User login
Mitchel is a reporter for MDedge based in the Philadelphia area. He started with the company in 1992, when it was International Medical News Group (IMNG), and has since covered a range of medical specialties. Mitchel trained as a virologist at Roswell Park Memorial Institute in Buffalo, and then worked briefly as a researcher at Boston Children's Hospital before pivoting to journalism as a AAAS Mass Media Fellow in 1980. His first reporting job was with Science Digest magazine, and from the mid-1980s to early-1990s he was a reporter with Medical World News. @mitchelzoler
Renal denervation proceeds as U.S. trial’s flaws emerge
PARIS – At least three different factors undermined the SYMPLICITY HTN-3 trial that earlier this year did not show a significant difference in blood pressure lowering between renal denervation and a sham-control procedure, most notably the failure of the vast majority of operators in the study to follow ablation instructions and produce thorough and reliable interruptions of sympathetic innervation of the kidneys, according to new data released by the trial’s investigators.
As the full range of problems with the U.S.-based SYMPLICITY HTN-3 trial, which had its main results reported in April (N. Engl. J. Med. 2014;370:1393-1401), became apparent in a report at the annual congress of the European Association of Percutaneous Cardiovascular Interventions, many top European practitioners and supporters of renal denervation voiced their belief that the treatment is an effective and safe option for many patients with true drug-resistant, severe hypertension.
The only qualifications they now add are that renal denervation is not easily performed and must be done carefully and in a more targeted way, with an ongoing need to find the patients best suited for treatment and the best methods for delivering treatment.
During the meeting, Dr. Felix Mahfoud, an interventional cardiologist at the University Hospital of Saarland in Homburg/Saar, Germany, joined with hypertension specialist Dr. Konstantinos Tsioufis of the University of Athens and Dr. William Wijns, codirector of EuroPCR, in an official statement from the meeting that despite the SYMPLICITY HTN-3 results they continued to support renal denervation as a treatment option for selected patients with drug-resistant, severe hypertension.
Their sentiment echoed another endorsement made a few weeks earlier for continued use and study of renal denervation from the European Society of Hypertension (ESH) in reaction to the SYMPLICITY HTN-3 results.

The ESH "sticks to its statement" from 2013 on using renal denervation in appropriate patients with treatment-resistant, severe hypertension (Eurointervention 2013;9:R58-R66), said Dr. Roland E. Schmieder, first author for the 2013 ESH position paper and a leader in European use of renal denervation.
"We need more studies to prove that renal denervation works, and in particular to get more precise information on which patients get the greatest benefit," Dr. Schmieder said in a separate talk at the meeting. For the time being, he said he was comfortable with routine use of renal denervation in patients with an office systolic BP of at least 160 mm Hg that remains at this level despite maximally tolerated treatment with at least three antihypertensive drugs, including a diuretic, the use endorsed by current European guidelines. It remains appropriate to investigate the impact of renal denervation on other disorders, such as heart failure, arrhythmia, metabolic syndrome, and depressed renal function, said Dr. Schmieder, professor and head of hypertension and vascular medicine research at University Hospital in Erlangen, Germany.
The problems with SYMPLICITY HTN-3
While much speculation swirled around what had gone wrong in the SYMPLICITY HTN-3 trial after researchers on the study gave their first report on the results early in the spring, the full extent of the study’s problems didn’t flesh out until a follow-up report during EuroPCR by coinvestigator Dr. David E. Kandzari. In his analysis, Dr. Kandzari highlighted three distinct problems with the trial that he and his associates identified in a series of post hoc analyses:
• The failure of a large minority of enrolled patients in both arms of the study to remain on a stable medical regimen during the 6 months of follow-up before the primary efficacy outcomes were measured.
• The inexplicably large reduction in BP among the sham-control patients, especially among African American patients, who made up a quarter of the trial’s population.
• The vastly incomplete nerve-ablation treatment that most patients received, treatments that usually failed to meet the standards specified in the trial’s protocol.
The background medical regimens that patients received proved unstable during SYMPLICITY HTN-3 even though the study design mandated that patients be on a stable regimen for at least 2 weeks before entering the study. Roughly 80% of enrolled patients in both the denervation and sham-control arms of the study had been on a stable regimen for at least 6 weeks before they entered. Despite that, during the 6 months of follow-up, 211 (39%) of patients in the study underwent a change in their medication regimen. The changes occurred at virtually identical rates in both study arms, and in more than two-thirds of cases were driven by medical necessity.

