User login
Reinforcing mesh at ostomy site prevents parastomal hernia
For patients undergoing elective permanent colostomy, prophylactic augmentation of the abdominal wall using mesh at the ostomy site prevents the development of parastomal hernia, according to a report published in the April issue of Annals of Surgery.
The incidence of parastomal hernia is expected to rise because of the increasing number of cancer patients surviving with a colostomy, and the rising number of obese patients who have increased tension on the abdominal wall because of their elevated intra-abdominal pressure and larger abdominal radius. Researchers in the Netherlands performed a prospective randomized study, the PREVENT trial, to assess whether augmenting the abdominal wall at the ostomy site, using a lightweight mesh, would be safe, feasible, and effective at preventing parastomal hernia. They reported their findings after 1 year of follow-up; the study will continue until longer-term results are available at 5 years.
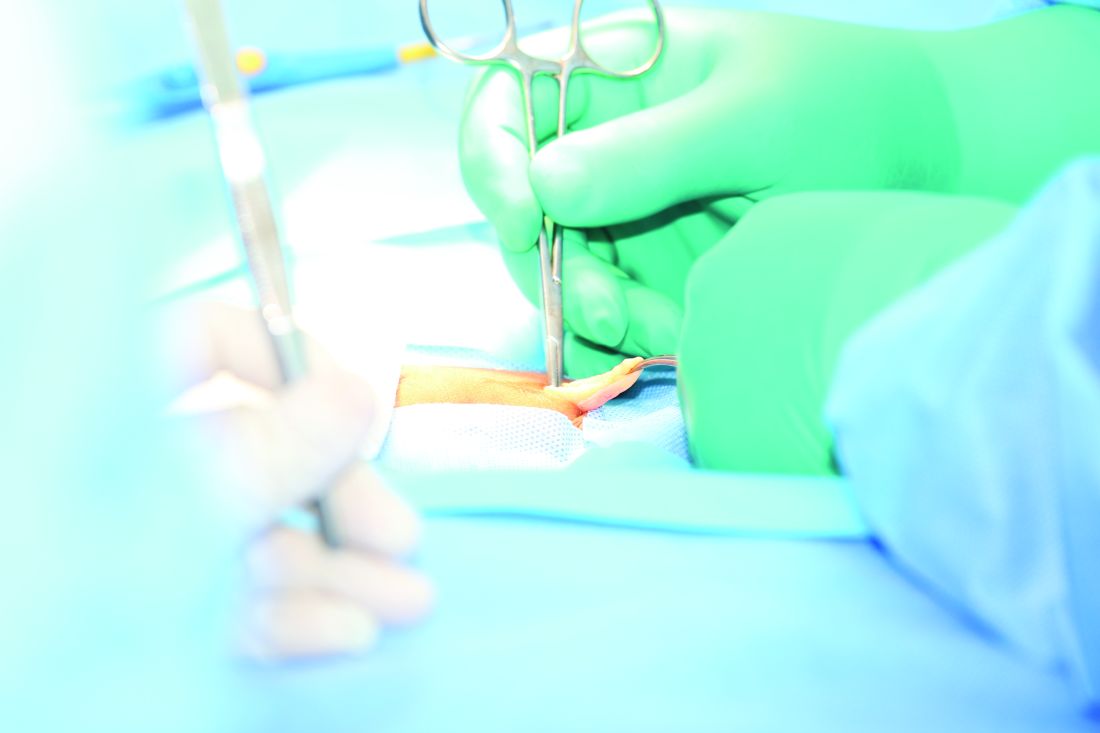
In the intervention group, a retromuscular space was created to accommodate the mesh by dissecting the muscle from the posterior fascia or peritoneum to the lateral border via a median laparotomy. An incision was made in the center of the mesh to allow passage of the colon, and the mesh was placed on the posterior rectus sheath and anchored laterally with two absorbable sutures. “On the medial side, the mesh was incorporated in the running suture closing the fascia, thus preventing contact between the mesh and the viscera,” the investigators said (Ann Surg. 2017;265:663-9).
The primary end point – the incidence of parastomal hernia at 1 year – occurred in 3 patients (4.5%) in the intervention group and 16 (24.2%) in the control group, a significant difference. There were no mesh-related complications such as infection, strictures, or adhesions. “The majority of the parastomal hernias that required surgical repair were in the control group, which supports the concept that if a hernia develops in a patient with mesh, it is smaller and less likely to cause complaints,” Dr. Brandsma and his associates said.
Significantly fewer patients in the mesh group (9%) than in the control group (21%) reported stoma-related complaints such as pain, leakage, and skin problems. Scores on measures of quality of life and pain severity were no different between the two study groups.
“Prophylactic augmentation of the abdominal wall with a retromuscular polypropylene mesh at the ostomy site is a safe and feasible procedure with no adverse events. It significantly reduces the incidence of parastomal hernia,” the investigators concluded.
This study was supported by Canisius Wilhelmina Hospital’s surgery research fund, the Netherlands Organization for Health Research and Development, and Covidien/Medtronic. Dr. Brandsma and his associates reported having no relevant financial disclosures.
For patients undergoing elective permanent colostomy, prophylactic augmentation of the abdominal wall using mesh at the ostomy site prevents the development of parastomal hernia, according to a report published in the April issue of Annals of Surgery.
The incidence of parastomal hernia is expected to rise because of the increasing number of cancer patients surviving with a colostomy, and the rising number of obese patients who have increased tension on the abdominal wall because of their elevated intra-abdominal pressure and larger abdominal radius. Researchers in the Netherlands performed a prospective randomized study, the PREVENT trial, to assess whether augmenting the abdominal wall at the ostomy site, using a lightweight mesh, would be safe, feasible, and effective at preventing parastomal hernia. They reported their findings after 1 year of follow-up; the study will continue until longer-term results are available at 5 years.
In the intervention group, a retromuscular space was created to accommodate the mesh by dissecting the muscle from the posterior fascia or peritoneum to the lateral border via a median laparotomy. An incision was made in the center of the mesh to allow passage of the colon, and the mesh was placed on the posterior rectus sheath and anchored laterally with two absorbable sutures. “On the medial side, the mesh was incorporated in the running suture closing the fascia, thus preventing contact between the mesh and the viscera,” the investigators said (Ann Surg. 2017;265:663-9).
The primary end point – the incidence of parastomal hernia at 1 year – occurred in 3 patients (4.5%) in the intervention group and 16 (24.2%) in the control group, a significant difference. There were no mesh-related complications such as infection, strictures, or adhesions. “The majority of the parastomal hernias that required surgical repair were in the control group, which supports the concept that if a hernia develops in a patient with mesh, it is smaller and less likely to cause complaints,” Dr. Brandsma and his associates said.
Significantly fewer patients in the mesh group (9%) than in the control group (21%) reported stoma-related complaints such as pain, leakage, and skin problems. Scores on measures of quality of life and pain severity were no different between the two study groups.
“Prophylactic augmentation of the abdominal wall with a retromuscular polypropylene mesh at the ostomy site is a safe and feasible procedure with no adverse events. It significantly reduces the incidence of parastomal hernia,” the investigators concluded.
This study was supported by Canisius Wilhelmina Hospital’s surgery research fund, the Netherlands Organization for Health Research and Development, and Covidien/Medtronic. Dr. Brandsma and his associates reported having no relevant financial disclosures.
For patients undergoing elective permanent colostomy, prophylactic augmentation of the abdominal wall using mesh at the ostomy site prevents the development of parastomal hernia, according to a report published in the April issue of Annals of Surgery.
The incidence of parastomal hernia is expected to rise because of the increasing number of cancer patients surviving with a colostomy, and the rising number of obese patients who have increased tension on the abdominal wall because of their elevated intra-abdominal pressure and larger abdominal radius. Researchers in the Netherlands performed a prospective randomized study, the PREVENT trial, to assess whether augmenting the abdominal wall at the ostomy site, using a lightweight mesh, would be safe, feasible, and effective at preventing parastomal hernia. They reported their findings after 1 year of follow-up; the study will continue until longer-term results are available at 5 years.
In the intervention group, a retromuscular space was created to accommodate the mesh by dissecting the muscle from the posterior fascia or peritoneum to the lateral border via a median laparotomy. An incision was made in the center of the mesh to allow passage of the colon, and the mesh was placed on the posterior rectus sheath and anchored laterally with two absorbable sutures. “On the medial side, the mesh was incorporated in the running suture closing the fascia, thus preventing contact between the mesh and the viscera,” the investigators said (Ann Surg. 2017;265:663-9).
The primary end point – the incidence of parastomal hernia at 1 year – occurred in 3 patients (4.5%) in the intervention group and 16 (24.2%) in the control group, a significant difference. There were no mesh-related complications such as infection, strictures, or adhesions. “The majority of the parastomal hernias that required surgical repair were in the control group, which supports the concept that if a hernia develops in a patient with mesh, it is smaller and less likely to cause complaints,” Dr. Brandsma and his associates said.
Significantly fewer patients in the mesh group (9%) than in the control group (21%) reported stoma-related complaints such as pain, leakage, and skin problems. Scores on measures of quality of life and pain severity were no different between the two study groups.
“Prophylactic augmentation of the abdominal wall with a retromuscular polypropylene mesh at the ostomy site is a safe and feasible procedure with no adverse events. It significantly reduces the incidence of parastomal hernia,” the investigators concluded.
This study was supported by Canisius Wilhelmina Hospital’s surgery research fund, the Netherlands Organization for Health Research and Development, and Covidien/Medtronic. Dr. Brandsma and his associates reported having no relevant financial disclosures.
FROM THE ANNALS OF SURGERY
Key clinical point: For patients undergoing permanent colostomy, prophylactic augmentation of the abdominal wall using mesh at the ostomy site prevents the development of parastomal hernia.
Major finding: The primary end point – the incidence of parastomal hernia at 1 year – occurred in 3 patients (4.5%) in the intervention group and 16 (24.2%) in the control group.
Data source: A prospective, multicenter, randomized cohort study comparing prophylactic mesh against standard care in 133 adults undergoing elective end-colostomy during a 3-year period.
Disclosures: This study was supported by Canisius Wilhelmina Hospital’s surgery research fund, the Netherlands Organization for Health Research and Development, and Covidien/Medtronic. Dr. Brandsma and his associates reported having no relevant financial disclosures.
Common gut yeast may exacerbate IBD
A ubiquitous yeast strain may play a role in exacerbating inflammatory bowel disease (IBD), an animal study showed.
While research has shown that the composition of gut microbiota in people with IBD is different from that of healthy people, most of the attention has been focused on bacteria. The roles of other microorganisms, including yeasts, are still poorly understood.
In research published in Science Translational Medicine, Tyson Chiaro, of the University of Utah, Salt Lake City, and his colleagues inoculated sterile mice with either of two fungal species: Rhodotorula aurantiaca – an environmentally acquired yeast found in milk and fruit juices – or Saccharomyces cerevisiae – Baker’s yeast – for which some people with Crohn’s disease have been shown to have elevated antibodies. The mice were inoculated gradually over a period of a week to mimic consumption of food enriched with yeast products. The researchers then treated the mice with drugs to induce colitislike symptoms and analyzed colon tissues for damage. Mr. Chiaro and his colleagues found that colonization with S. cerevisiae, but not with R. aurantiaca, aggravated colitis and resulted in epithelial damage leading to greater gut permeability (Sci Transl Med. 2017;9[380] pii: eaaf9044]).
Mr. Chiaro and his colleagues then investigated whether heat-killed S. cerevisiae also induced aggravated colitis and found that it did not, suggesting that a metabolically active organism was required to aggravate disease. Mr. Chiaro and his colleagues performed screens of fecal metabolites in the mice and found that S. cerevisiae colonization enhanced purine metabolism, resulting in increased uric acid production.
To test whether this purine pathway was aggravating colitis, the researchers blocked it with allopurinol (10 mg/kg). The S. cerevisiae– inoculated mice that were treated with allopurinol had reduced uric acid–levels and ameliorated colitis–symptoms. The results suggest that allopurinol might be of more clinical value in treating IBD than previously thought. The drug has been used in patients with Crohn’s disease to increase the efficacy of other IBD medications, and “many patients who received adjunctive allopurinol therapy were reported to have major clinical improvement,” Mr. Chiaro and his colleagues noted. The results “suggest that some of the improvement might come from preventing yeast-induced–uric acid buildup in the intestine. Thus, allopurinol treatment in some IBD patients with adverse reactions to yeast and high uric acid might be of therapeutic benefit and should be explored.”
Mr. Chiaro’s coauthors reported a variety of individual grant and fellowship awards, including from the National Institute of Allergy and Infectious Disease and the National Institutes of Health. None declared commercial conflicts of interest.
A ubiquitous yeast strain may play a role in exacerbating inflammatory bowel disease (IBD), an animal study showed.
While research has shown that the composition of gut microbiota in people with IBD is different from that of healthy people, most of the attention has been focused on bacteria. The roles of other microorganisms, including yeasts, are still poorly understood.
