User login
In utero Zika exposure can have delayed consequences
WASHINGTON – Evidence continues to mount that infants born to moms infected with Zika virus during pregnancy can have neurodevelopmental abnormalities as they age even if they showed no defects at birth, based on follow-up of 890 Colombian children tracked by epidemiologists from the U.S. Centers for Disease Control and Prevention.

Among the 890 neonates born to mothers apparently infected with Zika during pregnancy and followed for up to 2 years, 40 of the 852 (5%) without a detectable birth defect at delivery went on to show some type of neurodevelopmental sequelae during up to 24 months of age, Margaret Honein, PhD, said at an annual scientific meeting on infectious diseases.
In addition, among the children without birth defects at delivery who received follow-up examinations out to about 2 years, the incidence of “alerts” for possible neurodevelopmental issues was 15%-20% for each of the four domains studied (gross motor, fine motor, hearing and language, and personal and social functions), said Dr. Honein, an epidemiologist and chief of the birth defects branch of the CDC. In contrast, 17 of the 38 children (45%) followed who had identifiable birth defects at delivery also showed neurodevelopmental abnormalities when reexamined as long as 2 years after birth. These possible neurodevelopmental abnormalities, designated as alerts, were identified in comparison with a contemporaneous cohort of children born to uninfected mothers in the same regions of Colombia and assessed by the CDC researchers.
This cohort of children born to mothers who became infected with Zika virus during the 2016 Colombian epidemic will not undergo any planned, additional follow-up beyond the initial 2 years, Dr. Honein noted.

The findings she reported were consistent with observations from a much smaller cohort of 70 infants born to Colombian mothers infected with Zika virus while pregnant who had a normal head circumference and a normal clinical examination at delivery. When assessed once or twice 4-18 months after birth, these 70 infants showed an overall greater than one standard deviation (z-score) drop in their scores on the Warner Initial Developmental Evaluation of Adaptive and Functional Skills (WIDEA) metric by 12 months after birth and continuing out to 18 months, said Sarah B. Mulkey, MD, a fetal-neonatal neurologist at Children’s National Health System in Washington. These deficits were especially pronounced in the mobility and social cognition domains of the four-domain WIDEA metric. The social cognition domain is an important predictor of later problems with executive function and other neurologic disorders, Dr. Mulkey said while reporting her findings in a separate talk at the meeting. She acknowledged that the analysis was flawed by comparing the WIDEA outcomes of the Zika virus–exposed children to healthy children from either inner-city Chicago or Canada. Dr. Mulkey said that she and her associates plan to characterize a population of Zika virus–unexposed children in Colombia to use for future comparisons.
The study reported by Dr. Honein involved an enhanced surveillance program launched by the CDC in 2016 in three regions of Colombia and included 1,190 pregnancies accompanied by Zika symptoms in the mother and with a reported pregnancy outcome, including 1,185 live births. Nearly half of the Zika infections occurred during the first trimester, and 34% occurred during the second trimester. However, fewer than a third of the pregnant women underwent some type of laboratory testing to confirm their infection, either by serology or by a DNA-based assay, and of these 28% had a positive finding. Dr. Honein cautioned that many of the specimens that tested negative for Zika virus may have been false negatives.
The birth defects identified among the infants born from an apparently affected pregnancy included brain abnormalities, eye anomalies, and microcephaly, with 5% of the 1,185 live births showing one or more of these outcomes. The neurodevelopmental deficits identified during follow-up of 890 of the children out to 2 years included seizures; abnormalities of tone, movement, or swallowing; and impairments of vision or hearing.
WASHINGTON – Evidence continues to mount that infants born to moms infected with Zika virus during pregnancy can have neurodevelopmental abnormalities as they age even if they showed no defects at birth, based on follow-up of 890 Colombian children tracked by epidemiologists from the U.S. Centers for Disease Control and Prevention.
Among the 890 neonates born to mothers apparently infected with Zika during pregnancy and followed for up to 2 years, 40 of the 852 (5%) without a detectable birth defect at delivery went on to show some type of neurodevelopmental sequelae during up to 24 months of age, Margaret Honein, PhD, said at an annual scientific meeting on infectious diseases.
In addition, among the children without birth defects at delivery who received follow-up examinations out to about 2 years, the incidence of “alerts” for possible neurodevelopmental issues was 15%-20% for each of the four domains studied (gross motor, fine motor, hearing and language, and personal and social functions), said Dr. Honein, an epidemiologist and chief of the birth defects branch of the CDC. In contrast, 17 of the 38 children (45%) followed who had identifiable birth defects at delivery also showed neurodevelopmental abnormalities when reexamined as long as 2 years after birth. These possible neurodevelopmental abnormalities, designated as alerts, were identified in comparison with a contemporaneous cohort of children born to uninfected mothers in the same regions of Colombia and assessed by the CDC researchers.
This cohort of children born to mothers who became infected with Zika virus during the 2016 Colombian epidemic will not undergo any planned, additional follow-up beyond the initial 2 years, Dr. Honein noted.
The findings she reported were consistent with observations from a much smaller cohort of 70 infants born to Colombian mothers infected with Zika virus while pregnant who had a normal head circumference and a normal clinical examination at delivery. When assessed once or twice 4-18 months after birth, these 70 infants showed an overall greater than one standard deviation (z-score) drop in their scores on the Warner Initial Developmental Evaluation of Adaptive and Functional Skills (WIDEA) metric by 12 months after birth and continuing out to 18 months, said Sarah B. Mulkey, MD, a fetal-neonatal neurologist at Children’s National Health System in Washington. These deficits were especially pronounced in the mobility and social cognition domains of the four-domain WIDEA metric. The social cognition domain is an important predictor of later problems with executive function and other neurologic disorders, Dr. Mulkey said while reporting her findings in a separate talk at the meeting. She acknowledged that the analysis was flawed by comparing the WIDEA outcomes of the Zika virus–exposed children to healthy children from either inner-city Chicago or Canada. Dr. Mulkey said that she and her associates plan to characterize a population of Zika virus–unexposed children in Colombia to use for future comparisons.
The study reported by Dr. Honein involved an enhanced surveillance program launched by the CDC in 2016 in three regions of Colombia and included 1,190 pregnancies accompanied by Zika symptoms in the mother and with a reported pregnancy outcome, including 1,185 live births. Nearly half of the Zika infections occurred during the first trimester, and 34% occurred during the second trimester. However, fewer than a third of the pregnant women underwent some type of laboratory testing to confirm their infection, either by serology or by a DNA-based assay, and of these 28% had a positive finding. Dr. Honein cautioned that many of the specimens that tested negative for Zika virus may have been false negatives.
The birth defects identified among the infants born from an apparently affected pregnancy included brain abnormalities, eye anomalies, and microcephaly, with 5% of the 1,185 live births showing one or more of these outcomes. The neurodevelopmental deficits identified during follow-up of 890 of the children out to 2 years included seizures; abnormalities of tone, movement, or swallowing; and impairments of vision or hearing.
WASHINGTON – Evidence continues to mount that infants born to moms infected with Zika virus during pregnancy can have neurodevelopmental abnormalities as they age even if they showed no defects at birth, based on follow-up of 890 Colombian children tracked by epidemiologists from the U.S. Centers for Disease Control and Prevention.
Among the 890 neonates born to mothers apparently infected with Zika during pregnancy and followed for up to 2 years, 40 of the 852 (5%) without a detectable birth defect at delivery went on to show some type of neurodevelopmental sequelae during up to 24 months of age, Margaret Honein, PhD, said at an annual scientific meeting on infectious diseases.
In addition, among the children without birth defects at delivery who received follow-up examinations out to about 2 years, the incidence of “alerts” for possible neurodevelopmental issues was 15%-20% for each of the four domains studied (gross motor, fine motor, hearing and language, and personal and social functions), said Dr. Honein, an epidemiologist and chief of the birth defects branch of the CDC. In contrast, 17 of the 38 children (45%) followed who had identifiable birth defects at delivery also showed neurodevelopmental abnormalities when reexamined as long as 2 years after birth. These possible neurodevelopmental abnormalities, designated as alerts, were identified in comparison with a contemporaneous cohort of children born to uninfected mothers in the same regions of Colombia and assessed by the CDC researchers.
This cohort of children born to mothers who became infected with Zika virus during the 2016 Colombian epidemic will not undergo any planned, additional follow-up beyond the initial 2 years, Dr. Honein noted.
The findings she reported were consistent with observations from a much smaller cohort of 70 infants born to Colombian mothers infected with Zika virus while pregnant who had a normal head circumference and a normal clinical examination at delivery. When assessed once or twice 4-18 months after birth, these 70 infants showed an overall greater than one standard deviation (z-score) drop in their scores on the Warner Initial Developmental Evaluation of Adaptive and Functional Skills (WIDEA) metric by 12 months after birth and continuing out to 18 months, said Sarah B. Mulkey, MD, a fetal-neonatal neurologist at Children’s National Health System in Washington. These deficits were especially pronounced in the mobility and social cognition domains of the four-domain WIDEA metric. The social cognition domain is an important predictor of later problems with executive function and other neurologic disorders, Dr. Mulkey said while reporting her findings in a separate talk at the meeting. She acknowledged that the analysis was flawed by comparing the WIDEA outcomes of the Zika virus–exposed children to healthy children from either inner-city Chicago or Canada. Dr. Mulkey said that she and her associates plan to characterize a population of Zika virus–unexposed children in Colombia to use for future comparisons.
The study reported by Dr. Honein involved an enhanced surveillance program launched by the CDC in 2016 in three regions of Colombia and included 1,190 pregnancies accompanied by Zika symptoms in the mother and with a reported pregnancy outcome, including 1,185 live births. Nearly half of the Zika infections occurred during the first trimester, and 34% occurred during the second trimester. However, fewer than a third of the pregnant women underwent some type of laboratory testing to confirm their infection, either by serology or by a DNA-based assay, and of these 28% had a positive finding. Dr. Honein cautioned that many of the specimens that tested negative for Zika virus may have been false negatives.
The birth defects identified among the infants born from an apparently affected pregnancy included brain abnormalities, eye anomalies, and microcephaly, with 5% of the 1,185 live births showing one or more of these outcomes. The neurodevelopmental deficits identified during follow-up of 890 of the children out to 2 years included seizures; abnormalities of tone, movement, or swallowing; and impairments of vision or hearing.
REPORTING FROM ID WEEK 2019
Congenital heart disease in children linked to increased autism risk
A new study of children who were born with congenital heart disease (CHD) has found that they have increased odds of developing autism spectrum disorder.
“To our knowledge, this is the only study in which there has been a comparison between [autism spectrum disorder] and multiple CHD subtypes,” wrote Eric R. Sigmon, MD, of Emory University, Atlanta, and coauthors. “Our findings are consistent with previous studies of CHD developmental outcomes, which have shown an increased risk of developmental and academic delay after CHD diagnosis and treatment.” The study was published in Pediatrics.
To further investigate the association between CHD and autism, the researchers performed a case-control study using the Military Health System administrative database. They uncovered 8,760 cases of children with autism spectrum disorder and matched each one with three controls (n = 26,280). From that sample size, they identified 1,063 children with CHD: 401 in the autism spectrum disorder group and 662 in the control group.
Before analysis, children with autism spectrum disorder had an odds ratio of 1.85 of having any form of CHD, compared with controls (95% confidence interval, 1.63-2.10). After adjustment for covariates – including genetic syndromes, maternal age and morbidity, perinatal morbidity, and neonatal complications – the OR was 1.33 (95% CI, 1.16-1.52).
In the sensitivity analysis – which included only 593 children with CHD – the OR was a similar 1.32 (95% CI, 1.10-1.59).
Left heart obstructive lesion was significantly associated with autism spectrum disorder after covariate adjustment (OR, 1.42; 95% CI, 1.04-1.93), but the finding was no longer significant in the sensitivity analysis.
The authors noted the potential limitations of their study, including the general weaknesses of administrative data, which they attempted to counter with the sensitive analysis. In addition, they recognized that children with either autism spectrum disorder or CHD “tend to present for care more frequently,” which could have created an ascertainment bias.
In an accompanying editorial, Johanna Calderon, PhD, David C. Bellinger, PhD, and Jane W. Newburger, MD, MPH, stated that more work needs to be done to further quantify the relationship between CHD and autism spectrum disorder (Pediatrics. 2019 Oct 10. doi: 10.1542/peds.2019-2752). The three authors – all affiliated with Boston Children’s Hospital and Harvard Medical School, also in Boston – reiterated the acknowledgment from Dr. Sigmon and coauthors that the “etiologic pathways that might explain” the link between the two remains unknown. They also noted their surprise that autism spectrum disorder risk appears to be increased in children with modestly severe forms of CHD, stating that this finding required additional investigation.
“Despite the strengths of this study,” they wrote, “it raises more questions than answers.”
The study was funded by the Congressional Directed Medical Research Programs Autism Research Award. The authors reported no conflicts of interest.
SOURCE: Sigmon ER at al. Pediatrics. 2019 Oct 10. doi: 10.1542/peds.2018-4114.
