in the dermatologic community. In our case, symptoms appeared rapidly and resolved quickly after AZA was discontinued. Azathioprine-induced neutrophilic dermatosis presenting with EN should be recognized as a potential dermatologic manifestation of AZA hypersensitivity, which is a dose-dependent reaction even with normal TPMT levels. Rechallenge with AZA is not recommended due to the risk of a life-threatening anaphylactoid reaction.
To the Editor:
Azathioprine (AZA) hypersensitivity is an immunologically mediated reaction that presents within 1 to 4 weeks of drug initiation.1 Its cutaneous manifestations include Sweet syndrome, erythema nodosum (EN), and acute generalized exanthematous pustulosis, with 88% of cases presenting as neutrophilic dermatoses.2 Confirmation with cutaneous biopsy and cessation of medication is essential to prevent life-threatening anaphylactoid reactions.
A 58-year-old man with a history of Crohn disease was admitted with high fevers (>38.9°C); abdominal pain; diarrhea; and a nonpruritic “pimplelike” rash on the face, chest, and back with a tender nodule on the right leg of 5 days’ duration. Eight days prior to admission, he had started AZA for treatment of Crohn disease. In the hospital he received intravenous metronidazole for a presumed bowel infection; however, the lesions and symptoms did not resolve. Other medical history included psoriatic arthritis for which he was taking oral prednisone 50 mg daily; prednisone was continued during hospitalization.
Physical examination showed that the patient was alert and well appearing. On the face, upper chest and back (Figure 1), shoulders, and knees were fewer than 20 sparsely distributed, nontender, 3- to 4-mm pustules. The patient’s scalp, lower back, abdomen, arms, and feet were spared. There also was a solitary 3.5-cm, tender, erythematous nodule on the right lower leg (Figure 2). Blood tests revealed leukocytosis (15,000/mm3 [reference range, 4300–10,300/mm3]) with neutrophilia (90%) and an elevated C-reactive protein level of 173 mg/L (reference range, <10 mg/L). Liver function tests were normal. Thiopurine methyltransferase (TPMT) was on the low end of the reference range. Tissue culture of a shoulder pustule grew only Staphylococcus non-aureus. Blood cultures were negative. A 4-mm punch biopsy specimen from the right leg nodule revealed septal panniculitis with neutrophilic and granulomatous infiltrate consistent with EN.
A clinical diagnosis of AZA hypersensitivity was made. Antibiotics and AZA were discontinued and the patient’s lesions resolved within 6 days. Medication rechallenge was not attempted and the patient is now managed with infliximab.
Azathioprine is a well-known and commonly used drug for inflammatory bowel diseases, rheumatoid arthritis, and prevention of transplant rejection. Hypersensitivity is a lesser-known complication of AZA therapy, with most reactions occurring within 4 weeks of treatment initiation. A PubMed search of articles indexed for MEDLINE using the search terms azathioprine and hypersensitivity found only 67 documented cases of AZA hypersensitivity between 1986 and 2009.2 Common findings include fever, malaise, arthralgia, nausea, vomiting, diarrhea, headache, and neutrophilic dermatoses.
Previously reported cases of AZA hypersensitivity with cutaneous manifestations include Sweet syndrome (17.9%), small vessel vasculitis (10.4%), EN (4.4%), acute generalized exanthematous pustulosis (4.4%), and nonspecific cutaneous findings (11.9%).2 One other case reported AZA hypersensitivity presenting as EN with a neutrophilic pustular dermatosis.3 Although Sweet syndrome–like lesions, EN, and acute generalized exanthematous pustulosis have been reported in the context of inflammatory bowel disease, in this case the appearance of these symptoms within 1 week of AZA initiation and resolution after AZA discontinuation is highly suggestive of AZA hypersensitivity. Also, several reports have documented rapid (within a few hours) recurrence of symptoms on rechallenge with AZA.4-6 Moreover, cases of cutaneous AZA hypersensitivity reactions in patients with no history of inflammatory bowel diseases have been reported.6-8
As in this case, cutaneous AZA hypersensitivity can occur even in the setting of normal TPMT levels, suggesting that this phenomenon is a dose-independent reaction.2 Abnormal metabolism of AZA does not appear to be related to previously reported neutrophilic pustular dermatosis3,4 or EN.4 Although the mechanism of hypersensitivity is unclear, there is a report of a patient who developed AZA hypersensitivity but was able to tolerate 6-mercaptopurine, a metabolite of AZA. The authors suggested that the imidazole component of AZA might be responsible for hypersensitivity reactions.9
The differential diagnosis of a patient with these findings includes infectious, rheumatologic, neurologic, or autoimmune diseases, as well as septic shock. Hence, negative cultures and a failure to respond to antibiotics make infection less likely. An appropriate time course of AZA initiation, the development of rash, and a cutaneous biopsy can lead to prompt diagnosis and cessation of AZA.
Once AZA hypersensitivity is suspected, the drug should be discontinued and the reaction should resolve within 2 to 3 days2 and the skin lesions within 5 to 6 days.2,10 Medication rechallenge is contraindicated because AZA rarely has been associated with shock syndrome and hypotension.11-19
Azathioprine hypersensitivity is a serious yet still underrecognized condition in the dermatologic community. In our case, symptoms appeared rapidly and resolved quickly after AZA was discontinued. Azathioprine-induced neutrophilic dermatosis presenting with EN should be recognized as a potential dermatologic manifestation of AZA hypersensitivity, which is a dose-dependent reaction even with normal TPMT levels. Rechallenge with AZA is not recommended due to the risk of a life-threatening anaphylactoid reaction.
- Meggitt SJ, Anstey AV, Mohd Mustapa MF, et al. British Association of Dermatologists’ guidelines for the safe and effective prescribing of azathioprine 2011. Br J Dermatol. 2011;165:711-734.
- Bidinger JJ, Sky K, Battafarano DF, et al. The cutaneous and systemic manifestations of azathioprine hypersensitivity syndrome. J Am Acad Dermatol. 2011;65:184-191.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- De Fonclare AL, Khosrotehrani K, Aractingi S, et al. Erythema nodosum-like eruption as a manifestation of azathioprine hypersensitivity in patients with inflammatory bowel disease. Arch Dermatol. 2007;143:744-748.
- Jeurissen ME, Boerbooms AM, van de Putte LB, et al. Azathioprine induced fever, chills, rash, and hepatotoxicity in rheumatoid arthritis. Ann Rheum Dis. 1990;49:25-27.
- Goldenberg DL, Stor RA. Azathioprine hypersensitivity mimicking an acute exacerbation of dermatomyositis. J Rheumatol. 1975;2:346-349.
- Watts GF, Corston R. Hypersensitivity to azathioprine in myasthenia gravis. Postgrad Med J. 1984;60:362-363.
- El-Azhary RA, Brunner KL, Gibson LE. Sweet syndrome as a manifestation of azathioprine hypersensitivity. Mayo Clin Proc. 2008;83:1026-1030.
- Stetter M, Schmidl M, Krapf R. Azathioprine hypersensitivity mimicking Goodpasture’s syndrome. Am J Kidney Dis. 1994;23:874-877.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Cunningham T, Barraclough D, Muirdin K. Azathioprine induced shock. Br Med J. 1981;283:823-824.
- Elston GE, Johnston GA, Mortimer NJ, et al. Acute generalized exanthematous pustulosis associated with azathioprine hypersensitivity. Clin Exp Dermatol. 2007;32:52-53.
- Fields CL, Robinson JW, Roy TM, et al. Hypersensitivity reaction to azathioprine. South Med J. 1998;91:471-474.
- Keystone E, Schabas R. Hypotension with oliguria: a side effect of azathioprine. Arthritis Rheum. 1981;24:1453-1454.
- Rosenthal E. Azathioprine shock. Postgrad Med J. 1986;62:677-678.
- Sofat N, Houghton J, McHale J, et al. Azathioprine hypersensitivity. Ann Rheum Dis. 2001;60:719-720.
- Knowles SR, Gupta AK, Shear NH, et al. Azathioprine hypersensitivity-like reactions—a case report and a review of the literature. Clin Exp Dermatol. 1995;20:353-356.
- Demirtaş-Ertan G, Rowshani AT, ten Berge IJ. Azathioprine-induced shock in a patient suffering from undifferentiated erosive oligoarthritis. Neth J Med. 2006;64:124-126.
- Zaltzman M, Kallenbach J, Shapiro T, et al. Life-threatening hypotension associated with azathioprine therapy. a case report. S Afr Med J. 1984;65:306.
- Meggitt SJ, Anstey AV, Mohd Mustapa MF, et al. British Association of Dermatologists’ guidelines for the safe and effective prescribing of azathioprine 2011. Br J Dermatol. 2011;165:711-734.
- Bidinger JJ, Sky K, Battafarano DF, et al. The cutaneous and systemic manifestations of azathioprine hypersensitivity syndrome. J Am Acad Dermatol. 2011;65:184-191.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- De Fonclare AL, Khosrotehrani K, Aractingi S, et al. Erythema nodosum-like eruption as a manifestation of azathioprine hypersensitivity in patients with inflammatory bowel disease. Arch Dermatol. 2007;143:744-748.
- Jeurissen ME, Boerbooms AM, van de Putte LB, et al. Azathioprine induced fever, chills, rash, and hepatotoxicity in rheumatoid arthritis. Ann Rheum Dis. 1990;49:25-27.
- Goldenberg DL, Stor RA. Azathioprine hypersensitivity mimicking an acute exacerbation of dermatomyositis. J Rheumatol. 1975;2:346-349.
- Watts GF, Corston R. Hypersensitivity to azathioprine in myasthenia gravis. Postgrad Med J. 1984;60:362-363.
- El-Azhary RA, Brunner KL, Gibson LE. Sweet syndrome as a manifestation of azathioprine hypersensitivity. Mayo Clin Proc. 2008;83:1026-1030.
- Stetter M, Schmidl M, Krapf R. Azathioprine hypersensitivity mimicking Goodpasture’s syndrome. Am J Kidney Dis. 1994;23:874-877.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Cunningham T, Barraclough D, Muirdin K. Azathioprine induced shock. Br Med J. 1981;283:823-824.
- Elston GE, Johnston GA, Mortimer NJ, et al. Acute generalized exanthematous pustulosis associated with azathioprine hypersensitivity. Clin Exp Dermatol. 2007;32:52-53.
- Fields CL, Robinson JW, Roy TM, et al. Hypersensitivity reaction to azathioprine. South Med J. 1998;91:471-474.
- Keystone E, Schabas R. Hypotension with oliguria: a side effect of azathioprine. Arthritis Rheum. 1981;24:1453-1454.
- Rosenthal E. Azathioprine shock. Postgrad Med J. 1986;62:677-678.
- Sofat N, Houghton J, McHale J, et al. Azathioprine hypersensitivity. Ann Rheum Dis. 2001;60:719-720.
- Knowles SR, Gupta AK, Shear NH, et al. Azathioprine hypersensitivity-like reactions—a case report and a review of the literature. Clin Exp Dermatol. 1995;20:353-356.
- Demirtaş-Ertan G, Rowshani AT, ten Berge IJ. Azathioprine-induced shock in a patient suffering from undifferentiated erosive oligoarthritis. Neth J Med. 2006;64:124-126.
- Zaltzman M, Kallenbach J, Shapiro T, et al. Life-threatening hypotension associated with azathioprine therapy. a case report. S Afr Med J. 1984;65:306.
Practice Points
- Azathioprine is a well-known immunosuppressant for renal transplant recipients and inflammatory bowel disease with several off-label uses in dermatology including immunobullous dermatoses, neutrophilic dermatoses, and autoimmune connective tissue diseases.
- Azathioprine hypersensitivity is rare and can present with systemic symptoms of fever and a neutrophilic dermatosis, which is usually self-limited but can progress to an anaphylactoid reaction with multiorgan failure.
- If a more mild hypersensitivity reaction is appreciated, then a rechallenge is not recommended and should be avoided.
Resolution of Disseminated Granuloma Annulare With Removal of Surgical Hardware
To the Editor:
Disseminated granuloma annulare is a noninfectious granulomatous disease of unknown etiology. Reported precipitating factors include trauma, sun exposure, viral infection, vaccination, and malignancy.1 In contrast to a localized variant, disseminated granuloma annulare is associated with a later age of onset, longer duration, and recalcitrance to therapy.2 Although a variety of therapeutic approaches exist, there are limited efficacy data, which is complicated by the spontaneous, self-limited nature of the disease.3,4
A 47-year-old man presented with an eruption of a thick red plaque on the dorsal aspect of the left hand (Figure). The eruption began 6 weeks following fixation of a Galeazzi fracture of the right radius with a stainless steel volar plate. Subsequent to the initial eruption, similar indurated plaques developed on the left thenar area, bilateral axillae, and bilateral legs. A punch biopsy was conducted to rule out necrobiosis lipoidica diabeticorum and sarcoidosis as well as to histopathologically confirm the clinical diagnosis of disseminated granuloma annulare. Following diagnosis, the patient received topical clobetasol for application to the advancing borders of the plaques. At 4-month follow-up, additional plaques continued to develop. The patient was not interested in pursuing alternative courses of therapy and felt that the implantation of surgical hardware was the cause. To the best of our knowledge, there have been no reports of precipitation of disseminated granuloma annulare in response to surgical hardware. Given the time course of onset of the eruption it was plausible that the hardware was the inciting event. The orthopedist thought that the fracture had healed sufficiently to remove the volar plate. The patient elected to have the hardware removed to potentially resolve or arrest the progression of the plaques. Resolution of the plaques was observed by the patient 2 weeks following surgical removal of the volar plate. At 4 months following hardware removal, the patient only had 2 slightly pink, hyperpigmented lesions on the left hand in the areas most severely affected, with complete resolution of all other plaques. The patient was given topical clobetasol for the residual lesions.
