User login
How Low Should You Go? Optimizing BP in CKD
Q) I hear providers quote different numbers for target blood pressure in kidney patients. Which are correct?
The answer to this question starts with the word “meta-analysis”—but don’t stop reading! We’ll get down to the basics quickly. Determining the goal blood pressure (BP) for patients with chronic kidney disease (CKD) comes down to three questions.
1. Does the patient have diabetes? The National Kidney Foundation states that the goal BP for a patient with type 2 diabetes, CKD, and urine albumin > 30 mg/dL is < 140/90 mm Hg.1 This is in line with the JNC-8 recommendations for patients with hypertension and CKD, which do not take urine albumin level into consideration.2 It is important to recognize that while many patients with CKD do not have diabetes, those who do have a worse prognosis.3
2. Is the patient African-American? A meta-analysis of nine randomized clinical trials found that lowering BP to < 130/80 mm Hg was linked to a slower decline in glomerular filtration rate (GFR) in non-African-American patients.3 But this BP was not beneficial for African-American patients; in fact, it actually caused a faster decline in GFR.3 Therefore, target BP for African-American patients should be < 140/90 mm Hg.
3. Does the patient have significant albuminuria? An additional subgroup analysis for patients with high levels of proteinuria (defined as > 1 g/d) yielded inconclusive results.3 Patients with proteinuria > 1 g/d tended to have a slower decline in GFR with intensive BP control.3 Proteinuria > 0.5 g/d was correlated with a slowed progression to end-stage renal disease with intensive BP control.3 Again, these were trends and not statistically significant. So, for patients with high levels of proteinuria, it will not hurt to achieve a BP < 130/80 mm Hg, but there is no statistically significant difference between BP < 130/80 mm Hg and BP < 140/90 mm Hg.
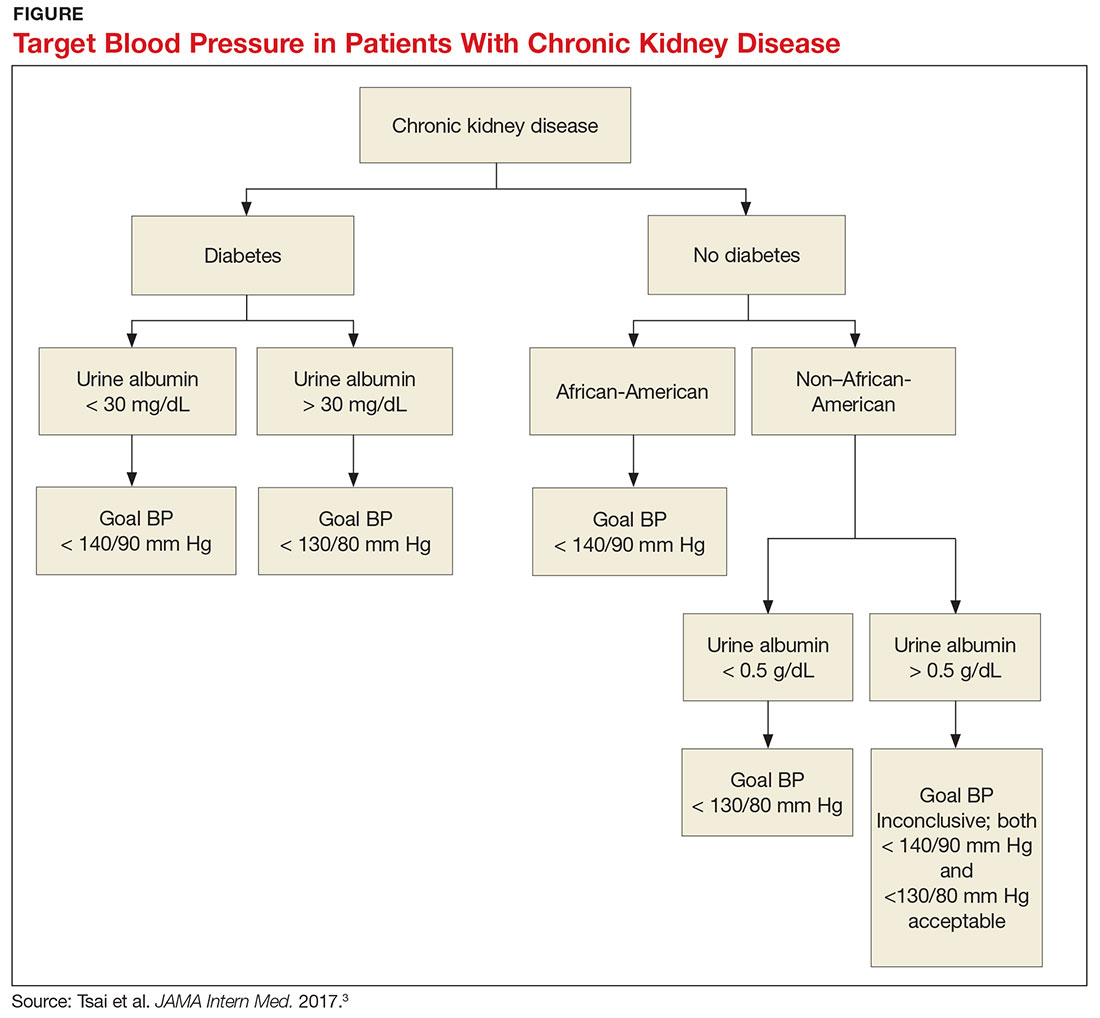
What, then, are the recommendations for an African-American patient with significant proteinuria? While not addressed directly in the analysis, the study results suggest that the goal should still be < 140/90 mm Hg, since the link between race and changes in GFR is statistically significant and the effects of proteinuria are not. Although the recommendations from this review are many, the main points are summarized in the Figure.—RC
Rebecca Clawson, MAT, PA-C
Instructor, PA Program, LSU Health Shreveport, Louisiana
1. Kidney Disease: Improving Global Outcomes (KDIGO) CKD work group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Inter Suppl. 2013;3(1):1-150.
2. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520.
3. Tsai WC, Wu HY, Peng YS, et al. Association of intensive blood pressure control and kidney disease progression in nondiabetic patients with chronic kidney disease: a systematic review and meta-analysis. JAMA Intern Med. 2017;177:792-799.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Zorica Kauric-Klein, APRN-BC, PhD, who is an Assistant Clinical Professor in the College of Nursing at Wayne State University in Detroit, and Rebecca Clawson, MAT, PA-C, who is an Instructor in the PA Program at LSU Health Shreveport in Louisiana.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Zorica Kauric-Klein, APRN-BC, PhD, who is an Assistant Clinical Professor in the College of Nursing at Wayne State University in Detroit, and Rebecca Clawson, MAT, PA-C, who is an Instructor in the PA Program at LSU Health Shreveport in Louisiana.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Zorica Kauric-Klein, APRN-BC, PhD, who is an Assistant Clinical Professor in the College of Nursing at Wayne State University in Detroit, and Rebecca Clawson, MAT, PA-C, who is an Instructor in the PA Program at LSU Health Shreveport in Louisiana.
Q) I hear providers quote different numbers for target blood pressure in kidney patients. Which are correct?
The answer to this question starts with the word “meta-analysis”—but don’t stop reading! We’ll get down to the basics quickly. Determining the goal blood pressure (BP) for patients with chronic kidney disease (CKD) comes down to three questions.
1. Does the patient have diabetes? The National Kidney Foundation states that the goal BP for a patient with type 2 diabetes, CKD, and urine albumin > 30 mg/dL is < 140/90 mm Hg.1 This is in line with the JNC-8 recommendations for patients with hypertension and CKD, which do not take urine albumin level into consideration.2 It is important to recognize that while many patients with CKD do not have diabetes, those who do have a worse prognosis.3
2. Is the patient African-American? A meta-analysis of nine randomized clinical trials found that lowering BP to < 130/80 mm Hg was linked to a slower decline in glomerular filtration rate (GFR) in non-African-American patients.3 But this BP was not beneficial for African-American patients; in fact, it actually caused a faster decline in GFR.3 Therefore, target BP for African-American patients should be < 140/90 mm Hg.
3. Does the patient have significant albuminuria? An additional subgroup analysis for patients with high levels of proteinuria (defined as > 1 g/d) yielded inconclusive results.3 Patients with proteinuria > 1 g/d tended to have a slower decline in GFR with intensive BP control.3 Proteinuria > 0.5 g/d was correlated with a slowed progression to end-stage renal disease with intensive BP control.3 Again, these were trends and not statistically significant. So, for patients with high levels of proteinuria, it will not hurt to achieve a BP < 130/80 mm Hg, but there is no statistically significant difference between BP < 130/80 mm Hg and BP < 140/90 mm Hg.
What, then, are the recommendations for an African-American patient with significant proteinuria? While not addressed directly in the analysis, the study results suggest that the goal should still be < 140/90 mm Hg, since the link between race and changes in GFR is statistically significant and the effects of proteinuria are not. Although the recommendations from this review are many, the main points are summarized in the Figure.—RC
Rebecca Clawson, MAT, PA-C
Instructor, PA Program, LSU Health Shreveport, Louisiana
Q) I hear providers quote different numbers for target blood pressure in kidney patients. Which are correct?
The answer to this question starts with the word “meta-analysis”—but don’t stop reading! We’ll get down to the basics quickly. Determining the goal blood pressure (BP) for patients with chronic kidney disease (CKD) comes down to three questions.
1. Does the patient have diabetes? The National Kidney Foundation states that the goal BP for a patient with type 2 diabetes, CKD, and urine albumin > 30 mg/dL is < 140/90 mm Hg.1 This is in line with the JNC-8 recommendations for patients with hypertension and CKD, which do not take urine albumin level into consideration.2 It is important to recognize that while many patients with CKD do not have diabetes, those who do have a worse prognosis.3
2. Is the patient African-American? A meta-analysis of nine randomized clinical trials found that lowering BP to < 130/80 mm Hg was linked to a slower decline in glomerular filtration rate (GFR) in non-African-American patients.3 But this BP was not beneficial for African-American patients; in fact, it actually caused a faster decline in GFR.3 Therefore, target BP for African-American patients should be < 140/90 mm Hg.
3. Does the patient have significant albuminuria? An additional subgroup analysis for patients with high levels of proteinuria (defined as > 1 g/d) yielded inconclusive results.3 Patients with proteinuria > 1 g/d tended to have a slower decline in GFR with intensive BP control.3 Proteinuria > 0.5 g/d was correlated with a slowed progression to end-stage renal disease with intensive BP control.3 Again, these were trends and not statistically significant. So, for patients with high levels of proteinuria, it will not hurt to achieve a BP < 130/80 mm Hg, but there is no statistically significant difference between BP < 130/80 mm Hg and BP < 140/90 mm Hg.
What, then, are the recommendations for an African-American patient with significant proteinuria? While not addressed directly in the analysis, the study results suggest that the goal should still be < 140/90 mm Hg, since the link between race and changes in GFR is statistically significant and the effects of proteinuria are not. Although the recommendations from this review are many, the main points are summarized in the Figure.—RC
Rebecca Clawson, MAT, PA-C
Instructor, PA Program, LSU Health Shreveport, Louisiana
1. Kidney Disease: Improving Global Outcomes (KDIGO) CKD work group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Inter Suppl. 2013;3(1):1-150.
2. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520.
3. Tsai WC, Wu HY, Peng YS, et al. Association of intensive blood pressure control and kidney disease progression in nondiabetic patients with chronic kidney disease: a systematic review and meta-analysis. JAMA Intern Med. 2017;177:792-799.
1. Kidney Disease: Improving Global Outcomes (KDIGO) CKD work group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Inter Suppl. 2013;3(1):1-150.
2. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520.
3. Tsai WC, Wu HY, Peng YS, et al. Association of intensive blood pressure control and kidney disease progression in nondiabetic patients with chronic kidney disease: a systematic review and meta-analysis. JAMA Intern Med. 2017;177:792-799.
Nocturia and sleep apnea
Author’s note: I have been writing “Myth of the Month” columns for the last several years. I will try to continue to write about myths when possible, but I would like to introduce a new column, “Pearl of the Month.” I want to share with you pearls that I have found really helpful in medical practice. Some of these will be new news, while some may be old news that may not be well known.
A 65-year-old man comes to a clinic concerned about frequent nocturia. He is getting up four times a night to urinate, and he has been urinating about every 5 hours during the day. He has been seen twice for this problem and was diagnosed with benign prostatic hyperplasia and started on tamsulosin.
He found a slight improvement when he started on 0.4 mg qhs, reducing his nocturia episodes from four to three. His dose was increased to 0.8 mg qhs, with no improvement in nocturia.
Exam today: BP, 140/94; pulse, 70. Rectal exam: Prostate is twice normal size without nodules. Labs: Na, 140; K, 4.0; glucose, 80; Ca, 9.6.
He is frustrated because he feels tired and sleepy from having to get up so often to urinate every night.
What is the best treatment/advice at this point?
A. Check hemoglobin A1C.
B. Start finasteride.
C. Switch tamsulosin to terazosin.
D. Evaluate for sleep apnea.
Umpei Yamamoto, MD, of Kyushu University Hospital, Japan, and colleagues studied the prevalence of sleep-disordered breathing among patients who presented to a urology clinic with nocturia and in those who visited a sleep apnea clinic with symptoms of excessive daytime sleepiness.1 Sleep-disordered breathing was found in 91% of the patients from the sleep apnea clinic and 70% of the patients from the urology clinic. The frequency of nocturia was reduced with continuous positive airway pressure (CPAP) in both groups in the patients who had not responded to conventional therapy or nocturia.
The symptom of nocturia as a symptom of sleep apnea might be even more common in women.2 Ozen K. Basoglu, MD, and Mehmet Sezai Tasbakan, MD, of Ege University, Izmir, Turkey, described clinical similarities and differences based on gender in a large group of patients with sleep apnea. Both men and women with sleep apnea had similar rates of excessive daytime sleepiness, snoring, and impaired concentration. Women had more frequent nocturia.
Nocturia especially should be considered a possible clue for the presence of sleep apnea in younger patients who have fewer other reasons to have nocturia. Takahiro Maeda, MD, of Keio University, Tokyo, and colleagues found that men younger than 50 years had more nocturnal urinations the worse their apnea-hypopnea index was.3 Overall in the study, 85% of the patients had a reduction in nighttime urination after CPAP therapy.
Treatment of sleep apnea has been shown in several studies to improve the nocturia that occurs in patients with sleep apnea. Hyoung Keun Park, MD, of Konkuk University, Seoul, and colleagues studied whether surgical intervention with uvulopalatopharyngoplasty (UPPP) reduced nocturia in patients with sleep apnea.4 In the study, there was a 73% success rate in treatment for sleep apnea with the UPPP surgery, and, among those who had successful surgeries, nocturia episodes decreased from 1.9 preoperatively to 0.7 postoperatively (P less than .001).
Minoru Miyazato, MD, PhD, of University of the Ryukyus, Okinawa, Japan, and colleagues looked at the effect of CPAP treatment on nighttime urine production in patients with obstructive sleep apnea.5 In this small study of 40 patients, mean nighttime voiding episodes decreased from 2.1 to 1.2 (P less than .01).

Pearl: Sleep apnea should be considered in the differential diagnosis of patients with nocturia, and treatment of sleep apnea may decrease nocturia.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at dpaauw@uw.edu.
References
1. Intern Med. 2016;55(8):901-5.
2. Sleep Breath. 2017 Feb 14. doi: 10.1007/s11325-017-1482-9.
3. Can Urol Assoc J. 2016 Jul-Aug;10(7-8):E241-5.
4. Int Neurourol J. 2016 Dec;20(4):329-34.
5. Neurourol Urodyn. 2017 Feb;36(2):376-9.
Author’s note: I have been writing “Myth of the Month” columns for the last several years. I will try to continue to write about myths when possible, but I would like to introduce a new column, “Pearl of the Month.” I want to share with you pearls that I have found really helpful in medical practice. Some of these will be new news, while some may be old news that may not be well known.
A 65-year-old man comes to a clinic concerned about frequent nocturia. He is getting up four times a night to urinate, and he has been urinating about every 5 hours during the day. He has been seen twice for this problem and was diagnosed with benign prostatic hyperplasia and started on tamsulosin.
He found a slight improvement when he started on 0.4 mg qhs, reducing his nocturia episodes from four to three. His dose was increased to 0.8 mg qhs, with no improvement in nocturia.
Exam today: BP, 140/94; pulse, 70. Rectal exam: Prostate is twice normal size without nodules. Labs: Na, 140; K, 4.0; glucose, 80; Ca, 9.6.
He is frustrated because he feels tired and sleepy from having to get up so often to urinate every night.
What is the best treatment/advice at this point?
A. Check hemoglobin A1C.
B. Start finasteride.
C. Switch tamsulosin to terazosin.
D. Evaluate for sleep apnea.
Umpei Yamamoto, MD, of Kyushu University Hospital, Japan, and colleagues studied the prevalence of sleep-disordered breathing among patients who presented to a urology clinic with nocturia and in those who visited a sleep apnea clinic with symptoms of excessive daytime sleepiness.1 Sleep-disordered breathing was found in 91% of the patients from the sleep apnea clinic and 70% of the patients from the urology clinic. The frequency of nocturia was reduced with continuous positive airway pressure (CPAP) in both groups in the patients who had not responded to conventional therapy or nocturia.
The symptom of nocturia as a symptom of sleep apnea might be even more common in women.2 Ozen K. Basoglu, MD, and Mehmet Sezai Tasbakan, MD, of Ege University, Izmir, Turkey, described clinical similarities and differences based on gender in a large group of patients with sleep apnea. Both men and women with sleep apnea had similar rates of excessive daytime sleepiness, snoring, and impaired concentration. Women had more frequent nocturia.
Nocturia especially should be considered a possible clue for the presence of sleep apnea in younger patients who have fewer other reasons to have nocturia. Takahiro Maeda, MD, of Keio University, Tokyo, and colleagues found that men younger than 50 years had more nocturnal urinations the worse their apnea-hypopnea index was.3 Overall in the study, 85% of the patients had a reduction in nighttime urination after CPAP therapy.
Treatment of sleep apnea has been shown in several studies to improve the nocturia that occurs in patients with sleep apnea. Hyoung Keun Park, MD, of Konkuk University, Seoul, and colleagues studied whether surgical intervention with uvulopalatopharyngoplasty (UPPP) reduced nocturia in patients with sleep apnea.4 In the study, there was a 73% success rate in treatment for sleep apnea with the UPPP surgery, and, among those who had successful surgeries, nocturia episodes decreased from 1.9 preoperatively to 0.7 postoperatively (P less than .001).
Minoru Miyazato, MD, PhD, of University of the Ryukyus, Okinawa, Japan, and colleagues looked at the effect of CPAP treatment on nighttime urine production in patients with obstructive sleep apnea.5 In this small study of 40 patients, mean nighttime voiding episodes decreased from 2.1 to 1.2 (P less than .01).
Pearl: Sleep apnea should be considered in the differential diagnosis of patients with nocturia, and treatment of sleep apnea may decrease nocturia.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at dpaauw@uw.edu.
References
1. Intern Med. 2016;55(8):901-5.
2. Sleep Breath. 2017 Feb 14. doi: 10.1007/s11325-017-1482-9.
3. Can Urol Assoc J. 2016 Jul-Aug;10(7-8):E241-5.
4. Int Neurourol J. 2016 Dec;20(4):329-34.
5. Neurourol Urodyn. 2017 Feb;36(2):376-9.
Author’s note: I have been writing “Myth of the Month” columns for the last several years. I will try to continue to write about myths when possible, but I would like to introduce a new column, “Pearl of the Month.” I want to share with you pearls that I have found really helpful in medical practice. Some of these will be new news, while some may be old news that may not be well known.
A 65-year-old man comes to a clinic concerned about frequent nocturia. He is getting up four times a night to urinate, and he has been urinating about every 5 hours during the day. He has been seen twice for this problem and was diagnosed with benign prostatic hyperplasia and started on tamsulosin.
He found a slight improvement when he started on 0.4 mg qhs, reducing his nocturia episodes from four to three. His dose was increased to 0.8 mg qhs, with no improvement in nocturia.
Exam today: BP, 140/94; pulse, 70. Rectal exam: Prostate is twice normal size without nodules. Labs: Na, 140; K, 4.0; glucose, 80; Ca, 9.6.
He is frustrated because he feels tired and sleepy from having to get up so often to urinate every night.
What is the best treatment/advice at this point?
A. Check hemoglobin A1C.
B. Start finasteride.
C. Switch tamsulosin to terazosin.
D. Evaluate for sleep apnea.
Umpei Yamamoto, MD, of Kyushu University Hospital, Japan, and colleagues studied the prevalence of sleep-disordered breathing among patients who presented to a urology clinic with nocturia and in those who visited a sleep apnea clinic with symptoms of excessive daytime sleepiness.1 Sleep-disordered breathing was found in 91% of the patients from the sleep apnea clinic and 70% of the patients from the urology clinic. The frequency of nocturia was reduced with continuous positive airway pressure (CPAP) in both groups in the patients who had not responded to conventional therapy or nocturia.
The symptom of nocturia as a symptom of sleep apnea might be even more common in women.2 Ozen K. Basoglu, MD, and Mehmet Sezai Tasbakan, MD, of Ege University, Izmir, Turkey, described clinical similarities and differences based on gender in a large group of patients with sleep apnea. Both men and women with sleep apnea had similar rates of excessive daytime sleepiness, snoring, and impaired concentration. Women had more frequent nocturia.
Nocturia especially should be considered a possible clue for the presence of sleep apnea in younger patients who have fewer other reasons to have nocturia. Takahiro Maeda, MD, of Keio University, Tokyo, and colleagues found that men younger than 50 years had more nocturnal urinations the worse their apnea-hypopnea index was.3 Overall in the study, 85% of the patients had a reduction in nighttime urination after CPAP therapy.
Treatment of sleep apnea has been shown in several studies to improve the nocturia that occurs in patients with sleep apnea. Hyoung Keun Park, MD, of Konkuk University, Seoul, and colleagues studied whether surgical intervention with uvulopalatopharyngoplasty (UPPP) reduced nocturia in patients with sleep apnea.4 In the study, there was a 73% success rate in treatment for sleep apnea with the UPPP surgery, and, among those who had successful surgeries, nocturia episodes decreased from 1.9 preoperatively to 0.7 postoperatively (P less than .001).
Minoru Miyazato, MD, PhD, of University of the Ryukyus, Okinawa, Japan, and colleagues looked at the effect of CPAP treatment on nighttime urine production in patients with obstructive sleep apnea.5 In this small study of 40 patients, mean nighttime voiding episodes decreased from 2.1 to 1.2 (P less than .01).
Pearl: Sleep apnea should be considered in the differential diagnosis of patients with nocturia, and treatment of sleep apnea may decrease nocturia.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at dpaauw@uw.edu.
References
1. Intern Med. 2016;55(8):901-5.
2. Sleep Breath. 2017 Feb 14. doi: 10.1007/s11325-017-1482-9.
3. Can Urol Assoc J. 2016 Jul-Aug;10(7-8):E241-5.
4. Int Neurourol J. 2016 Dec;20(4):329-34.
5. Neurourol Urodyn. 2017 Feb;36(2):376-9.
Transradial PCI in acute coronary syndrome causes less kidney damage
PARIS – Transradial-access percutaneous coronary intervention (PCI) in patients with acute coronary syndrome (ACS) results in a significantly lower risk of acute kidney injury (AKI), compared with the transfemoral approach, according to a new analysis from the large randomized MATRIX trial.
The results of this prespecified secondary subgroup analysis of MATRIX suggest it’s time to update the classic “five golden rules” for reduction of contrast medium–induced AKI by adding a sixth. “Use a transradial approach,” Bernardo Cortese, MD, said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.

He reported on 8,210 participants in the MATRIX trial (Minimizing Adverse Haemorrhagic Events by Transradial Access Site and Systemic Implementation of Angiox) who were randomized to transradial- or transfemoral-access PCI for non–ST-elevation MI or ST-elevation MI.
The primary results of the 78-site, four-country European study, previously published, showed that transradial PCI reduced the composite risk of death, MI, stroke, or major bleeding by 17%, compared with transfemoral PCI, a benefit mainly driven by a marked reduction in clinically important bleeding (Lancet. 2015 Jun 20;385[9986]:2465-76).
Left unanswered by the primary analysis was the question of whether transradial PCI in ACS patients also reduced AKI risk, as had previously been suggested by a meta-analysis of observational studies (Int J Cardiol. 2015 Jan 20;179:309-11). In designing the MATRIX trial, Dr. Cortese and the other investigators decided to address that issue separately in a prespecified secondary analysis known as AKI-MATRIX. For this purpose, AKI was defined as either a post-PCI in-hospital increase in serum creatinine level of more than 25%, compared with the preangiography baseline, or an absolute increase in serum creatinine of greater than 0.5 mg/dL.
AKI occurred in 15.4% of ACS patients who underwent PCI with transradial access and 17.3% of those randomized to transfemoral access, for a significant 13% relative risk reduction. This was accomplished without any increase in the volume of contrast media required. The average was 200 mL in both study groups.
The reduction in AKI achieved with transradial-access PCI was seen in all patient subgroups, including those at increased AKI risk because of an estimated glomerular filtration rate below 60 mL/min, age 75 or older, Killup class III or IV, or a Mehran score greater than 10.
Dr. Cortese proposed several possible mechanisms for the observed reduction in AKI seen with transradial-access PCI. The major factor in his view is that the transradial approach entails less bleeding, as earlier demonstrated in the primary analysis – and bleeding has been associated with impaired renal perfusion in several prior studies. Also, it’s plausible that the passage of the catheter across the renal arteries during the transfemoral approach dislodges atherosclerotic debris, which then travels down the renal vessels.