A new study of children who were born with congenital heart disease (CHD) has found that they have increased odds of developing autism spectrum disorder.
“To our knowledge, this is the only study in which there has been a comparison between [autism spectrum disorder] and multiple CHD subtypes,” wrote Eric R. Sigmon, MD, of Emory University, Atlanta, and coauthors. “Our findings are consistent with previous studies of CHD developmental outcomes, which have shown an increased risk of developmental and academic delay after CHD diagnosis and treatment.” The study was published in Pediatrics.
To further investigate the association between CHD and autism, the researchers performed a case-control study using the Military Health System administrative database. They uncovered 8,760 cases of children with autism spectrum disorder and matched each one with three controls (n = 26,280). From that sample size, they identified 1,063 children with CHD: 401 in the autism spectrum disorder group and 662 in the control group.
Before analysis, children with autism spectrum disorder had an odds ratio of 1.85 of having any form of CHD, compared with controls (95% confidence interval, 1.63-2.10). After adjustment for covariates – including genetic syndromes, maternal age and morbidity, perinatal morbidity, and neonatal complications – the OR was 1.33 (95% CI, 1.16-1.52).
In the sensitivity analysis – which included only 593 children with CHD – the OR was a similar 1.32 (95% CI, 1.10-1.59).
Left heart obstructive lesion was significantly associated with autism spectrum disorder after covariate adjustment (OR, 1.42; 95% CI, 1.04-1.93), but the finding was no longer significant in the sensitivity analysis.
The authors noted the potential limitations of their study, including the general weaknesses of administrative data, which they attempted to counter with the sensitive analysis. In addition, they recognized that children with either autism spectrum disorder or CHD “tend to present for care more frequently,” which could have created an ascertainment bias.
In an accompanying editorial, Johanna Calderon, PhD, David C. Bellinger, PhD, and Jane W. Newburger, MD, MPH, stated that more work needs to be done to further quantify the relationship between CHD and autism spectrum disorder (Pediatrics. 2019 Oct 10. doi: 10.1542/peds.2019-2752). The three authors – all affiliated with Boston Children’s Hospital and Harvard Medical School, also in Boston – reiterated the acknowledgment from Dr. Sigmon and coauthors that the “etiologic pathways that might explain” the link between the two remains unknown. They also noted their surprise that autism spectrum disorder risk appears to be increased in children with modestly severe forms of CHD, stating that this finding required additional investigation.
“Despite the strengths of this study,” they wrote, “it raises more questions than answers.”
The study was funded by the Congressional Directed Medical Research Programs Autism Research Award. The authors reported no conflicts of interest.
SOURCE: Sigmon ER at al. Pediatrics. 2019 Oct 10. doi: 10.1542/peds.2018-4114.
A new study of children who were born with congenital heart disease (CHD) has found that they have increased odds of developing autism spectrum disorder.
“To our knowledge, this is the only study in which there has been a comparison between [autism spectrum disorder] and multiple CHD subtypes,” wrote Eric R. Sigmon, MD, of Emory University, Atlanta, and coauthors. “Our findings are consistent with previous studies of CHD developmental outcomes, which have shown an increased risk of developmental and academic delay after CHD diagnosis and treatment.” The study was published in Pediatrics.
To further investigate the association between CHD and autism, the researchers performed a case-control study using the Military Health System administrative database. They uncovered 8,760 cases of children with autism spectrum disorder and matched each one with three controls (n = 26,280). From that sample size, they identified 1,063 children with CHD: 401 in the autism spectrum disorder group and 662 in the control group.
Before analysis, children with autism spectrum disorder had an odds ratio of 1.85 of having any form of CHD, compared with controls (95% confidence interval, 1.63-2.10). After adjustment for covariates – including genetic syndromes, maternal age and morbidity, perinatal morbidity, and neonatal complications – the OR was 1.33 (95% CI, 1.16-1.52).
In the sensitivity analysis – which included only 593 children with CHD – the OR was a similar 1.32 (95% CI, 1.10-1.59).
Left heart obstructive lesion was significantly associated with autism spectrum disorder after covariate adjustment (OR, 1.42; 95% CI, 1.04-1.93), but the finding was no longer significant in the sensitivity analysis.
The authors noted the potential limitations of their study, including the general weaknesses of administrative data, which they attempted to counter with the sensitive analysis. In addition, they recognized that children with either autism spectrum disorder or CHD “tend to present for care more frequently,” which could have created an ascertainment bias.
In an accompanying editorial, Johanna Calderon, PhD, David C. Bellinger, PhD, and Jane W. Newburger, MD, MPH, stated that more work needs to be done to further quantify the relationship between CHD and autism spectrum disorder (Pediatrics. 2019 Oct 10. doi: 10.1542/peds.2019-2752). The three authors – all affiliated with Boston Children’s Hospital and Harvard Medical School, also in Boston – reiterated the acknowledgment from Dr. Sigmon and coauthors that the “etiologic pathways that might explain” the link between the two remains unknown. They also noted their surprise that autism spectrum disorder risk appears to be increased in children with modestly severe forms of CHD, stating that this finding required additional investigation.
“Despite the strengths of this study,” they wrote, “it raises more questions than answers.”
The study was funded by the Congressional Directed Medical Research Programs Autism Research Award. The authors reported no conflicts of interest.
SOURCE: Sigmon ER at al. Pediatrics. 2019 Oct 10. doi: 10.1542/peds.2018-4114.
FROM PEDIATRICS
Key clinical point: Children born with congenital heart disease have higher odds of developing autism, especially with certain forms of CHD, such as atrial and ventricular septal defects.
Major finding: After sensitivity analysis, children with congenital heart disease had increased odds of autism, compared with controls (odds ratio, 1.32; 95% confidence interval, 1.10-1.59).
Study details: A case-control study of children enrolled in the U.S. Military Health System from 2001 to 2013.
Disclosures: The study was funded by the Congressional Directed Medical Research Programs Autism Research Award. The authors reported no conflicts of interest.
Source: Sigmon ER at al. Pediatrics. 2019 Oct 10. doi: 10.1542/peds.2018-4114.
Four genetic variants link psychotic experiences to multiple mental disorders
Four genetic variations appear to link psychotic experiences with other psychiatric disorders, including schizophrenia, major depressive disorder, bipolar disorder, and neurodevelopmental disorders, a large genetic study has concluded.
, reported Sophie E. Legge, PhD, and colleagues. Their study was published in JAMA Psychiatry.
Although it is informative, the study is unlikely to expand the knowledge of schizophrenia-specific genetics.
“Consistent with other studies, the heritability estimate (1.71%) was low, and given that the variance explained in our polygenic risk analysis was also low, the finding suggests that understanding the genetics of psychotic experiences is unlikely to have an important effect on understanding the genetics of schizophrenia specifically,” wrote Dr. Legge, of the MRC Center for Neuropsychiatric Genetics and Genomics in the division of psychological medicine and clinical neurosciences at Cardiff (Wales) University, and colleagues.
The team conducted a genomewide association study (GWAS) using data from 127,966 individuals in the U.K. Biobank. Of these, 6,123 reported any psychotic experience, 2,143 reported distressing psychotic experiences, and 3,337 reported multiple experiences. The remainder served as controls. At the time of the biobank data collection, the subjects were a mean of 64 years of age; 56% were women.
First psychotic experience occurred at a mean of almost 32 years of age, but about a third reported that the first episode occurred before age 20, or that psychotic experiences had been happening ever since they could remember. Another third reported their first experience between ages 40 and 76 years.
The investigators conducted three GWAS studies: one for any psychotic experience, one for distressing experiences, and one for multiple experiences.
No significant genetic associations were found among those with multiple psychotic experiences, the authors said.
But they did find four variants significantly associated with the other experience categories.
Two variants were associated with any psychotic experience. Those with rs10994278, an intronic variant within Ankyrin-3 (ANK3), were 16% more likely to have a psychotic experience (odds ratio, 1.16). Those with intergenic variant rs549656827 were 39% less likely (OR, 0.61). “The ANK3 gene encodes ankyrin-G, a protein that has been shown to regulate the assembly of voltage-gated sodium channels and is essential for normal synaptic function,” the authors said. “ANK3 is one of strongest and most replicated genes for bipolar disorder, and variants within ANK3 have also been associated in the Psychiatric Genomics Consortium cross-disorder GWAS, and in a rare variant analysis of autism spectrum disorder.”
Two variants were linked to distressing psychotic experiences: rs75459873, intronic to cannabinoid receptor 2 (CNR2), decreased the risk by 34% (OR, 0.66). Intergenic variant rs3849810 increased the risk by 12% (OR, 1.12).
“CNR2 encodes for CB2, one of two well-characterized cannabinoid receptors. Several lines of evidence have implicated the endocannabinoid system in psychiatric disorders, including schizophrenia and depression. The main psychoactive agent of cannabis, tetrahydrocannabinol, can cause acute psychotic symptoms and cognitive impairment. Given that cannabis use is strongly associated with psychotic experiences, we tested, but found no evidence for, a mediating or moderating effect of cannabis use on the association of rs75459873 and distressing psychotic experiences. However, while no evidence was found in this study, a mediating effect of cannabis use cannot be ruled out given the relatively low power of such analyses and the potential measurement error.”
Also, significant genetic correlations were found between any psychotic experiences and major depressive disorder, autism spectrum disorder, ADHD, and schizophrenia. However, the polygenic risk scores for schizophrenia, major depressive disorder, bipolar disorder, ADHD, and autism spectrum disorder, were low.
“We also considered individual psychotic symptoms and found that polygenic risk scores for schizophrenia, bipolar disorder, depression, and ADHD were more strongly associated with delusions of persecution than with the other psychotic symptoms.”
Those with distressing psychotic experiences tended to have more copy number variations (CNVs) associated with schizophrenia (OR, 2.04) and neurodevelopmental disorders (OR, 1.75). The team also found significant associations between distressing experiences and major depressive disorder, ADHD, autism spectrum disorder, and schizophrenia.
“We found particular enrichment of these [polygenic risk scores] in distressing psychotic experiences and for delusions of persecution,” they noted. “ ... All schizophrenia-associated [copy number variations] are also associated with neurodevelopmental disorders such as intellectual disability and autism.”
The study’s strengths include its large sample size. Among its limitations, the researchers said, are the study’s retrospective measurement of psychotic experiences based on self-report from a questionnaire that was online. Gathering the data in that way raised the likelihood of possible error, they said.
Dr. Legge reported having no disclosures.
SOURCE: Legge SE et al. JAMA Psychiatry. 2019 Sep 25. doi: 10.1001/jamapsychiatry.2019.2508.
The genetic links uncovered in this study offer an intriguing, but incomplete look at the risks of psychotic experiences and their complicated intertwinings with other mental disorders, wrote Albert R. Powers III, MD, PhD.
“Penetrance of the genes in question likely depends at least in part on environmental influences, some of which have been studied extensively,” he wrote. “Recently, some have proposed risk stratification by exposome – a composite score of relevant exposures that may increase risk for psychosis and is analogous to the polygenic risk score used [here].
“The combination of environmental and genetic composite scores may lead to improved insight into individualized pathways toward psychotic experiences, highlighting genetic vulnerabilities to specific stressors likely to lead to phenotypic expression. Ultimately, this will require a more sophisticated mapping between phenomenology and biology than currently exists.”
One approach would be to combine deep phenotyping and behavioral analyses in a framework that could link all relevant levels from symptoms to neurophysiology.
“One such framework is predictive processing theory, which is linked closely with the free energy principle and the Bayesian brain hypothesis and attempts to explain perceptual and cognitive phenomena as manifestations of a drive to maintain as accurate an internal model of one’s surroundings as possible by minimizing prediction errors. This relatively simple scheme makes specific – and, importantly, falsifiable – assessments of the mathematical signatures of neurotypical processes and the ways they might break down to produce specific psychiatric symptoms.”
Dr. Powers is an assistant professor at the department of psychiatry at Yale University, New Haven, Conn., and serves as medical director of the PRIME Psychosis Research Clinic at Yale. His comments came in an accompanying editorial (JAMA Psychiatry. 2019 Sep 25. doi: 10.1001/jamapsychiatry.2019.2391 ).
The genetic links uncovered in this study offer an intriguing, but incomplete look at the risks of psychotic experiences and their complicated intertwinings with other mental disorders, wrote Albert R. Powers III, MD, PhD.
“Penetrance of the genes in question likely depends at least in part on environmental influences, some of which have been studied extensively,” he wrote. “Recently, some have proposed risk stratification by exposome – a composite score of relevant exposures that may increase risk for psychosis and is analogous to the polygenic risk score used [here].
“The combination of environmental and genetic composite scores may lead to improved insight into individualized pathways toward psychotic experiences, highlighting genetic vulnerabilities to specific stressors likely to lead to phenotypic expression. Ultimately, this will require a more sophisticated mapping between phenomenology and biology than currently exists.”
One approach would be to combine deep phenotyping and behavioral analyses in a framework that could link all relevant levels from symptoms to neurophysiology.
