User login
Neonatal Consultations: Vascular Lumps, Bumps, and Tumors in the Neonate
Although most neonatal vascular lumps, bumps, and tumors are benign, proper diagnosis is important for prognosis and management. Therefore, knowledge of both common and rare conditions is important when evaluating a neonatal nodule. Differential diagnosis of neonatal vascular nodules must focus on important diagnostic clues that should prompt consideration and evaluation for less common and/or potentially threatening conditions. Infantile hemangioma (IH), congenital hemangioma (CH), venous malformation (VM), lymphatic malformation (LM), kaposiform hemangioendothelioma (KHE) and tufted angioma, and malignant tumors are reviewed here.
Infantile Hemangioma
Infantile hemangioma, a benign proliferation of capillaries, is the most common tumor of infancy with reported incidence of up to 5% in neonates.1 As such, suspicion for less common lesions is often predicated on identifying features that would be atypical for an IH. A superficial IH presents as a bright red papule, nodule, or plaque, while a deep IH presents as a flesh-colored to bluish nodule. Mixed IHs combine features of both superficial and deep lesions. The distribution may be focal or segmental, with segmental lesions encompassing a larger territory–like distribution and frequently displaying a thin, coarsely telangiectatic appearance.
Knowledge of the natural history of IH generally is crucial in differentiating it from other neonatal lesions. Infantile hemangiomas display a natural history that is distinct and predictable. They typically manifest within the first few weeks of life, though up to 30% present at birth with a premonitory mark, which may be a light red, pink, bluish, or vasoconstricted patch. Thus, mere presence of a lesion at birth is not the feature that distinguishes other congenital lesions from an IH. After initial appearance, IHs undergo a period of proliferation that occurs over 4 to 6 months in most patients. In some cases, areas of proliferation may be subtle, but nonetheless the presence of some areas of increased redness and/or volumetric growth generally is required to firmly establish the diagnosis of IH. Thereafter, IH will involute, a process that begins before 1 year of age in most cases and continues over years. Although IHs undergo involution, complete clearance may not occur, as nearly 70% will leave permanent residua such as fibrofatty masses or anetodermic skin.2 Nevertheless, the presence of a proliferative phase followed by a slower period of involution is a hallmark feature of the IH.
Biopsy and imaging rarely are required for establishing diagnosis of an IH. Histopathology showing a proliferation of capillaries with positive glucose transporter 1 (GLUT-1) staining is characteristic. Imaging with ultrasound reveals a fast-flow lesion. Apart from exceptionally rare cases, a cutaneous IH typically does not cross muscle fascia, and thus alternative diagnoses should be considered for a cutaneous lesion that demonstrates infiltration into nerve, bone, joint, or other deeper tissues. Most IHs do not require treatment; however, a small subset may be associated with complications and thus require intervention. Complications of IH may include impairment of function (eg, vision, feeding, respiratory), ulceration, and risk for permanent disfigurement. When treatment is indicated, the most commonly employed options during the proliferative phase are the topical beta-blocker timolol and the oral beta-blocker propranolol. In addition, certain IHs may be associated with either syndromic presentations and/or visceral involvement, thus requiring further workup (Table).

Congenital Hemangioma
A CH is an uncommon benign neonatal tumor that is distinct from an IH in behavior, biology, and treatment. Congenital hemangiomas may have a rapidly involuting course, referred to as RICH (rapidly involuting congenital hemangioma), or a noninvoluting course, referred to as NICH (noninvoluting congenital hemangioma). Partially involuting types also have been described.3 A RICH typically presents as a highly vascular, red-violaceous or bluish plaque, nodule, or large mass at birth. An NICH presents as a red-violaceous or bluish, coarsely telangiectatic patch, plaque, or nodule. A characteristic feature of the CH is the rim of vasoconstriction around the lesion, which is an important diagnostic clue (Figure 1). In contrast to IH, multifocal lesions are highly unlikely in CH, though it rarely has been reported.4
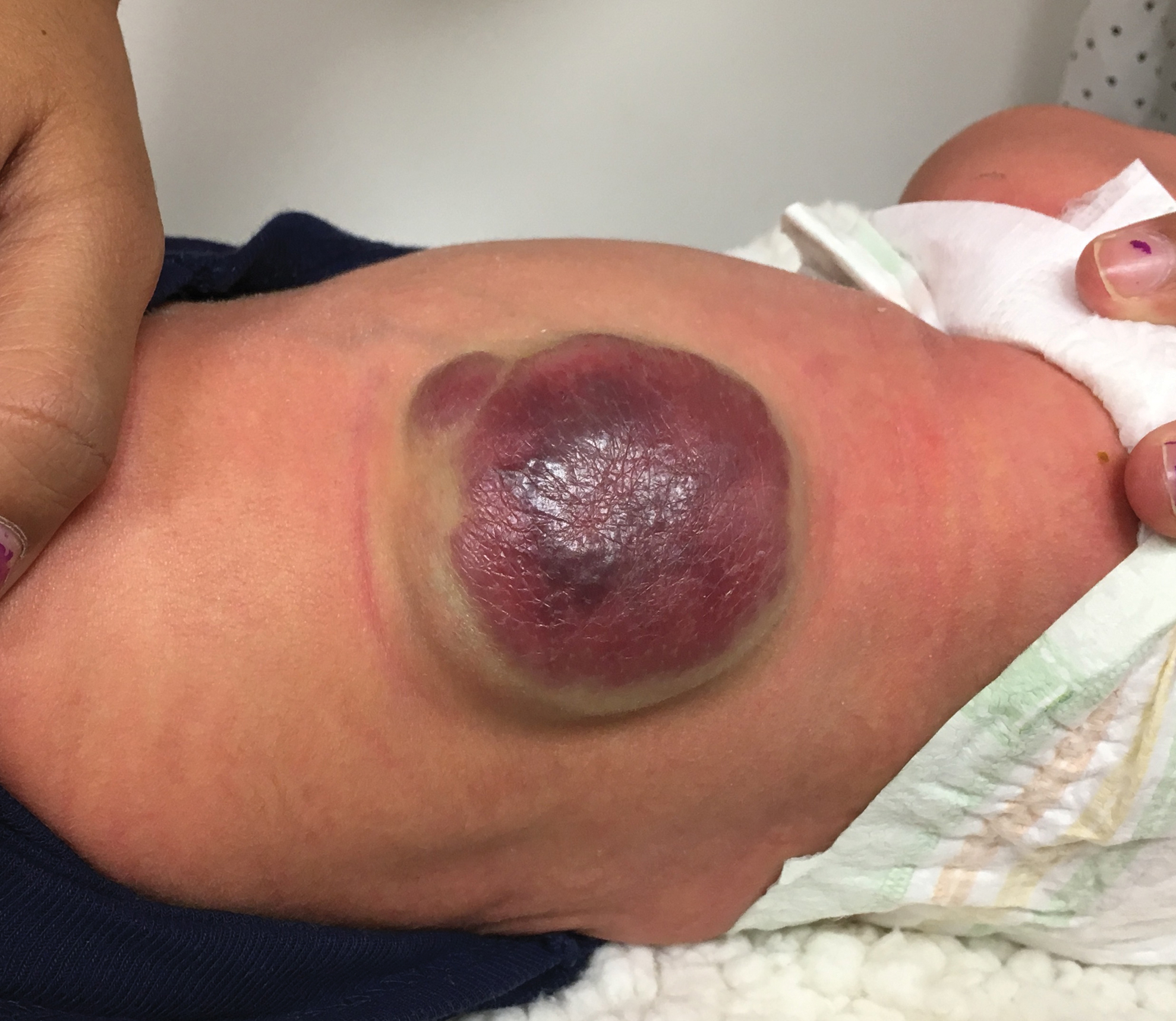
Regardless of subtype, CHs are fully developed at birth. Infantile hemangiomas, on the other hand, are either minimally present or not present at birth and thereafter proliferate. After birth, a RICH rapidly involutes over the first 9 to 12 months of life. This process generally is evident even in the first few weeks of life, which would not be expected of an IH and is therefore a major distinguishing factor. A NICH, on the other hand, is expected to be persistent, for the most part neither showing signs of proliferation nor involution.
Complications of CHs may include ulceration, functional impairment, or risk for permanent disfigurement depending on location. In addition, due to their fast-flow state and potential large size, some CHs may be complicated by high-output heart failure in the neonate. Distinguishing an IH from a CH is important not only for prognosis but also treatment. Beta-blocker therapy generally is not useful for CHs, and management usually is supportive in the neonatal period.
In the majority of cases, diagnosis can be achieved solely on clinical features. Biopsy with immunohistochemistry shows negative GLUT-1 staining, which will distinguish this lesion from an IH. At times, the highly vascular nature and/or striking size of a CH may lead some to consider the potential diagnosis of an arteriovenous malformation. However, soft-tissue arteriovenous malformations involving the skin are almost never fully developed in the neonatal period and generally take years to evolve from a quiescent state to a destructive lesion.
Venous Malformation
Venous malformations are congenital malformations of veins that may be apparent at birth or later. They appear as bluish to flesh-colored, compressible nodules or plaques. They tend to increase in size when the affected body part is in a dependent position, and this maneuver can be a helpful distinguishing clue. Although the majority of patients have a single lesion, multifocal involvement may occur uncommonly (Table). The diagnosis of VM usually is clinical, though at times, a VM may be difficult to distinguish from a purely deep IH. However, a VM will persist over time, growing in proportion to the patient. In addition, a VM displays low flow on ultrasound, a distinguishing feature from the fast-flow IH. Magnetic resonance imaging with and without contrast is the imaging study of choice. At times, cutaneous VMs will demonstrate infiltration into other tissue planes such as muscle and joint. Pain may occur secondary to thrombus formation within the malformation. In extensive lesions, intravascular coagulation may be notable, as reflected in elevated D-dimer and decreased fibrinogen levels. Treatment with sclerotherapy or surgery may be considered in select cases during infancy; however, in general, an asymptomatic VM may be observed early on in life.
Lymphatic Malformation
A lymphatic malformation (LM) is a congenital malformation of lymphatic vessels and may be further differentiated into microcystic, macrocystic, or mixed types depending on the size of the channels. An LM may present at birth or later and persists over time. Superficial microcystic LMs, synonymous with the term lymphangioma circumscriptum, characteristically appear as a group of clear and violaceous hemorrhagic vesicles on the skin. Deeper LMs appear as a tense or spongy, flesh-colored nodule or mass. Involvement of the head and neck is common. Complications frequently occur in LMs. Cutaneous LMs may ooze or bleed. Infection and hemorrhage into cysts may occur, which will cause acute pain, redness, swelling, and induration. Cervicofacial lesions may result in respiratory distress. Thus, the majority of LMs require treatment, though asymptomatic lesions may be observed in the neonate. An ultrasound will demonstrate a low-flow lesion, and magnetic resonance imaging is the diagnostic modality of choice for diagnosis and definition of extent.
KHE and Tufted Angioma
Kaposiform hemangioendothelioma is a rare, locally aggressive, vascular tumor that is frequently associated with a potentially life-threatening coagulopathy, Kasabach-Merritt phenomenon. Tufted angiomas are now understood to belong on a spectrum with KHEs, which usually present in the neonatal period or infancy as firm, red-violaceous plaques, nodules, or large tumors. Infiltration into nerve, muscle, and bone may occur. The firm/hard nature and deep violaceous appearance generally are initial clues that it is not an IH. Kasabach-Merritt phenomenon manifests as thrombocytopenia as well as low fibrinogen and elevated D-dimer levels. Thrombocytopenia is generally profound in Kasabach-Merritt phenomenon and results from platelet trapping within the vascular tumor. Given these potential complications, KHEs generally require immediate medical attention, and various treatment protocols including prednisone, vincristine, and sirolimus are utilized for complicated cases.5 The diagnosis may require biopsy to distinguish it from malignant tumors, particularly sarcomas.
Malignant Tumors
Various malignancies, including congenital leukemia, neuroblastoma, Langerhans cell histiocytosis, infantile fibrosarcoma, and rhabdomyosarcoma, rarely may present as cutaneous nodules or masses in a neonate mimicking hemangiomas or other vascular lesions (Figure 2). Neonates may present with multiple bluish papules and nodules resembling a blueberry muffin baby; multiple violaceous-red nodules; or a single red-violaceous, highly vascular–appearing mass mimicking hemangiomas. Malignant tumors may display vascularity on imaging, and thus the presence of vascular flow on ultrasound should not dissuade one from the possibility of a malignancy if other clinical features are atypical or unusual for a hemangioma. When a neonatal malignancy is suspected, a large punch biopsy or incisional biopsy is required for workup.
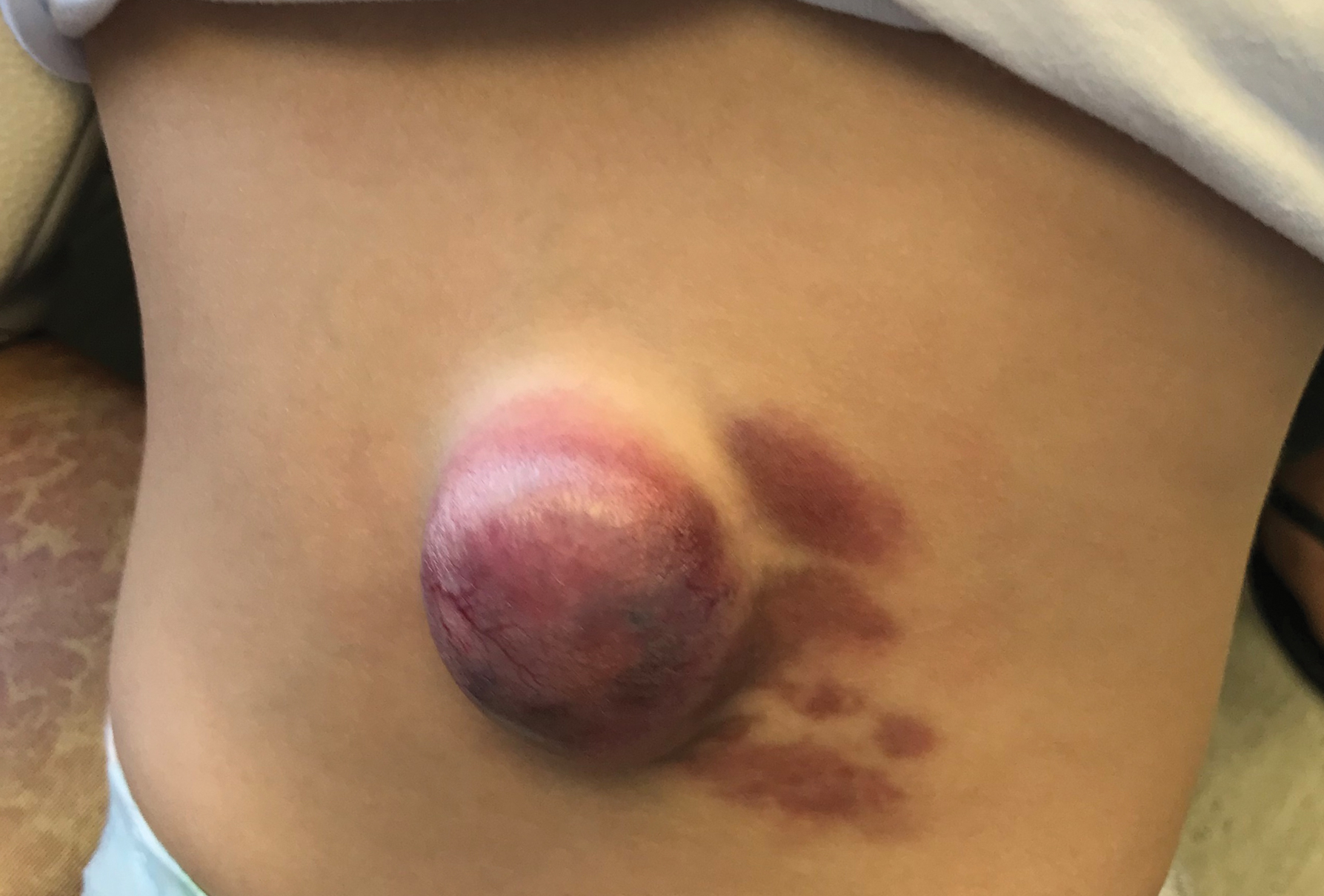
Final Thoughts
Although IHs are the most common vascular nodules in neonates and young infants, other conditions such as VMs, LMs, CHs, KHEs, and malignancy may occur less commonly. Identifying features that would be considered atypical for IH is crucial to recognize these less common possibilities.