In research published in Science Translational Medicine, Tyson Chiaro, of the University of Utah, Salt Lake City, and his colleagues inoculated sterile mice with either of two fungal species: Rhodotorula aurantiaca – an environmentally acquired yeast found in milk and fruit juices – or Saccharomyces cerevisiae – Baker’s yeast – for which some people with Crohn’s disease have been shown to have elevated antibodies. The mice were inoculated gradually over a period of a week to mimic consumption of food enriched with yeast products. The researchers then treated the mice with drugs to induce colitislike symptoms and analyzed colon tissues for damage. Mr. Chiaro and his colleagues found that colonization with S. cerevisiae, but not with R. aurantiaca, aggravated colitis and resulted in epithelial damage leading to greater gut permeability (Sci Transl Med. 2017;9[380] pii: eaaf9044]).
Mr. Chiaro and his colleagues then investigated whether heat-killed S. cerevisiae also induced aggravated colitis and found that it did not, suggesting that a metabolically active organism was required to aggravate disease. Mr. Chiaro and his colleagues performed screens of fecal metabolites in the mice and found that S. cerevisiae colonization enhanced purine metabolism, resulting in increased uric acid production.
To test whether this purine pathway was aggravating colitis, the researchers blocked it with allopurinol (10 mg/kg). The S. cerevisiae– inoculated mice that were treated with allopurinol had reduced uric acid–levels and ameliorated colitis–symptoms. The results suggest that allopurinol might be of more clinical value in treating IBD than previously thought. The drug has been used in patients with Crohn’s disease to increase the efficacy of other IBD medications, and “many patients who received adjunctive allopurinol therapy were reported to have major clinical improvement,” Mr. Chiaro and his colleagues noted. The results “suggest that some of the improvement might come from preventing yeast-induced–uric acid buildup in the intestine. Thus, allopurinol treatment in some IBD patients with adverse reactions to yeast and high uric acid might be of therapeutic benefit and should be explored.”
Mr. Chiaro’s coauthors reported a variety of individual grant and fellowship awards, including from the National Institute of Allergy and Infectious Disease and the National Institutes of Health. None declared commercial conflicts of interest.
A ubiquitous yeast strain may play a role in exacerbating inflammatory bowel disease (IBD), an animal study showed.
While research has shown that the composition of gut microbiota in people with IBD is different from that of healthy people, most of the attention has been focused on bacteria. The roles of other microorganisms, including yeasts, are still poorly understood.
In research published in Science Translational Medicine, Tyson Chiaro, of the University of Utah, Salt Lake City, and his colleagues inoculated sterile mice with either of two fungal species: Rhodotorula aurantiaca – an environmentally acquired yeast found in milk and fruit juices – or Saccharomyces cerevisiae – Baker’s yeast – for which some people with Crohn’s disease have been shown to have elevated antibodies. The mice were inoculated gradually over a period of a week to mimic consumption of food enriched with yeast products. The researchers then treated the mice with drugs to induce colitislike symptoms and analyzed colon tissues for damage. Mr. Chiaro and his colleagues found that colonization with S. cerevisiae, but not with R. aurantiaca, aggravated colitis and resulted in epithelial damage leading to greater gut permeability (Sci Transl Med. 2017;9[380] pii: eaaf9044]).
Mr. Chiaro and his colleagues then investigated whether heat-killed S. cerevisiae also induced aggravated colitis and found that it did not, suggesting that a metabolically active organism was required to aggravate disease. Mr. Chiaro and his colleagues performed screens of fecal metabolites in the mice and found that S. cerevisiae colonization enhanced purine metabolism, resulting in increased uric acid production.
To test whether this purine pathway was aggravating colitis, the researchers blocked it with allopurinol (10 mg/kg). The S. cerevisiae– inoculated mice that were treated with allopurinol had reduced uric acid–levels and ameliorated colitis–symptoms. The results suggest that allopurinol might be of more clinical value in treating IBD than previously thought. The drug has been used in patients with Crohn’s disease to increase the efficacy of other IBD medications, and “many patients who received adjunctive allopurinol therapy were reported to have major clinical improvement,” Mr. Chiaro and his colleagues noted. The results “suggest that some of the improvement might come from preventing yeast-induced–uric acid buildup in the intestine. Thus, allopurinol treatment in some IBD patients with adverse reactions to yeast and high uric acid might be of therapeutic benefit and should be explored.”
Mr. Chiaro’s coauthors reported a variety of individual grant and fellowship awards, including from the National Institute of Allergy and Infectious Disease and the National Institutes of Health. None declared commercial conflicts of interest.
FROM SCIENCE TRANSLATIONAL MEDICINE
Key clinical point:
Major finding: Sterile mice inoculated with S. cerevisiae and treated to induce colitis had more tissue damage than untreated mice or those treated with another type of yeast.
Data source: An experimental study on mice inoculated with one of two yeasts, then treated to induce colitislike symptoms.
Disclosures: Study authors had multiple sources of individual grant funding but no commercial conflicts of interest.
Norovirus reporting tool yields real-time outbreak data
NoroSTAT, the Centers for Disease Control and Prevention’s new program with which states can report norovirus outbreaks, yields more timely and more complete epidemiologic and laboratory data, which allows a faster and better-informed public health response to such outbreaks, according to a report published in the Morbidity and Mortality Weekly Report.
The CDC launched NoroSTAT (Norovirus Sentinel Testing and Tracking) in 2012 to permit the health departments in selected states to report specific epidemiologic and laboratory data regarding norovirus outbreaks more rapidly than usual – within 7 business days, said Minesh P. Shah, MD, of the Epidemic Intelligence Service and the division of viral diseases, CDC, Atlanta, and his associates.
NoroSTAT significantly reduced the median interval in reporting epidemiologic data concerning norovirus from 22 days to 2 days and significantly reduced the median interval in reporting relevant laboratory data from 21 days to 3 days. The percentage of reports submitted within 7 business days increased from 26% to 95% among the states participating in NoroSTAT, while remaining low – only 12%-13% – in nonparticipating states. The number of complete reports also increased substantially, from 87% to 99.9%, among the participating states.
These improvements likely result from NoroSTAT’s stringent reporting requirements and from the program’s ability “to enhance communication between epidemiologists and laboratorians in both state health departments and at CDC,” Dr. Shah and his associates said (MMWR Morbidity and Mortality Weekly Report. 2017 Feb 24;66:185-9).
NoroSTAT represents a key advancement in norovirus outbreak surveillance and has proved valuable in early identification and better characterization of outbreaks. It was expanded to include nine states in August 2016, the investigators added.
NoroSTAT, the Centers for Disease Control and Prevention’s new program with which states can report norovirus outbreaks, yields more timely and more complete epidemiologic and laboratory data, which allows a faster and better-informed public health response to such outbreaks, according to a report published in the Morbidity and Mortality Weekly Report.
The CDC launched NoroSTAT (Norovirus Sentinel Testing and Tracking) in 2012 to permit the health departments in selected states to report specific epidemiologic and laboratory data regarding norovirus outbreaks more rapidly than usual – within 7 business days, said Minesh P. Shah, MD, of the Epidemic Intelligence Service and the division of viral diseases, CDC, Atlanta, and his associates.
NoroSTAT significantly reduced the median interval in reporting epidemiologic data concerning norovirus from 22 days to 2 days and significantly reduced the median interval in reporting relevant laboratory data from 21 days to 3 days. The percentage of reports submitted within 7 business days increased from 26% to 95% among the states participating in NoroSTAT, while remaining low – only 12%-13% – in nonparticipating states. The number of complete reports also increased substantially, from 87% to 99.9%, among the participating states.
These improvements likely result from NoroSTAT’s stringent reporting requirements and from the program’s ability “to enhance communication between epidemiologists and laboratorians in both state health departments and at CDC,” Dr. Shah and his associates said (MMWR Morbidity and Mortality Weekly Report. 2017 Feb 24;66:185-9).
NoroSTAT represents a key advancement in norovirus outbreak surveillance and has proved valuable in early identification and better characterization of outbreaks. It was expanded to include nine states in August 2016, the investigators added.
NoroSTAT, the Centers for Disease Control and Prevention’s new program with which states can report norovirus outbreaks, yields more timely and more complete epidemiologic and laboratory data, which allows a faster and better-informed public health response to such outbreaks, according to a report published in the Morbidity and Mortality Weekly Report.
The CDC launched NoroSTAT (Norovirus Sentinel Testing and Tracking) in 2012 to permit the health departments in selected states to report specific epidemiologic and laboratory data regarding norovirus outbreaks more rapidly than usual – within 7 business days, said Minesh P. Shah, MD, of the Epidemic Intelligence Service and the division of viral diseases, CDC, Atlanta, and his associates.
NoroSTAT significantly reduced the median interval in reporting epidemiologic data concerning norovirus from 22 days to 2 days and significantly reduced the median interval in reporting relevant laboratory data from 21 days to 3 days. The percentage of reports submitted within 7 business days increased from 26% to 95% among the states participating in NoroSTAT, while remaining low – only 12%-13% – in nonparticipating states. The number of complete reports also increased substantially, from 87% to 99.9%, among the participating states.
These improvements likely result from NoroSTAT’s stringent reporting requirements and from the program’s ability “to enhance communication between epidemiologists and laboratorians in both state health departments and at CDC,” Dr. Shah and his associates said (MMWR Morbidity and Mortality Weekly Report. 2017 Feb 24;66:185-9).
NoroSTAT represents a key advancement in norovirus outbreak surveillance and has proved valuable in early identification and better characterization of outbreaks. It was expanded to include nine states in August 2016, the investigators added.
Key clinical point: NoroSTAT, the CDC’s new program with which states can report norovirus outbreaks, yields more timely and complete epidemiologic data.
Major finding: NoroSTAT significantly reduced the median interval in reporting epidemiologic data concerning norovirus from 22 days to 2 days and significantly reduced the median interval in reporting relevant laboratory data from 21 days to 3 days.
Data source: A comparison of epidemiologic and laboratory data reported by all 50 states for the 3 years before and the 3 years after NoroSTAT was implemented in 5 states.
Disclosures: This study was sponsored by the Centers for Disease Control and Prevention. No financial disclosures were provided.
Unique, multi-omic profile found in children with autism and functional GI disorders
The gut microbiomes of children with autism spectrum disorder (ASD) and functional gastrointestinal disorders (FGID) had significantly higher levels of several Clostridium species and lower concentrations of other bacteria compared with neurotypical children with and without FGIDs, which correlated with increases in inflammatory cytokines, decreased tryptophan, and increased serotonin, according to a small, single-center, cross-sectional study.
This “unique multi-omic profile [was] specific to ASD-FGID and ASD-FGID with abdominal pain,” wrote Ruth Ann Luna, PhD, of Texas Children’s Microbiome Center at Texas Children’s Hospital, Houston, and her associates. The report was published online in Cellular and Molecular Gastroenterology and Hepatology (doi: 10.1016/j.jcmgh.2016.11.008).
Children with ASD are at increased risk for FGIDs such as functional constipation, nonretentive fecal incontinence, functional abdominal pain, abdominal migraines, and irritable bowel syndrome, compared with their neurotypical peers. Changes in the gut microbiome can affect immunologic pathways and the balance between tryptophan and serotonin. This altered “microbial-gut-brain axis” has been reported in both ASD and FGID, suggesting “that altered gut-brain communications not only may play a role in the increased occurrence of FGIDs in ASD individuals, but could advance our understanding of potential risk factors for FGID in the ASD community,” the researchers wrote.
Previous studies of stool specimens have found higher levels of several species of Clostridium in pediatric ASD compared with neurotypical children. To confirm and expand on that work, the investigators examined microbial and neuroimmune markers in rectal biopsies and blood specimens from 14 children with ASD-FGID, 15 neurotypical children with FGID, and 6 asymptomatic neurotypical children. Participants were recruited from Nationwide Children’s Hospital in Columbus, Ohio. The researchers quantified microbial 16S ribosomal DNA community signatures, cytokines, chemokines, and serotonergic metabolites, and correlated results with parental responses to the Questionnaire on Pediatric Gastrointestinal Symptoms–Rome III version.
The ASD-FGID group had significantly higher numbers for ribosomal DNA sequences for Clostridium lituseburense (P = .002), Lachnoclostridium bolteae (P = .02), Lachnoclostridium hathewayi (P = .03), Clostridium aldenense (P = .04), and Oscillospira plautii (P = .04), compared with neurotypical children with and without FGID. Children with ASD-FGID also had significantly lower levels of Dorea formicigenerans (P = .006), Blautia luti (P = .02), and Sutterella species (P = .03). “Overall, our identification of clostridial species aligns with previous autism studies that have identified microbiome alterations,” the researchers noted.
