User login
Methotrexate coverage woes
Some things make my job much harder than it should be. One of them is prior authorizations.
At the start of every year, insurers release a new formulary, so prior authorizations are often required, even drugs that patients have been on for a long time. Everyday I spend anywhere from 10 to 30 minutes filling out prior authorization paperwork.
Sometimes, it’s not too difficult. If someone is getting a biologic for the first time, it usually means they’ve had an inadequate response to methotrexate. The form makes sure we’ve checked the patient’s TB status. Sometimes, it asks about a history of heart failure. These are reasonable questions designed to protect the patient.
Frequently, though, the questionnaires are more onerous, and for less altruistic motives. A prior authorization for celecoxib (Celebrex) requires that I list at least two other prescription NSAIDs that the patient has been on. I can guarantee that I do not know that information off the top of my head, so this requires some digging into the chart. If it’s a patient whose care I’ve assumed from someone else, or whose chart is several volumes thick, I won’t even know where to begin. Even worse is the pregabalin (Lyrica) prior authorization that asks about previous use of tricyclic antidepressants, cyclobenzaprine, SSRIs, gabapentin. When were they on these agents and for how long? What was the outcome of each failed medication?
These two examples do not ensure patient safety. They simply go directly to what the insurer’s bottom line is. They benefit no one but the insurer, and create lots of problems for everyone who has to abide by the insurer’s rules.
The worst of it, this year, has to be prior authorization for injectable methotrexate. I have recently run into a lot of problems with this one. For one patient who had been on weekly adalimumab (Humira) and subcutaneous methotrexate, I was asked to justify the combination with a journal article or two, because mechanistically the insurers reject methotrexate as having any role in therapy for patients already on weekly adalimumab, antidrug antibodies notwithstanding. I had inherited this patient from another rheumatologist, and she’d been on this regimen for over a decade; it was infuriating and frustrating to be asked to justify a regimen that the patient had not had problems with for the previous 10 years and that was working fine for her.
For another patient, I was asked to fax a letter to the pharmacy stating why the drug was necessary. Then I was asked to fax another letter to the insurer’s benefits manager. A day later, I was informed that there was no process of appeal for medications that were not on formulary. None whatsoever. The letters that they had asked me to write had no bearing and meant nothing to them. This was the very definition of a complete waste of time.
If I wanted the patient to be on subcutaneous methotrexate, the patient would have to pay out of pocket for the drug. I got on the phone with someone whose job was to repeatedly tell me that I had no options. She couldn’t direct me to anyone higher up than her. I understand that this automaton would have no intelligent answers for me, but I asked rhetorically if etanercept would be covered. She said yes. I then asked rhetorically if she knew how much more the biologic agent would cost the insurance company. I cut her off as she was about to look up the price of the drug.
I can think of hundreds of ways in which I could make better use of my time instead of playing a game I am forced to play against my will, a game that is shrouded in layers of bureaucracy over which I have very little control and has no benefit to my patients. There are days when the game is enough to make me want to throw in the towel.
Dr. Chan practices rheumatology in Pawtucket, R.I.
Some things make my job much harder than it should be. One of them is prior authorizations.
At the start of every year, insurers release a new formulary, so prior authorizations are often required, even drugs that patients have been on for a long time. Everyday I spend anywhere from 10 to 30 minutes filling out prior authorization paperwork.
Sometimes, it’s not too difficult. If someone is getting a biologic for the first time, it usually means they’ve had an inadequate response to methotrexate. The form makes sure we’ve checked the patient’s TB status. Sometimes, it asks about a history of heart failure. These are reasonable questions designed to protect the patient.
Frequently, though, the questionnaires are more onerous, and for less altruistic motives. A prior authorization for celecoxib (Celebrex) requires that I list at least two other prescription NSAIDs that the patient has been on. I can guarantee that I do not know that information off the top of my head, so this requires some digging into the chart. If it’s a patient whose care I’ve assumed from someone else, or whose chart is several volumes thick, I won’t even know where to begin. Even worse is the pregabalin (Lyrica) prior authorization that asks about previous use of tricyclic antidepressants, cyclobenzaprine, SSRIs, gabapentin. When were they on these agents and for how long? What was the outcome of each failed medication?
These two examples do not ensure patient safety. They simply go directly to what the insurer’s bottom line is. They benefit no one but the insurer, and create lots of problems for everyone who has to abide by the insurer’s rules.
The worst of it, this year, has to be prior authorization for injectable methotrexate. I have recently run into a lot of problems with this one. For one patient who had been on weekly adalimumab (Humira) and subcutaneous methotrexate, I was asked to justify the combination with a journal article or two, because mechanistically the insurers reject methotrexate as having any role in therapy for patients already on weekly adalimumab, antidrug antibodies notwithstanding. I had inherited this patient from another rheumatologist, and she’d been on this regimen for over a decade; it was infuriating and frustrating to be asked to justify a regimen that the patient had not had problems with for the previous 10 years and that was working fine for her.
For another patient, I was asked to fax a letter to the pharmacy stating why the drug was necessary. Then I was asked to fax another letter to the insurer’s benefits manager. A day later, I was informed that there was no process of appeal for medications that were not on formulary. None whatsoever. The letters that they had asked me to write had no bearing and meant nothing to them. This was the very definition of a complete waste of time.
If I wanted the patient to be on subcutaneous methotrexate, the patient would have to pay out of pocket for the drug. I got on the phone with someone whose job was to repeatedly tell me that I had no options. She couldn’t direct me to anyone higher up than her. I understand that this automaton would have no intelligent answers for me, but I asked rhetorically if etanercept would be covered. She said yes. I then asked rhetorically if she knew how much more the biologic agent would cost the insurance company. I cut her off as she was about to look up the price of the drug.
I can think of hundreds of ways in which I could make better use of my time instead of playing a game I am forced to play against my will, a game that is shrouded in layers of bureaucracy over which I have very little control and has no benefit to my patients. There are days when the game is enough to make me want to throw in the towel.
Dr. Chan practices rheumatology in Pawtucket, R.I.
Some things make my job much harder than it should be. One of them is prior authorizations.
At the start of every year, insurers release a new formulary, so prior authorizations are often required, even drugs that patients have been on for a long time. Everyday I spend anywhere from 10 to 30 minutes filling out prior authorization paperwork.
Sometimes, it’s not too difficult. If someone is getting a biologic for the first time, it usually means they’ve had an inadequate response to methotrexate. The form makes sure we’ve checked the patient’s TB status. Sometimes, it asks about a history of heart failure. These are reasonable questions designed to protect the patient.
Frequently, though, the questionnaires are more onerous, and for less altruistic motives. A prior authorization for celecoxib (Celebrex) requires that I list at least two other prescription NSAIDs that the patient has been on. I can guarantee that I do not know that information off the top of my head, so this requires some digging into the chart. If it’s a patient whose care I’ve assumed from someone else, or whose chart is several volumes thick, I won’t even know where to begin. Even worse is the pregabalin (Lyrica) prior authorization that asks about previous use of tricyclic antidepressants, cyclobenzaprine, SSRIs, gabapentin. When were they on these agents and for how long? What was the outcome of each failed medication?
These two examples do not ensure patient safety. They simply go directly to what the insurer’s bottom line is. They benefit no one but the insurer, and create lots of problems for everyone who has to abide by the insurer’s rules.
The worst of it, this year, has to be prior authorization for injectable methotrexate. I have recently run into a lot of problems with this one. For one patient who had been on weekly adalimumab (Humira) and subcutaneous methotrexate, I was asked to justify the combination with a journal article or two, because mechanistically the insurers reject methotrexate as having any role in therapy for patients already on weekly adalimumab, antidrug antibodies notwithstanding. I had inherited this patient from another rheumatologist, and she’d been on this regimen for over a decade; it was infuriating and frustrating to be asked to justify a regimen that the patient had not had problems with for the previous 10 years and that was working fine for her.
For another patient, I was asked to fax a letter to the pharmacy stating why the drug was necessary. Then I was asked to fax another letter to the insurer’s benefits manager. A day later, I was informed that there was no process of appeal for medications that were not on formulary. None whatsoever. The letters that they had asked me to write had no bearing and meant nothing to them. This was the very definition of a complete waste of time.
If I wanted the patient to be on subcutaneous methotrexate, the patient would have to pay out of pocket for the drug. I got on the phone with someone whose job was to repeatedly tell me that I had no options. She couldn’t direct me to anyone higher up than her. I understand that this automaton would have no intelligent answers for me, but I asked rhetorically if etanercept would be covered. She said yes. I then asked rhetorically if she knew how much more the biologic agent would cost the insurance company. I cut her off as she was about to look up the price of the drug.
I can think of hundreds of ways in which I could make better use of my time instead of playing a game I am forced to play against my will, a game that is shrouded in layers of bureaucracy over which I have very little control and has no benefit to my patients. There are days when the game is enough to make me want to throw in the towel.
Dr. Chan practices rheumatology in Pawtucket, R.I.

Consider Chikungunya virus in new-onset polyarthritis
Rheumatologists should consider the possibility of Chikungunya virus in patients who present with new symmetric polyarthritis, especially if they have just returned from an endemic region such as the Caribbean, according to researchers.
In their paper, first authors Dr. Jonathan J. Miner and Dr. Han-Xian Aw-Yeang and colleagues at Washington University, St. Louis, describe a cohort of 10 Americans who traveled to Haiti within a 20-day period in June 2014 and became infected with Chikungunya virus (CHIKV), an arthritogenic, mosquito-transmitted alphavirus (Arthritis Rheumatol. 2015 Jan. 20 [doi:10.1002/art.39027]).
The virus spread to the Caribbean in 2013 and the United States in 2014. Its acute phase of infection includes symptoms such as fever, headache, rash, arthralgia, arthritis, and myalgia.
The virus is likely to become a unique diagnostic challenge for rheumatologists as the arthritis symptoms mimic seronegative arthritis, the researchers said.
“Eight out of these patients would have met the 2010 ACR/EULAR criteria for RA [rheumatoid arthritis] if the initial fever, rash, and travel to the Caribbean had not been revealed,” they noted.
Most of the patients had joint pain and morning stiffness at least 8 weeks after initial infection, although some reported gradual improvement of their symptoms, the investigators reported.
One of the patients developed fever; diffuse arthritis; an erythematous, maculopapular rash; and severe symmetric polyarthritis that persisted more than 5 months post infection. She was treated with prednisolone but it exacerbated her joint pain and treatment was stopped.
The other patient detailed in the report presented with similar symptoms. His fever and rash resolved within 2 days but his arthritis symptoms persisted. He was treated with NSAIDs that provided only minimal relief.
To understand more about the immunologic parameters of the virus, the researchers used cytometry by time of flight to compare the peripheral mononuclear cells from CHIKV-infected patients with those of healthy controls and untreated patients with RA.
They discovered that naive, activated, and effector T killer– and T helper–cell populations occurred in similar percentages in the virus-infected patients and RA patients but not in the healthy controls.
The RA patients, however, had a higher expression of L-selectin expression in CD4+ T cells than did either virus-infected patients or healthy controls.
“These data suggest that lymphocyte phenotypes in patients with Chikungunya infection are similar to each other, with subtle but distinct trends that could potentially distinguish these two groups from healthy controls and from each other,” Dr. Miner and Dr. Aw-Yeang and their associates wrote.
Whether treatment with disease-modifying antirheumatic drugs or biologics used in RA is appropriate or effective is controversial in the absence of randomized controlled trials for CHIKV-related arthritis, they said.
“Immunosuppression in CHIKV-infected patients could be deleterious because viral RNA and antigens can be found in target tissues in the chronic phase in humans and experimental animals,” the authors wrote.
“Rheumatologists, even in non-CHIKV-endemic regions, need to consider CHIKV in their evaluation of symmetric polyarthritis lasting more than 6 weeks by obtaining a history of travel to CHIKV-endemic regions, which are likely to expand in the near future,” the investigators advised.
Patients who may have been exposed to the virus may need serologic testing before initiating immunosuppression, they said.
“Unfortunately, access to CHIKV testing is highly constrained at the current time as it is only available from the [Centers for Disease Control and Prevention] and research laboratories,” they added.
