User login
NIH announces Zika virus research priorities
The National Institutes of Health seeks applications for research on the Zika virus in reproduction, pregnancy, and the developing fetus and announced priorities for that research in a statement issued on Feb. 5.
“One of the highest priorities is to establish conclusively what role, if any, Zika virus has played in the marked increase in suspected microcephaly cases,” NIH officials said, noting that over 4,000 case of microcephaly have been reported in newborns in Brazil since October 2015. “It is possible that these microcephaly cases could have another cause, or that a contributing factor in addition to Zika virus – another virus, for example – could be leading to the condition.”Learning more about sexual transmission of the virus is also a priority. NIH is soliciting studies to determine if the virus is present in semen or vaginal secretions. Other studies “of interest” include whether infection with the virus – currently circulating in about 30 countries and territories – affects long-term fertility in both men and women and increases risk in subsequent pregnancies.
Current research can be modified, the statement points out, and may include modifying ongoing studies of pregnant women and infants to check tissue samples for the virus and evaluate the effects of exposure.
The full statement is available on the NIH website.
The National Institutes of Health seeks applications for research on the Zika virus in reproduction, pregnancy, and the developing fetus and announced priorities for that research in a statement issued on Feb. 5.
“One of the highest priorities is to establish conclusively what role, if any, Zika virus has played in the marked increase in suspected microcephaly cases,” NIH officials said, noting that over 4,000 case of microcephaly have been reported in newborns in Brazil since October 2015. “It is possible that these microcephaly cases could have another cause, or that a contributing factor in addition to Zika virus – another virus, for example – could be leading to the condition.”Learning more about sexual transmission of the virus is also a priority. NIH is soliciting studies to determine if the virus is present in semen or vaginal secretions. Other studies “of interest” include whether infection with the virus – currently circulating in about 30 countries and territories – affects long-term fertility in both men and women and increases risk in subsequent pregnancies.
Current research can be modified, the statement points out, and may include modifying ongoing studies of pregnant women and infants to check tissue samples for the virus and evaluate the effects of exposure.
The full statement is available on the NIH website.
The National Institutes of Health seeks applications for research on the Zika virus in reproduction, pregnancy, and the developing fetus and announced priorities for that research in a statement issued on Feb. 5.
“One of the highest priorities is to establish conclusively what role, if any, Zika virus has played in the marked increase in suspected microcephaly cases,” NIH officials said, noting that over 4,000 case of microcephaly have been reported in newborns in Brazil since October 2015. “It is possible that these microcephaly cases could have another cause, or that a contributing factor in addition to Zika virus – another virus, for example – could be leading to the condition.”Learning more about sexual transmission of the virus is also a priority. NIH is soliciting studies to determine if the virus is present in semen or vaginal secretions. Other studies “of interest” include whether infection with the virus – currently circulating in about 30 countries and territories – affects long-term fertility in both men and women and increases risk in subsequent pregnancies.
Current research can be modified, the statement points out, and may include modifying ongoing studies of pregnant women and infants to check tissue samples for the virus and evaluate the effects of exposure.
The full statement is available on the NIH website.
Wound-healing template approved for diabetic foot ulcers
A bilayer matrix used for dermal regeneration and first approved in 1996 as a treatment for third-degree burns is now approved as a treatment for diabetic foot ulcers.
The Integra Dermal Regeneration Template was approved for the new indication based on a study that showed that the matrix device “improved ulcer healing compared to standard diabetic foot ulcer care,” according to a Food and Drug Administration statement announcing the approval on Jan. 7. Specifically, the new indication is for treating “partial and full-thickness neuropathic diabetic foot ulcers that are greater than 6 weeks in duration, with no capsule, tendon or bone exposed, when used in conjunction with standard diabetic ulcer care.”
The product is a dermal-replacement layer that “consists of a porous, three-dimensional matrix, comprised of bovine collagen and chondroitin-6-sulfate,” with a temporary epidermal silicone layer “to provide immediate wound coverage and control moisture loss. … [It] provides an environment for new skin and tissue to regenerate and heal the wound,” according to the agency’s approval summary.
In a multicenter, randomized controlled study, 307 patients were first treated with 0.9% sodium chloride gel, a secondary dressing, and an offloading device for 2 weeks and were then randomized to a treatment or a control group that received continued treatment with the gel. After 16 weeks, 51% of those treated with the device and 32% of those in the control group had healed completely (P = .001). Among those whose wounds healed, the median time to healing was 43 days in the treatment group and 78 days in the control group.
More patients in the control group had severe adverse events (26.8% vs. 15.6%) and moderate adverse events (42.5% vs. 31.8%).The results of the study, funded and sponsored by the manufacturer, were recently published (Wound Repair Regen. 2015;23[6]:891-900).
The product is contraindicated in patients with bovine or chondroitin allergies and in patients with infected wounds.
The manufacturer, Integra LifeSciences, is marketing the device as Integra Omnigraft Dermal Regeneration Matrix for the diabetic foot ulcer indication.
A bilayer matrix used for dermal regeneration and first approved in 1996 as a treatment for third-degree burns is now approved as a treatment for diabetic foot ulcers.
The Integra Dermal Regeneration Template was approved for the new indication based on a study that showed that the matrix device “improved ulcer healing compared to standard diabetic foot ulcer care,” according to a Food and Drug Administration statement announcing the approval on Jan. 7. Specifically, the new indication is for treating “partial and full-thickness neuropathic diabetic foot ulcers that are greater than 6 weeks in duration, with no capsule, tendon or bone exposed, when used in conjunction with standard diabetic ulcer care.”
The product is a dermal-replacement layer that “consists of a porous, three-dimensional matrix, comprised of bovine collagen and chondroitin-6-sulfate,” with a temporary epidermal silicone layer “to provide immediate wound coverage and control moisture loss. … [It] provides an environment for new skin and tissue to regenerate and heal the wound,” according to the agency’s approval summary.
In a multicenter, randomized controlled study, 307 patients were first treated with 0.9% sodium chloride gel, a secondary dressing, and an offloading device for 2 weeks and were then randomized to a treatment or a control group that received continued treatment with the gel. After 16 weeks, 51% of those treated with the device and 32% of those in the control group had healed completely (P = .001). Among those whose wounds healed, the median time to healing was 43 days in the treatment group and 78 days in the control group.
More patients in the control group had severe adverse events (26.8% vs. 15.6%) and moderate adverse events (42.5% vs. 31.8%).The results of the study, funded and sponsored by the manufacturer, were recently published (Wound Repair Regen. 2015;23[6]:891-900).
The product is contraindicated in patients with bovine or chondroitin allergies and in patients with infected wounds.
The manufacturer, Integra LifeSciences, is marketing the device as Integra Omnigraft Dermal Regeneration Matrix for the diabetic foot ulcer indication.
A bilayer matrix used for dermal regeneration and first approved in 1996 as a treatment for third-degree burns is now approved as a treatment for diabetic foot ulcers.
The Integra Dermal Regeneration Template was approved for the new indication based on a study that showed that the matrix device “improved ulcer healing compared to standard diabetic foot ulcer care,” according to a Food and Drug Administration statement announcing the approval on Jan. 7. Specifically, the new indication is for treating “partial and full-thickness neuropathic diabetic foot ulcers that are greater than 6 weeks in duration, with no capsule, tendon or bone exposed, when used in conjunction with standard diabetic ulcer care.”