They also looked specifically at abdominal pain. Children with ASD-FGID and abdominal pain had significantly higher gut mucosal levels of Turicibacter sanguinis (P = .03), Clostridium aldenense (P = .004), Clostridium lituseburense (P = .003), Oscillospira plautii (P = .01), Clostridium disporicum (P = .049), and Clostridium tertium (P = .045) than did any other subgroup, the investigators found. Patients with both ASD-FGID and abdominal pain also had significantly higher levels of C. aldenense (P = .03), O. plautii (P = .04), Tyzzerella species (P = .045), and Parasutterella excrementihominis (P = .04) than did ASD-FGID patients without abdominal pain.
Both C. disporicum and C. tertium correlated with increases in the proinflammatory cytokines IL6 and interferon-gamma. Levels of these cytokines were highest in patients with ASD-FGID, and IL6 was highest of all among children with ASD-FGID with abdominal pain. Another proinflammatory cytokine, IL17A, also correlated with Clostridia species that were enriched in children with ASD-FGID. Both IL6 and IL17A have been implicated in autism-like phenotypes in rodents, the researchers noted. Several other cytokines also were linked to ASD-FGID, and abdominal pain correlated significantly with increases in MCP-1 (P = .03) and eotaxin (P = .03).
Gut mucosal levels of tryptophan were significantly lower among children with ASD-FGID compared with neurotypical children, either with (P = .006) or without (P = .009) FGID. In contrast, gut mucosal levels of 5-HIAA, the primary metabolite of serotonin, were significantly higher among children with ASD-FGID compared with neurotypical children (P = .01). Increased 5-HIAA also correlated significantly with abdominal pain (P = .04). Six species of bacteria correlated significantly with tryptophan or serotonin, implicating the gut microbiome in the serotonin pathway.
“Although these initial findings are correlative, these data form the framework for future studies targeting tryptophan-serotonin metabolism and inflammatory pathways in FGID in ASD,” the researchers concluded.
The U.S. Department of Health and Human Services funded the work. The investigators had no relevant disclosures.
Autism-spectrum disorder is a serious and increasingly prevalent developmental behavior disorder often accompanied and aggravated by a range of gastrointestinal and cognitive dysfunctions. Its etiology probably involves maternal diet and inflammatory events that alter central nervous system neurodevelopment critical to the cognition of social interaction. Candidate causal products of these events include the cytokines IL-6 and IL-17A, and certain bioactive amines, notably serotonin. Functional gastrointestinal disorders share these same molecules as biomarkers and disease modifiers, probably elicited in part by the intestinal microbiome. Hence, the comorbidity in ASD suggests these two disease processes are etiologically related.
The study by Luna and colleagues tightens the case for a microbial hub and serotonin and cytokine spokes in the gastrointestinal dysfunction of ASD: elevated mucosal tissue levels of select microbial taxa, mainly members of the genus Clostridium, and mucosal production of cytokines and serotonin-pathway bioamines associated with these and other select microbial species. Important and challenging questions loom ahead. What are the direct mucosal cell types and functions targeted of this network for the microbiota, and via what microbial products? Might they elicit epithelial or mucosal hematopoietic cell cytokine production that in turn causes mucosal bioamine secretion? And, what associated microbiota and products are just secondarily altered and not causally involved? The exciting study of Luna and colleagues raises confidence for this path ahead, and its promise for clarifying ASD pathogenesis and uncovering targetable elements for intervention.
Jonathan Braun, MD, PhD, is professor and chair of pathology and laboratory medicine, UCLA David Geffen School of Medicine, UCLA Health System, Los Angeles. He has no conflicts of interest.
Autism-spectrum disorder is a serious and increasingly prevalent developmental behavior disorder often accompanied and aggravated by a range of gastrointestinal and cognitive dysfunctions. Its etiology probably involves maternal diet and inflammatory events that alter central nervous system neurodevelopment critical to the cognition of social interaction. Candidate causal products of these events include the cytokines IL-6 and IL-17A, and certain bioactive amines, notably serotonin. Functional gastrointestinal disorders share these same molecules as biomarkers and disease modifiers, probably elicited in part by the intestinal microbiome. Hence, the comorbidity in ASD suggests these two disease processes are etiologically related.
The study by Luna and colleagues tightens the case for a microbial hub and serotonin and cytokine spokes in the gastrointestinal dysfunction of ASD: elevated mucosal tissue levels of select microbial taxa, mainly members of the genus Clostridium, and mucosal production of cytokines and serotonin-pathway bioamines associated with these and other select microbial species. Important and challenging questions loom ahead. What are the direct mucosal cell types and functions targeted of this network for the microbiota, and via what microbial products? Might they elicit epithelial or mucosal hematopoietic cell cytokine production that in turn causes mucosal bioamine secretion? And, what associated microbiota and products are just secondarily altered and not causally involved? The exciting study of Luna and colleagues raises confidence for this path ahead, and its promise for clarifying ASD pathogenesis and uncovering targetable elements for intervention.
Jonathan Braun, MD, PhD, is professor and chair of pathology and laboratory medicine, UCLA David Geffen School of Medicine, UCLA Health System, Los Angeles. He has no conflicts of interest.
Autism-spectrum disorder is a serious and increasingly prevalent developmental behavior disorder often accompanied and aggravated by a range of gastrointestinal and cognitive dysfunctions. Its etiology probably involves maternal diet and inflammatory events that alter central nervous system neurodevelopment critical to the cognition of social interaction. Candidate causal products of these events include the cytokines IL-6 and IL-17A, and certain bioactive amines, notably serotonin. Functional gastrointestinal disorders share these same molecules as biomarkers and disease modifiers, probably elicited in part by the intestinal microbiome. Hence, the comorbidity in ASD suggests these two disease processes are etiologically related.
The study by Luna and colleagues tightens the case for a microbial hub and serotonin and cytokine spokes in the gastrointestinal dysfunction of ASD: elevated mucosal tissue levels of select microbial taxa, mainly members of the genus Clostridium, and mucosal production of cytokines and serotonin-pathway bioamines associated with these and other select microbial species. Important and challenging questions loom ahead. What are the direct mucosal cell types and functions targeted of this network for the microbiota, and via what microbial products? Might they elicit epithelial or mucosal hematopoietic cell cytokine production that in turn causes mucosal bioamine secretion? And, what associated microbiota and products are just secondarily altered and not causally involved? The exciting study of Luna and colleagues raises confidence for this path ahead, and its promise for clarifying ASD pathogenesis and uncovering targetable elements for intervention.
Jonathan Braun, MD, PhD, is professor and chair of pathology and laboratory medicine, UCLA David Geffen School of Medicine, UCLA Health System, Los Angeles. He has no conflicts of interest.
The gut microbiomes of children with autism spectrum disorder (ASD) and functional gastrointestinal disorders (FGID) had significantly higher levels of several Clostridium species and lower concentrations of other bacteria compared with neurotypical children with and without FGIDs, which correlated with increases in inflammatory cytokines, decreased tryptophan, and increased serotonin, according to a small, single-center, cross-sectional study.
This “unique multi-omic profile [was] specific to ASD-FGID and ASD-FGID with abdominal pain,” wrote Ruth Ann Luna, PhD, of Texas Children’s Microbiome Center at Texas Children’s Hospital, Houston, and her associates. The report was published online in Cellular and Molecular Gastroenterology and Hepatology (doi: 10.1016/j.jcmgh.2016.11.008).
Children with ASD are at increased risk for FGIDs such as functional constipation, nonretentive fecal incontinence, functional abdominal pain, abdominal migraines, and irritable bowel syndrome, compared with their neurotypical peers. Changes in the gut microbiome can affect immunologic pathways and the balance between tryptophan and serotonin. This altered “microbial-gut-brain axis” has been reported in both ASD and FGID, suggesting “that altered gut-brain communications not only may play a role in the increased occurrence of FGIDs in ASD individuals, but could advance our understanding of potential risk factors for FGID in the ASD community,” the researchers wrote.
Previous studies of stool specimens have found higher levels of several species of Clostridium in pediatric ASD compared with neurotypical children. To confirm and expand on that work, the investigators examined microbial and neuroimmune markers in rectal biopsies and blood specimens from 14 children with ASD-FGID, 15 neurotypical children with FGID, and 6 asymptomatic neurotypical children. Participants were recruited from Nationwide Children’s Hospital in Columbus, Ohio. The researchers quantified microbial 16S ribosomal DNA community signatures, cytokines, chemokines, and serotonergic metabolites, and correlated results with parental responses to the Questionnaire on Pediatric Gastrointestinal Symptoms–Rome III version.
The ASD-FGID group had significantly higher numbers for ribosomal DNA sequences for Clostridium lituseburense (P = .002), Lachnoclostridium bolteae (P = .02), Lachnoclostridium hathewayi (P = .03), Clostridium aldenense (P = .04), and Oscillospira plautii (P = .04), compared with neurotypical children with and without FGID. Children with ASD-FGID also had significantly lower levels of Dorea formicigenerans (P = .006), Blautia luti (P = .02), and Sutterella species (P = .03). “Overall, our identification of clostridial species aligns with previous autism studies that have identified microbiome alterations,” the researchers noted.
They also looked specifically at abdominal pain. Children with ASD-FGID and abdominal pain had significantly higher gut mucosal levels of Turicibacter sanguinis (P = .03), Clostridium aldenense (P = .004), Clostridium lituseburense (P = .003), Oscillospira plautii (P = .01), Clostridium disporicum (P = .049), and Clostridium tertium (P = .045) than did any other subgroup, the investigators found. Patients with both ASD-FGID and abdominal pain also had significantly higher levels of C. aldenense (P = .03), O. plautii (P = .04), Tyzzerella species (P = .045), and Parasutterella excrementihominis (P = .04) than did ASD-FGID patients without abdominal pain.
Both C. disporicum and C. tertium correlated with increases in the proinflammatory cytokines IL6 and interferon-gamma. Levels of these cytokines were highest in patients with ASD-FGID, and IL6 was highest of all among children with ASD-FGID with abdominal pain. Another proinflammatory cytokine, IL17A, also correlated with Clostridia species that were enriched in children with ASD-FGID. Both IL6 and IL17A have been implicated in autism-like phenotypes in rodents, the researchers noted. Several other cytokines also were linked to ASD-FGID, and abdominal pain correlated significantly with increases in MCP-1 (P = .03) and eotaxin (P = .03).
Gut mucosal levels of tryptophan were significantly lower among children with ASD-FGID compared with neurotypical children, either with (P = .006) or without (P = .009) FGID. In contrast, gut mucosal levels of 5-HIAA, the primary metabolite of serotonin, were significantly higher among children with ASD-FGID compared with neurotypical children (P = .01). Increased 5-HIAA also correlated significantly with abdominal pain (P = .04). Six species of bacteria correlated significantly with tryptophan or serotonin, implicating the gut microbiome in the serotonin pathway.
“Although these initial findings are correlative, these data form the framework for future studies targeting tryptophan-serotonin metabolism and inflammatory pathways in FGID in ASD,” the researchers concluded.
The U.S. Department of Health and Human Services funded the work. The investigators had no relevant disclosures.
The gut microbiomes of children with autism spectrum disorder (ASD) and functional gastrointestinal disorders (FGID) had significantly higher levels of several Clostridium species and lower concentrations of other bacteria compared with neurotypical children with and without FGIDs, which correlated with increases in inflammatory cytokines, decreased tryptophan, and increased serotonin, according to a small, single-center, cross-sectional study.
This “unique multi-omic profile [was] specific to ASD-FGID and ASD-FGID with abdominal pain,” wrote Ruth Ann Luna, PhD, of Texas Children’s Microbiome Center at Texas Children’s Hospital, Houston, and her associates. The report was published online in Cellular and Molecular Gastroenterology and Hepatology (doi: 10.1016/j.jcmgh.2016.11.008).
Children with ASD are at increased risk for FGIDs such as functional constipation, nonretentive fecal incontinence, functional abdominal pain, abdominal migraines, and irritable bowel syndrome, compared with their neurotypical peers. Changes in the gut microbiome can affect immunologic pathways and the balance between tryptophan and serotonin. This altered “microbial-gut-brain axis” has been reported in both ASD and FGID, suggesting “that altered gut-brain communications not only may play a role in the increased occurrence of FGIDs in ASD individuals, but could advance our understanding of potential risk factors for FGID in the ASD community,” the researchers wrote.
Previous studies of stool specimens have found higher levels of several species of Clostridium in pediatric ASD compared with neurotypical children. To confirm and expand on that work, the investigators examined microbial and neuroimmune markers in rectal biopsies and blood specimens from 14 children with ASD-FGID, 15 neurotypical children with FGID, and 6 asymptomatic neurotypical children. Participants were recruited from Nationwide Children’s Hospital in Columbus, Ohio. The researchers quantified microbial 16S ribosomal DNA community signatures, cytokines, chemokines, and serotonergic metabolites, and correlated results with parental responses to the Questionnaire on Pediatric Gastrointestinal Symptoms–Rome III version.
The ASD-FGID group had significantly higher numbers for ribosomal DNA sequences for Clostridium lituseburense (P = .002), Lachnoclostridium bolteae (P = .02), Lachnoclostridium hathewayi (P = .03), Clostridium aldenense (P = .04), and Oscillospira plautii (P = .04), compared with neurotypical children with and without FGID. Children with ASD-FGID also had significantly lower levels of Dorea formicigenerans (P = .006), Blautia luti (P = .02), and Sutterella species (P = .03). “Overall, our identification of clostridial species aligns with previous autism studies that have identified microbiome alterations,” the researchers noted.
