User login
Make the Diagnosis - March 2020
The patient’s biopsy showed sparse and grouped and slightly enlarged atypical stained mononuclear cells in mostly perifollicular areas with focal epidermotropism. CD30 staining was positive. She responded to potent topical steroids.
The etiology of LyP is unknown. It is unclear whether the proliferation of T-cells is a benign and chronic disorder, or an indolent T-cell malignancy.
In addition, 10% of LyP cases are associated with anaplastic large-cell lymphoma, cutaneous T-cell lymphoma (mycosis fungoides), or Hodgkin lymphoma. Borderline cases are those that overlap LyP and lymphoma.
Patients typically present with crops of asymptomatic erythematous to brown papules that may become pustular, vesicular, or necrotic. Lesions tend to resolve within 2-8 weeks with or without scarring. The trunk and extremities are commonly affected. The condition tends to be chronic over months to years. The waxing and waning course is characteristic of LyP. Constitutional symptoms are generally absent in cases not associated with systemic disease.

Histopathologic examination reveals a dense wedge-shaped dermal infiltrate of atypical lymphocytes along with numerous eosinophils and neutrophils. Epidermotropism may be present and lymphocytes stain positive for CD30+. Vessels in the dermis may exhibit fibrin deposition and red blood cell extravasation. Histologically, LyP can be classified as Type A to E. These subtypes are determined by the size and type of atypical cells, location and amount of infiltrate, and staining of CD30 and CD8.
The differential diagnosis of LyP includes pityriasis lichenoides, anaplastic large cell lymphoma, cutaneous T-cell lymphoma, folliculitis, arthropod assault, Langerhans cell histiocytosis, and leukemia cutis. Treatment is symptomatic. Mild forms of LyP can many times be managed with superpotent topical corticosteroids. Bexarotene gel has been used for early lesions. For more widespread or persistent disease, intralesional corticosteroids, phototherapy (UVB or PUVA), tetracycline antibiotics, and methotrexate have been reported to be effective. Refractory cases may respond to interferon alpha or oral bexarotene. Routine evaluations are recommended as patients may be at increased risk for the development of lymphoma.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
The patient’s biopsy showed sparse and grouped and slightly enlarged atypical stained mononuclear cells in mostly perifollicular areas with focal epidermotropism. CD30 staining was positive. She responded to potent topical steroids.
The etiology of LyP is unknown. It is unclear whether the proliferation of T-cells is a benign and chronic disorder, or an indolent T-cell malignancy.
In addition, 10% of LyP cases are associated with anaplastic large-cell lymphoma, cutaneous T-cell lymphoma (mycosis fungoides), or Hodgkin lymphoma. Borderline cases are those that overlap LyP and lymphoma.
Patients typically present with crops of asymptomatic erythematous to brown papules that may become pustular, vesicular, or necrotic. Lesions tend to resolve within 2-8 weeks with or without scarring. The trunk and extremities are commonly affected. The condition tends to be chronic over months to years. The waxing and waning course is characteristic of LyP. Constitutional symptoms are generally absent in cases not associated with systemic disease.
Histopathologic examination reveals a dense wedge-shaped dermal infiltrate of atypical lymphocytes along with numerous eosinophils and neutrophils. Epidermotropism may be present and lymphocytes stain positive for CD30+. Vessels in the dermis may exhibit fibrin deposition and red blood cell extravasation. Histologically, LyP can be classified as Type A to E. These subtypes are determined by the size and type of atypical cells, location and amount of infiltrate, and staining of CD30 and CD8.
The differential diagnosis of LyP includes pityriasis lichenoides, anaplastic large cell lymphoma, cutaneous T-cell lymphoma, folliculitis, arthropod assault, Langerhans cell histiocytosis, and leukemia cutis. Treatment is symptomatic. Mild forms of LyP can many times be managed with superpotent topical corticosteroids. Bexarotene gel has been used for early lesions. For more widespread or persistent disease, intralesional corticosteroids, phototherapy (UVB or PUVA), tetracycline antibiotics, and methotrexate have been reported to be effective. Refractory cases may respond to interferon alpha or oral bexarotene. Routine evaluations are recommended as patients may be at increased risk for the development of lymphoma.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
The patient’s biopsy showed sparse and grouped and slightly enlarged atypical stained mononuclear cells in mostly perifollicular areas with focal epidermotropism. CD30 staining was positive. She responded to potent topical steroids.
The etiology of LyP is unknown. It is unclear whether the proliferation of T-cells is a benign and chronic disorder, or an indolent T-cell malignancy.
In addition, 10% of LyP cases are associated with anaplastic large-cell lymphoma, cutaneous T-cell lymphoma (mycosis fungoides), or Hodgkin lymphoma. Borderline cases are those that overlap LyP and lymphoma.
Patients typically present with crops of asymptomatic erythematous to brown papules that may become pustular, vesicular, or necrotic. Lesions tend to resolve within 2-8 weeks with or without scarring. The trunk and extremities are commonly affected. The condition tends to be chronic over months to years. The waxing and waning course is characteristic of LyP. Constitutional symptoms are generally absent in cases not associated with systemic disease.
Histopathologic examination reveals a dense wedge-shaped dermal infiltrate of atypical lymphocytes along with numerous eosinophils and neutrophils. Epidermotropism may be present and lymphocytes stain positive for CD30+. Vessels in the dermis may exhibit fibrin deposition and red blood cell extravasation. Histologically, LyP can be classified as Type A to E. These subtypes are determined by the size and type of atypical cells, location and amount of infiltrate, and staining of CD30 and CD8.
The differential diagnosis of LyP includes pityriasis lichenoides, anaplastic large cell lymphoma, cutaneous T-cell lymphoma, folliculitis, arthropod assault, Langerhans cell histiocytosis, and leukemia cutis. Treatment is symptomatic. Mild forms of LyP can many times be managed with superpotent topical corticosteroids. Bexarotene gel has been used for early lesions. For more widespread or persistent disease, intralesional corticosteroids, phototherapy (UVB or PUVA), tetracycline antibiotics, and methotrexate have been reported to be effective. Refractory cases may respond to interferon alpha or oral bexarotene. Routine evaluations are recommended as patients may be at increased risk for the development of lymphoma.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
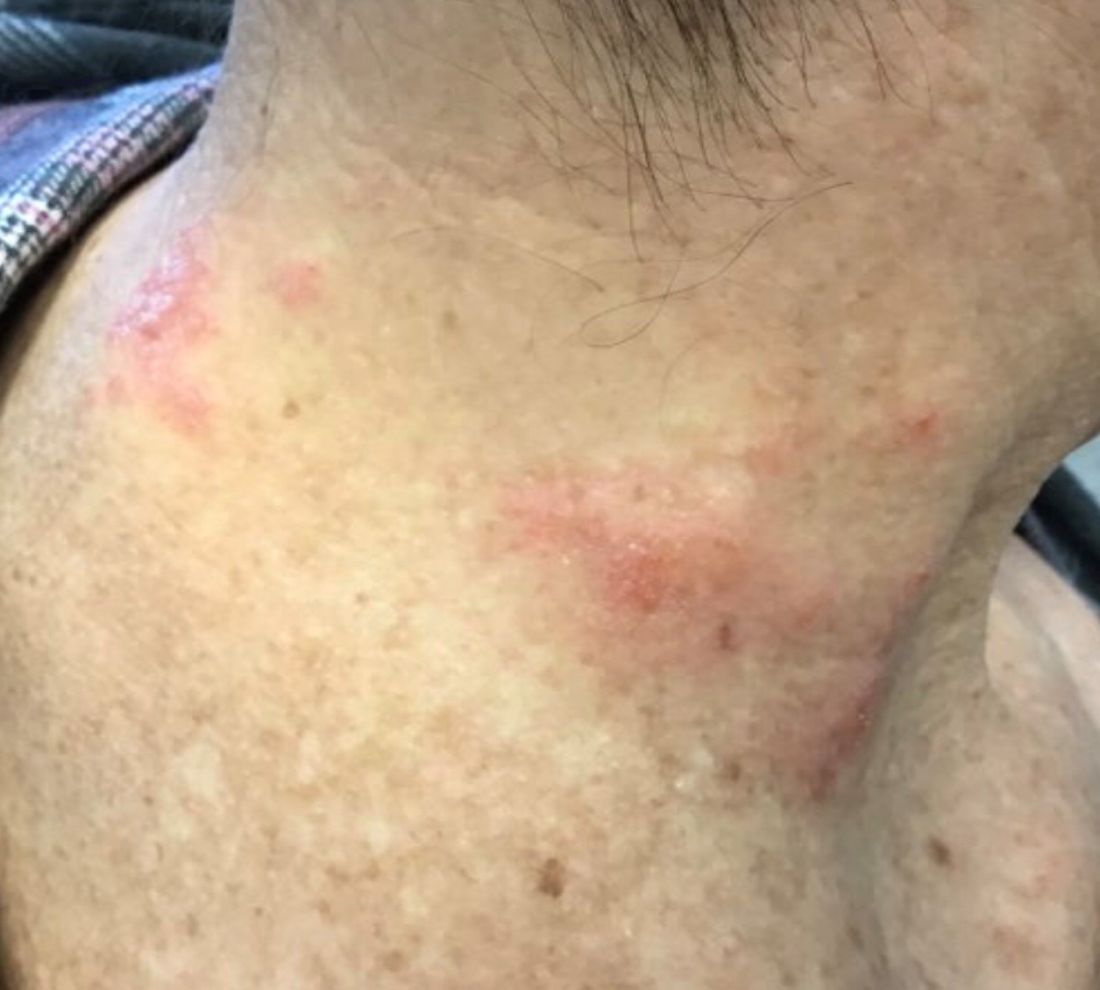
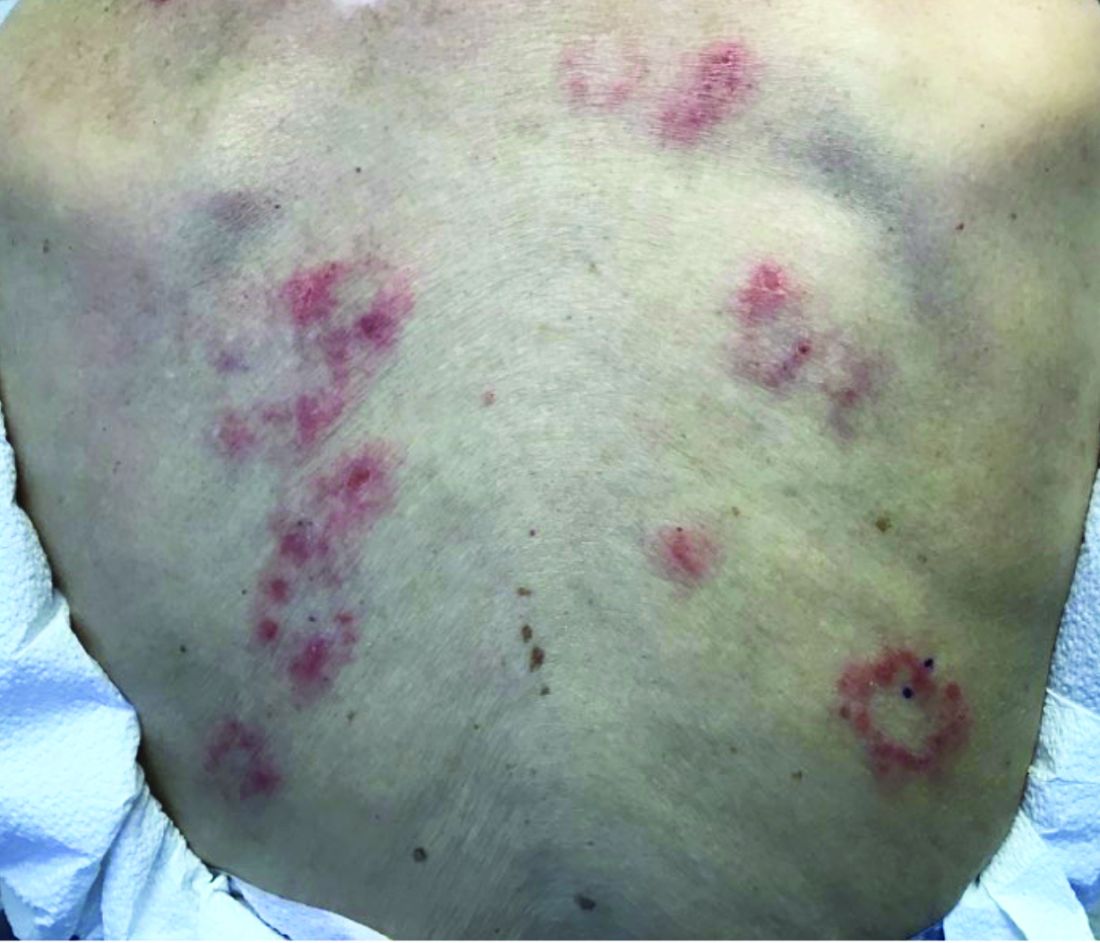
February 2020
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) is a type of cutaneous lupus erythematosus that may occur independently of or in combination with systemic lupus erythematosus. About 10%-15% of patients with SCLE will develop systemic lupus erythematosus. White females are more typically affected.