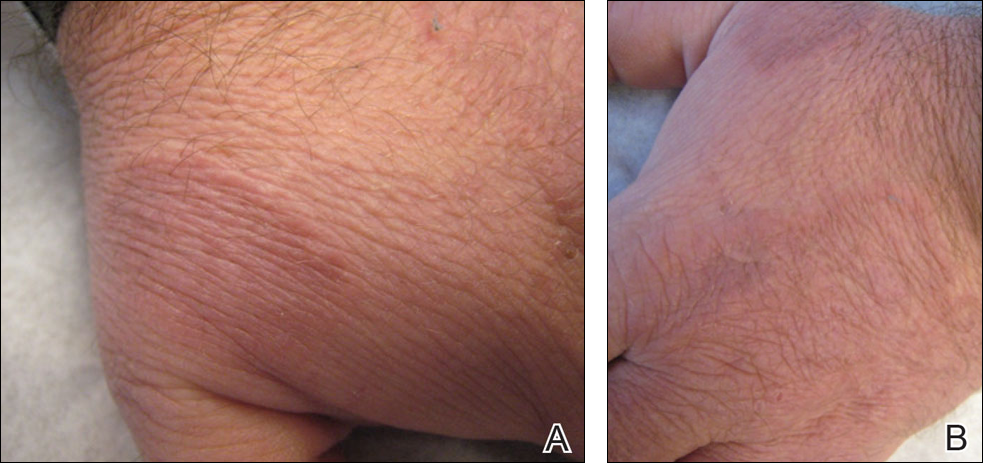
Precipitation and spontaneous resolution of disseminated granuloma annulare following the implantation and removal of surgical hardware is rare. Resolution following hardware removal is consistent with the theory that pathogenesis is due to a delayed-type hypersensitivity reaction to an inciting factor.5 Our case suggests that disseminated granuloma annulare may occur as a delayed-type hypersensitivity reaction to implanted surgical hardware, which should be considered in the etiology and potential therapeutic options for this disorder.
- Mills A, Chetty R. Auricular granuloma annulare. a consequence of trauma? Am J Dermatopathol. 1992;14:431-433.
- Dicken CH, Carrington SG, Winkelmann RK. Generalized granuloma annulare. Arch Dermatol. 1969;99:556-563.
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea [published online May 31, 2009]. Ann Dermatol. 2009:21:113-119
- Cyr PR. Diagnosis and management of granuloma annulare. Am Fam Physician. 2006;74:1729-1734.
- Buechner SA, Winkelmann RK, Banks PM. Identification of T-cell subpopulations in granuloma annulare. Arch Dermatol. 1983;119:125-128.
To the Editor:
Disseminated granuloma annulare is a noninfectious granulomatous disease of unknown etiology. Reported precipitating factors include trauma, sun exposure, viral infection, vaccination, and malignancy.1 In contrast to a localized variant, disseminated granuloma annulare is associated with a later age of onset, longer duration, and recalcitrance to therapy.2 Although a variety of therapeutic approaches exist, there are limited efficacy data, which is complicated by the spontaneous, self-limited nature of the disease.3,4
A 47-year-old man presented with an eruption of a thick red plaque on the dorsal aspect of the left hand (Figure). The eruption began 6 weeks following fixation of a Galeazzi fracture of the right radius with a stainless steel volar plate. Subsequent to the initial eruption, similar indurated plaques developed on the left thenar area, bilateral axillae, and bilateral legs. A punch biopsy was conducted to rule out necrobiosis lipoidica diabeticorum and sarcoidosis as well as to histopathologically confirm the clinical diagnosis of disseminated granuloma annulare. Following diagnosis, the patient received topical clobetasol for application to the advancing borders of the plaques. At 4-month follow-up, additional plaques continued to develop. The patient was not interested in pursuing alternative courses of therapy and felt that the implantation of surgical hardware was the cause. To the best of our knowledge, there have been no reports of precipitation of disseminated granuloma annulare in response to surgical hardware. Given the time course of onset of the eruption it was plausible that the hardware was the inciting event. The orthopedist thought that the fracture had healed sufficiently to remove the volar plate. The patient elected to have the hardware removed to potentially resolve or arrest the progression of the plaques. Resolution of the plaques was observed by the patient 2 weeks following surgical removal of the volar plate. At 4 months following hardware removal, the patient only had 2 slightly pink, hyperpigmented lesions on the left hand in the areas most severely affected, with complete resolution of all other plaques. The patient was given topical clobetasol for the residual lesions.
Precipitation and spontaneous resolution of disseminated granuloma annulare following the implantation and removal of surgical hardware is rare. Resolution following hardware removal is consistent with the theory that pathogenesis is due to a delayed-type hypersensitivity reaction to an inciting factor.5 Our case suggests that disseminated granuloma annulare may occur as a delayed-type hypersensitivity reaction to implanted surgical hardware, which should be considered in the etiology and potential therapeutic options for this disorder.
To the Editor:
Disseminated granuloma annulare is a noninfectious granulomatous disease of unknown etiology. Reported precipitating factors include trauma, sun exposure, viral infection, vaccination, and malignancy.1 In contrast to a localized variant, disseminated granuloma annulare is associated with a later age of onset, longer duration, and recalcitrance to therapy.2 Although a variety of therapeutic approaches exist, there are limited efficacy data, which is complicated by the spontaneous, self-limited nature of the disease.3,4
A 47-year-old man presented with an eruption of a thick red plaque on the dorsal aspect of the left hand (Figure). The eruption began 6 weeks following fixation of a Galeazzi fracture of the right radius with a stainless steel volar plate. Subsequent to the initial eruption, similar indurated plaques developed on the left thenar area, bilateral axillae, and bilateral legs. A punch biopsy was conducted to rule out necrobiosis lipoidica diabeticorum and sarcoidosis as well as to histopathologically confirm the clinical diagnosis of disseminated granuloma annulare. Following diagnosis, the patient received topical clobetasol for application to the advancing borders of the plaques. At 4-month follow-up, additional plaques continued to develop. The patient was not interested in pursuing alternative courses of therapy and felt that the implantation of surgical hardware was the cause. To the best of our knowledge, there have been no reports of precipitation of disseminated granuloma annulare in response to surgical hardware. Given the time course of onset of the eruption it was plausible that the hardware was the inciting event. The orthopedist thought that the fracture had healed sufficiently to remove the volar plate. The patient elected to have the hardware removed to potentially resolve or arrest the progression of the plaques. Resolution of the plaques was observed by the patient 2 weeks following surgical removal of the volar plate. At 4 months following hardware removal, the patient only had 2 slightly pink, hyperpigmented lesions on the left hand in the areas most severely affected, with complete resolution of all other plaques. The patient was given topical clobetasol for the residual lesions.
Precipitation and spontaneous resolution of disseminated granuloma annulare following the implantation and removal of surgical hardware is rare. Resolution following hardware removal is consistent with the theory that pathogenesis is due to a delayed-type hypersensitivity reaction to an inciting factor.5 Our case suggests that disseminated granuloma annulare may occur as a delayed-type hypersensitivity reaction to implanted surgical hardware, which should be considered in the etiology and potential therapeutic options for this disorder.
- Mills A, Chetty R. Auricular granuloma annulare. a consequence of trauma? Am J Dermatopathol. 1992;14:431-433.
- Dicken CH, Carrington SG, Winkelmann RK. Generalized granuloma annulare. Arch Dermatol. 1969;99:556-563.
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea [published online May 31, 2009]. Ann Dermatol. 2009:21:113-119
- Cyr PR. Diagnosis and management of granuloma annulare. Am Fam Physician. 2006;74:1729-1734.
- Buechner SA, Winkelmann RK, Banks PM. Identification of T-cell subpopulations in granuloma annulare. Arch Dermatol. 1983;119:125-128.
- Mills A, Chetty R. Auricular granuloma annulare. a consequence of trauma? Am J Dermatopathol. 1992;14:431-433.
- Dicken CH, Carrington SG, Winkelmann RK. Generalized granuloma annulare. Arch Dermatol. 1969;99:556-563.
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea [published online May 31, 2009]. Ann Dermatol. 2009:21:113-119
- Cyr PR. Diagnosis and management of granuloma annulare. Am Fam Physician. 2006;74:1729-1734.
- Buechner SA, Winkelmann RK, Banks PM. Identification of T-cell subpopulations in granuloma annulare. Arch Dermatol. 1983;119:125-128.
Practice Points
- Disseminated granuloma annulare may occur as a delayed-type hypersensitivity reaction to implanted surgical hardware.
- Resolution may occur following removal of surgical hardware.
Clinical Characteristics and HLA Alleles of a Family With Simultaneously Occurring Alopecia Areata
Alopecia areata (AA) presents as sudden, nonscarring, recurrent hair loss characterized by well-circumscribed hairless patches. Although AA may be observed on any hair-bearing areas of the body, the most commonly affected sites are the scalp, beard area, eyebrows, and eyelashes.1 The incidence of AA is 1% to 2% in the general population and it is more common in males than females younger than 40 years.2 Although the majority of patients present with self-limited and well-circumscribed hairless patches that resolve within 2 years, 7% to 10% display a chronic and severe prognosis.3
The etiopathogenesis of AA is not clearly understood, but its occurrence and progression can involve immune dysfunction, genetic predisposition, infections, and physical and psychological trauma.2 Alopecia areata is observed to occur sporadically in most patients. Family history has been found in 3% to 42% of cases, but simultaneous occurrence of AA in family members is rare.4 In this case series, we present 4 cases of active AA lesions occurring simultaneously in a family who also had associated psychologic disorders.
Case Series
Patient 1 (Proband)
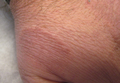
An 11-year-old boy presented with a 6-year history of ongoing AA with recurrent improvement and relapses on the scalp, eyebrows, and eyelashes. Various topical and oral medications had been prescribed by several outside dermatologists; however, these treatments provided minimal benefit and resulted in the recurrence of AA. Dermatologic examination revealed hair loss on the entire frontal, parietal, and temporal regions of the scalp, as well as half of the occipital region and one-third of the lateral side of the eyebrows (Figure 1). Psychological evaluation revealed introvert personality characteristics, lack of self-confidence, and signs of depression and anxiety.
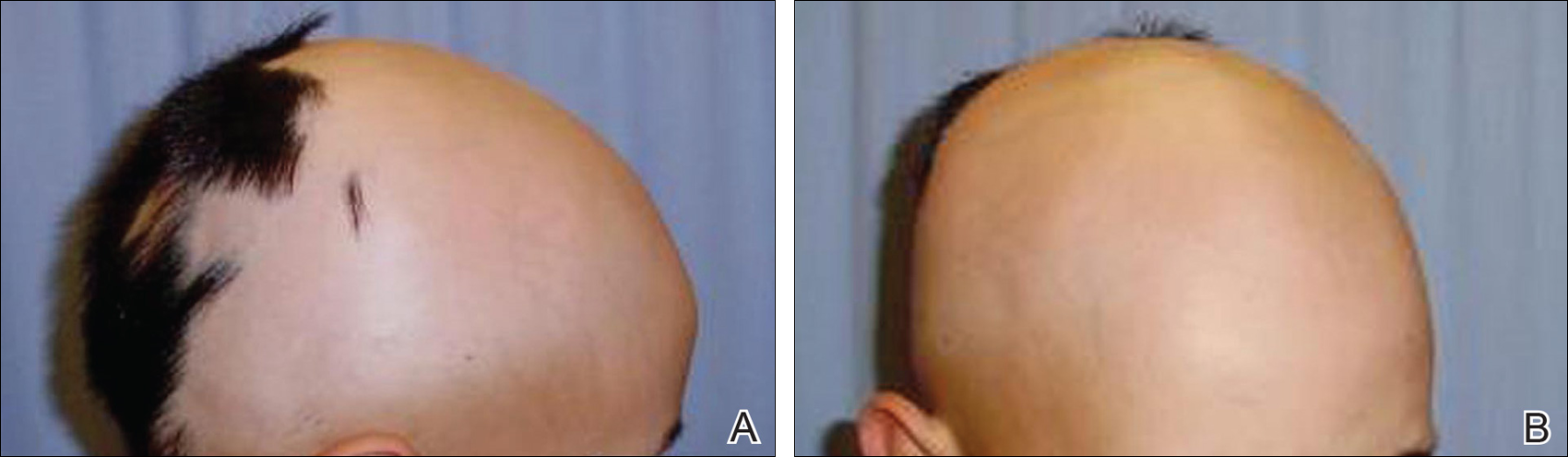
Patient 2 (Proband’s Father)
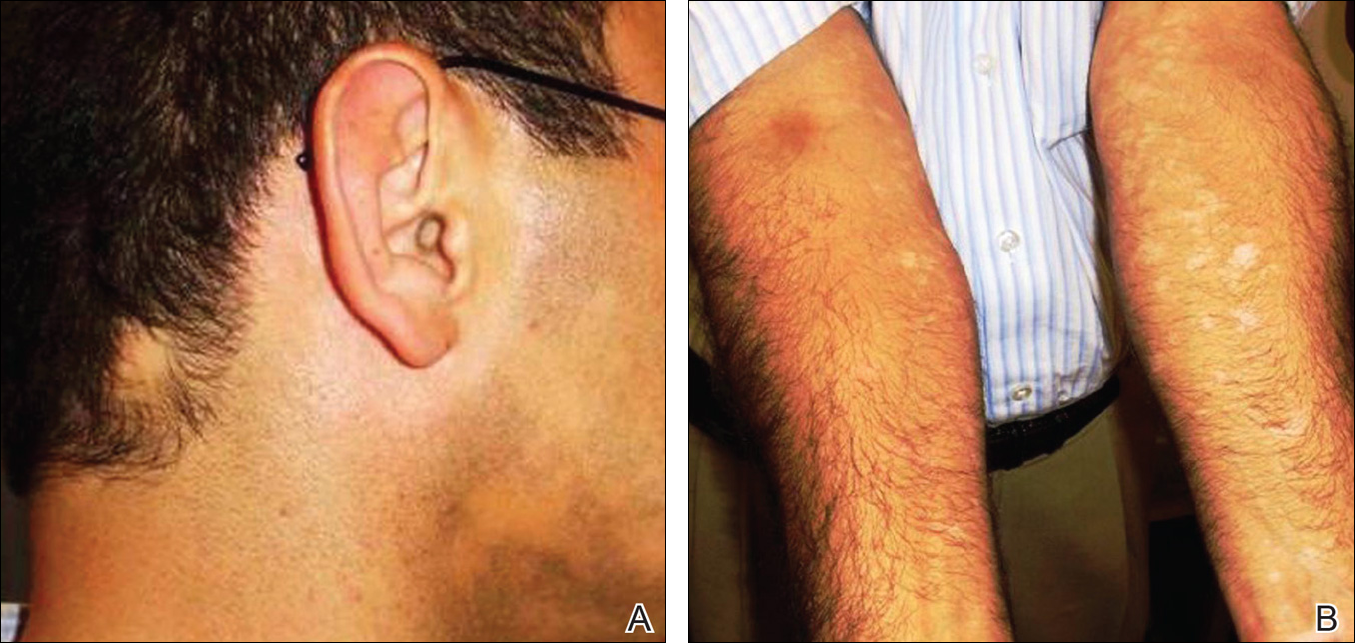
A 38-year-old man presented with a 16-year history of recurrent loss and regrowth of hair on the scalp and beard area and white spots on the penis and arms. He previously had not undergone any treatments. Dermatologic examination revealed well-circumscribed, 1- to 4-cm, hairless patches on the occipital region of the scalp and in the beard area (Figure 2A) and multiple, 2- to 10-mm, vitiliginous lesions on both forearms (Figure 2B) and the penis. The patient had been unemployed for 6 months. Psychological evaluation revealed obsessive-compulsive disorder and obsessive-compulsive personality disorder.