“One such framework is predictive processing theory, which is linked closely with the free energy principle and the Bayesian brain hypothesis and attempts to explain perceptual and cognitive phenomena as manifestations of a drive to maintain as accurate an internal model of one’s surroundings as possible by minimizing prediction errors. This relatively simple scheme makes specific – and, importantly, falsifiable – assessments of the mathematical signatures of neurotypical processes and the ways they might break down to produce specific psychiatric symptoms.”
Dr. Powers is an assistant professor at the department of psychiatry at Yale University, New Haven, Conn., and serves as medical director of the PRIME Psychosis Research Clinic at Yale. His comments came in an accompanying editorial (JAMA Psychiatry. 2019 Sep 25. doi: 10.1001/jamapsychiatry.2019.2391 ).
The genetic links uncovered in this study offer an intriguing, but incomplete look at the risks of psychotic experiences and their complicated intertwinings with other mental disorders, wrote Albert R. Powers III, MD, PhD.
“Penetrance of the genes in question likely depends at least in part on environmental influences, some of which have been studied extensively,” he wrote. “Recently, some have proposed risk stratification by exposome – a composite score of relevant exposures that may increase risk for psychosis and is analogous to the polygenic risk score used [here].
“The combination of environmental and genetic composite scores may lead to improved insight into individualized pathways toward psychotic experiences, highlighting genetic vulnerabilities to specific stressors likely to lead to phenotypic expression. Ultimately, this will require a more sophisticated mapping between phenomenology and biology than currently exists.”
One approach would be to combine deep phenotyping and behavioral analyses in a framework that could link all relevant levels from symptoms to neurophysiology.
“One such framework is predictive processing theory, which is linked closely with the free energy principle and the Bayesian brain hypothesis and attempts to explain perceptual and cognitive phenomena as manifestations of a drive to maintain as accurate an internal model of one’s surroundings as possible by minimizing prediction errors. This relatively simple scheme makes specific – and, importantly, falsifiable – assessments of the mathematical signatures of neurotypical processes and the ways they might break down to produce specific psychiatric symptoms.”
Dr. Powers is an assistant professor at the department of psychiatry at Yale University, New Haven, Conn., and serves as medical director of the PRIME Psychosis Research Clinic at Yale. His comments came in an accompanying editorial (JAMA Psychiatry. 2019 Sep 25. doi: 10.1001/jamapsychiatry.2019.2391 ).
Four genetic variations appear to link psychotic experiences with other psychiatric disorders, including schizophrenia, major depressive disorder, bipolar disorder, and neurodevelopmental disorders, a large genetic study has concluded.
, reported Sophie E. Legge, PhD, and colleagues. Their study was published in JAMA Psychiatry.
Although it is informative, the study is unlikely to expand the knowledge of schizophrenia-specific genetics.
“Consistent with other studies, the heritability estimate (1.71%) was low, and given that the variance explained in our polygenic risk analysis was also low, the finding suggests that understanding the genetics of psychotic experiences is unlikely to have an important effect on understanding the genetics of schizophrenia specifically,” wrote Dr. Legge, of the MRC Center for Neuropsychiatric Genetics and Genomics in the division of psychological medicine and clinical neurosciences at Cardiff (Wales) University, and colleagues.
The team conducted a genomewide association study (GWAS) using data from 127,966 individuals in the U.K. Biobank. Of these, 6,123 reported any psychotic experience, 2,143 reported distressing psychotic experiences, and 3,337 reported multiple experiences. The remainder served as controls. At the time of the biobank data collection, the subjects were a mean of 64 years of age; 56% were women.
First psychotic experience occurred at a mean of almost 32 years of age, but about a third reported that the first episode occurred before age 20, or that psychotic experiences had been happening ever since they could remember. Another third reported their first experience between ages 40 and 76 years.
The investigators conducted three GWAS studies: one for any psychotic experience, one for distressing experiences, and one for multiple experiences.
No significant genetic associations were found among those with multiple psychotic experiences, the authors said.
But they did find four variants significantly associated with the other experience categories.
Two variants were associated with any psychotic experience. Those with rs10994278, an intronic variant within Ankyrin-3 (ANK3), were 16% more likely to have a psychotic experience (odds ratio, 1.16). Those with intergenic variant rs549656827 were 39% less likely (OR, 0.61). “The ANK3 gene encodes ankyrin-G, a protein that has been shown to regulate the assembly of voltage-gated sodium channels and is essential for normal synaptic function,” the authors said. “ANK3 is one of strongest and most replicated genes for bipolar disorder, and variants within ANK3 have also been associated in the Psychiatric Genomics Consortium cross-disorder GWAS, and in a rare variant analysis of autism spectrum disorder.”
Two variants were linked to distressing psychotic experiences: rs75459873, intronic to cannabinoid receptor 2 (CNR2), decreased the risk by 34% (OR, 0.66). Intergenic variant rs3849810 increased the risk by 12% (OR, 1.12).
“CNR2 encodes for CB2, one of two well-characterized cannabinoid receptors. Several lines of evidence have implicated the endocannabinoid system in psychiatric disorders, including schizophrenia and depression. The main psychoactive agent of cannabis, tetrahydrocannabinol, can cause acute psychotic symptoms and cognitive impairment. Given that cannabis use is strongly associated with psychotic experiences, we tested, but found no evidence for, a mediating or moderating effect of cannabis use on the association of rs75459873 and distressing psychotic experiences. However, while no evidence was found in this study, a mediating effect of cannabis use cannot be ruled out given the relatively low power of such analyses and the potential measurement error.”
Also, significant genetic correlations were found between any psychotic experiences and major depressive disorder, autism spectrum disorder, ADHD, and schizophrenia. However, the polygenic risk scores for schizophrenia, major depressive disorder, bipolar disorder, ADHD, and autism spectrum disorder, were low.
“We also considered individual psychotic symptoms and found that polygenic risk scores for schizophrenia, bipolar disorder, depression, and ADHD were more strongly associated with delusions of persecution than with the other psychotic symptoms.”
Those with distressing psychotic experiences tended to have more copy number variations (CNVs) associated with schizophrenia (OR, 2.04) and neurodevelopmental disorders (OR, 1.75). The team also found significant associations between distressing experiences and major depressive disorder, ADHD, autism spectrum disorder, and schizophrenia.
“We found particular enrichment of these [polygenic risk scores] in distressing psychotic experiences and for delusions of persecution,” they noted. “ ... All schizophrenia-associated [copy number variations] are also associated with neurodevelopmental disorders such as intellectual disability and autism.”
The study’s strengths include its large sample size. Among its limitations, the researchers said, are the study’s retrospective measurement of psychotic experiences based on self-report from a questionnaire that was online. Gathering the data in that way raised the likelihood of possible error, they said.
Dr. Legge reported having no disclosures.
SOURCE: Legge SE et al. JAMA Psychiatry. 2019 Sep 25. doi: 10.1001/jamapsychiatry.2019.2508.
Four genetic variations appear to link psychotic experiences with other psychiatric disorders, including schizophrenia, major depressive disorder, bipolar disorder, and neurodevelopmental disorders, a large genetic study has concluded.
, reported Sophie E. Legge, PhD, and colleagues. Their study was published in JAMA Psychiatry.
Although it is informative, the study is unlikely to expand the knowledge of schizophrenia-specific genetics.
“Consistent with other studies, the heritability estimate (1.71%) was low, and given that the variance explained in our polygenic risk analysis was also low, the finding suggests that understanding the genetics of psychotic experiences is unlikely to have an important effect on understanding the genetics of schizophrenia specifically,” wrote Dr. Legge, of the MRC Center for Neuropsychiatric Genetics and Genomics in the division of psychological medicine and clinical neurosciences at Cardiff (Wales) University, and colleagues.
The team conducted a genomewide association study (GWAS) using data from 127,966 individuals in the U.K. Biobank. Of these, 6,123 reported any psychotic experience, 2,143 reported distressing psychotic experiences, and 3,337 reported multiple experiences. The remainder served as controls. At the time of the biobank data collection, the subjects were a mean of 64 years of age; 56% were women.
First psychotic experience occurred at a mean of almost 32 years of age, but about a third reported that the first episode occurred before age 20, or that psychotic experiences had been happening ever since they could remember. Another third reported their first experience between ages 40 and 76 years.
The investigators conducted three GWAS studies: one for any psychotic experience, one for distressing experiences, and one for multiple experiences.
No significant genetic associations were found among those with multiple psychotic experiences, the authors said.
But they did find four variants significantly associated with the other experience categories.
Two variants were associated with any psychotic experience. Those with rs10994278, an intronic variant within Ankyrin-3 (ANK3), were 16% more likely to have a psychotic experience (odds ratio, 1.16). Those with intergenic variant rs549656827 were 39% less likely (OR, 0.61). “The ANK3 gene encodes ankyrin-G, a protein that has been shown to regulate the assembly of voltage-gated sodium channels and is essential for normal synaptic function,” the authors said. “ANK3 is one of strongest and most replicated genes for bipolar disorder, and variants within ANK3 have also been associated in the Psychiatric Genomics Consortium cross-disorder GWAS, and in a rare variant analysis of autism spectrum disorder.”
Two variants were linked to distressing psychotic experiences: rs75459873, intronic to cannabinoid receptor 2 (CNR2), decreased the risk by 34% (OR, 0.66). Intergenic variant rs3849810 increased the risk by 12% (OR, 1.12).
“CNR2 encodes for CB2, one of two well-characterized cannabinoid receptors. Several lines of evidence have implicated the endocannabinoid system in psychiatric disorders, including schizophrenia and depression. The main psychoactive agent of cannabis, tetrahydrocannabinol, can cause acute psychotic symptoms and cognitive impairment. Given that cannabis use is strongly associated with psychotic experiences, we tested, but found no evidence for, a mediating or moderating effect of cannabis use on the association of rs75459873 and distressing psychotic experiences. However, while no evidence was found in this study, a mediating effect of cannabis use cannot be ruled out given the relatively low power of such analyses and the potential measurement error.”
Also, significant genetic correlations were found between any psychotic experiences and major depressive disorder, autism spectrum disorder, ADHD, and schizophrenia. However, the polygenic risk scores for schizophrenia, major depressive disorder, bipolar disorder, ADHD, and autism spectrum disorder, were low.
“We also considered individual psychotic symptoms and found that polygenic risk scores for schizophrenia, bipolar disorder, depression, and ADHD were more strongly associated with delusions of persecution than with the other psychotic symptoms.”
Those with distressing psychotic experiences tended to have more copy number variations (CNVs) associated with schizophrenia (OR, 2.04) and neurodevelopmental disorders (OR, 1.75). The team also found significant associations between distressing experiences and major depressive disorder, ADHD, autism spectrum disorder, and schizophrenia.
“We found particular enrichment of these [polygenic risk scores] in distressing psychotic experiences and for delusions of persecution,” they noted. “ ... All schizophrenia-associated [copy number variations] are also associated with neurodevelopmental disorders such as intellectual disability and autism.”
The study’s strengths include its large sample size. Among its limitations, the researchers said, are the study’s retrospective measurement of psychotic experiences based on self-report from a questionnaire that was online. Gathering the data in that way raised the likelihood of possible error, they said.
Dr. Legge reported having no disclosures.
SOURCE: Legge SE et al. JAMA Psychiatry. 2019 Sep 25. doi: 10.1001/jamapsychiatry.2019.2508.
FROM JAMA PSYCHIATRY
Osteoporosis, osteoarthritis risk high among cerebral palsy patients
compared with adults without the disorder, according to a study published in Bone.
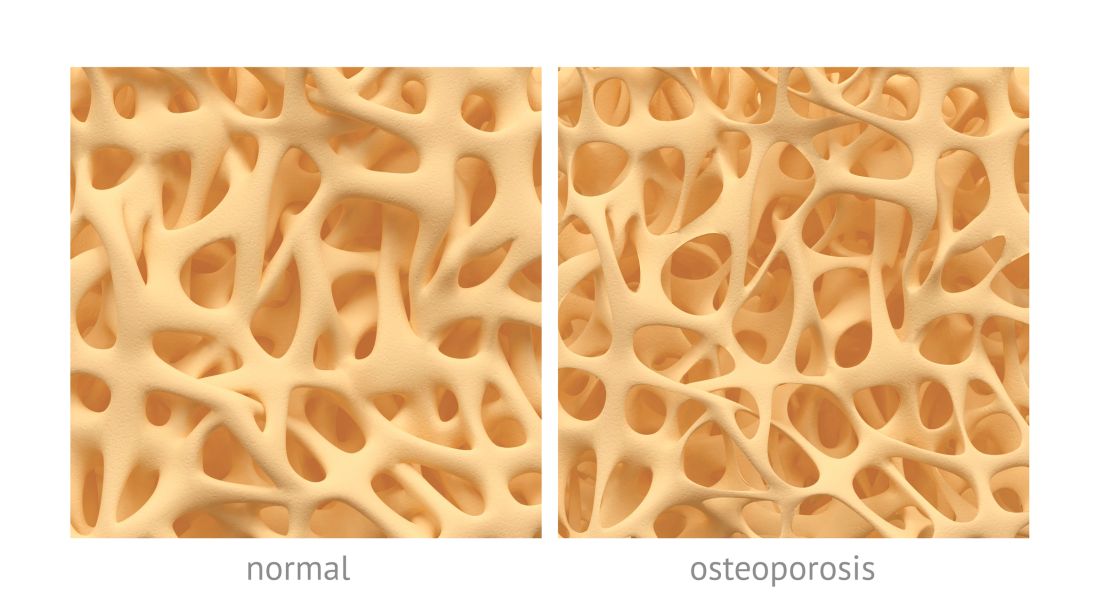
Neil E. O’Connell, PhD, of Brunel University London, and colleagues assessed the risks of osteoporosis, osteoarthritis, and inflammatory musculoskeletal diseases in a population-based cohort study that used data collected by the U.K. Clinical Practice Research Datalink during 1987-2015. The study included 1,705 patients with CP and 5,115 patients matched for age, sex, and general practices; data on smoking status and alcohol consumption for many of the patients also were gathered.