SCLE lesions often present as scaly, annular, or polycyclic scaly patches and plaques with central clearing. They may appear psoriasiform. They heal without atrophy or scarring but may leave dyspigmentation. Follicular plugging is absent. Lesions generally occur on sun exposed areas such as the neck, V of the chest, and upper extremities. Up to 75% of patients may exhibit associated symptoms such as photosensitivity, oral ulcers, and arthritis. Less than 20% of patients will develop internal disease, including nephritis and pulmonary disease. Symptoms of Sjögren’s syndrome and SCLE may overlap in some patients, and will portend higher risk for internal disease.
The differential diagnosis includes eczema, psoriasis, dermatophytosis, granuloma annulare, and erythema annulare centrifugum. Histology reveals epidermal atrophy and keratinocyte apoptosis, with a superficial and perivascular lymphohistiocytic infiltrate in the upper dermis. Interface changes at the dermal-epidermal junction can be seen. Direct immunofluorescence of lesional skin is positive in one-third of cases, often revealing granular deposits of IgG and IgM at the dermal-epidermal junction and around hair follicles (called the lupus-band test). Serology in SCLE may reveal a positive antinuclear antigen test, as well as positive Ro/SSA antigen. Other lupus serologies such as La/SSB, dsDNA, antihistone, and Sm antibodies may be positive, but are less commonly seen.
Several drugs may cause SCLE, such as hydrochlorothiazide, terbinafine, ACE inhibitors, NSAIDs, calcium-channel blockers, interferons, anticonvulsants, griseofulvin, penicillamine, spironolactone, tumor necrosis factor–alpha inhibitors, and statins. Discontinuing the offending medications may clear the lesions, but not always.
Treatment includes sunscreen and avoidance of sun exposure. Potent topical corticosteroids are helpful. If systemic treatment is indicated, antimalarials are first line.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) is a type of cutaneous lupus erythematosus that may occur independently of or in combination with systemic lupus erythematosus. About 10%-15% of patients with SCLE will develop systemic lupus erythematosus. White females are more typically affected.
SCLE lesions often present as scaly, annular, or polycyclic scaly patches and plaques with central clearing. They may appear psoriasiform. They heal without atrophy or scarring but may leave dyspigmentation. Follicular plugging is absent. Lesions generally occur on sun exposed areas such as the neck, V of the chest, and upper extremities. Up to 75% of patients may exhibit associated symptoms such as photosensitivity, oral ulcers, and arthritis. Less than 20% of patients will develop internal disease, including nephritis and pulmonary disease. Symptoms of Sjögren’s syndrome and SCLE may overlap in some patients, and will portend higher risk for internal disease.
The differential diagnosis includes eczema, psoriasis, dermatophytosis, granuloma annulare, and erythema annulare centrifugum. Histology reveals epidermal atrophy and keratinocyte apoptosis, with a superficial and perivascular lymphohistiocytic infiltrate in the upper dermis. Interface changes at the dermal-epidermal junction can be seen. Direct immunofluorescence of lesional skin is positive in one-third of cases, often revealing granular deposits of IgG and IgM at the dermal-epidermal junction and around hair follicles (called the lupus-band test). Serology in SCLE may reveal a positive antinuclear antigen test, as well as positive Ro/SSA antigen. Other lupus serologies such as La/SSB, dsDNA, antihistone, and Sm antibodies may be positive, but are less commonly seen.
Several drugs may cause SCLE, such as hydrochlorothiazide, terbinafine, ACE inhibitors, NSAIDs, calcium-channel blockers, interferons, anticonvulsants, griseofulvin, penicillamine, spironolactone, tumor necrosis factor–alpha inhibitors, and statins. Discontinuing the offending medications may clear the lesions, but not always.
Treatment includes sunscreen and avoidance of sun exposure. Potent topical corticosteroids are helpful. If systemic treatment is indicated, antimalarials are first line.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) is a type of cutaneous lupus erythematosus that may occur independently of or in combination with systemic lupus erythematosus. About 10%-15% of patients with SCLE will develop systemic lupus erythematosus. White females are more typically affected.
SCLE lesions often present as scaly, annular, or polycyclic scaly patches and plaques with central clearing. They may appear psoriasiform. They heal without atrophy or scarring but may leave dyspigmentation. Follicular plugging is absent. Lesions generally occur on sun exposed areas such as the neck, V of the chest, and upper extremities. Up to 75% of patients may exhibit associated symptoms such as photosensitivity, oral ulcers, and arthritis. Less than 20% of patients will develop internal disease, including nephritis and pulmonary disease. Symptoms of Sjögren’s syndrome and SCLE may overlap in some patients, and will portend higher risk for internal disease.
The differential diagnosis includes eczema, psoriasis, dermatophytosis, granuloma annulare, and erythema annulare centrifugum. Histology reveals epidermal atrophy and keratinocyte apoptosis, with a superficial and perivascular lymphohistiocytic infiltrate in the upper dermis. Interface changes at the dermal-epidermal junction can be seen. Direct immunofluorescence of lesional skin is positive in one-third of cases, often revealing granular deposits of IgG and IgM at the dermal-epidermal junction and around hair follicles (called the lupus-band test). Serology in SCLE may reveal a positive antinuclear antigen test, as well as positive Ro/SSA antigen. Other lupus serologies such as La/SSB, dsDNA, antihistone, and Sm antibodies may be positive, but are less commonly seen.
Several drugs may cause SCLE, such as hydrochlorothiazide, terbinafine, ACE inhibitors, NSAIDs, calcium-channel blockers, interferons, anticonvulsants, griseofulvin, penicillamine, spironolactone, tumor necrosis factor–alpha inhibitors, and statins. Discontinuing the offending medications may clear the lesions, but not always.
Treatment includes sunscreen and avoidance of sun exposure. Potent topical corticosteroids are helpful. If systemic treatment is indicated, antimalarials are first line.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
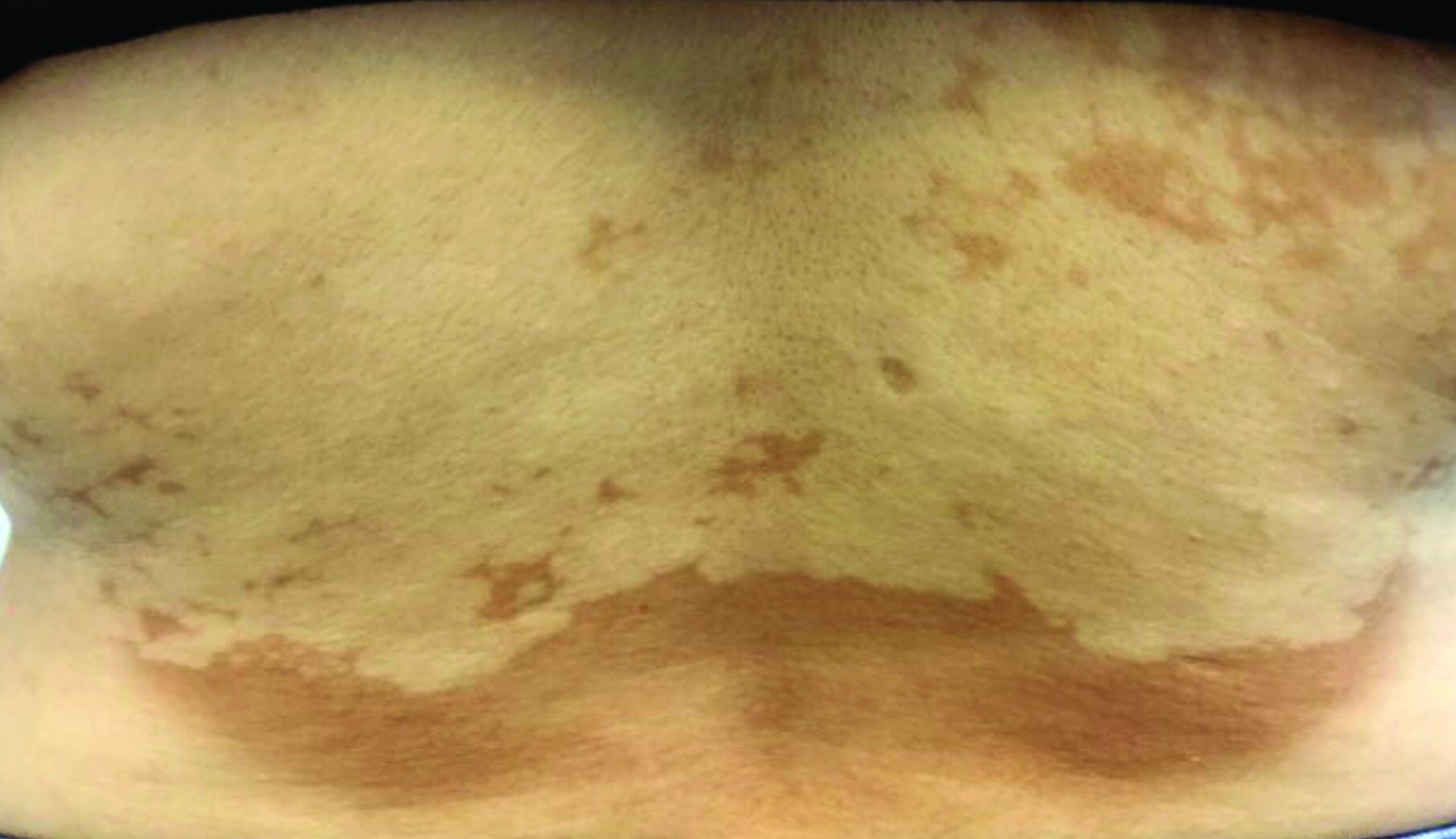
Asymptomatic hypopigmented macules and patches
, also known as Pityrosporum orbiculare or P. ovale. In its hyphal form, it produces skin lesions that appear as scaly, round or oval, hypopigmented, hyperpigmented, or pink macules or patches. Lesions are asymptomatic. The condition is more commonly seen in warm climates or during the summer months. Malassezia requires an oily environment for growth. Typically, TV appears in sebum-producing areas on the trunk. However, other sites may be affected such as the scalp, groin, and flexural areas. Infants may have facial lesions. Hypopigmentation may persist for months, even after lesions are treated, and takes time to resolve.