The five golden rules for preventing contrast media–induced AKI, he noted, are
1. Discontinue nephrotoxic drugs before the procedure.
2. Identify high-risk patients.
3. Hydrate them.
4. Choose an ideal contrast medium.
5. Adapt the dose of contrast medium to the patient’s specific situation.
Discussant Jacek Legutko, MD, PhD, of Jagiellonian University in Krakow, Poland, said the primary results of the MATRIX trial published in 2015 have had a major impact on Polish interventional cardiology, where transradial PCI is now used in 80% of PCIs. The AKI study results will reinforce this trend, he added.
“You have shown something opposite to what we’ve thought in the past, that maybe, with a radial approach, we would use more contrast medium, which is a risk factor for AKI. In your study – at least in ACS with very experienced transradial operators – there was no increase in contrast volume, and the risk of AKI decreased,” Dr. Legutko said.
Asked about the possibility that transradial PCI might be associated with an increased risk of embolization to the brain, much as the transfemoral approach might cause embolization to the kidneys, Dr. Cortese said there was no significant difference between the two AKI-MATRIX study arms in rates of transient ischemic attack or stroke.
“I did my first transradial PCI in 2003, and I haven’t seen any increase in these events or later dementia,” he added.
The prespecified secondary analysis of the MATRIX trial was conducted without commercial support. The presenter reported serving as a consultant to Abbott, AstraZeneca, Daiichi Sankyo, Eli Lilly, and Stentys.
Simultaneous with his presentation in Paris, the AKI-MATRIX study was published online at www.sciencedirect.com/science/article/pii/S0735109717368973.
PARIS – Transradial-access percutaneous coronary intervention (PCI) in patients with acute coronary syndrome (ACS) results in a significantly lower risk of acute kidney injury (AKI), compared with the transfemoral approach, according to a new analysis from the large randomized MATRIX trial.
The results of this prespecified secondary subgroup analysis of MATRIX suggest it’s time to update the classic “five golden rules” for reduction of contrast medium–induced AKI by adding a sixth. “Use a transradial approach,” Bernardo Cortese, MD, said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
He reported on 8,210 participants in the MATRIX trial (Minimizing Adverse Haemorrhagic Events by Transradial Access Site and Systemic Implementation of Angiox) who were randomized to transradial- or transfemoral-access PCI for non–ST-elevation MI or ST-elevation MI.
The primary results of the 78-site, four-country European study, previously published, showed that transradial PCI reduced the composite risk of death, MI, stroke, or major bleeding by 17%, compared with transfemoral PCI, a benefit mainly driven by a marked reduction in clinically important bleeding (Lancet. 2015 Jun 20;385[9986]:2465-76).
Left unanswered by the primary analysis was the question of whether transradial PCI in ACS patients also reduced AKI risk, as had previously been suggested by a meta-analysis of observational studies (Int J Cardiol. 2015 Jan 20;179:309-11). In designing the MATRIX trial, Dr. Cortese and the other investigators decided to address that issue separately in a prespecified secondary analysis known as AKI-MATRIX. For this purpose, AKI was defined as either a post-PCI in-hospital increase in serum creatinine level of more than 25%, compared with the preangiography baseline, or an absolute increase in serum creatinine of greater than 0.5 mg/dL.
AKI occurred in 15.4% of ACS patients who underwent PCI with transradial access and 17.3% of those randomized to transfemoral access, for a significant 13% relative risk reduction. This was accomplished without any increase in the volume of contrast media required. The average was 200 mL in both study groups.
The reduction in AKI achieved with transradial-access PCI was seen in all patient subgroups, including those at increased AKI risk because of an estimated glomerular filtration rate below 60 mL/min, age 75 or older, Killup class III or IV, or a Mehran score greater than 10.
Dr. Cortese proposed several possible mechanisms for the observed reduction in AKI seen with transradial-access PCI. The major factor in his view is that the transradial approach entails less bleeding, as earlier demonstrated in the primary analysis – and bleeding has been associated with impaired renal perfusion in several prior studies. Also, it’s plausible that the passage of the catheter across the renal arteries during the transfemoral approach dislodges atherosclerotic debris, which then travels down the renal vessels.
The five golden rules for preventing contrast media–induced AKI, he noted, are
1. Discontinue nephrotoxic drugs before the procedure.
2. Identify high-risk patients.
3. Hydrate them.
4. Choose an ideal contrast medium.
5. Adapt the dose of contrast medium to the patient’s specific situation.
Discussant Jacek Legutko, MD, PhD, of Jagiellonian University in Krakow, Poland, said the primary results of the MATRIX trial published in 2015 have had a major impact on Polish interventional cardiology, where transradial PCI is now used in 80% of PCIs. The AKI study results will reinforce this trend, he added.
“You have shown something opposite to what we’ve thought in the past, that maybe, with a radial approach, we would use more contrast medium, which is a risk factor for AKI. In your study – at least in ACS with very experienced transradial operators – there was no increase in contrast volume, and the risk of AKI decreased,” Dr. Legutko said.
Asked about the possibility that transradial PCI might be associated with an increased risk of embolization to the brain, much as the transfemoral approach might cause embolization to the kidneys, Dr. Cortese said there was no significant difference between the two AKI-MATRIX study arms in rates of transient ischemic attack or stroke.
“I did my first transradial PCI in 2003, and I haven’t seen any increase in these events or later dementia,” he added.
The prespecified secondary analysis of the MATRIX trial was conducted without commercial support. The presenter reported serving as a consultant to Abbott, AstraZeneca, Daiichi Sankyo, Eli Lilly, and Stentys.
Simultaneous with his presentation in Paris, the AKI-MATRIX study was published online at www.sciencedirect.com/science/article/pii/S0735109717368973.
PARIS – Transradial-access percutaneous coronary intervention (PCI) in patients with acute coronary syndrome (ACS) results in a significantly lower risk of acute kidney injury (AKI), compared with the transfemoral approach, according to a new analysis from the large randomized MATRIX trial.
The results of this prespecified secondary subgroup analysis of MATRIX suggest it’s time to update the classic “five golden rules” for reduction of contrast medium–induced AKI by adding a sixth. “Use a transradial approach,” Bernardo Cortese, MD, said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
He reported on 8,210 participants in the MATRIX trial (Minimizing Adverse Haemorrhagic Events by Transradial Access Site and Systemic Implementation of Angiox) who were randomized to transradial- or transfemoral-access PCI for non–ST-elevation MI or ST-elevation MI.
The primary results of the 78-site, four-country European study, previously published, showed that transradial PCI reduced the composite risk of death, MI, stroke, or major bleeding by 17%, compared with transfemoral PCI, a benefit mainly driven by a marked reduction in clinically important bleeding (Lancet. 2015 Jun 20;385[9986]:2465-76).
Left unanswered by the primary analysis was the question of whether transradial PCI in ACS patients also reduced AKI risk, as had previously been suggested by a meta-analysis of observational studies (Int J Cardiol. 2015 Jan 20;179:309-11). In designing the MATRIX trial, Dr. Cortese and the other investigators decided to address that issue separately in a prespecified secondary analysis known as AKI-MATRIX. For this purpose, AKI was defined as either a post-PCI in-hospital increase in serum creatinine level of more than 25%, compared with the preangiography baseline, or an absolute increase in serum creatinine of greater than 0.5 mg/dL.
AKI occurred in 15.4% of ACS patients who underwent PCI with transradial access and 17.3% of those randomized to transfemoral access, for a significant 13% relative risk reduction. This was accomplished without any increase in the volume of contrast media required. The average was 200 mL in both study groups.
The reduction in AKI achieved with transradial-access PCI was seen in all patient subgroups, including those at increased AKI risk because of an estimated glomerular filtration rate below 60 mL/min, age 75 or older, Killup class III or IV, or a Mehran score greater than 10.
Dr. Cortese proposed several possible mechanisms for the observed reduction in AKI seen with transradial-access PCI. The major factor in his view is that the transradial approach entails less bleeding, as earlier demonstrated in the primary analysis – and bleeding has been associated with impaired renal perfusion in several prior studies. Also, it’s plausible that the passage of the catheter across the renal arteries during the transfemoral approach dislodges atherosclerotic debris, which then travels down the renal vessels.
The five golden rules for preventing contrast media–induced AKI, he noted, are
1. Discontinue nephrotoxic drugs before the procedure.
2. Identify high-risk patients.
3. Hydrate them.
4. Choose an ideal contrast medium.
5. Adapt the dose of contrast medium to the patient’s specific situation.
Discussant Jacek Legutko, MD, PhD, of Jagiellonian University in Krakow, Poland, said the primary results of the MATRIX trial published in 2015 have had a major impact on Polish interventional cardiology, where transradial PCI is now used in 80% of PCIs. The AKI study results will reinforce this trend, he added.
“You have shown something opposite to what we’ve thought in the past, that maybe, with a radial approach, we would use more contrast medium, which is a risk factor for AKI. In your study – at least in ACS with very experienced transradial operators – there was no increase in contrast volume, and the risk of AKI decreased,” Dr. Legutko said.
Asked about the possibility that transradial PCI might be associated with an increased risk of embolization to the brain, much as the transfemoral approach might cause embolization to the kidneys, Dr. Cortese said there was no significant difference between the two AKI-MATRIX study arms in rates of transient ischemic attack or stroke.
“I did my first transradial PCI in 2003, and I haven’t seen any increase in these events or later dementia,” he added.
The prespecified secondary analysis of the MATRIX trial was conducted without commercial support. The presenter reported serving as a consultant to Abbott, AstraZeneca, Daiichi Sankyo, Eli Lilly, and Stentys.
Simultaneous with his presentation in Paris, the AKI-MATRIX study was published online at www.sciencedirect.com/science/article/pii/S0735109717368973.
AT EUROPCR
Key clinical point:
Major finding: Transradial-access PCI for ACS resulted in a 13% lower risk of acute kidney injury than the transfemoral approach.
Data source: A four-country European randomized trial of transradial- vs. transfemoral-access PCI in more than 8,200 patients with ACS.
Disclosures: This prespecified secondary analysis of the MATRIX trial was conducted without commercial support. The presenter reported serving as a consultant to Abbott, AstraZeneca, Daiichi Sankyo, Eli Lilly, and Stentys.
A 68-year-old man with a blue toe
A 68-year-old man presented with concern about a bluish toe. Several months earlier he had undergone total aortic arch replacement and coronary artery bypass grafting. Since then his renal function had declined and he had been losing weight.

He had hypercholesterolemia, hypertension, and a 20-pack-year smoking history. Physical examination confirmed that his right great toe was indeed bluish (Figure 1). Peripheral, neck, and abdominal vascular examinations were normal. Laboratory testing revealed:
- Serum creatinine concentration 5.15 mg/dL (reference range 0.61–1.04)
- C-reactive protein level 1.5 mg/dL (0–0.3)
- Eosinophil count 0.58 × 109/L (0–0.50)
- Serum complement level normal
- Urine sediment unremarkable.

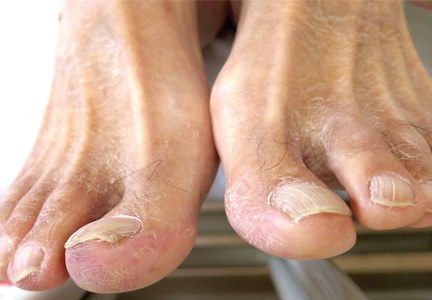
Transthoracic echocardiography revealed no evidence of vegetation, and a series of blood cultures were negative. The right toe was biopsied, and study revealed cholesterol clefts (Figure 2), confirming the diagnosis of cholesterol crystal embolism.
He was treated with prednisolone 20 mg/day, and his weight loss and renal function improved.
CHOLESTEROL CRYSTAL EMBOLISM
Cholesterol embolization typically occurs after arteriography, cardiac catheterization, vascular surgery, or anticoagulant use in men over age 55 with atherosclerosis.1 It presents with renal failure, abdominal pain, systemic symptoms, or, most commonly (in 88% of cases), skin findings.2
“Blue-toe syndrome,” characterized by tissue ischemia, is seen in 65% of patients.2 Lesions can appear anywhere on the body, but most commonly on the lower extremities. Most are painful due to ischemia. The condition can progress to necrosis.
Patients may have elevated C-reactive protein, hypocomplementemia (39%), and eosinophilia (80%).3,4 The diagnosis is confirmed only with histopathologic findings of intravascular cholesterol crystals, seen as cholesterol clefts.
The differential diagnosis includes contrast nephropathy and infectious endocarditis. However, contrast nephropathy begins to recover within several days and is not accompanied by skin lesions. Repeated blood cultures and echocardiography are useful to rule out infectious endocarditis.
Treatment includes managing cardiovascular risk factors and end-organ ischemia and preventing recurrent embolization. Surgical or endovascular treatment has been shown to be effective in decreasing the rate of further embolism.2 Corticosteroid therapy is assumed to control the secondary inflammation associated with cholesterol crystal embolism.1,5
- Paraskevas KI, Koutsias S, Mikhailidis DP, Giannoukas AD. Cholesterol crystal embolization: a possible complication of peripheral endovascular interventions. J Endovasc Ther 2008; 15:614–625.
- Jucgla A, Moreso F, Muniesa C, Moreno A, Vidaller A. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol 2006; 55:786–793.
- Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation 2010; 122:631–641.
- Lye WC, Cheah JS, Sinniah R. Renal cholesterol embolic disease. Case report and review of the literature. Am J Nephrol 1993; 13:489–493.
- Nakayama M, Izumaru K, Nagata M, et al. The effect of low-dose corticosteroids on short- and long-term renal outcome in patients with cholesterol crystal embolism. Ren Fail 2011; 33:298–306.
A 68-year-old man presented with concern about a bluish toe. Several months earlier he had undergone total aortic arch replacement and coronary artery bypass grafting. Since then his renal function had declined and he had been losing weight.
He had hypercholesterolemia, hypertension, and a 20-pack-year smoking history. Physical examination confirmed that his right great toe was indeed bluish (Figure 1). Peripheral, neck, and abdominal vascular examinations were normal. Laboratory testing revealed:
- Serum creatinine concentration 5.15 mg/dL (reference range 0.61–1.04)
- C-reactive protein level 1.5 mg/dL (0–0.3)
- Eosinophil count 0.58 × 109/L (0–0.50)
- Serum complement level normal
- Urine sediment unremarkable.
Transthoracic echocardiography revealed no evidence of vegetation, and a series of blood cultures were negative. The right toe was biopsied, and study revealed cholesterol clefts (Figure 2), confirming the diagnosis of cholesterol crystal embolism.
He was treated with prednisolone 20 mg/day, and his weight loss and renal function improved.
CHOLESTEROL CRYSTAL EMBOLISM
Cholesterol embolization typically occurs after arteriography, cardiac catheterization, vascular surgery, or anticoagulant use in men over age 55 with atherosclerosis.1 It presents with renal failure, abdominal pain, systemic symptoms, or, most commonly (in 88% of cases), skin findings.2
“Blue-toe syndrome,” characterized by tissue ischemia, is seen in 65% of patients.2 Lesions can appear anywhere on the body, but most commonly on the lower extremities. Most are painful due to ischemia. The condition can progress to necrosis.
Patients may have elevated C-reactive protein, hypocomplementemia (39%), and eosinophilia (80%).3,4 The diagnosis is confirmed only with histopathologic findings of intravascular cholesterol crystals, seen as cholesterol clefts.
The differential diagnosis includes contrast nephropathy and infectious endocarditis. However, contrast nephropathy begins to recover within several days and is not accompanied by skin lesions. Repeated blood cultures and echocardiography are useful to rule out infectious endocarditis.
Treatment includes managing cardiovascular risk factors and end-organ ischemia and preventing recurrent embolization. Surgical or endovascular treatment has been shown to be effective in decreasing the rate of further embolism.2 Corticosteroid therapy is assumed to control the secondary inflammation associated with cholesterol crystal embolism.1,5
A 68-year-old man presented with concern about a bluish toe. Several months earlier he had undergone total aortic arch replacement and coronary artery bypass grafting. Since then his renal function had declined and he had been losing weight.
He had hypercholesterolemia, hypertension, and a 20-pack-year smoking history. Physical examination confirmed that his right great toe was indeed bluish (Figure 1). Peripheral, neck, and abdominal vascular examinations were normal. Laboratory testing revealed:
- Serum creatinine concentration 5.15 mg/dL (reference range 0.61–1.04)
- C-reactive protein level 1.5 mg/dL (0–0.3)
- Eosinophil count 0.58 × 109/L (0–0.50)
- Serum complement level normal
- Urine sediment unremarkable.
Transthoracic echocardiography revealed no evidence of vegetation, and a series of blood cultures were negative. The right toe was biopsied, and study revealed cholesterol clefts (Figure 2), confirming the diagnosis of cholesterol crystal embolism.
He was treated with prednisolone 20 mg/day, and his weight loss and renal function improved.
CHOLESTEROL CRYSTAL EMBOLISM
Cholesterol embolization typically occurs after arteriography, cardiac catheterization, vascular surgery, or anticoagulant use in men over age 55 with atherosclerosis.1 It presents with renal failure, abdominal pain, systemic symptoms, or, most commonly (in 88% of cases), skin findings.2
“Blue-toe syndrome,” characterized by tissue ischemia, is seen in 65% of patients.2 Lesions can appear anywhere on the body, but most commonly on the lower extremities. Most are painful due to ischemia. The condition can progress to necrosis.
Patients may have elevated C-reactive protein, hypocomplementemia (39%), and eosinophilia (80%).3,4 The diagnosis is confirmed only with histopathologic findings of intravascular cholesterol crystals, seen as cholesterol clefts.
The differential diagnosis includes contrast nephropathy and infectious endocarditis. However, contrast nephropathy begins to recover within several days and is not accompanied by skin lesions. Repeated blood cultures and echocardiography are useful to rule out infectious endocarditis.
Treatment includes managing cardiovascular risk factors and end-organ ischemia and preventing recurrent embolization. Surgical or endovascular treatment has been shown to be effective in decreasing the rate of further embolism.2 Corticosteroid therapy is assumed to control the secondary inflammation associated with cholesterol crystal embolism.1,5
- Paraskevas KI, Koutsias S, Mikhailidis DP, Giannoukas AD. Cholesterol crystal embolization: a possible complication of peripheral endovascular interventions. J Endovasc Ther 2008; 15:614–625.
- Jucgla A, Moreso F, Muniesa C, Moreno A, Vidaller A. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol 2006; 55:786–793.
- Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation 2010; 122:631–641.
- Lye WC, Cheah JS, Sinniah R. Renal cholesterol embolic disease. Case report and review of the literature. Am J Nephrol 1993; 13:489–493.
- Nakayama M, Izumaru K, Nagata M, et al. The effect of low-dose corticosteroids on short- and long-term renal outcome in patients with cholesterol crystal embolism. Ren Fail 2011; 33:298–306.
- Paraskevas KI, Koutsias S, Mikhailidis DP, Giannoukas AD. Cholesterol crystal embolization: a possible complication of peripheral endovascular interventions. J Endovasc Ther 2008; 15:614–625.
- Jucgla A, Moreso F, Muniesa C, Moreno A, Vidaller A. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol 2006; 55:786–793.
- Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation 2010; 122:631–641.
- Lye WC, Cheah JS, Sinniah R. Renal cholesterol embolic disease. Case report and review of the literature. Am J Nephrol 1993; 13:489–493.
- Nakayama M, Izumaru K, Nagata M, et al. The effect of low-dose corticosteroids on short- and long-term renal outcome in patients with cholesterol crystal embolism. Ren Fail 2011; 33:298–306.
Autosomal dominant polycystic kidney disease and the heart and brain
Autosomal dominant polycystic kidney disease (ADPKD) has significant extrarenal manifestations. Hypertension is a common complication, arises early in the course of the disease, and is implicated in the development of left ventricular hypertrophy. Patients with ADPKD are also at risk of other cardiovascular complications (Table 1).
This article reviews the timely diagnosis of these common ADPKD complications and how to manage them.
ADPKD ACCOUNTS FOR 10% OF END-STAGE RENAL DISEASE

ADPKD is a genetic condition characterized by multiple renal cysts.1 Progressive enlargement of these cysts leads to a gradual decline in kidney function and eventually end-stage renal disease by the fifth or sixth decade of life.2 Worldwide, about 12.5 million people have ADPKD, and it accounts for about 10% of cases of end-stage renal disease.1,3,4
ADPKD has a variety of clinical presentations, including (in decreasing order of frequency) hypertension, flank pain, abdominal masses, urinary tract infection, renal failure, renal stones, and cerebrovascular accidents.2
Extrarenal complications are common and include hepatic cysts, hypertension, left ventricular hypertrophy, valvular heart disease, intracranial and extracranial aneurysms, pancreatic cysts, and diverticulosis.1–5
Less-common complications are dissection of the aorta and the internal carotid, vertebral, and iliac arteries6–10; aneurysm of the coronary, popliteal, and splenic arteries11–14; atrial myxoma15; cardiomyopathy16; pericardial effusion17; intracranial arterial dolichoectasia18; arachnoid cysts2; and intraoperative inferior vena cava syndrome (normally in ADPKD patients, pressure on the inferior vena cava results in compensatory sympathetic overactivity to maintain blood pressure), which occurs due to reduced sympathetic output under the influence of epidural or general anesthesia.19
Cardiovascular complications, especially cardiac hypertrophy and coronary artery disease, are now the leading cause of death in patients with ADPKD, as renal replacement therapy has improved and made death from end-stage renal disease less common.20,21
HYPERTENSION IN ADPKD
Hypertension is the most frequent initial presentation of ADPKD, occurring in 50% to 75% of cases and usually preceding the onset of renal failure.2,22 Hypertension is more common in male ADPKD patients, begins early in the course of the disease, and is diagnosed around the fourth decade of life.21
In a study in 2007, de Almeida et al23 used 24-hour ambulatory blood pressure monitoring early in the course of ADPKD and found significantly higher systolic, diastolic, and mean 24-hour blood pressures in ADPKD patients who had normal in-office blood pressure than in normotensive controls. In addition, nighttime systolic, nighttime diastolic, and nighttime mean blood pressures were significantly higher in the ADPKD group.
Hypertension is strongly associated with an accelerated decline in renal function to end-stage renal disease, development of left ventricular hypertrophy, and cardiovascular death.20,24
Although a prospective study25 showed a strong association between renal stones and hypertension in ADPKD, the relation between them is not clear. The incidence of renal stones is higher in hypertensive than in normotensive ADPKD patients, although evidence has to be established whether nephrolithiasis is a risk factor for hypertension or the other way around.25

Hypertension in ADPKD is multifactorial (Figure 1). The major factors associated with its development are increased activation of the renin-angiotensin-aldosterone system (RAAS); overexpression of endothelin receptor subtype A (ET-A) in cystic kidneys; increased production of endothelin 1 (ET-1); and sodium retention.26–31
The renin-angiotensin-aldosterone system
Activation of the RAAS plays a major role in the development and maintenance of hypertension in ADPKD. This is thought to be mainly due to progressive enlargement of renal cysts, which causes renal arteriolar attenuation and ischemia secondary to pressure effects, which in turn activates the RAAS.26,30,32–34 Two studies in patients with normal renal function found that cyst growth and increasing kidney volume have a strong relationship with the development of hypertension and declining kidney function.35,36
Ectopic secretion of RAAS components in polycystic kidneys has also been implicated in the development of hypertension, whereby renin, angiotensinogen, angiotensin-converting enzyme (ACE), angiotensin II, and angiotensin II receptors are produced in the epithelium of cysts and dilated tubules in polycystic kidneys.37–39 Proximal renal cysts and tubules produce ectopic angiotensinogen, which is converted to angiotensin I by renin in distal renal cysts. Angiotensin I is converted to angiotensin II by ACE in distal tubules, which in turn stimulates angiotensin II receptors, causing sodium and water retention in distal tubules.37 This may be responsible for hypertension in the initial stages; however, RAAS hyperactivity due to renal injury may predominate during later stages.37
Increased RAAS activity also increases sympathetic output, which in turn raises catecholamine levels and blood pressure.34 A study showed higher levels of plasma catecholamines in ADPKD hypertensive patients irrespective of renal function than in patients with essential hypertension.40
ET-A receptor and ET-1
A few studies have shown that in ADPKD patients, increased density of ET-A receptors and overproduction of ET-1, a potent vasoconstrictor, play a significant role in the development of hypertension and gradual loss of kidney function due to cyst enlargement and interstitial scarring.28,29 Ong et al29 found that expression of ET-A receptors is increased in smooth muscle cells of renal arteries, glomerular mesangial cells, and cyst epithelia in ADPKD.
Sodium retention
Studies in ADPKD patients with preserved renal function have linked high blood pressure to sodium retention and volume expansion.30,31,41 However, this phenomenon reverses when there is significant renal impairment in ADPKD.
As evidence of this, a study demonstrated significantly more natriuresis in patients with renal failure due to ADPKD than in patients with a similar degree of renal failure due to chronic glomerulonephritis.31 Moreover, another study found that the prevalence of hypertension is higher in ADPKD patients than in those with other nephropathies with preserved renal function, but this association reverses with significant decline in kidney function.22
MANAGING HYPERTENSION IN ADPKD
Early diagnosis of hypertension and effective control of it, even before ADPKD is diagnosed, is crucial to reduce cardiovascular mortality. Aggressive blood pressure control in the prehypertensive phase of ADPKD will also help reduce the incidence of left ventricular hypertrophy and mitral regurgitation and slow the progression of renal failure (Figure 2).

A meta-analysis42 revealed hypertension to be present in 20% of ADPKD patients younger than 21, and many of them were undiagnosed. This study also suggests that patients at risk of hypertension (ie, all patients with ADPKD) should be routinely screened for it.
