User login
Abatacept Fails to Provide Benefits in Relapsing MS
Abatacept does not reduce the number of new gadolinium-enhancing lesions on MRI in patients with relapsing-remitting multiple sclerosis (MS), according to results from the double-blind, randomized, placebo-controlled phase II ACCLAIM study published online ahead of print August 1 in Multiple Sclerosis.
In the ACCLAIM (A Cooperative Clinical Study of Abatacept in MS) study, 42 patients who received abatacept (Orencia) developed a mean of 0.43 new gadolinium-enhancing lesions by week 24, compared with 1.66 lesions for 20 placebo-treated patients. None of the secondary MRI end points (ie, lesion volume change and percent brain volume change) and clinical end points (ie, changes in MS Functional Composite score, Expanded Disability Status Scale [EDSS], and annualized relapse rate) at 24 weeks differed significantly between the groups. The rate of patients who met criteria for no evidence of disease activity or its components (ie, no EDSS progression, no clinical exacerbations, and no new gadolinium-enhancing MRI lesion) from week 8 and before week 28 did not differ between the groups.
During a 28-week extension phase in which the groups switched treatments, patients who switched from abatacept to placebo had a greater number of gadolinium-enhancing lesions than did those who switched from placebo to abatacept (1.25 vs 0.60, respectively), but the difference was not statistically significant.
Abatacept, which is approved for the treatment of rheumatoid arthritis and juvenile idiopathic arthritis, is a CTLA4 immunoglobulin fusion protein that inhibits the activation of T lymphocytes by blocking the CD28-B7 costimulatory pathway. It was thought to have potential to reduce immune-mediated disease activity in relapsing-remitting MS because T lymphocytes have been implicated in its pathogenesis.
The investigators closed enrollment for the trial early because of slow accrual. The 65 patients who were enrolled in the trial were about half of the population considered to be required (ie, 123) to demonstrate a treatment effect of 50% reduction of new gadolinium-enhancing MRI lesions.

—Jeff Evans
Suggested Reading
Khoury SJ, Rochon J, Ding L, et al. ACCLAIM: A randomized trial of abatacept (CTLA4-Ig) for relapsing-remitting multiple sclerosis. Mult Scler. 2016 Aug 1 [Epub ahead of print].
Abatacept does not reduce the number of new gadolinium-enhancing lesions on MRI in patients with relapsing-remitting multiple sclerosis (MS), according to results from the double-blind, randomized, placebo-controlled phase II ACCLAIM study published online ahead of print August 1 in Multiple Sclerosis.
In the ACCLAIM (A Cooperative Clinical Study of Abatacept in MS) study, 42 patients who received abatacept (Orencia) developed a mean of 0.43 new gadolinium-enhancing lesions by week 24, compared with 1.66 lesions for 20 placebo-treated patients. None of the secondary MRI end points (ie, lesion volume change and percent brain volume change) and clinical end points (ie, changes in MS Functional Composite score, Expanded Disability Status Scale [EDSS], and annualized relapse rate) at 24 weeks differed significantly between the groups. The rate of patients who met criteria for no evidence of disease activity or its components (ie, no EDSS progression, no clinical exacerbations, and no new gadolinium-enhancing MRI lesion) from week 8 and before week 28 did not differ between the groups.
During a 28-week extension phase in which the groups switched treatments, patients who switched from abatacept to placebo had a greater number of gadolinium-enhancing lesions than did those who switched from placebo to abatacept (1.25 vs 0.60, respectively), but the difference was not statistically significant.
Abatacept, which is approved for the treatment of rheumatoid arthritis and juvenile idiopathic arthritis, is a CTLA4 immunoglobulin fusion protein that inhibits the activation of T lymphocytes by blocking the CD28-B7 costimulatory pathway. It was thought to have potential to reduce immune-mediated disease activity in relapsing-remitting MS because T lymphocytes have been implicated in its pathogenesis.
The investigators closed enrollment for the trial early because of slow accrual. The 65 patients who were enrolled in the trial were about half of the population considered to be required (ie, 123) to demonstrate a treatment effect of 50% reduction of new gadolinium-enhancing MRI lesions.
—Jeff Evans
Abatacept does not reduce the number of new gadolinium-enhancing lesions on MRI in patients with relapsing-remitting multiple sclerosis (MS), according to results from the double-blind, randomized, placebo-controlled phase II ACCLAIM study published online ahead of print August 1 in Multiple Sclerosis.
In the ACCLAIM (A Cooperative Clinical Study of Abatacept in MS) study, 42 patients who received abatacept (Orencia) developed a mean of 0.43 new gadolinium-enhancing lesions by week 24, compared with 1.66 lesions for 20 placebo-treated patients. None of the secondary MRI end points (ie, lesion volume change and percent brain volume change) and clinical end points (ie, changes in MS Functional Composite score, Expanded Disability Status Scale [EDSS], and annualized relapse rate) at 24 weeks differed significantly between the groups. The rate of patients who met criteria for no evidence of disease activity or its components (ie, no EDSS progression, no clinical exacerbations, and no new gadolinium-enhancing MRI lesion) from week 8 and before week 28 did not differ between the groups.
During a 28-week extension phase in which the groups switched treatments, patients who switched from abatacept to placebo had a greater number of gadolinium-enhancing lesions than did those who switched from placebo to abatacept (1.25 vs 0.60, respectively), but the difference was not statistically significant.
Abatacept, which is approved for the treatment of rheumatoid arthritis and juvenile idiopathic arthritis, is a CTLA4 immunoglobulin fusion protein that inhibits the activation of T lymphocytes by blocking the CD28-B7 costimulatory pathway. It was thought to have potential to reduce immune-mediated disease activity in relapsing-remitting MS because T lymphocytes have been implicated in its pathogenesis.
The investigators closed enrollment for the trial early because of slow accrual. The 65 patients who were enrolled in the trial were about half of the population considered to be required (ie, 123) to demonstrate a treatment effect of 50% reduction of new gadolinium-enhancing MRI lesions.
—Jeff Evans
Suggested Reading
Khoury SJ, Rochon J, Ding L, et al. ACCLAIM: A randomized trial of abatacept (CTLA4-Ig) for relapsing-remitting multiple sclerosis. Mult Scler. 2016 Aug 1 [Epub ahead of print].
Suggested Reading
Khoury SJ, Rochon J, Ding L, et al. ACCLAIM: A randomized trial of abatacept (CTLA4-Ig) for relapsing-remitting multiple sclerosis. Mult Scler. 2016 Aug 1 [Epub ahead of print].

Cognitive impairment predicts worsening multiple sclerosis
Cognitive impairment in individuals at the time of multiple sclerosis diagnosis imparts a significantly higher risk for disability progression, transition to secondary progressive disease, and cortical thinning, according to results from a retrospective, 8-year, longitudinal observational study.
The results, note first author Marco Pitteri, PhD, of the University of Verona (Italy) and his colleagues, “confirm and extend previous studies showing that cognitive impairment could represent a clinical marker of more aggressive gray matter pathology,” and also identified cortical thinning as a neuroimaging correlate of cognitive impairment that was detected only at follow-up between the cognitively impaired and cognitively normal patients who were otherwise similar at baseline.

The study involved 78 consecutive patients with relapsing-remitting multiple sclerosis who underwent neurologic exams during 2015 at a single center in Italy. They all had a cognitive assessment and an MRI scan at the time of diagnosis and at least 8 years of clinical follow-up, including neurologic examination every 6 months. At the time of enrollment into the study, most patients were taking immunomodulatory therapy: 38 were treated with interferon beta-1a, 14 with interferon beta-1b, 15 with glatiramer acetate, and 6 with azathioprine, and 5 were untreated.
At baseline, 39 cognitively normal patients had a mean age of about 36 years, none had conversion to secondary progressive disease, and just 15% had disability progression on the Expanded Disability Status Scale (EDSS). Cortical thickness changed a mean of 3.6%.
The 26 patients with mild cognitive impairment (classified as failure of up to two tests on the Brief Repeatable Battery) were 38 years old at baseline, and only 4 transitioned to secondary progressive disease. Just over half experienced EDSS progression (an average of 0.85 points) and cortical thickness changed by an average of 17%.
The 13 remaining patients with severe cognitive impairment at baseline (three or more failed tests) were about 44 years of age on average and nearly all (92%) experienced EDSS progression, which averaged 1.31 points. Cortical thickness also changed a mean of 34%. Relapsing disease converted to secondary progressive disease in a total of six patients with severe cognitive impairment.
Patients with severe cognitive impairment at baseline had more severe white matter lesion volume at baseline than did others, but there was no difference in the accumulation of white matter lesions during follow-up between patients with normal cognition, mild cognitive impairment, and severe cognitive impairment, which corroborates previous studies. However, the predictive ability of early cognitive impairment in identifying patients with disability progression on the EDSS and cortical thinning “is in line with several previous longitudinal studies showing a strong relationship between gray matter damage and the development of cognitive impairment,” the authors wrote (Brain. 2012 Oct;135[Pt 10]:2952-61 and Neurology. 2013 Nov 12;81[20]:1759-67).
Although the administration of neuropsychological batteries is time consuming and requires expertise, the authors argued that their results “underline the need of using extended screening neuropsychological batteries instead of brief screening batteries, at least at the time of diagnosis, in order to increase the probability to detect even mild forms of cognitive impairment as early as possible in the disease course.”
Read the full paper online in Multiple Sclerosis (2016 Aug 15. doi: 10.1177/1352458516665496).
Cognitive impairment in individuals at the time of multiple sclerosis diagnosis imparts a significantly higher risk for disability progression, transition to secondary progressive disease, and cortical thinning, according to results from a retrospective, 8-year, longitudinal observational study.
The results, note first author Marco Pitteri, PhD, of the University of Verona (Italy) and his colleagues, “confirm and extend previous studies showing that cognitive impairment could represent a clinical marker of more aggressive gray matter pathology,” and also identified cortical thinning as a neuroimaging correlate of cognitive impairment that was detected only at follow-up between the cognitively impaired and cognitively normal patients who were otherwise similar at baseline.
The study involved 78 consecutive patients with relapsing-remitting multiple sclerosis who underwent neurologic exams during 2015 at a single center in Italy. They all had a cognitive assessment and an MRI scan at the time of diagnosis and at least 8 years of clinical follow-up, including neurologic examination every 6 months. At the time of enrollment into the study, most patients were taking immunomodulatory therapy: 38 were treated with interferon beta-1a, 14 with interferon beta-1b, 15 with glatiramer acetate, and 6 with azathioprine, and 5 were untreated.
