User login
Skin Scores: A Review of Clinical Scoring Systems in Dermatology
The practice of dermatology is rife with bedside tools: swabs, smears, and scoring systems. First popularized in specialties such as emergency medicine and internal medicine, clinical scoring systems are now emerging in dermatology. These evidence-based scores can be calculated quickly at the bedside—often through a free smartphone app—to help guide clinical decision-making regarding diagnosis, prognosis, and management. As with any medical tool, scoring systems have limitations and should be used as a supplement, not substitute, for one’s clinical judgement. This article reviews 4 clinical scoring systems practical for dermatology residents.
SCORTEN Prognosticates Cases of Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis
Perhaps the best-known scoring system in dermatology, the SCORTEN is widely used to predict hospital mortality from Stevens-Johnson syndrome/toxic epidermal necrolysis. The SCORTEN includes 7 variables of equal weight—age of 40 years or older, heart rate of 120 beats per minute or more, cancer/hematologic malignancy, involved body surface area (BSA) greater than 10%, serum urea greater than 10 mmol/L, serum bicarbonate less than 20 mmol/L, and serum glucose greater than 14 mmol/L—each contributing 1 point to the overall score if present.1 The involved BSA is defined as the sum of detached and detachable epidermis.1
The SCORTEN was developed and prospectively validated to be calculated at the end of the first 24 hours of admission; for this calculation, use the BSA affected at that time, and use the most abnormal values during the first 24 hours of admission for the other variables.1 In addition, a follow-up study including some of the original coauthors recommends recalculating the SCORTEN at the end of hospital day 3, having found that the score’s predictive value was better on this day than hospital days 1, 2, 4, or 5.2 Based on the original study, a SCORTEN of 0 to 1 corresponds to a mortality rate of 3.2%, 2 to 12.1%, 3 to 35.3%, 4 to 58.3%, and 5 or greater to 90.0%.1
Limitations of the SCORTEN include its ability to overestimate or underestimate mortality as demonstrated by 2 multi-institutional cohorts.3,4 Recently, the ABCD-10 score was developed as an alternative to the SCORTEN and was found to predict mortality similarly when validated in an internal cohort.5
PEST Screens for Psoriatic Arthritis
Dermatologists play an important role in screening for psoriatic arthritis, as an estimated 1 in 5 patients with psoriasis have psoriatic arthritis.6 To this end, several screening tools have been developed to help differentiate psoriatic arthritis from other arthritides. Joint guidelines from the American Academy of Dermatology and the National Psoriasis Foundation acknowledge that “. . . these screening tools have tended to perform less well when tested in groups of people other than those for which they were originally developed. As such, their usefulness in routine clinical practice remains controversial.”7 Nevertheless, the guidelines state, “[b]ecause screening and early detection of inflammatory arthritis are essential to optimize patient [quality of life] and reduce morbidity, providers may consider using a formal screening tool of their choice.”7
With these limitations in mind, I have found the Psoriasis Epidemiology Screening Tool (PEST) to be the most useful psoriatic arthritis screening tool. One study determined that the PEST has the best trade-off between sensitivity and specificity compared to 2 other psoriatic arthritis screening tools, the Psoriatic Arthritis Screening and Evaluation (PASE) and the Early Arthritis for Psoriatic Patients (EARP).8
The PEST is comprised of 5 questions: (1) Have you ever had a swollen joint (or joints)? (2) Has a doctor ever told you that you have arthritis? (3) Do your fingernails or toenails have holes or pits? (4) Have you had pain in your heel? (5) Have you had a finger or toe that was completely swollen and painful for no apparent reason? According to the PEST, a referral to a rheumatologist should be considered for patients answering yes to 3 or more questions, which is 97% sensitive and 79% specific for psoriatic arthritis.9 Patients who answer yes to fewer than 3 questions should still be referred to a rheumatologist if there is a strong clinical suspicion of psoriatic arthritis.10
The PEST can be accessed for free in 13 languages via the GRAPPA (Group for Research and Assessment of Psoriasis and Psoriatic Arthritis) app as well as downloaded for free from the National Psoriasis Foundation’s website (https://www.psoriasis.org/psa-screening/providers).
ALT-70 Differentiates Cellulitis From Pseudocellulitis
Overdiagnosing cellulitis in the United States has been estimated to result in up to 130,000 unnecessary hospitalizations and up to $515 million in avoidable health care spending.11 Dermatologists are in a unique position to help fix this issue. In one retrospective study of 1430 inpatient dermatology consultations, 74.32% of inpatients evaluated for presumed cellulitis by a dermatologist were instead diagnosed with a cellulitis mimicker (ie, pseudocellulitis), such as stasis dermatitis or contact dermatitis.12
The ALT-70 score was developed and prospectively validated to help differentiate lower extremity cellulitis from pseudocellulitis in adult patients in the emergency department (ED).13 In addition, the score has retrospectively been shown to function similarly in the inpatient setting when calculated at 24 and 48 hours after ED presentation.14 Although the ALT-70 score was designed for use by frontline clinicians prior to dermatology consultation, I also have found it helpful to calculate as a consultant, as it provides an objective measure of risk to communicate to the primary team in support of one diagnosis or another.
ALT-70 is an acronym for the score’s 4 variables: asymmetry, leukocytosis, tachycardia, and age of 70 years or older.15 If present, each variable confers a certain number of points to the final score: 3 points for asymmetry (defined as unilateral leg involvement), 1 point for leukocytosis (white blood cell count ≥10,000/μL), 1 point for tachycardia (≥90 beats per minute), and 2 points for age of 70 years or older. An ALT-70 score of 0 to 2 corresponds to an 83.3% or greater chance of pseudocellulitis, suggesting that the diagnosis of cellulitis be reconsidered. A score of 3 to 4 is indeterminate, and additional information such as a dermatology consultation should be pursued. A score of 5 to 7 corresponds to an 82.2% or greater chance of cellulitis, signifying that empiric treatment with antibiotics be considered.15
The ALT-70 score does not apply to cases involving areas other than the lower extremities; intravenous antibiotic use within 48 hours before ED presentation; surgery within the last 30 days; abscess; penetrating trauma; burn; or known history of osteomyelitis, diabetic ulcer, or indwelling hardware at the site of infection.15 The ALT-70 score is available for free via the MDCalc app and website (https://www.mdcalc.com/alt-70-score-cellulitis).
Mohs AUC Determines the Appropriateness of Mohs Micrographic Surgery
In 2012, the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and American Society for Mohs Surgery published appropriate use criteria (AUC) to guide the decision to pursue Mohs micrographic surgery (MMS) in the United States.16 Based on various tumor and patient characteristics, the Mohs AUC assign scores to 270 different clinical scenarios. A score of 1 to 3 signifies that MMS is inappropriate and generally not considered acceptable. A score 4 to 6 indicates that the appropriateness of MMS is uncertain. A score 7 to 9 means that MMS is appropriate and generally considered acceptable.16
Since publication, the Mohs AUC have been criticized for classifying most primary superficial basal cell carcinomas as appropriate for MMS17 (which an AUC coauthor18 and others19,20 have defended), excluding certain reasons for performing MMS (such as operating on multiple tumors on the same day),21 including counterintuitive scores,22 and omitting trials from Europe23 (which AUC coauthors also have defended24).
Final Thoughts
Scoring systems are emerging in dermatology as evidence-based bedside tools to help guide clinical decision-making. Despite their limitations, these scores have the potential to make a meaningful impact in dermatology as they have in other specialties.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Guegan S, Bastuji-Garin S, Poszepczynska-Guigne E, et al. Performance of the SCORTEN during the first five days of hospitalization to predict the prognosis of epidermal necrolysis. J Invest Dermatol. 2006;126:272-276.
- Micheletti RG, Chiesa-Fuxench Z, Noe MH, et al. Stevens-Johnson syndrome/toxic epidermal necrolysis: a multicenter retrospective study of 377 adult patients from the United States. J Invest Dermatol. 2018;138:2315-2321.
- Sekula P, Liss Y, Davidovici B, et al. Evaluation of SCORTEN on a cohort of patients with Stevens-Johnson syndrome and toxic epidermal necrolysis included in the RegiSCAR study. J Burn Care Res. 2011;32:237-245.
- Noe MH, Rosenbach M, Hubbard RA, et al. Development and validation of a risk prediction model for in-hospital mortality among patients with Stevens-Johnson syndrome/toxic epidermal necrolysis-ABCD-10. JAMA Dermatol. 2019;155:448-454.
- Alinaghi F, Calov M, Kristensen LE, et al. Prevalence of psoriatic arthritis in patients with psoriasis: a systematic review and meta-analysis of observational and clinical studies. J Am Acad Dermatol. 2019;80:251-265.e219.
- Elmets CA, Leonardi CL, Davis DMR, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with awareness and attention to comorbidities. J Am Acad Dermatol. 2019;80:1073-1113.
- Karreman MC, Weel A, van der Ven M, et al. Performance of screening tools for psoriatic arthritis: a cross-sectional study in primary care. Rheumatology (Oxford). 2017;56:597-602.
- Ibrahim GH, Buch MH, Lawson C, et al. Evaluation of an existing screening tool for psoriatic arthritis in people with psoriasis and the development of a new instrument: the Psoriasis Epidemiology Screening Tool (PEST) questionnaire. Clin Exp Rheumatol. 2009;27:469-474.
- Zhang A, Kurtzman DJB, Perez-Chada LM, et al. Psoriatic arthritis and the dermatologist: an approach to screening and clinical evaluation. Clin Dermatol. 2018;36:551-560.
- Weng QY, Raff AB, Cohen JM, et al. Costs and consequences associated with misdiagnosed lower extremity cellulitis. JAMA Dermatol. 2017;153:141-146.
- Strazzula L, Cotliar J, Fox LP, et al. Inpatient dermatology consultation aids diagnosis of cellulitis among hospitalized patients: a multi-institutional analysis. J Am Acad Dermatol. 2015;73:70-75.
- Li DG, Dewan AK, Xia FD, et al. The ALT-70 predictive model outperforms thermal imaging for the diagnosis of lower extremity cellulitis: a prospective evaluation. J Am Acad Dermatol. 2018;79:1076-1080.e1071.
- Singer S, Li DG, Gunasekera N, et al. The ALT-70 predictive model maintains predictive value at 24 and 48 hours after presentation [published online March 23, 2019]. J Am Acad Dermatol. doi:10.1016/j.jaad.2019.03.050.
- Raff AB, Weng QY, Cohen JM, et al. A predictive model for diagnosis of lower extremity cellulitis: a cross-sectional study. J Am Acad Dermatol. 2017;76:618-625.e2.
- Connolly SM, Baker DR, Coldiron BM, et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: a report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J Am Acad Dermatol. 2012;67:531-550.
- Steinman HK, Dixon A, Zachary CB. Reevaluating Mohs surgery appropriate use criteria for primary superficial basal cell carcinoma. JAMA Dermatol. 2018;154:755-756.
- Montuno MA, Coldiron BM. Mohs appropriate use criteria for superficial basal cell carcinoma. JAMA Dermatol. 2019;155:394-395.
- MacFarlane DF, Perlis C. Mohs appropriate use criteria for superficial basal cell carcinoma. JAMA Dermatol. 2019;155:395-396.
- Kantor J. Mohs appropriate use criteria for superficial basal cell carcinoma. JAMA Dermatol. 2019;155:395.
- Ruiz ES, Karia PS, Morgan FC, et al. Multiple Mohs micrographic surgery is the most common reason for divergence from the appropriate use criteria: a single institution retrospective cohort study. J Am Acad Dermatol. 2016;75:830-831.
- Croley JA, Joseph AK, Wagner RF Jr. Discrepancies in the Mohs Micrographic Surgery appropriate use criteria [published online December 23, 2018]. J Am Acad Dermatol. doi:10.1016/j.jaad.2018.11.064.
- Kelleners-Smeets NW, Mosterd K. Comment on 2012 appropriate use criteria for Mohs micrographic surgery. J Am Acad Dermatol. 2013;69:317-318.
- Connolly S, Baker D, Coldiron B, et al. Reply to “comment on 2012 appropriate use criteria for Mohs micrographic surgery.” J Am Acad Dermatol. 2013;69:318.
The practice of dermatology is rife with bedside tools: swabs, smears, and scoring systems. First popularized in specialties such as emergency medicine and internal medicine, clinical scoring systems are now emerging in dermatology. These evidence-based scores can be calculated quickly at the bedside—often through a free smartphone app—to help guide clinical decision-making regarding diagnosis, prognosis, and management. As with any medical tool, scoring systems have limitations and should be used as a supplement, not substitute, for one’s clinical judgement. This article reviews 4 clinical scoring systems practical for dermatology residents.
SCORTEN Prognosticates Cases of Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis
Perhaps the best-known scoring system in dermatology, the SCORTEN is widely used to predict hospital mortality from Stevens-Johnson syndrome/toxic epidermal necrolysis. The SCORTEN includes 7 variables of equal weight—age of 40 years or older, heart rate of 120 beats per minute or more, cancer/hematologic malignancy, involved body surface area (BSA) greater than 10%, serum urea greater than 10 mmol/L, serum bicarbonate less than 20 mmol/L, and serum glucose greater than 14 mmol/L—each contributing 1 point to the overall score if present.1 The involved BSA is defined as the sum of detached and detachable epidermis.1
The SCORTEN was developed and prospectively validated to be calculated at the end of the first 24 hours of admission; for this calculation, use the BSA affected at that time, and use the most abnormal values during the first 24 hours of admission for the other variables.1 In addition, a follow-up study including some of the original coauthors recommends recalculating the SCORTEN at the end of hospital day 3, having found that the score’s predictive value was better on this day than hospital days 1, 2, 4, or 5.2 Based on the original study, a SCORTEN of 0 to 1 corresponds to a mortality rate of 3.2%, 2 to 12.1%, 3 to 35.3%, 4 to 58.3%, and 5 or greater to 90.0%.1
Limitations of the SCORTEN include its ability to overestimate or underestimate mortality as demonstrated by 2 multi-institutional cohorts.3,4 Recently, the ABCD-10 score was developed as an alternative to the SCORTEN and was found to predict mortality similarly when validated in an internal cohort.5
PEST Screens for Psoriatic Arthritis
Dermatologists play an important role in screening for psoriatic arthritis, as an estimated 1 in 5 patients with psoriasis have psoriatic arthritis.6 To this end, several screening tools have been developed to help differentiate psoriatic arthritis from other arthritides. Joint guidelines from the American Academy of Dermatology and the National Psoriasis Foundation acknowledge that “. . . these screening tools have tended to perform less well when tested in groups of people other than those for which they were originally developed. As such, their usefulness in routine clinical practice remains controversial.”7 Nevertheless, the guidelines state, “[b]ecause screening and early detection of inflammatory arthritis are essential to optimize patient [quality of life] and reduce morbidity, providers may consider using a formal screening tool of their choice.”7
With these limitations in mind, I have found the Psoriasis Epidemiology Screening Tool (PEST) to be the most useful psoriatic arthritis screening tool. One study determined that the PEST has the best trade-off between sensitivity and specificity compared to 2 other psoriatic arthritis screening tools, the Psoriatic Arthritis Screening and Evaluation (PASE) and the Early Arthritis for Psoriatic Patients (EARP).8
The PEST is comprised of 5 questions: (1) Have you ever had a swollen joint (or joints)? (2) Has a doctor ever told you that you have arthritis? (3) Do your fingernails or toenails have holes or pits? (4) Have you had pain in your heel? (5) Have you had a finger or toe that was completely swollen and painful for no apparent reason? According to the PEST, a referral to a rheumatologist should be considered for patients answering yes to 3 or more questions, which is 97% sensitive and 79% specific for psoriatic arthritis.9 Patients who answer yes to fewer than 3 questions should still be referred to a rheumatologist if there is a strong clinical suspicion of psoriatic arthritis.10
The PEST can be accessed for free in 13 languages via the GRAPPA (Group for Research and Assessment of Psoriasis and Psoriatic Arthritis) app as well as downloaded for free from the National Psoriasis Foundation’s website (https://www.psoriasis.org/psa-screening/providers).
ALT-70 Differentiates Cellulitis From Pseudocellulitis
Overdiagnosing cellulitis in the United States has been estimated to result in up to 130,000 unnecessary hospitalizations and up to $515 million in avoidable health care spending.11 Dermatologists are in a unique position to help fix this issue. In one retrospective study of 1430 inpatient dermatology consultations, 74.32% of inpatients evaluated for presumed cellulitis by a dermatologist were instead diagnosed with a cellulitis mimicker (ie, pseudocellulitis), such as stasis dermatitis or contact dermatitis.12
The ALT-70 score was developed and prospectively validated to help differentiate lower extremity cellulitis from pseudocellulitis in adult patients in the emergency department (ED).13 In addition, the score has retrospectively been shown to function similarly in the inpatient setting when calculated at 24 and 48 hours after ED presentation.14 Although the ALT-70 score was designed for use by frontline clinicians prior to dermatology consultation, I also have found it helpful to calculate as a consultant, as it provides an objective measure of risk to communicate to the primary team in support of one diagnosis or another.
