User login
Five rules for evaluating melanonychia
WAIKOLOA, HAWAII – Many dermatologists find melanonychia to be intimidating. The clinical features are ambiguous, and the prospect of doing a painful nail apparatus biopsy can be daunting for the inexperienced. As a result, the biopsy gets delayed and melanoma of the nail is often initially a missed diagnosis, not uncommonly for years, with devastating consequences.

Here are five at the Hawaii Dermatology Seminar provided by the Global Academy for Medical Education/Skin Disease Education Foundation.
Rule #1: Always look beyond the nail
When a light-skinned person presents with more than one nail with pigmentation, the likelihood that one of them is melanoma is much less than if there is only one nail with melanonychia, according to Dr. Jellinek, a dermatologist in private practice in East Greenwich, R.I.
Also, be sure to look at the skin and mucosa. Consider the medications the patients may be taking: For example, cyclophosphamide (Cytoxan) is notorious for causing nail changes as a side effect. A past medical history of lichen planus, carpal tunnel syndrome, Addison disease, or other conditions may explain the melanonychia.
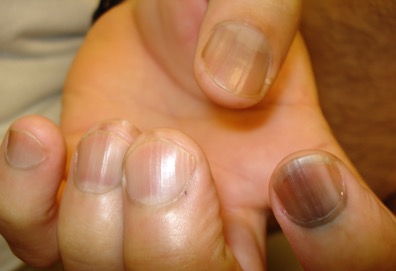
Laugier-Hunziker syndrome is a condition worth getting to know. It’s an acquired disorder characterized longitudinal melanonychia and other pigmentary changes, which may include diffuse hyperpigmentation of the orolabial mucosa, ocular pigment, and/or pigmented palmoplantar lesions. It’s said to be rare, but Dr. Jellinek disagrees.
“Learn this one if you don’t know it. I see a case about every 2 weeks. It’s not heritable and not associated with any other medical condition,” he said.
Rule #2: Your dermatoscope is great for nails
What Dr. Jellinek considers to be among the all-time best papers on the value of dermoscopy for nail pigmentation was authored by French investigators. They analyzed 148 consecutive cases of longitudinal melanonychia and concluded that the dermoscopic combination of a brown background coupled with irregular longitudinal lines in terms of color, spacing, diameter, and/or lack of parallelism strongly suggests melanoma. A micro-Hutchinson’s sign, while a rare finding, occurred only in melanoma, where it represented periungual spread of a radial growth phase malignancy (Arch Dermatol. 2002 Oct;138[10]:1327-33).
“I think nail dermoscopy is most helpful for subungual hemorrhage. I average one referral per week for hemorrhage under the nail. On dermoscopy it’s as if someone took paint and threw it at the nail. Purple to brown blood spots, with no background color. This should be a doorway diagnosis of hemorrhage,” Dr. Jellinek said.
Rule #3: Know when you don’t know
“This is really the key for me,” the dermatologist commented. “There are automatic cases for biopsy, and more commonly routine cases for reassurance. But the gray zone, when you know you don’t know, is the key decision making moment.”
When something just doesn’t feel right, there’s absolutely nothing wrong with getting a second opinion, he stressed.
“It’s worthwhile getting to know people whose opinions you trust. There’s a saying I like to teach our fellows: ‘Never worry alone.’ So if you’re worried about someone, listen to that inner voice. There’s no shame in getting a second opinion. It’s great! Patients are never upset, either. They feel really well taken care of,” he said.
Rule #4: Don’t wimp out when a biopsy is warranted
Many dermatologists hem and haw about doing a biopsy for a concerning lesion on the nail, when they wouldn’t hesitate to biopsy a similarly suspicious lesion on the face.
But it’s essential to biopsy the right area, he added. For longitudinal melanonychia, that’s the matrix. The nail plate is the wrong place; a biopsy obtained there will result in an inappropriate benign diagnosis.
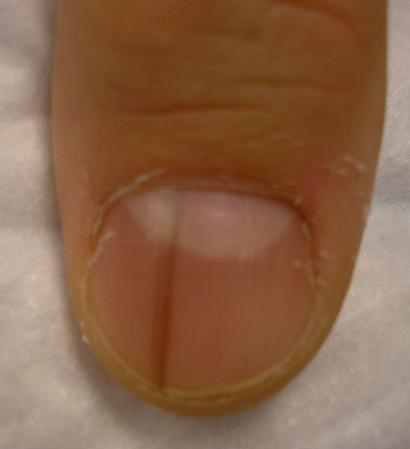
“The starter set is to do a punch biopsy. This is your gateway drug to the world of nail surgery. Lots of dermatologists are intimidated by nail surgery, but if you can do any minor surgery, you can do a punch of the matrix. All it takes is a little practice. And if all you can do is punch biopsies, you’re good for your career. If you can do that, you’re golden. There are people who’ve just done punch biopsies for their whole career and they don’t miss melanomas,” he said.
Step one is to undermine the proximal nail fold using a pediatric elevator, which costs only about $30. “If you’re going to do a lot of nail surgery, they’re really helpful,” he said.
There’s no need at all to evulse the nail. Just make oblique incisions in the proximal nail fold in order to reflect it and look at the matrix. A 3-mm punch is standard, directed right over the origin of the pigment. Resist the temptation to force or squeeze the specimen in order to extract it. Instead, use really fine-tipped scissors to nibble at the base of the specimen, then gently pull it out, making an effort to keep the nail plate attached to the digit and avoid getting it stuck up in the punch.
Rule #5: Have dermatopathologists extensively experienced with nail pathology on your Rolodex
The histopathologic findings present in early subungual melanoma in situ are often too subtle for general dermatopathologists to appreciate, in Dr. Jellinek’s experience. He cited other investigators’ study of 18 cases of subungual melanoma in situ, all marked by longitudinal melanonychia. Only half showed the classic giveaway on the original nail matrix biopsy, consisting of a significantly increased number of atypical melanocytes with marked nuclear atypia. Blatant pagetoid spread was infrequent. However, all 18 cases displayed a novel, more subtle, and previously undescribed finding: haphazard and uneven distribution of atypical solitary melanocytes with variably sized and shaped hyperchromatic nuclei (J Cutan Pathol. 2016 Jan;43[1]:41-52).
Dr. Jellinek reported having no financial conflicts regarding his presentation. SDEF/Global Academy for Medical Education and this news organization are owned by the same parent company.
WAIKOLOA, HAWAII – Many dermatologists find melanonychia to be intimidating. The clinical features are ambiguous, and the prospect of doing a painful nail apparatus biopsy can be daunting for the inexperienced. As a result, the biopsy gets delayed and melanoma of the nail is often initially a missed diagnosis, not uncommonly for years, with devastating consequences.
Here are five at the Hawaii Dermatology Seminar provided by the Global Academy for Medical Education/Skin Disease Education Foundation.
Rule #1: Always look beyond the nail
When a light-skinned person presents with more than one nail with pigmentation, the likelihood that one of them is melanoma is much less than if there is only one nail with melanonychia, according to Dr. Jellinek, a dermatologist in private practice in East Greenwich, R.I.
Also, be sure to look at the skin and mucosa. Consider the medications the patients may be taking: For example, cyclophosphamide (Cytoxan) is notorious for causing nail changes as a side effect. A past medical history of lichen planus, carpal tunnel syndrome, Addison disease, or other conditions may explain the melanonychia.
Laugier-Hunziker syndrome is a condition worth getting to know. It’s an acquired disorder characterized longitudinal melanonychia and other pigmentary changes, which may include diffuse hyperpigmentation of the orolabial mucosa, ocular pigment, and/or pigmented palmoplantar lesions. It’s said to be rare, but Dr. Jellinek disagrees.
“Learn this one if you don’t know it. I see a case about every 2 weeks. It’s not heritable and not associated with any other medical condition,” he said.
Rule #2: Your dermatoscope is great for nails
What Dr. Jellinek considers to be among the all-time best papers on the value of dermoscopy for nail pigmentation was authored by French investigators. They analyzed 148 consecutive cases of longitudinal melanonychia and concluded that the dermoscopic combination of a brown background coupled with irregular longitudinal lines in terms of color, spacing, diameter, and/or lack of parallelism strongly suggests melanoma. A micro-Hutchinson’s sign, while a rare finding, occurred only in melanoma, where it represented periungual spread of a radial growth phase malignancy (Arch Dermatol. 2002 Oct;138[10]:1327-33).
“I think nail dermoscopy is most helpful for subungual hemorrhage. I average one referral per week for hemorrhage under the nail. On dermoscopy it’s as if someone took paint and threw it at the nail. Purple to brown blood spots, with no background color. This should be a doorway diagnosis of hemorrhage,” Dr. Jellinek said.
Rule #3: Know when you don’t know
“This is really the key for me,” the dermatologist commented. “There are automatic cases for biopsy, and more commonly routine cases for reassurance. But the gray zone, when you know you don’t know, is the key decision making moment.”
When something just doesn’t feel right, there’s absolutely nothing wrong with getting a second opinion, he stressed.
“It’s worthwhile getting to know people whose opinions you trust. There’s a saying I like to teach our fellows: ‘Never worry alone.’ So if you’re worried about someone, listen to that inner voice. There’s no shame in getting a second opinion. It’s great! Patients are never upset, either. They feel really well taken care of,” he said.
Rule #4: Don’t wimp out when a biopsy is warranted
Many dermatologists hem and haw about doing a biopsy for a concerning lesion on the nail, when they wouldn’t hesitate to biopsy a similarly suspicious lesion on the face.
But it’s essential to biopsy the right area, he added. For longitudinal melanonychia, that’s the matrix. The nail plate is the wrong place; a biopsy obtained there will result in an inappropriate benign diagnosis.
“The starter set is to do a punch biopsy. This is your gateway drug to the world of nail surgery. Lots of dermatologists are intimidated by nail surgery, but if you can do any minor surgery, you can do a punch of the matrix. All it takes is a little practice. And if all you can do is punch biopsies, you’re good for your career. If you can do that, you’re golden. There are people who’ve just done punch biopsies for their whole career and they don’t miss melanomas,” he said.
Step one is to undermine the proximal nail fold using a pediatric elevator, which costs only about $30. “If you’re going to do a lot of nail surgery, they’re really helpful,” he said.
There’s no need at all to evulse the nail. Just make oblique incisions in the proximal nail fold in order to reflect it and look at the matrix. A 3-mm punch is standard, directed right over the origin of the pigment. Resist the temptation to force or squeeze the specimen in order to extract it. Instead, use really fine-tipped scissors to nibble at the base of the specimen, then gently pull it out, making an effort to keep the nail plate attached to the digit and avoid getting it stuck up in the punch.
Rule #5: Have dermatopathologists extensively experienced with nail pathology on your Rolodex
The histopathologic findings present in early subungual melanoma in situ are often too subtle for general dermatopathologists to appreciate, in Dr. Jellinek’s experience. He cited other investigators’ study of 18 cases of subungual melanoma in situ, all marked by longitudinal melanonychia. Only half showed the classic giveaway on the original nail matrix biopsy, consisting of a significantly increased number of atypical melanocytes with marked nuclear atypia. Blatant pagetoid spread was infrequent. However, all 18 cases displayed a novel, more subtle, and previously undescribed finding: haphazard and uneven distribution of atypical solitary melanocytes with variably sized and shaped hyperchromatic nuclei (J Cutan Pathol. 2016 Jan;43[1]:41-52).
Dr. Jellinek reported having no financial conflicts regarding his presentation. SDEF/Global Academy for Medical Education and this news organization are owned by the same parent company.
WAIKOLOA, HAWAII – Many dermatologists find melanonychia to be intimidating. The clinical features are ambiguous, and the prospect of doing a painful nail apparatus biopsy can be daunting for the inexperienced. As a result, the biopsy gets delayed and melanoma of the nail is often initially a missed diagnosis, not uncommonly for years, with devastating consequences.
Here are five at the Hawaii Dermatology Seminar provided by the Global Academy for Medical Education/Skin Disease Education Foundation.
Rule #1: Always look beyond the nail
When a light-skinned person presents with more than one nail with pigmentation, the likelihood that one of them is melanoma is much less than if there is only one nail with melanonychia, according to Dr. Jellinek, a dermatologist in private practice in East Greenwich, R.I.
Also, be sure to look at the skin and mucosa. Consider the medications the patients may be taking: For example, cyclophosphamide (Cytoxan) is notorious for causing nail changes as a side effect. A past medical history of lichen planus, carpal tunnel syndrome, Addison disease, or other conditions may explain the melanonychia.
Laugier-Hunziker syndrome is a condition worth getting to know. It’s an acquired disorder characterized longitudinal melanonychia and other pigmentary changes, which may include diffuse hyperpigmentation of the orolabial mucosa, ocular pigment, and/or pigmented palmoplantar lesions. It’s said to be rare, but Dr. Jellinek disagrees.
“Learn this one if you don’t know it. I see a case about every 2 weeks. It’s not heritable and not associated with any other medical condition,” he said.
Rule #2: Your dermatoscope is great for nails
What Dr. Jellinek considers to be among the all-time best papers on the value of dermoscopy for nail pigmentation was authored by French investigators. They analyzed 148 consecutive cases of longitudinal melanonychia and concluded that the dermoscopic combination of a brown background coupled with irregular longitudinal lines in terms of color, spacing, diameter, and/or lack of parallelism strongly suggests melanoma. A micro-Hutchinson’s sign, while a rare finding, occurred only in melanoma, where it represented periungual spread of a radial growth phase malignancy (Arch Dermatol. 2002 Oct;138[10]:1327-33).
“I think nail dermoscopy is most helpful for subungual hemorrhage. I average one referral per week for hemorrhage under the nail. On dermoscopy it’s as if someone took paint and threw it at the nail. Purple to brown blood spots, with no background color. This should be a doorway diagnosis of hemorrhage,” Dr. Jellinek said.
Rule #3: Know when you don’t know
“This is really the key for me,” the dermatologist commented. “There are automatic cases for biopsy, and more commonly routine cases for reassurance. But the gray zone, when you know you don’t know, is the key decision making moment.”
When something just doesn’t feel right, there’s absolutely nothing wrong with getting a second opinion, he stressed.
“It’s worthwhile getting to know people whose opinions you trust. There’s a saying I like to teach our fellows: ‘Never worry alone.’ So if you’re worried about someone, listen to that inner voice. There’s no shame in getting a second opinion. It’s great! Patients are never upset, either. They feel really well taken care of,” he said.
Rule #4: Don’t wimp out when a biopsy is warranted
Many dermatologists hem and haw about doing a biopsy for a concerning lesion on the nail, when they wouldn’t hesitate to biopsy a similarly suspicious lesion on the face.
But it’s essential to biopsy the right area, he added. For longitudinal melanonychia, that’s the matrix. The nail plate is the wrong place; a biopsy obtained there will result in an inappropriate benign diagnosis.
“The starter set is to do a punch biopsy. This is your gateway drug to the world of nail surgery. Lots of dermatologists are intimidated by nail surgery, but if you can do any minor surgery, you can do a punch of the matrix. All it takes is a little practice. And if all you can do is punch biopsies, you’re good for your career. If you can do that, you’re golden. There are people who’ve just done punch biopsies for their whole career and they don’t miss melanomas,” he said.
Step one is to undermine the proximal nail fold using a pediatric elevator, which costs only about $30. “If you’re going to do a lot of nail surgery, they’re really helpful,” he said.
There’s no need at all to evulse the nail. Just make oblique incisions in the proximal nail fold in order to reflect it and look at the matrix. A 3-mm punch is standard, directed right over the origin of the pigment. Resist the temptation to force or squeeze the specimen in order to extract it. Instead, use really fine-tipped scissors to nibble at the base of the specimen, then gently pull it out, making an effort to keep the nail plate attached to the digit and avoid getting it stuck up in the punch.
Rule #5: Have dermatopathologists extensively experienced with nail pathology on your Rolodex
The histopathologic findings present in early subungual melanoma in situ are often too subtle for general dermatopathologists to appreciate, in Dr. Jellinek’s experience. He cited other investigators’ study of 18 cases of subungual melanoma in situ, all marked by longitudinal melanonychia. Only half showed the classic giveaway on the original nail matrix biopsy, consisting of a significantly increased number of atypical melanocytes with marked nuclear atypia. Blatant pagetoid spread was infrequent. However, all 18 cases displayed a novel, more subtle, and previously undescribed finding: haphazard and uneven distribution of atypical solitary melanocytes with variably sized and shaped hyperchromatic nuclei (J Cutan Pathol. 2016 Jan;43[1]:41-52).
Dr. Jellinek reported having no financial conflicts regarding his presentation. SDEF/Global Academy for Medical Education and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM SDEF HAWAII DERMATOLOGY SEMINAR
Entrectinib exhibits activity in children with solid tumors
Entrectinib demonstrated “very promising” antitumor activity in children and adolescents with recurrent or refractory solid tumors, according to an investigator involved in a phase 1/1b trial.
Twelve of 29 patients enrolled in the trial have responded to entrectinib. All responders had fusions in genes targeted by the drug – NTRK1/2/3 (TRKA/B/C), ROS1, or ALK – or an ALK mutation.
Details of this study are scheduled to be presented at the annual meeting of the American Society of Clinical Oncology.
Giles W. Robinson, MD, of St. Jude Children’s Research Hospital in Memphis, Tenn., discussed the study during a press briefing in advance of the meeting.
“Entrectinib is an oral and potent inhibitor of the TRKA/B/C, ROS1, and ALK proteins, but it also penetrates into the brain to reach tumors in the brain and spine, which can be a hard area to get drugs to,” Dr. Robinson explained.
“Promising clinical activity was initially seen in the adult solid tumor patients with target rearrangements, and it was encouraging to see these patients also had responses when the tumors were located in their brains. And what got us really excited as pediatric oncologists was that a variety of pediatric cancers harbor these fusions and mutations within certain tumors.”
With this in mind, Dr. Robinson and colleagues conducted a phase 1/1b study (NCT02650401) of entrectinib in 29 patients with recurrent or refractory solid tumors, including central nervous system (CNS) tumors.
The patients’ median age was 7 years (range, 0-20 years), and roughly half of them were male (n = 15). Patients were diagnosed with neuroblastoma (n = 16), high-grade glioma (n = 5), inflammatory myofibroblastic tumors (n = 3), infantile fibrosarcoma (n = 2), CNS embryonal tumor (n = 1), melanoma (n = 1), and synovial sarcoma (n = 1).
In the dose-finding portion of the trial, patients received entrectinib at 250 mg/m2 (n = 3), 400 mg/m2 (n = 3), 550 mg/m2 (n = 7), or 750 mg/m2 (n = 3).
In the phase 1b portion, patients received entrectinib at 550 mg/m2 (n = 7) – the recommended dose – or 400 mg/m2 (n = 6) if they were unable to swallow intact capsules.
Dr. Robinson said entrectinib was “quite well tolerated” overall, but he did not present any data on adverse events. He did say dose-limiting toxicities included fatigue, elevated creatinine levels, dysgeusia resulting in loss of taste, weight gain, and, in one patient, pulmonary edema.
“Entrectinib produced striking, rapid, and durable responses in all children with refractory CNS and solid tumors that actually harbored these fusions in NTRK1/2/3, ROS1, or ALK,” Dr. Robinson said. “It also produced a significant response in one ALK-mutated neuroblastoma patient. [N]o responses were seen in tumors lacking aberrations in the target kinases.”
In all, 12 patients responded. The three complete responders had an ALK F1174L mutation, an ALK fusion, and an NTRK fusion, respectively. Five partial responders had NTRK fusions, three had ROS1 fusions, and one had an ALK fusion.
Three responders discontinued treatment. Ten patients were still receiving entrectinib at last follow-up, and 11 patients had died.
Progression-free survival was significantly longer among patients who had fusions than among those who did not (P less than .0001).
“To sum up, entrectinib really is very promising,” Dr. Robinson said. “It has very promising antitumor activity and progression-free survival but [only] in patients with target gene fusions.”
Dr. Robinson said this trial is ongoing, but it is now limited to patients with fusions targeted by entrectinib.
The trial is sponsored by Hoffman-La Roche Ltd. and supported by Alex’s Lemonade Stand Center of Excellence. Dr. Robinson has relationships with Lilly, Genentech/Roche, and Novartis.
SOURCE: Robinson GW et al. ASCO 2019. Abstract 10009.
Entrectinib demonstrated “very promising” antitumor activity in children and adolescents with recurrent or refractory solid tumors, according to an investigator involved in a phase 1/1b trial.
Twelve of 29 patients enrolled in the trial have responded to entrectinib. All responders had fusions in genes targeted by the drug – NTRK1/2/3 (TRKA/B/C), ROS1, or ALK – or an ALK mutation.
Details of this study are scheduled to be presented at the annual meeting of the American Society of Clinical Oncology.
Giles W. Robinson, MD, of St. Jude Children’s Research Hospital in Memphis, Tenn., discussed the study during a press briefing in advance of the meeting.
“Entrectinib is an oral and potent inhibitor of the TRKA/B/C, ROS1, and ALK proteins, but it also penetrates into the brain to reach tumors in the brain and spine, which can be a hard area to get drugs to,” Dr. Robinson explained.
“Promising clinical activity was initially seen in the adult solid tumor patients with target rearrangements, and it was encouraging to see these patients also had responses when the tumors were located in their brains. And what got us really excited as pediatric oncologists was that a variety of pediatric cancers harbor these fusions and mutations within certain tumors.”
With this in mind, Dr. Robinson and colleagues conducted a phase 1/1b study (NCT02650401) of entrectinib in 29 patients with recurrent or refractory solid tumors, including central nervous system (CNS) tumors.
The patients’ median age was 7 years (range, 0-20 years), and roughly half of them were male (n = 15). Patients were diagnosed with neuroblastoma (n = 16), high-grade glioma (n = 5), inflammatory myofibroblastic tumors (n = 3), infantile fibrosarcoma (n = 2), CNS embryonal tumor (n = 1), melanoma (n = 1), and synovial sarcoma (n = 1).
In the dose-finding portion of the trial, patients received entrectinib at 250 mg/m2 (n = 3), 400 mg/m2 (n = 3), 550 mg/m2 (n = 7), or 750 mg/m2 (n = 3).
In the phase 1b portion, patients received entrectinib at 550 mg/m2 (n = 7) – the recommended dose – or 400 mg/m2 (n = 6) if they were unable to swallow intact capsules.
Dr. Robinson said entrectinib was “quite well tolerated” overall, but he did not present any data on adverse events. He did say dose-limiting toxicities included fatigue, elevated creatinine levels, dysgeusia resulting in loss of taste, weight gain, and, in one patient, pulmonary edema.
“Entrectinib produced striking, rapid, and durable responses in all children with refractory CNS and solid tumors that actually harbored these fusions in NTRK1/2/3, ROS1, or ALK,” Dr. Robinson said. “It also produced a significant response in one ALK-mutated neuroblastoma patient. [N]o responses were seen in tumors lacking aberrations in the target kinases.”
In all, 12 patients responded. The three complete responders had an ALK F1174L mutation, an ALK fusion, and an NTRK fusion, respectively. Five partial responders had NTRK fusions, three had ROS1 fusions, and one had an ALK fusion.
Three responders discontinued treatment. Ten patients were still receiving entrectinib at last follow-up, and 11 patients had died.
Progression-free survival was significantly longer among patients who had fusions than among those who did not (P less than .0001).
“To sum up, entrectinib really is very promising,” Dr. Robinson said. “It has very promising antitumor activity and progression-free survival but [only] in patients with target gene fusions.”
Dr. Robinson said this trial is ongoing, but it is now limited to patients with fusions targeted by entrectinib.
The trial is sponsored by Hoffman-La Roche Ltd. and supported by Alex’s Lemonade Stand Center of Excellence. Dr. Robinson has relationships with Lilly, Genentech/Roche, and Novartis.
SOURCE: Robinson GW et al. ASCO 2019. Abstract 10009.
Entrectinib demonstrated “very promising” antitumor activity in children and adolescents with recurrent or refractory solid tumors, according to an investigator involved in a phase 1/1b trial.
Twelve of 29 patients enrolled in the trial have responded to entrectinib. All responders had fusions in genes targeted by the drug – NTRK1/2/3 (TRKA/B/C), ROS1, or ALK – or an ALK mutation.
Details of this study are scheduled to be presented at the annual meeting of the American Society of Clinical Oncology.
Giles W. Robinson, MD, of St. Jude Children’s Research Hospital in Memphis, Tenn., discussed the study during a press briefing in advance of the meeting.
“Entrectinib is an oral and potent inhibitor of the TRKA/B/C, ROS1, and ALK proteins, but it also penetrates into the brain to reach tumors in the brain and spine, which can be a hard area to get drugs to,” Dr. Robinson explained.
“Promising clinical activity was initially seen in the adult solid tumor patients with target rearrangements, and it was encouraging to see these patients also had responses when the tumors were located in their brains. And what got us really excited as pediatric oncologists was that a variety of pediatric cancers harbor these fusions and mutations within certain tumors.”
With this in mind, Dr. Robinson and colleagues conducted a phase 1/1b study (NCT02650401) of entrectinib in 29 patients with recurrent or refractory solid tumors, including central nervous system (CNS) tumors.
The patients’ median age was 7 years (range, 0-20 years), and roughly half of them were male (n = 15). Patients were diagnosed with neuroblastoma (n = 16), high-grade glioma (n = 5), inflammatory myofibroblastic tumors (n = 3), infantile fibrosarcoma (n = 2), CNS embryonal tumor (n = 1), melanoma (n = 1), and synovial sarcoma (n = 1).
In the dose-finding portion of the trial, patients received entrectinib at 250 mg/m2 (n = 3), 400 mg/m2 (n = 3), 550 mg/m2 (n = 7), or 750 mg/m2 (n = 3).
In the phase 1b portion, patients received entrectinib at 550 mg/m2 (n = 7) – the recommended dose – or 400 mg/m2 (n = 6) if they were unable to swallow intact capsules.
Dr. Robinson said entrectinib was “quite well tolerated” overall, but he did not present any data on adverse events. He did say dose-limiting toxicities included fatigue, elevated creatinine levels, dysgeusia resulting in loss of taste, weight gain, and, in one patient, pulmonary edema.
“Entrectinib produced striking, rapid, and durable responses in all children with refractory CNS and solid tumors that actually harbored these fusions in NTRK1/2/3, ROS1, or ALK,” Dr. Robinson said. “It also produced a significant response in one ALK-mutated neuroblastoma patient. [N]o responses were seen in tumors lacking aberrations in the target kinases.”
In all, 12 patients responded. The three complete responders had an ALK F1174L mutation, an ALK fusion, and an NTRK fusion, respectively. Five partial responders had NTRK fusions, three had ROS1 fusions, and one had an ALK fusion.
Three responders discontinued treatment. Ten patients were still receiving entrectinib at last follow-up, and 11 patients had died.
Progression-free survival was significantly longer among patients who had fusions than among those who did not (P less than .0001).
“To sum up, entrectinib really is very promising,” Dr. Robinson said. “It has very promising antitumor activity and progression-free survival but [only] in patients with target gene fusions.”
Dr. Robinson said this trial is ongoing, but it is now limited to patients with fusions targeted by entrectinib.
The trial is sponsored by Hoffman-La Roche Ltd. and supported by Alex’s Lemonade Stand Center of Excellence. Dr. Robinson has relationships with Lilly, Genentech/Roche, and Novartis.
SOURCE: Robinson GW et al. ASCO 2019. Abstract 10009.
REPORTING FROM ASCO 2019
Advanced Melanoma: Treatment After Progression on First-line Therapy
The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved objective response rates (ORRs), progression-free survival (PFS), and overall survival (OS) for patients with metastatic melanoma. This article reviews current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy. The selection of first-line therapy for metastatic melanoma is reviewed in a separate article.
Case Presentation
A 62-year-old man was diagnosed with stage IIA melanoma after undergoing wide local excision of a right scalp lesion (final staging was consistent with pT3aN0M0). After 3.5 years of follow-up, he developed symptoms of vertigo, diplopia, and recurrent falls prompting medical attention. Magnetic resonance imaging (MRI) brain revealed multiple supratentorial and infratentorial lesions concerning for intracranial metastases and computed tomography (CT) chest/abdomen/pelvis revealed a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He was treated with intravenous dexamethasone and further evaluation with an endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass revealed metastatic melanoma. The patient underwent whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy. Testing showed the melanoma was positive for a BRAF V600K mutation. He was started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially did well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass.
After 3 months of therapy, surveillance PET-CT notes increasing size and FDG avidity of the right lower lobe mass. MRI brain reveals resolution of several previously noted metastases, but with interval development of a new left frontal lobe mass concerning for progressive disease.
What is the general approach to treatment of metastatic melanoma after progression on first-line therapy?
Based on the current evidence, there is no definitive algorithm for the treatment of metastatic melanoma after progression on first-line therapy. Enrollment in clinical trials is encouraged to further elucidate the best sequencing of treatment. The current practice is to typically switch class of agents after progression on front-line therapy to either immunotherapy that has not yet been tried or to molecularly targeted therapy in patients harboring a BRAF V600 mutation. After further progression of disease, retreatment with a previously received agent is possible, and this may be combined with investigational therapies.
Immune Checkpoint Inhibitors in Progressive Disease
The 2 major populations of patients to consider are those with BRAF wild-type melanomas who progress on first-line immunotherapy and those with BRAF V600 mutation–positive melanoma who progress on molecularly targeted therapy with BRAF and MEK inhibitors. There is relatively limited data on the efficacy of immune checkpoint inhibition after progression on anti-programmed cell death 1 (PD-1) monotherapy. A small retrospective study of patients who progressed on anti-PD-1 monotherapy were treated with ipilimumab, with a 10% ORR and another 8% having stable disease for more than 6 months; however, 35% of patients experienced grade 3 to 5 immune-related adverse events.1 The only prospective data supports the efficacy of anti-PD-1 therapy after progression on ipilimumab, as supported by the CheckMate 037 trial (nivolumab versus chemotherapy)2 and KEYNOTE-002 trial (pembrolizumab versus chemotherapy)3,4; however, this is no longer applicable as ipilimumab is no longer given in the first-line setting and has been replaced by anti-PD-1 monotherapy or combination immunotherapy.
Another interesting facet of PD-1 monotherapy is the idea of treatment beyond progression. The concept of pseudoprogression—whereby patients receiving PD-1 inhibitors initially meet Response Evaluation Criteria in Solid Tumors (RECIST) criteria for progression, but then later go on to demonstrate significant decreases in tumor burden on subsequent imaging studies—has been described in melanoma patients receiving such immunotherapies. It is thought that pseudoprogression occurs due to either an initial delay in anti-tumor response to the immunotherapy or from the measured target lesion appearing larger due to surrounding immune/inflammatory infiltrate. In an analysis of individual patient data pooled from 8 multicenter clinical trials, 19% of patients were treated beyond initially documented RECIST progression and had subsequent imaging to evaluate the tumor burden; in these patients, the same target lesion later met RECIST criteria for response, with a greater than 30% reduction in tumor size. Furthermore, of the evaluable cohort, the median OS in patients who did receive treatment beyond progression was 24.4 months compared to 11.2 months in those who did not receive treatment beyond progression.5 While further randomized studies are warranted to characterize the potential benefit, the existing data suggests that selected patients who are doing well clinically despite evidence of radiographic progressive disease may benefit from continued treatment with PD-1 inhibitors.
Combination immunotherapy with both PD-1 and CTLA-4 blockade has been studied retrospectively in the second-line setting. A retrospective analysis of patients who had progressive disease on PD-1 inhibitor monotherapy compared the outcomes of patients who received just ipilimumab to those of patients who received both ipilimumab and nivolumab. The ORR (16% ipilimumab vs 21% combination group) and 1-year OS (54% vs 55%) were similar in both groups,6 and this demonstrated significantly less efficacy with combination therapy when compared to use in the first-line setting, albeit in a separate prospective trial.7 A multicenter, retrospective study by Tétu and colleagues compared outcomes with ipilimumab plus nivolumab across 3 groups that included previously untreated patients, patients who had progressed on single-agent immunotherapy, and patients who had progressed on prior molecularly targeted therapy.8 Despite clearly inferior efficacy in previously treated patients, the results support combination immunotherapy as a viable treatment option in the second-line setting. Outcomes are reported in Table 1 below. Of note, there is an ongoing phase 2 trial to assess the use of combined PD-1 and CTLA-4 inhibitors versus CTLA-4 inhibition alone after progression on first-line PD-1 inhibitor monotherapy (NCT03033576).

For patients with BRAF V600–mutation positive melanoma who progress on front-line molecularly targeted therapy, immune checkpoint inhibitor therapy with either anti-PD-1 monotherapy or combination anti-PD-1 and ipilimumab should be considered. The KEYNOTE-006 trial that demonstrated superiority of pembrolizumab compared to ipilimumab included patients who had received up to 1 prior systemic therapy that was not a PD-1 or CTLA-4 inhibitor, and subgroup analysis demonstrated efficacy with pembrolizumab in patients who had received prior treatment with a BRAF inhibitor.9 The retrospective analysis by Tétu et al (Table 1) noted efficacy of combination nivolumab and ipilimumab in patients treated with prior molecularly targeted therapy, as evidenced by an ORR of 35% and median OS of 16.5 months.8
A retrospective trial by Ackerman et al analyzed ORR, median PFS, and median OS from the time of commencement of BRAF inhibitor therapy (with or without a MEK inhibitor), and the comparison was made between those who received ipilimumab before or after molecularly targeted therapy. While ipilimumab is no longer the first-line immunotherapy agent used in advanced melanoma, the study did highlight some important concepts. First, ORRs to BRAF inhibitors were similar between the 2 treatment groups. The conclusions of the analysis were that there was no significant difference in median PFS or OS in regard to which therapy was given first, but median OS after BRAF inhibitors were discontinued was very short and patients had poor responses to ipilimumab after stopping a BRAF inhibitor. This highlights the concern that patients who have progressive disease on molecularly targeted therapy often have a poor performance status and undergo too rapid of a clinical decline to derive benefit from immunotherapy, which can often take weeks to months to take effect.10
A more recent retrospective study by Johnson et al compared efficacy outcomes in patients who received single-agent anti-PD-1 therapy prior to molecularly targeted therapy (BRAF inhibitor with or without MEK inhibitor) to those who received molecularly targeted therapy prior to anti-PD-1 therapy. The difference in median OS was not statistically significant (27.5 months with PD-1 inhibitor first vs 40.3 months with molecularly targeted therapy first). Both treatments demonstrated second-line efficacy, but outcomes were inferior to those reported when either type of therapy was used in the first-line setting. Interestingly, patients who were maintained on molecularly targeted therapy for more than 6 months prior to progression demonstrated an improved ORR to subsequent anti-PD-1 therapy (34% vs 15%).11
Molecularly Targeted Therapy in Progressive Disease
When melanoma patients with a BRAF V600 mutation are treated initially with immunotherapy and demonstrate progressive disease, molecularly targeted therapy with combined BRAF and MEK inhibition should be considered for second-line therapy. While there are no dedicated prospective trial results with BRAF/MEK inhibitors after progression on immune checkpoint inhibitors, for practical purposes, it may be reasonable to extrapolate outcomes from the currently available first-line studies.12-16 An ongoing study (NCT02224781) in which patients are randomized to receive ipilimumab/nivolumab followed by dabrafenib/trametinib at progression versus the reverse order is designed to help answer the question of optimal sequencing and timing of therapy. Johnson et al’s retrospective analysis of patients receiving single-agent anti-PD-1 therapy prior to molecularly targeted therapy compared to the reverse order concluded that there was no statistically significant difference in median OS.11 Ackerman et al’s retrospective study of patients who had received ipilimumab before or after molecularly targeted therapy noted similar response rates to molecularly targeted therapy in each treatment group.10
The issue of re-treatment with a BRAF/MEK inhibitor in a patient already progressing on targeted therapy is a more challenging situation, and currently available data suggests there is limited benefit. However, select patients may be considered for this approach. The combination of dabrafenib/trametinib demonstrated an ORR of approximately 15% in a cohort of patients who progressed on single-agent BRAF inhibitor therapy, with a suggestion that those patients who had previously derived benefit for more than 6 months may have a more favorable outcome.17
Based on the hypothesis that acquired resistance to BRAF/MEK inhibition may be reversible if the selective pressure of the medication is held for a period of time, a phase 2 trial analyzed outcomes with retreatment. The study included patients with BRAF V600–mutant melanoma who had progressed on prior BRAF inhibition (with or without MEK inhibitor) and required that they had been off of therapy for at least 12 weeks. Of the 25 patients who received dabrafenib plus trametinib as retreatment, 32% demonstrated a partial response and 40% had stable disease.18 While further studies are warranted, retreatment with molecularly targeted therapy may be a viable option, especially in light of the multiple approved BRAF and MEK inhibitor combinations.
