User login
Ice Pack–Induced Perniosis: A Rare and Underrecognized Association
Perniosis, or chilblain, is characterized by localized, tender, erythematous skin lesions that occur as an abnormal reaction to exposure to cold and damp conditions. Although the lesions favor the distal extremities, perniosis may present anywhere on the body. Lesions can develop within hours to days following exposure to temperature less than 10°C or damp environments with greater than 60% humidity.1 Acute cases may lead to pruritus and tenderness, whereas chronic cases may involve lesions that blister or ulcerate and can take weeks to heal. We report an unusual case of erythematous plaques arising on the buttocks of a 73-year-old woman using ice pack treatments for chronic low back pain.
Case Report
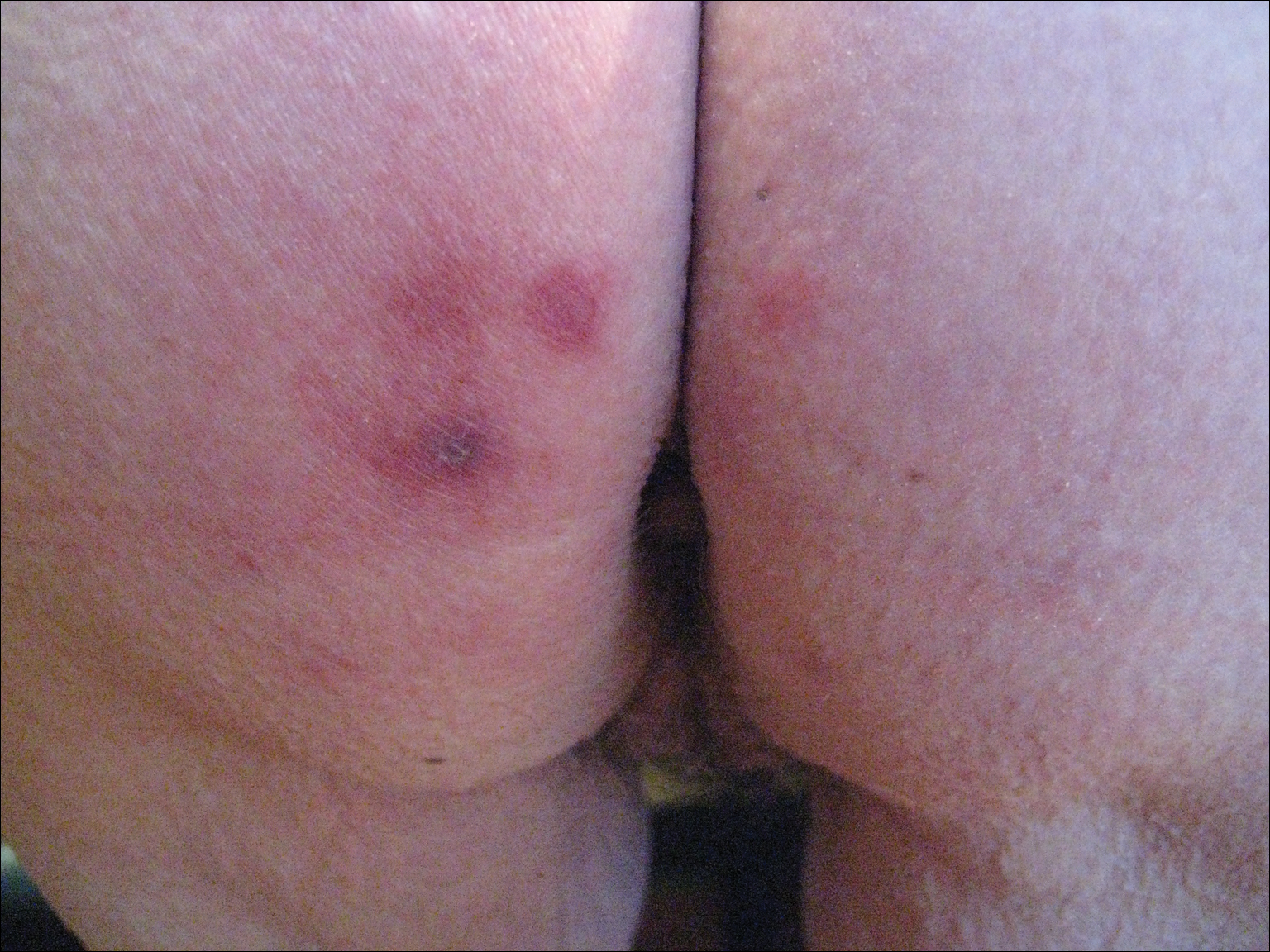
A 73-year-old woman presented with recurrent tender lesions on the buttocks of 5 years’ duration. Her medical history was remarkable for hypertension, hypothyroidism, and lumbar spinal fusion surgery 5 years prior. Physical examination revealed indurated erythematous plaques with areas of erosions on the left buttock with some involvement of the right buttock (Figure 1).
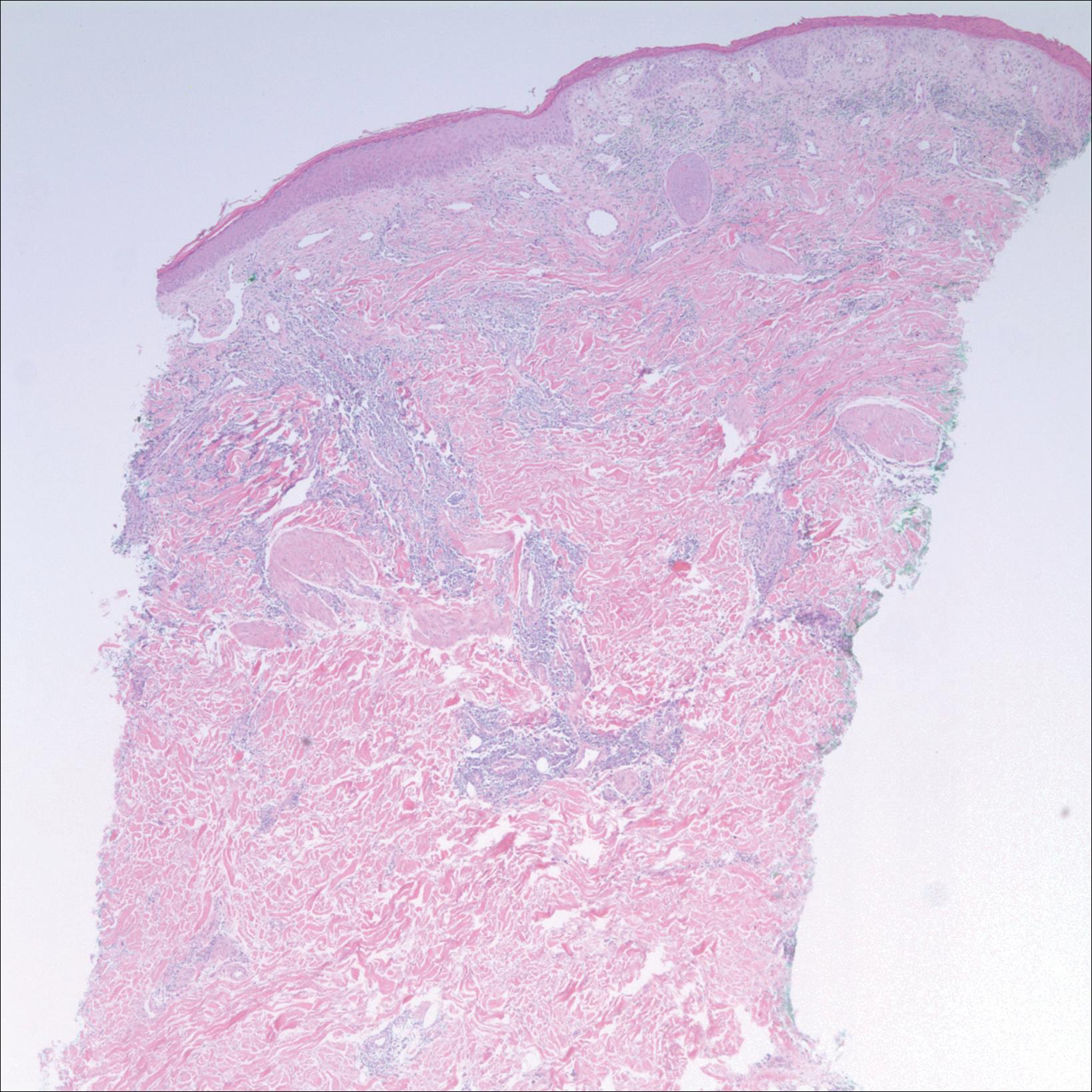
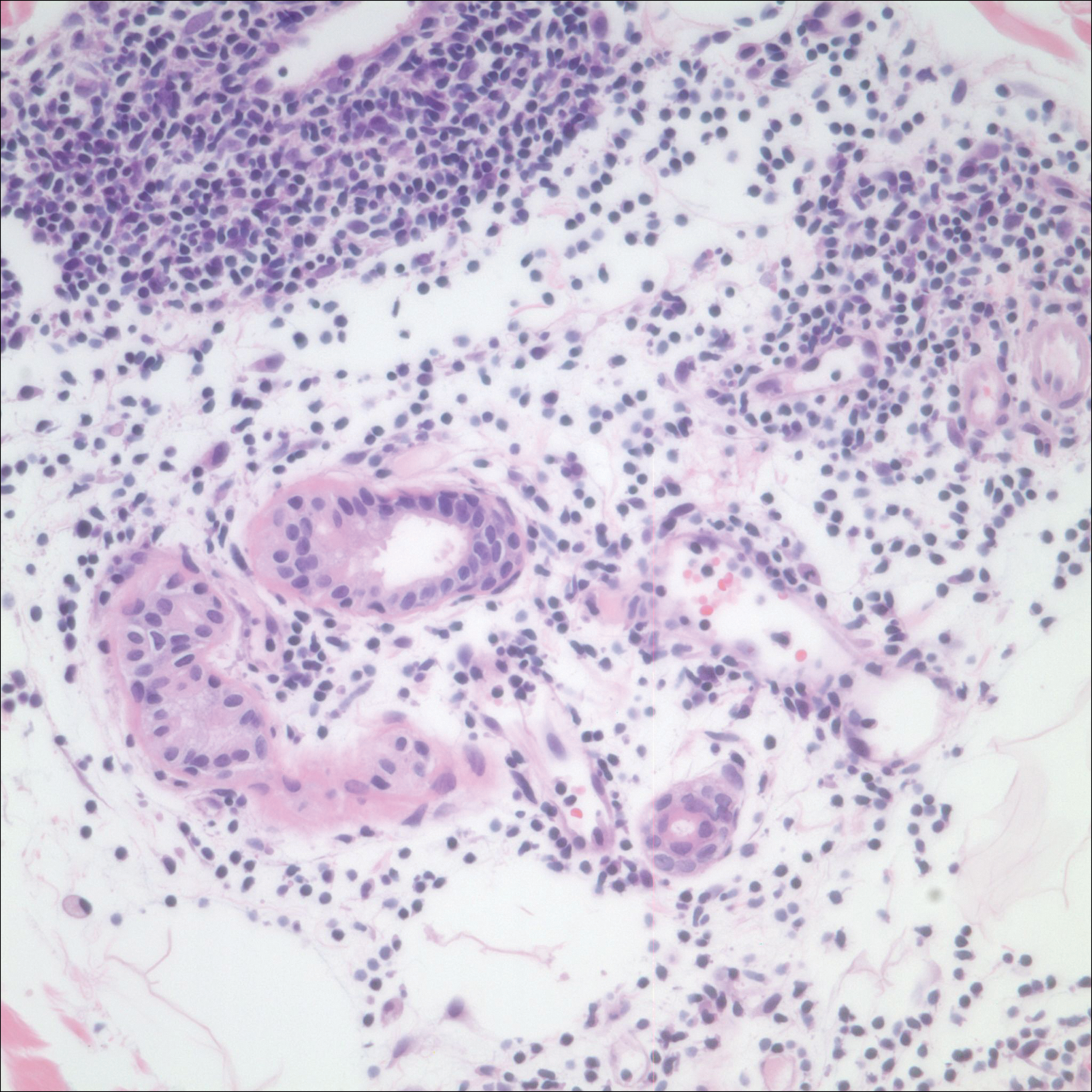
After a trial of oral valacyclovir for presumed herpes simplex infection provided no relief, a punch biopsy of the left buttock was performed, which revealed a cell-poor interface dermatitis with superficial and deep perivascular and periadnexal lymphocytic infiltrates (Figure 2). Perieccrine lymphocytes were present in a small portion of the reticular dermis (Figure 3). The patient revealed she had been sitting on ice packs for several hours daily since the lumbar spinal fusion surgery 5 years prior to alleviate chronic low back pain.
Based on the clinicopathologic correlation, a diagnosis of perniosis secondary to ice pack therapy was made. An evaluation for concomitant or underlying connective tissue disease (CTD) including a complete blood cell count with sedimentation rate, antinuclear antibodies (ANAs), serum protein electrophoresis, and serum levels of cryoglobulins and complement components was unremarkable. Our patient was treated with simple analgesia and was encouraged to avoid direct contact with ice packs for extended periods of time. Because of her low back pain, she continued to use ice packs but readjusted them sporadically and decreased frequency of use. She had complete resolution of the lesions at 6-month follow-up.
Comment
Perniosis is a self-limited condition, manifesting as erythematous plaques or nodules following exposure to cold and damp conditions. It was first reported in 1902 by Hochsinger2 as tender submental plaques occurring in children after exposure to cold weather. Since then, reports of perniosis have been described in equestrians and long-distance cyclists as well as in the context of other outdoor activities.3-5 In all cases, patients developed perniosis at sites of exposure to cold or damp conditions.
Perniosis arising in patients using ice pack therapy is a rare and recent phenomenon, with only 3 other known reported cases.6,7 In all cases, including ours, patients reported treating chronic low back pain with ice packs for more than 2 hours per day. Clinical presentations included erythematous to purpuric plaques with ulceration on the lower back or buttocks that reoccurred with subsequent use of ice packs. No concomitant CTD was reported.6
Much controversy exists as to whether idiopathic perniosis (IP) increases susceptibility to acquiring an autoimmune disease or if IP is a form of CTD that follows a more indolent course.8 In a prospective study of 33 patients with underlying IP, no patients developed lupus erythematosus (LE), with a median follow-up of 38 months.9 A study by Crowson and Magro8 revealed that 18 of 39 patients with perniotic lesions had an associated systemic disease including LE, human immunodeficiency virus, viral hepatitis, rheumatoid arthritis, cryofibrinogenemia, hypergammaglobulinemia, iritis, or Crohn disease. Of the 21 other patients who had no underlying CTD or systemic disease, 10 had a positive ANA test but no systemic symptoms; therefore, all 21 of these patients were classified as cases of IP.8
Cutaneous biopsy to distinguish between IP and autoimmune perniosis remains controversial; perniotic lesions and discoid LE share histopathologic features,9 as was evident with our case, which demonstrated overlapping findings of vacuolar change with superficial and deep perivascular and periadnexal lymphoid infiltrates. Typical features of IP include thrombosed capillaries in the papillary dermis and lymphocytic exocytosis localized to the acrosyringia, whereas secondary perniosis has superficial and deep perivascular and perieccrine lymphocytic infiltrates with vascular thrombosis in the reticular dermis. Vascular ectasia, dermal mucinosis, basement membrane zone thickening, and erythrocyte extravasation are not reliable and may be seen in both cases.8 One study revealed the only significant difference between both entities was the perieccrine distribution of lymphocytic infiltrate in cases of IP (P=.007), whereas an absence of perieccrine involvement was noted in autoimmune cases.9
Direct immunofluorescence (DIF) may help differentiate IP from autoimmune perniosis. In a prospective study by Viguier et al,9 6 of 9 patients with IP had negative DIF and 3 had slight nonspecific C3 immunoreactivity of dermal vessels. Conversely, in patients with autoimmune perniosis, positive DIF with the lupus band test was seen in 3 of 7 patients, all who had a positive ANA test9; however, positive ANA levels also were reported in patients with autoimmune perniosis but negative DIF, suggesting that DIF lacks specificity in diagnosing autoimmune perniosis.
Although histopathologic findings bear similarities to LE, there are no guidelines to suggest for or against laboratory testing for CTD in patients presenting with perniosis. Some investigators have suggested that any patient with clinical features suggestive of perniosis should undergo laboratory evaluation including a complete blood cell count and assessment for antibodies to Ro, ANA, rheumatoid factor, cryofibrinogens, and antiphospholipid antibodies.9 Serum protein electrophoresis and immunofixation electrophoresis may be done to exclude monoclonal gammopathy.
For idiopathic cases, treatment is aimed at limiting or removing cold exposure. Patients should be advised regarding the use of long-term ice pack use and the potential development of perniosis. For chronic perniosis lasting beyond several weeks, a combination of a slow taper of oral prednisone, hydroxychloroquine, and quinacrine has been successful in patients with persistent lesions despite making environmental modifications.3 Intralesional triamcinolone acetonide and nifedipine also have been effective in perniotic hand lesions.10
Conclusion
We report a rare case of perniosis on the buttocks that arose in a patient who utilized ice packs for treatment of chronic low back pain. Ice pack–induced perniosis may be an underreported entity. Histopathologic examination is nondescript, as overlapping features of perniosis and LE have been observed with no underlying CTD present. Correlation with patient history and clinical examination is paramount in diagnosis and management.
- Praminik T, Jha AK, Ghimire A. A retrospective study of cases with chilblains (perniosis) in Out Patient Department of Dermatology, Nepal Medical College and Teaching Hospital (NMCTH). Nepal Med Coll J. 2011;13:190-192.
- Hochsinger C. Acute perniosis in submental region of child [in German]. Monatsschr Kinderheilkd. 1902;1:323-327.
- Stewart CL, Adler DJ, Jacobson A, et al. Equestrian perniosis: a report of 2 cases and a review of the literature. Am J Dermatopathol. 2013;35:237-240.
- Neal AJ, Jarman AM, Bennett TG. Perniosis in a long-distance cyclist crossing Mongolia. J Travel Med. 2012;19:66-68.
- Price RD, Murdoch DR. Perniosis (chilblains) of the thigh: report of five cases including four following river crossings. High Alt Met Biol. 2001;2:535-538.
- West SA, McCalmont TH, North JP. Ice-pack dermatosis: a cold-induced dermatitis with similarities to cold panniculitis and perniosis that histopathologically resembles lupus. JAMA Dermatol. 2013;149:1314-1318.
- Haber JS, Ker KJ, Werth VP, et al. Ice‐pack dermatosis: a diagnositic pitfall for dermatopathologists that mimics lupus erythematosus. J Cutan Pathol. 2016;43:1-4.
- Crowson AN, Magro CM. Idiopathic perniosis and its mimics: a clinical and histological study of 38 cases. Hum Pathol. 1997;28:478-484.
- Viguier M, Pinguier L, Cavelier-Balloy B, et al. Clinical and histopathologic features and immunologic variables in patients with severe chilblains. a study of the relationship to lupus erythematosus. Medicine. 2001;80:180-188.
- Patra AK, Das AL, Ramadasan P. Diltiazem vs. nifedipine in chilblains: a clinical trial. Indian J Dermatol Venereol Leprol. 2003;69:209-211.
Perniosis, or chilblain, is characterized by localized, tender, erythematous skin lesions that occur as an abnormal reaction to exposure to cold and damp conditions. Although the lesions favor the distal extremities, perniosis may present anywhere on the body. Lesions can develop within hours to days following exposure to temperature less than 10°C or damp environments with greater than 60% humidity.1 Acute cases may lead to pruritus and tenderness, whereas chronic cases may involve lesions that blister or ulcerate and can take weeks to heal. We report an unusual case of erythematous plaques arising on the buttocks of a 73-year-old woman using ice pack treatments for chronic low back pain.
Case Report
A 73-year-old woman presented with recurrent tender lesions on the buttocks of 5 years’ duration. Her medical history was remarkable for hypertension, hypothyroidism, and lumbar spinal fusion surgery 5 years prior. Physical examination revealed indurated erythematous plaques with areas of erosions on the left buttock with some involvement of the right buttock (Figure 1).
After a trial of oral valacyclovir for presumed herpes simplex infection provided no relief, a punch biopsy of the left buttock was performed, which revealed a cell-poor interface dermatitis with superficial and deep perivascular and periadnexal lymphocytic infiltrates (Figure 2). Perieccrine lymphocytes were present in a small portion of the reticular dermis (Figure 3). The patient revealed she had been sitting on ice packs for several hours daily since the lumbar spinal fusion surgery 5 years prior to alleviate chronic low back pain.
Based on the clinicopathologic correlation, a diagnosis of perniosis secondary to ice pack therapy was made. An evaluation for concomitant or underlying connective tissue disease (CTD) including a complete blood cell count with sedimentation rate, antinuclear antibodies (ANAs), serum protein electrophoresis, and serum levels of cryoglobulins and complement components was unremarkable. Our patient was treated with simple analgesia and was encouraged to avoid direct contact with ice packs for extended periods of time. Because of her low back pain, she continued to use ice packs but readjusted them sporadically and decreased frequency of use. She had complete resolution of the lesions at 6-month follow-up.
Comment
Perniosis is a self-limited condition, manifesting as erythematous plaques or nodules following exposure to cold and damp conditions. It was first reported in 1902 by Hochsinger2 as tender submental plaques occurring in children after exposure to cold weather. Since then, reports of perniosis have been described in equestrians and long-distance cyclists as well as in the context of other outdoor activities.3-5 In all cases, patients developed perniosis at sites of exposure to cold or damp conditions.