Ambulatory blood pressure monitoring may play an important role in diagnosing hypertension early in the prehypertensive stage of ADPKD.23
Target blood pressures: No consensus
Two well-powered double-blind, placebo-controlled trials, known as HALT-PKD Study A and HALT-PKD Study B, tested the effects of 2 different blood pressure targets and of monotherapy with an ACE inhibitor vs combination therapy with an ACE inhibitor plus an angiotensin II receptor blocker (ARB) on renal function, total kidney volume, left ventricular mass index, and urinary albumin excretion in the early (estimated glomerular filtration rate [eGFR] > 60 mL/min) and late (eGFR 25–60 mL/min) stages of ADPKD, respectively.43,44
HALT-PKD Study A43 found that, in the early stages of ADPKD with preserved renal function, meticulous control of blood pressure (95–110/60–75 mm Hg) was strongly correlated with significant reductions in left ventricular mass index, albuminuria, and rate of total kidney volume growth without remarkable alteration in renal function compared with standard blood pressure control (120–130/70–80 mm Hg). However, no notable differences were observed between the ACE inhibitor and ACE inhibitor-plus-ARB groups.43
Despite the evidence, universal consensus guidelines are lacking, and the available guidelines on hypertension management have different blood pressure goals in patients with chronic kidney disease.
The eighth Joint National Committee guideline of 2014 recommends a blood pressure goal of less than 140/90 mm Hg in patients with diabetic and nondiabetic chronic kidney disease.45
The National Institute for Health and Care Excellence 2011 guideline recommends a blood pressure goal of less than 130/80 mm Hg in chronic kidney disease patients.46
The European Society of Hypertension and European Society of Cardiology joint 2013 guideline recommends a systolic blood pressure goal of less than 140 mm Hg in diabetic and nondiabetic patients with chronic kidney disease.47
The 2016 Kidney Health Australia-Caring for Australians With Renal Impairment guideline for diagnosis and management of ADPKD48 recommends a lower blood pressure goal of 96–110/60–75 mm Hg in patients with an eGFR greater than 60 mL/min/1.73 m2 who can tolerate it without side effects, which is based on the findings of HALT-PKD Study A.43
Helal et al recommend that blood pressure be controlled to less than 130/80 mm Hg, until there is more evidence for a safe and effective target blood pressure goal in ADPKD patients.49
We recommend a target blood pressure less than 110/75 mm Hg in hypertensive ADPKD patients with preserved renal function who can tolerate this level, and less than 130/80 mm Hg in ADPKD patients with stage 3 chronic kidney disease. These targets can be achieved with ACE inhibitor or ARB monotherapy.43,44 However, no studies have established the safest lower limit of target blood pressure in ADPKD.
ACE inhibitors, ARBs are mainstays
Mainstays of antihypertensive drug therapy in ADPKD are ACE inhibitors and ARBs.
HALT-PKD Study B44 demonstrated that, in the late stages of ADPKD, target blood pressure control (110–130/70–80 mm Hg) can be attained with ACE inhibitor monotherapy or with an ACE inhibitor plus an ARB, but the latter produced no additive benefit.
Patch et al,50 in a retrospective cohort study, showed that broadening the spectrum of antihypertensive therapy decreases mortality in ADPKD patients. Evaluating ADPKD patients from the UK General Practice Research Database between 1991 and 2008, they found a trend toward lower mortality rates as the number of antihypertensive drugs prescribed within 1 year increased. They also observed that the prescription of RAAS-blocking agents increased from 7% in 1991 to 46% in 2008.50
However, a 3-year prospective randomized double-blind study compared the effects of the ACE inhibitor ramipril and the beta-blocker metoprolol in hypertensive ADPKD patients.51 The results showed that effective blood pressure control could be achieved in both groups with no significant differences in left ventricular mass index, albuminuria, or kidney function.51
Treatment strategies
Lifestyle modification is the initial approach to the management of hypertension before starting drug therapy. Lifestyle changes include dietary salt restriction to less than 6 g/day, weight reduction, regular exercise, increased fluid intake (up to 3 L/day or to satisfy thirst), smoking cessation, and avoidance of caffeine.47–49
ACE inhibitors are first-line drugs in hypertensive ADPKD patients.
ARBs can also be considered, but there is no role for dual ACE inhibitor and ARB therapy.43,48 A study found ACE inhibitors to be more cost-effective and to decrease mortality rates to a greater extent than ARBs.52
Beta-blockers or calcium channel blockers should be considered instead if ACE inhibitors and ARBs are contraindicated, or as add-on drugs if ACE inhibitors and ARBs fail to reduce blood pressure adequately.48,49
Diuretics are third-line agents. Thiazides are preferred in ADPKD patients with normal renal function and loop diuretics in those with impaired renal function.49
LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Increased left ventricular mass is an indirect indicator of untreated hypertension, and it often goes unnoticed in patients with undiagnosed ADPKD. Left ventricular hypertrophy is associated with arrhythmias and heart failure, which contribute significantly to cardiovascular mortality and adverse renal outcomes.20,24
A 5-year randomized clinical trial by Cadnapaphornchai et al36 in ADPKD patients between 4 and 21 years of age showed strong correlations between hypertension, left ventricular mass index, and kidney volume and a negative correlation between left ventricular mass index and renal function.
Several factors are thought to contribute to left ventricular hypertrophy in ADPKD (Figure 1).
Hypertension. Two studies of 24-hour ambulatory blood pressure monitoring showed that nocturnal blood pressures decreased less in normotensive and hypertensive ADPKD patients than in normotensive and hypertensive controls.23,53 This persistent elevation of nocturnal blood pressure may contribute to the development and progression of left ventricular hypertrophy.
On the other hand, Valero et al54 reported that the left ventricular mass index was strongly associated with ambulatory systolic blood pressure rather than elevated nocturnal blood pressure in ADPKD patients compared with healthy controls.
FGF23. High levels of fibroblast growth factor 23 (FGF23) have been shown to be strongly associated with left ventricular hypertrophy in ADPKD. Experimental studies have shown that FGF23 is directly involved in the pathogenesis of left ventricular hypertrophy through stimulation of the calcineurin-nuclear factor of activated T cells pathway.
Faul et al55 induced cardiac hypertrophy in mice that were deficient in klotho (a transmembrane protein that increases FGF23 affinity for FGF receptors) by injecting FGF23 intravenously.
Yildiz et al56 observed higher levels of FGF23 in hypertensive and normotensive ADPKD patients with normal renal function than in healthy controls. They also found a lower elasticity index in the large and small arteries in normotensive and hypertensive ADPKD patients, which accounts for vascular dysfunction. High FGF23 levels may be responsible for the left ventricular hypertrophy seen in normotensive ADPKD patients with preserved renal function.
Polymorphisms in the ACE gene have been implicated in the development of cardiac hypertrophy in ADPKD.
Wanic-Kossowska et al57 studied the association between ACE gene polymorphisms and cardiovascular complications in ADPKD patients. They found a higher prevalence of the homozygous DD genotype among ADPKD patients with end-stage renal disease than in those in the early stages of chronic kidney disease in ADPKD. Also, the DD genotype has been shown to be more strongly associated with left ventricular hypertrophy and left ventricular dysfunction than other (II or ID) genotypes. These findings suggest that the DD genotype carries higher risk for the development of end-stage renal disease, left ventricular hypertrophy, and other cardiovascular complications.
MANAGING LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Preventing and halting progression of left ventricular hypertrophy primarily involves effective blood pressure control, especially in the early stages of ADPKD (Figure 2).
A 7-year prospective randomized trial in ADPKD patients with established hypertension and left ventricular hypertrophy proved that aggressive (< 120/80 mm Hg) compared with standard blood pressure control (135–140/85–90 mm Hg) significantly reduces left ventricular mass index. ACE inhibitors were preferred over calcium channel blockers.58
HALT-PKD Study A showed that a significant decrease in left ventricular mass index can be achieved by aggressive blood pressure control (95–110/60–75 mm Hg) with an ACE inhibitor alone or in combination with an ARB in the early stages of ADPKD with preserved renal function.43
A 5-year randomized clinical trial in children with borderline hypertension treated with an ACE inhibitor for effective control of blood pressure showed no change in left ventricular mass index or renal function.36,59
These results support starting ACE inhibitor therapy early in the disease process when blood pressure is still normal or borderline to prevent the progression of left ventricular hypertrophy or worsening kidney function.
Since FGF23 is directly involved in the causation of left ventricular hypertrophy, FGF receptors may be potential therapeutic targets to prevent left ventricular hypertrophy in ADPKD. An FGF receptor blocker was shown to decrease left ventricular hypertrophy in rats with chronic kidney disease without affecting blood pressure.55
INTRACRANIAL ANEURYSM IN ADPKD
Intracranial aneurysm is the most dangerous complication of ADPKD. When an aneurysm ruptures, the mortality rate is 4% to 7%, and 50% of survivors are left with residual neurologic deficits.5,60,61
In various studies, the prevalence of intracranial aneurysm in ADPKD ranged from 4% to 41.2%, compared with 1% in the general population.5,62,63 On follow-up ranging from 18 months to about 10 years, the incidence of new intracranial aneurysm was 2.6% to 13.3% in patients with previously normal findings on magnetic resonance angiography and 25% in patients with a history of intracranial aneurysm.62,64,65
The most common sites are the middle cerebral artery (45%), internal carotid artery (40.5%), and anterior communicating artery (35.1%).66 (The numbers add up to more than 100% because some patients have aneurysms in more than 1 site.) The mean size of a ruptured aneurysm was 6 mm per a recent systematic review.66 Intracranial aneurysms 6 mm or larger are at highest risk of rupture.66
SCREENING FOR INTRACRANIAL ANEURYSM
Timely screening and intervention for intracranial aneurysm is crucial to prevent death from intracranial hemorrhage.
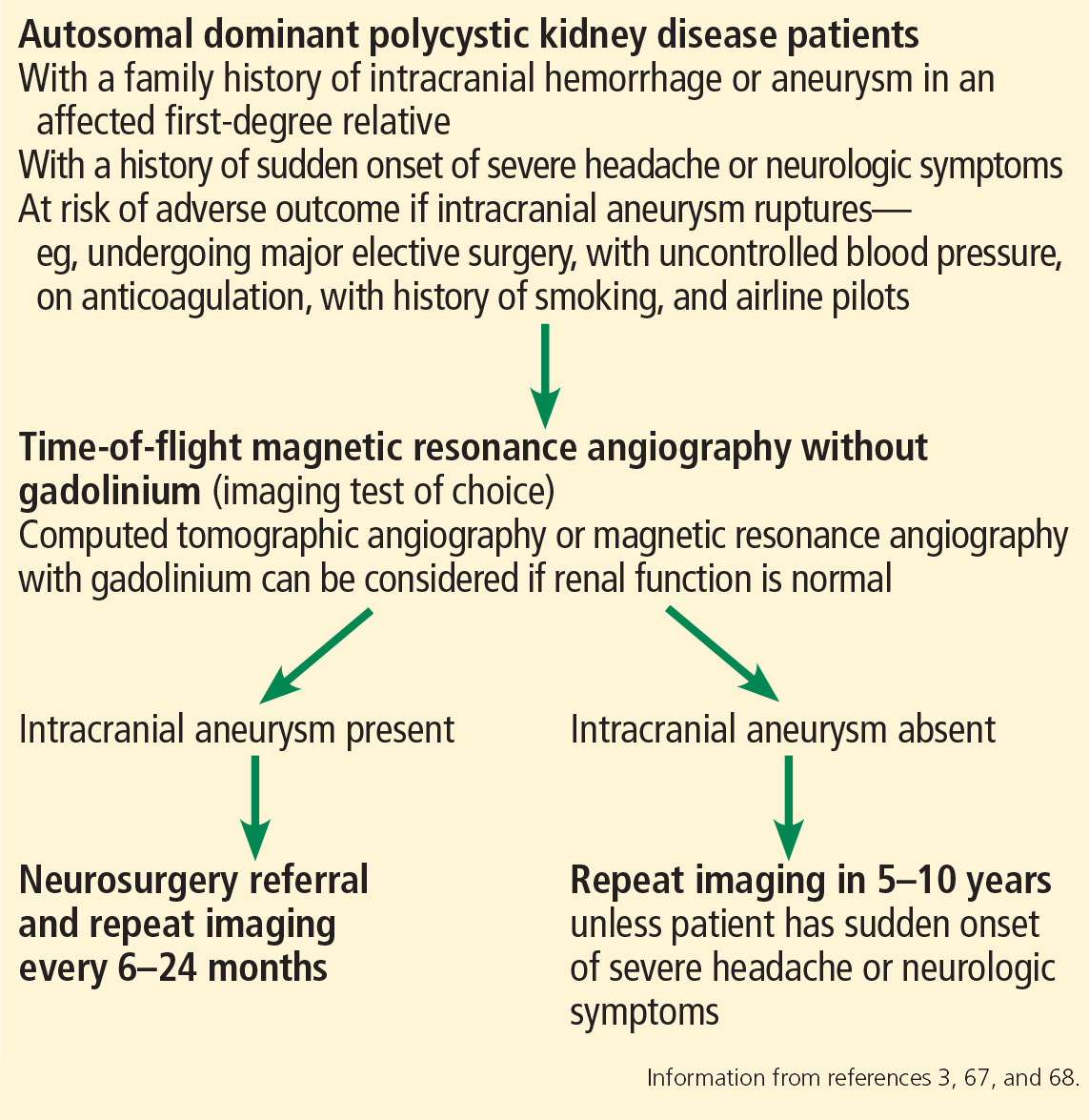
Currently, there are no standard guidelines for screening and follow-up of intracranial aneurysm in ADPKD patients. However, some recommendations are available from the ADPKD Kidney Disease Improving Global Outcomes Controversies Conference3 and Kidney Health Australia—Caring for Australasians With Renal Impairment ADPKD guidelines67 (Figure 3).
Imaging tests
Magnetic resonance angiography (MRA) with gadolinium enhancement and computed tomographic angiography (CTA) are recommended for screening in ADPKD patients with normal renal function,67 but time-of-flight MRA without gadolinium is the imaging test of choice because it is noninvasive and poses no risk of nephrotoxicity or contrast allergy.3,68 Further, gadolinium should be avoided in patients whose eGFR is 30 mL/min/1.73 m2 or less because of risk of nephrogenic systemic sclerosis and fibrosis.67,68
The sensitivity of time-of-flight MRA screening for intracranial aneurysm varies depending on the size of aneurysm; 67% for those less than 3 mm, 79% for those 3 to 5 mm, and 95% for those larger than 5 mm.69 The sensitivity of CTA screening is 95% for aneurysms larger than 7 mm and 53% for those measuring 2 mm.70,71 The specificity of CTA screening was reported to be 98.9% overall.71
When to screen
Screening for intracranial aneurysm is recommended at the time of ADPKD diagnosis for all high-risk patients, ie, those who have a family history of intracranial hemorrhage or aneurysm in an affected first-degree relative.67 It is also recommended for ADPKD patients with a history of sudden-onset severe headache or neurologic symptoms.67 A third group for whom screening is recommended is ADPKD patients who have no family history of intracranial aneurysm or hemorrhage but who are at risk of poor outcome if an intracranial aneurysm ruptures (eg, those undergoing major elective surgery, with uncontrolled blood pressure, on anticoagulation, with a history of or current smoking, and airline pilots).67
Patients found to have an intracranial aneurysm on screening should be referred to a neurosurgeon and should undergo repeat MRA or CTA imaging every 6 to 24 months.3 High-risk ADPKD patients with normal findings on initial screening should have repeat MRA or CTA screening in 5 to 10 years unless they suffer from sudden-onset severe headache or neurologic symptoms.65,67
Both smoking and high blood pressure increase the risk of formation and growth of intracranial aneurysm. Hence, meticulous control of blood pressure and smoking cessation are recommended in ADPKD patients.3,67
CARDIAC VALVULAR ABNORMALITIES IN ADPKD
Of the valvular abnormalities that complicate ADPKD, the more common ones are mitral valve prolapse and mitral and aortic regurgitation. The less common ones are tricuspid valve prolapse and tricuspid regurgitation.72–74
The pathophysiology underlying these valvular abnormalities is unclear. However, defective collagen synthesis and myxomatous degeneration have been demonstrated in histopathologic examination of affected valvular tissue.75 Also, ACE gene polymorphism, especially the DD genotype, has been shown to be associated with cardiac valvular calcifications and valvular insufficiency.57
Lumiaho et al72 found a higher prevalence of mitral valve prolapse, mitral regurgitation, and left ventricular hypertrophy in patients with ADPKD type 1 (due to abnormalities in PDK1) than in unaffected family members and healthy controls. The investigators speculated that mitral regurgitation is caused by the high blood pressure observed in ADPKD type 1 patients, since hypertension causes left ventricular hypertrophy and left ventricular dilatation. The severity of renal failure was related to mitral regurgitation but not mitral valve prolapse.
Similarly, Gabow et al24 showed that there is no significant relationship between mitral valve prolapse and progression of renal disease in ADPKD.
Interestingly, Fick et al20 found that mitral valve prolapse has no significant effect on cardiovascular mortality.
CORONARY ARTERY DISEASE AND ANEURYSM IN ADPKD
Atherosclerosis sets in early in ADPKD, resulting in coronary artery disease and adverse cardiovascular outcomes.
Coronary flow velocity reserve is the ability of coronary arteries to dilate in response to myocardial oxygen demand. Atherosclerosis decreases this reserve in ADPKD patients, as shown in several studies.
Turkmen et al, in a series of studies,76–78 found that ADPKD patients had significantly less coronary flow velocity reserve, thicker carotid intima media (a surrogate marker of atherosclerosis), and greater insulin resistance than healthy controls. These findings imply that atherosclerosis begins very early in the course of ADPKD and has remarkable effects on cardiovascular morbidity and mortality.76
Aneurysm. Although the risk of extracranial aneurysm is higher with ADPKD, coronary artery aneurysm is uncommon. The pathogenesis of coronary aneurysm has been linked to abnormal expression of the proteins polycystin 1 and polycystin 2 in vascular smooth muscle.11,79 The PKD1 and PKD2 genes encode polycystin 1 and 2, respectively, in ADPKD. These polycystins are also expressed in the liver, kidneys, and myocardium and are involved in the regulation of intracellular calcium, stretch-activated ion channels, and vascular smooth muscle cell proliferation and apoptosis.11,16 Abnormally expressed polycystin in ADPKD therefore has an impact on arterial wall integrity, resulting in focal medial defects in the vasculature that eventually develop into micro- and macroaneurysms.11
Hadimeri et al79 found a higher prevalence of coronary aneurysm and ectasia in ADPKD patients than in controls. Most coronary aneurysms are smaller than 1 cm; however, a coronary aneurysm measuring 4 cm in diameter was found at autopsy of an ADPKD patient.11
Spontaneous coronary artery dissection is very rare in the general population, but Bobrie et al reported a case of it in an ADPKD patient.9
ATRIAL MYXOMA, CARDIOMYOPATHY, AND PERICARDIAL EFFUSION IN ADPKD
Atrial myxoma in ADPKD patients has been described in 2 case reports.15,80 However, the association of atrial myxoma with ADPKD is poorly understood and may be a coincidental finding.
Cardiomyopathy in ADPKD has been linked to abnormalities in the intracellular calcium pathway, although a clear picture of its involvement has yet to be established.
Paavola et al16 described the pathophysiology of ADPKD-associated cardiomyopathy in PKD2 mutant zebrafish lacking polycystin 2. These mutants showed decreased cardiac output and had atrioventricular blocks. The findings were attributed to abnormal intracellular calcium cycling. These findings correlated well with the frequent finding of idiopathic dilated cardiomyopathy in ADPKD patients, especially with PKD2 mutations.16 Also, 2 cases of dilated cardiomyopathy in ADPKD have been reported and thought to be related to PKD2 mutations.81,82
Pericardial effusion. Even though the exact pathophysiology of pericardial effusion in ADPKD is unknown, it has been theorized to be related to defects in connective tissue and extracellular matrix due to PKD1 and PKD2 mutations. These abnormalities increase the compliance and impair the recoil capacity of connective tissue, which results in unusual distention of the parietal pericardium. This abnormal distention of the parietal pericardium together with increased extracellular volume may lead to pericardial effusion.17
Qian et al17 found a higher prevalence of pericardial effusion in ADPKD patients. It was generally asymptomatic, and the cause was attributed to these connective tissue and extracellular matrix abnormalities.
EMERGING THERAPIES AND TESTS
Recent trials have investigated the effects of vasopressin receptor antagonists, specifically V2 receptor blockers in ADPKD and its complications.
Tolvaptan has been shown to slow the rate of increase in cyst size and total kidney volume.83,84 Also, a correlation between kidney size and diastolic blood pressure has been observed in ADPKD patients.85 Reducing cyst volume may reduce pressure effects and decrease renal ischemia, which in turn may reduce RAAS activation; however, the evidence to support this hypothesis is poor. A major clinical trial of tolvaptan in ADPKD patients showed no effect on blood pressure control, but the drug slowed the rate of increase in total kidney volume and fall in eGFR.83
Endothelin receptor antagonists are also in the preliminary stage of development for use in ADPKD. The effects of acute blockade of the renal endothelial system with bosentan were investigated in animal models by Hocher et al.28 This study showed a greater reduction in mean arterial pressure after bosentan administration, resulting in significantly decreased GFR and renal blood flow. Nonetheless, the mean arterial pressure-lowering effect of bosentan was more marked than the reductions in GFR and renal blood flow.
Raina et al,86 in a pilot cross-sectional analysis, showed that urinary excretion of ET-1 is increased in ADPKD patients, and may serve as a surrogate marker for ET-1 in renal tissue and a noninvasive marker of early kidney injury.
- Ha SK, Park CH, Kna JS, et al. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Yonsei Med J 1997; 38:111–116.
- Romao EA, Moyses Neto M, Teixeira SR, Muglia VF, Vieira-Neto OM, Dantas M. Renal and extrarenal manifestations of autosomal dominant polycystic kidney disease. Brazilian J Med Biol Res 2006; 39:533–538.
- Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2015; 88:17–27.
- Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis 2001; 38:777–784.
- Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev 2013; 9:2–11.
- Silverio A, Prota C, Di Maio M, et al. Aortic dissection in patients with autosomal dominant polycystic kidney disease: a series of two cases and a review of the literature. Nephrology 2015; 20:229–235.
- Ramineni R, Daniel GK. Use of endovascular stent-graft repair for type B aortic dissection in polycystic kidney disease. J Invas Cardiol 2010; 22:E171–E174.
- Courtois A, Nusgens BV, Delvenne P, et al. Dissection of iliac artery in a patient with autosomal dominant polycystic kidney disease: a case report. Aorta 2013; 1:123–125.
- Bobrie G, Brunet-Bourgin F, Alamowitch S, et al. Spontaneous artery dissection: is it part of the spectrum of autosomal dominant polycystic kidney disease? Nephrol Dial Transplant 1998; 13:2138–2141.
- Minami T, Karube N, Sakamoto A. [Thoracic aortic dissection complicating autosomal dominant polycystic kidney disease; report of a case]. Kyobu Geka 2009; 62:924–927.
- Ohara K, Kimura T, Karasawa T, et al. A large coronary aneurysm and its probable precursor lesions in a patient with autosomal dominant polycystic kidney disease: an implication for the process of aneurysmogenesis. Pathol Int 2012; 62:758–762.
- Al-Hakim W, Goldsmith DJ. Bilateral popliteal aneurysms complicating adult polycystic kidney disease in a patient with a marfanoid habitus. Postgrad Med J 2003; 79:474–475.
- Kanagasundaram NS, Perry EP, Turney JH. Aneurysm of the splenic artery in a patient with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1999; 14:183–184.
- Kang YR, Ahn JH, Kim KH, Choi YM, Choi J, Park JR. Multiple cardiovascular manifestations in a patient with autosomal dominant polycystic kidney disease. J Cardiovasc Ultrasound 2014; 22:144–147.
- Iglesias D, Fraga AR, Arrizurieta E, et al. Atrial myxoma in a woman with autosomal dominant polycystic kidney disease type 2. Am J Kidney Dis 1997; 29:164–165.
- Paavola J, Schliffke S, Rossetti S, et al. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J Mol Cell Cardiol 2013; 58:199–208.
- Qian Q, Hartman RP, King BF, Torres VE. Increased occurrence of pericardial effusion in patients with autosomal dominant polycystic kidney disease.
- Schievink WI, Torres VE, Wiebers DO, Huston J 3rd. Intracranial arterial dolichoectasia in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1997; 8:1298–1303.
- Pierre SA, Jaeger MT, Siemens DR. Intra-operative inferior vena cava syndrome in a patient with autosomal dominant polycystic kidney disease. World J Urol 2006; 24:110–112.
- Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995; 5:2048–2056.
- Helal I, Reed B, Mettler P, et al. Prevalence of cardiovascular events in patients with autosomal dominant polycystic kidney disease. Am J Nephrol 2012; 36:362–370.
- Calabrese G, Vagelli G, Cristofano C, Barsotti G. Behaviour of arterial pressure in different stages of polycystic kidney disease. Nephron 1982; 32:207–208.
- de Almeida EA, de Oliveira EI, Lopes JA, Almeida AG, Lopes MG, Prata MM. Ambulatory blood pressure measurement in young normotensive patients with autosomal dominant polycystic kidney disease. Rev Port Cardiol 2007; 26:235–243.
- Gabow PA, Johnson AM, Kaehny WD, et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 41:1311–1319.
- Bajrami V, Idrizi A, Roshi E, Barbullushi M. Association between nephrolithiasis, hypertension and obesity in polycystic kidney disease. Open Access Maced J Med Sci 2016; 4:43–46.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 1990; 323:1091–1096.
- Nakamura T, Ebihara I, Fukui M, et al. Increased endothelin and endothelin receptor mRNA expression in polycystic kidneys of cpk mice. J Am Soc Nephrol 1993; 4:1064–1072.
