An 87-year-old woman was brought to the intensive care unit with worsening shortness of breath on exertion, fatigue, orthopnea, paroxysmal nocturnal dyspnea, lower extremity swelling, subjective fever, productive cough, and rhinorrhea over the last week. She reported no chest pain, lightheadedness, or palpitations. Her medical history included the following:
Cardiac arrest with recurrent ventricular tachycardia requiring an implanted cardioverter-defibrillator and amiodarone therapy
Hypothyroidism requiring levothyroxine
Asthma with a moderate obstructive pattern: forced expiratory volume in 1 second (FEV1) 60% of predicted, forced vital capacity (FVC) 2.06 L, FEV1/FVC 54%, diffusing capacity for carbon monoxide (DLCO) 72% of predicted with positive bronchodilator response
Long-standing essential thrombocythemia treated with hydroxyurea.
Before admission, she had been reliably taking guideline-directed heart failure therapy as well as amiodarone for her recurrent ventricular tachycardia. Her levothyroxine had recently been increased as well.
Physical examination. On admission, her blood pressure was 95/53 mm Hg, heart rate 73 beats per minute, temperature 36.7ºC (98.1ºF), and oxygen saturation 81% requiring supplemental oxygen 15 L/min by nonrebreather face mask. Physical examination revealed elevated jugular venous pressure, bibasilar crackles, lower extremity edema, and a grade 3 of 6 holosystolic murmur both at the left sternal border and at the apex radiating to the axilla. There was no evidence of wheezing or pulsus paradoxus.
Electrocardiography showed sinus rhythm and an old left bundle branch block.
Chest radiography showed cardiomegaly, bilateral pleural effusions, and pulmonary edema.
WHAT IS THE CAUSE OF HER SYMPTOMS?
1. Based on the available information, which of the following is the most likely cause of this patient’s clinical presentation?
Acute decompensated heart failure
Pulmonary embolism
Exacerbation of asthma
Exacerbation of chronic obstructive pulmonary disease (COPD)
Heart failure is a clinical diagnosis based on careful history-taking and physical examination. Major criteria include paroxysmal nocturnal dyspnea, orthopnea, elevated jugular venous pressure, pulmonary crackles, a third heart sound, cardiomegaly, pulmonary edema, and weight loss of more than 4.5 kg with diuretic therapy.1 N-terminal pro-B-type natriuretic peptide (NT-proBNP) is also an effective marker of acute decompensated heart failure in the proper clinical setting.2
Our patient’s elevated jugular venous pressure, bibasilar crackles, lower extremity edema, chest radiography findings consistent with pulmonary edema, markedly elevated NT-proBNP, history of orthopnea, paroxysmal nocturnal dyspnea, and dyspnea on exertion were most consistent with acute decompensated heart failure. Her cough and subjective fevers were thought to be due to an upper respiratory tract viral infection.
Pulmonary embolism causes pleuritic chest pain, dyspnea, and, occasionally, elevated troponin. The most common feature on electrocardiography is sinus tachycardia; nonspecific ST-segment and T-wave changes may also be seen.3
Although pulmonary embolism remained in the differential diagnosis, our patient’s lack of typical features of pulmonary embolism made this less likely.
Asthma is characterized by recurrent airflow obstruction and bronchial hyperresponsiveness.4 Asthma exacerbations present with wheezing, tachypnea, tachycardia, and pulsus paradoxus.5
Despite her previous asthma diagnosis, our patient’s lack of typical features of asthma exacerbation made this diagnosis unlikely.
COPD exacerbations present with increased dyspnea, cough, sputum production, wheezing, lung resonance to percussion, and distant heart sounds, and are characterized by airflow obstruction.6,7
Although our patient presented with cough and dyspnea, she had no history of COPD and her other signs and symptoms (elevated jugular venous pressure, elevated NT-proBNP, and peripheral edema) could not be explained by COPD exacerbation.
OUR PATIENT UNDERWENT FURTHER TESTING
Echocardiography revealed severe left ventricular enlargement, an ejection fraction of 20% (which was near her baseline value), diffuse regional wall-motion abnormalities, severe mitral regurgitation, and moderate tricuspid regurgitation consistent with an exacerbation of heart failure.
We considered the possibility that her heart failure symptoms might be due to precipitous up-titration of her levothyroxine dose, given her borderline-elevated free thyroxine (T4) and increase in cardiac index (currently 4.45 L/min/m2, previously 2.20 L/min/m2 by the left ventricular outflow tract velocity time integral method). However, given her reduced ejection fraction, this clinical presentation most likely represented an acute exacerbation of her chronic heart failure. Her subjective fevers were thought to be due to a viral infection of the upper respiratory tract. The macrocytic anemia and thrombocytopenia were thought to be a side effect of her long-standing treatment with hydroxyurea for essential thrombocythemia, although amiodarone has also been associated with cytopenia.8
Treatment was started with intravenous diuretics and positive pressure ventilation with oxygen supplementation. Her levothyroxine dose was reduced, and her hydroxyurea was stopped.
Figure 1. Chest computed tomography axial views demonstrated increased attenuation in the liver (A, arrow) and left pleural base (B, arrow). Evaluation of the right lung base revealed ground-glass opacities (C, arrow) and honeycombing (D, arrow). These findings were consistent with amiodarone pulmonary toxicity.
After aggressive diuresis, our patient returned to euvolemia. However, she had persistent fine crackles and hypoxia. She had no further fever, and her vital signs were otherwise stable. Her cytopenia improved with cessation of hydroxyurea. Chest computed tomography (CT) showed bibasilar ground-glass infiltrates with areas of interstitial fibrosis, high-attenuation pleural lesions, and increased liver attenuation (Figure 1).
Further testing for connective tissue disease and hypersensitivity pneumonitis was also done, and the results were negative. To exclude an atypical infection, bronchoalveolar lavage was performed; preliminary microbial testing was negative, and the white blood cell count in the lavage fluid was 90% macrophages (pigment-laden), 7% neutrophils, and 3% lymphocytes.
WHAT IS THE CAUSE OF HER PERSISTENT PULMONARY FINDINGS?
2. Given the CT findings and laboratory results, what is the most likely cause of our patient’s persistent crackles and hypoxia?
Heart failure with reduced ejection fraction
Bacterial pneumonia
Idiopathic pulmonary fibrosis
Amiodarone pulmonary toxicity
Heart failure with reduced ejection fraction can cause ground-glass opacities on CT due to increased pulmonary edema. Although our patient initially presented with acute decompensation of heart failure with reduced ejection fraction decompensation, she had returned to euvolemia after aggressive diuresis. Moreover, increased pleural and liver attenuation are not typically seen as a result of heart failure with reduced ejection fraction, making this diagnosis less likely.
Bacterial pneumonia typically presents with cough, fever, and purulent sputum production.9 Further evaluation usually reveals decreased breath sounds, dullness to percussion, and leukocytosis.10 Chest CT in bacterial pneumonia commonly shows a focal area of consolidation, which was not seen in our patient.11
Idiopathic pulmonary fibrosis usually presents with slowly progressive dyspnea and nonproductive cough.12 Physical examination usually reveals fine crackles and occasionally end-inspiratory “squeaks” if traction bronchiectasis is present.12 The diagnosis of idiopathic pulmonary fibrosis requires chest CT findings compatible with it (ie, basal fibrosis, reticular abnormalities, and honeycombing). However, it remains a diagnosis of exclusion and requires ruling out conditions known to cause pulmonary fibrosis such as hypersensitivity pneumonitis, connective tissue disease, and certain medications.12
Although idiopathic pulmonary fibrosis remained in the differential diagnosis, our patient remained on amiodarone, a known cause of pulmonary fibrosis.13 Similarly, the high-attenuation pleural lesions likely represented organizing pneumonia, which is more common in amiodarone pulmonary toxicity. And the ground-glass opacities made idiopathic pulmonary fibrosis unlikely, although they may be seen in an acute exacerbation of this disease.14 Thus, a diagnosis of idiopathic pulmonary fibrosis could not be made definitively.
Amiodarone pulmonary toxicity most commonly presents with acute to subacute cough and progressive dyspnea.13 Physical findings are similar to those in idiopathic pulmonary fibrosis and commonly include bibasilar crackles. Chest CT shows diffuse ground-glass opacities, reticular abnormalities, fibrosis, and increased attenuation of multiple organs, including the lungs, liver, and spleen.14 Bronchoalveolar lavage findings of lipid-laden macrophages suggest but do not definitively diagnose amiodarone pulmonary toxicity.15 And patients with acute amiodarone pulmonary toxicity may present with pigment-laden macrophages on bronchoalveolar lavage, as in our patient.16
Exclusion of hypersensitivity pneumonitis, connective tissue disease, and infection made our patient’s progressive dyspnea and chest CT findings of ground-glass opacities, fibrosis, and increased pulmonary and liver attenuation most consistent with amiodarone pulmonary toxicity.
Amiodarone was therefore discontinued. However, the test result of her lavage fluid for influenza A by polymerase chain reaction came back positive a few hours later.
WHAT IS THE NEXT STEP?
3. Given the positive influenza A polymerase chain reaction test, which of the following is the best next step in this patient’s management?
Surgical lung biopsy
Stop amiodarone and start supportive influenza management
Stop amiodarone and start dronedarone
Start an intravenous corticosteroid
Surgical lung biopsy is typically not required for diagnosis in patients with suspected amiodarone pulmonary toxicity. In addition, acute respiratory distress syndrome has been documented in patients who have undergone surgical biopsy for suspected amiodarone pulmonary toxicity.17
Thus, surgical biopsy is typically only done in cases of persistent symptoms despite withdrawal of amiodarone and initiation of steroid therapy.
Stopping amiodarone and starting supportive influenza management are the best next steps, as our patient’s fevers, cough, dyspnea, and laboratory test results were consistent with influenza.18 Moreover, CT findings of ground-glass opacities and reticular abnormalities can be seen in influenza.19
However, concomitant amiodarone pulmonary toxicity could not be ruled out, as CT showed increased lung and liver attenuation and fibrosis that could not be explained by influenza. And the elevation in aminotransferase levels more than 2 times the upper limit of normal and CT findings of increased liver attenuation suggested amiodarone hepatotoxicity. However, definitive diagnosis would require exclusion of other causes such as congestive hepatopathy, in some cases with liver biopsy.13
Our patient’s persistent hypoxia was thought to be due in part to influenza, and thus the best next step in management was to stop amiodarone and provide supportive care for influenza.
Dronedarone is an antiarrhythmic drug structurally and functionally similar to amiodarone. There are far fewer reports of pulmonary toxicity with dronedarone than with amiodarone.20 However, lack of data on dronedarone in amiodarone pulmonary toxicity, increased rates of hospitalization and death associated with dronedarone in patients like ours with advanced heart failure, and our patient’s previously implanted cardioverter-defibrillator for recurrent ventricular tachycardia all made dronedarone an undesirable alternative to amiodarone.21
Corticosteroids are useful in the treatment of amiodarone pulmonary toxicity when hypoxia and dyspnea are present at diagnosis.13 Our patient’s hypoxia and dyspnea were thought to be due in part to her acute influenza infection, and therefore corticosteroids were not used at the outset.
However, concomitant amiodarone pulmonary toxicity could not be excluded, and the elevation in aminotransferases of more than 2 times the upper limit of normal and CT findings of increased liver attenuation suggested amiodarone hepatotoxicity—though congestive hepatopathy remained in the differential diagnosis. Therefore, supportive therapy for influenza was instituted, and amiodarone was withheld. Her condition subsequently improved, and she was discharged.
FOLLOW-UP 1 MONTH LATER
At a follow-up visit 1 month later, our patient continued to have dyspnea and hypoxia. She did not have signs or symptoms consistent with decompensated heart failure.
Pulmonary function testing revealed the following values:
FEV1 0.69 L (56% of predicted)
FVC 1.08 L (64% of predicted)
Figure 2. In A, repeat chest computed tomography demonstrated increased liver attenuation (arrow); in B, it showed persistent ground-glass opacities (white arrow), increased pulmonary attenuation (black arrowhead), and worsening pleural effusions (black arrows). These findings supported the diagnosis of amiodarone pulmonary toxicity.
FEV1/FVC ratio 64%
DLCO 2.20 mL/min/mm Hg (12% of predicted).
Aminotransferase levels had also normalized. Repeat chest CT showed persistent bibasilar interstitial fibrotic changes, enlarging bilateral pleural effusions, and persistent peripheral ground-glass opacities (Figure 2).
WHAT FURTHER TREATMENT IS APPROPRIATE?
4. Given the chest CT findings, which of the following is the most appropriate treatment strategy for this patient?
No further management, continue to hold amiodarone
Corticosteroids
Repeat bronchoalveolar lavage
Intravenous antibiotics
No further management of amiodarone pulmonary toxicity would be appropriate if our patient did not have a high burden of symptoms. However, when patients with amiodarone pulmonary toxicity present with hypoxia and dyspnea, corticosteroids should be started.13 Our patient remained symptomatic after discontinuation of amiodarone and resolution of her influenza infection, and CT showed persistent signs of amiodarone pulmonary toxicity, which required further management.
Corticosteroids are useful in treating amiodarone pulmonary toxicity when hypoxia and dyspnea are present at diagnosis. Our patient’s persistent ground-glass opacities, fibrotic changes, and increased attenuation in multiple organs on CT, coupled with a confirmed reduction in FVC of greater than 15% and reduction in DLCO of greater than 20% after recovery from influenza, were most consistent with persistent amiodarone pulmonary toxicity.13
Although our patient’s amiodarone had been discontinued, the long half-life of the drug (45 days) allowed pulmonary toxicity to progress even after the drug was discontinued.22 Because our patient continued to have hypoxia and dyspnea on exertion, the most appropriate next step in management (in addition to managing her pleural effusions) was to start corticosteroids.
For amiodarone pulmonary toxicity, prednisone is typically started at 40 to 60 mg daily and can result in rapid improvement in symptoms.13 Tapering should be slow and may take several months.
Bronchoalveolar lavage is typically used in suspected cases of amiodarone pulmonary toxicity only to rule out an alternative diagnosis such as infection. Lipid-laden macrophages may be seen in the fluid. However, lipid-laden macrophages are not diagnostic of amiodarone pulmonary toxicity, as this finding may also be seen in patients taking amiodarone who do not develop pulmonary toxicity.15 Other findings on bronchoalveolar lavage in amiodarone pulmonary toxicity are nonspecific and are not diagnostically useful.13
Intravenous antibiotics are appropriate if bacterial pneumonia is suspected. However, bacterial pneumonia typically presents with cough, fever, purulent sputum production, and focal consolidation on chest imaging.9 Our patient’s CT findings of persistent peripheral ground-glass opacities and lack of cough, fever, or purulent sputum production were not consistent with bacterial pneumonia, and therefore intravenous antibiotics were not indicated.
CASE CONCLUSION
Given our patient’s persistent dyspnea, hypoxia, and chest CT findings consistent with amiodarone pulmonary toxicity, it was recommended that she start corticosteroids. However, before starting therapy, she suffered a femoral fracture that required surgical intervention. Around the time of the procedure, she had an ST-segment elevation myocardial infarction requiring vasopressor support and mechanical ventilation. At that time, the patient and family decided to pursue comfort measures, and she died peacefully.
MORE ABOUT AMIODARONE PULMONARY TOXICITY
Pulmonary toxicity is a well-described consequence of amiodarone therapy.23 Amiodarone carries a 2% risk of pulmonary toxicity.24 Although higher doses are more likely to cause pulmonary toxicity, lower doses also have been implicated.22,24 Preexisting pulmonary disease may predispose patients taking amiodarone to pulmonary toxicity; however, this is not uniformly seen.25
Mortality rates as high as 10% from amiodarone pulmonary toxicity have been reported. Thus, diligent surveillance for pulmonary toxicity with pulmonary function tests in patients taking amiodarone is mandatory. In particular, a reduction in FVC of greater than 15% or in DLCO of greater than 20% from baseline may be seen in amiodarone pulmonary toxicity.26
Amiodarone pulmonary toxicity can present at any time after the start of therapy, but it occurs most often after 6 to 12 months.13 Patients typically experience insidious dyspnea; however, presentation with acute to subacute cough and progressive dyspnea can occur, especially with high concentrations of supplemental oxygen with or without mechanical ventilation.12,27 Findings on physical examination include bibasilar crackles. CT chest findings include diffuse ground-glass opacities, reticular abnormalities, fibrosis, and increased attenuation in multiple organs, including the lung, liver, and spleen.14
The diagnosis of amiodarone pulmonary toxicity requires ruling out hypersensitivity pneumonitis, connective tissue disease, heart failure, and infection. Surgical biopsy and bronchoalveolar lavage are not commonly used to establish the diagnosis of amiodarone pulmonary toxicity, as surgical biopsy increases the risk of acute respiratory distress syndrome, and the results of bronchoalveolar lavage are usually nonspecific.13,15
Initial treatment involves discontinuing the amiodarone once the diagnosis is suspected. If patients have worsening hypoxia or dyspnea at the time of diagnosis, corticosteroids can be used. Prednisone is typically started at 40 to 60 mg daily and can result in rapid improvement in symptoms.13 Tapering of corticosteroids should occur slowly and may take several months.
References
McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Engl J Med 1971; 285(26):1441–1446. doi:10.1056/NEJM197112232852601
Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100(3):598–603. pmid:1909617
Baggish AL, Siebert U, Lainchbury JG, et al. A validated clinical and biochemical score for the diagnosis of acute heart failure: the ProBNP investigation of dyspnea in the emergency department (PRIDE) acute heart failure score. Am Heart J 2006; 151(1):48–54. doi:10.1016/j.ahj.2005.02.031
National Heart, Lung, and Blood Institute. National Asthma Education and Prevention Program. Expert panel report 3: Guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/sites/default/files/media/docs/asthgdln_1.pdf. Accessed August 3, 2018.
Brenner BE, Abraham E, Simon RR. Position and diaphoresis in acute asthma. Am J Med 1983; 74(6):1005–1009. pmid:6407304
Badgett RG, Tanaka DJ, Hunt DK, et al. Can moderate chronic obstructive pulmonary disease be diagnosed by historical and physical findings alone? Am J Med 1993; 94(2):188–196. pmid:8430714
Erie AJ, McClure RF, Wolanskyj AP. Amiodarone-induced bone marrow granulomas: an unusual cause of reversible pancytopenia. Hematol Rep 2010; 2(1):e6. doi:10.4081/hr.2010.e6
Marrie TJ. Community-acquired pneumonia. Clin Infect Dis 1994; 18(4):501–513. pmid:8038304
Metlay JP, Kapoor WN, Fine MJ. Does this patient have community-acquired pneumonia? Diagnosing pneumonia by history and physical examination. JAMA 1997; 278(17):1440–1445. pmid:9356004
Walker CM, Abbott GF, Greene RE, Shepard JA, Vummidi D, Digumarthy SR. Imaging pulmonary infection: classic signs and patterns. AJR Am J Roentgenol 2014; 202(3):479–492. doi:10.2214/AJR.13.11463
Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
Goldschlager N, Epstein AE, Naccarelli GV, et al; Practice Guidelines Sub-committee, North American Society of Pacing and Electrophysiology (HRS). A practical guide for clinicians who treat patients with amiodarone: 2007. Heart Rhythm 2007; 4(9):1250–1259. doi:10.1016/j.hrthm.2007.07.020
Martin WJ 2nd, Rosenow EC 3rd. Amiodarone pulmonary toxicity: recognition and pathogenesis (Part I). Chest 1988; 93(5):1067–1075. pmid:3282816
Iskandar SB, Abi-Saleh B, Keith RL, Byrd RP Jr, Roy TM. Amiodarone-induced alveolar hemorrhage. South Med J 2006; 99(4):383–387.
Van Mieghem W, Coolen L, Malysse I, Lacquet LM, Deneffe GJ, Demedts MG. Amiodarone and the development of ARDS after lung surgery. Chest 1994; 105(6):1642–1645. pmid:8205854
Nicholson KG. Clinical features of influenza. Semin Respir Infect 1992; 7(1):26–37. pmid:1609165
Muller NL, Franquet T, Lee KS, Silva CIS. Viruses, mycoplasma, and chlamydia. In: Imaging of Pulmonary Infections. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:94–114.
Stack S, Nguyen DV, Casto A, Ahuja N. Diffuse alveolar damage in a patient receiving dronedarone. Chest 2015; 147(4):e131–e133. doi:10.1378/chest.14-1849
De Ferrari GM, Dusi V. Drug safety evaluation of dronedarone in atrial fibrillation. Expert Opin Drug Saf 2012; 11(6):1023–1045. doi:10.1517/14740338.2012.722994
Okayasu K, Takeda Y, Kojima J, et al. Amiodarone pulmonary toxicity: a patient with three recurrences of pulmonary toxicity and consideration of the probable risk for relapse. Intern Med 2006; 45(22):1303–1307. pmid:17170505
Vorperian VR, Havighurst TC, Miller S, January CT. Adverse effects of low dose amiodarone: a meta-analysis. J Am Coll Cardiol 1997; 30(3):791–798. pmid:9283542
Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomised trials. Lancet 1997; 350(9089):1417–1424. pmid:9371164
Olshansky B, Sami M, Rubin A, et al; NHLBI AFFIRM Investigators. Use of amiodarone for atrial fibrillation in patients with preexisting pulmonary disease in the AFFIRM study. Am J Cardiol 2005; 95(3):404–405. doi:10.1016/j.amjcard.2004.09.044
Camus P. Interstitial lung disease from drugs, biologics, and radiation. In: Schwartz MI, King TE Jr, eds. Interstitial Lung Disease. 5th ed. Shelton, CT: People’s Medical Publishing House; 2011:637–644.
Wolkove N, Baltzan M. Amiodarone pulmonary toxicity. Can Respir J 2009; 16(2):43–48. doi:10.1155/2009/282540
An 87-year-old woman was brought to the intensive care unit with worsening shortness of breath on exertion, fatigue, orthopnea, paroxysmal nocturnal dyspnea, lower extremity swelling, subjective fever, productive cough, and rhinorrhea over the last week. She reported no chest pain, lightheadedness, or palpitations. Her medical history included the following:
Cardiac arrest with recurrent ventricular tachycardia requiring an implanted cardioverter-defibrillator and amiodarone therapy
Hypothyroidism requiring levothyroxine
Asthma with a moderate obstructive pattern: forced expiratory volume in 1 second (FEV1) 60% of predicted, forced vital capacity (FVC) 2.06 L, FEV1/FVC 54%, diffusing capacity for carbon monoxide (DLCO) 72% of predicted with positive bronchodilator response
Long-standing essential thrombocythemia treated with hydroxyurea.
Before admission, she had been reliably taking guideline-directed heart failure therapy as well as amiodarone for her recurrent ventricular tachycardia. Her levothyroxine had recently been increased as well.
Physical examination. On admission, her blood pressure was 95/53 mm Hg, heart rate 73 beats per minute, temperature 36.7ºC (98.1ºF), and oxygen saturation 81% requiring supplemental oxygen 15 L/min by nonrebreather face mask. Physical examination revealed elevated jugular venous pressure, bibasilar crackles, lower extremity edema, and a grade 3 of 6 holosystolic murmur both at the left sternal border and at the apex radiating to the axilla. There was no evidence of wheezing or pulsus paradoxus.
Electrocardiography showed sinus rhythm and an old left bundle branch block.
Chest radiography showed cardiomegaly, bilateral pleural effusions, and pulmonary edema.
WHAT IS THE CAUSE OF HER SYMPTOMS?
1. Based on the available information, which of the following is the most likely cause of this patient’s clinical presentation?
Acute decompensated heart failure
Pulmonary embolism
Exacerbation of asthma
Exacerbation of chronic obstructive pulmonary disease (COPD)
Heart failure is a clinical diagnosis based on careful history-taking and physical examination. Major criteria include paroxysmal nocturnal dyspnea, orthopnea, elevated jugular venous pressure, pulmonary crackles, a third heart sound, cardiomegaly, pulmonary edema, and weight loss of more than 4.5 kg with diuretic therapy.1 N-terminal pro-B-type natriuretic peptide (NT-proBNP) is also an effective marker of acute decompensated heart failure in the proper clinical setting.2
Our patient’s elevated jugular venous pressure, bibasilar crackles, lower extremity edema, chest radiography findings consistent with pulmonary edema, markedly elevated NT-proBNP, history of orthopnea, paroxysmal nocturnal dyspnea, and dyspnea on exertion were most consistent with acute decompensated heart failure. Her cough and subjective fevers were thought to be due to an upper respiratory tract viral infection.
Pulmonary embolism causes pleuritic chest pain, dyspnea, and, occasionally, elevated troponin. The most common feature on electrocardiography is sinus tachycardia; nonspecific ST-segment and T-wave changes may also be seen.3
Although pulmonary embolism remained in the differential diagnosis, our patient’s lack of typical features of pulmonary embolism made this less likely.
Asthma is characterized by recurrent airflow obstruction and bronchial hyperresponsiveness.4 Asthma exacerbations present with wheezing, tachypnea, tachycardia, and pulsus paradoxus.5
Despite her previous asthma diagnosis, our patient’s lack of typical features of asthma exacerbation made this diagnosis unlikely.
COPD exacerbations present with increased dyspnea, cough, sputum production, wheezing, lung resonance to percussion, and distant heart sounds, and are characterized by airflow obstruction.6,7
Although our patient presented with cough and dyspnea, she had no history of COPD and her other signs and symptoms (elevated jugular venous pressure, elevated NT-proBNP, and peripheral edema) could not be explained by COPD exacerbation.
OUR PATIENT UNDERWENT FURTHER TESTING
Echocardiography revealed severe left ventricular enlargement, an ejection fraction of 20% (which was near her baseline value), diffuse regional wall-motion abnormalities, severe mitral regurgitation, and moderate tricuspid regurgitation consistent with an exacerbation of heart failure.
We considered the possibility that her heart failure symptoms might be due to precipitous up-titration of her levothyroxine dose, given her borderline-elevated free thyroxine (T4) and increase in cardiac index (currently 4.45 L/min/m2, previously 2.20 L/min/m2 by the left ventricular outflow tract velocity time integral method). However, given her reduced ejection fraction, this clinical presentation most likely represented an acute exacerbation of her chronic heart failure. Her subjective fevers were thought to be due to a viral infection of the upper respiratory tract. The macrocytic anemia and thrombocytopenia were thought to be a side effect of her long-standing treatment with hydroxyurea for essential thrombocythemia, although amiodarone has also been associated with cytopenia.8
Treatment was started with intravenous diuretics and positive pressure ventilation with oxygen supplementation. Her levothyroxine dose was reduced, and her hydroxyurea was stopped.
Figure 1. Chest computed tomography axial views demonstrated increased attenuation in the liver (A, arrow) and left pleural base (B, arrow). Evaluation of the right lung base revealed ground-glass opacities (C, arrow) and honeycombing (D, arrow). These findings were consistent with amiodarone pulmonary toxicity.
After aggressive diuresis, our patient returned to euvolemia. However, she had persistent fine crackles and hypoxia. She had no further fever, and her vital signs were otherwise stable. Her cytopenia improved with cessation of hydroxyurea. Chest computed tomography (CT) showed bibasilar ground-glass infiltrates with areas of interstitial fibrosis, high-attenuation pleural lesions, and increased liver attenuation (Figure 1).
Further testing for connective tissue disease and hypersensitivity pneumonitis was also done, and the results were negative. To exclude an atypical infection, bronchoalveolar lavage was performed; preliminary microbial testing was negative, and the white blood cell count in the lavage fluid was 90% macrophages (pigment-laden), 7% neutrophils, and 3% lymphocytes.
WHAT IS THE CAUSE OF HER PERSISTENT PULMONARY FINDINGS?
2. Given the CT findings and laboratory results, what is the most likely cause of our patient’s persistent crackles and hypoxia?
Heart failure with reduced ejection fraction
Bacterial pneumonia
Idiopathic pulmonary fibrosis
Amiodarone pulmonary toxicity
Heart failure with reduced ejection fraction can cause ground-glass opacities on CT due to increased pulmonary edema. Although our patient initially presented with acute decompensation of heart failure with reduced ejection fraction decompensation, she had returned to euvolemia after aggressive diuresis. Moreover, increased pleural and liver attenuation are not typically seen as a result of heart failure with reduced ejection fraction, making this diagnosis less likely.
Bacterial pneumonia typically presents with cough, fever, and purulent sputum production.9 Further evaluation usually reveals decreased breath sounds, dullness to percussion, and leukocytosis.10 Chest CT in bacterial pneumonia commonly shows a focal area of consolidation, which was not seen in our patient.11
Idiopathic pulmonary fibrosis usually presents with slowly progressive dyspnea and nonproductive cough.12 Physical examination usually reveals fine crackles and occasionally end-inspiratory “squeaks” if traction bronchiectasis is present.12 The diagnosis of idiopathic pulmonary fibrosis requires chest CT findings compatible with it (ie, basal fibrosis, reticular abnormalities, and honeycombing). However, it remains a diagnosis of exclusion and requires ruling out conditions known to cause pulmonary fibrosis such as hypersensitivity pneumonitis, connective tissue disease, and certain medications.12
Although idiopathic pulmonary fibrosis remained in the differential diagnosis, our patient remained on amiodarone, a known cause of pulmonary fibrosis.13 Similarly, the high-attenuation pleural lesions likely represented organizing pneumonia, which is more common in amiodarone pulmonary toxicity. And the ground-glass opacities made idiopathic pulmonary fibrosis unlikely, although they may be seen in an acute exacerbation of this disease.14 Thus, a diagnosis of idiopathic pulmonary fibrosis could not be made definitively.
Amiodarone pulmonary toxicity most commonly presents with acute to subacute cough and progressive dyspnea.13 Physical findings are similar to those in idiopathic pulmonary fibrosis and commonly include bibasilar crackles. Chest CT shows diffuse ground-glass opacities, reticular abnormalities, fibrosis, and increased attenuation of multiple organs, including the lungs, liver, and spleen.14 Bronchoalveolar lavage findings of lipid-laden macrophages suggest but do not definitively diagnose amiodarone pulmonary toxicity.15 And patients with acute amiodarone pulmonary toxicity may present with pigment-laden macrophages on bronchoalveolar lavage, as in our patient.16
Exclusion of hypersensitivity pneumonitis, connective tissue disease, and infection made our patient’s progressive dyspnea and chest CT findings of ground-glass opacities, fibrosis, and increased pulmonary and liver attenuation most consistent with amiodarone pulmonary toxicity.
Amiodarone was therefore discontinued. However, the test result of her lavage fluid for influenza A by polymerase chain reaction came back positive a few hours later.
WHAT IS THE NEXT STEP?
3. Given the positive influenza A polymerase chain reaction test, which of the following is the best next step in this patient’s management?
Surgical lung biopsy
Stop amiodarone and start supportive influenza management
Stop amiodarone and start dronedarone
Start an intravenous corticosteroid
Surgical lung biopsy is typically not required for diagnosis in patients with suspected amiodarone pulmonary toxicity. In addition, acute respiratory distress syndrome has been documented in patients who have undergone surgical biopsy for suspected amiodarone pulmonary toxicity.17
Thus, surgical biopsy is typically only done in cases of persistent symptoms despite withdrawal of amiodarone and initiation of steroid therapy.
Stopping amiodarone and starting supportive influenza management are the best next steps, as our patient’s fevers, cough, dyspnea, and laboratory test results were consistent with influenza.18 Moreover, CT findings of ground-glass opacities and reticular abnormalities can be seen in influenza.19
However, concomitant amiodarone pulmonary toxicity could not be ruled out, as CT showed increased lung and liver attenuation and fibrosis that could not be explained by influenza. And the elevation in aminotransferase levels more than 2 times the upper limit of normal and CT findings of increased liver attenuation suggested amiodarone hepatotoxicity. However, definitive diagnosis would require exclusion of other causes such as congestive hepatopathy, in some cases with liver biopsy.13
Our patient’s persistent hypoxia was thought to be due in part to influenza, and thus the best next step in management was to stop amiodarone and provide supportive care for influenza.
Dronedarone is an antiarrhythmic drug structurally and functionally similar to amiodarone. There are far fewer reports of pulmonary toxicity with dronedarone than with amiodarone.20 However, lack of data on dronedarone in amiodarone pulmonary toxicity, increased rates of hospitalization and death associated with dronedarone in patients like ours with advanced heart failure, and our patient’s previously implanted cardioverter-defibrillator for recurrent ventricular tachycardia all made dronedarone an undesirable alternative to amiodarone.21
Corticosteroids are useful in the treatment of amiodarone pulmonary toxicity when hypoxia and dyspnea are present at diagnosis.13 Our patient’s hypoxia and dyspnea were thought to be due in part to her acute influenza infection, and therefore corticosteroids were not used at the outset.
However, concomitant amiodarone pulmonary toxicity could not be excluded, and the elevation in aminotransferases of more than 2 times the upper limit of normal and CT findings of increased liver attenuation suggested amiodarone hepatotoxicity—though congestive hepatopathy remained in the differential diagnosis. Therefore, supportive therapy for influenza was instituted, and amiodarone was withheld. Her condition subsequently improved, and she was discharged.
FOLLOW-UP 1 MONTH LATER
At a follow-up visit 1 month later, our patient continued to have dyspnea and hypoxia. She did not have signs or symptoms consistent with decompensated heart failure.
Pulmonary function testing revealed the following values:
FEV1 0.69 L (56% of predicted)
FVC 1.08 L (64% of predicted)
Figure 2. In A, repeat chest computed tomography demonstrated increased liver attenuation (arrow); in B, it showed persistent ground-glass opacities (white arrow), increased pulmonary attenuation (black arrowhead), and worsening pleural effusions (black arrows). These findings supported the diagnosis of amiodarone pulmonary toxicity.
FEV1/FVC ratio 64%
DLCO 2.20 mL/min/mm Hg (12% of predicted).
Aminotransferase levels had also normalized. Repeat chest CT showed persistent bibasilar interstitial fibrotic changes, enlarging bilateral pleural effusions, and persistent peripheral ground-glass opacities (Figure 2).
WHAT FURTHER TREATMENT IS APPROPRIATE?
4. Given the chest CT findings, which of the following is the most appropriate treatment strategy for this patient?
No further management, continue to hold amiodarone
Corticosteroids
Repeat bronchoalveolar lavage
Intravenous antibiotics
No further management of amiodarone pulmonary toxicity would be appropriate if our patient did not have a high burden of symptoms. However, when patients with amiodarone pulmonary toxicity present with hypoxia and dyspnea, corticosteroids should be started.13 Our patient remained symptomatic after discontinuation of amiodarone and resolution of her influenza infection, and CT showed persistent signs of amiodarone pulmonary toxicity, which required further management.
Corticosteroids are useful in treating amiodarone pulmonary toxicity when hypoxia and dyspnea are present at diagnosis. Our patient’s persistent ground-glass opacities, fibrotic changes, and increased attenuation in multiple organs on CT, coupled with a confirmed reduction in FVC of greater than 15% and reduction in DLCO of greater than 20% after recovery from influenza, were most consistent with persistent amiodarone pulmonary toxicity.13
Although our patient’s amiodarone had been discontinued, the long half-life of the drug (45 days) allowed pulmonary toxicity to progress even after the drug was discontinued.22 Because our patient continued to have hypoxia and dyspnea on exertion, the most appropriate next step in management (in addition to managing her pleural effusions) was to start corticosteroids.
For amiodarone pulmonary toxicity, prednisone is typically started at 40 to 60 mg daily and can result in rapid improvement in symptoms.13 Tapering should be slow and may take several months.
Bronchoalveolar lavage is typically used in suspected cases of amiodarone pulmonary toxicity only to rule out an alternative diagnosis such as infection. Lipid-laden macrophages may be seen in the fluid. However, lipid-laden macrophages are not diagnostic of amiodarone pulmonary toxicity, as this finding may also be seen in patients taking amiodarone who do not develop pulmonary toxicity.15 Other findings on bronchoalveolar lavage in amiodarone pulmonary toxicity are nonspecific and are not diagnostically useful.13
Intravenous antibiotics are appropriate if bacterial pneumonia is suspected. However, bacterial pneumonia typically presents with cough, fever, purulent sputum production, and focal consolidation on chest imaging.9 Our patient’s CT findings of persistent peripheral ground-glass opacities and lack of cough, fever, or purulent sputum production were not consistent with bacterial pneumonia, and therefore intravenous antibiotics were not indicated.
CASE CONCLUSION
Given our patient’s persistent dyspnea, hypoxia, and chest CT findings consistent with amiodarone pulmonary toxicity, it was recommended that she start corticosteroids. However, before starting therapy, she suffered a femoral fracture that required surgical intervention. Around the time of the procedure, she had an ST-segment elevation myocardial infarction requiring vasopressor support and mechanical ventilation. At that time, the patient and family decided to pursue comfort measures, and she died peacefully.
MORE ABOUT AMIODARONE PULMONARY TOXICITY
Pulmonary toxicity is a well-described consequence of amiodarone therapy.23 Amiodarone carries a 2% risk of pulmonary toxicity.24 Although higher doses are more likely to cause pulmonary toxicity, lower doses also have been implicated.22,24 Preexisting pulmonary disease may predispose patients taking amiodarone to pulmonary toxicity; however, this is not uniformly seen.25
Mortality rates as high as 10% from amiodarone pulmonary toxicity have been reported. Thus, diligent surveillance for pulmonary toxicity with pulmonary function tests in patients taking amiodarone is mandatory. In particular, a reduction in FVC of greater than 15% or in DLCO of greater than 20% from baseline may be seen in amiodarone pulmonary toxicity.26
Amiodarone pulmonary toxicity can present at any time after the start of therapy, but it occurs most often after 6 to 12 months.13 Patients typically experience insidious dyspnea; however, presentation with acute to subacute cough and progressive dyspnea can occur, especially with high concentrations of supplemental oxygen with or without mechanical ventilation.12,27 Findings on physical examination include bibasilar crackles. CT chest findings include diffuse ground-glass opacities, reticular abnormalities, fibrosis, and increased attenuation in multiple organs, including the lung, liver, and spleen.14
The diagnosis of amiodarone pulmonary toxicity requires ruling out hypersensitivity pneumonitis, connective tissue disease, heart failure, and infection. Surgical biopsy and bronchoalveolar lavage are not commonly used to establish the diagnosis of amiodarone pulmonary toxicity, as surgical biopsy increases the risk of acute respiratory distress syndrome, and the results of bronchoalveolar lavage are usually nonspecific.13,15
Initial treatment involves discontinuing the amiodarone once the diagnosis is suspected. If patients have worsening hypoxia or dyspnea at the time of diagnosis, corticosteroids can be used. Prednisone is typically started at 40 to 60 mg daily and can result in rapid improvement in symptoms.13 Tapering of corticosteroids should occur slowly and may take several months.