- Kanada KN, Merin MR, Munden A, et al. A prospective study of cutaneous findings in newborns in the United States: correlation with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161:240-245.
- Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136:E1060-E1104.
- Nasseri E, Piram M, McCuaig CC, et al. Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol. 2014;70:75-79.
- Blumenthal S, Stefanko N, Cossio M, et al. Multifocal congenital hemangioma: expanding the pathogenesis of “neonatal hemangiomatosis.” Pediatr Dermatol. 2019;36:720-722.
- Croteau SE, Gupta D. The clinical spectrum of kaposiform hemangioendothelioma and tufted angioma. Semin Cutan Med Surg. 2016;35:147-152.
Although most neonatal vascular lumps, bumps, and tumors are benign, proper diagnosis is important for prognosis and management. Therefore, knowledge of both common and rare conditions is important when evaluating a neonatal nodule. Differential diagnosis of neonatal vascular nodules must focus on important diagnostic clues that should prompt consideration and evaluation for less common and/or potentially threatening conditions. Infantile hemangioma (IH), congenital hemangioma (CH), venous malformation (VM), lymphatic malformation (LM), kaposiform hemangioendothelioma (KHE) and tufted angioma, and malignant tumors are reviewed here.
Infantile Hemangioma
Infantile hemangioma, a benign proliferation of capillaries, is the most common tumor of infancy with reported incidence of up to 5% in neonates.1 As such, suspicion for less common lesions is often predicated on identifying features that would be atypical for an IH. A superficial IH presents as a bright red papule, nodule, or plaque, while a deep IH presents as a flesh-colored to bluish nodule. Mixed IHs combine features of both superficial and deep lesions. The distribution may be focal or segmental, with segmental lesions encompassing a larger territory–like distribution and frequently displaying a thin, coarsely telangiectatic appearance.
Knowledge of the natural history of IH generally is crucial in differentiating it from other neonatal lesions. Infantile hemangiomas display a natural history that is distinct and predictable. They typically manifest within the first few weeks of life, though up to 30% present at birth with a premonitory mark, which may be a light red, pink, bluish, or vasoconstricted patch. Thus, mere presence of a lesion at birth is not the feature that distinguishes other congenital lesions from an IH. After initial appearance, IHs undergo a period of proliferation that occurs over 4 to 6 months in most patients. In some cases, areas of proliferation may be subtle, but nonetheless the presence of some areas of increased redness and/or volumetric growth generally is required to firmly establish the diagnosis of IH. Thereafter, IH will involute, a process that begins before 1 year of age in most cases and continues over years. Although IHs undergo involution, complete clearance may not occur, as nearly 70% will leave permanent residua such as fibrofatty masses or anetodermic skin.2 Nevertheless, the presence of a proliferative phase followed by a slower period of involution is a hallmark feature of the IH.
Biopsy and imaging rarely are required for establishing diagnosis of an IH. Histopathology showing a proliferation of capillaries with positive glucose transporter 1 (GLUT-1) staining is characteristic. Imaging with ultrasound reveals a fast-flow lesion. Apart from exceptionally rare cases, a cutaneous IH typically does not cross muscle fascia, and thus alternative diagnoses should be considered for a cutaneous lesion that demonstrates infiltration into nerve, bone, joint, or other deeper tissues. Most IHs do not require treatment; however, a small subset may be associated with complications and thus require intervention. Complications of IH may include impairment of function (eg, vision, feeding, respiratory), ulceration, and risk for permanent disfigurement. When treatment is indicated, the most commonly employed options during the proliferative phase are the topical beta-blocker timolol and the oral beta-blocker propranolol. In addition, certain IHs may be associated with either syndromic presentations and/or visceral involvement, thus requiring further workup (Table).
Congenital Hemangioma
A CH is an uncommon benign neonatal tumor that is distinct from an IH in behavior, biology, and treatment. Congenital hemangiomas may have a rapidly involuting course, referred to as RICH (rapidly involuting congenital hemangioma), or a noninvoluting course, referred to as NICH (noninvoluting congenital hemangioma). Partially involuting types also have been described.3 A RICH typically presents as a highly vascular, red-violaceous or bluish plaque, nodule, or large mass at birth. An NICH presents as a red-violaceous or bluish, coarsely telangiectatic patch, plaque, or nodule. A characteristic feature of the CH is the rim of vasoconstriction around the lesion, which is an important diagnostic clue (Figure 1). In contrast to IH, multifocal lesions are highly unlikely in CH, though it rarely has been reported.4
Regardless of subtype, CHs are fully developed at birth. Infantile hemangiomas, on the other hand, are either minimally present or not present at birth and thereafter proliferate. After birth, a RICH rapidly involutes over the first 9 to 12 months of life. This process generally is evident even in the first few weeks of life, which would not be expected of an IH and is therefore a major distinguishing factor. A NICH, on the other hand, is expected to be persistent, for the most part neither showing signs of proliferation nor involution.
Complications of CHs may include ulceration, functional impairment, or risk for permanent disfigurement depending on location. In addition, due to their fast-flow state and potential large size, some CHs may be complicated by high-output heart failure in the neonate. Distinguishing an IH from a CH is important not only for prognosis but also treatment. Beta-blocker therapy generally is not useful for CHs, and management usually is supportive in the neonatal period.
In the majority of cases, diagnosis can be achieved solely on clinical features. Biopsy with immunohistochemistry shows negative GLUT-1 staining, which will distinguish this lesion from an IH. At times, the highly vascular nature and/or striking size of a CH may lead some to consider the potential diagnosis of an arteriovenous malformation. However, soft-tissue arteriovenous malformations involving the skin are almost never fully developed in the neonatal period and generally take years to evolve from a quiescent state to a destructive lesion.
Venous Malformation
Venous malformations are congenital malformations of veins that may be apparent at birth or later. They appear as bluish to flesh-colored, compressible nodules or plaques. They tend to increase in size when the affected body part is in a dependent position, and this maneuver can be a helpful distinguishing clue. Although the majority of patients have a single lesion, multifocal involvement may occur uncommonly (Table). The diagnosis of VM usually is clinical, though at times, a VM may be difficult to distinguish from a purely deep IH. However, a VM will persist over time, growing in proportion to the patient. In addition, a VM displays low flow on ultrasound, a distinguishing feature from the fast-flow IH. Magnetic resonance imaging with and without contrast is the imaging study of choice. At times, cutaneous VMs will demonstrate infiltration into other tissue planes such as muscle and joint. Pain may occur secondary to thrombus formation within the malformation. In extensive lesions, intravascular coagulation may be notable, as reflected in elevated D-dimer and decreased fibrinogen levels. Treatment with sclerotherapy or surgery may be considered in select cases during infancy; however, in general, an asymptomatic VM may be observed early on in life.
Lymphatic Malformation
A lymphatic malformation (LM) is a congenital malformation of lymphatic vessels and may be further differentiated into microcystic, macrocystic, or mixed types depending on the size of the channels. An LM may present at birth or later and persists over time. Superficial microcystic LMs, synonymous with the term lymphangioma circumscriptum, characteristically appear as a group of clear and violaceous hemorrhagic vesicles on the skin. Deeper LMs appear as a tense or spongy, flesh-colored nodule or mass. Involvement of the head and neck is common. Complications frequently occur in LMs. Cutaneous LMs may ooze or bleed. Infection and hemorrhage into cysts may occur, which will cause acute pain, redness, swelling, and induration. Cervicofacial lesions may result in respiratory distress. Thus, the majority of LMs require treatment, though asymptomatic lesions may be observed in the neonate. An ultrasound will demonstrate a low-flow lesion, and magnetic resonance imaging is the diagnostic modality of choice for diagnosis and definition of extent.
KHE and Tufted Angioma
Kaposiform hemangioendothelioma is a rare, locally aggressive, vascular tumor that is frequently associated with a potentially life-threatening coagulopathy, Kasabach-Merritt phenomenon. Tufted angiomas are now understood to belong on a spectrum with KHEs, which usually present in the neonatal period or infancy as firm, red-violaceous plaques, nodules, or large tumors. Infiltration into nerve, muscle, and bone may occur. The firm/hard nature and deep violaceous appearance generally are initial clues that it is not an IH. Kasabach-Merritt phenomenon manifests as thrombocytopenia as well as low fibrinogen and elevated D-dimer levels. Thrombocytopenia is generally profound in Kasabach-Merritt phenomenon and results from platelet trapping within the vascular tumor. Given these potential complications, KHEs generally require immediate medical attention, and various treatment protocols including prednisone, vincristine, and sirolimus are utilized for complicated cases.5 The diagnosis may require biopsy to distinguish it from malignant tumors, particularly sarcomas.
Malignant Tumors
Various malignancies, including congenital leukemia, neuroblastoma, Langerhans cell histiocytosis, infantile fibrosarcoma, and rhabdomyosarcoma, rarely may present as cutaneous nodules or masses in a neonate mimicking hemangiomas or other vascular lesions (Figure 2). Neonates may present with multiple bluish papules and nodules resembling a blueberry muffin baby; multiple violaceous-red nodules; or a single red-violaceous, highly vascular–appearing mass mimicking hemangiomas. Malignant tumors may display vascularity on imaging, and thus the presence of vascular flow on ultrasound should not dissuade one from the possibility of a malignancy if other clinical features are atypical or unusual for a hemangioma. When a neonatal malignancy is suspected, a large punch biopsy or incisional biopsy is required for workup.
Final Thoughts
Although IHs are the most common vascular nodules in neonates and young infants, other conditions such as VMs, LMs, CHs, KHEs, and malignancy may occur less commonly. Identifying features that would be considered atypical for IH is crucial to recognize these less common possibilities.
Although most neonatal vascular lumps, bumps, and tumors are benign, proper diagnosis is important for prognosis and management. Therefore, knowledge of both common and rare conditions is important when evaluating a neonatal nodule. Differential diagnosis of neonatal vascular nodules must focus on important diagnostic clues that should prompt consideration and evaluation for less common and/or potentially threatening conditions. Infantile hemangioma (IH), congenital hemangioma (CH), venous malformation (VM), lymphatic malformation (LM), kaposiform hemangioendothelioma (KHE) and tufted angioma, and malignant tumors are reviewed here.
Infantile Hemangioma
Infantile hemangioma, a benign proliferation of capillaries, is the most common tumor of infancy with reported incidence of up to 5% in neonates.1 As such, suspicion for less common lesions is often predicated on identifying features that would be atypical for an IH. A superficial IH presents as a bright red papule, nodule, or plaque, while a deep IH presents as a flesh-colored to bluish nodule. Mixed IHs combine features of both superficial and deep lesions. The distribution may be focal or segmental, with segmental lesions encompassing a larger territory–like distribution and frequently displaying a thin, coarsely telangiectatic appearance.
Knowledge of the natural history of IH generally is crucial in differentiating it from other neonatal lesions. Infantile hemangiomas display a natural history that is distinct and predictable. They typically manifest within the first few weeks of life, though up to 30% present at birth with a premonitory mark, which may be a light red, pink, bluish, or vasoconstricted patch. Thus, mere presence of a lesion at birth is not the feature that distinguishes other congenital lesions from an IH. After initial appearance, IHs undergo a period of proliferation that occurs over 4 to 6 months in most patients. In some cases, areas of proliferation may be subtle, but nonetheless the presence of some areas of increased redness and/or volumetric growth generally is required to firmly establish the diagnosis of IH. Thereafter, IH will involute, a process that begins before 1 year of age in most cases and continues over years. Although IHs undergo involution, complete clearance may not occur, as nearly 70% will leave permanent residua such as fibrofatty masses or anetodermic skin.2 Nevertheless, the presence of a proliferative phase followed by a slower period of involution is a hallmark feature of the IH.
Biopsy and imaging rarely are required for establishing diagnosis of an IH. Histopathology showing a proliferation of capillaries with positive glucose transporter 1 (GLUT-1) staining is characteristic. Imaging with ultrasound reveals a fast-flow lesion. Apart from exceptionally rare cases, a cutaneous IH typically does not cross muscle fascia, and thus alternative diagnoses should be considered for a cutaneous lesion that demonstrates infiltration into nerve, bone, joint, or other deeper tissues. Most IHs do not require treatment; however, a small subset may be associated with complications and thus require intervention. Complications of IH may include impairment of function (eg, vision, feeding, respiratory), ulceration, and risk for permanent disfigurement. When treatment is indicated, the most commonly employed options during the proliferative phase are the topical beta-blocker timolol and the oral beta-blocker propranolol. In addition, certain IHs may be associated with either syndromic presentations and/or visceral involvement, thus requiring further workup (Table).
Congenital Hemangioma
A CH is an uncommon benign neonatal tumor that is distinct from an IH in behavior, biology, and treatment. Congenital hemangiomas may have a rapidly involuting course, referred to as RICH (rapidly involuting congenital hemangioma), or a noninvoluting course, referred to as NICH (noninvoluting congenital hemangioma). Partially involuting types also have been described.3 A RICH typically presents as a highly vascular, red-violaceous or bluish plaque, nodule, or large mass at birth. An NICH presents as a red-violaceous or bluish, coarsely telangiectatic patch, plaque, or nodule. A characteristic feature of the CH is the rim of vasoconstriction around the lesion, which is an important diagnostic clue (Figure 1). In contrast to IH, multifocal lesions are highly unlikely in CH, though it rarely has been reported.4
Regardless of subtype, CHs are fully developed at birth. Infantile hemangiomas, on the other hand, are either minimally present or not present at birth and thereafter proliferate. After birth, a RICH rapidly involutes over the first 9 to 12 months of life. This process generally is evident even in the first few weeks of life, which would not be expected of an IH and is therefore a major distinguishing factor. A NICH, on the other hand, is expected to be persistent, for the most part neither showing signs of proliferation nor involution.