The product is a dermal-replacement layer that “consists of a porous, three-dimensional matrix, comprised of bovine collagen and chondroitin-6-sulfate,” with a temporary epidermal silicone layer “to provide immediate wound coverage and control moisture loss. … [It] provides an environment for new skin and tissue to regenerate and heal the wound,” according to the agency’s approval summary.
In a multicenter, randomized controlled study, 307 patients were first treated with 0.9% sodium chloride gel, a secondary dressing, and an offloading device for 2 weeks and were then randomized to a treatment or a control group that received continued treatment with the gel. After 16 weeks, 51% of those treated with the device and 32% of those in the control group had healed completely (P = .001). Among those whose wounds healed, the median time to healing was 43 days in the treatment group and 78 days in the control group.
More patients in the control group had severe adverse events (26.8% vs. 15.6%) and moderate adverse events (42.5% vs. 31.8%).The results of the study, funded and sponsored by the manufacturer, were recently published (Wound Repair Regen. 2015;23[6]:891-900).
The product is contraindicated in patients with bovine or chondroitin allergies and in patients with infected wounds.
The manufacturer, Integra LifeSciences, is marketing the device as Integra Omnigraft Dermal Regeneration Matrix for the diabetic foot ulcer indication.
Insulin Resistance in 22% of Men With Acne
Young adult men with acne were more likely to have insulin resistance and to have higher fasting plasma glucose levels than were men of the same age who did not have acne, in a cross-sectional study of 20 to 32 year old men in India.
In a study published online in JAMA Dermatology, on Dec. 23 (doi: 10.1001/jamadermatol.2015.4499), Dr. Mohit Nagpal, of the Postgraduate Institute of Medical Education and Research, Chandigarh, India, and associates, wrote that “Insulin resistance may be a stage of prediabetes, and the patients may develop hyperinsulinemia or type 2 diabetes in the future. These patients should be followed up to determine whether they develop conditions associated with insulin resistance.”
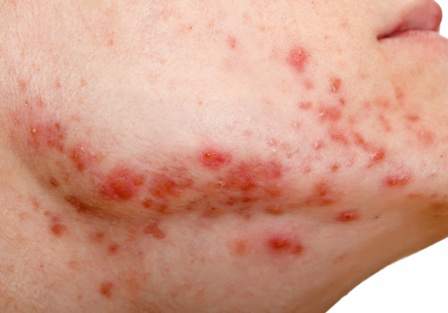
The researchers compared 100 men with acne, aged 20 to 32 years, with 100 age-matched men who did not have acne and were being treated for non-acne dermatoses; all were being treated at the Institute’s dermatology outpatient department. Insulin resistance, as defined by a Homeostasis Model Assessment–Insulin Resistance (HOMA-IR) value greater than 2.5, was present in 22% of those with acne, vs. 11% of those without acne, a significant difference (P = .036). Metabolic syndrome, based on criteria of the modified National Cholesterol Education Program’s Adult Treatment Panel III (NCEP-ATP III), was more common among those with acne (17% vs. 9%), but the difference was not significant (P = .09).
The mean diastolic and systolic blood pressure values were also significantly higher among those with acne, compared with controls, as were mean fasting plasma glucose levels.
When evaluated by acne severity (mild, moderate, severe, or very severe), there were no significant differences in the prevalence of insulin resistance or metabolic syndrome between the four groups. However, the mean body mass index and the mean weights among those with very severe acne were significantly higher than among those with mild acne (P = .04).
The cross-sectional design of the study was a limitation, the authors noted, and future studies will “follow up patients with acne to assess the development of clinical conditions associated with insulin resistance,” such as acanthosis nigricans and metabolic syndrome.
In an accompanying editorial, Dr. Rachel V. Reynolds of Beth Israel Deaconess Medical Center, Boston, wrote that this study, and another study published in the same issue regarding insulin resistance and polycystic ovary syndrome, “highlight the important role that the dermatologist plays in identifying and characterizing patients with common skin disorders who may be at risk for metabolic and androgen-mediated disease.” The study, “to our knowledge, [is] the largest cohort to date examining the prevalence of insulin resistance and metabolic syndrome in postadolescent males with acne of varying severity,” she added (doi: 10.1001/jamadermatol.2015.4500).
The authors of the study had no disclosures.
Young adult men with acne were more likely to have insulin resistance and to have higher fasting plasma glucose levels than were men of the same age who did not have acne, in a cross-sectional study of 20 to 32 year old men in India.
In a study published online in JAMA Dermatology, on Dec. 23 (doi: 10.1001/jamadermatol.2015.4499), Dr. Mohit Nagpal, of the Postgraduate Institute of Medical Education and Research, Chandigarh, India, and associates, wrote that “Insulin resistance may be a stage of prediabetes, and the patients may develop hyperinsulinemia or type 2 diabetes in the future. These patients should be followed up to determine whether they develop conditions associated with insulin resistance.”
The researchers compared 100 men with acne, aged 20 to 32 years, with 100 age-matched men who did not have acne and were being treated for non-acne dermatoses; all were being treated at the Institute’s dermatology outpatient department. Insulin resistance, as defined by a Homeostasis Model Assessment–Insulin Resistance (HOMA-IR) value greater than 2.5, was present in 22% of those with acne, vs. 11% of those without acne, a significant difference (P = .036). Metabolic syndrome, based on criteria of the modified National Cholesterol Education Program’s Adult Treatment Panel III (NCEP-ATP III), was more common among those with acne (17% vs. 9%), but the difference was not significant (P = .09).
The mean diastolic and systolic blood pressure values were also significantly higher among those with acne, compared with controls, as were mean fasting plasma glucose levels.
When evaluated by acne severity (mild, moderate, severe, or very severe), there were no significant differences in the prevalence of insulin resistance or metabolic syndrome between the four groups. However, the mean body mass index and the mean weights among those with very severe acne were significantly higher than among those with mild acne (P = .04).
The cross-sectional design of the study was a limitation, the authors noted, and future studies will “follow up patients with acne to assess the development of clinical conditions associated with insulin resistance,” such as acanthosis nigricans and metabolic syndrome.
In an accompanying editorial, Dr. Rachel V. Reynolds of Beth Israel Deaconess Medical Center, Boston, wrote that this study, and another study published in the same issue regarding insulin resistance and polycystic ovary syndrome, “highlight the important role that the dermatologist plays in identifying and characterizing patients with common skin disorders who may be at risk for metabolic and androgen-mediated disease.” The study, “to our knowledge, [is] the largest cohort to date examining the prevalence of insulin resistance and metabolic syndrome in postadolescent males with acne of varying severity,” she added (doi: 10.1001/jamadermatol.2015.4500).
The authors of the study had no disclosures.
Young adult men with acne were more likely to have insulin resistance and to have higher fasting plasma glucose levels than were men of the same age who did not have acne, in a cross-sectional study of 20 to 32 year old men in India.
In a study published online in JAMA Dermatology, on Dec. 23 (doi: 10.1001/jamadermatol.2015.4499), Dr. Mohit Nagpal, of the Postgraduate Institute of Medical Education and Research, Chandigarh, India, and associates, wrote that “Insulin resistance may be a stage of prediabetes, and the patients may develop hyperinsulinemia or type 2 diabetes in the future. These patients should be followed up to determine whether they develop conditions associated with insulin resistance.”