After adjustment for smoking status, alcohol consumption, and mean yearly general practice visits, investigators found evidence of significantly increased risk for osteoarthritis (hazard ratio, 1.54; 95% confidence interval, 1.17-2.02; P = .002) and osteoporosis (HR, 6.19; 95% CI, 3.37-11.39; P less than .001); they did not see increased risk for inflammatory musculoskeletal diseases (HR, 0.89; 95% CI, 0.45-1.75; P = .731).
One limitation of the study is the risk for residual confounding given the investigators could not account for mobility status or physical activity. Other limitations include potential incompleteness of diagnostic code lists, how identification of cases is depending on quality of original recording in the database, and that data regarding smoking status and alcohol consumption were missing for a substantial proportion of patients.
“Despite previous studies identifying a high prevalence of joint pain and functional deterioration among people with CP, there is a dearth of literature on the burden of musculoskeletal disorders in this population,” they wrote. “Further research is required into effective management of these conditions in adults with CP.”
This study was supported by an interdisciplinary award from Brunel University London’s Research Catalyst Fund. The authors declared no competing interests.
SOURCE: O’Connell NE et al. Bone. 2019 Aug;125:30-5.
compared with adults without the disorder, according to a study published in Bone.
Neil E. O’Connell, PhD, of Brunel University London, and colleagues assessed the risks of osteoporosis, osteoarthritis, and inflammatory musculoskeletal diseases in a population-based cohort study that used data collected by the U.K. Clinical Practice Research Datalink during 1987-2015. The study included 1,705 patients with CP and 5,115 patients matched for age, sex, and general practices; data on smoking status and alcohol consumption for many of the patients also were gathered.
After adjustment for smoking status, alcohol consumption, and mean yearly general practice visits, investigators found evidence of significantly increased risk for osteoarthritis (hazard ratio, 1.54; 95% confidence interval, 1.17-2.02; P = .002) and osteoporosis (HR, 6.19; 95% CI, 3.37-11.39; P less than .001); they did not see increased risk for inflammatory musculoskeletal diseases (HR, 0.89; 95% CI, 0.45-1.75; P = .731).
One limitation of the study is the risk for residual confounding given the investigators could not account for mobility status or physical activity. Other limitations include potential incompleteness of diagnostic code lists, how identification of cases is depending on quality of original recording in the database, and that data regarding smoking status and alcohol consumption were missing for a substantial proportion of patients.
“Despite previous studies identifying a high prevalence of joint pain and functional deterioration among people with CP, there is a dearth of literature on the burden of musculoskeletal disorders in this population,” they wrote. “Further research is required into effective management of these conditions in adults with CP.”
This study was supported by an interdisciplinary award from Brunel University London’s Research Catalyst Fund. The authors declared no competing interests.
SOURCE: O’Connell NE et al. Bone. 2019 Aug;125:30-5.
compared with adults without the disorder, according to a study published in Bone.
Neil E. O’Connell, PhD, of Brunel University London, and colleagues assessed the risks of osteoporosis, osteoarthritis, and inflammatory musculoskeletal diseases in a population-based cohort study that used data collected by the U.K. Clinical Practice Research Datalink during 1987-2015. The study included 1,705 patients with CP and 5,115 patients matched for age, sex, and general practices; data on smoking status and alcohol consumption for many of the patients also were gathered.
After adjustment for smoking status, alcohol consumption, and mean yearly general practice visits, investigators found evidence of significantly increased risk for osteoarthritis (hazard ratio, 1.54; 95% confidence interval, 1.17-2.02; P = .002) and osteoporosis (HR, 6.19; 95% CI, 3.37-11.39; P less than .001); they did not see increased risk for inflammatory musculoskeletal diseases (HR, 0.89; 95% CI, 0.45-1.75; P = .731).
One limitation of the study is the risk for residual confounding given the investigators could not account for mobility status or physical activity. Other limitations include potential incompleteness of diagnostic code lists, how identification of cases is depending on quality of original recording in the database, and that data regarding smoking status and alcohol consumption were missing for a substantial proportion of patients.
“Despite previous studies identifying a high prevalence of joint pain and functional deterioration among people with CP, there is a dearth of literature on the burden of musculoskeletal disorders in this population,” they wrote. “Further research is required into effective management of these conditions in adults with CP.”
This study was supported by an interdisciplinary award from Brunel University London’s Research Catalyst Fund. The authors declared no competing interests.
SOURCE: O’Connell NE et al. Bone. 2019 Aug;125:30-5.
FROM BONE
Patients with intellectual disability require nuanced care
SAN FRANCISCO – Some physicians are uncomfortable providing mental health care to patients with intellectual disability (ID) because many of the patients’ communications skills are limited. But many resources are available that can help.
In this video, Nita V. Bhatt, MD, MPH, interviews Julie P. Gentile, MD, about some of those resources and discusses how to approach psychiatric treatment interventions for patients with ID.
In addition to the DSM-5, Dr. Gentile said the National Association for the Dually Diagnosed has published the Diagnostic Manual – Intellectual Disability. Another resource is a practical reference manual originally proposed by one of Dr. Gentile’s residents.
“He came into my office for supervision one day and said, ‘You know, there’s all these nuances for psychiatric treatment in this patient population. So we should write a practice, quick reference manual to help clinicians who aren’t able to spend as much time concentrate on this patient population.’ ”
As a result, several residents and faculty members formed a team to produce an 18-chapter book published this year by Springer called the Guide to Intellectual Disabilities: A Clinical Handbook.
Dr. Bhatt is a staff psychiatrist at Twin Valley Behavioral Healthcare, the state psychiatric hospital in Columbus, Ohio. Dr. Gentile is professor and chair of the department of psychiatry at Wright State in Dayton. She is also serves as project director of Ohio’s Telepsychiatry Project for Intellectual Disability and has been awarded more than $7 million in grant funding to support her projects in the field of ID.
Dr. Gentile’s work has been funded by the Ohio Department of Developmental Disabilities and the Ohio Department of Mental Health and Addiction Services.
SAN FRANCISCO – Some physicians are uncomfortable providing mental health care to patients with intellectual disability (ID) because many of the patients’ communications skills are limited. But many resources are available that can help.
In this video, Nita V. Bhatt, MD, MPH, interviews Julie P. Gentile, MD, about some of those resources and discusses how to approach psychiatric treatment interventions for patients with ID.
In addition to the DSM-5, Dr. Gentile said the National Association for the Dually Diagnosed has published the Diagnostic Manual – Intellectual Disability. Another resource is a practical reference manual originally proposed by one of Dr. Gentile’s residents.
“He came into my office for supervision one day and said, ‘You know, there’s all these nuances for psychiatric treatment in this patient population. So we should write a practice, quick reference manual to help clinicians who aren’t able to spend as much time concentrate on this patient population.’ ”
As a result, several residents and faculty members formed a team to produce an 18-chapter book published this year by Springer called the Guide to Intellectual Disabilities: A Clinical Handbook.
Dr. Bhatt is a staff psychiatrist at Twin Valley Behavioral Healthcare, the state psychiatric hospital in Columbus, Ohio. Dr. Gentile is professor and chair of the department of psychiatry at Wright State in Dayton. She is also serves as project director of Ohio’s Telepsychiatry Project for Intellectual Disability and has been awarded more than $7 million in grant funding to support her projects in the field of ID.
Dr. Gentile’s work has been funded by the Ohio Department of Developmental Disabilities and the Ohio Department of Mental Health and Addiction Services.
SAN FRANCISCO – Some physicians are uncomfortable providing mental health care to patients with intellectual disability (ID) because many of the patients’ communications skills are limited. But many resources are available that can help.
In this video, Nita V. Bhatt, MD, MPH, interviews Julie P. Gentile, MD, about some of those resources and discusses how to approach psychiatric treatment interventions for patients with ID.
In addition to the DSM-5, Dr. Gentile said the National Association for the Dually Diagnosed has published the Diagnostic Manual – Intellectual Disability. Another resource is a practical reference manual originally proposed by one of Dr. Gentile’s residents.
“He came into my office for supervision one day and said, ‘You know, there’s all these nuances for psychiatric treatment in this patient population. So we should write a practice, quick reference manual to help clinicians who aren’t able to spend as much time concentrate on this patient population.’ ”
As a result, several residents and faculty members formed a team to produce an 18-chapter book published this year by Springer called the Guide to Intellectual Disabilities: A Clinical Handbook.
Dr. Bhatt is a staff psychiatrist at Twin Valley Behavioral Healthcare, the state psychiatric hospital in Columbus, Ohio. Dr. Gentile is professor and chair of the department of psychiatry at Wright State in Dayton. She is also serves as project director of Ohio’s Telepsychiatry Project for Intellectual Disability and has been awarded more than $7 million in grant funding to support her projects in the field of ID.
Dr. Gentile’s work has been funded by the Ohio Department of Developmental Disabilities and the Ohio Department of Mental Health and Addiction Services.
REPORTING FROM APA 2019

Neurodevelopmental concerns may emerge later in Zika-exposed infants
BALTIMORE – Most infants prenatally exposed to Zika showed relatively normal neurodevelopment if their fetal MRI and birth head circumference were normal, but others with similarly initial normal measures appeared to struggle with social cognition and mobility as they got older, according to a new study.

“I think we need to be cautious with saying that these children are normal when these normal-appearing children may not be doing as well as we think,” lead author Sarah Mulkey, MD, of Children’s National Health System and George Washington University, Washington, said in an interview. “While most children are showing fairly normal development, there are some children who are … becoming more abnormal over time.”
Dr. Mulkey shared her findings at the Pediatric Academic Societies annual meeting. She and her colleagues had previously published a prospective study of 82 Zika-exposed infants’ fetal brain MRIs. In their new study, they followed up with the 78 Colombian infants from that study whose fetal neuroimaging and birth head circumstance had been normal.
The researchers used the Alberta Infant Motor Scale (AIMS) and the Warner Initial Developmental Evaluation of Adaptive and Functional Skills (WIDEA) to evaluate 72 of the children, 34 of whom underwent assessment twice. Forty of the children were an average 5.7 months old when evaluated, and 66 were an average 13.5 months old.
As the children got older, their overall WIDEA z-score and their subscores in the social cognition domain and especially in the mobility domain trended downward. Three of the children had AIMS scores two standard deviations below normal, but the rest fell within the normal range.
Their WIDEA communication z-score hovered relatively close to the norm, but self-care also showed a very slight slope downward, albeit not as substantially as in the social cognition and mobility domains.
The younger a child is, the fewer skills they generally show related to neurocognitive development, Dr. Mulkey explained. But as they grow older and are expected to show more skills, it becomes more apparent where gaps and delays might exist.
“We can see that there are a lot of kids doing well, but some of these kids certainly are not,” she said. “Until children have a long time to develop, you really can’t see these changes unless you follow them long-term.”
The researchers also looked separately at a subgroup of 19 children (26%) whose cranial ultrasounds showed mild nonspecific findings. These findings – such as lenticulostriate vasculopathy, choroid plexus cysts, subependymal cysts and calcifications – do not usually indicate any problems, but they appeared in a quarter of this population, considerably more than the approximately 5% typically seen in the general population, Dr. Mulkey said.
Though the findings did not reach significance, infants in this subgroup tended to have a lower WIDEA mobility z-scores (P = .054) and lower AIMS scores (P = .26) than the Zika-exposed infants with normal cranial ultrasounds.
“Mild nonspecific cranial ultrasound findings may represent a mild injury” related to exposure to their mother’s Zika infection during pregnancy, the researchers suggested. “It may be a risk factor for the lower mobility outcome,” Dr. Mulkey said.
The researchers hope to continue later follow-ups as the children age.
The research was funded by the Thrasher Research Fund. Dr. Mulkey had no conflicts of interest.
BALTIMORE – Most infants prenatally exposed to Zika showed relatively normal neurodevelopment if their fetal MRI and birth head circumference were normal, but others with similarly initial normal measures appeared to struggle with social cognition and mobility as they got older, according to a new study.
