User login
Masterclass: First-episode psychosis with Dr. Henry A. Nasrallah
from the Psychopharmacology Update meeting in Cincinnati. Dr. Nasrallah is editor in chief of Current Psychiatry and is the Sydney W. Souers Endowed Chair and professor and chairman of the department of neurology and psychiatry and behavioral neuroscience at Saint Louis University.
If you would like to respond to any of Dr. Nasrallah’s comments in this Masterclass, email us at podcasts@mdedge.com.
Amazon
Apple Podcasts
Google Podcasts
Spotify
from the Psychopharmacology Update meeting in Cincinnati. Dr. Nasrallah is editor in chief of Current Psychiatry and is the Sydney W. Souers Endowed Chair and professor and chairman of the department of neurology and psychiatry and behavioral neuroscience at Saint Louis University.
If you would like to respond to any of Dr. Nasrallah’s comments in this Masterclass, email us at podcasts@mdedge.com.
Amazon
Apple Podcasts
Google Podcasts
Spotify
from the Psychopharmacology Update meeting in Cincinnati. Dr. Nasrallah is editor in chief of Current Psychiatry and is the Sydney W. Souers Endowed Chair and professor and chairman of the department of neurology and psychiatry and behavioral neuroscience at Saint Louis University.
If you would like to respond to any of Dr. Nasrallah’s comments in this Masterclass, email us at podcasts@mdedge.com.
Amazon
Apple Podcasts
Google Podcasts
Spotify
Antipsychotic use in young people tied to 80% increased risk of death
Children and young people who received antipsychotic doses higher than 50-mg chlorpromazine equivalents had an 80% increased risk of death at follow-up, compared with a control group, according to a study of young Medicaid enrollees who recently had begun medication.
“The study findings seem to reinforce existing guidelines for improving the outcomes of antipsychotic therapy in children and youths,” wrote lead author Wayne A. Ray, PhD, of the department of health policy at the Vanderbilt University in Nashville, Tenn., and his coauthors. Those guidelines include using “psychosocial interventions when possible, cardiometabolic assessment before treatment and monitoring after treatment, and limiting therapy to the lowest dose and shortest duration possible,” they wrote.
The study, published online in JAMA Psychiatry, analyzed children and young adults from Tennessee, aged 5-24 years, who were new medication users, and had been enrolled in Medicaid between 1999 and 2014.
They were split into three groups: a control group (189,361) with users primarily taking attention-deficit/hyperactivity disorder medications and antidepressants; a group (28,377) with users who received antipsychotic doses of 50 mg or less chlorpromazine equivalents; and a group (30,120) with users who received doses higher than 50-mg chlorpromazine equivalents.
At follow-up, the incidence of death in the higher-dose group was 146.2 per 100,000 person-years (95% confidence interval, 107.3-199.4 per 100,000 person-years), compared with 49.5 in the lower-dose group (95% CI, 24.8-99.0) and 54.5 in the control group (95% CI, 42.9-69.2). This difference was attributed to unexpected deaths, which accounted for 52.5% of deaths in the higher-dose group. No increased risk of death was noted for injuries or suicides. “The elevated risk persisted for unexpected deaths not due to overdose, with a 4.3-fold increased risk of death from cardiovascular or metabolic causes,” Dr. Ray and his coauthors wrote.
The authors shared potential limitations of their study, including a relatively small number of deaths during follow-up and subsequent statistical adjustment during analysis. They also recognized that their data did not factor in important characteristics such as body mass index and family history, and that a “single-state Medicaid cohort may limit the study’s generalizability.”
Nonetheless, they emphasized Medicaid’s relevance as coverage provider for an estimated 39% of U.S. children, along with noting that
“Further studies are needed that compare antipsychotic users and controls within more narrow comorbidity ranges or in analyses that include richer clinical data,” they wrote.
The study was supported by grants from the National Heart, Lung, and Blood Institute, and the National Institute for Child Health and Human Development. No conflicts of interest were reported.
SOURCE: Ray WA et al. JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3421.
This study by Wayne A. Ray, PhD, and his colleagues addresses the risks of antipsychotic use in childhood while highlighting the contradictions in how psychiatrically ill children are treated and medicated, according to Barbara Geller, MD, of the department of psychiatry at Washington University in St. Louis.
Before commenting on the study itself, Dr. Geller noted that child psychiatry is not a subspecialty that deals with “little patients and little problems,” despite that lingering perception among some. “Fifty percent of psychiatry disorders begin by age 14 years,” she wrote, “and childhood age at onset is a risk factor for a more severe longitudinal course in mood and other disorders.”
In addition, though it seems instinctually that antipsychotic medications would have lesser side effects on healthy children, that is not always the case. “The opposite is true for certain metabolic and endocrine effects,” she explained, “such as relatively greater weight gain and prolactin level elevation than adults and the onset of type 2 diabetes within the first year of treatment.”
When it came to the study, Dr. Geller posed questions about the findings, including whether an increase in unexpected deaths among the higher-dose group could be attributed to suicide. She also recommended that future investigations “examine outcomes within child, adolescent, and young adult age subgroups, as opposed to combining all youth 6 to 24 years old.”
That said, this research does probe depths that require continued exploration. “Results in the study by Ray et al. heighten the already increased caution about prescribing antipsychotics to children and adolescents,” she wrote, “and emphasize the need to consider situational triggers of psychopathology to avoid medicating the environment.”
These comments are adapted from an accompanying editorial (JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3409). No conflicts of interest were reported.
This study by Wayne A. Ray, PhD, and his colleagues addresses the risks of antipsychotic use in childhood while highlighting the contradictions in how psychiatrically ill children are treated and medicated, according to Barbara Geller, MD, of the department of psychiatry at Washington University in St. Louis.
Before commenting on the study itself, Dr. Geller noted that child psychiatry is not a subspecialty that deals with “little patients and little problems,” despite that lingering perception among some. “Fifty percent of psychiatry disorders begin by age 14 years,” she wrote, “and childhood age at onset is a risk factor for a more severe longitudinal course in mood and other disorders.”
In addition, though it seems instinctually that antipsychotic medications would have lesser side effects on healthy children, that is not always the case. “The opposite is true for certain metabolic and endocrine effects,” she explained, “such as relatively greater weight gain and prolactin level elevation than adults and the onset of type 2 diabetes within the first year of treatment.”
When it came to the study, Dr. Geller posed questions about the findings, including whether an increase in unexpected deaths among the higher-dose group could be attributed to suicide. She also recommended that future investigations “examine outcomes within child, adolescent, and young adult age subgroups, as opposed to combining all youth 6 to 24 years old.”
That said, this research does probe depths that require continued exploration. “Results in the study by Ray et al. heighten the already increased caution about prescribing antipsychotics to children and adolescents,” she wrote, “and emphasize the need to consider situational triggers of psychopathology to avoid medicating the environment.”
These comments are adapted from an accompanying editorial (JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3409). No conflicts of interest were reported.
This study by Wayne A. Ray, PhD, and his colleagues addresses the risks of antipsychotic use in childhood while highlighting the contradictions in how psychiatrically ill children are treated and medicated, according to Barbara Geller, MD, of the department of psychiatry at Washington University in St. Louis.
Before commenting on the study itself, Dr. Geller noted that child psychiatry is not a subspecialty that deals with “little patients and little problems,” despite that lingering perception among some. “Fifty percent of psychiatry disorders begin by age 14 years,” she wrote, “and childhood age at onset is a risk factor for a more severe longitudinal course in mood and other disorders.”
In addition, though it seems instinctually that antipsychotic medications would have lesser side effects on healthy children, that is not always the case. “The opposite is true for certain metabolic and endocrine effects,” she explained, “such as relatively greater weight gain and prolactin level elevation than adults and the onset of type 2 diabetes within the first year of treatment.”
When it came to the study, Dr. Geller posed questions about the findings, including whether an increase in unexpected deaths among the higher-dose group could be attributed to suicide. She also recommended that future investigations “examine outcomes within child, adolescent, and young adult age subgroups, as opposed to combining all youth 6 to 24 years old.”
That said, this research does probe depths that require continued exploration. “Results in the study by Ray et al. heighten the already increased caution about prescribing antipsychotics to children and adolescents,” she wrote, “and emphasize the need to consider situational triggers of psychopathology to avoid medicating the environment.”
These comments are adapted from an accompanying editorial (JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3409). No conflicts of interest were reported.
Children and young people who received antipsychotic doses higher than 50-mg chlorpromazine equivalents had an 80% increased risk of death at follow-up, compared with a control group, according to a study of young Medicaid enrollees who recently had begun medication.
“The study findings seem to reinforce existing guidelines for improving the outcomes of antipsychotic therapy in children and youths,” wrote lead author Wayne A. Ray, PhD, of the department of health policy at the Vanderbilt University in Nashville, Tenn., and his coauthors. Those guidelines include using “psychosocial interventions when possible, cardiometabolic assessment before treatment and monitoring after treatment, and limiting therapy to the lowest dose and shortest duration possible,” they wrote.
The study, published online in JAMA Psychiatry, analyzed children and young adults from Tennessee, aged 5-24 years, who were new medication users, and had been enrolled in Medicaid between 1999 and 2014.
They were split into three groups: a control group (189,361) with users primarily taking attention-deficit/hyperactivity disorder medications and antidepressants; a group (28,377) with users who received antipsychotic doses of 50 mg or less chlorpromazine equivalents; and a group (30,120) with users who received doses higher than 50-mg chlorpromazine equivalents.
At follow-up, the incidence of death in the higher-dose group was 146.2 per 100,000 person-years (95% confidence interval, 107.3-199.4 per 100,000 person-years), compared with 49.5 in the lower-dose group (95% CI, 24.8-99.0) and 54.5 in the control group (95% CI, 42.9-69.2). This difference was attributed to unexpected deaths, which accounted for 52.5% of deaths in the higher-dose group. No increased risk of death was noted for injuries or suicides. “The elevated risk persisted for unexpected deaths not due to overdose, with a 4.3-fold increased risk of death from cardiovascular or metabolic causes,” Dr. Ray and his coauthors wrote.
The authors shared potential limitations of their study, including a relatively small number of deaths during follow-up and subsequent statistical adjustment during analysis. They also recognized that their data did not factor in important characteristics such as body mass index and family history, and that a “single-state Medicaid cohort may limit the study’s generalizability.”
Nonetheless, they emphasized Medicaid’s relevance as coverage provider for an estimated 39% of U.S. children, along with noting that
“Further studies are needed that compare antipsychotic users and controls within more narrow comorbidity ranges or in analyses that include richer clinical data,” they wrote.
The study was supported by grants from the National Heart, Lung, and Blood Institute, and the National Institute for Child Health and Human Development. No conflicts of interest were reported.
SOURCE: Ray WA et al. JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3421.
Children and young people who received antipsychotic doses higher than 50-mg chlorpromazine equivalents had an 80% increased risk of death at follow-up, compared with a control group, according to a study of young Medicaid enrollees who recently had begun medication.
“The study findings seem to reinforce existing guidelines for improving the outcomes of antipsychotic therapy in children and youths,” wrote lead author Wayne A. Ray, PhD, of the department of health policy at the Vanderbilt University in Nashville, Tenn., and his coauthors. Those guidelines include using “psychosocial interventions when possible, cardiometabolic assessment before treatment and monitoring after treatment, and limiting therapy to the lowest dose and shortest duration possible,” they wrote.
The study, published online in JAMA Psychiatry, analyzed children and young adults from Tennessee, aged 5-24 years, who were new medication users, and had been enrolled in Medicaid between 1999 and 2014.
They were split into three groups: a control group (189,361) with users primarily taking attention-deficit/hyperactivity disorder medications and antidepressants; a group (28,377) with users who received antipsychotic doses of 50 mg or less chlorpromazine equivalents; and a group (30,120) with users who received doses higher than 50-mg chlorpromazine equivalents.
At follow-up, the incidence of death in the higher-dose group was 146.2 per 100,000 person-years (95% confidence interval, 107.3-199.4 per 100,000 person-years), compared with 49.5 in the lower-dose group (95% CI, 24.8-99.0) and 54.5 in the control group (95% CI, 42.9-69.2). This difference was attributed to unexpected deaths, which accounted for 52.5% of deaths in the higher-dose group. No increased risk of death was noted for injuries or suicides. “The elevated risk persisted for unexpected deaths not due to overdose, with a 4.3-fold increased risk of death from cardiovascular or metabolic causes,” Dr. Ray and his coauthors wrote.
The authors shared potential limitations of their study, including a relatively small number of deaths during follow-up and subsequent statistical adjustment during analysis. They also recognized that their data did not factor in important characteristics such as body mass index and family history, and that a “single-state Medicaid cohort may limit the study’s generalizability.”
Nonetheless, they emphasized Medicaid’s relevance as coverage provider for an estimated 39% of U.S. children, along with noting that
“Further studies are needed that compare antipsychotic users and controls within more narrow comorbidity ranges or in analyses that include richer clinical data,” they wrote.
The study was supported by grants from the National Heart, Lung, and Blood Institute, and the National Institute for Child Health and Human Development. No conflicts of interest were reported.
SOURCE: Ray WA et al. JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3421.
FROM JAMA PSYCHIATRY
Key clinical point: Children and youths who received higher doses of antipsychotic medication had an 80% increased risk of death, compared with those in a control group.
Major finding: The incidence of unexpected death was 76.8 per 100,000 person-years in the higher-dose group, compared with 17.9 per 100,000 person-years in the control group.
Study details: A retrospective cohort study of Medicaid-enrolled children and young adults from Tennessee, aged 5-24 years, who were new users of antipsychotic or control medications.
Disclosures: The study was supported by grants from the National Heart, Lung, and Blood Institute, and the National Institute for Child Health and Human Development. No conflicts of interest were reported.
Source: Ray WA et al. JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3421.
‘Error neuron’ EEG findings could open up future clinical applications
, and this activity can be tracked through a scalp EEG pattern called error-related negativity, according to findings from experiments carried out during intracranial EEG recordings of candidates for surgical treatment of epilepsy.

“Our results suggest that coordinated neural activity can serve as a substrate for information routing that enables the performance-monitoring system to communicate the need for behavioral control to other brain regions, including those that maintain flexible goal information, such as the lateral prefrontal cortex and the frontal polar cortex,” first author Zhongzheng Fu, a PhD student at the California Institute of Technology in Pasadena, Calif., and Cedars-Sinai Medical Center, Los Angeles, and his colleagues reported in Neuron.
The findings offer insights that could lead to treatments for conditions in which the important executive function task of error self-monitoring is unbalanced, such as obsessive-compulsive disorder and schizophrenia, the authors noted in a press release.
“We discovered that the activity of error neurons correlates with the size of the ERN [error-related negativity],” Mr. Fu said. “This identifies the brain area that causes the ERN and helps explain what it signifies. This new insight might allow doctors to use the ERN as a standard tool to diagnose mental diseases and monitor responses to treatment.”
Error neuron firing and intracranial ERN occurred first in pre-supplementary motor area (pre-SMA), then in the dorsal anterior cingulate cortex (dACC) about 50 ms later, with significant correlations between firing and intracranial ERN in both locations. In dACC, this activity, with error-integrating neuron responses, correlated with magnitude of post-error slowing (PES).
Previous research suggested a link between “the detection of self-generated errors, as reflected in the ERN, with changes in cognitive control, as exhibited behaviorally in PES,” the investigators wrote. “However, several electroencephalogram (EEG) studies have failed to find a significant relationship between PES and ERN.”
The present study involved intracranial EEG of 29 candidates for surgical treatment of epilepsy and scalp EEG of 12 control participants, with each modality measuring activity in the frontal cortex. Both cohorts performed a rapid version of the color-word Stroop task, in which the words “red,” “green,” or “blue” were printed either in corresponding or noncorresponding colors of red, green, or blue. Subjects were presented various color-word combinations while being asked to click one of three buttons indicating the color of the word as quickly as possible. The investigators monitored neuronal activity throughout, discarding responses that were too slow.
As found in previous trials, the subjects demonstrated the “Stroop effect,” which refers to a slower response when word and color are incongruent (224.9 ms difference; P less than .001). As anticipated, correct responses following correct responses were faster than were correct responses following erroneous responses, which defines PES.
In the intracranial EEG group, the investigators isolated 1,171 neurons, of which 618 were located in dACC and 553 in pre-SMA. Using a Poisson regression model and correlations with erroneous responses, the investigators identified 99 “type I” error neurons in dACC and 118 in pre-SMA, based on higher frequency of firing during erroneous responses than during correct responses. At a single-cell level, error neuron mean spike rates were highest when intracranial ERN amplitude was greatest, such that error neuron firing in dACC and pre-SMA had maximal likelihood ratios of 7.9 (P = .01) and 15.1 (P less than .001), respectively. The strength of correlation between intracranial ERN and error neuron firing rate was directly related to PES magnitude exclusively in the dACC (maximum likelihood ratio of 13.9; P = .015). In post-error trials, faster error-integrating neuron firing rates in dACC predicted greater PES (maximal likelihood ratio of 18.3; P less than .001).
The study was funded by the National Institutes of Health, the McKnight Endowment for Neuroscience, and the National Science Foundation. The authors declared no conflicts of interest.
SOURCE: Fu Z et al. Neuron. 2018 Dec 4. doi: 10.1016/j.neuron.2018.11.016
, and this activity can be tracked through a scalp EEG pattern called error-related negativity, according to findings from experiments carried out during intracranial EEG recordings of candidates for surgical treatment of epilepsy.
“Our results suggest that coordinated neural activity can serve as a substrate for information routing that enables the performance-monitoring system to communicate the need for behavioral control to other brain regions, including those that maintain flexible goal information, such as the lateral prefrontal cortex and the frontal polar cortex,” first author Zhongzheng Fu, a PhD student at the California Institute of Technology in Pasadena, Calif., and Cedars-Sinai Medical Center, Los Angeles, and his colleagues reported in Neuron.
The findings offer insights that could lead to treatments for conditions in which the important executive function task of error self-monitoring is unbalanced, such as obsessive-compulsive disorder and schizophrenia, the authors noted in a press release.
“We discovered that the activity of error neurons correlates with the size of the ERN [error-related negativity],” Mr. Fu said. “This identifies the brain area that causes the ERN and helps explain what it signifies. This new insight might allow doctors to use the ERN as a standard tool to diagnose mental diseases and monitor responses to treatment.”
Error neuron firing and intracranial ERN occurred first in pre-supplementary motor area (pre-SMA), then in the dorsal anterior cingulate cortex (dACC) about 50 ms later, with significant correlations between firing and intracranial ERN in both locations. In dACC, this activity, with error-integrating neuron responses, correlated with magnitude of post-error slowing (PES).
Previous research suggested a link between “the detection of self-generated errors, as reflected in the ERN, with changes in cognitive control, as exhibited behaviorally in PES,” the investigators wrote. “However, several electroencephalogram (EEG) studies have failed to find a significant relationship between PES and ERN.”
The present study involved intracranial EEG of 29 candidates for surgical treatment of epilepsy and scalp EEG of 12 control participants, with each modality measuring activity in the frontal cortex. Both cohorts performed a rapid version of the color-word Stroop task, in which the words “red,” “green,” or “blue” were printed either in corresponding or noncorresponding colors of red, green, or blue. Subjects were presented various color-word combinations while being asked to click one of three buttons indicating the color of the word as quickly as possible. The investigators monitored neuronal activity throughout, discarding responses that were too slow.
As found in previous trials, the subjects demonstrated the “Stroop effect,” which refers to a slower response when word and color are incongruent (224.9 ms difference; P less than .001). As anticipated, correct responses following correct responses were faster than were correct responses following erroneous responses, which defines PES.
In the intracranial EEG group, the investigators isolated 1,171 neurons, of which 618 were located in dACC and 553 in pre-SMA. Using a Poisson regression model and correlations with erroneous responses, the investigators identified 99 “type I” error neurons in dACC and 118 in pre-SMA, based on higher frequency of firing during erroneous responses than during correct responses. At a single-cell level, error neuron mean spike rates were highest when intracranial ERN amplitude was greatest, such that error neuron firing in dACC and pre-SMA had maximal likelihood ratios of 7.9 (P = .01) and 15.1 (P less than .001), respectively. The strength of correlation between intracranial ERN and error neuron firing rate was directly related to PES magnitude exclusively in the dACC (maximum likelihood ratio of 13.9; P = .015). In post-error trials, faster error-integrating neuron firing rates in dACC predicted greater PES (maximal likelihood ratio of 18.3; P less than .001).
The study was funded by the National Institutes of Health, the McKnight Endowment for Neuroscience, and the National Science Foundation. The authors declared no conflicts of interest.
SOURCE: Fu Z et al. Neuron. 2018 Dec 4. doi: 10.1016/j.neuron.2018.11.016
, and this activity can be tracked through a scalp EEG pattern called error-related negativity, according to findings from experiments carried out during intracranial EEG recordings of candidates for surgical treatment of epilepsy.
“Our results suggest that coordinated neural activity can serve as a substrate for information routing that enables the performance-monitoring system to communicate the need for behavioral control to other brain regions, including those that maintain flexible goal information, such as the lateral prefrontal cortex and the frontal polar cortex,” first author Zhongzheng Fu, a PhD student at the California Institute of Technology in Pasadena, Calif., and Cedars-Sinai Medical Center, Los Angeles, and his colleagues reported in Neuron.
The findings offer insights that could lead to treatments for conditions in which the important executive function task of error self-monitoring is unbalanced, such as obsessive-compulsive disorder and schizophrenia, the authors noted in a press release.
“We discovered that the activity of error neurons correlates with the size of the ERN [error-related negativity],” Mr. Fu said. “This identifies the brain area that causes the ERN and helps explain what it signifies. This new insight might allow doctors to use the ERN as a standard tool to diagnose mental diseases and monitor responses to treatment.”
Error neuron firing and intracranial ERN occurred first in pre-supplementary motor area (pre-SMA), then in the dorsal anterior cingulate cortex (dACC) about 50 ms later, with significant correlations between firing and intracranial ERN in both locations. In dACC, this activity, with error-integrating neuron responses, correlated with magnitude of post-error slowing (PES).
Previous research suggested a link between “the detection of self-generated errors, as reflected in the ERN, with changes in cognitive control, as exhibited behaviorally in PES,” the investigators wrote. “However, several electroencephalogram (EEG) studies have failed to find a significant relationship between PES and ERN.”
The present study involved intracranial EEG of 29 candidates for surgical treatment of epilepsy and scalp EEG of 12 control participants, with each modality measuring activity in the frontal cortex. Both cohorts performed a rapid version of the color-word Stroop task, in which the words “red,” “green,” or “blue” were printed either in corresponding or noncorresponding colors of red, green, or blue. Subjects were presented various color-word combinations while being asked to click one of three buttons indicating the color of the word as quickly as possible. The investigators monitored neuronal activity throughout, discarding responses that were too slow.
As found in previous trials, the subjects demonstrated the “Stroop effect,” which refers to a slower response when word and color are incongruent (224.9 ms difference; P less than .001). As anticipated, correct responses following correct responses were faster than were correct responses following erroneous responses, which defines PES.
In the intracranial EEG group, the investigators isolated 1,171 neurons, of which 618 were located in dACC and 553 in pre-SMA. Using a Poisson regression model and correlations with erroneous responses, the investigators identified 99 “type I” error neurons in dACC and 118 in pre-SMA, based on higher frequency of firing during erroneous responses than during correct responses. At a single-cell level, error neuron mean spike rates were highest when intracranial ERN amplitude was greatest, such that error neuron firing in dACC and pre-SMA had maximal likelihood ratios of 7.9 (P = .01) and 15.1 (P less than .001), respectively. The strength of correlation between intracranial ERN and error neuron firing rate was directly related to PES magnitude exclusively in the dACC (maximum likelihood ratio of 13.9; P = .015). In post-error trials, faster error-integrating neuron firing rates in dACC predicted greater PES (maximal likelihood ratio of 18.3; P less than .001).
The study was funded by the National Institutes of Health, the McKnight Endowment for Neuroscience, and the National Science Foundation. The authors declared no conflicts of interest.
SOURCE: Fu Z et al. Neuron. 2018 Dec 4. doi: 10.1016/j.neuron.2018.11.016
FROM NEURON
Treating negative symptoms of schizophrenia
In schizophrenia, negative symptoms such as social withdrawal, avoidance, lack of spontaneity and flow of conversation, reduced initiative, anhedonia, and blunted affect are among the most challenging to treat. These symptoms commonly persist after positive symptoms such as hallucinations, delusions, and paranoia have subsided. In an analysis of 20 pivotal placebo-controlled trials of second-generation antipsychotics (SGAs), almost 45% of patients who completed 6 weeks of treatment still had at least 1 residual negative symptom of at least moderate severity, and approximately 25% had 2 or more.1 Negative symptoms are viewed as being intrinsic to schizophrenia, and also as the result of extrapyramidal symptoms, depression, and psychosis.
Nearly a decade ago, the Schizophrenia Patient Outcomes Research Team (PORT) published its recommendations for psychopharmacologic and psychosocial treatments of schizophrenia. Unfortunately, due to insufficient evidence, there is still no proven treatment for negative symptoms.2-4 This is particularly problematic because negative symptoms are a major determinant of the poor social and vocational abilities of patients with schizophrenia.
This review focuses on treatments for negative symptoms of schizophrenia that have been evaluated since the PORT treatment recommendations were published and highlights those approaches that show promise.
_
The limitations of antipsychotics
Antipsychotics can both worsen and alleviate negative symptoms by reducing psychotic symptoms. Double-blind, placebo-controlled trials have found that most, if not all, antipsychotics are superior to placebo for treating negative symptoms in patients with acute psychosis.4 However, because these improvements occur in the early stages of treatment, concomitantly with improvement of psychotic symptoms, antipsychotics generally are not viewed as being very effective in the treatment of primary negative symptoms.4 Indeed, an examination of patients with prominent negative symptoms without prominent positive symptoms in the NEWMEDS cohort, which was extracted from 20 pivotal placebo-controlled trials of SGAs, revealed no clinically meaningful treatment effect on negative symptoms.1
There is evidence that antipsychotics can contribute to the development of apathy, flat affect, and other negative symptoms.5 Dopamine (D2)-blocking antipsychotics produce secondary negative symptoms that are not always easy to distinguish from primary negative symptoms.6 In a double-blind, placebo-controlled trial of single doses of risperidone, haloperidol, or placebo in healthy participants, the antipsychotics increased negative symptoms, particularly avolition/apathy.7 Another study found that chronic treatment with antipsychotics did not necessarily affect motivation in patients with schizophrenia.8
Adverse effects, such as anhedonia, often produce and enhance negative symptoms and therefore can limit the use of pharmacologic treatment options. Other adverse effects associated with specific antipsychotics include extrapyramidal symptoms, sedation, increased prolactin secretion, weight gain, and other metabolic abnormalities.
