User login
There is more to the TSH than a number

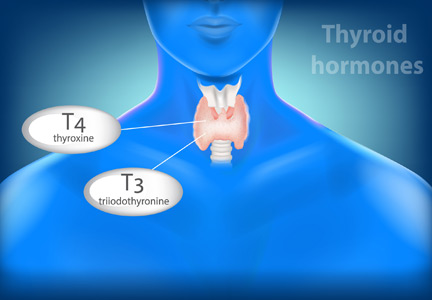
At a quick read, the messages from these articles may seem contradictory. But the biology is more complex in the setting of endogenous production of T4 by the thyroid gland, which is regulated by TSH, which in turn is regulated in a feedback loop by the thyroid-produced T4. In the setting of a fixed replacement dose of exogenous levothyroxine, the provided hormone affects the pituitary production of TSH, which likely will have no significant subsequent effect on the T4 level. Thus, the feedback control loop is far simpler.
There has not been a definitive study demonstrating that thyroxine supplementation in patients with subclinical hypothyroidism results in a superior clinical outcome. There are hints that this may be the case, and Azim and Nasr cite some of these studies. Recognizing a few markedly different physiologic reasons why the TSH can be slightly elevated and the T4 normal helps explain the lack of uniform clinical success with supplementation therapy and provides rationales for some management strategies.
Any biological variability in the responsiveness of the thyroid gland to TSH may affect the relationship between the levels of TSH and thyroid gland-released T4. In theory, if the thyroid receptor has decreased affinity for TSH, a higher TSH concentration will be needed to get the thyroid gland to secrete the level of T4 that the pituitary sensing mechanism deems normal for that individual. If the receptor affinity was decreased due to a gene polymorphism, this relationship between TSH and T4 may be stable, and providing exogenous T4 will result in a lower, “normalized” TSH level but may disrupt the thyroid-pituitary crosstalk and may even produce clinical hyperthyroidism.
A similar scenario exists in the setting of early thyroid gland failure, such as in Hashimoto thyroiditis. But in the latter scenario, the TSH-to-T4 production relationship may be unstable over time, for as additional thyroid gland is destroyed, T4 production will continue to decrease, the TSH will increase, and the thyroid gland may ultimately fail and hypothyroidism will occur. Hence the recommendation that in the setting of subclinical hypothyroidism and antiperoxidase antibodies, T4 and TSH levels should be monitored regularly in order to detect early true thyroid gland failure when the T4 level can no longer be maintained despite the increased stimulation of the gland by the elevated TSH. Analogous to this may be subclinical hypothyroidism in the elderly, in whom thyroid gland failure may develop, despite an increased TSH, from senescence rather than autoimmunity. What I am suggesting is that the natural history of all patients with subclinical hypothyroidism is not alike, and it thus should not be surprising that there does not seem to be a one-size-fits-all approach to management.
Symptoms in patients with subclinical hypothyroidism have not uniformly improved with T4 treatment compared with placebo. Notably, most patients with subclinical hypothyroidism experience no symptoms. But consider the extremely common symptom of fatigue, which can be present for a myriad of defined and undefined reasons. This symptom may often lead physicians to check the TSH and, if that is even slightly elevated, to also check the T4. It may also lead some physicians to routinely check the T4. Subclinical hypothyroidism is also quite common; thus, by chance alone or because of the circadian timing of checking the TSH, a slightly elevated TSH and fatigue may coexist and yet be unrelated.
Additionally, a positive biochemical response to thyroxine supplementation, such as a lowering of cholesterol, does not prove that the patient was clinically hypothyroid prior to supplementation, any more than lowering a patient’s blood glucose with insulin proves that the patient was diabetic. The management of subclinical hypothyroidism should be nuanced and based on both clinical and laboratory parameters.
- Nasr C. Is a serum TSH measurement sufficient to monitor the treatment of primary hypothyroidism? Cleve Clin J Med 2016; 83(8):571–573. doi:10.3949/ccjm.83a.15165
- Mandell BF. Trust the thyroid thermostat. Cleve Clin J Med 2016; 83(8):552–553. doi:10.3949/ccjm.83b.08016
At a quick read, the messages from these articles may seem contradictory. But the biology is more complex in the setting of endogenous production of T4 by the thyroid gland, which is regulated by TSH, which in turn is regulated in a feedback loop by the thyroid-produced T4. In the setting of a fixed replacement dose of exogenous levothyroxine, the provided hormone affects the pituitary production of TSH, which likely will have no significant subsequent effect on the T4 level. Thus, the feedback control loop is far simpler.
There has not been a definitive study demonstrating that thyroxine supplementation in patients with subclinical hypothyroidism results in a superior clinical outcome. There are hints that this may be the case, and Azim and Nasr cite some of these studies. Recognizing a few markedly different physiologic reasons why the TSH can be slightly elevated and the T4 normal helps explain the lack of uniform clinical success with supplementation therapy and provides rationales for some management strategies.
Any biological variability in the responsiveness of the thyroid gland to TSH may affect the relationship between the levels of TSH and thyroid gland-released T4. In theory, if the thyroid receptor has decreased affinity for TSH, a higher TSH concentration will be needed to get the thyroid gland to secrete the level of T4 that the pituitary sensing mechanism deems normal for that individual. If the receptor affinity was decreased due to a gene polymorphism, this relationship between TSH and T4 may be stable, and providing exogenous T4 will result in a lower, “normalized” TSH level but may disrupt the thyroid-pituitary crosstalk and may even produce clinical hyperthyroidism.
A similar scenario exists in the setting of early thyroid gland failure, such as in Hashimoto thyroiditis. But in the latter scenario, the TSH-to-T4 production relationship may be unstable over time, for as additional thyroid gland is destroyed, T4 production will continue to decrease, the TSH will increase, and the thyroid gland may ultimately fail and hypothyroidism will occur. Hence the recommendation that in the setting of subclinical hypothyroidism and antiperoxidase antibodies, T4 and TSH levels should be monitored regularly in order to detect early true thyroid gland failure when the T4 level can no longer be maintained despite the increased stimulation of the gland by the elevated TSH. Analogous to this may be subclinical hypothyroidism in the elderly, in whom thyroid gland failure may develop, despite an increased TSH, from senescence rather than autoimmunity. What I am suggesting is that the natural history of all patients with subclinical hypothyroidism is not alike, and it thus should not be surprising that there does not seem to be a one-size-fits-all approach to management.
Symptoms in patients with subclinical hypothyroidism have not uniformly improved with T4 treatment compared with placebo. Notably, most patients with subclinical hypothyroidism experience no symptoms. But consider the extremely common symptom of fatigue, which can be present for a myriad of defined and undefined reasons. This symptom may often lead physicians to check the TSH and, if that is even slightly elevated, to also check the T4. It may also lead some physicians to routinely check the T4. Subclinical hypothyroidism is also quite common; thus, by chance alone or because of the circadian timing of checking the TSH, a slightly elevated TSH and fatigue may coexist and yet be unrelated.
Additionally, a positive biochemical response to thyroxine supplementation, such as a lowering of cholesterol, does not prove that the patient was clinically hypothyroid prior to supplementation, any more than lowering a patient’s blood glucose with insulin proves that the patient was diabetic. The management of subclinical hypothyroidism should be nuanced and based on both clinical and laboratory parameters.
At a quick read, the messages from these articles may seem contradictory. But the biology is more complex in the setting of endogenous production of T4 by the thyroid gland, which is regulated by TSH, which in turn is regulated in a feedback loop by the thyroid-produced T4. In the setting of a fixed replacement dose of exogenous levothyroxine, the provided hormone affects the pituitary production of TSH, which likely will have no significant subsequent effect on the T4 level. Thus, the feedback control loop is far simpler.
There has not been a definitive study demonstrating that thyroxine supplementation in patients with subclinical hypothyroidism results in a superior clinical outcome. There are hints that this may be the case, and Azim and Nasr cite some of these studies. Recognizing a few markedly different physiologic reasons why the TSH can be slightly elevated and the T4 normal helps explain the lack of uniform clinical success with supplementation therapy and provides rationales for some management strategies.
Any biological variability in the responsiveness of the thyroid gland to TSH may affect the relationship between the levels of TSH and thyroid gland-released T4. In theory, if the thyroid receptor has decreased affinity for TSH, a higher TSH concentration will be needed to get the thyroid gland to secrete the level of T4 that the pituitary sensing mechanism deems normal for that individual. If the receptor affinity was decreased due to a gene polymorphism, this relationship between TSH and T4 may be stable, and providing exogenous T4 will result in a lower, “normalized” TSH level but may disrupt the thyroid-pituitary crosstalk and may even produce clinical hyperthyroidism.
A similar scenario exists in the setting of early thyroid gland failure, such as in Hashimoto thyroiditis. But in the latter scenario, the TSH-to-T4 production relationship may be unstable over time, for as additional thyroid gland is destroyed, T4 production will continue to decrease, the TSH will increase, and the thyroid gland may ultimately fail and hypothyroidism will occur. Hence the recommendation that in the setting of subclinical hypothyroidism and antiperoxidase antibodies, T4 and TSH levels should be monitored regularly in order to detect early true thyroid gland failure when the T4 level can no longer be maintained despite the increased stimulation of the gland by the elevated TSH. Analogous to this may be subclinical hypothyroidism in the elderly, in whom thyroid gland failure may develop, despite an increased TSH, from senescence rather than autoimmunity. What I am suggesting is that the natural history of all patients with subclinical hypothyroidism is not alike, and it thus should not be surprising that there does not seem to be a one-size-fits-all approach to management.
Symptoms in patients with subclinical hypothyroidism have not uniformly improved with T4 treatment compared with placebo. Notably, most patients with subclinical hypothyroidism experience no symptoms. But consider the extremely common symptom of fatigue, which can be present for a myriad of defined and undefined reasons. This symptom may often lead physicians to check the TSH and, if that is even slightly elevated, to also check the T4. It may also lead some physicians to routinely check the T4. Subclinical hypothyroidism is also quite common; thus, by chance alone or because of the circadian timing of checking the TSH, a slightly elevated TSH and fatigue may coexist and yet be unrelated.
Additionally, a positive biochemical response to thyroxine supplementation, such as a lowering of cholesterol, does not prove that the patient was clinically hypothyroid prior to supplementation, any more than lowering a patient’s blood glucose with insulin proves that the patient was diabetic. The management of subclinical hypothyroidism should be nuanced and based on both clinical and laboratory parameters.
- Nasr C. Is a serum TSH measurement sufficient to monitor the treatment of primary hypothyroidism? Cleve Clin J Med 2016; 83(8):571–573. doi:10.3949/ccjm.83a.15165
- Mandell BF. Trust the thyroid thermostat. Cleve Clin J Med 2016; 83(8):552–553. doi:10.3949/ccjm.83b.08016
- Nasr C. Is a serum TSH measurement sufficient to monitor the treatment of primary hypothyroidism? Cleve Clin J Med 2016; 83(8):571–573. doi:10.3949/ccjm.83a.15165
- Mandell BF. Trust the thyroid thermostat. Cleve Clin J Med 2016; 83(8):552–553. doi:10.3949/ccjm.83b.08016
MGUS: It’s about the protein, not just the marrow
In the past decade, it has been increasingly recognized that these clonally produced proteins—entire immunoglobulins or free light chains—may be directly pathogenic, independent of any pathologic effect of cellular clonal expansion and infiltration. Brouet class 1 cryoglobulinemia (in which a monoclonal paraprotein precipitates in cooler temperatures and acts as a source of complement, activating the immune complex) and light chain (usually lambda)-related amyloidosis have been recognized for much longer. But a newer concept, monoclonal gammopathy of renal significance (MGRS), has attracted significant attention and to some extent has modified our approach to patients with either known MGUS or unexplained chronic kidney disease.