Complications of CHs may include ulceration, functional impairment, or risk for permanent disfigurement depending on location. In addition, due to their fast-flow state and potential large size, some CHs may be complicated by high-output heart failure in the neonate. Distinguishing an IH from a CH is important not only for prognosis but also treatment. Beta-blocker therapy generally is not useful for CHs, and management usually is supportive in the neonatal period.
In the majority of cases, diagnosis can be achieved solely on clinical features. Biopsy with immunohistochemistry shows negative GLUT-1 staining, which will distinguish this lesion from an IH. At times, the highly vascular nature and/or striking size of a CH may lead some to consider the potential diagnosis of an arteriovenous malformation. However, soft-tissue arteriovenous malformations involving the skin are almost never fully developed in the neonatal period and generally take years to evolve from a quiescent state to a destructive lesion.
Venous Malformation
Venous malformations are congenital malformations of veins that may be apparent at birth or later. They appear as bluish to flesh-colored, compressible nodules or plaques. They tend to increase in size when the affected body part is in a dependent position, and this maneuver can be a helpful distinguishing clue. Although the majority of patients have a single lesion, multifocal involvement may occur uncommonly (Table). The diagnosis of VM usually is clinical, though at times, a VM may be difficult to distinguish from a purely deep IH. However, a VM will persist over time, growing in proportion to the patient. In addition, a VM displays low flow on ultrasound, a distinguishing feature from the fast-flow IH. Magnetic resonance imaging with and without contrast is the imaging study of choice. At times, cutaneous VMs will demonstrate infiltration into other tissue planes such as muscle and joint. Pain may occur secondary to thrombus formation within the malformation. In extensive lesions, intravascular coagulation may be notable, as reflected in elevated D-dimer and decreased fibrinogen levels. Treatment with sclerotherapy or surgery may be considered in select cases during infancy; however, in general, an asymptomatic VM may be observed early on in life.
Lymphatic Malformation
A lymphatic malformation (LM) is a congenital malformation of lymphatic vessels and may be further differentiated into microcystic, macrocystic, or mixed types depending on the size of the channels. An LM may present at birth or later and persists over time. Superficial microcystic LMs, synonymous with the term lymphangioma circumscriptum, characteristically appear as a group of clear and violaceous hemorrhagic vesicles on the skin. Deeper LMs appear as a tense or spongy, flesh-colored nodule or mass. Involvement of the head and neck is common. Complications frequently occur in LMs. Cutaneous LMs may ooze or bleed. Infection and hemorrhage into cysts may occur, which will cause acute pain, redness, swelling, and induration. Cervicofacial lesions may result in respiratory distress. Thus, the majority of LMs require treatment, though asymptomatic lesions may be observed in the neonate. An ultrasound will demonstrate a low-flow lesion, and magnetic resonance imaging is the diagnostic modality of choice for diagnosis and definition of extent.
KHE and Tufted Angioma
Kaposiform hemangioendothelioma is a rare, locally aggressive, vascular tumor that is frequently associated with a potentially life-threatening coagulopathy, Kasabach-Merritt phenomenon. Tufted angiomas are now understood to belong on a spectrum with KHEs, which usually present in the neonatal period or infancy as firm, red-violaceous plaques, nodules, or large tumors. Infiltration into nerve, muscle, and bone may occur. The firm/hard nature and deep violaceous appearance generally are initial clues that it is not an IH. Kasabach-Merritt phenomenon manifests as thrombocytopenia as well as low fibrinogen and elevated D-dimer levels. Thrombocytopenia is generally profound in Kasabach-Merritt phenomenon and results from platelet trapping within the vascular tumor. Given these potential complications, KHEs generally require immediate medical attention, and various treatment protocols including prednisone, vincristine, and sirolimus are utilized for complicated cases.5 The diagnosis may require biopsy to distinguish it from malignant tumors, particularly sarcomas.
Malignant Tumors
Various malignancies, including congenital leukemia, neuroblastoma, Langerhans cell histiocytosis, infantile fibrosarcoma, and rhabdomyosarcoma, rarely may present as cutaneous nodules or masses in a neonate mimicking hemangiomas or other vascular lesions (Figure 2). Neonates may present with multiple bluish papules and nodules resembling a blueberry muffin baby; multiple violaceous-red nodules; or a single red-violaceous, highly vascular–appearing mass mimicking hemangiomas. Malignant tumors may display vascularity on imaging, and thus the presence of vascular flow on ultrasound should not dissuade one from the possibility of a malignancy if other clinical features are atypical or unusual for a hemangioma. When a neonatal malignancy is suspected, a large punch biopsy or incisional biopsy is required for workup.
Final Thoughts
Although IHs are the most common vascular nodules in neonates and young infants, other conditions such as VMs, LMs, CHs, KHEs, and malignancy may occur less commonly. Identifying features that would be considered atypical for IH is crucial to recognize these less common possibilities.
- Kanada KN, Merin MR, Munden A, et al. A prospective study of cutaneous findings in newborns in the United States: correlation with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161:240-245.
- Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136:E1060-E1104.
- Nasseri E, Piram M, McCuaig CC, et al. Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol. 2014;70:75-79.
- Blumenthal S, Stefanko N, Cossio M, et al. Multifocal congenital hemangioma: expanding the pathogenesis of “neonatal hemangiomatosis.” Pediatr Dermatol. 2019;36:720-722.
- Croteau SE, Gupta D. The clinical spectrum of kaposiform hemangioendothelioma and tufted angioma. Semin Cutan Med Surg. 2016;35:147-152.
- Kanada KN, Merin MR, Munden A, et al. A prospective study of cutaneous findings in newborns in the United States: correlation with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161:240-245.
- Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136:E1060-E1104.
- Nasseri E, Piram M, McCuaig CC, et al. Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol. 2014;70:75-79.
- Blumenthal S, Stefanko N, Cossio M, et al. Multifocal congenital hemangioma: expanding the pathogenesis of “neonatal hemangiomatosis.” Pediatr Dermatol. 2019;36:720-722.
- Croteau SE, Gupta D. The clinical spectrum of kaposiform hemangioendothelioma and tufted angioma. Semin Cutan Med Surg. 2016;35:147-152.
Rare Pediatric Diseases Special Report 2019


CMT1A neuropathy improves with investigational drug PXT3003
AUSTIN, TEX. – , according to new research.
“The study has established for the first time that patients after up to 15 months of treatment had a statistically significant and clinically relevant disability improvement as illustrated by the change from baseline of their ONLS [Overall Neurology Limitations Scale] scale,” concluded Florian Thomas, MD, PhD, of Hackensack (N.J.) University Medical Center, and his associates at Pharnext. “PXT3003 with dose 4 has at least stabilized, even improved, the disease.”
The researchers presented their findings in a poster at the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine.
The PLEO-CMT study was an international, multicenter, randomized, double-blind, placebo-controlled, phase 3 trial that evaluated the efficacy and safety of PXT3003, an oral 3-drug combination, for CMT1A. CMT1A neuropathy, occurring in an estimated 1 in 5,000 people, is characterized by distal muscle atrophy that affects walking and causes stocking-glove sensory loss and lower quality of life.
The trial enrolled 323 patients, aged 16-65, who had mild to moderate CMT1A that had been genetically confirmed. The modified full set analysis (n = 235), which represented the main study analysis for the primary endpoint, included a placebo group of 87 participants while two other groups received one of two doses of the fixed-dose drug combination twice daily: Ninety-three participants received 3 mg baclofen, 0.35 mg naltrexone, and 105 mg sorbitol (dose 1), and 55 participants received twice that dose (dose 2).
The primary endpoint was mean change from baseline to 12 and 15 months on the ONLS. At baseline, 90% of patients had an ONLS score of 2-4, and the researchers determined an average 0.3 points reduction to be a clinically meaningful effect.
Secondary endpoints included the 10-meter walk test, the 9-hole peg test, and a subscore of Charcot-Marie-Tooth neuropathy score version 2 (CMTNSv2).
Only those taking the higher dose (dose 2) showed a clinically significant drop in ONLS, –0.37 points, compared with those taking placebo (P = .0008). The in-group change from baseline to 15 months in ONLS score for patients taking dose 2 showed a trend of improvement that did not reach significance (–0.20; P = .098).
Participants receiving dose 2 of PXT3003 also walked 0.47 seconds faster on the 10-meter walk test, compared with those receiving placebo (P = .016). No significant differences occurred in the other secondary endpoints, although nonsignificant trends of improvement occurred.
Treatment-emergent adverse events were similar across all three groups and led to trial withdrawal at similar rates for dose 1 (5.5%), dose 2 (5.3%), and placebo (5.9%). One serious adverse event, benign thyroid adenoma, led to trial withdrawal, but no serious adverse events occurred related to the treatment.
Pharnext funded the research. Dr. Thomas is a researcher with Pharnext and Acceleron and has received speaking or advisory board fees from Novartis, Acceleron, Sanofi, and Genentech. The other seven authors are employees of Pharnext.
SOURCE: Thomas F et al. AANEM 2019, Abstract 92.
AUSTIN, TEX. – , according to new research.
“The study has established for the first time that patients after up to 15 months of treatment had a statistically significant and clinically relevant disability improvement as illustrated by the change from baseline of their ONLS [Overall Neurology Limitations Scale] scale,” concluded Florian Thomas, MD, PhD, of Hackensack (N.J.) University Medical Center, and his associates at Pharnext. “PXT3003 with dose 4 has at least stabilized, even improved, the disease.”
The researchers presented their findings in a poster at the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine.
The PLEO-CMT study was an international, multicenter, randomized, double-blind, placebo-controlled, phase 3 trial that evaluated the efficacy and safety of PXT3003, an oral 3-drug combination, for CMT1A. CMT1A neuropathy, occurring in an estimated 1 in 5,000 people, is characterized by distal muscle atrophy that affects walking and causes stocking-glove sensory loss and lower quality of life.
The trial enrolled 323 patients, aged 16-65, who had mild to moderate CMT1A that had been genetically confirmed. The modified full set analysis (n = 235), which represented the main study analysis for the primary endpoint, included a placebo group of 87 participants while two other groups received one of two doses of the fixed-dose drug combination twice daily: Ninety-three participants received 3 mg baclofen, 0.35 mg naltrexone, and 105 mg sorbitol (dose 1), and 55 participants received twice that dose (dose 2).
The primary endpoint was mean change from baseline to 12 and 15 months on the ONLS. At baseline, 90% of patients had an ONLS score of 2-4, and the researchers determined an average 0.3 points reduction to be a clinically meaningful effect.
Secondary endpoints included the 10-meter walk test, the 9-hole peg test, and a subscore of Charcot-Marie-Tooth neuropathy score version 2 (CMTNSv2).
Only those taking the higher dose (dose 2) showed a clinically significant drop in ONLS, –0.37 points, compared with those taking placebo (P = .0008). The in-group change from baseline to 15 months in ONLS score for patients taking dose 2 showed a trend of improvement that did not reach significance (–0.20; P = .098).
Participants receiving dose 2 of PXT3003 also walked 0.47 seconds faster on the 10-meter walk test, compared with those receiving placebo (P = .016). No significant differences occurred in the other secondary endpoints, although nonsignificant trends of improvement occurred.
Treatment-emergent adverse events were similar across all three groups and led to trial withdrawal at similar rates for dose 1 (5.5%), dose 2 (5.3%), and placebo (5.9%). One serious adverse event, benign thyroid adenoma, led to trial withdrawal, but no serious adverse events occurred related to the treatment.
Pharnext funded the research. Dr. Thomas is a researcher with Pharnext and Acceleron and has received speaking or advisory board fees from Novartis, Acceleron, Sanofi, and Genentech. The other seven authors are employees of Pharnext.
SOURCE: Thomas F et al. AANEM 2019, Abstract 92.
AUSTIN, TEX. – , according to new research.
“The study has established for the first time that patients after up to 15 months of treatment had a statistically significant and clinically relevant disability improvement as illustrated by the change from baseline of their ONLS [Overall Neurology Limitations Scale] scale,” concluded Florian Thomas, MD, PhD, of Hackensack (N.J.) University Medical Center, and his associates at Pharnext. “PXT3003 with dose 4 has at least stabilized, even improved, the disease.”
The researchers presented their findings in a poster at the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine.
The PLEO-CMT study was an international, multicenter, randomized, double-blind, placebo-controlled, phase 3 trial that evaluated the efficacy and safety of PXT3003, an oral 3-drug combination, for CMT1A. CMT1A neuropathy, occurring in an estimated 1 in 5,000 people, is characterized by distal muscle atrophy that affects walking and causes stocking-glove sensory loss and lower quality of life.
The trial enrolled 323 patients, aged 16-65, who had mild to moderate CMT1A that had been genetically confirmed. The modified full set analysis (n = 235), which represented the main study analysis for the primary endpoint, included a placebo group of 87 participants while two other groups received one of two doses of the fixed-dose drug combination twice daily: Ninety-three participants received 3 mg baclofen, 0.35 mg naltrexone, and 105 mg sorbitol (dose 1), and 55 participants received twice that dose (dose 2).
The primary endpoint was mean change from baseline to 12 and 15 months on the ONLS. At baseline, 90% of patients had an ONLS score of 2-4, and the researchers determined an average 0.3 points reduction to be a clinically meaningful effect.
Secondary endpoints included the 10-meter walk test, the 9-hole peg test, and a subscore of Charcot-Marie-Tooth neuropathy score version 2 (CMTNSv2).
Only those taking the higher dose (dose 2) showed a clinically significant drop in ONLS, –0.37 points, compared with those taking placebo (P = .0008). The in-group change from baseline to 15 months in ONLS score for patients taking dose 2 showed a trend of improvement that did not reach significance (–0.20; P = .098).
Participants receiving dose 2 of PXT3003 also walked 0.47 seconds faster on the 10-meter walk test, compared with those receiving placebo (P = .016). No significant differences occurred in the other secondary endpoints, although nonsignificant trends of improvement occurred.
Treatment-emergent adverse events were similar across all three groups and led to trial withdrawal at similar rates for dose 1 (5.5%), dose 2 (5.3%), and placebo (5.9%). One serious adverse event, benign thyroid adenoma, led to trial withdrawal, but no serious adverse events occurred related to the treatment.
Pharnext funded the research. Dr. Thomas is a researcher with Pharnext and Acceleron and has received speaking or advisory board fees from Novartis, Acceleron, Sanofi, and Genentech. The other seven authors are employees of Pharnext.
SOURCE: Thomas F et al. AANEM 2019, Abstract 92.
REPORTING FROM AANEM 2019
Baricitinib may benefit patients with Aicardi-Goutières syndrome
CHARLOTTE, N.C. – Scores on a novel AGS scale improved, and skin and liver complications resolved in children with AGS who received treatment with baricitinib, according to results presented at the annual meeting of the Child Neurology Society.