The researchers compared 100 men with acne, aged 20 to 32 years, with 100 age-matched men who did not have acne and were being treated for non-acne dermatoses; all were being treated at the Institute’s dermatology outpatient department. Insulin resistance, as defined by a Homeostasis Model Assessment–Insulin Resistance (HOMA-IR) value greater than 2.5, was present in 22% of those with acne, vs. 11% of those without acne, a significant difference (P = .036). Metabolic syndrome, based on criteria of the modified National Cholesterol Education Program’s Adult Treatment Panel III (NCEP-ATP III), was more common among those with acne (17% vs. 9%), but the difference was not significant (P = .09).
The mean diastolic and systolic blood pressure values were also significantly higher among those with acne, compared with controls, as were mean fasting plasma glucose levels.
When evaluated by acne severity (mild, moderate, severe, or very severe), there were no significant differences in the prevalence of insulin resistance or metabolic syndrome between the four groups. However, the mean body mass index and the mean weights among those with very severe acne were significantly higher than among those with mild acne (P = .04).
The cross-sectional design of the study was a limitation, the authors noted, and future studies will “follow up patients with acne to assess the development of clinical conditions associated with insulin resistance,” such as acanthosis nigricans and metabolic syndrome.
In an accompanying editorial, Dr. Rachel V. Reynolds of Beth Israel Deaconess Medical Center, Boston, wrote that this study, and another study published in the same issue regarding insulin resistance and polycystic ovary syndrome, “highlight the important role that the dermatologist plays in identifying and characterizing patients with common skin disorders who may be at risk for metabolic and androgen-mediated disease.” The study, “to our knowledge, [is] the largest cohort to date examining the prevalence of insulin resistance and metabolic syndrome in postadolescent males with acne of varying severity,” she added (doi: 10.1001/jamadermatol.2015.4500).
The authors of the study had no disclosures.
FROM JAMA DERMATOLOGY
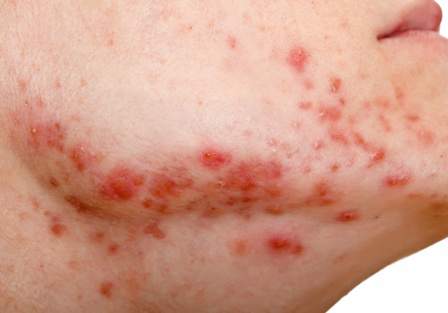
Insulin resistance in 22% of men with acne
Young adult men with acne were more likely to have insulin resistance and to have higher fasting plasma glucose levels than were men of the same age who did not have acne, in a cross-sectional study of 20 to 32 year old men in India.
In a study published online in JAMA Dermatology, on Dec. 23 (doi: 10.1001/jamadermatol.2015.4499), Dr. Mohit Nagpal, of the Postgraduate Institute of Medical Education and Research, Chandigarh, India, and associates, wrote that “Insulin resistance may be a stage of prediabetes, and the patients may develop hyperinsulinemia or type 2 diabetes in the future. These patients should be followed up to determine whether they develop conditions associated with insulin resistance.”

The researchers compared 100 men with acne, aged 20 to 32 years, with 100 age-matched men who did not have acne and were being treated for non-acne dermatoses; all were being treated at the Institute’s dermatology outpatient department. Insulin resistance, as defined by a Homeostasis Model Assessment–Insulin Resistance (HOMA-IR) value greater than 2.5, was present in 22% of those with acne, vs. 11% of those without acne, a significant difference (P = .036). Metabolic syndrome, based on criteria of the modified National Cholesterol Education Program’s Adult Treatment Panel III (NCEP-ATP III), was more common among those with acne (17% vs. 9%), but the difference was not significant (P = .09).
The mean diastolic and systolic blood pressure values were also significantly higher among those with acne, compared with controls, as were mean fasting plasma glucose levels.
When evaluated by acne severity (mild, moderate, severe, or very severe), there were no significant differences in the prevalence of insulin resistance or metabolic syndrome between the four groups. However, the mean body mass index and the mean weights among those with very severe acne were significantly higher than among those with mild acne (P = .04).
The cross-sectional design of the study was a limitation, the authors noted, and future studies will “follow up patients with acne to assess the development of clinical conditions associated with insulin resistance,” such as acanthosis nigricans and metabolic syndrome.
In an accompanying editorial, Dr. Rachel V. Reynolds of Beth Israel Deaconess Medical Center, Boston, wrote that this study, and another study published in the same issue regarding insulin resistance and polycystic ovary syndrome, “highlight the important role that the dermatologist plays in identifying and characterizing patients with common skin disorders who may be at risk for metabolic and androgen-mediated disease.” The study, “to our knowledge, [is] the largest cohort to date examining the prevalence of insulin resistance and metabolic syndrome in postadolescent males with acne of varying severity,” she added (doi: 10.1001/jamadermatol.2015.4500).
The authors of the study had no disclosures.
Young adult men with acne were more likely to have insulin resistance and to have higher fasting plasma glucose levels than were men of the same age who did not have acne, in a cross-sectional study of 20 to 32 year old men in India.
In a study published online in JAMA Dermatology, on Dec. 23 (doi: 10.1001/jamadermatol.2015.4499), Dr. Mohit Nagpal, of the Postgraduate Institute of Medical Education and Research, Chandigarh, India, and associates, wrote that “Insulin resistance may be a stage of prediabetes, and the patients may develop hyperinsulinemia or type 2 diabetes in the future. These patients should be followed up to determine whether they develop conditions associated with insulin resistance.”
The researchers compared 100 men with acne, aged 20 to 32 years, with 100 age-matched men who did not have acne and were being treated for non-acne dermatoses; all were being treated at the Institute’s dermatology outpatient department. Insulin resistance, as defined by a Homeostasis Model Assessment–Insulin Resistance (HOMA-IR) value greater than 2.5, was present in 22% of those with acne, vs. 11% of those without acne, a significant difference (P = .036). Metabolic syndrome, based on criteria of the modified National Cholesterol Education Program’s Adult Treatment Panel III (NCEP-ATP III), was more common among those with acne (17% vs. 9%), but the difference was not significant (P = .09).
The mean diastolic and systolic blood pressure values were also significantly higher among those with acne, compared with controls, as were mean fasting plasma glucose levels.
When evaluated by acne severity (mild, moderate, severe, or very severe), there were no significant differences in the prevalence of insulin resistance or metabolic syndrome between the four groups. However, the mean body mass index and the mean weights among those with very severe acne were significantly higher than among those with mild acne (P = .04).
The cross-sectional design of the study was a limitation, the authors noted, and future studies will “follow up patients with acne to assess the development of clinical conditions associated with insulin resistance,” such as acanthosis nigricans and metabolic syndrome.
In an accompanying editorial, Dr. Rachel V. Reynolds of Beth Israel Deaconess Medical Center, Boston, wrote that this study, and another study published in the same issue regarding insulin resistance and polycystic ovary syndrome, “highlight the important role that the dermatologist plays in identifying and characterizing patients with common skin disorders who may be at risk for metabolic and androgen-mediated disease.” The study, “to our knowledge, [is] the largest cohort to date examining the prevalence of insulin resistance and metabolic syndrome in postadolescent males with acne of varying severity,” she added (doi: 10.1001/jamadermatol.2015.4500).