The study was supported by the Barnes-Jewish Hospital Foundation and the Howard Hughes Medical Institute. The authors reported no conflicts of interest.
Rheumatologists should consider the possibility of Chikungunya virus in patients who present with new symmetric polyarthritis, especially if they have just returned from an endemic region such as the Caribbean, according to researchers.
In their paper, first authors Dr. Jonathan J. Miner and Dr. Han-Xian Aw-Yeang and colleagues at Washington University, St. Louis, describe a cohort of 10 Americans who traveled to Haiti within a 20-day period in June 2014 and became infected with Chikungunya virus (CHIKV), an arthritogenic, mosquito-transmitted alphavirus (Arthritis Rheumatol. 2015 Jan. 20 [doi:10.1002/art.39027]).
The virus spread to the Caribbean in 2013 and the United States in 2014. Its acute phase of infection includes symptoms such as fever, headache, rash, arthralgia, arthritis, and myalgia.
The virus is likely to become a unique diagnostic challenge for rheumatologists as the arthritis symptoms mimic seronegative arthritis, the researchers said.
“Eight out of these patients would have met the 2010 ACR/EULAR criteria for RA [rheumatoid arthritis] if the initial fever, rash, and travel to the Caribbean had not been revealed,” they noted.
Most of the patients had joint pain and morning stiffness at least 8 weeks after initial infection, although some reported gradual improvement of their symptoms, the investigators reported.
One of the patients developed fever; diffuse arthritis; an erythematous, maculopapular rash; and severe symmetric polyarthritis that persisted more than 5 months post infection. She was treated with prednisolone but it exacerbated her joint pain and treatment was stopped.
The other patient detailed in the report presented with similar symptoms. His fever and rash resolved within 2 days but his arthritis symptoms persisted. He was treated with NSAIDs that provided only minimal relief.
To understand more about the immunologic parameters of the virus, the researchers used cytometry by time of flight to compare the peripheral mononuclear cells from CHIKV-infected patients with those of healthy controls and untreated patients with RA.
They discovered that naive, activated, and effector T killer– and T helper–cell populations occurred in similar percentages in the virus-infected patients and RA patients but not in the healthy controls.
The RA patients, however, had a higher expression of L-selectin expression in CD4+ T cells than did either virus-infected patients or healthy controls.
“These data suggest that lymphocyte phenotypes in patients with Chikungunya infection are similar to each other, with subtle but distinct trends that could potentially distinguish these two groups from healthy controls and from each other,” Dr. Miner and Dr. Aw-Yeang and their associates wrote.
Whether treatment with disease-modifying antirheumatic drugs or biologics used in RA is appropriate or effective is controversial in the absence of randomized controlled trials for CHIKV-related arthritis, they said.
“Immunosuppression in CHIKV-infected patients could be deleterious because viral RNA and antigens can be found in target tissues in the chronic phase in humans and experimental animals,” the authors wrote.
“Rheumatologists, even in non-CHIKV-endemic regions, need to consider CHIKV in their evaluation of symmetric polyarthritis lasting more than 6 weeks by obtaining a history of travel to CHIKV-endemic regions, which are likely to expand in the near future,” the investigators advised.
Patients who may have been exposed to the virus may need serologic testing before initiating immunosuppression, they said.
“Unfortunately, access to CHIKV testing is highly constrained at the current time as it is only available from the [Centers for Disease Control and Prevention] and research laboratories,” they added.
The study was supported by the Barnes-Jewish Hospital Foundation and the Howard Hughes Medical Institute. The authors reported no conflicts of interest.
Rheumatologists should consider the possibility of Chikungunya virus in patients who present with new symmetric polyarthritis, especially if they have just returned from an endemic region such as the Caribbean, according to researchers.
In their paper, first authors Dr. Jonathan J. Miner and Dr. Han-Xian Aw-Yeang and colleagues at Washington University, St. Louis, describe a cohort of 10 Americans who traveled to Haiti within a 20-day period in June 2014 and became infected with Chikungunya virus (CHIKV), an arthritogenic, mosquito-transmitted alphavirus (Arthritis Rheumatol. 2015 Jan. 20 [doi:10.1002/art.39027]).
The virus spread to the Caribbean in 2013 and the United States in 2014. Its acute phase of infection includes symptoms such as fever, headache, rash, arthralgia, arthritis, and myalgia.
The virus is likely to become a unique diagnostic challenge for rheumatologists as the arthritis symptoms mimic seronegative arthritis, the researchers said.
“Eight out of these patients would have met the 2010 ACR/EULAR criteria for RA [rheumatoid arthritis] if the initial fever, rash, and travel to the Caribbean had not been revealed,” they noted.
Most of the patients had joint pain and morning stiffness at least 8 weeks after initial infection, although some reported gradual improvement of their symptoms, the investigators reported.
One of the patients developed fever; diffuse arthritis; an erythematous, maculopapular rash; and severe symmetric polyarthritis that persisted more than 5 months post infection. She was treated with prednisolone but it exacerbated her joint pain and treatment was stopped.
The other patient detailed in the report presented with similar symptoms. His fever and rash resolved within 2 days but his arthritis symptoms persisted. He was treated with NSAIDs that provided only minimal relief.
To understand more about the immunologic parameters of the virus, the researchers used cytometry by time of flight to compare the peripheral mononuclear cells from CHIKV-infected patients with those of healthy controls and untreated patients with RA.
They discovered that naive, activated, and effector T killer– and T helper–cell populations occurred in similar percentages in the virus-infected patients and RA patients but not in the healthy controls.
The RA patients, however, had a higher expression of L-selectin expression in CD4+ T cells than did either virus-infected patients or healthy controls.
“These data suggest that lymphocyte phenotypes in patients with Chikungunya infection are similar to each other, with subtle but distinct trends that could potentially distinguish these two groups from healthy controls and from each other,” Dr. Miner and Dr. Aw-Yeang and their associates wrote.
Whether treatment with disease-modifying antirheumatic drugs or biologics used in RA is appropriate or effective is controversial in the absence of randomized controlled trials for CHIKV-related arthritis, they said.
“Immunosuppression in CHIKV-infected patients could be deleterious because viral RNA and antigens can be found in target tissues in the chronic phase in humans and experimental animals,” the authors wrote.
“Rheumatologists, even in non-CHIKV-endemic regions, need to consider CHIKV in their evaluation of symmetric polyarthritis lasting more than 6 weeks by obtaining a history of travel to CHIKV-endemic regions, which are likely to expand in the near future,” the investigators advised.
Patients who may have been exposed to the virus may need serologic testing before initiating immunosuppression, they said.
“Unfortunately, access to CHIKV testing is highly constrained at the current time as it is only available from the [Centers for Disease Control and Prevention] and research laboratories,” they added.
The study was supported by the Barnes-Jewish Hospital Foundation and the Howard Hughes Medical Institute. The authors reported no conflicts of interest.
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point: The mosquito-spread Chikungunya virus mimics seronegative rheumatoid arthritis. Rheumatologists should consider this diagnosis in patients who present with arthritis symptoms after visiting an endemic area.
Major finding: Eight out of the 10 people confirmed to be infected with the CHIKV virus met the 2010 ACR/EULAR criteria for RA.
Source: An observational study of a group of 10 Americans who traveled to Haiti within a 20-day period in June 2014 and became infected with CHIKV.
Disclosures: The study was supported by the Barnes-Jewish Hospital Foundation and the Howard Hughes Medical Institute. The authors reported no conflicts of interest.
RA clinical remission possible with half etanercept dose
Maintaining clinical remission with half-dose etanercept is possible for many rheumatoid arthritis patients, according to a single-center, prospective cohort study.
Over 80% of rheumatoid arthritis (RA) patients in remission who were randomized to a half dose treatment of 25 mg of etanercept per week were still in remission after an average follow-up period of 3.6 years, Dr. Bernd Raffeiner and his colleagues at the University of Padova (Italy) reported (Clin. Exp. Rheumatol. 2014 Dec. 22).
The study randomized 323 RA patients to etanercept 25 mg weekly (group A, n = 159) or etanercept 25 mg bi-weekly (group B, n = 164). The patients had a mean age of 56 years and about 82% were female. had failed traditional synthetic disease-modifying antirheumatic drugs and then achieved remission on etanercept 25 mg bi-weekly (50 mg per week). The investigators defined remission as a 28-joint disease activity score using erythrocyte sedimentation rate of less than 2.6 for at least 12 months.
At the end of follow-up, 81.8% of the patients in group A maintained remission for a mean of 3.6 years.
These patients were slightly younger than those who failed dose reduction and were taking fewer nonsteroidal anti-inflammatory drugs. Disease activity prior to biologic therapy did not influence response to half-dose etanercept.
The data showed that half-dose etanercept halted radiographic damage as effectively as standard dose, the researchers said. About 80% of patients reached radiographic remission, and almost 20% showed minor radiographic changes. Just 1.5% of patients experienced a relevant progression among all groups.
Patients who flared during dose reduction and returned to full dose did not show a greater radiographic progression.
“Low dose etanercept seems to prevent radiographic damage even if full clinical response is not achieved,” they wrote.
Furthermore, most (75.9%) of the patients who went back onto full-dose treatment regained remission.
Etanercept has unique pharmacokinetics and pharmacodynamics that are different from other TNF-α inhibitors, which could make it more suitable for dose adjustment, the study authors suggested.
The findings have important economic implications, they said. In Italy, low-dose etanercept is the lowest priced biologic treatment protocol available that meets the present standards in RA therapy.
“The resources gained could be invested for candidate patients for TNF-α inhibitor or other biologic therapy, especially in situations where policy maker intervention sets limitations to contain ever more increasing healthcare costs,” they wrote.
There were no conflicts of interest declared.
Maintaining clinical remission with half-dose etanercept is possible for many rheumatoid arthritis patients, according to a single-center, prospective cohort study.
Over 80% of rheumatoid arthritis (RA) patients in remission who were randomized to a half dose treatment of 25 mg of etanercept per week were still in remission after an average follow-up period of 3.6 years, Dr. Bernd Raffeiner and his colleagues at the University of Padova (Italy) reported (Clin. Exp. Rheumatol. 2014 Dec. 22).
The study randomized 323 RA patients to etanercept 25 mg weekly (group A, n = 159) or etanercept 25 mg bi-weekly (group B, n = 164). The patients had a mean age of 56 years and about 82% were female. had failed traditional synthetic disease-modifying antirheumatic drugs and then achieved remission on etanercept 25 mg bi-weekly (50 mg per week). The investigators defined remission as a 28-joint disease activity score using erythrocyte sedimentation rate of less than 2.6 for at least 12 months.
At the end of follow-up, 81.8% of the patients in group A maintained remission for a mean of 3.6 years.
These patients were slightly younger than those who failed dose reduction and were taking fewer nonsteroidal anti-inflammatory drugs. Disease activity prior to biologic therapy did not influence response to half-dose etanercept.
The data showed that half-dose etanercept halted radiographic damage as effectively as standard dose, the researchers said. About 80% of patients reached radiographic remission, and almost 20% showed minor radiographic changes. Just 1.5% of patients experienced a relevant progression among all groups.
Patients who flared during dose reduction and returned to full dose did not show a greater radiographic progression.
“Low dose etanercept seems to prevent radiographic damage even if full clinical response is not achieved,” they wrote.
Furthermore, most (75.9%) of the patients who went back onto full-dose treatment regained remission.
Etanercept has unique pharmacokinetics and pharmacodynamics that are different from other TNF-α inhibitors, which could make it more suitable for dose adjustment, the study authors suggested.
The findings have important economic implications, they said. In Italy, low-dose etanercept is the lowest priced biologic treatment protocol available that meets the present standards in RA therapy.
“The resources gained could be invested for candidate patients for TNF-α inhibitor or other biologic therapy, especially in situations where policy maker intervention sets limitations to contain ever more increasing healthcare costs,” they wrote.
There were no conflicts of interest declared.
Maintaining clinical remission with half-dose etanercept is possible for many rheumatoid arthritis patients, according to a single-center, prospective cohort study.
Over 80% of rheumatoid arthritis (RA) patients in remission who were randomized to a half dose treatment of 25 mg of etanercept per week were still in remission after an average follow-up period of 3.6 years, Dr. Bernd Raffeiner and his colleagues at the University of Padova (Italy) reported (Clin. Exp. Rheumatol. 2014 Dec. 22).
