User login
Rituximab may outperform some other first-line multiple sclerosis treatments
Rituximab was associated with a lower drug discontinuation rate versus all other commonly prescribed disease-modifying treatments (DMTs) used as initial therapy for relapsing-remitting multiple sclerosis (RRMS) in a retrospective study of patient data from a Swedish multiple sclerosis registry.
In addition, relapse rates were lower with rituximab (Rituxan) than they were with injectable DMTs and dimethyl fumarate (Tecfidera), according to results of the study, which appeared online Jan. 8 in JAMA Neurology.
Anti-CD20 agents such as rituximab are “likely to become an additional treatment option” for RRMS, and off-label use of rituximab for this indication has “increased considerably” in Sweden in recent years, the investigators said. RRMS is not an approved indication for rituximab in the United States, whereas across Sweden “there is no difference in reimbursement policy ... because all DMTs are covered by the national health insurance, including off-label medications.”
The addition of new DMTs for RRMS has changed the treatment landscape recently, although in real-world practice, there is a lack of “detailed knowledge about how to tailor therapy,” the authors said. They noted that the majority of patients discontinue traditional first-line treatment with injectable DMTs (that is, interferon beta and glatiramer acetate) within 2 years, suggesting a need for better treatment options.
To evaluate the real-world effectiveness of rituximab in this setting, Dr. Granqvist and his colleagues selected patient registry data for two Swedish counties that 494 included who received a diagnosis of RRMS between January 1, 2012, and October 31, 2015.
The largest subset of patients (n = 215) received injectable DMTs, while the rest received rituximab (n = 120), dimethyl fumarate (n = 86), natalizumab (Tysabri; n = 50), fingolimod (Gilenya; n = 17), or another treatment (n = 6), according to data in the report.
The proportion of patients who stayed on treatment was significantly higher for rituximab versus all other DMTs, study authors found. Compared with rituximab, the hazard ratios for drug discontinuation after adjusting for covariates and propensity score were 11.4 (95% confidence interval, 4.7-27.4) for injectable DMTs, 15.1 (95% CI, 3.9-58.0) for dimethyl fumarate, 5.9 (95% CI, 1.5-23.4) for fingolimod, and 11.3 (95% CI 3.2-39.4) for natalizumab.
Rituximab-treated patients also had lower rates of clinical relapse, neuroradiologic disease activity, and adverse events, compared with injectable DMTs or dimethyl fumarate, according to the investigators.
In comparison with fingolimod and natalizumab, relapse rates and gadolinium-enhancing lesions with rituximab were less frequent, but the authors said those differences did not reach statistical significance in all analyses.
The study was funded by the Swedish Medical Research Council, among other sources. Study authors reported conflicts of interest related to Biogen, Novartis, and Genzyme.
SOURCE: Granqvist M et al. JAMA Neurol. 2018 Jan 8. doi: 10.1001/jamaneurol.2017.4011
Rituximab was associated with a lower drug discontinuation rate versus all other commonly prescribed disease-modifying treatments (DMTs) used as initial therapy for relapsing-remitting multiple sclerosis (RRMS) in a retrospective study of patient data from a Swedish multiple sclerosis registry.
In addition, relapse rates were lower with rituximab (Rituxan) than they were with injectable DMTs and dimethyl fumarate (Tecfidera), according to results of the study, which appeared online Jan. 8 in JAMA Neurology.
Anti-CD20 agents such as rituximab are “likely to become an additional treatment option” for RRMS, and off-label use of rituximab for this indication has “increased considerably” in Sweden in recent years, the investigators said. RRMS is not an approved indication for rituximab in the United States, whereas across Sweden “there is no difference in reimbursement policy ... because all DMTs are covered by the national health insurance, including off-label medications.”
The addition of new DMTs for RRMS has changed the treatment landscape recently, although in real-world practice, there is a lack of “detailed knowledge about how to tailor therapy,” the authors said. They noted that the majority of patients discontinue traditional first-line treatment with injectable DMTs (that is, interferon beta and glatiramer acetate) within 2 years, suggesting a need for better treatment options.
To evaluate the real-world effectiveness of rituximab in this setting, Dr. Granqvist and his colleagues selected patient registry data for two Swedish counties that 494 included who received a diagnosis of RRMS between January 1, 2012, and October 31, 2015.
The largest subset of patients (n = 215) received injectable DMTs, while the rest received rituximab (n = 120), dimethyl fumarate (n = 86), natalizumab (Tysabri; n = 50), fingolimod (Gilenya; n = 17), or another treatment (n = 6), according to data in the report.
The proportion of patients who stayed on treatment was significantly higher for rituximab versus all other DMTs, study authors found. Compared with rituximab, the hazard ratios for drug discontinuation after adjusting for covariates and propensity score were 11.4 (95% confidence interval, 4.7-27.4) for injectable DMTs, 15.1 (95% CI, 3.9-58.0) for dimethyl fumarate, 5.9 (95% CI, 1.5-23.4) for fingolimod, and 11.3 (95% CI 3.2-39.4) for natalizumab.
Rituximab-treated patients also had lower rates of clinical relapse, neuroradiologic disease activity, and adverse events, compared with injectable DMTs or dimethyl fumarate, according to the investigators.
In comparison with fingolimod and natalizumab, relapse rates and gadolinium-enhancing lesions with rituximab were less frequent, but the authors said those differences did not reach statistical significance in all analyses.
The study was funded by the Swedish Medical Research Council, among other sources. Study authors reported conflicts of interest related to Biogen, Novartis, and Genzyme.
SOURCE: Granqvist M et al. JAMA Neurol. 2018 Jan 8. doi: 10.1001/jamaneurol.2017.4011
Rituximab was associated with a lower drug discontinuation rate versus all other commonly prescribed disease-modifying treatments (DMTs) used as initial therapy for relapsing-remitting multiple sclerosis (RRMS) in a retrospective study of patient data from a Swedish multiple sclerosis registry.
In addition, relapse rates were lower with rituximab (Rituxan) than they were with injectable DMTs and dimethyl fumarate (Tecfidera), according to results of the study, which appeared online Jan. 8 in JAMA Neurology.
Anti-CD20 agents such as rituximab are “likely to become an additional treatment option” for RRMS, and off-label use of rituximab for this indication has “increased considerably” in Sweden in recent years, the investigators said. RRMS is not an approved indication for rituximab in the United States, whereas across Sweden “there is no difference in reimbursement policy ... because all DMTs are covered by the national health insurance, including off-label medications.”
The addition of new DMTs for RRMS has changed the treatment landscape recently, although in real-world practice, there is a lack of “detailed knowledge about how to tailor therapy,” the authors said. They noted that the majority of patients discontinue traditional first-line treatment with injectable DMTs (that is, interferon beta and glatiramer acetate) within 2 years, suggesting a need for better treatment options.
To evaluate the real-world effectiveness of rituximab in this setting, Dr. Granqvist and his colleagues selected patient registry data for two Swedish counties that 494 included who received a diagnosis of RRMS between January 1, 2012, and October 31, 2015.
The largest subset of patients (n = 215) received injectable DMTs, while the rest received rituximab (n = 120), dimethyl fumarate (n = 86), natalizumab (Tysabri; n = 50), fingolimod (Gilenya; n = 17), or another treatment (n = 6), according to data in the report.
The proportion of patients who stayed on treatment was significantly higher for rituximab versus all other DMTs, study authors found. Compared with rituximab, the hazard ratios for drug discontinuation after adjusting for covariates and propensity score were 11.4 (95% confidence interval, 4.7-27.4) for injectable DMTs, 15.1 (95% CI, 3.9-58.0) for dimethyl fumarate, 5.9 (95% CI, 1.5-23.4) for fingolimod, and 11.3 (95% CI 3.2-39.4) for natalizumab.
Rituximab-treated patients also had lower rates of clinical relapse, neuroradiologic disease activity, and adverse events, compared with injectable DMTs or dimethyl fumarate, according to the investigators.
In comparison with fingolimod and natalizumab, relapse rates and gadolinium-enhancing lesions with rituximab were less frequent, but the authors said those differences did not reach statistical significance in all analyses.
The study was funded by the Swedish Medical Research Council, among other sources. Study authors reported conflicts of interest related to Biogen, Novartis, and Genzyme.
SOURCE: Granqvist M et al. JAMA Neurol. 2018 Jan 8. doi: 10.1001/jamaneurol.2017.4011
FROM JAMA NEUROLOGY
Key clinical point:
Major finding: Rituximab-treated patients had significantly lower rates of discontinuation, compared with injectable DMTs, fingolimod, natalizumab, and dimethyl fumarate. Relapse rates with rituximab were lower than they were with injectable DMTs and dimethyl fumarate.
Data source: Retrospective cohort study of data from a Swedish multiple sclerosis registry that included 494 patients with a diagnosis of RRMS.
Disclosures: The study was funded by the Swedish Medical Research Council, among other sources. Study authors reported conflicts of interest related to Biogen, Novartis, and Genzyme.
Source: Granqvist M et al. JAMA Neurol. 2018 Jan 8. doi: 10.1001/jamaneurol.2017.4011
FDA cites manufacturer of autologous stem cells for regulatory, manufacturing missteps
for manufacturing processes that may compromise its safety and for failing to toe the regulatory line in marketing.
American CryoStem received an FDA warning letter Jan. 3 demanding that the company comply with best-manufacturing processes and obtain an investigational new drug application if it wishes to continue marketing ATCELL for its currently advertised clinical indications and administration routes. These include intravenous, intrathecal, or aerosol inhalation of the product for anoxic brain injury, Parkinson’s disease, amyotrophic lateral sclerosis, stroke, and multiple sclerosis.
“Please be advised that, to lawfully market a drug that is a biological product, a valid biologics license must be in effect,” noted the letter. “Such licenses are issued only after a showing that the product is safe, pure, and potent. While in the development stage, such products may be distributed for clinical use in humans only if the sponsor has an investigational new drug application (IND) in effect as specified by FDA regulations. ATCELL is not the subject of an approved biologics license application nor is there an IND in effect. Based on this information, we have determined that your actions have violated the Food, Drug, and Cosmetic Act and the Public Health Service Act.”
FDA inspectors conducted a site inspection of American CryoStem in Eatontown, N.J., last summer, during which they “documented evidence of significant deviations from current good manufacturing practice.” The agency then provided the company a chance to respond to these issues. The new warning letter discussed each complaint, noting that some were inadequately addressed, and demanded that the company take action within 15 working days or face potential legal process, including seizure and/or injunction.
American CryoStem is one of the first companies to experience increased scrutiny under FDA’s new commitment to regulate the rapid growth and development of regenerative medicine products, which include novel cellular therapies, with the aim of ensuring their safety and effectiveness.
The new policy is designed to support the potential of cellular rejuvenation medicine, while protecting patients from “unscrupulous actors” who might endanger public health with untested products, according to FDA Commissioner Scott Gottlieb, MD. As enthusiasm for stem cell treatments surges, so are reports of adverse events. The New England Journal of Medicine recently reported on three patients with age-related macular degeneration who were blinded by intravitreal injection of autologous adipose-derived stem cells (N Engl J Med. 2017;376:1047-53).
Under the new policy, cell- and tissue-based products could be exempt from FDA premarket review only if they are removed from and implanted back into the same patient in their original form, or if the products are “minimally manipulated.” ATCELL fulfills neither qualification, the FDA warning letter said.
“You process adipose tissue ... to isolate cellular components of adipose tissue, commonly referred to as stromal vascular fraction [SVF]. Such processing is more than minimal manipulation because [it alters] the original relevant characteristics of the [tissue] relating to its utility for reconstruction, repair, or replacement. Then you process the SVF by expanding it in cell culture to manufacture ATCELL. Such expansion also is more than minimal manipulation because it alters the original relevant characteristics of the tissue.”