Another concept that has been studied is treatment beyond disease progression with molecularly targeted therapy. In a retrospective analysis of patients who had progressed on a single-agent BRAF inhibitor, 39% of those patients were continued on the same BRAF inhibitor and compared to patients who received no subsequent therapy or changed to an alternative systemic therapy. In the multivariable analysis adjusting for other prognostic factors, continued treatment with the BRAF inhibitor was associated with prolonged OS.19
Case Conclusion
The patient is started on second-line therapy with nivolumab and ipilimumab and demonstrates a partial response. One year later he continues to feel well with decreased size of the intracranial and right lower lobe lesions, and without any interval development of new areas of metastatic disease.
Special Considerations
Intralesional Therapies
Talimogene laherparepvec (T-VEC) is a genetically modified herpesvirus-1 oncolytic virus that is injected into melanoma skin lesions and leads to the expression of granulocyte-macrophage colony-stimulating factor. While T-VEC is currently approved for local treatment of unresectable cutaneous, subcutaneous, or nodal recurrences,20 it has also been investigated in combination with other therapies for patients with advanced disease. In patients with previously treated melanoma, T-VEC plus ipilimumab demonstrated superior ORR to ipilimumab alone (39% vs 18%), and the tumor response was not limited to the injected lesions. The observation of systemic response suggests synergy between T-VEC and immune checkpoint blockade in enhancing the anti-tumor immune response.21 The phase 1b MASTERKEY-265 trial combining pembrolizumab and T-VEC led to an ORR of 62% and CR of 33%.22 A phase 3 trial comparing pembrolizumab plus T-VEC to pembrolizumab alone is ongoing (NCT02263508).
Melanoma Brain Metastases
The presence of brain metastases is a common event in patients with metastatic melanoma, and often confers a poor prognosis.23 The approach to the management of brain metastases should be multidisciplinary among medical oncology, neurosurgery, and radiation oncology providers, as treatment algorithms continue to rapidly evolve. Historically, there has been little prospective clinical trial data regarding optimal systemic therapy, and local therapies such as surgery or stereotactic radiation have long been the mainstay of therapy for intracranial disease.24 However, recent data with both immunotherapy and molecularly targeted therapy has demonstrated efficacy with intracranial metastases.
A recent trial of combined nivolumab and ipilimumab as frontline therapy in patients with asymptomatic melanoma brain metastases demonstrated a complete response rate of 26% and partial response rate of 30% in patients with a median follow-up of 14 months.25 In a separate study, ipilimumab plus nivolumab demonstrated better intracranial ORR when compared to nivolumab alone in asymptomatic, previously untreated patients. Outcomes were better in patients presenting with asymptomatic versus symptomatic brain metastases.26 Collectively, these results suggest that systemic immunotherapy alone may be adequate for patients with asymptomatic, previously untreated brain metastases.
For molecularly targeted therapy in patients with BRAF mutations and brain metastases, the BREAK-MB trial demonstrated that an intracranial response was attainable with dabrafenib regardless of whether the patient had previously received local therapy in the form of surgery or radiation.27 The COMBI-MB trial enhanced the preexisting data by testing the intracranial efficacy of dabrafenib plus trametinib in 4 different cohorts of patients, further supporting that systemic molecularly targeted therapy can provide significant intracranial activity in patients with both symptomatic and asymptomatic brain lesions and regardless of prior local therapy (Table 2).28

Conclusion
The treatment of advanced melanoma has been drastically improved over the past decade by the development and study of immune checkpoint inhibitors and molecularly targeted agents. There is still much to learn regarding the optimal combination and sequencing of therapies. Many of these trials are ongoing and will provide additional evidence to guide treatment decisions moving forward.
1. Bowyer S, Prithviraj P, Lorigan P, et al. Efficacy and toxicity of treatment with the anti-CTLA-4 antibody ipilimumab in patients with metastatic melanoma after prior anti-PD-1 therapy. Br J Cancer. 2016;114:1084-1089.
2. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-384.
3. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-918.
4. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
5. Beaver JA, Hazarika M, Mulkey F, et al. Patients with melanoma treated with an anti-PD-1 antibody beyond RECIST progression: a US Food and Drug Administration pooled analysis. Lancet Oncol. 2018;19:229-239.
6. Zimmer L, Apuri S, Eroglu Z, et al. Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur J Cancer. 2017;75:47-55.
7. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
8. Tétu P, Mangana J, Dummer R, et al. Benefit of the nivolumab and ipilimumab combination in pretreated advanced melanoma. Eur J Cancer. 2018;93:147-149.
9. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
10. Ackerman A, Klein O, McDermott D, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120:1695-1701.
11. Johnson DB, Pectasides E, Feld E, et al. Sequencing treatment in BRAFV600 mutant melanoma: anti-pd-1 before and after BRAF inhibition. J Immunother. 2017;40:31-35.
12. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
13. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
14. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-1260.
15. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
16. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
17. Johnson DB, Flaherty KT, Weber, JS et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32:3697-3704.
18. Schreuer M, Jansen Y, Planken S, et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAFV600-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017;18:464-472.
19. Chan MM, Haydu LE, Azer MW, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120:3142-3153.
20. Imlygic (talimogene laherparepvec) suspension for intralesional injection [package insert]. Thousand Oaks, CA: BioVex; 2015.
21. Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase ii study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36:1658-1667.
22. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2018;174:1031-1032.
23. Sampson JH, Carter Jr. JH, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
24. Yamamoto M, Serizawa T, Shuto T, et al. Stereotactic radiosurgery for patients with multiple brain metastases (JLGK0901): a multi-institutional prospective observational study. Lancet Oncol. 2014;15:387-395.
25. Tawbi HA, Forsyth PA, Hamid O, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379:722-730.
26. Long GV, Atkinson V, La S, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicenter randomised phase 2 study. Lancet Oncol. 2018;19:672-681.
27. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicenter, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087-1095.
28. Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicenter, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18:863-873.
The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved objective response rates (ORRs), progression-free survival (PFS), and overall survival (OS) for patients with metastatic melanoma. This article reviews current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy. The selection of first-line therapy for metastatic melanoma is reviewed in a separate article.
Case Presentation
A 62-year-old man was diagnosed with stage IIA melanoma after undergoing wide local excision of a right scalp lesion (final staging was consistent with pT3aN0M0). After 3.5 years of follow-up, he developed symptoms of vertigo, diplopia, and recurrent falls prompting medical attention. Magnetic resonance imaging (MRI) brain revealed multiple supratentorial and infratentorial lesions concerning for intracranial metastases and computed tomography (CT) chest/abdomen/pelvis revealed a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He was treated with intravenous dexamethasone and further evaluation with an endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass revealed metastatic melanoma. The patient underwent whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy. Testing showed the melanoma was positive for a BRAF V600K mutation. He was started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially did well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass.
After 3 months of therapy, surveillance PET-CT notes increasing size and FDG avidity of the right lower lobe mass. MRI brain reveals resolution of several previously noted metastases, but with interval development of a new left frontal lobe mass concerning for progressive disease.
What is the general approach to treatment of metastatic melanoma after progression on first-line therapy?
Based on the current evidence, there is no definitive algorithm for the treatment of metastatic melanoma after progression on first-line therapy. Enrollment in clinical trials is encouraged to further elucidate the best sequencing of treatment. The current practice is to typically switch class of agents after progression on front-line therapy to either immunotherapy that has not yet been tried or to molecularly targeted therapy in patients harboring a BRAF V600 mutation. After further progression of disease, retreatment with a previously received agent is possible, and this may be combined with investigational therapies.
Immune Checkpoint Inhibitors in Progressive Disease
The 2 major populations of patients to consider are those with BRAF wild-type melanomas who progress on first-line immunotherapy and those with BRAF V600 mutation–positive melanoma who progress on molecularly targeted therapy with BRAF and MEK inhibitors. There is relatively limited data on the efficacy of immune checkpoint inhibition after progression on anti-programmed cell death 1 (PD-1) monotherapy. A small retrospective study of patients who progressed on anti-PD-1 monotherapy were treated with ipilimumab, with a 10% ORR and another 8% having stable disease for more than 6 months; however, 35% of patients experienced grade 3 to 5 immune-related adverse events.1 The only prospective data supports the efficacy of anti-PD-1 therapy after progression on ipilimumab, as supported by the CheckMate 037 trial (nivolumab versus chemotherapy)2 and KEYNOTE-002 trial (pembrolizumab versus chemotherapy)3,4; however, this is no longer applicable as ipilimumab is no longer given in the first-line setting and has been replaced by anti-PD-1 monotherapy or combination immunotherapy.
Another interesting facet of PD-1 monotherapy is the idea of treatment beyond progression. The concept of pseudoprogression—whereby patients receiving PD-1 inhibitors initially meet Response Evaluation Criteria in Solid Tumors (RECIST) criteria for progression, but then later go on to demonstrate significant decreases in tumor burden on subsequent imaging studies—has been described in melanoma patients receiving such immunotherapies. It is thought that pseudoprogression occurs due to either an initial delay in anti-tumor response to the immunotherapy or from the measured target lesion appearing larger due to surrounding immune/inflammatory infiltrate. In an analysis of individual patient data pooled from 8 multicenter clinical trials, 19% of patients were treated beyond initially documented RECIST progression and had subsequent imaging to evaluate the tumor burden; in these patients, the same target lesion later met RECIST criteria for response, with a greater than 30% reduction in tumor size. Furthermore, of the evaluable cohort, the median OS in patients who did receive treatment beyond progression was 24.4 months compared to 11.2 months in those who did not receive treatment beyond progression.5 While further randomized studies are warranted to characterize the potential benefit, the existing data suggests that selected patients who are doing well clinically despite evidence of radiographic progressive disease may benefit from continued treatment with PD-1 inhibitors.
Combination immunotherapy with both PD-1 and CTLA-4 blockade has been studied retrospectively in the second-line setting. A retrospective analysis of patients who had progressive disease on PD-1 inhibitor monotherapy compared the outcomes of patients who received just ipilimumab to those of patients who received both ipilimumab and nivolumab. The ORR (16% ipilimumab vs 21% combination group) and 1-year OS (54% vs 55%) were similar in both groups,6 and this demonstrated significantly less efficacy with combination therapy when compared to use in the first-line setting, albeit in a separate prospective trial.7 A multicenter, retrospective study by Tétu and colleagues compared outcomes with ipilimumab plus nivolumab across 3 groups that included previously untreated patients, patients who had progressed on single-agent immunotherapy, and patients who had progressed on prior molecularly targeted therapy.8 Despite clearly inferior efficacy in previously treated patients, the results support combination immunotherapy as a viable treatment option in the second-line setting. Outcomes are reported in Table 1 below. Of note, there is an ongoing phase 2 trial to assess the use of combined PD-1 and CTLA-4 inhibitors versus CTLA-4 inhibition alone after progression on first-line PD-1 inhibitor monotherapy (NCT03033576).
For patients with BRAF V600–mutation positive melanoma who progress on front-line molecularly targeted therapy, immune checkpoint inhibitor therapy with either anti-PD-1 monotherapy or combination anti-PD-1 and ipilimumab should be considered. The KEYNOTE-006 trial that demonstrated superiority of pembrolizumab compared to ipilimumab included patients who had received up to 1 prior systemic therapy that was not a PD-1 or CTLA-4 inhibitor, and subgroup analysis demonstrated efficacy with pembrolizumab in patients who had received prior treatment with a BRAF inhibitor.9 The retrospective analysis by Tétu et al (Table 1) noted efficacy of combination nivolumab and ipilimumab in patients treated with prior molecularly targeted therapy, as evidenced by an ORR of 35% and median OS of 16.5 months.8
A retrospective trial by Ackerman et al analyzed ORR, median PFS, and median OS from the time of commencement of BRAF inhibitor therapy (with or without a MEK inhibitor), and the comparison was made between those who received ipilimumab before or after molecularly targeted therapy. While ipilimumab is no longer the first-line immunotherapy agent used in advanced melanoma, the study did highlight some important concepts. First, ORRs to BRAF inhibitors were similar between the 2 treatment groups. The conclusions of the analysis were that there was no significant difference in median PFS or OS in regard to which therapy was given first, but median OS after BRAF inhibitors were discontinued was very short and patients had poor responses to ipilimumab after stopping a BRAF inhibitor. This highlights the concern that patients who have progressive disease on molecularly targeted therapy often have a poor performance status and undergo too rapid of a clinical decline to derive benefit from immunotherapy, which can often take weeks to months to take effect.10
A more recent retrospective study by Johnson et al compared efficacy outcomes in patients who received single-agent anti-PD-1 therapy prior to molecularly targeted therapy (BRAF inhibitor with or without MEK inhibitor) to those who received molecularly targeted therapy prior to anti-PD-1 therapy. The difference in median OS was not statistically significant (27.5 months with PD-1 inhibitor first vs 40.3 months with molecularly targeted therapy first). Both treatments demonstrated second-line efficacy, but outcomes were inferior to those reported when either type of therapy was used in the first-line setting. Interestingly, patients who were maintained on molecularly targeted therapy for more than 6 months prior to progression demonstrated an improved ORR to subsequent anti-PD-1 therapy (34% vs 15%).11
Molecularly Targeted Therapy in Progressive Disease
When melanoma patients with a BRAF V600 mutation are treated initially with immunotherapy and demonstrate progressive disease, molecularly targeted therapy with combined BRAF and MEK inhibition should be considered for second-line therapy. While there are no dedicated prospective trial results with BRAF/MEK inhibitors after progression on immune checkpoint inhibitors, for practical purposes, it may be reasonable to extrapolate outcomes from the currently available first-line studies.12-16 An ongoing study (NCT02224781) in which patients are randomized to receive ipilimumab/nivolumab followed by dabrafenib/trametinib at progression versus the reverse order is designed to help answer the question of optimal sequencing and timing of therapy. Johnson et al’s retrospective analysis of patients receiving single-agent anti-PD-1 therapy prior to molecularly targeted therapy compared to the reverse order concluded that there was no statistically significant difference in median OS.11 Ackerman et al’s retrospective study of patients who had received ipilimumab before or after molecularly targeted therapy noted similar response rates to molecularly targeted therapy in each treatment group.10
The issue of re-treatment with a BRAF/MEK inhibitor in a patient already progressing on targeted therapy is a more challenging situation, and currently available data suggests there is limited benefit. However, select patients may be considered for this approach. The combination of dabrafenib/trametinib demonstrated an ORR of approximately 15% in a cohort of patients who progressed on single-agent BRAF inhibitor therapy, with a suggestion that those patients who had previously derived benefit for more than 6 months may have a more favorable outcome.17
Based on the hypothesis that acquired resistance to BRAF/MEK inhibition may be reversible if the selective pressure of the medication is held for a period of time, a phase 2 trial analyzed outcomes with retreatment. The study included patients with BRAF V600–mutant melanoma who had progressed on prior BRAF inhibition (with or without MEK inhibitor) and required that they had been off of therapy for at least 12 weeks. Of the 25 patients who received dabrafenib plus trametinib as retreatment, 32% demonstrated a partial response and 40% had stable disease.18 While further studies are warranted, retreatment with molecularly targeted therapy may be a viable option, especially in light of the multiple approved BRAF and MEK inhibitor combinations.
Another concept that has been studied is treatment beyond disease progression with molecularly targeted therapy. In a retrospective analysis of patients who had progressed on a single-agent BRAF inhibitor, 39% of those patients were continued on the same BRAF inhibitor and compared to patients who received no subsequent therapy or changed to an alternative systemic therapy. In the multivariable analysis adjusting for other prognostic factors, continued treatment with the BRAF inhibitor was associated with prolonged OS.19
Case Conclusion
The patient is started on second-line therapy with nivolumab and ipilimumab and demonstrates a partial response. One year later he continues to feel well with decreased size of the intracranial and right lower lobe lesions, and without any interval development of new areas of metastatic disease.
Special Considerations
Intralesional Therapies
Talimogene laherparepvec (T-VEC) is a genetically modified herpesvirus-1 oncolytic virus that is injected into melanoma skin lesions and leads to the expression of granulocyte-macrophage colony-stimulating factor. While T-VEC is currently approved for local treatment of unresectable cutaneous, subcutaneous, or nodal recurrences,20 it has also been investigated in combination with other therapies for patients with advanced disease. In patients with previously treated melanoma, T-VEC plus ipilimumab demonstrated superior ORR to ipilimumab alone (39% vs 18%), and the tumor response was not limited to the injected lesions. The observation of systemic response suggests synergy between T-VEC and immune checkpoint blockade in enhancing the anti-tumor immune response.21 The phase 1b MASTERKEY-265 trial combining pembrolizumab and T-VEC led to an ORR of 62% and CR of 33%.22 A phase 3 trial comparing pembrolizumab plus T-VEC to pembrolizumab alone is ongoing (NCT02263508).
Melanoma Brain Metastases
The presence of brain metastases is a common event in patients with metastatic melanoma, and often confers a poor prognosis.23 The approach to the management of brain metastases should be multidisciplinary among medical oncology, neurosurgery, and radiation oncology providers, as treatment algorithms continue to rapidly evolve. Historically, there has been little prospective clinical trial data regarding optimal systemic therapy, and local therapies such as surgery or stereotactic radiation have long been the mainstay of therapy for intracranial disease.24 However, recent data with both immunotherapy and molecularly targeted therapy has demonstrated efficacy with intracranial metastases.
A recent trial of combined nivolumab and ipilimumab as frontline therapy in patients with asymptomatic melanoma brain metastases demonstrated a complete response rate of 26% and partial response rate of 30% in patients with a median follow-up of 14 months.25 In a separate study, ipilimumab plus nivolumab demonstrated better intracranial ORR when compared to nivolumab alone in asymptomatic, previously untreated patients. Outcomes were better in patients presenting with asymptomatic versus symptomatic brain metastases.26 Collectively, these results suggest that systemic immunotherapy alone may be adequate for patients with asymptomatic, previously untreated brain metastases.
For molecularly targeted therapy in patients with BRAF mutations and brain metastases, the BREAK-MB trial demonstrated that an intracranial response was attainable with dabrafenib regardless of whether the patient had previously received local therapy in the form of surgery or radiation.27 The COMBI-MB trial enhanced the preexisting data by testing the intracranial efficacy of dabrafenib plus trametinib in 4 different cohorts of patients, further supporting that systemic molecularly targeted therapy can provide significant intracranial activity in patients with both symptomatic and asymptomatic brain lesions and regardless of prior local therapy (Table 2).28
Conclusion
The treatment of advanced melanoma has been drastically improved over the past decade by the development and study of immune checkpoint inhibitors and molecularly targeted agents. There is still much to learn regarding the optimal combination and sequencing of therapies. Many of these trials are ongoing and will provide additional evidence to guide treatment decisions moving forward.
The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved objective response rates (ORRs), progression-free survival (PFS), and overall survival (OS) for patients with metastatic melanoma. This article reviews current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy. The selection of first-line therapy for metastatic melanoma is reviewed in a separate article.
Case Presentation
A 62-year-old man was diagnosed with stage IIA melanoma after undergoing wide local excision of a right scalp lesion (final staging was consistent with pT3aN0M0). After 3.5 years of follow-up, he developed symptoms of vertigo, diplopia, and recurrent falls prompting medical attention. Magnetic resonance imaging (MRI) brain revealed multiple supratentorial and infratentorial lesions concerning for intracranial metastases and computed tomography (CT) chest/abdomen/pelvis revealed a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He was treated with intravenous dexamethasone and further evaluation with an endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass revealed metastatic melanoma. The patient underwent whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy. Testing showed the melanoma was positive for a BRAF V600K mutation. He was started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially did well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass.
After 3 months of therapy, surveillance PET-CT notes increasing size and FDG avidity of the right lower lobe mass. MRI brain reveals resolution of several previously noted metastases, but with interval development of a new left frontal lobe mass concerning for progressive disease.
What is the general approach to treatment of metastatic melanoma after progression on first-line therapy?
Based on the current evidence, there is no definitive algorithm for the treatment of metastatic melanoma after progression on first-line therapy. Enrollment in clinical trials is encouraged to further elucidate the best sequencing of treatment. The current practice is to typically switch class of agents after progression on front-line therapy to either immunotherapy that has not yet been tried or to molecularly targeted therapy in patients harboring a BRAF V600 mutation. After further progression of disease, retreatment with a previously received agent is possible, and this may be combined with investigational therapies.
Immune Checkpoint Inhibitors in Progressive Disease
The 2 major populations of patients to consider are those with BRAF wild-type melanomas who progress on first-line immunotherapy and those with BRAF V600 mutation–positive melanoma who progress on molecularly targeted therapy with BRAF and MEK inhibitors. There is relatively limited data on the efficacy of immune checkpoint inhibition after progression on anti-programmed cell death 1 (PD-1) monotherapy. A small retrospective study of patients who progressed on anti-PD-1 monotherapy were treated with ipilimumab, with a 10% ORR and another 8% having stable disease for more than 6 months; however, 35% of patients experienced grade 3 to 5 immune-related adverse events.1 The only prospective data supports the efficacy of anti-PD-1 therapy after progression on ipilimumab, as supported by the CheckMate 037 trial (nivolumab versus chemotherapy)2 and KEYNOTE-002 trial (pembrolizumab versus chemotherapy)3,4; however, this is no longer applicable as ipilimumab is no longer given in the first-line setting and has been replaced by anti-PD-1 monotherapy or combination immunotherapy.
Another interesting facet of PD-1 monotherapy is the idea of treatment beyond progression. The concept of pseudoprogression—whereby patients receiving PD-1 inhibitors initially meet Response Evaluation Criteria in Solid Tumors (RECIST) criteria for progression, but then later go on to demonstrate significant decreases in tumor burden on subsequent imaging studies—has been described in melanoma patients receiving such immunotherapies. It is thought that pseudoprogression occurs due to either an initial delay in anti-tumor response to the immunotherapy or from the measured target lesion appearing larger due to surrounding immune/inflammatory infiltrate. In an analysis of individual patient data pooled from 8 multicenter clinical trials, 19% of patients were treated beyond initially documented RECIST progression and had subsequent imaging to evaluate the tumor burden; in these patients, the same target lesion later met RECIST criteria for response, with a greater than 30% reduction in tumor size. Furthermore, of the evaluable cohort, the median OS in patients who did receive treatment beyond progression was 24.4 months compared to 11.2 months in those who did not receive treatment beyond progression.5 While further randomized studies are warranted to characterize the potential benefit, the existing data suggests that selected patients who are doing well clinically despite evidence of radiographic progressive disease may benefit from continued treatment with PD-1 inhibitors.
Combination immunotherapy with both PD-1 and CTLA-4 blockade has been studied retrospectively in the second-line setting. A retrospective analysis of patients who had progressive disease on PD-1 inhibitor monotherapy compared the outcomes of patients who received just ipilimumab to those of patients who received both ipilimumab and nivolumab. The ORR (16% ipilimumab vs 21% combination group) and 1-year OS (54% vs 55%) were similar in both groups,6 and this demonstrated significantly less efficacy with combination therapy when compared to use in the first-line setting, albeit in a separate prospective trial.7 A multicenter, retrospective study by Tétu and colleagues compared outcomes with ipilimumab plus nivolumab across 3 groups that included previously untreated patients, patients who had progressed on single-agent immunotherapy, and patients who had progressed on prior molecularly targeted therapy.8 Despite clearly inferior efficacy in previously treated patients, the results support combination immunotherapy as a viable treatment option in the second-line setting. Outcomes are reported in Table 1 below. Of note, there is an ongoing phase 2 trial to assess the use of combined PD-1 and CTLA-4 inhibitors versus CTLA-4 inhibition alone after progression on first-line PD-1 inhibitor monotherapy (NCT03033576).
For patients with BRAF V600–mutation positive melanoma who progress on front-line molecularly targeted therapy, immune checkpoint inhibitor therapy with either anti-PD-1 monotherapy or combination anti-PD-1 and ipilimumab should be considered. The KEYNOTE-006 trial that demonstrated superiority of pembrolizumab compared to ipilimumab included patients who had received up to 1 prior systemic therapy that was not a PD-1 or CTLA-4 inhibitor, and subgroup analysis demonstrated efficacy with pembrolizumab in patients who had received prior treatment with a BRAF inhibitor.9 The retrospective analysis by Tétu et al (Table 1) noted efficacy of combination nivolumab and ipilimumab in patients treated with prior molecularly targeted therapy, as evidenced by an ORR of 35% and median OS of 16.5 months.8
A retrospective trial by Ackerman et al analyzed ORR, median PFS, and median OS from the time of commencement of BRAF inhibitor therapy (with or without a MEK inhibitor), and the comparison was made between those who received ipilimumab before or after molecularly targeted therapy. While ipilimumab is no longer the first-line immunotherapy agent used in advanced melanoma, the study did highlight some important concepts. First, ORRs to BRAF inhibitors were similar between the 2 treatment groups. The conclusions of the analysis were that there was no significant difference in median PFS or OS in regard to which therapy was given first, but median OS after BRAF inhibitors were discontinued was very short and patients had poor responses to ipilimumab after stopping a BRAF inhibitor. This highlights the concern that patients who have progressive disease on molecularly targeted therapy often have a poor performance status and undergo too rapid of a clinical decline to derive benefit from immunotherapy, which can often take weeks to months to take effect.10
A more recent retrospective study by Johnson et al compared efficacy outcomes in patients who received single-agent anti-PD-1 therapy prior to molecularly targeted therapy (BRAF inhibitor with or without MEK inhibitor) to those who received molecularly targeted therapy prior to anti-PD-1 therapy. The difference in median OS was not statistically significant (27.5 months with PD-1 inhibitor first vs 40.3 months with molecularly targeted therapy first). Both treatments demonstrated second-line efficacy, but outcomes were inferior to those reported when either type of therapy was used in the first-line setting. Interestingly, patients who were maintained on molecularly targeted therapy for more than 6 months prior to progression demonstrated an improved ORR to subsequent anti-PD-1 therapy (34% vs 15%).11
Molecularly Targeted Therapy in Progressive Disease
When melanoma patients with a BRAF V600 mutation are treated initially with immunotherapy and demonstrate progressive disease, molecularly targeted therapy with combined BRAF and MEK inhibition should be considered for second-line therapy. While there are no dedicated prospective trial results with BRAF/MEK inhibitors after progression on immune checkpoint inhibitors, for practical purposes, it may be reasonable to extrapolate outcomes from the currently available first-line studies.12-16 An ongoing study (NCT02224781) in which patients are randomized to receive ipilimumab/nivolumab followed by dabrafenib/trametinib at progression versus the reverse order is designed to help answer the question of optimal sequencing and timing of therapy. Johnson et al’s retrospective analysis of patients receiving single-agent anti-PD-1 therapy prior to molecularly targeted therapy compared to the reverse order concluded that there was no statistically significant difference in median OS.11 Ackerman et al’s retrospective study of patients who had received ipilimumab before or after molecularly targeted therapy noted similar response rates to molecularly targeted therapy in each treatment group.10
The issue of re-treatment with a BRAF/MEK inhibitor in a patient already progressing on targeted therapy is a more challenging situation, and currently available data suggests there is limited benefit. However, select patients may be considered for this approach. The combination of dabrafenib/trametinib demonstrated an ORR of approximately 15% in a cohort of patients who progressed on single-agent BRAF inhibitor therapy, with a suggestion that those patients who had previously derived benefit for more than 6 months may have a more favorable outcome.17
Based on the hypothesis that acquired resistance to BRAF/MEK inhibition may be reversible if the selective pressure of the medication is held for a period of time, a phase 2 trial analyzed outcomes with retreatment. The study included patients with BRAF V600–mutant melanoma who had progressed on prior BRAF inhibition (with or without MEK inhibitor) and required that they had been off of therapy for at least 12 weeks. Of the 25 patients who received dabrafenib plus trametinib as retreatment, 32% demonstrated a partial response and 40% had stable disease.18 While further studies are warranted, retreatment with molecularly targeted therapy may be a viable option, especially in light of the multiple approved BRAF and MEK inhibitor combinations.
Another concept that has been studied is treatment beyond disease progression with molecularly targeted therapy. In a retrospective analysis of patients who had progressed on a single-agent BRAF inhibitor, 39% of those patients were continued on the same BRAF inhibitor and compared to patients who received no subsequent therapy or changed to an alternative systemic therapy. In the multivariable analysis adjusting for other prognostic factors, continued treatment with the BRAF inhibitor was associated with prolonged OS.19
Case Conclusion
The patient is started on second-line therapy with nivolumab and ipilimumab and demonstrates a partial response. One year later he continues to feel well with decreased size of the intracranial and right lower lobe lesions, and without any interval development of new areas of metastatic disease.
Special Considerations
Intralesional Therapies
Talimogene laherparepvec (T-VEC) is a genetically modified herpesvirus-1 oncolytic virus that is injected into melanoma skin lesions and leads to the expression of granulocyte-macrophage colony-stimulating factor. While T-VEC is currently approved for local treatment of unresectable cutaneous, subcutaneous, or nodal recurrences,20 it has also been investigated in combination with other therapies for patients with advanced disease. In patients with previously treated melanoma, T-VEC plus ipilimumab demonstrated superior ORR to ipilimumab alone (39% vs 18%), and the tumor response was not limited to the injected lesions. The observation of systemic response suggests synergy between T-VEC and immune checkpoint blockade in enhancing the anti-tumor immune response.21 The phase 1b MASTERKEY-265 trial combining pembrolizumab and T-VEC led to an ORR of 62% and CR of 33%.22 A phase 3 trial comparing pembrolizumab plus T-VEC to pembrolizumab alone is ongoing (NCT02263508).
Melanoma Brain Metastases
The presence of brain metastases is a common event in patients with metastatic melanoma, and often confers a poor prognosis.23 The approach to the management of brain metastases should be multidisciplinary among medical oncology, neurosurgery, and radiation oncology providers, as treatment algorithms continue to rapidly evolve. Historically, there has been little prospective clinical trial data regarding optimal systemic therapy, and local therapies such as surgery or stereotactic radiation have long been the mainstay of therapy for intracranial disease.24 However, recent data with both immunotherapy and molecularly targeted therapy has demonstrated efficacy with intracranial metastases.
A recent trial of combined nivolumab and ipilimumab as frontline therapy in patients with asymptomatic melanoma brain metastases demonstrated a complete response rate of 26% and partial response rate of 30% in patients with a median follow-up of 14 months.25 In a separate study, ipilimumab plus nivolumab demonstrated better intracranial ORR when compared to nivolumab alone in asymptomatic, previously untreated patients. Outcomes were better in patients presenting with asymptomatic versus symptomatic brain metastases.26 Collectively, these results suggest that systemic immunotherapy alone may be adequate for patients with asymptomatic, previously untreated brain metastases.
For molecularly targeted therapy in patients with BRAF mutations and brain metastases, the BREAK-MB trial demonstrated that an intracranial response was attainable with dabrafenib regardless of whether the patient had previously received local therapy in the form of surgery or radiation.27 The COMBI-MB trial enhanced the preexisting data by testing the intracranial efficacy of dabrafenib plus trametinib in 4 different cohorts of patients, further supporting that systemic molecularly targeted therapy can provide significant intracranial activity in patients with both symptomatic and asymptomatic brain lesions and regardless of prior local therapy (Table 2).28
Conclusion
The treatment of advanced melanoma has been drastically improved over the past decade by the development and study of immune checkpoint inhibitors and molecularly targeted agents. There is still much to learn regarding the optimal combination and sequencing of therapies. Many of these trials are ongoing and will provide additional evidence to guide treatment decisions moving forward.
1. Bowyer S, Prithviraj P, Lorigan P, et al. Efficacy and toxicity of treatment with the anti-CTLA-4 antibody ipilimumab in patients with metastatic melanoma after prior anti-PD-1 therapy. Br J Cancer. 2016;114:1084-1089.
2. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-384.
3. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-918.
4. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
5. Beaver JA, Hazarika M, Mulkey F, et al. Patients with melanoma treated with an anti-PD-1 antibody beyond RECIST progression: a US Food and Drug Administration pooled analysis. Lancet Oncol. 2018;19:229-239.
6. Zimmer L, Apuri S, Eroglu Z, et al. Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur J Cancer. 2017;75:47-55.
7. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
8. Tétu P, Mangana J, Dummer R, et al. Benefit of the nivolumab and ipilimumab combination in pretreated advanced melanoma. Eur J Cancer. 2018;93:147-149.
9. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
10. Ackerman A, Klein O, McDermott D, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120:1695-1701.
11. Johnson DB, Pectasides E, Feld E, et al. Sequencing treatment in BRAFV600 mutant melanoma: anti-pd-1 before and after BRAF inhibition. J Immunother. 2017;40:31-35.
12. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
13. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
14. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-1260.
15. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
16. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
17. Johnson DB, Flaherty KT, Weber, JS et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32:3697-3704.
18. Schreuer M, Jansen Y, Planken S, et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAFV600-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017;18:464-472.
19. Chan MM, Haydu LE, Azer MW, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120:3142-3153.
20. Imlygic (talimogene laherparepvec) suspension for intralesional injection [package insert]. Thousand Oaks, CA: BioVex; 2015.
21. Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase ii study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36:1658-1667.
22. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2018;174:1031-1032.
23. Sampson JH, Carter Jr. JH, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
24. Yamamoto M, Serizawa T, Shuto T, et al. Stereotactic radiosurgery for patients with multiple brain metastases (JLGK0901): a multi-institutional prospective observational study. Lancet Oncol. 2014;15:387-395.
25. Tawbi HA, Forsyth PA, Hamid O, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379:722-730.
26. Long GV, Atkinson V, La S, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicenter randomised phase 2 study. Lancet Oncol. 2018;19:672-681.
27. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicenter, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087-1095.
28. Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicenter, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18:863-873.
1. Bowyer S, Prithviraj P, Lorigan P, et al. Efficacy and toxicity of treatment with the anti-CTLA-4 antibody ipilimumab in patients with metastatic melanoma after prior anti-PD-1 therapy. Br J Cancer. 2016;114:1084-1089.
2. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-384.
3. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-918.
4. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
5. Beaver JA, Hazarika M, Mulkey F, et al. Patients with melanoma treated with an anti-PD-1 antibody beyond RECIST progression: a US Food and Drug Administration pooled analysis. Lancet Oncol. 2018;19:229-239.
