User login
High praise, condemnation for CMS Aduhelm coverage plan
Medicare has received a key endorsement of its plan to restrict payment for the controversial Alzheimer’s disease (AD) drug aducanumab (Aduhelm) – but also drew pleas from other groups for more generous reimbursement of the drug, as well as expected similar medications currently in development.
The Centers for Medicare & Medicaid Services received more than 9,900 comments on its plan, according to the current tally posted on its website. However, it is unclear when the final count will be available.
CMS intends to limit federal payment for monoclonal antibodies that target amyloid to clinical trials. Among supporters of this approach is the influential Medicare Payment Advisory Commission, an expert panel that helps Congress and CMS manage the federal health program.
Opponents of the CMS plan include several pharmaceutical companies. Patient and consumer groups, individuals, and lawmakers had mixed views.
CMS officials will weigh the feedback provided in the comments when setting a final coverage policy for aducanumab. It is expected the agency’s final decision will be announced on April 11.
Ongoing debate
The Food and Drug Administration’s unusual approach to clearing the drug for U.S. sales triggered a review of its management of the accelerated approval process by the Office of Inspector General for the Department of Health & Human Services.
The FDA granted an accelerated approval for aducanumab in June based on evidence that the drug clears amyloid in the brain. However, it is unclear whether clearing the protein from the brain results in clinical benefit.
Usually, accelerated approvals precede the completion of phase 3 drug trials, with the FDA allowing early access to a medicine while awaiting confirmatory trials.
In the case of aducanumab, results of the phase 3 confirmatory trials ENGAGE and EMERGE were available at the time of FDA approval. However, interpretation of the findings is controversial.
Biogen contends that the amyloid-clearing effect of the higher dose of aducanumab shown in EMERGE indicates the drug has clinical potential. However, others argue that amyloid clearance does not indicate clinical benefit.
Limiting Medicare coverage of aducanumab for treatment of AD means “the progression of disease, for nearly all beneficiaries, would continue unabated,” Biogen wrote in its comment to CMS.
Conflicting data
Supporters of the CMS plan have a different view of the trial data. They note the failure of aducanumab in the companion ENGAGE trial, while also questioning the magnitude of benefit suggested by even the most positive data cited for the drug in the EMERGE trial.
Both studies used the Clinical Dementia Rating-Sum of Boxes (CDR-SB) score, an 18-point scale measuring cognition and function.
In his comment to CMS, MedPAC chairman Michael E. Chernew, PhD, noted the change in CDR-SB score of 0.39 in EMERGE’s high-dose aducanumab group. CMS has described this as being “less than the 1-2 point change that has been suggested as a minimal clinically important difference,” Dr. Chernew wrote.
MedPAC does not normally comment on Medicare coverage decisions, but did so in this case because of its significance and because of the potential fiscal implications, he noted.
“Though there is only limited, conflicting data on Aduhelm’s clinical effectiveness, Medicare would pay a high price for the product,” Dr. Chernew wrote, pointing out the $28,200 annual U.S. price of the drug.
MedPAC thus endorsed the coverage-with-evidence-development (CED) pathway. Under this approach, Medicare would pay for these drugs when used in clinical trials that meet certain criteria.
Legal challenge?
In its comment to CMS, Biogen questioned the agency’s legal grounds for limiting coverage of aducanumab. A mandate on clinical trials as part of the CED proposal “runs afoul of the Administrative Procedure Act’s prohibition against arbitrary and capricious agency action,” Biogen said.
The drug company argued that its own planned follow-on studies would provide the kind of data Medicare officials want to see. It also argued for greater use of observational data, including real-world evidence, and of information from Medicare claims.
Roche’s Genentech, which is also developing antiamyloid drugs for AD, echoed some of Biogen’s concerns about the aducanumab plan.
CMS’ CED plan would be “unnecessarily restrictive and discouraging for patients living with this destructive disease,” David Burt, executive director for federal government affairs at Genentech, wrote in a comment to CMS.
CMS should clarify that the CED requirement would not apply to cases of FDA-approved antiamyloid therapies that have demonstrated “clinically meaningful improvement,” Mr. Burt added. He noted there are phase 3 trials of drugs in this class that could soon produce data.
CMS should “fully consider the broad ramifications and significant unintended consequences of prematurely placing unduly severe restrictions on the entire class of antiamyloid monoclonal antibodies,” Mr. Burt wrote.
Health care inequity
In its comment to CMS, Biogen also noted the Medicare proposal would “compound the already pervasive inequities in access to treatment and will ultimately prove highly detrimental to health equity.”
There are already concerns about the access of Black and Latinx patients to clinical trials. The planned CED approach would tightly restrict access to aducanumab, as well as expected follow-ons in the amyloid-directed monoclonal antibody (mAb) drug class, the company said.
“Many of the trial sites for Aduhelm, as well as for other amyloid-directed [monoclonal antibodies] are not hospital-based outpatient settings, but include infusion centers, private practices, and medical research centers,” Biogen wrote.
Patient groups such as UsAgainstAlzheimer’s told CMS the CED approach would worsen disparities, despite the aim of Medicare officials to increase participation of Black and Latinx patients in future testing.
“CMS will be hard-pressed to achieve diversity if such hospitals are the only locations where Medicare beneficiaries are able to access mAbs,” USAgainstAlzheimer’s wrote in a Feb. 10 comment.
In contrast, the nonprofit National Center for Health Research praised CMS for what it described as an effort to address a lack of representation of Black and Latinx patients in earlier aducanumab research.
However, the NCHR also suggested CMS revise its plan to mandate that clinical trials include patients who are representative of the national population diagnosed with AD.
“Rather than being concerned about the percentage of patients in specific racial and ethnic groups, we propose that CMS include sufficient numbers of patients in different racial, ethnic, and age groups to ensure that there is enough statistical power for subgroup analyses to determine safety and efficacy for each of the major demographic groups,” the NCHR wrote.
Patient health, Medicare at risk
On Feb. 8, a group of House Republican lawmakers asked CMS to reverse its stance. In a publicly released letter, Rep. Cathy McMorris Rodgers of Washington state, the ranking Republican on the House Energy and Commerce Committee, and colleagues urged broader coverage of aducanumab.
In the letter, the group emphasized the idea of aducanumab as a potential treatment for patients with Down syndrome who are at risk for AD.
“The link between Down Syndrome and AD is still being researched by scientists,” Rep. Rodgers and colleagues wrote.
“However, there appears to be a correlation between the additional 21st chromosome present in people with Down Syndrome and the chromosome’s gene that makes amyloid precursor proteins and can cause a build-up of the beta-amyloid plaques common amongst those with AD,” they add.
On the other hand, CMS garnered earlier support from influential Democrats. On Jan. 13, House Energy and Commerce Chairman Frank Pallone Jr (D-N.J.) and House Oversight and Reform Chairwoman Carolyn B. Maloney (D-N.Y.) released a letter praising CMS for its plan for covering aducanumab.
In addition to the HHS-OIG review of the FDA’s approval of the drug, the two House committees are in the midst of their own investigation of the agency’s decision to clear the drug.
“Any broader coverage determination before there is clarity on Aduhelm’s approval process and findings from the myriad ongoing investigations may put the health of millions of Alzheimer’s patients on the line and the financial stability of the nation’s health insurance program for American seniors at risk,” Rep. Pallone and Rep. Maloney wrote.
A version of this article first appeared on Medscape.com.
Medicare has received a key endorsement of its plan to restrict payment for the controversial Alzheimer’s disease (AD) drug aducanumab (Aduhelm) – but also drew pleas from other groups for more generous reimbursement of the drug, as well as expected similar medications currently in development.
The Centers for Medicare & Medicaid Services received more than 9,900 comments on its plan, according to the current tally posted on its website. However, it is unclear when the final count will be available.
CMS intends to limit federal payment for monoclonal antibodies that target amyloid to clinical trials. Among supporters of this approach is the influential Medicare Payment Advisory Commission, an expert panel that helps Congress and CMS manage the federal health program.
Opponents of the CMS plan include several pharmaceutical companies. Patient and consumer groups, individuals, and lawmakers had mixed views.
CMS officials will weigh the feedback provided in the comments when setting a final coverage policy for aducanumab. It is expected the agency’s final decision will be announced on April 11.
Ongoing debate
The Food and Drug Administration’s unusual approach to clearing the drug for U.S. sales triggered a review of its management of the accelerated approval process by the Office of Inspector General for the Department of Health & Human Services.
The FDA granted an accelerated approval for aducanumab in June based on evidence that the drug clears amyloid in the brain. However, it is unclear whether clearing the protein from the brain results in clinical benefit.
Usually, accelerated approvals precede the completion of phase 3 drug trials, with the FDA allowing early access to a medicine while awaiting confirmatory trials.
In the case of aducanumab, results of the phase 3 confirmatory trials ENGAGE and EMERGE were available at the time of FDA approval. However, interpretation of the findings is controversial.
Biogen contends that the amyloid-clearing effect of the higher dose of aducanumab shown in EMERGE indicates the drug has clinical potential. However, others argue that amyloid clearance does not indicate clinical benefit.
Limiting Medicare coverage of aducanumab for treatment of AD means “the progression of disease, for nearly all beneficiaries, would continue unabated,” Biogen wrote in its comment to CMS.
Conflicting data
Supporters of the CMS plan have a different view of the trial data. They note the failure of aducanumab in the companion ENGAGE trial, while also questioning the magnitude of benefit suggested by even the most positive data cited for the drug in the EMERGE trial.
Both studies used the Clinical Dementia Rating-Sum of Boxes (CDR-SB) score, an 18-point scale measuring cognition and function.
In his comment to CMS, MedPAC chairman Michael E. Chernew, PhD, noted the change in CDR-SB score of 0.39 in EMERGE’s high-dose aducanumab group. CMS has described this as being “less than the 1-2 point change that has been suggested as a minimal clinically important difference,” Dr. Chernew wrote.
MedPAC does not normally comment on Medicare coverage decisions, but did so in this case because of its significance and because of the potential fiscal implications, he noted.
“Though there is only limited, conflicting data on Aduhelm’s clinical effectiveness, Medicare would pay a high price for the product,” Dr. Chernew wrote, pointing out the $28,200 annual U.S. price of the drug.
MedPAC thus endorsed the coverage-with-evidence-development (CED) pathway. Under this approach, Medicare would pay for these drugs when used in clinical trials that meet certain criteria.
Legal challenge?
In its comment to CMS, Biogen questioned the agency’s legal grounds for limiting coverage of aducanumab. A mandate on clinical trials as part of the CED proposal “runs afoul of the Administrative Procedure Act’s prohibition against arbitrary and capricious agency action,” Biogen said.
The drug company argued that its own planned follow-on studies would provide the kind of data Medicare officials want to see. It also argued for greater use of observational data, including real-world evidence, and of information from Medicare claims.
Roche’s Genentech, which is also developing antiamyloid drugs for AD, echoed some of Biogen’s concerns about the aducanumab plan.
CMS’ CED plan would be “unnecessarily restrictive and discouraging for patients living with this destructive disease,” David Burt, executive director for federal government affairs at Genentech, wrote in a comment to CMS.
CMS should clarify that the CED requirement would not apply to cases of FDA-approved antiamyloid therapies that have demonstrated “clinically meaningful improvement,” Mr. Burt added. He noted there are phase 3 trials of drugs in this class that could soon produce data.
CMS should “fully consider the broad ramifications and significant unintended consequences of prematurely placing unduly severe restrictions on the entire class of antiamyloid monoclonal antibodies,” Mr. Burt wrote.
Health care inequity
In its comment to CMS, Biogen also noted the Medicare proposal would “compound the already pervasive inequities in access to treatment and will ultimately prove highly detrimental to health equity.”
There are already concerns about the access of Black and Latinx patients to clinical trials. The planned CED approach would tightly restrict access to aducanumab, as well as expected follow-ons in the amyloid-directed monoclonal antibody (mAb) drug class, the company said.
“Many of the trial sites for Aduhelm, as well as for other amyloid-directed [monoclonal antibodies] are not hospital-based outpatient settings, but include infusion centers, private practices, and medical research centers,” Biogen wrote.
Patient groups such as UsAgainstAlzheimer’s told CMS the CED approach would worsen disparities, despite the aim of Medicare officials to increase participation of Black and Latinx patients in future testing.
“CMS will be hard-pressed to achieve diversity if such hospitals are the only locations where Medicare beneficiaries are able to access mAbs,” USAgainstAlzheimer’s wrote in a Feb. 10 comment.
In contrast, the nonprofit National Center for Health Research praised CMS for what it described as an effort to address a lack of representation of Black and Latinx patients in earlier aducanumab research.
However, the NCHR also suggested CMS revise its plan to mandate that clinical trials include patients who are representative of the national population diagnosed with AD.
“Rather than being concerned about the percentage of patients in specific racial and ethnic groups, we propose that CMS include sufficient numbers of patients in different racial, ethnic, and age groups to ensure that there is enough statistical power for subgroup analyses to determine safety and efficacy for each of the major demographic groups,” the NCHR wrote.
Patient health, Medicare at risk
On Feb. 8, a group of House Republican lawmakers asked CMS to reverse its stance. In a publicly released letter, Rep. Cathy McMorris Rodgers of Washington state, the ranking Republican on the House Energy and Commerce Committee, and colleagues urged broader coverage of aducanumab.
In the letter, the group emphasized the idea of aducanumab as a potential treatment for patients with Down syndrome who are at risk for AD.
“The link between Down Syndrome and AD is still being researched by scientists,” Rep. Rodgers and colleagues wrote.
“However, there appears to be a correlation between the additional 21st chromosome present in people with Down Syndrome and the chromosome’s gene that makes amyloid precursor proteins and can cause a build-up of the beta-amyloid plaques common amongst those with AD,” they add.
On the other hand, CMS garnered earlier support from influential Democrats. On Jan. 13, House Energy and Commerce Chairman Frank Pallone Jr (D-N.J.) and House Oversight and Reform Chairwoman Carolyn B. Maloney (D-N.Y.) released a letter praising CMS for its plan for covering aducanumab.
In addition to the HHS-OIG review of the FDA’s approval of the drug, the two House committees are in the midst of their own investigation of the agency’s decision to clear the drug.
“Any broader coverage determination before there is clarity on Aduhelm’s approval process and findings from the myriad ongoing investigations may put the health of millions of Alzheimer’s patients on the line and the financial stability of the nation’s health insurance program for American seniors at risk,” Rep. Pallone and Rep. Maloney wrote.
A version of this article first appeared on Medscape.com.
Medicare has received a key endorsement of its plan to restrict payment for the controversial Alzheimer’s disease (AD) drug aducanumab (Aduhelm) – but also drew pleas from other groups for more generous reimbursement of the drug, as well as expected similar medications currently in development.
The Centers for Medicare & Medicaid Services received more than 9,900 comments on its plan, according to the current tally posted on its website. However, it is unclear when the final count will be available.
CMS intends to limit federal payment for monoclonal antibodies that target amyloid to clinical trials. Among supporters of this approach is the influential Medicare Payment Advisory Commission, an expert panel that helps Congress and CMS manage the federal health program.
Opponents of the CMS plan include several pharmaceutical companies. Patient and consumer groups, individuals, and lawmakers had mixed views.
CMS officials will weigh the feedback provided in the comments when setting a final coverage policy for aducanumab. It is expected the agency’s final decision will be announced on April 11.
Ongoing debate
The Food and Drug Administration’s unusual approach to clearing the drug for U.S. sales triggered a review of its management of the accelerated approval process by the Office of Inspector General for the Department of Health & Human Services.
The FDA granted an accelerated approval for aducanumab in June based on evidence that the drug clears amyloid in the brain. However, it is unclear whether clearing the protein from the brain results in clinical benefit.
Usually, accelerated approvals precede the completion of phase 3 drug trials, with the FDA allowing early access to a medicine while awaiting confirmatory trials.
In the case of aducanumab, results of the phase 3 confirmatory trials ENGAGE and EMERGE were available at the time of FDA approval. However, interpretation of the findings is controversial.
Biogen contends that the amyloid-clearing effect of the higher dose of aducanumab shown in EMERGE indicates the drug has clinical potential. However, others argue that amyloid clearance does not indicate clinical benefit.
Limiting Medicare coverage of aducanumab for treatment of AD means “the progression of disease, for nearly all beneficiaries, would continue unabated,” Biogen wrote in its comment to CMS.
Conflicting data
Supporters of the CMS plan have a different view of the trial data. They note the failure of aducanumab in the companion ENGAGE trial, while also questioning the magnitude of benefit suggested by even the most positive data cited for the drug in the EMERGE trial.
Both studies used the Clinical Dementia Rating-Sum of Boxes (CDR-SB) score, an 18-point scale measuring cognition and function.
In his comment to CMS, MedPAC chairman Michael E. Chernew, PhD, noted the change in CDR-SB score of 0.39 in EMERGE’s high-dose aducanumab group. CMS has described this as being “less than the 1-2 point change that has been suggested as a minimal clinically important difference,” Dr. Chernew wrote.
MedPAC does not normally comment on Medicare coverage decisions, but did so in this case because of its significance and because of the potential fiscal implications, he noted.
“Though there is only limited, conflicting data on Aduhelm’s clinical effectiveness, Medicare would pay a high price for the product,” Dr. Chernew wrote, pointing out the $28,200 annual U.S. price of the drug.
MedPAC thus endorsed the coverage-with-evidence-development (CED) pathway. Under this approach, Medicare would pay for these drugs when used in clinical trials that meet certain criteria.
Legal challenge?
In its comment to CMS, Biogen questioned the agency’s legal grounds for limiting coverage of aducanumab. A mandate on clinical trials as part of the CED proposal “runs afoul of the Administrative Procedure Act’s prohibition against arbitrary and capricious agency action,” Biogen said.
The drug company argued that its own planned follow-on studies would provide the kind of data Medicare officials want to see. It also argued for greater use of observational data, including real-world evidence, and of information from Medicare claims.
Roche’s Genentech, which is also developing antiamyloid drugs for AD, echoed some of Biogen’s concerns about the aducanumab plan.
CMS’ CED plan would be “unnecessarily restrictive and discouraging for patients living with this destructive disease,” David Burt, executive director for federal government affairs at Genentech, wrote in a comment to CMS.
CMS should clarify that the CED requirement would not apply to cases of FDA-approved antiamyloid therapies that have demonstrated “clinically meaningful improvement,” Mr. Burt added. He noted there are phase 3 trials of drugs in this class that could soon produce data.
CMS should “fully consider the broad ramifications and significant unintended consequences of prematurely placing unduly severe restrictions on the entire class of antiamyloid monoclonal antibodies,” Mr. Burt wrote.
Health care inequity
In its comment to CMS, Biogen also noted the Medicare proposal would “compound the already pervasive inequities in access to treatment and will ultimately prove highly detrimental to health equity.”
There are already concerns about the access of Black and Latinx patients to clinical trials. The planned CED approach would tightly restrict access to aducanumab, as well as expected follow-ons in the amyloid-directed monoclonal antibody (mAb) drug class, the company said.
“Many of the trial sites for Aduhelm, as well as for other amyloid-directed [monoclonal antibodies] are not hospital-based outpatient settings, but include infusion centers, private practices, and medical research centers,” Biogen wrote.
Patient groups such as UsAgainstAlzheimer’s told CMS the CED approach would worsen disparities, despite the aim of Medicare officials to increase participation of Black and Latinx patients in future testing.
“CMS will be hard-pressed to achieve diversity if such hospitals are the only locations where Medicare beneficiaries are able to access mAbs,” USAgainstAlzheimer’s wrote in a Feb. 10 comment.
In contrast, the nonprofit National Center for Health Research praised CMS for what it described as an effort to address a lack of representation of Black and Latinx patients in earlier aducanumab research.