An 87-year-old woman was brought to the intensive care unit with worsening shortness of breath on exertion, fatigue, orthopnea, paroxysmal nocturnal dyspnea, lower extremity swelling, subjective fever, productive cough, and rhinorrhea over the last week. She reported no chest pain, lightheadedness, or palpitations. Her medical history included the following:
Cardiac arrest with recurrent ventricular tachycardia requiring an implanted cardioverter-defibrillator and amiodarone therapy
Hypothyroidism requiring levothyroxine
Asthma with a moderate obstructive pattern: forced expiratory volume in 1 second (FEV1) 60% of predicted, forced vital capacity (FVC) 2.06 L, FEV1/FVC 54%, diffusing capacity for carbon monoxide (DLCO) 72% of predicted with positive bronchodilator response
Long-standing essential thrombocythemia treated with hydroxyurea.
Before admission, she had been reliably taking guideline-directed heart failure therapy as well as amiodarone for her recurrent ventricular tachycardia. Her levothyroxine had recently been increased as well.
Physical examination. On admission, her blood pressure was 95/53 mm Hg, heart rate 73 beats per minute, temperature 36.7ºC (98.1ºF), and oxygen saturation 81% requiring supplemental oxygen 15 L/min by nonrebreather face mask. Physical examination revealed elevated jugular venous pressure, bibasilar crackles, lower extremity edema, and a grade 3 of 6 holosystolic murmur both at the left sternal border and at the apex radiating to the axilla. There was no evidence of wheezing or pulsus paradoxus.
Electrocardiography showed sinus rhythm and an old left bundle branch block.
Chest radiography showed cardiomegaly, bilateral pleural effusions, and pulmonary edema.
WHAT IS THE CAUSE OF HER SYMPTOMS?
1. Based on the available information, which of the following is the most likely cause of this patient’s clinical presentation?
Acute decompensated heart failure
Pulmonary embolism
Exacerbation of asthma
Exacerbation of chronic obstructive pulmonary disease (COPD)
Heart failure is a clinical diagnosis based on careful history-taking and physical examination. Major criteria include paroxysmal nocturnal dyspnea, orthopnea, elevated jugular venous pressure, pulmonary crackles, a third heart sound, cardiomegaly, pulmonary edema, and weight loss of more than 4.5 kg with diuretic therapy.1 N-terminal pro-B-type natriuretic peptide (NT-proBNP) is also an effective marker of acute decompensated heart failure in the proper clinical setting.2
Our patient’s elevated jugular venous pressure, bibasilar crackles, lower extremity edema, chest radiography findings consistent with pulmonary edema, markedly elevated NT-proBNP, history of orthopnea, paroxysmal nocturnal dyspnea, and dyspnea on exertion were most consistent with acute decompensated heart failure. Her cough and subjective fevers were thought to be due to an upper respiratory tract viral infection.
Pulmonary embolism causes pleuritic chest pain, dyspnea, and, occasionally, elevated troponin. The most common feature on electrocardiography is sinus tachycardia; nonspecific ST-segment and T-wave changes may also be seen.3
Although pulmonary embolism remained in the differential diagnosis, our patient’s lack of typical features of pulmonary embolism made this less likely.
Asthma is characterized by recurrent airflow obstruction and bronchial hyperresponsiveness.4 Asthma exacerbations present with wheezing, tachypnea, tachycardia, and pulsus paradoxus.5
Despite her previous asthma diagnosis, our patient’s lack of typical features of asthma exacerbation made this diagnosis unlikely.
COPD exacerbations present with increased dyspnea, cough, sputum production, wheezing, lung resonance to percussion, and distant heart sounds, and are characterized by airflow obstruction.6,7
Although our patient presented with cough and dyspnea, she had no history of COPD and her other signs and symptoms (elevated jugular venous pressure, elevated NT-proBNP, and peripheral edema) could not be explained by COPD exacerbation.
OUR PATIENT UNDERWENT FURTHER TESTING
Echocardiography revealed severe left ventricular enlargement, an ejection fraction of 20% (which was near her baseline value), diffuse regional wall-motion abnormalities, severe mitral regurgitation, and moderate tricuspid regurgitation consistent with an exacerbation of heart failure.
We considered the possibility that her heart failure symptoms might be due to precipitous up-titration of her levothyroxine dose, given her borderline-elevated free thyroxine (T4) and increase in cardiac index (currently 4.45 L/min/m2, previously 2.20 L/min/m2 by the left ventricular outflow tract velocity time integral method). However, given her reduced ejection fraction, this clinical presentation most likely represented an acute exacerbation of her chronic heart failure. Her subjective fevers were thought to be due to a viral infection of the upper respiratory tract. The macrocytic anemia and thrombocytopenia were thought to be a side effect of her long-standing treatment with hydroxyurea for essential thrombocythemia, although amiodarone has also been associated with cytopenia.8
Treatment was started with intravenous diuretics and positive pressure ventilation with oxygen supplementation. Her levothyroxine dose was reduced, and her hydroxyurea was stopped.
Figure 1. Chest computed tomography axial views demonstrated increased attenuation in the liver (A, arrow) and left pleural base (B, arrow). Evaluation of the right lung base revealed ground-glass opacities (C, arrow) and honeycombing (D, arrow). These findings were consistent with amiodarone pulmonary toxicity.
After aggressive diuresis, our patient returned to euvolemia. However, she had persistent fine crackles and hypoxia. She had no further fever, and her vital signs were otherwise stable. Her cytopenia improved with cessation of hydroxyurea. Chest computed tomography (CT) showed bibasilar ground-glass infiltrates with areas of interstitial fibrosis, high-attenuation pleural lesions, and increased liver attenuation (Figure 1).
Further testing for connective tissue disease and hypersensitivity pneumonitis was also done, and the results were negative. To exclude an atypical infection, bronchoalveolar lavage was performed; preliminary microbial testing was negative, and the white blood cell count in the lavage fluid was 90% macrophages (pigment-laden), 7% neutrophils, and 3% lymphocytes.
WHAT IS THE CAUSE OF HER PERSISTENT PULMONARY FINDINGS?
2. Given the CT findings and laboratory results, what is the most likely cause of our patient’s persistent crackles and hypoxia?
Heart failure with reduced ejection fraction
Bacterial pneumonia
Idiopathic pulmonary fibrosis
Amiodarone pulmonary toxicity
Heart failure with reduced ejection fraction can cause ground-glass opacities on CT due to increased pulmonary edema. Although our patient initially presented with acute decompensation of heart failure with reduced ejection fraction decompensation, she had returned to euvolemia after aggressive diuresis. Moreover, increased pleural and liver attenuation are not typically seen as a result of heart failure with reduced ejection fraction, making this diagnosis less likely.
Bacterial pneumonia typically presents with cough, fever, and purulent sputum production.9 Further evaluation usually reveals decreased breath sounds, dullness to percussion, and leukocytosis.10 Chest CT in bacterial pneumonia commonly shows a focal area of consolidation, which was not seen in our patient.11
Idiopathic pulmonary fibrosis usually presents with slowly progressive dyspnea and nonproductive cough.12 Physical examination usually reveals fine crackles and occasionally end-inspiratory “squeaks” if traction bronchiectasis is present.12 The diagnosis of idiopathic pulmonary fibrosis requires chest CT findings compatible with it (ie, basal fibrosis, reticular abnormalities, and honeycombing). However, it remains a diagnosis of exclusion and requires ruling out conditions known to cause pulmonary fibrosis such as hypersensitivity pneumonitis, connective tissue disease, and certain medications.12
Although idiopathic pulmonary fibrosis remained in the differential diagnosis, our patient remained on amiodarone, a known cause of pulmonary fibrosis.13 Similarly, the high-attenuation pleural lesions likely represented organizing pneumonia, which is more common in amiodarone pulmonary toxicity. And the ground-glass opacities made idiopathic pulmonary fibrosis unlikely, although they may be seen in an acute exacerbation of this disease.14 Thus, a diagnosis of idiopathic pulmonary fibrosis could not be made definitively.
Amiodarone pulmonary toxicity most commonly presents with acute to subacute cough and progressive dyspnea.13 Physical findings are similar to those in idiopathic pulmonary fibrosis and commonly include bibasilar crackles. Chest CT shows diffuse ground-glass opacities, reticular abnormalities, fibrosis, and increased attenuation of multiple organs, including the lungs, liver, and spleen.14 Bronchoalveolar lavage findings of lipid-laden macrophages suggest but do not definitively diagnose amiodarone pulmonary toxicity.15 And patients with acute amiodarone pulmonary toxicity may present with pigment-laden macrophages on bronchoalveolar lavage, as in our patient.16
Exclusion of hypersensitivity pneumonitis, connective tissue disease, and infection made our patient’s progressive dyspnea and chest CT findings of ground-glass opacities, fibrosis, and increased pulmonary and liver attenuation most consistent with amiodarone pulmonary toxicity.
Amiodarone was therefore discontinued. However, the test result of her lavage fluid for influenza A by polymerase chain reaction came back positive a few hours later.
WHAT IS THE NEXT STEP?
3. Given the positive influenza A polymerase chain reaction test, which of the following is the best next step in this patient’s management?
Surgical lung biopsy
Stop amiodarone and start supportive influenza management
Stop amiodarone and start dronedarone
Start an intravenous corticosteroid
Surgical lung biopsy is typically not required for diagnosis in patients with suspected amiodarone pulmonary toxicity. In addition, acute respiratory distress syndrome has been documented in patients who have undergone surgical biopsy for suspected amiodarone pulmonary toxicity.17
Thus, surgical biopsy is typically only done in cases of persistent symptoms despite withdrawal of amiodarone and initiation of steroid therapy.
Stopping amiodarone and starting supportive influenza management are the best next steps, as our patient’s fevers, cough, dyspnea, and laboratory test results were consistent with influenza.18 Moreover, CT findings of ground-glass opacities and reticular abnormalities can be seen in influenza.19
However, concomitant amiodarone pulmonary toxicity could not be ruled out, as CT showed increased lung and liver attenuation and fibrosis that could not be explained by influenza. And the elevation in aminotransferase levels more than 2 times the upper limit of normal and CT findings of increased liver attenuation suggested amiodarone hepatotoxicity. However, definitive diagnosis would require exclusion of other causes such as congestive hepatopathy, in some cases with liver biopsy.13
Our patient’s persistent hypoxia was thought to be due in part to influenza, and thus the best next step in management was to stop amiodarone and provide supportive care for influenza.
Dronedarone is an antiarrhythmic drug structurally and functionally similar to amiodarone. There are far fewer reports of pulmonary toxicity with dronedarone than with amiodarone.20 However, lack of data on dronedarone in amiodarone pulmonary toxicity, increased rates of hospitalization and death associated with dronedarone in patients like ours with advanced heart failure, and our patient’s previously implanted cardioverter-defibrillator for recurrent ventricular tachycardia all made dronedarone an undesirable alternative to amiodarone.21
Corticosteroids are useful in the treatment of amiodarone pulmonary toxicity when hypoxia and dyspnea are present at diagnosis.13 Our patient’s hypoxia and dyspnea were thought to be due in part to her acute influenza infection, and therefore corticosteroids were not used at the outset.
However, concomitant amiodarone pulmonary toxicity could not be excluded, and the elevation in aminotransferases of more than 2 times the upper limit of normal and CT findings of increased liver attenuation suggested amiodarone hepatotoxicity—though congestive hepatopathy remained in the differential diagnosis. Therefore, supportive therapy for influenza was instituted, and amiodarone was withheld. Her condition subsequently improved, and she was discharged.
FOLLOW-UP 1 MONTH LATER
At a follow-up visit 1 month later, our patient continued to have dyspnea and hypoxia. She did not have signs or symptoms consistent with decompensated heart failure.
Pulmonary function testing revealed the following values:
FEV1 0.69 L (56% of predicted)
FVC 1.08 L (64% of predicted)
Figure 2. In A, repeat chest computed tomography demonstrated increased liver attenuation (arrow); in B, it showed persistent ground-glass opacities (white arrow), increased pulmonary attenuation (black arrowhead), and worsening pleural effusions (black arrows). These findings supported the diagnosis of amiodarone pulmonary toxicity.
FEV1/FVC ratio 64%
DLCO 2.20 mL/min/mm Hg (12% of predicted).
Aminotransferase levels had also normalized. Repeat chest CT showed persistent bibasilar interstitial fibrotic changes, enlarging bilateral pleural effusions, and persistent peripheral ground-glass opacities (Figure 2).
WHAT FURTHER TREATMENT IS APPROPRIATE?
4. Given the chest CT findings, which of the following is the most appropriate treatment strategy for this patient?
No further management, continue to hold amiodarone
Corticosteroids
Repeat bronchoalveolar lavage
Intravenous antibiotics
No further management of amiodarone pulmonary toxicity would be appropriate if our patient did not have a high burden of symptoms. However, when patients with amiodarone pulmonary toxicity present with hypoxia and dyspnea, corticosteroids should be started.13 Our patient remained symptomatic after discontinuation of amiodarone and resolution of her influenza infection, and CT showed persistent signs of amiodarone pulmonary toxicity, which required further management.
Corticosteroids are useful in treating amiodarone pulmonary toxicity when hypoxia and dyspnea are present at diagnosis. Our patient’s persistent ground-glass opacities, fibrotic changes, and increased attenuation in multiple organs on CT, coupled with a confirmed reduction in FVC of greater than 15% and reduction in DLCO of greater than 20% after recovery from influenza, were most consistent with persistent amiodarone pulmonary toxicity.13
Although our patient’s amiodarone had been discontinued, the long half-life of the drug (45 days) allowed pulmonary toxicity to progress even after the drug was discontinued.22 Because our patient continued to have hypoxia and dyspnea on exertion, the most appropriate next step in management (in addition to managing her pleural effusions) was to start corticosteroids.
For amiodarone pulmonary toxicity, prednisone is typically started at 40 to 60 mg daily and can result in rapid improvement in symptoms.13 Tapering should be slow and may take several months.
Bronchoalveolar lavage is typically used in suspected cases of amiodarone pulmonary toxicity only to rule out an alternative diagnosis such as infection. Lipid-laden macrophages may be seen in the fluid. However, lipid-laden macrophages are not diagnostic of amiodarone pulmonary toxicity, as this finding may also be seen in patients taking amiodarone who do not develop pulmonary toxicity.15 Other findings on bronchoalveolar lavage in amiodarone pulmonary toxicity are nonspecific and are not diagnostically useful.13
Intravenous antibiotics are appropriate if bacterial pneumonia is suspected. However, bacterial pneumonia typically presents with cough, fever, purulent sputum production, and focal consolidation on chest imaging.9 Our patient’s CT findings of persistent peripheral ground-glass opacities and lack of cough, fever, or purulent sputum production were not consistent with bacterial pneumonia, and therefore intravenous antibiotics were not indicated.
CASE CONCLUSION
Given our patient’s persistent dyspnea, hypoxia, and chest CT findings consistent with amiodarone pulmonary toxicity, it was recommended that she start corticosteroids. However, before starting therapy, she suffered a femoral fracture that required surgical intervention. Around the time of the procedure, she had an ST-segment elevation myocardial infarction requiring vasopressor support and mechanical ventilation. At that time, the patient and family decided to pursue comfort measures, and she died peacefully.
MORE ABOUT AMIODARONE PULMONARY TOXICITY
Pulmonary toxicity is a well-described consequence of amiodarone therapy.23 Amiodarone carries a 2% risk of pulmonary toxicity.24 Although higher doses are more likely to cause pulmonary toxicity, lower doses also have been implicated.22,24 Preexisting pulmonary disease may predispose patients taking amiodarone to pulmonary toxicity; however, this is not uniformly seen.25
Mortality rates as high as 10% from amiodarone pulmonary toxicity have been reported. Thus, diligent surveillance for pulmonary toxicity with pulmonary function tests in patients taking amiodarone is mandatory. In particular, a reduction in FVC of greater than 15% or in DLCO of greater than 20% from baseline may be seen in amiodarone pulmonary toxicity.26
Amiodarone pulmonary toxicity can present at any time after the start of therapy, but it occurs most often after 6 to 12 months.13 Patients typically experience insidious dyspnea; however, presentation with acute to subacute cough and progressive dyspnea can occur, especially with high concentrations of supplemental oxygen with or without mechanical ventilation.12,27 Findings on physical examination include bibasilar crackles. CT chest findings include diffuse ground-glass opacities, reticular abnormalities, fibrosis, and increased attenuation in multiple organs, including the lung, liver, and spleen.14
The diagnosis of amiodarone pulmonary toxicity requires ruling out hypersensitivity pneumonitis, connective tissue disease, heart failure, and infection. Surgical biopsy and bronchoalveolar lavage are not commonly used to establish the diagnosis of amiodarone pulmonary toxicity, as surgical biopsy increases the risk of acute respiratory distress syndrome, and the results of bronchoalveolar lavage are usually nonspecific.13,15
Initial treatment involves discontinuing the amiodarone once the diagnosis is suspected. If patients have worsening hypoxia or dyspnea at the time of diagnosis, corticosteroids can be used. Prednisone is typically started at 40 to 60 mg daily and can result in rapid improvement in symptoms.13 Tapering of corticosteroids should occur slowly and may take several months.
References
McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Engl J Med 1971; 285(26):1441–1446. doi:10.1056/NEJM197112232852601
Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100(3):598–603. pmid:1909617
Baggish AL, Siebert U, Lainchbury JG, et al. A validated clinical and biochemical score for the diagnosis of acute heart failure: the ProBNP investigation of dyspnea in the emergency department (PRIDE) acute heart failure score. Am Heart J 2006; 151(1):48–54. doi:10.1016/j.ahj.2005.02.031
National Heart, Lung, and Blood Institute. National Asthma Education and Prevention Program. Expert panel report 3: Guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/sites/default/files/media/docs/asthgdln_1.pdf. Accessed August 3, 2018.
Brenner BE, Abraham E, Simon RR. Position and diaphoresis in acute asthma. Am J Med 1983; 74(6):1005–1009. pmid:6407304
Badgett RG, Tanaka DJ, Hunt DK, et al. Can moderate chronic obstructive pulmonary disease be diagnosed by historical and physical findings alone? Am J Med 1993; 94(2):188–196. pmid:8430714
Erie AJ, McClure RF, Wolanskyj AP. Amiodarone-induced bone marrow granulomas: an unusual cause of reversible pancytopenia. Hematol Rep 2010; 2(1):e6. doi:10.4081/hr.2010.e6
Marrie TJ. Community-acquired pneumonia. Clin Infect Dis 1994; 18(4):501–513. pmid:8038304
Metlay JP, Kapoor WN, Fine MJ. Does this patient have community-acquired pneumonia? Diagnosing pneumonia by history and physical examination. JAMA 1997; 278(17):1440–1445. pmid:9356004
Walker CM, Abbott GF, Greene RE, Shepard JA, Vummidi D, Digumarthy SR. Imaging pulmonary infection: classic signs and patterns. AJR Am J Roentgenol 2014; 202(3):479–492. doi:10.2214/AJR.13.11463
Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
Goldschlager N, Epstein AE, Naccarelli GV, et al; Practice Guidelines Sub-committee, North American Society of Pacing and Electrophysiology (HRS). A practical guide for clinicians who treat patients with amiodarone: 2007. Heart Rhythm 2007; 4(9):1250–1259. doi:10.1016/j.hrthm.2007.07.020
Martin WJ 2nd, Rosenow EC 3rd. Amiodarone pulmonary toxicity: recognition and pathogenesis (Part I). Chest 1988; 93(5):1067–1075. pmid:3282816
Iskandar SB, Abi-Saleh B, Keith RL, Byrd RP Jr, Roy TM. Amiodarone-induced alveolar hemorrhage. South Med J 2006; 99(4):383–387.
Van Mieghem W, Coolen L, Malysse I, Lacquet LM, Deneffe GJ, Demedts MG. Amiodarone and the development of ARDS after lung surgery. Chest 1994; 105(6):1642–1645. pmid:8205854
Nicholson KG. Clinical features of influenza. Semin Respir Infect 1992; 7(1):26–37. pmid:1609165
Muller NL, Franquet T, Lee KS, Silva CIS. Viruses, mycoplasma, and chlamydia. In: Imaging of Pulmonary Infections. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:94–114.
Stack S, Nguyen DV, Casto A, Ahuja N. Diffuse alveolar damage in a patient receiving dronedarone. Chest 2015; 147(4):e131–e133. doi:10.1378/chest.14-1849
De Ferrari GM, Dusi V. Drug safety evaluation of dronedarone in atrial fibrillation. Expert Opin Drug Saf 2012; 11(6):1023–1045. doi:10.1517/14740338.2012.722994
Okayasu K, Takeda Y, Kojima J, et al. Amiodarone pulmonary toxicity: a patient with three recurrences of pulmonary toxicity and consideration of the probable risk for relapse. Intern Med 2006; 45(22):1303–1307. pmid:17170505
Vorperian VR, Havighurst TC, Miller S, January CT. Adverse effects of low dose amiodarone: a meta-analysis. J Am Coll Cardiol 1997; 30(3):791–798. pmid:9283542
Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomised trials. Lancet 1997; 350(9089):1417–1424. pmid:9371164
Olshansky B, Sami M, Rubin A, et al; NHLBI AFFIRM Investigators. Use of amiodarone for atrial fibrillation in patients with preexisting pulmonary disease in the AFFIRM study. Am J Cardiol 2005; 95(3):404–405. doi:10.1016/j.amjcard.2004.09.044
Camus P. Interstitial lung disease from drugs, biologics, and radiation. In: Schwartz MI, King TE Jr, eds. Interstitial Lung Disease. 5th ed. Shelton, CT: People’s Medical Publishing House; 2011:637–644.
Wolkove N, Baltzan M. Amiodarone pulmonary toxicity. Can Respir J 2009; 16(2):43–48. doi:10.1155/2009/282540
References
McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Engl J Med 1971; 285(26):1441–1446. doi:10.1056/NEJM197112232852601
Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100(3):598–603. pmid:1909617
Baggish AL, Siebert U, Lainchbury JG, et al. A validated clinical and biochemical score for the diagnosis of acute heart failure: the ProBNP investigation of dyspnea in the emergency department (PRIDE) acute heart failure score. Am Heart J 2006; 151(1):48–54. doi:10.1016/j.ahj.2005.02.031
National Heart, Lung, and Blood Institute. National Asthma Education and Prevention Program. Expert panel report 3: Guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/sites/default/files/media/docs/asthgdln_1.pdf. Accessed August 3, 2018.
Brenner BE, Abraham E, Simon RR. Position and diaphoresis in acute asthma. Am J Med 1983; 74(6):1005–1009. pmid:6407304
Badgett RG, Tanaka DJ, Hunt DK, et al. Can moderate chronic obstructive pulmonary disease be diagnosed by historical and physical findings alone? Am J Med 1993; 94(2):188–196. pmid:8430714
Erie AJ, McClure RF, Wolanskyj AP. Amiodarone-induced bone marrow granulomas: an unusual cause of reversible pancytopenia. Hematol Rep 2010; 2(1):e6. doi:10.4081/hr.2010.e6
Marrie TJ. Community-acquired pneumonia. Clin Infect Dis 1994; 18(4):501–513. pmid:8038304
Metlay JP, Kapoor WN, Fine MJ. Does this patient have community-acquired pneumonia? Diagnosing pneumonia by history and physical examination. JAMA 1997; 278(17):1440–1445. pmid:9356004
Walker CM, Abbott GF, Greene RE, Shepard JA, Vummidi D, Digumarthy SR. Imaging pulmonary infection: classic signs and patterns. AJR Am J Roentgenol 2014; 202(3):479–492. doi:10.2214/AJR.13.11463
Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
Goldschlager N, Epstein AE, Naccarelli GV, et al; Practice Guidelines Sub-committee, North American Society of Pacing and Electrophysiology (HRS). A practical guide for clinicians who treat patients with amiodarone: 2007. Heart Rhythm 2007; 4(9):1250–1259. doi:10.1016/j.hrthm.2007.07.020
Martin WJ 2nd, Rosenow EC 3rd. Amiodarone pulmonary toxicity: recognition and pathogenesis (Part I). Chest 1988; 93(5):1067–1075. pmid:3282816
Iskandar SB, Abi-Saleh B, Keith RL, Byrd RP Jr, Roy TM. Amiodarone-induced alveolar hemorrhage. South Med J 2006; 99(4):383–387.
Van Mieghem W, Coolen L, Malysse I, Lacquet LM, Deneffe GJ, Demedts MG. Amiodarone and the development of ARDS after lung surgery. Chest 1994; 105(6):1642–1645. pmid:8205854
Nicholson KG. Clinical features of influenza. Semin Respir Infect 1992; 7(1):26–37. pmid:1609165
Muller NL, Franquet T, Lee KS, Silva CIS. Viruses, mycoplasma, and chlamydia. In: Imaging of Pulmonary Infections. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:94–114.
Stack S, Nguyen DV, Casto A, Ahuja N. Diffuse alveolar damage in a patient receiving dronedarone. Chest 2015; 147(4):e131–e133. doi:10.1378/chest.14-1849
De Ferrari GM, Dusi V. Drug safety evaluation of dronedarone in atrial fibrillation. Expert Opin Drug Saf 2012; 11(6):1023–1045. doi:10.1517/14740338.2012.722994
Okayasu K, Takeda Y, Kojima J, et al. Amiodarone pulmonary toxicity: a patient with three recurrences of pulmonary toxicity and consideration of the probable risk for relapse. Intern Med 2006; 45(22):1303–1307. pmid:17170505
Vorperian VR, Havighurst TC, Miller S, January CT. Adverse effects of low dose amiodarone: a meta-analysis. J Am Coll Cardiol 1997; 30(3):791–798. pmid:9283542
Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomised trials. Lancet 1997; 350(9089):1417–1424. pmid:9371164
Olshansky B, Sami M, Rubin A, et al; NHLBI AFFIRM Investigators. Use of amiodarone for atrial fibrillation in patients with preexisting pulmonary disease in the AFFIRM study. Am J Cardiol 2005; 95(3):404–405. doi:10.1016/j.amjcard.2004.09.044
Camus P. Interstitial lung disease from drugs, biologics, and radiation. In: Schwartz MI, King TE Jr, eds. Interstitial Lung Disease. 5th ed. Shelton, CT: People’s Medical Publishing House; 2011:637–644.
Wolkove N, Baltzan M. Amiodarone pulmonary toxicity. Can Respir J 2009; 16(2):43–48. doi:10.1155/2009/282540
We live in the era of evidence-based medicine, so new interventions must meet criteria for both safety and efficacy before they are adopted. However, we have inherited many practices adopted before the current standards were in place, and we have not always been rigorous in reevaluating traditional remedies. A conservative belief in established practice or the influence of vested interests may account for this lack of rigor in reappraisal.1 Calcium and vitamin D supplements are possible examples of this phenomenon.
BONE METABOLISM IS TIGHTLY REGULATED
Bone is a connective tissue, its matrix composed principally of type 1 collagen, which provides tensile strength. Hydroxyapatite crystals, composed predominantly of calcium and phosphate, lie between the collagen fibers and provide compressive strength. In a tightly regulated process, osteoblasts lay down the collagenous matrix, and osteoclasts remove it. Mineralization of newly formed bone proceeds if normal levels of extracellular calcium and phosphate are present, in the absence of inhibitors of mineralization.
High calcium intake does not drive bone formation
The endocrine system is critical in maintaining normocalcemia. A decrease in calcium intake results in increased parathyroid hormone secretion, resulting in increased renal tubular calcium reabsorption, increased bone turnover (both formation and resorption), and increased activation of vitamin D leading to increased intestinal absorption of calcium. High calcium intake reverses these changes.
Reid IR, Bristow SM, Bolland MJ. Calcium supplements: benefits and risks. J Intern Med 2015; 278(4):354–368. Copyright 2015, The Association for the Publication of the Journal of Internal Medicine.
Figure 1. Absolute change in total body bone mineral content (BMC) over 5 years in normal postmenopausal women, as a function of each woman’s average calcium intake assessed at baseline and at year 5. The lines show the regression (with 95% confidence intervals) for this relationship (P = .53)
Thus, a normal serum calcium concentration can be maintained with calcium intake ranging from 200 to more than 2,000 mg/day, and rates of bone loss in postmenopausal women are unaffected by calcium intake (Figure 1).2
If calcium intake is very low, hypocalcemia and secondary hyperparathyroidism develop,3 and bone mineralization may be impaired. However, levels of calcium intake in Africa and in East and Southeast Asia are typically less than 400 mg/day,4 yet there is no evidence that these levels adversely affect skeletal health. In fact, fracture risk is lower in these regions than in North America, where calcium intake is several times greater.
Thus, some calcium intake is required to maintain circulating concentrations, but there is no mechanism by which high calcium intake can drive bone formation. Quite the opposite, in fact.
Vitamin D deficiency has little relationship with diet
Vitamin D is a biologically inactive secosteroid activated by hydroxylation in the liver and kidney to function as the key regulator of intestinal calcium absorption. As with calcium, its deficiency results in hypocalcemia and impaired bone mineralization.
Paradoxically, high levels of vitamin D stimulate bone resorption and inhibit bone mineralization in mice,5 and large doses increase bone resorption markers acutely in clinical studies.6 Thus, it is important to ensure an adequate vitamin D supply, but not an oversupply.
In the absence of supplements, most vitamin D is produced in the skin as a result of the action of ultraviolet light (from sunlight) on 7-dehydrocholesterol. Thus, vitamin D deficiency occurs in those deprived of skin exposure to sunlight (eg, due to veiling, living at high latitude, staying permanently indoors), but it has little relationship with diet.
ARE CALCIUM SUPPLEMENTS EFFECTIVE?
Calcium supplements are certainly biologically active. They transiently increase serum calcium concentrations, suppress parathyroid hormone, and reduce bone resorption.2 In the first year of use, they increase bone density by about 1% compared with placebo.7 However, longer use does not result in further bone density advantage over placebo,7 suggesting that the response simply reflects a decreased number of osteoclastic resorption sites and does not indicate a sustained change in bone balance.
A 1% difference in bone density would not be expected to reduce fracture risk, and a number of large, carefully conducted randomized controlled trials published over the last 15 years have failed to demonstrate antifracture efficacy for calcium.8–12 As a result, the US Preventive Services Task Force recommends against the routine use of calcium supplements in community-dwelling adults.13
In contrast, in a placebo-controlled trial published in 1992, Chapuy et al14 found that elderly women residing in nursing homes who received calcium and vitamin D supplements had fewer fractures. At 18 months, by intention-to-treat analysis, nonvertebral fractures had occurred in 160 (12%) of 1,387 women in the supplement group compared with 215 (15%) of 1,403 women in the placebo group (P < .001). However, these women were severely vitamin D-deficient (the mean serum 25-hydroxyvitamin D level at baseline in the placebo group was 13 ng/mL, normal range 15–50), to the extent that many must have had osteomalacia.
Thus, this study shows that calcium and vitamin D are effective in managing osteomalacia, but the subsequent trials8–12 did not observe any benefit in community-dwelling cohorts. Meta-analyses that pool the Chapuy study with community-based studies generally find that calcium with vitamin D is beneficial, but the heterogeneity of these populations means that such pooling is inappropriate.15
It is sometimes stated that calcium and vitamin D should always be given with osteoporosis medications because the efficacy of these drugs has only been demonstrated when coadministered with these supplements. This is incorrect. The addition of calcium to alendronate does not alter its effects on bone density,16 and the antifracture efficacy of both bisphosphonates17 and estrogen18,19 has been demonstrated in the absence of supplementation with calcium or vitamin D. The evidence that bisphosphonates prevent fractures in the absence of calcium supplements has recently been strengthened by the results of a randomized controlled trial comparing zoledronate with placebo in women over age 65 with osteopenia.20
ARE CALCIUM SUPPLEMENTS SAFE?
Calcium supplements often cause gastrointestinal symptoms, particularly constipation. They have been shown to double the risk of hospital admission due to abdominal symptoms.21 In the absence of clear evidence of benefit, these facts alone should militate against their routine use. Calcium supplements also cause hypercalcemia and hypercalciuria22 and increase the risk of renal calculi (by 17% in the Women’s Health Initiative8).
Over the last decade, evidence has emerged that calcium supplements may also increase the risk of myocardial infarction, and possibly stroke. This finding was not statistically significant in any single study, but is consistently present in meta-analyses.23
Evidence from the Women’s Health Initiative
When studies of calcium with vitamin D are added to these meta-analyses, the results are less consistent. This is because such meta-analyses are dominated by the Women's Health Initiative (because of its large size, with 36,282 participants). There have been 2 different analyses of this trial with respect to cardiovascular events.
When the Women’s Health Initiative as a whole was analyzed, there was no significant effect of calcium plus vitamin D on vascular end points. However, there is a significant interaction between body mass index and the effect of supplements, such that nonobese women demonstrated a 17% increase in myocardial infarction.24 This study was unusual in that it included women already taking calcium and vitamin D supplements.
Bolland MJ, Grey A, Avenell A, Gamble GD, Reid IR. Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women’s Health Initiative limited access dataset and meta-analysis. BMJ 2011; 342:d2040.
Figure 2. Effect of calcium supplements on cardiovascular events, with or without vitamin D. Data for 28,072 participants in 8 trials of calcium supplements with trial-level data, plus data for Women’s Health Initiative CaD study participants not taking calcium supplements at baseline.
There was a significant interaction between baseline use of supplements and the effects of the trial intervention on vascular events, justifying analyzing the supplement-naive individuals separately. In this group of 16,000 women, an increase in clinical myocardial infarction of 22% was found, similar to the findings with calcium supplements alone.25
Thus, there is consistent evidence that introducing a calcium supplement de novo increases the risk of myocardial infarction (Figure 2).16,25–31 We calculate that treating 1,000 patients with calcium or calcium plus vitamin D for 5 years would cause an additional 6 myocardial infarctions or strokes (number needed to harm 178) and prevent only 3 fractures (number needed to treat 302).25
ARE VITAMIN D SUPPLEMENTS EFFECTIVE?
Vitamin D is highly effective in treating osteomalacia, improving symptoms within days and increasing bone density by as much as 50% over 1 year.32,33 In contrast, randomized controlled trials of vitamin D supplements alone in people without osteomalacia have not shown increases in bone density or changes in fracture risk.34–37
Reid IR, Horne AM, Mihov B, et al. Effect of monthly high-dose vitamin D on bone density in community-dwelling older adults substudy of a randomized controlled trial. J Intern Med 2017; 282(5):452–460. Copyright 2017, Assoc for Publication of J Int Med
Figure 3. Changes in bone mineral density (BMD) from baseline to 2 years in the vitamin D and placebo groups of the Vitamin D Assessment study, according to baseline serum 25(OH)D (25-hydroxyvitamin D) concentrations. Data are mean ± 95% confidence intervals. P values are shown for between-group comparisons.
In 2017, my colleagues and I published a trial showing that vitamin D supplementation increases bone density by 2% to 3% in the spine and femoral neck in participants with baseline 25-hydroxyvitamin D levels below 30- nmol/L (12 ng/mL), but those starting above this level showed no effect (Figure 3).38 And a reanalysis of an earlier study confirmed this 30 nmol/L threshold for an effect of vitamin D on bone density.39 The finding of a clear-cut threshold for vitamin D effects is predicted by the physiologic considerations set out above.
Belief that higher levels of 25-hydroxyvitamin D are better is based on observational data. However, correlation does not prove causation, and it is likely that causation is reversed here. Those with better health are likely to spend more time exercising outdoors, are less likely to be obese, and are less likely to have inflammatory conditions; and as a result, they are more likely to have better vitamin D status. We should now be using trial-based definitions of vitamin D deficiency as opposed to thresholds derived from disease associations in observational studies.
Vitamin D supplements have also been suggested to benefit cardiovascular health and to reduce cancer risk, though current clinical trial data provide no support for these hypotheses.36,40 Other trials addressing these questions are ongoing.
ARE VITAMIN D SUPPLEMENTS SAFE?
The safety of vitamin D supplements has generally been assessed with respect to the incidence of hypercalcemia. On this basis, very high doses have been promoted. However, there is now evidence that doses of 4,000 IU/day, 60,000 IU/month, and 500,000 IU/year increase the risk of falls and fractures.41,42
The threshold for bone benefits discussed above (12 ng/mL) is easily exceeded with doses of vitamin D of 400 to 1,000 IU/day. At these levels, vitamin D supplements have no known adverse effects and can be widely endorsed for individuals at risk of deficiency. Supplement doses greater than 2,000 IU/day should be used only in exceptional circumstances, and with appropriate monitoring.
LITTLE USE FOR CALCIUM AND VITAMIN D SUPPLEMENTS
Extensive clinical trials have failed to demonstrate meaningful benefit from calcium supplements in the management of osteoporosis. Calcium supplements are often prescribed in patients who are receiving other treatments for osteoporosis, which may be justified with interventions that have the potential to cause hypocalcemia, but their coadministration with bisphosphonates has been shown to be unnecessary.
Calcium supplements commonly cause gastrointestinal symptoms that are sometimes severe and are likely to contribute to high levels of noncompliance with osteoporosis medications. They increase the risk of kidney stones,8 and there is reasonable evidence to suggest an adverse effect on vascular risk as well.23
Vitamin D deficiency is common in frail elderly people, particularly those with dark skin or living at high latitudes. Low doses of vitamin D are safe and highly effective in preventing osteomalacia. But vitamin D supplements are unnecessary in those who regularly have sun exposure. And high doses of vitamin D have no demonstrated advantage and have been shown to increase the risk of falls and fractures.
Our decision to prescribe calcium and vitamin D supplements should be based on evidence that is of the same quality as for any other intervention we prescribe. Current evidence suggests that there is little reason to prescribe calcium, and that vitamin D should be targeted at those at risk of 25-hydroxyvitamin D levels less than 12 ng/mL.