Continue to: Seeking new pharmacologic options
Seeking new pharmacologic options
The years since the PORT review have been filled with initial promise, a series of bitter disappointments, and a renewed spark of hope in the quest to treat negative symptoms in schizophrenia.
Compounds that have been abandoned. Since PORT, researchers have evaluated 5 major compounds that mainly targeted cognition and negative symptoms in patients with schizophrenia (Box9-17). Unfortunately, 4 of them failed to provide significant superiority over placebo, and 1 was withdrawn due to safety concerns.
Box
Since the Schizophrenia Patient Outcomes Research Team (PORT) treatment recommendations were published in 2010, many compounds have been investigated for treating negative symptoms of schizophrenia. Based on the findings of early research, further development of 5 of these has been abandoned.
Encenicline and TC-56199 were both α-7 nicotinic acetylcholine receptor agonists10; bitopertin and AMG 74711 were glycine reuptake inhibitors12; and pomaglumetad methionil13 was an amino acid analog drug that acts as a highly selective agonist for the metabotropic glutamate receptor.
Encenicline showed a treatment effect on negative symptoms in an add-on phase II study,14 but not in 2 subsequent phase III trials (NCT01716975, NCT01714661). TC-5619 showed a treatment effect in a 12-week phase II study of participants with persistent negative symptoms,15 but then failed in a subsequent study.9 Bitopertin showed a treatment effect on negative symptoms in 1 clinical trial,16 but the results were not replicated in a subsequent large multi-center trial.17 The AMG 747 development program was halted due to safety concerns.11 Finally, pomaglumetad methionil failed to meet its primary endpoint in a study of prominent negative symptoms and to show a treatment effect on psychotic symptoms in 2 pivotal phase III trials.13
Initial favorable results. Registered, robust trials of other compounds have had some initial favorable results that need to be replicated. These agents include:
- MIN-101 is a novel cyclic amide derivative.18 In a phase IIb 12-week study of MIN-101 monotherapy (32 mg, n = 78; 64 mg, n = 83) vs placebo (n = 83), both dose groups had significantly more improvement on the Positive and Negative Syndrome Scale (PANSS) negative factor score, which was the primary outcome measure, than placebo (32 mg/d; effect size = .45, P < .02, 64 mg/d; effect size = .57, P < .004) as well as on PANSS negative symptom score and other measures of negative symptoms.18
- Cariprazine is a D2 and D3 receptor partial agonist with high selectivity towards the D3 receptor19
- Minocycline is a broad-spectrum tetracyclic antibiotic displaying neuroprotective properties18,20,21
- Raloxifene is a selective estrogen receptor modulator for postmenopausal women22,23
- Pimavanserin, which was FDA-approved in 2016 for the treatment of Parkinson’s disease psychosis, is being tested in a large trial for adjunctive treatment of patients with negative symptoms of schizophrenia. This medication is a nondopaminergic antipsychotic that acts as a selective serotonin inverse agonist that preferentially targets 5-HT2A receptors while avoiding activity at common targets such as dopamine.24
All of these compounds except MIN-101 are currently available in the U.S. but have not been approved for the treatment of negative symptoms in patients with schizophrenia. MIN-101 is in phase III testing (NCT03397134).
Continue to: Nonpharmacologic treatments
Nonpharmacologic treatments
Recent studies of nonpharmacologic treatments for negative symptoms, including psychosocial approaches and noninvasive electromagnetic neurostimulation, have also been performed. The major psychosocial approaches that have been studied include social skills training (SST), cognitive-behavioral therapy (CBT) for psychosis, cognitive remediation, and family intervention. Some positive findings have been reported. A recent review of psychosocial treatments for negative symptoms in schizophrenia concluded that CBT and SST have the most empirical support, with some evidence even suggesting that gains from CBT are maintained as long as 6 months after treatment.25 Another review found that CBT was significantly more efficacious for reducing positive symptoms and SST in reducing negative symptoms.26
It remains unclear if a combined treatment approach provides improvements above and beyond those associated with each individual treatment modality. Motivation and Enhancement therapy (MOVE) is a potentially promising approach that combines environmental support, CBT, skills training, and other components in an attempt to address all domains of negative symptoms.27 Preliminary results from a randomized controlled trial examining 51 patients with clinically meaningful negative symptoms suggested that MOVE improves negative symptoms. However, the group differences were not significant until after 9 months of treatment and not for all negative symptom scales. A follow-up study has been completed, but the results are not yet available.28
Some small studies have suggested improvement of negative symptoms after noninvasive electromagnetic neurostimulation,29-31 but this has not been replicated in larger studies.32 In the last few years, there were several studies underway that could help clarify if there is a role for noninvasive electromagnetic neurostimulation in the treatment of negative symptoms in schizophrenia; however, results have not been reported at this time.33-35
_
Social skills training and combined interventions
Taken together, the data suggest that treating negative symptoms in schizophrenia remains a major challenge. Patients with negative symptoms are difficult to engage and motivate for treatment and there are no well-supported treatment options. Given the lack of evidence, it is not possible to synthesize this data into clear treatment recommendations. Because many of the negative symptoms are social in nature, it is perhaps not surprising that some evidence has emerged supporting the role of psychosocial approaches. Studies have pointed to the potential role of SST. It is believed to be beneficial as it targets participants’ social functioning by training verbal and nonverbal communication alongside perception and responses to social cues.36 Some evidence suggests that treatment packages that combine several psychosocial interventions (eg, family psychoeducation and skill training) achieve better outcomes than standalone interventions.37 Thus, psychosocial approaches appear to be potentially effective for the treatment of negative symptoms in patients with schizophrenia. In addition, because some antipsychotics has been shown to be associated with fewer negative symptoms than others, another treatment strategy could be to attempt the use of a different antipsychotic, or to revisit whether continued antipsychotic treatment is needed in the absence of positive symptoms.
Bottom Line
Treating negative symptoms in schizophrenia remains a major challenge. There is a lack of evidence for pharmacologic treatments; psychosocial approaches may be beneficial due to the social nature of many negative symptoms. Further, some evidence suggests that treatment packages that combine several psychosocial interventions may achieve better outcomes than standalone interventions.
Related Resource
Tandon R, Jibson M. Negative symptoms of schizophrenia: How to treat them most effectively. Current Psychiatry. 2002;1(9):36-42.
Drug Brand Names
Cariprazine • Vraylar
Haloperidol • Haldol
Minocycline • Dynacin, Minocin
Pimavanserin • Nuplazid
Raloxifene • Evista
Risperidone • Risperdal
1. Rabinowitz J, Werbeloff N, Caers I, et al. Negative symptoms in schizophrenia--the remarkable impact of inclusion definitions in clinical trials and their consequences. Schizophr Res. 2013;150(2-3):334-338.
2. Kreyenbuhl J, Buchanan RW, Dickerson FB, et al. The schizophrenia patient outcomes research team (PORT): updated treatment recommendations 2009. Schizophrenia bulletin. 2010;36(1):94-103.
3. Veerman SRT, Schulte PFJ, de Haan L. Treatment for negative symptoms in schizophrenia: a comprehensive review. Drugs. 2017.
4. Aleman A, Lincoln TM, Bruggeman R, et al. Treatment of negative symptoms: Where do we stand, and where do we go? Schizophr Res. 2017;186:55-62.
5. Awad AG. Subjective tolerability of antipsychotic medications and the emerging science of subjective tolerability disorders. Expert Rev Pharmacoecon Outcomes Res. 2010;10(1):1-4.
6. Kirkpatrick B. Recognizing primary vs secondary negative symptoms and apathy vs expression domains. J Clin Psychiatry. 2014;75(4):e09.
7. Artaloytia JF, Arango C, Lahti A, et al. Negative signs and symptoms secondary to antipsychotics: a double-blind, randomized trial of a single dose of placebo, haloperidol, and risperidone in healthy volunteers. Am J Psychiatry. 2006;163(3):488-493.
8. Fervaha G, Takeuchi H, Lee J, et al. Antipsychotics and amotivation. Neuropsychopharmacology. 2015;40(6):1539-1548.
9. Walling D, Marder SR, Kane J, et al. Phase 2 Trial of an alpha-7 nicotinic receptor agonist (TC-5619) in negative and cognitive symptoms of schizophrenia. Schizophr Bull. 2016;42(2):335-343.
10. Haig GM, Bain EE, Robieson WZ, et al. A randomized trial to assess the efficacy and safety of ABT-126, a selective alpha7 nicotinic acetylcholine receptor agonist, in the treatment of cognitive impairment in schizophrenia. Am J Psychiatry. 2016;173(8):827-835.
11. U.S. National Library of Medicing. ClinicalTrials.gov. 20110165: Study to evaluate the effect of AMG 747 on schizophrenia negative symptoms (study 165). https://clinicaltrials.gov/ct2/show/NCT01568229. Accessed July 1, 2017.
12. Bugarski-Kirola D, Blaettler T, Arango C, et al. Bitopertin in negative symptoms of schizophrenia-results from the phase III FlashLyte and DayLyte studies. Biol Psychiatry. 2017;82(1):8-16.
13. Stauffer VL, Millen BA, Andersen S, et al. Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr Res. 2013;150(2-3):434-441.
14. Keefe RS, Meltzer HA, Dgetluck N, et al. Randomized, double-blind, placebo-controlled study of encenicline, an alpha7 nicotinic acetylcholine receptor agonist, as a treatment for cognitive impairment in schizophrenia. Neuropsychopharmacology. 2015;40(13):3053-3060.
15. Lieberman JA, Dunbar G, Segreti AC, et al. A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology. 2013;38(6):968-975.
16. Umbricht D, Alberati D, Martin-Facklam M, et al. Effect of bitopertin, a glycine reuptake inhibitor, on negative symptoms of schizophrenia: a randomized, double-blind, proof-of-concept study. JAMA Psychiatry. 2014;71(6):637-646.
17. Kingwell K. Schizophrenia drug gets negative results for negative symptoms. Nat Rev Drug Discov. 2014;13(4):244-245.
18. Davidson M, Saoud J, Staner C, et al. Efficacy and safety of MIN-101: a 12-week randomized, double-blind, placebo-controlled trial of a new drug in development for the treatment of negative symptoms in schizophrenia. Am J Psychiatry. 2017;172(12):1195-1202.
19. Nemeth G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
20. Levkovitz Y, Mendlovich S, Riwkes S, et al. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry. 2010;71(2):138-149.
21. Chaudhry IB, Hallak J, Husain N, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J Psychopharmacology. 2012;26(9):1185-1193.
22. Usall J, Huerta-Ramos E, Labad J, et al. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a 24-week double-blind, randomized, parallel, placebo-controlled trial. Schizophr Bull. 2016;42(2):309-317.
23. Usall J, Huerta-Ramos E, Iniesta R, et al. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2011;72(11):1552-1557.
24. Acadia Pharmaceuticals. Pimavanserin - schizophrenia negative symptoms. http://www.acadia-pharm.com/pipeline/pimavanserin-schizophrenia-negative-symptoms/. Accessed July 23, 2017.
25. Elis O, Caponigro JM, Kring AM. Psychosocial treatments for negative symptoms in schizophrenia: current practices and future directions. Clin Psychol Rev. 2013;33(8):914-928.
26. Turner DT, van der Gaag M, Karyotaki E, et al. Psychological interventions for psychosis: a meta-analysis of comparative outcome studies. Am J Psychiatry. 2014;171(5):523-538.
27. Velligan DI, Roberts D, Mintz J, et al. A randomized pilot study of MOtiVation and Enhancement (MOVE) Training for negative symptoms in schizophrenia. Schizophr Res. 2015;165(2-3):175-180.
28. U.S. National Library of Medicing. ClinicalTrials.gov. Treatment Development Targeting Severe and Persistent Negative Symptoms (MOVE). https://clinicaltrials.gov/ct2/show/NCT01550666. Accessed July 20, 2017.
29. Rabany L, Deutsch L, Levkovitz Y. Double-blind, randomized sham controlled study of deep-TMS add-on treatment for negative symptoms and cognitive deficits in schizophrenia. J Psychopharmacology. 2014;28(7):686-690.
30. Bation R, Brunelin J, Saoud M, et al. Intermittent theta burst stimulation of the left dorsolateral prefrontal cortex for the treatment of persistent negative symptoms in schizophrenia. European Neuropsychopharmacology. 2015;25:S329-S30.
31. Li Z, Yin M, Lyu XL, et al. Delayed effect of repetitive transcranial magnetic stimulation (rTMS) on negative symptoms of schizophrenia: findings from a randomized controlled trial. Psychiatry Res. 2016;240:333-335.
32. Wobrock T, Guse B, Cordes J, et al. Left prefrontal high-frequency repetitive transcranial magnetic stimulation for the treatment of schizophrenia with predominant negative symptoms: a sham-controlled, randomized multicenter trial. Biol Psychiatry. 2015;77(11):979-988.
33. U.S. National Library of Medicing. ClinicalTrials.gov. Repetitive transcranial magnetic stimulation and intermittent theta burst (iTBS) in schizophrenia phase 2. https://clinicaltrials.gov/ct2/show/NCT01315587. Accessed July 18, 2017.
34. Treatment of Negative Symptoms and Schizophrenia (STICCS) Phase 1/2. https://clinicaltrials.gov/ct2/show/NCT02204787. Accessed July 15, 2017.
35. U.S. National Library of Medicing. ClinicalTrials.gov. Schizophrenia TreAtment With electRic Transcranial Stimulation (STARTS). https://clinicaltrials.gov/ct2/show/NCT02535676. Accessed July 10, 2017.
36. Bellack AS, Mueser KT, Gingerich S, Agresta J. Social skills training for schizophrenia. A step-by-step guide. New York, NY: Guilford Press; 1997:20-30.
37. Hogarty GE, Anderson CM, Reiss DJ, et al. Family psychoeducation, social skills training, and maintenance chemotherapy in the aftercare treatment of schizophrenia. I. one-year effects of a controlled study on relapse and expressed emotion. Arch Gen Psychiatry. 1986;43(7):633-642.
In schizophrenia, negative symptoms such as social withdrawal, avoidance, lack of spontaneity and flow of conversation, reduced initiative, anhedonia, and blunted affect are among the most challenging to treat. These symptoms commonly persist after positive symptoms such as hallucinations, delusions, and paranoia have subsided. In an analysis of 20 pivotal placebo-controlled trials of second-generation antipsychotics (SGAs), almost 45% of patients who completed 6 weeks of treatment still had at least 1 residual negative symptom of at least moderate severity, and approximately 25% had 2 or more.1 Negative symptoms are viewed as being intrinsic to schizophrenia, and also as the result of extrapyramidal symptoms, depression, and psychosis.
Nearly a decade ago, the Schizophrenia Patient Outcomes Research Team (PORT) published its recommendations for psychopharmacologic and psychosocial treatments of schizophrenia. Unfortunately, due to insufficient evidence, there is still no proven treatment for negative symptoms.2-4 This is particularly problematic because negative symptoms are a major determinant of the poor social and vocational abilities of patients with schizophrenia.
This review focuses on treatments for negative symptoms of schizophrenia that have been evaluated since the PORT treatment recommendations were published and highlights those approaches that show promise.
_
The limitations of antipsychotics
Antipsychotics can both worsen and alleviate negative symptoms by reducing psychotic symptoms. Double-blind, placebo-controlled trials have found that most, if not all, antipsychotics are superior to placebo for treating negative symptoms in patients with acute psychosis.4 However, because these improvements occur in the early stages of treatment, concomitantly with improvement of psychotic symptoms, antipsychotics generally are not viewed as being very effective in the treatment of primary negative symptoms.4 Indeed, an examination of patients with prominent negative symptoms without prominent positive symptoms in the NEWMEDS cohort, which was extracted from 20 pivotal placebo-controlled trials of SGAs, revealed no clinically meaningful treatment effect on negative symptoms.1
There is evidence that antipsychotics can contribute to the development of apathy, flat affect, and other negative symptoms.5 Dopamine (D2)-blocking antipsychotics produce secondary negative symptoms that are not always easy to distinguish from primary negative symptoms.6 In a double-blind, placebo-controlled trial of single doses of risperidone, haloperidol, or placebo in healthy participants, the antipsychotics increased negative symptoms, particularly avolition/apathy.7 Another study found that chronic treatment with antipsychotics did not necessarily affect motivation in patients with schizophrenia.8
Adverse effects, such as anhedonia, often produce and enhance negative symptoms and therefore can limit the use of pharmacologic treatment options. Other adverse effects associated with specific antipsychotics include extrapyramidal symptoms, sedation, increased prolactin secretion, weight gain, and other metabolic abnormalities.
Continue to: Seeking new pharmacologic options
Seeking new pharmacologic options
The years since the PORT review have been filled with initial promise, a series of bitter disappointments, and a renewed spark of hope in the quest to treat negative symptoms in schizophrenia.
Compounds that have been abandoned. Since PORT, researchers have evaluated 5 major compounds that mainly targeted cognition and negative symptoms in patients with schizophrenia (Box9-17). Unfortunately, 4 of them failed to provide significant superiority over placebo, and 1 was withdrawn due to safety concerns.
Box
Since the Schizophrenia Patient Outcomes Research Team (PORT) treatment recommendations were published in 2010, many compounds have been investigated for treating negative symptoms of schizophrenia. Based on the findings of early research, further development of 5 of these has been abandoned.
Encenicline and TC-56199 were both α-7 nicotinic acetylcholine receptor agonists10; bitopertin and AMG 74711 were glycine reuptake inhibitors12; and pomaglumetad methionil13 was an amino acid analog drug that acts as a highly selective agonist for the metabotropic glutamate receptor.
Encenicline showed a treatment effect on negative symptoms in an add-on phase II study,14 but not in 2 subsequent phase III trials (NCT01716975, NCT01714661). TC-5619 showed a treatment effect in a 12-week phase II study of participants with persistent negative symptoms,15 but then failed in a subsequent study.9 Bitopertin showed a treatment effect on negative symptoms in 1 clinical trial,16 but the results were not replicated in a subsequent large multi-center trial.17 The AMG 747 development program was halted due to safety concerns.11 Finally, pomaglumetad methionil failed to meet its primary endpoint in a study of prominent negative symptoms and to show a treatment effect on psychotic symptoms in 2 pivotal phase III trials.13
Initial favorable results. Registered, robust trials of other compounds have had some initial favorable results that need to be replicated. These agents include:
- MIN-101 is a novel cyclic amide derivative.18 In a phase IIb 12-week study of MIN-101 monotherapy (32 mg, n = 78; 64 mg, n = 83) vs placebo (n = 83), both dose groups had significantly more improvement on the Positive and Negative Syndrome Scale (PANSS) negative factor score, which was the primary outcome measure, than placebo (32 mg/d; effect size = .45, P < .02, 64 mg/d; effect size = .57, P < .004) as well as on PANSS negative symptom score and other measures of negative symptoms.18
- Cariprazine is a D2 and D3 receptor partial agonist with high selectivity towards the D3 receptor19
- Minocycline is a broad-spectrum tetracyclic antibiotic displaying neuroprotective properties18,20,21
- Raloxifene is a selective estrogen receptor modulator for postmenopausal women22,23
- Pimavanserin, which was FDA-approved in 2016 for the treatment of Parkinson’s disease psychosis, is being tested in a large trial for adjunctive treatment of patients with negative symptoms of schizophrenia. This medication is a nondopaminergic antipsychotic that acts as a selective serotonin inverse agonist that preferentially targets 5-HT2A receptors while avoiding activity at common targets such as dopamine.24
All of these compounds except MIN-101 are currently available in the U.S. but have not been approved for the treatment of negative symptoms in patients with schizophrenia. MIN-101 is in phase III testing (NCT03397134).
Continue to: Nonpharmacologic treatments
Nonpharmacologic treatments
Recent studies of nonpharmacologic treatments for negative symptoms, including psychosocial approaches and noninvasive electromagnetic neurostimulation, have also been performed. The major psychosocial approaches that have been studied include social skills training (SST), cognitive-behavioral therapy (CBT) for psychosis, cognitive remediation, and family intervention. Some positive findings have been reported. A recent review of psychosocial treatments for negative symptoms in schizophrenia concluded that CBT and SST have the most empirical support, with some evidence even suggesting that gains from CBT are maintained as long as 6 months after treatment.25 Another review found that CBT was significantly more efficacious for reducing positive symptoms and SST in reducing negative symptoms.26
It remains unclear if a combined treatment approach provides improvements above and beyond those associated with each individual treatment modality. Motivation and Enhancement therapy (MOVE) is a potentially promising approach that combines environmental support, CBT, skills training, and other components in an attempt to address all domains of negative symptoms.27 Preliminary results from a randomized controlled trial examining 51 patients with clinically meaningful negative symptoms suggested that MOVE improves negative symptoms. However, the group differences were not significant until after 9 months of treatment and not for all negative symptom scales. A follow-up study has been completed, but the results are not yet available.28
Some small studies have suggested improvement of negative symptoms after noninvasive electromagnetic neurostimulation,29-31 but this has not been replicated in larger studies.32 In the last few years, there were several studies underway that could help clarify if there is a role for noninvasive electromagnetic neurostimulation in the treatment of negative symptoms in schizophrenia; however, results have not been reported at this time.33-35
_
Social skills training and combined interventions
Taken together, the data suggest that treating negative symptoms in schizophrenia remains a major challenge. Patients with negative symptoms are difficult to engage and motivate for treatment and there are no well-supported treatment options. Given the lack of evidence, it is not possible to synthesize this data into clear treatment recommendations. Because many of the negative symptoms are social in nature, it is perhaps not surprising that some evidence has emerged supporting the role of psychosocial approaches. Studies have pointed to the potential role of SST. It is believed to be beneficial as it targets participants’ social functioning by training verbal and nonverbal communication alongside perception and responses to social cues.36 Some evidence suggests that treatment packages that combine several psychosocial interventions (eg, family psychoeducation and skill training) achieve better outcomes than standalone interventions.37 Thus, psychosocial approaches appear to be potentially effective for the treatment of negative symptoms in patients with schizophrenia. In addition, because some antipsychotics has been shown to be associated with fewer negative symptoms than others, another treatment strategy could be to attempt the use of a different antipsychotic, or to revisit whether continued antipsychotic treatment is needed in the absence of positive symptoms.
Bottom Line
Treating negative symptoms in schizophrenia remains a major challenge. There is a lack of evidence for pharmacologic treatments; psychosocial approaches may be beneficial due to the social nature of many negative symptoms. Further, some evidence suggests that treatment packages that combine several psychosocial interventions may achieve better outcomes than standalone interventions.
Related Resource
Tandon R, Jibson M. Negative symptoms of schizophrenia: How to treat them most effectively. Current Psychiatry. 2002;1(9):36-42.
Drug Brand Names
Cariprazine • Vraylar
Haloperidol • Haldol
Minocycline • Dynacin, Minocin
Pimavanserin • Nuplazid
Raloxifene • Evista
Risperidone • Risperdal
In schizophrenia, negative symptoms such as social withdrawal, avoidance, lack of spontaneity and flow of conversation, reduced initiative, anhedonia, and blunted affect are among the most challenging to treat. These symptoms commonly persist after positive symptoms such as hallucinations, delusions, and paranoia have subsided. In an analysis of 20 pivotal placebo-controlled trials of second-generation antipsychotics (SGAs), almost 45% of patients who completed 6 weeks of treatment still had at least 1 residual negative symptom of at least moderate severity, and approximately 25% had 2 or more.1 Negative symptoms are viewed as being intrinsic to schizophrenia, and also as the result of extrapyramidal symptoms, depression, and psychosis.
Nearly a decade ago, the Schizophrenia Patient Outcomes Research Team (PORT) published its recommendations for psychopharmacologic and psychosocial treatments of schizophrenia. Unfortunately, due to insufficient evidence, there is still no proven treatment for negative symptoms.2-4 This is particularly problematic because negative symptoms are a major determinant of the poor social and vocational abilities of patients with schizophrenia.
This review focuses on treatments for negative symptoms of schizophrenia that have been evaluated since the PORT treatment recommendations were published and highlights those approaches that show promise.
_
The limitations of antipsychotics
Antipsychotics can both worsen and alleviate negative symptoms by reducing psychotic symptoms. Double-blind, placebo-controlled trials have found that most, if not all, antipsychotics are superior to placebo for treating negative symptoms in patients with acute psychosis.4 However, because these improvements occur in the early stages of treatment, concomitantly with improvement of psychotic symptoms, antipsychotics generally are not viewed as being very effective in the treatment of primary negative symptoms.4 Indeed, an examination of patients with prominent negative symptoms without prominent positive symptoms in the NEWMEDS cohort, which was extracted from 20 pivotal placebo-controlled trials of SGAs, revealed no clinically meaningful treatment effect on negative symptoms.1
There is evidence that antipsychotics can contribute to the development of apathy, flat affect, and other negative symptoms.5 Dopamine (D2)-blocking antipsychotics produce secondary negative symptoms that are not always easy to distinguish from primary negative symptoms.6 In a double-blind, placebo-controlled trial of single doses of risperidone, haloperidol, or placebo in healthy participants, the antipsychotics increased negative symptoms, particularly avolition/apathy.7 Another study found that chronic treatment with antipsychotics did not necessarily affect motivation in patients with schizophrenia.8
Adverse effects, such as anhedonia, often produce and enhance negative symptoms and therefore can limit the use of pharmacologic treatment options. Other adverse effects associated with specific antipsychotics include extrapyramidal symptoms, sedation, increased prolactin secretion, weight gain, and other metabolic abnormalities.
Continue to: Seeking new pharmacologic options
Seeking new pharmacologic options
The years since the PORT review have been filled with initial promise, a series of bitter disappointments, and a renewed spark of hope in the quest to treat negative symptoms in schizophrenia.
Compounds that have been abandoned. Since PORT, researchers have evaluated 5 major compounds that mainly targeted cognition and negative symptoms in patients with schizophrenia (Box9-17). Unfortunately, 4 of them failed to provide significant superiority over placebo, and 1 was withdrawn due to safety concerns.
Box
Since the Schizophrenia Patient Outcomes Research Team (PORT) treatment recommendations were published in 2010, many compounds have been investigated for treating negative symptoms of schizophrenia. Based on the findings of early research, further development of 5 of these has been abandoned.
Encenicline and TC-56199 were both α-7 nicotinic acetylcholine receptor agonists10; bitopertin and AMG 74711 were glycine reuptake inhibitors12; and pomaglumetad methionil13 was an amino acid analog drug that acts as a highly selective agonist for the metabotropic glutamate receptor.