Finding MGUS still warrants a parsimonious evaluation for possible progression to myeloma or other proliferative disorder, as discussed by Khouri et al in this issue of the Journal. But it should also prompt a thoughtful assessment of renal function, including estimating the glomerular filtration rate and looking for proteinuria, hematuria, and unexplained glucosuria or inappropriate urine pH. While typical light chain-induced renal tubular injury is usually associated with high levels of proteins such as those seen with myeloma, other patterns of glomerular, vascular, and mixed renal disease are associated with deposition of proteins that, once considered in the differential diagnosis, warrant renal biopsy to diagnose and direct appropriate therapy. That MGUS and MGRS occur more frequently in older patients, who are already at greater risk of multiple common causes of kidney disease, complicates clinical decision-making.1 Some of these disorders are associated with other initially subtle or seemingly disconnected clinical symptoms such as polyneuropathy, rash, and carpal tunnel syndrome, but many are at least initially limited to the kidneys.
As we enter a new calendar year, we at the Journal send our best wishes to all of our readers, authors, and peer reviewers, and we thank you for sharing in our medical education ventures. I personally hope that we have added some joy, enthusiasm—and some knowledge—to your professional activities, and I hope that we all can participate in some way to refashion a more civil and peaceful world in 2019.
- Rosner MH, Edeani A, Yanagita M, et al. Paraprotein-related kidney disease: diagnosing and treating monoclonal gammopathy of renal significance. Clin J Am Soc Neph 2016; 11(12):2280–2287. doi:10.2215/CJN.02920316
In the past decade, it has been increasingly recognized that these clonally produced proteins—entire immunoglobulins or free light chains—may be directly pathogenic, independent of any pathologic effect of cellular clonal expansion and infiltration. Brouet class 1 cryoglobulinemia (in which a monoclonal paraprotein precipitates in cooler temperatures and acts as a source of complement, activating the immune complex) and light chain (usually lambda)-related amyloidosis have been recognized for much longer. But a newer concept, monoclonal gammopathy of renal significance (MGRS), has attracted significant attention and to some extent has modified our approach to patients with either known MGUS or unexplained chronic kidney disease.
Finding MGUS still warrants a parsimonious evaluation for possible progression to myeloma or other proliferative disorder, as discussed by Khouri et al in this issue of the Journal. But it should also prompt a thoughtful assessment of renal function, including estimating the glomerular filtration rate and looking for proteinuria, hematuria, and unexplained glucosuria or inappropriate urine pH. While typical light chain-induced renal tubular injury is usually associated with high levels of proteins such as those seen with myeloma, other patterns of glomerular, vascular, and mixed renal disease are associated with deposition of proteins that, once considered in the differential diagnosis, warrant renal biopsy to diagnose and direct appropriate therapy. That MGUS and MGRS occur more frequently in older patients, who are already at greater risk of multiple common causes of kidney disease, complicates clinical decision-making.1 Some of these disorders are associated with other initially subtle or seemingly disconnected clinical symptoms such as polyneuropathy, rash, and carpal tunnel syndrome, but many are at least initially limited to the kidneys.
As we enter a new calendar year, we at the Journal send our best wishes to all of our readers, authors, and peer reviewers, and we thank you for sharing in our medical education ventures. I personally hope that we have added some joy, enthusiasm—and some knowledge—to your professional activities, and I hope that we all can participate in some way to refashion a more civil and peaceful world in 2019.
In the past decade, it has been increasingly recognized that these clonally produced proteins—entire immunoglobulins or free light chains—may be directly pathogenic, independent of any pathologic effect of cellular clonal expansion and infiltration. Brouet class 1 cryoglobulinemia (in which a monoclonal paraprotein precipitates in cooler temperatures and acts as a source of complement, activating the immune complex) and light chain (usually lambda)-related amyloidosis have been recognized for much longer. But a newer concept, monoclonal gammopathy of renal significance (MGRS), has attracted significant attention and to some extent has modified our approach to patients with either known MGUS or unexplained chronic kidney disease.
Finding MGUS still warrants a parsimonious evaluation for possible progression to myeloma or other proliferative disorder, as discussed by Khouri et al in this issue of the Journal. But it should also prompt a thoughtful assessment of renal function, including estimating the glomerular filtration rate and looking for proteinuria, hematuria, and unexplained glucosuria or inappropriate urine pH. While typical light chain-induced renal tubular injury is usually associated with high levels of proteins such as those seen with myeloma, other patterns of glomerular, vascular, and mixed renal disease are associated with deposition of proteins that, once considered in the differential diagnosis, warrant renal biopsy to diagnose and direct appropriate therapy. That MGUS and MGRS occur more frequently in older patients, who are already at greater risk of multiple common causes of kidney disease, complicates clinical decision-making.1 Some of these disorders are associated with other initially subtle or seemingly disconnected clinical symptoms such as polyneuropathy, rash, and carpal tunnel syndrome, but many are at least initially limited to the kidneys.
As we enter a new calendar year, we at the Journal send our best wishes to all of our readers, authors, and peer reviewers, and we thank you for sharing in our medical education ventures. I personally hope that we have added some joy, enthusiasm—and some knowledge—to your professional activities, and I hope that we all can participate in some way to refashion a more civil and peaceful world in 2019.
- Rosner MH, Edeani A, Yanagita M, et al. Paraprotein-related kidney disease: diagnosing and treating monoclonal gammopathy of renal significance. Clin J Am Soc Neph 2016; 11(12):2280–2287. doi:10.2215/CJN.02920316
- Rosner MH, Edeani A, Yanagita M, et al. Paraprotein-related kidney disease: diagnosing and treating monoclonal gammopathy of renal significance. Clin J Am Soc Neph 2016; 11(12):2280–2287. doi:10.2215/CJN.02920316

A new reason to reconsider that antibiotic prescription: The microbiome
But, after the results of many recent studies, it turns out I should not have been so comfortable after all. This should not be a surprise. We should never be overly confident with our understanding of anything in clinical practice.
In this issue, Dr. Martin Blaser discusses his work, which supports the hypothesis that the currently increased prevalence of obesity and diabetes is at least in part due to reduced diversity in the gut microbiome. The increased exposure to antibiotics through prescriptions for women before and during pregnancy, as well as perhaps their exposure to antibiotics in the environment, results in changes to the gut and vaginal flora that influence the developing gut and likely other anatomic microbiomes in the neonate and infant. Fascinating research done in mice, utilizing fecal transfer experiments, is building an evidence trail to support the concept that the microbiome plays a major role in the development of childhood and adult obesity, and the gut microbiome is influenced by its exposure to antibiotics, perhaps given years earlier.
Knowledge of the gastrointestinal and other human microbiomes is exploding. I now wonder how many seemingly random clinical events associated with antibiotic use that were not understood and were easily dismissed as stochastic warrant formal study. Some of my patients with rheumatoid arthritis have described flares after eating certain foods and transient remissions or exacerbations after treatment with antibiotics. An epidemiologic study has linked the likelihood of developing childhood inflammatory bowel disease with exposure to antibiotics. Even more fascinating are observations that the microbiota composition (influenced by antibiotics) can influence the outcome of cardiac allografts in a murine model and the response of certain tumors to immune checkpoint inhibitors in murine and human studies. The mechanism may relate to the effects of the microbiome on immune cell activation and migration. Several disorders have been linked to specific bacteria in the gut microbiome, and others as diverse as cardiovascular events and the acute inflammatory response to monosodium urate crystals (gout) are affected by metabolites generated by bacteria in the gut.
The use of germ-free and antibiotic-treated mice in the laboratory, with selective repopulation of their gut microbiome with flora harvested from other strains of mice or selected humans, will continue to teach us much about the role that these microbes and other inhabitants play in controlling normal and disease-disrupted homeostasis. C difficile overgrowth after antibiotic exposure, and the successful treatment of refractory C difficile with fecal transplantation,1 was just the beginning.
The simple writing of a prescription for an antibiotic is a far more complicated and long-lasting affair than most of us have thought.
- Agito MD, Atreja A, Rizk MK. Fecal microbiota transplantation for recurrent C difficile infection: ready for prime time? Cleve Clin J Med 2013; 80(2):101–108. doi:10.3949/ccjm.80a.12110
But, after the results of many recent studies, it turns out I should not have been so comfortable after all. This should not be a surprise. We should never be overly confident with our understanding of anything in clinical practice.
In this issue, Dr. Martin Blaser discusses his work, which supports the hypothesis that the currently increased prevalence of obesity and diabetes is at least in part due to reduced diversity in the gut microbiome. The increased exposure to antibiotics through prescriptions for women before and during pregnancy, as well as perhaps their exposure to antibiotics in the environment, results in changes to the gut and vaginal flora that influence the developing gut and likely other anatomic microbiomes in the neonate and infant. Fascinating research done in mice, utilizing fecal transfer experiments, is building an evidence trail to support the concept that the microbiome plays a major role in the development of childhood and adult obesity, and the gut microbiome is influenced by its exposure to antibiotics, perhaps given years earlier.
Knowledge of the gastrointestinal and other human microbiomes is exploding. I now wonder how many seemingly random clinical events associated with antibiotic use that were not understood and were easily dismissed as stochastic warrant formal study. Some of my patients with rheumatoid arthritis have described flares after eating certain foods and transient remissions or exacerbations after treatment with antibiotics. An epidemiologic study has linked the likelihood of developing childhood inflammatory bowel disease with exposure to antibiotics. Even more fascinating are observations that the microbiota composition (influenced by antibiotics) can influence the outcome of cardiac allografts in a murine model and the response of certain tumors to immune checkpoint inhibitors in murine and human studies. The mechanism may relate to the effects of the microbiome on immune cell activation and migration. Several disorders have been linked to specific bacteria in the gut microbiome, and others as diverse as cardiovascular events and the acute inflammatory response to monosodium urate crystals (gout) are affected by metabolites generated by bacteria in the gut.
The use of germ-free and antibiotic-treated mice in the laboratory, with selective repopulation of their gut microbiome with flora harvested from other strains of mice or selected humans, will continue to teach us much about the role that these microbes and other inhabitants play in controlling normal and disease-disrupted homeostasis. C difficile overgrowth after antibiotic exposure, and the successful treatment of refractory C difficile with fecal transplantation,1 was just the beginning.
The simple writing of a prescription for an antibiotic is a far more complicated and long-lasting affair than most of us have thought.
But, after the results of many recent studies, it turns out I should not have been so comfortable after all. This should not be a surprise. We should never be overly confident with our understanding of anything in clinical practice.
In this issue, Dr. Martin Blaser discusses his work, which supports the hypothesis that the currently increased prevalence of obesity and diabetes is at least in part due to reduced diversity in the gut microbiome. The increased exposure to antibiotics through prescriptions for women before and during pregnancy, as well as perhaps their exposure to antibiotics in the environment, results in changes to the gut and vaginal flora that influence the developing gut and likely other anatomic microbiomes in the neonate and infant. Fascinating research done in mice, utilizing fecal transfer experiments, is building an evidence trail to support the concept that the microbiome plays a major role in the development of childhood and adult obesity, and the gut microbiome is influenced by its exposure to antibiotics, perhaps given years earlier.