The authors of the study had no disclosures.
Young adult men with acne were more likely to have insulin resistance and to have higher fasting plasma glucose levels than were men of the same age who did not have acne, in a cross-sectional study of 20 to 32 year old men in India.
In a study published online in JAMA Dermatology, on Dec. 23 (doi: 10.1001/jamadermatol.2015.4499), Dr. Mohit Nagpal, of the Postgraduate Institute of Medical Education and Research, Chandigarh, India, and associates, wrote that “Insulin resistance may be a stage of prediabetes, and the patients may develop hyperinsulinemia or type 2 diabetes in the future. These patients should be followed up to determine whether they develop conditions associated with insulin resistance.”
The researchers compared 100 men with acne, aged 20 to 32 years, with 100 age-matched men who did not have acne and were being treated for non-acne dermatoses; all were being treated at the Institute’s dermatology outpatient department. Insulin resistance, as defined by a Homeostasis Model Assessment–Insulin Resistance (HOMA-IR) value greater than 2.5, was present in 22% of those with acne, vs. 11% of those without acne, a significant difference (P = .036). Metabolic syndrome, based on criteria of the modified National Cholesterol Education Program’s Adult Treatment Panel III (NCEP-ATP III), was more common among those with acne (17% vs. 9%), but the difference was not significant (P = .09).
The mean diastolic and systolic blood pressure values were also significantly higher among those with acne, compared with controls, as were mean fasting plasma glucose levels.
When evaluated by acne severity (mild, moderate, severe, or very severe), there were no significant differences in the prevalence of insulin resistance or metabolic syndrome between the four groups. However, the mean body mass index and the mean weights among those with very severe acne were significantly higher than among those with mild acne (P = .04).
The cross-sectional design of the study was a limitation, the authors noted, and future studies will “follow up patients with acne to assess the development of clinical conditions associated with insulin resistance,” such as acanthosis nigricans and metabolic syndrome.
In an accompanying editorial, Dr. Rachel V. Reynolds of Beth Israel Deaconess Medical Center, Boston, wrote that this study, and another study published in the same issue regarding insulin resistance and polycystic ovary syndrome, “highlight the important role that the dermatologist plays in identifying and characterizing patients with common skin disorders who may be at risk for metabolic and androgen-mediated disease.” The study, “to our knowledge, [is] the largest cohort to date examining the prevalence of insulin resistance and metabolic syndrome in postadolescent males with acne of varying severity,” she added (doi: 10.1001/jamadermatol.2015.4500).
The authors of the study had no disclosures.
FROM JAMA DERMATOLOGY
Key clinical point: Acne in young men may be a sign of insulin resistance.
Major finding: 22% of the young men with acne had insulin resistance, compared with 11% of the age-matched controls, a significant difference (P = .036).
Data source: The cross-sectional study compared the prevalence of insulin resistance and metabolic syndrome in 100 men aged 20-32 years with acne and 100 age-matched controls without acne.
Disclosures: The authors had no disclosures.
FDA approves pembrolizumab as first-line advanced melanoma therapy
Pembrolizumab, the programmed death receptor-1 (PD-1)–blocking antibody, is now approved as a first-line treatment of unresectable or metastatic melanoma, based on the results of the KEYNOTE-006 study.
This is the second approval for an advanced melanoma indication for pembrolizumab (Keytruda), according to a statement issued by Merck, the manufacturer. Pembrolizumab was first approved in 2014 for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
The FDA-approved dose is 2 mg/kg every 3 weeks.
In KEYNOTE-006, a phase III study that compared pembrolizumab to ipilimumab (Yervoy), also a PD-1–blocking antibody, in 834 patients with unresectable or metastatic melanoma with progression of disease, overall survival and progression-free survival were significantly increased among those treated with pembrolizumab, compared with ipilimumab.
The results were reported at the annual meeting of the American Association for Cancer Research in April, and published simultaneously in the New England Journal of Medicine (2015; 372:2521-32).
Pembrolizumab, the programmed death receptor-1 (PD-1)–blocking antibody, is now approved as a first-line treatment of unresectable or metastatic melanoma, based on the results of the KEYNOTE-006 study.
This is the second approval for an advanced melanoma indication for pembrolizumab (Keytruda), according to a statement issued by Merck, the manufacturer. Pembrolizumab was first approved in 2014 for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
The FDA-approved dose is 2 mg/kg every 3 weeks.
In KEYNOTE-006, a phase III study that compared pembrolizumab to ipilimumab (Yervoy), also a PD-1–blocking antibody, in 834 patients with unresectable or metastatic melanoma with progression of disease, overall survival and progression-free survival were significantly increased among those treated with pembrolizumab, compared with ipilimumab.
The results were reported at the annual meeting of the American Association for Cancer Research in April, and published simultaneously in the New England Journal of Medicine (2015; 372:2521-32).
Pembrolizumab, the programmed death receptor-1 (PD-1)–blocking antibody, is now approved as a first-line treatment of unresectable or metastatic melanoma, based on the results of the KEYNOTE-006 study.
This is the second approval for an advanced melanoma indication for pembrolizumab (Keytruda), according to a statement issued by Merck, the manufacturer. Pembrolizumab was first approved in 2014 for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor.
The FDA-approved dose is 2 mg/kg every 3 weeks.
In KEYNOTE-006, a phase III study that compared pembrolizumab to ipilimumab (Yervoy), also a PD-1–blocking antibody, in 834 patients with unresectable or metastatic melanoma with progression of disease, overall survival and progression-free survival were significantly increased among those treated with pembrolizumab, compared with ipilimumab.
The results were reported at the annual meeting of the American Association for Cancer Research in April, and published simultaneously in the New England Journal of Medicine (2015; 372:2521-32).
‘Cytokine converter’ cells alleviate psoriasis in mice
Implantation of cells designed to sense the presence of proinflammatory cytokines and subsequently produce and deliver anti-inflammatory molecules prevented flares of psoriasis in mice, according to Lina Schukur of the department of Biosystems Science and Engineering, ETH Zurich, in Basel, Switzerland, and associates.
The investigators designed and engineered human cells that they explain “sequentially detected elevated TNF [tumor necrosis factor] and IL-22 [interleukin-22] levels from a psoriatic flare and, in response, produced therapeutic doses of IL-4 [interleukin-4] and IL-10 [interleukin-10].” TNF and IL-22 are proinflammatory cytokines, which have been found to be upregulated in patients with active psoriasis; and IL-4 and IL-10 are immunomodulatory cytokines that have been shown to induce rapid improvements in people with psoriasis in phase II clinical trials, they noted.
The researchers implanted these designer cells in mice with psoriasis-like lesions, which showed that this “antipsoriatic cytokine converter network” was able to improve skin lesions “and restore dermal tissue morphology.” They also found that the cells were responsive in blood samples from psoriasis patients, which suggested that the cytokine converter “is sufficiently sensitive to detect circulating TNF and IL-22 in humans.”
“These synthetic circuits, which program designer cells to process complex metabolic information, open the door to autonomously prevent, attenuate, or reset acute or chronic medical conditions without constant injections of drugs or cumbersome dosing schedules, and thus provide a new opportunity for personalized medicine,” the investigators wrote.