At baseline, 39 cognitively normal patients had a mean age of about 36 years, none had conversion to secondary progressive disease, and just 15% had disability progression on the Expanded Disability Status Scale (EDSS). Cortical thickness changed a mean of 3.6%.
The 26 patients with mild cognitive impairment (classified as failure of up to two tests on the Brief Repeatable Battery) were 38 years old at baseline, and only 4 transitioned to secondary progressive disease. Just over half experienced EDSS progression (an average of 0.85 points) and cortical thickness changed by an average of 17%.
The 13 remaining patients with severe cognitive impairment at baseline (three or more failed tests) were about 44 years of age on average and nearly all (92%) experienced EDSS progression, which averaged 1.31 points. Cortical thickness also changed a mean of 34%. Relapsing disease converted to secondary progressive disease in a total of six patients with severe cognitive impairment.
Patients with severe cognitive impairment at baseline had more severe white matter lesion volume at baseline than did others, but there was no difference in the accumulation of white matter lesions during follow-up between patients with normal cognition, mild cognitive impairment, and severe cognitive impairment, which corroborates previous studies. However, the predictive ability of early cognitive impairment in identifying patients with disability progression on the EDSS and cortical thinning “is in line with several previous longitudinal studies showing a strong relationship between gray matter damage and the development of cognitive impairment,” the authors wrote (Brain. 2012 Oct;135[Pt 10]:2952-61 and Neurology. 2013 Nov 12;81[20]:1759-67).
Although the administration of neuropsychological batteries is time consuming and requires expertise, the authors argued that their results “underline the need of using extended screening neuropsychological batteries instead of brief screening batteries, at least at the time of diagnosis, in order to increase the probability to detect even mild forms of cognitive impairment as early as possible in the disease course.”
Read the full paper online in Multiple Sclerosis (2016 Aug 15. doi: 10.1177/1352458516665496).
Cognitive impairment in individuals at the time of multiple sclerosis diagnosis imparts a significantly higher risk for disability progression, transition to secondary progressive disease, and cortical thinning, according to results from a retrospective, 8-year, longitudinal observational study.
The results, note first author Marco Pitteri, PhD, of the University of Verona (Italy) and his colleagues, “confirm and extend previous studies showing that cognitive impairment could represent a clinical marker of more aggressive gray matter pathology,” and also identified cortical thinning as a neuroimaging correlate of cognitive impairment that was detected only at follow-up between the cognitively impaired and cognitively normal patients who were otherwise similar at baseline.
The study involved 78 consecutive patients with relapsing-remitting multiple sclerosis who underwent neurologic exams during 2015 at a single center in Italy. They all had a cognitive assessment and an MRI scan at the time of diagnosis and at least 8 years of clinical follow-up, including neurologic examination every 6 months. At the time of enrollment into the study, most patients were taking immunomodulatory therapy: 38 were treated with interferon beta-1a, 14 with interferon beta-1b, 15 with glatiramer acetate, and 6 with azathioprine, and 5 were untreated.
At baseline, 39 cognitively normal patients had a mean age of about 36 years, none had conversion to secondary progressive disease, and just 15% had disability progression on the Expanded Disability Status Scale (EDSS). Cortical thickness changed a mean of 3.6%.
The 26 patients with mild cognitive impairment (classified as failure of up to two tests on the Brief Repeatable Battery) were 38 years old at baseline, and only 4 transitioned to secondary progressive disease. Just over half experienced EDSS progression (an average of 0.85 points) and cortical thickness changed by an average of 17%.
The 13 remaining patients with severe cognitive impairment at baseline (three or more failed tests) were about 44 years of age on average and nearly all (92%) experienced EDSS progression, which averaged 1.31 points. Cortical thickness also changed a mean of 34%. Relapsing disease converted to secondary progressive disease in a total of six patients with severe cognitive impairment.
Patients with severe cognitive impairment at baseline had more severe white matter lesion volume at baseline than did others, but there was no difference in the accumulation of white matter lesions during follow-up between patients with normal cognition, mild cognitive impairment, and severe cognitive impairment, which corroborates previous studies. However, the predictive ability of early cognitive impairment in identifying patients with disability progression on the EDSS and cortical thinning “is in line with several previous longitudinal studies showing a strong relationship between gray matter damage and the development of cognitive impairment,” the authors wrote (Brain. 2012 Oct;135[Pt 10]:2952-61 and Neurology. 2013 Nov 12;81[20]:1759-67).
Although the administration of neuropsychological batteries is time consuming and requires expertise, the authors argued that their results “underline the need of using extended screening neuropsychological batteries instead of brief screening batteries, at least at the time of diagnosis, in order to increase the probability to detect even mild forms of cognitive impairment as early as possible in the disease course.”
Read the full paper online in Multiple Sclerosis (2016 Aug 15. doi: 10.1177/1352458516665496).
FROM MULTIPLE SCLEROSIS

Harold Moses Jr, MD
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Abatacept fails to provide benefit in relapsing-remitting MS
Results from the double-blind, randomized, placebo-controlled phase II ACCLAIM study indicate that abatacept has no effect on reducing the number of new gadolinium-enhancing lesions on MRI in patients with relapsing-remitting multiple sclerosis, according to Samia J. Khoury, MD, of Brigham and Women’s Hospital, Boston, and her colleagues from the Immune Tolerance Network.
In the ACCLAIM (A Cooperative Clinical Study of Abatacept in Multiple Sclerosis) study, 42 patients who received abatacept (Orencia) developed a mean of 0.43 new gadolinium-enhancing lesions by week 24, compared with 1.66 for 20 placebo-treated patients (P = .87). None of the secondary MRI endpoints (lesion volume change and percent brain volume change) and clinical endpoints (changes in Multiple Sclerosis Functional Composite score, Expanded Disability Status Scale [EDSS], and annualized relapse rate) at 24 weeks differed significantly between the groups. The rate of patients who met criteria for no evidence of disease activity or its components (no EDSS progression, no clinical exacerbations, and no new gadolinium-enhancing MRI lesion) from week 8 and before week 28 did not differ between the groups.

During a 28-week extension phase in which the groups switched treatments, patients who switched from abatacept to placebo had a greater number of gadolinium-enhancing lesions than did those who switched from placebo to abatacept (1.25 vs. 0.60, respectively), but the difference was not statistically significant.
Abatacept, which is approved for the treatment of rheumatoid arthritis and juvenile idiopathic arthritis, is a CTLA4 immunoglobulin fusion protein that inhibits the activation of T lymphocytes by targeting the adaptive arm of the immune system by blocking the CD28-B7 costimulatory pathway. It was thought to have potential to reduce immune-mediated disease activity in relapsing-remitting multiple sclerosis because T lymphocytes have been implicated in its pathogenesis.
The investigators closed enrollment for the trial early because of slow accrual. The 65 total patients who enrolled in the trial were about half of the number designated in the trial design (n = 123) in order to demonstrate a treatment effect of 50% reduction of new gadolinium-enhancing MRI lesions.
The number of participants “was too small to demonstrate efficacy at the 50% level,” the investigators wrote, and “low numbers of new gadolinium-enhancing MRI lesions in the study population reduced the chances of demonstrating a treatment effect for abatacept.”
A prior phase II trial of abatacept that was stopped early due to safety events yielded inconclusive results because of an imbalance in the baseline disease activity of participants.
Read the full report online in Multiple Sclerosis Journal (Mult Scler J. 2016 Aug 1. doi: 10.1177/1352458516662727).
Results from the double-blind, randomized, placebo-controlled phase II ACCLAIM study indicate that abatacept has no effect on reducing the number of new gadolinium-enhancing lesions on MRI in patients with relapsing-remitting multiple sclerosis, according to Samia J. Khoury, MD, of Brigham and Women’s Hospital, Boston, and her colleagues from the Immune Tolerance Network.
In the ACCLAIM (A Cooperative Clinical Study of Abatacept in Multiple Sclerosis) study, 42 patients who received abatacept (Orencia) developed a mean of 0.43 new gadolinium-enhancing lesions by week 24, compared with 1.66 for 20 placebo-treated patients (P = .87). None of the secondary MRI endpoints (lesion volume change and percent brain volume change) and clinical endpoints (changes in Multiple Sclerosis Functional Composite score, Expanded Disability Status Scale [EDSS], and annualized relapse rate) at 24 weeks differed significantly between the groups. The rate of patients who met criteria for no evidence of disease activity or its components (no EDSS progression, no clinical exacerbations, and no new gadolinium-enhancing MRI lesion) from week 8 and before week 28 did not differ between the groups.
During a 28-week extension phase in which the groups switched treatments, patients who switched from abatacept to placebo had a greater number of gadolinium-enhancing lesions than did those who switched from placebo to abatacept (1.25 vs. 0.60, respectively), but the difference was not statistically significant.
Abatacept, which is approved for the treatment of rheumatoid arthritis and juvenile idiopathic arthritis, is a CTLA4 immunoglobulin fusion protein that inhibits the activation of T lymphocytes by targeting the adaptive arm of the immune system by blocking the CD28-B7 costimulatory pathway. It was thought to have potential to reduce immune-mediated disease activity in relapsing-remitting multiple sclerosis because T lymphocytes have been implicated in its pathogenesis.
The investigators closed enrollment for the trial early because of slow accrual. The 65 total patients who enrolled in the trial were about half of the number designated in the trial design (n = 123) in order to demonstrate a treatment effect of 50% reduction of new gadolinium-enhancing MRI lesions.
The number of participants “was too small to demonstrate efficacy at the 50% level,” the investigators wrote, and “low numbers of new gadolinium-enhancing MRI lesions in the study population reduced the chances of demonstrating a treatment effect for abatacept.”
A prior phase II trial of abatacept that was stopped early due to safety events yielded inconclusive results because of an imbalance in the baseline disease activity of participants.
Read the full report online in Multiple Sclerosis Journal (Mult Scler J. 2016 Aug 1. doi: 10.1177/1352458516662727).
Results from the double-blind, randomized, placebo-controlled phase II ACCLAIM study indicate that abatacept has no effect on reducing the number of new gadolinium-enhancing lesions on MRI in patients with relapsing-remitting multiple sclerosis, according to Samia J. Khoury, MD, of Brigham and Women’s Hospital, Boston, and her colleagues from the Immune Tolerance Network.