Encenicline showed a treatment effect on negative symptoms in an add-on phase II study,14 but not in 2 subsequent phase III trials (NCT01716975, NCT01714661). TC-5619 showed a treatment effect in a 12-week phase II study of participants with persistent negative symptoms,15 but then failed in a subsequent study.9 Bitopertin showed a treatment effect on negative symptoms in 1 clinical trial,16 but the results were not replicated in a subsequent large multi-center trial.17 The AMG 747 development program was halted due to safety concerns.11 Finally, pomaglumetad methionil failed to meet its primary endpoint in a study of prominent negative symptoms and to show a treatment effect on psychotic symptoms in 2 pivotal phase III trials.13
Initial favorable results. Registered, robust trials of other compounds have had some initial favorable results that need to be replicated. These agents include:
- MIN-101 is a novel cyclic amide derivative.18 In a phase IIb 12-week study of MIN-101 monotherapy (32 mg, n = 78; 64 mg, n = 83) vs placebo (n = 83), both dose groups had significantly more improvement on the Positive and Negative Syndrome Scale (PANSS) negative factor score, which was the primary outcome measure, than placebo (32 mg/d; effect size = .45, P < .02, 64 mg/d; effect size = .57, P < .004) as well as on PANSS negative symptom score and other measures of negative symptoms.18
- Cariprazine is a D2 and D3 receptor partial agonist with high selectivity towards the D3 receptor19
- Minocycline is a broad-spectrum tetracyclic antibiotic displaying neuroprotective properties18,20,21
- Raloxifene is a selective estrogen receptor modulator for postmenopausal women22,23
- Pimavanserin, which was FDA-approved in 2016 for the treatment of Parkinson’s disease psychosis, is being tested in a large trial for adjunctive treatment of patients with negative symptoms of schizophrenia. This medication is a nondopaminergic antipsychotic that acts as a selective serotonin inverse agonist that preferentially targets 5-HT2A receptors while avoiding activity at common targets such as dopamine.24
All of these compounds except MIN-101 are currently available in the U.S. but have not been approved for the treatment of negative symptoms in patients with schizophrenia. MIN-101 is in phase III testing (NCT03397134).
Continue to: Nonpharmacologic treatments
Nonpharmacologic treatments
Recent studies of nonpharmacologic treatments for negative symptoms, including psychosocial approaches and noninvasive electromagnetic neurostimulation, have also been performed. The major psychosocial approaches that have been studied include social skills training (SST), cognitive-behavioral therapy (CBT) for psychosis, cognitive remediation, and family intervention. Some positive findings have been reported. A recent review of psychosocial treatments for negative symptoms in schizophrenia concluded that CBT and SST have the most empirical support, with some evidence even suggesting that gains from CBT are maintained as long as 6 months after treatment.25 Another review found that CBT was significantly more efficacious for reducing positive symptoms and SST in reducing negative symptoms.26
It remains unclear if a combined treatment approach provides improvements above and beyond those associated with each individual treatment modality. Motivation and Enhancement therapy (MOVE) is a potentially promising approach that combines environmental support, CBT, skills training, and other components in an attempt to address all domains of negative symptoms.27 Preliminary results from a randomized controlled trial examining 51 patients with clinically meaningful negative symptoms suggested that MOVE improves negative symptoms. However, the group differences were not significant until after 9 months of treatment and not for all negative symptom scales. A follow-up study has been completed, but the results are not yet available.28
Some small studies have suggested improvement of negative symptoms after noninvasive electromagnetic neurostimulation,29-31 but this has not been replicated in larger studies.32 In the last few years, there were several studies underway that could help clarify if there is a role for noninvasive electromagnetic neurostimulation in the treatment of negative symptoms in schizophrenia; however, results have not been reported at this time.33-35
_
Social skills training and combined interventions
Taken together, the data suggest that treating negative symptoms in schizophrenia remains a major challenge. Patients with negative symptoms are difficult to engage and motivate for treatment and there are no well-supported treatment options. Given the lack of evidence, it is not possible to synthesize this data into clear treatment recommendations. Because many of the negative symptoms are social in nature, it is perhaps not surprising that some evidence has emerged supporting the role of psychosocial approaches. Studies have pointed to the potential role of SST. It is believed to be beneficial as it targets participants’ social functioning by training verbal and nonverbal communication alongside perception and responses to social cues.36 Some evidence suggests that treatment packages that combine several psychosocial interventions (eg, family psychoeducation and skill training) achieve better outcomes than standalone interventions.37 Thus, psychosocial approaches appear to be potentially effective for the treatment of negative symptoms in patients with schizophrenia. In addition, because some antipsychotics has been shown to be associated with fewer negative symptoms than others, another treatment strategy could be to attempt the use of a different antipsychotic, or to revisit whether continued antipsychotic treatment is needed in the absence of positive symptoms.
Bottom Line
Treating negative symptoms in schizophrenia remains a major challenge. There is a lack of evidence for pharmacologic treatments; psychosocial approaches may be beneficial due to the social nature of many negative symptoms. Further, some evidence suggests that treatment packages that combine several psychosocial interventions may achieve better outcomes than standalone interventions.
Related Resource
Tandon R, Jibson M. Negative symptoms of schizophrenia: How to treat them most effectively. Current Psychiatry. 2002;1(9):36-42.
Drug Brand Names
Cariprazine • Vraylar
Haloperidol • Haldol
Minocycline • Dynacin, Minocin
Pimavanserin • Nuplazid
Raloxifene • Evista
Risperidone • Risperdal
1. Rabinowitz J, Werbeloff N, Caers I, et al. Negative symptoms in schizophrenia--the remarkable impact of inclusion definitions in clinical trials and their consequences. Schizophr Res. 2013;150(2-3):334-338.
2. Kreyenbuhl J, Buchanan RW, Dickerson FB, et al. The schizophrenia patient outcomes research team (PORT): updated treatment recommendations 2009. Schizophrenia bulletin. 2010;36(1):94-103.
3. Veerman SRT, Schulte PFJ, de Haan L. Treatment for negative symptoms in schizophrenia: a comprehensive review. Drugs. 2017.
4. Aleman A, Lincoln TM, Bruggeman R, et al. Treatment of negative symptoms: Where do we stand, and where do we go? Schizophr Res. 2017;186:55-62.
5. Awad AG. Subjective tolerability of antipsychotic medications and the emerging science of subjective tolerability disorders. Expert Rev Pharmacoecon Outcomes Res. 2010;10(1):1-4.
6. Kirkpatrick B. Recognizing primary vs secondary negative symptoms and apathy vs expression domains. J Clin Psychiatry. 2014;75(4):e09.
7. Artaloytia JF, Arango C, Lahti A, et al. Negative signs and symptoms secondary to antipsychotics: a double-blind, randomized trial of a single dose of placebo, haloperidol, and risperidone in healthy volunteers. Am J Psychiatry. 2006;163(3):488-493.
8. Fervaha G, Takeuchi H, Lee J, et al. Antipsychotics and amotivation. Neuropsychopharmacology. 2015;40(6):1539-1548.
9. Walling D, Marder SR, Kane J, et al. Phase 2 Trial of an alpha-7 nicotinic receptor agonist (TC-5619) in negative and cognitive symptoms of schizophrenia. Schizophr Bull. 2016;42(2):335-343.
10. Haig GM, Bain EE, Robieson WZ, et al. A randomized trial to assess the efficacy and safety of ABT-126, a selective alpha7 nicotinic acetylcholine receptor agonist, in the treatment of cognitive impairment in schizophrenia. Am J Psychiatry. 2016;173(8):827-835.
11. U.S. National Library of Medicing. ClinicalTrials.gov. 20110165: Study to evaluate the effect of AMG 747 on schizophrenia negative symptoms (study 165). https://clinicaltrials.gov/ct2/show/NCT01568229. Accessed July 1, 2017.
12. Bugarski-Kirola D, Blaettler T, Arango C, et al. Bitopertin in negative symptoms of schizophrenia-results from the phase III FlashLyte and DayLyte studies. Biol Psychiatry. 2017;82(1):8-16.
13. Stauffer VL, Millen BA, Andersen S, et al. Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr Res. 2013;150(2-3):434-441.
14. Keefe RS, Meltzer HA, Dgetluck N, et al. Randomized, double-blind, placebo-controlled study of encenicline, an alpha7 nicotinic acetylcholine receptor agonist, as a treatment for cognitive impairment in schizophrenia. Neuropsychopharmacology. 2015;40(13):3053-3060.
15. Lieberman JA, Dunbar G, Segreti AC, et al. A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology. 2013;38(6):968-975.
16. Umbricht D, Alberati D, Martin-Facklam M, et al. Effect of bitopertin, a glycine reuptake inhibitor, on negative symptoms of schizophrenia: a randomized, double-blind, proof-of-concept study. JAMA Psychiatry. 2014;71(6):637-646.
17. Kingwell K. Schizophrenia drug gets negative results for negative symptoms. Nat Rev Drug Discov. 2014;13(4):244-245.
18. Davidson M, Saoud J, Staner C, et al. Efficacy and safety of MIN-101: a 12-week randomized, double-blind, placebo-controlled trial of a new drug in development for the treatment of negative symptoms in schizophrenia. Am J Psychiatry. 2017;172(12):1195-1202.
19. Nemeth G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
20. Levkovitz Y, Mendlovich S, Riwkes S, et al. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry. 2010;71(2):138-149.
21. Chaudhry IB, Hallak J, Husain N, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J Psychopharmacology. 2012;26(9):1185-1193.
22. Usall J, Huerta-Ramos E, Labad J, et al. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a 24-week double-blind, randomized, parallel, placebo-controlled trial. Schizophr Bull. 2016;42(2):309-317.
23. Usall J, Huerta-Ramos E, Iniesta R, et al. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2011;72(11):1552-1557.
24. Acadia Pharmaceuticals. Pimavanserin - schizophrenia negative symptoms. http://www.acadia-pharm.com/pipeline/pimavanserin-schizophrenia-negative-symptoms/. Accessed July 23, 2017.
25. Elis O, Caponigro JM, Kring AM. Psychosocial treatments for negative symptoms in schizophrenia: current practices and future directions. Clin Psychol Rev. 2013;33(8):914-928.
26. Turner DT, van der Gaag M, Karyotaki E, et al. Psychological interventions for psychosis: a meta-analysis of comparative outcome studies. Am J Psychiatry. 2014;171(5):523-538.
27. Velligan DI, Roberts D, Mintz J, et al. A randomized pilot study of MOtiVation and Enhancement (MOVE) Training for negative symptoms in schizophrenia. Schizophr Res. 2015;165(2-3):175-180.
28. U.S. National Library of Medicing. ClinicalTrials.gov. Treatment Development Targeting Severe and Persistent Negative Symptoms (MOVE). https://clinicaltrials.gov/ct2/show/NCT01550666. Accessed July 20, 2017.
29. Rabany L, Deutsch L, Levkovitz Y. Double-blind, randomized sham controlled study of deep-TMS add-on treatment for negative symptoms and cognitive deficits in schizophrenia. J Psychopharmacology. 2014;28(7):686-690.
30. Bation R, Brunelin J, Saoud M, et al. Intermittent theta burst stimulation of the left dorsolateral prefrontal cortex for the treatment of persistent negative symptoms in schizophrenia. European Neuropsychopharmacology. 2015;25:S329-S30.
31. Li Z, Yin M, Lyu XL, et al. Delayed effect of repetitive transcranial magnetic stimulation (rTMS) on negative symptoms of schizophrenia: findings from a randomized controlled trial. Psychiatry Res. 2016;240:333-335.
32. Wobrock T, Guse B, Cordes J, et al. Left prefrontal high-frequency repetitive transcranial magnetic stimulation for the treatment of schizophrenia with predominant negative symptoms: a sham-controlled, randomized multicenter trial. Biol Psychiatry. 2015;77(11):979-988.
33. U.S. National Library of Medicing. ClinicalTrials.gov. Repetitive transcranial magnetic stimulation and intermittent theta burst (iTBS) in schizophrenia phase 2. https://clinicaltrials.gov/ct2/show/NCT01315587. Accessed July 18, 2017.
34. Treatment of Negative Symptoms and Schizophrenia (STICCS) Phase 1/2. https://clinicaltrials.gov/ct2/show/NCT02204787. Accessed July 15, 2017.
35. U.S. National Library of Medicing. ClinicalTrials.gov. Schizophrenia TreAtment With electRic Transcranial Stimulation (STARTS). https://clinicaltrials.gov/ct2/show/NCT02535676. Accessed July 10, 2017.
36. Bellack AS, Mueser KT, Gingerich S, Agresta J. Social skills training for schizophrenia. A step-by-step guide. New York, NY: Guilford Press; 1997:20-30.
37. Hogarty GE, Anderson CM, Reiss DJ, et al. Family psychoeducation, social skills training, and maintenance chemotherapy in the aftercare treatment of schizophrenia. I. one-year effects of a controlled study on relapse and expressed emotion. Arch Gen Psychiatry. 1986;43(7):633-642.
1. Rabinowitz J, Werbeloff N, Caers I, et al. Negative symptoms in schizophrenia--the remarkable impact of inclusion definitions in clinical trials and their consequences. Schizophr Res. 2013;150(2-3):334-338.
2. Kreyenbuhl J, Buchanan RW, Dickerson FB, et al. The schizophrenia patient outcomes research team (PORT): updated treatment recommendations 2009. Schizophrenia bulletin. 2010;36(1):94-103.
3. Veerman SRT, Schulte PFJ, de Haan L. Treatment for negative symptoms in schizophrenia: a comprehensive review. Drugs. 2017.
4. Aleman A, Lincoln TM, Bruggeman R, et al. Treatment of negative symptoms: Where do we stand, and where do we go? Schizophr Res. 2017;186:55-62.
5. Awad AG. Subjective tolerability of antipsychotic medications and the emerging science of subjective tolerability disorders. Expert Rev Pharmacoecon Outcomes Res. 2010;10(1):1-4.
6. Kirkpatrick B. Recognizing primary vs secondary negative symptoms and apathy vs expression domains. J Clin Psychiatry. 2014;75(4):e09.
7. Artaloytia JF, Arango C, Lahti A, et al. Negative signs and symptoms secondary to antipsychotics: a double-blind, randomized trial of a single dose of placebo, haloperidol, and risperidone in healthy volunteers. Am J Psychiatry. 2006;163(3):488-493.
8. Fervaha G, Takeuchi H, Lee J, et al. Antipsychotics and amotivation. Neuropsychopharmacology. 2015;40(6):1539-1548.
9. Walling D, Marder SR, Kane J, et al. Phase 2 Trial of an alpha-7 nicotinic receptor agonist (TC-5619) in negative and cognitive symptoms of schizophrenia. Schizophr Bull. 2016;42(2):335-343.
10. Haig GM, Bain EE, Robieson WZ, et al. A randomized trial to assess the efficacy and safety of ABT-126, a selective alpha7 nicotinic acetylcholine receptor agonist, in the treatment of cognitive impairment in schizophrenia. Am J Psychiatry. 2016;173(8):827-835.
11. U.S. National Library of Medicing. ClinicalTrials.gov. 20110165: Study to evaluate the effect of AMG 747 on schizophrenia negative symptoms (study 165). https://clinicaltrials.gov/ct2/show/NCT01568229. Accessed July 1, 2017.
12. Bugarski-Kirola D, Blaettler T, Arango C, et al. Bitopertin in negative symptoms of schizophrenia-results from the phase III FlashLyte and DayLyte studies. Biol Psychiatry. 2017;82(1):8-16.
13. Stauffer VL, Millen BA, Andersen S, et al. Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr Res. 2013;150(2-3):434-441.
14. Keefe RS, Meltzer HA, Dgetluck N, et al. Randomized, double-blind, placebo-controlled study of encenicline, an alpha7 nicotinic acetylcholine receptor agonist, as a treatment for cognitive impairment in schizophrenia. Neuropsychopharmacology. 2015;40(13):3053-3060.
15. Lieberman JA, Dunbar G, Segreti AC, et al. A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology. 2013;38(6):968-975.
16. Umbricht D, Alberati D, Martin-Facklam M, et al. Effect of bitopertin, a glycine reuptake inhibitor, on negative symptoms of schizophrenia: a randomized, double-blind, proof-of-concept study. JAMA Psychiatry. 2014;71(6):637-646.
17. Kingwell K. Schizophrenia drug gets negative results for negative symptoms. Nat Rev Drug Discov. 2014;13(4):244-245.
18. Davidson M, Saoud J, Staner C, et al. Efficacy and safety of MIN-101: a 12-week randomized, double-blind, placebo-controlled trial of a new drug in development for the treatment of negative symptoms in schizophrenia. Am J Psychiatry. 2017;172(12):1195-1202.
19. Nemeth G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
20. Levkovitz Y, Mendlovich S, Riwkes S, et al. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry. 2010;71(2):138-149.
21. Chaudhry IB, Hallak J, Husain N, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J Psychopharmacology. 2012;26(9):1185-1193.
22. Usall J, Huerta-Ramos E, Labad J, et al. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a 24-week double-blind, randomized, parallel, placebo-controlled trial. Schizophr Bull. 2016;42(2):309-317.
23. Usall J, Huerta-Ramos E, Iniesta R, et al. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2011;72(11):1552-1557.
24. Acadia Pharmaceuticals. Pimavanserin - schizophrenia negative symptoms. http://www.acadia-pharm.com/pipeline/pimavanserin-schizophrenia-negative-symptoms/. Accessed July 23, 2017.
25. Elis O, Caponigro JM, Kring AM. Psychosocial treatments for negative symptoms in schizophrenia: current practices and future directions. Clin Psychol Rev. 2013;33(8):914-928.
26. Turner DT, van der Gaag M, Karyotaki E, et al. Psychological interventions for psychosis: a meta-analysis of comparative outcome studies. Am J Psychiatry. 2014;171(5):523-538.
27. Velligan DI, Roberts D, Mintz J, et al. A randomized pilot study of MOtiVation and Enhancement (MOVE) Training for negative symptoms in schizophrenia. Schizophr Res. 2015;165(2-3):175-180.
28. U.S. National Library of Medicing. ClinicalTrials.gov. Treatment Development Targeting Severe and Persistent Negative Symptoms (MOVE). https://clinicaltrials.gov/ct2/show/NCT01550666. Accessed July 20, 2017.
29. Rabany L, Deutsch L, Levkovitz Y. Double-blind, randomized sham controlled study of deep-TMS add-on treatment for negative symptoms and cognitive deficits in schizophrenia. J Psychopharmacology. 2014;28(7):686-690.
30. Bation R, Brunelin J, Saoud M, et al. Intermittent theta burst stimulation of the left dorsolateral prefrontal cortex for the treatment of persistent negative symptoms in schizophrenia. European Neuropsychopharmacology. 2015;25:S329-S30.
31. Li Z, Yin M, Lyu XL, et al. Delayed effect of repetitive transcranial magnetic stimulation (rTMS) on negative symptoms of schizophrenia: findings from a randomized controlled trial. Psychiatry Res. 2016;240:333-335.
32. Wobrock T, Guse B, Cordes J, et al. Left prefrontal high-frequency repetitive transcranial magnetic stimulation for the treatment of schizophrenia with predominant negative symptoms: a sham-controlled, randomized multicenter trial. Biol Psychiatry. 2015;77(11):979-988.
33. U.S. National Library of Medicing. ClinicalTrials.gov. Repetitive transcranial magnetic stimulation and intermittent theta burst (iTBS) in schizophrenia phase 2. https://clinicaltrials.gov/ct2/show/NCT01315587. Accessed July 18, 2017.
34. Treatment of Negative Symptoms and Schizophrenia (STICCS) Phase 1/2. https://clinicaltrials.gov/ct2/show/NCT02204787. Accessed July 15, 2017.
35. U.S. National Library of Medicing. ClinicalTrials.gov. Schizophrenia TreAtment With electRic Transcranial Stimulation (STARTS). https://clinicaltrials.gov/ct2/show/NCT02535676. Accessed July 10, 2017.
36. Bellack AS, Mueser KT, Gingerich S, Agresta J. Social skills training for schizophrenia. A step-by-step guide. New York, NY: Guilford Press; 1997:20-30.
37. Hogarty GE, Anderson CM, Reiss DJ, et al. Family psychoeducation, social skills training, and maintenance chemotherapy in the aftercare treatment of schizophrenia. I. one-year effects of a controlled study on relapse and expressed emotion. Arch Gen Psychiatry. 1986;43(7):633-642.

The daunting challenge of schizophrenia: Hundreds of biotypes and dozens of theories
Islands of knowledge in an ocean of ignorance. That summarizes the advances in unraveling the enigma of schizophrenia, arguably the most complex psychiatric brain disorder. The more breakthroughs are made, the more questions emerge.
Progress is definitely being made and the published literature, replete with new findings, is growing logarithmically. Particularly exciting are the recent advances in the etiology of schizophrenia, both genetic and environmental. Collaboration among geneticists around the world has enabled genome-wide association studies on almost 50,000 DNA samples and has revealed 3 genetic pathways to disrupted brain development, which lead to schizophrenia in early adulthood. Those genetic pathways include:
1. Susceptibility genes—more than 340 of them—are found significantly more often in patients with schizophrenia compared with the general population. These risk genes are scattered across all 23 pairs of chromosomes. They influence neurotransmitter functions, neuroplasticity, and immune regulation. The huge task that lies ahead is identifying what each of the risk genes disrupts in brain structure and/or function.
2. Copy number variants (CNVs), such as deletions (1 allele instead of the normal 2) or duplications (3 alleles), are much more frequent in patients with schizophrenia compared with the general population. That means too little or too much protein is made, which can disrupt the 4 stages of brain development (proliferation, migration, differentiation, and elimination).
3. de novo nonsense mutations, leading to complete absence of protein coding by the affected genes, with adverse ripple effects on brain development.
Approximately 10,000 genes (close to 50% of all 22,000 coding genes in the human genome) are involved in constructing the human brain. The latest estimate is that 79% of the hundreds of biotypes of schizophrenia are genetic in etiology.
In addition, multiple environmental factors can disrupt brain development and lead to schizophrenia. These include older paternal age (>45 years) at the time of conception, pregnancy complications (infections, gestational diabetes, vitamin D deficiency, hypoxia during delivery), childhood maltreatment (sexual or physical abuse or neglect) in the first 5 to 6 years of life, as well as migration and urbanicity (being born and raised in a large metropolitan area).
The bottom line: Schizophrenia is not only very complex, but also an extremely heterogeneous brain syndrome, both biologically and clinically. Psychiatric practitioners are fully cognizant of the extensive clinical variability in patients with schizophrenia, including the presence, absence, or severity of various signs and symptoms, such as insight, delusions, hallucinations, conceptual disorganization, bizarre behaviors, emotional withdrawal, agitation, depression, suicidality, anxiety, substance use, somatic concerns, hostility, idiosyncratic mannerisms, blunted affect, apathy, avolition, self-neglect, poor attention, memory impairment, and problems with decision-making, planning ahead, or organizing one’s life.
In addition, heterogeneity is encountered in such variables as age of onset, minor physical anomalies, soft neurologic signs, naturally occurring movement disorders, premorbid functioning, family history, general medical comorbidities, psychiatry comorbidities, structural brain abnormalities on neuroimaging, neurophysiological deviations (pre-pulse inhibition, p50, p300, N100, mismatch negativity, smooth pursuit eye movements), pituitary volume, rapidity and extent of response to antipsychotics, type and frequency of adverse effects, and functional disability or restoration of vocational functioning.
No wonder, then, given the daunting biologic and clinical heterogeneity of this complex brain syndrome, that myriad hypotheses have been proposed over the past century. The Table lists approximately 50 hypotheses, some discredited but others plausible and still viable. The most absurd hypotheses are the double bind theory of schizophrenia in the 1950s by Gregory Bateson et al, or the latent homosexuality theory of Freud. Some hypotheses may be related to a specific biotype within the schizophrenia syndrome (such as the megavitamin theory) that do not apply to other biotypes. Some of the hypotheses seem to be the product of the rich imagination of an enthusiastic researcher based on limited data.
Another consequence of the extensive heterogeneity of schizophrenia is the large number of “lab tests” that have been reported over the past few decades.1 Those hundreds of biomarkers probably mirror the biologies of the numerous disease subtypes within the schizophrenia syndrome. Some are blood tests, others neurophysiological or neuroimaging, others molecular or genetic, along with many postmortem tissue markers. Obviously, none of these “lab tests” can be used clinically because there would be an unacceptably large number of false positives and false negatives when applied to a heterogeneous sample of patients with schizophrenia.
Heterogeneity also represents a formidable challenge for researchers. Replication of a research finding by investigators across the world can be quite challenging because of the variable composition of biotypes in different countries. This heterogeneity also complicates FDA clinical trials by pharmaceutical companies seeking approval for a new drug to treat schizophrenia. The FDA requires use of DSM diagnostic criteria, which would include patients with similar clinical symptoms, but who can vary widely at the biological level. This results in failed clinical trials where only 20% or 30% of patients with schizophrenia show significant improvement compared with placebo. Given the advances in schizophrenia, a better strategy is to recruit participants who share a specific biomarker to assemble a biologically more homogeneous sample of schizophrenia. If the clinical trial is successful, the same biomarker can then be used by practitioners to predict response to the new drug. That would fulfill the aspirations of applying precision medicine in psychiatric practice.
The famous Eugen Bleuler (whose sister suffered from schizophrenia) was prescient when a century ago he published his classic book titled Dementia Praecox or the Group of Schizophrenias.2 His astute clinical observations led him to recognize the heterogeneity of the syndrome whose name he coined (schizophrenia, or disconnected thoughts). His conceptualization of schizophrenia as a spectrum of disorders of variable outcomes contrasted with that of Emil Kraepelin’s model,3 which regarded dementia praecox as a single, homogeneous, deteriorating disease. But neither Bleuler nor Kraepelin, both of whom relied on clinical observations without any biologic studies, could even imagine the spectacular complexity of the neurobiology of the schizophrenia syndrome and how difficult it is to identify its many biotypes. The monumental advances in neuroscience and neurogenetics, with their sophisticated methodologies, will eventually decipher the mysteries of this neuropsychiatric syndrome, which generates so many aberrations in thought, affect, mood, cognition, and behavior, often leading to severe functional disability among young adults, and untold anguish for their families.
To comment on this editorial or other topics of interest: henry.nasrallah@currentpsychiatry.com.
1. Nasrallah HA. Lab tests for psychiatric disorders: Few clinicians are aware of them. Current Psychiatry. 2013;12(2):5-7.
2. Bleuler E. Dementia praecox or the group of schizophrenias. New York, NY: International University Press; 1950.
3. Hippius H, Muller N. The work of Emil Kraepelin and his research group in Munich. Eur Arch Psychiatry Clin Neurosci. 2008;258(Suppl 2):3-11.
Islands of knowledge in an ocean of ignorance. That summarizes the advances in unraveling the enigma of schizophrenia, arguably the most complex psychiatric brain disorder. The more breakthroughs are made, the more questions emerge.