6. Zimmer L, Apuri S, Eroglu Z, et al. Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur J Cancer. 2017;75:47-55.
7. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
8. Tétu P, Mangana J, Dummer R, et al. Benefit of the nivolumab and ipilimumab combination in pretreated advanced melanoma. Eur J Cancer. 2018;93:147-149.
9. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
10. Ackerman A, Klein O, McDermott D, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120:1695-1701.
11. Johnson DB, Pectasides E, Feld E, et al. Sequencing treatment in BRAFV600 mutant melanoma: anti-pd-1 before and after BRAF inhibition. J Immunother. 2017;40:31-35.
12. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
13. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
14. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-1260.
15. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
16. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
17. Johnson DB, Flaherty KT, Weber, JS et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32:3697-3704.
18. Schreuer M, Jansen Y, Planken S, et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAFV600-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017;18:464-472.
19. Chan MM, Haydu LE, Azer MW, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120:3142-3153.
20. Imlygic (talimogene laherparepvec) suspension for intralesional injection [package insert]. Thousand Oaks, CA: BioVex; 2015.
21. Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase ii study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36:1658-1667.
22. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2018;174:1031-1032.
23. Sampson JH, Carter Jr. JH, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
24. Yamamoto M, Serizawa T, Shuto T, et al. Stereotactic radiosurgery for patients with multiple brain metastases (JLGK0901): a multi-institutional prospective observational study. Lancet Oncol. 2014;15:387-395.
25. Tawbi HA, Forsyth PA, Hamid O, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379:722-730.
26. Long GV, Atkinson V, La S, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicenter randomised phase 2 study. Lancet Oncol. 2018;19:672-681.
27. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicenter, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087-1095.
28. Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicenter, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18:863-873.
Patients lack confidence in their skin self-exam abilities
Elderly patients with a history of localized melanoma recognized the importance of skin self-examination (SSE), but expressed low confidence in their abilities to do it correctly, based on results of a small study. While many were willing to use teledermoscopy to assist them in self-exams, patients still preferred to rely on regularly scheduled exams by physicians.
“Low confidence in SSE is a key barrier to patient-led surveillance and to the use of digital technologies for SSE,” wrote Mbathio Dieng, PhD, of the University of Sydney and her coauthors in a study published in JAMA Dermatology.
The Australian researchers conducted semistructured interviews with 37 patients from Sydney. Patients’ median age was 67 years; 26 of 37 (70%) were men.
Barriers to SSE included a perceived lack of competence in self-exams; the patients said they doubted they would catch anything that needed to be caught.
As for digitally supported SSE, many patients expressed a willingness to consider tools that would assist in the self-assessment process. Several said they valued the additional reassurance between clinical visits and the ability to create their own action plan.
The authors acknowledged that they only recruited from a single tertiary center so the results are limited. However, in terms of steps forward, they noted that their study was one of the first “to assess the perceptions of patients with melanoma of the use of new digital technologies in surveillance for recurrent or new primary melanomas.”
The study was supported by a grant from the Australian National Health and Medical Research Council (NHMRC). One author reported receiving salary support from an Australian NHMRC fellowship; another received fellowships from the NHMRC and Cancer Institute New South Wales. Another author received scholarships and awards from the University of Sydney and the Sydney Catalyst Translational Cancer Research Centre.
SOURCE: Dieng M et al. JAMA Dermatol. 2019 May 15. doi: 10.1001/jamadermatol.2019.0434.
Elderly patients with a history of localized melanoma recognized the importance of skin self-examination (SSE), but expressed low confidence in their abilities to do it correctly, based on results of a small study. While many were willing to use teledermoscopy to assist them in self-exams, patients still preferred to rely on regularly scheduled exams by physicians.
“Low confidence in SSE is a key barrier to patient-led surveillance and to the use of digital technologies for SSE,” wrote Mbathio Dieng, PhD, of the University of Sydney and her coauthors in a study published in JAMA Dermatology.
The Australian researchers conducted semistructured interviews with 37 patients from Sydney. Patients’ median age was 67 years; 26 of 37 (70%) were men.
Barriers to SSE included a perceived lack of competence in self-exams; the patients said they doubted they would catch anything that needed to be caught.
As for digitally supported SSE, many patients expressed a willingness to consider tools that would assist in the self-assessment process. Several said they valued the additional reassurance between clinical visits and the ability to create their own action plan.
The authors acknowledged that they only recruited from a single tertiary center so the results are limited. However, in terms of steps forward, they noted that their study was one of the first “to assess the perceptions of patients with melanoma of the use of new digital technologies in surveillance for recurrent or new primary melanomas.”
The study was supported by a grant from the Australian National Health and Medical Research Council (NHMRC). One author reported receiving salary support from an Australian NHMRC fellowship; another received fellowships from the NHMRC and Cancer Institute New South Wales. Another author received scholarships and awards from the University of Sydney and the Sydney Catalyst Translational Cancer Research Centre.
SOURCE: Dieng M et al. JAMA Dermatol. 2019 May 15. doi: 10.1001/jamadermatol.2019.0434.
Elderly patients with a history of localized melanoma recognized the importance of skin self-examination (SSE), but expressed low confidence in their abilities to do it correctly, based on results of a small study. While many were willing to use teledermoscopy to assist them in self-exams, patients still preferred to rely on regularly scheduled exams by physicians.
“Low confidence in SSE is a key barrier to patient-led surveillance and to the use of digital technologies for SSE,” wrote Mbathio Dieng, PhD, of the University of Sydney and her coauthors in a study published in JAMA Dermatology.
The Australian researchers conducted semistructured interviews with 37 patients from Sydney. Patients’ median age was 67 years; 26 of 37 (70%) were men.
Barriers to SSE included a perceived lack of competence in self-exams; the patients said they doubted they would catch anything that needed to be caught.
As for digitally supported SSE, many patients expressed a willingness to consider tools that would assist in the self-assessment process. Several said they valued the additional reassurance between clinical visits and the ability to create their own action plan.
The authors acknowledged that they only recruited from a single tertiary center so the results are limited. However, in terms of steps forward, they noted that their study was one of the first “to assess the perceptions of patients with melanoma of the use of new digital technologies in surveillance for recurrent or new primary melanomas.”
The study was supported by a grant from the Australian National Health and Medical Research Council (NHMRC). One author reported receiving salary support from an Australian NHMRC fellowship; another received fellowships from the NHMRC and Cancer Institute New South Wales. Another author received scholarships and awards from the University of Sydney and the Sydney Catalyst Translational Cancer Research Centre.
SOURCE: Dieng M et al. JAMA Dermatol. 2019 May 15. doi: 10.1001/jamadermatol.2019.0434.
FROM JAMA DERMATOLOGY
Advanced Melanoma: First-line Therapy
Malignant melanoma is the most serious form of primary skin cancer and one of the only malignancies in which the incidence rate has been rising. It is estimated that in 2018 there were 91,270 newly diagnosed cases and 9320 deaths from advanced melanoma in the United States. Melanoma is the fifth most common cancer type in males and the sixth most common in females. Despite rising incidence rates, improvement in the treatment of advanced melanoma has resulted in declining death rates over the past decade.1 Although most melanoma is diagnosed at an early stage and can be cured with surgical excision, the prognosis for metastatic melanoma had been historically poor prior to recent advancements in treatment. Conventional chemotherapy treatment with dacarbazine or temozolomide resulted in response rates ranging from 7.5% to 12.1%, but without much impact on median overall survival (OS), with reported OS ranging from 6.4 to 7.8 months. Combination approaches with interferon alfa-2B and low-dose interleukin-2 resulted in improved response rates compared with traditional chemotherapy, but again without survival benefit.2
Immunotherapy in the form of high-dose interleukin-2 emerged as the first therapy to alter the natural history of advanced melanoma, with both improved response rates (objective response rate [ORR], 16%) and median OS (2 months), with some patients achieving durable responses lasting more than 30 months. However, significant systemic toxicity limited its application to carefully selected patients.3 The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved ORRs, progression-free survival (PFS), and OS for patients with metastatic melanoma.4-8
This review is the first of 2 articles focusing on the treatment and sequencing of therapies in advanced melanoma. Here, we review the selection of first-line therapy for metastatic melanoma. Current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy is discussed in a separate article.
Pathogenesis
The incidence of melanoma is strongly associated with ultraviolet light–mediated DNA damage related to sun exposure. Specifically, melanoma is associated to a greater degree with intense intermittent sun exposure and sunburn, but not associated with higher occupational exposure.9 Ultraviolet radiation can induce DNA damage by a number of mechanisms, and deficient DNA repair leads to somatic mutations that drive the progression from normal melanocyte to melanoma.10
The most commonly identified genetic mutations in cutaneous melanomas are alterations in the mitogen-activated protein kinase (MAPK) pathway. Typically, an extracellular growth factor causes dimerization of the growth factor receptor, which activates the intracellular RAS GTPase protein. Subsequently BRAF is phosphorylated within the kinase domain, which leads to downstream activation of the MEK and ERK kinases through phosphorylation. Activated ERK leads to phosphorylation of various cytoplasmic and nuclear targets, and the downstream effects of these changes promote cellular proliferation. While activation of this pathway usually requires phosphorylation of BRAF by RAS, mutations placing an acidic amino acid near the kinase domain mimics phosphorylation and leads to constitutive activation of the BRAF serine/threonine kinase in the absence of upstream signaling from extracellular growth factors mediated through RAS.11 One study of tumor samples of 71 patients with cutaneous melanoma detected NRAS mutations in 30% and BRAF mutations in 59% of all tumors tested. Of the BRAF mutation–positive tumors, 88% harbored the Val599Glu mutation, now commonly referred to as the BRAF V600E mutation. The same study demonstrated that the vast majority of BRAF mutations were seen in the primary tumor and were preserved when metastases were analyzed. Additionally, both NRAS and BRAF mutations were detected in the radial growth phase of the melanoma tumor. These findings indicate that alterations in the MAPK pathway occur early in the pathogenesis of advanced melanoma.11 Another group demonstrated that 66% of malignant melanoma tumor samples harbored BRAF mutations, of which 80% were specifically the V600E mutation. In vitro assays showed that the BRAF V600E–mutated kinase had greater than 10-fold kinase activity compared to wild-type BRAF, and that this kinase enhanced cellular proliferation even when upstream NRAS signaling was inhibited.12
The Cancer Genome Atlas Network performed a large analysis of tumor samples from 331 different melanoma patients and studied variations at the DNA, RNA, and protein levels. The study established a framework of 4 notable genomic subtypes, including mutant BRAF (52%), mutant RAS (28%), mutant NF1 (14%), and triple wild-type (6%). Additionally, mRNA transcriptomic analysis of overexpressed genes identified 3 different subclasses, which were labeled as “immune,” “keratin,” and “MITF-low.” The immune subclass was characterized by increased expression of proteins found in immune cells, immune signaling molecules, immune checkpoint proteins, cytokines, and chemokines, and correlated with increased lymphocyte invasion within the tumor. Interestingly, in the post-accession survival analysis, the “immune” transcriptomic subclass was statistically correlated with an improved prognosis.13 Having an understanding of the molecular pathogenesis of advanced melanoma helps to create a framework for understanding the mechanisms of current standard of care therapies for the disease.
Case Presentation
A 62-year-old Caucasian man with a history of well-controlled type 2 diabetes mellitus and hypertension is being followed by his dermatologist for surveillance of melanocytic nevi. On follow-up he is noted to have an asymmetrical melanocytic lesion over the right scalp with irregular borders and variegated color. He is asymptomatic and the remainder of physical examination is unremarkable, as he has no other concerning skin lesions and no cervical, axillary, or inguinal lymphadenopathy.
How is melanoma diagnosed?
Detailed discussion about diagnosis and staging will be deferred in this review of treatment of advanced melanoma. In brief, melanoma is best diagnosed by excisional biopsy and histopathology. Staging of melanoma is done according to the American Joint Committee on Cancer’s (AJCC) Cancer Staging Manual, 8th edition, using a TNM staging system that incorporates tumor thickness (Breslow depth); ulceration; number of involved regional lymph nodes; presence of in-transit, satellite, and/or microsatellite metastases; distant metastases; and serum lactate dehydrogenase level.14
Case Continued
The patient undergoes a wide excisional biopsy of the right scalp lesion, which is consistent with malignant melanoma. Pathology demonstrates a Breslow depth of 2.6 mm, 2 mitotic figures/mm2, and no evidence of ulceration. He subsequently undergoes wide local excision with 0/3 sentinel lymph nodes positive for malignancy. His final staging is consistent with pT3aN0M0, stage IIA melanoma.
He is seen in follow-up with medical oncology for the next 3.5 years without any evidence of disease recurrence. He then develops symptoms of vertigo, diplopia, and recurrent falls, prompting medical attention. Magnetic resonance imaging (MRI) brain reveals multiple supratentorial and infratentorial lesions concerning for intracranial metastases. Further imaging with computed tomography (CT) chest/abdomen/pelvis reveals a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He is admitted for treatment with intravenous dexamethasone and further evaluation with endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass, which reveals metastatic melanoma. Given the extent of his intracranial metastases, he is treated with whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy.
What is the general approach to first-line treatment for metastatic melanoma?
The past decade has brought an abundance of data supporting the use of immunotherapy with immune checkpoint inhibitors or molecularly targeted therapy with combined BRAF/MEK inhibitors in the first-line setting.4-8 After the diagnosis of metastatic melanoma has been made, molecular testing is recommended to determine the BRAF status of the tumor. Immunotherapy is the clear choice for first-line therapy in the absence of an activating BRAF V600 mutation. When a BRAF V600 mutation is present, current evidence supports the use of either immunotherapy or molecularly targeted therapy as first-line therapy.
To date, there have been no prospective clinical trials comparing the sequencing of immunotherapy and molecularly targeted therapy in the first-line setting. An ongoing clinical trial (NCT02224781) is comparing dabrafenib and trametinib followed by ipilimumab and nivolumab at time of progression to ipilimumab and nivolumab followed by dabrafenib and trametinib in patients with newly diagnosed stage III/IV BRAF V600 mutation–positive melanoma. The primary outcome measure is 2-year OS. Until completion of that trial, current practice regarding which type of therapy to use in the first-line setting is based on a number of factors including clinical characteristics and provider preferences.
Data suggest that immunotherapies can produce durable responses, especially after treatment completion or discontinuation, albeit at the expense of taking a longer time to achieve clinical benefit and the risk of potentially serious immune-related adverse effects. This idea of a durable, off-treatment response is highlighted by a study that followed 105 patients who had achieved a complete response (CR) and found that 24-month disease-free survival from the time of CR was 90.9% in all patients and 89.9% in the 67 patients who had discontinued pembrolizumab after attaining CR.15 BRAF/MEK inhibition has the potential for rapid clinical responses, though concerns exist about the development of resistance to therapy. The following sections explore the evidence supporting the use of these therapies.
Immunotherapy with Immune Checkpoint Inhibitors
Immunotherapy via immune checkpoint blockade has revolutionized the treatment of many solid tumors over the past decade. The promise of immunotherapy revolves around the potential for achieving a dynamic and durable systemic response against cancer by augmenting the antitumor effects of the immune system. T-cells are central to mounting a systemic antitumor response, and, in addition to antigen recognition, their function depends heavily on fine tuning between co-stimulatory and co-inhibitory signaling. The cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) expressed on T-cells was the first discovered co-inhibitory receptor of T-cell activation.16 Later, it was discovered that the programmed cell death 1 receptor (PD-1), expressed on T-cells, and its ligands PD-L1 and PD-L2, expressed on antigen presenting cells, tumor cells, or other cells in the tumor microenvironment, also served as a potent negative regulator of T-cell function.17
Together, these 2 signaling pathways help to maintain peripheral immune tolerance, whereby autoreactive T-cells that have escaped from the thymus are silenced to prevent autoimmunity. However, these pathways can also be utilized by cancer cells to escape immune surveillance. Monoclonal antibodies that inhibit the aforementioned co-inhibitory signaling pathways, and thus augment the immune response, have proven to be an effective anticancer therapy capable of producing profound and durable responses in certain malignancies.16,17
Ipilimumab
Ipilimumab is a monoclonal antibody that inhibits the function of the CTLA-4 co-inhibitory immune checkpoint. In a phase 3 randomized controlled trial of 676 patients with previously treated metastatic melanoma, ipilimumab at a dose of 3 mg/kg every 3 weeks for 4 cycles, with or without a gp100 peptide vaccine, resulted in an improved median OS of 10.0 and 10.1 months, respectively, compared to 6.4 months in those receiving the peptide vaccine alone, meeting the primary endpoint.4 Subsequently, a phase 3 trial of 502 patients with untreated metastatic melanoma compared ipilimumab at a dose of 10 mg/kg every 3 weeks for 4 cycles plus dacarbazine to dacarbazine plus placebo and found a significant increase in median OS (11.2 months vs 9.1 months), with no additive benefit of chemotherapy. There was a higher reported rate of grade 3 or 4 adverse events in this trial with ipilimumab dosed at 10 mg/kg, which was felt to be dose-related.18 These trials were the first to show improved OS with any systemic therapy in metastatic melanoma and led to US Food and Drug Administration approval of ipilimumab for this indication in 2011.
PD-1 Inhibitor Monotherapy
The PD-1 inhibitors nivolumab and pembrolizumab were initially approved for metastatic melanoma after progression on ipilimumab. In the phase 1 trial of patients with previously treated metastatic melanoma, nivolumab therapy resulted in an ORR of 28%.19 The subsequent phase 2 trial conducted in pretreated patients, including patients who had progressed on ipilimumab, confirmed a similar ORR of 31%, as well as a median PFS of 3.7 months and a median OS of 16.8 months. The estimated response duration in patients who did achieve a response to therapy was 2 years.20 A phase 3 trial (CheckMate 037) comparing nivolumab (n = 120) to investigator’s choice chemotherapy (n = 47) in those with melanoma refractory to ipilimumab demonstrated that nivolumab was superior for the primary endpoint of ORR (31.7% vs 10.6%), had less toxicity (5% rate of grade 3 or 4 adverse events versus 9%), and increased median duration of response (32 months vs 13 months).21
The phase 1 trial (KEYNOTE-001) testing the efficacy of pembrolizumab demonstrated an ORR of 33% in the total population of patients treated and an ORR of 45% in those who were treatment-naive. Additionally, the median OS was 23 months for the total population and 31 months for treatment-naive patients, with only 14% of patients experiencing a grade 3 or 4 adverse event.22 The KEYNOTE-002 phase 2 trial compared 2 different pembrolizumab doses (2 mg/kg and 10 mg/kg every 3 weeks) to investigator’s choice chemotherapy (paclitaxel plus carboplatin, paclitaxel, carboplatin, dacarbazine, or oral temozolomide) in 540 patients with advanced melanoma with documented progression on ipilimumab with or without prior progression on molecularly targeted therapy if positive for a BRAF V600 mutation. The final analysis demonstrated significantly improved ORR with pembrolizumab (22% at 2 mg/kg vs 26% at 10 mg/kg vs 4% chemotherapy) and significantly improved 24-month PFS (16% vs 22% vs 0.6%, respectively). There was a nonstatistically significant improvement in median OS (13.4 months vs 14.7 months vs 10 months), although 55% of the patients initially assigned to the chemotherapy arm crossed over and received pembrolizumab after documentation of progressive disease.23,24
Because PD-1 inhibition improved efficacy with less toxicity than chemotherapy when studied in progressive disease, subsequent studies focused on PD-1 inhibition in the frontline setting. CheckMate 066 was a phase 3 trial comparing nivolumab to dacarbazine as first-line therapy for 418 patients with untreated metastatic melanoma who did not have a BRAF mutation. For the primary end point of 1-year OS, nivolumab was superior to dacarbazine (72.9% vs 42.1%; hazard ratio [HR], 0.42; P < 0.001). Treatment with nivolumab also resulted in superior ORR (40% vs 14%) and PFS (5.1 months vs 2.2 months). Additionally, nivolumab therapy had a lower rate of grade 3 or 4 toxicity compared to dacarbazine (11.7% vs 17.6%).25
The KEYNOTE-006 trial compared 2 separate dosing schedules of pembrolizumab (10 mg/kg every 2 weeks versus every 3 weeks) to ipilimumab (3 mg/kg every 3 weeks for 4 cycles) in a 1:1:1 ratio in 834 patients with metastatic melanoma who had received up to 1 prior systemic therapy, but no prior CTLA-4 or PD-1 inhibitors. The first published data reported statistically significant outcomes for the co-primary end points of 6-month PFS (47.3% for pembrolizumab every 2 weeks vs 46.4% for pembrolizumab every 3 weeks vs 26.5% for ipilimumab; HR, 0.58 for both pembrolizumab groups compared to ipilimumab; P < 0.001) and 12-month OS (74.1% vs 68.4% vs 58.2%) with pembrolizumab compared to ipilimumab. Compared to ipilimumab, pembrolizumab every 2 weeks had a hazard ratio of 0.63 (P = 0.0005) and pembrolizumab every 3 weeks had a hazard ratio of 0.69 (P = 0.0036). The pembrolizumab groups was also had lower rates of grade 3 to 5 toxicity (13.3% vs 10.1% vs 19.9%).5 Updated outcomes demonstrated improved ORR compared to the first analysis (37% vs 36% vs 13%), and improved OS (median OS, not reached for the pembrolizumab groups vs 16.0 months for the ipilimumab group; HR, 0.68, P = 0.0009 for pembrolizumab every 2 weeks versus HR 0.68, P = 0.0008 for pembrolizumab every 3 weeks).26 In addition, 24-month OS was 55% in both pembrolizumab groups compared to 43% in the ipilimumab group. Grade 3 or 4 toxicity occurred less frequently with pembrolizumab (17% vs 17% vs 20%).
Further analysis from the KEYNOTE-006 trial data demonstrated improved ORR, PFS, and OS with pembrolizumab compared to ipilimumab in tumors positive for PD-L1 expression. For PD-L1-negative tumors, response rate was higher, and PFS and OS rates were similar with pembrolizumab compared to ipilimumab. Given that pembrolizumab was associated with similar survival outcomes in PD-L1-negative tumors and with less toxicity than ipilimumab, the superiority of PD-L1 inhibitors over ipilimumab was further supported, regardless of tumor PD-L1 status.27
In sum, PD-1 inhibition should be considered the first-line immunotherapy in advanced melanoma, either alone or in combination with ipilimumab, as discussed in the following section. There is no longer a role for ipilimumab monotherapy in the first-line setting, based on evidence from direct comparison to single-agent PD-1 inhibition in clinical trials that demonstrated superior efficacy and less serious toxicity with PD-1 inhibitors.5,26 The finding that ORR and OS outcomes with single-agent PD-1 inhibitors are higher in treatment-naive patients compared to those receiving prior therapies also supports this approach.22
Combination CTLA-4 and PD-1 Therapy
Despite the potential for durable responses, the majority of patients fail to respond to single-agent PD-1 therapy. Given that preclinical data had suggested the potential for synergy between dual inhibition of CTLA-4 and PD-1, clinical trials were designed to test this approach. The first randomized phase 2 trial that established superior efficacy with combination therapy was the CheckMate 069 trial comparing nivolumab plus ipilimumab to ipilimumab monotherapy. Combination therapy resulted in increased ORR (59% vs 11%), median PFS (not reached vs 3.0 months), 2-year PFS (51.3% vs 12.0%), and 2-year OS (63.8% vs 53.6%).28 Similarly, a phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and 1-year OS of 89%.29
The landmark phase 3 CheckMate 067 trial analyzed efficacy outcomes for 3 different treatment regimens including nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy in previously untreated patients with unresectable stage III or IV melanoma. The trial was powered to compare survival outcomes for both the combination therapy arm against ipilimumab and the nivolumab monotherapy arm against ipilimumab, but not to compare combination therapy to nivolumab monotherapy. The initial analysis demonstrated a median PFS of 11.5 months with combination therapy versus 6.9 months with nivolumab and 2.9 months with ipilimumab, as well as an ORR of 58% versus 44% and 19%, respectively (Table 1).6 The updated 3-year survival outcomes from CheckMate 067 were notable for superior median OS with combination therapy (not reached in combination vs 37.6 months for nivolumab vs 19.9 months ipilimumab), improved 3-year OS (58% vs 52% vs 34%), and improved 3-year PFS (39% vs 32% vs 10%).7 In the reported 4-year survival outcomes, median OS was not reached in the combination therapy group, and was 36.9 months in the nivolumab monotherapy group and 19.9 months in the ipilimumab monotherapy group. Rates of grade 3 or 4 adverse events were significantly higher in the combination therapy group, at 59% compared to 22% with nivolumab monotherapy and 28% with ipilimumab alone.30 The 3- and 4-year OS outcomes (58% and 54%, respectively) with combination therapy were the highest seen in any phase 3 trial for treatment of advanced melanoma, supporting its use as the best approved first-line therapy in those who can tolerate the potential toxicity of combination therapy7,30 The conclusions from this landmark trial were that both combination therapy and nivolumab monotherapy resulted in statistically significant improvement in OS compared to ipilimumab.

Toxicity Associated with Immune Checkpoint Inhibitors
While immune checkpoint inhibitors have revolutionized the treatment of many solid tumor malignancies, this new class of cancer therapy has brought about a new type of toxicity for clinicians to be aware of, termed immune-related adverse events (irAEs). As immune checkpoint inhibitors amplify the immune response against malignancy, they also increase the likelihood that autoreactive T-cells persist and proliferate within the circulation. Therefore, these therapies can result in almost any type of autoimmune side effect. The most commonly reported irAEs in large clinical trials studying CTLA-4 and PD-1 inhibitors include rash/pruritus, diarrhea/colitis, hepatitis, endocrinopathies (thyroiditis, hypophysitis, adrenalitis), and pneumonitis. Other more rare toxicities include pancreatitis, autoimmune hematologic toxicities, cardiac toxicity (myocarditis, heart failure), and neurologic toxicities (neuropathies, myasthenia gravis-like syndrome, Guillain-Barré syndrome). It has been observed that PD-1 inhibitors have a lower incidence of irAEs than CTLA-4 inhibitors, and that the combined use of PD-1 and CTLA-4 inhibitors is associated with a greater incidence of irAEs compared to monotherapy with either agent.31 Toxicities associated with ipilimumab have been noted to be dose dependent.18 Generally, these toxicities are treated with immunosuppression in the form of glucocorticoids and are often reversible.31 There are several published guidelines that include algorithms for the management of irAEs by organizations such as the National Comprehensive Cancer Network.32
For example, previously untreated patients treated with ipilimumab plus dacarbazine as compared to dacarbazine plus placebo had greater grade 3 or 4 adverse events (56.3% vs 27.5%), and 77.7% of patients experiencing an irAE of any grade.18 In the CheckMate 066 trial comparing frontline nivolumab to dacarbazine, nivolumab had a lower rate of grade 3 or 4 toxicity (11.7% vs 17.6%) and irAEs were relatively infrequent, with diarrhea and elevated alanine aminotransferase level each being the most prominent irAE (affecting 1.0% of patients).25 In the KEYNOTE-006 trial, irAEs seen in more than 1% of patients treated with pembrolizumab included colitis, hepatitis, hypothyroidism, and hyperthyroidism, whereas those occurring in more than 1% of patients treated with ipilimumab included colitis and hypophysitis. Overall, there were lower rates of grade 3 to 5 toxicity with the 2 pembrolizumab doses compared to ipilimumab (13.3% pembrolizumab every 2 weeks vs 10.1% pembrolizumab every 3 weeks vs 19.9% ipilimumab).5 In the CheckMate 067 trial comparing nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy, rates of treatment-related adverse events of any grade were higher in the combination group (96% combination vs 86% nivolumab vs 86% ipilimumab), as were rates of grade 3 or 4 adverse events (59% vs 21% vs 28%, respectively). The irAE profile was similar to that demonstrated in prior studies: rash/pruritus were the most common, and diarrhea/colitis, elevated aminotransferases, and endocrinopathies were among the more common irAEs.7
Alternative dosing strategies have been investigated in an effort to preserve efficacy and minimize toxicity. A phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and a 1-year OS of 89%. This combination led to 45% of patients having a grade 3 or 4 adverse event, 60% having irAEs of any grade, and only 27% having grade 3 or 4 irAEs.29 The CheckMate 067 trial studied the combination of nivolumab 1 mg/kg plus ipilimumab 3 mg/kg.6 The CheckMate 511 trial compared different combination dosing strategies (nivolumab 3 mg/kg + ipilimumab 1 mg/kg versus nivolumab 1 mg/kg + ipilimumab 3 mg/kg) to assess for safety benefit. In the results published in abstract form, the reduced ipilimumab dose (nivolumab 3 mg/kg + ipilimumab 1 mg/kg arm) resulted in significantly decreased grade 3 to 5 adverse events (33.9% vs 48.3%) without significant differences in ORR, PFS, or OS.33
The question about the efficacy of checkpoint inhibitors in patients who discontinue treatment due to irAEs has been raised, as one hypothesis suggests that such toxicities may also indicate that the antitumor immune response has been activated. In a retrospective pooled analysis of phase 2 and 3 trials where patients received combination therapy with ipilimumab and nivolumab and discontinued therapy during the induction phase due to irAEs, outcomes did not appear to be inferior. Median PFS was 8.4 months in those who discontinued therapy compared to 10.8 months in those who continued therapy, but this did not reach statistical significance. Median OS had not been reached in either group and ORR was actually higher in those who discontinued due to adverse events (58.3% vs 50.2%). While this retrospective analysis needs to be validated, it does suggest that patients likely derive antitumor benefit from immunotherapy even if they have to discontinue therapy due to irAEs. Of note, patients in this analysis were not trialed on nivolumab monotherapy after receiving immunosuppressive treatment for toxicity related to combination therapy, which has since been deemed a reasonable treatment option.34
Molecularly Targeted Therapy for Metastatic Melanoma
As previously mentioned, the MAPK pathway is frequently altered in metastatic melanoma and thus serves as a target for therapy. Mutations in BRAF can cause constitutive activation of the protein’s kinase function, which subsequently phosphorylates/activates MEK in the absence of extracellular growth signals and causes increased cellular proliferation. For the roughly half of patients diagnosed with metastatic melanoma who harbor a BRAF V600 mutation, molecularly targeted therapy with BRAF/MEK inhibitors has emerged as a standard of care treatment option. As such, all patients with advanced disease should be tested for BRAF mutations.
After early phase 1 studies of the BRAF inhibitor vemurafenib demonstrated successful inhibition of mutated BRAF,35 subsequent studies confirmed the benefit of BRAF targeted therapy. In the phase 3 randomized controlled BRIM-3 trial comparing vemurafenib with dacarbazine for treatment of 675 patients with previously untreated metastatic melanoma positive for a BRAF V600E mutation, the vemurafenib group had superior ORR and 6-month OS during the first analysis.36 In a subsequent analysis, median PFS and median OS were also superior with vemurafenib compared to dacarbazine, as vemurafenib had a median OS of 13.6 months compared to 9.7 months with dacarbazine (HR, 0.70; P = 0.0008).37 Dabrafenib was the next BRAF inhibitor to demonstrate clinical efficacy with superior PFS compared to dacarbazine.38
Despite tumor shrinkage in the majority of patients, the development of resistance to therapy was an issue early on. The development of acquired resistance emerged as a heterogeneous process, though many of the identified resistance mechanisms involved reactivation of the MAPK pathway.39 A phase 3 trial of 322 patients with metastatic melanoma comparing the MEK inhibitor trametinib as monotherapy against chemotherapy demonstrated a modest improvement in both median PFS and OS.40 As a result, subsequent efforts focused on a strategy of concurrent MEK inhibition as a means to overcome resistance to molecularly targeted monotherapy
At least 4 large phase 3 randomized controlled trials of combination therapy with BRAF plus MEK inhibitors showed an improved ORR, PFS, and OS when compared to BRAF inhibition alone. The COMBI-d trial comparing dabrafenib plus trametinib versus dabrafenib alone was the first to demonstrate the superiority of combined BRAF/MEK inhibition and made combination therapy the current standard of care for patients with metastatic melanoma and a BRAF V600 mutation. In the final analysis of this trial, 3-year PFS was 22% with combination therapy compared to 12% with dabrafenib alone, and 3-year OS was 44% compared to 32%.8,41,42 A second trial with the combination of dabrafenib and trametinib (COMBI-V) also demonstrated superior efficacy when compared to single-agent vemurafenib without increased toxicity.43 Subsequently, the combination of vemurafenib with MEK inhibitor cobimetinib demonstrated superiority compared to vemurafenib alone,44 followed by the newest combination encorafenib (BRAF inhibitor) and binimetinib (MEK inhibitor) proving superior to either vemurafenib or encorafenib alone.45,46
It is important to note that there have been no studies directly comparing the efficacy of the 3 approved BRAF/MEK inhibitor combinations, but the 3 different regimens have some differences in their toxicity profiles (Table 2). Of note, single-agent BRAF inhibition was associated with increased cutaneous toxicity, including secondary squamous cell carcinoma and keratoacanthoma,47 which was demonstrated to be driven by paradoxical activation of the MAPK pathway.48 The concerning cutaneous toxicities such as squamous cell carcinoma were substantially reduced by combination BRAF/MEK inhibitor therapy.47 Collectively, the higher efficacy along with manageable toxicity profile established combination BRAF/MEK inhibition as the preferred regimen for patients with BRAF-mutated metastatic melanoma who are being considered for molecularly targeted therapy. BRAF inhibitor monotherapy should only be used when there is a specific concern regarding the use of a MEK inhibitor in certain clinical circumstances.

Other driver mutations associated with metastatic melanoma such as NRAS-mutated tumors have proven more difficult to effectively treat with molecularly targeted therapy, with one study showing that the MEK inhibitor binimetinib resulted in a modest improvement in ORR and median PFS without OS benefit compared to dacarbazine.49 Several phase 2 trials involving metastatic melanoma harboring a c-Kit alteration have demonstrated some efficacy with the tyrosine kinase inhibitor imatinib. The largest phase 2 trial of 43 patients treated with imatinib resulted in a 53.5% disease control rate (23.3% partial response and 30.2% stable disease), with 9 of the 10 patients who achieved partial response having a mutation in either exon 11 or 13. Median PFS was 3.5 months and 1-year OS was 51.0%.50
Case Conclusion
Prior to initiation of systemic therapy, the patient’s melanoma is tested and is found to be positive for a BRAF V600K mutation. At his follow-up appointment, the patient continues to endorse generalized weakness, fatigue, issues with balance, and residual pulmonary symptoms after being treated for post-obstructive pneumonia. Given his current symptoms and extent of metastatic disease, immunotherapy is deferred and he is started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially does well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass. Further follow-up of this patient is presented in the second article in this 2-part review of advanced melanoma.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin. 2018;68:7-30.
2. Ives NJ, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2621 patients. J Clin Oncol. 2007;25:5426-34.
3. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105-16.
4. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23.
5. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
6. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
7. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345-1356.
8. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
9. Elwood JM, Jopson J. Melanoma and sun exposure: an overview of published studies. Int J Cancer. 1997;73:198-203.
10. Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 199;340:1341-1348.
11. Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483-8.
12. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54.
13. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681-96.
14. Gershenwald JE, Scolyer RA, Hess KR, et al. Melanoma staging: evidence-based changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer J Clin. 2017;67:472-492.
15. Robert C, Ribas A, Hamid O, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol. 2018;36:1668-1674.
16. Salama AKS, Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clin Cancer Res. 2011;17:4622-8.
17. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767-1778.
18. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
19. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
20. Topalian S, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020-30.
21. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-84.
22. Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315:1600-1609.
23. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-18.
24. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
25. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
26. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicenter, randomised, open-label phase 3 study (KEYNOTE-006). Lancet Oncol. 2017;390:1853-1862.
27. Carlino MS, Long GV, Schadendorf D, et al. Outcomes by line of therapy and programmed death ligand 1 expression in patients with advanced melanoma treated with pembrolizumab or ipilimumab in KEYNOTE-006. A randomised clinical trial. Eur J Cancer. 2018;101:236-243.
28. Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558-1568.
29. Long GV, Atkinson V, Cebon JS, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 2017;18:1202-10.
30. Hodi FS, Chiarion-Sileni V, Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480-1492.
31. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. 2016;2:1346-1353.
32. National Comprehensive Cancer Network. Management of immunotherapy-related toxicities (version 2.2019). www.nccn.org/professionals/physician_gls/pdf/immunotherapy.pdf. Accessed April 8, 2019.
33. Lebbé C, Meyer N, Mortier L, et al. Initial results from a phase IIIb/IV study evaluating two dosing regimens of nivolumab (NIVO) in combination with ipilimumab (IPI) in patients with advanced melanoma (CheckMate 511) [Abstract LBA47]. Ann Oncol. 2018;29:mdy424.057.
34. Schadendorf D, Wolchok JD, Hodi FS, et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase ii and iii trials. J Clin Oncol. 2017;35:3807-3814.
35. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
36. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
37. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
38. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicenter, open-label, phase 3 randomised controlled trial. Lancet Oncol. 2012;380:358-365.
39. Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965-1977.
40. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107-114.
41. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
42. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
43. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
44. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-260.
45. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
46. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
47. Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA. Dermatol 2015;151:1103-1109.
48. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
49. Dummer R, Schadendorf D, Ascierto P, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18:435-445.
50. Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904-2909.
Malignant melanoma is the most serious form of primary skin cancer and one of the only malignancies in which the incidence rate has been rising. It is estimated that in 2018 there were 91,270 newly diagnosed cases and 9320 deaths from advanced melanoma in the United States. Melanoma is the fifth most common cancer type in males and the sixth most common in females. Despite rising incidence rates, improvement in the treatment of advanced melanoma has resulted in declining death rates over the past decade.1 Although most melanoma is diagnosed at an early stage and can be cured with surgical excision, the prognosis for metastatic melanoma had been historically poor prior to recent advancements in treatment. Conventional chemotherapy treatment with dacarbazine or temozolomide resulted in response rates ranging from 7.5% to 12.1%, but without much impact on median overall survival (OS), with reported OS ranging from 6.4 to 7.8 months. Combination approaches with interferon alfa-2B and low-dose interleukin-2 resulted in improved response rates compared with traditional chemotherapy, but again without survival benefit.2
Immunotherapy in the form of high-dose interleukin-2 emerged as the first therapy to alter the natural history of advanced melanoma, with both improved response rates (objective response rate [ORR], 16%) and median OS (2 months), with some patients achieving durable responses lasting more than 30 months. However, significant systemic toxicity limited its application to carefully selected patients.3 The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved ORRs, progression-free survival (PFS), and OS for patients with metastatic melanoma.4-8
This review is the first of 2 articles focusing on the treatment and sequencing of therapies in advanced melanoma. Here, we review the selection of first-line therapy for metastatic melanoma. Current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy is discussed in a separate article.
Pathogenesis
The incidence of melanoma is strongly associated with ultraviolet light–mediated DNA damage related to sun exposure. Specifically, melanoma is associated to a greater degree with intense intermittent sun exposure and sunburn, but not associated with higher occupational exposure.9 Ultraviolet radiation can induce DNA damage by a number of mechanisms, and deficient DNA repair leads to somatic mutations that drive the progression from normal melanocyte to melanoma.10
The most commonly identified genetic mutations in cutaneous melanomas are alterations in the mitogen-activated protein kinase (MAPK) pathway. Typically, an extracellular growth factor causes dimerization of the growth factor receptor, which activates the intracellular RAS GTPase protein. Subsequently BRAF is phosphorylated within the kinase domain, which leads to downstream activation of the MEK and ERK kinases through phosphorylation. Activated ERK leads to phosphorylation of various cytoplasmic and nuclear targets, and the downstream effects of these changes promote cellular proliferation. While activation of this pathway usually requires phosphorylation of BRAF by RAS, mutations placing an acidic amino acid near the kinase domain mimics phosphorylation and leads to constitutive activation of the BRAF serine/threonine kinase in the absence of upstream signaling from extracellular growth factors mediated through RAS.11 One study of tumor samples of 71 patients with cutaneous melanoma detected NRAS mutations in 30% and BRAF mutations in 59% of all tumors tested. Of the BRAF mutation–positive tumors, 88% harbored the Val599Glu mutation, now commonly referred to as the BRAF V600E mutation. The same study demonstrated that the vast majority of BRAF mutations were seen in the primary tumor and were preserved when metastases were analyzed. Additionally, both NRAS and BRAF mutations were detected in the radial growth phase of the melanoma tumor. These findings indicate that alterations in the MAPK pathway occur early in the pathogenesis of advanced melanoma.11 Another group demonstrated that 66% of malignant melanoma tumor samples harbored BRAF mutations, of which 80% were specifically the V600E mutation. In vitro assays showed that the BRAF V600E–mutated kinase had greater than 10-fold kinase activity compared to wild-type BRAF, and that this kinase enhanced cellular proliferation even when upstream NRAS signaling was inhibited.12
The Cancer Genome Atlas Network performed a large analysis of tumor samples from 331 different melanoma patients and studied variations at the DNA, RNA, and protein levels. The study established a framework of 4 notable genomic subtypes, including mutant BRAF (52%), mutant RAS (28%), mutant NF1 (14%), and triple wild-type (6%). Additionally, mRNA transcriptomic analysis of overexpressed genes identified 3 different subclasses, which were labeled as “immune,” “keratin,” and “MITF-low.” The immune subclass was characterized by increased expression of proteins found in immune cells, immune signaling molecules, immune checkpoint proteins, cytokines, and chemokines, and correlated with increased lymphocyte invasion within the tumor. Interestingly, in the post-accession survival analysis, the “immune” transcriptomic subclass was statistically correlated with an improved prognosis.13 Having an understanding of the molecular pathogenesis of advanced melanoma helps to create a framework for understanding the mechanisms of current standard of care therapies for the disease.
Case Presentation
A 62-year-old Caucasian man with a history of well-controlled type 2 diabetes mellitus and hypertension is being followed by his dermatologist for surveillance of melanocytic nevi. On follow-up he is noted to have an asymmetrical melanocytic lesion over the right scalp with irregular borders and variegated color. He is asymptomatic and the remainder of physical examination is unremarkable, as he has no other concerning skin lesions and no cervical, axillary, or inguinal lymphadenopathy.
How is melanoma diagnosed?
Detailed discussion about diagnosis and staging will be deferred in this review of treatment of advanced melanoma. In brief, melanoma is best diagnosed by excisional biopsy and histopathology. Staging of melanoma is done according to the American Joint Committee on Cancer’s (AJCC) Cancer Staging Manual, 8th edition, using a TNM staging system that incorporates tumor thickness (Breslow depth); ulceration; number of involved regional lymph nodes; presence of in-transit, satellite, and/or microsatellite metastases; distant metastases; and serum lactate dehydrogenase level.14
Case Continued
The patient undergoes a wide excisional biopsy of the right scalp lesion, which is consistent with malignant melanoma. Pathology demonstrates a Breslow depth of 2.6 mm, 2 mitotic figures/mm2, and no evidence of ulceration. He subsequently undergoes wide local excision with 0/3 sentinel lymph nodes positive for malignancy. His final staging is consistent with pT3aN0M0, stage IIA melanoma.
He is seen in follow-up with medical oncology for the next 3.5 years without any evidence of disease recurrence. He then develops symptoms of vertigo, diplopia, and recurrent falls, prompting medical attention. Magnetic resonance imaging (MRI) brain reveals multiple supratentorial and infratentorial lesions concerning for intracranial metastases. Further imaging with computed tomography (CT) chest/abdomen/pelvis reveals a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He is admitted for treatment with intravenous dexamethasone and further evaluation with endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass, which reveals metastatic melanoma. Given the extent of his intracranial metastases, he is treated with whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy.
What is the general approach to first-line treatment for metastatic melanoma?
The past decade has brought an abundance of data supporting the use of immunotherapy with immune checkpoint inhibitors or molecularly targeted therapy with combined BRAF/MEK inhibitors in the first-line setting.4-8 After the diagnosis of metastatic melanoma has been made, molecular testing is recommended to determine the BRAF status of the tumor. Immunotherapy is the clear choice for first-line therapy in the absence of an activating BRAF V600 mutation. When a BRAF V600 mutation is present, current evidence supports the use of either immunotherapy or molecularly targeted therapy as first-line therapy.
To date, there have been no prospective clinical trials comparing the sequencing of immunotherapy and molecularly targeted therapy in the first-line setting. An ongoing clinical trial (NCT02224781) is comparing dabrafenib and trametinib followed by ipilimumab and nivolumab at time of progression to ipilimumab and nivolumab followed by dabrafenib and trametinib in patients with newly diagnosed stage III/IV BRAF V600 mutation–positive melanoma. The primary outcome measure is 2-year OS. Until completion of that trial, current practice regarding which type of therapy to use in the first-line setting is based on a number of factors including clinical characteristics and provider preferences.
Data suggest that immunotherapies can produce durable responses, especially after treatment completion or discontinuation, albeit at the expense of taking a longer time to achieve clinical benefit and the risk of potentially serious immune-related adverse effects. This idea of a durable, off-treatment response is highlighted by a study that followed 105 patients who had achieved a complete response (CR) and found that 24-month disease-free survival from the time of CR was 90.9% in all patients and 89.9% in the 67 patients who had discontinued pembrolizumab after attaining CR.15 BRAF/MEK inhibition has the potential for rapid clinical responses, though concerns exist about the development of resistance to therapy. The following sections explore the evidence supporting the use of these therapies.
Immunotherapy with Immune Checkpoint Inhibitors
Immunotherapy via immune checkpoint blockade has revolutionized the treatment of many solid tumors over the past decade. The promise of immunotherapy revolves around the potential for achieving a dynamic and durable systemic response against cancer by augmenting the antitumor effects of the immune system. T-cells are central to mounting a systemic antitumor response, and, in addition to antigen recognition, their function depends heavily on fine tuning between co-stimulatory and co-inhibitory signaling. The cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) expressed on T-cells was the first discovered co-inhibitory receptor of T-cell activation.16 Later, it was discovered that the programmed cell death 1 receptor (PD-1), expressed on T-cells, and its ligands PD-L1 and PD-L2, expressed on antigen presenting cells, tumor cells, or other cells in the tumor microenvironment, also served as a potent negative regulator of T-cell function.17
Together, these 2 signaling pathways help to maintain peripheral immune tolerance, whereby autoreactive T-cells that have escaped from the thymus are silenced to prevent autoimmunity. However, these pathways can also be utilized by cancer cells to escape immune surveillance. Monoclonal antibodies that inhibit the aforementioned co-inhibitory signaling pathways, and thus augment the immune response, have proven to be an effective anticancer therapy capable of producing profound and durable responses in certain malignancies.16,17
Ipilimumab
Ipilimumab is a monoclonal antibody that inhibits the function of the CTLA-4 co-inhibitory immune checkpoint. In a phase 3 randomized controlled trial of 676 patients with previously treated metastatic melanoma, ipilimumab at a dose of 3 mg/kg every 3 weeks for 4 cycles, with or without a gp100 peptide vaccine, resulted in an improved median OS of 10.0 and 10.1 months, respectively, compared to 6.4 months in those receiving the peptide vaccine alone, meeting the primary endpoint.4 Subsequently, a phase 3 trial of 502 patients with untreated metastatic melanoma compared ipilimumab at a dose of 10 mg/kg every 3 weeks for 4 cycles plus dacarbazine to dacarbazine plus placebo and found a significant increase in median OS (11.2 months vs 9.1 months), with no additive benefit of chemotherapy. There was a higher reported rate of grade 3 or 4 adverse events in this trial with ipilimumab dosed at 10 mg/kg, which was felt to be dose-related.18 These trials were the first to show improved OS with any systemic therapy in metastatic melanoma and led to US Food and Drug Administration approval of ipilimumab for this indication in 2011.
PD-1 Inhibitor Monotherapy
The PD-1 inhibitors nivolumab and pembrolizumab were initially approved for metastatic melanoma after progression on ipilimumab. In the phase 1 trial of patients with previously treated metastatic melanoma, nivolumab therapy resulted in an ORR of 28%.19 The subsequent phase 2 trial conducted in pretreated patients, including patients who had progressed on ipilimumab, confirmed a similar ORR of 31%, as well as a median PFS of 3.7 months and a median OS of 16.8 months. The estimated response duration in patients who did achieve a response to therapy was 2 years.20 A phase 3 trial (CheckMate 037) comparing nivolumab (n = 120) to investigator’s choice chemotherapy (n = 47) in those with melanoma refractory to ipilimumab demonstrated that nivolumab was superior for the primary endpoint of ORR (31.7% vs 10.6%), had less toxicity (5% rate of grade 3 or 4 adverse events versus 9%), and increased median duration of response (32 months vs 13 months).21
The phase 1 trial (KEYNOTE-001) testing the efficacy of pembrolizumab demonstrated an ORR of 33% in the total population of patients treated and an ORR of 45% in those who were treatment-naive. Additionally, the median OS was 23 months for the total population and 31 months for treatment-naive patients, with only 14% of patients experiencing a grade 3 or 4 adverse event.22 The KEYNOTE-002 phase 2 trial compared 2 different pembrolizumab doses (2 mg/kg and 10 mg/kg every 3 weeks) to investigator’s choice chemotherapy (paclitaxel plus carboplatin, paclitaxel, carboplatin, dacarbazine, or oral temozolomide) in 540 patients with advanced melanoma with documented progression on ipilimumab with or without prior progression on molecularly targeted therapy if positive for a BRAF V600 mutation. The final analysis demonstrated significantly improved ORR with pembrolizumab (22% at 2 mg/kg vs 26% at 10 mg/kg vs 4% chemotherapy) and significantly improved 24-month PFS (16% vs 22% vs 0.6%, respectively). There was a nonstatistically significant improvement in median OS (13.4 months vs 14.7 months vs 10 months), although 55% of the patients initially assigned to the chemotherapy arm crossed over and received pembrolizumab after documentation of progressive disease.23,24
Because PD-1 inhibition improved efficacy with less toxicity than chemotherapy when studied in progressive disease, subsequent studies focused on PD-1 inhibition in the frontline setting. CheckMate 066 was a phase 3 trial comparing nivolumab to dacarbazine as first-line therapy for 418 patients with untreated metastatic melanoma who did not have a BRAF mutation. For the primary end point of 1-year OS, nivolumab was superior to dacarbazine (72.9% vs 42.1%; hazard ratio [HR], 0.42; P < 0.001). Treatment with nivolumab also resulted in superior ORR (40% vs 14%) and PFS (5.1 months vs 2.2 months). Additionally, nivolumab therapy had a lower rate of grade 3 or 4 toxicity compared to dacarbazine (11.7% vs 17.6%).25
The KEYNOTE-006 trial compared 2 separate dosing schedules of pembrolizumab (10 mg/kg every 2 weeks versus every 3 weeks) to ipilimumab (3 mg/kg every 3 weeks for 4 cycles) in a 1:1:1 ratio in 834 patients with metastatic melanoma who had received up to 1 prior systemic therapy, but no prior CTLA-4 or PD-1 inhibitors. The first published data reported statistically significant outcomes for the co-primary end points of 6-month PFS (47.3% for pembrolizumab every 2 weeks vs 46.4% for pembrolizumab every 3 weeks vs 26.5% for ipilimumab; HR, 0.58 for both pembrolizumab groups compared to ipilimumab; P < 0.001) and 12-month OS (74.1% vs 68.4% vs 58.2%) with pembrolizumab compared to ipilimumab. Compared to ipilimumab, pembrolizumab every 2 weeks had a hazard ratio of 0.63 (P = 0.0005) and pembrolizumab every 3 weeks had a hazard ratio of 0.69 (P = 0.0036). The pembrolizumab groups was also had lower rates of grade 3 to 5 toxicity (13.3% vs 10.1% vs 19.9%).5 Updated outcomes demonstrated improved ORR compared to the first analysis (37% vs 36% vs 13%), and improved OS (median OS, not reached for the pembrolizumab groups vs 16.0 months for the ipilimumab group; HR, 0.68, P = 0.0009 for pembrolizumab every 2 weeks versus HR 0.68, P = 0.0008 for pembrolizumab every 3 weeks).26 In addition, 24-month OS was 55% in both pembrolizumab groups compared to 43% in the ipilimumab group. Grade 3 or 4 toxicity occurred less frequently with pembrolizumab (17% vs 17% vs 20%).
Further analysis from the KEYNOTE-006 trial data demonstrated improved ORR, PFS, and OS with pembrolizumab compared to ipilimumab in tumors positive for PD-L1 expression. For PD-L1-negative tumors, response rate was higher, and PFS and OS rates were similar with pembrolizumab compared to ipilimumab. Given that pembrolizumab was associated with similar survival outcomes in PD-L1-negative tumors and with less toxicity than ipilimumab, the superiority of PD-L1 inhibitors over ipilimumab was further supported, regardless of tumor PD-L1 status.27
In sum, PD-1 inhibition should be considered the first-line immunotherapy in advanced melanoma, either alone or in combination with ipilimumab, as discussed in the following section. There is no longer a role for ipilimumab monotherapy in the first-line setting, based on evidence from direct comparison to single-agent PD-1 inhibition in clinical trials that demonstrated superior efficacy and less serious toxicity with PD-1 inhibitors.5,26 The finding that ORR and OS outcomes with single-agent PD-1 inhibitors are higher in treatment-naive patients compared to those receiving prior therapies also supports this approach.22
Combination CTLA-4 and PD-1 Therapy
Despite the potential for durable responses, the majority of patients fail to respond to single-agent PD-1 therapy. Given that preclinical data had suggested the potential for synergy between dual inhibition of CTLA-4 and PD-1, clinical trials were designed to test this approach. The first randomized phase 2 trial that established superior efficacy with combination therapy was the CheckMate 069 trial comparing nivolumab plus ipilimumab to ipilimumab monotherapy. Combination therapy resulted in increased ORR (59% vs 11%), median PFS (not reached vs 3.0 months), 2-year PFS (51.3% vs 12.0%), and 2-year OS (63.8% vs 53.6%).28 Similarly, a phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and 1-year OS of 89%.29
The landmark phase 3 CheckMate 067 trial analyzed efficacy outcomes for 3 different treatment regimens including nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy in previously untreated patients with unresectable stage III or IV melanoma. The trial was powered to compare survival outcomes for both the combination therapy arm against ipilimumab and the nivolumab monotherapy arm against ipilimumab, but not to compare combination therapy to nivolumab monotherapy. The initial analysis demonstrated a median PFS of 11.5 months with combination therapy versus 6.9 months with nivolumab and 2.9 months with ipilimumab, as well as an ORR of 58% versus 44% and 19%, respectively (Table 1).6 The updated 3-year survival outcomes from CheckMate 067 were notable for superior median OS with combination therapy (not reached in combination vs 37.6 months for nivolumab vs 19.9 months ipilimumab), improved 3-year OS (58% vs 52% vs 34%), and improved 3-year PFS (39% vs 32% vs 10%).7 In the reported 4-year survival outcomes, median OS was not reached in the combination therapy group, and was 36.9 months in the nivolumab monotherapy group and 19.9 months in the ipilimumab monotherapy group. Rates of grade 3 or 4 adverse events were significantly higher in the combination therapy group, at 59% compared to 22% with nivolumab monotherapy and 28% with ipilimumab alone.30 The 3- and 4-year OS outcomes (58% and 54%, respectively) with combination therapy were the highest seen in any phase 3 trial for treatment of advanced melanoma, supporting its use as the best approved first-line therapy in those who can tolerate the potential toxicity of combination therapy7,30 The conclusions from this landmark trial were that both combination therapy and nivolumab monotherapy resulted in statistically significant improvement in OS compared to ipilimumab.
Toxicity Associated with Immune Checkpoint Inhibitors
While immune checkpoint inhibitors have revolutionized the treatment of many solid tumor malignancies, this new class of cancer therapy has brought about a new type of toxicity for clinicians to be aware of, termed immune-related adverse events (irAEs). As immune checkpoint inhibitors amplify the immune response against malignancy, they also increase the likelihood that autoreactive T-cells persist and proliferate within the circulation. Therefore, these therapies can result in almost any type of autoimmune side effect. The most commonly reported irAEs in large clinical trials studying CTLA-4 and PD-1 inhibitors include rash/pruritus, diarrhea/colitis, hepatitis, endocrinopathies (thyroiditis, hypophysitis, adrenalitis), and pneumonitis. Other more rare toxicities include pancreatitis, autoimmune hematologic toxicities, cardiac toxicity (myocarditis, heart failure), and neurologic toxicities (neuropathies, myasthenia gravis-like syndrome, Guillain-Barré syndrome). It has been observed that PD-1 inhibitors have a lower incidence of irAEs than CTLA-4 inhibitors, and that the combined use of PD-1 and CTLA-4 inhibitors is associated with a greater incidence of irAEs compared to monotherapy with either agent.31 Toxicities associated with ipilimumab have been noted to be dose dependent.18 Generally, these toxicities are treated with immunosuppression in the form of glucocorticoids and are often reversible.31 There are several published guidelines that include algorithms for the management of irAEs by organizations such as the National Comprehensive Cancer Network.32
For example, previously untreated patients treated with ipilimumab plus dacarbazine as compared to dacarbazine plus placebo had greater grade 3 or 4 adverse events (56.3% vs 27.5%), and 77.7% of patients experiencing an irAE of any grade.18 In the CheckMate 066 trial comparing frontline nivolumab to dacarbazine, nivolumab had a lower rate of grade 3 or 4 toxicity (11.7% vs 17.6%) and irAEs were relatively infrequent, with diarrhea and elevated alanine aminotransferase level each being the most prominent irAE (affecting 1.0% of patients).25 In the KEYNOTE-006 trial, irAEs seen in more than 1% of patients treated with pembrolizumab included colitis, hepatitis, hypothyroidism, and hyperthyroidism, whereas those occurring in more than 1% of patients treated with ipilimumab included colitis and hypophysitis. Overall, there were lower rates of grade 3 to 5 toxicity with the 2 pembrolizumab doses compared to ipilimumab (13.3% pembrolizumab every 2 weeks vs 10.1% pembrolizumab every 3 weeks vs 19.9% ipilimumab).5 In the CheckMate 067 trial comparing nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy, rates of treatment-related adverse events of any grade were higher in the combination group (96% combination vs 86% nivolumab vs 86% ipilimumab), as were rates of grade 3 or 4 adverse events (59% vs 21% vs 28%, respectively). The irAE profile was similar to that demonstrated in prior studies: rash/pruritus were the most common, and diarrhea/colitis, elevated aminotransferases, and endocrinopathies were among the more common irAEs.7
Alternative dosing strategies have been investigated in an effort to preserve efficacy and minimize toxicity. A phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and a 1-year OS of 89%. This combination led to 45% of patients having a grade 3 or 4 adverse event, 60% having irAEs of any grade, and only 27% having grade 3 or 4 irAEs.29 The CheckMate 067 trial studied the combination of nivolumab 1 mg/kg plus ipilimumab 3 mg/kg.6 The CheckMate 511 trial compared different combination dosing strategies (nivolumab 3 mg/kg + ipilimumab 1 mg/kg versus nivolumab 1 mg/kg + ipilimumab 3 mg/kg) to assess for safety benefit. In the results published in abstract form, the reduced ipilimumab dose (nivolumab 3 mg/kg + ipilimumab 1 mg/kg arm) resulted in significantly decreased grade 3 to 5 adverse events (33.9% vs 48.3%) without significant differences in ORR, PFS, or OS.33
The question about the efficacy of checkpoint inhibitors in patients who discontinue treatment due to irAEs has been raised, as one hypothesis suggests that such toxicities may also indicate that the antitumor immune response has been activated. In a retrospective pooled analysis of phase 2 and 3 trials where patients received combination therapy with ipilimumab and nivolumab and discontinued therapy during the induction phase due to irAEs, outcomes did not appear to be inferior. Median PFS was 8.4 months in those who discontinued therapy compared to 10.8 months in those who continued therapy, but this did not reach statistical significance. Median OS had not been reached in either group and ORR was actually higher in those who discontinued due to adverse events (58.3% vs 50.2%). While this retrospective analysis needs to be validated, it does suggest that patients likely derive antitumor benefit from immunotherapy even if they have to discontinue therapy due to irAEs. Of note, patients in this analysis were not trialed on nivolumab monotherapy after receiving immunosuppressive treatment for toxicity related to combination therapy, which has since been deemed a reasonable treatment option.34
Molecularly Targeted Therapy for Metastatic Melanoma
As previously mentioned, the MAPK pathway is frequently altered in metastatic melanoma and thus serves as a target for therapy. Mutations in BRAF can cause constitutive activation of the protein’s kinase function, which subsequently phosphorylates/activates MEK in the absence of extracellular growth signals and causes increased cellular proliferation. For the roughly half of patients diagnosed with metastatic melanoma who harbor a BRAF V600 mutation, molecularly targeted therapy with BRAF/MEK inhibitors has emerged as a standard of care treatment option. As such, all patients with advanced disease should be tested for BRAF mutations.
After early phase 1 studies of the BRAF inhibitor vemurafenib demonstrated successful inhibition of mutated BRAF,35 subsequent studies confirmed the benefit of BRAF targeted therapy. In the phase 3 randomized controlled BRIM-3 trial comparing vemurafenib with dacarbazine for treatment of 675 patients with previously untreated metastatic melanoma positive for a BRAF V600E mutation, the vemurafenib group had superior ORR and 6-month OS during the first analysis.36 In a subsequent analysis, median PFS and median OS were also superior with vemurafenib compared to dacarbazine, as vemurafenib had a median OS of 13.6 months compared to 9.7 months with dacarbazine (HR, 0.70; P = 0.0008).37 Dabrafenib was the next BRAF inhibitor to demonstrate clinical efficacy with superior PFS compared to dacarbazine.38
Despite tumor shrinkage in the majority of patients, the development of resistance to therapy was an issue early on. The development of acquired resistance emerged as a heterogeneous process, though many of the identified resistance mechanisms involved reactivation of the MAPK pathway.39 A phase 3 trial of 322 patients with metastatic melanoma comparing the MEK inhibitor trametinib as monotherapy against chemotherapy demonstrated a modest improvement in both median PFS and OS.40 As a result, subsequent efforts focused on a strategy of concurrent MEK inhibition as a means to overcome resistance to molecularly targeted monotherapy
At least 4 large phase 3 randomized controlled trials of combination therapy with BRAF plus MEK inhibitors showed an improved ORR, PFS, and OS when compared to BRAF inhibition alone. The COMBI-d trial comparing dabrafenib plus trametinib versus dabrafenib alone was the first to demonstrate the superiority of combined BRAF/MEK inhibition and made combination therapy the current standard of care for patients with metastatic melanoma and a BRAF V600 mutation. In the final analysis of this trial, 3-year PFS was 22% with combination therapy compared to 12% with dabrafenib alone, and 3-year OS was 44% compared to 32%.8,41,42 A second trial with the combination of dabrafenib and trametinib (COMBI-V) also demonstrated superior efficacy when compared to single-agent vemurafenib without increased toxicity.43 Subsequently, the combination of vemurafenib with MEK inhibitor cobimetinib demonstrated superiority compared to vemurafenib alone,44 followed by the newest combination encorafenib (BRAF inhibitor) and binimetinib (MEK inhibitor) proving superior to either vemurafenib or encorafenib alone.45,46
It is important to note that there have been no studies directly comparing the efficacy of the 3 approved BRAF/MEK inhibitor combinations, but the 3 different regimens have some differences in their toxicity profiles (Table 2). Of note, single-agent BRAF inhibition was associated with increased cutaneous toxicity, including secondary squamous cell carcinoma and keratoacanthoma,47 which was demonstrated to be driven by paradoxical activation of the MAPK pathway.48 The concerning cutaneous toxicities such as squamous cell carcinoma were substantially reduced by combination BRAF/MEK inhibitor therapy.47 Collectively, the higher efficacy along with manageable toxicity profile established combination BRAF/MEK inhibition as the preferred regimen for patients with BRAF-mutated metastatic melanoma who are being considered for molecularly targeted therapy. BRAF inhibitor monotherapy should only be used when there is a specific concern regarding the use of a MEK inhibitor in certain clinical circumstances.
Other driver mutations associated with metastatic melanoma such as NRAS-mutated tumors have proven more difficult to effectively treat with molecularly targeted therapy, with one study showing that the MEK inhibitor binimetinib resulted in a modest improvement in ORR and median PFS without OS benefit compared to dacarbazine.49 Several phase 2 trials involving metastatic melanoma harboring a c-Kit alteration have demonstrated some efficacy with the tyrosine kinase inhibitor imatinib. The largest phase 2 trial of 43 patients treated with imatinib resulted in a 53.5% disease control rate (23.3% partial response and 30.2% stable disease), with 9 of the 10 patients who achieved partial response having a mutation in either exon 11 or 13. Median PFS was 3.5 months and 1-year OS was 51.0%.50
Case Conclusion
Prior to initiation of systemic therapy, the patient’s melanoma is tested and is found to be positive for a BRAF V600K mutation. At his follow-up appointment, the patient continues to endorse generalized weakness, fatigue, issues with balance, and residual pulmonary symptoms after being treated for post-obstructive pneumonia. Given his current symptoms and extent of metastatic disease, immunotherapy is deferred and he is started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially does well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass. Further follow-up of this patient is presented in the second article in this 2-part review of advanced melanoma.
Malignant melanoma is the most serious form of primary skin cancer and one of the only malignancies in which the incidence rate has been rising. It is estimated that in 2018 there were 91,270 newly diagnosed cases and 9320 deaths from advanced melanoma in the United States. Melanoma is the fifth most common cancer type in males and the sixth most common in females. Despite rising incidence rates, improvement in the treatment of advanced melanoma has resulted in declining death rates over the past decade.1 Although most melanoma is diagnosed at an early stage and can be cured with surgical excision, the prognosis for metastatic melanoma had been historically poor prior to recent advancements in treatment. Conventional chemotherapy treatment with dacarbazine or temozolomide resulted in response rates ranging from 7.5% to 12.1%, but without much impact on median overall survival (OS), with reported OS ranging from 6.4 to 7.8 months. Combination approaches with interferon alfa-2B and low-dose interleukin-2 resulted in improved response rates compared with traditional chemotherapy, but again without survival benefit.2
Immunotherapy in the form of high-dose interleukin-2 emerged as the first therapy to alter the natural history of advanced melanoma, with both improved response rates (objective response rate [ORR], 16%) and median OS (2 months), with some patients achieving durable responses lasting more than 30 months. However, significant systemic toxicity limited its application to carefully selected patients.3 The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved ORRs, progression-free survival (PFS), and OS for patients with metastatic melanoma.4-8
This review is the first of 2 articles focusing on the treatment and sequencing of therapies in advanced melanoma. Here, we review the selection of first-line therapy for metastatic melanoma. Current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy is discussed in a separate article.
Pathogenesis
The incidence of melanoma is strongly associated with ultraviolet light–mediated DNA damage related to sun exposure. Specifically, melanoma is associated to a greater degree with intense intermittent sun exposure and sunburn, but not associated with higher occupational exposure.9 Ultraviolet radiation can induce DNA damage by a number of mechanisms, and deficient DNA repair leads to somatic mutations that drive the progression from normal melanocyte to melanoma.10
The most commonly identified genetic mutations in cutaneous melanomas are alterations in the mitogen-activated protein kinase (MAPK) pathway. Typically, an extracellular growth factor causes dimerization of the growth factor receptor, which activates the intracellular RAS GTPase protein. Subsequently BRAF is phosphorylated within the kinase domain, which leads to downstream activation of the MEK and ERK kinases through phosphorylation. Activated ERK leads to phosphorylation of various cytoplasmic and nuclear targets, and the downstream effects of these changes promote cellular proliferation. While activation of this pathway usually requires phosphorylation of BRAF by RAS, mutations placing an acidic amino acid near the kinase domain mimics phosphorylation and leads to constitutive activation of the BRAF serine/threonine kinase in the absence of upstream signaling from extracellular growth factors mediated through RAS.11 One study of tumor samples of 71 patients with cutaneous melanoma detected NRAS mutations in 30% and BRAF mutations in 59% of all tumors tested. Of the BRAF mutation–positive tumors, 88% harbored the Val599Glu mutation, now commonly referred to as the BRAF V600E mutation. The same study demonstrated that the vast majority of BRAF mutations were seen in the primary tumor and were preserved when metastases were analyzed. Additionally, both NRAS and BRAF mutations were detected in the radial growth phase of the melanoma tumor. These findings indicate that alterations in the MAPK pathway occur early in the pathogenesis of advanced melanoma.11 Another group demonstrated that 66% of malignant melanoma tumor samples harbored BRAF mutations, of which 80% were specifically the V600E mutation. In vitro assays showed that the BRAF V600E–mutated kinase had greater than 10-fold kinase activity compared to wild-type BRAF, and that this kinase enhanced cellular proliferation even when upstream NRAS signaling was inhibited.12
The Cancer Genome Atlas Network performed a large analysis of tumor samples from 331 different melanoma patients and studied variations at the DNA, RNA, and protein levels. The study established a framework of 4 notable genomic subtypes, including mutant BRAF (52%), mutant RAS (28%), mutant NF1 (14%), and triple wild-type (6%). Additionally, mRNA transcriptomic analysis of overexpressed genes identified 3 different subclasses, which were labeled as “immune,” “keratin,” and “MITF-low.” The immune subclass was characterized by increased expression of proteins found in immune cells, immune signaling molecules, immune checkpoint proteins, cytokines, and chemokines, and correlated with increased lymphocyte invasion within the tumor. Interestingly, in the post-accession survival analysis, the “immune” transcriptomic subclass was statistically correlated with an improved prognosis.13 Having an understanding of the molecular pathogenesis of advanced melanoma helps to create a framework for understanding the mechanisms of current standard of care therapies for the disease.
Case Presentation
A 62-year-old Caucasian man with a history of well-controlled type 2 diabetes mellitus and hypertension is being followed by his dermatologist for surveillance of melanocytic nevi. On follow-up he is noted to have an asymmetrical melanocytic lesion over the right scalp with irregular borders and variegated color. He is asymptomatic and the remainder of physical examination is unremarkable, as he has no other concerning skin lesions and no cervical, axillary, or inguinal lymphadenopathy.
How is melanoma diagnosed?
Detailed discussion about diagnosis and staging will be deferred in this review of treatment of advanced melanoma. In brief, melanoma is best diagnosed by excisional biopsy and histopathology. Staging of melanoma is done according to the American Joint Committee on Cancer’s (AJCC) Cancer Staging Manual, 8th edition, using a TNM staging system that incorporates tumor thickness (Breslow depth); ulceration; number of involved regional lymph nodes; presence of in-transit, satellite, and/or microsatellite metastases; distant metastases; and serum lactate dehydrogenase level.14
Case Continued
The patient undergoes a wide excisional biopsy of the right scalp lesion, which is consistent with malignant melanoma. Pathology demonstrates a Breslow depth of 2.6 mm, 2 mitotic figures/mm2, and no evidence of ulceration. He subsequently undergoes wide local excision with 0/3 sentinel lymph nodes positive for malignancy. His final staging is consistent with pT3aN0M0, stage IIA melanoma.