References
Grey A, Bolland M. Web of industry, advocacy, and academia in the management of osteoporosis. BMJ 2015; 351:h3170. doi:10.1136/bmj.h3170
Reid IR, Bristow SM, Bolland MJ. Calcium supplements: benefits and risks. J Intern Med 2015; 278(4):354–368. doi:10.1111/joim.12394
Bolland MJ, Grey AB, Ames RW, Horne AM, Gamble GD, Reid IR. Fat mass is an important predictor of parathyroid hormone levels in postmenopausal women. Bone 2006; 38(3):317–321. doi:10.1016/j.bone.2005.08.018
Lieben L, Masuyama R, Torrekens S, et al. Normocalcemia is maintained in mice under conditions of calcium malabsorption by vitamin D-induced inhibition of bone mineralization. J Clin Invest 2012; 122(5):1803–1815. doi:10.1172/JCI45890
Rossini M, Gatti D, Viapiana O, et al. Short-term effects on bone turnover markers of a single high dose of oral vitamin D3. J Clin Endocrinol Metab 2012; 97(4):E622–E626. doi:10.1210/jc.2011-2448
Tai V, Leung W, Grey A, Reid IR, Bolland MJ. Calcium intake and bone mineral density: systematic review and meta-analysis. BMJ 2015; 351:h4183. doi:10.1136/bmj.h4183
Jackson RD, LaCroix AZ, Gass M, et al; Women’s Health Initiative Investigators. Calcium plus vitamin D supplementation and the risk of fractures. N Engl J Med 2006; 354(7):669–683. doi:10.1056/NEJMoa055218
Grant AM, Avenell A, Campbell MK, et al; RECORD Trial Group. Oral vitamin D3 and calcium for secondary prevention of low-trauma fractures in elderly people (Randomised Evaluation of Calcium or vitamin D, RECORD): a randomised placebo-controlled trial. Lancet 2005; 365(9471):1621–1628. doi:10.1016/S0140-6736(05)63013-9
Prince RL, Devine A, Dhaliwal SS, Dick IM. Effects of calcium supplementation on clinical fracture and bone structure: results of a 5-year, double-blind, placebo-controlled trial in elderly women. Arch Intern Med 2006; 166(8):869–875. doi:10.1001/archinte.166.8.869
Reid IR, Mason B, Horne A, et al. Randomized controlled trial of calcium in healthy older women. Am J Med 2006; 119(9):777–785. doi:10.1016/j.amjmed.2006.02.038
Salovaara K, Tuppurainen M, Karkkainen M, et al. Effect of vitamin D-3 and calcium on fracture risk in 65-to 71-year-old women: a population-based 3-year randomized, controlled trial—the OSTPRE-FPS. J Bone Miner Res 2010; 25(7):1487–1495. doi:10.1002/jbmr.48
Moyer VA, US Preventive Services Task Force. Vitamin D and calcium supplementation to prevent fractures in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2013; 158(9):691–696. doi:10.7326/0003-4819-158-9-201305070-00603
Chapuy MC, Arlot ME, Duboeuf F, et al. Vitamin D3 and calcium to prevent hip fractures in elderly women. N Engl J Med 1992; 327(23):1637–1642. doi:10.1056/NEJM199212033272305
Tang BMP, Eslick GD, Nowson C, Smith C, Bensoussan A. Use of calcium or calcium in combination with vitamin D supplementation to prevent fractures and bone loss in people aged 50 years and older: a meta-analysis. Lancet 2007; 370(9588):657–666. doi:10.1016/S0140-6736(07)61342-7
Bonnick S, Broy S, Kaiser F, et al. Treatment with alendronate plus calcium, alendronate alone, or calcium alone for postmenopausal low bone mineral density. Curr Med Res Opin 2007; 23(6):1341–1349. doi:10.1185/030079907X188035
McCloskey EV, Beneton M, Charlesworth D, et al. Clodronate reduces the incidence of fractures in community-dwelling elderly women unselected for osteoporosis: results of a double-blind, placebo-controlled randomized study. J Bone Miner Res 2007; 22(1):135–141. doi:10.1359/jbmr.061008
Lindsay R, Hart DM, Forrest C, Baird C. Prevention of spinal osteoporosis in oophorectomised women. Lancet 1980; 2(8205):1151–1154. pmid:6107766
Cauley JA, Robbins J, Chen Z, et al; Women’s Health Initiative Investigators. Effects of estrogen plus progestin on risk of fracture and bone mineral density: the Women’s Health Initiative randomized trial. JAMA 2003; 290(13):1729–1738. doi:10.1001/jama.290.13.1729
Reid I, Horne A, Mihov B, et al. Abstracts of the ECTS Congress 2018: Zoledronate every 18 months for 6 years in osteopenic postmenopausal women reduces non-vertebral fractures and height loss. Calcif Tissue Int 2018; 102:S1-S159. doi:10.1007/s00223-018-0418-0
Lewis JR, Zhu K, Prince RL. Adverse events from calcium supplementation: relationship to errors in myocardial infarction self-reporting in randomized controlled trials of calcium supplementation. J Bone Miner Res 2012; 27(3):719–722. doi:10.1002/jbmr.1484
Gallagher JC, Smith LM, Yalamanchili V. Incidence of hypercalciuria and hypercalcemia during vitamin D and calcium supplementation in older women. Menopause 2014; 21(11):1173–1180. doi:10.1097/GME.0000000000000270
Reid IR, Bristow SM, Bolland MJ. Calcium and cardiovascular disease. Endocrinol Metab (Seoul) 2017; 32(3):339–349. doi:10.3803/EnM.2017.32.3.339
Hsia J, Heiss G, Ren H, et al; Women’s Health Initiative Investigators. Calcium/vitamin D supplementation and cardiovascular events. Circulation 2007; 115(7):846–854. doi:10.1161/CIRCULATIONAHA.106.673491
Bolland MJ, Grey A, Avenell A, Gamble GD, Reid IR. Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women’s Health Initiative limited access dataset and meta-analysis. BMJ 2011; 342:d2040. doi:10.1136/bmj.d2040
Baron JA, Beach M, Mandel JS, et al. Calcium supplements for the prevention of colorectal adenomas. Calcium Polyp Prevention Study Group. N Engl J Med 1999; 340(3):101–107. doi:10.1056/NEJM199901143400204
Bolland MJ, Barber PA, Doughty RN, et al. Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ 2008; 336(7638):262–266. doi:10.1136/bmj.39440.525752.BE
Lappe JM, Travers-Gustafson D, Davies KM, Recker RR, Heaney RP. Vitamin D and calcium supplementation reduces cancer risk: results of a randomized trial. Am J Clin Nutr 2007; 85(6):1586–1591. doi:10.1093/ajcn/85.6.1586
Reid IR, Ames R, Mason B, et al. Randomized controlled trial of calcium supplementation in healthy, non-osteoporotic, older men. Arch Intern Med 2008; 168(20):2276–2282. doi:10.1001/archinte.168.20.2276
Reid IR, Ames RW, Evans MC,Gamble GD, Sharpe SJ. Effect of calcium supplementation on bone loss in postmenopausal women. N Engl J Med 1993; 328(7):460–464. doi:10.1056/NEJM199302183280702
Reid IR, Ames RW, Evans MC, Gamble GD, Sharpe SJ. Long-term effects of calcium supplementation on bone loss and fractures in postmenopausal women: a randomized controlled trial. Am J Med 1995; 98(4):331–335. doi:10.1016/S0002-9343(99)80310-6
Al-Ali H, Fuleihan GE. Nutritional osteomalacia: substantial clinical improvement and gain in bone density posttherapy. J Clin Densitom 2000; 3(1):97–101. pmid:10745306
El-Desouki MI, Othman SM, Fouda MA. Bone mineral density and bone scintigraphy in adult Saudi female patients with osteomalacia. Saudi Med J 2004; 25(3):355–358.
Reid IR, Bolland MJ, Grey A. Effects of vitamin D supplements on bone mineral density: a systematic review and meta-analysis. Lancet 2014; 383(9912):146–155. doi:10.1016/S0140-6736(13)61647-5
Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev 2014; (4):CD000227. doi:10.1002/14651858.CD000227.pub4
Bolland MJ, Grey A, Gamble GD, Reid IR. The effect of vitamin D supplementation on skeletal, vascular, or cancer outcomes: a trial sequential meta-analysis. Lancet Diabetes Endocrinol 2014; 2(4):307–320. doi:10.1016/S2213-8587(13)70212-2
DIPART (Vitamin D Individual Patient Analysis of Randomized Trials) Group. Patient level pooled analysis of 68 500 patients from seven major vitamin D fracture trials in US and Europe. BMJ 2010; 340:b5463. doi:10.1136/bmj.b5463
Reid IR, Horne AM, Mihov B, et al. Effect of monthly high-dose vitamin D on bone density in community-dwelling older adults substudy of a randomized controlled trial. J Intern Med 2017; 282(5):452–460. doi:10.1111/joim.12651
MacDonald HM, Reid IR, Gamble GD, Fraser WD, Tang JC, Wood AD. 25-Hydroxyvitamin D threshold for the effects of vitamin D supplements on bone density secondary analysis of a randomized controlled trial. J Bone Miner Res 2018. Epub ahead of print. doi:10.1002/jbmr.3442
Scragg R, Stewart AW, Waayer D, et al. Effect of monthly high-dose vitamin D supplementation on cardiovascular disease in the vitamin D assessment study: a randomized clinical trial. JAMA Cardiol 2017; 2(6):608–616. doi:10.1001/jamacardio.2017.0175
Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA 2010; 303(18):1815–1822. doi:10.1001/jama.2010.594
Smith LM, Gallagher JC, Suiter C. Medium doses of daily vitamin D decrease falls and higher doses of daily vitamin D3 increase falls: a randomized clinical trial. J Steroid Biochem Mol Biol 2017; 173:317–322. doi:10.1016/j.jsbmb.2017.03.015
Ian R. Reid, MD University of Auckland, Auckland, New Zealand; Department of Endocrinology, Auckland District Health Board, Auckland, New Zealand
Address: Ian R. Reid, MD, Faculty of Medical and Health Sciences, University of Auckland, Private Bag 92019, Auckland, New Zealand; i.reid@auckland.ac.nz
Dr. Reid has disclosed consulting for Amgen and Merck and teaching and speaking for Amgen and Eli Lilly. He is supported by the Health Research Council of New Zealand.
Ian R. Reid, MD University of Auckland, Auckland, New Zealand; Department of Endocrinology, Auckland District Health Board, Auckland, New Zealand
Address: Ian R. Reid, MD, Faculty of Medical and Health Sciences, University of Auckland, Private Bag 92019, Auckland, New Zealand; i.reid@auckland.ac.nz
Dr. Reid has disclosed consulting for Amgen and Merck and teaching and speaking for Amgen and Eli Lilly. He is supported by the Health Research Council of New Zealand.
Author and Disclosure Information
Ian R. Reid, MD University of Auckland, Auckland, New Zealand; Department of Endocrinology, Auckland District Health Board, Auckland, New Zealand
Address: Ian R. Reid, MD, Faculty of Medical and Health Sciences, University of Auckland, Private Bag 92019, Auckland, New Zealand; i.reid@auckland.ac.nz
Dr. Reid has disclosed consulting for Amgen and Merck and teaching and speaking for Amgen and Eli Lilly. He is supported by the Health Research Council of New Zealand.
We live in the era of evidence-based medicine, so new interventions must meet criteria for both safety and efficacy before they are adopted. However, we have inherited many practices adopted before the current standards were in place, and we have not always been rigorous in reevaluating traditional remedies. A conservative belief in established practice or the influence of vested interests may account for this lack of rigor in reappraisal.1 Calcium and vitamin D supplements are possible examples of this phenomenon.
BONE METABOLISM IS TIGHTLY REGULATED
Bone is a connective tissue, its matrix composed principally of type 1 collagen, which provides tensile strength. Hydroxyapatite crystals, composed predominantly of calcium and phosphate, lie between the collagen fibers and provide compressive strength. In a tightly regulated process, osteoblasts lay down the collagenous matrix, and osteoclasts remove it. Mineralization of newly formed bone proceeds if normal levels of extracellular calcium and phosphate are present, in the absence of inhibitors of mineralization.
High calcium intake does not drive bone formation
The endocrine system is critical in maintaining normocalcemia. A decrease in calcium intake results in increased parathyroid hormone secretion, resulting in increased renal tubular calcium reabsorption, increased bone turnover (both formation and resorption), and increased activation of vitamin D leading to increased intestinal absorption of calcium. High calcium intake reverses these changes.
Reid IR, Bristow SM, Bolland MJ. Calcium supplements: benefits and risks. J Intern Med 2015; 278(4):354–368. Copyright 2015, The Association for the Publication of the Journal of Internal Medicine.
Figure 1. Absolute change in total body bone mineral content (BMC) over 5 years in normal postmenopausal women, as a function of each woman’s average calcium intake assessed at baseline and at year 5. The lines show the regression (with 95% confidence intervals) for this relationship (P = .53)
Thus, a normal serum calcium concentration can be maintained with calcium intake ranging from 200 to more than 2,000 mg/day, and rates of bone loss in postmenopausal women are unaffected by calcium intake (Figure 1).2
If calcium intake is very low, hypocalcemia and secondary hyperparathyroidism develop,3 and bone mineralization may be impaired. However, levels of calcium intake in Africa and in East and Southeast Asia are typically less than 400 mg/day,4 yet there is no evidence that these levels adversely affect skeletal health. In fact, fracture risk is lower in these regions than in North America, where calcium intake is several times greater.
Thus, some calcium intake is required to maintain circulating concentrations, but there is no mechanism by which high calcium intake can drive bone formation. Quite the opposite, in fact.
Vitamin D deficiency has little relationship with diet
Vitamin D is a biologically inactive secosteroid activated by hydroxylation in the liver and kidney to function as the key regulator of intestinal calcium absorption. As with calcium, its deficiency results in hypocalcemia and impaired bone mineralization.
Paradoxically, high levels of vitamin D stimulate bone resorption and inhibit bone mineralization in mice,5 and large doses increase bone resorption markers acutely in clinical studies.6 Thus, it is important to ensure an adequate vitamin D supply, but not an oversupply.
In the absence of supplements, most vitamin D is produced in the skin as a result of the action of ultraviolet light (from sunlight) on 7-dehydrocholesterol. Thus, vitamin D deficiency occurs in those deprived of skin exposure to sunlight (eg, due to veiling, living at high latitude, staying permanently indoors), but it has little relationship with diet.
ARE CALCIUM SUPPLEMENTS EFFECTIVE?
Calcium supplements are certainly biologically active. They transiently increase serum calcium concentrations, suppress parathyroid hormone, and reduce bone resorption.2 In the first year of use, they increase bone density by about 1% compared with placebo.7 However, longer use does not result in further bone density advantage over placebo,7 suggesting that the response simply reflects a decreased number of osteoclastic resorption sites and does not indicate a sustained change in bone balance.
A 1% difference in bone density would not be expected to reduce fracture risk, and a number of large, carefully conducted randomized controlled trials published over the last 15 years have failed to demonstrate antifracture efficacy for calcium.8–12 As a result, the US Preventive Services Task Force recommends against the routine use of calcium supplements in community-dwelling adults.13
In contrast, in a placebo-controlled trial published in 1992, Chapuy et al14 found that elderly women residing in nursing homes who received calcium and vitamin D supplements had fewer fractures. At 18 months, by intention-to-treat analysis, nonvertebral fractures had occurred in 160 (12%) of 1,387 women in the supplement group compared with 215 (15%) of 1,403 women in the placebo group (P < .001). However, these women were severely vitamin D-deficient (the mean serum 25-hydroxyvitamin D level at baseline in the placebo group was 13 ng/mL, normal range 15–50), to the extent that many must have had osteomalacia.
Thus, this study shows that calcium and vitamin D are effective in managing osteomalacia, but the subsequent trials8–12 did not observe any benefit in community-dwelling cohorts. Meta-analyses that pool the Chapuy study with community-based studies generally find that calcium with vitamin D is beneficial, but the heterogeneity of these populations means that such pooling is inappropriate.15
It is sometimes stated that calcium and vitamin D should always be given with osteoporosis medications because the efficacy of these drugs has only been demonstrated when coadministered with these supplements. This is incorrect. The addition of calcium to alendronate does not alter its effects on bone density,16 and the antifracture efficacy of both bisphosphonates17 and estrogen18,19 has been demonstrated in the absence of supplementation with calcium or vitamin D. The evidence that bisphosphonates prevent fractures in the absence of calcium supplements has recently been strengthened by the results of a randomized controlled trial comparing zoledronate with placebo in women over age 65 with osteopenia.20
ARE CALCIUM SUPPLEMENTS SAFE?
Calcium supplements often cause gastrointestinal symptoms, particularly constipation. They have been shown to double the risk of hospital admission due to abdominal symptoms.21 In the absence of clear evidence of benefit, these facts alone should militate against their routine use. Calcium supplements also cause hypercalcemia and hypercalciuria22 and increase the risk of renal calculi (by 17% in the Women’s Health Initiative8).
Over the last decade, evidence has emerged that calcium supplements may also increase the risk of myocardial infarction, and possibly stroke. This finding was not statistically significant in any single study, but is consistently present in meta-analyses.23
Evidence from the Women’s Health Initiative
When studies of calcium with vitamin D are added to these meta-analyses, the results are less consistent. This is because such meta-analyses are dominated by the Women's Health Initiative (because of its large size, with 36,282 participants). There have been 2 different analyses of this trial with respect to cardiovascular events.
When the Women’s Health Initiative as a whole was analyzed, there was no significant effect of calcium plus vitamin D on vascular end points. However, there is a significant interaction between body mass index and the effect of supplements, such that nonobese women demonstrated a 17% increase in myocardial infarction.24 This study was unusual in that it included women already taking calcium and vitamin D supplements.
Bolland MJ, Grey A, Avenell A, Gamble GD, Reid IR. Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women’s Health Initiative limited access dataset and meta-analysis. BMJ 2011; 342:d2040.
Figure 2. Effect of calcium supplements on cardiovascular events, with or without vitamin D. Data for 28,072 participants in 8 trials of calcium supplements with trial-level data, plus data for Women’s Health Initiative CaD study participants not taking calcium supplements at baseline.
There was a significant interaction between baseline use of supplements and the effects of the trial intervention on vascular events, justifying analyzing the supplement-naive individuals separately. In this group of 16,000 women, an increase in clinical myocardial infarction of 22% was found, similar to the findings with calcium supplements alone.25
Thus, there is consistent evidence that introducing a calcium supplement de novo increases the risk of myocardial infarction (Figure 2).16,25–31 We calculate that treating 1,000 patients with calcium or calcium plus vitamin D for 5 years would cause an additional 6 myocardial infarctions or strokes (number needed to harm 178) and prevent only 3 fractures (number needed to treat 302).25
ARE VITAMIN D SUPPLEMENTS EFFECTIVE?
Vitamin D is highly effective in treating osteomalacia, improving symptoms within days and increasing bone density by as much as 50% over 1 year.32,33 In contrast, randomized controlled trials of vitamin D supplements alone in people without osteomalacia have not shown increases in bone density or changes in fracture risk.34–37
Reid IR, Horne AM, Mihov B, et al. Effect of monthly high-dose vitamin D on bone density in community-dwelling older adults substudy of a randomized controlled trial. J Intern Med 2017; 282(5):452–460. Copyright 2017, Assoc for Publication of J Int Med
Figure 3. Changes in bone mineral density (BMD) from baseline to 2 years in the vitamin D and placebo groups of the Vitamin D Assessment study, according to baseline serum 25(OH)D (25-hydroxyvitamin D) concentrations. Data are mean ± 95% confidence intervals. P values are shown for between-group comparisons.
In 2017, my colleagues and I published a trial showing that vitamin D supplementation increases bone density by 2% to 3% in the spine and femoral neck in participants with baseline 25-hydroxyvitamin D levels below 30- nmol/L (12 ng/mL), but those starting above this level showed no effect (Figure 3).38 And a reanalysis of an earlier study confirmed this 30 nmol/L threshold for an effect of vitamin D on bone density.39 The finding of a clear-cut threshold for vitamin D effects is predicted by the physiologic considerations set out above.
Belief that higher levels of 25-hydroxyvitamin D are better is based on observational data. However, correlation does not prove causation, and it is likely that causation is reversed here. Those with better health are likely to spend more time exercising outdoors, are less likely to be obese, and are less likely to have inflammatory conditions; and as a result, they are more likely to have better vitamin D status. We should now be using trial-based definitions of vitamin D deficiency as opposed to thresholds derived from disease associations in observational studies.
Vitamin D supplements have also been suggested to benefit cardiovascular health and to reduce cancer risk, though current clinical trial data provide no support for these hypotheses.36,40 Other trials addressing these questions are ongoing.
ARE VITAMIN D SUPPLEMENTS SAFE?
The safety of vitamin D supplements has generally been assessed with respect to the incidence of hypercalcemia. On this basis, very high doses have been promoted. However, there is now evidence that doses of 4,000 IU/day, 60,000 IU/month, and 500,000 IU/year increase the risk of falls and fractures.41,42
The threshold for bone benefits discussed above (12 ng/mL) is easily exceeded with doses of vitamin D of 400 to 1,000 IU/day. At these levels, vitamin D supplements have no known adverse effects and can be widely endorsed for individuals at risk of deficiency. Supplement doses greater than 2,000 IU/day should be used only in exceptional circumstances, and with appropriate monitoring.
LITTLE USE FOR CALCIUM AND VITAMIN D SUPPLEMENTS
Extensive clinical trials have failed to demonstrate meaningful benefit from calcium supplements in the management of osteoporosis. Calcium supplements are often prescribed in patients who are receiving other treatments for osteoporosis, which may be justified with interventions that have the potential to cause hypocalcemia, but their coadministration with bisphosphonates has been shown to be unnecessary.
Calcium supplements commonly cause gastrointestinal symptoms that are sometimes severe and are likely to contribute to high levels of noncompliance with osteoporosis medications. They increase the risk of kidney stones,8 and there is reasonable evidence to suggest an adverse effect on vascular risk as well.23
Vitamin D deficiency is common in frail elderly people, particularly those with dark skin or living at high latitudes. Low doses of vitamin D are safe and highly effective in preventing osteomalacia. But vitamin D supplements are unnecessary in those who regularly have sun exposure. And high doses of vitamin D have no demonstrated advantage and have been shown to increase the risk of falls and fractures.
Our decision to prescribe calcium and vitamin D supplements should be based on evidence that is of the same quality as for any other intervention we prescribe. Current evidence suggests that there is little reason to prescribe calcium, and that vitamin D should be targeted at those at risk of 25-hydroxyvitamin D levels less than 12 ng/mL.
We live in the era of evidence-based medicine, so new interventions must meet criteria for both safety and efficacy before they are adopted. However, we have inherited many practices adopted before the current standards were in place, and we have not always been rigorous in reevaluating traditional remedies. A conservative belief in established practice or the influence of vested interests may account for this lack of rigor in reappraisal.1 Calcium and vitamin D supplements are possible examples of this phenomenon.
BONE METABOLISM IS TIGHTLY REGULATED
Bone is a connective tissue, its matrix composed principally of type 1 collagen, which provides tensile strength. Hydroxyapatite crystals, composed predominantly of calcium and phosphate, lie between the collagen fibers and provide compressive strength. In a tightly regulated process, osteoblasts lay down the collagenous matrix, and osteoclasts remove it. Mineralization of newly formed bone proceeds if normal levels of extracellular calcium and phosphate are present, in the absence of inhibitors of mineralization.
High calcium intake does not drive bone formation
The endocrine system is critical in maintaining normocalcemia. A decrease in calcium intake results in increased parathyroid hormone secretion, resulting in increased renal tubular calcium reabsorption, increased bone turnover (both formation and resorption), and increased activation of vitamin D leading to increased intestinal absorption of calcium. High calcium intake reverses these changes.
Reid IR, Bristow SM, Bolland MJ. Calcium supplements: benefits and risks. J Intern Med 2015; 278(4):354–368. Copyright 2015, The Association for the Publication of the Journal of Internal Medicine.
Figure 1. Absolute change in total body bone mineral content (BMC) over 5 years in normal postmenopausal women, as a function of each woman’s average calcium intake assessed at baseline and at year 5. The lines show the regression (with 95% confidence intervals) for this relationship (P = .53)
Thus, a normal serum calcium concentration can be maintained with calcium intake ranging from 200 to more than 2,000 mg/day, and rates of bone loss in postmenopausal women are unaffected by calcium intake (Figure 1).2
If calcium intake is very low, hypocalcemia and secondary hyperparathyroidism develop,3 and bone mineralization may be impaired. However, levels of calcium intake in Africa and in East and Southeast Asia are typically less than 400 mg/day,4 yet there is no evidence that these levels adversely affect skeletal health. In fact, fracture risk is lower in these regions than in North America, where calcium intake is several times greater.
Thus, some calcium intake is required to maintain circulating concentrations, but there is no mechanism by which high calcium intake can drive bone formation. Quite the opposite, in fact.
Vitamin D deficiency has little relationship with diet
Vitamin D is a biologically inactive secosteroid activated by hydroxylation in the liver and kidney to function as the key regulator of intestinal calcium absorption. As with calcium, its deficiency results in hypocalcemia and impaired bone mineralization.
Paradoxically, high levels of vitamin D stimulate bone resorption and inhibit bone mineralization in mice,5 and large doses increase bone resorption markers acutely in clinical studies.6 Thus, it is important to ensure an adequate vitamin D supply, but not an oversupply.
In the absence of supplements, most vitamin D is produced in the skin as a result of the action of ultraviolet light (from sunlight) on 7-dehydrocholesterol. Thus, vitamin D deficiency occurs in those deprived of skin exposure to sunlight (eg, due to veiling, living at high latitude, staying permanently indoors), but it has little relationship with diet.
ARE CALCIUM SUPPLEMENTS EFFECTIVE?
Calcium supplements are certainly biologically active. They transiently increase serum calcium concentrations, suppress parathyroid hormone, and reduce bone resorption.2 In the first year of use, they increase bone density by about 1% compared with placebo.7 However, longer use does not result in further bone density advantage over placebo,7 suggesting that the response simply reflects a decreased number of osteoclastic resorption sites and does not indicate a sustained change in bone balance.
A 1% difference in bone density would not be expected to reduce fracture risk, and a number of large, carefully conducted randomized controlled trials published over the last 15 years have failed to demonstrate antifracture efficacy for calcium.8–12 As a result, the US Preventive Services Task Force recommends against the routine use of calcium supplements in community-dwelling adults.13
In contrast, in a placebo-controlled trial published in 1992, Chapuy et al14 found that elderly women residing in nursing homes who received calcium and vitamin D supplements had fewer fractures. At 18 months, by intention-to-treat analysis, nonvertebral fractures had occurred in 160 (12%) of 1,387 women in the supplement group compared with 215 (15%) of 1,403 women in the placebo group (P < .001). However, these women were severely vitamin D-deficient (the mean serum 25-hydroxyvitamin D level at baseline in the placebo group was 13 ng/mL, normal range 15–50), to the extent that many must have had osteomalacia.
Thus, this study shows that calcium and vitamin D are effective in managing osteomalacia, but the subsequent trials8–12 did not observe any benefit in community-dwelling cohorts. Meta-analyses that pool the Chapuy study with community-based studies generally find that calcium with vitamin D is beneficial, but the heterogeneity of these populations means that such pooling is inappropriate.15
It is sometimes stated that calcium and vitamin D should always be given with osteoporosis medications because the efficacy of these drugs has only been demonstrated when coadministered with these supplements. This is incorrect. The addition of calcium to alendronate does not alter its effects on bone density,16 and the antifracture efficacy of both bisphosphonates17 and estrogen18,19 has been demonstrated in the absence of supplementation with calcium or vitamin D. The evidence that bisphosphonates prevent fractures in the absence of calcium supplements has recently been strengthened by the results of a randomized controlled trial comparing zoledronate with placebo in women over age 65 with osteopenia.20
ARE CALCIUM SUPPLEMENTS SAFE?
Calcium supplements often cause gastrointestinal symptoms, particularly constipation. They have been shown to double the risk of hospital admission due to abdominal symptoms.21 In the absence of clear evidence of benefit, these facts alone should militate against their routine use. Calcium supplements also cause hypercalcemia and hypercalciuria22 and increase the risk of renal calculi (by 17% in the Women’s Health Initiative8).
Over the last decade, evidence has emerged that calcium supplements may also increase the risk of myocardial infarction, and possibly stroke. This finding was not statistically significant in any single study, but is consistently present in meta-analyses.23
Evidence from the Women’s Health Initiative
When studies of calcium with vitamin D are added to these meta-analyses, the results are less consistent. This is because such meta-analyses are dominated by the Women's Health Initiative (because of its large size, with 36,282 participants). There have been 2 different analyses of this trial with respect to cardiovascular events.
When the Women’s Health Initiative as a whole was analyzed, there was no significant effect of calcium plus vitamin D on vascular end points. However, there is a significant interaction between body mass index and the effect of supplements, such that nonobese women demonstrated a 17% increase in myocardial infarction.24 This study was unusual in that it included women already taking calcium and vitamin D supplements.
Bolland MJ, Grey A, Avenell A, Gamble GD, Reid IR. Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women’s Health Initiative limited access dataset and meta-analysis. BMJ 2011; 342:d2040.
Figure 2. Effect of calcium supplements on cardiovascular events, with or without vitamin D. Data for 28,072 participants in 8 trials of calcium supplements with trial-level data, plus data for Women’s Health Initiative CaD study participants not taking calcium supplements at baseline.
There was a significant interaction between baseline use of supplements and the effects of the trial intervention on vascular events, justifying analyzing the supplement-naive individuals separately. In this group of 16,000 women, an increase in clinical myocardial infarction of 22% was found, similar to the findings with calcium supplements alone.25
Thus, there is consistent evidence that introducing a calcium supplement de novo increases the risk of myocardial infarction (Figure 2).16,25–31 We calculate that treating 1,000 patients with calcium or calcium plus vitamin D for 5 years would cause an additional 6 myocardial infarctions or strokes (number needed to harm 178) and prevent only 3 fractures (number needed to treat 302).25
ARE VITAMIN D SUPPLEMENTS EFFECTIVE?
Vitamin D is highly effective in treating osteomalacia, improving symptoms within days and increasing bone density by as much as 50% over 1 year.32,33 In contrast, randomized controlled trials of vitamin D supplements alone in people without osteomalacia have not shown increases in bone density or changes in fracture risk.34–37
Reid IR, Horne AM, Mihov B, et al. Effect of monthly high-dose vitamin D on bone density in community-dwelling older adults substudy of a randomized controlled trial. J Intern Med 2017; 282(5):452–460. Copyright 2017, Assoc for Publication of J Int Med
Figure 3. Changes in bone mineral density (BMD) from baseline to 2 years in the vitamin D and placebo groups of the Vitamin D Assessment study, according to baseline serum 25(OH)D (25-hydroxyvitamin D) concentrations. Data are mean ± 95% confidence intervals. P values are shown for between-group comparisons.
In 2017, my colleagues and I published a trial showing that vitamin D supplementation increases bone density by 2% to 3% in the spine and femoral neck in participants with baseline 25-hydroxyvitamin D levels below 30- nmol/L (12 ng/mL), but those starting above this level showed no effect (Figure 3).38 And a reanalysis of an earlier study confirmed this 30 nmol/L threshold for an effect of vitamin D on bone density.39 The finding of a clear-cut threshold for vitamin D effects is predicted by the physiologic considerations set out above.
Belief that higher levels of 25-hydroxyvitamin D are better is based on observational data. However, correlation does not prove causation, and it is likely that causation is reversed here. Those with better health are likely to spend more time exercising outdoors, are less likely to be obese, and are less likely to have inflammatory conditions; and as a result, they are more likely to have better vitamin D status. We should now be using trial-based definitions of vitamin D deficiency as opposed to thresholds derived from disease associations in observational studies.
Vitamin D supplements have also been suggested to benefit cardiovascular health and to reduce cancer risk, though current clinical trial data provide no support for these hypotheses.36,40 Other trials addressing these questions are ongoing.
ARE VITAMIN D SUPPLEMENTS SAFE?
The safety of vitamin D supplements has generally been assessed with respect to the incidence of hypercalcemia. On this basis, very high doses have been promoted. However, there is now evidence that doses of 4,000 IU/day, 60,000 IU/month, and 500,000 IU/year increase the risk of falls and fractures.41,42
The threshold for bone benefits discussed above (12 ng/mL) is easily exceeded with doses of vitamin D of 400 to 1,000 IU/day. At these levels, vitamin D supplements have no known adverse effects and can be widely endorsed for individuals at risk of deficiency. Supplement doses greater than 2,000 IU/day should be used only in exceptional circumstances, and with appropriate monitoring.
LITTLE USE FOR CALCIUM AND VITAMIN D SUPPLEMENTS
Extensive clinical trials have failed to demonstrate meaningful benefit from calcium supplements in the management of osteoporosis. Calcium supplements are often prescribed in patients who are receiving other treatments for osteoporosis, which may be justified with interventions that have the potential to cause hypocalcemia, but their coadministration with bisphosphonates has been shown to be unnecessary.
Calcium supplements commonly cause gastrointestinal symptoms that are sometimes severe and are likely to contribute to high levels of noncompliance with osteoporosis medications. They increase the risk of kidney stones,8 and there is reasonable evidence to suggest an adverse effect on vascular risk as well.23
Vitamin D deficiency is common in frail elderly people, particularly those with dark skin or living at high latitudes. Low doses of vitamin D are safe and highly effective in preventing osteomalacia. But vitamin D supplements are unnecessary in those who regularly have sun exposure. And high doses of vitamin D have no demonstrated advantage and have been shown to increase the risk of falls and fractures.
Our decision to prescribe calcium and vitamin D supplements should be based on evidence that is of the same quality as for any other intervention we prescribe. Current evidence suggests that there is little reason to prescribe calcium, and that vitamin D should be targeted at those at risk of 25-hydroxyvitamin D levels less than 12 ng/mL.
References
Grey A, Bolland M. Web of industry, advocacy, and academia in the management of osteoporosis. BMJ 2015; 351:h3170. doi:10.1136/bmj.h3170
Reid IR, Bristow SM, Bolland MJ. Calcium supplements: benefits and risks. J Intern Med 2015; 278(4):354–368. doi:10.1111/joim.12394
Bolland MJ, Grey AB, Ames RW, Horne AM, Gamble GD, Reid IR. Fat mass is an important predictor of parathyroid hormone levels in postmenopausal women. Bone 2006; 38(3):317–321. doi:10.1016/j.bone.2005.08.018
Lieben L, Masuyama R, Torrekens S, et al. Normocalcemia is maintained in mice under conditions of calcium malabsorption by vitamin D-induced inhibition of bone mineralization. J Clin Invest 2012; 122(5):1803–1815. doi:10.1172/JCI45890
Rossini M, Gatti D, Viapiana O, et al. Short-term effects on bone turnover markers of a single high dose of oral vitamin D3. J Clin Endocrinol Metab 2012; 97(4):E622–E626. doi:10.1210/jc.2011-2448
Tai V, Leung W, Grey A, Reid IR, Bolland MJ. Calcium intake and bone mineral density: systematic review and meta-analysis. BMJ 2015; 351:h4183. doi:10.1136/bmj.h4183
Jackson RD, LaCroix AZ, Gass M, et al; Women’s Health Initiative Investigators. Calcium plus vitamin D supplementation and the risk of fractures. N Engl J Med 2006; 354(7):669–683. doi:10.1056/NEJMoa055218
Grant AM, Avenell A, Campbell MK, et al; RECORD Trial Group. Oral vitamin D3 and calcium for secondary prevention of low-trauma fractures in elderly people (Randomised Evaluation of Calcium or vitamin D, RECORD): a randomised placebo-controlled trial. Lancet 2005; 365(9471):1621–1628. doi:10.1016/S0140-6736(05)63013-9
Prince RL, Devine A, Dhaliwal SS, Dick IM. Effects of calcium supplementation on clinical fracture and bone structure: results of a 5-year, double-blind, placebo-controlled trial in elderly women. Arch Intern Med 2006; 166(8):869–875. doi:10.1001/archinte.166.8.869
Reid IR, Mason B, Horne A, et al. Randomized controlled trial of calcium in healthy older women. Am J Med 2006; 119(9):777–785. doi:10.1016/j.amjmed.2006.02.038
Salovaara K, Tuppurainen M, Karkkainen M, et al. Effect of vitamin D-3 and calcium on fracture risk in 65-to 71-year-old women: a population-based 3-year randomized, controlled trial—the OSTPRE-FPS. J Bone Miner Res 2010; 25(7):1487–1495. doi:10.1002/jbmr.48
Moyer VA, US Preventive Services Task Force. Vitamin D and calcium supplementation to prevent fractures in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2013; 158(9):691–696. doi:10.7326/0003-4819-158-9-201305070-00603
Chapuy MC, Arlot ME, Duboeuf F, et al. Vitamin D3 and calcium to prevent hip fractures in elderly women. N Engl J Med 1992; 327(23):1637–1642. doi:10.1056/NEJM199212033272305
Tang BMP, Eslick GD, Nowson C, Smith C, Bensoussan A. Use of calcium or calcium in combination with vitamin D supplementation to prevent fractures and bone loss in people aged 50 years and older: a meta-analysis. Lancet 2007; 370(9588):657–666. doi:10.1016/S0140-6736(07)61342-7
Bonnick S, Broy S, Kaiser F, et al. Treatment with alendronate plus calcium, alendronate alone, or calcium alone for postmenopausal low bone mineral density. Curr Med Res Opin 2007; 23(6):1341–1349. doi:10.1185/030079907X188035
McCloskey EV, Beneton M, Charlesworth D, et al. Clodronate reduces the incidence of fractures in community-dwelling elderly women unselected for osteoporosis: results of a double-blind, placebo-controlled randomized study. J Bone Miner Res 2007; 22(1):135–141. doi:10.1359/jbmr.061008
Lindsay R, Hart DM, Forrest C, Baird C. Prevention of spinal osteoporosis in oophorectomised women. Lancet 1980; 2(8205):1151–1154. pmid:6107766
Cauley JA, Robbins J, Chen Z, et al; Women’s Health Initiative Investigators. Effects of estrogen plus progestin on risk of fracture and bone mineral density: the Women’s Health Initiative randomized trial. JAMA 2003; 290(13):1729–1738. doi:10.1001/jama.290.13.1729
Reid I, Horne A, Mihov B, et al. Abstracts of the ECTS Congress 2018: Zoledronate every 18 months for 6 years in osteopenic postmenopausal women reduces non-vertebral fractures and height loss. Calcif Tissue Int 2018; 102:S1-S159. doi:10.1007/s00223-018-0418-0
Lewis JR, Zhu K, Prince RL. Adverse events from calcium supplementation: relationship to errors in myocardial infarction self-reporting in randomized controlled trials of calcium supplementation. J Bone Miner Res 2012; 27(3):719–722. doi:10.1002/jbmr.1484
Gallagher JC, Smith LM, Yalamanchili V. Incidence of hypercalciuria and hypercalcemia during vitamin D and calcium supplementation in older women. Menopause 2014; 21(11):1173–1180. doi:10.1097/GME.0000000000000270
Reid IR, Bristow SM, Bolland MJ. Calcium and cardiovascular disease. Endocrinol Metab (Seoul) 2017; 32(3):339–349. doi:10.3803/EnM.2017.32.3.339
Hsia J, Heiss G, Ren H, et al; Women’s Health Initiative Investigators. Calcium/vitamin D supplementation and cardiovascular events. Circulation 2007; 115(7):846–854. doi:10.1161/CIRCULATIONAHA.106.673491
Bolland MJ, Grey A, Avenell A, Gamble GD, Reid IR. Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women’s Health Initiative limited access dataset and meta-analysis. BMJ 2011; 342:d2040. doi:10.1136/bmj.d2040
Baron JA, Beach M, Mandel JS, et al. Calcium supplements for the prevention of colorectal adenomas. Calcium Polyp Prevention Study Group. N Engl J Med 1999; 340(3):101–107. doi:10.1056/NEJM199901143400204
Bolland MJ, Barber PA, Doughty RN, et al. Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ 2008; 336(7638):262–266. doi:10.1136/bmj.39440.525752.BE
Lappe JM, Travers-Gustafson D, Davies KM, Recker RR, Heaney RP. Vitamin D and calcium supplementation reduces cancer risk: results of a randomized trial. Am J Clin Nutr 2007; 85(6):1586–1591. doi:10.1093/ajcn/85.6.1586
Reid IR, Ames R, Mason B, et al. Randomized controlled trial of calcium supplementation in healthy, non-osteoporotic, older men. Arch Intern Med 2008; 168(20):2276–2282. doi:10.1001/archinte.168.20.2276
Reid IR, Ames RW, Evans MC,Gamble GD, Sharpe SJ. Effect of calcium supplementation on bone loss in postmenopausal women. N Engl J Med 1993; 328(7):460–464. doi:10.1056/NEJM199302183280702
Reid IR, Ames RW, Evans MC, Gamble GD, Sharpe SJ. Long-term effects of calcium supplementation on bone loss and fractures in postmenopausal women: a randomized controlled trial. Am J Med 1995; 98(4):331–335. doi:10.1016/S0002-9343(99)80310-6
Al-Ali H, Fuleihan GE. Nutritional osteomalacia: substantial clinical improvement and gain in bone density posttherapy. J Clin Densitom 2000; 3(1):97–101. pmid:10745306
El-Desouki MI, Othman SM, Fouda MA. Bone mineral density and bone scintigraphy in adult Saudi female patients with osteomalacia. Saudi Med J 2004; 25(3):355–358.