An editor’s summary of the study states that, “in demonstrating that the converter cells were responsive to blood from psoriasis patients, the authors suggest that synthetic biology may be ready to autonomously flip therapeutic switches in people and later take on other diseases with defined disease indicators.”
The full study, published on Dec. 16, 2015, is available in Science Translational Medicine at http://stm.sciencemag.org/content/7/318/318ra201.
Implantation of cells designed to sense the presence of proinflammatory cytokines and subsequently produce and deliver anti-inflammatory molecules prevented flares of psoriasis in mice, according to Lina Schukur of the department of Biosystems Science and Engineering, ETH Zurich, in Basel, Switzerland, and associates.
The investigators designed and engineered human cells that they explain “sequentially detected elevated TNF [tumor necrosis factor] and IL-22 [interleukin-22] levels from a psoriatic flare and, in response, produced therapeutic doses of IL-4 [interleukin-4] and IL-10 [interleukin-10].” TNF and IL-22 are proinflammatory cytokines, which have been found to be upregulated in patients with active psoriasis; and IL-4 and IL-10 are immunomodulatory cytokines that have been shown to induce rapid improvements in people with psoriasis in phase II clinical trials, they noted.
The researchers implanted these designer cells in mice with psoriasis-like lesions, which showed that this “antipsoriatic cytokine converter network” was able to improve skin lesions “and restore dermal tissue morphology.” They also found that the cells were responsive in blood samples from psoriasis patients, which suggested that the cytokine converter “is sufficiently sensitive to detect circulating TNF and IL-22 in humans.”
“These synthetic circuits, which program designer cells to process complex metabolic information, open the door to autonomously prevent, attenuate, or reset acute or chronic medical conditions without constant injections of drugs or cumbersome dosing schedules, and thus provide a new opportunity for personalized medicine,” the investigators wrote.
An editor’s summary of the study states that, “in demonstrating that the converter cells were responsive to blood from psoriasis patients, the authors suggest that synthetic biology may be ready to autonomously flip therapeutic switches in people and later take on other diseases with defined disease indicators.”
The full study, published on Dec. 16, 2015, is available in Science Translational Medicine at http://stm.sciencemag.org/content/7/318/318ra201.
Implantation of cells designed to sense the presence of proinflammatory cytokines and subsequently produce and deliver anti-inflammatory molecules prevented flares of psoriasis in mice, according to Lina Schukur of the department of Biosystems Science and Engineering, ETH Zurich, in Basel, Switzerland, and associates.
The investigators designed and engineered human cells that they explain “sequentially detected elevated TNF [tumor necrosis factor] and IL-22 [interleukin-22] levels from a psoriatic flare and, in response, produced therapeutic doses of IL-4 [interleukin-4] and IL-10 [interleukin-10].” TNF and IL-22 are proinflammatory cytokines, which have been found to be upregulated in patients with active psoriasis; and IL-4 and IL-10 are immunomodulatory cytokines that have been shown to induce rapid improvements in people with psoriasis in phase II clinical trials, they noted.
The researchers implanted these designer cells in mice with psoriasis-like lesions, which showed that this “antipsoriatic cytokine converter network” was able to improve skin lesions “and restore dermal tissue morphology.” They also found that the cells were responsive in blood samples from psoriasis patients, which suggested that the cytokine converter “is sufficiently sensitive to detect circulating TNF and IL-22 in humans.”
“These synthetic circuits, which program designer cells to process complex metabolic information, open the door to autonomously prevent, attenuate, or reset acute or chronic medical conditions without constant injections of drugs or cumbersome dosing schedules, and thus provide a new opportunity for personalized medicine,” the investigators wrote.
An editor’s summary of the study states that, “in demonstrating that the converter cells were responsive to blood from psoriasis patients, the authors suggest that synthetic biology may be ready to autonomously flip therapeutic switches in people and later take on other diseases with defined disease indicators.”
The full study, published on Dec. 16, 2015, is available in Science Translational Medicine at http://stm.sciencemag.org/content/7/318/318ra201.
FROM SCIENCE TRANSLATIONAL MEDICINE
FDA approves treatment for chemotherapy ODs, life-threatening toxicities
Uridine triacetate, a pyrimidine analogue, has been approved for the emergency treatment of fluorouracil or capecitabine overdoses in adults and children, and for patients who develop “certain severe or life-threatening toxicities within 4 days of receiving” these treatments, the Food and Drug Administration announced on Dec. 11.

“Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents,” Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement. It will be marketed as Vistogard by Wellstat Therapeutics.
Uridine comes in an oral granule formulation that can be mixed into soft foods or, when necessary, administered via a nasogastric or gastrostomy tube, the prescribing information states. The indication is for use after an overdose “regardless of the presence of symptoms,” and for treating “early-onset, severe, or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration,” according to the prescribing information.
Uridine blocks cell damage and cell death caused by fluorouracil chemotherapy, according to the statement, which adds that it is up to the patient’s health care provider to “determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.”
Uridine was evaluated in two studies of 135 adults and children with cancer, treated with uridine for a fluorouracil or capecitabine overdose, or for early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving fluorouracil (not because of an overdose). Among those treated for an overdose, 97% were alive 30 days after treatment, and among those treated for early-onset severe or life-threatening toxicity, 89% were alive 30 days after treatment. In addition, 33% of the patients resumed chemotherapy within 30 days, according to the FDA statement. Diarrhea, vomiting, and nausea were the most common adverse events associated with treatment.
Uridine was granted orphan drug, priority review, and fast track designations.
Uridine triacetate, a pyrimidine analogue, has been approved for the emergency treatment of fluorouracil or capecitabine overdoses in adults and children, and for patients who develop “certain severe or life-threatening toxicities within 4 days of receiving” these treatments, the Food and Drug Administration announced on Dec. 11.
“Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents,” Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement. It will be marketed as Vistogard by Wellstat Therapeutics.
Uridine comes in an oral granule formulation that can be mixed into soft foods or, when necessary, administered via a nasogastric or gastrostomy tube, the prescribing information states. The indication is for use after an overdose “regardless of the presence of symptoms,” and for treating “early-onset, severe, or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration,” according to the prescribing information.
Uridine blocks cell damage and cell death caused by fluorouracil chemotherapy, according to the statement, which adds that it is up to the patient’s health care provider to “determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.”
Uridine was evaluated in two studies of 135 adults and children with cancer, treated with uridine for a fluorouracil or capecitabine overdose, or for early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving fluorouracil (not because of an overdose). Among those treated for an overdose, 97% were alive 30 days after treatment, and among those treated for early-onset severe or life-threatening toxicity, 89% were alive 30 days after treatment. In addition, 33% of the patients resumed chemotherapy within 30 days, according to the FDA statement. Diarrhea, vomiting, and nausea were the most common adverse events associated with treatment.
Uridine was granted orphan drug, priority review, and fast track designations.
Uridine triacetate, a pyrimidine analogue, has been approved for the emergency treatment of fluorouracil or capecitabine overdoses in adults and children, and for patients who develop “certain severe or life-threatening toxicities within 4 days of receiving” these treatments, the Food and Drug Administration announced on Dec. 11.
“Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents,” Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement. It will be marketed as Vistogard by Wellstat Therapeutics.