Perniosis arising in patients using ice pack therapy is a rare and recent phenomenon, with only 3 other known reported cases.6,7 In all cases, including ours, patients reported treating chronic low back pain with ice packs for more than 2 hours per day. Clinical presentations included erythematous to purpuric plaques with ulceration on the lower back or buttocks that reoccurred with subsequent use of ice packs. No concomitant CTD was reported.6
Much controversy exists as to whether idiopathic perniosis (IP) increases susceptibility to acquiring an autoimmune disease or if IP is a form of CTD that follows a more indolent course.8 In a prospective study of 33 patients with underlying IP, no patients developed lupus erythematosus (LE), with a median follow-up of 38 months.9 A study by Crowson and Magro8 revealed that 18 of 39 patients with perniotic lesions had an associated systemic disease including LE, human immunodeficiency virus, viral hepatitis, rheumatoid arthritis, cryofibrinogenemia, hypergammaglobulinemia, iritis, or Crohn disease. Of the 21 other patients who had no underlying CTD or systemic disease, 10 had a positive ANA test but no systemic symptoms; therefore, all 21 of these patients were classified as cases of IP.8
Cutaneous biopsy to distinguish between IP and autoimmune perniosis remains controversial; perniotic lesions and discoid LE share histopathologic features,9 as was evident with our case, which demonstrated overlapping findings of vacuolar change with superficial and deep perivascular and periadnexal lymphoid infiltrates. Typical features of IP include thrombosed capillaries in the papillary dermis and lymphocytic exocytosis localized to the acrosyringia, whereas secondary perniosis has superficial and deep perivascular and perieccrine lymphocytic infiltrates with vascular thrombosis in the reticular dermis. Vascular ectasia, dermal mucinosis, basement membrane zone thickening, and erythrocyte extravasation are not reliable and may be seen in both cases.8 One study revealed the only significant difference between both entities was the perieccrine distribution of lymphocytic infiltrate in cases of IP (P=.007), whereas an absence of perieccrine involvement was noted in autoimmune cases.9
Direct immunofluorescence (DIF) may help differentiate IP from autoimmune perniosis. In a prospective study by Viguier et al,9 6 of 9 patients with IP had negative DIF and 3 had slight nonspecific C3 immunoreactivity of dermal vessels. Conversely, in patients with autoimmune perniosis, positive DIF with the lupus band test was seen in 3 of 7 patients, all who had a positive ANA test9; however, positive ANA levels also were reported in patients with autoimmune perniosis but negative DIF, suggesting that DIF lacks specificity in diagnosing autoimmune perniosis.
Although histopathologic findings bear similarities to LE, there are no guidelines to suggest for or against laboratory testing for CTD in patients presenting with perniosis. Some investigators have suggested that any patient with clinical features suggestive of perniosis should undergo laboratory evaluation including a complete blood cell count and assessment for antibodies to Ro, ANA, rheumatoid factor, cryofibrinogens, and antiphospholipid antibodies.9 Serum protein electrophoresis and immunofixation electrophoresis may be done to exclude monoclonal gammopathy.
For idiopathic cases, treatment is aimed at limiting or removing cold exposure. Patients should be advised regarding the use of long-term ice pack use and the potential development of perniosis. For chronic perniosis lasting beyond several weeks, a combination of a slow taper of oral prednisone, hydroxychloroquine, and quinacrine has been successful in patients with persistent lesions despite making environmental modifications.3 Intralesional triamcinolone acetonide and nifedipine also have been effective in perniotic hand lesions.10
Conclusion
We report a rare case of perniosis on the buttocks that arose in a patient who utilized ice packs for treatment of chronic low back pain. Ice pack–induced perniosis may be an underreported entity. Histopathologic examination is nondescript, as overlapping features of perniosis and LE have been observed with no underlying CTD present. Correlation with patient history and clinical examination is paramount in diagnosis and management.
Perniosis, or chilblain, is characterized by localized, tender, erythematous skin lesions that occur as an abnormal reaction to exposure to cold and damp conditions. Although the lesions favor the distal extremities, perniosis may present anywhere on the body. Lesions can develop within hours to days following exposure to temperature less than 10°C or damp environments with greater than 60% humidity.1 Acute cases may lead to pruritus and tenderness, whereas chronic cases may involve lesions that blister or ulcerate and can take weeks to heal. We report an unusual case of erythematous plaques arising on the buttocks of a 73-year-old woman using ice pack treatments for chronic low back pain.
Case Report
A 73-year-old woman presented with recurrent tender lesions on the buttocks of 5 years’ duration. Her medical history was remarkable for hypertension, hypothyroidism, and lumbar spinal fusion surgery 5 years prior. Physical examination revealed indurated erythematous plaques with areas of erosions on the left buttock with some involvement of the right buttock (Figure 1).
After a trial of oral valacyclovir for presumed herpes simplex infection provided no relief, a punch biopsy of the left buttock was performed, which revealed a cell-poor interface dermatitis with superficial and deep perivascular and periadnexal lymphocytic infiltrates (Figure 2). Perieccrine lymphocytes were present in a small portion of the reticular dermis (Figure 3). The patient revealed she had been sitting on ice packs for several hours daily since the lumbar spinal fusion surgery 5 years prior to alleviate chronic low back pain.
Based on the clinicopathologic correlation, a diagnosis of perniosis secondary to ice pack therapy was made. An evaluation for concomitant or underlying connective tissue disease (CTD) including a complete blood cell count with sedimentation rate, antinuclear antibodies (ANAs), serum protein electrophoresis, and serum levels of cryoglobulins and complement components was unremarkable. Our patient was treated with simple analgesia and was encouraged to avoid direct contact with ice packs for extended periods of time. Because of her low back pain, she continued to use ice packs but readjusted them sporadically and decreased frequency of use. She had complete resolution of the lesions at 6-month follow-up.
Comment
Perniosis is a self-limited condition, manifesting as erythematous plaques or nodules following exposure to cold and damp conditions. It was first reported in 1902 by Hochsinger2 as tender submental plaques occurring in children after exposure to cold weather. Since then, reports of perniosis have been described in equestrians and long-distance cyclists as well as in the context of other outdoor activities.3-5 In all cases, patients developed perniosis at sites of exposure to cold or damp conditions.
Perniosis arising in patients using ice pack therapy is a rare and recent phenomenon, with only 3 other known reported cases.6,7 In all cases, including ours, patients reported treating chronic low back pain with ice packs for more than 2 hours per day. Clinical presentations included erythematous to purpuric plaques with ulceration on the lower back or buttocks that reoccurred with subsequent use of ice packs. No concomitant CTD was reported.6
Much controversy exists as to whether idiopathic perniosis (IP) increases susceptibility to acquiring an autoimmune disease or if IP is a form of CTD that follows a more indolent course.8 In a prospective study of 33 patients with underlying IP, no patients developed lupus erythematosus (LE), with a median follow-up of 38 months.9 A study by Crowson and Magro8 revealed that 18 of 39 patients with perniotic lesions had an associated systemic disease including LE, human immunodeficiency virus, viral hepatitis, rheumatoid arthritis, cryofibrinogenemia, hypergammaglobulinemia, iritis, or Crohn disease. Of the 21 other patients who had no underlying CTD or systemic disease, 10 had a positive ANA test but no systemic symptoms; therefore, all 21 of these patients were classified as cases of IP.8
Cutaneous biopsy to distinguish between IP and autoimmune perniosis remains controversial; perniotic lesions and discoid LE share histopathologic features,9 as was evident with our case, which demonstrated overlapping findings of vacuolar change with superficial and deep perivascular and periadnexal lymphoid infiltrates. Typical features of IP include thrombosed capillaries in the papillary dermis and lymphocytic exocytosis localized to the acrosyringia, whereas secondary perniosis has superficial and deep perivascular and perieccrine lymphocytic infiltrates with vascular thrombosis in the reticular dermis. Vascular ectasia, dermal mucinosis, basement membrane zone thickening, and erythrocyte extravasation are not reliable and may be seen in both cases.8 One study revealed the only significant difference between both entities was the perieccrine distribution of lymphocytic infiltrate in cases of IP (P=.007), whereas an absence of perieccrine involvement was noted in autoimmune cases.9
Direct immunofluorescence (DIF) may help differentiate IP from autoimmune perniosis. In a prospective study by Viguier et al,9 6 of 9 patients with IP had negative DIF and 3 had slight nonspecific C3 immunoreactivity of dermal vessels. Conversely, in patients with autoimmune perniosis, positive DIF with the lupus band test was seen in 3 of 7 patients, all who had a positive ANA test9; however, positive ANA levels also were reported in patients with autoimmune perniosis but negative DIF, suggesting that DIF lacks specificity in diagnosing autoimmune perniosis.
Although histopathologic findings bear similarities to LE, there are no guidelines to suggest for or against laboratory testing for CTD in patients presenting with perniosis. Some investigators have suggested that any patient with clinical features suggestive of perniosis should undergo laboratory evaluation including a complete blood cell count and assessment for antibodies to Ro, ANA, rheumatoid factor, cryofibrinogens, and antiphospholipid antibodies.9 Serum protein electrophoresis and immunofixation electrophoresis may be done to exclude monoclonal gammopathy.
For idiopathic cases, treatment is aimed at limiting or removing cold exposure. Patients should be advised regarding the use of long-term ice pack use and the potential development of perniosis. For chronic perniosis lasting beyond several weeks, a combination of a slow taper of oral prednisone, hydroxychloroquine, and quinacrine has been successful in patients with persistent lesions despite making environmental modifications.3 Intralesional triamcinolone acetonide and nifedipine also have been effective in perniotic hand lesions.10
Conclusion
We report a rare case of perniosis on the buttocks that arose in a patient who utilized ice packs for treatment of chronic low back pain. Ice pack–induced perniosis may be an underreported entity. Histopathologic examination is nondescript, as overlapping features of perniosis and LE have been observed with no underlying CTD present. Correlation with patient history and clinical examination is paramount in diagnosis and management.
- Praminik T, Jha AK, Ghimire A. A retrospective study of cases with chilblains (perniosis) in Out Patient Department of Dermatology, Nepal Medical College and Teaching Hospital (NMCTH). Nepal Med Coll J. 2011;13:190-192.
- Hochsinger C. Acute perniosis in submental region of child [in German]. Monatsschr Kinderheilkd. 1902;1:323-327.
- Stewart CL, Adler DJ, Jacobson A, et al. Equestrian perniosis: a report of 2 cases and a review of the literature. Am J Dermatopathol. 2013;35:237-240.
- Neal AJ, Jarman AM, Bennett TG. Perniosis in a long-distance cyclist crossing Mongolia. J Travel Med. 2012;19:66-68.
- Price RD, Murdoch DR. Perniosis (chilblains) of the thigh: report of five cases including four following river crossings. High Alt Met Biol. 2001;2:535-538.
- West SA, McCalmont TH, North JP. Ice-pack dermatosis: a cold-induced dermatitis with similarities to cold panniculitis and perniosis that histopathologically resembles lupus. JAMA Dermatol. 2013;149:1314-1318.
- Haber JS, Ker KJ, Werth VP, et al. Ice‐pack dermatosis: a diagnositic pitfall for dermatopathologists that mimics lupus erythematosus. J Cutan Pathol. 2016;43:1-4.
- Crowson AN, Magro CM. Idiopathic perniosis and its mimics: a clinical and histological study of 38 cases. Hum Pathol. 1997;28:478-484.
- Viguier M, Pinguier L, Cavelier-Balloy B, et al. Clinical and histopathologic features and immunologic variables in patients with severe chilblains. a study of the relationship to lupus erythematosus. Medicine. 2001;80:180-188.
- Patra AK, Das AL, Ramadasan P. Diltiazem vs. nifedipine in chilblains: a clinical trial. Indian J Dermatol Venereol Leprol. 2003;69:209-211.
- Praminik T, Jha AK, Ghimire A. A retrospective study of cases with chilblains (perniosis) in Out Patient Department of Dermatology, Nepal Medical College and Teaching Hospital (NMCTH). Nepal Med Coll J. 2011;13:190-192.
- Hochsinger C. Acute perniosis in submental region of child [in German]. Monatsschr Kinderheilkd. 1902;1:323-327.
- Stewart CL, Adler DJ, Jacobson A, et al. Equestrian perniosis: a report of 2 cases and a review of the literature. Am J Dermatopathol. 2013;35:237-240.
- Neal AJ, Jarman AM, Bennett TG. Perniosis in a long-distance cyclist crossing Mongolia. J Travel Med. 2012;19:66-68.
- Price RD, Murdoch DR. Perniosis (chilblains) of the thigh: report of five cases including four following river crossings. High Alt Met Biol. 2001;2:535-538.
- West SA, McCalmont TH, North JP. Ice-pack dermatosis: a cold-induced dermatitis with similarities to cold panniculitis and perniosis that histopathologically resembles lupus. JAMA Dermatol. 2013;149:1314-1318.
- Haber JS, Ker KJ, Werth VP, et al. Ice‐pack dermatosis: a diagnositic pitfall for dermatopathologists that mimics lupus erythematosus. J Cutan Pathol. 2016;43:1-4.
- Crowson AN, Magro CM. Idiopathic perniosis and its mimics: a clinical and histological study of 38 cases. Hum Pathol. 1997;28:478-484.
- Viguier M, Pinguier L, Cavelier-Balloy B, et al. Clinical and histopathologic features and immunologic variables in patients with severe chilblains. a study of the relationship to lupus erythematosus. Medicine. 2001;80:180-188.
- Patra AK, Das AL, Ramadasan P. Diltiazem vs. nifedipine in chilblains: a clinical trial. Indian J Dermatol Venereol Leprol. 2003;69:209-211.
Practice Points
- Ice pack-induced perniosis is a rare condition that can occur in patients using long-term ice pack therapy.
- This entity histopathologically mimics cutaneous lupus erythematosus and can present a diagnostic challenge.
- A thorough clinical history and awareness of this diagnosis is essential for diagnostic accuracy.
2018: A banner year for hematology drug approvals
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
SAN DIEGO – It was banner year for new hematology drug approvals, according to R. Angelo de Claro, MD, of the Food and Drug Administration.
, including 12 first-time approvals, 5 new biosimilars, and 15 new indications for previously approved drugs, Dr. de Claro, clinical team leader in the FDA’s division of hematology products in Silver Spring, Md., said during an overview of the approvals at the annual meeting of the American Society of Hematology.
These include six new approvals for first-line treatment, and eight for pediatric indications, he said.
Highlights were discussed at two ASH-FDA joint symposia at the meeting, including one focused on the malignant hematology approvals, and another on the nonmalignant hematology approvals. In a video interview, Dr. de Claro provides some additional insight into their importance and about what might lie ahead.
“I think what’s exciting is that you have drug development occurring in more common conditions such as chronic lymphocytic leukemia, as well as in rare conditions, including hairy cell leukemia – and the first-ever approval in hemophagocytic lymphohistiocytosis,” he said. “It’s been very busy at the FDA; stay tuned ... the year’s not done yet. There could be more coming and we certainly anticipate more applications in the future.”
Dr. de Claro is an FDA employee. He reported having no other relevant disclosures.
REPORTING FROM ASH 2018

Huntington’s research returns to Latin America, as scientists tread with care
BARRANQUILLA, COLOMBIA – “We don’t like to call them brigades. That sounds militant,” said neuropsychologist Johan Acosta-López, PhD.
Dr. Acosta-López, the head of cognitive neurosciences at Simón Bolivar University in this city on Colombia’s Atlantic coast, was among five Colombian clinicians – neurologists, psychiatrists, and neuropsychologists – stuffed into a car on their way to a conference hotel in July 2018.

The following day they would be joined by clinicians and researchers from North America, other Latin American countries, and Europe for a first-of-its kind meeting on Huntington’s disease (HD) in the region, sponsored by Factor-H, an HD charity working in Latin America.
Once the talks wrapped up, the researchers – clinicians and basic scientists – were invited to see patients at a hospital in a town an hour inland with a large concentration of HD families, most of them extremely poor. For some, the Factor-H–sponsored “brigade” would be their first hands-on experience with HD patients in a developing country.
There was some debate in the car about what to call such events: brigades, “integrated health days,” or clinics. Around here – where HD abounded but patients were weary of researchers – terminology mattered.
“We’ve had so many investigators arrive in this area – foreigners and Colombians – telling people ‘we’ve got this huge, great project that you’ll benefit from.’ And they take blood samples and never return,” Dr. Acosta-López said.

Even as a local investigator, Dr. Acosta-López has faced challenges getting a new study off the ground. Dr. Acosta-López and his colleagues are working under a grant from the Colombian government to recruit 241 presymptomatic subjects with confirmed genetic markers for HD, and evaluate them for cognitive and neurologic changes preceding disease onset.
It’s a cross-sectional study, and such studies are usually funded for a year. But the investigators knew it would take much more than a year to recruit patients here, and planned their study for 3 years. As of July, the team had been engaging with the community for 6 months but still didn’t have a single blood sample.
“We’ve had to convince everyone that this time is different,” he said, “and that means focusing on the social aspect” – setting up a legal-assistance program through the university to help families claim health benefits and a job-training program sponsored by local businesses.
It’s unusual for researchers to find themselves playing such extensive roles in coordinating social and economic support for their subjects. But with HD, it’s happening across Latin America, where researchers speak frequently of a “debt” owed to HD families in this region.
Huntington’s disease is a neurodegenerative disease caused by a genetic mutation in the huntingtin (HTT) gene, changing the normal protein it expresses in the body to a toxic form that damages cortical and basal ganglia neurons. It affects between 0.5 to 1 in 10,000 people worldwide, with higher prevalence in the United States, Europe, and Australia.
HD is inherited in an autosomal dominant pattern; a child of a parent with the mutation has a 50% chance of developing the disease. Patients develop cognitive symptoms that progress to dementia, along with the debilitating involuntary, dancelike movements that gave the disease the name by which it was formerly known: Huntington’s chorea.
In the 1980s and 1990s, several generations of Latin American HD families provided data that allowed for some of the greatest research advances in the disease – and they may represent a large share of the world’s HD cases. Yet, they continue to live in extreme poverty and have benefited little from the findings of the past 3 decades.
Without recognizing this and working to improve the families’ well-being, the researchers at the conference said it’s unlikely that promising therapies in the pipeline will ever reach the populations that need them the most.
Discovery in Venezuela

Some 8 hours by car from Barranquilla sits Lake Maracaibo, Venezuela, home to the largest known clusters of HD patients worldwide. The disease is believed to have come to the shores of Lake Maracaibo with a lone European immigrant – a Spanish sailor, many claim – at the end of the 18th century. Cases were first described in the 1950s by a young Venezuelan physician named Américo Negrette, MD.
Dr. Negrette’s findings were ignored by health officials in Venezuela and went unnoticed in the international research community until 1972, when a student of Dr. Negrette’s presented at an HD conference in Ohio. There he drew the attention of the American neuropsychologist Nancy S. Wexler, PhD. Dr. Wexler’s own mother had died of HD, and her father Milton, a noted psychoanalyst, founded the first research foundation dedicated to the disease.

While prevalence of HD in North America, Australia, and Europe is about 1 in 10,000, the region around Lake Maracaibo saw 70 times that rate at the time, thanks to high birth rates, geographic isolation, and extensive intermarriage within a handful of families. The families comprised mostly poor fishermen who lived in makeshift homes in towns ringing the lake.
In 1979, Dr. Wexler, with funding from the U.S. National Institutes of Health, began making annual research visits to Lake Maracaibo, and in 1983, the research group she coordinated, using data from blood and tissue samples donated by affected families, identified the location of the huntingtin gene on chromosome 4 (Nature. 1983 Nov 17;306[5940]:234-8). A decade later, the researchers isolated the mutant version of the gene and found it to be a triplet (CAG) expansion mutation, with more CAG repetitions associated with earlier age at disease onset (Cell. 1993 Mar 26;72[6]:971-83). Dr. Wexler and her colleagues’ findings led to the first genetic tests for HD.
Nationalist policies in Venezuela ended Dr. Wexler and her colleagues’ annual visits to Lake Maracaibo in 2002, along with the food, clothing, and medicines that the group routinely distributed to the families when they came.
Over 23 years, the researchers obtained data from some 18,000 individuals, but the families did not benefit in any durable way from the research. Local investigators with whom Dr. Wexler’s group collaborated lacked the resources and training to continue independently.
Access to medications is limited in Venezuela, and there is no institutional support for hundreds of HD patients living in extreme poverty, many of them descendants of the patients who contributed to the research and generation of these samples. The families’ biological material was sent to labs abroad, where investigators continue to derive findings from it today. Though genetic testing was performed on thousands at risk for the disease, few received access to their results through genetic counseling. A hospice established by Dr. Wexler’s foundation limped along until 2014, when it was finally shuttered.
Rebuilding bridges
A handful of families from the Lake Maracaibo towns attended the conference in Barranquilla. Their travel costs were picked up by Factor-H, which sponsored the event.
Ignacio Muñoz-Sanjuán, PhD, Factor-H’s founder and president, knew the families personally. He’s visited them regularly for years. In 2017, Dr. Muñoz-Sanjuán, a molecular biologist known affectionately in the HD community as “Nacho,” invited several to Rome for a meeting with Pope Francis, as part of an effort to raise awareness of HD and to request support from the Catholic Church for the Latin American families.

Humanitarian work is relatively new to Dr. Muñoz-Sanjuán, who’s spent his career in drug development. In addition to his unpaid work with Factor-H, he is vice president of biology with the CHDI Foundation, a Los Angeles–based nonprofit that funds drug research in HD. CHDI is reported to have about $100 million in annual funding – about triple the NIH budget in recent years for HD research. Its major donors are a group of investors who for years have remained anonymous and do not publicly discuss their philanthropy.
The Spanish-born Dr. Muñoz-Sanjuán had little direct experience with HD populations in Latin America until a few years ago, he said.
At a CHDI meeting in Brazil, he said, “I was talking with physicians and patient advocates from Latin America, telling them they had to be willing to be involved, that these communities with high prevalence had a lot to offer science,” Dr. Muñoz-Sanjuán said in an interview. “I was told that it was me who needed to understand the conditions in which HD patients lived. It completely put me on the spot.”