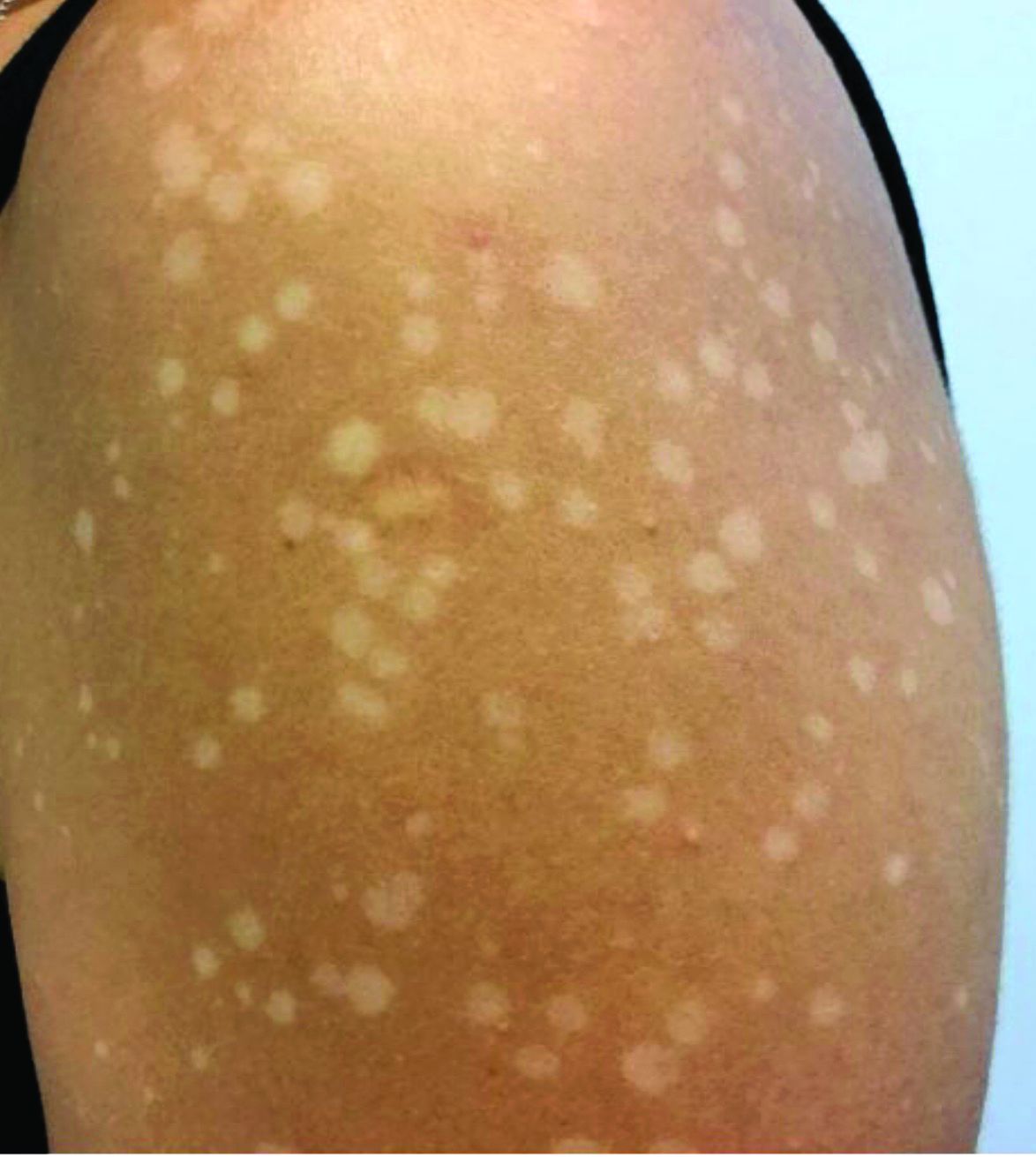
The differential diagnosis of hypopigmented lesions of tinea versicolor includes vitiligo, hypopigmented mycosis fungoides, progressive macular hypomelanosis (PMH), secondary syphilis, and pityriasis alba. Potassium hydroxide (KOH) preparations can be performed in the office for TV to reveal short, thick fungal hyphae with multiple spores, often referred to as “spaghetti and meatballs.” Use of a Wood’s light may aid in diagnosis. In TV, lesions may fluoresce yellow-green in adjacent follicles, unlike PMH, which characteristically show orange-red follicular fluorescence. A skin biopsy is necessary to rule out hypopgimented mycosis fungoides or syphilis. Histologically in TV, hyphae and spores will be present in the stratum corneum or in hair follicles. These are readily seen with PAS or GMS (Grocott methenamine silver) stains. There is usually no inflammation and skin appears “normal.” A biopsy was performed in this patient that revealed PAS positive hyphae.
Treatment for TV can be topical or systemic. Antifungal azole shampoo or creams, selenium sulfide shampoo, sulfur preparations, and allylamine creams have all been reported as useful treatments. Oral itraconazole or fluconazole are often given as systemic treatments. Monthly or weekly topical therapy may help prevent relapse.
This case and the photos were provided by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
, also known as Pityrosporum orbiculare or P. ovale. In its hyphal form, it produces skin lesions that appear as scaly, round or oval, hypopigmented, hyperpigmented, or pink macules or patches. Lesions are asymptomatic. The condition is more commonly seen in warm climates or during the summer months. Malassezia requires an oily environment for growth. Typically, TV appears in sebum-producing areas on the trunk. However, other sites may be affected such as the scalp, groin, and flexural areas. Infants may have facial lesions. Hypopigmentation may persist for months, even after lesions are treated, and takes time to resolve.
The differential diagnosis of hypopigmented lesions of tinea versicolor includes vitiligo, hypopigmented mycosis fungoides, progressive macular hypomelanosis (PMH), secondary syphilis, and pityriasis alba. Potassium hydroxide (KOH) preparations can be performed in the office for TV to reveal short, thick fungal hyphae with multiple spores, often referred to as “spaghetti and meatballs.” Use of a Wood’s light may aid in diagnosis. In TV, lesions may fluoresce yellow-green in adjacent follicles, unlike PMH, which characteristically show orange-red follicular fluorescence. A skin biopsy is necessary to rule out hypopgimented mycosis fungoides or syphilis. Histologically in TV, hyphae and spores will be present in the stratum corneum or in hair follicles. These are readily seen with PAS or GMS (Grocott methenamine silver) stains. There is usually no inflammation and skin appears “normal.” A biopsy was performed in this patient that revealed PAS positive hyphae.
Treatment for TV can be topical or systemic. Antifungal azole shampoo or creams, selenium sulfide shampoo, sulfur preparations, and allylamine creams have all been reported as useful treatments. Oral itraconazole or fluconazole are often given as systemic treatments. Monthly or weekly topical therapy may help prevent relapse.
This case and the photos were provided by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
, also known as Pityrosporum orbiculare or P. ovale. In its hyphal form, it produces skin lesions that appear as scaly, round or oval, hypopigmented, hyperpigmented, or pink macules or patches. Lesions are asymptomatic. The condition is more commonly seen in warm climates or during the summer months. Malassezia requires an oily environment for growth. Typically, TV appears in sebum-producing areas on the trunk. However, other sites may be affected such as the scalp, groin, and flexural areas. Infants may have facial lesions. Hypopigmentation may persist for months, even after lesions are treated, and takes time to resolve.
The differential diagnosis of hypopigmented lesions of tinea versicolor includes vitiligo, hypopigmented mycosis fungoides, progressive macular hypomelanosis (PMH), secondary syphilis, and pityriasis alba. Potassium hydroxide (KOH) preparations can be performed in the office for TV to reveal short, thick fungal hyphae with multiple spores, often referred to as “spaghetti and meatballs.” Use of a Wood’s light may aid in diagnosis. In TV, lesions may fluoresce yellow-green in adjacent follicles, unlike PMH, which characteristically show orange-red follicular fluorescence. A skin biopsy is necessary to rule out hypopgimented mycosis fungoides or syphilis. Histologically in TV, hyphae and spores will be present in the stratum corneum or in hair follicles. These are readily seen with PAS or GMS (Grocott methenamine silver) stains. There is usually no inflammation and skin appears “normal.” A biopsy was performed in this patient that revealed PAS positive hyphae.
Treatment for TV can be topical or systemic. Antifungal azole shampoo or creams, selenium sulfide shampoo, sulfur preparations, and allylamine creams have all been reported as useful treatments. Oral itraconazole or fluconazole are often given as systemic treatments. Monthly or weekly topical therapy may help prevent relapse.