ALT-70 is an acronym for the score’s 4 variables: asymmetry, leukocytosis, tachycardia, and age of 70 years or older.15 If present, each variable confers a certain number of points to the final score: 3 points for asymmetry (defined as unilateral leg involvement), 1 point for leukocytosis (white blood cell count ≥10,000/μL), 1 point for tachycardia (≥90 beats per minute), and 2 points for age of 70 years or older. An ALT-70 score of 0 to 2 corresponds to an 83.3% or greater chance of pseudocellulitis, suggesting that the diagnosis of cellulitis be reconsidered. A score of 3 to 4 is indeterminate, and additional information such as a dermatology consultation should be pursued. A score of 5 to 7 corresponds to an 82.2% or greater chance of cellulitis, signifying that empiric treatment with antibiotics be considered.15
The ALT-70 score does not apply to cases involving areas other than the lower extremities; intravenous antibiotic use within 48 hours before ED presentation; surgery within the last 30 days; abscess; penetrating trauma; burn; or known history of osteomyelitis, diabetic ulcer, or indwelling hardware at the site of infection.15 The ALT-70 score is available for free via the MDCalc app and website (https://www.mdcalc.com/alt-70-score-cellulitis).
Mohs AUC Determines the Appropriateness of Mohs Micrographic Surgery
In 2012, the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and American Society for Mohs Surgery published appropriate use criteria (AUC) to guide the decision to pursue Mohs micrographic surgery (MMS) in the United States.16 Based on various tumor and patient characteristics, the Mohs AUC assign scores to 270 different clinical scenarios. A score of 1 to 3 signifies that MMS is inappropriate and generally not considered acceptable. A score 4 to 6 indicates that the appropriateness of MMS is uncertain. A score 7 to 9 means that MMS is appropriate and generally considered acceptable.16
Since publication, the Mohs AUC have been criticized for classifying most primary superficial basal cell carcinomas as appropriate for MMS17 (which an AUC coauthor18 and others19,20 have defended), excluding certain reasons for performing MMS (such as operating on multiple tumors on the same day),21 including counterintuitive scores,22 and omitting trials from Europe23 (which AUC coauthors also have defended24).
Final Thoughts
Scoring systems are emerging in dermatology as evidence-based bedside tools to help guide clinical decision-making. Despite their limitations, these scores have the potential to make a meaningful impact in dermatology as they have in other specialties.
The practice of dermatology is rife with bedside tools: swabs, smears, and scoring systems. First popularized in specialties such as emergency medicine and internal medicine, clinical scoring systems are now emerging in dermatology. These evidence-based scores can be calculated quickly at the bedside—often through a free smartphone app—to help guide clinical decision-making regarding diagnosis, prognosis, and management. As with any medical tool, scoring systems have limitations and should be used as a supplement, not substitute, for one’s clinical judgement. This article reviews 4 clinical scoring systems practical for dermatology residents.
SCORTEN Prognosticates Cases of Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis
Perhaps the best-known scoring system in dermatology, the SCORTEN is widely used to predict hospital mortality from Stevens-Johnson syndrome/toxic epidermal necrolysis. The SCORTEN includes 7 variables of equal weight—age of 40 years or older, heart rate of 120 beats per minute or more, cancer/hematologic malignancy, involved body surface area (BSA) greater than 10%, serum urea greater than 10 mmol/L, serum bicarbonate less than 20 mmol/L, and serum glucose greater than 14 mmol/L—each contributing 1 point to the overall score if present.1 The involved BSA is defined as the sum of detached and detachable epidermis.1
The SCORTEN was developed and prospectively validated to be calculated at the end of the first 24 hours of admission; for this calculation, use the BSA affected at that time, and use the most abnormal values during the first 24 hours of admission for the other variables.1 In addition, a follow-up study including some of the original coauthors recommends recalculating the SCORTEN at the end of hospital day 3, having found that the score’s predictive value was better on this day than hospital days 1, 2, 4, or 5.2 Based on the original study, a SCORTEN of 0 to 1 corresponds to a mortality rate of 3.2%, 2 to 12.1%, 3 to 35.3%, 4 to 58.3%, and 5 or greater to 90.0%.1
Limitations of the SCORTEN include its ability to overestimate or underestimate mortality as demonstrated by 2 multi-institutional cohorts.3,4 Recently, the ABCD-10 score was developed as an alternative to the SCORTEN and was found to predict mortality similarly when validated in an internal cohort.5
PEST Screens for Psoriatic Arthritis
Dermatologists play an important role in screening for psoriatic arthritis, as an estimated 1 in 5 patients with psoriasis have psoriatic arthritis.6 To this end, several screening tools have been developed to help differentiate psoriatic arthritis from other arthritides. Joint guidelines from the American Academy of Dermatology and the National Psoriasis Foundation acknowledge that “. . . these screening tools have tended to perform less well when tested in groups of people other than those for which they were originally developed. As such, their usefulness in routine clinical practice remains controversial.”7 Nevertheless, the guidelines state, “[b]ecause screening and early detection of inflammatory arthritis are essential to optimize patient [quality of life] and reduce morbidity, providers may consider using a formal screening tool of their choice.”7
With these limitations in mind, I have found the Psoriasis Epidemiology Screening Tool (PEST) to be the most useful psoriatic arthritis screening tool. One study determined that the PEST has the best trade-off between sensitivity and specificity compared to 2 other psoriatic arthritis screening tools, the Psoriatic Arthritis Screening and Evaluation (PASE) and the Early Arthritis for Psoriatic Patients (EARP).8
The PEST is comprised of 5 questions: (1) Have you ever had a swollen joint (or joints)? (2) Has a doctor ever told you that you have arthritis? (3) Do your fingernails or toenails have holes or pits? (4) Have you had pain in your heel? (5) Have you had a finger or toe that was completely swollen and painful for no apparent reason? According to the PEST, a referral to a rheumatologist should be considered for patients answering yes to 3 or more questions, which is 97% sensitive and 79% specific for psoriatic arthritis.9 Patients who answer yes to fewer than 3 questions should still be referred to a rheumatologist if there is a strong clinical suspicion of psoriatic arthritis.10
The PEST can be accessed for free in 13 languages via the GRAPPA (Group for Research and Assessment of Psoriasis and Psoriatic Arthritis) app as well as downloaded for free from the National Psoriasis Foundation’s website (https://www.psoriasis.org/psa-screening/providers).
ALT-70 Differentiates Cellulitis From Pseudocellulitis
Overdiagnosing cellulitis in the United States has been estimated to result in up to 130,000 unnecessary hospitalizations and up to $515 million in avoidable health care spending.11 Dermatologists are in a unique position to help fix this issue. In one retrospective study of 1430 inpatient dermatology consultations, 74.32% of inpatients evaluated for presumed cellulitis by a dermatologist were instead diagnosed with a cellulitis mimicker (ie, pseudocellulitis), such as stasis dermatitis or contact dermatitis.12
The ALT-70 score was developed and prospectively validated to help differentiate lower extremity cellulitis from pseudocellulitis in adult patients in the emergency department (ED).13 In addition, the score has retrospectively been shown to function similarly in the inpatient setting when calculated at 24 and 48 hours after ED presentation.14 Although the ALT-70 score was designed for use by frontline clinicians prior to dermatology consultation, I also have found it helpful to calculate as a consultant, as it provides an objective measure of risk to communicate to the primary team in support of one diagnosis or another.
ALT-70 is an acronym for the score’s 4 variables: asymmetry, leukocytosis, tachycardia, and age of 70 years or older.15 If present, each variable confers a certain number of points to the final score: 3 points for asymmetry (defined as unilateral leg involvement), 1 point for leukocytosis (white blood cell count ≥10,000/μL), 1 point for tachycardia (≥90 beats per minute), and 2 points for age of 70 years or older. An ALT-70 score of 0 to 2 corresponds to an 83.3% or greater chance of pseudocellulitis, suggesting that the diagnosis of cellulitis be reconsidered. A score of 3 to 4 is indeterminate, and additional information such as a dermatology consultation should be pursued. A score of 5 to 7 corresponds to an 82.2% or greater chance of cellulitis, signifying that empiric treatment with antibiotics be considered.15
The ALT-70 score does not apply to cases involving areas other than the lower extremities; intravenous antibiotic use within 48 hours before ED presentation; surgery within the last 30 days; abscess; penetrating trauma; burn; or known history of osteomyelitis, diabetic ulcer, or indwelling hardware at the site of infection.15 The ALT-70 score is available for free via the MDCalc app and website (https://www.mdcalc.com/alt-70-score-cellulitis).
Mohs AUC Determines the Appropriateness of Mohs Micrographic Surgery
In 2012, the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and American Society for Mohs Surgery published appropriate use criteria (AUC) to guide the decision to pursue Mohs micrographic surgery (MMS) in the United States.16 Based on various tumor and patient characteristics, the Mohs AUC assign scores to 270 different clinical scenarios. A score of 1 to 3 signifies that MMS is inappropriate and generally not considered acceptable. A score 4 to 6 indicates that the appropriateness of MMS is uncertain. A score 7 to 9 means that MMS is appropriate and generally considered acceptable.16
Since publication, the Mohs AUC have been criticized for classifying most primary superficial basal cell carcinomas as appropriate for MMS17 (which an AUC coauthor18 and others19,20 have defended), excluding certain reasons for performing MMS (such as operating on multiple tumors on the same day),21 including counterintuitive scores,22 and omitting trials from Europe23 (which AUC coauthors also have defended24).
Final Thoughts
Scoring systems are emerging in dermatology as evidence-based bedside tools to help guide clinical decision-making. Despite their limitations, these scores have the potential to make a meaningful impact in dermatology as they have in other specialties.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Guegan S, Bastuji-Garin S, Poszepczynska-Guigne E, et al. Performance of the SCORTEN during the first five days of hospitalization to predict the prognosis of epidermal necrolysis. J Invest Dermatol. 2006;126:272-276.
- Micheletti RG, Chiesa-Fuxench Z, Noe MH, et al. Stevens-Johnson syndrome/toxic epidermal necrolysis: a multicenter retrospective study of 377 adult patients from the United States. J Invest Dermatol. 2018;138:2315-2321.
- Sekula P, Liss Y, Davidovici B, et al. Evaluation of SCORTEN on a cohort of patients with Stevens-Johnson syndrome and toxic epidermal necrolysis included in the RegiSCAR study. J Burn Care Res. 2011;32:237-245.
- Noe MH, Rosenbach M, Hubbard RA, et al. Development and validation of a risk prediction model for in-hospital mortality among patients with Stevens-Johnson syndrome/toxic epidermal necrolysis-ABCD-10. JAMA Dermatol. 2019;155:448-454.
- Alinaghi F, Calov M, Kristensen LE, et al. Prevalence of psoriatic arthritis in patients with psoriasis: a systematic review and meta-analysis of observational and clinical studies. J Am Acad Dermatol. 2019;80:251-265.e219.
- Elmets CA, Leonardi CL, Davis DMR, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with awareness and attention to comorbidities. J Am Acad Dermatol. 2019;80:1073-1113.
- Karreman MC, Weel A, van der Ven M, et al. Performance of screening tools for psoriatic arthritis: a cross-sectional study in primary care. Rheumatology (Oxford). 2017;56:597-602.
- Ibrahim GH, Buch MH, Lawson C, et al. Evaluation of an existing screening tool for psoriatic arthritis in people with psoriasis and the development of a new instrument: the Psoriasis Epidemiology Screening Tool (PEST) questionnaire. Clin Exp Rheumatol. 2009;27:469-474.
- Zhang A, Kurtzman DJB, Perez-Chada LM, et al. Psoriatic arthritis and the dermatologist: an approach to screening and clinical evaluation. Clin Dermatol. 2018;36:551-560.
- Weng QY, Raff AB, Cohen JM, et al. Costs and consequences associated with misdiagnosed lower extremity cellulitis. JAMA Dermatol. 2017;153:141-146.
- Strazzula L, Cotliar J, Fox LP, et al. Inpatient dermatology consultation aids diagnosis of cellulitis among hospitalized patients: a multi-institutional analysis. J Am Acad Dermatol. 2015;73:70-75.
- Li DG, Dewan AK, Xia FD, et al. The ALT-70 predictive model outperforms thermal imaging for the diagnosis of lower extremity cellulitis: a prospective evaluation. J Am Acad Dermatol. 2018;79:1076-1080.e1071.
- Singer S, Li DG, Gunasekera N, et al. The ALT-70 predictive model maintains predictive value at 24 and 48 hours after presentation [published online March 23, 2019]. J Am Acad Dermatol. doi:10.1016/j.jaad.2019.03.050.
- Raff AB, Weng QY, Cohen JM, et al. A predictive model for diagnosis of lower extremity cellulitis: a cross-sectional study. J Am Acad Dermatol. 2017;76:618-625.e2.
- Connolly SM, Baker DR, Coldiron BM, et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: a report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J Am Acad Dermatol. 2012;67:531-550.
- Steinman HK, Dixon A, Zachary CB. Reevaluating Mohs surgery appropriate use criteria for primary superficial basal cell carcinoma. JAMA Dermatol. 2018;154:755-756.
- Montuno MA, Coldiron BM. Mohs appropriate use criteria for superficial basal cell carcinoma. JAMA Dermatol. 2019;155:394-395.
- MacFarlane DF, Perlis C. Mohs appropriate use criteria for superficial basal cell carcinoma. JAMA Dermatol. 2019;155:395-396.
- Kantor J. Mohs appropriate use criteria for superficial basal cell carcinoma. JAMA Dermatol. 2019;155:395.
- Ruiz ES, Karia PS, Morgan FC, et al. Multiple Mohs micrographic surgery is the most common reason for divergence from the appropriate use criteria: a single institution retrospective cohort study. J Am Acad Dermatol. 2016;75:830-831.
- Croley JA, Joseph AK, Wagner RF Jr. Discrepancies in the Mohs Micrographic Surgery appropriate use criteria [published online December 23, 2018]. J Am Acad Dermatol. doi:10.1016/j.jaad.2018.11.064.
- Kelleners-Smeets NW, Mosterd K. Comment on 2012 appropriate use criteria for Mohs micrographic surgery. J Am Acad Dermatol. 2013;69:317-318.
- Connolly S, Baker D, Coldiron B, et al. Reply to “comment on 2012 appropriate use criteria for Mohs micrographic surgery.” J Am Acad Dermatol. 2013;69:318.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Guegan S, Bastuji-Garin S, Poszepczynska-Guigne E, et al. Performance of the SCORTEN during the first five days of hospitalization to predict the prognosis of epidermal necrolysis. J Invest Dermatol. 2006;126:272-276.
- Micheletti RG, Chiesa-Fuxench Z, Noe MH, et al. Stevens-Johnson syndrome/toxic epidermal necrolysis: a multicenter retrospective study of 377 adult patients from the United States. J Invest Dermatol. 2018;138:2315-2321.
- Sekula P, Liss Y, Davidovici B, et al. Evaluation of SCORTEN on a cohort of patients with Stevens-Johnson syndrome and toxic epidermal necrolysis included in the RegiSCAR study. J Burn Care Res. 2011;32:237-245.
- Noe MH, Rosenbach M, Hubbard RA, et al. Development and validation of a risk prediction model for in-hospital mortality among patients with Stevens-Johnson syndrome/toxic epidermal necrolysis-ABCD-10. JAMA Dermatol. 2019;155:448-454.
- Alinaghi F, Calov M, Kristensen LE, et al. Prevalence of psoriatic arthritis in patients with psoriasis: a systematic review and meta-analysis of observational and clinical studies. J Am Acad Dermatol. 2019;80:251-265.e219.
- Elmets CA, Leonardi CL, Davis DMR, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with awareness and attention to comorbidities. J Am Acad Dermatol. 2019;80:1073-1113.
- Karreman MC, Weel A, van der Ven M, et al. Performance of screening tools for psoriatic arthritis: a cross-sectional study in primary care. Rheumatology (Oxford). 2017;56:597-602.
- Ibrahim GH, Buch MH, Lawson C, et al. Evaluation of an existing screening tool for psoriatic arthritis in people with psoriasis and the development of a new instrument: the Psoriasis Epidemiology Screening Tool (PEST) questionnaire. Clin Exp Rheumatol. 2009;27:469-474.
- Zhang A, Kurtzman DJB, Perez-Chada LM, et al. Psoriatic arthritis and the dermatologist: an approach to screening and clinical evaluation. Clin Dermatol. 2018;36:551-560.
- Weng QY, Raff AB, Cohen JM, et al. Costs and consequences associated with misdiagnosed lower extremity cellulitis. JAMA Dermatol. 2017;153:141-146.
- Strazzula L, Cotliar J, Fox LP, et al. Inpatient dermatology consultation aids diagnosis of cellulitis among hospitalized patients: a multi-institutional analysis. J Am Acad Dermatol. 2015;73:70-75.
- Li DG, Dewan AK, Xia FD, et al. The ALT-70 predictive model outperforms thermal imaging for the diagnosis of lower extremity cellulitis: a prospective evaluation. J Am Acad Dermatol. 2018;79:1076-1080.e1071.