“I think we need to be cautious with saying that these children are normal when these normal-appearing children may not be doing as well as we think,” lead author Sarah Mulkey, MD, of Children’s National Health System and George Washington University, Washington, said in an interview. “While most children are showing fairly normal development, there are some children who are … becoming more abnormal over time.”
Dr. Mulkey shared her findings at the Pediatric Academic Societies annual meeting. She and her colleagues had previously published a prospective study of 82 Zika-exposed infants’ fetal brain MRIs. In their new study, they followed up with the 78 Colombian infants from that study whose fetal neuroimaging and birth head circumstance had been normal.
The researchers used the Alberta Infant Motor Scale (AIMS) and the Warner Initial Developmental Evaluation of Adaptive and Functional Skills (WIDEA) to evaluate 72 of the children, 34 of whom underwent assessment twice. Forty of the children were an average 5.7 months old when evaluated, and 66 were an average 13.5 months old.
As the children got older, their overall WIDEA z-score and their subscores in the social cognition domain and especially in the mobility domain trended downward. Three of the children had AIMS scores two standard deviations below normal, but the rest fell within the normal range.
Their WIDEA communication z-score hovered relatively close to the norm, but self-care also showed a very slight slope downward, albeit not as substantially as in the social cognition and mobility domains.
The younger a child is, the fewer skills they generally show related to neurocognitive development, Dr. Mulkey explained. But as they grow older and are expected to show more skills, it becomes more apparent where gaps and delays might exist.
“We can see that there are a lot of kids doing well, but some of these kids certainly are not,” she said. “Until children have a long time to develop, you really can’t see these changes unless you follow them long-term.”
The researchers also looked separately at a subgroup of 19 children (26%) whose cranial ultrasounds showed mild nonspecific findings. These findings – such as lenticulostriate vasculopathy, choroid plexus cysts, subependymal cysts and calcifications – do not usually indicate any problems, but they appeared in a quarter of this population, considerably more than the approximately 5% typically seen in the general population, Dr. Mulkey said.
Though the findings did not reach significance, infants in this subgroup tended to have a lower WIDEA mobility z-scores (P = .054) and lower AIMS scores (P = .26) than the Zika-exposed infants with normal cranial ultrasounds.
“Mild nonspecific cranial ultrasound findings may represent a mild injury” related to exposure to their mother’s Zika infection during pregnancy, the researchers suggested. “It may be a risk factor for the lower mobility outcome,” Dr. Mulkey said.
The researchers hope to continue later follow-ups as the children age.
The research was funded by the Thrasher Research Fund. Dr. Mulkey had no conflicts of interest.
BALTIMORE – Most infants prenatally exposed to Zika showed relatively normal neurodevelopment if their fetal MRI and birth head circumference were normal, but others with similarly initial normal measures appeared to struggle with social cognition and mobility as they got older, according to a new study.
“I think we need to be cautious with saying that these children are normal when these normal-appearing children may not be doing as well as we think,” lead author Sarah Mulkey, MD, of Children’s National Health System and George Washington University, Washington, said in an interview. “While most children are showing fairly normal development, there are some children who are … becoming more abnormal over time.”
Dr. Mulkey shared her findings at the Pediatric Academic Societies annual meeting. She and her colleagues had previously published a prospective study of 82 Zika-exposed infants’ fetal brain MRIs. In their new study, they followed up with the 78 Colombian infants from that study whose fetal neuroimaging and birth head circumstance had been normal.
The researchers used the Alberta Infant Motor Scale (AIMS) and the Warner Initial Developmental Evaluation of Adaptive and Functional Skills (WIDEA) to evaluate 72 of the children, 34 of whom underwent assessment twice. Forty of the children were an average 5.7 months old when evaluated, and 66 were an average 13.5 months old.
As the children got older, their overall WIDEA z-score and their subscores in the social cognition domain and especially in the mobility domain trended downward. Three of the children had AIMS scores two standard deviations below normal, but the rest fell within the normal range.
Their WIDEA communication z-score hovered relatively close to the norm, but self-care also showed a very slight slope downward, albeit not as substantially as in the social cognition and mobility domains.
The younger a child is, the fewer skills they generally show related to neurocognitive development, Dr. Mulkey explained. But as they grow older and are expected to show more skills, it becomes more apparent where gaps and delays might exist.
“We can see that there are a lot of kids doing well, but some of these kids certainly are not,” she said. “Until children have a long time to develop, you really can’t see these changes unless you follow them long-term.”
The researchers also looked separately at a subgroup of 19 children (26%) whose cranial ultrasounds showed mild nonspecific findings. These findings – such as lenticulostriate vasculopathy, choroid plexus cysts, subependymal cysts and calcifications – do not usually indicate any problems, but they appeared in a quarter of this population, considerably more than the approximately 5% typically seen in the general population, Dr. Mulkey said.
Though the findings did not reach significance, infants in this subgroup tended to have a lower WIDEA mobility z-scores (P = .054) and lower AIMS scores (P = .26) than the Zika-exposed infants with normal cranial ultrasounds.
“Mild nonspecific cranial ultrasound findings may represent a mild injury” related to exposure to their mother’s Zika infection during pregnancy, the researchers suggested. “It may be a risk factor for the lower mobility outcome,” Dr. Mulkey said.
The researchers hope to continue later follow-ups as the children age.
The research was funded by the Thrasher Research Fund. Dr. Mulkey had no conflicts of interest.
REPORTING FROM PAS 2019
Key clinical point: .
Major finding: Zika-exposed infants with normal fetal MRI neuroimaging showed increasingly lower mobility and social cognition skills as they approached their first birthday.
Study details: The findings are based on neurodevelopmental assessments of 72 Zika-exposed Colombian children at 4-18 months old.
Disclosures: The research was funded by the Thrasher Research Fund. Dr. Mulkey had no conflicts of interest.
Valproate, topiramate prescribed in young women despite known teratogenicity risks
results of a retrospective analysis suggest.

Topiramate, linked to increased risk of cleft palate and smaller-than-gestational-age newborns, was among the top three antiepileptic drugs (AEDs) prescribed to women 15-44 years of age in the population-based cohort study.
Valproate, linked to increases in both anatomic and behavioral teratogenicity, was less often prescribed, but nevertheless still prescribed in a considerable proportion of patients in the study, which looked at U.S. commercial, Medicare, and Medicaid claims data from 2009 to 2013.
Presence of comorbidities could be influencing whether or not a woman of childbearing age receives one of these AEDs, the investigators said. Specifically, they found valproate more often prescribed for women with epilepsy who also had mood or anxiety and dissociative disorder, while topiramate was more often prescribed in women with headaches or migraines.
Taken together, these findings suggest a lack of awareness of the teratogenic risks of valproate and topiramate, said the investigators, led by Hyunmi Kim, MD, PhD, MPH, of the department of neurology at Stanford (Calif.) University.
“To improve current practice, knowledge of the teratogenicity of certain AEDs should be disseminated to health care professionals and patients,” they wrote. The report is in JAMA Neurology.
The findings of Dr. Kim and her colleagues were based on data for 46,767 women of childbearing age: 8,003 incident (new) cases with a mean age of 27 years, and 38,764 prevalent cases with a mean age of 30 years.
Topiramate was the second- or third-most prescribed AED in the analyses, alongside levetiracetam and lamotrigine. In particular, topiramate prescriptions were found in incident cases receiving first-line monotherapy (15%), prevalent cases receiving first-line monotherapy (13%), and prevalent cases receiving polytherapy (29%).
Valproate was the fifth-most prescribed AED for incident and prevalent cases receiving first-line monotherapy (5% and 10%, respectively), and came in fourth place among prevalent cases receiving polytherapy (22%).
The somewhat lower rate of valproate prescriptions tracks with other recent analyses showing that valproate use decreased among women of childbearing age following recommendations against its use during pregnancy, according to Dr. Kim and her coauthors.
However, topiramate is another story: “Although the magnitude of risk and range of adverse reproductive outcomes associated with topiramate use appear substantially less than those associated with valproate, some reduction in the use of topiramate in this population might be expected after evidence emerged in 2008 of its association with cleft palate,” they said in their report.
UCB Pharma sponsored this study. Study authors reported disclosures related to UCB Pharma, Biogen, Eisai, SK Life Science, Brain Sentinel, UCB Pharma, and the University of Alabama at Birmingham.
SOURCE: Kim H et al. JAMA Neurol. 2019 Apr 1. doi: 10.1001/jamaneurol.2019.0447.
results of a retrospective analysis suggest.
Topiramate, linked to increased risk of cleft palate and smaller-than-gestational-age newborns, was among the top three antiepileptic drugs (AEDs) prescribed to women 15-44 years of age in the population-based cohort study.
Valproate, linked to increases in both anatomic and behavioral teratogenicity, was less often prescribed, but nevertheless still prescribed in a considerable proportion of patients in the study, which looked at U.S. commercial, Medicare, and Medicaid claims data from 2009 to 2013.
Presence of comorbidities could be influencing whether or not a woman of childbearing age receives one of these AEDs, the investigators said. Specifically, they found valproate more often prescribed for women with epilepsy who also had mood or anxiety and dissociative disorder, while topiramate was more often prescribed in women with headaches or migraines.
Taken together, these findings suggest a lack of awareness of the teratogenic risks of valproate and topiramate, said the investigators, led by Hyunmi Kim, MD, PhD, MPH, of the department of neurology at Stanford (Calif.) University.
“To improve current practice, knowledge of the teratogenicity of certain AEDs should be disseminated to health care professionals and patients,” they wrote. The report is in JAMA Neurology.
The findings of Dr. Kim and her colleagues were based on data for 46,767 women of childbearing age: 8,003 incident (new) cases with a mean age of 27 years, and 38,764 prevalent cases with a mean age of 30 years.
Topiramate was the second- or third-most prescribed AED in the analyses, alongside levetiracetam and lamotrigine. In particular, topiramate prescriptions were found in incident cases receiving first-line monotherapy (15%), prevalent cases receiving first-line monotherapy (13%), and prevalent cases receiving polytherapy (29%).
Valproate was the fifth-most prescribed AED for incident and prevalent cases receiving first-line monotherapy (5% and 10%, respectively), and came in fourth place among prevalent cases receiving polytherapy (22%).
The somewhat lower rate of valproate prescriptions tracks with other recent analyses showing that valproate use decreased among women of childbearing age following recommendations against its use during pregnancy, according to Dr. Kim and her coauthors.
However, topiramate is another story: “Although the magnitude of risk and range of adverse reproductive outcomes associated with topiramate use appear substantially less than those associated with valproate, some reduction in the use of topiramate in this population might be expected after evidence emerged in 2008 of its association with cleft palate,” they said in their report.
UCB Pharma sponsored this study. Study authors reported disclosures related to UCB Pharma, Biogen, Eisai, SK Life Science, Brain Sentinel, UCB Pharma, and the University of Alabama at Birmingham.
SOURCE: Kim H et al. JAMA Neurol. 2019 Apr 1. doi: 10.1001/jamaneurol.2019.0447.
results of a retrospective analysis suggest.
Topiramate, linked to increased risk of cleft palate and smaller-than-gestational-age newborns, was among the top three antiepileptic drugs (AEDs) prescribed to women 15-44 years of age in the population-based cohort study.
Valproate, linked to increases in both anatomic and behavioral teratogenicity, was less often prescribed, but nevertheless still prescribed in a considerable proportion of patients in the study, which looked at U.S. commercial, Medicare, and Medicaid claims data from 2009 to 2013.
Presence of comorbidities could be influencing whether or not a woman of childbearing age receives one of these AEDs, the investigators said. Specifically, they found valproate more often prescribed for women with epilepsy who also had mood or anxiety and dissociative disorder, while topiramate was more often prescribed in women with headaches or migraines.
Taken together, these findings suggest a lack of awareness of the teratogenic risks of valproate and topiramate, said the investigators, led by Hyunmi Kim, MD, PhD, MPH, of the department of neurology at Stanford (Calif.) University.
“To improve current practice, knowledge of the teratogenicity of certain AEDs should be disseminated to health care professionals and patients,” they wrote. The report is in JAMA Neurology.
The findings of Dr. Kim and her colleagues were based on data for 46,767 women of childbearing age: 8,003 incident (new) cases with a mean age of 27 years, and 38,764 prevalent cases with a mean age of 30 years.
Topiramate was the second- or third-most prescribed AED in the analyses, alongside levetiracetam and lamotrigine. In particular, topiramate prescriptions were found in incident cases receiving first-line monotherapy (15%), prevalent cases receiving first-line monotherapy (13%), and prevalent cases receiving polytherapy (29%).
Valproate was the fifth-most prescribed AED for incident and prevalent cases receiving first-line monotherapy (5% and 10%, respectively), and came in fourth place among prevalent cases receiving polytherapy (22%).
The somewhat lower rate of valproate prescriptions tracks with other recent analyses showing that valproate use decreased among women of childbearing age following recommendations against its use during pregnancy, according to Dr. Kim and her coauthors.