The study randomized 323 RA patients to etanercept 25 mg weekly (group A, n = 159) or etanercept 25 mg bi-weekly (group B, n = 164). The patients had a mean age of 56 years and about 82% were female. had failed traditional synthetic disease-modifying antirheumatic drugs and then achieved remission on etanercept 25 mg bi-weekly (50 mg per week). The investigators defined remission as a 28-joint disease activity score using erythrocyte sedimentation rate of less than 2.6 for at least 12 months.
At the end of follow-up, 81.8% of the patients in group A maintained remission for a mean of 3.6 years.
These patients were slightly younger than those who failed dose reduction and were taking fewer nonsteroidal anti-inflammatory drugs. Disease activity prior to biologic therapy did not influence response to half-dose etanercept.
The data showed that half-dose etanercept halted radiographic damage as effectively as standard dose, the researchers said. About 80% of patients reached radiographic remission, and almost 20% showed minor radiographic changes. Just 1.5% of patients experienced a relevant progression among all groups.
Patients who flared during dose reduction and returned to full dose did not show a greater radiographic progression.
“Low dose etanercept seems to prevent radiographic damage even if full clinical response is not achieved,” they wrote.
Furthermore, most (75.9%) of the patients who went back onto full-dose treatment regained remission.
Etanercept has unique pharmacokinetics and pharmacodynamics that are different from other TNF-α inhibitors, which could make it more suitable for dose adjustment, the study authors suggested.
The findings have important economic implications, they said. In Italy, low-dose etanercept is the lowest priced biologic treatment protocol available that meets the present standards in RA therapy.
“The resources gained could be invested for candidate patients for TNF-α inhibitor or other biologic therapy, especially in situations where policy maker intervention sets limitations to contain ever more increasing healthcare costs,” they wrote.
There were no conflicts of interest declared.
FROM CLINICAL AND EXPERIMENTAL RHEUMATOLOGY
Key clinical point: Half dose etanercept halts radiographic damage in RA as effectively as standard dose.
Major finding: A large majority (81.8%) of RA patients stayed in remission on half-dose etanercept (25 mg weekly).
Data source: A single-center, prospective, long-term follow-up study of 524 biologic-naïve RA patients who were randomized to half-dose etanercept (25 mg weekly) or 25 mg bi-weekly.
Disclosures: None declared.
MRI shows ongoing inflammation despite clinical remission in early RA
Two years of either triple therapy or treatment with tumor necrosis factor plus methotrexate failed to eliminate joint inflammation on MRI in a subcohort of patients with early rheumatoid arthritis from the randomized, double-blind Treatment of Early Aggressive Rheumatoid Arthritis (TEAR) trial.
In 118 patients with a mean age of 51 years, short disease duration, and severe disease at TEAR trial entry – 92% of whom were seropositive – only 29 had wrist pain, tenderness, or swelling at 2-year follow-up. However, all 118 patients had MRI evidence of residual joint inflammation after 2 years, and 78% had evidence of osteitis, Dr. Veena K. Ranganath of the University of California, Los Angeles, and her colleagues reported (Arthritis Care Res. 2015 Jan. 7 [doi:10.1002/acr.22541]).

Inflammation remained despite significant improvement of disease activity measures at the time of the MRI, compared with baseline (for example, 28-joint disease activity score using erythrocyte sedimentation rate [DAS28-ESR] decreased from 5.8 to 2.9). Total MRI inflammation scores were significantly lower in patients who met 2011 American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) Boolean remission criteria and remission by Chronic Disease Activity Index (CDAI), but not in those with DAS28-ESR remission, they noted.
The findings demonstrate that total MRI inflammatory scores are “best differentiated by the most stringent clinical remission criteria” – CDAI and 2011 ACR/EULAR Boolean Criteria, as opposed to DAS28-ESR (with a 2.6 cutpoint). Further, no differences were seen in damage or MRI inflammatory scores based on treatment regimen, which supports methotrexate-first recommendations for the TEAR trial, they said, noting that the long-term prognostic implications of the study findings are unclear because of short-follow-up, and that it remains unclear whether attainment of clinical remission warrants a drug holiday or cessation of RA treatment.
Thus, it is “ill-advised to discontinue therapy until future studies suggest otherwise,” they concluded, adding that this is particularly true given that prior published data suggest a link between osteitis – which occurred at a high rate in this study despite clinical remission – and future radiographic progression.
The TEAR trial was supported by Amgen. The current research was supported by a National Institutes of Health/National Center for Advancing Translational Science UCLA CTSI grant, and individual authors were supported by ACR/REF grants, a National Institutes of Health award, the Margaret J. Miller Endowed Professor of Research Chair, and the Agency for Healthcare Research and Quality.
Two years of either triple therapy or treatment with tumor necrosis factor plus methotrexate failed to eliminate joint inflammation on MRI in a subcohort of patients with early rheumatoid arthritis from the randomized, double-blind Treatment of Early Aggressive Rheumatoid Arthritis (TEAR) trial.
In 118 patients with a mean age of 51 years, short disease duration, and severe disease at TEAR trial entry – 92% of whom were seropositive – only 29 had wrist pain, tenderness, or swelling at 2-year follow-up. However, all 118 patients had MRI evidence of residual joint inflammation after 2 years, and 78% had evidence of osteitis, Dr. Veena K. Ranganath of the University of California, Los Angeles, and her colleagues reported (Arthritis Care Res. 2015 Jan. 7 [doi:10.1002/acr.22541]).
Inflammation remained despite significant improvement of disease activity measures at the time of the MRI, compared with baseline (for example, 28-joint disease activity score using erythrocyte sedimentation rate [DAS28-ESR] decreased from 5.8 to 2.9). Total MRI inflammation scores were significantly lower in patients who met 2011 American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) Boolean remission criteria and remission by Chronic Disease Activity Index (CDAI), but not in those with DAS28-ESR remission, they noted.
The findings demonstrate that total MRI inflammatory scores are “best differentiated by the most stringent clinical remission criteria” – CDAI and 2011 ACR/EULAR Boolean Criteria, as opposed to DAS28-ESR (with a 2.6 cutpoint). Further, no differences were seen in damage or MRI inflammatory scores based on treatment regimen, which supports methotrexate-first recommendations for the TEAR trial, they said, noting that the long-term prognostic implications of the study findings are unclear because of short-follow-up, and that it remains unclear whether attainment of clinical remission warrants a drug holiday or cessation of RA treatment.
Thus, it is “ill-advised to discontinue therapy until future studies suggest otherwise,” they concluded, adding that this is particularly true given that prior published data suggest a link between osteitis – which occurred at a high rate in this study despite clinical remission – and future radiographic progression.
The TEAR trial was supported by Amgen. The current research was supported by a National Institutes of Health/National Center for Advancing Translational Science UCLA CTSI grant, and individual authors were supported by ACR/REF grants, a National Institutes of Health award, the Margaret J. Miller Endowed Professor of Research Chair, and the Agency for Healthcare Research and Quality.
Two years of either triple therapy or treatment with tumor necrosis factor plus methotrexate failed to eliminate joint inflammation on MRI in a subcohort of patients with early rheumatoid arthritis from the randomized, double-blind Treatment of Early Aggressive Rheumatoid Arthritis (TEAR) trial.
In 118 patients with a mean age of 51 years, short disease duration, and severe disease at TEAR trial entry – 92% of whom were seropositive – only 29 had wrist pain, tenderness, or swelling at 2-year follow-up. However, all 118 patients had MRI evidence of residual joint inflammation after 2 years, and 78% had evidence of osteitis, Dr. Veena K. Ranganath of the University of California, Los Angeles, and her colleagues reported (Arthritis Care Res. 2015 Jan. 7 [doi:10.1002/acr.22541]).
Inflammation remained despite significant improvement of disease activity measures at the time of the MRI, compared with baseline (for example, 28-joint disease activity score using erythrocyte sedimentation rate [DAS28-ESR] decreased from 5.8 to 2.9). Total MRI inflammation scores were significantly lower in patients who met 2011 American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) Boolean remission criteria and remission by Chronic Disease Activity Index (CDAI), but not in those with DAS28-ESR remission, they noted.
The findings demonstrate that total MRI inflammatory scores are “best differentiated by the most stringent clinical remission criteria” – CDAI and 2011 ACR/EULAR Boolean Criteria, as opposed to DAS28-ESR (with a 2.6 cutpoint). Further, no differences were seen in damage or MRI inflammatory scores based on treatment regimen, which supports methotrexate-first recommendations for the TEAR trial, they said, noting that the long-term prognostic implications of the study findings are unclear because of short-follow-up, and that it remains unclear whether attainment of clinical remission warrants a drug holiday or cessation of RA treatment.
Thus, it is “ill-advised to discontinue therapy until future studies suggest otherwise,” they concluded, adding that this is particularly true given that prior published data suggest a link between osteitis – which occurred at a high rate in this study despite clinical remission – and future radiographic progression.
The TEAR trial was supported by Amgen. The current research was supported by a National Institutes of Health/National Center for Advancing Translational Science UCLA CTSI grant, and individual authors were supported by ACR/REF grants, a National Institutes of Health award, the Margaret J. Miller Endowed Professor of Research Chair, and the Agency for Healthcare Research and Quality.
FROM ARTHRITIS CARE & RESEARCH
Key clinical point: Until data suggest otherwise, treatment should continue despite clinical remission in early RA patients.
Major finding: Only 29 of 118 patients had symptoms, but all 118 had MRI evidence of inflammation.
Data source: A subcohort of 118 patients from the randomized, double-blind TEAR trial .
Disclosures: The TEAR trial was supported by Amgen. The current research was supported by a National Institutes of Health/National Center for Advancing Translational Science UCLA CTSI grant, and individual authors were supported by ACR/REF grants, a National Institutes of Health award, the Margaret J. Miller Endowed Professor of Research Chair, and the Agency for Healthcare Research and Quality.

Reducing abatacept feasible for poor-prognosis early RA patients
Patients with poor-prognosis early rheumatoid arthritis who have been on a regimen of abatacept for at least 1 year and have low disease activity scores can safely scale back their medication doses, according to the findings of a substudy of a phase III trial.
“Drug-free remission remains a therapeutic goal in RA [rheumatoid arthritis], [but] in established RA, withdrawal of biological therapy generally leads to loss of remission for the majority of patients,” wrote the study’s authors, led by Dr. René Westhovens of the Katholieke Universiteit in Leuven, Belgium. “However, dose reduction is a feasible strategy for some patients ... [and] in early RA, dose reduction is possible for the large majority of patients.”
Dr. Westhovens and his coinvestigators crafted a substudy from the existing AGREE (Abatacept study to gauge remission and joint damage progression in methotrexate-naive patients with early erosive rheumatoid arthritis) trial, using 35 of the 87 sites enrolling patients in the larger study. Substudy participants were required to have a 28-joint disease activity score with erythrocyte sedimentation rate (DAS28-ESR) of less than 2.6 after 2 years (701 days) in the AGREE trial (Ann. Rheum. Dis. 2014 Dec. 30 [doi:10.1136/annrheumdis-2014-206149]).
In total, 108 patients were randomized into two groups: Fifty-eight subjects stayed on the AGREE trial dose of about 10 mg/kg abatacept, and 50 subjects began a lower dose of about 5 mg/kg abatacept. Mean age of patients in these groups was 50-51 years, and 76%-82% were female. From the 10-mg/kg and 5-mg/kg groups, three and five patients, respectively, discontinued, while four per group returned to open-label abatacept. The primary outcome over 12 months was time to disease relapse, which was defined as additional disease-modifying antirheumatic drugs, at least two courses high-dose steroids, return to open-label abatacept at about 10 mg/kg, or a DAS28 (with C-reactive protein [CRP]) of at least 3.2 at two consecutive visits. At baseline, the mean DAS28-CRP score in each group was 2.1.