AGS is caused by various heritable disorders of the innate immunity that result in excessive interferon production. AGS characteristically manifests as an early-onset encephalopathy that causes intellectual and physical disability, but patients may have a wide range of clinical phenotypes. The disease may involve the skin, liver, lungs, heart, and other organs, as well as the brain.
A multisystem disorder
“The neurologic features, while they are the most compelling for us, are really only the tip of the iceberg,” said Adeline Vanderver, MD, program director of the leukodystrophy center, and the Jacob A. Kamens Endowed Chair in Neurologic Disorders and Translational Neurotherapeutics at Children’s Hospital of Philadelphia. “Nearly every single organ system in the body is affected, from either direct interferon injury or from a secondary vasculopathy related to the interferonopathy.”
Dr. Vanderver presented results from the compassionate use study, which assessed whether the JAK inhibitor baricitinib (Olumiant) may decrease interferon signaling in AGS and limit the morbidity of the disease.
The phase 1, open-label trial “included compassionate use of baricitinib in AGS under the argument that these children did not have time to wait for approval of the drug,” said Dr. Vanderver. In 2018, the Food and Drug Administration approved baricitinib for moderate to severe rheumatoid arthritis in adults with an inadequate response to methotrexate.
The phase 1 trial in AGS included 35 patients with mutation-defined AGS and evidence of inflammatory disease that could be targeted by JAK inhibition. The trial population was 36% female. The average age of disease onset was 0.8 years, and patients’ average age at treatment was 6.1 years. The investigators assessed safety and laboratory data every 3 months and conducted clinical assessments every 6 months.
The heterogeneity of AGS phenotypes within families and across genotypes makes treatment trials in this disorder a challenge, Dr. Vanderver said. Outcome measures may have ceiling or floor effects that fail to capture the range of severity of AGS symptoms. Dr. Vanderver and colleagues developed a novel AGS scale to capture the scope of neurologic function in patients with AGS
.
When the researchers applied the AGS scale to a historical cohort of patients, most had stable scores about 6 months after disease onset. “After the first 6 months of the disease, the disease tends to be much more static, as the children have sustained significant neurologic injury,” Dr. Vanderver said.
They applied the novel AGS scale post hoc as an exploratory endpoint in the phase 1 trial. In addition, parents recorded information in a diary about skin involvement, irritability, seizures, and fever. “Over time, we see a reduction, although not always a statistically significant reduction, in symptom burden,” Dr. Vanderver said. The AGS clinical diary scores reflect “what the parents were telling us – that they felt like their children were feeling better during treatment,” she said.
Several patients had skin conditions that improved with treatment. One patient with dermatitis or eczema had the skin abnormality resolve within 3 days. A patient with full-body panniculitis began healing for the first time after about a month of treatment. Seasonal variations and dose adjustments led to fluctuations in some of the skin conditions. Nevertheless, the results suggested significant improvement in skin manifestations in patients with AGS, Dr. Vanderver said.
Patients generally had stable AGS scale scores in the year before treatment, although a couple of patients who were closer to disease onset had precipitous decline in neurologic function, she said. “We had a statistically significant increase in that scale of neurologic function in our patients during the period of the study, even in patients who had sometimes had years of disease duration,” said Dr. Vanderver.
Dr. Vanderver cautioned that she does not want to overstate the changes in function. Patients with AGS may have less potential for recuperation, compared with patients with other conditions. “A child with significant disruptive CNS disease may not recuperate normal functioning,” Dr. Vanderver said, “but it can be clinically meaningful to families if children start having better head control, smile, communicate, even if they might not regain all their motor milestones.”
In addition, a small subset of patients who had potentially life threatening liver complications from the disease experienced rapid normalization and improvement of liver function. “This blockade can be important not just for neurologic function but also to maintain normal physiologic homeostasis of other organs that are affected by the interferonopathy,” Dr. Vanderver said.
Interferon signaling scores decreased in the days after starting treatment and subsequently leveled out.
Serious adverse events that occurred during the trial, such as hospitalizations, were attributable to AGS. One child died from unrecognized pulmonary hypertension, which is now known to be a complication of AGS but was not at the time.
Harnessing a side effect
The most significant and recurrent laboratory abnormality was thrombocytosis. “That is a known complication of this family of drugs that in many cases allowed us to improve previous treatment-resistant thrombocytopenia, so we kind of like that side effect in most cases, but in two cases it did ... result in dose adjustments, although we never had to stop the medication for that.”
The study offers proof of principle that AGS is treatable, Dr. Vanderver said. A phase 2 trial is enrolling patients closer to disease onset. Early treatment of AGS may remain a challenge until there is newborn screening for the disease, she said.
Dr. Vanderver receives grant and in-kind support for translational research without personal compensation from Eli Lilly, Takeda, Illumina, Biogen, Homology, and Ionis. In addition, Dr. Vanderver serves on the scientific advisory boards of the European Leukodystrophy Association and the United Leukodystrophy Foundation, as well as in an unpaid capacity for Takeda, Ionis, Biogen, and Illumina.
Eli Lilly provided support for the phase 1 study. In addition, the study received support from the AGS Association Americas Family Foundation, National Human Genome Research Institute, National Institute of Neurological Disorders and Stroke, and the Children’s Hospital of Philadelphia Research Institute.
SOURCE: Vanderver A et al. CNS 2019. Abstract PL1-6.
CHARLOTTE, N.C. – Scores on a novel AGS scale improved, and skin and liver complications resolved in children with AGS who received treatment with baricitinib, according to results presented at the annual meeting of the Child Neurology Society.
AGS is caused by various heritable disorders of the innate immunity that result in excessive interferon production. AGS characteristically manifests as an early-onset encephalopathy that causes intellectual and physical disability, but patients may have a wide range of clinical phenotypes. The disease may involve the skin, liver, lungs, heart, and other organs, as well as the brain.
A multisystem disorder
“The neurologic features, while they are the most compelling for us, are really only the tip of the iceberg,” said Adeline Vanderver, MD, program director of the leukodystrophy center, and the Jacob A. Kamens Endowed Chair in Neurologic Disorders and Translational Neurotherapeutics at Children’s Hospital of Philadelphia. “Nearly every single organ system in the body is affected, from either direct interferon injury or from a secondary vasculopathy related to the interferonopathy.”
Dr. Vanderver presented results from the compassionate use study, which assessed whether the JAK inhibitor baricitinib (Olumiant) may decrease interferon signaling in AGS and limit the morbidity of the disease.
The phase 1, open-label trial “included compassionate use of baricitinib in AGS under the argument that these children did not have time to wait for approval of the drug,” said Dr. Vanderver. In 2018, the Food and Drug Administration approved baricitinib for moderate to severe rheumatoid arthritis in adults with an inadequate response to methotrexate.
The phase 1 trial in AGS included 35 patients with mutation-defined AGS and evidence of inflammatory disease that could be targeted by JAK inhibition. The trial population was 36% female. The average age of disease onset was 0.8 years, and patients’ average age at treatment was 6.1 years. The investigators assessed safety and laboratory data every 3 months and conducted clinical assessments every 6 months.
The heterogeneity of AGS phenotypes within families and across genotypes makes treatment trials in this disorder a challenge, Dr. Vanderver said. Outcome measures may have ceiling or floor effects that fail to capture the range of severity of AGS symptoms. Dr. Vanderver and colleagues developed a novel AGS scale to capture the scope of neurologic function in patients with AGS
.
When the researchers applied the AGS scale to a historical cohort of patients, most had stable scores about 6 months after disease onset. “After the first 6 months of the disease, the disease tends to be much more static, as the children have sustained significant neurologic injury,” Dr. Vanderver said.
They applied the novel AGS scale post hoc as an exploratory endpoint in the phase 1 trial. In addition, parents recorded information in a diary about skin involvement, irritability, seizures, and fever. “Over time, we see a reduction, although not always a statistically significant reduction, in symptom burden,” Dr. Vanderver said. The AGS clinical diary scores reflect “what the parents were telling us – that they felt like their children were feeling better during treatment,” she said.
Several patients had skin conditions that improved with treatment. One patient with dermatitis or eczema had the skin abnormality resolve within 3 days. A patient with full-body panniculitis began healing for the first time after about a month of treatment. Seasonal variations and dose adjustments led to fluctuations in some of the skin conditions. Nevertheless, the results suggested significant improvement in skin manifestations in patients with AGS, Dr. Vanderver said.
Patients generally had stable AGS scale scores in the year before treatment, although a couple of patients who were closer to disease onset had precipitous decline in neurologic function, she said. “We had a statistically significant increase in that scale of neurologic function in our patients during the period of the study, even in patients who had sometimes had years of disease duration,” said Dr. Vanderver.
Dr. Vanderver cautioned that she does not want to overstate the changes in function. Patients with AGS may have less potential for recuperation, compared with patients with other conditions. “A child with significant disruptive CNS disease may not recuperate normal functioning,” Dr. Vanderver said, “but it can be clinically meaningful to families if children start having better head control, smile, communicate, even if they might not regain all their motor milestones.”
In addition, a small subset of patients who had potentially life threatening liver complications from the disease experienced rapid normalization and improvement of liver function. “This blockade can be important not just for neurologic function but also to maintain normal physiologic homeostasis of other organs that are affected by the interferonopathy,” Dr. Vanderver said.
Interferon signaling scores decreased in the days after starting treatment and subsequently leveled out.
Serious adverse events that occurred during the trial, such as hospitalizations, were attributable to AGS. One child died from unrecognized pulmonary hypertension, which is now known to be a complication of AGS but was not at the time.
Harnessing a side effect
The most significant and recurrent laboratory abnormality was thrombocytosis. “That is a known complication of this family of drugs that in many cases allowed us to improve previous treatment-resistant thrombocytopenia, so we kind of like that side effect in most cases, but in two cases it did ... result in dose adjustments, although we never had to stop the medication for that.”
The study offers proof of principle that AGS is treatable, Dr. Vanderver said. A phase 2 trial is enrolling patients closer to disease onset. Early treatment of AGS may remain a challenge until there is newborn screening for the disease, she said.
Dr. Vanderver receives grant and in-kind support for translational research without personal compensation from Eli Lilly, Takeda, Illumina, Biogen, Homology, and Ionis. In addition, Dr. Vanderver serves on the scientific advisory boards of the European Leukodystrophy Association and the United Leukodystrophy Foundation, as well as in an unpaid capacity for Takeda, Ionis, Biogen, and Illumina.
Eli Lilly provided support for the phase 1 study. In addition, the study received support from the AGS Association Americas Family Foundation, National Human Genome Research Institute, National Institute of Neurological Disorders and Stroke, and the Children’s Hospital of Philadelphia Research Institute.
SOURCE: Vanderver A et al. CNS 2019. Abstract PL1-6.
CHARLOTTE, N.C. – Scores on a novel AGS scale improved, and skin and liver complications resolved in children with AGS who received treatment with baricitinib, according to results presented at the annual meeting of the Child Neurology Society.
AGS is caused by various heritable disorders of the innate immunity that result in excessive interferon production. AGS characteristically manifests as an early-onset encephalopathy that causes intellectual and physical disability, but patients may have a wide range of clinical phenotypes. The disease may involve the skin, liver, lungs, heart, and other organs, as well as the brain.
A multisystem disorder
“The neurologic features, while they are the most compelling for us, are really only the tip of the iceberg,” said Adeline Vanderver, MD, program director of the leukodystrophy center, and the Jacob A. Kamens Endowed Chair in Neurologic Disorders and Translational Neurotherapeutics at Children’s Hospital of Philadelphia. “Nearly every single organ system in the body is affected, from either direct interferon injury or from a secondary vasculopathy related to the interferonopathy.”
Dr. Vanderver presented results from the compassionate use study, which assessed whether the JAK inhibitor baricitinib (Olumiant) may decrease interferon signaling in AGS and limit the morbidity of the disease.
The phase 1, open-label trial “included compassionate use of baricitinib in AGS under the argument that these children did not have time to wait for approval of the drug,” said Dr. Vanderver. In 2018, the Food and Drug Administration approved baricitinib for moderate to severe rheumatoid arthritis in adults with an inadequate response to methotrexate.
The phase 1 trial in AGS included 35 patients with mutation-defined AGS and evidence of inflammatory disease that could be targeted by JAK inhibition. The trial population was 36% female. The average age of disease onset was 0.8 years, and patients’ average age at treatment was 6.1 years. The investigators assessed safety and laboratory data every 3 months and conducted clinical assessments every 6 months.
The heterogeneity of AGS phenotypes within families and across genotypes makes treatment trials in this disorder a challenge, Dr. Vanderver said. Outcome measures may have ceiling or floor effects that fail to capture the range of severity of AGS symptoms. Dr. Vanderver and colleagues developed a novel AGS scale to capture the scope of neurologic function in patients with AGS
.
When the researchers applied the AGS scale to a historical cohort of patients, most had stable scores about 6 months after disease onset. “After the first 6 months of the disease, the disease tends to be much more static, as the children have sustained significant neurologic injury,” Dr. Vanderver said.
They applied the novel AGS scale post hoc as an exploratory endpoint in the phase 1 trial. In addition, parents recorded information in a diary about skin involvement, irritability, seizures, and fever. “Over time, we see a reduction, although not always a statistically significant reduction, in symptom burden,” Dr. Vanderver said. The AGS clinical diary scores reflect “what the parents were telling us – that they felt like their children were feeling better during treatment,” she said.
Several patients had skin conditions that improved with treatment. One patient with dermatitis or eczema had the skin abnormality resolve within 3 days. A patient with full-body panniculitis began healing for the first time after about a month of treatment. Seasonal variations and dose adjustments led to fluctuations in some of the skin conditions. Nevertheless, the results suggested significant improvement in skin manifestations in patients with AGS, Dr. Vanderver said.
Patients generally had stable AGS scale scores in the year before treatment, although a couple of patients who were closer to disease onset had precipitous decline in neurologic function, she said. “We had a statistically significant increase in that scale of neurologic function in our patients during the period of the study, even in patients who had sometimes had years of disease duration,” said Dr. Vanderver.
Dr. Vanderver cautioned that she does not want to overstate the changes in function. Patients with AGS may have less potential for recuperation, compared with patients with other conditions. “A child with significant disruptive CNS disease may not recuperate normal functioning,” Dr. Vanderver said, “but it can be clinically meaningful to families if children start having better head control, smile, communicate, even if they might not regain all their motor milestones.”
In addition, a small subset of patients who had potentially life threatening liver complications from the disease experienced rapid normalization and improvement of liver function. “This blockade can be important not just for neurologic function but also to maintain normal physiologic homeostasis of other organs that are affected by the interferonopathy,” Dr. Vanderver said.
Interferon signaling scores decreased in the days after starting treatment and subsequently leveled out.
Serious adverse events that occurred during the trial, such as hospitalizations, were attributable to AGS. One child died from unrecognized pulmonary hypertension, which is now known to be a complication of AGS but was not at the time.
Harnessing a side effect
The most significant and recurrent laboratory abnormality was thrombocytosis. “That is a known complication of this family of drugs that in many cases allowed us to improve previous treatment-resistant thrombocytopenia, so we kind of like that side effect in most cases, but in two cases it did ... result in dose adjustments, although we never had to stop the medication for that.”