- Singer S, Li DG, Gunasekera N, et al. The ALT-70 predictive model maintains predictive value at 24 and 48 hours after presentation [published online March 23, 2019]. J Am Acad Dermatol. doi:10.1016/j.jaad.2019.03.050.
- Raff AB, Weng QY, Cohen JM, et al. A predictive model for diagnosis of lower extremity cellulitis: a cross-sectional study. J Am Acad Dermatol. 2017;76:618-625.e2.
- Connolly SM, Baker DR, Coldiron BM, et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: a report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J Am Acad Dermatol. 2012;67:531-550.
- Steinman HK, Dixon A, Zachary CB. Reevaluating Mohs surgery appropriate use criteria for primary superficial basal cell carcinoma. JAMA Dermatol. 2018;154:755-756.
- Montuno MA, Coldiron BM. Mohs appropriate use criteria for superficial basal cell carcinoma. JAMA Dermatol. 2019;155:394-395.
- MacFarlane DF, Perlis C. Mohs appropriate use criteria for superficial basal cell carcinoma. JAMA Dermatol. 2019;155:395-396.
- Kantor J. Mohs appropriate use criteria for superficial basal cell carcinoma. JAMA Dermatol. 2019;155:395.
- Ruiz ES, Karia PS, Morgan FC, et al. Multiple Mohs micrographic surgery is the most common reason for divergence from the appropriate use criteria: a single institution retrospective cohort study. J Am Acad Dermatol. 2016;75:830-831.
- Croley JA, Joseph AK, Wagner RF Jr. Discrepancies in the Mohs Micrographic Surgery appropriate use criteria [published online December 23, 2018]. J Am Acad Dermatol. doi:10.1016/j.jaad.2018.11.064.
- Kelleners-Smeets NW, Mosterd K. Comment on 2012 appropriate use criteria for Mohs micrographic surgery. J Am Acad Dermatol. 2013;69:317-318.
- Connolly S, Baker D, Coldiron B, et al. Reply to “comment on 2012 appropriate use criteria for Mohs micrographic surgery.” J Am Acad Dermatol. 2013;69:318.
Resident Pearls
- Mortality from Stevens-Johnson syndrome/toxic epidermal necrolysis can be estimated by calculating the SCORTEN at the end of days 1 and 3 of hospitalization.
- The Psoriasis Epidemiology Screening Tool (PEST) assists with triaging which patients with psoriasis should be evaluated for psoriatic arthritis by a rheumatologist.
- The ALT-70 score is helpful to support one’s diagnosis of cellulitis or pseudocellulitis.
- The Mohs appropriate use criteria (AUC) score 270 different clinical scenarios as appropriate, uncertain, or inappropriate for Mohs micrographic surgery.

mIDH1 inhibitor ivosidenib improves progression-free survival in advanced cholangiocarcinoma
BARCELONA – Ivosidenib, a first-in-class, oral, small-molecule inhibitor of the mutant isocitrate dehydrogenase 1 (mIDH1) protein, significantly improved progression-free survival, compared with placebo, for the treatment of advanced cholangiocarcinoma in the global, randomized, phase 3 ClarIDHy trial.
A trend toward favorable overall survival was also seen in the pivotal double-blind trial, Ghassan K. Abou-Alfa, MD, of Memorial Sloan-Kettering Cancer Center, New York, reported at the European Society for Medical Oncology Congress.
Median progression-free survival (PFS) in 124 patients randomized to receive 500 mg of ivosidenib (IVO) once daily was 2.7 months, compared with 1.4 months in 61 patients who received placebo (hazard ratio, 0.37).
The primary study endpoint – PFS by central review – was reached, Dr. Abou-Alfa said, noting that the PFS rates at 6 and 12 months were 32% and 22% in the IVO arm, whereas none of the patients in the placebo arm were progression-free for 6 or more months.
Although the 1.3-month difference in PFS between the treatment and placebo arms “may seem short and some people may question whether this is clinically meaningful,” this outcome actually represents an important breakthrough for patients with a disease that has few treatment options, Angela Lamarca, MD, PhD, of the Christie NHS Foundation Trust, Manchester, England, explained in a press release about the study.
“A treatment that increases the chance of being free from progression by 32% at 6 months after starting treatment and that prolongs survival from 6 months with placebo to 10.8 months with ivosidenib, after adjusting for crossover, is definitely meaningful for our patients with cholangiocarcinoma and their families,” said Dr. Lamarca, representing the ESMO Press & Media Affairs Committee.
Indeed, the overall response rate in the IVO arm was 2.4%, representing three partial responses, and the stable disease rate was 50.8%. The overall response rate and stable disease rates in the placebo arm were 0% and 28%, Dr. Abou-Alfa said.
Median overall survival in the intent-to-treat population, including the 57% of placebo arm patients who crossed over to the treatment arm at the time of radiographic progression as allowed by study protocol, was 10.8 months, compared with 9.7 months in the placebo arm, respectively (HR, 0.69), Dr. Abou-Alfa said.
Crossover-adjusted overall survival was 6 months for the placebo arm (HR, 0.46) as assessed using rank-preserving structural failure time analysis, which suggested that the overall survival benefit would have been statistically significant had there been no crossover.
Study subjects had unresectable or metastatic mIDH1 cholangiocarcinoma, good performance status, and measurable disease. They had a median age of 62 years, and 68 were men. Most had intrahepatic disease (91%) and metastatic disease (92%), and 43% had two prior therapies, he noted.
They were randomized 2:1 to the treatment and placebo arms, respectively.
Since IVO targets mIDH1, which occurs in about 20% of patients with cholangiocarcinoma and results in production of the oncogenesis-promoting oncometabolite D-2-hydroxyglutarate, and since the IVO has shown encouraging activity in smaller prior studies, ClarIDHy was designed to evaluate it in the advanced mIDH1 cholangiocarcinoma setting because patients with this aggressive disease have generally poor prognosis and few treatment options beyond chemotherapy, he explained.
In addition to providing significant, clinically meaningful survival benefit, the treatment also was generally well tolerated, he noted.
Treatment-emergent adverse events occurring in more than 15% of patients in the IVO arm included nausea (32.1%), diarrhea (28.8%), fatigue (23.7%), cough (19.2%), abdominal pain (18.6%), ascites (18.6%), decreased appetite (17.3%), anemia (16.0%), and vomiting (16.0%). Grade 3 or higher adverse events were reported in 46% and 36% of the IVO and placebo patients, respectively.
The findings are notable, in part because of the unmet need and also because this is the first pivotal study demonstrating the clinical benefit of targeting mIDH1 in patients with advanced mIDH1 cholangiocarcinoma, he said, concluding that “these pivotal data demonstrate the clinical relevance and benefit of ivosidenib in mIDH1 cholangiocarcinoma ... [and] establish the role for genomic testing in this rare cancer with a high unmet need.”
He also said studies should investigate IVO in the first-line setting for IDH1-mutated cholangiocarcinoma, in addition to its use in combination therapy and as adjuvant therapy.
In the ESMO press release, Chris Verslype, MD, of University Hospital Leuven (Belgium) called the findings of this study unprecedented given the lack of treatment options in those who fail systemic therapy, which has led to very limited survival.
The findings are “very likely to change clinical practice” and “will, for sure, drive the further development of targeted therapy for the disease,” he said.
Despite being limited by the requirement that patients have good performance status after prior chemotherapy (which means the findings may not be representative of all patients), ClarIDHy is “still a strong study because of the randomization to placebo.”
“It showed a real effect,” he said.
ClarIDHy was funded by Agios Pharmaceuticals. Dr. Abou-Alfa reported both personal and institutional relationships with industry. These include advisory /consulting roles and research grants/funding from numerous pharmaceutical companies.
SOURCE: Abou-Alfa GK et al. ESMO 2019, Abstract LBA10-PR.
BARCELONA – Ivosidenib, a first-in-class, oral, small-molecule inhibitor of the mutant isocitrate dehydrogenase 1 (mIDH1) protein, significantly improved progression-free survival, compared with placebo, for the treatment of advanced cholangiocarcinoma in the global, randomized, phase 3 ClarIDHy trial.
A trend toward favorable overall survival was also seen in the pivotal double-blind trial, Ghassan K. Abou-Alfa, MD, of Memorial Sloan-Kettering Cancer Center, New York, reported at the European Society for Medical Oncology Congress.
Median progression-free survival (PFS) in 124 patients randomized to receive 500 mg of ivosidenib (IVO) once daily was 2.7 months, compared with 1.4 months in 61 patients who received placebo (hazard ratio, 0.37).
The primary study endpoint – PFS by central review – was reached, Dr. Abou-Alfa said, noting that the PFS rates at 6 and 12 months were 32% and 22% in the IVO arm, whereas none of the patients in the placebo arm were progression-free for 6 or more months.
Although the 1.3-month difference in PFS between the treatment and placebo arms “may seem short and some people may question whether this is clinically meaningful,” this outcome actually represents an important breakthrough for patients with a disease that has few treatment options, Angela Lamarca, MD, PhD, of the Christie NHS Foundation Trust, Manchester, England, explained in a press release about the study.
“A treatment that increases the chance of being free from progression by 32% at 6 months after starting treatment and that prolongs survival from 6 months with placebo to 10.8 months with ivosidenib, after adjusting for crossover, is definitely meaningful for our patients with cholangiocarcinoma and their families,” said Dr. Lamarca, representing the ESMO Press & Media Affairs Committee.
Indeed, the overall response rate in the IVO arm was 2.4%, representing three partial responses, and the stable disease rate was 50.8%. The overall response rate and stable disease rates in the placebo arm were 0% and 28%, Dr. Abou-Alfa said.
Median overall survival in the intent-to-treat population, including the 57% of placebo arm patients who crossed over to the treatment arm at the time of radiographic progression as allowed by study protocol, was 10.8 months, compared with 9.7 months in the placebo arm, respectively (HR, 0.69), Dr. Abou-Alfa said.
Crossover-adjusted overall survival was 6 months for the placebo arm (HR, 0.46) as assessed using rank-preserving structural failure time analysis, which suggested that the overall survival benefit would have been statistically significant had there been no crossover.
Study subjects had unresectable or metastatic mIDH1 cholangiocarcinoma, good performance status, and measurable disease. They had a median age of 62 years, and 68 were men. Most had intrahepatic disease (91%) and metastatic disease (92%), and 43% had two prior therapies, he noted.
They were randomized 2:1 to the treatment and placebo arms, respectively.
Since IVO targets mIDH1, which occurs in about 20% of patients with cholangiocarcinoma and results in production of the oncogenesis-promoting oncometabolite D-2-hydroxyglutarate, and since the IVO has shown encouraging activity in smaller prior studies, ClarIDHy was designed to evaluate it in the advanced mIDH1 cholangiocarcinoma setting because patients with this aggressive disease have generally poor prognosis and few treatment options beyond chemotherapy, he explained.
In addition to providing significant, clinically meaningful survival benefit, the treatment also was generally well tolerated, he noted.
Treatment-emergent adverse events occurring in more than 15% of patients in the IVO arm included nausea (32.1%), diarrhea (28.8%), fatigue (23.7%), cough (19.2%), abdominal pain (18.6%), ascites (18.6%), decreased appetite (17.3%), anemia (16.0%), and vomiting (16.0%). Grade 3 or higher adverse events were reported in 46% and 36% of the IVO and placebo patients, respectively.
The findings are notable, in part because of the unmet need and also because this is the first pivotal study demonstrating the clinical benefit of targeting mIDH1 in patients with advanced mIDH1 cholangiocarcinoma, he said, concluding that “these pivotal data demonstrate the clinical relevance and benefit of ivosidenib in mIDH1 cholangiocarcinoma ... [and] establish the role for genomic testing in this rare cancer with a high unmet need.”
He also said studies should investigate IVO in the first-line setting for IDH1-mutated cholangiocarcinoma, in addition to its use in combination therapy and as adjuvant therapy.
In the ESMO press release, Chris Verslype, MD, of University Hospital Leuven (Belgium) called the findings of this study unprecedented given the lack of treatment options in those who fail systemic therapy, which has led to very limited survival.
The findings are “very likely to change clinical practice” and “will, for sure, drive the further development of targeted therapy for the disease,” he said.
Despite being limited by the requirement that patients have good performance status after prior chemotherapy (which means the findings may not be representative of all patients), ClarIDHy is “still a strong study because of the randomization to placebo.”
“It showed a real effect,” he said.
ClarIDHy was funded by Agios Pharmaceuticals. Dr. Abou-Alfa reported both personal and institutional relationships with industry. These include advisory /consulting roles and research grants/funding from numerous pharmaceutical companies.
SOURCE: Abou-Alfa GK et al. ESMO 2019, Abstract LBA10-PR.
BARCELONA – Ivosidenib, a first-in-class, oral, small-molecule inhibitor of the mutant isocitrate dehydrogenase 1 (mIDH1) protein, significantly improved progression-free survival, compared with placebo, for the treatment of advanced cholangiocarcinoma in the global, randomized, phase 3 ClarIDHy trial.
A trend toward favorable overall survival was also seen in the pivotal double-blind trial, Ghassan K. Abou-Alfa, MD, of Memorial Sloan-Kettering Cancer Center, New York, reported at the European Society for Medical Oncology Congress.
Median progression-free survival (PFS) in 124 patients randomized to receive 500 mg of ivosidenib (IVO) once daily was 2.7 months, compared with 1.4 months in 61 patients who received placebo (hazard ratio, 0.37).
The primary study endpoint – PFS by central review – was reached, Dr. Abou-Alfa said, noting that the PFS rates at 6 and 12 months were 32% and 22% in the IVO arm, whereas none of the patients in the placebo arm were progression-free for 6 or more months.
Although the 1.3-month difference in PFS between the treatment and placebo arms “may seem short and some people may question whether this is clinically meaningful,” this outcome actually represents an important breakthrough for patients with a disease that has few treatment options, Angela Lamarca, MD, PhD, of the Christie NHS Foundation Trust, Manchester, England, explained in a press release about the study.
“A treatment that increases the chance of being free from progression by 32% at 6 months after starting treatment and that prolongs survival from 6 months with placebo to 10.8 months with ivosidenib, after adjusting for crossover, is definitely meaningful for our patients with cholangiocarcinoma and their families,” said Dr. Lamarca, representing the ESMO Press & Media Affairs Committee.
Indeed, the overall response rate in the IVO arm was 2.4%, representing three partial responses, and the stable disease rate was 50.8%. The overall response rate and stable disease rates in the placebo arm were 0% and 28%, Dr. Abou-Alfa said.
Median overall survival in the intent-to-treat population, including the 57% of placebo arm patients who crossed over to the treatment arm at the time of radiographic progression as allowed by study protocol, was 10.8 months, compared with 9.7 months in the placebo arm, respectively (HR, 0.69), Dr. Abou-Alfa said.
Crossover-adjusted overall survival was 6 months for the placebo arm (HR, 0.46) as assessed using rank-preserving structural failure time analysis, which suggested that the overall survival benefit would have been statistically significant had there been no crossover.
Study subjects had unresectable or metastatic mIDH1 cholangiocarcinoma, good performance status, and measurable disease. They had a median age of 62 years, and 68 were men. Most had intrahepatic disease (91%) and metastatic disease (92%), and 43% had two prior therapies, he noted.
They were randomized 2:1 to the treatment and placebo arms, respectively.
Since IVO targets mIDH1, which occurs in about 20% of patients with cholangiocarcinoma and results in production of the oncogenesis-promoting oncometabolite D-2-hydroxyglutarate, and since the IVO has shown encouraging activity in smaller prior studies, ClarIDHy was designed to evaluate it in the advanced mIDH1 cholangiocarcinoma setting because patients with this aggressive disease have generally poor prognosis and few treatment options beyond chemotherapy, he explained.
In addition to providing significant, clinically meaningful survival benefit, the treatment also was generally well tolerated, he noted.
Treatment-emergent adverse events occurring in more than 15% of patients in the IVO arm included nausea (32.1%), diarrhea (28.8%), fatigue (23.7%), cough (19.2%), abdominal pain (18.6%), ascites (18.6%), decreased appetite (17.3%), anemia (16.0%), and vomiting (16.0%). Grade 3 or higher adverse events were reported in 46% and 36% of the IVO and placebo patients, respectively.
The findings are notable, in part because of the unmet need and also because this is the first pivotal study demonstrating the clinical benefit of targeting mIDH1 in patients with advanced mIDH1 cholangiocarcinoma, he said, concluding that “these pivotal data demonstrate the clinical relevance and benefit of ivosidenib in mIDH1 cholangiocarcinoma ... [and] establish the role for genomic testing in this rare cancer with a high unmet need.”
He also said studies should investigate IVO in the first-line setting for IDH1-mutated cholangiocarcinoma, in addition to its use in combination therapy and as adjuvant therapy.
In the ESMO press release, Chris Verslype, MD, of University Hospital Leuven (Belgium) called the findings of this study unprecedented given the lack of treatment options in those who fail systemic therapy, which has led to very limited survival.