This case and the photos were provided by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
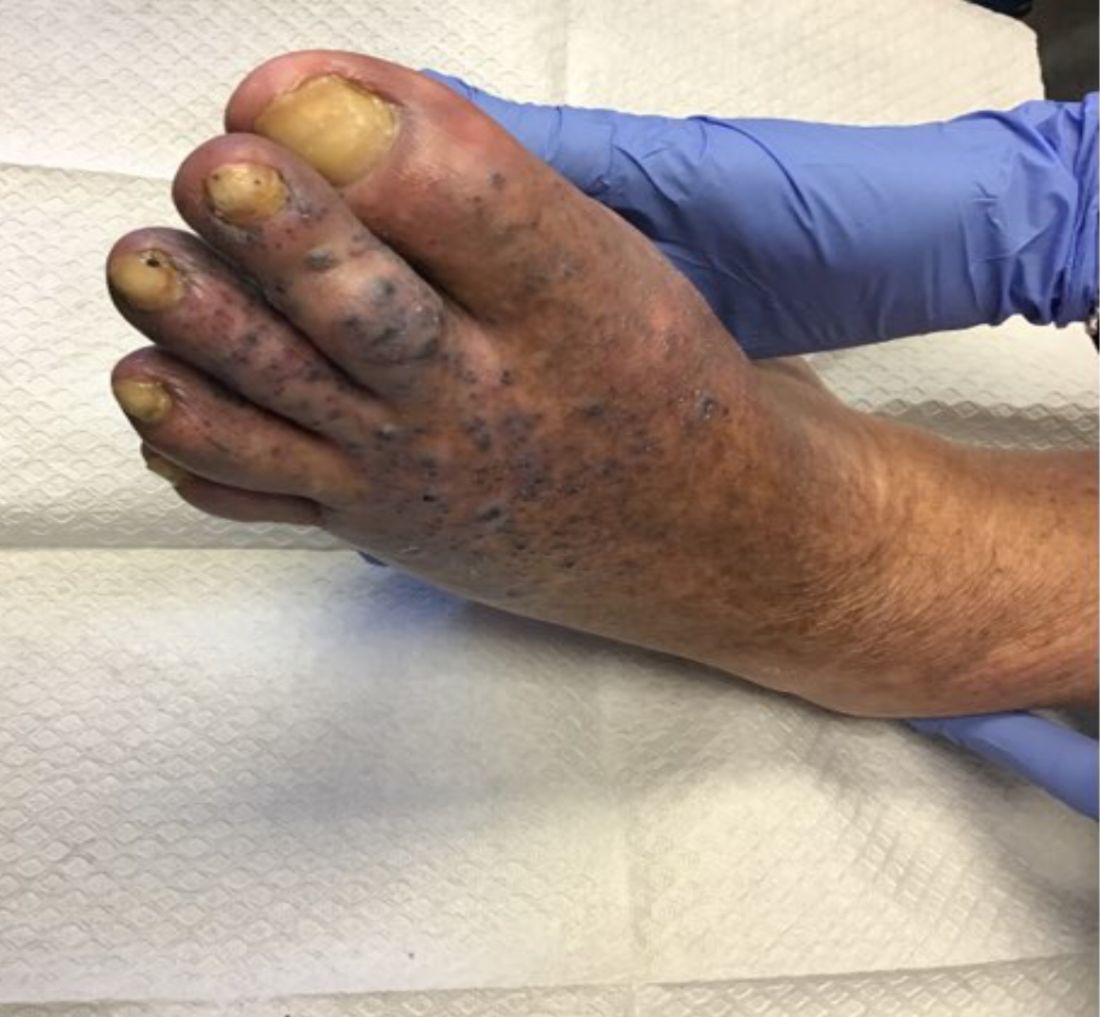
Violaceous papules on calf & foot
that usually presents on the distal lower extremities or feet as red to violaceous papules, patches, and plaques.
Clinically, lesions may look similar to Kaposi sarcoma (KS). It is considered to be a variant of stasis dermatitis or severe chronic venous stasis with a more exuberant vascular proliferation of preexisting vasculature. Although the exact etiology is unknown, it is thought that chronic edema, increased venous pressure, and tissue hypoxia may induce fibroblast and vascular proliferation.
It has also been described in association with vascular anomalies, such as Klippel-Trenaunay syndrome, Stewart-Bluefarb syndrome, and Prader-Labhart-Willi syndrome, and is caused by arteriovenous fistulae. Paralysis of lower extremities and amputation stumps are predisposing factors.
KS has four clinical variants: classic KS, African endemic KS, KS in immunocompromised patients, and AIDS-related epidemic KS. All types are caused by the human herpesvirus-8 (HHV-8). Violaceous lesions generally begin as macules and may progress to nodules or tumors.
Punch biopsies were performed in our patient. Histologically, thin-walled, dilated, capillary-like structures were present with a thin layer of surrounding pericytes with reactive fibrosis, hemorrhage, hemosiderin, and a scant chronic inflammatory cell infiltrate. The endothelial cells did not show atypia or mitotic activity. Endothelial cells were positive for CD31, CD34, and CD99. Pericytes and some of the endothelial cells were positive for actin and negative for D2-40, desmin, and HHV-8. In KS, vessels appear like slitlike or jagged spaces lined by spindled endothelial cells. Mild cytologic atypia is usually present. Endothelial cells are characteristically plump. A distinguishing feature in KS is the “promontory sign,” in which new vessels protrude into the vascular space. CD34 is usually negative and HHV-8 is positive.
Acroangiodermatitis of Mali may improve when the underlying venous insufficiency is addressed with compression stockings, pumps, or vascular intervention. Laser ablation of individual lesions has been described in the literature. Dapsone, oral erythromycin, and topical corticosteroids have been reported as helpful in some patients.
This case and these photos were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
that usually presents on the distal lower extremities or feet as red to violaceous papules, patches, and plaques.
Clinically, lesions may look similar to Kaposi sarcoma (KS). It is considered to be a variant of stasis dermatitis or severe chronic venous stasis with a more exuberant vascular proliferation of preexisting vasculature. Although the exact etiology is unknown, it is thought that chronic edema, increased venous pressure, and tissue hypoxia may induce fibroblast and vascular proliferation.
It has also been described in association with vascular anomalies, such as Klippel-Trenaunay syndrome, Stewart-Bluefarb syndrome, and Prader-Labhart-Willi syndrome, and is caused by arteriovenous fistulae. Paralysis of lower extremities and amputation stumps are predisposing factors.
KS has four clinical variants: classic KS, African endemic KS, KS in immunocompromised patients, and AIDS-related epidemic KS. All types are caused by the human herpesvirus-8 (HHV-8). Violaceous lesions generally begin as macules and may progress to nodules or tumors.
Punch biopsies were performed in our patient. Histologically, thin-walled, dilated, capillary-like structures were present with a thin layer of surrounding pericytes with reactive fibrosis, hemorrhage, hemosiderin, and a scant chronic inflammatory cell infiltrate. The endothelial cells did not show atypia or mitotic activity. Endothelial cells were positive for CD31, CD34, and CD99. Pericytes and some of the endothelial cells were positive for actin and negative for D2-40, desmin, and HHV-8. In KS, vessels appear like slitlike or jagged spaces lined by spindled endothelial cells. Mild cytologic atypia is usually present. Endothelial cells are characteristically plump. A distinguishing feature in KS is the “promontory sign,” in which new vessels protrude into the vascular space. CD34 is usually negative and HHV-8 is positive.
Acroangiodermatitis of Mali may improve when the underlying venous insufficiency is addressed with compression stockings, pumps, or vascular intervention. Laser ablation of individual lesions has been described in the literature. Dapsone, oral erythromycin, and topical corticosteroids have been reported as helpful in some patients.
This case and these photos were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
that usually presents on the distal lower extremities or feet as red to violaceous papules, patches, and plaques.
Clinically, lesions may look similar to Kaposi sarcoma (KS). It is considered to be a variant of stasis dermatitis or severe chronic venous stasis with a more exuberant vascular proliferation of preexisting vasculature. Although the exact etiology is unknown, it is thought that chronic edema, increased venous pressure, and tissue hypoxia may induce fibroblast and vascular proliferation.
It has also been described in association with vascular anomalies, such as Klippel-Trenaunay syndrome, Stewart-Bluefarb syndrome, and Prader-Labhart-Willi syndrome, and is caused by arteriovenous fistulae. Paralysis of lower extremities and amputation stumps are predisposing factors.
KS has four clinical variants: classic KS, African endemic KS, KS in immunocompromised patients, and AIDS-related epidemic KS. All types are caused by the human herpesvirus-8 (HHV-8). Violaceous lesions generally begin as macules and may progress to nodules or tumors.
Punch biopsies were performed in our patient. Histologically, thin-walled, dilated, capillary-like structures were present with a thin layer of surrounding pericytes with reactive fibrosis, hemorrhage, hemosiderin, and a scant chronic inflammatory cell infiltrate. The endothelial cells did not show atypia or mitotic activity. Endothelial cells were positive for CD31, CD34, and CD99. Pericytes and some of the endothelial cells were positive for actin and negative for D2-40, desmin, and HHV-8. In KS, vessels appear like slitlike or jagged spaces lined by spindled endothelial cells. Mild cytologic atypia is usually present. Endothelial cells are characteristically plump. A distinguishing feature in KS is the “promontory sign,” in which new vessels protrude into the vascular space. CD34 is usually negative and HHV-8 is positive.
Acroangiodermatitis of Mali may improve when the underlying venous insufficiency is addressed with compression stockings, pumps, or vascular intervention. Laser ablation of individual lesions has been described in the literature. Dapsone, oral erythromycin, and topical corticosteroids have been reported as helpful in some patients.
This case and these photos were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Intense stinging and burning, followed by the development of skin lesions minutes after exposure to a plant
Urticaria from stinging nettle
The stinging nettle (Urtica dioica) is a plant that grows in the United States, Eurasia, Northern Africa, and some parts of South America. It is commonly found in patches along hiking trails and near streams. The leaves are green with a characteristic tapered tip and bear tiny spines or hairs. The spines contain substances such as histamine, serotonin, and acetylcholine. Within seconds of contact with the stinging nettle, sharp stinging and burning will occur. Urticaria and pruritus may appear a few minutes later and may last up to 24 hours. The plant is eaten in some parts of the world and has been used as medicine.

The wood nettle (Laportea canadensis) is a relative of the stinging nettle that often grows in woodlands. Like the stinging nettle, the wood nettle leaves are covered with spines that sting when they come into contact with skin. However, the leaves are shorter and more oval shaped that the stinging nettle, and they lack the tapered tip that is characteristic for the stinging nettle. The reaction from the wood nettle is generally milder than that of the stinging nettle.
Plants can illicit different types of reactions in the skin: urticaria (immunologic and toxin mediated), irritant dermatitis (mechanical and chemical), phototoxic dermatitis (phytophotodermatitis), and allergic contact dermatitis. where anyone coming into contact with the hairs of the plant can be affected. Previous sensitization is not required. The reaction usually occurs immediately after exposure.
The allergic contact dermatitis seen with toxicodendron (poison ivy and poison sumac) appears 48 hours after exposure of a previously sensitized person to the plant. This type of delayed hypersensitivity reaction is known as cell-mediated hypersensitivity. Generally, no reaction is elicited upon the first exposure to the allergen. In fact, it may take years of exposure to allergens for someone to develop an allergic contact dermatitis.
The poison ivy plant can grow anywhere and is characteristically found in “leaves of three.” Skin reactions are often appear as linearly-arranged vesicles a few days after the exposure to the urushiol chemical in the sap of the plant. Poison sumac has red stems with 7-12 green, smooth leaves, and causes a similar skin reaction as poison ivy. It typically grows in wet areas.
Most stings are self-limited. Topical corticosteroid creams may be used if needed.
This case and photo were submitted by Susannah McClain, MD, of Three Rivers Dermatology in Coraopolis, Pa., and Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Urticaria from stinging nettle
The stinging nettle (Urtica dioica) is a plant that grows in the United States, Eurasia, Northern Africa, and some parts of South America. It is commonly found in patches along hiking trails and near streams. The leaves are green with a characteristic tapered tip and bear tiny spines or hairs. The spines contain substances such as histamine, serotonin, and acetylcholine. Within seconds of contact with the stinging nettle, sharp stinging and burning will occur. Urticaria and pruritus may appear a few minutes later and may last up to 24 hours. The plant is eaten in some parts of the world and has been used as medicine.
The wood nettle (Laportea canadensis) is a relative of the stinging nettle that often grows in woodlands. Like the stinging nettle, the wood nettle leaves are covered with spines that sting when they come into contact with skin. However, the leaves are shorter and more oval shaped that the stinging nettle, and they lack the tapered tip that is characteristic for the stinging nettle. The reaction from the wood nettle is generally milder than that of the stinging nettle.