However, the NCHR also suggested CMS revise its plan to mandate that clinical trials include patients who are representative of the national population diagnosed with AD.
“Rather than being concerned about the percentage of patients in specific racial and ethnic groups, we propose that CMS include sufficient numbers of patients in different racial, ethnic, and age groups to ensure that there is enough statistical power for subgroup analyses to determine safety and efficacy for each of the major demographic groups,” the NCHR wrote.
Patient health, Medicare at risk
On Feb. 8, a group of House Republican lawmakers asked CMS to reverse its stance. In a publicly released letter, Rep. Cathy McMorris Rodgers of Washington state, the ranking Republican on the House Energy and Commerce Committee, and colleagues urged broader coverage of aducanumab.
In the letter, the group emphasized the idea of aducanumab as a potential treatment for patients with Down syndrome who are at risk for AD.
“The link between Down Syndrome and AD is still being researched by scientists,” Rep. Rodgers and colleagues wrote.
“However, there appears to be a correlation between the additional 21st chromosome present in people with Down Syndrome and the chromosome’s gene that makes amyloid precursor proteins and can cause a build-up of the beta-amyloid plaques common amongst those with AD,” they add.
On the other hand, CMS garnered earlier support from influential Democrats. On Jan. 13, House Energy and Commerce Chairman Frank Pallone Jr (D-N.J.) and House Oversight and Reform Chairwoman Carolyn B. Maloney (D-N.Y.) released a letter praising CMS for its plan for covering aducanumab.
In addition to the HHS-OIG review of the FDA’s approval of the drug, the two House committees are in the midst of their own investigation of the agency’s decision to clear the drug.
“Any broader coverage determination before there is clarity on Aduhelm’s approval process and findings from the myriad ongoing investigations may put the health of millions of Alzheimer’s patients on the line and the financial stability of the nation’s health insurance program for American seniors at risk,” Rep. Pallone and Rep. Maloney wrote.
A version of this article first appeared on Medscape.com.
Biden’s FDA chief nominee narrowly wins Senate confirmation
On Feb. 15, Robert Califf, MD, narrowly won Senate confirmation to once again serve as the commissioner of the Food and Drug Administration, overcoming protest votes from lawmakers about abortion and opioid issues.

The Senate voted 50-46 in favor of Dr. Califf’s nomination. A cardiologist long affiliated with Duke University and a noted expert on clinical trials, Dr. Califf also led the FDA from February 2016 through January 2017.
In 2016, the Senate confirmed him as FDA chief in an 89-4 vote. At that time, Sen. Joe Manchin, D-WV, and a few other senators said they were concerned that Dr. Califf’s links to the drug industry would hamper his ability to regulate drugmakers, particularly in terms of rules on prescription painkillers.
Sen. Manchin also objected to Dr. Califf’s second nomination as FDA commissioner, as did several fellow Democrats, including Sen. Edward Markey of Massachusetts. In a statement issued after the Feb. 15 vote, Sen. Markey said he has “consistently raised concerns about the FDA’s egregious mishandling of opioid approvals and its role in enabling the current opioid epidemic.”
“To date, the FDA still has not implemented many of the reforms necessary to ensure that it is fulfilling its role as our nation’s top pharmaceutical cop on the beat,” Sen. Markey said. “I have not received any real commitment from Dr. Califf to truly reform the FDA or to learn from the failures that fueled this public health crisis.”
This time, Dr. Califf lost support among Republican senators due to objections raised by groups seeking to end women’s access to abortion. Susan B. Anthony List and National Right to Life asked senators in a January letter to oppose Dr. Califf’s nomination, citing their objections to how the FDA handled reporting of adverse events from abortions by medication during Dr. Califf’s Tenure.
But some Republicans supported Califf in the Tuesday vote. Sens. Roy Blunt of Missouri, Richard Burr of North Carolina, Susan Collins of Maine, Lisa Murkowski of Alaska, Mitt Romney of Utah, and Pat Toomey of Pennsylvania all voted in his favor.
On Feb. 14, Sen. Patty Murray, D-WA, chairwoman of the Senate Health, Education, Labor, and Pensions Committee, urged her colleagues to vote for Dr. Califf to give the FDA strong leadership to tackle urgent health needs such as the opioid crisis, youth tobacco use, antimicrobial resistance, and inequities in health care.
“At this critical moment, we need a trusted hand to lead the FDA,” she said in a floor speech. Dr. Califf’s previous service at the FDA and his years spent as a research scientist “give him the experience to take on this challenge.”
Separately, three former FDA commissioners on Feb. 15 published an opinion article that appeared in The Hill. Republican presidents nominated two of these former FDA chiefs: Scott Gottlieb, MD, and Mark McClellan, MD. The third, Margaret Hamburg, MD, was nominated by President Barack Obama, as was Dr. Califf for his first time as FDA chief.
There’s an urgent need for a confirmed leader at the FDA as the United States seeks to move beyond the pandemic, the former FDA chiefs wrote. The work ahead includes continued efforts with vaccines as well as efforts to bolster medical supply chains, they said.
Dr. Califf “knows how to advance the safe development and use of medical products and to bring a sound, science-based foundation to the FDA’s regulatory actions. Because of this, he has earned the confidence of FDA’s professional career staff, as well as a broad base of patient groups, academic experts, medical professionals, and public health organizations,” Dr. Gottlieb, Dr. Hamburg, and Dr. McClellan wrote.
The article also was signed by former Centers for Medicare and Medicaid Services Administrator Andy Slavitt, who served in the Obama administration.
Support of medical community
The American Heart Association issued a statement on Feb.15, congratulating Dr. Califf on his second confirmation after the Senate vote.
“With a distinguished career in public service and a long-time volunteer leader at the American Heart Association, Dr. Califf has honed his ability to communicate and build trust with diverse constituencies,” CEO Nancy Brown said in the statement. “He will use his experience as a cardiologist to safeguard the health and well-being of people throughout the country, and his background in research to prioritize science and evidence-based policymaking.”
Dr. Califf was also backed by the Association of American Medical Colleges, the American Academy of Pediatrics, the American Academy of Family Physicians, and the American College of Physicians when he was nominated for the role last year by President Joe Biden.
A version of this article first appeared on Medscape.com.
On Feb. 15, Robert Califf, MD, narrowly won Senate confirmation to once again serve as the commissioner of the Food and Drug Administration, overcoming protest votes from lawmakers about abortion and opioid issues.
The Senate voted 50-46 in favor of Dr. Califf’s nomination. A cardiologist long affiliated with Duke University and a noted expert on clinical trials, Dr. Califf also led the FDA from February 2016 through January 2017.
In 2016, the Senate confirmed him as FDA chief in an 89-4 vote. At that time, Sen. Joe Manchin, D-WV, and a few other senators said they were concerned that Dr. Califf’s links to the drug industry would hamper his ability to regulate drugmakers, particularly in terms of rules on prescription painkillers.
Sen. Manchin also objected to Dr. Califf’s second nomination as FDA commissioner, as did several fellow Democrats, including Sen. Edward Markey of Massachusetts. In a statement issued after the Feb. 15 vote, Sen. Markey said he has “consistently raised concerns about the FDA’s egregious mishandling of opioid approvals and its role in enabling the current opioid epidemic.”
“To date, the FDA still has not implemented many of the reforms necessary to ensure that it is fulfilling its role as our nation’s top pharmaceutical cop on the beat,” Sen. Markey said. “I have not received any real commitment from Dr. Califf to truly reform the FDA or to learn from the failures that fueled this public health crisis.”
This time, Dr. Califf lost support among Republican senators due to objections raised by groups seeking to end women’s access to abortion. Susan B. Anthony List and National Right to Life asked senators in a January letter to oppose Dr. Califf’s nomination, citing their objections to how the FDA handled reporting of adverse events from abortions by medication during Dr. Califf’s Tenure.
But some Republicans supported Califf in the Tuesday vote. Sens. Roy Blunt of Missouri, Richard Burr of North Carolina, Susan Collins of Maine, Lisa Murkowski of Alaska, Mitt Romney of Utah, and Pat Toomey of Pennsylvania all voted in his favor.
On Feb. 14, Sen. Patty Murray, D-WA, chairwoman of the Senate Health, Education, Labor, and Pensions Committee, urged her colleagues to vote for Dr. Califf to give the FDA strong leadership to tackle urgent health needs such as the opioid crisis, youth tobacco use, antimicrobial resistance, and inequities in health care.
“At this critical moment, we need a trusted hand to lead the FDA,” she said in a floor speech. Dr. Califf’s previous service at the FDA and his years spent as a research scientist “give him the experience to take on this challenge.”
Separately, three former FDA commissioners on Feb. 15 published an opinion article that appeared in The Hill. Republican presidents nominated two of these former FDA chiefs: Scott Gottlieb, MD, and Mark McClellan, MD. The third, Margaret Hamburg, MD, was nominated by President Barack Obama, as was Dr. Califf for his first time as FDA chief.
There’s an urgent need for a confirmed leader at the FDA as the United States seeks to move beyond the pandemic, the former FDA chiefs wrote. The work ahead includes continued efforts with vaccines as well as efforts to bolster medical supply chains, they said.
Dr. Califf “knows how to advance the safe development and use of medical products and to bring a sound, science-based foundation to the FDA’s regulatory actions. Because of this, he has earned the confidence of FDA’s professional career staff, as well as a broad base of patient groups, academic experts, medical professionals, and public health organizations,” Dr. Gottlieb, Dr. Hamburg, and Dr. McClellan wrote.
The article also was signed by former Centers for Medicare and Medicaid Services Administrator Andy Slavitt, who served in the Obama administration.
Support of medical community
The American Heart Association issued a statement on Feb.15, congratulating Dr. Califf on his second confirmation after the Senate vote.
“With a distinguished career in public service and a long-time volunteer leader at the American Heart Association, Dr. Califf has honed his ability to communicate and build trust with diverse constituencies,” CEO Nancy Brown said in the statement. “He will use his experience as a cardiologist to safeguard the health and well-being of people throughout the country, and his background in research to prioritize science and evidence-based policymaking.”
Dr. Califf was also backed by the Association of American Medical Colleges, the American Academy of Pediatrics, the American Academy of Family Physicians, and the American College of Physicians when he was nominated for the role last year by President Joe Biden.
A version of this article first appeared on Medscape.com.
On Feb. 15, Robert Califf, MD, narrowly won Senate confirmation to once again serve as the commissioner of the Food and Drug Administration, overcoming protest votes from lawmakers about abortion and opioid issues.
The Senate voted 50-46 in favor of Dr. Califf’s nomination. A cardiologist long affiliated with Duke University and a noted expert on clinical trials, Dr. Califf also led the FDA from February 2016 through January 2017.
In 2016, the Senate confirmed him as FDA chief in an 89-4 vote. At that time, Sen. Joe Manchin, D-WV, and a few other senators said they were concerned that Dr. Califf’s links to the drug industry would hamper his ability to regulate drugmakers, particularly in terms of rules on prescription painkillers.
Sen. Manchin also objected to Dr. Califf’s second nomination as FDA commissioner, as did several fellow Democrats, including Sen. Edward Markey of Massachusetts. In a statement issued after the Feb. 15 vote, Sen. Markey said he has “consistently raised concerns about the FDA’s egregious mishandling of opioid approvals and its role in enabling the current opioid epidemic.”
“To date, the FDA still has not implemented many of the reforms necessary to ensure that it is fulfilling its role as our nation’s top pharmaceutical cop on the beat,” Sen. Markey said. “I have not received any real commitment from Dr. Califf to truly reform the FDA or to learn from the failures that fueled this public health crisis.”
This time, Dr. Califf lost support among Republican senators due to objections raised by groups seeking to end women’s access to abortion. Susan B. Anthony List and National Right to Life asked senators in a January letter to oppose Dr. Califf’s nomination, citing their objections to how the FDA handled reporting of adverse events from abortions by medication during Dr. Califf’s Tenure.
But some Republicans supported Califf in the Tuesday vote. Sens. Roy Blunt of Missouri, Richard Burr of North Carolina, Susan Collins of Maine, Lisa Murkowski of Alaska, Mitt Romney of Utah, and Pat Toomey of Pennsylvania all voted in his favor.
On Feb. 14, Sen. Patty Murray, D-WA, chairwoman of the Senate Health, Education, Labor, and Pensions Committee, urged her colleagues to vote for Dr. Califf to give the FDA strong leadership to tackle urgent health needs such as the opioid crisis, youth tobacco use, antimicrobial resistance, and inequities in health care.
“At this critical moment, we need a trusted hand to lead the FDA,” she said in a floor speech. Dr. Califf’s previous service at the FDA and his years spent as a research scientist “give him the experience to take on this challenge.”
Separately, three former FDA commissioners on Feb. 15 published an opinion article that appeared in The Hill. Republican presidents nominated two of these former FDA chiefs: Scott Gottlieb, MD, and Mark McClellan, MD. The third, Margaret Hamburg, MD, was nominated by President Barack Obama, as was Dr. Califf for his first time as FDA chief.
There’s an urgent need for a confirmed leader at the FDA as the United States seeks to move beyond the pandemic, the former FDA chiefs wrote. The work ahead includes continued efforts with vaccines as well as efforts to bolster medical supply chains, they said.
Dr. Califf “knows how to advance the safe development and use of medical products and to bring a sound, science-based foundation to the FDA’s regulatory actions. Because of this, he has earned the confidence of FDA’s professional career staff, as well as a broad base of patient groups, academic experts, medical professionals, and public health organizations,” Dr. Gottlieb, Dr. Hamburg, and Dr. McClellan wrote.
The article also was signed by former Centers for Medicare and Medicaid Services Administrator Andy Slavitt, who served in the Obama administration.
Support of medical community
The American Heart Association issued a statement on Feb.15, congratulating Dr. Califf on his second confirmation after the Senate vote.
“With a distinguished career in public service and a long-time volunteer leader at the American Heart Association, Dr. Califf has honed his ability to communicate and build trust with diverse constituencies,” CEO Nancy Brown said in the statement. “He will use his experience as a cardiologist to safeguard the health and well-being of people throughout the country, and his background in research to prioritize science and evidence-based policymaking.”
Dr. Califf was also backed by the Association of American Medical Colleges, the American Academy of Pediatrics, the American Academy of Family Physicians, and the American College of Physicians when he was nominated for the role last year by President Joe Biden.
A version of this article first appeared on Medscape.com.
Appendectomy or antibiotics? Large trial helps decision-making
The presence of mineralized stool, known as appendicolith, was associated with a nearly twofold increased risk of undergoing appendectomy within 30 days of initiating antibiotics, write David Flum, MD, of the University of Washington, Seattle, and coauthors in a paper published in JAMA Surgery on Jan. 12, 2021.
But the surprise was the lack of an association between appendectomy and factors often presumed to be consistent with more severe appendicitis.
Physicians have had their own ideas about what factors make a patient more likely to need an appendectomy after an initial round of treatment with antibiotics, such as a high white blood cell count or a perforation seen on CT scan, Dr. Flum said in an interview. But the research didn’t support some of these theories.
“This is why we do the studies,” Dr. Flum said. “Sometimes we find out that our hunches were wrong.”
Dr. Flum and coauthors measured the association between different patient factors and disease severity and the need for appendectomy following a course of antibiotics. They used adjusted odds ratios to describe these relationships while accounting for other differences.
An OR of 1.0 – or when the confidence interval around an OR crosses 1 – signals that there is no association between that factor and appendectomy. Positive ORs with confidence intervals that exclude 1.0 suggest the factor was associated with appendectomy.
The OR was 1.99 for the presence of appendicolith, a finding with a 95% confidence interval of 1.28-3.10. The OR was 1.53 (95% CI, 1.01-2.31) for female sex.
But the OR was 1.14 (95% CI, 0.66-1.98) for perforation, abscess, or fat stranding.
The OR was 1.09 (95% CI, 1.00-1.18) for radiographic finding of a larger appendix, as measured by diameter.
And the OR was 1.03 (95% CI, 0.98-1.09) for having a higher white blood cell count, as measured by a 1,000-cells/mcL increase.
Appy or not?
This paper draws from the Comparison of Outcomes of Antibiotic Drugs and Appendectomy (CODA) trial (NCT02800785), for which top-line results were published in 2020 in the New England Journal of Medicine. In that paper, Dr. Flum and colleagues reported on results for 1,552 adults (414 with an appendicolith) who were evenly randomized to either antibiotics treatment or appendectomy. After 30 days, antibiotics were found to be noninferior to appendectomy, as reported by this news organization.
The federal Patient-Centered Outcomes Research Institute funded the CODA research. Dr. Flum said the National Institutes of Health had not appeared interested in funding a look at the different options available to patients experiencing appendicitis. Congress created PCORI as part of the Affordable Care Act of 2010, seeking to encourage researchers to study which treatments best serve patients through direct comparisons. Its support was critical for Dr. Flum and colleagues in seeking to help people weigh their options for treating appendicitis.
The CODA study “models what the patient’s experience is like, and this has not been the focus of NIH as much,” Dr. Flum said.
The CODA team has sought to make it easy for patients to consider what its findings and other research on appendicitis mean for them. They created an online decision-making tool, available at the aptly named http://www.appyornot.org/ website, which has videos in English and Spanish explaining patients’ options in simple terms. The website also asks questions about personal preferences, priorities, and resources to help them choose a treatment based on their individual situation.
Shift away from ‘paternalistic framing’
In the past, surgeons focused on the risk for patients from procedures, making the decisions for them about whether or not to proceed. There’s now a drive to shift away from this “paternalistic framing” toward shared decision-making, Dr. Flum said.
Surgeons need to have conversations with their patients about what’s happening in their lives as well as to assess their fears and concerns about treatment options, he said. These are aspects of patient care that were not covered in medical school or surgical training, but they lead to “less paternalistic” treatment. A patient’s decision about whether to choose surgery or antibiotics for appendicitis may hinge on factors such as insurance coverage, access to childcare, and the ability to miss days of work.
Dr. Flum said his fellow surgeons by and large have reacted well to the CODA team’s work.
“To their credit, the surgical community has embraced a healthy skepticism about the role of surgery,” Dr. Flum said.
The guidelines of the American College of Surgeons state that there is “high-quality evidence” that most patients with appendicitis can be managed with antibiotics instead of appendectomy (69% overall avoid appendectomy by 90 days, 75% of those without appendicolith, and 59% of those with appendicolith).
“Based on the surgeon’s judgment, patient preferences, and local resources (e.g., hospital staff, bed, and PPE supply availability) antibiotics are an acceptable first-line treatment, with appendectomy offered for those with worsening or recurrent symptoms,” the ACS guidelines say.
In an interview, Samir M. Fakhry, MD, vice president of HCA Center for Trauma and Acute Care Surgery Research in Nashville, Tenn., agreed with Dr. Flum about the shift taking place in medicine.
The CODA research, including the new paper in JAMA Surgery, makes it easier for physicians to work with patients and their families to reach decisions about how to treat appendicitis, Dr. Fakhry said.
These important discussions take time, he said, and patients must be allowed that time. Patients might feel misled, for example, if a surgeon pressed for appendectomy without explaining that a course of antibiotics may have served them well. Other patients may opt for surgery right away, especially in cases with appendicoliths, to avoid the potential for repeat episodes of medical care.
“You’ve got people who just want to get it done and over with. You’ve got people who want to avoid surgery no matter what,” Dr. Fakhry said. “It’s not just about the science and the data.”
This study was supported by a grant from PCORI. The authors reported having served as consultants or reviewers or have received fees for work outside of this paper from Stryker, Kerecis, Acera, Medline, Shriner’s Research Fund, UpToDate, and Tetraphase Pharmaceuticals Stryker.
A version of this article first appeared on Medscape.com.