The study offers proof of principle that AGS is treatable, Dr. Vanderver said. A phase 2 trial is enrolling patients closer to disease onset. Early treatment of AGS may remain a challenge until there is newborn screening for the disease, she said.
Dr. Vanderver receives grant and in-kind support for translational research without personal compensation from Eli Lilly, Takeda, Illumina, Biogen, Homology, and Ionis. In addition, Dr. Vanderver serves on the scientific advisory boards of the European Leukodystrophy Association and the United Leukodystrophy Foundation, as well as in an unpaid capacity for Takeda, Ionis, Biogen, and Illumina.
Eli Lilly provided support for the phase 1 study. In addition, the study received support from the AGS Association Americas Family Foundation, National Human Genome Research Institute, National Institute of Neurological Disorders and Stroke, and the Children’s Hospital of Philadelphia Research Institute.
SOURCE: Vanderver A et al. CNS 2019. Abstract PL1-6.
REPORTING FROM CNS 2019
Spotting immunodeficiency in the pediatric dermatology clinic
SEATTLE – Immunodeficiency in children can look much like eczematous dermatitis. Be aware of this potential diagnosis.

“Although it is important to know these are extremely rare conditions, you don’t want to miss them because you can literally change that child’s life,” Markus Boos, MD, an assistant professor of pediatrics at the University of Washington, Seattle, said in an interview at the annual Coastal Dermatology Symposium.
He outlined some key clinical features and patient history that can raise a potential red flag.
“ and you really spend time looking at the morphology and distribution of the rash,” Dr. Boos said.
The distribution of the rash also can be distinctive. For example, hyper-IgE syndrome shows up as little red pus bumps that are widespread, but specifically occur on the face and other areas that usually aren’t affected eczematous dermatitis. “You should really focus on that, and not just assume that because something [like eczematous dermatitis] is common, everything has to be that,” Dr. Boos said at the meeting, which was jointly presented by the University of Louisville and Global Academy for Medical Education.
He also warned about a false positive. You may be alerted to high eosinophil and high IgE levels determined by a primary care physician’s tests, but these aren’t necessarily a strong indicator of hyper-IgE syndrome, he said. “Many inflammatory conditions in children have high levels of both those, so they aren’t a distinguishing feature of any one of them. You can reassure a family that the child doesn’t necessarily have hyper-IgE syndrome. There’s this leap [people take] because it sounds like the name, but it’s not a very specific marker of that particular condition.”
Patient history of an immunodeficiency patient in general obviously can include a history of infections, although a high rate of ear infections is pretty typical among children. The key is to ask yourself: “At what point does it seem like something that is beyond normal?” Dr. Boos said. Infections that required hospitalizations or were invasive or required antibiotics all are potential clues. Other factors to consider include growth and development issues such as frequent diarrhea or failure to thrive, or family members with frequent infections or who died prematurely.
Hyper-IgE patients also may have a prominent forehead and chin, deep-set eyes, broad nose, thickened facial skin, or a high arched palate. These physical features become more prominent by adolescence. For a reference for physical features go to https://primaryimmune.org/about-primary-immunodeficiencies/specific-disease-types/hyper-ige-syndrome.
Clinical features of various immunodeficiencies include the following:
- Papulopustular eruption with frequent infections and musculoskeletal changes. This presentation is suggestive of autosomal dominant hyper-IgE syndrome. These children have a “heterozygous mutation in the gene encoding the transcription factor STAT3,” according to the Immune Deficiency Foundation.
- Severe atopy with extensive warts/molluscum/herpes simplex virus. This presentation is suggestive of autosomal recessive hyper-IgE syndrome. These children have “mutations and deletions in the DOCK8 gene,” the Immune Deficiency Foundation asserts.
- Diffusely red baby. Consider immunodeficiency if the patient also has experienced failure to thrive and/or diarrhea, or has a history of infection. High IgE levels are not a strong signal of hyper-IgE syndrome.
- Severe eczematous (or psoriasiform) dermatitis with chronic diarrhea, failure to thrive, and diabetes or hypothyroidism. This presentation is suggestive of IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked).
- Atopic dermatitis with bloody diarrhea, thrombocytopenia, recurrent ear infections. This presentation is indicative of Wiskott-Aldrich syndrome.
Dr. Boos is personally familiar with primary immunodeficiencies because he works closely with an immunology clinic, which also means he has a lot of support. Most clinicians diagnosing these patients don’t. If you find yourself with a case, “call in the troops,” he advised. You should be connected to a rheumatologist when there’s evidence of autoimmune disease, and hematologists or oncologists for the treatment, which requires a bone marrow transplant in the case of autosomal recessive hyper-IgE syndrome. Otherwise treatment is largely supportive for this immunodeficiency.
Having that network can be invaluable in managing what can be a very complicated patient. “If you ever feel uncomfortable making a decision about their care, discussing it with those other providers can give you some peace of mind,” he said.
Dr. Boos disclosed that he is a clinical researcher for Regeneron. This publication and Global Academy for Medical Education are owned by the same parent company.
SEATTLE – Immunodeficiency in children can look much like eczematous dermatitis. Be aware of this potential diagnosis.
“Although it is important to know these are extremely rare conditions, you don’t want to miss them because you can literally change that child’s life,” Markus Boos, MD, an assistant professor of pediatrics at the University of Washington, Seattle, said in an interview at the annual Coastal Dermatology Symposium.
He outlined some key clinical features and patient history that can raise a potential red flag.
“ and you really spend time looking at the morphology and distribution of the rash,” Dr. Boos said.
The distribution of the rash also can be distinctive. For example, hyper-IgE syndrome shows up as little red pus bumps that are widespread, but specifically occur on the face and other areas that usually aren’t affected eczematous dermatitis. “You should really focus on that, and not just assume that because something [like eczematous dermatitis] is common, everything has to be that,” Dr. Boos said at the meeting, which was jointly presented by the University of Louisville and Global Academy for Medical Education.
He also warned about a false positive. You may be alerted to high eosinophil and high IgE levels determined by a primary care physician’s tests, but these aren’t necessarily a strong indicator of hyper-IgE syndrome, he said. “Many inflammatory conditions in children have high levels of both those, so they aren’t a distinguishing feature of any one of them. You can reassure a family that the child doesn’t necessarily have hyper-IgE syndrome. There’s this leap [people take] because it sounds like the name, but it’s not a very specific marker of that particular condition.”
Patient history of an immunodeficiency patient in general obviously can include a history of infections, although a high rate of ear infections is pretty typical among children. The key is to ask yourself: “At what point does it seem like something that is beyond normal?” Dr. Boos said. Infections that required hospitalizations or were invasive or required antibiotics all are potential clues. Other factors to consider include growth and development issues such as frequent diarrhea or failure to thrive, or family members with frequent infections or who died prematurely.
Hyper-IgE patients also may have a prominent forehead and chin, deep-set eyes, broad nose, thickened facial skin, or a high arched palate. These physical features become more prominent by adolescence. For a reference for physical features go to https://primaryimmune.org/about-primary-immunodeficiencies/specific-disease-types/hyper-ige-syndrome.
Clinical features of various immunodeficiencies include the following:
- Papulopustular eruption with frequent infections and musculoskeletal changes. This presentation is suggestive of autosomal dominant hyper-IgE syndrome. These children have a “heterozygous mutation in the gene encoding the transcription factor STAT3,” according to the Immune Deficiency Foundation.
- Severe atopy with extensive warts/molluscum/herpes simplex virus. This presentation is suggestive of autosomal recessive hyper-IgE syndrome. These children have “mutations and deletions in the DOCK8 gene,” the Immune Deficiency Foundation asserts.
- Diffusely red baby. Consider immunodeficiency if the patient also has experienced failure to thrive and/or diarrhea, or has a history of infection. High IgE levels are not a strong signal of hyper-IgE syndrome.
- Severe eczematous (or psoriasiform) dermatitis with chronic diarrhea, failure to thrive, and diabetes or hypothyroidism. This presentation is suggestive of IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked).
- Atopic dermatitis with bloody diarrhea, thrombocytopenia, recurrent ear infections. This presentation is indicative of Wiskott-Aldrich syndrome.
Dr. Boos is personally familiar with primary immunodeficiencies because he works closely with an immunology clinic, which also means he has a lot of support. Most clinicians diagnosing these patients don’t. If you find yourself with a case, “call in the troops,” he advised. You should be connected to a rheumatologist when there’s evidence of autoimmune disease, and hematologists or oncologists for the treatment, which requires a bone marrow transplant in the case of autosomal recessive hyper-IgE syndrome. Otherwise treatment is largely supportive for this immunodeficiency.
Having that network can be invaluable in managing what can be a very complicated patient. “If you ever feel uncomfortable making a decision about their care, discussing it with those other providers can give you some peace of mind,” he said.
Dr. Boos disclosed that he is a clinical researcher for Regeneron. This publication and Global Academy for Medical Education are owned by the same parent company.
SEATTLE – Immunodeficiency in children can look much like eczematous dermatitis. Be aware of this potential diagnosis.
“Although it is important to know these are extremely rare conditions, you don’t want to miss them because you can literally change that child’s life,” Markus Boos, MD, an assistant professor of pediatrics at the University of Washington, Seattle, said in an interview at the annual Coastal Dermatology Symposium.
He outlined some key clinical features and patient history that can raise a potential red flag.
“ and you really spend time looking at the morphology and distribution of the rash,” Dr. Boos said.
The distribution of the rash also can be distinctive. For example, hyper-IgE syndrome shows up as little red pus bumps that are widespread, but specifically occur on the face and other areas that usually aren’t affected eczematous dermatitis. “You should really focus on that, and not just assume that because something [like eczematous dermatitis] is common, everything has to be that,” Dr. Boos said at the meeting, which was jointly presented by the University of Louisville and Global Academy for Medical Education.
He also warned about a false positive. You may be alerted to high eosinophil and high IgE levels determined by a primary care physician’s tests, but these aren’t necessarily a strong indicator of hyper-IgE syndrome, he said. “Many inflammatory conditions in children have high levels of both those, so they aren’t a distinguishing feature of any one of them. You can reassure a family that the child doesn’t necessarily have hyper-IgE syndrome. There’s this leap [people take] because it sounds like the name, but it’s not a very specific marker of that particular condition.”
Patient history of an immunodeficiency patient in general obviously can include a history of infections, although a high rate of ear infections is pretty typical among children. The key is to ask yourself: “At what point does it seem like something that is beyond normal?” Dr. Boos said. Infections that required hospitalizations or were invasive or required antibiotics all are potential clues. Other factors to consider include growth and development issues such as frequent diarrhea or failure to thrive, or family members with frequent infections or who died prematurely.
Hyper-IgE patients also may have a prominent forehead and chin, deep-set eyes, broad nose, thickened facial skin, or a high arched palate. These physical features become more prominent by adolescence. For a reference for physical features go to https://primaryimmune.org/about-primary-immunodeficiencies/specific-disease-types/hyper-ige-syndrome.
Clinical features of various immunodeficiencies include the following:
- Papulopustular eruption with frequent infections and musculoskeletal changes. This presentation is suggestive of autosomal dominant hyper-IgE syndrome. These children have a “heterozygous mutation in the gene encoding the transcription factor STAT3,” according to the Immune Deficiency Foundation.
- Severe atopy with extensive warts/molluscum/herpes simplex virus. This presentation is suggestive of autosomal recessive hyper-IgE syndrome. These children have “mutations and deletions in the DOCK8 gene,” the Immune Deficiency Foundation asserts.
- Diffusely red baby. Consider immunodeficiency if the patient also has experienced failure to thrive and/or diarrhea, or has a history of infection. High IgE levels are not a strong signal of hyper-IgE syndrome.
- Severe eczematous (or psoriasiform) dermatitis with chronic diarrhea, failure to thrive, and diabetes or hypothyroidism. This presentation is suggestive of IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked).
- Atopic dermatitis with bloody diarrhea, thrombocytopenia, recurrent ear infections. This presentation is indicative of Wiskott-Aldrich syndrome.
Dr. Boos is personally familiar with primary immunodeficiencies because he works closely with an immunology clinic, which also means he has a lot of support. Most clinicians diagnosing these patients don’t. If you find yourself with a case, “call in the troops,” he advised. You should be connected to a rheumatologist when there’s evidence of autoimmune disease, and hematologists or oncologists for the treatment, which requires a bone marrow transplant in the case of autosomal recessive hyper-IgE syndrome. Otherwise treatment is largely supportive for this immunodeficiency.
Having that network can be invaluable in managing what can be a very complicated patient. “If you ever feel uncomfortable making a decision about their care, discussing it with those other providers can give you some peace of mind,” he said.
Dr. Boos disclosed that he is a clinical researcher for Regeneron. This publication and Global Academy for Medical Education are owned by the same parent company.
EXPERT ANALYSIS FROM COASTAL DERM
Rituximab bests mycophenolate in pemphigus vulgaris
MADRID – Pascal Joly, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.

Not only did rituximab prove superior in terms of efficacy in the PEMPHIX trial, with a five times greater likelihood of achieving a complete remission lasting for at least 16 weeks while off oral corticosteroids than with mycophenolate mofetil in the 52-week study, but the total number of disease flares in the mycophenolate mofetil group was five times higher. Moreover, rituximab-treated patients received a markedly lower cumulative dose of prednisone as well.
“Rituximab has a superior overall benefit-risk profile, compared to mycophenolate mofetil, in patients with moderate to severe pemphigus vulgaris,” concluded Dr. Joly, professor of dermatology at the University of Rouen (France) and president of the French Society of Dermatology.
The study was undertaken because mycophenolate mofetil is commonly used as a corticosteroid-sparing drug in patients with pemphigus vulgaris, even though its efficacy for the treatment of this rare, severe autoimmune blistering disease is unproven, he explained.
In contrast, rituximab was approved by the Food and Drug Administration and European regulators for treatment of pemphigus vulgaris on the strength of the pivotal phase 3 Ritux 3 trial – also led by Dr. Joly – which demonstrated the superiority of this intravenously administered anti-CD20 monoclonal antibody plus short-term prednisone over high-dose corticosteroid monotherapy, which for decades had been the standard treatment despite its considerable toxicity burden (Lancet. 2017 May 20;389[10083]:2031-40).
An independently conducted analysis of Ritux 3 recently concluded that rituximab plus short-term prednisone was more effective than prednisone alone, with a lower risk of life-threatening, corticosteroid-related adverse events and less cumulative corticosteroid exposure (Br J Dermatol. 2019 Sep 5. doi: 10.1111/bjd.18482).
Also, an international panel of 93 pemphigus experts has declared that rituximab should be considered an evidence-based first-line therapy for moderate to-severe pemphigus (J Am Acad Dermatol. 2018 Feb 10. doi: 10.1016/j.jaad.2018.02.021).
The phase 3, placebo-controlled PEMPHIX trial randomized 135 patients with moderate or severe pemphigus at 49 academic medical centers in the United States and nine other countries to double-blind rituximab or mycophenolate mofetil on top of background oral prednisone at 1.0-1.5 mg/kg per day, with the steroid to be tapered and discontinued within 4-6 months.