He is seen in follow-up with medical oncology for the next 3.5 years without any evidence of disease recurrence. He then develops symptoms of vertigo, diplopia, and recurrent falls, prompting medical attention. Magnetic resonance imaging (MRI) brain reveals multiple supratentorial and infratentorial lesions concerning for intracranial metastases. Further imaging with computed tomography (CT) chest/abdomen/pelvis reveals a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He is admitted for treatment with intravenous dexamethasone and further evaluation with endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass, which reveals metastatic melanoma. Given the extent of his intracranial metastases, he is treated with whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy.
What is the general approach to first-line treatment for metastatic melanoma?
The past decade has brought an abundance of data supporting the use of immunotherapy with immune checkpoint inhibitors or molecularly targeted therapy with combined BRAF/MEK inhibitors in the first-line setting.4-8 After the diagnosis of metastatic melanoma has been made, molecular testing is recommended to determine the BRAF status of the tumor. Immunotherapy is the clear choice for first-line therapy in the absence of an activating BRAF V600 mutation. When a BRAF V600 mutation is present, current evidence supports the use of either immunotherapy or molecularly targeted therapy as first-line therapy.
To date, there have been no prospective clinical trials comparing the sequencing of immunotherapy and molecularly targeted therapy in the first-line setting. An ongoing clinical trial (NCT02224781) is comparing dabrafenib and trametinib followed by ipilimumab and nivolumab at time of progression to ipilimumab and nivolumab followed by dabrafenib and trametinib in patients with newly diagnosed stage III/IV BRAF V600 mutation–positive melanoma. The primary outcome measure is 2-year OS. Until completion of that trial, current practice regarding which type of therapy to use in the first-line setting is based on a number of factors including clinical characteristics and provider preferences.
Data suggest that immunotherapies can produce durable responses, especially after treatment completion or discontinuation, albeit at the expense of taking a longer time to achieve clinical benefit and the risk of potentially serious immune-related adverse effects. This idea of a durable, off-treatment response is highlighted by a study that followed 105 patients who had achieved a complete response (CR) and found that 24-month disease-free survival from the time of CR was 90.9% in all patients and 89.9% in the 67 patients who had discontinued pembrolizumab after attaining CR.15 BRAF/MEK inhibition has the potential for rapid clinical responses, though concerns exist about the development of resistance to therapy. The following sections explore the evidence supporting the use of these therapies.
Immunotherapy with Immune Checkpoint Inhibitors
Immunotherapy via immune checkpoint blockade has revolutionized the treatment of many solid tumors over the past decade. The promise of immunotherapy revolves around the potential for achieving a dynamic and durable systemic response against cancer by augmenting the antitumor effects of the immune system. T-cells are central to mounting a systemic antitumor response, and, in addition to antigen recognition, their function depends heavily on fine tuning between co-stimulatory and co-inhibitory signaling. The cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) expressed on T-cells was the first discovered co-inhibitory receptor of T-cell activation.16 Later, it was discovered that the programmed cell death 1 receptor (PD-1), expressed on T-cells, and its ligands PD-L1 and PD-L2, expressed on antigen presenting cells, tumor cells, or other cells in the tumor microenvironment, also served as a potent negative regulator of T-cell function.17
Together, these 2 signaling pathways help to maintain peripheral immune tolerance, whereby autoreactive T-cells that have escaped from the thymus are silenced to prevent autoimmunity. However, these pathways can also be utilized by cancer cells to escape immune surveillance. Monoclonal antibodies that inhibit the aforementioned co-inhibitory signaling pathways, and thus augment the immune response, have proven to be an effective anticancer therapy capable of producing profound and durable responses in certain malignancies.16,17
Ipilimumab
Ipilimumab is a monoclonal antibody that inhibits the function of the CTLA-4 co-inhibitory immune checkpoint. In a phase 3 randomized controlled trial of 676 patients with previously treated metastatic melanoma, ipilimumab at a dose of 3 mg/kg every 3 weeks for 4 cycles, with or without a gp100 peptide vaccine, resulted in an improved median OS of 10.0 and 10.1 months, respectively, compared to 6.4 months in those receiving the peptide vaccine alone, meeting the primary endpoint.4 Subsequently, a phase 3 trial of 502 patients with untreated metastatic melanoma compared ipilimumab at a dose of 10 mg/kg every 3 weeks for 4 cycles plus dacarbazine to dacarbazine plus placebo and found a significant increase in median OS (11.2 months vs 9.1 months), with no additive benefit of chemotherapy. There was a higher reported rate of grade 3 or 4 adverse events in this trial with ipilimumab dosed at 10 mg/kg, which was felt to be dose-related.18 These trials were the first to show improved OS with any systemic therapy in metastatic melanoma and led to US Food and Drug Administration approval of ipilimumab for this indication in 2011.
PD-1 Inhibitor Monotherapy
The PD-1 inhibitors nivolumab and pembrolizumab were initially approved for metastatic melanoma after progression on ipilimumab. In the phase 1 trial of patients with previously treated metastatic melanoma, nivolumab therapy resulted in an ORR of 28%.19 The subsequent phase 2 trial conducted in pretreated patients, including patients who had progressed on ipilimumab, confirmed a similar ORR of 31%, as well as a median PFS of 3.7 months and a median OS of 16.8 months. The estimated response duration in patients who did achieve a response to therapy was 2 years.20 A phase 3 trial (CheckMate 037) comparing nivolumab (n = 120) to investigator’s choice chemotherapy (n = 47) in those with melanoma refractory to ipilimumab demonstrated that nivolumab was superior for the primary endpoint of ORR (31.7% vs 10.6%), had less toxicity (5% rate of grade 3 or 4 adverse events versus 9%), and increased median duration of response (32 months vs 13 months).21
The phase 1 trial (KEYNOTE-001) testing the efficacy of pembrolizumab demonstrated an ORR of 33% in the total population of patients treated and an ORR of 45% in those who were treatment-naive. Additionally, the median OS was 23 months for the total population and 31 months for treatment-naive patients, with only 14% of patients experiencing a grade 3 or 4 adverse event.22 The KEYNOTE-002 phase 2 trial compared 2 different pembrolizumab doses (2 mg/kg and 10 mg/kg every 3 weeks) to investigator’s choice chemotherapy (paclitaxel plus carboplatin, paclitaxel, carboplatin, dacarbazine, or oral temozolomide) in 540 patients with advanced melanoma with documented progression on ipilimumab with or without prior progression on molecularly targeted therapy if positive for a BRAF V600 mutation. The final analysis demonstrated significantly improved ORR with pembrolizumab (22% at 2 mg/kg vs 26% at 10 mg/kg vs 4% chemotherapy) and significantly improved 24-month PFS (16% vs 22% vs 0.6%, respectively). There was a nonstatistically significant improvement in median OS (13.4 months vs 14.7 months vs 10 months), although 55% of the patients initially assigned to the chemotherapy arm crossed over and received pembrolizumab after documentation of progressive disease.23,24
Because PD-1 inhibition improved efficacy with less toxicity than chemotherapy when studied in progressive disease, subsequent studies focused on PD-1 inhibition in the frontline setting. CheckMate 066 was a phase 3 trial comparing nivolumab to dacarbazine as first-line therapy for 418 patients with untreated metastatic melanoma who did not have a BRAF mutation. For the primary end point of 1-year OS, nivolumab was superior to dacarbazine (72.9% vs 42.1%; hazard ratio [HR], 0.42; P < 0.001). Treatment with nivolumab also resulted in superior ORR (40% vs 14%) and PFS (5.1 months vs 2.2 months). Additionally, nivolumab therapy had a lower rate of grade 3 or 4 toxicity compared to dacarbazine (11.7% vs 17.6%).25
The KEYNOTE-006 trial compared 2 separate dosing schedules of pembrolizumab (10 mg/kg every 2 weeks versus every 3 weeks) to ipilimumab (3 mg/kg every 3 weeks for 4 cycles) in a 1:1:1 ratio in 834 patients with metastatic melanoma who had received up to 1 prior systemic therapy, but no prior CTLA-4 or PD-1 inhibitors. The first published data reported statistically significant outcomes for the co-primary end points of 6-month PFS (47.3% for pembrolizumab every 2 weeks vs 46.4% for pembrolizumab every 3 weeks vs 26.5% for ipilimumab; HR, 0.58 for both pembrolizumab groups compared to ipilimumab; P < 0.001) and 12-month OS (74.1% vs 68.4% vs 58.2%) with pembrolizumab compared to ipilimumab. Compared to ipilimumab, pembrolizumab every 2 weeks had a hazard ratio of 0.63 (P = 0.0005) and pembrolizumab every 3 weeks had a hazard ratio of 0.69 (P = 0.0036). The pembrolizumab groups was also had lower rates of grade 3 to 5 toxicity (13.3% vs 10.1% vs 19.9%).5 Updated outcomes demonstrated improved ORR compared to the first analysis (37% vs 36% vs 13%), and improved OS (median OS, not reached for the pembrolizumab groups vs 16.0 months for the ipilimumab group; HR, 0.68, P = 0.0009 for pembrolizumab every 2 weeks versus HR 0.68, P = 0.0008 for pembrolizumab every 3 weeks).26 In addition, 24-month OS was 55% in both pembrolizumab groups compared to 43% in the ipilimumab group. Grade 3 or 4 toxicity occurred less frequently with pembrolizumab (17% vs 17% vs 20%).
Further analysis from the KEYNOTE-006 trial data demonstrated improved ORR, PFS, and OS with pembrolizumab compared to ipilimumab in tumors positive for PD-L1 expression. For PD-L1-negative tumors, response rate was higher, and PFS and OS rates were similar with pembrolizumab compared to ipilimumab. Given that pembrolizumab was associated with similar survival outcomes in PD-L1-negative tumors and with less toxicity than ipilimumab, the superiority of PD-L1 inhibitors over ipilimumab was further supported, regardless of tumor PD-L1 status.27
In sum, PD-1 inhibition should be considered the first-line immunotherapy in advanced melanoma, either alone or in combination with ipilimumab, as discussed in the following section. There is no longer a role for ipilimumab monotherapy in the first-line setting, based on evidence from direct comparison to single-agent PD-1 inhibition in clinical trials that demonstrated superior efficacy and less serious toxicity with PD-1 inhibitors.5,26 The finding that ORR and OS outcomes with single-agent PD-1 inhibitors are higher in treatment-naive patients compared to those receiving prior therapies also supports this approach.22
Combination CTLA-4 and PD-1 Therapy
Despite the potential for durable responses, the majority of patients fail to respond to single-agent PD-1 therapy. Given that preclinical data had suggested the potential for synergy between dual inhibition of CTLA-4 and PD-1, clinical trials were designed to test this approach. The first randomized phase 2 trial that established superior efficacy with combination therapy was the CheckMate 069 trial comparing nivolumab plus ipilimumab to ipilimumab monotherapy. Combination therapy resulted in increased ORR (59% vs 11%), median PFS (not reached vs 3.0 months), 2-year PFS (51.3% vs 12.0%), and 2-year OS (63.8% vs 53.6%).28 Similarly, a phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and 1-year OS of 89%.29
The landmark phase 3 CheckMate 067 trial analyzed efficacy outcomes for 3 different treatment regimens including nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy in previously untreated patients with unresectable stage III or IV melanoma. The trial was powered to compare survival outcomes for both the combination therapy arm against ipilimumab and the nivolumab monotherapy arm against ipilimumab, but not to compare combination therapy to nivolumab monotherapy. The initial analysis demonstrated a median PFS of 11.5 months with combination therapy versus 6.9 months with nivolumab and 2.9 months with ipilimumab, as well as an ORR of 58% versus 44% and 19%, respectively (Table 1).6 The updated 3-year survival outcomes from CheckMate 067 were notable for superior median OS with combination therapy (not reached in combination vs 37.6 months for nivolumab vs 19.9 months ipilimumab), improved 3-year OS (58% vs 52% vs 34%), and improved 3-year PFS (39% vs 32% vs 10%).7 In the reported 4-year survival outcomes, median OS was not reached in the combination therapy group, and was 36.9 months in the nivolumab monotherapy group and 19.9 months in the ipilimumab monotherapy group. Rates of grade 3 or 4 adverse events were significantly higher in the combination therapy group, at 59% compared to 22% with nivolumab monotherapy and 28% with ipilimumab alone.30 The 3- and 4-year OS outcomes (58% and 54%, respectively) with combination therapy were the highest seen in any phase 3 trial for treatment of advanced melanoma, supporting its use as the best approved first-line therapy in those who can tolerate the potential toxicity of combination therapy7,30 The conclusions from this landmark trial were that both combination therapy and nivolumab monotherapy resulted in statistically significant improvement in OS compared to ipilimumab.
Toxicity Associated with Immune Checkpoint Inhibitors
While immune checkpoint inhibitors have revolutionized the treatment of many solid tumor malignancies, this new class of cancer therapy has brought about a new type of toxicity for clinicians to be aware of, termed immune-related adverse events (irAEs). As immune checkpoint inhibitors amplify the immune response against malignancy, they also increase the likelihood that autoreactive T-cells persist and proliferate within the circulation. Therefore, these therapies can result in almost any type of autoimmune side effect. The most commonly reported irAEs in large clinical trials studying CTLA-4 and PD-1 inhibitors include rash/pruritus, diarrhea/colitis, hepatitis, endocrinopathies (thyroiditis, hypophysitis, adrenalitis), and pneumonitis. Other more rare toxicities include pancreatitis, autoimmune hematologic toxicities, cardiac toxicity (myocarditis, heart failure), and neurologic toxicities (neuropathies, myasthenia gravis-like syndrome, Guillain-Barré syndrome). It has been observed that PD-1 inhibitors have a lower incidence of irAEs than CTLA-4 inhibitors, and that the combined use of PD-1 and CTLA-4 inhibitors is associated with a greater incidence of irAEs compared to monotherapy with either agent.31 Toxicities associated with ipilimumab have been noted to be dose dependent.18 Generally, these toxicities are treated with immunosuppression in the form of glucocorticoids and are often reversible.31 There are several published guidelines that include algorithms for the management of irAEs by organizations such as the National Comprehensive Cancer Network.32
For example, previously untreated patients treated with ipilimumab plus dacarbazine as compared to dacarbazine plus placebo had greater grade 3 or 4 adverse events (56.3% vs 27.5%), and 77.7% of patients experiencing an irAE of any grade.18 In the CheckMate 066 trial comparing frontline nivolumab to dacarbazine, nivolumab had a lower rate of grade 3 or 4 toxicity (11.7% vs 17.6%) and irAEs were relatively infrequent, with diarrhea and elevated alanine aminotransferase level each being the most prominent irAE (affecting 1.0% of patients).25 In the KEYNOTE-006 trial, irAEs seen in more than 1% of patients treated with pembrolizumab included colitis, hepatitis, hypothyroidism, and hyperthyroidism, whereas those occurring in more than 1% of patients treated with ipilimumab included colitis and hypophysitis. Overall, there were lower rates of grade 3 to 5 toxicity with the 2 pembrolizumab doses compared to ipilimumab (13.3% pembrolizumab every 2 weeks vs 10.1% pembrolizumab every 3 weeks vs 19.9% ipilimumab).5 In the CheckMate 067 trial comparing nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy, rates of treatment-related adverse events of any grade were higher in the combination group (96% combination vs 86% nivolumab vs 86% ipilimumab), as were rates of grade 3 or 4 adverse events (59% vs 21% vs 28%, respectively). The irAE profile was similar to that demonstrated in prior studies: rash/pruritus were the most common, and diarrhea/colitis, elevated aminotransferases, and endocrinopathies were among the more common irAEs.7
Alternative dosing strategies have been investigated in an effort to preserve efficacy and minimize toxicity. A phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and a 1-year OS of 89%. This combination led to 45% of patients having a grade 3 or 4 adverse event, 60% having irAEs of any grade, and only 27% having grade 3 or 4 irAEs.29 The CheckMate 067 trial studied the combination of nivolumab 1 mg/kg plus ipilimumab 3 mg/kg.6 The CheckMate 511 trial compared different combination dosing strategies (nivolumab 3 mg/kg + ipilimumab 1 mg/kg versus nivolumab 1 mg/kg + ipilimumab 3 mg/kg) to assess for safety benefit. In the results published in abstract form, the reduced ipilimumab dose (nivolumab 3 mg/kg + ipilimumab 1 mg/kg arm) resulted in significantly decreased grade 3 to 5 adverse events (33.9% vs 48.3%) without significant differences in ORR, PFS, or OS.33
The question about the efficacy of checkpoint inhibitors in patients who discontinue treatment due to irAEs has been raised, as one hypothesis suggests that such toxicities may also indicate that the antitumor immune response has been activated. In a retrospective pooled analysis of phase 2 and 3 trials where patients received combination therapy with ipilimumab and nivolumab and discontinued therapy during the induction phase due to irAEs, outcomes did not appear to be inferior. Median PFS was 8.4 months in those who discontinued therapy compared to 10.8 months in those who continued therapy, but this did not reach statistical significance. Median OS had not been reached in either group and ORR was actually higher in those who discontinued due to adverse events (58.3% vs 50.2%). While this retrospective analysis needs to be validated, it does suggest that patients likely derive antitumor benefit from immunotherapy even if they have to discontinue therapy due to irAEs. Of note, patients in this analysis were not trialed on nivolumab monotherapy after receiving immunosuppressive treatment for toxicity related to combination therapy, which has since been deemed a reasonable treatment option.34
Molecularly Targeted Therapy for Metastatic Melanoma
As previously mentioned, the MAPK pathway is frequently altered in metastatic melanoma and thus serves as a target for therapy. Mutations in BRAF can cause constitutive activation of the protein’s kinase function, which subsequently phosphorylates/activates MEK in the absence of extracellular growth signals and causes increased cellular proliferation. For the roughly half of patients diagnosed with metastatic melanoma who harbor a BRAF V600 mutation, molecularly targeted therapy with BRAF/MEK inhibitors has emerged as a standard of care treatment option. As such, all patients with advanced disease should be tested for BRAF mutations.
After early phase 1 studies of the BRAF inhibitor vemurafenib demonstrated successful inhibition of mutated BRAF,35 subsequent studies confirmed the benefit of BRAF targeted therapy. In the phase 3 randomized controlled BRIM-3 trial comparing vemurafenib with dacarbazine for treatment of 675 patients with previously untreated metastatic melanoma positive for a BRAF V600E mutation, the vemurafenib group had superior ORR and 6-month OS during the first analysis.36 In a subsequent analysis, median PFS and median OS were also superior with vemurafenib compared to dacarbazine, as vemurafenib had a median OS of 13.6 months compared to 9.7 months with dacarbazine (HR, 0.70; P = 0.0008).37 Dabrafenib was the next BRAF inhibitor to demonstrate clinical efficacy with superior PFS compared to dacarbazine.38
Despite tumor shrinkage in the majority of patients, the development of resistance to therapy was an issue early on. The development of acquired resistance emerged as a heterogeneous process, though many of the identified resistance mechanisms involved reactivation of the MAPK pathway.39 A phase 3 trial of 322 patients with metastatic melanoma comparing the MEK inhibitor trametinib as monotherapy against chemotherapy demonstrated a modest improvement in both median PFS and OS.40 As a result, subsequent efforts focused on a strategy of concurrent MEK inhibition as a means to overcome resistance to molecularly targeted monotherapy
At least 4 large phase 3 randomized controlled trials of combination therapy with BRAF plus MEK inhibitors showed an improved ORR, PFS, and OS when compared to BRAF inhibition alone. The COMBI-d trial comparing dabrafenib plus trametinib versus dabrafenib alone was the first to demonstrate the superiority of combined BRAF/MEK inhibition and made combination therapy the current standard of care for patients with metastatic melanoma and a BRAF V600 mutation. In the final analysis of this trial, 3-year PFS was 22% with combination therapy compared to 12% with dabrafenib alone, and 3-year OS was 44% compared to 32%.8,41,42 A second trial with the combination of dabrafenib and trametinib (COMBI-V) also demonstrated superior efficacy when compared to single-agent vemurafenib without increased toxicity.43 Subsequently, the combination of vemurafenib with MEK inhibitor cobimetinib demonstrated superiority compared to vemurafenib alone,44 followed by the newest combination encorafenib (BRAF inhibitor) and binimetinib (MEK inhibitor) proving superior to either vemurafenib or encorafenib alone.45,46
It is important to note that there have been no studies directly comparing the efficacy of the 3 approved BRAF/MEK inhibitor combinations, but the 3 different regimens have some differences in their toxicity profiles (Table 2). Of note, single-agent BRAF inhibition was associated with increased cutaneous toxicity, including secondary squamous cell carcinoma and keratoacanthoma,47 which was demonstrated to be driven by paradoxical activation of the MAPK pathway.48 The concerning cutaneous toxicities such as squamous cell carcinoma were substantially reduced by combination BRAF/MEK inhibitor therapy.47 Collectively, the higher efficacy along with manageable toxicity profile established combination BRAF/MEK inhibition as the preferred regimen for patients with BRAF-mutated metastatic melanoma who are being considered for molecularly targeted therapy. BRAF inhibitor monotherapy should only be used when there is a specific concern regarding the use of a MEK inhibitor in certain clinical circumstances.
Other driver mutations associated with metastatic melanoma such as NRAS-mutated tumors have proven more difficult to effectively treat with molecularly targeted therapy, with one study showing that the MEK inhibitor binimetinib resulted in a modest improvement in ORR and median PFS without OS benefit compared to dacarbazine.49 Several phase 2 trials involving metastatic melanoma harboring a c-Kit alteration have demonstrated some efficacy with the tyrosine kinase inhibitor imatinib. The largest phase 2 trial of 43 patients treated with imatinib resulted in a 53.5% disease control rate (23.3% partial response and 30.2% stable disease), with 9 of the 10 patients who achieved partial response having a mutation in either exon 11 or 13. Median PFS was 3.5 months and 1-year OS was 51.0%.50
Case Conclusion
Prior to initiation of systemic therapy, the patient’s melanoma is tested and is found to be positive for a BRAF V600K mutation. At his follow-up appointment, the patient continues to endorse generalized weakness, fatigue, issues with balance, and residual pulmonary symptoms after being treated for post-obstructive pneumonia. Given his current symptoms and extent of metastatic disease, immunotherapy is deferred and he is started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially does well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass. Further follow-up of this patient is presented in the second article in this 2-part review of advanced melanoma.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin. 2018;68:7-30.
2. Ives NJ, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2621 patients. J Clin Oncol. 2007;25:5426-34.
3. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105-16.
4. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23.
5. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
6. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
7. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345-1356.
8. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
9. Elwood JM, Jopson J. Melanoma and sun exposure: an overview of published studies. Int J Cancer. 1997;73:198-203.
10. Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 199;340:1341-1348.
11. Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483-8.
12. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54.
13. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681-96.
14. Gershenwald JE, Scolyer RA, Hess KR, et al. Melanoma staging: evidence-based changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer J Clin. 2017;67:472-492.
15. Robert C, Ribas A, Hamid O, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol. 2018;36:1668-1674.
16. Salama AKS, Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clin Cancer Res. 2011;17:4622-8.
17. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767-1778.
18. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
19. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
20. Topalian S, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020-30.
21. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-84.
22. Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315:1600-1609.
23. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-18.
24. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
25. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
26. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicenter, randomised, open-label phase 3 study (KEYNOTE-006). Lancet Oncol. 2017;390:1853-1862.
27. Carlino MS, Long GV, Schadendorf D, et al. Outcomes by line of therapy and programmed death ligand 1 expression in patients with advanced melanoma treated with pembrolizumab or ipilimumab in KEYNOTE-006. A randomised clinical trial. Eur J Cancer. 2018;101:236-243.
28. Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558-1568.
29. Long GV, Atkinson V, Cebon JS, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 2017;18:1202-10.
30. Hodi FS, Chiarion-Sileni V, Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480-1492.
31. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. 2016;2:1346-1353.
32. National Comprehensive Cancer Network. Management of immunotherapy-related toxicities (version 2.2019). www.nccn.org/professionals/physician_gls/pdf/immunotherapy.pdf. Accessed April 8, 2019.
33. Lebbé C, Meyer N, Mortier L, et al. Initial results from a phase IIIb/IV study evaluating two dosing regimens of nivolumab (NIVO) in combination with ipilimumab (IPI) in patients with advanced melanoma (CheckMate 511) [Abstract LBA47]. Ann Oncol. 2018;29:mdy424.057.
34. Schadendorf D, Wolchok JD, Hodi FS, et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase ii and iii trials. J Clin Oncol. 2017;35:3807-3814.
35. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
36. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
37. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
38. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicenter, open-label, phase 3 randomised controlled trial. Lancet Oncol. 2012;380:358-365.
39. Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965-1977.
40. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107-114.
41. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
42. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
43. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
44. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-260.
45. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
46. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
47. Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA. Dermatol 2015;151:1103-1109.
48. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
49. Dummer R, Schadendorf D, Ascierto P, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18:435-445.
50. Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904-2909.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin. 2018;68:7-30.
2. Ives NJ, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2621 patients. J Clin Oncol. 2007;25:5426-34.
3. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105-16.
4. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23.
5. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
6. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
7. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345-1356.
8. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
9. Elwood JM, Jopson J. Melanoma and sun exposure: an overview of published studies. Int J Cancer. 1997;73:198-203.
10. Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 199;340:1341-1348.
11. Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483-8.
12. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54.
13. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681-96.
14. Gershenwald JE, Scolyer RA, Hess KR, et al. Melanoma staging: evidence-based changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer J Clin. 2017;67:472-492.
15. Robert C, Ribas A, Hamid O, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol. 2018;36:1668-1674.
16. Salama AKS, Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clin Cancer Res. 2011;17:4622-8.
17. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767-1778.
18. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
19. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
20. Topalian S, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020-30.
21. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-84.
22. Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315:1600-1609.
23. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-18.
24. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
25. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
26. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicenter, randomised, open-label phase 3 study (KEYNOTE-006). Lancet Oncol. 2017;390:1853-1862.
27. Carlino MS, Long GV, Schadendorf D, et al. Outcomes by line of therapy and programmed death ligand 1 expression in patients with advanced melanoma treated with pembrolizumab or ipilimumab in KEYNOTE-006. A randomised clinical trial. Eur J Cancer. 2018;101:236-243.
28. Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558-1568.
29. Long GV, Atkinson V, Cebon JS, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 2017;18:1202-10.
30. Hodi FS, Chiarion-Sileni V, Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480-1492.
31. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. 2016;2:1346-1353.
32. National Comprehensive Cancer Network. Management of immunotherapy-related toxicities (version 2.2019). www.nccn.org/professionals/physician_gls/pdf/immunotherapy.pdf. Accessed April 8, 2019.
33. Lebbé C, Meyer N, Mortier L, et al. Initial results from a phase IIIb/IV study evaluating two dosing regimens of nivolumab (NIVO) in combination with ipilimumab (IPI) in patients with advanced melanoma (CheckMate 511) [Abstract LBA47]. Ann Oncol. 2018;29:mdy424.057.
34. Schadendorf D, Wolchok JD, Hodi FS, et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase ii and iii trials. J Clin Oncol. 2017;35:3807-3814.
35. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
36. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
37. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
38. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicenter, open-label, phase 3 randomised controlled trial. Lancet Oncol. 2012;380:358-365.
39. Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965-1977.
40. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107-114.
41. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
42. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
43. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
44. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-260.
45. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
46. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
47. Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA. Dermatol 2015;151:1103-1109.
48. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
49. Dummer R, Schadendorf D, Ascierto P, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18:435-445.
50. Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904-2909.
"Doctor, Do I Need a Skin Check?"
What does your patient need to know at the first visit?
A patient may be scheduled for a total-body skin examination (TBSE) through several routes: primary care referral, continued cancer screening for an at-risk patient or patient transfer, or patient-directed scheduling for general screening regardless of risk factors. At the patient's first visit, it is imperative that the course of the appointment is smooth and predictable for patient comfort and for a thorough and effective examination. The nurse initially solicits salient medical history, particularly personal and family history of skin cancer, current medications, and any acute concerns. The nurse then prepares the patient for the logistics of the TBSE, namely to undress, don a gown that ties and opens in the back, and be seated on the examination table. When I enter the room, the conversation commences with me seated across from the patient, reviewing specifics about his/her history and risk factors. Then the TBSE is executed from head to toe.
Do you broadly recommend TBSE?
Firstly, TBSE is a safe clinical tool, supported by data outlining a lack of notable patient morbidity during the examination, including psychosocial factors, and it is generally well-received by patients (Risica et al). In 2016, the US Preventative Services Task Force (USPSTF) outlined its recommendations regarding screening for skin cancer, concluding that there is insufficient evidence to broadly recommend TBSE. Unfortunately, USPSTF findings amassed data from all types of screenings, including those by nondermatologists, and did not extract specialty-specific benefits and risks to patients. The recommendation also did not outline the influence of TBSE on morbidity and mortality for at-risk groups. The guidelines target primary care practice trends; therefore, specialty societies such as the American Academy of Dermatology issued statements following the USPSTF recommendation outlining these salient clarifications, namely that TBSE detects melanoma and keratinocyte carcinomas earlier than in patients who are not screened. Randomized controlled trials to prove this observation are lacking, particularly because of the ethics of withholding screening from a prospective study group. However, in 2017, Johnson et al outlined the best available survival data in concert with the USPSTF statement to arrive at the most beneficial screening recommendations for patients, specifically targeting risk groups--those with a history of skin cancer, immunosuppression, indoor tanning and/or many blistering sunburns, and several other genetic parameters--for at least annual TBSE.
The technique and reproducibility of TBSE also are not standardized, though they seem to have been endearingly apprenticed but variably implemented through generations of dermatology residents going forward into practice. As it is, depending on patient body surface area, mobility, willingness to disrobe, and adornments (eg, tattoos, hair appliances), multiple factors can restrict full view of a patient's skin. Recently, Helm et al proposed standardizing the TBSE sequence to minimize omitted areas of the body, which may become an imperative tool for streamlined resident teaching and optimal screening encounters.
How do you keep patients compliant with TBSE?
During and following TBSE, I typically outline any lesions of concern and plan for further testing, screening, and behavioral prevention strategies. Frequency of TBSE and importance of compliance are discussed during the visit and reinforced at checkout where the appointment templates are established a year in advance for those with skin cancer. Further, for those with melanoma, their appointment slots are given priority status so that any cancellations or delays are rescheduled preferentially. Particularly during the discussion about TBSE frequency, I emphasize the comparison and importance of this visit akin to other recommended screenings, such as mammograms and colonoscopies, and that we, as dermatologists, are part of their cancer surveillance team.
What do you do if patients refuse your recommendations?
Some patients refuse a gown or removal of certain clothing items (eg, undergarments, socks, wigs). Some patients defer a yearly TBSE upon checkout and schedule an appointment only when a lesion of concern arises. My advice is not to shame patients and to take advantage of as much as the patient is able and comfortable to show us and be present for, welcoming that we have the opportunity to take care of them and screen for cancer in any capacity. In underserved or limited budget practice regions, lesion-directed examination vs TBSE may be the only screening method utilized and may even attract more patients to a screening facility (Hoorens et al).
In the opposite corner are those patients who deem the recommended TBSE interval as too infrequent, which poses a delicate dilemma. In my opinion, these situations present another cohort of risks. Namely, the patient may become (or continue to be) overly fixated on the small details of every skin lesion, and in my experience, they tend to develop the habit of expecting at least 1 biopsy at each visit, typically of a lesion of their choosing. Depending on the validity of this expectation vs my clinical examination, it can lead to a difficult discussion with the patient about oversampling lesions and the potential for many scars, copious reexcisions for ambiguous lesion pathology, and a trend away from prudent clinical care. In addition, multiple visits incur more patient co-pays and time away from school, work, or home. To ease the patient's mind, I advise to call our office for a more acute visit if there is a lesion of concern; I additionally recommend taking a smartphone photograph of a concerning lesion and monitoring it for changes or sending the photograph to our patient portal messaging system so we can evaluate its acuity.
What take-home advice do you give to patients?
As the visit ends, I further explain that home self-examination or examination by a partner between visits is intuitively a valuable screening adjunct for skin cancer. In 2018, the USPSTF recommended behavioral skin cancer prevention counseling and self-examination only for younger-age cohorts with fair skin (6 months to 24 years), but its utility in specialty practice must be qualified. The American Academy of Dermatology Association subsequently issued a statement to support safe sun-protective practices and diligent self-screening for changing lesions, as earlier detection and management of skin cancer can lead to decreased morbidity and mortality from these neoplasms.
Resources for Patients
American Academy of Dermatology's SPOT Skin Cancer
Centers for Disease Control and Prevention: What Screening Tests Are There?
Suggested Readings
AAD statement on USPSTF recommendation on skin cancer screening. Schaumburg, IL: American Academy of Dermatology; July 26, 2016. https://www.aad.org/media/news-releases/aad-statement-on-uspstf. Accessed April 26, 2019.
AADA responds to USPSTF recommendation on skin cancer prevention counseling. Rosemont, IL: American Academy of Dermatology Association; March 20, 2018. https://www.aad.org/media/news-releases/skin-cancer-prevention-counseling. Accessed April 26, 2019.
Helm MF, Hallock KK, Bisbee E, et al. Optimizing the total body skin exam: an observational cohort study [published online February 15, 2019]. J Am Acad Dermatol. doi:10.1016/j.jaad.2019.02.028.
Hoorens I, Vossaert K, Pil L, et al. Total-body examination vs lesion-directed skin cancer screening. JAMA Dermatol. 2016;152:27-34.
Johnson MM, Leachman SA, Aspinwall LG, et al. Skin cancer screening: recommendations for data-driven screening guidelines and a review of the US Preventive Services Task Force controversy. Melanoma Manag. 2017;4:13-37.
Risica PM, Matthews NH, Dionne L, et al. Psychosocial consequences of skin cancer screening. Prev Med Rep. 2018;10:310-316.
US Preventive Services Task Force, Bibbins-Domingo K, Grossman DC, et al. Screening for skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2016;316:429-435.
US Preventive Services Task Force, Grossman DC, Curry SJ, et al. Behavioral counseling to prevent skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2018;319:1134-1142.
What does your patient need to know at the first visit?
A patient may be scheduled for a total-body skin examination (TBSE) through several routes: primary care referral, continued cancer screening for an at-risk patient or patient transfer, or patient-directed scheduling for general screening regardless of risk factors. At the patient's first visit, it is imperative that the course of the appointment is smooth and predictable for patient comfort and for a thorough and effective examination. The nurse initially solicits salient medical history, particularly personal and family history of skin cancer, current medications, and any acute concerns. The nurse then prepares the patient for the logistics of the TBSE, namely to undress, don a gown that ties and opens in the back, and be seated on the examination table. When I enter the room, the conversation commences with me seated across from the patient, reviewing specifics about his/her history and risk factors. Then the TBSE is executed from head to toe.
Do you broadly recommend TBSE?