In the ACCLAIM (A Cooperative Clinical Study of Abatacept in Multiple Sclerosis) study, 42 patients who received abatacept (Orencia) developed a mean of 0.43 new gadolinium-enhancing lesions by week 24, compared with 1.66 for 20 placebo-treated patients (P = .87). None of the secondary MRI endpoints (lesion volume change and percent brain volume change) and clinical endpoints (changes in Multiple Sclerosis Functional Composite score, Expanded Disability Status Scale [EDSS], and annualized relapse rate) at 24 weeks differed significantly between the groups. The rate of patients who met criteria for no evidence of disease activity or its components (no EDSS progression, no clinical exacerbations, and no new gadolinium-enhancing MRI lesion) from week 8 and before week 28 did not differ between the groups.
During a 28-week extension phase in which the groups switched treatments, patients who switched from abatacept to placebo had a greater number of gadolinium-enhancing lesions than did those who switched from placebo to abatacept (1.25 vs. 0.60, respectively), but the difference was not statistically significant.
Abatacept, which is approved for the treatment of rheumatoid arthritis and juvenile idiopathic arthritis, is a CTLA4 immunoglobulin fusion protein that inhibits the activation of T lymphocytes by targeting the adaptive arm of the immune system by blocking the CD28-B7 costimulatory pathway. It was thought to have potential to reduce immune-mediated disease activity in relapsing-remitting multiple sclerosis because T lymphocytes have been implicated in its pathogenesis.
The investigators closed enrollment for the trial early because of slow accrual. The 65 total patients who enrolled in the trial were about half of the number designated in the trial design (n = 123) in order to demonstrate a treatment effect of 50% reduction of new gadolinium-enhancing MRI lesions.
The number of participants “was too small to demonstrate efficacy at the 50% level,” the investigators wrote, and “low numbers of new gadolinium-enhancing MRI lesions in the study population reduced the chances of demonstrating a treatment effect for abatacept.”
A prior phase II trial of abatacept that was stopped early due to safety events yielded inconclusive results because of an imbalance in the baseline disease activity of participants.
Read the full report online in Multiple Sclerosis Journal (Mult Scler J. 2016 Aug 1. doi: 10.1177/1352458516662727).
FROM MULTIPLE SCLEROSIS JOURNAL

Is Biotin an Effective Treatment for Progressive MS?
VANCOUVER—MD1003, a pharmaceutical-grade formulation of biotin, reverses disease progression in a significant proportion of patients with progressive multiple sclerosis (MS), according to research presented at the 68th Annual Meeting of the American Academy of Neurology. The drug is safe and remains effective throughout two years of treatment. Delayed treatment with MD1003 is beneficial, but the delay results in higher levels of disability, compared with immediate treatment.
The drug, however, does not significantly improve visual acuity, compared with placebo, in patients with relapsing-remitting or progressive MS and chronic optic neuropathy, said Ayman Tourbah, MD, PhD, Professor of Neurology at Centre Hospitalier Universitaire in Reims, France.

MD1003 and Disease Progression
To investigate MD1003’s effect on disease progression in patients with progressive MS, Dr. Tourbah and colleagues conducted a placebo-controlled, double-blind study. Eligible patients had primary or secondary progressive MS and an Expanded Disability Status Scale (EDSS) score between 4.5 and 7. In addition, participants had to have had disease progression in the previous two years without evidence of clinical or MRI inflammatory activity within the previous year.
In the first phase of the trial, which lasted for 12 months, patients were randomized 2:1 to MD1003 or placebo. A 12-month extension phase followed, in which all patients received MD1003. Patients and clinicians remained blinded to the treatment assignments of the double-blind phase. The trial’s primary end point was the proportion of patients that improved at month nine and had confirmed improvement at month 12 on EDSS or the Timed 25-Foot Walk, compared with the best baseline measures.
In all, 103 patients were randomized to MD1003, and 51 patients were randomized to placebo. Ninety-one patients from the MD1003 group entered the extension phase, along with 42 patients from the placebo group. In the extension phase, 17 patients originally assigned to MD1003 and four patients originally assigned to placebo discontinued treatment. The main reason for discontinuation in both arms was consent withdrawal.
At baseline, the researchers observed no major differences between treatment arms with regard to sex ratio, mean age, mean disease duration, and mean EDSS score. Slightly more patients in the MD1003 arm had primary progressive MS than in the placebo arm. Approximately 40% of patients were taking concomitant disease-modifying therapies.
In the double-blind phase, 12.6% of patients receiving MD1003 had improvement at month nine and confirmed improvement at month 12. No patient receiving placebo had these outcomes, and the difference between groups was statistically significant. In the extension phase, treatment efficacy was maintained among patients randomized to MD1003 and became apparent in patients who were switched from placebo to MD1003. The level of disability remained higher in the latter group, however.
The rate of adverse events was consistent from the trial’s double-blind phase through its extension phase. The most frequent side effects were infections and infestations and disorders involving the nervous, gastrointestinal, musculoskeletal, and connective tissue systems. Patients originally randomized to placebo did not have more adverse events after switching to MD1003. A few cases of apparent hyperthyroidism were encountered. These are known to be related to interferences between high doses of biotin and biotin-based laboratory tests. Two cases of neoplasm were reported in the extension phase and were not reported as related to the treatment.
“This is the first time that a drug has reversed the progression of the disease in a statistically significant proportion of patients,” said Dr. Tourbah. “Almost no progression was observed in patients treated with MD1003 for 24 months, and this has never been observed before. When we compare these results to other trials in progressive MS that involved more than 6,000 patients overall, this is clearly the best effect size ever observed.”
MD1003 and Visual Acuity
In a separate study, Dr. Tourbah and colleagues analyzed the efficacy of MD1003, compared with placebo, in patients with relapsing-remitting or progressive MS and visual loss related to chronic optic neuropathy. Eligible participants had at least one eye with confirmed visual acuity of 20/40 or less on a standard chart, and worsening of visual acuity within the previous three years. The investigators categorized patients as having progressive optic neuropathy (ie, progressive visual loss observed at two separate ophthalmologic examinations within the three years preceding inclusion) or nonprogressive optic neuropathy (ie, a fixed visual loss for six months or more following an episode of acute optic neuritis).
Patients were randomized 2:1 to MD1003 or placebo for six months. In a subsequent six-month extension phase, patients on placebo were switched to MD1003. Patients and clinicians remained blinded to the treatments that had been given in the double-blind phase. The primary end point was the mean change in visual acuity between month zero and month six in the eye with worse visual acuity at baseline and visual worsening within the previous three years. Dr. Tourbah and colleagues used the Early Treatment Diabetic Retinopathy Study chart at 100% to evaluate visual acuity.
In all, 65 patients were randomized to MD1003, and 28 patients to placebo. One patient did not enter the extension phase of the trial. Demographic characteristics were similar in the two treatment arms, but the majority of patients had nonprogressive chronic optic neuropathy, especially in the placebo arm. Slightly more patients randomized to MD1003 were taking disease-modifying therapies, compared with the placebo arm.
At six months, visual acuity improved in all patients. Improvement was more pronounced in patients receiving MD1003, but the difference between groups was not statistically significant. When the investigators examined only patients with nonprogressive chronic optic neuropathy, they found no difference between treatment groups at six months. When they examined participants with progressive chronic optic neuropathy, however, patients randomized to MD1003 had improved visual acuity at six months, while patients randomized to placebo had worsened visual acuity.
In the extension phase, patients who had improved on placebo continued to improve after switching to MD1003, and patients who had improved on MD1003 continued to improve. The researchers saw no differences in visual acuity between groups at 12 months. Among participants with nonprogressive chronic optic neuropathy, visual acuity improved in both treatment arms, and the researchers saw little difference between them at 12 months. Among patients with progressive chronic optic neuropathy, disease progression stopped in participants who switched from placebo to MD1003.
These results suggest that the treatment has no indication in patients with relapsing-remitting MS and show a trend toward efficacy in patients with progressive chronic optic neuropathy, which is consistent with the results of the MS-SPI study.
In addition, Dr. Tourbah and colleagues observed no major differences between treatment arms regarding the most frequent adverse events (ie, infections and infestations, nervous system disorders, and intestinal disorders). They noted, however, that more patients receiving MD1003 had relapses, compared with patients receiving placebo. “Whether MD1003 may trigger exacerbations in patients with relapsing-remitting MS deserves further investigation,” Dr. Tourbah concluded.
—Erik Greb
Suggested Reading
Sedel F, Bernard D, Mock DM, Tourbah A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology. 2015 Sep 5 [Epub ahead of print].
Sedel F, Papeix C, Bellanger A, et al. High doses of biotin in chronic progressive multiple sclerosis: a pilot study. Mult Scler Relat Disord. 2015;4(2):159-169.
VANCOUVER—MD1003, a pharmaceutical-grade formulation of biotin, reverses disease progression in a significant proportion of patients with progressive multiple sclerosis (MS), according to research presented at the 68th Annual Meeting of the American Academy of Neurology. The drug is safe and remains effective throughout two years of treatment. Delayed treatment with MD1003 is beneficial, but the delay results in higher levels of disability, compared with immediate treatment.
The drug, however, does not significantly improve visual acuity, compared with placebo, in patients with relapsing-remitting or progressive MS and chronic optic neuropathy, said Ayman Tourbah, MD, PhD, Professor of Neurology at Centre Hospitalier Universitaire in Reims, France.
MD1003 and Disease Progression
To investigate MD1003’s effect on disease progression in patients with progressive MS, Dr. Tourbah and colleagues conducted a placebo-controlled, double-blind study. Eligible patients had primary or secondary progressive MS and an Expanded Disability Status Scale (EDSS) score between 4.5 and 7. In addition, participants had to have had disease progression in the previous two years without evidence of clinical or MRI inflammatory activity within the previous year.
In the first phase of the trial, which lasted for 12 months, patients were randomized 2:1 to MD1003 or placebo. A 12-month extension phase followed, in which all patients received MD1003. Patients and clinicians remained blinded to the treatment assignments of the double-blind phase. The trial’s primary end point was the proportion of patients that improved at month nine and had confirmed improvement at month 12 on EDSS or the Timed 25-Foot Walk, compared with the best baseline measures.
In all, 103 patients were randomized to MD1003, and 51 patients were randomized to placebo. Ninety-one patients from the MD1003 group entered the extension phase, along with 42 patients from the placebo group. In the extension phase, 17 patients originally assigned to MD1003 and four patients originally assigned to placebo discontinued treatment. The main reason for discontinuation in both arms was consent withdrawal.
