User login
Alogliptin CV risk acceptable, FDA panel agrees
SILVER SPRING, MD.– Alogliptin’s cardiovascular risk profile is acceptably safe for high-risk patients with type 2 diabetes, a Food and Drug Administration advisory panel has unanimously agreed.
At a meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee on April 14, panelists reviewed the results of the results of a large cardiovascular outcomes study of the dipeptidyl peptidase-4 (DPP-4) inhibitor, the Examination of Cardiovascular Outcomes with Alogliptin versus Standard of Care (EXAMINE) study, an international, randomized, double-blind, placebo-controlled trial of about 5,400 people with type 2 diabetes and established cardiovascular disease.
The FDA is requiring CV outcomes trials for all type 2 diabetes drugs, including those in development, and issued a guidance for industry for these trials in December 2008, which states that the trials should evaluate the major adverse cardiovascular event (MACE) incidence in patients at increased risk for CVD and that a 30% or greater excess CV risk over placebo should be excluded to meet the criteria for acceptable CV safety.
Alogliptin, marketed as Nesina by Takeda Pharmaceutical USA, was approved in January 2013. It also is combined with metformin (Kazano) and with pioglitazone (Oseni). The EXAMINE study, which was underway at the time of approval, enrolled people who had an acute coronary syndrome event (acute MI or unstable angina requiring hospitalization) within 15-90 days of enrollment.
Over a median follow-up of 1.5 years, there was no increase in the MACE composite endpoint of CV death, nonfatal MI, and nonfatal stroke among those on alogliptin (11.3%), compared with those on placebo (11.8%), for a hazard ratio of 0.96 – meeting the FDA criteria for CV safety (N. Engl. J. Med. 2013;369:1327-35).
Some of the issues raised with the study included a lower-than-planned proportion of patients enrolled in the United States and Canada – less than 16% of the total – and a subgroup analysis showing that, in the North American patients, the MACE rate among those on alogliptin was elevated compared with placebo (HR, 1.28). The FDA reviewers said that the North American population had some characteristics that might explain this difference.
Another finding discussed was the higher number of hospitalizations for heart failure in patients on alogliptin (106) than for those on placebo (89), for a rate of 2.6 vs. 2.3 cases per 100 patient-years (HR, 1.19), according to the FDA. In the CV outcomes study of another DPP-4 inhibitor, saxagliptin, which the panel reviewed earlier in the day, there was a similar signal for an increased risk of heart failure hospitalizations associated with the drug.
Panelists, however, said they were not convinced that the geographic differences represented a real effect, which they said could be due to chance and did not affect the overall results – although they would have preferred to have more people from the United States enrolled in the study. Panelists also were not overly concerned about the risk of heart failure, but they pointed out that the risk of heart failure risk should be monitored in patients on the drug because of the signal seen with saxagliptin. Several panelists said that they suspected that heart failure may turn out to be a class effect. CV outcomes trials for the two other approved DPP-4 inhibitors – sitagliptin (Januvia) and linagliptin (Tradjenta) – are ongoing.
“It’s clear that the trial achieved its primary objective in a manner consistent with the 2008 guidelines,” said one of the panelists, Dr. Thomas Wang, director of the division of cardiovascular medicine at Vanderbilt University, Nashville, Tenn. Although there were some limitations of the study, such as the low proportion of U.S. participants and the relatively short duration of follow-up, “these limitations do not come close to negating the conclusion” regarding the cardiovascular risk profile, he commented.
In another vote, 13 of the 16 panelists said that the new safety information should be added to the alogliptin prescribing information. “This trial represented a huge effort and we know a lot about the outcomes related to this drug now,” and that information should be included in the label, said one of the panelists, Dr. Susan Heckbert, professor in the department of epidemiology, University of Washington, Seattle.
Three panelists voted not to add any information to the label. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest
SILVER SPRING, MD.– Alogliptin’s cardiovascular risk profile is acceptably safe for high-risk patients with type 2 diabetes, a Food and Drug Administration advisory panel has unanimously agreed.
At a meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee on April 14, panelists reviewed the results of the results of a large cardiovascular outcomes study of the dipeptidyl peptidase-4 (DPP-4) inhibitor, the Examination of Cardiovascular Outcomes with Alogliptin versus Standard of Care (EXAMINE) study, an international, randomized, double-blind, placebo-controlled trial of about 5,400 people with type 2 diabetes and established cardiovascular disease.
The FDA is requiring CV outcomes trials for all type 2 diabetes drugs, including those in development, and issued a guidance for industry for these trials in December 2008, which states that the trials should evaluate the major adverse cardiovascular event (MACE) incidence in patients at increased risk for CVD and that a 30% or greater excess CV risk over placebo should be excluded to meet the criteria for acceptable CV safety.
Alogliptin, marketed as Nesina by Takeda Pharmaceutical USA, was approved in January 2013. It also is combined with metformin (Kazano) and with pioglitazone (Oseni). The EXAMINE study, which was underway at the time of approval, enrolled people who had an acute coronary syndrome event (acute MI or unstable angina requiring hospitalization) within 15-90 days of enrollment.
Over a median follow-up of 1.5 years, there was no increase in the MACE composite endpoint of CV death, nonfatal MI, and nonfatal stroke among those on alogliptin (11.3%), compared with those on placebo (11.8%), for a hazard ratio of 0.96 – meeting the FDA criteria for CV safety (N. Engl. J. Med. 2013;369:1327-35).
Some of the issues raised with the study included a lower-than-planned proportion of patients enrolled in the United States and Canada – less than 16% of the total – and a subgroup analysis showing that, in the North American patients, the MACE rate among those on alogliptin was elevated compared with placebo (HR, 1.28). The FDA reviewers said that the North American population had some characteristics that might explain this difference.
Another finding discussed was the higher number of hospitalizations for heart failure in patients on alogliptin (106) than for those on placebo (89), for a rate of 2.6 vs. 2.3 cases per 100 patient-years (HR, 1.19), according to the FDA. In the CV outcomes study of another DPP-4 inhibitor, saxagliptin, which the panel reviewed earlier in the day, there was a similar signal for an increased risk of heart failure hospitalizations associated with the drug.
Panelists, however, said they were not convinced that the geographic differences represented a real effect, which they said could be due to chance and did not affect the overall results – although they would have preferred to have more people from the United States enrolled in the study. Panelists also were not overly concerned about the risk of heart failure, but they pointed out that the risk of heart failure risk should be monitored in patients on the drug because of the signal seen with saxagliptin. Several panelists said that they suspected that heart failure may turn out to be a class effect. CV outcomes trials for the two other approved DPP-4 inhibitors – sitagliptin (Januvia) and linagliptin (Tradjenta) – are ongoing.
“It’s clear that the trial achieved its primary objective in a manner consistent with the 2008 guidelines,” said one of the panelists, Dr. Thomas Wang, director of the division of cardiovascular medicine at Vanderbilt University, Nashville, Tenn. Although there were some limitations of the study, such as the low proportion of U.S. participants and the relatively short duration of follow-up, “these limitations do not come close to negating the conclusion” regarding the cardiovascular risk profile, he commented.
In another vote, 13 of the 16 panelists said that the new safety information should be added to the alogliptin prescribing information. “This trial represented a huge effort and we know a lot about the outcomes related to this drug now,” and that information should be included in the label, said one of the panelists, Dr. Susan Heckbert, professor in the department of epidemiology, University of Washington, Seattle.
Three panelists voted not to add any information to the label. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest
SILVER SPRING, MD.– Alogliptin’s cardiovascular risk profile is acceptably safe for high-risk patients with type 2 diabetes, a Food and Drug Administration advisory panel has unanimously agreed.
At a meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee on April 14, panelists reviewed the results of the results of a large cardiovascular outcomes study of the dipeptidyl peptidase-4 (DPP-4) inhibitor, the Examination of Cardiovascular Outcomes with Alogliptin versus Standard of Care (EXAMINE) study, an international, randomized, double-blind, placebo-controlled trial of about 5,400 people with type 2 diabetes and established cardiovascular disease.
The FDA is requiring CV outcomes trials for all type 2 diabetes drugs, including those in development, and issued a guidance for industry for these trials in December 2008, which states that the trials should evaluate the major adverse cardiovascular event (MACE) incidence in patients at increased risk for CVD and that a 30% or greater excess CV risk over placebo should be excluded to meet the criteria for acceptable CV safety.
Alogliptin, marketed as Nesina by Takeda Pharmaceutical USA, was approved in January 2013. It also is combined with metformin (Kazano) and with pioglitazone (Oseni). The EXAMINE study, which was underway at the time of approval, enrolled people who had an acute coronary syndrome event (acute MI or unstable angina requiring hospitalization) within 15-90 days of enrollment.
Over a median follow-up of 1.5 years, there was no increase in the MACE composite endpoint of CV death, nonfatal MI, and nonfatal stroke among those on alogliptin (11.3%), compared with those on placebo (11.8%), for a hazard ratio of 0.96 – meeting the FDA criteria for CV safety (N. Engl. J. Med. 2013;369:1327-35).
Some of the issues raised with the study included a lower-than-planned proportion of patients enrolled in the United States and Canada – less than 16% of the total – and a subgroup analysis showing that, in the North American patients, the MACE rate among those on alogliptin was elevated compared with placebo (HR, 1.28). The FDA reviewers said that the North American population had some characteristics that might explain this difference.
Another finding discussed was the higher number of hospitalizations for heart failure in patients on alogliptin (106) than for those on placebo (89), for a rate of 2.6 vs. 2.3 cases per 100 patient-years (HR, 1.19), according to the FDA. In the CV outcomes study of another DPP-4 inhibitor, saxagliptin, which the panel reviewed earlier in the day, there was a similar signal for an increased risk of heart failure hospitalizations associated with the drug.
Panelists, however, said they were not convinced that the geographic differences represented a real effect, which they said could be due to chance and did not affect the overall results – although they would have preferred to have more people from the United States enrolled in the study. Panelists also were not overly concerned about the risk of heart failure, but they pointed out that the risk of heart failure risk should be monitored in patients on the drug because of the signal seen with saxagliptin. Several panelists said that they suspected that heart failure may turn out to be a class effect. CV outcomes trials for the two other approved DPP-4 inhibitors – sitagliptin (Januvia) and linagliptin (Tradjenta) – are ongoing.
“It’s clear that the trial achieved its primary objective in a manner consistent with the 2008 guidelines,” said one of the panelists, Dr. Thomas Wang, director of the division of cardiovascular medicine at Vanderbilt University, Nashville, Tenn. Although there were some limitations of the study, such as the low proportion of U.S. participants and the relatively short duration of follow-up, “these limitations do not come close to negating the conclusion” regarding the cardiovascular risk profile, he commented.
In another vote, 13 of the 16 panelists said that the new safety information should be added to the alogliptin prescribing information. “This trial represented a huge effort and we know a lot about the outcomes related to this drug now,” and that information should be included in the label, said one of the panelists, Dr. Susan Heckbert, professor in the department of epidemiology, University of Washington, Seattle.
Three panelists voted not to add any information to the label. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest
AT AN FDA ADVISORY COMMITTEE MEETING
VIDEO: Treating heart failure congestion improved hyperglycemia
SAN DIEGO – Congestion secondary to advanced heart failure appears to cause type 2 diabetes in a significant subgroup of patients, based on suggestive findings from a series of recently reported studies, Dr. Maya Guglin said during an interview at the annual meeting of the American College of Cardiology.
Dr. Guglin reviewed convergent findings from several groups of patients who received left ventricular assist devices to treat advanced heart failure. The analyses all showed that many, though not a majority, of those patients also had type 2 diabetes. Soon after patients received an assist device, the type 2 diabetes uniformly improved – and in many cases, glycemic control normalized.
Dr. Guglin, who has named this condition “cardiogenic diabetes,” said that reducing congestion with an assist device or with diuretic treatment seems the best way to both reduce congestion and improve or resolve the diabetes.
Those treatments will “improve quality of life, reduce hospital admissions for heart failure, and also improve the course of diabetes,” said Dr. Guglin, professor of medicine and medical director of the ventricular assist device program at the University of Kentucky in Lexington.
Dr. Guglin had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
SAN DIEGO – Congestion secondary to advanced heart failure appears to cause type 2 diabetes in a significant subgroup of patients, based on suggestive findings from a series of recently reported studies, Dr. Maya Guglin said during an interview at the annual meeting of the American College of Cardiology.
Dr. Guglin reviewed convergent findings from several groups of patients who received left ventricular assist devices to treat advanced heart failure. The analyses all showed that many, though not a majority, of those patients also had type 2 diabetes. Soon after patients received an assist device, the type 2 diabetes uniformly improved – and in many cases, glycemic control normalized.
Dr. Guglin, who has named this condition “cardiogenic diabetes,” said that reducing congestion with an assist device or with diuretic treatment seems the best way to both reduce congestion and improve or resolve the diabetes.
Those treatments will “improve quality of life, reduce hospital admissions for heart failure, and also improve the course of diabetes,” said Dr. Guglin, professor of medicine and medical director of the ventricular assist device program at the University of Kentucky in Lexington.
Dr. Guglin had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
SAN DIEGO – Congestion secondary to advanced heart failure appears to cause type 2 diabetes in a significant subgroup of patients, based on suggestive findings from a series of recently reported studies, Dr. Maya Guglin said during an interview at the annual meeting of the American College of Cardiology.
Dr. Guglin reviewed convergent findings from several groups of patients who received left ventricular assist devices to treat advanced heart failure. The analyses all showed that many, though not a majority, of those patients also had type 2 diabetes. Soon after patients received an assist device, the type 2 diabetes uniformly improved – and in many cases, glycemic control normalized.
Dr. Guglin, who has named this condition “cardiogenic diabetes,” said that reducing congestion with an assist device or with diuretic treatment seems the best way to both reduce congestion and improve or resolve the diabetes.
Those treatments will “improve quality of life, reduce hospital admissions for heart failure, and also improve the course of diabetes,” said Dr. Guglin, professor of medicine and medical director of the ventricular assist device program at the University of Kentucky in Lexington.
Dr. Guglin had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM ACC 15

FDA panel reassured by saxagliptin’s CV safety data
SILVER SPRING, MD.– Results of a large postmarketing study evaluating the cardiovascular safety of saxagliptin in a high-risk population provided reassuring evidence about the drug’s overall cardiovascular risk, although a signal for heart failure hospitalizations associated with the drug was a concern, according to a Food and Drug Administration advisory panel.
At a meeting on April 14, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1, with one abstention, that the results of the SAVOR (Saxagliptin Assessment of Vascular Outcomes Recorded in Patients With Diabetes Mellitus) trial showed that the use of saxagliptin in patients with type 2 diabetes had an acceptable cardiovascular risk profile. However, the panelists were concerned about the signal for an increased risk of hospitalizations for heart failure associated with saxagliptin in the study, which they agreed should be studied further. Nearly all of the panelists also voted that the safety data, including the heart failure finding and an imbalance in all-cause mortality among those on saxagliptin arm of the study, be added to the drug’s prescribing information.

