User login
Hypertension in older adults: What is the target blood pressure?
We should aim for a standard office systolic pressure lower than 130 mm Hg in most adults age 65 and older if the patient can take multiple antihypertensive medications and be followed closely for adverse effects.
This recommendation is part of the 2017 hypertension guideline from the American College of Cardiology and American Heart Association.1 This new guideline advocates drug treatment of hypertension to a target less than 130/80 mm Hg for patients of all ages for secondary prevention of cardiovascular disease, and for primary prevention in those at high risk (ie, an estimated 10-year risk of atherosclerotic cardiovascular disease of 10% or higher). The target blood pressure for those at lower risk is less than 140/90 mm Hg.
There are multiple tools to estimate the 10-year risk. All tools incorporate major predictors such as age, blood pressure, cholesterol profile, and other markers, depending on the tool. Although risk increases with age, the tools are inaccurate once the patient is approximately 80 years of age.
The recommendation for older adults omits a target diastolic pressure, since treating elevated systolic pressure has more data supporting it than treating elevated diastolic blood pressure in older people. These recommendations apply only to older adults who can walk and are living in the community, not in an institution, and includes the subset of older adults who have mild cognitive impairment and frailty. The goals of treatment should be patient-centered.
DATA BEHIND THE GUIDELINE: THE SPRINT TRIAL
The Systolic Blood Pressure Intervention Trial (SPRINT)2 enrolled 9,361 patients who, to enter, had to be at least 50 years old (the mean age was 67.9), have a systolic blood pressure of 130 to 180 mm Hg (the mean was 139.7 mm Hg), and be at risk of cardiovascular disease due to chronic kidney disease, clinical or subclinical cardiovascular disease, a 10-year Framingham risk score of at least 15%, or age 75 or older. They had few comorbidities, and patients with diabetes mellitus or prior stroke were excluded. The objective was to see if intensive blood pressure treatment reduced the incidence of adverse cardiovascular outcomes compared with standard control.
The participants were randomized to either an intensive treatment goal of systolic pressure less than 120 mm Hg or a standard treatment goal of less than 140 mm Hg. Investigators chose drugs and doses according to their clinical judgment. The study protocol called for blood pressure measurement using an untended automated cuff, which probably resulted in systolic pressure readings 5 to 10 mm Hg lower than with typical methods used in the office.3
The intensive treatment group achieved a mean systolic pressure of 121.5 mm Hg, which required an average of 3 drugs. In contrast, the standard treatment group achieved a systolic pressure of 136.2 mm Hg, which required an average of 1.9 drugs.
Due to an absolute risk reduction in cardiovascular events and mortality, SPRINT was discontinued early after a median follow-up of 3.3 years. In the entire cohort, 61 patients needed to be treated intensively to prevent 1 cardiovascular event, and 90 needed to be treated intensively to prevent 1 death.2
Favorable outcomes in the oldest subgroup
The oldest patients in the SPRINT trial tolerated the intensive treatment as well as the youngest.2,4
Exploratory analysis of the subgroup of patients age 75 and older, who constituted 28% of the patients in the trial, demonstrated significant benefit from intensive treatment. In this subgroup, 27 patients needed to be treated aggressively (compared with standard treatment) to prevent 1 cardiovascular event, and 41 needed to be treated intensively to prevent 1 death.4 The lower numbers needing to be treated in the older subgroup than in the overall trial reflect the higher absolute risk in this older population.
Serious adverse events were more common with intensive treatment than with standard treatment in the subgroup of older patients who were frail.4 Emergency department visits or serious adverse events were more likely when gait speed (a measure of frailty) was missing from the medical record in the intensive treatment group compared with the standard treatment group. Hyponatremia (serum sodium level < 130 mmol/L) was more likely in the intensively treated group than in the standard treatment group. Although the rate of falls was higher in the oldest subgroup than in the overall SPRINT population, within this subgroup the rate of injurious falls resulting in an emergency department visit was lower with intensive treatment than with standard treatment (11.6% vs 14.1%, P = .04).4
Most of the oldest patients scored below the nominal cutoff for normal (26 points)5 on the 30-point Montreal Cognitive Assessment, and about one-quarter scored below 19, which may be consistent with a major neurocognitive disorder.6
The SPRINT investigators validated a frailty scale in the study patients and found that the most frail benefited from intensive blood pressure control, as did the slowest walkers.
SPRINT results do not apply to very frail, sick patients
For older patients with hypertension, a high burden of comorbidity, and a limited life expectancy, the 2017 guidelines defer treatment decisions to clinical judgment and patient preference.
There have been no randomized trials of blood pressure management for older adults with substantial comorbidities or dementia. The “frail” older adults in the SPRINT trial were still living in the community, without dementia. The intensively treated frail older adults had more serious adverse events than with standard treatment. Those who were documented as being unable to walk at the time of enrollment also had more serious adverse events. Institutionalized older adults and nonambulatory adults in the community would likely have even higher rates of serious adverse events with intensive treatment than the SPRINT patients, and there is concern for excessive adverse effects from intensive blood pressure control in more debilitated older patients.
DOES TREATING HIGH BLOOD PRESSURE PREVENT FRAILTY OR DEMENTIA?
Aging without frailty is an important goal of geriatric care and is likely related to cardiovascular health.7 An older adult who becomes slower physically or mentally, with diminished strength and energy, is less likely to be able to live independently.
Would treating systolic blood pressure to a target of 120 to 130 mm Hg reduce the risk of prefrailty or frailty? Unfortunately, the 3-year SPRINT follow-up of the adults age 75 and older did not show any effect of intensive treatment on gait speed or mobility limitation.8 It is possible that the early termination of the study limited outcomes.
Regarding cognition, the new guidelines say that lowering blood pressure in adults with hypertension to prevent cognitive decline and dementia is reasonable, giving it a class IIa (moderate) recommendation, but they do not offer a particular blood pressure target.
Two systematic reviews of randomized placebo-controlled trials9,10 suggested that pharmacologic treatment of hypertension reduces the progression of cognitive impairment. The trials did not use an intensive treatment goal.
The impact of intensive treatment of hypertension (to a target of 120–130 mm Hg) on the development or progression of cognitive impairment is not known at this time. The SPRINT Memory and Cognition in Decreased Hypertension analysis may shed light on the effect of intensive treatment of blood pressure on the incidence of dementia, although the early termination of SPRINT may limit its conclusions as well.
GOALS SHOULD BE PATIENT-CENTERED
The new hypertension guideline gives clinicians 2 things to think about when treating hypertensive, ambulatory, noninstitutionalized, nondemented older adults, including those age 75 and older:
- Older adults tolerate intensive blood pressure treatment as well as standard treatment. In particular, the fall rate is not increased and may even be less with intensive treatment.
- Older adults have better cardiovascular outcomes with blood pressure less than 130 mm Hg than with higher levels.
Adherence to the new guidelines would require many older adults without significant multimorbidity to take 3 drugs and undergo more frequent monitoring. This burden may align with the goals of care for many older adults. However, data do not exist to prove a benefit from intensive blood pressure control in debilitated elderly patients, and there may be harm. Lowering the medication burden may be a more important goal than lowering the pressure for this population. Blood pressure targets and hypertension management should reflect patient-centered goals of care.
- Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2017. Epub ahead of print.
- SPRINT Research Group; Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- Bakris GL. The implications of blood pressure measurement methods on treatment targets for blood pressure. Circulation 2016; 134:904–905.
- Williamson JD, Supiano MA, Applegate WB, et al; SPRINT Research Group. Intensive vs standard blood pressure control and cardiovascular disease outcomes in adults aged ≥ 75 years: a randomized clinical trial. JAMA 2016; 315:2673–2682.
- Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699.
- Borland E, Nagga K, Nilsson PM, Minthon L, Nilsson ED, Palmqvist S. The Montreal Cognitive Assessment: normative data from a large Swedish population-based cohort. J Alzheimers Dis 2017; 59:893–901.
- Graciani A, Garcia-Esquinas E, Lopez-Garcia E, Banegas JR, Rodriguez-Artalejo F. Ideal cardiovascular health and risk of frailty in older adults. Circ Cardiovasc Qual Outcomes 2016; 9:239–245.
- Odden MC, Peralta CA, Berlowitz DR, et al; Systolic Blood Pressure Intervention Trial (SPRINT) Research Group. Effect of intensive blood pressure control on gait speed and mobility limitation in adults 75 years or older: a randomized clinical trial. JAMA Intern Med 2017; 177:500–507.
- Tully PJ, Hanon O, Cosh S, Tzourio C. Diuretic antihypertensive drugs and incident dementia risk: a systematic review, meta-analysis and meta-regression of prospective studies. J Hypertens 2016; 34:1027–1035.
- Rouch L, Cestac P, Hanon O, et al. Antihypertensive drugs, prevention of cognitive decline and dementia: a systematic review of observational studies, randomized controlled trials and meta-analyses, with discussion of potential mechanisms. CNS Drugs 2015; 29:113–130.
We should aim for a standard office systolic pressure lower than 130 mm Hg in most adults age 65 and older if the patient can take multiple antihypertensive medications and be followed closely for adverse effects.
This recommendation is part of the 2017 hypertension guideline from the American College of Cardiology and American Heart Association.1 This new guideline advocates drug treatment of hypertension to a target less than 130/80 mm Hg for patients of all ages for secondary prevention of cardiovascular disease, and for primary prevention in those at high risk (ie, an estimated 10-year risk of atherosclerotic cardiovascular disease of 10% or higher). The target blood pressure for those at lower risk is less than 140/90 mm Hg.
There are multiple tools to estimate the 10-year risk. All tools incorporate major predictors such as age, blood pressure, cholesterol profile, and other markers, depending on the tool. Although risk increases with age, the tools are inaccurate once the patient is approximately 80 years of age.
The recommendation for older adults omits a target diastolic pressure, since treating elevated systolic pressure has more data supporting it than treating elevated diastolic blood pressure in older people. These recommendations apply only to older adults who can walk and are living in the community, not in an institution, and includes the subset of older adults who have mild cognitive impairment and frailty. The goals of treatment should be patient-centered.
DATA BEHIND THE GUIDELINE: THE SPRINT TRIAL
The Systolic Blood Pressure Intervention Trial (SPRINT)2 enrolled 9,361 patients who, to enter, had to be at least 50 years old (the mean age was 67.9), have a systolic blood pressure of 130 to 180 mm Hg (the mean was 139.7 mm Hg), and be at risk of cardiovascular disease due to chronic kidney disease, clinical or subclinical cardiovascular disease, a 10-year Framingham risk score of at least 15%, or age 75 or older. They had few comorbidities, and patients with diabetes mellitus or prior stroke were excluded. The objective was to see if intensive blood pressure treatment reduced the incidence of adverse cardiovascular outcomes compared with standard control.
The participants were randomized to either an intensive treatment goal of systolic pressure less than 120 mm Hg or a standard treatment goal of less than 140 mm Hg. Investigators chose drugs and doses according to their clinical judgment. The study protocol called for blood pressure measurement using an untended automated cuff, which probably resulted in systolic pressure readings 5 to 10 mm Hg lower than with typical methods used in the office.3
The intensive treatment group achieved a mean systolic pressure of 121.5 mm Hg, which required an average of 3 drugs. In contrast, the standard treatment group achieved a systolic pressure of 136.2 mm Hg, which required an average of 1.9 drugs.
Due to an absolute risk reduction in cardiovascular events and mortality, SPRINT was discontinued early after a median follow-up of 3.3 years. In the entire cohort, 61 patients needed to be treated intensively to prevent 1 cardiovascular event, and 90 needed to be treated intensively to prevent 1 death.2
Favorable outcomes in the oldest subgroup
The oldest patients in the SPRINT trial tolerated the intensive treatment as well as the youngest.2,4
Exploratory analysis of the subgroup of patients age 75 and older, who constituted 28% of the patients in the trial, demonstrated significant benefit from intensive treatment. In this subgroup, 27 patients needed to be treated aggressively (compared with standard treatment) to prevent 1 cardiovascular event, and 41 needed to be treated intensively to prevent 1 death.4 The lower numbers needing to be treated in the older subgroup than in the overall trial reflect the higher absolute risk in this older population.
Serious adverse events were more common with intensive treatment than with standard treatment in the subgroup of older patients who were frail.4 Emergency department visits or serious adverse events were more likely when gait speed (a measure of frailty) was missing from the medical record in the intensive treatment group compared with the standard treatment group. Hyponatremia (serum sodium level < 130 mmol/L) was more likely in the intensively treated group than in the standard treatment group. Although the rate of falls was higher in the oldest subgroup than in the overall SPRINT population, within this subgroup the rate of injurious falls resulting in an emergency department visit was lower with intensive treatment than with standard treatment (11.6% vs 14.1%, P = .04).4
Most of the oldest patients scored below the nominal cutoff for normal (26 points)5 on the 30-point Montreal Cognitive Assessment, and about one-quarter scored below 19, which may be consistent with a major neurocognitive disorder.6
The SPRINT investigators validated a frailty scale in the study patients and found that the most frail benefited from intensive blood pressure control, as did the slowest walkers.
SPRINT results do not apply to very frail, sick patients
For older patients with hypertension, a high burden of comorbidity, and a limited life expectancy, the 2017 guidelines defer treatment decisions to clinical judgment and patient preference.
There have been no randomized trials of blood pressure management for older adults with substantial comorbidities or dementia. The “frail” older adults in the SPRINT trial were still living in the community, without dementia. The intensively treated frail older adults had more serious adverse events than with standard treatment. Those who were documented as being unable to walk at the time of enrollment also had more serious adverse events. Institutionalized older adults and nonambulatory adults in the community would likely have even higher rates of serious adverse events with intensive treatment than the SPRINT patients, and there is concern for excessive adverse effects from intensive blood pressure control in more debilitated older patients.
DOES TREATING HIGH BLOOD PRESSURE PREVENT FRAILTY OR DEMENTIA?
Aging without frailty is an important goal of geriatric care and is likely related to cardiovascular health.7 An older adult who becomes slower physically or mentally, with diminished strength and energy, is less likely to be able to live independently.
Would treating systolic blood pressure to a target of 120 to 130 mm Hg reduce the risk of prefrailty or frailty? Unfortunately, the 3-year SPRINT follow-up of the adults age 75 and older did not show any effect of intensive treatment on gait speed or mobility limitation.8 It is possible that the early termination of the study limited outcomes.
Regarding cognition, the new guidelines say that lowering blood pressure in adults with hypertension to prevent cognitive decline and dementia is reasonable, giving it a class IIa (moderate) recommendation, but they do not offer a particular blood pressure target.
Two systematic reviews of randomized placebo-controlled trials9,10 suggested that pharmacologic treatment of hypertension reduces the progression of cognitive impairment. The trials did not use an intensive treatment goal.
The impact of intensive treatment of hypertension (to a target of 120–130 mm Hg) on the development or progression of cognitive impairment is not known at this time. The SPRINT Memory and Cognition in Decreased Hypertension analysis may shed light on the effect of intensive treatment of blood pressure on the incidence of dementia, although the early termination of SPRINT may limit its conclusions as well.
GOALS SHOULD BE PATIENT-CENTERED
The new hypertension guideline gives clinicians 2 things to think about when treating hypertensive, ambulatory, noninstitutionalized, nondemented older adults, including those age 75 and older:
- Older adults tolerate intensive blood pressure treatment as well as standard treatment. In particular, the fall rate is not increased and may even be less with intensive treatment.
- Older adults have better cardiovascular outcomes with blood pressure less than 130 mm Hg than with higher levels.
Adherence to the new guidelines would require many older adults without significant multimorbidity to take 3 drugs and undergo more frequent monitoring. This burden may align with the goals of care for many older adults. However, data do not exist to prove a benefit from intensive blood pressure control in debilitated elderly patients, and there may be harm. Lowering the medication burden may be a more important goal than lowering the pressure for this population. Blood pressure targets and hypertension management should reflect patient-centered goals of care.
We should aim for a standard office systolic pressure lower than 130 mm Hg in most adults age 65 and older if the patient can take multiple antihypertensive medications and be followed closely for adverse effects.
This recommendation is part of the 2017 hypertension guideline from the American College of Cardiology and American Heart Association.1 This new guideline advocates drug treatment of hypertension to a target less than 130/80 mm Hg for patients of all ages for secondary prevention of cardiovascular disease, and for primary prevention in those at high risk (ie, an estimated 10-year risk of atherosclerotic cardiovascular disease of 10% or higher). The target blood pressure for those at lower risk is less than 140/90 mm Hg.
There are multiple tools to estimate the 10-year risk. All tools incorporate major predictors such as age, blood pressure, cholesterol profile, and other markers, depending on the tool. Although risk increases with age, the tools are inaccurate once the patient is approximately 80 years of age.
The recommendation for older adults omits a target diastolic pressure, since treating elevated systolic pressure has more data supporting it than treating elevated diastolic blood pressure in older people. These recommendations apply only to older adults who can walk and are living in the community, not in an institution, and includes the subset of older adults who have mild cognitive impairment and frailty. The goals of treatment should be patient-centered.
DATA BEHIND THE GUIDELINE: THE SPRINT TRIAL
The Systolic Blood Pressure Intervention Trial (SPRINT)2 enrolled 9,361 patients who, to enter, had to be at least 50 years old (the mean age was 67.9), have a systolic blood pressure of 130 to 180 mm Hg (the mean was 139.7 mm Hg), and be at risk of cardiovascular disease due to chronic kidney disease, clinical or subclinical cardiovascular disease, a 10-year Framingham risk score of at least 15%, or age 75 or older. They had few comorbidities, and patients with diabetes mellitus or prior stroke were excluded. The objective was to see if intensive blood pressure treatment reduced the incidence of adverse cardiovascular outcomes compared with standard control.
The participants were randomized to either an intensive treatment goal of systolic pressure less than 120 mm Hg or a standard treatment goal of less than 140 mm Hg. Investigators chose drugs and doses according to their clinical judgment. The study protocol called for blood pressure measurement using an untended automated cuff, which probably resulted in systolic pressure readings 5 to 10 mm Hg lower than with typical methods used in the office.3
The intensive treatment group achieved a mean systolic pressure of 121.5 mm Hg, which required an average of 3 drugs. In contrast, the standard treatment group achieved a systolic pressure of 136.2 mm Hg, which required an average of 1.9 drugs.
Due to an absolute risk reduction in cardiovascular events and mortality, SPRINT was discontinued early after a median follow-up of 3.3 years. In the entire cohort, 61 patients needed to be treated intensively to prevent 1 cardiovascular event, and 90 needed to be treated intensively to prevent 1 death.2
Favorable outcomes in the oldest subgroup
The oldest patients in the SPRINT trial tolerated the intensive treatment as well as the youngest.2,4
Exploratory analysis of the subgroup of patients age 75 and older, who constituted 28% of the patients in the trial, demonstrated significant benefit from intensive treatment. In this subgroup, 27 patients needed to be treated aggressively (compared with standard treatment) to prevent 1 cardiovascular event, and 41 needed to be treated intensively to prevent 1 death.4 The lower numbers needing to be treated in the older subgroup than in the overall trial reflect the higher absolute risk in this older population.
Serious adverse events were more common with intensive treatment than with standard treatment in the subgroup of older patients who were frail.4 Emergency department visits or serious adverse events were more likely when gait speed (a measure of frailty) was missing from the medical record in the intensive treatment group compared with the standard treatment group. Hyponatremia (serum sodium level < 130 mmol/L) was more likely in the intensively treated group than in the standard treatment group. Although the rate of falls was higher in the oldest subgroup than in the overall SPRINT population, within this subgroup the rate of injurious falls resulting in an emergency department visit was lower with intensive treatment than with standard treatment (11.6% vs 14.1%, P = .04).4
Most of the oldest patients scored below the nominal cutoff for normal (26 points)5 on the 30-point Montreal Cognitive Assessment, and about one-quarter scored below 19, which may be consistent with a major neurocognitive disorder.6
The SPRINT investigators validated a frailty scale in the study patients and found that the most frail benefited from intensive blood pressure control, as did the slowest walkers.
SPRINT results do not apply to very frail, sick patients
For older patients with hypertension, a high burden of comorbidity, and a limited life expectancy, the 2017 guidelines defer treatment decisions to clinical judgment and patient preference.
There have been no randomized trials of blood pressure management for older adults with substantial comorbidities or dementia. The “frail” older adults in the SPRINT trial were still living in the community, without dementia. The intensively treated frail older adults had more serious adverse events than with standard treatment. Those who were documented as being unable to walk at the time of enrollment also had more serious adverse events. Institutionalized older adults and nonambulatory adults in the community would likely have even higher rates of serious adverse events with intensive treatment than the SPRINT patients, and there is concern for excessive adverse effects from intensive blood pressure control in more debilitated older patients.
DOES TREATING HIGH BLOOD PRESSURE PREVENT FRAILTY OR DEMENTIA?
Aging without frailty is an important goal of geriatric care and is likely related to cardiovascular health.7 An older adult who becomes slower physically or mentally, with diminished strength and energy, is less likely to be able to live independently.
Would treating systolic blood pressure to a target of 120 to 130 mm Hg reduce the risk of prefrailty or frailty? Unfortunately, the 3-year SPRINT follow-up of the adults age 75 and older did not show any effect of intensive treatment on gait speed or mobility limitation.8 It is possible that the early termination of the study limited outcomes.
Regarding cognition, the new guidelines say that lowering blood pressure in adults with hypertension to prevent cognitive decline and dementia is reasonable, giving it a class IIa (moderate) recommendation, but they do not offer a particular blood pressure target.
Two systematic reviews of randomized placebo-controlled trials9,10 suggested that pharmacologic treatment of hypertension reduces the progression of cognitive impairment. The trials did not use an intensive treatment goal.
The impact of intensive treatment of hypertension (to a target of 120–130 mm Hg) on the development or progression of cognitive impairment is not known at this time. The SPRINT Memory and Cognition in Decreased Hypertension analysis may shed light on the effect of intensive treatment of blood pressure on the incidence of dementia, although the early termination of SPRINT may limit its conclusions as well.
GOALS SHOULD BE PATIENT-CENTERED
The new hypertension guideline gives clinicians 2 things to think about when treating hypertensive, ambulatory, noninstitutionalized, nondemented older adults, including those age 75 and older:
- Older adults tolerate intensive blood pressure treatment as well as standard treatment. In particular, the fall rate is not increased and may even be less with intensive treatment.
- Older adults have better cardiovascular outcomes with blood pressure less than 130 mm Hg than with higher levels.
Adherence to the new guidelines would require many older adults without significant multimorbidity to take 3 drugs and undergo more frequent monitoring. This burden may align with the goals of care for many older adults. However, data do not exist to prove a benefit from intensive blood pressure control in debilitated elderly patients, and there may be harm. Lowering the medication burden may be a more important goal than lowering the pressure for this population. Blood pressure targets and hypertension management should reflect patient-centered goals of care.
- Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2017. Epub ahead of print.
- SPRINT Research Group; Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- Bakris GL. The implications of blood pressure measurement methods on treatment targets for blood pressure. Circulation 2016; 134:904–905.
- Williamson JD, Supiano MA, Applegate WB, et al; SPRINT Research Group. Intensive vs standard blood pressure control and cardiovascular disease outcomes in adults aged ≥ 75 years: a randomized clinical trial. JAMA 2016; 315:2673–2682.
- Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699.
- Borland E, Nagga K, Nilsson PM, Minthon L, Nilsson ED, Palmqvist S. The Montreal Cognitive Assessment: normative data from a large Swedish population-based cohort. J Alzheimers Dis 2017; 59:893–901.
- Graciani A, Garcia-Esquinas E, Lopez-Garcia E, Banegas JR, Rodriguez-Artalejo F. Ideal cardiovascular health and risk of frailty in older adults. Circ Cardiovasc Qual Outcomes 2016; 9:239–245.
- Odden MC, Peralta CA, Berlowitz DR, et al; Systolic Blood Pressure Intervention Trial (SPRINT) Research Group. Effect of intensive blood pressure control on gait speed and mobility limitation in adults 75 years or older: a randomized clinical trial. JAMA Intern Med 2017; 177:500–507.
- Tully PJ, Hanon O, Cosh S, Tzourio C. Diuretic antihypertensive drugs and incident dementia risk: a systematic review, meta-analysis and meta-regression of prospective studies. J Hypertens 2016; 34:1027–1035.
- Rouch L, Cestac P, Hanon O, et al. Antihypertensive drugs, prevention of cognitive decline and dementia: a systematic review of observational studies, randomized controlled trials and meta-analyses, with discussion of potential mechanisms. CNS Drugs 2015; 29:113–130.
- Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2017. Epub ahead of print.
- SPRINT Research Group; Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- Bakris GL. The implications of blood pressure measurement methods on treatment targets for blood pressure. Circulation 2016; 134:904–905.
- Williamson JD, Supiano MA, Applegate WB, et al; SPRINT Research Group. Intensive vs standard blood pressure control and cardiovascular disease outcomes in adults aged ≥ 75 years: a randomized clinical trial. JAMA 2016; 315:2673–2682.
- Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699.
- Borland E, Nagga K, Nilsson PM, Minthon L, Nilsson ED, Palmqvist S. The Montreal Cognitive Assessment: normative data from a large Swedish population-based cohort. J Alzheimers Dis 2017; 59:893–901.
- Graciani A, Garcia-Esquinas E, Lopez-Garcia E, Banegas JR, Rodriguez-Artalejo F. Ideal cardiovascular health and risk of frailty in older adults. Circ Cardiovasc Qual Outcomes 2016; 9:239–245.
- Odden MC, Peralta CA, Berlowitz DR, et al; Systolic Blood Pressure Intervention Trial (SPRINT) Research Group. Effect of intensive blood pressure control on gait speed and mobility limitation in adults 75 years or older: a randomized clinical trial. JAMA Intern Med 2017; 177:500–507.
- Tully PJ, Hanon O, Cosh S, Tzourio C. Diuretic antihypertensive drugs and incident dementia risk: a systematic review, meta-analysis and meta-regression of prospective studies. J Hypertens 2016; 34:1027–1035.
- Rouch L, Cestac P, Hanon O, et al. Antihypertensive drugs, prevention of cognitive decline and dementia: a systematic review of observational studies, randomized controlled trials and meta-analyses, with discussion of potential mechanisms. CNS Drugs 2015; 29:113–130.

A 67-year-old woman with bilateral hand numbness
A 67-year-old woman presents to the emergency department after 8 weeks of progressive numbness and tingling in both hands, involving all fingers. The numbness has increased in severity in the last 3 days. She also has occasional numbness around her mouth. She reports no numbness in her feet.
She says she underwent thyroid surgery twice for thyroid cancer 10 years ago. Her medical history also includes type 2 diabetes mellitus (diagnosed 1 year ago), hypertension, dyslipidemia, and diastolic heart failure (diagnosed 5 years ago).
Her current medications are:
- Metformin 1 g twice a day
- Candesartan 16 mg once a day
- Atorvastatin 20 mg once a day
- Furosemide 40 mg twice a day
- Levothyroxine 100 μg per day
- Calcium carbonate 1,500 mg twice a day
- A vitamin D tablet twice a day, which she has not taken for the last 2 months.
She admits she has not been taking her medications regularly because she has been feeling depressed.
On physical examination, she is alert and oriented but appears anxious. She is not in respiratory distress. Her blood pressure is 150/90 mm Hg and her pulse is 92 beats per minute and regular. There is a thyroidectomy scar on the anterior neck. Her jugular venous pressure is not elevated. Her heart sounds are normal without extra sounds. She has no pulmonary rales and no lower-extremity edema.
The Phalen test and Tinel test for carpal tunnel syndrome are negative in both hands. Using a Katz hand diagram, the patient reports tingling and numbness in all fingers, both palms, and the dorsum of both hands. Tapping the area over the facial nerve does not elicit twitching of the facial muscles (ie, no Chvostek sign), but compression of the upper arm elicits carpal spasm (ie, positive Trousseau sign). There is no evidence of motor weakness in her hands. The rest of the physical examination is unremarkable.
POSSIBLE CAUSES OF NUMBNESS
1. Based on the initial evaluation, which of the following is the most likely cause of our patient’s bilateral hand numbness?
- Hypocalcemia due to primary hypoparathyroidism
- Carpal tunnel syndrome due to primary hypothyroidism
- Diabetic peripheral neuropathy
- Vitamin B12 deficiency due to metformin
- Hypocalcemia due to low serum calcitonin
All the conditions above except low serum calcitonin can cause bilateral hand paresthesia. Our patient most likely has hypocalcemia due to primary hypoparathyroidism.
Hypocalcemia
In our patient, bilateral hand numbness and perioral numbness after stopping vitamin D and a positive Trousseau sign strongly suggest hypocalcemia. The classic physical findings in patients with hypocalcemia are the Trousseau sign and the Chvostek sign. The Trousseau sign is elicited by inflating a blood pressure cuff above the systolic blood pressure for 3 minutes and observing for ischemia-induced carpopedal spasm, wrist and metacarpophalangeal joint flexion, thumb adduction, and interphalangeal joint extension. The Chvostek sign is elicited by tapping over the area of the facial nerve below the zygoma in front of the tragus, resulting in ipsilateral twitching of facial muscles.
Although the Trousseau sign is more sensitive and specific than the Chvostek sign, neither is pathognomonic for hypocalcemia.1 The Chvostek sign has been reported to be negative in 30% of patients with hypocalcemia and positive in 10% of normocalcemic individuals.1 The Trousseau sign, however, is present in 94% of hypocalcemic patients vs 1% of normocalcemic individuals.2
Primary hypoparathyroidism secondary to thyroidectomy. Postsurgical hypoparathyroidism is the most common cause of primary hypoparathyroidism. It results from ischemic injury or accidental removal of the parathyroid glands during anterior neck surgery.3,4 The consequent hypocalcemia can be transient, intermittent, or permanent. Permanent postsurgical hypoparathyroidism is defined as persistent hypocalcemia with insufficient parathyroid hormone (PTH) for more than 12 months after neck surgery; however, some consider 6 months to be enough to define the condition.5–7
The incidence of postsurgical hypoparathyroidism varies considerably with the extent of thyroid surgery and the experience of the surgeon.6,8 In the hands of experienced surgeons, permanent hypoparathyroidism occurs in fewer than 1% of patients after total thyroidectomy, whereas the rate may be higher than 6% with less-experienced surgeons.5,9 Other risk factors for postsurgical hypoparathyroidism include female sex, autoimmune thyroid disease, pregnancy, and lactation.5
Pseudohypoparathyroidism is a group of disorders characterized by renal resistance to PTH, leading to hypocalcemia, hyperphosphatemia, and elevated serum PTH. It is also associated with phenotypic features such as short stature and short fourth metacarpal bones.
Calcitonin deficiency. Calcitonin is a polypeptide hormone secreted from the parafollicular (C) cells of the thyroid gland. After total thyroidectomy, calcitonin levels are expected to be reduced. However, the role of calcitonin in humans is unclear. One study has shown that calcitonin is possibly a vestigial hormone, given that no calcitonin-related disorders (excess or deficiency) have been reported in humans.10
Carpal tunnel syndrome due to hypothyroidism
Our patient also could have primary hypothyroidism as a result of thyroidectomy. Hypothyroidism can cause bilateral hand numbness due to carpal tunnel syndrome, which is mediated by mucopolysaccharide deposition and synovial membrane swelling.11 One study reported that 29% of patients with hypothyroidism had carpal tunnel syndrome.12 Symptoms of carpal tunnel syndrome in hypothyroid patients may occur despite thyroid replacement therapy.13

Carpal tunnel syndrome is a clinical diagnosis. Patients usually experience hand paresthesia in the distribution of the median nerve. Provocative physical tests for carpal tunnel syndrome include the Tinel test, the Phalen test, and the Katz hand diagram, which is considered the best of the 3 tests.14,15 Based on how the patient marks the location and type of symptoms on the diagram, carpal tunnel syndrome is rated as classic, probable, possible, or unlikely (Table 1).14,16,17 The sensitivity of a classic or probable diagram ranges from 64% to 80%, while the specificity ranges from 73% to 90%.14,15
Carpal tunnel syndrome is less likely to be the cause of our patient’s symptoms, as her Katz hand diagram indicates only “possible” carpal tunnel syndrome. Her perioral numbness and positive Trousseau sign make hypocalcemia a more likely cause.
Diabetic peripheral neuropathy
Sensory peripheral neuropathy is a recognized complication of diabetes mellitus. However, neuropathy in diabetic patients most commonly manifests initially as distal symmetrical ascending neuropathy starting in the lower extremities.18 Therefore, diabetic peripheral neuropathy is less likely in this patient since her symptoms are limited to her hands.
Vitamin B12 deficiency
Metformin-induced vitamin B12 deficiency is another possible cause of peripheral neuropathy. It might be secondary to metformin-induced changes in intrinsic factor levels and small-intestine motility with resultant bacterial overgrowth, as well as inhibition of vitamin B12 absorption in the terminal ileum.19
However, metformin-induced vitamin B12 deficiency is not the most likely cause of our patient’s neuropathy, since she has been taking this drug for only 1 year. Vitamin B12 deficiency with consequent peripheral neuropathy is more likely in patients taking metformin in high doses for 10 or more years.20
Laboratory results and electrocardiography

Table 2 shows the patient’s initial laboratory results. Of note, her serum calcium level is 5.7 mg/dL (reference range 8.9–10.1). Electrocardiography in the emergency department shows:
- Prolonged PR interval (23 msec)
- Wide QRS complexes (13 msec)
- Flat T waves
- Prolonged corrected QT interval (475 msec)
- Occasional premature ventricular complexes.
CLINICAL MANIFESTATIONS OF HYPOCALCEMIA
2. Which of the following is not a manifestation of hypocalcemia?
- Tonic-clonic seizures
- Cyanosis
- Cardiac ventricular arrhythmias
- Acute pancreatitis
- Depression

Hypocalcemia can cause a wide range of clinical manifestations (Table 3), the extent and severity of which depend on the severity of hypocalcemia and how quickly it develops. The more acute the hypocalcemia, the more severe the manifestations.21
Tetany can cause seizures
Hypocalcemia is characterized by neuromuscular hyperexcitability, manifested clinically by tetany.22 Manifestations of tetany are numerous and include acral paresthesia, perioral numbness, muscle cramps, carpopedal spasm, and seizures. Tetany is the hallmark of hypocalcemia regardless of etiology. However, certain causes are associated with peculiar clinical manifestations. For example, chronic primary hypoparathyroidism may be associated with basal ganglia calcifications that can result in parkinsonism, other extrapyramidal disorders, and dementia (Table 4).6

Airway spasm can be fatal
A serious manifestation of acute severe hypocalcemia is spasm of the glottis muscles, which may cause cyanosis and, if untreated, death.21
Ventricular arrhythmias
Another potential fatal complication of acute severe hypocalcemia is polymorphic ventricular tachycardia due to prolongation of the QT interval, which is readily identified with electrocardiography.23
Hypocalcemia does not cause pancreatitis
Hypercalcemia, rather than hypocalcemia, may cause acute pancreatitis.24 Conversely, acute pancreatitis may cause hypocalcemia due to precipitation of calcium in the abdominal cavity.25
Psychiatric manifestations
In addition to depression, hypocalcemia is associated with psychiatric manifestations including anxiety, confusion, and emotional instability.
STEPS TO DIAGNOSIS OF HYPOCALCEMIA
First step: Confirm true hypocalcemia
Calcium circulates in the blood in 3 forms: bound to albumin (40% to 45%), bound to anions (10% to 15%), and free (ionized) (45%). Although ionized calcium is the active form, most laboratories report total serum calcium.
Since changes in serum albumin concentration affect the total serum calcium level, it is imperative to correct the measured serum calcium to the serum albumin concentration. Each 1-g/dL decrease in serum albumin lowers the total serum calcium by 0.8 mg/dL. Thus:
Corrected serum calcium (mg/dL) =
measured total serum calcium (mg/dL) +
0.8 (4 − serum albumin [g/dL]).
If the patient’s serum calcium level remains low when corrected for serum albumin, he or she has true hypocalcemia, which implies a low ionized serum calcium. Conversely, pseudohypocalcemia means that the measured calcium level is low but the corrected serum calcium is normal.
Using this formula, our patient’s corrected calcium level is calculated as 5.7 + 0.8 (4 – 3.2) = 6.3 mg/dL, indicating true hypocalcemia.
PHOSPHATE IS OFTEN HIGH WHEN CALCIUM IS LOW
In addition to hypocalcemia, our patient has an elevated phosphate level (Table 2).
3. Which of the following hypocalcemic disorders is not associated with hyperphosphatemia?
- End-stage renal disease
- Primary hypoparathyroidism
- Pseudohypoparathyroidism
- Vitamin D3 deficiency
- Rhabdomyolysis
Vitamin D deficiency is not associated with hyperphosphatemia.
Second step in evaluating hypocalcemia: Check phosphate, magnesium, creatinine