"The pattern of drug changes challenges the notion of maximally tolerated therapy," Dr. Kandzari said during his report. "Can this [maximally tolerated therapy] be sustained in a randomized, controlled trial?" It also raised the issues of how trial design can better limit drug changes.
Even though it remains unclear why blood pressure reduction was so pronounced among the African Americans in the sham-control group, the impact of this unexpected effect substantially upended the trial’s endpoints. Among the 49 African Americans randomized to sham treatment, office-measured systolic pressure dropped by an average of 17.8 mm Hg, far exceeding the 8.6–mm Hg decline seen among the non–African Americans in the control arm and even exceeding the average 15.5–mm Hg drop in office systolic BP among African Americans treated with renal denervation.
"The absolute reduction in blood pressure by renal denervation in African Americans was identical to non–African Americans." The problem that arose "related more to what happened in the sham-control group of African Americans, who had a nearly 18–mm Hg reduction in blood pressure," said Dr. Kandzari, chief scientific officer and director of interventional cardiology at Piedmont Heart Institute in Atlanta.
The low rate at which patients assigned to receive renal denervation actually received the type of treatment spelled out in the study’s protocol may have been the biggest problem of all, although Dr. Kandzari stressed that, in his opinion "no single factor led to the neutral efficacy seen in the study."
The supplementary methods section of the SYMPLICITY HTN-3 report published in April explicitly called for patients to receive "4-6 ablations" per side, delivering them in a spiral, circumferential pattern starting distally in each renal artery. That meant each patient was to receive a minimum of eight total ablations.
But analysis of data recorded independently by the research nurse and by the proctor during each procedure, as well as cineangiography films made and submitted by the operator for each ablation, clearly showed that many patients did not receive the treatment that the protocol spelled out. Synthesis of the data collected by the three methods showed that about half of the 364 patients randomized to renal denervation received at least eight ablations, while the other half did not receive this minimum number.
The three separate sets of ablation records also contained information on whether ablations occurred in the anterior, posterior, superior, or inferior quadrants of each renal artery. Full circumferential ablation, what the protocol prescribed, required an ablation in at least one of each of these quadrants per side. What actually happened was that 253 patients (70%) received no circumferential ablations, 68 patients (19%) received circumferential ablation on just one side, and 19 patients (5%) received the bilateral circumferential ablations that the protocol called for. Data for the remaining 24 patients treated with renal denervation were not amenable to analysis for this parameter.
As might be expected, greater ablation number and completeness strongly linked with a robust blood pressure effect.
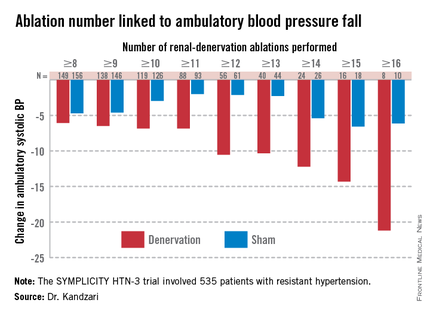
Among patients who received at least eight ablations, office systolic pressure fell by an average 13.1 mm Hg. But among the nine patients who received 16 or more ablations, the average systolic BP reduction at 6 months was 30.9 mm Hg. Among the 18 patients who received at least 15 ablations, the average systolic pressure reduction was 25.4 mm Hg. A very similar relationship occurred for BPs measured by ambulatory monitoring (see graphic), and the data also suggested a positive link between an increasing number of ablations and an increased effect on heart rate. The consistency of the association across all three measures lent further support to this as a real relationship, Dr. Kandzari noted.
Circumferentiality of the ablations showed a similar pattern. The average office systolic pressure fall in patients with no circumferential ablations was 14.2 mm Hg, and it was 16.1 mm Hg in patients who received just one circumferential ablation. But in the 19 patients who received circumferential ablations bilaterally, the average office systolic pressure reduction was 24.3 mm Hg, with a similar pattern seen for ambulatory measures as well as for home-based BP measurements.
"All patients randomized to renal denervation received renal denervation, but they may not have received it in a fashion that seemed to translate into a greater blood pressure reduction," Dr. Kandzari concluded.
Who to treat, where to treat, how to treat
"One result of the neutral HTN-3 result was a call to revisit the basic science behind renal denervation. The clinical enthusiasm had exceeded the science behind renal denervation," Dr. Kandzari observed.
Renal denervation’s many European advocates seem to agree, and have begun the process of determining characteristics of the best patients to receive renal denervation and where and how ablations are best delivered within the renal artery to achieve interruption of sympathetic innervation, although the targeting information they have right now is rudimentary.
"Probably most important is patient selection. You must be sure to get the right patient, one with high sympathetic activity, because the treatment lowers sympathetic activity," said Dr. Atul Pathak, an interventional cardiologist at Paul Sabatier University in Toulouse, France.
Some clues for patient selection have come from the Global SYMPLICITY Registry, which is enrolling patients treated with renal denervation at more than 200 experienced centers worldwide, many of them in Germany but also elsewhere in Europe, Australia, Canada, Korea, and other locations. Initial findings from the first 1,000 patients entered into the registry and followed for 6 months came out in March at the annual meeting of the American College of Cardiology, and Dr. Mahfoud presented new analyses of the data at EuroPCR.
"The major concern we had when we started renal denervation was its safety. I believe the safety issue is now answered," especially with the data collected in the global registry as well as in the SYMPLICITY HTN-3 trial, by far the largest trial completed for the procedure, said Dr. Thomas Zeller, professor and head of clinical and interventional angiology at the Heart Center in Bad Krozingen, Germany. "I was concerned that we might harm the renal arteries with long-lasting stenosis or embolic showers, but this does not happen, at least with the Symplicity catheter," he said during a talk at the meeting.
"The number of patients suitable for renal denervation is potentially much smaller than we initially expected. Real drug resistance is rare, poor adherence is common, and the Symplicity catheter is technically challenging and not effective in every patient. It is hard to rotate the catheter in the tortuous iliac arteries that some patients with hypertension have; the anatomic conditions of hypertension may not be suited to the Symplicity flex catheter," said Dr. Zeller, who added that he has performed renal denervations with the Symplicity catheter since 2009.
"We should focus on the patients that the HTN-3 trial identified as responders, including patients younger than 65, and patients on an aldosterone antagonist," he suggested in a talk at the meeting.
Finding the right patients and the right ablation targets
In the SYMPLICITY HTN-3 trial, 123 (23%) of the 535 patients remained severely hypertensive despite treatment with an aldosterone antagonist such as spironolactone at the time of entry into the study. In this subgroup, renal denervation produced an average 8.1–mm Hg additional reduction in office systolic BP compared with the average reduction seen among the sham-control patients, a much larger effect than the average 3.2–mm Hg incremental reduction by renal denervation over control seen in the patients who were not on an aldosterone antagonist at baseline, Dr. Manesh Patel reported in a talk at the meeting.
One possible explanation for this effect is that "these patients were resistant to an aldosterone antagonist and hence have a good chance of having high sympathetic activity," explained Dr. Patel, director of interventional cardiology at Duke University in Durham, N.C., and a coinvestigator on the SYMPLICITY HTN-3 trial. Another possibility is that "aldosterone antagonist use is a marker for patients who have been treated in a hypertension clinic to receive this fourth-line agent," and hence are more likely to have true drug-resistant hypertension, he added. More recent analyses of the HTN-3 results also showed that the 38% of patients who entered the study while on treatment with a vasodilator had absolutely no added benefit from renal denervation compared with the sham controls, while in the patients not on a vasodilator renal denervation produced an average 6.7–mm Hg reduction in office systolic BP compared with control patients, a statistically significant difference.
"We must accept that currently denervation is a ‘black box’ procedure. You deliver energy and you hope blood pressure goes down, but the main confounder is we are not sure if we have damaged the nerve fibers," Dr. Mahfoud said.
According to data he compiled, the depth of ablation penetration varies by device, with several devices including the Symplicity producing an ablation depth of 3 mm, while a few other systems produce ablation depths of 4 mm or even 6 mm.
Results from autopsy studies he analyzed suggested that afferent nerve density closer to the renal-artery lumen is highest in the distal section of the renal artery compared with the more proximal side, and that the posterior and anterior quadrants of the distal renal artery harbor a higher concentration of nerve fibers closer to the lumen than the superior and inferior quadrants.
This information begins to define the "sweet spot" for applying denervation energy, Dr. Mahfoud said. When he performs renal denervation today "we go even more distally, into the branches [off the distal renal arteries] if they are large enough" to accommodate the catheter. "Nerves are not equally distributed over the entire renal artery," and ideally this information should help guide ablation placements, he said.
The global divide in renal denervation use
The inability of the SYMPLICITY HTN-3 trial to prove the treatment’s efficacy has further divided use of renal denervation by geography. The technology remains unapproved for U.S. use, and will remain that way until another large, sham-controlled trial finishes and shows a clear benefit for BP reduction. In contrast, the procedure’s use in Europe seems on track to continue and grow further, although European thought leaders urge caution and further research to identify the best denervation techniques and optimal patients.
European leaders such as Dr. Mahfoud and Dr. Schmieder also see great promise in using renal denervation for other types of patients, such as those with heart failure or arrhythmias. Just one example of the wide-ranging effects examined for renal denervation was a report Dr. Mahfoud cited published earlier this year that focused on changes in left ventricular mass in 55 patients with resistant hypertension who underwent renal denervation. The results collected by Dr. Mahfoud and his associates showed that even when patients experienced little or no change in their systolic BP they often had substantial reductions in left ventricular mass (Eur. Heart J. 2014 March 6 [doi:10.1093/eurheartj/ehu093]).
"Reducing systolic blood pressure by 10 mm Hg [in patients with severe, drug-resistant hypertension] would have a massive impact, so renal denervation remains an important tool for potentially benefiting patients with uncontrolled hypertension," Dr. Wijns, codirector of the Cardiovascular Center in Aalst, Belgium, said in an interview.
But the renal denervation tool that is increasingly seen as important by the cardiovascular disease leadership in Europe will remain beyond the reach of U.S. physicians for some time to come.
The SYMPLICITY HTN-3 trial and the Global SYMPLICITY Registry were sponsored by Medtronic, which markets the Symplicity catheter. All of the sources for this article have received speaker fees, consulting fees, and/or research grants from Medtronic and numerous other medical device, drug, or biotechnology companies.
On Twitter @mitchelzoler
PARIS – At least three different factors undermined the SYMPLICITY HTN-3 trial that earlier this year did not show a significant difference in blood pressure lowering between renal denervation and a sham-control procedure, most notably the failure of the vast majority of operators in the study to follow ablation instructions and produce thorough and reliable interruptions of sympathetic innervation of the kidneys, according to new data released by the trial’s investigators.
As the full range of problems with the U.S.-based SYMPLICITY HTN-3 trial, which had its main results reported in April (N. Engl. J. Med. 2014;370:1393-1401), became apparent in a report at the annual congress of the European Association of Percutaneous Cardiovascular Interventions, many top European practitioners and supporters of renal denervation voiced their belief that the treatment is an effective and safe option for many patients with true drug-resistant, severe hypertension.
The only qualifications they now add are that renal denervation is not easily performed and must be done carefully and in a more targeted way, with an ongoing need to find the patients best suited for treatment and the best methods for delivering treatment.
During the meeting, Dr. Felix Mahfoud, an interventional cardiologist at the University Hospital of Saarland in Homburg/Saar, Germany, joined with hypertension specialist Dr. Konstantinos Tsioufis of the University of Athens and Dr. William Wijns, codirector of EuroPCR, in an official statement from the meeting that despite the SYMPLICITY HTN-3 results they continued to support renal denervation as a treatment option for selected patients with drug-resistant, severe hypertension.
Their sentiment echoed another endorsement made a few weeks earlier for continued use and study of renal denervation from the European Society of Hypertension (ESH) in reaction to the SYMPLICITY HTN-3 results.
The ESH "sticks to its statement" from 2013 on using renal denervation in appropriate patients with treatment-resistant, severe hypertension (Eurointervention 2013;9:R58-R66), said Dr. Roland E. Schmieder, first author for the 2013 ESH position paper and a leader in European use of renal denervation.
"We need more studies to prove that renal denervation works, and in particular to get more precise information on which patients get the greatest benefit," Dr. Schmieder said in a separate talk at the meeting. For the time being, he said he was comfortable with routine use of renal denervation in patients with an office systolic BP of at least 160 mm Hg that remains at this level despite maximally tolerated treatment with at least three antihypertensive drugs, including a diuretic, the use endorsed by current European guidelines. It remains appropriate to investigate the impact of renal denervation on other disorders, such as heart failure, arrhythmia, metabolic syndrome, and depressed renal function, said Dr. Schmieder, professor and head of hypertension and vascular medicine research at University Hospital in Erlangen, Germany.
The problems with SYMPLICITY HTN-3
While much speculation swirled around what had gone wrong in the SYMPLICITY HTN-3 trial after researchers on the study gave their first report on the results early in the spring, the full extent of the study’s problems didn’t flesh out until a follow-up report during EuroPCR by coinvestigator Dr. David E. Kandzari. In his analysis, Dr. Kandzari highlighted three distinct problems with the trial that he and his associates identified in a series of post hoc analyses:
• The failure of a large minority of enrolled patients in both arms of the study to remain on a stable medical regimen during the 6 months of follow-up before the primary efficacy outcomes were measured.
• The inexplicably large reduction in BP among the sham-control patients, especially among African American patients, who made up a quarter of the trial’s population.
• The vastly incomplete nerve-ablation treatment that most patients received, treatments that usually failed to meet the standards specified in the trial’s protocol.
The background medical regimens that patients received proved unstable during SYMPLICITY HTN-3 even though the study design mandated that patients be on a stable regimen for at least 2 weeks before entering the study. Roughly 80% of enrolled patients in both the denervation and sham-control arms of the study had been on a stable regimen for at least 6 weeks before they entered. Despite that, during the 6 months of follow-up, 211 (39%) of patients in the study underwent a change in their medication regimen. The changes occurred at virtually identical rates in both study arms, and in more than two-thirds of cases were driven by medical necessity.
"The pattern of drug changes challenges the notion of maximally tolerated therapy," Dr. Kandzari said during his report. "Can this [maximally tolerated therapy] be sustained in a randomized, controlled trial?" It also raised the issues of how trial design can better limit drug changes.
Even though it remains unclear why blood pressure reduction was so pronounced among the African Americans in the sham-control group, the impact of this unexpected effect substantially upended the trial’s endpoints. Among the 49 African Americans randomized to sham treatment, office-measured systolic pressure dropped by an average of 17.8 mm Hg, far exceeding the 8.6–mm Hg decline seen among the non–African Americans in the control arm and even exceeding the average 15.5–mm Hg drop in office systolic BP among African Americans treated with renal denervation.
"The absolute reduction in blood pressure by renal denervation in African Americans was identical to non–African Americans." The problem that arose "related more to what happened in the sham-control group of African Americans, who had a nearly 18–mm Hg reduction in blood pressure," said Dr. Kandzari, chief scientific officer and director of interventional cardiology at Piedmont Heart Institute in Atlanta.
The low rate at which patients assigned to receive renal denervation actually received the type of treatment spelled out in the study’s protocol may have been the biggest problem of all, although Dr. Kandzari stressed that, in his opinion "no single factor led to the neutral efficacy seen in the study."
The supplementary methods section of the SYMPLICITY HTN-3 report published in April explicitly called for patients to receive "4-6 ablations" per side, delivering them in a spiral, circumferential pattern starting distally in each renal artery. That meant each patient was to receive a minimum of eight total ablations.
But analysis of data recorded independently by the research nurse and by the proctor during each procedure, as well as cineangiography films made and submitted by the operator for each ablation, clearly showed that many patients did not receive the treatment that the protocol spelled out. Synthesis of the data collected by the three methods showed that about half of the 364 patients randomized to renal denervation received at least eight ablations, while the other half did not receive this minimum number.
The three separate sets of ablation records also contained information on whether ablations occurred in the anterior, posterior, superior, or inferior quadrants of each renal artery. Full circumferential ablation, what the protocol prescribed, required an ablation in at least one of each of these quadrants per side. What actually happened was that 253 patients (70%) received no circumferential ablations, 68 patients (19%) received circumferential ablation on just one side, and 19 patients (5%) received the bilateral circumferential ablations that the protocol called for. Data for the remaining 24 patients treated with renal denervation were not amenable to analysis for this parameter.
As might be expected, greater ablation number and completeness strongly linked with a robust blood pressure effect.
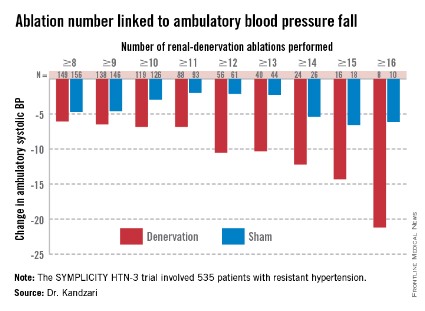
Among patients who received at least eight ablations, office systolic pressure fell by an average 13.1 mm Hg. But among the nine patients who received 16 or more ablations, the average systolic BP reduction at 6 months was 30.9 mm Hg. Among the 18 patients who received at least 15 ablations, the average systolic pressure reduction was 25.4 mm Hg. A very similar relationship occurred for BPs measured by ambulatory monitoring (see graphic), and the data also suggested a positive link between an increasing number of ablations and an increased effect on heart rate. The consistency of the association across all three measures lent further support to this as a real relationship, Dr. Kandzari noted.
Circumferentiality of the ablations showed a similar pattern. The average office systolic pressure fall in patients with no circumferential ablations was 14.2 mm Hg, and it was 16.1 mm Hg in patients who received just one circumferential ablation. But in the 19 patients who received circumferential ablations bilaterally, the average office systolic pressure reduction was 24.3 mm Hg, with a similar pattern seen for ambulatory measures as well as for home-based BP measurements.
"All patients randomized to renal denervation received renal denervation, but they may not have received it in a fashion that seemed to translate into a greater blood pressure reduction," Dr. Kandzari concluded.
Who to treat, where to treat, how to treat
"One result of the neutral HTN-3 result was a call to revisit the basic science behind renal denervation. The clinical enthusiasm had exceeded the science behind renal denervation," Dr. Kandzari observed.
Renal denervation’s many European advocates seem to agree, and have begun the process of determining characteristics of the best patients to receive renal denervation and where and how ablations are best delivered within the renal artery to achieve interruption of sympathetic innervation, although the targeting information they have right now is rudimentary.
"Probably most important is patient selection. You must be sure to get the right patient, one with high sympathetic activity, because the treatment lowers sympathetic activity," said Dr. Atul Pathak, an interventional cardiologist at Paul Sabatier University in Toulouse, France.
Some clues for patient selection have come from the Global SYMPLICITY Registry, which is enrolling patients treated with renal denervation at more than 200 experienced centers worldwide, many of them in Germany but also elsewhere in Europe, Australia, Canada, Korea, and other locations. Initial findings from the first 1,000 patients entered into the registry and followed for 6 months came out in March at the annual meeting of the American College of Cardiology, and Dr. Mahfoud presented new analyses of the data at EuroPCR.
"The major concern we had when we started renal denervation was its safety. I believe the safety issue is now answered," especially with the data collected in the global registry as well as in the SYMPLICITY HTN-3 trial, by far the largest trial completed for the procedure, said Dr. Thomas Zeller, professor and head of clinical and interventional angiology at the Heart Center in Bad Krozingen, Germany. "I was concerned that we might harm the renal arteries with long-lasting stenosis or embolic showers, but this does not happen, at least with the Symplicity catheter," he said during a talk at the meeting.