HD tends to strike during the most productive years of a person’s life, from the late 30s onward, keeping them from working and obliging family members to stop working to care for them. In a poor community, it can condemn a family to a state of extreme poverty for generations. Tetrabenazine (Xenazine), a medication to quiet chorea symptoms, is costly enough that many patients must do without it. Ensuring adequate calorie intake is difficult in HD patients, whose constant movements cause them to lose weight.
Dr. Muñoz-Sanjuán traveled to Colombia, Venezuela, and Brazil, meeting HD families and doctors like neurologist Gustavo Barrios, MD, of Hospital Occidente de Kennedy in Bogotá, Colombia. In a talk at the Barranquilla conference, Dr. Barrios related the experience of his first visit to El Dificil, a community in northern Colombia where some large HD families are forced to survive on the equivalent of $5 a day. “I had to confront not only the fact that these families were living with a terrible disease but in conditions of extreme deprivation,” he said. “My life as a doctor changed that day.”
Dr. Muñoz-Sanjuán helped form a Latin American HD network to involve clinicians like Dr. Barrios who worked with HD clusters, most of them poorly studied. “These are all neglected communities that share similar features,” Dr. Muñoz-Sanjuán said.
On Colombia’s Caribbean coast, for example, HD had been documented since the early 1990s, but genotyping was not performed until recently. Prevalence data are “virtually nonexistent” in Colombia, said Sonia Moreno, PhD, a neuropsychologist at the University of Antioquia in Medellin. In a pilot study presented this year at the CHDI Foundation’s Enroll-HD Congress, Dr. Moreno and her colleagues mined Colombian public health data for likely HD cases, and argued for the creation of a national registry.
In 2012, Dr. Muñoz-Sanjuán founded Factor-H with the aim of improving living conditions for Latin American communities with HD.

Factor-H does not receive funds from CHDI Foundation and instead relies on donations from individuals and companies; its annual budget is less than $200,000. But through contracts with local nongovernmental organizations, it has sponsored health clinics and ongoing food assistance, delivered shipments of medicines and clothing, and started a sponsorship program for young people in HD families, whose studies often are interrupted caring for sick parents. It hopes to build permanent support centers in Colombia and Venezuela where HD families can get their food and medical needs met.
“The traditional thinking in the HD research community is that we’re helping people by doing the legwork to make medicine – and that’s not necessarily enough. You need a more holistic approach,” Dr. Muñoz-Sanjuán said.
Lennie Pineda, MSc, who recently retired as a geneticist with the University of Zulia in Maracaibo, Venezuela, said that Dr. Muñoz-Sanjuán was viewed skeptically when he first visited, in part because of his biomedical research background.
Ms. Pineda, who worked with the region’s HD families her whole career, has been wary of past research efforts in Venezuela. In 2010, she published a paper critical of Dr. Wexler’s and his colleagues’ approach (Revista Redbioética/UNESCO. 2010;1[2]:50-61), particularly regarding issues of informed consent.
“I was very cold to Nacho,” she laughed. “We all looked at him suspiciously.”
Ms. Pineda said Dr. Muñoz-Sanjuán won her over with his interest and creativity in finding concrete ways improve the lives of families in the Lake Maracaibo towns.
In a talk at the conference, Edison Soto, a young man from San Luis, a town on Lake Maracaibo that is a key cluster of HD, said Dr. Muñoz-Sanjuán’s visits had reawakened hope among the families there. “For years, no one thought about us, and because of the situation in the country it’s been hard, really hard,” he said.
“Nacho’s smart,” Ms. Pineda said. “He’s not coming to build a research cohort, he’s coming with genuine intention to help. But if one day conditions are adequate to support investigation, and the people here are well informed and volunteer for a study with full consent, well, all the better,” she said.
Dr. Muñoz-Sanjuán acknowledged that his humanitarian work could be perceived as preparing the ground for future clinical trials.
“I’m not doing anything research oriented with Latin America,” he said. “I would never approach these communities and recommend they take part in a study or give samples, unless their conditions change significantly. But the idea of cross-contamination is a problem I might need to fix. There may come a day where I need to depersonalize Factor-H from me.”
A research platform, a novel agent
Though HD research in Latin America remains rife with challenges, a number of investigators at the conference talked optimistically about planned and ongoing HD studies in Latin America.
The biggest of these is ENROLL-HD, a long-term global observational study of families with HD that uses a standardized approach to data collection. The platform, launched in 2013, aims to enroll 20,000 participants for yearly (or more frequent) assessment. Data from ENROLL-HD will support a diverse range of studies on everything from biomarkers to genetic modifiers to quality of life measures in HD.
ENROLL-HD has opened study sites in Argentina, Chile, and Colombia, and plans to launch a site near Lima, Peru, that is home to an HD cluster. Venezuela is considered out of reach, at least for now.
In Barranquilla, Claudia Perandones, MD, PhD, a genetics researcher in Argentina who manages ENROLL-HD for Latin America and is a cofounder of Factor-H, explained why the kind of clusters seen in Latin America are so valuable scientifically.
The extended family groups share a disease haplotype, eat the same foods, and live in similar environments, Dr. Perandones noted. Because not all the variation in HD can be explained by the number of CAG repeats a patient has, having a large sample with a common haplotype would help researchers pinpoint other environmental and genetic factors that can modify the onset or progress of the disease.
Another key goal of ENROLL-HD, investigators say, is to speed recruitment into clinical trials as they arise. And for the first time in history, potentially game-changing therapies are being developed specifically for HD.
For the past 5 years the Swiss pharmaceutical giant Roche has worked with a smaller biotech firm, Ionis Pharmaceuticals, on an agent called RG6042, which was known until recently as IONIS-HTTRx. CHDI was extensively involved in the agent’s preclinical development, contributing some $10 million to get it off the ground.
RG6042 is an antisense oligonucleotide, delivered by spinal injection, which works by interrupting an mRNA signaling pathway to suppress production of mutant HTT (mHTT) protein in the brain. Antisense oligonucleotides, sometimes called gene silencing therapies, are a new and promising approach in neurodegenerative diseases. Two have received FDA approval to treat spinal muscular atrophy and Duchenne muscular dystrophy.
In April 2018, Roche announced positive results from phase 1/2a study in 46 HD patients in Europe and North America. Patients in that 13-week study saw significant (up to 60%) dose-dependent reductions of the mHTT in their cerebrospinal fluid; a post hoc analysis also found some evidence of functional improvement (Neurology. 2018;90[15 Supplement]:CT.002).
These encouraging findings led to Roche’s announcement of a global phase 3 randomized, controlled trial that is scheduled to begin enrolling in 2019. Roche hopes to randomize 660 patients with mild HD across 15 countries for the 2-year trial, called GENERATION-HD1.
Sites in Latin America are expected to include Argentina, Chile, and Colombia.
At the Barranquilla meeting, Daniel Ciriano, MD, Roche’s Argentina-based medical director for Latin America, extolled the company’s commitment to ethics and social welfare in the region. In recent years, Roche has increased its humanitarian commitments across Latin America, including helping rebuild a Chilean village after an earthquake and offering free breast cancer and kidney disease treatments.
RG6042 is only one of a number of promising approaches to HD. Other therapies in the pipeline include gene silencing delivered by viral vectors instead of repeated spinal injections, an oral drug that interrupts mHTT production, immunotherapies, and even CRISPR gene–editing techniques.
Little was said at the conference, however, about how Latin American HD communities might be able to afford RG6042 or any other therapy that emerges from the pipeline.
Dr. Muñoz-Sanjuán called the issue “a theme for future discussion.”
“This is an area that has to be handled carefully and not one we are heavily invested in yet, although it’s very important,” he said.
On the ground
Several of the European and North American scientists who presented in Barranquilla took pains to express their concern with the well-being of HD patients in Latin America and to demonstrate goodwill toward the local researchers and clinicians.
Hilal A. Lashuel, PhD, a molecular biologist working on the structure and behavior of the HTT protein, said his participation in the Factor-H event at the Vatican the year before had awakened him to “the real human part of HD,” and changed the way he does science.
Normally, Dr. Lashuel said, “we do research disconnected from the realities of the diseases we work with.”
“We need to not just to do research but [to ensure] that research is done right,” he said, which means also focusing on improving patients’ standard of living.
The room broke out in applause when Dr. Lashuel announced new internships for investigators from developing countries. He also presented a parting video from his research team at the École Polytechnique Fédérale de Lausanne (Switzerland), complete with music and affectionate messages in Spanish.
Pharmacologist Elena Cattaneo, PhD, a stem cell researcher long active in the HD community, and also a senator in Italy’s parliament, delivered a similarly warm, carefully choreographed video message from her laboratory at the University of Milan.
Just days later in the town of Juan de Acosta, an hour inland of Barranquilla, the same researchers sat down with patients and families who crowded the waiting room of the town’s only hospital, as the sun beat in through the windows and as mule carts, stray dogs, and buses passed by on the main drag outside.
The event had been titled a “brigade” after all, but the HD families did not seem to mind – and indeed so many showed up that a sign had to be placed on the door saying that no one who arrived after noon could be seen. Consults were not limited to HD-related matters, so families could be seen for any complaint.
HD was first documented in this town in the early 1990s, but much remains to be understood about the size of the cluster, the haplotype, and its relation to other clusters in Colombia or Venezuela. The families here share a handful of last names and likely share a common ancestor. In the early 19th century, the Barranquilla region was flooded with European migrants who reached the city by ship. (HD clusters in Latin America tend to be concentrated in coastal regions, possibly because of migration patterns.)
The waiting room of the hospital was loud with chatter. Small children played as their relatives waited for consults. Some showed the characteristic restless movements and emaciated bodies of people with advanced HD.
The foreign scientists were barred from taking any patient data out of the hospital or asking for samples. Even picture taking was prohibited. Instead they performed genetic counseling and neuropsychological tests; they sorted out differential diagnoses and advised on medications. Visiting Colombian and Venezuelan physicians did the same, while their assistants met with families in the waiting room, taking medical histories and sketching out basic genealogies.
Some of the foreign researchers reported fruitful interactions with patients, while others seemed perplexed by what they’d experienced. Alba di Pardo, PhD, a genetic epidemiologist at the Istituto Neurologico Mediterraneo Pozzilli (Italy), said she’d spent the morning doing genetic counseling with families and going over genealogies to assess risk. Yet, despite the fact that anyone with an HD parent has a 50% chance of developing the disease, some family members acted uninterested, she said.
Dr. di Pardo’s colleague at the Istituto, biologist Vittorio Maglione, PhD, reported having a similar experience. As he was counseling a young woman about her risk for HD, she scrolled indifferently through Facebook posts on her phone, he said.
On some level, Dr. Maglione said, he could understand patients’ reluctance to engage noting that, while there were many potential HD therapies to try, any new treatment paradigm for HD was many years away from a place like this – and potentially very costly. Dr. Maglione – along with Dr. di Pardo – is researching the SP1 axis, a sphingolipid pathway implicated in neurodegenerative such diseases as HD and which has potential as a drug target (Trends Pharmacol Sci. 2018;39[5]:468-80).
Psychologist Pedro Puentes Rozo of Simón Bolivar University, who is working with Dr. Acosta-López on the local cohort study of presymptomatic HD patients, said that, for most of the families in the clinic that day, any seeming indifference probably masked deeper fears. People already were well aware of their risk. “They’ve known about it forever, said Dr. Puentes Rozo, who has been working with this HD population for a decade. “But this is a catastrophic illness and can generate a lot of anxiety.”
Dr. Puentes Rozo said the group’s planned study, unlike studies in the past, would be conducted under strict “international ethical norms and standards.” Subjects would receive ongoing psychological support, and the researchers were working to establish a genetic counseling center so that people who want to know their status “can be prepared,” he said, and plan for their lives and families.
By fall 2018, the cohort study was underway. The group had sponsored several more hospital brigades – or “integrated health days” as they preferred to call them, at the hospital in Juan de Acosta, giving them a chance to work face to face with families.
They drew no blood during the clinics, as investigators in the past had done. Instead, they explained the study to patients, performed the initial screenings, and invited them to designated study appointments at the university. Legal assistance was up and running, and the jobs program would start in 2019.
Enrollment was climbing. And the group was steadily accumulating data.
BARRANQUILLA, COLOMBIA – “We don’t like to call them brigades. That sounds militant,” said neuropsychologist Johan Acosta-López, PhD.
Dr. Acosta-López, the head of cognitive neurosciences at Simón Bolivar University in this city on Colombia’s Atlantic coast, was among five Colombian clinicians – neurologists, psychiatrists, and neuropsychologists – stuffed into a car on their way to a conference hotel in July 2018.
The following day they would be joined by clinicians and researchers from North America, other Latin American countries, and Europe for a first-of-its kind meeting on Huntington’s disease (HD) in the region, sponsored by Factor-H, an HD charity working in Latin America.
Once the talks wrapped up, the researchers – clinicians and basic scientists – were invited to see patients at a hospital in a town an hour inland with a large concentration of HD families, most of them extremely poor. For some, the Factor-H–sponsored “brigade” would be their first hands-on experience with HD patients in a developing country.
There was some debate in the car about what to call such events: brigades, “integrated health days,” or clinics. Around here – where HD abounded but patients were weary of researchers – terminology mattered.
“We’ve had so many investigators arrive in this area – foreigners and Colombians – telling people ‘we’ve got this huge, great project that you’ll benefit from.’ And they take blood samples and never return,” Dr. Acosta-López said.
Even as a local investigator, Dr. Acosta-López has faced challenges getting a new study off the ground. Dr. Acosta-López and his colleagues are working under a grant from the Colombian government to recruit 241 presymptomatic subjects with confirmed genetic markers for HD, and evaluate them for cognitive and neurologic changes preceding disease onset.
It’s a cross-sectional study, and such studies are usually funded for a year. But the investigators knew it would take much more than a year to recruit patients here, and planned their study for 3 years. As of July, the team had been engaging with the community for 6 months but still didn’t have a single blood sample.
“We’ve had to convince everyone that this time is different,” he said, “and that means focusing on the social aspect” – setting up a legal-assistance program through the university to help families claim health benefits and a job-training program sponsored by local businesses.
It’s unusual for researchers to find themselves playing such extensive roles in coordinating social and economic support for their subjects. But with HD, it’s happening across Latin America, where researchers speak frequently of a “debt” owed to HD families in this region.
Huntington’s disease is a neurodegenerative disease caused by a genetic mutation in the huntingtin (HTT) gene, changing the normal protein it expresses in the body to a toxic form that damages cortical and basal ganglia neurons. It affects between 0.5 to 1 in 10,000 people worldwide, with higher prevalence in the United States, Europe, and Australia.
HD is inherited in an autosomal dominant pattern; a child of a parent with the mutation has a 50% chance of developing the disease. Patients develop cognitive symptoms that progress to dementia, along with the debilitating involuntary, dancelike movements that gave the disease the name by which it was formerly known: Huntington’s chorea.
In the 1980s and 1990s, several generations of Latin American HD families provided data that allowed for some of the greatest research advances in the disease – and they may represent a large share of the world’s HD cases. Yet, they continue to live in extreme poverty and have benefited little from the findings of the past 3 decades.
Without recognizing this and working to improve the families’ well-being, the researchers at the conference said it’s unlikely that promising therapies in the pipeline will ever reach the populations that need them the most.
Discovery in Venezuela
Some 8 hours by car from Barranquilla sits Lake Maracaibo, Venezuela, home to the largest known clusters of HD patients worldwide. The disease is believed to have come to the shores of Lake Maracaibo with a lone European immigrant – a Spanish sailor, many claim – at the end of the 18th century. Cases were first described in the 1950s by a young Venezuelan physician named Américo Negrette, MD.
Dr. Negrette’s findings were ignored by health officials in Venezuela and went unnoticed in the international research community until 1972, when a student of Dr. Negrette’s presented at an HD conference in Ohio. There he drew the attention of the American neuropsychologist Nancy S. Wexler, PhD. Dr. Wexler’s own mother had died of HD, and her father Milton, a noted psychoanalyst, founded the first research foundation dedicated to the disease.
While prevalence of HD in North America, Australia, and Europe is about 1 in 10,000, the region around Lake Maracaibo saw 70 times that rate at the time, thanks to high birth rates, geographic isolation, and extensive intermarriage within a handful of families. The families comprised mostly poor fishermen who lived in makeshift homes in towns ringing the lake.
In 1979, Dr. Wexler, with funding from the U.S. National Institutes of Health, began making annual research visits to Lake Maracaibo, and in 1983, the research group she coordinated, using data from blood and tissue samples donated by affected families, identified the location of the huntingtin gene on chromosome 4 (Nature. 1983 Nov 17;306[5940]:234-8). A decade later, the researchers isolated the mutant version of the gene and found it to be a triplet (CAG) expansion mutation, with more CAG repetitions associated with earlier age at disease onset (Cell. 1993 Mar 26;72[6]:971-83). Dr. Wexler and her colleagues’ findings led to the first genetic tests for HD.
Nationalist policies in Venezuela ended Dr. Wexler and her colleagues’ annual visits to Lake Maracaibo in 2002, along with the food, clothing, and medicines that the group routinely distributed to the families when they came.
Over 23 years, the researchers obtained data from some 18,000 individuals, but the families did not benefit in any durable way from the research. Local investigators with whom Dr. Wexler’s group collaborated lacked the resources and training to continue independently.
Access to medications is limited in Venezuela, and there is no institutional support for hundreds of HD patients living in extreme poverty, many of them descendants of the patients who contributed to the research and generation of these samples. The families’ biological material was sent to labs abroad, where investigators continue to derive findings from it today. Though genetic testing was performed on thousands at risk for the disease, few received access to their results through genetic counseling. A hospice established by Dr. Wexler’s foundation limped along until 2014, when it was finally shuttered.
Rebuilding bridges
A handful of families from the Lake Maracaibo towns attended the conference in Barranquilla. Their travel costs were picked up by Factor-H, which sponsored the event.
Ignacio Muñoz-Sanjuán, PhD, Factor-H’s founder and president, knew the families personally. He’s visited them regularly for years. In 2017, Dr. Muñoz-Sanjuán, a molecular biologist known affectionately in the HD community as “Nacho,” invited several to Rome for a meeting with Pope Francis, as part of an effort to raise awareness of HD and to request support from the Catholic Church for the Latin American families.
Humanitarian work is relatively new to Dr. Muñoz-Sanjuán, who’s spent his career in drug development. In addition to his unpaid work with Factor-H, he is vice president of biology with the CHDI Foundation, a Los Angeles–based nonprofit that funds drug research in HD. CHDI is reported to have about $100 million in annual funding – about triple the NIH budget in recent years for HD research. Its major donors are a group of investors who for years have remained anonymous and do not publicly discuss their philanthropy.
The Spanish-born Dr. Muñoz-Sanjuán had little direct experience with HD populations in Latin America until a few years ago, he said.
At a CHDI meeting in Brazil, he said, “I was talking with physicians and patient advocates from Latin America, telling them they had to be willing to be involved, that these communities with high prevalence had a lot to offer science,” Dr. Muñoz-Sanjuán said in an interview. “I was told that it was me who needed to understand the conditions in which HD patients lived. It completely put me on the spot.”
HD tends to strike during the most productive years of a person’s life, from the late 30s onward, keeping them from working and obliging family members to stop working to care for them. In a poor community, it can condemn a family to a state of extreme poverty for generations. Tetrabenazine (Xenazine), a medication to quiet chorea symptoms, is costly enough that many patients must do without it. Ensuring adequate calorie intake is difficult in HD patients, whose constant movements cause them to lose weight.
Dr. Muñoz-Sanjuán traveled to Colombia, Venezuela, and Brazil, meeting HD families and doctors like neurologist Gustavo Barrios, MD, of Hospital Occidente de Kennedy in Bogotá, Colombia. In a talk at the Barranquilla conference, Dr. Barrios related the experience of his first visit to El Dificil, a community in northern Colombia where some large HD families are forced to survive on the equivalent of $5 a day. “I had to confront not only the fact that these families were living with a terrible disease but in conditions of extreme deprivation,” he said. “My life as a doctor changed that day.”
Dr. Muñoz-Sanjuán helped form a Latin American HD network to involve clinicians like Dr. Barrios who worked with HD clusters, most of them poorly studied. “These are all neglected communities that share similar features,” Dr. Muñoz-Sanjuán said.
On Colombia’s Caribbean coast, for example, HD had been documented since the early 1990s, but genotyping was not performed until recently. Prevalence data are “virtually nonexistent” in Colombia, said Sonia Moreno, PhD, a neuropsychologist at the University of Antioquia in Medellin. In a pilot study presented this year at the CHDI Foundation’s Enroll-HD Congress, Dr. Moreno and her colleagues mined Colombian public health data for likely HD cases, and argued for the creation of a national registry.
In 2012, Dr. Muñoz-Sanjuán founded Factor-H with the aim of improving living conditions for Latin American communities with HD.
Factor-H does not receive funds from CHDI Foundation and instead relies on donations from individuals and companies; its annual budget is less than $200,000. But through contracts with local nongovernmental organizations, it has sponsored health clinics and ongoing food assistance, delivered shipments of medicines and clothing, and started a sponsorship program for young people in HD families, whose studies often are interrupted caring for sick parents. It hopes to build permanent support centers in Colombia and Venezuela where HD families can get their food and medical needs met.
“The traditional thinking in the HD research community is that we’re helping people by doing the legwork to make medicine – and that’s not necessarily enough. You need a more holistic approach,” Dr. Muñoz-Sanjuán said.
Lennie Pineda, MSc, who recently retired as a geneticist with the University of Zulia in Maracaibo, Venezuela, said that Dr. Muñoz-Sanjuán was viewed skeptically when he first visited, in part because of his biomedical research background.
Ms. Pineda, who worked with the region’s HD families her whole career, has been wary of past research efforts in Venezuela. In 2010, she published a paper critical of Dr. Wexler’s and his colleagues’ approach (Revista Redbioética/UNESCO. 2010;1[2]:50-61), particularly regarding issues of informed consent.