The findings are “very likely to change clinical practice” and “will, for sure, drive the further development of targeted therapy for the disease,” he said.
Despite being limited by the requirement that patients have good performance status after prior chemotherapy (which means the findings may not be representative of all patients), ClarIDHy is “still a strong study because of the randomization to placebo.”
“It showed a real effect,” he said.
ClarIDHy was funded by Agios Pharmaceuticals. Dr. Abou-Alfa reported both personal and institutional relationships with industry. These include advisory /consulting roles and research grants/funding from numerous pharmaceutical companies.
SOURCE: Abou-Alfa GK et al. ESMO 2019, Abstract LBA10-PR.
REPORTING FROM ESMO 2019
FDA approves rituximab to treat children with rare vasculitis
The Food and Drug Administration approved rituximab (Rituxan) by injection to treat granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) in children 2 years of age and older in combination with glucocorticoid treatment, according to an FDA news release.

These rare forms of vasculitis damage small blood vessels through inflammation and can lead to serious organ failure, including lungs and kidneys.
The Genentech drug received priority review and an orphan drug designation based on the results of a pediatric clinical trial of 25 patients aged 6-17 years with active GPA or MPA who were treated with rituximab in an international multicenter, open-label, uncontrolled study. Patients in the trial were also given methylprednisolone prior to starting treatment.
The trial consisted of a 6-month remission induction phase where patients were treated only with rituximab and glucocorticoids. In addition, patients who had not achieved remission could receive additional treatment, including other therapies, at the discretion of the investigator, according to the FDA. By 6 months, 14 of the patients were in remission, and after 18 months, all 25 patients were in remission.
Rituximab contains a boxed warning regarding increased risks of fatal infusion reactions, potentially fatal severe skin and mouth reactions, hepatitis B virus reactivation that may cause serious or lethal liver problems, and progressive multifocal leukoencephalopathy, a rare, potentially lethal brain infection.
The trial was conducted and sponsored by F. Hoffmann-La Roche, which owns Genentech.
The Food and Drug Administration approved rituximab (Rituxan) by injection to treat granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) in children 2 years of age and older in combination with glucocorticoid treatment, according to an FDA news release.
These rare forms of vasculitis damage small blood vessels through inflammation and can lead to serious organ failure, including lungs and kidneys.
The Genentech drug received priority review and an orphan drug designation based on the results of a pediatric clinical trial of 25 patients aged 6-17 years with active GPA or MPA who were treated with rituximab in an international multicenter, open-label, uncontrolled study. Patients in the trial were also given methylprednisolone prior to starting treatment.
The trial consisted of a 6-month remission induction phase where patients were treated only with rituximab and glucocorticoids. In addition, patients who had not achieved remission could receive additional treatment, including other therapies, at the discretion of the investigator, according to the FDA. By 6 months, 14 of the patients were in remission, and after 18 months, all 25 patients were in remission.
Rituximab contains a boxed warning regarding increased risks of fatal infusion reactions, potentially fatal severe skin and mouth reactions, hepatitis B virus reactivation that may cause serious or lethal liver problems, and progressive multifocal leukoencephalopathy, a rare, potentially lethal brain infection.
The trial was conducted and sponsored by F. Hoffmann-La Roche, which owns Genentech.
The Food and Drug Administration approved rituximab (Rituxan) by injection to treat granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) in children 2 years of age and older in combination with glucocorticoid treatment, according to an FDA news release.
These rare forms of vasculitis damage small blood vessels through inflammation and can lead to serious organ failure, including lungs and kidneys.
The Genentech drug received priority review and an orphan drug designation based on the results of a pediatric clinical trial of 25 patients aged 6-17 years with active GPA or MPA who were treated with rituximab in an international multicenter, open-label, uncontrolled study. Patients in the trial were also given methylprednisolone prior to starting treatment.
The trial consisted of a 6-month remission induction phase where patients were treated only with rituximab and glucocorticoids. In addition, patients who had not achieved remission could receive additional treatment, including other therapies, at the discretion of the investigator, according to the FDA. By 6 months, 14 of the patients were in remission, and after 18 months, all 25 patients were in remission.
Rituximab contains a boxed warning regarding increased risks of fatal infusion reactions, potentially fatal severe skin and mouth reactions, hepatitis B virus reactivation that may cause serious or lethal liver problems, and progressive multifocal leukoencephalopathy, a rare, potentially lethal brain infection.
The trial was conducted and sponsored by F. Hoffmann-La Roche, which owns Genentech.
Anakinra treatment for pediatric ‘cytokine storms’: Does one size fit all?
The biologic drug anakinra appears to be effective in treating children with secondary hemophagocytic lymphohistiocytosis (sHLH)/macrophage activation syndrome (MAS), a dangerous “cytokine storm” that can emerge from infections, cancer, and rheumatic diseases.
Children with systematic juvenile idiopathic arthritis (sJIA) and sHLH/MAS are especially good candidates for treatment with the interleukin-1 receptor antagonist anakinra (Kineret), in whom its safety and benefits have been more widely explored than in pediatric patients with sHLH/MAS related to non-sJIA underlying conditions.
In a study published in Arthritis & Rheumatology, Esraa Eloseily, MD, and colleagues at the University of Alabama at Birmingham, looked at hospitalization records for 44 children (mean age, 10 years; n = 25 females) with sHLH/MAS. The children in the study had heterogeneous underlying conditions including leukemias, infections, and rheumatic diseases. About one-third of patients had no known rheumatic or autoimmune disorder.
Dr. Eloseily and colleagues found that early initiation of anakinra (within 5 days of hospitalization) was significantly associated with improved survival across the cohort, for which mortality was 27%. Thrombocytopenia (less than 100,000/mcL) and STXBP2 mutations were both seen significantly associated with mortality.
Patients with blood cancers – even those in remission at the time of treatment – did poorly. None of the three patients in the cohort with leukemia survived.
Importantly, no deaths were seen among the 13 patients with underlying SJIA who were treated with anakinra, suggesting particular benefit for this patient group.
“In addition to the 10% risk of developing overt MAS as part of sJIA, another 30%-40% of sJIA patients may have occult or subclinical MAS during a disease flare that can eventually lead to overt MAS,” Dr. Eloseily and colleagues wrote. “This association of MAS with sJIA suggested that anakinra would also be a valuable treatment for sJIA-MAS.”
The investigators acknowledged that their study was limited by its retrospective design and “nonuniform approach to therapy, lack of treatment controls, and variable follow-up period.” The authors also acknowledged the potential for selection bias favoring anakinra use in patients who are less severely ill.
In a comment accompanying Dr. Eloseily and colleagues’ study, Sarah Nikiforow, MD, PhD, of the Dana-Farber Cancer Institute in Boston, and Nancy Berliner, MD, of Brigham & Women’s Hospital in Boston, urged clinicians not to interpret the study results as supporting anakinra as “a carte blanche approach to hyperinflammatory syndromes.”
While the study supported the use of anakinra in sJIA with MAS or sHLH, “we posit that patients [with sHLH/MAS] in sepsis, cytokine release syndrome following chimeric antigen receptor T-cell therapy, and other hyperinflammatory syndromes still require individualized approaches to therapy,” Dr. Nikiforow and Dr. Berliner wrote, adding that, “in several studies and anecdotally in our institutional practice, cytotoxic chemotherapy was/is preferred over biologic agents in patients with evidence of more severe inflammatory activity.”
Outside sJIA, Dr. Nikiforow and Dr. Berliner wrote, “early anakinra therapy should be extended to treatment of other forms of sHLH with extreme caution. Specifically, the authors’ suggestion that cytotoxic therapy should be ‘considered’ only after anakinra therapy may be dangerous for some patients.”
Two of Dr. Eloseily’s coinvestigators reported financial and research support from Sobi, the manufacturer of anakinra. No other conflicts of interest were reported.
SOURCES: Eloseily E et al. Arthritis Rheumatol. 2019 Sep 12. doi: 10.1002/art.41103; Nikiforow S, Berliner N. Arthritis Rheumatol. 2019 Sep 16. doi: 10.1002/art.41106.
The biologic drug anakinra appears to be effective in treating children with secondary hemophagocytic lymphohistiocytosis (sHLH)/macrophage activation syndrome (MAS), a dangerous “cytokine storm” that can emerge from infections, cancer, and rheumatic diseases.
Children with systematic juvenile idiopathic arthritis (sJIA) and sHLH/MAS are especially good candidates for treatment with the interleukin-1 receptor antagonist anakinra (Kineret), in whom its safety and benefits have been more widely explored than in pediatric patients with sHLH/MAS related to non-sJIA underlying conditions.
In a study published in Arthritis & Rheumatology, Esraa Eloseily, MD, and colleagues at the University of Alabama at Birmingham, looked at hospitalization records for 44 children (mean age, 10 years; n = 25 females) with sHLH/MAS. The children in the study had heterogeneous underlying conditions including leukemias, infections, and rheumatic diseases. About one-third of patients had no known rheumatic or autoimmune disorder.
Dr. Eloseily and colleagues found that early initiation of anakinra (within 5 days of hospitalization) was significantly associated with improved survival across the cohort, for which mortality was 27%. Thrombocytopenia (less than 100,000/mcL) and STXBP2 mutations were both seen significantly associated with mortality.
Patients with blood cancers – even those in remission at the time of treatment – did poorly. None of the three patients in the cohort with leukemia survived.
Importantly, no deaths were seen among the 13 patients with underlying SJIA who were treated with anakinra, suggesting particular benefit for this patient group.
“In addition to the 10% risk of developing overt MAS as part of sJIA, another 30%-40% of sJIA patients may have occult or subclinical MAS during a disease flare that can eventually lead to overt MAS,” Dr. Eloseily and colleagues wrote. “This association of MAS with sJIA suggested that anakinra would also be a valuable treatment for sJIA-MAS.”
The investigators acknowledged that their study was limited by its retrospective design and “nonuniform approach to therapy, lack of treatment controls, and variable follow-up period.” The authors also acknowledged the potential for selection bias favoring anakinra use in patients who are less severely ill.
In a comment accompanying Dr. Eloseily and colleagues’ study, Sarah Nikiforow, MD, PhD, of the Dana-Farber Cancer Institute in Boston, and Nancy Berliner, MD, of Brigham & Women’s Hospital in Boston, urged clinicians not to interpret the study results as supporting anakinra as “a carte blanche approach to hyperinflammatory syndromes.”
While the study supported the use of anakinra in sJIA with MAS or sHLH, “we posit that patients [with sHLH/MAS] in sepsis, cytokine release syndrome following chimeric antigen receptor T-cell therapy, and other hyperinflammatory syndromes still require individualized approaches to therapy,” Dr. Nikiforow and Dr. Berliner wrote, adding that, “in several studies and anecdotally in our institutional practice, cytotoxic chemotherapy was/is preferred over biologic agents in patients with evidence of more severe inflammatory activity.”
Outside sJIA, Dr. Nikiforow and Dr. Berliner wrote, “early anakinra therapy should be extended to treatment of other forms of sHLH with extreme caution. Specifically, the authors’ suggestion that cytotoxic therapy should be ‘considered’ only after anakinra therapy may be dangerous for some patients.”
Two of Dr. Eloseily’s coinvestigators reported financial and research support from Sobi, the manufacturer of anakinra. No other conflicts of interest were reported.
SOURCES: Eloseily E et al. Arthritis Rheumatol. 2019 Sep 12. doi: 10.1002/art.41103; Nikiforow S, Berliner N. Arthritis Rheumatol. 2019 Sep 16. doi: 10.1002/art.41106.
The biologic drug anakinra appears to be effective in treating children with secondary hemophagocytic lymphohistiocytosis (sHLH)/macrophage activation syndrome (MAS), a dangerous “cytokine storm” that can emerge from infections, cancer, and rheumatic diseases.
Children with systematic juvenile idiopathic arthritis (sJIA) and sHLH/MAS are especially good candidates for treatment with the interleukin-1 receptor antagonist anakinra (Kineret), in whom its safety and benefits have been more widely explored than in pediatric patients with sHLH/MAS related to non-sJIA underlying conditions.
In a study published in Arthritis & Rheumatology, Esraa Eloseily, MD, and colleagues at the University of Alabama at Birmingham, looked at hospitalization records for 44 children (mean age, 10 years; n = 25 females) with sHLH/MAS. The children in the study had heterogeneous underlying conditions including leukemias, infections, and rheumatic diseases. About one-third of patients had no known rheumatic or autoimmune disorder.
Dr. Eloseily and colleagues found that early initiation of anakinra (within 5 days of hospitalization) was significantly associated with improved survival across the cohort, for which mortality was 27%. Thrombocytopenia (less than 100,000/mcL) and STXBP2 mutations were both seen significantly associated with mortality.
Patients with blood cancers – even those in remission at the time of treatment – did poorly. None of the three patients in the cohort with leukemia survived.
Importantly, no deaths were seen among the 13 patients with underlying SJIA who were treated with anakinra, suggesting particular benefit for this patient group.
“In addition to the 10% risk of developing overt MAS as part of sJIA, another 30%-40% of sJIA patients may have occult or subclinical MAS during a disease flare that can eventually lead to overt MAS,” Dr. Eloseily and colleagues wrote. “This association of MAS with sJIA suggested that anakinra would also be a valuable treatment for sJIA-MAS.”
The investigators acknowledged that their study was limited by its retrospective design and “nonuniform approach to therapy, lack of treatment controls, and variable follow-up period.” The authors also acknowledged the potential for selection bias favoring anakinra use in patients who are less severely ill.
In a comment accompanying Dr. Eloseily and colleagues’ study, Sarah Nikiforow, MD, PhD, of the Dana-Farber Cancer Institute in Boston, and Nancy Berliner, MD, of Brigham & Women’s Hospital in Boston, urged clinicians not to interpret the study results as supporting anakinra as “a carte blanche approach to hyperinflammatory syndromes.”
While the study supported the use of anakinra in sJIA with MAS or sHLH, “we posit that patients [with sHLH/MAS] in sepsis, cytokine release syndrome following chimeric antigen receptor T-cell therapy, and other hyperinflammatory syndromes still require individualized approaches to therapy,” Dr. Nikiforow and Dr. Berliner wrote, adding that, “in several studies and anecdotally in our institutional practice, cytotoxic chemotherapy was/is preferred over biologic agents in patients with evidence of more severe inflammatory activity.”
Outside sJIA, Dr. Nikiforow and Dr. Berliner wrote, “early anakinra therapy should be extended to treatment of other forms of sHLH with extreme caution. Specifically, the authors’ suggestion that cytotoxic therapy should be ‘considered’ only after anakinra therapy may be dangerous for some patients.”
Two of Dr. Eloseily’s coinvestigators reported financial and research support from Sobi, the manufacturer of anakinra. No other conflicts of interest were reported.
SOURCES: Eloseily E et al. Arthritis Rheumatol. 2019 Sep 12. doi: 10.1002/art.41103; Nikiforow S, Berliner N. Arthritis Rheumatol. 2019 Sep 16. doi: 10.1002/art.41106.
FROM ARTHRITIS & RHEUMATOLOGY
USPSTF: Screening pregnant women for asymptomatic bacteriuria cuts pyelonephritis risk
according to new recommendations set forth by the United States Preventive Services Task Force (USPSTF).
However, the investigating committee reported, there is evidence against screening nonpregnant women and adult men. In fact, the committee found “adequate” evidence of potential harm associated with treating asymptomatic bacteriuria in adults of both sexes, including adverse effects of antibiotics and on the microbiome.
The new document downgrades from A to B the group’s prior recommendation that urine culture screening for asymptomatic bacteriuria should be performed among pregnant women at 12-16 weeks’ gestation or at their first prenatal visit. The USPSTF recommendation to not screen nonpregnant adults retained its D rating, Jerome A. Leis, MD and Christine Soong, MD said in an accompanying editorial.
“Not screening or treating asymptomatic bacteriuria in this population has long been an ironclad recommendation endorsed by the Infectious Diseases Society of America, as well as numerous professional societies as part of the Choosing Wisely campaign,” wrote Dr. Leis of Sunnybrook Health Sciences Centre, Toronto, and Dr. Soong of the University of Toronto. “Restating this steadfast and pervasive recommendation may seem unremarkable and almost pedantic, yet it remains stubbornly disregarded by clinicians across multiple settings.”
The new recommendations were based on a review of 19 studies involving almost 8,500 pregnant and nonpregnant women, as well as a small number of adult men. Most were carried out in the 1960s or 1970s. The most recent ones were published in 2002 and 2015. The dearth of more recent data may have limited some conclusions and certainly highlighted the need for more research, said Jillian T. Henderson, PhD, chair of the committee assigned to investigate the evidence.
“Few studies of asymptomatic bacteriuria screening or treatment in pregnant populations have been conducted in the past 40 years,” wrote Dr. Henderson of Kaiser Permanente Northwest, Portland, and associates. “Historical evidence established asymptomatic bacteriuria screening and treatment as standard obstetric practice in the United States.” But these trials typically were less rigorous than modern studies, and the results are out of touch with modern clinical settings and treatment protocols, the team noted.