"The number of patients suitable for renal denervation is potentially much smaller than we initially expected. Real drug resistance is rare, poor adherence is common, and the Symplicity catheter is technically challenging and not effective in every patient. It is hard to rotate the catheter in the tortuous iliac arteries that some patients with hypertension have; the anatomic conditions of hypertension may not be suited to the Symplicity flex catheter," said Dr. Zeller, who added that he has performed renal denervations with the Symplicity catheter since 2009.
"We should focus on the patients that the HTN-3 trial identified as responders, including patients younger than 65, and patients on an aldosterone antagonist," he suggested in a talk at the meeting.
Finding the right patients and the right ablation targets
In the SYMPLICITY HTN-3 trial, 123 (23%) of the 535 patients remained severely hypertensive despite treatment with an aldosterone antagonist such as spironolactone at the time of entry into the study. In this subgroup, renal denervation produced an average 8.1–mm Hg additional reduction in office systolic BP compared with the average reduction seen among the sham-control patients, a much larger effect than the average 3.2–mm Hg incremental reduction by renal denervation over control seen in the patients who were not on an aldosterone antagonist at baseline, Dr. Manesh Patel reported in a talk at the meeting.
One possible explanation for this effect is that "these patients were resistant to an aldosterone antagonist and hence have a good chance of having high sympathetic activity," explained Dr. Patel, director of interventional cardiology at Duke University in Durham, N.C., and a coinvestigator on the SYMPLICITY HTN-3 trial. Another possibility is that "aldosterone antagonist use is a marker for patients who have been treated in a hypertension clinic to receive this fourth-line agent," and hence are more likely to have true drug-resistant hypertension, he added. More recent analyses of the HTN-3 results also showed that the 38% of patients who entered the study while on treatment with a vasodilator had absolutely no added benefit from renal denervation compared with the sham controls, while in the patients not on a vasodilator renal denervation produced an average 6.7–mm Hg reduction in office systolic BP compared with control patients, a statistically significant difference.
"We must accept that currently denervation is a ‘black box’ procedure. You deliver energy and you hope blood pressure goes down, but the main confounder is we are not sure if we have damaged the nerve fibers," Dr. Mahfoud said.
According to data he compiled, the depth of ablation penetration varies by device, with several devices including the Symplicity producing an ablation depth of 3 mm, while a few other systems produce ablation depths of 4 mm or even 6 mm.
Results from autopsy studies he analyzed suggested that afferent nerve density closer to the renal-artery lumen is highest in the distal section of the renal artery compared with the more proximal side, and that the posterior and anterior quadrants of the distal renal artery harbor a higher concentration of nerve fibers closer to the lumen than the superior and inferior quadrants.
This information begins to define the "sweet spot" for applying denervation energy, Dr. Mahfoud said. When he performs renal denervation today "we go even more distally, into the branches [off the distal renal arteries] if they are large enough" to accommodate the catheter. "Nerves are not equally distributed over the entire renal artery," and ideally this information should help guide ablation placements, he said.
The global divide in renal denervation use
The inability of the SYMPLICITY HTN-3 trial to prove the treatment’s efficacy has further divided use of renal denervation by geography. The technology remains unapproved for U.S. use, and will remain that way until another large, sham-controlled trial finishes and shows a clear benefit for BP reduction. In contrast, the procedure’s use in Europe seems on track to continue and grow further, although European thought leaders urge caution and further research to identify the best denervation techniques and optimal patients.
European leaders such as Dr. Mahfoud and Dr. Schmieder also see great promise in using renal denervation for other types of patients, such as those with heart failure or arrhythmias. Just one example of the wide-ranging effects examined for renal denervation was a report Dr. Mahfoud cited published earlier this year that focused on changes in left ventricular mass in 55 patients with resistant hypertension who underwent renal denervation. The results collected by Dr. Mahfoud and his associates showed that even when patients experienced little or no change in their systolic BP they often had substantial reductions in left ventricular mass (Eur. Heart J. 2014 March 6 [doi:10.1093/eurheartj/ehu093]).
"Reducing systolic blood pressure by 10 mm Hg [in patients with severe, drug-resistant hypertension] would have a massive impact, so renal denervation remains an important tool for potentially benefiting patients with uncontrolled hypertension," Dr. Wijns, codirector of the Cardiovascular Center in Aalst, Belgium, said in an interview.
But the renal denervation tool that is increasingly seen as important by the cardiovascular disease leadership in Europe will remain beyond the reach of U.S. physicians for some time to come.
The SYMPLICITY HTN-3 trial and the Global SYMPLICITY Registry were sponsored by Medtronic, which markets the Symplicity catheter. All of the sources for this article have received speaker fees, consulting fees, and/or research grants from Medtronic and numerous other medical device, drug, or biotechnology companies.
On Twitter @mitchelzoler
PARIS – At least three different factors undermined the SYMPLICITY HTN-3 trial that earlier this year did not show a significant difference in blood pressure lowering between renal denervation and a sham-control procedure, most notably the failure of the vast majority of operators in the study to follow ablation instructions and produce thorough and reliable interruptions of sympathetic innervation of the kidneys, according to new data released by the trial’s investigators.
As the full range of problems with the U.S.-based SYMPLICITY HTN-3 trial, which had its main results reported in April (N. Engl. J. Med. 2014;370:1393-1401), became apparent in a report at the annual congress of the European Association of Percutaneous Cardiovascular Interventions, many top European practitioners and supporters of renal denervation voiced their belief that the treatment is an effective and safe option for many patients with true drug-resistant, severe hypertension.
The only qualifications they now add are that renal denervation is not easily performed and must be done carefully and in a more targeted way, with an ongoing need to find the patients best suited for treatment and the best methods for delivering treatment.
During the meeting, Dr. Felix Mahfoud, an interventional cardiologist at the University Hospital of Saarland in Homburg/Saar, Germany, joined with hypertension specialist Dr. Konstantinos Tsioufis of the University of Athens and Dr. William Wijns, codirector of EuroPCR, in an official statement from the meeting that despite the SYMPLICITY HTN-3 results they continued to support renal denervation as a treatment option for selected patients with drug-resistant, severe hypertension.
Their sentiment echoed another endorsement made a few weeks earlier for continued use and study of renal denervation from the European Society of Hypertension (ESH) in reaction to the SYMPLICITY HTN-3 results.
The ESH "sticks to its statement" from 2013 on using renal denervation in appropriate patients with treatment-resistant, severe hypertension (Eurointervention 2013;9:R58-R66), said Dr. Roland E. Schmieder, first author for the 2013 ESH position paper and a leader in European use of renal denervation.
"We need more studies to prove that renal denervation works, and in particular to get more precise information on which patients get the greatest benefit," Dr. Schmieder said in a separate talk at the meeting. For the time being, he said he was comfortable with routine use of renal denervation in patients with an office systolic BP of at least 160 mm Hg that remains at this level despite maximally tolerated treatment with at least three antihypertensive drugs, including a diuretic, the use endorsed by current European guidelines. It remains appropriate to investigate the impact of renal denervation on other disorders, such as heart failure, arrhythmia, metabolic syndrome, and depressed renal function, said Dr. Schmieder, professor and head of hypertension and vascular medicine research at University Hospital in Erlangen, Germany.
The problems with SYMPLICITY HTN-3
While much speculation swirled around what had gone wrong in the SYMPLICITY HTN-3 trial after researchers on the study gave their first report on the results early in the spring, the full extent of the study’s problems didn’t flesh out until a follow-up report during EuroPCR by coinvestigator Dr. David E. Kandzari. In his analysis, Dr. Kandzari highlighted three distinct problems with the trial that he and his associates identified in a series of post hoc analyses:
• The failure of a large minority of enrolled patients in both arms of the study to remain on a stable medical regimen during the 6 months of follow-up before the primary efficacy outcomes were measured.
• The inexplicably large reduction in BP among the sham-control patients, especially among African American patients, who made up a quarter of the trial’s population.
• The vastly incomplete nerve-ablation treatment that most patients received, treatments that usually failed to meet the standards specified in the trial’s protocol.
The background medical regimens that patients received proved unstable during SYMPLICITY HTN-3 even though the study design mandated that patients be on a stable regimen for at least 2 weeks before entering the study. Roughly 80% of enrolled patients in both the denervation and sham-control arms of the study had been on a stable regimen for at least 6 weeks before they entered. Despite that, during the 6 months of follow-up, 211 (39%) of patients in the study underwent a change in their medication regimen. The changes occurred at virtually identical rates in both study arms, and in more than two-thirds of cases were driven by medical necessity.
"The pattern of drug changes challenges the notion of maximally tolerated therapy," Dr. Kandzari said during his report. "Can this [maximally tolerated therapy] be sustained in a randomized, controlled trial?" It also raised the issues of how trial design can better limit drug changes.
Even though it remains unclear why blood pressure reduction was so pronounced among the African Americans in the sham-control group, the impact of this unexpected effect substantially upended the trial’s endpoints. Among the 49 African Americans randomized to sham treatment, office-measured systolic pressure dropped by an average of 17.8 mm Hg, far exceeding the 8.6–mm Hg decline seen among the non–African Americans in the control arm and even exceeding the average 15.5–mm Hg drop in office systolic BP among African Americans treated with renal denervation.
"The absolute reduction in blood pressure by renal denervation in African Americans was identical to non–African Americans." The problem that arose "related more to what happened in the sham-control group of African Americans, who had a nearly 18–mm Hg reduction in blood pressure," said Dr. Kandzari, chief scientific officer and director of interventional cardiology at Piedmont Heart Institute in Atlanta.
The low rate at which patients assigned to receive renal denervation actually received the type of treatment spelled out in the study’s protocol may have been the biggest problem of all, although Dr. Kandzari stressed that, in his opinion "no single factor led to the neutral efficacy seen in the study."
The supplementary methods section of the SYMPLICITY HTN-3 report published in April explicitly called for patients to receive "4-6 ablations" per side, delivering them in a spiral, circumferential pattern starting distally in each renal artery. That meant each patient was to receive a minimum of eight total ablations.
But analysis of data recorded independently by the research nurse and by the proctor during each procedure, as well as cineangiography films made and submitted by the operator for each ablation, clearly showed that many patients did not receive the treatment that the protocol spelled out. Synthesis of the data collected by the three methods showed that about half of the 364 patients randomized to renal denervation received at least eight ablations, while the other half did not receive this minimum number.
The three separate sets of ablation records also contained information on whether ablations occurred in the anterior, posterior, superior, or inferior quadrants of each renal artery. Full circumferential ablation, what the protocol prescribed, required an ablation in at least one of each of these quadrants per side. What actually happened was that 253 patients (70%) received no circumferential ablations, 68 patients (19%) received circumferential ablation on just one side, and 19 patients (5%) received the bilateral circumferential ablations that the protocol called for. Data for the remaining 24 patients treated with renal denervation were not amenable to analysis for this parameter.
As might be expected, greater ablation number and completeness strongly linked with a robust blood pressure effect.
Among patients who received at least eight ablations, office systolic pressure fell by an average 13.1 mm Hg. But among the nine patients who received 16 or more ablations, the average systolic BP reduction at 6 months was 30.9 mm Hg. Among the 18 patients who received at least 15 ablations, the average systolic pressure reduction was 25.4 mm Hg. A very similar relationship occurred for BPs measured by ambulatory monitoring (see graphic), and the data also suggested a positive link between an increasing number of ablations and an increased effect on heart rate. The consistency of the association across all three measures lent further support to this as a real relationship, Dr. Kandzari noted.
Circumferentiality of the ablations showed a similar pattern. The average office systolic pressure fall in patients with no circumferential ablations was 14.2 mm Hg, and it was 16.1 mm Hg in patients who received just one circumferential ablation. But in the 19 patients who received circumferential ablations bilaterally, the average office systolic pressure reduction was 24.3 mm Hg, with a similar pattern seen for ambulatory measures as well as for home-based BP measurements.
"All patients randomized to renal denervation received renal denervation, but they may not have received it in a fashion that seemed to translate into a greater blood pressure reduction," Dr. Kandzari concluded.
Who to treat, where to treat, how to treat
"One result of the neutral HTN-3 result was a call to revisit the basic science behind renal denervation. The clinical enthusiasm had exceeded the science behind renal denervation," Dr. Kandzari observed.
Renal denervation’s many European advocates seem to agree, and have begun the process of determining characteristics of the best patients to receive renal denervation and where and how ablations are best delivered within the renal artery to achieve interruption of sympathetic innervation, although the targeting information they have right now is rudimentary.
"Probably most important is patient selection. You must be sure to get the right patient, one with high sympathetic activity, because the treatment lowers sympathetic activity," said Dr. Atul Pathak, an interventional cardiologist at Paul Sabatier University in Toulouse, France.
Some clues for patient selection have come from the Global SYMPLICITY Registry, which is enrolling patients treated with renal denervation at more than 200 experienced centers worldwide, many of them in Germany but also elsewhere in Europe, Australia, Canada, Korea, and other locations. Initial findings from the first 1,000 patients entered into the registry and followed for 6 months came out in March at the annual meeting of the American College of Cardiology, and Dr. Mahfoud presented new analyses of the data at EuroPCR.
"The major concern we had when we started renal denervation was its safety. I believe the safety issue is now answered," especially with the data collected in the global registry as well as in the SYMPLICITY HTN-3 trial, by far the largest trial completed for the procedure, said Dr. Thomas Zeller, professor and head of clinical and interventional angiology at the Heart Center in Bad Krozingen, Germany. "I was concerned that we might harm the renal arteries with long-lasting stenosis or embolic showers, but this does not happen, at least with the Symplicity catheter," he said during a talk at the meeting.
"The number of patients suitable for renal denervation is potentially much smaller than we initially expected. Real drug resistance is rare, poor adherence is common, and the Symplicity catheter is technically challenging and not effective in every patient. It is hard to rotate the catheter in the tortuous iliac arteries that some patients with hypertension have; the anatomic conditions of hypertension may not be suited to the Symplicity flex catheter," said Dr. Zeller, who added that he has performed renal denervations with the Symplicity catheter since 2009.
"We should focus on the patients that the HTN-3 trial identified as responders, including patients younger than 65, and patients on an aldosterone antagonist," he suggested in a talk at the meeting.
Finding the right patients and the right ablation targets
In the SYMPLICITY HTN-3 trial, 123 (23%) of the 535 patients remained severely hypertensive despite treatment with an aldosterone antagonist such as spironolactone at the time of entry into the study. In this subgroup, renal denervation produced an average 8.1–mm Hg additional reduction in office systolic BP compared with the average reduction seen among the sham-control patients, a much larger effect than the average 3.2–mm Hg incremental reduction by renal denervation over control seen in the patients who were not on an aldosterone antagonist at baseline, Dr. Manesh Patel reported in a talk at the meeting.
One possible explanation for this effect is that "these patients were resistant to an aldosterone antagonist and hence have a good chance of having high sympathetic activity," explained Dr. Patel, director of interventional cardiology at Duke University in Durham, N.C., and a coinvestigator on the SYMPLICITY HTN-3 trial. Another possibility is that "aldosterone antagonist use is a marker for patients who have been treated in a hypertension clinic to receive this fourth-line agent," and hence are more likely to have true drug-resistant hypertension, he added. More recent analyses of the HTN-3 results also showed that the 38% of patients who entered the study while on treatment with a vasodilator had absolutely no added benefit from renal denervation compared with the sham controls, while in the patients not on a vasodilator renal denervation produced an average 6.7–mm Hg reduction in office systolic BP compared with control patients, a statistically significant difference.
"We must accept that currently denervation is a ‘black box’ procedure. You deliver energy and you hope blood pressure goes down, but the main confounder is we are not sure if we have damaged the nerve fibers," Dr. Mahfoud said.
According to data he compiled, the depth of ablation penetration varies by device, with several devices including the Symplicity producing an ablation depth of 3 mm, while a few other systems produce ablation depths of 4 mm or even 6 mm.
Results from autopsy studies he analyzed suggested that afferent nerve density closer to the renal-artery lumen is highest in the distal section of the renal artery compared with the more proximal side, and that the posterior and anterior quadrants of the distal renal artery harbor a higher concentration of nerve fibers closer to the lumen than the superior and inferior quadrants.
This information begins to define the "sweet spot" for applying denervation energy, Dr. Mahfoud said. When he performs renal denervation today "we go even more distally, into the branches [off the distal renal arteries] if they are large enough" to accommodate the catheter. "Nerves are not equally distributed over the entire renal artery," and ideally this information should help guide ablation placements, he said.
The global divide in renal denervation use
The inability of the SYMPLICITY HTN-3 trial to prove the treatment’s efficacy has further divided use of renal denervation by geography. The technology remains unapproved for U.S. use, and will remain that way until another large, sham-controlled trial finishes and shows a clear benefit for BP reduction. In contrast, the procedure’s use in Europe seems on track to continue and grow further, although European thought leaders urge caution and further research to identify the best denervation techniques and optimal patients.
European leaders such as Dr. Mahfoud and Dr. Schmieder also see great promise in using renal denervation for other types of patients, such as those with heart failure or arrhythmias. Just one example of the wide-ranging effects examined for renal denervation was a report Dr. Mahfoud cited published earlier this year that focused on changes in left ventricular mass in 55 patients with resistant hypertension who underwent renal denervation. The results collected by Dr. Mahfoud and his associates showed that even when patients experienced little or no change in their systolic BP they often had substantial reductions in left ventricular mass (Eur. Heart J. 2014 March 6 [doi:10.1093/eurheartj/ehu093]).
"Reducing systolic blood pressure by 10 mm Hg [in patients with severe, drug-resistant hypertension] would have a massive impact, so renal denervation remains an important tool for potentially benefiting patients with uncontrolled hypertension," Dr. Wijns, codirector of the Cardiovascular Center in Aalst, Belgium, said in an interview.
But the renal denervation tool that is increasingly seen as important by the cardiovascular disease leadership in Europe will remain beyond the reach of U.S. physicians for some time to come.
The SYMPLICITY HTN-3 trial and the Global SYMPLICITY Registry were sponsored by Medtronic, which markets the Symplicity catheter. All of the sources for this article have received speaker fees, consulting fees, and/or research grants from Medtronic and numerous other medical device, drug, or biotechnology companies.
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM EUROPCR 2014
Upcoming ESC revascularization guidelines cement heart team’s role
PARIS – A joint European Society of Cardiology and European Association for Cardio-Thoracic Surgery task force that will publish revised revascularization guidelines in late August gave a sneak peak of some important elements of the revision, including renewed endorsement of and a refinement to the heart team concept that was first introduced in the prior, 2010 version of the guidelines.
"One of the most important aspects of the 2010 guidelines was the introduction of the heart team (Eur. Heart J. 2010;31:2501-55) said Dr. Philippe H. Kolh. "In 2010, the heart team concept was still controversial, but I think now it is well accepted. We are further supporting and emphasizing the importance of the heart team," he said of the revised guidelines that will be released in August, during a session that previewed selected parts of the new guidelines at the annual congress of the European Association of Percutaneous Cardiovascular Interventions, an organization that also collaborated on the guidelines.