The proportion of subjects experiencing disease relapse did not differ significantly between the two groups: 18 out of 58 (31%) in the 10-mg/kg group, and 17 out of 50 (34%) in the 5-mg/kg group (hazard ratio, 0.87; 95% confidence interval, 0.45-1.69). That occurred despite an average steady-state trough serum concentration of 20.3-24.1 mcg/mL in the 10-mg/kg group and 8.8-12.0 mcg/mL in the 5-mg/kg group.
“Considering the potential to alter the course of disease in some patients with early RA, along with the safety and health economic benefits in avoiding unnecessary drug exposure, timely induction of biological agents (preferably in combination with methotrexate), followed by dose reduction, might be a therapeutic option in [appropriate] patients,” Dr. Westhovens and his associates wrote.
The AGREE study and related statistical analyses were funded and performed by Bristol-Myers Squibb. Several coauthors are affiliated with or employed by Bristol-Myers Squibb, and disclosed other potential conflicts of interest.
Patients with poor-prognosis early rheumatoid arthritis who have been on a regimen of abatacept for at least 1 year and have low disease activity scores can safely scale back their medication doses, according to the findings of a substudy of a phase III trial.
“Drug-free remission remains a therapeutic goal in RA [rheumatoid arthritis], [but] in established RA, withdrawal of biological therapy generally leads to loss of remission for the majority of patients,” wrote the study’s authors, led by Dr. René Westhovens of the Katholieke Universiteit in Leuven, Belgium. “However, dose reduction is a feasible strategy for some patients ... [and] in early RA, dose reduction is possible for the large majority of patients.”
Dr. Westhovens and his coinvestigators crafted a substudy from the existing AGREE (Abatacept study to gauge remission and joint damage progression in methotrexate-naive patients with early erosive rheumatoid arthritis) trial, using 35 of the 87 sites enrolling patients in the larger study. Substudy participants were required to have a 28-joint disease activity score with erythrocyte sedimentation rate (DAS28-ESR) of less than 2.6 after 2 years (701 days) in the AGREE trial (Ann. Rheum. Dis. 2014 Dec. 30 [doi:10.1136/annrheumdis-2014-206149]).
In total, 108 patients were randomized into two groups: Fifty-eight subjects stayed on the AGREE trial dose of about 10 mg/kg abatacept, and 50 subjects began a lower dose of about 5 mg/kg abatacept. Mean age of patients in these groups was 50-51 years, and 76%-82% were female. From the 10-mg/kg and 5-mg/kg groups, three and five patients, respectively, discontinued, while four per group returned to open-label abatacept. The primary outcome over 12 months was time to disease relapse, which was defined as additional disease-modifying antirheumatic drugs, at least two courses high-dose steroids, return to open-label abatacept at about 10 mg/kg, or a DAS28 (with C-reactive protein [CRP]) of at least 3.2 at two consecutive visits. At baseline, the mean DAS28-CRP score in each group was 2.1.
The proportion of subjects experiencing disease relapse did not differ significantly between the two groups: 18 out of 58 (31%) in the 10-mg/kg group, and 17 out of 50 (34%) in the 5-mg/kg group (hazard ratio, 0.87; 95% confidence interval, 0.45-1.69). That occurred despite an average steady-state trough serum concentration of 20.3-24.1 mcg/mL in the 10-mg/kg group and 8.8-12.0 mcg/mL in the 5-mg/kg group.
“Considering the potential to alter the course of disease in some patients with early RA, along with the safety and health economic benefits in avoiding unnecessary drug exposure, timely induction of biological agents (preferably in combination with methotrexate), followed by dose reduction, might be a therapeutic option in [appropriate] patients,” Dr. Westhovens and his associates wrote.
The AGREE study and related statistical analyses were funded and performed by Bristol-Myers Squibb. Several coauthors are affiliated with or employed by Bristol-Myers Squibb, and disclosed other potential conflicts of interest.
Patients with poor-prognosis early rheumatoid arthritis who have been on a regimen of abatacept for at least 1 year and have low disease activity scores can safely scale back their medication doses, according to the findings of a substudy of a phase III trial.
“Drug-free remission remains a therapeutic goal in RA [rheumatoid arthritis], [but] in established RA, withdrawal of biological therapy generally leads to loss of remission for the majority of patients,” wrote the study’s authors, led by Dr. René Westhovens of the Katholieke Universiteit in Leuven, Belgium. “However, dose reduction is a feasible strategy for some patients ... [and] in early RA, dose reduction is possible for the large majority of patients.”
Dr. Westhovens and his coinvestigators crafted a substudy from the existing AGREE (Abatacept study to gauge remission and joint damage progression in methotrexate-naive patients with early erosive rheumatoid arthritis) trial, using 35 of the 87 sites enrolling patients in the larger study. Substudy participants were required to have a 28-joint disease activity score with erythrocyte sedimentation rate (DAS28-ESR) of less than 2.6 after 2 years (701 days) in the AGREE trial (Ann. Rheum. Dis. 2014 Dec. 30 [doi:10.1136/annrheumdis-2014-206149]).
In total, 108 patients were randomized into two groups: Fifty-eight subjects stayed on the AGREE trial dose of about 10 mg/kg abatacept, and 50 subjects began a lower dose of about 5 mg/kg abatacept. Mean age of patients in these groups was 50-51 years, and 76%-82% were female. From the 10-mg/kg and 5-mg/kg groups, three and five patients, respectively, discontinued, while four per group returned to open-label abatacept. The primary outcome over 12 months was time to disease relapse, which was defined as additional disease-modifying antirheumatic drugs, at least two courses high-dose steroids, return to open-label abatacept at about 10 mg/kg, or a DAS28 (with C-reactive protein [CRP]) of at least 3.2 at two consecutive visits. At baseline, the mean DAS28-CRP score in each group was 2.1.
The proportion of subjects experiencing disease relapse did not differ significantly between the two groups: 18 out of 58 (31%) in the 10-mg/kg group, and 17 out of 50 (34%) in the 5-mg/kg group (hazard ratio, 0.87; 95% confidence interval, 0.45-1.69). That occurred despite an average steady-state trough serum concentration of 20.3-24.1 mcg/mL in the 10-mg/kg group and 8.8-12.0 mcg/mL in the 5-mg/kg group.
“Considering the potential to alter the course of disease in some patients with early RA, along with the safety and health economic benefits in avoiding unnecessary drug exposure, timely induction of biological agents (preferably in combination with methotrexate), followed by dose reduction, might be a therapeutic option in [appropriate] patients,” Dr. Westhovens and his associates wrote.
The AGREE study and related statistical analyses were funded and performed by Bristol-Myers Squibb. Several coauthors are affiliated with or employed by Bristol-Myers Squibb, and disclosed other potential conflicts of interest.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point: Patients with poor-prognosis early rheumatoid arthritis and DAS28-ESR of less than 2.6 after at least 1 year on abatacept (10 mg/kg) should have the option of reducing their dose.
Major finding: The proportion of subjects experiencing disease relapse did not differ significantly between the 10-mg/kg and the 5-mg/kg groups (31% vs. 34%, respectively; HR, 0.87; 95% confidence interval, 0.45-1.69).
Data source: A 1-year, multinational, randomized, double-blind, two-arm, parallel-dosing, exploratory substudy of 108 patients in the AGREE trial.
Disclosures: Several coauthors disclosed ties with Bristol-Myers Squibb, which markets abatacept and funded the study and related analyses.
Antibody elimination may mark radiographic outcomes in RA
The elimination of anticitrullinated vimentin antibodies during early antirheumatic therapy in rheumatoid arthritis patients may significantly increase the likelihood of positive radiographic outcomes, according to findings from a longitudinal cohort study.
“The development of serological tests for [autoantibodies recognizing citrullinated proteins (ACPA)] detection have improved the ability to diagnose rheumatoid arthritis [RA] at an early stage and to identify patients at increased risk of a severe disease course,” wrote lead author Dr. Alf Kastbom of the Karolinska Institute in Stockholm, and his associates (Ann. Rheum. Dis. 2014 Dec. 30 [doi:10.1136/annrheumdis-2014-205698]). “These clinically very useful tests do not, however, formally address the question of which specific citrullinated autoantigens are targeted.”
Dr. Kastbom and his coinvestigators longitudinally analyzed serum samples from 316 of 487 patients with early RA (symptoms < 1 year) who had participated in the Swedish Farmacotherapy (SWEFOT) trial, a randomized, nonblinded trial. These 316 patients had baseline and 3-month serum samples available that were analyzed for any antibodies against cyclic citrullinated peptides (CCP) and citrullinated peptides that were derived from vimentin (cVim), fibrinogen (cFib), and alpha-enolase (CEP-1).
For the first 3 months, all patients received methotrexate. Next, patients who were deemed “methotrexate monotherapy–inadequate responders” were subsequently randomized into cohorts that received add-on therapy with either sulfasalazine and hydroxychloroquine or infliximab. The investigators again assessed ACPA levels at 12 and 24 months after commencement of therapy in these subjects and used this 2-year follow-up period to compare proportions of antibody-positive patients and relative changes in antibody levels across ACPA specificities, as well as therapeutic responses and radiographic progressions.
Dr. Kastbom and his coauthors found that changes in antibody status within the first 3 months were most closely associated with radiographic progression of RA over the 2-year study period. Specifically, subjects who experienced significant decreases in anti-cVim antibodies in the first 3 months had lower rates of radiographic progression and lower total van der Heijde modified Sharp score (SHS) after 2 years than did those whose anti-cVim antibody levels remained positive (P = .012 and P = .015, respectively). Anti-cVim antibody results were also adjusted for baseline 28-joint disease activity score, baseline erosions, current smoking status, sex, and European League Against Rheumatism response at 3 months (P = .005 for SHS progression > 1 and P = .013 for SHS progression > 5, respectively).
However, no significant relationships with radiographic progression were found in the anti-cFib and anti-CEP-1 antibody seroreversions. Median antibody levels of all tested ACPAs declined during initial methotrexate therapy and after commencement of add-on therapy, but the investigators found no significant association between treatment regimen and radiographic progression.
“It is intriguing to see that our study clearly demonstrates an association between the early disappearance of specific anti-cVim antibodies and less radiological progression,” wrote Dr. Kastbom and his colleagues, adding that the findings of this study should “encourage further work to explore the prognostic potential of repeated measurements of different ACPA specificities in early RA.”
This study was financed by grants from European Union–funded FP7 projects and several Swedish institutions. Several coauthors also disclosed individual ties to various pharmaceutical companies.
The elimination of anticitrullinated vimentin antibodies during early antirheumatic therapy in rheumatoid arthritis patients may significantly increase the likelihood of positive radiographic outcomes, according to findings from a longitudinal cohort study.
“The development of serological tests for [autoantibodies recognizing citrullinated proteins (ACPA)] detection have improved the ability to diagnose rheumatoid arthritis [RA] at an early stage and to identify patients at increased risk of a severe disease course,” wrote lead author Dr. Alf Kastbom of the Karolinska Institute in Stockholm, and his associates (Ann. Rheum. Dis. 2014 Dec. 30 [doi:10.1136/annrheumdis-2014-205698]). “These clinically very useful tests do not, however, formally address the question of which specific citrullinated autoantigens are targeted.”
Dr. Kastbom and his coinvestigators longitudinally analyzed serum samples from 316 of 487 patients with early RA (symptoms < 1 year) who had participated in the Swedish Farmacotherapy (SWEFOT) trial, a randomized, nonblinded trial. These 316 patients had baseline and 3-month serum samples available that were analyzed for any antibodies against cyclic citrullinated peptides (CCP) and citrullinated peptides that were derived from vimentin (cVim), fibrinogen (cFib), and alpha-enolase (CEP-1).
For the first 3 months, all patients received methotrexate. Next, patients who were deemed “methotrexate monotherapy–inadequate responders” were subsequently randomized into cohorts that received add-on therapy with either sulfasalazine and hydroxychloroquine or infliximab. The investigators again assessed ACPA levels at 12 and 24 months after commencement of therapy in these subjects and used this 2-year follow-up period to compare proportions of antibody-positive patients and relative changes in antibody levels across ACPA specificities, as well as therapeutic responses and radiographic progressions.