The primary endpoint of the study was the proportion of patients in each study arm at week 52 who had achieved a sustained complete remission lasting for at least 16 weeks while off prednisone. The rate was 40.3% in the rituximab group and 9.5% with mycophenolate mofetil, for a 383% increased likelihood of sustained complete remission in the rituximab group.
In addition, all of the study’s secondary endpoints significantly favored rituximab. The median cumulative dose of corticosteroid was 2.7 g through 52 weeks in the rituximab arm, compared with 4 g with mycophenolate mofetil. The total number of disease flares over 52 weeks was 6 in the rituximab group and 44 in the mycophenolate arm. Five rituximab-treated patients experienced disease flares, as did 26 on mycophenolate. Thus, the likelihood of a flare was seven times lower with rituximab.
Scores on the Dermatology Life Quality Index improved by an average of 8.87 points from baseline to week 52 in the rituximab group versus 6 points with mycophenolate. And 62.7% of rituximab-treated patients had a week-52 Dermatology Life Quality Index score of 0, meaning no impact of the disease on their quality of life, compared with 25% of the mycophenolate group.
Dr. Joly characterized the safety profile of rituximab as “manageable, with acceptable tolerability.” About 9% of the rituximab group and 7.4% of mycophenolate-treated patients had one or more treatment-related adverse events, a nonsignificant difference. The rate of treatment-related serious infections was 3.0% with rituximab and 2.9% with mycophenolate. Serious infusion reactions leading to study withdrawal occurred in three patients on rituximab and one on mycophenolate. The rate of grade 3 or worse corticosteroid-related adverse events was 1.5% with rituximab and significantly greater at 7.4% with mycophenolate.
An additional 48-week safety assessment beyond the 52-week primary outcome is ongoing.
Asked what future role he sees for mycophenolate in pemphigus vulgaris, Dr. Joly replied that the only study in the literature that shows the drug outperforms placebo was seriously flawed. “In the future, it’s very likely that the indications for use of mycophenolate in pemphigus vulgaris will be fewer and fewer,” the dermatologist added.
In reply to a question about the merits of routine antibiotic prophylaxis against pneumocystis carinii pneumonia in patients taking rituximab for pemphigus vulgaris, Dr. Joly said the incidence isn’t sufficiently high to justify such practice. After all, he noted, there were no cases of pneumocystis carinii pneumonia in rituximab-treated patients in PEMPHIX and only one in Ritux 3.
EADV Scientific Programming Committee Chair Brigitte Dreno, MD, PhD, professor and head of dermatology at University Hospital in Nantes, France, inquired as to whether there’s a role for maintenance therapy in a potent rituximab-based treatment strategy such as utilized in PEMPHIX.
Definitely, Dr. Joly replied. However, further study is required to work out the best maintenance program.
“There are many arguments for maintenance therapy in these patients. For one, the frequency of relapses increases with the length of follow-up. Also, anti–desmoglein-specific T cells can still be detected after rituximab therapy, even in patients in complete remission. So there is a need for maintenance therapy, perhaps at months 6, 12, and 18, but the optimal regimen isn’t determined yet,” according to Dr. Joly.
PEMPHIX was sponsored by F. Hoffmann-La Roche. Dr. Joly reported serving as a consultant to Roche, Amgen, Principia Biopharma, and Argenx.
MADRID – Pascal Joly, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Not only did rituximab prove superior in terms of efficacy in the PEMPHIX trial, with a five times greater likelihood of achieving a complete remission lasting for at least 16 weeks while off oral corticosteroids than with mycophenolate mofetil in the 52-week study, but the total number of disease flares in the mycophenolate mofetil group was five times higher. Moreover, rituximab-treated patients received a markedly lower cumulative dose of prednisone as well.
“Rituximab has a superior overall benefit-risk profile, compared to mycophenolate mofetil, in patients with moderate to severe pemphigus vulgaris,” concluded Dr. Joly, professor of dermatology at the University of Rouen (France) and president of the French Society of Dermatology.
The study was undertaken because mycophenolate mofetil is commonly used as a corticosteroid-sparing drug in patients with pemphigus vulgaris, even though its efficacy for the treatment of this rare, severe autoimmune blistering disease is unproven, he explained.
In contrast, rituximab was approved by the Food and Drug Administration and European regulators for treatment of pemphigus vulgaris on the strength of the pivotal phase 3 Ritux 3 trial – also led by Dr. Joly – which demonstrated the superiority of this intravenously administered anti-CD20 monoclonal antibody plus short-term prednisone over high-dose corticosteroid monotherapy, which for decades had been the standard treatment despite its considerable toxicity burden (Lancet. 2017 May 20;389[10083]:2031-40).
An independently conducted analysis of Ritux 3 recently concluded that rituximab plus short-term prednisone was more effective than prednisone alone, with a lower risk of life-threatening, corticosteroid-related adverse events and less cumulative corticosteroid exposure (Br J Dermatol. 2019 Sep 5. doi: 10.1111/bjd.18482).
Also, an international panel of 93 pemphigus experts has declared that rituximab should be considered an evidence-based first-line therapy for moderate to-severe pemphigus (J Am Acad Dermatol. 2018 Feb 10. doi: 10.1016/j.jaad.2018.02.021).
The phase 3, placebo-controlled PEMPHIX trial randomized 135 patients with moderate or severe pemphigus at 49 academic medical centers in the United States and nine other countries to double-blind rituximab or mycophenolate mofetil on top of background oral prednisone at 1.0-1.5 mg/kg per day, with the steroid to be tapered and discontinued within 4-6 months.
The primary endpoint of the study was the proportion of patients in each study arm at week 52 who had achieved a sustained complete remission lasting for at least 16 weeks while off prednisone. The rate was 40.3% in the rituximab group and 9.5% with mycophenolate mofetil, for a 383% increased likelihood of sustained complete remission in the rituximab group.
In addition, all of the study’s secondary endpoints significantly favored rituximab. The median cumulative dose of corticosteroid was 2.7 g through 52 weeks in the rituximab arm, compared with 4 g with mycophenolate mofetil. The total number of disease flares over 52 weeks was 6 in the rituximab group and 44 in the mycophenolate arm. Five rituximab-treated patients experienced disease flares, as did 26 on mycophenolate. Thus, the likelihood of a flare was seven times lower with rituximab.
Scores on the Dermatology Life Quality Index improved by an average of 8.87 points from baseline to week 52 in the rituximab group versus 6 points with mycophenolate. And 62.7% of rituximab-treated patients had a week-52 Dermatology Life Quality Index score of 0, meaning no impact of the disease on their quality of life, compared with 25% of the mycophenolate group.
Dr. Joly characterized the safety profile of rituximab as “manageable, with acceptable tolerability.” About 9% of the rituximab group and 7.4% of mycophenolate-treated patients had one or more treatment-related adverse events, a nonsignificant difference. The rate of treatment-related serious infections was 3.0% with rituximab and 2.9% with mycophenolate. Serious infusion reactions leading to study withdrawal occurred in three patients on rituximab and one on mycophenolate. The rate of grade 3 or worse corticosteroid-related adverse events was 1.5% with rituximab and significantly greater at 7.4% with mycophenolate.
An additional 48-week safety assessment beyond the 52-week primary outcome is ongoing.
Asked what future role he sees for mycophenolate in pemphigus vulgaris, Dr. Joly replied that the only study in the literature that shows the drug outperforms placebo was seriously flawed. “In the future, it’s very likely that the indications for use of mycophenolate in pemphigus vulgaris will be fewer and fewer,” the dermatologist added.
In reply to a question about the merits of routine antibiotic prophylaxis against pneumocystis carinii pneumonia in patients taking rituximab for pemphigus vulgaris, Dr. Joly said the incidence isn’t sufficiently high to justify such practice. After all, he noted, there were no cases of pneumocystis carinii pneumonia in rituximab-treated patients in PEMPHIX and only one in Ritux 3.
EADV Scientific Programming Committee Chair Brigitte Dreno, MD, PhD, professor and head of dermatology at University Hospital in Nantes, France, inquired as to whether there’s a role for maintenance therapy in a potent rituximab-based treatment strategy such as utilized in PEMPHIX.
Definitely, Dr. Joly replied. However, further study is required to work out the best maintenance program.
“There are many arguments for maintenance therapy in these patients. For one, the frequency of relapses increases with the length of follow-up. Also, anti–desmoglein-specific T cells can still be detected after rituximab therapy, even in patients in complete remission. So there is a need for maintenance therapy, perhaps at months 6, 12, and 18, but the optimal regimen isn’t determined yet,” according to Dr. Joly.
PEMPHIX was sponsored by F. Hoffmann-La Roche. Dr. Joly reported serving as a consultant to Roche, Amgen, Principia Biopharma, and Argenx.
MADRID – Pascal Joly, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Not only did rituximab prove superior in terms of efficacy in the PEMPHIX trial, with a five times greater likelihood of achieving a complete remission lasting for at least 16 weeks while off oral corticosteroids than with mycophenolate mofetil in the 52-week study, but the total number of disease flares in the mycophenolate mofetil group was five times higher. Moreover, rituximab-treated patients received a markedly lower cumulative dose of prednisone as well.
“Rituximab has a superior overall benefit-risk profile, compared to mycophenolate mofetil, in patients with moderate to severe pemphigus vulgaris,” concluded Dr. Joly, professor of dermatology at the University of Rouen (France) and president of the French Society of Dermatology.
The study was undertaken because mycophenolate mofetil is commonly used as a corticosteroid-sparing drug in patients with pemphigus vulgaris, even though its efficacy for the treatment of this rare, severe autoimmune blistering disease is unproven, he explained.
In contrast, rituximab was approved by the Food and Drug Administration and European regulators for treatment of pemphigus vulgaris on the strength of the pivotal phase 3 Ritux 3 trial – also led by Dr. Joly – which demonstrated the superiority of this intravenously administered anti-CD20 monoclonal antibody plus short-term prednisone over high-dose corticosteroid monotherapy, which for decades had been the standard treatment despite its considerable toxicity burden (Lancet. 2017 May 20;389[10083]:2031-40).
An independently conducted analysis of Ritux 3 recently concluded that rituximab plus short-term prednisone was more effective than prednisone alone, with a lower risk of life-threatening, corticosteroid-related adverse events and less cumulative corticosteroid exposure (Br J Dermatol. 2019 Sep 5. doi: 10.1111/bjd.18482).
Also, an international panel of 93 pemphigus experts has declared that rituximab should be considered an evidence-based first-line therapy for moderate to-severe pemphigus (J Am Acad Dermatol. 2018 Feb 10. doi: 10.1016/j.jaad.2018.02.021).
The phase 3, placebo-controlled PEMPHIX trial randomized 135 patients with moderate or severe pemphigus at 49 academic medical centers in the United States and nine other countries to double-blind rituximab or mycophenolate mofetil on top of background oral prednisone at 1.0-1.5 mg/kg per day, with the steroid to be tapered and discontinued within 4-6 months.
The primary endpoint of the study was the proportion of patients in each study arm at week 52 who had achieved a sustained complete remission lasting for at least 16 weeks while off prednisone. The rate was 40.3% in the rituximab group and 9.5% with mycophenolate mofetil, for a 383% increased likelihood of sustained complete remission in the rituximab group.
In addition, all of the study’s secondary endpoints significantly favored rituximab. The median cumulative dose of corticosteroid was 2.7 g through 52 weeks in the rituximab arm, compared with 4 g with mycophenolate mofetil. The total number of disease flares over 52 weeks was 6 in the rituximab group and 44 in the mycophenolate arm. Five rituximab-treated patients experienced disease flares, as did 26 on mycophenolate. Thus, the likelihood of a flare was seven times lower with rituximab.
Scores on the Dermatology Life Quality Index improved by an average of 8.87 points from baseline to week 52 in the rituximab group versus 6 points with mycophenolate. And 62.7% of rituximab-treated patients had a week-52 Dermatology Life Quality Index score of 0, meaning no impact of the disease on their quality of life, compared with 25% of the mycophenolate group.
Dr. Joly characterized the safety profile of rituximab as “manageable, with acceptable tolerability.” About 9% of the rituximab group and 7.4% of mycophenolate-treated patients had one or more treatment-related adverse events, a nonsignificant difference. The rate of treatment-related serious infections was 3.0% with rituximab and 2.9% with mycophenolate. Serious infusion reactions leading to study withdrawal occurred in three patients on rituximab and one on mycophenolate. The rate of grade 3 or worse corticosteroid-related adverse events was 1.5% with rituximab and significantly greater at 7.4% with mycophenolate.
An additional 48-week safety assessment beyond the 52-week primary outcome is ongoing.
Asked what future role he sees for mycophenolate in pemphigus vulgaris, Dr. Joly replied that the only study in the literature that shows the drug outperforms placebo was seriously flawed. “In the future, it’s very likely that the indications for use of mycophenolate in pemphigus vulgaris will be fewer and fewer,” the dermatologist added.
In reply to a question about the merits of routine antibiotic prophylaxis against pneumocystis carinii pneumonia in patients taking rituximab for pemphigus vulgaris, Dr. Joly said the incidence isn’t sufficiently high to justify such practice. After all, he noted, there were no cases of pneumocystis carinii pneumonia in rituximab-treated patients in PEMPHIX and only one in Ritux 3.
EADV Scientific Programming Committee Chair Brigitte Dreno, MD, PhD, professor and head of dermatology at University Hospital in Nantes, France, inquired as to whether there’s a role for maintenance therapy in a potent rituximab-based treatment strategy such as utilized in PEMPHIX.
Definitely, Dr. Joly replied. However, further study is required to work out the best maintenance program.
“There are many arguments for maintenance therapy in these patients. For one, the frequency of relapses increases with the length of follow-up. Also, anti–desmoglein-specific T cells can still be detected after rituximab therapy, even in patients in complete remission. So there is a need for maintenance therapy, perhaps at months 6, 12, and 18, but the optimal regimen isn’t determined yet,” according to Dr. Joly.
PEMPHIX was sponsored by F. Hoffmann-La Roche. Dr. Joly reported serving as a consultant to Roche, Amgen, Principia Biopharma, and Argenx.
REPORTING FROM THE EADV CONGRESS
Adverse cytogenetics trump molecular risk in NPM1-mutated AML
A pooled analysis suggests adverse cytogenetics are a key factor negatively impacting outcomes in patients with NPM1mut/FLT3-ITDneg/low acute myeloid leukemia (AML).