Additionally, Dr. Henderson and coauthors said, rates of pyelonephritis were about 10 times higher then than they are now. In the more recent studies, pyelonephritis rates in control groups were 2.2% and 2.5%; in most of the older studies, control group rates ranged from 33% to 36%.
In commissioning the investigation, the task force looked at the following four questions:
Does screening improve health outcomes?
Neither of two studies involving 5,289 women, one from Spain and one from Turkey, addressed this question in nonpregnant women; however, studies that looked at pregnant women generally found that screening did reduce the risk of pyelonephritis by about 70%. The investigators cautioned that these studies were out of date and perhaps methodologically flawed.
The only study that looked at newborn outcomes found no difference in birth weights or premature births between the screened and unscreened cohorts.
No study examined this question in nonpregnant women or men.
What are the harms of such screening?
A single study of 372 pregnant women described potential prenatal and perinatal harms associated with screening and treatment. It found a slight increase in congenital abnormalities in the screened cohort (1.6%), compared with those who were not screened (1.1%). However, those who were not screened were presumably not prescribed antibiotics.
Does treatment of screening-detected asymptomatic bacteriuria improve health outcomes?
Twelve trials of pregnant women (2,377) addressed this issue. All but two were conducted in the 1960s and 1970s. Treatment varied widely; sulfonamides were the most common, including the now discarded sulfamethazine and sulfadimethoxine. Dosages and duration of treatment also were considerably higher and longer than current practice.
In all but one study, there were higher rates of pyelonephritis in the control group. A pooled risk analysis indicated that treatment reduced the risk of pyelonephritis by nearly 80% (relative risk, 0.24).
Seven studies found higher rates of low birth weight in infants born to mothers who were treated, but two studies reported a significant reduction in the risk of low birth weight.
Among the six trials that examined perinatal mortality, none found significant associations with treatment.
Five studies examined treatment in nonpregnant women with screening-detected asymptomatic bacteriuria, and one included men as well. Of the four that reported the rate of symptomatic infection or pyelonephritis, none found a significant difference between treatment and control groups. The single study that included men also found no significant difference between treatment and control groups.
Among the three studies that focused on older adults, there also were no significant between-group differences in outcomes.
What harms are associated with treatment of screening-detected asymptomatic bacteriuria?
Seven studies comprised pregnant women. Five reported congenital malformations in the intervention and control groups. Overall, there were very few cases of malformations, with more – although not significantly more – in the control groups.
Evidence related to other infant and maternal harms was “sparsely and inconsistently reported,” Dr. Henderson and coauthors noted, “and there was a lack of evidence on long-term neonatal outcomes after antibiotic treatment of asymptomatic bacteriuria in pregnancy.”
Two studies listed maternal adverse events associated with different treatments including vaginitis and diarrhea with ampicillin and rashes and nausea with nalidixic acid.
In terms of nonpregnant women and men, four studies reported adverse events. None occurred with nitrofurantoin or trimethoprim treatment; however, one study that included daily treatment with ofloxacin noted that 6% withdrew because of adverse events – vertigo and gastrointestinal symptoms.
Treatments didn’t affect hematocrit, bilirubin, serum urea, or nitrogen, although some studies found a slight reduction in serum creatinine.
Although there’s a need for additional research into this question, the new recommendations provide a good reason to further reduce unnecessary antibiotic exposure, Lindsey E. Nicolle, MD, wrote in a second commentary.
These updated recommendations “contribute to the evolution of management of asymptomatic bacteriuria in healthy women,” wrote Dr. Nicolle of the University of Manitoba, Winnipeg. “However, questions remain about the risks and benefits of universal screening for and treatment of asymptomatic bacteriuria in pregnant women in the context of current clinical practice. The effects of changes in fetal-maternal care, of low- compared with high-risk pregnancies, and of health care access need to be understood. In the short term, application of current diagnostic recommendations for identification of persistent symptomatic bacteriuria with a second urine culture may provide an immediate opportunity to limit unnecessary antimicrobial use for some pregnant women.”
No conflicts of interest were reported by the USPSTF authors, nor by Dr. Leis, Dr. Soong, or Dr. Nicolle. The USPSTF report was funded by the Agency for Healthcare Research and Quality.
SOURCES: U.S. Preventive Services Task Force. JAMA. 2019;322(12):1188-94; Henderson JT et al. JAMA. 2019;322(12):1195-205; Leis JA and Soong C. JAMA. 2019. doi: 10.1001/jamainternmed.2019.4515; Nicolle LE. JAMA. 2019;322(12):1152-4.
according to new recommendations set forth by the United States Preventive Services Task Force (USPSTF).
However, the investigating committee reported, there is evidence against screening nonpregnant women and adult men. In fact, the committee found “adequate” evidence of potential harm associated with treating asymptomatic bacteriuria in adults of both sexes, including adverse effects of antibiotics and on the microbiome.
The new document downgrades from A to B the group’s prior recommendation that urine culture screening for asymptomatic bacteriuria should be performed among pregnant women at 12-16 weeks’ gestation or at their first prenatal visit. The USPSTF recommendation to not screen nonpregnant adults retained its D rating, Jerome A. Leis, MD and Christine Soong, MD said in an accompanying editorial.
“Not screening or treating asymptomatic bacteriuria in this population has long been an ironclad recommendation endorsed by the Infectious Diseases Society of America, as well as numerous professional societies as part of the Choosing Wisely campaign,” wrote Dr. Leis of Sunnybrook Health Sciences Centre, Toronto, and Dr. Soong of the University of Toronto. “Restating this steadfast and pervasive recommendation may seem unremarkable and almost pedantic, yet it remains stubbornly disregarded by clinicians across multiple settings.”
The new recommendations were based on a review of 19 studies involving almost 8,500 pregnant and nonpregnant women, as well as a small number of adult men. Most were carried out in the 1960s or 1970s. The most recent ones were published in 2002 and 2015. The dearth of more recent data may have limited some conclusions and certainly highlighted the need for more research, said Jillian T. Henderson, PhD, chair of the committee assigned to investigate the evidence.
“Few studies of asymptomatic bacteriuria screening or treatment in pregnant populations have been conducted in the past 40 years,” wrote Dr. Henderson of Kaiser Permanente Northwest, Portland, and associates. “Historical evidence established asymptomatic bacteriuria screening and treatment as standard obstetric practice in the United States.” But these trials typically were less rigorous than modern studies, and the results are out of touch with modern clinical settings and treatment protocols, the team noted.
Additionally, Dr. Henderson and coauthors said, rates of pyelonephritis were about 10 times higher then than they are now. In the more recent studies, pyelonephritis rates in control groups were 2.2% and 2.5%; in most of the older studies, control group rates ranged from 33% to 36%.
In commissioning the investigation, the task force looked at the following four questions:
Does screening improve health outcomes?
Neither of two studies involving 5,289 women, one from Spain and one from Turkey, addressed this question in nonpregnant women; however, studies that looked at pregnant women generally found that screening did reduce the risk of pyelonephritis by about 70%. The investigators cautioned that these studies were out of date and perhaps methodologically flawed.
The only study that looked at newborn outcomes found no difference in birth weights or premature births between the screened and unscreened cohorts.
No study examined this question in nonpregnant women or men.
What are the harms of such screening?
A single study of 372 pregnant women described potential prenatal and perinatal harms associated with screening and treatment. It found a slight increase in congenital abnormalities in the screened cohort (1.6%), compared with those who were not screened (1.1%). However, those who were not screened were presumably not prescribed antibiotics.
Does treatment of screening-detected asymptomatic bacteriuria improve health outcomes?
Twelve trials of pregnant women (2,377) addressed this issue. All but two were conducted in the 1960s and 1970s. Treatment varied widely; sulfonamides were the most common, including the now discarded sulfamethazine and sulfadimethoxine. Dosages and duration of treatment also were considerably higher and longer than current practice.
In all but one study, there were higher rates of pyelonephritis in the control group. A pooled risk analysis indicated that treatment reduced the risk of pyelonephritis by nearly 80% (relative risk, 0.24).
Seven studies found higher rates of low birth weight in infants born to mothers who were treated, but two studies reported a significant reduction in the risk of low birth weight.
Among the six trials that examined perinatal mortality, none found significant associations with treatment.
Five studies examined treatment in nonpregnant women with screening-detected asymptomatic bacteriuria, and one included men as well. Of the four that reported the rate of symptomatic infection or pyelonephritis, none found a significant difference between treatment and control groups. The single study that included men also found no significant difference between treatment and control groups.
Among the three studies that focused on older adults, there also were no significant between-group differences in outcomes.
What harms are associated with treatment of screening-detected asymptomatic bacteriuria?
Seven studies comprised pregnant women. Five reported congenital malformations in the intervention and control groups. Overall, there were very few cases of malformations, with more – although not significantly more – in the control groups.
Evidence related to other infant and maternal harms was “sparsely and inconsistently reported,” Dr. Henderson and coauthors noted, “and there was a lack of evidence on long-term neonatal outcomes after antibiotic treatment of asymptomatic bacteriuria in pregnancy.”
Two studies listed maternal adverse events associated with different treatments including vaginitis and diarrhea with ampicillin and rashes and nausea with nalidixic acid.
In terms of nonpregnant women and men, four studies reported adverse events. None occurred with nitrofurantoin or trimethoprim treatment; however, one study that included daily treatment with ofloxacin noted that 6% withdrew because of adverse events – vertigo and gastrointestinal symptoms.
Treatments didn’t affect hematocrit, bilirubin, serum urea, or nitrogen, although some studies found a slight reduction in serum creatinine.
Although there’s a need for additional research into this question, the new recommendations provide a good reason to further reduce unnecessary antibiotic exposure, Lindsey E. Nicolle, MD, wrote in a second commentary.
These updated recommendations “contribute to the evolution of management of asymptomatic bacteriuria in healthy women,” wrote Dr. Nicolle of the University of Manitoba, Winnipeg. “However, questions remain about the risks and benefits of universal screening for and treatment of asymptomatic bacteriuria in pregnant women in the context of current clinical practice. The effects of changes in fetal-maternal care, of low- compared with high-risk pregnancies, and of health care access need to be understood. In the short term, application of current diagnostic recommendations for identification of persistent symptomatic bacteriuria with a second urine culture may provide an immediate opportunity to limit unnecessary antimicrobial use for some pregnant women.”
No conflicts of interest were reported by the USPSTF authors, nor by Dr. Leis, Dr. Soong, or Dr. Nicolle. The USPSTF report was funded by the Agency for Healthcare Research and Quality.
SOURCES: U.S. Preventive Services Task Force. JAMA. 2019;322(12):1188-94; Henderson JT et al. JAMA. 2019;322(12):1195-205; Leis JA and Soong C. JAMA. 2019. doi: 10.1001/jamainternmed.2019.4515; Nicolle LE. JAMA. 2019;322(12):1152-4.
according to new recommendations set forth by the United States Preventive Services Task Force (USPSTF).
However, the investigating committee reported, there is evidence against screening nonpregnant women and adult men. In fact, the committee found “adequate” evidence of potential harm associated with treating asymptomatic bacteriuria in adults of both sexes, including adverse effects of antibiotics and on the microbiome.
The new document downgrades from A to B the group’s prior recommendation that urine culture screening for asymptomatic bacteriuria should be performed among pregnant women at 12-16 weeks’ gestation or at their first prenatal visit. The USPSTF recommendation to not screen nonpregnant adults retained its D rating, Jerome A. Leis, MD and Christine Soong, MD said in an accompanying editorial.
“Not screening or treating asymptomatic bacteriuria in this population has long been an ironclad recommendation endorsed by the Infectious Diseases Society of America, as well as numerous professional societies as part of the Choosing Wisely campaign,” wrote Dr. Leis of Sunnybrook Health Sciences Centre, Toronto, and Dr. Soong of the University of Toronto. “Restating this steadfast and pervasive recommendation may seem unremarkable and almost pedantic, yet it remains stubbornly disregarded by clinicians across multiple settings.”
The new recommendations were based on a review of 19 studies involving almost 8,500 pregnant and nonpregnant women, as well as a small number of adult men. Most were carried out in the 1960s or 1970s. The most recent ones were published in 2002 and 2015. The dearth of more recent data may have limited some conclusions and certainly highlighted the need for more research, said Jillian T. Henderson, PhD, chair of the committee assigned to investigate the evidence.
“Few studies of asymptomatic bacteriuria screening or treatment in pregnant populations have been conducted in the past 40 years,” wrote Dr. Henderson of Kaiser Permanente Northwest, Portland, and associates. “Historical evidence established asymptomatic bacteriuria screening and treatment as standard obstetric practice in the United States.” But these trials typically were less rigorous than modern studies, and the results are out of touch with modern clinical settings and treatment protocols, the team noted.
Additionally, Dr. Henderson and coauthors said, rates of pyelonephritis were about 10 times higher then than they are now. In the more recent studies, pyelonephritis rates in control groups were 2.2% and 2.5%; in most of the older studies, control group rates ranged from 33% to 36%.
In commissioning the investigation, the task force looked at the following four questions:
Does screening improve health outcomes?
Neither of two studies involving 5,289 women, one from Spain and one from Turkey, addressed this question in nonpregnant women; however, studies that looked at pregnant women generally found that screening did reduce the risk of pyelonephritis by about 70%. The investigators cautioned that these studies were out of date and perhaps methodologically flawed.
The only study that looked at newborn outcomes found no difference in birth weights or premature births between the screened and unscreened cohorts.
No study examined this question in nonpregnant women or men.
What are the harms of such screening?
A single study of 372 pregnant women described potential prenatal and perinatal harms associated with screening and treatment. It found a slight increase in congenital abnormalities in the screened cohort (1.6%), compared with those who were not screened (1.1%). However, those who were not screened were presumably not prescribed antibiotics.
Does treatment of screening-detected asymptomatic bacteriuria improve health outcomes?
Twelve trials of pregnant women (2,377) addressed this issue. All but two were conducted in the 1960s and 1970s. Treatment varied widely; sulfonamides were the most common, including the now discarded sulfamethazine and sulfadimethoxine. Dosages and duration of treatment also were considerably higher and longer than current practice.
In all but one study, there were higher rates of pyelonephritis in the control group. A pooled risk analysis indicated that treatment reduced the risk of pyelonephritis by nearly 80% (relative risk, 0.24).
Seven studies found higher rates of low birth weight in infants born to mothers who were treated, but two studies reported a significant reduction in the risk of low birth weight.
Among the six trials that examined perinatal mortality, none found significant associations with treatment.
Five studies examined treatment in nonpregnant women with screening-detected asymptomatic bacteriuria, and one included men as well. Of the four that reported the rate of symptomatic infection or pyelonephritis, none found a significant difference between treatment and control groups. The single study that included men also found no significant difference between treatment and control groups.
Among the three studies that focused on older adults, there also were no significant between-group differences in outcomes.
What harms are associated with treatment of screening-detected asymptomatic bacteriuria?
Seven studies comprised pregnant women. Five reported congenital malformations in the intervention and control groups. Overall, there were very few cases of malformations, with more – although not significantly more – in the control groups.
Evidence related to other infant and maternal harms was “sparsely and inconsistently reported,” Dr. Henderson and coauthors noted, “and there was a lack of evidence on long-term neonatal outcomes after antibiotic treatment of asymptomatic bacteriuria in pregnancy.”
Two studies listed maternal adverse events associated with different treatments including vaginitis and diarrhea with ampicillin and rashes and nausea with nalidixic acid.
In terms of nonpregnant women and men, four studies reported adverse events. None occurred with nitrofurantoin or trimethoprim treatment; however, one study that included daily treatment with ofloxacin noted that 6% withdrew because of adverse events – vertigo and gastrointestinal symptoms.
Treatments didn’t affect hematocrit, bilirubin, serum urea, or nitrogen, although some studies found a slight reduction in serum creatinine.
Although there’s a need for additional research into this question, the new recommendations provide a good reason to further reduce unnecessary antibiotic exposure, Lindsey E. Nicolle, MD, wrote in a second commentary.
These updated recommendations “contribute to the evolution of management of asymptomatic bacteriuria in healthy women,” wrote Dr. Nicolle of the University of Manitoba, Winnipeg. “However, questions remain about the risks and benefits of universal screening for and treatment of asymptomatic bacteriuria in pregnant women in the context of current clinical practice. The effects of changes in fetal-maternal care, of low- compared with high-risk pregnancies, and of health care access need to be understood. In the short term, application of current diagnostic recommendations for identification of persistent symptomatic bacteriuria with a second urine culture may provide an immediate opportunity to limit unnecessary antimicrobial use for some pregnant women.”
No conflicts of interest were reported by the USPSTF authors, nor by Dr. Leis, Dr. Soong, or Dr. Nicolle. The USPSTF report was funded by the Agency for Healthcare Research and Quality.
SOURCES: U.S. Preventive Services Task Force. JAMA. 2019;322(12):1188-94; Henderson JT et al. JAMA. 2019;322(12):1195-205; Leis JA and Soong C. JAMA. 2019. doi: 10.1001/jamainternmed.2019.4515; Nicolle LE. JAMA. 2019;322(12):1152-4.