At baseline, the researchers observed no major differences between treatment arms with regard to sex ratio, mean age, mean disease duration, and mean EDSS score. Slightly more patients in the MD1003 arm had primary progressive MS than in the placebo arm. Approximately 40% of patients were taking concomitant disease-modifying therapies.
In the double-blind phase, 12.6% of patients receiving MD1003 had improvement at month nine and confirmed improvement at month 12. No patient receiving placebo had these outcomes, and the difference between groups was statistically significant. In the extension phase, treatment efficacy was maintained among patients randomized to MD1003 and became apparent in patients who were switched from placebo to MD1003. The level of disability remained higher in the latter group, however.
The rate of adverse events was consistent from the trial’s double-blind phase through its extension phase. The most frequent side effects were infections and infestations and disorders involving the nervous, gastrointestinal, musculoskeletal, and connective tissue systems. Patients originally randomized to placebo did not have more adverse events after switching to MD1003. A few cases of apparent hyperthyroidism were encountered. These are known to be related to interferences between high doses of biotin and biotin-based laboratory tests. Two cases of neoplasm were reported in the extension phase and were not reported as related to the treatment.
“This is the first time that a drug has reversed the progression of the disease in a statistically significant proportion of patients,” said Dr. Tourbah. “Almost no progression was observed in patients treated with MD1003 for 24 months, and this has never been observed before. When we compare these results to other trials in progressive MS that involved more than 6,000 patients overall, this is clearly the best effect size ever observed.”
MD1003 and Visual Acuity
In a separate study, Dr. Tourbah and colleagues analyzed the efficacy of MD1003, compared with placebo, in patients with relapsing-remitting or progressive MS and visual loss related to chronic optic neuropathy. Eligible participants had at least one eye with confirmed visual acuity of 20/40 or less on a standard chart, and worsening of visual acuity within the previous three years. The investigators categorized patients as having progressive optic neuropathy (ie, progressive visual loss observed at two separate ophthalmologic examinations within the three years preceding inclusion) or nonprogressive optic neuropathy (ie, a fixed visual loss for six months or more following an episode of acute optic neuritis).
Patients were randomized 2:1 to MD1003 or placebo for six months. In a subsequent six-month extension phase, patients on placebo were switched to MD1003. Patients and clinicians remained blinded to the treatments that had been given in the double-blind phase. The primary end point was the mean change in visual acuity between month zero and month six in the eye with worse visual acuity at baseline and visual worsening within the previous three years. Dr. Tourbah and colleagues used the Early Treatment Diabetic Retinopathy Study chart at 100% to evaluate visual acuity.
In all, 65 patients were randomized to MD1003, and 28 patients to placebo. One patient did not enter the extension phase of the trial. Demographic characteristics were similar in the two treatment arms, but the majority of patients had nonprogressive chronic optic neuropathy, especially in the placebo arm. Slightly more patients randomized to MD1003 were taking disease-modifying therapies, compared with the placebo arm.
At six months, visual acuity improved in all patients. Improvement was more pronounced in patients receiving MD1003, but the difference between groups was not statistically significant. When the investigators examined only patients with nonprogressive chronic optic neuropathy, they found no difference between treatment groups at six months. When they examined participants with progressive chronic optic neuropathy, however, patients randomized to MD1003 had improved visual acuity at six months, while patients randomized to placebo had worsened visual acuity.
In the extension phase, patients who had improved on placebo continued to improve after switching to MD1003, and patients who had improved on MD1003 continued to improve. The researchers saw no differences in visual acuity between groups at 12 months. Among participants with nonprogressive chronic optic neuropathy, visual acuity improved in both treatment arms, and the researchers saw little difference between them at 12 months. Among patients with progressive chronic optic neuropathy, disease progression stopped in participants who switched from placebo to MD1003.
These results suggest that the treatment has no indication in patients with relapsing-remitting MS and show a trend toward efficacy in patients with progressive chronic optic neuropathy, which is consistent with the results of the MS-SPI study.
In addition, Dr. Tourbah and colleagues observed no major differences between treatment arms regarding the most frequent adverse events (ie, infections and infestations, nervous system disorders, and intestinal disorders). They noted, however, that more patients receiving MD1003 had relapses, compared with patients receiving placebo. “Whether MD1003 may trigger exacerbations in patients with relapsing-remitting MS deserves further investigation,” Dr. Tourbah concluded.
—Erik Greb
VANCOUVER—MD1003, a pharmaceutical-grade formulation of biotin, reverses disease progression in a significant proportion of patients with progressive multiple sclerosis (MS), according to research presented at the 68th Annual Meeting of the American Academy of Neurology. The drug is safe and remains effective throughout two years of treatment. Delayed treatment with MD1003 is beneficial, but the delay results in higher levels of disability, compared with immediate treatment.
The drug, however, does not significantly improve visual acuity, compared with placebo, in patients with relapsing-remitting or progressive MS and chronic optic neuropathy, said Ayman Tourbah, MD, PhD, Professor of Neurology at Centre Hospitalier Universitaire in Reims, France.
MD1003 and Disease Progression
To investigate MD1003’s effect on disease progression in patients with progressive MS, Dr. Tourbah and colleagues conducted a placebo-controlled, double-blind study. Eligible patients had primary or secondary progressive MS and an Expanded Disability Status Scale (EDSS) score between 4.5 and 7. In addition, participants had to have had disease progression in the previous two years without evidence of clinical or MRI inflammatory activity within the previous year.
In the first phase of the trial, which lasted for 12 months, patients were randomized 2:1 to MD1003 or placebo. A 12-month extension phase followed, in which all patients received MD1003. Patients and clinicians remained blinded to the treatment assignments of the double-blind phase. The trial’s primary end point was the proportion of patients that improved at month nine and had confirmed improvement at month 12 on EDSS or the Timed 25-Foot Walk, compared with the best baseline measures.
In all, 103 patients were randomized to MD1003, and 51 patients were randomized to placebo. Ninety-one patients from the MD1003 group entered the extension phase, along with 42 patients from the placebo group. In the extension phase, 17 patients originally assigned to MD1003 and four patients originally assigned to placebo discontinued treatment. The main reason for discontinuation in both arms was consent withdrawal.
At baseline, the researchers observed no major differences between treatment arms with regard to sex ratio, mean age, mean disease duration, and mean EDSS score. Slightly more patients in the MD1003 arm had primary progressive MS than in the placebo arm. Approximately 40% of patients were taking concomitant disease-modifying therapies.
In the double-blind phase, 12.6% of patients receiving MD1003 had improvement at month nine and confirmed improvement at month 12. No patient receiving placebo had these outcomes, and the difference between groups was statistically significant. In the extension phase, treatment efficacy was maintained among patients randomized to MD1003 and became apparent in patients who were switched from placebo to MD1003. The level of disability remained higher in the latter group, however.
The rate of adverse events was consistent from the trial’s double-blind phase through its extension phase. The most frequent side effects were infections and infestations and disorders involving the nervous, gastrointestinal, musculoskeletal, and connective tissue systems. Patients originally randomized to placebo did not have more adverse events after switching to MD1003. A few cases of apparent hyperthyroidism were encountered. These are known to be related to interferences between high doses of biotin and biotin-based laboratory tests. Two cases of neoplasm were reported in the extension phase and were not reported as related to the treatment.
“This is the first time that a drug has reversed the progression of the disease in a statistically significant proportion of patients,” said Dr. Tourbah. “Almost no progression was observed in patients treated with MD1003 for 24 months, and this has never been observed before. When we compare these results to other trials in progressive MS that involved more than 6,000 patients overall, this is clearly the best effect size ever observed.”
MD1003 and Visual Acuity
In a separate study, Dr. Tourbah and colleagues analyzed the efficacy of MD1003, compared with placebo, in patients with relapsing-remitting or progressive MS and visual loss related to chronic optic neuropathy. Eligible participants had at least one eye with confirmed visual acuity of 20/40 or less on a standard chart, and worsening of visual acuity within the previous three years. The investigators categorized patients as having progressive optic neuropathy (ie, progressive visual loss observed at two separate ophthalmologic examinations within the three years preceding inclusion) or nonprogressive optic neuropathy (ie, a fixed visual loss for six months or more following an episode of acute optic neuritis).
Patients were randomized 2:1 to MD1003 or placebo for six months. In a subsequent six-month extension phase, patients on placebo were switched to MD1003. Patients and clinicians remained blinded to the treatments that had been given in the double-blind phase. The primary end point was the mean change in visual acuity between month zero and month six in the eye with worse visual acuity at baseline and visual worsening within the previous three years. Dr. Tourbah and colleagues used the Early Treatment Diabetic Retinopathy Study chart at 100% to evaluate visual acuity.
In all, 65 patients were randomized to MD1003, and 28 patients to placebo. One patient did not enter the extension phase of the trial. Demographic characteristics were similar in the two treatment arms, but the majority of patients had nonprogressive chronic optic neuropathy, especially in the placebo arm. Slightly more patients randomized to MD1003 were taking disease-modifying therapies, compared with the placebo arm.
At six months, visual acuity improved in all patients. Improvement was more pronounced in patients receiving MD1003, but the difference between groups was not statistically significant. When the investigators examined only patients with nonprogressive chronic optic neuropathy, they found no difference between treatment groups at six months. When they examined participants with progressive chronic optic neuropathy, however, patients randomized to MD1003 had improved visual acuity at six months, while patients randomized to placebo had worsened visual acuity.
In the extension phase, patients who had improved on placebo continued to improve after switching to MD1003, and patients who had improved on MD1003 continued to improve. The researchers saw no differences in visual acuity between groups at 12 months. Among participants with nonprogressive chronic optic neuropathy, visual acuity improved in both treatment arms, and the researchers saw little difference between them at 12 months. Among patients with progressive chronic optic neuropathy, disease progression stopped in participants who switched from placebo to MD1003.
These results suggest that the treatment has no indication in patients with relapsing-remitting MS and show a trend toward efficacy in patients with progressive chronic optic neuropathy, which is consistent with the results of the MS-SPI study.
In addition, Dr. Tourbah and colleagues observed no major differences between treatment arms regarding the most frequent adverse events (ie, infections and infestations, nervous system disorders, and intestinal disorders). They noted, however, that more patients receiving MD1003 had relapses, compared with patients receiving placebo. “Whether MD1003 may trigger exacerbations in patients with relapsing-remitting MS deserves further investigation,” Dr. Tourbah concluded.