“I was very cold to Nacho,” she laughed. “We all looked at him suspiciously.”
Ms. Pineda said Dr. Muñoz-Sanjuán won her over with his interest and creativity in finding concrete ways improve the lives of families in the Lake Maracaibo towns.
In a talk at the conference, Edison Soto, a young man from San Luis, a town on Lake Maracaibo that is a key cluster of HD, said Dr. Muñoz-Sanjuán’s visits had reawakened hope among the families there. “For years, no one thought about us, and because of the situation in the country it’s been hard, really hard,” he said.
“Nacho’s smart,” Ms. Pineda said. “He’s not coming to build a research cohort, he’s coming with genuine intention to help. But if one day conditions are adequate to support investigation, and the people here are well informed and volunteer for a study with full consent, well, all the better,” she said.
Dr. Muñoz-Sanjuán acknowledged that his humanitarian work could be perceived as preparing the ground for future clinical trials.
“I’m not doing anything research oriented with Latin America,” he said. “I would never approach these communities and recommend they take part in a study or give samples, unless their conditions change significantly. But the idea of cross-contamination is a problem I might need to fix. There may come a day where I need to depersonalize Factor-H from me.”
A research platform, a novel agent
Though HD research in Latin America remains rife with challenges, a number of investigators at the conference talked optimistically about planned and ongoing HD studies in Latin America.
The biggest of these is ENROLL-HD, a long-term global observational study of families with HD that uses a standardized approach to data collection. The platform, launched in 2013, aims to enroll 20,000 participants for yearly (or more frequent) assessment. Data from ENROLL-HD will support a diverse range of studies on everything from biomarkers to genetic modifiers to quality of life measures in HD.
ENROLL-HD has opened study sites in Argentina, Chile, and Colombia, and plans to launch a site near Lima, Peru, that is home to an HD cluster. Venezuela is considered out of reach, at least for now.
In Barranquilla, Claudia Perandones, MD, PhD, a genetics researcher in Argentina who manages ENROLL-HD for Latin America and is a cofounder of Factor-H, explained why the kind of clusters seen in Latin America are so valuable scientifically.
The extended family groups share a disease haplotype, eat the same foods, and live in similar environments, Dr. Perandones noted. Because not all the variation in HD can be explained by the number of CAG repeats a patient has, having a large sample with a common haplotype would help researchers pinpoint other environmental and genetic factors that can modify the onset or progress of the disease.
Another key goal of ENROLL-HD, investigators say, is to speed recruitment into clinical trials as they arise. And for the first time in history, potentially game-changing therapies are being developed specifically for HD.
For the past 5 years the Swiss pharmaceutical giant Roche has worked with a smaller biotech firm, Ionis Pharmaceuticals, on an agent called RG6042, which was known until recently as IONIS-HTTRx. CHDI was extensively involved in the agent’s preclinical development, contributing some $10 million to get it off the ground.
RG6042 is an antisense oligonucleotide, delivered by spinal injection, which works by interrupting an mRNA signaling pathway to suppress production of mutant HTT (mHTT) protein in the brain. Antisense oligonucleotides, sometimes called gene silencing therapies, are a new and promising approach in neurodegenerative diseases. Two have received FDA approval to treat spinal muscular atrophy and Duchenne muscular dystrophy.
In April 2018, Roche announced positive results from phase 1/2a study in 46 HD patients in Europe and North America. Patients in that 13-week study saw significant (up to 60%) dose-dependent reductions of the mHTT in their cerebrospinal fluid; a post hoc analysis also found some evidence of functional improvement (Neurology. 2018;90[15 Supplement]:CT.002).
These encouraging findings led to Roche’s announcement of a global phase 3 randomized, controlled trial that is scheduled to begin enrolling in 2019. Roche hopes to randomize 660 patients with mild HD across 15 countries for the 2-year trial, called GENERATION-HD1.
Sites in Latin America are expected to include Argentina, Chile, and Colombia.
At the Barranquilla meeting, Daniel Ciriano, MD, Roche’s Argentina-based medical director for Latin America, extolled the company’s commitment to ethics and social welfare in the region. In recent years, Roche has increased its humanitarian commitments across Latin America, including helping rebuild a Chilean village after an earthquake and offering free breast cancer and kidney disease treatments.
RG6042 is only one of a number of promising approaches to HD. Other therapies in the pipeline include gene silencing delivered by viral vectors instead of repeated spinal injections, an oral drug that interrupts mHTT production, immunotherapies, and even CRISPR gene–editing techniques.
Little was said at the conference, however, about how Latin American HD communities might be able to afford RG6042 or any other therapy that emerges from the pipeline.
Dr. Muñoz-Sanjuán called the issue “a theme for future discussion.”
“This is an area that has to be handled carefully and not one we are heavily invested in yet, although it’s very important,” he said.
On the ground
Several of the European and North American scientists who presented in Barranquilla took pains to express their concern with the well-being of HD patients in Latin America and to demonstrate goodwill toward the local researchers and clinicians.
Hilal A. Lashuel, PhD, a molecular biologist working on the structure and behavior of the HTT protein, said his participation in the Factor-H event at the Vatican the year before had awakened him to “the real human part of HD,” and changed the way he does science.
Normally, Dr. Lashuel said, “we do research disconnected from the realities of the diseases we work with.”
“We need to not just to do research but [to ensure] that research is done right,” he said, which means also focusing on improving patients’ standard of living.
The room broke out in applause when Dr. Lashuel announced new internships for investigators from developing countries. He also presented a parting video from his research team at the École Polytechnique Fédérale de Lausanne (Switzerland), complete with music and affectionate messages in Spanish.
Pharmacologist Elena Cattaneo, PhD, a stem cell researcher long active in the HD community, and also a senator in Italy’s parliament, delivered a similarly warm, carefully choreographed video message from her laboratory at the University of Milan.
Just days later in the town of Juan de Acosta, an hour inland of Barranquilla, the same researchers sat down with patients and families who crowded the waiting room of the town’s only hospital, as the sun beat in through the windows and as mule carts, stray dogs, and buses passed by on the main drag outside.
The event had been titled a “brigade” after all, but the HD families did not seem to mind – and indeed so many showed up that a sign had to be placed on the door saying that no one who arrived after noon could be seen. Consults were not limited to HD-related matters, so families could be seen for any complaint.
HD was first documented in this town in the early 1990s, but much remains to be understood about the size of the cluster, the haplotype, and its relation to other clusters in Colombia or Venezuela. The families here share a handful of last names and likely share a common ancestor. In the early 19th century, the Barranquilla region was flooded with European migrants who reached the city by ship. (HD clusters in Latin America tend to be concentrated in coastal regions, possibly because of migration patterns.)
The waiting room of the hospital was loud with chatter. Small children played as their relatives waited for consults. Some showed the characteristic restless movements and emaciated bodies of people with advanced HD.
The foreign scientists were barred from taking any patient data out of the hospital or asking for samples. Even picture taking was prohibited. Instead they performed genetic counseling and neuropsychological tests; they sorted out differential diagnoses and advised on medications. Visiting Colombian and Venezuelan physicians did the same, while their assistants met with families in the waiting room, taking medical histories and sketching out basic genealogies.
Some of the foreign researchers reported fruitful interactions with patients, while others seemed perplexed by what they’d experienced. Alba di Pardo, PhD, a genetic epidemiologist at the Istituto Neurologico Mediterraneo Pozzilli (Italy), said she’d spent the morning doing genetic counseling with families and going over genealogies to assess risk. Yet, despite the fact that anyone with an HD parent has a 50% chance of developing the disease, some family members acted uninterested, she said.
Dr. di Pardo’s colleague at the Istituto, biologist Vittorio Maglione, PhD, reported having a similar experience. As he was counseling a young woman about her risk for HD, she scrolled indifferently through Facebook posts on her phone, he said.
On some level, Dr. Maglione said, he could understand patients’ reluctance to engage noting that, while there were many potential HD therapies to try, any new treatment paradigm for HD was many years away from a place like this – and potentially very costly. Dr. Maglione – along with Dr. di Pardo – is researching the SP1 axis, a sphingolipid pathway implicated in neurodegenerative such diseases as HD and which has potential as a drug target (Trends Pharmacol Sci. 2018;39[5]:468-80).
Psychologist Pedro Puentes Rozo of Simón Bolivar University, who is working with Dr. Acosta-López on the local cohort study of presymptomatic HD patients, said that, for most of the families in the clinic that day, any seeming indifference probably masked deeper fears. People already were well aware of their risk. “They’ve known about it forever, said Dr. Puentes Rozo, who has been working with this HD population for a decade. “But this is a catastrophic illness and can generate a lot of anxiety.”
Dr. Puentes Rozo said the group’s planned study, unlike studies in the past, would be conducted under strict “international ethical norms and standards.” Subjects would receive ongoing psychological support, and the researchers were working to establish a genetic counseling center so that people who want to know their status “can be prepared,” he said, and plan for their lives and families.
By fall 2018, the cohort study was underway. The group had sponsored several more hospital brigades – or “integrated health days” as they preferred to call them, at the hospital in Juan de Acosta, giving them a chance to work face to face with families.
They drew no blood during the clinics, as investigators in the past had done. Instead, they explained the study to patients, performed the initial screenings, and invited them to designated study appointments at the university. Legal assistance was up and running, and the jobs program would start in 2019.
Enrollment was climbing. And the group was steadily accumulating data.
BARRANQUILLA, COLOMBIA – “We don’t like to call them brigades. That sounds militant,” said neuropsychologist Johan Acosta-López, PhD.
Dr. Acosta-López, the head of cognitive neurosciences at Simón Bolivar University in this city on Colombia’s Atlantic coast, was among five Colombian clinicians – neurologists, psychiatrists, and neuropsychologists – stuffed into a car on their way to a conference hotel in July 2018.
The following day they would be joined by clinicians and researchers from North America, other Latin American countries, and Europe for a first-of-its kind meeting on Huntington’s disease (HD) in the region, sponsored by Factor-H, an HD charity working in Latin America.
Once the talks wrapped up, the researchers – clinicians and basic scientists – were invited to see patients at a hospital in a town an hour inland with a large concentration of HD families, most of them extremely poor. For some, the Factor-H–sponsored “brigade” would be their first hands-on experience with HD patients in a developing country.
There was some debate in the car about what to call such events: brigades, “integrated health days,” or clinics. Around here – where HD abounded but patients were weary of researchers – terminology mattered.
“We’ve had so many investigators arrive in this area – foreigners and Colombians – telling people ‘we’ve got this huge, great project that you’ll benefit from.’ And they take blood samples and never return,” Dr. Acosta-López said.
Even as a local investigator, Dr. Acosta-López has faced challenges getting a new study off the ground. Dr. Acosta-López and his colleagues are working under a grant from the Colombian government to recruit 241 presymptomatic subjects with confirmed genetic markers for HD, and evaluate them for cognitive and neurologic changes preceding disease onset.
It’s a cross-sectional study, and such studies are usually funded for a year. But the investigators knew it would take much more than a year to recruit patients here, and planned their study for 3 years. As of July, the team had been engaging with the community for 6 months but still didn’t have a single blood sample.
“We’ve had to convince everyone that this time is different,” he said, “and that means focusing on the social aspect” – setting up a legal-assistance program through the university to help families claim health benefits and a job-training program sponsored by local businesses.
It’s unusual for researchers to find themselves playing such extensive roles in coordinating social and economic support for their subjects. But with HD, it’s happening across Latin America, where researchers speak frequently of a “debt” owed to HD families in this region.
Huntington’s disease is a neurodegenerative disease caused by a genetic mutation in the huntingtin (HTT) gene, changing the normal protein it expresses in the body to a toxic form that damages cortical and basal ganglia neurons. It affects between 0.5 to 1 in 10,000 people worldwide, with higher prevalence in the United States, Europe, and Australia.
HD is inherited in an autosomal dominant pattern; a child of a parent with the mutation has a 50% chance of developing the disease. Patients develop cognitive symptoms that progress to dementia, along with the debilitating involuntary, dancelike movements that gave the disease the name by which it was formerly known: Huntington’s chorea.
In the 1980s and 1990s, several generations of Latin American HD families provided data that allowed for some of the greatest research advances in the disease – and they may represent a large share of the world’s HD cases. Yet, they continue to live in extreme poverty and have benefited little from the findings of the past 3 decades.
Without recognizing this and working to improve the families’ well-being, the researchers at the conference said it’s unlikely that promising therapies in the pipeline will ever reach the populations that need them the most.
Discovery in Venezuela
Some 8 hours by car from Barranquilla sits Lake Maracaibo, Venezuela, home to the largest known clusters of HD patients worldwide. The disease is believed to have come to the shores of Lake Maracaibo with a lone European immigrant – a Spanish sailor, many claim – at the end of the 18th century. Cases were first described in the 1950s by a young Venezuelan physician named Américo Negrette, MD.
Dr. Negrette’s findings were ignored by health officials in Venezuela and went unnoticed in the international research community until 1972, when a student of Dr. Negrette’s presented at an HD conference in Ohio. There he drew the attention of the American neuropsychologist Nancy S. Wexler, PhD. Dr. Wexler’s own mother had died of HD, and her father Milton, a noted psychoanalyst, founded the first research foundation dedicated to the disease.
While prevalence of HD in North America, Australia, and Europe is about 1 in 10,000, the region around Lake Maracaibo saw 70 times that rate at the time, thanks to high birth rates, geographic isolation, and extensive intermarriage within a handful of families. The families comprised mostly poor fishermen who lived in makeshift homes in towns ringing the lake.
In 1979, Dr. Wexler, with funding from the U.S. National Institutes of Health, began making annual research visits to Lake Maracaibo, and in 1983, the research group she coordinated, using data from blood and tissue samples donated by affected families, identified the location of the huntingtin gene on chromosome 4 (Nature. 1983 Nov 17;306[5940]:234-8). A decade later, the researchers isolated the mutant version of the gene and found it to be a triplet (CAG) expansion mutation, with more CAG repetitions associated with earlier age at disease onset (Cell. 1993 Mar 26;72[6]:971-83). Dr. Wexler and her colleagues’ findings led to the first genetic tests for HD.
Nationalist policies in Venezuela ended Dr. Wexler and her colleagues’ annual visits to Lake Maracaibo in 2002, along with the food, clothing, and medicines that the group routinely distributed to the families when they came.
Over 23 years, the researchers obtained data from some 18,000 individuals, but the families did not benefit in any durable way from the research. Local investigators with whom Dr. Wexler’s group collaborated lacked the resources and training to continue independently.
Access to medications is limited in Venezuela, and there is no institutional support for hundreds of HD patients living in extreme poverty, many of them descendants of the patients who contributed to the research and generation of these samples. The families’ biological material was sent to labs abroad, where investigators continue to derive findings from it today. Though genetic testing was performed on thousands at risk for the disease, few received access to their results through genetic counseling. A hospice established by Dr. Wexler’s foundation limped along until 2014, when it was finally shuttered.
Rebuilding bridges
A handful of families from the Lake Maracaibo towns attended the conference in Barranquilla. Their travel costs were picked up by Factor-H, which sponsored the event.
Ignacio Muñoz-Sanjuán, PhD, Factor-H’s founder and president, knew the families personally. He’s visited them regularly for years. In 2017, Dr. Muñoz-Sanjuán, a molecular biologist known affectionately in the HD community as “Nacho,” invited several to Rome for a meeting with Pope Francis, as part of an effort to raise awareness of HD and to request support from the Catholic Church for the Latin American families.
Humanitarian work is relatively new to Dr. Muñoz-Sanjuán, who’s spent his career in drug development. In addition to his unpaid work with Factor-H, he is vice president of biology with the CHDI Foundation, a Los Angeles–based nonprofit that funds drug research in HD. CHDI is reported to have about $100 million in annual funding – about triple the NIH budget in recent years for HD research. Its major donors are a group of investors who for years have remained anonymous and do not publicly discuss their philanthropy.
The Spanish-born Dr. Muñoz-Sanjuán had little direct experience with HD populations in Latin America until a few years ago, he said.
At a CHDI meeting in Brazil, he said, “I was talking with physicians and patient advocates from Latin America, telling them they had to be willing to be involved, that these communities with high prevalence had a lot to offer science,” Dr. Muñoz-Sanjuán said in an interview. “I was told that it was me who needed to understand the conditions in which HD patients lived. It completely put me on the spot.”
HD tends to strike during the most productive years of a person’s life, from the late 30s onward, keeping them from working and obliging family members to stop working to care for them. In a poor community, it can condemn a family to a state of extreme poverty for generations. Tetrabenazine (Xenazine), a medication to quiet chorea symptoms, is costly enough that many patients must do without it. Ensuring adequate calorie intake is difficult in HD patients, whose constant movements cause them to lose weight.
Dr. Muñoz-Sanjuán traveled to Colombia, Venezuela, and Brazil, meeting HD families and doctors like neurologist Gustavo Barrios, MD, of Hospital Occidente de Kennedy in Bogotá, Colombia. In a talk at the Barranquilla conference, Dr. Barrios related the experience of his first visit to El Dificil, a community in northern Colombia where some large HD families are forced to survive on the equivalent of $5 a day. “I had to confront not only the fact that these families were living with a terrible disease but in conditions of extreme deprivation,” he said. “My life as a doctor changed that day.”
Dr. Muñoz-Sanjuán helped form a Latin American HD network to involve clinicians like Dr. Barrios who worked with HD clusters, most of them poorly studied. “These are all neglected communities that share similar features,” Dr. Muñoz-Sanjuán said.
On Colombia’s Caribbean coast, for example, HD had been documented since the early 1990s, but genotyping was not performed until recently. Prevalence data are “virtually nonexistent” in Colombia, said Sonia Moreno, PhD, a neuropsychologist at the University of Antioquia in Medellin. In a pilot study presented this year at the CHDI Foundation’s Enroll-HD Congress, Dr. Moreno and her colleagues mined Colombian public health data for likely HD cases, and argued for the creation of a national registry.
In 2012, Dr. Muñoz-Sanjuán founded Factor-H with the aim of improving living conditions for Latin American communities with HD.
Factor-H does not receive funds from CHDI Foundation and instead relies on donations from individuals and companies; its annual budget is less than $200,000. But through contracts with local nongovernmental organizations, it has sponsored health clinics and ongoing food assistance, delivered shipments of medicines and clothing, and started a sponsorship program for young people in HD families, whose studies often are interrupted caring for sick parents. It hopes to build permanent support centers in Colombia and Venezuela where HD families can get their food and medical needs met.
“The traditional thinking in the HD research community is that we’re helping people by doing the legwork to make medicine – and that’s not necessarily enough. You need a more holistic approach,” Dr. Muñoz-Sanjuán said.
Lennie Pineda, MSc, who recently retired as a geneticist with the University of Zulia in Maracaibo, Venezuela, said that Dr. Muñoz-Sanjuán was viewed skeptically when he first visited, in part because of his biomedical research background.
Ms. Pineda, who worked with the region’s HD families her whole career, has been wary of past research efforts in Venezuela. In 2010, she published a paper critical of Dr. Wexler’s and his colleagues’ approach (Revista Redbioética/UNESCO. 2010;1[2]:50-61), particularly regarding issues of informed consent.
“I was very cold to Nacho,” she laughed. “We all looked at him suspiciously.”
Ms. Pineda said Dr. Muñoz-Sanjuán won her over with his interest and creativity in finding concrete ways improve the lives of families in the Lake Maracaibo towns.
In a talk at the conference, Edison Soto, a young man from San Luis, a town on Lake Maracaibo that is a key cluster of HD, said Dr. Muñoz-Sanjuán’s visits had reawakened hope among the families there. “For years, no one thought about us, and because of the situation in the country it’s been hard, really hard,” he said.
“Nacho’s smart,” Ms. Pineda said. “He’s not coming to build a research cohort, he’s coming with genuine intention to help. But if one day conditions are adequate to support investigation, and the people here are well informed and volunteer for a study with full consent, well, all the better,” she said.
Dr. Muñoz-Sanjuán acknowledged that his humanitarian work could be perceived as preparing the ground for future clinical trials.
“I’m not doing anything research oriented with Latin America,” he said. “I would never approach these communities and recommend they take part in a study or give samples, unless their conditions change significantly. But the idea of cross-contamination is a problem I might need to fix. There may come a day where I need to depersonalize Factor-H from me.”
A research platform, a novel agent
Though HD research in Latin America remains rife with challenges, a number of investigators at the conference talked optimistically about planned and ongoing HD studies in Latin America.
The biggest of these is ENROLL-HD, a long-term global observational study of families with HD that uses a standardized approach to data collection. The platform, launched in 2013, aims to enroll 20,000 participants for yearly (or more frequent) assessment. Data from ENROLL-HD will support a diverse range of studies on everything from biomarkers to genetic modifiers to quality of life measures in HD.
ENROLL-HD has opened study sites in Argentina, Chile, and Colombia, and plans to launch a site near Lima, Peru, that is home to an HD cluster. Venezuela is considered out of reach, at least for now.
In Barranquilla, Claudia Perandones, MD, PhD, a genetics researcher in Argentina who manages ENROLL-HD for Latin America and is a cofounder of Factor-H, explained why the kind of clusters seen in Latin America are so valuable scientifically.
The extended family groups share a disease haplotype, eat the same foods, and live in similar environments, Dr. Perandones noted. Because not all the variation in HD can be explained by the number of CAG repeats a patient has, having a large sample with a common haplotype would help researchers pinpoint other environmental and genetic factors that can modify the onset or progress of the disease.
Another key goal of ENROLL-HD, investigators say, is to speed recruitment into clinical trials as they arise. And for the first time in history, potentially game-changing therapies are being developed specifically for HD.
For the past 5 years the Swiss pharmaceutical giant Roche has worked with a smaller biotech firm, Ionis Pharmaceuticals, on an agent called RG6042, which was known until recently as IONIS-HTTRx. CHDI was extensively involved in the agent’s preclinical development, contributing some $10 million to get it off the ground.