- Hocher B, Zart R, Schwarz A, et al. Renal endothelin system in polycystic kidney disease. J Am Soc Nephrol 1998; 9:1169–1177.
- Ong AC, Newby LJ, Dashwood MR. Expression and cellular localisation of renal endothelin-1 and endothelin receptor subtypes in autosomal-dominant polycystic kidney disease. Nephron Exper Nephrol 2003; 93:e80.
- Nash DA Jr. Hypertension in polycystic kidney disease without renal failure. Arch Intern Med 1977; 137:1571–1575.
- D’Angelo A, Mioni G, Ossi E, Lupo A, Valvo E, Maschio G. Alterations in renal tubular sodium and water transport in polycystic kidney disease. Clin Nephrol 1975; 3:99–105.
- Ettinger A, Kahn PC, Wise HM Jr. The importance of selective renal angiography in the diagnosis of polycystic disease. J Urol 1969; 102:156–161.
- Cornell SH. Angiography in polycystic disease of the kidneys. J Urol 1970; 103:24–26.
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2009; 20:1888–1893.
- Chapman AB, Guay-Woodford LM, Grantham JJ, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int 2003; 64:1035–1045.
- Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Prospective change in renal volume and function in children with ADPKD. Clin J Am Soc Nephrol 2009; 4:820–829.
- Loghman-Adham M, Soto CE, Inagami T, Cassis L. The intrarenal renin-angiotensin system in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol 2004; 287:F775–F788.
- Torres VE, Donovan KA, Scicli G, et al. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 42:364–373.
- Graham PC, Lindop GB. The anatomy of the renin-secreting cell in adult polycystic kidney disease. Kidney Int 1988; 33:1084–1090.
- Cerasola G, Vecchi M, Mule G, et al. Sympathetic activity and blood pressure pattern in autosomal dominant polycystic kidney disease hypertensives. Am J Nephrol 1998; 18:391–398.
- Valvo E, Gammaro L, Tessitore N, et al. Hypertension of polycystic kidney disease: mechanisms and hemodynamic alterations. Am J Nephrol 1985; 5:176–181.
- Marlais M, Cuthell O, Langan D, Dudley J, Sinha MD, Winyard PJ. Hypertension in autosomal dominant polycystic kidney disease: a meta-analysis. Arch Dis Child 2016; 101:1142–1147.
- Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2255–2266.
- Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2267–2276.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Ritchie LD, Campbell NC, Murchie P. New NICE guidelines for hypertension. BMJ 2011; 343:d5644.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- Rangan GK, Alexander SI, Campbell KL, et al. KHA-CARI guideline recommendations for the diagnosis and management of autosomal dominant polycystic kidney disease. Nephrology 2016; 21:705–716.
- Helal I, Al-Rowaie F, Abderrahim E, Kheder A. Update on pathogenesis, management, and treatment of hypertension in autosomal dominant polycystic kidney disease. Saudi J Kidney Dis Transpl 2017; 28:253–260.
- Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis 2011; 57:856–862.
- Zeltner R, Poliak R, Stiasny B, Schmieder RE, Schulze BD. Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2008; 23:573–579.
- Clark LA, Whitmire S, Patton S, Clark C, Blanchette CM, Howden R. Cost-effectiveness of angiotensin-converting enzyme inhibitors versus angiotensin II receptor blockers as first-line treatment in autosomal dominant polycystic kidney disease. J Med Econ 2017:1–17.
- Li Kam Wa TC, Macnicol AM, Watson ML. Ambulatory blood pressure in hypertensive patients with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1997; 12:2075–2080.
- Valero FA, Martinez-Vea A, Bardaji A, et al. Ambulatory blood pressure and left ventricular mass in normotensive patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1999; 10:1020–1026.
- Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011; 121:4393–4408.
- Yildiz A, Gul CB, Ersoy A, Asiltas B, Ermurat S, Dogan S, et al. Arterial dysfunction in early autosomal dominant polycystic kidney disease independent of fibroblast growth factor 23. Iranian J Kidney Dis 2014; 8:443–449.
- Wanic-Kossowska M, Posnik B, Kobelski M, et al. The polymorphism of the ACE gene affects left ventricular hypertrophy and causes disturbances in left ventricular systolic/diastolic function in patients with autosomal dominant polycystic kidney disease. ScientificWorldJournal 2014; 2014:707658.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Cadnapaphornchai MA. Hypertension in children with autosomal dominant polycystic kidney disease (ADPKD). Curr Hypertens Rev 2013; 9:21–26.
- Schievink WI, Prendergast V, Zabramski JM. Rupture of a previously documented small asymptomatic intracranial aneurysm in a patient with autosomal dominant polycystic kidney disease. Case report. J Neurosurg 1998; 89:479–482.
- Graf S, Schischma A, Eberhardt KE, Istel R, Stiasny B, Schulze BD. Intracranial aneurysms and dolichoectasia in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2002; 17:819–823.
- Nakajima F, Shibahara N, Arai M, Gohji K, Ueda H, Katsuoka Y. Intracranial aneurysms and autosomal dominant polycystic kidney disease: followup study by magnetic resonance angiography. J Urol 2000; 164:311–313.
- Wakabayashi T, Fujita S, Ohbora Y, Suyama T, Tamaki N, Matsumoto S. Polycystic kidney disease and intracranial aneurysms. Early angiographic diagnosis and early operation for the unruptured aneurysm. J Neurosurg 1983; 58:488–491.
- Belz MM, Fick-Brosnahan GM, Hughes RL, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63:1824–1830.
- Schrier RW, Belz MM, Johnson AM, et al. Repeat imaging for intracranial aneurysms in patients with autosomal dominant polycystic kidney disease with initially negative studies: a prospective ten-year follow-up. J Am Soc Nephrol 2004; 15:1023–1028.
- Cagnazzo F, Gambacciani C, Morganti R, Perrini P. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: prevalence, risk of rupture, and management. A systematic review. Acta Neurochirurgica 2017; 5:811–821.
- Lee VW, Dexter MA, Mai J, Vladica P, Lopez-Vargas P, Rangan GK. KHA-CARI autosomal dominant polycystic kidney disease guideline: management of intracranial aneurysms. Semin Nephrol 2015; 35:612–617.
- Rozenfeld MN, Ansari SA, Shaibani A, Russell EJ, Mohan P, Hurley MC. Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms? AJNR Am J Neuroradiol 2014; 35:3–9.
- Hiratsuka Y, Miki H, Kiriyama I, et al. Diagnosis of unruptured intracranial aneurysms: 3T MR angiography versus 64-channel multi-detector row CT angiography. Magn Reson Med Sci 2008; 7:169–178.
- van Gelder JM. Computed tomographic angiography for detecting cerebral aneurysms: implications of aneurysm size distribution for the sensitivity, specificity, and likelihood ratios. Neurosurgery 2003; 53:597–605.
- Villablanca JP, Jahan R, Hooshi P, et al. Detection and characterization of very small cerebral aneurysms by using 2D and 3D helical CT angiography. AJNR Am J Neuroradiol 2002; 23:1187–1198.
- Lumiaho A, Ikäheimo R, Miettinen R, et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis 2001; 38:1208–1216.
- Timio M, Monarca C, Pede S, Gentili S, Verdura C, Lolli S. The spectrum of cardiovascular abnormalities in autosomal dominant polycystic kidney disease: a 10-year follow-up in a five-generation kindred. Clin Nephrol 1992; 37:245–251.
- Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988; 319:907–912.
- Leier CV, Baker PB, Kilman JW, Wooley CF. Cardiovascular abnormalities associated with adult polycystic kidney disease. Ann Intern Med 1984; 100:683–688.
- Turkmen K, Oflaz H, Uslu B, et al. Coronary flow velocity reserve and carotid intima media thickness in patients with autosomal dominant polycystic kidney disease: from impaired tubules to impaired carotid and coronary arteries. Clin J Am Soc Nephrol 2008; 3:986–991.
- Turkmen K, Tufan F, Alpay N, et al. Insulin resistance and coronary flow velocity reserve in patients with autosomal dominant polycystic kidney disease. Intern Med J 2012; 42:146–153.
- Turkmen K, Tufan F, Selcuk E, Akpinar T, Oflaz H, Ecder T. Neutrophil-to-lymphocyte ratio, insulin resistance, and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Indian J Nephrol 2013; 23:34–40.
- Hadimeri H, Lamm C, Nyberg G. Coronary aneurysms in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1998; 9:837–841.
- Earle K, Hoffbrand BI. Adult dominant polycystic kidney disease and atrial myxoma. Nephron 1989; 52:197.
- Chung BM, Chong S, Lee W-S, Hwang S-N. Autosomal dominant polycystic kidney disease combined with intracranial aneurysm and dilated cardiomyopathy: a case report. J Korean Soc Radiol 2014; 71:84–88.
- Mariathasan DAL, Kumanan T. Adult polycystic kidney disease and idiopathic dilated cardiomyopathy: a rare genetic association. J Ceylon Coll Physicians 2016: 46:42–44.
- Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407–2018.
- Higashihara E, Torres VE, Chapman AB, et al. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol 2011; 6:2499–2507.
- Sans Atxer L, Roca-Cusachs A, Torra R, et al. Relationship between renal size and blood pressure profile in patients with autosomal dominant polycystic kidney disease without renal failure. Nefrologia 2010; 30:567–572. In Spanish.
- Raina R, Lou L, Berger B, et al. Relationship of urinary endothelin-1 with estimated glomerular filtration rate in autosomal dominant polycystic kidney disease: a pilot cross-sectional analysis. BMC Nephrol 2016; 17:22.
Autosomal dominant polycystic kidney disease (ADPKD) has significant extrarenal manifestations. Hypertension is a common complication, arises early in the course of the disease, and is implicated in the development of left ventricular hypertrophy. Patients with ADPKD are also at risk of other cardiovascular complications (Table 1).
This article reviews the timely diagnosis of these common ADPKD complications and how to manage them.
ADPKD ACCOUNTS FOR 10% OF END-STAGE RENAL DISEASE
ADPKD is a genetic condition characterized by multiple renal cysts.1 Progressive enlargement of these cysts leads to a gradual decline in kidney function and eventually end-stage renal disease by the fifth or sixth decade of life.2 Worldwide, about 12.5 million people have ADPKD, and it accounts for about 10% of cases of end-stage renal disease.1,3,4
ADPKD has a variety of clinical presentations, including (in decreasing order of frequency) hypertension, flank pain, abdominal masses, urinary tract infection, renal failure, renal stones, and cerebrovascular accidents.2
Extrarenal complications are common and include hepatic cysts, hypertension, left ventricular hypertrophy, valvular heart disease, intracranial and extracranial aneurysms, pancreatic cysts, and diverticulosis.1–5
Less-common complications are dissection of the aorta and the internal carotid, vertebral, and iliac arteries6–10; aneurysm of the coronary, popliteal, and splenic arteries11–14; atrial myxoma15; cardiomyopathy16; pericardial effusion17; intracranial arterial dolichoectasia18; arachnoid cysts2; and intraoperative inferior vena cava syndrome (normally in ADPKD patients, pressure on the inferior vena cava results in compensatory sympathetic overactivity to maintain blood pressure), which occurs due to reduced sympathetic output under the influence of epidural or general anesthesia.19
Cardiovascular complications, especially cardiac hypertrophy and coronary artery disease, are now the leading cause of death in patients with ADPKD, as renal replacement therapy has improved and made death from end-stage renal disease less common.20,21
HYPERTENSION IN ADPKD
Hypertension is the most frequent initial presentation of ADPKD, occurring in 50% to 75% of cases and usually preceding the onset of renal failure.2,22 Hypertension is more common in male ADPKD patients, begins early in the course of the disease, and is diagnosed around the fourth decade of life.21
In a study in 2007, de Almeida et al23 used 24-hour ambulatory blood pressure monitoring early in the course of ADPKD and found significantly higher systolic, diastolic, and mean 24-hour blood pressures in ADPKD patients who had normal in-office blood pressure than in normotensive controls. In addition, nighttime systolic, nighttime diastolic, and nighttime mean blood pressures were significantly higher in the ADPKD group.
Hypertension is strongly associated with an accelerated decline in renal function to end-stage renal disease, development of left ventricular hypertrophy, and cardiovascular death.20,24
Although a prospective study25 showed a strong association between renal stones and hypertension in ADPKD, the relation between them is not clear. The incidence of renal stones is higher in hypertensive than in normotensive ADPKD patients, although evidence has to be established whether nephrolithiasis is a risk factor for hypertension or the other way around.25
Hypertension in ADPKD is multifactorial (Figure 1). The major factors associated with its development are increased activation of the renin-angiotensin-aldosterone system (RAAS); overexpression of endothelin receptor subtype A (ET-A) in cystic kidneys; increased production of endothelin 1 (ET-1); and sodium retention.26–31
The renin-angiotensin-aldosterone system
Activation of the RAAS plays a major role in the development and maintenance of hypertension in ADPKD. This is thought to be mainly due to progressive enlargement of renal cysts, which causes renal arteriolar attenuation and ischemia secondary to pressure effects, which in turn activates the RAAS.26,30,32–34 Two studies in patients with normal renal function found that cyst growth and increasing kidney volume have a strong relationship with the development of hypertension and declining kidney function.35,36
Ectopic secretion of RAAS components in polycystic kidneys has also been implicated in the development of hypertension, whereby renin, angiotensinogen, angiotensin-converting enzyme (ACE), angiotensin II, and angiotensin II receptors are produced in the epithelium of cysts and dilated tubules in polycystic kidneys.37–39 Proximal renal cysts and tubules produce ectopic angiotensinogen, which is converted to angiotensin I by renin in distal renal cysts. Angiotensin I is converted to angiotensin II by ACE in distal tubules, which in turn stimulates angiotensin II receptors, causing sodium and water retention in distal tubules.37 This may be responsible for hypertension in the initial stages; however, RAAS hyperactivity due to renal injury may predominate during later stages.37
Increased RAAS activity also increases sympathetic output, which in turn raises catecholamine levels and blood pressure.34 A study showed higher levels of plasma catecholamines in ADPKD hypertensive patients irrespective of renal function than in patients with essential hypertension.40
ET-A receptor and ET-1
A few studies have shown that in ADPKD patients, increased density of ET-A receptors and overproduction of ET-1, a potent vasoconstrictor, play a significant role in the development of hypertension and gradual loss of kidney function due to cyst enlargement and interstitial scarring.28,29 Ong et al29 found that expression of ET-A receptors is increased in smooth muscle cells of renal arteries, glomerular mesangial cells, and cyst epithelia in ADPKD.
Sodium retention
Studies in ADPKD patients with preserved renal function have linked high blood pressure to sodium retention and volume expansion.30,31,41 However, this phenomenon reverses when there is significant renal impairment in ADPKD.
As evidence of this, a study demonstrated significantly more natriuresis in patients with renal failure due to ADPKD than in patients with a similar degree of renal failure due to chronic glomerulonephritis.31 Moreover, another study found that the prevalence of hypertension is higher in ADPKD patients than in those with other nephropathies with preserved renal function, but this association reverses with significant decline in kidney function.22
MANAGING HYPERTENSION IN ADPKD
Early diagnosis of hypertension and effective control of it, even before ADPKD is diagnosed, is crucial to reduce cardiovascular mortality. Aggressive blood pressure control in the prehypertensive phase of ADPKD will also help reduce the incidence of left ventricular hypertrophy and mitral regurgitation and slow the progression of renal failure (Figure 2).
A meta-analysis42 revealed hypertension to be present in 20% of ADPKD patients younger than 21, and many of them were undiagnosed. This study also suggests that patients at risk of hypertension (ie, all patients with ADPKD) should be routinely screened for it.
Ambulatory blood pressure monitoring may play an important role in diagnosing hypertension early in the prehypertensive stage of ADPKD.23
Target blood pressures: No consensus
Two well-powered double-blind, placebo-controlled trials, known as HALT-PKD Study A and HALT-PKD Study B, tested the effects of 2 different blood pressure targets and of monotherapy with an ACE inhibitor vs combination therapy with an ACE inhibitor plus an angiotensin II receptor blocker (ARB) on renal function, total kidney volume, left ventricular mass index, and urinary albumin excretion in the early (estimated glomerular filtration rate [eGFR] > 60 mL/min) and late (eGFR 25–60 mL/min) stages of ADPKD, respectively.43,44
HALT-PKD Study A43 found that, in the early stages of ADPKD with preserved renal function, meticulous control of blood pressure (95–110/60–75 mm Hg) was strongly correlated with significant reductions in left ventricular mass index, albuminuria, and rate of total kidney volume growth without remarkable alteration in renal function compared with standard blood pressure control (120–130/70–80 mm Hg). However, no notable differences were observed between the ACE inhibitor and ACE inhibitor-plus-ARB groups.43
Despite the evidence, universal consensus guidelines are lacking, and the available guidelines on hypertension management have different blood pressure goals in patients with chronic kidney disease.
The eighth Joint National Committee guideline of 2014 recommends a blood pressure goal of less than 140/90 mm Hg in patients with diabetic and nondiabetic chronic kidney disease.45
The National Institute for Health and Care Excellence 2011 guideline recommends a blood pressure goal of less than 130/80 mm Hg in chronic kidney disease patients.46
The European Society of Hypertension and European Society of Cardiology joint 2013 guideline recommends a systolic blood pressure goal of less than 140 mm Hg in diabetic and nondiabetic patients with chronic kidney disease.47
The 2016 Kidney Health Australia-Caring for Australians With Renal Impairment guideline for diagnosis and management of ADPKD48 recommends a lower blood pressure goal of 96–110/60–75 mm Hg in patients with an eGFR greater than 60 mL/min/1.73 m2 who can tolerate it without side effects, which is based on the findings of HALT-PKD Study A.43
Helal et al recommend that blood pressure be controlled to less than 130/80 mm Hg, until there is more evidence for a safe and effective target blood pressure goal in ADPKD patients.49
We recommend a target blood pressure less than 110/75 mm Hg in hypertensive ADPKD patients with preserved renal function who can tolerate this level, and less than 130/80 mm Hg in ADPKD patients with stage 3 chronic kidney disease. These targets can be achieved with ACE inhibitor or ARB monotherapy.43,44 However, no studies have established the safest lower limit of target blood pressure in ADPKD.
ACE inhibitors, ARBs are mainstays
Mainstays of antihypertensive drug therapy in ADPKD are ACE inhibitors and ARBs.
HALT-PKD Study B44 demonstrated that, in the late stages of ADPKD, target blood pressure control (110–130/70–80 mm Hg) can be attained with ACE inhibitor monotherapy or with an ACE inhibitor plus an ARB, but the latter produced no additive benefit.
Patch et al,50 in a retrospective cohort study, showed that broadening the spectrum of antihypertensive therapy decreases mortality in ADPKD patients. Evaluating ADPKD patients from the UK General Practice Research Database between 1991 and 2008, they found a trend toward lower mortality rates as the number of antihypertensive drugs prescribed within 1 year increased. They also observed that the prescription of RAAS-blocking agents increased from 7% in 1991 to 46% in 2008.50
However, a 3-year prospective randomized double-blind study compared the effects of the ACE inhibitor ramipril and the beta-blocker metoprolol in hypertensive ADPKD patients.51 The results showed that effective blood pressure control could be achieved in both groups with no significant differences in left ventricular mass index, albuminuria, or kidney function.51
Treatment strategies
Lifestyle modification is the initial approach to the management of hypertension before starting drug therapy. Lifestyle changes include dietary salt restriction to less than 6 g/day, weight reduction, regular exercise, increased fluid intake (up to 3 L/day or to satisfy thirst), smoking cessation, and avoidance of caffeine.47–49
ACE inhibitors are first-line drugs in hypertensive ADPKD patients.
ARBs can also be considered, but there is no role for dual ACE inhibitor and ARB therapy.43,48 A study found ACE inhibitors to be more cost-effective and to decrease mortality rates to a greater extent than ARBs.52
Beta-blockers or calcium channel blockers should be considered instead if ACE inhibitors and ARBs are contraindicated, or as add-on drugs if ACE inhibitors and ARBs fail to reduce blood pressure adequately.48,49
Diuretics are third-line agents. Thiazides are preferred in ADPKD patients with normal renal function and loop diuretics in those with impaired renal function.49
LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Increased left ventricular mass is an indirect indicator of untreated hypertension, and it often goes unnoticed in patients with undiagnosed ADPKD. Left ventricular hypertrophy is associated with arrhythmias and heart failure, which contribute significantly to cardiovascular mortality and adverse renal outcomes.20,24
A 5-year randomized clinical trial by Cadnapaphornchai et al36 in ADPKD patients between 4 and 21 years of age showed strong correlations between hypertension, left ventricular mass index, and kidney volume and a negative correlation between left ventricular mass index and renal function.
Several factors are thought to contribute to left ventricular hypertrophy in ADPKD (Figure 1).
Hypertension. Two studies of 24-hour ambulatory blood pressure monitoring showed that nocturnal blood pressures decreased less in normotensive and hypertensive ADPKD patients than in normotensive and hypertensive controls.23,53 This persistent elevation of nocturnal blood pressure may contribute to the development and progression of left ventricular hypertrophy.
On the other hand, Valero et al54 reported that the left ventricular mass index was strongly associated with ambulatory systolic blood pressure rather than elevated nocturnal blood pressure in ADPKD patients compared with healthy controls.
FGF23. High levels of fibroblast growth factor 23 (FGF23) have been shown to be strongly associated with left ventricular hypertrophy in ADPKD. Experimental studies have shown that FGF23 is directly involved in the pathogenesis of left ventricular hypertrophy through stimulation of the calcineurin-nuclear factor of activated T cells pathway.
Faul et al55 induced cardiac hypertrophy in mice that were deficient in klotho (a transmembrane protein that increases FGF23 affinity for FGF receptors) by injecting FGF23 intravenously.
Yildiz et al56 observed higher levels of FGF23 in hypertensive and normotensive ADPKD patients with normal renal function than in healthy controls. They also found a lower elasticity index in the large and small arteries in normotensive and hypertensive ADPKD patients, which accounts for vascular dysfunction. High FGF23 levels may be responsible for the left ventricular hypertrophy seen in normotensive ADPKD patients with preserved renal function.
Polymorphisms in the ACE gene have been implicated in the development of cardiac hypertrophy in ADPKD.
Wanic-Kossowska et al57 studied the association between ACE gene polymorphisms and cardiovascular complications in ADPKD patients. They found a higher prevalence of the homozygous DD genotype among ADPKD patients with end-stage renal disease than in those in the early stages of chronic kidney disease in ADPKD. Also, the DD genotype has been shown to be more strongly associated with left ventricular hypertrophy and left ventricular dysfunction than other (II or ID) genotypes. These findings suggest that the DD genotype carries higher risk for the development of end-stage renal disease, left ventricular hypertrophy, and other cardiovascular complications.
MANAGING LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Preventing and halting progression of left ventricular hypertrophy primarily involves effective blood pressure control, especially in the early stages of ADPKD (Figure 2).
A 7-year prospective randomized trial in ADPKD patients with established hypertension and left ventricular hypertrophy proved that aggressive (< 120/80 mm Hg) compared with standard blood pressure control (135–140/85–90 mm Hg) significantly reduces left ventricular mass index. ACE inhibitors were preferred over calcium channel blockers.58
HALT-PKD Study A showed that a significant decrease in left ventricular mass index can be achieved by aggressive blood pressure control (95–110/60–75 mm Hg) with an ACE inhibitor alone or in combination with an ARB in the early stages of ADPKD with preserved renal function.43
A 5-year randomized clinical trial in children with borderline hypertension treated with an ACE inhibitor for effective control of blood pressure showed no change in left ventricular mass index or renal function.36,59
These results support starting ACE inhibitor therapy early in the disease process when blood pressure is still normal or borderline to prevent the progression of left ventricular hypertrophy or worsening kidney function.
Since FGF23 is directly involved in the causation of left ventricular hypertrophy, FGF receptors may be potential therapeutic targets to prevent left ventricular hypertrophy in ADPKD. An FGF receptor blocker was shown to decrease left ventricular hypertrophy in rats with chronic kidney disease without affecting blood pressure.55
INTRACRANIAL ANEURYSM IN ADPKD
Intracranial aneurysm is the most dangerous complication of ADPKD. When an aneurysm ruptures, the mortality rate is 4% to 7%, and 50% of survivors are left with residual neurologic deficits.5,60,61
In various studies, the prevalence of intracranial aneurysm in ADPKD ranged from 4% to 41.2%, compared with 1% in the general population.5,62,63 On follow-up ranging from 18 months to about 10 years, the incidence of new intracranial aneurysm was 2.6% to 13.3% in patients with previously normal findings on magnetic resonance angiography and 25% in patients with a history of intracranial aneurysm.62,64,65
The most common sites are the middle cerebral artery (45%), internal carotid artery (40.5%), and anterior communicating artery (35.1%).66 (The numbers add up to more than 100% because some patients have aneurysms in more than 1 site.) The mean size of a ruptured aneurysm was 6 mm per a recent systematic review.66 Intracranial aneurysms 6 mm or larger are at highest risk of rupture.66
SCREENING FOR INTRACRANIAL ANEURYSM
Timely screening and intervention for intracranial aneurysm is crucial to prevent death from intracranial hemorrhage.
Currently, there are no standard guidelines for screening and follow-up of intracranial aneurysm in ADPKD patients. However, some recommendations are available from the ADPKD Kidney Disease Improving Global Outcomes Controversies Conference3 and Kidney Health Australia—Caring for Australasians With Renal Impairment ADPKD guidelines67 (Figure 3).