Progress is definitely being made and the published literature, replete with new findings, is growing logarithmically. Particularly exciting are the recent advances in the etiology of schizophrenia, both genetic and environmental. Collaboration among geneticists around the world has enabled genome-wide association studies on almost 50,000 DNA samples and has revealed 3 genetic pathways to disrupted brain development, which lead to schizophrenia in early adulthood. Those genetic pathways include:
1. Susceptibility genes—more than 340 of them—are found significantly more often in patients with schizophrenia compared with the general population. These risk genes are scattered across all 23 pairs of chromosomes. They influence neurotransmitter functions, neuroplasticity, and immune regulation. The huge task that lies ahead is identifying what each of the risk genes disrupts in brain structure and/or function.
2. Copy number variants (CNVs), such as deletions (1 allele instead of the normal 2) or duplications (3 alleles), are much more frequent in patients with schizophrenia compared with the general population. That means too little or too much protein is made, which can disrupt the 4 stages of brain development (proliferation, migration, differentiation, and elimination).
3. de novo nonsense mutations, leading to complete absence of protein coding by the affected genes, with adverse ripple effects on brain development.
Approximately 10,000 genes (close to 50% of all 22,000 coding genes in the human genome) are involved in constructing the human brain. The latest estimate is that 79% of the hundreds of biotypes of schizophrenia are genetic in etiology.
In addition, multiple environmental factors can disrupt brain development and lead to schizophrenia. These include older paternal age (>45 years) at the time of conception, pregnancy complications (infections, gestational diabetes, vitamin D deficiency, hypoxia during delivery), childhood maltreatment (sexual or physical abuse or neglect) in the first 5 to 6 years of life, as well as migration and urbanicity (being born and raised in a large metropolitan area).
The bottom line: Schizophrenia is not only very complex, but also an extremely heterogeneous brain syndrome, both biologically and clinically. Psychiatric practitioners are fully cognizant of the extensive clinical variability in patients with schizophrenia, including the presence, absence, or severity of various signs and symptoms, such as insight, delusions, hallucinations, conceptual disorganization, bizarre behaviors, emotional withdrawal, agitation, depression, suicidality, anxiety, substance use, somatic concerns, hostility, idiosyncratic mannerisms, blunted affect, apathy, avolition, self-neglect, poor attention, memory impairment, and problems with decision-making, planning ahead, or organizing one’s life.
In addition, heterogeneity is encountered in such variables as age of onset, minor physical anomalies, soft neurologic signs, naturally occurring movement disorders, premorbid functioning, family history, general medical comorbidities, psychiatry comorbidities, structural brain abnormalities on neuroimaging, neurophysiological deviations (pre-pulse inhibition, p50, p300, N100, mismatch negativity, smooth pursuit eye movements), pituitary volume, rapidity and extent of response to antipsychotics, type and frequency of adverse effects, and functional disability or restoration of vocational functioning.
No wonder, then, given the daunting biologic and clinical heterogeneity of this complex brain syndrome, that myriad hypotheses have been proposed over the past century. The Table lists approximately 50 hypotheses, some discredited but others plausible and still viable. The most absurd hypotheses are the double bind theory of schizophrenia in the 1950s by Gregory Bateson et al, or the latent homosexuality theory of Freud. Some hypotheses may be related to a specific biotype within the schizophrenia syndrome (such as the megavitamin theory) that do not apply to other biotypes. Some of the hypotheses seem to be the product of the rich imagination of an enthusiastic researcher based on limited data.
Another consequence of the extensive heterogeneity of schizophrenia is the large number of “lab tests” that have been reported over the past few decades.1 Those hundreds of biomarkers probably mirror the biologies of the numerous disease subtypes within the schizophrenia syndrome. Some are blood tests, others neurophysiological or neuroimaging, others molecular or genetic, along with many postmortem tissue markers. Obviously, none of these “lab tests” can be used clinically because there would be an unacceptably large number of false positives and false negatives when applied to a heterogeneous sample of patients with schizophrenia.
Heterogeneity also represents a formidable challenge for researchers. Replication of a research finding by investigators across the world can be quite challenging because of the variable composition of biotypes in different countries. This heterogeneity also complicates FDA clinical trials by pharmaceutical companies seeking approval for a new drug to treat schizophrenia. The FDA requires use of DSM diagnostic criteria, which would include patients with similar clinical symptoms, but who can vary widely at the biological level. This results in failed clinical trials where only 20% or 30% of patients with schizophrenia show significant improvement compared with placebo. Given the advances in schizophrenia, a better strategy is to recruit participants who share a specific biomarker to assemble a biologically more homogeneous sample of schizophrenia. If the clinical trial is successful, the same biomarker can then be used by practitioners to predict response to the new drug. That would fulfill the aspirations of applying precision medicine in psychiatric practice.
The famous Eugen Bleuler (whose sister suffered from schizophrenia) was prescient when a century ago he published his classic book titled Dementia Praecox or the Group of Schizophrenias.2 His astute clinical observations led him to recognize the heterogeneity of the syndrome whose name he coined (schizophrenia, or disconnected thoughts). His conceptualization of schizophrenia as a spectrum of disorders of variable outcomes contrasted with that of Emil Kraepelin’s model,3 which regarded dementia praecox as a single, homogeneous, deteriorating disease. But neither Bleuler nor Kraepelin, both of whom relied on clinical observations without any biologic studies, could even imagine the spectacular complexity of the neurobiology of the schizophrenia syndrome and how difficult it is to identify its many biotypes. The monumental advances in neuroscience and neurogenetics, with their sophisticated methodologies, will eventually decipher the mysteries of this neuropsychiatric syndrome, which generates so many aberrations in thought, affect, mood, cognition, and behavior, often leading to severe functional disability among young adults, and untold anguish for their families.
To comment on this editorial or other topics of interest: henry.nasrallah@currentpsychiatry.com.
Islands of knowledge in an ocean of ignorance. That summarizes the advances in unraveling the enigma of schizophrenia, arguably the most complex psychiatric brain disorder. The more breakthroughs are made, the more questions emerge.
Progress is definitely being made and the published literature, replete with new findings, is growing logarithmically. Particularly exciting are the recent advances in the etiology of schizophrenia, both genetic and environmental. Collaboration among geneticists around the world has enabled genome-wide association studies on almost 50,000 DNA samples and has revealed 3 genetic pathways to disrupted brain development, which lead to schizophrenia in early adulthood. Those genetic pathways include:
1. Susceptibility genes—more than 340 of them—are found significantly more often in patients with schizophrenia compared with the general population. These risk genes are scattered across all 23 pairs of chromosomes. They influence neurotransmitter functions, neuroplasticity, and immune regulation. The huge task that lies ahead is identifying what each of the risk genes disrupts in brain structure and/or function.
2. Copy number variants (CNVs), such as deletions (1 allele instead of the normal 2) or duplications (3 alleles), are much more frequent in patients with schizophrenia compared with the general population. That means too little or too much protein is made, which can disrupt the 4 stages of brain development (proliferation, migration, differentiation, and elimination).
3. de novo nonsense mutations, leading to complete absence of protein coding by the affected genes, with adverse ripple effects on brain development.
Approximately 10,000 genes (close to 50% of all 22,000 coding genes in the human genome) are involved in constructing the human brain. The latest estimate is that 79% of the hundreds of biotypes of schizophrenia are genetic in etiology.
In addition, multiple environmental factors can disrupt brain development and lead to schizophrenia. These include older paternal age (>45 years) at the time of conception, pregnancy complications (infections, gestational diabetes, vitamin D deficiency, hypoxia during delivery), childhood maltreatment (sexual or physical abuse or neglect) in the first 5 to 6 years of life, as well as migration and urbanicity (being born and raised in a large metropolitan area).
The bottom line: Schizophrenia is not only very complex, but also an extremely heterogeneous brain syndrome, both biologically and clinically. Psychiatric practitioners are fully cognizant of the extensive clinical variability in patients with schizophrenia, including the presence, absence, or severity of various signs and symptoms, such as insight, delusions, hallucinations, conceptual disorganization, bizarre behaviors, emotional withdrawal, agitation, depression, suicidality, anxiety, substance use, somatic concerns, hostility, idiosyncratic mannerisms, blunted affect, apathy, avolition, self-neglect, poor attention, memory impairment, and problems with decision-making, planning ahead, or organizing one’s life.
In addition, heterogeneity is encountered in such variables as age of onset, minor physical anomalies, soft neurologic signs, naturally occurring movement disorders, premorbid functioning, family history, general medical comorbidities, psychiatry comorbidities, structural brain abnormalities on neuroimaging, neurophysiological deviations (pre-pulse inhibition, p50, p300, N100, mismatch negativity, smooth pursuit eye movements), pituitary volume, rapidity and extent of response to antipsychotics, type and frequency of adverse effects, and functional disability or restoration of vocational functioning.
No wonder, then, given the daunting biologic and clinical heterogeneity of this complex brain syndrome, that myriad hypotheses have been proposed over the past century. The Table lists approximately 50 hypotheses, some discredited but others plausible and still viable. The most absurd hypotheses are the double bind theory of schizophrenia in the 1950s by Gregory Bateson et al, or the latent homosexuality theory of Freud. Some hypotheses may be related to a specific biotype within the schizophrenia syndrome (such as the megavitamin theory) that do not apply to other biotypes. Some of the hypotheses seem to be the product of the rich imagination of an enthusiastic researcher based on limited data.
Another consequence of the extensive heterogeneity of schizophrenia is the large number of “lab tests” that have been reported over the past few decades.1 Those hundreds of biomarkers probably mirror the biologies of the numerous disease subtypes within the schizophrenia syndrome. Some are blood tests, others neurophysiological or neuroimaging, others molecular or genetic, along with many postmortem tissue markers. Obviously, none of these “lab tests” can be used clinically because there would be an unacceptably large number of false positives and false negatives when applied to a heterogeneous sample of patients with schizophrenia.
Heterogeneity also represents a formidable challenge for researchers. Replication of a research finding by investigators across the world can be quite challenging because of the variable composition of biotypes in different countries. This heterogeneity also complicates FDA clinical trials by pharmaceutical companies seeking approval for a new drug to treat schizophrenia. The FDA requires use of DSM diagnostic criteria, which would include patients with similar clinical symptoms, but who can vary widely at the biological level. This results in failed clinical trials where only 20% or 30% of patients with schizophrenia show significant improvement compared with placebo. Given the advances in schizophrenia, a better strategy is to recruit participants who share a specific biomarker to assemble a biologically more homogeneous sample of schizophrenia. If the clinical trial is successful, the same biomarker can then be used by practitioners to predict response to the new drug. That would fulfill the aspirations of applying precision medicine in psychiatric practice.
The famous Eugen Bleuler (whose sister suffered from schizophrenia) was prescient when a century ago he published his classic book titled Dementia Praecox or the Group of Schizophrenias.2 His astute clinical observations led him to recognize the heterogeneity of the syndrome whose name he coined (schizophrenia, or disconnected thoughts). His conceptualization of schizophrenia as a spectrum of disorders of variable outcomes contrasted with that of Emil Kraepelin’s model,3 which regarded dementia praecox as a single, homogeneous, deteriorating disease. But neither Bleuler nor Kraepelin, both of whom relied on clinical observations without any biologic studies, could even imagine the spectacular complexity of the neurobiology of the schizophrenia syndrome and how difficult it is to identify its many biotypes. The monumental advances in neuroscience and neurogenetics, with their sophisticated methodologies, will eventually decipher the mysteries of this neuropsychiatric syndrome, which generates so many aberrations in thought, affect, mood, cognition, and behavior, often leading to severe functional disability among young adults, and untold anguish for their families.
To comment on this editorial or other topics of interest: henry.nasrallah@currentpsychiatry.com.
1. Nasrallah HA. Lab tests for psychiatric disorders: Few clinicians are aware of them. Current Psychiatry. 2013;12(2):5-7.
2. Bleuler E. Dementia praecox or the group of schizophrenias. New York, NY: International University Press; 1950.
3. Hippius H, Muller N. The work of Emil Kraepelin and his research group in Munich. Eur Arch Psychiatry Clin Neurosci. 2008;258(Suppl 2):3-11.
1. Nasrallah HA. Lab tests for psychiatric disorders: Few clinicians are aware of them. Current Psychiatry. 2013;12(2):5-7.
2. Bleuler E. Dementia praecox or the group of schizophrenias. New York, NY: International University Press; 1950.
3. Hippius H, Muller N. The work of Emil Kraepelin and his research group in Munich. Eur Arch Psychiatry Clin Neurosci. 2008;258(Suppl 2):3-11.

Risperidone extended-release injectable suspension
Oral antipsychotic nonadherence is a significant contributor to relapse in patients with schizophrenia spectrum disorders. Long-acting injectable (LAI) antipsychotics have been developed to provide sustained antipsychotic exposure, with evidence that use of LAIs significantly reduces hospitalization rates.1 One limiting factor in transitioning patients to certain LAIs is the need for prolonged oral coverage at the onset of treatment for agents that cannot be loaded. Nonadherence with this bridging oral therapy places the patient at risk for symptom exacerbation until effective antipsychotic plasma levels are achieved from the LAI.2 Although risperidone is one of the more widely used antipsychotics for treating schizophrenia, until recently the only available LAI preparation, risperidone microspheres (Risperdal Consta), required 3 weeks of oral coverage upon initiation.3

Clinical implications
Oral medication nonadherence remains a significant public health issue for patients with schizophrenia, with an estimated 50% of patients failing to achieve 80% adherence even when enrolled in clinical trials specifically designed to track adherence.5 Although LAI atypical antipsychotics have been available since the approval of Risperdal Consta, the LAI form of risperidone, and both LAI forms of aripiprazole, were not designed to be loaded. A 1-day initiation regimen for aripiprazole lauroxil has been developed to avoid the need for 3 weeks of oral medication coverage,6,7 but aripiprazole monohydrate and risperidone microspheres mandate oral bridging of 2 and 3 weeks, respectively.2 Because one of the primary indications for LAI antipsychotic therapy is oral medication nonadherence, this prolonged period of oral coverage creates a risk for symptom exacerbation when the bridging period occurs outside of a controlled setting, as is common when patients are discharged from inpatient hospitalization.
One solution to this problem has its antecedents in the development of the Atrigel biodegradable injectable polymer, which was designed to deliver prolonged medication exposure after subcutaneous injection.8 This biodegradable polymer drug delivery system suspends and dissolves the medication of interest (in this case, risperidone) in a poly DL-lactide-coglycolide gel and its biocompatible carrier.9 The viscous liquid undergoes a phase transition upon contact with tissue fluids after subcutaneous injection, resulting in an implant that releases risperidone in a controlled manner as it is resorbed. Importantly, the kinetic parameters of RBP-7000 are such that effective drug levels are seen within the first week without the need for oral coverage.10
Use in adults with schizophrenia. After establishing tolerability with oral risperidone, the recommended doses are 90 mg or 120 mg monthly, which correspond to oral daily risperidone doses of 3 mg or 4 mg. RBP-7000 must be administered as a subcutaneous abdominal injection by a health care professional. It is recommended that the patient be in the supine position for the injection and that the injection sites be rotated monthly among 4 quadrants in the abdominal region. The injection volumes for the 90 mg and 120 mg doses are 0.6 mL and 0.8 mL, respectively.10 As the gel implant becomes firmer, the patient will notice a lump for several weeks that will decrease in size over time. Patients should be advised not to rub or massage the injection site, and to be aware of the placement of any belts or clothing with waistbands.10
Pharmacologic profile, adverse reactions
Risperidone is an atypical antipsychotic that has been commercially available in the U.S. since December 29, 1993, and its adverse effect profile is well characterized. The most common adverse effects associated with risperidone include those related to dopamine D2 antagonism, metabolic adverse effects, and an increase in serum prolactin. In the 12-month long-term safety study of RBP-7000, 1-minute post-dose injection site pain scores (on a 100-point scale) were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following the last injection).10
Continue to: How the Atrigel system works
How the Atrigel system works. The Atrigel system was developed in the late 1980s and consists of a solution of a resorbable polymer in a biocompatible carrier.11 After in vivo administration (typically via subcutaneous injection), the polymer undergoes a phase change from a liquid to a formed implant (Figure 1). Being in liquid form, this system provides the advantage of placement by simple means, such as injection by syringes. The absorption rates of various polymers and the release rates for various drugs are tailored to the desired indication. Approved uses for Atrigel include the subgingival delivery of the antibiotic doxycycline for chronic adult periodontitis (approved September 1998), and the monthly subcutaneous injectable form of the anti-androgen leuprolide, which was approved in January 2002.8,12 Release periods up to 4 months have been achieved with Atrigel; 1 month is the most often desired release period. The biodegradable polymer used for RBP-7000 is designed to provide effective plasma drug levels during the first week of treatment, and sustained levels with a 1-month dosing interval. The small subcutaneous implant that is formed is gradually resorbed over the course of 1 month.
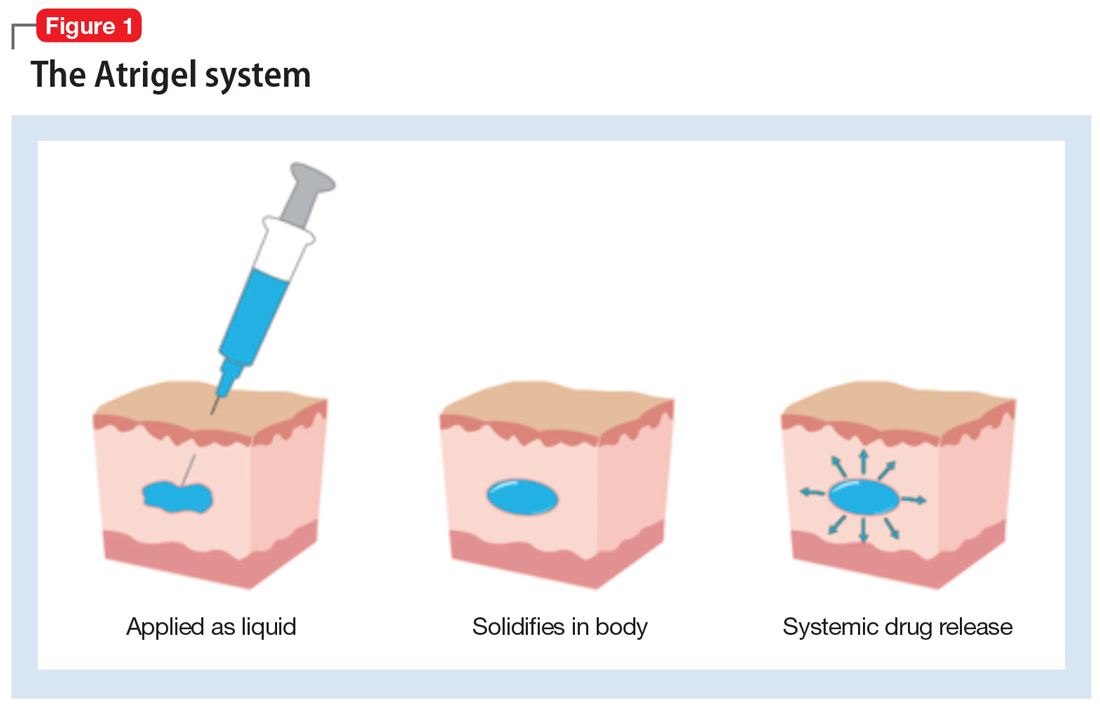
Pharmacokinetics. As with all LAI medications, the half-life with repeated dosing vastly exceeds that achieved with oral administration. Following oral administration, mean peak plasma levels of risperidone occur at 1 hour, and those for the active metabolite 9-OH risperidone occur at 3 hours.13 Oral risperidone has a mean half-life of 3 hours, while the active metabolite 9-OH risperidone has a mean half-life of 21 hours.14 Due to its longer half-life, the metabolite comprises 83% of the active drug levels at steady state.14 Although risperidone is susceptible to interactions via cytochrome P450 (CYP) inhibitors and inducers, particularly CYP2D6 (Table 210), the pharmacokinetics of the combined total of risperidone and 9-OH risperidone levels (deemed the active moiety) are similar in CYP2D6 extensive and poor metabolizers, with an overall mean elimination half-life of approximately 20 hours.13

The kinetics for RBP-7000 are markedly different than those for oral risperidone (Figure 215). After a single subcutaneous injection, RBP-7000 shows 2 absorption peaks for risperidone. The first lower peak occurs with a Tmax of 4 to 6 hours due to initial release of risperidone during the implant formation process; a second risperidone peak occurs after 10 to 14 days and is associated with slow release from the subcutaneous depot.9,16,17 For both 9-OH risperidone levels and the total active moiety (risperidone plus 9-OH risperidone levels), the median Tmax of the first peak ranges from 4 to 48 hours and the second peak ranges from 7 to 11 days. Following a single subcutaneous injection of RBP-7000, the apparent terminal half-life of risperidone ranges from 9 to 11 days, on average. The mean apparent terminal half-life of the active moiety ranges from 8 to 9 days.9,16,17 Based on population pharmacokinetic modeling, the 90 mg and 120 mg doses of RBP-7000 are estimated to provide drug exposure equivalent to 3 mg/d and 4 mg/d of oral risperidone, respectively.9,16,17
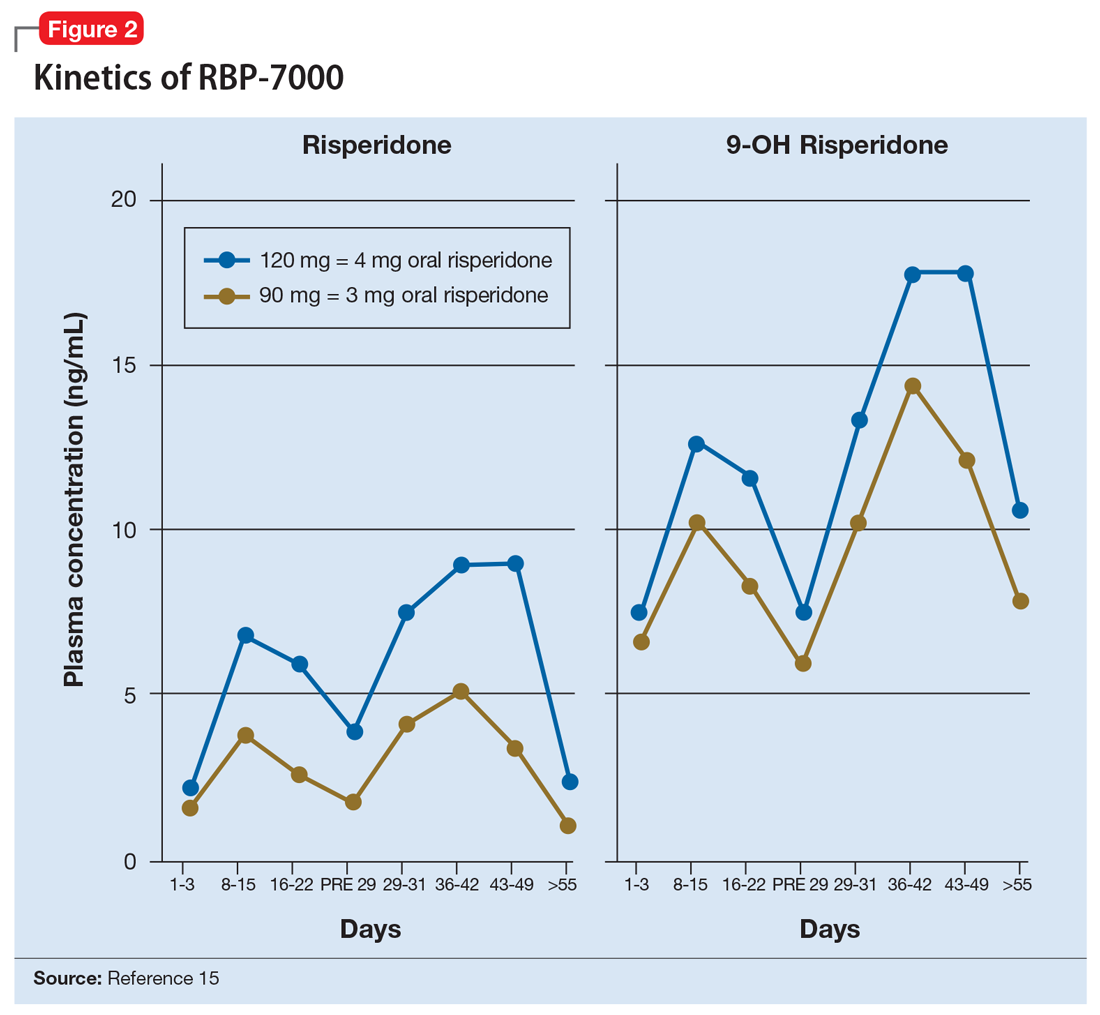
Continue to: Efficacy of RBP-7000
Efficacy of RBP-7000 was established in an 8-week, double-blind, placebo-controlled trial of adult patients experiencing an acute exacerbation of schizophrenia (age 18 to 55).4 Eligible participants had:
- An acute exacerbation of schizophrenia that occurred ≤8 weeks before the screening visit and would have benefited from psychiatric hospitalization or continued hospitalization
- Positive and Negative Syndrome Scale (PANSS) total score between 80 and 120 at visit 1 and a score of >4 on at least 2 of the following 4 items: hallucinatory behavior, delusions, conceptual disorganization, or suspiciousness/persecution
- The diagnosis of acute exacerbation of schizophrenia and PANSS total score were confirmed through an independent video-conference interview conducted by an experienced rater.
Participants were excluded if they:
- Experienced a ≥20% improvement in PANSS total score between the initial screening visit and the first injection
- had been treated at any time with clozapine for treatment-resistant schizophrenia
- had met DSM-IV-TR criteria for substance dependence (with the exception of nicotine or caffeine) before screening.
During the initial screening visit, participants received a 0.25-mg tablet of oral risperidone on 2 consecutive days to assess the tolerability of risperidone.
Outcome. Participants were randomized in a 1:1:1 manner to placebo (n = 112) or 1 of 2 monthly doses of RBP-7000: 90 mg (n = 111) or 120 mg (n = 114). Using the least squares means of repeated-measures changes from baseline in PANSS total scores, there was a significant improvement in the difference in PANSS total scores from baseline to the end of the study compared with placebo: 90-mg RBP-7000, -6.148 points (95% confidence interval [CI], -9.982 to -2.314, P = .0004); 120-mg RBP-7000, -7.237 points (95% CI, -11.045 to -3.429, P < .0001). The absolute change from baseline in PANSS total score was -15.367 points for the 90-mg dose and -16.456 points for the 120-mg dose.4 Completion rates across all 3 arms were comparable: placebo 70.6%, RBP-7000 90 mg 77.6%, and RBP-7000 120 mg 71.4%.