Reid IR, Bolland MJ, Grey A. Effects of vitamin D supplements on bone mineral density: a systematic review and meta-analysis. Lancet 2014; 383(9912):146–155. doi:10.1016/S0140-6736(13)61647-5
Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev 2014; (4):CD000227. doi:10.1002/14651858.CD000227.pub4
Bolland MJ, Grey A, Gamble GD, Reid IR. The effect of vitamin D supplementation on skeletal, vascular, or cancer outcomes: a trial sequential meta-analysis. Lancet Diabetes Endocrinol 2014; 2(4):307–320. doi:10.1016/S2213-8587(13)70212-2
DIPART (Vitamin D Individual Patient Analysis of Randomized Trials) Group. Patient level pooled analysis of 68 500 patients from seven major vitamin D fracture trials in US and Europe. BMJ 2010; 340:b5463. doi:10.1136/bmj.b5463
Reid IR, Horne AM, Mihov B, et al. Effect of monthly high-dose vitamin D on bone density in community-dwelling older adults substudy of a randomized controlled trial. J Intern Med 2017; 282(5):452–460. doi:10.1111/joim.12651
MacDonald HM, Reid IR, Gamble GD, Fraser WD, Tang JC, Wood AD. 25-Hydroxyvitamin D threshold for the effects of vitamin D supplements on bone density secondary analysis of a randomized controlled trial. J Bone Miner Res 2018. Epub ahead of print. doi:10.1002/jbmr.3442
Scragg R, Stewart AW, Waayer D, et al. Effect of monthly high-dose vitamin D supplementation on cardiovascular disease in the vitamin D assessment study: a randomized clinical trial. JAMA Cardiol 2017; 2(6):608–616. doi:10.1001/jamacardio.2017.0175
Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA 2010; 303(18):1815–1822. doi:10.1001/jama.2010.594
Smith LM, Gallagher JC, Suiter C. Medium doses of daily vitamin D decrease falls and higher doses of daily vitamin D3 increase falls: a randomized clinical trial. J Steroid Biochem Mol Biol 2017; 173:317–322. doi:10.1016/j.jsbmb.2017.03.015
References
Grey A, Bolland M. Web of industry, advocacy, and academia in the management of osteoporosis. BMJ 2015; 351:h3170. doi:10.1136/bmj.h3170
Reid IR, Bristow SM, Bolland MJ. Calcium supplements: benefits and risks. J Intern Med 2015; 278(4):354–368. doi:10.1111/joim.12394
Bolland MJ, Grey AB, Ames RW, Horne AM, Gamble GD, Reid IR. Fat mass is an important predictor of parathyroid hormone levels in postmenopausal women. Bone 2006; 38(3):317–321. doi:10.1016/j.bone.2005.08.018
Lieben L, Masuyama R, Torrekens S, et al. Normocalcemia is maintained in mice under conditions of calcium malabsorption by vitamin D-induced inhibition of bone mineralization. J Clin Invest 2012; 122(5):1803–1815. doi:10.1172/JCI45890
Rossini M, Gatti D, Viapiana O, et al. Short-term effects on bone turnover markers of a single high dose of oral vitamin D3. J Clin Endocrinol Metab 2012; 97(4):E622–E626. doi:10.1210/jc.2011-2448
Tai V, Leung W, Grey A, Reid IR, Bolland MJ. Calcium intake and bone mineral density: systematic review and meta-analysis. BMJ 2015; 351:h4183. doi:10.1136/bmj.h4183
Jackson RD, LaCroix AZ, Gass M, et al; Women’s Health Initiative Investigators. Calcium plus vitamin D supplementation and the risk of fractures. N Engl J Med 2006; 354(7):669–683. doi:10.1056/NEJMoa055218
Grant AM, Avenell A, Campbell MK, et al; RECORD Trial Group. Oral vitamin D3 and calcium for secondary prevention of low-trauma fractures in elderly people (Randomised Evaluation of Calcium or vitamin D, RECORD): a randomised placebo-controlled trial. Lancet 2005; 365(9471):1621–1628. doi:10.1016/S0140-6736(05)63013-9
Prince RL, Devine A, Dhaliwal SS, Dick IM. Effects of calcium supplementation on clinical fracture and bone structure: results of a 5-year, double-blind, placebo-controlled trial in elderly women. Arch Intern Med 2006; 166(8):869–875. doi:10.1001/archinte.166.8.869
Reid IR, Mason B, Horne A, et al. Randomized controlled trial of calcium in healthy older women. Am J Med 2006; 119(9):777–785. doi:10.1016/j.amjmed.2006.02.038
Salovaara K, Tuppurainen M, Karkkainen M, et al. Effect of vitamin D-3 and calcium on fracture risk in 65-to 71-year-old women: a population-based 3-year randomized, controlled trial—the OSTPRE-FPS. J Bone Miner Res 2010; 25(7):1487–1495. doi:10.1002/jbmr.48
Moyer VA, US Preventive Services Task Force. Vitamin D and calcium supplementation to prevent fractures in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2013; 158(9):691–696. doi:10.7326/0003-4819-158-9-201305070-00603
Chapuy MC, Arlot ME, Duboeuf F, et al. Vitamin D3 and calcium to prevent hip fractures in elderly women. N Engl J Med 1992; 327(23):1637–1642. doi:10.1056/NEJM199212033272305
Tang BMP, Eslick GD, Nowson C, Smith C, Bensoussan A. Use of calcium or calcium in combination with vitamin D supplementation to prevent fractures and bone loss in people aged 50 years and older: a meta-analysis. Lancet 2007; 370(9588):657–666. doi:10.1016/S0140-6736(07)61342-7
Bonnick S, Broy S, Kaiser F, et al. Treatment with alendronate plus calcium, alendronate alone, or calcium alone for postmenopausal low bone mineral density. Curr Med Res Opin 2007; 23(6):1341–1349. doi:10.1185/030079907X188035
McCloskey EV, Beneton M, Charlesworth D, et al. Clodronate reduces the incidence of fractures in community-dwelling elderly women unselected for osteoporosis: results of a double-blind, placebo-controlled randomized study. J Bone Miner Res 2007; 22(1):135–141. doi:10.1359/jbmr.061008
Lindsay R, Hart DM, Forrest C, Baird C. Prevention of spinal osteoporosis in oophorectomised women. Lancet 1980; 2(8205):1151–1154. pmid:6107766
Cauley JA, Robbins J, Chen Z, et al; Women’s Health Initiative Investigators. Effects of estrogen plus progestin on risk of fracture and bone mineral density: the Women’s Health Initiative randomized trial. JAMA 2003; 290(13):1729–1738. doi:10.1001/jama.290.13.1729
Reid I, Horne A, Mihov B, et al. Abstracts of the ECTS Congress 2018: Zoledronate every 18 months for 6 years in osteopenic postmenopausal women reduces non-vertebral fractures and height loss. Calcif Tissue Int 2018; 102:S1-S159. doi:10.1007/s00223-018-0418-0
Lewis JR, Zhu K, Prince RL. Adverse events from calcium supplementation: relationship to errors in myocardial infarction self-reporting in randomized controlled trials of calcium supplementation. J Bone Miner Res 2012; 27(3):719–722. doi:10.1002/jbmr.1484
Gallagher JC, Smith LM, Yalamanchili V. Incidence of hypercalciuria and hypercalcemia during vitamin D and calcium supplementation in older women. Menopause 2014; 21(11):1173–1180. doi:10.1097/GME.0000000000000270
Reid IR, Bristow SM, Bolland MJ. Calcium and cardiovascular disease. Endocrinol Metab (Seoul) 2017; 32(3):339–349. doi:10.3803/EnM.2017.32.3.339
Hsia J, Heiss G, Ren H, et al; Women’s Health Initiative Investigators. Calcium/vitamin D supplementation and cardiovascular events. Circulation 2007; 115(7):846–854. doi:10.1161/CIRCULATIONAHA.106.673491
Bolland MJ, Grey A, Avenell A, Gamble GD, Reid IR. Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women’s Health Initiative limited access dataset and meta-analysis. BMJ 2011; 342:d2040. doi:10.1136/bmj.d2040
Baron JA, Beach M, Mandel JS, et al. Calcium supplements for the prevention of colorectal adenomas. Calcium Polyp Prevention Study Group. N Engl J Med 1999; 340(3):101–107. doi:10.1056/NEJM199901143400204
Bolland MJ, Barber PA, Doughty RN, et al. Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ 2008; 336(7638):262–266. doi:10.1136/bmj.39440.525752.BE
Lappe JM, Travers-Gustafson D, Davies KM, Recker RR, Heaney RP. Vitamin D and calcium supplementation reduces cancer risk: results of a randomized trial. Am J Clin Nutr 2007; 85(6):1586–1591. doi:10.1093/ajcn/85.6.1586
Reid IR, Ames R, Mason B, et al. Randomized controlled trial of calcium supplementation in healthy, non-osteoporotic, older men. Arch Intern Med 2008; 168(20):2276–2282. doi:10.1001/archinte.168.20.2276
Reid IR, Ames RW, Evans MC,Gamble GD, Sharpe SJ. Effect of calcium supplementation on bone loss in postmenopausal women. N Engl J Med 1993; 328(7):460–464. doi:10.1056/NEJM199302183280702
Reid IR, Ames RW, Evans MC, Gamble GD, Sharpe SJ. Long-term effects of calcium supplementation on bone loss and fractures in postmenopausal women: a randomized controlled trial. Am J Med 1995; 98(4):331–335. doi:10.1016/S0002-9343(99)80310-6
Al-Ali H, Fuleihan GE. Nutritional osteomalacia: substantial clinical improvement and gain in bone density posttherapy. J Clin Densitom 2000; 3(1):97–101. pmid:10745306
El-Desouki MI, Othman SM, Fouda MA. Bone mineral density and bone scintigraphy in adult Saudi female patients with osteomalacia. Saudi Med J 2004; 25(3):355–358.
Reid IR, Bolland MJ, Grey A. Effects of vitamin D supplements on bone mineral density: a systematic review and meta-analysis. Lancet 2014; 383(9912):146–155. doi:10.1016/S0140-6736(13)61647-5
Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev 2014; (4):CD000227. doi:10.1002/14651858.CD000227.pub4
Bolland MJ, Grey A, Gamble GD, Reid IR. The effect of vitamin D supplementation on skeletal, vascular, or cancer outcomes: a trial sequential meta-analysis. Lancet Diabetes Endocrinol 2014; 2(4):307–320. doi:10.1016/S2213-8587(13)70212-2
DIPART (Vitamin D Individual Patient Analysis of Randomized Trials) Group. Patient level pooled analysis of 68 500 patients from seven major vitamin D fracture trials in US and Europe. BMJ 2010; 340:b5463. doi:10.1136/bmj.b5463
Reid IR, Horne AM, Mihov B, et al. Effect of monthly high-dose vitamin D on bone density in community-dwelling older adults substudy of a randomized controlled trial. J Intern Med 2017; 282(5):452–460. doi:10.1111/joim.12651
MacDonald HM, Reid IR, Gamble GD, Fraser WD, Tang JC, Wood AD. 25-Hydroxyvitamin D threshold for the effects of vitamin D supplements on bone density secondary analysis of a randomized controlled trial. J Bone Miner Res 2018. Epub ahead of print. doi:10.1002/jbmr.3442
Scragg R, Stewart AW, Waayer D, et al. Effect of monthly high-dose vitamin D supplementation on cardiovascular disease in the vitamin D assessment study: a randomized clinical trial. JAMA Cardiol 2017; 2(6):608–616. doi:10.1001/jamacardio.2017.0175
Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA 2010; 303(18):1815–1822. doi:10.1001/jama.2010.594
Smith LM, Gallagher JC, Suiter C. Medium doses of daily vitamin D decrease falls and higher doses of daily vitamin D3 increase falls: a randomized clinical trial. J Steroid Biochem Mol Biol 2017; 173:317–322. doi:10.1016/j.jsbmb.2017.03.015
To the Editor: We read with interest the article by Cikach et al on thoracic aortic aneurysm.1 For medical management of this condition, the authors emphasized controlling blood pressure and heart rate and also avoiding isometric exercises and heavy lifting. In addition to their recommendations, we believe there is plausible evidence to advise caution if fluoroquinolone antibiotics are used in this setting.
Three large population-based studies, from Canada,2 Taiwan,3 and Sweden,4 collectively demonstrated a significant 2-fold increase in the incidence of aortic aneurysm and dissection presenting within 60 days of fluoroquinolone use compared with other antibiotic exposure. Moreover, a longer duration of fluoroquinolone use was associated with a significantly higher incidence of aortic aneurysm and dissection.3
Mechanistically, fluoroquinolones have been shown to up-regulate production of several matrix metalloproteinases, including metalloproteinase 2, leading to degradation of type I collagen.2,5 Type I and type III are the dominant collagens in the aortic wall, and collagen degradation is implicated in aortic aneurysm formation and expansion.
Fluoroquinolones are widely prescribed in both outpatient and inpatient settings and are sometimes used for long durations in the geriatric population.2 It is possible that these drugs have a propensity to increase aortic aneurysm expansion and dissection in older patients who already have aortic aneurysm. Accordingly, this might make the risk-benefit ratio unfavorable for using these drugs in these situations, and other antibiotics should be used, if indicated.
Furthermore, if fluoroquinolones are used in patients with aortic aneurysm, perhaps imaging studies of the aneurysm should be done more frequently than once a year to detect accelerated aneurysm growth. Finally, physicians should be aware of the possibility of increased aortic aneurysm expansion and dissection with fluoroquinolone use.
References
Cikach F, Desai MY, Roselli EE, Kalahasti V. Thoracic aortic aneurysm: how to counsel, when to refer. Cleve Clin J Med 2018; 85(6):481–492. doi:10.3949/ccjm.85a.17039
Daneman N, Lu H, Redelmeier DA. Fluoroquinolones and collagen associated severe adverse events: a longitudinal cohort study. BMJ Open 2015; 5:e010077. doi:10.1136/bmjopen-2015-010077
Lee C-C, Lee MG, Chen Y-S, et al. Risk of aortic dissection and aortic aneurysm in patients taking oral fluoroquinolone. JAMA Intern Med 2015; 175:1839–1847. doi:10.1001/jamainternmed.2015.5389
Pasternak B, Inghammar M, Svanström H. Fluoroquinolone use and risk of aortic aneurysm and dissection: nationwide cohort study. BMJ Open 2018; 360:k678. doi:10.1136/bmj.k678
Tsai W-C, Hsu C-C, Chen CPC, et al. Ciprofloxacin up-regulates tendon cells to express matrix metalloproteinase-2 with degradation of type I collagen. J Orthop Res 2011; 29(1):67–73. doi:10.1002/jor.21196
To the Editor: We read with interest the article by Cikach et al on thoracic aortic aneurysm.1 For medical management of this condition, the authors emphasized controlling blood pressure and heart rate and also avoiding isometric exercises and heavy lifting. In addition to their recommendations, we believe there is plausible evidence to advise caution if fluoroquinolone antibiotics are used in this setting.
Three large population-based studies, from Canada,2 Taiwan,3 and Sweden,4 collectively demonstrated a significant 2-fold increase in the incidence of aortic aneurysm and dissection presenting within 60 days of fluoroquinolone use compared with other antibiotic exposure. Moreover, a longer duration of fluoroquinolone use was associated with a significantly higher incidence of aortic aneurysm and dissection.3
Mechanistically, fluoroquinolones have been shown to up-regulate production of several matrix metalloproteinases, including metalloproteinase 2, leading to degradation of type I collagen.2,5 Type I and type III are the dominant collagens in the aortic wall, and collagen degradation is implicated in aortic aneurysm formation and expansion.
Fluoroquinolones are widely prescribed in both outpatient and inpatient settings and are sometimes used for long durations in the geriatric population.2 It is possible that these drugs have a propensity to increase aortic aneurysm expansion and dissection in older patients who already have aortic aneurysm. Accordingly, this might make the risk-benefit ratio unfavorable for using these drugs in these situations, and other antibiotics should be used, if indicated.
Furthermore, if fluoroquinolones are used in patients with aortic aneurysm, perhaps imaging studies of the aneurysm should be done more frequently than once a year to detect accelerated aneurysm growth. Finally, physicians should be aware of the possibility of increased aortic aneurysm expansion and dissection with fluoroquinolone use.
To the Editor: We read with interest the article by Cikach et al on thoracic aortic aneurysm.1 For medical management of this condition, the authors emphasized controlling blood pressure and heart rate and also avoiding isometric exercises and heavy lifting. In addition to their recommendations, we believe there is plausible evidence to advise caution if fluoroquinolone antibiotics are used in this setting.
Three large population-based studies, from Canada,2 Taiwan,3 and Sweden,4 collectively demonstrated a significant 2-fold increase in the incidence of aortic aneurysm and dissection presenting within 60 days of fluoroquinolone use compared with other antibiotic exposure. Moreover, a longer duration of fluoroquinolone use was associated with a significantly higher incidence of aortic aneurysm and dissection.3
Mechanistically, fluoroquinolones have been shown to up-regulate production of several matrix metalloproteinases, including metalloproteinase 2, leading to degradation of type I collagen.2,5 Type I and type III are the dominant collagens in the aortic wall, and collagen degradation is implicated in aortic aneurysm formation and expansion.
Fluoroquinolones are widely prescribed in both outpatient and inpatient settings and are sometimes used for long durations in the geriatric population.2 It is possible that these drugs have a propensity to increase aortic aneurysm expansion and dissection in older patients who already have aortic aneurysm. Accordingly, this might make the risk-benefit ratio unfavorable for using these drugs in these situations, and other antibiotics should be used, if indicated.
Furthermore, if fluoroquinolones are used in patients with aortic aneurysm, perhaps imaging studies of the aneurysm should be done more frequently than once a year to detect accelerated aneurysm growth. Finally, physicians should be aware of the possibility of increased aortic aneurysm expansion and dissection with fluoroquinolone use.
References
Cikach F, Desai MY, Roselli EE, Kalahasti V. Thoracic aortic aneurysm: how to counsel, when to refer. Cleve Clin J Med 2018; 85(6):481–492. doi:10.3949/ccjm.85a.17039
Daneman N, Lu H, Redelmeier DA. Fluoroquinolones and collagen associated severe adverse events: a longitudinal cohort study. BMJ Open 2015; 5:e010077. doi:10.1136/bmjopen-2015-010077
Lee C-C, Lee MG, Chen Y-S, et al. Risk of aortic dissection and aortic aneurysm in patients taking oral fluoroquinolone. JAMA Intern Med 2015; 175:1839–1847. doi:10.1001/jamainternmed.2015.5389
Pasternak B, Inghammar M, Svanström H. Fluoroquinolone use and risk of aortic aneurysm and dissection: nationwide cohort study. BMJ Open 2018; 360:k678. doi:10.1136/bmj.k678
Tsai W-C, Hsu C-C, Chen CPC, et al. Ciprofloxacin up-regulates tendon cells to express matrix metalloproteinase-2 with degradation of type I collagen. J Orthop Res 2011; 29(1):67–73. doi:10.1002/jor.21196
References
Cikach F, Desai MY, Roselli EE, Kalahasti V. Thoracic aortic aneurysm: how to counsel, when to refer. Cleve Clin J Med 2018; 85(6):481–492. doi:10.3949/ccjm.85a.17039
Daneman N, Lu H, Redelmeier DA. Fluoroquinolones and collagen associated severe adverse events: a longitudinal cohort study. BMJ Open 2015; 5:e010077. doi:10.1136/bmjopen-2015-010077
Lee C-C, Lee MG, Chen Y-S, et al. Risk of aortic dissection and aortic aneurysm in patients taking oral fluoroquinolone. JAMA Intern Med 2015; 175:1839–1847. doi:10.1001/jamainternmed.2015.5389
Pasternak B, Inghammar M, Svanström H. Fluoroquinolone use and risk of aortic aneurysm and dissection: nationwide cohort study. BMJ Open 2018; 360:k678. doi:10.1136/bmj.k678
Tsai W-C, Hsu C-C, Chen CPC, et al. Ciprofloxacin up-regulates tendon cells to express matrix metalloproteinase-2 with degradation of type I collagen. J Orthop Res 2011; 29(1):67–73. doi:10.1002/jor.21196
In Reply: We thank Drs. Goldstein and Mascitelli for their comments regarding fluoroquinolones and thoracic aortic aneurysms. We acknowledge that fluoroquinolones (particularly ciprofloxacin) have been associated with a risk of aortic aneurysm and dissection based on large observational studies from Taiwan, Canada, and Sweden. Although all of the studies have shown an association between ciprofloxacin and aortic aneurysm, the causative role is not well established. In addition, the numbers of events were very small in these large cohorts of patients. In our large tertiary care practice at Cleveland Clinic, we have very few patients with aortic aneurysm or dissection who have used fluoroquinolones.
We recognize the association; however, our paper was intended to emphasize the more common causes and treatment options that primary care physicians are likely to encounter in routine practice.
We also thank Drs. Ayoubieh and MacCarrick for their comments about genetic counseling. We agree that genetic counseling is important, as is a detailed physical examination for subtle features of genetically mediated aortic aneurysm. In fact, we incorporate the physical examination when patients are seen at our aortic center so as to recognize the physical features. We do routinely recommend screening of first-degree relatives even without significant family history on an individual basis and make appropriate referrals for other conditions that can be seen in these patients. Our article, however, is primarily intended to emphasize the importance of referring these patients for more-focused care at a specialized center, where we incorporate all of the suggestions that were made.
In Reply: We thank Drs. Goldstein and Mascitelli for their comments regarding fluoroquinolones and thoracic aortic aneurysms. We acknowledge that fluoroquinolones (particularly ciprofloxacin) have been associated with a risk of aortic aneurysm and dissection based on large observational studies from Taiwan, Canada, and Sweden. Although all of the studies have shown an association between ciprofloxacin and aortic aneurysm, the causative role is not well established. In addition, the numbers of events were very small in these large cohorts of patients. In our large tertiary care practice at Cleveland Clinic, we have very few patients with aortic aneurysm or dissection who have used fluoroquinolones.
We recognize the association; however, our paper was intended to emphasize the more common causes and treatment options that primary care physicians are likely to encounter in routine practice.
We also thank Drs. Ayoubieh and MacCarrick for their comments about genetic counseling. We agree that genetic counseling is important, as is a detailed physical examination for subtle features of genetically mediated aortic aneurysm. In fact, we incorporate the physical examination when patients are seen at our aortic center so as to recognize the physical features. We do routinely recommend screening of first-degree relatives even without significant family history on an individual basis and make appropriate referrals for other conditions that can be seen in these patients. Our article, however, is primarily intended to emphasize the importance of referring these patients for more-focused care at a specialized center, where we incorporate all of the suggestions that were made.
In Reply: We thank Drs. Goldstein and Mascitelli for their comments regarding fluoroquinolones and thoracic aortic aneurysms. We acknowledge that fluoroquinolones (particularly ciprofloxacin) have been associated with a risk of aortic aneurysm and dissection based on large observational studies from Taiwan, Canada, and Sweden. Although all of the studies have shown an association between ciprofloxacin and aortic aneurysm, the causative role is not well established. In addition, the numbers of events were very small in these large cohorts of patients. In our large tertiary care practice at Cleveland Clinic, we have very few patients with aortic aneurysm or dissection who have used fluoroquinolones.
We recognize the association; however, our paper was intended to emphasize the more common causes and treatment options that primary care physicians are likely to encounter in routine practice.
We also thank Drs. Ayoubieh and MacCarrick for their comments about genetic counseling. We agree that genetic counseling is important, as is a detailed physical examination for subtle features of genetically mediated aortic aneurysm. In fact, we incorporate the physical examination when patients are seen at our aortic center so as to recognize the physical features. We do routinely recommend screening of first-degree relatives even without significant family history on an individual basis and make appropriate referrals for other conditions that can be seen in these patients. Our article, however, is primarily intended to emphasize the importance of referring these patients for more-focused care at a specialized center, where we incorporate all of the suggestions that were made.
Data suggest that antihypertensive agents may be disease-modifying therapies for cerebrovascular dementia.
CHICAGO—Lowering systolic blood pressure to a target of 120 mm Hg or less in people with cardiovascular risk factors reduces the risk of mild cognitive impairment (MCI) by 19% and reduces the risk of probable all-cause dementia by 17%, compared with achieving a less intensive target of lower than 140 mm Hg, according to research presented at AAIC 2018.
The class of antihypertensive did not affect the association. Generic drugs were as effective as branded drugs. Antihypertensive agents provided equal benefits to men, women, whites, blacks, and Hispanics. Furthermore, maintaining systolic blood pressure at 120 mm Hg or lower prevented MCI as well in patients older than 75 as it did in younger patients.
Jeff D. Williamson, MD, Chief of Geriatric Medicine at Wake Forest University in Winston-Salem, North Carolina, presented the results of the four-year SPRINT MIND study. Strict blood pressure control (ie, a systolic target of 120 mm Hg or lower) for 3.2 years reduced the incidence of MCI to a greater extent than any amyloid-targeting investigational drug has done.
“This is the first disease-modifying strategy to reduce the risk of MCI,” said Dr. Williamson. Although the effect of strict blood pressure control on the primary end point (ie, a 17% risk reduction for probable all-cause dementia) was not statistically significant, “it is comforting to see that the benefit went in the same direction and was of the same magnitude,” said Dr. Williamson. “Three years of treatment and 3.2 years of follow-up absolutely reduced the risk.”
Brain imaging underscored the clinical importance of this finding and showed its physiologic pathway. Participants who underwent strict blood pressure control had 18% fewer white matter hyperintensities after four years of follow-up than other participants.
The results may represent a step forward in a field that has seen few of them recently. Generic antihypertensive agents can be inexpensive. They are widely available and confer benefits not only on cardiovascular health, but on kidney health as well, said Dr. Williamson.
“Hypertension is a highly prevalent condition…. The 19% overall risk reduction for MCI will have a huge impact,” he added.
“The most we can say right now is that we are able to reduce risk,” said Maria Carrillo, PhD, Chief Scientific Officer of the Alzheimer’s Association, in an interview. “Reducing the risk of MCI by 19% will have a huge impact on dementia overall. Slowing down the disease progress is a disease modification, versus developing symptoms. So, if that is the definition we are using, then I would say yes, it is disease modifying,” for dementias arising from cerebrovascular pathology.
A Substudy of the SPRINT Trial
SPRINT MIND was a substudy of the Hypertension Systolic Blood Pressure Intervention Trial (SPRINT). It compared two strategies for managing hypertension in older adults. The intensive strategy had a target systolic blood pressure of less than 120 mm Hg, and the standard care strategy had a target of less than 140 mm Hg. SPRINT showed that intensive blood pressure control reduced the risk of cardiovascular events, stroke, and cardiovascular death by 30%. The study results helped inform the 2017 American Heart Association and American College of Cardiology clinical guidelines for treating high blood pressure.
The SPRINT MIND substudy examined whether intensive blood pressure management affected the risk of probable all-cause dementia or MCI or affected white matter lesion volume and brain volume.
The investigators examined data for 9,361 SPRINT subjects who were age 50 or older (mean age, 68) and had at least one cardiovascular risk factor. Approximately 30% of participants were black, and 10% were Hispanic. The primary outcome was incident probable dementia. Secondary outcomes were MCI and a composite of MCI and probable dementia.
In SPRINT, physicians could choose any appropriate antihypertensive regimen, but were encouraged to use drugs with the strongest evidence of cardiovascular benefit. These drugs included thiazide-type diuretics, loop diuretics, and beta-adrenergic blockers. About 90% of the drugs used during the study were generic.
Subjects were seen monthly for the first three months. During this time, medications were adjusted to achieve the target blood pressure. After the third month, subjects were examined every three months. Medications could be adjusted monthly.
Results Favored Intensive Treatment
At one year, mean systolic blood pressure was 121.4 mm Hg in the intensive-treatment group and 136.2 mm Hg in the standard treatment group. Treatment was stopped early after a median follow-up of 3.26 years because of the observed cardiovascular disease benefit.
The SPRINT MIND study did not meet its primary end point. Incident probable all-cause dementia occurred in 175 people in the standard care group and 147 people in the intensive treatment group. The difference between groups in the rate of 17% risk reduction was not statistically significant.
The results for both secondary end points were significant, however. Incident MCI occurred in 348 participants in the standard treatment group and 285 participants in the intensive treatment group. The difference indicated a statistically significant 19% risk reduction associated with intensive treatment. Furthermore, intensive treatment significantly reduced the risk of the combined secondary end point of MCI and probable dementia by 15%. In all, 463 participants in the standard care group met this end point, compared with 398 in the intensive care group.
The imaging study included 454 subjects who had brain MRI at baseline and at four years after randomization. Total brain volume did not change, said Ilya Nasrallah, MD, Assistant Professor of Radiology at the University of Pennsylvania in Philadelphia. Patients in the intensively managed group, however, had 18% lower white matter lesion load than those in the standard care group.
White matter lesions often indicate small-vessel disease, which is associated with vascular dementia and, perhaps, Alzheimer’s disease. Most patients with Alzheimer’s disease have a mixed dementia that includes a vascular component, said Dr. Carrillo.
David Knopman, MD
The Gravity of MCI
SPRINT MIND did not follow subjects for longer than four years or include follow-up for amyloid positivity or Alzheimer’s disease diagnosis. Nevertheless, preventing MCI is a significant achievement, according to David Knopman, MD, a consultant in the department of neurology at Mayo Clinic in Rochester, Minnesota.
“There is nothing benign about MCI,” said Dr. Knopman. “It is the first sign of overt cognitive dysfunction, and although the rate at which MCI progresses to dementia is slow, the appearance of it is just as important as the appearance of more severe dementia. To be able to see an effect in 3.2 years is quite remarkable. I think this study is going to change clinical practice for people in primary care, and the benefits at the population level are going to be substantial.”
Physicians may want to think about how the SPRINT MIND results might apply to younger patients with hypertension, whether they have other cardiovascular risk factors or not, said Dr. Williamson. “I will adhere to the guidelines we have and keep blood pressure at less than 130 mm Hg and certainly start treating people in their 50s, and probably in their 40s,” he concluded.
—Michele G. Sullivan
Suggested Reading
SPRINT Research Group, Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373(22):2103-2116.
Data suggest that antihypertensive agents may be disease-modifying therapies for cerebrovascular dementia.
Data suggest that antihypertensive agents may be disease-modifying therapies for cerebrovascular dementia.
CHICAGO—Lowering systolic blood pressure to a target of 120 mm Hg or less in people with cardiovascular risk factors reduces the risk of mild cognitive impairment (MCI) by 19% and reduces the risk of probable all-cause dementia by 17%, compared with achieving a less intensive target of lower than 140 mm Hg, according to research presented at AAIC 2018.
The class of antihypertensive did not affect the association. Generic drugs were as effective as branded drugs. Antihypertensive agents provided equal benefits to men, women, whites, blacks, and Hispanics. Furthermore, maintaining systolic blood pressure at 120 mm Hg or lower prevented MCI as well in patients older than 75 as it did in younger patients.
Jeff D. Williamson, MD, Chief of Geriatric Medicine at Wake Forest University in Winston-Salem, North Carolina, presented the results of the four-year SPRINT MIND study. Strict blood pressure control (ie, a systolic target of 120 mm Hg or lower) for 3.2 years reduced the incidence of MCI to a greater extent than any amyloid-targeting investigational drug has done.
“This is the first disease-modifying strategy to reduce the risk of MCI,” said Dr. Williamson. Although the effect of strict blood pressure control on the primary end point (ie, a 17% risk reduction for probable all-cause dementia) was not statistically significant, “it is comforting to see that the benefit went in the same direction and was of the same magnitude,” said Dr. Williamson. “Three years of treatment and 3.2 years of follow-up absolutely reduced the risk.”
Brain imaging underscored the clinical importance of this finding and showed its physiologic pathway. Participants who underwent strict blood pressure control had 18% fewer white matter hyperintensities after four years of follow-up than other participants.
The results may represent a step forward in a field that has seen few of them recently. Generic antihypertensive agents can be inexpensive. They are widely available and confer benefits not only on cardiovascular health, but on kidney health as well, said Dr. Williamson.
“Hypertension is a highly prevalent condition…. The 19% overall risk reduction for MCI will have a huge impact,” he added.
“The most we can say right now is that we are able to reduce risk,” said Maria Carrillo, PhD, Chief Scientific Officer of the Alzheimer’s Association, in an interview. “Reducing the risk of MCI by 19% will have a huge impact on dementia overall. Slowing down the disease progress is a disease modification, versus developing symptoms. So, if that is the definition we are using, then I would say yes, it is disease modifying,” for dementias arising from cerebrovascular pathology.
A Substudy of the SPRINT Trial
SPRINT MIND was a substudy of the Hypertension Systolic Blood Pressure Intervention Trial (SPRINT). It compared two strategies for managing hypertension in older adults. The intensive strategy had a target systolic blood pressure of less than 120 mm Hg, and the standard care strategy had a target of less than 140 mm Hg. SPRINT showed that intensive blood pressure control reduced the risk of cardiovascular events, stroke, and cardiovascular death by 30%. The study results helped inform the 2017 American Heart Association and American College of Cardiology clinical guidelines for treating high blood pressure.
The SPRINT MIND substudy examined whether intensive blood pressure management affected the risk of probable all-cause dementia or MCI or affected white matter lesion volume and brain volume.
The investigators examined data for 9,361 SPRINT subjects who were age 50 or older (mean age, 68) and had at least one cardiovascular risk factor. Approximately 30% of participants were black, and 10% were Hispanic. The primary outcome was incident probable dementia. Secondary outcomes were MCI and a composite of MCI and probable dementia.
In SPRINT, physicians could choose any appropriate antihypertensive regimen, but were encouraged to use drugs with the strongest evidence of cardiovascular benefit. These drugs included thiazide-type diuretics, loop diuretics, and beta-adrenergic blockers. About 90% of the drugs used during the study were generic.
Subjects were seen monthly for the first three months. During this time, medications were adjusted to achieve the target blood pressure. After the third month, subjects were examined every three months. Medications could be adjusted monthly.
Results Favored Intensive Treatment
At one year, mean systolic blood pressure was 121.4 mm Hg in the intensive-treatment group and 136.2 mm Hg in the standard treatment group. Treatment was stopped early after a median follow-up of 3.26 years because of the observed cardiovascular disease benefit.
The SPRINT MIND study did not meet its primary end point. Incident probable all-cause dementia occurred in 175 people in the standard care group and 147 people in the intensive treatment group. The difference between groups in the rate of 17% risk reduction was not statistically significant.
The results for both secondary end points were significant, however. Incident MCI occurred in 348 participants in the standard treatment group and 285 participants in the intensive treatment group. The difference indicated a statistically significant 19% risk reduction associated with intensive treatment. Furthermore, intensive treatment significantly reduced the risk of the combined secondary end point of MCI and probable dementia by 15%. In all, 463 participants in the standard care group met this end point, compared with 398 in the intensive care group.
The imaging study included 454 subjects who had brain MRI at baseline and at four years after randomization. Total brain volume did not change, said Ilya Nasrallah, MD, Assistant Professor of Radiology at the University of Pennsylvania in Philadelphia. Patients in the intensively managed group, however, had 18% lower white matter lesion load than those in the standard care group.