RG6042 is an antisense oligonucleotide, delivered by spinal injection, which works by interrupting an mRNA signaling pathway to suppress production of mutant HTT (mHTT) protein in the brain. Antisense oligonucleotides, sometimes called gene silencing therapies, are a new and promising approach in neurodegenerative diseases. Two have received FDA approval to treat spinal muscular atrophy and Duchenne muscular dystrophy.
In April 2018, Roche announced positive results from phase 1/2a study in 46 HD patients in Europe and North America. Patients in that 13-week study saw significant (up to 60%) dose-dependent reductions of the mHTT in their cerebrospinal fluid; a post hoc analysis also found some evidence of functional improvement (Neurology. 2018;90[15 Supplement]:CT.002).
These encouraging findings led to Roche’s announcement of a global phase 3 randomized, controlled trial that is scheduled to begin enrolling in 2019. Roche hopes to randomize 660 patients with mild HD across 15 countries for the 2-year trial, called GENERATION-HD1.
Sites in Latin America are expected to include Argentina, Chile, and Colombia.
At the Barranquilla meeting, Daniel Ciriano, MD, Roche’s Argentina-based medical director for Latin America, extolled the company’s commitment to ethics and social welfare in the region. In recent years, Roche has increased its humanitarian commitments across Latin America, including helping rebuild a Chilean village after an earthquake and offering free breast cancer and kidney disease treatments.
RG6042 is only one of a number of promising approaches to HD. Other therapies in the pipeline include gene silencing delivered by viral vectors instead of repeated spinal injections, an oral drug that interrupts mHTT production, immunotherapies, and even CRISPR gene–editing techniques.
Little was said at the conference, however, about how Latin American HD communities might be able to afford RG6042 or any other therapy that emerges from the pipeline.
Dr. Muñoz-Sanjuán called the issue “a theme for future discussion.”
“This is an area that has to be handled carefully and not one we are heavily invested in yet, although it’s very important,” he said.
On the ground
Several of the European and North American scientists who presented in Barranquilla took pains to express their concern with the well-being of HD patients in Latin America and to demonstrate goodwill toward the local researchers and clinicians.
Hilal A. Lashuel, PhD, a molecular biologist working on the structure and behavior of the HTT protein, said his participation in the Factor-H event at the Vatican the year before had awakened him to “the real human part of HD,” and changed the way he does science.
Normally, Dr. Lashuel said, “we do research disconnected from the realities of the diseases we work with.”
“We need to not just to do research but [to ensure] that research is done right,” he said, which means also focusing on improving patients’ standard of living.
The room broke out in applause when Dr. Lashuel announced new internships for investigators from developing countries. He also presented a parting video from his research team at the École Polytechnique Fédérale de Lausanne (Switzerland), complete with music and affectionate messages in Spanish.
Pharmacologist Elena Cattaneo, PhD, a stem cell researcher long active in the HD community, and also a senator in Italy’s parliament, delivered a similarly warm, carefully choreographed video message from her laboratory at the University of Milan.
Just days later in the town of Juan de Acosta, an hour inland of Barranquilla, the same researchers sat down with patients and families who crowded the waiting room of the town’s only hospital, as the sun beat in through the windows and as mule carts, stray dogs, and buses passed by on the main drag outside.
The event had been titled a “brigade” after all, but the HD families did not seem to mind – and indeed so many showed up that a sign had to be placed on the door saying that no one who arrived after noon could be seen. Consults were not limited to HD-related matters, so families could be seen for any complaint.
HD was first documented in this town in the early 1990s, but much remains to be understood about the size of the cluster, the haplotype, and its relation to other clusters in Colombia or Venezuela. The families here share a handful of last names and likely share a common ancestor. In the early 19th century, the Barranquilla region was flooded with European migrants who reached the city by ship. (HD clusters in Latin America tend to be concentrated in coastal regions, possibly because of migration patterns.)
The waiting room of the hospital was loud with chatter. Small children played as their relatives waited for consults. Some showed the characteristic restless movements and emaciated bodies of people with advanced HD.
The foreign scientists were barred from taking any patient data out of the hospital or asking for samples. Even picture taking was prohibited. Instead they performed genetic counseling and neuropsychological tests; they sorted out differential diagnoses and advised on medications. Visiting Colombian and Venezuelan physicians did the same, while their assistants met with families in the waiting room, taking medical histories and sketching out basic genealogies.
Some of the foreign researchers reported fruitful interactions with patients, while others seemed perplexed by what they’d experienced. Alba di Pardo, PhD, a genetic epidemiologist at the Istituto Neurologico Mediterraneo Pozzilli (Italy), said she’d spent the morning doing genetic counseling with families and going over genealogies to assess risk. Yet, despite the fact that anyone with an HD parent has a 50% chance of developing the disease, some family members acted uninterested, she said.
Dr. di Pardo’s colleague at the Istituto, biologist Vittorio Maglione, PhD, reported having a similar experience. As he was counseling a young woman about her risk for HD, she scrolled indifferently through Facebook posts on her phone, he said.
On some level, Dr. Maglione said, he could understand patients’ reluctance to engage noting that, while there were many potential HD therapies to try, any new treatment paradigm for HD was many years away from a place like this – and potentially very costly. Dr. Maglione – along with Dr. di Pardo – is researching the SP1 axis, a sphingolipid pathway implicated in neurodegenerative such diseases as HD and which has potential as a drug target (Trends Pharmacol Sci. 2018;39[5]:468-80).
Psychologist Pedro Puentes Rozo of Simón Bolivar University, who is working with Dr. Acosta-López on the local cohort study of presymptomatic HD patients, said that, for most of the families in the clinic that day, any seeming indifference probably masked deeper fears. People already were well aware of their risk. “They’ve known about it forever, said Dr. Puentes Rozo, who has been working with this HD population for a decade. “But this is a catastrophic illness and can generate a lot of anxiety.”
Dr. Puentes Rozo said the group’s planned study, unlike studies in the past, would be conducted under strict “international ethical norms and standards.” Subjects would receive ongoing psychological support, and the researchers were working to establish a genetic counseling center so that people who want to know their status “can be prepared,” he said, and plan for their lives and families.
By fall 2018, the cohort study was underway. The group had sponsored several more hospital brigades – or “integrated health days” as they preferred to call them, at the hospital in Juan de Acosta, giving them a chance to work face to face with families.
They drew no blood during the clinics, as investigators in the past had done. Instead, they explained the study to patients, performed the initial screenings, and invited them to designated study appointments at the university. Legal assistance was up and running, and the jobs program would start in 2019.
Enrollment was climbing. And the group was steadily accumulating data.
RELIEF: In Behçet’s, apremilast improves oral ulcers for up to 28 weeks
CHICAGO – Apremilast was effective and well tolerated for up to 28 weeks for the treatment of oral ulcers in patients with active Behçet’s disease, based on findings from the randomized, placebo-controlled, phase 3 RELIEF trial.
At baseline, mean oral ulcer counts were 4.2 in 104 patients randomized to receive the oral phosphodiesterase-4 inhibitor and 3.9 in 103 patients in the placebo group. Mean visual analog scale (VAS) pain scores were 61.2 and 60.8 in the two groups, respectively.
The primary study endpoint of area under the curve for total number of oral ulcers over a 12-week period (AUCWk0-12) – a measure that reflects the number of oral ulcers that occur over time and also accounts for the recurring-remitting course of oral ulcers – was achieved. AUCWk0-12 was significantly lower in the apremilast group than in the placebo group (129.54 vs. 222.14, respectively; P less than .0001), Gulen Hatemi, MD, reported at the annual meeting of the American College of Rheumatology.
From baseline to week 12, apremilast treatment also resulted in a significantly lower number of oral ulcers (mean of 1.1 vs. 2.0 for placebo at 12 weeks) and significantly reduced pain from oral ulcers at every visit from week 1 through week 12 of the study, compared with placebo (mean VAS score change from baseline, –40.7 vs. –15.9), said Dr. Hatemi, a professor of medicine at Istanbul University.
“The [12-week] complete response rate ... was 53% in the apremilast group and 22.3% in the placebo group. The [12-week] partial response rate ...was 76% in the apremilast group and 48% in the placebo group,” she said, adding that the efficacy of apremilast was sustained with continued treatment through 28 weeks.
Study participants were adults (mean age, 40 years) with active Behçet’s disease and three or more oral ulcers at randomization or two or more at screening and at randomization. All had been previously treated with at least one nonbiologic medication for oral ulcers and were allowed to have received previous biologic therapies for other disease manifestations. Those with active major organ involvement were excluded.
Treatment included a 30-mg dose of apremilast twice daily for 12 weeks or placebo. After 12 weeks, all patients received apremilast through at least 28 weeks of the 64-week study.
At the 28-week analysis, patients who were initially randomized to placebo and who switched to apremilast after week 12 had benefits comparable with those seen in those randomized to apremilast at the start of the study. A complete response was seen in 59% and 62% of patients in the groups, respectively, and a partial response was seen in 90% and 85%, respectively. Additionally, the mean change in the VAS score for oral ulcer pain in the groups at that time was –40.6 and –41.9, Dr. Hatemi said.
Apremilast was well tolerated in this study; the incidence of adverse events was comparable in the treatment and placebo groups during the 12-week placebo-controlled phase of the study – 78.8% and 71.8%, respectively. The most common events were diarrhea, nausea, headache, and upper respiratory tract infection, she said.
“These were generally mild to moderate, and only two patients had to discontinue the study due to gastrointestinal adverse events,” she said, noting that no new safety signals were observed.
Behçet’s disease is a chronic, relapsing, multisystem inflammatory disorder characterized by recurrent oral ulcers that can be disabling and have a substantial effect on quality of life. These findings, which include efficacy data up to 28 weeks and safety data for at least 100 patients exposed to apremilast for at least 1 year, demonstrate the efficacy of apremilast for the treatment oral ulcers in patients with Behçet’s disease, she said, noting that “the safety findings were consistent with the known safety profile of apremilast.”
The RELIEF study was supported by Celgene. Dr. Hatemi reported receiving grant/research support from Celgene and serving as a speaker for AbbVie, Mustafa Nevzet Pharmaceuticals, and UCB.
SOURCE: Hatemi G et al. Arthritis Rheumatol. 2018;70(Suppl 10), Abstract 2789.
CHICAGO – Apremilast was effective and well tolerated for up to 28 weeks for the treatment of oral ulcers in patients with active Behçet’s disease, based on findings from the randomized, placebo-controlled, phase 3 RELIEF trial.
At baseline, mean oral ulcer counts were 4.2 in 104 patients randomized to receive the oral phosphodiesterase-4 inhibitor and 3.9 in 103 patients in the placebo group. Mean visual analog scale (VAS) pain scores were 61.2 and 60.8 in the two groups, respectively.
The primary study endpoint of area under the curve for total number of oral ulcers over a 12-week period (AUCWk0-12) – a measure that reflects the number of oral ulcers that occur over time and also accounts for the recurring-remitting course of oral ulcers – was achieved. AUCWk0-12 was significantly lower in the apremilast group than in the placebo group (129.54 vs. 222.14, respectively; P less than .0001), Gulen Hatemi, MD, reported at the annual meeting of the American College of Rheumatology.
From baseline to week 12, apremilast treatment also resulted in a significantly lower number of oral ulcers (mean of 1.1 vs. 2.0 for placebo at 12 weeks) and significantly reduced pain from oral ulcers at every visit from week 1 through week 12 of the study, compared with placebo (mean VAS score change from baseline, –40.7 vs. –15.9), said Dr. Hatemi, a professor of medicine at Istanbul University.
“The [12-week] complete response rate ... was 53% in the apremilast group and 22.3% in the placebo group. The [12-week] partial response rate ...was 76% in the apremilast group and 48% in the placebo group,” she said, adding that the efficacy of apremilast was sustained with continued treatment through 28 weeks.
Study participants were adults (mean age, 40 years) with active Behçet’s disease and three or more oral ulcers at randomization or two or more at screening and at randomization. All had been previously treated with at least one nonbiologic medication for oral ulcers and were allowed to have received previous biologic therapies for other disease manifestations. Those with active major organ involvement were excluded.
Treatment included a 30-mg dose of apremilast twice daily for 12 weeks or placebo. After 12 weeks, all patients received apremilast through at least 28 weeks of the 64-week study.
At the 28-week analysis, patients who were initially randomized to placebo and who switched to apremilast after week 12 had benefits comparable with those seen in those randomized to apremilast at the start of the study. A complete response was seen in 59% and 62% of patients in the groups, respectively, and a partial response was seen in 90% and 85%, respectively. Additionally, the mean change in the VAS score for oral ulcer pain in the groups at that time was –40.6 and –41.9, Dr. Hatemi said.
Apremilast was well tolerated in this study; the incidence of adverse events was comparable in the treatment and placebo groups during the 12-week placebo-controlled phase of the study – 78.8% and 71.8%, respectively. The most common events were diarrhea, nausea, headache, and upper respiratory tract infection, she said.
“These were generally mild to moderate, and only two patients had to discontinue the study due to gastrointestinal adverse events,” she said, noting that no new safety signals were observed.
Behçet’s disease is a chronic, relapsing, multisystem inflammatory disorder characterized by recurrent oral ulcers that can be disabling and have a substantial effect on quality of life. These findings, which include efficacy data up to 28 weeks and safety data for at least 100 patients exposed to apremilast for at least 1 year, demonstrate the efficacy of apremilast for the treatment oral ulcers in patients with Behçet’s disease, she said, noting that “the safety findings were consistent with the known safety profile of apremilast.”
The RELIEF study was supported by Celgene. Dr. Hatemi reported receiving grant/research support from Celgene and serving as a speaker for AbbVie, Mustafa Nevzet Pharmaceuticals, and UCB.
SOURCE: Hatemi G et al. Arthritis Rheumatol. 2018;70(Suppl 10), Abstract 2789.
CHICAGO – Apremilast was effective and well tolerated for up to 28 weeks for the treatment of oral ulcers in patients with active Behçet’s disease, based on findings from the randomized, placebo-controlled, phase 3 RELIEF trial.
At baseline, mean oral ulcer counts were 4.2 in 104 patients randomized to receive the oral phosphodiesterase-4 inhibitor and 3.9 in 103 patients in the placebo group. Mean visual analog scale (VAS) pain scores were 61.2 and 60.8 in the two groups, respectively.
The primary study endpoint of area under the curve for total number of oral ulcers over a 12-week period (AUCWk0-12) – a measure that reflects the number of oral ulcers that occur over time and also accounts for the recurring-remitting course of oral ulcers – was achieved. AUCWk0-12 was significantly lower in the apremilast group than in the placebo group (129.54 vs. 222.14, respectively; P less than .0001), Gulen Hatemi, MD, reported at the annual meeting of the American College of Rheumatology.
From baseline to week 12, apremilast treatment also resulted in a significantly lower number of oral ulcers (mean of 1.1 vs. 2.0 for placebo at 12 weeks) and significantly reduced pain from oral ulcers at every visit from week 1 through week 12 of the study, compared with placebo (mean VAS score change from baseline, –40.7 vs. –15.9), said Dr. Hatemi, a professor of medicine at Istanbul University.
“The [12-week] complete response rate ... was 53% in the apremilast group and 22.3% in the placebo group. The [12-week] partial response rate ...was 76% in the apremilast group and 48% in the placebo group,” she said, adding that the efficacy of apremilast was sustained with continued treatment through 28 weeks.
Study participants were adults (mean age, 40 years) with active Behçet’s disease and three or more oral ulcers at randomization or two or more at screening and at randomization. All had been previously treated with at least one nonbiologic medication for oral ulcers and were allowed to have received previous biologic therapies for other disease manifestations. Those with active major organ involvement were excluded.
Treatment included a 30-mg dose of apremilast twice daily for 12 weeks or placebo. After 12 weeks, all patients received apremilast through at least 28 weeks of the 64-week study.
At the 28-week analysis, patients who were initially randomized to placebo and who switched to apremilast after week 12 had benefits comparable with those seen in those randomized to apremilast at the start of the study. A complete response was seen in 59% and 62% of patients in the groups, respectively, and a partial response was seen in 90% and 85%, respectively. Additionally, the mean change in the VAS score for oral ulcer pain in the groups at that time was –40.6 and –41.9, Dr. Hatemi said.
Apremilast was well tolerated in this study; the incidence of adverse events was comparable in the treatment and placebo groups during the 12-week placebo-controlled phase of the study – 78.8% and 71.8%, respectively. The most common events were diarrhea, nausea, headache, and upper respiratory tract infection, she said.
“These were generally mild to moderate, and only two patients had to discontinue the study due to gastrointestinal adverse events,” she said, noting that no new safety signals were observed.
Behçet’s disease is a chronic, relapsing, multisystem inflammatory disorder characterized by recurrent oral ulcers that can be disabling and have a substantial effect on quality of life. These findings, which include efficacy data up to 28 weeks and safety data for at least 100 patients exposed to apremilast for at least 1 year, demonstrate the efficacy of apremilast for the treatment oral ulcers in patients with Behçet’s disease, she said, noting that “the safety findings were consistent with the known safety profile of apremilast.”
The RELIEF study was supported by Celgene. Dr. Hatemi reported receiving grant/research support from Celgene and serving as a speaker for AbbVie, Mustafa Nevzet Pharmaceuticals, and UCB.
SOURCE: Hatemi G et al. Arthritis Rheumatol. 2018;70(Suppl 10), Abstract 2789.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point: Apremilast is safe and effective for treating oral ulcers in patients with Behçet’s disease.
Major finding: The AUCWk0-12 was significantly lower with apremilast (129.54) versus placebo (222.14).
Study details: A randomized, placebo-controlled, phase 3 study of 207 patients.
Disclosures: The RELIEF study was supported by Celgene. Dr. Hatemi reported receiving grant/research support from Celgene and serving as a speaker for AbbVie, Mustafa Nevzet Pharmaceuticals, and UCB.
Source: Hatemi G et al. Arthritis Rheumatol. 2018;70(Suppl 10), Abstract 2789
Firdapse approved: First treatment for rare autoimmune disorder
The Food and Drug Administration has approved amifampridine (Firdapse) as the first treatment for the rare autoimmune disorder known as Lambert-Eaton myasthenic syndrome, which causes the immune system to attack the neuromuscular junction and thereby disrupts the nerves’ ability to send signals to muscle cells. This causes fatigue and weakness in those affected, so they can experience difficulties with activities of daily living as a result.

The most common side effects included prickling sensation, upper respiratory tract infection, abdominal pain, and muscle spasms.
More information can be found in the FDA’s press announcement.
cpalmer@mdedge.com
The Food and Drug Administration has approved amifampridine (Firdapse) as the first treatment for the rare autoimmune disorder known as Lambert-Eaton myasthenic syndrome, which causes the immune system to attack the neuromuscular junction and thereby disrupts the nerves’ ability to send signals to muscle cells. This causes fatigue and weakness in those affected, so they can experience difficulties with activities of daily living as a result.
The most common side effects included prickling sensation, upper respiratory tract infection, abdominal pain, and muscle spasms.
More information can be found in the FDA’s press announcement.
cpalmer@mdedge.com
The Food and Drug Administration has approved amifampridine (Firdapse) as the first treatment for the rare autoimmune disorder known as Lambert-Eaton myasthenic syndrome, which causes the immune system to attack the neuromuscular junction and thereby disrupts the nerves’ ability to send signals to muscle cells. This causes fatigue and weakness in those affected, so they can experience difficulties with activities of daily living as a result.
The most common side effects included prickling sensation, upper respiratory tract infection, abdominal pain, and muscle spasms.
More information can be found in the FDA’s press announcement.
cpalmer@mdedge.com
FDA approves larotrectinib for cancers with NTRK gene fusions
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
Ichthyosiform Sarcoidosis and Systemic Involvement
Sarcoidosis is a multiorgan, systemic, granulomatous disease that most commonly affects the cutaneous, pulmonary, ocular, and cardiac organ systems. Cutaneous involvement occurs in approximately 20% to 35% of patients, with approximately 25% of patients demonstrating only dermatologic findings.1 Cutaneous sarcoidosis can have a highly variable presentation. Ichthyosiform sarcoidosis (IS) is a rare form of this disease that has been described as presenting as polygonal adherent scales.2 It often is associated with internal organ involvement. We present a case of IS without any organ system involvement at the time of diagnosis. A review of the English-language literature was performed to ascertain the internal organ associations most commonly reported with IS.
Case Report
A 66-year-old black woman presented to dermatology with dark scaly patches noted by her primary care physician to be present on both of the lower extremities. The patient believed they were present for at least 4 years. She described dark spots confined to the lower legs that had gradually increased in size. Review of systems was negative for fever, chills, night sweats, weight loss, vision changes, cough, dyspnea, and joint pains, and there was no history of either personal or familial cutaneous diseases.
Physical examination revealed cutaneous patches of thin white scale with a sharp edge in arciform patterns on the lower extremities. Several of these patches were hyperpigmented and xerotic in appearance (Figure 1). The patches were limited to the lower legs, with no other lesions noted.