They also looked specifically at abdominal pain. Children with ASD-FGID and abdominal pain had significantly higher gut mucosal levels of Turicibacter sanguinis (P = .03), Clostridium aldenense (P = .004), Clostridium lituseburense (P = .003), Oscillospira plautii (P = .01), Clostridium disporicum (P = .049), and Clostridium tertium (P = .045) than did any other subgroup, the investigators found. Patients with both ASD-FGID and abdominal pain also had significantly higher levels of C. aldenense (P = .03), O. plautii (P = .04), Tyzzerella species (P = .045), and Parasutterella excrementihominis (P = .04) than did ASD-FGID patients without abdominal pain.
Both C. disporicum and C. tertium correlated with increases in the proinflammatory cytokines IL6 and interferon-gamma. Levels of these cytokines were highest in patients with ASD-FGID, and IL6 was highest of all among children with ASD-FGID with abdominal pain. Another proinflammatory cytokine, IL17A, also correlated with Clostridia species that were enriched in children with ASD-FGID. Both IL6 and IL17A have been implicated in autism-like phenotypes in rodents, the researchers noted. Several other cytokines also were linked to ASD-FGID, and abdominal pain correlated significantly with increases in MCP-1 (P = .03) and eotaxin (P = .03).
Gut mucosal levels of tryptophan were significantly lower among children with ASD-FGID compared with neurotypical children, either with (P = .006) or without (P = .009) FGID. In contrast, gut mucosal levels of 5-HIAA, the primary metabolite of serotonin, were significantly higher among children with ASD-FGID compared with neurotypical children (P = .01). Increased 5-HIAA also correlated significantly with abdominal pain (P = .04). Six species of bacteria correlated significantly with tryptophan or serotonin, implicating the gut microbiome in the serotonin pathway.
“Although these initial findings are correlative, these data form the framework for future studies targeting tryptophan-serotonin metabolism and inflammatory pathways in FGID in ASD,” the researchers concluded.
The U.S. Department of Health and Human Services funded the work. The investigators had no relevant disclosures.
FROM CELLULAR AND MOLECULAR GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: The mucosal microbiome of children with comorbid autism spectrum disorder and functional gastrointestinal disorders significantly differed from that of neurotypical children with and without FGIDs, and these differences correlated with altered levels of inflammatory cytokines, tryptophan, and serotonin.
Major finding: Children with ASD-FGID had significant increases in Clostridium lituseburense (P = .002), Lachnoclostridium bolteae (P = .02), Lachnoclostridium hathewayi (P = .03), Clostridium aldenense (P = .04), and Oscillospira plautii (P = .04), and significant decreases in Dorea formicigenerans (P = .006), Blautia luti (P = .020), and Sutterella species (P = .025). The ASD-FGID phenotype was characterized by significantly lower gut levels of tryptophan, with higher levels of the serotonin metabolite 5-HIAA, and with several proinflammatory cytokines. Several bacterial species correlated with tryptophan, serotonin, or proinflammatory cytokines.
Data source: A single-center cross-sectional study of 14 children with ASD-FGID and 21 neurotypical children, of whom 15 had FGIDs.
Disclosures: The U.S. Department of Health and Human Services funded the work. The investigators had no relevant disclosures.
VIDEO: Infectious enteritis quadrupled short-term risk of IBS
More than 10% of patients developed irritable bowel syndrome (IBS) within a year after infectious enteritis, which gave them a more than fourfold greater risk than that of controls, according to a systematic review and meta-analysis of 45 studies.
“Protozoal and bacterial enteritis confer the greatest overall risk, although the magnitude of increased risk diminishes with time since exposure,” Fabiane B. Klem, MD, and Akhilesh Wadhwa, MD, of Mayo Clinic in Rochester, Minn., and their associates wrote in the April issue of Gastroenterology. Other significant risk factors for postinfectious IBS (PI-IBS) included female sex, clinically severe infections, antibiotic therapy, and comorbid psychological distress, they said.
Postinfectious IBS can last at least a decade after resolution of campylobacteriosis, shigellosis, salmonellosis, giardiasis, and norovirus infections, even when patients have no other risk factors for IBS, the researchers noted. To update and expand the most recent meta-analysis of this topic (Aliment Pharmacol Ther. 2007;26:535-44), the investigators searched Ovid Medline, EMBASE, Web of Science, and Cochrane Database of Systematic Reviews for studies published from 2006 through Aug. 31, 2015. This search yielded 45 studies, including 30 studies comparing infected patients with controls, who were usually matched by age, sex, and geographic location (Gastroenterology. 2017 Jan 6. doi: 10.1053/j.gastro.2016.12.039).
In all, 10.1% of patients with infectious enteritis developed IBS in the next 12 months (95% confidence interval, 7.2-14.1) – a 4.2-fold increase in risk, compared with that of controls (risk ratio, 4.2; 95% CI, 3.2-5.7). This risk subsequently dropped, but remained significantly elevated (RR, 2.3; 95% CI, 1.8-3.0), compared with controls. “Of patients with enteritis caused by protozoa or parasites, 41.9% developed IBS; of patients with enteritis caused by bacterial infection, 13.8% developed IBS,” the researchers emphasized. Patients with these infections remained at elevated risk of PI-IBS even after 1 year. Viral enteritis also significantly increased the risk of PI-IBS, but risk dropped to baseline levels after a year.
Among 10 pooled studies of IBS subtypes, 46% of patients had mixed IBS, 39% had diarrhea-predominant IBS, and 15% had constipation-predominant IBS. Female sex doubled the odds of PI-IBS (odds ratio, 2.2; 95% CI, 1.6-3.1) in 11 pooled studies. Significant clinical risk factors for PI-IBS included diarrhea lasting more than 7 days (eight studies; OR, 2.6; 95% CI, 1.5-4.6), bloody stool (four studies; OR, 1.9; 95% CI, 1.1-3.0), and antibiotic therapy during infectious enteritis (seven studies; OR, 1.7; 95% CI, 1.2-2.4).
Multiple reports linked PI-IBS to clinical psychological distress at the time of infectious enteritis. Specific risk factors included depression based on the Hospitalization Anxiety and Depression Scale (five studies; OR, 1.5; 95% CI, 1.2-1.9), anxiety based on the Hospital Anxiety and Depression Scale (four studies; OR, 2.0; 95% CI, 1.3-2.9), somatization (four studies; OR, 4.1; 95% CI, 2.7-6.0), and neuroticism (two studies; OR, 3.3; 95% CI, 1.6-6.6). Isolated studies also implicated hypochondriasis, extroversion, negative illness beliefs, stress, sleep disturbance, and adverse life events in the preceding year, the researchers said.
They found no evidence of publication bias, but noted a substantial amount of heterogeneity among studies. Also, some studies did not report multivariate analyses, so individual odds ratios might reflect “a conglomeration of factors,” they said.
The National Institutes of Health and the American Gastroenterological Association funded the work. The investigators reported having no conflicts of interest.
Source: American Gastroenterological Association
The phenomenon of IBS developing after a bout of gastroenteritis (postinfectious [PI]–IBS) was first reported in 1950 and subsequently elaborated by studies from Oxford (Q J Med. 1962;123:307-22), Sheffield (Gut. 1999;44:400-6), and Nottingham (BMJ 1997;314:779-82; Gut. 2000;47:804-11). It has proven to be a fertile area for research, which is the basis for this excellent meta-analysis.
The authors identified 45 studies, 29 in the last decade including a total of 21,421 participants with exposure to gastroenteritis. The pooled prevalence for PI-IBS was 11.5% (95% CI, 8.2%-15.8%) but with considerable heterogeneity, which the authors attempted to explain by a number of subgroup analyses. The authors report that protozoal infection seems to have a higher rate of PI-IBS than bacterial or viral infection, though some caution is warranted, since these figures rely on reports from just one outbreak of giardiasis in Bergen, Norway (Scand J Gastroenterol. 2012;47:956-61). However, if true, this might suggest that a different immune response could be responsible, a feature which others have suggested might predispose particular individuals to PI-IBS (Gut. 2016;65[8]1279-88).

The meta-analysis confirms the consistent increased risk in female patients (odds ratio, 1.69), anxiety (OR, 1.97), and somatization (greatest RR, 4.05), all common risks for the development of IBS but not specific to PI-IBS. Initial disease severity indicators, including bloody stool and more than 7 days of initial illness, which might indicate the severity of underlying damage to the gut, were shown to be significant risk factors. Animal studies of acute infection, particularly parasitic infestation, indicate that significant changes can be seen in both nerve and muscle, but routine histology in PI-IBS patients is normal. Infection produces a striking increase in gut permeability (Gut 2000;47:804-11), a feature of IBS whose molecular basis has been demonstrated by a series of elegant studies (Gut. 2017 Jan 12 [Epub ahead of print]; Gut. 2015;64:1379-88) demonstrating altered tight junctions and immune activation in IBS with diarrhea. The authors found treatment with antibiotics increased the risk of PI-IBS but whether this is attributable to confounding by indication is unclear.
This meta-analysis indicates that PI-IBS also potentially is the most common cause of IBS, given that both the Centers for Disease Control and Prevention in the United States and community surveys in the United Kingdom (BMJ. 1999;318:1046-50) indicate that gastroenteritis affects around 1 in 5 of the population each year. If the incidence of PI-IBS is around 10%, modeling suggests PI-IBS could account for the majority of new cases (J Neurogastroenterol Motil. 2012;18:200-4).
Dr. Robin Spiller is professor of gastroenterology, NIHR Nottingham Digestive Diseases Biomedical Research Unit, Nottingham Digestive Diseases Centre, University of Nottingham, England. He has no relevant conflicts of interest.
The phenomenon of IBS developing after a bout of gastroenteritis (postinfectious [PI]–IBS) was first reported in 1950 and subsequently elaborated by studies from Oxford (Q J Med. 1962;123:307-22), Sheffield (Gut. 1999;44:400-6), and Nottingham (BMJ 1997;314:779-82; Gut. 2000;47:804-11). It has proven to be a fertile area for research, which is the basis for this excellent meta-analysis.
The authors identified 45 studies, 29 in the last decade including a total of 21,421 participants with exposure to gastroenteritis. The pooled prevalence for PI-IBS was 11.5% (95% CI, 8.2%-15.8%) but with considerable heterogeneity, which the authors attempted to explain by a number of subgroup analyses. The authors report that protozoal infection seems to have a higher rate of PI-IBS than bacterial or viral infection, though some caution is warranted, since these figures rely on reports from just one outbreak of giardiasis in Bergen, Norway (Scand J Gastroenterol. 2012;47:956-61). However, if true, this might suggest that a different immune response could be responsible, a feature which others have suggested might predispose particular individuals to PI-IBS (Gut. 2016;65[8]1279-88).
The meta-analysis confirms the consistent increased risk in female patients (odds ratio, 1.69), anxiety (OR, 1.97), and somatization (greatest RR, 4.05), all common risks for the development of IBS but not specific to PI-IBS. Initial disease severity indicators, including bloody stool and more than 7 days of initial illness, which might indicate the severity of underlying damage to the gut, were shown to be significant risk factors. Animal studies of acute infection, particularly parasitic infestation, indicate that significant changes can be seen in both nerve and muscle, but routine histology in PI-IBS patients is normal. Infection produces a striking increase in gut permeability (Gut 2000;47:804-11), a feature of IBS whose molecular basis has been demonstrated by a series of elegant studies (Gut. 2017 Jan 12 [Epub ahead of print]; Gut. 2015;64:1379-88) demonstrating altered tight junctions and immune activation in IBS with diarrhea. The authors found treatment with antibiotics increased the risk of PI-IBS but whether this is attributable to confounding by indication is unclear.
This meta-analysis indicates that PI-IBS also potentially is the most common cause of IBS, given that both the Centers for Disease Control and Prevention in the United States and community surveys in the United Kingdom (BMJ. 1999;318:1046-50) indicate that gastroenteritis affects around 1 in 5 of the population each year. If the incidence of PI-IBS is around 10%, modeling suggests PI-IBS could account for the majority of new cases (J Neurogastroenterol Motil. 2012;18:200-4).
Dr. Robin Spiller is professor of gastroenterology, NIHR Nottingham Digestive Diseases Biomedical Research Unit, Nottingham Digestive Diseases Centre, University of Nottingham, England. He has no relevant conflicts of interest.
The phenomenon of IBS developing after a bout of gastroenteritis (postinfectious [PI]–IBS) was first reported in 1950 and subsequently elaborated by studies from Oxford (Q J Med. 1962;123:307-22), Sheffield (Gut. 1999;44:400-6), and Nottingham (BMJ 1997;314:779-82; Gut. 2000;47:804-11). It has proven to be a fertile area for research, which is the basis for this excellent meta-analysis.