White matter lesions often indicate small-vessel disease, which is associated with vascular dementia and, perhaps, Alzheimer’s disease. Most patients with Alzheimer’s disease have a mixed dementia that includes a vascular component, said Dr. Carrillo.
David Knopman, MD
The Gravity of MCI
SPRINT MIND did not follow subjects for longer than four years or include follow-up for amyloid positivity or Alzheimer’s disease diagnosis. Nevertheless, preventing MCI is a significant achievement, according to David Knopman, MD, a consultant in the department of neurology at Mayo Clinic in Rochester, Minnesota.
“There is nothing benign about MCI,” said Dr. Knopman. “It is the first sign of overt cognitive dysfunction, and although the rate at which MCI progresses to dementia is slow, the appearance of it is just as important as the appearance of more severe dementia. To be able to see an effect in 3.2 years is quite remarkable. I think this study is going to change clinical practice for people in primary care, and the benefits at the population level are going to be substantial.”
Physicians may want to think about how the SPRINT MIND results might apply to younger patients with hypertension, whether they have other cardiovascular risk factors or not, said Dr. Williamson. “I will adhere to the guidelines we have and keep blood pressure at less than 130 mm Hg and certainly start treating people in their 50s, and probably in their 40s,” he concluded.
—Michele G. Sullivan
Suggested Reading
SPRINT Research Group, Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373(22):2103-2116.
CHICAGO—Lowering systolic blood pressure to a target of 120 mm Hg or less in people with cardiovascular risk factors reduces the risk of mild cognitive impairment (MCI) by 19% and reduces the risk of probable all-cause dementia by 17%, compared with achieving a less intensive target of lower than 140 mm Hg, according to research presented at AAIC 2018.
The class of antihypertensive did not affect the association. Generic drugs were as effective as branded drugs. Antihypertensive agents provided equal benefits to men, women, whites, blacks, and Hispanics. Furthermore, maintaining systolic blood pressure at 120 mm Hg or lower prevented MCI as well in patients older than 75 as it did in younger patients.
Jeff D. Williamson, MD, Chief of Geriatric Medicine at Wake Forest University in Winston-Salem, North Carolina, presented the results of the four-year SPRINT MIND study. Strict blood pressure control (ie, a systolic target of 120 mm Hg or lower) for 3.2 years reduced the incidence of MCI to a greater extent than any amyloid-targeting investigational drug has done.
“This is the first disease-modifying strategy to reduce the risk of MCI,” said Dr. Williamson. Although the effect of strict blood pressure control on the primary end point (ie, a 17% risk reduction for probable all-cause dementia) was not statistically significant, “it is comforting to see that the benefit went in the same direction and was of the same magnitude,” said Dr. Williamson. “Three years of treatment and 3.2 years of follow-up absolutely reduced the risk.”
Brain imaging underscored the clinical importance of this finding and showed its physiologic pathway. Participants who underwent strict blood pressure control had 18% fewer white matter hyperintensities after four years of follow-up than other participants.
The results may represent a step forward in a field that has seen few of them recently. Generic antihypertensive agents can be inexpensive. They are widely available and confer benefits not only on cardiovascular health, but on kidney health as well, said Dr. Williamson.
“Hypertension is a highly prevalent condition…. The 19% overall risk reduction for MCI will have a huge impact,” he added.
“The most we can say right now is that we are able to reduce risk,” said Maria Carrillo, PhD, Chief Scientific Officer of the Alzheimer’s Association, in an interview. “Reducing the risk of MCI by 19% will have a huge impact on dementia overall. Slowing down the disease progress is a disease modification, versus developing symptoms. So, if that is the definition we are using, then I would say yes, it is disease modifying,” for dementias arising from cerebrovascular pathology.
A Substudy of the SPRINT Trial
SPRINT MIND was a substudy of the Hypertension Systolic Blood Pressure Intervention Trial (SPRINT). It compared two strategies for managing hypertension in older adults. The intensive strategy had a target systolic blood pressure of less than 120 mm Hg, and the standard care strategy had a target of less than 140 mm Hg. SPRINT showed that intensive blood pressure control reduced the risk of cardiovascular events, stroke, and cardiovascular death by 30%. The study results helped inform the 2017 American Heart Association and American College of Cardiology clinical guidelines for treating high blood pressure.
The SPRINT MIND substudy examined whether intensive blood pressure management affected the risk of probable all-cause dementia or MCI or affected white matter lesion volume and brain volume.
The investigators examined data for 9,361 SPRINT subjects who were age 50 or older (mean age, 68) and had at least one cardiovascular risk factor. Approximately 30% of participants were black, and 10% were Hispanic. The primary outcome was incident probable dementia. Secondary outcomes were MCI and a composite of MCI and probable dementia.
In SPRINT, physicians could choose any appropriate antihypertensive regimen, but were encouraged to use drugs with the strongest evidence of cardiovascular benefit. These drugs included thiazide-type diuretics, loop diuretics, and beta-adrenergic blockers. About 90% of the drugs used during the study were generic.
Subjects were seen monthly for the first three months. During this time, medications were adjusted to achieve the target blood pressure. After the third month, subjects were examined every three months. Medications could be adjusted monthly.
Results Favored Intensive Treatment
At one year, mean systolic blood pressure was 121.4 mm Hg in the intensive-treatment group and 136.2 mm Hg in the standard treatment group. Treatment was stopped early after a median follow-up of 3.26 years because of the observed cardiovascular disease benefit.
The SPRINT MIND study did not meet its primary end point. Incident probable all-cause dementia occurred in 175 people in the standard care group and 147 people in the intensive treatment group. The difference between groups in the rate of 17% risk reduction was not statistically significant.
The results for both secondary end points were significant, however. Incident MCI occurred in 348 participants in the standard treatment group and 285 participants in the intensive treatment group. The difference indicated a statistically significant 19% risk reduction associated with intensive treatment. Furthermore, intensive treatment significantly reduced the risk of the combined secondary end point of MCI and probable dementia by 15%. In all, 463 participants in the standard care group met this end point, compared with 398 in the intensive care group.
The imaging study included 454 subjects who had brain MRI at baseline and at four years after randomization. Total brain volume did not change, said Ilya Nasrallah, MD, Assistant Professor of Radiology at the University of Pennsylvania in Philadelphia. Patients in the intensively managed group, however, had 18% lower white matter lesion load than those in the standard care group.
White matter lesions often indicate small-vessel disease, which is associated with vascular dementia and, perhaps, Alzheimer’s disease. Most patients with Alzheimer’s disease have a mixed dementia that includes a vascular component, said Dr. Carrillo.
David Knopman, MD
The Gravity of MCI
SPRINT MIND did not follow subjects for longer than four years or include follow-up for amyloid positivity or Alzheimer’s disease diagnosis. Nevertheless, preventing MCI is a significant achievement, according to David Knopman, MD, a consultant in the department of neurology at Mayo Clinic in Rochester, Minnesota.
“There is nothing benign about MCI,” said Dr. Knopman. “It is the first sign of overt cognitive dysfunction, and although the rate at which MCI progresses to dementia is slow, the appearance of it is just as important as the appearance of more severe dementia. To be able to see an effect in 3.2 years is quite remarkable. I think this study is going to change clinical practice for people in primary care, and the benefits at the population level are going to be substantial.”
Physicians may want to think about how the SPRINT MIND results might apply to younger patients with hypertension, whether they have other cardiovascular risk factors or not, said Dr. Williamson. “I will adhere to the guidelines we have and keep blood pressure at less than 130 mm Hg and certainly start treating people in their 50s, and probably in their 40s,” he concluded.
—Michele G. Sullivan
Suggested Reading
SPRINT Research Group, Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373(22):2103-2116.
Biosimilars are copies of FDA-approved biologic drugs (those generally derived from a living organism) that cannot be identical to the reference drug but demonstrate a high similarity to it. As patents on the reference drugs expire, biosimilars are being developed to increase competition in the marketplace to reduce costs and improve patient access to therapy. Although the US Food and Drug Administration (FDA) has no regulatory power over drug prices, it recently announced efforts to streamline the biosimilar approval process to facilitate access to therapies and curb the associated skyrocketing costs.
Several biosimilars have been approved by the agency in recent years, and earlier this year they were joined by 2 more: the approval in May of epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for all indications of the reference product (epoetin alfa; Epogen/Procrit, Amgen), including the treatment of anemia caused by myelosuppressive chemotherapy, when there is a minimum of 2 additional months of planned chemotherapy;1 and the June approval of pegfilgrastim-jmdb (Fulphila, Mylan and Biocon) for the treatment of patients undergoing myelosuppressive chemotherapy to help reduce the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells).2 The reference product for pegfilgrastim-jmdb is pegfilgrastim (Neulasta, Amgen).
The approval of both biosimilars was based on a review of a body of evidence that included structural and functional characterization, animal study data, human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical immunogenicity data, and other clinical safety and efficacy data. This evidence established that the biosimilars were highly similar to the already FDA-approved reference products, with no clinically relevant differences.
Biocon and Mylan-GmBH, which jointly developed pegfilgrastim-jmdb, originally filed for approval in 2017; and Hospira Inc, a Pfizer company that developed epoetin alfa-epbx, filed for the first time in 2015. They subsequently received complete response letters from the FDA, twice in the case of the epoetin alfa biosimilar, rejecting their approval. For pegfilgrastim-jmdb, the complete response letter was related to a pending update of the Biologic License Application as the result of requalification activities taken because of modifications at their manufacturing plant. For epoetin alfa-epbx, the FDA expressed concerns relating to a manufacturing facility. The companies addressed the concerns in the complete response letters and submitted corrective and preventive action plans.3,4
Pegfilgrastim-jmdb
The results from a phase 3, multicenter, randomized, double-blind parallel-group trial of pegfilgrastim-jmdb compared with European Union-approved pegfilgrastim were published in 2016. Chemotherapy and radiation-naïve patients with newly diagnosed breast cancer (n = 194) received the biosimilar or reference product every 3 weeks for 6 cycles. The primary endpoint was duration of severe neutropenia in cycle 1, defined as days with absolute neutrophil count <0.5 x 109/L. The mean standard deviation was 1.2 [0.93] in the pegfilgrastim-jmdb arm and 1.2 [1.10] in the EU-pegfilgrastim arm, and the 95% confidence interval of least squares means differences was within the -1 day, +1 day range, indicating equivalency.5
A characterization and similarity assessment of pegfilgrastim-jmdb compared with US- and EU-approved pegfilgrastim was presented at the 2018 Annual Meeting of the American Society of Clinical Oncology. G-CSF receptor (G-CSFR) binding was assessed by surface plasmon resonance and potency was measured by in vitro stimulated proliferation in a mouse myelogenous leukemia cell line. In vivo rodent studies were also performed and included a PD study with a single dose of up to 3 mg/kg.6
There was high similarity in the structure, molecular mass, impurities and functional activity of the biosimilar and reference products, as well as similar G-CSFR binding and equivalent relative potency. Neutrophil and leukocyte counts were increased to a similar degree, and toxicology and drug kinetics were also comparable.
The recommended dose of pegfilgrastim-jmdb is a 6 mg/0.6 ml injection in a single-dose prefilled syringe for manual use only, administered subcutaneously once per chemotherapy cycle. The prescribing information also has dosing guidelines for administration in pediatric patients who weigh less than 45 kg. Pegfilgrastim-jmdb should not be administered between 14 days before and 24 hours after administration of chemotherapy.
The prescribing information details warnings and precautions relating to splenic rupture, acute respiratory distress syndrome (ARDS), serious allergic reactions, potential for severe/fatal sickle cell crises in patients with sickle cell disorders, glomerulonephritis, leukocytosis, capillary leak syndrome, and the potential for tumor growth or recurrence.7
Patients should be evaluated for an enlarged spleen or splenic rupture if they report upper left abdominal or shoulder pain. Patients who develop fever and lung infiltrates or respiratory distress should be evaluated for ARDS and treatment discontinued if a diagnosis is confirmed. Pegfilgrastim-jmdb should be permanently discontinued in patients who develop serious allergic reactions and should not be used in patients with a history of serious allergic reactions to pegfilgrastim or filgrastim products.
Dose-reduction or interruption should be considered in patients who develop glomerulonephritis. Complete blood counts should be monitored throughout treatment. Patients should be monitored closely for capillary leak syndrome and treated with standard therapy. Pegfilgrastim-jmdb is marketed as Fulphila.
Epoetin alfa-epbx
Epoetin alfa-epbx was evaluated in 2 clinical trials in healthy individuals. The EPOE-12-02 trial established the PK and PD following a single subcutaneous dose of 100 U/kg in 81 participants. The EPOE-14-1 study was designed to determine the PK and PD of multiple doses of subcutaneous 100 U/kg 3 times weekly for 3 weeks in 129 participants. Both studies met prespecified criteria for PK and PD similarity to US-approved epoetin alfa, including geometric mean of area under the curve (AUC)0-120h, AUC0-inf, Cmax (maximum serum concentration achieved by a drug in a specified area of the body) and Emax (maximum response achievable for a drug dose).
Evidence of efficacy and safety were also evaluated using pooled data from EPOE-10-13 and EPOE-10-01, conducted in patients with chronic kidney disease, which was considered the most sensitive population in which to evaluate clinically meaningful differences between the biosimilar and reference product.8,9
There were no clinically meaningful differences in efficacy and a similar adverse event profile. The most common side effects include high blood pressure, joint pain, muscle spasm, fever, dizziness, respiratory infection, and cough, among others.
The recommended dose of epoetin alfa-epbx, which is marketed as Retacrit, is 40,000 Units weekly or 150 U/kg 3 times weekly in adults and 600 U/kg intravenously weekly in pediatric patients aged 5 years or younger. Epoetin alfa-epbx comes with a boxed warning to alert health care providers to the increased risks of death, heart problems, stroke, and tumor growth, or recurrence. The prescribing information also details warnings and precautions relating to these risks, as well as hypertension, seizures, lack or loss of hemoglobin response, pure red cell aplasia, serious allergic reactions, and severe cutaneous reactions.9
Blood pressure should be appropriately controlled before treatment initiation, treatment should be reduced or withheld if it becomes uncontrollable, and patients should be advised of the importance of compliance with anti-hypertensive medication and dietary restrictions. Patients should be monitored closely for premonitory neurologic symptoms and advised to contact their provider in the event of new-onset seizures, premonitory symptoms, or change in seizure frequency.
The prescribing information has dosing recommendations for lack or loss of hemoglobin response to epoetin alfa-epbx. If severe anemia or low reticulocyte count occur, treatment should be withheld and patients evaluated for neutralizing antibodies to erythropoietin and, in the event that PRCA is confirmed, treatment should be permanently discontinued. Treatment should be immediately and permanently discontinued for serious allergic reactions or severe cutaneous reactions.
References
1. US Food and Drug Administration website. FDA approves first epoetin alfa biosimilar for the treatment of anemia. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607703.htm. Updated May 15, 2018. Accessed June 22, 2018.
2. US Food and Drug Administration website. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Updated June 4, 2018. Accessed June 22, 2018.
3. Reuters. BRIEF – Biocon says US FDA issues complete response letter for proposed biosimilar pegfilgrastim. https://www.reuters.com/article/brief-biocon-says-us-fda-issued-complete/brief-biocon-says-us-fda-issued-complete-response-letter-for-proposed-biosimilar-pegfilgrastim-idUSFWN1MK0Q1. Updated October 9, 2017. Accessed June 22, 2018.
5. Waller CF, Blakeley C, Pennella E. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):1433O.
6. Sankaran PV, Palanivelu DV, Nair R, et al. Characterization and similarity assessment of a pegfilgrastim biosimilar MYL-1401H. J Clin Oncol. 2018;36(suppl; abstr e19028).
7. Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use. Prescribing information. Mylan GmBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Released June 2018. Accessed June 22, 2018.
8. US Food and Drug Administration website. ‘Epoetin Hospira,’ a proposed biosimilar to US-licensed Epogen/Procrit. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Updated May 25, 2017. Accessed June 22, 2018.
9. Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use. Prescribing information. Pfizer. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Released May 2018. Accessed June 22, 2018.
Biosimilars are copies of FDA-approved biologic drugs (those generally derived from a living organism) that cannot be identical to the reference drug but demonstrate a high similarity to it. As patents on the reference drugs expire, biosimilars are being developed to increase competition in the marketplace to reduce costs and improve patient access to therapy. Although the US Food and Drug Administration (FDA) has no regulatory power over drug prices, it recently announced efforts to streamline the biosimilar approval process to facilitate access to therapies and curb the associated skyrocketing costs.
Several biosimilars have been approved by the agency in recent years, and earlier this year they were joined by 2 more: the approval in May of epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for all indications of the reference product (epoetin alfa; Epogen/Procrit, Amgen), including the treatment of anemia caused by myelosuppressive chemotherapy, when there is a minimum of 2 additional months of planned chemotherapy;1 and the June approval of pegfilgrastim-jmdb (Fulphila, Mylan and Biocon) for the treatment of patients undergoing myelosuppressive chemotherapy to help reduce the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells).2 The reference product for pegfilgrastim-jmdb is pegfilgrastim (Neulasta, Amgen).
The approval of both biosimilars was based on a review of a body of evidence that included structural and functional characterization, animal study data, human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical immunogenicity data, and other clinical safety and efficacy data. This evidence established that the biosimilars were highly similar to the already FDA-approved reference products, with no clinically relevant differences.
Biocon and Mylan-GmBH, which jointly developed pegfilgrastim-jmdb, originally filed for approval in 2017; and Hospira Inc, a Pfizer company that developed epoetin alfa-epbx, filed for the first time in 2015. They subsequently received complete response letters from the FDA, twice in the case of the epoetin alfa biosimilar, rejecting their approval. For pegfilgrastim-jmdb, the complete response letter was related to a pending update of the Biologic License Application as the result of requalification activities taken because of modifications at their manufacturing plant. For epoetin alfa-epbx, the FDA expressed concerns relating to a manufacturing facility. The companies addressed the concerns in the complete response letters and submitted corrective and preventive action plans.3,4
Pegfilgrastim-jmdb
The results from a phase 3, multicenter, randomized, double-blind parallel-group trial of pegfilgrastim-jmdb compared with European Union-approved pegfilgrastim were published in 2016. Chemotherapy and radiation-naïve patients with newly diagnosed breast cancer (n = 194) received the biosimilar or reference product every 3 weeks for 6 cycles. The primary endpoint was duration of severe neutropenia in cycle 1, defined as days with absolute neutrophil count <0.5 x 109/L. The mean standard deviation was 1.2 [0.93] in the pegfilgrastim-jmdb arm and 1.2 [1.10] in the EU-pegfilgrastim arm, and the 95% confidence interval of least squares means differences was within the -1 day, +1 day range, indicating equivalency.5
A characterization and similarity assessment of pegfilgrastim-jmdb compared with US- and EU-approved pegfilgrastim was presented at the 2018 Annual Meeting of the American Society of Clinical Oncology. G-CSF receptor (G-CSFR) binding was assessed by surface plasmon resonance and potency was measured by in vitro stimulated proliferation in a mouse myelogenous leukemia cell line. In vivo rodent studies were also performed and included a PD study with a single dose of up to 3 mg/kg.6
There was high similarity in the structure, molecular mass, impurities and functional activity of the biosimilar and reference products, as well as similar G-CSFR binding and equivalent relative potency. Neutrophil and leukocyte counts were increased to a similar degree, and toxicology and drug kinetics were also comparable.
The recommended dose of pegfilgrastim-jmdb is a 6 mg/0.6 ml injection in a single-dose prefilled syringe for manual use only, administered subcutaneously once per chemotherapy cycle. The prescribing information also has dosing guidelines for administration in pediatric patients who weigh less than 45 kg. Pegfilgrastim-jmdb should not be administered between 14 days before and 24 hours after administration of chemotherapy.
The prescribing information details warnings and precautions relating to splenic rupture, acute respiratory distress syndrome (ARDS), serious allergic reactions, potential for severe/fatal sickle cell crises in patients with sickle cell disorders, glomerulonephritis, leukocytosis, capillary leak syndrome, and the potential for tumor growth or recurrence.7
Patients should be evaluated for an enlarged spleen or splenic rupture if they report upper left abdominal or shoulder pain. Patients who develop fever and lung infiltrates or respiratory distress should be evaluated for ARDS and treatment discontinued if a diagnosis is confirmed. Pegfilgrastim-jmdb should be permanently discontinued in patients who develop serious allergic reactions and should not be used in patients with a history of serious allergic reactions to pegfilgrastim or filgrastim products.
Dose-reduction or interruption should be considered in patients who develop glomerulonephritis. Complete blood counts should be monitored throughout treatment. Patients should be monitored closely for capillary leak syndrome and treated with standard therapy. Pegfilgrastim-jmdb is marketed as Fulphila.
Epoetin alfa-epbx
Epoetin alfa-epbx was evaluated in 2 clinical trials in healthy individuals. The EPOE-12-02 trial established the PK and PD following a single subcutaneous dose of 100 U/kg in 81 participants. The EPOE-14-1 study was designed to determine the PK and PD of multiple doses of subcutaneous 100 U/kg 3 times weekly for 3 weeks in 129 participants. Both studies met prespecified criteria for PK and PD similarity to US-approved epoetin alfa, including geometric mean of area under the curve (AUC)0-120h, AUC0-inf, Cmax (maximum serum concentration achieved by a drug in a specified area of the body) and Emax (maximum response achievable for a drug dose).
Evidence of efficacy and safety were also evaluated using pooled data from EPOE-10-13 and EPOE-10-01, conducted in patients with chronic kidney disease, which was considered the most sensitive population in which to evaluate clinically meaningful differences between the biosimilar and reference product.8,9
There were no clinically meaningful differences in efficacy and a similar adverse event profile. The most common side effects include high blood pressure, joint pain, muscle spasm, fever, dizziness, respiratory infection, and cough, among others.
The recommended dose of epoetin alfa-epbx, which is marketed as Retacrit, is 40,000 Units weekly or 150 U/kg 3 times weekly in adults and 600 U/kg intravenously weekly in pediatric patients aged 5 years or younger. Epoetin alfa-epbx comes with a boxed warning to alert health care providers to the increased risks of death, heart problems, stroke, and tumor growth, or recurrence. The prescribing information also details warnings and precautions relating to these risks, as well as hypertension, seizures, lack or loss of hemoglobin response, pure red cell aplasia, serious allergic reactions, and severe cutaneous reactions.9
Blood pressure should be appropriately controlled before treatment initiation, treatment should be reduced or withheld if it becomes uncontrollable, and patients should be advised of the importance of compliance with anti-hypertensive medication and dietary restrictions. Patients should be monitored closely for premonitory neurologic symptoms and advised to contact their provider in the event of new-onset seizures, premonitory symptoms, or change in seizure frequency.
The prescribing information has dosing recommendations for lack or loss of hemoglobin response to epoetin alfa-epbx. If severe anemia or low reticulocyte count occur, treatment should be withheld and patients evaluated for neutralizing antibodies to erythropoietin and, in the event that PRCA is confirmed, treatment should be permanently discontinued. Treatment should be immediately and permanently discontinued for serious allergic reactions or severe cutaneous reactions.
Biosimilars are copies of FDA-approved biologic drugs (those generally derived from a living organism) that cannot be identical to the reference drug but demonstrate a high similarity to it. As patents on the reference drugs expire, biosimilars are being developed to increase competition in the marketplace to reduce costs and improve patient access to therapy. Although the US Food and Drug Administration (FDA) has no regulatory power over drug prices, it recently announced efforts to streamline the biosimilar approval process to facilitate access to therapies and curb the associated skyrocketing costs.
Several biosimilars have been approved by the agency in recent years, and earlier this year they were joined by 2 more: the approval in May of epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for all indications of the reference product (epoetin alfa; Epogen/Procrit, Amgen), including the treatment of anemia caused by myelosuppressive chemotherapy, when there is a minimum of 2 additional months of planned chemotherapy;1 and the June approval of pegfilgrastim-jmdb (Fulphila, Mylan and Biocon) for the treatment of patients undergoing myelosuppressive chemotherapy to help reduce the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells).2 The reference product for pegfilgrastim-jmdb is pegfilgrastim (Neulasta, Amgen).
The approval of both biosimilars was based on a review of a body of evidence that included structural and functional characterization, animal study data, human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical immunogenicity data, and other clinical safety and efficacy data. This evidence established that the biosimilars were highly similar to the already FDA-approved reference products, with no clinically relevant differences.
Biocon and Mylan-GmBH, which jointly developed pegfilgrastim-jmdb, originally filed for approval in 2017; and Hospira Inc, a Pfizer company that developed epoetin alfa-epbx, filed for the first time in 2015. They subsequently received complete response letters from the FDA, twice in the case of the epoetin alfa biosimilar, rejecting their approval. For pegfilgrastim-jmdb, the complete response letter was related to a pending update of the Biologic License Application as the result of requalification activities taken because of modifications at their manufacturing plant. For epoetin alfa-epbx, the FDA expressed concerns relating to a manufacturing facility. The companies addressed the concerns in the complete response letters and submitted corrective and preventive action plans.3,4
Pegfilgrastim-jmdb
The results from a phase 3, multicenter, randomized, double-blind parallel-group trial of pegfilgrastim-jmdb compared with European Union-approved pegfilgrastim were published in 2016. Chemotherapy and radiation-naïve patients with newly diagnosed breast cancer (n = 194) received the biosimilar or reference product every 3 weeks for 6 cycles. The primary endpoint was duration of severe neutropenia in cycle 1, defined as days with absolute neutrophil count <0.5 x 109/L. The mean standard deviation was 1.2 [0.93] in the pegfilgrastim-jmdb arm and 1.2 [1.10] in the EU-pegfilgrastim arm, and the 95% confidence interval of least squares means differences was within the -1 day, +1 day range, indicating equivalency.5
A characterization and similarity assessment of pegfilgrastim-jmdb compared with US- and EU-approved pegfilgrastim was presented at the 2018 Annual Meeting of the American Society of Clinical Oncology. G-CSF receptor (G-CSFR) binding was assessed by surface plasmon resonance and potency was measured by in vitro stimulated proliferation in a mouse myelogenous leukemia cell line. In vivo rodent studies were also performed and included a PD study with a single dose of up to 3 mg/kg.6
There was high similarity in the structure, molecular mass, impurities and functional activity of the biosimilar and reference products, as well as similar G-CSFR binding and equivalent relative potency. Neutrophil and leukocyte counts were increased to a similar degree, and toxicology and drug kinetics were also comparable.
The recommended dose of pegfilgrastim-jmdb is a 6 mg/0.6 ml injection in a single-dose prefilled syringe for manual use only, administered subcutaneously once per chemotherapy cycle. The prescribing information also has dosing guidelines for administration in pediatric patients who weigh less than 45 kg. Pegfilgrastim-jmdb should not be administered between 14 days before and 24 hours after administration of chemotherapy.
The prescribing information details warnings and precautions relating to splenic rupture, acute respiratory distress syndrome (ARDS), serious allergic reactions, potential for severe/fatal sickle cell crises in patients with sickle cell disorders, glomerulonephritis, leukocytosis, capillary leak syndrome, and the potential for tumor growth or recurrence.7
Patients should be evaluated for an enlarged spleen or splenic rupture if they report upper left abdominal or shoulder pain. Patients who develop fever and lung infiltrates or respiratory distress should be evaluated for ARDS and treatment discontinued if a diagnosis is confirmed. Pegfilgrastim-jmdb should be permanently discontinued in patients who develop serious allergic reactions and should not be used in patients with a history of serious allergic reactions to pegfilgrastim or filgrastim products.
Dose-reduction or interruption should be considered in patients who develop glomerulonephritis. Complete blood counts should be monitored throughout treatment. Patients should be monitored closely for capillary leak syndrome and treated with standard therapy. Pegfilgrastim-jmdb is marketed as Fulphila.
Epoetin alfa-epbx
Epoetin alfa-epbx was evaluated in 2 clinical trials in healthy individuals. The EPOE-12-02 trial established the PK and PD following a single subcutaneous dose of 100 U/kg in 81 participants. The EPOE-14-1 study was designed to determine the PK and PD of multiple doses of subcutaneous 100 U/kg 3 times weekly for 3 weeks in 129 participants. Both studies met prespecified criteria for PK and PD similarity to US-approved epoetin alfa, including geometric mean of area under the curve (AUC)0-120h, AUC0-inf, Cmax (maximum serum concentration achieved by a drug in a specified area of the body) and Emax (maximum response achievable for a drug dose).
Evidence of efficacy and safety were also evaluated using pooled data from EPOE-10-13 and EPOE-10-01, conducted in patients with chronic kidney disease, which was considered the most sensitive population in which to evaluate clinically meaningful differences between the biosimilar and reference product.8,9
There were no clinically meaningful differences in efficacy and a similar adverse event profile. The most common side effects include high blood pressure, joint pain, muscle spasm, fever, dizziness, respiratory infection, and cough, among others.
The recommended dose of epoetin alfa-epbx, which is marketed as Retacrit, is 40,000 Units weekly or 150 U/kg 3 times weekly in adults and 600 U/kg intravenously weekly in pediatric patients aged 5 years or younger. Epoetin alfa-epbx comes with a boxed warning to alert health care providers to the increased risks of death, heart problems, stroke, and tumor growth, or recurrence. The prescribing information also details warnings and precautions relating to these risks, as well as hypertension, seizures, lack or loss of hemoglobin response, pure red cell aplasia, serious allergic reactions, and severe cutaneous reactions.9
Blood pressure should be appropriately controlled before treatment initiation, treatment should be reduced or withheld if it becomes uncontrollable, and patients should be advised of the importance of compliance with anti-hypertensive medication and dietary restrictions. Patients should be monitored closely for premonitory neurologic symptoms and advised to contact their provider in the event of new-onset seizures, premonitory symptoms, or change in seizure frequency.
The prescribing information has dosing recommendations for lack or loss of hemoglobin response to epoetin alfa-epbx. If severe anemia or low reticulocyte count occur, treatment should be withheld and patients evaluated for neutralizing antibodies to erythropoietin and, in the event that PRCA is confirmed, treatment should be permanently discontinued. Treatment should be immediately and permanently discontinued for serious allergic reactions or severe cutaneous reactions.
References
1. US Food and Drug Administration website. FDA approves first epoetin alfa biosimilar for the treatment of anemia. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607703.htm. Updated May 15, 2018. Accessed June 22, 2018.
2. US Food and Drug Administration website. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Updated June 4, 2018. Accessed June 22, 2018.
3. Reuters. BRIEF – Biocon says US FDA issues complete response letter for proposed biosimilar pegfilgrastim. https://www.reuters.com/article/brief-biocon-says-us-fda-issued-complete/brief-biocon-says-us-fda-issued-complete-response-letter-for-proposed-biosimilar-pegfilgrastim-idUSFWN1MK0Q1. Updated October 9, 2017. Accessed June 22, 2018.
5. Waller CF, Blakeley C, Pennella E. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):1433O.
6. Sankaran PV, Palanivelu DV, Nair R, et al. Characterization and similarity assessment of a pegfilgrastim biosimilar MYL-1401H. J Clin Oncol. 2018;36(suppl; abstr e19028).
7. Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use. Prescribing information. Mylan GmBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Released June 2018. Accessed June 22, 2018.
8. US Food and Drug Administration website. ‘Epoetin Hospira,’ a proposed biosimilar to US-licensed Epogen/Procrit. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Updated May 25, 2017. Accessed June 22, 2018.
9. Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use. Prescribing information. Pfizer. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Released May 2018. Accessed June 22, 2018.
References
1. US Food and Drug Administration website. FDA approves first epoetin alfa biosimilar for the treatment of anemia. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607703.htm. Updated May 15, 2018. Accessed June 22, 2018.
2. US Food and Drug Administration website. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Updated June 4, 2018. Accessed June 22, 2018.
3. Reuters. BRIEF – Biocon says US FDA issues complete response letter for proposed biosimilar pegfilgrastim. https://www.reuters.com/article/brief-biocon-says-us-fda-issued-complete/brief-biocon-says-us-fda-issued-complete-response-letter-for-proposed-biosimilar-pegfilgrastim-idUSFWN1MK0Q1. Updated October 9, 2017. Accessed June 22, 2018.
5. Waller CF, Blakeley C, Pennella E. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):1433O.
6. Sankaran PV, Palanivelu DV, Nair R, et al. Characterization and similarity assessment of a pegfilgrastim biosimilar MYL-1401H. J Clin Oncol. 2018;36(suppl; abstr e19028).
7. Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use. Prescribing information. Mylan GmBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Released June 2018. Accessed June 22, 2018.
8. US Food and Drug Administration website. ‘Epoetin Hospira,’ a proposed biosimilar to US-licensed Epogen/Procrit. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Updated May 25, 2017. Accessed June 22, 2018.
9. Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use. Prescribing information. Pfizer. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Released May 2018. Accessed June 22, 2018.
Issue
The Journal of Community and Supportive Oncology - 16(5)
Issue
The Journal of Community and Supportive Oncology - 16(5)
An estimated 72,000 drug overdose deaths occurred in 2017 in the United States, making it the worst year on record, according to preliminary data released by the Centers for Disease Control and Prevention.
The record high was driven by a sharp increase in deaths attributed to synthetic opioids, such as fentanyl and tramadol, data published on the agency’s website show.
The provisional counts are based on death records sent to the CDC’s National Center for Health Statistics from state vital registration offices in all 50 states and the District of Columbia, reported lead author Farida B. Ahmad, MPH, of the division of vital statistics at the NCHS. Overall, the predicted number of drug overdose deaths has climbed steadily, from 54,207 in November 2015 to 66,012 in November 2016, and to 72,287 in November 2017, according to an interactive chart accessible on the website.
Deaths attributable to synthetic opioids have climbed faster than any other drug class, soaring from just 9,983 in 2015 to 20,310 in 2016, and to 29,418 in 2017.
The next-largest category, heroin-related deaths, increased from 13,407 in 2015 to 16,012 in 2016, but appeared to plateau at 15,959 in 2017. However, the CDC cautioned that flat or declining numbers could be attributable to incomplete data, true decreases in deaths, or some combination of the two. “True declines or plateaus in the numbers of drug overdose deaths across the U.S. cannot be ascertained until final data become available.”
Cocaine-related deaths were fewer in number but appear to have risen substantially to the point where the number of deaths now nearly rival that of heroin. The number of deaths was 7,106 in 2015, 10,868 in 2016, and 14,614 in 2017.
The count of drug overdose deaths varied by state. Nebraska had the sharpest increase in predicted deaths between January 2017 and January 2018, coming in at 33.3%, though the absolute numbers of cases were low (114 through January 2017 and 152 through January 2018). North Carolina also showed substantial increases (from 2,053 to 2,515 cases, 22.5%), as did New Jersey (2,219 to 2,687 cases, 21.1%), the CDC data showed.
Provisional data will be updated on a monthly basis as additional records are received, the CDC said.
An estimated 72,000 drug overdose deaths occurred in 2017 in the United States, making it the worst year on record, according to preliminary data released by the Centers for Disease Control and Prevention.
The record high was driven by a sharp increase in deaths attributed to synthetic opioids, such as fentanyl and tramadol, data published on the agency’s website show.
The provisional counts are based on death records sent to the CDC’s National Center for Health Statistics from state vital registration offices in all 50 states and the District of Columbia, reported lead author Farida B. Ahmad, MPH, of the division of vital statistics at the NCHS. Overall, the predicted number of drug overdose deaths has climbed steadily, from 54,207 in November 2015 to 66,012 in November 2016, and to 72,287 in November 2017, according to an interactive chart accessible on the website.
Deaths attributable to synthetic opioids have climbed faster than any other drug class, soaring from just 9,983 in 2015 to 20,310 in 2016, and to 29,418 in 2017.
The next-largest category, heroin-related deaths, increased from 13,407 in 2015 to 16,012 in 2016, but appeared to plateau at 15,959 in 2017. However, the CDC cautioned that flat or declining numbers could be attributable to incomplete data, true decreases in deaths, or some combination of the two. “True declines or plateaus in the numbers of drug overdose deaths across the U.S. cannot be ascertained until final data become available.”
Cocaine-related deaths were fewer in number but appear to have risen substantially to the point where the number of deaths now nearly rival that of heroin. The number of deaths was 7,106 in 2015, 10,868 in 2016, and 14,614 in 2017.
The count of drug overdose deaths varied by state. Nebraska had the sharpest increase in predicted deaths between January 2017 and January 2018, coming in at 33.3%, though the absolute numbers of cases were low (114 through January 2017 and 152 through January 2018). North Carolina also showed substantial increases (from 2,053 to 2,515 cases, 22.5%), as did New Jersey (2,219 to 2,687 cases, 21.1%), the CDC data showed.
Provisional data will be updated on a monthly basis as additional records are received, the CDC said.
An estimated 72,000 drug overdose deaths occurred in 2017 in the United States, making it the worst year on record, according to preliminary data released by the Centers for Disease Control and Prevention.
The record high was driven by a sharp increase in deaths attributed to synthetic opioids, such as fentanyl and tramadol, data published on the agency’s website show.
The provisional counts are based on death records sent to the CDC’s National Center for Health Statistics from state vital registration offices in all 50 states and the District of Columbia, reported lead author Farida B. Ahmad, MPH, of the division of vital statistics at the NCHS. Overall, the predicted number of drug overdose deaths has climbed steadily, from 54,207 in November 2015 to 66,012 in November 2016, and to 72,287 in November 2017, according to an interactive chart accessible on the website.
Deaths attributable to synthetic opioids have climbed faster than any other drug class, soaring from just 9,983 in 2015 to 20,310 in 2016, and to 29,418 in 2017.
The next-largest category, heroin-related deaths, increased from 13,407 in 2015 to 16,012 in 2016, but appeared to plateau at 15,959 in 2017. However, the CDC cautioned that flat or declining numbers could be attributable to incomplete data, true decreases in deaths, or some combination of the two. “True declines or plateaus in the numbers of drug overdose deaths across the U.S. cannot be ascertained until final data become available.”
Cocaine-related deaths were fewer in number but appear to have risen substantially to the point where the number of deaths now nearly rival that of heroin. The number of deaths was 7,106 in 2015, 10,868 in 2016, and 14,614 in 2017.