Approved in 2009, saxagliptin, a dipeptidyl peptidase–4 (DPP4) inhibitor, is indicated “as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus in multiple clinical settings” and is marketed as Onglyza by AstraZeneca. A fixed-dose combination of saxagliptin with extended-release metformin (Kombiglyze XR) was approved in 2010. The FDA panel votes apply to both products.
The FDA is requiring that manufacturers conduct CV outcomes trials for all type 2 diabetes drugs in patients at high risk for cardiovascular disease (CVD), and in December 2008, issued a guidance document for industry, outlining the requirements for these studies, which included showing that the CVD risk, based on a major adverse cardiovascular event (MACE) endpoint, is not increased by more than 30% compared to placebo.
AstraZeneca conducted SAVOR, a prospective, randomized, double-blind, placebo-controlled study of nearly 16,500 people with type 2 diabetes, who had or were at risk of CVD, comparing saxagliptin to placebo, on top of standard treatment (N. Engl. J. Med. 2013;369:1317-26). After a median follow-up of about 2 years, the incidence of MACE (a composite of cardiovascular death, nonfatal MI, and nonfatal ischemic stroke) was identical in both groups, at 7.4%, with a hazard ratio of 1.0, meeting the FDA criteria for cardiovascular safety as outlined in the guidance, according to the company. Saxagliptin was not superior to placebo in reducing the MACE events, another primary objective of the study.
Unexpectedly, hospitalization for heart failure (HF), a component of the secondary composite endpoint, was increased among those treated with saxagliptin (hazard ratio, 1.27). There was also a “numerical imbalance” in all-cause mortality between groups, with 17 deaths in the saxagliptin patients vs. 11 in the placebo group (HR, 1.11).
However, deaths caused by heart failure were balanced between those on saxagliptin and those on placebo, and no possible mechanism for the HF finding could be identified, according to AstraZeneca, which is planning a mechanistic study to evaluate the effects of saxagliptin on volume, neurohormonal changes, and cardiac function. The company has proposed that the HF finding be addressed by adding this information to the prescribing information.
The cardiovascular safety results were reassuring, said Dr. Morris Schambelan, a panelist and professor emeritus of medicine in the division of endocrinology at the University of California, San Francisco, but like others on the committee, added that he believes the heart failure signal was real and that providing clinicians with information in the drug’s label to help predict those at higher risk in would be helpful.
Several panelists were somewhat concerned about the all-cause mortality finding, which is a strong endpoint, and therefore cannot be ruled out, they said. But they also pointed out that follow-up was relatively short for a drug that is used long term and that the results should be applied cautiously to patients at lower risk than were those enrolled in the trial.
The FDA usually follows the recommendations of its advisory panel members. Panelists had no relevant disclosures.
CV outcomes trials for other approved DPP4 inhibitors approved for type 2 diabetes – sitagliptin (Januvia) and linagliptin (Tradjenta) – are ongoing. The panel review of the CV outcomes trial for another DPP4 inhibitor approved, alogliptin (Nesina), followed the meeting on saxagliptin.
SILVER SPRING, MD.– Results of a large postmarketing study evaluating the cardiovascular safety of saxagliptin in a high-risk population provided reassuring evidence about the drug’s overall cardiovascular risk, although a signal for heart failure hospitalizations associated with the drug was a concern, according to a Food and Drug Administration advisory panel.
At a meeting on April 14, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1, with one abstention, that the results of the SAVOR (Saxagliptin Assessment of Vascular Outcomes Recorded in Patients With Diabetes Mellitus) trial showed that the use of saxagliptin in patients with type 2 diabetes had an acceptable cardiovascular risk profile. However, the panelists were concerned about the signal for an increased risk of hospitalizations for heart failure associated with saxagliptin in the study, which they agreed should be studied further. Nearly all of the panelists also voted that the safety data, including the heart failure finding and an imbalance in all-cause mortality among those on saxagliptin arm of the study, be added to the drug’s prescribing information.
Approved in 2009, saxagliptin, a dipeptidyl peptidase–4 (DPP4) inhibitor, is indicated “as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus in multiple clinical settings” and is marketed as Onglyza by AstraZeneca. A fixed-dose combination of saxagliptin with extended-release metformin (Kombiglyze XR) was approved in 2010. The FDA panel votes apply to both products.
The FDA is requiring that manufacturers conduct CV outcomes trials for all type 2 diabetes drugs in patients at high risk for cardiovascular disease (CVD), and in December 2008, issued a guidance document for industry, outlining the requirements for these studies, which included showing that the CVD risk, based on a major adverse cardiovascular event (MACE) endpoint, is not increased by more than 30% compared to placebo.
AstraZeneca conducted SAVOR, a prospective, randomized, double-blind, placebo-controlled study of nearly 16,500 people with type 2 diabetes, who had or were at risk of CVD, comparing saxagliptin to placebo, on top of standard treatment (N. Engl. J. Med. 2013;369:1317-26). After a median follow-up of about 2 years, the incidence of MACE (a composite of cardiovascular death, nonfatal MI, and nonfatal ischemic stroke) was identical in both groups, at 7.4%, with a hazard ratio of 1.0, meeting the FDA criteria for cardiovascular safety as outlined in the guidance, according to the company. Saxagliptin was not superior to placebo in reducing the MACE events, another primary objective of the study.
Unexpectedly, hospitalization for heart failure (HF), a component of the secondary composite endpoint, was increased among those treated with saxagliptin (hazard ratio, 1.27). There was also a “numerical imbalance” in all-cause mortality between groups, with 17 deaths in the saxagliptin patients vs. 11 in the placebo group (HR, 1.11).
However, deaths caused by heart failure were balanced between those on saxagliptin and those on placebo, and no possible mechanism for the HF finding could be identified, according to AstraZeneca, which is planning a mechanistic study to evaluate the effects of saxagliptin on volume, neurohormonal changes, and cardiac function. The company has proposed that the HF finding be addressed by adding this information to the prescribing information.
The cardiovascular safety results were reassuring, said Dr. Morris Schambelan, a panelist and professor emeritus of medicine in the division of endocrinology at the University of California, San Francisco, but like others on the committee, added that he believes the heart failure signal was real and that providing clinicians with information in the drug’s label to help predict those at higher risk in would be helpful.
Several panelists were somewhat concerned about the all-cause mortality finding, which is a strong endpoint, and therefore cannot be ruled out, they said. But they also pointed out that follow-up was relatively short for a drug that is used long term and that the results should be applied cautiously to patients at lower risk than were those enrolled in the trial.
The FDA usually follows the recommendations of its advisory panel members. Panelists had no relevant disclosures.
CV outcomes trials for other approved DPP4 inhibitors approved for type 2 diabetes – sitagliptin (Januvia) and linagliptin (Tradjenta) – are ongoing. The panel review of the CV outcomes trial for another DPP4 inhibitor approved, alogliptin (Nesina), followed the meeting on saxagliptin.
SILVER SPRING, MD.– Results of a large postmarketing study evaluating the cardiovascular safety of saxagliptin in a high-risk population provided reassuring evidence about the drug’s overall cardiovascular risk, although a signal for heart failure hospitalizations associated with the drug was a concern, according to a Food and Drug Administration advisory panel.
At a meeting on April 14, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1, with one abstention, that the results of the SAVOR (Saxagliptin Assessment of Vascular Outcomes Recorded in Patients With Diabetes Mellitus) trial showed that the use of saxagliptin in patients with type 2 diabetes had an acceptable cardiovascular risk profile. However, the panelists were concerned about the signal for an increased risk of hospitalizations for heart failure associated with saxagliptin in the study, which they agreed should be studied further. Nearly all of the panelists also voted that the safety data, including the heart failure finding and an imbalance in all-cause mortality among those on saxagliptin arm of the study, be added to the drug’s prescribing information.
Approved in 2009, saxagliptin, a dipeptidyl peptidase–4 (DPP4) inhibitor, is indicated “as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus in multiple clinical settings” and is marketed as Onglyza by AstraZeneca. A fixed-dose combination of saxagliptin with extended-release metformin (Kombiglyze XR) was approved in 2010. The FDA panel votes apply to both products.
The FDA is requiring that manufacturers conduct CV outcomes trials for all type 2 diabetes drugs in patients at high risk for cardiovascular disease (CVD), and in December 2008, issued a guidance document for industry, outlining the requirements for these studies, which included showing that the CVD risk, based on a major adverse cardiovascular event (MACE) endpoint, is not increased by more than 30% compared to placebo.
AstraZeneca conducted SAVOR, a prospective, randomized, double-blind, placebo-controlled study of nearly 16,500 people with type 2 diabetes, who had or were at risk of CVD, comparing saxagliptin to placebo, on top of standard treatment (N. Engl. J. Med. 2013;369:1317-26). After a median follow-up of about 2 years, the incidence of MACE (a composite of cardiovascular death, nonfatal MI, and nonfatal ischemic stroke) was identical in both groups, at 7.4%, with a hazard ratio of 1.0, meeting the FDA criteria for cardiovascular safety as outlined in the guidance, according to the company. Saxagliptin was not superior to placebo in reducing the MACE events, another primary objective of the study.
Unexpectedly, hospitalization for heart failure (HF), a component of the secondary composite endpoint, was increased among those treated with saxagliptin (hazard ratio, 1.27). There was also a “numerical imbalance” in all-cause mortality between groups, with 17 deaths in the saxagliptin patients vs. 11 in the placebo group (HR, 1.11).
However, deaths caused by heart failure were balanced between those on saxagliptin and those on placebo, and no possible mechanism for the HF finding could be identified, according to AstraZeneca, which is planning a mechanistic study to evaluate the effects of saxagliptin on volume, neurohormonal changes, and cardiac function. The company has proposed that the HF finding be addressed by adding this information to the prescribing information.
The cardiovascular safety results were reassuring, said Dr. Morris Schambelan, a panelist and professor emeritus of medicine in the division of endocrinology at the University of California, San Francisco, but like others on the committee, added that he believes the heart failure signal was real and that providing clinicians with information in the drug’s label to help predict those at higher risk in would be helpful.
Several panelists were somewhat concerned about the all-cause mortality finding, which is a strong endpoint, and therefore cannot be ruled out, they said. But they also pointed out that follow-up was relatively short for a drug that is used long term and that the results should be applied cautiously to patients at lower risk than were those enrolled in the trial.
The FDA usually follows the recommendations of its advisory panel members. Panelists had no relevant disclosures.
CV outcomes trials for other approved DPP4 inhibitors approved for type 2 diabetes – sitagliptin (Januvia) and linagliptin (Tradjenta) – are ongoing. The panel review of the CV outcomes trial for another DPP4 inhibitor approved, alogliptin (Nesina), followed the meeting on saxagliptin.
AT AN FDA ADVISORY COMMITTEE MEETING

Heart failure spurs 'cardiogenic diabetes'
SAN DIEGO – A series of reports in 2014 from several independent groups implicated heart failure as a trigger of type 2 diabetes; findings from several of the analyses also suggested that relief of congestion can result in rapid resolution of the diabetes.
The best way to manage new-onset diabetes in heart failure patients is to “minimize the congestion,” and to “try to achieve as good control of the heart failure as possible,” said Dr. Maya Guglin during a talk at the annual meeting of the American College of Cardiology, in which she laid out the evidence for this newly recognized form of type 2 diabetes. In a review she published in 2014, Dr. Guglin coined the term “cardiogenic diabetes” to describe the condition (Heart Fail. Rev. 2014;19:595-602).

Dr. Guglin traced the data trail for cardiogenic diabetes starting in a 2011 retrospective study of 15 patients with advanced heart failure who received a left ventricular assist device (LVAD) at Columbia University in New York (Eur. J. Heart Fail. 2011;13:195-9). These 15, about a third of the 43 total LVAD recipients at Columbia at the time, had been diagnosed with type 2 diabetes for an average of 6 years before receiving the device. Just before they got their device, their average hemoglobin A1c (HbA1c) level was 7.7%, and their average fasting plasma glucose level was 158 mg/dL. An average of 4 months later, their mean HbA1c had dropped to 6%, and their mean fasting glucose had fallen to 104 mg/dL. Six patients were completely off any diabetes medication. All this occurred while patients had a small increase in their body mass index, which Dr. Guglin attributed to their better physical condition and improved appetite.
Last year, another four reports appeared from four independent, U.S. heart failure groups with results that mirrored the Columbia experience. Dr. Guglin and her associates at the University of Kentucky, Lexington, reported their experience with 50 patients who received an LVAD during 2002-2012 and had type 2 diabetes just before they received a device, with an average HbA1c of 7.6%. Three months after LVAD placement, their average HbA1c had dropped to 5.7%, and 9-12 months after device placement, their average HbA1c level was 5.3% (ASAIO J. 2014;60:290-3). As in the Columbia series, these improvements in hyperglycemia occurred without any significant change in body mass index.
Dr. Guglin also cited similar findings in 50 LVAD patients treated at the University of Rochester (N.Y.)(ASAIO J. 2014;60:675-80), 28 LVAD patients at Penn State Medical College in Hershey, Pa. (Heart Surg. Forum 2014;17:E98-102), and 66 LVAD patients from the University of Illinois in Chicago (Eur. J. Heart Fail. 2014;16:1120-4). In these reports type 2 diabetes existed in roughly a quarter to a third of patients with advanced heart failure who qualified for an LVAD just prior to the time they received the device.
Dr. Guglin also cited two epidemiologic analyses with complementary findings on the risk for incident diabetes faced by heart failure patients. She and her associates reviewed data from 3,165 elderly Americans free from diabetes enrolled in the Cardiovascular Health Study. This cohort included 80 patients with heart failure and 3,085 without heart failure. During 3-4 years of follow-up, 6% of the heart failure patients developed new-onset diabetes, and an additional 10% developed new-onset impaired fasting glucose. In contrast, these incidence rates were 1.5% and 5%, respectively, in the enrollees without heart failure at baseline. In an analysis that controlled for several demographic and biomedical factors, heart failure linked with a statistically significant, 2.4-fold increased risk for the development of diabetes (Cardiology 2014;129:84-92).
And a Danish nationwide cohort study of more than 99,000 residents discharged from a first-time hospitalization for heart failure during 1997-2010 showed a statistically significant link between heart failure severity and an increased rate of development of incident diabetes using diuretic treatment dosage as a surrogate measure of heart failure severity (Diabetologia 2014;57:1595-1600).
The apparent impact of LVAD placement on type 2 diabetes contrasts with what happens in patients who receive a heart transplant, where this association has not been seen. Dr. Guglin suggested that may be because of the immunosuppression with steroids that heart transplant recipients receive, treatment that also prevents diabetes resolution, she said.
“It all boils down to congestion,” Dr. Guglin said in an interview. “Control congestion as much as possible to control the diabetes.”
Dr. Guglin had no relevant financial disclosures.
On Twitter @mitchelzoler
SAN DIEGO – A series of reports in 2014 from several independent groups implicated heart failure as a trigger of type 2 diabetes; findings from several of the analyses also suggested that relief of congestion can result in rapid resolution of the diabetes.
The best way to manage new-onset diabetes in heart failure patients is to “minimize the congestion,” and to “try to achieve as good control of the heart failure as possible,” said Dr. Maya Guglin during a talk at the annual meeting of the American College of Cardiology, in which she laid out the evidence for this newly recognized form of type 2 diabetes. In a review she published in 2014, Dr. Guglin coined the term “cardiogenic diabetes” to describe the condition (Heart Fail. Rev. 2014;19:595-602).
Dr. Guglin traced the data trail for cardiogenic diabetes starting in a 2011 retrospective study of 15 patients with advanced heart failure who received a left ventricular assist device (LVAD) at Columbia University in New York (Eur. J. Heart Fail. 2011;13:195-9). These 15, about a third of the 43 total LVAD recipients at Columbia at the time, had been diagnosed with type 2 diabetes for an average of 6 years before receiving the device. Just before they got their device, their average hemoglobin A1c (HbA1c) level was 7.7%, and their average fasting plasma glucose level was 158 mg/dL. An average of 4 months later, their mean HbA1c had dropped to 6%, and their mean fasting glucose had fallen to 104 mg/dL. Six patients were completely off any diabetes medication. All this occurred while patients had a small increase in their body mass index, which Dr. Guglin attributed to their better physical condition and improved appetite.
Last year, another four reports appeared from four independent, U.S. heart failure groups with results that mirrored the Columbia experience. Dr. Guglin and her associates at the University of Kentucky, Lexington, reported their experience with 50 patients who received an LVAD during 2002-2012 and had type 2 diabetes just before they received a device, with an average HbA1c of 7.6%. Three months after LVAD placement, their average HbA1c had dropped to 5.7%, and 9-12 months after device placement, their average HbA1c level was 5.3% (ASAIO J. 2014;60:290-3). As in the Columbia series, these improvements in hyperglycemia occurred without any significant change in body mass index.
Dr. Guglin also cited similar findings in 50 LVAD patients treated at the University of Rochester (N.Y.)(ASAIO J. 2014;60:675-80), 28 LVAD patients at Penn State Medical College in Hershey, Pa. (Heart Surg. Forum 2014;17:E98-102), and 66 LVAD patients from the University of Illinois in Chicago (Eur. J. Heart Fail. 2014;16:1120-4). In these reports type 2 diabetes existed in roughly a quarter to a third of patients with advanced heart failure who qualified for an LVAD just prior to the time they received the device.
Dr. Guglin also cited two epidemiologic analyses with complementary findings on the risk for incident diabetes faced by heart failure patients. She and her associates reviewed data from 3,165 elderly Americans free from diabetes enrolled in the Cardiovascular Health Study. This cohort included 80 patients with heart failure and 3,085 without heart failure. During 3-4 years of follow-up, 6% of the heart failure patients developed new-onset diabetes, and an additional 10% developed new-onset impaired fasting glucose. In contrast, these incidence rates were 1.5% and 5%, respectively, in the enrollees without heart failure at baseline. In an analysis that controlled for several demographic and biomedical factors, heart failure linked with a statistically significant, 2.4-fold increased risk for the development of diabetes (Cardiology 2014;129:84-92).
And a Danish nationwide cohort study of more than 99,000 residents discharged from a first-time hospitalization for heart failure during 1997-2010 showed a statistically significant link between heart failure severity and an increased rate of development of incident diabetes using diuretic treatment dosage as a surrogate measure of heart failure severity (Diabetologia 2014;57:1595-1600).
The apparent impact of LVAD placement on type 2 diabetes contrasts with what happens in patients who receive a heart transplant, where this association has not been seen. Dr. Guglin suggested that may be because of the immunosuppression with steroids that heart transplant recipients receive, treatment that also prevents diabetes resolution, she said.
“It all boils down to congestion,” Dr. Guglin said in an interview. “Control congestion as much as possible to control the diabetes.”
Dr. Guglin had no relevant financial disclosures.
On Twitter @mitchelzoler
SAN DIEGO – A series of reports in 2014 from several independent groups implicated heart failure as a trigger of type 2 diabetes; findings from several of the analyses also suggested that relief of congestion can result in rapid resolution of the diabetes.
The best way to manage new-onset diabetes in heart failure patients is to “minimize the congestion,” and to “try to achieve as good control of the heart failure as possible,” said Dr. Maya Guglin during a talk at the annual meeting of the American College of Cardiology, in which she laid out the evidence for this newly recognized form of type 2 diabetes. In a review she published in 2014, Dr. Guglin coined the term “cardiogenic diabetes” to describe the condition (Heart Fail. Rev. 2014;19:595-602).
Dr. Guglin traced the data trail for cardiogenic diabetes starting in a 2011 retrospective study of 15 patients with advanced heart failure who received a left ventricular assist device (LVAD) at Columbia University in New York (Eur. J. Heart Fail. 2011;13:195-9). These 15, about a third of the 43 total LVAD recipients at Columbia at the time, had been diagnosed with type 2 diabetes for an average of 6 years before receiving the device. Just before they got their device, their average hemoglobin A1c (HbA1c) level was 7.7%, and their average fasting plasma glucose level was 158 mg/dL. An average of 4 months later, their mean HbA1c had dropped to 6%, and their mean fasting glucose had fallen to 104 mg/dL. Six patients were completely off any diabetes medication. All this occurred while patients had a small increase in their body mass index, which Dr. Guglin attributed to their better physical condition and improved appetite.
Last year, another four reports appeared from four independent, U.S. heart failure groups with results that mirrored the Columbia experience. Dr. Guglin and her associates at the University of Kentucky, Lexington, reported their experience with 50 patients who received an LVAD during 2002-2012 and had type 2 diabetes just before they received a device, with an average HbA1c of 7.6%. Three months after LVAD placement, their average HbA1c had dropped to 5.7%, and 9-12 months after device placement, their average HbA1c level was 5.3% (ASAIO J. 2014;60:290-3). As in the Columbia series, these improvements in hyperglycemia occurred without any significant change in body mass index.
Dr. Guglin also cited similar findings in 50 LVAD patients treated at the University of Rochester (N.Y.)(ASAIO J. 2014;60:675-80), 28 LVAD patients at Penn State Medical College in Hershey, Pa. (Heart Surg. Forum 2014;17:E98-102), and 66 LVAD patients from the University of Illinois in Chicago (Eur. J. Heart Fail. 2014;16:1120-4). In these reports type 2 diabetes existed in roughly a quarter to a third of patients with advanced heart failure who qualified for an LVAD just prior to the time they received the device.
Dr. Guglin also cited two epidemiologic analyses with complementary findings on the risk for incident diabetes faced by heart failure patients. She and her associates reviewed data from 3,165 elderly Americans free from diabetes enrolled in the Cardiovascular Health Study. This cohort included 80 patients with heart failure and 3,085 without heart failure. During 3-4 years of follow-up, 6% of the heart failure patients developed new-onset diabetes, and an additional 10% developed new-onset impaired fasting glucose. In contrast, these incidence rates were 1.5% and 5%, respectively, in the enrollees without heart failure at baseline. In an analysis that controlled for several demographic and biomedical factors, heart failure linked with a statistically significant, 2.4-fold increased risk for the development of diabetes (Cardiology 2014;129:84-92).
And a Danish nationwide cohort study of more than 99,000 residents discharged from a first-time hospitalization for heart failure during 1997-2010 showed a statistically significant link between heart failure severity and an increased rate of development of incident diabetes using diuretic treatment dosage as a surrogate measure of heart failure severity (Diabetologia 2014;57:1595-1600).
The apparent impact of LVAD placement on type 2 diabetes contrasts with what happens in patients who receive a heart transplant, where this association has not been seen. Dr. Guglin suggested that may be because of the immunosuppression with steroids that heart transplant recipients receive, treatment that also prevents diabetes resolution, she said.
“It all boils down to congestion,” Dr. Guglin said in an interview. “Control congestion as much as possible to control the diabetes.”
Dr. Guglin had no relevant financial disclosures.
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM ACC 2015