The presence of mineralized stool, known as appendicolith, was associated with a nearly twofold increased risk of undergoing appendectomy within 30 days of initiating antibiotics, write David Flum, MD, of the University of Washington, Seattle, and coauthors in a paper published in JAMA Surgery on Jan. 12, 2021.
But the surprise was the lack of an association between appendectomy and factors often presumed to be consistent with more severe appendicitis.
Physicians have had their own ideas about what factors make a patient more likely to need an appendectomy after an initial round of treatment with antibiotics, such as a high white blood cell count or a perforation seen on CT scan, Dr. Flum said in an interview. But the research didn’t support some of these theories.
“This is why we do the studies,” Dr. Flum said. “Sometimes we find out that our hunches were wrong.”
Dr. Flum and coauthors measured the association between different patient factors and disease severity and the need for appendectomy following a course of antibiotics. They used adjusted odds ratios to describe these relationships while accounting for other differences.
An OR of 1.0 – or when the confidence interval around an OR crosses 1 – signals that there is no association between that factor and appendectomy. Positive ORs with confidence intervals that exclude 1.0 suggest the factor was associated with appendectomy.
The OR was 1.99 for the presence of appendicolith, a finding with a 95% confidence interval of 1.28-3.10. The OR was 1.53 (95% CI, 1.01-2.31) for female sex.
But the OR was 1.14 (95% CI, 0.66-1.98) for perforation, abscess, or fat stranding.
The OR was 1.09 (95% CI, 1.00-1.18) for radiographic finding of a larger appendix, as measured by diameter.
And the OR was 1.03 (95% CI, 0.98-1.09) for having a higher white blood cell count, as measured by a 1,000-cells/mcL increase.
Appy or not?
This paper draws from the Comparison of Outcomes of Antibiotic Drugs and Appendectomy (CODA) trial (NCT02800785), for which top-line results were published in 2020 in the New England Journal of Medicine. In that paper, Dr. Flum and colleagues reported on results for 1,552 adults (414 with an appendicolith) who were evenly randomized to either antibiotics treatment or appendectomy. After 30 days, antibiotics were found to be noninferior to appendectomy, as reported by this news organization.
The federal Patient-Centered Outcomes Research Institute funded the CODA research. Dr. Flum said the National Institutes of Health had not appeared interested in funding a look at the different options available to patients experiencing appendicitis. Congress created PCORI as part of the Affordable Care Act of 2010, seeking to encourage researchers to study which treatments best serve patients through direct comparisons. Its support was critical for Dr. Flum and colleagues in seeking to help people weigh their options for treating appendicitis.
The CODA study “models what the patient’s experience is like, and this has not been the focus of NIH as much,” Dr. Flum said.
The CODA team has sought to make it easy for patients to consider what its findings and other research on appendicitis mean for them. They created an online decision-making tool, available at the aptly named http://www.appyornot.org/ website, which has videos in English and Spanish explaining patients’ options in simple terms. The website also asks questions about personal preferences, priorities, and resources to help them choose a treatment based on their individual situation.
Shift away from ‘paternalistic framing’
In the past, surgeons focused on the risk for patients from procedures, making the decisions for them about whether or not to proceed. There’s now a drive to shift away from this “paternalistic framing” toward shared decision-making, Dr. Flum said.
Surgeons need to have conversations with their patients about what’s happening in their lives as well as to assess their fears and concerns about treatment options, he said. These are aspects of patient care that were not covered in medical school or surgical training, but they lead to “less paternalistic” treatment. A patient’s decision about whether to choose surgery or antibiotics for appendicitis may hinge on factors such as insurance coverage, access to childcare, and the ability to miss days of work.
Dr. Flum said his fellow surgeons by and large have reacted well to the CODA team’s work.
“To their credit, the surgical community has embraced a healthy skepticism about the role of surgery,” Dr. Flum said.
The guidelines of the American College of Surgeons state that there is “high-quality evidence” that most patients with appendicitis can be managed with antibiotics instead of appendectomy (69% overall avoid appendectomy by 90 days, 75% of those without appendicolith, and 59% of those with appendicolith).
“Based on the surgeon’s judgment, patient preferences, and local resources (e.g., hospital staff, bed, and PPE supply availability) antibiotics are an acceptable first-line treatment, with appendectomy offered for those with worsening or recurrent symptoms,” the ACS guidelines say.
In an interview, Samir M. Fakhry, MD, vice president of HCA Center for Trauma and Acute Care Surgery Research in Nashville, Tenn., agreed with Dr. Flum about the shift taking place in medicine.
The CODA research, including the new paper in JAMA Surgery, makes it easier for physicians to work with patients and their families to reach decisions about how to treat appendicitis, Dr. Fakhry said.
These important discussions take time, he said, and patients must be allowed that time. Patients might feel misled, for example, if a surgeon pressed for appendectomy without explaining that a course of antibiotics may have served them well. Other patients may opt for surgery right away, especially in cases with appendicoliths, to avoid the potential for repeat episodes of medical care.
“You’ve got people who just want to get it done and over with. You’ve got people who want to avoid surgery no matter what,” Dr. Fakhry said. “It’s not just about the science and the data.”
This study was supported by a grant from PCORI. The authors reported having served as consultants or reviewers or have received fees for work outside of this paper from Stryker, Kerecis, Acera, Medline, Shriner’s Research Fund, UpToDate, and Tetraphase Pharmaceuticals Stryker.
A version of this article first appeared on Medscape.com.
The presence of mineralized stool, known as appendicolith, was associated with a nearly twofold increased risk of undergoing appendectomy within 30 days of initiating antibiotics, write David Flum, MD, of the University of Washington, Seattle, and coauthors in a paper published in JAMA Surgery on Jan. 12, 2021.
But the surprise was the lack of an association between appendectomy and factors often presumed to be consistent with more severe appendicitis.
Physicians have had their own ideas about what factors make a patient more likely to need an appendectomy after an initial round of treatment with antibiotics, such as a high white blood cell count or a perforation seen on CT scan, Dr. Flum said in an interview. But the research didn’t support some of these theories.
“This is why we do the studies,” Dr. Flum said. “Sometimes we find out that our hunches were wrong.”
Dr. Flum and coauthors measured the association between different patient factors and disease severity and the need for appendectomy following a course of antibiotics. They used adjusted odds ratios to describe these relationships while accounting for other differences.
An OR of 1.0 – or when the confidence interval around an OR crosses 1 – signals that there is no association between that factor and appendectomy. Positive ORs with confidence intervals that exclude 1.0 suggest the factor was associated with appendectomy.
The OR was 1.99 for the presence of appendicolith, a finding with a 95% confidence interval of 1.28-3.10. The OR was 1.53 (95% CI, 1.01-2.31) for female sex.
But the OR was 1.14 (95% CI, 0.66-1.98) for perforation, abscess, or fat stranding.
The OR was 1.09 (95% CI, 1.00-1.18) for radiographic finding of a larger appendix, as measured by diameter.
And the OR was 1.03 (95% CI, 0.98-1.09) for having a higher white blood cell count, as measured by a 1,000-cells/mcL increase.
Appy or not?
This paper draws from the Comparison of Outcomes of Antibiotic Drugs and Appendectomy (CODA) trial (NCT02800785), for which top-line results were published in 2020 in the New England Journal of Medicine. In that paper, Dr. Flum and colleagues reported on results for 1,552 adults (414 with an appendicolith) who were evenly randomized to either antibiotics treatment or appendectomy. After 30 days, antibiotics were found to be noninferior to appendectomy, as reported by this news organization.
The federal Patient-Centered Outcomes Research Institute funded the CODA research. Dr. Flum said the National Institutes of Health had not appeared interested in funding a look at the different options available to patients experiencing appendicitis. Congress created PCORI as part of the Affordable Care Act of 2010, seeking to encourage researchers to study which treatments best serve patients through direct comparisons. Its support was critical for Dr. Flum and colleagues in seeking to help people weigh their options for treating appendicitis.
The CODA study “models what the patient’s experience is like, and this has not been the focus of NIH as much,” Dr. Flum said.
The CODA team has sought to make it easy for patients to consider what its findings and other research on appendicitis mean for them. They created an online decision-making tool, available at the aptly named http://www.appyornot.org/ website, which has videos in English and Spanish explaining patients’ options in simple terms. The website also asks questions about personal preferences, priorities, and resources to help them choose a treatment based on their individual situation.
Shift away from ‘paternalistic framing’
In the past, surgeons focused on the risk for patients from procedures, making the decisions for them about whether or not to proceed. There’s now a drive to shift away from this “paternalistic framing” toward shared decision-making, Dr. Flum said.
Surgeons need to have conversations with their patients about what’s happening in their lives as well as to assess their fears and concerns about treatment options, he said. These are aspects of patient care that were not covered in medical school or surgical training, but they lead to “less paternalistic” treatment. A patient’s decision about whether to choose surgery or antibiotics for appendicitis may hinge on factors such as insurance coverage, access to childcare, and the ability to miss days of work.
Dr. Flum said his fellow surgeons by and large have reacted well to the CODA team’s work.
“To their credit, the surgical community has embraced a healthy skepticism about the role of surgery,” Dr. Flum said.
The guidelines of the American College of Surgeons state that there is “high-quality evidence” that most patients with appendicitis can be managed with antibiotics instead of appendectomy (69% overall avoid appendectomy by 90 days, 75% of those without appendicolith, and 59% of those with appendicolith).
“Based on the surgeon’s judgment, patient preferences, and local resources (e.g., hospital staff, bed, and PPE supply availability) antibiotics are an acceptable first-line treatment, with appendectomy offered for those with worsening or recurrent symptoms,” the ACS guidelines say.
In an interview, Samir M. Fakhry, MD, vice president of HCA Center for Trauma and Acute Care Surgery Research in Nashville, Tenn., agreed with Dr. Flum about the shift taking place in medicine.
The CODA research, including the new paper in JAMA Surgery, makes it easier for physicians to work with patients and their families to reach decisions about how to treat appendicitis, Dr. Fakhry said.
These important discussions take time, he said, and patients must be allowed that time. Patients might feel misled, for example, if a surgeon pressed for appendectomy without explaining that a course of antibiotics may have served them well. Other patients may opt for surgery right away, especially in cases with appendicoliths, to avoid the potential for repeat episodes of medical care.
“You’ve got people who just want to get it done and over with. You’ve got people who want to avoid surgery no matter what,” Dr. Fakhry said. “It’s not just about the science and the data.”
This study was supported by a grant from PCORI. The authors reported having served as consultants or reviewers or have received fees for work outside of this paper from Stryker, Kerecis, Acera, Medline, Shriner’s Research Fund, UpToDate, and Tetraphase Pharmaceuticals Stryker.
A version of this article first appeared on Medscape.com.
FROM JAMA SURGERY
Medicare intends to limit payment for controversial Alzheimer’s drug
, federal officials announced Jan. 11.
On Dec. 20, 2021, Biogen announced a plan to reduce the annual U.S. cost of the drug by 50% – from $56,000 to $28,200 – as Centers for Medicare & Medicaid Services officials were deciding on Medicare’s coverage policy for the medication.
In making its proposed coverage decision, the CMS announced it will pay for aducanumab, a monoclonal antibody, under its coverage-with-evidence-development (CED) mechanism. In making its decision, the CMS approached aducanumab as the first of a potential new class of monoclonal antibodies for the treatment of Alzheimer’s disease. Food and Drug Administration–approved drugs in this class would be covered for those with Medicare only if they are enrolled in qualifying clinical trials, the CMS said. The agency will accept public comments on this decision for 30 days.
In a statement, CMS Administrator Chiquita Brooks-LaSure said the agency is “committed to providing the American public with a clear, trusted, evidence-based decision that is made only after a thorough analysis of public feedback on the benefits and risks of coverage for Medicare patients.”
As previously reported, the FDA approved aducanumab on June 7, 2021, via an accelerated approval process. The approval, which set off a firestorm of controversy that included resignations of three FDA Peripheral and Central Nervous System Drugs Advisory Committee panel members, was granted based on the medication’s ability to reduce beta-amyloid plaque.
Under the accelerated approval mechanism, Biogen still must deliver solid scientific proof that aducanumab has clinically significant disease-modifying effects. However, the final evidence won’t be in any time soon. In its approval letter, the FDA set a 2030 deadline for a final report on this research.
‘Unusual but appropriate’ step
The Medicare decision marks something of a shift in the agency’s approach to paying for medications. On a call with reporters, Tamara Syrek Jensen, JD, director of CMS’ Coverage and Analysis Group, admitted that the agency had taken an “unusual but appropriate” step in trying to set a national policy regarding payment for a drug.
On the same call, Lee Fleisher, MD, CMS’ chief medical officer, addressed the challenges presented by aducanumab, given the serious need for treatments for Alzheimer’s disease. “As a practicing physician, I cannot overemphasize the need to understand the risks and benefits of a given treatment in order to better inform patients and their families,” Dr. Fleisher said. “We do know based on some of the evidence that there may be potential promise with this treatment. That’s why it is critical for us to pursue additional scientific evidence.”
The coverage-with-evidence program will allow Medicare to aid in gathering data, while protecting patients, Dr. Fleisher noted.
“CMS is using its authority provided by Congress to determine if the drug is considered reasonable and necessary, meaning that the benefits of improvement of cognition outweigh the harms in the Medicare population,” Dr. Fleisher said.
Biogen disappointed
Cambridge, Mass.–based Biogen urged the CMS to reconsider its approach to payment for aducanumab. In a statement, the company said Medicare should cover “the class of amyloid-directed therapies with the populations studied in the respective clinical trials and guided by expert recommendations for appropriate use.
“We believe Alzheimer’s patients should have access consistent with other therapies with FDA accelerated approval,” Biogen said in the release.
In the company’s view, the CED approach will “significantly limit patient access to an FDA-approved treatment, especially for underserved patients as evidenced in other CED determinations.
“CEDs can take months to years to initiate, and hundreds of Alzheimer’s patients – the majority of whom are Medicare beneficiaries – are progressing each day from mild to moderate disease stages, where treatment may no longer be an option,” Biogen said.
Drug makers had been worried about CMS opting for CED even before the draft decision was unveiled.
Others weigh in
BIO, the trade group for biotechnology companies, urged the CMS to provide access to aducanumab without excess restrictions.
There already are concerns among drug makers about CMS efforts “to impose new coverage barriers – and, in particular, coverage with evidence development,” Crystal Kuntz, vice president of policy and research at BIO, and Andy Cosgrove, the organization’s senior director for policy and research, noted in a July 2021 comment about the aducanumab review.
Medicare should instead continue to provide access to medicines for indications that the FDA has approved, with additional flexibility for off-label indications of cancer drugs, they noted. “We believe this should continue to be the case, to ensure that vulnerable Medicare beneficiaries have necessary access to life-altering and lifesaving medications,” the BIO officials wrote.
However, the CMS also received many pleas from physicians asking the agency to limit use of aducanumab at least until there is evidence that it produces a significant clinical benefit.
In a press release, Howard Fillit, MD, cofounder and chief science officer of the Alzheimer’s Drug Discovery Foundation, applauded the decision, describing it as “the right call.
“This decision supports conducting additional clinical trials, which are needed to obtain further insights into the clinical efficacy and safety profile of this drug in real-world populations. This decision has implications for other drugs in this class in late-stage development. If these trials show more clear and robust clinical efficacy, then it is possible the FDA will give these amyloid monoclonal antibodies full approval, and Medicare would be likely to provide full payment,” he added.
A version of this article first appeared on Medscape.com.
, federal officials announced Jan. 11.
On Dec. 20, 2021, Biogen announced a plan to reduce the annual U.S. cost of the drug by 50% – from $56,000 to $28,200 – as Centers for Medicare & Medicaid Services officials were deciding on Medicare’s coverage policy for the medication.
In making its proposed coverage decision, the CMS announced it will pay for aducanumab, a monoclonal antibody, under its coverage-with-evidence-development (CED) mechanism. In making its decision, the CMS approached aducanumab as the first of a potential new class of monoclonal antibodies for the treatment of Alzheimer’s disease. Food and Drug Administration–approved drugs in this class would be covered for those with Medicare only if they are enrolled in qualifying clinical trials, the CMS said. The agency will accept public comments on this decision for 30 days.
In a statement, CMS Administrator Chiquita Brooks-LaSure said the agency is “committed to providing the American public with a clear, trusted, evidence-based decision that is made only after a thorough analysis of public feedback on the benefits and risks of coverage for Medicare patients.”
As previously reported, the FDA approved aducanumab on June 7, 2021, via an accelerated approval process. The approval, which set off a firestorm of controversy that included resignations of three FDA Peripheral and Central Nervous System Drugs Advisory Committee panel members, was granted based on the medication’s ability to reduce beta-amyloid plaque.
Under the accelerated approval mechanism, Biogen still must deliver solid scientific proof that aducanumab has clinically significant disease-modifying effects. However, the final evidence won’t be in any time soon. In its approval letter, the FDA set a 2030 deadline for a final report on this research.
‘Unusual but appropriate’ step
The Medicare decision marks something of a shift in the agency’s approach to paying for medications. On a call with reporters, Tamara Syrek Jensen, JD, director of CMS’ Coverage and Analysis Group, admitted that the agency had taken an “unusual but appropriate” step in trying to set a national policy regarding payment for a drug.
On the same call, Lee Fleisher, MD, CMS’ chief medical officer, addressed the challenges presented by aducanumab, given the serious need for treatments for Alzheimer’s disease. “As a practicing physician, I cannot overemphasize the need to understand the risks and benefits of a given treatment in order to better inform patients and their families,” Dr. Fleisher said. “We do know based on some of the evidence that there may be potential promise with this treatment. That’s why it is critical for us to pursue additional scientific evidence.”
The coverage-with-evidence program will allow Medicare to aid in gathering data, while protecting patients, Dr. Fleisher noted.
“CMS is using its authority provided by Congress to determine if the drug is considered reasonable and necessary, meaning that the benefits of improvement of cognition outweigh the harms in the Medicare population,” Dr. Fleisher said.
Biogen disappointed
Cambridge, Mass.–based Biogen urged the CMS to reconsider its approach to payment for aducanumab. In a statement, the company said Medicare should cover “the class of amyloid-directed therapies with the populations studied in the respective clinical trials and guided by expert recommendations for appropriate use.
“We believe Alzheimer’s patients should have access consistent with other therapies with FDA accelerated approval,” Biogen said in the release.
In the company’s view, the CED approach will “significantly limit patient access to an FDA-approved treatment, especially for underserved patients as evidenced in other CED determinations.
“CEDs can take months to years to initiate, and hundreds of Alzheimer’s patients – the majority of whom are Medicare beneficiaries – are progressing each day from mild to moderate disease stages, where treatment may no longer be an option,” Biogen said.
Drug makers had been worried about CMS opting for CED even before the draft decision was unveiled.
Others weigh in
BIO, the trade group for biotechnology companies, urged the CMS to provide access to aducanumab without excess restrictions.
There already are concerns among drug makers about CMS efforts “to impose new coverage barriers – and, in particular, coverage with evidence development,” Crystal Kuntz, vice president of policy and research at BIO, and Andy Cosgrove, the organization’s senior director for policy and research, noted in a July 2021 comment about the aducanumab review.
Medicare should instead continue to provide access to medicines for indications that the FDA has approved, with additional flexibility for off-label indications of cancer drugs, they noted. “We believe this should continue to be the case, to ensure that vulnerable Medicare beneficiaries have necessary access to life-altering and lifesaving medications,” the BIO officials wrote.
However, the CMS also received many pleas from physicians asking the agency to limit use of aducanumab at least until there is evidence that it produces a significant clinical benefit.
In a press release, Howard Fillit, MD, cofounder and chief science officer of the Alzheimer’s Drug Discovery Foundation, applauded the decision, describing it as “the right call.