—Erik Greb
Suggested Reading
Sedel F, Bernard D, Mock DM, Tourbah A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology. 2015 Sep 5 [Epub ahead of print].
Sedel F, Papeix C, Bellanger A, et al. High doses of biotin in chronic progressive multiple sclerosis: a pilot study. Mult Scler Relat Disord. 2015;4(2):159-169.
Suggested Reading
Sedel F, Bernard D, Mock DM, Tourbah A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology. 2015 Sep 5 [Epub ahead of print].
Sedel F, Papeix C, Bellanger A, et al. High doses of biotin in chronic progressive multiple sclerosis: a pilot study. Mult Scler Relat Disord. 2015;4(2):159-169.

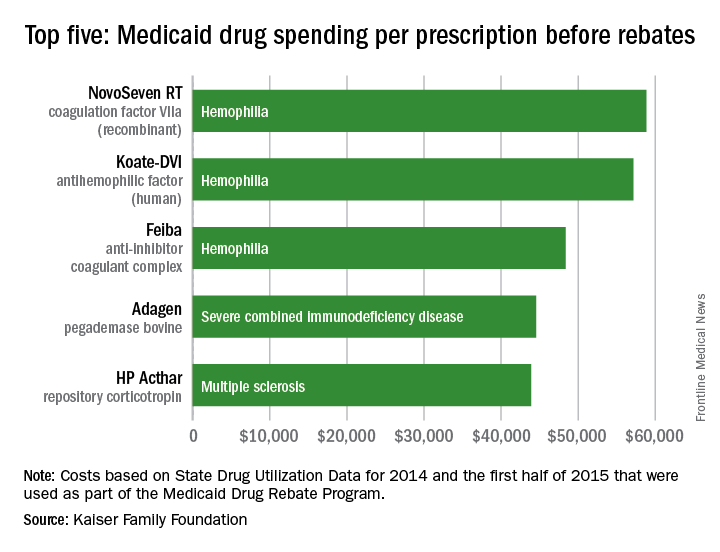
Hemophilia drugs top Medicaid spending per prescription
Medicaid’s three most expensive drugs by spending per prescription were for treatment of hemophilia, with the cost leader coming in at almost $59,000, according to an analysis by the Kaiser Family Foundation covering 2014 and the first half of 2015.
The trio of hemophilia drugs was topped by NovoSeven RT (coagulation factor VIIa [recombinant]), with Koate-DVI (antihemophilic factor [human]) second at $57,000 per prescription and Feiba (anti-inhibitor coagulant complex) third at $48,000 for each prescription.
None of the Medicaid costs include rebates since those data are unavailable to the public, Kaiser noted.
The fourth and fifth most expensive drugs were Adagen (pegademase bovine), which is used in the treatment of severe combined immunodeficiency disease associated with a deficiency of adenosine deaminase and cost Medicaid $45,000 per prescription, and the multiple sclerosis drug HP Acthar (repository corticotropin), which went for almost $44,000 a prescription, Kaiser said in its analysis, which used State Drug Utilization Data that are part of the Medicaid Drug Rebate Program.

Medicaid’s three most expensive drugs by spending per prescription were for treatment of hemophilia, with the cost leader coming in at almost $59,000, according to an analysis by the Kaiser Family Foundation covering 2014 and the first half of 2015.
The trio of hemophilia drugs was topped by NovoSeven RT (coagulation factor VIIa [recombinant]), with Koate-DVI (antihemophilic factor [human]) second at $57,000 per prescription and Feiba (anti-inhibitor coagulant complex) third at $48,000 for each prescription.
None of the Medicaid costs include rebates since those data are unavailable to the public, Kaiser noted.
The fourth and fifth most expensive drugs were Adagen (pegademase bovine), which is used in the treatment of severe combined immunodeficiency disease associated with a deficiency of adenosine deaminase and cost Medicaid $45,000 per prescription, and the multiple sclerosis drug HP Acthar (repository corticotropin), which went for almost $44,000 a prescription, Kaiser said in its analysis, which used State Drug Utilization Data that are part of the Medicaid Drug Rebate Program.
Medicaid’s three most expensive drugs by spending per prescription were for treatment of hemophilia, with the cost leader coming in at almost $59,000, according to an analysis by the Kaiser Family Foundation covering 2014 and the first half of 2015.
The trio of hemophilia drugs was topped by NovoSeven RT (coagulation factor VIIa [recombinant]), with Koate-DVI (antihemophilic factor [human]) second at $57,000 per prescription and Feiba (anti-inhibitor coagulant complex) third at $48,000 for each prescription.
None of the Medicaid costs include rebates since those data are unavailable to the public, Kaiser noted.
The fourth and fifth most expensive drugs were Adagen (pegademase bovine), which is used in the treatment of severe combined immunodeficiency disease associated with a deficiency of adenosine deaminase and cost Medicaid $45,000 per prescription, and the multiple sclerosis drug HP Acthar (repository corticotropin), which went for almost $44,000 a prescription, Kaiser said in its analysis, which used State Drug Utilization Data that are part of the Medicaid Drug Rebate Program.
FDA accepting comments on draft guidelines on compounding law
The Food and Drug Administration is currently accepting public comments on the agency’s proposed plans to implement a law that will restrict compounding of human drug products.
A statement issued by the FDA provides links to two draft guidances that describe how the agency “would implement provisions of federal law that restrict compounding human drug products that are essentially copies of commercially available or approved drug products.” One draft guidance and the legal restrictions referenced therein are relevant to physicians and pharmacists, as well as state-licensed pharmacies or federal facilities that compound drugs, according to the FDA. The other guidance applies to outsourcing facilities.
Although compounded drug products, such as a medication made without a dye for a patient allergic to that dye, or a medication made into liquid form for a patient who cannot swallow a pill, “may benefit certain patients whose medical needs cannot be met by a commercially available or an FDA-approved drug product,” the FDA statement said. “Taking compounded drug products that are essentially copies of a commercially available or approved drug needlessly exposes patients to drug products that FDA has not evaluated for safety, effectiveness, and quality. In addition, the compounded drugs may not have been produced according to appropriate quality standards. Such compounding would also undermine the new drug approval and over-the-counter drug monograph systems in the United States.”
The statement refers to serious adverse events, including infections and deaths that have resulted from “poor-quality” compounded drugs.
Written or electronic comments can be submitted until Oct. 11, and information on submitting comments is available at regulations.gov.
The Food and Drug Administration is currently accepting public comments on the agency’s proposed plans to implement a law that will restrict compounding of human drug products.
A statement issued by the FDA provides links to two draft guidances that describe how the agency “would implement provisions of federal law that restrict compounding human drug products that are essentially copies of commercially available or approved drug products.” One draft guidance and the legal restrictions referenced therein are relevant to physicians and pharmacists, as well as state-licensed pharmacies or federal facilities that compound drugs, according to the FDA. The other guidance applies to outsourcing facilities.
Although compounded drug products, such as a medication made without a dye for a patient allergic to that dye, or a medication made into liquid form for a patient who cannot swallow a pill, “may benefit certain patients whose medical needs cannot be met by a commercially available or an FDA-approved drug product,” the FDA statement said. “Taking compounded drug products that are essentially copies of a commercially available or approved drug needlessly exposes patients to drug products that FDA has not evaluated for safety, effectiveness, and quality. In addition, the compounded drugs may not have been produced according to appropriate quality standards. Such compounding would also undermine the new drug approval and over-the-counter drug monograph systems in the United States.”
The statement refers to serious adverse events, including infections and deaths that have resulted from “poor-quality” compounded drugs.
Written or electronic comments can be submitted until Oct. 11, and information on submitting comments is available at regulations.gov.
The Food and Drug Administration is currently accepting public comments on the agency’s proposed plans to implement a law that will restrict compounding of human drug products.
A statement issued by the FDA provides links to two draft guidances that describe how the agency “would implement provisions of federal law that restrict compounding human drug products that are essentially copies of commercially available or approved drug products.” One draft guidance and the legal restrictions referenced therein are relevant to physicians and pharmacists, as well as state-licensed pharmacies or federal facilities that compound drugs, according to the FDA. The other guidance applies to outsourcing facilities.
Although compounded drug products, such as a medication made without a dye for a patient allergic to that dye, or a medication made into liquid form for a patient who cannot swallow a pill, “may benefit certain patients whose medical needs cannot be met by a commercially available or an FDA-approved drug product,” the FDA statement said. “Taking compounded drug products that are essentially copies of a commercially available or approved drug needlessly exposes patients to drug products that FDA has not evaluated for safety, effectiveness, and quality. In addition, the compounded drugs may not have been produced according to appropriate quality standards. Such compounding would also undermine the new drug approval and over-the-counter drug monograph systems in the United States.”
The statement refers to serious adverse events, including infections and deaths that have resulted from “poor-quality” compounded drugs.
Written or electronic comments can be submitted until Oct. 11, and information on submitting comments is available at regulations.gov.
Peter Chin, MD
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Siponimod shows promise through 24 months in relapsing-remitting MS
Once-daily oral siponimod was associated with sustained effects on MRI outcomes at 24 months in patients with relapsing-remitting multiple sclerosis in a dose-blinded extension of the phase II study.
Disease activity was low, “with some evidence of greater benefit associated with siponimod at 10-mg, 2-mg, and 1.25-mg doses than with siponimod at 0.25-mg and 0.5-mg doses. No new safety signals emerged, and dose titration at treatment initiation mitigated cardiac effects,” Ludwig Kappos, MD, of University Hospital Basel (Switzerland), and his associates wrote in JAMA Neurology. “With similar efficacy but lower rates of lymphopenia relative to the 10-mg dose, siponimod 2 mg, has been chosen for further development,” they wrote.

Siponimod (BAF312) is a selective sphingosine 1-phosphate receptor (S1P1,5) modulator that was evaluated for up to 6 months at five doses in the phase II BOLD (BAF312 on MRI Lesion Given Once Daily) study. Patients with relapsing-remitting MS who received 10 mg siponimod had up to 80% reductions in MRI combined unique active lesions (CUALs, or gadolinium-enhancing T1 lesions and/or new and newly enlarging T2 lesions, without double counting), compared with the placebo group. The 2-mg and 0.5-mg dose cohorts had reductions of 72% and 50%, respectively. The current study was a 24-month, dose-blinded extension phase that included 185 participants (73% of 252 eligible patients), of whom 33 patients received 10 mg siponimod, 29 received 2 mg, 43 received 1.25 mg, 29 received 0.5 mg, and 50 received 0.25 mg (JAMA Neurol. 2016 July 5. doi: 10.1001/jamaneurol.2016.1451).