Ventricular gel injections improve advanced heart failure
SAN DIEGO – Beefing up a sick left ventricle via a set of injections of an inert alginate hydrogel resulted in significantly improved functional capacity, compared with optimal medical therapy through 6 months of follow-up in patients with advanced heart failure in the randomized AUGMENT-HF trial.
Investigators also noted “an interesting and striking reduction” in hospitalizations for worsening heart failure in the group that received left ventricular (LV) augmentation with the material, known as Algisyl-LVR, Dr. Stefan D. Anker reported at the annual meeting of the American College of Cardiology.

Indeed, among 78 patients with advanced heart failure randomized to hydrogel injections plus optimal medical therapy or to optimal medical therapy alone, there were 14 hospitalizations for worsening heart failure in eight controls, compared with 5 hospitalizations in four patients in the LV augmentation group. The between-group difference is large, but the number of hospitalizations is still small. AUGMENT-HF will continue for 2 years of follow-up.
“This gives us hope for the future,” said Dr. Anker, professor of cardiology and cachexia research at Charité Medical School, Berlin.
In addition, based upon the favorable 6-month study results, planning is underway for a larger, pivotal phase III U.S. trial of Algisyl-LVR, classified as a medical device, to start later this year.
At present, surgeons implant the hydrogel through a minithoracotomy. The procedure involves 10-20 injections totaling 4-5 mL of the inert, permanent material, which is placed as a ring of beads along a circumferential line at the left ventricular midwall.
“We make the wall thicker and the cavity of the ventricle a little smaller, thereby reducing wall stress. We basically try to change the physics of the pump action of the heart to improve patient status and perhaps patient outcome,” Dr. Anker explained.
Surgeons say it’s an easily learned procedure. The surgical morbidity and mortality seen in AUGMENT-HF were deemed acceptable by investigators and the study sponsor, so this new therapy will initially be developed as a surgical procedure. But it’s certainly a treatment that lends itself to delivery by percutaneous catheter in the future, according to the cardiologist.
Study participants had moderate to severe heart failure, with an average LV ejection fraction of 25%. Most were New York Heart Association (NYHA) functional class III.
The primary study endpoint was change in peak oxygen uptake (VO2) at 6 months from a baseline of 12.2 mL/kg/min. The value improved to 13.5 mL/kg/min in the LV augmentation group, compared with 12.4 mL/kg/min in controls, a between-group difference that Dr. Anker characterized as clinically relevant. He noted that one of the study’s strengths was that each peak VO2 result was the average of two tests performed on the same occasion, a method that markedly improves test reproducibility.
Also, 6-minute walk distance improved in the LV augmentation group by a mean of 84.7 meters from a baseline 280 meters, while decreasing by 15.4 meters in controls.
“This is quite a positive result rarely seen with other therapies. For everybody involved, this was a very positive finding,” Dr. Anker said.
Among controls, NYHC class stayed steady over the course of 6 months while showing a 0.9-class improvement in the LV augmentation group.
Heart failure etiology – ischemic versus nonischemic – had no bearing on LV augmentation’s effectiveness. Baseline 6-minute walk distance did, though. Patients with a baseline walk distance of less than 287 meters experienced a much larger treatment effect: a mean 2.42 mL/kg/min greater improvement from baseline to 6 months with LV augmentation than in controls, as compared with a nonsignificant 0.4 mL/kg/min advantage among patients who covered more than 287 meters at baseline.
The mean procedure time was 80 minutes, with 190 minutes of anesthesia time. Patients spent an average of 2 days in the ICU.
Three deaths occurred in the surgical group within the first 30 days. Excluding the index hospitalization, there were 22 major adverse cardiovascular events in the control group and 9 in the LV augmentation group. Among these were three cardiovascular deaths in each study arm, for a total of six deaths through 6 months in the LV augmentation patients. However, with additional study follow-up beyond the 6 months presented at ACC 15, mortality has evened out in the two groups, according to Dr. Anker. Sustained ventricular tachycardia occurred in four controls and one patient who received LV augmentation. Several audience members expressed surprise at the low arrhythmia rate in the LV augmentation group, but Dr. Anker’s coinvestigator Dr. Douglas L. Mann explained that the implantation doesn’t create an isthmus, thus there is no nidus for arrhythmia formation.
“No arrhythmia signal has been seen. There is actually a reduction in both atrial and ventricular arrhythmias,” said Dr. Mann, professor of internal medicine and chief of the division of cardiovascular medicine at Washington University in St. Louis.
The AUGMENT-HF trial was sponsored by LoneStar Heart. Dr. Anker reported having no financial relationship with LoneStar, although he serves as a consultant to half a dozen other health care companies.
SAN DIEGO – Beefing up a sick left ventricle via a set of injections of an inert alginate hydrogel resulted in significantly improved functional capacity, compared with optimal medical therapy through 6 months of follow-up in patients with advanced heart failure in the randomized AUGMENT-HF trial.
Investigators also noted “an interesting and striking reduction” in hospitalizations for worsening heart failure in the group that received left ventricular (LV) augmentation with the material, known as Algisyl-LVR, Dr. Stefan D. Anker reported at the annual meeting of the American College of Cardiology.
Indeed, among 78 patients with advanced heart failure randomized to hydrogel injections plus optimal medical therapy or to optimal medical therapy alone, there were 14 hospitalizations for worsening heart failure in eight controls, compared with 5 hospitalizations in four patients in the LV augmentation group. The between-group difference is large, but the number of hospitalizations is still small. AUGMENT-HF will continue for 2 years of follow-up.
“This gives us hope for the future,” said Dr. Anker, professor of cardiology and cachexia research at Charité Medical School, Berlin.
In addition, based upon the favorable 6-month study results, planning is underway for a larger, pivotal phase III U.S. trial of Algisyl-LVR, classified as a medical device, to start later this year.
At present, surgeons implant the hydrogel through a minithoracotomy. The procedure involves 10-20 injections totaling 4-5 mL of the inert, permanent material, which is placed as a ring of beads along a circumferential line at the left ventricular midwall.
“We make the wall thicker and the cavity of the ventricle a little smaller, thereby reducing wall stress. We basically try to change the physics of the pump action of the heart to improve patient status and perhaps patient outcome,” Dr. Anker explained.
Surgeons say it’s an easily learned procedure. The surgical morbidity and mortality seen in AUGMENT-HF were deemed acceptable by investigators and the study sponsor, so this new therapy will initially be developed as a surgical procedure. But it’s certainly a treatment that lends itself to delivery by percutaneous catheter in the future, according to the cardiologist.
Study participants had moderate to severe heart failure, with an average LV ejection fraction of 25%. Most were New York Heart Association (NYHA) functional class III.
The primary study endpoint was change in peak oxygen uptake (VO2) at 6 months from a baseline of 12.2 mL/kg/min. The value improved to 13.5 mL/kg/min in the LV augmentation group, compared with 12.4 mL/kg/min in controls, a between-group difference that Dr. Anker characterized as clinically relevant. He noted that one of the study’s strengths was that each peak VO2 result was the average of two tests performed on the same occasion, a method that markedly improves test reproducibility.
Also, 6-minute walk distance improved in the LV augmentation group by a mean of 84.7 meters from a baseline 280 meters, while decreasing by 15.4 meters in controls.
“This is quite a positive result rarely seen with other therapies. For everybody involved, this was a very positive finding,” Dr. Anker said.
Among controls, NYHC class stayed steady over the course of 6 months while showing a 0.9-class improvement in the LV augmentation group.
Heart failure etiology – ischemic versus nonischemic – had no bearing on LV augmentation’s effectiveness. Baseline 6-minute walk distance did, though. Patients with a baseline walk distance of less than 287 meters experienced a much larger treatment effect: a mean 2.42 mL/kg/min greater improvement from baseline to 6 months with LV augmentation than in controls, as compared with a nonsignificant 0.4 mL/kg/min advantage among patients who covered more than 287 meters at baseline.
The mean procedure time was 80 minutes, with 190 minutes of anesthesia time. Patients spent an average of 2 days in the ICU.
Three deaths occurred in the surgical group within the first 30 days. Excluding the index hospitalization, there were 22 major adverse cardiovascular events in the control group and 9 in the LV augmentation group. Among these were three cardiovascular deaths in each study arm, for a total of six deaths through 6 months in the LV augmentation patients. However, with additional study follow-up beyond the 6 months presented at ACC 15, mortality has evened out in the two groups, according to Dr. Anker. Sustained ventricular tachycardia occurred in four controls and one patient who received LV augmentation. Several audience members expressed surprise at the low arrhythmia rate in the LV augmentation group, but Dr. Anker’s coinvestigator Dr. Douglas L. Mann explained that the implantation doesn’t create an isthmus, thus there is no nidus for arrhythmia formation.
“No arrhythmia signal has been seen. There is actually a reduction in both atrial and ventricular arrhythmias,” said Dr. Mann, professor of internal medicine and chief of the division of cardiovascular medicine at Washington University in St. Louis.
The AUGMENT-HF trial was sponsored by LoneStar Heart. Dr. Anker reported having no financial relationship with LoneStar, although he serves as a consultant to half a dozen other health care companies.
SAN DIEGO – Beefing up a sick left ventricle via a set of injections of an inert alginate hydrogel resulted in significantly improved functional capacity, compared with optimal medical therapy through 6 months of follow-up in patients with advanced heart failure in the randomized AUGMENT-HF trial.
Investigators also noted “an interesting and striking reduction” in hospitalizations for worsening heart failure in the group that received left ventricular (LV) augmentation with the material, known as Algisyl-LVR, Dr. Stefan D. Anker reported at the annual meeting of the American College of Cardiology.
Indeed, among 78 patients with advanced heart failure randomized to hydrogel injections plus optimal medical therapy or to optimal medical therapy alone, there were 14 hospitalizations for worsening heart failure in eight controls, compared with 5 hospitalizations in four patients in the LV augmentation group. The between-group difference is large, but the number of hospitalizations is still small. AUGMENT-HF will continue for 2 years of follow-up.
“This gives us hope for the future,” said Dr. Anker, professor of cardiology and cachexia research at Charité Medical School, Berlin.
In addition, based upon the favorable 6-month study results, planning is underway for a larger, pivotal phase III U.S. trial of Algisyl-LVR, classified as a medical device, to start later this year.
At present, surgeons implant the hydrogel through a minithoracotomy. The procedure involves 10-20 injections totaling 4-5 mL of the inert, permanent material, which is placed as a ring of beads along a circumferential line at the left ventricular midwall.
“We make the wall thicker and the cavity of the ventricle a little smaller, thereby reducing wall stress. We basically try to change the physics of the pump action of the heart to improve patient status and perhaps patient outcome,” Dr. Anker explained.
Surgeons say it’s an easily learned procedure. The surgical morbidity and mortality seen in AUGMENT-HF were deemed acceptable by investigators and the study sponsor, so this new therapy will initially be developed as a surgical procedure. But it’s certainly a treatment that lends itself to delivery by percutaneous catheter in the future, according to the cardiologist.
Study participants had moderate to severe heart failure, with an average LV ejection fraction of 25%. Most were New York Heart Association (NYHA) functional class III.
The primary study endpoint was change in peak oxygen uptake (VO2) at 6 months from a baseline of 12.2 mL/kg/min. The value improved to 13.5 mL/kg/min in the LV augmentation group, compared with 12.4 mL/kg/min in controls, a between-group difference that Dr. Anker characterized as clinically relevant. He noted that one of the study’s strengths was that each peak VO2 result was the average of two tests performed on the same occasion, a method that markedly improves test reproducibility.
Also, 6-minute walk distance improved in the LV augmentation group by a mean of 84.7 meters from a baseline 280 meters, while decreasing by 15.4 meters in controls.
“This is quite a positive result rarely seen with other therapies. For everybody involved, this was a very positive finding,” Dr. Anker said.
Among controls, NYHC class stayed steady over the course of 6 months while showing a 0.9-class improvement in the LV augmentation group.
Heart failure etiology – ischemic versus nonischemic – had no bearing on LV augmentation’s effectiveness. Baseline 6-minute walk distance did, though. Patients with a baseline walk distance of less than 287 meters experienced a much larger treatment effect: a mean 2.42 mL/kg/min greater improvement from baseline to 6 months with LV augmentation than in controls, as compared with a nonsignificant 0.4 mL/kg/min advantage among patients who covered more than 287 meters at baseline.
The mean procedure time was 80 minutes, with 190 minutes of anesthesia time. Patients spent an average of 2 days in the ICU.
Three deaths occurred in the surgical group within the first 30 days. Excluding the index hospitalization, there were 22 major adverse cardiovascular events in the control group and 9 in the LV augmentation group. Among these were three cardiovascular deaths in each study arm, for a total of six deaths through 6 months in the LV augmentation patients. However, with additional study follow-up beyond the 6 months presented at ACC 15, mortality has evened out in the two groups, according to Dr. Anker. Sustained ventricular tachycardia occurred in four controls and one patient who received LV augmentation. Several audience members expressed surprise at the low arrhythmia rate in the LV augmentation group, but Dr. Anker’s coinvestigator Dr. Douglas L. Mann explained that the implantation doesn’t create an isthmus, thus there is no nidus for arrhythmia formation.
“No arrhythmia signal has been seen. There is actually a reduction in both atrial and ventricular arrhythmias,” said Dr. Mann, professor of internal medicine and chief of the division of cardiovascular medicine at Washington University in St. Louis.
The AUGMENT-HF trial was sponsored by LoneStar Heart. Dr. Anker reported having no financial relationship with LoneStar, although he serves as a consultant to half a dozen other health care companies.
AT ACC 15
Key clinical point: Left ventricular augmentation achieved through injections of an inert hydrogel shows promise as a novel therapy for advanced heart failure.
Major finding: Mean peak VO2 improved over the course of 6 months from a baseline of 12.2 mL/kg/min to 13.5 mL/kg/min in patients on optimal medical therapy who underwent the left ventricular augmentation procedure, compared with a 6-month value of 12.4 mL/kg/min in controls on optimal medical therapy alone.
Data source: AUGMENT-HF, a randomized, prospective, 78-patient clinical trial conducted in five countries.
Disclosures: AUGMENT-HF was sponsored by LoneStar Heart. The presenter reported having no financial relationship with LoneStar, although he serves as a consultant to half a dozen other health care companies.