The authors identified 45 studies, 29 in the last decade including a total of 21,421 participants with exposure to gastroenteritis. The pooled prevalence for PI-IBS was 11.5% (95% CI, 8.2%-15.8%) but with considerable heterogeneity, which the authors attempted to explain by a number of subgroup analyses. The authors report that protozoal infection seems to have a higher rate of PI-IBS than bacterial or viral infection, though some caution is warranted, since these figures rely on reports from just one outbreak of giardiasis in Bergen, Norway (Scand J Gastroenterol. 2012;47:956-61). However, if true, this might suggest that a different immune response could be responsible, a feature which others have suggested might predispose particular individuals to PI-IBS (Gut. 2016;65[8]1279-88).
The meta-analysis confirms the consistent increased risk in female patients (odds ratio, 1.69), anxiety (OR, 1.97), and somatization (greatest RR, 4.05), all common risks for the development of IBS but not specific to PI-IBS. Initial disease severity indicators, including bloody stool and more than 7 days of initial illness, which might indicate the severity of underlying damage to the gut, were shown to be significant risk factors. Animal studies of acute infection, particularly parasitic infestation, indicate that significant changes can be seen in both nerve and muscle, but routine histology in PI-IBS patients is normal. Infection produces a striking increase in gut permeability (Gut 2000;47:804-11), a feature of IBS whose molecular basis has been demonstrated by a series of elegant studies (Gut. 2017 Jan 12 [Epub ahead of print]; Gut. 2015;64:1379-88) demonstrating altered tight junctions and immune activation in IBS with diarrhea. The authors found treatment with antibiotics increased the risk of PI-IBS but whether this is attributable to confounding by indication is unclear.
This meta-analysis indicates that PI-IBS also potentially is the most common cause of IBS, given that both the Centers for Disease Control and Prevention in the United States and community surveys in the United Kingdom (BMJ. 1999;318:1046-50) indicate that gastroenteritis affects around 1 in 5 of the population each year. If the incidence of PI-IBS is around 10%, modeling suggests PI-IBS could account for the majority of new cases (J Neurogastroenterol Motil. 2012;18:200-4).
Dr. Robin Spiller is professor of gastroenterology, NIHR Nottingham Digestive Diseases Biomedical Research Unit, Nottingham Digestive Diseases Centre, University of Nottingham, England. He has no relevant conflicts of interest.
More than 10% of patients developed irritable bowel syndrome (IBS) within a year after infectious enteritis, which gave them a more than fourfold greater risk than that of controls, according to a systematic review and meta-analysis of 45 studies.
“Protozoal and bacterial enteritis confer the greatest overall risk, although the magnitude of increased risk diminishes with time since exposure,” Fabiane B. Klem, MD, and Akhilesh Wadhwa, MD, of Mayo Clinic in Rochester, Minn., and their associates wrote in the April issue of Gastroenterology. Other significant risk factors for postinfectious IBS (PI-IBS) included female sex, clinically severe infections, antibiotic therapy, and comorbid psychological distress, they said.
Postinfectious IBS can last at least a decade after resolution of campylobacteriosis, shigellosis, salmonellosis, giardiasis, and norovirus infections, even when patients have no other risk factors for IBS, the researchers noted. To update and expand the most recent meta-analysis of this topic (Aliment Pharmacol Ther. 2007;26:535-44), the investigators searched Ovid Medline, EMBASE, Web of Science, and Cochrane Database of Systematic Reviews for studies published from 2006 through Aug. 31, 2015. This search yielded 45 studies, including 30 studies comparing infected patients with controls, who were usually matched by age, sex, and geographic location (Gastroenterology. 2017 Jan 6. doi: 10.1053/j.gastro.2016.12.039).
In all, 10.1% of patients with infectious enteritis developed IBS in the next 12 months (95% confidence interval, 7.2-14.1) – a 4.2-fold increase in risk, compared with that of controls (risk ratio, 4.2; 95% CI, 3.2-5.7). This risk subsequently dropped, but remained significantly elevated (RR, 2.3; 95% CI, 1.8-3.0), compared with controls. “Of patients with enteritis caused by protozoa or parasites, 41.9% developed IBS; of patients with enteritis caused by bacterial infection, 13.8% developed IBS,” the researchers emphasized. Patients with these infections remained at elevated risk of PI-IBS even after 1 year. Viral enteritis also significantly increased the risk of PI-IBS, but risk dropped to baseline levels after a year.
Among 10 pooled studies of IBS subtypes, 46% of patients had mixed IBS, 39% had diarrhea-predominant IBS, and 15% had constipation-predominant IBS. Female sex doubled the odds of PI-IBS (odds ratio, 2.2; 95% CI, 1.6-3.1) in 11 pooled studies. Significant clinical risk factors for PI-IBS included diarrhea lasting more than 7 days (eight studies; OR, 2.6; 95% CI, 1.5-4.6), bloody stool (four studies; OR, 1.9; 95% CI, 1.1-3.0), and antibiotic therapy during infectious enteritis (seven studies; OR, 1.7; 95% CI, 1.2-2.4).
Multiple reports linked PI-IBS to clinical psychological distress at the time of infectious enteritis. Specific risk factors included depression based on the Hospitalization Anxiety and Depression Scale (five studies; OR, 1.5; 95% CI, 1.2-1.9), anxiety based on the Hospital Anxiety and Depression Scale (four studies; OR, 2.0; 95% CI, 1.3-2.9), somatization (four studies; OR, 4.1; 95% CI, 2.7-6.0), and neuroticism (two studies; OR, 3.3; 95% CI, 1.6-6.6). Isolated studies also implicated hypochondriasis, extroversion, negative illness beliefs, stress, sleep disturbance, and adverse life events in the preceding year, the researchers said.
They found no evidence of publication bias, but noted a substantial amount of heterogeneity among studies. Also, some studies did not report multivariate analyses, so individual odds ratios might reflect “a conglomeration of factors,” they said.
The National Institutes of Health and the American Gastroenterological Association funded the work. The investigators reported having no conflicts of interest.
Source: American Gastroenterological Association
More than 10% of patients developed irritable bowel syndrome (IBS) within a year after infectious enteritis, which gave them a more than fourfold greater risk than that of controls, according to a systematic review and meta-analysis of 45 studies.
“Protozoal and bacterial enteritis confer the greatest overall risk, although the magnitude of increased risk diminishes with time since exposure,” Fabiane B. Klem, MD, and Akhilesh Wadhwa, MD, of Mayo Clinic in Rochester, Minn., and their associates wrote in the April issue of Gastroenterology. Other significant risk factors for postinfectious IBS (PI-IBS) included female sex, clinically severe infections, antibiotic therapy, and comorbid psychological distress, they said.
Postinfectious IBS can last at least a decade after resolution of campylobacteriosis, shigellosis, salmonellosis, giardiasis, and norovirus infections, even when patients have no other risk factors for IBS, the researchers noted. To update and expand the most recent meta-analysis of this topic (Aliment Pharmacol Ther. 2007;26:535-44), the investigators searched Ovid Medline, EMBASE, Web of Science, and Cochrane Database of Systematic Reviews for studies published from 2006 through Aug. 31, 2015. This search yielded 45 studies, including 30 studies comparing infected patients with controls, who were usually matched by age, sex, and geographic location (Gastroenterology. 2017 Jan 6. doi: 10.1053/j.gastro.2016.12.039).
In all, 10.1% of patients with infectious enteritis developed IBS in the next 12 months (95% confidence interval, 7.2-14.1) – a 4.2-fold increase in risk, compared with that of controls (risk ratio, 4.2; 95% CI, 3.2-5.7). This risk subsequently dropped, but remained significantly elevated (RR, 2.3; 95% CI, 1.8-3.0), compared with controls. “Of patients with enteritis caused by protozoa or parasites, 41.9% developed IBS; of patients with enteritis caused by bacterial infection, 13.8% developed IBS,” the researchers emphasized. Patients with these infections remained at elevated risk of PI-IBS even after 1 year. Viral enteritis also significantly increased the risk of PI-IBS, but risk dropped to baseline levels after a year.
Among 10 pooled studies of IBS subtypes, 46% of patients had mixed IBS, 39% had diarrhea-predominant IBS, and 15% had constipation-predominant IBS. Female sex doubled the odds of PI-IBS (odds ratio, 2.2; 95% CI, 1.6-3.1) in 11 pooled studies. Significant clinical risk factors for PI-IBS included diarrhea lasting more than 7 days (eight studies; OR, 2.6; 95% CI, 1.5-4.6), bloody stool (four studies; OR, 1.9; 95% CI, 1.1-3.0), and antibiotic therapy during infectious enteritis (seven studies; OR, 1.7; 95% CI, 1.2-2.4).
Multiple reports linked PI-IBS to clinical psychological distress at the time of infectious enteritis. Specific risk factors included depression based on the Hospitalization Anxiety and Depression Scale (five studies; OR, 1.5; 95% CI, 1.2-1.9), anxiety based on the Hospital Anxiety and Depression Scale (four studies; OR, 2.0; 95% CI, 1.3-2.9), somatization (four studies; OR, 4.1; 95% CI, 2.7-6.0), and neuroticism (two studies; OR, 3.3; 95% CI, 1.6-6.6). Isolated studies also implicated hypochondriasis, extroversion, negative illness beliefs, stress, sleep disturbance, and adverse life events in the preceding year, the researchers said.
They found no evidence of publication bias, but noted a substantial amount of heterogeneity among studies. Also, some studies did not report multivariate analyses, so individual odds ratios might reflect “a conglomeration of factors,” they said.
The National Institutes of Health and the American Gastroenterological Association funded the work. The investigators reported having no conflicts of interest.
Source: American Gastroenterological Association
FROM GASTROENTEROLOGY
Key clinical point.
Major finding: A total of 10.1% of patients with infectious enteritis developed IBS in the next 12 months, a 4.2-fold increase in risk, compared with that of controls.
Data source: A systematic review and meta-analysis of 45 studies.
Disclosures: The National Institutes of Health and the American Gastroenterological Association funded the work. The investigators reported having no conflicts of interest.
Gluten-free diets related to high levels of arsenic, mercury
Individuals who adopt a gluten-free diet are putting themselves at risk for uncommonly high levels of arsenic and mercury, according to the findings of a recent study published in Epidemiology.
“Despite [less than] 1% of Americans having diagnosed celiac disease, an estimated 25% of American consumers reported consuming gluten-free food in 2015, a 67% increase from 2013,” wrote the authors of the study, led by Maria Argos, PhD, of the University of Illinois at Chicago. “Despite such a dramatic shift in the diet of many Americans, little is known about how gluten-free diets might affect exposure to toxic metals found in certain foods,” they noted.
Dr. Argos and her colleagues analyzed data collected from the National Health and Nutrition Examination Survey (NHANES), which included self-reported questionnaires in which subjects indicated what type of diet they were on, if any. Data on those who indicated that they followed a gluten-free diet were analyzed to determine if their urinary and blood biomarkers indicated any exposure to toxic metals. A total of 7,471 subjects from the NHANES were included in the analysis.
“We accounted for the complex sampling design of NHANES [by] using Taylor series linearization and sampling weights, per the NHANES analytic guidelines, to ensure unbiased and nationally representative estimates,” the authors explained. 
A total of 73 subjects identified themselves as following a gluten-free diet. Within this group, the mean total arsenic level in urine was found to be 12.1 mcg/L, compared to 7.8 mcg/L for the other 7,398 subjects. Levels of dimethylarsinic acid averaged 5.3 mcg/L for those who were gluten-free, but only 3.7 for everyone else, while cadmium and lead levels were also slightly higher for gluten-free individuals: 0.18 mcg/L vs. 0.16 mcg/L, and 0.40 mcg/L vs. 0.37 mcg/L, respectively.
Blood analyses showed that total mercury levels were also substantially higher in the gluten-free group, at a mean of 1.3 mcg/L compared to 0.8 mcg/L. While cadmium levels were the same between the two – both showed a mean level of 0.29 mcg/L – lead measured 1.1 mcg/dL and inorganic mercury measured 0.30 mcg/L, compared to 0.96 mcg/L and 0.28 mcg/L in everyone else, respectively.
Geometric mean ratios showed that total arsenic, total arsenic 1, and total mercury levels had the largest disparity between the two groups. Total arsenic registered a 1.5 (95% CI, 1.2-2.0), total mercury a 1.7 (95% CI, 1.1-2.4), and total arsenic 1 a 1.9 (95% CI 1.3-2.6), meaning the gluten-free group had nearly double the risk for higher levels than those on other diets.
“These findings may have important health implications since the health effects of low-level arsenic and mercury exposure from food sources are uncertain but may increase the risk for cancer and other chronic diseases,” Dr. Argos and her coauthors concluded, adding that “future studies are needed to more fully examine exposure to toxic metals from consuming gluten-free foods.”
The study was funded by a grant from the National Institutes of Health. Dr. Argos and her coauthors did not report any relevant financial disclosures.