Firstly, TBSE is a safe clinical tool, supported by data outlining a lack of notable patient morbidity during the examination, including psychosocial factors, and it is generally well-received by patients (Risica et al). In 2016, the US Preventative Services Task Force (USPSTF) outlined its recommendations regarding screening for skin cancer, concluding that there is insufficient evidence to broadly recommend TBSE. Unfortunately, USPSTF findings amassed data from all types of screenings, including those by nondermatologists, and did not extract specialty-specific benefits and risks to patients. The recommendation also did not outline the influence of TBSE on morbidity and mortality for at-risk groups. The guidelines target primary care practice trends; therefore, specialty societies such as the American Academy of Dermatology issued statements following the USPSTF recommendation outlining these salient clarifications, namely that TBSE detects melanoma and keratinocyte carcinomas earlier than in patients who are not screened. Randomized controlled trials to prove this observation are lacking, particularly because of the ethics of withholding screening from a prospective study group. However, in 2017, Johnson et al outlined the best available survival data in concert with the USPSTF statement to arrive at the most beneficial screening recommendations for patients, specifically targeting risk groups--those with a history of skin cancer, immunosuppression, indoor tanning and/or many blistering sunburns, and several other genetic parameters--for at least annual TBSE.
The technique and reproducibility of TBSE also are not standardized, though they seem to have been endearingly apprenticed but variably implemented through generations of dermatology residents going forward into practice. As it is, depending on patient body surface area, mobility, willingness to disrobe, and adornments (eg, tattoos, hair appliances), multiple factors can restrict full view of a patient's skin. Recently, Helm et al proposed standardizing the TBSE sequence to minimize omitted areas of the body, which may become an imperative tool for streamlined resident teaching and optimal screening encounters.
How do you keep patients compliant with TBSE?
During and following TBSE, I typically outline any lesions of concern and plan for further testing, screening, and behavioral prevention strategies. Frequency of TBSE and importance of compliance are discussed during the visit and reinforced at checkout where the appointment templates are established a year in advance for those with skin cancer. Further, for those with melanoma, their appointment slots are given priority status so that any cancellations or delays are rescheduled preferentially. Particularly during the discussion about TBSE frequency, I emphasize the comparison and importance of this visit akin to other recommended screenings, such as mammograms and colonoscopies, and that we, as dermatologists, are part of their cancer surveillance team.
What do you do if patients refuse your recommendations?
Some patients refuse a gown or removal of certain clothing items (eg, undergarments, socks, wigs). Some patients defer a yearly TBSE upon checkout and schedule an appointment only when a lesion of concern arises. My advice is not to shame patients and to take advantage of as much as the patient is able and comfortable to show us and be present for, welcoming that we have the opportunity to take care of them and screen for cancer in any capacity. In underserved or limited budget practice regions, lesion-directed examination vs TBSE may be the only screening method utilized and may even attract more patients to a screening facility (Hoorens et al).
In the opposite corner are those patients who deem the recommended TBSE interval as too infrequent, which poses a delicate dilemma. In my opinion, these situations present another cohort of risks. Namely, the patient may become (or continue to be) overly fixated on the small details of every skin lesion, and in my experience, they tend to develop the habit of expecting at least 1 biopsy at each visit, typically of a lesion of their choosing. Depending on the validity of this expectation vs my clinical examination, it can lead to a difficult discussion with the patient about oversampling lesions and the potential for many scars, copious reexcisions for ambiguous lesion pathology, and a trend away from prudent clinical care. In addition, multiple visits incur more patient co-pays and time away from school, work, or home. To ease the patient's mind, I advise to call our office for a more acute visit if there is a lesion of concern; I additionally recommend taking a smartphone photograph of a concerning lesion and monitoring it for changes or sending the photograph to our patient portal messaging system so we can evaluate its acuity.
What take-home advice do you give to patients?
As the visit ends, I further explain that home self-examination or examination by a partner between visits is intuitively a valuable screening adjunct for skin cancer. In 2018, the USPSTF recommended behavioral skin cancer prevention counseling and self-examination only for younger-age cohorts with fair skin (6 months to 24 years), but its utility in specialty practice must be qualified. The American Academy of Dermatology Association subsequently issued a statement to support safe sun-protective practices and diligent self-screening for changing lesions, as earlier detection and management of skin cancer can lead to decreased morbidity and mortality from these neoplasms.
Resources for Patients
American Academy of Dermatology's SPOT Skin Cancer
Centers for Disease Control and Prevention: What Screening Tests Are There?
What does your patient need to know at the first visit?
A patient may be scheduled for a total-body skin examination (TBSE) through several routes: primary care referral, continued cancer screening for an at-risk patient or patient transfer, or patient-directed scheduling for general screening regardless of risk factors. At the patient's first visit, it is imperative that the course of the appointment is smooth and predictable for patient comfort and for a thorough and effective examination. The nurse initially solicits salient medical history, particularly personal and family history of skin cancer, current medications, and any acute concerns. The nurse then prepares the patient for the logistics of the TBSE, namely to undress, don a gown that ties and opens in the back, and be seated on the examination table. When I enter the room, the conversation commences with me seated across from the patient, reviewing specifics about his/her history and risk factors. Then the TBSE is executed from head to toe.
Do you broadly recommend TBSE?
Firstly, TBSE is a safe clinical tool, supported by data outlining a lack of notable patient morbidity during the examination, including psychosocial factors, and it is generally well-received by patients (Risica et al). In 2016, the US Preventative Services Task Force (USPSTF) outlined its recommendations regarding screening for skin cancer, concluding that there is insufficient evidence to broadly recommend TBSE. Unfortunately, USPSTF findings amassed data from all types of screenings, including those by nondermatologists, and did not extract specialty-specific benefits and risks to patients. The recommendation also did not outline the influence of TBSE on morbidity and mortality for at-risk groups. The guidelines target primary care practice trends; therefore, specialty societies such as the American Academy of Dermatology issued statements following the USPSTF recommendation outlining these salient clarifications, namely that TBSE detects melanoma and keratinocyte carcinomas earlier than in patients who are not screened. Randomized controlled trials to prove this observation are lacking, particularly because of the ethics of withholding screening from a prospective study group. However, in 2017, Johnson et al outlined the best available survival data in concert with the USPSTF statement to arrive at the most beneficial screening recommendations for patients, specifically targeting risk groups--those with a history of skin cancer, immunosuppression, indoor tanning and/or many blistering sunburns, and several other genetic parameters--for at least annual TBSE.
The technique and reproducibility of TBSE also are not standardized, though they seem to have been endearingly apprenticed but variably implemented through generations of dermatology residents going forward into practice. As it is, depending on patient body surface area, mobility, willingness to disrobe, and adornments (eg, tattoos, hair appliances), multiple factors can restrict full view of a patient's skin. Recently, Helm et al proposed standardizing the TBSE sequence to minimize omitted areas of the body, which may become an imperative tool for streamlined resident teaching and optimal screening encounters.
How do you keep patients compliant with TBSE?
During and following TBSE, I typically outline any lesions of concern and plan for further testing, screening, and behavioral prevention strategies. Frequency of TBSE and importance of compliance are discussed during the visit and reinforced at checkout where the appointment templates are established a year in advance for those with skin cancer. Further, for those with melanoma, their appointment slots are given priority status so that any cancellations or delays are rescheduled preferentially. Particularly during the discussion about TBSE frequency, I emphasize the comparison and importance of this visit akin to other recommended screenings, such as mammograms and colonoscopies, and that we, as dermatologists, are part of their cancer surveillance team.
What do you do if patients refuse your recommendations?
Some patients refuse a gown or removal of certain clothing items (eg, undergarments, socks, wigs). Some patients defer a yearly TBSE upon checkout and schedule an appointment only when a lesion of concern arises. My advice is not to shame patients and to take advantage of as much as the patient is able and comfortable to show us and be present for, welcoming that we have the opportunity to take care of them and screen for cancer in any capacity. In underserved or limited budget practice regions, lesion-directed examination vs TBSE may be the only screening method utilized and may even attract more patients to a screening facility (Hoorens et al).
In the opposite corner are those patients who deem the recommended TBSE interval as too infrequent, which poses a delicate dilemma. In my opinion, these situations present another cohort of risks. Namely, the patient may become (or continue to be) overly fixated on the small details of every skin lesion, and in my experience, they tend to develop the habit of expecting at least 1 biopsy at each visit, typically of a lesion of their choosing. Depending on the validity of this expectation vs my clinical examination, it can lead to a difficult discussion with the patient about oversampling lesions and the potential for many scars, copious reexcisions for ambiguous lesion pathology, and a trend away from prudent clinical care. In addition, multiple visits incur more patient co-pays and time away from school, work, or home. To ease the patient's mind, I advise to call our office for a more acute visit if there is a lesion of concern; I additionally recommend taking a smartphone photograph of a concerning lesion and monitoring it for changes or sending the photograph to our patient portal messaging system so we can evaluate its acuity.
What take-home advice do you give to patients?
As the visit ends, I further explain that home self-examination or examination by a partner between visits is intuitively a valuable screening adjunct for skin cancer. In 2018, the USPSTF recommended behavioral skin cancer prevention counseling and self-examination only for younger-age cohorts with fair skin (6 months to 24 years), but its utility in specialty practice must be qualified. The American Academy of Dermatology Association subsequently issued a statement to support safe sun-protective practices and diligent self-screening for changing lesions, as earlier detection and management of skin cancer can lead to decreased morbidity and mortality from these neoplasms.
Resources for Patients
American Academy of Dermatology's SPOT Skin Cancer
Centers for Disease Control and Prevention: What Screening Tests Are There?
Suggested Readings
AAD statement on USPSTF recommendation on skin cancer screening. Schaumburg, IL: American Academy of Dermatology; July 26, 2016. https://www.aad.org/media/news-releases/aad-statement-on-uspstf. Accessed April 26, 2019.
AADA responds to USPSTF recommendation on skin cancer prevention counseling. Rosemont, IL: American Academy of Dermatology Association; March 20, 2018. https://www.aad.org/media/news-releases/skin-cancer-prevention-counseling. Accessed April 26, 2019.
Helm MF, Hallock KK, Bisbee E, et al. Optimizing the total body skin exam: an observational cohort study [published online February 15, 2019]. J Am Acad Dermatol. doi:10.1016/j.jaad.2019.02.028.
Hoorens I, Vossaert K, Pil L, et al. Total-body examination vs lesion-directed skin cancer screening. JAMA Dermatol. 2016;152:27-34.
Johnson MM, Leachman SA, Aspinwall LG, et al. Skin cancer screening: recommendations for data-driven screening guidelines and a review of the US Preventive Services Task Force controversy. Melanoma Manag. 2017;4:13-37.
Risica PM, Matthews NH, Dionne L, et al. Psychosocial consequences of skin cancer screening. Prev Med Rep. 2018;10:310-316.
US Preventive Services Task Force, Bibbins-Domingo K, Grossman DC, et al. Screening for skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2016;316:429-435.
US Preventive Services Task Force, Grossman DC, Curry SJ, et al. Behavioral counseling to prevent skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2018;319:1134-1142.
Suggested Readings
AAD statement on USPSTF recommendation on skin cancer screening. Schaumburg, IL: American Academy of Dermatology; July 26, 2016. https://www.aad.org/media/news-releases/aad-statement-on-uspstf. Accessed April 26, 2019.
AADA responds to USPSTF recommendation on skin cancer prevention counseling. Rosemont, IL: American Academy of Dermatology Association; March 20, 2018. https://www.aad.org/media/news-releases/skin-cancer-prevention-counseling. Accessed April 26, 2019.
Helm MF, Hallock KK, Bisbee E, et al. Optimizing the total body skin exam: an observational cohort study [published online February 15, 2019]. J Am Acad Dermatol. doi:10.1016/j.jaad.2019.02.028.
Hoorens I, Vossaert K, Pil L, et al. Total-body examination vs lesion-directed skin cancer screening. JAMA Dermatol. 2016;152:27-34.
Johnson MM, Leachman SA, Aspinwall LG, et al. Skin cancer screening: recommendations for data-driven screening guidelines and a review of the US Preventive Services Task Force controversy. Melanoma Manag. 2017;4:13-37.
Risica PM, Matthews NH, Dionne L, et al. Psychosocial consequences of skin cancer screening. Prev Med Rep. 2018;10:310-316.
US Preventive Services Task Force, Bibbins-Domingo K, Grossman DC, et al. Screening for skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2016;316:429-435.
US Preventive Services Task Force, Grossman DC, Curry SJ, et al. Behavioral counseling to prevent skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2018;319:1134-1142.
Topical Chemotherapy for Numerous Superficial Basal Cell Carcinomas Years After Isolated Limb Perfusion for Melanoma
Isolated limb perfusion (ILP) for the adjuvant treatment of melanoma involves isolating the blood flow of a limb from the rest of the body to allow for high concentrations of chemotherapeutic agents locally. Chemotherapy with nitrogen mustard is the preferred chemotherapeutic agent in ILP for the adjuvant treatment of locally advanced melanoma.1 Systemic exposure to nitrogen mustard has shown to be carcinogenic, and its topical application has been associated with the development of actinic keratosis, basal cell carcinoma (BCC), and squamous cell carcinoma.2,3 However, the long-term effects of ILP with nitrogen mustard are not well defined. In 1998, one of the authors (R.L.M.) described a patient with melanoma of the left leg that was treated with ILP with nitrogen mustard who subsequently developed numerous BCCs on the same leg.4 This same patient has since been successfully managed with only topical chemotherapeutic agents for the last 21 years.
An 86-year-old man with a history of melanoma underwent wide resection, lymph node dissection, and adjuvant ILP with nitrogen mustard for the treatment of melanoma of the medial left thigh approximately 50 years ago. He denied any prior radiation treatment. He subsequently presented years later to our dermatology clinic with many biopsy-proven superficial and nodular BCCs of the left leg over the course of the last 30 years. On physical examination, the patient had several pink papules and macules on the left lower leg (Figure). The patient had previously undergone multiple invasive excisions with grafting for the treatment of BCCs by a plastic surgeon prior to presentation to our clinic but has since had many years of control under our care with only topical chemotherapeutic agents. His current medication regimen consists of 5-fluorouracil twice daily, which he tolerates without serious side effects. He also has used imiquimod in the past.
Isolated limb perfusion was first described by Creech et al5 in 1958. Chemotherapy in ILP is designed to maximize limb perfusion while minimizing systemic absorption.1 Meta
Topical use of nitrogen mustard has been linked to the development of nonmelanoma skin cancer (NMSC)2,3; however, a 30-year population-based study found no significant increase in secondary malignancies, including NMSC or melanoma, following use of topical nitrogen mustard.6 There also have been reported cases of secondary cancers following ILP reported in the literature, including pleomorphic sarcoma and Merkel cell carcinoma.7 We hypothesize that our patient’s exposure to nitrogen mustard during ILP led to the development of numerous BCCs, but further research is necessary to confirm this relationship.
Treatment modalities for NMSC include surgical excision with defined margins, Mohs micrographic surgery, radiotherapy, electrodesiccation and curettage, cryotherapy, photodynamic therapy, and topical therapy. Our patient experienced such a high volume of superficial BCCs that the decision was made to avoid frequent surgical procedures and to treat with topical chemotherapeutic agents. He had an excellent response to topical 5-fluorouracil, and the treatment has been well tolerated. This case is valuable for clinicians, as it demonstrates that topical chemotherapy can be a well-tolerated option for patients who present with frequent superficial BCCs to prevent numerous invasive surgical treatments.
- Benckhuijsen C, Kroon BB, van Geel AN, et al. Regional perfusion treatment with melphalan for melanoma in a limb: an evaluation of drug kinetics. Eur J Surg Oncol. 1988;14:157-163.
- Abel EA, Sendagorta E, Hoppe RT. Cutaneous malignancies and metastatic squamous cell carcinoma following topical therapy for mycosis fungoides. J Am Acad Dermatol. 1986;14:1029-1038.
- Lee LA, Fritz KA, Golitz L, et al. Second cutaneous malignancies in patients with mycosis fungoides treated with topical nitrogen mustard. J Am Acad Dermatol. 1982;7:590-598.
- Lamb PM, Menaker GM, Moy RL. Multiple basal cell carcinomas of the limb after adjuvant treatment of melanoma with isolated limb perfusion. J Am Acad Dermatol. 1998;38:767-768.
- Creech O Jr, Krementz ET, Ryan RF, et al. Chemotherapy of cancer: regional perfusion utilizing an extracorporal circuit. Ann Surg. 1958;148:616-632.
- Lindahl L, Fenger-Grøn M, Iversen L. Secondary cancers, comorbidities and mortality associated with nitrogen mustard therapy in patients with mycosis fungoides: a 30-year population-based cohort study. Br J Dermatol. 2014;170:699-704.
- Lenormand C, Pelletier C, Goeldel AL, et al. Second malignant neoplasm occurring years after hyperthermic isolated limb perfusion for melanoma. Arch Dermatol. 2010;146:319-321.
Isolated limb perfusion (ILP) for the adjuvant treatment of melanoma involves isolating the blood flow of a limb from the rest of the body to allow for high concentrations of chemotherapeutic agents locally. Chemotherapy with nitrogen mustard is the preferred chemotherapeutic agent in ILP for the adjuvant treatment of locally advanced melanoma.1 Systemic exposure to nitrogen mustard has shown to be carcinogenic, and its topical application has been associated with the development of actinic keratosis, basal cell carcinoma (BCC), and squamous cell carcinoma.2,3 However, the long-term effects of ILP with nitrogen mustard are not well defined. In 1998, one of the authors (R.L.M.) described a patient with melanoma of the left leg that was treated with ILP with nitrogen mustard who subsequently developed numerous BCCs on the same leg.4 This same patient has since been successfully managed with only topical chemotherapeutic agents for the last 21 years.
An 86-year-old man with a history of melanoma underwent wide resection, lymph node dissection, and adjuvant ILP with nitrogen mustard for the treatment of melanoma of the medial left thigh approximately 50 years ago. He denied any prior radiation treatment. He subsequently presented years later to our dermatology clinic with many biopsy-proven superficial and nodular BCCs of the left leg over the course of the last 30 years. On physical examination, the patient had several pink papules and macules on the left lower leg (Figure). The patient had previously undergone multiple invasive excisions with grafting for the treatment of BCCs by a plastic surgeon prior to presentation to our clinic but has since had many years of control under our care with only topical chemotherapeutic agents. His current medication regimen consists of 5-fluorouracil twice daily, which he tolerates without serious side effects. He also has used imiquimod in the past.
Isolated limb perfusion was first described by Creech et al5 in 1958. Chemotherapy in ILP is designed to maximize limb perfusion while minimizing systemic absorption.1 Meta
Topical use of nitrogen mustard has been linked to the development of nonmelanoma skin cancer (NMSC)2,3; however, a 30-year population-based study found no significant increase in secondary malignancies, including NMSC or melanoma, following use of topical nitrogen mustard.6 There also have been reported cases of secondary cancers following ILP reported in the literature, including pleomorphic sarcoma and Merkel cell carcinoma.7 We hypothesize that our patient’s exposure to nitrogen mustard during ILP led to the development of numerous BCCs, but further research is necessary to confirm this relationship.
Treatment modalities for NMSC include surgical excision with defined margins, Mohs micrographic surgery, radiotherapy, electrodesiccation and curettage, cryotherapy, photodynamic therapy, and topical therapy. Our patient experienced such a high volume of superficial BCCs that the decision was made to avoid frequent surgical procedures and to treat with topical chemotherapeutic agents. He had an excellent response to topical 5-fluorouracil, and the treatment has been well tolerated. This case is valuable for clinicians, as it demonstrates that topical chemotherapy can be a well-tolerated option for patients who present with frequent superficial BCCs to prevent numerous invasive surgical treatments.
Isolated limb perfusion (ILP) for the adjuvant treatment of melanoma involves isolating the blood flow of a limb from the rest of the body to allow for high concentrations of chemotherapeutic agents locally. Chemotherapy with nitrogen mustard is the preferred chemotherapeutic agent in ILP for the adjuvant treatment of locally advanced melanoma.1 Systemic exposure to nitrogen mustard has shown to be carcinogenic, and its topical application has been associated with the development of actinic keratosis, basal cell carcinoma (BCC), and squamous cell carcinoma.2,3 However, the long-term effects of ILP with nitrogen mustard are not well defined. In 1998, one of the authors (R.L.M.) described a patient with melanoma of the left leg that was treated with ILP with nitrogen mustard who subsequently developed numerous BCCs on the same leg.4 This same patient has since been successfully managed with only topical chemotherapeutic agents for the last 21 years.
An 86-year-old man with a history of melanoma underwent wide resection, lymph node dissection, and adjuvant ILP with nitrogen mustard for the treatment of melanoma of the medial left thigh approximately 50 years ago. He denied any prior radiation treatment. He subsequently presented years later to our dermatology clinic with many biopsy-proven superficial and nodular BCCs of the left leg over the course of the last 30 years. On physical examination, the patient had several pink papules and macules on the left lower leg (Figure). The patient had previously undergone multiple invasive excisions with grafting for the treatment of BCCs by a plastic surgeon prior to presentation to our clinic but has since had many years of control under our care with only topical chemotherapeutic agents. His current medication regimen consists of 5-fluorouracil twice daily, which he tolerates without serious side effects. He also has used imiquimod in the past.
Isolated limb perfusion was first described by Creech et al5 in 1958. Chemotherapy in ILP is designed to maximize limb perfusion while minimizing systemic absorption.1 Meta
Topical use of nitrogen mustard has been linked to the development of nonmelanoma skin cancer (NMSC)2,3; however, a 30-year population-based study found no significant increase in secondary malignancies, including NMSC or melanoma, following use of topical nitrogen mustard.6 There also have been reported cases of secondary cancers following ILP reported in the literature, including pleomorphic sarcoma and Merkel cell carcinoma.7 We hypothesize that our patient’s exposure to nitrogen mustard during ILP led to the development of numerous BCCs, but further research is necessary to confirm this relationship.
Treatment modalities for NMSC include surgical excision with defined margins, Mohs micrographic surgery, radiotherapy, electrodesiccation and curettage, cryotherapy, photodynamic therapy, and topical therapy. Our patient experienced such a high volume of superficial BCCs that the decision was made to avoid frequent surgical procedures and to treat with topical chemotherapeutic agents. He had an excellent response to topical 5-fluorouracil, and the treatment has been well tolerated. This case is valuable for clinicians, as it demonstrates that topical chemotherapy can be a well-tolerated option for patients who present with frequent superficial BCCs to prevent numerous invasive surgical treatments.
- Benckhuijsen C, Kroon BB, van Geel AN, et al. Regional perfusion treatment with melphalan for melanoma in a limb: an evaluation of drug kinetics. Eur J Surg Oncol. 1988;14:157-163.
- Abel EA, Sendagorta E, Hoppe RT. Cutaneous malignancies and metastatic squamous cell carcinoma following topical therapy for mycosis fungoides. J Am Acad Dermatol. 1986;14:1029-1038.
- Lee LA, Fritz KA, Golitz L, et al. Second cutaneous malignancies in patients with mycosis fungoides treated with topical nitrogen mustard. J Am Acad Dermatol. 1982;7:590-598.
- Lamb PM, Menaker GM, Moy RL. Multiple basal cell carcinomas of the limb after adjuvant treatment of melanoma with isolated limb perfusion. J Am Acad Dermatol. 1998;38:767-768.
- Creech O Jr, Krementz ET, Ryan RF, et al. Chemotherapy of cancer: regional perfusion utilizing an extracorporal circuit. Ann Surg. 1958;148:616-632.
- Lindahl L, Fenger-Grøn M, Iversen L. Secondary cancers, comorbidities and mortality associated with nitrogen mustard therapy in patients with mycosis fungoides: a 30-year population-based cohort study. Br J Dermatol. 2014;170:699-704.
- Lenormand C, Pelletier C, Goeldel AL, et al. Second malignant neoplasm occurring years after hyperthermic isolated limb perfusion for melanoma. Arch Dermatol. 2010;146:319-321.
- Benckhuijsen C, Kroon BB, van Geel AN, et al. Regional perfusion treatment with melphalan for melanoma in a limb: an evaluation of drug kinetics. Eur J Surg Oncol. 1988;14:157-163.
- Abel EA, Sendagorta E, Hoppe RT. Cutaneous malignancies and metastatic squamous cell carcinoma following topical therapy for mycosis fungoides. J Am Acad Dermatol. 1986;14:1029-1038.
- Lee LA, Fritz KA, Golitz L, et al. Second cutaneous malignancies in patients with mycosis fungoides treated with topical nitrogen mustard. J Am Acad Dermatol. 1982;7:590-598.
- Lamb PM, Menaker GM, Moy RL. Multiple basal cell carcinomas of the limb after adjuvant treatment of melanoma with isolated limb perfusion. J Am Acad Dermatol. 1998;38:767-768.
- Creech O Jr, Krementz ET, Ryan RF, et al. Chemotherapy of cancer: regional perfusion utilizing an extracorporal circuit. Ann Surg. 1958;148:616-632.
- Lindahl L, Fenger-Grøn M, Iversen L. Secondary cancers, comorbidities and mortality associated with nitrogen mustard therapy in patients with mycosis fungoides: a 30-year population-based cohort study. Br J Dermatol. 2014;170:699-704.
- Lenormand C, Pelletier C, Goeldel AL, et al. Second malignant neoplasm occurring years after hyperthermic isolated limb perfusion for melanoma. Arch Dermatol. 2010;146:319-321.
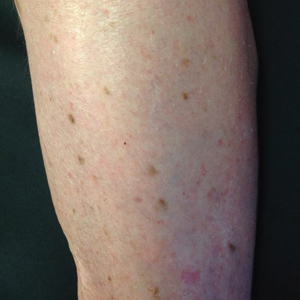
Dermoscopic Patterns of Acral Melanocytic Lesions in Skin of Color
Acral lentiginous melanoma (ALM) is a rare subtype of melanoma that occurs on the palms, soles, and nail apparatus. Unlike more common types of melanoma, ALM occurs on sun-protected areas of the skin and has distinct clinical, histologic, and genetic features. Acral lentiginous melanoma accounts for a larger proportion of melanomas in individuals with skin of color and has a worse prognosis and recurrence rate than other forms of melanoma.
Population Trends in Skin of Color
Much of the literature on malignant melanoma historically has involved non-Hispanic white patients, but the incidence in lighter-skinned populations has been increasing steadily over the last few decades.1 Although ALM can occur in any race, it disproportionately affects skin of color populations; ALM accounts for only 0.8% to 1% of all melanomas in white populations, but it constitutes 4% to 58% of melanomas in ethnic populations and is the most common melanoma subtype among black Americans.2-5 Acral lentiginous melanoma also is associated with a worse prognosis compared to other subtypes, which may indicate a more aggressive biological nature6 but also may point toward socioeconomic and cultural barriers (eg, low income or education levels, lack of insurance, lower health literacy), leading to disparities in access to care and diagnosis at advanced stages.5
Similarly, the distribution of acral melanocytic nevi appears to demonstrate an association with ethnicity and skin pigmentation. Although skin of color patients have fewer nevi than non-Hispanic whites, the proportion of acral melanocytic nevi tends to be greater.6,7 Given its grim prognosis, accurately differentiating ALM from acral nevi is of utmost importance.
Diagnostic Challenges of Acral Lesions
Due to the unique nature of the surfaces of acral sites, melanocytic lesions on the palms, soles, and nail apparatus present many diagnostic challenges. It can be difficult to distinguish acral melanoma from benign lesions using the naked eye alone. Volar surfaces are characterized by the presence of dermatoglyphics, and pigment deposition along ridges and furrows create particular dermoscopic patterns exclusive to these sites.8 Thus, dermoscopy can be useful on acral surfaces, but the dermoscopic features are different from those on the rest of the body and must be learned separately.
In addition, nearly half of patients are unaware of their acral lesions.6 Acral surfaces may not always be examined by clinicians during total-body skin examinations, leading to further possibility of overlooking a lesion. Obtaining biopsies on glabrous skin or nails also is challenging because they can be more painful and hemostasis can be more difficult, especially in the nail. Acral melanomas also may be amelanotic, including those at subungual sites.
Dermoscopic Patterns of Acral Volar Skin
Dermoscopy is a useful noninvasive tool for distinguishing between benign and malignant acral melanocytic lesions, and its efficacy in improving diagnostic accuracy and decreasing unnecessary biopsies is well-established in the literature.13,14 Acral dermoscopy allows for visualization of pigment along the dermatoglyphics that constitute the characteristic dermoscopic patterns.
Acral Lentiginous Melanoma
The hallmark dermoscopic pattern and most important finding of ALM is the parallel ridge pattern, characterized by parallel linear pigmentation along the ridges of dermatoglyphics. In the early phases of malignancy, the pattern appears light brown and involves most of the lesion; as the tumor develops, increasing melanin production results in focal areas of the parallel ridge pattern with darker bands.15,16 The sensitivity and specificity of a parallel ridge pattern for diagnosing early ALM has been shown to be 86% and 99%, respectively.15,16
A pattern of irregular diffuse pigmentation also can be observed in more advanced ALM. Dermoscopy may reveal a structureless pattern (ie, lack of identifiable structures or patterns) in a background of tan-black coloration due to more exuberant melanocyte proliferation along the epidermis.15 Sensitivity and specificity of this dermoscopic finding for invasive lesions is high at 94% and 97%, respectively.16,17 Interestingly, once ALM lesions have advanced even further, conventional melanoma-associated structures (ie, blue-white veil, polymorphous blood vessels, ulceration, irregular dots/globules or streaks) or atypical forms of typically benign acral dermoscopic patterns may be observed.15
Per a 3-step diagnostic algorithm created by Koga and Saida,18 a suspected acral lesion should first be evaluated for a parallel ridge pattern to determine the need for biopsy, as it is seen in approximately two-thirds of ALMs.19 If no parallel ridge pattern is observed, the lesion should then be checked for any of the typical dermoscopic patterns seen in benign acral nevi (eg, parallel furrow, latticelike, or fibrillar patterns).18 The maximum diameter should be measured only if the lesion does not exhibit any of the typical dermoscopic patterns. If the lesion’s diameter is greater than 7 mm in diameter, it should be biopsied; if the diameter is less than 7 mm, it should have regular clinical and dermoscopic follow-up.18
In 2015, Lallas et al20 developed the BRAAFF checklist, a scoring system of 6 variables: blotches, ridge pattern, asymmetry of structures, asymmetry of colors, parallel furrow pattern, and fibrillar pattern. The checklist also was shown to substantially improve diagnostic accuracy of dermoscopy for ALM, with sensitivity and specificity at 93.1% and 86.7%, respectively.20
Acquired Acral Nevi
Three classic dermoscopic patterns are associated with acquired acral nevi: parallel furrow pattern, latticelike pattern, and fibrillar pattern.15,21 Approximately three-quarters of all acquired acral nevi exhibit one of these patterns, roughly half exhibiting parallel furrow with tan-brown bandlike pigmentation along dermatoglyphic grooves.16,17
Latticelike patterns also are characterized by brown parallel lines along the sulci of dermatoglyphics but additionally have multiple intersecting lines. Thus, this pattern can be considered a variant of the parallel furrow pattern.15 The crisscross markings can be predominantly found in the plantar arch.22 This dermoscopic pattern comprises 15% to 25% of all acral nevi.21
Fibrillar pattern accounts for 10% to 20% of all acral melanocytic nevi.21 Dermoscopically, these lesions demonstrate parallel filamentous streaks that cross dermatoglyphics obliquely. The fibrillar pattern is predominantly found on weight-bearing areas of the sole,22 which likely is explained by pressure causing slanting of melanin columns in the horny layer.23 The fibrillar pattern has been shown to be the benign acral dermoscopic pattern that is most commonly misdiagnosed, with higher reported rates of biopsy.24
Acral Congenital Melanocytic Nevi
Congenital melanocytic nevi (CMN) present at birth or appear during the first few weeks of life. Congenital melanocytic nevi can vary widely in size, shape, and color, and they are occasionally biopsied in cases of larger diameter or dermoscopic atypia to differentiate from melanoma.25 Congenital melanocytic nevi also can occur on acral volar surfaces. Possible dermoscopic patterns include parallel furrow or fibrillar patterns as well as a crista dotted pattern, defined as evenly spaced dots/globules on the ridges near the openings of eccrine ducts.26 A more commonly observed dermoscopic pattern in acral CMN is a combination of the crista dotted and parallel furrow patterns, known as the peas-in-a-pod pattern. Changes in the clinical appearance and dermoscopic features of an acral CMN are possible over time; some lesions also may fade with age.26
Final Thoughts
Acral lentiginous melanoma is a rare but potentially aggressive melanoma subtype that accounts for a larger proportion of melanomas in patients with skin of color than in white patients. Dermoscopy of acral volar skin provides invaluable diagnostic information and allows for better management of acral melanocytic lesions. Dermoscopic patterns such as the parallel ridge, parallel furrow, latticelike, fibrillar, and peas-in-a-pod patterns are unique to acral sites and can be used to differentiate between ALMs, acquired nevi, or CMNs.
- Whiteman DC, Green AC, Olsen CM. The growing burden of invasive melanoma: projections of incidence rates and numbers of new cases in six susceptible populations through 2031. J Invest Dermatol. 2016;136:1161-1171.
- Bradford PT, Goldstein AM, McMaster ML, et al. Acral lentiginous melanoma: incidence and survival patterns in the United States, 1986-2005. Arch Dermatol. 2009;145:427-434.
- Nakamura Y, Fujisawa Y. Diagnosis and management of acral lentiginous melanoma. Curr Treat Options Oncol. 2018;19:42.
- Cormier JN, Xing Y, Ding M, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006;166:1907-1914.
- Wang Y, Zhao Y, Ma S. Racial differences in six major subtypes of melanoma: descriptive epidemiology. BMC Cancer. 2016;16:691.
- Madankumar R, Gumaste PV, Martires K, et al. Acral melanocytic lesions in the United States: prevalence, awareness, and dermoscopic patterns in skin-of-color and non-Hispanic white patients. J Am Acad Dermatol. 2016;74:724.e1-730.e1.
- Palicka GA, Rhodes AR. Acral melanocytic nevi: prevalence and distribution of gross morphologic features in white and black adults. Arch Dermatol. 2010;146:1085-1094.
- Thomas L, Phan A, Pralong P, et al. Special locations dermoscopy: facial, acral, and nail. Dermatol Clin. 2013;31:615-624.
- Gong HZ, Zheng HY, Li J. Amelanotic melanoma [published online January 21, 2019]. Melanoma Res. doi:10.1097/CMR.0000000000000571.
- Ise M, Yasuda F, Konohana I, et al. Acral melanoma with hyperkeratosis mimicking a pigmented wart. Dermatol Pract Concept. 2013;3:37-39.
- Serarslan G, Akçaly CM, Atik E. Acral lentiginous melanoma misdiagnosed as tinea pedis: a case report. Int J Dermatol. 2004;43:37-38.
- Gumaste P, Penn L, Cohen N, et al. Acral lentiginous melanoma of the foot misdiagnosed as a traumatic ulcer. a cautionary case. J Am Podiatr Med Assoc. 2015;105:189-194.
- Carli P, de Giorgi V, Chiarugi A, et al. Addition of dermoscopy to conventional naked-eye examination in melanoma screening: a randomized study. J Am Acad Dermatol. 2004;50:683-689.
- Carli P, de Giorgi V, Crocetti E, et al. Improvement of malignant/benign ratio in excised melanocytic lesions in the ‘dermoscopy era’: a retrospective study 1997-2001. Br J Dermatol. 2004;150:687-692.
- Saida T, Koga H, Uhara H. Key points in dermoscopic differentiation between early acral melanoma and acral nevus. J Dermatol. 2011;38:25-34.