However, topiramate is another story: “Although the magnitude of risk and range of adverse reproductive outcomes associated with topiramate use appear substantially less than those associated with valproate, some reduction in the use of topiramate in this population might be expected after evidence emerged in 2008 of its association with cleft palate,” they said in their report.
UCB Pharma sponsored this study. Study authors reported disclosures related to UCB Pharma, Biogen, Eisai, SK Life Science, Brain Sentinel, UCB Pharma, and the University of Alabama at Birmingham.
SOURCE: Kim H et al. JAMA Neurol. 2019 Apr 1. doi: 10.1001/jamaneurol.2019.0447.
FROM JAMA NEUROLOGY
Key clinical point: Both valproate and topiramate are prescribed relatively often in women of childbearing age despite known teratogenic risks.
Major finding: Topiramate was the second- or third-most prescribed AED in the analyses. Valproate was the fifth-most prescribed AED for incident and prevalent cases receiving first-line monotherapy.
Study details: Retrospective cohort study including nearly 47,000 women of childbearing age enrolled in claims databases between 2009 and 2013.
Disclosures: UCB Pharma sponsored the study. Study authors reported disclosures related to UCB Pharma, Biogen, Eisai, SK Life Science, Brain Sentinel, UCB Pharma, and the University of Alabama at Birmingham.
Source: Kim H et al. JAMA Neurol. 2019 Apr 1. doi: 10.1001/jamaneurol.2019.0447.
Prenatal, postnatal neuroimaging IDs most Zika-related brain injuries
Prenatal ultrasound can identify most abnormalities in fetuses exposed to Zika virus during pregnancy, and neuroimaging after birth can detect infant exposure in cases that appeared normal on prenatal ultrasound, according to research published in JAMA Pediatrics.
“Absence of prolonged maternal viremia did not have predictive associations with normal fetal or neonatal brain imaging,” Sarah B. Mulkey, MD, PhD, from the division of fetal and transitional medicine at Children’s National Health System, in Washington, and her colleagues wrote. “Postnatal imaging can detect changes not seen on fetal imaging, supporting the current CDC [Centers for Disease Control and Prevention] recommendation for postnatal cranial [ultrasound].”
Dr. Mulkey and her colleagues performed a prospective cohort analysis of 82 pregnant women from Colombia and the United States who had clinical evidence of probable exposure to the Zika virus through travel (U.S. cases, 2 patients), physician referral, or community cases during June 2016-June 2017. Pregnant women underwent fetal MRI or ultrasound during the second or third trimesters between 4 weeks and 10 weeks after symptom onset, with infants undergoing brain MRI and cranial ultrasound after birth.
Of those 82 pregnancies, there were 80 live births, 1 case of termination because of severe fetal brain abnormalities, and 1 near-term fetal death of unknown cause. There was one death 3 days after birth and one instance of neurosurgical intervention from encephalocele. The researchers found 3 of 82 cases (4%) displayed fetal abnormalities from MRI, which consisted of 2 cases of heterotopias and malformations in cortical development and 1 case with parietal encephalocele, Chiari II malformation, and microcephaly. One infant had a normal ultrasound despite abnormalities displayed on fetal MRI.
After birth, of the 79 infants with normal ultrasound results, 53 infants underwent a postnatal brain MRI and Dr. Mulkey and her associates found 7 cases with mild abnormalities (13%). There were 57 infants who underwent cranial ultrasound, which yielded 21 cases of lenticulostriate vasculopathy, choroid plexus cysts, germinolytic/subependymal cysts, and/or calcification; these were poorly characterized by MRI.
“Normal fetal imaging had predictive associations with normal postnatal imaging or mild postnatal imaging findings unlikely to be of significant clinical consequence,” they said.
Nonetheless, “there is a need for long-term follow-up to assess the neurodevelopmental significance of these early neuroimaging findings, both normal and abnormal; such studies are in progress,” Dr. Mulkey and her colleagues said.
The researchers noted the timing of maternal infections and symptoms as well as the Zika testing, ultrasound, and MRI performance, technique during fetal MRI, and incomplete prenatal testing in the cohort as limitations in the study.
This study was funded in part by Children’s National Health System and by a philanthropic gift from the Ikaria Healthcare Fund. Dr. Mulkey received research support from the Thrasher Research Fund and is supported by awards from the National Institutes of Health National Center for Advancing Translational Sciences. The other authors reported no relevant conflicts of interest.
SOURCE: Mulkey SB et al. JAMA Pediatr. 2018 Nov. 26. doi: 10.1001/jamapediatrics.2018.4138.
While the study by Mulkey et al. adds to the body of evidence of prenatal and postnatal brain abnormalities, there are still many unanswered questions about the Zika virus and how to handle its unique diagnostic and clinical challenges, Margaret A. Honein, PhD, MPH, and Denise J. Jamieson, MD, MPH, wrote in a related editorial.
For example, Centers for Disease Control and Prevention recommendations state that infants with possible Zika exposure should receive an ophthalmologic and ultrasonographic examination at 1 month, and if the hearing test used otoacoustic emissions methods only, an automated auditory brainstem response test should be administered. While Mulkey et al. examined brain abnormalities in utero and in infants, it is not clear whether all CDC guidelines were followed in these cases.
In addition, because there is no reliable way to determine whether infants acquired Zika virus through the mother or through vertical transmission, assessing the proportion of congenitally infected infants or vertical-transmission infected infants who have neurodevelopmental disabilities and defects is not possible, they said. More longitudinal studies are needed to study the effects of the Zika virus and to prepare for the next outbreak.
“Zika was affecting pregnant women and their infants years before its teratogenic effect was recognized, and Zika will remain a serious risk to pregnant women and their infants until we have a safe vaccine that can fully prevent Zika virus infection during pregnancy,” they said. “Until then, ongoing public health efforts are essential to protect mothers and babies from this threat and ensure all disabilities associated with Zika virus infection are promptly identified, so that timely interventions can be provided.”
Dr. Honein is from the National Center on Birth Defects and Developmental Disabilities at the Centers for Disease Control and Prevention, and Dr. Jamieson is from the department of gynecology & obstetrics at Emory University School of Medicine, Atlanta. These comments summarize their editorial in response to Mulkey et al. (JAMA Pediatr. 2018 Nov. 26. doi: 10.1001/jamapediatrics.2018.4164). They reported no relevant conflicts of interest.
While the study by Mulkey et al. adds to the body of evidence of prenatal and postnatal brain abnormalities, there are still many unanswered questions about the Zika virus and how to handle its unique diagnostic and clinical challenges, Margaret A. Honein, PhD, MPH, and Denise J. Jamieson, MD, MPH, wrote in a related editorial.
For example, Centers for Disease Control and Prevention recommendations state that infants with possible Zika exposure should receive an ophthalmologic and ultrasonographic examination at 1 month, and if the hearing test used otoacoustic emissions methods only, an automated auditory brainstem response test should be administered. While Mulkey et al. examined brain abnormalities in utero and in infants, it is not clear whether all CDC guidelines were followed in these cases.
In addition, because there is no reliable way to determine whether infants acquired Zika virus through the mother or through vertical transmission, assessing the proportion of congenitally infected infants or vertical-transmission infected infants who have neurodevelopmental disabilities and defects is not possible, they said. More longitudinal studies are needed to study the effects of the Zika virus and to prepare for the next outbreak.
“Zika was affecting pregnant women and their infants years before its teratogenic effect was recognized, and Zika will remain a serious risk to pregnant women and their infants until we have a safe vaccine that can fully prevent Zika virus infection during pregnancy,” they said. “Until then, ongoing public health efforts are essential to protect mothers and babies from this threat and ensure all disabilities associated with Zika virus infection are promptly identified, so that timely interventions can be provided.”
Dr. Honein is from the National Center on Birth Defects and Developmental Disabilities at the Centers for Disease Control and Prevention, and Dr. Jamieson is from the department of gynecology & obstetrics at Emory University School of Medicine, Atlanta. These comments summarize their editorial in response to Mulkey et al. (JAMA Pediatr. 2018 Nov. 26. doi: 10.1001/jamapediatrics.2018.4164). They reported no relevant conflicts of interest.
While the study by Mulkey et al. adds to the body of evidence of prenatal and postnatal brain abnormalities, there are still many unanswered questions about the Zika virus and how to handle its unique diagnostic and clinical challenges, Margaret A. Honein, PhD, MPH, and Denise J. Jamieson, MD, MPH, wrote in a related editorial.
For example, Centers for Disease Control and Prevention recommendations state that infants with possible Zika exposure should receive an ophthalmologic and ultrasonographic examination at 1 month, and if the hearing test used otoacoustic emissions methods only, an automated auditory brainstem response test should be administered. While Mulkey et al. examined brain abnormalities in utero and in infants, it is not clear whether all CDC guidelines were followed in these cases.
In addition, because there is no reliable way to determine whether infants acquired Zika virus through the mother or through vertical transmission, assessing the proportion of congenitally infected infants or vertical-transmission infected infants who have neurodevelopmental disabilities and defects is not possible, they said. More longitudinal studies are needed to study the effects of the Zika virus and to prepare for the next outbreak.
“Zika was affecting pregnant women and their infants years before its teratogenic effect was recognized, and Zika will remain a serious risk to pregnant women and their infants until we have a safe vaccine that can fully prevent Zika virus infection during pregnancy,” they said. “Until then, ongoing public health efforts are essential to protect mothers and babies from this threat and ensure all disabilities associated with Zika virus infection are promptly identified, so that timely interventions can be provided.”
Dr. Honein is from the National Center on Birth Defects and Developmental Disabilities at the Centers for Disease Control and Prevention, and Dr. Jamieson is from the department of gynecology & obstetrics at Emory University School of Medicine, Atlanta. These comments summarize their editorial in response to Mulkey et al. (JAMA Pediatr. 2018 Nov. 26. doi: 10.1001/jamapediatrics.2018.4164). They reported no relevant conflicts of interest.
Prenatal ultrasound can identify most abnormalities in fetuses exposed to Zika virus during pregnancy, and neuroimaging after birth can detect infant exposure in cases that appeared normal on prenatal ultrasound, according to research published in JAMA Pediatrics.
“Absence of prolonged maternal viremia did not have predictive associations with normal fetal or neonatal brain imaging,” Sarah B. Mulkey, MD, PhD, from the division of fetal and transitional medicine at Children’s National Health System, in Washington, and her colleagues wrote. “Postnatal imaging can detect changes not seen on fetal imaging, supporting the current CDC [Centers for Disease Control and Prevention] recommendation for postnatal cranial [ultrasound].”
Dr. Mulkey and her colleagues performed a prospective cohort analysis of 82 pregnant women from Colombia and the United States who had clinical evidence of probable exposure to the Zika virus through travel (U.S. cases, 2 patients), physician referral, or community cases during June 2016-June 2017. Pregnant women underwent fetal MRI or ultrasound during the second or third trimesters between 4 weeks and 10 weeks after symptom onset, with infants undergoing brain MRI and cranial ultrasound after birth.
Of those 82 pregnancies, there were 80 live births, 1 case of termination because of severe fetal brain abnormalities, and 1 near-term fetal death of unknown cause. There was one death 3 days after birth and one instance of neurosurgical intervention from encephalocele. The researchers found 3 of 82 cases (4%) displayed fetal abnormalities from MRI, which consisted of 2 cases of heterotopias and malformations in cortical development and 1 case with parietal encephalocele, Chiari II malformation, and microcephaly. One infant had a normal ultrasound despite abnormalities displayed on fetal MRI.
After birth, of the 79 infants with normal ultrasound results, 53 infants underwent a postnatal brain MRI and Dr. Mulkey and her associates found 7 cases with mild abnormalities (13%). There were 57 infants who underwent cranial ultrasound, which yielded 21 cases of lenticulostriate vasculopathy, choroid plexus cysts, germinolytic/subependymal cysts, and/or calcification; these were poorly characterized by MRI.
“Normal fetal imaging had predictive associations with normal postnatal imaging or mild postnatal imaging findings unlikely to be of significant clinical consequence,” they said.
Nonetheless, “there is a need for long-term follow-up to assess the neurodevelopmental significance of these early neuroimaging findings, both normal and abnormal; such studies are in progress,” Dr. Mulkey and her colleagues said.
The researchers noted the timing of maternal infections and symptoms as well as the Zika testing, ultrasound, and MRI performance, technique during fetal MRI, and incomplete prenatal testing in the cohort as limitations in the study.
This study was funded in part by Children’s National Health System and by a philanthropic gift from the Ikaria Healthcare Fund. Dr. Mulkey received research support from the Thrasher Research Fund and is supported by awards from the National Institutes of Health National Center for Advancing Translational Sciences. The other authors reported no relevant conflicts of interest.
SOURCE: Mulkey SB et al. JAMA Pediatr. 2018 Nov. 26. doi: 10.1001/jamapediatrics.2018.4138.
Prenatal ultrasound can identify most abnormalities in fetuses exposed to Zika virus during pregnancy, and neuroimaging after birth can detect infant exposure in cases that appeared normal on prenatal ultrasound, according to research published in JAMA Pediatrics.