Plants can illicit different types of reactions in the skin: urticaria (immunologic and toxin mediated), irritant dermatitis (mechanical and chemical), phototoxic dermatitis (phytophotodermatitis), and allergic contact dermatitis. where anyone coming into contact with the hairs of the plant can be affected. Previous sensitization is not required. The reaction usually occurs immediately after exposure.
The allergic contact dermatitis seen with toxicodendron (poison ivy and poison sumac) appears 48 hours after exposure of a previously sensitized person to the plant. This type of delayed hypersensitivity reaction is known as cell-mediated hypersensitivity. Generally, no reaction is elicited upon the first exposure to the allergen. In fact, it may take years of exposure to allergens for someone to develop an allergic contact dermatitis.
The poison ivy plant can grow anywhere and is characteristically found in “leaves of three.” Skin reactions are often appear as linearly-arranged vesicles a few days after the exposure to the urushiol chemical in the sap of the plant. Poison sumac has red stems with 7-12 green, smooth leaves, and causes a similar skin reaction as poison ivy. It typically grows in wet areas.
Most stings are self-limited. Topical corticosteroid creams may be used if needed.
This case and photo were submitted by Susannah McClain, MD, of Three Rivers Dermatology in Coraopolis, Pa., and Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Urticaria from stinging nettle
The stinging nettle (Urtica dioica) is a plant that grows in the United States, Eurasia, Northern Africa, and some parts of South America. It is commonly found in patches along hiking trails and near streams. The leaves are green with a characteristic tapered tip and bear tiny spines or hairs. The spines contain substances such as histamine, serotonin, and acetylcholine. Within seconds of contact with the stinging nettle, sharp stinging and burning will occur. Urticaria and pruritus may appear a few minutes later and may last up to 24 hours. The plant is eaten in some parts of the world and has been used as medicine.
The wood nettle (Laportea canadensis) is a relative of the stinging nettle that often grows in woodlands. Like the stinging nettle, the wood nettle leaves are covered with spines that sting when they come into contact with skin. However, the leaves are shorter and more oval shaped that the stinging nettle, and they lack the tapered tip that is characteristic for the stinging nettle. The reaction from the wood nettle is generally milder than that of the stinging nettle.
Plants can illicit different types of reactions in the skin: urticaria (immunologic and toxin mediated), irritant dermatitis (mechanical and chemical), phototoxic dermatitis (phytophotodermatitis), and allergic contact dermatitis. where anyone coming into contact with the hairs of the plant can be affected. Previous sensitization is not required. The reaction usually occurs immediately after exposure.
The allergic contact dermatitis seen with toxicodendron (poison ivy and poison sumac) appears 48 hours after exposure of a previously sensitized person to the plant. This type of delayed hypersensitivity reaction is known as cell-mediated hypersensitivity. Generally, no reaction is elicited upon the first exposure to the allergen. In fact, it may take years of exposure to allergens for someone to develop an allergic contact dermatitis.
The poison ivy plant can grow anywhere and is characteristically found in “leaves of three.” Skin reactions are often appear as linearly-arranged vesicles a few days after the exposure to the urushiol chemical in the sap of the plant. Poison sumac has red stems with 7-12 green, smooth leaves, and causes a similar skin reaction as poison ivy. It typically grows in wet areas.
Most stings are self-limited. Topical corticosteroid creams may be used if needed.
This case and photo were submitted by Susannah McClain, MD, of Three Rivers Dermatology in Coraopolis, Pa., and Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.

An asymptomatic reddish-brown plaque in a healthy adult man
Chromosomal translocation abnormalities in the tumor cells involving chromosomes 17 and 22 resulting in the fusion gene COL1A1-PDGFB have been reported in DFSP. This translocation causes an overproduction of the protein platelet-derived growth factor, resulting in tumor growth.
Lesions are most common in the trunk and proximal extremities. Less commonly, the head and neck may be involved. Lesions present as painless slow-growing red-brown nodules that may become painful as they enlarge. The differential diagnosis for early DFSP includes large dermatofibroma, keloid, dermatomyofibroma, and morphea. DFSP in childhood tends to appear more atrophic. It may be difficult to diagnose DFSP if the initial biopsy is superficial. If clinical suspicion is high, rebiopsy, ideally into the fat, is recommended.
Histologically, there is a cellular proliferation of thin spindled fibroblasts and collagen in the dermis that extend into the fat, often in a multilayered pattern. Adnexal structures can be obliterated. Fibroblasts may form a cartwheel or storiform pattern. There is mild cytologic atypia. Fibrosarcomatous change may signal increased risk of metastasis. CD34 is often positive and factor XIIIa is negative, unlike in dermatofibroma, which is opposite. Forms of DFSP that can be seen histologically include atrophic DFSP (flat rather than nodular), myxoid DFSP, and pigmented DFSP (also known as Bednar tumor).
DFSP can have irregular shapes with extensions into the fat. Subsequently, DFSP has a high recurrence rate with traditional surgical removal. Mohs surgery is now the treatment of choice. Recurrent tumors should be resected. As metastasis is rare, further work-up is not routinely indicated unless history and physical examination warrant it. Imatinib mesylate (Gleevec), which targets the platelet-derived growth factor receptor, has been tried with patients with inoperable or metastatic DFSP with some success. Radiation may also be used as an adjuvant after surgery. Regular follow-up exams with examination of the surgical site for possible recurrence should be performed every 6-12 months.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Chromosomal translocation abnormalities in the tumor cells involving chromosomes 17 and 22 resulting in the fusion gene COL1A1-PDGFB have been reported in DFSP. This translocation causes an overproduction of the protein platelet-derived growth factor, resulting in tumor growth.
Lesions are most common in the trunk and proximal extremities. Less commonly, the head and neck may be involved. Lesions present as painless slow-growing red-brown nodules that may become painful as they enlarge. The differential diagnosis for early DFSP includes large dermatofibroma, keloid, dermatomyofibroma, and morphea. DFSP in childhood tends to appear more atrophic. It may be difficult to diagnose DFSP if the initial biopsy is superficial. If clinical suspicion is high, rebiopsy, ideally into the fat, is recommended.
Histologically, there is a cellular proliferation of thin spindled fibroblasts and collagen in the dermis that extend into the fat, often in a multilayered pattern. Adnexal structures can be obliterated. Fibroblasts may form a cartwheel or storiform pattern. There is mild cytologic atypia. Fibrosarcomatous change may signal increased risk of metastasis. CD34 is often positive and factor XIIIa is negative, unlike in dermatofibroma, which is opposite. Forms of DFSP that can be seen histologically include atrophic DFSP (flat rather than nodular), myxoid DFSP, and pigmented DFSP (also known as Bednar tumor).
DFSP can have irregular shapes with extensions into the fat. Subsequently, DFSP has a high recurrence rate with traditional surgical removal. Mohs surgery is now the treatment of choice. Recurrent tumors should be resected. As metastasis is rare, further work-up is not routinely indicated unless history and physical examination warrant it. Imatinib mesylate (Gleevec), which targets the platelet-derived growth factor receptor, has been tried with patients with inoperable or metastatic DFSP with some success. Radiation may also be used as an adjuvant after surgery. Regular follow-up exams with examination of the surgical site for possible recurrence should be performed every 6-12 months.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Chromosomal translocation abnormalities in the tumor cells involving chromosomes 17 and 22 resulting in the fusion gene COL1A1-PDGFB have been reported in DFSP. This translocation causes an overproduction of the protein platelet-derived growth factor, resulting in tumor growth.
Lesions are most common in the trunk and proximal extremities. Less commonly, the head and neck may be involved. Lesions present as painless slow-growing red-brown nodules that may become painful as they enlarge. The differential diagnosis for early DFSP includes large dermatofibroma, keloid, dermatomyofibroma, and morphea. DFSP in childhood tends to appear more atrophic. It may be difficult to diagnose DFSP if the initial biopsy is superficial. If clinical suspicion is high, rebiopsy, ideally into the fat, is recommended.
Histologically, there is a cellular proliferation of thin spindled fibroblasts and collagen in the dermis that extend into the fat, often in a multilayered pattern. Adnexal structures can be obliterated. Fibroblasts may form a cartwheel or storiform pattern. There is mild cytologic atypia. Fibrosarcomatous change may signal increased risk of metastasis. CD34 is often positive and factor XIIIa is negative, unlike in dermatofibroma, which is opposite. Forms of DFSP that can be seen histologically include atrophic DFSP (flat rather than nodular), myxoid DFSP, and pigmented DFSP (also known as Bednar tumor).