- Ishihara Y, Saida T, Miyazaki A, et al. Early acral melanoma in situ: correlation between the parallel ridge pattern on dermoscopy and microscopic features. Am J Dermatopathol. 2006;28:21-27.
- Saida T, Miyazaki A, Oguchi S, et al. Significance of dermoscopic patterns in detecting malignant melanoma on acral volar skin: results of a multicenter study in Japan. Arch Dermatol. 2004;140:1233-1238.
- Koga H, Saida T. Revised 3-step dermoscopic algorithm for the management of acral melanocytic lesions. Arch Dermatol. 2011;147:741-743.
- Lallas A, Sgouros D, Zalaudek I, et al. Palmar and plantar melanomas differ for sex prevalence and tumor thickness but not for dermoscopic patterns. Melanoma Res. 2014;24:83-87.
- Lallas A, Kyrgidis A, Koga H, et al. The BRAAFF checklist: a new dermoscopic algorithm for diagnosing acral melanoma. Br J Dermatol. 2015;173:1041-1049.
- Saida T, Koga H. Dermoscopic patterns of acral melanocytic nevi: their variations, changes, and significance. Arch Dermatol. 2007;143:1423-1426.
- Miyazaki A, Saida T, Koga H, et al. Anatomical and histopathological correlates of the dermoscopic patterns seen in melanocytic nevi on the sole: a retrospective study. J Am Acad Dermatol. 2005;53:230-236.
- Watanabe S, Sawada M, Ishizaki S, et al. Comparison of dermatoscopic images of acral lentiginous melanoma and acral melanocytic nevus occurring on body weight-bearing areas. Dermatol Pract Concept. 2014;4:47-50.
- Costello CM, Ghanavatian S, Temkit M, et al. Educational and practice gaps in the management of volar melanocytic lesions. J Eur Acad Dermatol Venereol. 2018;32:1450-1455.
- Alikhan A, Ibrahimi OA, Eisen DB. Congenital melanocytic nevi: where are we now? part I. clinical presentation, epidemiology, pathogenesis, histology, malignant transformation, and neurocutaneous melanosis. J Am Acad Dermatol. 2012;67:495.e1-495.e17; quiz 512-514.
- Minagawa A, Koga H, Saida T. Dermoscopic characteristics of congenital melanocytic nevi affecting acral volar skin. Arch Dermatol. 2011;147:809-813.
Acral lentiginous melanoma (ALM) is a rare subtype of melanoma that occurs on the palms, soles, and nail apparatus. Unlike more common types of melanoma, ALM occurs on sun-protected areas of the skin and has distinct clinical, histologic, and genetic features. Acral lentiginous melanoma accounts for a larger proportion of melanomas in individuals with skin of color and has a worse prognosis and recurrence rate than other forms of melanoma.
Population Trends in Skin of Color
Much of the literature on malignant melanoma historically has involved non-Hispanic white patients, but the incidence in lighter-skinned populations has been increasing steadily over the last few decades.1 Although ALM can occur in any race, it disproportionately affects skin of color populations; ALM accounts for only 0.8% to 1% of all melanomas in white populations, but it constitutes 4% to 58% of melanomas in ethnic populations and is the most common melanoma subtype among black Americans.2-5 Acral lentiginous melanoma also is associated with a worse prognosis compared to other subtypes, which may indicate a more aggressive biological nature6 but also may point toward socioeconomic and cultural barriers (eg, low income or education levels, lack of insurance, lower health literacy), leading to disparities in access to care and diagnosis at advanced stages.5
Similarly, the distribution of acral melanocytic nevi appears to demonstrate an association with ethnicity and skin pigmentation. Although skin of color patients have fewer nevi than non-Hispanic whites, the proportion of acral melanocytic nevi tends to be greater.6,7 Given its grim prognosis, accurately differentiating ALM from acral nevi is of utmost importance.
Diagnostic Challenges of Acral Lesions
Due to the unique nature of the surfaces of acral sites, melanocytic lesions on the palms, soles, and nail apparatus present many diagnostic challenges. It can be difficult to distinguish acral melanoma from benign lesions using the naked eye alone. Volar surfaces are characterized by the presence of dermatoglyphics, and pigment deposition along ridges and furrows create particular dermoscopic patterns exclusive to these sites.8 Thus, dermoscopy can be useful on acral surfaces, but the dermoscopic features are different from those on the rest of the body and must be learned separately.
In addition, nearly half of patients are unaware of their acral lesions.6 Acral surfaces may not always be examined by clinicians during total-body skin examinations, leading to further possibility of overlooking a lesion. Obtaining biopsies on glabrous skin or nails also is challenging because they can be more painful and hemostasis can be more difficult, especially in the nail. Acral melanomas also may be amelanotic, including those at subungual sites.
Dermoscopic Patterns of Acral Volar Skin
Dermoscopy is a useful noninvasive tool for distinguishing between benign and malignant acral melanocytic lesions, and its efficacy in improving diagnostic accuracy and decreasing unnecessary biopsies is well-established in the literature.13,14 Acral dermoscopy allows for visualization of pigment along the dermatoglyphics that constitute the characteristic dermoscopic patterns.
Acral Lentiginous Melanoma
The hallmark dermoscopic pattern and most important finding of ALM is the parallel ridge pattern, characterized by parallel linear pigmentation along the ridges of dermatoglyphics. In the early phases of malignancy, the pattern appears light brown and involves most of the lesion; as the tumor develops, increasing melanin production results in focal areas of the parallel ridge pattern with darker bands.15,16 The sensitivity and specificity of a parallel ridge pattern for diagnosing early ALM has been shown to be 86% and 99%, respectively.15,16
A pattern of irregular diffuse pigmentation also can be observed in more advanced ALM. Dermoscopy may reveal a structureless pattern (ie, lack of identifiable structures or patterns) in a background of tan-black coloration due to more exuberant melanocyte proliferation along the epidermis.15 Sensitivity and specificity of this dermoscopic finding for invasive lesions is high at 94% and 97%, respectively.16,17 Interestingly, once ALM lesions have advanced even further, conventional melanoma-associated structures (ie, blue-white veil, polymorphous blood vessels, ulceration, irregular dots/globules or streaks) or atypical forms of typically benign acral dermoscopic patterns may be observed.15
Per a 3-step diagnostic algorithm created by Koga and Saida,18 a suspected acral lesion should first be evaluated for a parallel ridge pattern to determine the need for biopsy, as it is seen in approximately two-thirds of ALMs.19 If no parallel ridge pattern is observed, the lesion should then be checked for any of the typical dermoscopic patterns seen in benign acral nevi (eg, parallel furrow, latticelike, or fibrillar patterns).18 The maximum diameter should be measured only if the lesion does not exhibit any of the typical dermoscopic patterns. If the lesion’s diameter is greater than 7 mm in diameter, it should be biopsied; if the diameter is less than 7 mm, it should have regular clinical and dermoscopic follow-up.18
In 2015, Lallas et al20 developed the BRAAFF checklist, a scoring system of 6 variables: blotches, ridge pattern, asymmetry of structures, asymmetry of colors, parallel furrow pattern, and fibrillar pattern. The checklist also was shown to substantially improve diagnostic accuracy of dermoscopy for ALM, with sensitivity and specificity at 93.1% and 86.7%, respectively.20
Acquired Acral Nevi
Three classic dermoscopic patterns are associated with acquired acral nevi: parallel furrow pattern, latticelike pattern, and fibrillar pattern.15,21 Approximately three-quarters of all acquired acral nevi exhibit one of these patterns, roughly half exhibiting parallel furrow with tan-brown bandlike pigmentation along dermatoglyphic grooves.16,17
Latticelike patterns also are characterized by brown parallel lines along the sulci of dermatoglyphics but additionally have multiple intersecting lines. Thus, this pattern can be considered a variant of the parallel furrow pattern.15 The crisscross markings can be predominantly found in the plantar arch.22 This dermoscopic pattern comprises 15% to 25% of all acral nevi.21
Fibrillar pattern accounts for 10% to 20% of all acral melanocytic nevi.21 Dermoscopically, these lesions demonstrate parallel filamentous streaks that cross dermatoglyphics obliquely. The fibrillar pattern is predominantly found on weight-bearing areas of the sole,22 which likely is explained by pressure causing slanting of melanin columns in the horny layer.23 The fibrillar pattern has been shown to be the benign acral dermoscopic pattern that is most commonly misdiagnosed, with higher reported rates of biopsy.24
Acral Congenital Melanocytic Nevi
Congenital melanocytic nevi (CMN) present at birth or appear during the first few weeks of life. Congenital melanocytic nevi can vary widely in size, shape, and color, and they are occasionally biopsied in cases of larger diameter or dermoscopic atypia to differentiate from melanoma.25 Congenital melanocytic nevi also can occur on acral volar surfaces. Possible dermoscopic patterns include parallel furrow or fibrillar patterns as well as a crista dotted pattern, defined as evenly spaced dots/globules on the ridges near the openings of eccrine ducts.26 A more commonly observed dermoscopic pattern in acral CMN is a combination of the crista dotted and parallel furrow patterns, known as the peas-in-a-pod pattern. Changes in the clinical appearance and dermoscopic features of an acral CMN are possible over time; some lesions also may fade with age.26
Final Thoughts
Acral lentiginous melanoma is a rare but potentially aggressive melanoma subtype that accounts for a larger proportion of melanomas in patients with skin of color than in white patients. Dermoscopy of acral volar skin provides invaluable diagnostic information and allows for better management of acral melanocytic lesions. Dermoscopic patterns such as the parallel ridge, parallel furrow, latticelike, fibrillar, and peas-in-a-pod patterns are unique to acral sites and can be used to differentiate between ALMs, acquired nevi, or CMNs.
Acral lentiginous melanoma (ALM) is a rare subtype of melanoma that occurs on the palms, soles, and nail apparatus. Unlike more common types of melanoma, ALM occurs on sun-protected areas of the skin and has distinct clinical, histologic, and genetic features. Acral lentiginous melanoma accounts for a larger proportion of melanomas in individuals with skin of color and has a worse prognosis and recurrence rate than other forms of melanoma.
Population Trends in Skin of Color
Much of the literature on malignant melanoma historically has involved non-Hispanic white patients, but the incidence in lighter-skinned populations has been increasing steadily over the last few decades.1 Although ALM can occur in any race, it disproportionately affects skin of color populations; ALM accounts for only 0.8% to 1% of all melanomas in white populations, but it constitutes 4% to 58% of melanomas in ethnic populations and is the most common melanoma subtype among black Americans.2-5 Acral lentiginous melanoma also is associated with a worse prognosis compared to other subtypes, which may indicate a more aggressive biological nature6 but also may point toward socioeconomic and cultural barriers (eg, low income or education levels, lack of insurance, lower health literacy), leading to disparities in access to care and diagnosis at advanced stages.5
Similarly, the distribution of acral melanocytic nevi appears to demonstrate an association with ethnicity and skin pigmentation. Although skin of color patients have fewer nevi than non-Hispanic whites, the proportion of acral melanocytic nevi tends to be greater.6,7 Given its grim prognosis, accurately differentiating ALM from acral nevi is of utmost importance.
Diagnostic Challenges of Acral Lesions
Due to the unique nature of the surfaces of acral sites, melanocytic lesions on the palms, soles, and nail apparatus present many diagnostic challenges. It can be difficult to distinguish acral melanoma from benign lesions using the naked eye alone. Volar surfaces are characterized by the presence of dermatoglyphics, and pigment deposition along ridges and furrows create particular dermoscopic patterns exclusive to these sites.8 Thus, dermoscopy can be useful on acral surfaces, but the dermoscopic features are different from those on the rest of the body and must be learned separately.
In addition, nearly half of patients are unaware of their acral lesions.6 Acral surfaces may not always be examined by clinicians during total-body skin examinations, leading to further possibility of overlooking a lesion. Obtaining biopsies on glabrous skin or nails also is challenging because they can be more painful and hemostasis can be more difficult, especially in the nail. Acral melanomas also may be amelanotic, including those at subungual sites.
Dermoscopic Patterns of Acral Volar Skin
Dermoscopy is a useful noninvasive tool for distinguishing between benign and malignant acral melanocytic lesions, and its efficacy in improving diagnostic accuracy and decreasing unnecessary biopsies is well-established in the literature.13,14 Acral dermoscopy allows for visualization of pigment along the dermatoglyphics that constitute the characteristic dermoscopic patterns.
Acral Lentiginous Melanoma
The hallmark dermoscopic pattern and most important finding of ALM is the parallel ridge pattern, characterized by parallel linear pigmentation along the ridges of dermatoglyphics. In the early phases of malignancy, the pattern appears light brown and involves most of the lesion; as the tumor develops, increasing melanin production results in focal areas of the parallel ridge pattern with darker bands.15,16 The sensitivity and specificity of a parallel ridge pattern for diagnosing early ALM has been shown to be 86% and 99%, respectively.15,16
A pattern of irregular diffuse pigmentation also can be observed in more advanced ALM. Dermoscopy may reveal a structureless pattern (ie, lack of identifiable structures or patterns) in a background of tan-black coloration due to more exuberant melanocyte proliferation along the epidermis.15 Sensitivity and specificity of this dermoscopic finding for invasive lesions is high at 94% and 97%, respectively.16,17 Interestingly, once ALM lesions have advanced even further, conventional melanoma-associated structures (ie, blue-white veil, polymorphous blood vessels, ulceration, irregular dots/globules or streaks) or atypical forms of typically benign acral dermoscopic patterns may be observed.15
Per a 3-step diagnostic algorithm created by Koga and Saida,18 a suspected acral lesion should first be evaluated for a parallel ridge pattern to determine the need for biopsy, as it is seen in approximately two-thirds of ALMs.19 If no parallel ridge pattern is observed, the lesion should then be checked for any of the typical dermoscopic patterns seen in benign acral nevi (eg, parallel furrow, latticelike, or fibrillar patterns).18 The maximum diameter should be measured only if the lesion does not exhibit any of the typical dermoscopic patterns. If the lesion’s diameter is greater than 7 mm in diameter, it should be biopsied; if the diameter is less than 7 mm, it should have regular clinical and dermoscopic follow-up.18
In 2015, Lallas et al20 developed the BRAAFF checklist, a scoring system of 6 variables: blotches, ridge pattern, asymmetry of structures, asymmetry of colors, parallel furrow pattern, and fibrillar pattern. The checklist also was shown to substantially improve diagnostic accuracy of dermoscopy for ALM, with sensitivity and specificity at 93.1% and 86.7%, respectively.20
Acquired Acral Nevi
Three classic dermoscopic patterns are associated with acquired acral nevi: parallel furrow pattern, latticelike pattern, and fibrillar pattern.15,21 Approximately three-quarters of all acquired acral nevi exhibit one of these patterns, roughly half exhibiting parallel furrow with tan-brown bandlike pigmentation along dermatoglyphic grooves.16,17
Latticelike patterns also are characterized by brown parallel lines along the sulci of dermatoglyphics but additionally have multiple intersecting lines. Thus, this pattern can be considered a variant of the parallel furrow pattern.15 The crisscross markings can be predominantly found in the plantar arch.22 This dermoscopic pattern comprises 15% to 25% of all acral nevi.21
Fibrillar pattern accounts for 10% to 20% of all acral melanocytic nevi.21 Dermoscopically, these lesions demonstrate parallel filamentous streaks that cross dermatoglyphics obliquely. The fibrillar pattern is predominantly found on weight-bearing areas of the sole,22 which likely is explained by pressure causing slanting of melanin columns in the horny layer.23 The fibrillar pattern has been shown to be the benign acral dermoscopic pattern that is most commonly misdiagnosed, with higher reported rates of biopsy.24
Acral Congenital Melanocytic Nevi
Congenital melanocytic nevi (CMN) present at birth or appear during the first few weeks of life. Congenital melanocytic nevi can vary widely in size, shape, and color, and they are occasionally biopsied in cases of larger diameter or dermoscopic atypia to differentiate from melanoma.25 Congenital melanocytic nevi also can occur on acral volar surfaces. Possible dermoscopic patterns include parallel furrow or fibrillar patterns as well as a crista dotted pattern, defined as evenly spaced dots/globules on the ridges near the openings of eccrine ducts.26 A more commonly observed dermoscopic pattern in acral CMN is a combination of the crista dotted and parallel furrow patterns, known as the peas-in-a-pod pattern. Changes in the clinical appearance and dermoscopic features of an acral CMN are possible over time; some lesions also may fade with age.26
Final Thoughts
Acral lentiginous melanoma is a rare but potentially aggressive melanoma subtype that accounts for a larger proportion of melanomas in patients with skin of color than in white patients. Dermoscopy of acral volar skin provides invaluable diagnostic information and allows for better management of acral melanocytic lesions. Dermoscopic patterns such as the parallel ridge, parallel furrow, latticelike, fibrillar, and peas-in-a-pod patterns are unique to acral sites and can be used to differentiate between ALMs, acquired nevi, or CMNs.
- Whiteman DC, Green AC, Olsen CM. The growing burden of invasive melanoma: projections of incidence rates and numbers of new cases in six susceptible populations through 2031. J Invest Dermatol. 2016;136:1161-1171.
- Bradford PT, Goldstein AM, McMaster ML, et al. Acral lentiginous melanoma: incidence and survival patterns in the United States, 1986-2005. Arch Dermatol. 2009;145:427-434.
- Nakamura Y, Fujisawa Y. Diagnosis and management of acral lentiginous melanoma. Curr Treat Options Oncol. 2018;19:42.
- Cormier JN, Xing Y, Ding M, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006;166:1907-1914.
- Wang Y, Zhao Y, Ma S. Racial differences in six major subtypes of melanoma: descriptive epidemiology. BMC Cancer. 2016;16:691.
- Madankumar R, Gumaste PV, Martires K, et al. Acral melanocytic lesions in the United States: prevalence, awareness, and dermoscopic patterns in skin-of-color and non-Hispanic white patients. J Am Acad Dermatol. 2016;74:724.e1-730.e1.
- Palicka GA, Rhodes AR. Acral melanocytic nevi: prevalence and distribution of gross morphologic features in white and black adults. Arch Dermatol. 2010;146:1085-1094.
- Thomas L, Phan A, Pralong P, et al. Special locations dermoscopy: facial, acral, and nail. Dermatol Clin. 2013;31:615-624.
- Gong HZ, Zheng HY, Li J. Amelanotic melanoma [published online January 21, 2019]. Melanoma Res. doi:10.1097/CMR.0000000000000571.
- Ise M, Yasuda F, Konohana I, et al. Acral melanoma with hyperkeratosis mimicking a pigmented wart. Dermatol Pract Concept. 2013;3:37-39.
- Serarslan G, Akçaly CM, Atik E. Acral lentiginous melanoma misdiagnosed as tinea pedis: a case report. Int J Dermatol. 2004;43:37-38.
- Gumaste P, Penn L, Cohen N, et al. Acral lentiginous melanoma of the foot misdiagnosed as a traumatic ulcer. a cautionary case. J Am Podiatr Med Assoc. 2015;105:189-194.
- Carli P, de Giorgi V, Chiarugi A, et al. Addition of dermoscopy to conventional naked-eye examination in melanoma screening: a randomized study. J Am Acad Dermatol. 2004;50:683-689.
- Carli P, de Giorgi V, Crocetti E, et al. Improvement of malignant/benign ratio in excised melanocytic lesions in the ‘dermoscopy era’: a retrospective study 1997-2001. Br J Dermatol. 2004;150:687-692.
- Saida T, Koga H, Uhara H. Key points in dermoscopic differentiation between early acral melanoma and acral nevus. J Dermatol. 2011;38:25-34.
- Ishihara Y, Saida T, Miyazaki A, et al. Early acral melanoma in situ: correlation between the parallel ridge pattern on dermoscopy and microscopic features. Am J Dermatopathol. 2006;28:21-27.
- Saida T, Miyazaki A, Oguchi S, et al. Significance of dermoscopic patterns in detecting malignant melanoma on acral volar skin: results of a multicenter study in Japan. Arch Dermatol. 2004;140:1233-1238.
- Koga H, Saida T. Revised 3-step dermoscopic algorithm for the management of acral melanocytic lesions. Arch Dermatol. 2011;147:741-743.
- Lallas A, Sgouros D, Zalaudek I, et al. Palmar and plantar melanomas differ for sex prevalence and tumor thickness but not for dermoscopic patterns. Melanoma Res. 2014;24:83-87.
- Lallas A, Kyrgidis A, Koga H, et al. The BRAAFF checklist: a new dermoscopic algorithm for diagnosing acral melanoma. Br J Dermatol. 2015;173:1041-1049.
- Saida T, Koga H. Dermoscopic patterns of acral melanocytic nevi: their variations, changes, and significance. Arch Dermatol. 2007;143:1423-1426.
- Miyazaki A, Saida T, Koga H, et al. Anatomical and histopathological correlates of the dermoscopic patterns seen in melanocytic nevi on the sole: a retrospective study. J Am Acad Dermatol. 2005;53:230-236.
- Watanabe S, Sawada M, Ishizaki S, et al. Comparison of dermatoscopic images of acral lentiginous melanoma and acral melanocytic nevus occurring on body weight-bearing areas. Dermatol Pract Concept. 2014;4:47-50.
- Costello CM, Ghanavatian S, Temkit M, et al. Educational and practice gaps in the management of volar melanocytic lesions. J Eur Acad Dermatol Venereol. 2018;32:1450-1455.
- Alikhan A, Ibrahimi OA, Eisen DB. Congenital melanocytic nevi: where are we now? part I. clinical presentation, epidemiology, pathogenesis, histology, malignant transformation, and neurocutaneous melanosis. J Am Acad Dermatol. 2012;67:495.e1-495.e17; quiz 512-514.
- Minagawa A, Koga H, Saida T. Dermoscopic characteristics of congenital melanocytic nevi affecting acral volar skin. Arch Dermatol. 2011;147:809-813.
- Whiteman DC, Green AC, Olsen CM. The growing burden of invasive melanoma: projections of incidence rates and numbers of new cases in six susceptible populations through 2031. J Invest Dermatol. 2016;136:1161-1171.
- Bradford PT, Goldstein AM, McMaster ML, et al. Acral lentiginous melanoma: incidence and survival patterns in the United States, 1986-2005. Arch Dermatol. 2009;145:427-434.
- Nakamura Y, Fujisawa Y. Diagnosis and management of acral lentiginous melanoma. Curr Treat Options Oncol. 2018;19:42.
- Cormier JN, Xing Y, Ding M, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006;166:1907-1914.
- Wang Y, Zhao Y, Ma S. Racial differences in six major subtypes of melanoma: descriptive epidemiology. BMC Cancer. 2016;16:691.
- Madankumar R, Gumaste PV, Martires K, et al. Acral melanocytic lesions in the United States: prevalence, awareness, and dermoscopic patterns in skin-of-color and non-Hispanic white patients. J Am Acad Dermatol. 2016;74:724.e1-730.e1.
- Palicka GA, Rhodes AR. Acral melanocytic nevi: prevalence and distribution of gross morphologic features in white and black adults. Arch Dermatol. 2010;146:1085-1094.
- Thomas L, Phan A, Pralong P, et al. Special locations dermoscopy: facial, acral, and nail. Dermatol Clin. 2013;31:615-624.
- Gong HZ, Zheng HY, Li J. Amelanotic melanoma [published online January 21, 2019]. Melanoma Res. doi:10.1097/CMR.0000000000000571.
- Ise M, Yasuda F, Konohana I, et al. Acral melanoma with hyperkeratosis mimicking a pigmented wart. Dermatol Pract Concept. 2013;3:37-39.
- Serarslan G, Akçaly CM, Atik E. Acral lentiginous melanoma misdiagnosed as tinea pedis: a case report. Int J Dermatol. 2004;43:37-38.
- Gumaste P, Penn L, Cohen N, et al. Acral lentiginous melanoma of the foot misdiagnosed as a traumatic ulcer. a cautionary case. J Am Podiatr Med Assoc. 2015;105:189-194.
- Carli P, de Giorgi V, Chiarugi A, et al. Addition of dermoscopy to conventional naked-eye examination in melanoma screening: a randomized study. J Am Acad Dermatol. 2004;50:683-689.
- Carli P, de Giorgi V, Crocetti E, et al. Improvement of malignant/benign ratio in excised melanocytic lesions in the ‘dermoscopy era’: a retrospective study 1997-2001. Br J Dermatol. 2004;150:687-692.
- Saida T, Koga H, Uhara H. Key points in dermoscopic differentiation between early acral melanoma and acral nevus. J Dermatol. 2011;38:25-34.
- Ishihara Y, Saida T, Miyazaki A, et al. Early acral melanoma in situ: correlation between the parallel ridge pattern on dermoscopy and microscopic features. Am J Dermatopathol. 2006;28:21-27.
- Saida T, Miyazaki A, Oguchi S, et al. Significance of dermoscopic patterns in detecting malignant melanoma on acral volar skin: results of a multicenter study in Japan. Arch Dermatol. 2004;140:1233-1238.
- Koga H, Saida T. Revised 3-step dermoscopic algorithm for the management of acral melanocytic lesions. Arch Dermatol. 2011;147:741-743.
- Lallas A, Sgouros D, Zalaudek I, et al. Palmar and plantar melanomas differ for sex prevalence and tumor thickness but not for dermoscopic patterns. Melanoma Res. 2014;24:83-87.
- Lallas A, Kyrgidis A, Koga H, et al. The BRAAFF checklist: a new dermoscopic algorithm for diagnosing acral melanoma. Br J Dermatol. 2015;173:1041-1049.
- Saida T, Koga H. Dermoscopic patterns of acral melanocytic nevi: their variations, changes, and significance. Arch Dermatol. 2007;143:1423-1426.
- Miyazaki A, Saida T, Koga H, et al. Anatomical and histopathological correlates of the dermoscopic patterns seen in melanocytic nevi on the sole: a retrospective study. J Am Acad Dermatol. 2005;53:230-236.
- Watanabe S, Sawada M, Ishizaki S, et al. Comparison of dermatoscopic images of acral lentiginous melanoma and acral melanocytic nevus occurring on body weight-bearing areas. Dermatol Pract Concept. 2014;4:47-50.
- Costello CM, Ghanavatian S, Temkit M, et al. Educational and practice gaps in the management of volar melanocytic lesions. J Eur Acad Dermatol Venereol. 2018;32:1450-1455.
- Alikhan A, Ibrahimi OA, Eisen DB. Congenital melanocytic nevi: where are we now? part I. clinical presentation, epidemiology, pathogenesis, histology, malignant transformation, and neurocutaneous melanosis. J Am Acad Dermatol. 2012;67:495.e1-495.e17; quiz 512-514.
- Minagawa A, Koga H, Saida T. Dermoscopic characteristics of congenital melanocytic nevi affecting acral volar skin. Arch Dermatol. 2011;147:809-813.
Practice Points
- Dermatologists should be familiar with common dermoscopic patterns seen at acral sites in patients with skin of color as well as the most up-to-date diagnostic algorithms.
- Acral lentiginous melanoma should be strongly suspected if dermoscopy reveals a parallel ridge pattern or if dermoscopy of volar skin reveals a lack of typical dermoscopic patterns in lesions with a diameter greater than 7 mm.
The Dayanara Effect: Increasing Skin Cancer Awareness in the Hispanic Community
In February 2019, Dayanara Torres announced that she had been diagnosed with metastatic melanoma. Ms. Torres, a Puerto Rican–born former Miss Universe who has more than 1 million followers on Instagram (@dayanarapr), seemed an unlikely candidate for skin cancer, which often is associated with fair-skinned and light-eyed individuals. She shared the news of her diagnosis in an Instagram video that has now received more than 850,000 views. In the video, Ms. Torres described a new mole with uneven surface that had developed on her leg and noted that she had ignored it, even though it had been growing for years. Ultimately, she was diagnosed with melanoma that had already metastasized to regional lymph nodes in her leg. Ms. Torres concluded the video by urging fans and viewers to be mindful of new or changing skin lesions and to be aware of the seriousness of skin cancer. In March 2019, Ms. Torres posted a follow-up educational video on Instagram highlighting the features of melanoma that has now received more than 300,000 views.
Since her announcement, we have noticed that more Hispanic patients with concerns about skin cancer are presenting to our dermatology clinic, which is located in a highly diverse city (New Brunswick, New Jersey) with approximately 50% of residents identifying as Hispanic.1 Most Hispanic patients typically present to our dermatology clinic for non–skin cancer–related concerns, such as acne, rash, and dyschromia; however, following Ms. Torres’ announcement, many have cited her diagnosis of metastatic melanoma as a cause for concern and a motivating factor in having their skin examined. The diagnosis in a prominent celebrity and Hispanic woman has given a new face to metastatic melanoma.
Although melanoma most commonly occurs in white patients, Hispanic patients experience disproportionately greater morbidity and mortality when diagnosed with melanoma.2 Poor prognosis in patients with skin of color is multifactorial and may be due to poor use of sun protection, misconceptions about melanoma risk, atypical clinical presentation, impaired access to care, and delay in diagnosis. The Hispanic community encompasses a wide variety of individuals with varying levels of skin pigmentation and sun sensitivity.3 However, Hispanics report low levels of sun-protective behaviors. They also may have misconceptions that sunscreen is ineffective in preventing skin cancer and that little can be done to decrease the risk for developing skin cancer.4,5 Additionally, Hispanic patients often have lower perceptions of their personal risk for melanoma and report low rates of clinical and self-examinations compared to non-Hispanic white patients.6-8 Many Hispanic patients have reported that they were not instructed to perform self-examinations of their skin regularly by dermatologists or other providers and did not know the signs of skin cancer.7 Furthermore, a language barrier also may impede communication and education regarding melanoma risk.9
Similar to white patients, superficial spreading melanoma is the most common histologic subtype in Hispanic patients, followed by acral lentiginous melanoma, which is the most common subtype in black and Asian patients.2,4 Compared to non-Hispanic white patients, who most commonly present with truncal melanomas, Hispanic patients (particularly those from Puerto Rico, such as Ms. Torres) are more likely to present with melanoma on the lower extremities.4,10 Additionally, Hispanic patients have high rates of head, neck, and mucosal melanomas compared to all other racial and ethnic groups.2
Hispanic patients diagnosed with melanoma are more likely to present with thicker primary tumors, later stages of disease, and distant metastases compared to non-Hispanic white patients, all of which are associated with poor prognosis.2,4,11 Five-year survival rates for melanoma are lower in Hispanic patients compared to non-Hispanic white patients.12 Although the Hispanic community is diverse in socioeconomic and immigration status as well as occupation, lack of insurance also may contribute to decreased access to care, delayed diagnosis, and ultimately worse survival.
These disparities have spurred suggestions for increased education about skin cancer and the signs and symptoms of melanoma, encouragement of self-examinations, and routine clinical skin examinations for Hispanic patients by dermatologists and other providers.8 There is evidence that knowledge-based interventions, especially when presented in Spanish, produce statistically significant improvements in knowledge of skin cancer risk and sun-protective behavior among Hispanic patients.12 Similarly, we have observed that the videos shared by Ms. Torres regarding her melanoma diagnosis and the features of melanoma, in which she spoke in Spanish, have compelled many Hispanic patients to examine their own skin and have led to increased concern for skin cancer in this patient population. In our practice, we refer to the increase in spot checks and skin examinations requested by Hispanic patients as “The Dayanara Effect,” and we hypothesize that this same effect may be taking place throughout the dermatology community.
- New Brunswick, NJ. Data USA website. https://datausa.io/profile/geo/new-brunswick-nj. Accessed April 17, 2019.
- Higgins S, Nazemi A, Feinstein S, et al. Clinical presentations of melanoma in African Americans, Hispanics, and Asians [published online January 4, 2019]. Dermatol Surg. doi:10.1097/dss.0000000000001759.
- Robinson JK, Penedo FJ, Hay JL, et al. Recognizing Latinos’ range of skin pigment and phototypes to enhance skin cancer prevention [published online July 4, 2017]. Pigment Cell Melanoma Res. 2017;30:488-492.
- Garnett E, Townsend J, Steele B, et al. Characteristics, rates, and trends of melanoma incidence among Hispanics in the USA. Cancer Causes Control. 2016;27:647-659.
- Agbai ON, Buster K, Sanchez M, et al. Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol. 2014;70:748-762.
- Andreeva VA, Cockburn MG. Cutaneous melanoma and other skin cancer screening among Hispanics in the United States: a review of the evidence, disparities, and need for expanding the intervention and research agendas. Arch Dermatol. 2011;147:743-745.
- Roman C, Lugo-Somolinos A, Thomas N. Skin cancer knowledge and skin self-examinations in the Hispanic population of North Carolina: the patient’s perspective. JAMA Dermatol. 2013;149:103-104.
- Jaimes N, Oliveria S, Halpern A. A cautionary note on melanoma screening in the Hispanic/Latino population. JAMA Dermatol. 2013;149:396-397.
- Wich LG, Ma MW, Price LS, et al. Impact of socioeconomic status and sociodemographic factors on melanoma presentation among ethnic minorities. J Community Health. 2011;36:461-468.
- Rouhani P, Hu S, Kirsner RS. Melanoma in Hispanic and black Americans. Cancer Control. 2008;15:248-253.
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991.
- Kailas A, Botwin AL, Pritchett EN, et al. Assessing the effectiveness of knowledge-based interventions in increasing skin cancer awareness, knowledge, and protective behaviors in skin of color populations. Cutis. 2017;100:235-240.
In February 2019, Dayanara Torres announced that she had been diagnosed with metastatic melanoma. Ms. Torres, a Puerto Rican–born former Miss Universe who has more than 1 million followers on Instagram (@dayanarapr), seemed an unlikely candidate for skin cancer, which often is associated with fair-skinned and light-eyed individuals. She shared the news of her diagnosis in an Instagram video that has now received more than 850,000 views. In the video, Ms. Torres described a new mole with uneven surface that had developed on her leg and noted that she had ignored it, even though it had been growing for years. Ultimately, she was diagnosed with melanoma that had already metastasized to regional lymph nodes in her leg. Ms. Torres concluded the video by urging fans and viewers to be mindful of new or changing skin lesions and to be aware of the seriousness of skin cancer. In March 2019, Ms. Torres posted a follow-up educational video on Instagram highlighting the features of melanoma that has now received more than 300,000 views.
Since her announcement, we have noticed that more Hispanic patients with concerns about skin cancer are presenting to our dermatology clinic, which is located in a highly diverse city (New Brunswick, New Jersey) with approximately 50% of residents identifying as Hispanic.1 Most Hispanic patients typically present to our dermatology clinic for non–skin cancer–related concerns, such as acne, rash, and dyschromia; however, following Ms. Torres’ announcement, many have cited her diagnosis of metastatic melanoma as a cause for concern and a motivating factor in having their skin examined. The diagnosis in a prominent celebrity and Hispanic woman has given a new face to metastatic melanoma.