Average reductions in gadolinium-enhancing T1 lesion counts were sustained in the 10-mg, 2-mg, 1.25-mg, and 0.5-mg dose groups at month 24, the researchers reported. The 1.25-mg and 2-mg doses yielded the highest proportions of patients free from new MRI activity (58% for both dose groups) and free from new or newly enlarging T2 lesions (61% and 58%, respectively). New or newly enlarging T2 lesions were numerically lower at doses exceeding 0.25 mg. “There were no clear changes in normalized brain volume within or between any of the treatment groups,” the investigators added.
Dose titration during the first 10 days of treatment mitigated bradycardia and atrioventricular conduction effects. Rates of adverse events within dose groups ranged from 84% to 97% and did not show a trend with dose size. Serious adverse events affected nine patients (5%) and included one case each of otosclerosis, gastritis, anaphylaxis, acute pyelonephritis, femoral and ankle fractures, basal cell carcinoma, cervical neoplasm, and abortion. Thirteen patients (7%) required treatment interruptions because of adverse events, of which seven consisted of lymphopenia or decreased lymphocyte count at the 10-mg dose. Other adverse events leading to dose interruptions or adjustments included neutropenia, upper respiratory tract infection, elevated hepatic transaminases, and hypertension.
Novartis is developing siponimod and funded the study. Coinvestigators employed by Novartis participated in all aspects of the study, including interpretation of the data and manuscript submission. Dr. Kappos disclosed financial ties to Novartis and numerous other pharmaceutical companies, foundations, and societies.
Dr. Kappos and colleagues acknowledged some limitations including the decreasing proportion of patients with evaluable MRIs and low numbers within each dosing group. Furthermore, the variable time between the end of the BOLD study and start of the extension phase, and the lack of a control group, limit conclusive evidence statements. Nonetheless, the current data may be the most relevant to report the safety and efficacy of long-term siponimod use in relapsing-remitting MS.
The full cumulative dose effect of siponimod beginning from the initiation of the BOLD study cannot be completely evidenced because of variable and prolonged times between the end of the BOLD study and initiating the extension phase, the dose-titration escalation procedure, and limiting reporting to the extension phase. The low numbers of participants do not allow an assessment of dose-related adverse events. The observed adverse events may be expected based on the pharmacokinetic profile of siponimod.
The extension phase of clinical trials performed without a reference treatment arm can result in challenges for interpreting and contextualizing findings. Where possible, a potential approach would be to rerandomize patients prior to the start of an extension phase and maintain a placebo or comparator arm.
Edward R. Hammond, MD, PhD, MPH, is with AstraZeneca’s Medical Evidence and Observational Research Center in Gaithersburg, Md. These comments are based on his accompanying editorial (JAMA Neurol. 2016 July 5. doi: 10.1001/jamaneurol.2016.2284).
Dr. Kappos and colleagues acknowledged some limitations including the decreasing proportion of patients with evaluable MRIs and low numbers within each dosing group. Furthermore, the variable time between the end of the BOLD study and start of the extension phase, and the lack of a control group, limit conclusive evidence statements. Nonetheless, the current data may be the most relevant to report the safety and efficacy of long-term siponimod use in relapsing-remitting MS.
The full cumulative dose effect of siponimod beginning from the initiation of the BOLD study cannot be completely evidenced because of variable and prolonged times between the end of the BOLD study and initiating the extension phase, the dose-titration escalation procedure, and limiting reporting to the extension phase. The low numbers of participants do not allow an assessment of dose-related adverse events. The observed adverse events may be expected based on the pharmacokinetic profile of siponimod.
The extension phase of clinical trials performed without a reference treatment arm can result in challenges for interpreting and contextualizing findings. Where possible, a potential approach would be to rerandomize patients prior to the start of an extension phase and maintain a placebo or comparator arm.
Edward R. Hammond, MD, PhD, MPH, is with AstraZeneca’s Medical Evidence and Observational Research Center in Gaithersburg, Md. These comments are based on his accompanying editorial (JAMA Neurol. 2016 July 5. doi: 10.1001/jamaneurol.2016.2284).
Dr. Kappos and colleagues acknowledged some limitations including the decreasing proportion of patients with evaluable MRIs and low numbers within each dosing group. Furthermore, the variable time between the end of the BOLD study and start of the extension phase, and the lack of a control group, limit conclusive evidence statements. Nonetheless, the current data may be the most relevant to report the safety and efficacy of long-term siponimod use in relapsing-remitting MS.
The full cumulative dose effect of siponimod beginning from the initiation of the BOLD study cannot be completely evidenced because of variable and prolonged times between the end of the BOLD study and initiating the extension phase, the dose-titration escalation procedure, and limiting reporting to the extension phase. The low numbers of participants do not allow an assessment of dose-related adverse events. The observed adverse events may be expected based on the pharmacokinetic profile of siponimod.
The extension phase of clinical trials performed without a reference treatment arm can result in challenges for interpreting and contextualizing findings. Where possible, a potential approach would be to rerandomize patients prior to the start of an extension phase and maintain a placebo or comparator arm.
Edward R. Hammond, MD, PhD, MPH, is with AstraZeneca’s Medical Evidence and Observational Research Center in Gaithersburg, Md. These comments are based on his accompanying editorial (JAMA Neurol. 2016 July 5. doi: 10.1001/jamaneurol.2016.2284).
Once-daily oral siponimod was associated with sustained effects on MRI outcomes at 24 months in patients with relapsing-remitting multiple sclerosis in a dose-blinded extension of the phase II study.
Disease activity was low, “with some evidence of greater benefit associated with siponimod at 10-mg, 2-mg, and 1.25-mg doses than with siponimod at 0.25-mg and 0.5-mg doses. No new safety signals emerged, and dose titration at treatment initiation mitigated cardiac effects,” Ludwig Kappos, MD, of University Hospital Basel (Switzerland), and his associates wrote in JAMA Neurology. “With similar efficacy but lower rates of lymphopenia relative to the 10-mg dose, siponimod 2 mg, has been chosen for further development,” they wrote.
Siponimod (BAF312) is a selective sphingosine 1-phosphate receptor (S1P1,5) modulator that was evaluated for up to 6 months at five doses in the phase II BOLD (BAF312 on MRI Lesion Given Once Daily) study. Patients with relapsing-remitting MS who received 10 mg siponimod had up to 80% reductions in MRI combined unique active lesions (CUALs, or gadolinium-enhancing T1 lesions and/or new and newly enlarging T2 lesions, without double counting), compared with the placebo group. The 2-mg and 0.5-mg dose cohorts had reductions of 72% and 50%, respectively. The current study was a 24-month, dose-blinded extension phase that included 185 participants (73% of 252 eligible patients), of whom 33 patients received 10 mg siponimod, 29 received 2 mg, 43 received 1.25 mg, 29 received 0.5 mg, and 50 received 0.25 mg (JAMA Neurol. 2016 July 5. doi: 10.1001/jamaneurol.2016.1451).
Average reductions in gadolinium-enhancing T1 lesion counts were sustained in the 10-mg, 2-mg, 1.25-mg, and 0.5-mg dose groups at month 24, the researchers reported. The 1.25-mg and 2-mg doses yielded the highest proportions of patients free from new MRI activity (58% for both dose groups) and free from new or newly enlarging T2 lesions (61% and 58%, respectively). New or newly enlarging T2 lesions were numerically lower at doses exceeding 0.25 mg. “There were no clear changes in normalized brain volume within or between any of the treatment groups,” the investigators added.
Dose titration during the first 10 days of treatment mitigated bradycardia and atrioventricular conduction effects. Rates of adverse events within dose groups ranged from 84% to 97% and did not show a trend with dose size. Serious adverse events affected nine patients (5%) and included one case each of otosclerosis, gastritis, anaphylaxis, acute pyelonephritis, femoral and ankle fractures, basal cell carcinoma, cervical neoplasm, and abortion. Thirteen patients (7%) required treatment interruptions because of adverse events, of which seven consisted of lymphopenia or decreased lymphocyte count at the 10-mg dose. Other adverse events leading to dose interruptions or adjustments included neutropenia, upper respiratory tract infection, elevated hepatic transaminases, and hypertension.
Novartis is developing siponimod and funded the study. Coinvestigators employed by Novartis participated in all aspects of the study, including interpretation of the data and manuscript submission. Dr. Kappos disclosed financial ties to Novartis and numerous other pharmaceutical companies, foundations, and societies.
Once-daily oral siponimod was associated with sustained effects on MRI outcomes at 24 months in patients with relapsing-remitting multiple sclerosis in a dose-blinded extension of the phase II study.
Disease activity was low, “with some evidence of greater benefit associated with siponimod at 10-mg, 2-mg, and 1.25-mg doses than with siponimod at 0.25-mg and 0.5-mg doses. No new safety signals emerged, and dose titration at treatment initiation mitigated cardiac effects,” Ludwig Kappos, MD, of University Hospital Basel (Switzerland), and his associates wrote in JAMA Neurology. “With similar efficacy but lower rates of lymphopenia relative to the 10-mg dose, siponimod 2 mg, has been chosen for further development,” they wrote.
Siponimod (BAF312) is a selective sphingosine 1-phosphate receptor (S1P1,5) modulator that was evaluated for up to 6 months at five doses in the phase II BOLD (BAF312 on MRI Lesion Given Once Daily) study. Patients with relapsing-remitting MS who received 10 mg siponimod had up to 80% reductions in MRI combined unique active lesions (CUALs, or gadolinium-enhancing T1 lesions and/or new and newly enlarging T2 lesions, without double counting), compared with the placebo group. The 2-mg and 0.5-mg dose cohorts had reductions of 72% and 50%, respectively. The current study was a 24-month, dose-blinded extension phase that included 185 participants (73% of 252 eligible patients), of whom 33 patients received 10 mg siponimod, 29 received 2 mg, 43 received 1.25 mg, 29 received 0.5 mg, and 50 received 0.25 mg (JAMA Neurol. 2016 July 5. doi: 10.1001/jamaneurol.2016.1451).
Average reductions in gadolinium-enhancing T1 lesion counts were sustained in the 10-mg, 2-mg, 1.25-mg, and 0.5-mg dose groups at month 24, the researchers reported. The 1.25-mg and 2-mg doses yielded the highest proportions of patients free from new MRI activity (58% for both dose groups) and free from new or newly enlarging T2 lesions (61% and 58%, respectively). New or newly enlarging T2 lesions were numerically lower at doses exceeding 0.25 mg. “There were no clear changes in normalized brain volume within or between any of the treatment groups,” the investigators added.