Knowledge of the gastrointestinal and other human microbiomes is exploding. I now wonder how many seemingly random clinical events associated with antibiotic use that were not understood and were easily dismissed as stochastic warrant formal study. Some of my patients with rheumatoid arthritis have described flares after eating certain foods and transient remissions or exacerbations after treatment with antibiotics. An epidemiologic study has linked the likelihood of developing childhood inflammatory bowel disease with exposure to antibiotics. Even more fascinating are observations that the microbiota composition (influenced by antibiotics) can influence the outcome of cardiac allografts in a murine model and the response of certain tumors to immune checkpoint inhibitors in murine and human studies. The mechanism may relate to the effects of the microbiome on immune cell activation and migration. Several disorders have been linked to specific bacteria in the gut microbiome, and others as diverse as cardiovascular events and the acute inflammatory response to monosodium urate crystals (gout) are affected by metabolites generated by bacteria in the gut.
The use of germ-free and antibiotic-treated mice in the laboratory, with selective repopulation of their gut microbiome with flora harvested from other strains of mice or selected humans, will continue to teach us much about the role that these microbes and other inhabitants play in controlling normal and disease-disrupted homeostasis. C difficile overgrowth after antibiotic exposure, and the successful treatment of refractory C difficile with fecal transplantation,1 was just the beginning.
The simple writing of a prescription for an antibiotic is a far more complicated and long-lasting affair than most of us have thought.
- Agito MD, Atreja A, Rizk MK. Fecal microbiota transplantation for recurrent C difficile infection: ready for prime time? Cleve Clin J Med 2013; 80(2):101–108. doi:10.3949/ccjm.80a.12110
- Agito MD, Atreja A, Rizk MK. Fecal microbiota transplantation for recurrent C difficile infection: ready for prime time? Cleve Clin J Med 2013; 80(2):101–108. doi:10.3949/ccjm.80a.12110

A physician’s response to observational studies of opioid prescribing
Several months ago, we invited readers to submit short personalized commentaries on articles that changed the way they approach a specific clinical problem and the way they take care of patients. In this issue of the Journal, addiction specialist Charles Reznikoff, MD, discusses 3 observational studies that focused on how prescribing opioids for acute pain can lead to chronic opioid use and addiction, and how these studies have influenced his practice.
Although observational studies rank lower on the level-of-evidence scale than randomized controlled trials, they can intellectually stimulate and inform us in ways that lead us to modify how we deliver clinical care.
The initial prescribing of pain medications and the management of patients with chronic pain are currently under intense scrutiny, and are the topic of much discussion in the United States. The opioid epidemic has spilled over into all aspects of daily life, far beyond the medical community. But since we physicians are the only legal and regulated source of narcotics and other pain medications, we are under the microscope—and rightly so.
We, our patients, the pharmaceutical industry, legislators, and the law enforcement community struggle to navigate a complex maze, one with moving walls. Not long ago, physicians were told that we were not attentive enough to our patients’ suffering and needed to do better at relieving it. “Pain” became a vital sign and a recorded metric of quality care. Some excellent changes evolved from this focus, such as increased emphasis on postoperative regional and local pain control. But pain measurements continue to be recorded at every outpatient visit, an almost mindless requirement.
Recently, a patient with lupus nephritis whom I was seeing for blood pressure management reported a pain level of 8 on a scale of 10. I confess that I usually don’t even look at these metrics, but for whatever reason I saw her answer. I asked her about it. She had burned her finger while cooking and said, “I had no idea what number to pick. I picked 8. It’s no big deal.”
But the ongoing emphasis on this metric may lead some patients to expect total pain relief, a problematic expectation in those with chronic pain syndromes such as fibromyalgia. As Dr. Reznikoff points out, a large proportion of patients report they have chronic pain, and many (but clearly not all) suffer from recognized or masked chronic anxiety and depression disorders1 that may well influence how they use pain medications.
Thus, while physicians indeed are on the front lines of offering initial prescriptions for pain medications, we remain betwixt and between in the challenges of responding to the immediate needs of our patients while trying to predict the long-term effects of our prescription on the individual patient and of our prescribing patterns on society in general.
I again welcome your submissions describing how individual publications have affected your personal approach to managing patients and specific diseases. We will publish selected contributions in print and online.
- Tsang A, Von Korff M, Lee S, et al. Common chronic pain conditions in developed and developing countries: gender and age differences and comorbidity with depression-anxiety disorders. J Pain 2008; 9(10):883–891. doi:10.1016/j.jpain.2008.05.005
Several months ago, we invited readers to submit short personalized commentaries on articles that changed the way they approach a specific clinical problem and the way they take care of patients. In this issue of the Journal, addiction specialist Charles Reznikoff, MD, discusses 3 observational studies that focused on how prescribing opioids for acute pain can lead to chronic opioid use and addiction, and how these studies have influenced his practice.
Although observational studies rank lower on the level-of-evidence scale than randomized controlled trials, they can intellectually stimulate and inform us in ways that lead us to modify how we deliver clinical care.
The initial prescribing of pain medications and the management of patients with chronic pain are currently under intense scrutiny, and are the topic of much discussion in the United States. The opioid epidemic has spilled over into all aspects of daily life, far beyond the medical community. But since we physicians are the only legal and regulated source of narcotics and other pain medications, we are under the microscope—and rightly so.
We, our patients, the pharmaceutical industry, legislators, and the law enforcement community struggle to navigate a complex maze, one with moving walls. Not long ago, physicians were told that we were not attentive enough to our patients’ suffering and needed to do better at relieving it. “Pain” became a vital sign and a recorded metric of quality care. Some excellent changes evolved from this focus, such as increased emphasis on postoperative regional and local pain control. But pain measurements continue to be recorded at every outpatient visit, an almost mindless requirement.
Recently, a patient with lupus nephritis whom I was seeing for blood pressure management reported a pain level of 8 on a scale of 10. I confess that I usually don’t even look at these metrics, but for whatever reason I saw her answer. I asked her about it. She had burned her finger while cooking and said, “I had no idea what number to pick. I picked 8. It’s no big deal.”
But the ongoing emphasis on this metric may lead some patients to expect total pain relief, a problematic expectation in those with chronic pain syndromes such as fibromyalgia. As Dr. Reznikoff points out, a large proportion of patients report they have chronic pain, and many (but clearly not all) suffer from recognized or masked chronic anxiety and depression disorders1 that may well influence how they use pain medications.
Thus, while physicians indeed are on the front lines of offering initial prescriptions for pain medications, we remain betwixt and between in the challenges of responding to the immediate needs of our patients while trying to predict the long-term effects of our prescription on the individual patient and of our prescribing patterns on society in general.
I again welcome your submissions describing how individual publications have affected your personal approach to managing patients and specific diseases. We will publish selected contributions in print and online.
Several months ago, we invited readers to submit short personalized commentaries on articles that changed the way they approach a specific clinical problem and the way they take care of patients. In this issue of the Journal, addiction specialist Charles Reznikoff, MD, discusses 3 observational studies that focused on how prescribing opioids for acute pain can lead to chronic opioid use and addiction, and how these studies have influenced his practice.
Although observational studies rank lower on the level-of-evidence scale than randomized controlled trials, they can intellectually stimulate and inform us in ways that lead us to modify how we deliver clinical care.
The initial prescribing of pain medications and the management of patients with chronic pain are currently under intense scrutiny, and are the topic of much discussion in the United States. The opioid epidemic has spilled over into all aspects of daily life, far beyond the medical community. But since we physicians are the only legal and regulated source of narcotics and other pain medications, we are under the microscope—and rightly so.
We, our patients, the pharmaceutical industry, legislators, and the law enforcement community struggle to navigate a complex maze, one with moving walls. Not long ago, physicians were told that we were not attentive enough to our patients’ suffering and needed to do better at relieving it. “Pain” became a vital sign and a recorded metric of quality care. Some excellent changes evolved from this focus, such as increased emphasis on postoperative regional and local pain control. But pain measurements continue to be recorded at every outpatient visit, an almost mindless requirement.
Recently, a patient with lupus nephritis whom I was seeing for blood pressure management reported a pain level of 8 on a scale of 10. I confess that I usually don’t even look at these metrics, but for whatever reason I saw her answer. I asked her about it. She had burned her finger while cooking and said, “I had no idea what number to pick. I picked 8. It’s no big deal.”
But the ongoing emphasis on this metric may lead some patients to expect total pain relief, a problematic expectation in those with chronic pain syndromes such as fibromyalgia. As Dr. Reznikoff points out, a large proportion of patients report they have chronic pain, and many (but clearly not all) suffer from recognized or masked chronic anxiety and depression disorders1 that may well influence how they use pain medications.
Thus, while physicians indeed are on the front lines of offering initial prescriptions for pain medications, we remain betwixt and between in the challenges of responding to the immediate needs of our patients while trying to predict the long-term effects of our prescription on the individual patient and of our prescribing patterns on society in general.
I again welcome your submissions describing how individual publications have affected your personal approach to managing patients and specific diseases. We will publish selected contributions in print and online.
- Tsang A, Von Korff M, Lee S, et al. Common chronic pain conditions in developed and developing countries: gender and age differences and comorbidity with depression-anxiety disorders. J Pain 2008; 9(10):883–891. doi:10.1016/j.jpain.2008.05.005
- Tsang A, Von Korff M, Lee S, et al. Common chronic pain conditions in developed and developing countries: gender and age differences and comorbidity with depression-anxiety disorders. J Pain 2008; 9(10):883–891. doi:10.1016/j.jpain.2008.05.005
Small fibers, large impact
The details about an individual’s search for information tell us a lot about healthcare concerns and uncertainty across the medical universe. For nearly a decade, one of the most “clicked on” papers we have published in the Journal has been a review of small fiber neuropathy—a clinical entity with a prevalence of perhaps 1 in 1,000 to 2,000 people and, to my knowledge, no associated walkathons or arm bracelets. Yet it certainly piques the interest of clinicians from many specialties far broader than neurology. In this issue of the Journal, Dr. Jinny Tavee updates her 2009 review and provides us with a clinical overview of the disorder and the opportunity to assess how much further we need to more fully understand its management and associated comorbid conditions.
The wide interest in this disorder plugs into our current seeming epidemic of patients with chronic pain. It seems that almost half of my new patients have issues related to chronic pain that are not directly explained by active inflammation or anatomic damage. Many of these patients have diffuse body pains with associated fatigue and sleep disorders and are diagnosed with fibromyalgia. But others describe pain with a burning and tingling quality that seems of neurologic origin, yet their neurologic examination is normal. A few describe a predominantly distal symmetric stocking-and-glove distribution, but most do not. In some patients these pains are spatially random and evanescent, which to me are usually the hardest to fathom. Nerve conduction studies, when performed, are unrevealing.
A number of systemic autoimmune disorders, as discussed by Dr. Tavee in her article, are suggested to have an association with these symptoms. Given the chronicity and the frustrating nature of the symptoms, it is no surprise that a panoply of immune serologies are frequently ordered. Invariably, since serologies (eg, ANA, SSA, SSB, rheumatoid factor) are not specific for any single entity, some will return as positive. The strength of many of these associations is weak; even when the clinical diagnosis of lupus, for example, is definite, treatment of the underlying disease does not necessarily improve the dysesthetic pain. In an alternative scenario, the small fiber neuropathy is ascribed to a systemic autoimmune disorder that has been diagnosed because an autoantibody has been detected, but this rarely helps the patient and may in fact worsen symptoms by increasing anxiety and concern over having a systemic disease such as Sjögren syndrome or lupus (both of which sound terrible when reviewed on the Internet).
Some patients describe autonomic symptoms. Given the biologic basis that has been defined for this entity, it is no surprise that some patients have marked symptoms of decreased exocrine gland function, gastrointestinal dysmotility, and orthostasis. These symptoms may not be recognized unless specifically sought out when interviewing the patient.