Dr. Kastbom and his coauthors found that changes in antibody status within the first 3 months were most closely associated with radiographic progression of RA over the 2-year study period. Specifically, subjects who experienced significant decreases in anti-cVim antibodies in the first 3 months had lower rates of radiographic progression and lower total van der Heijde modified Sharp score (SHS) after 2 years than did those whose anti-cVim antibody levels remained positive (P = .012 and P = .015, respectively). Anti-cVim antibody results were also adjusted for baseline 28-joint disease activity score, baseline erosions, current smoking status, sex, and European League Against Rheumatism response at 3 months (P = .005 for SHS progression > 1 and P = .013 for SHS progression > 5, respectively).
However, no significant relationships with radiographic progression were found in the anti-cFib and anti-CEP-1 antibody seroreversions. Median antibody levels of all tested ACPAs declined during initial methotrexate therapy and after commencement of add-on therapy, but the investigators found no significant association between treatment regimen and radiographic progression.
“It is intriguing to see that our study clearly demonstrates an association between the early disappearance of specific anti-cVim antibodies and less radiological progression,” wrote Dr. Kastbom and his colleagues, adding that the findings of this study should “encourage further work to explore the prognostic potential of repeated measurements of different ACPA specificities in early RA.”
This study was financed by grants from European Union–funded FP7 projects and several Swedish institutions. Several coauthors also disclosed individual ties to various pharmaceutical companies.
The elimination of anticitrullinated vimentin antibodies during early antirheumatic therapy in rheumatoid arthritis patients may significantly increase the likelihood of positive radiographic outcomes, according to findings from a longitudinal cohort study.
“The development of serological tests for [autoantibodies recognizing citrullinated proteins (ACPA)] detection have improved the ability to diagnose rheumatoid arthritis [RA] at an early stage and to identify patients at increased risk of a severe disease course,” wrote lead author Dr. Alf Kastbom of the Karolinska Institute in Stockholm, and his associates (Ann. Rheum. Dis. 2014 Dec. 30 [doi:10.1136/annrheumdis-2014-205698]). “These clinically very useful tests do not, however, formally address the question of which specific citrullinated autoantigens are targeted.”
Dr. Kastbom and his coinvestigators longitudinally analyzed serum samples from 316 of 487 patients with early RA (symptoms < 1 year) who had participated in the Swedish Farmacotherapy (SWEFOT) trial, a randomized, nonblinded trial. These 316 patients had baseline and 3-month serum samples available that were analyzed for any antibodies against cyclic citrullinated peptides (CCP) and citrullinated peptides that were derived from vimentin (cVim), fibrinogen (cFib), and alpha-enolase (CEP-1).
For the first 3 months, all patients received methotrexate. Next, patients who were deemed “methotrexate monotherapy–inadequate responders” were subsequently randomized into cohorts that received add-on therapy with either sulfasalazine and hydroxychloroquine or infliximab. The investigators again assessed ACPA levels at 12 and 24 months after commencement of therapy in these subjects and used this 2-year follow-up period to compare proportions of antibody-positive patients and relative changes in antibody levels across ACPA specificities, as well as therapeutic responses and radiographic progressions.
Dr. Kastbom and his coauthors found that changes in antibody status within the first 3 months were most closely associated with radiographic progression of RA over the 2-year study period. Specifically, subjects who experienced significant decreases in anti-cVim antibodies in the first 3 months had lower rates of radiographic progression and lower total van der Heijde modified Sharp score (SHS) after 2 years than did those whose anti-cVim antibody levels remained positive (P = .012 and P = .015, respectively). Anti-cVim antibody results were also adjusted for baseline 28-joint disease activity score, baseline erosions, current smoking status, sex, and European League Against Rheumatism response at 3 months (P = .005 for SHS progression > 1 and P = .013 for SHS progression > 5, respectively).
However, no significant relationships with radiographic progression were found in the anti-cFib and anti-CEP-1 antibody seroreversions. Median antibody levels of all tested ACPAs declined during initial methotrexate therapy and after commencement of add-on therapy, but the investigators found no significant association between treatment regimen and radiographic progression.
“It is intriguing to see that our study clearly demonstrates an association between the early disappearance of specific anti-cVim antibodies and less radiological progression,” wrote Dr. Kastbom and his colleagues, adding that the findings of this study should “encourage further work to explore the prognostic potential of repeated measurements of different ACPA specificities in early RA.”
This study was financed by grants from European Union–funded FP7 projects and several Swedish institutions. Several coauthors also disclosed individual ties to various pharmaceutical companies.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point: Early disappearance of anti-cVim antibodies can lead to better radiological outcomes in patients in the early stages of rheumatoid arthritis.
Major finding: Anti-cVim antibody seroreversion during the first 3 months of treatment was associated with significantly less 2-year radiographic progression, compared with patients who remained antibody positive.
Data source: A longitudinal cohort study of 316 participants in the randomized, nonblinded SWEFOT trial.
Disclosures: This study was financed by grants from European Union–funded FP7 projects and several Swedish institutions. Several coauthors also disclosed individual ties to various pharmaceutical companies.
Rx for specialists: Know how ACA affects patients’ ability to pay for meds
ORLANDO – Despite recent, significant shifts in health care coverage thanks to the Affordable Care Act, many specialists are unaware of how patients pay for pricey prescriptions such as biologics.
One reason is that clinicians just haven’t been paying enough attention, according to Dr. Kim L. Isaacs, codirector of the multidiscipline treatment and research center for inflammatory bowel disease at the University of North Carolina.
“It gets complicated because we’re taking care of patients, so it’s hard to think about the financial end of things as well, and it lands on the patient’s lap,” Dr. Isaacs said in an interview after her presentation, “Navigating the Affordable Care Act,” at a conference on inflammatory bowel diseases sponsored by the Crohn’s and Colitis Foundation of America.
But not knowing how and if chronically ill patients can afford to pay for their medications can impact their compliance, and even their disease states.
Dr. Isaacs compared the histories of two patients. The first one purchased through the ACA’s health insurance marketplace provision, and regardless of her preexisting condition, she was able to receive and afford treatment for her Crohn’s disease for the first time in a decade. The second patient purchased ACA-sanctioned insurance but it still wasn’t enough to cover the costs of her care.
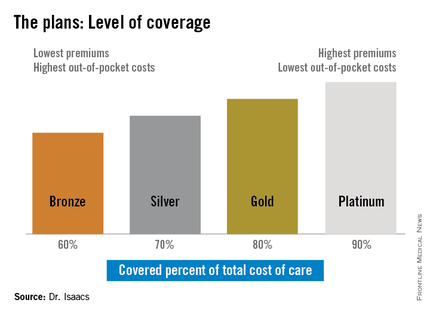
“She told me she had thought the Silver Plan would work for her, but that she still couldn’t afford her medications, and I was thinking, ‘What’s a Silver Plan?’ ” Dr. Isaacs said. “I didn’t have a clue.”
The discrepancy between the patients’ plans prompted Dr. Isaacs to investigate whether what she was prescribing was practical under her patients’ various levels of coverage. She discovered that under the ACA, the individual mandate requiring all Americans to purchase some form of health insurance means many have turned to a variety of state- and federally-sponsored health insurance marketplaces that offer coverage plans ranging from Bronze to Platinum.
“If you ask [most specialists] what the Gold Plan is, most of them won’t have heard of it, they don’t know,” she said.

But she said that physicians need to be aware of whether the drugs they are prescribing are on the formulary used by their patients’ respective plans since the different insurance providers that back the various plans often don’t cover the same medications.
In addition, the so-called “donut hole,” the annually adjusted gap in prescription drug coverage for Medicare patients, is not scheduled to close until 2020. While the gap exists, Medicare patients have a set amount of annual drug coverage after which the patient shares a substantial portion of the cost until the following year when the process begins again.
“For our IBD patients, this is very important because for some of our more expensive drugs, there may be a period of time [annually] when the drugs are not covered,” Dr. Isaacs said.
The same could be true for other specialties such as oncology, rheumatology, and neurology, where disease states require high-cost specialty drugs; however, some patient advocacy groups will assist patients in paying for their treatment, she said.
Patients who purchase their health plans through the government-sponsored insurance marketplaces are usually eligible for subsidies, depending on their income and where they fall in relation to the federally set poverty level, said Dr. Isaacs.
Another important, basic point, she added, is that providers need to ensure that they are on their patients’ chosen plans. “If you’re not, then you need to be sure there is someone else who is on the plan who can take care of your patient.”
Dr. Isaacs reported she has affiliations with AbbVie, Janssen, Millennium, Takeda, UCB, and others.
On Twitter @whitneymcknight
ORLANDO – Despite recent, significant shifts in health care coverage thanks to the Affordable Care Act, many specialists are unaware of how patients pay for pricey prescriptions such as biologics.
One reason is that clinicians just haven’t been paying enough attention, according to Dr. Kim L. Isaacs, codirector of the multidiscipline treatment and research center for inflammatory bowel disease at the University of North Carolina.
“It gets complicated because we’re taking care of patients, so it’s hard to think about the financial end of things as well, and it lands on the patient’s lap,” Dr. Isaacs said in an interview after her presentation, “Navigating the Affordable Care Act,” at a conference on inflammatory bowel diseases sponsored by the Crohn’s and Colitis Foundation of America.
But not knowing how and if chronically ill patients can afford to pay for their medications can impact their compliance, and even their disease states.
Dr. Isaacs compared the histories of two patients. The first one purchased through the ACA’s health insurance marketplace provision, and regardless of her preexisting condition, she was able to receive and afford treatment for her Crohn’s disease for the first time in a decade. The second patient purchased ACA-sanctioned insurance but it still wasn’t enough to cover the costs of her care.
“She told me she had thought the Silver Plan would work for her, but that she still couldn’t afford her medications, and I was thinking, ‘What’s a Silver Plan?’ ” Dr. Isaacs said. “I didn’t have a clue.”
The discrepancy between the patients’ plans prompted Dr. Isaacs to investigate whether what she was prescribing was practical under her patients’ various levels of coverage. She discovered that under the ACA, the individual mandate requiring all Americans to purchase some form of health insurance means many have turned to a variety of state- and federally-sponsored health insurance marketplaces that offer coverage plans ranging from Bronze to Platinum.
“If you ask [most specialists] what the Gold Plan is, most of them won’t have heard of it, they don’t know,” she said.
But she said that physicians need to be aware of whether the drugs they are prescribing are on the formulary used by their patients’ respective plans since the different insurance providers that back the various plans often don’t cover the same medications.
In addition, the so-called “donut hole,” the annually adjusted gap in prescription drug coverage for Medicare patients, is not scheduled to close until 2020. While the gap exists, Medicare patients have a set amount of annual drug coverage after which the patient shares a substantial portion of the cost until the following year when the process begins again.
“For our IBD patients, this is very important because for some of our more expensive drugs, there may be a period of time [annually] when the drugs are not covered,” Dr. Isaacs said.
The same could be true for other specialties such as oncology, rheumatology, and neurology, where disease states require high-cost specialty drugs; however, some patient advocacy groups will assist patients in paying for their treatment, she said.
Patients who purchase their health plans through the government-sponsored insurance marketplaces are usually eligible for subsidies, depending on their income and where they fall in relation to the federally set poverty level, said Dr. Isaacs.
Another important, basic point, she added, is that providers need to ensure that they are on their patients’ chosen plans. “If you’re not, then you need to be sure there is someone else who is on the plan who can take care of your patient.”
Dr. Isaacs reported she has affiliations with AbbVie, Janssen, Millennium, Takeda, UCB, and others.
On Twitter @whitneymcknight
ORLANDO – Despite recent, significant shifts in health care coverage thanks to the Affordable Care Act, many specialists are unaware of how patients pay for pricey prescriptions such as biologics.
One reason is that clinicians just haven’t been paying enough attention, according to Dr. Kim L. Isaacs, codirector of the multidiscipline treatment and research center for inflammatory bowel disease at the University of North Carolina.
“It gets complicated because we’re taking care of patients, so it’s hard to think about the financial end of things as well, and it lands on the patient’s lap,” Dr. Isaacs said in an interview after her presentation, “Navigating the Affordable Care Act,” at a conference on inflammatory bowel diseases sponsored by the Crohn’s and Colitis Foundation of America.
But not knowing how and if chronically ill patients can afford to pay for their medications can impact their compliance, and even their disease states.