Uridine comes in an oral granule formulation that can be mixed into soft foods or, when necessary, administered via a nasogastric or gastrostomy tube, the prescribing information states. The indication is for use after an overdose “regardless of the presence of symptoms,” and for treating “early-onset, severe, or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration,” according to the prescribing information.
Uridine blocks cell damage and cell death caused by fluorouracil chemotherapy, according to the statement, which adds that it is up to the patient’s health care provider to “determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.”
Uridine was evaluated in two studies of 135 adults and children with cancer, treated with uridine for a fluorouracil or capecitabine overdose, or for early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving fluorouracil (not because of an overdose). Among those treated for an overdose, 97% were alive 30 days after treatment, and among those treated for early-onset severe or life-threatening toxicity, 89% were alive 30 days after treatment. In addition, 33% of the patients resumed chemotherapy within 30 days, according to the FDA statement. Diarrhea, vomiting, and nausea were the most common adverse events associated with treatment.
Uridine was granted orphan drug, priority review, and fast track designations.
Necitumumab approved as first-line treatment of metastatic squamous NSCLC
Necitumumab, an epidermal growth factor receptor (EGFR) antagonist, combined with two forms of chemotherapy, has been approved as a first-line treatment of patients with metastatic squamous non–small cell lung cancer (NSCLC). the Food and Drug Administration announced on Nov. 24.
Necitumumab is a “monoclonal antibody that blocks activity of EGFR, a protein commonly found on squamous NSCLC tumors,” the FDA statement said. The statement points put that necitumumab “was not found to be an effective treatment” in patients with nonsquamous NSCLC.
Eli Lilly will market necitumumab as Portrazza. It is administered as an intravenous infusion over 60 minutes on days 1 and 8 of each 3-week cycle.
“Today’s approval provides certain patients with squamous cell lung cancer a new option that may extend survival,” Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA Center for Drug Evaluation and Research, said in the statement.
Necitumumab was evaluated in multicenter, randomized, open-label study of 1,093 people with advanced squamous NSCLC, treated with gemcitabine and cisplatin with or without necitumumab. Median overall survival, the primary endpoint, was 11.5 months in the necitumumab-treated arm vs. 9.9 months among controls, a 1.6 month difference that was statistically significant.
At a July meeting of the FDA’s Oncologic Drugs Advisory Committee, 11 of the 12 panelists agreed that the efficacy and safety results of this study supported a “positive benefit risk assessment” of necitumumab combined with gemcitabine and cisplatin for this group of patients – despite a modest effect on overall survival, a very small but statistically significant effect on progression-free survival, and some safety concerns, the main question they were asked by the FDA to discuss.
Rash and hypomagnesemia are the most common adverse events associated with necitumumab, according to the FDA.
The prescribing information includes a boxed warning regarding the risk of cardiopulmonary arrest and hypomagnesemia associated with treatment, and points out that 83% of the patients treated with necitumumab, gemcitabine, and cisplatin developed hypomagnesemia, which was severe in 20%; and that the rate of cardiopulmonary arrest and/or sudden death occurred in 3% of those on the combination.
Serious adverse events associated with necitumumab should be reported to the FDA’s MedWatch program.
Necitumumab, an epidermal growth factor receptor (EGFR) antagonist, combined with two forms of chemotherapy, has been approved as a first-line treatment of patients with metastatic squamous non–small cell lung cancer (NSCLC). the Food and Drug Administration announced on Nov. 24.
Necitumumab is a “monoclonal antibody that blocks activity of EGFR, a protein commonly found on squamous NSCLC tumors,” the FDA statement said. The statement points put that necitumumab “was not found to be an effective treatment” in patients with nonsquamous NSCLC.
Eli Lilly will market necitumumab as Portrazza. It is administered as an intravenous infusion over 60 minutes on days 1 and 8 of each 3-week cycle.
“Today’s approval provides certain patients with squamous cell lung cancer a new option that may extend survival,” Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA Center for Drug Evaluation and Research, said in the statement.
Necitumumab was evaluated in multicenter, randomized, open-label study of 1,093 people with advanced squamous NSCLC, treated with gemcitabine and cisplatin with or without necitumumab. Median overall survival, the primary endpoint, was 11.5 months in the necitumumab-treated arm vs. 9.9 months among controls, a 1.6 month difference that was statistically significant.
At a July meeting of the FDA’s Oncologic Drugs Advisory Committee, 11 of the 12 panelists agreed that the efficacy and safety results of this study supported a “positive benefit risk assessment” of necitumumab combined with gemcitabine and cisplatin for this group of patients – despite a modest effect on overall survival, a very small but statistically significant effect on progression-free survival, and some safety concerns, the main question they were asked by the FDA to discuss.
Rash and hypomagnesemia are the most common adverse events associated with necitumumab, according to the FDA.
The prescribing information includes a boxed warning regarding the risk of cardiopulmonary arrest and hypomagnesemia associated with treatment, and points out that 83% of the patients treated with necitumumab, gemcitabine, and cisplatin developed hypomagnesemia, which was severe in 20%; and that the rate of cardiopulmonary arrest and/or sudden death occurred in 3% of those on the combination.
Serious adverse events associated with necitumumab should be reported to the FDA’s MedWatch program.
Necitumumab, an epidermal growth factor receptor (EGFR) antagonist, combined with two forms of chemotherapy, has been approved as a first-line treatment of patients with metastatic squamous non–small cell lung cancer (NSCLC). the Food and Drug Administration announced on Nov. 24.
Necitumumab is a “monoclonal antibody that blocks activity of EGFR, a protein commonly found on squamous NSCLC tumors,” the FDA statement said. The statement points put that necitumumab “was not found to be an effective treatment” in patients with nonsquamous NSCLC.
Eli Lilly will market necitumumab as Portrazza. It is administered as an intravenous infusion over 60 minutes on days 1 and 8 of each 3-week cycle.
“Today’s approval provides certain patients with squamous cell lung cancer a new option that may extend survival,” Dr. Richard Pazdur, director of the office of hematology and oncology products in the FDA Center for Drug Evaluation and Research, said in the statement.
Necitumumab was evaluated in multicenter, randomized, open-label study of 1,093 people with advanced squamous NSCLC, treated with gemcitabine and cisplatin with or without necitumumab. Median overall survival, the primary endpoint, was 11.5 months in the necitumumab-treated arm vs. 9.9 months among controls, a 1.6 month difference that was statistically significant.
At a July meeting of the FDA’s Oncologic Drugs Advisory Committee, 11 of the 12 panelists agreed that the efficacy and safety results of this study supported a “positive benefit risk assessment” of necitumumab combined with gemcitabine and cisplatin for this group of patients – despite a modest effect on overall survival, a very small but statistically significant effect on progression-free survival, and some safety concerns, the main question they were asked by the FDA to discuss.
Rash and hypomagnesemia are the most common adverse events associated with necitumumab, according to the FDA.
The prescribing information includes a boxed warning regarding the risk of cardiopulmonary arrest and hypomagnesemia associated with treatment, and points out that 83% of the patients treated with necitumumab, gemcitabine, and cisplatin developed hypomagnesemia, which was severe in 20%; and that the rate of cardiopulmonary arrest and/or sudden death occurred in 3% of those on the combination.