CoreValve receives first TAVR valve-in-valve indication
The U.S. Food and Drug Administration on March 30 expanded its approved use of the CoreValve transcatheter aortic-valve replacement (TAVR) system to include patients who already have undergone aortic valve replacement and need a second valve replacement done as a valve-in-valve placement.
With this action, CoreValve became the first TAVR system to receive U.S. approval for valve-in-valve use. The CoreValve System received FDA approval for TAVR performed on native aortic valves in January 2014 in patients at “extreme risk,” and in June 2014 for those at “high risk,” for surgical aortic valve replacement.* Valve-in-valve TAVR is only feasible in patients with a failing bioprosthetic aortic valve: It is not an option for patients with a failing mechanical aortic valve.

“The CoreValve System offers a less-invasive treatment option for a significant number of patients with failed tissue aortic valves whose medical teams determine that the risks associated with repeat open-heart surgery are high or extremely high,” Dr. William H. Maisel, deputy center director for science and chief scientist in the FDA’s Center for Devices and Radiological Health, said in a written statement. “The approval is an important expansion of the authorized use of the transcatheter aortic valve replacement technology.”
The CoreValve, which is designed to sit in a supra-annular location 12 mm above the aortic valve annulus, is well suited for valve-in-valve replacement because the only portion of the CoreValve that actually fills the annular space and the ring of the existing valve is the CoreValve’s sealer. This results in a tight seal that produces less paravalvular leak than when the sealer sits in a native annulus that is often deformed with calcium, noted Dr. Michael J. Reardon, professor of cardiothoracic surgery at Methodist Hospital in Houston.
In addition, because the sealer exerts pressure on the old valve ring in the annulus instead of on myocardium, placing the CoreValve as a valve-in-valve produces much less conduction disruption and results in fewer patients who need a pacemaker following TAVR, he said.

The CoreValve as a valve-in-valve “works quite well, and is not hard to position,” said Dr. Reardon, who added that he has now performed several valve-in-valve TAVRs using the CoreValve.
Similar TAVR procedures are usually not possible using the balloon-expandable SAPIEN System because the SAPIEN valve is designed to sit directly in the annulus and, in most patients, the existing valve ring does not provide enough space to accommodate a SAPIEN valve.
Dr. Reardon anticipates that many U.S. patients now in their 80s with a failing bioprosthetic aortic valve will be interested in nonsurgical TAVR replacement. These patients often do not want conventional open-heart surgery, he said in an interview.
To evaluate the safety and efficacy of the CoreValve System for aortic valve-in-valve replacement, the FDA reviewed clinical data collected from a U.S. clinical trial with 143 patients, an agency representative said in the statement. In the clinical trial, the estimated rate of 30-day survival without major stroke was 96%, and 89% after 6 months. “This compares well to the corresponding rates reported previously for trial participants who received the same device to replace their own, native diseased or damaged aortic valve,” the agency’s statement said.
According to the agency, aortic valve-in-valve use of the CoreValve System should be limited to patients who need replacement of a failed tissue aortic valve but are at extreme or high risk of death or serious complications from traditional open-heart surgery. A decision as to whether the product and procedure are appropriate for a patient “should involve careful evaluation by the patient’s heart medical team, including a cardiologist and a cardiac surgeon.”
The FDA said that the CoreValve System should not be used in patients who have any infection, have a mechanical aortic heart valve, cannot tolerate anticoagulant drugs, or have sensitivity to titanium, nickel, or contrast media.
Dr. Maisel had no disclosures. Dr. Reardon has served as an advisor to Medtronic, the company that markets the CoreValve.
On Twitter @mitchelzoler
*Correction, 4/1/2015: An earlier version of this article misstated the device’s approval history.
The U.S. Food and Drug Administration on March 30 expanded its approved use of the CoreValve transcatheter aortic-valve replacement (TAVR) system to include patients who already have undergone aortic valve replacement and need a second valve replacement done as a valve-in-valve placement.
With this action, CoreValve became the first TAVR system to receive U.S. approval for valve-in-valve use. The CoreValve System received FDA approval for TAVR performed on native aortic valves in January 2014 in patients at “extreme risk,” and in June 2014 for those at “high risk,” for surgical aortic valve replacement.* Valve-in-valve TAVR is only feasible in patients with a failing bioprosthetic aortic valve: It is not an option for patients with a failing mechanical aortic valve.
“The CoreValve System offers a less-invasive treatment option for a significant number of patients with failed tissue aortic valves whose medical teams determine that the risks associated with repeat open-heart surgery are high or extremely high,” Dr. William H. Maisel, deputy center director for science and chief scientist in the FDA’s Center for Devices and Radiological Health, said in a written statement. “The approval is an important expansion of the authorized use of the transcatheter aortic valve replacement technology.”
The CoreValve, which is designed to sit in a supra-annular location 12 mm above the aortic valve annulus, is well suited for valve-in-valve replacement because the only portion of the CoreValve that actually fills the annular space and the ring of the existing valve is the CoreValve’s sealer. This results in a tight seal that produces less paravalvular leak than when the sealer sits in a native annulus that is often deformed with calcium, noted Dr. Michael J. Reardon, professor of cardiothoracic surgery at Methodist Hospital in Houston.
In addition, because the sealer exerts pressure on the old valve ring in the annulus instead of on myocardium, placing the CoreValve as a valve-in-valve produces much less conduction disruption and results in fewer patients who need a pacemaker following TAVR, he said.
The CoreValve as a valve-in-valve “works quite well, and is not hard to position,” said Dr. Reardon, who added that he has now performed several valve-in-valve TAVRs using the CoreValve.
Similar TAVR procedures are usually not possible using the balloon-expandable SAPIEN System because the SAPIEN valve is designed to sit directly in the annulus and, in most patients, the existing valve ring does not provide enough space to accommodate a SAPIEN valve.
Dr. Reardon anticipates that many U.S. patients now in their 80s with a failing bioprosthetic aortic valve will be interested in nonsurgical TAVR replacement. These patients often do not want conventional open-heart surgery, he said in an interview.
To evaluate the safety and efficacy of the CoreValve System for aortic valve-in-valve replacement, the FDA reviewed clinical data collected from a U.S. clinical trial with 143 patients, an agency representative said in the statement. In the clinical trial, the estimated rate of 30-day survival without major stroke was 96%, and 89% after 6 months. “This compares well to the corresponding rates reported previously for trial participants who received the same device to replace their own, native diseased or damaged aortic valve,” the agency’s statement said.
According to the agency, aortic valve-in-valve use of the CoreValve System should be limited to patients who need replacement of a failed tissue aortic valve but are at extreme or high risk of death or serious complications from traditional open-heart surgery. A decision as to whether the product and procedure are appropriate for a patient “should involve careful evaluation by the patient’s heart medical team, including a cardiologist and a cardiac surgeon.”
The FDA said that the CoreValve System should not be used in patients who have any infection, have a mechanical aortic heart valve, cannot tolerate anticoagulant drugs, or have sensitivity to titanium, nickel, or contrast media.
Dr. Maisel had no disclosures. Dr. Reardon has served as an advisor to Medtronic, the company that markets the CoreValve.
On Twitter @mitchelzoler
*Correction, 4/1/2015: An earlier version of this article misstated the device’s approval history.
The U.S. Food and Drug Administration on March 30 expanded its approved use of the CoreValve transcatheter aortic-valve replacement (TAVR) system to include patients who already have undergone aortic valve replacement and need a second valve replacement done as a valve-in-valve placement.
With this action, CoreValve became the first TAVR system to receive U.S. approval for valve-in-valve use. The CoreValve System received FDA approval for TAVR performed on native aortic valves in January 2014 in patients at “extreme risk,” and in June 2014 for those at “high risk,” for surgical aortic valve replacement.* Valve-in-valve TAVR is only feasible in patients with a failing bioprosthetic aortic valve: It is not an option for patients with a failing mechanical aortic valve.
“The CoreValve System offers a less-invasive treatment option for a significant number of patients with failed tissue aortic valves whose medical teams determine that the risks associated with repeat open-heart surgery are high or extremely high,” Dr. William H. Maisel, deputy center director for science and chief scientist in the FDA’s Center for Devices and Radiological Health, said in a written statement. “The approval is an important expansion of the authorized use of the transcatheter aortic valve replacement technology.”
The CoreValve, which is designed to sit in a supra-annular location 12 mm above the aortic valve annulus, is well suited for valve-in-valve replacement because the only portion of the CoreValve that actually fills the annular space and the ring of the existing valve is the CoreValve’s sealer. This results in a tight seal that produces less paravalvular leak than when the sealer sits in a native annulus that is often deformed with calcium, noted Dr. Michael J. Reardon, professor of cardiothoracic surgery at Methodist Hospital in Houston.
In addition, because the sealer exerts pressure on the old valve ring in the annulus instead of on myocardium, placing the CoreValve as a valve-in-valve produces much less conduction disruption and results in fewer patients who need a pacemaker following TAVR, he said.
The CoreValve as a valve-in-valve “works quite well, and is not hard to position,” said Dr. Reardon, who added that he has now performed several valve-in-valve TAVRs using the CoreValve.
Similar TAVR procedures are usually not possible using the balloon-expandable SAPIEN System because the SAPIEN valve is designed to sit directly in the annulus and, in most patients, the existing valve ring does not provide enough space to accommodate a SAPIEN valve.
Dr. Reardon anticipates that many U.S. patients now in their 80s with a failing bioprosthetic aortic valve will be interested in nonsurgical TAVR replacement. These patients often do not want conventional open-heart surgery, he said in an interview.
To evaluate the safety and efficacy of the CoreValve System for aortic valve-in-valve replacement, the FDA reviewed clinical data collected from a U.S. clinical trial with 143 patients, an agency representative said in the statement. In the clinical trial, the estimated rate of 30-day survival without major stroke was 96%, and 89% after 6 months. “This compares well to the corresponding rates reported previously for trial participants who received the same device to replace their own, native diseased or damaged aortic valve,” the agency’s statement said.
According to the agency, aortic valve-in-valve use of the CoreValve System should be limited to patients who need replacement of a failed tissue aortic valve but are at extreme or high risk of death or serious complications from traditional open-heart surgery. A decision as to whether the product and procedure are appropriate for a patient “should involve careful evaluation by the patient’s heart medical team, including a cardiologist and a cardiac surgeon.”
The FDA said that the CoreValve System should not be used in patients who have any infection, have a mechanical aortic heart valve, cannot tolerate anticoagulant drugs, or have sensitivity to titanium, nickel, or contrast media.
Dr. Maisel had no disclosures. Dr. Reardon has served as an advisor to Medtronic, the company that markets the CoreValve.
On Twitter @mitchelzoler
*Correction, 4/1/2015: An earlier version of this article misstated the device’s approval history.

LEGACY: Weight loss markedly improves atrial fibrillation
SAN DIEGO – Maintenance of long-term weight loss in obese or overweight individuals with atrial fibrillation was associated with nearly a sixfold greater likelihood of freedom from recurrent AF during nearly 5 years of active follow-up in the LEGACY study.
“The effect was dose dependent. The most important finding is that 46% of patients with at least a 10% weight loss were free from AF without the use of drugs or ablation procedures through nearly 5 years, versus 22% of those with 3%-9% weight loss, and just 13% with less than a 3% weight loss,” Dr. Rajeev K. Pathak said at the annual meeting of the American College of Cardiology.

LEGACY (Long-Term Effect of Goal-Directed Weight Management in an Atrial Fibrillation Cohort: A Long-Term Follow-Up Study) included 355 overweight or obese participants with paroxsymal or persistent AF who were offered the chance to participate in a dedicated weight-loss clinic. Regular participation in this clinic proved to be a key factor in losing weight, keeping it off, and reducing AF burden; the more frequently patients attended the quarterly sessions the better the outcomes, noted Dr. Pathak, a cardiologist and electrophysiology fellow at the University of Adelaide (Australia).
Of the 355 participants, 135 lost at least 10% of their body weight, 103 had a 3%-9% drop in body weight, and 117 had less than a 3% loss or a weight gain.
Year-by-year weight trends had a significant impact on outcome. The 141 patients with linear weight loss had a 76% AF-free rate with or without the use of drugs or ablation, compared with a 59% in the 179 patients with weight fluctuations and the 38% rate in those with no loss or a weight gain. Atrial fibrillation status was assessed by 7-day Holter monitoring at least annually.
Weight fluctuation – defined as a 1% or greater change in weight between two consecutive annual follow-ups – dampened the benefits conferred by weight loss. Patients who experienced more than a 5% weight fluctuation were 2.2-fold more likely to experience AF recurrence than were those without weight fluctuation.
Patients with a sustained 10% weight loss were 5.6-fold more likely to achieve long-term freedom from AF than were patients with lesser or no weight loss. The goal was 10% weight loss rather than a body mass index of 25 kg/m2 or less because once AF patients get down to a BMI below 27 kg/m2 the incremental benefit of each additional 1 BMI point in terms of freedom from AF becomes much smaller, said Dr. Pathak.
Weight loss also showed a dose-dependent effect on various cardiovascular risk factors. For example, mean systolic blood pressure fell by 18 mm Hg in subjects with at least a 10% weight loss, by 10 mm Hg with a 3%-9% loss, and by 7 mm Hg with a lesser weight loss. Triglycerides, LDL cholesterol, and glycemic control improved in similar fashion. In addition, weight loss showed beneficial effects on cardiac structure, with dose-dependent reductions in left atrial volume indexed for body surface area as well as interventricular septal thickness, Dr. Pathak continued.
He described the weight loss clinic as a “very simple” structured motivational and goal-directed program with face-to-face counseling.
“We have one patient, one physician, no props. We sit with the patient, discuss areas we can improve, then we devise a low-carb, low-fat, high-protein diet in consultation with the patient. Patients maintain a lifestyle journal where they log their meals and exercise. We prescribe at least 200 minutes of moderate-intensity activity per week. Eating is a behavioral pattern, and we have found this approach to be a very effective behavioral tool. Because the plan is developed in consultation with the patient, we’ve found patients tend to adhere to what they have planned,” he explained.