Dr. Isaacs compared the histories of two patients. The first one purchased through the ACA’s health insurance marketplace provision, and regardless of her preexisting condition, she was able to receive and afford treatment for her Crohn’s disease for the first time in a decade. The second patient purchased ACA-sanctioned insurance but it still wasn’t enough to cover the costs of her care.
“She told me she had thought the Silver Plan would work for her, but that she still couldn’t afford her medications, and I was thinking, ‘What’s a Silver Plan?’ ” Dr. Isaacs said. “I didn’t have a clue.”
The discrepancy between the patients’ plans prompted Dr. Isaacs to investigate whether what she was prescribing was practical under her patients’ various levels of coverage. She discovered that under the ACA, the individual mandate requiring all Americans to purchase some form of health insurance means many have turned to a variety of state- and federally-sponsored health insurance marketplaces that offer coverage plans ranging from Bronze to Platinum.
“If you ask [most specialists] what the Gold Plan is, most of them won’t have heard of it, they don’t know,” she said.
But she said that physicians need to be aware of whether the drugs they are prescribing are on the formulary used by their patients’ respective plans since the different insurance providers that back the various plans often don’t cover the same medications.
In addition, the so-called “donut hole,” the annually adjusted gap in prescription drug coverage for Medicare patients, is not scheduled to close until 2020. While the gap exists, Medicare patients have a set amount of annual drug coverage after which the patient shares a substantial portion of the cost until the following year when the process begins again.
“For our IBD patients, this is very important because for some of our more expensive drugs, there may be a period of time [annually] when the drugs are not covered,” Dr. Isaacs said.
The same could be true for other specialties such as oncology, rheumatology, and neurology, where disease states require high-cost specialty drugs; however, some patient advocacy groups will assist patients in paying for their treatment, she said.
Patients who purchase their health plans through the government-sponsored insurance marketplaces are usually eligible for subsidies, depending on their income and where they fall in relation to the federally set poverty level, said Dr. Isaacs.
Another important, basic point, she added, is that providers need to ensure that they are on their patients’ chosen plans. “If you’re not, then you need to be sure there is someone else who is on the plan who can take care of your patient.”
Dr. Isaacs reported she has affiliations with AbbVie, Janssen, Millennium, Takeda, UCB, and others.
On Twitter @whitneymcknight
EXPERT ANALYSIS FROM 2014 ADVANCES IN IBD
Number, not level, of positive antibodies predicts mortality in early RA
Seropositivity for both rheumatoid factor and anti-citrullinated protein antibody was associated with increased mortality, compared with single positivity or seronegativity in patients with early inflammatory arthritis in two large, independent observational cohorts.
However, higher levels of the antibodies did not appear to be associated with early deaths as the influence of increasing antibody level was not consistent in the two cohorts, Dr. Jennifer H. Humphreys of the University of Manchester (England) and her colleagues reported (Arthritis Res. Ther. 2014 Dec. 4 [doi:10.1186/s13075-014-0483-3]).

Among 3,053 patients in the Norfolk Arthritis Register (NOAR) cohort and 1,909 in the Leiden Early Arthritis Clinic (EAC) cohort, double antibody positivity – which occurred in 35% and 42% of patients in the cohorts, respectively – was associated with excess mortality (adjusted hazard ratios, 1.35 and 1.57). However, when antibody status was negative, low-positive, or high-positive, findings in one cohort were not replicated in the other. For example, there was a marked difference in rheumatoid factor high and low positivity in the NOAR cohort (adjusted HRs of 0.80 and 1.49, respectively), but not in the EAC cohort (adjusted HRs of 1.62 and 1.63, respectively), the investigators said.
Median patient age at symptom onset was 56 years in NOAR and 54 years in EAC, and 65% and 63% of subjects in the cohorts, respectively, were women. They were followed for a mean of 11.8 years and 8.5 years, until death or censor date, and analyses were adjusted for age, sex, smoking status, inflammatory markers, and year of enrollment.
The findings suggest that in patients presenting with early rheumatoid arthritis, mortality risk may be better assessed by the number of positive antibodies than by the antibody levels, the investigators concluded.
NOAR is funded by Arthritis Research UK grants, as is Dr. Humphreys. The current study was also funded by the European Community Seventh Framework Program FP7 and a Vidi grant of the Netherlands Organization for Scientific Research. The authors reported having no other disclosures.
Seropositivity for both rheumatoid factor and anti-citrullinated protein antibody was associated with increased mortality, compared with single positivity or seronegativity in patients with early inflammatory arthritis in two large, independent observational cohorts.
However, higher levels of the antibodies did not appear to be associated with early deaths as the influence of increasing antibody level was not consistent in the two cohorts, Dr. Jennifer H. Humphreys of the University of Manchester (England) and her colleagues reported (Arthritis Res. Ther. 2014 Dec. 4 [doi:10.1186/s13075-014-0483-3]).
Among 3,053 patients in the Norfolk Arthritis Register (NOAR) cohort and 1,909 in the Leiden Early Arthritis Clinic (EAC) cohort, double antibody positivity – which occurred in 35% and 42% of patients in the cohorts, respectively – was associated with excess mortality (adjusted hazard ratios, 1.35 and 1.57). However, when antibody status was negative, low-positive, or high-positive, findings in one cohort were not replicated in the other. For example, there was a marked difference in rheumatoid factor high and low positivity in the NOAR cohort (adjusted HRs of 0.80 and 1.49, respectively), but not in the EAC cohort (adjusted HRs of 1.62 and 1.63, respectively), the investigators said.
Median patient age at symptom onset was 56 years in NOAR and 54 years in EAC, and 65% and 63% of subjects in the cohorts, respectively, were women. They were followed for a mean of 11.8 years and 8.5 years, until death or censor date, and analyses were adjusted for age, sex, smoking status, inflammatory markers, and year of enrollment.
The findings suggest that in patients presenting with early rheumatoid arthritis, mortality risk may be better assessed by the number of positive antibodies than by the antibody levels, the investigators concluded.
NOAR is funded by Arthritis Research UK grants, as is Dr. Humphreys. The current study was also funded by the European Community Seventh Framework Program FP7 and a Vidi grant of the Netherlands Organization for Scientific Research. The authors reported having no other disclosures.
Seropositivity for both rheumatoid factor and anti-citrullinated protein antibody was associated with increased mortality, compared with single positivity or seronegativity in patients with early inflammatory arthritis in two large, independent observational cohorts.
However, higher levels of the antibodies did not appear to be associated with early deaths as the influence of increasing antibody level was not consistent in the two cohorts, Dr. Jennifer H. Humphreys of the University of Manchester (England) and her colleagues reported (Arthritis Res. Ther. 2014 Dec. 4 [doi:10.1186/s13075-014-0483-3]).
Among 3,053 patients in the Norfolk Arthritis Register (NOAR) cohort and 1,909 in the Leiden Early Arthritis Clinic (EAC) cohort, double antibody positivity – which occurred in 35% and 42% of patients in the cohorts, respectively – was associated with excess mortality (adjusted hazard ratios, 1.35 and 1.57). However, when antibody status was negative, low-positive, or high-positive, findings in one cohort were not replicated in the other. For example, there was a marked difference in rheumatoid factor high and low positivity in the NOAR cohort (adjusted HRs of 0.80 and 1.49, respectively), but not in the EAC cohort (adjusted HRs of 1.62 and 1.63, respectively), the investigators said.
Median patient age at symptom onset was 56 years in NOAR and 54 years in EAC, and 65% and 63% of subjects in the cohorts, respectively, were women. They were followed for a mean of 11.8 years and 8.5 years, until death or censor date, and analyses were adjusted for age, sex, smoking status, inflammatory markers, and year of enrollment.
The findings suggest that in patients presenting with early rheumatoid arthritis, mortality risk may be better assessed by the number of positive antibodies than by the antibody levels, the investigators concluded.
NOAR is funded by Arthritis Research UK grants, as is Dr. Humphreys. The current study was also funded by the European Community Seventh Framework Program FP7 and a Vidi grant of the Netherlands Organization for Scientific Research. The authors reported having no other disclosures.
FROM ARTHRITIS RESEARCH & THERAPY
Key clinical point: Dual rheumatoid factor and anti-citrullinated protein antibody positivity may be better than antibody levels for predicting mortality risk in patients with early inflammatory arthritis.
Major finding: Double antibody positivity was associated with excess mortality in the NOAR and EAC cohorts (adjusted hazard ratios, 1.35 and 1.57).
Data source: Two observational cohorts totaling 4,962 patients.
Disclosures: NOAR is funded by Arthritis Research UK grants, as is Dr. Humphreys. The current study was also funded by the European Community Seventh Framework Program FP7 and a Vidi grant of the Netherlands Organization for Scientific Research. The authors reported having no other disclosures.

FDA issues new pregnancy/lactation drug label standards
The U.S. Food and Drug Administration has issued a final rule requiring content and format changes to pregnancy and lactation labeling information for prescription drugs and biologic products.
The long-awaited “Content and Format of Labeling for Human Prescription Drug and Biological Products; Requirements for Pregnancy and Lactation Labeling, or the Pregnancy and Lactation Labeling Rule”(PLLR) is part of broad effort by the FDA to improve the content and format of prescription drug labeling. The PLLR, which finalizes many of the provisions in a proposed rule issued in May 2008 after input from numerous stakeholders, calls for replacement of the current A, B, C, D, and X drug classification system with more detailed information about the risks and benefits of use during pregnancy and breastfeeding.
The rule will take effect June 30, 2015.

Under the PLLR, labels will be required to include three detailed subsections entitled Pregnancy, Lactation, and Females and Males of Reproductive Potential. Each will include a risk summary, a discussion of the supporting data, and relevant information to help providers make prescribing and counseling decisions, according to the FDA. If no data are available to guide decision making, this must be stated.
The Pregnancy subsection combines the existing Pregnancy and Labor and Delivery subsections, and will address use of the drug during pregnancy as well as provide information about relevant registries that collect and maintain data on the use of the product in pregnant women. The Lactation subsection replaces the existing Nursing Mothers subsection, and will include information about use of the product during breastfeeding, including the amount of drug in breast milk and potential effects on the breastfed child. The new Females and Males of Reproductive Potential subsection will address pregnancy testing, contraception, and fertility issues as they relate to use of the product.
The existing A, B, C, D, and X categories were frequently misinterpreted as a grading system, giving an over simplified view of product risk, according to Dr. Sandra Kweder, deputy director of the Office of New Drugs in the FDA’s Center for Drug Evaluation and Research.
The new, more detailed approach to labeling will better address the complex risk-benefit considerations inherent in prescribing decisions during pregnancy and lactation, she said during a press briefing.
“I’m excited because clinicians will, going forward, be able to rely on FDA-approved drug labeling for comprehensive, chronically relevant, and user-friendly information in this part of labeling – something that has been missing for many years,” she said, noting that the changes are particularly important, given that the more than 6 million women who become pregnant each year in the United States take an average of 3-5 different prescription products during the course of their pregnancy and while breastfeeding.
“It is our hope that this new system will help their health care professionals and these women as they discuss treatment options,” she said.
Importantly, the PLLR ensures that more robust and informative data about drugs will be provided than ever before – and in a manner that speaks directly to the concerns that are common among providers, she said.
In addition to the elimination of the letter categories and the addition of the three new subsections, the use of standardized risk statement also was eliminated, as these had the same limitations as the letter categories. A section on inadvertent exposure also was eliminated due to redundancy, as the risk would be the same as with intentional exposure.
The rule also requires that labels be updated as they become outdated.
Much of the information that will be included on the new labels, which will be phased in for existing drugs and required immediately for drugs approved after June 30, 2015, was already included, but was scattered and difficult to find. The new formatting requirements provides for consistency across labels by pulling this information together in one place, Dr. Kweder said.
In an official statement, the American College of Obstetricians and Gynecologists applauded the rule for “taking needed steps to increase understanding about the effect of prescription medicine on women during pregnancy and lactation.”
“The FDA’s updated method of presenting information about both risk and benefit will improve the ability of all physicians to treat their pregnant and breastfeeding patients, as well as women who may become pregnant. It will also help more women to understand and take part in their health care decision making,” according to the ACOG statement, which also noted that the organization hopes the new content on prescription drug and biological product labels will “provide added incentives for clinical research as well as participation in patient registries.”