Serious adverse events associated with necitumumab should be reported to the FDA’s MedWatch program.
Nivolumab approved as first-line, single agent for advanced melanoma
Nivolumab, a programmed death receptor–1 (PD-1) blocking antibody, has been approved as a single agent for the first-line treatment of patients with BRAF V600 wild-type unresectable or metastatic melanoma, the manufacturer has announced.
The approval is based on the results of a phase III study, CheckMate 066, which found a significant overall survival (OS) benefit for nivolumab, compared with chemotherapy, as a first-line treatment of patients with BRAF wild-type advanced melanoma, according to a press release issued by Bristol Myers Squibb (BMS) on Nov. 24.

The company markets nivolumab as Opdivo, which was first approved by the Food and Drug Administration in December 2014 for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab (Yervoy) treatment and, if BRAF V600 mutation positive, a BRAF inhibitor.
OS was the primary endpoint in the CheckMate 066 trial, which compared treatment with nivolumab (3mg/kg administered intravenously every 2 weeks) to dacarbazine (1,000 mg/m2 administered intravenously every 3 weeks) administered in 418 treatment-naive patients with BRAF wild-type unresectable or metastatic melanoma. In an interim analysis, the median OS for those on dacarbazine was 10.8 months, but was not reached for those on nivolumab (hazard ratio, 0.42; P less than .0001), according to BMS.
The study was stopped early in 2014, after the OS results were noted, the company statement said. At the time the study was designed, ipilimumab had not yet been approved, and dacarbazine was chosen as the comparator because “it represented the standard of care in many regions outside of the U.S.,” the statement added.
Other results included a significantly greater median PFS among those on nivolumab vs. dacarbazine (5.1 months vs. 2.2 months).
In the study, 36% of nivolumab-treated patients had serious adverse reactions; and 41% had grade 3 and 4 adverse reactions, the most common included an increase in gamma-glutamyl transferase in almost 4%, and diarrhea in 3.4%. In addition, 7% stopped treatment permanently because of adverse events, and treatment had to be interrupted in 26%.
The most common adverse reactions reported by those on nivolumab included fatigue, in 49% (vs. 39% of those on dacarbazine) and musculoskeletal pain in 32% (vs. 25%). Rash and pruritus were reported by 28% and 23%, respectively, among those on nivolumab vs. 12% for both rash and pruritus among those on dacarbazine, BMS said.
Nivolumab, a programmed death receptor–1 (PD-1) blocking antibody, has been approved as a single agent for the first-line treatment of patients with BRAF V600 wild-type unresectable or metastatic melanoma, the manufacturer has announced.
The approval is based on the results of a phase III study, CheckMate 066, which found a significant overall survival (OS) benefit for nivolumab, compared with chemotherapy, as a first-line treatment of patients with BRAF wild-type advanced melanoma, according to a press release issued by Bristol Myers Squibb (BMS) on Nov. 24.
The company markets nivolumab as Opdivo, which was first approved by the Food and Drug Administration in December 2014 for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab (Yervoy) treatment and, if BRAF V600 mutation positive, a BRAF inhibitor.
OS was the primary endpoint in the CheckMate 066 trial, which compared treatment with nivolumab (3mg/kg administered intravenously every 2 weeks) to dacarbazine (1,000 mg/m2 administered intravenously every 3 weeks) administered in 418 treatment-naive patients with BRAF wild-type unresectable or metastatic melanoma. In an interim analysis, the median OS for those on dacarbazine was 10.8 months, but was not reached for those on nivolumab (hazard ratio, 0.42; P less than .0001), according to BMS.
The study was stopped early in 2014, after the OS results were noted, the company statement said. At the time the study was designed, ipilimumab had not yet been approved, and dacarbazine was chosen as the comparator because “it represented the standard of care in many regions outside of the U.S.,” the statement added.
Other results included a significantly greater median PFS among those on nivolumab vs. dacarbazine (5.1 months vs. 2.2 months).
In the study, 36% of nivolumab-treated patients had serious adverse reactions; and 41% had grade 3 and 4 adverse reactions, the most common included an increase in gamma-glutamyl transferase in almost 4%, and diarrhea in 3.4%. In addition, 7% stopped treatment permanently because of adverse events, and treatment had to be interrupted in 26%.
The most common adverse reactions reported by those on nivolumab included fatigue, in 49% (vs. 39% of those on dacarbazine) and musculoskeletal pain in 32% (vs. 25%). Rash and pruritus were reported by 28% and 23%, respectively, among those on nivolumab vs. 12% for both rash and pruritus among those on dacarbazine, BMS said.
Nivolumab, a programmed death receptor–1 (PD-1) blocking antibody, has been approved as a single agent for the first-line treatment of patients with BRAF V600 wild-type unresectable or metastatic melanoma, the manufacturer has announced.
The approval is based on the results of a phase III study, CheckMate 066, which found a significant overall survival (OS) benefit for nivolumab, compared with chemotherapy, as a first-line treatment of patients with BRAF wild-type advanced melanoma, according to a press release issued by Bristol Myers Squibb (BMS) on Nov. 24.
The company markets nivolumab as Opdivo, which was first approved by the Food and Drug Administration in December 2014 for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab (Yervoy) treatment and, if BRAF V600 mutation positive, a BRAF inhibitor.
OS was the primary endpoint in the CheckMate 066 trial, which compared treatment with nivolumab (3mg/kg administered intravenously every 2 weeks) to dacarbazine (1,000 mg/m2 administered intravenously every 3 weeks) administered in 418 treatment-naive patients with BRAF wild-type unresectable or metastatic melanoma. In an interim analysis, the median OS for those on dacarbazine was 10.8 months, but was not reached for those on nivolumab (hazard ratio, 0.42; P less than .0001), according to BMS.
The study was stopped early in 2014, after the OS results were noted, the company statement said. At the time the study was designed, ipilimumab had not yet been approved, and dacarbazine was chosen as the comparator because “it represented the standard of care in many regions outside of the U.S.,” the statement added.
Other results included a significantly greater median PFS among those on nivolumab vs. dacarbazine (5.1 months vs. 2.2 months).
In the study, 36% of nivolumab-treated patients had serious adverse reactions; and 41% had grade 3 and 4 adverse reactions, the most common included an increase in gamma-glutamyl transferase in almost 4%, and diarrhea in 3.4%. In addition, 7% stopped treatment permanently because of adverse events, and treatment had to be interrupted in 26%.
The most common adverse reactions reported by those on nivolumab included fatigue, in 49% (vs. 39% of those on dacarbazine) and musculoskeletal pain in 32% (vs. 25%). Rash and pruritus were reported by 28% and 23%, respectively, among those on nivolumab vs. 12% for both rash and pruritus among those on dacarbazine, BMS said.

Topical calcipotriene-betamethasone combination approved for plaque psoriasis
A combination of calcipotriene and betamethasone dipropionate in a foam formulation has been approved by the Food and Drug Administration for the topical treatment of plaque psoriasis in adults, according to the manufacturer.
The product will be marketed as Enstilar (calcipotriene and betamethasone dipropionate) foam, 0.005%/0.064%, by Leo Pharma.
Each gram contains 52.2 mcg calcipotriene hydrate (equivalent to 50 mcg of calcipotriene) and 0.643 mg of betamethasone dipropionate (equivalent to 0.5 mg of betamethasone); it is applied to affected areas once a day for up to 4 weeks, according to the prescribing information, which states that patient should not use more than 60 g every 4 days.