Given the chronicity and sometimes the vagaries of symptoms, it is often comforting for patients to get an actual diagnosis. Dr. Tavee notes the relative simplicity of diagnostic procedures. But determining the clinical implications of the results may not be straightforward, and devising a fully and uniformly effective therapeutic approach eludes us still. As she points out, a multidisciplinary approach to therapy and diagnosis can be quite helpful.
The details about an individual’s search for information tell us a lot about healthcare concerns and uncertainty across the medical universe. For nearly a decade, one of the most “clicked on” papers we have published in the Journal has been a review of small fiber neuropathy—a clinical entity with a prevalence of perhaps 1 in 1,000 to 2,000 people and, to my knowledge, no associated walkathons or arm bracelets. Yet it certainly piques the interest of clinicians from many specialties far broader than neurology. In this issue of the Journal, Dr. Jinny Tavee updates her 2009 review and provides us with a clinical overview of the disorder and the opportunity to assess how much further we need to more fully understand its management and associated comorbid conditions.
The wide interest in this disorder plugs into our current seeming epidemic of patients with chronic pain. It seems that almost half of my new patients have issues related to chronic pain that are not directly explained by active inflammation or anatomic damage. Many of these patients have diffuse body pains with associated fatigue and sleep disorders and are diagnosed with fibromyalgia. But others describe pain with a burning and tingling quality that seems of neurologic origin, yet their neurologic examination is normal. A few describe a predominantly distal symmetric stocking-and-glove distribution, but most do not. In some patients these pains are spatially random and evanescent, which to me are usually the hardest to fathom. Nerve conduction studies, when performed, are unrevealing.
A number of systemic autoimmune disorders, as discussed by Dr. Tavee in her article, are suggested to have an association with these symptoms. Given the chronicity and the frustrating nature of the symptoms, it is no surprise that a panoply of immune serologies are frequently ordered. Invariably, since serologies (eg, ANA, SSA, SSB, rheumatoid factor) are not specific for any single entity, some will return as positive. The strength of many of these associations is weak; even when the clinical diagnosis of lupus, for example, is definite, treatment of the underlying disease does not necessarily improve the dysesthetic pain. In an alternative scenario, the small fiber neuropathy is ascribed to a systemic autoimmune disorder that has been diagnosed because an autoantibody has been detected, but this rarely helps the patient and may in fact worsen symptoms by increasing anxiety and concern over having a systemic disease such as Sjögren syndrome or lupus (both of which sound terrible when reviewed on the Internet).
Some patients describe autonomic symptoms. Given the biologic basis that has been defined for this entity, it is no surprise that some patients have marked symptoms of decreased exocrine gland function, gastrointestinal dysmotility, and orthostasis. These symptoms may not be recognized unless specifically sought out when interviewing the patient.
Given the chronicity and sometimes the vagaries of symptoms, it is often comforting for patients to get an actual diagnosis. Dr. Tavee notes the relative simplicity of diagnostic procedures. But determining the clinical implications of the results may not be straightforward, and devising a fully and uniformly effective therapeutic approach eludes us still. As she points out, a multidisciplinary approach to therapy and diagnosis can be quite helpful.
The details about an individual’s search for information tell us a lot about healthcare concerns and uncertainty across the medical universe. For nearly a decade, one of the most “clicked on” papers we have published in the Journal has been a review of small fiber neuropathy—a clinical entity with a prevalence of perhaps 1 in 1,000 to 2,000 people and, to my knowledge, no associated walkathons or arm bracelets. Yet it certainly piques the interest of clinicians from many specialties far broader than neurology. In this issue of the Journal, Dr. Jinny Tavee updates her 2009 review and provides us with a clinical overview of the disorder and the opportunity to assess how much further we need to more fully understand its management and associated comorbid conditions.
The wide interest in this disorder plugs into our current seeming epidemic of patients with chronic pain. It seems that almost half of my new patients have issues related to chronic pain that are not directly explained by active inflammation or anatomic damage. Many of these patients have diffuse body pains with associated fatigue and sleep disorders and are diagnosed with fibromyalgia. But others describe pain with a burning and tingling quality that seems of neurologic origin, yet their neurologic examination is normal. A few describe a predominantly distal symmetric stocking-and-glove distribution, but most do not. In some patients these pains are spatially random and evanescent, which to me are usually the hardest to fathom. Nerve conduction studies, when performed, are unrevealing.
A number of systemic autoimmune disorders, as discussed by Dr. Tavee in her article, are suggested to have an association with these symptoms. Given the chronicity and the frustrating nature of the symptoms, it is no surprise that a panoply of immune serologies are frequently ordered. Invariably, since serologies (eg, ANA, SSA, SSB, rheumatoid factor) are not specific for any single entity, some will return as positive. The strength of many of these associations is weak; even when the clinical diagnosis of lupus, for example, is definite, treatment of the underlying disease does not necessarily improve the dysesthetic pain. In an alternative scenario, the small fiber neuropathy is ascribed to a systemic autoimmune disorder that has been diagnosed because an autoantibody has been detected, but this rarely helps the patient and may in fact worsen symptoms by increasing anxiety and concern over having a systemic disease such as Sjögren syndrome or lupus (both of which sound terrible when reviewed on the Internet).
Some patients describe autonomic symptoms. Given the biologic basis that has been defined for this entity, it is no surprise that some patients have marked symptoms of decreased exocrine gland function, gastrointestinal dysmotility, and orthostasis. These symptoms may not be recognized unless specifically sought out when interviewing the patient.
Given the chronicity and sometimes the vagaries of symptoms, it is often comforting for patients to get an actual diagnosis. Dr. Tavee notes the relative simplicity of diagnostic procedures. But determining the clinical implications of the results may not be straightforward, and devising a fully and uniformly effective therapeutic approach eludes us still. As she points out, a multidisciplinary approach to therapy and diagnosis can be quite helpful.
We can learn a lot from drug adverse effects
Some drugs exhibit a dose-toxicity relationship that is sufficiently predictable to permit drug-level monitoring to limit renal or other toxicity. Others cause ocular or marrow toxicity that can be limited by weight-based dosing and careful monitoring. With azathioprine, some toxicity can be predicted by assessing the activity of the enzyme thiopurine methyltransferase, which metabolizes the drug. Other approaches to using pharmacogenomics have included HLA-B locus haplotyping to detect increased risk of immune-mediated toxicities of drugs such as carbamazepine and allopurinol. Both of these drugs exhibit serious systemic toxicities that are incompletely understood, but these are nascent and significant steps toward providing personalized (precision) medical care.
Adverse effects of some drugs may result from their intracellular effects, which are only partially predictable by drug levels or dosing. Colchicine, hydroxychloroquine, and amiodarone all affect intracellular vacuolar transport and lysosomal processing. Yet, although the footprints of drug effect can be seen in many histopathology samples, only some patients—but maybe more than currently recognized—suffer cardiac and skeletal muscle vacuolar myopathy, axonal neuropathies, or pulmonary or retinal cell toxicity from these drugs. But distinguishing the histopathologic footprints of drug exposure and the biologic effect from true drug toxicity with organ damage is not always straightforward.
Rare adverse effects may only become apparent with frequent use of a drug in the general community. These often remain mechanistically unexplained: Why can minoxidil cause pericardial effusions or a nonsteroidal anti-inflammatory drug cause aseptic meningitis? Some effects may be due to altering of the unique balance of biochemical pathways in a given patient, leading to unexpected drug metabolism with generation of toxic metabolites.
More interesting to me are effects that are seemingly off-target biologic outcomes caused by disrupting normal physiologic homeostasis and stimulating counterregulatory pathways in such a way that unexpected biologic effects occur. Angioedema and cough in some patients who have taken angiotensin-converting enzyme inhibitors are examples, but why the disturbed control mechanisms lead to these effects in only occasional patients is still incompletely elucidated.
Two additional classes of drugs with unique systemic adverse effects are discussed in this issue of the Journal. The “flulike” syndrome after bisphosphonate treatment, presumably resulting from selective cytokine release by macrophages that have ingested certain bisphosphonates, is a not uncommon and significant annoyance to many patients, and in my experience it is a reason patients discontinue the treatment. Lim and Bolster describe the reaction and their approach to its management, and comment on the fairly obscure pathway that may explain its occurrence. Again, it is not clear to me why only relatively few patients experience the reaction. Is there a genetic predisposition? Or is it influenced by the patient’s baseline “inflammatory tone,” as influenced by the state of his or her microbiome or other still uncharacterized factors? And why does this reaction often diminish with repeated dosing of the drug?
Most striking is the description and discussion by Khan et al of the management of autoimmune colitis after administration of immune checkpoint inhibitor anticancer therapies. These drugs represent important advances in the therapy of various cancers. They are novel in that they are not specific to tumor type, although certain drugs within this new class of immunotherapy seem to exhibit more dramatic and enduring responses against one type of cancer than against another. These therapies are not directly tumor-reactive, but act by down-regulating the normal “brakes” or checkpoints of the immune system that normally play a role in reigning in the immune-inflammatory system response to infection once the offending infective agent is neutralized. These checkpoints have also been thought to limit the development of autoimmunity. Many cancers seem to capitalize on the activation of these brakes to evade tumor immunity. That these checkpoint therapies are so effective in some patients with heretofore unresponsive cancers is obviously a major advance. But equally striking is the scientific proof of the immunologic concept that by inhibiting these normal brakes on inflammation there is loss of normal regulation of the immune response and autoimmunity ensues unchecked. Khan et al discuss the colitis that can occur with these therapies, but a host of fascinating and potentially life-threatening organ-specific complications can be invoked by the checkpoint inhibitors, including hypophysitis, myositis, nephritis, and pneumonitis. What determines which patient will suffer these immune complications, which organs will be affected in a given patient, and the relationships between preexisting autoimmune disease, antitumor response, and these autoimmune complications are still being unraveled.
If you have not yet encountered patients with these complications in your practice, it is quite likely you will. The topic is worth reading about now (see the review by June et al1), and we will provide additional reviews in the future.
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med 2017; 23(5):540–547. doi:10.1038/nm.4321. Correction in Nat Med 2017; 23(8):1004. doi:10.1038/nm0817-1004b
Some drugs exhibit a dose-toxicity relationship that is sufficiently predictable to permit drug-level monitoring to limit renal or other toxicity. Others cause ocular or marrow toxicity that can be limited by weight-based dosing and careful monitoring. With azathioprine, some toxicity can be predicted by assessing the activity of the enzyme thiopurine methyltransferase, which metabolizes the drug. Other approaches to using pharmacogenomics have included HLA-B locus haplotyping to detect increased risk of immune-mediated toxicities of drugs such as carbamazepine and allopurinol. Both of these drugs exhibit serious systemic toxicities that are incompletely understood, but these are nascent and significant steps toward providing personalized (precision) medical care.
Adverse effects of some drugs may result from their intracellular effects, which are only partially predictable by drug levels or dosing. Colchicine, hydroxychloroquine, and amiodarone all affect intracellular vacuolar transport and lysosomal processing. Yet, although the footprints of drug effect can be seen in many histopathology samples, only some patients—but maybe more than currently recognized—suffer cardiac and skeletal muscle vacuolar myopathy, axonal neuropathies, or pulmonary or retinal cell toxicity from these drugs. But distinguishing the histopathologic footprints of drug exposure and the biologic effect from true drug toxicity with organ damage is not always straightforward.
Rare adverse effects may only become apparent with frequent use of a drug in the general community. These often remain mechanistically unexplained: Why can minoxidil cause pericardial effusions or a nonsteroidal anti-inflammatory drug cause aseptic meningitis? Some effects may be due to altering of the unique balance of biochemical pathways in a given patient, leading to unexpected drug metabolism with generation of toxic metabolites.