Discussant Dr. Bernard J. Gersh praised LEGACY as “a really wonderful study – very important.
“There are years of epidemiologic evidence suggesting that obesity contributes in a major way to the epidemic of atrial fibrillation, and you’ve taken it a step further,” added Dr. Gersh, professor of medicine at the Mayo Clinic in Rochester, Minn.
Playing devil’s advocate, he asked whether the observed reduction in AF might have nothing to do with weight loss, but could be explained simply by LEGACY perhaps having enrolled a highly compliant group of patients who agreed to attend a clinic and were more willing to take their medications.
Dr. Pathak rejected the compliance factor as an explanation for the results. He noted that while cardiovascular risk factors improved with greater weight loss, the need for antihypertensive, lipid-lowering, and antiarrhythmic medications decreased.
“The effect can’t possibly be due to increased compliance with medications, but rather it’s a true effect of the weight loss itself. So I think this is a true clinical effect and not an epiphenomenon,” he replied.
Dr. Pathak added that he and his coinvestigators are organizing a randomized controlled confirmatory study.

Dr. Prediman K. Shah, who chaired a press conference where the LEGACY study was highlighted, said the study provided him with one of the major take-home lessons from ACC 15.
“We can argue about the mechanism of atrial fibrillation till kingdom come, but the fact is that the association is very strong that weight loss is associated with a reduced burden of atrial fibrillation, and with a very robust magnitude of benefit. That’s one of the messages that I’ll take home with me from this meeting: The next time I see my fat patient with atrial fibrillation, I’m putting him on a weight-reducing diet as the first approach,” declared Dr. Shah, professor of medicine at UCLA and director of the Oppenheimer Atherosclerosis Research Center at Cedars-Sinai Medical Center in Los Angeles.
Dr. Pathak reported having no financial conflicts regarding this study, which was supported by university funds.
Simultaneously with Dr. Pathak’s presentation at ACC 15, the LEGACY study was published online (J. Am. Coll. Cardiol. 2015 [doi: 10.1016/j.jacc.2015.03.002])
SAN DIEGO – Maintenance of long-term weight loss in obese or overweight individuals with atrial fibrillation was associated with nearly a sixfold greater likelihood of freedom from recurrent AF during nearly 5 years of active follow-up in the LEGACY study.
“The effect was dose dependent. The most important finding is that 46% of patients with at least a 10% weight loss were free from AF without the use of drugs or ablation procedures through nearly 5 years, versus 22% of those with 3%-9% weight loss, and just 13% with less than a 3% weight loss,” Dr. Rajeev K. Pathak said at the annual meeting of the American College of Cardiology.
LEGACY (Long-Term Effect of Goal-Directed Weight Management in an Atrial Fibrillation Cohort: A Long-Term Follow-Up Study) included 355 overweight or obese participants with paroxsymal or persistent AF who were offered the chance to participate in a dedicated weight-loss clinic. Regular participation in this clinic proved to be a key factor in losing weight, keeping it off, and reducing AF burden; the more frequently patients attended the quarterly sessions the better the outcomes, noted Dr. Pathak, a cardiologist and electrophysiology fellow at the University of Adelaide (Australia).
Of the 355 participants, 135 lost at least 10% of their body weight, 103 had a 3%-9% drop in body weight, and 117 had less than a 3% loss or a weight gain.
Year-by-year weight trends had a significant impact on outcome. The 141 patients with linear weight loss had a 76% AF-free rate with or without the use of drugs or ablation, compared with a 59% in the 179 patients with weight fluctuations and the 38% rate in those with no loss or a weight gain. Atrial fibrillation status was assessed by 7-day Holter monitoring at least annually.
Weight fluctuation – defined as a 1% or greater change in weight between two consecutive annual follow-ups – dampened the benefits conferred by weight loss. Patients who experienced more than a 5% weight fluctuation were 2.2-fold more likely to experience AF recurrence than were those without weight fluctuation.
Patients with a sustained 10% weight loss were 5.6-fold more likely to achieve long-term freedom from AF than were patients with lesser or no weight loss. The goal was 10% weight loss rather than a body mass index of 25 kg/m2 or less because once AF patients get down to a BMI below 27 kg/m2 the incremental benefit of each additional 1 BMI point in terms of freedom from AF becomes much smaller, said Dr. Pathak.
Weight loss also showed a dose-dependent effect on various cardiovascular risk factors. For example, mean systolic blood pressure fell by 18 mm Hg in subjects with at least a 10% weight loss, by 10 mm Hg with a 3%-9% loss, and by 7 mm Hg with a lesser weight loss. Triglycerides, LDL cholesterol, and glycemic control improved in similar fashion. In addition, weight loss showed beneficial effects on cardiac structure, with dose-dependent reductions in left atrial volume indexed for body surface area as well as interventricular septal thickness, Dr. Pathak continued.
He described the weight loss clinic as a “very simple” structured motivational and goal-directed program with face-to-face counseling.
“We have one patient, one physician, no props. We sit with the patient, discuss areas we can improve, then we devise a low-carb, low-fat, high-protein diet in consultation with the patient. Patients maintain a lifestyle journal where they log their meals and exercise. We prescribe at least 200 minutes of moderate-intensity activity per week. Eating is a behavioral pattern, and we have found this approach to be a very effective behavioral tool. Because the plan is developed in consultation with the patient, we’ve found patients tend to adhere to what they have planned,” he explained.
Discussant Dr. Bernard J. Gersh praised LEGACY as “a really wonderful study – very important.
“There are years of epidemiologic evidence suggesting that obesity contributes in a major way to the epidemic of atrial fibrillation, and you’ve taken it a step further,” added Dr. Gersh, professor of medicine at the Mayo Clinic in Rochester, Minn.
Playing devil’s advocate, he asked whether the observed reduction in AF might have nothing to do with weight loss, but could be explained simply by LEGACY perhaps having enrolled a highly compliant group of patients who agreed to attend a clinic and were more willing to take their medications.
Dr. Pathak rejected the compliance factor as an explanation for the results. He noted that while cardiovascular risk factors improved with greater weight loss, the need for antihypertensive, lipid-lowering, and antiarrhythmic medications decreased.
“The effect can’t possibly be due to increased compliance with medications, but rather it’s a true effect of the weight loss itself. So I think this is a true clinical effect and not an epiphenomenon,” he replied.
Dr. Pathak added that he and his coinvestigators are organizing a randomized controlled confirmatory study.
Dr. Prediman K. Shah, who chaired a press conference where the LEGACY study was highlighted, said the study provided him with one of the major take-home lessons from ACC 15.
“We can argue about the mechanism of atrial fibrillation till kingdom come, but the fact is that the association is very strong that weight loss is associated with a reduced burden of atrial fibrillation, and with a very robust magnitude of benefit. That’s one of the messages that I’ll take home with me from this meeting: The next time I see my fat patient with atrial fibrillation, I’m putting him on a weight-reducing diet as the first approach,” declared Dr. Shah, professor of medicine at UCLA and director of the Oppenheimer Atherosclerosis Research Center at Cedars-Sinai Medical Center in Los Angeles.
Dr. Pathak reported having no financial conflicts regarding this study, which was supported by university funds.
Simultaneously with Dr. Pathak’s presentation at ACC 15, the LEGACY study was published online (J. Am. Coll. Cardiol. 2015 [doi: 10.1016/j.jacc.2015.03.002])
SAN DIEGO – Maintenance of long-term weight loss in obese or overweight individuals with atrial fibrillation was associated with nearly a sixfold greater likelihood of freedom from recurrent AF during nearly 5 years of active follow-up in the LEGACY study.
“The effect was dose dependent. The most important finding is that 46% of patients with at least a 10% weight loss were free from AF without the use of drugs or ablation procedures through nearly 5 years, versus 22% of those with 3%-9% weight loss, and just 13% with less than a 3% weight loss,” Dr. Rajeev K. Pathak said at the annual meeting of the American College of Cardiology.
LEGACY (Long-Term Effect of Goal-Directed Weight Management in an Atrial Fibrillation Cohort: A Long-Term Follow-Up Study) included 355 overweight or obese participants with paroxsymal or persistent AF who were offered the chance to participate in a dedicated weight-loss clinic. Regular participation in this clinic proved to be a key factor in losing weight, keeping it off, and reducing AF burden; the more frequently patients attended the quarterly sessions the better the outcomes, noted Dr. Pathak, a cardiologist and electrophysiology fellow at the University of Adelaide (Australia).
Of the 355 participants, 135 lost at least 10% of their body weight, 103 had a 3%-9% drop in body weight, and 117 had less than a 3% loss or a weight gain.
Year-by-year weight trends had a significant impact on outcome. The 141 patients with linear weight loss had a 76% AF-free rate with or without the use of drugs or ablation, compared with a 59% in the 179 patients with weight fluctuations and the 38% rate in those with no loss or a weight gain. Atrial fibrillation status was assessed by 7-day Holter monitoring at least annually.
Weight fluctuation – defined as a 1% or greater change in weight between two consecutive annual follow-ups – dampened the benefits conferred by weight loss. Patients who experienced more than a 5% weight fluctuation were 2.2-fold more likely to experience AF recurrence than were those without weight fluctuation.
Patients with a sustained 10% weight loss were 5.6-fold more likely to achieve long-term freedom from AF than were patients with lesser or no weight loss. The goal was 10% weight loss rather than a body mass index of 25 kg/m2 or less because once AF patients get down to a BMI below 27 kg/m2 the incremental benefit of each additional 1 BMI point in terms of freedom from AF becomes much smaller, said Dr. Pathak.
Weight loss also showed a dose-dependent effect on various cardiovascular risk factors. For example, mean systolic blood pressure fell by 18 mm Hg in subjects with at least a 10% weight loss, by 10 mm Hg with a 3%-9% loss, and by 7 mm Hg with a lesser weight loss. Triglycerides, LDL cholesterol, and glycemic control improved in similar fashion. In addition, weight loss showed beneficial effects on cardiac structure, with dose-dependent reductions in left atrial volume indexed for body surface area as well as interventricular septal thickness, Dr. Pathak continued.
He described the weight loss clinic as a “very simple” structured motivational and goal-directed program with face-to-face counseling.
“We have one patient, one physician, no props. We sit with the patient, discuss areas we can improve, then we devise a low-carb, low-fat, high-protein diet in consultation with the patient. Patients maintain a lifestyle journal where they log their meals and exercise. We prescribe at least 200 minutes of moderate-intensity activity per week. Eating is a behavioral pattern, and we have found this approach to be a very effective behavioral tool. Because the plan is developed in consultation with the patient, we’ve found patients tend to adhere to what they have planned,” he explained.
Discussant Dr. Bernard J. Gersh praised LEGACY as “a really wonderful study – very important.
“There are years of epidemiologic evidence suggesting that obesity contributes in a major way to the epidemic of atrial fibrillation, and you’ve taken it a step further,” added Dr. Gersh, professor of medicine at the Mayo Clinic in Rochester, Minn.
Playing devil’s advocate, he asked whether the observed reduction in AF might have nothing to do with weight loss, but could be explained simply by LEGACY perhaps having enrolled a highly compliant group of patients who agreed to attend a clinic and were more willing to take their medications.
Dr. Pathak rejected the compliance factor as an explanation for the results. He noted that while cardiovascular risk factors improved with greater weight loss, the need for antihypertensive, lipid-lowering, and antiarrhythmic medications decreased.
“The effect can’t possibly be due to increased compliance with medications, but rather it’s a true effect of the weight loss itself. So I think this is a true clinical effect and not an epiphenomenon,” he replied.
Dr. Pathak added that he and his coinvestigators are organizing a randomized controlled confirmatory study.
Dr. Prediman K. Shah, who chaired a press conference where the LEGACY study was highlighted, said the study provided him with one of the major take-home lessons from ACC 15.
“We can argue about the mechanism of atrial fibrillation till kingdom come, but the fact is that the association is very strong that weight loss is associated with a reduced burden of atrial fibrillation, and with a very robust magnitude of benefit. That’s one of the messages that I’ll take home with me from this meeting: The next time I see my fat patient with atrial fibrillation, I’m putting him on a weight-reducing diet as the first approach,” declared Dr. Shah, professor of medicine at UCLA and director of the Oppenheimer Atherosclerosis Research Center at Cedars-Sinai Medical Center in Los Angeles.
Dr. Pathak reported having no financial conflicts regarding this study, which was supported by university funds.
Simultaneously with Dr. Pathak’s presentation at ACC 15, the LEGACY study was published online (J. Am. Coll. Cardiol. 2015 [doi: 10.1016/j.jacc.2015.03.002])
AT ACC 15
Key clinical point: Nearly half of overweight or obese patients with atrial fibrillation who achieved at least a 10% sustained weight loss remained arrhythmia free without resort to antiarrhythmic drugs or ablation procedures during nearly 5 years of follow-up.
Major finding: The weight-loss-associated freedom from recurrent AF was accompanied by improvements in cardiac structure as well as improved cardiovascular risk factors despite lesser use of risk factor-modifying drugs.
Data source: An observational study of 355 overweight or obese patients with atrial fibrillation who agreed to participate in a weight loss clinic.
Disclosures: The LEGACY study was supported by university funds. The presenter reported no financial conflicts.

Anticipation runs high for coming megatrials in general cardiology
SAN DIEGO – Preventive cardiovascular medicine will get “a big boost” from ongoing major randomized controlled trials due to report results during the next several years, Dr. Eric D. Peterson predicted at the annual meeting of the American College of Cardiology.
He presented an overview of eagerly anticipated clinical trials approaching completion in three of the hottest areas of research in general cardiology: new cardiovascular prevention agents and strategies; innovative health policy initiatives; and optimal antithrombotic regimens across the range of chronic coronary artery disease, peripheral vascular disease, patients with atrial fibrillation who’ve undergone percutaneous coronary intervention, and cerebrovascular disease.