The vitamin D analog-corticosteroid combination product was compared with treatment with the two individual components and/or a vehicle only, in more than 700 patients with plaque psoriasis in two studies. At baseline, about two-thirds of the patients had moderate disease, 14%-15% had mild disease, and 10% had moderately severe disease, based on a 5-point Investigator’s Global Assessment scale. The proportion of patients who were considered clear or almost clear at 4 weeks, based on IGA scale results, was the efficacy endpoint defining treatment success. Among patients with mild disease at baseline, treatment was considered successful if they were considered clear at 4 weeks.
In one study of 302 patients, 45% treated with the combination product had achieved treatment success at week 4 vs. 30.7% of those treated with betamethasone and 14.9% treated with calcipotriene, according to the prescribing information. In the second study, 323 patients were treated with the combination product and 103 received the vehicle. At 4 weeks, 53.3% of those treated with the foam met the efficacy endpoint vs. 4.8% of those in the vehicle group.
Application site irritation, application site pruritus, folliculitis, skin hypopigmentation, hypercalcemia, urticaria, and psoriasis exacerbations were among the adverse reactions reported in fewer than 1% of those treated with the combination product. The warnings and precautions section of the prescribing information includes a statement noting that the product contains propellants that are flammable, and that patients should be advised to “avoid fire, flame, or smoking during and immediately after using this product.”
This approval is for an adult population. As a postmarketing commitment, the FDA is requiring that the company conduct a study in pediatric patients with plaque psoriasis, according to the agency’s approval letter, dated Oct. 16. That study is described as an open-label study to assess the effect of the product on calcium metabolism in pediatric patients aged 12-16 years, with plaque psoriasis of the scalp and body. In a subgroup of these children, pharmacokinetics and hypothalamic-pituitary axis suppression also will be assessed, the letter says.
Serious adverse events associated with this product should be reported to the FDA’s MedWatch program at 800-332-1088, or http://www.fda.gov/Safety/MedWatch/default.htm.
A combination of calcipotriene and betamethasone dipropionate in a foam formulation has been approved by the Food and Drug Administration for the topical treatment of plaque psoriasis in adults, according to the manufacturer.
The product will be marketed as Enstilar (calcipotriene and betamethasone dipropionate) foam, 0.005%/0.064%, by Leo Pharma.
Each gram contains 52.2 mcg calcipotriene hydrate (equivalent to 50 mcg of calcipotriene) and 0.643 mg of betamethasone dipropionate (equivalent to 0.5 mg of betamethasone); it is applied to affected areas once a day for up to 4 weeks, according to the prescribing information, which states that patient should not use more than 60 g every 4 days.
The vitamin D analog-corticosteroid combination product was compared with treatment with the two individual components and/or a vehicle only, in more than 700 patients with plaque psoriasis in two studies. At baseline, about two-thirds of the patients had moderate disease, 14%-15% had mild disease, and 10% had moderately severe disease, based on a 5-point Investigator’s Global Assessment scale. The proportion of patients who were considered clear or almost clear at 4 weeks, based on IGA scale results, was the efficacy endpoint defining treatment success. Among patients with mild disease at baseline, treatment was considered successful if they were considered clear at 4 weeks.
In one study of 302 patients, 45% treated with the combination product had achieved treatment success at week 4 vs. 30.7% of those treated with betamethasone and 14.9% treated with calcipotriene, according to the prescribing information. In the second study, 323 patients were treated with the combination product and 103 received the vehicle. At 4 weeks, 53.3% of those treated with the foam met the efficacy endpoint vs. 4.8% of those in the vehicle group.
Application site irritation, application site pruritus, folliculitis, skin hypopigmentation, hypercalcemia, urticaria, and psoriasis exacerbations were among the adverse reactions reported in fewer than 1% of those treated with the combination product. The warnings and precautions section of the prescribing information includes a statement noting that the product contains propellants that are flammable, and that patients should be advised to “avoid fire, flame, or smoking during and immediately after using this product.”
This approval is for an adult population. As a postmarketing commitment, the FDA is requiring that the company conduct a study in pediatric patients with plaque psoriasis, according to the agency’s approval letter, dated Oct. 16. That study is described as an open-label study to assess the effect of the product on calcium metabolism in pediatric patients aged 12-16 years, with plaque psoriasis of the scalp and body. In a subgroup of these children, pharmacokinetics and hypothalamic-pituitary axis suppression also will be assessed, the letter says.
Serious adverse events associated with this product should be reported to the FDA’s MedWatch program at 800-332-1088, or http://www.fda.gov/Safety/MedWatch/default.htm.
A combination of calcipotriene and betamethasone dipropionate in a foam formulation has been approved by the Food and Drug Administration for the topical treatment of plaque psoriasis in adults, according to the manufacturer.
The product will be marketed as Enstilar (calcipotriene and betamethasone dipropionate) foam, 0.005%/0.064%, by Leo Pharma.
Each gram contains 52.2 mcg calcipotriene hydrate (equivalent to 50 mcg of calcipotriene) and 0.643 mg of betamethasone dipropionate (equivalent to 0.5 mg of betamethasone); it is applied to affected areas once a day for up to 4 weeks, according to the prescribing information, which states that patient should not use more than 60 g every 4 days.
The vitamin D analog-corticosteroid combination product was compared with treatment with the two individual components and/or a vehicle only, in more than 700 patients with plaque psoriasis in two studies. At baseline, about two-thirds of the patients had moderate disease, 14%-15% had mild disease, and 10% had moderately severe disease, based on a 5-point Investigator’s Global Assessment scale. The proportion of patients who were considered clear or almost clear at 4 weeks, based on IGA scale results, was the efficacy endpoint defining treatment success. Among patients with mild disease at baseline, treatment was considered successful if they were considered clear at 4 weeks.
In one study of 302 patients, 45% treated with the combination product had achieved treatment success at week 4 vs. 30.7% of those treated with betamethasone and 14.9% treated with calcipotriene, according to the prescribing information. In the second study, 323 patients were treated with the combination product and 103 received the vehicle. At 4 weeks, 53.3% of those treated with the foam met the efficacy endpoint vs. 4.8% of those in the vehicle group.
Application site irritation, application site pruritus, folliculitis, skin hypopigmentation, hypercalcemia, urticaria, and psoriasis exacerbations were among the adverse reactions reported in fewer than 1% of those treated with the combination product. The warnings and precautions section of the prescribing information includes a statement noting that the product contains propellants that are flammable, and that patients should be advised to “avoid fire, flame, or smoking during and immediately after using this product.”
This approval is for an adult population. As a postmarketing commitment, the FDA is requiring that the company conduct a study in pediatric patients with plaque psoriasis, according to the agency’s approval letter, dated Oct. 16. That study is described as an open-label study to assess the effect of the product on calcium metabolism in pediatric patients aged 12-16 years, with plaque psoriasis of the scalp and body. In a subgroup of these children, pharmacokinetics and hypothalamic-pituitary axis suppression also will be assessed, the letter says.
Serious adverse events associated with this product should be reported to the FDA’s MedWatch program at 800-332-1088, or http://www.fda.gov/Safety/MedWatch/default.htm.