DFSP can have irregular shapes with extensions into the fat. Subsequently, DFSP has a high recurrence rate with traditional surgical removal. Mohs surgery is now the treatment of choice. Recurrent tumors should be resected. As metastasis is rare, further work-up is not routinely indicated unless history and physical examination warrant it. Imatinib mesylate (Gleevec), which targets the platelet-derived growth factor receptor, has been tried with patients with inoperable or metastatic DFSP with some success. Radiation may also be used as an adjuvant after surgery. Regular follow-up exams with examination of the surgical site for possible recurrence should be performed every 6-12 months.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
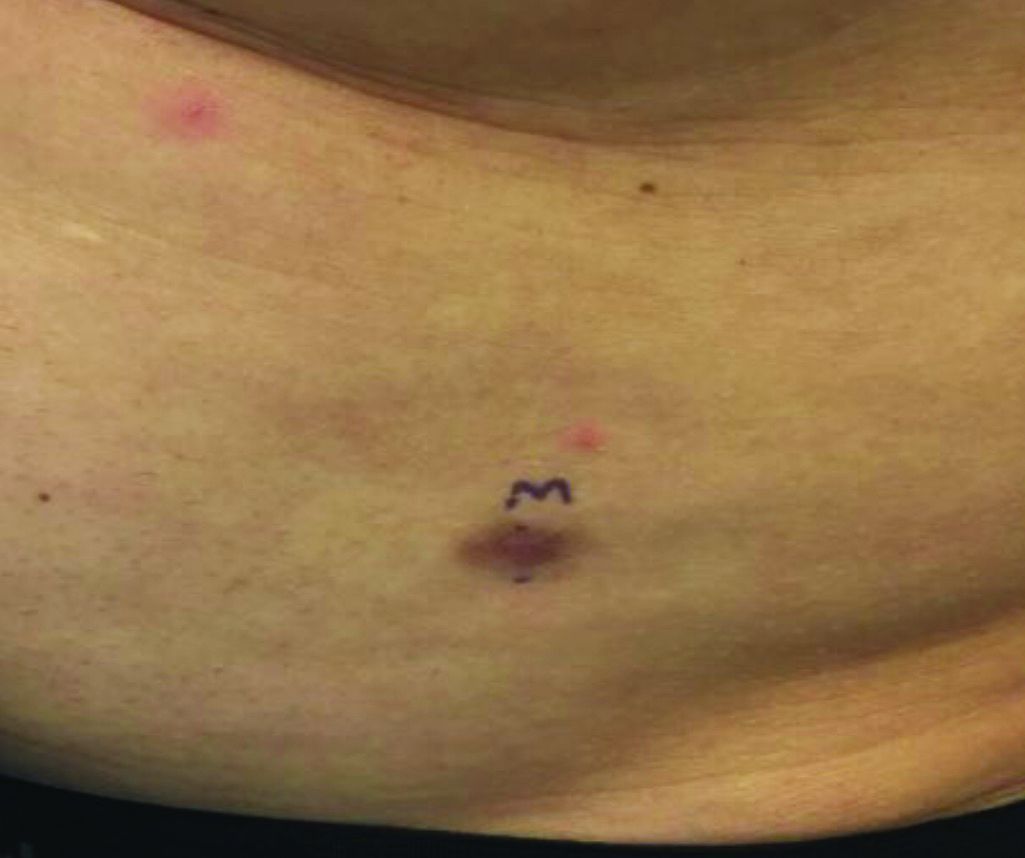
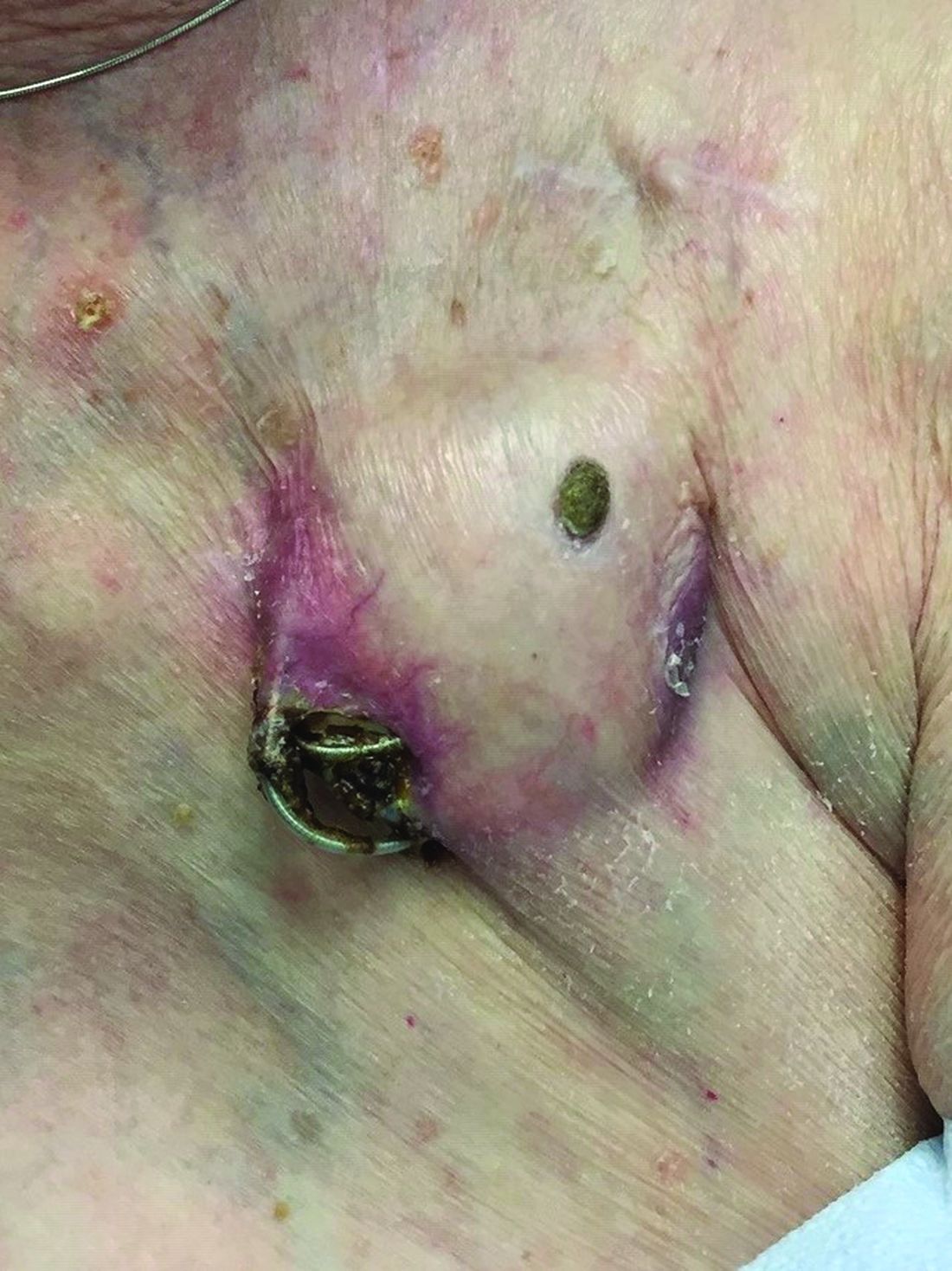
An 89-year-old woman presented with an ulceration overlying a cardiac pacemaker
Cardiac implantable electronic devices (CIEDs) – cardiac pacemakers and implantable cardioverter defibrillators –are an established treatment for the management of cardiac dysrhythmias in millions of patients. Complications occur in up to 15%, some of which may present first to the dermatologist.
The differential (caused by local venous obstruction and pressure dermatitis), and impending skin erosion/device extrusion.
Erosion and extrusion is a major complication with significant morbidity and mortality. The two main causes are pressure necrosis and infection. Pressure necrosis is influenced by the size of the device, complexity of the connections, and technical skill with which the pacemaker chest wall pocket is created.
After extrusion, the pacemaker should be considered contaminated and removed, and the necrotic tissue debrided. If infected, a prolonged course of appropriate antibiotic therapy is indicated. A bacterial culture in the patient presented here was negative.
Pocket infection of CIEDs is rare and may manifest as erythema, tenderness, drainage, erosion, or pruritus above the site of the pacemaker, along with systemic symptoms and signs, including fever, chills, or malaise. Some may have just the systemic symptoms. Fewer than half of patients with CIED infection present within 1 year of their last procedure.
Ruptured epidermal cysts usually manifest as acute swelling, inflammation, and tenderness of previously long-standing asymptomatic epidermal cysts. There may be drainage of malodorous keratinous and purulent debris. They are typically not infected. Treatment includes incision and drainage for fluctuant lesions or intralesional corticosteroid injection for early, nonfluctuant cases.
Allergic contact dermatitis to metal may be seen with implantable devices. Patch testing to various metal allergens can be helpful in determining if any allergy is present.
This case and photo were submitted by Michael Stierstorfer, MD, East Penn Dermatology, North Wales, Pa.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Cardiac implantable electronic devices (CIEDs) – cardiac pacemakers and implantable cardioverter defibrillators –are an established treatment for the management of cardiac dysrhythmias in millions of patients. Complications occur in up to 15%, some of which may present first to the dermatologist.
The differential (caused by local venous obstruction and pressure dermatitis), and impending skin erosion/device extrusion.
Erosion and extrusion is a major complication with significant morbidity and mortality. The two main causes are pressure necrosis and infection. Pressure necrosis is influenced by the size of the device, complexity of the connections, and technical skill with which the pacemaker chest wall pocket is created.
After extrusion, the pacemaker should be considered contaminated and removed, and the necrotic tissue debrided. If infected, a prolonged course of appropriate antibiotic therapy is indicated. A bacterial culture in the patient presented here was negative.
Pocket infection of CIEDs is rare and may manifest as erythema, tenderness, drainage, erosion, or pruritus above the site of the pacemaker, along with systemic symptoms and signs, including fever, chills, or malaise. Some may have just the systemic symptoms. Fewer than half of patients with CIED infection present within 1 year of their last procedure.
Ruptured epidermal cysts usually manifest as acute swelling, inflammation, and tenderness of previously long-standing asymptomatic epidermal cysts. There may be drainage of malodorous keratinous and purulent debris. They are typically not infected. Treatment includes incision and drainage for fluctuant lesions or intralesional corticosteroid injection for early, nonfluctuant cases.
Allergic contact dermatitis to metal may be seen with implantable devices. Patch testing to various metal allergens can be helpful in determining if any allergy is present.
This case and photo were submitted by Michael Stierstorfer, MD, East Penn Dermatology, North Wales, Pa.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Cardiac implantable electronic devices (CIEDs) – cardiac pacemakers and implantable cardioverter defibrillators –are an established treatment for the management of cardiac dysrhythmias in millions of patients. Complications occur in up to 15%, some of which may present first to the dermatologist.
The differential (caused by local venous obstruction and pressure dermatitis), and impending skin erosion/device extrusion.
Erosion and extrusion is a major complication with significant morbidity and mortality. The two main causes are pressure necrosis and infection. Pressure necrosis is influenced by the size of the device, complexity of the connections, and technical skill with which the pacemaker chest wall pocket is created.
After extrusion, the pacemaker should be considered contaminated and removed, and the necrotic tissue debrided. If infected, a prolonged course of appropriate antibiotic therapy is indicated. A bacterial culture in the patient presented here was negative.
Pocket infection of CIEDs is rare and may manifest as erythema, tenderness, drainage, erosion, or pruritus above the site of the pacemaker, along with systemic symptoms and signs, including fever, chills, or malaise. Some may have just the systemic symptoms. Fewer than half of patients with CIED infection present within 1 year of their last procedure.
Ruptured epidermal cysts usually manifest as acute swelling, inflammation, and tenderness of previously long-standing asymptomatic epidermal cysts. There may be drainage of malodorous keratinous and purulent debris. They are typically not infected. Treatment includes incision and drainage for fluctuant lesions or intralesional corticosteroid injection for early, nonfluctuant cases.
Allergic contact dermatitis to metal may be seen with implantable devices. Patch testing to various metal allergens can be helpful in determining if any allergy is present.
This case and photo were submitted by Michael Stierstorfer, MD, East Penn Dermatology, North Wales, Pa.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
A 72-year-old white male with a history of psoriatic arthritis presented with a 1-year history of multiple, intermittently pruritic papules on his face and trunk
Scleromyxedema
area, characteristically involving the glabella and ears. In some patients, the skin may be intensely pruritic, but this is not a universal finding and varies considerably among patients. In addition to affecting the skin, scleromyxedema has variable multisystem effects on the gastrointestinal tract, and musculoskeletal, pulmonary, cardiovascular, renal, and central nervous systems. The most common symptoms are proximal muscle weakness, dysphagia, and dyspnea on exertion. Scleromyxedema can also be associated with a paraproteinemia, mainly immunoglobulin G-lambda type.
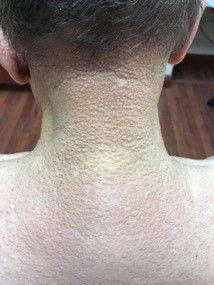
Scleromyxedema shares some features with other cutaneous diseases, and the main differential diagnosis includes localized scleromyxedema, also known as lichen myxedematosus. Lichen myxedematosus presents with waxy, firm papules and plaques. Systemic involvement and monoclonal gammopathy are characteristically absent. Scleroderma differs given the increase in fibrosis of cutaneous lesions, a higher percentage of Raynaud’s phenomenon, prominent lung disease, and autoantibodies.