The major causes of hypocalcemia can be categorized according to the serum phosphate level: high vs normal or low (Table 5).
High-phosphate, low-calcium states. In the absence of concurrent end-stage renal disease and an excessive phosphate load, primary hypoparathyroidism is the most likely cause of hypocalcemia associated with hyperphosphatemia.
PTH increases serum ionized calcium by26,27:
- Increasing bone resorption
- Increasing reabsorption of calcium from the distal renal tubules
- Increasing the activity of 1-alpha-hydroxylase, responsible for conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 (the most biologically active vitamin D metabolite); 1,25-dihydroxyvitamin D increases the absorption of calcium and phosphate from the intestine.
Conversely, PTH decreases reabsorption of phosphate from proximal renal tubules, resulting in hypophosphatemia. Therefore, low serum PTH (primary hypoparathyroidism) or a PTH-resistant state (pseudohypoparathyroidism) results in hypocalcemia and hyperphosphatemia.26,27
Both end-stage renal disease and rhabdomyolysis are associated with high serum phosphate levels. The kidney normally excretes excess dietary phosphate to maintain phosphate homeostasis; however, this is impaired in end-stage renal disease, leading to hyperphosphatemia. In rhabdomyolysis, it is mainly the transcellular shift of phosphate into the extracellular space from myocyte injury that raises phosphate levels.
Normal- or low-phosphate, low calcium states. Hypocalcemia can also result from vitamin D deficiency, but this cause is associated with a low or normal serum phosphate level. In such cases, hypocalcemia causes secondary hyperparathyroidism with consequent renal phosphate loss and, thus, hypophosphatemia.27
Third step: Check serum intact PTH and 25-hydroxyvitamin D levels
Hypocalcemia stimulates secretion of PTH. Therefore, hypocalcemia with elevated serum PTH is caused by disorders that do not impair PTH secretion, including chronic renal failure and vitamin D deficiency (Table 5). Conversely, hypocalcemia with low or normal serum PTH levels suggests primary hypoparathyroidism.
Our patient’s serum PTH level is 20 ng/mL, which is within the reference range. This does not discount the diagnosis of primary hypoparathyroidism. Although most patients with primary hypoparathyroidism have low or undetectable serum PTH levels, some have normal PTH levels if some degree of PTH production is preserved.5,7,28–30 In these patients, the remaining functioning parathyroid tissue is not enough to maintain a normal serum calcium level, resulting in hypocalcemia. As a result, hypocalcemia stimulates the remaining parathyroid tissue to its maximum output, producing PTH levels usually within the lower or middle-normal range.30 In such patients, the terms parathyroid insufficiency and relative primary hypoparathyroidism are more precise than primary hypoparathyroidism.
Postsurgical hypoparathyroidism with an inappropriately normal PTH level is usually seen in patients with disorders that impair intestinal calcium absorption or bone resorption.31 In our patient’s case, the “normal” serum PTH level is likely due to maximal stimulation of remaining functioning parathyroid tissue by severe hypocalcemia, which is a result of her discontinuation of calcium and calcitriol therapy and her vitamin D deficiency.
CASE RESUMED: NO RESPONSE TO INTRAVENOUS CALCIUM GLUCONATE
The patient is given 2 10-mL ampules of 10% calcium gluconate diluted in 100 mL of 5% dextrose in water over 20 minutes intravenously. Electrocardiographic monitoring is continued. Two hours later, her measured serum calcium is only 5.8 mg/dL, with no improvement in her symptoms.
A continuous infusion of calcium gluconate is started: 12 ampules of calcium gluconate are added to 380 mL of 5% dextrose in water and infused at 40 mL/hour (infused rate of elemental calcium = 1.3 mg/kg/hour); 3 hours later, her measured serum calcium level is still only 5.8 mg//dL; at 6 hours it is 5.9 mg/dL, and her symptoms have not improved.
4. Which of the following is the most appropriate next step?
- Change the calcium gluconate to calcium chloride
- Increase the infusion rate to 1.5 mg of elemental calcium/kg/hour
- Give a bolus of 2 10-mL ampules of 10% calcium gluconate intravenously over 1 minute
- Give additional oral calcium tablets
- Check the serum magnesium level
Treatment of hypocalcemia can involve intravenous or oral calcium therapy.
Intravenous calcium is indicated for patients with any of the following6,32:
- Moderate to severe neuromuscular irritability (eg, acral paresthesia, carpopedal spasm, prolonged QT interval, seizures, laryngospasm, bronchospasm)
- Acute hypocalcemia with corrected serum calcium level less than 7.6 mg/dL, even if the patient is asymptomatic
- Cardiac failure.
One 10-mL ampule of 10% calcium gluconate contains 93 mg of elemental calcium; 1 or 2 ampules are typically diluted in 50 to 100 mL of 5% dextrose in water and infused slowly over 15 to 20 minutes. Rapid administration of intravenous calcium is contraindicated, as it may produce cardiac arrhythmias and possibly cardiac arrest. Therefore, intravenous calcium should be given slowly while continuing electrocardiographic monitoring.33
Since the effect of 1 ampule of calcium gluconate lasts only 2 to 3 hours, most patients with symptomatic hypocalcemia require continuous intravenous calcium infusion. The recommended dose of infused elemental calcium is 0.5 to 1.5 mg/kg/hour.34 Several ampules are added to 500 to 1,000 mL of 5% dextrose in water or 0.9% normal saline and infused at a rate appropriate for the patient’s corrected calcium and symptoms.
Oral calcium and vitamin D supplements can be given initially to patients with a corrected serum calcium level of 7.6 mg/dL or greater, with or without mild symptoms, if they can tolerate oral intake. However, this is not the treatment of choice for resistant acute hypocalcemia, as in this case.
Calcium chloride has no advantages over calcium gluconate. Further, it can be associated with local irritation and may result in tissue necrosis if extravasation occurs.35
Increasing the infusion rate of calcium gluconate to the maximum recommended dose may improve the patient’s ionized calcium level and symptoms somewhat. However, it is not the best option for this patient, given that she did not respond to 2 ampules of calcium gluconate followed by continuous infusion of 1.3 mg/kg/hour for 6 hours.
Calcium gluconate bolus. Similarly, giving the patient an additional 2 ampules of calcium gluconate over 1 minute would not be recommended, as rapid administration of intravenous calcium gluconate (eg, over 1 minute) is contraindicated.
Check magnesium
If hypocalcemia persists despite intravenous calcium therapy, as in our patient, further investigation or action is required. An important cause of persistent hypocalcemia is severe hypomagnesemia. Severe hypomagnesemia (serum magnesium < 0.8 mg/dL) causes resistant hypocalcemia by several mechanisms:
- Inducing PTH resistance32,36,37
- Decreasing PTH secretion32,36
- Decreasing calcitriol production.
The decrease in calcitriol production is a direct effect of hypomagnesemia, but it is also an indirect effect of low PTH secretion, which inhibits the enzyme 1-alpha-hydroxylase. Thus, conversion of 25-hydroxyvitamin D3 to calcitriol is impaired, leading to low calcitriol production.
Our patient could have hypomagnesemia due to furosemide use and uncontrolled diabetes mellitus. Hypocalcemia resistant to calcium therapy may occasionally respond to magnesium therapy even if the serum magnesium level is normal. This may be due to depleted intracellular magnesium salt levels.6,38 Rarely, severe hypermagnesemia can also be associated with hypocalcemia due to inhibition of PTH secretion.37,39
CASE RESUMED
Our patient’s serum magnesium level is 0.6 mg/dL (reference range 1.7–2.4 mg/dL). She is given 2 g of magnesium sulfate in 60 mL of 0.9% normal saline infused over 1 hour, followed by a continuous infusion of magnesium sulfate (12 g diluted in 250 mL of 0.9% normal saline, infused over 24 hours). On repeat testing 4 hours later, her serum magnesium level is 0.7 mg/dL, and at 8 hours later it is 0.9 mg/dL. She is subsequently started on oral magnesium oxide 600 mg per day. The magnesium sulfate infusion is continued for another 24 hours.
PREVENTING HYPERCALCIURIA
Patients with low PTH (primary hypoparathyroidism) may have hypercalciuria due to decreased renal tubular calcium reabsorption. Two important measures can minimize hypercalciuria in such patients:
- Keeping the serum calcium level in the low-normal range4,5,40
- Giving a thiazide diuretic (eg, hydrochlorothiazide 12.5–50 mg daily) with a low-salt diet.41,42
A thiazide diuretic is usually started once the 24-hour urine calcium reaches 250 mg.6 Thiazides are thought to enhance both proximal and distal renal tubular calcium reabsorption.43,44
PRIMARY HYPOPARATHYROIDISM: LONG-TERM MANAGEMENT
Long-term management of primary hypoparathyroidism requires calcium and vitamin D supplementation.
Calcium supplements. The most commonly prescribed calcium preparations are calcium carbonate and calcium citrate (containing 40% and 20% elemental calcium, respectively). Calcium carbonate, which is less expensive than calcium citrate, binds with phosphate intake and requires an acidic environment for absorption, and so it is better absorbed when taken with meals. Because calcium citrate does not require an acidic environment for absorption, it is the calcium preparation of choice for patients on proton pump inhibitors, or patients with achlorhydria or constipation.45 Calcium doses vary widely, with most hypoparathyroid patients requiring 1 to 2 g of elemental calcium daily.6
Vitamin D supplements. To promote intestinal absorption, calcium is combined with vitamin D in a fixed-dose preparation given in divided doses.46 Calcitriol (1,25-dihydroxyvitamin D3) is the most active metabolite of vitamin D, with rapid onset and offset of action, and it is the preferred form of vitamin D therapy for patients with hypoparathyroidism. If calcitriol is not available or is not affordable, alphacalcidol (1-alpha-hydroxyvitamin D3) is another option. This is a synthetic analogue of vitamin D that is already hyroxylated at the C1 position. After oral intake, it is hydroxylated in the liver to form calcitriol.
Since renal production of calcitriol is PTH-dependent, in hypoparathyroidism the conversion of 25-hydroxyvitamin D3 to calcitriol is limited. Therefore, vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol) are not the preferred forms of vitamin D for such patients. However, either can be added to calcitriol, as they may have extraskeletal benefits.7
CASE CONCLUDED
Our patient presented with primary parathyroid insufficiency associated with vitamin D deficiency. Therefore, in addition to calcitriol and calcium combined with vitamin D in a fixed-dose preparation, her management included vitamin D3 for her vitamin D deficiency.
She was discharged on these medications. At a follow-up visit 3 weeks later, her measured serum calcium level was 8.6 mg/dL. She reported gradual resolution of her symptoms. She was also referred to a psychiatrist for her depression.
TAKE-HOME POINTS
- Hypocalcemia causes neuromuscular excitability, manifested clinically by tetany.
- Common causes of hypocalcemia include vitamin D deficiency, hypomagnesemia, renal failure, and primary hypoparathyroidism.
- The first step in evaluating hypocalcemia is to correct the measured serum calcium to the serum albumin concentration.
- Laboratory testing for hypocalcemia should include serum phosphorus, magnesium, creatinine, PTH, and 25-hydroxyvitamin D3.
- Primary hypoparathyroidism is characterized by hypocalcemia, hyperphosphatemia, and low serum PTH.
- Moderate to severe manifestations of hypo-
calcemia and acute hypocalcemia (< 7.6 mg/dL), even if asymptomatic, warrant intravenous calcium therapy. - Correction of hypomagnesemia is essential to treat hypocalcemia, especially if resistant to intravenous calcium therapy.
- The goal of chronic management of primary hypoparathyroidism includes correcting the serum calcium level to a low-normal range, the serum phosphorus level to an upper-normal range, and prevention of hypercalciuria.
Acknowledgments: The authors wish to thank Mr. Michael Edward Tierney of the School of Medicine, University of Sydney, Australia, for his linguistic editing of the manuscript.
- Jesus JE, Landry A. Images in clinical medicine. Chvostek’s and Trousseau’s signs. N Engl J Med 2012; 367:e15.
- Urbano FL. Signs of hypocalcemia: Chvostek’s and Trousseau’s. Hosp Physician 2000; 36:43–45.
- Chisthi MM, Nair RS, Kuttanchettiyar KG, Yadev I. Mechanisms behind post-thyroidectomy hypocalcemia: interplay of calcitonin, parathormone, and albumin—a prospective study. J Invest Surg 2017; 30:217–225.
- Shoback DM, Bilezikian JP, Costa AG, et al. Presentation of hypoparathyroidism: etiologies and clinical features. J Clin Endocrinol Metab 2016; 101:2300–2312.
- Stack BC Jr, Bimston DN, Bodenner DL, et al. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: postoperative hypoparathyroidism—definitions and management. Endocr Pract 2015; 21:674–685.
- Shoback D. Clinical practice. Hypoparathyroidism. N Engl J Med 2008; 359:391–403.
- Abate EG, Clarke BL. Review of hypoparathyroidism. Front Endocrinol (Lausanne) 2017; 7:172.
- Coimbra C, Monteiro F, Oliveira P, Ribeiro L, de Almeida MG, Condé A. Hypoparathyroidism following thyroidectomy: predictive factors. Acta Otorrinolaringol Esp 2017; 68:106–111.
- Thomusch O, Machens A, Sekulla C, Ukkat J, Brauckhoff M, Dralle H. The impact of surgical technique on postoperative hypoparathyroidism in bilateral thyroid surgery: a multivariate analysis of 5846 consecutive patients. Surgery 2003; 133:180–185.
- Hirsch PF, Lester GE, Talmage RV. Calcitonin, an enigmatic hormone: does it have a function? J Musculoskelet Neuronal Interact 2001; 1:299–305.
- Karne SS, Bhalerao NS. Carpal tunnel syndrome in hypothyroidism. J Clin Diagn Res 2016; 10:OC36–OC38.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Palumbo CF, Szabo RM, Olmsted SL. The effects of hypothyroidism and thyroid replacement on the development of carpal tunnel syndrome. J Hand Surg Am 2000; 25:734–739.
- Katz JN, Stirrat CR, Larson MG, Fossel AH, Eaton HM, Liang MH. A self-administered hand symptom diagram for the diagnosis and epidemiologic study of carpal tunnel syndrome. J Rheumatol 1990; 17:1495–1498.
- Katz JN, Stirrat CR. A self-administered hand diagram for the diagnosis of carpal tunnel syndrome. J Hand Surg Am 1990; 15:360–363.
- Calfee RP, Dale AM, Ryan D, Descatha A, Franzblau A, Evanoff B. Performance of simplified scoring systems for hand diagrams in carpal tunnel syndrome screening. J Hand Surg Am 2012; 37:10–17.
- D’Arcy CA, McGee S. The rational clinical examination. Does this patient have carpal tunnel syndrome? JAMA 2000; 283:3110–3117.
- Marchettini P, Lacerenza M, Mauri E, Marangoni C. Painful peripheral neuropathies. Curr Neuropharmacol 2006; 4:175–181.
- Kibirige D, Mwebaze R. Vitamin B12 deficiency among patients with diabetes mellitus: is routine screening and supplementation justified? J Diabetes Metab Disord 2013;12:17.
- Akinlade KS, Agbebaku SO, Rahamon SK, Balogun WO. Vitamin B12 levels in patients with type 2 diabetes mellitus on metformin. Ann Ib Postgrad Med 2015; 13:79–83.
- Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am 1993; 22:363–375.
- Macefield G, Burke D. Paraesthesiae and tetany induced by voluntary hyperventilation. Increased excitability of human cutaneous and motor axons. Brain 1991; 114:527–540.
- Benoit SR, Mendelsohn AB, Nourjah P, Staffa JA, Graham DJ. Risk factors for prolonged QTc among US adults: Third National Health and Nutrition Examination Survey. Eur J Cardiovasc Prev Rehabil 2005; 12:363–368.
- Khoo TK, Vege SS, Abu-Lebdeh HS, Ryu E, Nadeem S, Wermers RA. Acute pancreatitis in primary hyperparathyroidism: a population-based study. J Clin Endocrinol Metab 2009; 94:2115–2118.
- McKay C, Beastall GH, Imrie CW, Baxter JN. Circulating intact parathyroid hormone levels in acute pancreatitis. Br J Surg 1994; 81:357–360.
- Talmage RV, Mobley HT. Calcium homeostasis: reassessment of the actions of parathyroid hormone. Gen Comp Endocrinol 2008; 156:1–8.
- Friedman PA, Gesek FA. Calcium transport in renal epithelial cells. Am J Physiol 1993; 264:F181–F198.
- Jensen PV, Jelstrup SM, Homøe P. Long-term outcomes after total thyroidectomy. Dan Med J 2015; 62:A5156.
- Ritter K, Elfenbein D, Schneider DF, Chen H, Sippel RS. Hypoparathyroidism after total thyroidectomy: incidence and resolution. J Surg Res 2015; 197:348–353.
- Promberger R, Ott J, Kober F, Karik M, Freissmuth M, Hermann M. Normal parathyroid hormone levels do not exclude permanent hypoparathyroidism after thyroidectomy. Thyroid 2011; 21:145–150.
- Lorente-Poch L, Sancho JJ, Muñoz-Nova JL, Sánchez-Velázquez P, Sitges-Serra A. Defining the syndromes of parathyroid failure after total thyroidectomy. Gland Surgery 2015; 4:82–90.
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ 2008; 336:1298–1302.
- Tohme JF, Bilezikian JP. Diagnosis and treatment of hypocalcemic emergencies. Endocrinologist 1996; 6:10–18.
- Carroll R, Matfin G. Endocrine and metabolic emergencies: hypocalcaemia. Ther Adv Endocrinol Metab 2010; 1:29–33.
- Kim MP, Raho VJ, Mak J, Kaynar AM. Skin and soft tissue necrosis from calcium chloride in a deicer. J Emerg Med 2007; 32:41–44.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20:3–17.
- Cholst IN, Steinberg SF, Tropper PJ, Fox HE, Segre GV, Bilezikian JP. The influence of hypermagnesemia on serum calcium and parathyroid hormone levels in human subjects. N Engl J Med 1984; 310:1221–1225.
- Ryzen E, Nelson TA, Rude RK. Low blood mononuclear cell magnesium content and hypocalcemia in normomagnesemic patients. West J Med 1987; 147:549–553.
- Koontz SL, Friedman SA, Schwartz ML. Symptomatic hypocalcemia after tocolytic therapy with magnesium sulfate and nifedipine. Am J Obstet Gynecol 2004; 190:1773–1776.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Porter RH, Cox BG, Heaney D, Hostetter TH, Stinebaugh BJ, Suki WN. Treatment of hypoparathyroid patients with chlorthalidone. N Engl J Med 1978; 298:577–581.
- Clarke BL, Brown EM, Collins MT, et al. Epidemiology and diagnosis of hypoparathyroidism. J Clin Endocrinol Metab 2016; 101:2284–2299.
- Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 2005; 115:1651–1658.
- Costanzo LS. Localization of diuretic action in microperfused rat distal tubules: Ca and Na transport. Am J Physiol 1985; 248:F527–F535.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Scotti A, Bianchini C, Abbiati G, Marzo A. Absorption of calcium administered alone or in fixed combination with vitamin D to healthy volunteers. Arzneimittelforschung 2001; 51:493–500.
A 67-year-old woman presents to the emergency department after 8 weeks of progressive numbness and tingling in both hands, involving all fingers. The numbness has increased in severity in the last 3 days. She also has occasional numbness around her mouth. She reports no numbness in her feet.
She says she underwent thyroid surgery twice for thyroid cancer 10 years ago. Her medical history also includes type 2 diabetes mellitus (diagnosed 1 year ago), hypertension, dyslipidemia, and diastolic heart failure (diagnosed 5 years ago).
Her current medications are:
- Metformin 1 g twice a day
- Candesartan 16 mg once a day
- Atorvastatin 20 mg once a day
- Furosemide 40 mg twice a day
- Levothyroxine 100 μg per day
- Calcium carbonate 1,500 mg twice a day
- A vitamin D tablet twice a day, which she has not taken for the last 2 months.
She admits she has not been taking her medications regularly because she has been feeling depressed.
On physical examination, she is alert and oriented but appears anxious. She is not in respiratory distress. Her blood pressure is 150/90 mm Hg and her pulse is 92 beats per minute and regular. There is a thyroidectomy scar on the anterior neck. Her jugular venous pressure is not elevated. Her heart sounds are normal without extra sounds. She has no pulmonary rales and no lower-extremity edema.
The Phalen test and Tinel test for carpal tunnel syndrome are negative in both hands. Using a Katz hand diagram, the patient reports tingling and numbness in all fingers, both palms, and the dorsum of both hands. Tapping the area over the facial nerve does not elicit twitching of the facial muscles (ie, no Chvostek sign), but compression of the upper arm elicits carpal spasm (ie, positive Trousseau sign). There is no evidence of motor weakness in her hands. The rest of the physical examination is unremarkable.
POSSIBLE CAUSES OF NUMBNESS
1. Based on the initial evaluation, which of the following is the most likely cause of our patient’s bilateral hand numbness?
- Hypocalcemia due to primary hypoparathyroidism
- Carpal tunnel syndrome due to primary hypothyroidism
- Diabetic peripheral neuropathy
- Vitamin B12 deficiency due to metformin
- Hypocalcemia due to low serum calcitonin
All the conditions above except low serum calcitonin can cause bilateral hand paresthesia. Our patient most likely has hypocalcemia due to primary hypoparathyroidism.
Hypocalcemia
In our patient, bilateral hand numbness and perioral numbness after stopping vitamin D and a positive Trousseau sign strongly suggest hypocalcemia. The classic physical findings in patients with hypocalcemia are the Trousseau sign and the Chvostek sign. The Trousseau sign is elicited by inflating a blood pressure cuff above the systolic blood pressure for 3 minutes and observing for ischemia-induced carpopedal spasm, wrist and metacarpophalangeal joint flexion, thumb adduction, and interphalangeal joint extension. The Chvostek sign is elicited by tapping over the area of the facial nerve below the zygoma in front of the tragus, resulting in ipsilateral twitching of facial muscles.
Although the Trousseau sign is more sensitive and specific than the Chvostek sign, neither is pathognomonic for hypocalcemia.1 The Chvostek sign has been reported to be negative in 30% of patients with hypocalcemia and positive in 10% of normocalcemic individuals.1 The Trousseau sign, however, is present in 94% of hypocalcemic patients vs 1% of normocalcemic individuals.2
Primary hypoparathyroidism secondary to thyroidectomy. Postsurgical hypoparathyroidism is the most common cause of primary hypoparathyroidism. It results from ischemic injury or accidental removal of the parathyroid glands during anterior neck surgery.3,4 The consequent hypocalcemia can be transient, intermittent, or permanent. Permanent postsurgical hypoparathyroidism is defined as persistent hypocalcemia with insufficient parathyroid hormone (PTH) for more than 12 months after neck surgery; however, some consider 6 months to be enough to define the condition.5–7
The incidence of postsurgical hypoparathyroidism varies considerably with the extent of thyroid surgery and the experience of the surgeon.6,8 In the hands of experienced surgeons, permanent hypoparathyroidism occurs in fewer than 1% of patients after total thyroidectomy, whereas the rate may be higher than 6% with less-experienced surgeons.5,9 Other risk factors for postsurgical hypoparathyroidism include female sex, autoimmune thyroid disease, pregnancy, and lactation.5
Pseudohypoparathyroidism is a group of disorders characterized by renal resistance to PTH, leading to hypocalcemia, hyperphosphatemia, and elevated serum PTH. It is also associated with phenotypic features such as short stature and short fourth metacarpal bones.
Calcitonin deficiency. Calcitonin is a polypeptide hormone secreted from the parafollicular (C) cells of the thyroid gland. After total thyroidectomy, calcitonin levels are expected to be reduced. However, the role of calcitonin in humans is unclear. One study has shown that calcitonin is possibly a vestigial hormone, given that no calcitonin-related disorders (excess or deficiency) have been reported in humans.10
Carpal tunnel syndrome due to hypothyroidism
Our patient also could have primary hypothyroidism as a result of thyroidectomy. Hypothyroidism can cause bilateral hand numbness due to carpal tunnel syndrome, which is mediated by mucopolysaccharide deposition and synovial membrane swelling.11 One study reported that 29% of patients with hypothyroidism had carpal tunnel syndrome.12 Symptoms of carpal tunnel syndrome in hypothyroid patients may occur despite thyroid replacement therapy.13
Carpal tunnel syndrome is a clinical diagnosis. Patients usually experience hand paresthesia in the distribution of the median nerve. Provocative physical tests for carpal tunnel syndrome include the Tinel test, the Phalen test, and the Katz hand diagram, which is considered the best of the 3 tests.14,15 Based on how the patient marks the location and type of symptoms on the diagram, carpal tunnel syndrome is rated as classic, probable, possible, or unlikely (Table 1).14,16,17 The sensitivity of a classic or probable diagram ranges from 64% to 80%, while the specificity ranges from 73% to 90%.14,15
Carpal tunnel syndrome is less likely to be the cause of our patient’s symptoms, as her Katz hand diagram indicates only “possible” carpal tunnel syndrome. Her perioral numbness and positive Trousseau sign make hypocalcemia a more likely cause.
Diabetic peripheral neuropathy
Sensory peripheral neuropathy is a recognized complication of diabetes mellitus. However, neuropathy in diabetic patients most commonly manifests initially as distal symmetrical ascending neuropathy starting in the lower extremities.18 Therefore, diabetic peripheral neuropathy is less likely in this patient since her symptoms are limited to her hands.
Vitamin B12 deficiency
Metformin-induced vitamin B12 deficiency is another possible cause of peripheral neuropathy. It might be secondary to metformin-induced changes in intrinsic factor levels and small-intestine motility with resultant bacterial overgrowth, as well as inhibition of vitamin B12 absorption in the terminal ileum.19
However, metformin-induced vitamin B12 deficiency is not the most likely cause of our patient’s neuropathy, since she has been taking this drug for only 1 year. Vitamin B12 deficiency with consequent peripheral neuropathy is more likely in patients taking metformin in high doses for 10 or more years.20
Laboratory results and electrocardiography
Table 2 shows the patient’s initial laboratory results. Of note, her serum calcium level is 5.7 mg/dL (reference range 8.9–10.1). Electrocardiography in the emergency department shows:
- Prolonged PR interval (23 msec)
- Wide QRS complexes (13 msec)
- Flat T waves
- Prolonged corrected QT interval (475 msec)
- Occasional premature ventricular complexes.
CLINICAL MANIFESTATIONS OF HYPOCALCEMIA
2. Which of the following is not a manifestation of hypocalcemia?
- Tonic-clonic seizures
- Cyanosis
- Cardiac ventricular arrhythmias
- Acute pancreatitis
- Depression
Hypocalcemia can cause a wide range of clinical manifestations (Table 3), the extent and severity of which depend on the severity of hypocalcemia and how quickly it develops. The more acute the hypocalcemia, the more severe the manifestations.21
Tetany can cause seizures
Hypocalcemia is characterized by neuromuscular hyperexcitability, manifested clinically by tetany.22 Manifestations of tetany are numerous and include acral paresthesia, perioral numbness, muscle cramps, carpopedal spasm, and seizures. Tetany is the hallmark of hypocalcemia regardless of etiology. However, certain causes are associated with peculiar clinical manifestations. For example, chronic primary hypoparathyroidism may be associated with basal ganglia calcifications that can result in parkinsonism, other extrapyramidal disorders, and dementia (Table 4).6
Airway spasm can be fatal
A serious manifestation of acute severe hypocalcemia is spasm of the glottis muscles, which may cause cyanosis and, if untreated, death.21
Ventricular arrhythmias
Another potential fatal complication of acute severe hypocalcemia is polymorphic ventricular tachycardia due to prolongation of the QT interval, which is readily identified with electrocardiography.23
Hypocalcemia does not cause pancreatitis
Hypercalcemia, rather than hypocalcemia, may cause acute pancreatitis.24 Conversely, acute pancreatitis may cause hypocalcemia due to precipitation of calcium in the abdominal cavity.25
Psychiatric manifestations
In addition to depression, hypocalcemia is associated with psychiatric manifestations including anxiety, confusion, and emotional instability.
STEPS TO DIAGNOSIS OF HYPOCALCEMIA
First step: Confirm true hypocalcemia
Calcium circulates in the blood in 3 forms: bound to albumin (40% to 45%), bound to anions (10% to 15%), and free (ionized) (45%). Although ionized calcium is the active form, most laboratories report total serum calcium.
Since changes in serum albumin concentration affect the total serum calcium level, it is imperative to correct the measured serum calcium to the serum albumin concentration. Each 1-g/dL decrease in serum albumin lowers the total serum calcium by 0.8 mg/dL. Thus:
Corrected serum calcium (mg/dL) =
measured total serum calcium (mg/dL) +
0.8 (4 − serum albumin [g/dL]).
If the patient’s serum calcium level remains low when corrected for serum albumin, he or she has true hypocalcemia, which implies a low ionized serum calcium. Conversely, pseudohypocalcemia means that the measured calcium level is low but the corrected serum calcium is normal.
Using this formula, our patient’s corrected calcium level is calculated as 5.7 + 0.8 (4 – 3.2) = 6.3 mg/dL, indicating true hypocalcemia.
PHOSPHATE IS OFTEN HIGH WHEN CALCIUM IS LOW
In addition to hypocalcemia, our patient has an elevated phosphate level (Table 2).
3. Which of the following hypocalcemic disorders is not associated with hyperphosphatemia?
- End-stage renal disease
- Primary hypoparathyroidism
- Pseudohypoparathyroidism
- Vitamin D3 deficiency
- Rhabdomyolysis
Vitamin D deficiency is not associated with hyperphosphatemia.
Second step in evaluating hypocalcemia: Check phosphate, magnesium, creatinine
The major causes of hypocalcemia can be categorized according to the serum phosphate level: high vs normal or low (Table 5).
High-phosphate, low-calcium states. In the absence of concurrent end-stage renal disease and an excessive phosphate load, primary hypoparathyroidism is the most likely cause of hypocalcemia associated with hyperphosphatemia.
PTH increases serum ionized calcium by26,27:
- Increasing bone resorption
- Increasing reabsorption of calcium from the distal renal tubules
- Increasing the activity of 1-alpha-hydroxylase, responsible for conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 (the most biologically active vitamin D metabolite); 1,25-dihydroxyvitamin D increases the absorption of calcium and phosphate from the intestine.
Conversely, PTH decreases reabsorption of phosphate from proximal renal tubules, resulting in hypophosphatemia. Therefore, low serum PTH (primary hypoparathyroidism) or a PTH-resistant state (pseudohypoparathyroidism) results in hypocalcemia and hyperphosphatemia.26,27
Both end-stage renal disease and rhabdomyolysis are associated with high serum phosphate levels. The kidney normally excretes excess dietary phosphate to maintain phosphate homeostasis; however, this is impaired in end-stage renal disease, leading to hyperphosphatemia. In rhabdomyolysis, it is mainly the transcellular shift of phosphate into the extracellular space from myocyte injury that raises phosphate levels.
Normal- or low-phosphate, low calcium states. Hypocalcemia can also result from vitamin D deficiency, but this cause is associated with a low or normal serum phosphate level. In such cases, hypocalcemia causes secondary hyperparathyroidism with consequent renal phosphate loss and, thus, hypophosphatemia.27
Third step: Check serum intact PTH and 25-hydroxyvitamin D levels
Hypocalcemia stimulates secretion of PTH. Therefore, hypocalcemia with elevated serum PTH is caused by disorders that do not impair PTH secretion, including chronic renal failure and vitamin D deficiency (Table 5). Conversely, hypocalcemia with low or normal serum PTH levels suggests primary hypoparathyroidism.
Our patient’s serum PTH level is 20 ng/mL, which is within the reference range. This does not discount the diagnosis of primary hypoparathyroidism. Although most patients with primary hypoparathyroidism have low or undetectable serum PTH levels, some have normal PTH levels if some degree of PTH production is preserved.5,7,28–30 In these patients, the remaining functioning parathyroid tissue is not enough to maintain a normal serum calcium level, resulting in hypocalcemia. As a result, hypocalcemia stimulates the remaining parathyroid tissue to its maximum output, producing PTH levels usually within the lower or middle-normal range.30 In such patients, the terms parathyroid insufficiency and relative primary hypoparathyroidism are more precise than primary hypoparathyroidism.
Postsurgical hypoparathyroidism with an inappropriately normal PTH level is usually seen in patients with disorders that impair intestinal calcium absorption or bone resorption.31 In our patient’s case, the “normal” serum PTH level is likely due to maximal stimulation of remaining functioning parathyroid tissue by severe hypocalcemia, which is a result of her discontinuation of calcium and calcitriol therapy and her vitamin D deficiency.
CASE RESUMED: NO RESPONSE TO INTRAVENOUS CALCIUM GLUCONATE
The patient is given 2 10-mL ampules of 10% calcium gluconate diluted in 100 mL of 5% dextrose in water over 20 minutes intravenously. Electrocardiographic monitoring is continued. Two hours later, her measured serum calcium is only 5.8 mg/dL, with no improvement in her symptoms.
A continuous infusion of calcium gluconate is started: 12 ampules of calcium gluconate are added to 380 mL of 5% dextrose in water and infused at 40 mL/hour (infused rate of elemental calcium = 1.3 mg/kg/hour); 3 hours later, her measured serum calcium level is still only 5.8 mg//dL; at 6 hours it is 5.9 mg/dL, and her symptoms have not improved.
4. Which of the following is the most appropriate next step?
- Change the calcium gluconate to calcium chloride
- Increase the infusion rate to 1.5 mg of elemental calcium/kg/hour
- Give a bolus of 2 10-mL ampules of 10% calcium gluconate intravenously over 1 minute
- Give additional oral calcium tablets
- Check the serum magnesium level
Treatment of hypocalcemia can involve intravenous or oral calcium therapy.
Intravenous calcium is indicated for patients with any of the following6,32:
- Moderate to severe neuromuscular irritability (eg, acral paresthesia, carpopedal spasm, prolonged QT interval, seizures, laryngospasm, bronchospasm)
- Acute hypocalcemia with corrected serum calcium level less than 7.6 mg/dL, even if the patient is asymptomatic
- Cardiac failure.
One 10-mL ampule of 10% calcium gluconate contains 93 mg of elemental calcium; 1 or 2 ampules are typically diluted in 50 to 100 mL of 5% dextrose in water and infused slowly over 15 to 20 minutes. Rapid administration of intravenous calcium is contraindicated, as it may produce cardiac arrhythmias and possibly cardiac arrest. Therefore, intravenous calcium should be given slowly while continuing electrocardiographic monitoring.33
Since the effect of 1 ampule of calcium gluconate lasts only 2 to 3 hours, most patients with symptomatic hypocalcemia require continuous intravenous calcium infusion. The recommended dose of infused elemental calcium is 0.5 to 1.5 mg/kg/hour.34 Several ampules are added to 500 to 1,000 mL of 5% dextrose in water or 0.9% normal saline and infused at a rate appropriate for the patient’s corrected calcium and symptoms.
Oral calcium and vitamin D supplements can be given initially to patients with a corrected serum calcium level of 7.6 mg/dL or greater, with or without mild symptoms, if they can tolerate oral intake. However, this is not the treatment of choice for resistant acute hypocalcemia, as in this case.
Calcium chloride has no advantages over calcium gluconate. Further, it can be associated with local irritation and may result in tissue necrosis if extravasation occurs.35
Increasing the infusion rate of calcium gluconate to the maximum recommended dose may improve the patient’s ionized calcium level and symptoms somewhat. However, it is not the best option for this patient, given that she did not respond to 2 ampules of calcium gluconate followed by continuous infusion of 1.3 mg/kg/hour for 6 hours.
Calcium gluconate bolus. Similarly, giving the patient an additional 2 ampules of calcium gluconate over 1 minute would not be recommended, as rapid administration of intravenous calcium gluconate (eg, over 1 minute) is contraindicated.
Check magnesium
If hypocalcemia persists despite intravenous calcium therapy, as in our patient, further investigation or action is required. An important cause of persistent hypocalcemia is severe hypomagnesemia. Severe hypomagnesemia (serum magnesium < 0.8 mg/dL) causes resistant hypocalcemia by several mechanisms:
- Inducing PTH resistance32,36,37
- Decreasing PTH secretion32,36
- Decreasing calcitriol production.
The decrease in calcitriol production is a direct effect of hypomagnesemia, but it is also an indirect effect of low PTH secretion, which inhibits the enzyme 1-alpha-hydroxylase. Thus, conversion of 25-hydroxyvitamin D3 to calcitriol is impaired, leading to low calcitriol production.
Our patient could have hypomagnesemia due to furosemide use and uncontrolled diabetes mellitus. Hypocalcemia resistant to calcium therapy may occasionally respond to magnesium therapy even if the serum magnesium level is normal. This may be due to depleted intracellular magnesium salt levels.6,38 Rarely, severe hypermagnesemia can also be associated with hypocalcemia due to inhibition of PTH secretion.37,39
CASE RESUMED
Our patient’s serum magnesium level is 0.6 mg/dL (reference range 1.7–2.4 mg/dL). She is given 2 g of magnesium sulfate in 60 mL of 0.9% normal saline infused over 1 hour, followed by a continuous infusion of magnesium sulfate (12 g diluted in 250 mL of 0.9% normal saline, infused over 24 hours). On repeat testing 4 hours later, her serum magnesium level is 0.7 mg/dL, and at 8 hours later it is 0.9 mg/dL. She is subsequently started on oral magnesium oxide 600 mg per day. The magnesium sulfate infusion is continued for another 24 hours.
PREVENTING HYPERCALCIURIA
Patients with low PTH (primary hypoparathyroidism) may have hypercalciuria due to decreased renal tubular calcium reabsorption. Two important measures can minimize hypercalciuria in such patients:
- Keeping the serum calcium level in the low-normal range4,5,40
- Giving a thiazide diuretic (eg, hydrochlorothiazide 12.5–50 mg daily) with a low-salt diet.41,42
A thiazide diuretic is usually started once the 24-hour urine calcium reaches 250 mg.6 Thiazides are thought to enhance both proximal and distal renal tubular calcium reabsorption.43,44
PRIMARY HYPOPARATHYROIDISM: LONG-TERM MANAGEMENT
Long-term management of primary hypoparathyroidism requires calcium and vitamin D supplementation.
Calcium supplements. The most commonly prescribed calcium preparations are calcium carbonate and calcium citrate (containing 40% and 20% elemental calcium, respectively). Calcium carbonate, which is less expensive than calcium citrate, binds with phosphate intake and requires an acidic environment for absorption, and so it is better absorbed when taken with meals. Because calcium citrate does not require an acidic environment for absorption, it is the calcium preparation of choice for patients on proton pump inhibitors, or patients with achlorhydria or constipation.45 Calcium doses vary widely, with most hypoparathyroid patients requiring 1 to 2 g of elemental calcium daily.6
Vitamin D supplements. To promote intestinal absorption, calcium is combined with vitamin D in a fixed-dose preparation given in divided doses.46 Calcitriol (1,25-dihydroxyvitamin D3) is the most active metabolite of vitamin D, with rapid onset and offset of action, and it is the preferred form of vitamin D therapy for patients with hypoparathyroidism. If calcitriol is not available or is not affordable, alphacalcidol (1-alpha-hydroxyvitamin D3) is another option. This is a synthetic analogue of vitamin D that is already hyroxylated at the C1 position. After oral intake, it is hydroxylated in the liver to form calcitriol.
Since renal production of calcitriol is PTH-dependent, in hypoparathyroidism the conversion of 25-hydroxyvitamin D3 to calcitriol is limited. Therefore, vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol) are not the preferred forms of vitamin D for such patients. However, either can be added to calcitriol, as they may have extraskeletal benefits.7
CASE CONCLUDED
Our patient presented with primary parathyroid insufficiency associated with vitamin D deficiency. Therefore, in addition to calcitriol and calcium combined with vitamin D in a fixed-dose preparation, her management included vitamin D3 for her vitamin D deficiency.
She was discharged on these medications. At a follow-up visit 3 weeks later, her measured serum calcium level was 8.6 mg/dL. She reported gradual resolution of her symptoms. She was also referred to a psychiatrist for her depression.
TAKE-HOME POINTS
- Hypocalcemia causes neuromuscular excitability, manifested clinically by tetany.
- Common causes of hypocalcemia include vitamin D deficiency, hypomagnesemia, renal failure, and primary hypoparathyroidism.
- The first step in evaluating hypocalcemia is to correct the measured serum calcium to the serum albumin concentration.
- Laboratory testing for hypocalcemia should include serum phosphorus, magnesium, creatinine, PTH, and 25-hydroxyvitamin D3.
- Primary hypoparathyroidism is characterized by hypocalcemia, hyperphosphatemia, and low serum PTH.
- Moderate to severe manifestations of hypo-
calcemia and acute hypocalcemia (< 7.6 mg/dL), even if asymptomatic, warrant intravenous calcium therapy. - Correction of hypomagnesemia is essential to treat hypocalcemia, especially if resistant to intravenous calcium therapy.
- The goal of chronic management of primary hypoparathyroidism includes correcting the serum calcium level to a low-normal range, the serum phosphorus level to an upper-normal range, and prevention of hypercalciuria.
Acknowledgments: The authors wish to thank Mr. Michael Edward Tierney of the School of Medicine, University of Sydney, Australia, for his linguistic editing of the manuscript.
A 67-year-old woman presents to the emergency department after 8 weeks of progressive numbness and tingling in both hands, involving all fingers. The numbness has increased in severity in the last 3 days. She also has occasional numbness around her mouth. She reports no numbness in her feet.
She says she underwent thyroid surgery twice for thyroid cancer 10 years ago. Her medical history also includes type 2 diabetes mellitus (diagnosed 1 year ago), hypertension, dyslipidemia, and diastolic heart failure (diagnosed 5 years ago).
Her current medications are:
- Metformin 1 g twice a day
- Candesartan 16 mg once a day
- Atorvastatin 20 mg once a day
- Furosemide 40 mg twice a day
- Levothyroxine 100 μg per day
- Calcium carbonate 1,500 mg twice a day
- A vitamin D tablet twice a day, which she has not taken for the last 2 months.
She admits she has not been taking her medications regularly because she has been feeling depressed.
On physical examination, she is alert and oriented but appears anxious. She is not in respiratory distress. Her blood pressure is 150/90 mm Hg and her pulse is 92 beats per minute and regular. There is a thyroidectomy scar on the anterior neck. Her jugular venous pressure is not elevated. Her heart sounds are normal without extra sounds. She has no pulmonary rales and no lower-extremity edema.
The Phalen test and Tinel test for carpal tunnel syndrome are negative in both hands. Using a Katz hand diagram, the patient reports tingling and numbness in all fingers, both palms, and the dorsum of both hands. Tapping the area over the facial nerve does not elicit twitching of the facial muscles (ie, no Chvostek sign), but compression of the upper arm elicits carpal spasm (ie, positive Trousseau sign). There is no evidence of motor weakness in her hands. The rest of the physical examination is unremarkable.
POSSIBLE CAUSES OF NUMBNESS
1. Based on the initial evaluation, which of the following is the most likely cause of our patient’s bilateral hand numbness?
- Hypocalcemia due to primary hypoparathyroidism
- Carpal tunnel syndrome due to primary hypothyroidism
- Diabetic peripheral neuropathy
- Vitamin B12 deficiency due to metformin
- Hypocalcemia due to low serum calcitonin
All the conditions above except low serum calcitonin can cause bilateral hand paresthesia. Our patient most likely has hypocalcemia due to primary hypoparathyroidism.
Hypocalcemia
In our patient, bilateral hand numbness and perioral numbness after stopping vitamin D and a positive Trousseau sign strongly suggest hypocalcemia. The classic physical findings in patients with hypocalcemia are the Trousseau sign and the Chvostek sign. The Trousseau sign is elicited by inflating a blood pressure cuff above the systolic blood pressure for 3 minutes and observing for ischemia-induced carpopedal spasm, wrist and metacarpophalangeal joint flexion, thumb adduction, and interphalangeal joint extension. The Chvostek sign is elicited by tapping over the area of the facial nerve below the zygoma in front of the tragus, resulting in ipsilateral twitching of facial muscles.
Although the Trousseau sign is more sensitive and specific than the Chvostek sign, neither is pathognomonic for hypocalcemia.1 The Chvostek sign has been reported to be negative in 30% of patients with hypocalcemia and positive in 10% of normocalcemic individuals.1 The Trousseau sign, however, is present in 94% of hypocalcemic patients vs 1% of normocalcemic individuals.2
Primary hypoparathyroidism secondary to thyroidectomy. Postsurgical hypoparathyroidism is the most common cause of primary hypoparathyroidism. It results from ischemic injury or accidental removal of the parathyroid glands during anterior neck surgery.3,4 The consequent hypocalcemia can be transient, intermittent, or permanent. Permanent postsurgical hypoparathyroidism is defined as persistent hypocalcemia with insufficient parathyroid hormone (PTH) for more than 12 months after neck surgery; however, some consider 6 months to be enough to define the condition.5–7
The incidence of postsurgical hypoparathyroidism varies considerably with the extent of thyroid surgery and the experience of the surgeon.6,8 In the hands of experienced surgeons, permanent hypoparathyroidism occurs in fewer than 1% of patients after total thyroidectomy, whereas the rate may be higher than 6% with less-experienced surgeons.5,9 Other risk factors for postsurgical hypoparathyroidism include female sex, autoimmune thyroid disease, pregnancy, and lactation.5
Pseudohypoparathyroidism is a group of disorders characterized by renal resistance to PTH, leading to hypocalcemia, hyperphosphatemia, and elevated serum PTH. It is also associated with phenotypic features such as short stature and short fourth metacarpal bones.
Calcitonin deficiency. Calcitonin is a polypeptide hormone secreted from the parafollicular (C) cells of the thyroid gland. After total thyroidectomy, calcitonin levels are expected to be reduced. However, the role of calcitonin in humans is unclear. One study has shown that calcitonin is possibly a vestigial hormone, given that no calcitonin-related disorders (excess or deficiency) have been reported in humans.10
Carpal tunnel syndrome due to hypothyroidism
Our patient also could have primary hypothyroidism as a result of thyroidectomy. Hypothyroidism can cause bilateral hand numbness due to carpal tunnel syndrome, which is mediated by mucopolysaccharide deposition and synovial membrane swelling.11 One study reported that 29% of patients with hypothyroidism had carpal tunnel syndrome.12 Symptoms of carpal tunnel syndrome in hypothyroid patients may occur despite thyroid replacement therapy.13
Carpal tunnel syndrome is a clinical diagnosis. Patients usually experience hand paresthesia in the distribution of the median nerve. Provocative physical tests for carpal tunnel syndrome include the Tinel test, the Phalen test, and the Katz hand diagram, which is considered the best of the 3 tests.14,15 Based on how the patient marks the location and type of symptoms on the diagram, carpal tunnel syndrome is rated as classic, probable, possible, or unlikely (Table 1).14,16,17 The sensitivity of a classic or probable diagram ranges from 64% to 80%, while the specificity ranges from 73% to 90%.14,15
Carpal tunnel syndrome is less likely to be the cause of our patient’s symptoms, as her Katz hand diagram indicates only “possible” carpal tunnel syndrome. Her perioral numbness and positive Trousseau sign make hypocalcemia a more likely cause.
Diabetic peripheral neuropathy
Sensory peripheral neuropathy is a recognized complication of diabetes mellitus. However, neuropathy in diabetic patients most commonly manifests initially as distal symmetrical ascending neuropathy starting in the lower extremities.18 Therefore, diabetic peripheral neuropathy is less likely in this patient since her symptoms are limited to her hands.
Vitamin B12 deficiency
Metformin-induced vitamin B12 deficiency is another possible cause of peripheral neuropathy. It might be secondary to metformin-induced changes in intrinsic factor levels and small-intestine motility with resultant bacterial overgrowth, as well as inhibition of vitamin B12 absorption in the terminal ileum.19
However, metformin-induced vitamin B12 deficiency is not the most likely cause of our patient’s neuropathy, since she has been taking this drug for only 1 year. Vitamin B12 deficiency with consequent peripheral neuropathy is more likely in patients taking metformin in high doses for 10 or more years.20
Laboratory results and electrocardiography
Table 2 shows the patient’s initial laboratory results. Of note, her serum calcium level is 5.7 mg/dL (reference range 8.9–10.1). Electrocardiography in the emergency department shows:
- Prolonged PR interval (23 msec)
- Wide QRS complexes (13 msec)
- Flat T waves
- Prolonged corrected QT interval (475 msec)
- Occasional premature ventricular complexes.
CLINICAL MANIFESTATIONS OF HYPOCALCEMIA
2. Which of the following is not a manifestation of hypocalcemia?
- Tonic-clonic seizures
- Cyanosis
- Cardiac ventricular arrhythmias
- Acute pancreatitis
- Depression
Hypocalcemia can cause a wide range of clinical manifestations (Table 3), the extent and severity of which depend on the severity of hypocalcemia and how quickly it develops. The more acute the hypocalcemia, the more severe the manifestations.21
Tetany can cause seizures
Hypocalcemia is characterized by neuromuscular hyperexcitability, manifested clinically by tetany.22 Manifestations of tetany are numerous and include acral paresthesia, perioral numbness, muscle cramps, carpopedal spasm, and seizures. Tetany is the hallmark of hypocalcemia regardless of etiology. However, certain causes are associated with peculiar clinical manifestations. For example, chronic primary hypoparathyroidism may be associated with basal ganglia calcifications that can result in parkinsonism, other extrapyramidal disorders, and dementia (Table 4).6
Airway spasm can be fatal
A serious manifestation of acute severe hypocalcemia is spasm of the glottis muscles, which may cause cyanosis and, if untreated, death.21
Ventricular arrhythmias
Another potential fatal complication of acute severe hypocalcemia is polymorphic ventricular tachycardia due to prolongation of the QT interval, which is readily identified with electrocardiography.23
Hypocalcemia does not cause pancreatitis
Hypercalcemia, rather than hypocalcemia, may cause acute pancreatitis.24 Conversely, acute pancreatitis may cause hypocalcemia due to precipitation of calcium in the abdominal cavity.25
Psychiatric manifestations
In addition to depression, hypocalcemia is associated with psychiatric manifestations including anxiety, confusion, and emotional instability.
STEPS TO DIAGNOSIS OF HYPOCALCEMIA
First step: Confirm true hypocalcemia
Calcium circulates in the blood in 3 forms: bound to albumin (40% to 45%), bound to anions (10% to 15%), and free (ionized) (45%). Although ionized calcium is the active form, most laboratories report total serum calcium.
Since changes in serum albumin concentration affect the total serum calcium level, it is imperative to correct the measured serum calcium to the serum albumin concentration. Each 1-g/dL decrease in serum albumin lowers the total serum calcium by 0.8 mg/dL. Thus:
Corrected serum calcium (mg/dL) =
measured total serum calcium (mg/dL) +
0.8 (4 − serum albumin [g/dL]).
If the patient’s serum calcium level remains low when corrected for serum albumin, he or she has true hypocalcemia, which implies a low ionized serum calcium. Conversely, pseudohypocalcemia means that the measured calcium level is low but the corrected serum calcium is normal.
Using this formula, our patient’s corrected calcium level is calculated as 5.7 + 0.8 (4 – 3.2) = 6.3 mg/dL, indicating true hypocalcemia.
PHOSPHATE IS OFTEN HIGH WHEN CALCIUM IS LOW
In addition to hypocalcemia, our patient has an elevated phosphate level (Table 2).
3. Which of the following hypocalcemic disorders is not associated with hyperphosphatemia?
- End-stage renal disease
- Primary hypoparathyroidism
- Pseudohypoparathyroidism
- Vitamin D3 deficiency
- Rhabdomyolysis
Vitamin D deficiency is not associated with hyperphosphatemia.
Second step in evaluating hypocalcemia: Check phosphate, magnesium, creatinine
The major causes of hypocalcemia can be categorized according to the serum phosphate level: high vs normal or low (Table 5).
High-phosphate, low-calcium states. In the absence of concurrent end-stage renal disease and an excessive phosphate load, primary hypoparathyroidism is the most likely cause of hypocalcemia associated with hyperphosphatemia.
PTH increases serum ionized calcium by26,27:
- Increasing bone resorption
- Increasing reabsorption of calcium from the distal renal tubules
- Increasing the activity of 1-alpha-hydroxylase, responsible for conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 (the most biologically active vitamin D metabolite); 1,25-dihydroxyvitamin D increases the absorption of calcium and phosphate from the intestine.
Conversely, PTH decreases reabsorption of phosphate from proximal renal tubules, resulting in hypophosphatemia. Therefore, low serum PTH (primary hypoparathyroidism) or a PTH-resistant state (pseudohypoparathyroidism) results in hypocalcemia and hyperphosphatemia.26,27
Both end-stage renal disease and rhabdomyolysis are associated with high serum phosphate levels. The kidney normally excretes excess dietary phosphate to maintain phosphate homeostasis; however, this is impaired in end-stage renal disease, leading to hyperphosphatemia. In rhabdomyolysis, it is mainly the transcellular shift of phosphate into the extracellular space from myocyte injury that raises phosphate levels.
Normal- or low-phosphate, low calcium states. Hypocalcemia can also result from vitamin D deficiency, but this cause is associated with a low or normal serum phosphate level. In such cases, hypocalcemia causes secondary hyperparathyroidism with consequent renal phosphate loss and, thus, hypophosphatemia.27
Third step: Check serum intact PTH and 25-hydroxyvitamin D levels
Hypocalcemia stimulates secretion of PTH. Therefore, hypocalcemia with elevated serum PTH is caused by disorders that do not impair PTH secretion, including chronic renal failure and vitamin D deficiency (Table 5). Conversely, hypocalcemia with low or normal serum PTH levels suggests primary hypoparathyroidism.
Our patient’s serum PTH level is 20 ng/mL, which is within the reference range. This does not discount the diagnosis of primary hypoparathyroidism. Although most patients with primary hypoparathyroidism have low or undetectable serum PTH levels, some have normal PTH levels if some degree of PTH production is preserved.5,7,28–30 In these patients, the remaining functioning parathyroid tissue is not enough to maintain a normal serum calcium level, resulting in hypocalcemia. As a result, hypocalcemia stimulates the remaining parathyroid tissue to its maximum output, producing PTH levels usually within the lower or middle-normal range.30 In such patients, the terms parathyroid insufficiency and relative primary hypoparathyroidism are more precise than primary hypoparathyroidism.
Postsurgical hypoparathyroidism with an inappropriately normal PTH level is usually seen in patients with disorders that impair intestinal calcium absorption or bone resorption.31 In our patient’s case, the “normal” serum PTH level is likely due to maximal stimulation of remaining functioning parathyroid tissue by severe hypocalcemia, which is a result of her discontinuation of calcium and calcitriol therapy and her vitamin D deficiency.
CASE RESUMED: NO RESPONSE TO INTRAVENOUS CALCIUM GLUCONATE
The patient is given 2 10-mL ampules of 10% calcium gluconate diluted in 100 mL of 5% dextrose in water over 20 minutes intravenously. Electrocardiographic monitoring is continued. Two hours later, her measured serum calcium is only 5.8 mg/dL, with no improvement in her symptoms.
A continuous infusion of calcium gluconate is started: 12 ampules of calcium gluconate are added to 380 mL of 5% dextrose in water and infused at 40 mL/hour (infused rate of elemental calcium = 1.3 mg/kg/hour); 3 hours later, her measured serum calcium level is still only 5.8 mg//dL; at 6 hours it is 5.9 mg/dL, and her symptoms have not improved.
4. Which of the following is the most appropriate next step?
- Change the calcium gluconate to calcium chloride
- Increase the infusion rate to 1.5 mg of elemental calcium/kg/hour
- Give a bolus of 2 10-mL ampules of 10% calcium gluconate intravenously over 1 minute
- Give additional oral calcium tablets
- Check the serum magnesium level
Treatment of hypocalcemia can involve intravenous or oral calcium therapy.
Intravenous calcium is indicated for patients with any of the following6,32:
- Moderate to severe neuromuscular irritability (eg, acral paresthesia, carpopedal spasm, prolonged QT interval, seizures, laryngospasm, bronchospasm)
- Acute hypocalcemia with corrected serum calcium level less than 7.6 mg/dL, even if the patient is asymptomatic
- Cardiac failure.
One 10-mL ampule of 10% calcium gluconate contains 93 mg of elemental calcium; 1 or 2 ampules are typically diluted in 50 to 100 mL of 5% dextrose in water and infused slowly over 15 to 20 minutes. Rapid administration of intravenous calcium is contraindicated, as it may produce cardiac arrhythmias and possibly cardiac arrest. Therefore, intravenous calcium should be given slowly while continuing electrocardiographic monitoring.33
Since the effect of 1 ampule of calcium gluconate lasts only 2 to 3 hours, most patients with symptomatic hypocalcemia require continuous intravenous calcium infusion. The recommended dose of infused elemental calcium is 0.5 to 1.5 mg/kg/hour.34 Several ampules are added to 500 to 1,000 mL of 5% dextrose in water or 0.9% normal saline and infused at a rate appropriate for the patient’s corrected calcium and symptoms.
Oral calcium and vitamin D supplements can be given initially to patients with a corrected serum calcium level of 7.6 mg/dL or greater, with or without mild symptoms, if they can tolerate oral intake. However, this is not the treatment of choice for resistant acute hypocalcemia, as in this case.
Calcium chloride has no advantages over calcium gluconate. Further, it can be associated with local irritation and may result in tissue necrosis if extravasation occurs.35
Increasing the infusion rate of calcium gluconate to the maximum recommended dose may improve the patient’s ionized calcium level and symptoms somewhat. However, it is not the best option for this patient, given that she did not respond to 2 ampules of calcium gluconate followed by continuous infusion of 1.3 mg/kg/hour for 6 hours.
Calcium gluconate bolus. Similarly, giving the patient an additional 2 ampules of calcium gluconate over 1 minute would not be recommended, as rapid administration of intravenous calcium gluconate (eg, over 1 minute) is contraindicated.
Check magnesium
If hypocalcemia persists despite intravenous calcium therapy, as in our patient, further investigation or action is required. An important cause of persistent hypocalcemia is severe hypomagnesemia. Severe hypomagnesemia (serum magnesium < 0.8 mg/dL) causes resistant hypocalcemia by several mechanisms:
- Inducing PTH resistance32,36,37
- Decreasing PTH secretion32,36
- Decreasing calcitriol production.
The decrease in calcitriol production is a direct effect of hypomagnesemia, but it is also an indirect effect of low PTH secretion, which inhibits the enzyme 1-alpha-hydroxylase. Thus, conversion of 25-hydroxyvitamin D3 to calcitriol is impaired, leading to low calcitriol production.
Our patient could have hypomagnesemia due to furosemide use and uncontrolled diabetes mellitus. Hypocalcemia resistant to calcium therapy may occasionally respond to magnesium therapy even if the serum magnesium level is normal. This may be due to depleted intracellular magnesium salt levels.6,38 Rarely, severe hypermagnesemia can also be associated with hypocalcemia due to inhibition of PTH secretion.37,39
CASE RESUMED
Our patient’s serum magnesium level is 0.6 mg/dL (reference range 1.7–2.4 mg/dL). She is given 2 g of magnesium sulfate in 60 mL of 0.9% normal saline infused over 1 hour, followed by a continuous infusion of magnesium sulfate (12 g diluted in 250 mL of 0.9% normal saline, infused over 24 hours). On repeat testing 4 hours later, her serum magnesium level is 0.7 mg/dL, and at 8 hours later it is 0.9 mg/dL. She is subsequently started on oral magnesium oxide 600 mg per day. The magnesium sulfate infusion is continued for another 24 hours.
PREVENTING HYPERCALCIURIA
Patients with low PTH (primary hypoparathyroidism) may have hypercalciuria due to decreased renal tubular calcium reabsorption. Two important measures can minimize hypercalciuria in such patients:
- Keeping the serum calcium level in the low-normal range4,5,40
- Giving a thiazide diuretic (eg, hydrochlorothiazide 12.5–50 mg daily) with a low-salt diet.41,42
A thiazide diuretic is usually started once the 24-hour urine calcium reaches 250 mg.6 Thiazides are thought to enhance both proximal and distal renal tubular calcium reabsorption.43,44
PRIMARY HYPOPARATHYROIDISM: LONG-TERM MANAGEMENT
Long-term management of primary hypoparathyroidism requires calcium and vitamin D supplementation.
Calcium supplements. The most commonly prescribed calcium preparations are calcium carbonate and calcium citrate (containing 40% and 20% elemental calcium, respectively). Calcium carbonate, which is less expensive than calcium citrate, binds with phosphate intake and requires an acidic environment for absorption, and so it is better absorbed when taken with meals. Because calcium citrate does not require an acidic environment for absorption, it is the calcium preparation of choice for patients on proton pump inhibitors, or patients with achlorhydria or constipation.45 Calcium doses vary widely, with most hypoparathyroid patients requiring 1 to 2 g of elemental calcium daily.6
Vitamin D supplements. To promote intestinal absorption, calcium is combined with vitamin D in a fixed-dose preparation given in divided doses.46 Calcitriol (1,25-dihydroxyvitamin D3) is the most active metabolite of vitamin D, with rapid onset and offset of action, and it is the preferred form of vitamin D therapy for patients with hypoparathyroidism. If calcitriol is not available or is not affordable, alphacalcidol (1-alpha-hydroxyvitamin D3) is another option. This is a synthetic analogue of vitamin D that is already hyroxylated at the C1 position. After oral intake, it is hydroxylated in the liver to form calcitriol.
Since renal production of calcitriol is PTH-dependent, in hypoparathyroidism the conversion of 25-hydroxyvitamin D3 to calcitriol is limited. Therefore, vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol) are not the preferred forms of vitamin D for such patients. However, either can be added to calcitriol, as they may have extraskeletal benefits.7
CASE CONCLUDED
Our patient presented with primary parathyroid insufficiency associated with vitamin D deficiency. Therefore, in addition to calcitriol and calcium combined with vitamin D in a fixed-dose preparation, her management included vitamin D3 for her vitamin D deficiency.
She was discharged on these medications. At a follow-up visit 3 weeks later, her measured serum calcium level was 8.6 mg/dL. She reported gradual resolution of her symptoms. She was also referred to a psychiatrist for her depression.
TAKE-HOME POINTS
- Hypocalcemia causes neuromuscular excitability, manifested clinically by tetany.
- Common causes of hypocalcemia include vitamin D deficiency, hypomagnesemia, renal failure, and primary hypoparathyroidism.
- The first step in evaluating hypocalcemia is to correct the measured serum calcium to the serum albumin concentration.
- Laboratory testing for hypocalcemia should include serum phosphorus, magnesium, creatinine, PTH, and 25-hydroxyvitamin D3.
- Primary hypoparathyroidism is characterized by hypocalcemia, hyperphosphatemia, and low serum PTH.
- Moderate to severe manifestations of hypo-
calcemia and acute hypocalcemia (< 7.6 mg/dL), even if asymptomatic, warrant intravenous calcium therapy. - Correction of hypomagnesemia is essential to treat hypocalcemia, especially if resistant to intravenous calcium therapy.
- The goal of chronic management of primary hypoparathyroidism includes correcting the serum calcium level to a low-normal range, the serum phosphorus level to an upper-normal range, and prevention of hypercalciuria.
Acknowledgments: The authors wish to thank Mr. Michael Edward Tierney of the School of Medicine, University of Sydney, Australia, for his linguistic editing of the manuscript.
- Jesus JE, Landry A. Images in clinical medicine. Chvostek’s and Trousseau’s signs. N Engl J Med 2012; 367:e15.
- Urbano FL. Signs of hypocalcemia: Chvostek’s and Trousseau’s. Hosp Physician 2000; 36:43–45.
- Chisthi MM, Nair RS, Kuttanchettiyar KG, Yadev I. Mechanisms behind post-thyroidectomy hypocalcemia: interplay of calcitonin, parathormone, and albumin—a prospective study. J Invest Surg 2017; 30:217–225.
- Shoback DM, Bilezikian JP, Costa AG, et al. Presentation of hypoparathyroidism: etiologies and clinical features. J Clin Endocrinol Metab 2016; 101:2300–2312.
- Stack BC Jr, Bimston DN, Bodenner DL, et al. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: postoperative hypoparathyroidism—definitions and management. Endocr Pract 2015; 21:674–685.
- Shoback D. Clinical practice. Hypoparathyroidism. N Engl J Med 2008; 359:391–403.
- Abate EG, Clarke BL. Review of hypoparathyroidism. Front Endocrinol (Lausanne) 2017; 7:172.
- Coimbra C, Monteiro F, Oliveira P, Ribeiro L, de Almeida MG, Condé A. Hypoparathyroidism following thyroidectomy: predictive factors. Acta Otorrinolaringol Esp 2017; 68:106–111.
- Thomusch O, Machens A, Sekulla C, Ukkat J, Brauckhoff M, Dralle H. The impact of surgical technique on postoperative hypoparathyroidism in bilateral thyroid surgery: a multivariate analysis of 5846 consecutive patients. Surgery 2003; 133:180–185.
- Hirsch PF, Lester GE, Talmage RV. Calcitonin, an enigmatic hormone: does it have a function? J Musculoskelet Neuronal Interact 2001; 1:299–305.
- Karne SS, Bhalerao NS. Carpal tunnel syndrome in hypothyroidism. J Clin Diagn Res 2016; 10:OC36–OC38.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Palumbo CF, Szabo RM, Olmsted SL. The effects of hypothyroidism and thyroid replacement on the development of carpal tunnel syndrome. J Hand Surg Am 2000; 25:734–739.
- Katz JN, Stirrat CR, Larson MG, Fossel AH, Eaton HM, Liang MH. A self-administered hand symptom diagram for the diagnosis and epidemiologic study of carpal tunnel syndrome. J Rheumatol 1990; 17:1495–1498.
- Katz JN, Stirrat CR. A self-administered hand diagram for the diagnosis of carpal tunnel syndrome. J Hand Surg Am 1990; 15:360–363.
- Calfee RP, Dale AM, Ryan D, Descatha A, Franzblau A, Evanoff B. Performance of simplified scoring systems for hand diagrams in carpal tunnel syndrome screening. J Hand Surg Am 2012; 37:10–17.
- D’Arcy CA, McGee S. The rational clinical examination. Does this patient have carpal tunnel syndrome? JAMA 2000; 283:3110–3117.
- Marchettini P, Lacerenza M, Mauri E, Marangoni C. Painful peripheral neuropathies. Curr Neuropharmacol 2006; 4:175–181.
- Kibirige D, Mwebaze R. Vitamin B12 deficiency among patients with diabetes mellitus: is routine screening and supplementation justified? J Diabetes Metab Disord 2013;12:17.
- Akinlade KS, Agbebaku SO, Rahamon SK, Balogun WO. Vitamin B12 levels in patients with type 2 diabetes mellitus on metformin. Ann Ib Postgrad Med 2015; 13:79–83.
- Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am 1993; 22:363–375.
- Macefield G, Burke D. Paraesthesiae and tetany induced by voluntary hyperventilation. Increased excitability of human cutaneous and motor axons. Brain 1991; 114:527–540.
- Benoit SR, Mendelsohn AB, Nourjah P, Staffa JA, Graham DJ. Risk factors for prolonged QTc among US adults: Third National Health and Nutrition Examination Survey. Eur J Cardiovasc Prev Rehabil 2005; 12:363–368.
- Khoo TK, Vege SS, Abu-Lebdeh HS, Ryu E, Nadeem S, Wermers RA. Acute pancreatitis in primary hyperparathyroidism: a population-based study. J Clin Endocrinol Metab 2009; 94:2115–2118.
- McKay C, Beastall GH, Imrie CW, Baxter JN. Circulating intact parathyroid hormone levels in acute pancreatitis. Br J Surg 1994; 81:357–360.
- Talmage RV, Mobley HT. Calcium homeostasis: reassessment of the actions of parathyroid hormone. Gen Comp Endocrinol 2008; 156:1–8.
- Friedman PA, Gesek FA. Calcium transport in renal epithelial cells. Am J Physiol 1993; 264:F181–F198.
- Jensen PV, Jelstrup SM, Homøe P. Long-term outcomes after total thyroidectomy. Dan Med J 2015; 62:A5156.
- Ritter K, Elfenbein D, Schneider DF, Chen H, Sippel RS. Hypoparathyroidism after total thyroidectomy: incidence and resolution. J Surg Res 2015; 197:348–353.
- Promberger R, Ott J, Kober F, Karik M, Freissmuth M, Hermann M. Normal parathyroid hormone levels do not exclude permanent hypoparathyroidism after thyroidectomy. Thyroid 2011; 21:145–150.
- Lorente-Poch L, Sancho JJ, Muñoz-Nova JL, Sánchez-Velázquez P, Sitges-Serra A. Defining the syndromes of parathyroid failure after total thyroidectomy. Gland Surgery 2015; 4:82–90.
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ 2008; 336:1298–1302.
- Tohme JF, Bilezikian JP. Diagnosis and treatment of hypocalcemic emergencies. Endocrinologist 1996; 6:10–18.
- Carroll R, Matfin G. Endocrine and metabolic emergencies: hypocalcaemia. Ther Adv Endocrinol Metab 2010; 1:29–33.
- Kim MP, Raho VJ, Mak J, Kaynar AM. Skin and soft tissue necrosis from calcium chloride in a deicer. J Emerg Med 2007; 32:41–44.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20:3–17.
- Cholst IN, Steinberg SF, Tropper PJ, Fox HE, Segre GV, Bilezikian JP. The influence of hypermagnesemia on serum calcium and parathyroid hormone levels in human subjects. N Engl J Med 1984; 310:1221–1225.
- Ryzen E, Nelson TA, Rude RK. Low blood mononuclear cell magnesium content and hypocalcemia in normomagnesemic patients. West J Med 1987; 147:549–553.
- Koontz SL, Friedman SA, Schwartz ML. Symptomatic hypocalcemia after tocolytic therapy with magnesium sulfate and nifedipine. Am J Obstet Gynecol 2004; 190:1773–1776.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Porter RH, Cox BG, Heaney D, Hostetter TH, Stinebaugh BJ, Suki WN. Treatment of hypoparathyroid patients with chlorthalidone. N Engl J Med 1978; 298:577–581.
- Clarke BL, Brown EM, Collins MT, et al. Epidemiology and diagnosis of hypoparathyroidism. J Clin Endocrinol Metab 2016; 101:2284–2299.
- Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 2005; 115:1651–1658.
- Costanzo LS. Localization of diuretic action in microperfused rat distal tubules: Ca and Na transport. Am J Physiol 1985; 248:F527–F535.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Scotti A, Bianchini C, Abbiati G, Marzo A. Absorption of calcium administered alone or in fixed combination with vitamin D to healthy volunteers. Arzneimittelforschung 2001; 51:493–500.
- Jesus JE, Landry A. Images in clinical medicine. Chvostek’s and Trousseau’s signs. N Engl J Med 2012; 367:e15.
- Urbano FL. Signs of hypocalcemia: Chvostek’s and Trousseau’s. Hosp Physician 2000; 36:43–45.
- Chisthi MM, Nair RS, Kuttanchettiyar KG, Yadev I. Mechanisms behind post-thyroidectomy hypocalcemia: interplay of calcitonin, parathormone, and albumin—a prospective study. J Invest Surg 2017; 30:217–225.
- Shoback DM, Bilezikian JP, Costa AG, et al. Presentation of hypoparathyroidism: etiologies and clinical features. J Clin Endocrinol Metab 2016; 101:2300–2312.
- Stack BC Jr, Bimston DN, Bodenner DL, et al. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: postoperative hypoparathyroidism—definitions and management. Endocr Pract 2015; 21:674–685.
- Shoback D. Clinical practice. Hypoparathyroidism. N Engl J Med 2008; 359:391–403.
- Abate EG, Clarke BL. Review of hypoparathyroidism. Front Endocrinol (Lausanne) 2017; 7:172.
- Coimbra C, Monteiro F, Oliveira P, Ribeiro L, de Almeida MG, Condé A. Hypoparathyroidism following thyroidectomy: predictive factors. Acta Otorrinolaringol Esp 2017; 68:106–111.
- Thomusch O, Machens A, Sekulla C, Ukkat J, Brauckhoff M, Dralle H. The impact of surgical technique on postoperative hypoparathyroidism in bilateral thyroid surgery: a multivariate analysis of 5846 consecutive patients. Surgery 2003; 133:180–185.
- Hirsch PF, Lester GE, Talmage RV. Calcitonin, an enigmatic hormone: does it have a function? J Musculoskelet Neuronal Interact 2001; 1:299–305.
- Karne SS, Bhalerao NS. Carpal tunnel syndrome in hypothyroidism. J Clin Diagn Res 2016; 10:OC36–OC38.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Palumbo CF, Szabo RM, Olmsted SL. The effects of hypothyroidism and thyroid replacement on the development of carpal tunnel syndrome. J Hand Surg Am 2000; 25:734–739.
- Katz JN, Stirrat CR, Larson MG, Fossel AH, Eaton HM, Liang MH. A self-administered hand symptom diagram for the diagnosis and epidemiologic study of carpal tunnel syndrome. J Rheumatol 1990; 17:1495–1498.
- Katz JN, Stirrat CR. A self-administered hand diagram for the diagnosis of carpal tunnel syndrome. J Hand Surg Am 1990; 15:360–363.
- Calfee RP, Dale AM, Ryan D, Descatha A, Franzblau A, Evanoff B. Performance of simplified scoring systems for hand diagrams in carpal tunnel syndrome screening. J Hand Surg Am 2012; 37:10–17.
- D’Arcy CA, McGee S. The rational clinical examination. Does this patient have carpal tunnel syndrome? JAMA 2000; 283:3110–3117.
- Marchettini P, Lacerenza M, Mauri E, Marangoni C. Painful peripheral neuropathies. Curr Neuropharmacol 2006; 4:175–181.
- Kibirige D, Mwebaze R. Vitamin B12 deficiency among patients with diabetes mellitus: is routine screening and supplementation justified? J Diabetes Metab Disord 2013;12:17.
- Akinlade KS, Agbebaku SO, Rahamon SK, Balogun WO. Vitamin B12 levels in patients with type 2 diabetes mellitus on metformin. Ann Ib Postgrad Med 2015; 13:79–83.
- Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am 1993; 22:363–375.
- Macefield G, Burke D. Paraesthesiae and tetany induced by voluntary hyperventilation. Increased excitability of human cutaneous and motor axons. Brain 1991; 114:527–540.
- Benoit SR, Mendelsohn AB, Nourjah P, Staffa JA, Graham DJ. Risk factors for prolonged QTc among US adults: Third National Health and Nutrition Examination Survey. Eur J Cardiovasc Prev Rehabil 2005; 12:363–368.
- Khoo TK, Vege SS, Abu-Lebdeh HS, Ryu E, Nadeem S, Wermers RA. Acute pancreatitis in primary hyperparathyroidism: a population-based study. J Clin Endocrinol Metab 2009; 94:2115–2118.
- McKay C, Beastall GH, Imrie CW, Baxter JN. Circulating intact parathyroid hormone levels in acute pancreatitis. Br J Surg 1994; 81:357–360.
- Talmage RV, Mobley HT. Calcium homeostasis: reassessment of the actions of parathyroid hormone. Gen Comp Endocrinol 2008; 156:1–8.
- Friedman PA, Gesek FA. Calcium transport in renal epithelial cells. Am J Physiol 1993; 264:F181–F198.
- Jensen PV, Jelstrup SM, Homøe P. Long-term outcomes after total thyroidectomy. Dan Med J 2015; 62:A5156.
- Ritter K, Elfenbein D, Schneider DF, Chen H, Sippel RS. Hypoparathyroidism after total thyroidectomy: incidence and resolution. J Surg Res 2015; 197:348–353.
- Promberger R, Ott J, Kober F, Karik M, Freissmuth M, Hermann M. Normal parathyroid hormone levels do not exclude permanent hypoparathyroidism after thyroidectomy. Thyroid 2011; 21:145–150.
- Lorente-Poch L, Sancho JJ, Muñoz-Nova JL, Sánchez-Velázquez P, Sitges-Serra A. Defining the syndromes of parathyroid failure after total thyroidectomy. Gland Surgery 2015; 4:82–90.
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ 2008; 336:1298–1302.
- Tohme JF, Bilezikian JP. Diagnosis and treatment of hypocalcemic emergencies. Endocrinologist 1996; 6:10–18.
- Carroll R, Matfin G. Endocrine and metabolic emergencies: hypocalcaemia. Ther Adv Endocrinol Metab 2010; 1:29–33.
- Kim MP, Raho VJ, Mak J, Kaynar AM. Skin and soft tissue necrosis from calcium chloride in a deicer. J Emerg Med 2007; 32:41–44.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20:3–17.
- Cholst IN, Steinberg SF, Tropper PJ, Fox HE, Segre GV, Bilezikian JP. The influence of hypermagnesemia on serum calcium and parathyroid hormone levels in human subjects. N Engl J Med 1984; 310:1221–1225.
- Ryzen E, Nelson TA, Rude RK. Low blood mononuclear cell magnesium content and hypocalcemia in normomagnesemic patients. West J Med 1987; 147:549–553.
- Koontz SL, Friedman SA, Schwartz ML. Symptomatic hypocalcemia after tocolytic therapy with magnesium sulfate and nifedipine. Am J Obstet Gynecol 2004; 190:1773–1776.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Porter RH, Cox BG, Heaney D, Hostetter TH, Stinebaugh BJ, Suki WN. Treatment of hypoparathyroid patients with chlorthalidone. N Engl J Med 1978; 298:577–581.
- Clarke BL, Brown EM, Collins MT, et al. Epidemiology and diagnosis of hypoparathyroidism. J Clin Endocrinol Metab 2016; 101:2284–2299.
- Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 2005; 115:1651–1658.
- Costanzo LS. Localization of diuretic action in microperfused rat distal tubules: Ca and Na transport. Am J Physiol 1985; 248:F527–F535.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Scotti A, Bianchini C, Abbiati G, Marzo A. Absorption of calcium administered alone or in fixed combination with vitamin D to healthy volunteers. Arzneimittelforschung 2001; 51:493–500.
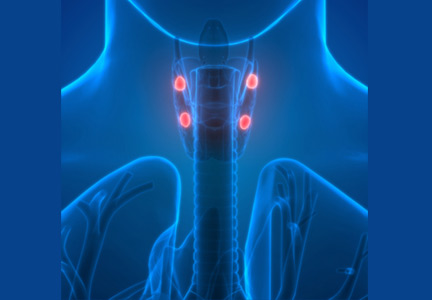
Ascites from intraperitoneal urine leakage after pelvic radiation
A 44-year-old woman was admitted to the hospital for the second time in 2 months with acute onset of severe abdominal pain. She had a history of cervical cancer treated with total hysterectomy with bilateral salpingo-oophorectomy, chemotherapy, and radiotherapy at age 38.
 and irregularity in the bladder wall (arrow).")
. Right, cystography showed intraperitoneal leakage of contrast medium from the bladder (white arrow).")
LONG-TERM EFFECTS OF RADIATION ON THE BLADDER
Urinary ascites from intraperitoneal urine leakage is a rare but clinically important sequel to bladder fistula or bladder wall rupture. Fistula or rupture can be caused by pelvic irradiation, blunt trauma, or surgical procedures, but may also be spontaneous.2
When the total radiation dose to the bladder exceeds 60 Gy, radiation cystitis may occur, leading to bladder fistula.3 Effects of radiation on the bladder are usually seen within 2 to 4 years3 but may occur long after the completion of radiation therapy—10 years2 or even 30 to 40 years later.4 Therefore, ascites of unknown origin in a patient with a history of pelvic radiation therapy should lead to an evaluation for late radiation cystitis and urinary ascites from bladder rupture.
- Ramcharan K, Poon-King TM, Indar R. Spontaneous intraperitoneal rupture of a neurogenic bladder; the importance of ascitic fluid urea and electrolytes in diagnosis. Postgrad Med J 1987; 63:999–1000.
- Matsumura M, Ando N, Kumabe A, Dhaliwal G. Pseudo-renal failure: bladder rupture with urinary ascites. BMJ Case Rep 2015; pii:bcr2015212671.
- Shi F, Wang T, Wang J, et al. Peritoneal bladder fistula following radiotherapy for cervical cancer: a case report. Oncol Lett 2016; 12:2008–2010.
- Hayashi W, Nishino T, Namie S, Obata Y, Furukawa M, Kohno S. Spontaneous bladder rupture diagnosis based on urinary appearance of mesothelial cells: a case report. J Med Case Rep 2014; 8:46.
A 44-year-old woman was admitted to the hospital for the second time in 2 months with acute onset of severe abdominal pain. She had a history of cervical cancer treated with total hysterectomy with bilateral salpingo-oophorectomy, chemotherapy, and radiotherapy at age 38.
LONG-TERM EFFECTS OF RADIATION ON THE BLADDER
Urinary ascites from intraperitoneal urine leakage is a rare but clinically important sequel to bladder fistula or bladder wall rupture. Fistula or rupture can be caused by pelvic irradiation, blunt trauma, or surgical procedures, but may also be spontaneous.2
When the total radiation dose to the bladder exceeds 60 Gy, radiation cystitis may occur, leading to bladder fistula.3 Effects of radiation on the bladder are usually seen within 2 to 4 years3 but may occur long after the completion of radiation therapy—10 years2 or even 30 to 40 years later.4 Therefore, ascites of unknown origin in a patient with a history of pelvic radiation therapy should lead to an evaluation for late radiation cystitis and urinary ascites from bladder rupture.
A 44-year-old woman was admitted to the hospital for the second time in 2 months with acute onset of severe abdominal pain. She had a history of cervical cancer treated with total hysterectomy with bilateral salpingo-oophorectomy, chemotherapy, and radiotherapy at age 38.
LONG-TERM EFFECTS OF RADIATION ON THE BLADDER
Urinary ascites from intraperitoneal urine leakage is a rare but clinically important sequel to bladder fistula or bladder wall rupture. Fistula or rupture can be caused by pelvic irradiation, blunt trauma, or surgical procedures, but may also be spontaneous.2
When the total radiation dose to the bladder exceeds 60 Gy, radiation cystitis may occur, leading to bladder fistula.3 Effects of radiation on the bladder are usually seen within 2 to 4 years3 but may occur long after the completion of radiation therapy—10 years2 or even 30 to 40 years later.4 Therefore, ascites of unknown origin in a patient with a history of pelvic radiation therapy should lead to an evaluation for late radiation cystitis and urinary ascites from bladder rupture.
- Ramcharan K, Poon-King TM, Indar R. Spontaneous intraperitoneal rupture of a neurogenic bladder; the importance of ascitic fluid urea and electrolytes in diagnosis. Postgrad Med J 1987; 63:999–1000.
- Matsumura M, Ando N, Kumabe A, Dhaliwal G. Pseudo-renal failure: bladder rupture with urinary ascites. BMJ Case Rep 2015; pii:bcr2015212671.
- Shi F, Wang T, Wang J, et al. Peritoneal bladder fistula following radiotherapy for cervical cancer: a case report. Oncol Lett 2016; 12:2008–2010.
- Hayashi W, Nishino T, Namie S, Obata Y, Furukawa M, Kohno S. Spontaneous bladder rupture diagnosis based on urinary appearance of mesothelial cells: a case report. J Med Case Rep 2014; 8:46.
- Ramcharan K, Poon-King TM, Indar R. Spontaneous intraperitoneal rupture of a neurogenic bladder; the importance of ascitic fluid urea and electrolytes in diagnosis. Postgrad Med J 1987; 63:999–1000.
- Matsumura M, Ando N, Kumabe A, Dhaliwal G. Pseudo-renal failure: bladder rupture with urinary ascites. BMJ Case Rep 2015; pii:bcr2015212671.
- Shi F, Wang T, Wang J, et al. Peritoneal bladder fistula following radiotherapy for cervical cancer: a case report. Oncol Lett 2016; 12:2008–2010.
- Hayashi W, Nishino T, Namie S, Obata Y, Furukawa M, Kohno S. Spontaneous bladder rupture diagnosis based on urinary appearance of mesothelial cells: a case report. J Med Case Rep 2014; 8:46.
How should electrolyte abnormalities be managed in patients with chronic kidney disease?
Case
A 55-year-old woman with diabetes, hypertension, and chronic kidney disease (CKD) stage 4 is admitted to the hospital for treatment of left lower-extremity cellulitis. Laboratory studies on admission show a creatinine level of 2.5 mg/dL (glomerular filtration rate [GFR] is 20 mL/min per 1.73 m2; baseline creatinine is between 2.2 and 2.6 mg/dL), potassium level of 3.0 mEq/L, magnesium level of 1.5 mEq/L, bicarbonate level of 18 mEq/L, phosphate level of 6.5 mg/dL, and calcium level of 7.5 mg/dL.
She is put on renally dosed vancomycin to treat her cellulitis. As the hospitalist, how should you manage her multiple electrolyte abnormalities?
Overview of the issue