Christina Chambers, Ph.D., professor of pediatrics and director of clinical research for the department of pediatrics at the University of California, San Diego, also praised the new labeling rule, noting in an interview that “the final rule has been long awaited by many who work in the field of counseling pregnant and breastfeeding women about risks and safety of prescription medications, such as counselors with organizations like MotherToBaby, a service of the Organization of Teratology Information Specialists, which provides information about medication and other exposures during pregnancy and breastfeeding, and which was involved in development of the final rule.
“The MotherToBaby counselors located throughout the United States who answer questions about medication exposures for hundreds of women every day, have struggled for years with trying to explain the not-so-useful A, B, C, D, X pregnancy categories to patients and providers alike who commonly misinterpret their meaning. The new label format is much more content rich and evidence-based, and encompasses the larger picture of the safety data in the context of treatment (or lack of treatment) of the maternal condition. This is a huge step forward – and will make even more clear how critical the need is for more human pregnancy data for all medications likely to be used by women of reproductive age,” she said.
Dr. Chambers is the program director for MotherToBaby California, and director of the MotherToBaby research center at the University of California, San Diego. She reported having no relevant disclosures.
The U.S. Food and Drug Administration has issued a final rule requiring content and format changes to pregnancy and lactation labeling information for prescription drugs and biologic products.
The long-awaited “Content and Format of Labeling for Human Prescription Drug and Biological Products; Requirements for Pregnancy and Lactation Labeling, or the Pregnancy and Lactation Labeling Rule”(PLLR) is part of broad effort by the FDA to improve the content and format of prescription drug labeling. The PLLR, which finalizes many of the provisions in a proposed rule issued in May 2008 after input from numerous stakeholders, calls for replacement of the current A, B, C, D, and X drug classification system with more detailed information about the risks and benefits of use during pregnancy and breastfeeding.
The rule will take effect June 30, 2015.
Under the PLLR, labels will be required to include three detailed subsections entitled Pregnancy, Lactation, and Females and Males of Reproductive Potential. Each will include a risk summary, a discussion of the supporting data, and relevant information to help providers make prescribing and counseling decisions, according to the FDA. If no data are available to guide decision making, this must be stated.
The Pregnancy subsection combines the existing Pregnancy and Labor and Delivery subsections, and will address use of the drug during pregnancy as well as provide information about relevant registries that collect and maintain data on the use of the product in pregnant women. The Lactation subsection replaces the existing Nursing Mothers subsection, and will include information about use of the product during breastfeeding, including the amount of drug in breast milk and potential effects on the breastfed child. The new Females and Males of Reproductive Potential subsection will address pregnancy testing, contraception, and fertility issues as they relate to use of the product.
The existing A, B, C, D, and X categories were frequently misinterpreted as a grading system, giving an over simplified view of product risk, according to Dr. Sandra Kweder, deputy director of the Office of New Drugs in the FDA’s Center for Drug Evaluation and Research.
The new, more detailed approach to labeling will better address the complex risk-benefit considerations inherent in prescribing decisions during pregnancy and lactation, she said during a press briefing.
“I’m excited because clinicians will, going forward, be able to rely on FDA-approved drug labeling for comprehensive, chronically relevant, and user-friendly information in this part of labeling – something that has been missing for many years,” she said, noting that the changes are particularly important, given that the more than 6 million women who become pregnant each year in the United States take an average of 3-5 different prescription products during the course of their pregnancy and while breastfeeding.
“It is our hope that this new system will help their health care professionals and these women as they discuss treatment options,” she said.
Importantly, the PLLR ensures that more robust and informative data about drugs will be provided than ever before – and in a manner that speaks directly to the concerns that are common among providers, she said.
In addition to the elimination of the letter categories and the addition of the three new subsections, the use of standardized risk statement also was eliminated, as these had the same limitations as the letter categories. A section on inadvertent exposure also was eliminated due to redundancy, as the risk would be the same as with intentional exposure.
The rule also requires that labels be updated as they become outdated.
Much of the information that will be included on the new labels, which will be phased in for existing drugs and required immediately for drugs approved after June 30, 2015, was already included, but was scattered and difficult to find. The new formatting requirements provides for consistency across labels by pulling this information together in one place, Dr. Kweder said.
In an official statement, the American College of Obstetricians and Gynecologists applauded the rule for “taking needed steps to increase understanding about the effect of prescription medicine on women during pregnancy and lactation.”
“The FDA’s updated method of presenting information about both risk and benefit will improve the ability of all physicians to treat their pregnant and breastfeeding patients, as well as women who may become pregnant. It will also help more women to understand and take part in their health care decision making,” according to the ACOG statement, which also noted that the organization hopes the new content on prescription drug and biological product labels will “provide added incentives for clinical research as well as participation in patient registries.”
Christina Chambers, Ph.D., professor of pediatrics and director of clinical research for the department of pediatrics at the University of California, San Diego, also praised the new labeling rule, noting in an interview that “the final rule has been long awaited by many who work in the field of counseling pregnant and breastfeeding women about risks and safety of prescription medications, such as counselors with organizations like MotherToBaby, a service of the Organization of Teratology Information Specialists, which provides information about medication and other exposures during pregnancy and breastfeeding, and which was involved in development of the final rule.
“The MotherToBaby counselors located throughout the United States who answer questions about medication exposures for hundreds of women every day, have struggled for years with trying to explain the not-so-useful A, B, C, D, X pregnancy categories to patients and providers alike who commonly misinterpret their meaning. The new label format is much more content rich and evidence-based, and encompasses the larger picture of the safety data in the context of treatment (or lack of treatment) of the maternal condition. This is a huge step forward – and will make even more clear how critical the need is for more human pregnancy data for all medications likely to be used by women of reproductive age,” she said.
Dr. Chambers is the program director for MotherToBaby California, and director of the MotherToBaby research center at the University of California, San Diego. She reported having no relevant disclosures.
The U.S. Food and Drug Administration has issued a final rule requiring content and format changes to pregnancy and lactation labeling information for prescription drugs and biologic products.
The long-awaited “Content and Format of Labeling for Human Prescription Drug and Biological Products; Requirements for Pregnancy and Lactation Labeling, or the Pregnancy and Lactation Labeling Rule”(PLLR) is part of broad effort by the FDA to improve the content and format of prescription drug labeling. The PLLR, which finalizes many of the provisions in a proposed rule issued in May 2008 after input from numerous stakeholders, calls for replacement of the current A, B, C, D, and X drug classification system with more detailed information about the risks and benefits of use during pregnancy and breastfeeding.
The rule will take effect June 30, 2015.
Under the PLLR, labels will be required to include three detailed subsections entitled Pregnancy, Lactation, and Females and Males of Reproductive Potential. Each will include a risk summary, a discussion of the supporting data, and relevant information to help providers make prescribing and counseling decisions, according to the FDA. If no data are available to guide decision making, this must be stated.
The Pregnancy subsection combines the existing Pregnancy and Labor and Delivery subsections, and will address use of the drug during pregnancy as well as provide information about relevant registries that collect and maintain data on the use of the product in pregnant women. The Lactation subsection replaces the existing Nursing Mothers subsection, and will include information about use of the product during breastfeeding, including the amount of drug in breast milk and potential effects on the breastfed child. The new Females and Males of Reproductive Potential subsection will address pregnancy testing, contraception, and fertility issues as they relate to use of the product.
The existing A, B, C, D, and X categories were frequently misinterpreted as a grading system, giving an over simplified view of product risk, according to Dr. Sandra Kweder, deputy director of the Office of New Drugs in the FDA’s Center for Drug Evaluation and Research.
The new, more detailed approach to labeling will better address the complex risk-benefit considerations inherent in prescribing decisions during pregnancy and lactation, she said during a press briefing.
“I’m excited because clinicians will, going forward, be able to rely on FDA-approved drug labeling for comprehensive, chronically relevant, and user-friendly information in this part of labeling – something that has been missing for many years,” she said, noting that the changes are particularly important, given that the more than 6 million women who become pregnant each year in the United States take an average of 3-5 different prescription products during the course of their pregnancy and while breastfeeding.
“It is our hope that this new system will help their health care professionals and these women as they discuss treatment options,” she said.
Importantly, the PLLR ensures that more robust and informative data about drugs will be provided than ever before – and in a manner that speaks directly to the concerns that are common among providers, she said.
In addition to the elimination of the letter categories and the addition of the three new subsections, the use of standardized risk statement also was eliminated, as these had the same limitations as the letter categories. A section on inadvertent exposure also was eliminated due to redundancy, as the risk would be the same as with intentional exposure.
The rule also requires that labels be updated as they become outdated.
Much of the information that will be included on the new labels, which will be phased in for existing drugs and required immediately for drugs approved after June 30, 2015, was already included, but was scattered and difficult to find. The new formatting requirements provides for consistency across labels by pulling this information together in one place, Dr. Kweder said.
In an official statement, the American College of Obstetricians and Gynecologists applauded the rule for “taking needed steps to increase understanding about the effect of prescription medicine on women during pregnancy and lactation.”
“The FDA’s updated method of presenting information about both risk and benefit will improve the ability of all physicians to treat their pregnant and breastfeeding patients, as well as women who may become pregnant. It will also help more women to understand and take part in their health care decision making,” according to the ACOG statement, which also noted that the organization hopes the new content on prescription drug and biological product labels will “provide added incentives for clinical research as well as participation in patient registries.”
Christina Chambers, Ph.D., professor of pediatrics and director of clinical research for the department of pediatrics at the University of California, San Diego, also praised the new labeling rule, noting in an interview that “the final rule has been long awaited by many who work in the field of counseling pregnant and breastfeeding women about risks and safety of prescription medications, such as counselors with organizations like MotherToBaby, a service of the Organization of Teratology Information Specialists, which provides information about medication and other exposures during pregnancy and breastfeeding, and which was involved in development of the final rule.
“The MotherToBaby counselors located throughout the United States who answer questions about medication exposures for hundreds of women every day, have struggled for years with trying to explain the not-so-useful A, B, C, D, X pregnancy categories to patients and providers alike who commonly misinterpret their meaning. The new label format is much more content rich and evidence-based, and encompasses the larger picture of the safety data in the context of treatment (or lack of treatment) of the maternal condition. This is a huge step forward – and will make even more clear how critical the need is for more human pregnancy data for all medications likely to be used by women of reproductive age,” she said.
Dr. Chambers is the program director for MotherToBaby California, and director of the MotherToBaby research center at the University of California, San Diego. She reported having no relevant disclosures.

Tapering TNFi’s safe and much cheaper in RA
BOSTON – Tapering but perhaps not completely stopping tumor necrosis factor inhibitors can result in good control of rheumatoid arthritis with considerable cost savings, according to investigators in two studies.
Dr. Noortje van Herwaarden and colleagues from Sint Maartenskliniek in Nijmegen, the Netherlands, conducted a randomized, open-label study of dose-reduction vs. usual care in 180 patients with stable, well-controlled rheumatoid arthritis (RA) on a tumor necrosis factor inhibitor (TNFi) – either adalimumab (Humira) or etanercept (Enbrel) – plus in some cases a stable disease-modifying antirheumatic drug (DMARD) or glucocorticoid.

“This is the first study to demonstrate that disease activity–guided tapering of adalimumab and etanercept in RA patients doing well is noninferior to usual care regarding major flare,” Dr. van Herwaarden said at the annual meeting of the American College of Rheumatology.
She and her colleagues randomly assigned the patients on a 2:1 basis to TNFi dose reduction or usual care. Dose reduction was accomplished with a stepwise increase in the interval between injections every 3 months until flare. Flare was defined as an increase greater than 1.2 in the 28-joint Disease Activity Score calculated using C-reactive protein (DAS28-CRP) or a DAS28-CRP increase greater than 0.6 with a current DAS28-CRP score of 3.2 or greater, compared with baseline. Follow-up was for 18 months with visits every 3 months or any time a patient reported more symptoms.