The count of drug overdose deaths varied by state. Nebraska had the sharpest increase in predicted deaths between January 2017 and January 2018, coming in at 33.3%, though the absolute numbers of cases were low (114 through January 2017 and 152 through January 2018). North Carolina also showed substantial increases (from 2,053 to 2,515 cases, 22.5%), as did New Jersey (2,219 to 2,687 cases, 21.1%), the CDC data showed.
Provisional data will be updated on a monthly basis as additional records are received, the CDC said.
The prevalence of opioid use disorder more than quadrupled among pregnant women at labor and delivery from 1999 to 2014, according to the Centers for Disease Control and Prevention.
The national prevalence of opioid use disorder increased by 333% as it went from 1.5 cases per 1,000 delivery hospitalizations in 1999 to 6.5 cases per 1,000 in 2014. At the state level, there were significant increases in all 28 states with data available for at least 3 consecutive years during the study period, Sarah C. Haight, MPH, and her associates at the CDC in Atlanta said in the Morbidity and Mortality Weekly Report.
Average annual rate changes for those states ranged from a low of 0.01 per 1,000 delivery hospitalizations per year in California to 5.37 per year in Vermont, with the national rate change coming in at 0.39 per year. Of the 14 states with data available in 1999, Iowa had the lowest rate at 0.1 per 1,000 deliveries and Maryland had the highest at 8.2. In 2014, when data were available for 26 states and the District of Columbia, the highest rate was Vermont’s 48.6 per 1,000 deliveries and the lowest was 0.7 in Washington, D.C., the investigators reported.
Although “increasing trends might represent actual increases in prevalence or improved screening and diagnosis,” Ms. Haight and her associates added that “these estimates also correlate with state opioid prescribing rates in the general population. West Virginia, for example, had a prescribing rate estimated at 138 opioid prescriptions per 100 persons in 2012.”
“These findings illustrate the devastating impact of the opioid epidemic on families across the U.S., including on the very youngest,” said CDC Director Robert R. Redfield, MD. “Untreated opioid use disorder during pregnancy can lead to heartbreaking results. Each case represents a mother, a child, and a family in need of continued treatment and support.”
Data for the analysis came from the Agency for Healthcare Research and Quality’s National Inpatient Sample and State Inpatient Databases.
The prevalence of opioid use disorder more than quadrupled among pregnant women at labor and delivery from 1999 to 2014, according to the Centers for Disease Control and Prevention.
The national prevalence of opioid use disorder increased by 333% as it went from 1.5 cases per 1,000 delivery hospitalizations in 1999 to 6.5 cases per 1,000 in 2014. At the state level, there were significant increases in all 28 states with data available for at least 3 consecutive years during the study period, Sarah C. Haight, MPH, and her associates at the CDC in Atlanta said in the Morbidity and Mortality Weekly Report.
Average annual rate changes for those states ranged from a low of 0.01 per 1,000 delivery hospitalizations per year in California to 5.37 per year in Vermont, with the national rate change coming in at 0.39 per year. Of the 14 states with data available in 1999, Iowa had the lowest rate at 0.1 per 1,000 deliveries and Maryland had the highest at 8.2. In 2014, when data were available for 26 states and the District of Columbia, the highest rate was Vermont’s 48.6 per 1,000 deliveries and the lowest was 0.7 in Washington, D.C., the investigators reported.
Although “increasing trends might represent actual increases in prevalence or improved screening and diagnosis,” Ms. Haight and her associates added that “these estimates also correlate with state opioid prescribing rates in the general population. West Virginia, for example, had a prescribing rate estimated at 138 opioid prescriptions per 100 persons in 2012.”
“These findings illustrate the devastating impact of the opioid epidemic on families across the U.S., including on the very youngest,” said CDC Director Robert R. Redfield, MD. “Untreated opioid use disorder during pregnancy can lead to heartbreaking results. Each case represents a mother, a child, and a family in need of continued treatment and support.”
Data for the analysis came from the Agency for Healthcare Research and Quality’s National Inpatient Sample and State Inpatient Databases.
The prevalence of opioid use disorder more than quadrupled among pregnant women at labor and delivery from 1999 to 2014, according to the Centers for Disease Control and Prevention.
The national prevalence of opioid use disorder increased by 333% as it went from 1.5 cases per 1,000 delivery hospitalizations in 1999 to 6.5 cases per 1,000 in 2014. At the state level, there were significant increases in all 28 states with data available for at least 3 consecutive years during the study period, Sarah C. Haight, MPH, and her associates at the CDC in Atlanta said in the Morbidity and Mortality Weekly Report.
Average annual rate changes for those states ranged from a low of 0.01 per 1,000 delivery hospitalizations per year in California to 5.37 per year in Vermont, with the national rate change coming in at 0.39 per year. Of the 14 states with data available in 1999, Iowa had the lowest rate at 0.1 per 1,000 deliveries and Maryland had the highest at 8.2. In 2014, when data were available for 26 states and the District of Columbia, the highest rate was Vermont’s 48.6 per 1,000 deliveries and the lowest was 0.7 in Washington, D.C., the investigators reported.
Although “increasing trends might represent actual increases in prevalence or improved screening and diagnosis,” Ms. Haight and her associates added that “these estimates also correlate with state opioid prescribing rates in the general population. West Virginia, for example, had a prescribing rate estimated at 138 opioid prescriptions per 100 persons in 2012.”
“These findings illustrate the devastating impact of the opioid epidemic on families across the U.S., including on the very youngest,” said CDC Director Robert R. Redfield, MD. “Untreated opioid use disorder during pregnancy can lead to heartbreaking results. Each case represents a mother, a child, and a family in need of continued treatment and support.”
Data for the analysis came from the Agency for Healthcare Research and Quality’s National Inpatient Sample and State Inpatient Databases.
The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
ROLE OF THE INTERNIST
With kidney disease common and on the increase,1 nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists—including prescribing and managing phosphorus binders in patients with kidney disease.
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in 3 parts:
Phosphorus balance
The interplay of hormones, including fibroblast growth factor 23 (FGF23)
The mechanism of hyperphosphatemia in kidney disease.
Phosphorus balance
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism, and cell signaling. It exists primarily in the form of inorganic phosphate.
Net phosphorus balance depends on dietary phosphorus intake, gastrointestinal absorption, renal function, and flux between extracellular and intracellular (skeletal) pools (Table 1).
An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.2
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).3
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10% to 20% is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration.4 Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.5
Interplay of hormones
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.6
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.7
Figure 1. Hormonal regulation of calcium and phosphorus. Serum calcium and phosphorus balance is maintained by a tight interplay between parathyroid hormone (PTH), vitamin D, and fibroblast growth factor 23 (FGF23).Parathyroid hormone, in contrast, has a mixed effect. It increases renal excretion of phosphorus on one hand but increases phosphorus release from bone into the serum on the other. The latter is accomplished by increasing both bone resorption (directly) and intestinal absorption (indirectly, via stimulation of calcitriol) of phosphorus.8
FGF23 inhibits parathyroid hormone and calcitriol. Parathyroid hormone stimulates both FGF23 and calcitriol, whereas calcitriol inhibits parathyroid hormone. The complex interplay between these hormones is shown in Figure 1 and Table 2.
Mechanism of hyperphosphatemia in kidney disease
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
Decreased calcitriol due to its inhibition by FGF239
Hypocalcemia due to decreased calcitriol (leading to decreased intestinal calcium absorption) and calcium binding of retained phosphorus
Elevated parathyroid hormone due to low calcitriol levels (lack of inhibitory feedback by calcitriol), hyperphosphatemia, and hypocalcemia (direct parathyroid hormone stimulation).
Figure 2. Pathophysiologic processes of hyperphosphatemia. As the glomerular filtration rate (GFR) drops, the serum inorganic phosphorus (Pi) level spikes and prompts a series of responses that include stepwise increases in fibroblast growth factor 23 (FGF23), decreases in calcitriol (1,25 D), and increases in parathyroid hormone (PTH).
As chronic kidney disease progresses, the glomerular filtration rate falls, the phosphorus level rises, and the above sequence of events is repeated and accentuated, which leads to correction of the phosphorus levels. However, once the glomerular filtration rate falls below 25 to 40 mL/min/1.73 m2, these response mechanisms no longer suffice and the phosphorus level stays elevated.10 This is illustrated in Figure 2.
As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
HYPERPHOSPHATEMIA MAY LEAD TO VASCULAR CALCIFICATION
Elevated serum phosphorus levels (normal range 2.48–4.65 mg/dL in adults11) are associated with cardiovascular calcification and subsequent increases in mortality and morbidity rates. Elevations in serum phosphorus and calcium levels are associated with progression in vascular calcification12 and likely account for the accelerated vascular calcification that is seen in kidney disease.13
Hyperphosphatemia has been identified as an independent risk factor for death in patients with end-stage renal disease,14 but that relationship is less clear in patients with chronic kidney disease. A study in patients with chronic kidney disease and not on dialysis found a lower mortality rate in those who were prescribed phosphorus binders,15 but the study was criticized for limitations in its design.
Hyperphosphatemia can also lead to adverse effects on bone health due to complications such as renal osteodystrophy.
However, in its 2017 update, the Kidney Disease: Improving Global Outcomes (KDIGO) program only “suggests” lowering elevated phosphorus levels “toward” the normal range in patients with chronic kidney disease stages G3a through G5D, ie, those with glomerular filtration rates less than 60 mL/min/1.73 m2, including those on dialysis. The recommendation is graded 2C, ie, weak, based on low-quality evidence (https://kdigo.org/guidelines/ckd-mbd).
DIETARY RESTRICTION OF PHOSPHORUS
Diet is the major source of phosphorus intake. The average daily phosphorus consumption is 20 mg/kg, or 1,400 mg, and protein is the major source of dietary phosphorus.
In patients with stage 4 or 5 chronic kidney disease, the Kidney Disease Outcomes Quality Initiative recommends limiting protein intake to 0.6 mg/kg/day.16 However, in patients on hemodialysis, they recommend increasing protein intake to 1.1 mg/kg/day while limiting phosphorus intake to about 800 to 1,000 mg/day. This poses a challenge, as limiting phosphorus intake can reduce protein intake.
The phosphorus-to-protein ratio reflects the phosphorus content of protein-rich foods. A phosphorus-to-protein ratio of less than 10 mg/g helps to balance adequate protein intake while preventing hyperphosphatemia.17 Egg whites, for example, have a phosphorus-to-protein ratio of 2 mg/g (Table 3).
Sources of protein can be broadly classified as plant-based or animal-based. Animal protein contains organic phosphorus, which is easily absorbed.18 Plant protein may not be absorbed as easily.
Moe et al19 studied the importance of the protein source of phosphorus after 7 days of controlled diets. Despite equivalent protein and phosphorus concentrations in the vegetarian and meat-based diets, participants on the vegetarian diet had lower serum phosphorus levels, a trend toward lower 24-hour urinary phosphorus excretion, and significantly lower FGF23 levels than those on the meat-based diet. This suggests that a vegetarian diet may have advantages in terms of preventing hyperphosphatemia.
Another measure to reduce phosphorus absorption from meat is to boil it, which reduces the phosphorus content by 50%.20
Processed foods containing additives and preservatives are very high in phosphorus21 and should be avoided, particularly as there is no mandate to label phosphorus content in food.
PHOSPHORUS AND DIALYSIS
Although hemodialysis removes phosphorus, it does not remove enough to keep levels within normal limits. Indeed, even when patients adhere to a daily phosphorus limit of 1,000 mg, phosphorus accumulates. If 70% of the phosphorus in the diet is absorbed, this is 4,500 to 5,000 mg in a week. A 4-hour hemodialysis session will remove only 1,000 mg of phosphorus, which equals about 3,000 mg for patients undergoing dialysis 3 times a week,22 far less than phosphorus absorption.
In patients on continuous ambulatory peritoneal dialysis, a daily regimen of 4 exchanges of 2 L per exchange removes about 200 mg of phosphorus per day. In a 2012 study, patients on nocturnal dialysis or home dialysis involving longer session length had greater lowering of phosphorus levels than patients undergoing routine hemodialysis.23
Hence, phosphorus binders are often necessary in patients on routine hemodialysis or peritoneal dialysis.
PHOSPHORUS BINDERS
Phosphorus binders reduce serum phosphorus levels by binding with ingested phosphorus in the gastrointestinal tract and forming insoluble complexes that are not absorbed. For this reason they are much more effective when taken with meals. Phosphorus binders come in different formulations: pills, capsules, chewable tablets, liquids, and even powders that can be sprinkled on food.
The potency of each binder is quantified by its “phosphorus binder equivalent dose,” ie, its binding capacity compared with that of calcium carbonate as a reference.24
Phosphorus binders are broadly divided into those that contain calcium and those that do not.
Calcium-containing binders
The 2 most commonly used preparations are calcium carbonate (eg, Tums) and calcium acetate (eg, Phoslo). While these are relatively safe, some studies suggest that their use can lead to accelerated vascular calcification.25
According to KDIGO,26 calcium-containing binders should be avoided in hypercalcemia and adynamic bone disease. Additionally, the daily elemental calcium intake from binders should be limited to 1,500 mg, with a total daily intake that does not exceed 2,000 mg.
The elemental calcium content of calcium carbonate is about 40% of its weight (eg, 200 mg of elemental calcium in a 500-mg tablet of Tums), while the elemental calcium content of calcium acetate is about 25%. Therefore, a patient who needs 6 g of calcium carbonate for efficacy will be ingesting 2.4 g of elemental calcium per day, and that exceeds the recommended daily maximum. The main advantage of calcium carbonate is its low cost and easy availability. Commonly reported side effects include nausea and constipation.
A less commonly used calcium-based binder is calcium citrate (eg, Calcitrate). It should, however, be avoided in chronic kidney disease because of the risk of aluminum accumulation. Calcium citrate can enhance intestinal absorption of aluminum from dietary sources, as aluminum can form complexes with citrate.27
Calcium-free binders
There are several calcium-free binders. Some are based on metals such as aluminum, magnesium, iron, and lanthanum; others, such as sevelamer, are resin-based.
Aluminum- and magnesium-based binders are generally not used long-term in kidney disease because of the toxicity associated with aluminum and magnesium accumulation. However, aluminum hydroxide has an off-label use as a phosphorus binder in the acute setting, particularly when serum phosphorus levels are above 7 mg/dL.28 The dose is 300 to 600 mg 3 times daily with meals for a maximum of 4 weeks.
Sevelamer. Approved by the US Food and Drug Administration (FDA) in 1998, sevelamer acts by trapping phosphorus through ion exchange and hydrogen binding. It has the advantage of being calcium-free, which makes it particularly desirable in patients with hypercalcemia.
The Renagel in New Dialysis25 and Treat-To-Goal29 studies were randomized controlled trials that looked at the effects of sevelamer vs calcium-based binders on the risk of vascular calcification. The primary end points were serum phosphorus and calcium levels, while the secondary end points were coronary artery calcification on computed tomography and thoracic vertebral bone density. Both studies demonstrated a higher risk of vascular calcification with the calcium-based binders.
Another possible benefit of sevelamer is an improvement in lipid profile. Sevelamer lowers total cholesterol and low-density lipoprotein cholesterol levels without affecting high-density lipoprotein cholesterol or triglyceride levels.30 This is likely due to its bile acid-binding effect.31 Sevelamer has also been shown to lower C-reactive protein levels.32 While the cardiovascular profile appears to be improved with the treatment, there are no convincing data to confirm that those properties translate to a proven independent survival benefit.
The Calcium Acetate Renagel Evaluation33 was a randomized controlled study comparing sevelamer and calcium acetate. The authors attempted to control for the lipid-lowering effects of sevelamer by giving atorvastatin to all patients in both groups who had a low-density lipoprotein level greater than 70 mg/dL. The study found sevelamer to be not inferior to calcium acetate in terms of mortality and coronary calcification.
Further studies such as the Brazilian Renagel and Calcium trial34 and the Dialysis Clinical Outcomes Revisited trial failed to show a significant long-term benefit of sevelamer over calcium-based binders. However, a secondary statistical analysis of the latter study showed possible benefit of sevelamer over calcium acetate among those age 65 and older.35
To understand how sevelamer could affect vascular calcification, Yilmaz et al36 compared the effects of sevelamer vs calcium acetate on FGF23 and fetuin A levels. Fetuin A is an important inhibitor of vascular calcification and is progressively diminished in kidney disease, leading to accelerated calcification.37 Patients on sevelamer had higher levels of fetuin A than their counterparts on calcium acetate.37 The authors proposed increased fetuin A levels as a mechanism for decreased vascular calcification.
In summary, some studies suggest that sevelamer may offer the advantage of decreasing vascular calcification, but the data are mixed and do not provide a solid answer. The main disadvantages of sevelamer are a high pill burden and side effects of nausea and dyspepsia.
Lanthanum, a metallic element, was approved as a phosphorus binder by the FDA in 2008. It comes as a chewable tablet and offers the advantage of requiring the patient to take fewer pills than sevelamer and calcium-based binders.
Lanthanum is minimally absorbed and is eliminated mainly by the hepatobiliary pathway. There were initial concerns regarding possible toxicity from accumulation. However, a study looking at 10-year data on lanthanum use showed no evidence of serious toxicity or accumulation.38 The most commonly reported side effects were nausea and diarrhea. A disadvantage of lanthanum is its relatively high cost (Table 4).
Sucroferric oxyhydroxide comes as a chewable tablet. It was approved by the FDA in 2013. Although each tablet contains 500 mg of iron, it has not been shown to improve iron markers. In terms of phosphorus-lowering ability, it has been shown to be noninferior to sevelamer.39 Advantages include a significantly lower pill burden. Disadvantages include gastrointestinal side effects such as diarrhea and nausea and the drug’s high cost.
Ferric citrate was approved by the FDA in 2014, and 1 g delivers 210 mg of elemental iron. The main advantage of ferric citrate is its ability to increase iron markers. The phase 3 trial that demonstrated its efficacy as a binder showed an increase in ferritin compared with the active control.40 The study also showed a decrease in the need to use intravenous iron and erythropoesis-stimulating agents. This was thought to be due to improved iron stores, leading to decreased erythropoietin resistance.41
The mean number of ferric citrate tablets needed to achieve the desired phosphorus-lowering effect was 8 per day, containing 1,680 mg of iron. In comparison, oral ferrous sulfate typically provides 210 mg of iron per day.42
Disadvantages of ferric citrate include high pill burden, high cost, and gastrointestinal side effects such as nausea and constipation.
Chitosan binds salivary phosphorus. It can potentially be used, but it is not approved, and its efficacy in lowering serum phosphorus remains unclear.43
CHOOSING THE APPROPRIATE PHOSPHORUS BINDER
The choice of phosphorus binder is based on the patient’s serum calcium level and iron stores and on the drug’s side effect profile, iron pill burden, and cost. Involving patients in the choice after discussing potential side effects, pill burden, and cost is important for shared decision-making and could play a role in improving adherence.
Phosphorus binders are a major portion of the pill burden in patients with end-stage renal disease, possibly affecting patient adherence. The cost of phosphorus binders is estimated at half a billion dollars annually, underlining the significant economic impact of phosphorus control.11
Calcium-based binders should be the first choice when there is secondary hyperparathyroidism without hypercalcemia. There is no clear evidence regarding the benefit of correcting hypocalcemia, but KDIGO recommends keeping the serum calcium level within the reference range. KDIGO also recommends restricting calcium-based binders in persistent hypercalcemia, arterial calcification, and adynamic bone disease. This recommendation is largely based on expert opinion.
Noncalcium-based binders, which in theory might prevent vascular calcification, should be considered for patients with at least 1 of the following44:
Complicated diabetes mellitus
Vascular or valvular calcification
Persistent inflammation.
Noncalcium-based binders are also preferred in low bone-turnover states such as adynamic bone disease, as elevated calcium can inhibit parathyroid hormone.
However, the advantage of noncalcium-based binders regarding vascular calcification is largely theoretical and has not been proven clinically. Indeed, there are data comparing long-term outcomes of the different classes of phosphorus binders, but studies were limited by short follow-up, and individual studies have lacked power to detect statistical significance between two classes of binders on long-term outcomes. Meta-analyses have provided conflicting data, with some suggesting better outcomes with sevelamer than with calcium-based binders, and with others failing to show any difference.45
Because iron deficiency is common in kidney disease, ferric citrate, which can improve iron markers, may be a suitable option, provided its cost is covered by insurance.
SPECIAL CIRCUMSTANCES FOR THE USE OF PHOSPHORUS BINDERS
Tumor lysis syndrome
Tumor lysis syndrome occurs when tumor cells release their contents into the bloodstream, either spontaneously or in response to therapy, leading to the characteristic findings of hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.46 Phosphorus binders in conjunction with intravenous hydration are used to treat hyperphosphatemia, but evidence about their efficacy in this setting is limited.
Hypocalcemia in tumor lysis syndrome is usually not treated unless symptomatic, as the calcium-phosphorus product can increase, leading to calcium phosphate crystallization. When the calcium-phosphorus product is greater than 60, there is a higher risk of calcium phosphate deposition in the renal tubules that can lead to acute renal failure in tumor lysis syndrome.47 To lower the risk of calcium phosphate crystallization, calcium-based binders should be avoided in tumor lysis syndrome.
Total parenteral nutrition
Since patients on total parenteral nutrition do not eat, phosphorus binders are considered ineffective; there are no concrete data showing that phosphorus binders are effective in these patients.48 In patients with kidney disease, the phosphorus content in the parenteral nutrition formulation must be reduced.
Pregnancy
Data on phosphorus binders in pregnancy are limited. Calcium can cross the placenta. Calcium carbonate can be used in pregnancy, and fetal harm is not expected if calcium concentrations are within normal limits.49 Calcium acetate, sevelamer, and lanthanum are considered pregnancy category C drugs. Patients with advanced chronic kidney disease and end-stage renal disease who become pregnant must receive specialized obstetric care for high-risk pregnancy.
FUTURE DIRECTIONS
Future therapies may target FGF23 and other inflammatory markers that are up-regulated in renal hyperparathyroidism. However, trials studying these markers are needed to provide a better understanding of their role in bone mineral and cardiovascular health and in overall long-term outcomes. Additionally, randomized controlled trials are needed to study long-term nonsurrogate outcomes such as reduction in cardiovascular disease and rates of overall mortality.
References
Collins AJ, Foley RN, Herzog C, et al. US renal data system 2012 annual data report. Am J Kidney Dis 2013; 61(1 suppl 1):A7,e1–476. doi:10.1053/j.ajkd.2012.11.031
Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia Na/phosphate cotransporter. Annu Rev Nutr 2005; 25:197–214. doi:10.1146/annurev.nutr.25.050304.092642
Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol 2012; 303(3):F321–F327. doi:10.1152/ajprenal.00093.2012
Block GA, Ix JH, Ketteler M, et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62(3):457–473. doi:10.1053/j.ajkd.2013.03.042
Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1):131–155. doi:10.1152/physrev.00002.2011
Nissenson RA, Juppner H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th ed. Ames, IA: Wiley-Blackwell; 2013:208–214.
Chauhan V, Kelepouris E, Chauhan N, Vaid M. Current concepts and management strategies in chronic kidney disease-mineral and bone disorder. South Med J 2012; 105(9):479–485. doi:10.1097/SMJ.0b013e318261f7fe
Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest 1968; 47(8):1865–1874. doi:10.1172/JCI105877
Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 2016; 11(6):1088–1100. doi:10.2215/CJN.11901115
Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15(8):2208–2218. doi:10.1097/01.ASN.0000133041.27682.A2
Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31(4):607–617. pmid:9531176
Bhandari SK, Liu IA, Kujubu DA, et al. Use of phosphorus binders among non-dialysis chronic kidney disease patients and mortality outcomes. Am J Nephrol 2017; 45(5):431–441. doi:10.1159/000474959
Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(6 suppl 2):S1–S140. pmid:10895784
Streja E, Lau WL, Goldstein L, et al. Hyperphosphatemia is a combined function of high serum PTH and high dietary protein intake in dialysis patients. Kidney Int Suppl (2011) 2013; 3(5):462–468. doi:10.1038/kisup.2013.96
Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 2010; 5(3):519–530. doi:10.2215/CJN.06080809
Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
Cupisti A, Comar F, Benini O, et al. Effect of boiling on dietary phosphate and nitrogen intake. J Ren Nutr 2006; 16(1):36–40. doi:10.1053/j.jrn.2005.10.005
Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Semin Dial 2003; 16(3):186–188. pmid:12753675
Hou SH, Zhao J, Ellman CF, et al. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 1991; 18(2):217–224. pmid:1867178
Daugirdas JT, Chertow GM, Larive B, et al; Frequent Hemodialysis Network (FHN) Trial Group. Effects of frequent hemodialysis on measures of CKD mineral and bone disorder. J Am Soc Nephrol 2012; 23(4):727–738. doi:10.1681/ASN.2011070688
Daugirdas JT, Finn WF, Emmett M, Chertow GM; Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial 2011; 24(1):41–49. doi:10.1111/j.1525-139X.2011.00849.x
Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2005; 68(4):1815–1824. doi:10.1111/j.1523-1755.2005.00600.x
National Kidney Foundation. KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–S201. pmid:14520607
Nolan CR, Califano JR, Butzin CA. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kidney Int 1990; 38(5):937–941. pmid:2266679
Schucker JJ, Ward KE. Hyperphosphatemia and phosphate binders. Am J Health Syst Pharm 2005; 62(22):2355–2361. doi:10.2146/ajhp050198
Chertow GM, Burke SK, Raggi P; Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62(1):245–252. doi:10.1046/j.1523-1755.2002.00434.x
Chertow GM, Burke SK, Dillon MA, Slatopolsky E. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14(12):2907–2914. pmid:10570096
Yamada K, Fujimoto S, Tokura T, et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27(4):361–365. pmid:16060120
Qunibi W, Moustafa M, Muenz LR, et al; CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008; 51(6):952–965. doi:10.1053/j.ajkd.2008.02.298
Barreto DV, Barreto Fde C, de Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification—results from the BRIC study. Nephron Clin Pract 2008; 110(4):c273–c283. doi:10.1159/000170783
Cozzolino M, Mazzaferro S, Brandenburg V. The treatment of hyperphosphataemia in CKD: calcium-based or calcium-free phosphate binders? Nephrol Dial Transplant 2011; 26(2):402–407. doi:10.1093/ndt/gfq691
Yilmaz MI, Sonmez A, Saglam M, et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59(2):177–185. doi:10.1053/j.ajkd.2011.11.007
Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
Hutchison AJ, Wilson RJ, Garafola S, Copley JB. Lanthanum carbonate: safety data after 10 years. Nephrology (Carlton) 2016; 21(12):987–994. doi:10.1111/nep.12864
Floege J, Covic AC, Ketteler M, et al; PA21 Study Group. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int 2014; 86(3):638–647. doi:10.1038/ki.2014.58
Lewis JB, Sika M, Koury MJ, et al; Collaborative Study Group. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol 2015; 26(2):493–503. doi:10.1681/ASN.2014020212
Liu K, Kaffes AJ. Iron deficiency anemia: a review of diagnosis, investigation and management. Eur J Gastroenterol Hepatol 2012; 24(2):109–116. doi:10.1097/MEG.0b013e32834f3140
Shah HH, Hazzan AD, Fishbane S. Novel iron-based phosphate binders in patients with chronic kidney disease. Curr Opin Nephrol Hypertens 2015; 24(4):330–335. doi:10.1097/MNH.0000000000000128
Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol 2009; 20(3):460–462. doi:10.1681/ASN.2009010067
Raggi P, Vukicevic S, Moysés RM, Wesseling K, Spiegel DM. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 2010; 5(suppl 1):S31–S40. doi:10.2215/CJN.05880809
Airy M, Winkelmayer WC, Navaneethan SD. Phosphate binders: the evidence gap persists. Am J Kidney Dis 2016; 68(5):667–670. doi:10.1053/j.ajkd.2016.08.008
Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med 2011; 364(19):1844–1854. doi:10.1056/NEJMra0904569
Van den Berg H, Reintsema AM. Renal tubular damage in rasburicase: risks of alkalinisation. Ann Oncol 2004; 15(1):175–176. pmid:14679140
Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011; 96(1):53–58. doi:10.1210/jc.2010-2704
Arjun Sekar, MD Associates in Kidney Care, Des Moines, IA
Taranpreet Kaur, MD Department of Nephrology and Hypertension, Cleveland Clinic
Joseph V. Nally, Jr., MD Department of Nephrology and Hypertension, Cleveland Clinic; Clinical Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Hernan Rincon-Choles, MD Department of Nephrology and Hypertension, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Stacey Jolly, MD, MAS, FACP Department of Internal Medicine, Cleveland Clinic; Associate Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Georges N. Nakhoul, MD Director, Center for Chronic Kidney Disease, Department of Nephrology and Hypertension, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Arjun Sekar, MD, Associates in Kidney Care, 411 Laurel Street, Suite 2350, Des Moines, IA 50314; arjun_sekar@hotmail.com
phosphorus, phosphate, end-stage renal disease, kidney disease, hyperphosphatemia, phosphorus binders, calciphylaxis, Arun Sekar, T. Kaur, Joseph Nally, H. Rincon-Choles, S. Jolly, Georges Nakhoul
Arjun Sekar, MD Associates in Kidney Care, Des Moines, IA
Taranpreet Kaur, MD Department of Nephrology and Hypertension, Cleveland Clinic
Joseph V. Nally, Jr., MD Department of Nephrology and Hypertension, Cleveland Clinic; Clinical Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Hernan Rincon-Choles, MD Department of Nephrology and Hypertension, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Stacey Jolly, MD, MAS, FACP Department of Internal Medicine, Cleveland Clinic; Associate Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Georges N. Nakhoul, MD Director, Center for Chronic Kidney Disease, Department of Nephrology and Hypertension, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Arjun Sekar, MD, Associates in Kidney Care, 411 Laurel Street, Suite 2350, Des Moines, IA 50314; arjun_sekar@hotmail.com
Author and Disclosure Information
Arjun Sekar, MD Associates in Kidney Care, Des Moines, IA
Taranpreet Kaur, MD Department of Nephrology and Hypertension, Cleveland Clinic
Joseph V. Nally, Jr., MD Department of Nephrology and Hypertension, Cleveland Clinic; Clinical Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Hernan Rincon-Choles, MD Department of Nephrology and Hypertension, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Stacey Jolly, MD, MAS, FACP Department of Internal Medicine, Cleveland Clinic; Associate Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Georges N. Nakhoul, MD Director, Center for Chronic Kidney Disease, Department of Nephrology and Hypertension, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Arjun Sekar, MD, Associates in Kidney Care, 411 Laurel Street, Suite 2350, Des Moines, IA 50314; arjun_sekar@hotmail.com
The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
ROLE OF THE INTERNIST
With kidney disease common and on the increase,1 nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists—including prescribing and managing phosphorus binders in patients with kidney disease.
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in 3 parts:
Phosphorus balance
The interplay of hormones, including fibroblast growth factor 23 (FGF23)
The mechanism of hyperphosphatemia in kidney disease.
Phosphorus balance
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism, and cell signaling. It exists primarily in the form of inorganic phosphate.
Net phosphorus balance depends on dietary phosphorus intake, gastrointestinal absorption, renal function, and flux between extracellular and intracellular (skeletal) pools (Table 1).
An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.2
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).3
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10% to 20% is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration.4 Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.5
Interplay of hormones
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.6
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.7
Figure 1. Hormonal regulation of calcium and phosphorus. Serum calcium and phosphorus balance is maintained by a tight interplay between parathyroid hormone (PTH), vitamin D, and fibroblast growth factor 23 (FGF23).Parathyroid hormone, in contrast, has a mixed effect. It increases renal excretion of phosphorus on one hand but increases phosphorus release from bone into the serum on the other. The latter is accomplished by increasing both bone resorption (directly) and intestinal absorption (indirectly, via stimulation of calcitriol) of phosphorus.8
FGF23 inhibits parathyroid hormone and calcitriol. Parathyroid hormone stimulates both FGF23 and calcitriol, whereas calcitriol inhibits parathyroid hormone. The complex interplay between these hormones is shown in Figure 1 and Table 2.
Mechanism of hyperphosphatemia in kidney disease
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
Decreased calcitriol due to its inhibition by FGF239
Hypocalcemia due to decreased calcitriol (leading to decreased intestinal calcium absorption) and calcium binding of retained phosphorus
Elevated parathyroid hormone due to low calcitriol levels (lack of inhibitory feedback by calcitriol), hyperphosphatemia, and hypocalcemia (direct parathyroid hormone stimulation).
Figure 2. Pathophysiologic processes of hyperphosphatemia. As the glomerular filtration rate (GFR) drops, the serum inorganic phosphorus (Pi) level spikes and prompts a series of responses that include stepwise increases in fibroblast growth factor 23 (FGF23), decreases in calcitriol (1,25 D), and increases in parathyroid hormone (PTH).
As chronic kidney disease progresses, the glomerular filtration rate falls, the phosphorus level rises, and the above sequence of events is repeated and accentuated, which leads to correction of the phosphorus levels. However, once the glomerular filtration rate falls below 25 to 40 mL/min/1.73 m2, these response mechanisms no longer suffice and the phosphorus level stays elevated.10 This is illustrated in Figure 2.
As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
HYPERPHOSPHATEMIA MAY LEAD TO VASCULAR CALCIFICATION
Elevated serum phosphorus levels (normal range 2.48–4.65 mg/dL in adults11) are associated with cardiovascular calcification and subsequent increases in mortality and morbidity rates. Elevations in serum phosphorus and calcium levels are associated with progression in vascular calcification12 and likely account for the accelerated vascular calcification that is seen in kidney disease.13
Hyperphosphatemia has been identified as an independent risk factor for death in patients with end-stage renal disease,14 but that relationship is less clear in patients with chronic kidney disease. A study in patients with chronic kidney disease and not on dialysis found a lower mortality rate in those who were prescribed phosphorus binders,15 but the study was criticized for limitations in its design.
Hyperphosphatemia can also lead to adverse effects on bone health due to complications such as renal osteodystrophy.
However, in its 2017 update, the Kidney Disease: Improving Global Outcomes (KDIGO) program only “suggests” lowering elevated phosphorus levels “toward” the normal range in patients with chronic kidney disease stages G3a through G5D, ie, those with glomerular filtration rates less than 60 mL/min/1.73 m2, including those on dialysis. The recommendation is graded 2C, ie, weak, based on low-quality evidence (https://kdigo.org/guidelines/ckd-mbd).
DIETARY RESTRICTION OF PHOSPHORUS
Diet is the major source of phosphorus intake. The average daily phosphorus consumption is 20 mg/kg, or 1,400 mg, and protein is the major source of dietary phosphorus.
In patients with stage 4 or 5 chronic kidney disease, the Kidney Disease Outcomes Quality Initiative recommends limiting protein intake to 0.6 mg/kg/day.16 However, in patients on hemodialysis, they recommend increasing protein intake to 1.1 mg/kg/day while limiting phosphorus intake to about 800 to 1,000 mg/day. This poses a challenge, as limiting phosphorus intake can reduce protein intake.
The phosphorus-to-protein ratio reflects the phosphorus content of protein-rich foods. A phosphorus-to-protein ratio of less than 10 mg/g helps to balance adequate protein intake while preventing hyperphosphatemia.17 Egg whites, for example, have a phosphorus-to-protein ratio of 2 mg/g (Table 3).
Sources of protein can be broadly classified as plant-based or animal-based. Animal protein contains organic phosphorus, which is easily absorbed.18 Plant protein may not be absorbed as easily.
Moe et al19 studied the importance of the protein source of phosphorus after 7 days of controlled diets. Despite equivalent protein and phosphorus concentrations in the vegetarian and meat-based diets, participants on the vegetarian diet had lower serum phosphorus levels, a trend toward lower 24-hour urinary phosphorus excretion, and significantly lower FGF23 levels than those on the meat-based diet. This suggests that a vegetarian diet may have advantages in terms of preventing hyperphosphatemia.
Another measure to reduce phosphorus absorption from meat is to boil it, which reduces the phosphorus content by 50%.20
Processed foods containing additives and preservatives are very high in phosphorus21 and should be avoided, particularly as there is no mandate to label phosphorus content in food.
PHOSPHORUS AND DIALYSIS
Although hemodialysis removes phosphorus, it does not remove enough to keep levels within normal limits. Indeed, even when patients adhere to a daily phosphorus limit of 1,000 mg, phosphorus accumulates. If 70% of the phosphorus in the diet is absorbed, this is 4,500 to 5,000 mg in a week. A 4-hour hemodialysis session will remove only 1,000 mg of phosphorus, which equals about 3,000 mg for patients undergoing dialysis 3 times a week,22 far less than phosphorus absorption.
In patients on continuous ambulatory peritoneal dialysis, a daily regimen of 4 exchanges of 2 L per exchange removes about 200 mg of phosphorus per day. In a 2012 study, patients on nocturnal dialysis or home dialysis involving longer session length had greater lowering of phosphorus levels than patients undergoing routine hemodialysis.23
Hence, phosphorus binders are often necessary in patients on routine hemodialysis or peritoneal dialysis.
PHOSPHORUS BINDERS
Phosphorus binders reduce serum phosphorus levels by binding with ingested phosphorus in the gastrointestinal tract and forming insoluble complexes that are not absorbed. For this reason they are much more effective when taken with meals. Phosphorus binders come in different formulations: pills, capsules, chewable tablets, liquids, and even powders that can be sprinkled on food.
The potency of each binder is quantified by its “phosphorus binder equivalent dose,” ie, its binding capacity compared with that of calcium carbonate as a reference.24
Phosphorus binders are broadly divided into those that contain calcium and those that do not.
Calcium-containing binders
The 2 most commonly used preparations are calcium carbonate (eg, Tums) and calcium acetate (eg, Phoslo). While these are relatively safe, some studies suggest that their use can lead to accelerated vascular calcification.25
According to KDIGO,26 calcium-containing binders should be avoided in hypercalcemia and adynamic bone disease. Additionally, the daily elemental calcium intake from binders should be limited to 1,500 mg, with a total daily intake that does not exceed 2,000 mg.
The elemental calcium content of calcium carbonate is about 40% of its weight (eg, 200 mg of elemental calcium in a 500-mg tablet of Tums), while the elemental calcium content of calcium acetate is about 25%. Therefore, a patient who needs 6 g of calcium carbonate for efficacy will be ingesting 2.4 g of elemental calcium per day, and that exceeds the recommended daily maximum. The main advantage of calcium carbonate is its low cost and easy availability. Commonly reported side effects include nausea and constipation.