Dr. Peterson is executive director of the Duke Clinical Research Institute (DCRI), a major player in some of these studies. To gain additional perspective regarding what’s in store, he consulted a who’s who list of eminent American cardiology clinical trialists as to what’s going to be big in 2016-2017 and shortly beyond.
One thing’s clear looking out at the research horizon: Megatrials enrolling tens of thousands of patients to answer key clinical questions are not a thing of the past.
Far from it.
Cardiovascular prevention
Exciting pivotal phase III cardiovascular prevention trials are well underway in the areas of lipid modification, diabetes, and hypertension. In addition, the possible preventive effect of vitamin D and omega-3 free fatty acid supplementation seen in hypothesis-generating epidemiologic studies is finally being put to a definitive test in a 26,000-subject randomized trial known as VITAL (the Vitamin D and omega-3 trial).
Lipids: All eyes are on ongoing pivotal phase III randomized clinical outcome trials of three monoclonal antibodies that inhibit proprotein convertase subtilisin kexin type 9 (PCSK9): the 22,500-patient FOURIER trial of evolocumab, which includes 9,000 patients older than 65 years; the 18,000-patient ODYSSEY Outcomes trial of alirocumab; and the SPIRE-1 and SPIRE-2 trials of bococizumab totaling 18,300 patients.
In phase II studies, these agents have generated enormous excitement, because they safely achieve unprecedented LDL lowering, with early hints of improved clinical outcomes beyond what’s achievable with today’s drugs, noted Dr. Peterson, professor of medicine at Duke University in Durham, N.C.
CETP inhibitors: The cholesteryl ester transfer protein inhibitors torcetrapib and dalcetrapib flamed out in clinical trials because of safety concerns and lack of clinical benefit. But pivotal phase III trials of two other CETP inhibitors are well underway: anacetrapib, which is the focus of the 30,000-patient REVEAL trial; and evacetrapib, featured in the 11,000-patient ACCELERATE trial. The CETP inhibitors simultaneously boost HDL while reducing LDL.
Diabetes: The search continues for new drugs that not only enhance diabetes control but also improve cardiovascular outcomes, or at the very least are safe in diabetes patients with coronary disease.
Next up is the nearly 15,000-patient Trial Evaluating Cardiovascular Outcomes with Sitagliptin (TECOS), coordinated by the DCRI and University of Oxford. It’s due to be presented in June at the annual meeting of the American Diabetes Association in Boston. Trials of other dipeptidyl peptidase-4 inhibitors have shown evidence of an increased risk of heart failure, specifically with saxagliptin in the SAVOR-TIMI 53 trial and alogliptin in EXAMINE, so a lot is riding on TECOS.
The glucagon-like peptide-1 (GLP-1) agonists are under study in cardiovascular event-driven randomized clinical trials collectively totaling more than 33,000 patients with type 2 diabetes. The Exenatide Study of Cardiovascular Event Lowering (EXSCEL), also coordinated by the DCRI and University of Oxford, includes 14,000 randomized patients. Other major trials of GLP-1 agonists are ELIXA (lixisenatide), LEADER (liraglutide), and REWIND (dulaglutide).
In addition, ongoing phase III trials of sodium glucose cotransporter 2 (SGLT-2) inhibitors with cardiovascular endpoints include the Canagliflozin Cardiovascular Assessment Study (CANVAS) and a 7,000-patient study of empagliflozin known as EMPA-REG OUTCOME.
Hypertension: The Systolic Blood Pressure Intervention Trial (SPRINT) is an NIH-funded, randomized trial designed to finally answer a question bothering physicians for years: What’s the right amount of blood pressure lowering?
Nearly 9,400 high-risk patients with clinical or subclinical cardiovascular disease have been randomized to a target systolic blood pressure of less than 120 mm Hg, compared with less than 140 mm Hg. The primary composite endpoint is cardiovascular death, nonfatal MI or stroke, or hospitalization for acute coronary syndrome. Secondary endpoints focus on cognitive impairment, dementia, and rates of progression of chronic kidney disease. Results are expected in 2018.
Vitamin D: The VITAL trial, also sponsored by the NIH, has randomized 25,875 initially healthy men and women to 2,000 IU of vitamin D3 per day or placebo, and further randomized them to 1 g/day of omega-3 fatty acids or placebo. Key endpoints are total cancers, MI, stroke, and cardiovascular death. Results are coming in 2017.
Optimal antithrombotic therapy
Chronic coronary artery disease: It’s remarkable that physicians still don’t know the right dose of aspirin to use, even though the drug has been around since 1897. The answer is finally on the way. It will come from the ADAPTABLE eTrial, the first comparative effectiveness research project funded by the Patient-Centered Outcomes Research Institute (PCORI).
Twenty thousand U.S. patients with known atherosclerotic cardiovascular disease and at least one extra risk factor will be randomized to daily aspirin at 81 mg or 325 mg and followed for a maximum of 30 months. The primary efficacy endpoint is all-cause mortality, nonfatal MI, or nonfatal stroke. The primary safety endpoint is major bleeding.
This is a groundbreaking innovative study: It’s a 20,000-patient trial budgeted at a mere $10 million – peanuts in the world of megatrials. By comparison, the NIH-funded SPRINT hypertension trial includes less than half as many patients, yet the price tag is $114 million.
The bargain-basement cost of the aspirin trial is possible because follow-up will be via electronic medical records and claims data provided every 3 months by 29 health data networks participating in PCORI’s National Patient-Centered Clinical Research Network (PCORnet). The aspirin study is sort of a shakedown cruise for an exciting new cost-effective approach to conducting comparative effectiveness research.
“This study could be a game changer,” Dr. Peterson declared. “Whether you think the research question is important or not, the ability to utilize electronic health records in long-term patient follow-up in randomized trials will be revolutionary in terms of answering key clinical questions cost effectively.”
Peripheral vascular disease: In search of the antithrombotic regimen that optimizes limb salvage while minimizing vascular event rate, the EUCLID trial has randomized 13,500 patients with symptomatic peripheral artery disease to ticagrelor at 90 mg BID or clopidogrel at 75 mg/day. Follow-up will be for 18 months, with the primary endpoint a composite of cardiovascular death, nonfatal MI, or ischemic stroke.
Atrial fibrillation patients who undergo coronary stent placement: The 2,100-patient PIONEER AF-PCI study, now recruiting, is evaluating different combinations of rivaroxaban in various dosing regimens coupled with clopidogrel, warfarin, and/or aspirin.
Stroke: The SOCRATES trial is recruiting 9,600 patients with acute ischemic stroke or high-risk transient ischemic attack to be randomized within 24 hours to ticagrelor at 90 mg BID or aspirin at 100 mg/day. As with PIONEER, results from SOCRATES are anticipated next year.
Health policy and implementation
The ARTEMIS study poses the question of whether providing a guideline-directed medication gratis will improve patient adherence and/or clinical outcomes.
Some 9,000 high-cardiovascular-risk patients at 300 sites are being randomized to free ticagrelor or clopidogrel – no copay – for 1 year or to usual care. Outcomes include choice of medication, adherence, and major adverse cardiovascular events.
“If this works, there’s the possibility that insurers can be persuaded to pay for medications and basically give them away as a means of ultimately resulting in better outcomes for patients and lower costs,” Dr. Peterson said.
IMPACT AF is an international quality improvement project aimed at improving the use of oral anticoagulation therapy in high-risk patients with atrial fibrillation in Argentina, Brazil, China, India, and Romania. Participating centers will be randomized to receive a provider education and feedback program or not. The primary outcome is a change from baseline through year 1 in the proportion of patients with atrial fibrillation taking an oral anticoagulant.
In summing up the state of research in general cardiology, Dr. Peterson observed, “The way we do trials is changing. The size of the studies is changing. But ultimately, we will find better ways to treat patients, and perhaps we can find better ways to encourage patients to take their medications long term.”
Dr. Peterson reported serving as a consultant to AstraZeneca Boehringer Ingelheim, Genentech, Janssen, and Sanofi, and receiving research funding from Eli Lilly and Janssen.
SAN DIEGO – Preventive cardiovascular medicine will get “a big boost” from ongoing major randomized controlled trials due to report results during the next several years, Dr. Eric D. Peterson predicted at the annual meeting of the American College of Cardiology.
He presented an overview of eagerly anticipated clinical trials approaching completion in three of the hottest areas of research in general cardiology: new cardiovascular prevention agents and strategies; innovative health policy initiatives; and optimal antithrombotic regimens across the range of chronic coronary artery disease, peripheral vascular disease, patients with atrial fibrillation who’ve undergone percutaneous coronary intervention, and cerebrovascular disease.
Dr. Peterson is executive director of the Duke Clinical Research Institute (DCRI), a major player in some of these studies. To gain additional perspective regarding what’s in store, he consulted a who’s who list of eminent American cardiology clinical trialists as to what’s going to be big in 2016-2017 and shortly beyond.
One thing’s clear looking out at the research horizon: Megatrials enrolling tens of thousands of patients to answer key clinical questions are not a thing of the past.
Far from it.
Cardiovascular prevention
Exciting pivotal phase III cardiovascular prevention trials are well underway in the areas of lipid modification, diabetes, and hypertension. In addition, the possible preventive effect of vitamin D and omega-3 free fatty acid supplementation seen in hypothesis-generating epidemiologic studies is finally being put to a definitive test in a 26,000-subject randomized trial known as VITAL (the Vitamin D and omega-3 trial).
Lipids: All eyes are on ongoing pivotal phase III randomized clinical outcome trials of three monoclonal antibodies that inhibit proprotein convertase subtilisin kexin type 9 (PCSK9): the 22,500-patient FOURIER trial of evolocumab, which includes 9,000 patients older than 65 years; the 18,000-patient ODYSSEY Outcomes trial of alirocumab; and the SPIRE-1 and SPIRE-2 trials of bococizumab totaling 18,300 patients.
In phase II studies, these agents have generated enormous excitement, because they safely achieve unprecedented LDL lowering, with early hints of improved clinical outcomes beyond what’s achievable with today’s drugs, noted Dr. Peterson, professor of medicine at Duke University in Durham, N.C.
CETP inhibitors: The cholesteryl ester transfer protein inhibitors torcetrapib and dalcetrapib flamed out in clinical trials because of safety concerns and lack of clinical benefit. But pivotal phase III trials of two other CETP inhibitors are well underway: anacetrapib, which is the focus of the 30,000-patient REVEAL trial; and evacetrapib, featured in the 11,000-patient ACCELERATE trial. The CETP inhibitors simultaneously boost HDL while reducing LDL.
Diabetes: The search continues for new drugs that not only enhance diabetes control but also improve cardiovascular outcomes, or at the very least are safe in diabetes patients with coronary disease.
Next up is the nearly 15,000-patient Trial Evaluating Cardiovascular Outcomes with Sitagliptin (TECOS), coordinated by the DCRI and University of Oxford. It’s due to be presented in June at the annual meeting of the American Diabetes Association in Boston. Trials of other dipeptidyl peptidase-4 inhibitors have shown evidence of an increased risk of heart failure, specifically with saxagliptin in the SAVOR-TIMI 53 trial and alogliptin in EXAMINE, so a lot is riding on TECOS.
The glucagon-like peptide-1 (GLP-1) agonists are under study in cardiovascular event-driven randomized clinical trials collectively totaling more than 33,000 patients with type 2 diabetes. The Exenatide Study of Cardiovascular Event Lowering (EXSCEL), also coordinated by the DCRI and University of Oxford, includes 14,000 randomized patients. Other major trials of GLP-1 agonists are ELIXA (lixisenatide), LEADER (liraglutide), and REWIND (dulaglutide).
In addition, ongoing phase III trials of sodium glucose cotransporter 2 (SGLT-2) inhibitors with cardiovascular endpoints include the Canagliflozin Cardiovascular Assessment Study (CANVAS) and a 7,000-patient study of empagliflozin known as EMPA-REG OUTCOME.
Hypertension: The Systolic Blood Pressure Intervention Trial (SPRINT) is an NIH-funded, randomized trial designed to finally answer a question bothering physicians for years: What’s the right amount of blood pressure lowering?
Nearly 9,400 high-risk patients with clinical or subclinical cardiovascular disease have been randomized to a target systolic blood pressure of less than 120 mm Hg, compared with less than 140 mm Hg. The primary composite endpoint is cardiovascular death, nonfatal MI or stroke, or hospitalization for acute coronary syndrome. Secondary endpoints focus on cognitive impairment, dementia, and rates of progression of chronic kidney disease. Results are expected in 2018.
Vitamin D: The VITAL trial, also sponsored by the NIH, has randomized 25,875 initially healthy men and women to 2,000 IU of vitamin D3 per day or placebo, and further randomized them to 1 g/day of omega-3 fatty acids or placebo. Key endpoints are total cancers, MI, stroke, and cardiovascular death. Results are coming in 2017.
Optimal antithrombotic therapy
Chronic coronary artery disease: It’s remarkable that physicians still don’t know the right dose of aspirin to use, even though the drug has been around since 1897. The answer is finally on the way. It will come from the ADAPTABLE eTrial, the first comparative effectiveness research project funded by the Patient-Centered Outcomes Research Institute (PCORI).
Twenty thousand U.S. patients with known atherosclerotic cardiovascular disease and at least one extra risk factor will be randomized to daily aspirin at 81 mg or 325 mg and followed for a maximum of 30 months. The primary efficacy endpoint is all-cause mortality, nonfatal MI, or nonfatal stroke. The primary safety endpoint is major bleeding.
This is a groundbreaking innovative study: It’s a 20,000-patient trial budgeted at a mere $10 million – peanuts in the world of megatrials. By comparison, the NIH-funded SPRINT hypertension trial includes less than half as many patients, yet the price tag is $114 million.
The bargain-basement cost of the aspirin trial is possible because follow-up will be via electronic medical records and claims data provided every 3 months by 29 health data networks participating in PCORI’s National Patient-Centered Clinical Research Network (PCORnet). The aspirin study is sort of a shakedown cruise for an exciting new cost-effective approach to conducting comparative effectiveness research.
“This study could be a game changer,” Dr. Peterson declared. “Whether you think the research question is important or not, the ability to utilize electronic health records in long-term patient follow-up in randomized trials will be revolutionary in terms of answering key clinical questions cost effectively.”
Peripheral vascular disease: In search of the antithrombotic regimen that optimizes limb salvage while minimizing vascular event rate, the EUCLID trial has randomized 13,500 patients with symptomatic peripheral artery disease to ticagrelor at 90 mg BID or clopidogrel at 75 mg/day. Follow-up will be for 18 months, with the primary endpoint a composite of cardiovascular death, nonfatal MI, or ischemic stroke.
Atrial fibrillation patients who undergo coronary stent placement: The 2,100-patient PIONEER AF-PCI study, now recruiting, is evaluating different combinations of rivaroxaban in various dosing regimens coupled with clopidogrel, warfarin, and/or aspirin.
Stroke: The SOCRATES trial is recruiting 9,600 patients with acute ischemic stroke or high-risk transient ischemic attack to be randomized within 24 hours to ticagrelor at 90 mg BID or aspirin at 100 mg/day. As with PIONEER, results from SOCRATES are anticipated next year.
Health policy and implementation
The ARTEMIS study poses the question of whether providing a guideline-directed medication gratis will improve patient adherence and/or clinical outcomes.
Some 9,000 high-cardiovascular-risk patients at 300 sites are being randomized to free ticagrelor or clopidogrel – no copay – for 1 year or to usual care. Outcomes include choice of medication, adherence, and major adverse cardiovascular events.
“If this works, there’s the possibility that insurers can be persuaded to pay for medications and basically give them away as a means of ultimately resulting in better outcomes for patients and lower costs,” Dr. Peterson said.
IMPACT AF is an international quality improvement project aimed at improving the use of oral anticoagulation therapy in high-risk patients with atrial fibrillation in Argentina, Brazil, China, India, and Romania. Participating centers will be randomized to receive a provider education and feedback program or not. The primary outcome is a change from baseline through year 1 in the proportion of patients with atrial fibrillation taking an oral anticoagulant.
In summing up the state of research in general cardiology, Dr. Peterson observed, “The way we do trials is changing. The size of the studies is changing. But ultimately, we will find better ways to treat patients, and perhaps we can find better ways to encourage patients to take their medications long term.”
Dr. Peterson reported serving as a consultant to AstraZeneca Boehringer Ingelheim, Genentech, Janssen, and Sanofi, and receiving research funding from Eli Lilly and Janssen.
SAN DIEGO – Preventive cardiovascular medicine will get “a big boost” from ongoing major randomized controlled trials due to report results during the next several years, Dr. Eric D. Peterson predicted at the annual meeting of the American College of Cardiology.
He presented an overview of eagerly anticipated clinical trials approaching completion in three of the hottest areas of research in general cardiology: new cardiovascular prevention agents and strategies; innovative health policy initiatives; and optimal antithrombotic regimens across the range of chronic coronary artery disease, peripheral vascular disease, patients with atrial fibrillation who’ve undergone percutaneous coronary intervention, and cerebrovascular disease.
Dr. Peterson is executive director of the Duke Clinical Research Institute (DCRI), a major player in some of these studies. To gain additional perspective regarding what’s in store, he consulted a who’s who list of eminent American cardiology clinical trialists as to what’s going to be big in 2016-2017 and shortly beyond.
One thing’s clear looking out at the research horizon: Megatrials enrolling tens of thousands of patients to answer key clinical questions are not a thing of the past.
Far from it.
Cardiovascular prevention
Exciting pivotal phase III cardiovascular prevention trials are well underway in the areas of lipid modification, diabetes, and hypertension. In addition, the possible preventive effect of vitamin D and omega-3 free fatty acid supplementation seen in hypothesis-generating epidemiologic studies is finally being put to a definitive test in a 26,000-subject randomized trial known as VITAL (the Vitamin D and omega-3 trial).
Lipids: All eyes are on ongoing pivotal phase III randomized clinical outcome trials of three monoclonal antibodies that inhibit proprotein convertase subtilisin kexin type 9 (PCSK9): the 22,500-patient FOURIER trial of evolocumab, which includes 9,000 patients older than 65 years; the 18,000-patient ODYSSEY Outcomes trial of alirocumab; and the SPIRE-1 and SPIRE-2 trials of bococizumab totaling 18,300 patients.
In phase II studies, these agents have generated enormous excitement, because they safely achieve unprecedented LDL lowering, with early hints of improved clinical outcomes beyond what’s achievable with today’s drugs, noted Dr. Peterson, professor of medicine at Duke University in Durham, N.C.
CETP inhibitors: The cholesteryl ester transfer protein inhibitors torcetrapib and dalcetrapib flamed out in clinical trials because of safety concerns and lack of clinical benefit. But pivotal phase III trials of two other CETP inhibitors are well underway: anacetrapib, which is the focus of the 30,000-patient REVEAL trial; and evacetrapib, featured in the 11,000-patient ACCELERATE trial. The CETP inhibitors simultaneously boost HDL while reducing LDL.
Diabetes: The search continues for new drugs that not only enhance diabetes control but also improve cardiovascular outcomes, or at the very least are safe in diabetes patients with coronary disease.
Next up is the nearly 15,000-patient Trial Evaluating Cardiovascular Outcomes with Sitagliptin (TECOS), coordinated by the DCRI and University of Oxford. It’s due to be presented in June at the annual meeting of the American Diabetes Association in Boston. Trials of other dipeptidyl peptidase-4 inhibitors have shown evidence of an increased risk of heart failure, specifically with saxagliptin in the SAVOR-TIMI 53 trial and alogliptin in EXAMINE, so a lot is riding on TECOS.
The glucagon-like peptide-1 (GLP-1) agonists are under study in cardiovascular event-driven randomized clinical trials collectively totaling more than 33,000 patients with type 2 diabetes. The Exenatide Study of Cardiovascular Event Lowering (EXSCEL), also coordinated by the DCRI and University of Oxford, includes 14,000 randomized patients. Other major trials of GLP-1 agonists are ELIXA (lixisenatide), LEADER (liraglutide), and REWIND (dulaglutide).
In addition, ongoing phase III trials of sodium glucose cotransporter 2 (SGLT-2) inhibitors with cardiovascular endpoints include the Canagliflozin Cardiovascular Assessment Study (CANVAS) and a 7,000-patient study of empagliflozin known as EMPA-REG OUTCOME.
Hypertension: The Systolic Blood Pressure Intervention Trial (SPRINT) is an NIH-funded, randomized trial designed to finally answer a question bothering physicians for years: What’s the right amount of blood pressure lowering?
Nearly 9,400 high-risk patients with clinical or subclinical cardiovascular disease have been randomized to a target systolic blood pressure of less than 120 mm Hg, compared with less than 140 mm Hg. The primary composite endpoint is cardiovascular death, nonfatal MI or stroke, or hospitalization for acute coronary syndrome. Secondary endpoints focus on cognitive impairment, dementia, and rates of progression of chronic kidney disease. Results are expected in 2018.
Vitamin D: The VITAL trial, also sponsored by the NIH, has randomized 25,875 initially healthy men and women to 2,000 IU of vitamin D3 per day or placebo, and further randomized them to 1 g/day of omega-3 fatty acids or placebo. Key endpoints are total cancers, MI, stroke, and cardiovascular death. Results are coming in 2017.
Optimal antithrombotic therapy
Chronic coronary artery disease: It’s remarkable that physicians still don’t know the right dose of aspirin to use, even though the drug has been around since 1897. The answer is finally on the way. It will come from the ADAPTABLE eTrial, the first comparative effectiveness research project funded by the Patient-Centered Outcomes Research Institute (PCORI).
Twenty thousand U.S. patients with known atherosclerotic cardiovascular disease and at least one extra risk factor will be randomized to daily aspirin at 81 mg or 325 mg and followed for a maximum of 30 months. The primary efficacy endpoint is all-cause mortality, nonfatal MI, or nonfatal stroke. The primary safety endpoint is major bleeding.
This is a groundbreaking innovative study: It’s a 20,000-patient trial budgeted at a mere $10 million – peanuts in the world of megatrials. By comparison, the NIH-funded SPRINT hypertension trial includes less than half as many patients, yet the price tag is $114 million.
The bargain-basement cost of the aspirin trial is possible because follow-up will be via electronic medical records and claims data provided every 3 months by 29 health data networks participating in PCORI’s National Patient-Centered Clinical Research Network (PCORnet). The aspirin study is sort of a shakedown cruise for an exciting new cost-effective approach to conducting comparative effectiveness research.
“This study could be a game changer,” Dr. Peterson declared. “Whether you think the research question is important or not, the ability to utilize electronic health records in long-term patient follow-up in randomized trials will be revolutionary in terms of answering key clinical questions cost effectively.”
Peripheral vascular disease: In search of the antithrombotic regimen that optimizes limb salvage while minimizing vascular event rate, the EUCLID trial has randomized 13,500 patients with symptomatic peripheral artery disease to ticagrelor at 90 mg BID or clopidogrel at 75 mg/day. Follow-up will be for 18 months, with the primary endpoint a composite of cardiovascular death, nonfatal MI, or ischemic stroke.
Atrial fibrillation patients who undergo coronary stent placement: The 2,100-patient PIONEER AF-PCI study, now recruiting, is evaluating different combinations of rivaroxaban in various dosing regimens coupled with clopidogrel, warfarin, and/or aspirin.
Stroke: The SOCRATES trial is recruiting 9,600 patients with acute ischemic stroke or high-risk transient ischemic attack to be randomized within 24 hours to ticagrelor at 90 mg BID or aspirin at 100 mg/day. As with PIONEER, results from SOCRATES are anticipated next year.
Health policy and implementation
The ARTEMIS study poses the question of whether providing a guideline-directed medication gratis will improve patient adherence and/or clinical outcomes.
Some 9,000 high-cardiovascular-risk patients at 300 sites are being randomized to free ticagrelor or clopidogrel – no copay – for 1 year or to usual care. Outcomes include choice of medication, adherence, and major adverse cardiovascular events.
“If this works, there’s the possibility that insurers can be persuaded to pay for medications and basically give them away as a means of ultimately resulting in better outcomes for patients and lower costs,” Dr. Peterson said.
IMPACT AF is an international quality improvement project aimed at improving the use of oral anticoagulation therapy in high-risk patients with atrial fibrillation in Argentina, Brazil, China, India, and Romania. Participating centers will be randomized to receive a provider education and feedback program or not. The primary outcome is a change from baseline through year 1 in the proportion of patients with atrial fibrillation taking an oral anticoagulant.
In summing up the state of research in general cardiology, Dr. Peterson observed, “The way we do trials is changing. The size of the studies is changing. But ultimately, we will find better ways to treat patients, and perhaps we can find better ways to encourage patients to take their medications long term.”
Dr. Peterson reported serving as a consultant to AstraZeneca Boehringer Ingelheim, Genentech, Janssen, and Sanofi, and receiving research funding from Eli Lilly and Janssen.
EXPERT ANALYSIS FROM ACC 15