FROM JAMA
Juvenile dermatomyositis derails growth and pubertal development
Children with juvenile dermatomyositis showed significant growth failure and pubertal delay, based on data from a longitudinal cohort study.

“Both the inflammatory activity of this severe chronic rheumatic disease and the well-known side effects of corticosteroid treatment may interfere with normal growth and pubertal development of children,” wrote Ellen Nordal, MD, of the University Hospital of Northern Norway, Tromsø, and colleagues.
The goal in treating juvenile dermatomyositis (JDM) is to achieve inactive disease and prevent permanent damage, but long-term data on growth and puberty in JDM patients are limited, they wrote.
In a study published in Arthritis Care & Research, the investigators reviewed data from 196 children and followed them for 2 years. The patients were part of the Paediatric Rheumatology International Trials Organisation (PRINTO) observational cohort study.
Overall, the researchers identified growth failure, height deflection, and/or delayed puberty in 94 children (48%) at the last study visit.
Growth failure was present at baseline in 17% of girls and 10% of boys. Over the 2-year study period, height deflection increased to 25% of girls and 31% of boys, but this change was not significant. Height deflection was defined as a change in the height z score of less than –0.25 per year from baseline. However, body mass index increased significantly from baseline during the study.
Catch-up growth had occurred by the final study visit in some patients, based on parent-adjusted z scores over time. Girls with a disease duration of 12 months or more showed no catch-up growth at 2 years and had significantly lower parent-adjusted height z scores.
In addition, the researchers observed a delay in the onset of puberty (including pubertal tempo and menarche) in approximately 36% of both boys and girls. However, neither growth failure nor height deflection was significantly associated with delayed puberty in either sex.
“In follow-up, clinicians should therefore be aware of both the pubertal development and the growth of the child, assess the milestones of development, and ensure that the children reach as much as possible of their genetic potential,” the researchers wrote.
The study participants were younger than 18 years at study enrollment, and all were in an active disease phase, defined as needing to start or receive a major dose increase of corticosteroids and/or immunosuppressants. Patients were assessed at baseline, at 6 months and/or at 12 months, and during a final visit at approximately 26 months. During the study, approximately half of the participants (50.5%) received methotrexate, 30 (15.3%) received cyclosporine A, 10 (5.1%) received cyclophosphamide, and 27 (13.8%) received intravenous immunoglobulin.
The study findings were limited by several factors, including the short follow-up period for assessing pubertal development and the inability to analyze any impact of corticosteroid use prior to the study, the researchers noted. However, “the overall frequency of growth failure was not significantly higher at the final study visit 2 years after baseline, indicating that the very high doses of corticosteroid treatment given during the study period is reasonably well tolerated with regards to growth,” they wrote. But monitoring remains essential, especially for children with previous growth failure or with disease onset early in pubertal development.
The study was supported by the European Union, Helse Nord Research grants, and by IRCCS Istituto Giannina Gaslini. Five authors of the study reported financial relationships with pharmaceutical companies.
SOURCE: Nordal E et al. Arthritis Care Res. 2019 Sep 10. doi: 10.1002/acr.24065.
Children with juvenile dermatomyositis showed significant growth failure and pubertal delay, based on data from a longitudinal cohort study.
“Both the inflammatory activity of this severe chronic rheumatic disease and the well-known side effects of corticosteroid treatment may interfere with normal growth and pubertal development of children,” wrote Ellen Nordal, MD, of the University Hospital of Northern Norway, Tromsø, and colleagues.
The goal in treating juvenile dermatomyositis (JDM) is to achieve inactive disease and prevent permanent damage, but long-term data on growth and puberty in JDM patients are limited, they wrote.
In a study published in Arthritis Care & Research, the investigators reviewed data from 196 children and followed them for 2 years. The patients were part of the Paediatric Rheumatology International Trials Organisation (PRINTO) observational cohort study.
Overall, the researchers identified growth failure, height deflection, and/or delayed puberty in 94 children (48%) at the last study visit.
Growth failure was present at baseline in 17% of girls and 10% of boys. Over the 2-year study period, height deflection increased to 25% of girls and 31% of boys, but this change was not significant. Height deflection was defined as a change in the height z score of less than –0.25 per year from baseline. However, body mass index increased significantly from baseline during the study.
Catch-up growth had occurred by the final study visit in some patients, based on parent-adjusted z scores over time. Girls with a disease duration of 12 months or more showed no catch-up growth at 2 years and had significantly lower parent-adjusted height z scores.
In addition, the researchers observed a delay in the onset of puberty (including pubertal tempo and menarche) in approximately 36% of both boys and girls. However, neither growth failure nor height deflection was significantly associated with delayed puberty in either sex.
“In follow-up, clinicians should therefore be aware of both the pubertal development and the growth of the child, assess the milestones of development, and ensure that the children reach as much as possible of their genetic potential,” the researchers wrote.
The study participants were younger than 18 years at study enrollment, and all were in an active disease phase, defined as needing to start or receive a major dose increase of corticosteroids and/or immunosuppressants. Patients were assessed at baseline, at 6 months and/or at 12 months, and during a final visit at approximately 26 months. During the study, approximately half of the participants (50.5%) received methotrexate, 30 (15.3%) received cyclosporine A, 10 (5.1%) received cyclophosphamide, and 27 (13.8%) received intravenous immunoglobulin.
The study findings were limited by several factors, including the short follow-up period for assessing pubertal development and the inability to analyze any impact of corticosteroid use prior to the study, the researchers noted. However, “the overall frequency of growth failure was not significantly higher at the final study visit 2 years after baseline, indicating that the very high doses of corticosteroid treatment given during the study period is reasonably well tolerated with regards to growth,” they wrote. But monitoring remains essential, especially for children with previous growth failure or with disease onset early in pubertal development.
The study was supported by the European Union, Helse Nord Research grants, and by IRCCS Istituto Giannina Gaslini. Five authors of the study reported financial relationships with pharmaceutical companies.
SOURCE: Nordal E et al. Arthritis Care Res. 2019 Sep 10. doi: 10.1002/acr.24065.
Children with juvenile dermatomyositis showed significant growth failure and pubertal delay, based on data from a longitudinal cohort study.
“Both the inflammatory activity of this severe chronic rheumatic disease and the well-known side effects of corticosteroid treatment may interfere with normal growth and pubertal development of children,” wrote Ellen Nordal, MD, of the University Hospital of Northern Norway, Tromsø, and colleagues.
The goal in treating juvenile dermatomyositis (JDM) is to achieve inactive disease and prevent permanent damage, but long-term data on growth and puberty in JDM patients are limited, they wrote.
In a study published in Arthritis Care & Research, the investigators reviewed data from 196 children and followed them for 2 years. The patients were part of the Paediatric Rheumatology International Trials Organisation (PRINTO) observational cohort study.
Overall, the researchers identified growth failure, height deflection, and/or delayed puberty in 94 children (48%) at the last study visit.
Growth failure was present at baseline in 17% of girls and 10% of boys. Over the 2-year study period, height deflection increased to 25% of girls and 31% of boys, but this change was not significant. Height deflection was defined as a change in the height z score of less than –0.25 per year from baseline. However, body mass index increased significantly from baseline during the study.
Catch-up growth had occurred by the final study visit in some patients, based on parent-adjusted z scores over time. Girls with a disease duration of 12 months or more showed no catch-up growth at 2 years and had significantly lower parent-adjusted height z scores.
In addition, the researchers observed a delay in the onset of puberty (including pubertal tempo and menarche) in approximately 36% of both boys and girls. However, neither growth failure nor height deflection was significantly associated with delayed puberty in either sex.
“In follow-up, clinicians should therefore be aware of both the pubertal development and the growth of the child, assess the milestones of development, and ensure that the children reach as much as possible of their genetic potential,” the researchers wrote.
The study participants were younger than 18 years at study enrollment, and all were in an active disease phase, defined as needing to start or receive a major dose increase of corticosteroids and/or immunosuppressants. Patients were assessed at baseline, at 6 months and/or at 12 months, and during a final visit at approximately 26 months. During the study, approximately half of the participants (50.5%) received methotrexate, 30 (15.3%) received cyclosporine A, 10 (5.1%) received cyclophosphamide, and 27 (13.8%) received intravenous immunoglobulin.
The study findings were limited by several factors, including the short follow-up period for assessing pubertal development and the inability to analyze any impact of corticosteroid use prior to the study, the researchers noted. However, “the overall frequency of growth failure was not significantly higher at the final study visit 2 years after baseline, indicating that the very high doses of corticosteroid treatment given during the study period is reasonably well tolerated with regards to growth,” they wrote. But monitoring remains essential, especially for children with previous growth failure or with disease onset early in pubertal development.
The study was supported by the European Union, Helse Nord Research grants, and by IRCCS Istituto Giannina Gaslini. Five authors of the study reported financial relationships with pharmaceutical companies.
SOURCE: Nordal E et al. Arthritis Care Res. 2019 Sep 10. doi: 10.1002/acr.24065.
FROM ARTHRITIS CARE & RESEARCH
New genotype of S. pyrogenes found in rise of scarlet fever in U.K.
A new Streptococcus pyogenes genotype (designated M1UK) emerged in 2014 in England causing an increase in scarlet fever “unprecedented in modern times.” Researchers discovered that this new genotype became dominant during this increased period of scarlet fever. This new genotype was characterized by an increased production of streptococcal pyrogenic exotoxin A (SpeA, also known as scarlet fever or erythrogenic toxin A) compared to previous isolates, according to a report in The Lancet Infectious Diseases.
_web.jpg)
The researchers analyzed changes in S. pyogenes emm1 genotypes sampled from scarlet fever and invasive disease cases in 2014-2016. The emm1 gene encodes the cell surface M virulence protein and is used for serotyping S. pyogenes isolates. Using regional (northwest London) and national (England and Wales) data, they compared genomes of 135 noninvasive and 552 invasive emm1 isolates from 2009-2016 with 2,800 global emm1 sequences.
During the increase in scarlet fever and invasive disease, emm1 S. pyogenes upper respiratory tract isolates increased significantly in northwest London during the March to May periods over 3 years from 5% of isolates in 2014 to 19% isolates in 2015 to 33% isolates in 2016. Similarly, invasive emm1 isolates collected nationally in the same period increased from 31% of isolates in 2015 to 42% in 2016 (P less than .0001). Sequences of emm1 isolates from 2009-2016 showed emergence of a new emm1 lineage (designated M1UK), which could be genotypically distinguished from pandemic emm1 isolates (M1global) by 27 single-nucleotide polymorphisms. In addition, the median SpeA protein concentration was 9 times greater among M1UK isolates than among M1global isolates. By 2016, M1UK expanded nationally to comprise 84% of all emm1 genomes tested. Dataset analysis also identified single M1UK isolates present in Denmark and the United States.
“The expansion of such a lineage within the community reservoir of S. pyogenes might be sufficient to explain England’s recent increase in invasive infection. Further research to assess the likely effects of M1UK on infection transmissibility, treatment response, disease burden, and severity is required, coupled with consideration of public health interventions to limit transmission where appropriate,” Dr. Lynskey and colleagues concluded.
The authors reported that they had no disclosures.
SOURCE: Linskey NN et al. Lancet Infect Dis. 2019. doi: 10.1016/S1473-3099(19)30446-3.
A new Streptococcus pyogenes genotype (designated M1UK) emerged in 2014 in England causing an increase in scarlet fever “unprecedented in modern times.” Researchers discovered that this new genotype became dominant during this increased period of scarlet fever. This new genotype was characterized by an increased production of streptococcal pyrogenic exotoxin A (SpeA, also known as scarlet fever or erythrogenic toxin A) compared to previous isolates, according to a report in The Lancet Infectious Diseases.
The researchers analyzed changes in S. pyogenes emm1 genotypes sampled from scarlet fever and invasive disease cases in 2014-2016. The emm1 gene encodes the cell surface M virulence protein and is used for serotyping S. pyogenes isolates. Using regional (northwest London) and national (England and Wales) data, they compared genomes of 135 noninvasive and 552 invasive emm1 isolates from 2009-2016 with 2,800 global emm1 sequences.
During the increase in scarlet fever and invasive disease, emm1 S. pyogenes upper respiratory tract isolates increased significantly in northwest London during the March to May periods over 3 years from 5% of isolates in 2014 to 19% isolates in 2015 to 33% isolates in 2016. Similarly, invasive emm1 isolates collected nationally in the same period increased from 31% of isolates in 2015 to 42% in 2016 (P less than .0001). Sequences of emm1 isolates from 2009-2016 showed emergence of a new emm1 lineage (designated M1UK), which could be genotypically distinguished from pandemic emm1 isolates (M1global) by 27 single-nucleotide polymorphisms. In addition, the median SpeA protein concentration was 9 times greater among M1UK isolates than among M1global isolates. By 2016, M1UK expanded nationally to comprise 84% of all emm1 genomes tested. Dataset analysis also identified single M1UK isolates present in Denmark and the United States.
“The expansion of such a lineage within the community reservoir of S. pyogenes might be sufficient to explain England’s recent increase in invasive infection. Further research to assess the likely effects of M1UK on infection transmissibility, treatment response, disease burden, and severity is required, coupled with consideration of public health interventions to limit transmission where appropriate,” Dr. Lynskey and colleagues concluded.
The authors reported that they had no disclosures.
SOURCE: Linskey NN et al. Lancet Infect Dis. 2019. doi: 10.1016/S1473-3099(19)30446-3.
A new Streptococcus pyogenes genotype (designated M1UK) emerged in 2014 in England causing an increase in scarlet fever “unprecedented in modern times.” Researchers discovered that this new genotype became dominant during this increased period of scarlet fever. This new genotype was characterized by an increased production of streptococcal pyrogenic exotoxin A (SpeA, also known as scarlet fever or erythrogenic toxin A) compared to previous isolates, according to a report in The Lancet Infectious Diseases.
The researchers analyzed changes in S. pyogenes emm1 genotypes sampled from scarlet fever and invasive disease cases in 2014-2016. The emm1 gene encodes the cell surface M virulence protein and is used for serotyping S. pyogenes isolates. Using regional (northwest London) and national (England and Wales) data, they compared genomes of 135 noninvasive and 552 invasive emm1 isolates from 2009-2016 with 2,800 global emm1 sequences.
During the increase in scarlet fever and invasive disease, emm1 S. pyogenes upper respiratory tract isolates increased significantly in northwest London during the March to May periods over 3 years from 5% of isolates in 2014 to 19% isolates in 2015 to 33% isolates in 2016. Similarly, invasive emm1 isolates collected nationally in the same period increased from 31% of isolates in 2015 to 42% in 2016 (P less than .0001). Sequences of emm1 isolates from 2009-2016 showed emergence of a new emm1 lineage (designated M1UK), which could be genotypically distinguished from pandemic emm1 isolates (M1global) by 27 single-nucleotide polymorphisms. In addition, the median SpeA protein concentration was 9 times greater among M1UK isolates than among M1global isolates. By 2016, M1UK expanded nationally to comprise 84% of all emm1 genomes tested. Dataset analysis also identified single M1UK isolates present in Denmark and the United States.
“The expansion of such a lineage within the community reservoir of S. pyogenes might be sufficient to explain England’s recent increase in invasive infection. Further research to assess the likely effects of M1UK on infection transmissibility, treatment response, disease burden, and severity is required, coupled with consideration of public health interventions to limit transmission where appropriate,” Dr. Lynskey and colleagues concluded.
The authors reported that they had no disclosures.
SOURCE: Linskey NN et al. Lancet Infect Dis. 2019. doi: 10.1016/S1473-3099(19)30446-3.
FROM THE LANCET INFECTIOUS DISEASES
Key clinical point: An Streptococcus pyrogenes isolate with increased scarlet fever toxin production has become dominant.
Major finding: By 2016, M1UK expanded nationally to constitute 84% of all emm1 genomes tested.
Study details: Genomic comparison of 135 noninvasive and 552 invasive emm1 isolates from 2009-2016 with 2,800 global emm1 sequences.
Disclosures: The authors reported that they had no disclosures.
Source: Linskey NN et al. Lancet Infect Dis. 2019. doi: 10.1016/S1473-3099(19)30446-3.
Expert spotlights telltale clinical signs of xeroderma pigmentosum
AUSTIN, TEX. – If a child presents with acute (XP), a rare autosomal recessive disorder.

Other telltale symptoms of XP include the presence of skin cancer at an early age and a large number of skin cancers.
At the annual meeting of the Society for Pediatric Dermatology, John J. DiGiovanna, MD, described XP as a disorder of genomic instability, which has no cure. It’s caused by a mutation in genes XPA through XPG and the XP variant (XPV) gene. “The genome controls our genes, and UV rays damage DNA,” said Dr. DiGiovanna, who is a senior research physician at the National Cancer Institute’s Laboratory of Cancer and Biology and Genetics, Bethesda, Md. “This damage from UV radiation is similar to damage from chemical agents that form DNA adducts, such as cigarette smoke and certain chemotherapy agents such as cisplatinum.”