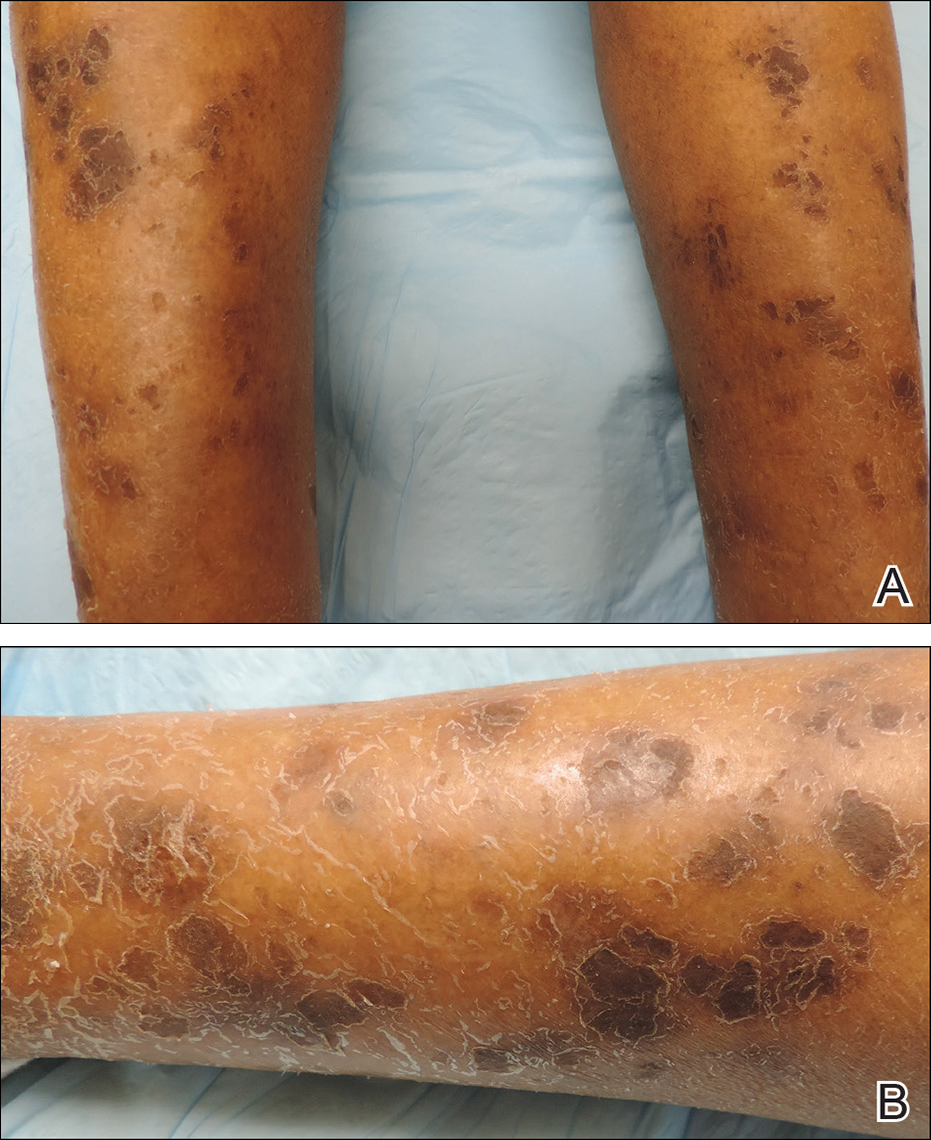
A punch biopsy of the skin on the right lower leg was performed. Histopathologic analysis showed epidermal compact hyperkeratosis with deep granulomatous infiltration into the subcutaneous tissue (Figures 2A and 2B). At high power, these granulomas were noted to be noncaseating naked granulomas composed of epithelioid histiocytes surrounded by sparse lymphocytic inflammation (Figure 2C). Special stains including acid-fast bacilli, Fite, and periodic acid–Schiff were negative. The diagnosis of IS was made based on clinical presentation and primarily by histopathologic analysis.
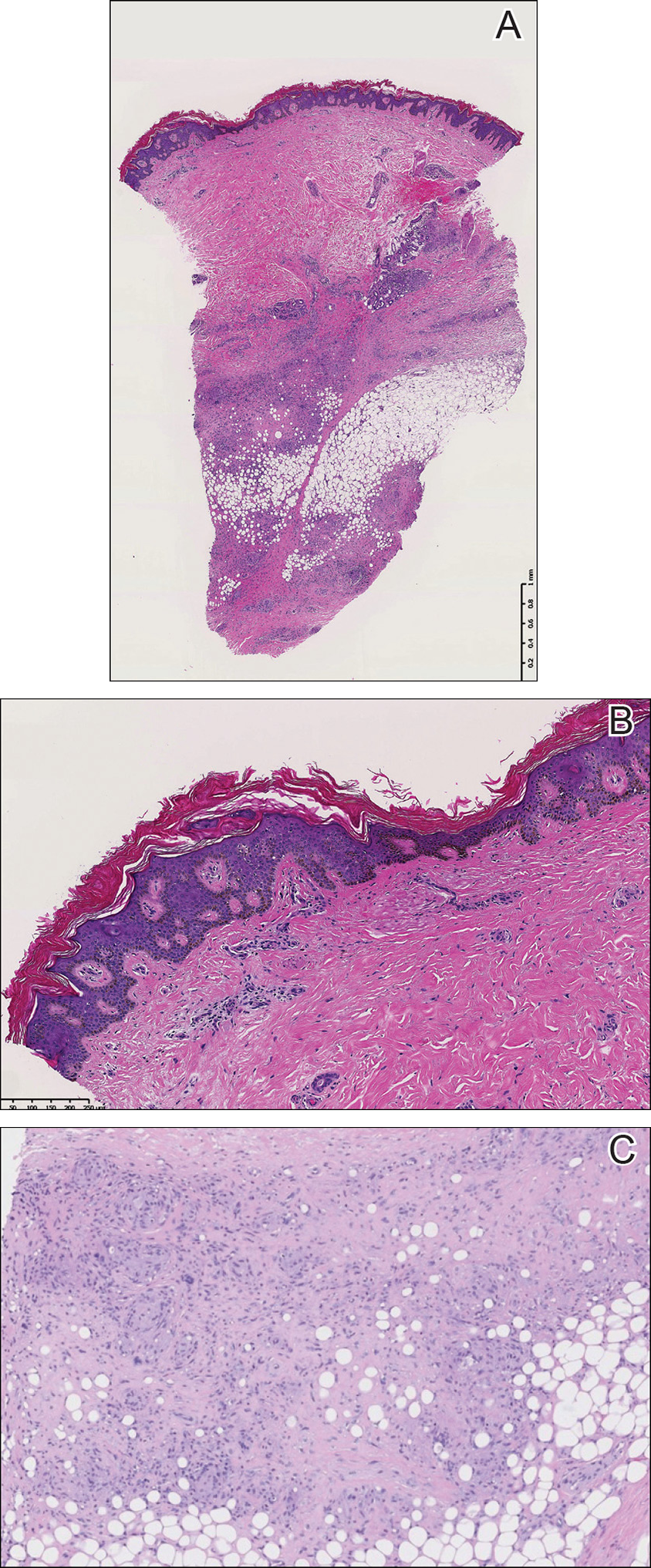
The patient’s cutaneous lesions were treated with fluocinonide ointment 0.05% twice daily. Although she did not notice a dramatic improvement in the plaques, they stabilized in size. Her primary care physician was notified and advised to begin a workup for involvement of other organ systems by sarcoidosis. Her initial evaluation, which included a chest radiograph and electrocardiogram, were unremarkable. Despite multiple attempts to persuade the patient to return for further follow-up, neither dermatology nor her primary care physician were able to complete a full workup.
Comment
Etiology
Although there are several theories regarding the etiology of sarcoidosis, the exact cause remains unknown. The body’s immune response, infectious agents, genetics, and the environment have all been thought to play a role. It has been well established that helper T cell (TH1) production of interferon and increased levels of tumor necrosis factor propagate the inflammatory response seen in sarcoidosis.3 More recently, TH17 cells have been found in cutaneous lesions, bronchoalveolar lavage samples, and the blood of patients with sarcoidosis, especially in those with active disease progression.3 Infectious agents such as mycobacteria and propionibacteria DNA or RNA also have been found in sarcoid samples.4 Several HLA-DRB1 variants have been associated with an increased incidence of sarcoidosis.5
Presentation
Characteristic dermatologic findings of sarcoidosis include macules, papules, nodules, and plaques located on the face, especially the nose, cheeks, and ears, and on the shins or ankles, as well as similar lesions around tattoos or scars. Sarcoid lesions also have been described as angiolupoid, lichenoid, annular, verrucous, ulcerative, and psoriasiform. Here we present an example of the uncommon type, ichthyosiform. Ichthyosiform sarcoidosis is a rare variant described primarily in dark-skinned individuals, a finding supported by both our case and prior reports. Most reported cases have described IS lesions as having a pasted-on appearance, with adherent centers on the extensor surfaces of the lower extremities, head, and/or neck.6 Our case follows this descriptive pattern previously reported with adherent patches limited to the lower extremities.
Histopathology
The key histopathologic finding is the presence of noncaseating granulomas on biopsy. Sarcoid “specific” lesions rest on the identification of the noncaseating granulomas, while “nonspecific” lesions such as erythema nodosum fail to demonstrate this finding.1
Systemic Involvement
The IS type is believed to be an excellent marker for systemic disease, with approximately 95% of reported cases having some form of systemic illness.6 Acquired ichthyosis should warrant further investigation for systemic disease. Early recognition could be beneficial for the patient because the ichthyosiform type is believed to precede the diagnosis of systemic disease in most cases by a median of 3 months.6
The most common site of internal sarcoid involvement is the lungs, but the lymph nodes, eyes, liver, spleen, heart, and central nervous system also can be involved. Patients can present with nonspecific symptoms such as erythema nodosum in the skin, dyspnea, cough, chest pain, vision changes, enlarged lymph nodes, headaches, joint pain, fever, fatigue, weight loss, and malaise. According to a PubMed search of articles indexed for MEDLINE using the term ichthyosiform sarcoidosis, 16 cases have been reported in the English-language literature (eTable).1,6-14 Of these 16 cases, 3 involved men and 13 involved women. The median age of a patient diagnosed with IS was 37 years. The respiratory system was found to be the most common organ system involved (14 of 16 patients), with hilar adenopathy and restrictive lung disease being the most common findings. Neurologic findings and hepatic involvement also were seen in 3 and 3 patients, respectively. Eight of 16 cases had an elevated serum angiotensin-converting enzyme level. Details of systemic involvement in other cases of IS are listed in the eTable.
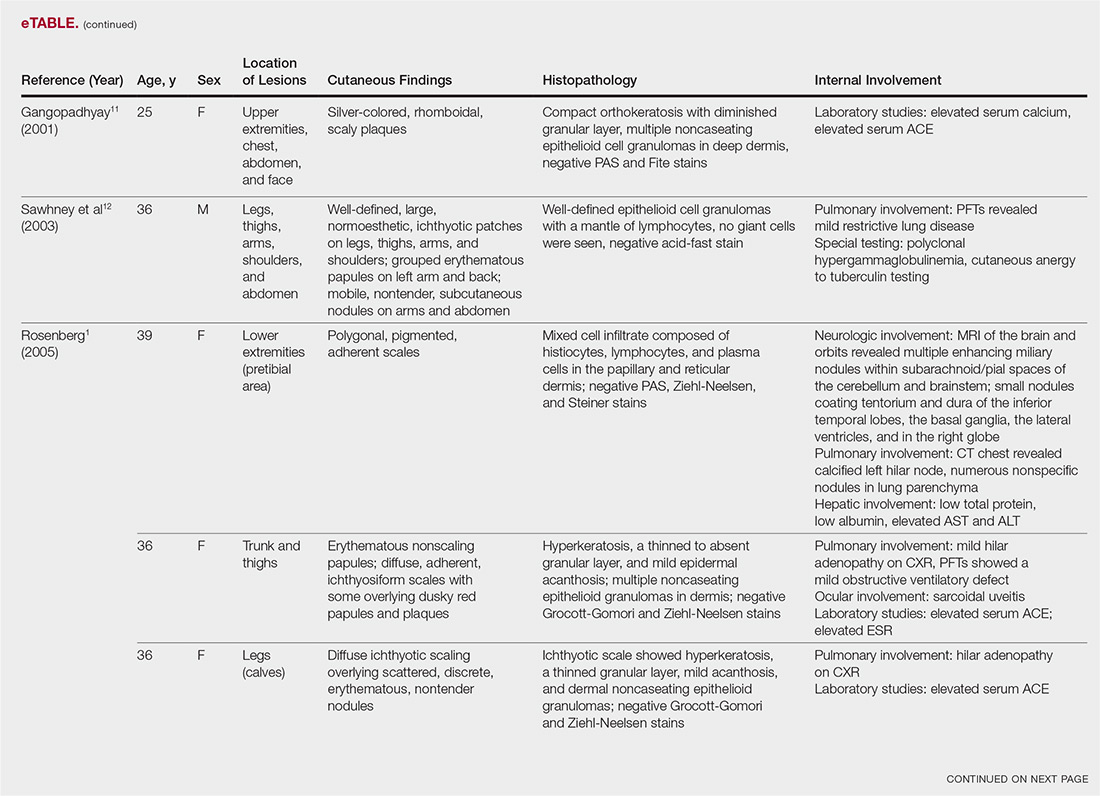
Management
Most patients are given topical corticosteroids for their cutaneous lesions, but patients with systemic involvement will likely need some type of systemic immunosuppressive therapy to control their disease. Systemic therapy often is warranted in IS because of reports of rapid progression. Our case differs from these prior reports in the relative stability of the disease at the last patient encounter. Systemic treatment commonly includes oral corticosteroids such as prednisone. Other options, such as hydroxychloroquine, methotrexate, azathioprine, pentoxifylline, thalidomide, cyclophosphamide, cyclosporine, and infliximab, can be considered if other treatments fail.13 Ichthyosiform sarcoidosis patients should continue to have regular follow-up to monitor for disease progression.
Differential
When evaluating an acquired ichthyosis, dermatologists can consider other associations such as Hodgkin disease, hypothyroidism, multiple myeloma, carcinomatosis, and chronic malnutrition.1 Skin biopsy demonstrating granuloma formation also is not specific for sarcoidosis. Other etiologies, such as autoimmune diseases, immunodeficiency disorders, infections, foreign body granulomas, neoplasms, and drug reactions, should be considered.15 All patients with acquired ichthyosis should undergo a thorough evaluation for internal involvement.
Conclusion
We presented a case of IS, a rare type of sarcoidosis commonly associated with further internal involvement of the respiratory, nervous, or hepatic organ systems. Recognition of an acquired form of ichthyosis and its potential disease associations, including sarcoidosis, is important to improve early detection of any internal disease, allowing prompt initiation of treatment.
- Rosenberg B. Ichthyosiform sarcoidosis. Dermatol Online J. 2005;11:15.
- Banse-Kupin L, Pelachyk JM. Ichthyosiform sarcoidosis: report of two cases and review of the literature. J Am Acad Dermatol. 1987;17:616-620.
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416.
- Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36:561-568.
- Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569-584.
- Kelley BP, George DE, LeLeux TM, et al. Ichthyosiform sarcoidosis: a case report and review of the literature. Dermatol Online J. 2010;16:5.
- Kauh YC, Goody HE, Luscombe HA. Ichthyosiform sarcoidosis. Arch Dermatol. 1978;114:100-101.
- Matsuoka LY, LeVine M, Glasser S, et al. Ichthyosiform sarcoid. Cutis. 1980;25:188-189.
- Matarasso SL, Bruce S. Ichthyosiform sarcoidosis: report of a case. Cutis. 1991;47:405-408.
- Feind-Koopmans AG, Lucker GP, van de Kerkhof PC. Acquired ichthyosiform erythroderma and sarcoidosis. J Am Acad Dermatol. 1996;35:826-828.
- Gangopadhyay AK. Ichthyosiform sarcoidosis. Indian J Dermatol Venereol Leprol. 2001;67:91-92.
- Sawhney M, Sharma YK, Gera V, et al. Ichthyosiform sarcoidosis following chemotherapy of Hodgkin’s disease. Indian J Dermatol Venereol Leprol. 2003;69:220-222.
- Ghosh UC, Ghosh SK, Hazra K, et al. Ichthyosiform sarcoidosis revisited. Indian J Dermatol Venereol Leprol. 2013;79:795-798.
- Miura T, Kato Y, Yamamoto T. Ichthyosiform sarcoidosis: report of three cases from Japan and literature review. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:392-397.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
Sarcoidosis is a multiorgan, systemic, granulomatous disease that most commonly affects the cutaneous, pulmonary, ocular, and cardiac organ systems. Cutaneous involvement occurs in approximately 20% to 35% of patients, with approximately 25% of patients demonstrating only dermatologic findings.1 Cutaneous sarcoidosis can have a highly variable presentation. Ichthyosiform sarcoidosis (IS) is a rare form of this disease that has been described as presenting as polygonal adherent scales.2 It often is associated with internal organ involvement. We present a case of IS without any organ system involvement at the time of diagnosis. A review of the English-language literature was performed to ascertain the internal organ associations most commonly reported with IS.
Case Report
A 66-year-old black woman presented to dermatology with dark scaly patches noted by her primary care physician to be present on both of the lower extremities. The patient believed they were present for at least 4 years. She described dark spots confined to the lower legs that had gradually increased in size. Review of systems was negative for fever, chills, night sweats, weight loss, vision changes, cough, dyspnea, and joint pains, and there was no history of either personal or familial cutaneous diseases.
Physical examination revealed cutaneous patches of thin white scale with a sharp edge in arciform patterns on the lower extremities. Several of these patches were hyperpigmented and xerotic in appearance (Figure 1). The patches were limited to the lower legs, with no other lesions noted.
A punch biopsy of the skin on the right lower leg was performed. Histopathologic analysis showed epidermal compact hyperkeratosis with deep granulomatous infiltration into the subcutaneous tissue (Figures 2A and 2B). At high power, these granulomas were noted to be noncaseating naked granulomas composed of epithelioid histiocytes surrounded by sparse lymphocytic inflammation (Figure 2C). Special stains including acid-fast bacilli, Fite, and periodic acid–Schiff were negative. The diagnosis of IS was made based on clinical presentation and primarily by histopathologic analysis.
The patient’s cutaneous lesions were treated with fluocinonide ointment 0.05% twice daily. Although she did not notice a dramatic improvement in the plaques, they stabilized in size. Her primary care physician was notified and advised to begin a workup for involvement of other organ systems by sarcoidosis. Her initial evaluation, which included a chest radiograph and electrocardiogram, were unremarkable. Despite multiple attempts to persuade the patient to return for further follow-up, neither dermatology nor her primary care physician were able to complete a full workup.
Comment
Etiology
Although there are several theories regarding the etiology of sarcoidosis, the exact cause remains unknown. The body’s immune response, infectious agents, genetics, and the environment have all been thought to play a role. It has been well established that helper T cell (TH1) production of interferon and increased levels of tumor necrosis factor propagate the inflammatory response seen in sarcoidosis.3 More recently, TH17 cells have been found in cutaneous lesions, bronchoalveolar lavage samples, and the blood of patients with sarcoidosis, especially in those with active disease progression.3 Infectious agents such as mycobacteria and propionibacteria DNA or RNA also have been found in sarcoid samples.4 Several HLA-DRB1 variants have been associated with an increased incidence of sarcoidosis.5
Presentation
Characteristic dermatologic findings of sarcoidosis include macules, papules, nodules, and plaques located on the face, especially the nose, cheeks, and ears, and on the shins or ankles, as well as similar lesions around tattoos or scars. Sarcoid lesions also have been described as angiolupoid, lichenoid, annular, verrucous, ulcerative, and psoriasiform. Here we present an example of the uncommon type, ichthyosiform. Ichthyosiform sarcoidosis is a rare variant described primarily in dark-skinned individuals, a finding supported by both our case and prior reports. Most reported cases have described IS lesions as having a pasted-on appearance, with adherent centers on the extensor surfaces of the lower extremities, head, and/or neck.6 Our case follows this descriptive pattern previously reported with adherent patches limited to the lower extremities.
Histopathology
The key histopathologic finding is the presence of noncaseating granulomas on biopsy. Sarcoid “specific” lesions rest on the identification of the noncaseating granulomas, while “nonspecific” lesions such as erythema nodosum fail to demonstrate this finding.1
Systemic Involvement
The IS type is believed to be an excellent marker for systemic disease, with approximately 95% of reported cases having some form of systemic illness.6 Acquired ichthyosis should warrant further investigation for systemic disease. Early recognition could be beneficial for the patient because the ichthyosiform type is believed to precede the diagnosis of systemic disease in most cases by a median of 3 months.6
The most common site of internal sarcoid involvement is the lungs, but the lymph nodes, eyes, liver, spleen, heart, and central nervous system also can be involved. Patients can present with nonspecific symptoms such as erythema nodosum in the skin, dyspnea, cough, chest pain, vision changes, enlarged lymph nodes, headaches, joint pain, fever, fatigue, weight loss, and malaise. According to a PubMed search of articles indexed for MEDLINE using the term ichthyosiform sarcoidosis, 16 cases have been reported in the English-language literature (eTable).1,6-14 Of these 16 cases, 3 involved men and 13 involved women. The median age of a patient diagnosed with IS was 37 years. The respiratory system was found to be the most common organ system involved (14 of 16 patients), with hilar adenopathy and restrictive lung disease being the most common findings. Neurologic findings and hepatic involvement also were seen in 3 and 3 patients, respectively. Eight of 16 cases had an elevated serum angiotensin-converting enzyme level. Details of systemic involvement in other cases of IS are listed in the eTable.
Management
Most patients are given topical corticosteroids for their cutaneous lesions, but patients with systemic involvement will likely need some type of systemic immunosuppressive therapy to control their disease. Systemic therapy often is warranted in IS because of reports of rapid progression. Our case differs from these prior reports in the relative stability of the disease at the last patient encounter. Systemic treatment commonly includes oral corticosteroids such as prednisone. Other options, such as hydroxychloroquine, methotrexate, azathioprine, pentoxifylline, thalidomide, cyclophosphamide, cyclosporine, and infliximab, can be considered if other treatments fail.13 Ichthyosiform sarcoidosis patients should continue to have regular follow-up to monitor for disease progression.