The revision also calls on each institution where operators perform revascularization to establish local protocols to guide the choice in routine cases between percutaneous coronary interventions (PCIs) or coronary artery bypass grafting (CABG), said Dr. Kolh, a cardiac surgeon at University Hospital in Liège, Belgium, and cochairman of the guideline-writing panel.
"The 2010 guidelines produced a misconception that every patient needs to be discussed by a heart team; the 2014 revision makes it clear that the heart team should develop institutional protocols for appropriate revascularization strategies for different types of patients. So if a patient has single-vessel disease, you can go ahead and do PCI and not wait for a heart-team decision," said Dr. Ulf Landmesser, professor and head of the acute cardiology clinic at University Hospital, Zurich, and a member of the 2014 panel. "Hopefully, it will now be clear that the heart team only needs to discuss complex patients that involve difficult decisions, and that institutional protocols can handle routine cases," Dr. Landmesser said.
The revision comes at a time when "the competition today is not so much between CABG and PCI; the more burning question is who should have revascularization, and how do patients get to the cath lab," noted Dr. Spencer B. King III, an interventional cardiologist at St. Joseph’s Medical Group in Atlanta who was invited to the session to comment on the new revision.

Results from a new meta-analysis highlight the critical role of revascularization relative to medical therapy alone in improving outcomes of patients with coronary artery disease. This finding is especially relevant in 2014, because it marks the 50th anniversary of the launch of revascularization with the first successful CABG performed, observed Dr. Stephan Windecker, professor and chief of cardiology at University Hospital in Bern, Switzerland, and cochairman of the guidelines-writing panel.
He presented an analysis of results from 100 randomized, controlled trials that compared some form of revascularization against medical therapy in 93,553 randomized patients followed for more than 260,000 patient-years. The results showed that CABG cut the rate of all-cause mortality by 20%, compared with medical therapy, a statistically significant difference, and that treatment with new-generation drug-eluting stents produced a significant reduction of more than 25%, according to an as-yet unpublished report by members of the European Myocardial Revascularization Collaborative. Dr. Windecker also noted that all the recommendations in the new revision were approved with 100% consensus by the panel, which included cardiac surgeons, interventional cardiologists, and noninterventional cardiologists in equal numbers.
The session highlighted several other notable new elements in the revised guidelines, although Dr. Windecker stressed several times during the session that everything presented remained pending until the final version is released later this summer. The changes include:
• An "upgrade" of the recommendation for PCI use in patients with left main disease and a SYNTAX score of 23-32 to a IIa, "should be considered" class recommendation, boosted from class III "not recommended" status in 2010. Five-year outcomes from the SYNTAX trial showed "no difference in outcomes between PCI and CABG, a major reason to upgrade the recommendation for PCI," said Dr. Landmesser (Lancet 2013;381:629-38). "The guidelines put a lot of weight on SYNTAX score."
• When performing PCI in patients with non–ST-elevation myocardial infarction (NSTEMI), bivalirudin (Angiomax) is recommended exclusively as the anticoagulant to use during and immediately following PCI – with unfractionated heparin recommended only for patients who cannot receive bivalirudin – based on bivalirudin’s proven reduced risk for causing major bleeds, said Dr. Franz-Josef Neumann, professor and director of the University Heart Center in Bad Krozingen, Germany.
• But for patients with ST-elevation MI (STEMI) undergoing primary PCI, unfractionated heparin received the only unqualified, level I recommendation for anticoagulation, with bivalirudin receiving a level IIa, "should be considered" recommendation. This repositioning of the two options occurred, based to some extent on yet unpublished results from a very large, single-center study in Liverpool, HEAT-PPCI, reported at the annual meeting of the American College of Cardiology meeting in March that showed unfractionated heparin outperformed bivalirudin for 28-day outcomes, Dr. Neumann said. "I was very pleased and sort of amazed that results from HEAT-PPCI jumped into the guidelines, and it’s not even published yet. That [recommendation] will have an impact, I suspect," commented Dr. King.
• For patients with either STEMI or NSTEMI, the preferred antiplatelet P2Y12 inhibitors are prasugrel (Effient) and ticagrelor (Brilinta), with clopidogrel reduced to a back-up role "only when prasugrel or ticagrelor are not available," said Dr. Neumann. "I was a little surprised that clopidogrel has fallen off the charts. With the new stents having a low stent thrombosis rate, U.S. physicians tend to stick with clopidogrel; there has been more of a shift in Europe," commented Dr. King. "For elective cases, we still have a clear statement in favor of clopidogrel," countered Dr. Neumann. "It is only for higher risk, acute coronary syndrome and STEMI patients where the guidelines recommend the new agents."
Dr. Kolh said that he has received honoraria from Astra Zeneca and Braun, and research support from Edwards. Dr. Landmesser said that he had no disclosures. Dr. King said that he had no disclosures. Dr. Windecker said that he had received honoraria from, had been a consultant to, or had been a speaker for nine companies and had received research grants from seven companies. Dr. Neumann said that his institution had received research grants from 15 companies.
On Twitter @mitchelzoler
PARIS – A joint European Society of Cardiology and European Association for Cardio-Thoracic Surgery task force that will publish revised revascularization guidelines in late August gave a sneak peak of some important elements of the revision, including renewed endorsement of and a refinement to the heart team concept that was first introduced in the prior, 2010 version of the guidelines.
"One of the most important aspects of the 2010 guidelines was the introduction of the heart team (Eur. Heart J. 2010;31:2501-55) said Dr. Philippe H. Kolh. "In 2010, the heart team concept was still controversial, but I think now it is well accepted. We are further supporting and emphasizing the importance of the heart team," he said of the revised guidelines that will be released in August, during a session that previewed selected parts of the new guidelines at the annual congress of the European Association of Percutaneous Cardiovascular Interventions, an organization that also collaborated on the guidelines.
The revision also calls on each institution where operators perform revascularization to establish local protocols to guide the choice in routine cases between percutaneous coronary interventions (PCIs) or coronary artery bypass grafting (CABG), said Dr. Kolh, a cardiac surgeon at University Hospital in Liège, Belgium, and cochairman of the guideline-writing panel.
"The 2010 guidelines produced a misconception that every patient needs to be discussed by a heart team; the 2014 revision makes it clear that the heart team should develop institutional protocols for appropriate revascularization strategies for different types of patients. So if a patient has single-vessel disease, you can go ahead and do PCI and not wait for a heart-team decision," said Dr. Ulf Landmesser, professor and head of the acute cardiology clinic at University Hospital, Zurich, and a member of the 2014 panel. "Hopefully, it will now be clear that the heart team only needs to discuss complex patients that involve difficult decisions, and that institutional protocols can handle routine cases," Dr. Landmesser said.
The revision comes at a time when "the competition today is not so much between CABG and PCI; the more burning question is who should have revascularization, and how do patients get to the cath lab," noted Dr. Spencer B. King III, an interventional cardiologist at St. Joseph’s Medical Group in Atlanta who was invited to the session to comment on the new revision.
Results from a new meta-analysis highlight the critical role of revascularization relative to medical therapy alone in improving outcomes of patients with coronary artery disease. This finding is especially relevant in 2014, because it marks the 50th anniversary of the launch of revascularization with the first successful CABG performed, observed Dr. Stephan Windecker, professor and chief of cardiology at University Hospital in Bern, Switzerland, and cochairman of the guidelines-writing panel.
He presented an analysis of results from 100 randomized, controlled trials that compared some form of revascularization against medical therapy in 93,553 randomized patients followed for more than 260,000 patient-years. The results showed that CABG cut the rate of all-cause mortality by 20%, compared with medical therapy, a statistically significant difference, and that treatment with new-generation drug-eluting stents produced a significant reduction of more than 25%, according to an as-yet unpublished report by members of the European Myocardial Revascularization Collaborative. Dr. Windecker also noted that all the recommendations in the new revision were approved with 100% consensus by the panel, which included cardiac surgeons, interventional cardiologists, and noninterventional cardiologists in equal numbers.
The session highlighted several other notable new elements in the revised guidelines, although Dr. Windecker stressed several times during the session that everything presented remained pending until the final version is released later this summer. The changes include:
• An "upgrade" of the recommendation for PCI use in patients with left main disease and a SYNTAX score of 23-32 to a IIa, "should be considered" class recommendation, boosted from class III "not recommended" status in 2010. Five-year outcomes from the SYNTAX trial showed "no difference in outcomes between PCI and CABG, a major reason to upgrade the recommendation for PCI," said Dr. Landmesser (Lancet 2013;381:629-38). "The guidelines put a lot of weight on SYNTAX score."
• When performing PCI in patients with non–ST-elevation myocardial infarction (NSTEMI), bivalirudin (Angiomax) is recommended exclusively as the anticoagulant to use during and immediately following PCI – with unfractionated heparin recommended only for patients who cannot receive bivalirudin – based on bivalirudin’s proven reduced risk for causing major bleeds, said Dr. Franz-Josef Neumann, professor and director of the University Heart Center in Bad Krozingen, Germany.
• But for patients with ST-elevation MI (STEMI) undergoing primary PCI, unfractionated heparin received the only unqualified, level I recommendation for anticoagulation, with bivalirudin receiving a level IIa, "should be considered" recommendation. This repositioning of the two options occurred, based to some extent on yet unpublished results from a very large, single-center study in Liverpool, HEAT-PPCI, reported at the annual meeting of the American College of Cardiology meeting in March that showed unfractionated heparin outperformed bivalirudin for 28-day outcomes, Dr. Neumann said. "I was very pleased and sort of amazed that results from HEAT-PPCI jumped into the guidelines, and it’s not even published yet. That [recommendation] will have an impact, I suspect," commented Dr. King.
• For patients with either STEMI or NSTEMI, the preferred antiplatelet P2Y12 inhibitors are prasugrel (Effient) and ticagrelor (Brilinta), with clopidogrel reduced to a back-up role "only when prasugrel or ticagrelor are not available," said Dr. Neumann. "I was a little surprised that clopidogrel has fallen off the charts. With the new stents having a low stent thrombosis rate, U.S. physicians tend to stick with clopidogrel; there has been more of a shift in Europe," commented Dr. King. "For elective cases, we still have a clear statement in favor of clopidogrel," countered Dr. Neumann. "It is only for higher risk, acute coronary syndrome and STEMI patients where the guidelines recommend the new agents."
Dr. Kolh said that he has received honoraria from Astra Zeneca and Braun, and research support from Edwards. Dr. Landmesser said that he had no disclosures. Dr. King said that he had no disclosures. Dr. Windecker said that he had received honoraria from, had been a consultant to, or had been a speaker for nine companies and had received research grants from seven companies. Dr. Neumann said that his institution had received research grants from 15 companies.
On Twitter @mitchelzoler
PARIS – A joint European Society of Cardiology and European Association for Cardio-Thoracic Surgery task force that will publish revised revascularization guidelines in late August gave a sneak peak of some important elements of the revision, including renewed endorsement of and a refinement to the heart team concept that was first introduced in the prior, 2010 version of the guidelines.
"One of the most important aspects of the 2010 guidelines was the introduction of the heart team (Eur. Heart J. 2010;31:2501-55) said Dr. Philippe H. Kolh. "In 2010, the heart team concept was still controversial, but I think now it is well accepted. We are further supporting and emphasizing the importance of the heart team," he said of the revised guidelines that will be released in August, during a session that previewed selected parts of the new guidelines at the annual congress of the European Association of Percutaneous Cardiovascular Interventions, an organization that also collaborated on the guidelines.
The revision also calls on each institution where operators perform revascularization to establish local protocols to guide the choice in routine cases between percutaneous coronary interventions (PCIs) or coronary artery bypass grafting (CABG), said Dr. Kolh, a cardiac surgeon at University Hospital in Liège, Belgium, and cochairman of the guideline-writing panel.
"The 2010 guidelines produced a misconception that every patient needs to be discussed by a heart team; the 2014 revision makes it clear that the heart team should develop institutional protocols for appropriate revascularization strategies for different types of patients. So if a patient has single-vessel disease, you can go ahead and do PCI and not wait for a heart-team decision," said Dr. Ulf Landmesser, professor and head of the acute cardiology clinic at University Hospital, Zurich, and a member of the 2014 panel. "Hopefully, it will now be clear that the heart team only needs to discuss complex patients that involve difficult decisions, and that institutional protocols can handle routine cases," Dr. Landmesser said.
The revision comes at a time when "the competition today is not so much between CABG and PCI; the more burning question is who should have revascularization, and how do patients get to the cath lab," noted Dr. Spencer B. King III, an interventional cardiologist at St. Joseph’s Medical Group in Atlanta who was invited to the session to comment on the new revision.
Results from a new meta-analysis highlight the critical role of revascularization relative to medical therapy alone in improving outcomes of patients with coronary artery disease. This finding is especially relevant in 2014, because it marks the 50th anniversary of the launch of revascularization with the first successful CABG performed, observed Dr. Stephan Windecker, professor and chief of cardiology at University Hospital in Bern, Switzerland, and cochairman of the guidelines-writing panel.
He presented an analysis of results from 100 randomized, controlled trials that compared some form of revascularization against medical therapy in 93,553 randomized patients followed for more than 260,000 patient-years. The results showed that CABG cut the rate of all-cause mortality by 20%, compared with medical therapy, a statistically significant difference, and that treatment with new-generation drug-eluting stents produced a significant reduction of more than 25%, according to an as-yet unpublished report by members of the European Myocardial Revascularization Collaborative. Dr. Windecker also noted that all the recommendations in the new revision were approved with 100% consensus by the panel, which included cardiac surgeons, interventional cardiologists, and noninterventional cardiologists in equal numbers.
The session highlighted several other notable new elements in the revised guidelines, although Dr. Windecker stressed several times during the session that everything presented remained pending until the final version is released later this summer. The changes include:
• An "upgrade" of the recommendation for PCI use in patients with left main disease and a SYNTAX score of 23-32 to a IIa, "should be considered" class recommendation, boosted from class III "not recommended" status in 2010. Five-year outcomes from the SYNTAX trial showed "no difference in outcomes between PCI and CABG, a major reason to upgrade the recommendation for PCI," said Dr. Landmesser (Lancet 2013;381:629-38). "The guidelines put a lot of weight on SYNTAX score."
• When performing PCI in patients with non–ST-elevation myocardial infarction (NSTEMI), bivalirudin (Angiomax) is recommended exclusively as the anticoagulant to use during and immediately following PCI – with unfractionated heparin recommended only for patients who cannot receive bivalirudin – based on bivalirudin’s proven reduced risk for causing major bleeds, said Dr. Franz-Josef Neumann, professor and director of the University Heart Center in Bad Krozingen, Germany.
• But for patients with ST-elevation MI (STEMI) undergoing primary PCI, unfractionated heparin received the only unqualified, level I recommendation for anticoagulation, with bivalirudin receiving a level IIa, "should be considered" recommendation. This repositioning of the two options occurred, based to some extent on yet unpublished results from a very large, single-center study in Liverpool, HEAT-PPCI, reported at the annual meeting of the American College of Cardiology meeting in March that showed unfractionated heparin outperformed bivalirudin for 28-day outcomes, Dr. Neumann said. "I was very pleased and sort of amazed that results from HEAT-PPCI jumped into the guidelines, and it’s not even published yet. That [recommendation] will have an impact, I suspect," commented Dr. King.
• For patients with either STEMI or NSTEMI, the preferred antiplatelet P2Y12 inhibitors are prasugrel (Effient) and ticagrelor (Brilinta), with clopidogrel reduced to a back-up role "only when prasugrel or ticagrelor are not available," said Dr. Neumann. "I was a little surprised that clopidogrel has fallen off the charts. With the new stents having a low stent thrombosis rate, U.S. physicians tend to stick with clopidogrel; there has been more of a shift in Europe," commented Dr. King. "For elective cases, we still have a clear statement in favor of clopidogrel," countered Dr. Neumann. "It is only for higher risk, acute coronary syndrome and STEMI patients where the guidelines recommend the new agents."
Dr. Kolh said that he has received honoraria from Astra Zeneca and Braun, and research support from Edwards. Dr. Landmesser said that he had no disclosures. Dr. King said that he had no disclosures. Dr. Windecker said that he had received honoraria from, had been a consultant to, or had been a speaker for nine companies and had received research grants from seven companies. Dr. Neumann said that his institution had received research grants from 15 companies.
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM EUROPCR 2014

Paclitaxel-eluting balloon shows high claudication efficacy
PARIS – A drug-eluting balloon produced a stentlike rate of primary patency and need for target vessel revascularization in a multinational, controlled trial with 331 patients with claudication.
The results showed that the paclitaxel-eluting angioplasty balloon used in the study, the IN.PACT model made by Medtronic, has the "potential to become the standard of care" for treating stenoses in the superficial femoral and popliteal arteries, Dr. Marianne Brodmann said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions. After 1 year, the rate of clinically driven target-vessel revascularizations was 2% in the 220 patients treated with the drug-eluting balloon and 21% in 111 control patients treated with plain balloon angioplasty, a statistically significant difference, reported Dr. Brodmann, professor of angiology at the Medical University of Graz (Austria).*

The results seen in this trial contrast with results from studies of other types of drug-eluting balloons in these arteries, said Dr. Marc Bosiers, head of the department of vascular surgery at St. Blasius Hospital in Dendermonde, Belgium, and a coinvestigator in the study. "What we’ve learned from this trial, if you look at the results from other trials [of drug-eluting balloons], is that not all drug-eluting balloons are equal, just as not all stents are equal," Dr. Bosiers said.
The IN.PACT SFA Trial enrolled 150 patients at 13 centers in Europe and 181 patients at 44 U.S. centers. All patients were adults with Rutherford stage 2, 3, or 4 disease; claudication and rest pain; and a single or closely tandem lesion in the superficial femoral or popliteal arteries with a total length of no more than 18 cm. Their average age was 68, and about 40% had diabetes. The trial protocol allowed provisional stenting, which occurred in 7% of the patients treated with a paclitaxel-eluting balloon and in 13% of those treated with a plain balloon. The average lesion length treated was about 9 cm in both arms of the study.
The study’s primary endpoint was the rate of primary patency at 12 months, defined as freedom from clinically driven target-vessel revascularization and freedom from restenosis assessed by Doppler ultrasound at 12 months, which was 82% in patients treated with the drug-eluting balloon and 52% among patients in the control arm, a statistically significant difference.
The study’s primary safety endpoint was the combined rate of procedure- and device-related death at 30 days, freedom from target-limb major amputation at 1 year, and freedom from clinically driven target-vessel revascularization at 1 year, which occurred in 96% of patients treated with the paclitaxel-eluting balloon and in 77% of the control patients, a statistically significant difference.
These outcomes included "the lowest target-vessel revascularization rates and the highest patency rates ever reported" in this setting, and provide "robust, level 1 evidence" for the safety and efficacy of the paclitaxel-eluting balloon for this indication, Dr. Brodmann concluded.
If restenosis were to occur in the target vessel following treatment with the paclitaxel-eluting balloon, it would be possible to retreat the same vessel with a second paclitaxel-eluting balloon, although that scenario was not tested in the trial, Dr. Brodmann said in an interview. The paclitaxel essentially disappears within a few months of treatment, which should allow safe retreatment.
A written statement from Medtronic said that the company has an application pending with the Food and Drug Administration for U.S. marketing approval of the IN.PACT balloon for this indication. The balloon has been available in Europe since 2009.
The IN.PACT SFA Trial was sponsored by Medtronic, which markets the IN.PACT drug-eluting balloon. Dr. Brodmann said she is a consultant to Medtronic. Dr. Bosiers said that he had no disclosures.
On Twitter @mitchelzoler
*Correction, 6/24/2014: A previous version of this article misstated the number of patients in the control arm, the number of study centers, the name of the study, and two references to the device.

|
Mitchel L. Zoler/Frontline Medical News
|
The results from this trial change the way we think about treating stenoses in the superficial femoral and popliteal arteries. These results are probably the first to show with such robust, level 1 evidence that a drug-eluting balloon works at least as well as the best stent available today.
The results mean that the concept of "leave nothing behind" when treating vascular disease in the superficial femoral artery will be the best approach going forward. The 82% 1-year patency rate and the 2.4% rate of clinically driven target-vessel revascularizations were absolutely outstanding results.
Dr. Alberto Cremonesi, director of the interventional cardioangiology unit at Villa Maria Cecilia Hospital in Cotignola-Ravenna, Italy, made these comments in an interview. He said he had no relevant financial disclosures.
|
|
Mitchel L. Zoler/Frontline Medical News
|
The results from this trial change the way we think about treating stenoses in the superficial femoral and popliteal arteries. These results are probably the first to show with such robust, level 1 evidence that a drug-eluting balloon works at least as well as the best stent available today.
The results mean that the concept of "leave nothing behind" when treating vascular disease in the superficial femoral artery will be the best approach going forward. The 82% 1-year patency rate and the 2.4% rate of clinically driven target-vessel revascularizations were absolutely outstanding results.
Dr. Alberto Cremonesi, director of the interventional cardioangiology unit at Villa Maria Cecilia Hospital in Cotignola-Ravenna, Italy, made these comments in an interview. He said he had no relevant financial disclosures.
|
|
Mitchel L. Zoler/Frontline Medical News
|
The results from this trial change the way we think about treating stenoses in the superficial femoral and popliteal arteries. These results are probably the first to show with such robust, level 1 evidence that a drug-eluting balloon works at least as well as the best stent available today.
The results mean that the concept of "leave nothing behind" when treating vascular disease in the superficial femoral artery will be the best approach going forward. The 82% 1-year patency rate and the 2.4% rate of clinically driven target-vessel revascularizations were absolutely outstanding results.
Dr. Alberto Cremonesi, director of the interventional cardioangiology unit at Villa Maria Cecilia Hospital in Cotignola-Ravenna, Italy, made these comments in an interview. He said he had no relevant financial disclosures.
PARIS – A drug-eluting balloon produced a stentlike rate of primary patency and need for target vessel revascularization in a multinational, controlled trial with 331 patients with claudication.
The results showed that the paclitaxel-eluting angioplasty balloon used in the study, the IN.PACT model made by Medtronic, has the "potential to become the standard of care" for treating stenoses in the superficial femoral and popliteal arteries, Dr. Marianne Brodmann said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions. After 1 year, the rate of clinically driven target-vessel revascularizations was 2% in the 220 patients treated with the drug-eluting balloon and 21% in 111 control patients treated with plain balloon angioplasty, a statistically significant difference, reported Dr. Brodmann, professor of angiology at the Medical University of Graz (Austria).*
The results seen in this trial contrast with results from studies of other types of drug-eluting balloons in these arteries, said Dr. Marc Bosiers, head of the department of vascular surgery at St. Blasius Hospital in Dendermonde, Belgium, and a coinvestigator in the study. "What we’ve learned from this trial, if you look at the results from other trials [of drug-eluting balloons], is that not all drug-eluting balloons are equal, just as not all stents are equal," Dr. Bosiers said.
The IN.PACT SFA Trial enrolled 150 patients at 13 centers in Europe and 181 patients at 44 U.S. centers. All patients were adults with Rutherford stage 2, 3, or 4 disease; claudication and rest pain; and a single or closely tandem lesion in the superficial femoral or popliteal arteries with a total length of no more than 18 cm. Their average age was 68, and about 40% had diabetes. The trial protocol allowed provisional stenting, which occurred in 7% of the patients treated with a paclitaxel-eluting balloon and in 13% of those treated with a plain balloon. The average lesion length treated was about 9 cm in both arms of the study.
The study’s primary endpoint was the rate of primary patency at 12 months, defined as freedom from clinically driven target-vessel revascularization and freedom from restenosis assessed by Doppler ultrasound at 12 months, which was 82% in patients treated with the drug-eluting balloon and 52% among patients in the control arm, a statistically significant difference.
The study’s primary safety endpoint was the combined rate of procedure- and device-related death at 30 days, freedom from target-limb major amputation at 1 year, and freedom from clinically driven target-vessel revascularization at 1 year, which occurred in 96% of patients treated with the paclitaxel-eluting balloon and in 77% of the control patients, a statistically significant difference.
These outcomes included "the lowest target-vessel revascularization rates and the highest patency rates ever reported" in this setting, and provide "robust, level 1 evidence" for the safety and efficacy of the paclitaxel-eluting balloon for this indication, Dr. Brodmann concluded.
If restenosis were to occur in the target vessel following treatment with the paclitaxel-eluting balloon, it would be possible to retreat the same vessel with a second paclitaxel-eluting balloon, although that scenario was not tested in the trial, Dr. Brodmann said in an interview. The paclitaxel essentially disappears within a few months of treatment, which should allow safe retreatment.
A written statement from Medtronic said that the company has an application pending with the Food and Drug Administration for U.S. marketing approval of the IN.PACT balloon for this indication. The balloon has been available in Europe since 2009.
The IN.PACT SFA Trial was sponsored by Medtronic, which markets the IN.PACT drug-eluting balloon. Dr. Brodmann said she is a consultant to Medtronic. Dr. Bosiers said that he had no disclosures.
On Twitter @mitchelzoler
*Correction, 6/24/2014: A previous version of this article misstated the number of patients in the control arm, the number of study centers, the name of the study, and two references to the device.
PARIS – A drug-eluting balloon produced a stentlike rate of primary patency and need for target vessel revascularization in a multinational, controlled trial with 331 patients with claudication.
The results showed that the paclitaxel-eluting angioplasty balloon used in the study, the IN.PACT model made by Medtronic, has the "potential to become the standard of care" for treating stenoses in the superficial femoral and popliteal arteries, Dr. Marianne Brodmann said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions. After 1 year, the rate of clinically driven target-vessel revascularizations was 2% in the 220 patients treated with the drug-eluting balloon and 21% in 111 control patients treated with plain balloon angioplasty, a statistically significant difference, reported Dr. Brodmann, professor of angiology at the Medical University of Graz (Austria).*
The results seen in this trial contrast with results from studies of other types of drug-eluting balloons in these arteries, said Dr. Marc Bosiers, head of the department of vascular surgery at St. Blasius Hospital in Dendermonde, Belgium, and a coinvestigator in the study. "What we’ve learned from this trial, if you look at the results from other trials [of drug-eluting balloons], is that not all drug-eluting balloons are equal, just as not all stents are equal," Dr. Bosiers said.
The IN.PACT SFA Trial enrolled 150 patients at 13 centers in Europe and 181 patients at 44 U.S. centers. All patients were adults with Rutherford stage 2, 3, or 4 disease; claudication and rest pain; and a single or closely tandem lesion in the superficial femoral or popliteal arteries with a total length of no more than 18 cm. Their average age was 68, and about 40% had diabetes. The trial protocol allowed provisional stenting, which occurred in 7% of the patients treated with a paclitaxel-eluting balloon and in 13% of those treated with a plain balloon. The average lesion length treated was about 9 cm in both arms of the study.
The study’s primary endpoint was the rate of primary patency at 12 months, defined as freedom from clinically driven target-vessel revascularization and freedom from restenosis assessed by Doppler ultrasound at 12 months, which was 82% in patients treated with the drug-eluting balloon and 52% among patients in the control arm, a statistically significant difference.
The study’s primary safety endpoint was the combined rate of procedure- and device-related death at 30 days, freedom from target-limb major amputation at 1 year, and freedom from clinically driven target-vessel revascularization at 1 year, which occurred in 96% of patients treated with the paclitaxel-eluting balloon and in 77% of the control patients, a statistically significant difference.
These outcomes included "the lowest target-vessel revascularization rates and the highest patency rates ever reported" in this setting, and provide "robust, level 1 evidence" for the safety and efficacy of the paclitaxel-eluting balloon for this indication, Dr. Brodmann concluded.
If restenosis were to occur in the target vessel following treatment with the paclitaxel-eluting balloon, it would be possible to retreat the same vessel with a second paclitaxel-eluting balloon, although that scenario was not tested in the trial, Dr. Brodmann said in an interview. The paclitaxel essentially disappears within a few months of treatment, which should allow safe retreatment.
A written statement from Medtronic said that the company has an application pending with the Food and Drug Administration for U.S. marketing approval of the IN.PACT balloon for this indication. The balloon has been available in Europe since 2009.
The IN.PACT SFA Trial was sponsored by Medtronic, which markets the IN.PACT drug-eluting balloon. Dr. Brodmann said she is a consultant to Medtronic. Dr. Bosiers said that he had no disclosures.
On Twitter @mitchelzoler
*Correction, 6/24/2014: A previous version of this article misstated the number of patients in the control arm, the number of study centers, the name of the study, and two references to the device.
AT EUROPCR 2014
Key clinical point: A drug-eluting balloon produced stentlike patency after 1 year in superficial femoral and popliteal arteries.
Major finding: Angioplasty with a paclitaxel-eluting balloon produced a 1-year 82% primary patency rate, compared with 52% in controls.
Data source: A multicenter, randomized controlled trial with 331 patients with claudication and rest pain treated at 57 international sites.
Disclosures: The IN.PACT SFA Trial was sponsored by Medtronic, which markets the IN.PACT drug-eluting balloon. Dr. Brodmann said she is a consultant to Medtronic. Dr. Bosiers said he had no disclosures.