XP patients present with or without acute burning after minimal sun exposure, while children with both subtypes develop “freckling” by the time they reach 2 years of age. Dr. DiGiovanna pointed out that lentigo maligna lesions associated with XP resemble freckles at first glance, yet they vary in size, intensity, and border. Meanwhile, freckles in healthy patients are similar in size, are light tan in color, and have a regular border.
“The burning with minimal sun exposure that occurs during childhood leads to pigmentary changes, atrophy, xerosis, and telangiectasias,” he said. A follow-up analysis of 106 XP patients admitted to the National Institutes of Health between 1971 and 2009 found that patients were diagnosed with their first nonmelanoma skin cancer at a median age of 9 years, compared with age 67 among those in the general population (J Med Genet 2011 Mar;48[3]:168-76). “This is a 58-year decrease in age at risk, which is a 10,000-fold increase in skin cancer,” said Dr. DiGiovanna, who was one of the study authors.
Melanoma also occurs at an earlier age among XP patients – a median age of 22 years, compared with a median of 55 years in the general population. “In the general population, melanoma occurs at a younger age than nonmelanoma skin cancer, while in the XP population, melanoma occurs at an older age,” he said. “This is giving us a good biologic lesson that the melanoma induction mechanism must be different from nonmelanoma skin cancer.”
He recalled one XP patient who was followed by NIH researchers for 4 decades. She worked in a doctor’s office and drove a car, but developed progressive neurologic degeneration and died at the age of 40. “This was not due to unrepaired UV damage, but there are other agents which damage other neurons,” Dr. DiGiovanna explained. “Over time, what you get is a decrease in brain volume, an increase in the brain ventricles, and a loss of brain tissue. At postmortem examination, her brain was of infantile size, compared with that of an equivalent 40-year-old. This is a disease of neuronal loss, and it’s progressive. Only about 20%-25% of XP patients experience neural degeneration.”
Management of XP involves strict sun avoidance, including use of a portable UV meter and many layers of UV protection, including application of sunscreen, wearing protective clothing, sunglasses, hats, and face shields, and the use of UV-blocking window film, LED lights, and a vitamin D diet or oral supplementation. Affected individuals also require frequent skin monitoring by the patients and their family members, frequent dermatologic exams by clinicians, biopsy of suspicious lesions, removal of any skin cancers found, field treatments with agents such as 5-fluorouracil and imiquimod, and chemoprevention with oral retinoids for patients who are actively developing large numbers of new lesions (N Engl J Med. 1988 Jun23;318[25]:1633-7).
“Probably the most important thing you can do is refer them to patient support groups,” Dr. DiGiovanna said. “They are present in many countries and can help them manage the day-to-day issues of their condition.” Support groups based in North America include the XP Family Support Group, XP Society, and XP Grupo Luz De Esperanza.
Dr. DiGiovanna reported having no financial disclosures.
AUSTIN, TEX. – If a child presents with acute (XP), a rare autosomal recessive disorder.
Other telltale symptoms of XP include the presence of skin cancer at an early age and a large number of skin cancers.
At the annual meeting of the Society for Pediatric Dermatology, John J. DiGiovanna, MD, described XP as a disorder of genomic instability, which has no cure. It’s caused by a mutation in genes XPA through XPG and the XP variant (XPV) gene. “The genome controls our genes, and UV rays damage DNA,” said Dr. DiGiovanna, who is a senior research physician at the National Cancer Institute’s Laboratory of Cancer and Biology and Genetics, Bethesda, Md. “This damage from UV radiation is similar to damage from chemical agents that form DNA adducts, such as cigarette smoke and certain chemotherapy agents such as cisplatinum.”
XP patients present with or without acute burning after minimal sun exposure, while children with both subtypes develop “freckling” by the time they reach 2 years of age. Dr. DiGiovanna pointed out that lentigo maligna lesions associated with XP resemble freckles at first glance, yet they vary in size, intensity, and border. Meanwhile, freckles in healthy patients are similar in size, are light tan in color, and have a regular border.
“The burning with minimal sun exposure that occurs during childhood leads to pigmentary changes, atrophy, xerosis, and telangiectasias,” he said. A follow-up analysis of 106 XP patients admitted to the National Institutes of Health between 1971 and 2009 found that patients were diagnosed with their first nonmelanoma skin cancer at a median age of 9 years, compared with age 67 among those in the general population (J Med Genet 2011 Mar;48[3]:168-76). “This is a 58-year decrease in age at risk, which is a 10,000-fold increase in skin cancer,” said Dr. DiGiovanna, who was one of the study authors.
Melanoma also occurs at an earlier age among XP patients – a median age of 22 years, compared with a median of 55 years in the general population. “In the general population, melanoma occurs at a younger age than nonmelanoma skin cancer, while in the XP population, melanoma occurs at an older age,” he said. “This is giving us a good biologic lesson that the melanoma induction mechanism must be different from nonmelanoma skin cancer.”
He recalled one XP patient who was followed by NIH researchers for 4 decades. She worked in a doctor’s office and drove a car, but developed progressive neurologic degeneration and died at the age of 40. “This was not due to unrepaired UV damage, but there are other agents which damage other neurons,” Dr. DiGiovanna explained. “Over time, what you get is a decrease in brain volume, an increase in the brain ventricles, and a loss of brain tissue. At postmortem examination, her brain was of infantile size, compared with that of an equivalent 40-year-old. This is a disease of neuronal loss, and it’s progressive. Only about 20%-25% of XP patients experience neural degeneration.”
Management of XP involves strict sun avoidance, including use of a portable UV meter and many layers of UV protection, including application of sunscreen, wearing protective clothing, sunglasses, hats, and face shields, and the use of UV-blocking window film, LED lights, and a vitamin D diet or oral supplementation. Affected individuals also require frequent skin monitoring by the patients and their family members, frequent dermatologic exams by clinicians, biopsy of suspicious lesions, removal of any skin cancers found, field treatments with agents such as 5-fluorouracil and imiquimod, and chemoprevention with oral retinoids for patients who are actively developing large numbers of new lesions (N Engl J Med. 1988 Jun23;318[25]:1633-7).
“Probably the most important thing you can do is refer them to patient support groups,” Dr. DiGiovanna said. “They are present in many countries and can help them manage the day-to-day issues of their condition.” Support groups based in North America include the XP Family Support Group, XP Society, and XP Grupo Luz De Esperanza.
Dr. DiGiovanna reported having no financial disclosures.
AUSTIN, TEX. – If a child presents with acute (XP), a rare autosomal recessive disorder.
Other telltale symptoms of XP include the presence of skin cancer at an early age and a large number of skin cancers.
At the annual meeting of the Society for Pediatric Dermatology, John J. DiGiovanna, MD, described XP as a disorder of genomic instability, which has no cure. It’s caused by a mutation in genes XPA through XPG and the XP variant (XPV) gene. “The genome controls our genes, and UV rays damage DNA,” said Dr. DiGiovanna, who is a senior research physician at the National Cancer Institute’s Laboratory of Cancer and Biology and Genetics, Bethesda, Md. “This damage from UV radiation is similar to damage from chemical agents that form DNA adducts, such as cigarette smoke and certain chemotherapy agents such as cisplatinum.”
XP patients present with or without acute burning after minimal sun exposure, while children with both subtypes develop “freckling” by the time they reach 2 years of age. Dr. DiGiovanna pointed out that lentigo maligna lesions associated with XP resemble freckles at first glance, yet they vary in size, intensity, and border. Meanwhile, freckles in healthy patients are similar in size, are light tan in color, and have a regular border.
“The burning with minimal sun exposure that occurs during childhood leads to pigmentary changes, atrophy, xerosis, and telangiectasias,” he said. A follow-up analysis of 106 XP patients admitted to the National Institutes of Health between 1971 and 2009 found that patients were diagnosed with their first nonmelanoma skin cancer at a median age of 9 years, compared with age 67 among those in the general population (J Med Genet 2011 Mar;48[3]:168-76). “This is a 58-year decrease in age at risk, which is a 10,000-fold increase in skin cancer,” said Dr. DiGiovanna, who was one of the study authors.
Melanoma also occurs at an earlier age among XP patients – a median age of 22 years, compared with a median of 55 years in the general population. “In the general population, melanoma occurs at a younger age than nonmelanoma skin cancer, while in the XP population, melanoma occurs at an older age,” he said. “This is giving us a good biologic lesson that the melanoma induction mechanism must be different from nonmelanoma skin cancer.”
He recalled one XP patient who was followed by NIH researchers for 4 decades. She worked in a doctor’s office and drove a car, but developed progressive neurologic degeneration and died at the age of 40. “This was not due to unrepaired UV damage, but there are other agents which damage other neurons,” Dr. DiGiovanna explained. “Over time, what you get is a decrease in brain volume, an increase in the brain ventricles, and a loss of brain tissue. At postmortem examination, her brain was of infantile size, compared with that of an equivalent 40-year-old. This is a disease of neuronal loss, and it’s progressive. Only about 20%-25% of XP patients experience neural degeneration.”
Management of XP involves strict sun avoidance, including use of a portable UV meter and many layers of UV protection, including application of sunscreen, wearing protective clothing, sunglasses, hats, and face shields, and the use of UV-blocking window film, LED lights, and a vitamin D diet or oral supplementation. Affected individuals also require frequent skin monitoring by the patients and their family members, frequent dermatologic exams by clinicians, biopsy of suspicious lesions, removal of any skin cancers found, field treatments with agents such as 5-fluorouracil and imiquimod, and chemoprevention with oral retinoids for patients who are actively developing large numbers of new lesions (N Engl J Med. 1988 Jun23;318[25]:1633-7).
“Probably the most important thing you can do is refer them to patient support groups,” Dr. DiGiovanna said. “They are present in many countries and can help them manage the day-to-day issues of their condition.” Support groups based in North America include the XP Family Support Group, XP Society, and XP Grupo Luz De Esperanza.
Dr. DiGiovanna reported having no financial disclosures.
REPORTING FROM SPD 2019
Painful and Pruritic Erosions on the Back
The Diagnosis: Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare blistering disease that affects patients with systemic lupus erythematosus (SLE). Our patient had a several-year history of SLE and was being managed by a rheumatologist. She was taking hydroxychloroquine at the time of the flare. Although BSLE tends to present in those with SLE that has already been diagnosed, BSLE has been reported as a possible initial manifestation of SLE.1
Bullous systemic lupus erythematosus is estimated to occur in less than 5% of patients with SLE and is more common in black women between the second and third decades of life,2 though it also can be seen in the pediatric population.3 The lesions of BSLE usually present as subepidermal blisters often located on the face, neck, and arms on an erythematous or possibly urticarial base. Although non-BSLE vesiculobullous eruptions may be seen in patients with SLE, BSLE is differentiated from these other eruptions by its appearance on sun-exposed and non-sun-exposed areas of the body, while other vesiculobullous eruptions associated with SLE typically are limited to sun-exposed sites.4
Due to its clinical presentation overlapping with several vesiculobullous conditions, a set of diagnostic criteria have been suggested for BSLE, including the following: (1) fulfillment of the American Rheumatism Association's criteria for SLE5; (2) a new-onset vesiculobullous eruption, primarily on sun-exposed skin; (3) histology showing a subepidermal blister with a predominantly neutrophilic infiltrate; (4) presence of IgG, IgA, IgM, and C3 at the basement membrane zone; (5) evidence of antibodies to type VII collagen; and (6) immunoelectron microscopy showing codistribution of immunoglobulin deposits with anchoring fibrils/type VII collagen. To meet the diagnosis of type I BSLE, all 6 criteria must be satisfied. To meet the diagnosis of type II BSLE, only criteria 1 to 4 need to be satisfied.6
Patients with BSLE may be presumed to have a different but clinically similar vesiculobullous condition (eg, bullous pemphigoid, cutaneous manifestations of SLE) and may be started on systemic corticosteroids. However, BSLE patients often do not show great improvement while on corticosteroids and may even flare shortly after beginning systemic corticosteroid treatment. The current treatment of choice for BSLE is dapsone, a sulfa drug that is thought to exhibit its anti-inflammatory properties via the inhibition of the alternative pathway of the complement system and through the inhibition of polymorphonuclear leukocyte functions.7 A response to dapsone helps differentiate BSLE from histopathologically and immunopathologically identical conditions such as epidermolysis bullosa acquisita.4 Bullous systemic lupus erythematosus can be differentiated from dermatitis herpetiformis with the presence of antigliadin and antitissue transglutaminase antibodies, which are found in the latter. Additionally, BSLE may show the presence of IgG and IgM deposition in addition to IgA deposition, as opposed to dermatitis herpetiformis where only IgA is found.8 The presence of these additional antibody depositions also help differentiate BSLE from linear IgA bullous dermatosis (LABD), as LABD will only have IgA depositions and often presents with an annular, crown of jewels-like appearance. Finally, there is a well-described phenomenon of LABD being drug induced, particularly after a course of vancomycin,9 and such an association with vancomycin has not been documented for BSLE.
Our patient was diagnosed with BSLE following the flare approximately 1.5 years prior to the current presentation. She had been started on dapsone 75 mg daily at that time and was taking 75 mg at the time of presentation. She was admitted and treated as an inpatient with high-dose (1 mg/kg) intravenous prednisone due to the extensive current flare.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Miziara ID, Mahmoud A, Chagury AA, et al. Bullous systemic lupus erythematosus: case report. Int Arch Otorhinolaryngol. 2013;17:344-346.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Grover C, Khurana A, Sharma S, et al. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58:492.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of RheumatologyClassification Criteria for Systemic Lupus Erythematosus [published online August 6, 2019]. Arthritis Rheumatol. 2019;71:1400-1412.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S.
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:6.
- Barbosa WS, Rodarte CM, Guerra JG, et al. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 suppl 1):S92-S95.
- Yordanova I, Valtchev V, Gospodinov D, et al. IgA linear bullous dermatosis in childhood. J IMAB. 2015;21:1012-1014.
The Diagnosis: Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare blistering disease that affects patients with systemic lupus erythematosus (SLE). Our patient had a several-year history of SLE and was being managed by a rheumatologist. She was taking hydroxychloroquine at the time of the flare. Although BSLE tends to present in those with SLE that has already been diagnosed, BSLE has been reported as a possible initial manifestation of SLE.1
Bullous systemic lupus erythematosus is estimated to occur in less than 5% of patients with SLE and is more common in black women between the second and third decades of life,2 though it also can be seen in the pediatric population.3 The lesions of BSLE usually present as subepidermal blisters often located on the face, neck, and arms on an erythematous or possibly urticarial base. Although non-BSLE vesiculobullous eruptions may be seen in patients with SLE, BSLE is differentiated from these other eruptions by its appearance on sun-exposed and non-sun-exposed areas of the body, while other vesiculobullous eruptions associated with SLE typically are limited to sun-exposed sites.4
Due to its clinical presentation overlapping with several vesiculobullous conditions, a set of diagnostic criteria have been suggested for BSLE, including the following: (1) fulfillment of the American Rheumatism Association's criteria for SLE5; (2) a new-onset vesiculobullous eruption, primarily on sun-exposed skin; (3) histology showing a subepidermal blister with a predominantly neutrophilic infiltrate; (4) presence of IgG, IgA, IgM, and C3 at the basement membrane zone; (5) evidence of antibodies to type VII collagen; and (6) immunoelectron microscopy showing codistribution of immunoglobulin deposits with anchoring fibrils/type VII collagen. To meet the diagnosis of type I BSLE, all 6 criteria must be satisfied. To meet the diagnosis of type II BSLE, only criteria 1 to 4 need to be satisfied.6
Patients with BSLE may be presumed to have a different but clinically similar vesiculobullous condition (eg, bullous pemphigoid, cutaneous manifestations of SLE) and may be started on systemic corticosteroids. However, BSLE patients often do not show great improvement while on corticosteroids and may even flare shortly after beginning systemic corticosteroid treatment. The current treatment of choice for BSLE is dapsone, a sulfa drug that is thought to exhibit its anti-inflammatory properties via the inhibition of the alternative pathway of the complement system and through the inhibition of polymorphonuclear leukocyte functions.7 A response to dapsone helps differentiate BSLE from histopathologically and immunopathologically identical conditions such as epidermolysis bullosa acquisita.4 Bullous systemic lupus erythematosus can be differentiated from dermatitis herpetiformis with the presence of antigliadin and antitissue transglutaminase antibodies, which are found in the latter. Additionally, BSLE may show the presence of IgG and IgM deposition in addition to IgA deposition, as opposed to dermatitis herpetiformis where only IgA is found.8 The presence of these additional antibody depositions also help differentiate BSLE from linear IgA bullous dermatosis (LABD), as LABD will only have IgA depositions and often presents with an annular, crown of jewels-like appearance. Finally, there is a well-described phenomenon of LABD being drug induced, particularly after a course of vancomycin,9 and such an association with vancomycin has not been documented for BSLE.