MOOD-HF: SSRI ineffective in heart failure with depression
SAN DIEGO – Patients with major depressive disorder and heart failure with reduced ejection fraction fared no better with a selective serotonin reuptake inhibitor than with placebo in terms of heart failure outcomes or improved psychological well being in MOOD-HF, the first large randomized trial to examine the issue.
“MOOD-HF does not provide a rationale for the use of escitalopram in patients with systolic heart failure and comorbid depression. MOOD-HF suggests, however, that optimal heart failure management resulting in improved signs and symptoms might possibly be a means to ameliorate comorbid depression,” Dr. Christiane E. Angermann said at the annual meeting of the American College of Cardiology.

Moreover, exploratory secondary analysis suggested “a very subtle long-term effect of the antidepressant that may not be so favorable” in patients with more severe heart failure, she added.
“It seems that some of our patients take antidepressants in vain, and we should care more about the heart failure medications,” said Dr. Angermann, chairman of the MOOD-HF trial and director of clinical research at the Comprehensive Heart Failure Center at the University of Wurzburg (Germany).
MOOD-HF was an ambitious clinical trial conducted at 16 German academic medical centers. The study began with the screening – via the Patient Health Questionnaire–9 – of more than 11,000 patients who had heart failure with reduced ejection fraction. A psychiatrist using the Structured Clinical Interview for Depression eventually diagnosed a subgroup as having major depressive disorder. Ultimately, 372 patients with an average left ventricular ejection fraction of 35% were randomized in double-blind fashion to escitalopram (Lexapro) or placebo for a planned 24 months.
The study rationale stemmed from the well-established finding that depression is three to five times more common in heart failure patients than in the general population, and that the severity of depression in these patients shows a dose-response relationship with their risks of all-cause mortality and heart failure hospitalization. The primary study hypothesis in MOOD-HF was that treating the comorbid depression would result in a reduced risk of all-cause mortality and unplanned hospitalization for any cause.
“Never before had an ethics committee been persuaded to allow patients with major depressive disorder to be on a placebo for 2 years,” said Dr. Angermann. “It took a lot of discussion with the psychiatrists, addressing whether these were depressed patients with comorbid heart failure or patients with heart failure and comorbid depression. We ended up agreeing that since these patients mostly die from heart failure, therefore this is the main disease. And since there was no evidence from other cardiovascular studies that antidepressants were of use, we were allowed to do the study,” she explained.
The trial was halted early for futility at the request of the data safety monitoring board after a median of 18 months. At that point, the primary composite endpoint of death or unplanned hospitalization had occurred in 116 patients in the escitalopram group and 119 on placebo.
A key secondary endpoint – improvement in the Montgomery-Asberg Depression Rating Scale (MADRS) at 12 weeks – was also a virtual dead heat. From a baseline score of 21, both the escitalopram and placebo groups showed a clinically meaningful improvement of roughly 9 points. The improvement in MADRS scores in the control group piqued investigators’ interest because no psychotherapy or counseling was involved in the trial.
The target dose of escitalopram was 20 mg/day, with uptitration to that goal taking place over the first 12 weeks. The mean dose of the antidepressant at week 12 was 13.7 mg/day. During the course of the study, however, the target dose dropped to 10 mg/day in patients over age 65 in accordance with updated regulatory guidance regarding possible proarrhythmic QTc prolongation in older patients. As it turned out, QTc prolongation was not an issue in MOOD-HF. Indeed, there was no difference between the two groups in terms of any serious adverse events. Adherence to the study medication was good, Dr. Angermann continued.
While the dose of escitalopram was being uptitrated during the first 12 weeks, so was guideline-directed medical therapy for heart failure in both study arms. The patients improved from being at 56% of the recommended dose of ACE inhibitor/angiotensin receptor blocker therapy at baseline to 64%, and from 58% of the target dose of beta-blocker therapy to 63%. That, rather than the antidepressant, appeared to be responsible for the improvement in heart failure, she said.
Both study arms showed similar improvements over time in New York Heart Association functional class and left ventricular ejection fraction. But levels of N-terminal pro-B-type natriuretic peptide (NT-proBNP) – an important biomarker of heart failure severity – improved only in the placebo-treated controls. The same held true for reduction in left ventricular end diastolic diameter.
“This suggests a somewhat attenuated improvement in heart failure in the escitalopram group,” the cardiologist observed.
The investigators conducted a post hoc analysis to generate a hypothesis-generating risk score for the primary outcome. One point each was given for age greater than 62.6 years, a baseline NT-proBNP level in excess of 807 ng/L, a heart rate greater than 68 bpm, being New York Heart Association class III/IV, and having a left ventricular end diastolic diameter in excess of 59 mm. These were thresholds that identified patients in the top half of the study population for each of these variables.
In patients with a risk score of 0-2, escitalopram was associated with a highly significant 33% reduction in the risk of death or unplanned hospitalization compared with placebo. However, in patients with a score of 3-5, the escitalopram group had a 52% greater risk of the primary endpoint than controls.
“This suggests a heterogeneous pathophysiology and an impact of individual disease profiles on the effects of the study medication,” Dr. Angermann said.
MOOD-HF was funded chiefly by the German Ministry of Education and Research and the University of Wurzburg. Lundbeck provided the escitalopram gratis. Dr. Angermann reported serving as a consultant to ResMed, Novartis, and Vifor and receiving research grants from Lundbeck, Alere, and Boehringer Ingelheim.
SAN DIEGO – Patients with major depressive disorder and heart failure with reduced ejection fraction fared no better with a selective serotonin reuptake inhibitor than with placebo in terms of heart failure outcomes or improved psychological well being in MOOD-HF, the first large randomized trial to examine the issue.
“MOOD-HF does not provide a rationale for the use of escitalopram in patients with systolic heart failure and comorbid depression. MOOD-HF suggests, however, that optimal heart failure management resulting in improved signs and symptoms might possibly be a means to ameliorate comorbid depression,” Dr. Christiane E. Angermann said at the annual meeting of the American College of Cardiology.
Moreover, exploratory secondary analysis suggested “a very subtle long-term effect of the antidepressant that may not be so favorable” in patients with more severe heart failure, she added.
“It seems that some of our patients take antidepressants in vain, and we should care more about the heart failure medications,” said Dr. Angermann, chairman of the MOOD-HF trial and director of clinical research at the Comprehensive Heart Failure Center at the University of Wurzburg (Germany).
MOOD-HF was an ambitious clinical trial conducted at 16 German academic medical centers. The study began with the screening – via the Patient Health Questionnaire–9 – of more than 11,000 patients who had heart failure with reduced ejection fraction. A psychiatrist using the Structured Clinical Interview for Depression eventually diagnosed a subgroup as having major depressive disorder. Ultimately, 372 patients with an average left ventricular ejection fraction of 35% were randomized in double-blind fashion to escitalopram (Lexapro) or placebo for a planned 24 months.
The study rationale stemmed from the well-established finding that depression is three to five times more common in heart failure patients than in the general population, and that the severity of depression in these patients shows a dose-response relationship with their risks of all-cause mortality and heart failure hospitalization. The primary study hypothesis in MOOD-HF was that treating the comorbid depression would result in a reduced risk of all-cause mortality and unplanned hospitalization for any cause.
“Never before had an ethics committee been persuaded to allow patients with major depressive disorder to be on a placebo for 2 years,” said Dr. Angermann. “It took a lot of discussion with the psychiatrists, addressing whether these were depressed patients with comorbid heart failure or patients with heart failure and comorbid depression. We ended up agreeing that since these patients mostly die from heart failure, therefore this is the main disease. And since there was no evidence from other cardiovascular studies that antidepressants were of use, we were allowed to do the study,” she explained.
The trial was halted early for futility at the request of the data safety monitoring board after a median of 18 months. At that point, the primary composite endpoint of death or unplanned hospitalization had occurred in 116 patients in the escitalopram group and 119 on placebo.
A key secondary endpoint – improvement in the Montgomery-Asberg Depression Rating Scale (MADRS) at 12 weeks – was also a virtual dead heat. From a baseline score of 21, both the escitalopram and placebo groups showed a clinically meaningful improvement of roughly 9 points. The improvement in MADRS scores in the control group piqued investigators’ interest because no psychotherapy or counseling was involved in the trial.
The target dose of escitalopram was 20 mg/day, with uptitration to that goal taking place over the first 12 weeks. The mean dose of the antidepressant at week 12 was 13.7 mg/day. During the course of the study, however, the target dose dropped to 10 mg/day in patients over age 65 in accordance with updated regulatory guidance regarding possible proarrhythmic QTc prolongation in older patients. As it turned out, QTc prolongation was not an issue in MOOD-HF. Indeed, there was no difference between the two groups in terms of any serious adverse events. Adherence to the study medication was good, Dr. Angermann continued.
While the dose of escitalopram was being uptitrated during the first 12 weeks, so was guideline-directed medical therapy for heart failure in both study arms. The patients improved from being at 56% of the recommended dose of ACE inhibitor/angiotensin receptor blocker therapy at baseline to 64%, and from 58% of the target dose of beta-blocker therapy to 63%. That, rather than the antidepressant, appeared to be responsible for the improvement in heart failure, she said.
Both study arms showed similar improvements over time in New York Heart Association functional class and left ventricular ejection fraction. But levels of N-terminal pro-B-type natriuretic peptide (NT-proBNP) – an important biomarker of heart failure severity – improved only in the placebo-treated controls. The same held true for reduction in left ventricular end diastolic diameter.
“This suggests a somewhat attenuated improvement in heart failure in the escitalopram group,” the cardiologist observed.
The investigators conducted a post hoc analysis to generate a hypothesis-generating risk score for the primary outcome. One point each was given for age greater than 62.6 years, a baseline NT-proBNP level in excess of 807 ng/L, a heart rate greater than 68 bpm, being New York Heart Association class III/IV, and having a left ventricular end diastolic diameter in excess of 59 mm. These were thresholds that identified patients in the top half of the study population for each of these variables.
In patients with a risk score of 0-2, escitalopram was associated with a highly significant 33% reduction in the risk of death or unplanned hospitalization compared with placebo. However, in patients with a score of 3-5, the escitalopram group had a 52% greater risk of the primary endpoint than controls.
“This suggests a heterogeneous pathophysiology and an impact of individual disease profiles on the effects of the study medication,” Dr. Angermann said.
MOOD-HF was funded chiefly by the German Ministry of Education and Research and the University of Wurzburg. Lundbeck provided the escitalopram gratis. Dr. Angermann reported serving as a consultant to ResMed, Novartis, and Vifor and receiving research grants from Lundbeck, Alere, and Boehringer Ingelheim.
SAN DIEGO – Patients with major depressive disorder and heart failure with reduced ejection fraction fared no better with a selective serotonin reuptake inhibitor than with placebo in terms of heart failure outcomes or improved psychological well being in MOOD-HF, the first large randomized trial to examine the issue.
“MOOD-HF does not provide a rationale for the use of escitalopram in patients with systolic heart failure and comorbid depression. MOOD-HF suggests, however, that optimal heart failure management resulting in improved signs and symptoms might possibly be a means to ameliorate comorbid depression,” Dr. Christiane E. Angermann said at the annual meeting of the American College of Cardiology.
Moreover, exploratory secondary analysis suggested “a very subtle long-term effect of the antidepressant that may not be so favorable” in patients with more severe heart failure, she added.
“It seems that some of our patients take antidepressants in vain, and we should care more about the heart failure medications,” said Dr. Angermann, chairman of the MOOD-HF trial and director of clinical research at the Comprehensive Heart Failure Center at the University of Wurzburg (Germany).
MOOD-HF was an ambitious clinical trial conducted at 16 German academic medical centers. The study began with the screening – via the Patient Health Questionnaire–9 – of more than 11,000 patients who had heart failure with reduced ejection fraction. A psychiatrist using the Structured Clinical Interview for Depression eventually diagnosed a subgroup as having major depressive disorder. Ultimately, 372 patients with an average left ventricular ejection fraction of 35% were randomized in double-blind fashion to escitalopram (Lexapro) or placebo for a planned 24 months.
The study rationale stemmed from the well-established finding that depression is three to five times more common in heart failure patients than in the general population, and that the severity of depression in these patients shows a dose-response relationship with their risks of all-cause mortality and heart failure hospitalization. The primary study hypothesis in MOOD-HF was that treating the comorbid depression would result in a reduced risk of all-cause mortality and unplanned hospitalization for any cause.
“Never before had an ethics committee been persuaded to allow patients with major depressive disorder to be on a placebo for 2 years,” said Dr. Angermann. “It took a lot of discussion with the psychiatrists, addressing whether these were depressed patients with comorbid heart failure or patients with heart failure and comorbid depression. We ended up agreeing that since these patients mostly die from heart failure, therefore this is the main disease. And since there was no evidence from other cardiovascular studies that antidepressants were of use, we were allowed to do the study,” she explained.
The trial was halted early for futility at the request of the data safety monitoring board after a median of 18 months. At that point, the primary composite endpoint of death or unplanned hospitalization had occurred in 116 patients in the escitalopram group and 119 on placebo.
A key secondary endpoint – improvement in the Montgomery-Asberg Depression Rating Scale (MADRS) at 12 weeks – was also a virtual dead heat. From a baseline score of 21, both the escitalopram and placebo groups showed a clinically meaningful improvement of roughly 9 points. The improvement in MADRS scores in the control group piqued investigators’ interest because no psychotherapy or counseling was involved in the trial.
The target dose of escitalopram was 20 mg/day, with uptitration to that goal taking place over the first 12 weeks. The mean dose of the antidepressant at week 12 was 13.7 mg/day. During the course of the study, however, the target dose dropped to 10 mg/day in patients over age 65 in accordance with updated regulatory guidance regarding possible proarrhythmic QTc prolongation in older patients. As it turned out, QTc prolongation was not an issue in MOOD-HF. Indeed, there was no difference between the two groups in terms of any serious adverse events. Adherence to the study medication was good, Dr. Angermann continued.
While the dose of escitalopram was being uptitrated during the first 12 weeks, so was guideline-directed medical therapy for heart failure in both study arms. The patients improved from being at 56% of the recommended dose of ACE inhibitor/angiotensin receptor blocker therapy at baseline to 64%, and from 58% of the target dose of beta-blocker therapy to 63%. That, rather than the antidepressant, appeared to be responsible for the improvement in heart failure, she said.
Both study arms showed similar improvements over time in New York Heart Association functional class and left ventricular ejection fraction. But levels of N-terminal pro-B-type natriuretic peptide (NT-proBNP) – an important biomarker of heart failure severity – improved only in the placebo-treated controls. The same held true for reduction in left ventricular end diastolic diameter.
“This suggests a somewhat attenuated improvement in heart failure in the escitalopram group,” the cardiologist observed.
The investigators conducted a post hoc analysis to generate a hypothesis-generating risk score for the primary outcome. One point each was given for age greater than 62.6 years, a baseline NT-proBNP level in excess of 807 ng/L, a heart rate greater than 68 bpm, being New York Heart Association class III/IV, and having a left ventricular end diastolic diameter in excess of 59 mm. These were thresholds that identified patients in the top half of the study population for each of these variables.
In patients with a risk score of 0-2, escitalopram was associated with a highly significant 33% reduction in the risk of death or unplanned hospitalization compared with placebo. However, in patients with a score of 3-5, the escitalopram group had a 52% greater risk of the primary endpoint than controls.
“This suggests a heterogeneous pathophysiology and an impact of individual disease profiles on the effects of the study medication,” Dr. Angermann said.
MOOD-HF was funded chiefly by the German Ministry of Education and Research and the University of Wurzburg. Lundbeck provided the escitalopram gratis. Dr. Angermann reported serving as a consultant to ResMed, Novartis, and Vifor and receiving research grants from Lundbeck, Alere, and Boehringer Ingelheim.
AT ACC 15
Key clinical point: Prescribing escitalopram in patients with major depressive disorder and heart failure with reduced ejection fraction did not improve either disease any more than did placebo.
Major finding: The study was stopped for reasons of futility at a median of 18.6 months into a planned 24-month course of treatment.
Data source: MOOD-HF, a randomized, double-blind, placebo-controlled, prospective, 16-center study involving 372 patients with major depressive disorder and heart failure.
Disclosures: The trial was funded chiefly by the German Ministry of Education and Research and the University of Wurzburg. Lundbeck provided the escitalopram gratis. The presenter disclosed ties with Lundbeck, Alere, Boehringer Ingelheim, ResMed, Novartis, and Vifor.