CKD is also an important risk factor for acute kidney injury. Additionally, rates of readmission for CKD patients are higher than those without CKD. Given that CKD is a “silent disease” that many patients do not realize they have, it is very possible that the first documentation of CKD could happen during an acute hospitalization.
Among the various manifestations of CKD, electrolyte abnormalities are the most likely ones hospitalists will run into.
Overview of the data
Hypokalemia and Hypomagnesemia
Hypokalemia (potassium levels less than 3.5 mEq/L) is not as common as hyperkalemia (potassium levels greater than 5.0 mEq/L) in CKD, which is the result of impaired renal excretion of potassium. Hypokalemia can occur as a result of GI losses, urinary losses, or decreased intake and can be worsened by the use of certain drugs, such as non–K-sparing diuretics.
In the setting of diuretic use involving thiazides and loop diuretics, hypokalemia is dose and sodium-intake dependent. Potassium deficiency worsens the effects of detrimental sodium excess, which plays a role in hypertension and its associated complications. Potassium also has a protective vascular effect, which is a major reason why potassium should be kept normal in patients with CKD.
Acutely, hypokalemia can cause arrhythmias, ileus, and paralysis, which are all indications for immediate repletion. In these cases, hypokalemia must be repleted carefully in small increments (some suggest 20 mEq doses), and the patient must be monitored frequently to avoid hyperkalemia. If patients are persistently hypokalemic, several options can be considered based on the underlying cause. Dietary modifications with foods rich in potassium (containing 250mg/100g) can be suggested. Daily potassium chloride supplementation can be used in those on diuretic therapy who have hypokalemia and metabolic alkalosis (bicarbonate levels greater than 30 mEq/L). Alkalinizing salts, containing citrate or bicarbonate, can be used in hypokalemia without metabolic alkalosis. Initiation of angiotensin-converting-enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), beta-adrenergic blockers, and K-sparing diuretics can be used as well.
Potassium supplementation and K-sparing diuretics should be used with extreme caution in CKD 3 and 4 given the risk of overcorrection. If potassium supplements or drugs to raise serum potassium are initiated in house, potassium should be rechecked within a week. These treatments should be avoided in individuals with diabetes, who are at highest risk for hyperkalemia given hyporeninemic hypoaldosteronism (type IV renal tubular acidosis).