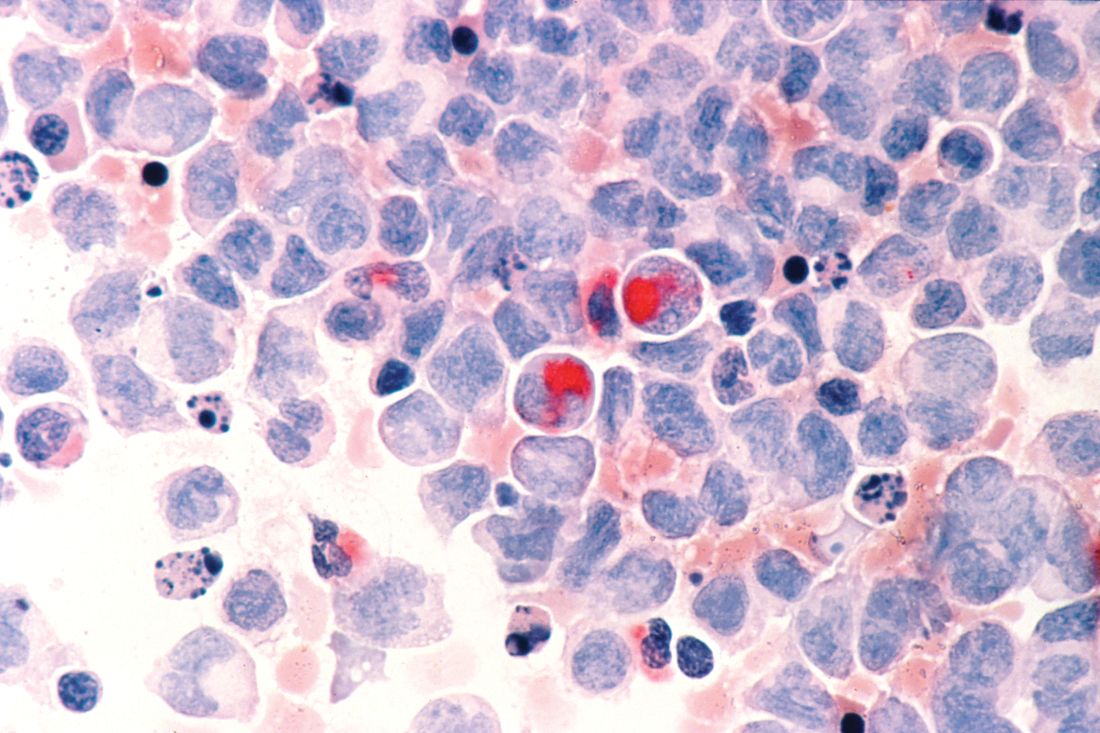
In patients with adverse chromosomal abnormalities, NPM1 mutational status was found not to confer a favorable outcome. The findings suggest cytogenetic risk outweighs molecular risk in patients with NPM1 mutations and the FLT3-ITDneg/low genotype.
“Patients carrying adverse-risk cytogenetics shared a virtually identical unfavorable outcome, regardless of whether the otherwise beneficial NPM1mut/FLT3-ITDneg/low status was present. The type of the adverse chromosomal abnormality did not seem to influence this effect, although low numbers might obscure detection of heterogeneity among individual aberrations,” Linus Angenendt, MD, of University Hospital Munster (Germany) and colleagues, wrote in the Journal of Clinical Oncology.
The researchers retrospectively analyzed 2,426 patients with NPM1mut/FLT3-ITDneg/low AML. Of these, 17.6% had an abnormal karyotype, and 3.4% had adverse-risk chromosomal aberrations.
Prior to analysis, individual patient data were pooled from nine international AML study group registries or treatment centers.
After analysis, the researchers found that adverse cytogenetics were associated with inferior complete remission rates (66.3%), compared with in patients with normal karyotype or intermediate-risk cytogenetic abnormalities (87.7% and 86.0%, respectively; P less than .001). The complete remission rates for the NPM1mut/FLT3-ITDneg/low AML adverse cytogenetics group was similar to patients with NPM1wt/FLT3-ITDneg/low and adverse cytogenetic abnormalities (66.3% vs. 57.5%).
Five-year event-free survival rates and overall survival rates were also lower in patients with NPM1mut/FLT3-ITDneg/low AML and adverse cytogenetics, compared with patients with normal karyotype or intermediate-risk cytogenetic abnormalities (P less than .001).
“Even though the combination of an NPM1 mutation with these abnormalities is rare, the prognostic effect of adverse cytogenetics in NPM1mut AML has important implications for postremission treatment decisions, in particular, the current recommendation that patients who are NPM1mut/FLT3-ITDneg/low not receive allogeneic hematopoietic stem cell transplantation (HSCT), given their presumed low risk of relapse might be altered if the adverse karyotype increased the risk,” they wrote.
The type of chromosomal aberration did not appear to impact this effect, but the small sample size may have hindered the ability to detect a difference between different abnormalities, the researchers noted.
One key limitation of the study was the retrospective design. As a result, in patients with an abnormal karyotype, some genetic analyses could have been underutilized.
“These results demand additional validation within prospective trials,” the researchers concluded.
The study was funded by the University of Munster Medical School, the German Research Foundation, the French government, the Ministry of Health of the Czech Republic, and others. The authors reported financial affiliations with numerous pharmaceutical companies.
SOURCE: Angenendt L et al. J Clin Oncol. 2019 Oct 10;37(29):2632-42.
A pooled analysis suggests adverse cytogenetics are a key factor negatively impacting outcomes in patients with NPM1mut/FLT3-ITDneg/low acute myeloid leukemia (AML).
In patients with adverse chromosomal abnormalities, NPM1 mutational status was found not to confer a favorable outcome. The findings suggest cytogenetic risk outweighs molecular risk in patients with NPM1 mutations and the FLT3-ITDneg/low genotype.
“Patients carrying adverse-risk cytogenetics shared a virtually identical unfavorable outcome, regardless of whether the otherwise beneficial NPM1mut/FLT3-ITDneg/low status was present. The type of the adverse chromosomal abnormality did not seem to influence this effect, although low numbers might obscure detection of heterogeneity among individual aberrations,” Linus Angenendt, MD, of University Hospital Munster (Germany) and colleagues, wrote in the Journal of Clinical Oncology.
The researchers retrospectively analyzed 2,426 patients with NPM1mut/FLT3-ITDneg/low AML. Of these, 17.6% had an abnormal karyotype, and 3.4% had adverse-risk chromosomal aberrations.
Prior to analysis, individual patient data were pooled from nine international AML study group registries or treatment centers.
After analysis, the researchers found that adverse cytogenetics were associated with inferior complete remission rates (66.3%), compared with in patients with normal karyotype or intermediate-risk cytogenetic abnormalities (87.7% and 86.0%, respectively; P less than .001). The complete remission rates for the NPM1mut/FLT3-ITDneg/low AML adverse cytogenetics group was similar to patients with NPM1wt/FLT3-ITDneg/low and adverse cytogenetic abnormalities (66.3% vs. 57.5%).
Five-year event-free survival rates and overall survival rates were also lower in patients with NPM1mut/FLT3-ITDneg/low AML and adverse cytogenetics, compared with patients with normal karyotype or intermediate-risk cytogenetic abnormalities (P less than .001).
“Even though the combination of an NPM1 mutation with these abnormalities is rare, the prognostic effect of adverse cytogenetics in NPM1mut AML has important implications for postremission treatment decisions, in particular, the current recommendation that patients who are NPM1mut/FLT3-ITDneg/low not receive allogeneic hematopoietic stem cell transplantation (HSCT), given their presumed low risk of relapse might be altered if the adverse karyotype increased the risk,” they wrote.
The type of chromosomal aberration did not appear to impact this effect, but the small sample size may have hindered the ability to detect a difference between different abnormalities, the researchers noted.
One key limitation of the study was the retrospective design. As a result, in patients with an abnormal karyotype, some genetic analyses could have been underutilized.
“These results demand additional validation within prospective trials,” the researchers concluded.
The study was funded by the University of Munster Medical School, the German Research Foundation, the French government, the Ministry of Health of the Czech Republic, and others. The authors reported financial affiliations with numerous pharmaceutical companies.
SOURCE: Angenendt L et al. J Clin Oncol. 2019 Oct 10;37(29):2632-42.
A pooled analysis suggests adverse cytogenetics are a key factor negatively impacting outcomes in patients with NPM1mut/FLT3-ITDneg/low acute myeloid leukemia (AML).
In patients with adverse chromosomal abnormalities, NPM1 mutational status was found not to confer a favorable outcome. The findings suggest cytogenetic risk outweighs molecular risk in patients with NPM1 mutations and the FLT3-ITDneg/low genotype.
“Patients carrying adverse-risk cytogenetics shared a virtually identical unfavorable outcome, regardless of whether the otherwise beneficial NPM1mut/FLT3-ITDneg/low status was present. The type of the adverse chromosomal abnormality did not seem to influence this effect, although low numbers might obscure detection of heterogeneity among individual aberrations,” Linus Angenendt, MD, of University Hospital Munster (Germany) and colleagues, wrote in the Journal of Clinical Oncology.
The researchers retrospectively analyzed 2,426 patients with NPM1mut/FLT3-ITDneg/low AML. Of these, 17.6% had an abnormal karyotype, and 3.4% had adverse-risk chromosomal aberrations.
Prior to analysis, individual patient data were pooled from nine international AML study group registries or treatment centers.
After analysis, the researchers found that adverse cytogenetics were associated with inferior complete remission rates (66.3%), compared with in patients with normal karyotype or intermediate-risk cytogenetic abnormalities (87.7% and 86.0%, respectively; P less than .001). The complete remission rates for the NPM1mut/FLT3-ITDneg/low AML adverse cytogenetics group was similar to patients with NPM1wt/FLT3-ITDneg/low and adverse cytogenetic abnormalities (66.3% vs. 57.5%).
Five-year event-free survival rates and overall survival rates were also lower in patients with NPM1mut/FLT3-ITDneg/low AML and adverse cytogenetics, compared with patients with normal karyotype or intermediate-risk cytogenetic abnormalities (P less than .001).
“Even though the combination of an NPM1 mutation with these abnormalities is rare, the prognostic effect of adverse cytogenetics in NPM1mut AML has important implications for postremission treatment decisions, in particular, the current recommendation that patients who are NPM1mut/FLT3-ITDneg/low not receive allogeneic hematopoietic stem cell transplantation (HSCT), given their presumed low risk of relapse might be altered if the adverse karyotype increased the risk,” they wrote.
The type of chromosomal aberration did not appear to impact this effect, but the small sample size may have hindered the ability to detect a difference between different abnormalities, the researchers noted.
One key limitation of the study was the retrospective design. As a result, in patients with an abnormal karyotype, some genetic analyses could have been underutilized.
“These results demand additional validation within prospective trials,” the researchers concluded.
The study was funded by the University of Munster Medical School, the German Research Foundation, the French government, the Ministry of Health of the Czech Republic, and others. The authors reported financial affiliations with numerous pharmaceutical companies.
SOURCE: Angenendt L et al. J Clin Oncol. 2019 Oct 10;37(29):2632-42.
REPORTING FROM THE JOURNAL OF CLINICAL ONCOLOGY
Neurologists publish consensus statement on stridor in MSA
The statement was published Oct. 1 in Neurology. In addition to reviewing the literature on the topic and providing recommendations, the authors described several areas for future research.
MSA is a rare neurodegenerative disorder that entails autonomic failure, cerebellar ataxia, and parkinsonism. Laryngeal stridor has a high positive predictive value in the diagnosis of MSA, but consensus about its definition and clinical implications had not been established previously. The Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) delle Scienze Neurologiche di Bologna (Italy) convened a consensus conference of experts in 2017 to determine diagnostic criteria for stridor in MSA, define its prognostic value, suggest treatment options, and indicate subjects for future research. The neurologists reviewed studies of any design that reported original data. They based their statements on 34 published articles, most of which were class III or IV.
The authors defined stridor in MSA as “a strained, high-pitched, harsh respiratory sound, mainly inspiratory, caused by laryngeal dysfunction leading to narrowing of the rima glottidis.” Stridor may occur exclusively during sleep or during sleep and wakefulness. It may be recognized during a clinical examination, through witness report, or through an audio recording. Neurologists may consider laryngoscopy to exclude mechanical lesions or functional vocal cord abnormalities related to other neurologic conditions, wrote the authors. Drug-induced sleep endoscopy and video polysomnography also may be considered.
Whether stridor, or certain features of stridor, affects survival in MSA is uncertain. “Stridor within 3 years of motor or autonomic symptom onset may shorten survival,” according to the statement. “However, identification of stridor onset may be difficult.” Moreover, stridor during wakefulness is considered to reflect a more advanced stage of disease, compared with stridor during sleep. Although stridor can be distressing for the patient and his or her caregivers, its influence on health-related quality of life has yet to be determined, according to the statement.
Continuous positive airway pressure (CPAP) during sleep can be a useful symptomatic treatment and should be considered a first-line therapy for stridor, wrote the authors. Tracheostomy, another effective symptomatic treatment, bypasses upper-airway obstruction at the larynx. “Persistent and severe stridor may require tracheostomy,” according to the statement. It is not certain whether CPAP improves survival in patients with MSA and stridor, and tracheostomy may improve survival. The literature contains insufficient evidence about whether minimally invasive procedures or botulinum toxin injections are effective symptomatic treatments for stridor, wrote the authors.
During their review of the literature, the authors identified what they considered to be several research gaps. The diagnosis of stridor remains challenging, and investigators should develop a questionnaire for detecting stridor, they wrote. A smartphone application also could be developed to recognize stridor automatically. “The relationship between stridor and other breathing disorders (i.e., central apneas and breathing rate abnormalities) and their respective contributions to disease prognosis and survival should be determined through a multicenter prospective study,” according to the statement. Finally, randomized controlled trials comparing CPAP and tracheostomy for various degrees of stridor could guide physicians’ choice of treatment.
The IRCCS funded the study. One of the authors is a section editor for Neurology, and other authors reported receiving honoraria from various companies such as Novartis, Sanofi, and UCB.
The statement was published Oct. 1 in Neurology. In addition to reviewing the literature on the topic and providing recommendations, the authors described several areas for future research.
MSA is a rare neurodegenerative disorder that entails autonomic failure, cerebellar ataxia, and parkinsonism. Laryngeal stridor has a high positive predictive value in the diagnosis of MSA, but consensus about its definition and clinical implications had not been established previously. The Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) delle Scienze Neurologiche di Bologna (Italy) convened a consensus conference of experts in 2017 to determine diagnostic criteria for stridor in MSA, define its prognostic value, suggest treatment options, and indicate subjects for future research. The neurologists reviewed studies of any design that reported original data. They based their statements on 34 published articles, most of which were class III or IV.
The authors defined stridor in MSA as “a strained, high-pitched, harsh respiratory sound, mainly inspiratory, caused by laryngeal dysfunction leading to narrowing of the rima glottidis.” Stridor may occur exclusively during sleep or during sleep and wakefulness. It may be recognized during a clinical examination, through witness report, or through an audio recording. Neurologists may consider laryngoscopy to exclude mechanical lesions or functional vocal cord abnormalities related to other neurologic conditions, wrote the authors. Drug-induced sleep endoscopy and video polysomnography also may be considered.
Whether stridor, or certain features of stridor, affects survival in MSA is uncertain. “Stridor within 3 years of motor or autonomic symptom onset may shorten survival,” according to the statement. “However, identification of stridor onset may be difficult.” Moreover, stridor during wakefulness is considered to reflect a more advanced stage of disease, compared with stridor during sleep. Although stridor can be distressing for the patient and his or her caregivers, its influence on health-related quality of life has yet to be determined, according to the statement.