Patient 3 (Proband’s Mother)
A 32-year-old woman presented with a 3-year history of chronic AA. She previously had not undergone any treatments. Dermatologic examination revealed 2 well-circumscribed, 3- to 4-cm patches of hair loss on the occipital and left temporal regions of the scalp (Figure 3). Psychological evaluation revealed obsessive-compulsive personality disorder and depression. The patient did not have any autoimmune diseases.

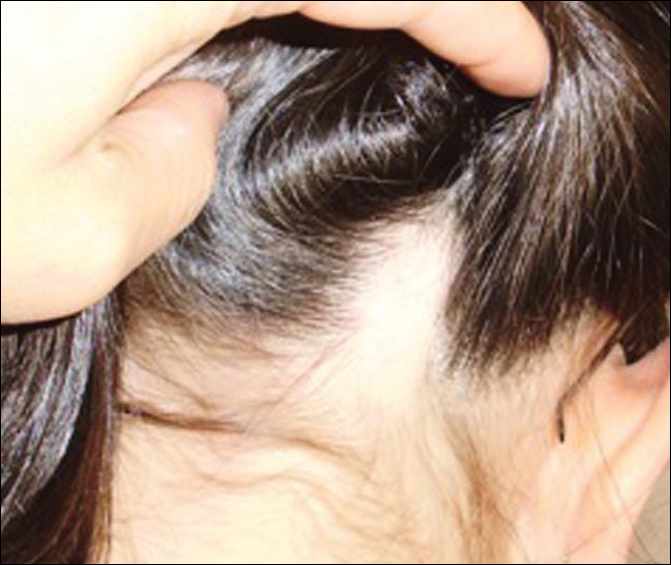
Patient 4 (Proband’s Sister)
A 10-year-old girl presented with a 6-year history of recurrent, self-limited AA on various areas of scalp. She previously had not undergone any treatments. Dermatologic examination revealed a 3-cm hairless patch on the occipital region of the scalp (Figure 4). Psychiatric evaluation revealed narcissistic personality disorder, anxiety, and lack of self-confidence.
Laboratory Evaluation and HLA Antigen DNA Typing
Laboratory testing including complete blood cell count; liver, kidney, and thyroid function; and vitamin B12, zinc, folic acid, and fasting blood sugar levels were performed in all patients.
HLA antigen DNA typing was performed by polymerase chain reaction with sequence-specific primers in all patients after informed consent was obtained.
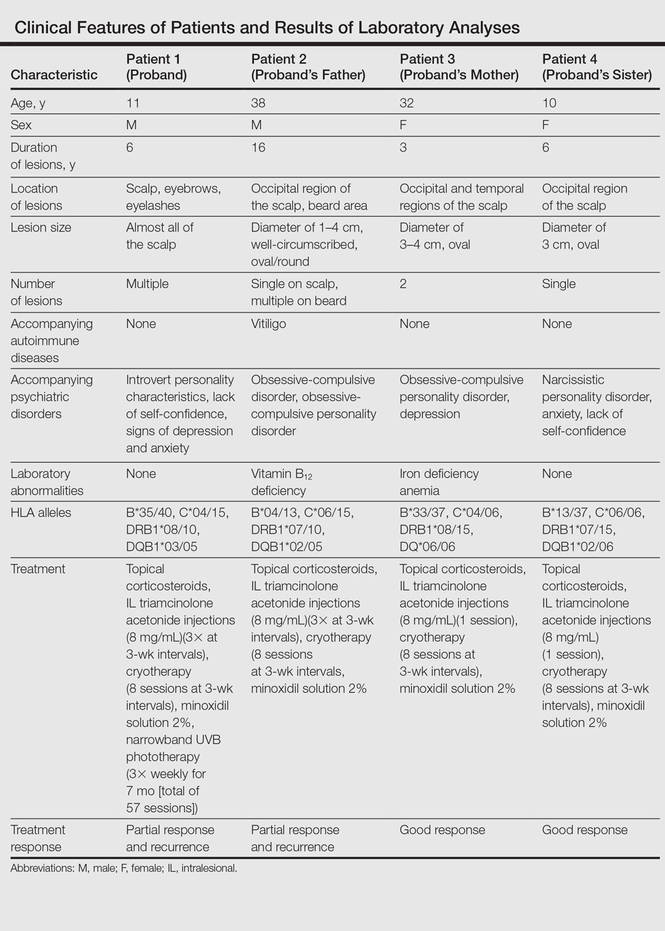
Clinical and laboratory examinations revealed no symptoms or findings of Epstein-Barr virus and cytomegalovirus infections, cicatricial alopecia, or connective tissue diseases in any of the patients. HLA antigen DNA typing revealed the following HLA alleles: B*35/40, C*04/15, DRB1*08/10, and DQB1*03/05 in patient 1; B*04/13, C*06/15, DRB1*07/10, and DQB1*02/05 in patient 2; B*33/37, C*04/06, DRB1*08/15, and DQ*06/06 in patient 3; B*13/37, C*06/06, DRB1*07/15, and DQB1*02/06 in patient 4.
Laboratory testing revealed vitamin B12 deficiency in patient 2 and iron deficiency anemia in patient 3; all other laboratory tests were within reference range. Antithyroglobulin and antithyroid peroxidase autoantibodies were all negative. Clinical features and laboratory analyses for all patients are summarized in the Table.
Treatment
All patients were recommended psychiatric therapy and started on dermatologic treatments. Topical corticosteroids, intralesional triamcinolone acetonide (8 mg/mL) injections into areas of hair loss, 8 total sessions of cryotherapy administered at 3-week intervals, and minoxidil solution 2% were administered respectively to all 4 patients. Alopecia areata in patients 3 and 4 completely regressed; however, no benefit was observed in patients 1 and 2 after 1 year of treatment. Because there was no response to the prior interventions, patient 1 was started on treatment with cyclosporine 2.5 mg/kg twice daily. However, therapy was discontinued after 1 month and treatment with narrowband UVB (3 times per week for 7 months [total of 57 sessions]) and topical corticosteroids were initiated (Table). The patient partially benefited from these regimens and recurrence was observed during the course of the treatment.
Although it was recommended that all 4 patients undergo psychiatric treatment and follow-up regularly with a psychiatrist, the patients declined. After approximately 1 year of dermatologic treatment, all 4 patients were lost to follow-up.
Comment
The etiopathogenesis of AA is unclear, but there is strong evidence suggesting that it is a T-cell–mediated autoimmune disease targeting the hair follicles. Common association of AA with autoimmune diseases such as vitiligo and thyroiditis support the immunological origin of the disease.3 In our case, patient 2 had AA along with vitiligo, but no associated autoimmune diseases (eg, vitiligo, diabetes mellitus, pernicious anemia, thyroid diseases) were noted in the other patients. Genetic and environmental factors are known to be influential as much as immune dysfunction in the etiology of AA.2
The presence of family history in 20% of patients supports the genetic predisposition of AA.4 In a genetic study by Martinez-Mir et al,5 susceptibility loci for AA were demonstrated on chromosomes 6, 10, 16, and 18. HLA antigen alleles, which provide predisposition to AA, have been investigated and associations with many different HLA antigens have been described for AA. In these studies, a relationship between AA and HLA class I antigens was not determined. Notable results mainly focused on HLA class II antigens.6-8 Colombe et al7 and Marques Da Costa et al8 demonstrated that long-lasting alopecia totalis or alopecia universalis (AT/AU) patients had a strong relationship with HLA-DRB1*1104; DRB1*04/05 was reported to be the most frequent HLA group among all patients with AA.6-10 In contrast, we did not detect these alleles in our patients. Colombe et al7,11 noted that HLA-DQB1*03 is a marker for both patch-type AA and AT/AU. Colombe et al10 showed that HLA-DQB1*03 was present in more than 80% of patients (N=286) with long-lasting AA. Barahmani et al9 confirmed a strong association between HLA-DQB1*0301, DRB1*1104, and AT/AU. In our patients, we detected HLA-DQB1*03/05 in patient 1 who had the earliest onset and most severe presentation of AA. In some studies, HLA-DRB1*03 was found to be less frequent in patients with AA, and this allele was suggested to be a protective factor.6,12 However, this allele was not detected in any of our patients.
The association of HLA alleles and AA has been investigated in Turkish patients with AA.13-15 Akar et al13 and Kavak et al14 detected that the frequency of HLA-DQB1*03 allele was remarkably higher in patients with AA than in healthy controls. These results were consistent with Colombe et al.10 On the other hand, Kavak et al14 reported that the frequency of HLA-DR16 was decreased in the patient group with AA. In another study, the frequency of HLA-B62 was increased in patients with AA compared to healthy controls.15 The HLA-DQB1*03 allele was found to be associated with AA in only patient 1 in our case series, and HLA alleles were not commonly shared among the 4 patients. Additionally, lack of consanguinity between patients 2 and 3 (the parents) also suggested that genetic factors were not involved in our familial cases.
Blaumeiser et al16 reported a lifetime risk of 7.4% in parents and 7.1% in siblings of 206 AA patients; however, because these studies investigated the presence of AA in any given life period of the family members, their results do not reflect frequency of simultaneous AA presence within one family. In a literature search using PubMed, Google Scholar, and other national databases for the terms alopecia areata as well as family, sibling, concurrently, concomitant, co-existent, and simultaneously, only 2 cases involving a husband and wife and 1 case of 2 siblings who concurrently had AA have been previously reported.17,18 Simultaneous presence of AA in more than 3 members of the same family is rare, and these cases have been observed in different generations and time periods.19 Among our patients, despite different age of onset and duration, AA was simultaneously present in the entire family.
Moreover, Rodriguez et al20 reported that the concordance rate of AA in identical twins was 42% and dizygotic twins was 10%. Environmental factors and infections also have been implicated in the etiology of AA. Infections caused by viruses such as cytomegalovirus and Epstein-Barr virus have been thought to be potential triggering factors; however, no evidence has been found.21,22 The clinical and laboratory examinations in our study did not reveal any presence and/or history of any known infectious disease, and there was no history of contact with water infected by acrylamide or a similar chemical.
Various life events and intense psychological stress may play an important role in triggering AA. Depression, hysteria, psychopathic deviance, psychasthenia, schizophrenia, anxiety, health concerns, bizarre thoughts, and family problems were found to be more frequent in patients with AA than healthy controls.23 The most common psychological disorders associated with AA are generalized anxiety disorder, major depressive disorder, adjustment disorders, and phobias.1,24 Ruiz-Doblado et al25 determined the presence of psychiatric comorbidities in 66% (21/32) of AA cases. Chu et al26 reported that the differences in ages of onset of AA revealed differences in psychiatric comorbidities. The risk for depression was higher in patients with AA younger than 20 years. An increased rate of anxiety was detected with patients with an onset of AA between the ages of 20 and 39 years. Obsessive-compulsive disorder and anxiety were more common in patients aged 40 to 59 years. Interestingly, the investigators also observed that approximately 50% of psychiatric disorders occurred prior to onset of AA.26 One study showed higher rates of stressful life events in children than in controls.27 Ghanizadeh24 reported at least 1 psychiatric disorder in 78% (11/14) of children and adolescents with AA. In the same study, obsessive-compulsive disorder was found to be the second common condition following major depression in AA.24
In our patients, psychiatric evaluations revealed obsessive-compulsive personality disorder in patients 2 and 3, depression in patient 3, and symptoms of anxiety with a lack of self-confidence in patients 1 and 4. Psychiatric disorders affecting the entire family may stem from unemployment of the father. Similar to the results noted in prior studies, depression, the most commonly associated psychiatric disorder of AA, was present in 2 of 4 patients. Obsessive-compulsive disorder, the second most common psychiatric disorder among AA patients, was present in patients 2 and 3. These results indicate that AA may be associated with shared stressful events and psychiatric disorders. Therefore, in addition to dermatologic treatment, it was recommended that all patients undergo psychiatric treatment and follow-up regularly with a psychiatrist; however, the patients declined. At the end of a 1-year treatment period and follow-up, resistance to therapy with minimal recovery followed by a rapid recurrence was determined in patients 1 and 2.