Furthermore, the letter noted, at least one of the components used in the clonal expansion process is investigational and not intended for human use. The manufacturer of that component, which was not named, “indicates the following: ‘Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to human and animals.”
The FDA also took exception with several equipment and lab safety issues. ATCELL was being created in areas that had no clean space designation – a serious concern, the letter said.
“American CryoStem’s unvalidated processes, inadequately controlled environment, lack of control of components used in production, and lack of sufficient and validated product testing ... pose a significant risk that ATCELL may be contaminated with microorganisms or have other serious product quality defects ... Because the product is administered to humans by various higher risk routes of administration, including intravenously, intrathecally, and by aerosol inhalation, if contaminated, its use could cause a range of adverse events, from infections to death.”
FDA also expressed concerns over a lack of consistent quality control testing of each batch and questioned whether the company’s method of shipping ATCELL to clinicians had been adequately validated.
Finally, the agency raised concerns that ATCELL, while it is labeled as being for research purposes only, may harm patients indirectly by preventing them from seeking timely treatment with proven therapies.
“ATCELL is intended to treat a variety of serious or life-threatening diseases or conditions, all of which are non-homologous uses,” the warning letter noted. “Such uses raise potential significant safety concerns because there is less basis on which to predict the product’s behavior in the recipient, and use of these unapproved products may cause users to delay or discontinue medical treatments that have been found safe and effective.”
SOURCE: FDA warning letter
for manufacturing processes that may compromise its safety and for failing to toe the regulatory line in marketing.
American CryoStem received an FDA warning letter Jan. 3 demanding that the company comply with best-manufacturing processes and obtain an investigational new drug application if it wishes to continue marketing ATCELL for its currently advertised clinical indications and administration routes. These include intravenous, intrathecal, or aerosol inhalation of the product for anoxic brain injury, Parkinson’s disease, amyotrophic lateral sclerosis, stroke, and multiple sclerosis.
“Please be advised that, to lawfully market a drug that is a biological product, a valid biologics license must be in effect,” noted the letter. “Such licenses are issued only after a showing that the product is safe, pure, and potent. While in the development stage, such products may be distributed for clinical use in humans only if the sponsor has an investigational new drug application (IND) in effect as specified by FDA regulations. ATCELL is not the subject of an approved biologics license application nor is there an IND in effect. Based on this information, we have determined that your actions have violated the Food, Drug, and Cosmetic Act and the Public Health Service Act.”
FDA inspectors conducted a site inspection of American CryoStem in Eatontown, N.J., last summer, during which they “documented evidence of significant deviations from current good manufacturing practice.” The agency then provided the company a chance to respond to these issues. The new warning letter discussed each complaint, noting that some were inadequately addressed, and demanded that the company take action within 15 working days or face potential legal process, including seizure and/or injunction.
American CryoStem is one of the first companies to experience increased scrutiny under FDA’s new commitment to regulate the rapid growth and development of regenerative medicine products, which include novel cellular therapies, with the aim of ensuring their safety and effectiveness.
The new policy is designed to support the potential of cellular rejuvenation medicine, while protecting patients from “unscrupulous actors” who might endanger public health with untested products, according to FDA Commissioner Scott Gottlieb, MD. As enthusiasm for stem cell treatments surges, so are reports of adverse events. The New England Journal of Medicine recently reported on three patients with age-related macular degeneration who were blinded by intravitreal injection of autologous adipose-derived stem cells (N Engl J Med. 2017;376:1047-53).
Under the new policy, cell- and tissue-based products could be exempt from FDA premarket review only if they are removed from and implanted back into the same patient in their original form, or if the products are “minimally manipulated.” ATCELL fulfills neither qualification, the FDA warning letter said.
“You process adipose tissue ... to isolate cellular components of adipose tissue, commonly referred to as stromal vascular fraction [SVF]. Such processing is more than minimal manipulation because [it alters] the original relevant characteristics of the [tissue] relating to its utility for reconstruction, repair, or replacement. Then you process the SVF by expanding it in cell culture to manufacture ATCELL. Such expansion also is more than minimal manipulation because it alters the original relevant characteristics of the tissue.”
Furthermore, the letter noted, at least one of the components used in the clonal expansion process is investigational and not intended for human use. The manufacturer of that component, which was not named, “indicates the following: ‘Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to human and animals.”
The FDA also took exception with several equipment and lab safety issues. ATCELL was being created in areas that had no clean space designation – a serious concern, the letter said.
“American CryoStem’s unvalidated processes, inadequately controlled environment, lack of control of components used in production, and lack of sufficient and validated product testing ... pose a significant risk that ATCELL may be contaminated with microorganisms or have other serious product quality defects ... Because the product is administered to humans by various higher risk routes of administration, including intravenously, intrathecally, and by aerosol inhalation, if contaminated, its use could cause a range of adverse events, from infections to death.”
FDA also expressed concerns over a lack of consistent quality control testing of each batch and questioned whether the company’s method of shipping ATCELL to clinicians had been adequately validated.
Finally, the agency raised concerns that ATCELL, while it is labeled as being for research purposes only, may harm patients indirectly by preventing them from seeking timely treatment with proven therapies.
“ATCELL is intended to treat a variety of serious or life-threatening diseases or conditions, all of which are non-homologous uses,” the warning letter noted. “Such uses raise potential significant safety concerns because there is less basis on which to predict the product’s behavior in the recipient, and use of these unapproved products may cause users to delay or discontinue medical treatments that have been found safe and effective.”
SOURCE: FDA warning letter
for manufacturing processes that may compromise its safety and for failing to toe the regulatory line in marketing.
American CryoStem received an FDA warning letter Jan. 3 demanding that the company comply with best-manufacturing processes and obtain an investigational new drug application if it wishes to continue marketing ATCELL for its currently advertised clinical indications and administration routes. These include intravenous, intrathecal, or aerosol inhalation of the product for anoxic brain injury, Parkinson’s disease, amyotrophic lateral sclerosis, stroke, and multiple sclerosis.
“Please be advised that, to lawfully market a drug that is a biological product, a valid biologics license must be in effect,” noted the letter. “Such licenses are issued only after a showing that the product is safe, pure, and potent. While in the development stage, such products may be distributed for clinical use in humans only if the sponsor has an investigational new drug application (IND) in effect as specified by FDA regulations. ATCELL is not the subject of an approved biologics license application nor is there an IND in effect. Based on this information, we have determined that your actions have violated the Food, Drug, and Cosmetic Act and the Public Health Service Act.”
FDA inspectors conducted a site inspection of American CryoStem in Eatontown, N.J., last summer, during which they “documented evidence of significant deviations from current good manufacturing practice.” The agency then provided the company a chance to respond to these issues. The new warning letter discussed each complaint, noting that some were inadequately addressed, and demanded that the company take action within 15 working days or face potential legal process, including seizure and/or injunction.
American CryoStem is one of the first companies to experience increased scrutiny under FDA’s new commitment to regulate the rapid growth and development of regenerative medicine products, which include novel cellular therapies, with the aim of ensuring their safety and effectiveness.
The new policy is designed to support the potential of cellular rejuvenation medicine, while protecting patients from “unscrupulous actors” who might endanger public health with untested products, according to FDA Commissioner Scott Gottlieb, MD. As enthusiasm for stem cell treatments surges, so are reports of adverse events. The New England Journal of Medicine recently reported on three patients with age-related macular degeneration who were blinded by intravitreal injection of autologous adipose-derived stem cells (N Engl J Med. 2017;376:1047-53).
Under the new policy, cell- and tissue-based products could be exempt from FDA premarket review only if they are removed from and implanted back into the same patient in their original form, or if the products are “minimally manipulated.” ATCELL fulfills neither qualification, the FDA warning letter said.
“You process adipose tissue ... to isolate cellular components of adipose tissue, commonly referred to as stromal vascular fraction [SVF]. Such processing is more than minimal manipulation because [it alters] the original relevant characteristics of the [tissue] relating to its utility for reconstruction, repair, or replacement. Then you process the SVF by expanding it in cell culture to manufacture ATCELL. Such expansion also is more than minimal manipulation because it alters the original relevant characteristics of the tissue.”
Furthermore, the letter noted, at least one of the components used in the clonal expansion process is investigational and not intended for human use. The manufacturer of that component, which was not named, “indicates the following: ‘Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to human and animals.”
The FDA also took exception with several equipment and lab safety issues. ATCELL was being created in areas that had no clean space designation – a serious concern, the letter said.
“American CryoStem’s unvalidated processes, inadequately controlled environment, lack of control of components used in production, and lack of sufficient and validated product testing ... pose a significant risk that ATCELL may be contaminated with microorganisms or have other serious product quality defects ... Because the product is administered to humans by various higher risk routes of administration, including intravenously, intrathecally, and by aerosol inhalation, if contaminated, its use could cause a range of adverse events, from infections to death.”
FDA also expressed concerns over a lack of consistent quality control testing of each batch and questioned whether the company’s method of shipping ATCELL to clinicians had been adequately validated.
Finally, the agency raised concerns that ATCELL, while it is labeled as being for research purposes only, may harm patients indirectly by preventing them from seeking timely treatment with proven therapies.
“ATCELL is intended to treat a variety of serious or life-threatening diseases or conditions, all of which are non-homologous uses,” the warning letter noted. “Such uses raise potential significant safety concerns because there is less basis on which to predict the product’s behavior in the recipient, and use of these unapproved products may cause users to delay or discontinue medical treatments that have been found safe and effective.”
SOURCE: FDA warning letter
MRI Reveals Lymphatic Vessels in Dura
Researchers have visualized lymphatic vessels in the dura mater of humans on MRI, according to a short report published October 3, 2017, in eLife. They also have identified lymphatic vessels in brain tissue samples using immunostaining. The results suggest that the vessels could act as a pipeline between the brain and the immune system.
“Overall, our data clearly and consistently demonstrate the existence of lymphatic vessels within the dura mater of human and nonhuman primates,” said Daniel S. Reich, MD, PhD, Senior Investigator at the NINDS, and colleagues. “The ability to image the meningeal lymphatics noninvasively immediately suggests the possibility of studying potential abnormalities” in neurologic disorders, they said.

A Fundamental Shift
In most of the body, lymphatic vessels transport immune cells and waste products from organs to the bloodstream, but the brain was thought not to have lymphatic vessels. In 2015, however, researchers found evidence of the brain’s lymphatic system in the dura of mice. Dr. Reich saw a presentation by an author of one the mouse studies, Jonathan Kipnis, PhD, Chair of the Department of Neuroscience at the University of Virginia in Charlottesville, and “was completely surprised.”
“In medical school, we were taught that the brain has no lymphatic system,” Dr. Reich said. “After Dr. Kipnis’s talk, I thought maybe we could find it in human brains.”
Dr. Reich and colleagues scanned the brains of five healthy volunteers who had been injected with gadobutrol, a dye used during MRI scans to visualize brain blood vessels. Gadobutrol that had leaked out of blood vessels in the dura as part of a normal process collected inside lymphatic vessels in the dura and showed up as bright white lines on MRI. “We watched people’s brains drain fluid into these vessels,” said Dr. Reich. When they repeated the experiment using a different dye that leaks much less out of blood vessels (ie, gadofosveset), the lymphatic vessels did not appear on imaging.
Similar findings were observed in monkeys.
The lymphatic vessels had been difficult to identify because they resemble blood vessels, which are far more numerous, the researchers said.