There is emerging evidence that hypomagnesemia can play a part in progression to end-stage renal disease. In the setting of cardiovascular disease, which often co-exists with CKD, the risk of hypomagnesemia precipitating arrhythmia necessitates repletion to a normal level. Any of the magnesium salts and antacids can be used for treatment. K-sparing diuretics are also magnesium sparing. An important side effect of magnesium repletion is diarrhea, which can potentiate electrolyte losses and reduce long-term adherence rates.
Metabolic acidosis
Acid-base balance is maintained by the kidney through urinary excretion of hydrogen ions both as titratable acids and ammonium. In CKD, renal excretion of the daily acid load is impaired, primarily from decreased ammonium excretion caused by there being too few functioning nephrons.
Metabolic acidosis in CKD is defined as a serum bicarbonate concentration of persistently less than 22 mEq/L. The overall prevalence of metabolic acidosis in cases of CKD that don’t require dialysis is about 15% and increases linearly with a decline in GFR. In the Chronic Renal Insufficiency Cohort study, 7%, 13%, and 37% of participants with CKD stages 2, 3, and 4 respectively had metabolic acidosis.
Metabolic acidosis has a variety of adverse outcomes, including bone demineralization, increased protein catabolism and muscle wasting, impaired cardiac function, impaired glucose homeostasis, and systemic inflammation. Additionally, multiple studies have shown an association between metabolic acidosis and progression of CKD and increased mortality.
The 2013 Kidney Disease Improving Global Outcomes (KDIGO) guidelines recommend maintaining the serum bicarbonate level within the reference range (23-29 mEq/L) with alkali therapy. Options include sodium bicarbonate or sodium citrate (which is rapidly metabolized to bicarbonate) in doses of 0.5-1.0 mEq/kg once per day. Sodium bicarbonate is inexpensive; however, it can lead to gastrointestinal upset as the bicarbonate is converted into CO2 in the stomach. This side effect is usually self-limited and improves with time. Typical starting doses are 650 mg twice a day if the serum bicarbonate level is 19-21 mEq/L or 1300 mg twice a day if the serum bicarbonate level is less than or equal to 18 mEq/L.
Sodium citrate can be used if gastrointestinal upset occurs, although caution should be used in those on aluminum binders or with liver disease. Alkali treatment should be started when bicarbonate levels are persistently low (for weeks or months) or if very low (less than or equal to 18 mEq/L) without an acute reversible cause. After patients have begun therapy, they should be monitored for the development of worsening hypertension or edema caused by sodium-mediated fluid retention, although this rarely occurs.
Hyperphosphatemia and Hypocalcemia
Hyperphosphatemia (phosphate levels greater than 4.6 mg/dL) develops early in CKD because of a reduced filtered-phosphate load. Hypocalcemia and hyperphosphatemia can lead to secondary hyperparathyroidism. Given that hyperphosphatemia has been associated with an increased mortality among patients with CKD, treatment is warranted, but the optimal phosphorus range is unknown. According to the KDIGO guidelines, the goal phosphorus level is less than 4.5 mg/dL in patients with CKD who are not on dialysis.
Treatment includes dietary restriction to 900 mg/day and phosphate binders. There is a high phosphate load in processed foods and colas because of food additives. It is therefore recommended to reduce consumption of these foods while encouraging consumption of meat and eggs, which offer additional nutritional value. Those who have failed dietary restrictions should be put on a phosphate binder, either calcium containing (calcium carbonate, calcium acetate) or non–calcium containing (Sevelamer, lanthanum). Non–calcium-containing binders are recommended for patients with hypercalcemia (levels greater than 9.5 mg/dL). There is some data that suggests that non–calcium-containing binders are superior to calcium-containing binders in terms of vascular disease outcomes, but non–calcium-containing binders are sometimes difficult to obtain because of cost and insurance coverage.
Hypocalcemia (calcium levels below 8.4 mg/dL) occurs in the setting of late stage untreated CKD because of decreased GI uptake of calcium from diet in the context of vitamin D deficiency (less than 30 ng/mL) in addition to hyperphosphatemia. Phosphate and vitamin D correction is preferred to calcium supplementation because hyperphosphatemia and vitamin D deficiency occur earlier in CKD. Phosphate reduction is described above.
Regarding vitamin D deficiency, it is recommended to start supplementation with either vitamin D2 or D3. Doses should be adjusted if GFR is less than 30 mL/min per 1.73 m2. It is important to monitor for hypercalcemia, which can also occur in CKD in this context, because it has also been associated with increased morbidity and mortality. If calcium levels are greater than 10.2 mg/dL, all vitamin D supplementation should be discontinued.
Back to the case
Our patient who was admitted for cellulitis has concomitant hypokalemia, hypomagnesemia, acidosis, and hyperphosphatemia with related hypocalcemia. She revealed that her diet was poor prior to her admission for her infection. She was given 20 mEq of potassium orally and placed on a potassium rich diet until potassium levels normalized. She was also given magnesium oxide orally on the first and second day of admission, with repeat levels that were normal. Her acidosis was treated with sodium bicarbonate – 1,300 mg orally twice daily. For her hyperphosphatemia and hypocalcemia, she was placed on phosphate restriction with nutritional counseling with plans to follow up as an outpatient to determine need for phosphate binders. In addition, vitamin D levels were checked, and she was started on repletion for vitamin D deficiency (27 ng/mL). Daily BMP, magnesium, and phosphorus were checked while in house until they were normal for 2 days, and follow-up lab work was requested with her nephrology appointment, which was scheduled for within 1 week.
Bottom line
Electrolyte abnormalities in CKD are numerous and have multiple adverse clinical outcomes. Early intervention and management, especially of metabolic acidosis and hyperphosphatemia, can have a significant effect, including prevention of progression of CKD and possibly reduced mortality.
Dr. Daya, Dr. Apgar, and Dr. Eniasivam are assistant clinical professors in the division of hospital medicine at the University of California, San Francisco.
References
1. Coresh J et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007 Nov 7;298(17):2038-47.
2. Dhondup T et al. Electrolyte and acid-base disorders in chronic kidney disease and end-stage kidney failure. Blood Purif. 2017;43(1-3):179-188.
3. K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis. 2004 May;43(5 Suppl 1):S1-290.
4. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl (2011). 2013 Jan;3(1):1–150.
5. Sakaguchi Y et al. Hypomagnesemia in type 2 diabetic nephropathy: A novel predictor of end-stage renal disease. Diabetes Care. 2012 Jul;35(7):1591-7.
6. Palmer SC et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: A systematic review and meta-analysis. JAMA. 2011 Mar 16;305(11):1119-27.
7. Patel L et al. Sevelamer versus calcium-based binders for treatment of hyperphosphatemia in CKD: A meta-analysis of randomized controlled trials. Clin J Am Soc Nephrol. 2016 Feb 5;11(2):232-44.
8. Raphael KL. Approach to the treatment of chronic metabolic acidosis in CKD. Am J Kidney Dis. 2016 Apr;67(4):696-702.
9. Raphael KL et al. Prevalence of and risk factors for reduced serum bicarbonate in chronic kidney disease. Nephrology (Carlton). 2014 Oct;19(10):648-54.
Additional reading
1. Chapter 3: Management of Progression and Complications of CKD. Kidney Int Suppl (2011). 2013 Jan:3(1):73-90.
2. Raphael KL. Approach to the treatment of chronic metabolic acidosis in CKD. Am J Kidney Dis. 2016 Apr;67(4):696-702.
Quiz
A 75-year-old male with hypertension and CKD Stage 4 is admitted to the hospital for a hip fracture following a fall. Laboratory studies on admission show a potassium level of 3.2 mEq/L, vitamin D level of 45 ng/mL, bicarbonate level of 17 mEq/L, phosphate level of 5.0 mg/dL, and calcium level of 10.3 mg/dL.
What electrolyte replacements should be initiated?
A. Dietary restriction of phosphate, sodium bicarbonate, potassium chloride, and vitamin D.
B. Non–calcium-containing phosphate binder, vitamin D, and potassium chloride.
C. Calcium-containing phosphate binder and sodium bicarbonate.
D. Non–calcium-containing phosphate binder, sodium bicarbonate, and potassium chloride.
Answer: D. Given the patient’s hypokalemia, potassium supplementation should be considered. Additionally, given his hyperphosphatemia and hypercalcemia, a non–calcium-containing phosphate binder like Sevelamer should be started. His metabolic acidosis should be corrected with sodium bicarbonate. There is no indication to supplement vitamin D based on his current lab values.
Key Points
- Identify and treat underlying causes of hypokalemia and hypomagnesemia.
- Do not hesitate to treat metabolic acidosis in CKD.
- Manage hyperphosphatemia and hypocalcemia by ordering appropriate lab studies and providing nutritional consultation with outpatient nephrology follow-up as indicated.
Case
A 55-year-old woman with diabetes, hypertension, and chronic kidney disease (CKD) stage 4 is admitted to the hospital for treatment of left lower-extremity cellulitis. Laboratory studies on admission show a creatinine level of 2.5 mg/dL (glomerular filtration rate [GFR] is 20 mL/min per 1.73 m2; baseline creatinine is between 2.2 and 2.6 mg/dL), potassium level of 3.0 mEq/L, magnesium level of 1.5 mEq/L, bicarbonate level of 18 mEq/L, phosphate level of 6.5 mg/dL, and calcium level of 7.5 mg/dL.
She is put on renally dosed vancomycin to treat her cellulitis. As the hospitalist, how should you manage her multiple electrolyte abnormalities?
Overview of the issue
CKD is also an important risk factor for acute kidney injury. Additionally, rates of readmission for CKD patients are higher than those without CKD. Given that CKD is a “silent disease” that many patients do not realize they have, it is very possible that the first documentation of CKD could happen during an acute hospitalization.
Among the various manifestations of CKD, electrolyte abnormalities are the most likely ones hospitalists will run into.
Overview of the data
Hypokalemia and Hypomagnesemia
Hypokalemia (potassium levels less than 3.5 mEq/L) is not as common as hyperkalemia (potassium levels greater than 5.0 mEq/L) in CKD, which is the result of impaired renal excretion of potassium. Hypokalemia can occur as a result of GI losses, urinary losses, or decreased intake and can be worsened by the use of certain drugs, such as non–K-sparing diuretics.
In the setting of diuretic use involving thiazides and loop diuretics, hypokalemia is dose and sodium-intake dependent. Potassium deficiency worsens the effects of detrimental sodium excess, which plays a role in hypertension and its associated complications. Potassium also has a protective vascular effect, which is a major reason why potassium should be kept normal in patients with CKD.
Acutely, hypokalemia can cause arrhythmias, ileus, and paralysis, which are all indications for immediate repletion. In these cases, hypokalemia must be repleted carefully in small increments (some suggest 20 mEq doses), and the patient must be monitored frequently to avoid hyperkalemia. If patients are persistently hypokalemic, several options can be considered based on the underlying cause. Dietary modifications with foods rich in potassium (containing 250mg/100g) can be suggested. Daily potassium chloride supplementation can be used in those on diuretic therapy who have hypokalemia and metabolic alkalosis (bicarbonate levels greater than 30 mEq/L). Alkalinizing salts, containing citrate or bicarbonate, can be used in hypokalemia without metabolic alkalosis. Initiation of angiotensin-converting-enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), beta-adrenergic blockers, and K-sparing diuretics can be used as well.
Potassium supplementation and K-sparing diuretics should be used with extreme caution in CKD 3 and 4 given the risk of overcorrection. If potassium supplements or drugs to raise serum potassium are initiated in house, potassium should be rechecked within a week. These treatments should be avoided in individuals with diabetes, who are at highest risk for hyperkalemia given hyporeninemic hypoaldosteronism (type IV renal tubular acidosis).
There is emerging evidence that hypomagnesemia can play a part in progression to end-stage renal disease. In the setting of cardiovascular disease, which often co-exists with CKD, the risk of hypomagnesemia precipitating arrhythmia necessitates repletion to a normal level. Any of the magnesium salts and antacids can be used for treatment. K-sparing diuretics are also magnesium sparing. An important side effect of magnesium repletion is diarrhea, which can potentiate electrolyte losses and reduce long-term adherence rates.
Metabolic acidosis
Acid-base balance is maintained by the kidney through urinary excretion of hydrogen ions both as titratable acids and ammonium. In CKD, renal excretion of the daily acid load is impaired, primarily from decreased ammonium excretion caused by there being too few functioning nephrons.
Metabolic acidosis in CKD is defined as a serum bicarbonate concentration of persistently less than 22 mEq/L. The overall prevalence of metabolic acidosis in cases of CKD that don’t require dialysis is about 15% and increases linearly with a decline in GFR. In the Chronic Renal Insufficiency Cohort study, 7%, 13%, and 37% of participants with CKD stages 2, 3, and 4 respectively had metabolic acidosis.
Metabolic acidosis has a variety of adverse outcomes, including bone demineralization, increased protein catabolism and muscle wasting, impaired cardiac function, impaired glucose homeostasis, and systemic inflammation. Additionally, multiple studies have shown an association between metabolic acidosis and progression of CKD and increased mortality.
The 2013 Kidney Disease Improving Global Outcomes (KDIGO) guidelines recommend maintaining the serum bicarbonate level within the reference range (23-29 mEq/L) with alkali therapy. Options include sodium bicarbonate or sodium citrate (which is rapidly metabolized to bicarbonate) in doses of 0.5-1.0 mEq/kg once per day. Sodium bicarbonate is inexpensive; however, it can lead to gastrointestinal upset as the bicarbonate is converted into CO2 in the stomach. This side effect is usually self-limited and improves with time. Typical starting doses are 650 mg twice a day if the serum bicarbonate level is 19-21 mEq/L or 1300 mg twice a day if the serum bicarbonate level is less than or equal to 18 mEq/L.
Sodium citrate can be used if gastrointestinal upset occurs, although caution should be used in those on aluminum binders or with liver disease. Alkali treatment should be started when bicarbonate levels are persistently low (for weeks or months) or if very low (less than or equal to 18 mEq/L) without an acute reversible cause. After patients have begun therapy, they should be monitored for the development of worsening hypertension or edema caused by sodium-mediated fluid retention, although this rarely occurs.
Hyperphosphatemia and Hypocalcemia
Hyperphosphatemia (phosphate levels greater than 4.6 mg/dL) develops early in CKD because of a reduced filtered-phosphate load. Hypocalcemia and hyperphosphatemia can lead to secondary hyperparathyroidism. Given that hyperphosphatemia has been associated with an increased mortality among patients with CKD, treatment is warranted, but the optimal phosphorus range is unknown. According to the KDIGO guidelines, the goal phosphorus level is less than 4.5 mg/dL in patients with CKD who are not on dialysis.
Treatment includes dietary restriction to 900 mg/day and phosphate binders. There is a high phosphate load in processed foods and colas because of food additives. It is therefore recommended to reduce consumption of these foods while encouraging consumption of meat and eggs, which offer additional nutritional value. Those who have failed dietary restrictions should be put on a phosphate binder, either calcium containing (calcium carbonate, calcium acetate) or non–calcium containing (Sevelamer, lanthanum). Non–calcium-containing binders are recommended for patients with hypercalcemia (levels greater than 9.5 mg/dL). There is some data that suggests that non–calcium-containing binders are superior to calcium-containing binders in terms of vascular disease outcomes, but non–calcium-containing binders are sometimes difficult to obtain because of cost and insurance coverage.
Hypocalcemia (calcium levels below 8.4 mg/dL) occurs in the setting of late stage untreated CKD because of decreased GI uptake of calcium from diet in the context of vitamin D deficiency (less than 30 ng/mL) in addition to hyperphosphatemia. Phosphate and vitamin D correction is preferred to calcium supplementation because hyperphosphatemia and vitamin D deficiency occur earlier in CKD. Phosphate reduction is described above.
Regarding vitamin D deficiency, it is recommended to start supplementation with either vitamin D2 or D3. Doses should be adjusted if GFR is less than 30 mL/min per 1.73 m2. It is important to monitor for hypercalcemia, which can also occur in CKD in this context, because it has also been associated with increased morbidity and mortality. If calcium levels are greater than 10.2 mg/dL, all vitamin D supplementation should be discontinued.
Back to the case
Our patient who was admitted for cellulitis has concomitant hypokalemia, hypomagnesemia, acidosis, and hyperphosphatemia with related hypocalcemia. She revealed that her diet was poor prior to her admission for her infection. She was given 20 mEq of potassium orally and placed on a potassium rich diet until potassium levels normalized. She was also given magnesium oxide orally on the first and second day of admission, with repeat levels that were normal. Her acidosis was treated with sodium bicarbonate – 1,300 mg orally twice daily. For her hyperphosphatemia and hypocalcemia, she was placed on phosphate restriction with nutritional counseling with plans to follow up as an outpatient to determine need for phosphate binders. In addition, vitamin D levels were checked, and she was started on repletion for vitamin D deficiency (27 ng/mL). Daily BMP, magnesium, and phosphorus were checked while in house until they were normal for 2 days, and follow-up lab work was requested with her nephrology appointment, which was scheduled for within 1 week.
Bottom line
Electrolyte abnormalities in CKD are numerous and have multiple adverse clinical outcomes. Early intervention and management, especially of metabolic acidosis and hyperphosphatemia, can have a significant effect, including prevention of progression of CKD and possibly reduced mortality.
Dr. Daya, Dr. Apgar, and Dr. Eniasivam are assistant clinical professors in the division of hospital medicine at the University of California, San Francisco.
References
1. Coresh J et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007 Nov 7;298(17):2038-47.
2. Dhondup T et al. Electrolyte and acid-base disorders in chronic kidney disease and end-stage kidney failure. Blood Purif. 2017;43(1-3):179-188.
3. K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis. 2004 May;43(5 Suppl 1):S1-290.
4. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl (2011). 2013 Jan;3(1):1–150.
5. Sakaguchi Y et al. Hypomagnesemia in type 2 diabetic nephropathy: A novel predictor of end-stage renal disease. Diabetes Care. 2012 Jul;35(7):1591-7.
6. Palmer SC et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: A systematic review and meta-analysis. JAMA. 2011 Mar 16;305(11):1119-27.
7. Patel L et al. Sevelamer versus calcium-based binders for treatment of hyperphosphatemia in CKD: A meta-analysis of randomized controlled trials. Clin J Am Soc Nephrol. 2016 Feb 5;11(2):232-44.
8. Raphael KL. Approach to the treatment of chronic metabolic acidosis in CKD. Am J Kidney Dis. 2016 Apr;67(4):696-702.
9. Raphael KL et al. Prevalence of and risk factors for reduced serum bicarbonate in chronic kidney disease. Nephrology (Carlton). 2014 Oct;19(10):648-54.
Additional reading
1. Chapter 3: Management of Progression and Complications of CKD. Kidney Int Suppl (2011). 2013 Jan:3(1):73-90.
2. Raphael KL. Approach to the treatment of chronic metabolic acidosis in CKD. Am J Kidney Dis. 2016 Apr;67(4):696-702.
Quiz
A 75-year-old male with hypertension and CKD Stage 4 is admitted to the hospital for a hip fracture following a fall. Laboratory studies on admission show a potassium level of 3.2 mEq/L, vitamin D level of 45 ng/mL, bicarbonate level of 17 mEq/L, phosphate level of 5.0 mg/dL, and calcium level of 10.3 mg/dL.
What electrolyte replacements should be initiated?
A. Dietary restriction of phosphate, sodium bicarbonate, potassium chloride, and vitamin D.
B. Non–calcium-containing phosphate binder, vitamin D, and potassium chloride.
C. Calcium-containing phosphate binder and sodium bicarbonate.
D. Non–calcium-containing phosphate binder, sodium bicarbonate, and potassium chloride.
Answer: D. Given the patient’s hypokalemia, potassium supplementation should be considered. Additionally, given his hyperphosphatemia and hypercalcemia, a non–calcium-containing phosphate binder like Sevelamer should be started. His metabolic acidosis should be corrected with sodium bicarbonate. There is no indication to supplement vitamin D based on his current lab values.
Key Points
- Identify and treat underlying causes of hypokalemia and hypomagnesemia.
- Do not hesitate to treat metabolic acidosis in CKD.
- Manage hyperphosphatemia and hypocalcemia by ordering appropriate lab studies and providing nutritional consultation with outpatient nephrology follow-up as indicated.
Case
A 55-year-old woman with diabetes, hypertension, and chronic kidney disease (CKD) stage 4 is admitted to the hospital for treatment of left lower-extremity cellulitis. Laboratory studies on admission show a creatinine level of 2.5 mg/dL (glomerular filtration rate [GFR] is 20 mL/min per 1.73 m2; baseline creatinine is between 2.2 and 2.6 mg/dL), potassium level of 3.0 mEq/L, magnesium level of 1.5 mEq/L, bicarbonate level of 18 mEq/L, phosphate level of 6.5 mg/dL, and calcium level of 7.5 mg/dL.
She is put on renally dosed vancomycin to treat her cellulitis. As the hospitalist, how should you manage her multiple electrolyte abnormalities?
Overview of the issue
CKD is also an important risk factor for acute kidney injury. Additionally, rates of readmission for CKD patients are higher than those without CKD. Given that CKD is a “silent disease” that many patients do not realize they have, it is very possible that the first documentation of CKD could happen during an acute hospitalization.
Among the various manifestations of CKD, electrolyte abnormalities are the most likely ones hospitalists will run into.
Overview of the data
Hypokalemia and Hypomagnesemia
Hypokalemia (potassium levels less than 3.5 mEq/L) is not as common as hyperkalemia (potassium levels greater than 5.0 mEq/L) in CKD, which is the result of impaired renal excretion of potassium. Hypokalemia can occur as a result of GI losses, urinary losses, or decreased intake and can be worsened by the use of certain drugs, such as non–K-sparing diuretics.
In the setting of diuretic use involving thiazides and loop diuretics, hypokalemia is dose and sodium-intake dependent. Potassium deficiency worsens the effects of detrimental sodium excess, which plays a role in hypertension and its associated complications. Potassium also has a protective vascular effect, which is a major reason why potassium should be kept normal in patients with CKD.
Acutely, hypokalemia can cause arrhythmias, ileus, and paralysis, which are all indications for immediate repletion. In these cases, hypokalemia must be repleted carefully in small increments (some suggest 20 mEq doses), and the patient must be monitored frequently to avoid hyperkalemia. If patients are persistently hypokalemic, several options can be considered based on the underlying cause. Dietary modifications with foods rich in potassium (containing 250mg/100g) can be suggested. Daily potassium chloride supplementation can be used in those on diuretic therapy who have hypokalemia and metabolic alkalosis (bicarbonate levels greater than 30 mEq/L). Alkalinizing salts, containing citrate or bicarbonate, can be used in hypokalemia without metabolic alkalosis. Initiation of angiotensin-converting-enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), beta-adrenergic blockers, and K-sparing diuretics can be used as well.
Potassium supplementation and K-sparing diuretics should be used with extreme caution in CKD 3 and 4 given the risk of overcorrection. If potassium supplements or drugs to raise serum potassium are initiated in house, potassium should be rechecked within a week. These treatments should be avoided in individuals with diabetes, who are at highest risk for hyperkalemia given hyporeninemic hypoaldosteronism (type IV renal tubular acidosis).
There is emerging evidence that hypomagnesemia can play a part in progression to end-stage renal disease. In the setting of cardiovascular disease, which often co-exists with CKD, the risk of hypomagnesemia precipitating arrhythmia necessitates repletion to a normal level. Any of the magnesium salts and antacids can be used for treatment. K-sparing diuretics are also magnesium sparing. An important side effect of magnesium repletion is diarrhea, which can potentiate electrolyte losses and reduce long-term adherence rates.
Metabolic acidosis
Acid-base balance is maintained by the kidney through urinary excretion of hydrogen ions both as titratable acids and ammonium. In CKD, renal excretion of the daily acid load is impaired, primarily from decreased ammonium excretion caused by there being too few functioning nephrons.
Metabolic acidosis in CKD is defined as a serum bicarbonate concentration of persistently less than 22 mEq/L. The overall prevalence of metabolic acidosis in cases of CKD that don’t require dialysis is about 15% and increases linearly with a decline in GFR. In the Chronic Renal Insufficiency Cohort study, 7%, 13%, and 37% of participants with CKD stages 2, 3, and 4 respectively had metabolic acidosis.
Metabolic acidosis has a variety of adverse outcomes, including bone demineralization, increased protein catabolism and muscle wasting, impaired cardiac function, impaired glucose homeostasis, and systemic inflammation. Additionally, multiple studies have shown an association between metabolic acidosis and progression of CKD and increased mortality.
The 2013 Kidney Disease Improving Global Outcomes (KDIGO) guidelines recommend maintaining the serum bicarbonate level within the reference range (23-29 mEq/L) with alkali therapy. Options include sodium bicarbonate or sodium citrate (which is rapidly metabolized to bicarbonate) in doses of 0.5-1.0 mEq/kg once per day. Sodium bicarbonate is inexpensive; however, it can lead to gastrointestinal upset as the bicarbonate is converted into CO2 in the stomach. This side effect is usually self-limited and improves with time. Typical starting doses are 650 mg twice a day if the serum bicarbonate level is 19-21 mEq/L or 1300 mg twice a day if the serum bicarbonate level is less than or equal to 18 mEq/L.
Sodium citrate can be used if gastrointestinal upset occurs, although caution should be used in those on aluminum binders or with liver disease. Alkali treatment should be started when bicarbonate levels are persistently low (for weeks or months) or if very low (less than or equal to 18 mEq/L) without an acute reversible cause. After patients have begun therapy, they should be monitored for the development of worsening hypertension or edema caused by sodium-mediated fluid retention, although this rarely occurs.
Hyperphosphatemia and Hypocalcemia
Hyperphosphatemia (phosphate levels greater than 4.6 mg/dL) develops early in CKD because of a reduced filtered-phosphate load. Hypocalcemia and hyperphosphatemia can lead to secondary hyperparathyroidism. Given that hyperphosphatemia has been associated with an increased mortality among patients with CKD, treatment is warranted, but the optimal phosphorus range is unknown. According to the KDIGO guidelines, the goal phosphorus level is less than 4.5 mg/dL in patients with CKD who are not on dialysis.
Treatment includes dietary restriction to 900 mg/day and phosphate binders. There is a high phosphate load in processed foods and colas because of food additives. It is therefore recommended to reduce consumption of these foods while encouraging consumption of meat and eggs, which offer additional nutritional value. Those who have failed dietary restrictions should be put on a phosphate binder, either calcium containing (calcium carbonate, calcium acetate) or non–calcium containing (Sevelamer, lanthanum). Non–calcium-containing binders are recommended for patients with hypercalcemia (levels greater than 9.5 mg/dL). There is some data that suggests that non–calcium-containing binders are superior to calcium-containing binders in terms of vascular disease outcomes, but non–calcium-containing binders are sometimes difficult to obtain because of cost and insurance coverage.
Hypocalcemia (calcium levels below 8.4 mg/dL) occurs in the setting of late stage untreated CKD because of decreased GI uptake of calcium from diet in the context of vitamin D deficiency (less than 30 ng/mL) in addition to hyperphosphatemia. Phosphate and vitamin D correction is preferred to calcium supplementation because hyperphosphatemia and vitamin D deficiency occur earlier in CKD. Phosphate reduction is described above.
Regarding vitamin D deficiency, it is recommended to start supplementation with either vitamin D2 or D3. Doses should be adjusted if GFR is less than 30 mL/min per 1.73 m2. It is important to monitor for hypercalcemia, which can also occur in CKD in this context, because it has also been associated with increased morbidity and mortality. If calcium levels are greater than 10.2 mg/dL, all vitamin D supplementation should be discontinued.
Back to the case
Our patient who was admitted for cellulitis has concomitant hypokalemia, hypomagnesemia, acidosis, and hyperphosphatemia with related hypocalcemia. She revealed that her diet was poor prior to her admission for her infection. She was given 20 mEq of potassium orally and placed on a potassium rich diet until potassium levels normalized. She was also given magnesium oxide orally on the first and second day of admission, with repeat levels that were normal. Her acidosis was treated with sodium bicarbonate – 1,300 mg orally twice daily. For her hyperphosphatemia and hypocalcemia, she was placed on phosphate restriction with nutritional counseling with plans to follow up as an outpatient to determine need for phosphate binders. In addition, vitamin D levels were checked, and she was started on repletion for vitamin D deficiency (27 ng/mL). Daily BMP, magnesium, and phosphorus were checked while in house until they were normal for 2 days, and follow-up lab work was requested with her nephrology appointment, which was scheduled for within 1 week.
Bottom line
Electrolyte abnormalities in CKD are numerous and have multiple adverse clinical outcomes. Early intervention and management, especially of metabolic acidosis and hyperphosphatemia, can have a significant effect, including prevention of progression of CKD and possibly reduced mortality.
Dr. Daya, Dr. Apgar, and Dr. Eniasivam are assistant clinical professors in the division of hospital medicine at the University of California, San Francisco.
References
1. Coresh J et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007 Nov 7;298(17):2038-47.
2. Dhondup T et al. Electrolyte and acid-base disorders in chronic kidney disease and end-stage kidney failure. Blood Purif. 2017;43(1-3):179-188.
3. K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis. 2004 May;43(5 Suppl 1):S1-290.
4. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl (2011). 2013 Jan;3(1):1–150.
5. Sakaguchi Y et al. Hypomagnesemia in type 2 diabetic nephropathy: A novel predictor of end-stage renal disease. Diabetes Care. 2012 Jul;35(7):1591-7.
6. Palmer SC et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: A systematic review and meta-analysis. JAMA. 2011 Mar 16;305(11):1119-27.
7. Patel L et al. Sevelamer versus calcium-based binders for treatment of hyperphosphatemia in CKD: A meta-analysis of randomized controlled trials. Clin J Am Soc Nephrol. 2016 Feb 5;11(2):232-44.
8. Raphael KL. Approach to the treatment of chronic metabolic acidosis in CKD. Am J Kidney Dis. 2016 Apr;67(4):696-702.
9. Raphael KL et al. Prevalence of and risk factors for reduced serum bicarbonate in chronic kidney disease. Nephrology (Carlton). 2014 Oct;19(10):648-54.
Additional reading
1. Chapter 3: Management of Progression and Complications of CKD. Kidney Int Suppl (2011). 2013 Jan:3(1):73-90.
2. Raphael KL. Approach to the treatment of chronic metabolic acidosis in CKD. Am J Kidney Dis. 2016 Apr;67(4):696-702.
Quiz
A 75-year-old male with hypertension and CKD Stage 4 is admitted to the hospital for a hip fracture following a fall. Laboratory studies on admission show a potassium level of 3.2 mEq/L, vitamin D level of 45 ng/mL, bicarbonate level of 17 mEq/L, phosphate level of 5.0 mg/dL, and calcium level of 10.3 mg/dL.
What electrolyte replacements should be initiated?
A. Dietary restriction of phosphate, sodium bicarbonate, potassium chloride, and vitamin D.
B. Non–calcium-containing phosphate binder, vitamin D, and potassium chloride.
C. Calcium-containing phosphate binder and sodium bicarbonate.
D. Non–calcium-containing phosphate binder, sodium bicarbonate, and potassium chloride.
Answer: D. Given the patient’s hypokalemia, potassium supplementation should be considered. Additionally, given his hyperphosphatemia and hypercalcemia, a non–calcium-containing phosphate binder like Sevelamer should be started. His metabolic acidosis should be corrected with sodium bicarbonate. There is no indication to supplement vitamin D based on his current lab values.
Key Points
- Identify and treat underlying causes of hypokalemia and hypomagnesemia.
- Do not hesitate to treat metabolic acidosis in CKD.
- Manage hyperphosphatemia and hypocalcemia by ordering appropriate lab studies and providing nutritional consultation with outpatient nephrology follow-up as indicated.
Can case management cut hypertension’s consequences?
MONTREAL – who received phone calls, care coordination, and coaching from a nurse case manager, according to a retrospective population-based cohort study of almost 85,000 patients with hypertension.
The reduction yielded a hazard ratio for all-cause mortality of 0.504, with a number needed to treat (NNT) of 25 (P less than .05).