Continuous positive airway pressure (CPAP) during sleep can be a useful symptomatic treatment and should be considered a first-line therapy for stridor, wrote the authors. Tracheostomy, another effective symptomatic treatment, bypasses upper-airway obstruction at the larynx. “Persistent and severe stridor may require tracheostomy,” according to the statement. It is not certain whether CPAP improves survival in patients with MSA and stridor, and tracheostomy may improve survival. The literature contains insufficient evidence about whether minimally invasive procedures or botulinum toxin injections are effective symptomatic treatments for stridor, wrote the authors.
During their review of the literature, the authors identified what they considered to be several research gaps. The diagnosis of stridor remains challenging, and investigators should develop a questionnaire for detecting stridor, they wrote. A smartphone application also could be developed to recognize stridor automatically. “The relationship between stridor and other breathing disorders (i.e., central apneas and breathing rate abnormalities) and their respective contributions to disease prognosis and survival should be determined through a multicenter prospective study,” according to the statement. Finally, randomized controlled trials comparing CPAP and tracheostomy for various degrees of stridor could guide physicians’ choice of treatment.
The IRCCS funded the study. One of the authors is a section editor for Neurology, and other authors reported receiving honoraria from various companies such as Novartis, Sanofi, and UCB.
The statement was published Oct. 1 in Neurology. In addition to reviewing the literature on the topic and providing recommendations, the authors described several areas for future research.
MSA is a rare neurodegenerative disorder that entails autonomic failure, cerebellar ataxia, and parkinsonism. Laryngeal stridor has a high positive predictive value in the diagnosis of MSA, but consensus about its definition and clinical implications had not been established previously. The Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) delle Scienze Neurologiche di Bologna (Italy) convened a consensus conference of experts in 2017 to determine diagnostic criteria for stridor in MSA, define its prognostic value, suggest treatment options, and indicate subjects for future research. The neurologists reviewed studies of any design that reported original data. They based their statements on 34 published articles, most of which were class III or IV.
The authors defined stridor in MSA as “a strained, high-pitched, harsh respiratory sound, mainly inspiratory, caused by laryngeal dysfunction leading to narrowing of the rima glottidis.” Stridor may occur exclusively during sleep or during sleep and wakefulness. It may be recognized during a clinical examination, through witness report, or through an audio recording. Neurologists may consider laryngoscopy to exclude mechanical lesions or functional vocal cord abnormalities related to other neurologic conditions, wrote the authors. Drug-induced sleep endoscopy and video polysomnography also may be considered.
Whether stridor, or certain features of stridor, affects survival in MSA is uncertain. “Stridor within 3 years of motor or autonomic symptom onset may shorten survival,” according to the statement. “However, identification of stridor onset may be difficult.” Moreover, stridor during wakefulness is considered to reflect a more advanced stage of disease, compared with stridor during sleep. Although stridor can be distressing for the patient and his or her caregivers, its influence on health-related quality of life has yet to be determined, according to the statement.
Continuous positive airway pressure (CPAP) during sleep can be a useful symptomatic treatment and should be considered a first-line therapy for stridor, wrote the authors. Tracheostomy, another effective symptomatic treatment, bypasses upper-airway obstruction at the larynx. “Persistent and severe stridor may require tracheostomy,” according to the statement. It is not certain whether CPAP improves survival in patients with MSA and stridor, and tracheostomy may improve survival. The literature contains insufficient evidence about whether minimally invasive procedures or botulinum toxin injections are effective symptomatic treatments for stridor, wrote the authors.
During their review of the literature, the authors identified what they considered to be several research gaps. The diagnosis of stridor remains challenging, and investigators should develop a questionnaire for detecting stridor, they wrote. A smartphone application also could be developed to recognize stridor automatically. “The relationship between stridor and other breathing disorders (i.e., central apneas and breathing rate abnormalities) and their respective contributions to disease prognosis and survival should be determined through a multicenter prospective study,” according to the statement. Finally, randomized controlled trials comparing CPAP and tracheostomy for various degrees of stridor could guide physicians’ choice of treatment.
The IRCCS funded the study. One of the authors is a section editor for Neurology, and other authors reported receiving honoraria from various companies such as Novartis, Sanofi, and UCB.
FROM NEUROLOGY
FDA approves afamelanotide for treatment of rare condition with light-induced pain
The Food and Drug Administration has approved , a rare condition that causes extremely painful reactions when skin is exposed to light, according to an FDA announcement.

This is the first treatment approved to help patients with this condition increase their exposure to light, according to the release.
Afamelanotide, administered in a subcutaneous implant, is a melanocortin-1 receptor (MC1-R) agonist, which “increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources,” the release says.
Approval is based on a pair of parallel-group clinical trials that compared the number of hours spent in sunlight in the treatment and placebo groups. The first trial enrolled 93 patients; 48 received afamelanotide. The treated patients spent a median of 61 hours in total over 180 days in direct sunlight between 10 a.m. and 6 p.m. on days with no pain, compared with 41 hours for patients taking placebo.
The second trial assessed the total number of hours over 270 days spent outdoors between 10 a.m. and 3 p.m. on days with no pain for which “most of the day” was spent in direct sunlight. In this study, 38 patients treated with afamelanotide spent a median total of 6 hours, compared with 0.75 hours among the remaining 36 who were taking a placebo.
The most common side effects include implant site reaction, nausea, and oropharyngeal pain. The implant should be administered only by trained professionals. Because afamelanotide may cause skin darkening, it’s recommended that patients should undergo twice-yearly skin examinations. Patients are also encouraged to maintain sun protection measures to help prevent phototoxic reactions.
“Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible,” said Julie Beitz, MD, director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III in the FDA release.
The Food and Drug Administration has approved , a rare condition that causes extremely painful reactions when skin is exposed to light, according to an FDA announcement.
This is the first treatment approved to help patients with this condition increase their exposure to light, according to the release.
Afamelanotide, administered in a subcutaneous implant, is a melanocortin-1 receptor (MC1-R) agonist, which “increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources,” the release says.
Approval is based on a pair of parallel-group clinical trials that compared the number of hours spent in sunlight in the treatment and placebo groups. The first trial enrolled 93 patients; 48 received afamelanotide. The treated patients spent a median of 61 hours in total over 180 days in direct sunlight between 10 a.m. and 6 p.m. on days with no pain, compared with 41 hours for patients taking placebo.
The second trial assessed the total number of hours over 270 days spent outdoors between 10 a.m. and 3 p.m. on days with no pain for which “most of the day” was spent in direct sunlight. In this study, 38 patients treated with afamelanotide spent a median total of 6 hours, compared with 0.75 hours among the remaining 36 who were taking a placebo.
The most common side effects include implant site reaction, nausea, and oropharyngeal pain. The implant should be administered only by trained professionals. Because afamelanotide may cause skin darkening, it’s recommended that patients should undergo twice-yearly skin examinations. Patients are also encouraged to maintain sun protection measures to help prevent phototoxic reactions.
“Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible,” said Julie Beitz, MD, director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III in the FDA release.
The Food and Drug Administration has approved , a rare condition that causes extremely painful reactions when skin is exposed to light, according to an FDA announcement.
This is the first treatment approved to help patients with this condition increase their exposure to light, according to the release.
Afamelanotide, administered in a subcutaneous implant, is a melanocortin-1 receptor (MC1-R) agonist, which “increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources,” the release says.
Approval is based on a pair of parallel-group clinical trials that compared the number of hours spent in sunlight in the treatment and placebo groups. The first trial enrolled 93 patients; 48 received afamelanotide. The treated patients spent a median of 61 hours in total over 180 days in direct sunlight between 10 a.m. and 6 p.m. on days with no pain, compared with 41 hours for patients taking placebo.
The second trial assessed the total number of hours over 270 days spent outdoors between 10 a.m. and 3 p.m. on days with no pain for which “most of the day” was spent in direct sunlight. In this study, 38 patients treated with afamelanotide spent a median total of 6 hours, compared with 0.75 hours among the remaining 36 who were taking a placebo.
The most common side effects include implant site reaction, nausea, and oropharyngeal pain. The implant should be administered only by trained professionals. Because afamelanotide may cause skin darkening, it’s recommended that patients should undergo twice-yearly skin examinations. Patients are also encouraged to maintain sun protection measures to help prevent phototoxic reactions.
“Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible,” said Julie Beitz, MD, director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III in the FDA release.
C-Path and NORD team up to speed development of treatments for rare disorders
ROCKVILLE, MD – according to information provided at a launch event held Sept. 18, 2019.
By integrating data in a regulatory-grade format suitable for analytics, the RDCA-DAP hopes to accelerate the understanding of disease progression – including source of variability to optimize the characterization of subpopulations – develop clinical outcome measures and biomarkers, facilitate the development of mathematical models of disease, and promote innovative clinical trial designs.
The RDCA-DAP works as a database that will house patient-level data from a variety of sources, including clinical trials, longitudinal observational studies, patient registries, and other sources, such as real-world data collected from electronic health records across a wide range of rare diseases from all over the world. Data will then be made available to researchers to help speed the development of new treatments.
“The database and analytics we are creating will enable us to obtain new insight into these rare diseases,” C-Path President and CEO Joseph Scheeren, PharmD, said at the launch event. “Not only within specific diseases, but also we hope across related diseases.”

The key to the platform’s success will be data. “We need access to clinical data from the industry, patient groups, and academia,” Dr. Scheeren said. “Even more so, the RDCA-DAP will need to incorporate data from many sources. In addition to data from clinical trials conducted by industry and academia, we will want to access data from patients, hospitals, and any organization that can provide data.”
Dr. Scheeren said the data will take many forms, including numeric data, images, genomic information, and other forms of clinical information.
“The database will be able to handle these diverse datasets,” he said, adding that C-Path is preparing to be able to analyze the data sets “with the most sophisticated tools available.”
Dr. Scheeren made a call for all interested stakeholders with rare disease data to contribute to the platform.

NORD President and CEO Peter Saltonstall echoed that call. “We need data. We are accepting it immediately.”
Janet Woodcock, MD, director of the FDA Center for Drug Evaluation and Research, applauded the new platform. “I think foundations and patient advocacy groups and others that have been trying to help in this space have realized that simply funding basic research, although it is necessary and really important, it is not enough to get those therapies in the hands of doctors and patients,” she said. “You have to enable translation of research for that disease.”

Dr. Woodcock noted that the cures may not even come from that basic research, but rather from “left field” using research into cancer or another disease state, something that will be enabled by the disease-agnostic platform being created by C-Path and NORD. She said that the platform will not only put all the data in one spot, but will help to create a standardized set of disease definitions to help make the data useful across all research.
ROCKVILLE, MD – according to information provided at a launch event held Sept. 18, 2019.
By integrating data in a regulatory-grade format suitable for analytics, the RDCA-DAP hopes to accelerate the understanding of disease progression – including source of variability to optimize the characterization of subpopulations – develop clinical outcome measures and biomarkers, facilitate the development of mathematical models of disease, and promote innovative clinical trial designs.
The RDCA-DAP works as a database that will house patient-level data from a variety of sources, including clinical trials, longitudinal observational studies, patient registries, and other sources, such as real-world data collected from electronic health records across a wide range of rare diseases from all over the world. Data will then be made available to researchers to help speed the development of new treatments.
“The database and analytics we are creating will enable us to obtain new insight into these rare diseases,” C-Path President and CEO Joseph Scheeren, PharmD, said at the launch event. “Not only within specific diseases, but also we hope across related diseases.”
The key to the platform’s success will be data. “We need access to clinical data from the industry, patient groups, and academia,” Dr. Scheeren said. “Even more so, the RDCA-DAP will need to incorporate data from many sources. In addition to data from clinical trials conducted by industry and academia, we will want to access data from patients, hospitals, and any organization that can provide data.”
Dr. Scheeren said the data will take many forms, including numeric data, images, genomic information, and other forms of clinical information.
“The database will be able to handle these diverse datasets,” he said, adding that C-Path is preparing to be able to analyze the data sets “with the most sophisticated tools available.”
Dr. Scheeren made a call for all interested stakeholders with rare disease data to contribute to the platform.
NORD President and CEO Peter Saltonstall echoed that call. “We need data. We are accepting it immediately.”
Janet Woodcock, MD, director of the FDA Center for Drug Evaluation and Research, applauded the new platform. “I think foundations and patient advocacy groups and others that have been trying to help in this space have realized that simply funding basic research, although it is necessary and really important, it is not enough to get those therapies in the hands of doctors and patients,” she said. “You have to enable translation of research for that disease.”
Dr. Woodcock noted that the cures may not even come from that basic research, but rather from “left field” using research into cancer or another disease state, something that will be enabled by the disease-agnostic platform being created by C-Path and NORD. She said that the platform will not only put all the data in one spot, but will help to create a standardized set of disease definitions to help make the data useful across all research.
ROCKVILLE, MD – according to information provided at a launch event held Sept. 18, 2019.
By integrating data in a regulatory-grade format suitable for analytics, the RDCA-DAP hopes to accelerate the understanding of disease progression – including source of variability to optimize the characterization of subpopulations – develop clinical outcome measures and biomarkers, facilitate the development of mathematical models of disease, and promote innovative clinical trial designs.
The RDCA-DAP works as a database that will house patient-level data from a variety of sources, including clinical trials, longitudinal observational studies, patient registries, and other sources, such as real-world data collected from electronic health records across a wide range of rare diseases from all over the world. Data will then be made available to researchers to help speed the development of new treatments.
“The database and analytics we are creating will enable us to obtain new insight into these rare diseases,” C-Path President and CEO Joseph Scheeren, PharmD, said at the launch event. “Not only within specific diseases, but also we hope across related diseases.”
The key to the platform’s success will be data. “We need access to clinical data from the industry, patient groups, and academia,” Dr. Scheeren said. “Even more so, the RDCA-DAP will need to incorporate data from many sources. In addition to data from clinical trials conducted by industry and academia, we will want to access data from patients, hospitals, and any organization that can provide data.”
Dr. Scheeren said the data will take many forms, including numeric data, images, genomic information, and other forms of clinical information.
“The database will be able to handle these diverse datasets,” he said, adding that C-Path is preparing to be able to analyze the data sets “with the most sophisticated tools available.”
Dr. Scheeren made a call for all interested stakeholders with rare disease data to contribute to the platform.
NORD President and CEO Peter Saltonstall echoed that call. “We need data. We are accepting it immediately.”
Janet Woodcock, MD, director of the FDA Center for Drug Evaluation and Research, applauded the new platform. “I think foundations and patient advocacy groups and others that have been trying to help in this space have realized that simply funding basic research, although it is necessary and really important, it is not enough to get those therapies in the hands of doctors and patients,” she said. “You have to enable translation of research for that disease.”
Dr. Woodcock noted that the cures may not even come from that basic research, but rather from “left field” using research into cancer or another disease state, something that will be enabled by the disease-agnostic platform being created by C-Path and NORD. She said that the platform will not only put all the data in one spot, but will help to create a standardized set of disease definitions to help make the data useful across all research.