Stem-cell transplants growing routine for severe scleroderma
PARIS – Autologous stem-cell transplantation has emerged as an effective and feasible treatment for selected patients with severe systemic sclerosis who inadequately respond to conventional treatments, said two U.S. experts.
In addition, convincing evidence documenting the overall beneficial effect of autologous stem-cell transplantation will soon appear in a published report from the Autologous Stem Cell Transplantation International Scleroderma (ASTIS) trial, Dr. Dinesh Khanna said in a talk at the annual European Congress of Rheumatology.

In his talk, Dr. Khanna gave a short preview of the ASTIS results that he said would appear in a medical journal in the next few weeks. Those results show that among 79 scleroderma patients randomized to treatment with autologous stem-cell transplant, 8 (10%) died from treatment-related causes, compared with none in the control arm of patients who received conventional treatment with cyclophosphamide. But during follow-up, a total of 16 of the 79 (20%) stem-cell transplant patients died, compared with 24 of the 77 (31%) control patients, results that showed an overall mortality benefit from stem-cell transplantation.
The transplanted patients also showed "large improvements" in their skin score and "substantial improvements" in their forced vital capacity and in their activity as measured by the Health Assessment Questionnaire Disability Index (HAQ-DI), noted Dr. Khanna, director of the scleroderma program at the University of Michigan in Ann Arbor.
"Before and after treatment, patients look completely different. Some patients [treated with stem-cell transplants] you can’t tell that they had scleroderma. But you need to select the right patients," he cautioned. "There is high mortality early, but if you select patients correctly the benefits will outweigh the risks. This is what we now offer to patients" who don’t respond to conventional treatments and are suitable for transplantation, Dr. Khanna said.
Researchers had previously reported these ASTIS results during the EULAR 2012 meeting.
Stem-cell treatment is appropriate for "the 10%-15% of patients with scleroderma who are the worst," commented Dr. Daniel Furst, a professor in rheumatology at the University of California, Los Angeles. "The next step is to move this treatment from the worst patients to those who are less severe. In some centers in Europe, stem-cell transplantation is becoming widely used, even for patients with only skin symptoms," Dr. Furst said in an interview.
A major attraction of stem-cell transplantation is that over time it produces regression of fibrosis and collagen deposits and healing of prior organ damage. But the treatment also carries the risk of causing substantial immunosuppression while the immune system repopulates, leaving patients vulnerable to infections and other complications.
The upcoming publication of the ASTIS findings should further cement the role of stem-cell transplantation in scleroderma management, but "I’m more of a skeptic. I would like to see it reproduced" in a second trial, Dr. Furst said. Specifically, he means the North American–based Scleroderma: Cyclophosphamide or Transplantation (SCOT) trial now in progress. A positive result in SCOT is still needed to convince many insurers to cover the cost of stem-cell transplantation, Dr. Furst said. For example, when enrolling patients into SCOT, insurers were willing to reimburse the treatment of about a quarter of the patients who otherwise qualified for enrollment, he said.
Dr. Khanna has received research grants from 14 different drug companies. Dr. Furst has been a consultant to or speaker for 11 different drug companies, and has received research grants from 10 different companies.
mzoler@frontlinemedcom.com On Twitter @mitchelzoler
PARIS – Autologous stem-cell transplantation has emerged as an effective and feasible treatment for selected patients with severe systemic sclerosis who inadequately respond to conventional treatments, said two U.S. experts.
In addition, convincing evidence documenting the overall beneficial effect of autologous stem-cell transplantation will soon appear in a published report from the Autologous Stem Cell Transplantation International Scleroderma (ASTIS) trial, Dr. Dinesh Khanna said in a talk at the annual European Congress of Rheumatology.
In his talk, Dr. Khanna gave a short preview of the ASTIS results that he said would appear in a medical journal in the next few weeks. Those results show that among 79 scleroderma patients randomized to treatment with autologous stem-cell transplant, 8 (10%) died from treatment-related causes, compared with none in the control arm of patients who received conventional treatment with cyclophosphamide. But during follow-up, a total of 16 of the 79 (20%) stem-cell transplant patients died, compared with 24 of the 77 (31%) control patients, results that showed an overall mortality benefit from stem-cell transplantation.
The transplanted patients also showed "large improvements" in their skin score and "substantial improvements" in their forced vital capacity and in their activity as measured by the Health Assessment Questionnaire Disability Index (HAQ-DI), noted Dr. Khanna, director of the scleroderma program at the University of Michigan in Ann Arbor.
"Before and after treatment, patients look completely different. Some patients [treated with stem-cell transplants] you can’t tell that they had scleroderma. But you need to select the right patients," he cautioned. "There is high mortality early, but if you select patients correctly the benefits will outweigh the risks. This is what we now offer to patients" who don’t respond to conventional treatments and are suitable for transplantation, Dr. Khanna said.
Researchers had previously reported these ASTIS results during the EULAR 2012 meeting.
Stem-cell treatment is appropriate for "the 10%-15% of patients with scleroderma who are the worst," commented Dr. Daniel Furst, a professor in rheumatology at the University of California, Los Angeles. "The next step is to move this treatment from the worst patients to those who are less severe. In some centers in Europe, stem-cell transplantation is becoming widely used, even for patients with only skin symptoms," Dr. Furst said in an interview.
A major attraction of stem-cell transplantation is that over time it produces regression of fibrosis and collagen deposits and healing of prior organ damage. But the treatment also carries the risk of causing substantial immunosuppression while the immune system repopulates, leaving patients vulnerable to infections and other complications.
The upcoming publication of the ASTIS findings should further cement the role of stem-cell transplantation in scleroderma management, but "I’m more of a skeptic. I would like to see it reproduced" in a second trial, Dr. Furst said. Specifically, he means the North American–based Scleroderma: Cyclophosphamide or Transplantation (SCOT) trial now in progress. A positive result in SCOT is still needed to convince many insurers to cover the cost of stem-cell transplantation, Dr. Furst said. For example, when enrolling patients into SCOT, insurers were willing to reimburse the treatment of about a quarter of the patients who otherwise qualified for enrollment, he said.
Dr. Khanna has received research grants from 14 different drug companies. Dr. Furst has been a consultant to or speaker for 11 different drug companies, and has received research grants from 10 different companies.
mzoler@frontlinemedcom.com On Twitter @mitchelzoler
PARIS – Autologous stem-cell transplantation has emerged as an effective and feasible treatment for selected patients with severe systemic sclerosis who inadequately respond to conventional treatments, said two U.S. experts.
In addition, convincing evidence documenting the overall beneficial effect of autologous stem-cell transplantation will soon appear in a published report from the Autologous Stem Cell Transplantation International Scleroderma (ASTIS) trial, Dr. Dinesh Khanna said in a talk at the annual European Congress of Rheumatology.
In his talk, Dr. Khanna gave a short preview of the ASTIS results that he said would appear in a medical journal in the next few weeks. Those results show that among 79 scleroderma patients randomized to treatment with autologous stem-cell transplant, 8 (10%) died from treatment-related causes, compared with none in the control arm of patients who received conventional treatment with cyclophosphamide. But during follow-up, a total of 16 of the 79 (20%) stem-cell transplant patients died, compared with 24 of the 77 (31%) control patients, results that showed an overall mortality benefit from stem-cell transplantation.
The transplanted patients also showed "large improvements" in their skin score and "substantial improvements" in their forced vital capacity and in their activity as measured by the Health Assessment Questionnaire Disability Index (HAQ-DI), noted Dr. Khanna, director of the scleroderma program at the University of Michigan in Ann Arbor.
"Before and after treatment, patients look completely different. Some patients [treated with stem-cell transplants] you can’t tell that they had scleroderma. But you need to select the right patients," he cautioned. "There is high mortality early, but if you select patients correctly the benefits will outweigh the risks. This is what we now offer to patients" who don’t respond to conventional treatments and are suitable for transplantation, Dr. Khanna said.
Researchers had previously reported these ASTIS results during the EULAR 2012 meeting.
Stem-cell treatment is appropriate for "the 10%-15% of patients with scleroderma who are the worst," commented Dr. Daniel Furst, a professor in rheumatology at the University of California, Los Angeles. "The next step is to move this treatment from the worst patients to those who are less severe. In some centers in Europe, stem-cell transplantation is becoming widely used, even for patients with only skin symptoms," Dr. Furst said in an interview.
A major attraction of stem-cell transplantation is that over time it produces regression of fibrosis and collagen deposits and healing of prior organ damage. But the treatment also carries the risk of causing substantial immunosuppression while the immune system repopulates, leaving patients vulnerable to infections and other complications.
The upcoming publication of the ASTIS findings should further cement the role of stem-cell transplantation in scleroderma management, but "I’m more of a skeptic. I would like to see it reproduced" in a second trial, Dr. Furst said. Specifically, he means the North American–based Scleroderma: Cyclophosphamide or Transplantation (SCOT) trial now in progress. A positive result in SCOT is still needed to convince many insurers to cover the cost of stem-cell transplantation, Dr. Furst said. For example, when enrolling patients into SCOT, insurers were willing to reimburse the treatment of about a quarter of the patients who otherwise qualified for enrollment, he said.
Dr. Khanna has received research grants from 14 different drug companies. Dr. Furst has been a consultant to or speaker for 11 different drug companies, and has received research grants from 10 different companies.
mzoler@frontlinemedcom.com On Twitter @mitchelzoler
EXPERT ANALYSIS FROM THE EULAR CONGRESS 2014
Key clinical point: Use of autologous stem-cell transplants is spreading for intractable, severe scleroderma as the evidence base expands.
Major finding: Patients who were randomized to autologous stem-cell transplant showed a survival benefit, compared with control patients who received conventional treatment with cyclophosphamide. Transplanted patients also showed "large improvements" in their skin score and "substantial improvements" in forced vital capacity and HAQ-DI.
Data source: Results from 79 patients in the ASTIS trial who were randomized to stem-cell transplant or to cyclophosphamide.
Disclosures: Dr. Khanna has received research grants from 14 different drug companies. Dr. Furst has been a consultant to or speaker for 11 different drug companies, and has received research grants from 10 different companies.

FVC inadequate when assessing scleroderma lung disease
PARIS – The traditional way to assess the status of interstitial lung disease in patients with systemic sclerosis, forced vital capacity, may not be the best way, based on a new analysis of 83 patients enrolled in a scleroderma-treatment trial.
"A structural, physiologic, and patient-oriented composite outcome may be a more comprehensive measure of treatment response" for patients with systemic sclerosis (SSc) and interstitial lung disease, Dr. Elizabeth Volkmann said at the annual European Congress of Rheumatology. "The most robust" association seen in her analysis did not include forced vital capacity (FVC) but instead focused on the Transition Dyspnea Index (TDI), the scleroderma modified Health Assessment Questionnaire Disability Index (HAQ-DI), and quantitative, serial assessment of high-resolution CT (HRCT) images of the patient’s lungs, said Dr. Volkmann, a rheumatologist at the University of California, Los Angeles.

Although her main goal in this analysis was to identify the best assessment of lung disease in SSc patients enrolled in clinical trials, the findings also have implications for managing patients with SSc, also known as scleroderma, in routine practice, Dr. Volkmann said in an interview.
Many physicians "rely solely on FVC for following patients, and I think this may not be the best measure. Now that we have great imaging options we should use them. And the strongest correlates [in the new analysis] were with the HAQ-DI, a measure of what patients can do, and the TDI, in which patients say how much their disease has progressed. They are both patient oriented and tell you how the patient is doing," Dr. Volkmann said.
"The three were more robust and comprehensive than FVC," which can be influenced by many factors and has a variability of 10%. Dr. Volkmann conceded that the quantitative assessment of annual HRCT scans done in the study is not widely available, but she said that visual assessment of HRCT scans highly correlates with quantitative assessment and hence likely makes a reasonable substitute.
A senior collaborator on the study, Dr. Daniel Furst, said that in his opinion it was premature to completely abandon FVC for assessing SSc patients, but it was clearly useful to add the two patient-oriented questionnaires and HRCT imaging.
"We haven’t discarded FVC, but we’ve added the other things," he said in an interview. "Five years ago we only did FVC, 3 years ago we added the scleroderma HAQ-DI," and now he and his associates also use annual HRCT imaging as well as the TDI. The two questionnaires are administered every 3-6 months, said Dr. Furst, professor of rheumatology at UCLA.
"Using all four of these tools is not being widely done" right now in U.S. rheumatology practice. "I really think it’s a step forward." However, Dr. Furst also cautioned that for adoption into routine practice he would like to see evidence documenting that this approach has a positive impact on patient outcomes.