“These results could fundamentally change the way we think about how the brain and immune system interrelate,” said Walter J. Koroshetz, MD, NINDS director.
Meningeal Lymphatic Network
MRI showed collection of interstitial gadolinium within dural lymphatic vessels in all five of the healthy volunteers (ages 28 to 53, three women) and all three of the common marmoset monkeys studied. The vessels had a maximum apparent diameter of approximately 1 mm. “Our results suggest that in the dura, similar to many other organs throughout the body, small intravascular molecules extravasate into the interstitium and then, under a hydrostatic pressure gradient, collect into lymphatic capillaries through a loose lymphatic endothelium,” the researchers said. “On 3D rendering of subtraction MRI images, dural lymphatics are seen running parallel to the dural venous sinuses, especially the superior sagittal and straight sinuses, and alongside branches of the middle meningeal artery. The topography of the meningeal lymphatics fits with the previously described network in rodents.”
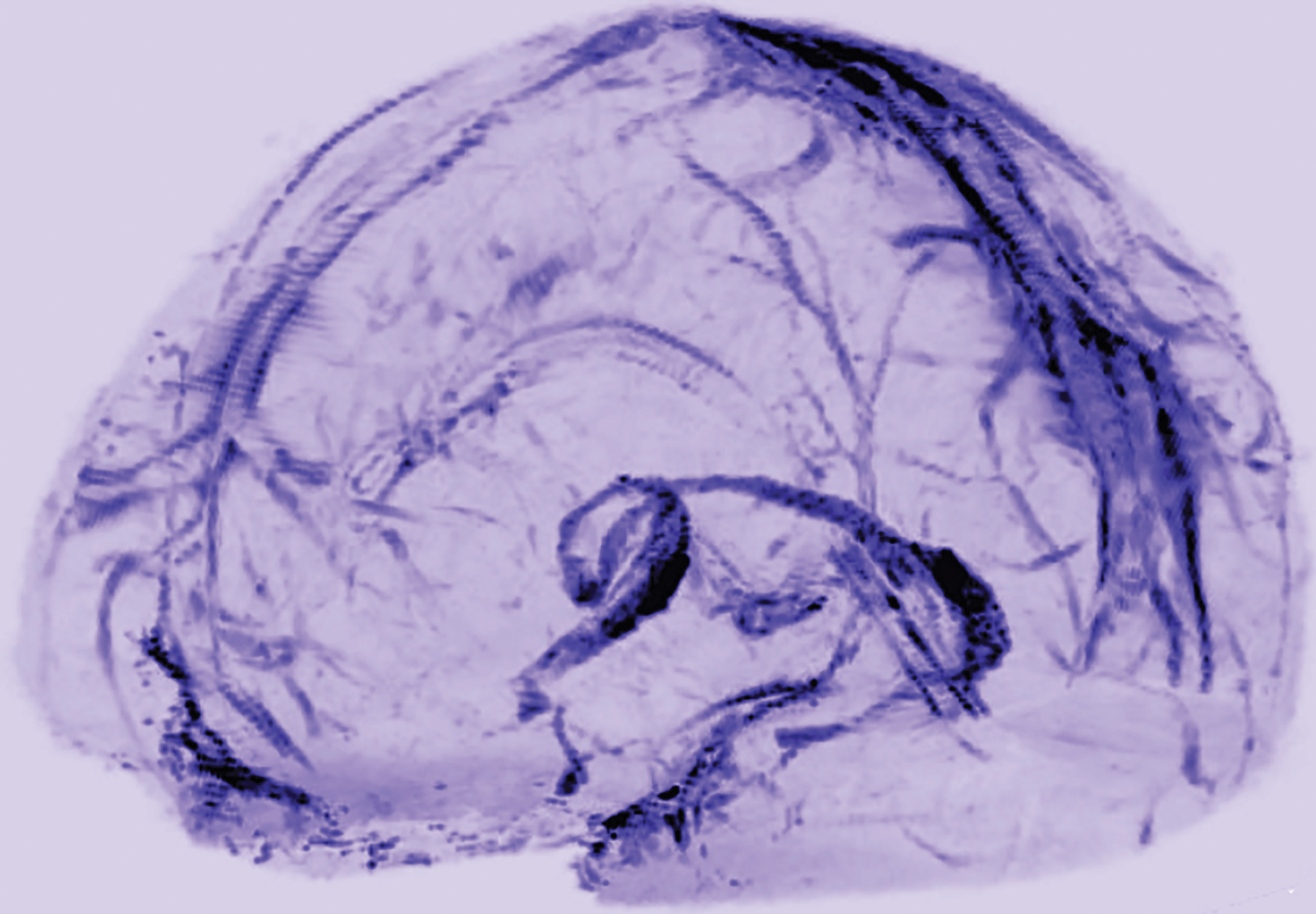
Although MRI shows large, slow-flow lymphatic ducts, “blind-ending and small lymphatic capillaries, clearly seen by histopathology, are unlikely to be revealed by MRI,” the researchers noted. In addition, they “could not prove whether dural lymphatic vessels drain immune cells, CSF, or other substances from the brain to deep cervical lymph nodes” or assess any link with the glymphatic system. “A comprehensive map of the meningeal lymphatic network would have implications for unraveling the ways in which the meningeal lymphatics participate in waste clearance and in immune cell trafficking within the CNS,” the researchers said.
Neuropathologic evaluation focused on dura samples from two formalin-fixed brains (from patients ages 60 and 77 with longstanding progressive multiple sclerosis) and from a 33-year-old patient with refractory epilepsy undergoing anterior temporal lobectomy.
Future studies may examine the role that dural lymphatics play in inflammatory pathologic conditions. The researchers have observed “clusters of extravascular CD3+ lymphocytes and CD68+ phagocytic meningeal macrophages … in the dura of several multiple sclerosis autopsies, confirming intense immune cell trafficking and communication.” Furthermore, “lymphatic dysfunction might impair waste clearance in neurodegenerative diseases and aging, in line with the recently captured deposition of β-amyloid in human dura in elderly people,” the researchers said.
—Jake Remaly
Suggested Reading
Absinta M, Ha SK, Nair G, et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife. 2017 Oct 3;6:e29738.
Researchers have visualized lymphatic vessels in the dura mater of humans on MRI, according to a short report published October 3, 2017, in eLife. They also have identified lymphatic vessels in brain tissue samples using immunostaining. The results suggest that the vessels could act as a pipeline between the brain and the immune system.
“Overall, our data clearly and consistently demonstrate the existence of lymphatic vessels within the dura mater of human and nonhuman primates,” said Daniel S. Reich, MD, PhD, Senior Investigator at the NINDS, and colleagues. “The ability to image the meningeal lymphatics noninvasively immediately suggests the possibility of studying potential abnormalities” in neurologic disorders, they said.
A Fundamental Shift
In most of the body, lymphatic vessels transport immune cells and waste products from organs to the bloodstream, but the brain was thought not to have lymphatic vessels. In 2015, however, researchers found evidence of the brain’s lymphatic system in the dura of mice. Dr. Reich saw a presentation by an author of one the mouse studies, Jonathan Kipnis, PhD, Chair of the Department of Neuroscience at the University of Virginia in Charlottesville, and “was completely surprised.”
“In medical school, we were taught that the brain has no lymphatic system,” Dr. Reich said. “After Dr. Kipnis’s talk, I thought maybe we could find it in human brains.”
Dr. Reich and colleagues scanned the brains of five healthy volunteers who had been injected with gadobutrol, a dye used during MRI scans to visualize brain blood vessels. Gadobutrol that had leaked out of blood vessels in the dura as part of a normal process collected inside lymphatic vessels in the dura and showed up as bright white lines on MRI. “We watched people’s brains drain fluid into these vessels,” said Dr. Reich. When they repeated the experiment using a different dye that leaks much less out of blood vessels (ie, gadofosveset), the lymphatic vessels did not appear on imaging.
Similar findings were observed in monkeys.
The lymphatic vessels had been difficult to identify because they resemble blood vessels, which are far more numerous, the researchers said.
“These results could fundamentally change the way we think about how the brain and immune system interrelate,” said Walter J. Koroshetz, MD, NINDS director.
Meningeal Lymphatic Network
MRI showed collection of interstitial gadolinium within dural lymphatic vessels in all five of the healthy volunteers (ages 28 to 53, three women) and all three of the common marmoset monkeys studied. The vessels had a maximum apparent diameter of approximately 1 mm. “Our results suggest that in the dura, similar to many other organs throughout the body, small intravascular molecules extravasate into the interstitium and then, under a hydrostatic pressure gradient, collect into lymphatic capillaries through a loose lymphatic endothelium,” the researchers said. “On 3D rendering of subtraction MRI images, dural lymphatics are seen running parallel to the dural venous sinuses, especially the superior sagittal and straight sinuses, and alongside branches of the middle meningeal artery. The topography of the meningeal lymphatics fits with the previously described network in rodents.”
Although MRI shows large, slow-flow lymphatic ducts, “blind-ending and small lymphatic capillaries, clearly seen by histopathology, are unlikely to be revealed by MRI,” the researchers noted. In addition, they “could not prove whether dural lymphatic vessels drain immune cells, CSF, or other substances from the brain to deep cervical lymph nodes” or assess any link with the glymphatic system. “A comprehensive map of the meningeal lymphatic network would have implications for unraveling the ways in which the meningeal lymphatics participate in waste clearance and in immune cell trafficking within the CNS,” the researchers said.
Neuropathologic evaluation focused on dura samples from two formalin-fixed brains (from patients ages 60 and 77 with longstanding progressive multiple sclerosis) and from a 33-year-old patient with refractory epilepsy undergoing anterior temporal lobectomy.
Future studies may examine the role that dural lymphatics play in inflammatory pathologic conditions. The researchers have observed “clusters of extravascular CD3+ lymphocytes and CD68+ phagocytic meningeal macrophages … in the dura of several multiple sclerosis autopsies, confirming intense immune cell trafficking and communication.” Furthermore, “lymphatic dysfunction might impair waste clearance in neurodegenerative diseases and aging, in line with the recently captured deposition of β-amyloid in human dura in elderly people,” the researchers said.
—Jake Remaly
Suggested Reading
Absinta M, Ha SK, Nair G, et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife. 2017 Oct 3;6:e29738.
Researchers have visualized lymphatic vessels in the dura mater of humans on MRI, according to a short report published October 3, 2017, in eLife. They also have identified lymphatic vessels in brain tissue samples using immunostaining. The results suggest that the vessels could act as a pipeline between the brain and the immune system.
“Overall, our data clearly and consistently demonstrate the existence of lymphatic vessels within the dura mater of human and nonhuman primates,” said Daniel S. Reich, MD, PhD, Senior Investigator at the NINDS, and colleagues. “The ability to image the meningeal lymphatics noninvasively immediately suggests the possibility of studying potential abnormalities” in neurologic disorders, they said.
A Fundamental Shift
In most of the body, lymphatic vessels transport immune cells and waste products from organs to the bloodstream, but the brain was thought not to have lymphatic vessels. In 2015, however, researchers found evidence of the brain’s lymphatic system in the dura of mice. Dr. Reich saw a presentation by an author of one the mouse studies, Jonathan Kipnis, PhD, Chair of the Department of Neuroscience at the University of Virginia in Charlottesville, and “was completely surprised.”
“In medical school, we were taught that the brain has no lymphatic system,” Dr. Reich said. “After Dr. Kipnis’s talk, I thought maybe we could find it in human brains.”