Individuals who adopt a gluten-free diet are putting themselves at risk for uncommonly high levels of arsenic and mercury, according to the findings of a recent study published in Epidemiology.
“Despite [less than] 1% of Americans having diagnosed celiac disease, an estimated 25% of American consumers reported consuming gluten-free food in 2015, a 67% increase from 2013,” wrote the authors of the study, led by Maria Argos, PhD, of the University of Illinois at Chicago. “Despite such a dramatic shift in the diet of many Americans, little is known about how gluten-free diets might affect exposure to toxic metals found in certain foods,” they noted.
Dr. Argos and her colleagues analyzed data collected from the National Health and Nutrition Examination Survey (NHANES), which included self-reported questionnaires in which subjects indicated what type of diet they were on, if any. Data on those who indicated that they followed a gluten-free diet were analyzed to determine if their urinary and blood biomarkers indicated any exposure to toxic metals. A total of 7,471 subjects from the NHANES were included in the analysis.
“We accounted for the complex sampling design of NHANES [by] using Taylor series linearization and sampling weights, per the NHANES analytic guidelines, to ensure unbiased and nationally representative estimates,” the authors explained.
A total of 73 subjects identified themselves as following a gluten-free diet. Within this group, the mean total arsenic level in urine was found to be 12.1 mcg/L, compared to 7.8 mcg/L for the other 7,398 subjects. Levels of dimethylarsinic acid averaged 5.3 mcg/L for those who were gluten-free, but only 3.7 for everyone else, while cadmium and lead levels were also slightly higher for gluten-free individuals: 0.18 mcg/L vs. 0.16 mcg/L, and 0.40 mcg/L vs. 0.37 mcg/L, respectively.
Blood analyses showed that total mercury levels were also substantially higher in the gluten-free group, at a mean of 1.3 mcg/L compared to 0.8 mcg/L. While cadmium levels were the same between the two – both showed a mean level of 0.29 mcg/L – lead measured 1.1 mcg/dL and inorganic mercury measured 0.30 mcg/L, compared to 0.96 mcg/L and 0.28 mcg/L in everyone else, respectively.
Geometric mean ratios showed that total arsenic, total arsenic 1, and total mercury levels had the largest disparity between the two groups. Total arsenic registered a 1.5 (95% CI, 1.2-2.0), total mercury a 1.7 (95% CI, 1.1-2.4), and total arsenic 1 a 1.9 (95% CI 1.3-2.6), meaning the gluten-free group had nearly double the risk for higher levels than those on other diets.
“These findings may have important health implications since the health effects of low-level arsenic and mercury exposure from food sources are uncertain but may increase the risk for cancer and other chronic diseases,” Dr. Argos and her coauthors concluded, adding that “future studies are needed to more fully examine exposure to toxic metals from consuming gluten-free foods.”
The study was funded by a grant from the National Institutes of Health. Dr. Argos and her coauthors did not report any relevant financial disclosures.
Individuals who adopt a gluten-free diet are putting themselves at risk for uncommonly high levels of arsenic and mercury, according to the findings of a recent study published in Epidemiology.
“Despite [less than] 1% of Americans having diagnosed celiac disease, an estimated 25% of American consumers reported consuming gluten-free food in 2015, a 67% increase from 2013,” wrote the authors of the study, led by Maria Argos, PhD, of the University of Illinois at Chicago. “Despite such a dramatic shift in the diet of many Americans, little is known about how gluten-free diets might affect exposure to toxic metals found in certain foods,” they noted.
Dr. Argos and her colleagues analyzed data collected from the National Health and Nutrition Examination Survey (NHANES), which included self-reported questionnaires in which subjects indicated what type of diet they were on, if any. Data on those who indicated that they followed a gluten-free diet were analyzed to determine if their urinary and blood biomarkers indicated any exposure to toxic metals. A total of 7,471 subjects from the NHANES were included in the analysis.
“We accounted for the complex sampling design of NHANES [by] using Taylor series linearization and sampling weights, per the NHANES analytic guidelines, to ensure unbiased and nationally representative estimates,” the authors explained.
A total of 73 subjects identified themselves as following a gluten-free diet. Within this group, the mean total arsenic level in urine was found to be 12.1 mcg/L, compared to 7.8 mcg/L for the other 7,398 subjects. Levels of dimethylarsinic acid averaged 5.3 mcg/L for those who were gluten-free, but only 3.7 for everyone else, while cadmium and lead levels were also slightly higher for gluten-free individuals: 0.18 mcg/L vs. 0.16 mcg/L, and 0.40 mcg/L vs. 0.37 mcg/L, respectively.
Blood analyses showed that total mercury levels were also substantially higher in the gluten-free group, at a mean of 1.3 mcg/L compared to 0.8 mcg/L. While cadmium levels were the same between the two – both showed a mean level of 0.29 mcg/L – lead measured 1.1 mcg/dL and inorganic mercury measured 0.30 mcg/L, compared to 0.96 mcg/L and 0.28 mcg/L in everyone else, respectively.
Geometric mean ratios showed that total arsenic, total arsenic 1, and total mercury levels had the largest disparity between the two groups. Total arsenic registered a 1.5 (95% CI, 1.2-2.0), total mercury a 1.7 (95% CI, 1.1-2.4), and total arsenic 1 a 1.9 (95% CI 1.3-2.6), meaning the gluten-free group had nearly double the risk for higher levels than those on other diets.
“These findings may have important health implications since the health effects of low-level arsenic and mercury exposure from food sources are uncertain but may increase the risk for cancer and other chronic diseases,” Dr. Argos and her coauthors concluded, adding that “future studies are needed to more fully examine exposure to toxic metals from consuming gluten-free foods.”
The study was funded by a grant from the National Institutes of Health. Dr. Argos and her coauthors did not report any relevant financial disclosures.
FROM EPIDEMIOLOGY
Key clinical point:
Major finding: Total arsenic and mercury levels were higher in those that self-reported being on gluten-free diet than in those who weren’t: 12.1 mcg/L vs. 7.8 mcg/L for arsenic and 1.3 mcg/L vs. 0.8 mcg/L for mercury.
Data source: Retrospective analysis of data from the National Health and Nutrition Examination Survey during 2009-2014.
Disclosures: The National Institutes of Health funded the study. The authors had no relevant financial disclosures.
Fecal microbiota transplantation a decolonization treatment option
The use of fecal microbiota transplantation is an option to eradicate highly drug-resistant enteric bacteria carriage, according to results from a small pilot study conducted by French investigators.
“A rapid and dramatic emergence of highly drug-resistant enteric bacteria (HDREB), i.e., carbapenem-resistant Enterobacteriaceae (CRE) and vancomycin-resistant enterococci (VRE), is occurring worldwide,” researchers led by Benjamin Davido, MD, of the infectious diseases unit at Raymond Poincaré Teaching Hospital, Garches, France, wrote in a study published online Feb. 1 in the Journal of Hospital Infection. “Patients carrying these bacteria are at risk of developing severe infections due to these bacteria; these infections are associated with a high mortality rate, partially because of inappropriate antimicrobial treatment.”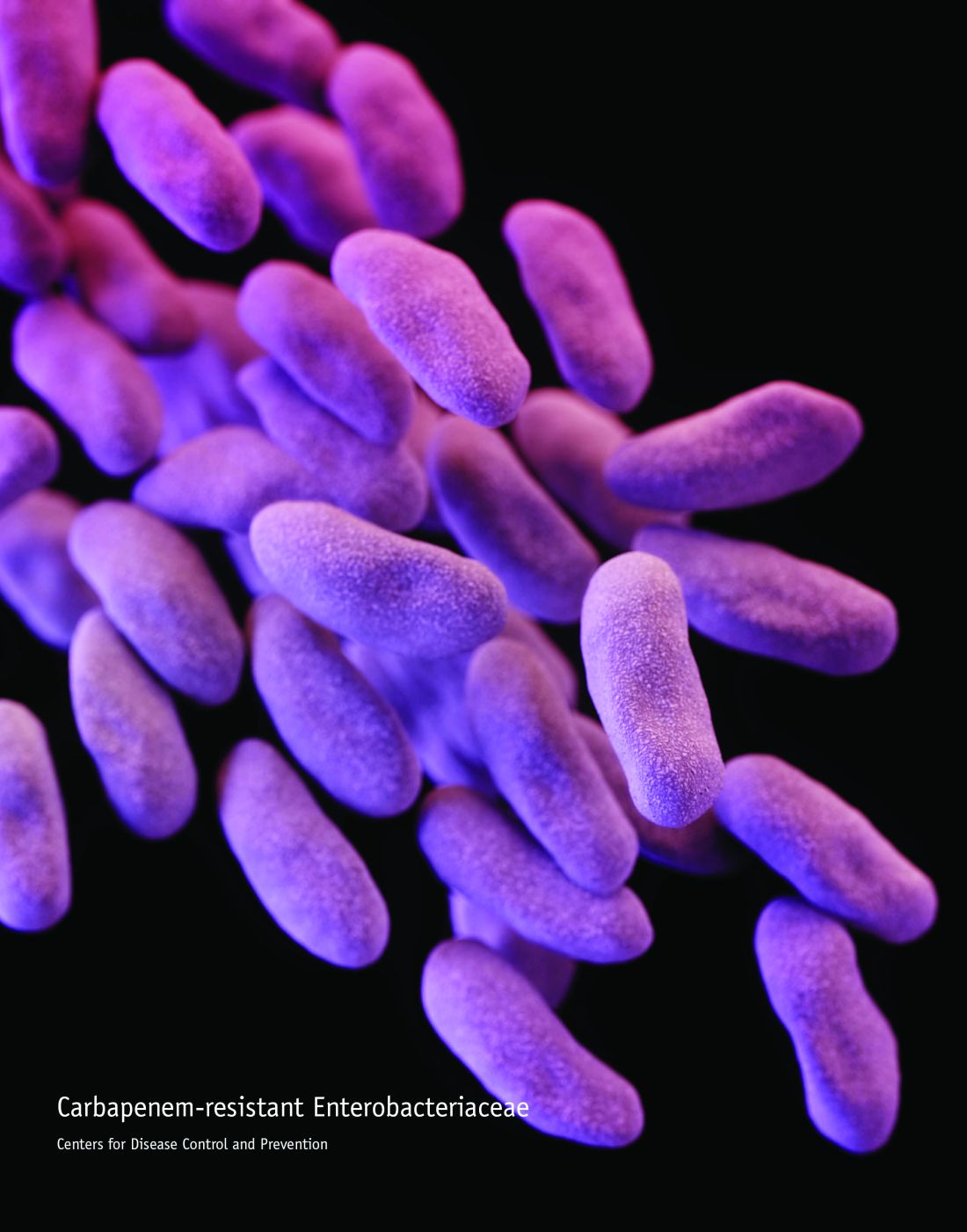
Citing recent studies that have demonstrated the efficacy of fecal microbiota transplant (FMT) as an accepted therapy to prevent recurrent Clostridium difficile infection, Dr. Davido and his associates prospectively identified eight different case reports of FMT used in adults for intestinal decolonization from extended-spectrum beta-lactamase (ESBL)–producing Enterobacteriaceae, VRE, or methicillin-resistant Staphylococcus aureus. Patients on immunosuppressive agents were excluded from the study, as were those taking antibiotics at the time of FMT (J Hosp Infect. 2017 Feb. 1. doi: 10.1016/j.jhin.2017.02.001).
The protocol for the procedure involved insertion of a nasoduodenal tube the day before FMT in order to perform a bowel lavage with XPrep solution. FMT was performed using a frozen preparation of fecal microbiota from a universal donor who was previously screened for potential diseases. The main outcome of interest was time to successful decolonization following FMT, which was determined by at least two consecutive negative rectal swabs at a 1-week interval during a follow-up of 3 months.
The mean age of the eight patients was 70 years, their median Charlson comorbidity index score was 5, and the median duration of carriage of HDREB before FMT was 83 days. The researchers observed that 1 month after FMT, two patients were free from CRE colonization, while one more patient was free from VRE after 3 months. Five patients received antibiotic during follow-up. Among them, one patient was decolonized at 1 month, while another was decolonized at 3 months. One patient with cirrhosis and persistent VRE carriage died 3 months after FMT from ascetic fluid infection and VRE bacteremia. No other adverse events were reported.
“In a context where no other efficient strategy is available, our first results show that FMT seems to be safe, with an impact on CRE decolonization at 1 month and on VRE decolonization at 3 months,” the researchers concluded. “Of particular importance, there was no recolonization after the intervention, which is contrary to decolonization with antibiotics. However, our study has important limitations in that the sample size was very small, it was nonrandomized, and follow-up was limited to a 3-month period.” They noted that at least five trials are underway to investigate the impact of FMT on multidrug resistant organism bacterial decolonization. The researchers reported having no financial disclosures.
The use of fecal microbiota transplantation is an option to eradicate highly drug-resistant enteric bacteria carriage, according to results from a small pilot study conducted by French investigators.