Service utilization also decreased for those participating in the intervention, compared with those receiving usual care: Hospitalizations fell by 7 per 100 patient-years, with greater reductions seen in emergency department, specialist, and primary care utilization (P less than .05 for all).
At the time the study was conceived, the Hong Kong Hospital Authority cared for about 200,000 hypertensive patients, of whom more than 40% hadn’t achieved the target blood pressure of less than 140/90 mm Hg, said Dr. Yu.
Dr. Yu of the department of family medicine and primary care at the University of Hong Kong said there are challenges in bringing more hypertension patients into good blood pressure control in Hong Kong. These include the “idiosyncratic practice” of some frontline physicians, who also often have limited time for patient consultations and only limited access to the services of allied health professionals to help them in their work. Patient adherence, she said, is also an issue.
To tackle these persistent high rates of patients whose blood pressures remained too high, Dr. Yu and her colleagues at the Hospital Authority launched the Risk Assessment and Management Program – Hypertension (RAMP-HT) in 2011. The program, she said, is an “evidence-based, structured, protocol-driven, multidisciplinary program” that includes risk assessment and screening for complications, and uses a risk-guided management approach.
Patients in RAMP-HT received interventions according to a matrix for risk management of patients with hypertension. Patients with a blood pressure between 140/90 and 160/100 mm Hg who were assessed as being low and medium risk according to the Joint British Societies guidelines for cardiovascular risk continued to receive management from their primary care physician. High-risk patients with blood pressure in this range also received a statin if their low-density lipoprotein cholesterol level was suboptimal.
Patients whose blood pressure was at least 160/100 mm Hg were followed by a RAMP-HT nurse. For those with this degree of blood pressure elevation who were already on at least three antihypertensive medications, specialty appointments were also arranged.
Other targeted interventions were also available to participants, including the services of dietitians and physical therapists for those with a body mass index (BMI) of at least 27.5 kg/m2; smoking cessation and mental health services were also available, as appropriate.
After 3 years, those participating in the RAMP-HT program (n = 79,116) were compared with those in the usual care group (n = 43,901). In both arms, adult patients with complete data and without preexisting cardiovascular disease, diabetes, or end-stage renal disease were included. In each group, about 58% of participants were female, and the mean age was about 65 years.
Primary outcome measures included the incidence of cardiovascular disease, an outcome that included coronary heart disease, heart failure, and stroke; end-stage renal disease; and all-cause mortality. The significant reductions in these measures for the RAMP-HT group remained after multivariable analysis accounted for sex, age, smoking status, renal function, lipid values, BMI, comorbidities, and antihypertensive and lipid-lowering medication use.
The reduced care utilization seen among RAMP-HT participants also persisted after multivariable analysis for these potential confounders.
Dr. Yu said the systematic, protocol-driven program was a primary strength of RAMP-HT. The key to the program was use of nurses to provide patient education and physicians and allied health resources only as needed, she said; the program reinforced the importance of self-management and adherence because patients heard a unified message from many different health care professionals.
However, lifestyle factors such as diet and exercise weren’t tracked, and the retrospective study design introduced the potential for some bias, she said. In ongoing work, the long-term efficacy and cost-effectiveness of the RAMP-HT program are being tracked.
Dr. Yu reported that the study was funded by the Hong Kong Health and Medical Research Fund. She reported no relevant conflicts of interest.
SOURCE: Yu, Esther et al. NAPCRG 2017, Abstract HY33.
MONTREAL – who received phone calls, care coordination, and coaching from a nurse case manager, according to a retrospective population-based cohort study of almost 85,000 patients with hypertension.
The reduction yielded a hazard ratio for all-cause mortality of 0.504, with a number needed to treat (NNT) of 25 (P less than .05).
Service utilization also decreased for those participating in the intervention, compared with those receiving usual care: Hospitalizations fell by 7 per 100 patient-years, with greater reductions seen in emergency department, specialist, and primary care utilization (P less than .05 for all).
At the time the study was conceived, the Hong Kong Hospital Authority cared for about 200,000 hypertensive patients, of whom more than 40% hadn’t achieved the target blood pressure of less than 140/90 mm Hg, said Dr. Yu.
Dr. Yu of the department of family medicine and primary care at the University of Hong Kong said there are challenges in bringing more hypertension patients into good blood pressure control in Hong Kong. These include the “idiosyncratic practice” of some frontline physicians, who also often have limited time for patient consultations and only limited access to the services of allied health professionals to help them in their work. Patient adherence, she said, is also an issue.
To tackle these persistent high rates of patients whose blood pressures remained too high, Dr. Yu and her colleagues at the Hospital Authority launched the Risk Assessment and Management Program – Hypertension (RAMP-HT) in 2011. The program, she said, is an “evidence-based, structured, protocol-driven, multidisciplinary program” that includes risk assessment and screening for complications, and uses a risk-guided management approach.
Patients in RAMP-HT received interventions according to a matrix for risk management of patients with hypertension. Patients with a blood pressure between 140/90 and 160/100 mm Hg who were assessed as being low and medium risk according to the Joint British Societies guidelines for cardiovascular risk continued to receive management from their primary care physician. High-risk patients with blood pressure in this range also received a statin if their low-density lipoprotein cholesterol level was suboptimal.
Patients whose blood pressure was at least 160/100 mm Hg were followed by a RAMP-HT nurse. For those with this degree of blood pressure elevation who were already on at least three antihypertensive medications, specialty appointments were also arranged.
Other targeted interventions were also available to participants, including the services of dietitians and physical therapists for those with a body mass index (BMI) of at least 27.5 kg/m2; smoking cessation and mental health services were also available, as appropriate.
After 3 years, those participating in the RAMP-HT program (n = 79,116) were compared with those in the usual care group (n = 43,901). In both arms, adult patients with complete data and without preexisting cardiovascular disease, diabetes, or end-stage renal disease were included. In each group, about 58% of participants were female, and the mean age was about 65 years.
Primary outcome measures included the incidence of cardiovascular disease, an outcome that included coronary heart disease, heart failure, and stroke; end-stage renal disease; and all-cause mortality. The significant reductions in these measures for the RAMP-HT group remained after multivariable analysis accounted for sex, age, smoking status, renal function, lipid values, BMI, comorbidities, and antihypertensive and lipid-lowering medication use.
The reduced care utilization seen among RAMP-HT participants also persisted after multivariable analysis for these potential confounders.
Dr. Yu said the systematic, protocol-driven program was a primary strength of RAMP-HT. The key to the program was use of nurses to provide patient education and physicians and allied health resources only as needed, she said; the program reinforced the importance of self-management and adherence because patients heard a unified message from many different health care professionals.
However, lifestyle factors such as diet and exercise weren’t tracked, and the retrospective study design introduced the potential for some bias, she said. In ongoing work, the long-term efficacy and cost-effectiveness of the RAMP-HT program are being tracked.
Dr. Yu reported that the study was funded by the Hong Kong Health and Medical Research Fund. She reported no relevant conflicts of interest.
SOURCE: Yu, Esther et al. NAPCRG 2017, Abstract HY33.
MONTREAL – who received phone calls, care coordination, and coaching from a nurse case manager, according to a retrospective population-based cohort study of almost 85,000 patients with hypertension.
The reduction yielded a hazard ratio for all-cause mortality of 0.504, with a number needed to treat (NNT) of 25 (P less than .05).
Service utilization also decreased for those participating in the intervention, compared with those receiving usual care: Hospitalizations fell by 7 per 100 patient-years, with greater reductions seen in emergency department, specialist, and primary care utilization (P less than .05 for all).
At the time the study was conceived, the Hong Kong Hospital Authority cared for about 200,000 hypertensive patients, of whom more than 40% hadn’t achieved the target blood pressure of less than 140/90 mm Hg, said Dr. Yu.
Dr. Yu of the department of family medicine and primary care at the University of Hong Kong said there are challenges in bringing more hypertension patients into good blood pressure control in Hong Kong. These include the “idiosyncratic practice” of some frontline physicians, who also often have limited time for patient consultations and only limited access to the services of allied health professionals to help them in their work. Patient adherence, she said, is also an issue.
To tackle these persistent high rates of patients whose blood pressures remained too high, Dr. Yu and her colleagues at the Hospital Authority launched the Risk Assessment and Management Program – Hypertension (RAMP-HT) in 2011. The program, she said, is an “evidence-based, structured, protocol-driven, multidisciplinary program” that includes risk assessment and screening for complications, and uses a risk-guided management approach.
Patients in RAMP-HT received interventions according to a matrix for risk management of patients with hypertension. Patients with a blood pressure between 140/90 and 160/100 mm Hg who were assessed as being low and medium risk according to the Joint British Societies guidelines for cardiovascular risk continued to receive management from their primary care physician. High-risk patients with blood pressure in this range also received a statin if their low-density lipoprotein cholesterol level was suboptimal.
Patients whose blood pressure was at least 160/100 mm Hg were followed by a RAMP-HT nurse. For those with this degree of blood pressure elevation who were already on at least three antihypertensive medications, specialty appointments were also arranged.
Other targeted interventions were also available to participants, including the services of dietitians and physical therapists for those with a body mass index (BMI) of at least 27.5 kg/m2; smoking cessation and mental health services were also available, as appropriate.
After 3 years, those participating in the RAMP-HT program (n = 79,116) were compared with those in the usual care group (n = 43,901). In both arms, adult patients with complete data and without preexisting cardiovascular disease, diabetes, or end-stage renal disease were included. In each group, about 58% of participants were female, and the mean age was about 65 years.
Primary outcome measures included the incidence of cardiovascular disease, an outcome that included coronary heart disease, heart failure, and stroke; end-stage renal disease; and all-cause mortality. The significant reductions in these measures for the RAMP-HT group remained after multivariable analysis accounted for sex, age, smoking status, renal function, lipid values, BMI, comorbidities, and antihypertensive and lipid-lowering medication use.
The reduced care utilization seen among RAMP-HT participants also persisted after multivariable analysis for these potential confounders.
Dr. Yu said the systematic, protocol-driven program was a primary strength of RAMP-HT. The key to the program was use of nurses to provide patient education and physicians and allied health resources only as needed, she said; the program reinforced the importance of self-management and adherence because patients heard a unified message from many different health care professionals.
However, lifestyle factors such as diet and exercise weren’t tracked, and the retrospective study design introduced the potential for some bias, she said. In ongoing work, the long-term efficacy and cost-effectiveness of the RAMP-HT program are being tracked.
Dr. Yu reported that the study was funded by the Hong Kong Health and Medical Research Fund. She reported no relevant conflicts of interest.
SOURCE: Yu, Esther et al. NAPCRG 2017, Abstract HY33.
REPORTING FROM NAPCRG 2017
Key clinical point:
Major finding: The hazard ratio for all-cause mortality was 0.504 for patients in the intervention arm.
Study details: A retrospective population-based cohort study of almost 85,000 Hong Kong patients with hypertension.
Disclosures: The study was funded by the Hong Kong Health and Medical Research Fund. Dr. Yu reported no relevant financial disclosures.
Source: Yu E et al. NAPCRG 2017, Abstract HY33.
Tamsulosin for Patients With Ureteral Stones?

A 54-year-old man presents to the emergency department (ED) with acute-onset left flank pain that radiates to the groin. CT of the abdomen/pelvis without contrast reveals a 7-mm distal ureteral stone. He is deemed an appropriate candidate for outpatient management. In addition to pain medications, should you prescribe tamsulosin?
According to the most recent National Health and Nutrition Examination Survey, the population prevalence of kidney stones is 8.8%, with a self-reported prevalence of 10.6% in men and 7.1% in women.2 Most ureteral stones can be treated in the outpatient setting with oral hydration, antiemetics, and pain control with NSAIDs as firstline treatment and opioids as a second-line option.3
In addition, α-blockers are used for medical expulsive therapy (MET). In fact, the European Association of Urology guideline on urolithiasis states that MET may accelerate passage of ureteral stones.3
Recently, however, uncertainty has surrounded the effectiveness of the α-blocker tamsulosin. Two systematic reviews (limited by heterogeneity because some of the studies lacked a placebo control and blinding) concluded that α-blockers increased stone passage within one to six weeks when compared with placebo or no additional therapy.4,5 However, a recent large, multicenter RCT revealed no difference between tamsulosin and nifedipine, or either one compared with placebo, at decreasing the need for further treatment to achieve stone passage within four weeks.6
STUDY SUMMARY
Results broken down by stone size
This meta-analysis, comprising eight double-blind RCTs, examined the effect of oral tamsulosin (0.4 mg/d; average course, 28 d) on distal ureteral stone passage in adult patients (N = 1,384).1 A subgroup analysis comparing stone size (< 5 mm and 5-10 mm) was also conducted to determine whether size modified the effect of tamsulosin.
The eight selected studies were published between 2009 and 2015; the trials were conducted in multiple countries, in ED and outpatient urology settings. The main outcome measure was the risk difference (RD) in stone passage between the tamsulosin group and placebo group after follow-up imaging at three weeks with CT or plain film radiographs.
Tamsulosin helps some, but not all. The pooled risk for stone passage was higher in the tamsulosin group than in the placebo group (85% vs 66%; RD, 17%), but significant heterogeneity existed across the trials (I2, 80.2%). Subgroup analysis by stone size (< 5 mm vs 5-10 mm) revealed that, compared to placebo, tamsulosin was beneficial for larger stones (6 trials, N = 514; RD, 22%; number needed to treat, 5) but not for smaller stones (4 trials, N = 533; RD, –0.3%). The 5-to-10–mm subgroup had a less heterogeneous population of studies than did the < 5-mm subgroup (I2, 33% and 0% respectively).
In terms of adverse events, tamsulosin did not increase the risk for dizziness (RD, 0.2%) or postural hypotension (RD, 0.1%), compared with placebo.
WHAT’S NEW
Increased passage of larger stones
This meta-analysis included only double-blind RCTs; prior meta-analyses did not. Also, this review included the SUSPEND (Spontaneous Urinary Stone Passage Enabled by Drugs) trial, an RCT discussed in a previous PURL (Clinician Reviews. 2016;26[4]:20,44), which recommended against the use of α-blockers tamsulosin and nifedipine for ureteral stones measuring < 10 mm.6,7
But the subgroup analysis in this review went one step further by examining passage rates by stone size (< 5 mm vs 5-10 mm) and revealing that passage of larger stones increased with tamsulosin use. The different results based on stone size may explain the recent uncertainty as to whether tamsulosin improves the rate of stone passage.
CAVEATS
What about proximal or XL stones?
Only distal stones were included in seven of the eight trials in this analysis. Thus, this meta-analysis was unable to determine the effect on more proximal stones. Also, it’s unclear if the drug provides any benefit with stones > 10 mm in size.
CHALLENGES TO IMPLEMENTATION
None worth mentioning
We see no challenges to implementation of this recommendation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2018; 67[1]:37-38).
1. Wang RC, Smith-Bindman R, Whitaker E, et al. Effect of tamsulosin on stone passage for ureteral stones: a systematic review and meta-analysis. Ann Emerg Med. 2017;69(3):353-361.
2. Scales CD Jr, Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62(1): 160-165.
3. Türk C, Petrik A, Sarica K, et al. EAU guidelines on diagnosis and conservative management of urolithiasis. Eur Urol. 2016;69(3):468-474.
4. Hollingsworth JM, Canales BK, Rogers MAM, et al. Alpha blockers for treatment of ureteric stones: systematic review and meta-analysis. BMJ. 2016;355:i6112.
5. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014:CD008509.
6. Pickard R, Starr K, MacLennan G, et al. Medical expulsion therapy in adults with ureteric colic: a multicentre, randomized, placebo-controlled trial. Lancet. 2015; 386(9991):341-349.
7. Slattengren AH, Prasad S, Jarrett JB. Kidney stones? It’s time to rethink those meds. J Fam Pract. 2016;65(2): 118-120.
A 54-year-old man presents to the emergency department (ED) with acute-onset left flank pain that radiates to the groin. CT of the abdomen/pelvis without contrast reveals a 7-mm distal ureteral stone. He is deemed an appropriate candidate for outpatient management. In addition to pain medications, should you prescribe tamsulosin?
According to the most recent National Health and Nutrition Examination Survey, the population prevalence of kidney stones is 8.8%, with a self-reported prevalence of 10.6% in men and 7.1% in women.2 Most ureteral stones can be treated in the outpatient setting with oral hydration, antiemetics, and pain control with NSAIDs as firstline treatment and opioids as a second-line option.3
In addition, α-blockers are used for medical expulsive therapy (MET). In fact, the European Association of Urology guideline on urolithiasis states that MET may accelerate passage of ureteral stones.3
Recently, however, uncertainty has surrounded the effectiveness of the α-blocker tamsulosin. Two systematic reviews (limited by heterogeneity because some of the studies lacked a placebo control and blinding) concluded that α-blockers increased stone passage within one to six weeks when compared with placebo or no additional therapy.4,5 However, a recent large, multicenter RCT revealed no difference between tamsulosin and nifedipine, or either one compared with placebo, at decreasing the need for further treatment to achieve stone passage within four weeks.6
STUDY SUMMARY
Results broken down by stone size
This meta-analysis, comprising eight double-blind RCTs, examined the effect of oral tamsulosin (0.4 mg/d; average course, 28 d) on distal ureteral stone passage in adult patients (N = 1,384).1 A subgroup analysis comparing stone size (< 5 mm and 5-10 mm) was also conducted to determine whether size modified the effect of tamsulosin.
The eight selected studies were published between 2009 and 2015; the trials were conducted in multiple countries, in ED and outpatient urology settings. The main outcome measure was the risk difference (RD) in stone passage between the tamsulosin group and placebo group after follow-up imaging at three weeks with CT or plain film radiographs.
Tamsulosin helps some, but not all. The pooled risk for stone passage was higher in the tamsulosin group than in the placebo group (85% vs 66%; RD, 17%), but significant heterogeneity existed across the trials (I2, 80.2%). Subgroup analysis by stone size (< 5 mm vs 5-10 mm) revealed that, compared to placebo, tamsulosin was beneficial for larger stones (6 trials, N = 514; RD, 22%; number needed to treat, 5) but not for smaller stones (4 trials, N = 533; RD, –0.3%). The 5-to-10–mm subgroup had a less heterogeneous population of studies than did the < 5-mm subgroup (I2, 33% and 0% respectively).
In terms of adverse events, tamsulosin did not increase the risk for dizziness (RD, 0.2%) or postural hypotension (RD, 0.1%), compared with placebo.
WHAT’S NEW
Increased passage of larger stones
This meta-analysis included only double-blind RCTs; prior meta-analyses did not. Also, this review included the SUSPEND (Spontaneous Urinary Stone Passage Enabled by Drugs) trial, an RCT discussed in a previous PURL (Clinician Reviews. 2016;26[4]:20,44), which recommended against the use of α-blockers tamsulosin and nifedipine for ureteral stones measuring < 10 mm.6,7
But the subgroup analysis in this review went one step further by examining passage rates by stone size (< 5 mm vs 5-10 mm) and revealing that passage of larger stones increased with tamsulosin use. The different results based on stone size may explain the recent uncertainty as to whether tamsulosin improves the rate of stone passage.
CAVEATS
What about proximal or XL stones?
Only distal stones were included in seven of the eight trials in this analysis. Thus, this meta-analysis was unable to determine the effect on more proximal stones. Also, it’s unclear if the drug provides any benefit with stones > 10 mm in size.
CHALLENGES TO IMPLEMENTATION
None worth mentioning
We see no challenges to implementation of this recommendation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2018; 67[1]:37-38).
A 54-year-old man presents to the emergency department (ED) with acute-onset left flank pain that radiates to the groin. CT of the abdomen/pelvis without contrast reveals a 7-mm distal ureteral stone. He is deemed an appropriate candidate for outpatient management. In addition to pain medications, should you prescribe tamsulosin?
According to the most recent National Health and Nutrition Examination Survey, the population prevalence of kidney stones is 8.8%, with a self-reported prevalence of 10.6% in men and 7.1% in women.2 Most ureteral stones can be treated in the outpatient setting with oral hydration, antiemetics, and pain control with NSAIDs as firstline treatment and opioids as a second-line option.3
In addition, α-blockers are used for medical expulsive therapy (MET). In fact, the European Association of Urology guideline on urolithiasis states that MET may accelerate passage of ureteral stones.3
Recently, however, uncertainty has surrounded the effectiveness of the α-blocker tamsulosin. Two systematic reviews (limited by heterogeneity because some of the studies lacked a placebo control and blinding) concluded that α-blockers increased stone passage within one to six weeks when compared with placebo or no additional therapy.4,5 However, a recent large, multicenter RCT revealed no difference between tamsulosin and nifedipine, or either one compared with placebo, at decreasing the need for further treatment to achieve stone passage within four weeks.6
STUDY SUMMARY
Results broken down by stone size
This meta-analysis, comprising eight double-blind RCTs, examined the effect of oral tamsulosin (0.4 mg/d; average course, 28 d) on distal ureteral stone passage in adult patients (N = 1,384).1 A subgroup analysis comparing stone size (< 5 mm and 5-10 mm) was also conducted to determine whether size modified the effect of tamsulosin.
The eight selected studies were published between 2009 and 2015; the trials were conducted in multiple countries, in ED and outpatient urology settings. The main outcome measure was the risk difference (RD) in stone passage between the tamsulosin group and placebo group after follow-up imaging at three weeks with CT or plain film radiographs.
Tamsulosin helps some, but not all. The pooled risk for stone passage was higher in the tamsulosin group than in the placebo group (85% vs 66%; RD, 17%), but significant heterogeneity existed across the trials (I2, 80.2%). Subgroup analysis by stone size (< 5 mm vs 5-10 mm) revealed that, compared to placebo, tamsulosin was beneficial for larger stones (6 trials, N = 514; RD, 22%; number needed to treat, 5) but not for smaller stones (4 trials, N = 533; RD, –0.3%). The 5-to-10–mm subgroup had a less heterogeneous population of studies than did the < 5-mm subgroup (I2, 33% and 0% respectively).
In terms of adverse events, tamsulosin did not increase the risk for dizziness (RD, 0.2%) or postural hypotension (RD, 0.1%), compared with placebo.
WHAT’S NEW
Increased passage of larger stones
This meta-analysis included only double-blind RCTs; prior meta-analyses did not. Also, this review included the SUSPEND (Spontaneous Urinary Stone Passage Enabled by Drugs) trial, an RCT discussed in a previous PURL (Clinician Reviews. 2016;26[4]:20,44), which recommended against the use of α-blockers tamsulosin and nifedipine for ureteral stones measuring < 10 mm.6,7
But the subgroup analysis in this review went one step further by examining passage rates by stone size (< 5 mm vs 5-10 mm) and revealing that passage of larger stones increased with tamsulosin use. The different results based on stone size may explain the recent uncertainty as to whether tamsulosin improves the rate of stone passage.
CAVEATS
What about proximal or XL stones?
Only distal stones were included in seven of the eight trials in this analysis. Thus, this meta-analysis was unable to determine the effect on more proximal stones. Also, it’s unclear if the drug provides any benefit with stones > 10 mm in size.
CHALLENGES TO IMPLEMENTATION
None worth mentioning
We see no challenges to implementation of this recommendation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2018; 67[1]:37-38).
1. Wang RC, Smith-Bindman R, Whitaker E, et al. Effect of tamsulosin on stone passage for ureteral stones: a systematic review and meta-analysis. Ann Emerg Med. 2017;69(3):353-361.
2. Scales CD Jr, Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62(1): 160-165.
3. Türk C, Petrik A, Sarica K, et al. EAU guidelines on diagnosis and conservative management of urolithiasis. Eur Urol. 2016;69(3):468-474.
4. Hollingsworth JM, Canales BK, Rogers MAM, et al. Alpha blockers for treatment of ureteric stones: systematic review and meta-analysis. BMJ. 2016;355:i6112.
5. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014:CD008509.
6. Pickard R, Starr K, MacLennan G, et al. Medical expulsion therapy in adults with ureteric colic: a multicentre, randomized, placebo-controlled trial. Lancet. 2015; 386(9991):341-349.
7. Slattengren AH, Prasad S, Jarrett JB. Kidney stones? It’s time to rethink those meds. J Fam Pract. 2016;65(2): 118-120.
1. Wang RC, Smith-Bindman R, Whitaker E, et al. Effect of tamsulosin on stone passage for ureteral stones: a systematic review and meta-analysis. Ann Emerg Med. 2017;69(3):353-361.
2. Scales CD Jr, Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62(1): 160-165.
3. Türk C, Petrik A, Sarica K, et al. EAU guidelines on diagnosis and conservative management of urolithiasis. Eur Urol. 2016;69(3):468-474.
4. Hollingsworth JM, Canales BK, Rogers MAM, et al. Alpha blockers for treatment of ureteric stones: systematic review and meta-analysis. BMJ. 2016;355:i6112.
5. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014:CD008509.
6. Pickard R, Starr K, MacLennan G, et al. Medical expulsion therapy in adults with ureteric colic: a multicentre, randomized, placebo-controlled trial. Lancet. 2015; 386(9991):341-349.
7. Slattengren AH, Prasad S, Jarrett JB. Kidney stones? It’s time to rethink those meds. J Fam Pract. 2016;65(2): 118-120.

Kidney transplant: New opportunities and challenges
Much has improved in renal transplantation over the past 20 years. The focus has shifted to using stronger immunotherapy rather than trying to minimize it. There has been increasing recognition of infection and ways to prevent and treat it. Induction therapy now has greater emphasis so that maintenance therapy can be eased, with the aim of reducing long-term toxicity. Perhaps the biggest change is the practice of screening for donor-specific antibodies at the time of transplant so that predictable problems can be prevented or better handled if they occur. Such advances have helped patients directly and by extending the life of their transplanted organs.
LONGER SURVIVAL
As early as the 1990s, it was recognized that kidney transplant offers a survival advantage for patients with end-stage renal disease over maintenance on dialysis.1 Although the risk of death is higher immediately after transplant, within a few months it becomes much lower than for patients on dialysis. Survival varies according to the health of the patient and the quality of the transplanted organ.
In general, patients who obtain the greatest benefit from transplants in terms of years of life gained are those with diabetes, especially those who are younger. Those ages 20 to 39 live about 8 years on dialysis vs 25 years after transplant.
CONTRAINDICATIONS TO TRANSPLANT

There are multiple contraindications to a solitary kidney transplant (Table 1), including smoking. Most transplant centers require that smokers quit before transplant. Long-standing smokers almost double their risk of a cardiac event after transplant and double their rate of malignancy. Active smoking at the time of transplant is associated with twice the risk of death by 10 years after transplant compared with that of nonsmokers.2 Cotinine testing can detect whether a patient is an active smoker.
WAITING-LIST CONSIDERATIONS
Organs are scarce
The number of patients on the kidney waiting list has increased rapidly in the last few decades, while the number of transplants performed each year has remained about the same. In 2016, about 100,000 patients were on the list, but only about 19,000 transplants were performed.3 Wait times, especially for deceased-donor organs, have increased to about 6 years, varying by blood type and geographic region.
Waiting-list placement
Placement on the waiting list for a deceased-donor kidney transplant occurs when a patient has an estimated glomerular filtration rate (GFR) of 20 mL/min/1.73 m2 or less, although referral to the list can be made earlier. Early listing remains advantageous, as total time on the list will be counted before starting dialysis. “Preemptive transplant” means the patient had no dialysis before transplant; this applies to about 10% of transplant recipients. These patients tend to fare the best and are usually recipients of a living-donor organ.
Most patients do not receive a transplant until the GFR is less than 15 mL/min/1.73 m2.
Since 2014, wait time has been measured from the beginning of dialysis rather than the date of waiting-list placement in patients who are listed after starting dialysis therapy. This approach is more fair but sometimes introduces problems. A patient who did not previously know about the list may suddenly jump to the head of the line after 10 years of dialysis, by which time comorbidities associated with long-term dialysis make the patient less likely to gain as much benefit from a transplant as people lower on the list. Time on dialysis, or “dialysis vintage,” predicts patient and kidney survival after transplant, with reduced survival associated with increasing time on dialysis.4
Shorter wait for a suboptimal kidney
The aging population has increased the number of older patients being listed for transplant, presenting multiple challenges. Patients age 65 or older have a 50% chance of dying before they receive a transplant during a 5-year wait.
A patient may shorten the wait by joining the list for a suboptimal organ. All deceased-donor organs are given a Kidney Donor Profile Index score, which predicts the longevity of an organ after transplant. The score is determined by donor age, kidney function based on the serum creatinine at the time of death, and other donor factors.
A kidney with a score higher than 85% is likely to function longer than only 15% of available kidneys. Patients who receive a kidney with that score have a longer period of risk of death soon after transplant and a slightly higher risk of death in the long term than patients who receive a healthier kidney, although on average they still do better than patients on dialysis.5
Older patients should be encouraged to sign up for both the regular waiting list and the suboptimal kidney waiting list to reduce the risk of dying before they get a kidney.
LIVING-DONOR ORGAN TRANSPLANT
Many advantages
Living-donor organ transplant is associated with a better survival rate than deceased-donor organ transplant, and the advantage becomes greater over time. At 1 year, patient survival is more than 90% in both groups, but by 5 years about 80% of patients with a living-donor organ are still alive vs only about 65% of patients with a deceased-donor organ.
The waiting time for a living-donor transplant may be only weeks to months, rather than years. Because increasing time on dialysis predicts worse patient and graft survival after transplant, the shorter wait time is a big advantage. In addition, because the donor and recipient are typically in adjacent operating rooms, the organ sustains less ischemic damage. In general, the kidney quality is better from healthy donors, resulting in superior function early on and longer graft survival by an average of 4 years. If the living donor is related to the recipient, human leukocyte antigen matching also tends to be better and predicts better outcomes.
Special challenges
Opting for a living-donor organ also entails special challenges. In addition to the ethical issues surrounding living-donor organ donation, an appropriate donor must be found. Donors must be highly motivated and pass physical, laboratory, and psychological evaluations.
For older patients, if the donor is a spouse or close friend, he or she is also likely to be older, making the organ less viable than one from a younger person. Even an adult child may not be an ideal donor if there is a family propensity to kidney disease, such as diabetic nephropathy. No test is available to determine the risk for future diabetes, but it is known to run in families.
POTENT IMMUNOSUPPRESSION
Induction therapy
Induction therapy with antithymocyte globulin or basiliximab provides intense immunosuppression to prevent acute rejection during the early posttransplant period.
Antithymocyte globulin is a potent agent that contains antibodies directed at T cells, B cells, neutrophils, platelets, adhesion molecules, and complement. It binds T cells and removes them from circulation by opsonization in splenic and lymphoid tissue. The immunosuppressive effect is sustained for at least 2 to 3 months after a series of injections (dosage 1.5 mg/kg/day, usually for 4 to 10 doses). Antithymocyte globulin is also used to treat acute rejection, especially high-grade rejection for which steroid therapy is likely to be insufficient.
Basiliximab consists of antibodies to the interleukin 2 (IL-2) receptor of T cells. Binding to T cells prevents their activation rather than removing them from circulation. The drug prevents rejection, with 30% relative reduction in early studies compared with placebo. However, it is ineffective in reversing established rejection. Dosage is 20 mg at day 0 and day 4, which provides receptor saturation for 30 to 45 days.
Basiliximab is also sometimes used off-label for patients who need to discontinue a calcineurin inhibitor (ie, tacrolimus or cyclosporine). In such cases, normal therapy is put on hold while basiliximab is given for 1 or 2 doses. Case series have been reported for this use, particularly for patients with a heart and liver transplant who develop acute kidney injury while hospitalized.6,7
Antithymocyte globulin is more effective but also more risky. Brennan et al8 randomized 278 transplant recipients to either antithymocyte globulin or basiliximab. Patients in the antithymocyte globulin group had a 16% rejection rate vs 26% in the basiliximab group.
Antithymocyte globulin therapy is associated with multiple adverse effects, including fever and chills, pulmonary edema, and long-standing immunosuppressive effects such as increased risk of lymphoma and cytomegalovirus (CMV) infection. Basiliximab side-effect profiles are similar to those of placebo.
Maintenance therapy
The calcineurin inhibitors cyclosporine and tacrolimus remain the standard of care in kidney transplant despite multiple drug interactions and side effects that include renal toxicity and fibrosis. Cyclosporine and tacrolimus both bind intracellular immunophilins and thereby prevent transcription of IL-2 and production of T cells. The drugs work similarly but have different binding sites. Cyclosporine has largely been replaced by tacrolimus because its reliability of dosing and higher potency are associated with lower rejection rates.
Tacrolimus is typically given twice daily (1–6 mg/dose). Twelve-hour trough levels are followed (target: 8–12 ng/mL early on, then 5–8 ng/mL after 3 months posttransplant). Side effects include hypertension and hypercholesterolemia, but less so than with cyclosporine. On the other hand, hyperglycemia tends to be worse with tacrolimus than with cyclosporine, and combining tacrolimus with steroids frequently leads to diabetes. Tacrolimus can also cause acute and chronic renal failure, especially at high drug levels, as well as neurotoxicity, tremors, and hair loss.

Cyclosporine, tacrolimus, and sirolimus (not a calcineurin inhibitor) are metabolized through the same cytochrome P450 pathway (CYP3A4), so they have common drug interactions (Table 2).
Mycophenolate mofetil is typically used as an adjunct therapy (500–1,000 mg twice daily). It is also used for other kidney diseases before transplant, including lupus nephritis. Transplanted kidney rejection rates with mycophenolate mofetil with steroids are about 40%, so the drug is not potent enough to be used without a calcineurin inhibitor.
Side effects include gastrointestinal toxicity in up to 20% of patients, and leukopenia, which is associated with viral infections.
CORONARY ARTERY DISEASE IS COMMON WITH DIALYSIS
Coronary artery disease is highly associated with end-stage kidney disease and occurs in as many as 85% of older patients with diabetes on dialysis. Although patients with end-stage kidney disease tend to have more numerous and severe atherosclerotic lesions compared with the general population, justifying aggressive management, cardiac care tends to be conservative in patients on dialysis.9
Death from acute myocardial infarction occurs in about 20% to 30% of patients on dialysis vs about 2% of patients with normal renal function. Five years after myocardial infarction, survival is only about 30% in patients on dialysis.9
There are many explanations for excess coronary artery disease in patients on dialysis. In addition to the traditional cardiovascular risk factors of diabetes, hypertension, and preexisting coronary artery disease, patients are in a proinflammatory uremic state and have high levels of phosphorus and fibroblast growth factor 23 that contribute to vascular calcification. Almost all patients have high homocysteine levels and hemodynamic instability, particularly if they are on hemodialysis.
Pretransplant evaluation for heart disease
Patients on the kidney transplant waiting list are screened aggressively for heart disease. A history of myocardial infarction usually results in removal from the list. All patients have an initial electrocardiogram and echocardiogram. Thallium or echocardiographic stress testing is used for patients who are age 50 and older, have diabetes, or have had dialysis for many years. Patients with evidence of ischemia undergo catheterization.
Patients are also screened with computed tomography before transplant. Because the kidney is typically anastomosed to the iliac artery and vein, heavy calcification of the iliac artery can make the procedure too difficult to perform.
Reduced long-term risk of myocardial infarction after transplant
Kasiske et al10 analyzed data from more than 50,000 patients from the US Renal Data System and found that, for about the first year after transplant, patients who underwent kidney transplant were more likely to have a myocardial infarction than those on dialysis. After that, they fared better than patients who remained on dialysis. Those with a living-donor transplant were less likely at all times to have a myocardial infarction than those with a deceased-donor transplant. By 3 years after transplant, the relative risk of having a myocardial infarction was 0.89 for deceased-donor organ recipients and 0.69 for living-donor recipients compared with patients on the waiting list.10
INFECTIOUS COMPLICATIONS IN KIDNEY RECIPIENTS