Tolerability. In the 8-week phase III efficacy trial of RBP-7000, adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo were weight gain (placebo 3.4%, 90 mg 13.0%, 120 mg 12.8%) and sedation (placebo 0%, 90 mg 7.0%, 120 mg 7.7%).10 Compared with baseline, participants had a mean weight gain at the end of the study of 2.83 kg in the placebo group, 5.15 kg in the 90-mg RBP-7000 group, and 4.69 kg in the 120-mg RBP-7000 group. There were no clinically significant differences at study endpoint in glucose and lipid parameters. Consistent with the known effects of risperidone, there were increases in mean prolactin levels during the 8-week study, the effects of which were greater for women. For men, mean prolactin levels from baseline to study end were: placebo: 9.8 ± 7.9 vs 9.9 ± 8.0 ng/mL; 90 mg: 8.9 ± 6.9 vs 22.4 ± 11.2 ng/mL; and 120 mg: 8.2 ± 5.2 vs 31.3 ± 14.8 ng/mL. For women, mean prolactin levels from baseline to study end were: placebo: 12.8 ± 11.7 vs 10.4 ± 8.0 ng/mL; 90 mg: 7.7 ± 5.3 vs 60.3 ± 46.9 ng/mL; and 120 mg: 10.9 ± 8.6 vs 85.5 ± 55.1 ng/mL. In the pivotal study, discontinuations due to adverse events were low across all treatment groups: 2.5% for placebo vs 0% for 90 mg and 1.7% for 120 mg.4 There was no single adverse reaction leading to discontinuation that occurred at a rate of ≥2% and greater than placebo in patients treated with RBP-7000.10 There were no clinically relevant differences in mean changes from baseline in corrected QT, QRS, and PR intervals, and in heart rate. Similarly, in the 12-month, long-term safety study, there were no clinically relevant changes in mean electrocardiography interval values from baseline to post-dose assessments.10
Using a 100-point visual analog scale (VAS), injection site pain scores 1 minute after the first dose decreased from a mean of 27 to the range of 3 to 7 for scores obtained 30 to 60 minutes post-dose. In the 12-month long-term safety study, 1-minute post-dose injection site pain VAS scores were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following last injection).10
Clinical considerations
Unique properties. RBP-7000 uses the established Atrigel system to provide effective antipsychotic levels in the first week of treatment, without the need for bridging oral coverage or a second loading injection. The abdominal subcutaneous injection volume is relatively small (0.6 mL or 0.8 mL).
Why Rx? The reasons to prescribe RBP-7000 for adult patients with schizophrenia include:
- no oral coverage required at the initiation of treatment
- effective plasma active moiety levels are seen within the first week without the need for a second loading injection
- monthly injection schedule.
Dosing. The recommended dosage of RBP-7000 is 90 mg or 120 mg once monthly, equivalent to 3 mg/d or 4 mg/d of oral risperidone, respectively. Oral risperidone tolerability should be established before the first injection. No oral risperidone coverage is required. RBP-7000 has not been studied in patients with renal or hepatic impairment and should be used with caution in these patients. Prior to initiating treatment in these patients, it is advised to carefully titrate up to at least 3 mg/d of oral risperidone. If a patient can tolerate 3 mg/d of oral risperidone and is psychiatrically stable, then the 90-mg dose of RBP-7000 can be considered.10
Contraindications. The only contraindications for RBP-7000 are known hypersensitivity to risperidone, paliperidone (9-OH risperidone), or other components of the injection.
Bottom Line
RBP-7000 (Perseris) is the second long-acting injectable (LAI) form of risperidone approved in the U.S. Unlike risperidone microspheres (Consta), RBP-7000 does not require any oral risperidone coverage at the beginning of therapy, provides effective drug levels within the first week of treatment with a single injection, and uses a monthly dosing interval. RBP-7000 does not require loading upon initiation. The monthly injection is <1 mL, is administered in abdominal subcutaneous tissue, and uses the Atrigel system.
Related Resource
- Carpenter J, Wong KK. Long-acting injectable antipsychotics: What to do about missed doses. Current Psychiatry. 2018;17(7):10-12,14-19,56.
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Carbatrol, Tegretol
Doxycycline • Atridox
Leuprolide acetate injectable suspension • Eligard
Paliperidone palmitate • Invega Sustenna
Risperidone • Risperdal
Risperidone extended-release injectable suspension • Perseris
Risperidone long-acting injection • Risperdal Consta
1. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
2. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
3. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
4. Nasser AF, Henderson DC, Fava M, et al. Efficacy, safety, and tolerability of RBP-7000 once-monthly risperidone for the treatment of acute schizophrenia: an 8-week, randomized, double-blind, placebo-controlled, multicenter phase 3 study. J Clin Psychopharmacol. 2016;36(2):130-140.
5. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
6. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
7. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
8. Southard GL, Dunn RL, Garrett S. The drug delivery and biomaterial attributes of the ATRIGEL technology in the treatment of periodontal disease. Expert Opin Investig Drugs. 1998;7(9):1483-1491.
9. Gomeni R, Heidbreder C, Fudala PJ, Nasser AF. A model-based approach to characterize the population pharmacokinetics and the relationship between the pharmacokinetic and safety profiles of RBP-7000, a new, long-acting, sustained-released formulation of risperidone. J Clin Pharmacol. 2013;53(10):1010-1019.
10. Perseris [package insert]. North Chesterfield, VA: Indivior Inc; 2018.
11. Malik K, Singh I, Nagpal M, et al. Atrigel: a potential parenteral controlled drug delivery system. Der Pharmacia Sinica. 2010;1(1):74-81.
12. Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003;61(2 Suppl 1):25-31.
13. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
14. de Leon J, Wynn G, Sandson NB. The pharmacokinetics of paliperidone versus risperidone. Psychosomatics. 2010;51(1):80-88.
15. Ivaturi V, Gopalakrishnan M, Gobburu JVS, et al. Exposure-response analysis after subcutaneous administration of RBP-7000, a once-a-month long-acting Atrigel formulation of risperidone. Br J Clin Pharmacol. 2017;83(7):1476-1498.
16. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetics and prediction of dopamine D2 receptor occupancy after multiple doses of RBP-7000, a new sustained-release formulation of risperidone, in schizophrenia patients on stable oral risperidone treatment. Clin Pharmacokinet. 2014;53(6):533-543.
17. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetic modeling and simulation to guide dose selection for RBP-7000, a new sustained-release formulation of risperidone. J Clin Pharmacol. 2015;55(1):93-103.
Oral antipsychotic nonadherence is a significant contributor to relapse in patients with schizophrenia spectrum disorders. Long-acting injectable (LAI) antipsychotics have been developed to provide sustained antipsychotic exposure, with evidence that use of LAIs significantly reduces hospitalization rates.1 One limiting factor in transitioning patients to certain LAIs is the need for prolonged oral coverage at the onset of treatment for agents that cannot be loaded. Nonadherence with this bridging oral therapy places the patient at risk for symptom exacerbation until effective antipsychotic plasma levels are achieved from the LAI.2 Although risperidone is one of the more widely used antipsychotics for treating schizophrenia, until recently the only available LAI preparation, risperidone microspheres (Risperdal Consta), required 3 weeks of oral coverage upon initiation.3
Clinical implications
Oral medication nonadherence remains a significant public health issue for patients with schizophrenia, with an estimated 50% of patients failing to achieve 80% adherence even when enrolled in clinical trials specifically designed to track adherence.5 Although LAI atypical antipsychotics have been available since the approval of Risperdal Consta, the LAI form of risperidone, and both LAI forms of aripiprazole, were not designed to be loaded. A 1-day initiation regimen for aripiprazole lauroxil has been developed to avoid the need for 3 weeks of oral medication coverage,6,7 but aripiprazole monohydrate and risperidone microspheres mandate oral bridging of 2 and 3 weeks, respectively.2 Because one of the primary indications for LAI antipsychotic therapy is oral medication nonadherence, this prolonged period of oral coverage creates a risk for symptom exacerbation when the bridging period occurs outside of a controlled setting, as is common when patients are discharged from inpatient hospitalization.
One solution to this problem has its antecedents in the development of the Atrigel biodegradable injectable polymer, which was designed to deliver prolonged medication exposure after subcutaneous injection.8 This biodegradable polymer drug delivery system suspends and dissolves the medication of interest (in this case, risperidone) in a poly DL-lactide-coglycolide gel and its biocompatible carrier.9 The viscous liquid undergoes a phase transition upon contact with tissue fluids after subcutaneous injection, resulting in an implant that releases risperidone in a controlled manner as it is resorbed. Importantly, the kinetic parameters of RBP-7000 are such that effective drug levels are seen within the first week without the need for oral coverage.10
Use in adults with schizophrenia. After establishing tolerability with oral risperidone, the recommended doses are 90 mg or 120 mg monthly, which correspond to oral daily risperidone doses of 3 mg or 4 mg. RBP-7000 must be administered as a subcutaneous abdominal injection by a health care professional. It is recommended that the patient be in the supine position for the injection and that the injection sites be rotated monthly among 4 quadrants in the abdominal region. The injection volumes for the 90 mg and 120 mg doses are 0.6 mL and 0.8 mL, respectively.10 As the gel implant becomes firmer, the patient will notice a lump for several weeks that will decrease in size over time. Patients should be advised not to rub or massage the injection site, and to be aware of the placement of any belts or clothing with waistbands.10
Pharmacologic profile, adverse reactions
Risperidone is an atypical antipsychotic that has been commercially available in the U.S. since December 29, 1993, and its adverse effect profile is well characterized. The most common adverse effects associated with risperidone include those related to dopamine D2 antagonism, metabolic adverse effects, and an increase in serum prolactin. In the 12-month long-term safety study of RBP-7000, 1-minute post-dose injection site pain scores (on a 100-point scale) were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following the last injection).10
Continue to: How the Atrigel system works
How the Atrigel system works. The Atrigel system was developed in the late 1980s and consists of a solution of a resorbable polymer in a biocompatible carrier.11 After in vivo administration (typically via subcutaneous injection), the polymer undergoes a phase change from a liquid to a formed implant (Figure 1). Being in liquid form, this system provides the advantage of placement by simple means, such as injection by syringes. The absorption rates of various polymers and the release rates for various drugs are tailored to the desired indication. Approved uses for Atrigel include the subgingival delivery of the antibiotic doxycycline for chronic adult periodontitis (approved September 1998), and the monthly subcutaneous injectable form of the anti-androgen leuprolide, which was approved in January 2002.8,12 Release periods up to 4 months have been achieved with Atrigel; 1 month is the most often desired release period. The biodegradable polymer used for RBP-7000 is designed to provide effective plasma drug levels during the first week of treatment, and sustained levels with a 1-month dosing interval. The small subcutaneous implant that is formed is gradually resorbed over the course of 1 month.
Pharmacokinetics. As with all LAI medications, the half-life with repeated dosing vastly exceeds that achieved with oral administration. Following oral administration, mean peak plasma levels of risperidone occur at 1 hour, and those for the active metabolite 9-OH risperidone occur at 3 hours.13 Oral risperidone has a mean half-life of 3 hours, while the active metabolite 9-OH risperidone has a mean half-life of 21 hours.14 Due to its longer half-life, the metabolite comprises 83% of the active drug levels at steady state.14 Although risperidone is susceptible to interactions via cytochrome P450 (CYP) inhibitors and inducers, particularly CYP2D6 (Table 210), the pharmacokinetics of the combined total of risperidone and 9-OH risperidone levels (deemed the active moiety) are similar in CYP2D6 extensive and poor metabolizers, with an overall mean elimination half-life of approximately 20 hours.13
The kinetics for RBP-7000 are markedly different than those for oral risperidone (Figure 215). After a single subcutaneous injection, RBP-7000 shows 2 absorption peaks for risperidone. The first lower peak occurs with a Tmax of 4 to 6 hours due to initial release of risperidone during the implant formation process; a second risperidone peak occurs after 10 to 14 days and is associated with slow release from the subcutaneous depot.9,16,17 For both 9-OH risperidone levels and the total active moiety (risperidone plus 9-OH risperidone levels), the median Tmax of the first peak ranges from 4 to 48 hours and the second peak ranges from 7 to 11 days. Following a single subcutaneous injection of RBP-7000, the apparent terminal half-life of risperidone ranges from 9 to 11 days, on average. The mean apparent terminal half-life of the active moiety ranges from 8 to 9 days.9,16,17 Based on population pharmacokinetic modeling, the 90 mg and 120 mg doses of RBP-7000 are estimated to provide drug exposure equivalent to 3 mg/d and 4 mg/d of oral risperidone, respectively.9,16,17
Continue to: Efficacy of RBP-7000
Efficacy of RBP-7000 was established in an 8-week, double-blind, placebo-controlled trial of adult patients experiencing an acute exacerbation of schizophrenia (age 18 to 55).4 Eligible participants had:
- An acute exacerbation of schizophrenia that occurred ≤8 weeks before the screening visit and would have benefited from psychiatric hospitalization or continued hospitalization
- Positive and Negative Syndrome Scale (PANSS) total score between 80 and 120 at visit 1 and a score of >4 on at least 2 of the following 4 items: hallucinatory behavior, delusions, conceptual disorganization, or suspiciousness/persecution
- The diagnosis of acute exacerbation of schizophrenia and PANSS total score were confirmed through an independent video-conference interview conducted by an experienced rater.
Participants were excluded if they:
- Experienced a ≥20% improvement in PANSS total score between the initial screening visit and the first injection
- had been treated at any time with clozapine for treatment-resistant schizophrenia
- had met DSM-IV-TR criteria for substance dependence (with the exception of nicotine or caffeine) before screening.
During the initial screening visit, participants received a 0.25-mg tablet of oral risperidone on 2 consecutive days to assess the tolerability of risperidone.
Outcome. Participants were randomized in a 1:1:1 manner to placebo (n = 112) or 1 of 2 monthly doses of RBP-7000: 90 mg (n = 111) or 120 mg (n = 114). Using the least squares means of repeated-measures changes from baseline in PANSS total scores, there was a significant improvement in the difference in PANSS total scores from baseline to the end of the study compared with placebo: 90-mg RBP-7000, -6.148 points (95% confidence interval [CI], -9.982 to -2.314, P = .0004); 120-mg RBP-7000, -7.237 points (95% CI, -11.045 to -3.429, P < .0001). The absolute change from baseline in PANSS total score was -15.367 points for the 90-mg dose and -16.456 points for the 120-mg dose.4 Completion rates across all 3 arms were comparable: placebo 70.6%, RBP-7000 90 mg 77.6%, and RBP-7000 120 mg 71.4%.
Tolerability. In the 8-week phase III efficacy trial of RBP-7000, adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo were weight gain (placebo 3.4%, 90 mg 13.0%, 120 mg 12.8%) and sedation (placebo 0%, 90 mg 7.0%, 120 mg 7.7%).10 Compared with baseline, participants had a mean weight gain at the end of the study of 2.83 kg in the placebo group, 5.15 kg in the 90-mg RBP-7000 group, and 4.69 kg in the 120-mg RBP-7000 group. There were no clinically significant differences at study endpoint in glucose and lipid parameters. Consistent with the known effects of risperidone, there were increases in mean prolactin levels during the 8-week study, the effects of which were greater for women. For men, mean prolactin levels from baseline to study end were: placebo: 9.8 ± 7.9 vs 9.9 ± 8.0 ng/mL; 90 mg: 8.9 ± 6.9 vs 22.4 ± 11.2 ng/mL; and 120 mg: 8.2 ± 5.2 vs 31.3 ± 14.8 ng/mL. For women, mean prolactin levels from baseline to study end were: placebo: 12.8 ± 11.7 vs 10.4 ± 8.0 ng/mL; 90 mg: 7.7 ± 5.3 vs 60.3 ± 46.9 ng/mL; and 120 mg: 10.9 ± 8.6 vs 85.5 ± 55.1 ng/mL. In the pivotal study, discontinuations due to adverse events were low across all treatment groups: 2.5% for placebo vs 0% for 90 mg and 1.7% for 120 mg.4 There was no single adverse reaction leading to discontinuation that occurred at a rate of ≥2% and greater than placebo in patients treated with RBP-7000.10 There were no clinically relevant differences in mean changes from baseline in corrected QT, QRS, and PR intervals, and in heart rate. Similarly, in the 12-month, long-term safety study, there were no clinically relevant changes in mean electrocardiography interval values from baseline to post-dose assessments.10
Using a 100-point visual analog scale (VAS), injection site pain scores 1 minute after the first dose decreased from a mean of 27 to the range of 3 to 7 for scores obtained 30 to 60 minutes post-dose. In the 12-month long-term safety study, 1-minute post-dose injection site pain VAS scores were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following last injection).10
Clinical considerations
Unique properties. RBP-7000 uses the established Atrigel system to provide effective antipsychotic levels in the first week of treatment, without the need for bridging oral coverage or a second loading injection. The abdominal subcutaneous injection volume is relatively small (0.6 mL or 0.8 mL).
Why Rx? The reasons to prescribe RBP-7000 for adult patients with schizophrenia include:
- no oral coverage required at the initiation of treatment
- effective plasma active moiety levels are seen within the first week without the need for a second loading injection
- monthly injection schedule.
Dosing. The recommended dosage of RBP-7000 is 90 mg or 120 mg once monthly, equivalent to 3 mg/d or 4 mg/d of oral risperidone, respectively. Oral risperidone tolerability should be established before the first injection. No oral risperidone coverage is required. RBP-7000 has not been studied in patients with renal or hepatic impairment and should be used with caution in these patients. Prior to initiating treatment in these patients, it is advised to carefully titrate up to at least 3 mg/d of oral risperidone. If a patient can tolerate 3 mg/d of oral risperidone and is psychiatrically stable, then the 90-mg dose of RBP-7000 can be considered.10
Contraindications. The only contraindications for RBP-7000 are known hypersensitivity to risperidone, paliperidone (9-OH risperidone), or other components of the injection.
Bottom Line
RBP-7000 (Perseris) is the second long-acting injectable (LAI) form of risperidone approved in the U.S. Unlike risperidone microspheres (Consta), RBP-7000 does not require any oral risperidone coverage at the beginning of therapy, provides effective drug levels within the first week of treatment with a single injection, and uses a monthly dosing interval. RBP-7000 does not require loading upon initiation. The monthly injection is <1 mL, is administered in abdominal subcutaneous tissue, and uses the Atrigel system.
Related Resource
- Carpenter J, Wong KK. Long-acting injectable antipsychotics: What to do about missed doses. Current Psychiatry. 2018;17(7):10-12,14-19,56.
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Carbatrol, Tegretol
Doxycycline • Atridox
Leuprolide acetate injectable suspension • Eligard
Paliperidone palmitate • Invega Sustenna
Risperidone • Risperdal
Risperidone extended-release injectable suspension • Perseris
Risperidone long-acting injection • Risperdal Consta
Oral antipsychotic nonadherence is a significant contributor to relapse in patients with schizophrenia spectrum disorders. Long-acting injectable (LAI) antipsychotics have been developed to provide sustained antipsychotic exposure, with evidence that use of LAIs significantly reduces hospitalization rates.1 One limiting factor in transitioning patients to certain LAIs is the need for prolonged oral coverage at the onset of treatment for agents that cannot be loaded. Nonadherence with this bridging oral therapy places the patient at risk for symptom exacerbation until effective antipsychotic plasma levels are achieved from the LAI.2 Although risperidone is one of the more widely used antipsychotics for treating schizophrenia, until recently the only available LAI preparation, risperidone microspheres (Risperdal Consta), required 3 weeks of oral coverage upon initiation.3
Clinical implications
Oral medication nonadherence remains a significant public health issue for patients with schizophrenia, with an estimated 50% of patients failing to achieve 80% adherence even when enrolled in clinical trials specifically designed to track adherence.5 Although LAI atypical antipsychotics have been available since the approval of Risperdal Consta, the LAI form of risperidone, and both LAI forms of aripiprazole, were not designed to be loaded. A 1-day initiation regimen for aripiprazole lauroxil has been developed to avoid the need for 3 weeks of oral medication coverage,6,7 but aripiprazole monohydrate and risperidone microspheres mandate oral bridging of 2 and 3 weeks, respectively.2 Because one of the primary indications for LAI antipsychotic therapy is oral medication nonadherence, this prolonged period of oral coverage creates a risk for symptom exacerbation when the bridging period occurs outside of a controlled setting, as is common when patients are discharged from inpatient hospitalization.
One solution to this problem has its antecedents in the development of the Atrigel biodegradable injectable polymer, which was designed to deliver prolonged medication exposure after subcutaneous injection.8 This biodegradable polymer drug delivery system suspends and dissolves the medication of interest (in this case, risperidone) in a poly DL-lactide-coglycolide gel and its biocompatible carrier.9 The viscous liquid undergoes a phase transition upon contact with tissue fluids after subcutaneous injection, resulting in an implant that releases risperidone in a controlled manner as it is resorbed. Importantly, the kinetic parameters of RBP-7000 are such that effective drug levels are seen within the first week without the need for oral coverage.10
Use in adults with schizophrenia. After establishing tolerability with oral risperidone, the recommended doses are 90 mg or 120 mg monthly, which correspond to oral daily risperidone doses of 3 mg or 4 mg. RBP-7000 must be administered as a subcutaneous abdominal injection by a health care professional. It is recommended that the patient be in the supine position for the injection and that the injection sites be rotated monthly among 4 quadrants in the abdominal region. The injection volumes for the 90 mg and 120 mg doses are 0.6 mL and 0.8 mL, respectively.10 As the gel implant becomes firmer, the patient will notice a lump for several weeks that will decrease in size over time. Patients should be advised not to rub or massage the injection site, and to be aware of the placement of any belts or clothing with waistbands.10
Pharmacologic profile, adverse reactions
Risperidone is an atypical antipsychotic that has been commercially available in the U.S. since December 29, 1993, and its adverse effect profile is well characterized. The most common adverse effects associated with risperidone include those related to dopamine D2 antagonism, metabolic adverse effects, and an increase in serum prolactin. In the 12-month long-term safety study of RBP-7000, 1-minute post-dose injection site pain scores (on a 100-point scale) were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following the last injection).10
Continue to: How the Atrigel system works
How the Atrigel system works. The Atrigel system was developed in the late 1980s and consists of a solution of a resorbable polymer in a biocompatible carrier.11 After in vivo administration (typically via subcutaneous injection), the polymer undergoes a phase change from a liquid to a formed implant (Figure 1). Being in liquid form, this system provides the advantage of placement by simple means, such as injection by syringes. The absorption rates of various polymers and the release rates for various drugs are tailored to the desired indication. Approved uses for Atrigel include the subgingival delivery of the antibiotic doxycycline for chronic adult periodontitis (approved September 1998), and the monthly subcutaneous injectable form of the anti-androgen leuprolide, which was approved in January 2002.8,12 Release periods up to 4 months have been achieved with Atrigel; 1 month is the most often desired release period. The biodegradable polymer used for RBP-7000 is designed to provide effective plasma drug levels during the first week of treatment, and sustained levels with a 1-month dosing interval. The small subcutaneous implant that is formed is gradually resorbed over the course of 1 month.
Pharmacokinetics. As with all LAI medications, the half-life with repeated dosing vastly exceeds that achieved with oral administration. Following oral administration, mean peak plasma levels of risperidone occur at 1 hour, and those for the active metabolite 9-OH risperidone occur at 3 hours.13 Oral risperidone has a mean half-life of 3 hours, while the active metabolite 9-OH risperidone has a mean half-life of 21 hours.14 Due to its longer half-life, the metabolite comprises 83% of the active drug levels at steady state.14 Although risperidone is susceptible to interactions via cytochrome P450 (CYP) inhibitors and inducers, particularly CYP2D6 (Table 210), the pharmacokinetics of the combined total of risperidone and 9-OH risperidone levels (deemed the active moiety) are similar in CYP2D6 extensive and poor metabolizers, with an overall mean elimination half-life of approximately 20 hours.13
The kinetics for RBP-7000 are markedly different than those for oral risperidone (Figure 215). After a single subcutaneous injection, RBP-7000 shows 2 absorption peaks for risperidone. The first lower peak occurs with a Tmax of 4 to 6 hours due to initial release of risperidone during the implant formation process; a second risperidone peak occurs after 10 to 14 days and is associated with slow release from the subcutaneous depot.9,16,17 For both 9-OH risperidone levels and the total active moiety (risperidone plus 9-OH risperidone levels), the median Tmax of the first peak ranges from 4 to 48 hours and the second peak ranges from 7 to 11 days. Following a single subcutaneous injection of RBP-7000, the apparent terminal half-life of risperidone ranges from 9 to 11 days, on average. The mean apparent terminal half-life of the active moiety ranges from 8 to 9 days.9,16,17 Based on population pharmacokinetic modeling, the 90 mg and 120 mg doses of RBP-7000 are estimated to provide drug exposure equivalent to 3 mg/d and 4 mg/d of oral risperidone, respectively.9,16,17
Continue to: Efficacy of RBP-7000
Efficacy of RBP-7000 was established in an 8-week, double-blind, placebo-controlled trial of adult patients experiencing an acute exacerbation of schizophrenia (age 18 to 55).4 Eligible participants had:
- An acute exacerbation of schizophrenia that occurred ≤8 weeks before the screening visit and would have benefited from psychiatric hospitalization or continued hospitalization
- Positive and Negative Syndrome Scale (PANSS) total score between 80 and 120 at visit 1 and a score of >4 on at least 2 of the following 4 items: hallucinatory behavior, delusions, conceptual disorganization, or suspiciousness/persecution
- The diagnosis of acute exacerbation of schizophrenia and PANSS total score were confirmed through an independent video-conference interview conducted by an experienced rater.
Participants were excluded if they:
- Experienced a ≥20% improvement in PANSS total score between the initial screening visit and the first injection
- had been treated at any time with clozapine for treatment-resistant schizophrenia
- had met DSM-IV-TR criteria for substance dependence (with the exception of nicotine or caffeine) before screening.
During the initial screening visit, participants received a 0.25-mg tablet of oral risperidone on 2 consecutive days to assess the tolerability of risperidone.
Outcome. Participants were randomized in a 1:1:1 manner to placebo (n = 112) or 1 of 2 monthly doses of RBP-7000: 90 mg (n = 111) or 120 mg (n = 114). Using the least squares means of repeated-measures changes from baseline in PANSS total scores, there was a significant improvement in the difference in PANSS total scores from baseline to the end of the study compared with placebo: 90-mg RBP-7000, -6.148 points (95% confidence interval [CI], -9.982 to -2.314, P = .0004); 120-mg RBP-7000, -7.237 points (95% CI, -11.045 to -3.429, P < .0001). The absolute change from baseline in PANSS total score was -15.367 points for the 90-mg dose and -16.456 points for the 120-mg dose.4 Completion rates across all 3 arms were comparable: placebo 70.6%, RBP-7000 90 mg 77.6%, and RBP-7000 120 mg 71.4%.