Differential
When evaluating an acquired ichthyosis, dermatologists can consider other associations such as Hodgkin disease, hypothyroidism, multiple myeloma, carcinomatosis, and chronic malnutrition.1 Skin biopsy demonstrating granuloma formation also is not specific for sarcoidosis. Other etiologies, such as autoimmune diseases, immunodeficiency disorders, infections, foreign body granulomas, neoplasms, and drug reactions, should be considered.15 All patients with acquired ichthyosis should undergo a thorough evaluation for internal involvement.
Conclusion
We presented a case of IS, a rare type of sarcoidosis commonly associated with further internal involvement of the respiratory, nervous, or hepatic organ systems. Recognition of an acquired form of ichthyosis and its potential disease associations, including sarcoidosis, is important to improve early detection of any internal disease, allowing prompt initiation of treatment.
Sarcoidosis is a multiorgan, systemic, granulomatous disease that most commonly affects the cutaneous, pulmonary, ocular, and cardiac organ systems. Cutaneous involvement occurs in approximately 20% to 35% of patients, with approximately 25% of patients demonstrating only dermatologic findings.1 Cutaneous sarcoidosis can have a highly variable presentation. Ichthyosiform sarcoidosis (IS) is a rare form of this disease that has been described as presenting as polygonal adherent scales.2 It often is associated with internal organ involvement. We present a case of IS without any organ system involvement at the time of diagnosis. A review of the English-language literature was performed to ascertain the internal organ associations most commonly reported with IS.
Case Report
A 66-year-old black woman presented to dermatology with dark scaly patches noted by her primary care physician to be present on both of the lower extremities. The patient believed they were present for at least 4 years. She described dark spots confined to the lower legs that had gradually increased in size. Review of systems was negative for fever, chills, night sweats, weight loss, vision changes, cough, dyspnea, and joint pains, and there was no history of either personal or familial cutaneous diseases.
Physical examination revealed cutaneous patches of thin white scale with a sharp edge in arciform patterns on the lower extremities. Several of these patches were hyperpigmented and xerotic in appearance (Figure 1). The patches were limited to the lower legs, with no other lesions noted.
A punch biopsy of the skin on the right lower leg was performed. Histopathologic analysis showed epidermal compact hyperkeratosis with deep granulomatous infiltration into the subcutaneous tissue (Figures 2A and 2B). At high power, these granulomas were noted to be noncaseating naked granulomas composed of epithelioid histiocytes surrounded by sparse lymphocytic inflammation (Figure 2C). Special stains including acid-fast bacilli, Fite, and periodic acid–Schiff were negative. The diagnosis of IS was made based on clinical presentation and primarily by histopathologic analysis.
The patient’s cutaneous lesions were treated with fluocinonide ointment 0.05% twice daily. Although she did not notice a dramatic improvement in the plaques, they stabilized in size. Her primary care physician was notified and advised to begin a workup for involvement of other organ systems by sarcoidosis. Her initial evaluation, which included a chest radiograph and electrocardiogram, were unremarkable. Despite multiple attempts to persuade the patient to return for further follow-up, neither dermatology nor her primary care physician were able to complete a full workup.
Comment
Etiology
Although there are several theories regarding the etiology of sarcoidosis, the exact cause remains unknown. The body’s immune response, infectious agents, genetics, and the environment have all been thought to play a role. It has been well established that helper T cell (TH1) production of interferon and increased levels of tumor necrosis factor propagate the inflammatory response seen in sarcoidosis.3 More recently, TH17 cells have been found in cutaneous lesions, bronchoalveolar lavage samples, and the blood of patients with sarcoidosis, especially in those with active disease progression.3 Infectious agents such as mycobacteria and propionibacteria DNA or RNA also have been found in sarcoid samples.4 Several HLA-DRB1 variants have been associated with an increased incidence of sarcoidosis.5
Presentation
Characteristic dermatologic findings of sarcoidosis include macules, papules, nodules, and plaques located on the face, especially the nose, cheeks, and ears, and on the shins or ankles, as well as similar lesions around tattoos or scars. Sarcoid lesions also have been described as angiolupoid, lichenoid, annular, verrucous, ulcerative, and psoriasiform. Here we present an example of the uncommon type, ichthyosiform. Ichthyosiform sarcoidosis is a rare variant described primarily in dark-skinned individuals, a finding supported by both our case and prior reports. Most reported cases have described IS lesions as having a pasted-on appearance, with adherent centers on the extensor surfaces of the lower extremities, head, and/or neck.6 Our case follows this descriptive pattern previously reported with adherent patches limited to the lower extremities.
Histopathology
The key histopathologic finding is the presence of noncaseating granulomas on biopsy. Sarcoid “specific” lesions rest on the identification of the noncaseating granulomas, while “nonspecific” lesions such as erythema nodosum fail to demonstrate this finding.1
Systemic Involvement
The IS type is believed to be an excellent marker for systemic disease, with approximately 95% of reported cases having some form of systemic illness.6 Acquired ichthyosis should warrant further investigation for systemic disease. Early recognition could be beneficial for the patient because the ichthyosiform type is believed to precede the diagnosis of systemic disease in most cases by a median of 3 months.6
The most common site of internal sarcoid involvement is the lungs, but the lymph nodes, eyes, liver, spleen, heart, and central nervous system also can be involved. Patients can present with nonspecific symptoms such as erythema nodosum in the skin, dyspnea, cough, chest pain, vision changes, enlarged lymph nodes, headaches, joint pain, fever, fatigue, weight loss, and malaise. According to a PubMed search of articles indexed for MEDLINE using the term ichthyosiform sarcoidosis, 16 cases have been reported in the English-language literature (eTable).1,6-14 Of these 16 cases, 3 involved men and 13 involved women. The median age of a patient diagnosed with IS was 37 years. The respiratory system was found to be the most common organ system involved (14 of 16 patients), with hilar adenopathy and restrictive lung disease being the most common findings. Neurologic findings and hepatic involvement also were seen in 3 and 3 patients, respectively. Eight of 16 cases had an elevated serum angiotensin-converting enzyme level. Details of systemic involvement in other cases of IS are listed in the eTable.
Management
Most patients are given topical corticosteroids for their cutaneous lesions, but patients with systemic involvement will likely need some type of systemic immunosuppressive therapy to control their disease. Systemic therapy often is warranted in IS because of reports of rapid progression. Our case differs from these prior reports in the relative stability of the disease at the last patient encounter. Systemic treatment commonly includes oral corticosteroids such as prednisone. Other options, such as hydroxychloroquine, methotrexate, azathioprine, pentoxifylline, thalidomide, cyclophosphamide, cyclosporine, and infliximab, can be considered if other treatments fail.13 Ichthyosiform sarcoidosis patients should continue to have regular follow-up to monitor for disease progression.
Differential
When evaluating an acquired ichthyosis, dermatologists can consider other associations such as Hodgkin disease, hypothyroidism, multiple myeloma, carcinomatosis, and chronic malnutrition.1 Skin biopsy demonstrating granuloma formation also is not specific for sarcoidosis. Other etiologies, such as autoimmune diseases, immunodeficiency disorders, infections, foreign body granulomas, neoplasms, and drug reactions, should be considered.15 All patients with acquired ichthyosis should undergo a thorough evaluation for internal involvement.
Conclusion
We presented a case of IS, a rare type of sarcoidosis commonly associated with further internal involvement of the respiratory, nervous, or hepatic organ systems. Recognition of an acquired form of ichthyosis and its potential disease associations, including sarcoidosis, is important to improve early detection of any internal disease, allowing prompt initiation of treatment.
- Rosenberg B. Ichthyosiform sarcoidosis. Dermatol Online J. 2005;11:15.
- Banse-Kupin L, Pelachyk JM. Ichthyosiform sarcoidosis: report of two cases and review of the literature. J Am Acad Dermatol. 1987;17:616-620.
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416.
- Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36:561-568.
- Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569-584.
- Kelley BP, George DE, LeLeux TM, et al. Ichthyosiform sarcoidosis: a case report and review of the literature. Dermatol Online J. 2010;16:5.
- Kauh YC, Goody HE, Luscombe HA. Ichthyosiform sarcoidosis. Arch Dermatol. 1978;114:100-101.
- Matsuoka LY, LeVine M, Glasser S, et al. Ichthyosiform sarcoid. Cutis. 1980;25:188-189.
- Matarasso SL, Bruce S. Ichthyosiform sarcoidosis: report of a case. Cutis. 1991;47:405-408.
- Feind-Koopmans AG, Lucker GP, van de Kerkhof PC. Acquired ichthyosiform erythroderma and sarcoidosis. J Am Acad Dermatol. 1996;35:826-828.
- Gangopadhyay AK. Ichthyosiform sarcoidosis. Indian J Dermatol Venereol Leprol. 2001;67:91-92.
- Sawhney M, Sharma YK, Gera V, et al. Ichthyosiform sarcoidosis following chemotherapy of Hodgkin’s disease. Indian J Dermatol Venereol Leprol. 2003;69:220-222.
- Ghosh UC, Ghosh SK, Hazra K, et al. Ichthyosiform sarcoidosis revisited. Indian J Dermatol Venereol Leprol. 2013;79:795-798.
- Miura T, Kato Y, Yamamoto T. Ichthyosiform sarcoidosis: report of three cases from Japan and literature review. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:392-397.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Rosenberg B. Ichthyosiform sarcoidosis. Dermatol Online J. 2005;11:15.
- Banse-Kupin L, Pelachyk JM. Ichthyosiform sarcoidosis: report of two cases and review of the literature. J Am Acad Dermatol. 1987;17:616-620.
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416.
- Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36:561-568.
- Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569-584.
- Kelley BP, George DE, LeLeux TM, et al. Ichthyosiform sarcoidosis: a case report and review of the literature. Dermatol Online J. 2010;16:5.
- Kauh YC, Goody HE, Luscombe HA. Ichthyosiform sarcoidosis. Arch Dermatol. 1978;114:100-101.
- Matsuoka LY, LeVine M, Glasser S, et al. Ichthyosiform sarcoid. Cutis. 1980;25:188-189.
- Matarasso SL, Bruce S. Ichthyosiform sarcoidosis: report of a case. Cutis. 1991;47:405-408.
- Feind-Koopmans AG, Lucker GP, van de Kerkhof PC. Acquired ichthyosiform erythroderma and sarcoidosis. J Am Acad Dermatol. 1996;35:826-828.
- Gangopadhyay AK. Ichthyosiform sarcoidosis. Indian J Dermatol Venereol Leprol. 2001;67:91-92.
- Sawhney M, Sharma YK, Gera V, et al. Ichthyosiform sarcoidosis following chemotherapy of Hodgkin’s disease. Indian J Dermatol Venereol Leprol. 2003;69:220-222.
- Ghosh UC, Ghosh SK, Hazra K, et al. Ichthyosiform sarcoidosis revisited. Indian J Dermatol Venereol Leprol. 2013;79:795-798.
- Miura T, Kato Y, Yamamoto T. Ichthyosiform sarcoidosis: report of three cases from Japan and literature review. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:392-397.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
Practice Points
- Ichthyosiform sarcoidosis is a rare form of sarcoidosis that presents as polygonal adherent scales.
- Ichthyosiform sarcoidosis is commonly associated with pulmonary, neurologic, and hepatic involvement.
- Acquired ichthyosis should warrant further investigation for systemic disease.
CDC: No medical therapy can yet be recommended for acute flaccid myelitis
The updated guidance on managing acute flaccid myelitis is unlikely to relieve the frustrations of physicians struggling to treat the condition.
![]()
After reviewing the extant data on the baffling disorder, the Centers for Disease Control and Prevention found no evidence that corticosteroids, interferon, antivirals, or any other immunologic or biologic therapy is an effective treatment.
All of the treatments mentioned in the guidance have been used anecdotally, and often for cases proven to be associated with enterovirus-related cases. However, there are no well validated studies confirming benefit for any of these approaches, the agency said in its clinical management document.
Acute flaccid myelitis (AFM) has stricken 90 patients in the United States this year and another 252 cases are being investigated, according to new data from the CDC. The number of confirmed cases is triple that seen in 2017. Whether the disease is an infectious or autoimmune process, or something else entirely, remains unknown.
In response to the outbreak – the largest since 2014 – an expert panel of 4 CDC staff physicians reviewed the literature to find what, if any, treatments were effective; another 14 external experts provided input on the recommendations. At this point, nothing can be officially recommended, the agency said.
Corticosteroids
Corticosteroids should not be administered to most patients with AFM. In addition to “a theoretical concern” about the potential adverse effects of these drugs in acute infections, there is some hard evidence that they are associated with worse outcomes in enteroviral neuroinvasive diseases, particularly those caused by EV-71.
This observation, following a 2012 outbreak in Cambodia, led a World Health Organization commission to conclude that corticosteroids were contraindicated in the management of EV-71–associated neuroinvasive disease. This year, there has been an uptick in EV-A71-associated neurologic disease.
The CDC did hedge its advice on corticosteroids a bit in the setting of AFM, however. “There may be theoretical benefit for steroids in the setting of severe cord swelling or long tract signs suggesting white matter involvement, where steroids may salvage tissue that may be harmed due to an ongoing immune/inflammatory response. While AFM is clinically and radiographically defined by the predominance of gray matter damage in the spinal cord, some patients may have some white matter involvement. It is not clear if these different patterns are important relative to therapeutic considerations.”
Nevertheless, the agency does not recommend corticosteroid use for these patients. “The possible benefits of the use of corticosteroids to manage spinal cord edema or white matter involvement in AFM should be balanced with the possible harm due to immunosuppression in the setting of possible viral infection.”
IVIG
While IVIG holds some theoretical benefit for AFM, there are no high-level human data, the guidelines state. The treatment is generally safe and well tolerated, but the few reports of its use in AFM did not show clear benefit. These include two case series. One suggested an acute improvement of neurologic status, but no long-term resolution of deficits. The other indicated neither significant improvement nor deterioration.
However, current practice at Children’s Hospital of Philadelphia is to initiate IVIG therapy at AFM diagnosis in hopes of boosting humoral immunity.
Nevertheless, the CDC said, “For IVIG to modify disease in an active viral infectious process, early administration is likely required, and possibly prior to exposure,” and the treatment cannot be recommended.
Plasma exchange
Plasma exchange in combination with IVIG and corticosteroids was ineffective in a case series of four Argentinian children, although a single case published last year found that the combination was associated with significant improvement. However, there are not enough data to recommend this approach.
Fluoxetine
Fluoxetine’s antiviral potential turned up in a high-throughput screening project to identify novel compounds with antiviral efficacy against enteroviruses. In 2012, researchers from the University of California, Los Angeles, tested more than 1,000 compounds and found that the SSRI is a potent inhibitor of coxsackievirus. A later project at the National Institutes of Health replicated this finding, and determined that fluoxetine inhibited several enteroviruses, including the AFM suspect, EV-D68.
Fluoxetine concentrates more highly in the central nervous system than it does in plasma, but its antiviral properties have nothing to do with neurotransmitter activity. Rather, it appears to inhibit protein 2C, a highly conserved nonstructural protein that’s crucial to the assembly of RNA into virion particles.
In early November, a retrospective study examined fluoxetine’s use in 30 AFM patients, compared with 26 who did not receive it. The primary outcome was change in summative limb strength score. The study did little to clarify any benefit, however. The authors concluded that fluoxetine was preferentially given to patients with EDV-68 infections. They had more severe impairment at nadir, and at the last follow-up of about 1 year, they had worse outcomes.
“There is no clear human evidence for efficacy of fluoxetine in the treatment of AFM based on a single retrospective evaluation conducted in patients with AFM, and data from a mouse model also did not support efficacy,” the CDC said.
Antiviral medications
The CDC is quite clear on its recommendation that these drugs are not indicated in AFM, since it is not yet proven to be an infectious process.
“Any guidance regarding antiviral medications should be interpreted with great caution, given the unknowns about the pathogenesis of this illness at present ... Testing has been conducted at CDC for antiviral activity of compounds pleconaril, pocapavir, and vapendavir and none have significant activity against currently circulating strains of EV-D68 at clinically relevant concentrations.”
Interferon
There is some anecdotal evidence that interferon alpha-2b was beneficial in treating a polio-like syndrome associated with West Nile virus and Saint Louis encephalitis. “Although there are limited in vitro, animal, and anecdotal human data suggesting activity of some interferons against viral infections, sufficient data are lacking in the setting of AFM,” the agency said. “There is no indication that interferon should be used for the treatment of AFM, and there is concern about the potential for harm from the use of interferon given the immunomodulatory effects in the setting of possible ongoing viral replication.”
SOURCE: CDC Acute Flaccid Myelitis: Interim Considerations for Clinical Management
The updated guidance on managing acute flaccid myelitis is unlikely to relieve the frustrations of physicians struggling to treat the condition.
![]()
After reviewing the extant data on the baffling disorder, the Centers for Disease Control and Prevention found no evidence that corticosteroids, interferon, antivirals, or any other immunologic or biologic therapy is an effective treatment.
All of the treatments mentioned in the guidance have been used anecdotally, and often for cases proven to be associated with enterovirus-related cases. However, there are no well validated studies confirming benefit for any of these approaches, the agency said in its clinical management document.
Acute flaccid myelitis (AFM) has stricken 90 patients in the United States this year and another 252 cases are being investigated, according to new data from the CDC. The number of confirmed cases is triple that seen in 2017. Whether the disease is an infectious or autoimmune process, or something else entirely, remains unknown.
In response to the outbreak – the largest since 2014 – an expert panel of 4 CDC staff physicians reviewed the literature to find what, if any, treatments were effective; another 14 external experts provided input on the recommendations. At this point, nothing can be officially recommended, the agency said.
Corticosteroids
Corticosteroids should not be administered to most patients with AFM. In addition to “a theoretical concern” about the potential adverse effects of these drugs in acute infections, there is some hard evidence that they are associated with worse outcomes in enteroviral neuroinvasive diseases, particularly those caused by EV-71.
This observation, following a 2012 outbreak in Cambodia, led a World Health Organization commission to conclude that corticosteroids were contraindicated in the management of EV-71–associated neuroinvasive disease. This year, there has been an uptick in EV-A71-associated neurologic disease.
The CDC did hedge its advice on corticosteroids a bit in the setting of AFM, however. “There may be theoretical benefit for steroids in the setting of severe cord swelling or long tract signs suggesting white matter involvement, where steroids may salvage tissue that may be harmed due to an ongoing immune/inflammatory response. While AFM is clinically and radiographically defined by the predominance of gray matter damage in the spinal cord, some patients may have some white matter involvement. It is not clear if these different patterns are important relative to therapeutic considerations.”
Nevertheless, the agency does not recommend corticosteroid use for these patients. “The possible benefits of the use of corticosteroids to manage spinal cord edema or white matter involvement in AFM should be balanced with the possible harm due to immunosuppression in the setting of possible viral infection.”
IVIG
While IVIG holds some theoretical benefit for AFM, there are no high-level human data, the guidelines state. The treatment is generally safe and well tolerated, but the few reports of its use in AFM did not show clear benefit. These include two case series. One suggested an acute improvement of neurologic status, but no long-term resolution of deficits. The other indicated neither significant improvement nor deterioration.
However, current practice at Children’s Hospital of Philadelphia is to initiate IVIG therapy at AFM diagnosis in hopes of boosting humoral immunity.
Nevertheless, the CDC said, “For IVIG to modify disease in an active viral infectious process, early administration is likely required, and possibly prior to exposure,” and the treatment cannot be recommended.
Plasma exchange
Plasma exchange in combination with IVIG and corticosteroids was ineffective in a case series of four Argentinian children, although a single case published last year found that the combination was associated with significant improvement. However, there are not enough data to recommend this approach.
Fluoxetine
Fluoxetine’s antiviral potential turned up in a high-throughput screening project to identify novel compounds with antiviral efficacy against enteroviruses. In 2012, researchers from the University of California, Los Angeles, tested more than 1,000 compounds and found that the SSRI is a potent inhibitor of coxsackievirus. A later project at the National Institutes of Health replicated this finding, and determined that fluoxetine inhibited several enteroviruses, including the AFM suspect, EV-D68.
Fluoxetine concentrates more highly in the central nervous system than it does in plasma, but its antiviral properties have nothing to do with neurotransmitter activity. Rather, it appears to inhibit protein 2C, a highly conserved nonstructural protein that’s crucial to the assembly of RNA into virion particles.
In early November, a retrospective study examined fluoxetine’s use in 30 AFM patients, compared with 26 who did not receive it. The primary outcome was change in summative limb strength score. The study did little to clarify any benefit, however. The authors concluded that fluoxetine was preferentially given to patients with EDV-68 infections. They had more severe impairment at nadir, and at the last follow-up of about 1 year, they had worse outcomes.
“There is no clear human evidence for efficacy of fluoxetine in the treatment of AFM based on a single retrospective evaluation conducted in patients with AFM, and data from a mouse model also did not support efficacy,” the CDC said.
Antiviral medications
The CDC is quite clear on its recommendation that these drugs are not indicated in AFM, since it is not yet proven to be an infectious process.
“Any guidance regarding antiviral medications should be interpreted with great caution, given the unknowns about the pathogenesis of this illness at present ... Testing has been conducted at CDC for antiviral activity of compounds pleconaril, pocapavir, and vapendavir and none have significant activity against currently circulating strains of EV-D68 at clinically relevant concentrations.”
Interferon
There is some anecdotal evidence that interferon alpha-2b was beneficial in treating a polio-like syndrome associated with West Nile virus and Saint Louis encephalitis. “Although there are limited in vitro, animal, and anecdotal human data suggesting activity of some interferons against viral infections, sufficient data are lacking in the setting of AFM,” the agency said. “There is no indication that interferon should be used for the treatment of AFM, and there is concern about the potential for harm from the use of interferon given the immunomodulatory effects in the setting of possible ongoing viral replication.”
SOURCE: CDC Acute Flaccid Myelitis: Interim Considerations for Clinical Management
The updated guidance on managing acute flaccid myelitis is unlikely to relieve the frustrations of physicians struggling to treat the condition.