On histopathologic review, scleromyxedema is associated with papular and mucin deposition, and increased fibroblast proliferation. The punch biopsy of the exhibited patient demonstrated a dome-shaped dermal nodule composed of fibroblasts in an edematous stroma. An Alcian blue stain highlighted increased mucin in the dermis and S-100 staining highlighted rare cells in the dermis.
The treatment of choice for scleromyxedema varies but includes intravenous immunoglobulins, systemic glucocorticosteroids, thalidomide, or immunosuppressant medications. In this patient, an IgG paraproteinemia was found on serum protein electrophoresis. The patient was evaluated by hematology-oncology, and no underlying myeloproliferative or dysplastic disease was found. The patient was started on intravenous immunoglobulin infusions with near complete resolution of his eruption and arthritic symptoms.
This case and photo were submitted by Jennifer Maldonado, a medical student at Nova Southeastern University, Ft. Lauderdale, Fla., and Kate Oberlin, MD, and Brian Morrison, MD, of the department of dermatology and cutaneous surgery, University of Miami; and Michelle Demory Beckler, PhD, of the College of Osteopathic Medicine and the department of microbiology, College of Medical Sciences, Nova Southeastern University.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Scleromyxedema
area, characteristically involving the glabella and ears. In some patients, the skin may be intensely pruritic, but this is not a universal finding and varies considerably among patients. In addition to affecting the skin, scleromyxedema has variable multisystem effects on the gastrointestinal tract, and musculoskeletal, pulmonary, cardiovascular, renal, and central nervous systems. The most common symptoms are proximal muscle weakness, dysphagia, and dyspnea on exertion. Scleromyxedema can also be associated with a paraproteinemia, mainly immunoglobulin G-lambda type.
Scleromyxedema shares some features with other cutaneous diseases, and the main differential diagnosis includes localized scleromyxedema, also known as lichen myxedematosus. Lichen myxedematosus presents with waxy, firm papules and plaques. Systemic involvement and monoclonal gammopathy are characteristically absent. Scleroderma differs given the increase in fibrosis of cutaneous lesions, a higher percentage of Raynaud’s phenomenon, prominent lung disease, and autoantibodies.
On histopathologic review, scleromyxedema is associated with papular and mucin deposition, and increased fibroblast proliferation. The punch biopsy of the exhibited patient demonstrated a dome-shaped dermal nodule composed of fibroblasts in an edematous stroma. An Alcian blue stain highlighted increased mucin in the dermis and S-100 staining highlighted rare cells in the dermis.
The treatment of choice for scleromyxedema varies but includes intravenous immunoglobulins, systemic glucocorticosteroids, thalidomide, or immunosuppressant medications. In this patient, an IgG paraproteinemia was found on serum protein electrophoresis. The patient was evaluated by hematology-oncology, and no underlying myeloproliferative or dysplastic disease was found. The patient was started on intravenous immunoglobulin infusions with near complete resolution of his eruption and arthritic symptoms.
This case and photo were submitted by Jennifer Maldonado, a medical student at Nova Southeastern University, Ft. Lauderdale, Fla., and Kate Oberlin, MD, and Brian Morrison, MD, of the department of dermatology and cutaneous surgery, University of Miami; and Michelle Demory Beckler, PhD, of the College of Osteopathic Medicine and the department of microbiology, College of Medical Sciences, Nova Southeastern University.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Scleromyxedema
area, characteristically involving the glabella and ears. In some patients, the skin may be intensely pruritic, but this is not a universal finding and varies considerably among patients. In addition to affecting the skin, scleromyxedema has variable multisystem effects on the gastrointestinal tract, and musculoskeletal, pulmonary, cardiovascular, renal, and central nervous systems. The most common symptoms are proximal muscle weakness, dysphagia, and dyspnea on exertion. Scleromyxedema can also be associated with a paraproteinemia, mainly immunoglobulin G-lambda type.
Scleromyxedema shares some features with other cutaneous diseases, and the main differential diagnosis includes localized scleromyxedema, also known as lichen myxedematosus. Lichen myxedematosus presents with waxy, firm papules and plaques. Systemic involvement and monoclonal gammopathy are characteristically absent. Scleroderma differs given the increase in fibrosis of cutaneous lesions, a higher percentage of Raynaud’s phenomenon, prominent lung disease, and autoantibodies.
On histopathologic review, scleromyxedema is associated with papular and mucin deposition, and increased fibroblast proliferation. The punch biopsy of the exhibited patient demonstrated a dome-shaped dermal nodule composed of fibroblasts in an edematous stroma. An Alcian blue stain highlighted increased mucin in the dermis and S-100 staining highlighted rare cells in the dermis.
The treatment of choice for scleromyxedema varies but includes intravenous immunoglobulins, systemic glucocorticosteroids, thalidomide, or immunosuppressant medications. In this patient, an IgG paraproteinemia was found on serum protein electrophoresis. The patient was evaluated by hematology-oncology, and no underlying myeloproliferative or dysplastic disease was found. The patient was started on intravenous immunoglobulin infusions with near complete resolution of his eruption and arthritic symptoms.
This case and photo were submitted by Jennifer Maldonado, a medical student at Nova Southeastern University, Ft. Lauderdale, Fla., and Kate Oberlin, MD, and Brian Morrison, MD, of the department of dermatology and cutaneous surgery, University of Miami; and Michelle Demory Beckler, PhD, of the College of Osteopathic Medicine and the department of microbiology, College of Medical Sciences, Nova Southeastern University.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
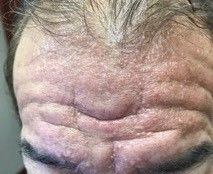
A 60-year-old white male presented with a painful nodule on the right lateral thigh that had been present for years
Benign tumors consisting of glomus cells may be subdivided into two types: glomus tumors and glomuvenous malformations or glomangiomas. Glomus cells are modified smooth muscle cells that normally line the Sucquet-Hoyer canal, an arteriovenous fistula that is involved in temperature regulation in the digits.
. In women, lesions more frequently occur on the fingers (especially nail beds). Glomus tumors are firm subcutaneous nodules, often skin colored or bluish in color. Subungual tumors tend to appear bluish under the nail plate. Lesions are extremely tender or painful, with worse pain upon palpation. Occasionally, nontender lesions can be seen.
In children, multiple nontender lesions are called glomangiomas or glomuvenous malformations. They may be sporadic or can be inherited in an autosomal dominant fashion due to a mutation in glomulin on chromosome 1p21-p22. Multiple lesions may be scattered or grouped, often in a segmental distribution. Congenital lesions tend to be large, blue-purple in color with a cobblestone appearance. They are more superficial than venous malformations.
Histologically, a proliferation of blood vessels surrounded by glomus cells is seen. Glomus cells appear as monotonous cells with a dense, round nucleus and abundant pink cytoplasm. Glomus cells can also be appreciated single-filing through pale stroma, resembling strings of black pearls. Glomus cells stain positive for smooth muscle actin and vimentin.
The painful tumor differential diagnosis has been described in the literature by the mnemonic “LEND AN EGG:” leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomus tumor, and granular cell tumor.
The malignant counterpart is glomangiosarcoma, which is a rare tumor. These lesions are often large and deeply located on the extremities. Histologically, sarcomatous areas are mixed with areas of benign glomus tumor.
Surgical excision is the treatment of choice for solitary glomus tumors to provide pain relief. Subungual tumors are more challenging due to their small size, but may be excised as well. Glomuvenous malformations may require different treatment modalities, such as surgery and laser, due to their larger size.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at www.mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Benign tumors consisting of glomus cells may be subdivided into two types: glomus tumors and glomuvenous malformations or glomangiomas. Glomus cells are modified smooth muscle cells that normally line the Sucquet-Hoyer canal, an arteriovenous fistula that is involved in temperature regulation in the digits.
. In women, lesions more frequently occur on the fingers (especially nail beds). Glomus tumors are firm subcutaneous nodules, often skin colored or bluish in color. Subungual tumors tend to appear bluish under the nail plate. Lesions are extremely tender or painful, with worse pain upon palpation. Occasionally, nontender lesions can be seen.
In children, multiple nontender lesions are called glomangiomas or glomuvenous malformations. They may be sporadic or can be inherited in an autosomal dominant fashion due to a mutation in glomulin on chromosome 1p21-p22. Multiple lesions may be scattered or grouped, often in a segmental distribution. Congenital lesions tend to be large, blue-purple in color with a cobblestone appearance. They are more superficial than venous malformations.
Histologically, a proliferation of blood vessels surrounded by glomus cells is seen. Glomus cells appear as monotonous cells with a dense, round nucleus and abundant pink cytoplasm. Glomus cells can also be appreciated single-filing through pale stroma, resembling strings of black pearls. Glomus cells stain positive for smooth muscle actin and vimentin.
The painful tumor differential diagnosis has been described in the literature by the mnemonic “LEND AN EGG:” leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomus tumor, and granular cell tumor.
The malignant counterpart is glomangiosarcoma, which is a rare tumor. These lesions are often large and deeply located on the extremities. Histologically, sarcomatous areas are mixed with areas of benign glomus tumor.
Surgical excision is the treatment of choice for solitary glomus tumors to provide pain relief. Subungual tumors are more challenging due to their small size, but may be excised as well. Glomuvenous malformations may require different treatment modalities, such as surgery and laser, due to their larger size.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at www.mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Benign tumors consisting of glomus cells may be subdivided into two types: glomus tumors and glomuvenous malformations or glomangiomas. Glomus cells are modified smooth muscle cells that normally line the Sucquet-Hoyer canal, an arteriovenous fistula that is involved in temperature regulation in the digits.
. In women, lesions more frequently occur on the fingers (especially nail beds). Glomus tumors are firm subcutaneous nodules, often skin colored or bluish in color. Subungual tumors tend to appear bluish under the nail plate. Lesions are extremely tender or painful, with worse pain upon palpation. Occasionally, nontender lesions can be seen.
In children, multiple nontender lesions are called glomangiomas or glomuvenous malformations. They may be sporadic or can be inherited in an autosomal dominant fashion due to a mutation in glomulin on chromosome 1p21-p22. Multiple lesions may be scattered or grouped, often in a segmental distribution. Congenital lesions tend to be large, blue-purple in color with a cobblestone appearance. They are more superficial than venous malformations.
Histologically, a proliferation of blood vessels surrounded by glomus cells is seen. Glomus cells appear as monotonous cells with a dense, round nucleus and abundant pink cytoplasm. Glomus cells can also be appreciated single-filing through pale stroma, resembling strings of black pearls. Glomus cells stain positive for smooth muscle actin and vimentin.
The painful tumor differential diagnosis has been described in the literature by the mnemonic “LEND AN EGG:” leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomus tumor, and granular cell tumor.
The malignant counterpart is glomangiosarcoma, which is a rare tumor. These lesions are often large and deeply located on the extremities. Histologically, sarcomatous areas are mixed with areas of benign glomus tumor.
Surgical excision is the treatment of choice for solitary glomus tumors to provide pain relief. Subungual tumors are more challenging due to their small size, but may be excised as well. Glomuvenous malformations may require different treatment modalities, such as surgery and laser, due to their larger size.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at www.mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
A 60-year-old white male presented with a painful nodule on the right lateral thigh that had been present for years. It has slowly been increasing in size over time.