Although melanoma most commonly occurs in white patients, Hispanic patients experience disproportionately greater morbidity and mortality when diagnosed with melanoma.2 Poor prognosis in patients with skin of color is multifactorial and may be due to poor use of sun protection, misconceptions about melanoma risk, atypical clinical presentation, impaired access to care, and delay in diagnosis. The Hispanic community encompasses a wide variety of individuals with varying levels of skin pigmentation and sun sensitivity.3 However, Hispanics report low levels of sun-protective behaviors. They also may have misconceptions that sunscreen is ineffective in preventing skin cancer and that little can be done to decrease the risk for developing skin cancer.4,5 Additionally, Hispanic patients often have lower perceptions of their personal risk for melanoma and report low rates of clinical and self-examinations compared to non-Hispanic white patients.6-8 Many Hispanic patients have reported that they were not instructed to perform self-examinations of their skin regularly by dermatologists or other providers and did not know the signs of skin cancer.7 Furthermore, a language barrier also may impede communication and education regarding melanoma risk.9
Similar to white patients, superficial spreading melanoma is the most common histologic subtype in Hispanic patients, followed by acral lentiginous melanoma, which is the most common subtype in black and Asian patients.2,4 Compared to non-Hispanic white patients, who most commonly present with truncal melanomas, Hispanic patients (particularly those from Puerto Rico, such as Ms. Torres) are more likely to present with melanoma on the lower extremities.4,10 Additionally, Hispanic patients have high rates of head, neck, and mucosal melanomas compared to all other racial and ethnic groups.2
Hispanic patients diagnosed with melanoma are more likely to present with thicker primary tumors, later stages of disease, and distant metastases compared to non-Hispanic white patients, all of which are associated with poor prognosis.2,4,11 Five-year survival rates for melanoma are lower in Hispanic patients compared to non-Hispanic white patients.12 Although the Hispanic community is diverse in socioeconomic and immigration status as well as occupation, lack of insurance also may contribute to decreased access to care, delayed diagnosis, and ultimately worse survival.
These disparities have spurred suggestions for increased education about skin cancer and the signs and symptoms of melanoma, encouragement of self-examinations, and routine clinical skin examinations for Hispanic patients by dermatologists and other providers.8 There is evidence that knowledge-based interventions, especially when presented in Spanish, produce statistically significant improvements in knowledge of skin cancer risk and sun-protective behavior among Hispanic patients.12 Similarly, we have observed that the videos shared by Ms. Torres regarding her melanoma diagnosis and the features of melanoma, in which she spoke in Spanish, have compelled many Hispanic patients to examine their own skin and have led to increased concern for skin cancer in this patient population. In our practice, we refer to the increase in spot checks and skin examinations requested by Hispanic patients as “The Dayanara Effect,” and we hypothesize that this same effect may be taking place throughout the dermatology community.
In February 2019, Dayanara Torres announced that she had been diagnosed with metastatic melanoma. Ms. Torres, a Puerto Rican–born former Miss Universe who has more than 1 million followers on Instagram (@dayanarapr), seemed an unlikely candidate for skin cancer, which often is associated with fair-skinned and light-eyed individuals. She shared the news of her diagnosis in an Instagram video that has now received more than 850,000 views. In the video, Ms. Torres described a new mole with uneven surface that had developed on her leg and noted that she had ignored it, even though it had been growing for years. Ultimately, she was diagnosed with melanoma that had already metastasized to regional lymph nodes in her leg. Ms. Torres concluded the video by urging fans and viewers to be mindful of new or changing skin lesions and to be aware of the seriousness of skin cancer. In March 2019, Ms. Torres posted a follow-up educational video on Instagram highlighting the features of melanoma that has now received more than 300,000 views.
Since her announcement, we have noticed that more Hispanic patients with concerns about skin cancer are presenting to our dermatology clinic, which is located in a highly diverse city (New Brunswick, New Jersey) with approximately 50% of residents identifying as Hispanic.1 Most Hispanic patients typically present to our dermatology clinic for non–skin cancer–related concerns, such as acne, rash, and dyschromia; however, following Ms. Torres’ announcement, many have cited her diagnosis of metastatic melanoma as a cause for concern and a motivating factor in having their skin examined. The diagnosis in a prominent celebrity and Hispanic woman has given a new face to metastatic melanoma.
Although melanoma most commonly occurs in white patients, Hispanic patients experience disproportionately greater morbidity and mortality when diagnosed with melanoma.2 Poor prognosis in patients with skin of color is multifactorial and may be due to poor use of sun protection, misconceptions about melanoma risk, atypical clinical presentation, impaired access to care, and delay in diagnosis. The Hispanic community encompasses a wide variety of individuals with varying levels of skin pigmentation and sun sensitivity.3 However, Hispanics report low levels of sun-protective behaviors. They also may have misconceptions that sunscreen is ineffective in preventing skin cancer and that little can be done to decrease the risk for developing skin cancer.4,5 Additionally, Hispanic patients often have lower perceptions of their personal risk for melanoma and report low rates of clinical and self-examinations compared to non-Hispanic white patients.6-8 Many Hispanic patients have reported that they were not instructed to perform self-examinations of their skin regularly by dermatologists or other providers and did not know the signs of skin cancer.7 Furthermore, a language barrier also may impede communication and education regarding melanoma risk.9
Similar to white patients, superficial spreading melanoma is the most common histologic subtype in Hispanic patients, followed by acral lentiginous melanoma, which is the most common subtype in black and Asian patients.2,4 Compared to non-Hispanic white patients, who most commonly present with truncal melanomas, Hispanic patients (particularly those from Puerto Rico, such as Ms. Torres) are more likely to present with melanoma on the lower extremities.4,10 Additionally, Hispanic patients have high rates of head, neck, and mucosal melanomas compared to all other racial and ethnic groups.2
Hispanic patients diagnosed with melanoma are more likely to present with thicker primary tumors, later stages of disease, and distant metastases compared to non-Hispanic white patients, all of which are associated with poor prognosis.2,4,11 Five-year survival rates for melanoma are lower in Hispanic patients compared to non-Hispanic white patients.12 Although the Hispanic community is diverse in socioeconomic and immigration status as well as occupation, lack of insurance also may contribute to decreased access to care, delayed diagnosis, and ultimately worse survival.
These disparities have spurred suggestions for increased education about skin cancer and the signs and symptoms of melanoma, encouragement of self-examinations, and routine clinical skin examinations for Hispanic patients by dermatologists and other providers.8 There is evidence that knowledge-based interventions, especially when presented in Spanish, produce statistically significant improvements in knowledge of skin cancer risk and sun-protective behavior among Hispanic patients.12 Similarly, we have observed that the videos shared by Ms. Torres regarding her melanoma diagnosis and the features of melanoma, in which she spoke in Spanish, have compelled many Hispanic patients to examine their own skin and have led to increased concern for skin cancer in this patient population. In our practice, we refer to the increase in spot checks and skin examinations requested by Hispanic patients as “The Dayanara Effect,” and we hypothesize that this same effect may be taking place throughout the dermatology community.
- New Brunswick, NJ. Data USA website. https://datausa.io/profile/geo/new-brunswick-nj. Accessed April 17, 2019.
- Higgins S, Nazemi A, Feinstein S, et al. Clinical presentations of melanoma in African Americans, Hispanics, and Asians [published online January 4, 2019]. Dermatol Surg. doi:10.1097/dss.0000000000001759.
- Robinson JK, Penedo FJ, Hay JL, et al. Recognizing Latinos’ range of skin pigment and phototypes to enhance skin cancer prevention [published online July 4, 2017]. Pigment Cell Melanoma Res. 2017;30:488-492.
- Garnett E, Townsend J, Steele B, et al. Characteristics, rates, and trends of melanoma incidence among Hispanics in the USA. Cancer Causes Control. 2016;27:647-659.
- Agbai ON, Buster K, Sanchez M, et al. Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol. 2014;70:748-762.
- Andreeva VA, Cockburn MG. Cutaneous melanoma and other skin cancer screening among Hispanics in the United States: a review of the evidence, disparities, and need for expanding the intervention and research agendas. Arch Dermatol. 2011;147:743-745.
- Roman C, Lugo-Somolinos A, Thomas N. Skin cancer knowledge and skin self-examinations in the Hispanic population of North Carolina: the patient’s perspective. JAMA Dermatol. 2013;149:103-104.
- Jaimes N, Oliveria S, Halpern A. A cautionary note on melanoma screening in the Hispanic/Latino population. JAMA Dermatol. 2013;149:396-397.
- Wich LG, Ma MW, Price LS, et al. Impact of socioeconomic status and sociodemographic factors on melanoma presentation among ethnic minorities. J Community Health. 2011;36:461-468.
- Rouhani P, Hu S, Kirsner RS. Melanoma in Hispanic and black Americans. Cancer Control. 2008;15:248-253.
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991.
- Kailas A, Botwin AL, Pritchett EN, et al. Assessing the effectiveness of knowledge-based interventions in increasing skin cancer awareness, knowledge, and protective behaviors in skin of color populations. Cutis. 2017;100:235-240.
- New Brunswick, NJ. Data USA website. https://datausa.io/profile/geo/new-brunswick-nj. Accessed April 17, 2019.
- Higgins S, Nazemi A, Feinstein S, et al. Clinical presentations of melanoma in African Americans, Hispanics, and Asians [published online January 4, 2019]. Dermatol Surg. doi:10.1097/dss.0000000000001759.
- Robinson JK, Penedo FJ, Hay JL, et al. Recognizing Latinos’ range of skin pigment and phototypes to enhance skin cancer prevention [published online July 4, 2017]. Pigment Cell Melanoma Res. 2017;30:488-492.
- Garnett E, Townsend J, Steele B, et al. Characteristics, rates, and trends of melanoma incidence among Hispanics in the USA. Cancer Causes Control. 2016;27:647-659.
- Agbai ON, Buster K, Sanchez M, et al. Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol. 2014;70:748-762.
- Andreeva VA, Cockburn MG. Cutaneous melanoma and other skin cancer screening among Hispanics in the United States: a review of the evidence, disparities, and need for expanding the intervention and research agendas. Arch Dermatol. 2011;147:743-745.
- Roman C, Lugo-Somolinos A, Thomas N. Skin cancer knowledge and skin self-examinations in the Hispanic population of North Carolina: the patient’s perspective. JAMA Dermatol. 2013;149:103-104.
- Jaimes N, Oliveria S, Halpern A. A cautionary note on melanoma screening in the Hispanic/Latino population. JAMA Dermatol. 2013;149:396-397.
- Wich LG, Ma MW, Price LS, et al. Impact of socioeconomic status and sociodemographic factors on melanoma presentation among ethnic minorities. J Community Health. 2011;36:461-468.
- Rouhani P, Hu S, Kirsner RS. Melanoma in Hispanic and black Americans. Cancer Control. 2008;15:248-253.
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991.
- Kailas A, Botwin AL, Pritchett EN, et al. Assessing the effectiveness of knowledge-based interventions in increasing skin cancer awareness, knowledge, and protective behaviors in skin of color populations. Cutis. 2017;100:235-240.
Systemic therapies are impacting melanoma’s prognostic factors
As systemic therapies for advanced melanoma increase, some historical prognostic factors continue to hold true while refined and novel risk factors are emerging, according to the results of three studies published in JAMA Dermatology.
Among the most prominent findings of those studies are that ulceration, mitotic index, and head and neck location in localized disease were predictive of early recurrence; time to recurrence was not associated with survival in unresectable stage IV melanoma; and certain gene markers may be linked with particular types of metastasis.
The first study, conducted by Lena A. von Schuckmann, MBBS, of the University of Queensland School of Public Health in Australia, and colleagues, evaluated the risk of early melanoma recurrence in patients with localized disease.
“With the introduction of targeted and immune therapies for treatment of metastatic melanoma, including possible adjuvant therapy, a detailed understanding of the risk of melanoma recurrence may assist clinicians to advise patients with a primary tumor at high risk of disease metastasis,” the researchers wrote (JAMA Dermatol. 2019 May 1. doi: 10.1001/jamadermatol.2019.0440).
They conducted a prospective cohort study of 700 patients with high-risk, category T1b to T4b cutaneous melanoma, refined from an initial recruitment population of 1,254 individuals. Using self-administered patient questionnaires in conjunction with histologic, imaging, and clinical data over the course of 2 years, the investigators looked for factors that predicted recurrence.
Of 700 patients, 94 (13.4%) had disease recurrence, most often (70.2%) locoregional recurrence. Independent predictors of recurrence included mitotic rate greater than 3/mm2, thickness, ulceration, and primary tumor location on the head or neck.
Patients with negative single lymph node biopsy (SLNB) were less likely to have recurrence than were those who did not undergo SLNB. Among 64 patients whose locoregional disease was excised, 37 (57.8%) were disease free at 2 years, whereas 7 patients (10.9%) had new locoregional disease and 20 patients (31.3%) developed a new distant recurrence.
“[O]ur data appear to support the recommendation for careful scar and regional skin and lymph node examination during patient follow-up,” the investigators concluded, alluding to the relatively high rate of locoregional recurrence. “Subsequent recurrences occurring at distant sites were more likely to involve multiple organs, which is consistent with other studies.”
The second melanoma article, investigating associations between time to relapse and survival, was authored by Anaïs Vallet, MD, of Hôpital Saint-Louis, Paris, and colleagues.
“Although the kinetics of metastatic disease seem to be correlated with patient survival, the first relapse is not predictable, and data from the literature on the topic are controversial,” they wrote (JAMA Dermatol. 2019 May 1. doi: 10.1001/jamadermatol.2019.0425). “We hypothesized that the progression of the metastatic disease would be associated with the time from primary excision to the first distant recurrence of melanoma.”
To test this hypothesis, the investigators analyzed data from 638 patients with unresectable stage III or IV melanoma. Inclusion required first-line treatment with chemotherapy, targeted therapies, or immunotherapies. The interval between primary excision and distant disease recurrence, measured as a categorical and continuous variable, was compared with overall survival and progression-free survival. The analysis revealed no associations between time to recurrence and either survival measure, even when stratified by treatment.
“Now that immunotherapies and targeted therapies have been approved in the adjuvant setting for patients with stage III disease, it would be interesting to analyze recurrence-free survival and [progression-free survival] in relapsing patients who previously received adjuvant therapies,” the investigators wrote.
The third study was conducted by Laura Calomarde-Rees, MD, of Instituto Valenciano de Oncología, València, Spain, and colleagues.
“Our aim was to identify risk factors associated with lymphatic (locoregional metastasis) or hematogenous (distant metastasis) progression because these have not been studied separately to date in patients with localized melanoma,” they wrote (JAMA Dermatol. 2019 May 1. doi: 10.1001/jamadermatol.2019.0069).
The retrospective study involved 1,177 patients with stage I to II melanoma. Multiple disease variables were evaluated in the context of each type of metastasis, including age, sex, tumor location, and others.
The investigators found locoregional spread was most often associated with vascular invasion (hazard ratio [HR], 3.2), greater Breslow thickness (HR, 5.4; thickness greater than 4 mm), acral location (HR, 2.4), head/neck location (HR, 1.7), and age greater than 55 years (HR, 1.9).
Distant metastasis was most often associated with greater Breslow thickness (HR, 10.4; thickness greater than 4 mm), TERT promoter mutations (HR, 2.9), BRAF mutations (HR, 1.9), and absence of regression (HR, 0.1).
“Risk factors for lymphatic and hematogenous metastasis differ,” the investigators concluded. “A greater understanding of the clinical, histopathologic, and molecular factors involved could help to identify patients with an increased risk of recurrence and guide the design of individualized follow-up programs and adjuvant targeted therapies.”
Dr. von Schuckmann and colleagues disclosed study funding from the National Health and Medical Research Council and other relationships with the Norwegian Cancer Society project. Dr. Vallet and colleagues reported study support from French National Cancer Institute, MSD, BMS, Roche, and Novartis; and additional relationships with Incyte, Amgen, Pfizer, and others. Dr. Calomarde-Rees and colleagues disclosed no conflicts of interest.
Effective systemic treatments have forever altered the kinetics of events in advanced melanoma, with the natural history outcome curves of this cancer now highly dependent on treatment response. The relapse-free survival (RFS) curves of patients with stage IIC/III melanoma with and without adjuvant dabrafenib/trametinib demonstrate the contemporary natural history of frequent early recurrences with few late events in the placebo groups and of fewer and later recurrences, with an as yet unknown frequency of late events, in the treated groups.
The three reported studies examine melanoma disease kinetics and speak to this new paradigm.
The first study, conducted by von Schuckmann and colleagues, looked for factors associated with disease recurrence within 2 years of treatment for localized, T1b to T4b melanoma.
Factors were not surprising and included the presence of ulceration, an increased mitotic index, increasing T stage, and location on the head and neck. Also, the study had two major limitations. First, the patient population was a heterogenous one – 442 patients with clinical T1b to T4b did not undergo sentinel lymph node biopsy (SLNB), 213 patients with pathologic T1b to T4b had a negative SLNB result, and 38 patients had at least stage IIIa disease after a positive SLNB finding. Second, as the 8th edition of the AJCC [American Joint Committee on Cancer] Staging Manual notes, most events in this cohort of patients are expected to occur after the 2-year threshold described in this prospective study and the disease can recur as much as 10 years after effective treatment.
The second study, conducted by Vallet and colleagues, analyzed data from patients with unresectable stage IV melanoma to determine if time to distant recurrence after excision of antecedent primary melanoma was associated with survival.
Time to first distant metastasis was not related to Breslow thickness or to tumor stage at the start of therapy; however, the data analysis did not include multiple patient variables that might have shed more light on predictive factors.
As systemic therapy increasingly leads to stabilization of previously progressive disease as well as durable complete remissions in melanoma patients, best response to immunotherapy or targeted therapy is likely to emerge as a much more important predictor than the time it took for stage IV melanoma to become apparent. There has been some thoughtful interest in exploring the prognostic value of establishing the kinetics of stage IV melanoma as a prognostic factor prior to initiating therapy, but there has been limited uptake of this approach by the melanoma oncology community.
In the third study, Calomarde-Rees and colleagues explored associations between melanoma disease characteristics and hematogenous or lymphatic metastases.
The anticipated findings include the observed association between tumor thickness and risk of recurrence, either lymphatic or hematogenous. As satellitosis is already considered a criterion for stage III in AJCC8, these two observations serve as internal controls that validate the credibility of the data set.
The researchers’ most intriguing finding is the strong potential association of the combination of both MAPK (either BRAF or NRAS) and TERT promoter mutations with poor survival, as demonstrated in their second prognostic model (hazard ratio, 5.7). This finding warrants cautious interpretation as the authors clearly acknowledge that a minority of the study population underwent detailed assessments of the mutation status of each gene.
While the biologic behavior of melanoma is likely to be much more complex than the mutation of one or two genes, the potential interaction of the mutated TERT promoter gene is provocative, especially in the context of a recent article suggesting a role for monitoring BRAF and TERT circulating free DNA as an indicator of response to systemic therapy and outcome.
Multigene panels are being developed, but it remains to be demonstrated whether any of these highly discrepant gene profiles will outperform optimized contemporary multivariable individual patient risk prediction models across the prognostic spectrum of melanoma.
Daniel G. Coit, MD is a surgical oncologist at Memorial Sloan Kettering Cancer Center in New York. He made his remarks in an editorial in JAMA Dermatology (2019 May 1. doi: 10.1001/jamadermatol.2019.0200). Dr. Coit disclosed that he has no relevant financial conflicts of interest.
Effective systemic treatments have forever altered the kinetics of events in advanced melanoma, with the natural history outcome curves of this cancer now highly dependent on treatment response. The relapse-free survival (RFS) curves of patients with stage IIC/III melanoma with and without adjuvant dabrafenib/trametinib demonstrate the contemporary natural history of frequent early recurrences with few late events in the placebo groups and of fewer and later recurrences, with an as yet unknown frequency of late events, in the treated groups.
The three reported studies examine melanoma disease kinetics and speak to this new paradigm.
The first study, conducted by von Schuckmann and colleagues, looked for factors associated with disease recurrence within 2 years of treatment for localized, T1b to T4b melanoma.
Factors were not surprising and included the presence of ulceration, an increased mitotic index, increasing T stage, and location on the head and neck. Also, the study had two major limitations. First, the patient population was a heterogenous one – 442 patients with clinical T1b to T4b did not undergo sentinel lymph node biopsy (SLNB), 213 patients with pathologic T1b to T4b had a negative SLNB result, and 38 patients had at least stage IIIa disease after a positive SLNB finding. Second, as the 8th edition of the AJCC [American Joint Committee on Cancer] Staging Manual notes, most events in this cohort of patients are expected to occur after the 2-year threshold described in this prospective study and the disease can recur as much as 10 years after effective treatment.
The second study, conducted by Vallet and colleagues, analyzed data from patients with unresectable stage IV melanoma to determine if time to distant recurrence after excision of antecedent primary melanoma was associated with survival.
Time to first distant metastasis was not related to Breslow thickness or to tumor stage at the start of therapy; however, the data analysis did not include multiple patient variables that might have shed more light on predictive factors.
As systemic therapy increasingly leads to stabilization of previously progressive disease as well as durable complete remissions in melanoma patients, best response to immunotherapy or targeted therapy is likely to emerge as a much more important predictor than the time it took for stage IV melanoma to become apparent. There has been some thoughtful interest in exploring the prognostic value of establishing the kinetics of stage IV melanoma as a prognostic factor prior to initiating therapy, but there has been limited uptake of this approach by the melanoma oncology community.
In the third study, Calomarde-Rees and colleagues explored associations between melanoma disease characteristics and hematogenous or lymphatic metastases.
The anticipated findings include the observed association between tumor thickness and risk of recurrence, either lymphatic or hematogenous. As satellitosis is already considered a criterion for stage III in AJCC8, these two observations serve as internal controls that validate the credibility of the data set.
The researchers’ most intriguing finding is the strong potential association of the combination of both MAPK (either BRAF or NRAS) and TERT promoter mutations with poor survival, as demonstrated in their second prognostic model (hazard ratio, 5.7). This finding warrants cautious interpretation as the authors clearly acknowledge that a minority of the study population underwent detailed assessments of the mutation status of each gene.
While the biologic behavior of melanoma is likely to be much more complex than the mutation of one or two genes, the potential interaction of the mutated TERT promoter gene is provocative, especially in the context of a recent article suggesting a role for monitoring BRAF and TERT circulating free DNA as an indicator of response to systemic therapy and outcome.
Multigene panels are being developed, but it remains to be demonstrated whether any of these highly discrepant gene profiles will outperform optimized contemporary multivariable individual patient risk prediction models across the prognostic spectrum of melanoma.
Daniel G. Coit, MD is a surgical oncologist at Memorial Sloan Kettering Cancer Center in New York. He made his remarks in an editorial in JAMA Dermatology (2019 May 1. doi: 10.1001/jamadermatol.2019.0200). Dr. Coit disclosed that he has no relevant financial conflicts of interest.
Effective systemic treatments have forever altered the kinetics of events in advanced melanoma, with the natural history outcome curves of this cancer now highly dependent on treatment response. The relapse-free survival (RFS) curves of patients with stage IIC/III melanoma with and without adjuvant dabrafenib/trametinib demonstrate the contemporary natural history of frequent early recurrences with few late events in the placebo groups and of fewer and later recurrences, with an as yet unknown frequency of late events, in the treated groups.
The three reported studies examine melanoma disease kinetics and speak to this new paradigm.
The first study, conducted by von Schuckmann and colleagues, looked for factors associated with disease recurrence within 2 years of treatment for localized, T1b to T4b melanoma.
Factors were not surprising and included the presence of ulceration, an increased mitotic index, increasing T stage, and location on the head and neck. Also, the study had two major limitations. First, the patient population was a heterogenous one – 442 patients with clinical T1b to T4b did not undergo sentinel lymph node biopsy (SLNB), 213 patients with pathologic T1b to T4b had a negative SLNB result, and 38 patients had at least stage IIIa disease after a positive SLNB finding. Second, as the 8th edition of the AJCC [American Joint Committee on Cancer] Staging Manual notes, most events in this cohort of patients are expected to occur after the 2-year threshold described in this prospective study and the disease can recur as much as 10 years after effective treatment.
The second study, conducted by Vallet and colleagues, analyzed data from patients with unresectable stage IV melanoma to determine if time to distant recurrence after excision of antecedent primary melanoma was associated with survival.
Time to first distant metastasis was not related to Breslow thickness or to tumor stage at the start of therapy; however, the data analysis did not include multiple patient variables that might have shed more light on predictive factors.
As systemic therapy increasingly leads to stabilization of previously progressive disease as well as durable complete remissions in melanoma patients, best response to immunotherapy or targeted therapy is likely to emerge as a much more important predictor than the time it took for stage IV melanoma to become apparent. There has been some thoughtful interest in exploring the prognostic value of establishing the kinetics of stage IV melanoma as a prognostic factor prior to initiating therapy, but there has been limited uptake of this approach by the melanoma oncology community.
In the third study, Calomarde-Rees and colleagues explored associations between melanoma disease characteristics and hematogenous or lymphatic metastases.
The anticipated findings include the observed association between tumor thickness and risk of recurrence, either lymphatic or hematogenous. As satellitosis is already considered a criterion for stage III in AJCC8, these two observations serve as internal controls that validate the credibility of the data set.
The researchers’ most intriguing finding is the strong potential association of the combination of both MAPK (either BRAF or NRAS) and TERT promoter mutations with poor survival, as demonstrated in their second prognostic model (hazard ratio, 5.7). This finding warrants cautious interpretation as the authors clearly acknowledge that a minority of the study population underwent detailed assessments of the mutation status of each gene.
While the biologic behavior of melanoma is likely to be much more complex than the mutation of one or two genes, the potential interaction of the mutated TERT promoter gene is provocative, especially in the context of a recent article suggesting a role for monitoring BRAF and TERT circulating free DNA as an indicator of response to systemic therapy and outcome.
Multigene panels are being developed, but it remains to be demonstrated whether any of these highly discrepant gene profiles will outperform optimized contemporary multivariable individual patient risk prediction models across the prognostic spectrum of melanoma.
Daniel G. Coit, MD is a surgical oncologist at Memorial Sloan Kettering Cancer Center in New York. He made his remarks in an editorial in JAMA Dermatology (2019 May 1. doi: 10.1001/jamadermatol.2019.0200). Dr. Coit disclosed that he has no relevant financial conflicts of interest.
As systemic therapies for advanced melanoma increase, some historical prognostic factors continue to hold true while refined and novel risk factors are emerging, according to the results of three studies published in JAMA Dermatology.
Among the most prominent findings of those studies are that ulceration, mitotic index, and head and neck location in localized disease were predictive of early recurrence; time to recurrence was not associated with survival in unresectable stage IV melanoma; and certain gene markers may be linked with particular types of metastasis.
The first study, conducted by Lena A. von Schuckmann, MBBS, of the University of Queensland School of Public Health in Australia, and colleagues, evaluated the risk of early melanoma recurrence in patients with localized disease.
“With the introduction of targeted and immune therapies for treatment of metastatic melanoma, including possible adjuvant therapy, a detailed understanding of the risk of melanoma recurrence may assist clinicians to advise patients with a primary tumor at high risk of disease metastasis,” the researchers wrote (JAMA Dermatol. 2019 May 1. doi: 10.1001/jamadermatol.2019.0440).
They conducted a prospective cohort study of 700 patients with high-risk, category T1b to T4b cutaneous melanoma, refined from an initial recruitment population of 1,254 individuals. Using self-administered patient questionnaires in conjunction with histologic, imaging, and clinical data over the course of 2 years, the investigators looked for factors that predicted recurrence.
Of 700 patients, 94 (13.4%) had disease recurrence, most often (70.2%) locoregional recurrence. Independent predictors of recurrence included mitotic rate greater than 3/mm2, thickness, ulceration, and primary tumor location on the head or neck.
Patients with negative single lymph node biopsy (SLNB) were less likely to have recurrence than were those who did not undergo SLNB. Among 64 patients whose locoregional disease was excised, 37 (57.8%) were disease free at 2 years, whereas 7 patients (10.9%) had new locoregional disease and 20 patients (31.3%) developed a new distant recurrence.
“[O]ur data appear to support the recommendation for careful scar and regional skin and lymph node examination during patient follow-up,” the investigators concluded, alluding to the relatively high rate of locoregional recurrence. “Subsequent recurrences occurring at distant sites were more likely to involve multiple organs, which is consistent with other studies.”
The second melanoma article, investigating associations between time to relapse and survival, was authored by Anaïs Vallet, MD, of Hôpital Saint-Louis, Paris, and colleagues.
“Although the kinetics of metastatic disease seem to be correlated with patient survival, the first relapse is not predictable, and data from the literature on the topic are controversial,” they wrote (JAMA Dermatol. 2019 May 1. doi: 10.1001/jamadermatol.2019.0425). “We hypothesized that the progression of the metastatic disease would be associated with the time from primary excision to the first distant recurrence of melanoma.”
To test this hypothesis, the investigators analyzed data from 638 patients with unresectable stage III or IV melanoma. Inclusion required first-line treatment with chemotherapy, targeted therapies, or immunotherapies. The interval between primary excision and distant disease recurrence, measured as a categorical and continuous variable, was compared with overall survival and progression-free survival. The analysis revealed no associations between time to recurrence and either survival measure, even when stratified by treatment.
“Now that immunotherapies and targeted therapies have been approved in the adjuvant setting for patients with stage III disease, it would be interesting to analyze recurrence-free survival and [progression-free survival] in relapsing patients who previously received adjuvant therapies,” the investigators wrote.
The third study was conducted by Laura Calomarde-Rees, MD, of Instituto Valenciano de Oncología, València, Spain, and colleagues.
“Our aim was to identify risk factors associated with lymphatic (locoregional metastasis) or hematogenous (distant metastasis) progression because these have not been studied separately to date in patients with localized melanoma,” they wrote (JAMA Dermatol. 2019 May 1. doi: 10.1001/jamadermatol.2019.0069).
The retrospective study involved 1,177 patients with stage I to II melanoma. Multiple disease variables were evaluated in the context of each type of metastasis, including age, sex, tumor location, and others.
The investigators found locoregional spread was most often associated with vascular invasion (hazard ratio [HR], 3.2), greater Breslow thickness (HR, 5.4; thickness greater than 4 mm), acral location (HR, 2.4), head/neck location (HR, 1.7), and age greater than 55 years (HR, 1.9).
Distant metastasis was most often associated with greater Breslow thickness (HR, 10.4; thickness greater than 4 mm), TERT promoter mutations (HR, 2.9), BRAF mutations (HR, 1.9), and absence of regression (HR, 0.1).
“Risk factors for lymphatic and hematogenous metastasis differ,” the investigators concluded. “A greater understanding of the clinical, histopathologic, and molecular factors involved could help to identify patients with an increased risk of recurrence and guide the design of individualized follow-up programs and adjuvant targeted therapies.”
Dr. von Schuckmann and colleagues disclosed study funding from the National Health and Medical Research Council and other relationships with the Norwegian Cancer Society project. Dr. Vallet and colleagues reported study support from French National Cancer Institute, MSD, BMS, Roche, and Novartis; and additional relationships with Incyte, Amgen, Pfizer, and others. Dr. Calomarde-Rees and colleagues disclosed no conflicts of interest.
As systemic therapies for advanced melanoma increase, some historical prognostic factors continue to hold true while refined and novel risk factors are emerging, according to the results of three studies published in JAMA Dermatology.
Among the most prominent findings of those studies are that ulceration, mitotic index, and head and neck location in localized disease were predictive of early recurrence; time to recurrence was not associated with survival in unresectable stage IV melanoma; and certain gene markers may be linked with particular types of metastasis.
The first study, conducted by Lena A. von Schuckmann, MBBS, of the University of Queensland School of Public Health in Australia, and colleagues, evaluated the risk of early melanoma recurrence in patients with localized disease.
“With the introduction of targeted and immune therapies for treatment of metastatic melanoma, including possible adjuvant therapy, a detailed understanding of the risk of melanoma recurrence may assist clinicians to advise patients with a primary tumor at high risk of disease metastasis,” the researchers wrote (JAMA Dermatol. 2019 May 1. doi: 10.1001/jamadermatol.2019.0440).
They conducted a prospective cohort study of 700 patients with high-risk, category T1b to T4b cutaneous melanoma, refined from an initial recruitment population of 1,254 individuals. Using self-administered patient questionnaires in conjunction with histologic, imaging, and clinical data over the course of 2 years, the investigators looked for factors that predicted recurrence.
Of 700 patients, 94 (13.4%) had disease recurrence, most often (70.2%) locoregional recurrence. Independent predictors of recurrence included mitotic rate greater than 3/mm2, thickness, ulceration, and primary tumor location on the head or neck.
Patients with negative single lymph node biopsy (SLNB) were less likely to have recurrence than were those who did not undergo SLNB. Among 64 patients whose locoregional disease was excised, 37 (57.8%) were disease free at 2 years, whereas 7 patients (10.9%) had new locoregional disease and 20 patients (31.3%) developed a new distant recurrence.
“[O]ur data appear to support the recommendation for careful scar and regional skin and lymph node examination during patient follow-up,” the investigators concluded, alluding to the relatively high rate of locoregional recurrence. “Subsequent recurrences occurring at distant sites were more likely to involve multiple organs, which is consistent with other studies.”
The second melanoma article, investigating associations between time to relapse and survival, was authored by Anaïs Vallet, MD, of Hôpital Saint-Louis, Paris, and colleagues.
“Although the kinetics of metastatic disease seem to be correlated with patient survival, the first relapse is not predictable, and data from the literature on the topic are controversial,” they wrote (JAMA Dermatol. 2019 May 1. doi: 10.1001/jamadermatol.2019.0425). “We hypothesized that the progression of the metastatic disease would be associated with the time from primary excision to the first distant recurrence of melanoma.”
To test this hypothesis, the investigators analyzed data from 638 patients with unresectable stage III or IV melanoma. Inclusion required first-line treatment with chemotherapy, targeted therapies, or immunotherapies. The interval between primary excision and distant disease recurrence, measured as a categorical and continuous variable, was compared with overall survival and progression-free survival. The analysis revealed no associations between time to recurrence and either survival measure, even when stratified by treatment.
“Now that immunotherapies and targeted therapies have been approved in the adjuvant setting for patients with stage III disease, it would be interesting to analyze recurrence-free survival and [progression-free survival] in relapsing patients who previously received adjuvant therapies,” the investigators wrote.
The third study was conducted by Laura Calomarde-Rees, MD, of Instituto Valenciano de Oncología, València, Spain, and colleagues.
“Our aim was to identify risk factors associated with lymphatic (locoregional metastasis) or hematogenous (distant metastasis) progression because these have not been studied separately to date in patients with localized melanoma,” they wrote (JAMA Dermatol. 2019 May 1. doi: 10.1001/jamadermatol.2019.0069).
The retrospective study involved 1,177 patients with stage I to II melanoma. Multiple disease variables were evaluated in the context of each type of metastasis, including age, sex, tumor location, and others.
The investigators found locoregional spread was most often associated with vascular invasion (hazard ratio [HR], 3.2), greater Breslow thickness (HR, 5.4; thickness greater than 4 mm), acral location (HR, 2.4), head/neck location (HR, 1.7), and age greater than 55 years (HR, 1.9).
Distant metastasis was most often associated with greater Breslow thickness (HR, 10.4; thickness greater than 4 mm), TERT promoter mutations (HR, 2.9), BRAF mutations (HR, 1.9), and absence of regression (HR, 0.1).
“Risk factors for lymphatic and hematogenous metastasis differ,” the investigators concluded. “A greater understanding of the clinical, histopathologic, and molecular factors involved could help to identify patients with an increased risk of recurrence and guide the design of individualized follow-up programs and adjuvant targeted therapies.”
Dr. von Schuckmann and colleagues disclosed study funding from the National Health and Medical Research Council and other relationships with the Norwegian Cancer Society project. Dr. Vallet and colleagues reported study support from French National Cancer Institute, MSD, BMS, Roche, and Novartis; and additional relationships with Incyte, Amgen, Pfizer, and others. Dr. Calomarde-Rees and colleagues disclosed no conflicts of interest.
FROM JAMA DERMATOLOGY