“This decision supports conducting additional clinical trials, which are needed to obtain further insights into the clinical efficacy and safety profile of this drug in real-world populations. This decision has implications for other drugs in this class in late-stage development. If these trials show more clear and robust clinical efficacy, then it is possible the FDA will give these amyloid monoclonal antibodies full approval, and Medicare would be likely to provide full payment,” he added.
A version of this article first appeared on Medscape.com.
, federal officials announced Jan. 11.
On Dec. 20, 2021, Biogen announced a plan to reduce the annual U.S. cost of the drug by 50% – from $56,000 to $28,200 – as Centers for Medicare & Medicaid Services officials were deciding on Medicare’s coverage policy for the medication.
In making its proposed coverage decision, the CMS announced it will pay for aducanumab, a monoclonal antibody, under its coverage-with-evidence-development (CED) mechanism. In making its decision, the CMS approached aducanumab as the first of a potential new class of monoclonal antibodies for the treatment of Alzheimer’s disease. Food and Drug Administration–approved drugs in this class would be covered for those with Medicare only if they are enrolled in qualifying clinical trials, the CMS said. The agency will accept public comments on this decision for 30 days.
In a statement, CMS Administrator Chiquita Brooks-LaSure said the agency is “committed to providing the American public with a clear, trusted, evidence-based decision that is made only after a thorough analysis of public feedback on the benefits and risks of coverage for Medicare patients.”
As previously reported, the FDA approved aducanumab on June 7, 2021, via an accelerated approval process. The approval, which set off a firestorm of controversy that included resignations of three FDA Peripheral and Central Nervous System Drugs Advisory Committee panel members, was granted based on the medication’s ability to reduce beta-amyloid plaque.
Under the accelerated approval mechanism, Biogen still must deliver solid scientific proof that aducanumab has clinically significant disease-modifying effects. However, the final evidence won’t be in any time soon. In its approval letter, the FDA set a 2030 deadline for a final report on this research.
‘Unusual but appropriate’ step
The Medicare decision marks something of a shift in the agency’s approach to paying for medications. On a call with reporters, Tamara Syrek Jensen, JD, director of CMS’ Coverage and Analysis Group, admitted that the agency had taken an “unusual but appropriate” step in trying to set a national policy regarding payment for a drug.
On the same call, Lee Fleisher, MD, CMS’ chief medical officer, addressed the challenges presented by aducanumab, given the serious need for treatments for Alzheimer’s disease. “As a practicing physician, I cannot overemphasize the need to understand the risks and benefits of a given treatment in order to better inform patients and their families,” Dr. Fleisher said. “We do know based on some of the evidence that there may be potential promise with this treatment. That’s why it is critical for us to pursue additional scientific evidence.”
The coverage-with-evidence program will allow Medicare to aid in gathering data, while protecting patients, Dr. Fleisher noted.
“CMS is using its authority provided by Congress to determine if the drug is considered reasonable and necessary, meaning that the benefits of improvement of cognition outweigh the harms in the Medicare population,” Dr. Fleisher said.
Biogen disappointed
Cambridge, Mass.–based Biogen urged the CMS to reconsider its approach to payment for aducanumab. In a statement, the company said Medicare should cover “the class of amyloid-directed therapies with the populations studied in the respective clinical trials and guided by expert recommendations for appropriate use.
“We believe Alzheimer’s patients should have access consistent with other therapies with FDA accelerated approval,” Biogen said in the release.
In the company’s view, the CED approach will “significantly limit patient access to an FDA-approved treatment, especially for underserved patients as evidenced in other CED determinations.
“CEDs can take months to years to initiate, and hundreds of Alzheimer’s patients – the majority of whom are Medicare beneficiaries – are progressing each day from mild to moderate disease stages, where treatment may no longer be an option,” Biogen said.
Drug makers had been worried about CMS opting for CED even before the draft decision was unveiled.
Others weigh in
BIO, the trade group for biotechnology companies, urged the CMS to provide access to aducanumab without excess restrictions.
There already are concerns among drug makers about CMS efforts “to impose new coverage barriers – and, in particular, coverage with evidence development,” Crystal Kuntz, vice president of policy and research at BIO, and Andy Cosgrove, the organization’s senior director for policy and research, noted in a July 2021 comment about the aducanumab review.
Medicare should instead continue to provide access to medicines for indications that the FDA has approved, with additional flexibility for off-label indications of cancer drugs, they noted. “We believe this should continue to be the case, to ensure that vulnerable Medicare beneficiaries have necessary access to life-altering and lifesaving medications,” the BIO officials wrote.
However, the CMS also received many pleas from physicians asking the agency to limit use of aducanumab at least until there is evidence that it produces a significant clinical benefit.
In a press release, Howard Fillit, MD, cofounder and chief science officer of the Alzheimer’s Drug Discovery Foundation, applauded the decision, describing it as “the right call.
“This decision supports conducting additional clinical trials, which are needed to obtain further insights into the clinical efficacy and safety profile of this drug in real-world populations. This decision has implications for other drugs in this class in late-stage development. If these trials show more clear and robust clinical efficacy, then it is possible the FDA will give these amyloid monoclonal antibodies full approval, and Medicare would be likely to provide full payment,” he added.
A version of this article first appeared on Medscape.com.
Study finds sharp drop in opioid scripts among most specialties
The volume of prescription opioids dispensed at retail pharmacies in the United States dropped by 21% in recent years amid efforts to reduce unnecessary use of the painkillers, but the rate of decline varied greatly among types of patients and by type of clinician, a study found.
In a brief report published by Annals of Internal Medicine, researchers from the nonprofit RAND Corp reported an analysis of opioid prescriptions from two periods, 2008-2009 and 2017-2018.
The researchers sought to assess total opioid use rather than simply track the number of pills dispensed. So they used days’ supply and total daily dose to calculate per capita morphine milligram equivalents (MME) for opioid prescriptions, write Bradley D. Stein, MD, PhD, MPH, the study’s lead author and a senior physician researcher at RAND Corp, and his coauthors in their paper.
For the study, the researchers used data from the consulting firm IQVIA, which they say covers about 90% of U.S. prescriptions. Total opioid volume per capita by prescriptions filled in retail pharmacies decreased from 951.4 MME in 2008-2009 to 749.3 MME in 2017-2018, Dr. Stein’s group found.
(In 2020, IQVIA separately said that prescription opioid use per adult in this country rose from an average of 16 pills, or 134 MMEs, in 1992 to a peak of about 55 pills a person, or 790 MMEs, in 2011. By 2019, opioid use per adult had declined to 29 pills and 366 MMEs per capita.)
The RAND report found substantial variation in opioid volume by type of insurance, including a 41.5% decline (636.5 MME to 372.6 MME) among people covered by commercial health plans. That exceeded the 27.7% drop seen for people enrolled in Medicaid (646.8 MME to 467.7 MME). The decline was smaller (17.5%; 2,780.2 MME to 2,294.2 MME) for those on Medicare, who as a group used the most opioids.
‘Almost functions as a Rorschach test’
The causes of the decline are easy to guess, although definitive conclusions are impossible, Dr. Stein told this news organization.
Significant work has been done in recent years to change attitudes about opioid prescriptions by physicians, researchers, and lawmakers. Aggressive promotion of prescription painkillers, particularly Purdue Pharma’s OxyContin, in the 1990s, is widely cited as the triggering event for the national opioid crisis.
In response, states created databases known as prescription drug monitoring programs. The Centers for Disease Control and Prevention in 2016 issued guidelines intended to curb unnecessary use of opioids. The guidelines noted that other medicines could treat chronic pain without raising the risk of addiction. The Choosing Wisely campaign, run by a foundation of the American Board of Internal Medicine, also offered recommendations about limiting use of opioids. And insurers have restricted access to opioids through the prior authorization process. As a result, researchers will make their own guesses at the causes of the decline in opioid prescriptions, based on their own experiences and research interests, Dr. Stein said.
“It almost functions as a Rorschach test,” he said.
Dr. Stein’s group also looked at trends among medical specialties. They found the largest reduction between 2008-2009 and 2017-2018 among emergency physicians (70.5% drop from 99,254.5 MME to 29,234.3 MME), psychiatrists (67.2% drop from 50,464.3 MME to 16,533.0 MME) and oncologists (59.5% drop from 51,731.2 MME to 20,941.4).
Among surgeons, the RAND researchers found a drop of 49.3% from 220,764.6 to 111,904.4. Among dentists, they found a drop of 41.3% from 22,345.3 to 13,126.1.
Among pain specialists, they found a drop of 15.4% from 1,020,808.4 MME to 863,140.7 MME.
Among adult primary care clinicians, Dr. Stein and his colleagues found a drop of 40% from 651,489.4 MME in 2008-2009 to 390,841.0 MME in 2017-2018.
However, one of the groups tracked in the study increased the volume of opioid prescriptions written: advanced practice providers, among whom scripts for the drugs rose 22.7%, from 112,873.9 MME to 138,459.3 MME.
Dr. Stein said he suspects that this gain reflects a change in the nature of the practice of primary care, with nurse practitioners and physician assistants taking more active roles in treatment of patients. Some of the reduction seen among primary care clinicians who treat adults may reflect a shift in which medical personnel in a practice write the opioid prescriptions.
Still, the trends in general seen by Dr. Stein and coauthors are encouraging, even if further study of these patterns is needed, he said.
“This is one of those papers that I think potentially raises as many questions as it provides answers for,” he said.
What’s missing
Maya Hambright, MD, a family medicine physician in New York’s Hudson Valley, who has been working mainly in addiction in response to the opioid overdose crisis, observed that the drop in total prescribed volume of prescription painkillers does not necessarily translate into a reduction in use of opioids
“No one is taking fewer opioids,” Dr. Hambright told this news organization. “I can say that comfortably. They are just getting them from other sources.”
CDC data support Dr. Hambright’s view.
An estimated 100,306 people in the United States died of a drug overdose in the 12 months that ended in April 2021, an increase of 28.5% from the 78,056 deaths during the same period the year before, according to the CDC.
Dr. Hambright said more physicians need to be involved in prescribing medication-assisted treatment (MAT).
The federal government has in the past year loosened restrictions on a requirement, known as an X waiver. Certain clinicians have been exempted from training requirements, as explained in the frequently asked questions page on the Substance Abuse and Mental Health Services Administration website.
SAMHSA says legislation is required to eliminate the waiver. As of Dec. 30, 2021, more than half of the members of the U.S. House of Representatives were listed as sponsors of the Mainstreaming Addiction Treatment (MAT) Act (HR 1384), which would end the need for X waivers. The bill has the backing of 187 Democrats and 43 Republicans.
At this time, too many physicians shy away from offering MAT, Dr. Hambright said.
“People are still scared of it,” she said. “People don’t want to deal with addicts.”
But Dr. Hambright said it’s well worth the initial time invested in having the needed conversations with patients about MAT.
“Afterwards, it’s so straightforward. People feel better. They’re healthier. It’s amazing,” she said. “You’re changing lives.”
The research was supported by grants from the National Institutes of Health. Dr. Stein and coauthors reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The volume of prescription opioids dispensed at retail pharmacies in the United States dropped by 21% in recent years amid efforts to reduce unnecessary use of the painkillers, but the rate of decline varied greatly among types of patients and by type of clinician, a study found.
In a brief report published by Annals of Internal Medicine, researchers from the nonprofit RAND Corp reported an analysis of opioid prescriptions from two periods, 2008-2009 and 2017-2018.
The researchers sought to assess total opioid use rather than simply track the number of pills dispensed. So they used days’ supply and total daily dose to calculate per capita morphine milligram equivalents (MME) for opioid prescriptions, write Bradley D. Stein, MD, PhD, MPH, the study’s lead author and a senior physician researcher at RAND Corp, and his coauthors in their paper.
For the study, the researchers used data from the consulting firm IQVIA, which they say covers about 90% of U.S. prescriptions. Total opioid volume per capita by prescriptions filled in retail pharmacies decreased from 951.4 MME in 2008-2009 to 749.3 MME in 2017-2018, Dr. Stein’s group found.
(In 2020, IQVIA separately said that prescription opioid use per adult in this country rose from an average of 16 pills, or 134 MMEs, in 1992 to a peak of about 55 pills a person, or 790 MMEs, in 2011. By 2019, opioid use per adult had declined to 29 pills and 366 MMEs per capita.)
The RAND report found substantial variation in opioid volume by type of insurance, including a 41.5% decline (636.5 MME to 372.6 MME) among people covered by commercial health plans. That exceeded the 27.7% drop seen for people enrolled in Medicaid (646.8 MME to 467.7 MME). The decline was smaller (17.5%; 2,780.2 MME to 2,294.2 MME) for those on Medicare, who as a group used the most opioids.
‘Almost functions as a Rorschach test’
The causes of the decline are easy to guess, although definitive conclusions are impossible, Dr. Stein told this news organization.
Significant work has been done in recent years to change attitudes about opioid prescriptions by physicians, researchers, and lawmakers. Aggressive promotion of prescription painkillers, particularly Purdue Pharma’s OxyContin, in the 1990s, is widely cited as the triggering event for the national opioid crisis.
In response, states created databases known as prescription drug monitoring programs. The Centers for Disease Control and Prevention in 2016 issued guidelines intended to curb unnecessary use of opioids. The guidelines noted that other medicines could treat chronic pain without raising the risk of addiction. The Choosing Wisely campaign, run by a foundation of the American Board of Internal Medicine, also offered recommendations about limiting use of opioids. And insurers have restricted access to opioids through the prior authorization process. As a result, researchers will make their own guesses at the causes of the decline in opioid prescriptions, based on their own experiences and research interests, Dr. Stein said.
“It almost functions as a Rorschach test,” he said.
Dr. Stein’s group also looked at trends among medical specialties. They found the largest reduction between 2008-2009 and 2017-2018 among emergency physicians (70.5% drop from 99,254.5 MME to 29,234.3 MME), psychiatrists (67.2% drop from 50,464.3 MME to 16,533.0 MME) and oncologists (59.5% drop from 51,731.2 MME to 20,941.4).
Among surgeons, the RAND researchers found a drop of 49.3% from 220,764.6 to 111,904.4. Among dentists, they found a drop of 41.3% from 22,345.3 to 13,126.1.
Among pain specialists, they found a drop of 15.4% from 1,020,808.4 MME to 863,140.7 MME.
Among adult primary care clinicians, Dr. Stein and his colleagues found a drop of 40% from 651,489.4 MME in 2008-2009 to 390,841.0 MME in 2017-2018.
However, one of the groups tracked in the study increased the volume of opioid prescriptions written: advanced practice providers, among whom scripts for the drugs rose 22.7%, from 112,873.9 MME to 138,459.3 MME.
Dr. Stein said he suspects that this gain reflects a change in the nature of the practice of primary care, with nurse practitioners and physician assistants taking more active roles in treatment of patients. Some of the reduction seen among primary care clinicians who treat adults may reflect a shift in which medical personnel in a practice write the opioid prescriptions.
Still, the trends in general seen by Dr. Stein and coauthors are encouraging, even if further study of these patterns is needed, he said.
“This is one of those papers that I think potentially raises as many questions as it provides answers for,” he said.
What’s missing
Maya Hambright, MD, a family medicine physician in New York’s Hudson Valley, who has been working mainly in addiction in response to the opioid overdose crisis, observed that the drop in total prescribed volume of prescription painkillers does not necessarily translate into a reduction in use of opioids
“No one is taking fewer opioids,” Dr. Hambright told this news organization. “I can say that comfortably. They are just getting them from other sources.”
CDC data support Dr. Hambright’s view.
An estimated 100,306 people in the United States died of a drug overdose in the 12 months that ended in April 2021, an increase of 28.5% from the 78,056 deaths during the same period the year before, according to the CDC.
Dr. Hambright said more physicians need to be involved in prescribing medication-assisted treatment (MAT).
The federal government has in the past year loosened restrictions on a requirement, known as an X waiver. Certain clinicians have been exempted from training requirements, as explained in the frequently asked questions page on the Substance Abuse and Mental Health Services Administration website.
SAMHSA says legislation is required to eliminate the waiver. As of Dec. 30, 2021, more than half of the members of the U.S. House of Representatives were listed as sponsors of the Mainstreaming Addiction Treatment (MAT) Act (HR 1384), which would end the need for X waivers. The bill has the backing of 187 Democrats and 43 Republicans.
At this time, too many physicians shy away from offering MAT, Dr. Hambright said.
“People are still scared of it,” she said. “People don’t want to deal with addicts.”
But Dr. Hambright said it’s well worth the initial time invested in having the needed conversations with patients about MAT.
“Afterwards, it’s so straightforward. People feel better. They’re healthier. It’s amazing,” she said. “You’re changing lives.”
The research was supported by grants from the National Institutes of Health. Dr. Stein and coauthors reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The volume of prescription opioids dispensed at retail pharmacies in the United States dropped by 21% in recent years amid efforts to reduce unnecessary use of the painkillers, but the rate of decline varied greatly among types of patients and by type of clinician, a study found.
In a brief report published by Annals of Internal Medicine, researchers from the nonprofit RAND Corp reported an analysis of opioid prescriptions from two periods, 2008-2009 and 2017-2018.
The researchers sought to assess total opioid use rather than simply track the number of pills dispensed. So they used days’ supply and total daily dose to calculate per capita morphine milligram equivalents (MME) for opioid prescriptions, write Bradley D. Stein, MD, PhD, MPH, the study’s lead author and a senior physician researcher at RAND Corp, and his coauthors in their paper.
For the study, the researchers used data from the consulting firm IQVIA, which they say covers about 90% of U.S. prescriptions. Total opioid volume per capita by prescriptions filled in retail pharmacies decreased from 951.4 MME in 2008-2009 to 749.3 MME in 2017-2018, Dr. Stein’s group found.
(In 2020, IQVIA separately said that prescription opioid use per adult in this country rose from an average of 16 pills, or 134 MMEs, in 1992 to a peak of about 55 pills a person, or 790 MMEs, in 2011. By 2019, opioid use per adult had declined to 29 pills and 366 MMEs per capita.)
The RAND report found substantial variation in opioid volume by type of insurance, including a 41.5% decline (636.5 MME to 372.6 MME) among people covered by commercial health plans. That exceeded the 27.7% drop seen for people enrolled in Medicaid (646.8 MME to 467.7 MME). The decline was smaller (17.5%; 2,780.2 MME to 2,294.2 MME) for those on Medicare, who as a group used the most opioids.
‘Almost functions as a Rorschach test’
The causes of the decline are easy to guess, although definitive conclusions are impossible, Dr. Stein told this news organization.
Significant work has been done in recent years to change attitudes about opioid prescriptions by physicians, researchers, and lawmakers. Aggressive promotion of prescription painkillers, particularly Purdue Pharma’s OxyContin, in the 1990s, is widely cited as the triggering event for the national opioid crisis.
In response, states created databases known as prescription drug monitoring programs. The Centers for Disease Control and Prevention in 2016 issued guidelines intended to curb unnecessary use of opioids. The guidelines noted that other medicines could treat chronic pain without raising the risk of addiction. The Choosing Wisely campaign, run by a foundation of the American Board of Internal Medicine, also offered recommendations about limiting use of opioids. And insurers have restricted access to opioids through the prior authorization process. As a result, researchers will make their own guesses at the causes of the decline in opioid prescriptions, based on their own experiences and research interests, Dr. Stein said.
“It almost functions as a Rorschach test,” he said.
Dr. Stein’s group also looked at trends among medical specialties. They found the largest reduction between 2008-2009 and 2017-2018 among emergency physicians (70.5% drop from 99,254.5 MME to 29,234.3 MME), psychiatrists (67.2% drop from 50,464.3 MME to 16,533.0 MME) and oncologists (59.5% drop from 51,731.2 MME to 20,941.4).