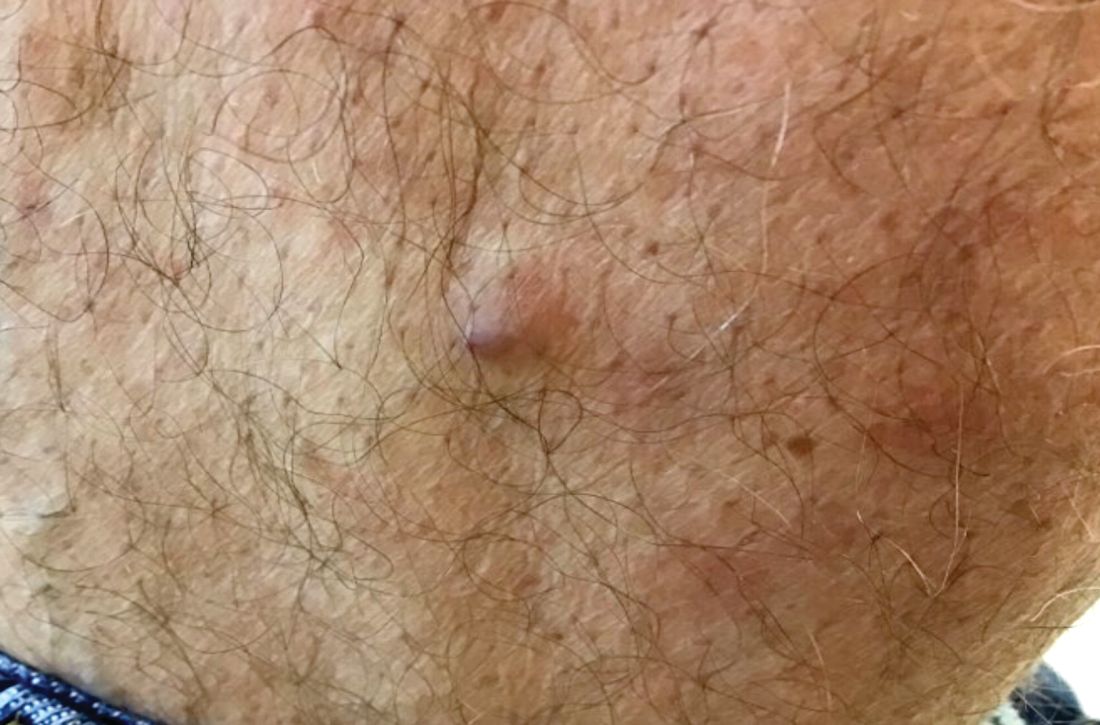
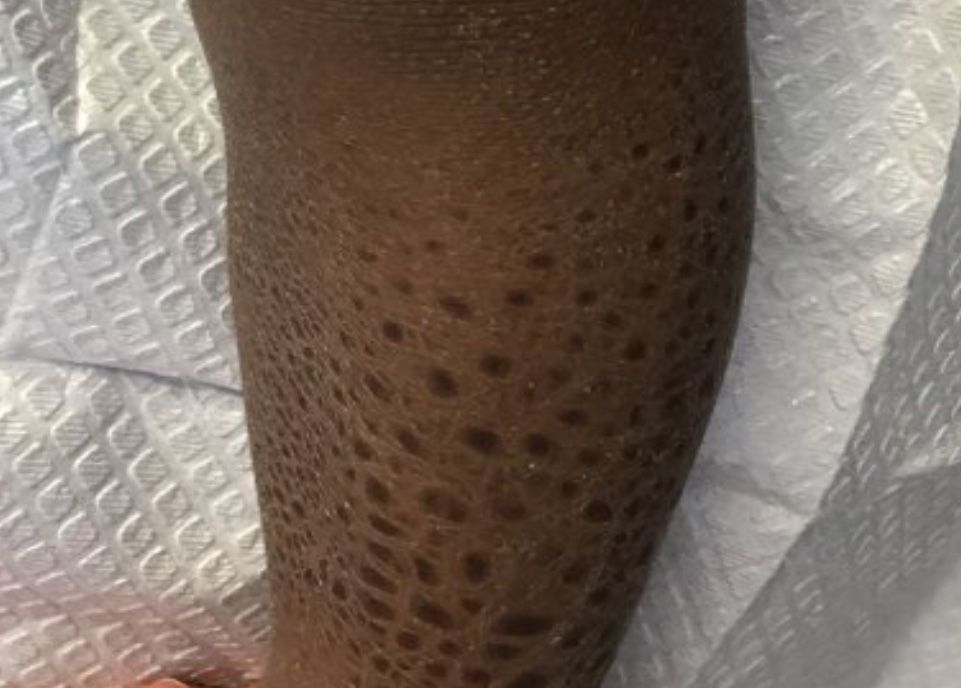
A 13-month-old, healthy black male presented with a 6-month history of dry, scaly skin on the body
Ichthyosis vulgaris
Ichthyoses describe a group of disorders of cornification in which the epidermis differentiates abnormally, leading to generalized scaling of the skin. Ichthyosis is derived from the Greek word for fish, “ichthys.” Ichthyosis vulgaris is the most common of these conditions and often presents in early childhood during the first year of life. It is inherited in an autosomal-dominant pattern. Skin is dry and scaly over the entire body, although the antecubital and popliteal fossa may be uninvolved. The scalp may be involved as well. Atopy and keratosis pilaris may be associated. By adulthood, symptoms tend to abate.
X-linked ichthyosis is an X-linked recessive trait, in which males are affected and mothers are carriers. The condition is caused by a deficiency of steroid sulfatase. This deficiency can result in low levels of estrogen during pregnancy in the mother of an affected fetus, hampering labor progression, and often requiring C-section. Children usually present before 3 months of age. Scales are large and dark. The antecubital and popliteal fossa are usually spared. The neck almost always is involved, coining the term “dirty neck disease.” Corneal opacities are present upon ophthalmologic examination. There is an increased risk of cryptorchidism and testicular cancer. Skin symptoms tend to worsen into adulthood.
Lamellar ichthyosis generally occurs at birth with a striking collodion-type membrane covering the body and underlying erythroderma, which then desquamates. Ectropion is usually present as well. Resulting scales are large and gray-brown. Lamellar ichthyosis is inherited in an autosomal recessive pattern. Mutations in transglutaminase 1 (TGM1), ALOXE3, ALOX12B, and ABCA12 genes have been implicated in this disorder.
Acquired ichthyosis can appear clinically similar to ichthyosis vulgaris. It occurs in patients with systemic diseases such as Hodgkin disease, non-Hodgkin lymphoma, mycosis fungoides, multiple myeloma, hypothyroidism, sarcoidosis, AIDS, and others.
improve hyperkeratosis. Urea-containing products can be helpful. Salicylic acid may be used but merit caution in children because of salicylate toxicity. Oral and topical retinoid can be helpful in lamellar ichthyosis.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews.com. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Ichthyosis vulgaris
Ichthyoses describe a group of disorders of cornification in which the epidermis differentiates abnormally, leading to generalized scaling of the skin. Ichthyosis is derived from the Greek word for fish, “ichthys.” Ichthyosis vulgaris is the most common of these conditions and often presents in early childhood during the first year of life. It is inherited in an autosomal-dominant pattern. Skin is dry and scaly over the entire body, although the antecubital and popliteal fossa may be uninvolved. The scalp may be involved as well. Atopy and keratosis pilaris may be associated. By adulthood, symptoms tend to abate.
X-linked ichthyosis is an X-linked recessive trait, in which males are affected and mothers are carriers. The condition is caused by a deficiency of steroid sulfatase. This deficiency can result in low levels of estrogen during pregnancy in the mother of an affected fetus, hampering labor progression, and often requiring C-section. Children usually present before 3 months of age. Scales are large and dark. The antecubital and popliteal fossa are usually spared. The neck almost always is involved, coining the term “dirty neck disease.” Corneal opacities are present upon ophthalmologic examination. There is an increased risk of cryptorchidism and testicular cancer. Skin symptoms tend to worsen into adulthood.
Lamellar ichthyosis generally occurs at birth with a striking collodion-type membrane covering the body and underlying erythroderma, which then desquamates. Ectropion is usually present as well. Resulting scales are large and gray-brown. Lamellar ichthyosis is inherited in an autosomal recessive pattern. Mutations in transglutaminase 1 (TGM1), ALOXE3, ALOX12B, and ABCA12 genes have been implicated in this disorder.
Acquired ichthyosis can appear clinically similar to ichthyosis vulgaris. It occurs in patients with systemic diseases such as Hodgkin disease, non-Hodgkin lymphoma, mycosis fungoides, multiple myeloma, hypothyroidism, sarcoidosis, AIDS, and others.
improve hyperkeratosis. Urea-containing products can be helpful. Salicylic acid may be used but merit caution in children because of salicylate toxicity. Oral and topical retinoid can be helpful in lamellar ichthyosis.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews.com. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Ichthyosis vulgaris
Ichthyoses describe a group of disorders of cornification in which the epidermis differentiates abnormally, leading to generalized scaling of the skin. Ichthyosis is derived from the Greek word for fish, “ichthys.” Ichthyosis vulgaris is the most common of these conditions and often presents in early childhood during the first year of life. It is inherited in an autosomal-dominant pattern. Skin is dry and scaly over the entire body, although the antecubital and popliteal fossa may be uninvolved. The scalp may be involved as well. Atopy and keratosis pilaris may be associated. By adulthood, symptoms tend to abate.
X-linked ichthyosis is an X-linked recessive trait, in which males are affected and mothers are carriers. The condition is caused by a deficiency of steroid sulfatase. This deficiency can result in low levels of estrogen during pregnancy in the mother of an affected fetus, hampering labor progression, and often requiring C-section. Children usually present before 3 months of age. Scales are large and dark. The antecubital and popliteal fossa are usually spared. The neck almost always is involved, coining the term “dirty neck disease.” Corneal opacities are present upon ophthalmologic examination. There is an increased risk of cryptorchidism and testicular cancer. Skin symptoms tend to worsen into adulthood.
Lamellar ichthyosis generally occurs at birth with a striking collodion-type membrane covering the body and underlying erythroderma, which then desquamates. Ectropion is usually present as well. Resulting scales are large and gray-brown. Lamellar ichthyosis is inherited in an autosomal recessive pattern. Mutations in transglutaminase 1 (TGM1), ALOXE3, ALOX12B, and ABCA12 genes have been implicated in this disorder.
Acquired ichthyosis can appear clinically similar to ichthyosis vulgaris. It occurs in patients with systemic diseases such as Hodgkin disease, non-Hodgkin lymphoma, mycosis fungoides, multiple myeloma, hypothyroidism, sarcoidosis, AIDS, and others.
improve hyperkeratosis. Urea-containing products can be helpful. Salicylic acid may be used but merit caution in children because of salicylate toxicity. Oral and topical retinoid can be helpful in lamellar ichthyosis.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews.com. To submit a case for possible publication, send an email to dermnews@mdedge.com.
A 13-month-old, healthy black male presented with a 6-month history of dry, scaly skin on the body, including scalp and extremities. His neck was unaffected. His mother reports an uneventful pregnancy and natural childbirth. He had been prescribed triamcinolone in the past for eczema.