Dr. Reich and colleagues scanned the brains of five healthy volunteers who had been injected with gadobutrol, a dye used during MRI scans to visualize brain blood vessels. Gadobutrol that had leaked out of blood vessels in the dura as part of a normal process collected inside lymphatic vessels in the dura and showed up as bright white lines on MRI. “We watched people’s brains drain fluid into these vessels,” said Dr. Reich. When they repeated the experiment using a different dye that leaks much less out of blood vessels (ie, gadofosveset), the lymphatic vessels did not appear on imaging.
Similar findings were observed in monkeys.
The lymphatic vessels had been difficult to identify because they resemble blood vessels, which are far more numerous, the researchers said.
“These results could fundamentally change the way we think about how the brain and immune system interrelate,” said Walter J. Koroshetz, MD, NINDS director.
Meningeal Lymphatic Network
MRI showed collection of interstitial gadolinium within dural lymphatic vessels in all five of the healthy volunteers (ages 28 to 53, three women) and all three of the common marmoset monkeys studied. The vessels had a maximum apparent diameter of approximately 1 mm. “Our results suggest that in the dura, similar to many other organs throughout the body, small intravascular molecules extravasate into the interstitium and then, under a hydrostatic pressure gradient, collect into lymphatic capillaries through a loose lymphatic endothelium,” the researchers said. “On 3D rendering of subtraction MRI images, dural lymphatics are seen running parallel to the dural venous sinuses, especially the superior sagittal and straight sinuses, and alongside branches of the middle meningeal artery. The topography of the meningeal lymphatics fits with the previously described network in rodents.”
Although MRI shows large, slow-flow lymphatic ducts, “blind-ending and small lymphatic capillaries, clearly seen by histopathology, are unlikely to be revealed by MRI,” the researchers noted. In addition, they “could not prove whether dural lymphatic vessels drain immune cells, CSF, or other substances from the brain to deep cervical lymph nodes” or assess any link with the glymphatic system. “A comprehensive map of the meningeal lymphatic network would have implications for unraveling the ways in which the meningeal lymphatics participate in waste clearance and in immune cell trafficking within the CNS,” the researchers said.
Neuropathologic evaluation focused on dura samples from two formalin-fixed brains (from patients ages 60 and 77 with longstanding progressive multiple sclerosis) and from a 33-year-old patient with refractory epilepsy undergoing anterior temporal lobectomy.
Future studies may examine the role that dural lymphatics play in inflammatory pathologic conditions. The researchers have observed “clusters of extravascular CD3+ lymphocytes and CD68+ phagocytic meningeal macrophages … in the dura of several multiple sclerosis autopsies, confirming intense immune cell trafficking and communication.” Furthermore, “lymphatic dysfunction might impair waste clearance in neurodegenerative diseases and aging, in line with the recently captured deposition of β-amyloid in human dura in elderly people,” the researchers said.
—Jake Remaly
Suggested Reading
Absinta M, Ha SK, Nair G, et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife. 2017 Oct 3;6:e29738.
Ibudilast May Slow Brain Atrophy in Progressive MS
PARIS—Ibudilast decreases the rate of brain atrophy, compared with placebo, in patients with progressive multiple sclerosis (MS), according to top-line results presented at the Seventh Joint ECTRIMS–ACTRIMS Meeting. The drug appears to be safe and tolerable.
Ibudilast is an orally bioavailable small-molecule drug that is available in Japan for treating asthma and post stroke dizziness. In a phase II trial that included patients with relapsing-remitting MS, ibudilast slowed brain atrophy progression in a dose-dependent manner. In addition, animal models suggest that the drug may promote neuroprotection.
Examining Ibudilast in a Phase II Trial
Robert Fox, MD, a neurologist at the Cleveland Clinic, and colleagues conducted a phase II clinical trial at 28 sites to analyze ibudilast’s effects in patients with primary or secondary progressive MS. Eligible patients were between ages 18 and 65, had a maximum Expanded Disability Status Scale (EDSS) score of 6.5, had disability progression in the previous two years, and were allowed to receive concurrent treatment with interferon beta or glatiramer acetate.

Patients were randomized in equal groups to placebo or ibudilast. The intervention group received 60 mg/day, 80 mg/day, or 100 mg/day of ibudilast as tolerated. Randomization was stratified by primary or secondary progressive MS and by whether the subjects received injectable disease-modifying therapies. For the first three months of treatment, patients presented for monthly safety evaluations. From the third month onward, patients returned quarterly for safety assessments and every six months for efficacy assessments, which included clinical disability and 3-T MRI measures. The treatment period was two years.
The trial’s primary end points were brain atrophy, as measured by brain parenchymal fraction; safety; and tolerability. Secondary outcomes included magnetization transfer ratio (MTR), diffusion tensor imaging (DTI), optical coherence tomography of the retinal nerve fiber layer, and cortical atrophy.
Researchers Observed Gastrointestinal Side Effects
The investigators enrolled 255 patients into the study. The population’s mean age was approximately 56, and mean EDSS was about 5. The study sample had approximately equal numbers of patients with primary progressive MS and secondary progressive MS. Eleven participants withdrew from the study before the first efficacy assessment, and another 24 participants discontinued the trial before week 96. The trial’s retention rate was 86%.
In the primary analysis, ibudilast was associated with a 48% reduction in the progression of brain atrophy over two years, compared with placebo. A per-protocol sensitivity analysis confirmed that ibudilast reduced the progression of brain atrophy by half, compared with placebo.
Treatment-related adverse events included gastrointestinal side effects such as nausea, vomiting, diarrhea, and abdominal pain. Patients treated with ibudilast also had higher rates of rash, depression, and fatigue than did controls. The rate of serious adverse events was similar in both groups. There were no related and unanticipated serious adverse events.
There was no significant difference in the rates of treatment discontinuation, early study termination, and early drug withdrawal between the two study arms. Approximately 25% of controls discontinued treatment during the study, compared with 30% of the ibudilast group.
Compared with placebo, ibudilast was associated with a significant slowing of the progression of MTR in normal-appearing brain tissue and normal-appearing gray matter. The researchers did not find a significant difference between treatment groups on DTI. Additional analyses of the secondary and tertiary outcomes are forthcoming, said Dr. Fox.
—Erik Greb
Suggested Reading
Barkhof F, Hulst HE, Drulovic J, et al. Ibudilast in relapsing-remitting multiple sclerosis: a neuroprotectant? Neurology. 2010;74(13):1033-1040.
Fox RJ, Coffey CS, Cudkowicz ME, et al. Design, rationale, and baseline characteristics of the randomized double-blind phase II clinical trial of ibudilast in progressive multiple sclerosis. Contemp Clin Trials. 2016;50:166-177.
PARIS—Ibudilast decreases the rate of brain atrophy, compared with placebo, in patients with progressive multiple sclerosis (MS), according to top-line results presented at the Seventh Joint ECTRIMS–ACTRIMS Meeting. The drug appears to be safe and tolerable.
Ibudilast is an orally bioavailable small-molecule drug that is available in Japan for treating asthma and post stroke dizziness. In a phase II trial that included patients with relapsing-remitting MS, ibudilast slowed brain atrophy progression in a dose-dependent manner. In addition, animal models suggest that the drug may promote neuroprotection.
Examining Ibudilast in a Phase II Trial
Robert Fox, MD, a neurologist at the Cleveland Clinic, and colleagues conducted a phase II clinical trial at 28 sites to analyze ibudilast’s effects in patients with primary or secondary progressive MS. Eligible patients were between ages 18 and 65, had a maximum Expanded Disability Status Scale (EDSS) score of 6.5, had disability progression in the previous two years, and were allowed to receive concurrent treatment with interferon beta or glatiramer acetate.
Patients were randomized in equal groups to placebo or ibudilast. The intervention group received 60 mg/day, 80 mg/day, or 100 mg/day of ibudilast as tolerated. Randomization was stratified by primary or secondary progressive MS and by whether the subjects received injectable disease-modifying therapies. For the first three months of treatment, patients presented for monthly safety evaluations. From the third month onward, patients returned quarterly for safety assessments and every six months for efficacy assessments, which included clinical disability and 3-T MRI measures. The treatment period was two years.
The trial’s primary end points were brain atrophy, as measured by brain parenchymal fraction; safety; and tolerability. Secondary outcomes included magnetization transfer ratio (MTR), diffusion tensor imaging (DTI), optical coherence tomography of the retinal nerve fiber layer, and cortical atrophy.
Researchers Observed Gastrointestinal Side Effects
The investigators enrolled 255 patients into the study. The population’s mean age was approximately 56, and mean EDSS was about 5. The study sample had approximately equal numbers of patients with primary progressive MS and secondary progressive MS. Eleven participants withdrew from the study before the first efficacy assessment, and another 24 participants discontinued the trial before week 96. The trial’s retention rate was 86%.
In the primary analysis, ibudilast was associated with a 48% reduction in the progression of brain atrophy over two years, compared with placebo. A per-protocol sensitivity analysis confirmed that ibudilast reduced the progression of brain atrophy by half, compared with placebo.
Treatment-related adverse events included gastrointestinal side effects such as nausea, vomiting, diarrhea, and abdominal pain. Patients treated with ibudilast also had higher rates of rash, depression, and fatigue than did controls. The rate of serious adverse events was similar in both groups. There were no related and unanticipated serious adverse events.
There was no significant difference in the rates of treatment discontinuation, early study termination, and early drug withdrawal between the two study arms. Approximately 25% of controls discontinued treatment during the study, compared with 30% of the ibudilast group.
Compared with placebo, ibudilast was associated with a significant slowing of the progression of MTR in normal-appearing brain tissue and normal-appearing gray matter. The researchers did not find a significant difference between treatment groups on DTI. Additional analyses of the secondary and tertiary outcomes are forthcoming, said Dr. Fox.
—Erik Greb
Suggested Reading
Barkhof F, Hulst HE, Drulovic J, et al. Ibudilast in relapsing-remitting multiple sclerosis: a neuroprotectant? Neurology. 2010;74(13):1033-1040.
Fox RJ, Coffey CS, Cudkowicz ME, et al. Design, rationale, and baseline characteristics of the randomized double-blind phase II clinical trial of ibudilast in progressive multiple sclerosis. Contemp Clin Trials. 2016;50:166-177.
PARIS—Ibudilast decreases the rate of brain atrophy, compared with placebo, in patients with progressive multiple sclerosis (MS), according to top-line results presented at the Seventh Joint ECTRIMS–ACTRIMS Meeting. The drug appears to be safe and tolerable.
Ibudilast is an orally bioavailable small-molecule drug that is available in Japan for treating asthma and post stroke dizziness. In a phase II trial that included patients with relapsing-remitting MS, ibudilast slowed brain atrophy progression in a dose-dependent manner. In addition, animal models suggest that the drug may promote neuroprotection.
Examining Ibudilast in a Phase II Trial
Robert Fox, MD, a neurologist at the Cleveland Clinic, and colleagues conducted a phase II clinical trial at 28 sites to analyze ibudilast’s effects in patients with primary or secondary progressive MS. Eligible patients were between ages 18 and 65, had a maximum Expanded Disability Status Scale (EDSS) score of 6.5, had disability progression in the previous two years, and were allowed to receive concurrent treatment with interferon beta or glatiramer acetate.
Patients were randomized in equal groups to placebo or ibudilast. The intervention group received 60 mg/day, 80 mg/day, or 100 mg/day of ibudilast as tolerated. Randomization was stratified by primary or secondary progressive MS and by whether the subjects received injectable disease-modifying therapies. For the first three months of treatment, patients presented for monthly safety evaluations. From the third month onward, patients returned quarterly for safety assessments and every six months for efficacy assessments, which included clinical disability and 3-T MRI measures. The treatment period was two years.