“Absence of prolonged maternal viremia did not have predictive associations with normal fetal or neonatal brain imaging,” Sarah B. Mulkey, MD, PhD, from the division of fetal and transitional medicine at Children’s National Health System, in Washington, and her colleagues wrote. “Postnatal imaging can detect changes not seen on fetal imaging, supporting the current CDC [Centers for Disease Control and Prevention] recommendation for postnatal cranial [ultrasound].”
Dr. Mulkey and her colleagues performed a prospective cohort analysis of 82 pregnant women from Colombia and the United States who had clinical evidence of probable exposure to the Zika virus through travel (U.S. cases, 2 patients), physician referral, or community cases during June 2016-June 2017. Pregnant women underwent fetal MRI or ultrasound during the second or third trimesters between 4 weeks and 10 weeks after symptom onset, with infants undergoing brain MRI and cranial ultrasound after birth.
Of those 82 pregnancies, there were 80 live births, 1 case of termination because of severe fetal brain abnormalities, and 1 near-term fetal death of unknown cause. There was one death 3 days after birth and one instance of neurosurgical intervention from encephalocele. The researchers found 3 of 82 cases (4%) displayed fetal abnormalities from MRI, which consisted of 2 cases of heterotopias and malformations in cortical development and 1 case with parietal encephalocele, Chiari II malformation, and microcephaly. One infant had a normal ultrasound despite abnormalities displayed on fetal MRI.
After birth, of the 79 infants with normal ultrasound results, 53 infants underwent a postnatal brain MRI and Dr. Mulkey and her associates found 7 cases with mild abnormalities (13%). There were 57 infants who underwent cranial ultrasound, which yielded 21 cases of lenticulostriate vasculopathy, choroid plexus cysts, germinolytic/subependymal cysts, and/or calcification; these were poorly characterized by MRI.
“Normal fetal imaging had predictive associations with normal postnatal imaging or mild postnatal imaging findings unlikely to be of significant clinical consequence,” they said.
Nonetheless, “there is a need for long-term follow-up to assess the neurodevelopmental significance of these early neuroimaging findings, both normal and abnormal; such studies are in progress,” Dr. Mulkey and her colleagues said.
The researchers noted the timing of maternal infections and symptoms as well as the Zika testing, ultrasound, and MRI performance, technique during fetal MRI, and incomplete prenatal testing in the cohort as limitations in the study.
This study was funded in part by Children’s National Health System and by a philanthropic gift from the Ikaria Healthcare Fund. Dr. Mulkey received research support from the Thrasher Research Fund and is supported by awards from the National Institutes of Health National Center for Advancing Translational Sciences. The other authors reported no relevant conflicts of interest.
SOURCE: Mulkey SB et al. JAMA Pediatr. 2018 Nov. 26. doi: 10.1001/jamapediatrics.2018.4138.
FROM JAMA PEDIATRICS
Key clinical point:
Major finding: In 82 pregnant women, prenatal neuroimaging identified fetal abnormalities in 3 cases, while postnatal neuroimaging in 53 of the remaining 79 cases yielded an additional 7 cases with mild abnormalities.
Study details: A prospective longitudinal cohort study of 82 pregnant women with clinical evidence of probable Zika infection in Colombia and the United States.
Disclosures: This study was funded in part by Children’s National Health System and by a philanthropic gift from the Ikaria Healthcare Fund. Dr Mulkey received research support from the Thrasher Research Fund and is supported by awards from the National Institutes of Health National Center for Advancing Translational Sciences. The other authors reported no relevant conflicts of interest.
Source: Mulkey SB et al. JAMA Pediatr. 2018 Nov. 26; doi: 10.1001/jamapediatrics.2018.4138.
Childhood Neuromuscular Disease as a Metaphor for the Scientific Advances in Child Neurology of the Last Quarter Century
John B. Bodensteiner, MD
Dr. Bodensteiner is Director of the American Board of Psychiatry and Neurology, Founding Editor and Senior Associate Editor of Seminars in Pediatric Neurology, and Senior Associate Editor of the Journal of Child Neurology.
The impetus for this article is the celebration of 25 years of publication of Neurology Reviews. Child neurology has advanced so much in these years that it is hardly recognizable from the previous state of our understanding. Many components of the discipline include, but are not limited to, epilepsy, headache, demyelinating diseases, autoimmune diseases, neoplastic, neonatal, neuromuscular disease, and developmental conditions. My interest has been related to the neuromuscular diseases of childhood. Thus, I have chosen to describe some of the ways the advances in basic medical science and our understanding of the genetic and molecular mechanisms of disease have altered the way we think about these conditions. For the first time, this knowledge has allowed us to identify targets and techniques for effective intervention and disease modification.
Myotonic Muscular Dystrophy
One example of how the advancement in the discipline of genetics has changed our understanding of disease would be myotonic muscular dystrophy (MyoD1). When I started seeing patients in the mid 1960s, MyoD1, particularly as seen in children, was recognized as an example of autosomal dominant inheritance. The tendency for subsequent generations to be more severely affected than their parents is a phenomenon known as anticipation. Anticipation was recognized, though not explained, with the understanding of the genetic mechanisms of the time. The fact that when the disease affected an infant or newborn it was almost always inherited from the mother was also well established, though the reason for this was also not understood. Actually, during the late 1970s and early 1980s the existence of the phenomenon of anticipation was widely questioned and some publications proposed that anticipation was just an artifact of observation due to the fact that we (medical clinicians) were getting better and thus identifying the disease earlier than in past years. The recognition of the importance of repeated segments of DNA in the pathogenesis of human disease and the subsequent development of the technology to study the phenomenon allowed recognition of this previously unappreciated mechanism of genetic disease causation. This new genetic mechanism explained how anticipation could occur and confirmed it as a real phenomenon. Expanded numbers of tandem repeats probably allowed the explanation of the difference in gene transmission related to the parental gender as well.1

Genetic Insights
Advancements in the understanding of genetic mechanisms of disease have made it possible to begin to identify the molecular mechanisms operating in many previously incomprehensible diseases. The development of laboratory techniques necessary for the identification of these genetic abnormalities has brought the recognition of these disease mechanisms into clinical practice. Twenty-five years ago, the ability to identify alterations in DNA sequences, the presence of deletions/duplications and expansion of trinucleotide repeats, as well as the determination (and recognition of the significance) of gene copy number, was not available to the clinician. This advancement in genetic understanding has, in turn, allowed the identification of therapeutic targets in the hope of being able to moderate or eliminate the consequence of the defective gene.
Spinal Muscular Atrophy
In the last few years, the use of relatively small molecules such as oligonucleotides to alter the transcription of mutated RNA to produce a functioning protein has been developed. Because they are small molecules, they can be delivered with relative ease to the desired site. This technology has been applied to the treatment of spinal muscular atrophy (SMA) with considerable success.
Survival motor neuron gene product is essential for the health of the anterior horn cells of the spinal cord of the central nervous system. In the human genome there are two copies of the survival motor neuron gene, labeled SMN1 and SMN2. SMN1 normally produces a functional protein that is stable and necessary for the anterior horn survival. All patients with SMA have mutations in SMN1 that result in the gene being inactive. The protein product of SMN2 is usually truncated and not very stable, though it has some function. The severity of the resulting disease is influenced by the number of copies of the SMN2 gene present, and thus the available amount of partially functional SMN2 product. The oligonucleotide, in this case delivered via lumbar puncture, serves to alter the splicing of the protein product from SMN2, allowing the production of the more effective and stable protein with properties more similar to the SMN1 protein, thus ameliorating the effect of the SMN1 mutation. The patient, however, requires the administration of the therapeutic oligonucleotide indefinitely.
Gene Editing
The Gold Ring of therapeutic intervention, being able to actually correct the gene defect in any given disease, is still a goal of medical science. For the first time, this is a realistic aspiration due to the identification, development, and application of the gene editing tool Cas9 and CRISPR-Cas9 techniques.2 This new technology allows the production of a custom piece of DNA (cassette) that can repair the defective gene if incorporated into the DNA sequence of the host. The techniques necessary to introduce the therapeutic cassette to the affected cell and encourage the incorporation of the new material into the DNA have largely been developed already. The hurdle has always been the difficulty producing the therapeutic cassette for the given defect. The application of the CRISPR-Cas9 technology offers the promise of being able to produce a custom cassette specific for the given mutation, and thus potentially correcting the mutation involved in a wide variety of diseases.
The last quarter century has seen the expansion of the genetic techniques to allow the recognition and diagnosis of diseases that had resisted definitive diagnosis up to this time. These techniques have also led to the uncovering of disease mechanisms previously opaque to our understanding. The next quarter century promises to produce an explosion of therapeutic possibilities on a scale previously unimaginable. Despite the considerable initial expense of these new therapies, I believe they will be of incalculable value going forward.
References
1. de Koning AP, Gu W, Castoe TA, et al. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7(12):e1002384.
2. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9(1):1911.
John B. Bodensteiner, MD
Dr. Bodensteiner is Director of the American Board of Psychiatry and Neurology, Founding Editor and Senior Associate Editor of Seminars in Pediatric Neurology, and Senior Associate Editor of the Journal of Child Neurology.
The impetus for this article is the celebration of 25 years of publication of Neurology Reviews. Child neurology has advanced so much in these years that it is hardly recognizable from the previous state of our understanding. Many components of the discipline include, but are not limited to, epilepsy, headache, demyelinating diseases, autoimmune diseases, neoplastic, neonatal, neuromuscular disease, and developmental conditions. My interest has been related to the neuromuscular diseases of childhood. Thus, I have chosen to describe some of the ways the advances in basic medical science and our understanding of the genetic and molecular mechanisms of disease have altered the way we think about these conditions. For the first time, this knowledge has allowed us to identify targets and techniques for effective intervention and disease modification.
Myotonic Muscular Dystrophy
One example of how the advancement in the discipline of genetics has changed our understanding of disease would be myotonic muscular dystrophy (MyoD1). When I started seeing patients in the mid 1960s, MyoD1, particularly as seen in children, was recognized as an example of autosomal dominant inheritance. The tendency for subsequent generations to be more severely affected than their parents is a phenomenon known as anticipation. Anticipation was recognized, though not explained, with the understanding of the genetic mechanisms of the time. The fact that when the disease affected an infant or newborn it was almost always inherited from the mother was also well established, though the reason for this was also not understood. Actually, during the late 1970s and early 1980s the existence of the phenomenon of anticipation was widely questioned and some publications proposed that anticipation was just an artifact of observation due to the fact that we (medical clinicians) were getting better and thus identifying the disease earlier than in past years. The recognition of the importance of repeated segments of DNA in the pathogenesis of human disease and the subsequent development of the technology to study the phenomenon allowed recognition of this previously unappreciated mechanism of genetic disease causation. This new genetic mechanism explained how anticipation could occur and confirmed it as a real phenomenon. Expanded numbers of tandem repeats probably allowed the explanation of the difference in gene transmission related to the parental gender as well.1
Genetic Insights
Advancements in the understanding of genetic mechanisms of disease have made it possible to begin to identify the molecular mechanisms operating in many previously incomprehensible diseases. The development of laboratory techniques necessary for the identification of these genetic abnormalities has brought the recognition of these disease mechanisms into clinical practice. Twenty-five years ago, the ability to identify alterations in DNA sequences, the presence of deletions/duplications and expansion of trinucleotide repeats, as well as the determination (and recognition of the significance) of gene copy number, was not available to the clinician. This advancement in genetic understanding has, in turn, allowed the identification of therapeutic targets in the hope of being able to moderate or eliminate the consequence of the defective gene.
Spinal Muscular Atrophy
In the last few years, the use of relatively small molecules such as oligonucleotides to alter the transcription of mutated RNA to produce a functioning protein has been developed. Because they are small molecules, they can be delivered with relative ease to the desired site. This technology has been applied to the treatment of spinal muscular atrophy (SMA) with considerable success.
Survival motor neuron gene product is essential for the health of the anterior horn cells of the spinal cord of the central nervous system. In the human genome there are two copies of the survival motor neuron gene, labeled SMN1 and SMN2. SMN1 normally produces a functional protein that is stable and necessary for the anterior horn survival. All patients with SMA have mutations in SMN1 that result in the gene being inactive. The protein product of SMN2 is usually truncated and not very stable, though it has some function. The severity of the resulting disease is influenced by the number of copies of the SMN2 gene present, and thus the available amount of partially functional SMN2 product. The oligonucleotide, in this case delivered via lumbar puncture, serves to alter the splicing of the protein product from SMN2, allowing the production of the more effective and stable protein with properties more similar to the SMN1 protein, thus ameliorating the effect of the SMN1 mutation. The patient, however, requires the administration of the therapeutic oligonucleotide indefinitely.
Gene Editing
The Gold Ring of therapeutic intervention, being able to actually correct the gene defect in any given disease, is still a goal of medical science. For the first time, this is a realistic aspiration due to the identification, development, and application of the gene editing tool Cas9 and CRISPR-Cas9 techniques.2 This new technology allows the production of a custom piece of DNA (cassette) that can repair the defective gene if incorporated into the DNA sequence of the host. The techniques necessary to introduce the therapeutic cassette to the affected cell and encourage the incorporation of the new material into the DNA have largely been developed already. The hurdle has always been the difficulty producing the therapeutic cassette for the given defect. The application of the CRISPR-Cas9 technology offers the promise of being able to produce a custom cassette specific for the given mutation, and thus potentially correcting the mutation involved in a wide variety of diseases.