Tolerability. In the 8-week phase III efficacy trial of RBP-7000, adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo were weight gain (placebo 3.4%, 90 mg 13.0%, 120 mg 12.8%) and sedation (placebo 0%, 90 mg 7.0%, 120 mg 7.7%).10 Compared with baseline, participants had a mean weight gain at the end of the study of 2.83 kg in the placebo group, 5.15 kg in the 90-mg RBP-7000 group, and 4.69 kg in the 120-mg RBP-7000 group. There were no clinically significant differences at study endpoint in glucose and lipid parameters. Consistent with the known effects of risperidone, there were increases in mean prolactin levels during the 8-week study, the effects of which were greater for women. For men, mean prolactin levels from baseline to study end were: placebo: 9.8 ± 7.9 vs 9.9 ± 8.0 ng/mL; 90 mg: 8.9 ± 6.9 vs 22.4 ± 11.2 ng/mL; and 120 mg: 8.2 ± 5.2 vs 31.3 ± 14.8 ng/mL. For women, mean prolactin levels from baseline to study end were: placebo: 12.8 ± 11.7 vs 10.4 ± 8.0 ng/mL; 90 mg: 7.7 ± 5.3 vs 60.3 ± 46.9 ng/mL; and 120 mg: 10.9 ± 8.6 vs 85.5 ± 55.1 ng/mL. In the pivotal study, discontinuations due to adverse events were low across all treatment groups: 2.5% for placebo vs 0% for 90 mg and 1.7% for 120 mg.4 There was no single adverse reaction leading to discontinuation that occurred at a rate of ≥2% and greater than placebo in patients treated with RBP-7000.10 There were no clinically relevant differences in mean changes from baseline in corrected QT, QRS, and PR intervals, and in heart rate. Similarly, in the 12-month, long-term safety study, there were no clinically relevant changes in mean electrocardiography interval values from baseline to post-dose assessments.10
Using a 100-point visual analog scale (VAS), injection site pain scores 1 minute after the first dose decreased from a mean of 27 to the range of 3 to 7 for scores obtained 30 to 60 minutes post-dose. In the 12-month long-term safety study, 1-minute post-dose injection site pain VAS scores were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following last injection).10
Clinical considerations
Unique properties. RBP-7000 uses the established Atrigel system to provide effective antipsychotic levels in the first week of treatment, without the need for bridging oral coverage or a second loading injection. The abdominal subcutaneous injection volume is relatively small (0.6 mL or 0.8 mL).
Why Rx? The reasons to prescribe RBP-7000 for adult patients with schizophrenia include:
- no oral coverage required at the initiation of treatment
- effective plasma active moiety levels are seen within the first week without the need for a second loading injection
- monthly injection schedule.
Dosing. The recommended dosage of RBP-7000 is 90 mg or 120 mg once monthly, equivalent to 3 mg/d or 4 mg/d of oral risperidone, respectively. Oral risperidone tolerability should be established before the first injection. No oral risperidone coverage is required. RBP-7000 has not been studied in patients with renal or hepatic impairment and should be used with caution in these patients. Prior to initiating treatment in these patients, it is advised to carefully titrate up to at least 3 mg/d of oral risperidone. If a patient can tolerate 3 mg/d of oral risperidone and is psychiatrically stable, then the 90-mg dose of RBP-7000 can be considered.10
Contraindications. The only contraindications for RBP-7000 are known hypersensitivity to risperidone, paliperidone (9-OH risperidone), or other components of the injection.
Bottom Line
RBP-7000 (Perseris) is the second long-acting injectable (LAI) form of risperidone approved in the U.S. Unlike risperidone microspheres (Consta), RBP-7000 does not require any oral risperidone coverage at the beginning of therapy, provides effective drug levels within the first week of treatment with a single injection, and uses a monthly dosing interval. RBP-7000 does not require loading upon initiation. The monthly injection is <1 mL, is administered in abdominal subcutaneous tissue, and uses the Atrigel system.
Related Resource
- Carpenter J, Wong KK. Long-acting injectable antipsychotics: What to do about missed doses. Current Psychiatry. 2018;17(7):10-12,14-19,56.
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Carbatrol, Tegretol
Doxycycline • Atridox
Leuprolide acetate injectable suspension • Eligard
Paliperidone palmitate • Invega Sustenna
Risperidone • Risperdal
Risperidone extended-release injectable suspension • Perseris
Risperidone long-acting injection • Risperdal Consta
1. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
2. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
3. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
4. Nasser AF, Henderson DC, Fava M, et al. Efficacy, safety, and tolerability of RBP-7000 once-monthly risperidone for the treatment of acute schizophrenia: an 8-week, randomized, double-blind, placebo-controlled, multicenter phase 3 study. J Clin Psychopharmacol. 2016;36(2):130-140.
5. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
6. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
7. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
8. Southard GL, Dunn RL, Garrett S. The drug delivery and biomaterial attributes of the ATRIGEL technology in the treatment of periodontal disease. Expert Opin Investig Drugs. 1998;7(9):1483-1491.
9. Gomeni R, Heidbreder C, Fudala PJ, Nasser AF. A model-based approach to characterize the population pharmacokinetics and the relationship between the pharmacokinetic and safety profiles of RBP-7000, a new, long-acting, sustained-released formulation of risperidone. J Clin Pharmacol. 2013;53(10):1010-1019.
10. Perseris [package insert]. North Chesterfield, VA: Indivior Inc; 2018.
11. Malik K, Singh I, Nagpal M, et al. Atrigel: a potential parenteral controlled drug delivery system. Der Pharmacia Sinica. 2010;1(1):74-81.
12. Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003;61(2 Suppl 1):25-31.
13. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
14. de Leon J, Wynn G, Sandson NB. The pharmacokinetics of paliperidone versus risperidone. Psychosomatics. 2010;51(1):80-88.
15. Ivaturi V, Gopalakrishnan M, Gobburu JVS, et al. Exposure-response analysis after subcutaneous administration of RBP-7000, a once-a-month long-acting Atrigel formulation of risperidone. Br J Clin Pharmacol. 2017;83(7):1476-1498.
16. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetics and prediction of dopamine D2 receptor occupancy after multiple doses of RBP-7000, a new sustained-release formulation of risperidone, in schizophrenia patients on stable oral risperidone treatment. Clin Pharmacokinet. 2014;53(6):533-543.
17. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetic modeling and simulation to guide dose selection for RBP-7000, a new sustained-release formulation of risperidone. J Clin Pharmacol. 2015;55(1):93-103.
1. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
2. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
3. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
4. Nasser AF, Henderson DC, Fava M, et al. Efficacy, safety, and tolerability of RBP-7000 once-monthly risperidone for the treatment of acute schizophrenia: an 8-week, randomized, double-blind, placebo-controlled, multicenter phase 3 study. J Clin Psychopharmacol. 2016;36(2):130-140.
5. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
6. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
7. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
8. Southard GL, Dunn RL, Garrett S. The drug delivery and biomaterial attributes of the ATRIGEL technology in the treatment of periodontal disease. Expert Opin Investig Drugs. 1998;7(9):1483-1491.
9. Gomeni R, Heidbreder C, Fudala PJ, Nasser AF. A model-based approach to characterize the population pharmacokinetics and the relationship between the pharmacokinetic and safety profiles of RBP-7000, a new, long-acting, sustained-released formulation of risperidone. J Clin Pharmacol. 2013;53(10):1010-1019.
10. Perseris [package insert]. North Chesterfield, VA: Indivior Inc; 2018.
11. Malik K, Singh I, Nagpal M, et al. Atrigel: a potential parenteral controlled drug delivery system. Der Pharmacia Sinica. 2010;1(1):74-81.
12. Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003;61(2 Suppl 1):25-31.
13. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
14. de Leon J, Wynn G, Sandson NB. The pharmacokinetics of paliperidone versus risperidone. Psychosomatics. 2010;51(1):80-88.
15. Ivaturi V, Gopalakrishnan M, Gobburu JVS, et al. Exposure-response analysis after subcutaneous administration of RBP-7000, a once-a-month long-acting Atrigel formulation of risperidone. Br J Clin Pharmacol. 2017;83(7):1476-1498.
16. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetics and prediction of dopamine D2 receptor occupancy after multiple doses of RBP-7000, a new sustained-release formulation of risperidone, in schizophrenia patients on stable oral risperidone treatment. Clin Pharmacokinet. 2014;53(6):533-543.
17. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetic modeling and simulation to guide dose selection for RBP-7000, a new sustained-release formulation of risperidone. J Clin Pharmacol. 2015;55(1):93-103.
Catatonia: Recognition, management, and prevention of complications
Mr. W, age 50, who has been diagnosed with hypertension and catatonia associated with schizophrenia, is brought to the emergency department by his case manager for evaluation of increasing disorganization, inability to function, and nonadherence to medications. He has not been bathing, eating, or drinking. During the admission interview, he is mute, and is noted to have purposeless activity, alternating between rocking from leg to leg to pacing in circles. At times Mr. W holds a rigid, prayer-type posture with his arms. Negativism is present, primarily opposition to interviewer requests.
Previously stable on
On the inpatient psychiatry unit, Mr. W continues to be mute, staying in bed except to use the bathroom. He refuses all food and fluids. The team initiates subcutaneous

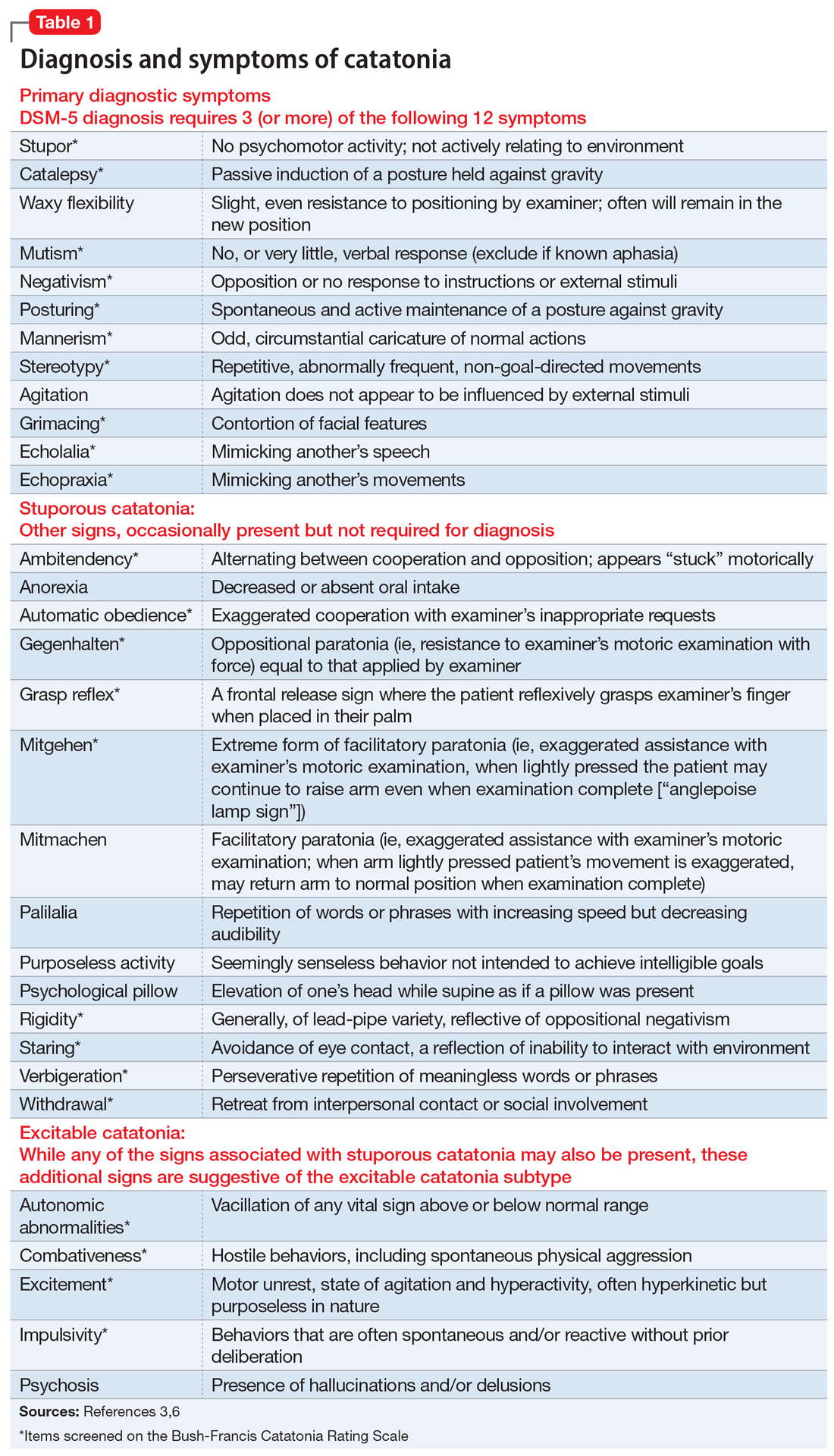
Continue to: Medical complications can be fatal
Medical complications can be fatal

Treatment usually starts with lorazepam
Benzodiazepines are a first-line option for the management of catatonia.2,5 Controversy exists as to effectiveness of different routes of administration. Generally, IV lorazepam is preferred due to its ease of administration, fast onset, and longer duration of action.1 Some inpatient psychiatric units are unable to administer IV benzodiazepines; in these scenarios, IM administration is preferred to oral benzodiazepines.
The initial lorazepam challenge dose should be 2 mg. A positive response to the lorazepam challenge often confirms the catatonia diagnosis.2,7 This challenge should be followed by maintenance doses ranging from 6 to 8 mg/d in divided doses (3 or 4 times a day). Higher doses (up to 24 mg/d) are sometimes used.2,5,8 A recent case report described catatonia remission using lorazepam, 28 mg/d, after unsuccessful ECT.9 The lorazepam dose prior to ECT was 8 mg/d.9 Response is usually seen within 3 to 7 days of an adequate dose.2,8 Parenteral lorazepam typically is continued for several days before converting to oral lorazepam.1 Approximately 70% to 80% of patients with catatonia will show improvement in symptoms with lorazepam.2,7,8
The optimal duration of benzodiazepine treatment is unclear.2 In some cases, once remission of the underlying illness is achieved, benzodiazepines are discontinued.2 However, in other cases, symptoms of catatonia may emerge when lorazepam is tapered, therefore suggesting the need for a longer duration of treatment.2 Despite this high rate of improvement, many patients ultimately receive ECT due to unsustained response or to prevent future episodes of catatonia.
A recent review of 60 Turkish patients with catatonia found 91.7% (n = 55) received oral lorazepam (up to 15 mg/d) as the first-line therapy.7 Improvement was seen in 23.7% (n = 13) of patients treated with lorazepam, yet 70% (n = 42) showed either no response or partial response, and ultimately received ECT in combination with lorazepam.7 The lower improvement rate seen in this review may be secondary to the use of oral lorazepam instead of parenteral, or may highlight the frequency in which patients ultimately go on to receive ECT.
Continue to: ECT
ECT. If high doses of benzodiazepines are not effective within 48 to 72 hours, ECT should be considered.1,7 ECT should be considered sooner for patients with life-threatening catatonia or those who present with excited features or malignant catatonia.1,2,7 In patients with catatonia, ECT response rates range from 80% to 100%.2,7 Unal et al7 reported a 100% response rate if ECT was used as the first-line treatment (n = 5), and a 92.9% (n = 39) response rate after adding ECT to lorazepam. Lorazepam may interfere with the seizure threshold, but if indicated, this medication can be continued.2 A minimum of 6 ECT treatments are suggested; however, as many as 20 treatments have been needed.1 Mr. W required a total of 18 ECT treatments. In some cases, maintenance ECT may be required.2
Antipsychotics. Discontinuation of antipsychotics is generally encouraged in patients presenting with catatonia.2,7,8 Antipsychotics carry a risk of potentially worsening catatonia, conversion to malignant catatonia, or precipitation of NMS; therefore, carefully weigh the risks vs benefits.1,2 If catatonia is secondary to psychosis, as in Mr. W’s case, antipsychotics may be considered once catatonia improves.2 If an antipsychotic is warranted, consider aripiprazole (because of its D2 partial agonist activity) or low-dose olanzapine.1,2 If catatonia is secondary to clozapine withdrawal, the initial therapy should be clozapine re-initiation.1 Although high-potency agents, such as haloperidol and risperidone, typically are not preferred, risperidone was restarted for Mr. W because of his history of response to and tolerability of this medication during a previous catatonic episode.
Other treatments. In a recent review, Beach et al1 described the use of additional agents, mostly in a small number of positive case reports, for managing catatonia. These included:
- zolpidem (zolpidem 10 mg as a challenge test, and doses of ≤40 mg/d)
- the N-methyl-
D -aspartic acid antagonists amantadine (100 to 600 mg/d) or memantine (5 to 20 mg/d) - carbidopa/levodopa
- methylphenidate
- antiepileptics (eg, carbamazepine, topiramate, and divalproex sodium)
- anticholinergics.1,2
Lithium has been used in attempts to prevent recurrent catatonia with limited success.2 There are also a few reports of using transcranial magnetic stimulation (TMS) to manage catatonia.1
Beach et al1 proposed a treatment algorithm in which IV lorazepam (Step 1) and ECT (Step 2) remain the preferred treatments. Next, for Step 3 consider a glutamate antagonist (amantadine or memantine), followed by an antiepileptic (Step 4), and lastly an atypical antipsychotic (aripiprazole, olanzapine, or clozapine) in combination with lorazepam (Step 5).
When indicated, don’t delay ECT
Initial management of catatonia is with a benzodiazepine challenge. Ultimately, the gold-standard treatment of catatonia that does not improve with benzodiazepines is ECT, and ECT should be implemented as soon as it is clear that pharmacotherapy is less than fully effective. Consider ECT initially in life-threatening cases and for patients with malignant catatonia. Although additional agents and TMS have been explored, these should be reserved for patients who fail to respond to, or who are not candidates for, benzodiazepines or ECT.
CASE CONTINUED
After 5 ECT treatments, Mr. W says a few words, but he communicates primarily with gestures (primarily waving people away). After 10 to 12 ECT treatments, Mr. W becomes more interactive and conversant, and his nutrition improves; however, he still exhibits symptoms of catatonia and is not at baseline. He undergoes a total of 18 ECT treatments. Antipsychotics were initially discontinued; however, given Mr. W’s improvement with ECT and the presence of auditory hallucinations, oral risperidone is restarted and titrated to 2 mg, 2 times a day, and he is transitioned back to paliperidone palmitate before he is discharged. Lorazepam is tapered and discontinued. Mr. W is discharged back to his nursing home and is interactive (laughing and joking with family) and attending to his activities of daily living. Unfortunately, Mr. W did not followup with the recommendation for maintenance ECT, and adherence to paliperidone palmitate injections is unknown. Mr. W presented to our facility again 6 months later with symptoms of catatonia and ultimately transferred to a state hospital.
Related Resources
- Fink M, Taylor MA. Catatonia: A clinician’s guide to diagnosis and treatment. New York, NY: Cambridge University Press; 2006. • Carroll BT, Spiegel DR. Catatonia on the consultation liaison service and other clinical settings. Hauppauge, NY: Nova Science Pub Inc.; 2016.
- Benarous X, Raffin M, Ferrafiat V, et al. Catatonia in children and adolescents: new perspectives. Schizophr Res. 2018;200:56-67.
- Malignant Hyperthermia Association of the United States. What is NMSIS? http://www.mhaus.org/nmsis/about-us/ what-is-nmsis/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Asenapine • Saphris
Carbamazepine • Carbatrol, Tegretol
Carbidopa/Levodopa • Sinemet
Citalopram • Celexa
Clozapine • Clozaril
Divalproex Sodium • Depakote
Enoxaparin • Lovenox
Fluoxetine • Prozac
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Lurasidone • Latuda
Memantine • Namenda
Methylphenidate • Concerta, Ritalin
Mirtazapine • Remeron
Olanzapine • Zyprexa
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone long-acting injection • Risperdal Consta
Topiramate • Topamax
Zolpidem • Ambien
1. Beach SR, Gomez-Bernal F, Huffman JC, et al. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1-19.
2. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:1-6.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome: focus on treatment and rechallenge. Ann Pharmacother. 2016;50(11):973-981.
5. Ohi K, Kuwata A, Shimada T, et al. Response to benzodiazepines and clinical course in malignant catatonia associated with schizophrenia: a case report. Medicine (Baltimore). 2017;96(16):e6566. doi: 10.1097/MD.0000000000006566.
6. Bush G, Fink M, Petrides G, et al. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
7. Unal A, Altindag A, Demir B, et al. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J ECT. 2017;33(4):290-293.
8. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1182-1183.
9. van der Markt A, Heller HM, van Exel E. A woman with catatonia, what to do after ECT fails: a case report. J ECT. 2016;32(3):e6-7. doi: 10.1097/YCT.0000000000000290.
Mr. W, age 50, who has been diagnosed with hypertension and catatonia associated with schizophrenia, is brought to the emergency department by his case manager for evaluation of increasing disorganization, inability to function, and nonadherence to medications. He has not been bathing, eating, or drinking. During the admission interview, he is mute, and is noted to have purposeless activity, alternating between rocking from leg to leg to pacing in circles. At times Mr. W holds a rigid, prayer-type posture with his arms. Negativism is present, primarily opposition to interviewer requests.
Previously stable on
On the inpatient psychiatry unit, Mr. W continues to be mute, staying in bed except to use the bathroom. He refuses all food and fluids. The team initiates subcutaneous
Continue to: Medical complications can be fatal
Medical complications can be fatal
Treatment usually starts with lorazepam
Benzodiazepines are a first-line option for the management of catatonia.2,5 Controversy exists as to effectiveness of different routes of administration. Generally, IV lorazepam is preferred due to its ease of administration, fast onset, and longer duration of action.1 Some inpatient psychiatric units are unable to administer IV benzodiazepines; in these scenarios, IM administration is preferred to oral benzodiazepines.
The initial lorazepam challenge dose should be 2 mg. A positive response to the lorazepam challenge often confirms the catatonia diagnosis.2,7 This challenge should be followed by maintenance doses ranging from 6 to 8 mg/d in divided doses (3 or 4 times a day). Higher doses (up to 24 mg/d) are sometimes used.2,5,8 A recent case report described catatonia remission using lorazepam, 28 mg/d, after unsuccessful ECT.9 The lorazepam dose prior to ECT was 8 mg/d.9 Response is usually seen within 3 to 7 days of an adequate dose.2,8 Parenteral lorazepam typically is continued for several days before converting to oral lorazepam.1 Approximately 70% to 80% of patients with catatonia will show improvement in symptoms with lorazepam.2,7,8
The optimal duration of benzodiazepine treatment is unclear.2 In some cases, once remission of the underlying illness is achieved, benzodiazepines are discontinued.2 However, in other cases, symptoms of catatonia may emerge when lorazepam is tapered, therefore suggesting the need for a longer duration of treatment.2 Despite this high rate of improvement, many patients ultimately receive ECT due to unsustained response or to prevent future episodes of catatonia.
A recent review of 60 Turkish patients with catatonia found 91.7% (n = 55) received oral lorazepam (up to 15 mg/d) as the first-line therapy.7 Improvement was seen in 23.7% (n = 13) of patients treated with lorazepam, yet 70% (n = 42) showed either no response or partial response, and ultimately received ECT in combination with lorazepam.7 The lower improvement rate seen in this review may be secondary to the use of oral lorazepam instead of parenteral, or may highlight the frequency in which patients ultimately go on to receive ECT.
Continue to: ECT
ECT. If high doses of benzodiazepines are not effective within 48 to 72 hours, ECT should be considered.1,7 ECT should be considered sooner for patients with life-threatening catatonia or those who present with excited features or malignant catatonia.1,2,7 In patients with catatonia, ECT response rates range from 80% to 100%.2,7 Unal et al7 reported a 100% response rate if ECT was used as the first-line treatment (n = 5), and a 92.9% (n = 39) response rate after adding ECT to lorazepam. Lorazepam may interfere with the seizure threshold, but if indicated, this medication can be continued.2 A minimum of 6 ECT treatments are suggested; however, as many as 20 treatments have been needed.1 Mr. W required a total of 18 ECT treatments. In some cases, maintenance ECT may be required.2
Antipsychotics. Discontinuation of antipsychotics is generally encouraged in patients presenting with catatonia.2,7,8 Antipsychotics carry a risk of potentially worsening catatonia, conversion to malignant catatonia, or precipitation of NMS; therefore, carefully weigh the risks vs benefits.1,2 If catatonia is secondary to psychosis, as in Mr. W’s case, antipsychotics may be considered once catatonia improves.2 If an antipsychotic is warranted, consider aripiprazole (because of its D2 partial agonist activity) or low-dose olanzapine.1,2 If catatonia is secondary to clozapine withdrawal, the initial therapy should be clozapine re-initiation.1 Although high-potency agents, such as haloperidol and risperidone, typically are not preferred, risperidone was restarted for Mr. W because of his history of response to and tolerability of this medication during a previous catatonic episode.
Other treatments. In a recent review, Beach et al1 described the use of additional agents, mostly in a small number of positive case reports, for managing catatonia. These included:
- zolpidem (zolpidem 10 mg as a challenge test, and doses of ≤40 mg/d)
- the N-methyl-
D -aspartic acid antagonists amantadine (100 to 600 mg/d) or memantine (5 to 20 mg/d) - carbidopa/levodopa
- methylphenidate
- antiepileptics (eg, carbamazepine, topiramate, and divalproex sodium)
- anticholinergics.1,2
Lithium has been used in attempts to prevent recurrent catatonia with limited success.2 There are also a few reports of using transcranial magnetic stimulation (TMS) to manage catatonia.1
Beach et al1 proposed a treatment algorithm in which IV lorazepam (Step 1) and ECT (Step 2) remain the preferred treatments. Next, for Step 3 consider a glutamate antagonist (amantadine or memantine), followed by an antiepileptic (Step 4), and lastly an atypical antipsychotic (aripiprazole, olanzapine, or clozapine) in combination with lorazepam (Step 5).
When indicated, don’t delay ECT
Initial management of catatonia is with a benzodiazepine challenge. Ultimately, the gold-standard treatment of catatonia that does not improve with benzodiazepines is ECT, and ECT should be implemented as soon as it is clear that pharmacotherapy is less than fully effective. Consider ECT initially in life-threatening cases and for patients with malignant catatonia. Although additional agents and TMS have been explored, these should be reserved for patients who fail to respond to, or who are not candidates for, benzodiazepines or ECT.