The trial’s primary end points were brain atrophy, as measured by brain parenchymal fraction; safety; and tolerability. Secondary outcomes included magnetization transfer ratio (MTR), diffusion tensor imaging (DTI), optical coherence tomography of the retinal nerve fiber layer, and cortical atrophy.
Researchers Observed Gastrointestinal Side Effects
The investigators enrolled 255 patients into the study. The population’s mean age was approximately 56, and mean EDSS was about 5. The study sample had approximately equal numbers of patients with primary progressive MS and secondary progressive MS. Eleven participants withdrew from the study before the first efficacy assessment, and another 24 participants discontinued the trial before week 96. The trial’s retention rate was 86%.
In the primary analysis, ibudilast was associated with a 48% reduction in the progression of brain atrophy over two years, compared with placebo. A per-protocol sensitivity analysis confirmed that ibudilast reduced the progression of brain atrophy by half, compared with placebo.
Treatment-related adverse events included gastrointestinal side effects such as nausea, vomiting, diarrhea, and abdominal pain. Patients treated with ibudilast also had higher rates of rash, depression, and fatigue than did controls. The rate of serious adverse events was similar in both groups. There were no related and unanticipated serious adverse events.
There was no significant difference in the rates of treatment discontinuation, early study termination, and early drug withdrawal between the two study arms. Approximately 25% of controls discontinued treatment during the study, compared with 30% of the ibudilast group.
Compared with placebo, ibudilast was associated with a significant slowing of the progression of MTR in normal-appearing brain tissue and normal-appearing gray matter. The researchers did not find a significant difference between treatment groups on DTI. Additional analyses of the secondary and tertiary outcomes are forthcoming, said Dr. Fox.
—Erik Greb
Suggested Reading
Barkhof F, Hulst HE, Drulovic J, et al. Ibudilast in relapsing-remitting multiple sclerosis: a neuroprotectant? Neurology. 2010;74(13):1033-1040.
Fox RJ, Coffey CS, Cudkowicz ME, et al. Design, rationale, and baseline characteristics of the randomized double-blind phase II clinical trial of ibudilast in progressive multiple sclerosis. Contemp Clin Trials. 2016;50:166-177.
FDA: Gadolinium retention prompts new GBCA class warning, safety measures
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.

Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
sworcester@frontlinemedcom.com
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
sworcester@frontlinemedcom.com
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
sworcester@frontlinemedcom.com
FDA: Gadolinium retention prompts new GBCA class warning, safety measures
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
Ozanimod Is Superior to Interferon in Relapsing-Remitting MS
PARIS—Ozanimod is superior to interferon beta-1a on clinical and MRI measures in patients with relapsing-remitting multiple sclerosis (MS), according to the results of two studies presented at the Seventh Joint ECTRIMS–ACTRIMS Meeting. The drug could become a safe and effective oral medication for this population, said the investigators.
Scientists at the Scripps Research Institute developed ozanimod, a selective sphingosine 1-phosphate 1 and 5 receptor modulator. “Receptor specificity for subtypes 1 and 5 is anticipated to preserve efficacy, but to lessen off-target treatment effects, as compared to nonselective sphingosine 1-phosphate receptor modulators,” said Jeffrey Cohen, MD, a neurologist at the Cleveland Clinic and an investigator on both of the studies.

The SUNBEAM Study
SUNBEAM was the first of the two trials of ozanimod to be presented at the meeting. Eligible participants were between ages 18 and 55, had relapsing-remitting MS, had had at least one documented relapse in the previous year, had not had a relapse in the previous 30 days, and had no history of cardiac conditions. The trial randomized participants to 0.5 mg/day of oral ozanimod, 1 mg/day of oral ozanimod, or weekly intramuscular injections of interferon beta-1a (30 µg). Patients in the ozanimod groups underwent a seven-day dose escalation as an initiation.
The trial’s primary end point was annualized relapse rate. Among the secondary end points were new and enlarging T2 lesions from baseline to month 12 and T1, gadolinium-enhancing lesions at month 12. The investigators carried out a prespecified evaluation of disability progression, based on Expanded Disability Status Scale (EDSS), using a pooled analysis of SUNBEAM and RADIANCE, the other phase III study.
The researchers enrolled 1,346 patients in 20 countries. The treatment groups were well balanced at baseline. Participants’ mean age was 36, and 66% of the population was female. Mean EDSS score at baseline was 2.6, mean number of relapses in the prior year was 1.3, and 47% of participants had gadolinium-enhancing lesions. In addition, 31% of participants previously had been treated with disease-modifying therapy.
The mean treatment duration in SUNBEAM was 13.6 months. The annualized relapse rate was reduced by 31% with 0.5 mg/day of ozanimod and by 48% with 1 mg/day of ozanimod, compared with interferon beta-1a. Overall, ozanimod reduced MRI activity by 63%, compared with interferon beta-1a. The number of new or enlarging T2 lesions and the adjusted mean number of gadolinium-enhancing lesions at month 12 were significantly reduced in the ozanimod groups, compared with the interferon group. The investigators observed a consistent dose response to ozanimod for all efficacy end points.
The majority of treatment-emergent adverse events were mild, and serious adverse events were uncommon. The rate of adverse events was similar for all treatment groups. The rate of discontinuation due to adverse events also was low and similar between treatment groups. The researchers observed no first-dose, clinically relevant cases of bradycardia.
The RADIANCE Trial
The eligibility criteria for the RADIANCE trial were the same as those of the SUNBEAM study. Participants were randomized to 0.5-mg/day or 1-mg/day doses of ozanimod or to 30 µg/week of interferon beta-1a. The primary end point in RADIANCE was annualized relapse rate at two years. Secondary end points included measures of MRI lesion activity, brain volume loss over two years, and a pooled analysis of confirmed disability worsening. Disability was assessed during a clinical evaluation every three months and at the time of potential relapse.
The investigators enrolled 1,313 patients into the study, and their baseline characteristics were similar across treatment groups. Participants’ mean age was 36, and 67% of the population was female. Mean EDSS score at baseline was 2.5, and mean number of relapses in the prior year was 1.3. About 43% of patients had gadolinium-enhancing lesions at baseline, and 29% previously had received disease-modifying therapy. Overall, 86% of the participants completed the trial, and the discontinuation rate was similar in all treatment arms.
The 0.5-mg dose of ozanimod was associated with a 21% reduction in annualized relapse rate, compared with interferon, and the 1-mg dose of ozanimod was associated with a 38% reduction in this measure. The adjusted mean number of new or enlarging T2 lesions per scan was reduced by 42% with ozanimod 1 mg and by 35% with 0.5 mg of ozanimod, compared with interferon. The adjusted mean number of gadolinium-enhancing lesions at 24 months also was reduced by 53% with 1 mg of ozanimod and by 47% with 0.5 mg of ozanimod, compared with interferon.
In addition, the study demonstrated an approximately 25% reduction in whole-brain volume loss in the two ozanimod arms, compared with the interferon group. Ozanimod also reduced segmented cortical gray volume loss and segmented thalamic volume loss to a similar extent, compared with interferon.
In the pooled analysis of both trials, the rate of confirmed disability worsening was low. The investigators did not observe any difference in this outcome between the three treatment arms.
Ozanimod was well tolerated. Liver enzyme elevations associated with the drug tended to be mild and resolved despite continuation of study drug. The drug was associated with a minimal change in heart rate, and the researchers observed no cases of second-degree or higher heart block. “It appears that the first-dose escalation protocol effectively mitigated cardiac effects,” said Dr. Cohen.
Celgene, the company developing the drug, expects to submit the compound for FDA approval and introduce it to the market by the end of 2018, according to a press release.
—Erik Greb
Suggested Reading
Cohen JA, Arnold DL, Comi G, et al. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(4):373-381.
Sørensen PS. Ozanimod: a better or just another S1P receptor modulator? Lancet Neurol. 2016;15(4):345-347.
PARIS—Ozanimod is superior to interferon beta-1a on clinical and MRI measures in patients with relapsing-remitting multiple sclerosis (MS), according to the results of two studies presented at the Seventh Joint ECTRIMS–ACTRIMS Meeting. The drug could become a safe and effective oral medication for this population, said the investigators.
Scientists at the Scripps Research Institute developed ozanimod, a selective sphingosine 1-phosphate 1 and 5 receptor modulator. “Receptor specificity for subtypes 1 and 5 is anticipated to preserve efficacy, but to lessen off-target treatment effects, as compared to nonselective sphingosine 1-phosphate receptor modulators,” said Jeffrey Cohen, MD, a neurologist at the Cleveland Clinic and an investigator on both of the studies.
The SUNBEAM Study
SUNBEAM was the first of the two trials of ozanimod to be presented at the meeting. Eligible participants were between ages 18 and 55, had relapsing-remitting MS, had had at least one documented relapse in the previous year, had not had a relapse in the previous 30 days, and had no history of cardiac conditions. The trial randomized participants to 0.5 mg/day of oral ozanimod, 1 mg/day of oral ozanimod, or weekly intramuscular injections of interferon beta-1a (30 µg). Patients in the ozanimod groups underwent a seven-day dose escalation as an initiation.
The trial’s primary end point was annualized relapse rate. Among the secondary end points were new and enlarging T2 lesions from baseline to month 12 and T1, gadolinium-enhancing lesions at month 12. The investigators carried out a prespecified evaluation of disability progression, based on Expanded Disability Status Scale (EDSS), using a pooled analysis of SUNBEAM and RADIANCE, the other phase III study.
The researchers enrolled 1,346 patients in 20 countries. The treatment groups were well balanced at baseline. Participants’ mean age was 36, and 66% of the population was female. Mean EDSS score at baseline was 2.6, mean number of relapses in the prior year was 1.3, and 47% of participants had gadolinium-enhancing lesions. In addition, 31% of participants previously had been treated with disease-modifying therapy.
The mean treatment duration in SUNBEAM was 13.6 months. The annualized relapse rate was reduced by 31% with 0.5 mg/day of ozanimod and by 48% with 1 mg/day of ozanimod, compared with interferon beta-1a. Overall, ozanimod reduced MRI activity by 63%, compared with interferon beta-1a. The number of new or enlarging T2 lesions and the adjusted mean number of gadolinium-enhancing lesions at month 12 were significantly reduced in the ozanimod groups, compared with the interferon group. The investigators observed a consistent dose response to ozanimod for all efficacy end points.
The majority of treatment-emergent adverse events were mild, and serious adverse events were uncommon. The rate of adverse events was similar for all treatment groups. The rate of discontinuation due to adverse events also was low and similar between treatment groups. The researchers observed no first-dose, clinically relevant cases of bradycardia.
The RADIANCE Trial
The eligibility criteria for the RADIANCE trial were the same as those of the SUNBEAM study. Participants were randomized to 0.5-mg/day or 1-mg/day doses of ozanimod or to 30 µg/week of interferon beta-1a. The primary end point in RADIANCE was annualized relapse rate at two years. Secondary end points included measures of MRI lesion activity, brain volume loss over two years, and a pooled analysis of confirmed disability worsening. Disability was assessed during a clinical evaluation every three months and at the time of potential relapse.
The investigators enrolled 1,313 patients into the study, and their baseline characteristics were similar across treatment groups. Participants’ mean age was 36, and 67% of the population was female. Mean EDSS score at baseline was 2.5, and mean number of relapses in the prior year was 1.3. About 43% of patients had gadolinium-enhancing lesions at baseline, and 29% previously had received disease-modifying therapy. Overall, 86% of the participants completed the trial, and the discontinuation rate was similar in all treatment arms.