Dr. Volkmann’s analysis involved the 158 U.S. patients enrolled in the first Scleroderma Lung Study, run in 2000-2004 at 13 U.S. centers to compare treatment with oral cyclophosphamide against placebo in patients with active SSC and interstitial lung disease (N. Engl. J. Med. 2006;354:2655-66). Of the 158 patients enrolled, 125 had an HRCT scan at baseline, and among those, 83 also had a HRCT scan after 12 months. These 83 patients formed the basis for Dr. Volkmann’s analysis, including 41 patients randomized to cyclophosphamide treatment and 42 randomized to placebo.
Multivariate analysis identified the HAQ-DI, TDI, FVC, and a quantitative lung fibrosis score derived from analysis of the serial HRCT images as collectively predicting best the outcomes of these patients. A second model that eliminated FVC was "slightly stronger," and both of these combined assessments were each "more robust than FVC alone," Dr. Volkmann said.
Dr. Volkmann said that she had no disclosures. Dr. Furst said that he had no relevant disclosures.
On Twitter @mitchelzoler
PARIS – The traditional way to assess the status of interstitial lung disease in patients with systemic sclerosis, forced vital capacity, may not be the best way, based on a new analysis of 83 patients enrolled in a scleroderma-treatment trial.
"A structural, physiologic, and patient-oriented composite outcome may be a more comprehensive measure of treatment response" for patients with systemic sclerosis (SSc) and interstitial lung disease, Dr. Elizabeth Volkmann said at the annual European Congress of Rheumatology. "The most robust" association seen in her analysis did not include forced vital capacity (FVC) but instead focused on the Transition Dyspnea Index (TDI), the scleroderma modified Health Assessment Questionnaire Disability Index (HAQ-DI), and quantitative, serial assessment of high-resolution CT (HRCT) images of the patient’s lungs, said Dr. Volkmann, a rheumatologist at the University of California, Los Angeles.
Although her main goal in this analysis was to identify the best assessment of lung disease in SSc patients enrolled in clinical trials, the findings also have implications for managing patients with SSc, also known as scleroderma, in routine practice, Dr. Volkmann said in an interview.
Many physicians "rely solely on FVC for following patients, and I think this may not be the best measure. Now that we have great imaging options we should use them. And the strongest correlates [in the new analysis] were with the HAQ-DI, a measure of what patients can do, and the TDI, in which patients say how much their disease has progressed. They are both patient oriented and tell you how the patient is doing," Dr. Volkmann said.
"The three were more robust and comprehensive than FVC," which can be influenced by many factors and has a variability of 10%. Dr. Volkmann conceded that the quantitative assessment of annual HRCT scans done in the study is not widely available, but she said that visual assessment of HRCT scans highly correlates with quantitative assessment and hence likely makes a reasonable substitute.
A senior collaborator on the study, Dr. Daniel Furst, said that in his opinion it was premature to completely abandon FVC for assessing SSc patients, but it was clearly useful to add the two patient-oriented questionnaires and HRCT imaging.
"We haven’t discarded FVC, but we’ve added the other things," he said in an interview. "Five years ago we only did FVC, 3 years ago we added the scleroderma HAQ-DI," and now he and his associates also use annual HRCT imaging as well as the TDI. The two questionnaires are administered every 3-6 months, said Dr. Furst, professor of rheumatology at UCLA.
"Using all four of these tools is not being widely done" right now in U.S. rheumatology practice. "I really think it’s a step forward." However, Dr. Furst also cautioned that for adoption into routine practice he would like to see evidence documenting that this approach has a positive impact on patient outcomes.
Dr. Volkmann’s analysis involved the 158 U.S. patients enrolled in the first Scleroderma Lung Study, run in 2000-2004 at 13 U.S. centers to compare treatment with oral cyclophosphamide against placebo in patients with active SSC and interstitial lung disease (N. Engl. J. Med. 2006;354:2655-66). Of the 158 patients enrolled, 125 had an HRCT scan at baseline, and among those, 83 also had a HRCT scan after 12 months. These 83 patients formed the basis for Dr. Volkmann’s analysis, including 41 patients randomized to cyclophosphamide treatment and 42 randomized to placebo.
Multivariate analysis identified the HAQ-DI, TDI, FVC, and a quantitative lung fibrosis score derived from analysis of the serial HRCT images as collectively predicting best the outcomes of these patients. A second model that eliminated FVC was "slightly stronger," and both of these combined assessments were each "more robust than FVC alone," Dr. Volkmann said.
Dr. Volkmann said that she had no disclosures. Dr. Furst said that he had no relevant disclosures.
On Twitter @mitchelzoler
PARIS – The traditional way to assess the status of interstitial lung disease in patients with systemic sclerosis, forced vital capacity, may not be the best way, based on a new analysis of 83 patients enrolled in a scleroderma-treatment trial.
"A structural, physiologic, and patient-oriented composite outcome may be a more comprehensive measure of treatment response" for patients with systemic sclerosis (SSc) and interstitial lung disease, Dr. Elizabeth Volkmann said at the annual European Congress of Rheumatology. "The most robust" association seen in her analysis did not include forced vital capacity (FVC) but instead focused on the Transition Dyspnea Index (TDI), the scleroderma modified Health Assessment Questionnaire Disability Index (HAQ-DI), and quantitative, serial assessment of high-resolution CT (HRCT) images of the patient’s lungs, said Dr. Volkmann, a rheumatologist at the University of California, Los Angeles.
Although her main goal in this analysis was to identify the best assessment of lung disease in SSc patients enrolled in clinical trials, the findings also have implications for managing patients with SSc, also known as scleroderma, in routine practice, Dr. Volkmann said in an interview.
Many physicians "rely solely on FVC for following patients, and I think this may not be the best measure. Now that we have great imaging options we should use them. And the strongest correlates [in the new analysis] were with the HAQ-DI, a measure of what patients can do, and the TDI, in which patients say how much their disease has progressed. They are both patient oriented and tell you how the patient is doing," Dr. Volkmann said.
"The three were more robust and comprehensive than FVC," which can be influenced by many factors and has a variability of 10%. Dr. Volkmann conceded that the quantitative assessment of annual HRCT scans done in the study is not widely available, but she said that visual assessment of HRCT scans highly correlates with quantitative assessment and hence likely makes a reasonable substitute.
A senior collaborator on the study, Dr. Daniel Furst, said that in his opinion it was premature to completely abandon FVC for assessing SSc patients, but it was clearly useful to add the two patient-oriented questionnaires and HRCT imaging.
"We haven’t discarded FVC, but we’ve added the other things," he said in an interview. "Five years ago we only did FVC, 3 years ago we added the scleroderma HAQ-DI," and now he and his associates also use annual HRCT imaging as well as the TDI. The two questionnaires are administered every 3-6 months, said Dr. Furst, professor of rheumatology at UCLA.
"Using all four of these tools is not being widely done" right now in U.S. rheumatology practice. "I really think it’s a step forward." However, Dr. Furst also cautioned that for adoption into routine practice he would like to see evidence documenting that this approach has a positive impact on patient outcomes.
Dr. Volkmann’s analysis involved the 158 U.S. patients enrolled in the first Scleroderma Lung Study, run in 2000-2004 at 13 U.S. centers to compare treatment with oral cyclophosphamide against placebo in patients with active SSC and interstitial lung disease (N. Engl. J. Med. 2006;354:2655-66). Of the 158 patients enrolled, 125 had an HRCT scan at baseline, and among those, 83 also had a HRCT scan after 12 months. These 83 patients formed the basis for Dr. Volkmann’s analysis, including 41 patients randomized to cyclophosphamide treatment and 42 randomized to placebo.
Multivariate analysis identified the HAQ-DI, TDI, FVC, and a quantitative lung fibrosis score derived from analysis of the serial HRCT images as collectively predicting best the outcomes of these patients. A second model that eliminated FVC was "slightly stronger," and both of these combined assessments were each "more robust than FVC alone," Dr. Volkmann said.
Dr. Volkmann said that she had no disclosures. Dr. Furst said that he had no relevant disclosures.
On Twitter @mitchelzoler
AT THE EULAR CONGRESS 2014
Key clinical point: Patient-oriented questionnaires and lung imaging are key tools to assess systemic sclerosis patients with interstitial lung disease.
Major finding: Combined assessment by patient-oriented questionnaires, CT imaging, and forced vital capacity better predicted patient outcomes than did FVC alone.
Data source: Retrospective analysis of data collected from 83 systemic sclerosis patients enrolled at any of 13 U.S. centers.
Disclosures: Dr. Volkmann said that she had no disclosures. Dr. Furst said that he had no relevant disclosures.

U.S. cell-based flu vaccine plant gets FDA approval
The Food and Drug Administration on June 17 approved the first U.S. facility to make seasonal influenza vaccine using cultured-cell–based technology, giving another layer of approval to the vaccine-making capabilities of a North Carolina plant that opened in January 2012.
The plant, in Holly Springs, N.C., resulted from a partnership of the Novartis Corp., which owns it, and the federal Biomedical Advanced Research and Development Authority, a branch of the Department of Health and Human Services.

The HHS goal was to boost capabilities for U.S.-based production of influenza vaccine, specifically cell-based vaccine production using cultured canine kidney cells. The facility, which would produce as much as 200 million doses of influenza vaccine for a pandemic, would receive FDA approval for a pandemic vaccine on a case by case basis as its production began to scale up.
The FDA’s action in June licensed the seasonal influenza vaccine made by the cell-culture process. The facility can produce at least 50 million doses of seasonal influenza vaccine each season, according to the announcement.
The Holly Springs cell-culture facility was seen by experts as an important way to broaden the scope of U.S. influenza vaccine production while also hastening the availability of significant amounts of vaccine for a pandemic, cutting the lag to vaccine appearance by perhaps as much as 1 month. But one expert criticized the facility as being inevitably archaic by producing a conventional, hemagglutinin-based vaccine that is "inadequate for providing the kind of immunologic protection we need," Michael T. Osterholm, Ph.D., said when the facility first opened.
"Just because you can make more of this [vaccine] faster, what does that mean? It’s better than nothing, absolutely, but that doesn’t mean it will be the answer for a very serious pandemic," said Dr. Osterholm, director of the center for infectious disease research and policy at the University of Minnesota in Minneapolis.
On Twitter @mitchelzoler
The Food and Drug Administration on June 17 approved the first U.S. facility to make seasonal influenza vaccine using cultured-cell–based technology, giving another layer of approval to the vaccine-making capabilities of a North Carolina plant that opened in January 2012.
The plant, in Holly Springs, N.C., resulted from a partnership of the Novartis Corp., which owns it, and the federal Biomedical Advanced Research and Development Authority, a branch of the Department of Health and Human Services.
The HHS goal was to boost capabilities for U.S.-based production of influenza vaccine, specifically cell-based vaccine production using cultured canine kidney cells. The facility, which would produce as much as 200 million doses of influenza vaccine for a pandemic, would receive FDA approval for a pandemic vaccine on a case by case basis as its production began to scale up.
The FDA’s action in June licensed the seasonal influenza vaccine made by the cell-culture process. The facility can produce at least 50 million doses of seasonal influenza vaccine each season, according to the announcement.
The Holly Springs cell-culture facility was seen by experts as an important way to broaden the scope of U.S. influenza vaccine production while also hastening the availability of significant amounts of vaccine for a pandemic, cutting the lag to vaccine appearance by perhaps as much as 1 month. But one expert criticized the facility as being inevitably archaic by producing a conventional, hemagglutinin-based vaccine that is "inadequate for providing the kind of immunologic protection we need," Michael T. Osterholm, Ph.D., said when the facility first opened.
"Just because you can make more of this [vaccine] faster, what does that mean? It’s better than nothing, absolutely, but that doesn’t mean it will be the answer for a very serious pandemic," said Dr. Osterholm, director of the center for infectious disease research and policy at the University of Minnesota in Minneapolis.
On Twitter @mitchelzoler
The Food and Drug Administration on June 17 approved the first U.S. facility to make seasonal influenza vaccine using cultured-cell–based technology, giving another layer of approval to the vaccine-making capabilities of a North Carolina plant that opened in January 2012.
The plant, in Holly Springs, N.C., resulted from a partnership of the Novartis Corp., which owns it, and the federal Biomedical Advanced Research and Development Authority, a branch of the Department of Health and Human Services.
The HHS goal was to boost capabilities for U.S.-based production of influenza vaccine, specifically cell-based vaccine production using cultured canine kidney cells. The facility, which would produce as much as 200 million doses of influenza vaccine for a pandemic, would receive FDA approval for a pandemic vaccine on a case by case basis as its production began to scale up.
The FDA’s action in June licensed the seasonal influenza vaccine made by the cell-culture process. The facility can produce at least 50 million doses of seasonal influenza vaccine each season, according to the announcement.
The Holly Springs cell-culture facility was seen by experts as an important way to broaden the scope of U.S. influenza vaccine production while also hastening the availability of significant amounts of vaccine for a pandemic, cutting the lag to vaccine appearance by perhaps as much as 1 month. But one expert criticized the facility as being inevitably archaic by producing a conventional, hemagglutinin-based vaccine that is "inadequate for providing the kind of immunologic protection we need," Michael T. Osterholm, Ph.D., said when the facility first opened.
"Just because you can make more of this [vaccine] faster, what does that mean? It’s better than nothing, absolutely, but that doesn’t mean it will be the answer for a very serious pandemic," said Dr. Osterholm, director of the center for infectious disease research and policy at the University of Minnesota in Minneapolis.
On Twitter @mitchelzoler

Gut inflammation linked to worsening spondyloarthritis
Spondyloarthritis patients with microscopic gut inflammation had to start anti–tumor necrosis factor treatment sooner in the course of their disease than patients without gut inflammation in a prospective, observational study of 63 Belgian patients with the axial form of spondyloarthritis, the peripheral form, or both.
The finding "is in line with the hypothesis that in SpA [spondyloarthritis] gut inflammation may drive disease," Dr. Heleen Cypers said at the annual European Congress of Rheumatology.