Conclusion
This report demonstrated that familial AA was strongly associated with psychological disorders that were detected in all patients. In our patients, HLA alleles did not seem to have a role in the development of familial AA. These results suggest that HLA was not associated with AA triggered by psychological stress. We believe that psychological disorders and stressful life events may play an important role in the occurrence of AA and lead to the development of resistance against treatment in familial and resistant AA cases.
- García-Hernández MJ, Ruiz-Doblado S, Rodriguez-Pichardo A, et al. Alopecia areata, stress and psychiatric disorders: a review. J Dermatol. 1999;26:625-632.
- Bhat YJ, Manzoor S, Khan AR, et al. Trace element levels in alopecia areata. Indian J Dermatol Venereol Leprol. 2009;75:29-31.
- Alexis AF, Dudda-Subramanya R, Sinha AA. Alopecia areata: autoimmune basis of hair loss. Eur J Dermatol. 2004;14:364-370.
- Green J, Sinclair RD. Genetics of alopecia areata. Australas J Dermatol. 2000;41:213-218.
- Martinez-Mir A, Zlotogorski A, Gordon D, et al.Genomewide scan for linkage reveals evidence of several susceptibility loci for alopecia areata. Am J Hum Genet. 2007;80:316-328.
- Entz P, Blaumeiser B, Betz RC, et al. Investigation of the HLA-DRB1 locus in alopecia areata. Eur J Dermatol. 2006;16:363-367.
- Colombe BW, Price VH, Khoury EL, et al. HLA class II alleles in long-standing alopecia totalis/alopecia universalis and long-standing patchy alopecia areata differentiate these two clinical groups. J Invest Dermatol. 1995;104(suppl 5):4-5.
- Marques Da Costa C, Dupont E, Van der Cruys M, et al. Earlier occurrence of severe alopecia areata in HLA-DRB1*11-positive patients. Dermatology. 2006;213:12-14.
- Barahmani N, de Andrade M, Slusser JP, et al. Human leukocyte antigen class II alleles are associated with risk of alopecia areata. J Invest Dermatol. 2008;128:240-243.
- Colombe BW, Lou CD, Price VH. The genetic basis of alopecia areata: HLA associations with patchy alopecia areata versus alopecia totalis and alopecia universalis. J Investig Dermatol Symp Proc. 1999;4:216-219.
- Colombe BW, Price VH, Khoury EL, et al. HLA class II antigen associations help to define two types of alopecia areata. J Am Acad Dermatol. 1995;33(5, pt 1):757-764.
- Broniarczyk-Dyła G, Prusińska-Bratoś M, Dubla-Berner M, et al. The protective role of the HLA-DR locus in patients with various clinical types of alopecia areata. Arch Immunol Ther Exp (Warsz). 2002;50:333-336.
- Akar A, Orkunuglu E, Sengul A, et al. HLA class II alleles in patients with alopecia areata. Eur J Dermatol. 2002;12:236-239.
- Kavak A, Baykal C, Ozarmagan G, et al. HLA in alopecia areata. Int J Dermatol. 2000;30:589-592.
- Aliagaoglu C, Pirim I, Atasoy M, et al. Association between alopecia areata and HLA class I and II in Turkey. J Dermatol. 2005;32:711-714.
- Blaumeiser B, Goot I, Fimmers R, et al. Familial aggregation of alopecia areata. J Am Acad Dermatol. 2006;54:627-632.
- Zalka AD, Byarlay JA, Goldsmith LA. Alopecia a deux: simultaneous occurrence of alopecia in a husband and wife. Arch Dermatol. 1994;130:390-392.
- Menon R, Kiran C. Concomitant presentation of alopecia areata in siblings: a rare occurrence. Int J Trichology. 2012;4:86-88.
- Valsecchi R, Vicari O, Frigeni A, et al. Familial alopecia areata-genetic susceptibility or coincidence? Acta Derm Venereol (Stockh). 1985;65:175-177.
- Rodriguez TA, Fernandes KE, Dresser KL, et al. Concordance rate of alopecia areata in identical twins supports both genetic and environmental factors. J Am Acad Dermatol. 2010;62:525-527.
- Rodriguez TA, Duvic M. Onset of alopecia areata after Epstein Barr virus infectious mononucleosis. J Am Acad Dermatol. 2008;59:137-139.
- Offidani A, Amerio P, Bernardini ML, et al. Role of cytomegalovirus replication in alopecia areata pathogenesis. J Cutan Med Surg. 2000;4:63-65.
- Alfani S, Antinone V, Mozzetta A, et al. Psychological status of patients with alopecia areata. Acta Derm Venereol. 2012;92:304-306.
- Ghanizadeh A. Comorbidity of psychiatric disorders in children and adolescents with alopecia areata in a child and adolescent psychiatry clinical sample. Int J Dermatol. 2008;47:1118-1120.
- Ruiz-Doblado S, Carrizosa A, Garcia-Hernandez MJ. Alopecia areata: psychiatric comorbidity and adjustment to illness. Int J Dermatol. 2003;42:434-437.
- Chu SY, Chen YJ, Tseng WC, et al. Psychiatric comorbidities in patients with alopecia areata in Taiwan: a case-control study. Br J Dermatol. 2012;166:525-531.
- Manolache L, Petrescu-Seceleanu D, Benea V. Alopecia areata and stressful events in children. J Eur Acad Dermatol Venereol. 2009;23:107-109.
Alopecia areata (AA) presents as sudden, nonscarring, recurrent hair loss characterized by well-circumscribed hairless patches. Although AA may be observed on any hair-bearing areas of the body, the most commonly affected sites are the scalp, beard area, eyebrows, and eyelashes.1 The incidence of AA is 1% to 2% in the general population and it is more common in males than females younger than 40 years.2 Although the majority of patients present with self-limited and well-circumscribed hairless patches that resolve within 2 years, 7% to 10% display a chronic and severe prognosis.3
The etiopathogenesis of AA is not clearly understood, but its occurrence and progression can involve immune dysfunction, genetic predisposition, infections, and physical and psychological trauma.2 Alopecia areata is observed to occur sporadically in most patients. Family history has been found in 3% to 42% of cases, but simultaneous occurrence of AA in family members is rare.4 In this case series, we present 4 cases of active AA lesions occurring simultaneously in a family who also had associated psychologic disorders.
Case Series
Patient 1 (Proband)
An 11-year-old boy presented with a 6-year history of ongoing AA with recurrent improvement and relapses on the scalp, eyebrows, and eyelashes. Various topical and oral medications had been prescribed by several outside dermatologists; however, these treatments provided minimal benefit and resulted in the recurrence of AA. Dermatologic examination revealed hair loss on the entire frontal, parietal, and temporal regions of the scalp, as well as half of the occipital region and one-third of the lateral side of the eyebrows (Figure 1). Psychological evaluation revealed introvert personality characteristics, lack of self-confidence, and signs of depression and anxiety.
Patient 2 (Proband’s Father)
A 38-year-old man presented with a 16-year history of recurrent loss and regrowth of hair on the scalp and beard area and white spots on the penis and arms. He previously had not undergone any treatments. Dermatologic examination revealed well-circumscribed, 1- to 4-cm, hairless patches on the occipital region of the scalp and in the beard area (Figure 2A) and multiple, 2- to 10-mm, vitiliginous lesions on both forearms (Figure 2B) and the penis. The patient had been unemployed for 6 months. Psychological evaluation revealed obsessive-compulsive disorder and obsessive-compulsive personality disorder.
Patient 3 (Proband’s Mother)
A 32-year-old woman presented with a 3-year history of chronic AA. She previously had not undergone any treatments. Dermatologic examination revealed 2 well-circumscribed, 3- to 4-cm patches of hair loss on the occipital and left temporal regions of the scalp (Figure 3). Psychological evaluation revealed obsessive-compulsive personality disorder and depression. The patient did not have any autoimmune diseases.
Patient 4 (Proband’s Sister)
A 10-year-old girl presented with a 6-year history of recurrent, self-limited AA on various areas of scalp. She previously had not undergone any treatments. Dermatologic examination revealed a 3-cm hairless patch on the occipital region of the scalp (Figure 4). Psychiatric evaluation revealed narcissistic personality disorder, anxiety, and lack of self-confidence.
Laboratory Evaluation and HLA Antigen DNA Typing
Laboratory testing including complete blood cell count; liver, kidney, and thyroid function; and vitamin B12, zinc, folic acid, and fasting blood sugar levels were performed in all patients.
HLA antigen DNA typing was performed by polymerase chain reaction with sequence-specific primers in all patients after informed consent was obtained.
Clinical and laboratory examinations revealed no symptoms or findings of Epstein-Barr virus and cytomegalovirus infections, cicatricial alopecia, or connective tissue diseases in any of the patients. HLA antigen DNA typing revealed the following HLA alleles: B*35/40, C*04/15, DRB1*08/10, and DQB1*03/05 in patient 1; B*04/13, C*06/15, DRB1*07/10, and DQB1*02/05 in patient 2; B*33/37, C*04/06, DRB1*08/15, and DQ*06/06 in patient 3; B*13/37, C*06/06, DRB1*07/15, and DQB1*02/06 in patient 4.
Laboratory testing revealed vitamin B12 deficiency in patient 2 and iron deficiency anemia in patient 3; all other laboratory tests were within reference range. Antithyroglobulin and antithyroid peroxidase autoantibodies were all negative. Clinical features and laboratory analyses for all patients are summarized in the Table.
Treatment
All patients were recommended psychiatric therapy and started on dermatologic treatments. Topical corticosteroids, intralesional triamcinolone acetonide (8 mg/mL) injections into areas of hair loss, 8 total sessions of cryotherapy administered at 3-week intervals, and minoxidil solution 2% were administered respectively to all 4 patients. Alopecia areata in patients 3 and 4 completely regressed; however, no benefit was observed in patients 1 and 2 after 1 year of treatment. Because there was no response to the prior interventions, patient 1 was started on treatment with cyclosporine 2.5 mg/kg twice daily. However, therapy was discontinued after 1 month and treatment with narrowband UVB (3 times per week for 7 months [total of 57 sessions]) and topical corticosteroids were initiated (Table). The patient partially benefited from these regimens and recurrence was observed during the course of the treatment.
Although it was recommended that all 4 patients undergo psychiatric treatment and follow-up regularly with a psychiatrist, the patients declined. After approximately 1 year of dermatologic treatment, all 4 patients were lost to follow-up.
Comment
The etiopathogenesis of AA is unclear, but there is strong evidence suggesting that it is a T-cell–mediated autoimmune disease targeting the hair follicles. Common association of AA with autoimmune diseases such as vitiligo and thyroiditis support the immunological origin of the disease.3 In our case, patient 2 had AA along with vitiligo, but no associated autoimmune diseases (eg, vitiligo, diabetes mellitus, pernicious anemia, thyroid diseases) were noted in the other patients. Genetic and environmental factors are known to be influential as much as immune dysfunction in the etiology of AA.2
The presence of family history in 20% of patients supports the genetic predisposition of AA.4 In a genetic study by Martinez-Mir et al,5 susceptibility loci for AA were demonstrated on chromosomes 6, 10, 16, and 18. HLA antigen alleles, which provide predisposition to AA, have been investigated and associations with many different HLA antigens have been described for AA. In these studies, a relationship between AA and HLA class I antigens was not determined. Notable results mainly focused on HLA class II antigens.6-8 Colombe et al7 and Marques Da Costa et al8 demonstrated that long-lasting alopecia totalis or alopecia universalis (AT/AU) patients had a strong relationship with HLA-DRB1*1104; DRB1*04/05 was reported to be the most frequent HLA group among all patients with AA.6-10 In contrast, we did not detect these alleles in our patients. Colombe et al7,11 noted that HLA-DQB1*03 is a marker for both patch-type AA and AT/AU. Colombe et al10 showed that HLA-DQB1*03 was present in more than 80% of patients (N=286) with long-lasting AA. Barahmani et al9 confirmed a strong association between HLA-DQB1*0301, DRB1*1104, and AT/AU. In our patients, we detected HLA-DQB1*03/05 in patient 1 who had the earliest onset and most severe presentation of AA. In some studies, HLA-DRB1*03 was found to be less frequent in patients with AA, and this allele was suggested to be a protective factor.6,12 However, this allele was not detected in any of our patients.
The association of HLA alleles and AA has been investigated in Turkish patients with AA.13-15 Akar et al13 and Kavak et al14 detected that the frequency of HLA-DQB1*03 allele was remarkably higher in patients with AA than in healthy controls. These results were consistent with Colombe et al.10 On the other hand, Kavak et al14 reported that the frequency of HLA-DR16 was decreased in the patient group with AA. In another study, the frequency of HLA-B62 was increased in patients with AA compared to healthy controls.15 The HLA-DQB1*03 allele was found to be associated with AA in only patient 1 in our case series, and HLA alleles were not commonly shared among the 4 patients. Additionally, lack of consanguinity between patients 2 and 3 (the parents) also suggested that genetic factors were not involved in our familial cases.
Blaumeiser et al16 reported a lifetime risk of 7.4% in parents and 7.1% in siblings of 206 AA patients; however, because these studies investigated the presence of AA in any given life period of the family members, their results do not reflect frequency of simultaneous AA presence within one family. In a literature search using PubMed, Google Scholar, and other national databases for the terms alopecia areata as well as family, sibling, concurrently, concomitant, co-existent, and simultaneously, only 2 cases involving a husband and wife and 1 case of 2 siblings who concurrently had AA have been previously reported.17,18 Simultaneous presence of AA in more than 3 members of the same family is rare, and these cases have been observed in different generations and time periods.19 Among our patients, despite different age of onset and duration, AA was simultaneously present in the entire family.