Kidney recipients are prone to many common and uncommon infections (Table 3). All potential recipients are tested pretransplant for hepatitis B, hepatitis C, human immunodeficiency virus, syphilis, and tuberculosis. A positive result does not necessarily rule out transplant.
The following viral serology tests are also done before transplant:
Epstein-Barr virus (antibodies are positive in about 90% of adults)
CMV (about 70% of adults are seropositive)
Varicella zoster (seronegative patients should be given live-attenuated varicella vaccine).
Risk of transmission of these viruses relates to the serostatus of the donor and recipient before transplant. If a donor is positive for viral antibodies but the recipient is not (a so-called “mismatch”), risk is higher after transplant.
Hepatitis C
Patients with hepatitis C fare better if they get a transplant than if they remain on dialysis, although their posttransplant course is worse compared with transplant patients who do not have hepatitis. Some patients develop accelerated liver disease after kidney transplant. Hepatitis C-related kidney disease—membranous proliferative glomerulonephritis—also occurs, as do comorbidities such as diabetes.
Careful evaluation is warranted before transplant, including liver imaging, alpha-fetoprotein testing, and liver biopsy to evaluate for hepatocellular carcinoma. A patient with advanced fibrosis or cirrhosis may not be a candidate for kidney transplant alone but could possibly receive a combined kidney and liver transplant.
There is a need to determine the best time to treat hepatitis C infection. Patients with advanced liver disease or hepatitis C-related kidney disease would likely benefit from early treatment. However, delaying treatment could shorten the wait time for a deceased-donor organ positive for hepatitis C. Transplant candidates with active hepatitis C are uniquely considered to accept hepatitis C-positive kidneys, which are often discarded, and may only wait weeks for such a transplant. The shortened kidney survival associated with a hepatitis C-positive kidney may no longer be true with the new antiviral hepatitis C therapy, which has been shown to be effective post-transplant.
Hepatitis B
No cure is available for hepatitis B infection, but it can be well controlled with antiviral therapy. Patients with hepatitis B infection may be candidates for transplant, but they should be stable on antiviral therapy (lamivudine, entecavir, or tenofovir) to eliminate the viral load before transplant, and therapy should be continued afterward. Liver imaging, alpha-fetoprotein levels, and biopsy are recommended for evaluation. All hepatitis B- negative patients should be vaccinated before transplant.
Organs from living or deceased donors that test positive for hepatitis B core antibody, indicating prior exposure, can be considered for transplant in a patient who tests positive for hepatitis B surface antibody, indicating successful vaccination or prior exposure in the recipient. But donors must have negative surface antigen and polymerase chain reaction (PCR) tests that indicate no active hepatitis B infection.
Cytomegalovirus
CMV typically does not appear until prophylactic therapy is stopped. Classic symptoms are fever, leukopenia, and diarrhea. Infection can involve any organ, and patients may present with hepatitis, pancreatitis or, less commonly, pneumonitis.
Patients who are negative for CMV before transplant and receive a donor-positive organ are at the highest risk. Patients who are CMV IgG-positive are considered to be at intermediate risk, regardless of the donor status. Patients who are negative for CMV and receive a donor-negative organ are at the lowest risk and do not need prophylaxis with valganciclovir.
CMV infection is diagnosed by PCR testing of the blood or immunostaining in tissue biopsy. Occasionally, blood testing is negative in the face of tissue-based disease.
BK virus
BK is a polyoma virus and a common virus associated with kidney transplant. Viremia is seen in about 18% of patients, whereas actual kidney disease associated with a higher level of virus is seen in fewer than 10% of patients. Most people are exposed to BK virus, often in childhood, and it can remain indolent in the bladder and uroepithelium.
Patients can develop BK nephropathy after exposure to transplant immunosuppression.11 Posttransplant monitoring protocols typically include PCR testing for BK virus at 1, 3, 6, and 12 months. No agent has been identified to specifically treat BK virus. The general strategy is to minimize immunosuppressive therapy by reducing or eliminating mycophenolate mofetil. Fortunately, BK virus does not tend to recur, and patients can have a low-level viremia (< 10,000 copies/mL) persisting over months or even years but often without clinical consequences.
The appearance of BK virus on biopsy can mimic acute rejection. Before BK viral nephropathy was a recognized entity, patients would have been diagnosed with acute rejection and may have been put on high-dose steroids, which would have worsened the BK infection.
Posttransplant lymphoproliferative disorder
Posttransplant lymphoproliferative disorder is most often associated with Epstein-Barr virus and usually involves a large, diffuse B-cell lymphoma. Burkitt lymphoma and plasma cell neoplasms also can occur less commonly.
The condition is about 30 times more common in patients after transplant than in the general population, and it is the third most common malignancy in transplant patients after skin and cervical cancers. About 80% of the cases occur early after transplant, within the first year.
Patients typically have a marked elevation in viral load of Epstein-Barr virus, although a negative viral load does not rule it out. A patient who is serologically negative for Epstein-Barr virus receiving a donor-positive kidney is at highest risk; this situation is most often seen in the pediatric population. Potent induction therapies (eg, antilymphocyte antibody therapy) are also associated with posttransplant lymphoproliferative disorder.
Patients typically present with fever of unknown origin with no localizing signs or symptoms. Mass lesions can be challenging to find; positron emission tomography may be helpful. The culprit is usually a focal mass, ulcer (especially in the gastrointestinal tract), or infiltrate (commonly localized to the allograft). Multifocal or disseminated disease can also occur, including lymphoma or with central nervous system, gastrointestinal, or pulmonary involvement.
Biopsy of the affected site is required for histopathology and Epstein-Barr virus markers. PCR blood testing is often positive for Epstein-Barr virus.
Typical antiviral therapy does not eliminate Epstein-Barr virus. In early polyclonal viral proliferation, the first goal is to reduce immunosuppressive therapy. Rituximab alone may also help in polymorphic cases. With disease that is clearly monomorphic and has transformed to a true malignancy, cytotoxic chemotherapy is also required. “R-CHOP,” a combination therapy consisting of rituximab with cyclophosphamide, doxorubicin, vincristine, and prednisone, is usually used. Radiation therapy may help in some cases.
Cryptococcal infection
Previously seen in patients with acquired immune deficiency syndrome, cryptococcal infection is now most commonly encountered in patients with solid-organ transplants. Vilchez et al12 found a 1% incidence in a series of more than 5,000 patients who had received an organ transplant.
Immunosuppression likely conveys risk, but because cryptococcal infection is acquired, environmental exposure also plays a role. It tends to appear more than 6 months after transplant, indicating that its cause is a primary infection by spore inhalation rather than by reactivation or transmission from the donor organ.13 Bird exposure is a risk factor for cryptococcal infection. One case identified the same strain of Cryptococcus in a kidney transplant recipient and the family’s pet cockatoo.14
Cryptococcal infection typically starts as pneumonia, which may be subclinical. The infection can then disseminate, with meningitis presenting with headache and mental status changes being the most concerning complication. The death rate is about 50% in most series of patients with meningitis. Skin and soft-tissue manifestations may also occur in 10% to 15% of cases and can be nodular, ulcerative, or cellulitic.
More than 75% of fungal infections requiring hospitalization in US patients who have undergone transplant are attributed to either Candida, Aspergillus, or Cryptococcus species.15 Risk of fungal infection is increased with diabetes, duration of pretransplant dialysis, tacrolimus therapy, or rejection treatment.
- Wolfe RA, Ashby VB, Milford EL, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med 1999; 341:1725–1730.
- Kasiske BL, Klinger D. Cigarette smoking in renal transplant recipients. J Am Soc Nephrol 2000; 11:753–759.
- United Network for Organ Sharing. Transplant trends. https://transplantpro.org/technology/transplant-trends/#waitlists_by_organ. Accessed December 13, 2017.
- Meier-Kriesche HU, Kaplan B. Waiting time on dialysis as the strongest modifiable risk factor for renal transplant outcomes: a paired donor kidney analysis. Transplantation 2002; 74:1377–1381.
- Ojo AO, Hanson JA, Meier-Kriesche H, et al. Survival in recipients of marginal cadaveric donor kidneys compared with other recipients and wait-listed transplant candidates. J Am Soc Nephrol 2001; 12:589–597.
- Alonso P. Sanchez-Lazaro I, Almenar L, et al. Use of a “CNI holidays” strategy in acute renal dysfunction late after heart transplant. Report of two cases. Heart Int 2014; 9:74–77.
- Cantarovich M, Metrakos P, Giannetti N, Cecere R, Barkun J, Tchervenkov J. Anti-CD25 monoclonal antibody coverage allows for calcineurin inhibitor “holiday” in solid organ transplant patients with acute renal dysfunction. Transplantation 2002; 73:1169–1172.
- Brennan DC, Daller JA, Lake KD, Cibrik D, Del Castillo D; Thymoglobulin Induction Study Group. Rabbit antithymocyte globulin versus basiliximab in renal transplantation. N Engl J Med 2006; 355:1967–1977.
- McCullough PA. Evaluation and treatment of coronary artery disease in patients with end-stage renal disease. Kidney Int 2005; 67:S51–S58.
- Kasiske BL, Maclean JR, Snyder JJ. Acute myocardial infarction and kidney transplantation. J Am Soc Nephrol 2006; 17:900–907.
- Bohl DL, Storch GA, Ryschkewitsch C, et al. Donor origin of BK virus in renal transplantation and role of HLA C7 in susceptibility to sustained BK viremia. Am J Transplant 2005; 5:2213–2221.
- Vilchez RA, Fung J, Kusne S. Cryptococcosis in organ transplant recipients: an overview. Am J Transplant 2002; 2:575–580.
- Vilchez R, Shapiro R, McCurry K, et al. Longitudinal study of cryptococcosis in adult solid-organ transplant recipients. Transpl Int 2003; 16:336–340.
- Nosanchuk JD, Shoham S, Fries BC, Shapiro DS, Levitz SM, Casadevall A. Evidence of zoonotic transmission of Cryptococcus neoformans from a pet cockatoo to an immunocompromised patient. Ann Intern Med 2000; 132:205–208.
- Abbott KC, Hypolite I, Poropatich RK, et al. Hospitalizations for fungal infections after renal transplantation in the United States. Transpl Infect Dis 2001; 3:203–211.
Much has improved in renal transplantation over the past 20 years. The focus has shifted to using stronger immunotherapy rather than trying to minimize it. There has been increasing recognition of infection and ways to prevent and treat it. Induction therapy now has greater emphasis so that maintenance therapy can be eased, with the aim of reducing long-term toxicity. Perhaps the biggest change is the practice of screening for donor-specific antibodies at the time of transplant so that predictable problems can be prevented or better handled if they occur. Such advances have helped patients directly and by extending the life of their transplanted organs.
LONGER SURVIVAL
As early as the 1990s, it was recognized that kidney transplant offers a survival advantage for patients with end-stage renal disease over maintenance on dialysis.1 Although the risk of death is higher immediately after transplant, within a few months it becomes much lower than for patients on dialysis. Survival varies according to the health of the patient and the quality of the transplanted organ.
In general, patients who obtain the greatest benefit from transplants in terms of years of life gained are those with diabetes, especially those who are younger. Those ages 20 to 39 live about 8 years on dialysis vs 25 years after transplant.
CONTRAINDICATIONS TO TRANSPLANT
There are multiple contraindications to a solitary kidney transplant (Table 1), including smoking. Most transplant centers require that smokers quit before transplant. Long-standing smokers almost double their risk of a cardiac event after transplant and double their rate of malignancy. Active smoking at the time of transplant is associated with twice the risk of death by 10 years after transplant compared with that of nonsmokers.2 Cotinine testing can detect whether a patient is an active smoker.
WAITING-LIST CONSIDERATIONS
Organs are scarce
The number of patients on the kidney waiting list has increased rapidly in the last few decades, while the number of transplants performed each year has remained about the same. In 2016, about 100,000 patients were on the list, but only about 19,000 transplants were performed.3 Wait times, especially for deceased-donor organs, have increased to about 6 years, varying by blood type and geographic region.
Waiting-list placement
Placement on the waiting list for a deceased-donor kidney transplant occurs when a patient has an estimated glomerular filtration rate (GFR) of 20 mL/min/1.73 m2 or less, although referral to the list can be made earlier. Early listing remains advantageous, as total time on the list will be counted before starting dialysis. “Preemptive transplant” means the patient had no dialysis before transplant; this applies to about 10% of transplant recipients. These patients tend to fare the best and are usually recipients of a living-donor organ.
Most patients do not receive a transplant until the GFR is less than 15 mL/min/1.73 m2.
Since 2014, wait time has been measured from the beginning of dialysis rather than the date of waiting-list placement in patients who are listed after starting dialysis therapy. This approach is more fair but sometimes introduces problems. A patient who did not previously know about the list may suddenly jump to the head of the line after 10 years of dialysis, by which time comorbidities associated with long-term dialysis make the patient less likely to gain as much benefit from a transplant as people lower on the list. Time on dialysis, or “dialysis vintage,” predicts patient and kidney survival after transplant, with reduced survival associated with increasing time on dialysis.4
Shorter wait for a suboptimal kidney
The aging population has increased the number of older patients being listed for transplant, presenting multiple challenges. Patients age 65 or older have a 50% chance of dying before they receive a transplant during a 5-year wait.
A patient may shorten the wait by joining the list for a suboptimal organ. All deceased-donor organs are given a Kidney Donor Profile Index score, which predicts the longevity of an organ after transplant. The score is determined by donor age, kidney function based on the serum creatinine at the time of death, and other donor factors.
A kidney with a score higher than 85% is likely to function longer than only 15% of available kidneys. Patients who receive a kidney with that score have a longer period of risk of death soon after transplant and a slightly higher risk of death in the long term than patients who receive a healthier kidney, although on average they still do better than patients on dialysis.5
Older patients should be encouraged to sign up for both the regular waiting list and the suboptimal kidney waiting list to reduce the risk of dying before they get a kidney.
LIVING-DONOR ORGAN TRANSPLANT
Many advantages
Living-donor organ transplant is associated with a better survival rate than deceased-donor organ transplant, and the advantage becomes greater over time. At 1 year, patient survival is more than 90% in both groups, but by 5 years about 80% of patients with a living-donor organ are still alive vs only about 65% of patients with a deceased-donor organ.
The waiting time for a living-donor transplant may be only weeks to months, rather than years. Because increasing time on dialysis predicts worse patient and graft survival after transplant, the shorter wait time is a big advantage. In addition, because the donor and recipient are typically in adjacent operating rooms, the organ sustains less ischemic damage. In general, the kidney quality is better from healthy donors, resulting in superior function early on and longer graft survival by an average of 4 years. If the living donor is related to the recipient, human leukocyte antigen matching also tends to be better and predicts better outcomes.
Special challenges
Opting for a living-donor organ also entails special challenges. In addition to the ethical issues surrounding living-donor organ donation, an appropriate donor must be found. Donors must be highly motivated and pass physical, laboratory, and psychological evaluations.
For older patients, if the donor is a spouse or close friend, he or she is also likely to be older, making the organ less viable than one from a younger person. Even an adult child may not be an ideal donor if there is a family propensity to kidney disease, such as diabetic nephropathy. No test is available to determine the risk for future diabetes, but it is known to run in families.
POTENT IMMUNOSUPPRESSION
Induction therapy
Induction therapy with antithymocyte globulin or basiliximab provides intense immunosuppression to prevent acute rejection during the early posttransplant period.
Antithymocyte globulin is a potent agent that contains antibodies directed at T cells, B cells, neutrophils, platelets, adhesion molecules, and complement. It binds T cells and removes them from circulation by opsonization in splenic and lymphoid tissue. The immunosuppressive effect is sustained for at least 2 to 3 months after a series of injections (dosage 1.5 mg/kg/day, usually for 4 to 10 doses). Antithymocyte globulin is also used to treat acute rejection, especially high-grade rejection for which steroid therapy is likely to be insufficient.
Basiliximab consists of antibodies to the interleukin 2 (IL-2) receptor of T cells. Binding to T cells prevents their activation rather than removing them from circulation. The drug prevents rejection, with 30% relative reduction in early studies compared with placebo. However, it is ineffective in reversing established rejection. Dosage is 20 mg at day 0 and day 4, which provides receptor saturation for 30 to 45 days.
Basiliximab is also sometimes used off-label for patients who need to discontinue a calcineurin inhibitor (ie, tacrolimus or cyclosporine). In such cases, normal therapy is put on hold while basiliximab is given for 1 or 2 doses. Case series have been reported for this use, particularly for patients with a heart and liver transplant who develop acute kidney injury while hospitalized.6,7
Antithymocyte globulin is more effective but also more risky. Brennan et al8 randomized 278 transplant recipients to either antithymocyte globulin or basiliximab. Patients in the antithymocyte globulin group had a 16% rejection rate vs 26% in the basiliximab group.
Antithymocyte globulin therapy is associated with multiple adverse effects, including fever and chills, pulmonary edema, and long-standing immunosuppressive effects such as increased risk of lymphoma and cytomegalovirus (CMV) infection. Basiliximab side-effect profiles are similar to those of placebo.
Maintenance therapy
The calcineurin inhibitors cyclosporine and tacrolimus remain the standard of care in kidney transplant despite multiple drug interactions and side effects that include renal toxicity and fibrosis. Cyclosporine and tacrolimus both bind intracellular immunophilins and thereby prevent transcription of IL-2 and production of T cells. The drugs work similarly but have different binding sites. Cyclosporine has largely been replaced by tacrolimus because its reliability of dosing and higher potency are associated with lower rejection rates.
Tacrolimus is typically given twice daily (1–6 mg/dose). Twelve-hour trough levels are followed (target: 8–12 ng/mL early on, then 5–8 ng/mL after 3 months posttransplant). Side effects include hypertension and hypercholesterolemia, but less so than with cyclosporine. On the other hand, hyperglycemia tends to be worse with tacrolimus than with cyclosporine, and combining tacrolimus with steroids frequently leads to diabetes. Tacrolimus can also cause acute and chronic renal failure, especially at high drug levels, as well as neurotoxicity, tremors, and hair loss.
Cyclosporine, tacrolimus, and sirolimus (not a calcineurin inhibitor) are metabolized through the same cytochrome P450 pathway (CYP3A4), so they have common drug interactions (Table 2).
Mycophenolate mofetil is typically used as an adjunct therapy (500–1,000 mg twice daily). It is also used for other kidney diseases before transplant, including lupus nephritis. Transplanted kidney rejection rates with mycophenolate mofetil with steroids are about 40%, so the drug is not potent enough to be used without a calcineurin inhibitor.
Side effects include gastrointestinal toxicity in up to 20% of patients, and leukopenia, which is associated with viral infections.
CORONARY ARTERY DISEASE IS COMMON WITH DIALYSIS
Coronary artery disease is highly associated with end-stage kidney disease and occurs in as many as 85% of older patients with diabetes on dialysis. Although patients with end-stage kidney disease tend to have more numerous and severe atherosclerotic lesions compared with the general population, justifying aggressive management, cardiac care tends to be conservative in patients on dialysis.9
Death from acute myocardial infarction occurs in about 20% to 30% of patients on dialysis vs about 2% of patients with normal renal function. Five years after myocardial infarction, survival is only about 30% in patients on dialysis.9
There are many explanations for excess coronary artery disease in patients on dialysis. In addition to the traditional cardiovascular risk factors of diabetes, hypertension, and preexisting coronary artery disease, patients are in a proinflammatory uremic state and have high levels of phosphorus and fibroblast growth factor 23 that contribute to vascular calcification. Almost all patients have high homocysteine levels and hemodynamic instability, particularly if they are on hemodialysis.
Pretransplant evaluation for heart disease
Patients on the kidney transplant waiting list are screened aggressively for heart disease. A history of myocardial infarction usually results in removal from the list. All patients have an initial electrocardiogram and echocardiogram. Thallium or echocardiographic stress testing is used for patients who are age 50 and older, have diabetes, or have had dialysis for many years. Patients with evidence of ischemia undergo catheterization.
Patients are also screened with computed tomography before transplant. Because the kidney is typically anastomosed to the iliac artery and vein, heavy calcification of the iliac artery can make the procedure too difficult to perform.
Reduced long-term risk of myocardial infarction after transplant
Kasiske et al10 analyzed data from more than 50,000 patients from the US Renal Data System and found that, for about the first year after transplant, patients who underwent kidney transplant were more likely to have a myocardial infarction than those on dialysis. After that, they fared better than patients who remained on dialysis. Those with a living-donor transplant were less likely at all times to have a myocardial infarction than those with a deceased-donor transplant. By 3 years after transplant, the relative risk of having a myocardial infarction was 0.89 for deceased-donor organ recipients and 0.69 for living-donor recipients compared with patients on the waiting list.10
INFECTIOUS COMPLICATIONS IN KIDNEY RECIPIENTS
Kidney recipients are prone to many common and uncommon infections (Table 3). All potential recipients are tested pretransplant for hepatitis B, hepatitis C, human immunodeficiency virus, syphilis, and tuberculosis. A positive result does not necessarily rule out transplant.
The following viral serology tests are also done before transplant:
Epstein-Barr virus (antibodies are positive in about 90% of adults)
CMV (about 70% of adults are seropositive)
Varicella zoster (seronegative patients should be given live-attenuated varicella vaccine).
Risk of transmission of these viruses relates to the serostatus of the donor and recipient before transplant. If a donor is positive for viral antibodies but the recipient is not (a so-called “mismatch”), risk is higher after transplant.
Hepatitis C
Patients with hepatitis C fare better if they get a transplant than if they remain on dialysis, although their posttransplant course is worse compared with transplant patients who do not have hepatitis. Some patients develop accelerated liver disease after kidney transplant. Hepatitis C-related kidney disease—membranous proliferative glomerulonephritis—also occurs, as do comorbidities such as diabetes.
Careful evaluation is warranted before transplant, including liver imaging, alpha-fetoprotein testing, and liver biopsy to evaluate for hepatocellular carcinoma. A patient with advanced fibrosis or cirrhosis may not be a candidate for kidney transplant alone but could possibly receive a combined kidney and liver transplant.
There is a need to determine the best time to treat hepatitis C infection. Patients with advanced liver disease or hepatitis C-related kidney disease would likely benefit from early treatment. However, delaying treatment could shorten the wait time for a deceased-donor organ positive for hepatitis C. Transplant candidates with active hepatitis C are uniquely considered to accept hepatitis C-positive kidneys, which are often discarded, and may only wait weeks for such a transplant. The shortened kidney survival associated with a hepatitis C-positive kidney may no longer be true with the new antiviral hepatitis C therapy, which has been shown to be effective post-transplant.
Hepatitis B
No cure is available for hepatitis B infection, but it can be well controlled with antiviral therapy. Patients with hepatitis B infection may be candidates for transplant, but they should be stable on antiviral therapy (lamivudine, entecavir, or tenofovir) to eliminate the viral load before transplant, and therapy should be continued afterward. Liver imaging, alpha-fetoprotein levels, and biopsy are recommended for evaluation. All hepatitis B- negative patients should be vaccinated before transplant.
Organs from living or deceased donors that test positive for hepatitis B core antibody, indicating prior exposure, can be considered for transplant in a patient who tests positive for hepatitis B surface antibody, indicating successful vaccination or prior exposure in the recipient. But donors must have negative surface antigen and polymerase chain reaction (PCR) tests that indicate no active hepatitis B infection.
Cytomegalovirus
CMV typically does not appear until prophylactic therapy is stopped. Classic symptoms are fever, leukopenia, and diarrhea. Infection can involve any organ, and patients may present with hepatitis, pancreatitis or, less commonly, pneumonitis.
Patients who are negative for CMV before transplant and receive a donor-positive organ are at the highest risk. Patients who are CMV IgG-positive are considered to be at intermediate risk, regardless of the donor status. Patients who are negative for CMV and receive a donor-negative organ are at the lowest risk and do not need prophylaxis with valganciclovir.
CMV infection is diagnosed by PCR testing of the blood or immunostaining in tissue biopsy. Occasionally, blood testing is negative in the face of tissue-based disease.
BK virus
BK is a polyoma virus and a common virus associated with kidney transplant. Viremia is seen in about 18% of patients, whereas actual kidney disease associated with a higher level of virus is seen in fewer than 10% of patients. Most people are exposed to BK virus, often in childhood, and it can remain indolent in the bladder and uroepithelium.
Patients can develop BK nephropathy after exposure to transplant immunosuppression.11 Posttransplant monitoring protocols typically include PCR testing for BK virus at 1, 3, 6, and 12 months. No agent has been identified to specifically treat BK virus. The general strategy is to minimize immunosuppressive therapy by reducing or eliminating mycophenolate mofetil. Fortunately, BK virus does not tend to recur, and patients can have a low-level viremia (< 10,000 copies/mL) persisting over months or even years but often without clinical consequences.
The appearance of BK virus on biopsy can mimic acute rejection. Before BK viral nephropathy was a recognized entity, patients would have been diagnosed with acute rejection and may have been put on high-dose steroids, which would have worsened the BK infection.
Posttransplant lymphoproliferative disorder
Posttransplant lymphoproliferative disorder is most often associated with Epstein-Barr virus and usually involves a large, diffuse B-cell lymphoma. Burkitt lymphoma and plasma cell neoplasms also can occur less commonly.
The condition is about 30 times more common in patients after transplant than in the general population, and it is the third most common malignancy in transplant patients after skin and cervical cancers. About 80% of the cases occur early after transplant, within the first year.
Patients typically have a marked elevation in viral load of Epstein-Barr virus, although a negative viral load does not rule it out. A patient who is serologically negative for Epstein-Barr virus receiving a donor-positive kidney is at highest risk; this situation is most often seen in the pediatric population. Potent induction therapies (eg, antilymphocyte antibody therapy) are also associated with posttransplant lymphoproliferative disorder.
Patients typically present with fever of unknown origin with no localizing signs or symptoms. Mass lesions can be challenging to find; positron emission tomography may be helpful. The culprit is usually a focal mass, ulcer (especially in the gastrointestinal tract), or infiltrate (commonly localized to the allograft). Multifocal or disseminated disease can also occur, including lymphoma or with central nervous system, gastrointestinal, or pulmonary involvement.
Biopsy of the affected site is required for histopathology and Epstein-Barr virus markers. PCR blood testing is often positive for Epstein-Barr virus.
Typical antiviral therapy does not eliminate Epstein-Barr virus. In early polyclonal viral proliferation, the first goal is to reduce immunosuppressive therapy. Rituximab alone may also help in polymorphic cases. With disease that is clearly monomorphic and has transformed to a true malignancy, cytotoxic chemotherapy is also required. “R-CHOP,” a combination therapy consisting of rituximab with cyclophosphamide, doxorubicin, vincristine, and prednisone, is usually used. Radiation therapy may help in some cases.
Cryptococcal infection
Previously seen in patients with acquired immune deficiency syndrome, cryptococcal infection is now most commonly encountered in patients with solid-organ transplants. Vilchez et al12 found a 1% incidence in a series of more than 5,000 patients who had received an organ transplant.
Immunosuppression likely conveys risk, but because cryptococcal infection is acquired, environmental exposure also plays a role. It tends to appear more than 6 months after transplant, indicating that its cause is a primary infection by spore inhalation rather than by reactivation or transmission from the donor organ.13 Bird exposure is a risk factor for cryptococcal infection. One case identified the same strain of Cryptococcus in a kidney transplant recipient and the family’s pet cockatoo.14
Cryptococcal infection typically starts as pneumonia, which may be subclinical. The infection can then disseminate, with meningitis presenting with headache and mental status changes being the most concerning complication. The death rate is about 50% in most series of patients with meningitis. Skin and soft-tissue manifestations may also occur in 10% to 15% of cases and can be nodular, ulcerative, or cellulitic.
More than 75% of fungal infections requiring hospitalization in US patients who have undergone transplant are attributed to either Candida, Aspergillus, or Cryptococcus species.15 Risk of fungal infection is increased with diabetes, duration of pretransplant dialysis, tacrolimus therapy, or rejection treatment.
Much has improved in renal transplantation over the past 20 years. The focus has shifted to using stronger immunotherapy rather than trying to minimize it. There has been increasing recognition of infection and ways to prevent and treat it. Induction therapy now has greater emphasis so that maintenance therapy can be eased, with the aim of reducing long-term toxicity. Perhaps the biggest change is the practice of screening for donor-specific antibodies at the time of transplant so that predictable problems can be prevented or better handled if they occur. Such advances have helped patients directly and by extending the life of their transplanted organs.
LONGER SURVIVAL
As early as the 1990s, it was recognized that kidney transplant offers a survival advantage for patients with end-stage renal disease over maintenance on dialysis.1 Although the risk of death is higher immediately after transplant, within a few months it becomes much lower than for patients on dialysis. Survival varies according to the health of the patient and the quality of the transplanted organ.
In general, patients who obtain the greatest benefit from transplants in terms of years of life gained are those with diabetes, especially those who are younger. Those ages 20 to 39 live about 8 years on dialysis vs 25 years after transplant.
CONTRAINDICATIONS TO TRANSPLANT
There are multiple contraindications to a solitary kidney transplant (Table 1), including smoking. Most transplant centers require that smokers quit before transplant. Long-standing smokers almost double their risk of a cardiac event after transplant and double their rate of malignancy. Active smoking at the time of transplant is associated with twice the risk of death by 10 years after transplant compared with that of nonsmokers.2 Cotinine testing can detect whether a patient is an active smoker.
WAITING-LIST CONSIDERATIONS
Organs are scarce
The number of patients on the kidney waiting list has increased rapidly in the last few decades, while the number of transplants performed each year has remained about the same. In 2016, about 100,000 patients were on the list, but only about 19,000 transplants were performed.3 Wait times, especially for deceased-donor organs, have increased to about 6 years, varying by blood type and geographic region.
Waiting-list placement
Placement on the waiting list for a deceased-donor kidney transplant occurs when a patient has an estimated glomerular filtration rate (GFR) of 20 mL/min/1.73 m2 or less, although referral to the list can be made earlier. Early listing remains advantageous, as total time on the list will be counted before starting dialysis. “Preemptive transplant” means the patient had no dialysis before transplant; this applies to about 10% of transplant recipients. These patients tend to fare the best and are usually recipients of a living-donor organ.
Most patients do not receive a transplant until the GFR is less than 15 mL/min/1.73 m2.
Since 2014, wait time has been measured from the beginning of dialysis rather than the date of waiting-list placement in patients who are listed after starting dialysis therapy. This approach is more fair but sometimes introduces problems. A patient who did not previously know about the list may suddenly jump to the head of the line after 10 years of dialysis, by which time comorbidities associated with long-term dialysis make the patient less likely to gain as much benefit from a transplant as people lower on the list. Time on dialysis, or “dialysis vintage,” predicts patient and kidney survival after transplant, with reduced survival associated with increasing time on dialysis.4
Shorter wait for a suboptimal kidney
The aging population has increased the number of older patients being listed for transplant, presenting multiple challenges. Patients age 65 or older have a 50% chance of dying before they receive a transplant during a 5-year wait.
A patient may shorten the wait by joining the list for a suboptimal organ. All deceased-donor organs are given a Kidney Donor Profile Index score, which predicts the longevity of an organ after transplant. The score is determined by donor age, kidney function based on the serum creatinine at the time of death, and other donor factors.
A kidney with a score higher than 85% is likely to function longer than only 15% of available kidneys. Patients who receive a kidney with that score have a longer period of risk of death soon after transplant and a slightly higher risk of death in the long term than patients who receive a healthier kidney, although on average they still do better than patients on dialysis.5
Older patients should be encouraged to sign up for both the regular waiting list and the suboptimal kidney waiting list to reduce the risk of dying before they get a kidney.
LIVING-DONOR ORGAN TRANSPLANT
Many advantages
Living-donor organ transplant is associated with a better survival rate than deceased-donor organ transplant, and the advantage becomes greater over time. At 1 year, patient survival is more than 90% in both groups, but by 5 years about 80% of patients with a living-donor organ are still alive vs only about 65% of patients with a deceased-donor organ.
The waiting time for a living-donor transplant may be only weeks to months, rather than years. Because increasing time on dialysis predicts worse patient and graft survival after transplant, the shorter wait time is a big advantage. In addition, because the donor and recipient are typically in adjacent operating rooms, the organ sustains less ischemic damage. In general, the kidney quality is better from healthy donors, resulting in superior function early on and longer graft survival by an average of 4 years. If the living donor is related to the recipient, human leukocyte antigen matching also tends to be better and predicts better outcomes.
Special challenges
Opting for a living-donor organ also entails special challenges. In addition to the ethical issues surrounding living-donor organ donation, an appropriate donor must be found. Donors must be highly motivated and pass physical, laboratory, and psychological evaluations.
For older patients, if the donor is a spouse or close friend, he or she is also likely to be older, making the organ less viable than one from a younger person. Even an adult child may not be an ideal donor if there is a family propensity to kidney disease, such as diabetic nephropathy. No test is available to determine the risk for future diabetes, but it is known to run in families.
POTENT IMMUNOSUPPRESSION
Induction therapy
Induction therapy with antithymocyte globulin or basiliximab provides intense immunosuppression to prevent acute rejection during the early posttransplant period.
Antithymocyte globulin is a potent agent that contains antibodies directed at T cells, B cells, neutrophils, platelets, adhesion molecules, and complement. It binds T cells and removes them from circulation by opsonization in splenic and lymphoid tissue. The immunosuppressive effect is sustained for at least 2 to 3 months after a series of injections (dosage 1.5 mg/kg/day, usually for 4 to 10 doses). Antithymocyte globulin is also used to treat acute rejection, especially high-grade rejection for which steroid therapy is likely to be insufficient.
Basiliximab consists of antibodies to the interleukin 2 (IL-2) receptor of T cells. Binding to T cells prevents their activation rather than removing them from circulation. The drug prevents rejection, with 30% relative reduction in early studies compared with placebo. However, it is ineffective in reversing established rejection. Dosage is 20 mg at day 0 and day 4, which provides receptor saturation for 30 to 45 days.
Basiliximab is also sometimes used off-label for patients who need to discontinue a calcineurin inhibitor (ie, tacrolimus or cyclosporine). In such cases, normal therapy is put on hold while basiliximab is given for 1 or 2 doses. Case series have been reported for this use, particularly for patients with a heart and liver transplant who develop acute kidney injury while hospitalized.6,7
Antithymocyte globulin is more effective but also more risky. Brennan et al8 randomized 278 transplant recipients to either antithymocyte globulin or basiliximab. Patients in the antithymocyte globulin group had a 16% rejection rate vs 26% in the basiliximab group.
Antithymocyte globulin therapy is associated with multiple adverse effects, including fever and chills, pulmonary edema, and long-standing immunosuppressive effects such as increased risk of lymphoma and cytomegalovirus (CMV) infection. Basiliximab side-effect profiles are similar to those of placebo.
Maintenance therapy
The calcineurin inhibitors cyclosporine and tacrolimus remain the standard of care in kidney transplant despite multiple drug interactions and side effects that include renal toxicity and fibrosis. Cyclosporine and tacrolimus both bind intracellular immunophilins and thereby prevent transcription of IL-2 and production of T cells. The drugs work similarly but have different binding sites. Cyclosporine has largely been replaced by tacrolimus because its reliability of dosing and higher potency are associated with lower rejection rates.
Tacrolimus is typically given twice daily (1–6 mg/dose). Twelve-hour trough levels are followed (target: 8–12 ng/mL early on, then 5–8 ng/mL after 3 months posttransplant). Side effects include hypertension and hypercholesterolemia, but less so than with cyclosporine. On the other hand, hyperglycemia tends to be worse with tacrolimus than with cyclosporine, and combining tacrolimus with steroids frequently leads to diabetes. Tacrolimus can also cause acute and chronic renal failure, especially at high drug levels, as well as neurotoxicity, tremors, and hair loss.
Cyclosporine, tacrolimus, and sirolimus (not a calcineurin inhibitor) are metabolized through the same cytochrome P450 pathway (CYP3A4), so they have common drug interactions (Table 2).
Mycophenolate mofetil is typically used as an adjunct therapy (500–1,000 mg twice daily). It is also used for other kidney diseases before transplant, including lupus nephritis. Transplanted kidney rejection rates with mycophenolate mofetil with steroids are about 40%, so the drug is not potent enough to be used without a calcineurin inhibitor.
Side effects include gastrointestinal toxicity in up to 20% of patients, and leukopenia, which is associated with viral infections.
CORONARY ARTERY DISEASE IS COMMON WITH DIALYSIS
Coronary artery disease is highly associated with end-stage kidney disease and occurs in as many as 85% of older patients with diabetes on dialysis. Although patients with end-stage kidney disease tend to have more numerous and severe atherosclerotic lesions compared with the general population, justifying aggressive management, cardiac care tends to be conservative in patients on dialysis.9
Death from acute myocardial infarction occurs in about 20% to 30% of patients on dialysis vs about 2% of patients with normal renal function. Five years after myocardial infarction, survival is only about 30% in patients on dialysis.9
There are many explanations for excess coronary artery disease in patients on dialysis. In addition to the traditional cardiovascular risk factors of diabetes, hypertension, and preexisting coronary artery disease, patients are in a proinflammatory uremic state and have high levels of phosphorus and fibroblast growth factor 23 that contribute to vascular calcification. Almost all patients have high homocysteine levels and hemodynamic instability, particularly if they are on hemodialysis.
Pretransplant evaluation for heart disease
Patients on the kidney transplant waiting list are screened aggressively for heart disease. A history of myocardial infarction usually results in removal from the list. All patients have an initial electrocardiogram and echocardiogram. Thallium or echocardiographic stress testing is used for patients who are age 50 and older, have diabetes, or have had dialysis for many years. Patients with evidence of ischemia undergo catheterization.
Patients are also screened with computed tomography before transplant. Because the kidney is typically anastomosed to the iliac artery and vein, heavy calcification of the iliac artery can make the procedure too difficult to perform.
Reduced long-term risk of myocardial infarction after transplant
Kasiske et al10 analyzed data from more than 50,000 patients from the US Renal Data System and found that, for about the first year after transplant, patients who underwent kidney transplant were more likely to have a myocardial infarction than those on dialysis. After that, they fared better than patients who remained on dialysis. Those with a living-donor transplant were less likely at all times to have a myocardial infarction than those with a deceased-donor transplant. By 3 years after transplant, the relative risk of having a myocardial infarction was 0.89 for deceased-donor organ recipients and 0.69 for living-donor recipients compared with patients on the waiting list.10
INFECTIOUS COMPLICATIONS IN KIDNEY RECIPIENTS
Kidney recipients are prone to many common and uncommon infections (Table 3). All potential recipients are tested pretransplant for hepatitis B, hepatitis C, human immunodeficiency virus, syphilis, and tuberculosis. A positive result does not necessarily rule out transplant.
The following viral serology tests are also done before transplant:
Epstein-Barr virus (antibodies are positive in about 90% of adults)
CMV (about 70% of adults are seropositive)
Varicella zoster (seronegative patients should be given live-attenuated varicella vaccine).
Risk of transmission of these viruses relates to the serostatus of the donor and recipient before transplant. If a donor is positive for viral antibodies but the recipient is not (a so-called “mismatch”), risk is higher after transplant.
Hepatitis C
Patients with hepatitis C fare better if they get a transplant than if they remain on dialysis, although their posttransplant course is worse compared with transplant patients who do not have hepatitis. Some patients develop accelerated liver disease after kidney transplant. Hepatitis C-related kidney disease—membranous proliferative glomerulonephritis—also occurs, as do comorbidities such as diabetes.
Careful evaluation is warranted before transplant, including liver imaging, alpha-fetoprotein testing, and liver biopsy to evaluate for hepatocellular carcinoma. A patient with advanced fibrosis or cirrhosis may not be a candidate for kidney transplant alone but could possibly receive a combined kidney and liver transplant.
There is a need to determine the best time to treat hepatitis C infection. Patients with advanced liver disease or hepatitis C-related kidney disease would likely benefit from early treatment. However, delaying treatment could shorten the wait time for a deceased-donor organ positive for hepatitis C. Transplant candidates with active hepatitis C are uniquely considered to accept hepatitis C-positive kidneys, which are often discarded, and may only wait weeks for such a transplant. The shortened kidney survival associated with a hepatitis C-positive kidney may no longer be true with the new antiviral hepatitis C therapy, which has been shown to be effective post-transplant.
Hepatitis B
No cure is available for hepatitis B infection, but it can be well controlled with antiviral therapy. Patients with hepatitis B infection may be candidates for transplant, but they should be stable on antiviral therapy (lamivudine, entecavir, or tenofovir) to eliminate the viral load before transplant, and therapy should be continued afterward. Liver imaging, alpha-fetoprotein levels, and biopsy are recommended for evaluation. All hepatitis B- negative patients should be vaccinated before transplant.
Organs from living or deceased donors that test positive for hepatitis B core antibody, indicating prior exposure, can be considered for transplant in a patient who tests positive for hepatitis B surface antibody, indicating successful vaccination or prior exposure in the recipient. But donors must have negative surface antigen and polymerase chain reaction (PCR) tests that indicate no active hepatitis B infection.
Cytomegalovirus
CMV typically does not appear until prophylactic therapy is stopped. Classic symptoms are fever, leukopenia, and diarrhea. Infection can involve any organ, and patients may present with hepatitis, pancreatitis or, less commonly, pneumonitis.
Patients who are negative for CMV before transplant and receive a donor-positive organ are at the highest risk. Patients who are CMV IgG-positive are considered to be at intermediate risk, regardless of the donor status. Patients who are negative for CMV and receive a donor-negative organ are at the lowest risk and do not need prophylaxis with valganciclovir.
CMV infection is diagnosed by PCR testing of the blood or immunostaining in tissue biopsy. Occasionally, blood testing is negative in the face of tissue-based disease.
BK virus
BK is a polyoma virus and a common virus associated with kidney transplant. Viremia is seen in about 18% of patients, whereas actual kidney disease associated with a higher level of virus is seen in fewer than 10% of patients. Most people are exposed to BK virus, often in childhood, and it can remain indolent in the bladder and uroepithelium.
Patients can develop BK nephropathy after exposure to transplant immunosuppression.11 Posttransplant monitoring protocols typically include PCR testing for BK virus at 1, 3, 6, and 12 months. No agent has been identified to specifically treat BK virus. The general strategy is to minimize immunosuppressive therapy by reducing or eliminating mycophenolate mofetil. Fortunately, BK virus does not tend to recur, and patients can have a low-level viremia (< 10,000 copies/mL) persisting over months or even years but often without clinical consequences.
The appearance of BK virus on biopsy can mimic acute rejection. Before BK viral nephropathy was a recognized entity, patients would have been diagnosed with acute rejection and may have been put on high-dose steroids, which would have worsened the BK infection.
Posttransplant lymphoproliferative disorder
Posttransplant lymphoproliferative disorder is most often associated with Epstein-Barr virus and usually involves a large, diffuse B-cell lymphoma. Burkitt lymphoma and plasma cell neoplasms also can occur less commonly.
The condition is about 30 times more common in patients after transplant than in the general population, and it is the third most common malignancy in transplant patients after skin and cervical cancers. About 80% of the cases occur early after transplant, within the first year.
Patients typically have a marked elevation in viral load of Epstein-Barr virus, although a negative viral load does not rule it out. A patient who is serologically negative for Epstein-Barr virus receiving a donor-positive kidney is at highest risk; this situation is most often seen in the pediatric population. Potent induction therapies (eg, antilymphocyte antibody therapy) are also associated with posttransplant lymphoproliferative disorder.
Patients typically present with fever of unknown origin with no localizing signs or symptoms. Mass lesions can be challenging to find; positron emission tomography may be helpful. The culprit is usually a focal mass, ulcer (especially in the gastrointestinal tract), or infiltrate (commonly localized to the allograft). Multifocal or disseminated disease can also occur, including lymphoma or with central nervous system, gastrointestinal, or pulmonary involvement.
Biopsy of the affected site is required for histopathology and Epstein-Barr virus markers. PCR blood testing is often positive for Epstein-Barr virus.
Typical antiviral therapy does not eliminate Epstein-Barr virus. In early polyclonal viral proliferation, the first goal is to reduce immunosuppressive therapy. Rituximab alone may also help in polymorphic cases. With disease that is clearly monomorphic and has transformed to a true malignancy, cytotoxic chemotherapy is also required. “R-CHOP,” a combination therapy consisting of rituximab with cyclophosphamide, doxorubicin, vincristine, and prednisone, is usually used. Radiation therapy may help in some cases.
Cryptococcal infection
Previously seen in patients with acquired immune deficiency syndrome, cryptococcal infection is now most commonly encountered in patients with solid-organ transplants. Vilchez et al12 found a 1% incidence in a series of more than 5,000 patients who had received an organ transplant.
Immunosuppression likely conveys risk, but because cryptococcal infection is acquired, environmental exposure also plays a role. It tends to appear more than 6 months after transplant, indicating that its cause is a primary infection by spore inhalation rather than by reactivation or transmission from the donor organ.13 Bird exposure is a risk factor for cryptococcal infection. One case identified the same strain of Cryptococcus in a kidney transplant recipient and the family’s pet cockatoo.14
Cryptococcal infection typically starts as pneumonia, which may be subclinical. The infection can then disseminate, with meningitis presenting with headache and mental status changes being the most concerning complication. The death rate is about 50% in most series of patients with meningitis. Skin and soft-tissue manifestations may also occur in 10% to 15% of cases and can be nodular, ulcerative, or cellulitic.
More than 75% of fungal infections requiring hospitalization in US patients who have undergone transplant are attributed to either Candida, Aspergillus, or Cryptococcus species.15 Risk of fungal infection is increased with diabetes, duration of pretransplant dialysis, tacrolimus therapy, or rejection treatment.
- Wolfe RA, Ashby VB, Milford EL, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med 1999; 341:1725–1730.
- Kasiske BL, Klinger D. Cigarette smoking in renal transplant recipients. J Am Soc Nephrol 2000; 11:753–759.
- United Network for Organ Sharing. Transplant trends. https://transplantpro.org/technology/transplant-trends/#waitlists_by_organ. Accessed December 13, 2017.
- Meier-Kriesche HU, Kaplan B. Waiting time on dialysis as the strongest modifiable risk factor for renal transplant outcomes: a paired donor kidney analysis. Transplantation 2002; 74:1377–1381.
- Ojo AO, Hanson JA, Meier-Kriesche H, et al. Survival in recipients of marginal cadaveric donor kidneys compared with other recipients and wait-listed transplant candidates. J Am Soc Nephrol 2001; 12:589–597.
- Alonso P. Sanchez-Lazaro I, Almenar L, et al. Use of a “CNI holidays” strategy in acute renal dysfunction late after heart transplant. Report of two cases. Heart Int 2014; 9:74–77.
- Cantarovich M, Metrakos P, Giannetti N, Cecere R, Barkun J, Tchervenkov J. Anti-CD25 monoclonal antibody coverage allows for calcineurin inhibitor “holiday” in solid organ transplant patients with acute renal dysfunction. Transplantation 2002; 73:1169–1172.
- Brennan DC, Daller JA, Lake KD, Cibrik D, Del Castillo D; Thymoglobulin Induction Study Group. Rabbit antithymocyte globulin versus basiliximab in renal transplantation. N Engl J Med 2006; 355:1967–1977.
- McCullough PA. Evaluation and treatment of coronary artery disease in patients with end-stage renal disease. Kidney Int 2005; 67:S51–S58.
- Kasiske BL, Maclean JR, Snyder JJ. Acute myocardial infarction and kidney transplantation. J Am Soc Nephrol 2006; 17:900–907.
- Bohl DL, Storch GA, Ryschkewitsch C, et al. Donor origin of BK virus in renal transplantation and role of HLA C7 in susceptibility to sustained BK viremia. Am J Transplant 2005; 5:2213–2221.
- Vilchez RA, Fung J, Kusne S. Cryptococcosis in organ transplant recipients: an overview. Am J Transplant 2002; 2:575–580.
- Vilchez R, Shapiro R, McCurry K, et al. Longitudinal study of cryptococcosis in adult solid-organ transplant recipients. Transpl Int 2003; 16:336–340.
- Nosanchuk JD, Shoham S, Fries BC, Shapiro DS, Levitz SM, Casadevall A. Evidence of zoonotic transmission of Cryptococcus neoformans from a pet cockatoo to an immunocompromised patient. Ann Intern Med 2000; 132:205–208.
- Abbott KC, Hypolite I, Poropatich RK, et al. Hospitalizations for fungal infections after renal transplantation in the United States. Transpl Infect Dis 2001; 3:203–211.
- Wolfe RA, Ashby VB, Milford EL, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med 1999; 341:1725–1730.
- Kasiske BL, Klinger D. Cigarette smoking in renal transplant recipients. J Am Soc Nephrol 2000; 11:753–759.
- United Network for Organ Sharing. Transplant trends. https://transplantpro.org/technology/transplant-trends/#waitlists_by_organ. Accessed December 13, 2017.
- Meier-Kriesche HU, Kaplan B. Waiting time on dialysis as the strongest modifiable risk factor for renal transplant outcomes: a paired donor kidney analysis. Transplantation 2002; 74:1377–1381.
- Ojo AO, Hanson JA, Meier-Kriesche H, et al. Survival in recipients of marginal cadaveric donor kidneys compared with other recipients and wait-listed transplant candidates. J Am Soc Nephrol 2001; 12:589–597.
- Alonso P. Sanchez-Lazaro I, Almenar L, et al. Use of a “CNI holidays” strategy in acute renal dysfunction late after heart transplant. Report of two cases. Heart Int 2014; 9:74–77.
- Cantarovich M, Metrakos P, Giannetti N, Cecere R, Barkun J, Tchervenkov J. Anti-CD25 monoclonal antibody coverage allows for calcineurin inhibitor “holiday” in solid organ transplant patients with acute renal dysfunction. Transplantation 2002; 73:1169–1172.
- Brennan DC, Daller JA, Lake KD, Cibrik D, Del Castillo D; Thymoglobulin Induction Study Group. Rabbit antithymocyte globulin versus basiliximab in renal transplantation. N Engl J Med 2006; 355:1967–1977.
- McCullough PA. Evaluation and treatment of coronary artery disease in patients with end-stage renal disease. Kidney Int 2005; 67:S51–S58.
- Kasiske BL, Maclean JR, Snyder JJ. Acute myocardial infarction and kidney transplantation. J Am Soc Nephrol 2006; 17:900–907.
- Bohl DL, Storch GA, Ryschkewitsch C, et al. Donor origin of BK virus in renal transplantation and role of HLA C7 in susceptibility to sustained BK viremia. Am J Transplant 2005; 5:2213–2221.
- Vilchez RA, Fung J, Kusne S. Cryptococcosis in organ transplant recipients: an overview. Am J Transplant 2002; 2:575–580.
- Vilchez R, Shapiro R, McCurry K, et al. Longitudinal study of cryptococcosis in adult solid-organ transplant recipients. Transpl Int 2003; 16:336–340.
- Nosanchuk JD, Shoham S, Fries BC, Shapiro DS, Levitz SM, Casadevall A. Evidence of zoonotic transmission of Cryptococcus neoformans from a pet cockatoo to an immunocompromised patient. Ann Intern Med 2000; 132:205–208.
- Abbott KC, Hypolite I, Poropatich RK, et al. Hospitalizations for fungal infections after renal transplantation in the United States. Transpl Infect Dis 2001; 3:203–211.
KEY POINTS
- Kidney transplant improves survival and long-term outcomes in patients with renal failure.
- Before transplant, patients should be carefully evaluated for cardiovascular and infectious disease risk.
- Potent immunosuppression is required to maintain a successful kidney transplant.
- After transplant, patients must be monitored for recurrent disease, side effects of immunosuppression, and opportunistic infections.
A 75-year-old with abdominal pain, hypoxia, and weak pulses in the left leg
A 75-year-old man presented to the emergency department for evaluation of abdominal pain. He had stage 3 chronic obstructive pulmonary disease (COPD), with a forced expiratory volume in 1 second of 33%.
PREVIOUS HOSPITALIZATION
Aside from his COPD, he had been healthy until 1 month earlier, when he had been hospitalized because of shortness of breath and chest pressure with exertion. His troponin T level had been elevated, peaking at 0.117 ng/mL (reference range 0–0.029).
Left heart catheterization had shown no significant coronary artery disease. A myocardial bridge of the distal left anterior descending coronary artery had been seen, so that the artery appeared to be narrowed by 50% to 60% with ventricular contraction. But this was not thought to have been the cause of his presentation.
On discharge, he required oxygen 4 L/min by nasal cannula. Previously, he had not needed supplemental oxygen.
CURRENT PRESENTATION
The patient described persistent and severe periumbilical abdominal pain during the previous day. It was not associated with eating, and he denied diarrhea, constipation, hematemesis, hematochezia, bright red blood per rectum, or melena. He continued to describe persistent shortness of breath and pleuritic chest pain. His vital signs were as follows:
- Heart rate 104 beats per minute
- Respiratory rate 16 to 20 breaths per minute
- Blood pressure 101–142/62–84 mm Hg
- Oxygen saturation 78% on room air.