![]()
After reviewing the extant data on the baffling disorder, the Centers for Disease Control and Prevention found no evidence that corticosteroids, interferon, antivirals, or any other immunologic or biologic therapy is an effective treatment.
All of the treatments mentioned in the guidance have been used anecdotally, and often for cases proven to be associated with enterovirus-related cases. However, there are no well validated studies confirming benefit for any of these approaches, the agency said in its clinical management document.
Acute flaccid myelitis (AFM) has stricken 90 patients in the United States this year and another 252 cases are being investigated, according to new data from the CDC. The number of confirmed cases is triple that seen in 2017. Whether the disease is an infectious or autoimmune process, or something else entirely, remains unknown.
In response to the outbreak – the largest since 2014 – an expert panel of 4 CDC staff physicians reviewed the literature to find what, if any, treatments were effective; another 14 external experts provided input on the recommendations. At this point, nothing can be officially recommended, the agency said.
Corticosteroids
Corticosteroids should not be administered to most patients with AFM. In addition to “a theoretical concern” about the potential adverse effects of these drugs in acute infections, there is some hard evidence that they are associated with worse outcomes in enteroviral neuroinvasive diseases, particularly those caused by EV-71.
This observation, following a 2012 outbreak in Cambodia, led a World Health Organization commission to conclude that corticosteroids were contraindicated in the management of EV-71–associated neuroinvasive disease. This year, there has been an uptick in EV-A71-associated neurologic disease.
The CDC did hedge its advice on corticosteroids a bit in the setting of AFM, however. “There may be theoretical benefit for steroids in the setting of severe cord swelling or long tract signs suggesting white matter involvement, where steroids may salvage tissue that may be harmed due to an ongoing immune/inflammatory response. While AFM is clinically and radiographically defined by the predominance of gray matter damage in the spinal cord, some patients may have some white matter involvement. It is not clear if these different patterns are important relative to therapeutic considerations.”
Nevertheless, the agency does not recommend corticosteroid use for these patients. “The possible benefits of the use of corticosteroids to manage spinal cord edema or white matter involvement in AFM should be balanced with the possible harm due to immunosuppression in the setting of possible viral infection.”
IVIG
While IVIG holds some theoretical benefit for AFM, there are no high-level human data, the guidelines state. The treatment is generally safe and well tolerated, but the few reports of its use in AFM did not show clear benefit. These include two case series. One suggested an acute improvement of neurologic status, but no long-term resolution of deficits. The other indicated neither significant improvement nor deterioration.
However, current practice at Children’s Hospital of Philadelphia is to initiate IVIG therapy at AFM diagnosis in hopes of boosting humoral immunity.
Nevertheless, the CDC said, “For IVIG to modify disease in an active viral infectious process, early administration is likely required, and possibly prior to exposure,” and the treatment cannot be recommended.
Plasma exchange
Plasma exchange in combination with IVIG and corticosteroids was ineffective in a case series of four Argentinian children, although a single case published last year found that the combination was associated with significant improvement. However, there are not enough data to recommend this approach.
Fluoxetine
Fluoxetine’s antiviral potential turned up in a high-throughput screening project to identify novel compounds with antiviral efficacy against enteroviruses. In 2012, researchers from the University of California, Los Angeles, tested more than 1,000 compounds and found that the SSRI is a potent inhibitor of coxsackievirus. A later project at the National Institutes of Health replicated this finding, and determined that fluoxetine inhibited several enteroviruses, including the AFM suspect, EV-D68.
Fluoxetine concentrates more highly in the central nervous system than it does in plasma, but its antiviral properties have nothing to do with neurotransmitter activity. Rather, it appears to inhibit protein 2C, a highly conserved nonstructural protein that’s crucial to the assembly of RNA into virion particles.
In early November, a retrospective study examined fluoxetine’s use in 30 AFM patients, compared with 26 who did not receive it. The primary outcome was change in summative limb strength score. The study did little to clarify any benefit, however. The authors concluded that fluoxetine was preferentially given to patients with EDV-68 infections. They had more severe impairment at nadir, and at the last follow-up of about 1 year, they had worse outcomes.
“There is no clear human evidence for efficacy of fluoxetine in the treatment of AFM based on a single retrospective evaluation conducted in patients with AFM, and data from a mouse model also did not support efficacy,” the CDC said.
Antiviral medications
The CDC is quite clear on its recommendation that these drugs are not indicated in AFM, since it is not yet proven to be an infectious process.
“Any guidance regarding antiviral medications should be interpreted with great caution, given the unknowns about the pathogenesis of this illness at present ... Testing has been conducted at CDC for antiviral activity of compounds pleconaril, pocapavir, and vapendavir and none have significant activity against currently circulating strains of EV-D68 at clinically relevant concentrations.”
Interferon
There is some anecdotal evidence that interferon alpha-2b was beneficial in treating a polio-like syndrome associated with West Nile virus and Saint Louis encephalitis. “Although there are limited in vitro, animal, and anecdotal human data suggesting activity of some interferons against viral infections, sufficient data are lacking in the setting of AFM,” the agency said. “There is no indication that interferon should be used for the treatment of AFM, and there is concern about the potential for harm from the use of interferon given the immunomodulatory effects in the setting of possible ongoing viral replication.”
SOURCE: CDC Acute Flaccid Myelitis: Interim Considerations for Clinical Management
AEDs strongly linked to rare serious skin reactions
Antiepileptic drugs were found to be linked with almost ninefold increased odds for two adverse skin reactions, Steven‐Johnson syndrome and toxic epidermal necrolysis, compared with non-AED medication classes in an analysis of adverse-event data from the Food and Drug Administration Adverse Event Reporting System.
Researchers at the University of Rhode Island College of Pharmacy in Kingston, who performed the retrospective study, also found that six drugs within the antiepileptic drug (AED) class had a reporting odds ratio estimate of more than 20, compared with other non-AEDs.
“Although several antiepileptic drugs have been associated with Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), the class effect and impact of other AEDs are not well described,” Eric P. Borrelli, PharmD, and his colleagues reported in Epilepsia.
The investigators examined rates of SJS and TEN for several AEDs using adverse event data from the Food and Drug Administration Adverse Event Reporting System between July 2014 and December 2017. The study investigators examined 198 adverse reaction reports related to AEDs, which was greater than any other drug class.
Overall, AEDs as a group had a reporting odds ratio risk estimate of 8.7 (95% confidence interval, 7.5-10.2), compared with non-AEDs. Similarly, the proportional reporting ratio was found to be 8.7 (95% CI, 7.5-10.2) in the AED group.
Within the class, the medications with the highest risk were zonisamide, rufinamide, and clorazepate, which had about 70-, 60-, and 56-fold higher odds for SJS and TEN, compared with all other medications. Other high-risk AEDs in the group included lamotrigine (reporting odds ratio, 53.0), carbamazepine (reporting OR, 24.5), and phenytoin (reporting OR, 26.3).
“Greater than 90% of SJS [and] TEN reactions associated with AEDs occur within the first 2 months of treatment initiation, although some AEDs have been associated with such reactions during long‐term use,” the researchers wrote.
The authors acknowledged that measures of prevalence and incidence could not be determined from these data since the number of patients taking AEDs is unknown.
“Increased awareness of this risk among both prescribers and patients, particularly variations in risk among different AEDs, along with education on early recognition of SJS [and] TEN signs [and] symptoms, may help mitigate the number and severity of these adverse events,” the researchers concluded.
The study was partially funded by the Department of Veterans Affairs. One coauthor reported receiving research funding from Pfizer, Merck (Cubist), and The Medicines Company.
SOURCE: Borrelli EP et al. Epilepsia. 2018 Nov 5. doi: 10.1111/epi.14591.
Antiepileptic drugs were found to be linked with almost ninefold increased odds for two adverse skin reactions, Steven‐Johnson syndrome and toxic epidermal necrolysis, compared with non-AED medication classes in an analysis of adverse-event data from the Food and Drug Administration Adverse Event Reporting System.
Researchers at the University of Rhode Island College of Pharmacy in Kingston, who performed the retrospective study, also found that six drugs within the antiepileptic drug (AED) class had a reporting odds ratio estimate of more than 20, compared with other non-AEDs.
“Although several antiepileptic drugs have been associated with Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), the class effect and impact of other AEDs are not well described,” Eric P. Borrelli, PharmD, and his colleagues reported in Epilepsia.
The investigators examined rates of SJS and TEN for several AEDs using adverse event data from the Food and Drug Administration Adverse Event Reporting System between July 2014 and December 2017. The study investigators examined 198 adverse reaction reports related to AEDs, which was greater than any other drug class.
Overall, AEDs as a group had a reporting odds ratio risk estimate of 8.7 (95% confidence interval, 7.5-10.2), compared with non-AEDs. Similarly, the proportional reporting ratio was found to be 8.7 (95% CI, 7.5-10.2) in the AED group.
Within the class, the medications with the highest risk were zonisamide, rufinamide, and clorazepate, which had about 70-, 60-, and 56-fold higher odds for SJS and TEN, compared with all other medications. Other high-risk AEDs in the group included lamotrigine (reporting odds ratio, 53.0), carbamazepine (reporting OR, 24.5), and phenytoin (reporting OR, 26.3).
“Greater than 90% of SJS [and] TEN reactions associated with AEDs occur within the first 2 months of treatment initiation, although some AEDs have been associated with such reactions during long‐term use,” the researchers wrote.
The authors acknowledged that measures of prevalence and incidence could not be determined from these data since the number of patients taking AEDs is unknown.
“Increased awareness of this risk among both prescribers and patients, particularly variations in risk among different AEDs, along with education on early recognition of SJS [and] TEN signs [and] symptoms, may help mitigate the number and severity of these adverse events,” the researchers concluded.
The study was partially funded by the Department of Veterans Affairs. One coauthor reported receiving research funding from Pfizer, Merck (Cubist), and The Medicines Company.
SOURCE: Borrelli EP et al. Epilepsia. 2018 Nov 5. doi: 10.1111/epi.14591.
Antiepileptic drugs were found to be linked with almost ninefold increased odds for two adverse skin reactions, Steven‐Johnson syndrome and toxic epidermal necrolysis, compared with non-AED medication classes in an analysis of adverse-event data from the Food and Drug Administration Adverse Event Reporting System.
Researchers at the University of Rhode Island College of Pharmacy in Kingston, who performed the retrospective study, also found that six drugs within the antiepileptic drug (AED) class had a reporting odds ratio estimate of more than 20, compared with other non-AEDs.
“Although several antiepileptic drugs have been associated with Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), the class effect and impact of other AEDs are not well described,” Eric P. Borrelli, PharmD, and his colleagues reported in Epilepsia.
The investigators examined rates of SJS and TEN for several AEDs using adverse event data from the Food and Drug Administration Adverse Event Reporting System between July 2014 and December 2017. The study investigators examined 198 adverse reaction reports related to AEDs, which was greater than any other drug class.
Overall, AEDs as a group had a reporting odds ratio risk estimate of 8.7 (95% confidence interval, 7.5-10.2), compared with non-AEDs. Similarly, the proportional reporting ratio was found to be 8.7 (95% CI, 7.5-10.2) in the AED group.
Within the class, the medications with the highest risk were zonisamide, rufinamide, and clorazepate, which had about 70-, 60-, and 56-fold higher odds for SJS and TEN, compared with all other medications. Other high-risk AEDs in the group included lamotrigine (reporting odds ratio, 53.0), carbamazepine (reporting OR, 24.5), and phenytoin (reporting OR, 26.3).
“Greater than 90% of SJS [and] TEN reactions associated with AEDs occur within the first 2 months of treatment initiation, although some AEDs have been associated with such reactions during long‐term use,” the researchers wrote.
The authors acknowledged that measures of prevalence and incidence could not be determined from these data since the number of patients taking AEDs is unknown.
“Increased awareness of this risk among both prescribers and patients, particularly variations in risk among different AEDs, along with education on early recognition of SJS [and] TEN signs [and] symptoms, may help mitigate the number and severity of these adverse events,” the researchers concluded.
The study was partially funded by the Department of Veterans Affairs. One coauthor reported receiving research funding from Pfizer, Merck (Cubist), and The Medicines Company.
SOURCE: Borrelli EP et al. Epilepsia. 2018 Nov 5. doi: 10.1111/epi.14591.
FROM EPILEPSIA
Key clinical point:
Major finding: The reporting odds ratio risk estimate for SJS and TEN in the AED group was 8.7 (95% CI, 7.5-10.2), compared with non-AEDs.
Study details: A retrospective analysis of 198 adverse reaction reports from the Food and Drug Administration Adverse Event Reporting System.
Disclosures: The study was partially funded by the Department of Veterans Affairs. One coauthor reported receiving research funding from Pfizer, Merck (Cubist), and The Medicines Company.
Source: Borrelli EP et al. Epilepsia. 2018 Nov 5. doi: 10.1111/epi.14591.
Rituximab shines in pediatric vasculitis
CHICAGO – Rituximab demonstrated a high degree of effectiveness with no safety surprises in the first-ever major clinical trial of the potent B-cell inhibitor conducted in pediatric patients with newly diagnosed or relapsing granulomatosis with polyangiitis or microscopic polyangiitis, Jennifer Cooper, MD, PharmD, said at the annual meeting of the American College of Rheumatology.

“This is exciting news. We know that rituximab is very effective in treating adults with GPA and MPA, both in terms of inducing remission and even for maintenance therapy for this rare and severe disease. A lot of pediatric rheumatologists would like to have access to rituximab. Some are even using it for this condition. But until now there were no data in children. I hope this study improves access to rituximab for pediatric patients with ANCA-associated vasculitis,” said Dr. Cooper, a pediatric rheumatologist at the University of Colorado, Denver.
She presented the results of the PePRS (Pediatric Polyangiitis and Rituximab Study), a phase 2a, single-arm, open-label, long-term study of 25 patients in six countries. She anticipates the results will be practice changing, given that pediatric GPA and MPA are recognized as severe systemic autoimmune disorders with a high unmet need for new therapies.
“I don’t believe there are plans for a randomized, controlled trial. Since we now have the pharmacokinetic and safety data in children, we’ll hopefully be able to use extrapolation and our exploratory efficacy endpoints to gain a pediatric indication, or at least a label update for these patients based on this study,” Dr. Cooper continued.
A total of 19 patients had GPA and 6 had MPA, with a median disease duration of 6 months at study entry. All received three pulsed doses of methylprednisolone during the screening period. Then for induction remission they got four once-weekly intravenous infusions of rituximab (Rituxan) at 375 mg/m2 as well as oral corticosteroids, which were tapered from 1 mg/kg per day to 0.2 mg/kg per day over the course of the first 6 months. After that, two-thirds of patients received additional rituximab at their provider’s discretion.
The remission rate by 6 months as defined by Pediatric Vasculitis Activity Score (PVAS) criteria was 56%. At 12 months, the rate was 92%, and at 18 months the rate of remission and sustained disease control was 100%. The mean and median durations of remission were 72 and 56 weeks, respectively.
These results in pediatric patients are comparable to those seen in adults in the landmark RAVE (Rituximab in ANCA-Associated Vasculitis) trial (N Engl J Med. 2013 Aug 1;369[5]:417-27), where 64% of patients reached remission at 6 months, Dr. Cooper noted.
All 25 patients were able to tolerate the four rituximab infusions for remission induction. The main side effect was infusion-related reactions. Overall, 32% of patients experienced such reactions in response to their first infusion, 20% with the second, 12% with the third, and only 8% with the fourth.
Eight patients withdrew from the study after 18 months, mostly because of transfer to adult care. The remaining participants were followed for as long as 4.5 years.
F. Hoffmann-La Roche and Genentech sponsored the PePRS study. Dr. Cooper was a Genentech clinical research fellow at the time.
SOURCE: Brogan P at al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract L04.
CHICAGO – Rituximab demonstrated a high degree of effectiveness with no safety surprises in the first-ever major clinical trial of the potent B-cell inhibitor conducted in pediatric patients with newly diagnosed or relapsing granulomatosis with polyangiitis or microscopic polyangiitis, Jennifer Cooper, MD, PharmD, said at the annual meeting of the American College of Rheumatology.
“This is exciting news. We know that rituximab is very effective in treating adults with GPA and MPA, both in terms of inducing remission and even for maintenance therapy for this rare and severe disease. A lot of pediatric rheumatologists would like to have access to rituximab. Some are even using it for this condition. But until now there were no data in children. I hope this study improves access to rituximab for pediatric patients with ANCA-associated vasculitis,” said Dr. Cooper, a pediatric rheumatologist at the University of Colorado, Denver.
She presented the results of the PePRS (Pediatric Polyangiitis and Rituximab Study), a phase 2a, single-arm, open-label, long-term study of 25 patients in six countries. She anticipates the results will be practice changing, given that pediatric GPA and MPA are recognized as severe systemic autoimmune disorders with a high unmet need for new therapies.
“I don’t believe there are plans for a randomized, controlled trial. Since we now have the pharmacokinetic and safety data in children, we’ll hopefully be able to use extrapolation and our exploratory efficacy endpoints to gain a pediatric indication, or at least a label update for these patients based on this study,” Dr. Cooper continued.
A total of 19 patients had GPA and 6 had MPA, with a median disease duration of 6 months at study entry. All received three pulsed doses of methylprednisolone during the screening period. Then for induction remission they got four once-weekly intravenous infusions of rituximab (Rituxan) at 375 mg/m2 as well as oral corticosteroids, which were tapered from 1 mg/kg per day to 0.2 mg/kg per day over the course of the first 6 months. After that, two-thirds of patients received additional rituximab at their provider’s discretion.
The remission rate by 6 months as defined by Pediatric Vasculitis Activity Score (PVAS) criteria was 56%. At 12 months, the rate was 92%, and at 18 months the rate of remission and sustained disease control was 100%. The mean and median durations of remission were 72 and 56 weeks, respectively.
These results in pediatric patients are comparable to those seen in adults in the landmark RAVE (Rituximab in ANCA-Associated Vasculitis) trial (N Engl J Med. 2013 Aug 1;369[5]:417-27), where 64% of patients reached remission at 6 months, Dr. Cooper noted.
All 25 patients were able to tolerate the four rituximab infusions for remission induction. The main side effect was infusion-related reactions. Overall, 32% of patients experienced such reactions in response to their first infusion, 20% with the second, 12% with the third, and only 8% with the fourth.
Eight patients withdrew from the study after 18 months, mostly because of transfer to adult care. The remaining participants were followed for as long as 4.5 years.
F. Hoffmann-La Roche and Genentech sponsored the PePRS study. Dr. Cooper was a Genentech clinical research fellow at the time.
SOURCE: Brogan P at al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract L04.
CHICAGO – Rituximab demonstrated a high degree of effectiveness with no safety surprises in the first-ever major clinical trial of the potent B-cell inhibitor conducted in pediatric patients with newly diagnosed or relapsing granulomatosis with polyangiitis or microscopic polyangiitis, Jennifer Cooper, MD, PharmD, said at the annual meeting of the American College of Rheumatology.
“This is exciting news. We know that rituximab is very effective in treating adults with GPA and MPA, both in terms of inducing remission and even for maintenance therapy for this rare and severe disease. A lot of pediatric rheumatologists would like to have access to rituximab. Some are even using it for this condition. But until now there were no data in children. I hope this study improves access to rituximab for pediatric patients with ANCA-associated vasculitis,” said Dr. Cooper, a pediatric rheumatologist at the University of Colorado, Denver.
She presented the results of the PePRS (Pediatric Polyangiitis and Rituximab Study), a phase 2a, single-arm, open-label, long-term study of 25 patients in six countries. She anticipates the results will be practice changing, given that pediatric GPA and MPA are recognized as severe systemic autoimmune disorders with a high unmet need for new therapies.
“I don’t believe there are plans for a randomized, controlled trial. Since we now have the pharmacokinetic and safety data in children, we’ll hopefully be able to use extrapolation and our exploratory efficacy endpoints to gain a pediatric indication, or at least a label update for these patients based on this study,” Dr. Cooper continued.
A total of 19 patients had GPA and 6 had MPA, with a median disease duration of 6 months at study entry. All received three pulsed doses of methylprednisolone during the screening period. Then for induction remission they got four once-weekly intravenous infusions of rituximab (Rituxan) at 375 mg/m2 as well as oral corticosteroids, which were tapered from 1 mg/kg per day to 0.2 mg/kg per day over the course of the first 6 months. After that, two-thirds of patients received additional rituximab at their provider’s discretion.
The remission rate by 6 months as defined by Pediatric Vasculitis Activity Score (PVAS) criteria was 56%. At 12 months, the rate was 92%, and at 18 months the rate of remission and sustained disease control was 100%. The mean and median durations of remission were 72 and 56 weeks, respectively.
These results in pediatric patients are comparable to those seen in adults in the landmark RAVE (Rituximab in ANCA-Associated Vasculitis) trial (N Engl J Med. 2013 Aug 1;369[5]:417-27), where 64% of patients reached remission at 6 months, Dr. Cooper noted.
All 25 patients were able to tolerate the four rituximab infusions for remission induction. The main side effect was infusion-related reactions. Overall, 32% of patients experienced such reactions in response to their first infusion, 20% with the second, 12% with the third, and only 8% with the fourth.
Eight patients withdrew from the study after 18 months, mostly because of transfer to adult care. The remaining participants were followed for as long as 4.5 years.
F. Hoffmann-La Roche and Genentech sponsored the PePRS study. Dr. Cooper was a Genentech clinical research fellow at the time.
SOURCE: Brogan P at al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract L04.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point:
Major finding: The first clinical trial of rituximab for newly diagnosed or relapsing GPA or MPA in pediatric patients showed remission rates of 56%, 92%, and 100% by months 6, 12, and 18, respectively.
Study details: The PePRS study was a phase 2a, single-arm, open-label, long-term study of 25 patients in six countries.
Disclosures: F. Hoffmann-La Roche and Genentech sponsored the study. The presenter was a Genentech clinical research fellow at the time.
Source: Brogan P at al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract L04.