Imaging tests
Magnetic resonance angiography (MRA) with gadolinium enhancement and computed tomographic angiography (CTA) are recommended for screening in ADPKD patients with normal renal function,67 but time-of-flight MRA without gadolinium is the imaging test of choice because it is noninvasive and poses no risk of nephrotoxicity or contrast allergy.3,68 Further, gadolinium should be avoided in patients whose eGFR is 30 mL/min/1.73 m2 or less because of risk of nephrogenic systemic sclerosis and fibrosis.67,68
The sensitivity of time-of-flight MRA screening for intracranial aneurysm varies depending on the size of aneurysm; 67% for those less than 3 mm, 79% for those 3 to 5 mm, and 95% for those larger than 5 mm.69 The sensitivity of CTA screening is 95% for aneurysms larger than 7 mm and 53% for those measuring 2 mm.70,71 The specificity of CTA screening was reported to be 98.9% overall.71
When to screen
Screening for intracranial aneurysm is recommended at the time of ADPKD diagnosis for all high-risk patients, ie, those who have a family history of intracranial hemorrhage or aneurysm in an affected first-degree relative.67 It is also recommended for ADPKD patients with a history of sudden-onset severe headache or neurologic symptoms.67 A third group for whom screening is recommended is ADPKD patients who have no family history of intracranial aneurysm or hemorrhage but who are at risk of poor outcome if an intracranial aneurysm ruptures (eg, those undergoing major elective surgery, with uncontrolled blood pressure, on anticoagulation, with a history of or current smoking, and airline pilots).67
Patients found to have an intracranial aneurysm on screening should be referred to a neurosurgeon and should undergo repeat MRA or CTA imaging every 6 to 24 months.3 High-risk ADPKD patients with normal findings on initial screening should have repeat MRA or CTA screening in 5 to 10 years unless they suffer from sudden-onset severe headache or neurologic symptoms.65,67
Both smoking and high blood pressure increase the risk of formation and growth of intracranial aneurysm. Hence, meticulous control of blood pressure and smoking cessation are recommended in ADPKD patients.3,67
CARDIAC VALVULAR ABNORMALITIES IN ADPKD
Of the valvular abnormalities that complicate ADPKD, the more common ones are mitral valve prolapse and mitral and aortic regurgitation. The less common ones are tricuspid valve prolapse and tricuspid regurgitation.72–74
The pathophysiology underlying these valvular abnormalities is unclear. However, defective collagen synthesis and myxomatous degeneration have been demonstrated in histopathologic examination of affected valvular tissue.75 Also, ACE gene polymorphism, especially the DD genotype, has been shown to be associated with cardiac valvular calcifications and valvular insufficiency.57
Lumiaho et al72 found a higher prevalence of mitral valve prolapse, mitral regurgitation, and left ventricular hypertrophy in patients with ADPKD type 1 (due to abnormalities in PDK1) than in unaffected family members and healthy controls. The investigators speculated that mitral regurgitation is caused by the high blood pressure observed in ADPKD type 1 patients, since hypertension causes left ventricular hypertrophy and left ventricular dilatation. The severity of renal failure was related to mitral regurgitation but not mitral valve prolapse.
Similarly, Gabow et al24 showed that there is no significant relationship between mitral valve prolapse and progression of renal disease in ADPKD.
Interestingly, Fick et al20 found that mitral valve prolapse has no significant effect on cardiovascular mortality.
CORONARY ARTERY DISEASE AND ANEURYSM IN ADPKD
Atherosclerosis sets in early in ADPKD, resulting in coronary artery disease and adverse cardiovascular outcomes.
Coronary flow velocity reserve is the ability of coronary arteries to dilate in response to myocardial oxygen demand. Atherosclerosis decreases this reserve in ADPKD patients, as shown in several studies.
Turkmen et al, in a series of studies,76–78 found that ADPKD patients had significantly less coronary flow velocity reserve, thicker carotid intima media (a surrogate marker of atherosclerosis), and greater insulin resistance than healthy controls. These findings imply that atherosclerosis begins very early in the course of ADPKD and has remarkable effects on cardiovascular morbidity and mortality.76
Aneurysm. Although the risk of extracranial aneurysm is higher with ADPKD, coronary artery aneurysm is uncommon. The pathogenesis of coronary aneurysm has been linked to abnormal expression of the proteins polycystin 1 and polycystin 2 in vascular smooth muscle.11,79 The PKD1 and PKD2 genes encode polycystin 1 and 2, respectively, in ADPKD. These polycystins are also expressed in the liver, kidneys, and myocardium and are involved in the regulation of intracellular calcium, stretch-activated ion channels, and vascular smooth muscle cell proliferation and apoptosis.11,16 Abnormally expressed polycystin in ADPKD therefore has an impact on arterial wall integrity, resulting in focal medial defects in the vasculature that eventually develop into micro- and macroaneurysms.11
Hadimeri et al79 found a higher prevalence of coronary aneurysm and ectasia in ADPKD patients than in controls. Most coronary aneurysms are smaller than 1 cm; however, a coronary aneurysm measuring 4 cm in diameter was found at autopsy of an ADPKD patient.11
Spontaneous coronary artery dissection is very rare in the general population, but Bobrie et al reported a case of it in an ADPKD patient.9
ATRIAL MYXOMA, CARDIOMYOPATHY, AND PERICARDIAL EFFUSION IN ADPKD
Atrial myxoma in ADPKD patients has been described in 2 case reports.15,80 However, the association of atrial myxoma with ADPKD is poorly understood and may be a coincidental finding.
Cardiomyopathy in ADPKD has been linked to abnormalities in the intracellular calcium pathway, although a clear picture of its involvement has yet to be established.
Paavola et al16 described the pathophysiology of ADPKD-associated cardiomyopathy in PKD2 mutant zebrafish lacking polycystin 2. These mutants showed decreased cardiac output and had atrioventricular blocks. The findings were attributed to abnormal intracellular calcium cycling. These findings correlated well with the frequent finding of idiopathic dilated cardiomyopathy in ADPKD patients, especially with PKD2 mutations.16 Also, 2 cases of dilated cardiomyopathy in ADPKD have been reported and thought to be related to PKD2 mutations.81,82
Pericardial effusion. Even though the exact pathophysiology of pericardial effusion in ADPKD is unknown, it has been theorized to be related to defects in connective tissue and extracellular matrix due to PKD1 and PKD2 mutations. These abnormalities increase the compliance and impair the recoil capacity of connective tissue, which results in unusual distention of the parietal pericardium. This abnormal distention of the parietal pericardium together with increased extracellular volume may lead to pericardial effusion.17
Qian et al17 found a higher prevalence of pericardial effusion in ADPKD patients. It was generally asymptomatic, and the cause was attributed to these connective tissue and extracellular matrix abnormalities.
EMERGING THERAPIES AND TESTS
Recent trials have investigated the effects of vasopressin receptor antagonists, specifically V2 receptor blockers in ADPKD and its complications.
Tolvaptan has been shown to slow the rate of increase in cyst size and total kidney volume.83,84 Also, a correlation between kidney size and diastolic blood pressure has been observed in ADPKD patients.85 Reducing cyst volume may reduce pressure effects and decrease renal ischemia, which in turn may reduce RAAS activation; however, the evidence to support this hypothesis is poor. A major clinical trial of tolvaptan in ADPKD patients showed no effect on blood pressure control, but the drug slowed the rate of increase in total kidney volume and fall in eGFR.83
Endothelin receptor antagonists are also in the preliminary stage of development for use in ADPKD. The effects of acute blockade of the renal endothelial system with bosentan were investigated in animal models by Hocher et al.28 This study showed a greater reduction in mean arterial pressure after bosentan administration, resulting in significantly decreased GFR and renal blood flow. Nonetheless, the mean arterial pressure-lowering effect of bosentan was more marked than the reductions in GFR and renal blood flow.
Raina et al,86 in a pilot cross-sectional analysis, showed that urinary excretion of ET-1 is increased in ADPKD patients, and may serve as a surrogate marker for ET-1 in renal tissue and a noninvasive marker of early kidney injury.
Autosomal dominant polycystic kidney disease (ADPKD) has significant extrarenal manifestations. Hypertension is a common complication, arises early in the course of the disease, and is implicated in the development of left ventricular hypertrophy. Patients with ADPKD are also at risk of other cardiovascular complications (Table 1).
This article reviews the timely diagnosis of these common ADPKD complications and how to manage them.
ADPKD ACCOUNTS FOR 10% OF END-STAGE RENAL DISEASE
ADPKD is a genetic condition characterized by multiple renal cysts.1 Progressive enlargement of these cysts leads to a gradual decline in kidney function and eventually end-stage renal disease by the fifth or sixth decade of life.2 Worldwide, about 12.5 million people have ADPKD, and it accounts for about 10% of cases of end-stage renal disease.1,3,4
ADPKD has a variety of clinical presentations, including (in decreasing order of frequency) hypertension, flank pain, abdominal masses, urinary tract infection, renal failure, renal stones, and cerebrovascular accidents.2
Extrarenal complications are common and include hepatic cysts, hypertension, left ventricular hypertrophy, valvular heart disease, intracranial and extracranial aneurysms, pancreatic cysts, and diverticulosis.1–5
Less-common complications are dissection of the aorta and the internal carotid, vertebral, and iliac arteries6–10; aneurysm of the coronary, popliteal, and splenic arteries11–14; atrial myxoma15; cardiomyopathy16; pericardial effusion17; intracranial arterial dolichoectasia18; arachnoid cysts2; and intraoperative inferior vena cava syndrome (normally in ADPKD patients, pressure on the inferior vena cava results in compensatory sympathetic overactivity to maintain blood pressure), which occurs due to reduced sympathetic output under the influence of epidural or general anesthesia.19
Cardiovascular complications, especially cardiac hypertrophy and coronary artery disease, are now the leading cause of death in patients with ADPKD, as renal replacement therapy has improved and made death from end-stage renal disease less common.20,21
HYPERTENSION IN ADPKD
Hypertension is the most frequent initial presentation of ADPKD, occurring in 50% to 75% of cases and usually preceding the onset of renal failure.2,22 Hypertension is more common in male ADPKD patients, begins early in the course of the disease, and is diagnosed around the fourth decade of life.21
In a study in 2007, de Almeida et al23 used 24-hour ambulatory blood pressure monitoring early in the course of ADPKD and found significantly higher systolic, diastolic, and mean 24-hour blood pressures in ADPKD patients who had normal in-office blood pressure than in normotensive controls. In addition, nighttime systolic, nighttime diastolic, and nighttime mean blood pressures were significantly higher in the ADPKD group.
Hypertension is strongly associated with an accelerated decline in renal function to end-stage renal disease, development of left ventricular hypertrophy, and cardiovascular death.20,24
Although a prospective study25 showed a strong association between renal stones and hypertension in ADPKD, the relation between them is not clear. The incidence of renal stones is higher in hypertensive than in normotensive ADPKD patients, although evidence has to be established whether nephrolithiasis is a risk factor for hypertension or the other way around.25
Hypertension in ADPKD is multifactorial (Figure 1). The major factors associated with its development are increased activation of the renin-angiotensin-aldosterone system (RAAS); overexpression of endothelin receptor subtype A (ET-A) in cystic kidneys; increased production of endothelin 1 (ET-1); and sodium retention.26–31
The renin-angiotensin-aldosterone system
Activation of the RAAS plays a major role in the development and maintenance of hypertension in ADPKD. This is thought to be mainly due to progressive enlargement of renal cysts, which causes renal arteriolar attenuation and ischemia secondary to pressure effects, which in turn activates the RAAS.26,30,32–34 Two studies in patients with normal renal function found that cyst growth and increasing kidney volume have a strong relationship with the development of hypertension and declining kidney function.35,36
Ectopic secretion of RAAS components in polycystic kidneys has also been implicated in the development of hypertension, whereby renin, angiotensinogen, angiotensin-converting enzyme (ACE), angiotensin II, and angiotensin II receptors are produced in the epithelium of cysts and dilated tubules in polycystic kidneys.37–39 Proximal renal cysts and tubules produce ectopic angiotensinogen, which is converted to angiotensin I by renin in distal renal cysts. Angiotensin I is converted to angiotensin II by ACE in distal tubules, which in turn stimulates angiotensin II receptors, causing sodium and water retention in distal tubules.37 This may be responsible for hypertension in the initial stages; however, RAAS hyperactivity due to renal injury may predominate during later stages.37
Increased RAAS activity also increases sympathetic output, which in turn raises catecholamine levels and blood pressure.34 A study showed higher levels of plasma catecholamines in ADPKD hypertensive patients irrespective of renal function than in patients with essential hypertension.40
ET-A receptor and ET-1
A few studies have shown that in ADPKD patients, increased density of ET-A receptors and overproduction of ET-1, a potent vasoconstrictor, play a significant role in the development of hypertension and gradual loss of kidney function due to cyst enlargement and interstitial scarring.28,29 Ong et al29 found that expression of ET-A receptors is increased in smooth muscle cells of renal arteries, glomerular mesangial cells, and cyst epithelia in ADPKD.
Sodium retention
Studies in ADPKD patients with preserved renal function have linked high blood pressure to sodium retention and volume expansion.30,31,41 However, this phenomenon reverses when there is significant renal impairment in ADPKD.
As evidence of this, a study demonstrated significantly more natriuresis in patients with renal failure due to ADPKD than in patients with a similar degree of renal failure due to chronic glomerulonephritis.31 Moreover, another study found that the prevalence of hypertension is higher in ADPKD patients than in those with other nephropathies with preserved renal function, but this association reverses with significant decline in kidney function.22
MANAGING HYPERTENSION IN ADPKD
Early diagnosis of hypertension and effective control of it, even before ADPKD is diagnosed, is crucial to reduce cardiovascular mortality. Aggressive blood pressure control in the prehypertensive phase of ADPKD will also help reduce the incidence of left ventricular hypertrophy and mitral regurgitation and slow the progression of renal failure (Figure 2).
A meta-analysis42 revealed hypertension to be present in 20% of ADPKD patients younger than 21, and many of them were undiagnosed. This study also suggests that patients at risk of hypertension (ie, all patients with ADPKD) should be routinely screened for it.
Ambulatory blood pressure monitoring may play an important role in diagnosing hypertension early in the prehypertensive stage of ADPKD.23
Target blood pressures: No consensus
Two well-powered double-blind, placebo-controlled trials, known as HALT-PKD Study A and HALT-PKD Study B, tested the effects of 2 different blood pressure targets and of monotherapy with an ACE inhibitor vs combination therapy with an ACE inhibitor plus an angiotensin II receptor blocker (ARB) on renal function, total kidney volume, left ventricular mass index, and urinary albumin excretion in the early (estimated glomerular filtration rate [eGFR] > 60 mL/min) and late (eGFR 25–60 mL/min) stages of ADPKD, respectively.43,44
HALT-PKD Study A43 found that, in the early stages of ADPKD with preserved renal function, meticulous control of blood pressure (95–110/60–75 mm Hg) was strongly correlated with significant reductions in left ventricular mass index, albuminuria, and rate of total kidney volume growth without remarkable alteration in renal function compared with standard blood pressure control (120–130/70–80 mm Hg). However, no notable differences were observed between the ACE inhibitor and ACE inhibitor-plus-ARB groups.43
Despite the evidence, universal consensus guidelines are lacking, and the available guidelines on hypertension management have different blood pressure goals in patients with chronic kidney disease.
The eighth Joint National Committee guideline of 2014 recommends a blood pressure goal of less than 140/90 mm Hg in patients with diabetic and nondiabetic chronic kidney disease.45
The National Institute for Health and Care Excellence 2011 guideline recommends a blood pressure goal of less than 130/80 mm Hg in chronic kidney disease patients.46
The European Society of Hypertension and European Society of Cardiology joint 2013 guideline recommends a systolic blood pressure goal of less than 140 mm Hg in diabetic and nondiabetic patients with chronic kidney disease.47
The 2016 Kidney Health Australia-Caring for Australians With Renal Impairment guideline for diagnosis and management of ADPKD48 recommends a lower blood pressure goal of 96–110/60–75 mm Hg in patients with an eGFR greater than 60 mL/min/1.73 m2 who can tolerate it without side effects, which is based on the findings of HALT-PKD Study A.43
Helal et al recommend that blood pressure be controlled to less than 130/80 mm Hg, until there is more evidence for a safe and effective target blood pressure goal in ADPKD patients.49
We recommend a target blood pressure less than 110/75 mm Hg in hypertensive ADPKD patients with preserved renal function who can tolerate this level, and less than 130/80 mm Hg in ADPKD patients with stage 3 chronic kidney disease. These targets can be achieved with ACE inhibitor or ARB monotherapy.43,44 However, no studies have established the safest lower limit of target blood pressure in ADPKD.
ACE inhibitors, ARBs are mainstays
Mainstays of antihypertensive drug therapy in ADPKD are ACE inhibitors and ARBs.
HALT-PKD Study B44 demonstrated that, in the late stages of ADPKD, target blood pressure control (110–130/70–80 mm Hg) can be attained with ACE inhibitor monotherapy or with an ACE inhibitor plus an ARB, but the latter produced no additive benefit.
Patch et al,50 in a retrospective cohort study, showed that broadening the spectrum of antihypertensive therapy decreases mortality in ADPKD patients. Evaluating ADPKD patients from the UK General Practice Research Database between 1991 and 2008, they found a trend toward lower mortality rates as the number of antihypertensive drugs prescribed within 1 year increased. They also observed that the prescription of RAAS-blocking agents increased from 7% in 1991 to 46% in 2008.50
However, a 3-year prospective randomized double-blind study compared the effects of the ACE inhibitor ramipril and the beta-blocker metoprolol in hypertensive ADPKD patients.51 The results showed that effective blood pressure control could be achieved in both groups with no significant differences in left ventricular mass index, albuminuria, or kidney function.51
Treatment strategies
Lifestyle modification is the initial approach to the management of hypertension before starting drug therapy. Lifestyle changes include dietary salt restriction to less than 6 g/day, weight reduction, regular exercise, increased fluid intake (up to 3 L/day or to satisfy thirst), smoking cessation, and avoidance of caffeine.47–49
ACE inhibitors are first-line drugs in hypertensive ADPKD patients.
ARBs can also be considered, but there is no role for dual ACE inhibitor and ARB therapy.43,48 A study found ACE inhibitors to be more cost-effective and to decrease mortality rates to a greater extent than ARBs.52
Beta-blockers or calcium channel blockers should be considered instead if ACE inhibitors and ARBs are contraindicated, or as add-on drugs if ACE inhibitors and ARBs fail to reduce blood pressure adequately.48,49
Diuretics are third-line agents. Thiazides are preferred in ADPKD patients with normal renal function and loop diuretics in those with impaired renal function.49
LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Increased left ventricular mass is an indirect indicator of untreated hypertension, and it often goes unnoticed in patients with undiagnosed ADPKD. Left ventricular hypertrophy is associated with arrhythmias and heart failure, which contribute significantly to cardiovascular mortality and adverse renal outcomes.20,24
A 5-year randomized clinical trial by Cadnapaphornchai et al36 in ADPKD patients between 4 and 21 years of age showed strong correlations between hypertension, left ventricular mass index, and kidney volume and a negative correlation between left ventricular mass index and renal function.
Several factors are thought to contribute to left ventricular hypertrophy in ADPKD (Figure 1).
Hypertension. Two studies of 24-hour ambulatory blood pressure monitoring showed that nocturnal blood pressures decreased less in normotensive and hypertensive ADPKD patients than in normotensive and hypertensive controls.23,53 This persistent elevation of nocturnal blood pressure may contribute to the development and progression of left ventricular hypertrophy.
On the other hand, Valero et al54 reported that the left ventricular mass index was strongly associated with ambulatory systolic blood pressure rather than elevated nocturnal blood pressure in ADPKD patients compared with healthy controls.
FGF23. High levels of fibroblast growth factor 23 (FGF23) have been shown to be strongly associated with left ventricular hypertrophy in ADPKD. Experimental studies have shown that FGF23 is directly involved in the pathogenesis of left ventricular hypertrophy through stimulation of the calcineurin-nuclear factor of activated T cells pathway.
Faul et al55 induced cardiac hypertrophy in mice that were deficient in klotho (a transmembrane protein that increases FGF23 affinity for FGF receptors) by injecting FGF23 intravenously.
Yildiz et al56 observed higher levels of FGF23 in hypertensive and normotensive ADPKD patients with normal renal function than in healthy controls. They also found a lower elasticity index in the large and small arteries in normotensive and hypertensive ADPKD patients, which accounts for vascular dysfunction. High FGF23 levels may be responsible for the left ventricular hypertrophy seen in normotensive ADPKD patients with preserved renal function.
Polymorphisms in the ACE gene have been implicated in the development of cardiac hypertrophy in ADPKD.
Wanic-Kossowska et al57 studied the association between ACE gene polymorphisms and cardiovascular complications in ADPKD patients. They found a higher prevalence of the homozygous DD genotype among ADPKD patients with end-stage renal disease than in those in the early stages of chronic kidney disease in ADPKD. Also, the DD genotype has been shown to be more strongly associated with left ventricular hypertrophy and left ventricular dysfunction than other (II or ID) genotypes. These findings suggest that the DD genotype carries higher risk for the development of end-stage renal disease, left ventricular hypertrophy, and other cardiovascular complications.
MANAGING LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Preventing and halting progression of left ventricular hypertrophy primarily involves effective blood pressure control, especially in the early stages of ADPKD (Figure 2).
A 7-year prospective randomized trial in ADPKD patients with established hypertension and left ventricular hypertrophy proved that aggressive (< 120/80 mm Hg) compared with standard blood pressure control (135–140/85–90 mm Hg) significantly reduces left ventricular mass index. ACE inhibitors were preferred over calcium channel blockers.58
HALT-PKD Study A showed that a significant decrease in left ventricular mass index can be achieved by aggressive blood pressure control (95–110/60–75 mm Hg) with an ACE inhibitor alone or in combination with an ARB in the early stages of ADPKD with preserved renal function.43
A 5-year randomized clinical trial in children with borderline hypertension treated with an ACE inhibitor for effective control of blood pressure showed no change in left ventricular mass index or renal function.36,59
These results support starting ACE inhibitor therapy early in the disease process when blood pressure is still normal or borderline to prevent the progression of left ventricular hypertrophy or worsening kidney function.
Since FGF23 is directly involved in the causation of left ventricular hypertrophy, FGF receptors may be potential therapeutic targets to prevent left ventricular hypertrophy in ADPKD. An FGF receptor blocker was shown to decrease left ventricular hypertrophy in rats with chronic kidney disease without affecting blood pressure.55
INTRACRANIAL ANEURYSM IN ADPKD
Intracranial aneurysm is the most dangerous complication of ADPKD. When an aneurysm ruptures, the mortality rate is 4% to 7%, and 50% of survivors are left with residual neurologic deficits.5,60,61
In various studies, the prevalence of intracranial aneurysm in ADPKD ranged from 4% to 41.2%, compared with 1% in the general population.5,62,63 On follow-up ranging from 18 months to about 10 years, the incidence of new intracranial aneurysm was 2.6% to 13.3% in patients with previously normal findings on magnetic resonance angiography and 25% in patients with a history of intracranial aneurysm.62,64,65
The most common sites are the middle cerebral artery (45%), internal carotid artery (40.5%), and anterior communicating artery (35.1%).66 (The numbers add up to more than 100% because some patients have aneurysms in more than 1 site.) The mean size of a ruptured aneurysm was 6 mm per a recent systematic review.66 Intracranial aneurysms 6 mm or larger are at highest risk of rupture.66
SCREENING FOR INTRACRANIAL ANEURYSM
Timely screening and intervention for intracranial aneurysm is crucial to prevent death from intracranial hemorrhage.
Currently, there are no standard guidelines for screening and follow-up of intracranial aneurysm in ADPKD patients. However, some recommendations are available from the ADPKD Kidney Disease Improving Global Outcomes Controversies Conference3 and Kidney Health Australia—Caring for Australasians With Renal Impairment ADPKD guidelines67 (Figure 3).
Imaging tests
Magnetic resonance angiography (MRA) with gadolinium enhancement and computed tomographic angiography (CTA) are recommended for screening in ADPKD patients with normal renal function,67 but time-of-flight MRA without gadolinium is the imaging test of choice because it is noninvasive and poses no risk of nephrotoxicity or contrast allergy.3,68 Further, gadolinium should be avoided in patients whose eGFR is 30 mL/min/1.73 m2 or less because of risk of nephrogenic systemic sclerosis and fibrosis.67,68
The sensitivity of time-of-flight MRA screening for intracranial aneurysm varies depending on the size of aneurysm; 67% for those less than 3 mm, 79% for those 3 to 5 mm, and 95% for those larger than 5 mm.69 The sensitivity of CTA screening is 95% for aneurysms larger than 7 mm and 53% for those measuring 2 mm.70,71 The specificity of CTA screening was reported to be 98.9% overall.71
When to screen
Screening for intracranial aneurysm is recommended at the time of ADPKD diagnosis for all high-risk patients, ie, those who have a family history of intracranial hemorrhage or aneurysm in an affected first-degree relative.67 It is also recommended for ADPKD patients with a history of sudden-onset severe headache or neurologic symptoms.67 A third group for whom screening is recommended is ADPKD patients who have no family history of intracranial aneurysm or hemorrhage but who are at risk of poor outcome if an intracranial aneurysm ruptures (eg, those undergoing major elective surgery, with uncontrolled blood pressure, on anticoagulation, with a history of or current smoking, and airline pilots).67
Patients found to have an intracranial aneurysm on screening should be referred to a neurosurgeon and should undergo repeat MRA or CTA imaging every 6 to 24 months.3 High-risk ADPKD patients with normal findings on initial screening should have repeat MRA or CTA screening in 5 to 10 years unless they suffer from sudden-onset severe headache or neurologic symptoms.65,67
Both smoking and high blood pressure increase the risk of formation and growth of intracranial aneurysm. Hence, meticulous control of blood pressure and smoking cessation are recommended in ADPKD patients.3,67
CARDIAC VALVULAR ABNORMALITIES IN ADPKD
Of the valvular abnormalities that complicate ADPKD, the more common ones are mitral valve prolapse and mitral and aortic regurgitation. The less common ones are tricuspid valve prolapse and tricuspid regurgitation.72–74
The pathophysiology underlying these valvular abnormalities is unclear. However, defective collagen synthesis and myxomatous degeneration have been demonstrated in histopathologic examination of affected valvular tissue.75 Also, ACE gene polymorphism, especially the DD genotype, has been shown to be associated with cardiac valvular calcifications and valvular insufficiency.57
Lumiaho et al72 found a higher prevalence of mitral valve prolapse, mitral regurgitation, and left ventricular hypertrophy in patients with ADPKD type 1 (due to abnormalities in PDK1) than in unaffected family members and healthy controls. The investigators speculated that mitral regurgitation is caused by the high blood pressure observed in ADPKD type 1 patients, since hypertension causes left ventricular hypertrophy and left ventricular dilatation. The severity of renal failure was related to mitral regurgitation but not mitral valve prolapse.