Moreover, Rodriguez et al20 reported that the concordance rate of AA in identical twins was 42% and dizygotic twins was 10%. Environmental factors and infections also have been implicated in the etiology of AA. Infections caused by viruses such as cytomegalovirus and Epstein-Barr virus have been thought to be potential triggering factors; however, no evidence has been found.21,22 The clinical and laboratory examinations in our study did not reveal any presence and/or history of any known infectious disease, and there was no history of contact with water infected by acrylamide or a similar chemical.
Various life events and intense psychological stress may play an important role in triggering AA. Depression, hysteria, psychopathic deviance, psychasthenia, schizophrenia, anxiety, health concerns, bizarre thoughts, and family problems were found to be more frequent in patients with AA than healthy controls.23 The most common psychological disorders associated with AA are generalized anxiety disorder, major depressive disorder, adjustment disorders, and phobias.1,24 Ruiz-Doblado et al25 determined the presence of psychiatric comorbidities in 66% (21/32) of AA cases. Chu et al26 reported that the differences in ages of onset of AA revealed differences in psychiatric comorbidities. The risk for depression was higher in patients with AA younger than 20 years. An increased rate of anxiety was detected with patients with an onset of AA between the ages of 20 and 39 years. Obsessive-compulsive disorder and anxiety were more common in patients aged 40 to 59 years. Interestingly, the investigators also observed that approximately 50% of psychiatric disorders occurred prior to onset of AA.26 One study showed higher rates of stressful life events in children than in controls.27 Ghanizadeh24 reported at least 1 psychiatric disorder in 78% (11/14) of children and adolescents with AA. In the same study, obsessive-compulsive disorder was found to be the second common condition following major depression in AA.24
In our patients, psychiatric evaluations revealed obsessive-compulsive personality disorder in patients 2 and 3, depression in patient 3, and symptoms of anxiety with a lack of self-confidence in patients 1 and 4. Psychiatric disorders affecting the entire family may stem from unemployment of the father. Similar to the results noted in prior studies, depression, the most commonly associated psychiatric disorder of AA, was present in 2 of 4 patients. Obsessive-compulsive disorder, the second most common psychiatric disorder among AA patients, was present in patients 2 and 3. These results indicate that AA may be associated with shared stressful events and psychiatric disorders. Therefore, in addition to dermatologic treatment, it was recommended that all patients undergo psychiatric treatment and follow-up regularly with a psychiatrist; however, the patients declined. At the end of a 1-year treatment period and follow-up, resistance to therapy with minimal recovery followed by a rapid recurrence was determined in patients 1 and 2.
Conclusion
This report demonstrated that familial AA was strongly associated with psychological disorders that were detected in all patients. In our patients, HLA alleles did not seem to have a role in the development of familial AA. These results suggest that HLA was not associated with AA triggered by psychological stress. We believe that psychological disorders and stressful life events may play an important role in the occurrence of AA and lead to the development of resistance against treatment in familial and resistant AA cases.
Alopecia areata (AA) presents as sudden, nonscarring, recurrent hair loss characterized by well-circumscribed hairless patches. Although AA may be observed on any hair-bearing areas of the body, the most commonly affected sites are the scalp, beard area, eyebrows, and eyelashes.1 The incidence of AA is 1% to 2% in the general population and it is more common in males than females younger than 40 years.2 Although the majority of patients present with self-limited and well-circumscribed hairless patches that resolve within 2 years, 7% to 10% display a chronic and severe prognosis.3
The etiopathogenesis of AA is not clearly understood, but its occurrence and progression can involve immune dysfunction, genetic predisposition, infections, and physical and psychological trauma.2 Alopecia areata is observed to occur sporadically in most patients. Family history has been found in 3% to 42% of cases, but simultaneous occurrence of AA in family members is rare.4 In this case series, we present 4 cases of active AA lesions occurring simultaneously in a family who also had associated psychologic disorders.
Case Series
Patient 1 (Proband)
An 11-year-old boy presented with a 6-year history of ongoing AA with recurrent improvement and relapses on the scalp, eyebrows, and eyelashes. Various topical and oral medications had been prescribed by several outside dermatologists; however, these treatments provided minimal benefit and resulted in the recurrence of AA. Dermatologic examination revealed hair loss on the entire frontal, parietal, and temporal regions of the scalp, as well as half of the occipital region and one-third of the lateral side of the eyebrows (Figure 1). Psychological evaluation revealed introvert personality characteristics, lack of self-confidence, and signs of depression and anxiety.
Patient 2 (Proband’s Father)
A 38-year-old man presented with a 16-year history of recurrent loss and regrowth of hair on the scalp and beard area and white spots on the penis and arms. He previously had not undergone any treatments. Dermatologic examination revealed well-circumscribed, 1- to 4-cm, hairless patches on the occipital region of the scalp and in the beard area (Figure 2A) and multiple, 2- to 10-mm, vitiliginous lesions on both forearms (Figure 2B) and the penis. The patient had been unemployed for 6 months. Psychological evaluation revealed obsessive-compulsive disorder and obsessive-compulsive personality disorder.
Patient 3 (Proband’s Mother)
A 32-year-old woman presented with a 3-year history of chronic AA. She previously had not undergone any treatments. Dermatologic examination revealed 2 well-circumscribed, 3- to 4-cm patches of hair loss on the occipital and left temporal regions of the scalp (Figure 3). Psychological evaluation revealed obsessive-compulsive personality disorder and depression. The patient did not have any autoimmune diseases.
Patient 4 (Proband’s Sister)
A 10-year-old girl presented with a 6-year history of recurrent, self-limited AA on various areas of scalp. She previously had not undergone any treatments. Dermatologic examination revealed a 3-cm hairless patch on the occipital region of the scalp (Figure 4). Psychiatric evaluation revealed narcissistic personality disorder, anxiety, and lack of self-confidence.
Laboratory Evaluation and HLA Antigen DNA Typing
Laboratory testing including complete blood cell count; liver, kidney, and thyroid function; and vitamin B12, zinc, folic acid, and fasting blood sugar levels were performed in all patients.
HLA antigen DNA typing was performed by polymerase chain reaction with sequence-specific primers in all patients after informed consent was obtained.
Clinical and laboratory examinations revealed no symptoms or findings of Epstein-Barr virus and cytomegalovirus infections, cicatricial alopecia, or connective tissue diseases in any of the patients. HLA antigen DNA typing revealed the following HLA alleles: B*35/40, C*04/15, DRB1*08/10, and DQB1*03/05 in patient 1; B*04/13, C*06/15, DRB1*07/10, and DQB1*02/05 in patient 2; B*33/37, C*04/06, DRB1*08/15, and DQ*06/06 in patient 3; B*13/37, C*06/06, DRB1*07/15, and DQB1*02/06 in patient 4.
Laboratory testing revealed vitamin B12 deficiency in patient 2 and iron deficiency anemia in patient 3; all other laboratory tests were within reference range. Antithyroglobulin and antithyroid peroxidase autoantibodies were all negative. Clinical features and laboratory analyses for all patients are summarized in the Table.
Treatment
All patients were recommended psychiatric therapy and started on dermatologic treatments. Topical corticosteroids, intralesional triamcinolone acetonide (8 mg/mL) injections into areas of hair loss, 8 total sessions of cryotherapy administered at 3-week intervals, and minoxidil solution 2% were administered respectively to all 4 patients. Alopecia areata in patients 3 and 4 completely regressed; however, no benefit was observed in patients 1 and 2 after 1 year of treatment. Because there was no response to the prior interventions, patient 1 was started on treatment with cyclosporine 2.5 mg/kg twice daily. However, therapy was discontinued after 1 month and treatment with narrowband UVB (3 times per week for 7 months [total of 57 sessions]) and topical corticosteroids were initiated (Table). The patient partially benefited from these regimens and recurrence was observed during the course of the treatment.
Although it was recommended that all 4 patients undergo psychiatric treatment and follow-up regularly with a psychiatrist, the patients declined. After approximately 1 year of dermatologic treatment, all 4 patients were lost to follow-up.
Comment
The etiopathogenesis of AA is unclear, but there is strong evidence suggesting that it is a T-cell–mediated autoimmune disease targeting the hair follicles. Common association of AA with autoimmune diseases such as vitiligo and thyroiditis support the immunological origin of the disease.3 In our case, patient 2 had AA along with vitiligo, but no associated autoimmune diseases (eg, vitiligo, diabetes mellitus, pernicious anemia, thyroid diseases) were noted in the other patients. Genetic and environmental factors are known to be influential as much as immune dysfunction in the etiology of AA.2
The presence of family history in 20% of patients supports the genetic predisposition of AA.4 In a genetic study by Martinez-Mir et al,5 susceptibility loci for AA were demonstrated on chromosomes 6, 10, 16, and 18. HLA antigen alleles, which provide predisposition to AA, have been investigated and associations with many different HLA antigens have been described for AA. In these studies, a relationship between AA and HLA class I antigens was not determined. Notable results mainly focused on HLA class II antigens.6-8 Colombe et al7 and Marques Da Costa et al8 demonstrated that long-lasting alopecia totalis or alopecia universalis (AT/AU) patients had a strong relationship with HLA-DRB1*1104; DRB1*04/05 was reported to be the most frequent HLA group among all patients with AA.6-10 In contrast, we did not detect these alleles in our patients. Colombe et al7,11 noted that HLA-DQB1*03 is a marker for both patch-type AA and AT/AU. Colombe et al10 showed that HLA-DQB1*03 was present in more than 80% of patients (N=286) with long-lasting AA. Barahmani et al9 confirmed a strong association between HLA-DQB1*0301, DRB1*1104, and AT/AU. In our patients, we detected HLA-DQB1*03/05 in patient 1 who had the earliest onset and most severe presentation of AA. In some studies, HLA-DRB1*03 was found to be less frequent in patients with AA, and this allele was suggested to be a protective factor.6,12 However, this allele was not detected in any of our patients.
The association of HLA alleles and AA has been investigated in Turkish patients with AA.13-15 Akar et al13 and Kavak et al14 detected that the frequency of HLA-DQB1*03 allele was remarkably higher in patients with AA than in healthy controls. These results were consistent with Colombe et al.10 On the other hand, Kavak et al14 reported that the frequency of HLA-DR16 was decreased in the patient group with AA. In another study, the frequency of HLA-B62 was increased in patients with AA compared to healthy controls.15 The HLA-DQB1*03 allele was found to be associated with AA in only patient 1 in our case series, and HLA alleles were not commonly shared among the 4 patients. Additionally, lack of consanguinity between patients 2 and 3 (the parents) also suggested that genetic factors were not involved in our familial cases.
Blaumeiser et al16 reported a lifetime risk of 7.4% in parents and 7.1% in siblings of 206 AA patients; however, because these studies investigated the presence of AA in any given life period of the family members, their results do not reflect frequency of simultaneous AA presence within one family. In a literature search using PubMed, Google Scholar, and other national databases for the terms alopecia areata as well as family, sibling, concurrently, concomitant, co-existent, and simultaneously, only 2 cases involving a husband and wife and 1 case of 2 siblings who concurrently had AA have been previously reported.17,18 Simultaneous presence of AA in more than 3 members of the same family is rare, and these cases have been observed in different generations and time periods.19 Among our patients, despite different age of onset and duration, AA was simultaneously present in the entire family.
Moreover, Rodriguez et al20 reported that the concordance rate of AA in identical twins was 42% and dizygotic twins was 10%. Environmental factors and infections also have been implicated in the etiology of AA. Infections caused by viruses such as cytomegalovirus and Epstein-Barr virus have been thought to be potential triggering factors; however, no evidence has been found.21,22 The clinical and laboratory examinations in our study did not reveal any presence and/or history of any known infectious disease, and there was no history of contact with water infected by acrylamide or a similar chemical.
Various life events and intense psychological stress may play an important role in triggering AA. Depression, hysteria, psychopathic deviance, psychasthenia, schizophrenia, anxiety, health concerns, bizarre thoughts, and family problems were found to be more frequent in patients with AA than healthy controls.23 The most common psychological disorders associated with AA are generalized anxiety disorder, major depressive disorder, adjustment disorders, and phobias.1,24 Ruiz-Doblado et al25 determined the presence of psychiatric comorbidities in 66% (21/32) of AA cases. Chu et al26 reported that the differences in ages of onset of AA revealed differences in psychiatric comorbidities. The risk for depression was higher in patients with AA younger than 20 years. An increased rate of anxiety was detected with patients with an onset of AA between the ages of 20 and 39 years. Obsessive-compulsive disorder and anxiety were more common in patients aged 40 to 59 years. Interestingly, the investigators also observed that approximately 50% of psychiatric disorders occurred prior to onset of AA.26 One study showed higher rates of stressful life events in children than in controls.27 Ghanizadeh24 reported at least 1 psychiatric disorder in 78% (11/14) of children and adolescents with AA. In the same study, obsessive-compulsive disorder was found to be the second common condition following major depression in AA.24
In our patients, psychiatric evaluations revealed obsessive-compulsive personality disorder in patients 2 and 3, depression in patient 3, and symptoms of anxiety with a lack of self-confidence in patients 1 and 4. Psychiatric disorders affecting the entire family may stem from unemployment of the father. Similar to the results noted in prior studies, depression, the most commonly associated psychiatric disorder of AA, was present in 2 of 4 patients. Obsessive-compulsive disorder, the second most common psychiatric disorder among AA patients, was present in patients 2 and 3. These results indicate that AA may be associated with shared stressful events and psychiatric disorders. Therefore, in addition to dermatologic treatment, it was recommended that all patients undergo psychiatric treatment and follow-up regularly with a psychiatrist; however, the patients declined. At the end of a 1-year treatment period and follow-up, resistance to therapy with minimal recovery followed by a rapid recurrence was determined in patients 1 and 2.