The last quarter century has seen the expansion of the genetic techniques to allow the recognition and diagnosis of diseases that had resisted definitive diagnosis up to this time. These techniques have also led to the uncovering of disease mechanisms previously opaque to our understanding. The next quarter century promises to produce an explosion of therapeutic possibilities on a scale previously unimaginable. Despite the considerable initial expense of these new therapies, I believe they will be of incalculable value going forward.
References
1. de Koning AP, Gu W, Castoe TA, et al. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7(12):e1002384.
2. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9(1):1911.
John B. Bodensteiner, MD
Dr. Bodensteiner is Director of the American Board of Psychiatry and Neurology, Founding Editor and Senior Associate Editor of Seminars in Pediatric Neurology, and Senior Associate Editor of the Journal of Child Neurology.
The impetus for this article is the celebration of 25 years of publication of Neurology Reviews. Child neurology has advanced so much in these years that it is hardly recognizable from the previous state of our understanding. Many components of the discipline include, but are not limited to, epilepsy, headache, demyelinating diseases, autoimmune diseases, neoplastic, neonatal, neuromuscular disease, and developmental conditions. My interest has been related to the neuromuscular diseases of childhood. Thus, I have chosen to describe some of the ways the advances in basic medical science and our understanding of the genetic and molecular mechanisms of disease have altered the way we think about these conditions. For the first time, this knowledge has allowed us to identify targets and techniques for effective intervention and disease modification.
Myotonic Muscular Dystrophy
One example of how the advancement in the discipline of genetics has changed our understanding of disease would be myotonic muscular dystrophy (MyoD1). When I started seeing patients in the mid 1960s, MyoD1, particularly as seen in children, was recognized as an example of autosomal dominant inheritance. The tendency for subsequent generations to be more severely affected than their parents is a phenomenon known as anticipation. Anticipation was recognized, though not explained, with the understanding of the genetic mechanisms of the time. The fact that when the disease affected an infant or newborn it was almost always inherited from the mother was also well established, though the reason for this was also not understood. Actually, during the late 1970s and early 1980s the existence of the phenomenon of anticipation was widely questioned and some publications proposed that anticipation was just an artifact of observation due to the fact that we (medical clinicians) were getting better and thus identifying the disease earlier than in past years. The recognition of the importance of repeated segments of DNA in the pathogenesis of human disease and the subsequent development of the technology to study the phenomenon allowed recognition of this previously unappreciated mechanism of genetic disease causation. This new genetic mechanism explained how anticipation could occur and confirmed it as a real phenomenon. Expanded numbers of tandem repeats probably allowed the explanation of the difference in gene transmission related to the parental gender as well.1
Genetic Insights
Advancements in the understanding of genetic mechanisms of disease have made it possible to begin to identify the molecular mechanisms operating in many previously incomprehensible diseases. The development of laboratory techniques necessary for the identification of these genetic abnormalities has brought the recognition of these disease mechanisms into clinical practice. Twenty-five years ago, the ability to identify alterations in DNA sequences, the presence of deletions/duplications and expansion of trinucleotide repeats, as well as the determination (and recognition of the significance) of gene copy number, was not available to the clinician. This advancement in genetic understanding has, in turn, allowed the identification of therapeutic targets in the hope of being able to moderate or eliminate the consequence of the defective gene.
Spinal Muscular Atrophy
In the last few years, the use of relatively small molecules such as oligonucleotides to alter the transcription of mutated RNA to produce a functioning protein has been developed. Because they are small molecules, they can be delivered with relative ease to the desired site. This technology has been applied to the treatment of spinal muscular atrophy (SMA) with considerable success.
Survival motor neuron gene product is essential for the health of the anterior horn cells of the spinal cord of the central nervous system. In the human genome there are two copies of the survival motor neuron gene, labeled SMN1 and SMN2. SMN1 normally produces a functional protein that is stable and necessary for the anterior horn survival. All patients with SMA have mutations in SMN1 that result in the gene being inactive. The protein product of SMN2 is usually truncated and not very stable, though it has some function. The severity of the resulting disease is influenced by the number of copies of the SMN2 gene present, and thus the available amount of partially functional SMN2 product. The oligonucleotide, in this case delivered via lumbar puncture, serves to alter the splicing of the protein product from SMN2, allowing the production of the more effective and stable protein with properties more similar to the SMN1 protein, thus ameliorating the effect of the SMN1 mutation. The patient, however, requires the administration of the therapeutic oligonucleotide indefinitely.
Gene Editing
The Gold Ring of therapeutic intervention, being able to actually correct the gene defect in any given disease, is still a goal of medical science. For the first time, this is a realistic aspiration due to the identification, development, and application of the gene editing tool Cas9 and CRISPR-Cas9 techniques.2 This new technology allows the production of a custom piece of DNA (cassette) that can repair the defective gene if incorporated into the DNA sequence of the host. The techniques necessary to introduce the therapeutic cassette to the affected cell and encourage the incorporation of the new material into the DNA have largely been developed already. The hurdle has always been the difficulty producing the therapeutic cassette for the given defect. The application of the CRISPR-Cas9 technology offers the promise of being able to produce a custom cassette specific for the given mutation, and thus potentially correcting the mutation involved in a wide variety of diseases.
The last quarter century has seen the expansion of the genetic techniques to allow the recognition and diagnosis of diseases that had resisted definitive diagnosis up to this time. These techniques have also led to the uncovering of disease mechanisms previously opaque to our understanding. The next quarter century promises to produce an explosion of therapeutic possibilities on a scale previously unimaginable. Despite the considerable initial expense of these new therapies, I believe they will be of incalculable value going forward.
References
1. de Koning AP, Gu W, Castoe TA, et al. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7(12):e1002384.
2. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9(1):1911.
Caffeine for apnea of prematurity found safe, effective at 11 years
Caffeine for apnea of prematurity was neurobehaviorally safe and significantly improved fine motor coordination, visuomotor integration, visual perception, and visuospatial organization at 11-year follow-up, according to the results of a double-blind, randomized, controlled trial.
“There was little evidence for differences between the caffeine and placebo groups on tests of general intelligence, attention, executive function, and behavior. This highlights the long-term safety and efficacy of caffeine therapy for apnea of prematurity in very-low-birth-weight neonates,” wrote Ines M. Mürner-Lavanchy, PhD, of Monash University, Clayton, Australia, and her associates. The Caffeine for Apnea of Prematurity (CAP) trial, the first to assess long-term neurobehavioral outcomes of neonatal caffeine therapy, was published online April 11 in Pediatrics.
Neonatal caffeine therapy significantly lowered the risk of death before 18 months, cerebral palsy, cognitive delay, severe hearing loss, and bilateral blindness, as has been reported (N Engl J Med. 2007;357:1893-902). By 5 years, caffeine no longer showed significant benefits, apart from improved motor performance, Dr. Mürner-Lavanchy and her associates noted.
At 11 years, available data from 870 patients showed generally similar neurobehavioral outcomes between groups, although the caffeine group scored higher on most scales. The most apparent benefits included visuomotor integration (mean difference from placebo, 1.8; 95% confidence interval, 0.0-3.7; P less than .05), visual perception (2.0; 95% CI, 0.3-3.8; P = .02), fine motor coordination (2.9; 95% CI, 0.7-5.1; P = .01), and Rey Complex Figure copy accuracy, a measure of visuospatial organization (1.2; 95% CI, 0.4-2.0; P = .003).
Eleven-year follow-up data were missing for 22% of patients, but their birth characteristics and childhood outcomes resembled those of patients with available data, the investigators said. “Therefore, we are confident that the outcomes of the whole cohort are reflected in the present results with sufficient accuracy.”
The Canadian Institutes of Health Research provided funding. The investigators reported having no relevant conflicts of interest.
SOURCE: Mürner-Lavanchy IM et al. Pediatrics. 2018 Apr 11. doi: 10.1542/peds.2017-4047.
Caffeine for apnea of prematurity was neurobehaviorally safe and significantly improved fine motor coordination, visuomotor integration, visual perception, and visuospatial organization at 11-year follow-up, according to the results of a double-blind, randomized, controlled trial.
“There was little evidence for differences between the caffeine and placebo groups on tests of general intelligence, attention, executive function, and behavior. This highlights the long-term safety and efficacy of caffeine therapy for apnea of prematurity in very-low-birth-weight neonates,” wrote Ines M. Mürner-Lavanchy, PhD, of Monash University, Clayton, Australia, and her associates. The Caffeine for Apnea of Prematurity (CAP) trial, the first to assess long-term neurobehavioral outcomes of neonatal caffeine therapy, was published online April 11 in Pediatrics.
Neonatal caffeine therapy significantly lowered the risk of death before 18 months, cerebral palsy, cognitive delay, severe hearing loss, and bilateral blindness, as has been reported (N Engl J Med. 2007;357:1893-902). By 5 years, caffeine no longer showed significant benefits, apart from improved motor performance, Dr. Mürner-Lavanchy and her associates noted.
At 11 years, available data from 870 patients showed generally similar neurobehavioral outcomes between groups, although the caffeine group scored higher on most scales. The most apparent benefits included visuomotor integration (mean difference from placebo, 1.8; 95% confidence interval, 0.0-3.7; P less than .05), visual perception (2.0; 95% CI, 0.3-3.8; P = .02), fine motor coordination (2.9; 95% CI, 0.7-5.1; P = .01), and Rey Complex Figure copy accuracy, a measure of visuospatial organization (1.2; 95% CI, 0.4-2.0; P = .003).
Eleven-year follow-up data were missing for 22% of patients, but their birth characteristics and childhood outcomes resembled those of patients with available data, the investigators said. “Therefore, we are confident that the outcomes of the whole cohort are reflected in the present results with sufficient accuracy.”
The Canadian Institutes of Health Research provided funding. The investigators reported having no relevant conflicts of interest.
SOURCE: Mürner-Lavanchy IM et al. Pediatrics. 2018 Apr 11. doi: 10.1542/peds.2017-4047.
Caffeine for apnea of prematurity was neurobehaviorally safe and significantly improved fine motor coordination, visuomotor integration, visual perception, and visuospatial organization at 11-year follow-up, according to the results of a double-blind, randomized, controlled trial.
“There was little evidence for differences between the caffeine and placebo groups on tests of general intelligence, attention, executive function, and behavior. This highlights the long-term safety and efficacy of caffeine therapy for apnea of prematurity in very-low-birth-weight neonates,” wrote Ines M. Mürner-Lavanchy, PhD, of Monash University, Clayton, Australia, and her associates. The Caffeine for Apnea of Prematurity (CAP) trial, the first to assess long-term neurobehavioral outcomes of neonatal caffeine therapy, was published online April 11 in Pediatrics.
Neonatal caffeine therapy significantly lowered the risk of death before 18 months, cerebral palsy, cognitive delay, severe hearing loss, and bilateral blindness, as has been reported (N Engl J Med. 2007;357:1893-902). By 5 years, caffeine no longer showed significant benefits, apart from improved motor performance, Dr. Mürner-Lavanchy and her associates noted.
At 11 years, available data from 870 patients showed generally similar neurobehavioral outcomes between groups, although the caffeine group scored higher on most scales. The most apparent benefits included visuomotor integration (mean difference from placebo, 1.8; 95% confidence interval, 0.0-3.7; P less than .05), visual perception (2.0; 95% CI, 0.3-3.8; P = .02), fine motor coordination (2.9; 95% CI, 0.7-5.1; P = .01), and Rey Complex Figure copy accuracy, a measure of visuospatial organization (1.2; 95% CI, 0.4-2.0; P = .003).
Eleven-year follow-up data were missing for 22% of patients, but their birth characteristics and childhood outcomes resembled those of patients with available data, the investigators said. “Therefore, we are confident that the outcomes of the whole cohort are reflected in the present results with sufficient accuracy.”
The Canadian Institutes of Health Research provided funding. The investigators reported having no relevant conflicts of interest.
SOURCE: Mürner-Lavanchy IM et al. Pediatrics. 2018 Apr 11. doi: 10.1542/peds.2017-4047.
FROM PEDIATRICS
Key clinical point:
Major finding: At 11 years, the caffeine group outperformed the placebo group on measures of fine motor coordination (P = .01), visuomotor integration (P less than .05), visual perception (P = .02), and visuospatial organization (P = .003).
Study details: The Caffeine for Apnea of Prematurity (CAP) trial, a double-blind, multicenter, randomized, placebo-controlled trial of 870 very-low-birth-weight infants (500-1,250 g).
Disclosures: The Canadian Institutes of Health Research provided funding. The investigators reported having no relevant conflicts of interest.
Source: Pediatrics. 2018 Apr 11. doi: 10.1542/peds.2017-4047.