His laboratory findings on presentation are shown in Table 1, and his electrocardiogram is shown in Figure 1.
WHAT DOES HIS ELECTROCARDIOGRAM SHOW?
1. Which of the following is the most accurate description of this patient’s electrocardiogram?
- Sinus tachycardia, peaked P waves (P pulmonale) in lead II, and T-wave inversions in the right precordial leads
- Sinus tachycardia and left bundle branch block
- Sinus tachycardia and poor R-wave progression
- Sinus tachycardia and ST elevation in the precordial leads
Our patient’s electrocardiogram shows sinus tachycardia, P pulmonale, T-wave inversion in the right precordial leads (V1–V3), and biphasic T waves in lead V4,, which suggest right ventricular strain.
The rhythm most commonly seen in patients with pulmonary embolism is sinus tachycardia, followed by nonspecific ST-segment or T-wave abnormalities. In one series of patients with acute pulmonary embolism, the classic findings of P pulmonale, right ventricular hypertrophy, right axis deviation, and right bundle branch block were rare (< 6%).1 Thus, these classic findings are not sensitive for the diagnosis of pulmonary embolism, and their absence does not rule it out.
Further studies for our patient
 with a chest pulmonary embolism protocol showed filling defects.")
Transthoracic echocardiography was performed to look for evidence of right ventricular strain secondary to the pulmonary embolism.
ECHOCARDIOGRAPHIC SIGNS OF PULMONARY EMBOLISM
2. Which of the following findings on transthoracic echocardiography would not suggest acute pulmonary embolism?
- Midright ventricular wall hypokinesis with apical sparing
- Severe tricuspid regurgitation
- Left ventricular dilation
- Lack of respiratory variation of the inferior vena cava
- Septal wall motion toward the left ventricle
Left ventricular dilation does not suggest acute pulmonary embolism. Echocardiograms of patients with acute submassive pulmonary embolism typically show evidence of right ventricular strain, such as the other entities listed above (midright ventricular hypokinesis with apical sparing, severe tricuspid regurgitation, lack of respiratory variation of the inferior vena cava, and septal wall motion toward the left ventricle).
The degree of right ventricular dysfunction is related to the extent of acute pulmonary vascular occlusion and aids in risk-stratification of patients with acute pulmonary embolism. Midright ventricular wall hypokinesis with apical sparing has been termed the McConnell sign.2
In our patient, transthoracic echocardiography showed:
- Normal left ventricular ejection fraction
- Mild diastolic dysfunction
- Right ventricular dilation with moderately decreased right ventricular systolic function and apical sparing
- Right ventricular systolic pressure 54 mm Hg, consistent with moderate pulmonary hypertension
- Right atrial pressure 10 mm Hg
- No inspiratory collapse of a dilated inferior vena cava
- Mild tricuspid valve regurgitation.
CLASSIFICATION OF ACUTE PULMONARY EMBOLISM
3. Given the above information, how would you classify the patient’s pulmonary embolism?
- Massive
- Submassive
- Low-risk
- Clinically stable
The patient’s pulmonary embolism is submassive.

Historically, the classification of pulmonary embolism was determined by the angiographic thrombus burden. However, this has limited utility because clinical factors (eg, hypotension on initial presentation) have been shown to be better predictors of short-term mortality risk.3
Our patient is characterized as having a submassive pulmonary embolism based on elevated biomarkers (troponin T, N-terminal pro-B-type natriuretic peptide) and right ventricular dysfunction in the absence of hypotension.
ULTRASONOGRAPHY FOR DIAGNOSIS OF DEEP VEIN THROMBOSIS

Venous duplex ultrasonography has become the standard for diagnosis of lower extremity deep vein thrombosis. However, its quality and diagnostic accuracy depend on the skill of the person performing the examination. It is further limited by certain patient characteristics, including severe obesity, edema, and wounds and dressings at the site being examined.5
Our patient underwent duplex ultrasonography of the lower extremities, which demonstrated acute proximal and calf deep vein thrombosis in the right femoral, popliteal, and peroneal veins and no deep vein thrombosis in the left leg.
RISK STRATIFICATION IN ACUTE PULMONARY EMBOLISM
Multiple models exist to estimate the risk of complications in patients with acute pulmonary embolism.
The Bova score6 is based on the following factors:
- Systolic blood pressure 90–100 mm Hg (2 points) (patients with systolic blood pressure lower than 90 mm Hg were excluded from the study from which this score was derived)
- Cardiac troponin elevation (2 points)
- Right ventricular dysfunction on echocardiography or computed tomography (2 points)
- Heart rate 100 beats/min or greater (1 point).
A total score of 0, 1, or 2 (stage I) denotes low risk, 3 or 4 points (stage II) intermediate risk, and more than 4 points (stage III) high risk.
The PESI score (Pulmonary Embolism Severity Index)7 is based on:
- Age (1 point per year)
- Sex (10 points for being male)
- Heart rate 110 per minute or greater (20 points)
- Cancer (30 points)
- Heart failure (10 points)
- Chronic lung disease (10 points)
- Systolic blood pressure less than 100 mm Hg (30 points)
- Respiratory rate at least 30 per minute (20 points)
- Temperature less than 36ºC (20 points)
- Altered mental status (60 points)
- Arterial oxygen saturation less than 90% (20 points).
The total score is broken down into 5 classes: I (< 65 points), II (65–85), III (86–105), IV (106–125), and V (> 126). Classes I and II are low risk, and the higher ones are high risk.
The simplified PESI score8 was developed to more rapidly risk-stratify patients and has been found to be similar to the PESI score in prognostic accuracy. Patients get 1 point for each of the following:
- Age over 80
- Cancer
- Chronic cardiopulmonary disease (heart failure or chronic lung disease)
- Heart rate 110 per minute or greater
- Systolic blood pressure less than 100 mm Hg
- Arterial oxygen saturation less than 90%.
A total score of 0 is low risk; anything higher is high risk.
Back to our patient
Our patient had proximal and calf deep vein thrombosis of the right leg, bilateral submassive pulmonary emboli with associated biomarker elevation and right ventricular dysfunction, and left renal artery thrombosis with infarction. Using the PESI score, his risk of death in the next 30 days was 13.7% and his 30-day risk of a complicated course was 27%. Using the Bova score, his 30-day risk of death was 15.5% and his 30-day risk of a complicated course was 29.2%.6,7
Notably, the patient’s right ventricular function had also been impaired on the echocardiogram performed during his admission 1 month previously. On transthoracic echocardiography during the current admission, the patient was found to have a similar degree of right ventricular dysfunction. This finding, along with the oxygen requirement that developed during the earlier admission, suggested that his pulmonary embolism may have been subacute and that the diagnosis may have been missed during the earlier hospital stay.
The patient was treated with unfractionated heparin. After the hospital’s multidisciplinary pulmonary embolism response team discussed and weighed the above factors, they recommended to not pursue thrombolytic therapy or inferior vena cava filter placement.
Of note, the patient’s pulses in the left lower extremity continued to be weak but palpable, and the left leg was cooler to touch than the right leg.
ASSESSING PERIPHERAL ARTERY DISEASE
4. How should the finding of weak pulses in this patient’s left leg be initially investigated?
- Computed tomographic angiography with runoff
- Ankle-brachial indices with pulse-volume recordings
- Arterial duplex ultrasonography
- Magnetic resonance angiography of the lower extremities
The ankle-brachial index is the initial diagnostic test for assessment of pulse abnormalities and for diagnosis of lower-extremity peripheral artery disease. It is calculated by dividing the higher of the ankle systolic pressures (posterior tibial or dorsalis pedis) by the higher of the 2 brachial pressures (left or right).9 Normal values are between 1.00 and 1.40.
Ankle-brachial indices in our patient
Our patient underwent measurement of his brachial, dorsalis pedis, and posterior tibial artery systolic pressures using blood pressure cuffs and continuous-wave Doppler. Ankle pulse-volume recordings were also obtained.

Given the patient’s poor renal function and concern for acute renal infarction, we thought it best to avoid iodinated or gadolinium contrast, such as with magnetic resonance or computed tomographic angiography.
Segmental leg pressures and pulse-volume recordings can be performed to help localize the level of arterial disease in the extremities, but were not done in this case because of the extensive deep vein thrombosis in the right leg.10,11
Arterial ultrasonography in our patient
Arterial duplex ultrasonography was performed to help determine the location of arterial disease. It showed patent arteries in the right leg. In the left lower extremity there was slow, monophasic blood flow in the distal superficial femoral artery. The popliteal artery was occluded. The posterior tibial artery was occluded at the origin, with reconstitution distally. The peroneal artery was occluded throughout. The anterior tibial artery was patent throughout. The ultrasonographic findings were thought to be suspicious for arterial thromboembolism.
WHAT CAN CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
5. Given that the patient had both arterial thrombosis (renal artery, lower-extremity arteries) and venous thromboembolism (deep vein thrombosis and pulmonary embolism), which of the following would be included in the differential diagnosis?
- Antiphospholipid antibody syndrome
- Protein C or protein S deficiency
- Malignancy
- Paradoxical embolization
- Factor V Leiden mutation
Correct answers include antiphospholipid antibody syndrome, malignancy, and paradoxical embolization.
The differential diagnosis for concomitant venous and arterial thrombosis is broad,12 and includes the following:
- Structural factors: patent foramen ovale, popliteal artery aneurysm
- Malignancy
- Inflammatory diseases: Behçet disease, Buerger disease, inflammatory bowel disease, antiphospholipid antibody syndrome, elevated lipoprotein(a), elevated homocysteine
- Hematologic diseases: myelodysplastic syndrome, disseminated intravascular coagulation, paroxysmal nocturnal hemoglobinuria, heparin-induced thrombocytopenia.
Traditional risk factors for venous thromboembolism include protein C deficiency, protein S deficiency, factor V Leiden mutation, the prothrombin G20210A gene mutation, and others. These are relatively minor risk factors for venous thrombosis and do not pose a risk for arterial thrombosis.12 In contrast, antiphospholipid antibody syndrome and malignancy pose a risk for both venous and arterial thrombosis. Paradoxical embolism is a mechanism by which arterial thrombosis (emboli) can develop in the setting of existing venous thrombosis.12
Our patient underwent testing for antiphospholipid antibodies and lupus anticoagulant, and he was encouraged to undergo age-appropriate cancer screening as an outpatient.12
ANTIPHOSPHOLIPID ANTIBODY SYNDROME
Antiphospholipid antibody syndrome is defined by both clinical and laboratory criteria. Clinical symptoms include vascular thrombosis (arterial, venous, or both) and pregnancy-related complications.13
Laboratory criteria require the presence of antiphospholipid antibodies or lupus anticoagulant. These must be confirmed with repeat testing in 12 weeks. Antiphospholipid antibodies are detected by an enzyme-linked immunosorbent assay; laboratory assessment for the presence of lupus anticoagulant is a stepwise process and relies on 4 criteria:
- There should be prolongation of a phospholipid-dependent clotting test (eg, activated partial thromboplastin time, dilute Russell viper venom time test).
- There must be evidence of an inhibitory activity with mixing study.
- The inhibitor must exhibit phospholipid dependence; that is, with more phospholipid there is shortening of clotting time.
- Specific inhibitors must be excluded, including factor VIII and anticoagulant drugs such as heparin.14–17