Conclusion
This report demonstrated that familial AA was strongly associated with psychological disorders that were detected in all patients. In our patients, HLA alleles did not seem to have a role in the development of familial AA. These results suggest that HLA was not associated with AA triggered by psychological stress. We believe that psychological disorders and stressful life events may play an important role in the occurrence of AA and lead to the development of resistance against treatment in familial and resistant AA cases.
- García-Hernández MJ, Ruiz-Doblado S, Rodriguez-Pichardo A, et al. Alopecia areata, stress and psychiatric disorders: a review. J Dermatol. 1999;26:625-632.
- Bhat YJ, Manzoor S, Khan AR, et al. Trace element levels in alopecia areata. Indian J Dermatol Venereol Leprol. 2009;75:29-31.
- Alexis AF, Dudda-Subramanya R, Sinha AA. Alopecia areata: autoimmune basis of hair loss. Eur J Dermatol. 2004;14:364-370.
- Green J, Sinclair RD. Genetics of alopecia areata. Australas J Dermatol. 2000;41:213-218.
- Martinez-Mir A, Zlotogorski A, Gordon D, et al.Genomewide scan for linkage reveals evidence of several susceptibility loci for alopecia areata. Am J Hum Genet. 2007;80:316-328.
- Entz P, Blaumeiser B, Betz RC, et al. Investigation of the HLA-DRB1 locus in alopecia areata. Eur J Dermatol. 2006;16:363-367.
- Colombe BW, Price VH, Khoury EL, et al. HLA class II alleles in long-standing alopecia totalis/alopecia universalis and long-standing patchy alopecia areata differentiate these two clinical groups. J Invest Dermatol. 1995;104(suppl 5):4-5.
- Marques Da Costa C, Dupont E, Van der Cruys M, et al. Earlier occurrence of severe alopecia areata in HLA-DRB1*11-positive patients. Dermatology. 2006;213:12-14.
- Barahmani N, de Andrade M, Slusser JP, et al. Human leukocyte antigen class II alleles are associated with risk of alopecia areata. J Invest Dermatol. 2008;128:240-243.
- Colombe BW, Lou CD, Price VH. The genetic basis of alopecia areata: HLA associations with patchy alopecia areata versus alopecia totalis and alopecia universalis. J Investig Dermatol Symp Proc. 1999;4:216-219.
- Colombe BW, Price VH, Khoury EL, et al. HLA class II antigen associations help to define two types of alopecia areata. J Am Acad Dermatol. 1995;33(5, pt 1):757-764.
- Broniarczyk-Dyła G, Prusińska-Bratoś M, Dubla-Berner M, et al. The protective role of the HLA-DR locus in patients with various clinical types of alopecia areata. Arch Immunol Ther Exp (Warsz). 2002;50:333-336.
- Akar A, Orkunuglu E, Sengul A, et al. HLA class II alleles in patients with alopecia areata. Eur J Dermatol. 2002;12:236-239.
- Kavak A, Baykal C, Ozarmagan G, et al. HLA in alopecia areata. Int J Dermatol. 2000;30:589-592.
- Aliagaoglu C, Pirim I, Atasoy M, et al. Association between alopecia areata and HLA class I and II in Turkey. J Dermatol. 2005;32:711-714.
- Blaumeiser B, Goot I, Fimmers R, et al. Familial aggregation of alopecia areata. J Am Acad Dermatol. 2006;54:627-632.
- Zalka AD, Byarlay JA, Goldsmith LA. Alopecia a deux: simultaneous occurrence of alopecia in a husband and wife. Arch Dermatol. 1994;130:390-392.
- Menon R, Kiran C. Concomitant presentation of alopecia areata in siblings: a rare occurrence. Int J Trichology. 2012;4:86-88.
- Valsecchi R, Vicari O, Frigeni A, et al. Familial alopecia areata-genetic susceptibility or coincidence? Acta Derm Venereol (Stockh). 1985;65:175-177.
- Rodriguez TA, Fernandes KE, Dresser KL, et al. Concordance rate of alopecia areata in identical twins supports both genetic and environmental factors. J Am Acad Dermatol. 2010;62:525-527.
- Rodriguez TA, Duvic M. Onset of alopecia areata after Epstein Barr virus infectious mononucleosis. J Am Acad Dermatol. 2008;59:137-139.
- Offidani A, Amerio P, Bernardini ML, et al. Role of cytomegalovirus replication in alopecia areata pathogenesis. J Cutan Med Surg. 2000;4:63-65.
- Alfani S, Antinone V, Mozzetta A, et al. Psychological status of patients with alopecia areata. Acta Derm Venereol. 2012;92:304-306.
- Ghanizadeh A. Comorbidity of psychiatric disorders in children and adolescents with alopecia areata in a child and adolescent psychiatry clinical sample. Int J Dermatol. 2008;47:1118-1120.
- Ruiz-Doblado S, Carrizosa A, Garcia-Hernandez MJ. Alopecia areata: psychiatric comorbidity and adjustment to illness. Int J Dermatol. 2003;42:434-437.
- Chu SY, Chen YJ, Tseng WC, et al. Psychiatric comorbidities in patients with alopecia areata in Taiwan: a case-control study. Br J Dermatol. 2012;166:525-531.
- Manolache L, Petrescu-Seceleanu D, Benea V. Alopecia areata and stressful events in children. J Eur Acad Dermatol Venereol. 2009;23:107-109.
- García-Hernández MJ, Ruiz-Doblado S, Rodriguez-Pichardo A, et al. Alopecia areata, stress and psychiatric disorders: a review. J Dermatol. 1999;26:625-632.
- Bhat YJ, Manzoor S, Khan AR, et al. Trace element levels in alopecia areata. Indian J Dermatol Venereol Leprol. 2009;75:29-31.
- Alexis AF, Dudda-Subramanya R, Sinha AA. Alopecia areata: autoimmune basis of hair loss. Eur J Dermatol. 2004;14:364-370.
- Green J, Sinclair RD. Genetics of alopecia areata. Australas J Dermatol. 2000;41:213-218.
- Martinez-Mir A, Zlotogorski A, Gordon D, et al.Genomewide scan for linkage reveals evidence of several susceptibility loci for alopecia areata. Am J Hum Genet. 2007;80:316-328.
- Entz P, Blaumeiser B, Betz RC, et al. Investigation of the HLA-DRB1 locus in alopecia areata. Eur J Dermatol. 2006;16:363-367.
- Colombe BW, Price VH, Khoury EL, et al. HLA class II alleles in long-standing alopecia totalis/alopecia universalis and long-standing patchy alopecia areata differentiate these two clinical groups. J Invest Dermatol. 1995;104(suppl 5):4-5.
- Marques Da Costa C, Dupont E, Van der Cruys M, et al. Earlier occurrence of severe alopecia areata in HLA-DRB1*11-positive patients. Dermatology. 2006;213:12-14.
- Barahmani N, de Andrade M, Slusser JP, et al. Human leukocyte antigen class II alleles are associated with risk of alopecia areata. J Invest Dermatol. 2008;128:240-243.
- Colombe BW, Lou CD, Price VH. The genetic basis of alopecia areata: HLA associations with patchy alopecia areata versus alopecia totalis and alopecia universalis. J Investig Dermatol Symp Proc. 1999;4:216-219.
- Colombe BW, Price VH, Khoury EL, et al. HLA class II antigen associations help to define two types of alopecia areata. J Am Acad Dermatol. 1995;33(5, pt 1):757-764.
- Broniarczyk-Dyła G, Prusińska-Bratoś M, Dubla-Berner M, et al. The protective role of the HLA-DR locus in patients with various clinical types of alopecia areata. Arch Immunol Ther Exp (Warsz). 2002;50:333-336.
- Akar A, Orkunuglu E, Sengul A, et al. HLA class II alleles in patients with alopecia areata. Eur J Dermatol. 2002;12:236-239.
- Kavak A, Baykal C, Ozarmagan G, et al. HLA in alopecia areata. Int J Dermatol. 2000;30:589-592.
- Aliagaoglu C, Pirim I, Atasoy M, et al. Association between alopecia areata and HLA class I and II in Turkey. J Dermatol. 2005;32:711-714.
- Blaumeiser B, Goot I, Fimmers R, et al. Familial aggregation of alopecia areata. J Am Acad Dermatol. 2006;54:627-632.
- Zalka AD, Byarlay JA, Goldsmith LA. Alopecia a deux: simultaneous occurrence of alopecia in a husband and wife. Arch Dermatol. 1994;130:390-392.
- Menon R, Kiran C. Concomitant presentation of alopecia areata in siblings: a rare occurrence. Int J Trichology. 2012;4:86-88.
- Valsecchi R, Vicari O, Frigeni A, et al. Familial alopecia areata-genetic susceptibility or coincidence? Acta Derm Venereol (Stockh). 1985;65:175-177.
- Rodriguez TA, Fernandes KE, Dresser KL, et al. Concordance rate of alopecia areata in identical twins supports both genetic and environmental factors. J Am Acad Dermatol. 2010;62:525-527.
- Rodriguez TA, Duvic M. Onset of alopecia areata after Epstein Barr virus infectious mononucleosis. J Am Acad Dermatol. 2008;59:137-139.
- Offidani A, Amerio P, Bernardini ML, et al. Role of cytomegalovirus replication in alopecia areata pathogenesis. J Cutan Med Surg. 2000;4:63-65.
- Alfani S, Antinone V, Mozzetta A, et al. Psychological status of patients with alopecia areata. Acta Derm Venereol. 2012;92:304-306.
- Ghanizadeh A. Comorbidity of psychiatric disorders in children and adolescents with alopecia areata in a child and adolescent psychiatry clinical sample. Int J Dermatol. 2008;47:1118-1120.
- Ruiz-Doblado S, Carrizosa A, Garcia-Hernandez MJ. Alopecia areata: psychiatric comorbidity and adjustment to illness. Int J Dermatol. 2003;42:434-437.
- Chu SY, Chen YJ, Tseng WC, et al. Psychiatric comorbidities in patients with alopecia areata in Taiwan: a case-control study. Br J Dermatol. 2012;166:525-531.
- Manolache L, Petrescu-Seceleanu D, Benea V. Alopecia areata and stressful events in children. J Eur Acad Dermatol Venereol. 2009;23:107-109.
Practice Points
- The etiopathogenesis of alopecia areata (AA) is not clearly understood, but its occurrence and progression can involve immune dysfunction, genetic predisposition, infections, and physical and psychological trauma.
- Alopecia areata is observed to occur sporadically in most patients. Simultaneous presence of AA in more than 3 members of the same family is rare, and these cases have been observed in different generations and time periods.
- HLA antigen alleles, which provide predisposition to AA, have been investigated, and associations with many different HLA antigens have been described for AA. In previous studies, HLA-DQB1*03 allele was reported as the most common HLA allele in patients with AA.
- Psychological disorders and shared stressful life events may play an important role in the occurrence of AA and lead to the development of resistance against treatment in familial and resistant AA cases.
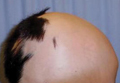
Cutaneous T-Cell Lymphoma in a Patient With Celiac Disease
To the Editor:
Mycosis fungoides (MF) is the most common form of a heterogeneous group of non-Hodgkin lymphomas known as cutaneous T-cell lymphomas. Celiac disease (CD) is associated with increased risk for development of enteropathy-associated T-cell lymphoma and other intraintestinal and extraintestinal non-Hodgkin lymphomas, but a firm association between CD and MF has not been established.1 The first and second cases of concomitant MF and CD were reported in 1985 and 2009 by Coulson and Sanderson2 and Moreira et al,3 respectively. Two other reports of celiac-associated dermatitis herpetiformis and MF exist.4,5 We report a patient with a unique constellation of MF, CD, and Sjögren syndrome (SS).
A 54-year-old woman presented with a worsening nonpruritic, slightly tender, eczematous patch on the back of 19 years’ duration. She had a history of SS diagnosed by salivary gland biopsy. She also had a diagnosis of CD confirmed with positive antigliadin IgA antibodies, with a dramatic improvement in symptoms on a gluten-free diet (GFD) after having abdominal pain and diarrhea for many years. She had no evidence of dermatitis herpetiformis. Recently, more red-brown areas of confluent light pink erythema without clear-cut borders had appeared on the axillae, trunk, and thigh (Figure). The patient also noted new lesions and more erythema of the patches when not adhering to a GFD. A biopsy specimen from the left side of the lateral trunk revealed a bandlike lymphocytic infiltrate with irregular nuclear contours displaying epidermotropism with a few Pautrier microabscesses. Immunohistochemistry showed strong CD3 and CD4 positivity with loss of CD7 and scattered CD8 staining. Peripheral blood flow cytometry showed no aberrant cell populations. The patient was diagnosed with MF stage IB and treated with topical corticosteroids and natural light with improvement.
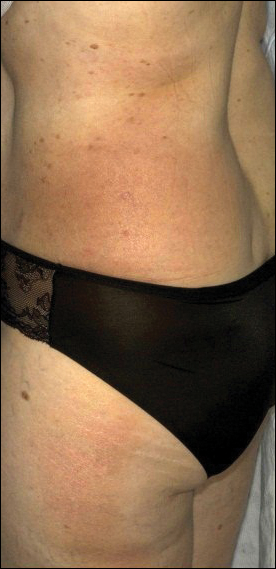
It has been hypothesized that early MF is an autoimmune process caused by dysregulation of a lymphocytic reaction against chronic exogenous or endogenous antigens.4,5 The association of MF with CD supports the possibility of lymphocytic stimulation by a persistent antigen (ie, gluten) in the gastrointestinal tract. Porter et al4 suggested that in susceptible individuals, the resulting clonal T cells may migrate into the epidermis, causing MF. This theory also is supported by the finding that adherence to a GFD leads to decreased risk for malignancy and morbidity.6 In our patient, the chronic autoimmune stimulation in SS could be a factor in the pathogenesis of MF. Additionally, SS, CD, and MF are all strongly associated with increased incidence of specific but different HLA class II antigens. Mycosis fungoides is associated with HLA-DR5 and DQB1*03 alleles, CD with HLA-DQ2 and DQ8, and SS with HLA-DR15 and DR3. We do not know the HLA type of our patient, but she likely possessed multiple alleles, leading to the unique aggregation of diseases.