The 0.5-mg dose of ozanimod was associated with a 21% reduction in annualized relapse rate, compared with interferon, and the 1-mg dose of ozanimod was associated with a 38% reduction in this measure. The adjusted mean number of new or enlarging T2 lesions per scan was reduced by 42% with ozanimod 1 mg and by 35% with 0.5 mg of ozanimod, compared with interferon. The adjusted mean number of gadolinium-enhancing lesions at 24 months also was reduced by 53% with 1 mg of ozanimod and by 47% with 0.5 mg of ozanimod, compared with interferon.
In addition, the study demonstrated an approximately 25% reduction in whole-brain volume loss in the two ozanimod arms, compared with the interferon group. Ozanimod also reduced segmented cortical gray volume loss and segmented thalamic volume loss to a similar extent, compared with interferon.
In the pooled analysis of both trials, the rate of confirmed disability worsening was low. The investigators did not observe any difference in this outcome between the three treatment arms.
Ozanimod was well tolerated. Liver enzyme elevations associated with the drug tended to be mild and resolved despite continuation of study drug. The drug was associated with a minimal change in heart rate, and the researchers observed no cases of second-degree or higher heart block. “It appears that the first-dose escalation protocol effectively mitigated cardiac effects,” said Dr. Cohen.
Celgene, the company developing the drug, expects to submit the compound for FDA approval and introduce it to the market by the end of 2018, according to a press release.
—Erik Greb
Suggested Reading
Cohen JA, Arnold DL, Comi G, et al. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(4):373-381.
Sørensen PS. Ozanimod: a better or just another S1P receptor modulator? Lancet Neurol. 2016;15(4):345-347.
PARIS—Ozanimod is superior to interferon beta-1a on clinical and MRI measures in patients with relapsing-remitting multiple sclerosis (MS), according to the results of two studies presented at the Seventh Joint ECTRIMS–ACTRIMS Meeting. The drug could become a safe and effective oral medication for this population, said the investigators.
Scientists at the Scripps Research Institute developed ozanimod, a selective sphingosine 1-phosphate 1 and 5 receptor modulator. “Receptor specificity for subtypes 1 and 5 is anticipated to preserve efficacy, but to lessen off-target treatment effects, as compared to nonselective sphingosine 1-phosphate receptor modulators,” said Jeffrey Cohen, MD, a neurologist at the Cleveland Clinic and an investigator on both of the studies.
The SUNBEAM Study
SUNBEAM was the first of the two trials of ozanimod to be presented at the meeting. Eligible participants were between ages 18 and 55, had relapsing-remitting MS, had had at least one documented relapse in the previous year, had not had a relapse in the previous 30 days, and had no history of cardiac conditions. The trial randomized participants to 0.5 mg/day of oral ozanimod, 1 mg/day of oral ozanimod, or weekly intramuscular injections of interferon beta-1a (30 µg). Patients in the ozanimod groups underwent a seven-day dose escalation as an initiation.
The trial’s primary end point was annualized relapse rate. Among the secondary end points were new and enlarging T2 lesions from baseline to month 12 and T1, gadolinium-enhancing lesions at month 12. The investigators carried out a prespecified evaluation of disability progression, based on Expanded Disability Status Scale (EDSS), using a pooled analysis of SUNBEAM and RADIANCE, the other phase III study.
The researchers enrolled 1,346 patients in 20 countries. The treatment groups were well balanced at baseline. Participants’ mean age was 36, and 66% of the population was female. Mean EDSS score at baseline was 2.6, mean number of relapses in the prior year was 1.3, and 47% of participants had gadolinium-enhancing lesions. In addition, 31% of participants previously had been treated with disease-modifying therapy.
The mean treatment duration in SUNBEAM was 13.6 months. The annualized relapse rate was reduced by 31% with 0.5 mg/day of ozanimod and by 48% with 1 mg/day of ozanimod, compared with interferon beta-1a. Overall, ozanimod reduced MRI activity by 63%, compared with interferon beta-1a. The number of new or enlarging T2 lesions and the adjusted mean number of gadolinium-enhancing lesions at month 12 were significantly reduced in the ozanimod groups, compared with the interferon group. The investigators observed a consistent dose response to ozanimod for all efficacy end points.
The majority of treatment-emergent adverse events were mild, and serious adverse events were uncommon. The rate of adverse events was similar for all treatment groups. The rate of discontinuation due to adverse events also was low and similar between treatment groups. The researchers observed no first-dose, clinically relevant cases of bradycardia.
The RADIANCE Trial
The eligibility criteria for the RADIANCE trial were the same as those of the SUNBEAM study. Participants were randomized to 0.5-mg/day or 1-mg/day doses of ozanimod or to 30 µg/week of interferon beta-1a. The primary end point in RADIANCE was annualized relapse rate at two years. Secondary end points included measures of MRI lesion activity, brain volume loss over two years, and a pooled analysis of confirmed disability worsening. Disability was assessed during a clinical evaluation every three months and at the time of potential relapse.
The investigators enrolled 1,313 patients into the study, and their baseline characteristics were similar across treatment groups. Participants’ mean age was 36, and 67% of the population was female. Mean EDSS score at baseline was 2.5, and mean number of relapses in the prior year was 1.3. About 43% of patients had gadolinium-enhancing lesions at baseline, and 29% previously had received disease-modifying therapy. Overall, 86% of the participants completed the trial, and the discontinuation rate was similar in all treatment arms.
The 0.5-mg dose of ozanimod was associated with a 21% reduction in annualized relapse rate, compared with interferon, and the 1-mg dose of ozanimod was associated with a 38% reduction in this measure. The adjusted mean number of new or enlarging T2 lesions per scan was reduced by 42% with ozanimod 1 mg and by 35% with 0.5 mg of ozanimod, compared with interferon. The adjusted mean number of gadolinium-enhancing lesions at 24 months also was reduced by 53% with 1 mg of ozanimod and by 47% with 0.5 mg of ozanimod, compared with interferon.
In addition, the study demonstrated an approximately 25% reduction in whole-brain volume loss in the two ozanimod arms, compared with the interferon group. Ozanimod also reduced segmented cortical gray volume loss and segmented thalamic volume loss to a similar extent, compared with interferon.
In the pooled analysis of both trials, the rate of confirmed disability worsening was low. The investigators did not observe any difference in this outcome between the three treatment arms.
Ozanimod was well tolerated. Liver enzyme elevations associated with the drug tended to be mild and resolved despite continuation of study drug. The drug was associated with a minimal change in heart rate, and the researchers observed no cases of second-degree or higher heart block. “It appears that the first-dose escalation protocol effectively mitigated cardiac effects,” said Dr. Cohen.
Celgene, the company developing the drug, expects to submit the compound for FDA approval and introduce it to the market by the end of 2018, according to a press release.
—Erik Greb
Suggested Reading
Cohen JA, Arnold DL, Comi G, et al. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(4):373-381.
Sørensen PS. Ozanimod: a better or just another S1P receptor modulator? Lancet Neurol. 2016;15(4):345-347.
2017 MS Highlights: The Year in Review

Revised McDonald Criteria Allow Substitution for Dissemination in Time
PARIS—Recommended revisions to the 2010 McDonald diagnostic criteria for multiple sclerosis (MS) include changes that are intended to enable neurologists to diagnose MS sooner in patients with a high likelihood of the disease. One addition allows the use of CSF-specific oligoclonal bands in lieu of demonstration of dissemination in time to make a diagnosis of MS in patients with a clinically isolated syndrome and demonstration of dissemination in space clinically or by MRI. Symptomatic and cortical lesions also may satisfy diagnostic criteria, according to the recommendations. In addition, the revised criteria include guidance to reduce the risk of misdiagnosing MS.

Jeffrey A. Cohen, MD, a neurologist with the Cleveland Clinic’s Mellen Center for MS Treatment and Research, presented the 2017 proposed revisions at the Seventh Joint ECTRIMS–ACTRIMS Meeting.
Dr. Cohen and Alan J. Thompson, MD, consultant neurologist at the National Hospital for Neurology and Neurosurgery in London, cochaired the International Panel on Diagnosis of MS, which drafted the new recommendations. The panel convened in November 2016 and May 2017. The meetings were organized under the International Advisory Committee on Clinical Trials in MS and supported by the US National MS Society and the European Committee for Treatment and Research in MS (ECTRIMS). The panel’s recommendations have been submitted for publication and are in the late stages of revision, Dr. Cohen said.
Facilitate Diagnosis
New data regarding the utility of MRI, CSF, and other tests in the diagnostic process motivated neurologists to reconvene the panel. In addition, “there has been increasing recognition of the continued frequency and potential consequences of misdiagnosis of MS,” Dr. Cohen said.
“We felt that the 2010 McDonald criteria overall performed well,” he said. “We did not anticipate making major changes to the criteria. But we sought to simplify and clarify some of the components of the 2010 criteria. We wanted to facilitate the ability to make the diagnosis of MS in patients who had a high likelihood of the disease but were not currently diagnosable by the 2010 criteria. We wanted to preserve the specificity of the 2010 criteria but promote their appropriate application … to reduce the risk of misdiagnosis.” Finally, the panel “wanted to ensure that any proposed changes did not weaken the existing criteria and were supported by reasonable evidence,” he said. Dr. Cohen highlighted five of the panel’s key revisions.
Recommended Changes
First and probably most controversially, “we propose that in a patient with a typical clinically isolated syndrome, and with fulfillment by either clinical or MRI criteria for dissemination in space, that the presence of CSF-specific oligoclonal bands now allows for diagnosis of MS,” Dr. Cohen said. “It does not represent demonstration of dissemination in time per se, but it allows substitution for demonstration of dissemination in time.”
A second recommendation is that symptomatic and asymptomatic MRI lesions can be considered in the determination of dissemination in space and time. In the 2010 criteria, a symptomatic lesion in a patient with a brainstem or spinal cord syndrome could not be included as MRI evidence of dissemination in time and space.
Third, in addition to juxtacortical lesions, cortical lesions can demonstrate dissemination in space. Neurologists’ ability to detect purely cortical MRI
Fourth, the criteria for primary progressive MS now allow the inclusion of symptomatic and cortical lesions as evidence of the disease. These criteria otherwise have not changed.
Finally, the panel recommends that neurologists determine a provisional disease course, as specified by Lublin et al, at the time of diagnosis and then periodically reevaluate the provisional course based on accumulated evidence.
Avoiding Misdiagnosis
“Much of our discussion started with the issue of misdiagnosis and differential diagnosis in MS,” Dr. Cohen said. “The potential differential diagnosis of MS is quite broad, and misdiagnosis remains an issue even today with advancements in MRI and other testing.” Solomon et al found that neuromyelitis optica spectrum disorders (NMOSDs) were the disorders most commonly misdiagnosed as MS. Physicians also misdiagnosed common conditions like migraine as MS. “Misdiagnosis may have harmful consequences, including inappropriate institution of disease therapy,” Dr. Cohen said.
Neurologists now recognize that aquaporin 4–related NMOSD is a distinct disorder from MS. “However, if you think back to the time that the 2010 criteria were developed, the relationship between NMOSD and MS was not quite as clear. Substantial data have been published since that time,” Dr. Cohen said. “We agreed that the McDonald criteria and the formal criteria for NMOSD largely distinguish the two diseases. However, there may be cases in which there is some uncertainty. Our recommendation is that … the possibility of NMOSD should be considered in all patients being evaluated for MS,” and any patient with features suggesting NMOSD should undergo aquaporin 4 testing.