“A rapid and dramatic emergence of highly drug-resistant enteric bacteria (HDREB), i.e., carbapenem-resistant Enterobacteriaceae (CRE) and vancomycin-resistant enterococci (VRE), is occurring worldwide,” researchers led by Benjamin Davido, MD, of the infectious diseases unit at Raymond Poincaré Teaching Hospital, Garches, France, wrote in a study published online Feb. 1 in the Journal of Hospital Infection. “Patients carrying these bacteria are at risk of developing severe infections due to these bacteria; these infections are associated with a high mortality rate, partially because of inappropriate antimicrobial treatment.”
Citing recent studies that have demonstrated the efficacy of fecal microbiota transplant (FMT) as an accepted therapy to prevent recurrent Clostridium difficile infection, Dr. Davido and his associates prospectively identified eight different case reports of FMT used in adults for intestinal decolonization from extended-spectrum beta-lactamase (ESBL)–producing Enterobacteriaceae, VRE, or methicillin-resistant Staphylococcus aureus. Patients on immunosuppressive agents were excluded from the study, as were those taking antibiotics at the time of FMT (J Hosp Infect. 2017 Feb. 1. doi: 10.1016/j.jhin.2017.02.001).
The protocol for the procedure involved insertion of a nasoduodenal tube the day before FMT in order to perform a bowel lavage with XPrep solution. FMT was performed using a frozen preparation of fecal microbiota from a universal donor who was previously screened for potential diseases. The main outcome of interest was time to successful decolonization following FMT, which was determined by at least two consecutive negative rectal swabs at a 1-week interval during a follow-up of 3 months.
The mean age of the eight patients was 70 years, their median Charlson comorbidity index score was 5, and the median duration of carriage of HDREB before FMT was 83 days. The researchers observed that 1 month after FMT, two patients were free from CRE colonization, while one more patient was free from VRE after 3 months. Five patients received antibiotic during follow-up. Among them, one patient was decolonized at 1 month, while another was decolonized at 3 months. One patient with cirrhosis and persistent VRE carriage died 3 months after FMT from ascetic fluid infection and VRE bacteremia. No other adverse events were reported.
“In a context where no other efficient strategy is available, our first results show that FMT seems to be safe, with an impact on CRE decolonization at 1 month and on VRE decolonization at 3 months,” the researchers concluded. “Of particular importance, there was no recolonization after the intervention, which is contrary to decolonization with antibiotics. However, our study has important limitations in that the sample size was very small, it was nonrandomized, and follow-up was limited to a 3-month period.” They noted that at least five trials are underway to investigate the impact of FMT on multidrug resistant organism bacterial decolonization. The researchers reported having no financial disclosures.
The use of fecal microbiota transplantation is an option to eradicate highly drug-resistant enteric bacteria carriage, according to results from a small pilot study conducted by French investigators.
“A rapid and dramatic emergence of highly drug-resistant enteric bacteria (HDREB), i.e., carbapenem-resistant Enterobacteriaceae (CRE) and vancomycin-resistant enterococci (VRE), is occurring worldwide,” researchers led by Benjamin Davido, MD, of the infectious diseases unit at Raymond Poincaré Teaching Hospital, Garches, France, wrote in a study published online Feb. 1 in the Journal of Hospital Infection. “Patients carrying these bacteria are at risk of developing severe infections due to these bacteria; these infections are associated with a high mortality rate, partially because of inappropriate antimicrobial treatment.”
Citing recent studies that have demonstrated the efficacy of fecal microbiota transplant (FMT) as an accepted therapy to prevent recurrent Clostridium difficile infection, Dr. Davido and his associates prospectively identified eight different case reports of FMT used in adults for intestinal decolonization from extended-spectrum beta-lactamase (ESBL)–producing Enterobacteriaceae, VRE, or methicillin-resistant Staphylococcus aureus. Patients on immunosuppressive agents were excluded from the study, as were those taking antibiotics at the time of FMT (J Hosp Infect. 2017 Feb. 1. doi: 10.1016/j.jhin.2017.02.001).
The protocol for the procedure involved insertion of a nasoduodenal tube the day before FMT in order to perform a bowel lavage with XPrep solution. FMT was performed using a frozen preparation of fecal microbiota from a universal donor who was previously screened for potential diseases. The main outcome of interest was time to successful decolonization following FMT, which was determined by at least two consecutive negative rectal swabs at a 1-week interval during a follow-up of 3 months.
The mean age of the eight patients was 70 years, their median Charlson comorbidity index score was 5, and the median duration of carriage of HDREB before FMT was 83 days. The researchers observed that 1 month after FMT, two patients were free from CRE colonization, while one more patient was free from VRE after 3 months. Five patients received antibiotic during follow-up. Among them, one patient was decolonized at 1 month, while another was decolonized at 3 months. One patient with cirrhosis and persistent VRE carriage died 3 months after FMT from ascetic fluid infection and VRE bacteremia. No other adverse events were reported.
“In a context where no other efficient strategy is available, our first results show that FMT seems to be safe, with an impact on CRE decolonization at 1 month and on VRE decolonization at 3 months,” the researchers concluded. “Of particular importance, there was no recolonization after the intervention, which is contrary to decolonization with antibiotics. However, our study has important limitations in that the sample size was very small, it was nonrandomized, and follow-up was limited to a 3-month period.” They noted that at least five trials are underway to investigate the impact of FMT on multidrug resistant organism bacterial decolonization. The researchers reported having no financial disclosures.
FROM THE JOURNAL OF HOSPITAL INFECTION
Key clinical point:
Major finding: Of eight patients who underwent fecal microbiota transplantation (FMT) for carbapenem-resistant Enterobacteriaceae (CRE) and vancomycin-resistant enterococci (VRE) digestive tract decolonization, three achieved decolonization after 3 months.
Data source: A prospective study of eight cases of FMT used in adults for intestinal decolonization from drug-resistant enteric bacteria carriage.
Disclosures: The researchers reported having no financial disclosures.
Peripheral B cells reflect intestinal damage in celiac disease
B-cell gene expression in the peripheral blood strongly correlates with the extent of gluten-induced damage to the intestinal mucosa in patients with celiac disease, according to a report in Cellular and Molecular Gastroenterology and Hepatology.
If this finding from a single-center cohort study is validated in other patient populations, the B-cell signature may become a useful, minimally invasive tool for diagnosing celiac disease. Eventually, if biomarkers are developed from the peripheral B-cell signature, a simple blood test could be used for monitoring changes in gut inflammation over time as well as treatment response, said Mitchell E. Garber, PhD, of Alvine Pharmaceuticals, San Carlos, Calif., and the department of chemistry at Stanford University, and his associates.
However, it would be “premature but intriguing” to speculate about using this discovery to devise new, B cell–centered treatments for celiac disease, they added.
Noting that an inflammatory, gluten-induced immune response in the gut can be reflected in the peripheral blood, the investigators assessed whether a 6-week gluten challenge would induce damage to the small intestine that would show up in B-cell gene expression detected in blood samples. They assigned 73 patients at a single medical center in Finland to follow their usual gluten-free diets but to ingest an additional 6 g (20 patients), 3 g (26 patients), or 1.5 g (27 patients) of wheat gluten with a meal once per day for the study period.
The study participants (median age, 59 years; range, 23-74 years) underwent small-bowel biopsies obtained from the descending duodenum at baseline and after the gluten challenge. Damage to the intestinal mucosa was assessed by measuring the ratio of the height of the intestinal villi to the depth of the proliferative crypts at the base of the villi (villi height to crypt depth, or Vh:Cd). In celiac disease, gluten blunts the projection of the villi and causes hypertrophy or elongation of the crypts, resulting in flattened mucosa and a Vh:Cd approaching zero.
The study participants showed a wide variation in mucosal damage from the gluten exposure, with some patients showing no change and relatively healthy mucosa, and others showing extensive change and nearly flattened mucosa. The mucosal damage did not differ by gluten dose.
The largest change in Vh:Cd occurred in three patients who transitioned from relatively healthy intestinal mucosa (Vh:Cd 3.1) at baseline to nearly flat mucosa (Vh:Cd 0.2) after gluten exposure.
Patients with undamaged gut mucosa showed a relative increase in B-cell gene expression during the study period, while those who had increasing damage showed a relative decrease in B-cell expression. “The peripheral B cell therefore tracked with oral tolerance across the full spectrum of intestinal damage, from no change to a nearly flat mucosa,” Dr. Garber and his associates said (Cell Molec Gastroenterol Hepatol. 2017. doi: 10.1016/j.jcmgh.2017.01.011).
“The net increase in B-cell gene expression from baseline to 6 weeks in patients with little to no intestinal damage [suggests] that these individuals may have mounted a B-cell immune response to maintain mucosal homeostasis and circumvent inflammation,” they added.
Further study is needed to determine whether peripheral B cells are a marker that indicates tolerance to gluten but doesn’t play a functional role in the inflammatory process, or whether B cells may actually promote immune tolerance, the investigators said.
This study was funded by Alvine Pharmaceuticals, the American Recovery and Reinvestment Act, Tampere (Finland) University Hospital, the Academy of Finland Research Council for Health, and the U.S. National Institutes of Health. Dr. Garber reported ties to Alvine Pharmaceuticals, and his associates reported ties to ImmusanT, Celimmune, and ImmunogenX.
B-cell gene expression in the peripheral blood strongly correlates with the extent of gluten-induced damage to the intestinal mucosa in patients with celiac disease, according to a report in Cellular and Molecular Gastroenterology and Hepatology.
If this finding from a single-center cohort study is validated in other patient populations, the B-cell signature may become a useful, minimally invasive tool for diagnosing celiac disease. Eventually, if biomarkers are developed from the peripheral B-cell signature, a simple blood test could be used for monitoring changes in gut inflammation over time as well as treatment response, said Mitchell E. Garber, PhD, of Alvine Pharmaceuticals, San Carlos, Calif., and the department of chemistry at Stanford University, and his associates.
However, it would be “premature but intriguing” to speculate about using this discovery to devise new, B cell–centered treatments for celiac disease, they added.
Noting that an inflammatory, gluten-induced immune response in the gut can be reflected in the peripheral blood, the investigators assessed whether a 6-week gluten challenge would induce damage to the small intestine that would show up in B-cell gene expression detected in blood samples. They assigned 73 patients at a single medical center in Finland to follow their usual gluten-free diets but to ingest an additional 6 g (20 patients), 3 g (26 patients), or 1.5 g (27 patients) of wheat gluten with a meal once per day for the study period.
The study participants (median age, 59 years; range, 23-74 years) underwent small-bowel biopsies obtained from the descending duodenum at baseline and after the gluten challenge. Damage to the intestinal mucosa was assessed by measuring the ratio of the height of the intestinal villi to the depth of the proliferative crypts at the base of the villi (villi height to crypt depth, or Vh:Cd). In celiac disease, gluten blunts the projection of the villi and causes hypertrophy or elongation of the crypts, resulting in flattened mucosa and a Vh:Cd approaching zero.
The study participants showed a wide variation in mucosal damage from the gluten exposure, with some patients showing no change and relatively healthy mucosa, and others showing extensive change and nearly flattened mucosa. The mucosal damage did not differ by gluten dose.
The largest change in Vh:Cd occurred in three patients who transitioned from relatively healthy intestinal mucosa (Vh:Cd 3.1) at baseline to nearly flat mucosa (Vh:Cd 0.2) after gluten exposure.
Patients with undamaged gut mucosa showed a relative increase in B-cell gene expression during the study period, while those who had increasing damage showed a relative decrease in B-cell expression. “The peripheral B cell therefore tracked with oral tolerance across the full spectrum of intestinal damage, from no change to a nearly flat mucosa,” Dr. Garber and his associates said (Cell Molec Gastroenterol Hepatol. 2017. doi: 10.1016/j.jcmgh.2017.01.011).
“The net increase in B-cell gene expression from baseline to 6 weeks in patients with little to no intestinal damage [suggests] that these individuals may have mounted a B-cell immune response to maintain mucosal homeostasis and circumvent inflammation,” they added.
Further study is needed to determine whether peripheral B cells are a marker that indicates tolerance to gluten but doesn’t play a functional role in the inflammatory process, or whether B cells may actually promote immune tolerance, the investigators said.
This study was funded by Alvine Pharmaceuticals, the American Recovery and Reinvestment Act, Tampere (Finland) University Hospital, the Academy of Finland Research Council for Health, and the U.S. National Institutes of Health. Dr. Garber reported ties to Alvine Pharmaceuticals, and his associates reported ties to ImmusanT, Celimmune, and ImmunogenX.
B-cell gene expression in the peripheral blood strongly correlates with the extent of gluten-induced damage to the intestinal mucosa in patients with celiac disease, according to a report in Cellular and Molecular Gastroenterology and Hepatology.
If this finding from a single-center cohort study is validated in other patient populations, the B-cell signature may become a useful, minimally invasive tool for diagnosing celiac disease. Eventually, if biomarkers are developed from the peripheral B-cell signature, a simple blood test could be used for monitoring changes in gut inflammation over time as well as treatment response, said Mitchell E. Garber, PhD, of Alvine Pharmaceuticals, San Carlos, Calif., and the department of chemistry at Stanford University, and his associates.