Furthermore, studies have shown that lymphocytes in CD patients display impaired regulatory T-cell function, causing increased incidence of autoimmune diseases and malignancy.7,8 By this theory, the occurrence of MF in patients is facilitated by the inability of CD lymphocytes to control the abnormal T-cell proliferation in the skin. Interestingly, the finding of SS in our patient supports the possibility of impaired regulatory T-cell function.
Although the occurrence of both MF and CD in our patient could be coincidental, the possibility of correlation must be considered as more cases are documented.
- Catassi C, Fabiani E, Corrao G, et al; Italian Working Group on Coeliac Disease and Non-Hodgkin’s-Lymphoma. Risk of non-Hodgkin Lymphoma in celiac disease. JAMA. 2002;287:1413-1419.
- Coulson IH, Sanderson KV. T-cell lymphoma presenting as tumour d’emblée mycosis fungoides associated with coeliac disease. J R Soc Med. 1985;78(suppl 11):23-24.
- Moreira AI, Menezes N, Varela P, et al. Primary cutaneous peripheral T cell lymphoma and celiac disease [in Portuguese]. Rev Assoc Med Bras. 2009;55:253-256.
- Porter WM, Dawe SA, Bunker CB. Dermatitis herpetiformis and cutaneous T-cell lymphoma. Clin Exp Dermatol. 2001;26:304-305.
- Sun G, Berthelot C, Duvic M. A second case of dermatitis herpetiformis and cutaneous T-cell lymphoma. Clin Exp Dermatol. 2008;33:506-507.
- Holmes GK, Prior P, Lane MR, et al. Malignancy in coeliac disease—effect of a gluten free diet. Gut. 1989;30:333-338.
- Granzotto M, dal Bo S, Quaglia S, et al. Regulatory T-cell function is impaired in celiac disease. Dig Dis Sci. 2009;54:1513-1519.
- Roychoudhuri R, Hirahara K, Mousavi K, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis [published online June 2, 2013]. Nature. 2013;498:506-510.
To the Editor:
Mycosis fungoides (MF) is the most common form of a heterogeneous group of non-Hodgkin lymphomas known as cutaneous T-cell lymphomas. Celiac disease (CD) is associated with increased risk for development of enteropathy-associated T-cell lymphoma and other intraintestinal and extraintestinal non-Hodgkin lymphomas, but a firm association between CD and MF has not been established.1 The first and second cases of concomitant MF and CD were reported in 1985 and 2009 by Coulson and Sanderson2 and Moreira et al,3 respectively. Two other reports of celiac-associated dermatitis herpetiformis and MF exist.4,5 We report a patient with a unique constellation of MF, CD, and Sjögren syndrome (SS).
A 54-year-old woman presented with a worsening nonpruritic, slightly tender, eczematous patch on the back of 19 years’ duration. She had a history of SS diagnosed by salivary gland biopsy. She also had a diagnosis of CD confirmed with positive antigliadin IgA antibodies, with a dramatic improvement in symptoms on a gluten-free diet (GFD) after having abdominal pain and diarrhea for many years. She had no evidence of dermatitis herpetiformis. Recently, more red-brown areas of confluent light pink erythema without clear-cut borders had appeared on the axillae, trunk, and thigh (Figure). The patient also noted new lesions and more erythema of the patches when not adhering to a GFD. A biopsy specimen from the left side of the lateral trunk revealed a bandlike lymphocytic infiltrate with irregular nuclear contours displaying epidermotropism with a few Pautrier microabscesses. Immunohistochemistry showed strong CD3 and CD4 positivity with loss of CD7 and scattered CD8 staining. Peripheral blood flow cytometry showed no aberrant cell populations. The patient was diagnosed with MF stage IB and treated with topical corticosteroids and natural light with improvement.
It has been hypothesized that early MF is an autoimmune process caused by dysregulation of a lymphocytic reaction against chronic exogenous or endogenous antigens.4,5 The association of MF with CD supports the possibility of lymphocytic stimulation by a persistent antigen (ie, gluten) in the gastrointestinal tract. Porter et al4 suggested that in susceptible individuals, the resulting clonal T cells may migrate into the epidermis, causing MF. This theory also is supported by the finding that adherence to a GFD leads to decreased risk for malignancy and morbidity.6 In our patient, the chronic autoimmune stimulation in SS could be a factor in the pathogenesis of MF. Additionally, SS, CD, and MF are all strongly associated with increased incidence of specific but different HLA class II antigens. Mycosis fungoides is associated with HLA-DR5 and DQB1*03 alleles, CD with HLA-DQ2 and DQ8, and SS with HLA-DR15 and DR3. We do not know the HLA type of our patient, but she likely possessed multiple alleles, leading to the unique aggregation of diseases.
Furthermore, studies have shown that lymphocytes in CD patients display impaired regulatory T-cell function, causing increased incidence of autoimmune diseases and malignancy.7,8 By this theory, the occurrence of MF in patients is facilitated by the inability of CD lymphocytes to control the abnormal T-cell proliferation in the skin. Interestingly, the finding of SS in our patient supports the possibility of impaired regulatory T-cell function.
Although the occurrence of both MF and CD in our patient could be coincidental, the possibility of correlation must be considered as more cases are documented.
To the Editor:
Mycosis fungoides (MF) is the most common form of a heterogeneous group of non-Hodgkin lymphomas known as cutaneous T-cell lymphomas. Celiac disease (CD) is associated with increased risk for development of enteropathy-associated T-cell lymphoma and other intraintestinal and extraintestinal non-Hodgkin lymphomas, but a firm association between CD and MF has not been established.1 The first and second cases of concomitant MF and CD were reported in 1985 and 2009 by Coulson and Sanderson2 and Moreira et al,3 respectively. Two other reports of celiac-associated dermatitis herpetiformis and MF exist.4,5 We report a patient with a unique constellation of MF, CD, and Sjögren syndrome (SS).
A 54-year-old woman presented with a worsening nonpruritic, slightly tender, eczematous patch on the back of 19 years’ duration. She had a history of SS diagnosed by salivary gland biopsy. She also had a diagnosis of CD confirmed with positive antigliadin IgA antibodies, with a dramatic improvement in symptoms on a gluten-free diet (GFD) after having abdominal pain and diarrhea for many years. She had no evidence of dermatitis herpetiformis. Recently, more red-brown areas of confluent light pink erythema without clear-cut borders had appeared on the axillae, trunk, and thigh (Figure). The patient also noted new lesions and more erythema of the patches when not adhering to a GFD. A biopsy specimen from the left side of the lateral trunk revealed a bandlike lymphocytic infiltrate with irregular nuclear contours displaying epidermotropism with a few Pautrier microabscesses. Immunohistochemistry showed strong CD3 and CD4 positivity with loss of CD7 and scattered CD8 staining. Peripheral blood flow cytometry showed no aberrant cell populations. The patient was diagnosed with MF stage IB and treated with topical corticosteroids and natural light with improvement.
It has been hypothesized that early MF is an autoimmune process caused by dysregulation of a lymphocytic reaction against chronic exogenous or endogenous antigens.4,5 The association of MF with CD supports the possibility of lymphocytic stimulation by a persistent antigen (ie, gluten) in the gastrointestinal tract. Porter et al4 suggested that in susceptible individuals, the resulting clonal T cells may migrate into the epidermis, causing MF. This theory also is supported by the finding that adherence to a GFD leads to decreased risk for malignancy and morbidity.6 In our patient, the chronic autoimmune stimulation in SS could be a factor in the pathogenesis of MF. Additionally, SS, CD, and MF are all strongly associated with increased incidence of specific but different HLA class II antigens. Mycosis fungoides is associated with HLA-DR5 and DQB1*03 alleles, CD with HLA-DQ2 and DQ8, and SS with HLA-DR15 and DR3. We do not know the HLA type of our patient, but she likely possessed multiple alleles, leading to the unique aggregation of diseases.
Furthermore, studies have shown that lymphocytes in CD patients display impaired regulatory T-cell function, causing increased incidence of autoimmune diseases and malignancy.7,8 By this theory, the occurrence of MF in patients is facilitated by the inability of CD lymphocytes to control the abnormal T-cell proliferation in the skin. Interestingly, the finding of SS in our patient supports the possibility of impaired regulatory T-cell function.
Although the occurrence of both MF and CD in our patient could be coincidental, the possibility of correlation must be considered as more cases are documented.
- Catassi C, Fabiani E, Corrao G, et al; Italian Working Group on Coeliac Disease and Non-Hodgkin’s-Lymphoma. Risk of non-Hodgkin Lymphoma in celiac disease. JAMA. 2002;287:1413-1419.
- Coulson IH, Sanderson KV. T-cell lymphoma presenting as tumour d’emblée mycosis fungoides associated with coeliac disease. J R Soc Med. 1985;78(suppl 11):23-24.
- Moreira AI, Menezes N, Varela P, et al. Primary cutaneous peripheral T cell lymphoma and celiac disease [in Portuguese]. Rev Assoc Med Bras. 2009;55:253-256.
- Porter WM, Dawe SA, Bunker CB. Dermatitis herpetiformis and cutaneous T-cell lymphoma. Clin Exp Dermatol. 2001;26:304-305.
- Sun G, Berthelot C, Duvic M. A second case of dermatitis herpetiformis and cutaneous T-cell lymphoma. Clin Exp Dermatol. 2008;33:506-507.
- Holmes GK, Prior P, Lane MR, et al. Malignancy in coeliac disease—effect of a gluten free diet. Gut. 1989;30:333-338.
- Granzotto M, dal Bo S, Quaglia S, et al. Regulatory T-cell function is impaired in celiac disease. Dig Dis Sci. 2009;54:1513-1519.
- Roychoudhuri R, Hirahara K, Mousavi K, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis [published online June 2, 2013]. Nature. 2013;498:506-510.
- Catassi C, Fabiani E, Corrao G, et al; Italian Working Group on Coeliac Disease and Non-Hodgkin’s-Lymphoma. Risk of non-Hodgkin Lymphoma in celiac disease. JAMA. 2002;287:1413-1419.
- Coulson IH, Sanderson KV. T-cell lymphoma presenting as tumour d’emblée mycosis fungoides associated with coeliac disease. J R Soc Med. 1985;78(suppl 11):23-24.
- Moreira AI, Menezes N, Varela P, et al. Primary cutaneous peripheral T cell lymphoma and celiac disease [in Portuguese]. Rev Assoc Med Bras. 2009;55:253-256.
- Porter WM, Dawe SA, Bunker CB. Dermatitis herpetiformis and cutaneous T-cell lymphoma. Clin Exp Dermatol. 2001;26:304-305.
- Sun G, Berthelot C, Duvic M. A second case of dermatitis herpetiformis and cutaneous T-cell lymphoma. Clin Exp Dermatol. 2008;33:506-507.
- Holmes GK, Prior P, Lane MR, et al. Malignancy in coeliac disease—effect of a gluten free diet. Gut. 1989;30:333-338.
- Granzotto M, dal Bo S, Quaglia S, et al. Regulatory T-cell function is impaired in celiac disease. Dig Dis Sci. 2009;54:1513-1519.
- Roychoudhuri R, Hirahara K, Mousavi K, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis [published online June 2, 2013]. Nature. 2013;498:506-510.
Practice Points
- Mycosis fungoides, the most common type of cutaneous T-cell lymphoma, is an entity for which the pathogenesis is largely unknown.
- Our case and other cases of celiac disease and mycosis fungoides seem to support the immunologic hypothesis of lymphocytic stimulation by a persistent antigen.
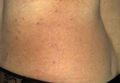
Nonpainful Ulcerations on the Nose and Forehead
The Diagnosis: Trigeminal Trophic Syndrome
Trigeminal trophic syndrome (TTS) is a rare condition occurring after injury to the sensory roots of the trigeminal nerve. Trigeminal trophic syndrome was first described in 1933 by Loveman1 as a complication of ablative treatment of trigeminal neuralgia; however, it has been observed with lesions to the central or peripheral nervous system that damage sensory components of the trigeminal nerve. In our patient, the cause was likely an infarction of the posterior inferior cerebellar artery, which supplies both the cerebellum and the trigeminal nuclei in the brain stem.
Other possible causes of TTS include injury from herpes zoster ophthalmicus; vertebrobasilar insufficiency; trauma; Mycobacterium leprae neuritis; spinal cord degeneration; syringobulbia; or tumors such as astrocytomas, meningiomas, and acoustic neuromas.2 The most typical presenting manifestation is a unilateral, crescent-shaped ulceration of the nasal ala, specifically where cartilage is lacking.3 Less frequently, it affects the upper lip, cheeks, forehead, scalp, and ear. It characteristically spares the nasal tip, which derives sensory innervation from the medial branch of the anterior ethmoidal nerve.4 The affected dermatomal distribution can show anesthesia or paresthesia, promoting the urge to touch, pick, or rub the area.
Histology of the TTS lesion is nondiagnostic, usually showing a mixed dermal inflammation without evidence of infection, vasculitis, or malignancy. In our patient, Gram stain, Grocott-Gomori methenamine-silver stain, and immunohistochemical stains to rule out leukemia or lymphoma were all negative. In addition, flow cytometry of the forehead tissue also was normal. Tests for human immunodeficiency virus, herpes simplex virus types 1 and 2, hepatitis, syphilis, histoplasmosis, blastomycosis, coccidioides, and tuberculosis were negative.During the biopsy, the patient was noted to have anesthesia of the skin. He also admitted to facial manipulation and was noted to have blood and tissue under the fingernails on mornings when the lesion was left uncovered overnight.