In general, neurologists should recognize that the McDonald criteria originally were developed to make the diagnosis of MS in patients who have a high likelihood of the disease, not to differentiate MS from other disorders, he said. “Historical events being taken to represent a prior attack should be interpreted with caution if there is no corroborating objective evidence,” Dr. Cohen said. “In cases in which the diagnosis of MS is uncertain, further testing should be pursued. And in some cases, a clinician may want to postpone making a diagnosis pending the accumulation of sufficient data.” CSF testing and spinal MRI are not required to make the diagnosis of MS, but there should be a low threshold for obtaining them.
While data generally support the validity of the 2010 McDonald criteria in geographically diverse populations, children, and older individuals, neurologists should address potentially relevant alternative diagnoses that may be more common in these and other populations (eg, patients with comorbidities), such as infections and nutritional deficiencies.
MAGNIMS Proposal
Several proposals generated discussion during the panel meetings but were not adopted, primarily because the evidence did not justify changing the current criteria. For instance, the 2016 MAGNIMS MRI criteria propose that the number of acquired periventricular lesions be increased from one to three to provide additional specificity. “We reviewed those data … and felt that the modest increase in specificity did not justify making the change,” Dr. Cohen said. The panel also discussed incorporating optic nerve involvement into the criteria, but this proposal was not included in the update.
“The 2017 revisions further refine the well-established McDonald criteria,” Dr. Cohen said. “The appropriate application of the criteria is critical to avoid misdiagnosis. Fundamentally, MS remains a clinical diagnosis. … It requires rigorous synthesis of clinical, imaging, and laboratory data by a clinician with expertise in MS.”
Making a Diagnosis Sooner
Neurologists have been diagnosing MS in patients sooner. At the same time, misdiagnosing other conditions as MS can have profound consequences, including the potentially serious side effects of disease-modifying therapy, said Jeremy Chataway, PhD, consultant neurologist at the National Hospital for Neurology and Neurosurgery, London, in a lecture about the application of the proposed criteria. He described cases in which the 2017 proposed criteria—by allowing the inclusion of a symptomatic spinal cord lesion, or by substituting CSF-specific oligoclonal bands in place of dissemination in time—would have allowed neurologists to diagnose MS sooner. In one case, a patient might have received a diagnosis of MS about two years earlier.
Sometimes a diagnosis of MS “is obvious,” Dr. Chataway said. “Sometimes it is hard, even with advanced MRI. You can see the gradation that is required … to get us to the correct diagnosis.”
—Jake Remaly
Suggested Reading
Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292-302.
Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: A multicenter study. Neurology. 2016;87(13):1393-1399.
PARIS—Recommended revisions to the 2010 McDonald diagnostic criteria for multiple sclerosis (MS) include changes that are intended to enable neurologists to diagnose MS sooner in patients with a high likelihood of the disease. One addition allows the use of CSF-specific oligoclonal bands in lieu of demonstration of dissemination in time to make a diagnosis of MS in patients with a clinically isolated syndrome and demonstration of dissemination in space clinically or by MRI. Symptomatic and cortical lesions also may satisfy diagnostic criteria, according to the recommendations. In addition, the revised criteria include guidance to reduce the risk of misdiagnosing MS.
Jeffrey A. Cohen, MD, a neurologist with the Cleveland Clinic’s Mellen Center for MS Treatment and Research, presented the 2017 proposed revisions at the Seventh Joint ECTRIMS–ACTRIMS Meeting.
Dr. Cohen and Alan J. Thompson, MD, consultant neurologist at the National Hospital for Neurology and Neurosurgery in London, cochaired the International Panel on Diagnosis of MS, which drafted the new recommendations. The panel convened in November 2016 and May 2017. The meetings were organized under the International Advisory Committee on Clinical Trials in MS and supported by the US National MS Society and the European Committee for Treatment and Research in MS (ECTRIMS). The panel’s recommendations have been submitted for publication and are in the late stages of revision, Dr. Cohen said.
Facilitate Diagnosis
New data regarding the utility of MRI, CSF, and other tests in the diagnostic process motivated neurologists to reconvene the panel. In addition, “there has been increasing recognition of the continued frequency and potential consequences of misdiagnosis of MS,” Dr. Cohen said.
“We felt that the 2010 McDonald criteria overall performed well,” he said. “We did not anticipate making major changes to the criteria. But we sought to simplify and clarify some of the components of the 2010 criteria. We wanted to facilitate the ability to make the diagnosis of MS in patients who had a high likelihood of the disease but were not currently diagnosable by the 2010 criteria. We wanted to preserve the specificity of the 2010 criteria but promote their appropriate application … to reduce the risk of misdiagnosis.” Finally, the panel “wanted to ensure that any proposed changes did not weaken the existing criteria and were supported by reasonable evidence,” he said. Dr. Cohen highlighted five of the panel’s key revisions.
Recommended Changes
First and probably most controversially, “we propose that in a patient with a typical clinically isolated syndrome, and with fulfillment by either clinical or MRI criteria for dissemination in space, that the presence of CSF-specific oligoclonal bands now allows for diagnosis of MS,” Dr. Cohen said. “It does not represent demonstration of dissemination in time per se, but it allows substitution for demonstration of dissemination in time.”
A second recommendation is that symptomatic and asymptomatic MRI lesions can be considered in the determination of dissemination in space and time. In the 2010 criteria, a symptomatic lesion in a patient with a brainstem or spinal cord syndrome could not be included as MRI evidence of dissemination in time and space.
Third, in addition to juxtacortical lesions, cortical lesions can demonstrate dissemination in space. Neurologists’ ability to detect purely cortical MRI
Fourth, the criteria for primary progressive MS now allow the inclusion of symptomatic and cortical lesions as evidence of the disease. These criteria otherwise have not changed.
Finally, the panel recommends that neurologists determine a provisional disease course, as specified by Lublin et al, at the time of diagnosis and then periodically reevaluate the provisional course based on accumulated evidence.
Avoiding Misdiagnosis
“Much of our discussion started with the issue of misdiagnosis and differential diagnosis in MS,” Dr. Cohen said. “The potential differential diagnosis of MS is quite broad, and misdiagnosis remains an issue even today with advancements in MRI and other testing.” Solomon et al found that neuromyelitis optica spectrum disorders (NMOSDs) were the disorders most commonly misdiagnosed as MS. Physicians also misdiagnosed common conditions like migraine as MS. “Misdiagnosis may have harmful consequences, including inappropriate institution of disease therapy,” Dr. Cohen said.
Neurologists now recognize that aquaporin 4–related NMOSD is a distinct disorder from MS. “However, if you think back to the time that the 2010 criteria were developed, the relationship between NMOSD and MS was not quite as clear. Substantial data have been published since that time,” Dr. Cohen said. “We agreed that the McDonald criteria and the formal criteria for NMOSD largely distinguish the two diseases. However, there may be cases in which there is some uncertainty. Our recommendation is that … the possibility of NMOSD should be considered in all patients being evaluated for MS,” and any patient with features suggesting NMOSD should undergo aquaporin 4 testing.
In general, neurologists should recognize that the McDonald criteria originally were developed to make the diagnosis of MS in patients who have a high likelihood of the disease, not to differentiate MS from other disorders, he said. “Historical events being taken to represent a prior attack should be interpreted with caution if there is no corroborating objective evidence,” Dr. Cohen said. “In cases in which the diagnosis of MS is uncertain, further testing should be pursued. And in some cases, a clinician may want to postpone making a diagnosis pending the accumulation of sufficient data.” CSF testing and spinal MRI are not required to make the diagnosis of MS, but there should be a low threshold for obtaining them.
While data generally support the validity of the 2010 McDonald criteria in geographically diverse populations, children, and older individuals, neurologists should address potentially relevant alternative diagnoses that may be more common in these and other populations (eg, patients with comorbidities), such as infections and nutritional deficiencies.
MAGNIMS Proposal
Several proposals generated discussion during the panel meetings but were not adopted, primarily because the evidence did not justify changing the current criteria. For instance, the 2016 MAGNIMS MRI criteria propose that the number of acquired periventricular lesions be increased from one to three to provide additional specificity. “We reviewed those data … and felt that the modest increase in specificity did not justify making the change,” Dr. Cohen said. The panel also discussed incorporating optic nerve involvement into the criteria, but this proposal was not included in the update.
“The 2017 revisions further refine the well-established McDonald criteria,” Dr. Cohen said. “The appropriate application of the criteria is critical to avoid misdiagnosis. Fundamentally, MS remains a clinical diagnosis. … It requires rigorous synthesis of clinical, imaging, and laboratory data by a clinician with expertise in MS.”
Making a Diagnosis Sooner
Neurologists have been diagnosing MS in patients sooner. At the same time, misdiagnosing other conditions as MS can have profound consequences, including the potentially serious side effects of disease-modifying therapy, said Jeremy Chataway, PhD, consultant neurologist at the National Hospital for Neurology and Neurosurgery, London, in a lecture about the application of the proposed criteria. He described cases in which the 2017 proposed criteria—by allowing the inclusion of a symptomatic spinal cord lesion, or by substituting CSF-specific oligoclonal bands in place of dissemination in time—would have allowed neurologists to diagnose MS sooner. In one case, a patient might have received a diagnosis of MS about two years earlier.
Sometimes a diagnosis of MS “is obvious,” Dr. Chataway said. “Sometimes it is hard, even with advanced MRI. You can see the gradation that is required … to get us to the correct diagnosis.”
—Jake Remaly
Suggested Reading
Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292-302.
Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: A multicenter study. Neurology. 2016;87(13):1393-1399.
PARIS—Recommended revisions to the 2010 McDonald diagnostic criteria for multiple sclerosis (MS) include changes that are intended to enable neurologists to diagnose MS sooner in patients with a high likelihood of the disease. One addition allows the use of CSF-specific oligoclonal bands in lieu of demonstration of dissemination in time to make a diagnosis of MS in patients with a clinically isolated syndrome and demonstration of dissemination in space clinically or by MRI. Symptomatic and cortical lesions also may satisfy diagnostic criteria, according to the recommendations. In addition, the revised criteria include guidance to reduce the risk of misdiagnosing MS.
Jeffrey A. Cohen, MD, a neurologist with the Cleveland Clinic’s Mellen Center for MS Treatment and Research, presented the 2017 proposed revisions at the Seventh Joint ECTRIMS–ACTRIMS Meeting.
Dr. Cohen and Alan J. Thompson, MD, consultant neurologist at the National Hospital for Neurology and Neurosurgery in London, cochaired the International Panel on Diagnosis of MS, which drafted the new recommendations. The panel convened in November 2016 and May 2017. The meetings were organized under the International Advisory Committee on Clinical Trials in MS and supported by the US National MS Society and the European Committee for Treatment and Research in MS (ECTRIMS). The panel’s recommendations have been submitted for publication and are in the late stages of revision, Dr. Cohen said.
Facilitate Diagnosis
New data regarding the utility of MRI, CSF, and other tests in the diagnostic process motivated neurologists to reconvene the panel. In addition, “there has been increasing recognition of the continued frequency and potential consequences of misdiagnosis of MS,” Dr. Cohen said.
“We felt that the 2010 McDonald criteria overall performed well,” he said. “We did not anticipate making major changes to the criteria. But we sought to simplify and clarify some of the components of the 2010 criteria. We wanted to facilitate the ability to make the diagnosis of MS in patients who had a high likelihood of the disease but were not currently diagnosable by the 2010 criteria. We wanted to preserve the specificity of the 2010 criteria but promote their appropriate application … to reduce the risk of misdiagnosis.” Finally, the panel “wanted to ensure that any proposed changes did not weaken the existing criteria and were supported by reasonable evidence,” he said. Dr. Cohen highlighted five of the panel’s key revisions.