A less commonly used calcium-based binder is calcium citrate (eg, Calcitrate). It should, however, be avoided in chronic kidney disease because of the risk of aluminum accumulation. Calcium citrate can enhance intestinal absorption of aluminum from dietary sources, as aluminum can form complexes with citrate.27
Calcium-free binders
There are several calcium-free binders. Some are based on metals such as aluminum, magnesium, iron, and lanthanum; others, such as sevelamer, are resin-based.
Aluminum- and magnesium-based binders are generally not used long-term in kidney disease because of the toxicity associated with aluminum and magnesium accumulation. However, aluminum hydroxide has an off-label use as a phosphorus binder in the acute setting, particularly when serum phosphorus levels are above 7 mg/dL.28 The dose is 300 to 600 mg 3 times daily with meals for a maximum of 4 weeks.
Sevelamer. Approved by the US Food and Drug Administration (FDA) in 1998, sevelamer acts by trapping phosphorus through ion exchange and hydrogen binding. It has the advantage of being calcium-free, which makes it particularly desirable in patients with hypercalcemia.
The Renagel in New Dialysis25 and Treat-To-Goal29 studies were randomized controlled trials that looked at the effects of sevelamer vs calcium-based binders on the risk of vascular calcification. The primary end points were serum phosphorus and calcium levels, while the secondary end points were coronary artery calcification on computed tomography and thoracic vertebral bone density. Both studies demonstrated a higher risk of vascular calcification with the calcium-based binders.
Another possible benefit of sevelamer is an improvement in lipid profile. Sevelamer lowers total cholesterol and low-density lipoprotein cholesterol levels without affecting high-density lipoprotein cholesterol or triglyceride levels.30 This is likely due to its bile acid-binding effect.31 Sevelamer has also been shown to lower C-reactive protein levels.32 While the cardiovascular profile appears to be improved with the treatment, there are no convincing data to confirm that those properties translate to a proven independent survival benefit.
The Calcium Acetate Renagel Evaluation33 was a randomized controlled study comparing sevelamer and calcium acetate. The authors attempted to control for the lipid-lowering effects of sevelamer by giving atorvastatin to all patients in both groups who had a low-density lipoprotein level greater than 70 mg/dL. The study found sevelamer to be not inferior to calcium acetate in terms of mortality and coronary calcification.
Further studies such as the Brazilian Renagel and Calcium trial34 and the Dialysis Clinical Outcomes Revisited trial failed to show a significant long-term benefit of sevelamer over calcium-based binders. However, a secondary statistical analysis of the latter study showed possible benefit of sevelamer over calcium acetate among those age 65 and older.35
To understand how sevelamer could affect vascular calcification, Yilmaz et al36 compared the effects of sevelamer vs calcium acetate on FGF23 and fetuin A levels. Fetuin A is an important inhibitor of vascular calcification and is progressively diminished in kidney disease, leading to accelerated calcification.37 Patients on sevelamer had higher levels of fetuin A than their counterparts on calcium acetate.37 The authors proposed increased fetuin A levels as a mechanism for decreased vascular calcification.
In summary, some studies suggest that sevelamer may offer the advantage of decreasing vascular calcification, but the data are mixed and do not provide a solid answer. The main disadvantages of sevelamer are a high pill burden and side effects of nausea and dyspepsia.
Lanthanum, a metallic element, was approved as a phosphorus binder by the FDA in 2008. It comes as a chewable tablet and offers the advantage of requiring the patient to take fewer pills than sevelamer and calcium-based binders.
Lanthanum is minimally absorbed and is eliminated mainly by the hepatobiliary pathway. There were initial concerns regarding possible toxicity from accumulation. However, a study looking at 10-year data on lanthanum use showed no evidence of serious toxicity or accumulation.38 The most commonly reported side effects were nausea and diarrhea. A disadvantage of lanthanum is its relatively high cost (Table 4).
Sucroferric oxyhydroxide comes as a chewable tablet. It was approved by the FDA in 2013. Although each tablet contains 500 mg of iron, it has not been shown to improve iron markers. In terms of phosphorus-lowering ability, it has been shown to be noninferior to sevelamer.39 Advantages include a significantly lower pill burden. Disadvantages include gastrointestinal side effects such as diarrhea and nausea and the drug’s high cost.
Ferric citrate was approved by the FDA in 2014, and 1 g delivers 210 mg of elemental iron. The main advantage of ferric citrate is its ability to increase iron markers. The phase 3 trial that demonstrated its efficacy as a binder showed an increase in ferritin compared with the active control.40 The study also showed a decrease in the need to use intravenous iron and erythropoesis-stimulating agents. This was thought to be due to improved iron stores, leading to decreased erythropoietin resistance.41
The mean number of ferric citrate tablets needed to achieve the desired phosphorus-lowering effect was 8 per day, containing 1,680 mg of iron. In comparison, oral ferrous sulfate typically provides 210 mg of iron per day.42
Disadvantages of ferric citrate include high pill burden, high cost, and gastrointestinal side effects such as nausea and constipation.
Chitosan binds salivary phosphorus. It can potentially be used, but it is not approved, and its efficacy in lowering serum phosphorus remains unclear.43
CHOOSING THE APPROPRIATE PHOSPHORUS BINDER
The choice of phosphorus binder is based on the patient’s serum calcium level and iron stores and on the drug’s side effect profile, iron pill burden, and cost. Involving patients in the choice after discussing potential side effects, pill burden, and cost is important for shared decision-making and could play a role in improving adherence.
Phosphorus binders are a major portion of the pill burden in patients with end-stage renal disease, possibly affecting patient adherence. The cost of phosphorus binders is estimated at half a billion dollars annually, underlining the significant economic impact of phosphorus control.11
Calcium-based binders should be the first choice when there is secondary hyperparathyroidism without hypercalcemia. There is no clear evidence regarding the benefit of correcting hypocalcemia, but KDIGO recommends keeping the serum calcium level within the reference range. KDIGO also recommends restricting calcium-based binders in persistent hypercalcemia, arterial calcification, and adynamic bone disease. This recommendation is largely based on expert opinion.
Noncalcium-based binders, which in theory might prevent vascular calcification, should be considered for patients with at least 1 of the following44:
Complicated diabetes mellitus
Vascular or valvular calcification
Persistent inflammation.
Noncalcium-based binders are also preferred in low bone-turnover states such as adynamic bone disease, as elevated calcium can inhibit parathyroid hormone.
However, the advantage of noncalcium-based binders regarding vascular calcification is largely theoretical and has not been proven clinically. Indeed, there are data comparing long-term outcomes of the different classes of phosphorus binders, but studies were limited by short follow-up, and individual studies have lacked power to detect statistical significance between two classes of binders on long-term outcomes. Meta-analyses have provided conflicting data, with some suggesting better outcomes with sevelamer than with calcium-based binders, and with others failing to show any difference.45
Because iron deficiency is common in kidney disease, ferric citrate, which can improve iron markers, may be a suitable option, provided its cost is covered by insurance.
SPECIAL CIRCUMSTANCES FOR THE USE OF PHOSPHORUS BINDERS
Tumor lysis syndrome
Tumor lysis syndrome occurs when tumor cells release their contents into the bloodstream, either spontaneously or in response to therapy, leading to the characteristic findings of hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.46 Phosphorus binders in conjunction with intravenous hydration are used to treat hyperphosphatemia, but evidence about their efficacy in this setting is limited.
Hypocalcemia in tumor lysis syndrome is usually not treated unless symptomatic, as the calcium-phosphorus product can increase, leading to calcium phosphate crystallization. When the calcium-phosphorus product is greater than 60, there is a higher risk of calcium phosphate deposition in the renal tubules that can lead to acute renal failure in tumor lysis syndrome.47 To lower the risk of calcium phosphate crystallization, calcium-based binders should be avoided in tumor lysis syndrome.
Total parenteral nutrition
Since patients on total parenteral nutrition do not eat, phosphorus binders are considered ineffective; there are no concrete data showing that phosphorus binders are effective in these patients.48 In patients with kidney disease, the phosphorus content in the parenteral nutrition formulation must be reduced.
Pregnancy
Data on phosphorus binders in pregnancy are limited. Calcium can cross the placenta. Calcium carbonate can be used in pregnancy, and fetal harm is not expected if calcium concentrations are within normal limits.49 Calcium acetate, sevelamer, and lanthanum are considered pregnancy category C drugs. Patients with advanced chronic kidney disease and end-stage renal disease who become pregnant must receive specialized obstetric care for high-risk pregnancy.
FUTURE DIRECTIONS
Future therapies may target FGF23 and other inflammatory markers that are up-regulated in renal hyperparathyroidism. However, trials studying these markers are needed to provide a better understanding of their role in bone mineral and cardiovascular health and in overall long-term outcomes. Additionally, randomized controlled trials are needed to study long-term nonsurrogate outcomes such as reduction in cardiovascular disease and rates of overall mortality.
The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
ROLE OF THE INTERNIST
With kidney disease common and on the increase,1 nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists—including prescribing and managing phosphorus binders in patients with kidney disease.
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in 3 parts:
Phosphorus balance
The interplay of hormones, including fibroblast growth factor 23 (FGF23)
The mechanism of hyperphosphatemia in kidney disease.
Phosphorus balance
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism, and cell signaling. It exists primarily in the form of inorganic phosphate.
Net phosphorus balance depends on dietary phosphorus intake, gastrointestinal absorption, renal function, and flux between extracellular and intracellular (skeletal) pools (Table 1).
An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.2
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).3
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10% to 20% is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration.4 Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.5
Interplay of hormones
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.6
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.7
Figure 1. Hormonal regulation of calcium and phosphorus. Serum calcium and phosphorus balance is maintained by a tight interplay between parathyroid hormone (PTH), vitamin D, and fibroblast growth factor 23 (FGF23).Parathyroid hormone, in contrast, has a mixed effect. It increases renal excretion of phosphorus on one hand but increases phosphorus release from bone into the serum on the other. The latter is accomplished by increasing both bone resorption (directly) and intestinal absorption (indirectly, via stimulation of calcitriol) of phosphorus.8
FGF23 inhibits parathyroid hormone and calcitriol. Parathyroid hormone stimulates both FGF23 and calcitriol, whereas calcitriol inhibits parathyroid hormone. The complex interplay between these hormones is shown in Figure 1 and Table 2.
Mechanism of hyperphosphatemia in kidney disease
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
Decreased calcitriol due to its inhibition by FGF239
Hypocalcemia due to decreased calcitriol (leading to decreased intestinal calcium absorption) and calcium binding of retained phosphorus
Elevated parathyroid hormone due to low calcitriol levels (lack of inhibitory feedback by calcitriol), hyperphosphatemia, and hypocalcemia (direct parathyroid hormone stimulation).
Figure 2. Pathophysiologic processes of hyperphosphatemia. As the glomerular filtration rate (GFR) drops, the serum inorganic phosphorus (Pi) level spikes and prompts a series of responses that include stepwise increases in fibroblast growth factor 23 (FGF23), decreases in calcitriol (1,25 D), and increases in parathyroid hormone (PTH).
As chronic kidney disease progresses, the glomerular filtration rate falls, the phosphorus level rises, and the above sequence of events is repeated and accentuated, which leads to correction of the phosphorus levels. However, once the glomerular filtration rate falls below 25 to 40 mL/min/1.73 m2, these response mechanisms no longer suffice and the phosphorus level stays elevated.10 This is illustrated in Figure 2.
As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
HYPERPHOSPHATEMIA MAY LEAD TO VASCULAR CALCIFICATION
Elevated serum phosphorus levels (normal range 2.48–4.65 mg/dL in adults11) are associated with cardiovascular calcification and subsequent increases in mortality and morbidity rates. Elevations in serum phosphorus and calcium levels are associated with progression in vascular calcification12 and likely account for the accelerated vascular calcification that is seen in kidney disease.13
Hyperphosphatemia has been identified as an independent risk factor for death in patients with end-stage renal disease,14 but that relationship is less clear in patients with chronic kidney disease. A study in patients with chronic kidney disease and not on dialysis found a lower mortality rate in those who were prescribed phosphorus binders,15 but the study was criticized for limitations in its design.
Hyperphosphatemia can also lead to adverse effects on bone health due to complications such as renal osteodystrophy.
However, in its 2017 update, the Kidney Disease: Improving Global Outcomes (KDIGO) program only “suggests” lowering elevated phosphorus levels “toward” the normal range in patients with chronic kidney disease stages G3a through G5D, ie, those with glomerular filtration rates less than 60 mL/min/1.73 m2, including those on dialysis. The recommendation is graded 2C, ie, weak, based on low-quality evidence (https://kdigo.org/guidelines/ckd-mbd).
DIETARY RESTRICTION OF PHOSPHORUS
Diet is the major source of phosphorus intake. The average daily phosphorus consumption is 20 mg/kg, or 1,400 mg, and protein is the major source of dietary phosphorus.
In patients with stage 4 or 5 chronic kidney disease, the Kidney Disease Outcomes Quality Initiative recommends limiting protein intake to 0.6 mg/kg/day.16 However, in patients on hemodialysis, they recommend increasing protein intake to 1.1 mg/kg/day while limiting phosphorus intake to about 800 to 1,000 mg/day. This poses a challenge, as limiting phosphorus intake can reduce protein intake.
The phosphorus-to-protein ratio reflects the phosphorus content of protein-rich foods. A phosphorus-to-protein ratio of less than 10 mg/g helps to balance adequate protein intake while preventing hyperphosphatemia.17 Egg whites, for example, have a phosphorus-to-protein ratio of 2 mg/g (Table 3).
Sources of protein can be broadly classified as plant-based or animal-based. Animal protein contains organic phosphorus, which is easily absorbed.18 Plant protein may not be absorbed as easily.
Moe et al19 studied the importance of the protein source of phosphorus after 7 days of controlled diets. Despite equivalent protein and phosphorus concentrations in the vegetarian and meat-based diets, participants on the vegetarian diet had lower serum phosphorus levels, a trend toward lower 24-hour urinary phosphorus excretion, and significantly lower FGF23 levels than those on the meat-based diet. This suggests that a vegetarian diet may have advantages in terms of preventing hyperphosphatemia.
Another measure to reduce phosphorus absorption from meat is to boil it, which reduces the phosphorus content by 50%.20
Processed foods containing additives and preservatives are very high in phosphorus21 and should be avoided, particularly as there is no mandate to label phosphorus content in food.
PHOSPHORUS AND DIALYSIS
Although hemodialysis removes phosphorus, it does not remove enough to keep levels within normal limits. Indeed, even when patients adhere to a daily phosphorus limit of 1,000 mg, phosphorus accumulates. If 70% of the phosphorus in the diet is absorbed, this is 4,500 to 5,000 mg in a week. A 4-hour hemodialysis session will remove only 1,000 mg of phosphorus, which equals about 3,000 mg for patients undergoing dialysis 3 times a week,22 far less than phosphorus absorption.
In patients on continuous ambulatory peritoneal dialysis, a daily regimen of 4 exchanges of 2 L per exchange removes about 200 mg of phosphorus per day. In a 2012 study, patients on nocturnal dialysis or home dialysis involving longer session length had greater lowering of phosphorus levels than patients undergoing routine hemodialysis.23
Hence, phosphorus binders are often necessary in patients on routine hemodialysis or peritoneal dialysis.
PHOSPHORUS BINDERS
Phosphorus binders reduce serum phosphorus levels by binding with ingested phosphorus in the gastrointestinal tract and forming insoluble complexes that are not absorbed. For this reason they are much more effective when taken with meals. Phosphorus binders come in different formulations: pills, capsules, chewable tablets, liquids, and even powders that can be sprinkled on food.
The potency of each binder is quantified by its “phosphorus binder equivalent dose,” ie, its binding capacity compared with that of calcium carbonate as a reference.24
Phosphorus binders are broadly divided into those that contain calcium and those that do not.
Calcium-containing binders
The 2 most commonly used preparations are calcium carbonate (eg, Tums) and calcium acetate (eg, Phoslo). While these are relatively safe, some studies suggest that their use can lead to accelerated vascular calcification.25
According to KDIGO,26 calcium-containing binders should be avoided in hypercalcemia and adynamic bone disease. Additionally, the daily elemental calcium intake from binders should be limited to 1,500 mg, with a total daily intake that does not exceed 2,000 mg.
The elemental calcium content of calcium carbonate is about 40% of its weight (eg, 200 mg of elemental calcium in a 500-mg tablet of Tums), while the elemental calcium content of calcium acetate is about 25%. Therefore, a patient who needs 6 g of calcium carbonate for efficacy will be ingesting 2.4 g of elemental calcium per day, and that exceeds the recommended daily maximum. The main advantage of calcium carbonate is its low cost and easy availability. Commonly reported side effects include nausea and constipation.
A less commonly used calcium-based binder is calcium citrate (eg, Calcitrate). It should, however, be avoided in chronic kidney disease because of the risk of aluminum accumulation. Calcium citrate can enhance intestinal absorption of aluminum from dietary sources, as aluminum can form complexes with citrate.27
Calcium-free binders
There are several calcium-free binders. Some are based on metals such as aluminum, magnesium, iron, and lanthanum; others, such as sevelamer, are resin-based.
Aluminum- and magnesium-based binders are generally not used long-term in kidney disease because of the toxicity associated with aluminum and magnesium accumulation. However, aluminum hydroxide has an off-label use as a phosphorus binder in the acute setting, particularly when serum phosphorus levels are above 7 mg/dL.28 The dose is 300 to 600 mg 3 times daily with meals for a maximum of 4 weeks.
Sevelamer. Approved by the US Food and Drug Administration (FDA) in 1998, sevelamer acts by trapping phosphorus through ion exchange and hydrogen binding. It has the advantage of being calcium-free, which makes it particularly desirable in patients with hypercalcemia.
The Renagel in New Dialysis25 and Treat-To-Goal29 studies were randomized controlled trials that looked at the effects of sevelamer vs calcium-based binders on the risk of vascular calcification. The primary end points were serum phosphorus and calcium levels, while the secondary end points were coronary artery calcification on computed tomography and thoracic vertebral bone density. Both studies demonstrated a higher risk of vascular calcification with the calcium-based binders.
Another possible benefit of sevelamer is an improvement in lipid profile. Sevelamer lowers total cholesterol and low-density lipoprotein cholesterol levels without affecting high-density lipoprotein cholesterol or triglyceride levels.30 This is likely due to its bile acid-binding effect.31 Sevelamer has also been shown to lower C-reactive protein levels.32 While the cardiovascular profile appears to be improved with the treatment, there are no convincing data to confirm that those properties translate to a proven independent survival benefit.
The Calcium Acetate Renagel Evaluation33 was a randomized controlled study comparing sevelamer and calcium acetate. The authors attempted to control for the lipid-lowering effects of sevelamer by giving atorvastatin to all patients in both groups who had a low-density lipoprotein level greater than 70 mg/dL. The study found sevelamer to be not inferior to calcium acetate in terms of mortality and coronary calcification.
Further studies such as the Brazilian Renagel and Calcium trial34 and the Dialysis Clinical Outcomes Revisited trial failed to show a significant long-term benefit of sevelamer over calcium-based binders. However, a secondary statistical analysis of the latter study showed possible benefit of sevelamer over calcium acetate among those age 65 and older.35
To understand how sevelamer could affect vascular calcification, Yilmaz et al36 compared the effects of sevelamer vs calcium acetate on FGF23 and fetuin A levels. Fetuin A is an important inhibitor of vascular calcification and is progressively diminished in kidney disease, leading to accelerated calcification.37 Patients on sevelamer had higher levels of fetuin A than their counterparts on calcium acetate.37 The authors proposed increased fetuin A levels as a mechanism for decreased vascular calcification.
In summary, some studies suggest that sevelamer may offer the advantage of decreasing vascular calcification, but the data are mixed and do not provide a solid answer. The main disadvantages of sevelamer are a high pill burden and side effects of nausea and dyspepsia.
Lanthanum, a metallic element, was approved as a phosphorus binder by the FDA in 2008. It comes as a chewable tablet and offers the advantage of requiring the patient to take fewer pills than sevelamer and calcium-based binders.
Lanthanum is minimally absorbed and is eliminated mainly by the hepatobiliary pathway. There were initial concerns regarding possible toxicity from accumulation. However, a study looking at 10-year data on lanthanum use showed no evidence of serious toxicity or accumulation.38 The most commonly reported side effects were nausea and diarrhea. A disadvantage of lanthanum is its relatively high cost (Table 4).
Sucroferric oxyhydroxide comes as a chewable tablet. It was approved by the FDA in 2013. Although each tablet contains 500 mg of iron, it has not been shown to improve iron markers. In terms of phosphorus-lowering ability, it has been shown to be noninferior to sevelamer.39 Advantages include a significantly lower pill burden. Disadvantages include gastrointestinal side effects such as diarrhea and nausea and the drug’s high cost.
Ferric citrate was approved by the FDA in 2014, and 1 g delivers 210 mg of elemental iron. The main advantage of ferric citrate is its ability to increase iron markers. The phase 3 trial that demonstrated its efficacy as a binder showed an increase in ferritin compared with the active control.40 The study also showed a decrease in the need to use intravenous iron and erythropoesis-stimulating agents. This was thought to be due to improved iron stores, leading to decreased erythropoietin resistance.41
The mean number of ferric citrate tablets needed to achieve the desired phosphorus-lowering effect was 8 per day, containing 1,680 mg of iron. In comparison, oral ferrous sulfate typically provides 210 mg of iron per day.42
Disadvantages of ferric citrate include high pill burden, high cost, and gastrointestinal side effects such as nausea and constipation.
Chitosan binds salivary phosphorus. It can potentially be used, but it is not approved, and its efficacy in lowering serum phosphorus remains unclear.43
CHOOSING THE APPROPRIATE PHOSPHORUS BINDER
The choice of phosphorus binder is based on the patient’s serum calcium level and iron stores and on the drug’s side effect profile, iron pill burden, and cost. Involving patients in the choice after discussing potential side effects, pill burden, and cost is important for shared decision-making and could play a role in improving adherence.
Phosphorus binders are a major portion of the pill burden in patients with end-stage renal disease, possibly affecting patient adherence. The cost of phosphorus binders is estimated at half a billion dollars annually, underlining the significant economic impact of phosphorus control.11
Calcium-based binders should be the first choice when there is secondary hyperparathyroidism without hypercalcemia. There is no clear evidence regarding the benefit of correcting hypocalcemia, but KDIGO recommends keeping the serum calcium level within the reference range. KDIGO also recommends restricting calcium-based binders in persistent hypercalcemia, arterial calcification, and adynamic bone disease. This recommendation is largely based on expert opinion.
Noncalcium-based binders, which in theory might prevent vascular calcification, should be considered for patients with at least 1 of the following44:
Complicated diabetes mellitus
Vascular or valvular calcification
Persistent inflammation.
Noncalcium-based binders are also preferred in low bone-turnover states such as adynamic bone disease, as elevated calcium can inhibit parathyroid hormone.
However, the advantage of noncalcium-based binders regarding vascular calcification is largely theoretical and has not been proven clinically. Indeed, there are data comparing long-term outcomes of the different classes of phosphorus binders, but studies were limited by short follow-up, and individual studies have lacked power to detect statistical significance between two classes of binders on long-term outcomes. Meta-analyses have provided conflicting data, with some suggesting better outcomes with sevelamer than with calcium-based binders, and with others failing to show any difference.45
Because iron deficiency is common in kidney disease, ferric citrate, which can improve iron markers, may be a suitable option, provided its cost is covered by insurance.
SPECIAL CIRCUMSTANCES FOR THE USE OF PHOSPHORUS BINDERS
Tumor lysis syndrome
Tumor lysis syndrome occurs when tumor cells release their contents into the bloodstream, either spontaneously or in response to therapy, leading to the characteristic findings of hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.46 Phosphorus binders in conjunction with intravenous hydration are used to treat hyperphosphatemia, but evidence about their efficacy in this setting is limited.
Hypocalcemia in tumor lysis syndrome is usually not treated unless symptomatic, as the calcium-phosphorus product can increase, leading to calcium phosphate crystallization. When the calcium-phosphorus product is greater than 60, there is a higher risk of calcium phosphate deposition in the renal tubules that can lead to acute renal failure in tumor lysis syndrome.47 To lower the risk of calcium phosphate crystallization, calcium-based binders should be avoided in tumor lysis syndrome.
Total parenteral nutrition
Since patients on total parenteral nutrition do not eat, phosphorus binders are considered ineffective; there are no concrete data showing that phosphorus binders are effective in these patients.48 In patients with kidney disease, the phosphorus content in the parenteral nutrition formulation must be reduced.
Pregnancy
Data on phosphorus binders in pregnancy are limited. Calcium can cross the placenta. Calcium carbonate can be used in pregnancy, and fetal harm is not expected if calcium concentrations are within normal limits.49 Calcium acetate, sevelamer, and lanthanum are considered pregnancy category C drugs. Patients with advanced chronic kidney disease and end-stage renal disease who become pregnant must receive specialized obstetric care for high-risk pregnancy.
FUTURE DIRECTIONS
Future therapies may target FGF23 and other inflammatory markers that are up-regulated in renal hyperparathyroidism. However, trials studying these markers are needed to provide a better understanding of their role in bone mineral and cardiovascular health and in overall long-term outcomes. Additionally, randomized controlled trials are needed to study long-term nonsurrogate outcomes such as reduction in cardiovascular disease and rates of overall mortality.
References
Collins AJ, Foley RN, Herzog C, et al. US renal data system 2012 annual data report. Am J Kidney Dis 2013; 61(1 suppl 1):A7,e1–476. doi:10.1053/j.ajkd.2012.11.031
Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia Na/phosphate cotransporter. Annu Rev Nutr 2005; 25:197–214. doi:10.1146/annurev.nutr.25.050304.092642
Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol 2012; 303(3):F321–F327. doi:10.1152/ajprenal.00093.2012
Block GA, Ix JH, Ketteler M, et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62(3):457–473. doi:10.1053/j.ajkd.2013.03.042
Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1):131–155. doi:10.1152/physrev.00002.2011
Nissenson RA, Juppner H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th ed. Ames, IA: Wiley-Blackwell; 2013:208–214.
Chauhan V, Kelepouris E, Chauhan N, Vaid M. Current concepts and management strategies in chronic kidney disease-mineral and bone disorder. South Med J 2012; 105(9):479–485. doi:10.1097/SMJ.0b013e318261f7fe
Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest 1968; 47(8):1865–1874. doi:10.1172/JCI105877
Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 2016; 11(6):1088–1100. doi:10.2215/CJN.11901115
Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15(8):2208–2218. doi:10.1097/01.ASN.0000133041.27682.A2
Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31(4):607–617. pmid:9531176
Bhandari SK, Liu IA, Kujubu DA, et al. Use of phosphorus binders among non-dialysis chronic kidney disease patients and mortality outcomes. Am J Nephrol 2017; 45(5):431–441. doi:10.1159/000474959
Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(6 suppl 2):S1–S140. pmid:10895784
Streja E, Lau WL, Goldstein L, et al. Hyperphosphatemia is a combined function of high serum PTH and high dietary protein intake in dialysis patients. Kidney Int Suppl (2011) 2013; 3(5):462–468. doi:10.1038/kisup.2013.96
Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 2010; 5(3):519–530. doi:10.2215/CJN.06080809
Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
Cupisti A, Comar F, Benini O, et al. Effect of boiling on dietary phosphate and nitrogen intake. J Ren Nutr 2006; 16(1):36–40. doi:10.1053/j.jrn.2005.10.005
Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Semin Dial 2003; 16(3):186–188. pmid:12753675
Hou SH, Zhao J, Ellman CF, et al. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 1991; 18(2):217–224. pmid:1867178
Daugirdas JT, Chertow GM, Larive B, et al; Frequent Hemodialysis Network (FHN) Trial Group. Effects of frequent hemodialysis on measures of CKD mineral and bone disorder. J Am Soc Nephrol 2012; 23(4):727–738. doi:10.1681/ASN.2011070688
Daugirdas JT, Finn WF, Emmett M, Chertow GM; Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial 2011; 24(1):41–49. doi:10.1111/j.1525-139X.2011.00849.x
Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2005; 68(4):1815–1824. doi:10.1111/j.1523-1755.2005.00600.x
National Kidney Foundation. KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–S201. pmid:14520607
Nolan CR, Califano JR, Butzin CA. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kidney Int 1990; 38(5):937–941. pmid:2266679
Schucker JJ, Ward KE. Hyperphosphatemia and phosphate binders. Am J Health Syst Pharm 2005; 62(22):2355–2361. doi:10.2146/ajhp050198
Chertow GM, Burke SK, Raggi P; Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62(1):245–252. doi:10.1046/j.1523-1755.2002.00434.x
Chertow GM, Burke SK, Dillon MA, Slatopolsky E. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14(12):2907–2914. pmid:10570096
Yamada K, Fujimoto S, Tokura T, et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27(4):361–365. pmid:16060120
Qunibi W, Moustafa M, Muenz LR, et al; CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008; 51(6):952–965. doi:10.1053/j.ajkd.2008.02.298
Barreto DV, Barreto Fde C, de Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification—results from the BRIC study. Nephron Clin Pract 2008; 110(4):c273–c283. doi:10.1159/000170783
Cozzolino M, Mazzaferro S, Brandenburg V. The treatment of hyperphosphataemia in CKD: calcium-based or calcium-free phosphate binders? Nephrol Dial Transplant 2011; 26(2):402–407. doi:10.1093/ndt/gfq691
Yilmaz MI, Sonmez A, Saglam M, et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59(2):177–185. doi:10.1053/j.ajkd.2011.11.007
Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
Hutchison AJ, Wilson RJ, Garafola S, Copley JB. Lanthanum carbonate: safety data after 10 years. Nephrology (Carlton) 2016; 21(12):987–994. doi:10.1111/nep.12864
Floege J, Covic AC, Ketteler M, et al; PA21 Study Group. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int 2014; 86(3):638–647. doi:10.1038/ki.2014.58
Lewis JB, Sika M, Koury MJ, et al; Collaborative Study Group. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol 2015; 26(2):493–503. doi:10.1681/ASN.2014020212
Liu K, Kaffes AJ. Iron deficiency anemia: a review of diagnosis, investigation and management. Eur J Gastroenterol Hepatol 2012; 24(2):109–116. doi:10.1097/MEG.0b013e32834f3140
Shah HH, Hazzan AD, Fishbane S. Novel iron-based phosphate binders in patients with chronic kidney disease. Curr Opin Nephrol Hypertens 2015; 24(4):330–335. doi:10.1097/MNH.0000000000000128
Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol 2009; 20(3):460–462. doi:10.1681/ASN.2009010067
Raggi P, Vukicevic S, Moysés RM, Wesseling K, Spiegel DM. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 2010; 5(suppl 1):S31–S40. doi:10.2215/CJN.05880809
Airy M, Winkelmayer WC, Navaneethan SD. Phosphate binders: the evidence gap persists. Am J Kidney Dis 2016; 68(5):667–670. doi:10.1053/j.ajkd.2016.08.008
Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med 2011; 364(19):1844–1854. doi:10.1056/NEJMra0904569
Van den Berg H, Reintsema AM. Renal tubular damage in rasburicase: risks of alkalinisation. Ann Oncol 2004; 15(1):175–176. pmid:14679140
Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011; 96(1):53–58. doi:10.1210/jc.2010-2704
References
Collins AJ, Foley RN, Herzog C, et al. US renal data system 2012 annual data report. Am J Kidney Dis 2013; 61(1 suppl 1):A7,e1–476. doi:10.1053/j.ajkd.2012.11.031
Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia Na/phosphate cotransporter. Annu Rev Nutr 2005; 25:197–214. doi:10.1146/annurev.nutr.25.050304.092642
Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol 2012; 303(3):F321–F327. doi:10.1152/ajprenal.00093.2012
Block GA, Ix JH, Ketteler M, et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62(3):457–473. doi:10.1053/j.ajkd.2013.03.042
Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1):131–155. doi:10.1152/physrev.00002.2011
Nissenson RA, Juppner H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th ed. Ames, IA: Wiley-Blackwell; 2013:208–214.
Chauhan V, Kelepouris E, Chauhan N, Vaid M. Current concepts and management strategies in chronic kidney disease-mineral and bone disorder. South Med J 2012; 105(9):479–485. doi:10.1097/SMJ.0b013e318261f7fe
Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest 1968; 47(8):1865–1874. doi:10.1172/JCI105877
Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 2016; 11(6):1088–1100. doi:10.2215/CJN.11901115
Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15(8):2208–2218. doi:10.1097/01.ASN.0000133041.27682.A2
Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31(4):607–617. pmid:9531176
Bhandari SK, Liu IA, Kujubu DA, et al. Use of phosphorus binders among non-dialysis chronic kidney disease patients and mortality outcomes. Am J Nephrol 2017; 45(5):431–441. doi:10.1159/000474959
Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(6 suppl 2):S1–S140. pmid:10895784
Streja E, Lau WL, Goldstein L, et al. Hyperphosphatemia is a combined function of high serum PTH and high dietary protein intake in dialysis patients. Kidney Int Suppl (2011) 2013; 3(5):462–468. doi:10.1038/kisup.2013.96
Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 2010; 5(3):519–530. doi:10.2215/CJN.06080809
Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
Cupisti A, Comar F, Benini O, et al. Effect of boiling on dietary phosphate and nitrogen intake. J Ren Nutr 2006; 16(1):36–40. doi:10.1053/j.jrn.2005.10.005
Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Semin Dial 2003; 16(3):186–188. pmid:12753675
Hou SH, Zhao J, Ellman CF, et al. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 1991; 18(2):217–224. pmid:1867178
Daugirdas JT, Chertow GM, Larive B, et al; Frequent Hemodialysis Network (FHN) Trial Group. Effects of frequent hemodialysis on measures of CKD mineral and bone disorder. J Am Soc Nephrol 2012; 23(4):727–738. doi:10.1681/ASN.2011070688
Daugirdas JT, Finn WF, Emmett M, Chertow GM; Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial 2011; 24(1):41–49. doi:10.1111/j.1525-139X.2011.00849.x
Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2005; 68(4):1815–1824. doi:10.1111/j.1523-1755.2005.00600.x
National Kidney Foundation. KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–S201. pmid:14520607
Nolan CR, Califano JR, Butzin CA. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kidney Int 1990; 38(5):937–941. pmid:2266679
Schucker JJ, Ward KE. Hyperphosphatemia and phosphate binders. Am J Health Syst Pharm 2005; 62(22):2355–2361. doi:10.2146/ajhp050198
Chertow GM, Burke SK, Raggi P; Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62(1):245–252. doi:10.1046/j.1523-1755.2002.00434.x
Chertow GM, Burke SK, Dillon MA, Slatopolsky E. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14(12):2907–2914. pmid:10570096
Yamada K, Fujimoto S, Tokura T, et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27(4):361–365. pmid:16060120
Qunibi W, Moustafa M, Muenz LR, et al; CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008; 51(6):952–965. doi:10.1053/j.ajkd.2008.02.298
Barreto DV, Barreto Fde C, de Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification—results from the BRIC study. Nephron Clin Pract 2008; 110(4):c273–c283. doi:10.1159/000170783
Cozzolino M, Mazzaferro S, Brandenburg V. The treatment of hyperphosphataemia in CKD: calcium-based or calcium-free phosphate binders? Nephrol Dial Transplant 2011; 26(2):402–407. doi:10.1093/ndt/gfq691
Yilmaz MI, Sonmez A, Saglam M, et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59(2):177–185. doi:10.1053/j.ajkd.2011.11.007
Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
Hutchison AJ, Wilson RJ, Garafola S, Copley JB. Lanthanum carbonate: safety data after 10 years. Nephrology (Carlton) 2016; 21(12):987–994. doi:10.1111/nep.12864
Floege J, Covic AC, Ketteler M, et al; PA21 Study Group. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int 2014; 86(3):638–647. doi:10.1038/ki.2014.58
Lewis JB, Sika M, Koury MJ, et al; Collaborative Study Group. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol 2015; 26(2):493–503. doi:10.1681/ASN.2014020212
Liu K, Kaffes AJ. Iron deficiency anemia: a review of diagnosis, investigation and management. Eur J Gastroenterol Hepatol 2012; 24(2):109–116. doi:10.1097/MEG.0b013e32834f3140
Shah HH, Hazzan AD, Fishbane S. Novel iron-based phosphate binders in patients with chronic kidney disease. Curr Opin Nephrol Hypertens 2015; 24(4):330–335. doi:10.1097/MNH.0000000000000128
Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol 2009; 20(3):460–462. doi:10.1681/ASN.2009010067
Raggi P, Vukicevic S, Moysés RM, Wesseling K, Spiegel DM. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 2010; 5(suppl 1):S31–S40. doi:10.2215/CJN.05880809
Airy M, Winkelmayer WC, Navaneethan SD. Phosphate binders: the evidence gap persists. Am J Kidney Dis 2016; 68(5):667–670. doi:10.1053/j.ajkd.2016.08.008
Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med 2011; 364(19):1844–1854. doi:10.1056/NEJMra0904569
Van den Berg H, Reintsema AM. Renal tubular damage in rasburicase: risks of alkalinisation. Ann Oncol 2004; 15(1):175–176. pmid:14679140
Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011; 96(1):53–58. doi:10.1210/jc.2010-2704
Phosphorus binders: The new and the old, and how to choose
Display Headline
Phosphorus binders: The new and the old, and how to choose
Legacy Keywords
phosphorus, phosphate, end-stage renal disease, kidney disease, hyperphosphatemia, phosphorus binders, calciphylaxis, Arun Sekar, T. Kaur, Joseph Nally, H. Rincon-Choles, S. Jolly, Georges Nakhoul
Legacy Keywords
phosphorus, phosphate, end-stage renal disease, kidney disease, hyperphosphatemia, phosphorus binders, calciphylaxis, Arun Sekar, T. Kaur, Joseph Nally, H. Rincon-Choles, S. Jolly, Georges Nakhoul
Serum phosphorus is maintained within normal levels in a tightly regulated system involving interplay between organs, hormones, diet, and other factors.
Dietary phosphorus comes mainly from protein, so restricting phosphorus without introducing protein deficiency is difficult. Food with a low phosphorus-to-protein ratio and plant-based sources of protein may be preferable.