Among surgeons, the RAND researchers found a drop of 49.3% from 220,764.6 to 111,904.4. Among dentists, they found a drop of 41.3% from 22,345.3 to 13,126.1.
Among pain specialists, they found a drop of 15.4% from 1,020,808.4 MME to 863,140.7 MME.
Among adult primary care clinicians, Dr. Stein and his colleagues found a drop of 40% from 651,489.4 MME in 2008-2009 to 390,841.0 MME in 2017-2018.
However, one of the groups tracked in the study increased the volume of opioid prescriptions written: advanced practice providers, among whom scripts for the drugs rose 22.7%, from 112,873.9 MME to 138,459.3 MME.
Dr. Stein said he suspects that this gain reflects a change in the nature of the practice of primary care, with nurse practitioners and physician assistants taking more active roles in treatment of patients. Some of the reduction seen among primary care clinicians who treat adults may reflect a shift in which medical personnel in a practice write the opioid prescriptions.
Still, the trends in general seen by Dr. Stein and coauthors are encouraging, even if further study of these patterns is needed, he said.
“This is one of those papers that I think potentially raises as many questions as it provides answers for,” he said.
What’s missing
Maya Hambright, MD, a family medicine physician in New York’s Hudson Valley, who has been working mainly in addiction in response to the opioid overdose crisis, observed that the drop in total prescribed volume of prescription painkillers does not necessarily translate into a reduction in use of opioids
“No one is taking fewer opioids,” Dr. Hambright told this news organization. “I can say that comfortably. They are just getting them from other sources.”
CDC data support Dr. Hambright’s view.
An estimated 100,306 people in the United States died of a drug overdose in the 12 months that ended in April 2021, an increase of 28.5% from the 78,056 deaths during the same period the year before, according to the CDC.
Dr. Hambright said more physicians need to be involved in prescribing medication-assisted treatment (MAT).
The federal government has in the past year loosened restrictions on a requirement, known as an X waiver. Certain clinicians have been exempted from training requirements, as explained in the frequently asked questions page on the Substance Abuse and Mental Health Services Administration website.
SAMHSA says legislation is required to eliminate the waiver. As of Dec. 30, 2021, more than half of the members of the U.S. House of Representatives were listed as sponsors of the Mainstreaming Addiction Treatment (MAT) Act (HR 1384), which would end the need for X waivers. The bill has the backing of 187 Democrats and 43 Republicans.
At this time, too many physicians shy away from offering MAT, Dr. Hambright said.
“People are still scared of it,” she said. “People don’t want to deal with addicts.”
But Dr. Hambright said it’s well worth the initial time invested in having the needed conversations with patients about MAT.
“Afterwards, it’s so straightforward. People feel better. They’re healthier. It’s amazing,” she said. “You’re changing lives.”
The research was supported by grants from the National Institutes of Health. Dr. Stein and coauthors reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Medicaid implements waivers for some clinical trial coverage
Federal officials will allow some flexibility in meeting new requirements on covering the costs of clinical trials for people enrolled in Medicaid, seeking to accommodate states where legislatures will not meet in time to make needed changes in rules.
Congress in 2020 ordered U.S. states to have their Medicaid programs cover expenses related to participation in certain clinical trials, a move that was hailed by the American Society of Clinical Oncology (ASCO) and other groups as a boost to trials as well as to patients with serious illness who have lower incomes.
The mandate went into effect on Jan. 1, but the Centers for Medicare & Medicaid Services will allow accommodations in terms of implementation time for states that have not yet been able to make needed legislative changes, Daniel Tsai, deputy administrator and director of the Center for Medicaid and CHIP Services, wrote in a Dec. 7 letter. Mr. Tsai’s letter doesn’t mention specific states. The CMS did not immediately respond to a request seeking information on the states expected to apply for waivers.
Medicaid has in recent years been a rare large U.S. insurance program that does not cover the costs of clinical trials. The Affordable Care Act of 2010 mandated this coverage for people in private insurance plans. The federal government in 2000 decided that Medicare would do so.
‘A hidden opportunity’
A perspective article last May in the New England Journal of Medicine referred to the new Medicaid mandate on clinical trials as a “hidden opportunity,” referring to its genesis as an add-on in a massive federal spending package enacted in December 2020.
In the article, Samuel U. Takvorian, MD, MSHP, of the University of Pennsylvania, Philadelphia, and coauthors noted that rates of participation in clinical trials remain low for racial and ethnic minority groups, due in part to the lack of Medicaid coverage.
“For example, non-Hispanic White patients are nearly twice as likely as Black patients and three times as likely as Hispanic patients to enroll in cancer clinical trials – a gap that has widened over time,” Dr. Takvorian and coauthors wrote. “Inequities in enrollment have also manifested during the COVID-19 pandemic, which has disproportionately affected non-White patients, without their commensurate representation in trials of COVID-19 therapeutics.”
In October, researchers from the Arthur G. James Cancer Hospital and Ohio State University, Columbus, published results of a retrospective study of patients with stage I-IV pancreatic cancer that also found inequities in enrollment. Mariam F. Eskander, MD, MPH, and coauthors reported what they found by examining records for 1,127 patients (0.4%) enrolled in clinical trials and 301,340 (99.6%) who did not enroll. They found that enrollment in trials increased over the study period, but not for Black patients or patients on Medicaid.
In an interview, Dr. Eskander said the new Medicaid policy will remove a major obstacle to participation in clinical trials. An oncologist, Dr. Eskander said she is looking forward to being able to help more of her patients get access to experimental medicines and treatments.
But that may not be enough to draw more people with low incomes into these studies, said Dr. Eskander, who is now at Rutgers Cancer Institute of New Jersey in New Brunswick. She urges greater use of patient navigators to help people on Medicaid understand the resources available to them, as well as broad use of Medicaid’s nonemergency medical transportation (NEMT) benefit.
“Some patients will be offered clinical trial enrollment and some will accept, but I really worry about the challenges low-income people face with things like transportation and getting time off work,” she said.
A version of this article first appeared on Medscape.com.
Federal officials will allow some flexibility in meeting new requirements on covering the costs of clinical trials for people enrolled in Medicaid, seeking to accommodate states where legislatures will not meet in time to make needed changes in rules.
Congress in 2020 ordered U.S. states to have their Medicaid programs cover expenses related to participation in certain clinical trials, a move that was hailed by the American Society of Clinical Oncology (ASCO) and other groups as a boost to trials as well as to patients with serious illness who have lower incomes.
The mandate went into effect on Jan. 1, but the Centers for Medicare & Medicaid Services will allow accommodations in terms of implementation time for states that have not yet been able to make needed legislative changes, Daniel Tsai, deputy administrator and director of the Center for Medicaid and CHIP Services, wrote in a Dec. 7 letter. Mr. Tsai’s letter doesn’t mention specific states. The CMS did not immediately respond to a request seeking information on the states expected to apply for waivers.
Medicaid has in recent years been a rare large U.S. insurance program that does not cover the costs of clinical trials. The Affordable Care Act of 2010 mandated this coverage for people in private insurance plans. The federal government in 2000 decided that Medicare would do so.
‘A hidden opportunity’
A perspective article last May in the New England Journal of Medicine referred to the new Medicaid mandate on clinical trials as a “hidden opportunity,” referring to its genesis as an add-on in a massive federal spending package enacted in December 2020.
In the article, Samuel U. Takvorian, MD, MSHP, of the University of Pennsylvania, Philadelphia, and coauthors noted that rates of participation in clinical trials remain low for racial and ethnic minority groups, due in part to the lack of Medicaid coverage.
“For example, non-Hispanic White patients are nearly twice as likely as Black patients and three times as likely as Hispanic patients to enroll in cancer clinical trials – a gap that has widened over time,” Dr. Takvorian and coauthors wrote. “Inequities in enrollment have also manifested during the COVID-19 pandemic, which has disproportionately affected non-White patients, without their commensurate representation in trials of COVID-19 therapeutics.”
In October, researchers from the Arthur G. James Cancer Hospital and Ohio State University, Columbus, published results of a retrospective study of patients with stage I-IV pancreatic cancer that also found inequities in enrollment. Mariam F. Eskander, MD, MPH, and coauthors reported what they found by examining records for 1,127 patients (0.4%) enrolled in clinical trials and 301,340 (99.6%) who did not enroll. They found that enrollment in trials increased over the study period, but not for Black patients or patients on Medicaid.
In an interview, Dr. Eskander said the new Medicaid policy will remove a major obstacle to participation in clinical trials. An oncologist, Dr. Eskander said she is looking forward to being able to help more of her patients get access to experimental medicines and treatments.
But that may not be enough to draw more people with low incomes into these studies, said Dr. Eskander, who is now at Rutgers Cancer Institute of New Jersey in New Brunswick. She urges greater use of patient navigators to help people on Medicaid understand the resources available to them, as well as broad use of Medicaid’s nonemergency medical transportation (NEMT) benefit.
“Some patients will be offered clinical trial enrollment and some will accept, but I really worry about the challenges low-income people face with things like transportation and getting time off work,” she said.
A version of this article first appeared on Medscape.com.
Federal officials will allow some flexibility in meeting new requirements on covering the costs of clinical trials for people enrolled in Medicaid, seeking to accommodate states where legislatures will not meet in time to make needed changes in rules.
Congress in 2020 ordered U.S. states to have their Medicaid programs cover expenses related to participation in certain clinical trials, a move that was hailed by the American Society of Clinical Oncology (ASCO) and other groups as a boost to trials as well as to patients with serious illness who have lower incomes.
The mandate went into effect on Jan. 1, but the Centers for Medicare & Medicaid Services will allow accommodations in terms of implementation time for states that have not yet been able to make needed legislative changes, Daniel Tsai, deputy administrator and director of the Center for Medicaid and CHIP Services, wrote in a Dec. 7 letter. Mr. Tsai’s letter doesn’t mention specific states. The CMS did not immediately respond to a request seeking information on the states expected to apply for waivers.
Medicaid has in recent years been a rare large U.S. insurance program that does not cover the costs of clinical trials. The Affordable Care Act of 2010 mandated this coverage for people in private insurance plans. The federal government in 2000 decided that Medicare would do so.
‘A hidden opportunity’
A perspective article last May in the New England Journal of Medicine referred to the new Medicaid mandate on clinical trials as a “hidden opportunity,” referring to its genesis as an add-on in a massive federal spending package enacted in December 2020.
In the article, Samuel U. Takvorian, MD, MSHP, of the University of Pennsylvania, Philadelphia, and coauthors noted that rates of participation in clinical trials remain low for racial and ethnic minority groups, due in part to the lack of Medicaid coverage.
“For example, non-Hispanic White patients are nearly twice as likely as Black patients and three times as likely as Hispanic patients to enroll in cancer clinical trials – a gap that has widened over time,” Dr. Takvorian and coauthors wrote. “Inequities in enrollment have also manifested during the COVID-19 pandemic, which has disproportionately affected non-White patients, without their commensurate representation in trials of COVID-19 therapeutics.”
In October, researchers from the Arthur G. James Cancer Hospital and Ohio State University, Columbus, published results of a retrospective study of patients with stage I-IV pancreatic cancer that also found inequities in enrollment. Mariam F. Eskander, MD, MPH, and coauthors reported what they found by examining records for 1,127 patients (0.4%) enrolled in clinical trials and 301,340 (99.6%) who did not enroll. They found that enrollment in trials increased over the study period, but not for Black patients or patients on Medicaid.
In an interview, Dr. Eskander said the new Medicaid policy will remove a major obstacle to participation in clinical trials. An oncologist, Dr. Eskander said she is looking forward to being able to help more of her patients get access to experimental medicines and treatments.
But that may not be enough to draw more people with low incomes into these studies, said Dr. Eskander, who is now at Rutgers Cancer Institute of New Jersey in New Brunswick. She urges greater use of patient navigators to help people on Medicaid understand the resources available to them, as well as broad use of Medicaid’s nonemergency medical transportation (NEMT) benefit.
“Some patients will be offered clinical trial enrollment and some will accept, but I really worry about the challenges low-income people face with things like transportation and getting time off work,” she said.
A version of this article first appeared on Medscape.com.
Califf plans work on opioids, accelerated approvals on return to FDA
Robert M. Califf, MD, plans to take a close look at federal policies on opioid prescriptions in his expected second turn as the top U.S. regulator of medical products, as well as keep closer tabs on the performance of drugs cleared with accelerated approvals.

Dr. Califf on Tuesday fielded questions at a Senate hearing about his nomination by President Joe Biden to serve as administrator of the U.S. Food and Drug Administration, a role in which he served in the Obama administration. He also spoke about the need to bolster the nation’s ability to maintain an adequate supply of key medical products, including drugs.
Members of the Senate Health, Education, Labor and Pensions Committee, which is handling Dr. Califf’s nomination, were largely cordial and supportive during the hearing. Sen. Patty Murray (D-Wash.), the committee chair, and the panel’s top Republican, Sen. Richard Burr of North Carolina, addressed Dr. Califf during the hearing as if he would soon serve again as the FDA’s leader. Both were among the senators who voted 89-4 to confirm Dr. Califf in a February 2016 vote.
Dr. Califf “was previously confirmed to lead FDA in an overwhelming bipartisan vote, and I look forward to working with him again to ensure FDA continues to protect families across the country, uphold the gold standard of safety and effectiveness, and put science and data first,” Sen. Murray said.
Less enthusiastic about Dr. Califf was Sen. Bernie Sanders (I-VT), who was among the seven senators who did not vote on Dr. Califf’s nomination in 2016.
Sen. Sanders objected in 2016 to Dr. Califf’s ties to the pharmaceutical industry, and he did so again Tuesday. A noted leader in conducting clinical trials, Dr. Califf has worked with many drugmakers. But at the hearing, Dr. Califf said he concurs with Sen. Sanders on an idea strongly opposed by the pharmaceutical industry.
In response to Sen. Sanders’ question, Dr. Califf said he already is “on record as being in favor of Medicare negotiating with the industry on prices.”
The FDA would not take direct part in negotiations, as this work would be handled by the Centers for Medicare & Medicaid Services. Democrats want to give Medicare some negotiating authority through their sweeping Build Back Better Act.
People in the United States are dismayed over both the cost of prescription drugs and the widespread distribution of prescription painkillers that helped fuel the current opioid epidemic, Sen. Sanders told Dr. Califf. Many people will be concerned about an FDA commissioner who has benefited from close ties to the industry, Sen. Sanders said.
“How are they going to believe that you’re going to be an independent and strong voice against this enormously powerful, special interest?” Sen. Sanders asked.
“I’m totally with you on the concept that the price of pharmaceuticals is way too high in this country,” Dr. Califf said in reply.
Dr. Califf was paid $2.7 million in salary and bonus by Verily Life Sciences, the biomedical research organization operated by Alphabet, parent company of Google, according to his federal financial disclosure. He also reported holding board positions with pharmaceutical companies AmyriAD and Centessa Pharmaceuticals.
Bloomberg Government reported that Dr. Califf has ties to about 16 other research organizations and biotech companies. Bloomberg Government also said that, in his earlier FDA service, Dr. Califf kept a whiteboard in his office that listed all the activities and projects that required his recusal, citing as a source Howard Sklamberg, who was a deputy commissioner under Dr. Califf.
“He was very, very, very careful,” Mr. Sklamberg, who’s now an attorney at Arnold & Porter LLP, told Bloomberg Government.
‘Work to do’ on opioids
Senators looped back repeatedly to the topic of opioids during Dr. Califf’s hearing, reflecting deep concerns about the FDA’s efforts to warn of the risks of prescription painkillers.
There were an estimated 100,306 drug overdose deaths in the United States in the 12 months ending in April, an increase of 28.5% from the 78,056 deaths during the same period the year before, according to the Centers for Disease Control and Prevention.
Dr. Califf said he plans to focus on what information the FDA conveys to the public about the risks of prescription painkillers, including a look at what the labels for these products say.
“I am committed to do a comprehensive review of the status of opioids, early in my tenure,” Dr. Califf said.
Dr. Califf indicated that physicians are still too quick to provide excess doses of these medicines, despite years of efforts to restrain their use. He said he knows relatives who were given 30-day prescriptions for opioids after minor surgery.
“So I know we have work to do,” Dr. Califf said.
Concerns about the FDA’s previous work in managing opioids has led to protests from a few Democratic senators about the prospect of President Biden nominating the acting FDA commissioner, Janet Woodcock, MD, for the permanent post.
At the hearing, Sen. Ben Ray Luján (D-NM) raised the case of the FDA’s approval of the powerful Zohydro painkiller. The agency approved that drug despite an 11-2 vote against it by the FDA’s Anesthetic and Analgesic Drug Products Advisory Committee.
Sen. Luján asked Dr. Califf what he would do if an FDA advisory committee voted “overwhelmingly” against recommending approval of a medicine, as happened in the Zohydro case.
While not mentioned by Sen. Luján in this exchange during the hearing with Dr. Califf, the FDA staff’s rejection of recommendations of advisory committees has been a growing concern among researchers.
The agency last year approved aducanumab (Aduhelm, Biogen), a drug for Alzheimer’s disease, dismissing the advice of its Peripheral and Central Nervous System Drugs Advisory Committee. That decision triggered the resignation of several members of the panel. The FDA staff also earlier rejected the conclusion the majority of members of the same advisory committee offered in 2016 on eteplirsen (Exondys 51, Sarepta), a drug for Duchenne muscular dystrophy.
Dr. Califf told Sen. Luján he had done recent research into how often the FDA staff does not concur with the recommendations of an advisory committee. He said the FDA takes a different course of action in about 25% of cases. In about three-quarters of those cases, the FDA staff opts for a “more stringent” approach regarding allowing the public access to the drug, as opposed to a more generous one as seen in the Zohydro, Aduhelm, and Exondys 51 cases.
Still, Dr. Califf said that when there’s an 11-2 advisory committee vote against recommendation of a product, “the leaders at FDA really need to take a close look” at what’s happening.
Question on accelerated approvals
The FDA’s approval of aducanumab drew attention to a debate already underway about conditional clearances known as accelerated approvals.
The FDA has used this path since the 1990s to speed access to drugs for serious conditions. The trade-off for early access is that the agency sometimes makes the wrong call based on initial findings, and clears a medicine later found not to benefit patients as expected.
The FDA’s cancer division is in the midst of public efforts to address cases where drugmakers have not been able to deliver studies that support accelerated approvals of their oncology drugs. In addition, the Office of Inspector General of the U.S. Department of Health & Human Services announced in August that it is reviewing the FDA’s handling of the accelerated approval process.
At Tuesday’s hearing, Sen. Burr grilled Dr. Califf about how he would respond to calls to change how the FDA handles the accelerated-approval process.
“Can you commit to me and to patients who may rely on cutting-edge treatments that you will not support efforts to narrow this pathway or raise the bar for drugs to be approved under those pathways?” Burr asked Califf.
Dr. Califf responded by saying he was “a fan of accelerated approval – for the right conditions.”
Earlier, in his opening statement, Dr. Califf had said his mother benefited directly from the accelerated approval of new drugs for multiple myeloma. Dr. Califf told Sen. Burr that he had spent “countless hours with patient groups” and understands the need to speed the approval of medicines for serious diseases.
But the FDA also has to make sure it holds up its end of the bargain struck with accelerated approvals. This involves checking on how these medicines work once they are marketed.
“We’re accepting that there’s more uncertainty,” Dr. Califf said. “That means we’ve got to have a better system to evaluate these products as they’re used on the market. And I think there are ways that we can do that now. Technology is making this possible in ways that it just was not possible before.”