More interesting to me are effects that are seemingly off-target biologic outcomes caused by disrupting normal physiologic homeostasis and stimulating counterregulatory pathways in such a way that unexpected biologic effects occur. Angioedema and cough in some patients who have taken angiotensin-converting enzyme inhibitors are examples, but why the disturbed control mechanisms lead to these effects in only occasional patients is still incompletely elucidated.
Two additional classes of drugs with unique systemic adverse effects are discussed in this issue of the Journal. The “flulike” syndrome after bisphosphonate treatment, presumably resulting from selective cytokine release by macrophages that have ingested certain bisphosphonates, is a not uncommon and significant annoyance to many patients, and in my experience it is a reason patients discontinue the treatment. Lim and Bolster describe the reaction and their approach to its management, and comment on the fairly obscure pathway that may explain its occurrence. Again, it is not clear to me why only relatively few patients experience the reaction. Is there a genetic predisposition? Or is it influenced by the patient’s baseline “inflammatory tone,” as influenced by the state of his or her microbiome or other still uncharacterized factors? And why does this reaction often diminish with repeated dosing of the drug?
Most striking is the description and discussion by Khan et al of the management of autoimmune colitis after administration of immune checkpoint inhibitor anticancer therapies. These drugs represent important advances in the therapy of various cancers. They are novel in that they are not specific to tumor type, although certain drugs within this new class of immunotherapy seem to exhibit more dramatic and enduring responses against one type of cancer than against another. These therapies are not directly tumor-reactive, but act by down-regulating the normal “brakes” or checkpoints of the immune system that normally play a role in reigning in the immune-inflammatory system response to infection once the offending infective agent is neutralized. These checkpoints have also been thought to limit the development of autoimmunity. Many cancers seem to capitalize on the activation of these brakes to evade tumor immunity. That these checkpoint therapies are so effective in some patients with heretofore unresponsive cancers is obviously a major advance. But equally striking is the scientific proof of the immunologic concept that by inhibiting these normal brakes on inflammation there is loss of normal regulation of the immune response and autoimmunity ensues unchecked. Khan et al discuss the colitis that can occur with these therapies, but a host of fascinating and potentially life-threatening organ-specific complications can be invoked by the checkpoint inhibitors, including hypophysitis, myositis, nephritis, and pneumonitis. What determines which patient will suffer these immune complications, which organs will be affected in a given patient, and the relationships between preexisting autoimmune disease, antitumor response, and these autoimmune complications are still being unraveled.
If you have not yet encountered patients with these complications in your practice, it is quite likely you will. The topic is worth reading about now (see the review by June et al1), and we will provide additional reviews in the future.
Some drugs exhibit a dose-toxicity relationship that is sufficiently predictable to permit drug-level monitoring to limit renal or other toxicity. Others cause ocular or marrow toxicity that can be limited by weight-based dosing and careful monitoring. With azathioprine, some toxicity can be predicted by assessing the activity of the enzyme thiopurine methyltransferase, which metabolizes the drug. Other approaches to using pharmacogenomics have included HLA-B locus haplotyping to detect increased risk of immune-mediated toxicities of drugs such as carbamazepine and allopurinol. Both of these drugs exhibit serious systemic toxicities that are incompletely understood, but these are nascent and significant steps toward providing personalized (precision) medical care.
Adverse effects of some drugs may result from their intracellular effects, which are only partially predictable by drug levels or dosing. Colchicine, hydroxychloroquine, and amiodarone all affect intracellular vacuolar transport and lysosomal processing. Yet, although the footprints of drug effect can be seen in many histopathology samples, only some patients—but maybe more than currently recognized—suffer cardiac and skeletal muscle vacuolar myopathy, axonal neuropathies, or pulmonary or retinal cell toxicity from these drugs. But distinguishing the histopathologic footprints of drug exposure and the biologic effect from true drug toxicity with organ damage is not always straightforward.
Rare adverse effects may only become apparent with frequent use of a drug in the general community. These often remain mechanistically unexplained: Why can minoxidil cause pericardial effusions or a nonsteroidal anti-inflammatory drug cause aseptic meningitis? Some effects may be due to altering of the unique balance of biochemical pathways in a given patient, leading to unexpected drug metabolism with generation of toxic metabolites.
More interesting to me are effects that are seemingly off-target biologic outcomes caused by disrupting normal physiologic homeostasis and stimulating counterregulatory pathways in such a way that unexpected biologic effects occur. Angioedema and cough in some patients who have taken angiotensin-converting enzyme inhibitors are examples, but why the disturbed control mechanisms lead to these effects in only occasional patients is still incompletely elucidated.
Two additional classes of drugs with unique systemic adverse effects are discussed in this issue of the Journal. The “flulike” syndrome after bisphosphonate treatment, presumably resulting from selective cytokine release by macrophages that have ingested certain bisphosphonates, is a not uncommon and significant annoyance to many patients, and in my experience it is a reason patients discontinue the treatment. Lim and Bolster describe the reaction and their approach to its management, and comment on the fairly obscure pathway that may explain its occurrence. Again, it is not clear to me why only relatively few patients experience the reaction. Is there a genetic predisposition? Or is it influenced by the patient’s baseline “inflammatory tone,” as influenced by the state of his or her microbiome or other still uncharacterized factors? And why does this reaction often diminish with repeated dosing of the drug?
Most striking is the description and discussion by Khan et al of the management of autoimmune colitis after administration of immune checkpoint inhibitor anticancer therapies. These drugs represent important advances in the therapy of various cancers. They are novel in that they are not specific to tumor type, although certain drugs within this new class of immunotherapy seem to exhibit more dramatic and enduring responses against one type of cancer than against another. These therapies are not directly tumor-reactive, but act by down-regulating the normal “brakes” or checkpoints of the immune system that normally play a role in reigning in the immune-inflammatory system response to infection once the offending infective agent is neutralized. These checkpoints have also been thought to limit the development of autoimmunity. Many cancers seem to capitalize on the activation of these brakes to evade tumor immunity. That these checkpoint therapies are so effective in some patients with heretofore unresponsive cancers is obviously a major advance. But equally striking is the scientific proof of the immunologic concept that by inhibiting these normal brakes on inflammation there is loss of normal regulation of the immune response and autoimmunity ensues unchecked. Khan et al discuss the colitis that can occur with these therapies, but a host of fascinating and potentially life-threatening organ-specific complications can be invoked by the checkpoint inhibitors, including hypophysitis, myositis, nephritis, and pneumonitis. What determines which patient will suffer these immune complications, which organs will be affected in a given patient, and the relationships between preexisting autoimmune disease, antitumor response, and these autoimmune complications are still being unraveled.
If you have not yet encountered patients with these complications in your practice, it is quite likely you will. The topic is worth reading about now (see the review by June et al1), and we will provide additional reviews in the future.
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med 2017; 23(5):540–547. doi:10.1038/nm.4321. Correction in Nat Med 2017; 23(8):1004. doi:10.1038/nm0817-1004b
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med 2017; 23(5):540–547. doi:10.1038/nm.4321. Correction in Nat Med 2017; 23(8):1004. doi:10.1038/nm0817-1004b

The bias of word choice and the interpretation of laboratory tests
In the current sociopolitical environment in the United States, the slogan “words matter” has become a battle cry for several groups and causes, emphasizing that our choice of words can influence the way we assess a specific person or situation. We are not immune to the subliminal bias of words, even as we evaluate such seemingly objective components of clinical management as laboratory test results.
Several years ago, I was supervising teaching rounds on a general medicine service. It was the first rounds of the month, and the patients were relatively new to the residents and totally unknown to me. One patient was an elderly man with weight loss, fatigue, weakness, and a history of excessive alcohol ingestion. His family had corroborated the last detail, but he had stopped drinking a long time before his admission. He had normal creatinine, minimal anemia, and markedly elevated and unexplained “liver function tests.” Liver biopsy was planned.
As we entered his room, we saw a gaunt man struggle to rise from the bedside chair to get back into bed. He rocked several times and then pushed himself up from the chair using his arms. Then, after a few short steps, he plopped back into bed and greeted us. His breakfast tray was untouched at the bedside. I introduced myself, we chatted for a short while as I examined him in front of our team, and we left.
In the hallway I asked, “Who would like to get an additional blood test before we do a liver biopsy?” Without waiting for a response I asked a second question, “What exactly are liver function tests?”
Words do matter, and they influence the way we analyze clinical scenarios. It could be argued that a complete and careful history would have established that our patient’s fatigue and weakness were due to proximal muscle weakness and not general asthenia, and that detailed questioning would have revealed that his weight loss was mainly from difficulty in swallowing without a sense of choking and coughing. But faced with an elderly man, a likely explanation for liver disease, and markedly elevated aspartate and alanine aminotransferase (AST and ALT) levels, there was premature closure of the diagnosis, and the decision was made to obtain a liver biopsy—which our hepatology consultants surely would not have done. I believe that a major contributor to the premature diagnosis was the choice of the words “liver function tests” and the default assumption that elevated serum levels of these enzymes always reflect liver disease.
Aminotransferases are fairly ubiquitous, likely present in various concentrations in all cells in our body. AST exists in mitochondrial and cytosolic forms, and ALT in the cytosol. The concentration of ALT is higher in the liver than in other organs, and its enzymatic activity is suppressed by hepatic exposure to alcohol. Both enzymes are present in muscle, and although AST is more abundant in cells other than hepatocytes, the longer serum half-life of ALT may result in roughly equal serum levels in the setting of chronic muscle injury such as myositis (the true diagnosis in our weak patient).
While a meticulous history and examination would indeed have led to the diagnosis of muscle disease in this man, they alone could not have determined whether he had coexistent liver and muscle disease. And this is a real challenge when acute muscle toxicity and liver toxicity are equally possible (eg, statin or immune checkpoint autoimmune tissue damage, or after significant trauma).
There are many nuances in the interpretation of even the most common laboratory tests. In this issue of the Journal, Agganis et al discuss liver enzymes (a term slightly more acceptable to me than liver function tests). In future issues, we will address the interpretation of other laboratory tests.
In the current sociopolitical environment in the United States, the slogan “words matter” has become a battle cry for several groups and causes, emphasizing that our choice of words can influence the way we assess a specific person or situation. We are not immune to the subliminal bias of words, even as we evaluate such seemingly objective components of clinical management as laboratory test results.
Several years ago, I was supervising teaching rounds on a general medicine service. It was the first rounds of the month, and the patients were relatively new to the residents and totally unknown to me. One patient was an elderly man with weight loss, fatigue, weakness, and a history of excessive alcohol ingestion. His family had corroborated the last detail, but he had stopped drinking a long time before his admission. He had normal creatinine, minimal anemia, and markedly elevated and unexplained “liver function tests.” Liver biopsy was planned.
As we entered his room, we saw a gaunt man struggle to rise from the bedside chair to get back into bed. He rocked several times and then pushed himself up from the chair using his arms. Then, after a few short steps, he plopped back into bed and greeted us. His breakfast tray was untouched at the bedside. I introduced myself, we chatted for a short while as I examined him in front of our team, and we left.
In the hallway I asked, “Who would like to get an additional blood test before we do a liver biopsy?” Without waiting for a response I asked a second question, “What exactly are liver function tests?”
Words do matter, and they influence the way we analyze clinical scenarios. It could be argued that a complete and careful history would have established that our patient’s fatigue and weakness were due to proximal muscle weakness and not general asthenia, and that detailed questioning would have revealed that his weight loss was mainly from difficulty in swallowing without a sense of choking and coughing. But faced with an elderly man, a likely explanation for liver disease, and markedly elevated aspartate and alanine aminotransferase (AST and ALT) levels, there was premature closure of the diagnosis, and the decision was made to obtain a liver biopsy—which our hepatology consultants surely would not have done. I believe that a major contributor to the premature diagnosis was the choice of the words “liver function tests” and the default assumption that elevated serum levels of these enzymes always reflect liver disease.