CASE CONTINUED
After 5 ECT treatments, Mr. W says a few words, but he communicates primarily with gestures (primarily waving people away). After 10 to 12 ECT treatments, Mr. W becomes more interactive and conversant, and his nutrition improves; however, he still exhibits symptoms of catatonia and is not at baseline. He undergoes a total of 18 ECT treatments. Antipsychotics were initially discontinued; however, given Mr. W’s improvement with ECT and the presence of auditory hallucinations, oral risperidone is restarted and titrated to 2 mg, 2 times a day, and he is transitioned back to paliperidone palmitate before he is discharged. Lorazepam is tapered and discontinued. Mr. W is discharged back to his nursing home and is interactive (laughing and joking with family) and attending to his activities of daily living. Unfortunately, Mr. W did not followup with the recommendation for maintenance ECT, and adherence to paliperidone palmitate injections is unknown. Mr. W presented to our facility again 6 months later with symptoms of catatonia and ultimately transferred to a state hospital.
Related Resources
- Fink M, Taylor MA. Catatonia: A clinician’s guide to diagnosis and treatment. New York, NY: Cambridge University Press; 2006. • Carroll BT, Spiegel DR. Catatonia on the consultation liaison service and other clinical settings. Hauppauge, NY: Nova Science Pub Inc.; 2016.
- Benarous X, Raffin M, Ferrafiat V, et al. Catatonia in children and adolescents: new perspectives. Schizophr Res. 2018;200:56-67.
- Malignant Hyperthermia Association of the United States. What is NMSIS? http://www.mhaus.org/nmsis/about-us/ what-is-nmsis/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Asenapine • Saphris
Carbamazepine • Carbatrol, Tegretol
Carbidopa/Levodopa • Sinemet
Citalopram • Celexa
Clozapine • Clozaril
Divalproex Sodium • Depakote
Enoxaparin • Lovenox
Fluoxetine • Prozac
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Lurasidone • Latuda
Memantine • Namenda
Methylphenidate • Concerta, Ritalin
Mirtazapine • Remeron
Olanzapine • Zyprexa
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone long-acting injection • Risperdal Consta
Topiramate • Topamax
Zolpidem • Ambien
Mr. W, age 50, who has been diagnosed with hypertension and catatonia associated with schizophrenia, is brought to the emergency department by his case manager for evaluation of increasing disorganization, inability to function, and nonadherence to medications. He has not been bathing, eating, or drinking. During the admission interview, he is mute, and is noted to have purposeless activity, alternating between rocking from leg to leg to pacing in circles. At times Mr. W holds a rigid, prayer-type posture with his arms. Negativism is present, primarily opposition to interviewer requests.
Previously stable on
On the inpatient psychiatry unit, Mr. W continues to be mute, staying in bed except to use the bathroom. He refuses all food and fluids. The team initiates subcutaneous
Continue to: Medical complications can be fatal
Medical complications can be fatal
Treatment usually starts with lorazepam
Benzodiazepines are a first-line option for the management of catatonia.2,5 Controversy exists as to effectiveness of different routes of administration. Generally, IV lorazepam is preferred due to its ease of administration, fast onset, and longer duration of action.1 Some inpatient psychiatric units are unable to administer IV benzodiazepines; in these scenarios, IM administration is preferred to oral benzodiazepines.
The initial lorazepam challenge dose should be 2 mg. A positive response to the lorazepam challenge often confirms the catatonia diagnosis.2,7 This challenge should be followed by maintenance doses ranging from 6 to 8 mg/d in divided doses (3 or 4 times a day). Higher doses (up to 24 mg/d) are sometimes used.2,5,8 A recent case report described catatonia remission using lorazepam, 28 mg/d, after unsuccessful ECT.9 The lorazepam dose prior to ECT was 8 mg/d.9 Response is usually seen within 3 to 7 days of an adequate dose.2,8 Parenteral lorazepam typically is continued for several days before converting to oral lorazepam.1 Approximately 70% to 80% of patients with catatonia will show improvement in symptoms with lorazepam.2,7,8
The optimal duration of benzodiazepine treatment is unclear.2 In some cases, once remission of the underlying illness is achieved, benzodiazepines are discontinued.2 However, in other cases, symptoms of catatonia may emerge when lorazepam is tapered, therefore suggesting the need for a longer duration of treatment.2 Despite this high rate of improvement, many patients ultimately receive ECT due to unsustained response or to prevent future episodes of catatonia.
A recent review of 60 Turkish patients with catatonia found 91.7% (n = 55) received oral lorazepam (up to 15 mg/d) as the first-line therapy.7 Improvement was seen in 23.7% (n = 13) of patients treated with lorazepam, yet 70% (n = 42) showed either no response or partial response, and ultimately received ECT in combination with lorazepam.7 The lower improvement rate seen in this review may be secondary to the use of oral lorazepam instead of parenteral, or may highlight the frequency in which patients ultimately go on to receive ECT.
Continue to: ECT
ECT. If high doses of benzodiazepines are not effective within 48 to 72 hours, ECT should be considered.1,7 ECT should be considered sooner for patients with life-threatening catatonia or those who present with excited features or malignant catatonia.1,2,7 In patients with catatonia, ECT response rates range from 80% to 100%.2,7 Unal et al7 reported a 100% response rate if ECT was used as the first-line treatment (n = 5), and a 92.9% (n = 39) response rate after adding ECT to lorazepam. Lorazepam may interfere with the seizure threshold, but if indicated, this medication can be continued.2 A minimum of 6 ECT treatments are suggested; however, as many as 20 treatments have been needed.1 Mr. W required a total of 18 ECT treatments. In some cases, maintenance ECT may be required.2
Antipsychotics. Discontinuation of antipsychotics is generally encouraged in patients presenting with catatonia.2,7,8 Antipsychotics carry a risk of potentially worsening catatonia, conversion to malignant catatonia, or precipitation of NMS; therefore, carefully weigh the risks vs benefits.1,2 If catatonia is secondary to psychosis, as in Mr. W’s case, antipsychotics may be considered once catatonia improves.2 If an antipsychotic is warranted, consider aripiprazole (because of its D2 partial agonist activity) or low-dose olanzapine.1,2 If catatonia is secondary to clozapine withdrawal, the initial therapy should be clozapine re-initiation.1 Although high-potency agents, such as haloperidol and risperidone, typically are not preferred, risperidone was restarted for Mr. W because of his history of response to and tolerability of this medication during a previous catatonic episode.
Other treatments. In a recent review, Beach et al1 described the use of additional agents, mostly in a small number of positive case reports, for managing catatonia. These included:
- zolpidem (zolpidem 10 mg as a challenge test, and doses of ≤40 mg/d)
- the N-methyl-
D -aspartic acid antagonists amantadine (100 to 600 mg/d) or memantine (5 to 20 mg/d) - carbidopa/levodopa
- methylphenidate
- antiepileptics (eg, carbamazepine, topiramate, and divalproex sodium)
- anticholinergics.1,2
Lithium has been used in attempts to prevent recurrent catatonia with limited success.2 There are also a few reports of using transcranial magnetic stimulation (TMS) to manage catatonia.1
Beach et al1 proposed a treatment algorithm in which IV lorazepam (Step 1) and ECT (Step 2) remain the preferred treatments. Next, for Step 3 consider a glutamate antagonist (amantadine or memantine), followed by an antiepileptic (Step 4), and lastly an atypical antipsychotic (aripiprazole, olanzapine, or clozapine) in combination with lorazepam (Step 5).
When indicated, don’t delay ECT
Initial management of catatonia is with a benzodiazepine challenge. Ultimately, the gold-standard treatment of catatonia that does not improve with benzodiazepines is ECT, and ECT should be implemented as soon as it is clear that pharmacotherapy is less than fully effective. Consider ECT initially in life-threatening cases and for patients with malignant catatonia. Although additional agents and TMS have been explored, these should be reserved for patients who fail to respond to, or who are not candidates for, benzodiazepines or ECT.
CASE CONTINUED
After 5 ECT treatments, Mr. W says a few words, but he communicates primarily with gestures (primarily waving people away). After 10 to 12 ECT treatments, Mr. W becomes more interactive and conversant, and his nutrition improves; however, he still exhibits symptoms of catatonia and is not at baseline. He undergoes a total of 18 ECT treatments. Antipsychotics were initially discontinued; however, given Mr. W’s improvement with ECT and the presence of auditory hallucinations, oral risperidone is restarted and titrated to 2 mg, 2 times a day, and he is transitioned back to paliperidone palmitate before he is discharged. Lorazepam is tapered and discontinued. Mr. W is discharged back to his nursing home and is interactive (laughing and joking with family) and attending to his activities of daily living. Unfortunately, Mr. W did not followup with the recommendation for maintenance ECT, and adherence to paliperidone palmitate injections is unknown. Mr. W presented to our facility again 6 months later with symptoms of catatonia and ultimately transferred to a state hospital.
Related Resources
- Fink M, Taylor MA. Catatonia: A clinician’s guide to diagnosis and treatment. New York, NY: Cambridge University Press; 2006. • Carroll BT, Spiegel DR. Catatonia on the consultation liaison service and other clinical settings. Hauppauge, NY: Nova Science Pub Inc.; 2016.
- Benarous X, Raffin M, Ferrafiat V, et al. Catatonia in children and adolescents: new perspectives. Schizophr Res. 2018;200:56-67.
- Malignant Hyperthermia Association of the United States. What is NMSIS? http://www.mhaus.org/nmsis/about-us/ what-is-nmsis/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Asenapine • Saphris
Carbamazepine • Carbatrol, Tegretol
Carbidopa/Levodopa • Sinemet
Citalopram • Celexa
Clozapine • Clozaril
Divalproex Sodium • Depakote
Enoxaparin • Lovenox
Fluoxetine • Prozac
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Lurasidone • Latuda
Memantine • Namenda
Methylphenidate • Concerta, Ritalin
Mirtazapine • Remeron
Olanzapine • Zyprexa
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone long-acting injection • Risperdal Consta
Topiramate • Topamax
Zolpidem • Ambien
1. Beach SR, Gomez-Bernal F, Huffman JC, et al. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1-19.
2. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:1-6.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome: focus on treatment and rechallenge. Ann Pharmacother. 2016;50(11):973-981.
5. Ohi K, Kuwata A, Shimada T, et al. Response to benzodiazepines and clinical course in malignant catatonia associated with schizophrenia: a case report. Medicine (Baltimore). 2017;96(16):e6566. doi: 10.1097/MD.0000000000006566.
6. Bush G, Fink M, Petrides G, et al. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
7. Unal A, Altindag A, Demir B, et al. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J ECT. 2017;33(4):290-293.
8. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1182-1183.
9. van der Markt A, Heller HM, van Exel E. A woman with catatonia, what to do after ECT fails: a case report. J ECT. 2016;32(3):e6-7. doi: 10.1097/YCT.0000000000000290.
1. Beach SR, Gomez-Bernal F, Huffman JC, et al. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1-19.
2. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:1-6.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome: focus on treatment and rechallenge. Ann Pharmacother. 2016;50(11):973-981.
5. Ohi K, Kuwata A, Shimada T, et al. Response to benzodiazepines and clinical course in malignant catatonia associated with schizophrenia: a case report. Medicine (Baltimore). 2017;96(16):e6566. doi: 10.1097/MD.0000000000006566.
6. Bush G, Fink M, Petrides G, et al. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
7. Unal A, Altindag A, Demir B, et al. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J ECT. 2017;33(4):290-293.
8. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1182-1183.
9. van der Markt A, Heller HM, van Exel E. A woman with catatonia, what to do after ECT fails: a case report. J ECT. 2016;32(3):e6-7. doi: 10.1097/YCT.0000000000000290.
Why is loxapine overlooked, underprescribed for psychosis?
I have always tried to practice common sense psychiatry, however, sometimes it seems I am alone in this pursuit. My best example is the minimal prescribing of loxapine (Adasuve) for treating the problem of psychosis, most notably schizophrenia.

Mind you, neither I nor anyone in family own stock in any pharmaceutical companies. I don’t lecture for them, so I have no conflicts in writing about this observation – which I hope will improve patient care, thereby saving lives and making a difference.
Everyone should be familiar with the evolution of atypical antipsychotics and how these medications are touted as “second-generation” classes of medication advertised as superior to the older, first-generation antipsychotics. However, as we get more experience with the second-generation atypical antipsychotics, we are learning that they have problematic side effects of their own. For example, they are associated with metabolic syndrome, so they cause weight gain, hyperglycemia, increased risk of stroke, sudden cardiac death, blood clots, and diabetes. Maybe these problems are so endemic in the low-income, African American population I treat that I am overly sensitive to trying to prevent these medical disorders while treating a patient’s mental illness. However, my public health leanings have long caused me to think that low-income African Americans are the canary in America’s health status coal mine, as it seems that what hits this group first eventually will hit the majority population. Accordingly, it seems to me that it is well advised to pay attention to this group’s well-being, physical health, and mental health challenges.
Everyone also should be aware that clozapine (Clozaril) had been dubbed the first atypical antipsychotic. But, in some regard, that designation might be given to thioridazine – although some maintain that the ratio of serotonergic to dopamine effects is not strong enough to earn that title. Unfortunately, both thioridazine and clozapine have serious side effects. Thioridazine is associated with severe cardiac arrhythmias, and clozapine has been associated with the aforementioned side effects of atypical antipsychotics but also can cause life-threatening agranulocytosis, necessitating regular white blood cell counts to monitor for this possibility.
, which belongs class of medication known as dibenzodiazepines – a class that is extraordinarily similar to dibenzoxazepine. The late William Glazer, MD, a distinguished psychopharmacologist long affiliated with Yale University, New Haven, Conn., even suggested that loxapine might behave as an atypical antipsychotic (J Clin Psychiatry. 1999;60 Suppl 10:42-6). Extensive clinical experience with loxapine suggests the same but with some key differences from the standard atypical antipsychotics regarding its side-effect profile.
First, loxapine, despite being chemically related to clozapine, does not cause agranulocytosis, so the need for white blood cell monitoring is not necessary. Second, I have not seen the problematic metabolic syndrome caused by standard atypical antipsychotic medication. It amazes me when I see patients on aripiprazole, clozapine, olanzapine, quetiapine, risperidone, or ziprasidone who also have diabetes and are on metformin – especially when the development of the patients’ diabetes can be traced back to when they were put on an atypical antipsychotic. I often find myself taking patients off their atypical antipsychotic and putting them on loxapine, resulting in gradual weight loss while maintaining the patients’ stable mental status and absence of psychotic symptoms.
It seems to me that if clozapine and loxapine are so similar (they both bind to serotonin and dopamine receptors), loxapine should be the first drug of choice for the treatment of psychotic symptoms. It acts like an atypical but without the problems of weight gain, hyperglycemia, increased risk of stroke, sudden cardiac death, blood clots, and diabetes that the atypicals may cause. Most of the hundreds of patients with psychotic symptoms I have treated over the past 40 years are on the low dose of loxapine 25 mg at bedtime (although the prescribing information on loxapine says it has to be given at least twice a day, as the half life of the medication is only 4 hours). In some rare instances, I prescribe a total of 50 mg at bedtime.
So, not prescribing loxapine does not make sense to me – other than the medication is generic and so it is not being marketed aggressively by people who make money from prescribing medication and are practicing money, not medicine. The other possibility is that most psychiatrists might not know the connection between clozapine and loxapine, so I thought I should use my influence (what little I have) to inform.
Dr. Bell is staff psychiatrist at Jackson Park Hospital’s surgical-medical/psychiatric inpatient unit in Chicago; and chairman of the department of psychiatry at Windsor University, St. Kitts. He also is clinical professor emeritus, department of psychiatry, University of Illinois at Chicago; former president/CEO of Community Mental Health Council; and former director of the Institute for Juvenile Research (the birthplace of child psychiatry), all in Chicago.
I have always tried to practice common sense psychiatry, however, sometimes it seems I am alone in this pursuit. My best example is the minimal prescribing of loxapine (Adasuve) for treating the problem of psychosis, most notably schizophrenia.
Mind you, neither I nor anyone in family own stock in any pharmaceutical companies. I don’t lecture for them, so I have no conflicts in writing about this observation – which I hope will improve patient care, thereby saving lives and making a difference.
Everyone should be familiar with the evolution of atypical antipsychotics and how these medications are touted as “second-generation” classes of medication advertised as superior to the older, first-generation antipsychotics. However, as we get more experience with the second-generation atypical antipsychotics, we are learning that they have problematic side effects of their own. For example, they are associated with metabolic syndrome, so they cause weight gain, hyperglycemia, increased risk of stroke, sudden cardiac death, blood clots, and diabetes. Maybe these problems are so endemic in the low-income, African American population I treat that I am overly sensitive to trying to prevent these medical disorders while treating a patient’s mental illness. However, my public health leanings have long caused me to think that low-income African Americans are the canary in America’s health status coal mine, as it seems that what hits this group first eventually will hit the majority population. Accordingly, it seems to me that it is well advised to pay attention to this group’s well-being, physical health, and mental health challenges.
Everyone also should be aware that clozapine (Clozaril) had been dubbed the first atypical antipsychotic. But, in some regard, that designation might be given to thioridazine – although some maintain that the ratio of serotonergic to dopamine effects is not strong enough to earn that title. Unfortunately, both thioridazine and clozapine have serious side effects. Thioridazine is associated with severe cardiac arrhythmias, and clozapine has been associated with the aforementioned side effects of atypical antipsychotics but also can cause life-threatening agranulocytosis, necessitating regular white blood cell counts to monitor for this possibility.
, which belongs class of medication known as dibenzodiazepines – a class that is extraordinarily similar to dibenzoxazepine. The late William Glazer, MD, a distinguished psychopharmacologist long affiliated with Yale University, New Haven, Conn., even suggested that loxapine might behave as an atypical antipsychotic (J Clin Psychiatry. 1999;60 Suppl 10:42-6). Extensive clinical experience with loxapine suggests the same but with some key differences from the standard atypical antipsychotics regarding its side-effect profile.
First, loxapine, despite being chemically related to clozapine, does not cause agranulocytosis, so the need for white blood cell monitoring is not necessary. Second, I have not seen the problematic metabolic syndrome caused by standard atypical antipsychotic medication. It amazes me when I see patients on aripiprazole, clozapine, olanzapine, quetiapine, risperidone, or ziprasidone who also have diabetes and are on metformin – especially when the development of the patients’ diabetes can be traced back to when they were put on an atypical antipsychotic. I often find myself taking patients off their atypical antipsychotic and putting them on loxapine, resulting in gradual weight loss while maintaining the patients’ stable mental status and absence of psychotic symptoms.
It seems to me that if clozapine and loxapine are so similar (they both bind to serotonin and dopamine receptors), loxapine should be the first drug of choice for the treatment of psychotic symptoms. It acts like an atypical but without the problems of weight gain, hyperglycemia, increased risk of stroke, sudden cardiac death, blood clots, and diabetes that the atypicals may cause. Most of the hundreds of patients with psychotic symptoms I have treated over the past 40 years are on the low dose of loxapine 25 mg at bedtime (although the prescribing information on loxapine says it has to be given at least twice a day, as the half life of the medication is only 4 hours). In some rare instances, I prescribe a total of 50 mg at bedtime.
So, not prescribing loxapine does not make sense to me – other than the medication is generic and so it is not being marketed aggressively by people who make money from prescribing medication and are practicing money, not medicine. The other possibility is that most psychiatrists might not know the connection between clozapine and loxapine, so I thought I should use my influence (what little I have) to inform.
Dr. Bell is staff psychiatrist at Jackson Park Hospital’s surgical-medical/psychiatric inpatient unit in Chicago; and chairman of the department of psychiatry at Windsor University, St. Kitts. He also is clinical professor emeritus, department of psychiatry, University of Illinois at Chicago; former president/CEO of Community Mental Health Council; and former director of the Institute for Juvenile Research (the birthplace of child psychiatry), all in Chicago.
I have always tried to practice common sense psychiatry, however, sometimes it seems I am alone in this pursuit. My best example is the minimal prescribing of loxapine (Adasuve) for treating the problem of psychosis, most notably schizophrenia.
Mind you, neither I nor anyone in family own stock in any pharmaceutical companies. I don’t lecture for them, so I have no conflicts in writing about this observation – which I hope will improve patient care, thereby saving lives and making a difference.
Everyone should be familiar with the evolution of atypical antipsychotics and how these medications are touted as “second-generation” classes of medication advertised as superior to the older, first-generation antipsychotics. However, as we get more experience with the second-generation atypical antipsychotics, we are learning that they have problematic side effects of their own. For example, they are associated with metabolic syndrome, so they cause weight gain, hyperglycemia, increased risk of stroke, sudden cardiac death, blood clots, and diabetes. Maybe these problems are so endemic in the low-income, African American population I treat that I am overly sensitive to trying to prevent these medical disorders while treating a patient’s mental illness. However, my public health leanings have long caused me to think that low-income African Americans are the canary in America’s health status coal mine, as it seems that what hits this group first eventually will hit the majority population. Accordingly, it seems to me that it is well advised to pay attention to this group’s well-being, physical health, and mental health challenges.
Everyone also should be aware that clozapine (Clozaril) had been dubbed the first atypical antipsychotic. But, in some regard, that designation might be given to thioridazine – although some maintain that the ratio of serotonergic to dopamine effects is not strong enough to earn that title. Unfortunately, both thioridazine and clozapine have serious side effects. Thioridazine is associated with severe cardiac arrhythmias, and clozapine has been associated with the aforementioned side effects of atypical antipsychotics but also can cause life-threatening agranulocytosis, necessitating regular white blood cell counts to monitor for this possibility.
, which belongs class of medication known as dibenzodiazepines – a class that is extraordinarily similar to dibenzoxazepine. The late William Glazer, MD, a distinguished psychopharmacologist long affiliated with Yale University, New Haven, Conn., even suggested that loxapine might behave as an atypical antipsychotic (J Clin Psychiatry. 1999;60 Suppl 10:42-6). Extensive clinical experience with loxapine suggests the same but with some key differences from the standard atypical antipsychotics regarding its side-effect profile.
First, loxapine, despite being chemically related to clozapine, does not cause agranulocytosis, so the need for white blood cell monitoring is not necessary. Second, I have not seen the problematic metabolic syndrome caused by standard atypical antipsychotic medication. It amazes me when I see patients on aripiprazole, clozapine, olanzapine, quetiapine, risperidone, or ziprasidone who also have diabetes and are on metformin – especially when the development of the patients’ diabetes can be traced back to when they were put on an atypical antipsychotic. I often find myself taking patients off their atypical antipsychotic and putting them on loxapine, resulting in gradual weight loss while maintaining the patients’ stable mental status and absence of psychotic symptoms.
It seems to me that if clozapine and loxapine are so similar (they both bind to serotonin and dopamine receptors), loxapine should be the first drug of choice for the treatment of psychotic symptoms. It acts like an atypical but without the problems of weight gain, hyperglycemia, increased risk of stroke, sudden cardiac death, blood clots, and diabetes that the atypicals may cause. Most of the hundreds of patients with psychotic symptoms I have treated over the past 40 years are on the low dose of loxapine 25 mg at bedtime (although the prescribing information on loxapine says it has to be given at least twice a day, as the half life of the medication is only 4 hours). In some rare instances, I prescribe a total of 50 mg at bedtime.
So, not prescribing loxapine does not make sense to me – other than the medication is generic and so it is not being marketed aggressively by people who make money from prescribing medication and are practicing money, not medicine. The other possibility is that most psychiatrists might not know the connection between clozapine and loxapine, so I thought I should use my influence (what little I have) to inform.
Dr. Bell is staff psychiatrist at Jackson Park Hospital’s surgical-medical/psychiatric inpatient unit in Chicago; and chairman of the department of psychiatry at Windsor University, St. Kitts. He also is clinical professor emeritus, department of psychiatry, University of Illinois at Chicago; former president/CEO of Community Mental Health Council; and former director of the Institute for Juvenile Research (the birthplace of child psychiatry), all in Chicago.
Delusional infestation: not so rare
PARIS – Ever wonder, when encountering an occasional patient afflicted with delusional infestation, just how common this mental disorder is?
John J. Kohorst, MD, and his coinvestigators at the Mayo Clinic in Rochester, Minn., have the evidence-based answer.
The age- and sex-adjusted point prevalence of delusional infestation among Olmsted County, Minn., residents on the final day of 2010 was 27.3 cases per 100,000 person-years, he reported at the annual congress of the European Academy of Dermatology and Venereology.
“This is the than previously suspected,” according to the dermatologist.
He and his coinvestigators retrospectively analyzed data from the Rochester Epidemiology Project. They identified 22 female and 13 male county residents with a firm diagnosis of delusional infestation, also known as delusional parasitosis. This disorder is marked by a patient’s fixed false belief that they are infested with insects, worms, or other pathogens.
The prevalence was similar in men and women. The most striking study finding was how heavily age-dependent delusional infestation was. Before age 40, the prevalence was a mere 1.2 cases per 100,000 person-years. Among 40- to 59-year-old Olmsted County residents, it was 35/100,000, jumping to 64.5/100,000 in the 60- to 79-year-old age bracket, then doubling to 130.1 cases per 100,000 person-years in individuals aged 80 or older.
Dr. Kohorst reported having no financial conflicts regarding his study, conducted free of commercial support.
PARIS – Ever wonder, when encountering an occasional patient afflicted with delusional infestation, just how common this mental disorder is?
John J. Kohorst, MD, and his coinvestigators at the Mayo Clinic in Rochester, Minn., have the evidence-based answer.
The age- and sex-adjusted point prevalence of delusional infestation among Olmsted County, Minn., residents on the final day of 2010 was 27.3 cases per 100,000 person-years, he reported at the annual congress of the European Academy of Dermatology and Venereology.
“This is the than previously suspected,” according to the dermatologist.
He and his coinvestigators retrospectively analyzed data from the Rochester Epidemiology Project. They identified 22 female and 13 male county residents with a firm diagnosis of delusional infestation, also known as delusional parasitosis. This disorder is marked by a patient’s fixed false belief that they are infested with insects, worms, or other pathogens.
The prevalence was similar in men and women. The most striking study finding was how heavily age-dependent delusional infestation was. Before age 40, the prevalence was a mere 1.2 cases per 100,000 person-years. Among 40- to 59-year-old Olmsted County residents, it was 35/100,000, jumping to 64.5/100,000 in the 60- to 79-year-old age bracket, then doubling to 130.1 cases per 100,000 person-years in individuals aged 80 or older.
Dr. Kohorst reported having no financial conflicts regarding his study, conducted free of commercial support.
PARIS – Ever wonder, when encountering an occasional patient afflicted with delusional infestation, just how common this mental disorder is?
John J. Kohorst, MD, and his coinvestigators at the Mayo Clinic in Rochester, Minn., have the evidence-based answer.
The age- and sex-adjusted point prevalence of delusional infestation among Olmsted County, Minn., residents on the final day of 2010 was 27.3 cases per 100,000 person-years, he reported at the annual congress of the European Academy of Dermatology and Venereology.
“This is the than previously suspected,” according to the dermatologist.