Dose titration during the first 10 days of treatment mitigated bradycardia and atrioventricular conduction effects. Rates of adverse events within dose groups ranged from 84% to 97% and did not show a trend with dose size. Serious adverse events affected nine patients (5%) and included one case each of otosclerosis, gastritis, anaphylaxis, acute pyelonephritis, femoral and ankle fractures, basal cell carcinoma, cervical neoplasm, and abortion. Thirteen patients (7%) required treatment interruptions because of adverse events, of which seven consisted of lymphopenia or decreased lymphocyte count at the 10-mg dose. Other adverse events leading to dose interruptions or adjustments included neutropenia, upper respiratory tract infection, elevated hepatic transaminases, and hypertension.
Novartis is developing siponimod and funded the study. Coinvestigators employed by Novartis participated in all aspects of the study, including interpretation of the data and manuscript submission. Dr. Kappos disclosed financial ties to Novartis and numerous other pharmaceutical companies, foundations, and societies.
FROM JAMA NEUROLOGY
Key clinical point: Siponimod was associated with sustained efficacy at 24 months among patients with relapsing-remitting multiple sclerosis.
Major finding: The proportion of patients free from new MRI activity was highest (58%) in the 1.25-mg and 2-mg dose groups. There were no new safety signals, and dose reduction during initiation mitigated cardiac adverse effects. Lymphopenia was more common at the 10-mg dose than at lower doses.
Data source: A dose-blinded extension of 184 patients from the phase II BOLD study.
Disclosures: Novartis funded the study. Coinvestigators employed by Novartis participated in all aspects of the study, including interpretation of the data and manuscript submission. Dr. Kappos disclosed financial ties to Novartis and numerous other pharmaceutical companies, foundations, and societies.

Can Stem Cells Halt Progression of MS?
NATIONAL HARBOR, MD—High-dose immunosuppressive therapy and autologous hematopoietic cell transplantation induces a high rate of remission among patients with highly active relapsing-remitting multiple sclerosis (MS), according to data presented at the 2016 CMSC Annual Meeting. Furthermore, that remission is sustained for five years without maintenance therapy. "We saw about 70% long-term, disease-free survival at five years," said Richard A. Nash, MD, a physician at Colorado Blood Cancer Institute in Denver and a HALT-MS investigator.

Richard A. Nash, MD
“What we studied was high-dose therapy, followed by transplant, for treatment of patients with poor-prognosis MS,” said Dr. Nash on behalf of his study collaborators. Their hypothesis was that intensive immunosuppressive therapy followed by transplant would arrest MS disease activity. Their phase II multicenter study had a prospective, open-label, single-arm design. There were three centers involved—Baylor College of Medicine in Houston, the Fred Hutchinson Cancer Research Center in Seattle, and the Ohio State University MS Center in Columbus.
The aim of the study was to determine the five-year durability of disease stabilization in patients with MS after high-dose therapy and autologous transplantation. An interim analysis at three years was published in JAMA Neurology.
The primary end point was event-free survival after transplant in the five-year period of follow-up. The end point included relapse, defined as neurologic signs or symptoms lasting more than 48 hours; MRI abnormalities at more than 12 months after transplant; progression in disability after six months post-transplant (as measured by Expanded Disability Status Scale or EDSS); and mortality. The end point was similar to no evidence of disease activity (NEDA), said Dr. Nash.
Patients who were eligible for the study were age 18 to 60, met McDonald criteria, and had had MS for less than 15 years. Patients had relapsing-remitting MS with cumulative disability or progressive-relapsing MS. EDSS score had to be between 3.0 and 5.5. Patients had to have T2 abnormalities consistent with MS and two or more relapses within 18 months on therapy, with an EDSS increase of more than 0.5. “I don’t think we had anyone in the study that met the second criterion, which was relapse on therapy with an EDSS increase of more than 1.0 and one separate event with gadolinium-enhancing lesions on MRI,” Dr. Nash said. A panel of two neurologists and one transplant physician reviewed the patients.
There were 25 patients in the HALT-MS trial. Median age at mobilization was 37 (range, 26 to 52). The study cohort was mostly female (17 female, 8 male). Baseline EDSS score was 4.5 (range, 3.0 to 5.5). Median disease duration was 4.9 years (range, seven months to 12 years). Therapies that patients had failed prior to study entry included interferon beta-1a (22 patients), interferon beta-1b (one patient), glatiramer acetate (18 patients), mitoxantrone (eight patients), natalizumab (six patients), and other therapies (11 patients).
James D. Bowen, MD, of the Swedish Neuroscience Institute in Seattle and a HALT-MS investigator, described the study intervention. “The protocol starts with mobilization of stem cells. Patients receive prednisone during this time so that the CSF is not vulnerable to MS attack. They then undergo leukapheresis to collect stem cells, which are further concentrated with a CD34 selection procedure that allows us to purify them.” Most patients with cancer require four or five pheresis sessions, Dr. Bowen noted, but patients with MS generally have healthy bone marrow, so most of the participants in HALT-MS only required two sessions. A few required three pheresis sessions.
“For our transplant protocol, we used BEAM plus ATG, which is a five-drug cocktail—BCNU, etoposide, Ara C, melphalan, and rabbit antithymocyte globulin,” said Dr. Bowen. “Immediately following that, transplant of the stem cells was done. After that, they got granulocyte-colony stimulating factor (G-CSF) to rev up the bone marrow, and we cover that with prednisone at days seven to 21 to avoid MS attacks precipitated by graft syndrome.”
Twenty-four patients went on to transplant. One of the patients had a pulmonary embolus and was not deemed a good candidate for transplantation. The rate of event-free survival, the primary end point, was 69.2%. Seven patients met the primary end point: two by progression of their disease, three by relapse of their disease, and two patients met end points much later in the follow-up, by changes in their MRI at 3.5 to 4.0 years.
The rate of five-year relapse-free survival was 86.9%. Two patients had changes on MRI. The two patients met primary end points and had their disease activity about 3.5 years after transplant. “There was an early event as well, at one year, but the patient who had MRI changes at one year had already met a primary end point by relapsing at about six months,” Dr. Nash said. The rate of disease progression-free survival was 91%.
Regarding change from baseline in T1 and T2 lesion volume, “we had very low incidence, except for two patients who at about 3.5 to 4.0 years after transplant became positive,” Dr. Nash reported. No other patients had development of significant lesions. The T2 lesion volume actually decreased over time and remained decreased through the five years of follow-up. Starting at about three years after transplantation, the researchers noted stability of brain volume.
The researchers recorded three deaths that were considered to be unrelated to the transplant. Two deaths were thought to be possibly related to progression of MS, and one death was in a patient who had evidence of asthma prior to transplant. “That patient was seen by pulmonary medicine, and it was thought that the patient was reasonable to go on to transplant, but the patient had persistent problems after transplant and died at about four years after the transplant,” Dr. Nash said.
The transplant itself appeared to be well tolerated. “There were few serious complications other than what we might expect from a transplant,” Dr. Nash said. It was highly effective for inducing sustained remission for highly active patients with relapsing-remitting MS through five years, and these patients have not received any maintenance therapy since the transplant. MRI lesion volume was reduced, and the brain volume stabilized at years three through five.
The HALT-MS study was sponsored by the National Institute of Allergy & Infectious Diseases (NIAID) and run by the Immune Tolerance Network.
—Glenn S. Williams
Suggested Reading
Bowen JD, Kraft GH, Wundes A, et al. Autologous hematopoietic cell transplantation following high-dose immunosuppressive therapy for advanced multiple sclerosis: long-term results. Bone Marrow Transplant. 2012;47(7):946-951.
Nash RA, Bowen JD, McSweeney PA, et al. High-dose immunosuppressive therapy and autologous peripheral blood stem cell transplantation for severe multiple sclerosis. Blood. 2003;102(7):2364-2372.
Nash RA, Hutton GJ, Racke MK, et al. High-dose immunosuppressive therapy and autologous hematopoietic cell transplantation for relapsing-remitting multiple sclerosis (HALT-MS): a 3-year interim report. JAMA Neurol. 2015;72(2):159-169.
NATIONAL HARBOR, MD—High-dose immunosuppressive therapy and autologous hematopoietic cell transplantation induces a high rate of remission among patients with highly active relapsing-remitting multiple sclerosis (MS), according to data presented at the 2016 CMSC Annual Meeting. Furthermore, that remission is sustained for five years without maintenance therapy. "We saw about 70% long-term, disease-free survival at five years," said Richard A. Nash, MD, a physician at Colorado Blood Cancer Institute in Denver and a HALT-MS investigator.
Richard A. Nash, MD
“What we studied was high-dose therapy, followed by transplant, for treatment of patients with poor-prognosis MS,” said Dr. Nash on behalf of his study collaborators. Their hypothesis was that intensive immunosuppressive therapy followed by transplant would arrest MS disease activity. Their phase II multicenter study had a prospective, open-label, single-arm design. There were three centers involved—Baylor College of Medicine in Houston, the Fred Hutchinson Cancer Research Center in Seattle, and the Ohio State University MS Center in Columbus.
The aim of the study was to determine the five-year durability of disease stabilization in patients with MS after high-dose therapy and autologous transplantation. An interim analysis at three years was published in JAMA Neurology.
The primary end point was event-free survival after transplant in the five-year period of follow-up. The end point included relapse, defined as neurologic signs or symptoms lasting more than 48 hours; MRI abnormalities at more than 12 months after transplant; progression in disability after six months post-transplant (as measured by Expanded Disability Status Scale or EDSS); and mortality. The end point was similar to no evidence of disease activity (NEDA), said Dr. Nash.
Patients who were eligible for the study were age 18 to 60, met McDonald criteria, and had had MS for less than 15 years. Patients had relapsing-remitting MS with cumulative disability or progressive-relapsing MS. EDSS score had to be between 3.0 and 5.5. Patients had to have T2 abnormalities consistent with MS and two or more relapses within 18 months on therapy, with an EDSS increase of more than 0.5. “I don’t think we had anyone in the study that met the second criterion, which was relapse on therapy with an EDSS increase of more than 1.0 and one separate event with gadolinium-enhancing lesions on MRI,” Dr. Nash said. A panel of two neurologists and one transplant physician reviewed the patients.