Similarly, Gabow et al24 showed that there is no significant relationship between mitral valve prolapse and progression of renal disease in ADPKD.
Interestingly, Fick et al20 found that mitral valve prolapse has no significant effect on cardiovascular mortality.
CORONARY ARTERY DISEASE AND ANEURYSM IN ADPKD
Atherosclerosis sets in early in ADPKD, resulting in coronary artery disease and adverse cardiovascular outcomes.
Coronary flow velocity reserve is the ability of coronary arteries to dilate in response to myocardial oxygen demand. Atherosclerosis decreases this reserve in ADPKD patients, as shown in several studies.
Turkmen et al, in a series of studies,76–78 found that ADPKD patients had significantly less coronary flow velocity reserve, thicker carotid intima media (a surrogate marker of atherosclerosis), and greater insulin resistance than healthy controls. These findings imply that atherosclerosis begins very early in the course of ADPKD and has remarkable effects on cardiovascular morbidity and mortality.76
Aneurysm. Although the risk of extracranial aneurysm is higher with ADPKD, coronary artery aneurysm is uncommon. The pathogenesis of coronary aneurysm has been linked to abnormal expression of the proteins polycystin 1 and polycystin 2 in vascular smooth muscle.11,79 The PKD1 and PKD2 genes encode polycystin 1 and 2, respectively, in ADPKD. These polycystins are also expressed in the liver, kidneys, and myocardium and are involved in the regulation of intracellular calcium, stretch-activated ion channels, and vascular smooth muscle cell proliferation and apoptosis.11,16 Abnormally expressed polycystin in ADPKD therefore has an impact on arterial wall integrity, resulting in focal medial defects in the vasculature that eventually develop into micro- and macroaneurysms.11
Hadimeri et al79 found a higher prevalence of coronary aneurysm and ectasia in ADPKD patients than in controls. Most coronary aneurysms are smaller than 1 cm; however, a coronary aneurysm measuring 4 cm in diameter was found at autopsy of an ADPKD patient.11
Spontaneous coronary artery dissection is very rare in the general population, but Bobrie et al reported a case of it in an ADPKD patient.9
ATRIAL MYXOMA, CARDIOMYOPATHY, AND PERICARDIAL EFFUSION IN ADPKD
Atrial myxoma in ADPKD patients has been described in 2 case reports.15,80 However, the association of atrial myxoma with ADPKD is poorly understood and may be a coincidental finding.
Cardiomyopathy in ADPKD has been linked to abnormalities in the intracellular calcium pathway, although a clear picture of its involvement has yet to be established.
Paavola et al16 described the pathophysiology of ADPKD-associated cardiomyopathy in PKD2 mutant zebrafish lacking polycystin 2. These mutants showed decreased cardiac output and had atrioventricular blocks. The findings were attributed to abnormal intracellular calcium cycling. These findings correlated well with the frequent finding of idiopathic dilated cardiomyopathy in ADPKD patients, especially with PKD2 mutations.16 Also, 2 cases of dilated cardiomyopathy in ADPKD have been reported and thought to be related to PKD2 mutations.81,82
Pericardial effusion. Even though the exact pathophysiology of pericardial effusion in ADPKD is unknown, it has been theorized to be related to defects in connective tissue and extracellular matrix due to PKD1 and PKD2 mutations. These abnormalities increase the compliance and impair the recoil capacity of connective tissue, which results in unusual distention of the parietal pericardium. This abnormal distention of the parietal pericardium together with increased extracellular volume may lead to pericardial effusion.17
Qian et al17 found a higher prevalence of pericardial effusion in ADPKD patients. It was generally asymptomatic, and the cause was attributed to these connective tissue and extracellular matrix abnormalities.
EMERGING THERAPIES AND TESTS
Recent trials have investigated the effects of vasopressin receptor antagonists, specifically V2 receptor blockers in ADPKD and its complications.
Tolvaptan has been shown to slow the rate of increase in cyst size and total kidney volume.83,84 Also, a correlation between kidney size and diastolic blood pressure has been observed in ADPKD patients.85 Reducing cyst volume may reduce pressure effects and decrease renal ischemia, which in turn may reduce RAAS activation; however, the evidence to support this hypothesis is poor. A major clinical trial of tolvaptan in ADPKD patients showed no effect on blood pressure control, but the drug slowed the rate of increase in total kidney volume and fall in eGFR.83
Endothelin receptor antagonists are also in the preliminary stage of development for use in ADPKD. The effects of acute blockade of the renal endothelial system with bosentan were investigated in animal models by Hocher et al.28 This study showed a greater reduction in mean arterial pressure after bosentan administration, resulting in significantly decreased GFR and renal blood flow. Nonetheless, the mean arterial pressure-lowering effect of bosentan was more marked than the reductions in GFR and renal blood flow.
Raina et al,86 in a pilot cross-sectional analysis, showed that urinary excretion of ET-1 is increased in ADPKD patients, and may serve as a surrogate marker for ET-1 in renal tissue and a noninvasive marker of early kidney injury.
- Ha SK, Park CH, Kna JS, et al. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Yonsei Med J 1997; 38:111–116.
- Romao EA, Moyses Neto M, Teixeira SR, Muglia VF, Vieira-Neto OM, Dantas M. Renal and extrarenal manifestations of autosomal dominant polycystic kidney disease. Brazilian J Med Biol Res 2006; 39:533–538.
- Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2015; 88:17–27.
- Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis 2001; 38:777–784.
- Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev 2013; 9:2–11.
- Silverio A, Prota C, Di Maio M, et al. Aortic dissection in patients with autosomal dominant polycystic kidney disease: a series of two cases and a review of the literature. Nephrology 2015; 20:229–235.
- Ramineni R, Daniel GK. Use of endovascular stent-graft repair for type B aortic dissection in polycystic kidney disease. J Invas Cardiol 2010; 22:E171–E174.
- Courtois A, Nusgens BV, Delvenne P, et al. Dissection of iliac artery in a patient with autosomal dominant polycystic kidney disease: a case report. Aorta 2013; 1:123–125.
- Bobrie G, Brunet-Bourgin F, Alamowitch S, et al. Spontaneous artery dissection: is it part of the spectrum of autosomal dominant polycystic kidney disease? Nephrol Dial Transplant 1998; 13:2138–2141.
- Minami T, Karube N, Sakamoto A. [Thoracic aortic dissection complicating autosomal dominant polycystic kidney disease; report of a case]. Kyobu Geka 2009; 62:924–927.
- Ohara K, Kimura T, Karasawa T, et al. A large coronary aneurysm and its probable precursor lesions in a patient with autosomal dominant polycystic kidney disease: an implication for the process of aneurysmogenesis. Pathol Int 2012; 62:758–762.
- Al-Hakim W, Goldsmith DJ. Bilateral popliteal aneurysms complicating adult polycystic kidney disease in a patient with a marfanoid habitus. Postgrad Med J 2003; 79:474–475.
- Kanagasundaram NS, Perry EP, Turney JH. Aneurysm of the splenic artery in a patient with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1999; 14:183–184.
- Kang YR, Ahn JH, Kim KH, Choi YM, Choi J, Park JR. Multiple cardiovascular manifestations in a patient with autosomal dominant polycystic kidney disease. J Cardiovasc Ultrasound 2014; 22:144–147.
- Iglesias D, Fraga AR, Arrizurieta E, et al. Atrial myxoma in a woman with autosomal dominant polycystic kidney disease type 2. Am J Kidney Dis 1997; 29:164–165.
- Paavola J, Schliffke S, Rossetti S, et al. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J Mol Cell Cardiol 2013; 58:199–208.
- Qian Q, Hartman RP, King BF, Torres VE. Increased occurrence of pericardial effusion in patients with autosomal dominant polycystic kidney disease.
- Schievink WI, Torres VE, Wiebers DO, Huston J 3rd. Intracranial arterial dolichoectasia in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1997; 8:1298–1303.
- Pierre SA, Jaeger MT, Siemens DR. Intra-operative inferior vena cava syndrome in a patient with autosomal dominant polycystic kidney disease. World J Urol 2006; 24:110–112.
- Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995; 5:2048–2056.
- Helal I, Reed B, Mettler P, et al. Prevalence of cardiovascular events in patients with autosomal dominant polycystic kidney disease. Am J Nephrol 2012; 36:362–370.
- Calabrese G, Vagelli G, Cristofano C, Barsotti G. Behaviour of arterial pressure in different stages of polycystic kidney disease. Nephron 1982; 32:207–208.
- de Almeida EA, de Oliveira EI, Lopes JA, Almeida AG, Lopes MG, Prata MM. Ambulatory blood pressure measurement in young normotensive patients with autosomal dominant polycystic kidney disease. Rev Port Cardiol 2007; 26:235–243.
- Gabow PA, Johnson AM, Kaehny WD, et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 41:1311–1319.
- Bajrami V, Idrizi A, Roshi E, Barbullushi M. Association between nephrolithiasis, hypertension and obesity in polycystic kidney disease. Open Access Maced J Med Sci 2016; 4:43–46.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 1990; 323:1091–1096.
- Nakamura T, Ebihara I, Fukui M, et al. Increased endothelin and endothelin receptor mRNA expression in polycystic kidneys of cpk mice. J Am Soc Nephrol 1993; 4:1064–1072.
- Hocher B, Zart R, Schwarz A, et al. Renal endothelin system in polycystic kidney disease. J Am Soc Nephrol 1998; 9:1169–1177.
- Ong AC, Newby LJ, Dashwood MR. Expression and cellular localisation of renal endothelin-1 and endothelin receptor subtypes in autosomal-dominant polycystic kidney disease. Nephron Exper Nephrol 2003; 93:e80.
- Nash DA Jr. Hypertension in polycystic kidney disease without renal failure. Arch Intern Med 1977; 137:1571–1575.
- D’Angelo A, Mioni G, Ossi E, Lupo A, Valvo E, Maschio G. Alterations in renal tubular sodium and water transport in polycystic kidney disease. Clin Nephrol 1975; 3:99–105.
- Ettinger A, Kahn PC, Wise HM Jr. The importance of selective renal angiography in the diagnosis of polycystic disease. J Urol 1969; 102:156–161.
- Cornell SH. Angiography in polycystic disease of the kidneys. J Urol 1970; 103:24–26.
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2009; 20:1888–1893.
- Chapman AB, Guay-Woodford LM, Grantham JJ, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int 2003; 64:1035–1045.
- Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Prospective change in renal volume and function in children with ADPKD. Clin J Am Soc Nephrol 2009; 4:820–829.
- Loghman-Adham M, Soto CE, Inagami T, Cassis L. The intrarenal renin-angiotensin system in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol 2004; 287:F775–F788.
- Torres VE, Donovan KA, Scicli G, et al. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 42:364–373.
- Graham PC, Lindop GB. The anatomy of the renin-secreting cell in adult polycystic kidney disease. Kidney Int 1988; 33:1084–1090.
- Cerasola G, Vecchi M, Mule G, et al. Sympathetic activity and blood pressure pattern in autosomal dominant polycystic kidney disease hypertensives. Am J Nephrol 1998; 18:391–398.
- Valvo E, Gammaro L, Tessitore N, et al. Hypertension of polycystic kidney disease: mechanisms and hemodynamic alterations. Am J Nephrol 1985; 5:176–181.
- Marlais M, Cuthell O, Langan D, Dudley J, Sinha MD, Winyard PJ. Hypertension in autosomal dominant polycystic kidney disease: a meta-analysis. Arch Dis Child 2016; 101:1142–1147.
- Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2255–2266.
- Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2267–2276.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Ritchie LD, Campbell NC, Murchie P. New NICE guidelines for hypertension. BMJ 2011; 343:d5644.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- Rangan GK, Alexander SI, Campbell KL, et al. KHA-CARI guideline recommendations for the diagnosis and management of autosomal dominant polycystic kidney disease. Nephrology 2016; 21:705–716.
- Helal I, Al-Rowaie F, Abderrahim E, Kheder A. Update on pathogenesis, management, and treatment of hypertension in autosomal dominant polycystic kidney disease. Saudi J Kidney Dis Transpl 2017; 28:253–260.
- Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis 2011; 57:856–862.
- Zeltner R, Poliak R, Stiasny B, Schmieder RE, Schulze BD. Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2008; 23:573–579.
- Clark LA, Whitmire S, Patton S, Clark C, Blanchette CM, Howden R. Cost-effectiveness of angiotensin-converting enzyme inhibitors versus angiotensin II receptor blockers as first-line treatment in autosomal dominant polycystic kidney disease. J Med Econ 2017:1–17.
- Li Kam Wa TC, Macnicol AM, Watson ML. Ambulatory blood pressure in hypertensive patients with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1997; 12:2075–2080.
- Valero FA, Martinez-Vea A, Bardaji A, et al. Ambulatory blood pressure and left ventricular mass in normotensive patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1999; 10:1020–1026.
- Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011; 121:4393–4408.
- Yildiz A, Gul CB, Ersoy A, Asiltas B, Ermurat S, Dogan S, et al. Arterial dysfunction in early autosomal dominant polycystic kidney disease independent of fibroblast growth factor 23. Iranian J Kidney Dis 2014; 8:443–449.
- Wanic-Kossowska M, Posnik B, Kobelski M, et al. The polymorphism of the ACE gene affects left ventricular hypertrophy and causes disturbances in left ventricular systolic/diastolic function in patients with autosomal dominant polycystic kidney disease. ScientificWorldJournal 2014; 2014:707658.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Cadnapaphornchai MA. Hypertension in children with autosomal dominant polycystic kidney disease (ADPKD). Curr Hypertens Rev 2013; 9:21–26.
- Schievink WI, Prendergast V, Zabramski JM. Rupture of a previously documented small asymptomatic intracranial aneurysm in a patient with autosomal dominant polycystic kidney disease. Case report. J Neurosurg 1998; 89:479–482.
- Graf S, Schischma A, Eberhardt KE, Istel R, Stiasny B, Schulze BD. Intracranial aneurysms and dolichoectasia in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2002; 17:819–823.
- Nakajima F, Shibahara N, Arai M, Gohji K, Ueda H, Katsuoka Y. Intracranial aneurysms and autosomal dominant polycystic kidney disease: followup study by magnetic resonance angiography. J Urol 2000; 164:311–313.
- Wakabayashi T, Fujita S, Ohbora Y, Suyama T, Tamaki N, Matsumoto S. Polycystic kidney disease and intracranial aneurysms. Early angiographic diagnosis and early operation for the unruptured aneurysm. J Neurosurg 1983; 58:488–491.
- Belz MM, Fick-Brosnahan GM, Hughes RL, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63:1824–1830.
- Schrier RW, Belz MM, Johnson AM, et al. Repeat imaging for intracranial aneurysms in patients with autosomal dominant polycystic kidney disease with initially negative studies: a prospective ten-year follow-up. J Am Soc Nephrol 2004; 15:1023–1028.
- Cagnazzo F, Gambacciani C, Morganti R, Perrini P. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: prevalence, risk of rupture, and management. A systematic review. Acta Neurochirurgica 2017; 5:811–821.
- Lee VW, Dexter MA, Mai J, Vladica P, Lopez-Vargas P, Rangan GK. KHA-CARI autosomal dominant polycystic kidney disease guideline: management of intracranial aneurysms. Semin Nephrol 2015; 35:612–617.
- Rozenfeld MN, Ansari SA, Shaibani A, Russell EJ, Mohan P, Hurley MC. Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms? AJNR Am J Neuroradiol 2014; 35:3–9.
- Hiratsuka Y, Miki H, Kiriyama I, et al. Diagnosis of unruptured intracranial aneurysms: 3T MR angiography versus 64-channel multi-detector row CT angiography. Magn Reson Med Sci 2008; 7:169–178.
- van Gelder JM. Computed tomographic angiography for detecting cerebral aneurysms: implications of aneurysm size distribution for the sensitivity, specificity, and likelihood ratios. Neurosurgery 2003; 53:597–605.
- Villablanca JP, Jahan R, Hooshi P, et al. Detection and characterization of very small cerebral aneurysms by using 2D and 3D helical CT angiography. AJNR Am J Neuroradiol 2002; 23:1187–1198.
- Lumiaho A, Ikäheimo R, Miettinen R, et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis 2001; 38:1208–1216.
- Timio M, Monarca C, Pede S, Gentili S, Verdura C, Lolli S. The spectrum of cardiovascular abnormalities in autosomal dominant polycystic kidney disease: a 10-year follow-up in a five-generation kindred. Clin Nephrol 1992; 37:245–251.
- Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988; 319:907–912.
- Leier CV, Baker PB, Kilman JW, Wooley CF. Cardiovascular abnormalities associated with adult polycystic kidney disease. Ann Intern Med 1984; 100:683–688.
- Turkmen K, Oflaz H, Uslu B, et al. Coronary flow velocity reserve and carotid intima media thickness in patients with autosomal dominant polycystic kidney disease: from impaired tubules to impaired carotid and coronary arteries. Clin J Am Soc Nephrol 2008; 3:986–991.
- Turkmen K, Tufan F, Alpay N, et al. Insulin resistance and coronary flow velocity reserve in patients with autosomal dominant polycystic kidney disease. Intern Med J 2012; 42:146–153.
- Turkmen K, Tufan F, Selcuk E, Akpinar T, Oflaz H, Ecder T. Neutrophil-to-lymphocyte ratio, insulin resistance, and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Indian J Nephrol 2013; 23:34–40.
- Hadimeri H, Lamm C, Nyberg G. Coronary aneurysms in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1998; 9:837–841.
- Earle K, Hoffbrand BI. Adult dominant polycystic kidney disease and atrial myxoma. Nephron 1989; 52:197.
- Chung BM, Chong S, Lee W-S, Hwang S-N. Autosomal dominant polycystic kidney disease combined with intracranial aneurysm and dilated cardiomyopathy: a case report. J Korean Soc Radiol 2014; 71:84–88.
- Mariathasan DAL, Kumanan T. Adult polycystic kidney disease and idiopathic dilated cardiomyopathy: a rare genetic association. J Ceylon Coll Physicians 2016: 46:42–44.
- Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407–2018.
- Higashihara E, Torres VE, Chapman AB, et al. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol 2011; 6:2499–2507.
- Sans Atxer L, Roca-Cusachs A, Torra R, et al. Relationship between renal size and blood pressure profile in patients with autosomal dominant polycystic kidney disease without renal failure. Nefrologia 2010; 30:567–572. In Spanish.
- Raina R, Lou L, Berger B, et al. Relationship of urinary endothelin-1 with estimated glomerular filtration rate in autosomal dominant polycystic kidney disease: a pilot cross-sectional analysis. BMC Nephrol 2016; 17:22.
- Ha SK, Park CH, Kna JS, et al. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Yonsei Med J 1997; 38:111–116.
- Romao EA, Moyses Neto M, Teixeira SR, Muglia VF, Vieira-Neto OM, Dantas M. Renal and extrarenal manifestations of autosomal dominant polycystic kidney disease. Brazilian J Med Biol Res 2006; 39:533–538.
- Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2015; 88:17–27.
- Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis 2001; 38:777–784.
- Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev 2013; 9:2–11.
- Silverio A, Prota C, Di Maio M, et al. Aortic dissection in patients with autosomal dominant polycystic kidney disease: a series of two cases and a review of the literature. Nephrology 2015; 20:229–235.
- Ramineni R, Daniel GK. Use of endovascular stent-graft repair for type B aortic dissection in polycystic kidney disease. J Invas Cardiol 2010; 22:E171–E174.
- Courtois A, Nusgens BV, Delvenne P, et al. Dissection of iliac artery in a patient with autosomal dominant polycystic kidney disease: a case report. Aorta 2013; 1:123–125.
- Bobrie G, Brunet-Bourgin F, Alamowitch S, et al. Spontaneous artery dissection: is it part of the spectrum of autosomal dominant polycystic kidney disease? Nephrol Dial Transplant 1998; 13:2138–2141.
- Minami T, Karube N, Sakamoto A. [Thoracic aortic dissection complicating autosomal dominant polycystic kidney disease; report of a case]. Kyobu Geka 2009; 62:924–927.
- Ohara K, Kimura T, Karasawa T, et al. A large coronary aneurysm and its probable precursor lesions in a patient with autosomal dominant polycystic kidney disease: an implication for the process of aneurysmogenesis. Pathol Int 2012; 62:758–762.
- Al-Hakim W, Goldsmith DJ. Bilateral popliteal aneurysms complicating adult polycystic kidney disease in a patient with a marfanoid habitus. Postgrad Med J 2003; 79:474–475.
- Kanagasundaram NS, Perry EP, Turney JH. Aneurysm of the splenic artery in a patient with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1999; 14:183–184.
- Kang YR, Ahn JH, Kim KH, Choi YM, Choi J, Park JR. Multiple cardiovascular manifestations in a patient with autosomal dominant polycystic kidney disease. J Cardiovasc Ultrasound 2014; 22:144–147.
- Iglesias D, Fraga AR, Arrizurieta E, et al. Atrial myxoma in a woman with autosomal dominant polycystic kidney disease type 2. Am J Kidney Dis 1997; 29:164–165.
- Paavola J, Schliffke S, Rossetti S, et al. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J Mol Cell Cardiol 2013; 58:199–208.
- Qian Q, Hartman RP, King BF, Torres VE. Increased occurrence of pericardial effusion in patients with autosomal dominant polycystic kidney disease.
- Schievink WI, Torres VE, Wiebers DO, Huston J 3rd. Intracranial arterial dolichoectasia in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1997; 8:1298–1303.
- Pierre SA, Jaeger MT, Siemens DR. Intra-operative inferior vena cava syndrome in a patient with autosomal dominant polycystic kidney disease. World J Urol 2006; 24:110–112.
- Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995; 5:2048–2056.
- Helal I, Reed B, Mettler P, et al. Prevalence of cardiovascular events in patients with autosomal dominant polycystic kidney disease. Am J Nephrol 2012; 36:362–370.
- Calabrese G, Vagelli G, Cristofano C, Barsotti G. Behaviour of arterial pressure in different stages of polycystic kidney disease. Nephron 1982; 32:207–208.
- de Almeida EA, de Oliveira EI, Lopes JA, Almeida AG, Lopes MG, Prata MM. Ambulatory blood pressure measurement in young normotensive patients with autosomal dominant polycystic kidney disease. Rev Port Cardiol 2007; 26:235–243.
- Gabow PA, Johnson AM, Kaehny WD, et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 41:1311–1319.
- Bajrami V, Idrizi A, Roshi E, Barbullushi M. Association between nephrolithiasis, hypertension and obesity in polycystic kidney disease. Open Access Maced J Med Sci 2016; 4:43–46.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 1990; 323:1091–1096.
- Nakamura T, Ebihara I, Fukui M, et al. Increased endothelin and endothelin receptor mRNA expression in polycystic kidneys of cpk mice. J Am Soc Nephrol 1993; 4:1064–1072.
- Hocher B, Zart R, Schwarz A, et al. Renal endothelin system in polycystic kidney disease. J Am Soc Nephrol 1998; 9:1169–1177.
- Ong AC, Newby LJ, Dashwood MR. Expression and cellular localisation of renal endothelin-1 and endothelin receptor subtypes in autosomal-dominant polycystic kidney disease. Nephron Exper Nephrol 2003; 93:e80.
- Nash DA Jr. Hypertension in polycystic kidney disease without renal failure. Arch Intern Med 1977; 137:1571–1575.
- D’Angelo A, Mioni G, Ossi E, Lupo A, Valvo E, Maschio G. Alterations in renal tubular sodium and water transport in polycystic kidney disease. Clin Nephrol 1975; 3:99–105.
- Ettinger A, Kahn PC, Wise HM Jr. The importance of selective renal angiography in the diagnosis of polycystic disease. J Urol 1969; 102:156–161.
- Cornell SH. Angiography in polycystic disease of the kidneys. J Urol 1970; 103:24–26.
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2009; 20:1888–1893.
- Chapman AB, Guay-Woodford LM, Grantham JJ, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int 2003; 64:1035–1045.
- Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Prospective change in renal volume and function in children with ADPKD. Clin J Am Soc Nephrol 2009; 4:820–829.
- Loghman-Adham M, Soto CE, Inagami T, Cassis L. The intrarenal renin-angiotensin system in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol 2004; 287:F775–F788.
- Torres VE, Donovan KA, Scicli G, et al. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 42:364–373.
- Graham PC, Lindop GB. The anatomy of the renin-secreting cell in adult polycystic kidney disease. Kidney Int 1988; 33:1084–1090.
- Cerasola G, Vecchi M, Mule G, et al. Sympathetic activity and blood pressure pattern in autosomal dominant polycystic kidney disease hypertensives. Am J Nephrol 1998; 18:391–398.