Treatment of TTS often can be difficult, especially if patients do not admit to wound manipulation or if manipulation occurs during sleep. Prevention of further ulceration can sometimes be achieved by occlusive dressing alone with mupirocin. Physical barriers can be supplemented with medications such as amitriptyline, carbamazepine, diazepam, chlorpromazine, and gabapentin to attempt to reduce paresthesia.2 Psychiatric consultation can sometimes be necessary and helpful.3 Additionally, negative pressure wound therapy has been used with success in the pediatric setting when digital manipulation is difficult to control.5 More aggressive treatments include transcutaneous electrical stimulation, stellate ganglionectomy, radiotherapy, and innervated rotation flaps.6,7
In summary, recognition of this relatively rare cause of unilateral facial erosions is critical, both to prevent counterproductive treatments such as steroids and to initiate measures to prevent further trauma to the lesion.
- Loveman A. An unusual dermatosis following section of the fifth cranial nerve. Arch Dermatol Syph. 1933;28:369-375.
- Monrad SU, Terrell JE, Aronoff DM. The trigeminal trophic syndrome: an unusual cause of nasal ulceration. J Am Acad Dermatol. 2004;50:949-952.
- Finlay AY. Trigeminal trophic syndrome. Arch Dermatol. 1979;115:1118.
- Setyadi HG, Cohen PR, Schulze KE, et al. Trigeminal trophic syndrome. South Med J. 2007;100:43-48.
- Fredeking AE, Silverman RA. Successful treatment of trigeminal trophic syndrome in a 6-year-old boy with negative pressure wound therapy. Arch Dermatol. 2008;144:984-986.
- Sadeghi P, Papay FA, Vidimos AT. Trigeminal trophic syndrome—report of four cases and review of the literature. Dermatol Surg. 2004;30:807-812.
- Willis M, Shockley W, Mobley SR. Treatment options in trigeminal trophic syndrome: a multi-institutional case series. Laryngoscope. 2011;121:712-716.
The Diagnosis: Trigeminal Trophic Syndrome
Trigeminal trophic syndrome (TTS) is a rare condition occurring after injury to the sensory roots of the trigeminal nerve. Trigeminal trophic syndrome was first described in 1933 by Loveman1 as a complication of ablative treatment of trigeminal neuralgia; however, it has been observed with lesions to the central or peripheral nervous system that damage sensory components of the trigeminal nerve. In our patient, the cause was likely an infarction of the posterior inferior cerebellar artery, which supplies both the cerebellum and the trigeminal nuclei in the brain stem.
Other possible causes of TTS include injury from herpes zoster ophthalmicus; vertebrobasilar insufficiency; trauma; Mycobacterium leprae neuritis; spinal cord degeneration; syringobulbia; or tumors such as astrocytomas, meningiomas, and acoustic neuromas.2 The most typical presenting manifestation is a unilateral, crescent-shaped ulceration of the nasal ala, specifically where cartilage is lacking.3 Less frequently, it affects the upper lip, cheeks, forehead, scalp, and ear. It characteristically spares the nasal tip, which derives sensory innervation from the medial branch of the anterior ethmoidal nerve.4 The affected dermatomal distribution can show anesthesia or paresthesia, promoting the urge to touch, pick, or rub the area.
Histology of the TTS lesion is nondiagnostic, usually showing a mixed dermal inflammation without evidence of infection, vasculitis, or malignancy. In our patient, Gram stain, Grocott-Gomori methenamine-silver stain, and immunohistochemical stains to rule out leukemia or lymphoma were all negative. In addition, flow cytometry of the forehead tissue also was normal. Tests for human immunodeficiency virus, herpes simplex virus types 1 and 2, hepatitis, syphilis, histoplasmosis, blastomycosis, coccidioides, and tuberculosis were negative.During the biopsy, the patient was noted to have anesthesia of the skin. He also admitted to facial manipulation and was noted to have blood and tissue under the fingernails on mornings when the lesion was left uncovered overnight.
Treatment of TTS often can be difficult, especially if patients do not admit to wound manipulation or if manipulation occurs during sleep. Prevention of further ulceration can sometimes be achieved by occlusive dressing alone with mupirocin. Physical barriers can be supplemented with medications such as amitriptyline, carbamazepine, diazepam, chlorpromazine, and gabapentin to attempt to reduce paresthesia.2 Psychiatric consultation can sometimes be necessary and helpful.3 Additionally, negative pressure wound therapy has been used with success in the pediatric setting when digital manipulation is difficult to control.5 More aggressive treatments include transcutaneous electrical stimulation, stellate ganglionectomy, radiotherapy, and innervated rotation flaps.6,7
In summary, recognition of this relatively rare cause of unilateral facial erosions is critical, both to prevent counterproductive treatments such as steroids and to initiate measures to prevent further trauma to the lesion.
The Diagnosis: Trigeminal Trophic Syndrome
Trigeminal trophic syndrome (TTS) is a rare condition occurring after injury to the sensory roots of the trigeminal nerve. Trigeminal trophic syndrome was first described in 1933 by Loveman1 as a complication of ablative treatment of trigeminal neuralgia; however, it has been observed with lesions to the central or peripheral nervous system that damage sensory components of the trigeminal nerve. In our patient, the cause was likely an infarction of the posterior inferior cerebellar artery, which supplies both the cerebellum and the trigeminal nuclei in the brain stem.
Other possible causes of TTS include injury from herpes zoster ophthalmicus; vertebrobasilar insufficiency; trauma; Mycobacterium leprae neuritis; spinal cord degeneration; syringobulbia; or tumors such as astrocytomas, meningiomas, and acoustic neuromas.2 The most typical presenting manifestation is a unilateral, crescent-shaped ulceration of the nasal ala, specifically where cartilage is lacking.3 Less frequently, it affects the upper lip, cheeks, forehead, scalp, and ear. It characteristically spares the nasal tip, which derives sensory innervation from the medial branch of the anterior ethmoidal nerve.4 The affected dermatomal distribution can show anesthesia or paresthesia, promoting the urge to touch, pick, or rub the area.
Histology of the TTS lesion is nondiagnostic, usually showing a mixed dermal inflammation without evidence of infection, vasculitis, or malignancy. In our patient, Gram stain, Grocott-Gomori methenamine-silver stain, and immunohistochemical stains to rule out leukemia or lymphoma were all negative. In addition, flow cytometry of the forehead tissue also was normal. Tests for human immunodeficiency virus, herpes simplex virus types 1 and 2, hepatitis, syphilis, histoplasmosis, blastomycosis, coccidioides, and tuberculosis were negative.During the biopsy, the patient was noted to have anesthesia of the skin. He also admitted to facial manipulation and was noted to have blood and tissue under the fingernails on mornings when the lesion was left uncovered overnight.
Treatment of TTS often can be difficult, especially if patients do not admit to wound manipulation or if manipulation occurs during sleep. Prevention of further ulceration can sometimes be achieved by occlusive dressing alone with mupirocin. Physical barriers can be supplemented with medications such as amitriptyline, carbamazepine, diazepam, chlorpromazine, and gabapentin to attempt to reduce paresthesia.2 Psychiatric consultation can sometimes be necessary and helpful.3 Additionally, negative pressure wound therapy has been used with success in the pediatric setting when digital manipulation is difficult to control.5 More aggressive treatments include transcutaneous electrical stimulation, stellate ganglionectomy, radiotherapy, and innervated rotation flaps.6,7
In summary, recognition of this relatively rare cause of unilateral facial erosions is critical, both to prevent counterproductive treatments such as steroids and to initiate measures to prevent further trauma to the lesion.
- Loveman A. An unusual dermatosis following section of the fifth cranial nerve. Arch Dermatol Syph. 1933;28:369-375.
- Monrad SU, Terrell JE, Aronoff DM. The trigeminal trophic syndrome: an unusual cause of nasal ulceration. J Am Acad Dermatol. 2004;50:949-952.
- Finlay AY. Trigeminal trophic syndrome. Arch Dermatol. 1979;115:1118.
- Setyadi HG, Cohen PR, Schulze KE, et al. Trigeminal trophic syndrome. South Med J. 2007;100:43-48.
- Fredeking AE, Silverman RA. Successful treatment of trigeminal trophic syndrome in a 6-year-old boy with negative pressure wound therapy. Arch Dermatol. 2008;144:984-986.
- Sadeghi P, Papay FA, Vidimos AT. Trigeminal trophic syndrome—report of four cases and review of the literature. Dermatol Surg. 2004;30:807-812.
- Willis M, Shockley W, Mobley SR. Treatment options in trigeminal trophic syndrome: a multi-institutional case series. Laryngoscope. 2011;121:712-716.
- Loveman A. An unusual dermatosis following section of the fifth cranial nerve. Arch Dermatol Syph. 1933;28:369-375.
- Monrad SU, Terrell JE, Aronoff DM. The trigeminal trophic syndrome: an unusual cause of nasal ulceration. J Am Acad Dermatol. 2004;50:949-952.
- Finlay AY. Trigeminal trophic syndrome. Arch Dermatol. 1979;115:1118.
- Setyadi HG, Cohen PR, Schulze KE, et al. Trigeminal trophic syndrome. South Med J. 2007;100:43-48.
- Fredeking AE, Silverman RA. Successful treatment of trigeminal trophic syndrome in a 6-year-old boy with negative pressure wound therapy. Arch Dermatol. 2008;144:984-986.
- Sadeghi P, Papay FA, Vidimos AT. Trigeminal trophic syndrome—report of four cases and review of the literature. Dermatol Surg. 2004;30:807-812.
- Willis M, Shockley W, Mobley SR. Treatment options in trigeminal trophic syndrome: a multi-institutional case series. Laryngoscope. 2011;121:712-716.
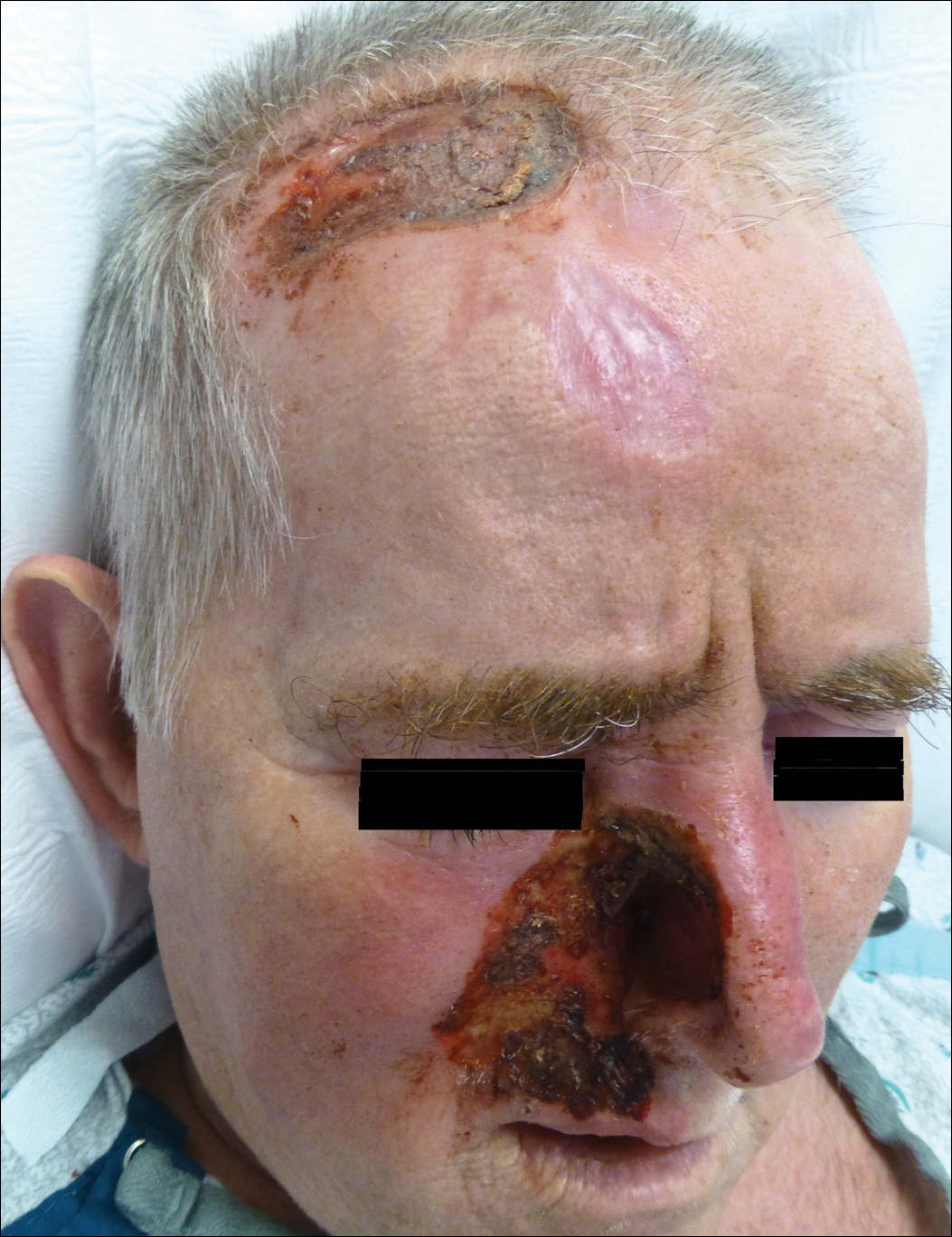
A 57-year-old man with a history of multiple cerebrovascular accidents was transferred from an outside hospital to our inpatient rheumatology service with nonpainful erosions of the forehead and nasal ala of 6 months’ duration. The patient reported that he initially developed a sore on the nose months prior to presentation with worsening sensations of itching and tingling on the forehead and nose. He also noted a headache and gradual loss of vision in the right eye. The patient was immunocompetent and denied arthralgia or any other skin lesions.