Recommended Changes
First and probably most controversially, “we propose that in a patient with a typical clinically isolated syndrome, and with fulfillment by either clinical or MRI criteria for dissemination in space, that the presence of CSF-specific oligoclonal bands now allows for diagnosis of MS,” Dr. Cohen said. “It does not represent demonstration of dissemination in time per se, but it allows substitution for demonstration of dissemination in time.”
A second recommendation is that symptomatic and asymptomatic MRI lesions can be considered in the determination of dissemination in space and time. In the 2010 criteria, a symptomatic lesion in a patient with a brainstem or spinal cord syndrome could not be included as MRI evidence of dissemination in time and space.
Third, in addition to juxtacortical lesions, cortical lesions can demonstrate dissemination in space. Neurologists’ ability to detect purely cortical MRI
Fourth, the criteria for primary progressive MS now allow the inclusion of symptomatic and cortical lesions as evidence of the disease. These criteria otherwise have not changed.
Finally, the panel recommends that neurologists determine a provisional disease course, as specified by Lublin et al, at the time of diagnosis and then periodically reevaluate the provisional course based on accumulated evidence.
Avoiding Misdiagnosis
“Much of our discussion started with the issue of misdiagnosis and differential diagnosis in MS,” Dr. Cohen said. “The potential differential diagnosis of MS is quite broad, and misdiagnosis remains an issue even today with advancements in MRI and other testing.” Solomon et al found that neuromyelitis optica spectrum disorders (NMOSDs) were the disorders most commonly misdiagnosed as MS. Physicians also misdiagnosed common conditions like migraine as MS. “Misdiagnosis may have harmful consequences, including inappropriate institution of disease therapy,” Dr. Cohen said.
Neurologists now recognize that aquaporin 4–related NMOSD is a distinct disorder from MS. “However, if you think back to the time that the 2010 criteria were developed, the relationship between NMOSD and MS was not quite as clear. Substantial data have been published since that time,” Dr. Cohen said. “We agreed that the McDonald criteria and the formal criteria for NMOSD largely distinguish the two diseases. However, there may be cases in which there is some uncertainty. Our recommendation is that … the possibility of NMOSD should be considered in all patients being evaluated for MS,” and any patient with features suggesting NMOSD should undergo aquaporin 4 testing.
In general, neurologists should recognize that the McDonald criteria originally were developed to make the diagnosis of MS in patients who have a high likelihood of the disease, not to differentiate MS from other disorders, he said. “Historical events being taken to represent a prior attack should be interpreted with caution if there is no corroborating objective evidence,” Dr. Cohen said. “In cases in which the diagnosis of MS is uncertain, further testing should be pursued. And in some cases, a clinician may want to postpone making a diagnosis pending the accumulation of sufficient data.” CSF testing and spinal MRI are not required to make the diagnosis of MS, but there should be a low threshold for obtaining them.
While data generally support the validity of the 2010 McDonald criteria in geographically diverse populations, children, and older individuals, neurologists should address potentially relevant alternative diagnoses that may be more common in these and other populations (eg, patients with comorbidities), such as infections and nutritional deficiencies.
MAGNIMS Proposal
Several proposals generated discussion during the panel meetings but were not adopted, primarily because the evidence did not justify changing the current criteria. For instance, the 2016 MAGNIMS MRI criteria propose that the number of acquired periventricular lesions be increased from one to three to provide additional specificity. “We reviewed those data … and felt that the modest increase in specificity did not justify making the change,” Dr. Cohen said. The panel also discussed incorporating optic nerve involvement into the criteria, but this proposal was not included in the update.
“The 2017 revisions further refine the well-established McDonald criteria,” Dr. Cohen said. “The appropriate application of the criteria is critical to avoid misdiagnosis. Fundamentally, MS remains a clinical diagnosis. … It requires rigorous synthesis of clinical, imaging, and laboratory data by a clinician with expertise in MS.”
Making a Diagnosis Sooner
Neurologists have been diagnosing MS in patients sooner. At the same time, misdiagnosing other conditions as MS can have profound consequences, including the potentially serious side effects of disease-modifying therapy, said Jeremy Chataway, PhD, consultant neurologist at the National Hospital for Neurology and Neurosurgery, London, in a lecture about the application of the proposed criteria. He described cases in which the 2017 proposed criteria—by allowing the inclusion of a symptomatic spinal cord lesion, or by substituting CSF-specific oligoclonal bands in place of dissemination in time—would have allowed neurologists to diagnose MS sooner. In one case, a patient might have received a diagnosis of MS about two years earlier.
Sometimes a diagnosis of MS “is obvious,” Dr. Chataway said. “Sometimes it is hard, even with advanced MRI. You can see the gradation that is required … to get us to the correct diagnosis.”
—Jake Remaly
Suggested Reading
Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292-302.
Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: A multicenter study. Neurology. 2016;87(13):1393-1399.
Durable Improvements in Clinical Outcomes With Alemtuzumab: Seven-Year Follow-Up
PARIS—Clinical efficacy of alemtuzumab was maintained for seven years in patients who had inadequate response to prior t

Alemtuzumab Treatment Then
In the CARE-MS II trial, alemtuzumab significantly improved clinical outcomes, compared with subcutaneous interferon beta-1a, over two years in patients with active RRMS and an inadequate response to prior therapy. Durable efficacy of alemtuzumab was demonstrated over six years in a completed extension study in the absence of continuous treatment. Patients in the CARE-MS II study received two courses of alemtuzumab 12 mg/day (five days of therapy at baseline and three days of therapy 12 months later). In the extension study, patients could receive as-needed alemtuzumab retreatment (12 mg/day on three consecutive days at least 12 months after a previous course for relapse or MRI activity) or other disease-modifying therapy per investigator discretion. Patients completing at least 48 months of the extension could enroll in the five-year TOPAZ study for further long-term evaluation.
Alemtuzumab Treatment Now
The goal of the TOPAZ study was to evaluate the seven-year efficacy and safety of alemtuzumab in patients with RRMS who received alemtuzumab in the CARE-MS II trial. In TOPAZ, patients could receive alemtuzumab retreatment 12 months or more after a previous course or other disease-modifying therapy at any time point (both per investigator discretion). MRI scans were performed annually. Annualized relapse rate, six-month confirmed disability worsening, six-month confirmed disability improvement, no evidence of disease activity (NEDA), and adverse events were analyzed in TOPAZ.
In total, 338 of 393 (86%) CARE-MS II patients who entered the extension remained in the study until the end of year six and then entered TOPAZ; 317 (94%) remained in the study through year seven. Annualized relapse rate remained low (0.14 at year seven), and the proportion of patients with stable or improved Expanded Disability Status Scale score remained high (73% at year seven). Through year seven, 69% of patients were free from six-month confirmed disability worsening, 44% achieved six-month confirmed disability improvement, and the majority achieved NEDA each year. These effects were achieved with 47% of patients receiving no additional treatment (alemtuzumab or other disease-modifying treatment) after their initial two courses of alemtuzumab. Incidences of overall adverse events, infusion-associated reactions, and infections decreased over time and were reduced, compared with those in the two-year core study. The incidence of thyroid adverse events incidence peaked at year three and then declined.
PARIS—Clinical efficacy of alemtuzumab was maintained for seven years in patients who had inadequate response to prior t
Alemtuzumab Treatment Then
In the CARE-MS II trial, alemtuzumab significantly improved clinical outcomes, compared with subcutaneous interferon beta-1a, over two years in patients with active RRMS and an inadequate response to prior therapy. Durable efficacy of alemtuzumab was demonstrated over six years in a completed extension study in the absence of continuous treatment. Patients in the CARE-MS II study received two courses of alemtuzumab 12 mg/day (five days of therapy at baseline and three days of therapy 12 months later). In the extension study, patients could receive as-needed alemtuzumab retreatment (12 mg/day on three consecutive days at least 12 months after a previous course for relapse or MRI activity) or other disease-modifying therapy per investigator discretion. Patients completing at least 48 months of the extension could enroll in the five-year TOPAZ study for further long-term evaluation.
Alemtuzumab Treatment Now
The goal of the TOPAZ study was to evaluate the seven-year efficacy and safety of alemtuzumab in patients with RRMS who received alemtuzumab in the CARE-MS II trial. In TOPAZ, patients could receive alemtuzumab retreatment 12 months or more after a previous course or other disease-modifying therapy at any time point (both per investigator discretion). MRI scans were performed annually. Annualized relapse rate, six-month confirmed disability worsening, six-month confirmed disability improvement, no evidence of disease activity (NEDA), and adverse events were analyzed in TOPAZ.
In total, 338 of 393 (86%) CARE-MS II patients who entered the extension remained in the study until the end of year six and then entered TOPAZ; 317 (94%) remained in the study through year seven. Annualized relapse rate remained low (0.14 at year seven), and the proportion of patients with stable or improved Expanded Disability Status Scale score remained high (73% at year seven). Through year seven, 69% of patients were free from six-month confirmed disability worsening, 44% achieved six-month confirmed disability improvement, and the majority achieved NEDA each year. These effects were achieved with 47% of patients receiving no additional treatment (alemtuzumab or other disease-modifying treatment) after their initial two courses of alemtuzumab. Incidences of overall adverse events, infusion-associated reactions, and infections decreased over time and were reduced, compared with those in the two-year core study. The incidence of thyroid adverse events incidence peaked at year three and then declined.
PARIS—Clinical efficacy of alemtuzumab was maintained for seven years in patients who had inadequate response to prior t
Alemtuzumab Treatment Then
In the CARE-MS II trial, alemtuzumab significantly improved clinical outcomes, compared with subcutaneous interferon beta-1a, over two years in patients with active RRMS and an inadequate response to prior therapy. Durable efficacy of alemtuzumab was demonstrated over six years in a completed extension study in the absence of continuous treatment. Patients in the CARE-MS II study received two courses of alemtuzumab 12 mg/day (five days of therapy at baseline and three days of therapy 12 months later). In the extension study, patients could receive as-needed alemtuzumab retreatment (12 mg/day on three consecutive days at least 12 months after a previous course for relapse or MRI activity) or other disease-modifying therapy per investigator discretion. Patients completing at least 48 months of the extension could enroll in the five-year TOPAZ study for further long-term evaluation.
Alemtuzumab Treatment Now
The goal of the TOPAZ study was to evaluate the seven-year efficacy and safety of alemtuzumab in patients with RRMS who received alemtuzumab in the CARE-MS II trial. In TOPAZ, patients could receive alemtuzumab retreatment 12 months or more after a previous course or other disease-modifying therapy at any time point (both per investigator discretion). MRI scans were performed annually. Annualized relapse rate, six-month confirmed disability worsening, six-month confirmed disability improvement, no evidence of disease activity (NEDA), and adverse events were analyzed in TOPAZ.
In total, 338 of 393 (86%) CARE-MS II patients who entered the extension remained in the study until the end of year six and then entered TOPAZ; 317 (94%) remained in the study through year seven. Annualized relapse rate remained low (0.14 at year seven), and the proportion of patients with stable or improved Expanded Disability Status Scale score remained high (73% at year seven). Through year seven, 69% of patients were free from six-month confirmed disability worsening, 44% achieved six-month confirmed disability improvement, and the majority achieved NEDA each year. These effects were achieved with 47% of patients receiving no additional treatment (alemtuzumab or other disease-modifying treatment) after their initial two courses of alemtuzumab. Incidences of overall adverse events, infusion-associated reactions, and infections decreased over time and were reduced, compared with those in the two-year core study. The incidence of thyroid adverse events incidence peaked at year three and then declined.