Aminotransferases are fairly ubiquitous, likely present in various concentrations in all cells in our body. AST exists in mitochondrial and cytosolic forms, and ALT in the cytosol. The concentration of ALT is higher in the liver than in other organs, and its enzymatic activity is suppressed by hepatic exposure to alcohol. Both enzymes are present in muscle, and although AST is more abundant in cells other than hepatocytes, the longer serum half-life of ALT may result in roughly equal serum levels in the setting of chronic muscle injury such as myositis (the true diagnosis in our weak patient).
While a meticulous history and examination would indeed have led to the diagnosis of muscle disease in this man, they alone could not have determined whether he had coexistent liver and muscle disease. And this is a real challenge when acute muscle toxicity and liver toxicity are equally possible (eg, statin or immune checkpoint autoimmune tissue damage, or after significant trauma).
There are many nuances in the interpretation of even the most common laboratory tests. In this issue of the Journal, Agganis et al discuss liver enzymes (a term slightly more acceptable to me than liver function tests). In future issues, we will address the interpretation of other laboratory tests.
In the current sociopolitical environment in the United States, the slogan “words matter” has become a battle cry for several groups and causes, emphasizing that our choice of words can influence the way we assess a specific person or situation. We are not immune to the subliminal bias of words, even as we evaluate such seemingly objective components of clinical management as laboratory test results.
Several years ago, I was supervising teaching rounds on a general medicine service. It was the first rounds of the month, and the patients were relatively new to the residents and totally unknown to me. One patient was an elderly man with weight loss, fatigue, weakness, and a history of excessive alcohol ingestion. His family had corroborated the last detail, but he had stopped drinking a long time before his admission. He had normal creatinine, minimal anemia, and markedly elevated and unexplained “liver function tests.” Liver biopsy was planned.
As we entered his room, we saw a gaunt man struggle to rise from the bedside chair to get back into bed. He rocked several times and then pushed himself up from the chair using his arms. Then, after a few short steps, he plopped back into bed and greeted us. His breakfast tray was untouched at the bedside. I introduced myself, we chatted for a short while as I examined him in front of our team, and we left.
In the hallway I asked, “Who would like to get an additional blood test before we do a liver biopsy?” Without waiting for a response I asked a second question, “What exactly are liver function tests?”
Words do matter, and they influence the way we analyze clinical scenarios. It could be argued that a complete and careful history would have established that our patient’s fatigue and weakness were due to proximal muscle weakness and not general asthenia, and that detailed questioning would have revealed that his weight loss was mainly from difficulty in swallowing without a sense of choking and coughing. But faced with an elderly man, a likely explanation for liver disease, and markedly elevated aspartate and alanine aminotransferase (AST and ALT) levels, there was premature closure of the diagnosis, and the decision was made to obtain a liver biopsy—which our hepatology consultants surely would not have done. I believe that a major contributor to the premature diagnosis was the choice of the words “liver function tests” and the default assumption that elevated serum levels of these enzymes always reflect liver disease.
Aminotransferases are fairly ubiquitous, likely present in various concentrations in all cells in our body. AST exists in mitochondrial and cytosolic forms, and ALT in the cytosol. The concentration of ALT is higher in the liver than in other organs, and its enzymatic activity is suppressed by hepatic exposure to alcohol. Both enzymes are present in muscle, and although AST is more abundant in cells other than hepatocytes, the longer serum half-life of ALT may result in roughly equal serum levels in the setting of chronic muscle injury such as myositis (the true diagnosis in our weak patient).
While a meticulous history and examination would indeed have led to the diagnosis of muscle disease in this man, they alone could not have determined whether he had coexistent liver and muscle disease. And this is a real challenge when acute muscle toxicity and liver toxicity are equally possible (eg, statin or immune checkpoint autoimmune tissue damage, or after significant trauma).
There are many nuances in the interpretation of even the most common laboratory tests. In this issue of the Journal, Agganis et al discuss liver enzymes (a term slightly more acceptable to me than liver function tests). In future issues, we will address the interpretation of other laboratory tests.

How well do we understand calcium and vitamin D?
With so much emphasis on clinical trials, evidence-based joint decision-making, and comparative-benefit studies when choosing treatment, the growth of the supplement market is a strong comment on the perceived and often real failings of traditional therapies. It also reflects our apparent failure as a profession to educate ourselves and the public about the difference between anecdote-based belief and clinical trial-based confidence, the difference between evidence and innuendo, and, equally important, the limitations of applying population-based clinical trial data to an individual patient.
Which brings me to the discussion of calcium and vitamin D supplementation by Drs. Kilim and Rosen in this issue of the Journal. We know a lot about calcium homeostasis and the role vitamin D plays in regulating circulating calcium levels. Only a small fraction of the calcium in the body circulates (most is in our skeleton), and likely only about 1% is truly exchangeable. But the circulating free calcium level is tightly controlled, as the function of our neuromuscular system, brain, and heart depend on keeping intra- and extracellular calcium levels within precise limits. If necessary, our bodies maintain stable levels of circulating free calcium at the expense of leaching calcium from our bones, placing us at risk of potentially fatal fractures. Thus was born the concept of guaranteeing adequate calcium stores through calcium supplementation.
Control of the free calcium level is not simple. There are several interrelated sensing and modulatory pathways, eg:
- Gut absorption, which is affected by the total gut load of calcium, intestinal integrity, and the specific ingested foodstuffs, and likely by our microbiome
- The parathyroid hormone (PTH) level, which directly or indirectly affects calcium absorption, the calcium-phosphate ratio, and thus, extraskeletal calcium localization and bone calcium content
- Vitamin D, with its many effects after interorgan multistep activation.
Despite this knowledge of calcium metabolism and the intricate cross-talk between the different pathways, I do not believe we truly understand how to determine the amount of dietary and supplemental calcium or vitamin D that is ideal for a given patient. I also do not believe we know with certainty what is the “normal” or ideal 25-hydroxyvitamin D level in that same patient: note the different ranges of normal proposed by different expert working groups.
How do we know when there is insufficient calcium in our diet? The total or free circulating calcium level is far too insensitive and, as noted above, complex homeostatic mechanisms are always trying to maintain an appropriate physiologic calcium level, whatever the intake. Urinary calcium excretion does not necessarily equal the ingested calcium load; there are too many factors influencing renal calcium excretion. Gastrointestinal excretion is also variable and complex. We know when the vitamin D level is functionally much too low, as the PTH level begins to rise; but the “normal” PTH range is wide, and the slope of the relationship between vitamin D and PTH is affected by many factors.
Additionally, accumulating information suggests that vitamin D metabolites significantly affect immune regulation and the onset and expression of a number of organ-specific and systemic autoimmune disorders. Further, the ideal 25-hydroxyvitamin D level for a healthy immune system is not known. Does the body have to compromise something in reconciling the target for a healthy skeleton and the target for a healthy immune system, which may conceivably be different? But importantly, this new knowledge does not imply that vitamin D supplementation will reduce the pain and symptoms from inflammatory arthritis or noninflammatory fibromyalgia.
Despite these many areas of uncertainty, Kilim and Rosen focus on bone health, summarize the wealth of accumulated data, and provide practical management advice we can use in the clinic. But if we struggle so much to know the correct way to manage calcium and vitamin D supplementation in our patients, it is little surprise that most of us struggle even more when asked about using supplements for which the biology is far less understood. As a medical community, we need to uniformly address this lack of understanding wherever it exists. Believing in a supplement or a treatment is not the same as understanding it or having strong evidence about its efficacy and safety.
With so much emphasis on clinical trials, evidence-based joint decision-making, and comparative-benefit studies when choosing treatment, the growth of the supplement market is a strong comment on the perceived and often real failings of traditional therapies. It also reflects our apparent failure as a profession to educate ourselves and the public about the difference between anecdote-based belief and clinical trial-based confidence, the difference between evidence and innuendo, and, equally important, the limitations of applying population-based clinical trial data to an individual patient.
Which brings me to the discussion of calcium and vitamin D supplementation by Drs. Kilim and Rosen in this issue of the Journal. We know a lot about calcium homeostasis and the role vitamin D plays in regulating circulating calcium levels. Only a small fraction of the calcium in the body circulates (most is in our skeleton), and likely only about 1% is truly exchangeable. But the circulating free calcium level is tightly controlled, as the function of our neuromuscular system, brain, and heart depend on keeping intra- and extracellular calcium levels within precise limits. If necessary, our bodies maintain stable levels of circulating free calcium at the expense of leaching calcium from our bones, placing us at risk of potentially fatal fractures. Thus was born the concept of guaranteeing adequate calcium stores through calcium supplementation.
Control of the free calcium level is not simple. There are several interrelated sensing and modulatory pathways, eg:
- Gut absorption, which is affected by the total gut load of calcium, intestinal integrity, and the specific ingested foodstuffs, and likely by our microbiome
- The parathyroid hormone (PTH) level, which directly or indirectly affects calcium absorption, the calcium-phosphate ratio, and thus, extraskeletal calcium localization and bone calcium content
- Vitamin D, with its many effects after interorgan multistep activation.
Despite this knowledge of calcium metabolism and the intricate cross-talk between the different pathways, I do not believe we truly understand how to determine the amount of dietary and supplemental calcium or vitamin D that is ideal for a given patient. I also do not believe we know with certainty what is the “normal” or ideal 25-hydroxyvitamin D level in that same patient: note the different ranges of normal proposed by different expert working groups.
How do we know when there is insufficient calcium in our diet? The total or free circulating calcium level is far too insensitive and, as noted above, complex homeostatic mechanisms are always trying to maintain an appropriate physiologic calcium level, whatever the intake. Urinary calcium excretion does not necessarily equal the ingested calcium load; there are too many factors influencing renal calcium excretion. Gastrointestinal excretion is also variable and complex. We know when the vitamin D level is functionally much too low, as the PTH level begins to rise; but the “normal” PTH range is wide, and the slope of the relationship between vitamin D and PTH is affected by many factors.
Additionally, accumulating information suggests that vitamin D metabolites significantly affect immune regulation and the onset and expression of a number of organ-specific and systemic autoimmune disorders. Further, the ideal 25-hydroxyvitamin D level for a healthy immune system is not known. Does the body have to compromise something in reconciling the target for a healthy skeleton and the target for a healthy immune system, which may conceivably be different? But importantly, this new knowledge does not imply that vitamin D supplementation will reduce the pain and symptoms from inflammatory arthritis or noninflammatory fibromyalgia.
Despite these many areas of uncertainty, Kilim and Rosen focus on bone health, summarize the wealth of accumulated data, and provide practical management advice we can use in the clinic. But if we struggle so much to know the correct way to manage calcium and vitamin D supplementation in our patients, it is little surprise that most of us struggle even more when asked about using supplements for which the biology is far less understood. As a medical community, we need to uniformly address this lack of understanding wherever it exists. Believing in a supplement or a treatment is not the same as understanding it or having strong evidence about its efficacy and safety.
With so much emphasis on clinical trials, evidence-based joint decision-making, and comparative-benefit studies when choosing treatment, the growth of the supplement market is a strong comment on the perceived and often real failings of traditional therapies. It also reflects our apparent failure as a profession to educate ourselves and the public about the difference between anecdote-based belief and clinical trial-based confidence, the difference between evidence and innuendo, and, equally important, the limitations of applying population-based clinical trial data to an individual patient.