SpA patients with microscopic gut inflammation initiated anti–tumor necrosis factor (TNF) therapy roughly twice as quickly as those without gut inflammation at each time period examined – at 6, 12, and 18 months after their initial diagnosis. After 18 months, 56% of patients with inflammation were on treatment with an anti-TNF agent, compared with almost 29% of the followed patients without microscopic gut inflammation, said Dr. Cypers, a rheumatologist at Ghent (Belgium) University.
The link between microscopic gut inflammation and a faster need to start on biologic treatment occurred at nearly identical rates in both the subset of 11 patients with acute gut inflammation and the subset of 21 patients with chronic inflammation.
Previous research by Dr. Cypers’ group had shown that chronic inflammation in the gut was associated with more extensive bone marrow edema of the sacroiliac joints, a higher risk of progression to ankylosing spondylitis, and a higher risk of developing Crohn’s disease. No information existed on the impact of gut inflammation on therapeutic decision-making in SpA, however.
The current study followed the Ghent Inflammatory Arthritis and Spondylitis Cohort (GIANT) of 63 newly diagnosed patients who underwent an ileocolonoscopy at baseline. Patients were not taking anti-TNF agents and were followed for 18 months. The decision to start anti-TNF treatment was made by a rheumatologist who was unaware of the patient’s gut history.
SpA patients who had microscopic gut inflammation at baseline were more likely to start anti-TNF treatment sooner than were patients without gut inflammation. During follow-up, just under a third of patients (9 of 31) with normal gut histology started anti-TNFs, compared with over half (18 of 32) of the patients with gut inflammation at baseline.
In most cases, anti-TNF drugs were started because first-line therapy had failed, Dr. Cypers reported. Her analysis also showed that initiation of an anti-TNF drug during follow-up was significantly linked with a higher level of C-reactive protein at baseline.
According to Dr. Cypers, the findings support the hypothesis that bowel inflammation in SpA induces a more chronic, persevering disease.
"The findings identify gut inflammation as a marker of a poor prognosis and have relevance for therapeutic decision-making in daily clinical practice. It is conceivable that assessment of gut inflammation will be included in future models for risk stratification of SpA," she said in an interview.
The underlying mechanisms explaining the link are still under investigation, but several studies have suggested that inflammation in the gut may be a triggering factor for SpA development. Interactions in the intestinal immune system also may play an important role, she said.
Future research by the group will focus on ruling out the interdependency of all of these factors, predicting response to biologic therapy based on gut histology, and determining whether therapies aiming at treating microscopic gut inflammation can affect the evolution of the disease.
Dr. Cypers said that she and her associates had no disclosures.
Spondyloarthritis patients with microscopic gut inflammation had to start anti–tumor necrosis factor treatment sooner in the course of their disease than patients without gut inflammation in a prospective, observational study of 63 Belgian patients with the axial form of spondyloarthritis, the peripheral form, or both.
The finding "is in line with the hypothesis that in SpA [spondyloarthritis] gut inflammation may drive disease," Dr. Heleen Cypers said at the annual European Congress of Rheumatology.
SpA patients with microscopic gut inflammation initiated anti–tumor necrosis factor (TNF) therapy roughly twice as quickly as those without gut inflammation at each time period examined – at 6, 12, and 18 months after their initial diagnosis. After 18 months, 56% of patients with inflammation were on treatment with an anti-TNF agent, compared with almost 29% of the followed patients without microscopic gut inflammation, said Dr. Cypers, a rheumatologist at Ghent (Belgium) University.
The link between microscopic gut inflammation and a faster need to start on biologic treatment occurred at nearly identical rates in both the subset of 11 patients with acute gut inflammation and the subset of 21 patients with chronic inflammation.
Previous research by Dr. Cypers’ group had shown that chronic inflammation in the gut was associated with more extensive bone marrow edema of the sacroiliac joints, a higher risk of progression to ankylosing spondylitis, and a higher risk of developing Crohn’s disease. No information existed on the impact of gut inflammation on therapeutic decision-making in SpA, however.
The current study followed the Ghent Inflammatory Arthritis and Spondylitis Cohort (GIANT) of 63 newly diagnosed patients who underwent an ileocolonoscopy at baseline. Patients were not taking anti-TNF agents and were followed for 18 months. The decision to start anti-TNF treatment was made by a rheumatologist who was unaware of the patient’s gut history.
SpA patients who had microscopic gut inflammation at baseline were more likely to start anti-TNF treatment sooner than were patients without gut inflammation. During follow-up, just under a third of patients (9 of 31) with normal gut histology started anti-TNFs, compared with over half (18 of 32) of the patients with gut inflammation at baseline.
In most cases, anti-TNF drugs were started because first-line therapy had failed, Dr. Cypers reported. Her analysis also showed that initiation of an anti-TNF drug during follow-up was significantly linked with a higher level of C-reactive protein at baseline.
According to Dr. Cypers, the findings support the hypothesis that bowel inflammation in SpA induces a more chronic, persevering disease.
"The findings identify gut inflammation as a marker of a poor prognosis and have relevance for therapeutic decision-making in daily clinical practice. It is conceivable that assessment of gut inflammation will be included in future models for risk stratification of SpA," she said in an interview.
The underlying mechanisms explaining the link are still under investigation, but several studies have suggested that inflammation in the gut may be a triggering factor for SpA development. Interactions in the intestinal immune system also may play an important role, she said.
Future research by the group will focus on ruling out the interdependency of all of these factors, predicting response to biologic therapy based on gut histology, and determining whether therapies aiming at treating microscopic gut inflammation can affect the evolution of the disease.
Dr. Cypers said that she and her associates had no disclosures.
Spondyloarthritis patients with microscopic gut inflammation had to start anti–tumor necrosis factor treatment sooner in the course of their disease than patients without gut inflammation in a prospective, observational study of 63 Belgian patients with the axial form of spondyloarthritis, the peripheral form, or both.
The finding "is in line with the hypothesis that in SpA [spondyloarthritis] gut inflammation may drive disease," Dr. Heleen Cypers said at the annual European Congress of Rheumatology.
SpA patients with microscopic gut inflammation initiated anti–tumor necrosis factor (TNF) therapy roughly twice as quickly as those without gut inflammation at each time period examined – at 6, 12, and 18 months after their initial diagnosis. After 18 months, 56% of patients with inflammation were on treatment with an anti-TNF agent, compared with almost 29% of the followed patients without microscopic gut inflammation, said Dr. Cypers, a rheumatologist at Ghent (Belgium) University.
The link between microscopic gut inflammation and a faster need to start on biologic treatment occurred at nearly identical rates in both the subset of 11 patients with acute gut inflammation and the subset of 21 patients with chronic inflammation.
Previous research by Dr. Cypers’ group had shown that chronic inflammation in the gut was associated with more extensive bone marrow edema of the sacroiliac joints, a higher risk of progression to ankylosing spondylitis, and a higher risk of developing Crohn’s disease. No information existed on the impact of gut inflammation on therapeutic decision-making in SpA, however.
The current study followed the Ghent Inflammatory Arthritis and Spondylitis Cohort (GIANT) of 63 newly diagnosed patients who underwent an ileocolonoscopy at baseline. Patients were not taking anti-TNF agents and were followed for 18 months. The decision to start anti-TNF treatment was made by a rheumatologist who was unaware of the patient’s gut history.
SpA patients who had microscopic gut inflammation at baseline were more likely to start anti-TNF treatment sooner than were patients without gut inflammation. During follow-up, just under a third of patients (9 of 31) with normal gut histology started anti-TNFs, compared with over half (18 of 32) of the patients with gut inflammation at baseline.
In most cases, anti-TNF drugs were started because first-line therapy had failed, Dr. Cypers reported. Her analysis also showed that initiation of an anti-TNF drug during follow-up was significantly linked with a higher level of C-reactive protein at baseline.
According to Dr. Cypers, the findings support the hypothesis that bowel inflammation in SpA induces a more chronic, persevering disease.
"The findings identify gut inflammation as a marker of a poor prognosis and have relevance for therapeutic decision-making in daily clinical practice. It is conceivable that assessment of gut inflammation will be included in future models for risk stratification of SpA," she said in an interview.
The underlying mechanisms explaining the link are still under investigation, but several studies have suggested that inflammation in the gut may be a triggering factor for SpA development. Interactions in the intestinal immune system also may play an important role, she said.
Future research by the group will focus on ruling out the interdependency of all of these factors, predicting response to biologic therapy based on gut histology, and determining whether therapies aiming at treating microscopic gut inflammation can affect the evolution of the disease.
Dr. Cypers said that she and her associates had no disclosures.
FROM THE EULAR CONGRESS 2014
Major finding: Among patients with newly diagnosed spondyloarthritis, microscopic gut inflammation at baseline was linked to a two-fold higher rate of anti-TNF treatment initiation.
Data source: A prospective, observational study of 63 Belgian patients with newly diagnosed spondyloarthritis who were followed for 18 months.
Disclosures: Dr. Cypers said that she and her associates had no disclosures.

Sarilumab shown safe, effective for RA in phase III trial
PARIS – Rheumatoid arthritis patients who received the interleukin-6–blocking drug sarilumab plus methotrexate had significantly better short-term and 1-year outcomes than did patients on methotrexate alone in a phase III trial with 1,197 patients.
Sarilumab treatment also was relatively safe, with no new safety concerns arising from the study, Dr. Mark Genovese said at the annual European Congress of Rheumatology.

"Both sarilumab-dose groups showed a statistically significant improvement, compared to the placebo group, thereby demonstrating that, in this study, sarilumab plus methotrexate improved the signs and symptoms of rheumatoid arthritis [RA], improved physical function, and inhibited progression of joint damage in adult patients with active RA and inadequate response to methotrexate. Importantly, in this first phase III study with this agent, we did not see any unique safety issues," Dr. Genovese said in an interview.
The SARIL-RA-MOBILITY trial is the first study to finish from a panel of four phase III studies of sarilumab in patients with RA. The three other phase III trials currently in progress have enrolled a total of about 1,400 patients. A submission to the Food and Drug Administration for marketing approval of sarilumab for patients with RA will await completion of these three additional studies, said Dr. Genovese, professor of medicine and cochief of the division of immunology and rheumatology at Stanford (Calif.) University.
The trial enrolled patients with RA and an inadequate response to methotrexate alone at more than 200 centers in more than 30 countries, including the United States. The study randomized patients to treatment with methotrexate plus 150 mg sarilumab administered as a subcutaneous injection every 2 weeks, 200 mg injected every 2 weeks, or placebo.
The study had three primary efficacy endpoints:
• The percentage of patients achieving an American College of Rheumatology 20 response after 24 weeks of treatment, which was achieved by 58% of 400 patients who received 150 mg sarilumab, 66% of 399 patients who received the 200-mg dose, and 33% of the 398 patients who received placebo injections.
• The average change from baseline in the Health Assessment Questionnaire disability index after 16 weeks of treatment, which fell by 0.53 in patients who received the 150-mg dose of sarilumab, by 0.55 in those who received 200-mg sarilumab injections, and by 0.29 in those who received placebo.
• The average change from baseline in the van der Heijde–modified total Sharp score after 52 weeks of treatment, which increased by 0.90 among patients on the lower sarilumab dose, increased by 0.25 in patients on the higher dose, and increased by 2.78 among patients who received placebo.
For all three endpoints, the differences between each of the sarilumab groups and the placebo controls were statistically significant as well as clinically meaningful. "We are encouraged by the potential of sarilumab to offer a new alternative in the treatment armamentarium for patients suffering from RA," Dr. Genovese said. When the two companies developing sarilumab submit their approval application, they will likely initially seek indications for treating adult patients with moderately to severely active RA who had an inadequate response to disease-modifying antirheumatic drugs (DMARDs) or tumor necrosis factor–alpha inhibitors, he predicted.
Serious adverse events during 52 weeks of follow-up occurred in 23 placebo patients, 38 patients on the lower sarilumab dose, and 46 on the higher dose. The most common serious adverse event was infection, which occurred in 10 patients on placebo, 11 patients on low-dose sarilumab, and 17 on the higher dose. No serious infections occurred in patients whose neutrophil level fell below 1,000/mcL.
"The number of serious adverse events thus far remains low, and it will be important to see the integrated safety data across the phase III program before making any judgments regarding the ideal dose and the benefit-to-safety risk ratio of any single dose," Dr. Genovese said.
"Among patients treated with sarilumab, a dose-dependent decrease in mean neutrophil counts was observed. Serious infections were not associated with grades 3 and 4 neutropenia in this study. Increases in mean LDL cholesterol level and transaminases were observed. These safety findings were consistent with those observed in prior investigational studies with sarilumab. The choice of doses will ultimately depend on the integrated safety data across the program and the performance of each dose across a broad range of patient populations," he said.
Sarilumab is the first agent tested from the class of interleukin (IL)-6 blockers. "IL-6 is a key proinflammatory cytokine that impacts inflammation, bone, and metabolism," Dr. Genovese noted.
"Blockade of IL-6 should provide significant improvements in all these areas. This study has demonstrated that inhibition of IL-6 can reduce the signs and symptoms of RA and help to stop the destruction of joints in patients with RA. Additional phase III studies of sarilumab are underway and should provide additional evidence that anti–IL-6 receptor therapy with sarilumab may be a future option for RA patients. It is possibly too early to say if there are any special advantages or opportunities from the strategy of IL-6 blockage, compared with any other class of biologic DMARD," he said.
The trial was sponsored by Sanofi and Regeneron, the companies developing the drug. Dr. Genovese said that he has been a consultant to and received research support from Sanofi. Several coauthors are employees of Sanofi or Regeneron.
On Twitter @mitchelzoler
PARIS – Rheumatoid arthritis patients who received the interleukin-6–blocking drug sarilumab plus methotrexate had significantly better short-term and 1-year outcomes than did patients on methotrexate alone in a phase III trial with 1,197 patients.
Sarilumab treatment also was relatively safe, with no new safety concerns arising from the study, Dr. Mark Genovese said at the annual European Congress of Rheumatology.
"Both sarilumab-dose groups showed a statistically significant improvement, compared to the placebo group, thereby demonstrating that, in this study, sarilumab plus methotrexate improved the signs and symptoms of rheumatoid arthritis [RA], improved physical function, and inhibited progression of joint damage in adult patients with active RA and inadequate response to methotrexate. Importantly, in this first phase III study with this agent, we did not see any unique safety issues," Dr. Genovese said in an interview.
The SARIL-RA-MOBILITY trial is the first study to finish from a panel of four phase III studies of sarilumab in patients with RA. The three other phase III trials currently in progress have enrolled a total of about 1,400 patients. A submission to the Food and Drug Administration for marketing approval of sarilumab for patients with RA will await completion of these three additional studies, said Dr. Genovese, professor of medicine and cochief of the division of immunology and rheumatology at Stanford (Calif.) University.
The trial enrolled patients with RA and an inadequate response to methotrexate alone at more than 200 centers in more than 30 countries, including the United States. The study randomized patients to treatment with methotrexate plus 150 mg sarilumab administered as a subcutaneous injection every 2 weeks, 200 mg injected every 2 weeks, or placebo.
The study had three primary efficacy endpoints:
• The percentage of patients achieving an American College of Rheumatology 20 response after 24 weeks of treatment, which was achieved by 58% of 400 patients who received 150 mg sarilumab, 66% of 399 patients who received the 200-mg dose, and 33% of the 398 patients who received placebo injections.
• The average change from baseline in the Health Assessment Questionnaire disability index after 16 weeks of treatment, which fell by 0.53 in patients who received the 150-mg dose of sarilumab, by 0.55 in those who received 200-mg sarilumab injections, and by 0.29 in those who received placebo.
• The average change from baseline in the van der Heijde–modified total Sharp score after 52 weeks of treatment, which increased by 0.90 among patients on the lower sarilumab dose, increased by 0.25 in patients on the higher dose, and increased by 2.78 among patients who received placebo.
For all three endpoints, the differences between each of the sarilumab groups and the placebo controls were statistically significant as well as clinically meaningful. "We are encouraged by the potential of sarilumab to offer a new alternative in the treatment armamentarium for patients suffering from RA," Dr. Genovese said. When the two companies developing sarilumab submit their approval application, they will likely initially seek indications for treating adult patients with moderately to severely active RA who had an inadequate response to disease-modifying antirheumatic drugs (DMARDs) or tumor necrosis factor–alpha inhibitors, he predicted.
Serious adverse events during 52 weeks of follow-up occurred in 23 placebo patients, 38 patients on the lower sarilumab dose, and 46 on the higher dose. The most common serious adverse event was infection, which occurred in 10 patients on placebo, 11 patients on low-dose sarilumab, and 17 on the higher dose. No serious infections occurred in patients whose neutrophil level fell below 1,000/mcL.
"The number of serious adverse events thus far remains low, and it will be important to see the integrated safety data across the phase III program before making any judgments regarding the ideal dose and the benefit-to-safety risk ratio of any single dose," Dr. Genovese said.
"Among patients treated with sarilumab, a dose-dependent decrease in mean neutrophil counts was observed. Serious infections were not associated with grades 3 and 4 neutropenia in this study. Increases in mean LDL cholesterol level and transaminases were observed. These safety findings were consistent with those observed in prior investigational studies with sarilumab. The choice of doses will ultimately depend on the integrated safety data across the program and the performance of each dose across a broad range of patient populations," he said.
Sarilumab is the first agent tested from the class of interleukin (IL)-6 blockers. "IL-6 is a key proinflammatory cytokine that impacts inflammation, bone, and metabolism," Dr. Genovese noted.
"Blockade of IL-6 should provide significant improvements in all these areas. This study has demonstrated that inhibition of IL-6 can reduce the signs and symptoms of RA and help to stop the destruction of joints in patients with RA. Additional phase III studies of sarilumab are underway and should provide additional evidence that anti–IL-6 receptor therapy with sarilumab may be a future option for RA patients. It is possibly too early to say if there are any special advantages or opportunities from the strategy of IL-6 blockage, compared with any other class of biologic DMARD," he said.
The trial was sponsored by Sanofi and Regeneron, the companies developing the drug. Dr. Genovese said that he has been a consultant to and received research support from Sanofi. Several coauthors are employees of Sanofi or Regeneron.
On Twitter @mitchelzoler
PARIS – Rheumatoid arthritis patients who received the interleukin-6–blocking drug sarilumab plus methotrexate had significantly better short-term and 1-year outcomes than did patients on methotrexate alone in a phase III trial with 1,197 patients.
Sarilumab treatment also was relatively safe, with no new safety concerns arising from the study, Dr. Mark Genovese said at the annual European Congress of Rheumatology.
"Both sarilumab-dose groups showed a statistically significant improvement, compared to the placebo group, thereby demonstrating that, in this study, sarilumab plus methotrexate improved the signs and symptoms of rheumatoid arthritis [RA], improved physical function, and inhibited progression of joint damage in adult patients with active RA and inadequate response to methotrexate. Importantly, in this first phase III study with this agent, we did not see any unique safety issues," Dr. Genovese said in an interview.
The SARIL-RA-MOBILITY trial is the first study to finish from a panel of four phase III studies of sarilumab in patients with RA. The three other phase III trials currently in progress have enrolled a total of about 1,400 patients. A submission to the Food and Drug Administration for marketing approval of sarilumab for patients with RA will await completion of these three additional studies, said Dr. Genovese, professor of medicine and cochief of the division of immunology and rheumatology at Stanford (Calif.) University.
The trial enrolled patients with RA and an inadequate response to methotrexate alone at more than 200 centers in more than 30 countries, including the United States. The study randomized patients to treatment with methotrexate plus 150 mg sarilumab administered as a subcutaneous injection every 2 weeks, 200 mg injected every 2 weeks, or placebo.
The study had three primary efficacy endpoints:
• The percentage of patients achieving an American College of Rheumatology 20 response after 24 weeks of treatment, which was achieved by 58% of 400 patients who received 150 mg sarilumab, 66% of 399 patients who received the 200-mg dose, and 33% of the 398 patients who received placebo injections.
• The average change from baseline in the Health Assessment Questionnaire disability index after 16 weeks of treatment, which fell by 0.53 in patients who received the 150-mg dose of sarilumab, by 0.55 in those who received 200-mg sarilumab injections, and by 0.29 in those who received placebo.
• The average change from baseline in the van der Heijde–modified total Sharp score after 52 weeks of treatment, which increased by 0.90 among patients on the lower sarilumab dose, increased by 0.25 in patients on the higher dose, and increased by 2.78 among patients who received placebo.
For all three endpoints, the differences between each of the sarilumab groups and the placebo controls were statistically significant as well as clinically meaningful. "We are encouraged by the potential of sarilumab to offer a new alternative in the treatment armamentarium for patients suffering from RA," Dr. Genovese said. When the two companies developing sarilumab submit their approval application, they will likely initially seek indications for treating adult patients with moderately to severely active RA who had an inadequate response to disease-modifying antirheumatic drugs (DMARDs) or tumor necrosis factor–alpha inhibitors, he predicted.
Serious adverse events during 52 weeks of follow-up occurred in 23 placebo patients, 38 patients on the lower sarilumab dose, and 46 on the higher dose. The most common serious adverse event was infection, which occurred in 10 patients on placebo, 11 patients on low-dose sarilumab, and 17 on the higher dose. No serious infections occurred in patients whose neutrophil level fell below 1,000/mcL.
"The number of serious adverse events thus far remains low, and it will be important to see the integrated safety data across the phase III program before making any judgments regarding the ideal dose and the benefit-to-safety risk ratio of any single dose," Dr. Genovese said.
"Among patients treated with sarilumab, a dose-dependent decrease in mean neutrophil counts was observed. Serious infections were not associated with grades 3 and 4 neutropenia in this study. Increases in mean LDL cholesterol level and transaminases were observed. These safety findings were consistent with those observed in prior investigational studies with sarilumab. The choice of doses will ultimately depend on the integrated safety data across the program and the performance of each dose across a broad range of patient populations," he said.
Sarilumab is the first agent tested from the class of interleukin (IL)-6 blockers. "IL-6 is a key proinflammatory cytokine that impacts inflammation, bone, and metabolism," Dr. Genovese noted.
"Blockade of IL-6 should provide significant improvements in all these areas. This study has demonstrated that inhibition of IL-6 can reduce the signs and symptoms of RA and help to stop the destruction of joints in patients with RA. Additional phase III studies of sarilumab are underway and should provide additional evidence that anti–IL-6 receptor therapy with sarilumab may be a future option for RA patients. It is possibly too early to say if there are any special advantages or opportunities from the strategy of IL-6 blockage, compared with any other class of biologic DMARD," he said.
The trial was sponsored by Sanofi and Regeneron, the companies developing the drug. Dr. Genovese said that he has been a consultant to and received research support from Sanofi. Several coauthors are employees of Sanofi or Regeneron.
On Twitter @mitchelzoler
AT THE EULAR CONGRESS 2014
Key clinical point: Treatment with the interleukin-6–blocking drug sarilumab showed efficacy and relative safety in the first reported results from a phase III trial.
Major finding: After 24 weeks, 58%-66% of patients on sarilumab had an ACR 20 response, compared with 33% of placebo patients.
Data source: The SARIL-RA-MOBILITY trial, which enrolled 1,197 patients at more than 200 international centers.
Disclosures: SARIL-RA-MOBILITY was sponsored by Sanofi and Regeneron, the companies developing the drug. Dr. Genovese said that he has been a consultant to and received research support from Sanofi. Several coauthors are employees of Sanofi or Regeneron.