Worries about the medical supply chain
Sen. Susan Collins (R-Maine) asked Dr. Califf about the vulnerability of the U.S. medical system to disruptions of the supply chain. She raised concerns about China’s dominance in antibiotic manufacturing as an example. She asked if Congress could do more to encourage domestic manufacturing of medical supplies, such as by offering tax incentives.
Dr. Califf told Sen. Collins he shared her concern about the U.S. manufacturing of ingredients used in both branded and generic drugs. He said he recently has served on a committee of the National Academy of Medicine that is examining supply chain issues.
This committee will soon release a report with specific recommendations, Dr. Califf said.
“We don’t have enough competitive entities in what’s become sort of a commodity business” of drug manufacturing, Dr. Califf said. “So we need a number of steps to make the system more resilient.”
A version of this article first appeared on Medscape.com.
Robert M. Califf, MD, plans to take a close look at federal policies on opioid prescriptions in his expected second turn as the top U.S. regulator of medical products, as well as keep closer tabs on the performance of drugs cleared with accelerated approvals.
Dr. Califf on Tuesday fielded questions at a Senate hearing about his nomination by President Joe Biden to serve as administrator of the U.S. Food and Drug Administration, a role in which he served in the Obama administration. He also spoke about the need to bolster the nation’s ability to maintain an adequate supply of key medical products, including drugs.
Members of the Senate Health, Education, Labor and Pensions Committee, which is handling Dr. Califf’s nomination, were largely cordial and supportive during the hearing. Sen. Patty Murray (D-Wash.), the committee chair, and the panel’s top Republican, Sen. Richard Burr of North Carolina, addressed Dr. Califf during the hearing as if he would soon serve again as the FDA’s leader. Both were among the senators who voted 89-4 to confirm Dr. Califf in a February 2016 vote.
Dr. Califf “was previously confirmed to lead FDA in an overwhelming bipartisan vote, and I look forward to working with him again to ensure FDA continues to protect families across the country, uphold the gold standard of safety and effectiveness, and put science and data first,” Sen. Murray said.
Less enthusiastic about Dr. Califf was Sen. Bernie Sanders (I-VT), who was among the seven senators who did not vote on Dr. Califf’s nomination in 2016.
Sen. Sanders objected in 2016 to Dr. Califf’s ties to the pharmaceutical industry, and he did so again Tuesday. A noted leader in conducting clinical trials, Dr. Califf has worked with many drugmakers. But at the hearing, Dr. Califf said he concurs with Sen. Sanders on an idea strongly opposed by the pharmaceutical industry.
In response to Sen. Sanders’ question, Dr. Califf said he already is “on record as being in favor of Medicare negotiating with the industry on prices.”
The FDA would not take direct part in negotiations, as this work would be handled by the Centers for Medicare & Medicaid Services. Democrats want to give Medicare some negotiating authority through their sweeping Build Back Better Act.
People in the United States are dismayed over both the cost of prescription drugs and the widespread distribution of prescription painkillers that helped fuel the current opioid epidemic, Sen. Sanders told Dr. Califf. Many people will be concerned about an FDA commissioner who has benefited from close ties to the industry, Sen. Sanders said.
“How are they going to believe that you’re going to be an independent and strong voice against this enormously powerful, special interest?” Sen. Sanders asked.
“I’m totally with you on the concept that the price of pharmaceuticals is way too high in this country,” Dr. Califf said in reply.
Dr. Califf was paid $2.7 million in salary and bonus by Verily Life Sciences, the biomedical research organization operated by Alphabet, parent company of Google, according to his federal financial disclosure. He also reported holding board positions with pharmaceutical companies AmyriAD and Centessa Pharmaceuticals.
Bloomberg Government reported that Dr. Califf has ties to about 16 other research organizations and biotech companies. Bloomberg Government also said that, in his earlier FDA service, Dr. Califf kept a whiteboard in his office that listed all the activities and projects that required his recusal, citing as a source Howard Sklamberg, who was a deputy commissioner under Dr. Califf.
“He was very, very, very careful,” Mr. Sklamberg, who’s now an attorney at Arnold & Porter LLP, told Bloomberg Government.
‘Work to do’ on opioids
Senators looped back repeatedly to the topic of opioids during Dr. Califf’s hearing, reflecting deep concerns about the FDA’s efforts to warn of the risks of prescription painkillers.
There were an estimated 100,306 drug overdose deaths in the United States in the 12 months ending in April, an increase of 28.5% from the 78,056 deaths during the same period the year before, according to the Centers for Disease Control and Prevention.
Dr. Califf said he plans to focus on what information the FDA conveys to the public about the risks of prescription painkillers, including a look at what the labels for these products say.
“I am committed to do a comprehensive review of the status of opioids, early in my tenure,” Dr. Califf said.
Dr. Califf indicated that physicians are still too quick to provide excess doses of these medicines, despite years of efforts to restrain their use. He said he knows relatives who were given 30-day prescriptions for opioids after minor surgery.
“So I know we have work to do,” Dr. Califf said.
Concerns about the FDA’s previous work in managing opioids has led to protests from a few Democratic senators about the prospect of President Biden nominating the acting FDA commissioner, Janet Woodcock, MD, for the permanent post.
At the hearing, Sen. Ben Ray Luján (D-NM) raised the case of the FDA’s approval of the powerful Zohydro painkiller. The agency approved that drug despite an 11-2 vote against it by the FDA’s Anesthetic and Analgesic Drug Products Advisory Committee.
Sen. Luján asked Dr. Califf what he would do if an FDA advisory committee voted “overwhelmingly” against recommending approval of a medicine, as happened in the Zohydro case.
While not mentioned by Sen. Luján in this exchange during the hearing with Dr. Califf, the FDA staff’s rejection of recommendations of advisory committees has been a growing concern among researchers.
The agency last year approved aducanumab (Aduhelm, Biogen), a drug for Alzheimer’s disease, dismissing the advice of its Peripheral and Central Nervous System Drugs Advisory Committee. That decision triggered the resignation of several members of the panel. The FDA staff also earlier rejected the conclusion the majority of members of the same advisory committee offered in 2016 on eteplirsen (Exondys 51, Sarepta), a drug for Duchenne muscular dystrophy.
Dr. Califf told Sen. Luján he had done recent research into how often the FDA staff does not concur with the recommendations of an advisory committee. He said the FDA takes a different course of action in about 25% of cases. In about three-quarters of those cases, the FDA staff opts for a “more stringent” approach regarding allowing the public access to the drug, as opposed to a more generous one as seen in the Zohydro, Aduhelm, and Exondys 51 cases.
Still, Dr. Califf said that when there’s an 11-2 advisory committee vote against recommendation of a product, “the leaders at FDA really need to take a close look” at what’s happening.
Question on accelerated approvals
The FDA’s approval of aducanumab drew attention to a debate already underway about conditional clearances known as accelerated approvals.
The FDA has used this path since the 1990s to speed access to drugs for serious conditions. The trade-off for early access is that the agency sometimes makes the wrong call based on initial findings, and clears a medicine later found not to benefit patients as expected.
The FDA’s cancer division is in the midst of public efforts to address cases where drugmakers have not been able to deliver studies that support accelerated approvals of their oncology drugs. In addition, the Office of Inspector General of the U.S. Department of Health & Human Services announced in August that it is reviewing the FDA’s handling of the accelerated approval process.
At Tuesday’s hearing, Sen. Burr grilled Dr. Califf about how he would respond to calls to change how the FDA handles the accelerated-approval process.
“Can you commit to me and to patients who may rely on cutting-edge treatments that you will not support efforts to narrow this pathway or raise the bar for drugs to be approved under those pathways?” Burr asked Califf.
Dr. Califf responded by saying he was “a fan of accelerated approval – for the right conditions.”
Earlier, in his opening statement, Dr. Califf had said his mother benefited directly from the accelerated approval of new drugs for multiple myeloma. Dr. Califf told Sen. Burr that he had spent “countless hours with patient groups” and understands the need to speed the approval of medicines for serious diseases.
But the FDA also has to make sure it holds up its end of the bargain struck with accelerated approvals. This involves checking on how these medicines work once they are marketed.
“We’re accepting that there’s more uncertainty,” Dr. Califf said. “That means we’ve got to have a better system to evaluate these products as they’re used on the market. And I think there are ways that we can do that now. Technology is making this possible in ways that it just was not possible before.”
Worries about the medical supply chain
Sen. Susan Collins (R-Maine) asked Dr. Califf about the vulnerability of the U.S. medical system to disruptions of the supply chain. She raised concerns about China’s dominance in antibiotic manufacturing as an example. She asked if Congress could do more to encourage domestic manufacturing of medical supplies, such as by offering tax incentives.
Dr. Califf told Sen. Collins he shared her concern about the U.S. manufacturing of ingredients used in both branded and generic drugs. He said he recently has served on a committee of the National Academy of Medicine that is examining supply chain issues.
This committee will soon release a report with specific recommendations, Dr. Califf said.
“We don’t have enough competitive entities in what’s become sort of a commodity business” of drug manufacturing, Dr. Califf said. “So we need a number of steps to make the system more resilient.”
A version of this article first appeared on Medscape.com.
Robert M. Califf, MD, plans to take a close look at federal policies on opioid prescriptions in his expected second turn as the top U.S. regulator of medical products, as well as keep closer tabs on the performance of drugs cleared with accelerated approvals.
Dr. Califf on Tuesday fielded questions at a Senate hearing about his nomination by President Joe Biden to serve as administrator of the U.S. Food and Drug Administration, a role in which he served in the Obama administration. He also spoke about the need to bolster the nation’s ability to maintain an adequate supply of key medical products, including drugs.
Members of the Senate Health, Education, Labor and Pensions Committee, which is handling Dr. Califf’s nomination, were largely cordial and supportive during the hearing. Sen. Patty Murray (D-Wash.), the committee chair, and the panel’s top Republican, Sen. Richard Burr of North Carolina, addressed Dr. Califf during the hearing as if he would soon serve again as the FDA’s leader. Both were among the senators who voted 89-4 to confirm Dr. Califf in a February 2016 vote.
Dr. Califf “was previously confirmed to lead FDA in an overwhelming bipartisan vote, and I look forward to working with him again to ensure FDA continues to protect families across the country, uphold the gold standard of safety and effectiveness, and put science and data first,” Sen. Murray said.
Less enthusiastic about Dr. Califf was Sen. Bernie Sanders (I-VT), who was among the seven senators who did not vote on Dr. Califf’s nomination in 2016.
Sen. Sanders objected in 2016 to Dr. Califf’s ties to the pharmaceutical industry, and he did so again Tuesday. A noted leader in conducting clinical trials, Dr. Califf has worked with many drugmakers. But at the hearing, Dr. Califf said he concurs with Sen. Sanders on an idea strongly opposed by the pharmaceutical industry.
In response to Sen. Sanders’ question, Dr. Califf said he already is “on record as being in favor of Medicare negotiating with the industry on prices.”
The FDA would not take direct part in negotiations, as this work would be handled by the Centers for Medicare & Medicaid Services. Democrats want to give Medicare some negotiating authority through their sweeping Build Back Better Act.
People in the United States are dismayed over both the cost of prescription drugs and the widespread distribution of prescription painkillers that helped fuel the current opioid epidemic, Sen. Sanders told Dr. Califf. Many people will be concerned about an FDA commissioner who has benefited from close ties to the industry, Sen. Sanders said.
“How are they going to believe that you’re going to be an independent and strong voice against this enormously powerful, special interest?” Sen. Sanders asked.
“I’m totally with you on the concept that the price of pharmaceuticals is way too high in this country,” Dr. Califf said in reply.
Dr. Califf was paid $2.7 million in salary and bonus by Verily Life Sciences, the biomedical research organization operated by Alphabet, parent company of Google, according to his federal financial disclosure. He also reported holding board positions with pharmaceutical companies AmyriAD and Centessa Pharmaceuticals.
Bloomberg Government reported that Dr. Califf has ties to about 16 other research organizations and biotech companies. Bloomberg Government also said that, in his earlier FDA service, Dr. Califf kept a whiteboard in his office that listed all the activities and projects that required his recusal, citing as a source Howard Sklamberg, who was a deputy commissioner under Dr. Califf.
“He was very, very, very careful,” Mr. Sklamberg, who’s now an attorney at Arnold & Porter LLP, told Bloomberg Government.
‘Work to do’ on opioids
Senators looped back repeatedly to the topic of opioids during Dr. Califf’s hearing, reflecting deep concerns about the FDA’s efforts to warn of the risks of prescription painkillers.
There were an estimated 100,306 drug overdose deaths in the United States in the 12 months ending in April, an increase of 28.5% from the 78,056 deaths during the same period the year before, according to the Centers for Disease Control and Prevention.
Dr. Califf said he plans to focus on what information the FDA conveys to the public about the risks of prescription painkillers, including a look at what the labels for these products say.
“I am committed to do a comprehensive review of the status of opioids, early in my tenure,” Dr. Califf said.
Dr. Califf indicated that physicians are still too quick to provide excess doses of these medicines, despite years of efforts to restrain their use. He said he knows relatives who were given 30-day prescriptions for opioids after minor surgery.
“So I know we have work to do,” Dr. Califf said.
Concerns about the FDA’s previous work in managing opioids has led to protests from a few Democratic senators about the prospect of President Biden nominating the acting FDA commissioner, Janet Woodcock, MD, for the permanent post.
At the hearing, Sen. Ben Ray Luján (D-NM) raised the case of the FDA’s approval of the powerful Zohydro painkiller. The agency approved that drug despite an 11-2 vote against it by the FDA’s Anesthetic and Analgesic Drug Products Advisory Committee.
Sen. Luján asked Dr. Califf what he would do if an FDA advisory committee voted “overwhelmingly” against recommending approval of a medicine, as happened in the Zohydro case.
While not mentioned by Sen. Luján in this exchange during the hearing with Dr. Califf, the FDA staff’s rejection of recommendations of advisory committees has been a growing concern among researchers.
The agency last year approved aducanumab (Aduhelm, Biogen), a drug for Alzheimer’s disease, dismissing the advice of its Peripheral and Central Nervous System Drugs Advisory Committee. That decision triggered the resignation of several members of the panel. The FDA staff also earlier rejected the conclusion the majority of members of the same advisory committee offered in 2016 on eteplirsen (Exondys 51, Sarepta), a drug for Duchenne muscular dystrophy.
Dr. Califf told Sen. Luján he had done recent research into how often the FDA staff does not concur with the recommendations of an advisory committee. He said the FDA takes a different course of action in about 25% of cases. In about three-quarters of those cases, the FDA staff opts for a “more stringent” approach regarding allowing the public access to the drug, as opposed to a more generous one as seen in the Zohydro, Aduhelm, and Exondys 51 cases.
Still, Dr. Califf said that when there’s an 11-2 advisory committee vote against recommendation of a product, “the leaders at FDA really need to take a close look” at what’s happening.
Question on accelerated approvals
The FDA’s approval of aducanumab drew attention to a debate already underway about conditional clearances known as accelerated approvals.
The FDA has used this path since the 1990s to speed access to drugs for serious conditions. The trade-off for early access is that the agency sometimes makes the wrong call based on initial findings, and clears a medicine later found not to benefit patients as expected.
The FDA’s cancer division is in the midst of public efforts to address cases where drugmakers have not been able to deliver studies that support accelerated approvals of their oncology drugs. In addition, the Office of Inspector General of the U.S. Department of Health & Human Services announced in August that it is reviewing the FDA’s handling of the accelerated approval process.
At Tuesday’s hearing, Sen. Burr grilled Dr. Califf about how he would respond to calls to change how the FDA handles the accelerated-approval process.
“Can you commit to me and to patients who may rely on cutting-edge treatments that you will not support efforts to narrow this pathway or raise the bar for drugs to be approved under those pathways?” Burr asked Califf.
Dr. Califf responded by saying he was “a fan of accelerated approval – for the right conditions.”
Earlier, in his opening statement, Dr. Califf had said his mother benefited directly from the accelerated approval of new drugs for multiple myeloma. Dr. Califf told Sen. Burr that he had spent “countless hours with patient groups” and understands the need to speed the approval of medicines for serious diseases.
But the FDA also has to make sure it holds up its end of the bargain struck with accelerated approvals. This involves checking on how these medicines work once they are marketed.
“We’re accepting that there’s more uncertainty,” Dr. Califf said. “That means we’ve got to have a better system to evaluate these products as they’re used on the market. And I think there are ways that we can do that now. Technology is making this possible in ways that it just was not possible before.”
Worries about the medical supply chain
Sen. Susan Collins (R-Maine) asked Dr. Califf about the vulnerability of the U.S. medical system to disruptions of the supply chain. She raised concerns about China’s dominance in antibiotic manufacturing as an example. She asked if Congress could do more to encourage domestic manufacturing of medical supplies, such as by offering tax incentives.
Dr. Califf told Sen. Collins he shared her concern about the U.S. manufacturing of ingredients used in both branded and generic drugs. He said he recently has served on a committee of the National Academy of Medicine that is examining supply chain issues.
This committee will soon release a report with specific recommendations, Dr. Califf said.
“We don’t have enough competitive entities in what’s become sort of a commodity business” of drug manufacturing, Dr. Califf said. “So we need a number of steps to make the system more resilient.”
A version of this article first appeared on Medscape.com.
Medicare insulin negotiations seen saving $17 billion
Medicare could have saved more than $16.7 billion on three kinds of insulin products from 2011 to 2017 if it had secured the same discounts other federal health programs get through negotiations, House Democrats argue in a new report.
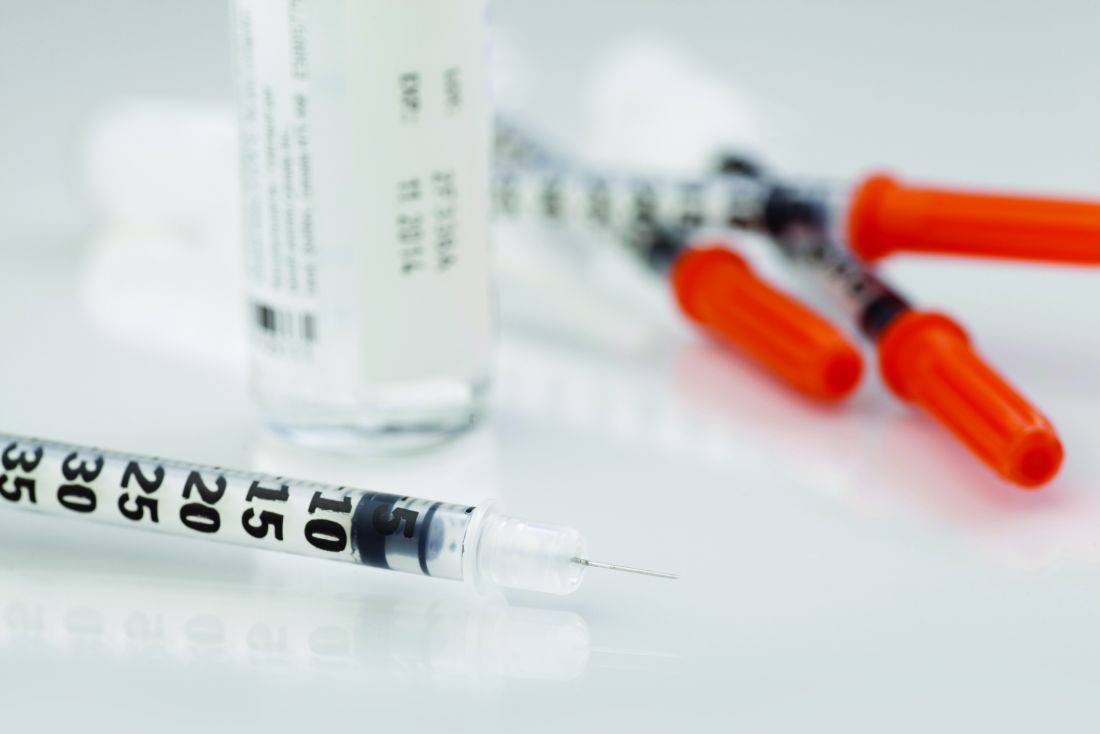
On Dec. 10, Democrats on the House Committee on Oversight and Reform released a final majority staff report, which they say is the culmination of an almost 3-year investigation into pharmaceutical pricing and business practices. The report draws from 1.5 million pages of internal company documents, the committee says.