- Valvo E, Gammaro L, Tessitore N, et al. Hypertension of polycystic kidney disease: mechanisms and hemodynamic alterations. Am J Nephrol 1985; 5:176–181.
- Marlais M, Cuthell O, Langan D, Dudley J, Sinha MD, Winyard PJ. Hypertension in autosomal dominant polycystic kidney disease: a meta-analysis. Arch Dis Child 2016; 101:1142–1147.
- Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2255–2266.
- Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2267–2276.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Ritchie LD, Campbell NC, Murchie P. New NICE guidelines for hypertension. BMJ 2011; 343:d5644.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- Rangan GK, Alexander SI, Campbell KL, et al. KHA-CARI guideline recommendations for the diagnosis and management of autosomal dominant polycystic kidney disease. Nephrology 2016; 21:705–716.
- Helal I, Al-Rowaie F, Abderrahim E, Kheder A. Update on pathogenesis, management, and treatment of hypertension in autosomal dominant polycystic kidney disease. Saudi J Kidney Dis Transpl 2017; 28:253–260.
- Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis 2011; 57:856–862.
- Zeltner R, Poliak R, Stiasny B, Schmieder RE, Schulze BD. Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2008; 23:573–579.
- Clark LA, Whitmire S, Patton S, Clark C, Blanchette CM, Howden R. Cost-effectiveness of angiotensin-converting enzyme inhibitors versus angiotensin II receptor blockers as first-line treatment in autosomal dominant polycystic kidney disease. J Med Econ 2017:1–17.
- Li Kam Wa TC, Macnicol AM, Watson ML. Ambulatory blood pressure in hypertensive patients with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1997; 12:2075–2080.
- Valero FA, Martinez-Vea A, Bardaji A, et al. Ambulatory blood pressure and left ventricular mass in normotensive patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1999; 10:1020–1026.
- Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011; 121:4393–4408.
- Yildiz A, Gul CB, Ersoy A, Asiltas B, Ermurat S, Dogan S, et al. Arterial dysfunction in early autosomal dominant polycystic kidney disease independent of fibroblast growth factor 23. Iranian J Kidney Dis 2014; 8:443–449.
- Wanic-Kossowska M, Posnik B, Kobelski M, et al. The polymorphism of the ACE gene affects left ventricular hypertrophy and causes disturbances in left ventricular systolic/diastolic function in patients with autosomal dominant polycystic kidney disease. ScientificWorldJournal 2014; 2014:707658.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Cadnapaphornchai MA. Hypertension in children with autosomal dominant polycystic kidney disease (ADPKD). Curr Hypertens Rev 2013; 9:21–26.
- Schievink WI, Prendergast V, Zabramski JM. Rupture of a previously documented small asymptomatic intracranial aneurysm in a patient with autosomal dominant polycystic kidney disease. Case report. J Neurosurg 1998; 89:479–482.
- Graf S, Schischma A, Eberhardt KE, Istel R, Stiasny B, Schulze BD. Intracranial aneurysms and dolichoectasia in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2002; 17:819–823.
- Nakajima F, Shibahara N, Arai M, Gohji K, Ueda H, Katsuoka Y. Intracranial aneurysms and autosomal dominant polycystic kidney disease: followup study by magnetic resonance angiography. J Urol 2000; 164:311–313.
- Wakabayashi T, Fujita S, Ohbora Y, Suyama T, Tamaki N, Matsumoto S. Polycystic kidney disease and intracranial aneurysms. Early angiographic diagnosis and early operation for the unruptured aneurysm. J Neurosurg 1983; 58:488–491.
- Belz MM, Fick-Brosnahan GM, Hughes RL, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63:1824–1830.
- Schrier RW, Belz MM, Johnson AM, et al. Repeat imaging for intracranial aneurysms in patients with autosomal dominant polycystic kidney disease with initially negative studies: a prospective ten-year follow-up. J Am Soc Nephrol 2004; 15:1023–1028.
- Cagnazzo F, Gambacciani C, Morganti R, Perrini P. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: prevalence, risk of rupture, and management. A systematic review. Acta Neurochirurgica 2017; 5:811–821.
- Lee VW, Dexter MA, Mai J, Vladica P, Lopez-Vargas P, Rangan GK. KHA-CARI autosomal dominant polycystic kidney disease guideline: management of intracranial aneurysms. Semin Nephrol 2015; 35:612–617.
- Rozenfeld MN, Ansari SA, Shaibani A, Russell EJ, Mohan P, Hurley MC. Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms? AJNR Am J Neuroradiol 2014; 35:3–9.
- Hiratsuka Y, Miki H, Kiriyama I, et al. Diagnosis of unruptured intracranial aneurysms: 3T MR angiography versus 64-channel multi-detector row CT angiography. Magn Reson Med Sci 2008; 7:169–178.
- van Gelder JM. Computed tomographic angiography for detecting cerebral aneurysms: implications of aneurysm size distribution for the sensitivity, specificity, and likelihood ratios. Neurosurgery 2003; 53:597–605.
- Villablanca JP, Jahan R, Hooshi P, et al. Detection and characterization of very small cerebral aneurysms by using 2D and 3D helical CT angiography. AJNR Am J Neuroradiol 2002; 23:1187–1198.
- Lumiaho A, Ikäheimo R, Miettinen R, et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis 2001; 38:1208–1216.
- Timio M, Monarca C, Pede S, Gentili S, Verdura C, Lolli S. The spectrum of cardiovascular abnormalities in autosomal dominant polycystic kidney disease: a 10-year follow-up in a five-generation kindred. Clin Nephrol 1992; 37:245–251.
- Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988; 319:907–912.
- Leier CV, Baker PB, Kilman JW, Wooley CF. Cardiovascular abnormalities associated with adult polycystic kidney disease. Ann Intern Med 1984; 100:683–688.
- Turkmen K, Oflaz H, Uslu B, et al. Coronary flow velocity reserve and carotid intima media thickness in patients with autosomal dominant polycystic kidney disease: from impaired tubules to impaired carotid and coronary arteries. Clin J Am Soc Nephrol 2008; 3:986–991.
- Turkmen K, Tufan F, Alpay N, et al. Insulin resistance and coronary flow velocity reserve in patients with autosomal dominant polycystic kidney disease. Intern Med J 2012; 42:146–153.
- Turkmen K, Tufan F, Selcuk E, Akpinar T, Oflaz H, Ecder T. Neutrophil-to-lymphocyte ratio, insulin resistance, and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Indian J Nephrol 2013; 23:34–40.
- Hadimeri H, Lamm C, Nyberg G. Coronary aneurysms in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1998; 9:837–841.
- Earle K, Hoffbrand BI. Adult dominant polycystic kidney disease and atrial myxoma. Nephron 1989; 52:197.
- Chung BM, Chong S, Lee W-S, Hwang S-N. Autosomal dominant polycystic kidney disease combined with intracranial aneurysm and dilated cardiomyopathy: a case report. J Korean Soc Radiol 2014; 71:84–88.
- Mariathasan DAL, Kumanan T. Adult polycystic kidney disease and idiopathic dilated cardiomyopathy: a rare genetic association. J Ceylon Coll Physicians 2016: 46:42–44.
- Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407–2018.
- Higashihara E, Torres VE, Chapman AB, et al. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol 2011; 6:2499–2507.
- Sans Atxer L, Roca-Cusachs A, Torra R, et al. Relationship between renal size and blood pressure profile in patients with autosomal dominant polycystic kidney disease without renal failure. Nefrologia 2010; 30:567–572. In Spanish.
- Raina R, Lou L, Berger B, et al. Relationship of urinary endothelin-1 with estimated glomerular filtration rate in autosomal dominant polycystic kidney disease: a pilot cross-sectional analysis. BMC Nephrol 2016; 17:22.
KEY POINTS
- Hypertension and left ventricular hypertrophy are common complications of ADPKD.
- Cardiovascular disease is a major cause of morbidity and death in ADPKD.
- Early diagnosis and aggressive management of high blood pressure, specifically with agents that block the renin-angiotensin-aldosterone system, are necessary to prevent left ventricular hypertrophy and progression of renal failure in ADPKD.
- Timely screening and intervention for intracranial aneurysm would lessen the rates of morbidity and death from intracranial hemorrhage.
AKI doubles risk of death for those with acute pancreatitis
CHICAGO – Acute kidney injury (AKI) doubles the risk of death among patients hospitalized for acute pancreatitis, Kalpit Devani, MD, reported at the annual Digestive Disease Week®.
This severe complication of acute pancreatitis also significantly increases the length of stay and drives up hospital costs, said Dr. Devani of East Tennessee State University, Johnson City. Fortunately, although the risks associated with it remain high, death from AKI in the setting of acute pancreatitis has decreased significantly, from a high of 17% in 2002 to 6.4% in 2012, Dr. Devani determined in his database review.
“Increasing awareness and prompt diagnosis of AKI could be the reason for the increasing trend of prevalence of AKI in acute pancreatitis patients,” he said in an interview. “Decreasing mortality can be related to adherence to recent advances in the management approach of acute pancreatitis, such as early (within 24 hours) and aggressive intravenous hydration and early enteral feeding.”
Dr. Devani examined these trends in data extracted from the National Inpatient Sample, 2002-2012. During that 10-year period, almost 3.5 million adults were hospitalized for acute pancreatitis. These patients were a mean of 53 years old, and half were women. Their mean length of stay was just over 5 days, at a mean cost of about $12,000. Of these, 273,687 (7.9%) also developed AKI.
There were some significant differences between those who did and did not develop AKI. AKI patients were significantly older (61 vs. 53 years), and less likely to be women (43% vs. 51%). They had a higher Charlson Comorbidity Index score (1.49 vs. 0.84). They were also significantly more likely to develop a number of complications, including systemic inflammatory response syndrome (2% vs. 0.4%), septic shock (6% vs. 0.3%), sepsis (8.7% vs. 1.4%), acute respiratory failure (18% vs. 2%), and electrolyte disorder (72% vs. 30%).
Not surprisingly, their length of stay was significantly longer (10 vs. 5 days), as was hospitalization cost ($25,923 vs. $10,889). Mortality was much higher, at almost 9% vs. 0.7%.
In a propensity matching analysis, Dr. Devani matched 53,000 pairs of acute pancreatitis patients with and without AKI. This determined that those with AKI faced a doubling in the risk of in-hospital mortality.
He also examined temporal trends with regard to the complication. The rate of diagnosed AKI in hospitalized acute pancreatitis cases rose dramatically, from 4% in 2002 to 11.6% in 2012. However, mortality in acute pancreatitis patients decreased among both those with AKI (17%-6%) and those without (1%-0.4%).
The mean length of stay in patients with AKI and pancreatitis likewise fell, from 14.8 to 8.6 days. Not surprisingly, total hospitalization cost for these patients fell as well ($42,975-$20,716).
Among pancreatitis patients without AKI, length of stay and costs declined, although not as dramatically as they did among AKI patients (6-5 days; $13,654-$10,895).
Dr. Devani had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
CHICAGO – Acute kidney injury (AKI) doubles the risk of death among patients hospitalized for acute pancreatitis, Kalpit Devani, MD, reported at the annual Digestive Disease Week®.
This severe complication of acute pancreatitis also significantly increases the length of stay and drives up hospital costs, said Dr. Devani of East Tennessee State University, Johnson City. Fortunately, although the risks associated with it remain high, death from AKI in the setting of acute pancreatitis has decreased significantly, from a high of 17% in 2002 to 6.4% in 2012, Dr. Devani determined in his database review.
“Increasing awareness and prompt diagnosis of AKI could be the reason for the increasing trend of prevalence of AKI in acute pancreatitis patients,” he said in an interview. “Decreasing mortality can be related to adherence to recent advances in the management approach of acute pancreatitis, such as early (within 24 hours) and aggressive intravenous hydration and early enteral feeding.”
Dr. Devani examined these trends in data extracted from the National Inpatient Sample, 2002-2012. During that 10-year period, almost 3.5 million adults were hospitalized for acute pancreatitis. These patients were a mean of 53 years old, and half were women. Their mean length of stay was just over 5 days, at a mean cost of about $12,000. Of these, 273,687 (7.9%) also developed AKI.
There were some significant differences between those who did and did not develop AKI. AKI patients were significantly older (61 vs. 53 years), and less likely to be women (43% vs. 51%). They had a higher Charlson Comorbidity Index score (1.49 vs. 0.84). They were also significantly more likely to develop a number of complications, including systemic inflammatory response syndrome (2% vs. 0.4%), septic shock (6% vs. 0.3%), sepsis (8.7% vs. 1.4%), acute respiratory failure (18% vs. 2%), and electrolyte disorder (72% vs. 30%).
Not surprisingly, their length of stay was significantly longer (10 vs. 5 days), as was hospitalization cost ($25,923 vs. $10,889). Mortality was much higher, at almost 9% vs. 0.7%.
In a propensity matching analysis, Dr. Devani matched 53,000 pairs of acute pancreatitis patients with and without AKI. This determined that those with AKI faced a doubling in the risk of in-hospital mortality.
He also examined temporal trends with regard to the complication. The rate of diagnosed AKI in hospitalized acute pancreatitis cases rose dramatically, from 4% in 2002 to 11.6% in 2012. However, mortality in acute pancreatitis patients decreased among both those with AKI (17%-6%) and those without (1%-0.4%).
The mean length of stay in patients with AKI and pancreatitis likewise fell, from 14.8 to 8.6 days. Not surprisingly, total hospitalization cost for these patients fell as well ($42,975-$20,716).
Among pancreatitis patients without AKI, length of stay and costs declined, although not as dramatically as they did among AKI patients (6-5 days; $13,654-$10,895).
Dr. Devani had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
CHICAGO – Acute kidney injury (AKI) doubles the risk of death among patients hospitalized for acute pancreatitis, Kalpit Devani, MD, reported at the annual Digestive Disease Week®.
This severe complication of acute pancreatitis also significantly increases the length of stay and drives up hospital costs, said Dr. Devani of East Tennessee State University, Johnson City. Fortunately, although the risks associated with it remain high, death from AKI in the setting of acute pancreatitis has decreased significantly, from a high of 17% in 2002 to 6.4% in 2012, Dr. Devani determined in his database review.
“Increasing awareness and prompt diagnosis of AKI could be the reason for the increasing trend of prevalence of AKI in acute pancreatitis patients,” he said in an interview. “Decreasing mortality can be related to adherence to recent advances in the management approach of acute pancreatitis, such as early (within 24 hours) and aggressive intravenous hydration and early enteral feeding.”
Dr. Devani examined these trends in data extracted from the National Inpatient Sample, 2002-2012. During that 10-year period, almost 3.5 million adults were hospitalized for acute pancreatitis. These patients were a mean of 53 years old, and half were women. Their mean length of stay was just over 5 days, at a mean cost of about $12,000. Of these, 273,687 (7.9%) also developed AKI.
There were some significant differences between those who did and did not develop AKI. AKI patients were significantly older (61 vs. 53 years), and less likely to be women (43% vs. 51%). They had a higher Charlson Comorbidity Index score (1.49 vs. 0.84). They were also significantly more likely to develop a number of complications, including systemic inflammatory response syndrome (2% vs. 0.4%), septic shock (6% vs. 0.3%), sepsis (8.7% vs. 1.4%), acute respiratory failure (18% vs. 2%), and electrolyte disorder (72% vs. 30%).
Not surprisingly, their length of stay was significantly longer (10 vs. 5 days), as was hospitalization cost ($25,923 vs. $10,889). Mortality was much higher, at almost 9% vs. 0.7%.
In a propensity matching analysis, Dr. Devani matched 53,000 pairs of acute pancreatitis patients with and without AKI. This determined that those with AKI faced a doubling in the risk of in-hospital mortality.
He also examined temporal trends with regard to the complication. The rate of diagnosed AKI in hospitalized acute pancreatitis cases rose dramatically, from 4% in 2002 to 11.6% in 2012. However, mortality in acute pancreatitis patients decreased among both those with AKI (17%-6%) and those without (1%-0.4%).
The mean length of stay in patients with AKI and pancreatitis likewise fell, from 14.8 to 8.6 days. Not surprisingly, total hospitalization cost for these patients fell as well ($42,975-$20,716).
Among pancreatitis patients without AKI, length of stay and costs declined, although not as dramatically as they did among AKI patients (6-5 days; $13,654-$10,895).
Dr. Devani had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
AT DDW
Key clinical point:
Major finding: Mortality among those with AKI was 9% vs. 0.7% among those without. After controlling for confounders, the risk of death was doubled.
Data source: A 10-year National Inpatient Sample database review comprising 3.5 million patients with pancreatitis.
Disclosures: Dr. Devani had no financial disclosures.
Alternating therapy in renal cell carcinoma fails to show an advantage
There was no efficacy or safety advantage for alternating everolimus with pazopanib over pazopanib alone in patients with metastatic or locally advanced clear cell renal cell carcinoma (ccRCC), according to a newly published randomized trial.
The study hypothesis was that alternating the two drugs would improve outcomes and reduce toxicity, but differences between arms for the major outcomes were not clinically significant, according to results of a multicenter trial led by Geert A. Cirkel, MD, of the department of medical oncology, University Medical Center, Utrecht, the Netherlands.
Investigators randomized 101 patients with histologically confirmed ccRCC to receive 8 weeks of pazopanib in a daily dose of 800 mg alternated with 8 weeks of everolimus in a daily dose of 10 mg or 800 mg per day of continuous pazopanib. Patients remained on either regimen until disease progression.
Median time until first progression or death was 7.4 months for the experimental alternating arm versus 9.4 months for the control arm of continuous single-agent pazopanib (P = .37), Dr. Cirkel and associates reported (JAMA Onc. 2017 Apr 1. doi: 10.1001/jamaoncol.2016.5202).
Progression-free survival after starting on a second-line therapy was 20.2 months for the alternating treatment vs. 14.5 months for the control, but the confidence intervals were wide, and the difference was not significant (P = .86).
There was no apparent toxicity or tolerability advantage from alternating therapy. Nearly 40% of patients in both arms required pazopanib dose reductions, while 14% in the alternating arm also required an everolimus dose reduction. The incidence of serious adverse events possibly related to treatment was comparable between arms.
Quality of life was measured with several tools, including the Functional Assessment of Cancer Therapy – Kidney Symptom Index Disease-Related Symptoms (FKSI-DRS), but no significant differences between treatment arms were observed in any measure.
Current guidelines recommend pazopanib, which is a tyrosine kinase inhibitor of the vascular endothelial growth factor receptor, as a first-line therapy in ccRCC. Everolimus, an inhibitor of mammalian target of rapamycin, is recommended in the second-line setting. Noting that resistance to pazopanib has been shown to be reversible after a period of withdrawal in experimental studies, the authors had speculated an on-off strategy with everolimus might better preserve the efficacy of pazopanib, providing longer periods of disease control. They had also hypothesized that the cumulative adverse events might be less if the drugs were sequenced, allowing recovery from each set of drug-specific adverse events.
Several potential explanations were offered for the lack of improved efficacy from alternating everolimus with pazopanib. For one, the improved activity of pazopanib after withdrawal in experimental models was observed after drug-free periods. The authors questioned whether a period of tumor regrowth may be needed in order to overcome pazopanib resistance.
The study may still have supported the use of an alternating regimen if the alternating therapy had led to a significantly improved quality of life, but the authors found none, a outcome that they characterized as unexpected. They concluded that there are no apparent advantages for the alternating regimen of pazopanib and everolimus relative to pazopanib alone.
There was no efficacy or safety advantage for alternating everolimus with pazopanib over pazopanib alone in patients with metastatic or locally advanced clear cell renal cell carcinoma (ccRCC), according to a newly published randomized trial.
The study hypothesis was that alternating the two drugs would improve outcomes and reduce toxicity, but differences between arms for the major outcomes were not clinically significant, according to results of a multicenter trial led by Geert A. Cirkel, MD, of the department of medical oncology, University Medical Center, Utrecht, the Netherlands.
Investigators randomized 101 patients with histologically confirmed ccRCC to receive 8 weeks of pazopanib in a daily dose of 800 mg alternated with 8 weeks of everolimus in a daily dose of 10 mg or 800 mg per day of continuous pazopanib. Patients remained on either regimen until disease progression.
Median time until first progression or death was 7.4 months for the experimental alternating arm versus 9.4 months for the control arm of continuous single-agent pazopanib (P = .37), Dr. Cirkel and associates reported (JAMA Onc. 2017 Apr 1. doi: 10.1001/jamaoncol.2016.5202).
Progression-free survival after starting on a second-line therapy was 20.2 months for the alternating treatment vs. 14.5 months for the control, but the confidence intervals were wide, and the difference was not significant (P = .86).
There was no apparent toxicity or tolerability advantage from alternating therapy. Nearly 40% of patients in both arms required pazopanib dose reductions, while 14% in the alternating arm also required an everolimus dose reduction. The incidence of serious adverse events possibly related to treatment was comparable between arms.
Quality of life was measured with several tools, including the Functional Assessment of Cancer Therapy – Kidney Symptom Index Disease-Related Symptoms (FKSI-DRS), but no significant differences between treatment arms were observed in any measure.
Current guidelines recommend pazopanib, which is a tyrosine kinase inhibitor of the vascular endothelial growth factor receptor, as a first-line therapy in ccRCC. Everolimus, an inhibitor of mammalian target of rapamycin, is recommended in the second-line setting. Noting that resistance to pazopanib has been shown to be reversible after a period of withdrawal in experimental studies, the authors had speculated an on-off strategy with everolimus might better preserve the efficacy of pazopanib, providing longer periods of disease control. They had also hypothesized that the cumulative adverse events might be less if the drugs were sequenced, allowing recovery from each set of drug-specific adverse events.
Several potential explanations were offered for the lack of improved efficacy from alternating everolimus with pazopanib. For one, the improved activity of pazopanib after withdrawal in experimental models was observed after drug-free periods. The authors questioned whether a period of tumor regrowth may be needed in order to overcome pazopanib resistance.
The study may still have supported the use of an alternating regimen if the alternating therapy had led to a significantly improved quality of life, but the authors found none, a outcome that they characterized as unexpected. They concluded that there are no apparent advantages for the alternating regimen of pazopanib and everolimus relative to pazopanib alone.
There was no efficacy or safety advantage for alternating everolimus with pazopanib over pazopanib alone in patients with metastatic or locally advanced clear cell renal cell carcinoma (ccRCC), according to a newly published randomized trial.
The study hypothesis was that alternating the two drugs would improve outcomes and reduce toxicity, but differences between arms for the major outcomes were not clinically significant, according to results of a multicenter trial led by Geert A. Cirkel, MD, of the department of medical oncology, University Medical Center, Utrecht, the Netherlands.
Investigators randomized 101 patients with histologically confirmed ccRCC to receive 8 weeks of pazopanib in a daily dose of 800 mg alternated with 8 weeks of everolimus in a daily dose of 10 mg or 800 mg per day of continuous pazopanib. Patients remained on either regimen until disease progression.
Median time until first progression or death was 7.4 months for the experimental alternating arm versus 9.4 months for the control arm of continuous single-agent pazopanib (P = .37), Dr. Cirkel and associates reported (JAMA Onc. 2017 Apr 1. doi: 10.1001/jamaoncol.2016.5202).
Progression-free survival after starting on a second-line therapy was 20.2 months for the alternating treatment vs. 14.5 months for the control, but the confidence intervals were wide, and the difference was not significant (P = .86).
There was no apparent toxicity or tolerability advantage from alternating therapy. Nearly 40% of patients in both arms required pazopanib dose reductions, while 14% in the alternating arm also required an everolimus dose reduction. The incidence of serious adverse events possibly related to treatment was comparable between arms.
Quality of life was measured with several tools, including the Functional Assessment of Cancer Therapy – Kidney Symptom Index Disease-Related Symptoms (FKSI-DRS), but no significant differences between treatment arms were observed in any measure.
Current guidelines recommend pazopanib, which is a tyrosine kinase inhibitor of the vascular endothelial growth factor receptor, as a first-line therapy in ccRCC. Everolimus, an inhibitor of mammalian target of rapamycin, is recommended in the second-line setting. Noting that resistance to pazopanib has been shown to be reversible after a period of withdrawal in experimental studies, the authors had speculated an on-off strategy with everolimus might better preserve the efficacy of pazopanib, providing longer periods of disease control. They had also hypothesized that the cumulative adverse events might be less if the drugs were sequenced, allowing recovery from each set of drug-specific adverse events.
Several potential explanations were offered for the lack of improved efficacy from alternating everolimus with pazopanib. For one, the improved activity of pazopanib after withdrawal in experimental models was observed after drug-free periods. The authors questioned whether a period of tumor regrowth may be needed in order to overcome pazopanib resistance.
The study may still have supported the use of an alternating regimen if the alternating therapy had led to a significantly improved quality of life, but the authors found none, a outcome that they characterized as unexpected. They concluded that there are no apparent advantages for the alternating regimen of pazopanib and everolimus relative to pazopanib alone.
FROM JAMA ONCOLOGY
Key clinical point:
Major finding: The median time to progression or death was 7.4 months for the combination versus 9.4 months for pazopanib alone (P = .37).
Data source: Randomized, multicenter controlled trial.
Disclosures: The principal investigator Dr. Cirkel reports travel expenses from Novartis, which, along with GlaxoSmithKline, provided funding for this study.
Topical imiquimod boosted response to intradermal hepatitis B vaccine
VIENNA – Topical imiquimod appeared to enhance the immunogenicity of an intradermal hepatitis B vaccine in patients on renal replacement therapy.
Patients on hemodialysis or peritoneal dialysis who got the combination developed significantly higher seroprotection and antibody levels than those who got either the typical intramuscular vaccination or an intradermal vaccination on unprepared skin, Ivan Fan-Ngai Hung, MD, said at the European Conference on Clinical Microbiology and Infectious Diseases. By 1 year, the protection and titers did begin to fall, but they still remained significantly higher than in the two comparator groups, said Dr. Hung, a clinical professor at the University of Hong Kong.