Which brings me to the discussion of calcium and vitamin D supplementation by Drs. Kilim and Rosen in this issue of the Journal. We know a lot about calcium homeostasis and the role vitamin D plays in regulating circulating calcium levels. Only a small fraction of the calcium in the body circulates (most is in our skeleton), and likely only about 1% is truly exchangeable. But the circulating free calcium level is tightly controlled, as the function of our neuromuscular system, brain, and heart depend on keeping intra- and extracellular calcium levels within precise limits. If necessary, our bodies maintain stable levels of circulating free calcium at the expense of leaching calcium from our bones, placing us at risk of potentially fatal fractures. Thus was born the concept of guaranteeing adequate calcium stores through calcium supplementation.
Control of the free calcium level is not simple. There are several interrelated sensing and modulatory pathways, eg:
- Gut absorption, which is affected by the total gut load of calcium, intestinal integrity, and the specific ingested foodstuffs, and likely by our microbiome
- The parathyroid hormone (PTH) level, which directly or indirectly affects calcium absorption, the calcium-phosphate ratio, and thus, extraskeletal calcium localization and bone calcium content
- Vitamin D, with its many effects after interorgan multistep activation.
Despite this knowledge of calcium metabolism and the intricate cross-talk between the different pathways, I do not believe we truly understand how to determine the amount of dietary and supplemental calcium or vitamin D that is ideal for a given patient. I also do not believe we know with certainty what is the “normal” or ideal 25-hydroxyvitamin D level in that same patient: note the different ranges of normal proposed by different expert working groups.
How do we know when there is insufficient calcium in our diet? The total or free circulating calcium level is far too insensitive and, as noted above, complex homeostatic mechanisms are always trying to maintain an appropriate physiologic calcium level, whatever the intake. Urinary calcium excretion does not necessarily equal the ingested calcium load; there are too many factors influencing renal calcium excretion. Gastrointestinal excretion is also variable and complex. We know when the vitamin D level is functionally much too low, as the PTH level begins to rise; but the “normal” PTH range is wide, and the slope of the relationship between vitamin D and PTH is affected by many factors.
Additionally, accumulating information suggests that vitamin D metabolites significantly affect immune regulation and the onset and expression of a number of organ-specific and systemic autoimmune disorders. Further, the ideal 25-hydroxyvitamin D level for a healthy immune system is not known. Does the body have to compromise something in reconciling the target for a healthy skeleton and the target for a healthy immune system, which may conceivably be different? But importantly, this new knowledge does not imply that vitamin D supplementation will reduce the pain and symptoms from inflammatory arthritis or noninflammatory fibromyalgia.
Despite these many areas of uncertainty, Kilim and Rosen focus on bone health, summarize the wealth of accumulated data, and provide practical management advice we can use in the clinic. But if we struggle so much to know the correct way to manage calcium and vitamin D supplementation in our patients, it is little surprise that most of us struggle even more when asked about using supplements for which the biology is far less understood. As a medical community, we need to uniformly address this lack of understanding wherever it exists. Believing in a supplement or a treatment is not the same as understanding it or having strong evidence about its efficacy and safety.

Training physician leaders to save the health system…and us
Lerman and Jameson1 recently called for expanding formal leadership development of physicians and increasing the reach of effective physician leaders. From the perspective of physicians, these are certainly good goals. We should have leaders in white coats who understand the joy of practicing medicine and can rally for our missions, challenges, and passions as doctors, clinical educators, and researchers.
But the rationale for most of their argument seems to stem from the perceived need to train physician leaders to promote the survival of increasingly complex health systems and strategically guide individual institutions. This seems also to be the focus of many health systems as they search for and hire physicians for various system leadership positions. But only in the final sentence of their thoughtful essay do Lerman and Jameson mention “patient and physician satisfaction and improved clinical outcomes,” and then only as a byproduct of achieving “organizational efficiency.”1
The financial success (ie, survival) of a health system and the emotional well-being and clinical skills of its physicians are clearly interrelated and cannot be completely dichotomized, and I am not implying that Lerman and Jameson have done so. But trying to promote both can create potential for mission malalignment. Achieving an appropriate balance between these two goals, which at a tactical level are not always congruent, is critical. Training leaders in the skills to achieve alignment with the organization’s culture and priorities may make it easier to administer and guide the course of a medical system with apparent physician leadership, but it still may not well represent the human constituents. The recent trend for health systems to establish new committees charged with improving physician wellness is a statement of problem recognition, but whether their existence can accomplish “mission alignment” remains to be seen.
Swensen et al2 from the Mayo Clinic have highlighted the need to focus on the needs of the physician, and not primarily on those of the institution, to reduce the epidemic of physician burnout. A physician leader who is wonderfully trained in organizational psychology, communication skills, and performance metrics analysis, but who lacks a deep and authentic understanding of the joys of practicing medicine and delivering care to the sick, of the need to stoke the individual physician’s intellectual curiosity and provide time for reflection to clinicians working in their institutions, is a unidirectional institutional leader, and thus a leader in name alone. We need to be careful about grooming young physician leaders at too early a stage. Having the book knowledge of a physician is not the same as having the heart of one.
We have appropriately morphed away from the academic curriculum vitae as the only ticket to titled organizational leadership. But we need to look closely at our choice of physician leaders and be careful that the physician component of that terminology is not defined by degree alone. Just as the number of first-authored publications and surgical procedures performed should not suffice for selection to leadership positions, nor should participation in leadership courses or the ability to respond as desired in a behavioral interview be viewed as adequate for selection as a leader of physicians. Administrators with a tailored MBA degree, extensive and real job experience in a clinical center, and participation in a seminar on physician behavior can also be successful supervisors and (maybe) leaders in a medical center.
But if we really care about our clinical staff, organizational leadership must include physicians in positions of influence who are true advocates for clinicians and patients, and not primarily caretakers of the health system and enforcers of performance metrics. Although (some of) the latter are absolutely necessary, we need more than that from our physician leaders. We need them to reflect and support our perspective as well as the institution’s needs. We need them to understand and defend the need to maintain the joy of practicing medicine. How can we as physicians really take care of others if there is no one to take care of us?3
Note: this discussion is not new. We had a dialogue on this topic in the Journal almost 10 years ago!4
- Lerman C, Jameson JL. Leadership development in medicine. N Engl J Med 2018; 378(20):1862–1863. doi:10.1056/NEJMp1801610
- Swensen S, Kabcenell A, Shanafelt T. Physician-organization collaboration reduces physician burnout and promotes engagement: the Mayo Clinic experience. J Healthcare Manag 2016; 61(2):105–127. pmid:27111930
- Shem S. The House of God: A Novel. New York: R. Marek Publishers, 1978.
- Longworth D. A medical center is not a hospital: reflections of a department chair still in the game. Cleve Clin J Med 2008; 75(12):832–834. doi:10.3949/ccjm.75a.08094
Lerman and Jameson1 recently called for expanding formal leadership development of physicians and increasing the reach of effective physician leaders. From the perspective of physicians, these are certainly good goals. We should have leaders in white coats who understand the joy of practicing medicine and can rally for our missions, challenges, and passions as doctors, clinical educators, and researchers.
But the rationale for most of their argument seems to stem from the perceived need to train physician leaders to promote the survival of increasingly complex health systems and strategically guide individual institutions. This seems also to be the focus of many health systems as they search for and hire physicians for various system leadership positions. But only in the final sentence of their thoughtful essay do Lerman and Jameson mention “patient and physician satisfaction and improved clinical outcomes,” and then only as a byproduct of achieving “organizational efficiency.”1
The financial success (ie, survival) of a health system and the emotional well-being and clinical skills of its physicians are clearly interrelated and cannot be completely dichotomized, and I am not implying that Lerman and Jameson have done so. But trying to promote both can create potential for mission malalignment. Achieving an appropriate balance between these two goals, which at a tactical level are not always congruent, is critical. Training leaders in the skills to achieve alignment with the organization’s culture and priorities may make it easier to administer and guide the course of a medical system with apparent physician leadership, but it still may not well represent the human constituents. The recent trend for health systems to establish new committees charged with improving physician wellness is a statement of problem recognition, but whether their existence can accomplish “mission alignment” remains to be seen.
Swensen et al2 from the Mayo Clinic have highlighted the need to focus on the needs of the physician, and not primarily on those of the institution, to reduce the epidemic of physician burnout. A physician leader who is wonderfully trained in organizational psychology, communication skills, and performance metrics analysis, but who lacks a deep and authentic understanding of the joys of practicing medicine and delivering care to the sick, of the need to stoke the individual physician’s intellectual curiosity and provide time for reflection to clinicians working in their institutions, is a unidirectional institutional leader, and thus a leader in name alone. We need to be careful about grooming young physician leaders at too early a stage. Having the book knowledge of a physician is not the same as having the heart of one.
We have appropriately morphed away from the academic curriculum vitae as the only ticket to titled organizational leadership. But we need to look closely at our choice of physician leaders and be careful that the physician component of that terminology is not defined by degree alone. Just as the number of first-authored publications and surgical procedures performed should not suffice for selection to leadership positions, nor should participation in leadership courses or the ability to respond as desired in a behavioral interview be viewed as adequate for selection as a leader of physicians. Administrators with a tailored MBA degree, extensive and real job experience in a clinical center, and participation in a seminar on physician behavior can also be successful supervisors and (maybe) leaders in a medical center.
But if we really care about our clinical staff, organizational leadership must include physicians in positions of influence who are true advocates for clinicians and patients, and not primarily caretakers of the health system and enforcers of performance metrics. Although (some of) the latter are absolutely necessary, we need more than that from our physician leaders. We need them to reflect and support our perspective as well as the institution’s needs. We need them to understand and defend the need to maintain the joy of practicing medicine. How can we as physicians really take care of others if there is no one to take care of us?3
Note: this discussion is not new. We had a dialogue on this topic in the Journal almost 10 years ago!4
Lerman and Jameson1 recently called for expanding formal leadership development of physicians and increasing the reach of effective physician leaders. From the perspective of physicians, these are certainly good goals. We should have leaders in white coats who understand the joy of practicing medicine and can rally for our missions, challenges, and passions as doctors, clinical educators, and researchers.
But the rationale for most of their argument seems to stem from the perceived need to train physician leaders to promote the survival of increasingly complex health systems and strategically guide individual institutions. This seems also to be the focus of many health systems as they search for and hire physicians for various system leadership positions. But only in the final sentence of their thoughtful essay do Lerman and Jameson mention “patient and physician satisfaction and improved clinical outcomes,” and then only as a byproduct of achieving “organizational efficiency.”1
The financial success (ie, survival) of a health system and the emotional well-being and clinical skills of its physicians are clearly interrelated and cannot be completely dichotomized, and I am not implying that Lerman and Jameson have done so. But trying to promote both can create potential for mission malalignment. Achieving an appropriate balance between these two goals, which at a tactical level are not always congruent, is critical. Training leaders in the skills to achieve alignment with the organization’s culture and priorities may make it easier to administer and guide the course of a medical system with apparent physician leadership, but it still may not well represent the human constituents. The recent trend for health systems to establish new committees charged with improving physician wellness is a statement of problem recognition, but whether their existence can accomplish “mission alignment” remains to be seen.
Swensen et al2 from the Mayo Clinic have highlighted the need to focus on the needs of the physician, and not primarily on those of the institution, to reduce the epidemic of physician burnout. A physician leader who is wonderfully trained in organizational psychology, communication skills, and performance metrics analysis, but who lacks a deep and authentic understanding of the joys of practicing medicine and delivering care to the sick, of the need to stoke the individual physician’s intellectual curiosity and provide time for reflection to clinicians working in their institutions, is a unidirectional institutional leader, and thus a leader in name alone. We need to be careful about grooming young physician leaders at too early a stage. Having the book knowledge of a physician is not the same as having the heart of one.