Documents from insulin makers Eli Lilly, Novo Nordisk, and Sanofi indicate these firms “raised their prices in lockstep in order to maintain ‘pricing parity’,” with senior executives encouraging the practice, the committee staff writes in the report.
“In a discussion among Novo Nordisk employees about an Eli Lilly price increase for a different diabetes product on Dec. 24, 2015, a Novo Nordisk pricing analyst remarked, ‘[M]aybe Sanofi will wait until tomorrow morning to announce their price increase ... that’s all I want for Christmas,’” the report states.
House Democrats are seeking to use the report findings to aid their Senate colleagues’ attempt to pass the sweeping Build Back Better bill, which includes many provisions addressing drug costs.
It’s still unclear when the Senate will act on the measure. The House passed the Build Back Better bill, 220-213, in November. It includes a provision that would allow Medicare to negotiate the prices of certain drugs covered by Part D pharmacy plans.
That would mark a reversal of the stance taken when Congress created the pharmacy benefit in a 2003 law, which left negotiations to insurers that cover Part D plans.
Republicans have long argued insurers get the best deals on drugs for people on Medicare. Democrats say this approach sacrifices much of Medicare’s bargaining clout, scattering it among plans.
“This fight has been going on since the Medicare Part D legislation which gave away the store” to drugmakers, said Speaker Nancy Pelosi (D-CA) at a Dec. 10 press conference about the House Oversight report. “And they got used to having the store to themselves.”
The Endocrine Society is urging the Senate to protect the insulin affordability provisions included in the Build Back Better Act and move quickly to pass this crucial legislation.
“We implore all Senators to ensure these provisions are not scaled back. The Build Back Better Act represents the best opportunity to address the price of insulin. Millions of Americans cannot wait any longer for a solution,” it said in a statement issued Dec. 14.
Better deals for military, medicaid programs
Medicare is unusual among federal programs in that it doesn’t directly leverage its clout to lower drug costs.
Total Part D expenditures were approximately $105 billion last year, according to Medicare’s board of trustees. This spending is divided among the many insurers that run Part D plans, which then make a myriad of decisions about formularies and other factors that affect pricing.
For drugs administered by clinicians, and thus covered by Medicare Part B, the program pays a premium of the reported average sales price. Part B drug spending was $39 billion in 2019, an increase of about 11.6% from the previous year, according to the Medicare Payment Advisory Commission.
In contrast, federal law calls for steep reductions in drug prices for people on Medicaid.
The Department of Veterans Affairs (VA) and the Defense Department (DoD)’s Tricare program use several bargaining strategies to lower prices. To control costs, VA and DoD often use formularies of preferred drugs, steer patients to lower-cost drugs, and buy drugs in large volumes, “all of which increase their leverage with drug manufacturers,” the staff of the Congressional Budget Office (CBO) wrote in a Feb. 2021 report.
The CBO report examines how those different federal agencies’ approaches played out in terms of prices, net of applicable rebates, and discounts of 176 top-selling brand-name drugs in Medicare Part D.
The average price for this group of drugs was $118 in Medicaid. And for VA and DoD, the average prices were $190 and $184, respectively, for drugs dispensed at the agencies’ medical facilities or by mail.
But for Medicare Part D, the average price was $343, CBO said in the report, which was one of the sources consulted by House Oversight staff when developing their report released on Dec. 10.
Insulin still of interest, 100 years after its discovery
The House Oversight report runs to almost 270 pages. It addresses several issues with drug prices, including strategies pharmaceutical companies have used to thwart generic competition. On Monday, the trade group America’s Health Insurance Plans separately released its own report looking at patents and delays to the introduction of generic drugs.
Yet, much of the debate on drug prices has focused on one of the oldest widely produced prescription drugs, insulin.
Even with the allowance of generic competition for the essential medicine, branded versions of insulin have been some of the costliest products for Medicare in recent years. Eli Lilly, Novo Nordisk, and Sanofi dominate the insulin market.
Medicare Part D spent about $2.5 billion in 2019 on Sanofi’s Lantus Solostar insulin, or about $2,585 per person in the program using it. The program also paid about $1.1 billion for another form of Lantus, or about $2,746 per patient.
Medicare Part D also spent about $1.84 billion in 2019 on Novo Nordisk’s NovoLog FlexPen, or about $3,063 per person.
Medicare Part D’s drug spending dashboard also lists eight versions of Lilly’s Humalog, with combined 2019 spending of more than $2 billion. The cost per patient in Medicare Part D ranges from $5,619 to $1,462.
“Over the past 20 years, they have repeatedly and dramatically raised the list prices of their rapid-acting and long-acting insulins and reaped billions of dollars in revenues,” write the House Oversight staff in their report.
Republicans on the House Oversight and Reform Committee disagree with their Democratic colleagues on many points in the debate on drug prices, but they also looked at insulin as a cause for concern.
GOP members of the committee released a separate report on Dec. 10. They call for greater clarity into the role middlemen in the drug-supply chain – known as pharmaceutical benefit managers – may play in the rising costs of medicines. The GOP report notes that there are bills pending in the House that would seek to steer any discounts offered on insulin within the supply chain toward consumers (Insulin Price Reduction Act H.R. 4906, Insulin Cost Reduction Act H.R. 5623).
Democratic staff in the committee’s report seek to draw attention to how manufacturers priced their insulin products, including the comment by the Novo Nordisk employee about wishing for a price hike for a competitor’s product.
In a statement provided to this news organization, Novo Nordisk said the committee’s report reflects “a limited picture of the efforts put forth by our company and other companies to manage formulary access.”
“This glimpse into the complexity of pricing, formularies, and the health care system demonstrates why Novo Nordisk continues to advocate for comprehensive solutions,” Denmark’s Novo Nordisk said in the statement.
$35 a month for insulin?
Paris-based Sanofi said it makes insulin-pricing decisions independently from competitors. Sanofi said the net price of its insulins has declined by 53% since 2012, arguing the high prices charged to patients reflect decisions made elsewhere in the supply chain.
“Over the same period, the net price for commercial and Medicare Part D plans of Lantus has fallen 44.9%, while average out-of-pocket costs for patients with commercial insurance and Medicare Part D has risen approximately 82%,” Sanofi said.
“For all the focus on the growth of list prices, today, the average net price of Lantus is below 2006 levels. That is why we support policy reforms to require health plans to share negotiated savings with patients by requiring patient cost-sharing be tied to the net prices.”
Indianapolis-based Lilly offered a similar response in a statement to this news organization.
“Lilly, like other companies, monitors competitor list-price changes that are available through publicly available services,” the company said. “However, any changes we make to our list prices are independent decisions, and to the extent they consider competitors they are informed only through publicly available data.”
Despite rising insurance deductibles, the average monthly out-of-pocket cost for Lilly insulin has dropped 27% to $28.05 over the past 4 years, the company said in an interview. Lilly also noted that there are “several affordability options now available” allowing people to purchase their monthly prescription of its insulin for $35, “whether they are uninsured or use commercial insurance, Medicaid, or a participating Medicare Part D plan.”
In 2020, Lilly had announced that people with commercial insurance and those without insurance would be able to get monthly prescriptions of Lilly insulin for $35.
The Build Back Better Act would require insurers, including Medicare Part D plans and private group or individual health plans, to charge patient cost-sharing of no more than $35 per month for insulin products, said the staff of the nonprofit Kaiser Family Foundation (KFF) in a review of the bill.
“Private group or individual plans would not be required to cover all insulin products, just one of each dosage form (vial, pen) and insulin type (rapid-acting, short-acting, intermediate-acting, and long-acting), for no more than $35,” the KFF staff state in the report.
People enrolled in Medicare can already choose to enroll in a Part D plan participating in a federal test program that can secure certain insulin products for them at a monthly copayment of $35. In 2022, a total of 2,159 Part D plans will participate in this model, a 32% increase in participating plans since 2021, KFF said.
A version of this article first appeared on Medscape.com.
Medicare could have saved more than $16.7 billion on three kinds of insulin products from 2011 to 2017 if it had secured the same discounts other federal health programs get through negotiations, House Democrats argue in a new report.
On Dec. 10, Democrats on the House Committee on Oversight and Reform released a final majority staff report, which they say is the culmination of an almost 3-year investigation into pharmaceutical pricing and business practices. The report draws from 1.5 million pages of internal company documents, the committee says.
Documents from insulin makers Eli Lilly, Novo Nordisk, and Sanofi indicate these firms “raised their prices in lockstep in order to maintain ‘pricing parity’,” with senior executives encouraging the practice, the committee staff writes in the report.
“In a discussion among Novo Nordisk employees about an Eli Lilly price increase for a different diabetes product on Dec. 24, 2015, a Novo Nordisk pricing analyst remarked, ‘[M]aybe Sanofi will wait until tomorrow morning to announce their price increase ... that’s all I want for Christmas,’” the report states.
House Democrats are seeking to use the report findings to aid their Senate colleagues’ attempt to pass the sweeping Build Back Better bill, which includes many provisions addressing drug costs.
It’s still unclear when the Senate will act on the measure. The House passed the Build Back Better bill, 220-213, in November. It includes a provision that would allow Medicare to negotiate the prices of certain drugs covered by Part D pharmacy plans.
That would mark a reversal of the stance taken when Congress created the pharmacy benefit in a 2003 law, which left negotiations to insurers that cover Part D plans.
Republicans have long argued insurers get the best deals on drugs for people on Medicare. Democrats say this approach sacrifices much of Medicare’s bargaining clout, scattering it among plans.
“This fight has been going on since the Medicare Part D legislation which gave away the store” to drugmakers, said Speaker Nancy Pelosi (D-CA) at a Dec. 10 press conference about the House Oversight report. “And they got used to having the store to themselves.”
The Endocrine Society is urging the Senate to protect the insulin affordability provisions included in the Build Back Better Act and move quickly to pass this crucial legislation.
“We implore all Senators to ensure these provisions are not scaled back. The Build Back Better Act represents the best opportunity to address the price of insulin. Millions of Americans cannot wait any longer for a solution,” it said in a statement issued Dec. 14.
Better deals for military, medicaid programs
Medicare is unusual among federal programs in that it doesn’t directly leverage its clout to lower drug costs.
Total Part D expenditures were approximately $105 billion last year, according to Medicare’s board of trustees. This spending is divided among the many insurers that run Part D plans, which then make a myriad of decisions about formularies and other factors that affect pricing.
For drugs administered by clinicians, and thus covered by Medicare Part B, the program pays a premium of the reported average sales price. Part B drug spending was $39 billion in 2019, an increase of about 11.6% from the previous year, according to the Medicare Payment Advisory Commission.
In contrast, federal law calls for steep reductions in drug prices for people on Medicaid.
The Department of Veterans Affairs (VA) and the Defense Department (DoD)’s Tricare program use several bargaining strategies to lower prices. To control costs, VA and DoD often use formularies of preferred drugs, steer patients to lower-cost drugs, and buy drugs in large volumes, “all of which increase their leverage with drug manufacturers,” the staff of the Congressional Budget Office (CBO) wrote in a Feb. 2021 report.
The CBO report examines how those different federal agencies’ approaches played out in terms of prices, net of applicable rebates, and discounts of 176 top-selling brand-name drugs in Medicare Part D.
The average price for this group of drugs was $118 in Medicaid. And for VA and DoD, the average prices were $190 and $184, respectively, for drugs dispensed at the agencies’ medical facilities or by mail.
But for Medicare Part D, the average price was $343, CBO said in the report, which was one of the sources consulted by House Oversight staff when developing their report released on Dec. 10.
Insulin still of interest, 100 years after its discovery
The House Oversight report runs to almost 270 pages. It addresses several issues with drug prices, including strategies pharmaceutical companies have used to thwart generic competition. On Monday, the trade group America’s Health Insurance Plans separately released its own report looking at patents and delays to the introduction of generic drugs.
Yet, much of the debate on drug prices has focused on one of the oldest widely produced prescription drugs, insulin.
Even with the allowance of generic competition for the essential medicine, branded versions of insulin have been some of the costliest products for Medicare in recent years. Eli Lilly, Novo Nordisk, and Sanofi dominate the insulin market.
Medicare Part D spent about $2.5 billion in 2019 on Sanofi’s Lantus Solostar insulin, or about $2,585 per person in the program using it. The program also paid about $1.1 billion for another form of Lantus, or about $2,746 per patient.
Medicare Part D also spent about $1.84 billion in 2019 on Novo Nordisk’s NovoLog FlexPen, or about $3,063 per person.
Medicare Part D’s drug spending dashboard also lists eight versions of Lilly’s Humalog, with combined 2019 spending of more than $2 billion. The cost per patient in Medicare Part D ranges from $5,619 to $1,462.
“Over the past 20 years, they have repeatedly and dramatically raised the list prices of their rapid-acting and long-acting insulins and reaped billions of dollars in revenues,” write the House Oversight staff in their report.
Republicans on the House Oversight and Reform Committee disagree with their Democratic colleagues on many points in the debate on drug prices, but they also looked at insulin as a cause for concern.
GOP members of the committee released a separate report on Dec. 10. They call for greater clarity into the role middlemen in the drug-supply chain – known as pharmaceutical benefit managers – may play in the rising costs of medicines. The GOP report notes that there are bills pending in the House that would seek to steer any discounts offered on insulin within the supply chain toward consumers (Insulin Price Reduction Act H.R. 4906, Insulin Cost Reduction Act H.R. 5623).
Democratic staff in the committee’s report seek to draw attention to how manufacturers priced their insulin products, including the comment by the Novo Nordisk employee about wishing for a price hike for a competitor’s product.
In a statement provided to this news organization, Novo Nordisk said the committee’s report reflects “a limited picture of the efforts put forth by our company and other companies to manage formulary access.”
“This glimpse into the complexity of pricing, formularies, and the health care system demonstrates why Novo Nordisk continues to advocate for comprehensive solutions,” Denmark’s Novo Nordisk said in the statement.
$35 a month for insulin?
Paris-based Sanofi said it makes insulin-pricing decisions independently from competitors. Sanofi said the net price of its insulins has declined by 53% since 2012, arguing the high prices charged to patients reflect decisions made elsewhere in the supply chain.
“Over the same period, the net price for commercial and Medicare Part D plans of Lantus has fallen 44.9%, while average out-of-pocket costs for patients with commercial insurance and Medicare Part D has risen approximately 82%,” Sanofi said.
“For all the focus on the growth of list prices, today, the average net price of Lantus is below 2006 levels. That is why we support policy reforms to require health plans to share negotiated savings with patients by requiring patient cost-sharing be tied to the net prices.”
Indianapolis-based Lilly offered a similar response in a statement to this news organization.
“Lilly, like other companies, monitors competitor list-price changes that are available through publicly available services,” the company said. “However, any changes we make to our list prices are independent decisions, and to the extent they consider competitors they are informed only through publicly available data.”
Despite rising insurance deductibles, the average monthly out-of-pocket cost for Lilly insulin has dropped 27% to $28.05 over the past 4 years, the company said in an interview. Lilly also noted that there are “several affordability options now available” allowing people to purchase their monthly prescription of its insulin for $35, “whether they are uninsured or use commercial insurance, Medicaid, or a participating Medicare Part D plan.”
In 2020, Lilly had announced that people with commercial insurance and those without insurance would be able to get monthly prescriptions of Lilly insulin for $35.
The Build Back Better Act would require insurers, including Medicare Part D plans and private group or individual health plans, to charge patient cost-sharing of no more than $35 per month for insulin products, said the staff of the nonprofit Kaiser Family Foundation (KFF) in a review of the bill.
“Private group or individual plans would not be required to cover all insulin products, just one of each dosage form (vial, pen) and insulin type (rapid-acting, short-acting, intermediate-acting, and long-acting), for no more than $35,” the KFF staff state in the report.
People enrolled in Medicare can already choose to enroll in a Part D plan participating in a federal test program that can secure certain insulin products for them at a monthly copayment of $35. In 2022, a total of 2,159 Part D plans will participate in this model, a 32% increase in participating plans since 2021, KFF said.
A version of this article first appeared on Medscape.com.
Medicare could have saved more than $16.7 billion on three kinds of insulin products from 2011 to 2017 if it had secured the same discounts other federal health programs get through negotiations, House Democrats argue in a new report.
On Dec. 10, Democrats on the House Committee on Oversight and Reform released a final majority staff report, which they say is the culmination of an almost 3-year investigation into pharmaceutical pricing and business practices. The report draws from 1.5 million pages of internal company documents, the committee says.
Documents from insulin makers Eli Lilly, Novo Nordisk, and Sanofi indicate these firms “raised their prices in lockstep in order to maintain ‘pricing parity’,” with senior executives encouraging the practice, the committee staff writes in the report.
“In a discussion among Novo Nordisk employees about an Eli Lilly price increase for a different diabetes product on Dec. 24, 2015, a Novo Nordisk pricing analyst remarked, ‘[M]aybe Sanofi will wait until tomorrow morning to announce their price increase ... that’s all I want for Christmas,’” the report states.
House Democrats are seeking to use the report findings to aid their Senate colleagues’ attempt to pass the sweeping Build Back Better bill, which includes many provisions addressing drug costs.
It’s still unclear when the Senate will act on the measure. The House passed the Build Back Better bill, 220-213, in November. It includes a provision that would allow Medicare to negotiate the prices of certain drugs covered by Part D pharmacy plans.
That would mark a reversal of the stance taken when Congress created the pharmacy benefit in a 2003 law, which left negotiations to insurers that cover Part D plans.
Republicans have long argued insurers get the best deals on drugs for people on Medicare. Democrats say this approach sacrifices much of Medicare’s bargaining clout, scattering it among plans.
“This fight has been going on since the Medicare Part D legislation which gave away the store” to drugmakers, said Speaker Nancy Pelosi (D-CA) at a Dec. 10 press conference about the House Oversight report. “And they got used to having the store to themselves.”
The Endocrine Society is urging the Senate to protect the insulin affordability provisions included in the Build Back Better Act and move quickly to pass this crucial legislation.
“We implore all Senators to ensure these provisions are not scaled back. The Build Back Better Act represents the best opportunity to address the price of insulin. Millions of Americans cannot wait any longer for a solution,” it said in a statement issued Dec. 14.
Better deals for military, medicaid programs
Medicare is unusual among federal programs in that it doesn’t directly leverage its clout to lower drug costs.
Total Part D expenditures were approximately $105 billion last year, according to Medicare’s board of trustees. This spending is divided among the many insurers that run Part D plans, which then make a myriad of decisions about formularies and other factors that affect pricing.
For drugs administered by clinicians, and thus covered by Medicare Part B, the program pays a premium of the reported average sales price. Part B drug spending was $39 billion in 2019, an increase of about 11.6% from the previous year, according to the Medicare Payment Advisory Commission.
In contrast, federal law calls for steep reductions in drug prices for people on Medicaid.
The Department of Veterans Affairs (VA) and the Defense Department (DoD)’s Tricare program use several bargaining strategies to lower prices. To control costs, VA and DoD often use formularies of preferred drugs, steer patients to lower-cost drugs, and buy drugs in large volumes, “all of which increase their leverage with drug manufacturers,” the staff of the Congressional Budget Office (CBO) wrote in a Feb. 2021 report.