Clinically, one should consider antiphospholipid antibody syndrome in patients who have arterial thrombosis, a history of pregnancy morbidity, or unexplained prolongation of activated partial thromboplastin time.13
Antiphospholipid antibodies may be present in up to a quarter of patients with venous thromboembolism, but it is persistent positivity of antibody assays that is associated with increased future risk of venous thromboembolism.19 Of note, the risk of venous thromboembolism in patients with confirmed antiphospholipid antibody syndrome is 10 times higher than in the general population.20
ANTIPHOSPHOLIPID ANTIBODIES ARE NOT ALL THE SAME
6. Which of the following antiphospholipid antibodies have not been associated with an increased thrombotic risk?
- Anti-beta-2-glycoprotein I IgG
- Lupus anticoagulant
- Antiphosphatidylserine
- Anticardiolipin IgM
- Anticardiolipin IgG
The correct answer is antiphosphatidylserine.15
Antiphospholipid antibodies are directed against a portion of select plasma proteins that are uncovered upon phospholipid binding. While lupus anticoagulant, anti-beta-2-glycoprotein I, and anticardiolipin antibodies are associated with thrombosis, antiprothrombin antibodies (including antiprothrombin and antiphosphatidylserine antibodies) are not.15,21
PARADOXICAL EMBOLISM
Patent foramen ovale, a communication between the right and left atrium in the interatrial septum, is associated with an increased risk of paradoxical embolization. The prevalence of patent foramen ovale is estimated to be 27% to 29% in the general population.22 Noncerebral systemic paradoxical embolism occurs less frequently than cerebral embolism, accounting for approximately 5% to 10% of paradoxical emboli.22
To evaluate for patent foramen ovale, transthoracic echocardiography is performed with a bubble (agitated saline contrast) study to assess for interatrial shunting. Transesophageal echocardiography or transcranial Doppler bubble studies may also be performed.
Although patent foramen ovale is most commonly associated with cerebral embolism, peripheral emboli can occur. Some research suggests that this may be a more common cause of arterial thromboembolism in younger patients. There have also been reports of other sites of systemic embolization, including the renal artery.12
Back to our patient
Initial antiphospholipid antibody testing was positive for lupus anticoagulant. Anticardiolipin and anti-beta-2-glycoprotein I antibodies were not detected.
Transesophageal echocardiography revealed a patent foramen ovale with a highly mobile atrial septum (atrial septal aneurysm).
The patient was treated with intravenous unfractionated heparin with bridging to warfarin with a target international normalized ratio (INR) of 2 to 3. His renal artery infarction and his lower-extremity arterial thromboembolic event were conservatively managed. His respiratory status improved, and he no longer required supplemental oxygen. His creatinine peaked at 1.7 mg/dL during his admission and improved to 1.2 mg/dL before he was discharged.
At follow-up, repeat echocardiography showed that his right ventricular systolic pressure had improved (decreased) to 37 mm Hg from 54 mm Hg. Repeat confirmatory testing was positive for lupus anticoagulant 12 weeks later. He has been maintained on warfarin with an INR goal of 2 to 3 as well as low-dose aspirin with plans for long-term anticoagulation. We decided to keep the patient on anticoagulation indefinitely with warfarin; he was not a candidate for a direct oral anticoagulant, given limited data on the use of these agents in the setting of lupus anticoagulant and antiphospholipid antibody syndrome.
SUMMARY OF CASE
In summary, this patient was a 75-year-old man with COPD who presented with abdominal pain. He was noted to have a left renal infarction, extensive unprovoked lower-extremity deep vein thrombosis with pulmonary emboli, and lower limb arterial thromboembolism.
He also had an underlying hypercoagulable state—antiphospholipid antibody syndrome—that predisposed him to both arterial and venous thrombosis. He was ultimately found to have a patent foramen ovale, which further increased the risk of arterial thrombosis by facilitating paradoxical embolization of venous thrombi. It is not certain whether the renal infarction and leg artery thrombi were due to paradoxical embolism or to in situ thrombosis, but we believe that it was most likely paradoxical embolization.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100:598–603.
- Alsoos F, Khaddam A. Echocardiographic evaluation methods for right ventricular function. J Echocardiogr 2015; 13:43–51.
- Jaff MR, McMurtry MS, Archer SL, et al; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; American Heart Association Council on Peripheral Vascular Disease; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 2011; 123:1788–1830.
- Heit JA, Silverstein MD, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ 3rd. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med 2000; 160:809–815.
- Gornik HL, Sharma AM. Duplex ultrasound in the diagnosis of lower-extremity deep venous thrombosis. Circulation 2014; 129:917–921.
- Fernández C, Bova C, Sanchez O, et al. Validation of a model for identification of patients at intermediate to high risk for complications associated with acute symptomatic pulmonary embolism. Chest 2015; 148:211–218.
- Aujesky D, Perrier A, Roy PM, et al. Validation of a clinical prognostic model to identify low-risk patients with pulmonary embolism. J Intern Med 2007; 261:597–604.
- Jiménez D, Aujesky D, Moores L, et al; RIETE Investigators. Simplification of the pulmonary embolism severity index for prognostication in patients with acute symptomatic pulmonary embolism. Arch Intern Med 2010; 170:1383–1389.
- Kim ES, Wattanakit K, Gornik HL. Using the ankle-brachial index to diagnose peripheral artery disease and assess cardiovascular risk. Cleve Clin J Med 2012; 79:651–661.
- Jaff MR. Lower extremity arterial disease. Diagnostic aspects. Cardiol Clin 2002; 20:491–500.
- Rooke TW, Hirsch AT, Misra S, et al; American College of Cardiology Foundation Task Force; American Heart Association Task Force. Management of patients with peripheral artery disease (compilation of 2005 and 2011 ACCF/AHA Guideline Recommendations): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:1555–1570.
- Lichtin A, Bartholomew J. The coagulation consult: a case-based guide. New York, NY: Springer; 2014.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
- Miyakis S, Lockshin M, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Nichols WL, Kottke-Marchant K, Ledford-Kraemer MR, Homburger HA, Cardel LK. Lupus anticoagulants, antiphospholipid antibodies, and antiphospholipid syndrome. In: Kottke-Marchant K, Davis BH, editors. Laboratory Hematology Practice. Hoboken, New Jersey: Blackwell Publishing, Ltd.; 2012:509–525.
- Houghton DE, Moll S. Antiphospholipid antibodies. Vasc Med 2017; 22:545–550.
- Roldan V, Lecumberri R, Muñoz-Torrero JFS, et al; RIETE Investigators. Thrombophilia testing in patients with venous thromboembolism. Findings from the RIETE registry. Thromb Res 2009; 124:174–177.
- Wahl DG, Guillemin F, de Maistre E, Perret-Guillaume C, Lecompte T, Thibaut G. Meta-analysis of the risk of venous thrombosis in individuals with antiphospholipid antibodies without underlying autoimmune disease or previous thrombosis. Lupus 1998; 7:15–22.
- Love PE, Santoro SA. Antiphospholipid antibodies: anticardiolipin and the lupus anticoagulant in systemic lupus erythematosus (SLE) and in non-SLE disorders. Prevalence and clinical significance. Ann Intern Med 1990; 112:682–698.
- Thompson T, Evans W. Paradoxical embolism. QJM 1930; os-23:135–150.
A 75-year-old man presented to the emergency department for evaluation of abdominal pain. He had stage 3 chronic obstructive pulmonary disease (COPD), with a forced expiratory volume in 1 second of 33%.
PREVIOUS HOSPITALIZATION
Aside from his COPD, he had been healthy until 1 month earlier, when he had been hospitalized because of shortness of breath and chest pressure with exertion. His troponin T level had been elevated, peaking at 0.117 ng/mL (reference range 0–0.029).
Left heart catheterization had shown no significant coronary artery disease. A myocardial bridge of the distal left anterior descending coronary artery had been seen, so that the artery appeared to be narrowed by 50% to 60% with ventricular contraction. But this was not thought to have been the cause of his presentation.
On discharge, he required oxygen 4 L/min by nasal cannula. Previously, he had not needed supplemental oxygen.
CURRENT PRESENTATION
The patient described persistent and severe periumbilical abdominal pain during the previous day. It was not associated with eating, and he denied diarrhea, constipation, hematemesis, hematochezia, bright red blood per rectum, or melena. He continued to describe persistent shortness of breath and pleuritic chest pain. His vital signs were as follows:
- Heart rate 104 beats per minute
- Respiratory rate 16 to 20 breaths per minute
- Blood pressure 101–142/62–84 mm Hg
- Oxygen saturation 78% on room air.
His laboratory findings on presentation are shown in Table 1, and his electrocardiogram is shown in Figure 1.
WHAT DOES HIS ELECTROCARDIOGRAM SHOW?
1. Which of the following is the most accurate description of this patient’s electrocardiogram?
- Sinus tachycardia, peaked P waves (P pulmonale) in lead II, and T-wave inversions in the right precordial leads
- Sinus tachycardia and left bundle branch block
- Sinus tachycardia and poor R-wave progression
- Sinus tachycardia and ST elevation in the precordial leads
Our patient’s electrocardiogram shows sinus tachycardia, P pulmonale, T-wave inversion in the right precordial leads (V1–V3), and biphasic T waves in lead V4,, which suggest right ventricular strain.
The rhythm most commonly seen in patients with pulmonary embolism is sinus tachycardia, followed by nonspecific ST-segment or T-wave abnormalities. In one series of patients with acute pulmonary embolism, the classic findings of P pulmonale, right ventricular hypertrophy, right axis deviation, and right bundle branch block were rare (< 6%).1 Thus, these classic findings are not sensitive for the diagnosis of pulmonary embolism, and their absence does not rule it out.
Further studies for our patient
Transthoracic echocardiography was performed to look for evidence of right ventricular strain secondary to the pulmonary embolism.
ECHOCARDIOGRAPHIC SIGNS OF PULMONARY EMBOLISM
2. Which of the following findings on transthoracic echocardiography would not suggest acute pulmonary embolism?
- Midright ventricular wall hypokinesis with apical sparing
- Severe tricuspid regurgitation
- Left ventricular dilation
- Lack of respiratory variation of the inferior vena cava
- Septal wall motion toward the left ventricle
Left ventricular dilation does not suggest acute pulmonary embolism. Echocardiograms of patients with acute submassive pulmonary embolism typically show evidence of right ventricular strain, such as the other entities listed above (midright ventricular hypokinesis with apical sparing, severe tricuspid regurgitation, lack of respiratory variation of the inferior vena cava, and septal wall motion toward the left ventricle).
The degree of right ventricular dysfunction is related to the extent of acute pulmonary vascular occlusion and aids in risk-stratification of patients with acute pulmonary embolism. Midright ventricular wall hypokinesis with apical sparing has been termed the McConnell sign.2
In our patient, transthoracic echocardiography showed:
- Normal left ventricular ejection fraction
- Mild diastolic dysfunction
- Right ventricular dilation with moderately decreased right ventricular systolic function and apical sparing
- Right ventricular systolic pressure 54 mm Hg, consistent with moderate pulmonary hypertension
- Right atrial pressure 10 mm Hg
- No inspiratory collapse of a dilated inferior vena cava
- Mild tricuspid valve regurgitation.
CLASSIFICATION OF ACUTE PULMONARY EMBOLISM
3. Given the above information, how would you classify the patient’s pulmonary embolism?
- Massive
- Submassive
- Low-risk
- Clinically stable
The patient’s pulmonary embolism is submassive.
Historically, the classification of pulmonary embolism was determined by the angiographic thrombus burden. However, this has limited utility because clinical factors (eg, hypotension on initial presentation) have been shown to be better predictors of short-term mortality risk.3
Our patient is characterized as having a submassive pulmonary embolism based on elevated biomarkers (troponin T, N-terminal pro-B-type natriuretic peptide) and right ventricular dysfunction in the absence of hypotension.
ULTRASONOGRAPHY FOR DIAGNOSIS OF DEEP VEIN THROMBOSIS
Venous duplex ultrasonography has become the standard for diagnosis of lower extremity deep vein thrombosis. However, its quality and diagnostic accuracy depend on the skill of the person performing the examination. It is further limited by certain patient characteristics, including severe obesity, edema, and wounds and dressings at the site being examined.5
Our patient underwent duplex ultrasonography of the lower extremities, which demonstrated acute proximal and calf deep vein thrombosis in the right femoral, popliteal, and peroneal veins and no deep vein thrombosis in the left leg.
RISK STRATIFICATION IN ACUTE PULMONARY EMBOLISM
Multiple models exist to estimate the risk of complications in patients with acute pulmonary embolism.
The Bova score6 is based on the following factors:
- Systolic blood pressure 90–100 mm Hg (2 points) (patients with systolic blood pressure lower than 90 mm Hg were excluded from the study from which this score was derived)
- Cardiac troponin elevation (2 points)
- Right ventricular dysfunction on echocardiography or computed tomography (2 points)
- Heart rate 100 beats/min or greater (1 point).
A total score of 0, 1, or 2 (stage I) denotes low risk, 3 or 4 points (stage II) intermediate risk, and more than 4 points (stage III) high risk.
The PESI score (Pulmonary Embolism Severity Index)7 is based on:
- Age (1 point per year)
- Sex (10 points for being male)
- Heart rate 110 per minute or greater (20 points)
- Cancer (30 points)
- Heart failure (10 points)
- Chronic lung disease (10 points)
- Systolic blood pressure less than 100 mm Hg (30 points)
- Respiratory rate at least 30 per minute (20 points)
- Temperature less than 36ºC (20 points)
- Altered mental status (60 points)
- Arterial oxygen saturation less than 90% (20 points).
The total score is broken down into 5 classes: I (< 65 points), II (65–85), III (86–105), IV (106–125), and V (> 126). Classes I and II are low risk, and the higher ones are high risk.
The simplified PESI score8 was developed to more rapidly risk-stratify patients and has been found to be similar to the PESI score in prognostic accuracy. Patients get 1 point for each of the following:
- Age over 80
- Cancer
- Chronic cardiopulmonary disease (heart failure or chronic lung disease)
- Heart rate 110 per minute or greater
- Systolic blood pressure less than 100 mm Hg
- Arterial oxygen saturation less than 90%.
A total score of 0 is low risk; anything higher is high risk.
Back to our patient
Our patient had proximal and calf deep vein thrombosis of the right leg, bilateral submassive pulmonary emboli with associated biomarker elevation and right ventricular dysfunction, and left renal artery thrombosis with infarction. Using the PESI score, his risk of death in the next 30 days was 13.7% and his 30-day risk of a complicated course was 27%. Using the Bova score, his 30-day risk of death was 15.5% and his 30-day risk of a complicated course was 29.2%.6,7
Notably, the patient’s right ventricular function had also been impaired on the echocardiogram performed during his admission 1 month previously. On transthoracic echocardiography during the current admission, the patient was found to have a similar degree of right ventricular dysfunction. This finding, along with the oxygen requirement that developed during the earlier admission, suggested that his pulmonary embolism may have been subacute and that the diagnosis may have been missed during the earlier hospital stay.
The patient was treated with unfractionated heparin. After the hospital’s multidisciplinary pulmonary embolism response team discussed and weighed the above factors, they recommended to not pursue thrombolytic therapy or inferior vena cava filter placement.
Of note, the patient’s pulses in the left lower extremity continued to be weak but palpable, and the left leg was cooler to touch than the right leg.
ASSESSING PERIPHERAL ARTERY DISEASE
4. How should the finding of weak pulses in this patient’s left leg be initially investigated?
- Computed tomographic angiography with runoff
- Ankle-brachial indices with pulse-volume recordings
- Arterial duplex ultrasonography
- Magnetic resonance angiography of the lower extremities
The ankle-brachial index is the initial diagnostic test for assessment of pulse abnormalities and for diagnosis of lower-extremity peripheral artery disease. It is calculated by dividing the higher of the ankle systolic pressures (posterior tibial or dorsalis pedis) by the higher of the 2 brachial pressures (left or right).9 Normal values are between 1.00 and 1.40.
Ankle-brachial indices in our patient
Our patient underwent measurement of his brachial, dorsalis pedis, and posterior tibial artery systolic pressures using blood pressure cuffs and continuous-wave Doppler. Ankle pulse-volume recordings were also obtained.
Given the patient’s poor renal function and concern for acute renal infarction, we thought it best to avoid iodinated or gadolinium contrast, such as with magnetic resonance or computed tomographic angiography.
Segmental leg pressures and pulse-volume recordings can be performed to help localize the level of arterial disease in the extremities, but were not done in this case because of the extensive deep vein thrombosis in the right leg.10,11
Arterial ultrasonography in our patient
Arterial duplex ultrasonography was performed to help determine the location of arterial disease. It showed patent arteries in the right leg. In the left lower extremity there was slow, monophasic blood flow in the distal superficial femoral artery. The popliteal artery was occluded. The posterior tibial artery was occluded at the origin, with reconstitution distally. The peroneal artery was occluded throughout. The anterior tibial artery was patent throughout. The ultrasonographic findings were thought to be suspicious for arterial thromboembolism.
WHAT CAN CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
5. Given that the patient had both arterial thrombosis (renal artery, lower-extremity arteries) and venous thromboembolism (deep vein thrombosis and pulmonary embolism), which of the following would be included in the differential diagnosis?
- Antiphospholipid antibody syndrome
- Protein C or protein S deficiency
- Malignancy
- Paradoxical embolization
- Factor V Leiden mutation
Correct answers include antiphospholipid antibody syndrome, malignancy, and paradoxical embolization.
The differential diagnosis for concomitant venous and arterial thrombosis is broad,12 and includes the following:
- Structural factors: patent foramen ovale, popliteal artery aneurysm
- Malignancy
- Inflammatory diseases: Behçet disease, Buerger disease, inflammatory bowel disease, antiphospholipid antibody syndrome, elevated lipoprotein(a), elevated homocysteine
- Hematologic diseases: myelodysplastic syndrome, disseminated intravascular coagulation, paroxysmal nocturnal hemoglobinuria, heparin-induced thrombocytopenia.
Traditional risk factors for venous thromboembolism include protein C deficiency, protein S deficiency, factor V Leiden mutation, the prothrombin G20210A gene mutation, and others. These are relatively minor risk factors for venous thrombosis and do not pose a risk for arterial thrombosis.12 In contrast, antiphospholipid antibody syndrome and malignancy pose a risk for both venous and arterial thrombosis. Paradoxical embolism is a mechanism by which arterial thrombosis (emboli) can develop in the setting of existing venous thrombosis.12
Our patient underwent testing for antiphospholipid antibodies and lupus anticoagulant, and he was encouraged to undergo age-appropriate cancer screening as an outpatient.12
ANTIPHOSPHOLIPID ANTIBODY SYNDROME
Antiphospholipid antibody syndrome is defined by both clinical and laboratory criteria. Clinical symptoms include vascular thrombosis (arterial, venous, or both) and pregnancy-related complications.13
Laboratory criteria require the presence of antiphospholipid antibodies or lupus anticoagulant. These must be confirmed with repeat testing in 12 weeks. Antiphospholipid antibodies are detected by an enzyme-linked immunosorbent assay; laboratory assessment for the presence of lupus anticoagulant is a stepwise process and relies on 4 criteria:
- There should be prolongation of a phospholipid-dependent clotting test (eg, activated partial thromboplastin time, dilute Russell viper venom time test).
- There must be evidence of an inhibitory activity with mixing study.
- The inhibitor must exhibit phospholipid dependence; that is, with more phospholipid there is shortening of clotting time.
- Specific inhibitors must be excluded, including factor VIII and anticoagulant drugs such as heparin.14–17
Clinically, one should consider antiphospholipid antibody syndrome in patients who have arterial thrombosis, a history of pregnancy morbidity, or unexplained prolongation of activated partial thromboplastin time.13
Antiphospholipid antibodies may be present in up to a quarter of patients with venous thromboembolism, but it is persistent positivity of antibody assays that is associated with increased future risk of venous thromboembolism.19 Of note, the risk of venous thromboembolism in patients with confirmed antiphospholipid antibody syndrome is 10 times higher than in the general population.20
ANTIPHOSPHOLIPID ANTIBODIES ARE NOT ALL THE SAME
6. Which of the following antiphospholipid antibodies have not been associated with an increased thrombotic risk?
- Anti-beta-2-glycoprotein I IgG
- Lupus anticoagulant
- Antiphosphatidylserine
- Anticardiolipin IgM
- Anticardiolipin IgG
The correct answer is antiphosphatidylserine.15
Antiphospholipid antibodies are directed against a portion of select plasma proteins that are uncovered upon phospholipid binding. While lupus anticoagulant, anti-beta-2-glycoprotein I, and anticardiolipin antibodies are associated with thrombosis, antiprothrombin antibodies (including antiprothrombin and antiphosphatidylserine antibodies) are not.15,21
PARADOXICAL EMBOLISM
Patent foramen ovale, a communication between the right and left atrium in the interatrial septum, is associated with an increased risk of paradoxical embolization. The prevalence of patent foramen ovale is estimated to be 27% to 29% in the general population.22 Noncerebral systemic paradoxical embolism occurs less frequently than cerebral embolism, accounting for approximately 5% to 10% of paradoxical emboli.22
To evaluate for patent foramen ovale, transthoracic echocardiography is performed with a bubble (agitated saline contrast) study to assess for interatrial shunting. Transesophageal echocardiography or transcranial Doppler bubble studies may also be performed.
Although patent foramen ovale is most commonly associated with cerebral embolism, peripheral emboli can occur. Some research suggests that this may be a more common cause of arterial thromboembolism in younger patients. There have also been reports of other sites of systemic embolization, including the renal artery.12
Back to our patient
Initial antiphospholipid antibody testing was positive for lupus anticoagulant. Anticardiolipin and anti-beta-2-glycoprotein I antibodies were not detected.
Transesophageal echocardiography revealed a patent foramen ovale with a highly mobile atrial septum (atrial septal aneurysm).
The patient was treated with intravenous unfractionated heparin with bridging to warfarin with a target international normalized ratio (INR) of 2 to 3. His renal artery infarction and his lower-extremity arterial thromboembolic event were conservatively managed. His respiratory status improved, and he no longer required supplemental oxygen. His creatinine peaked at 1.7 mg/dL during his admission and improved to 1.2 mg/dL before he was discharged.
At follow-up, repeat echocardiography showed that his right ventricular systolic pressure had improved (decreased) to 37 mm Hg from 54 mm Hg. Repeat confirmatory testing was positive for lupus anticoagulant 12 weeks later. He has been maintained on warfarin with an INR goal of 2 to 3 as well as low-dose aspirin with plans for long-term anticoagulation. We decided to keep the patient on anticoagulation indefinitely with warfarin; he was not a candidate for a direct oral anticoagulant, given limited data on the use of these agents in the setting of lupus anticoagulant and antiphospholipid antibody syndrome.
SUMMARY OF CASE
In summary, this patient was a 75-year-old man with COPD who presented with abdominal pain. He was noted to have a left renal infarction, extensive unprovoked lower-extremity deep vein thrombosis with pulmonary emboli, and lower limb arterial thromboembolism.
He also had an underlying hypercoagulable state—antiphospholipid antibody syndrome—that predisposed him to both arterial and venous thrombosis. He was ultimately found to have a patent foramen ovale, which further increased the risk of arterial thrombosis by facilitating paradoxical embolization of venous thrombi. It is not certain whether the renal infarction and leg artery thrombi were due to paradoxical embolism or to in situ thrombosis, but we believe that it was most likely paradoxical embolization.
A 75-year-old man presented to the emergency department for evaluation of abdominal pain. He had stage 3 chronic obstructive pulmonary disease (COPD), with a forced expiratory volume in 1 second of 33%.
PREVIOUS HOSPITALIZATION
Aside from his COPD, he had been healthy until 1 month earlier, when he had been hospitalized because of shortness of breath and chest pressure with exertion. His troponin T level had been elevated, peaking at 0.117 ng/mL (reference range 0–0.029).
Left heart catheterization had shown no significant coronary artery disease. A myocardial bridge of the distal left anterior descending coronary artery had been seen, so that the artery appeared to be narrowed by 50% to 60% with ventricular contraction. But this was not thought to have been the cause of his presentation.
On discharge, he required oxygen 4 L/min by nasal cannula. Previously, he had not needed supplemental oxygen.
CURRENT PRESENTATION
The patient described persistent and severe periumbilical abdominal pain during the previous day. It was not associated with eating, and he denied diarrhea, constipation, hematemesis, hematochezia, bright red blood per rectum, or melena. He continued to describe persistent shortness of breath and pleuritic chest pain. His vital signs were as follows:
- Heart rate 104 beats per minute
- Respiratory rate 16 to 20 breaths per minute
- Blood pressure 101–142/62–84 mm Hg
- Oxygen saturation 78% on room air.
His laboratory findings on presentation are shown in Table 1, and his electrocardiogram is shown in Figure 1.
WHAT DOES HIS ELECTROCARDIOGRAM SHOW?
1. Which of the following is the most accurate description of this patient’s electrocardiogram?
- Sinus tachycardia, peaked P waves (P pulmonale) in lead II, and T-wave inversions in the right precordial leads
- Sinus tachycardia and left bundle branch block
- Sinus tachycardia and poor R-wave progression
- Sinus tachycardia and ST elevation in the precordial leads
Our patient’s electrocardiogram shows sinus tachycardia, P pulmonale, T-wave inversion in the right precordial leads (V1–V3), and biphasic T waves in lead V4,, which suggest right ventricular strain.
The rhythm most commonly seen in patients with pulmonary embolism is sinus tachycardia, followed by nonspecific ST-segment or T-wave abnormalities. In one series of patients with acute pulmonary embolism, the classic findings of P pulmonale, right ventricular hypertrophy, right axis deviation, and right bundle branch block were rare (< 6%).1 Thus, these classic findings are not sensitive for the diagnosis of pulmonary embolism, and their absence does not rule it out.
Further studies for our patient
Transthoracic echocardiography was performed to look for evidence of right ventricular strain secondary to the pulmonary embolism.
ECHOCARDIOGRAPHIC SIGNS OF PULMONARY EMBOLISM
2. Which of the following findings on transthoracic echocardiography would not suggest acute pulmonary embolism?
- Midright ventricular wall hypokinesis with apical sparing
- Severe tricuspid regurgitation
- Left ventricular dilation
- Lack of respiratory variation of the inferior vena cava
- Septal wall motion toward the left ventricle
Left ventricular dilation does not suggest acute pulmonary embolism. Echocardiograms of patients with acute submassive pulmonary embolism typically show evidence of right ventricular strain, such as the other entities listed above (midright ventricular hypokinesis with apical sparing, severe tricuspid regurgitation, lack of respiratory variation of the inferior vena cava, and septal wall motion toward the left ventricle).
The degree of right ventricular dysfunction is related to the extent of acute pulmonary vascular occlusion and aids in risk-stratification of patients with acute pulmonary embolism. Midright ventricular wall hypokinesis with apical sparing has been termed the McConnell sign.2
In our patient, transthoracic echocardiography showed:
- Normal left ventricular ejection fraction
- Mild diastolic dysfunction
- Right ventricular dilation with moderately decreased right ventricular systolic function and apical sparing
- Right ventricular systolic pressure 54 mm Hg, consistent with moderate pulmonary hypertension
- Right atrial pressure 10 mm Hg
- No inspiratory collapse of a dilated inferior vena cava
- Mild tricuspid valve regurgitation.
CLASSIFICATION OF ACUTE PULMONARY EMBOLISM
3. Given the above information, how would you classify the patient’s pulmonary embolism?
- Massive
- Submassive
- Low-risk
- Clinically stable
The patient’s pulmonary embolism is submassive.
Historically, the classification of pulmonary embolism was determined by the angiographic thrombus burden. However, this has limited utility because clinical factors (eg, hypotension on initial presentation) have been shown to be better predictors of short-term mortality risk.3
Our patient is characterized as having a submassive pulmonary embolism based on elevated biomarkers (troponin T, N-terminal pro-B-type natriuretic peptide) and right ventricular dysfunction in the absence of hypotension.
ULTRASONOGRAPHY FOR DIAGNOSIS OF DEEP VEIN THROMBOSIS
Venous duplex ultrasonography has become the standard for diagnosis of lower extremity deep vein thrombosis. However, its quality and diagnostic accuracy depend on the skill of the person performing the examination. It is further limited by certain patient characteristics, including severe obesity, edema, and wounds and dressings at the site being examined.5
Our patient underwent duplex ultrasonography of the lower extremities, which demonstrated acute proximal and calf deep vein thrombosis in the right femoral, popliteal, and peroneal veins and no deep vein thrombosis in the left leg.
RISK STRATIFICATION IN ACUTE PULMONARY EMBOLISM
Multiple models exist to estimate the risk of complications in patients with acute pulmonary embolism.
The Bova score6 is based on the following factors:
- Systolic blood pressure 90–100 mm Hg (2 points) (patients with systolic blood pressure lower than 90 mm Hg were excluded from the study from which this score was derived)
- Cardiac troponin elevation (2 points)
- Right ventricular dysfunction on echocardiography or computed tomography (2 points)
- Heart rate 100 beats/min or greater (1 point).
A total score of 0, 1, or 2 (stage I) denotes low risk, 3 or 4 points (stage II) intermediate risk, and more than 4 points (stage III) high risk.
The PESI score (Pulmonary Embolism Severity Index)7 is based on:
- Age (1 point per year)
- Sex (10 points for being male)
- Heart rate 110 per minute or greater (20 points)
- Cancer (30 points)
- Heart failure (10 points)
- Chronic lung disease (10 points)
- Systolic blood pressure less than 100 mm Hg (30 points)
- Respiratory rate at least 30 per minute (20 points)
- Temperature less than 36ºC (20 points)
- Altered mental status (60 points)
- Arterial oxygen saturation less than 90% (20 points).
The total score is broken down into 5 classes: I (< 65 points), II (65–85), III (86–105), IV (106–125), and V (> 126). Classes I and II are low risk, and the higher ones are high risk.
The simplified PESI score8 was developed to more rapidly risk-stratify patients and has been found to be similar to the PESI score in prognostic accuracy. Patients get 1 point for each of the following:
- Age over 80
- Cancer
- Chronic cardiopulmonary disease (heart failure or chronic lung disease)
- Heart rate 110 per minute or greater
- Systolic blood pressure less than 100 mm Hg
- Arterial oxygen saturation less than 90%.
A total score of 0 is low risk; anything higher is high risk.
Back to our patient
Our patient had proximal and calf deep vein thrombosis of the right leg, bilateral submassive pulmonary emboli with associated biomarker elevation and right ventricular dysfunction, and left renal artery thrombosis with infarction. Using the PESI score, his risk of death in the next 30 days was 13.7% and his 30-day risk of a complicated course was 27%. Using the Bova score, his 30-day risk of death was 15.5% and his 30-day risk of a complicated course was 29.2%.6,7
Notably, the patient’s right ventricular function had also been impaired on the echocardiogram performed during his admission 1 month previously. On transthoracic echocardiography during the current admission, the patient was found to have a similar degree of right ventricular dysfunction. This finding, along with the oxygen requirement that developed during the earlier admission, suggested that his pulmonary embolism may have been subacute and that the diagnosis may have been missed during the earlier hospital stay.
The patient was treated with unfractionated heparin. After the hospital’s multidisciplinary pulmonary embolism response team discussed and weighed the above factors, they recommended to not pursue thrombolytic therapy or inferior vena cava filter placement.
Of note, the patient’s pulses in the left lower extremity continued to be weak but palpable, and the left leg was cooler to touch than the right leg.
ASSESSING PERIPHERAL ARTERY DISEASE
4. How should the finding of weak pulses in this patient’s left leg be initially investigated?
- Computed tomographic angiography with runoff
- Ankle-brachial indices with pulse-volume recordings
- Arterial duplex ultrasonography
- Magnetic resonance angiography of the lower extremities
The ankle-brachial index is the initial diagnostic test for assessment of pulse abnormalities and for diagnosis of lower-extremity peripheral artery disease. It is calculated by dividing the higher of the ankle systolic pressures (posterior tibial or dorsalis pedis) by the higher of the 2 brachial pressures (left or right).9 Normal values are between 1.00 and 1.40.
Ankle-brachial indices in our patient
Our patient underwent measurement of his brachial, dorsalis pedis, and posterior tibial artery systolic pressures using blood pressure cuffs and continuous-wave Doppler. Ankle pulse-volume recordings were also obtained.
Given the patient’s poor renal function and concern for acute renal infarction, we thought it best to avoid iodinated or gadolinium contrast, such as with magnetic resonance or computed tomographic angiography.
Segmental leg pressures and pulse-volume recordings can be performed to help localize the level of arterial disease in the extremities, but were not done in this case because of the extensive deep vein thrombosis in the right leg.10,11
Arterial ultrasonography in our patient
Arterial duplex ultrasonography was performed to help determine the location of arterial disease. It showed patent arteries in the right leg. In the left lower extremity there was slow, monophasic blood flow in the distal superficial femoral artery. The popliteal artery was occluded. The posterior tibial artery was occluded at the origin, with reconstitution distally. The peroneal artery was occluded throughout. The anterior tibial artery was patent throughout. The ultrasonographic findings were thought to be suspicious for arterial thromboembolism.
WHAT CAN CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
5. Given that the patient had both arterial thrombosis (renal artery, lower-extremity arteries) and venous thromboembolism (deep vein thrombosis and pulmonary embolism), which of the following would be included in the differential diagnosis?
- Antiphospholipid antibody syndrome
- Protein C or protein S deficiency
- Malignancy
- Paradoxical embolization
- Factor V Leiden mutation
Correct answers include antiphospholipid antibody syndrome, malignancy, and paradoxical embolization.
The differential diagnosis for concomitant venous and arterial thrombosis is broad,12 and includes the following:
- Structural factors: patent foramen ovale, popliteal artery aneurysm
- Malignancy
- Inflammatory diseases: Behçet disease, Buerger disease, inflammatory bowel disease, antiphospholipid antibody syndrome, elevated lipoprotein(a), elevated homocysteine
- Hematologic diseases: myelodysplastic syndrome, disseminated intravascular coagulation, paroxysmal nocturnal hemoglobinuria, heparin-induced thrombocytopenia.
Traditional risk factors for venous thromboembolism include protein C deficiency, protein S deficiency, factor V Leiden mutation, the prothrombin G20210A gene mutation, and others. These are relatively minor risk factors for venous thrombosis and do not pose a risk for arterial thrombosis.12 In contrast, antiphospholipid antibody syndrome and malignancy pose a risk for both venous and arterial thrombosis. Paradoxical embolism is a mechanism by which arterial thrombosis (emboli) can develop in the setting of existing venous thrombosis.12
Our patient underwent testing for antiphospholipid antibodies and lupus anticoagulant, and he was encouraged to undergo age-appropriate cancer screening as an outpatient.12
ANTIPHOSPHOLIPID ANTIBODY SYNDROME
Antiphospholipid antibody syndrome is defined by both clinical and laboratory criteria. Clinical symptoms include vascular thrombosis (arterial, venous, or both) and pregnancy-related complications.13
Laboratory criteria require the presence of antiphospholipid antibodies or lupus anticoagulant. These must be confirmed with repeat testing in 12 weeks. Antiphospholipid antibodies are detected by an enzyme-linked immunosorbent assay; laboratory assessment for the presence of lupus anticoagulant is a stepwise process and relies on 4 criteria:
- There should be prolongation of a phospholipid-dependent clotting test (eg, activated partial thromboplastin time, dilute Russell viper venom time test).
- There must be evidence of an inhibitory activity with mixing study.
- The inhibitor must exhibit phospholipid dependence; that is, with more phospholipid there is shortening of clotting time.
- Specific inhibitors must be excluded, including factor VIII and anticoagulant drugs such as heparin.14–17
Clinically, one should consider antiphospholipid antibody syndrome in patients who have arterial thrombosis, a history of pregnancy morbidity, or unexplained prolongation of activated partial thromboplastin time.13
Antiphospholipid antibodies may be present in up to a quarter of patients with venous thromboembolism, but it is persistent positivity of antibody assays that is associated with increased future risk of venous thromboembolism.19 Of note, the risk of venous thromboembolism in patients with confirmed antiphospholipid antibody syndrome is 10 times higher than in the general population.20
ANTIPHOSPHOLIPID ANTIBODIES ARE NOT ALL THE SAME
6. Which of the following antiphospholipid antibodies have not been associated with an increased thrombotic risk?
- Anti-beta-2-glycoprotein I IgG
- Lupus anticoagulant
- Antiphosphatidylserine
- Anticardiolipin IgM
- Anticardiolipin IgG
The correct answer is antiphosphatidylserine.15
Antiphospholipid antibodies are directed against a portion of select plasma proteins that are uncovered upon phospholipid binding. While lupus anticoagulant, anti-beta-2-glycoprotein I, and anticardiolipin antibodies are associated with thrombosis, antiprothrombin antibodies (including antiprothrombin and antiphosphatidylserine antibodies) are not.15,21
PARADOXICAL EMBOLISM
Patent foramen ovale, a communication between the right and left atrium in the interatrial septum, is associated with an increased risk of paradoxical embolization. The prevalence of patent foramen ovale is estimated to be 27% to 29% in the general population.22 Noncerebral systemic paradoxical embolism occurs less frequently than cerebral embolism, accounting for approximately 5% to 10% of paradoxical emboli.22
To evaluate for patent foramen ovale, transthoracic echocardiography is performed with a bubble (agitated saline contrast) study to assess for interatrial shunting. Transesophageal echocardiography or transcranial Doppler bubble studies may also be performed.
Although patent foramen ovale is most commonly associated with cerebral embolism, peripheral emboli can occur. Some research suggests that this may be a more common cause of arterial thromboembolism in younger patients. There have also been reports of other sites of systemic embolization, including the renal artery.12
Back to our patient
Initial antiphospholipid antibody testing was positive for lupus anticoagulant. Anticardiolipin and anti-beta-2-glycoprotein I antibodies were not detected.
Transesophageal echocardiography revealed a patent foramen ovale with a highly mobile atrial septum (atrial septal aneurysm).
The patient was treated with intravenous unfractionated heparin with bridging to warfarin with a target international normalized ratio (INR) of 2 to 3. His renal artery infarction and his lower-extremity arterial thromboembolic event were conservatively managed. His respiratory status improved, and he no longer required supplemental oxygen. His creatinine peaked at 1.7 mg/dL during his admission and improved to 1.2 mg/dL before he was discharged.
At follow-up, repeat echocardiography showed that his right ventricular systolic pressure had improved (decreased) to 37 mm Hg from 54 mm Hg. Repeat confirmatory testing was positive for lupus anticoagulant 12 weeks later. He has been maintained on warfarin with an INR goal of 2 to 3 as well as low-dose aspirin with plans for long-term anticoagulation. We decided to keep the patient on anticoagulation indefinitely with warfarin; he was not a candidate for a direct oral anticoagulant, given limited data on the use of these agents in the setting of lupus anticoagulant and antiphospholipid antibody syndrome.
SUMMARY OF CASE
In summary, this patient was a 75-year-old man with COPD who presented with abdominal pain. He was noted to have a left renal infarction, extensive unprovoked lower-extremity deep vein thrombosis with pulmonary emboli, and lower limb arterial thromboembolism.
He also had an underlying hypercoagulable state—antiphospholipid antibody syndrome—that predisposed him to both arterial and venous thrombosis. He was ultimately found to have a patent foramen ovale, which further increased the risk of arterial thrombosis by facilitating paradoxical embolization of venous thrombi. It is not certain whether the renal infarction and leg artery thrombi were due to paradoxical embolism or to in situ thrombosis, but we believe that it was most likely paradoxical embolization.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100:598–603.
- Alsoos F, Khaddam A. Echocardiographic evaluation methods for right ventricular function. J Echocardiogr 2015; 13:43–51.
- Jaff MR, McMurtry MS, Archer SL, et al; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; American Heart Association Council on Peripheral Vascular Disease; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 2011; 123:1788–1830.
- Heit JA, Silverstein MD, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ 3rd. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med 2000; 160:809–815.
- Gornik HL, Sharma AM. Duplex ultrasound in the diagnosis of lower-extremity deep venous thrombosis. Circulation 2014; 129:917–921.
- Fernández C, Bova C, Sanchez O, et al. Validation of a model for identification of patients at intermediate to high risk for complications associated with acute symptomatic pulmonary embolism. Chest 2015; 148:211–218.
- Aujesky D, Perrier A, Roy PM, et al. Validation of a clinical prognostic model to identify low-risk patients with pulmonary embolism. J Intern Med 2007; 261:597–604.
- Jiménez D, Aujesky D, Moores L, et al; RIETE Investigators. Simplification of the pulmonary embolism severity index for prognostication in patients with acute symptomatic pulmonary embolism. Arch Intern Med 2010; 170:1383–1389.
- Kim ES, Wattanakit K, Gornik HL. Using the ankle-brachial index to diagnose peripheral artery disease and assess cardiovascular risk. Cleve Clin J Med 2012; 79:651–661.
- Jaff MR. Lower extremity arterial disease. Diagnostic aspects. Cardiol Clin 2002; 20:491–500.
- Rooke TW, Hirsch AT, Misra S, et al; American College of Cardiology Foundation Task Force; American Heart Association Task Force. Management of patients with peripheral artery disease (compilation of 2005 and 2011 ACCF/AHA Guideline Recommendations): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:1555–1570.
- Lichtin A, Bartholomew J. The coagulation consult: a case-based guide. New York, NY: Springer; 2014.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
- Miyakis S, Lockshin M, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Nichols WL, Kottke-Marchant K, Ledford-Kraemer MR, Homburger HA, Cardel LK. Lupus anticoagulants, antiphospholipid antibodies, and antiphospholipid syndrome. In: Kottke-Marchant K, Davis BH, editors. Laboratory Hematology Practice. Hoboken, New Jersey: Blackwell Publishing, Ltd.; 2012:509–525.
- Houghton DE, Moll S. Antiphospholipid antibodies. Vasc Med 2017; 22:545–550.
- Roldan V, Lecumberri R, Muñoz-Torrero JFS, et al; RIETE Investigators. Thrombophilia testing in patients with venous thromboembolism. Findings from the RIETE registry. Thromb Res 2009; 124:174–177.
- Wahl DG, Guillemin F, de Maistre E, Perret-Guillaume C, Lecompte T, Thibaut G. Meta-analysis of the risk of venous thrombosis in individuals with antiphospholipid antibodies without underlying autoimmune disease or previous thrombosis. Lupus 1998; 7:15–22.
- Love PE, Santoro SA. Antiphospholipid antibodies: anticardiolipin and the lupus anticoagulant in systemic lupus erythematosus (SLE) and in non-SLE disorders. Prevalence and clinical significance. Ann Intern Med 1990; 112:682–698.
- Thompson T, Evans W. Paradoxical embolism. QJM 1930; os-23:135–150.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100:598–603.
- Alsoos F, Khaddam A. Echocardiographic evaluation methods for right ventricular function. J Echocardiogr 2015; 13:43–51.
- Jaff MR, McMurtry MS, Archer SL, et al; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; American Heart Association Council on Peripheral Vascular Disease; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 2011; 123:1788–1830.
- Heit JA, Silverstein MD, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ 3rd. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med 2000; 160:809–815.
- Gornik HL, Sharma AM. Duplex ultrasound in the diagnosis of lower-extremity deep venous thrombosis. Circulation 2014; 129:917–921.
- Fernández C, Bova C, Sanchez O, et al. Validation of a model for identification of patients at intermediate to high risk for complications associated with acute symptomatic pulmonary embolism. Chest 2015; 148:211–218.
- Aujesky D, Perrier A, Roy PM, et al. Validation of a clinical prognostic model to identify low-risk patients with pulmonary embolism. J Intern Med 2007; 261:597–604.
- Jiménez D, Aujesky D, Moores L, et al; RIETE Investigators. Simplification of the pulmonary embolism severity index for prognostication in patients with acute symptomatic pulmonary embolism. Arch Intern Med 2010; 170:1383–1389.
- Kim ES, Wattanakit K, Gornik HL. Using the ankle-brachial index to diagnose peripheral artery disease and assess cardiovascular risk. Cleve Clin J Med 2012; 79:651–661.
- Jaff MR. Lower extremity arterial disease. Diagnostic aspects. Cardiol Clin 2002; 20:491–500.
- Rooke TW, Hirsch AT, Misra S, et al; American College of Cardiology Foundation Task Force; American Heart Association Task Force. Management of patients with peripheral artery disease (compilation of 2005 and 2011 ACCF/AHA Guideline Recommendations): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:1555–1570.
- Lichtin A, Bartholomew J. The coagulation consult: a case-based guide. New York, NY: Springer; 2014.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
- Miyakis S, Lockshin M, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Nichols WL, Kottke-Marchant K, Ledford-Kraemer MR, Homburger HA, Cardel LK. Lupus anticoagulants, antiphospholipid antibodies, and antiphospholipid syndrome. In: Kottke-Marchant K, Davis BH, editors. Laboratory Hematology Practice. Hoboken, New Jersey: Blackwell Publishing, Ltd.; 2012:509–525.
- Houghton DE, Moll S. Antiphospholipid antibodies. Vasc Med 2017; 22:545–550.
- Roldan V, Lecumberri R, Muñoz-Torrero JFS, et al; RIETE Investigators. Thrombophilia testing in patients with venous thromboembolism. Findings from the RIETE registry. Thromb Res 2009; 124:174–177.
- Wahl DG, Guillemin F, de Maistre E, Perret-Guillaume C, Lecompte T, Thibaut G. Meta-analysis of the risk of venous thrombosis in individuals with antiphospholipid antibodies without underlying autoimmune disease or previous thrombosis. Lupus 1998; 7:15–22.
- Love PE, Santoro SA. Antiphospholipid antibodies: anticardiolipin and the lupus anticoagulant in systemic lupus erythematosus (SLE) and in non-SLE disorders. Prevalence and clinical significance. Ann Intern Med 1990; 112:682–698.
- Thompson T, Evans W. Paradoxical embolism. QJM 1930; os-23:135–150.
Contrast nephropathy after computed tomography
Clinical question: Do rates of acute kidney injury (AKI), renal replacement therapy (RRT), or mortality differ between adults receiving contrast-enhanced computed tomography (CT) versus those receiving noncontrast CT?
Background: Published estimates regarding the risk of postcontrast complications are highly variable and recent data show that the risk of postcontrast AKI may be lower than previously suggested.
Study design: Systematic review and meta-analysis..
Setting: Noninterventional studies assessing differences in AKI, new RRT, or mortality among adults who received contrast-enhanced CT, compared with those receiving noncontrast CT.
Synopsis: A search among six databases and Google Scholar from inception through 2016 yielded 28 observational studies meeting inclusion criteria that included 107,335 participants. Twenty-six assessed AKI, 13 assessed need for RRT, and 9 assessed all-cause mortality. Compared with noncontrast CT, contrast-enhanced CT was not significantly associated with AKI (odds ratio, 0.94; 95% confidence interval, 0.83-1.07), RRT (OR, 0.83; 95% CI 0.59-1.16), or all-cause mortality (OR, 1.0; 95% CI 0.73-1.36). The overall risk of bias ranged from low to serious among the included studies. Studies were observational in nature, they were conducted in multiple settings (for example, ICU, emergency department), and the baseline characteristics of included patients were highly variable.
Bottom line: This meta-analysis observed no difference in adverse events between patients receiving contrast-enhanced CT versus those receiving noncontrast CT but should be interpreted with caution given the observational nature of the studies and differing characteristics of the included patients and study settings.
Citation: Aycock RD et al. Acute kidney injury after computed tomography: a meta-analysis. Ann Emerg Med. 2017 Aug 12. doi: 10.1016/j.annemergmed.2017.06.041
Dr. Simonetti is a hospitalist at the University of Colorado School of Medicine.
Clinical question: Do rates of acute kidney injury (AKI), renal replacement therapy (RRT), or mortality differ between adults receiving contrast-enhanced computed tomography (CT) versus those receiving noncontrast CT?
Background: Published estimates regarding the risk of postcontrast complications are highly variable and recent data show that the risk of postcontrast AKI may be lower than previously suggested.
Study design: Systematic review and meta-analysis..
Setting: Noninterventional studies assessing differences in AKI, new RRT, or mortality among adults who received contrast-enhanced CT, compared with those receiving noncontrast CT.
Synopsis: A search among six databases and Google Scholar from inception through 2016 yielded 28 observational studies meeting inclusion criteria that included 107,335 participants. Twenty-six assessed AKI, 13 assessed need for RRT, and 9 assessed all-cause mortality. Compared with noncontrast CT, contrast-enhanced CT was not significantly associated with AKI (odds ratio, 0.94; 95% confidence interval, 0.83-1.07), RRT (OR, 0.83; 95% CI 0.59-1.16), or all-cause mortality (OR, 1.0; 95% CI 0.73-1.36). The overall risk of bias ranged from low to serious among the included studies. Studies were observational in nature, they were conducted in multiple settings (for example, ICU, emergency department), and the baseline characteristics of included patients were highly variable.
Bottom line: This meta-analysis observed no difference in adverse events between patients receiving contrast-enhanced CT versus those receiving noncontrast CT but should be interpreted with caution given the observational nature of the studies and differing characteristics of the included patients and study settings.
Citation: Aycock RD et al. Acute kidney injury after computed tomography: a meta-analysis. Ann Emerg Med. 2017 Aug 12. doi: 10.1016/j.annemergmed.2017.06.041
Dr. Simonetti is a hospitalist at the University of Colorado School of Medicine.
Clinical question: Do rates of acute kidney injury (AKI), renal replacement therapy (RRT), or mortality differ between adults receiving contrast-enhanced computed tomography (CT) versus those receiving noncontrast CT?
Background: Published estimates regarding the risk of postcontrast complications are highly variable and recent data show that the risk of postcontrast AKI may be lower than previously suggested.
Study design: Systematic review and meta-analysis..
Setting: Noninterventional studies assessing differences in AKI, new RRT, or mortality among adults who received contrast-enhanced CT, compared with those receiving noncontrast CT.
Synopsis: A search among six databases and Google Scholar from inception through 2016 yielded 28 observational studies meeting inclusion criteria that included 107,335 participants. Twenty-six assessed AKI, 13 assessed need for RRT, and 9 assessed all-cause mortality. Compared with noncontrast CT, contrast-enhanced CT was not significantly associated with AKI (odds ratio, 0.94; 95% confidence interval, 0.83-1.07), RRT (OR, 0.83; 95% CI 0.59-1.16), or all-cause mortality (OR, 1.0; 95% CI 0.73-1.36). The overall risk of bias ranged from low to serious among the included studies. Studies were observational in nature, they were conducted in multiple settings (for example, ICU, emergency department), and the baseline characteristics of included patients were highly variable.
Bottom line: This meta-analysis observed no difference in adverse events between patients receiving contrast-enhanced CT versus those receiving noncontrast CT but should be interpreted with caution given the observational nature of the studies and differing characteristics of the included patients and study settings.
Citation: Aycock RD et al. Acute kidney injury after computed tomography: a meta-analysis. Ann Emerg Med. 2017 Aug 12. doi: 10.1016/j.annemergmed.2017.06.041
Dr. Simonetti is a hospitalist at the University of Colorado School of Medicine.
EHR application doubles hypertension recognition rate
Hypertension discovery in pediatric patients more than doubled for physicians using a clinical decision support (CDS) tool connected to the EHR, results of a study found.
Elyse O. Kharbanda, MD, MPH, a researcher at the HealthPartners Institute, Minneapolis, and her fellow investigators assert that using such a tool will help rectify the trend of underreported hypertension in adolescents, which remains a serious concern despite providers’ routinely taking blood pressure measurements during outpatient visits.
“Among patients with multiple visits, electronic health records should contain sufficient information to diagnose hypertension,” Dr. Kharbanda and her associates reported in their article published in Pediatrics. “However, even when EHRs are configured to display BP percentiles, information on the patterns of BP percentiles over time, previous diagnoses, and medications is not presented in a format that is useful for clinicians.”
With TeenBP, providers are first prompted to take an initial BP reading, as well as height and weight measurements.
If the first measure is above the 95th percentile, the CDS requests an additional reading, which is then averaged with the first. If average of the two is above or within the 95th percentile, the provider is notified and sent a list of recommendations, including a diagnosis of hypertension, lipid screening, and nutrition referral.
The 2-year trial included 522 pediatric patients with incident hypertension; the data were gathered from 20 primary care clinics within one health system between April 2014 and April 2016.

The rate of clinical recognition of patients’ hypertension in the clinics utilizing the CDS tool was more than double the rate seen in the clinics that weren’t (55% and 21%, respectively; P less than .001).
More of the children seen in CDS clinics were referred to dietitians or weight loss programs, compared with those seen in the control clinics (17% and 4%, respectively; P = .001).
Those who used the tool reported high levels of satisfaction, which is likely partly because investigators consulted physicians to help design the application.
“The CDS tool was based on the guidelines for BP management in children and adolescents in effect at the time of the study with local input from clinical and operational leaders within the medical group, and thus it contained the so-called right information,” according to Dr. Kharbanda and her fellow investigators.
Of the 55 physicians who remembered using the tool, 92% thought is was useful in identifying hypertension, 94% considered the CDS a good use of time, and 95% believed is was a useful shared-decision making tool.
When designing TeenBP, investigators tailored the application to the work flow and culture of the health system used for the study, which may limit the generalizability of the findings.
The study was funded by the National Institutes of Health. Dr. Kharbanda and her associates reported no relevant financial disclosures.
ezimmerman@frontlinemedcom.com
Source: Kharbanda EO et al. Pediatrics. 2018. doi: 10.1542/peds.2017- 2954.
The rate of children and adolescents with elevated blood pressure going unrecognized is an increasingly concerning issue – one that is complicated because physicians lack a simple, single BP value system. Some have tried to fill the gap, creating simplified tables of BP values or automated displays of BP values in EHRs, but having a table that only takes age and sex into consideration for screening does not cover the complexities needed to identify hypertension.
Previous studies have looked into utilizing a clinical decision support (CDS) application, which has great potential as a digital multitool to improve quality of care, increase efficiency, and reduce medical errors. However, to be an effective CDS, it must fill the “CDS Five Rights” framework. This guideline states that a CDS tool needs to provide: “the right information, to the right people, through the right channels, in the right intervention formats, at the right points in the work flow.”
The TeenBP CDS developed by Kharbanda et al. fulfills these requirements and goes beyond any CDS previously designed. Even so, 45% of children with elevated BP or hypertension were not recognized, emphasizing the need for additional strategies outside of relying on new technology.
Visit summaries should be given to parents with BP readings so that they can monitor their children’s levels, for example.
Recognition of abnormal BP in teens is the first step toward preventing cardiovascular disease as an adult, and hopefully, the development of new tools, including this CDS, will help physicians find those children who have been overlooked.
Ari H. Pollack, MD, MSIM, is a pediatric nephrologist at the Seattle Children’s Hospital and an assistant professor of pediatrics at the University of Washington, Seattle. Joseph T. Flynn, MD, MS, is the division chief of nephrology in prenatal diagnosis and treatment at the Seattle Children’s Hospital and a professor of pediatrics at the same university. Dr. Pollack and Dr. Flynn reported no relevant financial disclosures in their commentary in Pediatrics (2018. doi: 10.1542/peds.2017-3756).
The rate of children and adolescents with elevated blood pressure going unrecognized is an increasingly concerning issue – one that is complicated because physicians lack a simple, single BP value system. Some have tried to fill the gap, creating simplified tables of BP values or automated displays of BP values in EHRs, but having a table that only takes age and sex into consideration for screening does not cover the complexities needed to identify hypertension.
Previous studies have looked into utilizing a clinical decision support (CDS) application, which has great potential as a digital multitool to improve quality of care, increase efficiency, and reduce medical errors. However, to be an effective CDS, it must fill the “CDS Five Rights” framework. This guideline states that a CDS tool needs to provide: “the right information, to the right people, through the right channels, in the right intervention formats, at the right points in the work flow.”
The TeenBP CDS developed by Kharbanda et al. fulfills these requirements and goes beyond any CDS previously designed. Even so, 45% of children with elevated BP or hypertension were not recognized, emphasizing the need for additional strategies outside of relying on new technology.
Visit summaries should be given to parents with BP readings so that they can monitor their children’s levels, for example.
Recognition of abnormal BP in teens is the first step toward preventing cardiovascular disease as an adult, and hopefully, the development of new tools, including this CDS, will help physicians find those children who have been overlooked.
Ari H. Pollack, MD, MSIM, is a pediatric nephrologist at the Seattle Children’s Hospital and an assistant professor of pediatrics at the University of Washington, Seattle. Joseph T. Flynn, MD, MS, is the division chief of nephrology in prenatal diagnosis and treatment at the Seattle Children’s Hospital and a professor of pediatrics at the same university. Dr. Pollack and Dr. Flynn reported no relevant financial disclosures in their commentary in Pediatrics (2018. doi: 10.1542/peds.2017-3756).
The rate of children and adolescents with elevated blood pressure going unrecognized is an increasingly concerning issue – one that is complicated because physicians lack a simple, single BP value system. Some have tried to fill the gap, creating simplified tables of BP values or automated displays of BP values in EHRs, but having a table that only takes age and sex into consideration for screening does not cover the complexities needed to identify hypertension.
Previous studies have looked into utilizing a clinical decision support (CDS) application, which has great potential as a digital multitool to improve quality of care, increase efficiency, and reduce medical errors. However, to be an effective CDS, it must fill the “CDS Five Rights” framework. This guideline states that a CDS tool needs to provide: “the right information, to the right people, through the right channels, in the right intervention formats, at the right points in the work flow.”
The TeenBP CDS developed by Kharbanda et al. fulfills these requirements and goes beyond any CDS previously designed. Even so, 45% of children with elevated BP or hypertension were not recognized, emphasizing the need for additional strategies outside of relying on new technology.
Visit summaries should be given to parents with BP readings so that they can monitor their children’s levels, for example.
Recognition of abnormal BP in teens is the first step toward preventing cardiovascular disease as an adult, and hopefully, the development of new tools, including this CDS, will help physicians find those children who have been overlooked.
Ari H. Pollack, MD, MSIM, is a pediatric nephrologist at the Seattle Children’s Hospital and an assistant professor of pediatrics at the University of Washington, Seattle. Joseph T. Flynn, MD, MS, is the division chief of nephrology in prenatal diagnosis and treatment at the Seattle Children’s Hospital and a professor of pediatrics at the same university. Dr. Pollack and Dr. Flynn reported no relevant financial disclosures in their commentary in Pediatrics (2018. doi: 10.1542/peds.2017-3756).
Hypertension discovery in pediatric patients more than doubled for physicians using a clinical decision support (CDS) tool connected to the EHR, results of a study found.
Elyse O. Kharbanda, MD, MPH, a researcher at the HealthPartners Institute, Minneapolis, and her fellow investigators assert that using such a tool will help rectify the trend of underreported hypertension in adolescents, which remains a serious concern despite providers’ routinely taking blood pressure measurements during outpatient visits.
“Among patients with multiple visits, electronic health records should contain sufficient information to diagnose hypertension,” Dr. Kharbanda and her associates reported in their article published in Pediatrics. “However, even when EHRs are configured to display BP percentiles, information on the patterns of BP percentiles over time, previous diagnoses, and medications is not presented in a format that is useful for clinicians.”
With TeenBP, providers are first prompted to take an initial BP reading, as well as height and weight measurements.
If the first measure is above the 95th percentile, the CDS requests an additional reading, which is then averaged with the first. If average of the two is above or within the 95th percentile, the provider is notified and sent a list of recommendations, including a diagnosis of hypertension, lipid screening, and nutrition referral.
The 2-year trial included 522 pediatric patients with incident hypertension; the data were gathered from 20 primary care clinics within one health system between April 2014 and April 2016.
The rate of clinical recognition of patients’ hypertension in the clinics utilizing the CDS tool was more than double the rate seen in the clinics that weren’t (55% and 21%, respectively; P less than .001).
More of the children seen in CDS clinics were referred to dietitians or weight loss programs, compared with those seen in the control clinics (17% and 4%, respectively; P = .001).
Those who used the tool reported high levels of satisfaction, which is likely partly because investigators consulted physicians to help design the application.
“The CDS tool was based on the guidelines for BP management in children and adolescents in effect at the time of the study with local input from clinical and operational leaders within the medical group, and thus it contained the so-called right information,” according to Dr. Kharbanda and her fellow investigators.
Of the 55 physicians who remembered using the tool, 92% thought is was useful in identifying hypertension, 94% considered the CDS a good use of time, and 95% believed is was a useful shared-decision making tool.
When designing TeenBP, investigators tailored the application to the work flow and culture of the health system used for the study, which may limit the generalizability of the findings.
The study was funded by the National Institutes of Health. Dr. Kharbanda and her associates reported no relevant financial disclosures.
ezimmerman@frontlinemedcom.com
Source: Kharbanda EO et al. Pediatrics. 2018. doi: 10.1542/peds.2017- 2954.
Hypertension discovery in pediatric patients more than doubled for physicians using a clinical decision support (CDS) tool connected to the EHR, results of a study found.
Elyse O. Kharbanda, MD, MPH, a researcher at the HealthPartners Institute, Minneapolis, and her fellow investigators assert that using such a tool will help rectify the trend of underreported hypertension in adolescents, which remains a serious concern despite providers’ routinely taking blood pressure measurements during outpatient visits.
“Among patients with multiple visits, electronic health records should contain sufficient information to diagnose hypertension,” Dr. Kharbanda and her associates reported in their article published in Pediatrics. “However, even when EHRs are configured to display BP percentiles, information on the patterns of BP percentiles over time, previous diagnoses, and medications is not presented in a format that is useful for clinicians.”
With TeenBP, providers are first prompted to take an initial BP reading, as well as height and weight measurements.
If the first measure is above the 95th percentile, the CDS requests an additional reading, which is then averaged with the first. If average of the two is above or within the 95th percentile, the provider is notified and sent a list of recommendations, including a diagnosis of hypertension, lipid screening, and nutrition referral.
The 2-year trial included 522 pediatric patients with incident hypertension; the data were gathered from 20 primary care clinics within one health system between April 2014 and April 2016.
The rate of clinical recognition of patients’ hypertension in the clinics utilizing the CDS tool was more than double the rate seen in the clinics that weren’t (55% and 21%, respectively; P less than .001).
More of the children seen in CDS clinics were referred to dietitians or weight loss programs, compared with those seen in the control clinics (17% and 4%, respectively; P = .001).
Those who used the tool reported high levels of satisfaction, which is likely partly because investigators consulted physicians to help design the application.
“The CDS tool was based on the guidelines for BP management in children and adolescents in effect at the time of the study with local input from clinical and operational leaders within the medical group, and thus it contained the so-called right information,” according to Dr. Kharbanda and her fellow investigators.
Of the 55 physicians who remembered using the tool, 92% thought is was useful in identifying hypertension, 94% considered the CDS a good use of time, and 95% believed is was a useful shared-decision making tool.
When designing TeenBP, investigators tailored the application to the work flow and culture of the health system used for the study, which may limit the generalizability of the findings.
The study was funded by the National Institutes of Health. Dr. Kharbanda and her associates reported no relevant financial disclosures.
ezimmerman@frontlinemedcom.com
Source: Kharbanda EO et al. Pediatrics. 2018. doi: 10.1542/peds.2017- 2954.
FROM PEDIATRICS
Key clinical point: Using a clinical decision support (CDS) tool doubles the rate of hypertension detection in children.
Major finding: Providers in clinics that used CDS recognized hypertension in 55% of patients, compared with 21% of patients in usual care (P less than .001).
Study details: Cluster-randomized trial of 522 pediatric patients across 20 primary care clinics who received care between April 2014 and April 2016.
Disclosures: The study was funded by the National Institutes of Health. The investigators reported no relevant financial disclosures.
Source: Kharbanda EO et al. Pediatrics. 2018. doi: 10.1542/peds.2017- 2954.