Dr. Hung and his colleagues have been investigating imiquimod’s immunogenicity-boosting potential for several years. Their initial murine work with an H1N1 influenza virus appeared in 2014 (Clin Vaccine Immunol. 2014 Apr;21[4]: 570-9). The investigators intraperitoneally immunized mice with a monovalent A(H1N1) vaccine combined with imiquimod (VIC) then intranasally inoculated them with a lethal dose of the virus. When compared with mice who received only vaccine, only imiquimod, or only placebo, the VIC group showed significantly greater and significantly longer survival. Virus-specific serum immunoglobulin M, IgG, and neutralizing antibodies were all significantly higher.
The investigators theorized that imiquimod, a Toll-like receptor 7 agonist, plays several key roles in boosting immune response, including inducing the differentiation and migration of dendritic cells, enhancing B cell differentiation, and increasing long-term B cell memory.
Within the past 2 years, the group has advanced to human influenza trials in healthy young adults and elders with comorbidities.
Both studies employed a 5% imiquimod cream delivering 250 mg of the drug. It was applied at the injection site 5 minutes before vaccination. In the elder study, 90% of the 91 subjects who got the combination achieved seroconversion, compared with 13% of those who got an intramuscular injection and 39% of those who got an intradermal injection plus placebo cream. The geometric mean titers went up faster and stayed elevated longer. The better immunogenicity was associated with fewer hospitalizations for influenza or pneumonia (Clin Infect Dis. 2014;59[9]:1246-55).
The immunogenicity findings were similar in the study of 160 healthy young people. This study had a surprising twist too, Dr. Hung said in his talk. Not only did the combination significantly improve immunogenicity against the vaccine influenza strains, it increased immunogenicity against the nonvaccine strains, especially the antigenically drifted H3N2 strain of 2015, which was not included in the 2013-2014 recommended vaccine (Lancet Inf Dis. 2016 Feb;16(2):209-18).
The study Dr. Hung presented in Vienna was an interim analysis of the first to apply this technique to a hepatitis B vaccine. It enrolled 69 patients (51 on peritoneal dialysis and 18 on hemodialysis). They were a mean 65 years old. All received 10 mcg of the Sci-B-Vac at baseline, 1 month, and 6 months. Vaccine was delivered in a trineedle unit designed for shallow intradermal penetration (MicronJet600; NanoPass Technologies) Group IQ received topical imiquimod along with the intradermal vaccine. Group ID received a placebo cream and the intradermal vaccine. Group IM received a placebo cream and an intramuscular vaccination.
Anti–hepatitis B titers were measured at baseline and at 1, 3, 6, and 12 months. The primary outcome was seroprotection at 1 month. The secondary outcomes were seroprotection at 3, 6, and 12 months; anti–hepatitis B antibody titer; and safety.
By 1 month, seroprotection was already significantly higher in the IQ group than in the ID and IM groups (60% vs. 50% and 38%, respectively).
By 3 months, the seroprotection rate in group IQ had risen to 85%. It remained elevated there at 6 months then tailed off to about 70% by 12 months. The ID and IM groups followed this same rising and falling curve but remained significantly lower at all time points. At 12 months, seroprotection was similar in both these groups – about 40%.
The anti–hepatitis B antibody titers told a similar story. Titers in the IQ group rose more rapidly and sharply, to 544 mIU/mL at 6 months and 566 mIU/mL at 12 months. The ID group also experienced a strong response, rising to 489 mIU/mL at 6 months. However, by 12 months, titer levels had dropped to 170 mIU/mL.
Titers in the IM group barely moved at all during the entire follow-up period, never rising above 21 mIU/mL.
There were no differences in systemic reactions among the three groups, but those who got the intradermal vaccines reported slightly more swelling and induration at the injection site.
“Since this is an interim analysis, we cannot determine long-term protection or antibody titers,” Dr. Hung cautioned. “However, we are starting a similar study in elderly patients and also one for those who are on low-dose immunosuppressants. We believe this regimen will also work for them.”
Dr. Hung has been on advisory boards for Pfizer and Gilead Sciences.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
VIENNA – Topical imiquimod appeared to enhance the immunogenicity of an intradermal hepatitis B vaccine in patients on renal replacement therapy.
Patients on hemodialysis or peritoneal dialysis who got the combination developed significantly higher seroprotection and antibody levels than those who got either the typical intramuscular vaccination or an intradermal vaccination on unprepared skin, Ivan Fan-Ngai Hung, MD, said at the European Conference on Clinical Microbiology and Infectious Diseases. By 1 year, the protection and titers did begin to fall, but they still remained significantly higher than in the two comparator groups, said Dr. Hung, a clinical professor at the University of Hong Kong.
Dr. Hung and his colleagues have been investigating imiquimod’s immunogenicity-boosting potential for several years. Their initial murine work with an H1N1 influenza virus appeared in 2014 (Clin Vaccine Immunol. 2014 Apr;21[4]: 570-9). The investigators intraperitoneally immunized mice with a monovalent A(H1N1) vaccine combined with imiquimod (VIC) then intranasally inoculated them with a lethal dose of the virus. When compared with mice who received only vaccine, only imiquimod, or only placebo, the VIC group showed significantly greater and significantly longer survival. Virus-specific serum immunoglobulin M, IgG, and neutralizing antibodies were all significantly higher.
The investigators theorized that imiquimod, a Toll-like receptor 7 agonist, plays several key roles in boosting immune response, including inducing the differentiation and migration of dendritic cells, enhancing B cell differentiation, and increasing long-term B cell memory.
Within the past 2 years, the group has advanced to human influenza trials in healthy young adults and elders with comorbidities.
Both studies employed a 5% imiquimod cream delivering 250 mg of the drug. It was applied at the injection site 5 minutes before vaccination. In the elder study, 90% of the 91 subjects who got the combination achieved seroconversion, compared with 13% of those who got an intramuscular injection and 39% of those who got an intradermal injection plus placebo cream. The geometric mean titers went up faster and stayed elevated longer. The better immunogenicity was associated with fewer hospitalizations for influenza or pneumonia (Clin Infect Dis. 2014;59[9]:1246-55).
The immunogenicity findings were similar in the study of 160 healthy young people. This study had a surprising twist too, Dr. Hung said in his talk. Not only did the combination significantly improve immunogenicity against the vaccine influenza strains, it increased immunogenicity against the nonvaccine strains, especially the antigenically drifted H3N2 strain of 2015, which was not included in the 2013-2014 recommended vaccine (Lancet Inf Dis. 2016 Feb;16(2):209-18).
The study Dr. Hung presented in Vienna was an interim analysis of the first to apply this technique to a hepatitis B vaccine. It enrolled 69 patients (51 on peritoneal dialysis and 18 on hemodialysis). They were a mean 65 years old. All received 10 mcg of the Sci-B-Vac at baseline, 1 month, and 6 months. Vaccine was delivered in a trineedle unit designed for shallow intradermal penetration (MicronJet600; NanoPass Technologies) Group IQ received topical imiquimod along with the intradermal vaccine. Group ID received a placebo cream and the intradermal vaccine. Group IM received a placebo cream and an intramuscular vaccination.
Anti–hepatitis B titers were measured at baseline and at 1, 3, 6, and 12 months. The primary outcome was seroprotection at 1 month. The secondary outcomes were seroprotection at 3, 6, and 12 months; anti–hepatitis B antibody titer; and safety.
By 1 month, seroprotection was already significantly higher in the IQ group than in the ID and IM groups (60% vs. 50% and 38%, respectively).
By 3 months, the seroprotection rate in group IQ had risen to 85%. It remained elevated there at 6 months then tailed off to about 70% by 12 months. The ID and IM groups followed this same rising and falling curve but remained significantly lower at all time points. At 12 months, seroprotection was similar in both these groups – about 40%.
The anti–hepatitis B antibody titers told a similar story. Titers in the IQ group rose more rapidly and sharply, to 544 mIU/mL at 6 months and 566 mIU/mL at 12 months. The ID group also experienced a strong response, rising to 489 mIU/mL at 6 months. However, by 12 months, titer levels had dropped to 170 mIU/mL.
Titers in the IM group barely moved at all during the entire follow-up period, never rising above 21 mIU/mL.
There were no differences in systemic reactions among the three groups, but those who got the intradermal vaccines reported slightly more swelling and induration at the injection site.
“Since this is an interim analysis, we cannot determine long-term protection or antibody titers,” Dr. Hung cautioned. “However, we are starting a similar study in elderly patients and also one for those who are on low-dose immunosuppressants. We believe this regimen will also work for them.”
Dr. Hung has been on advisory boards for Pfizer and Gilead Sciences.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
VIENNA – Topical imiquimod appeared to enhance the immunogenicity of an intradermal hepatitis B vaccine in patients on renal replacement therapy.
Patients on hemodialysis or peritoneal dialysis who got the combination developed significantly higher seroprotection and antibody levels than those who got either the typical intramuscular vaccination or an intradermal vaccination on unprepared skin, Ivan Fan-Ngai Hung, MD, said at the European Conference on Clinical Microbiology and Infectious Diseases. By 1 year, the protection and titers did begin to fall, but they still remained significantly higher than in the two comparator groups, said Dr. Hung, a clinical professor at the University of Hong Kong.
Dr. Hung and his colleagues have been investigating imiquimod’s immunogenicity-boosting potential for several years. Their initial murine work with an H1N1 influenza virus appeared in 2014 (Clin Vaccine Immunol. 2014 Apr;21[4]: 570-9). The investigators intraperitoneally immunized mice with a monovalent A(H1N1) vaccine combined with imiquimod (VIC) then intranasally inoculated them with a lethal dose of the virus. When compared with mice who received only vaccine, only imiquimod, or only placebo, the VIC group showed significantly greater and significantly longer survival. Virus-specific serum immunoglobulin M, IgG, and neutralizing antibodies were all significantly higher.
The investigators theorized that imiquimod, a Toll-like receptor 7 agonist, plays several key roles in boosting immune response, including inducing the differentiation and migration of dendritic cells, enhancing B cell differentiation, and increasing long-term B cell memory.
Within the past 2 years, the group has advanced to human influenza trials in healthy young adults and elders with comorbidities.
Both studies employed a 5% imiquimod cream delivering 250 mg of the drug. It was applied at the injection site 5 minutes before vaccination. In the elder study, 90% of the 91 subjects who got the combination achieved seroconversion, compared with 13% of those who got an intramuscular injection and 39% of those who got an intradermal injection plus placebo cream. The geometric mean titers went up faster and stayed elevated longer. The better immunogenicity was associated with fewer hospitalizations for influenza or pneumonia (Clin Infect Dis. 2014;59[9]:1246-55).
The immunogenicity findings were similar in the study of 160 healthy young people. This study had a surprising twist too, Dr. Hung said in his talk. Not only did the combination significantly improve immunogenicity against the vaccine influenza strains, it increased immunogenicity against the nonvaccine strains, especially the antigenically drifted H3N2 strain of 2015, which was not included in the 2013-2014 recommended vaccine (Lancet Inf Dis. 2016 Feb;16(2):209-18).
The study Dr. Hung presented in Vienna was an interim analysis of the first to apply this technique to a hepatitis B vaccine. It enrolled 69 patients (51 on peritoneal dialysis and 18 on hemodialysis). They were a mean 65 years old. All received 10 mcg of the Sci-B-Vac at baseline, 1 month, and 6 months. Vaccine was delivered in a trineedle unit designed for shallow intradermal penetration (MicronJet600; NanoPass Technologies) Group IQ received topical imiquimod along with the intradermal vaccine. Group ID received a placebo cream and the intradermal vaccine. Group IM received a placebo cream and an intramuscular vaccination.
Anti–hepatitis B titers were measured at baseline and at 1, 3, 6, and 12 months. The primary outcome was seroprotection at 1 month. The secondary outcomes were seroprotection at 3, 6, and 12 months; anti–hepatitis B antibody titer; and safety.
By 1 month, seroprotection was already significantly higher in the IQ group than in the ID and IM groups (60% vs. 50% and 38%, respectively).
By 3 months, the seroprotection rate in group IQ had risen to 85%. It remained elevated there at 6 months then tailed off to about 70% by 12 months. The ID and IM groups followed this same rising and falling curve but remained significantly lower at all time points. At 12 months, seroprotection was similar in both these groups – about 40%.
The anti–hepatitis B antibody titers told a similar story. Titers in the IQ group rose more rapidly and sharply, to 544 mIU/mL at 6 months and 566 mIU/mL at 12 months. The ID group also experienced a strong response, rising to 489 mIU/mL at 6 months. However, by 12 months, titer levels had dropped to 170 mIU/mL.
Titers in the IM group barely moved at all during the entire follow-up period, never rising above 21 mIU/mL.
There were no differences in systemic reactions among the three groups, but those who got the intradermal vaccines reported slightly more swelling and induration at the injection site.
“Since this is an interim analysis, we cannot determine long-term protection or antibody titers,” Dr. Hung cautioned. “However, we are starting a similar study in elderly patients and also one for those who are on low-dose immunosuppressants. We believe this regimen will also work for them.”
Dr. Hung has been on advisory boards for Pfizer and Gilead Sciences.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
AT ECCMID 2017
Key clinical point:
Major finding: By 1 month, seroprotection was significantly higher in the imiquimod group than in those who got an intradermal vaccination without imiquimod and those who had an intramuscular injection only (60% vs. 50% and 38%, respectively).
Data source: The four-armed randomized study comprised 69 patients on hemodialysis or peritoneal dialysis.
Disclosures: Dr. Hung has served on advisory boards for Pfizer and Gilead Sciences.
Alpha blockers may facilitate the expulsion of larger ureteric stones
CLINICAL QUESTION: Are alpha blockers efficacious in patients with ureteric stones?
STUDY DESIGN: Systematic review & meta-analysis.
SETTING: Randomized controlled trials (RCTs); most conducted in Europe and Asia.
SYNOPSIS: Fifty-five unique RCTs (5,990 subjects) examining alpha blockers as the main treatment of ureteric stones versus placebo or control were included regardless of language and publication status.
Treatment with alpha blockers resulted in a 49% greater likelihood of stone passage (RR, 1.49; CI, 1.39-1.61) with a number needed to treat of four. A priori subgroup analysis revealed treatment was only beneficial in patients with larger stones (5mm or greater) independent of stone location or type of alpha blocker.
Secondary outcomes included reduced time to stone passage, fewer episodes of pain, decreased risk of surgical intervention, and lower risk of hospital admission with alpha blocker treatment without an increase in serious adverse events.
The meta-analysis was limited by the overall lack of methodological rigor of and clinical heterogeneity between the pooled studies.
BOTTOM LINE: Based on available evidence, it is reasonable to utilize an alpha blocker as medical expulsive therapy in patients with larger ureteric stones.
CITATIONS: Hollingsworth JM, Canales BK, Rogers MA, et al. Alpha blockers for treatment of ureteric stones: systematic review and meta-analysis. BMJ. 2016;355:i6112.
Dr. Ecker is the assistant director of education, Division of Hospital Medicine, University of Colorado School of Medicine, Aurora.
CLINICAL QUESTION: Are alpha blockers efficacious in patients with ureteric stones?
STUDY DESIGN: Systematic review & meta-analysis.
SETTING: Randomized controlled trials (RCTs); most conducted in Europe and Asia.
SYNOPSIS: Fifty-five unique RCTs (5,990 subjects) examining alpha blockers as the main treatment of ureteric stones versus placebo or control were included regardless of language and publication status.
Treatment with alpha blockers resulted in a 49% greater likelihood of stone passage (RR, 1.49; CI, 1.39-1.61) with a number needed to treat of four. A priori subgroup analysis revealed treatment was only beneficial in patients with larger stones (5mm or greater) independent of stone location or type of alpha blocker.
Secondary outcomes included reduced time to stone passage, fewer episodes of pain, decreased risk of surgical intervention, and lower risk of hospital admission with alpha blocker treatment without an increase in serious adverse events.
The meta-analysis was limited by the overall lack of methodological rigor of and clinical heterogeneity between the pooled studies.
BOTTOM LINE: Based on available evidence, it is reasonable to utilize an alpha blocker as medical expulsive therapy in patients with larger ureteric stones.
CITATIONS: Hollingsworth JM, Canales BK, Rogers MA, et al. Alpha blockers for treatment of ureteric stones: systematic review and meta-analysis. BMJ. 2016;355:i6112.
Dr. Ecker is the assistant director of education, Division of Hospital Medicine, University of Colorado School of Medicine, Aurora.
CLINICAL QUESTION: Are alpha blockers efficacious in patients with ureteric stones?
STUDY DESIGN: Systematic review & meta-analysis.
SETTING: Randomized controlled trials (RCTs); most conducted in Europe and Asia.
SYNOPSIS: Fifty-five unique RCTs (5,990 subjects) examining alpha blockers as the main treatment of ureteric stones versus placebo or control were included regardless of language and publication status.
Treatment with alpha blockers resulted in a 49% greater likelihood of stone passage (RR, 1.49; CI, 1.39-1.61) with a number needed to treat of four. A priori subgroup analysis revealed treatment was only beneficial in patients with larger stones (5mm or greater) independent of stone location or type of alpha blocker.
Secondary outcomes included reduced time to stone passage, fewer episodes of pain, decreased risk of surgical intervention, and lower risk of hospital admission with alpha blocker treatment without an increase in serious adverse events.
The meta-analysis was limited by the overall lack of methodological rigor of and clinical heterogeneity between the pooled studies.
BOTTOM LINE: Based on available evidence, it is reasonable to utilize an alpha blocker as medical expulsive therapy in patients with larger ureteric stones.
CITATIONS: Hollingsworth JM, Canales BK, Rogers MA, et al. Alpha blockers for treatment of ureteric stones: systematic review and meta-analysis. BMJ. 2016;355:i6112.
Dr. Ecker is the assistant director of education, Division of Hospital Medicine, University of Colorado School of Medicine, Aurora.
UTI predictors identified in infants under 3 months of age
SAN FRANCISCO – Serum leukocytosis, pyuria, and urine leukocyte esterase have been identified as predictors of urinary tract infection (UTI) in infants younger than 90 days of age, with males being more likely to develop UTI than females.
The chance of the infection declined in older infants.
“Really young infants have a less well developed immune system, which may increase their susceptibility to UTIs,” said Sarah Berman, DO, a pediatric resident at Winthrop-University Hospital, Mineola, N.Y.

In 2011, the American Academy of Pediatrics issued “Urinary Tract Infection: Clinical Practice Guidelines for the Diagnosis and Management of the Initial UTI in Febrile Infants and Children 2 to 24 Months” (Pediatrics. 2011. doi: 10.1542/peds.2011-1330) concerning the diagnosis and management of the presence of bacteria in urine – a possible warning sign of UTI, but the AAP guidelines did not consider infants in the first 2 months of life.
To address this gap, the Winthrop-University Hospital researchers reviewed the medical records for all infants younger than 90 days of age diagnosed with UTI, fever, or viral illness from the beginning of 2009 to September 2015. Residence in neonatal intensive care, known congenital abnormalities, and prior antibiotic use were grounds for exclusion. Standard growth-based definitions of UTI and bacteriuria were used.
Of the 315 mainly white or Hispanic patients, 73 had a diagnosis of UTI and 261 did not. Both groups had the same mean age of 45 days. Fever within 24 hours of admission was more prevalent in those without UTI than those with (57% vs. 41%; P = .035). Those with UTI were significantly more likely (all P lesser than .001) to display serum leukocytosis (35% vs. 12%), pyuria (71% vs. 12%), and urine detection of leukocyte esterase (87% vs. 14%) and nitrite (19% vs. 0%).
In a univariate analysis, factors that were associated with UTI included serum leukocytosis, white blood cells in the urine, and urine leukocyte esterase, with fever within 24 hours of admission associated with reduced chance of UTI. A multivariate analysis that accounted for age, gender, and gestational age identified serum leukocytosis, pyruria, urine leukocyte esterase, and male sex as predictors of UTI, with increased odds ratio of 6.25, 3.19, 28, and 3.88, respectively.
The reduced likelihood of UTI in those who developed a fever within 24 hours of admission held up in the multivariate analysis (odds ratio 0.34).
Validation of the findings will require a prospective study, which is in the planning stage. If the findings bear out, the result could be improved diagnosis of UTI from birth onward, according to Dr. Berman.
Dr. Berman reported having no relevant financial disclosures.
SAN FRANCISCO – Serum leukocytosis, pyuria, and urine leukocyte esterase have been identified as predictors of urinary tract infection (UTI) in infants younger than 90 days of age, with males being more likely to develop UTI than females.
The chance of the infection declined in older infants.
“Really young infants have a less well developed immune system, which may increase their susceptibility to UTIs,” said Sarah Berman, DO, a pediatric resident at Winthrop-University Hospital, Mineola, N.Y.
In 2011, the American Academy of Pediatrics issued “Urinary Tract Infection: Clinical Practice Guidelines for the Diagnosis and Management of the Initial UTI in Febrile Infants and Children 2 to 24 Months” (Pediatrics. 2011. doi: 10.1542/peds.2011-1330) concerning the diagnosis and management of the presence of bacteria in urine – a possible warning sign of UTI, but the AAP guidelines did not consider infants in the first 2 months of life.
To address this gap, the Winthrop-University Hospital researchers reviewed the medical records for all infants younger than 90 days of age diagnosed with UTI, fever, or viral illness from the beginning of 2009 to September 2015. Residence in neonatal intensive care, known congenital abnormalities, and prior antibiotic use were grounds for exclusion. Standard growth-based definitions of UTI and bacteriuria were used.
Of the 315 mainly white or Hispanic patients, 73 had a diagnosis of UTI and 261 did not. Both groups had the same mean age of 45 days. Fever within 24 hours of admission was more prevalent in those without UTI than those with (57% vs. 41%; P = .035). Those with UTI were significantly more likely (all P lesser than .001) to display serum leukocytosis (35% vs. 12%), pyuria (71% vs. 12%), and urine detection of leukocyte esterase (87% vs. 14%) and nitrite (19% vs. 0%).
In a univariate analysis, factors that were associated with UTI included serum leukocytosis, white blood cells in the urine, and urine leukocyte esterase, with fever within 24 hours of admission associated with reduced chance of UTI. A multivariate analysis that accounted for age, gender, and gestational age identified serum leukocytosis, pyruria, urine leukocyte esterase, and male sex as predictors of UTI, with increased odds ratio of 6.25, 3.19, 28, and 3.88, respectively.
The reduced likelihood of UTI in those who developed a fever within 24 hours of admission held up in the multivariate analysis (odds ratio 0.34).
Validation of the findings will require a prospective study, which is in the planning stage. If the findings bear out, the result could be improved diagnosis of UTI from birth onward, according to Dr. Berman.
Dr. Berman reported having no relevant financial disclosures.
SAN FRANCISCO – Serum leukocytosis, pyuria, and urine leukocyte esterase have been identified as predictors of urinary tract infection (UTI) in infants younger than 90 days of age, with males being more likely to develop UTI than females.
The chance of the infection declined in older infants.
“Really young infants have a less well developed immune system, which may increase their susceptibility to UTIs,” said Sarah Berman, DO, a pediatric resident at Winthrop-University Hospital, Mineola, N.Y.
In 2011, the American Academy of Pediatrics issued “Urinary Tract Infection: Clinical Practice Guidelines for the Diagnosis and Management of the Initial UTI in Febrile Infants and Children 2 to 24 Months” (Pediatrics. 2011. doi: 10.1542/peds.2011-1330) concerning the diagnosis and management of the presence of bacteria in urine – a possible warning sign of UTI, but the AAP guidelines did not consider infants in the first 2 months of life.
To address this gap, the Winthrop-University Hospital researchers reviewed the medical records for all infants younger than 90 days of age diagnosed with UTI, fever, or viral illness from the beginning of 2009 to September 2015. Residence in neonatal intensive care, known congenital abnormalities, and prior antibiotic use were grounds for exclusion. Standard growth-based definitions of UTI and bacteriuria were used.
Of the 315 mainly white or Hispanic patients, 73 had a diagnosis of UTI and 261 did not. Both groups had the same mean age of 45 days. Fever within 24 hours of admission was more prevalent in those without UTI than those with (57% vs. 41%; P = .035). Those with UTI were significantly more likely (all P lesser than .001) to display serum leukocytosis (35% vs. 12%), pyuria (71% vs. 12%), and urine detection of leukocyte esterase (87% vs. 14%) and nitrite (19% vs. 0%).
In a univariate analysis, factors that were associated with UTI included serum leukocytosis, white blood cells in the urine, and urine leukocyte esterase, with fever within 24 hours of admission associated with reduced chance of UTI. A multivariate analysis that accounted for age, gender, and gestational age identified serum leukocytosis, pyruria, urine leukocyte esterase, and male sex as predictors of UTI, with increased odds ratio of 6.25, 3.19, 28, and 3.88, respectively.
The reduced likelihood of UTI in those who developed a fever within 24 hours of admission held up in the multivariate analysis (odds ratio 0.34).
Validation of the findings will require a prospective study, which is in the planning stage. If the findings bear out, the result could be improved diagnosis of UTI from birth onward, according to Dr. Berman.
Dr. Berman reported having no relevant financial disclosures.
AT PAS 17
Key clinical point: Pyuria, urine leukocyte esterase, and serum leukocytosis appear to be predictors of urinary tract infection in infants younger than 90 days of age.
Major finding: A multivariate analysis identified serum leukocytosis, pyruria, and urine leukocyte esterase as predictors of UTI, with increased odds ratio of 6.25, 3.19, and 28, respectively.
Data source: Retrospective analysis of medical records from one hospital.
Disclosures: Dr. Berman reported having no relevant financial disclosures.