However, it would be “premature but intriguing” to speculate about using this discovery to devise new, B cell–centered treatments for celiac disease, they added.
Noting that an inflammatory, gluten-induced immune response in the gut can be reflected in the peripheral blood, the investigators assessed whether a 6-week gluten challenge would induce damage to the small intestine that would show up in B-cell gene expression detected in blood samples. They assigned 73 patients at a single medical center in Finland to follow their usual gluten-free diets but to ingest an additional 6 g (20 patients), 3 g (26 patients), or 1.5 g (27 patients) of wheat gluten with a meal once per day for the study period.
The study participants (median age, 59 years; range, 23-74 years) underwent small-bowel biopsies obtained from the descending duodenum at baseline and after the gluten challenge. Damage to the intestinal mucosa was assessed by measuring the ratio of the height of the intestinal villi to the depth of the proliferative crypts at the base of the villi (villi height to crypt depth, or Vh:Cd). In celiac disease, gluten blunts the projection of the villi and causes hypertrophy or elongation of the crypts, resulting in flattened mucosa and a Vh:Cd approaching zero.
The study participants showed a wide variation in mucosal damage from the gluten exposure, with some patients showing no change and relatively healthy mucosa, and others showing extensive change and nearly flattened mucosa. The mucosal damage did not differ by gluten dose.
The largest change in Vh:Cd occurred in three patients who transitioned from relatively healthy intestinal mucosa (Vh:Cd 3.1) at baseline to nearly flat mucosa (Vh:Cd 0.2) after gluten exposure.
Patients with undamaged gut mucosa showed a relative increase in B-cell gene expression during the study period, while those who had increasing damage showed a relative decrease in B-cell expression. “The peripheral B cell therefore tracked with oral tolerance across the full spectrum of intestinal damage, from no change to a nearly flat mucosa,” Dr. Garber and his associates said (Cell Molec Gastroenterol Hepatol. 2017. doi: 10.1016/j.jcmgh.2017.01.011).
“The net increase in B-cell gene expression from baseline to 6 weeks in patients with little to no intestinal damage [suggests] that these individuals may have mounted a B-cell immune response to maintain mucosal homeostasis and circumvent inflammation,” they added.
Further study is needed to determine whether peripheral B cells are a marker that indicates tolerance to gluten but doesn’t play a functional role in the inflammatory process, or whether B cells may actually promote immune tolerance, the investigators said.
This study was funded by Alvine Pharmaceuticals, the American Recovery and Reinvestment Act, Tampere (Finland) University Hospital, the Academy of Finland Research Council for Health, and the U.S. National Institutes of Health. Dr. Garber reported ties to Alvine Pharmaceuticals, and his associates reported ties to ImmusanT, Celimmune, and ImmunogenX.
FROM CELLULAR AND MOLECULAR GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: B-cell gene expression in peripheral blood strongly correlates with the extent of gluten-induced intestinal damage in patients with celiac disease.
Major finding: Patients with undamaged gut mucosa showed a relative increase in B-cell gene expression during the study period, while those who had increasing damage showed a relative decrease in B-cell expression.
Data source: A single-center cohort study involving 73 adults with celiac disease who underwent a 6-week gluten challenge.
Disclosures: This study was funded by Alvine Pharmaceuticals, the American Recovery and Reinvestment Act, Tampere (Finland) University Hospital, the Academy of Finland Research Council for Health, and the U.S. National Institutes of Health. Dr. Garber reported ties to Alvine Pharmaceuticals, and his associates reported ties to ImmusanT, Celimmune, and ImmunogenX.
Lower C. difficile mortality with vancomycin than metronidazole
Treating Clostridium difficile infection with vancomycin achieves the same recurrence rates as does treatment with metronidazole, but with a significantly lower 30-day mortality, new research suggests.
A retrospective, propensity-matched cohort study examined U.S. Department of Veterans Affairs health care system data from 47,471 patients with C. difficile infection who were treated with either vancomycin or metronidazole, according to a report published online Feb. 6 in JAMA Internal Medicine.
“Current guidelines recommend metronidazole hydrochloride as initial therapy for most cases of mild to moderate CDI [Clostridium difficile infection],” wrote Vanessa W. Stevens, PhD, of Veterans Affairs Salt Lake City Health Care System, and her coauthors. “Although an early clinical trial found no difference in cure rates between vancomycin hydrochloride and metronidazole, subsequent observational data and clinical trials suggest that metronidazole is inferior to vancomycin for primary clinical cure, especially in severe cases.”
Their study found patients treated with vancomycin had a similar risk of recurrence compared with those treated with metronidazole (relative risk, 0.98; 95% confidence interval, 0.87 to 1.10), with an overall recurrence rate of 16%.
However, patients treated with vancomycin had a 14% reduction in 30-day mortality compared to the metronidazole-treated group. This was after adjustment for factors such as comorbidity scores, hospitalization history, receipt of chemotherapy, receipt of immunosuppressive medication or proton pump inhibitor therapy in the prior 30-days, or antibiotic use on the day of diagnosis.
The 30-day mortality was not significantly different among patients with mild to moderate CDI, but there was a significant 21% reduction among patients with severe infection. The number needed to treat to prevent one death among patients with severe infection was 25 (JAMA Intern Med. 2017 Feb 6. doi: 10.1001/jamainternmed.2016.9045).
“This is the largest study to date to compare vancomycin and metronidazole in a real-world setting and one of the few studies focused on downstream outcomes of CDI,” researchers reported.
The authors noted that despite strong evidence and guidelines supporting the use of vancomycin for severe CDI – and the fact that 42% of episodes in the study were classified as severe – only 4%-6% of patients were prescribed vancomycin.
“Although the excess treatment costs of vancomycin relative to metronidazole and the concern for vancomycin-resistant Enterococcus will likely remain barriers, improved clinical cure and mortality rates may warrant reconsideration of current prescribing practices,” they wrote. “One approach to minimizing the effects of increasing vancomycin use is to target vancomycin treatment to patients with severe disease.”
The study was supported by researcher grants from the U.S. Department of Veterans Affairs. No conflicts of interest were declared.
AGA Resource
AGA offers patient education materials to help you discuss C. diff with your patients at http://www.gastro.org/patient-care/conditions-diseases/clostridium-difficile-infection.
Treating Clostridium difficile infection with vancomycin achieves the same recurrence rates as does treatment with metronidazole, but with a significantly lower 30-day mortality, new research suggests.
A retrospective, propensity-matched cohort study examined U.S. Department of Veterans Affairs health care system data from 47,471 patients with C. difficile infection who were treated with either vancomycin or metronidazole, according to a report published online Feb. 6 in JAMA Internal Medicine.
“Current guidelines recommend metronidazole hydrochloride as initial therapy for most cases of mild to moderate CDI [Clostridium difficile infection],” wrote Vanessa W. Stevens, PhD, of Veterans Affairs Salt Lake City Health Care System, and her coauthors. “Although an early clinical trial found no difference in cure rates between vancomycin hydrochloride and metronidazole, subsequent observational data and clinical trials suggest that metronidazole is inferior to vancomycin for primary clinical cure, especially in severe cases.”
Their study found patients treated with vancomycin had a similar risk of recurrence compared with those treated with metronidazole (relative risk, 0.98; 95% confidence interval, 0.87 to 1.10), with an overall recurrence rate of 16%.
However, patients treated with vancomycin had a 14% reduction in 30-day mortality compared to the metronidazole-treated group. This was after adjustment for factors such as comorbidity scores, hospitalization history, receipt of chemotherapy, receipt of immunosuppressive medication or proton pump inhibitor therapy in the prior 30-days, or antibiotic use on the day of diagnosis.
The 30-day mortality was not significantly different among patients with mild to moderate CDI, but there was a significant 21% reduction among patients with severe infection. The number needed to treat to prevent one death among patients with severe infection was 25 (JAMA Intern Med. 2017 Feb 6. doi: 10.1001/jamainternmed.2016.9045).
“This is the largest study to date to compare vancomycin and metronidazole in a real-world setting and one of the few studies focused on downstream outcomes of CDI,” researchers reported.
The authors noted that despite strong evidence and guidelines supporting the use of vancomycin for severe CDI – and the fact that 42% of episodes in the study were classified as severe – only 4%-6% of patients were prescribed vancomycin.
“Although the excess treatment costs of vancomycin relative to metronidazole and the concern for vancomycin-resistant Enterococcus will likely remain barriers, improved clinical cure and mortality rates may warrant reconsideration of current prescribing practices,” they wrote. “One approach to minimizing the effects of increasing vancomycin use is to target vancomycin treatment to patients with severe disease.”
The study was supported by researcher grants from the U.S. Department of Veterans Affairs. No conflicts of interest were declared.
AGA Resource
AGA offers patient education materials to help you discuss C. diff with your patients at http://www.gastro.org/patient-care/conditions-diseases/clostridium-difficile-infection.
Treating Clostridium difficile infection with vancomycin achieves the same recurrence rates as does treatment with metronidazole, but with a significantly lower 30-day mortality, new research suggests.
A retrospective, propensity-matched cohort study examined U.S. Department of Veterans Affairs health care system data from 47,471 patients with C. difficile infection who were treated with either vancomycin or metronidazole, according to a report published online Feb. 6 in JAMA Internal Medicine.
“Current guidelines recommend metronidazole hydrochloride as initial therapy for most cases of mild to moderate CDI [Clostridium difficile infection],” wrote Vanessa W. Stevens, PhD, of Veterans Affairs Salt Lake City Health Care System, and her coauthors. “Although an early clinical trial found no difference in cure rates between vancomycin hydrochloride and metronidazole, subsequent observational data and clinical trials suggest that metronidazole is inferior to vancomycin for primary clinical cure, especially in severe cases.”
Their study found patients treated with vancomycin had a similar risk of recurrence compared with those treated with metronidazole (relative risk, 0.98; 95% confidence interval, 0.87 to 1.10), with an overall recurrence rate of 16%.
However, patients treated with vancomycin had a 14% reduction in 30-day mortality compared to the metronidazole-treated group. This was after adjustment for factors such as comorbidity scores, hospitalization history, receipt of chemotherapy, receipt of immunosuppressive medication or proton pump inhibitor therapy in the prior 30-days, or antibiotic use on the day of diagnosis.
The 30-day mortality was not significantly different among patients with mild to moderate CDI, but there was a significant 21% reduction among patients with severe infection. The number needed to treat to prevent one death among patients with severe infection was 25 (JAMA Intern Med. 2017 Feb 6. doi: 10.1001/jamainternmed.2016.9045).
“This is the largest study to date to compare vancomycin and metronidazole in a real-world setting and one of the few studies focused on downstream outcomes of CDI,” researchers reported.
The authors noted that despite strong evidence and guidelines supporting the use of vancomycin for severe CDI – and the fact that 42% of episodes in the study were classified as severe – only 4%-6% of patients were prescribed vancomycin.
“Although the excess treatment costs of vancomycin relative to metronidazole and the concern for vancomycin-resistant Enterococcus will likely remain barriers, improved clinical cure and mortality rates may warrant reconsideration of current prescribing practices,” they wrote. “One approach to minimizing the effects of increasing vancomycin use is to target vancomycin treatment to patients with severe disease.”
The study was supported by researcher grants from the U.S. Department of Veterans Affairs. No conflicts of interest were declared.
AGA Resource
AGA offers patient education materials to help you discuss C. diff with your patients at http://www.gastro.org/patient-care/conditions-diseases/clostridium-difficile-infection.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Vancomycin therapy is associated with significantly lower 30-day mortality from severe Clostridium difficile infection compared to metronidazole.
Major finding: Treatment with vancomycin prevented one death per 25 patients with severe C. difficile infection.
Data source: A retrospective, propensity-matched cohort study of 47,471 patients with C. difficile infection.
Disclosures: The study was supported by researcher grants from the U.S. Department of Veterans Affairs. No conflicts of interest were declared.
FDA opens abbreviated approval pathway for interchangeable biosimilars
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique treatments.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference product is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such biosimilars have come on the market, at an average price of about 30% less than the reference product. Prices have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive treatments. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer treatments (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those products to make up for the money they were losing on the Russian market.

“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive treatments, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).

“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the product was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American Gastroenterological Association has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing treatments, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer products are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the treatment maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older product, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique treatments.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference product is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such biosimilars have come on the market, at an average price of about 30% less than the reference product. Prices have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive treatments. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer treatments (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those products to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive treatments, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the product was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American Gastroenterological Association has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing treatments, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer products are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the treatment maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older product, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique treatments.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference product is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such biosimilars have come on the market, at an average price of about 30% less than the reference product. Prices have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive treatments. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer treatments (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those products to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive treatments, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the product was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American Gastroenterological Association has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing treatments, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer products are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the treatment maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older product, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
msullivan@frontlinemedcom.com
On Twitter @alz_gal