Biosimilars match infliximab, etanercept for RA treatment
*PARIS - Two different biosimilar drugs, one a mimic of infliximab, the other modeled on etanercept, closely matched the efficacy and safety of their brand-name counterparts in a pair of separate, randomized, controlled studies.
In one study, treatment with infliximab and a biosimilar agent showed virtually identical efficacy and safety performance at several time points during the first 16 weeks of treatment of 189 patients with rheumatoid arthritis in a head-to-head, randomized comparison done at 23 centers in India.
In the second study, an agent produced to match etanercept showed very similar efficacy and safety to the branded formulation in a total of 233 randomized patients treated for 24 weeks at several centers in Korea.
The second study run in India was the "first clinical trial of a biosimilar infliximab to demonstrate and report kinetics of response to treatment at multiple time points prior to the plateau phase," which starts at about weeks 16, Dr. Jonathan Kay said at the annual European Congress of Rheumatology. It provides "convincing evidence of therapeutic equivalence," and also serves as a "new paradigm for comparative effectiveness testing of biosimilars," said Dr. Kay, a professor of medicine at the University of Massachusetts in Worcester.

Dr. Kay and his colleagues randomized 62 patients with active rheumatoid arthritis (RA) on stable doses of methotrexate to infliximab (Remicade) and 127 patients to BOW015, the biosimilar under study. They measured the proportion of responders at 2, 6, 14, and 16 weeks, with the study’s primary endpoint the percentage of patients showing an American College of Rheumatology 20 (ACR20) response at 16 weeks. The proportion of responders at each of these four time points was virtually identical, Dr. Kay reported. At 16 weeks, the percent of ACR20 responders was 86% in the infliximab arm and 90% in the biosimilar arm. The percent of ACR50 and ACR70 responders was also virtually the same in both treatment arms at 16 weeks, and the percent of ACR20 responders was virtually identical in the two treatment arms at weeks 2, 6, and 14.
The safety profiles of the two drugs also showed no statistically significant differences for any parameter measured except for the incidence of skin disorders, which occurred in one patient treated with the biosimilar and four patients treated with infliximab, a statistically significant difference.
At the same session, Dr. Sang-Cheol Bae of the Hanyang University Hospital for Rheumatic Diseases, Seoul, South Korea, presented findings from a randomized, controlled trial that compared the etanercept biosimilar HD203 with etanercept (Enbrel). Dr. Bae and his colleagues randomized 115 patients with active rheumatoid arthritis to HD203 and 118 patients to etanercept, both in combination with methotrexate, with the primary endpoint set as the proportion of patients achieving ACR20 by week 24. No statistically significant differences were seen between the patient groups for this primary endpoint, and equivalence in efficacy was demonstrated within predefined margins. The percent of ACR20 responders at 24 weeks was 79% in the biosimilar arm and 76% with etanercept.

Secondary analyses showed that the ACR50 responses occurred in 65% of the patients on the biosimilar at 24 weeks and in 53% of patients on etanercept, a statistically significant difference; and after 48 weeks of treatment, the ACR50 rate was 68% in the biosimilar arm and 55% with etanercept, also a statistically significant difference. The two study groups showed no significant difference in the rate of ACR70 responders after 24 or 48 weeks, and also no statistically significant difference in the rate of ACR20 responders after 48 weeks.
The results also showed that the biosimilar was "well tolerated," with a safety profile "comparable" with etanercept, Dr. Bae said.
Dr. Kay has received research funding and/or consulting income from AbbVie, Amgen, Bristol-Myers Squibb, Eli Lilly, Epirus, Genentech, Hospira, Janssen, Pfizer, Roche, and UCB. Dr. Bae acknowledged receiving consulting income and/or research support from Abbott, Bristol-Myers Squibb, Eisai, GlaxoSmithKline, Hanwha Chemical, Merck Serono, MSD, and Pfizer.
* This story was updated 6/13/14.
*PARIS - Two different biosimilar drugs, one a mimic of infliximab, the other modeled on etanercept, closely matched the efficacy and safety of their brand-name counterparts in a pair of separate, randomized, controlled studies.
In one study, treatment with infliximab and a biosimilar agent showed virtually identical efficacy and safety performance at several time points during the first 16 weeks of treatment of 189 patients with rheumatoid arthritis in a head-to-head, randomized comparison done at 23 centers in India.
In the second study, an agent produced to match etanercept showed very similar efficacy and safety to the branded formulation in a total of 233 randomized patients treated for 24 weeks at several centers in Korea.
The second study run in India was the "first clinical trial of a biosimilar infliximab to demonstrate and report kinetics of response to treatment at multiple time points prior to the plateau phase," which starts at about weeks 16, Dr. Jonathan Kay said at the annual European Congress of Rheumatology. It provides "convincing evidence of therapeutic equivalence," and also serves as a "new paradigm for comparative effectiveness testing of biosimilars," said Dr. Kay, a professor of medicine at the University of Massachusetts in Worcester.
Dr. Kay and his colleagues randomized 62 patients with active rheumatoid arthritis (RA) on stable doses of methotrexate to infliximab (Remicade) and 127 patients to BOW015, the biosimilar under study. They measured the proportion of responders at 2, 6, 14, and 16 weeks, with the study’s primary endpoint the percentage of patients showing an American College of Rheumatology 20 (ACR20) response at 16 weeks. The proportion of responders at each of these four time points was virtually identical, Dr. Kay reported. At 16 weeks, the percent of ACR20 responders was 86% in the infliximab arm and 90% in the biosimilar arm. The percent of ACR50 and ACR70 responders was also virtually the same in both treatment arms at 16 weeks, and the percent of ACR20 responders was virtually identical in the two treatment arms at weeks 2, 6, and 14.
The safety profiles of the two drugs also showed no statistically significant differences for any parameter measured except for the incidence of skin disorders, which occurred in one patient treated with the biosimilar and four patients treated with infliximab, a statistically significant difference.
At the same session, Dr. Sang-Cheol Bae of the Hanyang University Hospital for Rheumatic Diseases, Seoul, South Korea, presented findings from a randomized, controlled trial that compared the etanercept biosimilar HD203 with etanercept (Enbrel). Dr. Bae and his colleagues randomized 115 patients with active rheumatoid arthritis to HD203 and 118 patients to etanercept, both in combination with methotrexate, with the primary endpoint set as the proportion of patients achieving ACR20 by week 24. No statistically significant differences were seen between the patient groups for this primary endpoint, and equivalence in efficacy was demonstrated within predefined margins. The percent of ACR20 responders at 24 weeks was 79% in the biosimilar arm and 76% with etanercept.
Secondary analyses showed that the ACR50 responses occurred in 65% of the patients on the biosimilar at 24 weeks and in 53% of patients on etanercept, a statistically significant difference; and after 48 weeks of treatment, the ACR50 rate was 68% in the biosimilar arm and 55% with etanercept, also a statistically significant difference. The two study groups showed no significant difference in the rate of ACR70 responders after 24 or 48 weeks, and also no statistically significant difference in the rate of ACR20 responders after 48 weeks.
The results also showed that the biosimilar was "well tolerated," with a safety profile "comparable" with etanercept, Dr. Bae said.
Dr. Kay has received research funding and/or consulting income from AbbVie, Amgen, Bristol-Myers Squibb, Eli Lilly, Epirus, Genentech, Hospira, Janssen, Pfizer, Roche, and UCB. Dr. Bae acknowledged receiving consulting income and/or research support from Abbott, Bristol-Myers Squibb, Eisai, GlaxoSmithKline, Hanwha Chemical, Merck Serono, MSD, and Pfizer.
* This story was updated 6/13/14.
*PARIS - Two different biosimilar drugs, one a mimic of infliximab, the other modeled on etanercept, closely matched the efficacy and safety of their brand-name counterparts in a pair of separate, randomized, controlled studies.
In one study, treatment with infliximab and a biosimilar agent showed virtually identical efficacy and safety performance at several time points during the first 16 weeks of treatment of 189 patients with rheumatoid arthritis in a head-to-head, randomized comparison done at 23 centers in India.
In the second study, an agent produced to match etanercept showed very similar efficacy and safety to the branded formulation in a total of 233 randomized patients treated for 24 weeks at several centers in Korea.
The second study run in India was the "first clinical trial of a biosimilar infliximab to demonstrate and report kinetics of response to treatment at multiple time points prior to the plateau phase," which starts at about weeks 16, Dr. Jonathan Kay said at the annual European Congress of Rheumatology. It provides "convincing evidence of therapeutic equivalence," and also serves as a "new paradigm for comparative effectiveness testing of biosimilars," said Dr. Kay, a professor of medicine at the University of Massachusetts in Worcester.
Dr. Kay and his colleagues randomized 62 patients with active rheumatoid arthritis (RA) on stable doses of methotrexate to infliximab (Remicade) and 127 patients to BOW015, the biosimilar under study. They measured the proportion of responders at 2, 6, 14, and 16 weeks, with the study’s primary endpoint the percentage of patients showing an American College of Rheumatology 20 (ACR20) response at 16 weeks. The proportion of responders at each of these four time points was virtually identical, Dr. Kay reported. At 16 weeks, the percent of ACR20 responders was 86% in the infliximab arm and 90% in the biosimilar arm. The percent of ACR50 and ACR70 responders was also virtually the same in both treatment arms at 16 weeks, and the percent of ACR20 responders was virtually identical in the two treatment arms at weeks 2, 6, and 14.
The safety profiles of the two drugs also showed no statistically significant differences for any parameter measured except for the incidence of skin disorders, which occurred in one patient treated with the biosimilar and four patients treated with infliximab, a statistically significant difference.
At the same session, Dr. Sang-Cheol Bae of the Hanyang University Hospital for Rheumatic Diseases, Seoul, South Korea, presented findings from a randomized, controlled trial that compared the etanercept biosimilar HD203 with etanercept (Enbrel). Dr. Bae and his colleagues randomized 115 patients with active rheumatoid arthritis to HD203 and 118 patients to etanercept, both in combination with methotrexate, with the primary endpoint set as the proportion of patients achieving ACR20 by week 24. No statistically significant differences were seen between the patient groups for this primary endpoint, and equivalence in efficacy was demonstrated within predefined margins. The percent of ACR20 responders at 24 weeks was 79% in the biosimilar arm and 76% with etanercept.
Secondary analyses showed that the ACR50 responses occurred in 65% of the patients on the biosimilar at 24 weeks and in 53% of patients on etanercept, a statistically significant difference; and after 48 weeks of treatment, the ACR50 rate was 68% in the biosimilar arm and 55% with etanercept, also a statistically significant difference. The two study groups showed no significant difference in the rate of ACR70 responders after 24 or 48 weeks, and also no statistically significant difference in the rate of ACR20 responders after 48 weeks.
The results also showed that the biosimilar was "well tolerated," with a safety profile "comparable" with etanercept, Dr. Bae said.
Dr. Kay has received research funding and/or consulting income from AbbVie, Amgen, Bristol-Myers Squibb, Eli Lilly, Epirus, Genentech, Hospira, Janssen, Pfizer, Roche, and UCB. Dr. Bae acknowledged receiving consulting income and/or research support from Abbott, Bristol-Myers Squibb, Eisai, GlaxoSmithKline, Hanwha Chemical, Merck Serono, MSD, and Pfizer.
* This story was updated 6/13/14.
AT THE EULAR CONGRESS 2014
Key clinical point: Two biosimilar agents showed nearly identical efficacy, safety to
infliximab and etanercept in a pair of randomized trials.
Major finding: An ACR20 response was seen in 86% of the infliximab arm and 90% in the biosimilar arm in the first study and 79% in the biosimilar arm and 76% with etanercept in the second study.
Data source: Two randomized, controlled studies, one with 189 patients, the other with 233 patients.
Disclosures: Dr. Kay has received research funding and/or consulting income from AbbVie, Amgen, Bristol-Myers Squibb, Eli Lilly, Epirus, Genentech, Hospira, Janssen, Pfizer, Roche, and UCB. Dr. Bae acknowledged receiving consulting income and/or research support from Abbott, Bristol-Myers Squibb, Eisai, GlaxoSmithKline, Hanwha Chemical, Merck Serono, MSD, and Pfizer.

VIDEO: In SYMPLICITY, operator inexperience produced incomplete ablations
PARIS – A central problem with the SYMPLICITY HTN-3 trial of renal denervation was that the operators were inexperienced with the denervation procedure – and thus a majority of the renal denervations they carried out were not properly performed and left intact too many of the nerves that surround the renal arteries, commented Dr. Richard R. Heuser during the annual congress of the European Association of Percutaneous Cardiovascular Intervention.
"If you don’t get to the nerves and denervate them, you’re not going to have an effect," said Dr. Heuser, professor of medicine at the University of Arizona and chief of cardiology at St. Luke’s Medical Center in Phoenix.
Another limitation of the SYMPLICITY HTN-3 trial (N. Engl. J. Med. 2014;370:1393-1401) was that 25% of its enrolled patients were African Americans, and the trial’s results suggest that this patient subgroup does not respond to renal denervation with a significant fall in blood pressure.
In a video interview at the meeting, Dr. Heuser discussed the trial's potential limitations and renal denervation’s post-SYMPLICITY future.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
PARIS – A central problem with the SYMPLICITY HTN-3 trial of renal denervation was that the operators were inexperienced with the denervation procedure – and thus a majority of the renal denervations they carried out were not properly performed and left intact too many of the nerves that surround the renal arteries, commented Dr. Richard R. Heuser during the annual congress of the European Association of Percutaneous Cardiovascular Intervention.
"If you don’t get to the nerves and denervate them, you’re not going to have an effect," said Dr. Heuser, professor of medicine at the University of Arizona and chief of cardiology at St. Luke’s Medical Center in Phoenix.
Another limitation of the SYMPLICITY HTN-3 trial (N. Engl. J. Med. 2014;370:1393-1401) was that 25% of its enrolled patients were African Americans, and the trial’s results suggest that this patient subgroup does not respond to renal denervation with a significant fall in blood pressure.
In a video interview at the meeting, Dr. Heuser discussed the trial's potential limitations and renal denervation’s post-SYMPLICITY future.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
PARIS – A central problem with the SYMPLICITY HTN-3 trial of renal denervation was that the operators were inexperienced with the denervation procedure – and thus a majority of the renal denervations they carried out were not properly performed and left intact too many of the nerves that surround the renal arteries, commented Dr. Richard R. Heuser during the annual congress of the European Association of Percutaneous Cardiovascular Intervention.
"If you don’t get to the nerves and denervate them, you’re not going to have an effect," said Dr. Heuser, professor of medicine at the University of Arizona and chief of cardiology at St. Luke’s Medical Center in Phoenix.
Another limitation of the SYMPLICITY HTN-3 trial (N. Engl. J. Med. 2014;370:1393-1401) was that 25% of its enrolled patients were African Americans, and the trial’s results suggest that this patient subgroup does not respond to renal denervation with a significant fall in blood pressure.
In a video interview at the meeting, Dr. Heuser discussed the trial's potential limitations and renal denervation’s post-SYMPLICITY future.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
AT EUROPCR 2014