Our patient was diagnosed with BSLE following the flare approximately 1.5 years prior to the current presentation. She had been started on dapsone 75 mg daily at that time and was taking 75 mg at the time of presentation. She was admitted and treated as an inpatient with high-dose (1 mg/kg) intravenous prednisone due to the extensive current flare.
The Diagnosis: Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare blistering disease that affects patients with systemic lupus erythematosus (SLE). Our patient had a several-year history of SLE and was being managed by a rheumatologist. She was taking hydroxychloroquine at the time of the flare. Although BSLE tends to present in those with SLE that has already been diagnosed, BSLE has been reported as a possible initial manifestation of SLE.1
Bullous systemic lupus erythematosus is estimated to occur in less than 5% of patients with SLE and is more common in black women between the second and third decades of life,2 though it also can be seen in the pediatric population.3 The lesions of BSLE usually present as subepidermal blisters often located on the face, neck, and arms on an erythematous or possibly urticarial base. Although non-BSLE vesiculobullous eruptions may be seen in patients with SLE, BSLE is differentiated from these other eruptions by its appearance on sun-exposed and non-sun-exposed areas of the body, while other vesiculobullous eruptions associated with SLE typically are limited to sun-exposed sites.4
Due to its clinical presentation overlapping with several vesiculobullous conditions, a set of diagnostic criteria have been suggested for BSLE, including the following: (1) fulfillment of the American Rheumatism Association's criteria for SLE5; (2) a new-onset vesiculobullous eruption, primarily on sun-exposed skin; (3) histology showing a subepidermal blister with a predominantly neutrophilic infiltrate; (4) presence of IgG, IgA, IgM, and C3 at the basement membrane zone; (5) evidence of antibodies to type VII collagen; and (6) immunoelectron microscopy showing codistribution of immunoglobulin deposits with anchoring fibrils/type VII collagen. To meet the diagnosis of type I BSLE, all 6 criteria must be satisfied. To meet the diagnosis of type II BSLE, only criteria 1 to 4 need to be satisfied.6
Patients with BSLE may be presumed to have a different but clinically similar vesiculobullous condition (eg, bullous pemphigoid, cutaneous manifestations of SLE) and may be started on systemic corticosteroids. However, BSLE patients often do not show great improvement while on corticosteroids and may even flare shortly after beginning systemic corticosteroid treatment. The current treatment of choice for BSLE is dapsone, a sulfa drug that is thought to exhibit its anti-inflammatory properties via the inhibition of the alternative pathway of the complement system and through the inhibition of polymorphonuclear leukocyte functions.7 A response to dapsone helps differentiate BSLE from histopathologically and immunopathologically identical conditions such as epidermolysis bullosa acquisita.4 Bullous systemic lupus erythematosus can be differentiated from dermatitis herpetiformis with the presence of antigliadin and antitissue transglutaminase antibodies, which are found in the latter. Additionally, BSLE may show the presence of IgG and IgM deposition in addition to IgA deposition, as opposed to dermatitis herpetiformis where only IgA is found.8 The presence of these additional antibody depositions also help differentiate BSLE from linear IgA bullous dermatosis (LABD), as LABD will only have IgA depositions and often presents with an annular, crown of jewels-like appearance. Finally, there is a well-described phenomenon of LABD being drug induced, particularly after a course of vancomycin,9 and such an association with vancomycin has not been documented for BSLE.
Our patient was diagnosed with BSLE following the flare approximately 1.5 years prior to the current presentation. She had been started on dapsone 75 mg daily at that time and was taking 75 mg at the time of presentation. She was admitted and treated as an inpatient with high-dose (1 mg/kg) intravenous prednisone due to the extensive current flare.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Miziara ID, Mahmoud A, Chagury AA, et al. Bullous systemic lupus erythematosus: case report. Int Arch Otorhinolaryngol. 2013;17:344-346.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Grover C, Khurana A, Sharma S, et al. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58:492.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of RheumatologyClassification Criteria for Systemic Lupus Erythematosus [published online August 6, 2019]. Arthritis Rheumatol. 2019;71:1400-1412.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S.
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:6.
- Barbosa WS, Rodarte CM, Guerra JG, et al. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 suppl 1):S92-S95.
- Yordanova I, Valtchev V, Gospodinov D, et al. IgA linear bullous dermatosis in childhood. J IMAB. 2015;21:1012-1014.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Miziara ID, Mahmoud A, Chagury AA, et al. Bullous systemic lupus erythematosus: case report. Int Arch Otorhinolaryngol. 2013;17:344-346.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Grover C, Khurana A, Sharma S, et al. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58:492.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of RheumatologyClassification Criteria for Systemic Lupus Erythematosus [published online August 6, 2019]. Arthritis Rheumatol. 2019;71:1400-1412.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S.
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:6.
- Barbosa WS, Rodarte CM, Guerra JG, et al. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 suppl 1):S92-S95.
- Yordanova I, Valtchev V, Gospodinov D, et al. IgA linear bullous dermatosis in childhood. J IMAB. 2015;21:1012-1014.
A 51-year-old black woman presented to the dermatology clinic with painful and pruritic erosions on the back, abdomen, neck, and arms of approximately 2 months' duration. The lesions started on the back and spread in a cephalocaudal manner. The patient denied any new changes in medication. Physical examination revealed large erosions with mild weeping of serosanguineous fluid on the back, abdomen, neck, and upper extremities. A few tense bullae were present on the dorsal aspect of the right hand. She had experienced a similar flare approximately 1.5 years prior to the current presentation. At that time, 2 shave biopsies from vesiculobullous lesions on the right side of the neck were sent for hematoxylin and eosin staining and direct immunofluorescence. Biopsy results showed a subepidermal blister that extended along the course of the hair follicle and was associated with an infiltrate of neutrophilic granulocytes that also extended along the course of the hair follicle. Direct immunofluorescence showed IgG and C3 deposition in the basement membrane zone extending along the floor of the blister where the epidermis was separated from the dermis.
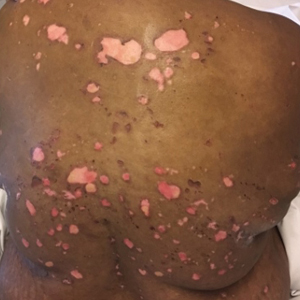
Can we eradicate malaria by 2050?
A new report by members of the Lancet Commission on Malaria Eradication has called for ending malaria in Africa within a generation, specifically aiming at the year 2050.
The Lancet Commission on Malaria Eradication is a joint endeavor between The Lancet and the University of California, San Francisco, and was convened in 2017 to consider the feasibility and affordability of malaria eradication, as well as to identify priority actions for the achievement of the goal. Eradication was considered “a necessary one given the never-ending struggle against drug and insecticide resistance and the social and economic costs associated with a failure to eradicate.”
Between 2000 and 2017, the worldwide annual incidence of malaria declined by 36%, and the annual death rate declined by 60%, according to the report. In 2007, Bill and Melinda Gates proposed that controlling malaria was not enough and complete eradication was the only scientifically and ethically defensible objective. This goal was adopted by the World Health Organization and other interested parties, and by 2015, global strategies and a potential timeline for eradication were developed.
“Global progress has stalled since 2015 and the malaria community is now at a critical moment, faced with a decision to either temper its ambitions as it did in 1969 or recommit to an eradication goal,” according to the report.
In the report, the authors used new modeling analysis to estimate plausible scenarios for the distribution and intensity of malaria in 2030 and 2050. Socioeconomic and environmental trends, together with enhanced access to high-quality diagnosis, treatment, and vector control, could lead to a “world largely free of malaria” by 2050, but with pockets of low-level transmission persisting across a belt of Africa.
Current statistics lend weight to the promise of eventual eradication, according to the report.
Between 2000 and 2017, 20 countries – constituting about one-fifth of the 106 malaria-endemic countries in 2000 – eliminated malaria transmission within their borders, reporting zero indigenous malaria cases for at least 1 year. However, this was counterbalanced by the fact that between 2015 and 2017, 55 countries had an increase in cases, and 38 countries had an increase in deaths.
“The good news is that 38 countries had incidences of fewer than ten cases per 1,000 population in 2017, with 25 countries reporting fewer than one case per 1,000 population. The same 38 countries reported just 5% of total malaria deaths. Nearly all of these low-burden countries are actively working towards national and regional elimination goals of 2030 or earlier,” according to the report.
The analysis undertaken for the report consisted of the following four steps:
1. Development of a machine-learning model to capture associations between malaria endemicity data and a wide range of socioeconomic and environmental geospatial covariates.
2. Mapping of covariate estimates to the years 2030 and 2050 on the basis of projected global trends.
3. Application of the associations learned in the first step to projected covariates generated in the second step to estimate the possible future global landscape of malaria endemicity.
4. Use of a mathematical transmission model to explore the potential effect of differing levels of malaria interventions.
The report indicates that an annual spending of $6 billion or more is required, while the current global expenditure is approximately $4.3 billion. An additional investment of $2 billion per year is necessary, with a quarter of the funds coming from increased development assistance from external donors and the rest from government health spending in malaria-endemic countries, according to the report.
However, other areas of concern remain, including the current lack of effective and widely deployable outdoor biting technologies, though these are expected to be available within the next decade, according to the report.
In terms of the modeling used in the report, the authors noted that past performance does not “capture the effect of mass drug administration or mass chemoprevention because these interventions are either relatively new or have yet to be applied widely. These underestimates might be counteracted by the absence of drug or insecticide resistance from our projections,which result in overly optimistic estimates for the continued efficacy of current tools.”
The commission was launched in October 2017 by the Global Health Group at the University of California, San Francisco. The commission built on the 2010 Lancet Malaria Elimination Series, “which evaluated the operational, technical, and financial requirements for malaria elimination and helped shape and build early support for the eradication agenda,” according to the report.
SOURCE: Feachem RGA et al. Lancet. 2019 Sept 8. doi: 10.1016/S0140-6736(19)31139-0.
A new report by members of the Lancet Commission on Malaria Eradication has called for ending malaria in Africa within a generation, specifically aiming at the year 2050.
The Lancet Commission on Malaria Eradication is a joint endeavor between The Lancet and the University of California, San Francisco, and was convened in 2017 to consider the feasibility and affordability of malaria eradication, as well as to identify priority actions for the achievement of the goal. Eradication was considered “a necessary one given the never-ending struggle against drug and insecticide resistance and the social and economic costs associated with a failure to eradicate.”
Between 2000 and 2017, the worldwide annual incidence of malaria declined by 36%, and the annual death rate declined by 60%, according to the report. In 2007, Bill and Melinda Gates proposed that controlling malaria was not enough and complete eradication was the only scientifically and ethically defensible objective. This goal was adopted by the World Health Organization and other interested parties, and by 2015, global strategies and a potential timeline for eradication were developed.
“Global progress has stalled since 2015 and the malaria community is now at a critical moment, faced with a decision to either temper its ambitions as it did in 1969 or recommit to an eradication goal,” according to the report.
In the report, the authors used new modeling analysis to estimate plausible scenarios for the distribution and intensity of malaria in 2030 and 2050. Socioeconomic and environmental trends, together with enhanced access to high-quality diagnosis, treatment, and vector control, could lead to a “world largely free of malaria” by 2050, but with pockets of low-level transmission persisting across a belt of Africa.
Current statistics lend weight to the promise of eventual eradication, according to the report.
Between 2000 and 2017, 20 countries – constituting about one-fifth of the 106 malaria-endemic countries in 2000 – eliminated malaria transmission within their borders, reporting zero indigenous malaria cases for at least 1 year. However, this was counterbalanced by the fact that between 2015 and 2017, 55 countries had an increase in cases, and 38 countries had an increase in deaths.
“The good news is that 38 countries had incidences of fewer than ten cases per 1,000 population in 2017, with 25 countries reporting fewer than one case per 1,000 population. The same 38 countries reported just 5% of total malaria deaths. Nearly all of these low-burden countries are actively working towards national and regional elimination goals of 2030 or earlier,” according to the report.
The analysis undertaken for the report consisted of the following four steps:
1. Development of a machine-learning model to capture associations between malaria endemicity data and a wide range of socioeconomic and environmental geospatial covariates.
2. Mapping of covariate estimates to the years 2030 and 2050 on the basis of projected global trends.
3. Application of the associations learned in the first step to projected covariates generated in the second step to estimate the possible future global landscape of malaria endemicity.
4. Use of a mathematical transmission model to explore the potential effect of differing levels of malaria interventions.
The report indicates that an annual spending of $6 billion or more is required, while the current global expenditure is approximately $4.3 billion. An additional investment of $2 billion per year is necessary, with a quarter of the funds coming from increased development assistance from external donors and the rest from government health spending in malaria-endemic countries, according to the report.
However, other areas of concern remain, including the current lack of effective and widely deployable outdoor biting technologies, though these are expected to be available within the next decade, according to the report.
In terms of the modeling used in the report, the authors noted that past performance does not “capture the effect of mass drug administration or mass chemoprevention because these interventions are either relatively new or have yet to be applied widely. These underestimates might be counteracted by the absence of drug or insecticide resistance from our projections,which result in overly optimistic estimates for the continued efficacy of current tools.”
The commission was launched in October 2017 by the Global Health Group at the University of California, San Francisco. The commission built on the 2010 Lancet Malaria Elimination Series, “which evaluated the operational, technical, and financial requirements for malaria elimination and helped shape and build early support for the eradication agenda,” according to the report.
SOURCE: Feachem RGA et al. Lancet. 2019 Sept 8. doi: 10.1016/S0140-6736(19)31139-0.
A new report by members of the Lancet Commission on Malaria Eradication has called for ending malaria in Africa within a generation, specifically aiming at the year 2050.
The Lancet Commission on Malaria Eradication is a joint endeavor between The Lancet and the University of California, San Francisco, and was convened in 2017 to consider the feasibility and affordability of malaria eradication, as well as to identify priority actions for the achievement of the goal. Eradication was considered “a necessary one given the never-ending struggle against drug and insecticide resistance and the social and economic costs associated with a failure to eradicate.”
Between 2000 and 2017, the worldwide annual incidence of malaria declined by 36%, and the annual death rate declined by 60%, according to the report. In 2007, Bill and Melinda Gates proposed that controlling malaria was not enough and complete eradication was the only scientifically and ethically defensible objective. This goal was adopted by the World Health Organization and other interested parties, and by 2015, global strategies and a potential timeline for eradication were developed.
“Global progress has stalled since 2015 and the malaria community is now at a critical moment, faced with a decision to either temper its ambitions as it did in 1969 or recommit to an eradication goal,” according to the report.
In the report, the authors used new modeling analysis to estimate plausible scenarios for the distribution and intensity of malaria in 2030 and 2050. Socioeconomic and environmental trends, together with enhanced access to high-quality diagnosis, treatment, and vector control, could lead to a “world largely free of malaria” by 2050, but with pockets of low-level transmission persisting across a belt of Africa.
Current statistics lend weight to the promise of eventual eradication, according to the report.
Between 2000 and 2017, 20 countries – constituting about one-fifth of the 106 malaria-endemic countries in 2000 – eliminated malaria transmission within their borders, reporting zero indigenous malaria cases for at least 1 year. However, this was counterbalanced by the fact that between 2015 and 2017, 55 countries had an increase in cases, and 38 countries had an increase in deaths.
“The good news is that 38 countries had incidences of fewer than ten cases per 1,000 population in 2017, with 25 countries reporting fewer than one case per 1,000 population. The same 38 countries reported just 5% of total malaria deaths. Nearly all of these low-burden countries are actively working towards national and regional elimination goals of 2030 or earlier,” according to the report.
The analysis undertaken for the report consisted of the following four steps:
1. Development of a machine-learning model to capture associations between malaria endemicity data and a wide range of socioeconomic and environmental geospatial covariates.
2. Mapping of covariate estimates to the years 2030 and 2050 on the basis of projected global trends.
3. Application of the associations learned in the first step to projected covariates generated in the second step to estimate the possible future global landscape of malaria endemicity.
4. Use of a mathematical transmission model to explore the potential effect of differing levels of malaria interventions.
The report indicates that an annual spending of $6 billion or more is required, while the current global expenditure is approximately $4.3 billion. An additional investment of $2 billion per year is necessary, with a quarter of the funds coming from increased development assistance from external donors and the rest from government health spending in malaria-endemic countries, according to the report.
However, other areas of concern remain, including the current lack of effective and widely deployable outdoor biting technologies, though these are expected to be available within the next decade, according to the report.
In terms of the modeling used in the report, the authors noted that past performance does not “capture the effect of mass drug administration or mass chemoprevention because these interventions are either relatively new or have yet to be applied widely. These underestimates might be counteracted by the absence of drug or insecticide resistance from our projections,which result in overly optimistic estimates for the continued efficacy of current tools.”
The commission was launched in October 2017 by the Global Health Group at the University of California, San Francisco. The commission built on the 2010 Lancet Malaria Elimination Series, “which evaluated the operational, technical, and financial requirements for malaria elimination and helped shape and build early support for the eradication agenda,” according to the report.
SOURCE: Feachem RGA et al. Lancet. 2019 Sept 8. doi: 10.1016/S0140-6736(19)31139-0.
FROM THE LANCET