Although dialysis removes phosphorus, it usually does not remove enough, and many patients require phosphorus-binding drugs.
Selection of an appropriate binder should consider serum calcium levels, pill burden, serum iron stores, and cost.
Disallow All Ads
Content Gating
No Gating (article Unlocked/Free)
Alternative CME
Disqus Comments
Default
Gate On Date
Un-Gate On Date
Consolidated Pubs: Do Not Show Source Publication Logo
Phosphorus is essential for life. However, both low and high levels of phosphorus in the body have consequences, and its concentration in the blood is tightly regulated through dietary absorption, bone flux, and renal excretion and is influenced by calcitriol (1,25 hydroxyvitamin D3), parathyroid hormone, and fibroblast growth factor 23 (FGF23).
Sekar et al,1 in this issue of the Journal, provide an extensive review of the pathophysiology of phosphorus metabolism and strategies to control phosphorus levels in patients with hyperphosphatemia and end-stage kidney disease.
PHOSPHORUS OR PHOSPHATE?
What's in a name? That which we call a rose By any other word would smell as sweet. —Shakespeare, Romeo and Juliet
The terms phosphate and phosphorus are often used interchangeably, though most writers still prefer phosphate over phosphorus.
The serum concentrations of phosphate and phosphorus are the same when expressed in millimoles per liter, as every mole of phosphate contains 1 mole of phosphorus, but not the same when expressed in milligrams per deciliter.2 The molecular weight of phosphorus is 30.97, whereas the molecular weight of the phosphate ion (PO43–) is 94.97—more than 3 times higher. Therefore, using these terms interchangeably in this context can lead to numerical error.3
Phosphorus, being highly reactive, does not exist by itself in nature and is typically present as phosphates in biologic systems. When describing phosphorus metabolism, the term phosphates should ideally be used because phosphates are the actual participants in the bodily processes. But in the clinical laboratory, all methods that measure serum phosphorus in fact measure inorganic phosphate and are expressed in terms of milligrams of phosphorus per deciliter rather than milligrams of phosphate per deciliter, and using these 2 terms interchangeably in clinical practice should not be of concern.4
THE PROBLEM
US adults typically ingest 1,200 mg of phosphorus each day, and about 60% to 70% of the ingested phosphorus is absorbed both by passive paracellular diffusion via tight junctions and by active transcellular transport via sodium-phosphate cotransport. The kidneys must excrete the same amount daily to maintain a steady state. As kidney function declines, phosphorus accumulates in the blood, leading to hyperphosphatemia.
Hyperphosphatemia is often asymptomatic, but it can cause generalized itching, red eyes, and adverse effects on the bone and parathyroid glands. Higher serum phosphorus levels have been shown to be associated with vascular calcification,5 cardiovascular events, and higher all-cause mortality rates in the general population,6 in patients with diabetes,7 and in those with chronic kidney disease.8 This association between higher serum phosphorus levels and the all-cause mortality rate led to the assumption that lowering serum phosphorus levels in these patients could reduce the rates of cardiovascular events and death, and to efforts to correct hyperphosphatemia.
Research into FGF23 continues, especially its role in cardiovascular complications of chronic kidney disease, as both phosphorus and FGF23 levels are elevated in chronic kidney disease and are implicated in poor clinical outcomes in these patients. However, both FGF23 and parathyroid hormone levels rise early in the course of kidney disease, long before overt hyperphosphatemia develops. Further, FGF23 rises earlier than parathyroid hormone and has been found to be an independent risk factor for cardiovascular events and death from any cause in end-stage kidney disease.9
Whether hyperphosphatemia is the culprit or merely an epiphenomenon of metabolic complications of chronic kidney disease is still unclear, as more molecules are being identified in the complex process of cardiovascular calcification.10
However, one thing is clear: vascular calcification is not just a simple precipitation of calcium and phosphorus. Instead, it is an active process that involves many regulators of mineral metabolism.10 The complex nature of this process is likely one of the reasons that evidence is conflicting11 about the benefits of phosphorus binders in terms of cardiovascular events or all-cause mortality in these patients.
STRATEGIES TO CONTROL HYPERPHOSPHATEMIA
Reducing intake
Dietary phosphorus restriction is the first step in controlling serum phosphorus. But reducing phosphorus intake while otherwise trying to optimize the nutritional status can be challenging.
The recommended daily protein intake is 1.0 to 1.2 g/kg. But phosphorus is typically found in foods rich in proteins, and restricting protein severely can compromise nutritional status and may be as bad as elevated phosphate levels in terms of outcomes.
Although plant-based foods contain more phosphate per gram of protein (ie, they have a higher ratio of phosphorus to protein) than animal-based foods, the bioavailability of phosphorus from plant foods is lower. Phosphorus in plant-based foods is mainly in the form of phytate. Humans cannot hydrolyze phytate because we lack the phytase enzyme; hence, the phosphorus in plant-based foods is not well absorbed. Therefore, a vegetarian diet may be preferable and beneficial in patients with chronic kidney disease. A small study in humans showed that a vegetarian diet resulted in lower serum phosphorus and FGF23 levels, but the study was limited by its small sample size.12
Patients should be advised to avoid foods that have a high phosphate content, such as processed foods, fast foods, and cola beverages, which often have phosphate-based food additives.
Further, one should be cautious about using supplements with healthy-sounding names. A case in point is “vitamin water”: 12 oz of this fruit punch-flavored beverage contains 392 mg of phosphorus,13 and this alone would require 12 to 15 phosphate binder tablets to bind its phosphorus content.
In addition, many prescription drugs have significant amounts of phosphorus, and this is often unrecognized.
Sherman et al14 reviewed 200 of the most commonly prescribed drugs in dialysis patients and found that 23 (11.5%) of the drug labels listed phosphorus-containing ingredients, but the actual amount of phosphorus was not listed. The phosphorus content ranged from 1.4 mg (clonidine 0.2 mg, Blue Point Laboratories, Dublin, Ireland) to 111.5 mg (paroxetine 40 mg, GlaxoSmith Kline, Philadelphia, PA). The phosphorus content was inconsistent and varied with the dose of the agent, type of formulation (tablet or syrup), branded or generic formulation, and manufacturer.
Branded lisinopril (Merck, Kenilworth, NJ) had 21.4 mg of phosphorus per 10-mg dose, while a generic product (Blue Point Laboratories, Dublin, Ireland) had 32.6 mg. Different brands of generic amlodipine 10 mg varied in their phosphorus content from 8.6 mg (Lupin Pharmaceuticals, Mumbai, India) to 27.8 mg (Greenstone LLC, Peapack, NJ) to 40.1 mg (Qualitest Pharmaceuticals, Huntsville, AL. Rena-Vite (Cypress Pharmaceuticals, Madison, MS), a multivitamin marketed to patients with kidney disease, had 37.7 mg of phosphorus per tablet. Thus, just to bind the phosphorus content of these 3 tablets (lisinopril, amlodipine, and Rena-Vite), a patient could need at least 3 to 4 extra doses of phosphate binder.
The phosphate content of medications should be considered when prescribing. For example, Reno Caps (Nnodum Pharmaceuticals, Cincinnati, OH), another vitamin supplement, has only 1.7 mg of phosphorus per tablet and should be considered, especially in patients with poorly controlled serum phosphorus levels. However, the challenge is that medication labels do not provide the phosphorus content.
Reducing phosphorus absorption
Because so many foods contain phosphorus, dietary efforts alone are often insufficient to control serum phosphorus levels, and most patients require additional strategies, eg, phosphorus binders (Table 1).
Although these agents reduce serum phosphorus and help reduce symptoms, an important quality-of-life measure, it is uncertain whether they improve clinical outcomes.11 To date, no specific phosphorus binder offers a survival benefit over placebo.11
Based on the limited and conflicting evidence, the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines, recently updated, suggest that oral phosphorus binders should be used in patients with hyperphosphatemia to lower serum phosphorus levels toward the normal range.15 They further recommend not exceeding 1,500 mg of elemental calcium per day if a calcium-based binder is used, and they recommend avoiding calcium-based binders in patients with hypercalcemia, adynamic bone disease, or vascular calcification.
Phosphorus binders may account for up to 50% of the daily pill burden and may contribute to poor medication adherence.16 Dialysis patients need to take a lot of these drugs: by weight, 5 to 6 pounds per year.
These drugs can bind and interfere with the absorption of other vital medications and so should be taken with meals and separately from other medications.
Figure 1. A stepwise approach to the management of hyperphosphatemia and selection of phosphorus binder.
At present, there is insufficient evidence to recommend one binder over the other, and the selection of phosphorus binder should be individualized for each patient, taking into consideration the stage of chronic kidney disease, degree of hyperphosphatemia, concomitant anemia, presence of vascular calcification, use of other medications, side effects, cost to the individual, and pill burden. A stepwise, opinion-based, clinical approach to the selection of the phosphorus binders in patients with hyperphosphatemia is presented in Figure 1.
Removing phosphorus
Removal of phosphorus by adequate dialysis or kidney transplant is the final strategy.
New agents under study
To improve phosphorus control, other agents that inhibit absorption of phosphate are being investigated.
Nicotinamide reduces expression of the sodium-phosphorus cotransporter NTP2b. Its use in combination with a low-phosphorus diet and phosphorus binders may maximize reductions in phosphorus absorption and is being studied in the CKD Optimal Management With Binders and Nicotinamide (COMBINE) study.
Tenapanor, an inhibitor of the sodium-hydrogen transporter NHE3, has been shown in animal studies to increase fecal phosphate excretion and decrease urinary phosphate excretion17 but requires further evaluation.
References
Sekar A, Kaur T, Nally JV Jr, Rincon-Choles H, Jolly S, Nakhoul G. Phosphorus binders: the new and the old, and how to choose. Cleve Clin J Med 2018; 85(8):629–638. doi:10.3949/ccjm.85a.17054
Young DS. "Phosphorus" or "phosphate." Ann Intern Med 1980; 93(4):631. pmid:7436198
Bartter FC. Reporting of phosphate and phosphorus plasma values. Am J Med 1981; 71(5):848. pmid:7304659.
Iheagwara OS, Ing TS, Kjellstrand CM, Lew SQ. Phosphorus, phosphorous, and phosphate. Hemodial Int 2013; 17(4):479–482. doi:10.1111/hdi.12010
Adeney KL, Siscovick DS, Ix JH, et al. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J Am Soc Nephrol 2009; 20(2):381–387. doi:10.1681/ASN.2008040349
Dhingra R, Sullivan LM, Fox CS, et al. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med 2007; 167(9):879–885. doi:10.1001/archinte.167.9.879
Chonchol M, Dale R, Schrier RW, Estacio R. Serum phosphorus and cardiovascular mortality in type 2 diabetes. Am J Med 2009; 122(4):380–386. doi:10.1016/j.amjmed.2008.09.039
Covic A, Kothawala P, Bernal M, Robbins S, Chalian A, Goldsmith D. Systematic review of the evidence underlying the association between mineral metabolism disturbances and risk of all-cause mortality, cardiovascular mortality and cardiovascular events in chronic kidney disease. Nephrol Dial Transplant 2009; 24(5):1506–1523. doi:10.1093/ndt/gfn613
Gutiérrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008; 359(6):584–592. doi:10.1056/NEJMoa0706130
Palmer SC, Gardner S, Tonelli M, et al. Phosphate-binding agents in adults with CKD: a network meta-analysis of randomized trials. Am J Kidney Dis 2016; 68(5):691–702. doi:10.1053/j.ajkd.2016.05.015
Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
Moser M, White K, Henry B, et al. Phosphorus content of popular beverages. Am J Kidney Dis 2015; 65(6):969–971. doi:10.1053/j.ajkd.2015.02.330
Sherman RA, Ravella S, Kapoian T. A dearth of data: the problem of phosphorus in prescription medications. Kidney Int 2015; 87(6):1097–1099. doi:10.1038/ki.2015.67
KDIGO 2017 clinical practice guideline update for diagnosis, evaluation, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Supplements 2017; 7(1 suppl): 1–59. www.kisupplements.org/article/S2157-1716(17)30001-1/pdf. Accessed June 5, 2018.
Fissell RB, Karaboyas A, Bieber BA, et al. Phosphate binder pill burden, patient-reported non-adherence, and mineral bone disorder markers: findings from the DOPPS. Hemodial Int 2016; 20(1):38–49. doi:10.1111/hdi.12315
Labonté ED, Carreras CW, Leadbetter MR, et al. Gastrointestinal inhibition of sodium-hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol 2015; 26(5):1138–1149. doi:10.1681/ASN.2014030317
Malvinder S. Parmar, MB, MS, FRCPC, FASN Professor of Medicine, Division of Clinical Sciences, Northern Ontario School of Medicine, Sudbury and Thunder Bay, ON, Canada; Director, Internal Medicine, Timmins and District Hospital, Timmins, ON, Canada
Address: Malvinder S. Parmar, MB, MS, Internal Medicine, Timmins and District Hospital, 700 Ross Ave. East, Timmins, Ontario P4N 8P2 Canada; Wittykidney@outlook.com
Malvinder S. Parmar, MB, MS, FRCPC, FASN Professor of Medicine, Division of Clinical Sciences, Northern Ontario School of Medicine, Sudbury and Thunder Bay, ON, Canada; Director, Internal Medicine, Timmins and District Hospital, Timmins, ON, Canada
Address: Malvinder S. Parmar, MB, MS, Internal Medicine, Timmins and District Hospital, 700 Ross Ave. East, Timmins, Ontario P4N 8P2 Canada; Wittykidney@outlook.com
Author and Disclosure Information
Malvinder S. Parmar, MB, MS, FRCPC, FASN Professor of Medicine, Division of Clinical Sciences, Northern Ontario School of Medicine, Sudbury and Thunder Bay, ON, Canada; Director, Internal Medicine, Timmins and District Hospital, Timmins, ON, Canada
Address: Malvinder S. Parmar, MB, MS, Internal Medicine, Timmins and District Hospital, 700 Ross Ave. East, Timmins, Ontario P4N 8P2 Canada; Wittykidney@outlook.com
Phosphorus is essential for life. However, both low and high levels of phosphorus in the body have consequences, and its concentration in the blood is tightly regulated through dietary absorption, bone flux, and renal excretion and is influenced by calcitriol (1,25 hydroxyvitamin D3), parathyroid hormone, and fibroblast growth factor 23 (FGF23).
Sekar et al,1 in this issue of the Journal, provide an extensive review of the pathophysiology of phosphorus metabolism and strategies to control phosphorus levels in patients with hyperphosphatemia and end-stage kidney disease.
PHOSPHORUS OR PHOSPHATE?
What's in a name? That which we call a rose By any other word would smell as sweet. —Shakespeare, Romeo and Juliet
The terms phosphate and phosphorus are often used interchangeably, though most writers still prefer phosphate over phosphorus.
The serum concentrations of phosphate and phosphorus are the same when expressed in millimoles per liter, as every mole of phosphate contains 1 mole of phosphorus, but not the same when expressed in milligrams per deciliter.2 The molecular weight of phosphorus is 30.97, whereas the molecular weight of the phosphate ion (PO43–) is 94.97—more than 3 times higher. Therefore, using these terms interchangeably in this context can lead to numerical error.3
Phosphorus, being highly reactive, does not exist by itself in nature and is typically present as phosphates in biologic systems. When describing phosphorus metabolism, the term phosphates should ideally be used because phosphates are the actual participants in the bodily processes. But in the clinical laboratory, all methods that measure serum phosphorus in fact measure inorganic phosphate and are expressed in terms of milligrams of phosphorus per deciliter rather than milligrams of phosphate per deciliter, and using these 2 terms interchangeably in clinical practice should not be of concern.4
THE PROBLEM
US adults typically ingest 1,200 mg of phosphorus each day, and about 60% to 70% of the ingested phosphorus is absorbed both by passive paracellular diffusion via tight junctions and by active transcellular transport via sodium-phosphate cotransport. The kidneys must excrete the same amount daily to maintain a steady state. As kidney function declines, phosphorus accumulates in the blood, leading to hyperphosphatemia.
Hyperphosphatemia is often asymptomatic, but it can cause generalized itching, red eyes, and adverse effects on the bone and parathyroid glands. Higher serum phosphorus levels have been shown to be associated with vascular calcification,5 cardiovascular events, and higher all-cause mortality rates in the general population,6 in patients with diabetes,7 and in those with chronic kidney disease.8 This association between higher serum phosphorus levels and the all-cause mortality rate led to the assumption that lowering serum phosphorus levels in these patients could reduce the rates of cardiovascular events and death, and to efforts to correct hyperphosphatemia.
Research into FGF23 continues, especially its role in cardiovascular complications of chronic kidney disease, as both phosphorus and FGF23 levels are elevated in chronic kidney disease and are implicated in poor clinical outcomes in these patients. However, both FGF23 and parathyroid hormone levels rise early in the course of kidney disease, long before overt hyperphosphatemia develops. Further, FGF23 rises earlier than parathyroid hormone and has been found to be an independent risk factor for cardiovascular events and death from any cause in end-stage kidney disease.9
Whether hyperphosphatemia is the culprit or merely an epiphenomenon of metabolic complications of chronic kidney disease is still unclear, as more molecules are being identified in the complex process of cardiovascular calcification.10
However, one thing is clear: vascular calcification is not just a simple precipitation of calcium and phosphorus. Instead, it is an active process that involves many regulators of mineral metabolism.10 The complex nature of this process is likely one of the reasons that evidence is conflicting11 about the benefits of phosphorus binders in terms of cardiovascular events or all-cause mortality in these patients.
STRATEGIES TO CONTROL HYPERPHOSPHATEMIA
Reducing intake
Dietary phosphorus restriction is the first step in controlling serum phosphorus. But reducing phosphorus intake while otherwise trying to optimize the nutritional status can be challenging.
The recommended daily protein intake is 1.0 to 1.2 g/kg. But phosphorus is typically found in foods rich in proteins, and restricting protein severely can compromise nutritional status and may be as bad as elevated phosphate levels in terms of outcomes.
Although plant-based foods contain more phosphate per gram of protein (ie, they have a higher ratio of phosphorus to protein) than animal-based foods, the bioavailability of phosphorus from plant foods is lower. Phosphorus in plant-based foods is mainly in the form of phytate. Humans cannot hydrolyze phytate because we lack the phytase enzyme; hence, the phosphorus in plant-based foods is not well absorbed. Therefore, a vegetarian diet may be preferable and beneficial in patients with chronic kidney disease. A small study in humans showed that a vegetarian diet resulted in lower serum phosphorus and FGF23 levels, but the study was limited by its small sample size.12
Patients should be advised to avoid foods that have a high phosphate content, such as processed foods, fast foods, and cola beverages, which often have phosphate-based food additives.
Further, one should be cautious about using supplements with healthy-sounding names. A case in point is “vitamin water”: 12 oz of this fruit punch-flavored beverage contains 392 mg of phosphorus,13 and this alone would require 12 to 15 phosphate binder tablets to bind its phosphorus content.
In addition, many prescription drugs have significant amounts of phosphorus, and this is often unrecognized.
Sherman et al14 reviewed 200 of the most commonly prescribed drugs in dialysis patients and found that 23 (11.5%) of the drug labels listed phosphorus-containing ingredients, but the actual amount of phosphorus was not listed. The phosphorus content ranged from 1.4 mg (clonidine 0.2 mg, Blue Point Laboratories, Dublin, Ireland) to 111.5 mg (paroxetine 40 mg, GlaxoSmith Kline, Philadelphia, PA). The phosphorus content was inconsistent and varied with the dose of the agent, type of formulation (tablet or syrup), branded or generic formulation, and manufacturer.
Branded lisinopril (Merck, Kenilworth, NJ) had 21.4 mg of phosphorus per 10-mg dose, while a generic product (Blue Point Laboratories, Dublin, Ireland) had 32.6 mg. Different brands of generic amlodipine 10 mg varied in their phosphorus content from 8.6 mg (Lupin Pharmaceuticals, Mumbai, India) to 27.8 mg (Greenstone LLC, Peapack, NJ) to 40.1 mg (Qualitest Pharmaceuticals, Huntsville, AL. Rena-Vite (Cypress Pharmaceuticals, Madison, MS), a multivitamin marketed to patients with kidney disease, had 37.7 mg of phosphorus per tablet. Thus, just to bind the phosphorus content of these 3 tablets (lisinopril, amlodipine, and Rena-Vite), a patient could need at least 3 to 4 extra doses of phosphate binder.
The phosphate content of medications should be considered when prescribing. For example, Reno Caps (Nnodum Pharmaceuticals, Cincinnati, OH), another vitamin supplement, has only 1.7 mg of phosphorus per tablet and should be considered, especially in patients with poorly controlled serum phosphorus levels. However, the challenge is that medication labels do not provide the phosphorus content.
Reducing phosphorus absorption
Because so many foods contain phosphorus, dietary efforts alone are often insufficient to control serum phosphorus levels, and most patients require additional strategies, eg, phosphorus binders (Table 1).
Although these agents reduce serum phosphorus and help reduce symptoms, an important quality-of-life measure, it is uncertain whether they improve clinical outcomes.11 To date, no specific phosphorus binder offers a survival benefit over placebo.11
Based on the limited and conflicting evidence, the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines, recently updated, suggest that oral phosphorus binders should be used in patients with hyperphosphatemia to lower serum phosphorus levels toward the normal range.15 They further recommend not exceeding 1,500 mg of elemental calcium per day if a calcium-based binder is used, and they recommend avoiding calcium-based binders in patients with hypercalcemia, adynamic bone disease, or vascular calcification.
Phosphorus binders may account for up to 50% of the daily pill burden and may contribute to poor medication adherence.16 Dialysis patients need to take a lot of these drugs: by weight, 5 to 6 pounds per year.
These drugs can bind and interfere with the absorption of other vital medications and so should be taken with meals and separately from other medications.
Figure 1. A stepwise approach to the management of hyperphosphatemia and selection of phosphorus binder.
At present, there is insufficient evidence to recommend one binder over the other, and the selection of phosphorus binder should be individualized for each patient, taking into consideration the stage of chronic kidney disease, degree of hyperphosphatemia, concomitant anemia, presence of vascular calcification, use of other medications, side effects, cost to the individual, and pill burden. A stepwise, opinion-based, clinical approach to the selection of the phosphorus binders in patients with hyperphosphatemia is presented in Figure 1.
Removing phosphorus
Removal of phosphorus by adequate dialysis or kidney transplant is the final strategy.
New agents under study
To improve phosphorus control, other agents that inhibit absorption of phosphate are being investigated.
Nicotinamide reduces expression of the sodium-phosphorus cotransporter NTP2b. Its use in combination with a low-phosphorus diet and phosphorus binders may maximize reductions in phosphorus absorption and is being studied in the CKD Optimal Management With Binders and Nicotinamide (COMBINE) study.
Tenapanor, an inhibitor of the sodium-hydrogen transporter NHE3, has been shown in animal studies to increase fecal phosphate excretion and decrease urinary phosphate excretion17 but requires further evaluation.
Phosphorus is essential for life. However, both low and high levels of phosphorus in the body have consequences, and its concentration in the blood is tightly regulated through dietary absorption, bone flux, and renal excretion and is influenced by calcitriol (1,25 hydroxyvitamin D3), parathyroid hormone, and fibroblast growth factor 23 (FGF23).
Sekar et al,1 in this issue of the Journal, provide an extensive review of the pathophysiology of phosphorus metabolism and strategies to control phosphorus levels in patients with hyperphosphatemia and end-stage kidney disease.
PHOSPHORUS OR PHOSPHATE?
What's in a name? That which we call a rose By any other word would smell as sweet. —Shakespeare, Romeo and Juliet
The terms phosphate and phosphorus are often used interchangeably, though most writers still prefer phosphate over phosphorus.
The serum concentrations of phosphate and phosphorus are the same when expressed in millimoles per liter, as every mole of phosphate contains 1 mole of phosphorus, but not the same when expressed in milligrams per deciliter.2 The molecular weight of phosphorus is 30.97, whereas the molecular weight of the phosphate ion (PO43–) is 94.97—more than 3 times higher. Therefore, using these terms interchangeably in this context can lead to numerical error.3
Phosphorus, being highly reactive, does not exist by itself in nature and is typically present as phosphates in biologic systems. When describing phosphorus metabolism, the term phosphates should ideally be used because phosphates are the actual participants in the bodily processes. But in the clinical laboratory, all methods that measure serum phosphorus in fact measure inorganic phosphate and are expressed in terms of milligrams of phosphorus per deciliter rather than milligrams of phosphate per deciliter, and using these 2 terms interchangeably in clinical practice should not be of concern.4
THE PROBLEM
US adults typically ingest 1,200 mg of phosphorus each day, and about 60% to 70% of the ingested phosphorus is absorbed both by passive paracellular diffusion via tight junctions and by active transcellular transport via sodium-phosphate cotransport. The kidneys must excrete the same amount daily to maintain a steady state. As kidney function declines, phosphorus accumulates in the blood, leading to hyperphosphatemia.
Hyperphosphatemia is often asymptomatic, but it can cause generalized itching, red eyes, and adverse effects on the bone and parathyroid glands. Higher serum phosphorus levels have been shown to be associated with vascular calcification,5 cardiovascular events, and higher all-cause mortality rates in the general population,6 in patients with diabetes,7 and in those with chronic kidney disease.8 This association between higher serum phosphorus levels and the all-cause mortality rate led to the assumption that lowering serum phosphorus levels in these patients could reduce the rates of cardiovascular events and death, and to efforts to correct hyperphosphatemia.
Research into FGF23 continues, especially its role in cardiovascular complications of chronic kidney disease, as both phosphorus and FGF23 levels are elevated in chronic kidney disease and are implicated in poor clinical outcomes in these patients. However, both FGF23 and parathyroid hormone levels rise early in the course of kidney disease, long before overt hyperphosphatemia develops. Further, FGF23 rises earlier than parathyroid hormone and has been found to be an independent risk factor for cardiovascular events and death from any cause in end-stage kidney disease.9
Whether hyperphosphatemia is the culprit or merely an epiphenomenon of metabolic complications of chronic kidney disease is still unclear, as more molecules are being identified in the complex process of cardiovascular calcification.10
However, one thing is clear: vascular calcification is not just a simple precipitation of calcium and phosphorus. Instead, it is an active process that involves many regulators of mineral metabolism.10 The complex nature of this process is likely one of the reasons that evidence is conflicting11 about the benefits of phosphorus binders in terms of cardiovascular events or all-cause mortality in these patients.
STRATEGIES TO CONTROL HYPERPHOSPHATEMIA
Reducing intake
Dietary phosphorus restriction is the first step in controlling serum phosphorus. But reducing phosphorus intake while otherwise trying to optimize the nutritional status can be challenging.
The recommended daily protein intake is 1.0 to 1.2 g/kg. But phosphorus is typically found in foods rich in proteins, and restricting protein severely can compromise nutritional status and may be as bad as elevated phosphate levels in terms of outcomes.
Although plant-based foods contain more phosphate per gram of protein (ie, they have a higher ratio of phosphorus to protein) than animal-based foods, the bioavailability of phosphorus from plant foods is lower. Phosphorus in plant-based foods is mainly in the form of phytate. Humans cannot hydrolyze phytate because we lack the phytase enzyme; hence, the phosphorus in plant-based foods is not well absorbed. Therefore, a vegetarian diet may be preferable and beneficial in patients with chronic kidney disease. A small study in humans showed that a vegetarian diet resulted in lower serum phosphorus and FGF23 levels, but the study was limited by its small sample size.12
Patients should be advised to avoid foods that have a high phosphate content, such as processed foods, fast foods, and cola beverages, which often have phosphate-based food additives.
Further, one should be cautious about using supplements with healthy-sounding names. A case in point is “vitamin water”: 12 oz of this fruit punch-flavored beverage contains 392 mg of phosphorus,13 and this alone would require 12 to 15 phosphate binder tablets to bind its phosphorus content.
In addition, many prescription drugs have significant amounts of phosphorus, and this is often unrecognized.
Sherman et al14 reviewed 200 of the most commonly prescribed drugs in dialysis patients and found that 23 (11.5%) of the drug labels listed phosphorus-containing ingredients, but the actual amount of phosphorus was not listed. The phosphorus content ranged from 1.4 mg (clonidine 0.2 mg, Blue Point Laboratories, Dublin, Ireland) to 111.5 mg (paroxetine 40 mg, GlaxoSmith Kline, Philadelphia, PA). The phosphorus content was inconsistent and varied with the dose of the agent, type of formulation (tablet or syrup), branded or generic formulation, and manufacturer.
Branded lisinopril (Merck, Kenilworth, NJ) had 21.4 mg of phosphorus per 10-mg dose, while a generic product (Blue Point Laboratories, Dublin, Ireland) had 32.6 mg. Different brands of generic amlodipine 10 mg varied in their phosphorus content from 8.6 mg (Lupin Pharmaceuticals, Mumbai, India) to 27.8 mg (Greenstone LLC, Peapack, NJ) to 40.1 mg (Qualitest Pharmaceuticals, Huntsville, AL. Rena-Vite (Cypress Pharmaceuticals, Madison, MS), a multivitamin marketed to patients with kidney disease, had 37.7 mg of phosphorus per tablet. Thus, just to bind the phosphorus content of these 3 tablets (lisinopril, amlodipine, and Rena-Vite), a patient could need at least 3 to 4 extra doses of phosphate binder.
The phosphate content of medications should be considered when prescribing. For example, Reno Caps (Nnodum Pharmaceuticals, Cincinnati, OH), another vitamin supplement, has only 1.7 mg of phosphorus per tablet and should be considered, especially in patients with poorly controlled serum phosphorus levels. However, the challenge is that medication labels do not provide the phosphorus content.
Reducing phosphorus absorption
Because so many foods contain phosphorus, dietary efforts alone are often insufficient to control serum phosphorus levels, and most patients require additional strategies, eg, phosphorus binders (Table 1).
Although these agents reduce serum phosphorus and help reduce symptoms, an important quality-of-life measure, it is uncertain whether they improve clinical outcomes.11 To date, no specific phosphorus binder offers a survival benefit over placebo.11
Based on the limited and conflicting evidence, the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines, recently updated, suggest that oral phosphorus binders should be used in patients with hyperphosphatemia to lower serum phosphorus levels toward the normal range.15 They further recommend not exceeding 1,500 mg of elemental calcium per day if a calcium-based binder is used, and they recommend avoiding calcium-based binders in patients with hypercalcemia, adynamic bone disease, or vascular calcification.
Phosphorus binders may account for up to 50% of the daily pill burden and may contribute to poor medication adherence.16 Dialysis patients need to take a lot of these drugs: by weight, 5 to 6 pounds per year.
These drugs can bind and interfere with the absorption of other vital medications and so should be taken with meals and separately from other medications.
Figure 1. A stepwise approach to the management of hyperphosphatemia and selection of phosphorus binder.
At present, there is insufficient evidence to recommend one binder over the other, and the selection of phosphorus binder should be individualized for each patient, taking into consideration the stage of chronic kidney disease, degree of hyperphosphatemia, concomitant anemia, presence of vascular calcification, use of other medications, side effects, cost to the individual, and pill burden. A stepwise, opinion-based, clinical approach to the selection of the phosphorus binders in patients with hyperphosphatemia is presented in Figure 1.
Removing phosphorus
Removal of phosphorus by adequate dialysis or kidney transplant is the final strategy.
New agents under study
To improve phosphorus control, other agents that inhibit absorption of phosphate are being investigated.
Nicotinamide reduces expression of the sodium-phosphorus cotransporter NTP2b. Its use in combination with a low-phosphorus diet and phosphorus binders may maximize reductions in phosphorus absorption and is being studied in the CKD Optimal Management With Binders and Nicotinamide (COMBINE) study.
Tenapanor, an inhibitor of the sodium-hydrogen transporter NHE3, has been shown in animal studies to increase fecal phosphate excretion and decrease urinary phosphate excretion17 but requires further evaluation.
References
Sekar A, Kaur T, Nally JV Jr, Rincon-Choles H, Jolly S, Nakhoul G. Phosphorus binders: the new and the old, and how to choose. Cleve Clin J Med 2018; 85(8):629–638. doi:10.3949/ccjm.85a.17054
Young DS. "Phosphorus" or "phosphate." Ann Intern Med 1980; 93(4):631. pmid:7436198
Bartter FC. Reporting of phosphate and phosphorus plasma values. Am J Med 1981; 71(5):848. pmid:7304659.
Iheagwara OS, Ing TS, Kjellstrand CM, Lew SQ. Phosphorus, phosphorous, and phosphate. Hemodial Int 2013; 17(4):479–482. doi:10.1111/hdi.12010
Adeney KL, Siscovick DS, Ix JH, et al. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J Am Soc Nephrol 2009; 20(2):381–387. doi:10.1681/ASN.2008040349
Dhingra R, Sullivan LM, Fox CS, et al. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med 2007; 167(9):879–885. doi:10.1001/archinte.167.9.879
Chonchol M, Dale R, Schrier RW, Estacio R. Serum phosphorus and cardiovascular mortality in type 2 diabetes. Am J Med 2009; 122(4):380–386. doi:10.1016/j.amjmed.2008.09.039
Covic A, Kothawala P, Bernal M, Robbins S, Chalian A, Goldsmith D. Systematic review of the evidence underlying the association between mineral metabolism disturbances and risk of all-cause mortality, cardiovascular mortality and cardiovascular events in chronic kidney disease. Nephrol Dial Transplant 2009; 24(5):1506–1523. doi:10.1093/ndt/gfn613
Gutiérrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008; 359(6):584–592. doi:10.1056/NEJMoa0706130
Palmer SC, Gardner S, Tonelli M, et al. Phosphate-binding agents in adults with CKD: a network meta-analysis of randomized trials. Am J Kidney Dis 2016; 68(5):691–702. doi:10.1053/j.ajkd.2016.05.015
Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
Moser M, White K, Henry B, et al. Phosphorus content of popular beverages. Am J Kidney Dis 2015; 65(6):969–971. doi:10.1053/j.ajkd.2015.02.330
Sherman RA, Ravella S, Kapoian T. A dearth of data: the problem of phosphorus in prescription medications. Kidney Int 2015; 87(6):1097–1099. doi:10.1038/ki.2015.67
KDIGO 2017 clinical practice guideline update for diagnosis, evaluation, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Supplements 2017; 7(1 suppl): 1–59. www.kisupplements.org/article/S2157-1716(17)30001-1/pdf. Accessed June 5, 2018.
Fissell RB, Karaboyas A, Bieber BA, et al. Phosphate binder pill burden, patient-reported non-adherence, and mineral bone disorder markers: findings from the DOPPS. Hemodial Int 2016; 20(1):38–49. doi:10.1111/hdi.12315
Labonté ED, Carreras CW, Leadbetter MR, et al. Gastrointestinal inhibition of sodium-hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol 2015; 26(5):1138–1149. doi:10.1681/ASN.2014030317
References
Sekar A, Kaur T, Nally JV Jr, Rincon-Choles H, Jolly S, Nakhoul G. Phosphorus binders: the new and the old, and how to choose. Cleve Clin J Med 2018; 85(8):629–638. doi:10.3949/ccjm.85a.17054
Young DS. "Phosphorus" or "phosphate." Ann Intern Med 1980; 93(4):631. pmid:7436198
Bartter FC. Reporting of phosphate and phosphorus plasma values. Am J Med 1981; 71(5):848. pmid:7304659.
Iheagwara OS, Ing TS, Kjellstrand CM, Lew SQ. Phosphorus, phosphorous, and phosphate. Hemodial Int 2013; 17(4):479–482. doi:10.1111/hdi.12010
Adeney KL, Siscovick DS, Ix JH, et al. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J Am Soc Nephrol 2009; 20(2):381–387. doi:10.1681/ASN.2008040349
Dhingra R, Sullivan LM, Fox CS, et al. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med 2007; 167(9):879–885. doi:10.1001/archinte.167.9.879
Chonchol M, Dale R, Schrier RW, Estacio R. Serum phosphorus and cardiovascular mortality in type 2 diabetes. Am J Med 2009; 122(4):380–386. doi:10.1016/j.amjmed.2008.09.039
Covic A, Kothawala P, Bernal M, Robbins S, Chalian A, Goldsmith D. Systematic review of the evidence underlying the association between mineral metabolism disturbances and risk of all-cause mortality, cardiovascular mortality and cardiovascular events in chronic kidney disease. Nephrol Dial Transplant 2009; 24(5):1506–1523. doi:10.1093/ndt/gfn613
Gutiérrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008; 359(6):584–592. doi:10.1056/NEJMoa0706130
Palmer SC, Gardner S, Tonelli M, et al. Phosphate-binding agents in adults with CKD: a network meta-analysis of randomized trials. Am J Kidney Dis 2016; 68(5):691–702. doi:10.1053/j.ajkd.2016.05.015
Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
Moser M, White K, Henry B, et al. Phosphorus content of popular beverages. Am J Kidney Dis 2015; 65(6):969–971. doi:10.1053/j.ajkd.2015.02.330
Sherman RA, Ravella S, Kapoian T. A dearth of data: the problem of phosphorus in prescription medications. Kidney Int 2015; 87(6):1097–1099. doi:10.1038/ki.2015.67
KDIGO 2017 clinical practice guideline update for diagnosis, evaluation, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Supplements 2017; 7(1 suppl): 1–59. www.kisupplements.org/article/S2157-1716(17)30001-1/pdf. Accessed June 5, 2018.
Fissell RB, Karaboyas A, Bieber BA, et al. Phosphate binder pill burden, patient-reported non-adherence, and mineral bone disorder markers: findings from the DOPPS. Hemodial Int 2016; 20(1):38–49. doi:10.1111/hdi.12315
Labonté ED, Carreras CW, Leadbetter MR, et al. Gastrointestinal inhibition of sodium-hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol 2015; 26(5):1138–1149. doi:10.1681/ASN.2014030317




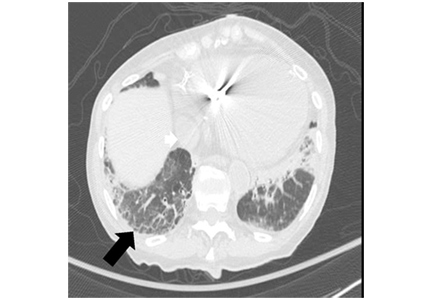
 over 5 years in normal postmenopausal women")

 from baseline to 2 years")