He and his coinvestigators retrospectively analyzed data from the Rochester Epidemiology Project. They identified 22 female and 13 male county residents with a firm diagnosis of delusional infestation, also known as delusional parasitosis. This disorder is marked by a patient’s fixed false belief that they are infested with insects, worms, or other pathogens.
The prevalence was similar in men and women. The most striking study finding was how heavily age-dependent delusional infestation was. Before age 40, the prevalence was a mere 1.2 cases per 100,000 person-years. Among 40- to 59-year-old Olmsted County residents, it was 35/100,000, jumping to 64.5/100,000 in the 60- to 79-year-old age bracket, then doubling to 130.1 cases per 100,000 person-years in individuals aged 80 or older.
Dr. Kohorst reported having no financial conflicts regarding his study, conducted free of commercial support.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Delusional infestation may be more common than previously suspected, particularly among older age groups.
Major finding: The age- and sex-adjusted point prevalence of delusional infestation among residents of one county in southeastern Minnesota is 27.3 cases per 100,000 person-years.
Study details: This was a retrospective analysis of data from the Rochester (Minn.) Epidemiology Project.
Disclosures: The presenter reported having no financial conflicts regarding his study, conducted free of commercial support.
Aripiprazole lauroxil nanocrystal suspension

Clinical implications
Nonadherence with oral antipsychotics is a common problem for patients with schizophrenia, one that is often underappreciated by clinicians.5 Whether one uses 70% or 80% as the measure of oral medication adherence, at least 50% of schizophrenia patients are nonadherent, with resultant increased risks for symptom exacerbation and hospitalization.5,6 Although 2 LAI forms of aripiprazole have been introduced over the past few years, neither was designed to be loaded, resulting in the need for 2 or 3 weeks of oral antipsychotic coverage following the first injectable dose.1 The primary reason for LAI antipsychotic therapy is oral medication nonadherence, and thus the need for 14 to 21 days of oral coverage at the outset of treatment creates a risk for symptom exacerbation if the patient is nonadherent with this oral bridging therapy which is needed to achieve the necessary serum concentrations until the long-acting formulation takes over.
One approach was to create a new form of AL using smaller nanomolecular particles rather than the micron-sized particles used for maintenance AL injections.3,4 This nanocrystal suspension is called Aristada Initio (AL

Use in adults with schizophrenia. After establishing tolerability with oral aripiprazole, AL
Continue to: Pharmacologic profile, adverse reactions
Pharmacologic profile, adverse reactions
Aripiprazole is a dopamine partial agonist atypical antipsychotic that has been commercially available in the United States since November 15, 2002, and its adverse effect profile is well characterized. The LAI formulation AL was approved on October 5, 2015. In the pivotal, 12-week, fixed-dose, placebo-controlled clinical trial of AL 441 mg or 882 mg monthly for adults with an acute exacerbation of schizophrenia, the only adverse effect that occurred in ≥5% of AL-treated patients and a rate at least twice that of placebo was akathisia (441 mg: 11%; 882 mg: 11%; placebo: 4%).10 Only 2 of 415 AL-treated patients discontinued the study due to akathisia. Injection-site reactions were reported by 4%, 5%, and 2% of patients treated with AL 441 mg, AL 882 mg, and placebo, respectively. Most of these were injection-site pain associated with the first injection, and decreased with each subsequent injection. Other injection-site reactions (induration, swelling, and redness) occurred at rates <1%.11
Having established that the range of plasma aripiprazole levels consistent with effective treatment is bounded by levels seen with AL 441 mg or 882 mg monthly, the FDA did not require additional efficacy studies for new AL doses provided that pharmacokinetic (PK) studies could demonstrate levels within the effective range. This is consistent with how new doses of other LAI antipsychotic preparations have been addressed in the past. For example, the 37.5 mg dose of risperidone microspheres was approved based on PK data, although the pivotal efficacy trials included doses of 25 mg, 50 mg, and 75 mg.12 Based on PK studies, AL doses of 662 mg monthly, 882 mg every 6 weeks, and 1,064 mg every 8 weeks were previously approved.13 The approval process for AL
Pharmacokinetic outcomes. A comparative phase 1 PK study was performed to evaluate initiation regimens: either 21 days of oral aripiprazole (15 mg/d) and one AL dose (n = 81) or one injection of AL
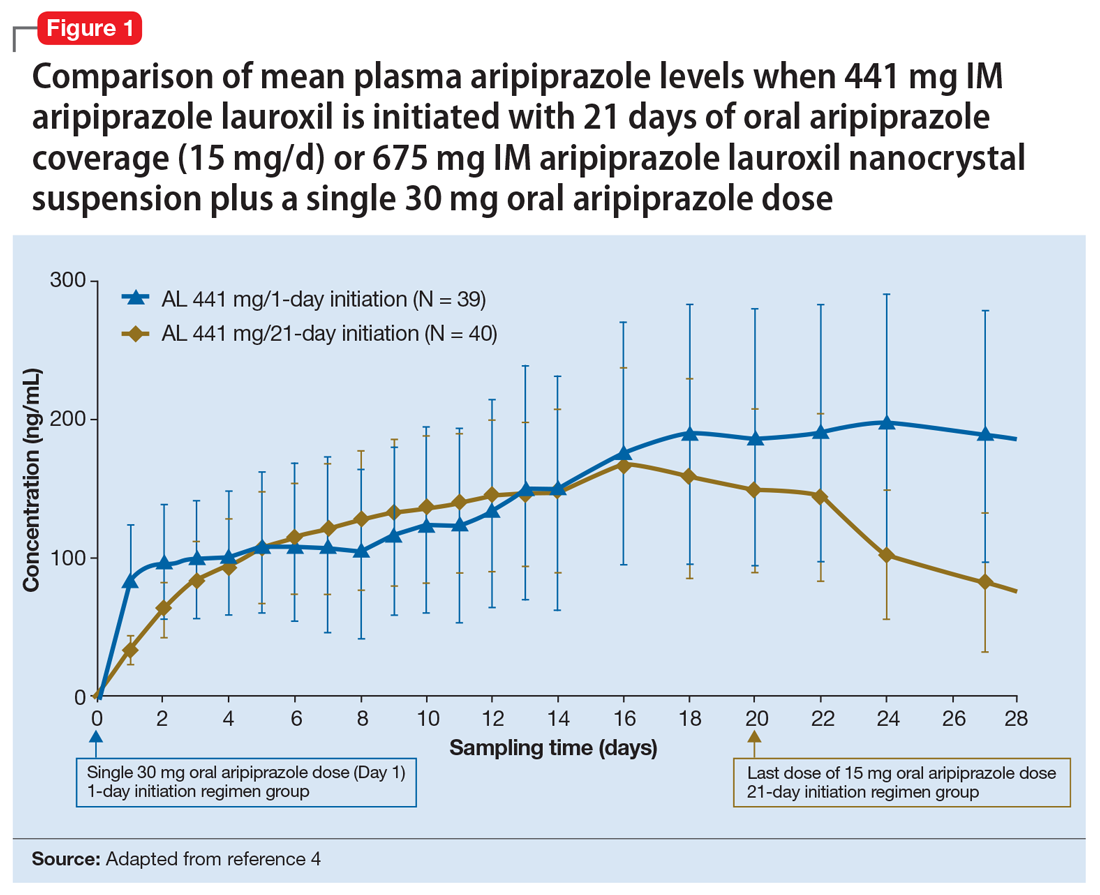
Tolerability. In PK studies, the safety profile and incidences of injection site reactions of AL
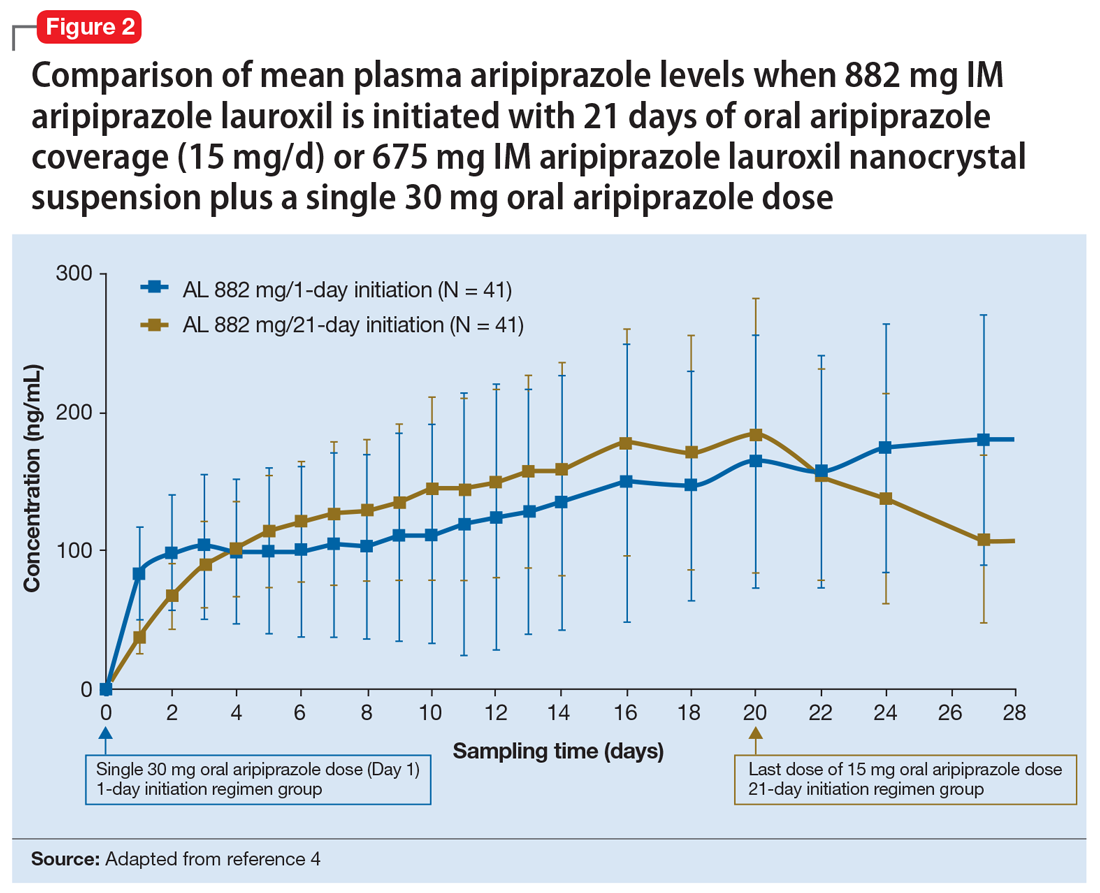
Continue to: Clinical considerations
Clinical considerations
AL
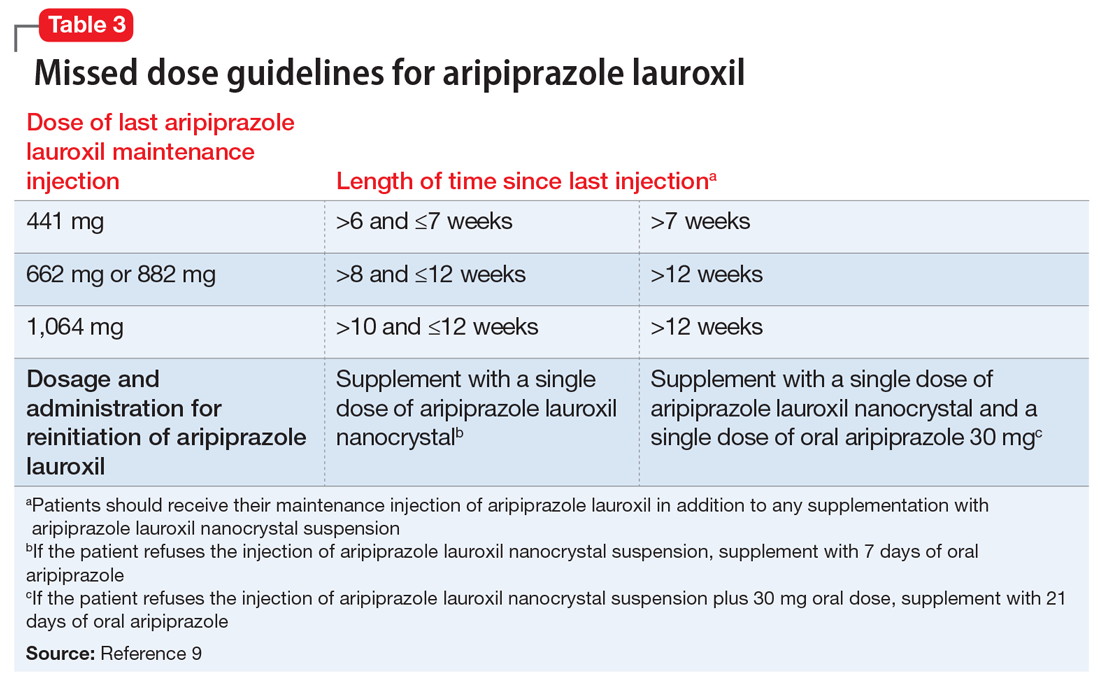
Unique properties. When combined with a single 30 mg oral dose, AL
Why Rx? The reasons to prescribe AL
- it obviates the need for 21 days of oral coverage previously required at the initiation of AL treatment
- clinically relevant plasma levels are seen within the first week when AL
ncd is combined with a single 30 mg oral aripiprazole dose - per the revised missed dose guidelines for AL, it can be used in those situations that previously demanded 7 days of oral coverage, and, when combined with a single 30 mg oral dose, can be used for resumption of therapy after prolonged absences that required 21 days of oral coverage. In all instances, the patient will also receive their usual maintenance dose of AL.
Dosing. There is only one dose available for AL
Contraindications. The only contraindication is a known hypersensitivity to aripiprazole.
Bottom Line
Aripiprazole lauroxil nanocrystal suspension (Aristada Initio) was specifically developed to obviate the need for 21 days of oral aripiprazole coverage when commencing treatment with aripiprazole lauroxil (Aristada). The plasma levels achieved when an injection of aripiprazole lauroxil nanocrystal suspension is combined with a single 30 mg oral dose are comparable to those achieved with 21 days of oral coverage. This initiation regimen, including a aripiprazole lauroxil nanocrystal injection and a 30 mg oral dose, should be administered on the same day as the maintenance aripiprazole lauroxil injection, although the latter can be administered on any of the next 10 days.
Related Resource
- Khan AY, Ovais DM. Long-acting injectable aripiprazole lauroxil for schizophrenia. Current Psychiatry. 2016;15(7):50-52,58.
Drug Brand Names
Aripiprazole lauroxil • Aristada
Aripiprazole lauroxil nanocrystal • Aristada Initio
Risperidone microspheres • Risperdal Consta
1. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
2. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
3. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
4. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
5. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatric Services. 2007;58(6):844-847.
6. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
7. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
8. Hard ML, Mills RJ, Sadler BM, et al. Aripiprazole lauroxil: pharmacokinetic profile of this long-acting injectable antipsychotic in persons with schizophrenia. J Clin Psychopharmacol. 2017;37(3):289-295.
9. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
10. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
11. Aristada [package insert]. Waltham, MA: Alkermes Inc; 2018.
12. Fleischhacker WW, Eerdekens M, Karcher K, et al. Treatment of schizophrenia with long-acting injectable risperidone: a 12-month open-label trial of the first long-acting second-generation antipsychotic. J Clin Psychiatry. 2003;64(10):1250-1257.
13. Hard ML, Mills RJ, Sadler BM, et al. Pharmacokinetic profile of a 2-month dose regimen of aripiprazole lauroxil: a phase I study and a population pharmacokinetic model. CNS Drugs. 2017;31(7):617-624.
Clinical implications
Nonadherence with oral antipsychotics is a common problem for patients with schizophrenia, one that is often underappreciated by clinicians.5 Whether one uses 70% or 80% as the measure of oral medication adherence, at least 50% of schizophrenia patients are nonadherent, with resultant increased risks for symptom exacerbation and hospitalization.5,6 Although 2 LAI forms of aripiprazole have been introduced over the past few years, neither was designed to be loaded, resulting in the need for 2 or 3 weeks of oral antipsychotic coverage following the first injectable dose.1 The primary reason for LAI antipsychotic therapy is oral medication nonadherence, and thus the need for 14 to 21 days of oral coverage at the outset of treatment creates a risk for symptom exacerbation if the patient is nonadherent with this oral bridging therapy which is needed to achieve the necessary serum concentrations until the long-acting formulation takes over.
One approach was to create a new form of AL using smaller nanomolecular particles rather than the micron-sized particles used for maintenance AL injections.3,4 This nanocrystal suspension is called Aristada Initio (AL
Use in adults with schizophrenia. After establishing tolerability with oral aripiprazole, AL
Continue to: Pharmacologic profile, adverse reactions
Pharmacologic profile, adverse reactions
Aripiprazole is a dopamine partial agonist atypical antipsychotic that has been commercially available in the United States since November 15, 2002, and its adverse effect profile is well characterized. The LAI formulation AL was approved on October 5, 2015. In the pivotal, 12-week, fixed-dose, placebo-controlled clinical trial of AL 441 mg or 882 mg monthly for adults with an acute exacerbation of schizophrenia, the only adverse effect that occurred in ≥5% of AL-treated patients and a rate at least twice that of placebo was akathisia (441 mg: 11%; 882 mg: 11%; placebo: 4%).10 Only 2 of 415 AL-treated patients discontinued the study due to akathisia. Injection-site reactions were reported by 4%, 5%, and 2% of patients treated with AL 441 mg, AL 882 mg, and placebo, respectively. Most of these were injection-site pain associated with the first injection, and decreased with each subsequent injection. Other injection-site reactions (induration, swelling, and redness) occurred at rates <1%.11
Having established that the range of plasma aripiprazole levels consistent with effective treatment is bounded by levels seen with AL 441 mg or 882 mg monthly, the FDA did not require additional efficacy studies for new AL doses provided that pharmacokinetic (PK) studies could demonstrate levels within the effective range. This is consistent with how new doses of other LAI antipsychotic preparations have been addressed in the past. For example, the 37.5 mg dose of risperidone microspheres was approved based on PK data, although the pivotal efficacy trials included doses of 25 mg, 50 mg, and 75 mg.12 Based on PK studies, AL doses of 662 mg monthly, 882 mg every 6 weeks, and 1,064 mg every 8 weeks were previously approved.13 The approval process for AL
Pharmacokinetic outcomes. A comparative phase 1 PK study was performed to evaluate initiation regimens: either 21 days of oral aripiprazole (15 mg/d) and one AL dose (n = 81) or one injection of AL
Tolerability. In PK studies, the safety profile and incidences of injection site reactions of AL
Continue to: Clinical considerations
Clinical considerations
AL
Unique properties. When combined with a single 30 mg oral dose, AL
Why Rx? The reasons to prescribe AL
- it obviates the need for 21 days of oral coverage previously required at the initiation of AL treatment
- clinically relevant plasma levels are seen within the first week when AL
ncd is combined with a single 30 mg oral aripiprazole dose - per the revised missed dose guidelines for AL, it can be used in those situations that previously demanded 7 days of oral coverage, and, when combined with a single 30 mg oral dose, can be used for resumption of therapy after prolonged absences that required 21 days of oral coverage. In all instances, the patient will also receive their usual maintenance dose of AL.
Dosing. There is only one dose available for AL
Contraindications. The only contraindication is a known hypersensitivity to aripiprazole.
Bottom Line
Aripiprazole lauroxil nanocrystal suspension (Aristada Initio) was specifically developed to obviate the need for 21 days of oral aripiprazole coverage when commencing treatment with aripiprazole lauroxil (Aristada). The plasma levels achieved when an injection of aripiprazole lauroxil nanocrystal suspension is combined with a single 30 mg oral dose are comparable to those achieved with 21 days of oral coverage. This initiation regimen, including a aripiprazole lauroxil nanocrystal injection and a 30 mg oral dose, should be administered on the same day as the maintenance aripiprazole lauroxil injection, although the latter can be administered on any of the next 10 days.
Related Resource
- Khan AY, Ovais DM. Long-acting injectable aripiprazole lauroxil for schizophrenia. Current Psychiatry. 2016;15(7):50-52,58.
Drug Brand Names
Aripiprazole lauroxil • Aristada
Aripiprazole lauroxil nanocrystal • Aristada Initio
Risperidone microspheres • Risperdal Consta
Clinical implications
Nonadherence with oral antipsychotics is a common problem for patients with schizophrenia, one that is often underappreciated by clinicians.5 Whether one uses 70% or 80% as the measure of oral medication adherence, at least 50% of schizophrenia patients are nonadherent, with resultant increased risks for symptom exacerbation and hospitalization.5,6 Although 2 LAI forms of aripiprazole have been introduced over the past few years, neither was designed to be loaded, resulting in the need for 2 or 3 weeks of oral antipsychotic coverage following the first injectable dose.1 The primary reason for LAI antipsychotic therapy is oral medication nonadherence, and thus the need for 14 to 21 days of oral coverage at the outset of treatment creates a risk for symptom exacerbation if the patient is nonadherent with this oral bridging therapy which is needed to achieve the necessary serum concentrations until the long-acting formulation takes over.
One approach was to create a new form of AL using smaller nanomolecular particles rather than the micron-sized particles used for maintenance AL injections.3,4 This nanocrystal suspension is called Aristada Initio (AL
Use in adults with schizophrenia. After establishing tolerability with oral aripiprazole, AL
Continue to: Pharmacologic profile, adverse reactions
Pharmacologic profile, adverse reactions
Aripiprazole is a dopamine partial agonist atypical antipsychotic that has been commercially available in the United States since November 15, 2002, and its adverse effect profile is well characterized. The LAI formulation AL was approved on October 5, 2015. In the pivotal, 12-week, fixed-dose, placebo-controlled clinical trial of AL 441 mg or 882 mg monthly for adults with an acute exacerbation of schizophrenia, the only adverse effect that occurred in ≥5% of AL-treated patients and a rate at least twice that of placebo was akathisia (441 mg: 11%; 882 mg: 11%; placebo: 4%).10 Only 2 of 415 AL-treated patients discontinued the study due to akathisia. Injection-site reactions were reported by 4%, 5%, and 2% of patients treated with AL 441 mg, AL 882 mg, and placebo, respectively. Most of these were injection-site pain associated with the first injection, and decreased with each subsequent injection. Other injection-site reactions (induration, swelling, and redness) occurred at rates <1%.11
Having established that the range of plasma aripiprazole levels consistent with effective treatment is bounded by levels seen with AL 441 mg or 882 mg monthly, the FDA did not require additional efficacy studies for new AL doses provided that pharmacokinetic (PK) studies could demonstrate levels within the effective range. This is consistent with how new doses of other LAI antipsychotic preparations have been addressed in the past. For example, the 37.5 mg dose of risperidone microspheres was approved based on PK data, although the pivotal efficacy trials included doses of 25 mg, 50 mg, and 75 mg.12 Based on PK studies, AL doses of 662 mg monthly, 882 mg every 6 weeks, and 1,064 mg every 8 weeks were previously approved.13 The approval process for AL
Pharmacokinetic outcomes. A comparative phase 1 PK study was performed to evaluate initiation regimens: either 21 days of oral aripiprazole (15 mg/d) and one AL dose (n = 81) or one injection of AL
Tolerability. In PK studies, the safety profile and incidences of injection site reactions of AL
Continue to: Clinical considerations
Clinical considerations
AL
Unique properties. When combined with a single 30 mg oral dose, AL
Why Rx? The reasons to prescribe AL
- it obviates the need for 21 days of oral coverage previously required at the initiation of AL treatment
- clinically relevant plasma levels are seen within the first week when AL
ncd is combined with a single 30 mg oral aripiprazole dose - per the revised missed dose guidelines for AL, it can be used in those situations that previously demanded 7 days of oral coverage, and, when combined with a single 30 mg oral dose, can be used for resumption of therapy after prolonged absences that required 21 days of oral coverage. In all instances, the patient will also receive their usual maintenance dose of AL.
Dosing. There is only one dose available for AL
Contraindications. The only contraindication is a known hypersensitivity to aripiprazole.
Bottom Line
Aripiprazole lauroxil nanocrystal suspension (Aristada Initio) was specifically developed to obviate the need for 21 days of oral aripiprazole coverage when commencing treatment with aripiprazole lauroxil (Aristada). The plasma levels achieved when an injection of aripiprazole lauroxil nanocrystal suspension is combined with a single 30 mg oral dose are comparable to those achieved with 21 days of oral coverage. This initiation regimen, including a aripiprazole lauroxil nanocrystal injection and a 30 mg oral dose, should be administered on the same day as the maintenance aripiprazole lauroxil injection, although the latter can be administered on any of the next 10 days.
Related Resource
- Khan AY, Ovais DM. Long-acting injectable aripiprazole lauroxil for schizophrenia. Current Psychiatry. 2016;15(7):50-52,58.
Drug Brand Names
Aripiprazole lauroxil • Aristada
Aripiprazole lauroxil nanocrystal • Aristada Initio
Risperidone microspheres • Risperdal Consta
1. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
2. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
3. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
4. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
5. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatric Services. 2007;58(6):844-847.
6. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
7. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
8. Hard ML, Mills RJ, Sadler BM, et al. Aripiprazole lauroxil: pharmacokinetic profile of this long-acting injectable antipsychotic in persons with schizophrenia. J Clin Psychopharmacol. 2017;37(3):289-295.
9. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
10. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
11. Aristada [package insert]. Waltham, MA: Alkermes Inc; 2018.
12. Fleischhacker WW, Eerdekens M, Karcher K, et al. Treatment of schizophrenia with long-acting injectable risperidone: a 12-month open-label trial of the first long-acting second-generation antipsychotic. J Clin Psychiatry. 2003;64(10):1250-1257.
13. Hard ML, Mills RJ, Sadler BM, et al. Pharmacokinetic profile of a 2-month dose regimen of aripiprazole lauroxil: a phase I study and a population pharmacokinetic model. CNS Drugs. 2017;31(7):617-624.
1. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
2. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
3. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
4. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
5. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatric Services. 2007;58(6):844-847.
6. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
7. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
8. Hard ML, Mills RJ, Sadler BM, et al. Aripiprazole lauroxil: pharmacokinetic profile of this long-acting injectable antipsychotic in persons with schizophrenia. J Clin Psychopharmacol. 2017;37(3):289-295.
9. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
10. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
11. Aristada [package insert]. Waltham, MA: Alkermes Inc; 2018.
12. Fleischhacker WW, Eerdekens M, Karcher K, et al. Treatment of schizophrenia with long-acting injectable risperidone: a 12-month open-label trial of the first long-acting second-generation antipsychotic. J Clin Psychiatry. 2003;64(10):1250-1257.
13. Hard ML, Mills RJ, Sadler BM, et al. Pharmacokinetic profile of a 2-month dose regimen of aripiprazole lauroxil: a phase I study and a population pharmacokinetic model. CNS Drugs. 2017;31(7):617-624.