There were 25 patients in the HALT-MS trial. Median age at mobilization was 37 (range, 26 to 52). The study cohort was mostly female (17 female, 8 male). Baseline EDSS score was 4.5 (range, 3.0 to 5.5). Median disease duration was 4.9 years (range, seven months to 12 years). Therapies that patients had failed prior to study entry included interferon beta-1a (22 patients), interferon beta-1b (one patient), glatiramer acetate (18 patients), mitoxantrone (eight patients), natalizumab (six patients), and other therapies (11 patients).
James D. Bowen, MD, of the Swedish Neuroscience Institute in Seattle and a HALT-MS investigator, described the study intervention. “The protocol starts with mobilization of stem cells. Patients receive prednisone during this time so that the CSF is not vulnerable to MS attack. They then undergo leukapheresis to collect stem cells, which are further concentrated with a CD34 selection procedure that allows us to purify them.” Most patients with cancer require four or five pheresis sessions, Dr. Bowen noted, but patients with MS generally have healthy bone marrow, so most of the participants in HALT-MS only required two sessions. A few required three pheresis sessions.
“For our transplant protocol, we used BEAM plus ATG, which is a five-drug cocktail—BCNU, etoposide, Ara C, melphalan, and rabbit antithymocyte globulin,” said Dr. Bowen. “Immediately following that, transplant of the stem cells was done. After that, they got granulocyte-colony stimulating factor (G-CSF) to rev up the bone marrow, and we cover that with prednisone at days seven to 21 to avoid MS attacks precipitated by graft syndrome.”
Twenty-four patients went on to transplant. One of the patients had a pulmonary embolus and was not deemed a good candidate for transplantation. The rate of event-free survival, the primary end point, was 69.2%. Seven patients met the primary end point: two by progression of their disease, three by relapse of their disease, and two patients met end points much later in the follow-up, by changes in their MRI at 3.5 to 4.0 years.
The rate of five-year relapse-free survival was 86.9%. Two patients had changes on MRI. The two patients met primary end points and had their disease activity about 3.5 years after transplant. “There was an early event as well, at one year, but the patient who had MRI changes at one year had already met a primary end point by relapsing at about six months,” Dr. Nash said. The rate of disease progression-free survival was 91%.
Regarding change from baseline in T1 and T2 lesion volume, “we had very low incidence, except for two patients who at about 3.5 to 4.0 years after transplant became positive,” Dr. Nash reported. No other patients had development of significant lesions. The T2 lesion volume actually decreased over time and remained decreased through the five years of follow-up. Starting at about three years after transplantation, the researchers noted stability of brain volume.
The researchers recorded three deaths that were considered to be unrelated to the transplant. Two deaths were thought to be possibly related to progression of MS, and one death was in a patient who had evidence of asthma prior to transplant. “That patient was seen by pulmonary medicine, and it was thought that the patient was reasonable to go on to transplant, but the patient had persistent problems after transplant and died at about four years after the transplant,” Dr. Nash said.
The transplant itself appeared to be well tolerated. “There were few serious complications other than what we might expect from a transplant,” Dr. Nash said. It was highly effective for inducing sustained remission for highly active patients with relapsing-remitting MS through five years, and these patients have not received any maintenance therapy since the transplant. MRI lesion volume was reduced, and the brain volume stabilized at years three through five.
The HALT-MS study was sponsored by the National Institute of Allergy & Infectious Diseases (NIAID) and run by the Immune Tolerance Network.
—Glenn S. Williams
NATIONAL HARBOR, MD—High-dose immunosuppressive therapy and autologous hematopoietic cell transplantation induces a high rate of remission among patients with highly active relapsing-remitting multiple sclerosis (MS), according to data presented at the 2016 CMSC Annual Meeting. Furthermore, that remission is sustained for five years without maintenance therapy. "We saw about 70% long-term, disease-free survival at five years," said Richard A. Nash, MD, a physician at Colorado Blood Cancer Institute in Denver and a HALT-MS investigator.
Richard A. Nash, MD
“What we studied was high-dose therapy, followed by transplant, for treatment of patients with poor-prognosis MS,” said Dr. Nash on behalf of his study collaborators. Their hypothesis was that intensive immunosuppressive therapy followed by transplant would arrest MS disease activity. Their phase II multicenter study had a prospective, open-label, single-arm design. There were three centers involved—Baylor College of Medicine in Houston, the Fred Hutchinson Cancer Research Center in Seattle, and the Ohio State University MS Center in Columbus.
The aim of the study was to determine the five-year durability of disease stabilization in patients with MS after high-dose therapy and autologous transplantation. An interim analysis at three years was published in JAMA Neurology.
The primary end point was event-free survival after transplant in the five-year period of follow-up. The end point included relapse, defined as neurologic signs or symptoms lasting more than 48 hours; MRI abnormalities at more than 12 months after transplant; progression in disability after six months post-transplant (as measured by Expanded Disability Status Scale or EDSS); and mortality. The end point was similar to no evidence of disease activity (NEDA), said Dr. Nash.
Patients who were eligible for the study were age 18 to 60, met McDonald criteria, and had had MS for less than 15 years. Patients had relapsing-remitting MS with cumulative disability or progressive-relapsing MS. EDSS score had to be between 3.0 and 5.5. Patients had to have T2 abnormalities consistent with MS and two or more relapses within 18 months on therapy, with an EDSS increase of more than 0.5. “I don’t think we had anyone in the study that met the second criterion, which was relapse on therapy with an EDSS increase of more than 1.0 and one separate event with gadolinium-enhancing lesions on MRI,” Dr. Nash said. A panel of two neurologists and one transplant physician reviewed the patients.
There were 25 patients in the HALT-MS trial. Median age at mobilization was 37 (range, 26 to 52). The study cohort was mostly female (17 female, 8 male). Baseline EDSS score was 4.5 (range, 3.0 to 5.5). Median disease duration was 4.9 years (range, seven months to 12 years). Therapies that patients had failed prior to study entry included interferon beta-1a (22 patients), interferon beta-1b (one patient), glatiramer acetate (18 patients), mitoxantrone (eight patients), natalizumab (six patients), and other therapies (11 patients).
James D. Bowen, MD, of the Swedish Neuroscience Institute in Seattle and a HALT-MS investigator, described the study intervention. “The protocol starts with mobilization of stem cells. Patients receive prednisone during this time so that the CSF is not vulnerable to MS attack. They then undergo leukapheresis to collect stem cells, which are further concentrated with a CD34 selection procedure that allows us to purify them.” Most patients with cancer require four or five pheresis sessions, Dr. Bowen noted, but patients with MS generally have healthy bone marrow, so most of the participants in HALT-MS only required two sessions. A few required three pheresis sessions.
“For our transplant protocol, we used BEAM plus ATG, which is a five-drug cocktail—BCNU, etoposide, Ara C, melphalan, and rabbit antithymocyte globulin,” said Dr. Bowen. “Immediately following that, transplant of the stem cells was done. After that, they got granulocyte-colony stimulating factor (G-CSF) to rev up the bone marrow, and we cover that with prednisone at days seven to 21 to avoid MS attacks precipitated by graft syndrome.”
Twenty-four patients went on to transplant. One of the patients had a pulmonary embolus and was not deemed a good candidate for transplantation. The rate of event-free survival, the primary end point, was 69.2%. Seven patients met the primary end point: two by progression of their disease, three by relapse of their disease, and two patients met end points much later in the follow-up, by changes in their MRI at 3.5 to 4.0 years.
The rate of five-year relapse-free survival was 86.9%. Two patients had changes on MRI. The two patients met primary end points and had their disease activity about 3.5 years after transplant. “There was an early event as well, at one year, but the patient who had MRI changes at one year had already met a primary end point by relapsing at about six months,” Dr. Nash said. The rate of disease progression-free survival was 91%.
Regarding change from baseline in T1 and T2 lesion volume, “we had very low incidence, except for two patients who at about 3.5 to 4.0 years after transplant became positive,” Dr. Nash reported. No other patients had development of significant lesions. The T2 lesion volume actually decreased over time and remained decreased through the five years of follow-up. Starting at about three years after transplantation, the researchers noted stability of brain volume.
The researchers recorded three deaths that were considered to be unrelated to the transplant. Two deaths were thought to be possibly related to progression of MS, and one death was in a patient who had evidence of asthma prior to transplant. “That patient was seen by pulmonary medicine, and it was thought that the patient was reasonable to go on to transplant, but the patient had persistent problems after transplant and died at about four years after the transplant,” Dr. Nash said.
The transplant itself appeared to be well tolerated. “There were few serious complications other than what we might expect from a transplant,” Dr. Nash said. It was highly effective for inducing sustained remission for highly active patients with relapsing-remitting MS through five years, and these patients have not received any maintenance therapy since the transplant. MRI lesion volume was reduced, and the brain volume stabilized at years three through five.
The HALT-MS study was sponsored by the National Institute of Allergy & Infectious Diseases (NIAID) and run by the Immune Tolerance Network.
—Glenn S. Williams
Suggested Reading
Bowen JD, Kraft GH, Wundes A, et al. Autologous hematopoietic cell transplantation following high-dose immunosuppressive therapy for advanced multiple sclerosis: long-term results. Bone Marrow Transplant. 2012;47(7):946-951.
Nash RA, Bowen JD, McSweeney PA, et al. High-dose immunosuppressive therapy and autologous peripheral blood stem cell transplantation for severe multiple sclerosis. Blood. 2003;102(7):2364-2372.
Nash RA, Hutton GJ, Racke MK, et al. High-dose immunosuppressive therapy and autologous hematopoietic cell transplantation for relapsing-remitting multiple sclerosis (HALT-MS): a 3-year interim report. JAMA Neurol. 2015;72(2):159-169.
Suggested Reading
Bowen JD, Kraft GH, Wundes A, et al. Autologous hematopoietic cell transplantation following high-dose immunosuppressive therapy for advanced multiple sclerosis: long-term results. Bone Marrow Transplant. 2012;47(7):946-951.
Nash RA, Bowen JD, McSweeney PA, et al. High-dose immunosuppressive therapy and autologous peripheral blood stem cell transplantation for severe multiple sclerosis. Blood. 2003;102(7):2364-2372.
Nash RA, Hutton GJ, Racke MK, et al. High-dose immunosuppressive therapy and autologous hematopoietic cell transplantation for relapsing-remitting multiple sclerosis (HALT-MS): a 3-year interim report. JAMA Neurol. 2015;72(2):159-169.