Catheter treatment for PAH outperforms medication
SAN DIEGO – Percutaneous pulmonary artery denervation for the treatment of pulmonary arterial hypertension safely resulted in significantly greater improvement in functional capacity and hemodynamics compared with medication, in a controlled before-and-after study.
A particularly noteworthy secondary finding in the study was that rehospitalizations during the first 6 months after pulmonary artery denervation (PADN) occurred just one-third as frequently as in the 6-month preprocedural period on standard medications, Dr. Shao-Liang Chen said at the annual meeting of the American College of Cardiology.
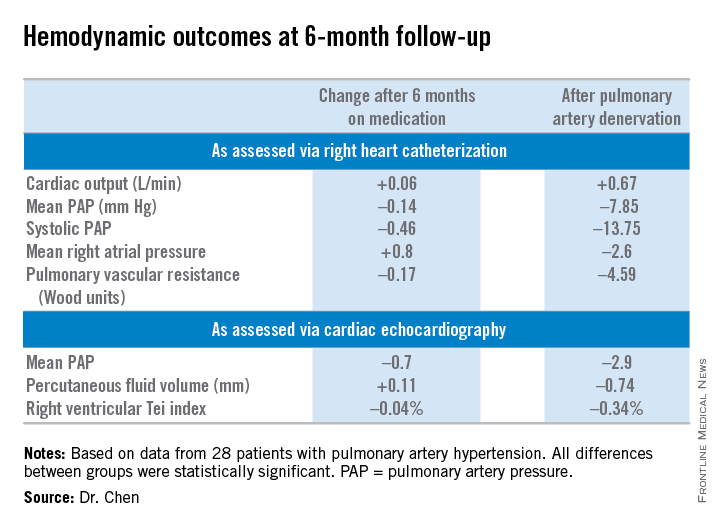
He and his coinvestigators, including Dr. Gregg W. Stone of Columbia University in New York, developed a percutaneous catheter-based method of destroying the pulmonary baroreceptor structure located at the bifurcation area of the middle pulmonary artery. Along the way, they redefined the understanding of the pathogenesis of pulmonary artery hypertension (PAH) by demonstrating that local sympathetic nerve activity plays a pivotal role in modulating the elevations of mean pulmonary artery pressure (mPAP) and pulmonary vascular resistance (PVR), which are the disease hallmarks.
Dr. Chen and coinvestigators previously reported the first-in-man study of PADN, which demonstrated safety and short-term efficacy (J. Am. Coll. Cardiol. 2013;62:1092-100). At ACC 15, Dr. Chen presented the findings of the new PADN-2 study, which expands upon the first study by including more patients and longer and more comprehensive follow-up.

The study comprised 28 patients with PAH, including 11 with idiopathic PAH and 8 with pulmonary hypertension caused by left ventricular disease. All of them underwent medication washout followed by right heart catheterization and echocardiography for baseline off-drug hemodynamic measurements as well as a 6-minute walk distance test of their functional capacity. Then they went back on medications for 6 months, after which they underwent repeat testing. Then their medications were discontinued and they underwent PADN. Six months after the procedure, still off medications, they were retested once again.
The primary study endpoint was change in 6-minute walk distance. After 6 months of medication it improved from 361 to 373 meters, a modest 3.9% gain over off-drug baseline. In contrast, 6-minute walk distance grew from 358 to 423 meters 6 months after PADN, a clinically important 23.9% improvement, reported Dr. Chen, a cardiologist at First Hospital of Nanjing (China) Medical University.
Multiple secondary hemodynamic endpoints also showed significantly greater improvement with PADN than medical therapy.
Twelve predefined clinical events – mostly involving worsening PAH – occurred during medical management, compared with three in the 6 months following PADN.
In addition, there were 12 hospitalizations during the 6 months on medical management compared with only 4 after the same patients underwent PADN. Health care costs averaged $35,000 per patient during the 6-month study period on medication compared with $6,000 per patient in the first 6 months after PADN.
There were no deaths, aneurysms, access site hematomas, or thrombotic events during either study period.
Further randomized, controlled trials are planned to explore the possibility that the benefits seen in the PADN-2 trial will result in reduced mortality in patients with PAH, according to Dr. Chen.
The PADN-2 trial was sponsored by Nanjing Medical University. Dr. Chen reported serving as a consultant to MicroPort.
SAN DIEGO – Percutaneous pulmonary artery denervation for the treatment of pulmonary arterial hypertension safely resulted in significantly greater improvement in functional capacity and hemodynamics compared with medication, in a controlled before-and-after study.
A particularly noteworthy secondary finding in the study was that rehospitalizations during the first 6 months after pulmonary artery denervation (PADN) occurred just one-third as frequently as in the 6-month preprocedural period on standard medications, Dr. Shao-Liang Chen said at the annual meeting of the American College of Cardiology.
He and his coinvestigators, including Dr. Gregg W. Stone of Columbia University in New York, developed a percutaneous catheter-based method of destroying the pulmonary baroreceptor structure located at the bifurcation area of the middle pulmonary artery. Along the way, they redefined the understanding of the pathogenesis of pulmonary artery hypertension (PAH) by demonstrating that local sympathetic nerve activity plays a pivotal role in modulating the elevations of mean pulmonary artery pressure (mPAP) and pulmonary vascular resistance (PVR), which are the disease hallmarks.
Dr. Chen and coinvestigators previously reported the first-in-man study of PADN, which demonstrated safety and short-term efficacy (J. Am. Coll. Cardiol. 2013;62:1092-100). At ACC 15, Dr. Chen presented the findings of the new PADN-2 study, which expands upon the first study by including more patients and longer and more comprehensive follow-up.
The study comprised 28 patients with PAH, including 11 with idiopathic PAH and 8 with pulmonary hypertension caused by left ventricular disease. All of them underwent medication washout followed by right heart catheterization and echocardiography for baseline off-drug hemodynamic measurements as well as a 6-minute walk distance test of their functional capacity. Then they went back on medications for 6 months, after which they underwent repeat testing. Then their medications were discontinued and they underwent PADN. Six months after the procedure, still off medications, they were retested once again.
The primary study endpoint was change in 6-minute walk distance. After 6 months of medication it improved from 361 to 373 meters, a modest 3.9% gain over off-drug baseline. In contrast, 6-minute walk distance grew from 358 to 423 meters 6 months after PADN, a clinically important 23.9% improvement, reported Dr. Chen, a cardiologist at First Hospital of Nanjing (China) Medical University.
Multiple secondary hemodynamic endpoints also showed significantly greater improvement with PADN than medical therapy.
Twelve predefined clinical events – mostly involving worsening PAH – occurred during medical management, compared with three in the 6 months following PADN.
In addition, there were 12 hospitalizations during the 6 months on medical management compared with only 4 after the same patients underwent PADN. Health care costs averaged $35,000 per patient during the 6-month study period on medication compared with $6,000 per patient in the first 6 months after PADN.
There were no deaths, aneurysms, access site hematomas, or thrombotic events during either study period.
Further randomized, controlled trials are planned to explore the possibility that the benefits seen in the PADN-2 trial will result in reduced mortality in patients with PAH, according to Dr. Chen.
The PADN-2 trial was sponsored by Nanjing Medical University. Dr. Chen reported serving as a consultant to MicroPort.
SAN DIEGO – Percutaneous pulmonary artery denervation for the treatment of pulmonary arterial hypertension safely resulted in significantly greater improvement in functional capacity and hemodynamics compared with medication, in a controlled before-and-after study.
A particularly noteworthy secondary finding in the study was that rehospitalizations during the first 6 months after pulmonary artery denervation (PADN) occurred just one-third as frequently as in the 6-month preprocedural period on standard medications, Dr. Shao-Liang Chen said at the annual meeting of the American College of Cardiology.
He and his coinvestigators, including Dr. Gregg W. Stone of Columbia University in New York, developed a percutaneous catheter-based method of destroying the pulmonary baroreceptor structure located at the bifurcation area of the middle pulmonary artery. Along the way, they redefined the understanding of the pathogenesis of pulmonary artery hypertension (PAH) by demonstrating that local sympathetic nerve activity plays a pivotal role in modulating the elevations of mean pulmonary artery pressure (mPAP) and pulmonary vascular resistance (PVR), which are the disease hallmarks.
Dr. Chen and coinvestigators previously reported the first-in-man study of PADN, which demonstrated safety and short-term efficacy (J. Am. Coll. Cardiol. 2013;62:1092-100). At ACC 15, Dr. Chen presented the findings of the new PADN-2 study, which expands upon the first study by including more patients and longer and more comprehensive follow-up.
The study comprised 28 patients with PAH, including 11 with idiopathic PAH and 8 with pulmonary hypertension caused by left ventricular disease. All of them underwent medication washout followed by right heart catheterization and echocardiography for baseline off-drug hemodynamic measurements as well as a 6-minute walk distance test of their functional capacity. Then they went back on medications for 6 months, after which they underwent repeat testing. Then their medications were discontinued and they underwent PADN. Six months after the procedure, still off medications, they were retested once again.
The primary study endpoint was change in 6-minute walk distance. After 6 months of medication it improved from 361 to 373 meters, a modest 3.9% gain over off-drug baseline. In contrast, 6-minute walk distance grew from 358 to 423 meters 6 months after PADN, a clinically important 23.9% improvement, reported Dr. Chen, a cardiologist at First Hospital of Nanjing (China) Medical University.
Multiple secondary hemodynamic endpoints also showed significantly greater improvement with PADN than medical therapy.
Twelve predefined clinical events – mostly involving worsening PAH – occurred during medical management, compared with three in the 6 months following PADN.
In addition, there were 12 hospitalizations during the 6 months on medical management compared with only 4 after the same patients underwent PADN. Health care costs averaged $35,000 per patient during the 6-month study period on medication compared with $6,000 per patient in the first 6 months after PADN.
There were no deaths, aneurysms, access site hematomas, or thrombotic events during either study period.
Further randomized, controlled trials are planned to explore the possibility that the benefits seen in the PADN-2 trial will result in reduced mortality in patients with PAH, according to Dr. Chen.
The PADN-2 trial was sponsored by Nanjing Medical University. Dr. Chen reported serving as a consultant to MicroPort.
AT ACC 15
Key clinical point: Pulmonary artery denervation for PAH showed promise in a small study.
Major finding: Six-minute walk distance improved by 3.9% after 6 months of medication, compared with 23.9% in testing done 6 months after percutaneous pulmonary artery denervation.
Data source: The PADN-2 trial, a prospective before-and-after study of 28 patients with pulmonary arterial hypertension.
Disclosures: Nanjing Medical University funded the study. The presenter reported serving as a consultant to MicroPort.