The CBO report examines how those different federal agencies’ approaches played out in terms of prices, net of applicable rebates, and discounts of 176 top-selling brand-name drugs in Medicare Part D.
The average price for this group of drugs was $118 in Medicaid. And for VA and DoD, the average prices were $190 and $184, respectively, for drugs dispensed at the agencies’ medical facilities or by mail.
But for Medicare Part D, the average price was $343, CBO said in the report, which was one of the sources consulted by House Oversight staff when developing their report released on Dec. 10.
Insulin still of interest, 100 years after its discovery
The House Oversight report runs to almost 270 pages. It addresses several issues with drug prices, including strategies pharmaceutical companies have used to thwart generic competition. On Monday, the trade group America’s Health Insurance Plans separately released its own report looking at patents and delays to the introduction of generic drugs.
Yet, much of the debate on drug prices has focused on one of the oldest widely produced prescription drugs, insulin.
Even with the allowance of generic competition for the essential medicine, branded versions of insulin have been some of the costliest products for Medicare in recent years. Eli Lilly, Novo Nordisk, and Sanofi dominate the insulin market.
Medicare Part D spent about $2.5 billion in 2019 on Sanofi’s Lantus Solostar insulin, or about $2,585 per person in the program using it. The program also paid about $1.1 billion for another form of Lantus, or about $2,746 per patient.
Medicare Part D also spent about $1.84 billion in 2019 on Novo Nordisk’s NovoLog FlexPen, or about $3,063 per person.
Medicare Part D’s drug spending dashboard also lists eight versions of Lilly’s Humalog, with combined 2019 spending of more than $2 billion. The cost per patient in Medicare Part D ranges from $5,619 to $1,462.
“Over the past 20 years, they have repeatedly and dramatically raised the list prices of their rapid-acting and long-acting insulins and reaped billions of dollars in revenues,” write the House Oversight staff in their report.
Republicans on the House Oversight and Reform Committee disagree with their Democratic colleagues on many points in the debate on drug prices, but they also looked at insulin as a cause for concern.
GOP members of the committee released a separate report on Dec. 10. They call for greater clarity into the role middlemen in the drug-supply chain – known as pharmaceutical benefit managers – may play in the rising costs of medicines. The GOP report notes that there are bills pending in the House that would seek to steer any discounts offered on insulin within the supply chain toward consumers (Insulin Price Reduction Act H.R. 4906, Insulin Cost Reduction Act H.R. 5623).
Democratic staff in the committee’s report seek to draw attention to how manufacturers priced their insulin products, including the comment by the Novo Nordisk employee about wishing for a price hike for a competitor’s product.
In a statement provided to this news organization, Novo Nordisk said the committee’s report reflects “a limited picture of the efforts put forth by our company and other companies to manage formulary access.”
“This glimpse into the complexity of pricing, formularies, and the health care system demonstrates why Novo Nordisk continues to advocate for comprehensive solutions,” Denmark’s Novo Nordisk said in the statement.
$35 a month for insulin?
Paris-based Sanofi said it makes insulin-pricing decisions independently from competitors. Sanofi said the net price of its insulins has declined by 53% since 2012, arguing the high prices charged to patients reflect decisions made elsewhere in the supply chain.
“Over the same period, the net price for commercial and Medicare Part D plans of Lantus has fallen 44.9%, while average out-of-pocket costs for patients with commercial insurance and Medicare Part D has risen approximately 82%,” Sanofi said.
“For all the focus on the growth of list prices, today, the average net price of Lantus is below 2006 levels. That is why we support policy reforms to require health plans to share negotiated savings with patients by requiring patient cost-sharing be tied to the net prices.”
Indianapolis-based Lilly offered a similar response in a statement to this news organization.
“Lilly, like other companies, monitors competitor list-price changes that are available through publicly available services,” the company said. “However, any changes we make to our list prices are independent decisions, and to the extent they consider competitors they are informed only through publicly available data.”
Despite rising insurance deductibles, the average monthly out-of-pocket cost for Lilly insulin has dropped 27% to $28.05 over the past 4 years, the company said in an interview. Lilly also noted that there are “several affordability options now available” allowing people to purchase their monthly prescription of its insulin for $35, “whether they are uninsured or use commercial insurance, Medicaid, or a participating Medicare Part D plan.”
In 2020, Lilly had announced that people with commercial insurance and those without insurance would be able to get monthly prescriptions of Lilly insulin for $35.
The Build Back Better Act would require insurers, including Medicare Part D plans and private group or individual health plans, to charge patient cost-sharing of no more than $35 per month for insulin products, said the staff of the nonprofit Kaiser Family Foundation (KFF) in a review of the bill.
“Private group or individual plans would not be required to cover all insulin products, just one of each dosage form (vial, pen) and insulin type (rapid-acting, short-acting, intermediate-acting, and long-acting), for no more than $35,” the KFF staff state in the report.
People enrolled in Medicare can already choose to enroll in a Part D plan participating in a federal test program that can secure certain insulin products for them at a monthly copayment of $35. In 2022, a total of 2,159 Part D plans will participate in this model, a 32% increase in participating plans since 2021, KFF said.
A version of this article first appeared on Medscape.com.
AMA, hospital group sue federal government over surprise billing law
which tilts toward using prevailing rates paid for services.
The American Hospital Association and American Medical Association said they will ask the U.S. District Court for the District of Columbia to try to prevent implementation of certain provisions of new federal rules on surprise bills. This court is often a venue for fights over federal rules. Also joining the suit are Nevada-based Renown Health, UMass Memorial Health, and two physicians based in North Carolina, AHA and AMA said.
Federal agencies, including the Department of Health & Human Services, in September had unveiled the rule on surprise medical bills that will take effect Jan. 1.
Under this rule, a key benchmark for payment disputes would be the qualifying payment amount (QPA), which is pegged to median contracted rates. In the dispute-resolution process outlined in the rule, there is a presumption that the QPA is the appropriate out-of-network rate.
The rule allows for exceptions in which the independent mediating organization handling the payment dispute resolution has “credible information” as to why the QPA is materially different from the appropriate out-of-network rate.
In the view of the federal agencies that issued the rule, this approach “encourages predictable outcomes,” which likely would reduce the number of disputes that go through the resolution process while also “providing equitable and clear standards” for cases to appropriately deviate from QPA. HHS was joined in issuing the rule by the Treasury and Labor Departments and the Office of Personnel Management.
AMA and AHA disagree with their view, seeing this approach as a boon for insurers at the expense of physicians and hospitals.
In a press release, they said the rule’s approach to surprise billing would “all but ensure that hospitals, physicians, and other providers will routinely be undercompensated by commercial insurers, and patients will have fewer choices for access to in-network services.”
The rule is part of the implementation of a federal law passed in December 2020, known as the No Surprises Act. In their statement, AHA and AMA said their legal challenge would not prevent “core patient protections’’ of that law from moving forward.
“No patient should fear receiving a surprise medical bill,” Rick Pollack, AHA president and chief executive, said in the statement. “That is why hospitals and health systems supported the No Surprises Act to protect patients and keep them out of the middle of disputes between providers and insurers. Congress carefully crafted the law with a balanced, patient-friendly approach and it should be implemented as intended.”
AMA President Gerald E. Harmon, MD, added the approach used in the rule on surprise billing could create “an unsustainable situation for physicians.”
“Our legal challenge urges regulators to ensure there is a fair and meaningful process to resolve disputes between health care providers and insurance companies,” Dr. Harmon said.
AHA and AMA included with their statement a link to a November letter from more than 150 members of Congress, who also objected to the approach taken in designing the independent dispute-resolution (IDR) process.
“This directive establishes a de facto benchmark rate, making the median in-network rate the default factor considered in the IDR process. This approach is contrary to statute and could incentivize insurance companies to set artificially low payment rates, which would narrow provider networks and jeopardize patient access to care – the exact opposite of the goal of the law,” wrote the members of Congress, including Rep. Raul Ruiz, MD, a California Democrat, and Rep. Larry Bucshon, MD, an Indiana Republican.
A version of this article first appeared on Medscape.com.
which tilts toward using prevailing rates paid for services.
The American Hospital Association and American Medical Association said they will ask the U.S. District Court for the District of Columbia to try to prevent implementation of certain provisions of new federal rules on surprise bills. This court is often a venue for fights over federal rules. Also joining the suit are Nevada-based Renown Health, UMass Memorial Health, and two physicians based in North Carolina, AHA and AMA said.
Federal agencies, including the Department of Health & Human Services, in September had unveiled the rule on surprise medical bills that will take effect Jan. 1.
Under this rule, a key benchmark for payment disputes would be the qualifying payment amount (QPA), which is pegged to median contracted rates. In the dispute-resolution process outlined in the rule, there is a presumption that the QPA is the appropriate out-of-network rate.
The rule allows for exceptions in which the independent mediating organization handling the payment dispute resolution has “credible information” as to why the QPA is materially different from the appropriate out-of-network rate.
In the view of the federal agencies that issued the rule, this approach “encourages predictable outcomes,” which likely would reduce the number of disputes that go through the resolution process while also “providing equitable and clear standards” for cases to appropriately deviate from QPA. HHS was joined in issuing the rule by the Treasury and Labor Departments and the Office of Personnel Management.
AMA and AHA disagree with their view, seeing this approach as a boon for insurers at the expense of physicians and hospitals.
In a press release, they said the rule’s approach to surprise billing would “all but ensure that hospitals, physicians, and other providers will routinely be undercompensated by commercial insurers, and patients will have fewer choices for access to in-network services.”
The rule is part of the implementation of a federal law passed in December 2020, known as the No Surprises Act. In their statement, AHA and AMA said their legal challenge would not prevent “core patient protections’’ of that law from moving forward.
“No patient should fear receiving a surprise medical bill,” Rick Pollack, AHA president and chief executive, said in the statement. “That is why hospitals and health systems supported the No Surprises Act to protect patients and keep them out of the middle of disputes between providers and insurers. Congress carefully crafted the law with a balanced, patient-friendly approach and it should be implemented as intended.”
AMA President Gerald E. Harmon, MD, added the approach used in the rule on surprise billing could create “an unsustainable situation for physicians.”
“Our legal challenge urges regulators to ensure there is a fair and meaningful process to resolve disputes between health care providers and insurance companies,” Dr. Harmon said.
AHA and AMA included with their statement a link to a November letter from more than 150 members of Congress, who also objected to the approach taken in designing the independent dispute-resolution (IDR) process.
“This directive establishes a de facto benchmark rate, making the median in-network rate the default factor considered in the IDR process. This approach is contrary to statute and could incentivize insurance companies to set artificially low payment rates, which would narrow provider networks and jeopardize patient access to care – the exact opposite of the goal of the law,” wrote the members of Congress, including Rep. Raul Ruiz, MD, a California Democrat, and Rep. Larry Bucshon, MD, an Indiana Republican.
A version of this article first appeared on Medscape.com.
which tilts toward using prevailing rates paid for services.
The American Hospital Association and American Medical Association said they will ask the U.S. District Court for the District of Columbia to try to prevent implementation of certain provisions of new federal rules on surprise bills. This court is often a venue for fights over federal rules. Also joining the suit are Nevada-based Renown Health, UMass Memorial Health, and two physicians based in North Carolina, AHA and AMA said.
Federal agencies, including the Department of Health & Human Services, in September had unveiled the rule on surprise medical bills that will take effect Jan. 1.
Under this rule, a key benchmark for payment disputes would be the qualifying payment amount (QPA), which is pegged to median contracted rates. In the dispute-resolution process outlined in the rule, there is a presumption that the QPA is the appropriate out-of-network rate.
The rule allows for exceptions in which the independent mediating organization handling the payment dispute resolution has “credible information” as to why the QPA is materially different from the appropriate out-of-network rate.
In the view of the federal agencies that issued the rule, this approach “encourages predictable outcomes,” which likely would reduce the number of disputes that go through the resolution process while also “providing equitable and clear standards” for cases to appropriately deviate from QPA. HHS was joined in issuing the rule by the Treasury and Labor Departments and the Office of Personnel Management.
AMA and AHA disagree with their view, seeing this approach as a boon for insurers at the expense of physicians and hospitals.
In a press release, they said the rule’s approach to surprise billing would “all but ensure that hospitals, physicians, and other providers will routinely be undercompensated by commercial insurers, and patients will have fewer choices for access to in-network services.”
The rule is part of the implementation of a federal law passed in December 2020, known as the No Surprises Act. In their statement, AHA and AMA said their legal challenge would not prevent “core patient protections’’ of that law from moving forward.
“No patient should fear receiving a surprise medical bill,” Rick Pollack, AHA president and chief executive, said in the statement. “That is why hospitals and health systems supported the No Surprises Act to protect patients and keep them out of the middle of disputes between providers and insurers. Congress carefully crafted the law with a balanced, patient-friendly approach and it should be implemented as intended.”
AMA President Gerald E. Harmon, MD, added the approach used in the rule on surprise billing could create “an unsustainable situation for physicians.”
“Our legal challenge urges regulators to ensure there is a fair and meaningful process to resolve disputes between health care providers and insurance companies,” Dr. Harmon said.
AHA and AMA included with their statement a link to a November letter from more than 150 members of Congress, who also objected to the approach taken in designing the independent dispute-resolution (IDR) process.
“This directive establishes a de facto benchmark rate, making the median in-network rate the default factor considered in the IDR process. This approach is contrary to statute and could incentivize insurance companies to set artificially low payment rates, which would narrow provider networks and jeopardize patient access to care – the exact opposite of the goal of the law,” wrote the members of Congress, including Rep. Raul Ruiz, MD, a California Democrat, and Rep. Larry Bucshon, MD, an Indiana Republican.
A version of this article first appeared on Medscape.com.
FDA puts clozapine REMS requirements on temporary hold
U.S. regulators have put some of the new clozapine risk evaluation and mitigation strategy (REMS) program on temporary hold because of start-up difficulties, including long telephone wait times.

In a Nov. 19 statement, the Food and Drug Administration announced it is temporarily suspending certain aspects of the program because of challenges reported by medical professionals who were trying to meet the original Nov. 15 deadline.
In response, the FDA has conceded that pharmacists can dispense clozapine without a REMS dispense authorization (RDA). Wholesalers can continue to ship clozapine to pharmacies and health care settings without confirming enrollment in the REMS, the FDA also said.
“We encourage pharmacists and prescribers to continue working with the clozapine REMS to complete certification and patient enrollment,” the FDA said in a statement.
In July, the FDA approved modifications to the clozapine REMS strategy. Clozapine is used to treat schizophrenia that is not well controlled with standard antipsychotics. It is also prescribed to patients with recurrent suicidal behavior associated with schizophrenia or schizoaffective disorder.
Although it is highly effective in some patients, it also carries serious risks. Specifically, it can decrease the neutrophil count, which can lead to severe neutropenia, serious infections, and death.
As a result, those taking the drug must undergo regular absolute neutrophil count (ANC) monitoring. Clozapine REMS is intended to maximize the benefits of the drug and minimize risk.
HCP frustration
, including a high call volume and long call wait times for stakeholders.
“We understand that this has caused frustration and has led to patient access issues for clozapine,” the FDA said in a statement.
“Continuity of care, patient access to clozapine, and patient safety are our highest priorities,” the FDA added. “We are working closely with the clozapine REMS program administrators to address these challenges and avoid interruptions in patient care.”
Abrupt discontinuation of clozapine can result in significant complications, the FDA said. The agency urged use of “clinical judgment” with respect to prescribing and dispensing clozapine to patients with an absolute neutrophil count within the acceptable range.
As previously reported by this news organization, the American Psychiatric Association and other national groups in a September letter asked the FDA to delay the implementation of a new REMS program until after Jan. 1, 2022.
A version of this article first appeared on Medscape.com.
U.S. regulators have put some of the new clozapine risk evaluation and mitigation strategy (REMS) program on temporary hold because of start-up difficulties, including long telephone wait times.
In a Nov. 19 statement, the Food and Drug Administration announced it is temporarily suspending certain aspects of the program because of challenges reported by medical professionals who were trying to meet the original Nov. 15 deadline.
In response, the FDA has conceded that pharmacists can dispense clozapine without a REMS dispense authorization (RDA). Wholesalers can continue to ship clozapine to pharmacies and health care settings without confirming enrollment in the REMS, the FDA also said.
“We encourage pharmacists and prescribers to continue working with the clozapine REMS to complete certification and patient enrollment,” the FDA said in a statement.
In July, the FDA approved modifications to the clozapine REMS strategy. Clozapine is used to treat schizophrenia that is not well controlled with standard antipsychotics. It is also prescribed to patients with recurrent suicidal behavior associated with schizophrenia or schizoaffective disorder.
Although it is highly effective in some patients, it also carries serious risks. Specifically, it can decrease the neutrophil count, which can lead to severe neutropenia, serious infections, and death.
As a result, those taking the drug must undergo regular absolute neutrophil count (ANC) monitoring. Clozapine REMS is intended to maximize the benefits of the drug and minimize risk.
HCP frustration
, including a high call volume and long call wait times for stakeholders.
“We understand that this has caused frustration and has led to patient access issues for clozapine,” the FDA said in a statement.
“Continuity of care, patient access to clozapine, and patient safety are our highest priorities,” the FDA added. “We are working closely with the clozapine REMS program administrators to address these challenges and avoid interruptions in patient care.”
Abrupt discontinuation of clozapine can result in significant complications, the FDA said. The agency urged use of “clinical judgment” with respect to prescribing and dispensing clozapine to patients with an absolute neutrophil count within the acceptable range.
As previously reported by this news organization, the American Psychiatric Association and other national groups in a September letter asked the FDA to delay the implementation of a new REMS program until after Jan. 1, 2022.
A version of this article first appeared on Medscape.com.
U.S. regulators have put some of the new clozapine risk evaluation and mitigation strategy (REMS) program on temporary hold because of start-up difficulties, including long telephone wait times.
In a Nov. 19 statement, the Food and Drug Administration announced it is temporarily suspending certain aspects of the program because of challenges reported by medical professionals who were trying to meet the original Nov. 15 deadline.
In response, the FDA has conceded that pharmacists can dispense clozapine without a REMS dispense authorization (RDA). Wholesalers can continue to ship clozapine to pharmacies and health care settings without confirming enrollment in the REMS, the FDA also said.
“We encourage pharmacists and prescribers to continue working with the clozapine REMS to complete certification and patient enrollment,” the FDA said in a statement.
In July, the FDA approved modifications to the clozapine REMS strategy. Clozapine is used to treat schizophrenia that is not well controlled with standard antipsychotics. It is also prescribed to patients with recurrent suicidal behavior associated with schizophrenia or schizoaffective disorder.
Although it is highly effective in some patients, it also carries serious risks. Specifically, it can decrease the neutrophil count, which can lead to severe neutropenia, serious infections, and death.
As a result, those taking the drug must undergo regular absolute neutrophil count (ANC) monitoring. Clozapine REMS is intended to maximize the benefits of the drug and minimize risk.
HCP frustration
, including a high call volume and long call wait times for stakeholders.
“We understand that this has caused frustration and has led to patient access issues for clozapine,” the FDA said in a statement.
“Continuity of care, patient access to clozapine, and patient safety are our highest priorities,” the FDA added. “We are working closely with the clozapine REMS program administrators to address these challenges and avoid interruptions in patient care.”
Abrupt discontinuation of clozapine can result in significant complications, the FDA said. The agency urged use of “clinical judgment” with respect to prescribing and dispensing clozapine to patients with an absolute neutrophil count within the acceptable range.
As previously reported by this news organization, the American Psychiatric Association and other national groups in a September letter asked the FDA to delay the implementation of a new REMS program until after Jan. 1, 2022.
A version of this article first appeared on Medscape.com.