We have appropriately morphed away from the academic curriculum vitae as the only ticket to titled organizational leadership. But we need to look closely at our choice of physician leaders and be careful that the physician component of that terminology is not defined by degree alone. Just as the number of first-authored publications and surgical procedures performed should not suffice for selection to leadership positions, nor should participation in leadership courses or the ability to respond as desired in a behavioral interview be viewed as adequate for selection as a leader of physicians. Administrators with a tailored MBA degree, extensive and real job experience in a clinical center, and participation in a seminar on physician behavior can also be successful supervisors and (maybe) leaders in a medical center.
But if we really care about our clinical staff, organizational leadership must include physicians in positions of influence who are true advocates for clinicians and patients, and not primarily caretakers of the health system and enforcers of performance metrics. Although (some of) the latter are absolutely necessary, we need more than that from our physician leaders. We need them to reflect and support our perspective as well as the institution’s needs. We need them to understand and defend the need to maintain the joy of practicing medicine. How can we as physicians really take care of others if there is no one to take care of us?3
Note: this discussion is not new. We had a dialogue on this topic in the Journal almost 10 years ago!4
- Lerman C, Jameson JL. Leadership development in medicine. N Engl J Med 2018; 378(20):1862–1863. doi:10.1056/NEJMp1801610
- Swensen S, Kabcenell A, Shanafelt T. Physician-organization collaboration reduces physician burnout and promotes engagement: the Mayo Clinic experience. J Healthcare Manag 2016; 61(2):105–127. pmid:27111930
- Shem S. The House of God: A Novel. New York: R. Marek Publishers, 1978.
- Longworth D. A medical center is not a hospital: reflections of a department chair still in the game. Cleve Clin J Med 2008; 75(12):832–834. doi:10.3949/ccjm.75a.08094
- Lerman C, Jameson JL. Leadership development in medicine. N Engl J Med 2018; 378(20):1862–1863. doi:10.1056/NEJMp1801610
- Swensen S, Kabcenell A, Shanafelt T. Physician-organization collaboration reduces physician burnout and promotes engagement: the Mayo Clinic experience. J Healthcare Manag 2016; 61(2):105–127. pmid:27111930
- Shem S. The House of God: A Novel. New York: R. Marek Publishers, 1978.
- Longworth D. A medical center is not a hospital: reflections of a department chair still in the game. Cleve Clin J Med 2008; 75(12):832–834. doi:10.3949/ccjm.75a.08094

The algorithm less traveled
To this day I remain impressed by the algorithmic nature of trauma management. A routine that to the internist could appear mindless and slavish was to the trauma physician a protocol designed to take no chances on missing a life-threatening complication in the heat of the moment. The trauma physician cannot afford to wait for a cognitively derived epiphany in a clinical setting that often rapidly unfolds as a series of “never-miss” scenarios. The appropriate algorithm, rigorously followed, offers the best chance of avoiding a catastrophe of omission. This was long before Atul Gawande published his Checklist Manifesto.
Reviewing the article by Sussman et al, “Eyes of the mimicker,” in this issue of the Journal got me thinking about the power of algorithmic thinking and practice in internal medicine, how the patient they describe specifically relates to my practice experiences over the years, and how important the context of where we practice and who we treat informs (and can misinform) our clinical reasoning. When I was a medical student at Bellevue Hospital in New York City (in the pre-HIV era), the rapid plasma reagin (RPR) was a routine blood test, as syphilis routinely earned its moniker as the “great imitator.” When I did my residency at the Hospital of the University of Pennsylvania, my ingrained habit of ordering this test was extinguished, along with my also previously learned habit of obtaining blood cultures in all patients who presented with new heart failure that was not explained by the electrocardiogram. These habits disappeared not because of arguments steeped in evidence-based medicine or an emphasis on Bayesian test-ordering, but because in Philadelphia at that time we were not seeing patients with occult syphilis and endocarditis with the same frequency as at Bellevue. Context can and should play a role in our diagnostic reasoning.
But I still remember the patient I saw in the Philadelphia emergency room, a second visit for a man in his 20s with a diffuse, mostly macular rash on his trunk, palms, and soles (visible when the light was turned up in his darkened room, as he felt uncomfortable with bright light), diffuse adenopathy, and enlarged doughy and minimally tender wrists and finger (metacarpophalangeal) joints. I recall wondering why no one had thought to obtain an RPR test on him the first time he had presented to the emergency room; if he had been at Bellevue, the test results would already have returned.
Without appropriate algorithms, things get missed. But using algorithms indiscriminately is cost-ineffective and can lead to cascades of inappropriate tests and interventions. Striking the appropriate balance is part of what comprises the writing of useful clinical care paths.
As I read the article by Sussman et al I wondered who first looked at the patient’s retinas and what initially prompted the testing that was ordered. The presentation was not typical of ocular syphilis, and I would guess that an ophthalmologist or infectious disease consultant evaluating the blurred vision observed the retinal findings, suspected the diagnosis, and ordered serologies, as well as other studies searching for infections and systemic autoimmune disorders that can also cause Roth spots. Gone are the days when internists (and residents) routinely examine the eyes as part of a full physical examination. I am certain an evidence-based study of this practice would find it time-ineffective and with inappropriately low sensitivity.
I don’t think the retinal examination will return to the internist’s checklist. Yet that is where the algorithm that led to this patient’s diagnosis likely began. One can “google” the causes of Roth spots, but as yet there is no app for demonstrating that they are present.
To this day I remain impressed by the algorithmic nature of trauma management. A routine that to the internist could appear mindless and slavish was to the trauma physician a protocol designed to take no chances on missing a life-threatening complication in the heat of the moment. The trauma physician cannot afford to wait for a cognitively derived epiphany in a clinical setting that often rapidly unfolds as a series of “never-miss” scenarios. The appropriate algorithm, rigorously followed, offers the best chance of avoiding a catastrophe of omission. This was long before Atul Gawande published his Checklist Manifesto.
Reviewing the article by Sussman et al, “Eyes of the mimicker,” in this issue of the Journal got me thinking about the power of algorithmic thinking and practice in internal medicine, how the patient they describe specifically relates to my practice experiences over the years, and how important the context of where we practice and who we treat informs (and can misinform) our clinical reasoning. When I was a medical student at Bellevue Hospital in New York City (in the pre-HIV era), the rapid plasma reagin (RPR) was a routine blood test, as syphilis routinely earned its moniker as the “great imitator.” When I did my residency at the Hospital of the University of Pennsylvania, my ingrained habit of ordering this test was extinguished, along with my also previously learned habit of obtaining blood cultures in all patients who presented with new heart failure that was not explained by the electrocardiogram. These habits disappeared not because of arguments steeped in evidence-based medicine or an emphasis on Bayesian test-ordering, but because in Philadelphia at that time we were not seeing patients with occult syphilis and endocarditis with the same frequency as at Bellevue. Context can and should play a role in our diagnostic reasoning.
But I still remember the patient I saw in the Philadelphia emergency room, a second visit for a man in his 20s with a diffuse, mostly macular rash on his trunk, palms, and soles (visible when the light was turned up in his darkened room, as he felt uncomfortable with bright light), diffuse adenopathy, and enlarged doughy and minimally tender wrists and finger (metacarpophalangeal) joints. I recall wondering why no one had thought to obtain an RPR test on him the first time he had presented to the emergency room; if he had been at Bellevue, the test results would already have returned.
Without appropriate algorithms, things get missed. But using algorithms indiscriminately is cost-ineffective and can lead to cascades of inappropriate tests and interventions. Striking the appropriate balance is part of what comprises the writing of useful clinical care paths.
As I read the article by Sussman et al I wondered who first looked at the patient’s retinas and what initially prompted the testing that was ordered. The presentation was not typical of ocular syphilis, and I would guess that an ophthalmologist or infectious disease consultant evaluating the blurred vision observed the retinal findings, suspected the diagnosis, and ordered serologies, as well as other studies searching for infections and systemic autoimmune disorders that can also cause Roth spots. Gone are the days when internists (and residents) routinely examine the eyes as part of a full physical examination. I am certain an evidence-based study of this practice would find it time-ineffective and with inappropriately low sensitivity.
I don’t think the retinal examination will return to the internist’s checklist. Yet that is where the algorithm that led to this patient’s diagnosis likely began. One can “google” the causes of Roth spots, but as yet there is no app for demonstrating that they are present.
Quite a while ago, when I used to moonlight as the medicine attending in a university medical center emergency department, I took a course, passed an exam, and became certified in advanced trauma life support. I guess that I am one of few board-certified rheumatologists to hold such certification, as there is little apparent clinical crossover between the management of patients with lupus or vasculitis and those with life-threatening trauma.
To this day I remain impressed by the algorithmic nature of trauma management. A routine that to the internist could appear mindless and slavish was to the trauma physician a protocol designed to take no chances on missing a life-threatening complication in the heat of the moment. The trauma physician cannot afford to wait for a cognitively derived epiphany in a clinical setting that often rapidly unfolds as a series of “never-miss” scenarios. The appropriate algorithm, rigorously followed, offers the best chance of avoiding a catastrophe of omission. This was long before Atul Gawande published his Checklist Manifesto.
Reviewing the article by Sussman et al, “Eyes of the mimicker,” in this issue of the Journal got me thinking about the power of algorithmic thinking and practice in internal medicine, how the patient they describe specifically relates to my practice experiences over the years, and how important the context of where we practice and who we treat informs (and can misinform) our clinical reasoning. When I was a medical student at Bellevue Hospital in New York City (in the pre-HIV era), the rapid plasma reagin (RPR) was a routine blood test, as syphilis routinely earned its moniker as the “great imitator.” When I did my residency at the Hospital of the University of Pennsylvania, my ingrained habit of ordering this test was extinguished, along with my also previously learned habit of obtaining blood cultures in all patients who presented with new heart failure that was not explained by the electrocardiogram. These habits disappeared not because of arguments steeped in evidence-based medicine or an emphasis on Bayesian test-ordering, but because in Philadelphia at that time we were not seeing patients with occult syphilis and endocarditis with the same frequency as at Bellevue. Context can and should play a role in our diagnostic reasoning.
But I still remember the patient I saw in the Philadelphia emergency room, a second visit for a man in his 20s with a diffuse, mostly macular rash on his trunk, palms, and soles (visible when the light was turned up in his darkened room, as he felt uncomfortable with bright light), diffuse adenopathy, and enlarged doughy and minimally tender wrists and finger (metacarpophalangeal) joints. I recall wondering why no one had thought to obtain an RPR test on him the first time he had presented to the emergency room; if he had been at Bellevue, the test results would already have returned.
Without appropriate algorithms, things get missed. But using algorithms indiscriminately is cost-ineffective and can lead to cascades of inappropriate tests and interventions. Striking the appropriate balance is part of what comprises the writing of useful clinical care paths.
As I read the article by Sussman et al I wondered who first looked at the patient’s retinas and what initially prompted the testing that was ordered. The presentation was not typical of ocular syphilis, and I would guess that an ophthalmologist or infectious disease consultant evaluating the blurred vision observed the retinal findings, suspected the diagnosis, and ordered serologies, as well as other studies searching for infections and systemic autoimmune disorders that can also cause Roth spots. Gone are the days when internists (and residents) routinely examine the eyes as part of a full physical examination. I am certain an evidence-based study of this practice would find it time-ineffective and with inappropriately low sensitivity.
I don’t think the retinal examination will return to the internist’s checklist. Yet that is where the algorithm that led to this patient’s diagnosis likely began. One can “google” the causes of Roth spots, but as yet there is no app for demonstrating that they are present.