Among 169 patients available for a per-protocol analysis, the cumulative incidence of major DAS28-CRP flares lasting more than 3 months (the primary endpoint) was 12% in patients randomized to dose reduction, compared with 10% in the usual-care group, a difference that was not statistically significant. The absolute difference was 2% and the 95% confidence interval of –12% to 12% fell below the prespecified margin of 20% for noninferiority of dose reduction, Dr. van Herwaarden said.
However, there was a significant difference in the cumulative incidence of short-term flares lasting 3 months or less, which occurred in 73% of patients on dose reduction, compared with 27% of those on usual care (P < .001).
Measures of disease activity, functioning, and quality of life were generally similar throughout the study, the investigators found.
In the dose-reduction group, 43% of patients successfully tapered the TNFi, and 20% stopped altogether, but for the remaining 37% of patients, no dose reduction was possible. There were no between-group differences in the use of DMARDs or glucocorticoids.
Minor radiographic progression
Looking at radiographic progression, the investigators found no differences in minimal clinically important change or smallest detectable change between the dose-reduction and usual-care groups. But significantly more patients in the dose-reduction group met stricter cutoff for progression of 0.5%, compared with the usual care group (32 patients vs. 15 patients, respectively). The total modified Sharp-van der Heijde score and joint-space narrowing scores also favored usual care, but the erosion score was not different between the groups.
Adverse events were similar between the groups, but there were more serious adverse events in the dose-reduction group, most of which were attributable to planned surgery among these patients, Dr. van Herwaarden said.
Mean total per-patient costs for the 18-month follow-up were almost $11,000 lower in the dose-reduction group ($15,728 vs. $26,576). The decremental cost-effectiveness ratio was calculated to be $488,116 – the amount that is saved when 1 quality-adjusted life-year (QALY) is lost. The difference is about five times higher than the widely accepted amount that society is willing to pay for 1 QALY gained, Dr. van Herwaarden said.
Reduce yes, stop no
In a separate study, Japanese researchers found that stopping a biological agent altogether in patients with RA in remission was associated with a high rate of failure.

The investigators looked at a Japanese rheumatic diseases registry data and found that among 31 patients who discontinued biological DMARDs while in remission in typical clinical practice, 71% by 1 year and 76% by 2 years had a treatment failure, defined as restart of the biological agent, glucocorticoid dose escalation, nonbiological DMARD addition, or loss of remission on the clinical disease activity index.
“Maintenance of disease activity after discontinuation is relatively difficult in established RA patients in the typical clinical practice setting. This may suggest that a reduction in dose intensity – either by prolongation of treatment intervals or by dose induction intervals – may be a more feasible option in established RA patients,” said Dr. Kazuki Yoshida, currently a research fellow in the section of clinical sciences in the division of rheumatology, immunology, and allergy at Brigham and Women’s Hospital in Boston.
Dr. van Herwaarden’s study was internally funded. Dr. Yoshida’s study was supported by a grant to Kameda Medical Center, Kamogawa City, Japan. Both physicians reported having no relevant disclosures.
BOSTON – Tapering but perhaps not completely stopping tumor necrosis factor inhibitors can result in good control of rheumatoid arthritis with considerable cost savings, according to investigators in two studies.
Dr. Noortje van Herwaarden and colleagues from Sint Maartenskliniek in Nijmegen, the Netherlands, conducted a randomized, open-label study of dose-reduction vs. usual care in 180 patients with stable, well-controlled rheumatoid arthritis (RA) on a tumor necrosis factor inhibitor (TNFi) – either adalimumab (Humira) or etanercept (Enbrel) – plus in some cases a stable disease-modifying antirheumatic drug (DMARD) or glucocorticoid.
“This is the first study to demonstrate that disease activity–guided tapering of adalimumab and etanercept in RA patients doing well is noninferior to usual care regarding major flare,” Dr. van Herwaarden said at the annual meeting of the American College of Rheumatology.
She and her colleagues randomly assigned the patients on a 2:1 basis to TNFi dose reduction or usual care. Dose reduction was accomplished with a stepwise increase in the interval between injections every 3 months until flare. Flare was defined as an increase greater than 1.2 in the 28-joint Disease Activity Score calculated using C-reactive protein (DAS28-CRP) or a DAS28-CRP increase greater than 0.6 with a current DAS28-CRP score of 3.2 or greater, compared with baseline. Follow-up was for 18 months with visits every 3 months or any time a patient reported more symptoms.
Among 169 patients available for a per-protocol analysis, the cumulative incidence of major DAS28-CRP flares lasting more than 3 months (the primary endpoint) was 12% in patients randomized to dose reduction, compared with 10% in the usual-care group, a difference that was not statistically significant. The absolute difference was 2% and the 95% confidence interval of –12% to 12% fell below the prespecified margin of 20% for noninferiority of dose reduction, Dr. van Herwaarden said.
However, there was a significant difference in the cumulative incidence of short-term flares lasting 3 months or less, which occurred in 73% of patients on dose reduction, compared with 27% of those on usual care (P < .001).
Measures of disease activity, functioning, and quality of life were generally similar throughout the study, the investigators found.
In the dose-reduction group, 43% of patients successfully tapered the TNFi, and 20% stopped altogether, but for the remaining 37% of patients, no dose reduction was possible. There were no between-group differences in the use of DMARDs or glucocorticoids.
Minor radiographic progression
Looking at radiographic progression, the investigators found no differences in minimal clinically important change or smallest detectable change between the dose-reduction and usual-care groups. But significantly more patients in the dose-reduction group met stricter cutoff for progression of 0.5%, compared with the usual care group (32 patients vs. 15 patients, respectively). The total modified Sharp-van der Heijde score and joint-space narrowing scores also favored usual care, but the erosion score was not different between the groups.
Adverse events were similar between the groups, but there were more serious adverse events in the dose-reduction group, most of which were attributable to planned surgery among these patients, Dr. van Herwaarden said.
Mean total per-patient costs for the 18-month follow-up were almost $11,000 lower in the dose-reduction group ($15,728 vs. $26,576). The decremental cost-effectiveness ratio was calculated to be $488,116 – the amount that is saved when 1 quality-adjusted life-year (QALY) is lost. The difference is about five times higher than the widely accepted amount that society is willing to pay for 1 QALY gained, Dr. van Herwaarden said.
Reduce yes, stop no
In a separate study, Japanese researchers found that stopping a biological agent altogether in patients with RA in remission was associated with a high rate of failure.
The investigators looked at a Japanese rheumatic diseases registry data and found that among 31 patients who discontinued biological DMARDs while in remission in typical clinical practice, 71% by 1 year and 76% by 2 years had a treatment failure, defined as restart of the biological agent, glucocorticoid dose escalation, nonbiological DMARD addition, or loss of remission on the clinical disease activity index.
“Maintenance of disease activity after discontinuation is relatively difficult in established RA patients in the typical clinical practice setting. This may suggest that a reduction in dose intensity – either by prolongation of treatment intervals or by dose induction intervals – may be a more feasible option in established RA patients,” said Dr. Kazuki Yoshida, currently a research fellow in the section of clinical sciences in the division of rheumatology, immunology, and allergy at Brigham and Women’s Hospital in Boston.
Dr. van Herwaarden’s study was internally funded. Dr. Yoshida’s study was supported by a grant to Kameda Medical Center, Kamogawa City, Japan. Both physicians reported having no relevant disclosures.
BOSTON – Tapering but perhaps not completely stopping tumor necrosis factor inhibitors can result in good control of rheumatoid arthritis with considerable cost savings, according to investigators in two studies.
Dr. Noortje van Herwaarden and colleagues from Sint Maartenskliniek in Nijmegen, the Netherlands, conducted a randomized, open-label study of dose-reduction vs. usual care in 180 patients with stable, well-controlled rheumatoid arthritis (RA) on a tumor necrosis factor inhibitor (TNFi) – either adalimumab (Humira) or etanercept (Enbrel) – plus in some cases a stable disease-modifying antirheumatic drug (DMARD) or glucocorticoid.
“This is the first study to demonstrate that disease activity–guided tapering of adalimumab and etanercept in RA patients doing well is noninferior to usual care regarding major flare,” Dr. van Herwaarden said at the annual meeting of the American College of Rheumatology.
She and her colleagues randomly assigned the patients on a 2:1 basis to TNFi dose reduction or usual care. Dose reduction was accomplished with a stepwise increase in the interval between injections every 3 months until flare. Flare was defined as an increase greater than 1.2 in the 28-joint Disease Activity Score calculated using C-reactive protein (DAS28-CRP) or a DAS28-CRP increase greater than 0.6 with a current DAS28-CRP score of 3.2 or greater, compared with baseline. Follow-up was for 18 months with visits every 3 months or any time a patient reported more symptoms.
Among 169 patients available for a per-protocol analysis, the cumulative incidence of major DAS28-CRP flares lasting more than 3 months (the primary endpoint) was 12% in patients randomized to dose reduction, compared with 10% in the usual-care group, a difference that was not statistically significant. The absolute difference was 2% and the 95% confidence interval of –12% to 12% fell below the prespecified margin of 20% for noninferiority of dose reduction, Dr. van Herwaarden said.
However, there was a significant difference in the cumulative incidence of short-term flares lasting 3 months or less, which occurred in 73% of patients on dose reduction, compared with 27% of those on usual care (P < .001).
Measures of disease activity, functioning, and quality of life were generally similar throughout the study, the investigators found.
In the dose-reduction group, 43% of patients successfully tapered the TNFi, and 20% stopped altogether, but for the remaining 37% of patients, no dose reduction was possible. There were no between-group differences in the use of DMARDs or glucocorticoids.
Minor radiographic progression
Looking at radiographic progression, the investigators found no differences in minimal clinically important change or smallest detectable change between the dose-reduction and usual-care groups. But significantly more patients in the dose-reduction group met stricter cutoff for progression of 0.5%, compared with the usual care group (32 patients vs. 15 patients, respectively). The total modified Sharp-van der Heijde score and joint-space narrowing scores also favored usual care, but the erosion score was not different between the groups.
Adverse events were similar between the groups, but there were more serious adverse events in the dose-reduction group, most of which were attributable to planned surgery among these patients, Dr. van Herwaarden said.
Mean total per-patient costs for the 18-month follow-up were almost $11,000 lower in the dose-reduction group ($15,728 vs. $26,576). The decremental cost-effectiveness ratio was calculated to be $488,116 – the amount that is saved when 1 quality-adjusted life-year (QALY) is lost. The difference is about five times higher than the widely accepted amount that society is willing to pay for 1 QALY gained, Dr. van Herwaarden said.
Reduce yes, stop no
In a separate study, Japanese researchers found that stopping a biological agent altogether in patients with RA in remission was associated with a high rate of failure.
The investigators looked at a Japanese rheumatic diseases registry data and found that among 31 patients who discontinued biological DMARDs while in remission in typical clinical practice, 71% by 1 year and 76% by 2 years had a treatment failure, defined as restart of the biological agent, glucocorticoid dose escalation, nonbiological DMARD addition, or loss of remission on the clinical disease activity index.
“Maintenance of disease activity after discontinuation is relatively difficult in established RA patients in the typical clinical practice setting. This may suggest that a reduction in dose intensity – either by prolongation of treatment intervals or by dose induction intervals – may be a more feasible option in established RA patients,” said Dr. Kazuki Yoshida, currently a research fellow in the section of clinical sciences in the division of rheumatology, immunology, and allergy at Brigham and Women’s Hospital in Boston.
Dr. van Herwaarden’s study was internally funded. Dr. Yoshida’s study was supported by a grant to Kameda Medical Center, Kamogawa City, Japan. Both physicians reported having no relevant disclosures.
AT THE ACR ANNUAL MEETING
Key clinical point: Tapering TNFi therapy based on RA activity can save money and doesn’t raise the rate of major flares or adverse events, but it does increase minor flares.
Major finding: The cumulative incidence of major DAS28-CRP flares lasting more than 3 months was 12% in patients randomized to dose reduction, compared with 10% in the usual care group.
Data source: Randomized, open-label study comparing dose-reduction vs. usual care in 180 patients with RA; Japanese registry study of 744 patients.
Disclosures: Dr. van Herwaarden’s study was internally funded. Dr. Yoshida’s study was supported by a grant to Kameda Medical Center, Kamogawa City, Japan. Both physicians reported having no relevant disclosures.
